Attached files
UNITED
STATES
SECURITIES
AND EXCHANGE COMMISSION
WASHINGTON,
D.C. 20549
———————
FORM
10-K
———————
☑
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
For
the Fiscal Year Ended December 31, 2017
OR
☐
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
For the
transition period from _____
to _____
Commission File
Number: 001-36498
———————
CELLULAR
BIOMEDICINE GROUP, INC.
(Exact name of registrant as specified in its charter)
———————
|
Delaware
|
|
86-1032927
|
|
State of Incorporation
|
|
IRS Employer Identification No.
|
19925
Stevens Creek Blvd., Suite 100
Cupertino,
California 95014
(Address of principal executive offices)
(408)
973-7884
(Registrant's telephone number)
Securities
registered pursuant to Section 12(b) of the Exchange
Act:
Common
Stock, par value $.001 per share
Securities
registered pursuant to Section 12(g) of the Exchange
Act:
None
Indicate by check
mark if the registrant is a well-known seasoned issuer, as defined
in Rule 405 of the Securities Act. ☐ Yes ☑
No
Indicate by check
mark if the registrant is not required to file reports pursuant to
Section 13 or Section 15(d) of the Act. ☐ Yes ☑
No
Indicate by check
mark whether the registrant (1) has filed all reports required to
be filed by Section 13 or 15(d) of the Securities Exchange Act of
1934 during the preceding 12 months (or for such shorter period
that the registrant was required to file such reports), and (2) has
been subject to such filing requirements for the past 90 days. Yes
☑ No ☐
Indicate by check
mark whether the registrant has submitted electronically and posted
on its corporate Web site, if any, every Interactive Data File
required to be submitted and posted pursuant to Rule 405 of
Regulation S-T during the preceding 12 months (or for such shorter
period that the registrant was required to submit and post such
files). Yes ☑ No ☐
Indicate by check
mark if disclosure of delinquent filers pursuant to Item 405 of
Regulation S-K is not contained herein, and will not be contained,
to the best of registrant's knowledge, in definitive proxy or
information statements incorporated by reference in Part III of
this Form 10-K or any amendment to this Form 10-K.
☐
Indicate
by check mark whether the registrant is a large accelerated filer,
an accelerated filer, a non-accelerated filer, smaller reporting
company, or an emerging growth company. See the definitions of
“large accelerated filer,” “accelerated
filer”, “smaller reporting company” and "emerging
growth company" in Rule 12b-2 of the Exchange Act. (Check
one):
|
Large accelerated filer
|
☐
|
Accelerated filer
|
☒
|
|
Non-accelerated
filer
|
☐ (Do not check if a smaller reporting
company)
|
Smaller reporting company
|
☐
|
|
|
|
Emerging
growth company
|
☐
|
If an
emerging growth company, indicate by check mark if the registrant
has elected not to use the extended transition period for complying
with any new or revised financial accounting standards provided
pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check
mark whether the registrant is a shell company (as defined in Rule
12b-2 of the Act). ☐ Yes ☑ No
State
the aggregate market value of the voting and non-voting common
equity held by non-affiliates computed by reference to the price at
which the common equity was last sold, or the average bid and asked
price of such common equity, as of the last business day of the
registrant's most recently completed second fiscal quarter –
$92,318,196 as of June 30, 2017.
Indicate the number
of shares outstanding of each of the registrant’s classes of
common stock, as of the latest practicable date: As of February 11,
2018, there were 17,430,762 shares and 17,003,968 shares of common
stock, par value $.001 per share issued and outstanding,
respectively.
Documents
Incorporated By Reference
THE
INFORMATION REQUIRED BY PART III OF THIS ANNUAL REPORT ON FORM
10-K, TO THE EXTENT NOT SET FORTH HEREIN, IS INCORPORATED BY
REFERENCE FROM THE REGISTRANT'S DEFINITIVE PROXY STATEMENT RELATING
TO THE ANNUAL MEETING OF STOCKHOLDERS, WHICH DEFINITIVE PROXY
STATEMENT SHALL BE FILED WITH THE SECURITIES AND EXCHANGE
COMMISSION WITHIN 120 DAYS AFTER THE END OF THE FISCAL YEAR TO
WHICH THIS ANNUAL REPORT ON FORM 10-K RELATES.
CELLULAR
BIOMEDICINE GROUP, INC.
ANNUAL
REPORT ON FORM 10-K
FOR
THE FISCAL YEAR ENDED DECEMBER 31, 2017
TABLE OF CONTENTS
|
|
|
|
Page
|
|
|
|
|
|
|
4
|
|
|
|
|
|
4
|
|
||
|
|
|
24
|
|
||
|
|
|
54
|
|
||
|
|
|
54
|
|
||
|
|
|
54
|
|
||
|
|
|
|
|
|
|
|
|
|
|
54
|
|
|
|
|
|
54
|
|
||
|
|
|
59
|
|
||
|
|
|
61
|
|
||
|
|
|
76
|
|
||
|
|
|
77
|
|
||
|
|
|
77
|
|
||
|
|
|
77
|
|
||
|
|
|
78
|
|
||
|
|
|
|
|
|
|
|
|
|
|
79
|
|
|
|
|
|
79
|
|
||
|
|
|
79
|
|
||
|
|
|
79
|
|
||
|
|
|
79
|
|
||
|
|
|
79
|
|
||
|
|
|
|
|
|
|
|
|
|
|
80
|
|
|
|
|
|
80
|
|
||
|
|
|
83
|
|
||
|
|
|
|
|
|
|
|
|
|
84
|
|
||
Cautionary Note Regarding Forward-looking Statements and Risk
Factors
This Annual Report on Form 10-K, or this Annual Report, may contain
“forward-looking statements” within the meaning of
Section 27A of the Securities Act of 1933, as amended, and Section
21E of the Securities Exchange Act of 1934, as amended, or the
Exchange Act and the Private Securities Litigation Reform Act of
1995, which are subject to the “safe harbor” created by
those sections. Our actual results could differ materially from
those anticipated in these forward-looking statements. This annual
report on Form 10-K of the Company may contain forward-looking
statements which reflect the Company's current views with respect
to future events and financial performance. The words "believe,"
"expect," "anticipate," "intends," "estimate," "forecast,"
"project," and similar expressions identify forward-looking
statements. All statements other than statements of historical fact
are statements that could be deemed to be forward-looking
statements, including plans, strategies and objectives of
management for future operations; proposed new products, services,
developments or industry rankings; future economic conditions or
performance; belief; and assumptions underlying any of the
foregoing. Although we believe that we have a reasonable basis for
each forward-looking statement contained in this report, we caution
you that these statements are based on a combination of facts and
factors currently known by us and our projections of the future,
about which we cannot be certain. Such "forward-looking statements"
are subject to risks and uncertainties set forth from time to time
in the Company's SEC reports and include, among others, the Risk
Factors set forth under Item 1A below.
The risks included herein are not exhaustive. This annual report on
Form 10-K filed with the SEC include additional factors which could
impact the Company's business and financial performance. Moreover,
the Company operates in a rapidly changing and competitive
environment. New risk factors emerge from time to time and it is
not possible for management to predict all such risk factors.
Further, it is not possible to assess the impact of all risk
factors on the Company's business or the extent to which any
factor, or combination of factors, may cause actual results to
differ materially from those contained in any forward-looking
statements. Forward-looking statements in this report include, but
are not limited to, statements about:
●
the success, cost
and timing of our product development activities and clinical
trials;
●
our ability and the
potential to successfully advance our technology platform to
improve the safety and effectiveness of our existing product
candidates;
●
the potential for
our identified research priorities to advance our cancer and
regenerative disease technologies;
●
our ability to
obtain drug designation or breakthrough status for our product
candidates and any other product candidates, or to obtain and
maintain regulatory approval of our product candidates, and any
related restrictions, limitations and/or warnings in the label of
an approved product candidate;
●
the ability to
generate or license additional intellectual property relating to
our product candidates;
●
regulatory
developments in China, United States and other foreign
countries;
●
the potential of
the technologies we have acquired, such as the acquisitions of the
technologies from AG, Blackbird, and the PLAGH (each as defined
below);
●
fluctuations in the
exchange rate between the U.S. dollars and the Chinese
Yuan;
●
the changes
associated with our move to the new Zhangjiang building in
Shanghai;
●
our plans to
continue to develop our manufacturing facilities.
Readers are cautioned not to place undue reliance on such
forward-looking statements as they speak only of the Company's
views as of the date the statement was made. The Company undertakes
no obligation to publicly update or revise any forward-looking
statements, whether as a result of new information, future events
or otherwise.
PART I
ITEM 1. BUSINESS.
As used
in this annual report, "we", "us", "our", "CBMG", "Company" or "our
company" refers to Cellular Biomedicine Group, Inc. and, unless the
context otherwise requires, all of its subsidiaries or deemed
controlled companies.
Overview
Cellular
Biomedicine Group, Inc. is a clinical stage biopharmaceutical
company, principally engaged in the development of therapies for
cancer and degenerative diseases utilizing proprietary cell-based
technologies. Our technology includes two major platforms: (i)
Immune cell therapy for treatment of a broad range of cancer
indications comprised of technologies in Chimeric Antigen Receptor
modified T cells (CAR-T), T-Cell Receptor (TCR), cancer vaccine,
and ex vivo expanded autologous Central Memory T Cells (Tcm), and
(ii) human adipose-derived mesenchymal progenitor cells (haMPC) for
treatment of joint and autoimmune diseases. CBMG’s Research
& Development facilities are based in China and the U.S., and
its manufacturing facilities are based in China in the cities of
Shanghai, Wuxi, and Beijing.
We are
focused on developing and marketing safe and effective cell-based
therapies based on our cellular platforms, to treat cancer,
orthopedic diseases and metabolic diseases. We have developed
proprietary technologies and know-hows in our cell therapy
platforms. Our primary target
market is Greater China. We believe that our cell-based therapies
will be able to help patients with high unmet medical needs. We
expect to carry out clinical studies leading to the eventual CFDA
approval of our products through Biologics License Application
(BLA) filings and authorized clinical centers throughout Greater
China.
We are
conducting clinical studies in China with our stem cell based
therapies to treat knee osteoarthritis (“KOA”). We have
completed a Phase IIb autologous haMPC KOA clinical study and
published its promising results. Led by Shanghai Renji Hospital,
one of the largest teaching hospitals in China, we have also
completed a Phase I clinical trial of our off-the-shelf allogeneic
haMPC (AlloJoinTM) therapy for
treating KOA patients. In addition, we have received an award of
$2.29 million grant from California Institute of Regenerative
Medicine’s (CIRM) and we have started manufacturing
AlloJoinTM
product in California to support preclinical and clinical studies
in the United States for the KOA indication.
We have
launched multiple clinical trials using our CAR-T products in
multiple indications such as Diffuse Large B-cell Lymphoma (DLBCL)
and Acute Lymphoblastic leukemia (ALL). We may also establish
partnerships with other companies for co-development in CAR-T,
TCR-T and stem cell based therapies. We are striving to build a
highly competitive research and development function, a
translational medicine unit, along with a well-established cellular
manufacturing capability and ample capacity, to support the
development of multiple assets in multiple indications. These
efforts will allow us to boost the Company's Immuno-Oncology
presence and pave the way for additional future
partnerships.
Corporate History
Cellular
Biomedicine Group, Inc. was incorporated in the State of Delaware
and its corporate headquarters is located at 19925 Stevens Creek
Blvd., Suite 100 in Cupertino, California. The Company is focusing
its resources on becoming a biotechnology company bringing
therapies to improve the health of patients in China.
Cellular
Biomedicine Group, Inc., a Delaware corporation (formerly known as
EastBridge Investment Group Corporation), was originally
incorporated in the State of Arizona on June 25, 2001. The
Company's principal activity through June 30, 2005 was to
manufacture mobile entertainment products.
In
2005, the Company decided to exit the mobile entertainment market
and dedicate its activities to providing investment related
services in Asia, with a strong focus on high GDP growth countries,
such as China. The Company concentrated its efforts in the Far East
(Hong Kong, mainland China, Australia) and in the United States and
sought to provide consulting services necessary for small to
medium-size companies to obtain capital to grow their business,
either to become public companies in the United States or to find
joint venture partners or raise capital to expand their
businesses.
On
February 6, 2013, the Company completed a merger to acquire
Cellular Biomedicine Group Ltd. In connection with the merger,
effective on March 5, 2013, the Company (formerly named
“EastBridge Investment Group Corporation”) changed its
name to “Cellular Biomedicine Group, Inc.” In addition
in March 2013 we changed our corporate headquarters from Arizona to
California.
From February 6,
2013 to June 23, 2014, we operated the Company in two separate
reportable segments: (i) Biomedicine Cell Therapy
(“Biomedicine”); and (ii) Financial Consulting
(“Consulting”). The Consulting segment was
conducted through EastBridge Sub. On June 23, 2014, the
Company announced the discontinuation of the Consulting segment as
it no longer fit into management’s long-term strategy and
vision. The Company is continuing to focus its resources
on becoming a biotechnology company bringing therapies to improve
the health of patients in China.
On
September 26, 2014, the Company completed its acquisition of
Beijing Agreen Biotechnology Co. Ltd. ("AG") and the U.S. patent
held by AG’s founder. AG is a biotech company with operations
in China, engaged in the development of treatments for cancerous
diseases utilizing proprietary cell technologies, which include
without limitation, preparation of subset T Cell and clonality
assay platform technology for treatment of a broad range of
cancers.
Recent Developments
In
January 2016, we launched a Phase I clinical trial of an
off-the-shelf allogeneic haMPC AlloJoin™ therapy for KOA (the
“Allogenic KOA Phase I Trial”) to evaluate the safety
and efficacy of AlloJoin™, an off-the-shelf allogeneic
adipose derived progenitor cell (haMPC) therapy for the treatment
of KOA.
On
March 23, 2016, the Company filed a Form S-3 Registration Statement
(the “S-3 Registration Statement”) with the SEC, which
was declared effective on June 17, 2016. The S-3 Registration
Statement contains three prospectuses:
●
Offering Prospectus. A base prospectus which covers the
offering, issuance and sale by us of up to $150,000,000 of our
common stock, preferred stock, debt securities, warrants, rights
and/or units;
●
Resale Prospectus. A prospectus to be used for the resale by
the selling stockholders of up to 3,824,395 shares of the Common
Stock; and
●
Sales Agreement Prospectus. A sales agreement prospectus
covering the offering, issuance and sale by the registrant of up to
a maximum aggregate offering price of $50,000,000 of the Common
Stock that may be issued and sold under a sales agreement with
Cantor Fitzgerald & Co.
On
August 5, 2016 we completed patient treatment for the Allogenic KOA
Phase I Trial. And on December 9, 2016 we announced interim 3-month
safety data from the Allogenic KOA Phase I Trial in China.
Preliminary results from interim analysis have demonstrated a
safety and tolerability profile of AlloJoinTM in the three doses
tested, and no serious adverse events (SAE) have been observed. The
trial has been completed and we are analyzing the results on
cartilage regeneration as well as recent development in the
competitive landscape.
On
November 29, 2016 we announced the approval and commencement of
patient enrollment in China for our CARD-1 (“CAR-T Against
DLBCL”) Phase I clinical trial of CD19 CAR-T therapy
utilizing our optimized proprietary C-CAR011 construct for the
treatment of patients with refractory DLBCL. The CARD-1 trial has
begun enrollment with more data expected to be available in the
first half of 2018.
On
December 9, 2016 we announced interim 3-month safety data from our
Phase I clinical trial in China for
AlloJoin™ off-the-shelf allogeneic stem cell therapy for
KOA. The preliminary data was presented in December 2016 at the
World Stem Cell Summit in West Palm Beach, Florida. The interim
analysis of the trial has preliminarily demonstrated a safety and
tolerability profile of AlloJoin™ in the three doses tested,
and adverse events (AE) are similar to that of our prior autologous
trials. No serious adverse events (SAE) have been
observed.
On
January 3, 2017, we announced the signing of a ten-year lease of an
113,038-square foot building located in the “Pharma
Valley” in Shanghai Zhangjiang High-Tech Park. The new
facility designed and built to GMP standards will consist of 40,000
square feet dedicated to advanced cell manufacturing. We
plan to invest an aggregate of approximately $35 million into the
Zhangjiang facility, of which $10 million will be spent to bring
the facility into compliance with current GMP standards and around
25 million will be spent on lease of this real estate. By the end
of 2017, the combination of new Zhangjiang facility, an expanded
Wuxi, and Beijing facilities, all meeting international GMP
standards, of the Company had provided 70,000 square feet for cell
manufacturing. The Company expects that it will be capable of
supporting clinical trials for five different CAR-T and stem
cell products simultaneously, or the ability to produce products to
treat approximately 10,000 cancer patients and 10,000 KOA patients
per year. To reach this capacity, we plan to hire an additional 60
R&D and Manufacturing personnel by end of 2018.
On January 9,
2017, we announced the commencement of patient enrollment in China
for our CALL-1 (“CAR-T against Acute Lymphoblastic
Leukemia”) Phase I clinical trial of CD19 CAR-T therapy
utilizing its optimized proprietary C-CAR011 construct for the
treatment of patients with relapsed or refractory (r/r) CD19+
B-cell ALL. The CALL-1 trial began enrollment with more data
expected to be available in the first half of 2018. Depending on
the Phase I CALL-1 results, CBMG expects to initiate a larger Phase
II clinical trial as soon as possible.
On
February 27, 2017 we announced the Company received a $2.29 million
grant from California Institute of Regenerative Medicine (CIRM) to
fund our off-the-shelf AlloJoinTM Allogeneic Stem
Cell Therapy for KOA in the United States. We are performing
pre-clinical studies at Children’s Hospital Los Angeles
(CHLA) and plan to file AlloJoinTM Phase
I clinical trial in the United States. On May 4, 2017, the Company
received $1.2 million from the CIRM grant, the first of four
disbursement totaling $2.29 million to fund our off-the-shelf
AlloJoin™ Allogeneic Stem Cell Therapy for KOA in the United
States.
On
March 30, 2017 we announced the completion of our newly expanded
30,000-square foot facility in Huishan High Tech Park in Wuxi,
China. 20,000 square feet of the Wuxi facility will be dedicated to
advanced stem cell culturing, centralized plasmid and viral
vector production, cell banking and development of
reagents.
On April 10,
2017, we announced a strategic research collaboration to co-develop
certain high-quality industrial control processes in CAR-T and stem
cell manufacturing with GE Healthcare Life Science. In connection
with the collaboration, a joint laboratory within CBMG’s new
Shanghai Zhangjiang facility designed and built to GMP standards
will be established and dedicated to the joint research and
development of a functionally integrated and automated
immunotherapy cell manufacturing system.
On May
15, 2017, we announced the addition of a new independent Phase
I clinical trial of the Company’s ongoing CARD-1 study in
patients with chemorefractory or refractory B cell Non-Hodgkin
Lymphoma (NHL). The Company and Shanghai Tongji Hospital (Tongji)
are conducting a single arm, non-randomized study to evaluate the
safety and efficacy of C-CAR011 (Anti-CD19 single-chain variable
fragment (scFv) (41BB-CD3f)) therapy in relapsed or refractory B
cell NHL patients. The trial will enroll 15 patients comprised of
DLBCL, Primary Mediastinal Large B-Cell Lymphoma (PMBCL) and
Follicular Lymphoma (FL).
On June
1, 2017, we announced our Board of Directors approved a new stock
repurchase program granting the company authority to repurchase up
to $10 million in common shares (the “2017 Stock
Repurchase Program”) through open market purchases pursuant
to a plan adopted in accordance with Rule 10b5-1 of the Securities
Exchange Act of 1934, as amended (the “Exchange Act”)
and in accordance with Rule 10b-18 of the Exchange Act. The 2017
Stock Repurchase Program contemplates repurchase shares of the
Company’scommon stock in the open market in accordance with
all applicable securities laws and regulations. From June to
December the Company repurchased a total of 426,794 shares at a
total cost of $3,977,929, or an average of
$9.32/share.
On June
20, 2017, we announced the establishment of an External Advisory
Board and the appointment of Michael A. Caligiuri, MD, as Chair of
the External Advisory Board to bring together experts from diverse
disciplines to provide knowledge and insight to help CBMG fulfill
its mission and build a network for development
opportunities.
On June
26, 2017, we announced the appointment of Dr. Xia Meng as Chief
Operating Officer for the Company.
On
November 4, 2017, we announced the grand opening of our Zhangjiang
facility. On the same day, we announced the
signing of a strategic partnership with Thermo Fisher Scientific
(China) Ltd. to build a joint Cell Therapy Technology Innovation
and Application Center (Center) at CBMG’s newly opened
Shanghai Zhangjiang facility.
On
December 28, 2017, the Company announced the closing of two private
placement transactions pursuant to which we sold an aggregate of
1,208,333 shares of the Company’s common stock to select key
executives and private investors at $12.00 per share, for total
aggregate gross proceeds of approximately $14.5
Million.
On
January 30, 2018 and February 5, 2018, the Company entered into
securities purchase agreements with certain investors pursuant to
which the Company agreed to sell, and the investors agreed to
purchase from the Company, an aggregate of 1,719,324 shares (the
“February 2018 Private Placement”) of the
Company’s common stock, par value $0.001 per share, at $17.80
per share, for total gross proceeds of approximately $30.6
million. The transaction closed on February 5,
2018. Pursuant to the
purchase agreement, the Investors have the right to nominate one
director to the board of directors of the Company to stand for
election at the 2018 Annual Meeting of Stockholders. Effective as
of the closing of the February 2018 Private Placement, Bosun S. Hau
was appointed as a non-executive Class III director of the
Company.
On
February 6, 2018, driven primarily by the Company’s strategy
move to expand its business operations in early diagnosis and
cancer intervention, Meng Xia transitioned from the role of Chief
Operating Officer to Head of the Early Diagnosis & Intervention
for the Company.
On
February 15, 2018, the Company obtained a 36 months exclusive
option with Augusta University to to negotiate a royalty-bearing,
exclusive license to the patent rights owned by the Augusta
University relating to an invention to identify novel alpha
fetoprotein specific T-cell receptors (TCR) for a hepatocellular
carcinoma ("HCC") immunotherapy. The Company plan to evaluate the
feasibility and opportunities of this novel alpha fetoprotein TCR
to redirect T Cells for the HCC indication.
In the
next 12 months, we aim to accomplish the following, though there
can be no assurances that we will be able to accomplish any of
these goals:
●
Bolster
R&D resources to fortify our intellectual properties portfolio
and scientific development. Continue to develop a competitive
immune cell therapy pipeline for CBMG. Seek
opportunities to file new patents in China and potentially the rest
of the world;
●
Continue to identify and evaluate advanced technologies and
seek partnerships to bolster our competitive edge in the cell
therapy field in China;
●
Submit to the CFDA an IND package for C-CAR011 in treating
patients with CD19+ B-cell malignancies.
●
Confirm
the safety and efficacy profile of C-CAR011 in China in refractory
aggressive DLBCL and to initiate a larger Phase II clinical trial
whenever feasible.
●
Confirm
the safety and efficacy of C-CAR011 in relapsed and refractory
(r/r) CD19+ B-cell Acute Lymphoblastic Leukemia (ALL) in China, and
/ to prepare for a follow up multicenter phase IIb
trial.
●
Initiate
an investigator sponsored phase I trial of anti-BCMA CART in adults
with relapsed/refractory multiple myeloma;
●
Implement
our GE Joint Technology Laboratory to develop control processes for
the manufacturing of CAR-T and Stem Cell Therapies;
●
Implement
steps to advance our Thermo Fisher joint Cell Therapy Technology
Innovation and Application Center;
●
Initiate
clinical study to support the BLA for Autologous and Allogeneic KOA
study in China;
●
Complete
Chemistry, Manufacturing and Controls (CMC), non-clinical and
preclinical study data package to prepare for Allogeneic KOA IND
filing in the United States;
●
Initiate
clinical study to support the BLA for Allogeneic KOA study in the
United States;
●
Evaluate new regenerative medicine technology platform for
other indications and review recent development in the competitive
landscape;
●
Evaluate our corporate development strategy on maintaining
the CAR-T and regenerative medicine dual technology platform;
and
●
Evaluate the feasibility and opportunities of novel Alpha
Fetoprotein Specific T Cell Receptors (TCR) to redirect T Cells for
a HCC Immunotherapy; and
●
Develop the new cancer diagnostics and intervention
business; and
●
Improve
liquidity and fortify our balance sheet by courting institutional
investors.
For the
years ended December 31, 2017, 2016 and 2015, we generated $0.3
million, $0.6 million and $2.5 million in technology consulting
service revenue, respectively. We expect our biopharmaceutical
business to generate revenues primarily from immune therapy and the
development of therapies for the treatment of KOA in the next three
to four years.
Our
operating expenses for year ended December 31, 2017 were in line
with management’s plans and expectations. We have a decrease
in total operating expenses of approximately $1.1 million for the
year ended December 31, 2017, as compared to the year ended
December 31, 2016, which is primarily attributable to an impairment
of investment of $4.6 million in 2016.
Corporate Structure
Our
current corporate structure is illustrated in the following
diagram:
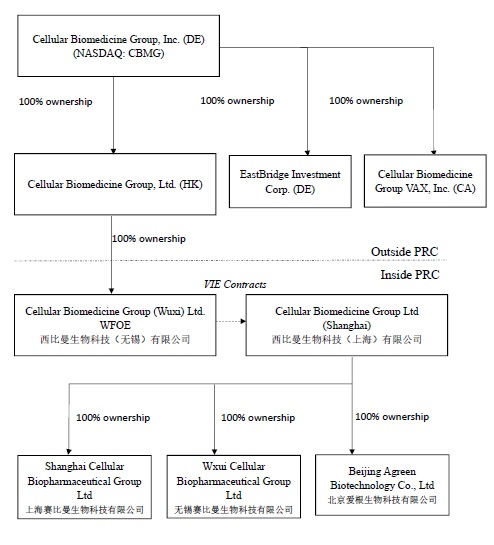
Currently we have
the following subsidiaries (including a controlled VIE
entity):
Eastbridge
Investment Corporation (“Eastbridge Sub”), a Delaware
corporation, is a wholly owned subsidiary of the
Company.
Cellular
Biomedicine Group VAX, Inc. (“CBMG VAX”), a California
corporation, is a wholly owned subsidiary of the
Company.
Cellular
Biomedicine Group HK Limited, a Hong Kong company limited by
shares, is a holding company and wholly owned subsidiary of the
Company.
Cellular
Biomedicine Group Ltd. (Wuxi), license number 320200400034410 (the
“WFOE”) is a wholly foreign-owned entity that is 100%
owned by Cellular Biomedicine Group HK Limited. This
entity’s legal name in Chinese translates to “Xi Biman
Biological Technology (Wuxi) Co. Ltd.” WFOE
controls and holds ownership rights in the business, assets and
operations of Cellular Biomedicine Group Ltd. (Shanghai)
(“CBMG Shanghai”) through variable interest entity
(VIE) agreements. We conduct certain biopharmaceutical
business activities through WFOE, including lab kit production and
research.
Cellular
Biomedicine Group Ltd. (Shanghai) license number 310104000501869
(“CBMG Shanghai”), is a PRC domestic corporation, which
we control and hold ownership rights in, through WFOE and the
above-mentioned VIE agreements. This entity’s
legal name in Chinese translates to “Xi Biman Biotech
(Shanghai) Co., Ltd.” We conduct certain
biopharmaceutical business activities through our controlled VIE
entity, CBMG Shanghai, including clinical trials and certain other
activities requiring a domestic license in the PRC. Mr.
Chen Mingzhe and Mr. Lu Junfeng together are the record holders of
all of the outstanding registered capital of CBMG Shanghai.
Mr. Chen and Mr. Lu are also directors of CBMG Shanghai
constituting the entire management of the same. Mr.
Chen and Mr. Lu receive no compensation for their roles as managers
of CBMG Shanghai.
Beijing
Agreen Biotechnology Co., Ltd. is a PRC domestic corporation and
wholly owned subsidiary of CBMG Shanghai.
Wuxi
Cellular Biopharmaceutical Group Ltd. was established on January
17, 2017 and it is a PRC domestic corporation and wholly owned
subsidiary of CBMG Shanghai.
Shanghai Cellular
Biopharmaceutical Group Ltd. was established on January 18, 2017
and it is a PRC domestic corporation and wholly owned subsidiary of
CBMG Shanghai.
Variable Interest Entity (VIE) Agreements
Through
our wholly foreign-owned entity and 100% subsidiary, Cellular
Biomedicine Group Ltd. (Wuxi), we control and have ownership rights
by means of a series of VIE agreements with CBMG Shanghai. The
shareholders of record for CBMG Shanghai were Cao Wei and Chen
Mingzhe, who together owned 100% of the equity interests in CBMG
Shanghai before October 26, 2016. On October 26, 2016, Cao Wei,
Chen Mingzhe and Lu Junfeng entered into an equity transfer
agreement and a supplementary agreement (“Equity Transfer
Agreement”), pursuant to which Cao Wei transferred his equity
interests in CBMG Shanghai to Chen Mingzhe and Lu Junfeng. As a
result of the transfer, each of Mr. Chen and Mr. Lu now owns a 50%
equity interest in CBMG Shanghai. On the same day, WFOE, CBMG
Shanghai, Cao Wei and Chen Mingzhe entered into a termination
agreement, pursuant to which, the series of VIE agreements executed
among the WFOE, CBMG Shanghai, Chen Mingzhe and Cao Wei were
terminated and a new set of VIE agreements were executed. The
following is a description of each of these VIE
agreements:
Exclusive Business Cooperation
Agreement. Through the WFOE, we are a party
to an exclusive business cooperation agreement dated October 26,
2016 with CBMG Shanghai, which provides that (i) the WFOE shall
exclusively provide CBMG Shanghai with complete technical support,
business support and related consulting services; (ii) without
prior written consent of the WFOE, CBMG Shanghai may not accept the
same or similar consultancy and/or services from any third party,
nor establish any similar cooperation relationship with any third
party regarding same matters during the term of the agreement;
(iii) CBMG Shanghai shall pay the WFOE service fees as calculated
based on the time of service rendered by the WFOE multiplying the
corresponding rate, plus an adjusted amount decided by the board of
the WFOE; and (iv) CBMG Shanghai grants to the WFOE an irrevocable
and exclusive option to purchase, at its sole discretion, any or
all of CBMG Shanghai’s assets at the lowest purchase price
permissible under PRC laws. The term of the agreement is
10 years, provided however the agreement may extended at the option
of the WFOE. Since this agreement permits the WFOE to determine the
service fee at its sole discretion, the agreement in
effect provides the WFOE with rights to all earnings of the
VIE.
Loan Agreement. Through the
WFOE, we are a party to a loan agreement with CBMG Shanghai, Lu
Junfeng and Chen Mingzhe dated October 26, 2016, in accordance with
which the WFOE agreed to provide an interest-free loan to CBMG
Shanghai. The term of the loan is 10 years, which may be
extended upon written consent of the parties. The method
of repayment of CBMG Shanghai shall be at the sole discretion of
the WFOE, including but not limited to an acquisition of CBMG
Shanghai in satisfaction of its loan obligations.
Exclusive Option Agreement with Lu
Junfeng. Through the WFOE, we are a party to an
option agreement with CBMG Shanghai and Lu Junfeng dated October
26, 2016, in accordance with which: (i) Lu Junfeng irrevocably
granted the WFOE an irrevocable and exclusive right to purchase, or
designate another person to purchase the entire equity interest in
CBMG Shanghai as then held by him, at an aggregate purchase price
to be determined; and (ii) any proceeds obtained by Lu Junfeng
through the above equity transfer in CBMG Shanghai shall be used
for the payment of the loan provided by the WFOE under the
aforementioned Loan Agreement.
Exclusive Option Agreement with Chen
Mingzhe. Through the WFOE, we are a party to an
exclusive option agreement with CBMG Shanghai and Chen Mingzhe
dated October 26, 2016, under which: (i) Chen Mingzhe irrevocably
granted the WFOE an irrevocable and exclusive right to purchase, or
designate another person to purchase the entire equity interest in
CBMG Shanghai for an aggregate purchase price to be determined; and
(ii) any proceeds obtained by Chen Mingzhe through the above equity
transfer in CBMG Shanghai shall be used for the payment of the loan
provided by the WFOE under the aforementioned Loan
Agreement.
Power of Attorney from Lu
Junfeng. Through the WFOE we are the recipient of
a power of attorney executed by Lu Junfeng on October 26, 2016, in
accordance with which Lu Junfeng authorized the WFOE to act on his
behalf as his exclusive agent with respect to all matters
concerning his equity interest in CBMG Shanghai, including without
limitation to attending the shareholder meetings of CBMG Shanghai,
exercising voting rights and designating and appointing senior
executives of CBMG Shanghai.
Power of Attorney from Chen
Mingzhe. Through the WFOE we are the recipient of
a power of attorney executed by Chen Mingzhe on October 26, 2016,
in accordance with which Chen Mingzhe authorized the WFOE to act on
his behalf as his exclusive agent with respect to all matters
concerning his equity interest in CBMG Shanghai, including without
limitation to attending the shareholders meetings of CBMG Shanghai,
exercising voting rights and designating and appointing senior
executives of CBMG Shanghai.
Equity Interest Pledge Agreement with Lu
Junfeng. Through the WFOE, we are a party to an
equity interest pledge agreement with CBMG Shanghai and Lu Junfeng
dated October 26, 2016, in accordance with which: (i) Lu Junfeng
pledged to the WFOE the entire equity interest he holds in CBMG
Shanghai as security for payment of the consulting and service fees
by CBMG Shanghai under the Exclusive Business Cooperation
Agreement; (ii) Lu Junfeng and CBMG Shanghai submitted all
necessary documents to ensure the registration of the Pledge of the
Equity Interest with the State Administration for Industry and
Commerce (“SAIC”), and the pledge became effective
on November 22, 2016; (iii) on the occurrence of any event of
default, unless it has been successfully resolved within 20 days
after the delivery of a rectification notice by the WFOE, the WFOE
may exercise its pledge rights at any time by a written notice to
Lu Junfeng.
Equity Interest Pledge Agreement with Chen
Mingzhe. Through the WFOE we are a party to
an equity interest pledge agreement with CBMG Shanghai and Chen
Mingzhe dated October 26, 2016, in accordance with which: (i) Chen
Mingzhe pledged to the WFOE the entire equity interest he holds in
CBMG Shanghai as security for payment of the consulting and service
fees by CBMG Shanghai under the Exclusive Business Cooperation
Agreement; (ii) Chen Mingzhe and CBMG Shanghai submitted all
necessary documents to ensure the registration of the Pledge of the
Equity Interest with SAIC, and the pledge became effective
on November 22, 2016; (iii) on the occurrence of any event of
default, unless it has been successfully resolved within 20 days
after the delivery of a rectification notice by the WFOE, the WFOE
may exercise its pledge rights at any time by a written notice to
Chen Mingzhe.
Our
relationship with our controlled VIE entity, CBMG Shanghai,
through the VIE agreements, is subject to various operational and
legal risks. Management believes that Mr. Chen and Mr.
Lu, as record holders of the VIE’s registered capital, have
no interest in acting contrary to the VIE
agreements. However, if Mr. Chen and Lu as shareholders
of the VIE entity were to reduce or eliminate their ownership of
the registered capital of the VIE entity, their interests may
diverge from that of CBMG and they may seek to act in a manner
contrary to the VIE agreements (for example by controlling the VIE
entity in such a way that is inconsistent with the directives of
CBMG management and the board; or causing non-payment by the VIE
entity of services fees). If such circumstances were to
occur the WFOE would have to assert control rights through the
powers of attorney and other VIE agreements, which would require
legal action through the PRC judicial system. While we
believe the VIE agreements are legally enforceable in the PRC,
there is a risk that enforcement of these agreements may involve
more extensive procedures and costs to enforce, in comparison to
direct equity ownership of the VIE entity. We believe
based on the advice of local counsel that the VIE agreements are
valid and in compliance with PRC laws presently in
effect. Notwithstanding the foregoing, if the applicable
PRC laws were to change or are interpreted by authorities in the
future in a manner which challenges or renders the VIE agreements
ineffective, the WFOE’s ability to control and obtain all
benefits (economic or otherwise) of ownership of the VIE entity
could be impaired or eliminated. In the event of
such future changes or new interpretations of PRC law, in an effort
to substantially preserve our rights we may have to either amend
our VIE agreements or enter into alternative arrangements which
comply with PRC laws as interpreted and then in
effect.
For
further discussion of risks associated with the above, please see
the section below titled “Risks Related to Our
Structure.”
BIOPHARMACEUTICAL BUSINESS
Our
biopharmaceutical business was founded in 2009 as a newly formed
specialty biomedicine company by a team of seasoned
Chinese-American executives, scientists and doctors. In 2010, we
established a facility designed and built to GMP standards in Wuxi,
and in 2012 we established a U.S. Food and Drug Administration
(“FDA”) GMP standard protocol-compliant manufacturing
facility in Shanghai. In October 2015, we opened a facility
designed and built to GMP standards in Beijing. In November 2017,
we announced the grand opening of our Zhangjiang facility in
Shanghai, of which 40,000 square feet was designed and built to GMP
standards and dedicated to advanced cell manufacturing. Our focus
has been to serve the rapidly growing health care market in China
by marketing and commercializing stem cell and immune cell
therapeutics, related tools and products from our patent-protected
homegrown and acquired cell technology, as well as by utilizing
exclusively in-licensed and other acquired intellectual
properties.
Our
current treatment focal points are cancer and other degenerative
diseases such as KOA.
Cancer. In the cancer field, with
the recent build-up of multiple cancer therapeutic technologies, we
have prioritized our clinical efforts on CAR-T. We are not actively
pursuing the fragmented Tcm technical services opportunities. On
November 29, 2016, we announced the approval and commencement of
patient enrollment in China for its CARD-1 (“CAR-T Against
DLBCL”) Phase I clinical trial utilizing its optimized
proprietary C-CAR011 construct of CD19 chimeric antigen receptor
T-cell (CAR-T) therapy for the treatment of patients with
refractory Diffuse Large B-cell Lymphoma (DLBCL). The CARD-1 trial
has begun enrollment with final data expected to be available in
the second half of 2018. On January 9, 2017 we announced the
approval and commencement of patient enrollment in China for its
CALL-1 (“CAR-T against Acute Lymphoblastic Leukemia”)
Phase I clinical trial utilizing its optimized proprietary C-CAR011
construct of CD19 chimeric antigen receptor T-cell
(“CAR-T”) therapy for the treatment of patients with
relapsed or refractory (r/r) CD19+ B-cell Acute Lymphoblastic
Leukemia (“ALL”). The CALL-1 trial has begun enrollment
with final data expected to be available in the second half of
2018. Depending on the Phase I CARD-1 and CALL-1 results, we expect
to initiate larger trials to confirm the safety and efficacy
profile and support BLA submission as soon as
practicable.
On May
15, 2017, we announced the addition of a new independent Phase I
clinical trial of the Company’s ongoing CARD-1 study in
patients with chemorefractory and aggressive DLBCL. Recruitment has
started on patients comprised of DLBCL, Primary Mediastinal Large
B-Cell Lymphoma (PMBCL) and Follicular Lymphoma (FL). Final data
for this single arm, non-randomized study to evaluate the safety
and efficacy of C- CAR011 (Anti-CD19 single-chain variable fragment
(scFv) (41BB-CD3f)) therapy in relapsed or refractory B cell
Non-Hodgkin Lymphoma (NHL) is expected in the first half of
2019.
KOA. In 2013, we completed a
Phase I/IIa clinical study, in China, for our Knee Osteoarthritis
(“KOA”) therapy named Re-Join®. The trial tested
the safety and efficacy of intra-articular injections of autologous
haMPCs in order to reduce inflammation and repair damaged joint
cartilage. The 6-month follow-up clinical data showed Re-Join®
therapy to be both safe and effective.
In Q2
of 2014, we completed patient enrollment for the Phase IIb clinical
trial of Re-Join® for KOA. The multi-center study enrolled 53
patients to participate in a randomized, single blind trial. We
published 48 weeks follow-up data of Phase I/IIa on December 5,
2014. The 48 week data indicated that patients have
reported a decrease in pain and a significant improvement in
mobility and flexibility, while the clinical data shows our
Re-Join® regenerative medicine treatment to be
safe. We announced the interim 24 week results for
Re-Join® on March 25, 2015 and released positive
Phase IIb 48 week follow-up data in January 2016, which shows the
primary and secondary endpoints of Re-Join® therapy group
having all improved significantly compared to their baseline, which
has confirmed some of the Company’s Phase I/IIa results. Our
Re-Join® human adipose-derived mesenchymal progenitor cell
(haMPC) therapy for KOA is an interventional therapy using
proprietary device, process, culture and medium:
●
Obtain adipose
(fat) tissue from the patient using our CFDA approved medical
device, the A-Stromal™ Kit;
●
Expand haMPCs using
our proprietary culture medium (serum-free and antibiotics-free);
and
●
Formulated for
ReJoin therapy using our proprietary formulation.
Our
process is distinguishable from sole Stromal Vascular Fraction
(SVF) therapy. The immunophenotype of our haMPCs exhibited multiple
biomarkers such as CD29+, CD73+, CD90+, CD49d+, HLA-I+, HLA-DR-,
Actin-, CD14-, CD34-, and CD45-. In contrast, SVF is
merely a heterogeneous fraction including preadipocytes,
endothelial cells, smooth muscle cells, pericytes, macrophages,
fibroblasts, and adipose-derived stem cells (ASCs).
In
January 2016, we launched the Allogeneic KOA Phase I Trial in China
to evaluate the safety and efficacy of AlloJoin™, an off-the
shelf allogeneic adipose derived progenitor cell (haMPC) therapy
for the treatment of KOA. On August 5, 2016 we completed patient
treatment for the Allogeneic KOA Phase I trial, and on December 9,
2016 we announced interim 3-month safety data from the Allogenic
KOA Phase I Trial in China. The interim analysis of the trial has
preliminarily demonstrated a safety and tolerability profile of
AlloJoin™ in the three doses tested, and no serious adverse
events (SAE) have been observed. The trial has been completed in
2017.
In
January 2015, we initiated patient recruitment in a phase II
clinical study, in China, of ReJoin (human adipose derived
mesenchymal progenitor cell or “haMPC”) in Cartilage
Damaged (“CD”) patients resulting from osteoarthritis
(“OA”) or sports injury, in further support of KOA
indication. The study is based on the same technology that has
shown significant efficacy in the treatment of Knee Osteoarthritis
(“KOA”), but requires two arthroscopic examinations and
the use of magnetic resonance imaging (“MRI”) to
further demonstrate the regenerative efficacy of ReJoin. Upon
further review of the protocol and the difficulty of getting
patients back for a second arthroscopic examination, we determined
to terminate the study.
The unique
lines of adult adipose-derived stem cells and the immune cell
therapies enable us to create multiple cell formulations in
treating specific medical conditions and diseases, as well as
applying single cell types in a specific treatment protocol. The
quality management systems of CBMG Shanghai and CBMG Wuxi were
issued a Certificate of ISO-9001:2008 by SGS /ANAB (ANSI-ASQ
National Accreditation Board). Our facility in Shanghai was issued
a Certificate of Compliance by ENV Services, Inc., an ISO
Inspection Service Provider that (i) its rooms 1-7, 10 are
certified to ISO Class 7 per ISO-14644 in accordance with cGMP;
(ii) its biological safety cabinets are certified per NSF/ANSI 49
and to ISO Class 5;and (iii) its instrumentation calibration has
been certified to perform in accordance with ANSI/NCSL Z-540-1 and
document in accordance with 10CFR21.Our facility in Shanghai was
issued a Testing Report by Shanghai Food and Drug Packaging
Material Control Center concluding that some testing items of the
cleanrooms are in compliance with the Good Manufacturing Practice
for Drugs (2010 Revision) of China. The cleanrooms in Beijing are
certified to meet the standard of CNAS L1669; and Wuxi has been
certified to meet the SHPMCC standard.
In
addition to standard protocols, we use proprietary processes and
procedures for manufacturing our cell lines, comprised
of:
●
Banking
processes that ensure cell preservation and viability;
●
DNA
identification for stem cell ownership; and
●
Bio-safety
testing at independently certified laboratories.
Recent
Developments in Cancer Cell Therapy
According to the
U.S. National Cancer Institute’s 2013 cancer topics research
update on CAR-T-Cells, excitement is growing for
immunotherapy—therapies that harness the power of a
patient’s immune system to combat their disease, or what some
in the research community are calling the “fifth
pillar” of cancer treatment.
One
approach to immunotherapy involves engineering patients’ own
immune cells to recognize and attack their tumors. And although
this approach, called adoptive cell transfer (ACT), has been
restricted to small clinical trials so far, treatments using these
engineered immune cells have generated some remarkable responses in
patients with advanced cancer. For example, in several early-stage
trials testing ACT in patients with ALL who had few if any
remaining treatment options, many patients’ cancers have
disappeared entirely. Several of these patients have remained
cancer free for extended periods.
Equally
promising results have been reported in several small clinical
trials involving patients with lymphoma. Although the lead
investigators cautioned that much more research is needed, the
results from the trials performed thus far indicate that
researchers can successfully alter patients’ T cells so that
they attack their cancer cells. As an example, we look
to Spectrum Pharmaceutical’s Folotyn approved in September
2009 for treatment of R/R peripheral T-cell lymphoma with approval
supported by a single arm trial observing an overall response rate
of 27% and median duration of response of 9.4 months. In addition,
CTI Therapeutics Pixuvri received a complete response letter in
April 2010 in R/R aggressive NHL in which a 37% overall response
rate and 5.5 month duration of response was observed.
ACT’s
building blocks are T cells, a type of immune cell collected from
the patient’s own blood. After collection, the T cells are
genetically engineered to produce special receptors on their
surface called chimeric antigen receptors (CARs). CARs are proteins
that allow the T cells to recognize a specific protein (antigen) on
tumor cells. These engineered CAR T cells are then grown in the
laboratory until they number in the billions. The expanded
population of CAR T cells is then infused into the patient. After
the infusion, if all goes as planned, the T cells multiply in the
patient’s body and, with guidance from their engineered
receptor, recognize and kill cancer cells that harbor the antigen
on their surfaces. This process builds on a similar form of ACT
pioneered from NCI’s Surgery Branch for patients with
advanced melanoma. According to
www.cancer.gov/.../research-updates/2013/CAR-T-Cells, in 2013
NCI’s Pediatric Oncology Branch commented that the CAR T
cells are much more potent than anything they can achieve with
other immune-based treatments being studied. Although investigators
working in this field caution that there is still much to learn
about CAR T-cell therapy, the early results from trials like these
have generated considerable optimism. Researchers opined that CAR
T-cell therapy eventually may become a standard therapy for some
B-cell malignancies like ALL and chronic lymphocytic
leukemia.
So
far, chimeric antigen receptor T cell therapy such as CD19 CAR-T,
have been tested in several hematological indications on patients
that are refractory/relapsing to chemotherapy, and many of them
have relapsed after stem cell transplantation. All of
these patients had very limited treatment option prior to CAR-T
therapy. CAR-T has shown positive clinical efficacy in
many of these patients. Some of have them lived for years post
CAR-T treatment.
Management believes
the remaining risk in monetizing cancer immune cell therapies is
concentrated in late stage clinical studies, speed-to-approval,
manufacturing and process optimization.
On July
2016, Juno Therapeutics, Inc. reported the death of patients
enrolled in the U.S. Phase II clinical trial of JCAR015 for the
treatment of relapsed or refractory B cell acute lymphoblastic
leukemia (B-ALL). The US FDA put the trial on hold and lifted the
hold within a week after Juno provided satisfactory explanation and
solution. Juno believes that the patient deaths were caused by the
use of Fludarabine preconditioning and they will use only
cyclophosphamide pre-conditioning in the future enrollment. The
trial was halted in November of 2016 after two more deaths occurred
after the trial resumed. The Company believes that its product and
study are distinguishable from Juno Therapeutics and plans to
continue to monitor any toxicities associated with the
study.
On
August 2017, the U.S. FDA approved Novartis’ CAR-T therapy on
relapsed or refractory (r/r) acute lymphoblastic leukemia (ALL),
the most common cancer in Children. Current treatments show a
rate of 80% remission using intensive chemotherapy. However, there
are almost no conventional treatments to help patients who have
relapsed. Novartis’ Tisagenlecleucel (Kymriah), a
CD19-targeted CAR-T therapy for children and adolescents with r/r
ALL has shown results of complete and long lasting remission, which
led the FDA to approve the
drug funded by Novartis and the first CAR-T therapy.
On
October 2017, the U.S. FDA approved Kite Pharmaceuticals’
(Gilead) CAR-T therapy for diffuse large B-cell lymphoma (DLBCL),
the most common type of non- Hodgkin
lymphoma (NHL) in adults. The initial results of axicabtagene
ciloleucel (Yescarta), Kite Pharma’s drug for
non-Hodgkin’s lymphoma, shows four out of seven patients
treated achieved complete remission, which continued after 12
months. The prognosis of high-grade chemo refractory NHL is dismal
with a medium survival time of a few weeks. Yescarta is a therapy
for patients who have not responded to or who have relapsed after
at least two other kinds of treatment.
In
December 2017, the Chinese government issued trial guidelines
concerning development and testing of cell therapy products in
China. Although these trial guidelines are not yet codified as
mandatory regulation, we believe they provide a measure of clarity
and a preliminary regulatory pathway for our cell therapy
operations in a still uncertain regulatory
environment.
Market
for Cell-Based Therapies
In
2013, U.S. sales of products which contain stem cells or progenitor
cells or which are used to concentrate autologous blood, bone
marrow or adipose tissues to yield concentrations of stem cells for
therapeutic use were, conservatively, valued at $236 million at the
hospital level. It is estimated that the orthopedics industry used
approximately 92% of the stem cell products.
The
forecast is that in the United States, shipments of treatments with
stem cells or instruments which concentrate stem cell preparations
for injection into painful joints will fuel an overall increase in
the use of stem cell based treatments and an increase to $5.7
billion in 2020, with key growth areas being Spinal Fusion, Sports
Medicine and Osteoarthritis of the joints. According to Centers for
Disease Control and Prevention. Prevalence of doctor-diagnosed
arthritis and arthritis-attributable activity limitation United
States. 2010-2012, Osteoarthritis (OA) is a chronic disease that is
characterized by degeneration of the articular cartilage,
hyperosteogeny, and ultimately, joint destruction that can affect
all of the joints. According to Dillon CF, Rasch EK, Gu Q et al.
Prevalence of knee osteoarthritis in the United States: Arthritis
Data from the Third National Health and Nutrition Examination
Survey 1991-94. J Rheumatol. 2006, the incidence of OA is 50% among
people over age 60 and 90% among people over age 65. KOA accounts
for the majority of total OA conditions and in adults, OA is the
second leading cause of work disability and the disability
incidence is high (53%). The costs of OA management have grown
exponentially over recent decades, accounting for up to 1% to 2.5%
of the gross national product of countries with aging populations,
including the U.S., Canada, the UK, France, and Australia.
According to the American Academy of Orthopedic Surgeons (AAOS),
the only pharmacologic therapies recommended for OA symptom
management are non-steroidal anti-inflammatory drugs (NSAIDs) and
tramadol (for patients with symptomatic osteoarthritis). Moreover,
there is no approved disease modification therapy for OA in the
world. Disease progression is a leading cause of hospitalization
and ultimately requires joint replacement surgery. In 2009, the
U.S. spent over $42 billion on replacement surgery for hip and knee
joints alone. International regulatory guidelines on clinical
investigation of medicinal products used in the treatment of OA
were updated in 2015, and clinical benefits (or trial outcomes) of
a disease modification therapy for KOA has been well defined and
recommended. Medicinal products used in the treatment of
osteoarthritis need to provide both a symptom relief effect for at
least 6 months and a structure modification effect to slow
cartilage degradation by at least 12 months. Symptom relief is
generally measured by a composite questionnaire Western Ontario and
McMaster Universities Osteoarthritis Index (WOMAC) score, and
structure modification is measured by MRI, or radiographic image as
accepted by international communities. The Company uses the WOMAC
as primary end point to demonstrate symptom relief, and MRI to
assess structure and regeneration benefits as a secondary
endpoint.
According to the
Foundation for the National Institutes of Health, there are 27
million Americans with Osteoarthritis (OA), and symptomatic Knee
Osteoarthritis (KOA) occurs in 13% of persons aged 60 and older.
The International Journal of Rheumatic Diseases, 2011 reports that
approximately 57 million people in China suffer from KOA. Currently
no treatment exists that can effectively preserve knee joint
cartilage or slow the progression of KOA. Current common drug-based
methods of management, including anti-inflammatory medications
(NSAIDs), only relieve symptoms and carry the risk of side effects.
Patients with KOA suffer from compromised mobility, leading to
sedentary lifestyles; doubling the risk of cardiovascular diseases,
diabetes, and obesity; and increasing the risk of all causes of
mortality, colon cancer, high blood pressure, osteoporosis, lipid
disorders, depression and anxiety. According to the Epidemiology of
Rheumatic Disease (Silman AJ, Hochberg MC. Oxford Univ. Press,
1993:257), 53% of patients with KOA will eventually become
disabled.
The
number of cell therapy companies that are currently in Phase 2 and
Phase 3 trials has been gathering momentum, and we anticipate that
new cellular therapy products will appear on the market within the
next several years.
Our
Strategy
The
majority of our biopharmaceutical business is in the development
stage. We intend to concentrate our business on cell therapies and
in the near-term, carrying our KOA stem cell therapy and cancer
immune cell therapies to commercialization.
We are
developing our business in cell therapeutics and capitalizing on
the increasing importance and promise that adult stem cells have in
regenerative medicine. Our most advanced candidate involves
adipose-derived mesenchymal stem cells to treat KOA. Based on
current estimates, we expect our biopharmaceutical business to
generate revenues primarily through the development of therapies
for the treatment of KOA within the next three to four
years.
Presently we have
two completed KOA cell therapy clinical studies in China, a Phase
IIb autologous study and a Phase I allogeneic study. If and when
either therapy obtains regulatory approval in the PRC, we will be
able to market and offer the therapy for clinical use in
China.
Our
strategy is to develop safe and effective cellular medicine
therapies for indications that represent a large unmet need in
China, based on technologies developed both in-house and obtained
through acquisition, licensing and collaboration arrangements
with other companies. Our near term objective is to pursue
successful clinical trials in China for our KOA
application. We intend to utilize our comprehensive cell
platform to support multiple cell lines to pursue multiple
therapies, both allogeneic and autologous. We intend to apply U.S.
Standard Operating Procedures ("SOPs") and protocols while
complying with Chinese regulations, while owning, developing and
executing our own clinical trial protocols. We plan to establish
domestic and international joint ventures or partnerships to set up
cell laboratories and/or research facilities, acquire technology or
in-license technology from outside of China, and build affiliations
with hospitals, to develop a commercialization path for our
therapies, once approved. We intend to use our first-mover
advantage in China, against a backdrop of enhanced regulation by
the central government, to differentiate ourselves from the
competition and establish a leading position in the China cell
therapeutic market. We also intend to out-license our
technologies to interested parties and are exploring the
feasibility of a U.S. allogeneic KOA clinical study with the
FDA.
With
the AG acquisition we intend to monetize AG’s U.S. and
Chinese intellectual property for immune cell therapy preparation
methodologies and patient immunity assessment by engaging with
prominent hospitals to conduct pre-clinical and clinical studies in
specific cancer indications. The T Cell clonality analysis
technology patent, together with AG’s other know-how for
immunity analysis, will enable the Company to establish an
immunoassay platform that is crucial for immunity evaluation of
patients with immune disorders as well as cancerous diseases that
are undergoing therapy. We will continue to
seek to empower hospitals' immune cell cancer therapy development
programs that help patients improve their quality of life and
improve their survival rate.
We
believe that few competitors in China are as well-equipped as we
are in the clinical trial development, diversified U.S. FDA
protocol compliant manufacturing facilities, regulatory compliance
and policy making participation, as well as a long-term presence in
the U.S. with U.S.-based management and investor base.
We
intend to continue our business development efforts by adding other
proven domestic and international biotechnology partners to
monetize the China health care market.
In
order to expedite fulfillment of patient treatment CBMG has been
actively developing technologies and products with a strong
intellectual properties protection, including haMPC, derived from
fat tissue, for the treatment of KOA and other indications.
CBMG’s acquisition of AG provides an enlarged opportunity to
expand the application of its cancer therapy-enabling technologies
and to initiate clinical trials with leading cancer
hospitals.
CBMG's
proprietary and patent-protected production processes and clinical
protocols enable us to produce raw material, manufacture cells, and
conduct cell banking and distribution. These protocols
include medical assessment to qualify each patient for treatment,
evaluation of each patient before and after a specific therapy,
cell transplantation methodologies including dosage, frequency and
the use of adjunct therapies, potential adverse effects and their
proper management. Applying our proprietary intellectual
property, we will be able to customize specialize formulations to
address complex diseases and debilitating conditions.
We
operate our manufacturing facilities under the design of the
standard good manufacturing practice ("GMP") conditions in the ISO
accredited laboratories standard. We employ an institutionalized
and proprietary process and quality management system to optimize
reproducibility and to hone our efficiency. Three facilities
designed and built to GMP in Beijing, Shanghai and Wuxi, China meet
international standards. With our integrated Plasmid, Viral
Vectors, and CAR-T cells Chemistry, Manufacturing, and
Controls processes as well as planned capacity expansion, we are
highly distinguishable with other companies in the cellular
medicine space.
In
total, our facilities have approximately 70,000 square feet of
space and are expected to have a capacity to provide therapies that
can treat approximately 10,000 cancer patients and 10,000 patients
per year.
Most
importantly, our most experienced team members have more than 20
years of relevant experience in China, European Union, and the
United States. All of these factors make CBMG a high quality cell
products manufacturer in China.
Our
Targeted Indications and Potential Therapies
Knee Osteoarthritis (KOA)
We are
currently pursuing two primary therapies for the treatment of KOA:
our Re-Join® therapy and our AlloJoinTM therapy.
We
completed the Phase I/IIa clinical trial for the
treatment of KOA. The trial tested the safety and efficacy of
intra-articular injections of autologous haMPCs in order to reduce
inflammation and repair damaged joint cartilage. The 6-month
follow-up clinical data showed Re-Join® therapy to be
both safe and effective.
In the
second quarter of 2014, we completed patient enrollment for the
Phase IIb clinical trial of Re-Join® for KOA. The multi-center
study has enrolled 53 patients to participate in a randomized,
single blind trial. We published 48 weeks follow-up data of Phase
I/IIa on December 5, 2014. The 48 weeks data indicated
that patients have reported a decrease in pain and a significant
improvement in mobility and flexibility, while the clinical data
shows our Re-Join® regenerative medicine treatment to be safe.
We announced positive Phase IIb 48-week follow-up data in January
2016, with statistical significant evidence that Re-Join®
enhanced cartilage regeneration, which concluded the planned phase
IIb trial.
In January 2016, we launched the Allogeneic KOA
Phase I Trial in China to
evaluate the safety and efficacy of AlloJoin™, an off-the
shelf haMPC therapy for the treatment of KOA. On August 5, 2016 we
completed patient treatment for the Allogeneic KOA Phase I trial. On August 5,
2016 we completed patient treatment for the Allogenic KOA Phase I
Trial, and on December 9, 2016, we announced interim 3-month safety
data from the Allogenic KOA Phase I Trial in China. The interim
analysis of the trial has preliminarily demonstrated a safety and
tolerability profile of AlloJoin™ in the three doses tested,
and no SAEs have been observed. The trial has been completed in
2017. We plan to
release the data in the second quarter of 2018.
Osteoarthritis is a
degenerative disease of the joints. KOA is one of the most common
types of osteoarthritis. Pathological manifestation of
osteoarthritis is primarily local inflammation caused by immune
response and subsequent damage of joints. Restoration of immune
response and joint tissues are the objective of
therapies.
According
to International Journal of
Rheumatic Diseases, 2011, 53% of KOA patients will
degenerate to the point of disability. Conventional treatment
usually involves invasive surgery with painful recovery and
physical therapy. As drug-based methods of management are
ineffective, the same journal estimates that some 1.5 million
patients with this disability will degenerate to the point of
requiring artificial joint replacement surgery every year. However,
only 40,000 patients will actually be able to undergo replacement
surgery, leaving the majority of patients to suffer from a
life-long disability due to lack of effective
treatment.
Adult
mesenchymal stem cells can currently be isolated from a variety of
adult human sources, such as liver, bone marrow, and adipose (fat)
tissue. We believe the advantages in using adipose tissue (as
opposed to bone marrow or blood) are that it is one of the richest
sources of pluripotent cells in the body, the easy and repeatable
access to fat via liposuction, and the simple cell isolation
procedures that can begin to take place even on-site with minor
equipment needs. The procedure we are testing for KOA involves
extracting a very small amount of fat using a minimally invasive
extraction process which takes up to 20 minutes, and leaves no
scarring. The haMPC cells are then processed and isolated on site,
and injected intra articularly into the knee joint with ultrasound
guidance.
These
haMPC cells are capable of differentiating into bone, cartilage,
tendon, skeletal muscle, and fat under the right conditions. As
such, haMPCs are an attractive focus for medical research and
clinical development. Importantly, we believe both allogeneic and
autologously sourced haMPCs may be used in the treatment of
disease. Numerous studies have provided preclinical data that
support the safety and efficacy of allogeneic and autologously
derived haMPC, offering a choice for those where factors such as
donor age and health are an issue.
Additionally,
certain disease treatment plans call for an initial infusion of
these cells in the form of SVF, an initial form of cell isolation
that can be completed and injected within ninety minutes of
receiving lipoaspirate. The therapeutic potential conferred by the
cocktail of ingredients present in the SVF is also evident, as it
is a rich source for preadipocytes, mesenchymal stem cells,
endothelial progenitor cells, T regulatory cells and
anti-inflammatory macrophages.
haMPCs
are currently being considered as a new and effective treatment for
osteoarthritis, with a huge potential
market. Osteoarthritis is one of the ten most disabling
diseases in developed countries. Worldwide estimates are that 9.6%
of men and 18.0% of women aged over 60 years have symptomatic
osteoarthritis. It is estimated that the global OA therapeutics
market was worth $4.4 billion in 2010 and is forecast to grow at a
compound annual growth rate (“CAGR”) of 3.8% to reach
$5.9 billion by 2018.
In
order to bring haMPC-based KOA therapy to market, our market
strategy is to: (a) establish regional laboratories that comply
with cGMP standards in Shanghai and Beijing that meet Chinese
regulatory approval; and (b) file joint applications with Class AAA
hospitals to use haMPCs to treat KOA in a clinical trial
setting.
Our
competitors are pursuing treatments for osteoarthritis with knee
cartilage implants. However, unlike their approach, our
KOA therapy is not surgically invasive – it uses a small
amount (30ml) of adipose tissue obtained via liposuction from the
patient, which is cultured and re-injected into the patient. The
injections are designed to induce the body’s secretion of
growth factors promoting immune response and regulation, and
regrowth of cartilage. The down-regulation of the patient’s
immune response is aimed at reducing and controlling inflammation
which is a central cause of KOA.
We
believe our proprietary method, subsequent haMPC proliferation and
processing know-how will enable haMPC therapy to be a low cost and
relatively safe and effective treatment for KOA. Additionally,
banked haMPCs can continue to be stored for additional use in the
future.
Immuno-oncology (I/o)
We
continue to fortify our cancer breakthrough technology platform
with I/o, programmed cell death and vaccine
technology.
Our
CAR-T platform is built on lenti-virial vector and
second-generation CAR design, which is used by most of the current
trials and studies. We rigorously select the patient population for
each asset and indication to allow the optimal path forward for
regulatory approval. We also fully integrate the state of art
translational medicine effort into each clinical study to aid in
dose selection, to confirm the mechanism of action and proof of
concept, and to identify the optimal targeting patient population
whenever appropriate. We plan to continue to grow our translational
medicine team and engage key opinion leaders to meet the
demand.
Solid
tumors pose more challenges than hematological
cancers. The patients are more heterogeneous, making it
difficult to have one drug to work effectively in the majority of
the patients in any cancer indication. The duration of
response is most likely shorter and patients are likely to relapse
even after initial positive clinical response. We will
continue our effort in developing cell based therapies to target
both hematological cancers and solid tumors.
Tumor Cell Specific Dendritic Cells (TC-DC)
Recent
scientific findings indicate the presence of special cells in
tumors that are responsible for cancer metastases and relapse.
Referred to as “cancer stem cells”, these cells make up
only a small portion of the tumor mass. The central concept behind
TC-DC therapy is to immunize against these cells. TC-DC therapy
takes a sample of the patient’s own purified and irradiated
cancer cells and combines them with specialized immune cells,
thereby ‘educating’ the immune cells to destroy the
cancer stem cells from which tumors arise. We believe
the selective targeting of cells that drive tumor growth would
allow for effective cancer treatment without the risks and side
effects of current therapies that also destroy healthy cells in the
body.
Our
strategy is, through the acquisition of AG and the technologies and
pre-clinical and clinical data of University of the South Florida
and PLAGH, to become an immune cell business leader in the China
cancer therapy market and specialty pharmaceutical market by
utilizing CBMG’s attractiveness as a Nasdaq listed company to
consolidate key China immune cell technology leaders with fortified
intellectual property and ramp up revenue with first mover’s
advantage in a safe and efficient manner. The Company
plans to accelerate cancer trials in China by using the knowledge
and experience gained from the Company’s ongoing KOA trials
and the recent, CAR-T and Tcm technologies. However, it remains
unclear if any of our clinical trials will qualify for
U.S.FDA-liked Fast Track designation as maintenance therapy in
subjects with advanced cancer who have limited options following
surgery and front-line platinum/taxane chemotherapy to improve
their progression-free survival. By applying U.S. SOP and protocols
and following authorized treatment plans in China, we believe we
are differentiated from our competition as we believe we have first
mover’s advantage and a fortified barrier to
entry. In addition, encouraged by the 2017 CIRM grant of
$2.29 million for our preclinical trial to replicate and validate
the manufacturing process and control system at the cGMP facility
located at Children’s Hospital Los Angeles to support the
filing of an IND with the FDA, we have begun to review
the feasibility of performing synergistic U.S. KOA clinical
trial.
Intellectual
Property
We have
built our intellectual property portfolio with a view towards
protecting our freedom of operation in China within our specialties
in the cellular biopharmaceutical field. Our portfolio contains
patents, trade secrets, and know-how.
The
production of stem cells for therapeutic use requires the ability
to purify and isolate these cells to an extremely high level of
purity. Accordingly, our portfolio is geared toward protecting our
proprietary process of purification, cell processing and related
steps in stem cell production. The combination of our patents and
trade secrets protects various aspects of our cell line production
methods and methods of use, including methods of purification,
extraction, freezing, preservation, processing and use in
treatment.
For our
haMPC therapy:
●
We
believe our intellectual property portfolio for haMPC is well-built
and abundant. It covers aspects of adipose stem cell medicine
production, including acquisition of human adipose tissue,
preservation, and storage, tissue, processing, stem cell
purification, expansion, and banking, formulation for
administration, and administration methods.
●
Our
portfolio also includes adipose derived cellular medicine
formulations and their applications in the potential treatment of
degenerative diseases and autoimmune diseases, including
osteoarthritis, rheumatoid arthritis, as well as potential
applications to anti-aging.
●
Our
haMPC intellectual property portfolio:
°
provides
coverage of all steps in the production process;
°
enables
achievement of high yields of Stromal Vascular Fraction (SVF), i.e.
stem cells derived from adipose tissue extracted by
liposuction;
°
makes adipose
tissue acquisition convenient and useful for purposes of cell
banking; and
°
employs
preservation techniques enabling long distance shipment of finished
cell medicine products.
For our
CAR-T and Tcm cancer immune cell therapy:
●
Our
recent amalgamation of technologies from AG and PLAGH in the cancer
cell therapy is comprehensive and well-rounded. It comprises of T
cell clonality, Chimeric Antigen Receptor T cell (CAR-T) therapy,
its recombinant expression vector CD19, CD20, CD30 and Human
Epidermal Growth Factor Receptor's (EGFR or HER1) Immuno-Oncology
patents applications, several preliminary clinical studies of
various CAR-T constructs targeting CD19-positive acute
lymphoblastic leukemia, CD20-positive lymphoma, CD30-positive
Hodgkin's lymphoma and EGFR-HER1-positive advanced lung cancer, and
Phase I/II clinical data of the aforementioned therapies and
manufacturing knowledge.
In
addition, our intellectual property portfolio covers various
aspects of other therapeutic categories including umbilical
cord-derived huMPC therapy, bone marrow-derived hbMPC
therapy.
Moreover, our
clinical trial protocols are proprietary, and we rely upon trade
secret laws for protection of these protocols.
We
intend to continue to vigorously pursue patent protection of the
technologies we develop, both in China and under the Patent
Cooperation Treaty (“PCT”). Additionally, we require
all of our employees to sign proprietary information and invention
agreements, and compartmentalize our trade secrets in order to
protect our confidential information.
Patents
The
following is a brief list of our patents, patent applications and
work in process as of December 31, 2017:
|
|
China Patents
|
U.S. Patents
|
EU Patents
|
Other International Patents
|
PCT
|
|
Work
in Process
|
7
|
-
|
-
|
-
|
-
|
|
Patents
Filed, Pending
|
25
|
2
|
1
|
2
|
2
|
|
Granted
|
23
|
2
|
1
|
-
|
-
|
|
Total
|
55
|
4
|
2
|
2
|
2
|
Generally, our
patents cover technology, methods, design and composition of and
relating to medical device kits used in collecting cell specimens,
cryopreservation of cells, purification, use of stem cells in a
range of potential therapies, adipose tissue extraction, cell
preservation and transportation, preparation of chimeric antigen
receptor, gene
detection and quality control.
Manufacturing
We
manufacture cells for our own research, testing and clinical
trials. We are scaling up and expanding our manufacturing capacity
to treat up to 10,000 CAR-T and 10,000 KOA patients per year when
our facilities are fully operational by end of 2018. Our facilities
are operated by a manufacturing and technology team with decades of
relevant experience in China, EU, and the U.S.
In any
precision setting, it is vital that all controlled environment
equipment meet certain design standards. We operate our
manufacturing facilities under good manufacturing practice ("GMP")
conditions in the ISO accredited laboratories standard. We employ
an institutionalized and proprietary process and quality management
system to optimize reproducibility and to hone our efficiency.
Three of our facilities designed and built to GMP in Beijing,
Shanghai and Wuxi, China meet international standards.
Specifically, our Shanghai cleanroom facility underwent rigorous
cleanroom certification since 2013. Our facility in Shanghai was
issued a Certificate of Compliance by ENV Services, Inc., an ISO
Inspection Service Provider, that (i) its rooms 1-7, 10 are
certified to ISO Class 7 per ISO-14644 in accordance with cGMP.
(ii) its biological safety cabinets are certified per NSF/ANSI 49
and to ISO Class 5. and (iii) its instrumentation calibration has
been certified to perform in accordance with ANSI/NCSL Z-540-1 and
document in accordance with 10CFR21. The cleanrooms in Beijing are
certified to meet the standard of CNAS L1669 and our Wuxi facility
has been certified to meet the SHPMCC standard. With our integrated
Plasmid, Viral Vectors, and CAR-T cells Chemistry, Manufacturing,
and Controls processes as well as planned capacity expansion, we
believe that are highly distinguishable with other companies in the
cellular medicine space.
In
January 2017, we leased a 113,038-square foot building located in
the “Pharma Valley” of Shanghai, the People’s
Republic of China. We are establishing 43,000 square foot
facilities there with 25 clean-rooms and equipped with 12
independent production lines to support clinical batch production
and commercial scale manufacturing. With above expansion, the
Company could support up to 10,000 patients with CAR-T therapy and
10,000 KOA patients with the stem cell therapy per
annum.
We have
built cell preparation and inspection laboratories that enable
the following mode of human body immune cell in-vitro culture
service to be provided: make cell preparation for human body venous
blood samples, after completion of the cell preparation, deliver
the immune cell agents to the customer; and provide immune function
evaluation for the patients in Jilin and several other hospitals in
China.
Competition
Many
companies operate in the cellular biopharmaceutical
field. In 2010, the FDA approved the first cell therapy
for Dendreon Corporation to apply an autologous cellular
immunotherapy for the treatment of a certain type of prostate
cancer. In May 2012 the Canadian authorities approved the
first stem cell drug and granted Osiris Therapeutics’
manufactured stem cell product for use in the pediatric
graft-versus-host disease. To date there are over thirty
publicly listed and several private cellular biopharmaceutical
focused companies outside of China with varying phases of clinical
trials addressing a variety of diseases. We compete with
these companies in bringing cellular therapies to the
market. However, our focus is to develop a core business
in the China market. This difference in focus places us in a
different competitive environment from other western companies with
respect to fund raising, clinical trials, collaborative
partnerships, and the markets in which we
compete.
The PRC
central government has a focused strategy to enable China to
compete effectively in certain designated areas of biotechnology
and the health sciences. Because of the aging population in
China, China’s Ministry of Science and Technology
(“MOST”) has targeted stem cell development as high
priority field, and development in this field has been intense in
the agencies under MOST. For example, the 973 Program has
funded a number of stem cell research projects such as
differentiation of human embryonic germ cells and the plasticity of
adult stem cells. Notwithstanding such governmental support,
China has had a highly fragmented cellular medicine
landscape. Shenzhen Beike Biotechnology Co. Ltd.
(“Beike”) and Union Stem Cell & Gene Engineering
Co., Ltd. (“Union Stem Cell”) are two large stem cell
companies in China. To the best of our knowledge, none of the
Chinese companies are utilizing our proposed international
manufacturing protocol and our unique technologies in conducting
what we believe will be fully compliant CFDA-sanctioned clinical
trials to commercialize cell therapies in China. Our
management believes that it is difficult for most of these Chinese
companies to turn their results into translational stem cell
science or commercially successful therapeutic products using
internationally acceptable standards.
We
compete globally with respect to the discovery and development of
new cell based therapies, and we also compete within China to bring
new therapies to market. The biotechnology industry,
namely in the areas of cell processing and manufacturing, clinical
development of cellular therapies and cell collection, processing
and storage, are characterized by rapidly evolving technology and
intense competition. Our competitors worldwide include
pharmaceutical, biopharmaceutical and biotechnology companies, as
well as numerous academic and research institutions and government
agencies engaged in drug discovery activities or funding, in the
U.S., Europe and Asia. Many of these companies are well-established
and possess technical, research and development, financial, and
sales and marketing resources significantly greater than ours. In
addition, many of our smaller potential competitors have formed
strategic collaborations, partnerships and other types of joint
ventures with larger, well established industry competitors that
afford these companies potential research and development and
commercialization advantages in the technology and therapeutic
areas currently being pursued by us. Academic
institutions, governmental agencies and other public and private
research organizations are also conducting and financing research
activities which may produce products directly competitive to those
being commercialized by us. Moreover, many of these competitors may
be able to obtain patent protection, obtain government (e.g. FDA)
and other regulatory approvals and begin commercial sales of their
products before us.
Our
primary competitors in the field of stem cell therapy for
osteoarthritis, and other indications include Beike, Cytori
Therapeutics Inc., Caladrius Biosciences, Inc. and
others. Among our competitors, to our knowledge, the
only ones based in and operating in Greater China are Beike, Lorem
Vascular, which has partnered with Cytori to commercialize
Cytori Cell Therapy for the cardiovascular, renal and diabetes
markets in China and Hong Kong, and OLife Bio, a
Medi-Post joint venture with JingYuan Bio in Taian, Shandong
Province, who plans to initiate clinical trial in China in
2016. Our primary competitors in the field of cancer
immune cell therapies include pharmaceutical, biotechnology
companies such as Northwest Biotherapeutics, Inc., Juno
Therapeutics, Inc., Kite Pharma, Inc., CARSgen, Sorrento
Therapeutics, Inc. and others. Among our competitors,
the ones based in and operating in Greater China are BeiGene,
Limited, CARsgen and China Oncology Focus Limited, which has
licensed Sorrento’s anti-PD-L1 monoclonal antibody for
Greater China. Other western big pharma and biotech companies in
the cancer immune cell therapies space are starting to make inroads
into China by partnering or seeking to partner with local
companies. For example, in April, 2016, Seattle-based Juno
Therapeutics, Inc started a new company with WuXi AppTec in China
named JW Biotechnology (Shanghai) Co., Ltd. Its mission is to build
China's leading cell therapy company by leveraging Juno's chimeric
antigen receptor (CAR) and T cell receptor (TCR) technologies
together with WuXi AppTec's R&D and manufacturing platform and
local expertise to develop novel cell-based immunotherapies for
patients with hematologic and solid organ cancers. In January 2017,
Shanghai Fosun Pharmaceutical created a joint venture with Santa
Monica-based Kite Pharma Inc. to develop, manufacture and
commercialize axicabtagene ciloleucel in China with the option to
include additional products, including two T cell receptor (TCR)
product candidates from Kite. Axicabtagene ciloleucel is Kite's
lead product candidate and is an investigational chimeric antigen
receptor (CAR) T-cell therapy under development for the treatment
of B-cell lymphomas and leukemias. In late 2017 Gilead acquired
Kite Pharma for $11.9 billion. On January 22, 2018 Celgene
announced that it had agreed to buy Juno Therapeutics for
approximately $9 billion.
The
CFDA has received six applications for CD19 chimeric antigen
receptor T cells cancer therapies from different companies. The
applicants are Nanjing Legend biotechnology, Chengdu Yinhe
Biological medicine, Shanghai HRAIN Biotechnology, Carsgene
Biomedicine (Shanghai), Biogene ANKE Cell Technology and Shanghai
Mingju Biotechnology.
Additionally, in
the general area of cell-based therapies for knee osteoarthritis
ailments, we potentially compete with a variety of companies, from
big pharma to specialty medical products or biotechnology
companies. Some of these, such as Abbvie, Merck KGaA, Sanofi, Teva,
GlaxosmithKline, Baxter, Johnson & Johnson, Sanumed, Medtronic
and Miltenyi Biotec, are well-established and have substantial
technical and financial resources compared to
ours. However, as cell-based products are only just
emerging as viable medical therapies, many of our more direct
competitors are smaller biotechnology and specialty medical
products companies comprised of Vericel Corporation, Regeneus Ltd.,
Advanced Cell Technology, Inc., Nuo Therapeutics, Inc., Arteriocyte
Medical Systems, Inc., ISTO technologies, Inc., Ember Therapeutics,
Athersys, Inc., Bioheart, Inc., Cytori Therapeutics, Inc., Harvest
Technologies Corporation, Mesoblast, Pluristem, Inc., TissueGene,
Inc. Medipost Co. Ltd. and others. There are also several non
cell-based, small molecule and peptide clinical trials targeting
knee osteoarthritis, and several other FDA approved treatment for
knee pain.
Some of
our competitors also work with adipose-derived stem
cells. To the best of our knowledge, none of these
companies are currently utilizing the same technologies as ours to
treat KOA, nor to our knowledge are any of these companies
conducting government-approved clinical trials in
China.
Some of
our targeted disease applications may compete with drugs from
traditional pharmaceutical or Traditional Chinese Medicine
companies. We believe that our chosen targeted disease
applications are not effectively in competition with the products
and therapies offered by traditional pharmaceutical or Traditional
Chinese Medicine companies.
We
believe we have a strategic advantage over our competitors based on
our ability to meet cGMP regulatory requirements, a capability
which we believe is possessed by few to none of our competitors in
China, in an industry in which meeting exacting standards and
achieving extremely high purity levels is crucial to
success. In addition, in comparison to the broader range
of cellar biopharmaceutical firms, we believe we have the
advantages of cost and expediency, and a first mover advantage with
respect to commercialization of cell therapy products and
treatments in the Greater China market.
Employees
As of
December 31, 2017, the total enrollment of full time employees of
CBMG is 125. Among these 125 professionals, 20 have PhD
degrees, 54 have postgraduate degrees and 42 have
undergraduate degrees. In other words, 93% of our
employees are well qualified professionals. As a biotech company,
93 out of our 125 employees have medical or biological scientific
credentials and qualifications.
Facilities
Our
corporate headquarters are located at 19925 Stevens Creek Blvd.,
Suite 100 in Cupertino, California. We currently pay rent for a
total of $268,000 per month for an aggregate of approximately
177,000 square feet of space to house our administration, research
and manufacturing facilities in Maryland and in the cities of Wuxi,
Beijing and Shanghai in China.
Certain
Tax Matters
Following the
completion of our merger with EastBridge Investment Group
Corporation (Delaware) on February 6, 2013, CBMG and its controlled
subsidiaries (the “CBMG Entities”) became a Controlled
Foreign Corporation (CFC) under U.S. Internal Revenue Code Section
957. As a result, the CBMG Entities are subject to anti-deferral
provisions within the U.S. federal income tax system that were
designed to limit deferral of taxable earnings otherwise achieved
by putting profit in low taxed offshore entities. While the CBMG
Entities are subject to review under such provisions, the CBMG
Entities’ earnings are from an active business and should not
be deemed to be distributions made to its U.S. parent
company.
On
December 22, 2017, the tax reform bill was passed (Tax Cut and Jobs
Act (H.R.1)) and reduced top corporate tax rate from 35% to 21%
effective from January 1, 2018. Pursuant to this new Act,
non-operating loss carry back period is eliminated and the loss
carry forward period was expanded from 20 years to an indefinite
period.
Pursuant to the
Corporate Income Tax Law of the PRC, all of the Company’s PRC
subsidiaries are liable to PRC CIT at a rate of 25% except for
Cellular Biomedicine Group Ltd. (Shanghai) (“CBMG
Shanghai”). According to Guoshuihan 2009 No. 203, if an
entity is certified as an “advanced and new technology
enterprise”, it is entitled to a preferential income tax rate
of 15%. CBMG Shanghai obtained the certificate of “advanced
and new technology enterprise” dated October 30, 2015 with an
effective period of three years and the provision for PRC corporate
income tax for CBMG Shanghai is calculated by applying the income
tax rate of 15% in 2017 (2016: 15%; 2015: 15%).
BIOPHARMACEUTICAL
REGULATION
PRC
Regulations
Our
cellular medicine business operates in a highly regulated
environment. In China, aside from provincial and local
licensing authorities, hospitals and their internal ethics and
utilization committees, and a system of institutional review boards
(“IRBs”) which in many cases have members appointed by
provincial authorities. With respect to cell therapies, however,
the Chinese regulatory infrastructure is less established and China
has not yet codified any mandatory regulations governing the
development of cell therapy products. In December 2017, the Chinese government
issued trial guidelines
concerning development and testing of cell therapy products, including stem cell treatments
and immune cell therapies such as CAR-T cell therapeutics. These
trial guidelines are not mandatory regulation but provide some
general principles and basic requirements for cell therapy products
in the areas of pharmaceutical
research, non-clinical research and clinical research. The cell
therapy products provided in the trial guideline refer to the
human-sourced living cell products which are used for human disease
therapy, whose source, operation and clinical trial process are in
line with ethics and whose research and registration application
are in line with regulations on pharmaceutical administration. The
competent authority of pharmaceutical administration is the CFDA.
It is further clarified by the CFDA that the non-registered
clinical trial data would be acceptable for drug registration on a
case by case basis, pending on the consistency of the samples used
for the clinical trial and the drug applied for registration, the
generation process of the clinical trial data, whether the data is
authentic, complete, accurate and traceable to the source, and the
inspection outcome of the CFDA on the clinical trial. Moreover, an
applicant of the clinical trial of the said cell therapyproducts
can propose the phases of the clinical trial and the trial plan by
itself (generally the trial can be divided into early stage
clinical trial phase and confirmatory clinical trial phase),
instead of the application of the traditional phases I, II and III
of a clinical trial. However, it remains unclear if
any of our clinical trials will be offered U.S.FDA-like Fast Track
designation as maintenance therapy in subjects with advanced cancer
who have limited options following surgery and front-line
platinum/taxane chemotherapy to improve their progression-free
survival. By applying U.S. standards and protocols and following
authorized treatment plans in China, we believe we are
differentiated from our competition as we believe we have first
mover’s advantage in an undeveloped industry. In
addition, we have begun to review the feasibility of
performing synergistic U.S. clinical studies.
PRC Operating Licenses
Our
business operations in China are subject to customary regulation
and licensing requirements under regulatory agencies including the
local Administration for Industry and Commerce, General
Administration of Quality Supervision, Inspection and Quarantine,
and the State Administration of Taxation, for each of our business
locations. Additionally, our clean room facilities and the use of
reagents is also regulated by local branches of the Ministry of
Environmental Protection. We are in good standing with respect to
each of our business operating licenses.
U.S. Government
Regulation
The
health care industry is one of the most highly regulated industries
in the United States. The federal government, individual state and
local governments, as well as private accreditation organizations,
oversee and monitor the activities of individuals and businesses
engaged in the development, manufacture and delivery of health care
products and services. Federal laws and regulations seek to protect
the health, safety, and welfare of the citizens of the United
States, as well as to prevent fraud and abuse associated with the
purchase of health care products and services with federal monies.
The relevant state and local laws and regulations similarly seek to
protect the health, safety, and welfare of the states’
citizens and prevent fraud and abuse. Accreditation organizations
help to establish and support industry standards and monitor new
developments.
HCT/P Regulations
Manufacturing
facilities that produce cellular therapies are subject to extensive
regulation by the U.S. FDA. In particular, U.S. FDA regulations set
forth requirements pertaining to establishments that manufacture
human cells, tissues, and cellular and tissue-based products
(“HCT/Ps”). Title 21, Code of Federal Regulations, Part
1271 (21 CFR Part 1271) provides for a unified registration and
listing system, donor-eligibility, current Good Tissue Practices
(“cGTP”), and other requirements that are intended to
prevent the introduction, transmission, and spread of communicable
diseases by HCT/Ps. While we currently have no plans to conduct
these activities within the United States, these regulations may be
relevant to us if in the future we become subject to them, or if
parallel rules are imposed on our operations in China.
We
currently collect, process, store and manufacture HCT/Ps, including
manufacturing cellular therapy products. We also collect, process,
and store HCT/Ps. Accordingly, we comply with cGTP and cGMP
guidelines that apply to biological products. Our management
believes that certain other requirements pertaining to biological
products, such as requirements pertaining to premarket approval, do
not currently apply to us because we are not currently
investigating, marketing or selling cellular therapy products in
the United States. If we change our business operations in the
future, the FDA requirements that apply to us may also
change.
Certain
state and local governments within the United States also regulate
cell-processing facilities by requiring them to obtain other
specific licenses. Certain states may also have enacted laws and
regulations, or may be considering laws and regulations, regarding
the use and marketing of stem cells or cell therapy products, such
as those derived from human embryos. While these laws and
regulations should not directly affect our business, they could
affect our future business. Presently we are not subject to any of
these state law requirements, because we do not conduct these
regulated activities within the United States.
Pharmaceutical and Biological Products
In the
United States, pharmaceutical and biological products, including
cellular therapies, are subject to extensive pre- and post-market
regulation by the FDA. The Federal Food, Drug, and Cosmetic Act
(“FD&C Act”), and other federal and state statutes
and regulations, govern, among other things, the research,
development, testing, manufacture, storage, recordkeeping,
approval, labeling, promotion and marketing, distribution,
post-approval monitoring and reporting, sampling, and import and
export of pharmaceutical products. Biological products are approved
for marketing under provisions of the Public Health Service Act, or
PHS Act. However, because most biological products also meet the
definition of “drugs” under the FD&C Act, they are
also subject to regulation under FD&C Act provisions. The PHS
Act requires the submission of a biologics license application
(“BLA”), rather than a New Drug Application ("NDA"),
for market authorization. However, the application process and
requirements for approval of BLAs are similar to those for NDAs,
and biologics are associated with similar approval risks and costs
as drugs. Presently we are not subject to any of these
requirements, because we do not conduct these regulated activities
within the United States. However, these regulations may
be relevant to us should we engage in these activities in the
United States in the future.
WHERE
YOU CAN FIND MORE INFORMATION
You are
advised to read this Form 10-K in conjunction with other reports
and documents that we file from time to time with the SEC. In
particular, please read our Quarterly Reports on Form 10-Q and
Current Reports on Form 8-K that we file from time to time. You may
obtain copies of these reports directly from us or from the SEC at
the SEC's Public Reference Room at 100 F. Street, N.E. Washington,
D.C. 20549, and you may obtain information about obtaining access
to the Reference Room by calling the SEC at 1-800-SEC-0330. In
addition, the SEC maintains information for electronic filers at
its website http://www.sec.gov.
ITEM
1A. Risk
Factors
RISKS
RELATED TO OUR COMPANY
We have a limited operating history and expect significant
operating losses for the next few years.
We are
a company with a limited operating history and have incurred
substantial losses and negative cash flow from operations through
the year ended December 31, 2017. Our cash flow from operations may
not be consistent from period to period, our biopharmaceutical
business has not yet generated substantial revenue, and we may
continue to incur losses and negative cash flow in future periods,
particularly within the next several years.
Our biopharmaceutical product development programs are based on
novel technologies and are inherently risky.
We are
subject to the risks of failure inherent in the development of
products based on new biomedical technologies. The novel nature of
these cell-based therapies creates significant challenges in regard
to product development and optimization, manufacturing, government
regulation, third party reimbursement, and market acceptance,
including the challenges of:
●
Educating
medical personnel regarding the application protocol;
●
Sourcing
clinical and commercial supplies for the materials used to
manufacture and process our product candidates;
●
Developing a
consistent and reliable process, while limiting contamination risks
regarding the application protocol;
●
Conditioning
patients with chemotherapy in conjunction with delivering immune
cell therapy treatment, which may increase the risk of adverse side
effects;
●
Obtaining
regulatory approval, as the Chinese Food and Drug Administration,
or CFDA, and other regulatory authorities have limited experience
with commercial development of cell-based therapies, and therefore
the pathway to regulatory approval may be more complex and require
more time than we anticipate; and
●
Establishing
sales and marketing capabilities upon obtaining any regulatory
approval to gain market acceptance of cell therapy.
These
challenges may prevent us from developing and commercializing
products on a timely or profitable basis or at all.
We face risks relating to the cell therapy industry, clinical
development and commercialization.
Cell
therapy is still a developing field and a significant global market
for our services has yet to emerge. Our cellular therapy candidates
are based on novel cell technologies that are inherently risky and
may not be understood or accepted by the marketplace. The current
market principally consists of providing manufacturing of cell and
tissue-based therapeutic products for clinical trials and
processing of stem cell products for therapeutic
programs.
The
degree of market acceptance of any future product candidates will
depend on a number of factors, including:
●
the
clinical safety and effectiveness of the product candidates, the
availability of alternative treatments and the perceived advantages
of the particular product candidates over alternative
treatments;
●
the
relative convenience and ease of administration of the product
candidates;
●
our
ability to separate the product candidates from the ethical
controversies and political barriers associated with stem cell
product candidates derived from human embryonic or fetal
tissue;
●
ethical concerns
that may arise regarding our commercial use of stem cells,
including adult stem cells, in the manufacture of the product
candidates;
●
the
frequency and severity of adverse events or other undesirable side
effects involving the product candidates or the products or product
candidates of others that are cell-based; and
●
the
cost of the products, the reimbursement policies of government and
third-party payors and our ability to obtain sufficient third-party
coverage or reimbursement.
Laws and the regulatory infrastructure governing cellular
biopharmaceuticals in China are relatively new and less established
in comparison to the U.S. and other countries; accordingly
regulation may be less stable and predictable than desired, and
regulatory changes may disrupt our commercialization
process.
In December 2017, the Chinese government
issued trial guidelines
concerning development and testing of cell therapy products, including stem cell treatments
and immune cell therapies such as CAR-T cell therapeutics. These
trial guidelines are not mandatory regulation but provide some
general principles and basic requirements for cell therapy products
in the areas of pharmaceutical
research, non-clinical research and clinical research. The cell
therapy products provided in the trial guideline refer to the
human-sourced living cell products which are used for human disease
therapy, whose source, operation and clinical trial process are in
line with ethics and whose research and registration application
are in line with regulations on pharmaceutical administration. The
competent authority of pharmaceutical administration is the CFDA.
It is further clarified by the CFDA that the non-registered
clinical trial data would be acceptable for drug registration on a
case by case basis, pending on the consistency of the samples used
for the clinical trial and the drug applied for registration, the
generation process of the clinical trial data, whether the data is
authentic, complete, accurate and traceable to the source, and the
inspection outcome of the CFDA on the clinical trial. Moreover, an
applicant of the clinical trial of the said cell therapyproducts
can propose the phases of the clinical trial and the trial plan by
itself (generally the trial can be divided into early stage
clinical trial phase and confirmatory clinical trial phase),
instead of the application of the traditional phases I, II and III
of a clinical trial. However, remains unclear if any of our
clinical trials will be offered U.S.FDA-like Fast Track designation
as maintenance therapy in subjects with advanced cancer who have
limited options following surgery and front-line platinum/taxane
chemotherapy to improve their progression-free survival. We do not
know if our animal studies documentation will be approved to
support trials in humans. We also do not know if our cell lines
will be accepted by the PRC health authorities. Thesefactors could
adversely affect the timing of the clinical trials, the timing of
receipt and reporting of clinical data, the timing of
Company-sponsored IND filings, and our ability to conduct future
planned clinical trials, and any of the above could have a material
adverse effect on our business.
CFDA’s regulations may limit our ability to develop, license,
manufacture and market our products and services.
Some or
all of our operations in China will be subject to oversight and
regulation by the CFDA and MOH. Government regulations, among other
things, cover the inspection of and controls over testing,
manufacturing, safety and environmental considerations, efficacy,
labeling, advertising, promotion, record keeping and sale and
distribution of pharmaceutical products. Such government
regulations may increase our costs and prevent or delay the
licensing, manufacturing and marketing of any of our products or
services. In the event we seek to license, manufacture, sell or
distribute new products or services, we likely will need approvals
from certain government agencies such as the future growth and
profitability of any operations in China would be contingent on
obtaining the requisite approvals. There can be no assurance that
we will obtain such approvals.
In
2003, the CFDA implemented new guidelines for the licensing of
pharmaceutical products. All existing manufacturers with licenses
were required to apply for the Good Manufacturing Practices
(“cGMP”) certifications. According to Good Manufacturing Practices for
Pharmaceutical Products (revised edition 2010), or the New
GMP Rules promulgated by the Ministry of Health of the PRC on
January 17, 2011 which became effective on March 1, 2011, all the
newly constructed manufacturing facilities of drug manufacture
enterprises in China shall comply with the requirements of the New
GMP Rules, which are stricter than the original GMP
standards.
In
addition, delays, product recalls or failures to receive approval
may be encountered based upon additional government regulation,
legislative changes, administrative action or changes in
governmental policy and interpretation applicable to the Chinese
pharmaceutical industry. Our pharmaceutical activities also may
subject us to government regulations with respect to product prices
and other marketing and promotional related activities. Government
regulations may substantially increase our costs for developing,
licensing, manufacturing and marketing any products or services,
which could have a material adverse effect on our business,
operating results and financial condition.
The
CFDA and other regulatory authorities in China have implemented a
series of new punitive and stringent measures regarding the
pharmaceuticals industry to redress certain past misconducts in the
industry and certain deficiencies in public health reform policies.
Given the nature and extent of such new enforcement measures, the
aggressive manner in which such enforcement is being conducted and
the fact that newly-constituted local level branches are encouraged
to issue such punishments and fines, there is the possibility of
large scale and significant penalties being levied on
manufacturers. These new measures may include fines, restriction
and suspension of operations and marketing and other unspecified
penalties. This new regulatory environment has added significantly
to the risks of our businesses in China and may have a material
adverse effect on our business, operating results and financial
condition.
Our technology platforms, including our CAR-T, Tcm, whether
preclinical or clinical, and the cancer vaccine technologies are
new approaches to cancer treatment that present significant
challenges.
We have
concentrated our research and development efforts on T cell
immunotherapy technology, and our future success in cancer
treatment is dependent on the successful development of T cell
immunotherapies in general and our CAR and vaccine technologies and
product candidates in particular. Our approach to cancer treatment
aims to alter T cells ex
vivo through genetic modification using viruses designed to
reengineer the T cells to recognize specific proteins on the
surface or inside cancer cells. Because this is a new approach to
cancer immunotherapy and cancer treatment generally, developing and
commercializing our product candidates subjects us to many
challenges.
We
cannot be sure that our T cell immunotherapy and vaccine
technologies will yield satisfactory products that are safe and
effective, scalable, or profitable. Additionally, because our
technology involves the genetic modification of patient cells
ex vivo using a virus, we
are subject to many of the challenges and risks that gene therapies
face, including regulatory requirements governing gene and cell
therapy products have changed frequently.
Moreover, public
perception of therapy safety issues, including adoption of new
therapeutics or novel approaches to treatment, may adversely
influence the willingness of subjects to participate in clinical
trials, or if approved, of physicians to subscribe to the novel
treatment mechanics. Physicians, hospitals and third-party payers
often are slow to adopt new products, technologies and treatment
practices that require additional upfront costs and training.
Physicians may not be willing to undergo training to adopt this
novel and personalized therapy, may decide the therapy is too
complex to adopt without appropriate training and may choose not to
administer the therapy. Based on these and other factors, hospitals
and payers may decide that the benefits of this new therapy do not
or will not outweigh its costs.
Our near term ability to generate significant product revenue
is dependent on the success of one or more of our CD19, CD22, CD30
and HER1, as well as CD40GVAX product candidates, each of which are
at an early-stage of development and will require significant
additional clinical testing before we can seek regulatory approval
and begin commercial sales.
Our
near term ability to generate significant product revenue is
highly dependent on our ability to obtain regulatory approval of
and successfully commercialize one or more of our CD19, CD20, CD30
and HER1, as well as CD40GVAX product candidates. All of these
products are in the early stages of development, have been tested
in a relatively small number of patients, and will require
additional clinical and nonclinical development, regulatory review
and approval in each jurisdiction in which we intend to market the
products, substantial investment, access to sufficient commercial
manufacturing capacity, and significant marketing efforts before we
can generate any revenue from product sales. Before obtaining
marketing approval from regulatory authorities for the sale of our
product candidates, we must conduct extensive clinical studies to
demonstrate the safety, purity, and potency of the product
candidates in humans. We cannot be certain that any of our product
candidates will be successful in clinical studies and they may not
receive regulatory approval even if they are successful in clinical
studies.
If our
products, once developed, encounter safety or efficacy
problems, developmental delays, regulatory issues, or other
problems, our development plans and business could be significantly
harmed. Further, competitors who are developing products with
similar technology may experience problems with their products that
could identify problems that would potentially harm our
business.
Third parties have sponsored and conducted all clinical trials of
our CD19, CD20, CD30 and HER1, as well as the CD40GVAX vaccine
product candidates so far, and our ability to influence the design
and conduct of such trials has been limited. We plan to assume
control over future clinical and regulatory development of the
CD20, CD30 and HER1, and may do so for other product candidates,
which will entail additional expenses and may be subject to delay.
Any failure by a third party to meet its obligations with respect
to the clinical and regulatory development of our product
candidates may delay or impair our ability to obtain regulatory
approval for our products and result in liability for our
company.
On
November 29, 2016 we announced the approval and commencement of
patient enrollment in China for our CARD-1 (“CAR-T Against
DLBCL”) Phase I clinical trial utilizing our optimized
proprietary C-CAR011 construct of CD19 CAR-T therapy for the
treatment of patients with refractory DLBCL. On January 9, 2017 we
announced the approval and commencement of patient enrollment in
China for our CALL-1 (“CAR-T against Acute Lymphoblastic
Leukemia”) Phase I clinical trial utilizing our optimized
proprietary C-CAR011 construct of CD19 CAR-T therapy for the
treatment of patients with relapsed or refractory (r/r) CD19+
B-cell ALL. We do not know if our Phase I CARD-1 or CALL-1 results
will justify initiation of larger Phase II clinical
trials.
To
date, we have yet to sponsor any clinical trials relating to our
CD20, CD30, HER1 and CD40GVAX product candidates or other product
candidates. Instead, faculty members at our third-party research
institution collaborators, or those institutions themselves, have
sponsored all clinical trials relating to these product candidates,
in each case under their own IRB with the respective regulatory
agency. We plan to assume control of the overall clinical and
regulatory development of CD19, CD20, CD30 and HER1 for future
clinical trials and obtain sponsorship of the INDs or file new
Company-sponsored INDs in China and/or the United States. We will
evaluate options to conducting the U.S. CD40LGVAX Trial and to
continuing the related IND with the Federal Drug Administration
(“FDA”). Failure to obtain, or delays in
obtaining, sponsorship of INDs or in filing new Company-sponsored
INDs for these or any other product candidates we determine to
advance could negatively affect the timing of our potential future
clinical trials. Such an impact on timing could increase research
and development costs and could delay or prevent obtaining
regulatory approval for our most advanced product candidates,
either of which could
have a material adverse effect on our business.
Further, even in
the event that the IND sponsorship is obtained for existing and new
INDs, it is possible that the CFDA or other regulatory agencies
will not accept any of the trials as providing adequate support for
future clinical trials, whether controlled by us or third parties,
for any of one or more reasons, including the safety, purity, and
potency of the product candidate, the degree of product
characterization, elements of the design or execution of the
previous trials or safety concerns, or other trial results. We may
also be subject to liabilities arising from any treatment-related
injuries or adverse effects in patients enrolled in these previous
trials. As a result, we may be subject to unforeseen third-party
claims and delays in our potential future clinical trials. We may
also be required to repeat in whole or in part clinical trials
previously conducted by our third-party research institution
collaborators, which will be expensive and delay the submission and
licensure or other regulatory approvals with respect to any of our
product candidates. Any such delay or liability could have a
material adverse effect on our business.
Moreover, although
we plan to assume control of the overall clinical and regulatory
development of CD19, CD20, CD30 and HER1 going forward, we have so
far been dependent on contractual arrangements with our third-party
research institution collaborators and will continue to be until we
assume control. To the extent that we do not use our own scientific
team to conduct trials, we are and will be dependent contractual
arrangements with third-party research institution collaborators
for ongoing and planned trials for our product candidates. Such
arrangements provide us certain information rights with respect to
the previous, planned, or ongoing trials, including access to and
the ability to use and reference the data, including for our own
regulatory filings, resulting from such trials. If our third-party
research institution collaborators breach these obligations, or if
the data prove to be inadequate compared to the first-hand
knowledge we might have gained had the completed trials been
Company-sponsored trials, then our ability to design and conduct
our planned corporate-sponsored clinical trials may be adversely
affected. Additionally, the regulatory agencies may disagree with
the sufficiency of our right to reference the preclinical,
manufacturing, or clinical data generated by these prior
investigator-sponsored trials, or our interpretation of
preclinical, manufacturing, or clinical data from these clinical
trials. If so, the regulatory agencies may require us to obtain and
submit additional preclinical, manufacturing, or clinical data
before we may begin our planned trials and/or may not accept such
additional data as adequate to begin our planned
trials.
Our CD19, CD20, CD30 and
HER1, as
well as the CD40GVAX product candidates are
biologics and the manufacture of our product candidates is complex
and we may encounter difficulties in production, particularly with
respect to process development or scaling-out of our manufacturing
capabilities. If we or any of our third-party manufacturers
encounter such difficulties, our ability to provide supply of our
product candidates for clinical trials or our products for
patients, if approved, could be delayed or stopped, or we may be
unable to maintain a commercially viable cost
structure.
Our immune cell CAR-T and vaccine product
candidates are biologics and the process of manufacturing our
products is complex, highly- regulated and subject to multiple
risks. The manufacture of our product candidates involves complex
processes, including harvesting T cells from patients, genetically
modifying the T cells ex vivo, multiplying the T cells to obtain the desired
dose, and ultimately infusing the T cells back into a
patient’s body. As a result of the complexities, the cost to
manufacture these biologics in general, and our genetically
modified cell product candidates in particular, is generally higher
than the adipose stem cell, and the manufacturing process is less
reliable and is more difficult to reproduce. Our manufacturing
process will be susceptible to product loss or failure due to
logistical issues associated with the collection of white blood
cells, or starting material, from the patient, shipping such
material to the manufacturing site, shipping the final product back
to the patient, and infusing the patient with the product,
manufacturing issues associated with the differences in patient
starting materials, interruptions in the manufacturing process,
contamination, equipment or reagent failure, improper installation
or operation of equipment, vendor or operator error, inconsistency
in cell growth, and variability in product characteristics. Even
minor deviations from normal manufacturing processes could result
in reduced production yields, product defects, and other supply
disruptions. If for any reason we lose a patient’s starting
material or later-developed product at any point in the process,
the manufacturing process for that patient will need to be
restarted and the resulting delay may adversely affect that
patient’s outcome. If microbial, viral, or other
contaminations are discovered in our product candidates or in the
manufacturing facilities in which our product candidates are made,
such manufacturing facilities may need to be closed for an extended
period of time to investigate and remedy the contamination. Because
our product candidates are manufactured for each particular
patient, we will be required to maintain a chain of identity with
respect to materials as they move from the patient to the
manufacturing facility, through the manufacturing process, and back
to the patient. Maintaining such a chain of identity is difficult
and complex, and failure to do so could result in adverse patient
outcomes, loss of product, or regulatory action including
withdrawal of our products from the market. Further, as product
candidates are developed through preclinical to late stage clinical
trials towards approval and commercialization, it is common that
various aspects of the development program, such as manufacturing
methods, are altered along the way in an effort to optimize
processes and results. Such changes carry the risk that they will
not achieve these intended objectives, and any of these changes
could cause our product candidates to perform differently and
affect the results of planned clinical trials or other future
clinical trials.
Although we
continue to develop our own manufacturing facilities to support our
clinical and commercial manufacturing activities, we may, in any
event, never be successful in developing our own manufacturing
facilities. We have not yet caused our product
candidates to be manufactured or processed on a commercial scale
and may not be able to do so for any of our product candidates.
Although our manufacturing and processing approach is based upon
the current approach undertaken by our third-party research
institution collaborators, we do not have experience in managing
the vaccine manufacturing process, and our process may be more
difficult or expensive than the approaches currently in use. We
will make changes as we work to optimize the manufacturing process,
and we cannot be sure that even minor changes in the process will
not result in significantly different CAR-T, stem cell or vaccine
that may not be as safe and effective as the current products
deployed by our third-party research institution collaborators. As
a result of these challenges, we may experience delays in our
clinical development and/or commercialization plans. The
manufacturing risks could delay or prevent the completion of our
clinical trials or the approval of any of our product candidates by
the FDA, CFDA or other regulatory authorities, result in higher
costs or adversely impact commercialization of our product
candidates. In addition, we will rely on third parties to perform
certain specification tests on our product candidates prior to
delivery to patients. If these tests are not appropriately done and
test data are not reliable, patients could be put at risk of
serious harm and the FDA, CFDA or other regulatory authorities
could require additional clinical trials or place significant
restrictions on our company until deficiencies are
remedied. We may ultimately be unable to reduce the cost
of goods for our product candidates to levels that will allow for
an attractive return on investment if and when those product
candidates are commercialized.
We rely heavily on third parties to conduct clinical trials on our
product candidates.
We
presently are party to, and expect that we will be required to
enter into, agreements with hospitals and other research partners
to perform clinical trials for us and to engage in sales, marketing
and distribution efforts for our products and product candidates we
may acquire in the future. We may be unable to establish or
maintain third-party relationships on a commercially reasonable
basis, if at all. In addition, these third parties may have similar
or more established relationships with our competitors or other
larger customers. Moreover, the loss for any reason of one or more
of these key partners could have a significant and adverse impact
on our business. If we are unable to obtain or retain third party
sales and marketing vendors on commercially acceptable terms, we
may not be able to commercialize our therapy products as planned
and we may experience delays in or suspension of our marketing
launch. Our dependence upon third parties may adversely affect our
ability to generate profits or acceptable profit margins and our
ability to develop and deliver such products on a timely and
competitive basis.
Outside scientists and their third-party research institutions on
whom we rely for research and development and early clinical
testing of our product candidates may have other commitments or
conflicts of interest, which could limit our access to their
expertise and harm our ability to leverage our technology
platform.
We
currently have limited internal research and development
capabilities and are currently conducting no independent clinical
trials with our, CD20, CD30, HER1 and CD40GVAX product candidates
or our other product candidates. We therefore rely at present on
our third-party research institution collaborators for both
capabilities.
The
outside scientists who conduct the clinical testing of our current
product candidates, and who conduct the research and development
upon which our product candidate pipeline depends, are not our
employees; rather they serve as either independent contractors or
the primary investigators under collaboration that we have with
their sponsoring academic or research institution. Such scientists
and collaborators may have other commitments that would limit their
availability to us. Although our scientific advisors generally
agree not to do competing work, if an actual or potential conflict
of interest between their work for us and their work for another
entity arises, we may lose their services. We are
currently evaluating the feasibility of conducting these trials
ourselves or commencing the trial in the United States or
elsewhere. These factors could adversely affect the timing of the
clinical trials, the timing of receipt and reporting of clinical
data, the timing of Company-sponsored IND filings, and our ability
to conduct future planned clinical trials. It is also possible that
some of our valuable proprietary knowledge may become publicly
known through these scientific advisors if they breach their
confidentiality agreements with us, which would cause competitive
harm to, and have a material adverse effect on our
business.
If we are unable to maintain our licenses, patents or other
intellectual property we could lose important protections that are
material to continuing our operations and our future
prospects.
We
operate in the highly technical field of development of
regenerative and immune cellular therapies. In addition to patents,
we rely in part on trademark, trade secret and protection to
protect our intellectual properties comprised of proprietary know
how, technology and processes. However, trade secrets are difficult
to protect. We have entered and expect to continue to enter into
confidentiality and intellectual property assignment agreements
with our employees, consultants, outside scientific
collaborators, sponsored researchers, affiliates, other advisors
and potential investors. These agreements generally require that
the other party keep confidential and not disclose to third parties
all confidential information developed by the party or made known
to the party by us. These agreements may also provide that
inventions conceived by the party in the course of rendering
services to us will be our exclusive property. However, these
agreements may be difficult to enforce, or can be breached and may
not effectively protect our intellectual property
rights.
In addition to contractual measures, we try to
protect the confidential nature of our proprietary information by
compartmentalize our intellectual properties as well as using other
security measures. Such physical and technology measures may not
provide adequate protection for our proprietary information. For
example, our security measures may not prevent an employee or
consultant with authorized access from misappropriating our trade
secrets and providing them to a competitor, and the recourse we
have available against such misconduct may be inadequate to
adequately protect our interests. Enforcing a claim that a party
illegally disclosed or misappropriated a trade secret can be
difficult, expensive and time consuming, and the outcome is
unpredictable. In addition, courts outside the United States may be
less willing to protect trade secrets. Furthermore, others may
independently develop our proprietary information in a manner that
could prevent legal recourse by us. If any of our confidential or
proprietary information, including our trade secrets and know how,
were to be disclosed or misappropriated, or if a competitor
independently developed any such information, our competitive
position could be harmed.
We may be unable to obtain or maintain patent protection for our
products and product candidates, which could have a material
adverse effect on our business.
Our
commercial success will depend, in part, on obtaining and
maintaining patent protection for new technologies, product
candidates, products and processes and successfully defending such
patents against third party challenges. To that end, we file or
acquire patent applications, and have been issued patents that are
intended to cover certain methods and uses relating to stem cells
and cancer immune cell therapies.
The
patent positions of biotechnology companies can be highly uncertain
and involve complex legal, scientific and factual questions and
recent court decisions have introduced significant uncertainty
regarding the strength of patents in the industry. Moreover, the
legal systems of some countries do not favor the aggressive
enforcement of patents and may not protect our intellectual
property rights to the same extent as they would, for instance,
under the laws of the United States. Any of the issued patents we
own or license may be challenged by third parties and held to be
invalid, unenforceable or with a narrower or different scope of
coverage that what we currently believe, effectively reducing or
eliminating protection we believed we had against competitors with
similar products or technologies. If we ultimately engage in and
lose any such patent disputes, we could be subject to competition
and/or significant liabilities, we could be required to enter into
third party licenses or we could be required to cease using the
disputed technology or product. In addition, even if such licenses
are available, the terms of any license requested by a third party
could be unacceptable to us.
The
claims of any current or future patents that may issue or be
licensed to us may not contain claims that are sufficiently broad
to prevent others from utilizing the covered technologies and thus
may provide us with little commercial protection against competing
products. Consequently, our competitors may independently develop
competing products that do not infringe our patents or other
intellectual property. To the extent a competitor can develop
similar products using a different chemistry, our patents and
patent applications may not prevent others from directly competing
with us. Product development and approval timelines for certain
products and therapies in our industry can require a significant
amount of time (i.e. many years). As such, it is possible that any
patents that may cover an approved product or therapy may have
expired at the time of commercialization or only have a short
remaining period of exclusivity, thereby reducing the commercial
advantages of the patent. In such case, we would then rely solely
on other forms of exclusivity which may provide less protection to
our competitive position.
Litigation relating to intellectual property is expensive, time
consuming and uncertain, and we may be unsuccessful in our efforts
to protect against infringement by third parties or defend
ourselves against claims of infringement.
To
protect our intellectual property, we may initiate litigation or
other proceedings. In general, intellectual property litigation is
costly, time-consuming, diverts the attention of management and
technical personnel and could result in substantial uncertainty
regarding our future viability, even if we ultimately
prevail. Some of our competitors may be able to sustain
the costs of such litigation or other proceedings more effectively
than can we because of their substantially greater financial
resources. The loss or narrowing of our intellectual property
protection, the inability to secure or enforce our intellectual
property rights or a finding that we have infringed the
intellectual property rights of a third party could limit our
ability to develop or market our products and services in the
future or adversely affect our revenues. Furthermore, any public
announcements related to such litigation or regulatory proceedings
could adversely affect the price of our common stock. Third parties
may allege that the research, development and commercialization
activities we conduct infringe patents or other proprietary rights
owned by such parties. This may turn out to be the case even though
we have conducted a search and analysis of third-party patent
rights and have determined that certain aspects of our research and
development and proposed products activities apparently do not
infringe on any third-party Chinese patent rights. If we are found
to have infringed the patents of a third party, we may be required
to pay substantial damages; we also may be required to seek from
such party a license, which may not be available on acceptable
terms, if at all, to continue our activities. A judicial finding or
infringement or the failure to obtain necessary licenses could
prevent us from commercializing our products, which would have a
material adverse effect on our business, operating results and
financial condition.
We will not seek to protect our intellectual property rights in all
jurisdictions throughout the world and we may not be able to
adequately enforce our intellectual property rights even in the
jurisdictions where we seek protection.
Filing,
prosecuting and defending patents on our product candidates in all
countries and jurisdictions throughout the world would be
impracticable and cost prohibitive, and our intellectual property
rights in some countries could be less extensive than those in the
People’s Republic of China or the United States, assuming
that rights are obtained in these jurisdiction. In addition, the
laws of some foreign countries may not protect all of our
intellectual properties.
If we are unable to protect the confidentiality of trade secrets,
our competitive position could be impaired.
A
significant amount of our technology, particularly with respect to
our proprietary manufacturing processes, is unpatented and is held
in the form of trade secrets. Our efforts to protect
these trade secrets are comprised of the use of confidentiality and
proprietary information agreement, physically secured
documentation, and knowledge segmentation among our staff. Even so,
improper use or disclosure of our confidential information could
occur and in such cases adequate remedies may be insufficient to
protect our competitive position or may not exist. The inadvertent
disclosure of our trade secrets could also impair our competitive
position.
PRC intellectual property law requires us to compensate our
employees for the intellectual property that they may help to
develop.
We have
entered and expect to continue to enter into confidentiality and
intellectual property assignment agreements with most of our
employees, consultants, outside scientific collaborators, sponsored
researchers, affiliates and other advisors. These agreements
generally require that the other party keep confidential and not
disclose to third parties all confidential information developed by
the party or made known to the party by us. These agreements may
also provide that inventions conceived by the party in the course
of rendering services to us will be our exclusive property.
However, these agreements may be difficult to enforce, or can be
breached and may not effectively protect our intellectual property
rights.
The PRC
laws codify a “reward/award” policy which entitles
employees to certain levels of compensation and bonus from their
service invention-creations for which their employers filed for
patent protection. In the absence of any contractual understanding,
the Implementing Rules of the
Patent Law require a minimum compensation and bonus to such
employees as below: bonus: (i) for each invention patent, a
one-time reward of no less than 3,000 RMB, or (ii) for each utility
model or design patent, a one-time reward of no less than 1,000
RMB, and compensation: (i) for each invention patent and utility
model, at least 2% of annual operating profits derived from the use
of the patent, (ii) for each design patent, at least 0.2% of annual
operating profits derived from the use of the design patent, and
(iii) at least 10% of royalties received from the licensing the
patent to a third party.
Although our bylaws
allow for us to issue bonuses to our employees, we have not
contractually limited the amount of compensation that we may pay
them for filing patents for their ideas, developments, discoveries
or inventions. As such, should any of our employees and consultants
who have not contractually agreed otherwise seek to enforce these
rights, we may be required to pay the statutorily mandated minimum
to our employees as required by this law. Our product candidates
are still in the clinical trial stage and as of the date of this
annual report, we have not derived any revenue from our
product-related patents. However, if and when we commercialize our
product candidates or therapies, or if we are required to pay our
employees any compensation for patents relating to our technical
services, such compensation could be substantial and may harm our
business prospects, financial condition and results of
operations.
Our technologies are at early stages of discovery and development,
and we may fail to develop any commercially acceptable or
profitable products.
We have
yet to develop any therapeutic products that have been approved for
marketing, and we do not expect to become profitable within the
next several years, but rather expect our biopharmaceutical
business to incur additional and increasing operating losses.
Before commercializing any therapeutic product in China, we may be
required to obtain regulatory approval from the MOH CFDA, local
regulatory authorities, and/or individual hospitals, and outside
China from equivalent foreign agencies after conducting extensive
preclinical studies and clinical trials that demonstrate that the
product candidate is safe and effective.
We may
elect to delay or discontinue studies or clinical trials based on
unfavorable results. Any product developed from, or based on, cell
technologies may fail to:
●
survive and
persist in the desired location;
●
provide the
intended therapeutic benefit;
●
engraft or
integrate into existing tissue in the desired manner;
or
●
achieve
therapeutic benefits equal to, or better than, the standard of
treatment at the time of testing.
In
addition, our therapeutic products may cause undesirable side
effects. Results of preclinical research in animals may not be
indicative of future clinical results in humans.
Ultimately if
regulatory authorities do not approve our products or if we fail to
maintain regulatory compliance, we would be unable to commercialize
our products, and our business and results of operations would be
harmed. Even if we do succeed in developing products, we will face
many potential obstacles such as the need to develop or obtain
manufacturing, marketing and distribution capabilities.
Furthermore, because transplantation of cells is a new form of
therapy, the marketplace may not accept any products we may
develop.
Most potential applications of our technology are
pre-commercialization, which subjects us to development and
marketing risks.
We are
in a relatively early stage on the path to commercialization with
many of our products. Successful development and market acceptance
of our products is subject to developmental risks, including
failure to achieve innovative solutions to problems during
development, ineffectiveness, lack of safety, unreliability,
failure to receive necessary regulatory clearances or approvals,
approval by hospital ethics committees and other governing bodies,
high commercial cost, preclusion or obsolescence resulting from
third parties’ proprietary rights or superior or equivalent
products, competition, and general economic conditions affecting
purchasing patterns. There is no assurance that we or our partners
will successfully develop and commercialize our products, or that
our competitors will not develop competing products, treatments or
technologies that are less expensive or superior. Failure to
successfully develop and market our products would have a
substantial negative effect on our results of operations and
financial condition.
Market acceptance of new technology such as ours can be difficult
to obtain.
New and
emerging cell therapy and cell banking technologies may have
difficulty or encounter significant delays in obtaining market
acceptance in some or all countries around the world due to the
novelty of our cell therapy and cell banking technologies.
Therefore, the market adoption of our cell therapy and cell banking
technologies may be slow and lengthy with no assurances that the
technology will be successfully adopted. The lack of market
adoption or reduced or minimal market adoption of cell therapy and
cell banking technologies may have a significant impact on our
ability to successfully sell our future product(s) or therapies
within China or in other countries. Our strategy depends in part on
the adoption of the therapies we may develop by state-owned
hospital systems in China, and the allocation of resources to new
technologies and treatment methods is largely dependent upon ethics
committees and governing bodies within the hospitals. Even if our
clinical trials are successful, there can be no assurance that
hospitals in China will adopt our technology and therapies as
readily as we may anticipate.
Future clinical trial results may differ significantly from our
expectations.
While
we have proceeded incrementally with our clinical trials in an
effort to gauge the risks of proceeding with larger and more
expensive trials, we cannot guarantee that we will not experience
negative results with larger and much more expensive clinical
trials than we have conducted to date. Poor results in our clinical
trials could result in substantial delays in commercialization,
substantial negative effects on the perception of our products, and
substantial additional costs. These risks are increased by our
reliance on third parties in the performance of many of the
clinical trial functions, including the clinical investigators,
hospitals, and other third party service providers.
If clinical trials of our technology fail to demonstrate safety and
efficacy to the satisfaction of the relevant regulatory
authorities, including the PRC’s State Food and Drug
Administration and the Ministry of Health, or do not otherwise
produce positive results, we may incur additional costs or
experience delays in completing, or ultimately be unable to
complete, the development and commercialization of such product
candidates.
Currently, a
regulatory structure has not been established to standardize the
approval process for products or therapies based on the technology
that exists or that is being developed in our field. Therefore we
must conduct, at our own expense, extensive clinical trials to
demonstrate the safety and efficacy of the product candidates in
humans, and then archive our results until such time as a new
regulatory regime is put in place. If and when this new regulatory
regime is adopted it may be easier or more difficult to navigate
than CBMG may anticipate, with the following potential
barriers:
●
regulators or
institutional review boards may not authorize us or our
investigators to commence clinical trials or conduct clinical
trials at a prospective trial site;
●
clinical trials
of product candidates may produce negative or inconclusive results,
and we may decide, or regulators may require us, to conduct
additional clinical trials or abandon product development programs
that we expect to be pursuing;
●
the
number of patients required for clinical trials of product
candidates may be larger than we anticipate, enrollment in these
clinical trials may be slower than we anticipate, or participants
may drop out of these clinical trials at a higher rate than we
anticipate;
●
third party
contractors may fail to comply with regulatory requirements or meet
their contractual obligations to us in a timely manner or at
all;
●
we
might have to suspend or terminate clinical trials of our product
candidates for various reasons, including a finding that the
participants are being exposed to unacceptable health
risks;
●
regulators or
institutional review boards may require that we or our
investigators suspend or terminate clinical research for various
reasons, including noncompliance with regulatory
requirements;
●
the
cost of clinical trials of our product candidates may be greater
than anticipated;
●
we
may be subject to a more complex regulatory process, since
cell-based therapies are relatively new and regulatory agencies
have less experience with them as compared to traditional
pharmaceutical products;
●
the
supply or quality of our product candidates or other materials
necessary to conduct clinical trials of these product candidates
may be insufficient or inadequate; and
●
our
product candidates may have undesirable side effects or other
unexpected characteristics, causing us or our investigators to halt
or terminate the trials.
We may
be unable to generate interest or meaningful revenue in out-license
our Intellectual Property.
The results of preclinical studies may not correlate with the
results of human clinical trials. In addition, early stage clinical
trial results do not ensure success in later stage clinical trials,
and interim trial results are not necessarily predictive of final
trial results.
To
date, we have not completed the development of any products through
regulatory approval. The results of preclinical studies in animals
may not be predictive of results in a clinical trial. Likewise, the
outcomes of early clinical trials may not be predictive of the
success of later clinical trials. New information regarding the
safety and efficacy of such product candidates may be less
favorable than the data observed to date. AG’s budding
technical service revenue in the Jilin Hospital should not be
relied upon as evidence that later or larger-scale clinical trials
will succeed. In addition, even if the trials are successfully
completed, we cannot guarantee that the CFDA will interpret the
results as we do, and more trials could be required before we
submit our product candidates for approval. To the extent that the
results of the trials are not satisfactory to the CFDA or other
foreign regulatory authorities for support of a marketing
application, approval of our product candidates may be
significantly delayed, or we may be required to expend significant
additional resources, which may not be available to us, to conduct
additional trials in support of potential approval of our product
candidates.
If we encounter difficulties enrolling patients in our clinical
trials, our clinical development activities could be delayed or
otherwise adversely affected.
We may
experience difficulties in patient enrollment in our clinical
trials for a variety of reasons. The timely completion of clinical
trials in accordance with their protocols depends, among other
things, on our ability to enroll a sufficient number of patients
who remain in the study until its conclusion. The enrollment of
patients depends on many factors, including:
●
the
patient eligibility criteria defined in the protocol;
●
the
size of the patient population required for analysis of the
trial’s primary endpoints;
●
the
proximity of patients to study sites;
●
the
design of the trial;
●
our
ability to recruit clinical trial investigators with the
appropriate competencies and experience;
●
our
ability to obtain and maintain patient consents; and
●
the
risk that patients enrolled in clinical trials will drop out of the
trials before completion.
In
addition, our clinical trials may compete with other clinical
trials for product candidates that are in the same therapeutic
areas as our product candidates, and this competition may reduce
the number and types of patients available to us, because some
patients who might have opted to enroll in our trials may instead
opt to enroll in a trial being conducted by one of our competitors.
Since the number of qualified clinical investigators is limited, we
expect to conduct some of our clinical trials at the same clinical
trial sites that some of our competitors use, which will reduce the
number of patients who are available for our clinical trials in
such clinical trial site. Moreover, because our product candidates
represent a departure from more commonly used methods for cancer
treatment, potential patients and their doctors may be inclined to
use conventional therapies, such as chemotherapy and or traditional
Chinese medicine, rather than enroll patients in any future
clinical trial.
Upon
commencing clinical trials, delays in patient enrollment may result
in increased costs or may affect the timing or outcome of the
planned clinical trials, which could prevent completion of these
trials and adversely affect our ability to advance the development
of our product candidates.
We are exposed to general
liability, non-clinical and clinical liability risks which could
place a substantial financial burden upon us, should lawsuits be
filed against us.
Our
business exposes us to potential liability risks that are inherent
in the testing, manufacturing and marketing of our therapies and
product candidates. We expect that such claims are likely to be
asserted against us at some point. In addition, the use in our
clinical trials of our therapies and products and the subsequent
sale of these therapies or product candidates by us or our
potential collaborators may cause us to bear a portion of or all
product liability risks. We currently have insurance coverage
relating to inventory, property plant and equipment and office
premises. The Company also purchased in insurance covering personal
injury, medical expenses and several clinical
trials. However, any claim under such insurance policies
may be subject to certain exceptions, and may not be honored fully,
in part, in a timely manner, or at all, and may not cover the full
extent of liability we may actually face. Therefore, a successful
liability claim or series of claims brought against us could have a
material adverse effect on our business, financial condition and
results of operations.
We currently have no CAR-T or KOA product marketing and sales
organization and have no experience in marketing such products. If
we are unable to establish product marketing and sales capabilities
or enter into agreements with third parties to market and sell our
product candidates, we may generate less product revenue than
expected.
We
currently have no CAR-T or KOA product sales, marketing or
distribution capabilities and have no experience in marketing
products. We intend to develop an in-house product marketing
organization and sales force, which will require significant
capital expenditures, management resources and time. We will have
to compete with other pharmaceutical and biotechnology companies to
recruit, hire, train and retain marketing and sales
personnel.
If we
are unable or decide not to establish internal sales, marketing and
distribution capabilities, we will pursue collaborative
arrangements regarding the sales and marketing of our products,
however, there can be no assurance that we will be able to
establish or maintain such collaborative arrangements, or if we are
able to do so, that they will have effective sales forces. Any
revenue we receive will depend upon the efforts of such third
parties, which may not be successful. We may have little or no
control over the marketing and sales efforts of such third parties
and our revenue from product sales may be lower than if we had
commercialized our product candidates ourselves. We also face
competition in our search for third parties to assist us with the
sales and marketing efforts of our product candidates. There can be
no assurance that we will be able to develop in-house sales and
distribution capabilities or establish or maintain relationships
with third-party collaborators to commercialize any product in
China or overseas.
Coverage and reimbursement may be limited or unavailable in certain
market segments for our product candidates, which could make it
difficult for us to sell our product candidates
profitably.
Successful sales of
our product candidates, if approved, depend on the availability of
adequate coverage and reimbursement from third-party payers. In
addition, because our product candidates represent new approaches
to the treatment of cancer, we cannot accurately estimate the
potential revenue from our product candidates.
Patients who are
provided medical treatment for their conditions generally rely on
third-party payers to reimburse all or part of the costs associated
with their treatment. Adequate coverage and reimbursement from
governmental healthcare programs and commercial payers is critical
to new product acceptance. In China, government authorities decide
which drugs and treatments they will cover and the amount of
reimbursement. Obtaining coverage and reimbursement approval of a
product from a government or other third-party payer is a
time-consuming and costly process that could require us to provide
to the payer supporting scientific, clinical and cost-effectiveness
data for the use of our products. Even if we obtain coverage for a
given product, the resulting reimbursement payment rates might not
be adequate for us to achieve or sustain profitability or may
require co-payments that patients find unacceptably high. Patients
are unlikely to use our product candidates unless coverage is
provided and reimbursement is adequate to cover a significant
portion of the cost of our product candidates. If we
obtain approval in one or more jurisdictions outside of China for
our product candidates, we will be subject to rules and regulations
in those jurisdictions. In some foreign countries, particularly
those in the EU, the pricing of biologics is subject to
governmental control. In these countries, pricing negotiations with
governmental authorities can take considerable time after obtaining
marketing approval of a product candidate. In addition, market
acceptance and sales of our product candidates will depend
significantly on the availability of adequate coverage and
reimbursement from third-party payers for our product candidates
and may be affected by existing and future health care reform
measures. The continuing efforts of the government,
insurance companies, managed care organizations and other payers of
healthcare services to contain or reduce costs of healthcare and/or
impose price controls may adversely affect:
●
the
demand for our product candidates, if we obtain regulatory
approval;
●
our
ability to set a price that we believe is fair for our
products;
●
our
ability to generate revenue and achieve or maintain
profitability;
●
the
level of taxes that we are required to pay; and
●
the
availability of capital.
Any
reduction in reimbursement from any government programs may result
in a similar reduction in payments from private payers, which may
adversely affect our future profitability.
Our product candidates may cause undesirable side effects or have
other properties that could interrupt our clinical development,
prevent or delay regulatory approval, and limit our commercial
value or result in significant negative consequences.
Undesirable
or unacceptable side effects caused by our product candidates could
cause us or regulatory authorities to delay, suspend or stop
clinical trials and could result in the delay or denial of
regulatory approval by the regulatory authorities. Results of our
trials could reveal unacceptable severe adverse effects or
unexpected characteristics.
There
have been reported patient deaths in Immune Cell therapies as a
result of factors comprised of cytokine release syndrome and
neurotoxicity. Immune Cell therapy treatment-related adverse side
effects could also affect patient recruitment or the ability of
enrolled subjects to complete the trial or result in potential
liability claims. In addition, these side effects may not be
recognized or properly managed by the treating medical staff, as
medical personnel do not normally encounter in the general patient
population toxicities resulting from personalized immune cell
therapy. We plan to conduct training for the medical personnel
using immune cell therapy to understand the adverse side effect
profile for our clinical trials and upon any commercialization of
any immune cell product candidates. Inability of the medical
personnel in recognizing or managing immune cell therapy’s
potential adverse side effects could result in patient deaths. Any
of these occurrences may harm our business, financial condition and
prospects significantly.
Our manufacturing facilities are subject to extensive government
regulation, and existing or future regulations may adversely affect
our current or future operations, increase our costs of operations,
or require us to make additional capital expenditures.
Environmental advocacy groups and regulatory
agencies in China have been focusing considerable attention on the
industries’ potential role in climate change. Stringent
government safety, environmental and bio-hazardous materials
disposal regulations at the city, provincial, and local level may
have substantial impact on our business and our third-party service
providers. A number of complex laws, rules, orders, and
interpretations govern environmental protection, health, safety,
land use, zoning, transportation, and related matters. The adoption
of laws and regulations to implement controls of bio-hazardous
material disposal and environmental compliance, including the
imposition of fees or taxes, could adversely affect the operations
with which we do business. Among other things, timeliness in
navigating the compliance of these regulations may restrict our
operations, our third-party service providers’ operations and
adversely affect our financial condition, results of operations,
and cash flows by imposing conditions including, but not limited to
new permits requirement, limitations or bans on disposal or
transportation of certain bio-hazardous materials or certain
categories of materials. The Company has completed the
environmental protection compliance for its new Zhangjiang
facility. The Company is in the process of applying for
(i) environmental protection
compliance for its new facility in Beijing and (ii) update of the
environmental protection permits for its Shanghai facility.
Technological and medical developments or improvements in
conventional therapies could render the use of cell therapy and our
services and planned products obsolete.
Advances in other
treatment methods or in disease prevention techniques could
significantly reduce or entirely eliminate the need for our cell
therapy services, planned products and therapeutic efforts. There
is no assurance that cell therapies will achieve the degree of
success envisioned by us in the treatment of disease. Nor is there
any assurance that new technological improvements or techniques
will not render obsolete the processes currently used by us, the
need for our services or our planned products. Additionally,
technological or medical developments may materially alter the
commercial viability of our technology or services, and require us
to incur significant costs to replace or modify equipment in which
we have a substantial investment. We are focused on novel cell
therapies, and if this field is substantially unsuccessful, this
could jeopardize our success or future results. The occurrence of
any of these factors may have a material adverse effect on our
business, operating results and financial condition.
We face significant competition from other Chinese biotechnology
and pharmaceutical companies, and our operating results will suffer
if we fail to compete effectively.
There
is intense competition and rapid innovation in the Chinese cell
therapy industry, and in the cancer immunotherapy space in
particular. Our competitors may be able to develop other herbal
medicine, compounds or drugs that are able to achieve similar or
better results. Our potential competitors are comprised of
traditional Chinese medicine companies, major multinational
pharmaceutical companies, established and new biotechnology
companies, specialty pharmaceutical companies, state-owned
enterprises, universities and other research institutions. Many of
our competitors have substantially greater scientific, financial,
technical and other resources, such as larger research and
development staff and experienced marketing and manufacturing
organizations and well-established sales forces. Smaller or
early-stage companies may also prove to be significant competitors,
particularly through collaborative arrangements with large,
established companies or are well funded by venture capitals.
Mergers and acquisitions in the biotechnology and pharmaceutical
industries may result in even more resources being concentrated in
our competitors. Competition may increase further as a result of
advances in the commercial applicability of technologies and
greater availability of capital for investment in these industries.
Our competitors, either alone or with collaborative partners, may
succeed in developing, acquiring or licensing on an exclusive basis
drug or biologic products that are more effective, safer, more
easily commercialized or less costly than our product candidates or
may develop proprietary technologies or secure patent protection
that we may need for the development of our technologies and
products. We believe the key competitive factors that will affect
the development and commercial success of our product candidates
are efficacy, safety, tolerability, reliability, and convenience of
use, price and reimbursement.
Even if
we obtain regulatory approval of our product candidates, the
availability and price of our competitors’ products could
limit the demand and the price we are able to charge for our
product candidates. We may not be able to implement our business
plan if the acceptance of our product candidates is inhibited by
price competition or the reluctance of doctors to switch from
existing methods of treatment to our product candidates, or if
doctors switch to other new drug or biologic products or choose to
reserve our product candidates for use in limited
circumstances.
We may be unable to attract or retain key employees for our
business if our share-based or other compensation programs cease to
be viewed as competitive and valuable benefits.
To be
competitive, we must attract, retain, and motivate executives and
other key employees. Hiring and retaining qualified executives,
scientists, technical staff, and professional staff are critical to
our business, and competition for experienced employees can be
intense. To help attract, retain, and motivate key employees, we
use share-based and other performance-based incentive awards such
as stock options, restricted stock units (RSUs) and cash bonuses.
If our share-based or other compensation programs cease to be
viewed as competitive and valuable benefits, our ability to
attract, retain, and motivate key employees could be weakened,
which could harm our results of operations.
There is a scarcity of experienced professionals in the field of
cell therapy and we may not be able to retain key officers or
employees or hire new key officers or employees needed to implement
our business strategy and develop our products. If we are unable to
retain or hire key officers or employees, we may be unable to grow
our biopharmaceutical business or implement our business strategy,
and the Company may be materially and adversely
affected.
Given
the specialized nature of cell therapy and the fact that it is a
young field, there is an inherent scarcity of experienced personnel
in the field. The Company is substantially dependent on the skills
and efforts of current senior management, as well as recently
acquired AG management and personnel, for their management,
operations and the implementation of their business strategy. As a
result of the difficulty in locating qualified new management, the
loss or incapacity of existing members of management or
unavailability of qualified management or as replacements for
management who resign or are terminated could adversely affect the
Company’s operations. The future success of the Company also
depends upon our ability to attract and retain additional qualified
personnel (including medical, scientific, technical, commercial,
business and administrative personnel) necessary to support our
anticipated growth, develop our business, perform our contractual
obligations to third parties and maintain appropriate licensure, on
acceptable terms. There can be no assurance that we will be
successful in attracting or retaining personnel required by us to
continue to grow our operations. The loss of a key employee, the
failure of a key employee to perform in his or her current position
or our inability to attract and retain skilled employees, as
needed, could result in our inability to grow our biopharmaceutical
business or implement our business strategy, or may have a material
adverse effect on our business, financial condition and operating
results.
We may fail to successfully integrate our acquired businesses,
operations and assets in the expected time frame, which may
adversely affect the combined company’s future
results.
We
believe that our acquisitions, including our CAR-T, Tcm and GVAX
technologies, will result in certain benefits, including certain
manufacturing, sales and distribution and operational
efficiencies. However, to realize these anticipated benefits,
our existing business and the acquired technologies must be
successfully combined. We may be unable to effectively
integrate the acquired technologies into our organization, make the
acquired technologies profitable, and may not succeed in managing
the acquired technologies. The process of integration of an
acquired technologies may subject us to a number of risks,
including:
●
Failure to
successfully manage relationships with hospitals, patients and
suppliers;
●
Demands on
management related to the increase in complexity of the company
after the acquisition;
●
Diversion of
management and scientists’ attention;
●
Potential
difficulties integrating and harmonizing large scale multi-site
clinical trials;
●
Difficulties in
the assimilation and retention of employees;
●
Exposure to
legal claims for activities of the acquired technologies;
and
●
Incurrence of
additional expenses in connection with the integration
process.
If the
acquired technologies is not successfully integrated into our
company, our business, financial condition and results of
operations could be materially adversely affected, as well as our
professional reputation. Furthermore, if we are unable to
successfully integrate the acquired technologies, or if there are
delays in implementing clinical trials using the acquired
technologies, the anticipated benefits of the acquisition may not
be realized fully or at all or may take longer to realize than
expected. Successful integration of the acquired technologies
will depend on our ability to manage large scale cancer clinical
trials and to realize opportunities in monetizing these
technologies.
We will need to grow the size of our organization, and we may
experience difficulties in managing this growth.
As our
development and commercialization plans and strategies develop, and
as we continue to expand operation as a public company, we expect
to grow our personnel needs in the managerial, operational, sales,
marketing, financial and other departments. Future growth would
impose significant added responsibilities on members of management,
including:
●
identifying,
recruiting, integrating, maintaining and motivating additional
employees;
●
managing our
internal development efforts effectively, including the clinical
trials and CFDA review process for our product candidates, while
complying with our contractual obligations to contractors and other
third parties; and
●
improving our
operational, financial and management controls, reporting systems
and procedures.
Our
future financial performance and our ability to commercialize our
product candidates will depend, in part, on our ability to
effectively manage any future growth, and our management may also
have to divert a disproportionate amount of its attention away from
day-to-day activities in order to devote a substantial amount of
time to managing these growth activities.
We
currently rely, and for the foreseeable future will continue to
rely, in substantial part on certain independent organizations such
as contract research organizations and hospitals to provide certain
services comprised of regulatory approval and clinical management.
There can be no assurance that the services of independent
organizations will continue to be available to us on a timely basis
when needed, or that we can find qualified replacements. In
addition, if we are unable to effectively manage our outsourced
activities or if the quality or accuracy of the services provided
by the independent organizations is compromised for any reason, our
clinical trials may be extended, delayed or terminated, and we may
not be able to obtain regulatory approval of our product candidates
or otherwise advance our business. If we are not able to
effectively expand our organization by hiring new employees, we may
not be able to successfully implement the tasks necessary to
further develop and commercialize our product candidates and,
accordingly, may not achieve our research, development and
commercialization goals.
We may form or seek strategic alliances or enter into licensing
arrangements in the future, and we may not realize the benefits of
such alliances or licensing arrangements.
We may
form or seek strategic alliances, create joint ventures or
collaborations or enter into licensing arrangements with third
parties that we believe will complement or augment our development
and commercialization efforts with respect to our product
candidates and any future product candidates that we may develop.
Any of these relationships may require us to incur non-recurring
and other charges, increase our near and long-term expenditures,
issue securities that dilute our existing stockholders or disrupt
our management and business. In addition, we face significant
competition in seeking appropriate strategic partners and the
negotiation process is time-consuming and complex. Moreover, we may
not be successful in our efforts to establish a strategic
partnership or other alternative arrangements for our product
candidates because they may be deemed to be at too early of a stage
of development for collaborative effort and third parties may not
view our product candidates as having the requisite potential to
demonstrate safety and efficacy. If we license products or
businesses, we may not be able to realize the benefit of such
transactions if we are unable to successfully integrate them with
our existing operations and company culture. We cannot be certain
that, following a strategic transaction or license, we will achieve
the revenue or specific net income that justifies such transaction.
Any delays in entering into new strategic partnership agreements
related to our product candidates could delay the development and
commercialization of our product candidates in certain geographies
for certain indications, which would harm our business prospects,
financial condition and results of operations.
We, our strategic partners and our customers conduct business in a
heavily regulated industry. If we or one or more of our strategic
partners or customers fail to comply with applicable current and
future laws and government regulations, our business and financial
results could be adversely affected.
The
healthcare industry is one of the most highly regulated industries.
Federal governments, individual state and local governments and
private accreditation organizations may oversee and monitor all the
activities of individuals and businesses engaged in the delivery of
health care products and services. Therefore, current laws, rules
and regulations could directly or indirectly negatively affect our
ability and the ability of our strategic partners and customers to
operate each of their businesses.
In addition,
as we expand into other parts of the world, we will need to comply
with the applicable laws and regulations in such foreign
jurisdictions. We have not yet thoroughly explored the requirements
or feasibility of such compliance. It is possible that we may not
be permitted to expand our business into one or more foreign
jurisdictions.
Although we intend
to conduct our business in compliance with applicable laws and
regulations, the laws and regulations affecting our business and
relationships are complex, and many aspects of such relationships
have not been the subject of judicial or regulatory interpretation.
Furthermore, the cell therapy industry is the topic of significant
government interest, and thus the laws and regulations applicable
to us and our strategic partners and customers and to their
business are subject to frequent change and/or reinterpretation and
there can be no assurance that the laws and regulations applicable
to us and our strategic partners and customers will not be amended
or interpreted in a manner that adversely affects our business,
financial condition, or operating results.
We anticipate that we will need substantial additional financing in
the future to continue our operations; if we are unable to raise
additional capital, as and when needed, or on acceptable terms, we
may be forced to delay, reduce or eliminate one or more of our
product or therapy development programs, cell therapy initiatives
or commercialization efforts and our business will be
harmed.
Our
current operating plan will require significant levels of
additional capital to fund, among other things, the continued
development of our cell therapy product or therapy candidates and
the operation, and expansion of our manufacturing operations to our
clinical development activities.
We have
launched the early phase of CAR-T and KOA clinical trials in China.
Supported by the CIRM grant, we plan to file for allogeneic KOA
pre-clinical trial application with the FDA in the second half of
2018, When approved by the FDA we plan to conduct allogeneic KOA
clinical trial in the United States. If these trials are
successful, we will require significant additional investment
capital over a multi-year period in order to conduct subsequent
phases, gain approval for these therapies by the MOH and CFDA, and
to commercialize these therapies, if ever. Subsequent phases may be
larger and more expensive than the Phase I trials. In order to
raise the necessary capital, we will need to raise additional money
in the capital markets, enter into collaboration agreements with
third parties or undertake some combination of these strategies. If
we are unsuccessful in these efforts, we may have no choice but to
delay or abandon the trials.
The
amount and timing of our future capital requirements also will
likely depend on many other factors, including:
●
the
scope, progress, results, costs, timing and outcomes of our other
cell therapy product or therapy candidates;
●
our
ability to enter into, or continue, any collaboration agreements
with third parties for our product or therapy candidates and the
timing and terms of any such agreements;
●
the
timing of and the costs involved in obtaining regulatory approvals
for our product or therapy candidates, a process which could be
particularly lengthy or complex given the lack of precedent for
cell therapy products in China; and
●
the
costs of maintaining, expanding and protecting our intellectual
property portfolio, including potential litigation costs and
liabilities.
To fund
clinical studies and support our future operations, we would likely
seek to raise capital through a variety of different public and/or
private financings vehicles. This could include, but not be limited
to, the use of loans or issuances of debt or equity securities in
public or private financings. If we raise capital
through the sale of equity, or securities convertible into equity,
it would result in dilution to our then existing
stockholders. Servicing the interest and principal
repayment obligations under debt facilities could divert funds that
would otherwise be available to support clinical or
commercialization activities. In certain cases, we also
may seek funding through collaborative arrangements, that would
likely require us to relinquish certain rights to our technology or
product or therapy candidates and share in the future revenues
associated with the partnered product or therapy.
Ultimately, we may
be unable to raise capital or enter into collaborative
relationships on terms that are acceptable to us, if at all. Our
inability to obtain necessary capital or financing to fund our
future operating needs could adversely affect our business, results
of operations and financial condition.
Failure to achieve and maintain effective internal controls in
accordance with Section 404 of the Sarbanes-Oxley Act could have a
material adverse effect on our business and operating
results.
It may
be time consuming, difficult and costly for us to develop and
implement the additional internal controls, processes and reporting
procedures required by the Sarbanes-Oxley Act. We may need to hire
additional financial reporting, internal auditing and other finance
staff in order to develop and implement appropriate additional
internal controls, processes and reporting procedures.
If we
fail to comply in a timely manner with the requirements of Section
404 of the Sarbanes-Oxley Act regarding internal controls over
financial reporting or to remedy any material weaknesses in our
internal controls that we may identify, such failure could result
in material misstatements in our financial statements, cause
investors to lose confidence in our reported financial information
and have a negative effect on the trading price of our common
stock.
In
connection with our on-going assessment of the effectiveness of our
internal control over financial reporting, we may discover
“material weaknesses” in our internal controls as
defined in standards established by the Public Company Accounting
Oversight Board (“PCAOB”). A material weakness is a
significant deficiency, or combination of significant deficiencies,
that results in more than a remote likelihood that a material
misstatement of the annual or interim financial statements will not
be prevented or detected. The PCAOB defines “significant
deficiency” as a deficiency that results in more than a
remote likelihood that a misstatement of the financial statements
that is more than inconsequential will not be prevented or
detected.
During
the year ended December 31, 2015, we made improvements in our
internal control and have remediated the deficiencies identified in
2014. In the event that future material weaknesses are
identified, we will attempt to employ qualified personnel and adopt
and implement policies and procedures to address any material
weaknesses we identify. However, the process of designing and
implementing effective internal controls is a continuous effort
that requires us to anticipate and react to changes in our business
and the economic and regulatory environments and to expend
significant resources to maintain a system of internal controls
that is adequate to satisfy our reporting obligations as a public
company.
Any
failure to complete our assessment of our internal control over
financial reporting, to remediate any material weaknesses that we
may identify or to implement new or improved controls, or
difficulties encountered in their implementation, could harm our
operating results, cause us to fail to meet our reporting
obligations or result in material misstatements in our financial
statements. Any such failure could also adversely affect the
results of the periodic management evaluations of our internal
controls and, in the case of a failure to remediate any material
weaknesses that we may identify, would adversely affect the annual
management reports regarding the effectiveness of our internal
control over financial reporting that are required under Section
404 of the Sarbanes-Oxley Act. Inadequate internal controls could
also cause investors to lose confidence in our reported financial
information, which could have a negative effect on the trading
price of our common stock.
The Company’s revenue may become subject to tightened
regulation that may affect the Company’s financial
condition.
Currently
we are not generating any meaningful revenue, which revenue is
currently primarily comprised of technical services relating to the
preparation of subset T Cell and clonality assay platform
technology for treatment of cancers. Nonetheless our revenue may be
subject to the risk of progressive regulatory actions by the PRC
government. From time to time there may also be adverse publicity
relating to the practice of cell therapy treatments in China, which
due to the sensitive and experimental nature of the treatment, may
trigger further governmental scrutiny. Any progressive regulatory
action in China arising out of such scrutiny may adversely affect
the Company’s financial condition or cash flows.
RISKS
RELATED TO OUR STRUCTURE
Our operations are subject to risks associated with emerging
markets.
The
Chinese economy is not well established and is only recently
emerging and growing as a significant market for consumer goods and
services. Accordingly, there is no assurance that the market will
continue to grow. Perceived risks associated with investing in
China, or a general disruption in the development of China’s
markets could materially and adversely affect the business,
operating results and financial condition of the
Company.
A substantial portion of our assets are currently located in the
PRC, and investors may not be able to enforce federal securities
laws or their other legal rights.
A
substantial portion of our assets are located in the PRC. As a
result, it may be difficult for investors in the U.S. to enforce
their legal rights, to effect service of process upon certain of
our directors or officers or to enforce judgments of U.S. courts
predicated upon civil liabilities and criminal penalties against
any of our directors and officers located outside of the
U.S.
The PRC government has the ability to exercise significant
influence and control over our operations in China.
In
recent years, the PRC government has implemented measures for
economic reform, the reduction of state ownership of productive
assets and the establishment of corporate governance practices in
business enterprises. However, many productive assets in China are
still owned by the PRC government. In addition, the government
continues to play a significant role in regulating industrial
development by imposing business regulations. It also exercises
significant control over the country’s economic growth
through the allocation of resources, controlling payment of foreign
currency-denominated obligations, setting monetary policy and
providing preferential treatment to particular industries or
companies.
There
can be no assurance that China’s economic, political or legal
systems will not develop in a way that becomes detrimental to our
business, results of operations and financial condition. Our
activities may be materially and adversely affected by changes in
China’s economic and social conditions and by changes in the
policies of the government, such as measures to control inflation,
changes in the rates or method of taxation and the imposition of
additional restrictions on currency conversion.
Additional factors
that we may experience in connection with having operations in
China that may adversely affect our business and results of
operations include:
●
our
inability to enforce or obtain a remedy under any material
agreements;
●
PRC
restrictions on foreign investment that could impair our ability to
conduct our business or acquire or contract with other entities in
the future;
●
restrictions on
currency exchange that may limit our ability to use cash flow most
effectively or to repatriate our investment;
●
fluctuations in
currency values;
●
cultural,
language and managerial differences that may reduce our overall
performance; and
●
political
instability in China.
Cultural, language and managerial differences may adversely affect
our overall performance.
We have
experienced difficulties in assimilating cultural, language and
managerial differences with our subsidiaries in China. Personnel
issues have developed in consolidating management teams from
different cultural backgrounds. In addition, language translation
issues from time to time have caused miscommunications. These
factors make the management of our operations in China more
difficult. Difficulties in coordinating the efforts of our
U.S.-based management team with our China-based management team may
cause our business, operating results and financial condition to be
materially and adversely affected.
We may not be able to enforce our rights in China given certain
features of its legal and judicial system.
China’s legal
and judicial system may negatively impact foreign investors. The
legal system in China is evolving rapidly, and enforcement of laws
is inconsistent. It may be impossible to obtain swift and equitable
enforcement of laws or enforcement of the judgment of one court by
a court of another jurisdiction. China’s legal system is
based on civil law or written statutes and a decision by one judge
does not set a legal precedent that must be followed by judges in
other cases. In addition, the interpretation of Chinese laws may
vary to reflect domestic political changes.
Since a
significant portion of our operations are presently based in China,
service of process on our business and officers may be difficult to
effect within the United States. Also, some of our assets are
located outside the United States and any judgment obtained in the
United States against us may not be enforceable outside the United
States.
There
are substantial uncertainties regarding the interpretation and
application to our business of PRC laws and regulations, since many
of the rules and regulations that companies face in China are not
made public. The effectiveness of newly enacted laws, regulations
or amendments may be delayed, resulting in detrimental reliance by
foreign investors. New laws and regulations that apply to future
businesses may be applied retroactively to existing businesses. We
cannot predict what effect the interpretations of existing or new
PRC laws or regulations may have on our
business.
Our operations in China are subject to government regulation that
limit or prohibit direct foreign investment, which may limit our
ability to control operations based in China.
The PRC
government has imposed regulations in various industries, including
medical research and the stem cell industry, that limit foreign
investors’ equity ownership or prohibit foreign investments
altogether in companies that operate in such industries. We are
currently structured as a U.S. corporation (Delaware) with
subsidiaries and controlled entities in China. As a result of these
regulations and the manner in which they may be applied or
enforced, our ability to control our existing operations based in
China may be limited or restricted.
If the
relevant Chinese authorities find us or any business combination to
be in violation of any laws or regulations, they would have broad
discretion in dealing with such violation, including, without
limitation: (i) levying fines; (ii) revoking our business and other
licenses; (iii) requiring that we restructure our ownership or
operations; and (iv) requiring that we discontinue any portion or
all of our business.
We may suffer losses if we cannot utilize our assets in
China.
The
Company’s Shanghai and Wuxi laboratory facilities were
originally intended for stem cell research and development, but has
been equipped to provide comprehensive cell manufacturing,
collection, processing and storage capabilities to provide cells
for clinical trials. If the Company does not determine to renew the
lease due to limitations on its utility under the new regulatory
initiatives in China or otherwise, the Company may incur certain
expenses in connection with returning the premises to the landlord.
Management believes it will be able to renew all leases without
difficulty.
Restrictions on currency exchange may limit our ability to utilize
our cash flow effectively.
Our
interests in China will be subject to China’s rules and
regulations on currency conversion. In particular, the initial
capitalization and operating expenses of the VIE (CBMG Shanghai)
are funded by our WFOE, Cellular Biomedicine Group Ltd. (Wuxi). In
China, the State Administration for Foreign Exchange (the
“SAFE”), regulates the conversion of the Chinese
Renminbi into foreign currencies and the conversion of foreign
currencies into Chinese Renminbi. Foreign investment enterprises
are allowed to open currency accounts including a “basic
account” and “capital account.” However,
conversion of currency in the “capital account,”
including capital items such as direct investments, loans, and
securities, require approval of the SAFE even though according to the
Notice of the State Administration
of Foreign Exchange on Reforming the Administration of the
Settlement of Foreign Exchange Capital of Foreign-invested
Enterprise promulgated on April 8, 2015, or the SAFE Notice
19, and Notice of the State
Administration of Foreign Exchange on Reforming and Regulating the
Policies for the Administration of Settlement of Foreign Exchange
under Capital Accounts promulgated on June 9, 2016, or the SAFE
Notice 16, foreign-invested enterprises are able to settle
foreign exchange capital at their discretion, Chinese banks
restricts foreign currency conversion for fear of “hot
money” going into China and may continue to limit our ability
to channel funds to the VIE entities for their operation. There can
be no assurance that the PRC regulatory authorities will not impose
further restrictions on the convertibility of the Chinese currency.
Future restrictions on currency exchanges may limit our ability to
use our cash flow for the distribution of dividends to our
stockholders or to fund operations we may have outside of China,
which could materially adversely affect our business and operating
results.
Fluctuations in the value of the Renminbi relative to the U.S.
dollar could affect our operating results.
We
prepare our financial statements in U.S. dollars, while our
underlying businesses operate in two currencies, U.S. dollars and
Chinese Renminbi. It is anticipated that our Chinese operations
will conduct their operations primarily in Renminbi and our U.S.
operations will conduct their operations in dollars. At the present
time, we do not expect to have significant cross currency
transactions that will be at risk to foreign currency exchange
rates. Nevertheless, the conversion of financial information using
a functional currency of Renminbi will be subject to risks related
to foreign currency exchange rate fluctuations. The value of
Renminbi against the U.S. dollar and other currencies may fluctuate
and is affected by, among other things, changes in China’s
political and economic conditions and supply and demand in local
markets. As we have significant operations in China, and will rely
principally on revenues earned in China, any significant
revaluation of the Renminbi could materially and adversely affect
our financial results. For example, to the extent that we need to
convert U.S. dollars we receive from an offering of our securities
into Renminbi for our operations, appreciation of the Renminbi
against the U.S. dollar could have a material adverse effect on our
business, financial condition and results of
operations.
Some of the laws and regulations governing our business in China
are vague and subject to risks of interpretation.
Some of
the PRC laws and regulations governing our business operations in
China are vague and their official interpretation and enforcement
may involve substantial uncertainty. These include, but are not
limited to, laws and regulations governing our business and the
enforcement and performance of our contractual arrangements in the
event of the imposition of statutory liens, death, bankruptcy and
criminal proceedings. Despite their uncertainty, we will be
required to comply.
New
laws and regulations that affect existing and proposed businesses
may be applied retroactively. Accordingly, the effectiveness of
newly enacted laws, regulations or amendments may not be clear. We
cannot predict what effect the interpretation of existing or new
PRC laws or regulations may have on our business.
The PRC government does not permit direct foreign investment in
stem cell research and development businesses. Accordingly, we
operate these businesses through local companies with which we have
contractual relationships but in which we do not have direct equity
ownership.
PRC
regulations prevent foreign companies from directly engaging in
stem cell-related research, development and commercial applications
in China. Therefore, to perform these activities, we conduct much
of our biopharmaceutical business operations in China through a
domestic variable interest entity, or VIE, a Chinese domestic
company controlled by the Chinese employees of the Company. Our
contractual arrangements may not be as effective in providing
control over these entities as direct ownership. For example, the
VIE could fail to take actions required for our business or fail to
conduct business in the manner we desire despite their contractual
obligation to do so. These companies are able to transact business
with parties not affiliated with us. If these companies fail to
perform under their agreements with us, we may have to rely on
legal remedies under PRC law, which may not be effective. In
addition, we cannot be certain that the individual equity owners of
the VIE would always act in our best interests, especially if they
have no other relationship with us.
Although other
foreign companies have used VIE structures similar to ours and such
arrangements are not uncommon in connection with business
operations of foreign companies in China in industry sectors in
which foreign direct investments are limited or prohibited,
recently there has been greater scrutiny by the business community
of the VIE structure and, additionally, the application of a VIE
structure to control companies in a sector in which foreign direct
investment is specifically prohibited carries increased
risks.
In
addition, the Ministry of Commerce (“MOFCOM”),
promulgated the Rules of Ministry
of Commerce on Implementation of Security Review System of Mergers
and Acquisitions of Domestic Enterprises by Foreign
Investors in August 2011, or the MOFCOM Security Review
Rules, to implement the Notice of
the General Office of the State Council on Establishing the
Security Review System for Mergers and Acquisitions of Domestic
Enterprises by Foreign Investors promulgated on February 3,
2011, or Circular No. 6. The MOFCOM Security Review Rules came into
effect on September 1, 2011 and replaced the Interim Provisions of the Ministry of Commerce
on Matters Relating to the Implementation of the Security Review
System for Mergers and Acquisitions of Domestic Enterprises by
Foreign Investors promulgated by MOFCOM in March 2011.
According to these circulars and rules, a security review is
required for mergers and acquisitions by foreign investors having
“national defense and
security” concerns and mergers and acquisitions by
which foreign investors may acquire the “de facto control” of
domestic enterprises having “national security”
concerns. In addition, when deciding whether a specific merger or
acquisition of a domestic enterprise by foreign investors is
subject to the security review, the MOFCOM will look into the
substance and actual impact of the transaction. The MOFCOM Security
Review Rules further prohibit foreign investors from bypassing the
security review requirement by structuring transactions through
proxies, trusts, indirect investments, leases, loans, control
through contractual arrangements or offshore transactions. There is
no explicit provision or official interpretation stating that our
business falls into the scope subject to the security review, and
there is no requirement for foreign investors in those mergers and
acquisitions transactions already completed prior to the
promulgation of Circular No. 6 to submit such transactions to
MOFCOM for security review. The enactment of the MOFCOM National
Security Review Rules specifically prohibits circumvention of the
rules through VIE arrangement in the area of foreign investment in
business of national security concern. Although we believe that our
business, judging from its scale, should not cause any concern for
national security review at its current state, there is no
assurance that MOFCOM would not apply the same concept of
anti-circumvention in the future to foreign investment in
prohibited areas through VIE structure, the same way that our
investment in China was structured.
Our relationship with our controlled VIE entity, CBMG Shanghai,
through the VIE agreements, is subject to various operational and
legal risks.
Management
believes the holders of the VIE’s registered capital, Messrs.
Chen Mingzhe and Lu Junfeng, have no interest in acting contrary to
the VIE agreements. However, if Messrs. Chen or Lu as
shareholders of the VIE entity were to reduce or eliminate their
ownership of the registered capital of the VIE entity, their
interests may diverge from that of CBMG and they may seek to act in
a manner contrary to the VIE agreements (for example by controlling
the VIE entity in such a way that is inconsistent with the
directives of CBMG management and the board; or causing non-payment
by the VIE entity of services fees). If such
circumstances were to occur the WFOE would have to assert control
rights through the powers of attorney, pledges and other VIE
agreements, which would require legal action through the PRC
judicial system. We believe based on the advice of local
counsel that the VIE agreements are valid and in compliance with
PRC laws presently in effect. However, there is a risk that the
enforcement of these agreements may involve more extensive
procedures and costs to enforce, in comparison to direct equity
ownership of the VIE entity. Notwithstanding the foregoing, if the
applicable PRC laws were to change or are interpreted by
authorities in the future in a manner which challenges or renders
the VIE agreements ineffective, the WFOE’s ability to control
and obtain all benefits (economic or otherwise) of ownership of the
VIE entity could be impaired or eliminated. In the
event of such future changes or new interpretations of PRC law, in
an effort to substantially preserve our rights, we may have to
either amend our VIE agreements or enter into alternative
arrangements which comply with PRC laws as interpreted and then in
effect.
Failure to comply with the U.S. Foreign Corrupt Practices Act could
subject us to penalties and other adverse
consequences.
We are
subject to the U.S. Foreign Corrupt Practices Act, which generally
prohibits U.S. companies from engaging in bribery or other
prohibited payments to foreign officials for the purpose of
obtaining or retaining business. Foreign companies, including some
that may compete with us, are not subject to these prohibitions.
Corruption, extortion, bribery, pay-offs, theft and other
fraudulent practices occur from time-to-time in the PRC. There can
be no assurance, however, that our employees or other agents will
not engage in such conduct for which we might be held responsible.
If our employees or other agents are found to have engaged in such
practices, we could suffer severe penalties and other consequences
that may have a material adverse effect on our business, financial
condition and results of operations.
If we make share compensation grants to persons who are PRC
citizens, they may be required to register with SAFE. We may also
face regulatory uncertainties that could restrict our ability to
adopt share compensation plans for our directors and employees and
other parties under PRC laws.
On
April 6, 2007, SAFE issued the “Operating Procedures for
Administration of Domestic Individuals Participating in the
Employee Stock Ownership Plan or Stock Option Plan of An Overseas
Listed Company, also known as Circular 78. On February 15, 2012, SAFE
promulgated the Circular on Relevant Issues Concerning Foreign
Exchange Administration for Domestic Individuals Participating in
an Employees Share Incentive Plan of an Overseas-Listed Company,
often known as Circular 7. Circular 7 has superseded Circular 78.
Under Circular 7, PRC resident individuals who participate in a
share incentive plan of an overseas listed company are required to
register with SAFE and complete certain other procedures. All such
participants need to retain a PRC agent through PRC subsidiary to
handle issues like foreign exchange registration, account opening,
funds transfer and remittance. Circular 7 further requires that an
offshore agent should also be designated to handle matters in
connection with the exercise or sale of share awards and proceeds
transferring for the share incentive plan participants. We have
obtained the SAFE approvals under Circular 7 for one PRC
subsidiary. If we or our PRC employees who have been granted stock
options fail to comply with these regulations, we or our PRC
employees who have been granted stock options may be subject to
fines and legal sanctions and will be unable to grant share
compensation to our PRC employees. In that case, our ability to
compensate our employees and directors through share compensation
would be hindered and our business operations may be adversely
affected.
The labor contract law and its implementation regulations may
increase our operating expenses and may materially and adversely
affect our business, financial condition and results of
operations.
Substantial
uncertainty of the PRC Labor Contract Law, or Labor Contract Law,
and the Implementation Regulation for the PRC Labor Contract Law,
or Implementation Regulation, remains as to their potential impact
on our business, financial condition and results of operations. The
implementation of the Labor Contract Law and the Implementation
Regulation may increase our operating expenses, in particular our
human resources costs and our administrative expenses. In addition,
as the interpretation and implementation of these regulations are
still evolving, we cannot assure you that our employment practices
will at all times be deemed to be in full compliance with the law.
In the event that we decide to significantly modify our employment
or labor policy or practice, or reduce the number of our sales
professionals, the Labor Contract Law and the Implementation
Regulation may limit our ability to effectuate the modifications or
changes in the manner that we believe to be most cost-efficient or
otherwise desirable, which could materially and adversely affect
our business, financial condition and results of operations. If we
are subject to severe penalties or incur significant liabilities in
connection with labor disputes or investigations, our business and
results of operations may be adversely affected.
If relations between the United States and China worsen, our stock
price may decrease and we may have difficulty accessing the U.S.
capital markets.
At
various times during recent years, the United States and China have
had disagreements over trade, economic and other policy issues.
Controversies may arise in the future between these two countries.
Any political or trade controversies between the United States and
China could adversely affect the market price of our common stock
and our and our clients' ability to access U.S. capital markets.
PRC regulations of loans to PRC entities and direct investment in
PRC entities by offshore holding companies may delay or prevent us
from using the proceeds of this offering to make loans or
additional capital contributions to our PRC
subsidiary.
We may
transfer funds to our PRC subsidiary or finance our PRC subsidiary
by means of shareholder loans or capital contributions. Any loans
from us to our PRC subsidiary, which is a foreign-invested
enterprise, is subject to a quota based on the statutory formulas
and there are two alternative applicable quotas: the difference
between the registered capital and total investment of the PRC
subsidiary; certain times of the net asset value of PRC subsidiary
(currently up to twice of the net assets value), and shall be
registered with the State Administration of Foreign Exchange, or
SAFE, or its local counterparts. Any capital contributions we make
to our PRC subsidiary shall be approved by or registered with (as
the case may be) the Ministry of Commerce or its local
counterparts. We may not be able to obtain these government
registrations or approvals on a timely basis, if at all. If we fail
to receive such registrations or approvals, our ability to provide
loans or capital contributions to our PRC subsidiary in a timely
manner may be negatively affected, which could materially and
adversely affect our liquidity and our ability to fund and expand
our business.
In
addition, registered capital of a foreign-invested company settled
in RMB converted from foreign currencies may only be used within
the business scope approved by the applicable governmental
authority. Foreign-invested companies may not change how they use
such capital without SAFE’s approval, and may not in any case
use such capital to repay RMB loans if proceeds of such loans have
not been utilized. Violations of these regulations may result in
severe penalties. These regulations may significantly limit our
ability to transfer the net proceeds from offshore offering and
subsequent offerings or financings to our PRC subsidiary, which may
adversely affect our liquidity and our ability to fund and expand
our business in China.
We may be subject to penalties, including restriction on our
ability to inject capital into our PRC subsidiary and our PRC
subsidiary’s ability to distribute profits to us, if our PRC
resident shareholders beneficial owners fail to comply with
relevant PRC foreign exchange rules.
The
Notice on Relevant Issues Concerning Foreign Exchange
Administration for PRC Residents to Engage in Financing and Inbound
Investment via Offshore Special Purpose Vehicles, often known as
Circular 75, was issued by SAFE in 2005. Circular 75 requires PRC
residents to register with the local SAFE branch in connection with
their establishment or control of any offshore special purpose
vehicle for the purpose of overseas equity financing involving a
roundtrip investment whereby the offshore special purpose vehicle
acquires or controls onshore assets or equity interests held by the
PRC residents. On July 4, 2014, SAFE issued the Notice on Relevant
Issues Concerning Foreign Exchange Administration for PRC Residents
to Engage in Outbound Investment and Financing and Inbound
Investment via Special Purpose Vehicles, or Circular 37, which has
superseded Circular 75. Under Circular 37 and other relevant
foreign exchange regulations, PRC residents who make, or have made,
prior to the implementation of these foreign exchange regulations,
direct or indirect investments in offshore companies are required
to register those investments with SAFE. In addition, any PRC
resident who is a direct or indirect shareholder of an offshore
company is also required to file or update the registration with
SAFE, with respect to that offshore company for any material change
involving its round-trip investment, capital variation, such as an
increase or decrease in capital, transfer or swap of shares,
merger, division, long-term equity or debt investment or the
creation of any security interest. If any PRC shareholder fails to
make the required registration or update the registration, the PRC
subsidiary of that offshore company may be prohibited from
distributing its profits and the proceeds from any reduction in
capital, share transfer or liquidation to that offshore company,
and that offshore company may also be prohibited from injecting
additional capital into its PRC subsidiary. Moreover, failure to
comply with the foreign exchange registration requirements
described above could result in liability under PRC laws for
evasion of applicable foreign exchange restrictions.
We cannot provide any assurance that all of
our shareholders and beneficial owners who are PRC residents have
fully complied or will obtain or update any applicable
registrations or have fully complied or will fully comply with
other requirements required by Circular 37 or other related rules
in a timely manner. The failure or inability of our shareholders
resident in China to comply with the registration requirements set
forth therein may subject them to fines and legal sanctions and may
also limit our ability to contribute additional capital into our
PRC subsidiaries, limit our PRC subsidiaries’ ability to
distribute profits and other proceeds to our
company or otherwise adversely affect
our business.
We and/or our Hong Kong subsidiary may be classified as a
“PRC resident enterprise” for PRC enterprise income tax
purposes. Such classification would likely result in unfavorable
tax consequences to us and our non-PRC shareholders and have a
material adverse effect on our results of operations and the value
of your investment.
The
Enterprise Income Tax Law provides that an enterprise established
outside China whose “de facto management body” is
located in China is considered a “PRC resident
enterprise” and will generally be subject to the uniform 25%
enterprise income tax on its global income. Under the
implementation rules of the Enterprise Income Tax Law, “de
facto management body” is defined as the organizational body
which effectively manages and controls the production and business
operation, personnel, accounting, properties and other aspects of
operations of an enterprise.”
Pursuant to the
Notice Regarding the Determination of Chinese-Controlled Offshore
Incorporated Enterprises as PRC Tax Resident Enterprises on the
Basis of De Facto Management Bodies, issued by the State
Administration of Taxation in 2009, a foreign enterprise controlled
by PRC enterprises or PRC enterprise groups is considered a PRC
resident enterprise if all of the following conditions are met:
(i) the senior management and core management departments in
charge of daily operations are located mainly within the PRC;
(ii) financial and human resources decisions are subject to
determination or approval by persons or bodies in the PRC;
(iii) major assets, accounting books, company seals and
minutes and files of board and shareholders’ meetings are
located or kept within the PRC; and (iv) at least half of the
enterprise’s directors with voting rights or senior
management reside within the PRC. Although the notice states that
these standards only apply to offshore enterprises that are
controlled by PRC enterprises or PRC enterprise groups, such
standards may reflect the general view of the State Administration
of Taxation in determining the tax residence of foreign
enterprises.
We
believe that neither our company nor our Hong Kong subsidiary is a
PRC resident enterprise because neither our company nor our Hong
Kong subsidiary meets all of the conditions enumerated. For
example, board and shareholders’ resolutions of our company
and our Hong Kong subsidiary are adopted in Hong Kong and the
minutes and related files are kept in Hong Kong. However, if the
PRC tax authorities were to disagree with our position, our company
and/or our Hong Kong subsidiary may be subject to PRC enterprise
income tax reporting obligations and to a 25% enterprise income tax
on our global taxable income, except for our income from dividends
received from our PRC subsidiary, which may be exempt from PRC tax.
If we and/or our Hong Kong subsidiary are treated as a PRC resident
enterprise, the 25% enterprise income tax may adversely affect our
ability to satisfy any of our cash needs.
In
addition, if we were to be classified as a PRC “resident
enterprise” for PRC enterprise income tax purpose, dividends
we pay to our non-PRC enterprise shareholders and gains derived by
our non-PRC shareholders from the sale of our shares and ADSs may
be become subject to a 10% PRC withholding tax. In addition, future
guidance may extend the withholding tax to dividends we pay to our
non-PRC individual shareholders and gains derived by such
shareholders from transferring our shares and ADSs. In addition to
the uncertainty in how the new “resident enterprise”
classification could apply, it is also possible that the rules may
change in the future, possibly with retroactive effect. If PRC
income tax were imposed on gains realized through the transfer of
our ADSs or ordinary shares or on dividends paid to our
non-resident shareholders, the value of your investment in our ADSs
or ordinary shares may be materially and adversely affected.
Any limitation on the ability of our PRC subsidiary to make
payments to us, or the tax implications of making payments to us,
could have a material adverse effect on our ability to conduct our
business or our financial condition.
We are
a holding company, and we rely principally on dividends and other
distributions from our PRC subsidiary for our cash needs, including
the funds necessary to pay dividends to our shareholders or service
any debt we may incur. Current PRC regulations permit our PRC
subsidiary to pay dividends only out of its accumulated profits, if
any, determined in accordance with PRC accounting standards and
regulations. In addition, our PRC subsidiary is required to set
aside at least 10% of its after tax profits each year, if any, to
fund certain statutory reserve funds until the aggregate amount of
such reserve funds reaches 50% of its registered capital. Apart
from these reserves, our PRC subsidiary may allocate a
discretionary portion of its after-tax profits to staff welfare and
bonus funds at its discretion. These reserves and funds are not
distributable as cash dividends. Furthermore, if our PRC subsidiary
incurs debt, the debt instruments may restrict its ability to pay
dividends or make other payments to us. We cannot assure you that
our PRC subsidiary will generate sufficient earnings and cash flows
in the near future to pay dividends or otherwise distribute
sufficient funds to enable us to meet our obligations, pay interest
and expenses or declare dividends.
Distributions made
by PRC companies to their offshore parents are generally subject to
a 10% withholding tax under the Enterprise Income Tax Law. Pursuant
to the Enterprise Income Tax Law and the Arrangement between the
Mainland of China and the Hong Kong Special Administrative Region
for the Avoidance of Double Taxation and the Prevention of Fiscal
Evasion with respect to Taxes on Income, the withholding tax rate
on dividends paid by our PRC subsidiary to our Hong Kong subsidiary
would generally be reduced to 5%, provided that our Hong Kong
subsidiary is the beneficial owner of the PRC sourced income. Our
PRC subsidiary has not obtained approval for a withholding tax rate
of 5% from the local tax authority and does not plan to obtain such
approval in the near future as we have not achieved
profitability. However, the Notice on How to Understand and
Determine the Beneficial Owners in a Tax Agreement, also known as
Circular 601, promulgated by the State Administration of Taxation
in 2009, provides guidance for determining whether a resident of a
contracting state is the “beneficial owner” of an item
of income under China’s tax treaties and similar
arrangements. According to Circular 601, a beneficial owner
generally must be engaged in substantive business activities. An
agent or conduit company will not be regarded as a beneficial owner
and, therefore, will not qualify for treaty benefits. For this
purpose, a conduit company is a company that is set up for the
purpose of avoiding or reducing taxes or transferring or
accumulating profits. Although our PRC subsidiary is wholly owned
by our Hong Kong subsidiary, we will not be able to enjoy the 5%
withholding tax rate with respect to any dividends or distributions
made by our PRC subsidiary to its parent company in Hong Kong if
our Hong Kong subsidiary is regarded as a “conduit
company.”
In
addition, if CBMG HK were deemed to be a PRC resident enterprise,
then any dividends payable by CBMG HK to CBMG Delaware Corporation
may become subject to PRC dividend withholding
tax.
A new
China taxation rule about the “beneficial owner” in a
tax agreement will be effective from April 1, 2018 which will
supersede Circular 601 and could affect the determination of
whether a resident of a contracting state is the “beneficial
owner” of an item of income under China’s tax treaties
and similar arrangements.
Restrictions on the remittance of RMB into and out of China and
governmental control of currency conversion may limit our ability
to pay dividends and other obligations, and affect the value of
your investment.
The PRC
government imposes controls on the convertibility of the RMB into
foreign currencies and the remittance of currency out of China. We
receive substantially all of our revenues in RMB and substantially
all of our cash inflows and outflows are denominated in RMB. Under
our current corporate structure, our revenues are primarily derived
from dividend payments from our subsidiary in China after it
receives payments from the VIE under various service and other
contractual arrangements. We may convert a portion of our revenues
into other currencies to meet our foreign currency obligations,
such as payments of dividends declared in respect of our ordinary
shares, if any. Shortages in the availability of foreign currency
may restrict the ability of our PRC subsidiary to remit sufficient
foreign currency to pay dividends or other payments to us, or
otherwise satisfy its foreign currency denominated
obligations.
Under
existing PRC foreign exchange regulations, payments of current
account items, including profit distributions, interest payments
and trade and service-related foreign exchange transactions, can be
made in foreign currencies without prior SAFE approval as long as
certain routine procedural requirements are fulfilled. Therefore,
our PRC subsidiary is allowed to pay dividends in foreign
currencies to us without prior SAFE approval by following certain
routine procedural requirements. However, approval from or
registration with competent government authorities is required
where the RMB is to be converted into foreign currency and remitted
out of China to pay capital expenses such as the repayment of loans
denominated in foreign currencies. The PRC government may at its
discretion restrict access to foreign currencies for current
account transactions in the future. If the foreign exchange control
system prevents us from obtaining sufficient foreign currencies to
satisfy our foreign currency demands, we may not be able to pay
dividends in foreign currencies to our shareholders, including the
U.S. shareholders.
Our financial condition and results of operations could be
materially and adversely affected if recent value added tax reforms
in the PRC become unfavorable to our PRC subsidiary or
VIE.
In
2012, China introduced a value added tax, or VAT, to replace the
previous 5% business tax. Our PRC subsidiary and the VIE have been
subject to VAT at a base rate of 6% since September 1, 2012.
The VIE’s subsidiary has been subject to VAT at a base rate
of 6% since July 1, 2013. Our financial condition and results
of operations could be materially and adversely affected if the
interpretation and enforcement of these tax rules become materially
unfavorable to our PRC subsidiary and VIE.
Failure to comply with PRC regulations regarding the registration
requirements for stock ownership plans or stock option plans may
subject PRC plan participants or us to fines and other legal or
administrative sanctions.
Under
SAFE regulations, PRC residents who participate in an employee
stock ownership plan or stock option plan in an overseas publicly
listed company are required to register with SAFE or its local
branch and complete certain other procedures. Participants of a
stock incentive plan who are PRC residents must retain a qualified
PRC agent, which could be a PRC subsidiary of such overseas
publicly listed company, to conduct the SAFE registration and other
procedures with respect to the stock incentive plan on behalf of
these participants. Such participants must also retain an overseas
entrusted institution to handle matters in connection with their
exercise or sale of stock options. In addition, the PRC agent is
required to amend the SAFE registration with respect to the stock
incentive plan if there is any material change to the stock
incentive plan, the PRC agent or the overseas entrusted institution
or other material changes.
We and
our PRC resident employees who participate in our share incentive
plans are subject to these regulations as our company is publicly
listed in the United States. We have obtained the SAFE approvals
regarding our PRC resident employees participating in our share
incentive plans. If we or any our PRC resident option grantees fail
to follow the compliance with above regulations, we or our PRC
resident option grantees may be subject to fines and other legal or
administrative sanctions.
Fluctuation in the value of the RMB may have a material adverse
effect on the value of your investment.
The
value of the RMB against the U.S. dollar and other currencies is
affected by changes in China’s political and economic
conditions and China’s foreign exchange policies, among other
things. On July 21, 2005, the PRC government changed its
decades-old policy of pegging the value of the RMB to the U.S.
dollar, and the RMB appreciated more than 20% against the U.S.
dollar over the following three years. Between July 2008 and June
2010, this appreciation halted and the exchange rate between the
RMB and the U.S. dollar remained within a narrow band. The PRC
government has allowed the RMB to appreciate slowly against the
U.S. dollar again, and it has appreciated more than 10% since June
2010. It is difficult to predict how market forces or PRC or U.S.
government policy may impact the exchange rate between the RMB and
the U.S. dollar in the future. In addition, there remains
significant international pressure on the PRC government to adopt a
substantial liberalization of its currency policy, which could
result in further appreciation in the value of the RMB against the
U.S. dollar. In 2015, due to the slow-down of China economic growth
rate and environment, RMB depreciated against the U.S. dollar from
third quarter.
Our
revenues and costs are mostly denominated in RMB, and a significant
portion of our financial assets are also denominated in RMB,
whereas our reporting currency is the U.S. dollar. Any significant
depreciation of the RMB may materially and adversely affect our
revenues, earnings and financial position as reported in U.S.
dollars. To the extent that we need to convert U.S. dollars we
received from this offering into RMB for our operations,
appreciation of the RMB against the U.S. dollar would have an
adverse effect on the RMB amount we would receive from the
conversion. Conversely, if we decide to convert our RMB into U.S.
dollars for the purpose of making payments for dividends on our
ordinary shares or for other business purposes, appreciation of the
U.S. dollar against the RMB would have a negative effect on the
U.S. dollar amount available to us.
PRC laws and regulations establish more complex procedures for some
acquisitions of Chinese companies by foreign investors, which could
make it more difficult for us to pursue growth through acquisitions
in China.
A
number of PRC laws and regulations, including the Regulations on
Mergers and Acquisitions of Domestic Enterprises by Foreign
Investors adopted by six PRC regulatory agencies in 2006, or the
M&A Rules, the Anti-monopoly Law, and the Rules of
Ministry of Commerce on Implementation of Security Review System of
Mergers and Acquisitions of Domestic Enterprises by Foreign
Investors promulgated by the Ministry of Commerce in August
2011, or the Security Review Rules, have established procedures and
requirements that are expected to make merger and acquisition
activities in China by foreign investors more time consuming and
complex. These include requirements in some instances that the
Ministry of Commerce be notified in advance of any change of
control transaction in which a foreign investor takes control of a
PRC domestic enterprise, or that the approval from the Ministry of
Commerce be obtained in circumstances where overseas companies
established or controlled by PRC enterprises or residents acquire
affiliated domestic companies. PRC laws and regulations also
require certain merger and acquisition transactions to be subject
to merger control review or security review.
The
Security Review Rules were formulated to implement the Notice of
the General Office of the State Council on Establishing the
Security Review System for Mergers and Acquisitions of Domestic
Enterprises by Foreign Investors, also known as Circular 6,
which was promulgated in 2011. Under these rules, a security review
is required for mergers and acquisitions by foreign investors
having “national defense and security” concerns and
mergers and acquisitions by which foreign investors may acquire the
“de facto control” of domestic enterprises have
“national security” concerns. In addition, when
deciding whether a specific merger or acquisition of a domestic
enterprise by foreign investors is subject to the security review,
the Ministry of Commerce will look into the substance and actual
impact of the transaction. The Security Review Rules further
prohibits foreign investors from bypassing the security review
requirement by structuring transactions through proxies, trusts,
indirect investments, leases, loans, control through contractual
arrangements or offshore transactions.
There
is no requirement for foreign investors in those mergers and
acquisitions transactions already completed prior to the
promulgation of Circular 6 to submit such transactions to the
Ministry of Commerce for security review. As we have already
obtained the “de facto control” over our affiliated PRC
entities prior to the effectiveness of these rules, we do not
believe we are required to submit our existing contractual
arrangements to the Ministry of Commerce for security
review.
However, as these rules are relatively new and
there is a lack of clear statutory interpretation on the
implementation of the same, there is no assurance that the Ministry
of Commerce will not apply these national security review-related
rules to the acquisition of equity interest in our PRC subsidiary.
If we are found to be in violation of the Security Review Rules and
other PRC laws and regulations with respect to the merger and
acquisition activities in China, or fail to obtain any of the
required approvals, the relevant regulatory authorities would have
broad discretion in dealing with such violation, including levying
fines, confiscating our income, revoking our PRC subsidiary’s
business or operating licenses, requiring us to restructure or
unwind the relevant ownership structure or operations. Any of these
actions could cause significant disruption to our business
operations and may materially and adversely affect our business,
financial condition and results of operations. Further, if the
business of any target company that we plan to acquire falls into
the ambit of security review, we may not be able to successfully
acquire such company either by equity or asset acquisition, capital
contribution or through any contractual arrangement. We may grow
our business in part by acquiring other companies operating in our
industry. Complying with the requirements of the relevant
regulations to complete such transactions could be time consuming,
and any required approval processes, including approval from the
Ministry of Commerce, may delay or inhibit our ability to complete
such transactions, which could affect our ability to expand our
business or maintain our market
share.
On July
30, 2017, MOFCOM issued the Interim Measures on Filing
Administration of Establishment and Changes of Foreign-Invested
Enterprises (2017 Revision) which came into force as of July 30,
2017. It is stipulated in the Interim Measures that the
transformation of a non-foreign invested enterprise into a foreign
invested enterprise through M&A, merger by absorption, foreign
investor’s strategic investment into non-foreign invested
listed company, etc. would no longer be subject to MOFCOM approval,
but instead would only need to undergo the simplified filing
procedures with MOFCOM, in case the business of the target
enterprise does not fall into the foreign investment negative list.
But, if any business of the target enterprise falls into the
foreign investment negative list, the complex procedures for an
acquisition of the target enterprise by foreign investors would be
still applicable.
The heightened scrutiny over acquisition transactions by the PRC
tax authorities may have a negative impact on our business
operations, our acquisition or restructuring strategy or the value
of your investment in us.
Pursuant to the
Notice on Strengthening Administration of Enterprise Income Tax for
Share Transfers by Non-PRC Resident Enterprises, or Circular 698,
issued by the State Administration of Taxation in
December 2009 with retroactive effect from January 1,
2008, where a non-PRC resident enterprise transfers the equity
interests of a PRC resident enterprise indirectly by disposition of
the equity interests of an overseas non-public holding company, or
an Indirect Transfer, and such overseas holding company is located
in a tax jurisdiction that: (i) has an effective tax rate of
less than 12.5% or (ii) does not impose income tax on
foreign income of its residents, the non-PRC resident
enterprise, being the transferor, must report to the competent tax
authority of the PRC resident enterprise this Indirect Transfer and
may be subject to PRC enterprise income tax of up to 10% of the
gains derived from the Indirect Transfer in certain
circumstances.
To
clarify the issues related to Circular 698, the State
Administration of Taxation released the Announcement of the State
Administration of Taxation on Several Issues Relating to the
Administration of Income Tax on Non-resident Enterprises in 2011,
known as Notice 24, and the Announcement on Issues Related to
Applications of Special Tax Treatment for Equity Transfer by
Non-resident Enterprises in 2013.
On
February 3, 2015, the State Administration of Taxation issued the
Announcement on Several Issues Concerning the Enterprise Income Tax
on Indirect Property Transfers by Non-PRC Resident Enterprises, or
Notice 7. Notice 7 introduces a new tax regime that is
significantly different from that under Circular 698. It superseded
the previous tax rules in relation to the offshore indirect equity
transfer, including those under Circular 698 as described above. It
extends the tax jurisdiction of State Administration of Taxation to
capture not only the Indirect Transfer but also the transactions
involving indirect transfer of (i) real properties in China and
(ii) assets of an “establishment or place” situated in
China, by a non-PRC resident enterprise through a disposition of
equity interests in an overseas holding company.
However, Notice 7
also brings uncertainties to the parties of the offshore indirect
transfers as the transferee and the transferor have to make
self-assessment on whether the transactions should be subject to
the corporate income tax and file or withhold the corporate income
tax accordingly. In addition, the PRC tax authorities have
discretion under Notice 7 to adjust the taxable capital gains based
on the difference between the fair value of the transferred equity
interests and the investment cost. We may pursue acquisitions in
the future that may involve complex corporate structures. If we are
considered as a non-PRC resident enterprise under the EIT Law and
if the PRC tax authorities make adjustments to the taxable income
of the transactions under Notice 7, our income tax expenses
associated with such potential acquisitions will be increased,
which may have an adverse effect on our financial condition and
results of operations.
We face certain risks relating to the real properties that we
lease.
We
primarily lease office and manufacturing space from third parties
for our operations in China. Any defects in lessors’ title to
the leased properties may disrupt our use of our offices, which may
in turn adversely affect our business operations. For example,
certain buildings and the underlying land are not allowed to be
used for industrial or commercial purposes without relevant
authorities’ approval, and the lease of such buildings to
companies like us may subject the lessor to pay premium fees to the
PRC government. We cannot assure you that the lessor has obtained
all or any of approvals from the relevant governmental authorities.
In addition, some of our lessors have not provided us with
documentation evidencing their title to the relevant leased
properties. We cannot assure you that title to these properties we
currently lease will not be challenged. In addition, we have not
registered any of our lease agreements with relevant PRC
governmental authorities as required by PRC law, and although
failure to do so does not in itself invalidate the leases, we may
not be able to defend these leases against bona fide third
parties.
As of
the date of this filing, we are not aware of any actions, claims or
investigations being contemplated by government authorities with
respect to the defects in our leased real properties or any
challenges by third parties to our use of these properties.
However, if third parties who purport to be property owners or
beneficiaries of the mortgaged properties challenge our right to
use the leased properties, we may not be able to protect our
leasehold interest and may be ordered to vacate the affected
premises, which could in turn materially and adversely affect our
business and operating results.
Our significant deposits in certain banks in China may be at risk
if these banks go bankrupt or otherwise do not have the liquidity
to pay us during our deposit period.
As of
December 31, 2017, we had approximately $22 million in cash and
bank deposits, such as time deposits, with large domestic banks in
China. Our remaining cash, cash equivalents and short-term
investments were held by financial institutions in the United
States and Hong Kong. The terms of these deposits are, in general,
up to twelve months. Historically, deposits in Chinese banks were
viewed as secure due to the state policy on protecting
depositors’ interests. However, the new Bankruptcy Law that
came into effect in 2007 contains an article expressly stating that
the State Council may promulgate implementation measures for the
bankruptcy of Chinese banks based on the Bankruptcy Law, so the law
contemplates the possibility that a Chinese bank may go bankrupt.
In addition, foreign banks have been gradually permitted to operate
in China since China’s accession to the World Trade
Organization and have become strong competitors of Chinese banks in
many respects, which may have increased the risk of bankruptcy or
illiquidity for Chinese banks, including those in which we have
deposits. In the event of bankruptcy or illiquidity of any one of
the banks which holds our deposits, we are unlikely to claim our
deposits back in full since we are unlikely to be classified as a
secured creditor based on PRC laws.
Our auditor, like other independent registered public accounting
firms operating in China, is not permitted to be subject to
inspection by Public Company Accounting Oversight Board, and
consequently investors may be deprived of the benefits of such
inspection.
Our
auditor, the independent registered public accounting firm that
issued the audit reports included elsewhere in this report, as an
auditor of companies that are traded publicly in the United States
and a firm registered with the Public Company Accounting Oversight
Board (United States), or PCAOB, is required by the laws of the
United States to undergo regular inspections by the PCAOB to assess
its compliance with the laws of the United States and applicable
professional standards. Our auditor is located in China and the
PCAOB is currently unable to conduct inspections on auditors in
China without the approval of the PRC authorities. Therefore, our
auditor, like other independent registered public accounting firms
operating in China, is currently not inspected by the
PCAOB.
In May
2013, the PCAOB announced that it has entered into a Memorandum of
Understanding (“MOU”) on Enforcement Cooperation with
the China Securities Regulatory Commission (the “CSRC”)
and the Ministry of Finance (the “MOF”). The MOU
establishes a cooperative framework between the parties for the
production and exchange of audit documents relevant to
investigations in both countries’ respective
jurisdictions. More specifically, it provides a mechanism for
the parties to request and receive from each other assistance in
obtaining documents and information in furtherance of their
investigative duties. In addition to developing enforcement
MOU, the PCAOB has been engaged in continuing discussions with the
CSRC and MOF to permit joint inspections in China of audit firms
that are registered with the PCAOB and audit Chinese companies that
trade on U.S. exchanges.
Inspections of
other firms that the PCAOB has conducted outside of China have
identified deficiencies in those firms’ audit procedures and
quality control procedures, and such deficiencies may be addressed
as part of the inspection process to improve future audit quality.
The inability of the PCAOB to conduct inspections of independent
registered public accounting firms operating in China makes it more
difficult to evaluate the effectiveness of our auditor’s
audit procedures or quality control procedures, and to the extent
that such inspections might have facilitated improvements in our
auditor’s audit procedures and quality control procedures,
investors may be deprived of such benefits.
On
November 18, 2016, the PCAOB issued its 2016 to 2020 Strategic Plan
on improving the quality of the audit for the protection and
benefits of investors, which revised the plan to update initiatives
relating to the PCAOB’s new standard-setting process,
planning for and adopting a permanent broker-dealer inspection
program, inspecting firms located in China, audit quality
indicators, monitoring and developing reports related to
independence and the business model of the firms, and business
continuity. This may eventually improve PCAOB’s ability to
conduct inspections of independent registered public accounting
firms operating in China.
RISKS
RELATED TO OUR COMMON STOCK
If we fail to meet all applicable Nasdaq Global Market requirements
and Nasdaq determines to delist our common stock, the delisting
could adversely affect the market liquidity of our common stock,
impair the value of your investment, adversely affect our ability
to raise needed funds and subject us to additional trading
restrictions and regulations.
Our
common stock trades on the Nasdaq Global Market. If we fail to
satisfy the continued listing requirements of The Nasdaq Global
Market, such as the corporate governance requirements or the
minimum closing bid price requirement, The Nasdaq Stock Market (or
Nasdaq) may take steps to de-list our common stock. Such a
de-listing would likely have a negative effect on the price of our
common stock and would impair your ability to sell or purchase our
common stock when you wish to do so. In the event of a de-listing,
we would take actions to restore our compliance with Nasdaq's
listing requirements, but we can provide no assurance that any such
action taken by us would allow our common stock to become listed
again, stabilize the market price or improve the liquidity of our
common stock, prevent our common stock from dropping below the
Nasdaq minimum bid price requirement or prevent future
non-compliance with Nasdaq's listing requirements.
If we
fail to meet all applicable Nasdaq requirements and Nasdaq delists
our securities from trading on its exchange, we expect our
securities could be quoted on the Over-The-Counter Bulletin Board
("OTCBB") or the "pink sheets." If this were to occur, we could
face significant material adverse consequences,
including:
●
a
limited availability of market quotations for our
securities;
●
reduced
liquidity for our securities;
●
a
determination that our common stock is "penny stock" which will
require brokers trading in our common stock to adhere to more
stringent rules and possibly result in a reduced level of trading
activity in the secondary trading market for our
securities;
●
a
limited amount of news and analyst coverage; and
●
a
decreased ability to issue additional securities or obtain
additional financing in the future.
Furthermore, The
National Securities Markets Improvement Act of 1996 ("NSMIA"),
which is a federal statute, prevents or preempts the states from
regulating the sale of certain securities, which are referred to as
"covered securities." Because our common stock is listed on Nasdaq,
they are covered securities for the purpose of NSMIA. If our
securities were no longer listed on Nasdaq and therefore not
"covered securities", we would be subject to regulation in each
state in which we offer our securities.
We do not intend to pay cash
dividends.
We do
not anticipate paying cash dividends on our common stock in the
foreseeable future. We may not have sufficient funds to legally pay
dividends. Even if funds are legally available to pay dividends, we
may nevertheless decide in our sole discretion not to pay
dividends. The declaration, payment and amount of any future
dividends will be made at the discretion of the board of directors,
and will depend upon, among other things, the results of our
operations, cash flows and financial condition, operating and
capital requirements, and other factors our board of directors may
consider relevant. There is no assurance that we will pay any
dividends in the future, and, if dividends are declared, there is
no assurance with respect to the amount of any such
dividend.
Our operating history and lack of profits could lead to wide
fluctuations in our share price. The market price for our common
shares is particularly volatile given our status as a relatively
unknown company with a small and thinly traded public
float.
The
market for our common shares is characterized by significant price
volatility when compared to seasoned issuers, and we expect that
our share price will continue to be more volatile than a seasoned
issuer for the indefinite future. The volatility in our share price
is attributable to a number of factors. First, as noted above, our
common shares are sporadically and thinly traded. As a consequence
of this lack of liquidity, the trading of relatively small
quantities of shares by our stockholders may disproportionately
influence the price of those shares in either direction. The price
for our shares could, for example, decline precipitously in the
event that a large number of our common shares are sold on the
market without commensurate demand, as compared to a seasoned
issuer which could better absorb those sales without adverse impact
on its share price. Secondly, we are a speculative or "risky"
investment due to our limited operating history and lack of profits
to date. As a consequence of this enhanced risk, more risk-adverse
investors may, under the fear of losing all or most of their
investment in the event of negative news or lack of progress, be
more inclined to sell their shares on the market more quickly and
at greater discounts than would be the case with the stock of a
seasoned issuer. Many of these factors are beyond our control and
may decrease the market price of our common shares, regardless of
our operating performance. We cannot make any predictions or
projections as to what the prevailing market price for our common
shares will be at any time, including as to whether our common
shares will sustain their current market prices, or as to what
effect that the sale of shares or the availability of common shares
for sale at any time will have on the prevailing market price.
ITEM 2.
PROPERTIES
Our
corporate headquarters are located at 19925 Stevens Creek Blvd.,
Suite 100 in Cupertino, California. On January 1, 2017, CBMG
Shanghai entered into a 10-year lease agreement with Shanghai
Chuangtong Industrial Development Co., Ltd., pursuant to which the
Company leased a 10,501.6 square meter building located in the
“Pharma Valley” of Shanghai, the People’s
Republic of China for research and development, manufacturing and
office space purposes. Subject to a 5-month rent-free renovation
period, the monthly rent for the first two years is determined by
floor and ranges from 3.7 yuan to 4.3 yuan per square meter per
day, for an aggregate monthly rent for the entire Property of
approximately 1.3 million yuan ($203,000). The term of the Lease is
10 years, starting from January 1, 2017 and ending on December 31,
2026 (the “Original Term”). During the Original Term,
the monthly rent will increase by 6% every two years. We currently
pay rent for a total of $268,000 per month for an aggregate of
approximately 177,000 square feet of space to house our
administration, research and manufacturing facilities in Maryland
and in the cities of Wuxi, Beijing and Shanghai in
China.
ITEM 3.
LEGAL
PROCEEDINGS
We are
currently not involved in any litigation that we believe could have
a materially adverse effect on our financial condition or results
of operations.
ITEM 4.
MINE SAFETY
DISCLOSURES
Not
applicable.
PART II
ITEM 5. MARKET FOR REGISTRANT'S COMMON STOCK, RELATED
STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY
SECURITIES.
Our
common stock is quoted on the Nasdaq Global Market under the symbol
"CBMG." Our stock was formerly quoted under the symbol
“EBIG.”
As of
February 11, 2018, there were 17,430,762 and 17,003,968 shares of
common stock of the Company issued and outstanding, respectively,
and there were approximately 1,700 stockholders of record of the
Company's common stock.
The
following table sets forth for the periods indicated the high and
low bid quotations for the Company's common stock. These quotations
represent inter-dealer quotations, without adjustment for retail
markup, markdown or commission and may not represent actual
transactions.
|
|
High
|
Low
|
|
Fiscal Year 2017
|
|
|
|
First
Quarter (January – March 2017)
|
$13.80
|
$10.05
|
|
Second
Quarter (April – June 2017)
|
$12.50
|
$5.05
|
|
Third
Quarter (July – September 2017)
|
$11.30
|
$8.30
|
|
Fourth
Quarter (October – December 2017)
|
$11.80
|
$9.25
|
|
|
|
|
|
Fiscal Year 2016
|
|
|
|
First
Quarter (January – March 2016)
|
$22.10
|
$10.44
|
|
Second
Quarter (April – June 2016)
|
$20.98
|
$11.07
|
|
Third
Quarter (July – September 2016)
|
$15.68
|
$11.85
|
|
Fourth
Quarter (October – December 2016)
|
$15.45
|
$11.00
|
Effective January
18, 2013, the Company completed its reincorporation from the State
of Arizona to the State of Delaware (the
“Reincorporation”). In connection with the
Reincorporation, shares of the former Arizona entity were exchanged
into shares of the Delaware entity at a ratio of 100 Arizona shares
for each 1 Delaware share, resulting in the same effect as a 1:100
reverse stock split. The Reincorporation became effective on
January 31, 2013. Please refer to the Current Report on Form 8-K,
filed by the Company on January 25, 2013. All values have been
retroactively adjusted.
Dividends
We did
not declare any cash dividends for the years ended December 31,
2017, 2016 and 2015. Our Board of Directors does not intend to
declare any dividends in the near future. The declaration, payment
and amount of any future dividends will be made at the discretion
of the Board of Directors, and will depend upon, among other
things, the results of our operations, cash flows and financial
condition, operating and capital requirements, and other factors as
the Board of Directors considers relevant. There is no assurance
that future dividends will be paid, and if dividends are paid,
there is no assurance with respect to the amount of any such
dividend.
Equity
Compensation Plans
2009 Stock Option Plan
During
the first quarter of 2009, the Company's Board of Directors
approved and adopted the 2009 Stock Option Plan (the "Plan") and
designated 100,000 of its common stock for issuance under the Plan
to employees, directors or consultants for the Company through
either the issuance of shares or stock option grants. Under the
terms of the Plan, stock option grants shall be made with exercise
prices not less than 100% of the fair market value of the shares of
common stock on the grant date. There are 4,593 shares available
for issuance under this plan as of December 31, 2017.
2011 Incentive Stock Option Plan (as amended)
During
the last quarter of 2011, the Company's Board of Directors approved
and adopted the 2011 Incentive Plan (the "2011 Plan") and
designated 300,000 of its no par common stock for issuance under
the 2011 Plan to employees, directors or consultants for the
Company through either the issuance of shares or stock option
grants. Under the terms of the 2011 Plan, stock option grants were
authorized to be made with exercise prices not less than 100% of
the fair market value of the shares of common stock on the grant
date. On November 30, 2012, the Company’s Board of Directors
approved the Amended and Restated 2011 Incentive Stock Option Plan
(the “Restated Plan”), which amended and restated the
2011 Plan to provide for the issuance of up to 780,000 (increasing
up to 1% per year) shares of common stock. The Restated Plan was
approved by our stockholders on
January 17, 2013. There are 25,777 shares available for issuance
under this plan as of December 31, 2017.
2013 Stock Incentive Plan
On August 29, 2013, the Company’s Board of
Directors adopted the Cellular Biomedicine Group, Inc. 2013 Stock
Incentive Plan (the “2013 Plan”) to attract and retain
the best available personnel, to provide additional incentive to
Employees, Directors and Consultants and to promote the success of
the Company’s business. The 2013 Plan was approved by our
stockholders on December 9, 2013. There are 47,714 shares
available for issuance under this plan as of December 31,
2017.
The
following summary describes the material features of the 2013
Plan. The summary, however, does not purport to be a
complete description of all the provisions of the 2013 Plan. The
following description is qualified in its entirety by reference to
the Plan.
Description of the 2013 Plan
The
purpose of the 2013 Plan is to attract and retain the best
available personnel, to provide additional incentive to employees,
directors and consultants and to promote the success of the
Company’s business. The Company has reserved up to
one million (1,000,000) of the authorized but unissued or
reacquired shares of common stock of the Company. The
Board or its appointed administrator has the power and authority to
grant awards and act as administrator thereunder to establish the
grant terms, including the grant price, vesting period and exercise
date.
Each sale or award of shares under the 2013 Plan
is made pursuant to the terms and conditions provided for in an
award agreement (an “Award
Agreement”) entered into
by the Company and the individual recipient. The
number of shares covered by each outstanding Award Agreement shall
be proportionately adjusted for (a) any increase or decrease in the
number of issued shares of common stock resulting from a stock
split, reverse stock split, stock dividend, combination or
reclassification of the common stock, or similar transaction
affecting the common stock or (b) any other increase or decrease in
the number of issued shares of common stock effected without
receipt of consideration by the Company.
Under
the 2013 Plan, the Board or its administrator have the
authority to: (i) to select the employees, directors and
consultants to whom awards may be granted from time to time
hereunder; (ii) to determine whether and to what extent awards
are granted; (iii) to determine the number of shares or the
amount of other consideration to be covered by each award granted;
(iv) to approve forms of Award Agreements for use under the
2013 Plan; (v) to determine the terms and conditions of any
award granted; (vi) to establish additional terms, conditions,
rules or procedures to accommodate the rules or laws of applicable
foreign jurisdictions and to afford grantees favorable treatment
under such rules or laws; provided, however, that no award shall be
granted under any such additional terms, conditions, rules or
procedures with terms or conditions which are inconsistent with the
provisions of the 2013 Plan; (vii) to amend the terms of any
outstanding award granted under the 2013 Plan, provided that any
amendment that would adversely affect the grantee’s rights
under an outstanding award shall not be made without the
grantee’s written consent; (viii) to construe and
interpret the terms of the 2013 Plan and awards, including without
limitation, any notice of award or Award Agreement, granted
pursuant to the 2013 Plan; (ix) to take such other action, not
inconsistent with the terms of the 2013 Plan, as the administrator
deems appropriate.
The
awards under the 2013 Plan other than Incentive Stock Options
(“ISOs”) may be granted to employees, directors and
consultants. ISOs may be granted only to Employees of
the Company, a parent or a subsidiary. An employee,
director or consultant who has been granted an award may, if
otherwise eligible, be granted additional awards. Awards
may be granted to such employees, directors or consultants who are
residing in foreign jurisdictions as the administrator may
determine from time to time. Options granted under the 2013 Plan
will be subject to the terms and conditions established by the
administrator. Under the terms of the 2013 Plan, the
exercise price of the options will not be less than the fair market
value (as determined under the 2013 Plan) of our common stock at
the time of grant. Options granted under the 2013 Plan will be
subject to such terms, including the exercise price and the
conditions and timing of exercise, as may be determined by the
administrator and specified in the applicable award agreement. The
maximum term of an option granted under the 2013 Plan will be ten
years from the date of grant. Payment in respect of the exercise of
an option may be made in cash, by certified or official bank check,
by money order or with shares, pursuant to a
“cashless” or “net issue” exercise, by
a combination thereof, or by such other method as the administrator
may determine to be appropriate and has been included in the terms
of the option.
The 2013 Plan may be amended, suspended or terminated by the
Board, or an administrator appointed by the Board, at any time and
for any reason.
2014 Stock Incentive Plan
On September 22, 2014, the Company’s Board
of Directors adopted the Cellular Biomedicine Group, Inc. 2014
Stock Incentive Plan (the “2014 Plan”) covering 1.2
million shares to attract and retain the best available personnel,
to provide additional incentive to Employees, Directors and
Consultants and to promote the success of the Company’s
business. The 2014 Plan was approved by our stockholders on
November 7, 2014. In 2017 the Company’s Board of
Directors approved the Amended and Restated 2014 Incentive Stock
Option Plan (the “Restated Plan”), which amended and
restated the 2014 Plan to increase the number of shares available
for issuance by 1,000,000 shares. The Restated Plan was approved by
our stockholders on April 28,
2017. There are 961,272 shares available for issuance under this
plan as of December 31, 2017.
The
following summary describes the material features of the 2014
Plan. The summary, however, does not purport to be a
complete description of all the provisions of the 2014 Plan. The
following description is qualified in its entirety by reference to
the Plan.
Description of the 2014 Plan
The
purpose of the 2014 Plan is to attract and retain the best
available personnel, to provide additional incentive to employees,
directors and consultants and to promote the success of the
Company’s business. The Company has reserved up to
1.2 million (1,200,000) of the authorized but unissued or
reacquired shares of common stock of the Company. The
Board or its appointed administrator has the power and authority to
grant awards and act as administrator thereunder to establish the
grant terms, including the grant price, vesting period and exercise
date.
Each sale or award of shares under the 2014 Plan
is made pursuant to the terms and conditions provided for in an
award agreement (an “Award
Agreement”) entered into
by the Company and the individual recipient. The
number of shares covered by each outstanding Award Agreement shall
be proportionately adjusted for (a) any increase or decrease in the
number of issued shares of common stock resulting from a stock
split, reverse stock split, stock dividend, combination or
reclassification of the common stock, or similar transaction
affecting the common stock or (b) any other increase or decrease in
the number of issued shares of common stock effected without
receipt of consideration by the Company.
Under
the 2014 Plan, the Board or its administrator have the
authority to: (i) to select the employees, directors and
consultants to whom awards may be granted from time to time
hereunder; (ii) to determine whether and to what extent awards
are granted; (iii) to determine the number of shares or the
amount of other consideration to be covered by each award granted;
(iv) to approve forms of Award Agreements for use under the
2014 Plan; (v) to determine the terms and conditions of any
award granted; (vi) to establish additional terms, conditions,
rules or procedures to accommodate the rules or laws of applicable
foreign jurisdictions and to afford grantees favorable treatment
under such rules or laws; provided, however, that no award shall be
granted under any such additional terms, conditions, rules or
procedures with terms or conditions which are inconsistent with the
provisions of the 2014 Plan; (vii) to amend the terms of any
outstanding award granted under the 2014 Plan, provided that any
amendment that would adversely affect the grantee’s rights
under an outstanding award shall not be made without the
grantee’s written consent; (viii) to construe and
interpret the terms of the 2014 Plan and awards, including without
limitation, any notice of award or Award Agreement, granted
pursuant to the 2014 Plan; (ix) to take such other action, not
inconsistent with the terms of the 2014 Plan, as the administrator
deems appropriate.
The
awards under the 2014 Plan other than Incentive Stock Options
(“ISOs”) may be granted to employees, directors and
consultants. ISOs may be granted only to Employees of
the Company, a parent or a subsidiary. An employee,
director or consultant who has been granted an award may, if
otherwise eligible, be granted additional awards. Awards
may be granted to such employees, directors or consultants who are
residing in foreign jurisdictions as the administrator may
determine from time to time. Options granted under the 2014 Plan
will be subject to the terms and conditions established by the
administrator. Under the terms of the 2014 Plan, the
exercise price of the options will not be less than the fair market
value (as determined under the 2013 Plan) of our common stock at
the time of grant. Options granted under the 2014 Plan will be
subject to such terms, including the exercise price and the
conditions and timing of exercise, as may be determined by the
administrator and specified in the applicable award agreement. The
maximum term of an option granted under the 2014 Plan will be ten
years from the date of grant. Payment in respect of the exercise of
an option may be made in cash, by certified or official bank check,
by money order or with shares, pursuant to a
“cashless” or “net issue” exercise, by
a combination thereof, or by such other method as the administrator
may determine to be appropriate and has been included in the terms
of the option.
The
2014 Plan may be amended, suspended or terminated by the Board, or
an administrator appointed by the Board, at any time and for any
reason.
All Equity Compensation Plans
The
following table presents securities authorized for issuance under
the Company’s equity compensation plans, as of December 31,
2017:
|
Plan
Category
|
|
|
|
|
|
Number of securities to be issued upon exercise of outstanding
options, warrants and rights (#)
|
Weighted-average
exercise price of outstanding options, warrants and rights
($)
|
Number of securities remaining available for future issuance under
equity compensation plans
|
|
Equity
compensation plans approved by stockholders
|
1,936,294
|
$11.28
|
1,039,356
|
|
Equity
compensation plans not approved by stockholders
|
-
|
-
|
-
|
|
Total
|
1,936,294
|
$11.28
|
1,039,356
|
Stock
Performance Graph
The
line graph that follows compares the cumulative total stockholder
return on our shares of common stock with the cumulative total
return of the Nasdaq Healthcare Index (^IXHC)* and the Russell 3000
Index (RUA)* Index for the five years ended December 31 2017. The
graph and table assume that $100 was invested on the last day of
trading for the fiscal year 2012 in each of our shares of common
stock, the Nasdaq Healthcare Index, and the Russell 3000 Index, and
that no dividends were paid. Cumulative total stockholder returns
for our shares of common stock, Nasdaq Healthcare Index, and the
Russell 3000 Index are based on our fiscal year, which is the same
as the calendar year.
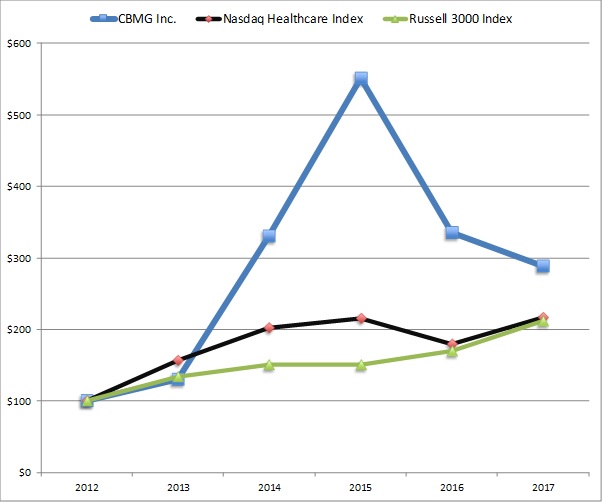
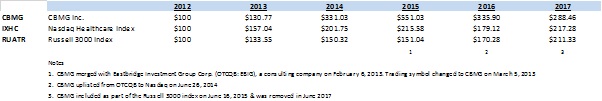
Transfer
Agent
The
Company’s transfer agent and Registrar for the common stock
is Corporate Stock Transfer, Inc. located in Denver,
Colorado.
Recent
Sales of Unregistered Securities
All
unregistered sales and issuances of equity securities for the year
ended December 31, 2017 were previously disclosed in a Form
8-K or Form 10-Q filed with the SEC.
ITEM 6.
SELECTED FINANCIAL
DATA
The following
tables set forth certain of our selected consolidated financial
data as of the dates and for the years indicated. Historical
results are not necessarily indicative of the results to be
expected for any future period.
The
following selected consolidated financial information was derived
from our fiscal year end consolidated financial statements. The
following information should be read in conjunction with those
statements and Item 7, “Management’s Discussion and
Analysis of Financial Condition and Results of Operations.”.
Our summary consolidated statement of operations and comprehensive
loss data for the fiscal years ended December 31, 2015, 2016 and
2017 and our summary consolidated balance sheet data as
of December 31, 2016 and 2017, as set forth below, are derived
from, and are qualified in their entirety by reference to, our
audited consolidated financial statements, including the notes
thereto, which are included in this Annual Report. The
summary balance sheet data as of December 31, 2015, 2014 and 2013,
and summary consolidated statement of operations and comprehensive
loss data for the fiscal years ended December 31, 2014 and 2013,
set forth below are derived from our audited consolidated financial
statements which are not included herein.
Our
consolidated financial statements are prepared and presented in
accordance with accounting principles generally accepted in the
United States, or U.S. GAAP.
Based on the fact that,
we were operating in two separate reportable segments: (i)
Biomedicine and (ii) Consulting, from February 6, 2013 to June 23,
2014 (when we decided to discontinue our Consulting segment),
the summary consolidated statement of operations and
comprehensive loss data for the fiscal years
ended December 31, 2014 and 2013 included the results of Consulting
segment, which were presented therein as “Loss on
discontinued operations, net of taxes”. The
Company’s results from continuing operations in the
summary consolidated statement of operations and comprehensive loss
data were
reflecting the operating results of Biomedicine segment. As
a result of prioritizing cancer
therapeutic technologies and focusing our clinical efforts on
developing CART technologies, Vaccine, Tcm and TCR clonality
technologies, we ceased the cooperation with the Jihua Hospital and
several agents in the second quarter of 2016, all of our revenues
was derived from cell therapy technology service since
then.
|
CELLULAR BIOMEDICINE GROUP, INC.
|
||||||||||||||
|
CONSOLIDATED STATEMENT OF OPERATIONS AND COMPREHENSIVE
LOSS
|
|
|
For the Year Ended
December 31,
|
||||
|
|
2017
|
2016
|
2015
|
2014
|
2013
|
|
Summary
Consolidated statement of operations
and comprehensive loss data:
|
|||||
|
|
|
|
|
|
|
|
Net
sales and revenue
|
$336,817
|
$627,930
|
$2,505,423
|
$564,377
|
$204,914
|
|
|
|
|
|
|
|
|
Operating
expenses:
|
|
|
|
|
|
|
Cost
of sales
|
162,218
|
860,417
|
1,880,331
|
242,215
|
296,212
|
|
General
and administrative
|
12,780,483
|
11,670,506
|
13,068,255
|
7,875,413
|
9,162,172
|
|
Selling
and marketing
|
360,766
|
425,040
|
709,151
|
314,894
|
58,275
|
|
Research
and development
|
14,609,917
|
11,475,587
|
7,573,228
|
3,146,499
|
2,041,872
|
|
Impairment
of investments
|
-
|
4,611,714
|
123,428
|
1,427,840
|
-
|
|
Total
operating expenses
|
27,913,384
|
29,043,264
|
23,354,393
|
13,006,861
|
11,558,531
|
|
Operating
loss
|
(27,576,567)
|
(28,415,334)
|
(20,848,970)
|
(12,442,484)
|
(11,353,617)
|
|
Other
income:
|
|
|
|
|
|
|
Interest
income
|
133,621
|
78,943
|
42,220
|
15,043
|
1,294
|
|
Other
income
|
1,955,086
|
132,108
|
630,428
|
71,982
|
(6,196)
|
|
Total
other income
|
2,088,707
|
211,051
|
672,648
|
87,025
|
(4,902)
|
|
Loss
from continuing operations before taxes
|
(25,487,860)
|
(28,204,283)
|
(20,176,322)
|
(12,355,459)
|
(11,358,519)
|
|
Income
taxes credit (provision)
|
(2,450)
|
(4,093)
|
728,601
|
-
|
-
|
|
Loss
from continuing operations
|
(25,490,310)
|
(28,208,376)
|
(19,447,721)
|
(12,355,459)
|
(11,358,519)
|
|
Loss
on discontinued operations, net of taxes
|
-
|
-
|
-
|
(3,119,152)
|
(2,438,514)
|
|
Net
loss
|
$(25,490,310)
|
$(28,208,376)
|
$(19,447,721)
|
$(15,474,611)
|
$(13,797,033)
|
|
Other comprehensive income (loss):
|
|
|
|
|
|
|
Cumulative
translation adjustment
|
967,189
|
(743,271)
|
(307,950)
|
15,254
|
78,650
|
|
Unrealized
gain (loss) on investments, net of tax
|
(240,000)
|
5,300,633
|
(1,376,540)
|
1,611,045
|
(198,200)
|
|
Reclassification
adjustments, net of tax, in connection with other-than-temporary
impairment of investments
|
-
|
(5,557,939)
|
-
|
-
|
-
|
|
Total
other comprehensive income (loss):
|
727,189
|
(1,000,577)
|
(1,684,490)
|
1,626,299
|
(119,550)
|
|
Comprehensive
loss
|
$(24,763,121)
|
$(29,208,953)
|
$(21,132,211)
|
$(13,848,312)
|
$(13,916,583)
|
|
|
|
|
|
|
|
|
Net
loss per share :
|
|
|
|
|
|
|
Basic
|
$(1.78)
|
$(2.09)
|
$(1.70)
|
$(1.79)
|
$(2.38)
|
|
Diluted
|
$(1.78)
|
$(2.09)
|
$(1.70)
|
$(1.79)
|
$(2.38)
|
|
Weighted average common shares outstanding:
|
|
|
|
||
|
Basic
|
14,345,604
|
13,507,408
|
11,472,306
|
8,627,094
|
5,792,888
|
|
Diluted
|
14,345,604
|
13,507,408
|
11,472,306
|
8,627,094
|
5,792,888
|
|
|
As of
December 31,
|
||||
|
|
2017
|
2016
|
2015
|
2014
|
2013
|
|
Summary Consolidated balance sheet
data:
|
|
|
|
|
|
|
Cash
and cash equivalents
|
$21,568,422
|
$39,252,432
|
$14,884,597
|
$14,770,584
|
$7,175,215
|
|
Current
working capital (1)
|
20,850,823
|
38,328,048
|
13,675,034
|
12,019,143
|
5,373,355
|
|
Total
assets
|
61,162,296
|
68,628,467
|
49,460,422
|
43,685,102
|
17,596,726
|
|
Other
non-current liabilities
|
183,649
|
370,477
|
76,229
|
452,689
|
-
|
|
Stockholders’
equity
|
57,302,526
|
65,893,954
|
46,364,936
|
39,156,091
|
15,395,073
|
(1) Current
working capital is the difference between total current assets and
total current liabilities.
ITEM 7.
MANAGEMENT'S DISCUSSION AND
ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF
OPERATIONS.
The
following is management's discussion and analysis of certain
significant factors that have affected our financial position and
operating results during the periods included in the accompanying
consolidated financial statements, as well as information relating
to the plans of our current management. This report includes
forward-looking statements. Generally, the words "believes,"
"anticipates," "may," "will," "should," "expect," "intend,"
"estimate," "continue," and similar expressions or the negative
thereof or comparable terminology are intended to identify
forward-looking statements. Such statements are subject to certain
risks and uncertainties, including the matters set forth in this
report or other reports or documents we file with the Securities
and Exchange Commission from time to time, which could cause actual
results or outcomes to differ materially from those projected.
Undue reliance should not be placed on these forward-looking
statements which speak only as of the date hereof. We undertake no
obligation to update these forward-looking statements.
The
following discussion and analysis should be read in conjunction
with our consolidated financial statements and the related notes
thereto and other financial information included in Item 8 of this
Annual Report on Form 10-K.
Critical
Accounting Policies and Estimates
We
prepare our consolidated financial statements in accordance with
accounting principles generally accepted in the United States of
America. The preparation of these financial statements requires the
use of estimates and assumptions that affect the reported amounts
of assets and liabilities and the disclosure of contingent assets
and liabilities at the date of the financial statements and the
reported amount of revenues and expenses during the reporting
period. Our management periodically evaluates the estimates and
judgments made. Management bases its estimates and judgments on
historical experience and on various factors that are believed to
be reasonable under the circumstances. Actual results may differ
from these estimates as a result of different assumptions or
conditions.
The
following summarizes critical estimates made by management in the
preparation of the consolidated financial statements.
Cash and Cash Equivalents
The
Company considers all highly liquid investments with an original
maturity of three months or less to be cash equivalents. As of
December 31, 2017 and 2016, respectively, cash and cash equivalents
include cash on hand and cash in the bank. At times, cash deposits
may exceed government-insured limits.
Accounts Receivable
Accounts receivable
represent amounts earned but not collected in connection with the
Company’s sales as of December 31, 2017 and 2016. Accounts
receivable are carried at their estimated collectible
amounts.
The
Company follows the allowance method of recognizing uncollectible
accounts receivable. The Company recognizes bad debt expense based
on specifically identified customers and invoices that are
anticipated to be uncollectable. At December 31, 2017 and 2016,
allowance of $10,789 and $10,163 was provided for debtors of
certain customers as those debts are unrecoverable from customers,
respectively.
Inventory
Inventories consist
of raw materials, work-in-process, semi-finished goods and finished
goods. Inventories are initially recognized at cost and
subsequently at the lower of cost and net realizable value under
first-in first-out method. Finished goods are comprised of direct
materials, direct labor, depreciation and manufacturing overhead.
Net realizable value is the estimated selling price, in the
ordinary course of business, less estimated costs to complete and
dispose. The Company regularly inspects the shelf life of prepared
finished goods and, if necessary, writes down their carrying value
based on their salability and expiration dates into cost of goods
sold.
Property, Plant and Equipment
Property, plant and
equipment are recorded at cost. Depreciation is provided for on the
straight-line method over the estimated useful lives of the assets
ranging from three to ten years and begins when the related assets
are placed in service. Maintenance and repairs that neither
materially add to the value of the property nor appreciably prolong
its life are charged to expense as incurred. Betterments or
renewals are capitalized when incurred. Plant, property and
equipment are reviewed each year to determine whether any events or
circumstances indicate that the carrying amount of the assets may
not be recoverable. We assess the recoverability of the asset by
comparing the projected undiscounted net cash flows associated with
the related assets over the estimated remaining life against the
respective carrying value.
Goodwill and Other Intangibles
Goodwill
represents the excess of the cost of assets acquired over the fair
value of the net assets at the date of acquisition. Intangible
assets represent the fair value of separately recognizable
intangible assets acquired in connection with the Company’s
business combinations. The Company evaluates its goodwill and other
intangibles for impairment on an annual basis or whenever events or
circumstances indicate that impairment may have occurred. As of
December 31, 2017, the goodwill is $7,678,789, which all derived
from the acquisition of Agreen.
As
stipulated in ASC 350-20-35-3A, an entity may assess qualitative
factors to determine whether it is more likely than not that the
fair value of a reporting unit is less than its carrying amount,
including goodwill, in which relevant triggering events and
circumstances should be assessed. During the year ended December
31, 2017, it considered no triggering event indicating that the
goodwill impairment test was required at the balance sheet date. In
addition, the Company’s market capitalization as at the
balance sheet date would fairly reflect the fair value of the
Company’s research and development efforts so as to provide
an indication of whether the goodwill is subject to the impairment
loss. Our market capitalization exceeds the carrying amount of net
assets (including goodwill) of the Company. No impairment loss of
goodwill is considered required as of December 31,
2017.
Other
intangibles mainly consists of knowhow, technologies, patent,
licenses acquired and purchased software. The Company reviews the
carrying value of long-lived assets to be held and used, including
other intangible assets subject to amortization, when events and
circumstances warrants such a review. The carrying value of a
long-lived asset is considered impaired when the anticipated
undiscounted cash flow from such asset is separately identifiable
and is less than its carrying value. No impairment is considered to
be required as of December 31, 2017.
The
Company is an expanding company with a short operating history,
accordingly, the Company faces some potential events and
uncertainties encountered by companies in the earlier stages of
development and expansion, such as: (1) continuing market
acceptance for our product extensions and our services; (2)
changing competitive conditions, technological advances or customer
preferences that could harm sales of our products or services; (3)
maintaining effective control of our costs and expenses. If the
Company is not able to meet the challenge of building our
businesses and managing our growth, the likely result would be
slowed growth, lower margins, additional operational costs and
lower income, and a risk of impairment charge of intangibles in
future filings.
Treasury Stock
The
treasury stock is recorded and carried at their repurchase cost.
The Company recorded the entire purchase price of the treasury
stock as a reduction of equity. A gain and or loss will be
determined when treasury stock is reissued or retired, and the
original issue price and book value of the stock do not enter into
the accounting. Additional paid-in capital from treasury stock is
credited for gains and debited for losses when treasury stock is
reissued at prices that differ from the repurchase
cost.
Government Grants
Government grants
are recognized in the balance sheet initially when there is
reasonable assurance that they will be received and that the
enterprise will comply with the conditions attached to them. When
the Company received the government grants but the conditions
attached to the grants have not been fulfilled, such government
grants are deferred and recorded as dferred income. The
reclassification of short-term or long-term liabilities is depended
on the management’s expectation of when the conditions
attached to the grant can be fulfilled. Grants that compensate the
Company for expenses incurred are recognized as other income in
statement of income on a systematic basis in the same periods in
which the expenses are incurred.
For the
year ended December 31, 2017 and 2016, the Company received
government grants of $1,905,213 and $422,839 for purpose of R&D
and related capital expenditure, respectively. Government subsidies
recognized as other income in the statement of income for the year
ended December 31, 2017 and 2016 were $2,077,486 and $78,542,
respectively.
Fair Value of Financial Instruments
Under
the FASB’s authoritative guidance on fair value measurements,
fair value is the price that would be received to sell an asset or
paid to transfer a liability in an orderly transaction between
market participants at the measurement date. In determining the
fair value, the Company uses various methods including market,
income and cost approaches. Based on these approaches, the Company
often utilizes certain assumptions that market participants would
use in pricing the asset or liability, including assumptions about
risk and the risks inherent in the inputs to the valuation
technique. These inputs can be readily observable, market
corroborated or generally unobservable inputs. The Company uses
valuation techniques that maximize the use of observable inputs and
minimize the use of unobservable inputs. Based on observability of
the inputs used in the valuation techniques, the Company is
required to provide the following information according to the fair
value hierarchy. The fair value hierarchy ranks the quality and
reliability of the information used to determine fair values.
Financial assets and liabilities carried at fair value are
classified and disclosed in one of the following three
categories:
Level
1: Valuations for assets and liabilities traded in active exchange
markets. Valuations are obtained from readily available pricing
sources for market transactions involving identical assets or
liabilities.
Level
2: Valuations for assets and liabilities traded in less active
dealer or broker markets. Valuations are obtained from third party
pricing services for identical or similar assets or
liabilities.
Level
3: Valuations for assets and liabilities that are derived from
other valuation methodologies, including option pricing models,
discounted cash flow models and similar techniques, and not based
on market exchange, dealer or broker traded transactions. Level 3
valuations incorporate certain unobservable assumptions and
projections in determining the fair value assigned to such
assets.
All
transfers between fair value hierarchy levels are recognized by the
Company at the end of each reporting period. In certain cases, the
inputs used to measure fair value may fall into different levels of
the fair value hierarchy. In such cases, an investment’s
level within the fair value hierarchy is based on the lowest level
of input that is significant to the fair value measurement in its
entirety requires judgment, and considers factors specific to the
investment. The inputs or methodology used for valuing financial
instruments are not necessarily an indication of the risks
associated with investment in those instruments.
The
carrying amounts of other financial instruments, including cash,
accounts receivable, accounts payable and accrued liabilities,
income tax payable and related party payable approximate fair value
due to their short maturities.
Investments
The
fair value of “investments” is dependent on the type of
investment, whether it is marketable or
non-marketable.
Marketable
securities held by the Company are held for an indefinite period of
time and thus are classified as available-for-sale securities. The
fair value is based on quoted market prices for the investment as
of the balance sheet date. Realized investment gains and losses are
included in the statement of operations, as are provisions for
other than temporary declines in the market value of available
for-sale securities. Unrealized gains and unrealized losses deemed
to be temporary are excluded from earnings (losses), net of
applicable taxes, as a component of other comprehensive income
(loss). Factors considered in judging whether an impairment is
other than temporary include the financial condition, business
prospects and creditworthiness of the issuer, the length of time
that fair value has been less than cost, the relative amount of
decline, and the Company’s ability and intent to hold the
investment until the fair value recovers.
Stock-Based Compensation
We
periodically use stock-based awards, consisting of shares of common
stock or stock options, to compensate officers, employees,
directors and consultants. Awards are expensed on a straight line
basis over the requisite service period based on the grant date
fair value, net of estimated forfeitures, if any.
Revenue Recognition
The
Company utilizes the guidance set forth in the FASB’s ASC
Topic 605, “Revenue Recognition”, regarding the
recognition, presentation and disclosure of revenue in its
financial statements. The Company recognizes revenue when pervasive
evidence of an arrangement exists, the price is fixed and
determinable, collection is reasonably assured and delivery of
products or services has been rendered.
Income Taxes
Income
taxes are accounted for using the asset and liability method as
prescribed by ASC 740 “Income Taxes”. Under this
method, deferred income tax assets and liabilities are recognized
for the future tax consequences attributable to temporary
differences between the financial statement carrying amounts of
existing assets and liabilities and their respective tax bases.
Deferred income tax assets and liabilities are measured using
enacted tax rates expected to apply to taxable income in the years
in which these temporary differences are expected to be recovered
or settled. The effect on deferred tax assets and liabilities of a
change in tax rates is recognized in income in the period that
includes the enactment date. A valuation allowance would be
provided for those deferred tax assets for which if it is more
likely than not that the related benefit will not be
realized.
While
we have optimistic plans for our business strategy, we determined
that a full valuation allowance was necessary against all net
deferred tax assets as of December 31, 2017 and 2016, given the
current and expected near term losses and the uncertainty with
respect to our ability to generate sufficient profits from our
business model.
Recent Accounting Pronouncements
Accounting pronouncements adopted during the year ended December
31, 2017
In April 2016, the Financial Accounting Standards
Board (“FASB”) issued Accounting Standards Update
(“ASU”) No. 2016-09, “Compensation—Stock
Compensation (Topic 718): Improvements to Employee Share-Based
Payment Accounting” (“ASU 2016-09”), which simplifies
several aspects of the accounting for employee share-based payment
transactions. The areas for simplification in ASU 2016-09 include
the income tax consequences, classification of awards as either
equity or liabilities, and classification on the statement of cash
flows. The amendments in this ASU will be effective for annual
periods beginning after December 15, 2016 and interim periods
within those annual periods. Early adoption is permitted. The
adoption of the ASU 2016-09 did not have a material impact on the
Company’s consolidated financial
statements.
Accounting pronouncements not yet effective
In February 2018, the FASB issued ASU No.
2018-02, “Income
Statement—Reporting Comprehensive Income (Topic 220):
Reclassification of Certain Tax Effects from Accumulated Other
Comprehensive Income” (“ASU 2018-02”), which provides
financial statement preparers with an option to reclassify stranded
tax effects within accumulated other comprehensive income to
retained earnings in each period in which the effect of the change
in the U.S. federal corporate income tax rate in the Tax Cuts and
Jobs Act (or portion thereof) is recorded. The amendments in this
ASU are effective for all entities for fiscal years beginning after
December 15, 2018, and interim periods within those fiscal years.
Early adoption of ASU 2018-02 is permitted, including adoption in
any interim period for the public business entities for reporting
periods for which financial statements have not yet been issued.
The amendments in this ASU should be applied either in the period
of adoption or retrospectively to each period (or periods) in which
the effect of the change in the U.S. federal corporate income tax
rate in the Tax Cuts and Jobs Act is recognized. We do not expect
the adoption of ASU 2018-02 to have a material impact on our
consolidated financial statements.
In July 2017, the FASB issued ASU No.
2017-11, “Earnings Per Share
(Topic 260); Distinguishing Liabilities from Equity (Topic 480);
Derivatives and Hedging (Topic 815): (Part I) Accounting for
Certain Financial Instruments with Down Round Features, (Part II)
Replacement of the Indefinite Deferral for Mandatorily Redeemable
Financial Instruments of Certain Nonpublic Entities and Certain
Mandatorily Redeemable Non-controlling Interests with a Scope
Exception” (“ASU
2017-11”), which addresses the complexity of accounting for
certain financial instruments with down round features. Down round
features are features of certain equity-linked instruments (or
embedded features) that result in the strike price being reduced on
the basis of the pricing of future equity offerings. Current
accounting guidance creates cost and complexity for entities that
issue financial instruments (such as warrants and convertible
instruments) with down round features that require fair value
measurement of the entire instrument or conversion option. The
amendments in Part I of this ASU are effective for public business
entities for fiscal years, and interim periods within those fiscal
years, beginning after December 15, 2018. The Company is currently
evaluating the impact of the adoption of ASU 2017-11 on its
consolidated financial statements.
In May 2017, the FASB issued ASU No.
2017-09, “Compensation—Stock
Compensation (Topic 718): Scope of Modification
Accounting” (“ASU
2017-09”), which provides guidance on determining which
changes to the terms and conditions of share-based payment awards
require an entity to apply modification accounting under Topic 718.
The amendments in this ASU are effective for all entities for
annual periods, and interim periods within those annual periods,
beginning after December 15, 2017. Early adoption is permitted,
including adoption in any interim period, for (1) public business
entities for reporting periods for which financial statements have
not yet been issued and (2) all other entities for reporting
periods for which financial statements have not yet been made
available for issuance. The amendments in this ASU should be
applied prospectively to an award modified on or after the adoption
date. We do not expect the adoption of ASU 2017-09 to have a
material impact on our consolidated financial
statements.
In February 2017, the FASB issued ASU No.
2017-05, “Other
Income—Gains and Losses from the Derecognition of
Nonfinancial Assets (Subtopic 610-20): Clarifying the Scope of
Asset Derecognition Guidance and Accounting for Partial Sales of
Nonfinancial Assets” (“ASU 2017-05”), which clarifies the
scope of the nonfinancial asset guidance in Subtopic 610-20. This
ASU also clarifies that the derecognition of all businesses and
nonprofit activities (except those related to conveyances of oil
and gas mineral rights or contracts with customers) should be
accounted for in accordance with the derecognition and
deconsolidation guidance in Subtopic 810-10. The amendments in this
ASU also provide guidance on the accounting for what often are
referred to as partial sales of nonfinancial assets within the
scope of Subtopic 610-20 and contributions of nonfinancial assets
to a joint venture or other non-controlled investee.
The amendments in this ASU are
effective for annual reporting reports beginning after December 15,
2017, including interim reporting periods within that reporting
period. Public entities may apply the guidance earlier but only as
of annual reporting periods beginning after December 15, 2016,
including interim reporting periods within that reporting period.
We do not expect the adoption of ASU 2017-05 to have a material
impact on our consolidated financial
statements.
In January 2017, the FASB issued ASU No.
2017-04, “Intangibles—Goodwill
and Other (Topic 350): Simplifying the Test for Goodwill
Impairment” (“ASU
2017-04”), which removes Step 2 from the goodwill impairment
test. An entity will apply a one-step quantitative test and record
the amount of goodwill impairment as the excess of a reporting
unit's carrying amount over its fair value, not to exceed the total
amount of goodwill allocated to the reporting unit. The new
guidance does not amend the optional qualitative assessment of
goodwill impairment. Public business entity that is a U.S.
Securities and Exchange Commission filer should adopt the
amendments in this ASU for its annual or any interim goodwill
impairment test in fiscal years beginning after December 15, 2019.
Early adoption is permitted for interim or annual goodwill
impairment tests performed on testing dates after January 1, 2017.
We are currently evaluating the impact of the adoption of ASU
2017-04 on our consolidated financial
statements.
In November 2016, the FASB issued ASU No.
2016-18, “Statement of Cash Flows
(Topic 230): Restricted Cash” (“ASU 2016-18”), which requires that a
statement of cash flows explain the change during the period in the
total of cash, cash equivalents, and amounts generally described as
restricted cash or restricted cash equivalents. Therefore, amounts
generally described as restricted cash and restricted cash
equivalents should be included with cash and cash equivalents when
reconciling the beginning-of-period and end-of-period total amounts
shown on the statement of cash flows. The amendments in this ASU do
not provide a definition of restricted cash or restricted cash
equivalents. The amendments in this ASU are effective for public
business entities for fiscal years beginning after December 15,
2017, and interim periods within those fiscal years. Early adoption
is permitted, including adoption in an interim period. We do not
expect the adoption of ASU 2016-18 to have a material impact on our
consolidated financial statements.
In August 2016, the FASB issued ASU No.
2016-15, “Statement of Cash Flows
(Topic 230): Classification of Certain Cash Receipts and Cash
Payments” (“ASU
2016-15”), which addresses the following eight specific cash
flow issues: debt prepayment or debt extinguishment costs;
settlement of zero-coupon debt instruments or other debt
instruments with coupon interest rates that are insignificant in
relation to the effective interest rate of the borrowing;
contingent consideration payments made after a business
combination; proceeds from the settlement of insurance claims;
proceeds from the settlement of corporate-owned life insurance
policies (including bank-owned life insurance policies;
distributions received from equity method investees; beneficial
interests in securitization transactions; and separately
identifiable cash flows and application of the predominance
principle. The amendments in this ASU are effective for public
business entities for fiscal years beginning after December 15,
2017, and interim periods within those fiscal years. Early adoption
is permitted, including adoption in an interim period. We do not
expect the adoption of ASU 2016-15 to have a material impact on our
consolidated financial statements.
In June 2016, the FASB issued ASU No.
2016-13, “Financial
Instruments—Credit Losses (Topic 326): Measurement of Credit
Losses on Financial Instruments” (“ASU 2016-13”). Financial
Instruments—Credit Losses (Topic 326) amends guideline on
reporting credit losses for assets held at amortized cost basis and
available-for-sale debt securities. For assets held at amortized
cost basis, Topic 326 eliminates the probable initial recognition
threshold in current GAAP and, instead, requires an entity to
reflect its current estimate of all expected credit losses. The
allowance for credit losses is a valuation account that is deducted
from the amortized cost basis of the financial assets to present
the net amount expected to be collected. For available-for-sale
debt securities, credit losses should be measured in a manner
similar to current GAAP, however Topic 326 will require that credit
losses be presented as an allowance rather than as a write-down.
ASU 2016-13 affects entities holding financial assets and net
investment in leases that are not accounted for at fair value
through net income. The amendments affect loans, debt securities,
trade receivables, net investments in leases, off balance sheet
credit exposures, reinsurance receivables, and any other financial
assets not excluded from the scope that have the contractual right
to receive cash. The amendments in this ASU will be effective for
fiscal years beginning after December 15, 2019, including interim
periods within those fiscal years. We are currently evaluating the
impact of the adoption of ASU 2016-13 on our consolidated financial
statements.
In February 2016, the FASB issued ASU No.
2016-02, “Leases (Topic
842)” (“ASU
2016-02”). The amendments in this update create Topic 842,
Leases, and supersede the leases requirements in Topic 840, Leases.
Topic 842 specifies the accounting for leases. The objective of
Topic 842 is to establish the principles that lessees and lessors
shall apply to report useful information to users of financial
statements about the amount, timing, and uncertainty of cash flows
arising from a lease. The main difference between Topic 842 and
Topic 840 is the recognition of lease assets and lease liabilities
for those leases classified as operating leases under Topic 840.
Topic 842 retains a distinction between finance leases and
operating leases. The classification criteria for distinguishing
between finance leases and operating leases are substantially
similar to the classification criteria for distinguishing between
capital leases and operating leases in the previous leases
guidance. The result of retaining a distinction between finance
leases and operating leases is that under the lessee accounting
model in Topic 842, the effect of leases in the statement of
comprehensive income and the statement of cash flows is largely
unchanged from previous GAAP. The amendments in ASU 2016-02 are
effective for fiscal years beginning after December 15, 2018,
including interim periods within those fiscal years for public
business entities. Early application of the amendments in ASU
2016-02 is permitted. We are currently evaluating the impact of the
adoption of ASU 2016-02 on our consolidated financial
statements.
In January 2016, the FASB issued ASU No.
2016-01, “Financial Instruments
– Overall (Subtopic 825-10): Recognition and Measurement of
Financial Assets and Financial Liabilities”
(“ASU 2016-01”). The
amendments in this update require all equity investments to be
measured at fair value with changes in the fair value recognized
through net income (other than those accounted for under equity
method of accounting or those that result in consolidation of the
investee). The amendments in this update also require an entity to
present separately in other comprehensive income the portion of the
total change in the fair value of a liability resulting from a
change in the instrument-specific credit risk when the entity has
elected to measure the liability at fair value in accordance with
the fair value option for financial instruments. In addition, the
amendments in this update eliminate the requirement for to disclose
the method(s) and significant assumptions used to estimate the fair
value that is required to be disclosed for financial instruments
measured at amortized cost on the balance sheet for public
entities. For public business entities, the amendments in ASU
2016-01 are effective for fiscal years beginning after December 15,
2017, including interim periods within those fiscal years. Except
for the early application guidance discussed in ASU 2016-01, early
adoption of the amendments in this update is not permitted. We do
not expect the adoption of ASU 2016-01 to have a material impact on
our consolidated financial statements.
In May 2014, the FASB issued ASU No.
2014-09, “Revenue from Contracts
with Customers (Topic 606)” (“ASU 2014-09”). ASU 2014-09
supersedes the revenue recognition requirements in “Revenue
Recognition (Topic 605)”, and requires entities to recognize
revenue when it transfers promised goods or services to customers
in an amount that reflects the consideration to which the entity
expects to be entitled to in exchange for those goods or services.
The FASB issued ASU No. 2015-14, “Revenue from Contracts
with Customers (Topic 606): Deferral of the Effective
Date” (“ASU
2015-14”) in August 2015. The amendments in ASU 2015-14 defer
the effective date of ASU 2014-09. Public business entities,
certain not-for-profit entities, and certain employee benefit plans
should apply the guidance in ASU 2014-09 to annual reporting
periods beginning after December 15, 2017, including interim
reporting periods within that reporting period. Earlier adoption is
permitted only as of annual reporting periods beginning after
December 15, 2016, including interim reporting periods within that
reporting period. Further to ASU 2014-09 and ASU 2015-14, the FASB
issued ASU No. 2016-08, “Revenue from Contracts
with Customers (Topic 606): Principal versus Agent Considerations
(Reporting Revenue Gross versus Net)” (“ASU 2016-08”) in March 2016, ASU No.
2016-10, “Revenue from Contracts
with Customers (Topic 606): Identifying Performance Obligations and
Licensing” (“ASU
2016-10”) in April 2016, ASU No. 2016-12, “Revenue from Contracts
with Customers (Topic 606): Narrow-Scope Improvements and Practical
Expedients” (“ASU
2016-12”), and ASU No. 2016-20, “Technical Corrections
and Improvements to Topic 606, Revenue from Contracts with
Customers” (“ASU
2016-20”), respectively. The amendments in ASU 2016-08
clarify the implementation guidance on principal versus agent
considerations, including indicators to assist an entity in
determining whether it controls a specified good or service before
it is transferred to the customers. ASU 2016-10 clarifies
guidelines related to identifying performance obligations and
licensing implementation guidance contained in the new revenue
recognition standard. The updates in ASU 2016-10 include targeted
improvements based on input the FASB received from the Transition
Resource Group for Revenue Recognition and other stakeholders. It
seeks to proactively address areas in which diversity in practice
potentially could arise, as well as to reduce the cost and
complexity of applying certain aspects of the guidance both at
implementation and on an ongoing basis. ASU 2016-12 addresses
narrow-scope improvements to the guidance on collectability,
non-cash consideration, and completed contracts at transition.
Additionally, the amendments in this ASU provide a practical
expedient for contract modifications at transition and an
accounting policy election related to the presentation of sales
taxes and other similar taxes collected from customers. The
amendments in ASU 2016-20 represents changes to make minor
corrections or minor improvements to the Codification that are not
expected to have a significant effect on current accounting
practice or create a significant administrative cost to most
entities. The effective date and transition requirements for ASU
2016-08, ASU 2016-10, ASU 2016-12 and ASU 2016-20 are the same as
ASU 2014-09. The Company will adopt ASC Topic
606, Revenue from Contracts with
Customers, using the modified
retrospective transition approach. Under this approach, ASC Topic
606 would apply to all new contracts initiated on or after January
1, 2018. For existing contracts that have remaining obligations as
of January 1, 2018, any difference between the recognition criteria
in these ASUs and the Company’s current revenue recognition
practices would be recognized using a cumulative effect adjustment
to the opening balance of accumulated deficit. The Company does not
anticipate the adoption of these ASUs to have material changes to
its revenue recognition practices nor material impact on its
consolidated financial statements.
Comparison of Year Ended December 31, 2017 to Years Ended December
31, 2016 and 2015
Although
the descriptions in the results of operations below reflect our
operating results as set forth in our Consolidated Statement of
Operations filed herewith, we are presenting consolidated pro forma
information below to reflect the impacts of the business
combination as if the transaction had occurred at the beginning of
the earliest period presented.
|
|
For the Year Ended December 31,
|
||
|
|
2017
|
2016
|
2015
|
|
Net
sales and revenue
|
$336,817
|
$627,930
|
$2,505,423
|
|
|
|
|
|
|
Operating
expenses:
|
|
|
|
|
Cost
of sales *
|
162,218
|
860,417
|
1,880,331
|
|
General
and administrative *
|
12,780,483
|
11,670,506
|
13,068,255
|
|
Selling
and marketing *
|
360,766
|
425,040
|
709,151
|
|
Research
and development *
|
14,609,917
|
11,475,587
|
7,573,228
|
|
Impairment
of investments
|
-
|
4,611,714
|
123,428
|
|
Total
operating expenses
|
27,913,384
|
29,043,264
|
23,354,393
|
|
Operating
loss
|
(27,576,567)
|
(28,415,334)
|
(20,848,970)
|
|
|
|
|
|
|
Other
income
|
|
|
|
|
Interest
income
|
133,621
|
78,943
|
42,220
|
|
Other
income
|
1,955,086
|
132,108
|
630,428
|
|
Total
other income
|
2,088,707
|
211,051
|
672,648
|
|
Loss
before taxes
|
(25,487,860)
|
(28,204,283)
|
(20,176,322)
|
|
|
|
|
|
|
Income
taxes credit (provision)
|
(2,450)
|
(4,093)
|
728,601
|
|
Net
loss
|
$(25,490,310)
|
$(28,208,376)
|
$(19,447,721)
|
|
Other
comprehensive income (loss):
|
|
|
|
|
Cumulative
translation adjustment
|
967,189
|
(743,271)
|
(307,950)
|
|
Unrealized
gain (loss) on investments, net of tax
|
(240,000)
|
5,300,633
|
(1,376,540)
|
|
Reclassification
adjustments, net of tax, in connection with other-than-temporary
impairment of investments
|
-
|
(5,557,939)
|
-
|
|
Total
other comprehensive income (loss):
|
727,189
|
(1,000,577)
|
(1,684,490)
|
|
|
|
|
|
|
Comprehensive
loss
|
$(24,763,121)
|
$(29,208,953)
|
$(21,132,211)
|
|
|
|
|
|
|
Net
loss per share:
|
|
|
|
|
Basic
|
$(1.78)
|
$(2.09)
|
$(1.70)
|
|
Diluted
|
$(1.78)
|
$(2.09)
|
$(1.70)
|
|
|
|
|
|
|
Weighted average common shares outstanding:
|
|
|
|
|
Basic
|
14,345,604
|
13,507,408
|
11,472,306
|
|
Diluted
|
14,345,604
|
13,507,408
|
11,472,306
|
|
|
|
|
|
|
* These
line items include the following amounts of non-cash, stock-based
compensation expense for the periods indicated:
|
|||
|
|
For
the Year Ended December 31,
|
||
|
|
2017
|
2016
|
2015
|
|
|
|
|
|
|
Cost
of sales
|
51,288
|
18,916
|
144,200
|
|
General
and administrative
|
2,935,798
|
3,110,237
|
4,948,375
|
|
Selling
and marketing
|
52,984
|
56,704
|
188,579
|
|
Research
and development
|
2,305,141
|
2,266,560
|
2,311,283
|
|
|
5,345,211
|
5,452,417
|
7,592,437
|
Segments
The
Company is engaged in the development of new treatments for
cancerous and degenerative diseases utilizing proprietary
cell-based technologies, which have been organized as one reporting
segment since they have similar nature and economic
characteristics. The Company’s principle operating decision
maker, the Chief Executive Officer, receives and reviews the result
of the operation for all major cell platforms as a whole when
making decisions about allocating resources and assessing
performance of the Company. In accordance with FASB ASC 280-10, the
Company is not required to report the segment
information.
Results
of Operations:
Revenues
|
|
|
|
|
2017 versus 2016
|
2016 versus 2015
|
||
|
|
2017
|
2016
|
2015
|
Change
|
Percent
|
Change
|
Percent
|
|
|
|
|
|
|
|
|
|
|
Year
ended December 31,
|
$336,817
|
$627,930
|
$2,505,423
|
$(291,113)
|
(46)%
|
$(1,877,493)
|
(75)%
|
Fiscal Year Ended December 31, 2017, Compared to Fiscal Year Ended
December 31, 2016
Majority of the
revenue was derived from cell therapy technology service for the
year ended December 31, 2017. The decrease in revenue is the result
of prioritizing cancer therapeutic technologies, and focusing our
clinical efforts on developing CAR-T technologies, Vaccine, Tcm and
TCR clonality technologies. Such decrease in revenue was also
attributable to the fact that the Company ceased its cooperation
with the Jihua Hospital and several agents in the second quarter of
2016 and were not actively pursuing the fragmented technical
services opportunities.
Fiscal Year Ended December 31, 2016, Compared to Fiscal Year Ended
December 31, 2015
All the
revenue was derived from cell therapy technology service for year
ended December 31, 2016. The decrease in revenue is the result of
prioritizing cancer therapeutic technologies and focusing our
clinical efforts on developing CART technologies, Vaccine, Tcm and
TCR clonality technologies. As a result of not focusing on the cell
therapy technology service revenue, in the second quarter of 2016
the Company ceased its cooperation with the Jihua Hospital and
several agents.
Cost of Sales
|
|
|
|
|
2017 versus 2016
|
2016 versus 2015
|
||
|
|
2017
|
2016
|
2015
|
Change
|
Percent
|
Change
|
Percent
|
|
|
|
|
|
|
|
|
|
|
Year
ended December 31,
|
$162,218
|
$860,417
|
$1,880,331
|
$(698,199)
|
(81)%
|
$(1,019,914)
|
(54)%
|
Fiscal Year Ended December 31, 2017, Compared to Fiscal Year Ended
December 31, 2016
The
cost of sales decreased in line with the sales. The cost of sales
in 2016 was mainly due to the high fixed cost of Beijing site.
Since there was no revenue from the Beijing site in 2017, the cost
of sales decreased significantly.
Fiscal Year Ended December 31, 2016, Compared to Fiscal Year Ended
December 31, 2015
The
cost of sales decreased in line with the sales. As fixed costs,
such as rental and staff costs etc., accounts for a majority of the
cost of sales, the cost of sales didn’t decrease as much as
sales.
General and Administrative Expenses
|
|
|
|
|
2017 versus 2016
|
2016 versus 2015
|
||
|
|
2017
|
2016
|
2015
|
Change
|
Percent
|
Change
|
Percent
|
|
|
|
|
|
|
|
|
|
|
Year
ended December 31,
|
$12,780,483
|
$11,670,506
|
$13,068,255
|
$1,109,977
|
10%
|
$(1,397,749)
|
(11)%
|
Fiscal Year Ended December 31, 2017, Compared to Fiscal Year Ended
December 31, 2016
Increased expenses
in 2017 was primarily attributed to below facts:
●
An
increase in rental expenses of $2,224,000, which mainly resulted
from the new leased plant located in the “Pharma
Valley” of Shanghai from January 1, 2017;
●
A
decrease in legal, audit and other professional fees of $478,000,
which mainly attributed to the Company’s registration
statements on Forms S-3 and S-8 filed in first half of 2016 that
led to large professional fees in 2016;
●
A
decrease in salary of $465,000; and
●
A
decrease in insurance fee of $171,000, which mainly resulted from
the decrease in premium for director and officer liability and
Company reimbursement insurance.
Fiscal Year Ended December 31, 2016, Compared to Fiscal Year Ended
December 31, 2015
Decreased expenses
in 2016 were primarily attributed to below facts:
●
A decrease in
stock-based compensation expense of $1,838,000, which primarily
resulted from: i) forfeiture of the options in connection with the
resignation of Wei Cao as the CEO of the Company in February 2016
and as director in May 2016. For further details please refer to
Item 15 Note 16-Commitments and Contingencies - Service Agreement
with Wei (William) Cao in this annual report; ii) the issuance of a
large amount of options in the first quarter of 2013, most of which
vested over 3 years. With the end of vesting periods, the
stock-based compensation expense decreased significantly in 2016;
and
●
Offset by an
increase in legal, audit and other professional fees of $383,000,
which mainly related to the Company’s filing of Registration
Statements on Forms S-3 and S-8 in 2016.
Sales and Marketing
Expenses
|
|
|
|
|
2017
versus 2016
|
2016
versus 2015
|
||
|
|
2017
|
2016
|
2015
|
Change
|
Percent
|
Change
|
Percent
|
|
|
|
|
|
|
|
|
|
|
Year
ended December 31,
|
$360,766
|
$425,040
|
$709,151
|
$(64,274)
|
(15)%
|
$(284,111)
|
(40)%
|
Fiscal Year Ended December 31, 2017, Compared to Fiscal Year Ended
December 31, 2016
The
selling and marketing expenses decrease was mainly attributed to
the decline in travelling expenses of $21,000, salary of $16,000,
entertainment expenses of $15,000 and marketing expenses of $14,000
in 2017.
Fiscal Year Ended December 31, 2016, Compared to Fiscal Year Ended
December 31, 2015
Decreased expenses
in 2016 were primarily attributed to below facts:
●
A decrease in
stock-based compensation of $132,000. This mainly resulted from the
fact that one sales vice president resigned in April 2016 and part
of her options were forfeited; and
●
A decrease in
travelling expenses of $51,000, a decrease in market analysis and
other professional fees of $68,000 and a decrease in staff cost of
$35,000 due to the Company ceased cooperation with hospitals and
agents in the second quarter of 2016.
Research and Development Expenses
|
|
|
|
|
2017 versus 2016
|
2016 versus 2015
|
||
|
|
2017
|
2016
|
2015
|
Change
|
Percent
|
Change
|
Percent
|
|
|
|
|
|
|
|
|
|
|
Year
ended December 31,
|
$14,609,917
|
$11,475,587
|
$7,573,228
|
$3,134,330
|
27%
|
$3,902,359
|
52%
|
Fiscal Year Ended December 31, 2017, Compared to Fiscal Year Ended
December 31, 2016
Research and
development costs increased by approximately $3,134,000 as compared
to the year ended December 31, 2016. The increase was primarily
attributed to the facts below:
●
An increase in
payroll expenses of $626,000 as a result of headcount increase and
payroll raise. Total headcount for our R&D team increased from
81 as of December 31, 2016 to 98 as of December 31,
2017;
●
An increase in raw
material consumption of $447,000;
●
An increase in
rental expenses of $1,514,000, which was mainly attributed to the
launching of R&D activities at our Beijing facility in the 2nd
quarter of 2016 and the lease of a GMP facility in the United
States to commence the KOA preclinical and clinical studies in
2017; and
●
An increase in
depreciation and amortization of $455,000, which was mainly
attributed to the purchase of our new equipment for immunotherapy
research and development.
Fiscal Year Ended December 31, 2016, Compared to Fiscal Year Ended
December 31, 2015
Research and
development costs increased by approximately $3,902,000 as compared
to the year ended December 31, 2015. The increase was primarily
attributed to the facts below:
●
An increase in
payroll expenses of $1,581,000 in line with the increase of our
immunotherapy research and development team. Total headcount of our
R&D team increased from 47 as of December 31, 2015 to 81 as of
December 31, 2016;
●
An increase in
depreciation and amortization of $568,000, which was mainly
attributed to the technology obtained from 301 Hospital in June
2015 and newly purchased equipment for immunotherapy research and
development;
●
An increase in
clinical studies expenditure of $675,000;
●
An increase in raw
material consumption of $697,000;
●
An increase in
rental expense of $210,000; and
●
An increase in
travelling expense of $59,000.
Impairment of Investments
|
|
|
|
|
2017 versus 2016
|
2016 versus 2015
|
||
|
|
2017
|
2016
|
2015
|
Change
|
Percent
|
Change
|
Percent
|
|
|
|
|
|
|
|
|
|
|
Year
ended December 31,
|
$-
|
$4,611,714
|
$123,428
|
$(4,611,714)
|
(100)%
|
$4,488,286
|
3636%
|
Fiscal Year Ended December 31, 2017, Compared to Fiscal Year Ended
December 31, 2016
No
impairment of investment was made in 2017. The impairment of
investments in 2016 is attributed to the recognition of other than
temporary impairment on the value of shares in
investments.
Fiscal Year Ended December 31, 2016, Compared to Fiscal Year Ended
December 31, 2015
The
impairment of investments in 2016 and 2015 is attributed to the
recognition of other than temporary impairment on the value of
shares in investments. The impairment on investments was primarily
attributed to the valuation loss for the stock investment in Arem
Pacific Corporation, which share price recently significantly
dropped. The stock of ARPC held by us are illiquid restricted
shares that are very thinly traded on the OTC Markets, we consider
that it indicates the likelihood that the impairment is
other-than-temporary.
Operating Loss
|
|
|
|
|
2017 versus 2016
|
2016 versus 2015
|
||
|
|
2017
|
2016
|
2015
|
Change
|
Percent
|
Change
|
Percent
|
|
|
|
|
|
|
|
|
|
|
Year
ended December 31,
|
$(27,576,567)
|
$(28,415,334)
|
$(20,848,970)
|
$838,767
|
(3)%
|
$(7,566,364)
|
36%
|
The
decrease in the operating loss for 2017 as compared to 2016 and the
increase compared to 2015 was primarily due to changes in revenues,
cost of sales, general and administrative expenses, research and
development expenses and impairment of investments, each of which
was described above.
Other Income
|
|
|
|
|
2017 versus 2016
|
2016 versus 2015
|
||
|
|
2017
|
2016
|
2015
|
Change
|
Percent
|
Change
|
Percent
|
|
|
|
|
|
|
|
|
|
|
Year
ended December 31,
|
$2,088,707
|
$211,051
|
$672,648
|
$1,877,656
|
890%
|
$(461,597)
|
(69)%
|
Fiscal Year Ended December 31, 2017, Compared to Fiscal Year Ended
December 31, 2016
Other
income, net for the year ended December 31, 2017 was primarily
government subsidy of $2,077,000, interest income of $134,000, and
netting of the net foreign exchange loss of $112,000.
Other
income, net for the year ended December 31, 2016 was primarily
interest income of $79,000, third party R&D subsidy of $40,000,
net foreign exchange gain of $90,000 and government subsidy of
$78,000, netting of the charity donation of $78,000.
Fiscal Year Ended December 31, 2016, Compared to Fiscal Year Ended
December 31, 2015
Other
income, net for the year ended December 31, 2016 was primarily
interest income of $79,000, third party R&D subsidy of $40,000,
net foreign exchange gain of $90,000 and government subsidy of
$78,000, netting of the charity donation of $78,000.
Other
income, net for the year ended December 31, 2015 was primarily a
decrease in fair value of accrued expenses for the acquisition of
intangible assets of $346,000, government subsidy income of
$233,000 and interest income of $42,000. On June 26, 2015, the
Company completed its acquisition of the certain license rights to
technology and know-how from Blackbird and entered into an
assignment and assumption agreement to acquire all of
Blackbird’s right, title and interest in and to the exclusive
worldwide license to a CD40LGVAX vaccine from the University of
South Florida. According to the Asset Purchase Agreement, by and
among the Company, Blackbird and its principals, 28,120 shares of
Company common stock were issued as part of the consideration of
this transaction. In addition, 18,747 shares of Company common
stock (equal to $700,000 based on the 20-day volume-weighted
average price of the Company’s stock on the closing date)
will be delivered to Blackbird on the 6 month anniversary of the
closing date upon satisfaction of certain conditions. Those shares
were finally issued in November 2015 with unanimous consent of the
Board. Above shares were revalued according to the fair market
value as of issuance date and resulted in the other income of
$346,000.
Income Tax (Expenses) Credit
|
|
|
|
|
2017 versus 2016
|
2016 versus 2015
|
||
|
|
2017
|
2016
|
2015
|
Change
|
Percent
|
Change
|
Percent
|
|
|
|
|
|
|
|
|
|
|
Year
ended December 31,
|
$(2,450)
|
$(4,093)
|
$728,601
|
$1,643
|
(40)%
|
$(732,694)
|
(101)%
|
Fiscal Year Ended December 31, 2017, Compared to Fiscal Year Ended
December 31, 2016
While
we have optimistic plans for our business strategy, we determined
that a valuation allowance was necessary given the current and
expected near term losses and the uncertainty with respect to our
ability to generate sufficient profits from our business model.
Therefore, we established a valuation allowance for deferred tax
assets other than the extent of the benefit from other
comprehensive income. Income tax expenses for the year ended
December 31, 2017 and 2016 all represent US state tax.
Fiscal Year Ended December 31, 2016, Compared to Fiscal Year Ended
December 31, 2015
While
we have optimistic plans for our business strategy, we determined
that a valuation allowance was necessary given the current and
expected near term losses and the uncertainty with respect to our
ability to generate sufficient profits from our business model.
Therefore, we established a valuation allowance for deferred tax
assets other than the extent of the benefit from other
comprehensive income.
Income
tax expenses for the year ended December 31, 2016 all represent US
state tax.
Income
tax expense in 2015 mainly included the current income tax credit
of $733,000 as tax losses incurred in U.S. group companies for year
ended December 31, 2015.
Net Loss
|
|
|
|
|
2017 versus 2016
|
2016 versus 2015
|
||
|
|
2017
|
2016
|
2015
|
Change
|
Percent
|
Change
|
Percent
|
|
|
|
|
|
|
|
|
|
|
Year
ended December 31,
|
$(25,490,310)
|
$(28,208,376)
|
$(19,447,721)
|
$2,718,066
|
(10)%
|
$(8,760,655)
|
45%
|
Changes
in net loss are primarily attributable to changes in operations of
our biomedicine segment which are described above.
Comprehensive Loss
|
|
|
|
|
2017 versus 2016
|
2016 versus 2015
|
||
|
|
2017
|
2016
|
2015
|
Change
|
Percent
|
Change
|
Percent
|
|
|
|
|
|
|
|
|
|
|
Year
ended December 31,
|
$(24,763,121)
|
$(29,208,953)
|
$(21,132,211)
|
$4,445,832
|
(15)%
|
$(8,076,742)
|
38%
|
Fiscal Year Ended December 31, 2017, Compared to Fiscal Year Ended
December 31, 2016
Comprehensive net
loss for 2017 includes unrealized loss on investments of
approximately $240,000 and a currency translation net gain of
approximately $967,000 combined with the changes in net loss. The
unrealized loss on investments was attributed to the valuation
change for the stock investment in ARPC.
Fiscal Year Ended December 31, 2016, Compared to Fiscal Year Ended
December 31, 2015
Comprehensive net
loss for 2016 includes unrealized net gain on investments of
approximately $5,301,000, reclassification adjustments, net of tax,
of approximately $5,558,000, in connection with
other-than-temporary impairment of investments and a currency
translation net loss of approximately $743,000 combined with the
changes in net loss. The unrealized gain and reclassification
adjustments on investments were primarily attributed to the
valuation change for the stock investment in ARPC.
Comprehensive loss
for the year ended December 31, 2015 included an unrecognized loss
on investments of approximately $1,377,000, and a currency
translation net loss of approximately $308,000 combined with the
changes in net income. The unrecognized loss on investments was
primarily attributed to the valuation loss for the stock investment
in Arem Pacific Corporation.
Share-Based Compensation
Share-based
compensation totaled $5.3 million in 2017 ($5.5 million in 2016 and
$7.6 million in 2015). Share-based compensation was included in
cost of sales and operating expenses.
As of
December 31, 2017, unrecognized share-based compensation costs and
the weighted average periods over which the costs are expected to
be recognized were as follows:
|
|
Shares
|
Unrealised Share-Based Compensation Costs
|
Weighted Average Period
|
|
Non-vested
stock options
|
592,249
|
$4,362,541
|
2.29
year
|
|
Non-vested
restricted stock
|
34,105
|
$385,331
|
1.37
year
|
Non-vested
restricted stock above doesn’t include restricted stock
awards (RSU) to be issued under long-term incentive plan. The
Company is authorized to deduct the amount of tax withholding from
the amount payable to the grantee upon settlement of the RSUs. The
Company will withhold from the total number of shares of Common
Stock the grantee is to receive a number of shares the value of
which is sufficient to satisfy any such withholding obligation at
the minimum applicable withholding rate.
LIQUIDITY
AND CAPITAL RESOURCES
We had
working capital of $20,850,823 as of December 31, 2017 compared to
$38,328,048 as of December 31, 2016. Our cash position decreased to
$21,568,422 at December 31, 2017 compared to $39,252,432 at
December 31, 2016, as we had an increase in cash used in operating
and investing activities, partially offset by cash inflow generated
from financing activities due to a private placement financing in
2017 for aggregate net proceeds of approximately
$14,496,000,
Net
cash provided by or used in operating, investing and financing
activities from continuing operations were as follows (in
thousands):
Net
cash used in operating activities was approximately $18,593,000,
$15,868,000 and $11,751,000 for the years ended December 31, 2017,
2016 and 2015, respectively. The following table reconciles net
loss to net cash used in operating activities:
|
|
|
|
|
2017 versus 2016
|
2016 versus 2015
|
|
For year ended December 31,
|
2017
|
2016
|
2015
|
Change
|
Change
|
|
Net
loss
|
$(25,490,310)
|
$(28,208,376)
|
$(19,447,721)
|
$2,718,066
|
$(8,760,655)
|
|
Income
statement reconciliation items
|
8,331,491
|
12,596,060
|
9,595,098
|
(4,264,569)
|
3,000,962
|
|
Changes
in operating assets, net
|
(1,434,573)
|
(255,419)
|
(1,898,475)
|
(1,179,154)
|
1,643,056
|
|
Net
cash used in operating activities
|
$(18,593,392)
|
$(15,867,735)
|
$(11,751,098)
|
$(2,725,657)
|
$(4,116,637)
|
The
2017 change in operating assets and liabilities was primarily due
to an increase in accounts receivable, other receivables and
long-term prepaid expenses as well as the decrease in accrued
expenses and non-current liabilities, netting of by the increase in
other current liabilities. The 2016 change in operating assets and
liabilities was primarily due to an increase in prepaid expenses
and long-term prepaid expenses, net of the decrease in accounts
receivable and inventory.
Net
cash used in investing activities was approximately $10,193,000,
$2,733,000 and $7,702,000 for the years ended December 31, 2017,
2016 and 2015, respectively. These amounts were the result of
acquisition of business, purchases of fixed assets and intangible
assets.
Cash
provided by financing activities was approximately $10,826,000,
$43,286,000 and $19,647,000 for the years ended December 31, 2017,
2016 and 2015, respectively. These amounts were mainly attributable
to the proceeds received from the issuance of common stock and
exercise of stock options, netting of by the cash used in
repurchase of treasury stock.
Liquidity
and Capital Requirements Outlook
Excluding any
potential TCR clinical development or sponsorship of a CD40LGVAX
Trial in the U.S. and other regions outside of China CD40LGVAX
Trial, we anticipate that the Company will require approximately
$45 million in cash to operate as planned in the coming 12 months.
Of this amount, approximately $36 million will be used to operate
our facilities and offices, including but not limited to payroll
expenses, rent and other operating costs, and to fund our research
and development as we continue to develop our products through the
clinical study process. Approximately $9 million will be used as
capital expenditure in machinery, equipment and facilities to
expand our immune cell therapy business and CAR-T research and
development, although we may revise these plans depending on the
changing circumstances of our biopharmaceutical
business.
We
expect to rely on current cash balances that we hold to provide for
these capital requirements. We do not intend to use, and will not
rely on our holdings in securities to fund our operations.
One of our stocks held, Arem Pacific Corporation, has a
declared effective S-1 prospectus which relates to the resale of up
to 13,694,711 shares of common stock, inclusive of the 8,000,000
shares held by the Company. However, the shares offered by this
filing may only be sold by the selling stockholders at $0.05 per
share until the shares are quoted on the OTCQB® tier of OTC
Markets or an exchange. Another one of our stocks held, Wonder
International Education & Investment Group Corporation
(“Wonder”), is no longer traded on any stock market.
We do not know whether we can liquidate our 8,000,000 shares
of Arem Pacific stock or the 2,057,131 shares of Wonder stock or
any of our other portfolio securities, or if liquidated,
whether the realized amount will be meaningful at all. As a result,
we have written down above stocks to their fair value.
On
April 15, 2016, the Company completed the second and final closing
of a financing transaction with Wuhan Dangdai Science &
Technology Industries Group Inc., pursuant to which the Company
sold to the Investor 2,006,842 shares of the Company’s common
stock, par value $0.001 per share, for approximately $38,130,000 in
gross proceeds. As previously disclosed in a Current Report on Form
8-K filed on February 10, 2016, the Company conducted the initial
closing of the financing on February 4, 2016. The aggregate gross
proceeds from both closings in the financing totaled approximately
$43,130,000. In the aggregate, 2,270,000 shares of Common Stock
were issued in the financing. On March 22, 2016, the Company filed
a registration statement on Form S-3 to offer and sell from time to
time, in one or more series, any of the securities of the Company,
for total gross proceeds up to $150,000,000. On June 17, 2016, the
SEC declared the S-3 effective; we have yet to utilize any of the
$150,000,000 registered under the S-3. On December 26, 2017, the
Company entered into a Share Purchase Agreement with two investors,
pursuant to which the Company agreed to sell and the two investors
agreed to purchase from the Company, an aggregate of 1,166,667
shares of the Company’s common stock, par value $0.001 per
share, at $12.00 per share, for total gross proceeds of
approximately $14,000,000. The transaction closed on December 28,
2017. Together with a private placement with three of its executive
officers on December 22, 2017, the Company raised an aggregate of
approximately $14.5 million in the two private placements in
December 2017. On January 30, 2018 and February 5, 2018, the
Company entered into Securities Purchase Agreements with certain
investors, pursuant to which the Company agreed to sell, and the
Investors agreed to purchase from the Company, an aggregate of
1,719,324 shares of the Company’s common stock, par value
$0.001 per share, at $17.80 per share, for total gross proceeds of
approximately $30.6 million. The February 2018 Private
Placement closed on February 5, 2018. As we continue to incur
losses, achieving profitability is dependent upon the successful
development of our cell therapy business and commercialization of
our technology in research and development phase, which is a number
of years in the future. Once that occurs, we will have to achieve a
level of revenues adequate to support our cost structure. We may
never achieve profitability, and unless and until we do, we will
continue to need to raise additional capital. Management intends to
fund future operations through additional private or public debt or
equity offerings, and may seek additional capital through
arrangements with strategic partners or from other
sources.
Our
medium to long term capital needs involve the further development
of our biopharmaceutical business, and may include, at
management’s discretion, new clinical trials for other
indications, strategic partnerships, joint ventures, acquisition of
licensing rights from new or current partners and/or expansion of
our research and development programs. Furthermore, as our
therapies pass through the clinical trial process and if they gain
regulatory approval, we expect to expend significant resources on
sales and marketing of our future products, services and
therapies.
In
order to finance our medium to long-term plans, we intend to rely
upon external financing. This financing may be in the form of
equity and or debt, in private placements and/or public offerings,
or arrangements with private lenders. Due to our short operating
history and our early stage of development, particularly in our
biopharmaceutical business, we may find it challenging to raise
capital on terms that are acceptable to us, or at all. Furthermore,
our negotiating position in the capital raising process may worsen
as we consume our existing resources. Investor interest in a
company such as ours is dependent on a wide array of factors,
including the state of regulation of our industry in China (e.g.
the policies of MOH and the CFDA), the U.S. and other countries,
political headwinds affecting our industry, the investment climate
for issuers involved in businesses located or conducted within
China, the risks associated with our corporate structure, risks
relating to our partners, licensed intellectual property, as well
as the condition of the global economy and financial markets in
general. Additional equity financing may be dilutive to our
stockholders; debt financing, if available, may involve significant
cash payment obligations and covenants that restrict our ability to
operate as a business; our stock price may not reach levels
necessary to induce option or warrant exercises; and asset sales
may not be possible on terms we consider acceptable. If we are
unable to raise the capital necessary to meet our medium- and
long-term liquidity needs, we may have to delay or discontinue
certain clinical trials, the licensing, acquisition and/or
development of cell therapy technologies, and/or the expansion of
our biopharmaceutical business; or we may have to raise funds on
terms that we consider unfavorable.
Off-Balance
Sheet Transactions
We do
not have any off-balance sheet arrangements except the lease and
capital commitment described in “Contractual
Obligations” below.
Contractual
Obligations
We
have various contractual obligations that will affect our
liquidity. The following table sets forth our contractual
obligations as of December 31, 2017.
|
|
Payments due by period
|
||||
|
Contractual Obligations
|
|
Less than
|
2-3
|
4-5
|
More than
|
|
|
Total
|
1 year
|
years
|
years
|
5 years
|
|
Capital Commitment
|
$1,444,449
|
$1,428,035
|
$16,414
|
$-
|
$-
|
|
Operating Lease Obligations
|
23,684,480
|
3,062,842
|
5,449,762
|
5,095,980
|
10,075,896
|
|
Total
|
$25,128,929
|
$4,490,877
|
$5,466,176
|
$5,095,980
|
$10,075,896
|
ITEM
7A. QUANTITATIVE AND
QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Exposure to credit,
liquidity, interest rate and currency risks arises in the normal
course of the Company’s business. The Company’s
exposure to these risks and the financial risk management policies
and practices used by the Company to manage these risks are
described below.
Credit
Risk
Credit
risk is the risk that one party to a financial instrument will
cause a financial loss for the other party by failing to discharge
an obligation. The Company’s credit risk is primarily
attributable to cash at bank and receivables etc. Exposure to these
credit risks are monitored by management on an ongoing
basis.
The
cash at bank of the Group is mainly held with well-known or state
owned financial institutions, such as HSBC, Bank of China and China
Merchant Bank etc. Management does not foresee any significant
credit risks from these deposits and does not expect that these
financial institutions may default and cause losses to the
Company.
In
respect of receivables, the Company does not obtain collateral from
customers. The Company’s exposure to credit risk is
influenced mainly by the individual characteristics of each
customer rather than the industry, country or area in which the
customers operate and therefore significant concentrations of
credit risk arise primarily when the Group has significant exposure
to individual customers. As of December 31, 2017, 82% of the total
accounts receivable was due from the largest customer of the
Company.
The
maximum exposure to credit risk is represented by the carrying
amount of each financial asset in the balance sheet.
Interest
Rate Risk
The
Company’s interest rate risk arises primarily from cash
deposited at banks and the Company doesn’t have any
interest-bearing long-term payable/ borrowing, therefore the
exposure to interest rate risk is limited.
Currency
Risk
The
Company is exposed to currency risk primarily from sales and
purchases which give rise to receivables, payables that are
denominated in a foreign currency (mainly RMB). The Company has
adopted USD as its functional currency, thus the fluctuation of
exchange rates between RMB and USD exposes the Company to currency
risk.
The
following table details the Company’s exposure as of December
31, 2017 to currency risk arising from recognised assets or
liabilities denominated in a currency other than the functional
currency of the entity to which they relate. For presentation
purposes, the amounts of the exposure are shown in USD translated
using the spot rate as of December 31, 2017. Differences resulting
from the translation of the financial statements of entities into
the Company’s presentation currency are
excluded.
|
|
Exposure to foreign currencies (Expressed in USD)
|
|
|
|
As of December 31, 2017
|
|
|
|
RMB
|
USD
|
|
Cash
and cash equivalents
|
2,604
|
372,330
|
|
Net
exposure arising from recognised assets and
liabilities
|
2,604
|
372,330
|
The
following table indicates the instantaneous change in the
Company’s net loss that would arise if foreign exchange rates
to which the Company has significant exposure at the end of the
reporting period had changed at that date, assuming all other risk
variables remained constant.
|
|
As of December 31, 2017
|
|
|
|
increase/(decrease)
in foreign exchange rates
|
Effect
on net loss (Expressed in USD)
|
|
RMB
(against USD)
|
5%
|
(18,486)
|
|
|
|
|
|
|
-5%
|
18,486
|
Results
of the analysis as presented in the above table represent an
aggregation of the instantaneous effects on each of the
Company’s subsidiaries’ net loss measured in the
respective functional currencies, translated into USD at the
exchange rate ruling at the end of the reporting period for
presentation purposes.
The
sensitivity analysis assumes that the change in foreign exchange
rates had been applied to re-measure those financial instruments
held by the Company which expose the Company to foreign currency
risk at the end of the reporting period, including inter-company
payables and receivables within the Company which are denominated
in a currency other than the functional currencies of the lender or
the borrower. The analysis excludes differences that would result
from the translation of the financial statements of subsidiaries
into the Company’s presentation currency.
ITEM 8.
FINANCIAL STATEMENTS AND
SUPPLEMENTARY DATA
Attached hereto and
filed as a part of this Annual Report on Form 10-K are our
Consolidated Financial Statements, beginning on page
F-1.
ITEM 9.
CHANGES IN AND DISAGREEMENTS WITH
ACCOUNTANTS ON ACCOUNTING AND FINANCIAL
DISCLOSURE
None.
ITEM
9A. CONTROLS AND
PROCEDURES
Evaluation of Disclosure Controls and
Procedures
We have
established disclosure controls and procedures, as such term is
defined in Rule 13a-15(e) under the Securities Exchange Act of
1934. Our disclosure controls and procedures are designed to ensure
that material information relating to us, including our
consolidated subsidiaries, is made known to our principal executive
officer and principal financial officer by others within our
organization. Under the supervision and with the participation of
our management, including our principal executive officer and
principal financial officer, we conducted an evaluation of the
effectiveness of our disclosure controls and procedures as of
December 31, 2017 to ensure that the information required to be
disclosed by us in the reports that we file or submit under the
Securities Exchange Act of 1934 is recorded, processed, summarized,
and reported within the time periods specified in the SEC’s
rules and forms. Disclosure controls and procedures include,
without limitation, controls and procedures designed to ensure that
information required to be disclosed by us in the reports that we
file or submit under the Securities Exchange Act of 1934 is
accumulated and communicated to our management, including our
principal executive officer and principal financial officer as
appropriate, to allow timely decisions regarding required
disclosure. Based on this evaluation, our principal executive
officer and principal financial officer concluded that our
disclosure controls and procedures were effective as of December
31, 2017.
Management’s Annual Report on Internal Control Over Financial
Reporting
Our
management is responsible for establishing and maintaining adequate
internal control over financial reporting. Under the supervision
and with the participation of our management, including our
principal executive officer and principal financial officer, we
conducted an evaluation of the effectiveness of our internal
control over financial reporting as of December 31, 2017, based on
the criteria established in Internal Control — Integrated
Framework (2013) issued by the Committee of Sponsoring
Organizations of the Treadway Commission (COSO). Based on this
evaluation, our management concluded that our internal control over
financial reporting was effective as of December 31, 2017. Our
internal control over financial reporting as of December 31, 2017,
has been audited and attested to by BDO China Shu Lun Pan Certified
Public Accountants LLP, or BDO China, an independent registered
public accounting firm, as stated in its report, which is included
herein.
Changes in Internal Control over Financial Reporting
During
the year ended December 31, 2017, there were no changes in our
internal control over financial reporting that materially affected,
or that are reasonably likely to materially affect, our internal
control over financial reporting.
ITEM
9B. OTHER
INFORMATION
UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF
PROCEEDS
As
previously disclosed on a Current Report on Form 8-K filed on June
1, 2017, the Company authorized a share repurchase program (the
“Share Repurchase Program”), pursuant to which the
Company may, from time to time, purchase shares of its common stock
for an aggregate purchase price not to exceed $10 million. The
table below summarizes purchases made by or on behalf of the
Company or affiliated purchasers as defined in Regulation S-K under
the Share Purchase Program.
|
Period
|
Total number of shares purchased
|
Average price paid per share
|
Total number of shares purchased as part of publicly announced
plans or programs
|
Maximum dollar value of shares that may yet be purchased under the
plans or programs
|
|
|
|
|
|
|
|
June
9, 2017 ~ June 30, 2017
|
170,169
|
$7.98
|
170,169
|
|
|
July
1, 2017 ~ September 30, 2017
|
114,156
|
$9.51
|
114,156
|
|
|
October
1, 2017 ~ December 31, 2017
|
142,469
|
$10.77
|
142,469
|
|
|
Total
|
426,794
|
$9.32
|
426,794
|
6,022,071
|
In
December 2017, the Company entered into Share Purchase Agreements
with two investors and three of its executive officers
(collectively, the “Purchasers”), pursuant to which the
Company sold to the Purchasers an aggregate of 1,208,334 shares
(the “Shares”) of the Company’s common stock at
$12.00 per share, for total gross proceeds of approximately
$14,500,000. The issuance to the two investors and the executive
officers was made in reliance on the exemption from registration
provided by Regulation S and Section 4(a)(2), respectively, under
the Securities Act.
PART III
Item 10. Directors, Executive Officers and
Corporate Governance
We
will file with the SEC a definitive Proxy Statement for our Annual
Meeting of Stockholders (the “2017 Proxy Statement”)
not later than 120 days after the fiscal year ended December 31,
2017. The information required by this item is incorporated herein
by reference to the information contained in the 2017 Proxy
Statement.
Item 11. Executive Compensation
The
information required by this item is incorporated herein by
reference to the information contained in the 2017 Proxy
Statement.
Item 12. Security Ownership of
Certain Beneficial Owners and Management and Related Stockholder
Matters
The
information required by this item is incorporated herein by
reference to the information contained in the 2017 Proxy
Statement.
Item 13. Certain Relationships and
Related Transactions, and Director Independence
The
information required by this item is incorporated herein by
reference to the information contained in the 2017 Proxy
Statement.
Item 14. Principal Accounting Fees
and Services
The
information required by this item is incorporated herein by
reference to the information contained in the 2017 Proxy
Statement.
PART IV
ITEM
15. EXHIBITS AND FINANCIAL
STATEMENT SCHEDULES.
|
Exhibit Number
|
|
Description
|
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
||
|
|
|
101.INS*
|
|
XBRL
Instance Document
|
|
101.SCH*
|
|
XBRL
Taxonomy Extension Schema Document
|
|
101.CAL*
|
|
XBRL
Taxonomy Extension Calculation Linkbase Document
|
|
101.DEF*
|
|
XBRL
Taxonomy Extension Definition Linkbase Document
|
|
101.LAB*
|
|
XBRL
Taxonomy Extension Label Linkbase Document
|
|
101.PRE*
|
|
XBRL
Taxonomy Extension Presentation Linkbase Document
|
* Filed
herewith.
———————
|
1.
|
Incorporated by
reference filed with the Registration Statement on Form 10-SB filed
with the Securities and Exchange Commission on October 30, 2006
(File No. 000-52282)
|
|
2.
|
Incorporated by
reference filed with the Registration Statement on Form 10-SB/A
filed with the Securities and Exchange Commission on February 27,
2007 (File No. 000-52282)
|
|
3.
|
Incorporated by
reference filed with the Registration Statement on Form S-8 filed
with the Securities and Exchange Commission on June 19, 2007 (File
No. 333-143878)
|
|
4.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on July 20, 2007 (File No.
000-52282)
|
|
5.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on September 25, 2007 (File No.
000-52282)
|
|
6.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on October 1, 2007 (File No.
000-52282)
|
|
7.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on October 9, 2007 (File No.
000-52282)
|
|
8.
|
Incorporated by
reference filed with the Registration Statement on Form S-8 filed
with the Securities and Exchange Commission on August 22, 2008
(File No. 333-153129)
|
|
9.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on October 22, 2008 (File No.
000-52282)
|
|
10.
|
Incorporated by
reference filed with the Registration Statement on Form S-8 filed
with the Securities and Exchange Commission on April 15, 2009 (File
No. 333-158583)
|
|
11.
|
Incorporated by
reference filed with the Form 8-K/A filed with the Securities and
Exchange Commission on December 12, 2013 (File No.
000-52282)
|
|
12.
|
Incorporated by
reference filed with the Form 10-K filed with the Securities and
Exchange Commission on April 15, 2010 (File No.
000-52282)
|
|
13.
|
Incorporated
by reference filed with the Form 8-K filed with the Securities and
Exchange Commission on July 14, 2010 (File No.
000-52282)
|
|
14.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on November 12, 2010 (File No.
000-52282
|
|
15.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on December 7, 2010 (File No.
000-52282)
|
|
16.
|
Incorporated by
reference filed with the Form 10-K filed with the Securities and
Exchange Commission on June 18, 2013 (File No.
000-52282)
|
|
17.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on November 20, 2012 (File No.
000-52282)
|
|
18.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on January 22, 2013 (File No.
000-52282)
|
|
19.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on February 4, 2013 (File No.
000-52282)
|
|
20.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on February 12, 2013 (File No.
000-52282)
|
|
21.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on January 3, 2012 (File No.
000-52282)
|
|
22.
|
Incorporated by
reference filed with the Registration Statement on Form S-8 filed
with the Securities and Exchange Commission on March 7, 2012 (File
No. 333-179974)
|
|
23.
|
Incorporated by
reference filed with the Form 10-K filed with the
Securities and Exchange Commission on April 4, 2013 (File
No. 000-52282)
|
|
24.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on December 16, 2013 (File No.
000-52282)
|
|
25.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on January 3, 2014 (File No.
000-52282)
|
|
26.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on October 2, 2014 (File No.
001-36498)
|
|
27.
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on July 2, 2015 (File No.
001-36498)
|
|
28.
|
Incorporated by
reference filed with Schedule 14A filed with the Securities and
Exchange Commission on November 21, 2013 (File No.
000-52282)
|
|
29.
|
Incorporated by
reference filed with Schedule 14A filed with the Securities and
Exchange Commission on September 23, 2014 (File No.
001-36498)
|
|
30
|
Incorporated by
reference filed with the Form 10-K filed with the Securities and
Exchange Commission on April 15, 2014 (File No.
000-52282).
|
|
31
|
Incorporated by
reference filed with the Form 10-K filed with the Securities and
Exchange Commission on March 31, 2015 (File No.
001-36498).
|
|
32
|
Incorporated by
reference filed with the Form 10-K filed with the Securities and
Exchange Commission on March 14, 2016 (File No.
001-36498).
|
|
33
|
Incorporated by
reference filed with the Form 10-Q filed with the Securities and
Exchange Commission on May 9, 2016 (File No.
001-36498).
|
|
34
|
Incorporated by
reference filed with the Form 10-Q filed with the Securities and
Exchange Commission on August 8, 2016 (File No.
001-36498).
|
|
35
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on February 4, 2016 (File No. 000-
36498).
|
|
36
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on April 15, 2016 (File No. 000-
36498).
|
|
37
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on November 15, 2016 (File No. 000-
36498).
|
|
38
|
Incorporated by
reference filed with Schedule 14A/A filed with the Securities and
Exchange Commission on March 23, 2017 (File No.
001-36498)
|
|
39
|
Incorporated by
reference filed with the Form 10-K filed with the Securities and
Exchange Commission on March 13, 2017 (File No.
001-36498).
|
|
40
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on January 8, 2013 (File No.
000-52282)
|
|
41
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on September 21, 2016 (File No.
000-36498).
|
|
42
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on December 21, 2017 (File No.
000-36498).
|
|
43
|
Incorporated by
reference filed with the Form 8-K filed with the Securities and
Exchange Commission on December 28, 2017 (File No.
000-36498).
|
Item 16. Form 10-K Summary
Not
applicable.
SIGNATURES
Pursuant to the
requirements of Section 13 or 15(d) of the Securities Exchange Act
of 1934, the registrant has duly caused this report to be signed on
its behalf by the undersigned, there unto duly
authorized.
|
|
Cellular Biomedicine Group, Inc.
|
|
|
|
|
|
|
|
|
Date:
March 5, 2018
|
By:
|
/s/
Bizou (Tony) Liu
|
|
|
|
|
Bizuo
(Tony) Liu
|
|
|
|
|
Chief
Executive Officer and Chief Financial Officer
(principal
executive officer and financial and accounting
officer)
|
|
|
|
|
|
|
Pursuant to the
requirements of the Exchange Act, this report has been signed below
by the following persons on behalf of the Company and in the
capacities and on the dates indicated.
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
/s/
Terry A. Belmont
|
|
Chairman of the
Board of Directors
|
|
March
5, 2018
|
|
Terry
A. Belmont
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
/s/
Bizuo (Tony) Liu
|
|
Chief
Executive Officer and Chief Financial Officer
|
|
March
5, 2018
|
|
Bizuo
(Tony) Liu
|
|
(principal
executive officer and financial and accounting
officer)
|
|
|
|
|
|
|
|
|
|
/s/ Wen
Tao (Steve) Liu
|
|
Director
|
|
March
5, 2018
|
|
Wen Tao
(Steve) Liu
|
|
|
|
|
|
|
|
|
|
|
|
/s/
Hansheng Zhou
|
|
Director
|
|
March
5, 2018
|
|
Hansheng
Zhou
|
|
|
|
|
|
|
|
|
|
|
|
/s/
Nadir Patel
|
|
Director
|
|
March
5, 2018
|
|
Nadir
Patel
|
|
|
|
|
|
|
|
|
|
|
|
/s/
Chun Kwok Alan Au
|
|
Director
|
|
March
5, 2018
|
|
Chun
Kwok Alan Au
|
|
|
|
|
|
|
|
|
|
|
|
/s/
Gang Ji
|
|
Director
|
|
March
5, 2018
|
|
Gang
Ji
|
|
|
|
|
|
/s/
Bosun S. Hau
|
|
Director
|
|
March
5, 2018
|
|
Bosun
S. Hau
|
|
|
|
|
CELLULAR
BIOMEDICINE GROUP, INC.
TABLE
OF CONTENTS
|
|
|
Page
|
|
|
|
|
|
F-2
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
F-4
|
|
|
|
|
|
|
|
|
|
|
|
F-5
|
|
|
|
|
|
|
|
|
|
|
|
F-6
|
|
|
|
|
|
|
|
|
|
|
|
F-7
|
|
|
|
|
|
|
|
|
|
|
|
F-8
|
|
|
REPORT OF INDEPENDENT
REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
Cellular Biomedicine Group, Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of
Cellular Biomedicine Group, Inc. and its subsidiaries and variable
interest entities (the “Company”) as of December 31,
2017 and 2016 and the related consolidated statements of operations
and comprehensive loss, changes in stockholders’ equity, and
cash flows for each of the three years in the period ended December
31, 2017, and the related notes (collectively referred to as the
“consolidated financial statements”). In our opinion,
the consolidated financial statements present fairly, in all
material respects, the financial position of the Company at
December 31, 2017 and 2016, and the results of its operations and
its cash flows for each of the three years in the period ended
December 31, 2017, in conformity with accounting principles
generally accepted in the United States of America.
We also have audited, in accordance with the standards of the
Public Company Accounting Oversight Board (United States)
(“PCAOB”), the Company’s internal control over
financial reporting as of December 31, 2017, based on criteria
established in Internal Control –
Integrated Framework (2013) issued by the Committee of Sponsoring
Organizations of the Treadway Commission (“COSO”) and
our report dated March 5, 2018 expressed an unqualified opinion
thereon.
Basis for Opinion
These consolidated financial statements are the responsibility of
the Company’s management. Our responsibility is to express an
opinion on the Company’s consolidated financial statements
based on our audits. We are a public accounting firm registered
with the PCAOB and are required to be independent with respect to
the Company in accordance with the U.S. federal securities laws and
the applicable rules and regulations of the Securities and Exchange
Commission and the PCAOB.
We conducted our audits in accordance with the standards of the
PCAOB. Those standards require that we plan and perform the audit
to obtain reasonable assurance about whether the consolidated
financial statements are free of material misstatement, whether due
to error or fraud.
Our audits included performing procedures to assess the risks of
material misstatement of the consolidated financial statements,
whether due to error or fraud, and performing procedures that
respond to those risks. Such procedures included examining, on a
test basis, evidence regarding the amounts and disclosures in the
consolidated financial statements. Our audits also included
evaluating the accounting principles used and significant estimates
made by management, as well as evaluating the overall presentation
of the consolidated financial statements. We believe that our
audits provide a reasonable basis for our opinion.
/s/ BDO China Shu Lun Pan Certified Public Accountants
LLP
We have served as the Company’s auditor since
2015.
Shenzhen, the People’s Republic of China
March 5, 2018
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING
FIRM
To the
Board of Directors and Stockholders of
Cellular
Biomedicine Group, Inc.
Opinion on Internal Control over Financial Reporting
We have audited the internal control over financial reporting of
Cellular Biomedicine Group, Inc. and its subsidiaries and variable
interest entities (the “Company”) as of December 31,
2017, based on criteria established in Internal Control - Integrated
Framework (2013) issued by the
Committee of Sponsoring Organizations of the Treadway Commission
(the “COSO criteria”). In our opinion, the Company
maintained, in all material respects, effective internal control
over financial reporting as of December 31, 2017, based on the COSO
criteria.
We also have audited, in accordance with the standards of the
Public Company Accounting Oversight Board (United States)
(“PCAOB”), the consolidated balance sheets of the
Company as of December 31, 2017 and 2016, the related consolidated
statements of operations and comprehensive loss, changes in
stockholders’ equity, and cash flows for each of the three
years in the period ended December 31, 2017, and the related notes
and our report dated March 5, 2018 expressed an unqualified opinion
thereon.
Basis for Opinion
The Company’s management is responsible for maintaining
effective internal control over financial reporting and for its
assessment of the effectiveness of internal control over financial
reporting, included in the accompanying Item 9A,
Controls and
Procedures, Management’s Annual Report on Internal Control
Over Financial Reporting. Our
responsibility is to express an opinion on the Company’s
internal control over financial reporting based on our audit. We
are a public accounting firm registered with the PCAOB and are
required to be independent with respect to the Company in
accordance with U.S. federal securities laws and the applicable
rules and regulations of the Securities and Exchange Commission and
the PCAOB.
We conducted our audit in accordance with the standards of the
PCAOB. Those standards require that we plan and perform the audit
to obtain reasonable assurance about whether effective internal
control over financial reporting was maintained in all material
respects. Our audit included obtaining an understanding of internal
control over financial reporting, assessing the risk that a
material weakness exists, and testing and evaluating the design and
operating effectiveness of internal control based on the assessed
risk. Our audit also included performing such other procedures as
we considered necessary in the circumstances. We believe that our
audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial
Reporting
A company’s internal control over financial reporting is a
process designed to provide reasonable assurance regarding the
reliability of financial reporting and the preparation of financial
statements for external purposes in accordance with generally
accepted accounting principles. A company’s internal control
over financial reporting includes those policies and procedures
that (1) pertain to the maintenance of records that, in reasonable
detail, accurately and fairly reflect the transactions and
dispositions of the assets of the company; (2) provide reasonable
assurance that transactions are recorded as necessary to permit
preparation of financial statements in accordance with generally
accepted accounting principles, and that receipts and expenditures
of the company are being made only in accordance with
authorizations of management and directors of the company; and (3)
provide reasonable assurance regarding prevention or timely
detection of unauthorized acquisition, use, or disposition of the
company’s assets that could have a material effect on the
financial statements.
Because of its inherent limitations, internal control over
financial reporting may not prevent or detect misstatements. Also,
projections of any evaluation of effectiveness to future periods
are subject to the risk that controls may become inadequate because
of changes in conditions, or that the degree of compliance with the
policies or procedures may deteriorate.
/s/ BDO China Shu Lun Pan Certified Public Accountants
LLP
Shenzhen, the People’s Republic of China
March 5, 2018
CELLULAR
BIOMEDICINE GROUP, INC.
CONSOLIDATED BALANCE
SHEETS
|
|
December 31,
|
December 31,
|
|
|
2017
|
2016
|
|
Assets
|
|
|
|
Cash
and cash equivalents
|
$21,568,422
|
$39,252,432
|
|
Accounts
receivable, less allowance for doubtful amounts of
$10,789
|
|
|
|
and
$10,163 as of December 31, 2017 and December 31, 2016,
respectively
|
202,887
|
39,974
|
|
Other
receivables
|
902,940
|
412,727
|
|
Prepaid
expenses
|
1,852,695
|
986,951
|
|
Total
current assets
|
24,526,944
|
40,692,084
|
|
|
|
|
|
Investments
|
269,424
|
509,424
|
|
Property,
plant and equipment, net
|
12,973,342
|
4,117,739
|
|
Goodwill
|
7,678,789
|
7,678,789
|
|
Intangibles,
net
|
12,419,692
|
14,092,581
|
|
Long-term
prepaid expenses and other assets
|
3,294,105
|
1,537,850
|
|
Total
assets (1)
|
$61,162,296
|
$68,628,467
|
|
|
|
|
|
Liabilities and Stockholders' Equity
|
|
|
|
|
|
|
|
Liabilities:
|
|
|
|
Accounts
payable
|
$225,287
|
$216,154
|
|
Accrued
expenses
|
1,097,327
|
1,168,787
|
|
Taxes
payable
|
28,875
|
28,875
|
|
Other
current liabilities
|
2,324,632
|
950,220
|
|
Total
current liabilities
|
3,676,121
|
2,364,036
|
|
|
|
|
|
Other
non-current liabilities
|
183,649
|
370,477
|
|
Total
liabilities (1)
|
3,859,770
|
2,734,513
|
|
Commitments
and Contingencies (note 13)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Preferred stock, par value $.001, 50,000,000 shares
|
|
|
|
authorized; none issued and outstanding as of
|
|
|
|
December 31, 2017 and 2016, respectively
|
-
|
-
|
|
|
|
|
|
Common stock, par value $.001, 300,000,000 shares
authorized;
|
|
|
|
15,615,558 and 14,281,378 issued; and 15,188,764 and 14,281,378
outstanding,
|
|
|
|
as of December 31, 2017 and 2016, respectively
|
15,616
|
14,281
|
|
Treasury stock at cost; 426,794 and nil shares of common
stock
|
(3,977,929)
|
-
|
|
as of December 31, 2017 and December 31, 2016,
respectively
|
|
|
|
Additional
paid in capital
|
172,691,339
|
152,543,052
|
|
Accumulated deficit
|
(111,036,997)
|
(85,546,687)
|
|
Accumulated other comprehensive loss
|
(389,503)
|
(1,116,692)
|
|
Total
stockholders' equity
|
57,302,526
|
65,893,954
|
|
|
|
|
|
Total
liabilities and stockholders' equity
|
$61,162,296
|
$68,628,467
|
|
(1)
|
The
Company’s consolidated assets as of December 31, 2017 and
2016 included $21,775,087 and $9,626,171, respectively, of assets
of variable interest entities, or VIEs, that can only be used to
settle obligations of the VIEs. Each of the following amounts
represent the balances as of December 31, 2017 and 2016,
respectively. These assets include cash and cash equivalents of
$2,337,173 and $4,021,992; other receivables of $773,384 and
$370,702; prepaid expenses of $1,750,509 and $777,445; property,
plant and equipment, net, of $12,477,315 and $2,398,576;
intangibles of $1,516,449 and $1,613,582; and long-term prepaid
expenses and other assets of $2,920,257 and $443,874. The
Company’s consolidated liabilities as of December 31, 2017
and 2016 included $2,688,520 and $1,372,391, respectively, of
liabilities of the VIEs whose creditors have no recourse to the
Company. These liabilities include accounts payable of $181,231 and
$161,825; other payables of $1,631,582 and $407,769; payroll
accrual of $682,248 and $792,706;deferred income of $9,810 and
nil;and other non-current liabilities of $183,649 and $10,091. See
further description in Note 4, Variable Interest
Entities.
|
The
accompanying notes are an integral part of these consolidated
financial statements.
CELLULAR BIOMEDICINE GROUP, INC.
CONSOLIDATED
STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
|
|
For the Year Ended
December 31,
|
||
|
|
2017
|
2016
|
2015
|
|
Net
sales and revenue
|
$336,817
|
$627,930
|
$2,505,423
|
|
|
|
|
|
|
Operating
expenses:
|
|
|
|
|
Cost
of sales
|
162,218
|
860,417
|
1,880,331
|
|
General
and administrative
|
12,780,483
|
11,670,506
|
13,068,255
|
|
Selling
and marketing
|
360,766
|
425,040
|
709,151
|
|
Research
and development
|
14,609,917
|
11,475,587
|
7,573,228
|
|
Impairment
of investments
|
-
|
4,611,714
|
123,428
|
|
Total operating expenses
|
27,913,384
|
29,043,264
|
23,354,393
|
|
Operating
loss
|
(27,576,567)
|
(28,415,334)
|
(20,848,970)
|
|
|
|
|
|
|
Other
income:
|
|
|
|
|
Interest
income
|
133,621
|
78,943
|
42,220
|
|
Other
income
|
1,955,086
|
132,108
|
630,428
|
|
Total other income
|
2,088,707
|
211,051
|
672,648
|
|
Loss
before taxes
|
(25,487,860)
|
(28,204,283)
|
(20,176,322)
|
|
|
|
|
|
|
Income
taxes (expenses) credit
|
(2,450)
|
(4,093)
|
728,601
|
|
|
|
|
|
|
|
|
|
|
|
Net
loss
|
$(25,490,310)
|
$(28,208,376)
|
$(19,447,721)
|
|
Other
comprehensive income (loss):
|
|
|
|
|
Cumulative
translation adjustment
|
967,189
|
(743,271)
|
(307,950)
|
|
Unrealized gain (loss) on investments, net of
tax
|
(240,000)
|
5,300,633
|
(1,376,540)
|
|
Reclassification adjustments, net of tax, in connection with
other-than-temporary impairment of investments
|
-
|
(5,557,939)
|
-
|
|
Total
other comprehensive income (loss):
|
727,189
|
(1,000,577)
|
(1,684,490)
|
|
|
|
|
|
|
Comprehensive
loss
|
$(24,763,121)
|
$(29,208,953)
|
$(21,132,211)
|
|
|
|
|
|
|
Net
loss per share
|
|
|
|
|
Basic
|
$(1.78)
|
$(2.09)
|
$(1.70)
|
|
Diluted
|
$(1.78)
|
$(2.09)
|
$(1.70)
|
|
|
|
|
|
|
Weighted
average common shares outstanding:
|
|
|
|
|
Basic
|
14,345,604
|
13,507,408
|
11,472,306
|
|
Diluted
|
14,345,604
|
13,507,408
|
11,472,306
|
The
accompanying notes are an integral part of these consolidated
financial statements.
CELLULAR BIOMEDICINE
GROUP, INC.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS'
EQUITY
|
|
|
|
|
|
|
|
|
|
Accumulated Other
|
|
|
|
Common Stock
|
Preferred Stock
|
Treasury Stock
|
Additional
|
Accumulated
|
Comprehensive
|
|
|||
|
|
Shares
|
Amount
|
Shares
|
Amount
|
Shares
|
Amount
|
Paid in Capital
|
Deficit
|
Income (Loss)
|
Total
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at
December 31, 2014
|
10,990,335
|
10,990
|
-
|
-
|
-
|
-
|
75,467,316
|
(37,890,590)
|
1,568,375
|
39,156,091
|
|
|
|
|
|
|
|
|
|
|
|
.
|
|
Common stock
issued with Private Placement
|
|
|
|
|
|
|
|
|
|
|
|
Memorandum
("PPM")
|
515,786
|
516
|
-
|
-
|
-
|
-
|
18,584,338
|
-
|
-
|
18,584,854
|
|
Common stock
issued for
acquisition
of intangible assets
|
46,867
|
47
|
-
|
-
|
-
|
-
|
1,481,415
|
|
|
1,481,462
|
|
Restricted
stock grants
|
6,253
|
6
|
-
|
-
|
-
|
-
|
410,314
|
-
|
-
|
410,320
|
|
Accrual of
stock options
|
-
|
-
|
-
|
-
|
-
|
-
|
7,182,117
|
-
|
-
|
7,182,117
|
|
Exercise of
stock options
|
152,404
|
152
|
-
|
-
|
-
|
-
|
682,151
|
-
|
-
|
682,303
|
|
Unrealized
loss on investments, net of tax
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(1,376,540)
|
(1,376,540)
|
|
Foreign
currency translation
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(307,950)
|
(307,950)
|
|
Net
loss
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(19,447,721)
|
-
|
(19,447,721)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at
December 31, 2015
|
11,711,645
|
11,711
|
-
|
-
|
-
|
-
|
103,807,651
|
(57,338,311)
|
(116,115)
|
46,364,936
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Common stock
issued with PPM and other financing
|
2,348,888
|
2,349
|
-
|
-
|
-
|
-
|
42,397,525
|
-
|
-
|
42,399,874
|
|
Restricted
stock grants
|
24,660
|
25
|
-
|
-
|
-
|
-
|
709,472
|
-
|
-
|
709,497
|
|
Accrual of
stock options
|
-
|
-
|
-
|
-
|
-
|
-
|
4,742,920
|
-
|
-
|
4,742,920
|
|
Exercise of
stock options
|
196,185
|
196
|
-
|
-
|
-
|
-
|
885,484
|
-
|
-
|
885,680
|
|
Unrealized
loss on investments, net of tax
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
5,300,633
|
5,300,633
|
|
Reclassification
adjustments, net of tax, in connection with other-than-temporary
impairment of investments
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(5,557,939)
|
(5,557,939)
|
|
Foreign
currency translation
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(743,271)
|
(743,271)
|
|
Net
loss
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(28,208,376)
|
-
|
(28,208,376)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at
December 31, 2016
|
14,281,378
|
14,281
|
-
|
-
|
-
|
-
|
152,543,052
|
(85,546,687)
|
(1,116,692)
|
65,893,954
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Common stock
issued with PPM
|
1,208,334
|
1,208
|
-
|
-
|
-
|
-
|
14,494,832
|
-
|
-
|
14,496,040
|
|
Restricted
stock grants
|
68,446
|
69
|
-
|
-
|
-
|
-
|
832,950
|
-
|
-
|
833,019
|
|
Accrual of
stock options
|
-
|
-
|
-
|
-
|
-
|
-
|
4,512,192
|
-
|
-
|
4,512,192
|
|
Exercise of
stock options
|
57,400
|
58
|
-
|
-
|
-
|
-
|
308,313
|
-
|
-
|
308,371
|
|
Treasury
stock purchase
|
-
|
-
|
-
|
-
|
(426,794)
|
(3,977,929)
|
-
|
-
|
-
|
(3,977,929)
|
|
Unrealized
loss on investments, net of tax
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(240,000)
|
(240,000)
|
|
Foreign
currency translation
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
967,189
|
967,189
|
|
Net
loss
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(25,490,310)
|
-
|
(25,490,310)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at
December 31, 2017
|
15,615,558
|
$15,616
|
-
|
$-
|
(426,794)
|
$(3,977,929)
|
$172,691,339
|
$(111,036,997)
|
$(389,503)
|
$57,302,526
|
The
accompanying notes are an integral part of these consolidated
financial statements.
CELLULAR BIOMEDICINE GROUP,
INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
|
For the Year Ended
|
||
|
|
December 31,
|
||
|
|
2017
|
2016
|
2015
|
|
|
|
|
|
|
CASH
FLOWS FROM OPERATING ACTIVITIES:
|
|
|
|
|
Net loss
|
$(25,490,310)
|
$(28,208,376)
|
$(19,447,721)
|
|
Adjustments to reconcile net loss to net cash
|
|
|
|
|
used in operating activities:
|
|
|
|
|
Depreciation
and amortization
|
2,985,963
|
2,635,001
|
2,094,644
|
|
Loss
on disposal of assets
|
317
|
2,156
|
1,444
|
|
Stock
based compensation expense
|
5,345,211
|
5,452,417
|
7,592,438
|
|
Other
than temporary impairment on investments
|
-
|
4,611,714
|
123,428
|
|
Realized
losses from sale of investments
|
-
|
-
|
5,178
|
|
(Reversal)
of inventory provision
|
-
|
(115,391)
|
123,848
|
|
Allowance
for doubtful account
|
-
|
10,163
|
-
|
|
Decrease
in fair value of accrued expenses for the acquisition of intangible
assets
|
-
|
-
|
(345,882)
|
|
Changes in operating assets and liabilities:
|
|
|
|
|
Accounts
receivable
|
(160,628)
|
537,155
|
(497,937)
|
|
Other
receivables
|
(467,985)
|
(156,672)
|
(143,711)
|
|
Inventory
|
-
|
514,734
|
(142,486)
|
|
Prepaid
expenses
|
(812,675)
|
(669,598)
|
181,679
|
|
Taxes
recoverable
|
-
|
150,082
|
(150,082)
|
|
Other
current assets
|
-
|
-
|
110,347
|
|
Long-term
prepaid expenses and other assets
|
(1,005,029)
|
(643,673)
|
(384,432)
|
|
Accounts
payable
|
(814)
|
(28,205)
|
(166,032)
|
|
Accrued
expenses
|
(118,968)
|
356,420
|
396,557
|
|
Advance
payable to related party
|
-
|
-
|
(30,216)
|
|
Other
current liabilities
|
1,339,866
|
(640,573)
|
113,919
|
|
Taxes
payable
|
-
|
28,875
|
(814,288)
|
|
Other
non-current liabilities
|
(208,340)
|
296,036
|
(371,793)
|
|
Net cash used in operating activities
|
(18,593,392)
|
(15,867,735)
|
(11,751,098)
|
|
|
|
|
|
|
CASH
FLOWS FROM INVESTING ACTIVITIES:
|
|
|
|
|
Acquisition of business, net of cash acquired
|
-
|
-
|
(1,568,627)
|
|
Proceed from sale of investments, net of issuance cost
paid
|
-
|
-
|
1,480
|
|
Purchases
of intangible assets
|
(23,734)
|
(56,519)
|
(4,260,420)
|
|
Purchases
of property, plant and equipment
|
(10,169,134)
|
(2,676,888)
|
(1,874,538)
|
|
Net cash used in investing activities
|
(10,192,868)
|
(2,733,407)
|
(7,702,105)
|
|
|
|
|
|
|
CASH
FLOWS FROM FINANCING ACTIVITIES:
|
|
|
|
|
Net
proceeds from the issuance of common stock
|
14,496,040
|
42,399,874
|
18,964,849
|
|
Proceeds
from exercise of stock options
|
308,371
|
885,680
|
682,303
|
|
Repurchase
of treasury stock
|
(3,977,929)
|
-
|
-
|
|
Net cash provided by financing activities
|
10,826,482
|
43,285,554
|
19,647,152
|
|
|
|
|
|
|
EFFECT
OF EXCHANGE RATE CHANGES ON CASH
|
275,768
|
(316,577)
|
(79,936)
|
|
|
|
|
|
|
INCREASE
IN CASH AND CASH EQUIVALENTS
|
(17,684,010)
|
24,367,835
|
114,013
|
|
CASH
AND CASH EQUIVALENTS, BEGINNING OF PERIOD
|
39,252,432
|
14,884,597
|
14,770,584
|
|
CASH
AND CASH EQUIVALENTS, END OF PERIOD
|
$21,568,422
|
$39,252,432
|
$14,884,597
|
|
|
|
|
|
|
SUPPLEMENTAL CASH FLOW INFORMATION
|
|
|
|
|
|
|
|
|
|
Cash
paid for income taxes
|
$2,450
|
$6,705
|
$108,075
|
|
|
|
|
|
|
Non-cash
investing activities
|
|
|
|
|
Acquisition of intangible assets through issuance of the Company's
stock
|
$-
|
$-
|
$1,481,462
|
The
accompanying notes are an integral part of these consolidated
financial statements.
F-7
CELLULAR BIOMEDICINE
GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
NOTE 1 – DESCRIPTION OF BUSINESS
As used
in this report, "we", "us", "our", "CBMG", "Company" or "our
company" refers to Cellular Biomedicine Group, Inc. and, unless the
context otherwise requires, all of its subsidiaries.
Overview
Cellular
Biomedicine Group, Inc. is a clinical stage biopharmaceutical
company, principally engaged in the development of therapies for
cancer and degenerative diseases utilizing proprietary cell-based
technologies. Our technology includes two major platforms: (i)
Immune cell therapy for treatment of a broad range of cancer
indications comprised of technologies in Chimeric Antigen Receptor
modified T cells (CAR-T), T-Cell Receptor (TCR), cancer vaccine,
and ex vivo expanded autologous Central Memory T Cells (Tcm), and
(ii) human adipose-derived mesenchymal progenitor cells (haMPC) for
treatment of joint and autoimmune diseases. CBMG’s Research
& Development are based in China and the U.S., and its
manufacturing facilities are based in China in the cities of
Shanghai, Wuxi, and Beijing.
We are
focused on developing and marketing safe and effective cell-based
therapies based on our cellular platforms, to treat cancer,
orthopedic diseases and metabolic diseases. We have developed
proprietary technologies and know-hows in our cell therapy
platforms. We are conducting clinical studies in China with our
stem cell based therapies to treat knee osteoarthritis
(“KOA”). We have completed a Phase IIb autologous haMPC
KOA clinical study and published its promising results. Led by
Shanghai Renji Hospital, one of the largest teaching hospitals in
China, we have also completed a Phase I clinical trial of our
off-the-shelf allogeneic haMPC (AlloJoinTM) therapy for
treating KOA patients. In addition, we have received an award of
$2.29 million grant from California Institute of Regenerative
Medicine’s (CIRM) and we have started manufacturing
AlloJoinTM
product in California to support preclinical and clinical studies
in the United States for the KOA indication.
Our
primary target market is Greater China. We believe that our
cell-based therapies will be able to help patients with high unmet
medical needs. We expect to carry out clinical studies leading to
the eventual CFDA approval of our products through Biologics
License Application (BLA) filings and authorized clinical centers
throughout Greater China.
We have
launched multiple clinical trials using our CAR-T products in
multiple indications such as DLBCL & ALL. We may also establish
partnerships with other companies for co-development in CAR-T,
TCR-T and stem cell based therapies. We are striving to build a
highly competitive research and development function, a
translational medicine unit, along with a well-established cellular
manufacturing capability and ample capacity, to support the
development of multiple assets in multiple indications. These
efforts will allow us to boost the Company's Immuno-Oncology
presence and pave the way for additional future
partnerships.
Corporate History
Cellular
Biomedicine Group, Inc., (formerly known as EastBridge Investment
Group Corporation) was originally incorporated in the State of
Arizona on June 25, 2001 under the name ATC Technology Corporation.
ATC Technology Corporation changed its corporate name to EastBridge
Investment Group Corporation in September 2005 and changed its
business focus to providing investment related services in
Asia.
On
November 13, 2012, EastBridge Investment Group Corporation, an
Arizona corporation (“EastBridge”), CBMG Acquisition
Limited, a British Virgin Islands company and the Company’s
wholly-owned subsidiary (“Merger Sub”) and Cellular
Biomedicine Group Ltd. (“CBMG BVI”), a British Virgin
Islands company, entered into a Merger Agreement, pursuant to which
CBMG BVI was the surviving entity in a merger with Merger Sub
whereby CBMG BVI became a wholly-owned subsidiary of the Company
(the “Merger”). The Merger was consummated on February
6, 2013 (the “Closing Date”).
Also in
connection with the Merger, the Company created a new Delaware
subsidiary named EastBridge Investment Corp. (“EastBridge
Sub”). Pursuant to a Contribution Agreement by and between
the Company and EastBridge Sub dated February 5, 2013, the Company
contributed all of its then current assets and liabilities to
EastBridge Sub which continued the business and operations of the
Company at the subsidiary level. A copy of the Contribution
Agreement is attached as Exhibit 10.1 to the Current Report on Form
8-K filed by the Company on February 12, 2013.
As a
result of the Merger, CBMG BVI and EastBridge Sub became the two
direct subsidiaries of the Company.
F-8
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
In
connection with the Merger, effective March 5, 2013, the Company
(formerly named “EastBridge Investment Group
Corporation”) changed its name to “Cellular Biomedicine
Group, Inc.” In addition in March 2013, the Company changed
its corporate headquarters to 530 University Avenue in Palo Alto,
California.
From
February 6, 2013 to June 23, 2014, we operated the Company in two
separate reportable segments: (i) Biomedicine Cell Therapy
(“Biomedicine”); and (ii) Financial Consulting
(“Consulting”). The Consulting segment was
conducted through EastBridge Sub. On June 23, 2014, the
Company announced the discontinuation of the Consulting segment as
it no longer fit into management’s long-term strategy and
vision. The Company is now focusing resources on
becoming a biotechnology company bringing therapies to improve the
health of patients in China.
On
September 26, 2014, the Company completed its acquisition of
Beijing Agreen Biotechnology Co. Ltd. ("AG") and the U.S. patent
held by AG’s founder. AG is a biotech company with operations
in China, engaged in the development of treatments for cancerous
diseases utilizing proprietary cell technologies.
At the
end of September, 2015, the Company moved its corporate
headquarters to 19925 Stevens Creek Blvd., Suite 100 in Cupertino,
California. The Company is a biopharmaceutical company focused on
developing therapies to improve the health of patients in
China.
NOTE 2 – BASIS OF PRESENTATION
The
consolidated financial statements include the financial statements
of the Company and all of its subsidiaries and variable interest
entities. All significant inter-company transactions and balances
are eliminated upon consolidation. The consolidated financial
statements have been prepared in accordance with the accounting
principles generally accepted in the United States of America
(“GAAP”).
NOTE 3 – SUMMARY OF SIGNIFICANT ACCOUNTING
POLICIES
Significant
accounting policies are as follows:
Principles of Consolidation
The
consolidated financial statements have been prepared in conformity
with GAAP, and reflect the accounts and operations of the Company
and its subsidiaries, beginning with the date of their respective
acquisition. In accordance with the provisions of Financial
Accounting Standards Board (“FASB”), Accounting
Standards Codification (“ASC”) Topic 810, or ASC
810, Consolidation,
the Company consolidates any variable interest entity, or VIE, of
which it is the primary beneficiary. The typical condition for a
controlling financial interest ownership is holding a majority of
the voting interests of an entity; however, a controlling financial
interest may also exist in entities, such as variable interest
entities, through arrangements that do not involve controlling
voting interests. ASC 810 requires a variable interest holder to
consolidate a VIE if that party has the power to direct the
activities of a VIE that most significantly impact the VIE’s
economic performance, and the obligation to absorb losses of the
VIE that could potentially be significant to the VIE or the right
to receive benefits from the VIE that could potentially be
significant to the VIE. The Company does not consolidate a VIE in
which it has a majority ownership interest when the Company is not
considered the primary beneficiary. The Company has determined that
it is the primary beneficiary in a VIE—refer to Note 4,
Variable Interest Entity. The Company evaluates its relationships
with the VIE on an ongoing basis to ensure that it continues to be
the primary beneficiary. All intercompany transactions and balances
have been eliminated in consolidation.
F-9
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
Use of Estimates
The
preparation of financial statements in conformity with GAAP
requires management to make estimates and assumptions that affect
the reported amounts of assets and liabilities and disclosure of
contingent assets and liabilities at the date of the financial
statements.
These
estimates and assumptions also affect the reported amounts of
revenues, costs and expenses during the reporting period.
Management evaluates these estimates and assumptions on a regular
basis. Significant accounting estimates reflected in the
Company’s consolidated financial statements include inventory
valuation, account receivable valuation, useful lives of property,
plant and equipment and acquired intangibles, the valuation
allowance for deferred income tax assets, valuation of goodwill,
valuation of long-lived assets and share-based compensation
expense. Actual results could materially differ from those
estimates.
Revenue Recognition
The
Company utilizes the guidance set forth in the FASB’s ASC
Topic 605, Revenue
Recognition, regarding the recognition, presentation and
disclosure of revenue in its financial statements. The Company
recognizes revenue when pervasive evidence of an arrangement
exists, the price is fixed and determinable, collection is
reasonably assured and delivery of products or services has been
rendered.
Cash and Cash Equivalents
The
Company considers all highly liquid investments with an original
maturity of three months or less to be cash equivalents. At
December 31, 2017 and 2016, respectively, cash and cash equivalents
include cash on hand and cash in the bank. At times, cash deposits
may exceed government-insured limits.
Accounts Receivable
Accounts receivable
represent amounts earned but not collected in connection with the
Company’s sales of goods or services as of December 31, 2017
and 2016. Account receivables are carried at their estimated
collectible amounts.
The
Company follows the allowance method of recognizing uncollectible
accounts receivable. The Company recognizes bad debt expense based
on specifically identified customers and invoices that are
anticipated to be uncollectable. At December 31, 2017 and 2016,
allowance of $10,789 and $10,163 was provided for debtors of
certain customers as those debts are unrecoverable from customers,
respectively.
Property, Plant and Equipment
Property, plant and
equipment are recorded at cost. Depreciation is provided for on the
straight-line method over the estimated useful lives of the assets
ranging from three to ten years and begins when the related assets
are placed in service. Maintenance and repairs that neither
materially add to the value of the property nor appreciably prolong
its life are charged to expense as incurred. Betterments or
renewals are capitalized when incurred. Plant, property and
equipment are reviewed each year to determine whether any events or
circumstances indicate that the carrying amount of the assets may
not be recoverable. We assess the recoverability of the asset by
comparing the projected undiscounted net cash flows associated with
the related assets over the estimated remaining life against the
respective carrying value.
For the
years ended December 31, 2017, 2016 and 2015, depreciation expense
was $1,195,705, $850,793 and $573,015, respectively.
Goodwill and Other Intangibles
Goodwill represents
the excess of the cost of assets acquired over the fair value of
the net assets at the date of acquisition. Intangible assets
represent the fair value of separately recognizable intangible
assets acquired in connection with the Company’s business
combinations. The Company evaluates its goodwill and other
intangibles for impairment on an annual basis or whenever events or
circumstances indicate that impairment may have
occurred.
The
carrying amount of the goodwill at December 31, 2017 and 2016
represents the cost arising from the business combinations in
previous years and no impairment on goodwill was recognized for the
years ended December 31, 2017 and 2016.
F-10
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
Treasury Stock
The
treasury stock is recorded and carried at their repurchase cost.
The Company recorded the entire purchase price of the treasury
stock as a reduction of equity. A gain or loss will be determined
when treasury stock is reissued or retired, and the original issue
price and book value of the stock do not enter into the accounting.
Additional paid-in capital from treasury stock is credited for
gains and debited for losses when treasury stock is reissued at
prices that differ from the repurchase cost.
Government Grants
Government grants
are recognized in the balance sheet initially when there is
reasonable assurance that they will be received and that the
enterprise will comply with the conditions attached to them. When
the Company received the government grants but the conditions
attached to the grants have not been fulfilled, such government
grants are deferred and recorded as deferred income. The
classification of short-term or long-term liabilities is depended
on the management’s expectation of when the conditions
attached to the grant can be fulfilled. Grants that compensate the
Company for expenses incurred are recognized as other income in
statement of income on a systematic basis in the same periods in
which the expenses are incurred.
For the
year ended December 31, 2017 and 2016, the Company received
government grants of $1,905,213 and $422,839 for purpose of R&D
and related capital expenditure, respectively. Government subsidies
recognized as other income in the statement of income for the year
ended December 31, 2017 and 2016 were $2,077,486 and $78,542,
respectively.
Valuation of long-lived asset
The
Company reviews the carrying value of long-lived assets to be held
and used, including other intangible assets subject to
amortization, when events and circumstances warrants such a review.
The carrying value of a long-lived asset is considered impaired
when the anticipated undiscounted cash flow from such asset is
separately identifiable and is less than its carrying value. In
that event, a loss is recognized based on the amount by which the
carrying value exceeds the fair market value of the long-lived
asset and intangible assets. Fair market value is determined
primarily using the anticipated cash flows discounted at a rate
commensurate with the risk involved. Losses on long-lived assets
and intangible assets to be disposed are determined in a similar
manner, except that fair market values are reduced for the cost to
dispose.
Income Taxes
Income
taxes are accounted for using the asset and liability method. Under
this method, deferred income tax assets and liabilities are
recognized for the future tax consequences attributable to
temporary differences between the financial statement carrying
amounts of existing assets and liabilities and their respective tax
bases. Deferred income tax assets and liabilities are measured
using enacted tax rates expected to apply to taxable income in the
years in which these temporary differences are expected to be
recovered or settled. The effect on deferred tax assets and
liabilities of a change in tax rates is recognized in income in the
period that includes the enactment date. A valuation allowance
would be provided for those deferred tax assets if it is more
likely than not that the related benefit will not be
realized.
A full
valuation allowance has been established against all net deferred
tax assets as of December 31, 2017 and 2016 based on estimates of
recoverability. While the Company has optimistic plans for its
business strategy, we determined that such a valuation allowance
was necessary given the current and expected near term losses and
the uncertainty with respect to the Company’s ability to
generate sufficient profits from its business model.
Share-Based Compensation
The
Company periodically uses stock-based awards, consisting of shares
of common stock and stock options, to compensate certain officers
and consultants. Shares are expensed on a straight line basis over
the requisite service period based on the grant date fair value,
net of estimated forfeitures, if any. We currently use the Black-Scholes option-pricing
model to estimate the fair value of our stock-based payment awards.
This model requires the input of highly subjective assumptions,
including the fair value of the underlying common stock, the
expected volatility of the price of our common stock, risk-free
interest rates, the expected term of the option and the expected
dividend yield of our common stock. These estimates involve
inherent uncertainties and the application of management’s
judgment. If factors change and different assumptions are used, our
stock-based compensation expense could be materially different in
the future. These assumptions are estimated as
follows:
F-11
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
|
●
|
Fair
Value of Our Common Stock — Our common stock is valued by
reference to the publicly-traded price of our common
stock.
|
|
●
|
Expected
Volatility — Prior to the Eastbridge merger, we did not have
a history of market prices for our common stock and since the
merger, we do not have what we consider a sufficiently active and
readily traded market for our common stock to use historical market
prices for our common stock to estimate volatility. Accordingly, we
estimate the expected stock price volatility for our common stock
by taking the median historical stock price volatility for industry
peers based on daily price observations over a period equivalent to
the expected term of the stock option grants. Industry peers
consist of other public companies in the stem cell industry similar
in size, stage of life cycle and financial leverage. We intend to
continue to consistently apply this process using the same or
similar public companies until a sufficient amount of historical
information regarding the volatility of our own common stock share
price becomes available.
|
|
●
|
Risk-Free
Interest Rate — The risk-free interest rate assumption is
based on observed interest rates appropriate for the expected terms
of our awards. The risk-free interest rate assumption is based on
the yields of U.S. Treasury securities with maturities similar to
the expected term of the options for each option
group.
|
|
●
|
Expected
Term — The expected term represents the period that our
stock-based awards are expected to be outstanding. The expected
terms of the awards are based on a simplified method which defines
the life as the average of the contractual term of the options and
the weighted-average vesting period for all open
tranches.
|
|
●
|
Expected
Dividend Yield — We have never declared or paid any cash
dividends and do not presently plan to pay cash dividends in the
foreseeable future. Consequently, we used an expected dividend
yield of zero.
|
In
addition to the assumptions used in the Black-Scholes
option-pricing model, the amount of stock option expense we
recognize in our consolidated statements of operations includes an
estimate of stock option forfeitures. We estimate our forfeiture
rate based on an analysis of our actual forfeitures and will
continue to evaluate the appropriateness of the forfeiture rate
based on actual forfeiture experience, analysis of employee
turnover and other factors. Changes in the estimated forfeiture
rate can have a significant impact on our stock-based compensation
expense as the cumulative effect of adjusting the rate is
recognized in the period the forfeiture estimate is changed. If a
revised forfeiture rate is higher than the previously estimated
forfeiture rate, an adjustment is made that will result in a
decrease to the stock-based compensation expense recognized in the
consolidated financial statements. If a revised forfeiture rate is
lower than the previously estimated forfeiture rate, an adjustment
is made that will result in an increase to the stock-based
compensation expense recognized in our consolidated financial
statements.
Fair Value of Financial Instruments
Under
the FASB’s authoritative guidance on fair value measurements,
fair value is the price that would be received to sell an asset or
paid to transfer a liability in an orderly transaction between
market participants at the measurement date. In determining the
fair value, the Company uses various methods including market,
income and cost approaches. Based on these approaches, the Company
often utilizes certain assumptions that market participants would
use in pricing the asset or liability, including assumptions about
risk and the risks inherent in the inputs to the valuation
technique. These inputs can be readily observable, market
corroborated or generally unobservable inputs. The Company uses
valuation techniques that maximize the use of observable inputs and
minimize the use of unobservable inputs. Based on observability of
the inputs used in the valuation techniques, the Company is
required to provide the following information according to the fair
value hierarchy. The fair value hierarchy ranks the quality and
reliability of the information used to determine fair values.
Financial assets and liabilities carried at fair value are
classified and disclosed in one of the following three
categories:
Level
1: Valuations for assets and liabilities traded in active exchange
markets. Valuations are obtained from readily available pricing
sources for market transactions involving identical assets or
liabilities.
Level
2: Valuations for assets and liabilities traded in less active
dealer or broker markets. Valuations are obtained from third party
pricing services for identical or similar assets or
liabilities.
Level
3: Valuations for assets and liabilities that are derived from
other valuation methodologies, including option pricing models,
discounted cash flow models and similar techniques, and not based
on market exchange, dealer or broker traded transactions. Level 3
valuations incorporate certain unobservable assumptions and
projections in determining the fair value assigned to such
assets.
F-12
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
All transfers
between fair value hierarchy levels are recognized by the Company
at the end of each reporting period. In certain cases, the inputs
used to measure fair value may fall into different levels of the
fair value hierarchy. In such cases, an investment’s level
within the fair value hierarchy is based on the lowest level of
input that is significant to the fair value measurement in its
entirety requires judgment, and considers factors specific to the
investment. The inputs or methodology used for valuing financial
instruments are not necessarily an indication of the risks
associated with investment in those instruments.
The
carrying amounts of other financial instruments, including cash,
accounts receivable, accounts payable and accrued liabilities,
income tax payable and related party payable approximate fair value
due to their short maturities.
Investments
The
fair value of “investments” is dependent on the type of
investment, whether it is marketable or
non-marketable.
Marketable
securities held by the Company are held for an indefinite period of
time and thus are classified as available-for-sale securities. The
fair value is based on quoted market prices for the investment as
of the balance sheet date. Realized investment gains and losses are
included in the statement of operations, as are provisions for
other than temporary declines in the market value of available
for-sale securities. Unrealized gains and unrealized losses deemed
to be temporary are excluded from earnings (losses), net of
applicable taxes, as a component of other comprehensive income
(loss). Factors considered in judging whether an impairment is
other than temporary include the financial condition, business
prospects and creditworthiness of the issuer, the length of time
that fair value has been less than cost, the relative amount of
decline, and the Company’s ability and intent to hold the
investment until the fair value recovers.
Basic and Diluted Net Loss Per Share
Diluted
net loss per share reflects potential dilution from the exercise or
conversion of securities into common stock. The dilutive effect of
the Company's share-based awards is computed using the treasury
stock method, which assumes that all share-based awards are
exercised and the hypothetical proceeds from exercise are used to
purchase common stock at the average market price during the
period. Share-based awards whose effects are anti-dilutive are
excluded from computing diluted net loss per share.
Foreign Currency Translation
The
Company's financial statements are presented in U.S. dollars ($),
which is the Company’s reporting currency, while some of the
Company’s subsidiaries’ functional currency is Chinese
Renminbi (RMB). Transactions in foreign currencies are initially
recorded at the functional currency rate ruling at the date of
transaction. Any differences between the initially recorded amount
and the settlement amount are recorded as a gain or loss on foreign
currency transaction in the consolidated statements of operations.
Monetary assets and liabilities denominated in foreign currency are
translated at the functional currency rate of exchange ruling at
the balance sheet date. Any differences are recorded as an
unrealized gain or loss on foreign currency translation in the
statements of operations and comprehensive loss. In accordance with
ASC 830, Foreign Currency Matters, the Company translates the
assets and liabilities into USD from RMB using the rate of exchange
prevailing at the applicable balance sheet date and the statements
of income and cash flows are translated at an average rate during
the reporting period. Adjustments resulting from the translation
are recorded in shareholders' equity as part of accumulated other
comprehensive income. The PRC government imposes significant
exchange restrictions on fund transfers out of the PRC that are not
related to business operations.
Comprehensive Loss
We
apply ASC No. 220, Comprehensive
Income (ASC 220). ASC 220 establishes standards for the
reporting and display of comprehensive income or loss, requiring
its components to be reported in a financial statement that is
displayed with the same prominence as other financial statements.
Our comprehensive loss was $24,763,121, $29,208,953 and $21,132,211
for the years ended December 31, 2017, 2016 and 2015,
respectively.
Segment Information
FASB
ASC Topic 280, “Segment Reporting” establishes
standards for reporting information about reportable segments.
Operating segments are defined as components of an enterprise about
which separate financial information is available that is evaluated
regularly by the chief operating decision maker, or decision-making
group in deciding how to allocate resources and in assessing
performance. Following the discontinuance of our consulting
business, we operate in a single reportable segment.
F-13
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
Recent Accounting Pronouncements
Accounting pronouncements adopted during the year ended December
31, 2017
In April 2016, the Financial Accounting Standards
Board (“FASB”) issued Accounting Standards Update
(“ASU”) No. 2016-09, “Compensation—Stock
Compensation (Topic 718): Improvements to Employee Share-Based
Payment Accounting” (“ASU 2016-09”), which simplifies
several aspects of the accounting for employee share-based payment
transactions. The areas for simplification in ASU 2016-09 include
the income tax consequences, classification of awards as either
equity or liabilities, and classification on the statement of cash
flows. The amendments in this ASU will be effective for annual
periods beginning after December 15, 2016 and interim periods
within those annual periods. Early adoption is permitted. The
adoption of the ASU 2016-09 did not have a material impact on the
Company’s consolidated financial
statements.
Accounting pronouncements not yet effective
In February 2018, the FASB issued ASU No.
2018-02, “Income
Statement—Reporting Comprehensive Income (Topic 220):
Reclassification of Certain Tax Effects from Accumulated Other
Comprehensive Income” (“ASU 2018-02”), which provides
financial statement preparers with an option to reclassify stranded
tax effects within accumulated other comprehensive income to
retained earnings in each period in which the effect of the change
in the U.S. federal corporate income tax rate in the Tax Cuts and
Jobs Act (or portion thereof) is recorded. The amendments in this
ASU are effective for all entities for fiscal years beginning after
December 15, 2018, and interim periods within those fiscal years.
Early adoption of ASU 2018-02 is permitted, including adoption in
any interim period for the public business entities for reporting
periods for which financial statements have not yet been issued.
The amendments in this ASU should be applied either in the period
of adoption or retrospectively to each period (or periods) in which
the effect of the change in the U.S. federal corporate income tax
rate in the Tax Cuts and Jobs Act is recognized. We do not expect
the adoption of ASU 2018-02 to have a material impact on our
consolidated financial statements.
In July 2017, the FASB issued ASU No.
2017-11, “Earnings Per Share
(Topic 260); Distinguishing Liabilities from Equity (Topic 480);
Derivatives and Hedging (Topic 815): (Part I) Accounting for
Certain Financial Instruments with Down Round Features, (Part II)
Replacement of the Indefinite Deferral for Mandatorily Redeemable
Financial Instruments of Certain Nonpublic Entities and Certain
Mandatorily Redeemable Non-controlling Interests with a Scope
Exception” (“ASU
2017-11”), which addresses the complexity of accounting for
certain financial instruments with down round features. Down round
features are features of certain equity-linked instruments (or
embedded features) that result in the strike price being reduced on
the basis of the pricing of future equity offerings. Current
accounting guidance creates cost and complexity for entities that
issue financial instruments (such as warrants and convertible
instruments) with down round features that require fair value
measurement of the entire instrument or conversion option. The
amendments in Part I of this ASU are effective for public business
entities for fiscal years, and interim periods within those fiscal
years, beginning after December 15, 2018. The Company is currently
evaluating the impact of the adoption of ASU 2017-11 on its
consolidated financial statements.
In May 2017, the FASB issued ASU No.
2017-09, “Compensation—Stock
Compensation (Topic 718): Scope of Modification
Accounting” (“ASU
2017-09”), which provides guidance on determining which
changes to the terms and conditions of share-based payment awards
require an entity to apply modification accounting under Topic 718.
The amendments in this ASU are effective for all entities for
annual periods, and interim periods within those annual periods,
beginning after December 15, 2017. Early adoption is permitted,
including adoption in any interim period, for (1) public business
entities for reporting periods for which financial statements have
not yet been issued and (2) all other entities for reporting
periods for which financial statements have not yet been made
available for issuance. The amendments in this ASU should be
applied prospectively to an award modified on or after the adoption
date. We do not expect the adoption of ASU 2017-09 to have a
material impact on our consolidated financial
statements.
In February 2017, the FASB issued ASU No.
2017-05, “Other
Income—Gains and Losses from the Derecognition of
Nonfinancial Assets (Subtopic 610-20): Clarifying the Scope of
Asset Derecognition Guidance and Accounting for Partial Sales of
Nonfinancial Assets” (“ASU 2017-05”), which clarifies the
scope of the nonfinancial asset guidance in Subtopic 610-20. This
ASU also clarifies that the derecognition of all businesses and
nonprofit activities (except those related to conveyances of oil
and gas mineral rights or contracts with customers) should be
accounted for in accordance with the derecognition and
deconsolidation guidance in Subtopic 810-10. The amendments in this
ASU also provide guidance on the accounting for what often are
referred to as partial sales of nonfinancial assets within the
scope of Subtopic 610-20 and contributions of nonfinancial assets
to a joint venture or other non-controlled investee. The amendments
in this ASU are effective for annual reporting reports beginning
after December 15, 2017, including interim reporting periods within
that reporting period. Public entities may apply the guidance
earlier but only as of annual reporting periods beginning after
December 15, 2016, including interim reporting periods within that
reporting period. We do not expect the adoption of ASU 2017-05 to
have a material impact on our consolidated financial
statements.
F-14
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
In January 2017, the FASB issued ASU No.
2017-04, “Intangibles—Goodwill
and Other (Topic 350): Simplifying the Test for Goodwill
Impairment” (“ASU
2017-04”), which removes Step 2 from the goodwill impairment
test. An entity will apply a one-step quantitative test and record
the amount of goodwill impairment as the excess of a reporting
unit's carrying amount over its fair value, not to exceed the total
amount of goodwill allocated to the reporting unit. The new
guidance does not amend the optional qualitative assessment of
goodwill impairment. Public business entity that is a U.S.
Securities and Exchange Commission filer should adopt the
amendments in this ASU for its annual or any interim goodwill
impairment test in fiscal years beginning after December 15, 2019.
Early adoption is permitted for interim or annual goodwill
impairment tests performed on testing dates after January 1, 2017.
We are currently evaluating the impact of the adoption of ASU
2017-04 on our consolidated financial
statements.
In November 2016, the FASB issued ASU No.
2016-18, “Statement of Cash Flows
(Topic 230): Restricted Cash” (“ASU 2016-18”), which requires that a
statement of cash flows explain the change during the period in the
total of cash, cash equivalents, and amounts generally described as
restricted cash or restricted cash equivalents. Therefore, amounts
generally described as restricted cash and restricted cash
equivalents should be included with cash and cash equivalents when
reconciling the beginning-of-period and end-of-period total amounts
shown on the statement of cash flows. The amendments in this ASU do
not provide a definition of restricted cash or restricted cash
equivalents. The amendments in this ASU are effective for public
business entities for fiscal years beginning after December 15,
2017, and interim periods within those fiscal years. Early adoption
is permitted, including adoption in an interim period. We do not
expect the adoption of ASU 2016-18 to have a material impact on our
consolidated financial statements.
In August 2016, the FASB issued ASU No.
2016-15, “Statement of Cash Flows
(Topic 230): Classification of Certain Cash Receipts and Cash
Payments” (“ASU
2016-15”), which addresses the following eight specific cash
flow issues: debt prepayment or debt extinguishment costs;
settlement of zero-coupon debt instruments or other debt
instruments with coupon interest rates that are insignificant in
relation to the effective interest rate of the borrowing;
contingent consideration payments made after a business
combination; proceeds from the settlement of insurance claims;
proceeds from the settlement of corporate-owned life insurance
policies (including bank-owned life insurance policies;
distributions received from equity method investees; beneficial
interests in securitization transactions; and separately
identifiable cash flows and application of the predominance
principle. The amendments in this ASU are effective for public
business entities for fiscal years beginning after December 15,
2017, and interim periods within those fiscal years. Early adoption
is permitted, including adoption in an interim period. We do not
expect the adoption of ASU 2016-15 to have a material impact on our
consolidated financial statements.
In June 2016, the FASB issued ASU No.
2016-13, “Financial
Instruments—Credit Losses (Topic 326): Measurement of Credit
Losses on Financial Instruments” (“ASU 2016-13”). Financial
Instruments—Credit Losses (Topic 326) amends guideline on
reporting credit losses for assets held at amortized cost basis and
available-for-sale debt securities. For assets held at amortized
cost basis, Topic 326 eliminates the probable initial recognition
threshold in current GAAP and, instead, requires an entity to
reflect its current estimate of all expected credit losses. The
allowance for credit losses is a valuation account that is deducted
from the amortized cost basis of the financial assets to present
the net amount expected to be collected. For available-for-sale
debt securities, credit losses should be measured in a manner
similar to current GAAP, however Topic 326 will require that credit
losses be presented as an allowance rather than as a write-down.
ASU 2016-13 affects entities holding financial assets and net
investment in leases that are not accounted for at fair value
through net income. The amendments affect loans, debt securities,
trade receivables, net investments in leases, off balance sheet
credit exposures, reinsurance receivables, and any other financial
assets not excluded from the scope that have the contractual right
to receive cash. The amendments in this ASU will be effective for
fiscal years beginning after December 15, 2019, including interim
periods within those fiscal years. We are currently evaluating the
impact of the adoption of ASU 2016-13 on our consolidated financial
statements.
In February 2016, the FASB issued ASU No.
2016-02, “Leases (Topic
842)” (“ASU
2016-02”). The amendments in this update create Topic 842,
Leases, and supersede the leases requirements in Topic 840, Leases.
Topic 842 specifies the accounting for leases. The objective of
Topic 842 is to establish the principles that lessees and lessors
shall apply to report useful information to users of financial
statements about the amount, timing, and uncertainty of cash flows
arising from a lease. The main difference between Topic 842 and
Topic 840 is the recognition of lease assets and lease liabilities
for those leases classified as operating leases under Topic 840.
Topic 842 retains a distinction between finance leases and
operating leases. The classification criteria for distinguishing
between finance leases and operating leases are substantially
similar to the classification criteria for distinguishing between
capital leases and operating leases in the previous leases
guidance. The result of retaining a distinction between finance
leases and operating leases is that under the lessee accounting
model in Topic 842, the effect of leases in the statement of
comprehensive income and the statement of cash flows is largely
unchanged from previous GAAP. The amendments in ASU 2016-02 are
effective for fiscal years beginning after December 15, 2018,
including interim periods within those fiscal years for public
business entities. Early application of the amendments in ASU
2016-02 is permitted. We are currently evaluating the impact of the
adoption of ASU 2016-02 on our consolidated financial
statements.
F-15
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
In January 2016, the FASB issued ASU No.
2016-01, “Financial Instruments
– Overall (Subtopic 825-10): Recognition and Measurement of
Financial Assets and Financial Liabilities”
(“ASU 2016-01”). The
amendments in this update require all equity investments to be
measured at fair value with changes in the fair value recognized
through net income (other than those accounted for under equity
method of accounting or those that result in consolidation of the
investee). The amendments in this update also require an entity to
present separately in other comprehensive income the portion of the
total change in the fair value of a liability resulting from a
change in the instrument-specific credit risk when the entity has
elected to measure the liability at fair value in accordance with
the fair value option for financial instruments. In addition, the
amendments in this update eliminate the requirement for to disclose
the method(s) and significant assumptions used to estimate the fair
value that is required to be disclosed for financial instruments
measured at amortized cost on the balance sheet for public
entities. For public business entities, the amendments in ASU
2016-01 are effective for fiscal years beginning after December 15,
2017, including interim periods within those fiscal years. Except
for the early application guidance discussed in ASU 2016-01, early
adoption of the amendments in this update is not permitted. We do
not expect the adoption of ASU 2016-01 to have a material impact on
our consolidated financial statements.
In May 2014, the FASB issued ASU No.
2014-09, “Revenue from Contracts
with Customers (Topic 606)” (“ASU 2014-09”). ASU 2014-09
supersedes the revenue recognition requirements in “Revenue
Recognition (Topic 605)”, and requires entities to recognize
revenue when it transfers promised goods or services to customers
in an amount that reflects the consideration to which the entity
expects to be entitled to in exchange for those goods or services.
The FASB issued ASU No. 2015-14, “Revenue from Contracts
with Customers (Topic 606): Deferral of the Effective
Date” (“ASU
2015-14”) in August 2015. The amendments in ASU 2015-14 defer
the effective date of ASU 2014-09. Public business entities,
certain not-for-profit entities, and certain employee benefit plans
should apply the guidance in ASU 2014-09 to annual reporting
periods beginning after December 15, 2017, including interim
reporting periods within that reporting period. Earlier adoption is
permitted only as of annual reporting periods beginning after
December 15, 2016, including interim reporting periods within that
reporting period. Further to ASU 2014-09 and ASU 2015-14, the FASB
issued ASU No. 2016-08, “Revenue from Contracts
with Customers (Topic 606): Principal versus Agent Considerations
(Reporting Revenue Gross versus Net)” (“ASU 2016-08”) in March 2016, ASU No.
2016-10, “Revenue from Contracts
with Customers (Topic 606): Identifying Performance Obligations and
Licensing” (“ASU
2016-10”) in April 2016, ASU No. 2016-12, “Revenue from Contracts
with Customers (Topic 606): Narrow-Scope Improvements and Practical
Expedients” (“ASU
2016-12”), and ASU No. 2016-20, “Technical Corrections
and Improvements to Topic 606, Revenue from Contracts with
Customers” (“ASU
2016-20”), respectively. The amendments in ASU 2016-08
clarify the implementation guidance on principal versus agent
considerations, including indicators to assist an entity in
determining whether it controls a specified good or service before
it is transferred to the customers. ASU 2016-10 clarifies
guidelines related to identifying performance obligations and
licensing implementation guidance contained in the new revenue
recognition standard. The updates in ASU 2016-10 include targeted
improvements based on input the FASB received from the Transition
Resource Group for Revenue Recognition and other stakeholders. It
seeks to proactively address areas in which diversity in practice
potentially could arise, as well as to reduce the cost and
complexity of applying certain aspects of the guidance both at
implementation and on an ongoing basis. ASU 2016-12 addresses
narrow-scope improvements to the guidance on collectability,
non-cash consideration, and completed contracts at transition.
Additionally, the amendments in this ASU provide a practical
expedient for contract modifications at transition and an
accounting policy election related to the presentation of sales
taxes and other similar taxes collected from customers. The
amendments in ASU 2016-20 represents changes to make minor
corrections or minor improvements to the Codification that are not
expected to have a significant effect on current accounting
practice or create a significant administrative cost to most
entities. The effective date and transition requirements for ASU
2016-08, ASU 2016-10, ASU 2016-12 and ASU 2016-20 are the same as
ASU 2014-09. The Company will adopt ASC Topic
606, Revenue from Contracts with
Customers, using the modified
retrospective transition approach. Under this approach, ASC Topic
606 would apply to all new contracts initiated on or after January
1, 2018. For existing contracts that have remaining obligations as
of January 1, 2018, any difference between the recognition criteria
in these ASUs and the Company’s current revenue recognition
practices would be recognized using a cumulative effect adjustment
to the opening balance of accumulated deficit. The Company does not
anticipate the adoption of these ASUs to have material changes to
its revenue recognition practices nor material impact on its
consolidated financial statements.
F-16
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
NOTE 4 – VARIABLE INTEREST ENTITY
VIEs are those entities
in which a company, through contractual arrangements, bears the
risk of, and enjoys the rewards normally associated with ownership
of the entity, and therefore the Company is the primary beneficiary
of the entity. Cellular Biomedicine Group Ltd (Shanghai)
(“CBMG Shanghai”) and its subsidiaries are variable
interest entities (VIEs), through which the Company conducts stem
cell and immune therapy research and clinical trials in China. The
registered shareholders of CBMG Shanghai are Lu Junfeng and Chen
Mingzhe, who together own 100% of the equity interests in CBMG
Shanghai. The initial capitalization and operating expenses of
CBMG Shanghai are funded by our wholly foreign-owned enterprise
(“WFOE”), Cellular Biomedicine Group Ltd. (Wuxi)
(“CBMG Wuxi”). The registered capital of CBMG Shanghai
is ten million RMB and was incorporated on October 19, 2011. AG was
100% acquired by CBMG Shanghai in September 2014. The registered
capital of AG is five million RMB and was incorporated on April 27,
2011. In 2017, CBMG Shanghai established two subsidiaries in Wuxi
and Shanghai. Wuxi Cellular Biopharmaceutical Group Ltd.
(“WX SBM”) was established on January 17, 2017 with
registered capital of RMB 20 million and wholly owned by CBMG
Shanghai. Shanghai Cellular Biopharmaceutical Group Ltd. (“SH
SBM”) was established on January 18, 2017 with registered
capital of RMB 100 million and wholly owned by CBMG Shanghai.
For the
year ended December 31, 2017, 2016 and 2015, 3%, 78% and 80% of the
Company revenue is derived from VIEs
respectively.
In February 2012, CBMG Wuxi provided financing to CBMG Shanghai in
the amount of $1,587,075 for working capital purposes. In
conjunction with the provided financing, exclusive option
agreements were executed granting CBMG Wuxi the irrevocable and
exclusive right to convert the unpaid portion of the provided
financing into equity interest of CBMG Shanghai at CBMG
Wuxi’s sole and absolute discretion. CBMG Wuxi and CBMG
Shanghai additionally executed a business cooperation agreement
whereby CBMG Wuxi is to provide CBMG Shanghai with technical and
business support, consulting services, and other commercial
services. The shareholders of CBMG Shanghai pledged their equity
interest in CBMG Shanghai as collateral in the event CBMG Shanghai
does not perform its obligations under the business cooperation
agreement.
The Company has determined it is the primary beneficiary of CBMG
Shanghai by reference to the power and benefits criterion under ASC
Topic 810, Consolidation. This determination was reached after
considering the financing provided by CBMG Wuxi to CBMG Shanghai is
convertible into equity interest of CBMG Shanghai and the business
cooperation agreement grants the Company and its officers the power
to manage and make decisions that affect the operation of CBMG
Shanghai.
There are substantial uncertainties regarding the interpretation,
application and enforcement of PRC laws and regulations, including
but not limited to the laws and regulations governing our business
or the enforcement and performance of our contractual arrangements.
See Risk Factors below regarding “Risks Related to Our
Structure”. The Company has not provided any guarantees
related to VIEs and no creditors of VIEs have recourse to the
general credit of the Company.
As the primary beneficiary of CBMG Shanghai and its subsidiaries,
the Company consolidates in its financial statements the financial
position, results of operations, and cash flows of CBMG Shanghai
and its subsidiaries, and all intercompany balances and
transactions between the Company and CBMG Shanghai and its
subsidiaries are eliminated in the consolidated financial
statements.
The Company has aggregated the financial information of CBMG
Shanghai and its subsidiaries in the table below. The aggregate
carrying value of assets and liabilities of CBMG Shanghai and its
subsidiaries (after elimination of intercompany transactions and
balances) in the Company’s consolidated balance sheets as of
December 31, 2017 and 2016 are as follows:
F-17
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
|
|
December 31,
|
December 31,
|
|
|
2017
|
2016
|
|
Assets
|
|
|
|
Cash
|
$2,337,173
|
$4,021,992
|
|
Other
receivables
|
773,384
|
370,702
|
|
Prepaid
expenses
|
1,750,509
|
777,445
|
|
Total
current assets
|
4,861,066
|
5,170,139
|
|
|
|
|
|
Property,
plant and equipment, net
|
12,477,315
|
2,398,576
|
|
Intangibles
|
1,516,449
|
1,613,582
|
|
Long-term
prepaid expenses and other assets
|
2,920,257
|
443,874
|
|
Total
assets
|
$21,775,087
|
$9,626,171
|
|
|
|
|
|
Liabilities
|
|
|
|
Accounts
payable
|
$181,231
|
$161,825
|
|
Other
payables
|
1,631,582
|
407,769
|
|
Payroll
accrual
|
682,248
|
792,706
|
|
Deferred
income
|
9,810
|
-
|
|
Total
current liabilities
|
$2,504,871
|
$1,362,300
|
|
|
|
|
|
Other
non-current liabilities
|
183,649
|
10,091
|
|
Total
liabilities
|
$2,688,520
|
$1,372,391
|
NOTE 5 – OTHER RECEIVABLES
The
Company pays deposits on various items relating to office expenses.
Management has classified these deposits as receivables as the
intention is to recover these deposits in less than 12 months. As
of December 31, 2017 and 2016 the amounts of other receivables was
$902,940 and $412,727, respectively.
F-18
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
NOTE 6 – PROPERTY, PLANT AND EQUIPMENT
As
of December 31, 2017 and 2016, property, plant and equipment,
carried at cost, consisted of the following:
|
|
December 31, 2017
|
December 31, 2016
|
|
|
|
|
|
Office
equipment
|
$105,114
|
$80,485
|
|
Manufacturing
equipment
|
4,781,936
|
3,347,458
|
|
Computer
equipment
|
233,539
|
162,769
|
|
Leasehold
improvements
|
4,196,589
|
1,912,573
|
|
Construction
in progress
|
7,498,272
|
1,172,433
|
|
|
|
|
|
|
16,815,450
|
6,675,718
|
|
Less:
accumulated depreciation
|
(3,842,108)
|
(2,557,979)
|
|
|
$12,973,342
|
$4,117,739
|
Depreciation
expense for the years ended December 31, 2017, 2016 and 2015 was
$1,195,705, $850,793 and $573,015, respectively.
NOTE 7 – INVESTMENTS
The
Company’s investments represent the investment in equity
securities listed in Over-The-Counter (“OTC”) markets
of the United States of America:
|
December 31, 2017
|
Adjusted Cost
|
Gross Unrealized Gains
|
Gross Unrealized Losses more than 12 months
|
Gross Unrealized Losses less than 12 months
|
Market or Fair Value
|
|
Equity
position in Alpha Lujo, Inc.
|
$251,388
|
$-
|
$(221,964)
|
$-
|
$29,424
|
|
Equity
position in Arem Pacific Corporation
|
480,000
|
-
|
-
|
(240,000)
|
240,000
|
|
Total
|
$731,388
|
$-
|
$(221,964)
|
$(240,000)
|
$269,424
|
|
December 31, 2016
|
Adjusted Cost
|
Gross Unrealized Gains
|
Gross Unrealized Losses more than 12 months
|
Gross Unrealized Losses less than 12 months
|
Market or Fair Value
|
|
Equity
position in Alpha Lujo, Inc.
|
$251,388
|
$-
|
$-
|
$(221,964)
|
$29,424
|
|
Equity
position in Arem Pacific Corporation
|
480,000
|
-
|
-
|
-
|
480,000
|
|
Total
|
$731,388
|
$-
|
$-
|
$(221,964)
|
$509,424
|
There
were no sale of investments for the year ended December 31, 2017
and 2016. Net proceeds from sale of investments for the year ended
December 31, 2015 was $1,480. Net realized losses from sale of
investments for the year ended December 31, 2017, 2016 and 2015 was
$nil, $nil and $5,178, respectively.
F-19
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
The
unrealized holding gain (loss) for the investments, net of tax that
were recognized in other comprehensive income for the year ended
December 31, 2017 was other comprehensive loss of $(240,000), as
compared to $5,300,633 and $(1,376,540) for the year ended December
31, 2016 and 2015, respectively. Reclassification adjustment of
$5,557,939 in connection with other-than-temporary impairment of
investments was recorded in other comprehensive income for the year
ended December 31, 2016. No adjustment was recorded in other
comprehensive income for the year ended December 31, 2017 and
2015.
The
Company tracks each investment with an unrealized loss and
evaluates them on an individual basis for other-than-temporary
impairments, including obtaining corroborating opinions from third
party sources, performing trend analysis and reviewing
management’s future plans. When investments have
declines determined by management to be other-than-temporary the
Company recognizes write downs through
earnings. Other-than-temporary impairment of investments
for the year ended December 31, 2017 was $nil, as compared with
$4,611,714 and $123,428 for the year ended December 31, 2016 and
2015, respectively.
NOTE 8 – FAIR VALUE ACCOUNTING
The Company has adopted ASC Topic 820, Fair
Value Measurement and Disclosure, which defines fair value,
establishes a framework for measuring fair value in GAAP, and
expands disclosures about fair value measurements. It does not
require any new fair value measurements, but provides guidance on
how to measure fair value by providing a fair value hierarchy used
to classify the source of the information. It establishes a
three-level valuation hierarchy of valuation techniques based on
observable and unobservable inputs, which may be used to measure
fair value and include the following:
Level
1 – Quoted prices in active markets for identical assets or
liabilities.
Level
2 – Inputs other than Level 1 that are observable, either
directly or indirectly, such as quoted prices for similar assets or
liabilities; quoted prices in markets that are not active; or other
inputs that are observable or can be corroborated by observable
market data for substantially the full term of the assets or
liabilities.
Level
3 – Unobservable inputs that are supported by little or no
market activity and that are significant to the fair value of the
assets or liabilities.
Classification
within the hierarchy is determined based on the lowest level of
input that is significant to the fair value
measurement.
The
carrying value of financial items of the Company including cash and
cash equivalents, accounts receivable, other receivables, accounts
payable and accrued liabilities, approximate their fair values due
to their short-term nature and are classified within Level 1 of the
fair value hierarchy. The Company’s investments are
classified within Level 2 of the fair value hierarchy because of
the insufficient volatility of the three stocks traded in OTC
market. The Company did not have any Level 3 financial instruments
as of December 31, 2017 and 2016.
F-20
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
Assets
measured at fair value within Level 2 on a recurring basis as of
December 31, 2017 and 2016 are summarized as follows:
|
|
As of December 31, 2017
|
|||
|
|
Fair Value Measurements at Reporting Date Using:
|
|||
|
|
|
Quoted Prices in
|
Significant Other
|
Significant
|
|
|
|
Active Markets for
|
Observable
|
Unobservable
|
|
|
|
Identical Assets
|
Inputs
|
Inputs
|
|
|
Total
|
(Level 1)
|
(Level 2)
|
(Level 3)
|
|
Assets:
|
|
|
|
|
|
Equity
position in Alpha Lujo, Inc.
|
$29,424
|
$-
|
$29,424
|
$-
|
|
Equity
position in Arem Pacific Corporation
|
240,000
|
-
|
240,000
|
-
|
|
|
$269,424
|
$-
|
$269,424
|
$-
|
|
|
As of December 31, 2016
|
|||
|
|
Fair Value Measurements at Reporting Date Using:
|
|||
|
|
|
Quoted Prices in
|
Significant Other
|
Significant
|
|
|
|
Active Markets for
|
Observable
|
Unobservable
|
|
|
|
Identical Assets
|
Inputs
|
Inputs
|
|
|
Total
|
(Level 1)
|
(Level 2)
|
(Level 3)
|
|
Assets:
|
|
|
|
|
|
Equity
position in Alpha Lujo, Inc.
|
$29,424
|
$-
|
$29,424
|
$-
|
|
Equity
position in Arem Pacific Corporation
|
480,000
|
-
|
480,000
|
-
|
|
|
$509,424
|
$-
|
$509,424
|
$-
|
No shares were acquired during the year ended
December 31, 2017 and 2016.
As
of December 31, 2017 and 2016, the Company holds 8,000,000 shares
in Arem Pacific Corporation, 2,942,350 shares in Alpha Lujo, Inc.
and 2,057,131 shares in Wonder International Education and
Investment Group Corporation. All available-for-sale
investments held by the Company at December 31, 2017 and 2016 have
been valued based on level 2 inputs due to the limited trading of
all three of these companies.
NOTE 9 – INTANGIBLE ASSETS
Intangible
assets that are subject to amortization are reviewed for potential
impairment whenever events or circumstances indicate that carrying
amounts may not be recoverable. Assets not subject to amortization
are tested for impairment at least annually. The Company evaluates
the continuing value of the intangibles at each balance sheet date
and records write-downs if the continuing value has become
impaired. An impairment is determined to exist if the anticipated
undiscounted future cash flow attributable to the asset is less
than its carrying value. The asset is then reduced to the net
present value of the anticipated future cash flow.
F-21
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
As
of December 31, 2017 and 2016, intangible assets, consisted of the
following:
|
Patents
& knowhow & license
|
|
|
|
|
December 31, 2017
|
December 31, 2016
|
|
Cost
basis
|
$17,674,431
|
$17,560,496
|
|
Less:
accumulated amortization
|
(5,325,113)
|
(3,539,617)
|
|
|
$12,349,318
|
$14,020,879
|
|
Software
|
|
|
|
|
December 31, 2017
|
December 31, 2016
|
|
Cost
basis
|
$158,273
|
$125,964
|
|
Less:
accumulated amortization
|
(87,899)
|
(54,262)
|
|
|
$70,374
|
$71,702
|
|
Total
intangibles, net
|
$12,419,692
|
$14,092,581
|
All software is provided by a third party
vendor, is not internally developed, and has an estimated useful
life of 5 years. Patents, knowhow and license are amortized using
an estimated useful life of five to ten years. Amortization expense for the years ended December
31, 2017, 2016 and 2015 was $1,790,258, $1,784,208 and $1,521,629,
respectively. Estimated amortization expense for each of the
ensuing years are as follows for the years ending December
31:
|
Years ending December 31,
|
Amount
|
|
2018
|
$1,788,814
|
|
2019
|
1,787,586
|
|
2020
|
1,784,335
|
|
2021
|
1,778,866
|
|
2022
and thereafter
|
5,280,091
|
|
|
$12,419,692
|
F-22
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
NOTE 10 – LEASES
The
Company leases facilities under non-cancellable operating lease
agreements. These facilities are located in the United
States, Hong Kong and China. The Company recognizes
rental expense on a straight-line basis over the life of the lease
period. Rent expense under operating leases for the year
ended December 31, 2017, 2016 and 2015 was approximately
$4,345,893, $1,043,968 and $1,043,833, respectively.
As
of December 31, 2017, the Company has the following future minimum
lease payments due under the foregoing lease
agreements:
|
Years ending December 31,
|
Amount
|
|
2018
|
$3,062,842
|
|
2019
|
2,822,735
|
|
2020
|
2,627,027
|
|
2021
|
2,547,990
|
|
2022
and thereafter
|
12,623,886
|
|
|
$23,684,480
|
NOTE 11 – RELATED PARTY TRANSACTIONS
As of
December 31, 2017 and 2016, accrued expenses included director fees
of $25,882 and $3,082 due to independent director Mr. Gang
Ji.
On
December 15, 2017, the Company entered into a Share Purchase
Agreement with three of its executive officers, pursuant to which
the Company agreed to sell, and the three executive officers agreed
to purchase an aggregate of 41,667 shares of the Company’s
common stock, par value $0.001 per share at $12.00 per share, for
total gross proceeds of approximately $500,000. The transaction
closed on December 22, 2017.
The
Company advanced petty cash to officers for business travel
purpose. As of December 31, 2017 and 2016, other
receivables due from officers for business travel purpose was
$8,531 and $nil, respectively.
NOTE 12 – EQUITY
ASC
Topic 505, “Equity”, paragraph 505-50-30-6
establishes that share-based payment transactions with nonemployees
shall be measured at the fair value of the consideration received
or the fair value of the equity instruments issued, whichever is
more reliably measurable.
In
March 2015, the Company closed a financing transaction pursuant to
which it sold 515,786 shares of the Company’s common stock to
selected investors at $38 per share, for total gross proceeds of
approximately $19,600,000. The shares were sold pursuant to
separate subscription agreements between the Company and each
investor. The Company incurred a finder fee of $979,992, equal to
5% of the gross proceeds from the investors that were introduced by
such finders, which was recorded as reduction in
equity.
On
June 26, 2015, the Company completed its acquisition of the certain
license rights to technology and know-how from Blackbird
BioFinance, LLC (“Blackbird”) and entered into an
assignment and assumption agreement to acquire all of
Blackbird’s right, title and interest in and to the exclusive
worldwide license to a CD40LGVAX vaccine from the University of
South Florida. According to the asset purchase agreement,
$1,050,500 in restricted common stock (based on the 20-day
volume-weighted average price of the Company’s stock on the
closing date) will be delivered to Blackbird at closing, thus
28,120 shares of Company common stock were issued as part of the
consideration of this transaction. In addition, 18,747 shares of
Company common stock (equal to $700,000 based on the 20-day
volume-weighted average price of the Company’s stock on the
closing date) would be delivered to Blackbird on the 6 month
anniversary of the closing date upon satisfaction of certain
conditions according to the agreements. Above shares were issued in
November 2015.
F-23
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
On
February 4, 2016, the Company conducted an initial closing of a
financing transaction (the “Financing”), pursuant to
which it sold an aggregate of 263,158 shares of the Company’s
common stock, par value $0.001 per share to Wuhan Dangdai Science
& Technology Industries Group Inc. (the “Investor”)
at $19.00 per share, for total gross proceeds of approximately
$5,000,000. The Investor agreed to purchase, in one or more
subsequent closings, up to an additional 2,006,842 shares on or
before April 15, 2016, for a potential aggregate additional raise
of $38,130,000. The Company had received the proceeds of $5,000,000
on February 4, 2016.
On
April 15, 2016, the Company completed the second and final closing
of the Financing with the Investor, pursuant to which the Company
sold to the Investor 2,006,842 shares of the Company’s Common
Stock, for approximately $38,130,000 in gross proceeds. The
aggregate gross proceeds from both closings in the Financing
totaled approximately $43,130,000. In the aggregate, 2,270,000
shares of Common Stock were issued in the Financing.
In
connection with the above Financing, the Company agreed to pay a
finder’s fee equal to 5% of the gross proceeds comprised of
(i) $657,628 from the gross proceeds of the Financing and (ii)
78,888 restricted shares of Common Stock based on the per share
purchase price in the Financing of $19 per share. On April 28,
2016, 78,888 shares of common stock were issued to the finder,
which was recorded against the equity.
On
December 15, 2017, the Company entered into a Share Purchase
Agreement with three of its executive officers, pursuant to which
the Company agreed to sell, and the three executive officers agreed
to purchase an aggregate of 41,667 shares of the Company’s
common stock, par value $0.001 per share at $12.00 per share, for
total gross proceeds of approximately $500,000. The transaction
closed on December 22, 2017.
On
December 26, 2017, the Company entered into a Share Purchase
Agreement with two investors, pursuant to which the Company agreed
to sell and the two investors agreed to purchase from the Company,
an aggregate of 1,166,667 shares of the Company’s common
stock, par value $0.001 per share, at $12.00 per share, for total
gross proceeds of approximately $14,000,000. The transaction closed
on December 28, 2017. Together with a private placement with three
of its executive officers on December 22, 2017, the Company raised
an aggregate of approximately $14.5 million in the two private
placements in December 2017.
During
the year ended December 31, 2017, 2016 and 2015, the Company
expensed $4,512,192, $4,742,920 and $7,182,118 associated with
unvested options awards and $833,019, $709,497 and $410,320
associated with restricted common stock issuances,
respectively.
During
the year ended December 31, 2017, 2016 and 2015, options for
57,400, 196,185 and 152,404 underlying shares were exercised,
57,400, 196,185 and 152,404 shares of the Company’s common
stock were issued accordingly.
During
the year ended December 31, 2017, 2016 and 2015, 68,446, 24,660 and
6,253 shares of the Company's restricted common stock were issued
to directors, employees and advisors respectively.
During
the year ended December 31, 2017, the Company repurchased 426,794
shares of the Company’s common stock with the total cost of
$3,977,929. There was no repurchase of stock in 2016. Details are
as follows:
|
Period
|
Total number of shares purchased
|
Average price paid per share
|
Total number of shares purchased as part of publicly announced
plans or programs
|
Maximum dollar value of shares that may yet be purchased under the
plans or programs
|
|
June
9, 2017 ~ June 30, 2017
|
170,169
|
$7.98
|
170,169
|
|
|
July
1, 2017 ~ September 30, 2017
|
114,156
|
$9.51
|
114,156
|
|
|
October
1, 2017 ~ December 31, 2017
|
142,469
|
$10.77
|
142,469
|
|
|
Total
|
426,794
|
$9.32
|
426,794
|
6,022,071
|
The Company's Board of Directors has approved a
new stock repurchase program granting the company authority to
repurchase up to $10 million in common shares (the "2017 Stock
Repurchase Program") through open market purchases pursuant to a
plan adopted in accordance with Rule 10b5-1 of the Securities
Exchange Act of 1934, as amended (the “Exchange
Act”)
and in accordance with Rule 10b-18 of the Exchange Act and was
announced on June 1, 2017.
F-24
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
NOTE 13 – COMMITMENTS AND CONTINGENCIES
Operating lease commitments
The
future minimum lease payment due under the executed operating lease
agreements as of December 31, 2017 was presented in Note 10 to the
consolidated financial statements.
Capital commitments
As of
December 31, 2017, the capital commitments of the Company are
summarized as follows:
|
|
December 31, 2017
|
|
Contracts
for acquisition of plant and equipment being or to be
executed
|
$1,444,449
|
F-25
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
NOTE 14 – STOCK BASED COMPENSATION
Our
stock-based compensation arrangements include grants of stock
options and restricted stock awards under the Stock Option Plan
(the “2009 Plan”,“2011 Plan”, “2013
Plan” and the “2014 Plan”), and certain awards
granted outside of these plans. The compensation cost that has been
charged against income related to stock options (including shares
issued for services and expense true-ups and reversals described in
Note 12) for the year ended December 31, 2017, 2016 and 2015 was
$4,512,192, $4,742,920 and
$7,182,118, respectively. The compensation cost that has
been charged against income related to restrict stock awards for
the year ended December 31, 2017, 2016 and 2015 was $833,019, $709,497 and $410,320,
respectively.
These
expenses are included in overhead, general and administrative
expense, selling and marketing expense as well as research and
development expenses in our Consolidated Statements of
Operations.
As of
December 31, 2017, there was $4,362,541 all unrecognized
compensation cost related to an aggregate of 592,249 of non-vested
stock option awards and $385,331 related to an aggregate of 34,105
of non-vested restricted stock awards. Restricted stock
awards under long-term incentive plan is not accounted for as
attendant could chose to surrender part of the restricted stock
awards for individual income tax payment purpose. These costs are
expected to be recognized over a weighted-average period of 2.29
years for the stock options awards and 1.37 years for the
restricted stock awards.
During
the year ended December 31, 2017, the Company issued an aggregate
of 547,793 options under the 2011 Plan, 2013 Plan and 2014 Plan to
officers, directors, employees and advisors. The grant date fair
value of these options was $4,600,926 using Black-Scholes option
valuation models with the following assumptions: grant date strike
price from $5.3 to $13.2, volatility 85.41% to 89.62%, expected
life 6.0 years, and risk-free rate of 1.86% to 2.29%. The Company
is expensing these options on a straight-line basis over the
requisite service period.
During
the year ended December 31, 2016, the Company issued an aggregate
of 309,382 options under the 2013 Plan and 2014 Plan to officers,
directors, employees and advisors. The grant date fair value of
these options was $3,811,362 using Black-Scholes option valuation
models with the following assumptions: grant date strike price from
$12.13 to $40, volatility 88.44% to 90.03%, expected life 6.0
years, and risk-free rate of 1.07% to 2.17%. The Company is
expensing these options on a straight-line basis over the requisite
service period.
During
the year ended December 31, 2015, the Company issued an aggregate
of 721,779 options under the 2013 Plan and 2014 Plan to officers,
directors and employees. The grant date fair value of these options
was $13,687,655 using Black-Scholes option valuation models with
the following assumptions: exercise price equal to the grant date
stock price of $12.91 to $38.4, volatility 88.41% to 99.27%,
expected life 6.0 years, and risk-free rate of 1.39% to 1.92%. The
Company is expensing these options on a straight-line basis over
the requisite service period.
F-26
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
The
following table summarizes stock option activity as of December 31,
2017 and 2016 and for the year ended December 31,
2017:
|
|
Number of Options
|
Weighted- Average Exercise Price
|
Weighted- Average Remaining Contractual Term (in
years)
|
Aggregate Intrinsic Value
|
|
Outstanding
at December 31, 2015
|
1,952,648
|
$12.42
|
7.8
|
$17,701,962
|
|
Grants
|
309,382
|
18.65
|
|
|
|
Forfeitures
|
(458,030)
|
19.45
|
|
|
|
Exercises
|
(196,185)
|
4.51
|
|
|
|
|
|
|
|
|
|
Outstanding
at December 31, 2016
|
1,607,815
|
$12.59
|
7.3
|
$6,355,072
|
|
Grants
|
547,793
|
11.34
|
|
|
|
Forfeitures
|
(206,019)
|
20.68
|
|
|
|
Exercises
|
(57,400)
|
5.37
|
|
|
|
Outstanding
at December 31, 2017
|
1,892,189
|
$11.54
|
7.0
|
$4,909,194
|
|
|
|
|
|
|
|
Vested
and exercisable at December 31, 2017
|
1,299,940
|
$10.52
|
6.1
|
$4,619,527
|
|
Exercise
|
Number of Options
|
|
|
Price
|
Outstanding
|
Exercisable
|
|
$3.00
- $4.95
|
185,547
|
185,547
|
|
$5.00
- $9.19
|
569,204
|
510,768
|
|
$9.20+
|
1,137,438
|
603,625
|
|
|
1,892,189
|
1,299,940
|
The
aggregate intrinsic value for stock options outstanding is defined
as the positive difference between the fair market value of our
common stock and the exercise price of the stock
options.
Cash
received from option exercises under all share-based payment
arrangements for the year ended December 31, 2017, 2016 and 2015
was $308,371, $885,680 and $682,303, respectively.
F-27
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
NOTE 15 – NET LOSS PER SHARE
Basic
and diluted net loss per common share is computed on the basis of
our weighted average number of common shares outstanding, as
determined by using the calculations outlined below:
|
|
For the Year Ended
|
||
|
|
December 31,
|
||
|
|
2017
|
2016
|
2015
|
|
Net
loss
|
$(25,490,310)
|
$(28,208,376)
|
$(19,447,721)
|
|
|
|
|
|
|
Weighted
average shares of common stock
|
14,345,604
|
13,507,408
|
11,472,306
|
|
Dilutive
effect of stock options
|
-
|
-
|
-
|
|
Restricted
stock vested not issued
|
-
|
-
|
-
|
|
Common
stock and common stock equivalents
|
14,345,604
|
13,507,408
|
11,472,306
|
|
|
|
|
|
|
Net
loss per basic share
|
$(1.78)
|
$(2.09)
|
$(1.70)
|
|
Net
loss per diluted share
|
$(1.78)
|
$(2.09)
|
$(1.70)
|
For
the year ended December 31, 2017, 2016 and 2015, the effect of
conversion and exercise of the Company’s outstanding options
are excluded from the calculations of dilutive net income (loss)
per share as their effects would have been anti-dilutive since the
Company had generated loss for the year ended December 31, 2017,
2016 and 2015.
NOTE 16 – INCOME TAXES
Income
taxes are accounted for under the asset and liability method.
Deferred tax assets and liabilities are recognized for the future
tax consequences attributable to differences between the financial
statement carrying amounts of existing assets and liabilities and
their respective tax bases and operating loss and tax credit
carry-forwards. Deferred tax assets and liabilities are measured
using enacted tax rates expected to apply to taxable income in the
years in which those temporary differences are expected to be
recovered or settled. The effect of a change in tax rates on
deferred tax assets and liabilities is recognized in income in the
period during which such rates are enacted.
The
Company considers all available evidence to determine whether it is
more likely than not that some portion or all of the deferred tax
assets will be realized. The ultimate realization of deferred tax
assets is dependent upon the generation of future taxable income
during the periods in which those temporary differences become
realizable. Management considers the scheduled reversal of deferred
tax liabilities (including the impact of available carryback and
carry-forward periods), and projected taxable income in assessing
the realizability of deferred tax assets. In making such judgments,
significant weight is given to evidence that can be objectively
verified. Based on all available evidence, in particular our
three-year historical cumulative losses, recent operating losses
and U.S. pre-tax loss for the year ended December 31, 2017, we
recorded a valuation allowance against our U.S. net deferred tax
assets. In order to fully realize the U.S. deferred tax assets, we
will need to generate sufficient taxable income in future periods
before the expiration of the deferred tax assets governed by the
tax code.
F-28
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
The following represent components
of the income tax (expense) credit for the
year ended December 31, 2017, 2016 and 2015:
|
|
For the Year Ended
|
||
|
|
December 31,
|
||
|
|
2017
|
2016
|
2015
|
|
Current:
|
|
|
|
|
US
federal
|
$-
|
$-
|
$733,158
|
|
US
state
|
(2,450)
|
(4,093)
|
(4,557)
|
|
Foreign
|
|
|
-
|
|
Total
current tax credit (expense)
|
$(2,450)
|
$(4,093)
|
$728,601
|
|
Deferred:
|
|
|
|
|
Federal
|
$-
|
$-
|
$-
|
|
State
|
-
|
-
|
-
|
|
Foreign
|
-
|
-
|
-
|
|
Total
deferred tax expense
|
$-
|
$-
|
$-
|
|
Total
income tax credit (expense)
|
$(2,450)
|
$(4,093)
|
$728,601
|
Tax
effects of temporary differences that give rise to significant
portions of the Company's deferred tax assets at December 31, 2017
and 2016 are presented below:
|
|
December 31,
|
December 31,
|
|
|
2017
|
2016
|
|
Deferred
tax assets:
|
|
|
|
Net
operating loss carry forwards (offshore)
|
$5,448,339
|
$3,827,747
|
|
Net
operating loss carry forwards (US)
|
3,864,824
|
4,496,655
|
|
Accruals
(offshore)
|
588,277
|
280,756
|
|
Accrued
compensation (US)
|
83,071
|
25,168
|
|
Stock-based
compensation (US)
|
2,315,801
|
3,018,905
|
|
Investments
(US)
|
1,893,532
|
3,673,382
|
|
Credits
(US)
|
217,329
|
97,504
|
|
Property
and equipment
|
123
|
-
|
|
Goodwill
& intangibles
|
49,653
|
33,079
|
|
Subtotal
|
14,460,949
|
15,453,196
|
|
Less:
valuation allowance
|
(14,460,949)
|
(15,452,737)
|
|
Total
deferred tax assets
|
-
|
459
|
|
|
|
|
|
Deferred
tax liabilities:
|
|
|
|
Property
and equipment
|
-
|
(459)
|
|
|
|
|
|
Subtotal
|
-
|
(459)
|
|
|
|
|
|
Net
deferred tax asset
|
$-
|
$-
|
In
each period since inception, the Company has recorded a valuation
allowance for the full amount of net deferred tax assets, as the
realization of deferred tax assets is uncertain. As a result, the
Company has not recorded any federal or state income tax benefit in
the consolidated statements of operations and comprehensive income
(loss).
F-29
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
On
December 22, 2017, US President signed tax reform bill (Tax Cut and
Jobs Act (H.R.1)) and reduced top corporate tax rate from 35% to
21% effective from January 1, 2018. Pursuant to this new Act,
non-operating loss carry back period is eliminated and an
indefinite carry forward period is permitted. As of December 31,
2017, the Company had net operating loss carryforwards of $16
million for U.S federal purposes, $6 million for U.S. state
purposes. As of December 31, 2017, the Company had net operating
loss carryforwards of $14 million for Chinese income tax purposes,
such losses are set to expire in 2022 for Chinese income tax
purposes. All deferred income tax expense is offset by changes in
the valuation allowance pertaining to the Company's existing net
operating loss carryforwards due to the unpredictability of future
profit streams prior to the expiration of the tax
losses. The Company's effective tax rate differs from
statutory rates of 21% for U.S. federal income tax purposes, 15% ~
25% for Chinese income tax purpose and 16.5% for Hong Kong income
tax purposes due to the effects of the valuation allowance and
certain permanent differences as it pertains to book-tax
differences in the value of client shares received for
services.
Pursuant to the Corporate Income Tax Law of the
PRC, all of the Company’s PRC subsidiaries are liable to PRC
Corporate Income Taxes (“CIT”) at a rate of 25% except
for Cellular Biomedicine Group
Ltd. (Shanghai) (“CBMG Shanghai”). According to
Guoshuihan 2009 No. 203, if an entity is certified as an
“advanced and new technology enterprise”, it is
entitled to a preferential income tax rate of 15%. CBMG Shanghai
obtained the certificate of “advanced and new technology
enterprise” dated October 30, 2015 with an effective period
of three years and the provision for PRC corporate income tax for
CBMG Shanghai is calculated by applying the income tax rate of 15%
from 2015. CBMG Shanghai will still apply the certificate of
“advanced and new technology enterprise” in
2018.
Income
tax expense for year ended December 31, 2017, 2016 and 2015
differed from the amounts computed by applying the statutory
federal income tax rate of 35% to pretax income (loss) as a result
of the following:
|
|
For the Year Ended
|
For the Year Ended
|
For the Year Ended
|
|
|
December 31, 2017
|
December 31, 2016
|
December 31, 2015
|
|
Effective
Tax Rate Reconciliation
|
|
|
|
|
Income
tax provision at statutory rate
|
35%
|
35%
|
35%
|
|
State
income taxes, net of federal benefit
|
0%
|
0%
|
0%
|
|
Rate
deduction
|
(20)%
|
0%
|
0%
|
|
Foreign
rate differential
|
(13)%
|
(9)%
|
(12)%
|
|
Other
permanent difference
|
(2)%
|
(2)%
|
(4)%
|
|
Change
in valuation allowance
|
0%
|
(24)%
|
(15)%
|
|
Total
tax credit (expense)
|
0%
|
0%
|
4%
|
NOTE 17 – COLLABORATION AGREEMENT
Part
of AG’s business includes a collaboration agreement to
establish and operate a biologic treatment center in the Jilin
province of China. Under the terms of the Collaboration
Agreement dated December 10, 2012 and its supplementary agreement
dated July 19, 2014 (the “Collaboration Agreement”),
AG’s collaborative partner (the “Partner”) funded
the development of the center and provides certain ongoing
services. In exchange, the Partner receives preferred
repayment of all funds that were invested in the development, 60%
of the net profits until all of the invested funds are repaid, and
40% of the net profits thereafter, and the rights to the physical
assets at the conclusion of the agreement. We accounted
for this transaction in accordance with ASC 808 Collaborative
Arrangements and have reflected all assets and liabilities of
the treatment center. With our recent build-up of
multiple cancer therapeutic technologies, we have prioritized our
clinical efforts on developing CAR-T technologies, Vaccine, Tcm and
TCR clonality technologies, and not actively pursuing the
fragmented technical services opportunities.
In
June 2016, the Company and the Partner agreed to terminate the
Collaboration Agreement and in July 2016 entered into a cooperation
termination agreement (the “Termination Agreement”)
with the Partner. In August 2016, in accordance with the
Termination Agreement, the Company paid $0.3 million (RMB2 million
equivalent) to settle all the liabilities with the Partner and
retain the ownership of all the assets under the Collaboration
Agreement.
F-30
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
NOTE 18 – SEGMENT INFORMATION
The
Company is engaged in the development of new treatments for
cancerous and degenerative diseases utilizing proprietary
cell-based technologies, which have been organized as one reporting
segment since they have similar nature and economic
characteristics. The Company’s principle operating decision
maker, the Chief Executive Officer, receives and reviews the result
of the operation for all major cell platforms as a whole when
making decisions about allocating resources and assessing
performance of the Company. In accordance with FASB ASC 280-10, the
Company is not required to report the segment
information.
NOTE 19 – SUBSEQUENT EVENTS
On
January 30, 2018 and February 5, 2018, the Company entered into
securities purchase agreements with certain investors pursuant to
which the Company agreed to sell, and the investors agreed to
purchase from the Company, an aggregate of 1,719,324 shares of the
Company’s common stock, par value $0.001 per share, at $17.80
per share, for total gross proceeds of approximately $30.6
million. The transaction closed on February 5,
2018.
Pursuant to the
Purchase Agreement, the Investors have the right to nominate one
director to the board of directors of the Company to stand for
election at the 2018 Annual Meeting of Stockholders. Effective as
of the closing of the February 2018 private placement, the Board
elected Bosun S. Hau as a nonexecutive Class III director of the
Company.
On
February 6, 2018, driven primarily by the Company’s strategy
move to expand its business operations in early diagnosis and
cancer intervention, Meng Xia transitioned from the role of Chief
Operating Officer to Head of the Early Diagnosis & Intervention
for the Company.
On
February 15, 2018, the Company obtained a 36 months exclusive
option with Augusta University to to negotiate a royalty-bearing,
exclusive license to the patent rights owned by the Augusta
University relating to an invention to identify novel alpha
fetoprotein specific T-cell receptors (TCR) for a hepatocellular
carcinoma immunotherapy ( “HCC”). The Company plan to
evaluate the feasibility and opportunities of this novel alpha
fetoprotein TCR to redirect T Cells for the HCC
indication.
F-31
CELLULAR BIOMEDICINE GROUP, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2017, 2016 AND 2015
NOTE 20 – UNAUDITED QUARTERLY FINANCIAL
INFORMATION
|
|
Year ended December 31, 2017
|
||||
|
|
Q4
|
Q3
|
Q2
|
Q1
|
Total
|
|
Selected Income Statement Data:
|
|
|
|
|
|
|
Net
sales and revenue
|
$68,691
|
$106,787
|
$62,914
|
$98,425
|
$336,817
|
|
Gross
Profit
|
37,266
|
51,493
|
24,817
|
61,023
|
174,599
|
|
Net
loss
|
(6,922,585)
|
(6,202,214)
|
(6,203,518)
|
(6,161,993)
|
(25,490,310)
|
|
Net
loss per share :
|
|
|
|
|
|
|
Basic
|
(0.48)
|
(0.43)
|
(0.43)
|
(0.43)
|
(1.78)
|
|
Diluted
|
(0.48)
|
(0.43)
|
(0.43)
|
(0.43)
|
(1.78)
|
|
|
Year ended December 31, 2016
|
||||
|
|
Q4
|
Q3
|
Q2
|
Q1
|
Total
|
|
Selected Income Statement Data:
|
|
|
|
|
|
|
Net
sales and revenue
|
$57,828
|
$10,012
|
$71,599
|
$488,491
|
$627,930
|
|
Gross
Profit/(Loss)
|
33,319
|
884
|
(251,988)
|
(14,702)
|
(232,487)
|
|
Net
loss
|
(6,139,761)
|
(10,661,220)
|
(7,197,282)
|
(4,210,113)
|
(28,208,376)
|
|
Net
loss per share :
|
|
|
|
|
|
|
Basic
|
(0.43)
|
(0.75)
|
(0.52)
|
(0.35)
|
(2.09)
|
|
Diluted
|
(0.43)
|
(0.75)
|
(0.52)
|
(0.35)
|
(2.09)
|
F-32