Attached files
| file | filename |
|---|---|
| 8-K - 8-K - EPIRUS Biopharmaceuticals, Inc. | a15-3051_38k.htm |
| EX-10.1 - EX-10.1 - EPIRUS Biopharmaceuticals, Inc. | a15-3051_3ex10d1.htm |
| EX-99.2 - EX-99.2 - EPIRUS Biopharmaceuticals, Inc. | a15-3051_3ex99d2.htm |
Exhibit 99.1
BUSINESS
Overview
We are a commercial-stage biopharmaceutical company focused on improving patient access to important biopharmaceuticals by developing, manufacturing, and commercializing biosimilar therapeutics, or biosimilars, in targeted geographies worldwide. We seek to build a sustainable, profitable biosimilar company with a pipeline of operationally synergistic monoclonal antibodies in inflammation and immunology.
Our lead product candidate is BOW015, a biosimilar version of Remicade® (infliximab). Remicade, marketed by Johnson & Johnson, Merck Schering and Mitsubishi Tanabe for the treatment of various inflammatory diseases, generated approximately $8.4 billion in global sales in 2013.
We have reported bioequivalence and efficacy data from a Phase 1 clinical trial in the United Kingdom and a Phase 3 clinical trial in India, both of which demonstrated the equivalence of BOW015 to Remicade. We recently announced positive 58 week follow up data from the Phase 3 trial. In this open label phase, we demonstrated that patients can be safely initiated and effectively maintained on BOW015 for 58 weeks, and that patients can be safely switched from Remicade to BOW015 and effectively maintained out to 58 weeks. We intend to initiate a global clinical program for BOW015 in North America and Europe in late 2015 or early 2016. We have been designing this clinical program in consultation with European regulatory bodies in order to obtain the data necessary to support eventual approval of BOW015 in North America and Europe.
In November 2014, we launched BOW015 in India, under the brand name InfimabTM, the first infliximab biosimilar in India, with our commercialization partner Ranbaxy Laboratories Limited, or Ranbaxy. We expect to utilize our existing regulatory data package to gain regulatory approval for BOW015 in additional countries.
Our pipeline of biosimilar product candidates also includes BOW050, a biosimilar version of Humira® (adalimumab), which is marketed by AbbVie and used to treat inflammatory diseases, and BOW070, a biosimilar version of Actemra® (tocilizumab), which is marketed by Genentech/Roche and used to treat inflammatory diseases. BOW050 and BOW070 are in preclinical development. Collectively, Remicade, Humira, and Actemra generated $20.5 billion in global sales in 2013, according to EvaluatePharma®. We are advancing existing development and commercialization partnerships for our product candidates in China and India, as well as in additional countries in Southeast Asia and North Africa. We are also pursuing development and commercial partnerships in the United States, Europe, Brazil, and elsewhere.
Biosimilars are highly similar versions of approved patented biological drug products, referred to as reference or innovator products. Based on projected global sales estimates from EvaluatePharma, more than $70 billion in major biologics sales is expected to face biosimilar competition as a result of patent expiries over the next decade. We are currently focused on developing biosimilars to therapeutic monoclonal antibodies, or MAbs. We seek to take advantage of a convergence of three trends shaping the global market for MAb biosimilars. First, the market for MAbs is large and growing, and comprises many of the top-selling therapeutics in the world. According to EvaluatePharma, sales of MAbs accounted for $61.8 billion globally in 2013 and grew at a compound annual growth rate of 14% from 2010 through 2013. Sixteen of these MAbs, representing $43.3 billion of global sales in 2013, are expected to lose patent protection globally by 2020, creating an opportunity for companies focusing on biosimilars to the referenced MAbs. Second, our commercial approach addresses the defined but evolving and diverse commercial and regulatory frameworks that exist globally for the introduction of biosimilars. These frameworks allow companies to develop and commercialize biosimilars at a cost that is expected to be less than that incurred by the innovators of the respective reference products. Third, MAbs are often very expensive. In many countries outside the United States, public and private payors
are seeking to lower the cost of biologics and improve patient access to these important medications. This favors biosimilar versions of biologics that are priced at a discount to the branded reference products.
We believe that successfully building a biosimilar business requires a clear path toward sustainability and profitability. In order to reach that objective, we have chosen to focus on markets that meet three important criteria: (1) a clear, precedent-driven regulatory pathway; (2) minimal exposure to potential patent encumbrances; and (3) a commercially viable path. We have developed distinct strategies to access each of these markets, which we have segmented into three types: Developed Markets, Local Production Markets, and Accessible Markets.
The Developed Markets, predominately North America and Europe, are expected to be the financial anchor of our business. In Europe, we intend to commercialize our products using a licensing or distribution model in conjunction with direct sales. This approach is designed to allow us to book top-line revenue and invest gradually in commercial infrastructure, thereby creating a sustainable, profitable enterprise. In North America, we anticipate seeking a commercial partner or alternative commercial model, which may include contracting directly with payors of other third-party entities. We plan to initiate a global clinical program for BOW015 in late 2015 or early 2016, after which we intend to pursue regulatory approval for BOW015 in North America and Europe with filings in 2017.
In Local Production Markets, which include China and Brazil, local authorities mandate or strongly encourage local production as a condition for regulatory and/or commercial acceptance. In these Local Production Markets, we intend to collaborate with local partners to enable in-country production of our products using our SCALETM manufacturing platform which enables turn-key, locally-based manufacturing of biosimilars. We believe that our SCALE platform provides us with a competitive advantage by giving us the ability to accelerate the entry of our biosimilar candidates into many emerging markets. In China, we have entered into an Exclusive License and Collaboration Agreement with Livzon Mabpharm Inc., or Livzon, for the global development and commercialization of certain antibodies or related biological compounds, including BOW015. We are also pursuing development and commercial partnerships in Brazil.
In Accessible Markets, in which our current regulatory data are expected to be sufficient for approval, we intend to commercialize through partnerships. We currently have an agreement to commercialize BOW015 in India with Ranbaxy. We launched BOW015, under the brand name InfimabTM, in India in November 2014. We are also actively pursuing access to additional markets through Ranbaxy and other potential licensing partners.
Products
Our product pipeline contains three products at different stages of development. The most advanced of these is BOW015 (infliximab), which has received marketing and manufacturing approval in India, and for which we have reported favorable Phase 1 and Phase 3 clinical data. The Phase 3 trial met its predefined primary endpoint and demonstrated comparability of BOW015 to Remicade, as measured by the standardized American College of Rheumatology 20% improvement scoring system, or ACR20, response, in severe rheumatoid arthritis patients. The study also showed no meaningful differences between BOW015 and Remicade with regard to safety or immunogenicity. Our other
pipeline products, BOW050 (adalimumab), a proposed biosimilar to Humira, and BOW070 (tocilizumab), a proposed biosimilar to Actemra, are in preclinical development.

* Emerging markets targeted for near-term MAA filings for BOW015 include: Indonesia, Mexico, Thailand, Korea, Argentina and Malaysia
BOW015 (Infliximab)
Our lead program is BOW015, a biosimilar version of Remicade. Remicade is a prescription product marketed globally by Johnson & Johnson, Merck Schering and Mitsubishi Tanabe for the treatment of inflammatory diseases including rheumatoid arthritis, Crohn’s Disease, ankylosing spondylitis, psoriatic arthritis and psoriasis. Sales of Remicade were $8.4 billion worldwide in 2013. We have conducted extensive bioanalytical and physicochemical comparisons of BOW015 to Remicade and have data from a Phase 1 study in the United Kingdom and a 189 patient Phase 3 double blind comparator study in India demonstrating bioequivalence, safety, quality and efficacy of BOW015. We launched BOW015 with our commercialization partner Ranbaxy, under the brand name InfimabTM, in November 2014. Infimab is the first infliximab biosimilar to be sold in India.
On September 23, 2014, we announced positive 58 week follow up data from an efficacy and safety trial comparing BOW015 to Remicade. The study consisted of a 16 week, double blinded, head to head comparison with Remicade for safety and efficacy followed by an open label phase where Remicade responders were switched to BOW015 and all patients were followed for the duration of the study. The study met its primary endpoint of ACR20 response, the American College of Rheumatology criteria for clinical improvement in patients with rheumatoid arthritis, indicating a 20% improvement across a series of diagnostic parameters. These patients were then followed out to week 58 in an open label phase of the trial, with BOW015 patients remaining on BOW015 and Remicade responders being switched to BOW015 for the remainder of the 58 weeks.
In the open-label phase, patients who continued on BOW015 were compared to patients who received four doses of Remicade, followed by a switch to four doses of BOW015. Immune responses as
well as overall safety and tolerability for BOW015 were comparable to the arm switched from Remicade to BOW015 and were consistent with the expected profile of Remicade. Further, ACR20 responses were durably maintained to 54 weeks from the week 16 primary endpoint previously reported. BOW015 was launched in India in November 2014 and is expected to launch in additional territories leveraging the Indian regulatory package.
BOW050 (Adalimumab)
We are currently developing and evaluating commercialization opportunities for BOW050 as a proposed biosimilar to Humira in a range of target markets, including potentially in collaboration with Livzon in China and with other partners in other target markets. Humira (marketed by AbbVie) is an inhibitor of TNF-α used to treat inflammatory diseases, including rheumatoid arthritis and certain other forms of adult and pediatric arthritis, ankylosing spondylitis, inflammatory bowel disease, and chronic psoriasis and psoriasis. According to EvaluatePharma, global sales of adalimumab in 2013 were $11 billion. Physicochemical characterization of BOW050 is ongoing and the product may enter clinical trials in 2016, providing for a path to filing and potential approval and commercialization subsequent to the time of Humira’s 2018 loss of exclusivity in Europe and other regions.
BOW070 (Tocilizumab)
We are in the comparability phase of development of BOW070 a proposed biosimilar version of Actemra (marketed by Genentech/Roche). Actemra is an immunosuppressive drug for the treatment of rheumatoid arthritis, polyarticular arthritis and systemic juvenile idiopathic arthritis. According to EvaluatePharma, global sales of Actemra in 2013 were $1.1 billion. Actemra is expected to lose exclusivity in 2019.
Management
Our senior management team has more than 180 years of collective experience in the biopharmaceutical industry. In addition, various members of our management team and our board of directors have worked for prominent biotechnology and pharmaceutical companies including Amgen, Biogen Idec, Pfizer, Wyeth (acquired by Pfizer), Genzyme (acquired by Sanofi), Shire, Cephalon, Millennium, Takeda, BioAssets (acquired by Cephalon), Cubist Pharmaceuticals (acquired by Merck), Invida (acquired by Menarini), Kythera, Therion Biologics and ToleRx. Our president and Chief Executive Officer, Amit Munshi, was a co-founder, and the chief business officer at Kythera Biopharmaceuticals, which underwent a successful initial public offering on NASDAQ in 2013. Mr. Munshi has more than 24 years of pharmaceutical and biotechnology experience in both the United States and internationally, including general management, product development, licensing and business development.
Strategy
We believe that successfully building a biosimilar business requires a clear path toward sustainability and profitability. In order to reach that objective, we have chosen to focus on markets that meet three important criteria: (1) clear, precedent-driven regulatory pathway; (2) minimal exposure to potential patent encumbrances; and (3) a commercially viable path. We have developed distinct strategies to access each of these markets and segmented them into three types: Developed Markets, Local Production Markets, and Accessible Markets. Our key operational priorities to execute on our strategy are as follows:
· In Developed Markets, including North America and Europe, we intend to commercialize our products using a licensing or distribution model in conjunction with direct sales. Europe has an existing regulatory approval pathway and a patent environment we believe offers a clear path
forward for our pipeline. From a commercial perspective, the European market for biosimilars is strengthened by the desire of governments to reduce overall healthcare expenditure. Through a combination of substitution rules, regional tenders and political pressure to introduce biosimilars, Europe represents a commercially tractable market. The United States biosimilar pathway is gaining clarity. We intend to initiate a global clinical program for BOW015 in late 2015 or early 2016, after which we intend to pursue regulatory approval for BOW015 in North America and Europe. Upon obtaining regulatory approval for BOW015 in Europe, we contemplate a “hybrid” commercial model which may include either a Europe- wide, two-brand co-marketing arrangement or a country-by-country partnership approach, whereby we retain exclusive commercial rights to a subset of European markets. We expect that either commercial approach, coupled with Europe’s existing regulatory framework and its approved biosimilar precedent will allow us to book top-line revenue and invest gradually in commercial infrastructure, thereby creating a sustainable, profitable enterprise. As the regulatory environment in the United States becomes clearer, we anticipate seeking a commercial partner or alternative commercial model for BOW015, which may include contracting directly with payors or other third-party entities.
· In Local Production Markets, we intend to collaborate with local partners to enable in-country production of our products using our SCALE manufacturing platform. In Local Production Markets, which include China and Brazil, local authorities mandate or strongly encourage local production, and additional clinical work above and beyond that submitted for Indian regulatory approval will likely be necessary to secure approval for BOW015. In these markets, several biologics have already seen their key patents expire. Furthermore, we expect to establish local commercialization partnerships in these, often tender driven, markets. Any necessary additional local studies would likely be supported by our current or future local partners. For instance, in China, we have entered into an Exclusive License and Collaboration Agreement with Livzon for the global development and commercialization of certain antibodies or related biological compounds, including BOW015. Finally, where appropriate, we will leverage our proprietary SCALE manufacturing technology to generate In Market, For MarketTM manufacturing solutions in these markets. While we do not have any current arrangements in Brazil, we are pursuing development and commercial partnerships there.
· In Accessible Markets, in which our current regulatory data are expected to be sufficient for approval, we intend to commercialize our lead product through licensing partnerships. These markets, which include certain countries in Latin America and Southeast Asia, are likely to reference and accept as the basis for approval the Indian BOW015 regulatory package and the current data set and also allow importation of BOW015 manufactured outside of such markets. These markets often present no innovator patent protection. In these markets, we intend to establish licensing and distribution arrangements with partners to generate near term revenue from sales of BOW015. We will seek to expand these markets by increasing penetration rates with discounted prices to innovator biologics. Under the terms of our existing license agreement, Ranbaxy is responsible for the commercialization of BOW015 in India and selected Southeast Asian and North African countries. In November 2014, we launched BOW015, under the brand name Infimab, in India with Ranbaxy. We are also actively pursuing access to additional markets through Ranbaxy and other potential licensing partners.
Our strategy for commercial success relies on tailored approaches to address the diversity of our target global markets. Additionally, we intend to leverage our development and commercial experience with BOW015 to both advance our pipeline and our overall direct sales infrastructure in Europe.
Developed Markets—North America and Europe
Europe has an established regulatory framework for biosimilars, and the EMA has approved Celltrion’s infliximab program under the trade names Inflectra/Remsima®. The initial launch of
Inflectra/Remsima, suggests that the legal landscape is conducive to the introduction of biosimilars. In general, the European patent landscape provides for far fewer patent extensions for manufacturing, method of use, or processes, as compared to the patent landscape in the United States. Finally, the European market for biosimilars is strengthened by the desire of governments to reduce overall healthcare expenditure. Through a combination of substitution rules, regional tenders and political pressure to introduce biosimilars, Europe represents a commercially tractable market.
In Europe, we intend to commercialize our products using a licensing and/or distribution model in conjunction with direct sales. By doing so, we believe we will be able to establish a broad geographic footprint and commercial presence while also building scalable commercial infrastructure to enable a staged growth plan for direct sales. Specifically, we contemplate a hybrid commercial model which may include either a Europe-wide, two-brand co-marketing arrangement or a country-by-country partnership approach, whereby we retain exclusive commercial rights to a subset of European markets. In either scenario, we believe the most reasonable course toward building a sustainable, viable enterprise is to be in a position to book top-line revenue and build commercial infrastructure over time. The specifics of the partnership(s) will be driven by the careful assessment and identification of partners, their capabilities, our long range plans and other factors which will dictate success.
As the regulatory environment in the United States becomes clearer, we anticipate seeking a commercial partnership or alternative commercial model, which may include contracting directly with payors or other third-party entities. We are continuing to build a technical package for submission to North American and European regulatory authorities. In late 2015 or early 2016, we intend to initiate a global clinical program to demonstrate similarity of BOW015 to Remicade in one or more selected indications to support biosimilar registration in Developed Markets.
Local Production Markets—China and Brazil
In Local Production Markets, governments either mandate or have a strong preference for local manufacture and supply of pharmaceutical products and have implemented frameworks and/or established various incentives for such local production. These incentives may include facilitating access to funding, acceleration of the regulatory review process, improved or preferential access to government tenders and direct or indirect trade barriers on imported products. In these markets, there is a clear regulatory and patent landscape. Also, the commercial opportunity is substantial, tractable and protectable. In most of these markets, the transference of product manufacturing is rewarded by government incentives, access to tenders, and ability to restrict competition from imported products.
We intend to leverage two approaches to facilitate In Market, For Market production. First, in countries where manufacturing know-how or infrastructure is not already present, we intend to assist a local partner in developing the requisite know-how and infrastructure by providing access to our SCALE manufacturing technology platform. SCALE is an integrated platform for multi-use disposable biological manufacturing that features a small footprint, flexible scalability and minimal infrastructure requirements. We intend to offer this SCALE platform to partners who lack the necessary expertise, infrastructure, and/or know-how for manufacturing biologic products to global standards. Implementation of SCALE entails the custom-fit of modular, single-use, disposable manufacturing suites. The modular nature of SCALE allows us to work with selected local partners to incorporate the SCALE manufacturing platform in a straightforward fashion within a build-out or expansion of manufacturing facilities by such local partners. Multiple biologic products can be manufactured in a single SCALE enabled facility. With this approach, we can capture the value of being an in-market producer of biosimilar pharmaceuticals in various important markets that currently lack biologics manufacturing infrastructure and expertise. Second, where the selected partner possesses or has access to local manufacturing infrastructure and/or know-how, we may leverage such existing infrastructure by providing appropriate technology transfer and technical expertise and training to our local partner in
order to accelerate In Market, For Market manufacturing of products covered by our agreement with the local partner.
In China, the central and provincial governments encourage local production of biopharmaceuticals. In March 2014, we entered into a binding term sheet for the negotiation of a collaboration with Livzon, a subsidiary of Livzon Pharmaceutical Group Inc. In September 2014, we entered into an Exclusive License and Collaboration Agreement with Livzon for the global development and commercialization of certain antibodies or related biological compounds, including BOW015. Livzon is a fully integrated pharmaceutical company based in Guangdong Province, China, with over 5,000 employees, multiple production facilities across China, and approximately $730 million in revenue from over 200 marketed products across a range of therapeutic areas. Livzon is focused on monoclonal antibody development and production, leveraging single use disposable systems that we expect will be compatible with the optimized processes we are currently developing with our partners for BOW015.
In Brazil, the government directly purchases a significant portion of all biopharmaceutical products. The Brazilian Ministry of Health has initiated the Productive Development Policy, or PDP, to establish a formalized pathway to access this public market. The PDP is a collaborative arrangement between public and private entities, and requires a full transfer of product and manufacturing technology into Brazil to gain access to the public markets. We are also pursuing development and commercial partnerships in Brazil.
Accessible Markets—Latin America, Southeast Asia, India
In the Accessible Markets, including India, Southeast Asia, and Latin America, regulatory frameworks are clear and patent environments allow for freedom to operate. From a commercial perspective, innovator drugs have had limited market penetration in these markets due, in part, to the relatively high cost of these branded products. Further, as evidenced by several products already launched, biosimilars may actually be able to significantly expand the accessible patient populations in these markets. The commercial focus in these markets is market development and expansion. As further discussed below, in March 2014, our manufacturing partner, RLS, obtained manufacturing and marketing approval on our behalf in India for BOW015 as a treatment for rheumatoid arthritis. In September 2014, again with RLS, we received final manufacturing clearance from the Drug Controller General of India, or DCGI. We are currently involved in a dispute with RLS regarding the terms of our contractual agreement and in the event that we are unable to satisfactorily resolve this dispute, we may need to establish alternative sources for the manufacturing, marketing and sale of BOW015. In January 2014, we entered into an agreement with Ranbaxy to commercialize BOW015 in India and other selected Asian and North African markets, pending marketing authorization in those jurisdictions. These markets, including India, do not require that BOW015 be manufactured within the applicable country. We launched BOW015, under the brand name Infimab, in India in November 2014. We will be responsible for any additional development activities required by Indian regulatory authorities. Ranbaxy is responsible for all marketing and commercialization activities with respect to BOW015 in India, as well as any costs associated with development, regulatory filings and marketing and commercialization in the additional countries covered by the agreement. Under the terms of the agreement, we will supply Ranbaxy with commercial products, and Ranbaxy will be required to make payments to us upon achievement of certain development and sales milestones for BOW015, as well as to pay us a royalty on net sales of BOW015 in all territories covered by the agreement.
In addition to India and the other countries covered by our agreement with Ranbaxy, we believe that our existing regulatory dossier for BOW015 will be sufficient to achieve regulatory approval in a range of South and Central American countries. We expect to grant rights to commercialize
BOW015 in these countries, pending receipt of marketing authorization, through a licensing structure similar to the approach taken in India.
Industry Overview
Biosimilars Definition
Biosimilars are highly similar versions of approved biological drug products, referred to as reference or innovator products. Because a biosimilar product may reference existing information regarding the structure, safety, and efficacy of a previously approved reference product, a biosimilar product application emphasizes analytical characterization to demonstrate similarity between the proposed biosimilar and the reference product. In addition, preclinical and clinical studies may be required to support an application for approval. Biosimilars are often characterized as fitting within one of two categories: first generation, less complex biologics; and second generation, more complex biologics, including fusion proteins and monoclonal antibodies, or MAbs.
Both first and second generation biosimilars are significantly more complex and difficult to characterize, manufacture and develop than small molecule generics. For example, the first generation biological drug, Epogen (epoetin alfa) has a molecular weight that is 25x greater than that of small molecule drug Lipitor (atorvastatin calcium). The second generation MAb biologics—e.g., Remicade (infliximab) and Humira (adalimumab)—are, in turn, nearly 5x larger than the first generation biologicals. MAb biosimilars are complex to manufacture in part because they require the use of living organisms to produce them, and this introduces challenges in manufacturing and production on a commercial scale. Glycosylation (complex carbohydrate branches that are added by the cellular machinery) and other forms of molecular modification are hallmarks of proteins produced in living cells, in particular mammalian cells. Compared to first generation biologics, MAbs are not only larger but also have greater structural complexity, including complex glycosylation patterns which are critical for the function and activity of the molecule. MAb biosimilars therefore must be rigorously and accurately characterized to establish their biosimilarity to reference biologics in terms of glycosylation patterns, or glycoforms, and other important molecular modifications. Biosimilars—both first and second generation—also require significantly more clinical testing and regulatory review than small molecule generics, as described in more detail below.
The manufacturing, clinical and regulatory complexity and challenges of developing “second generation” biosimilars create barriers to market entry. As such, these “second generation” biosimilars can usually be sold at relatively higher prices, and with better margins, than small molecule generics and first generation biosimilars.
Regulatory Aspects of Biosimilars
Similar to other follow-on product opportunities, product development proceeds differently with biosimilars than with innovative biologic candidates. This is a result of abbreviated development requirements for the approval of biosimilars as compared with innovative biologic products. Because the structure/function and target characteristics of the reference biologic are already known, a biosimilar product application emphasizes analytical characterization to demonstrate similarity between the biosimilar and the reference biological product, which regulators have already determined to be safe and effective.
Early Reduction of Risk in Drug Development
In the development of novel pharmaceuticals and biologic products, the question of whether or not a product will ever reach the point of being commercially viable remains largely unanswered. As such, the innovator company assumes significant risk through the completion of Phase 3 trials, in which the drug or biologic therapeutic is administered to a sufficient number of subjects to make a definitive
determination of a drug’s safety and efficacy. Comprehensive Phase 3 trials are conducted for novel biologics at significant cost and exposure for the innovator company. In contrast, a large proportion of de- risking occurs much earlier in the development process of biosimilars through preclinical analytic and safety testing. As the antibody, its target, and its mechanism of action have already been validated clinically by the reference product sponsor, and the dose, regimen, and indications are already known, early testing provides greater insight into the future regulatory success for biosimilars. Preclinical and early clinical studies required to demonstrate comparability to the originator drug’s pharmacokinetic/pharmacodynamic (PK/PD), safety, and potency profile may provide early indications of a product’s eventual regulatory outcome.
The regulatory standards applicable to establish such biosimilarity vary by jurisdiction. Over the last 10 years, many jurisdictions globally have established formal regulatory regimes for review and approval of biosimilar products, but these regimes are at differing stages of development, with limited harmonization among jurisdictions.
Technical Complexity of Biosimilars
Historically, biologics have not yielded readily to the development of generic versions. Because biologics are structurally far larger and more complex than small molecule drugs, their manufacturing processes and requirements are correspondingly more complex as well. Biologics are produced through a technologically challenging five-step process:
· Scientists isolate and identify the genetic code of the protein they want to produce.
· This genetic code is inserted into living cells (bacteria, yeast, or cultured mammalian cells). Once inside the cell, the genetic code instructs the cell to produce the protein, or biotech medicine, which will later be used to treat a specific disease. Mammalian cells are the most complex of the various cell lines and generate complexities in protein structure through various biochemical modifications.
· These genetically modified cells (known as cell lines) are carefully selected and cultured over time in bioreactor tanks, surrounded by nutrients designed to encourage protein production. The specific conditions for production determine important physical attributes of the protein—such as glycosylation (complex carbohydrate branches that are added by the cellular machinery)—for its function. The structure and function of the final product is therefore highly sensitive to the specific conditions under which it is generated.
· The protein is then isolated from the cells and the nutrients through a sequence of purification processes.
· Finally, the isolated protein is packaged, typically after a number of steps to ensure sterility and stability, into sterile vials for use by doctors and patients.
Because the structure and function of a biologic, such as a monoclonal antibody, are directly linked to its production process in living systems, companies that intend to develop a biosimilar must go through all five steps above, from isolation and identification of the target protein and creation of a proprietary cell line, to final packaging of the drug product. Furthermore, companies must go to great lengths to characterize their molecules relative to the reference biologic. They must also prove to regulators that the proposed biosimilar is highly similar to the original in terms of safety and efficacy through a combination of laboratory and clinical studies. In summary, biosimilars must be shown to be comparable to their reference biologics in terms of structure, purity, safety and efficacy.
Importantly, just as there is variation among individual human beings at the biochemical level, there is also natural variation among biologic molecules created by cell systems, because they are derived directly from living systems. Even originator biologics are characterized by inherent structural
and functional variability. This leads to a range of profiles and performance for the innovator molecules, themselves, even among batches produced at the same facility. As a result, “identical” copies of biologics such as antibodies are not the objective for biosimilars. Instead, biosimilars must fall within a range of values across important structural and functional parameters compared to those of the reference drug. This range of similarity is particularly relevant to a MAb’s glycosylation pattern.
Barriers to Entry
The high technical and performance standards that every biosimilar must achieve to gain regulatory approval provide two advantages to biosimilar developers and manufacturers, such as our company. First, such standards create a significant barrier to entry in the form of both regulatory and clinical hurdles that a company must overcome to bring biosimilar products to market, thus increasing the complexity and related costs for potential competitors. Second, these high standards are expensive to achieve, thus the limited number of producers who can meet these standards can then command pricing for biosimilar products that, while lower than the pricing for the reference product, is still high enough to generate meaningful revenues and profits.
Competition
Based on our market analysis, we may be subject to competition for BOW015 in various jurisdictions from three groups.
(1) Johnson & Johnson developed the reference product, Remicade, along with its partners Merck Schering and Mitsubishi Tanabe, which are responsible for sales outside the United States. Remicade is one of the longest established biologics, supported by extensive safety and efficacy data and widespread use in multiple indications. We expect that Remicade will continue to retain a significant market share in its current markets and that Johnson & Johnson will seek to defend its market share against biosimilar entry, which may include reduction in prices and other incentives.
(2) Celltrion (Korea) has developed a biosimilar infliximab product, marketed as Remsima, which is being commercialized through various partners worldwide. Celltrion has partnered with Hospira, Inc. to co-commercialize its biosimilar infliximab product in European and other markets and has received regulatory approval for this product in all of Remicade’s approved indications in such European markets under two different brands. Hospira plans to commercialize the product under the name Inflectra® and Celltrion plans to commercialize the brand under its existing Remsima® mark. Celltrion’s product has launched in certain areas of central and eastern Europe in 2014 and is anticipated to be launched in other areas of Europe in 2015 and at that time will be the first biosimilar infliximab to be launched in a major market. It is likely that Celltrion will seek approval for and launch Remsima in the United States upon expiration of patent protection on Remicade in 2018.
(3) We are aware that other companies, including Pfizer, Samsung Biologics, and Nichi-Iko Pharmaceuticals, are in earlier stages of development and may become competitors for our biologic products, including BOW015, in various markets over time. There is limited and conflicting publicly available data on potential competitive molecules. We cannot currently predict if and when potential competitors will launch in our target markets.
Clinical Development of BOW015 (Infliximab)
We have conducted extensive bioanalytical and physicochemical comparisons of BOW015 to Remicade and have data from a Phase 1 study in the United Kingdom and a 189-patient Phase 3 double blind comparator study in India demonstrating bioequivalence, safety and efficacy of BOW015. The Phase 3 study in India met the primary endpoint of efficacy at an interim analysis conducted at
16 weeks and finished its open label phase with a 54-week end-point in the third quarter of 2014. In March 2014, BOW015 was granted manufacturing and marketing approval in India through our manufacturing partner RLS as a treatment for rheumatoid arthritis. Final manufacturing site authorization was received in July 2014. The approval was based on the 16- week, double blind safety and efficacy data comparing BOW015 against Remicade as the active comparator and reference product, in accordance with the required regulatory process in India. We launched BOW015, under the brand name Infimab, in India in November 2014 and intend to launch in additional territories thereafter by leveraging our existing regulatory data package. We expect to commence a global clinical program in late 2015 or early 2016 prior to seeking marketing authorization for and launching BOW015 in North America and Europe.
Since BOW015 is a biosimilar molecule, its characterization requires comparative analysis to the reference product, Remicade. The BOW015 drug substance and drug product manufacturing processes have been designed in conjunction with our manufacturing partners to make the final BOW015 drug product comparable to that of Remicade, and with comparable safety and efficacy in accordance with regulatory requirements.
We have produced a data package to demonstrate biosimilarity of BOW015 to Remicade. Its comparability data set, developed under a comparability and characterization protocol, addresses the physicochemical, biochemical and biological properties of infliximab and has been designed to assess biosimilarity between the reference product and BOW015. Critical Quality Attributes, or CQAs, are physical, chemical, biological or microbiological properties or characteristics that should be within appropriate limits to ensure the desired product quality. The CQAs, of infliximab have been identified based on the mechanism of action, clinical experience, impact/risk assessment of production processes and the assessed ranges of specific attribute data generated by analysis of multiple lots of Remicade. The CQAs are supported by Annex I of the Summary of Product Characteristics of the Remicade European Public Assessment Report. Full side-by-side characterization of BOW015 and Remicade, including all CQAs for infliximab, has been completed. The data set includes all known attributes that have the potential to impact safety, potency and efficacy.
The types of assays used to assess biosimilarity include the following:
· Physicochemical. These are assays that measure the physical and chemical structure of the molecule.
· In vitro biochemical. These are assays that measure the interaction of the molecule with other molecules (e.g. target binding).
· In vitro biological. These are assays that measure the interaction of the molecule with biological media (e.g. cellular or animal test systems).
These three levels of characterization are complementary and correlations are drawn from the individual and aggregate findings. For example, glycosylation heterogeneity (a physicochemical attribute) has direct impact on FcYRIIIa binding (a biochemical attribute), which in turn drives ADCC (antigen-dependent cellular cytotoxicity) activity (a biological attribute). In this way, multiple data sets support and confirm each other and the biosimilarity of BOW015 to Remicade.
The assessment of CQAs demonstrated comparability between BOW015 and Remicade. Both BOW015 and Remicade are produced using similar manufacturing processes. Minor differences between BOW015 and Remicade in the non-critical quality attributes may be consequences of differing manufacturing technologies and have not demonstrated adverse impact on the biology and efficacy of BOW015 in either in vitro or clinical studies.
We have also conducted multiple preclinical studies on BOW015. These studies include:
· Single dose toxicity of BOW015 in Swiss albino mice
· Single dose toxicity of BOW015 in Wistar rats
· Repeat dose (4-week) study in Wistar rats
· Repeat dose (4-week) study in New Zealand White Rabbits
· Skin sensitization study of BOW015 in Dunkin Hartley Guinea Pigs (Maximization Test)
We believe that BOW015 has yielded satisfactory results in each of the preclinical studies to support regulatory filings in each of the additional markets we are seeking to target. Infliximab binds selectively to human and chimpanzee tumor necrosis factor-alpha (TNF-α), and thus additional preclinical studies in non-relevant species were not required by Medicines and Healthcare Products Regulatory Agency (UK) prior to initiating Phase 1.
BOW015 Phase 1 Study
Our Phase 1 bioequivalence study was conducted in the United Kingdom in 2012 under the authority of the Medicines and Healthcare Products Regulatory Agency. The primary objective of the study was to compare the pharmacokinetics of infliximab administered by intravenous infusion. The secondary objectives of the study were to assess (i) the safety and tolerability and (ii) immunogenicity of BOW015 compared to Remicade. The study was conducted at a single clinical site and compared the safety and pharmacokinetic profile of BOW015 to Remicade after a single intravenous dose. The two drugs were considered to be similar if at various timepoints the concentrations of the drugs were comparable and were within the specific statistical parameters of 80%-125%. The study design and criteria for success were based on standard bioequivalence requirements.
Eighty-four healthy volunteers were randomized one-to-one and given either BOW015 or Remicade via intravenous infusion at a dose level of 5mg/kg with a 12-week follow-up period. The study was to detect bioequivalence at 90% confidence interval of BOW015 to Remicade. Out of the 84 subjects, 43 evaluable subjects received the test product BOW015 and 41 subjects received the reference product Remicade.
The profile of BOW015 and Remicade is shown in the graph below. The pre- defined pharmacokinetic values for the maximum height of the drug concentration as well as the pattern of elimination are similar. Thus, the study demonstrated similarity in PK profiles between BOW015 and the reference product Remicade. A single severe adverse event was reported in one of the patients receiving Remicade. This was considered by the investigator as unlikely to be related to the experimental protocol.
No relevant differences in immunogenicity test results between the two treatment groups were observed, nor were any differences observed between the two groups in safety or tolerability.
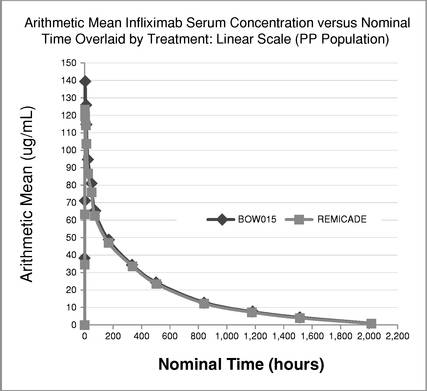
Serum concentration of BOW015 as compared to Remicade in Phase 1 Trial
BOW015 Phase 3 Study
We conducted a randomized, double-blind, active comparator Phase 3 study in India of the efficacy and safety of BOW015 in patients with severe, active rheumatoid arthritis on stable doses of methotrexate. The study randomized subjects to the two treatment arms in a 2:1 allocation. Out of 189 total subjects, 127 were given BOW015 and 62 were given Remicade during the first 16 weeks of the study. The primary endpoint of the study was equivalence of both arms on the standardized American College of Rheumatology 20% improvement (ACR20) scoring system—a composite scoring system that includes objective laboratory measures as well as physician and patient assessments of well- being. Secondary endpoints included the ACR50 and ACR70 (50% and 70% improvement respectively) and the various components of the ACR20 scoring system. From week 22, BOW015 responders were administered BOW015 in an open-label phase for the study duration of 54 weeks, while Remicade responders were crossed over into the open label phase and switched to BOW015 for the study duration of 54 weeks. Non-responders immediately entered a three-month follow-up phase.
Both BOW015 and Remicade were administered at a dose of 3mg/kg given as an intravenous infusion at week 0, followed with similar doses at weeks 2, 6 and 14. Subjects were assessed at week 16 and responders were able to enter an open-label phase. In the open-label phase, subjects received BOW015 at a dose of 3mg/kg given as an intravenous infusion at weeks 22, 30, 38 and 46 and were be followed up at Weeks 54 and 58. Subjects who were non-responders at week 16 entered a follow-up phase for immunogenicity, PK and safety for an additional 3 months.
The 16-week data showed that patients responded to BOW015 at a rate of 89.8% ACR20 compared to an 86.4% ACR20 response rate to Remicade. This outcome met its pre-specified statistical endpoint and was within a 15% equivalence margin at a 95% confidence interval. The results met the
23% equivalence margin authorities required for approval by the Indian regulatory authorities. There was no difference reported in safety or immunogenicity between the treatment groups. There was also no reported difference between the groups on the secondary endpoints.
We measured the patients’ responses on an ACR20 scoring system to BOW015 and Remicade at multiple time points. The data suggest that BOW015 and Remicade patients responded similarly at all time points up to the final 16 week efficacy endpoint.

Comparison of BOW015 and Remicade at multiple time points
In September 2014 we announced 58 week data, which demonstrated therapeutic equivalence to Remicade and confirmed the safety of switching from Remicade to BOW015. In the open-label phase, patients who continued on BOW015 were compared to patients who received four doses of Remicade, followed by a switch to four doses of BOW015. Immune responses as well as overall safety and tolerability for BOW015 were comparable to the arm switched from Remicade to BOW015 and were
consistent with the expected profile of Remicade. Further, ACR20 responses were durably maintained to 54 weeks from the week 16 primary endpoint previously reported.
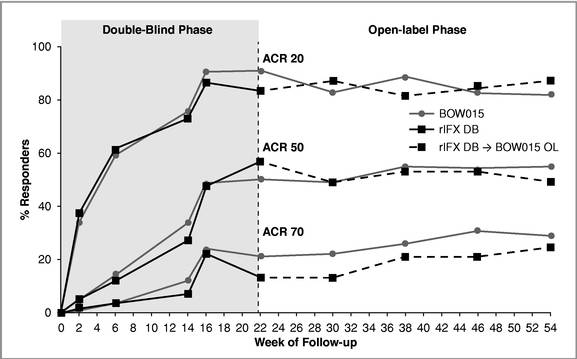
Equivalence in ACR20 Response Rates—Double Blind (DB) and Open Label (OL) Phase
License and Acquisition Agreements
Ranbaxy Laboratories Limited
In January 2014, we entered into a license agreement with Ranbaxy, pursuant to which we granted to Ranbaxy exclusive rights under our intellectual property and regulatory materials relating to BOW015, to develop and commercialize BOW015 in India and certain other countries in Asia and North Africa. Pursuant to the agreement, Ranbaxy has agreed to distribute and sell BOW015 in India. Under our agreement with Ranbaxy, we will be responsible, through RLS, for supplying BOW015 to Ranbaxy for sale in the licensed territory.
Under our agreement with Ranbaxy, Ranbaxy paid us an up-front payment of $0.5 million, and will be required to make payments to us upon the achievement of certain regulatory and commercialization milestones of up to $1 million in the aggregate. Ranbaxy is also required to make payments to us upon the achievement of specified levels of aggregate gross sales of BOW015 in the licensed territory totaling up to $10 million in the aggregate, and to pay to us a royalty on net sales of BOW015 at a percentage in the mid to high teens, subject to reductions in certain circumstances. Ranbaxy’s obligation to pay us royalties will expire 20 years following the first commercial sale of BOW015 in the licensed territory. On September 9, 2014, we amended the license agreement to revise the royalty tiers for net sales based on final product sale forecasts and supply costs in the territory. Under the amendment, we also revised Ranbaxy’s obligations to achieve specified minimum annual sales targets in any given country such that these obligations will now apply following the final grant of approval by the applicable regulatory authority for BOW015 for all indications for which the innovator product has received approvals in such country.
Our agreement with Ranbaxy will remain in force, absent earlier termination, for 20 years following the first commercial sale of BOW015 in the licensed territory. Either party may terminate the
agreement on 60 days’ notice for the other party’s uncured material breach or insolvency, or upon 30 days’ notice in the event that our rights in the BOW015 cell line under our agreement with Catalent are terminated, and we may terminate the agreement upon 45 days’ notice in the event of a patent challenge brought by Ranbaxy in relation to any patents licensed under the agreement. Ranbaxy may also terminate the agreement on 60 days’ notice to us in the event of certain failures relating to the qualification of the manufacturing facility for production of BOW015, unless we provide an appropriate plan to remedy such issues, and use commercially reasonable efforts to execute such plan, in which case the cure period for such manufacturing related breaches is extended for two years following the date of the remedial plan. If we fail to cure the breaches within the two-year period, and Ranbaxy elects to terminate the agreement, we may, under certain circumstances, be required to pay specified amounts in damages to Ranbaxy as Ranbaxy’s sole remedy for the breach.
Livzon Mabpharm Inc.
In September 2014, we entered into an Exclusive License and Collaboration Agreement with Livzon for the global development and commercialization of certain antibodies or related biological compounds, including BOW015.
Under the agreement, we and Livzon granted each other, in the other party’s territory, exclusive, royalty-bearing licenses under certain patent rights and know-how to develop, manufacture and commercialize BOW015 and up to four additional compounds chosen by mutual agreement of the parties, which we refer to as the Collaboration Compounds. Livzon’s territory consists of China, Hong Kong, Macau and Taiwan, and our territory contains the rest of the world. We share pre-clinical development expenses with Livzon for each Collaboration Compound based on certain factors specific to each such compound. Each party bears the responsibility and expenses for clinical development and commercialization of Collaboration Compounds in its territory. Livzon will be the preferred supplier of each Collaboration Compound for pre-clinical, clinical, and commercialization purposes, subject to Livzon’s satisfaction of certain performance criteria.
In consideration for the license granted to Livzon to develop and commercialize BOW015, we are eligible to receive from Livzon a milestone payment of $2.5 million upon the achievement of a specified regulatory milestone We are also eligible to receive from Livzon tiered royalties at a percentage in the low to high single digits based on net sales of BOW015 products in the Livzon Territory. Any future Collaboration Compounds have cross-milestone and royalty obligations in amounts to be mutually agreed upon at a later date.
Moksha8 Pharmaceuticals, Inc.
In December 2010, we entered into a Revenue and Negotiation Rights Agreement, which we refer to as the Moksha8 Revenue Agreement, with Moksha8 to settle an outstanding promissory note issued by us to Moksha8 in May 2009. The Moksha8 Revenue Agreement provided for certain milestone and royalty payments to be made to Moksha8 based upon future licensing revenues and worldwide net sales of products that are based on the assets acquired from Moksha8. These products, which we refer to collectively as the Products, include BOW015, and may include biosimilar adalimumab and biosimilar rituximab, if the biosimilar products we develop are derived from the assets we acquired from Moksha8.
In September and October 2014, we entered into amendments to the Moksha8 Revenue Agreement to terminate our payment obligations with respect to products that are biosimilar to infliximab, which includes BOW015, in exchange for our payment of $1.4 million in two installments. As a result, the foregoing milestone and regulatory payment obligations will no longer apply to our commercialization of BOW015.
Catalent Pharma Solutions, LLC
In January 2009, Moksha8 entered into a cell line sale agreement, which we refer to as the Cell Line Agreement, with Catalent Pharma Solutions, or Catalent, for the acquisition of a gene expression cell line for BOW015, which we refer to as the GPEx Cell Line, developed by Catalent for Moksha8 pursuant to a separate development and manufacturing agreement dated July 14, 2008. The Cell Line Agreement was assigned to us on May 14, 2009, and was amended July 31, 2009 to revise certain payment-related terms. Under the terms of the Cell Line Agreement, we exercised an option to acquire all rights in the GPEx Cell Line for development and commercialization of products using the GPEx Cell Line, namely BOW015, subject to certain obligations to make contingent payments to Catalent.
We paid Catalent $0.1 million on execution of the Cell Line Agreement, and paid a further $0.1 million upon exercise of our option to complete the purchase of the GPEx Cell Line. We are required to make additional payments to Catalent of up to $0.7 million in the aggregate upon the achievement of certain development and regulatory milestones. In March 2013, we paid $0.2 million to Catalent upon the occurrence of certain clinical trial events for BOW015. Pursuant to the agreement, we will be obligated to pay an additional $0.5 million upon the achievement of certain development and regulatory milestones. In addition, we will be required to pay a contingent sale fee in the form of royalties on worldwide net sales of BOW015 and any other product manufactured using the GPEx Cell Line at a percentage in the very low single digits for a period of 20 years following the first commercial sale of such product, and thereafter at a rate of less than one percent.
Either we or Catalent may terminate the Cell Line Agreement on 60 days’ notice for the other party’s material breach of the agreement, or for the other party’s insolvency, and in the event of Catalent’s termination for our material breach of the Cell Line Agreement, our ownership rights in the GPEx Cell Line would revert to Catalent. If we terminate the Cell Line Agreement for Catalent’s breach, we will retain ownership of the GPEX Cell Line, but our payment obligations to Catalent will terminate.
Bioceros B.V.
In April 2013, we entered into a license agreement, which we refer to as the Bioceros Agreement, with Bioceros B.V., or Bioceros, pursuant to which Bioceros granted us a non-exclusive license under its rights in the cell lines and associated intellectual property relating to trastuzumab, bevacizumab and rituximab to permit us to manufacture and commercialize antibody products incorporating the licensed antibodies worldwide, provided that solely with respect to trastuzumab, our licensed territory does not include China, Macau and Hong Kong. Bioceros also granted us the right to receive the tangible embodiments of the cell line and all related know-how for trastuzumab, and an option, subject to payment of certain option fees, to evaluate and receive the tangible embodiments of the cell lines and know-how relating to rituximab and bevacizumab. We exercised our option in relation to bevacizumab in June 2013, and subsequently Bioceros transferred the cell line for bevacizumab to us. The Bioceros Agreement was amended in June 2013 to modify certain payment provisions, and to provide for the performance of certain preclinical services by Bioceros, in each case in relation to bevacizumab.
In October 2013, we entered into a second license agreement with Bioceros, which we refer to as the Second Bioceros Agreement, on substantially similar terms to those of the April 2013 Bioceros Agreement, pursuant to which Bioceros granted us a non-exclusive license under its rights in the cell line and associated intellectual property relating to adalimumab, to permit us to manufacture and commercialize antibody products incorporating adalimumab worldwide except in Japan. We collectively refer to the initial Bioceros Agreement and the Second Bioceros Agreement as the Bioceros Agreements.
Under the Bioceros Agreement, we paid to Bioceros an up-front payment of $0.3 million as a license issuance fee applicable to the rights in trastuzumab, and have paid a further $0.4 million in the
aggregate as a result of the exercise of our option in June 2013 with respect to bevacizumab. Under the Second Bioceros Agreement, we paid Bioceros an up-front payment of $0.07 million upon execution of the Second Bioceros Agreement, and in the first quarter of 2014, paid a further up-front payment of $0.3 million as a license issuance fee applicable to the rights in adalimumab. Under each of the Bioceros Agreements, we are also required to pay Bioceros a tiered annual license maintenance fee, and will be required to make certain payments to Bioceros upon the achievement of specified regulatory milestones for the products covered by the Bioceros Agreement, totaling up to $1.2 million in the aggregate for all potential products across both agreements. Upon commercialization of products covered by the rights licensed under the Bioceros Agreements, we will be required to pay, on a product-by-product basis, a royalty on net sales at a rate of less than one percent, subject to a specified minimum and maximum annual royalty amount under each agreement. Our obligation to pay royalties to Bioceros under each of the Bioceros Agreements will expire, on a product-by-product basis, on the date that is 10 years following the first commercial sale of each such product. We are required to use commercially reasonable efforts to develop and commercialize products in the licensed territory.
Absent earlier termination, the Bioceros Agreements will remain in force until the expiration of all payment obligations under the applicable agreement. Either we or Bioceros may terminate either of the Bioceros Agreements on 30 days’ notice for the uncured material breach by the other party, or immediately upon the other party’s insolvency. We may terminate each of the Bioceros Agreements, on an antibody asset-by-antibody asset basis for any reason on 60 days’ notice to Bioceros, and Bioceros may terminate the agreements in the event that we challenge any Bioceros patents included in Bioceros’s proprietary antibody production platform.
Manufacturing
We currently manufacture BOW015 through a manufacturing and supply agreement with RLS, which we refer to as the RLS Agreement. In order to migrate from traditional stainless steel manufacturing to single use disposable systems for our SCALE process, we also have an agreement with Fujifilm Diosynth Biotechnologies U.S.A., or Fujifilm, for BOW015 process development with a view toward establishing Fujifilm as a source of future clinical and commercial supply of BOW015. We believe that our relationship with Fujifilm will allow us to expand future capacity and provide a back-up secondary manufacturing site. We are also currently working with our manufacturing partners to establish appropriate arrangements for the necessary scale-up of manufacturing operations for longer term commercial supply in markets where we or our licensees are developing and commercializing products. For markets requiring in-country manufacturing, we expect to work with local partners to deploy our SCALE manufacturing platform in whole or in part to enable our In Market, For Market solution. We expect that this strategy will provide us with multiple sourcing options to enable uninterrupted product supply to our partners and therefore patients, and to meet the needs of countries requiring locally-based manufacturing.
In December 2014, RLS exercised its three-year termination for- convenience right with respect to the RLS Agreement, which will cause the agreement to terminate in December 2017. In addition, in January 2015, RLS informed us of its intention to initiate a dispute proceeding under the RLS Agreement alleging that the agreement grants RLS exclusive global supply rights for BOW015, and that the terms of our collaboration agreements with Fujifilm and Livzon (described above) violate the RLS Agreement. These collaboration agreements provide, in the case of Fujifilm, for process development and scale up of manufacturing processes for BOW015 to a capacity that may be sufficient to fulfill future requirements for clinical and commercial supply, and, in the case of Livzon, for a grant of rights, under certain circumstances, to supply BOW015 in the future in certain markets. We disagree with these allegations. RLS’ delivery of a formal dispute notice will trigger a contractual 30-day period of negotiation between executives of RLS and Epirus to attempt to resolve the dispute. If the dispute is not resolved during this period, RLS may then file an arbitration proceeding to determine the outcome
of the dispute. Following any such period of arbitration, if the arbitrator were to decide in favor of RLS, we would have 30 days to cure the alleged breach, after which time RLS may exercise its right to terminate the agreement. RLS has indicated that it will continue to supply us with BOW015 during any such periods of negotiation and arbitration. We believe that a number of alternative BOW015 contract manufacturing sources are available to replace RLS as our manufacturer of BOW015. In connection with obtaining new sources of BOW015, it will also be necessary to obtain the proper regulatory approvals in India. We intend to continue to actively pursue alternative BOW015 manufacturing arrangements in order to have such arrangement in place in the event we do not prevail in a dispute proceeding with RLS and with the intent of limiting any potential disruption to our business. Ultimately, if our dispute with RLS is not resolved and an arbitrator makes a determination in favor of RLS, RLS may terminate the RLS Agreement and/or refuse to provide us with an adequate supply of BOW015. We believe that although this result could cause a temporary disruption to our supply of BOW015 in India, any such disruption in this market would not have a meaningful effect on our business given our focus on the development and commercialization of biosimilars in the broader global market.
SCALE Manufacturing Platform and In Market, For Market Solution
Manufacturing of biologics is currently shifting away from traditional methods involving steel bioreactors to small, single-use bioreactors. Until recently, production facilities relied on the use of relatively inflexible, hard-piped equipment including large, stainless steel bioreactors and tanks to manufacture product intermediates and buffers. However, there is an increasing trend towards the adoption of single-use technologies across the manufacturing process.
There are several key advantages to single-use technologies. These include:
· Reduced capital costs for plant construction;
· Reduced risk for product cross-contamination in a multiproduct facility;
· Increased flexibility to change rapidly from one product to another;
· Lower utility costs due to reduced need for steaming-in-place sterilization; and
· Reduced time for a new facility to become operational.
Manufacturing using single-use bioreactors is at the heart of SCALE, our solution for manufacturing biosimilars in emerging markets. The modular nature of the SCALE manufacturing facilities enables our business plan of building manufacturing facilities to suit the local requirements of our partners. Investment into a SCALE facility can range from $20 million to $40 million for a facility that, once up and running, could produce up to 150 kilograms per year of biologic material. Multiple biologic products can easily be manufactured in a single SCALE facility. Single-use bioreactors enable smaller production runs and facilitate operation of a multi-product facility that is well suited to the evolving market for biologics. Once the market demand exceeds the production output of a single bioreactor, additional equipment can easily be ordered and installed.
Intellectual Property
We currently own trademark registrations in the United States to the marks “SCALE” and “In Market, For Market.” As a company focused on biosimilars, we do not own any product related patents.
In January 2009, our predecessor, Moksha8, acquired rights in and title to the GPEx Cell Line from Catalent Pharma Solutions, LLC, or Catalent, for the gene expression product M80015 (renamed BOW015), subject to our obligation to pay certain milestones and royalties on net sales to Catalent with respect to the development and commercialization of BOW015. Moksha8 assigned their agreement with Catalent to us on May 14, 2009. Please see “Business—License and Acquisition Agreements” for a more detailed description of our rights and obligations with respect to BOW015.
We are the non-exclusive licensee, under two separate license agreements with Bioceros, to certain proprietary cell lines applicable to our pipeline MAb biosimilar products, including the cell lines for BOW050 (adalimumab).
Government Regulation
We and our partners are subject to a variety of laws and regulations governing the development, manufacture, marketing, and distribution of biosimilars. Regulatory authorities around the world regulate, among other things, the research and development, testing, manufacture, quality control, safety, purity, potency, labeling, storage, record keeping, approval, advertising and promotion, distribution, post-approval monitoring and reporting, sampling, and import and export of our products and product candidates. The regulatory requirements and approval processes vary from country to country, and the various regulatory regimes are at various stages of maturity depending on the jurisdiction.
United States
FDA
The Biologics Price Competition and Innovation Act, or BPCIA, enacted in 2010, established an abbreviated approval pathway for biosimilars in the United States. The law defines biosimilars as products that are highly similar to biologics already licensed by the FDA pursuant to Biologic License Applications, or BLAs, notwithstanding minor differences in clinically inactive components, and that have no clinically meaningful differences from the reference product in terms of safety, purity and potency. A biosimilar application submitted pursuant to the BPCIA must contain information demonstrating biosimilarity based upon the following, unless the FDA determines otherwise:
· data derived from analytical studies, demonstrating that the proposed biosimilar product is highly similar to the approved product notwithstanding minor differences in clinically inactive components;
· animal studies (including an assessment of toxicity); and
· a clinical study or studies (including an assessment of immunogenicity and pharmacokinetics or pharmacodynamics), sufficient to demonstrate safety, purity and potency in one or more conditions for which the reference product is licensed and intended to be used.
In addition, a biosimilar application must include information demonstrating (1) sameness of strength, dosage form, route of administration and mechanism(s) of action with the reference product (where known), (2) approval of the reference product for the condition(s) of use prescribed, recommended or suggested in the labeling proposed for the biosimilar product, and (3) appropriate manufacturing, processing, packing, and holding facilities that meet the standards designed to ensure a safe, pure and potent medicine. The FDA will approve a biosimilar application based on a finding of biosimilarity with the reference product.
A pending biosimilar application or a supplement to an approved biosimilar application may seek an FDA determination that the proposed or approved product is “interchangeable” with the reference product. FDA will determine that the product is interchangeable with the reference product if the application includes sufficient information to show that the product is biosimilar to the reference product and that it can be expected to produce the same clinical result as the reference product in any given patient. If the product may be administered more than once to a patient, the applicant must demonstrate that the risk in terms of safety or diminished efficacy of alternating or switching between the biosimilar and reference product is not greater than the risk of using the reference product without such alternation or switch. The determination of interchangeability means that the biosimilar product
may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product, which is also subject to state laws.
The timing of final FDA approval of a biosimilar for commercial distribution depends on a variety of factors, including whether the reference product is entitled to one or more of a number of exclusivity protections available for reference products under the BPCIA that may delay submission and approval of biosimilar applications. The law prohibits the submission of a biosimilar application until four years after the date on which the reference product was first licensed, and delays the approval of a biosimilar application from becoming effective until twelve years after the date on which the reference product was first licensed. The first-licensure exclusivity provisions are not triggered by a supplement to the original application for the reference product, or by the submission of an entirely new BLA filed by the same sponsor or manufacturer of the reference product for certain changes made to the reference product. Such changes include (1) a non-structural change that results in a new indication, route of administration, dosing schedule, dosage form, delivery system, delivery device or strength, and (2) a structural change that does not result in a change in safety, purity or potency. In addition, as in the case of applications for approval of small molecule drugs in the United States, BLAs may be entitled to other periods of exclusivity. For example, a reference product designated for a rare disease or condition (an “orphan drug”) may be entitled to seven years of market exclusivity, in which case no product that is biosimilar to the reference product may be approved until either the end of the twelve year period provided under the BPCIA or the end of the seven year orphan exclusivity period, whichever occurs later. In certain circumstances, a regulatory exclusivity period can extend beyond the life of a patent, and thus block biosimilar applications from being approved on or after the patent expiration date. In addition, the FDA may under certain circumstances extend the exclusivity period for the reference product by an additional six months if the FDA requests, and the manufacturer undertakes, studies on the effect of its product in children, referred to as a pediatric extension.
The first biosimilar determined to be interchangeable with a particular reference product for any condition of use is also protected by a period of exclusivity that delays an FDA determination that a second or subsequent biosimilar product is interchangeable with that reference product for a period of time generally ranging from 12 to 42 months from approval or as determined by a number of patent litigation triggers. Specifically, this exclusivity period extends until the earlier of: (1) one year after the first commercial marketing of the first interchangeable product; (2) 18 months after resolution of a patent infringement suit instituted under 42 U.S.C. § 262(l)(6) against the applicant that submitted the application for the first interchangeable product, based on a final court decision regarding all of the patents in the litigation or dismissal of the litigation with or without prejudice; (3) 42 months after approval of the first interchangeable product, if a patent infringement suit instituted under 42 U.S.C. § 262(l)(6) against the applicant that submitted the application for the first interchangeable product is still ongoing; or (4) 18 months after approval of the first interchangeable product if the applicant that submitted the application for the first interchangeable product has not been sued under 42 U.S.C. § 262(l)(6).
After obtaining regulatory approval of a product, manufacturers may be required to comply with a number of post-approval requirements. For example, as a condition of approval of a BLA or a biosimilar application, the FDA may require post marketing testing and surveillance to monitor the product’s safety or efficacy. In addition, the holder of an approved BLA or biosimilar application is required to report certain adverse reactions and production problems involving its product to the FDA to provide updated safety and efficacy information and to comply with requirements concerning advertising and promotional labeling for the product. Quality control and manufacturing procedures must also continue to conform to cGMPs after approval, and the FDA periodically inspects manufacturing facilities to assess compliance with cGMPs.
The BPCIA also establishes a detailed framework for addressing potential patent disputes between biosimilar product sponsors and reference product sponsors. The biosimilar pathway approval process
does not require patents to be listed in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations (referred to as the “Orange Book”), and companies submitting biosimilar applications are not required to submit patent certifications. However, once the biosimilar applicant has received notification that the FDA has accepted its application for review, the BPCIA establishes a 20-day time frame within which time the biosimilar applicant must provide a copy of the application, along with any other information that describes the manufacturing processes for the biosimilar product, to the reference product sponsor’s in-house counsel, the reference product sponsor’s outside counsel, and/or a representative of the owner of a patent exclusively licensed to the reference product sponsor with respect to the reference product who has retained a right to assert the patent or participate in litigation. The copy of the application and any other information provided are considered “confidential information,” and recipients are generally prohibited from disclosing anything contained therein and from using the information for any purposes other than to determine whether a patent infringement claim may reasonably be asserted. The reference product sponsor then has sixty days within which to provide the biosimilar applicant with a list of patents for which it believes a patent infringement claim could reasonably be brought. The reference product sponsor may also choose to designate patents that it would be willing to license to the biosimilar applicant.
Subsequently, the parties must engage in a back and forth negotiation regarding which patents will be part of the anticipated litigation, during which time the FDA continues to review the biosimilar application. Unlike in the context of applications for small molecule generics submitted under the Hatch-Waxman Act, the BPCIA does not require the FDA to stay approval of a follow-on biologic application for 30 months once patent litigation has been initiated.
In September 2014, the FDA published the “Purple Book,” which lists biological products licensed by the FDA pursuant to approved BLAs and identifies the date of licensure and whether the FDA has evaluated the biological product for reference product exclusivity. The Purple Book will also enable users to identify whether a biological product licensed as a follow-on biologic has been determined by the FDA to be biosimilar to or interchangeable with a reference biological product. Biosimilar and interchangeable biological products will be listed under the reference product to which biosimilarity or interchangeability was demonstrated.
To date, the FDA has published six draft guidance documents regarding implementation of the BPCIA regulatory pathway. These draft guidances focus on scientific considerations, quality considerations and clinical pharmacology data related to demonstrating biosimilarity; meetings between the FDA and biosimilar product sponsors; requests for reference product exclusivity; and common questions and answers regarding implementation of the BPCIA. However, none of the draft guidances addresses the standards for interchangeability.
The FDA is still in the process of implementing the BPCIA, and no biosimilar product has yet been approved by the FDA under the BPCIA pathway. However, on January 7, 2015, the FDA’s Oncologic Drugs Advisory Committee unanimously recommended that the FDA approve Sandoz’s biosimilar version of Amgen’s Neupogen (filgrastim) for all of the reference product’s approved indications. The FDA is not required to follow the recommendations of its advisory committees, but frequently does so.
Coverage and Reimbursement
The commercial success of our biosimilar product candidates and our ability to commercialize those products successfully if approved will depend in part on the extent to which governmental payor programs at the federal and state levels, including Medicare and Medicaid, private health insurers and other third-party payors provide adequate coverage and reimbursement. These third-party payors generally develop their own policies as to which drugs they will pay for and the reimbursement levels for the drugs. For example, governmental programs in the United States often require manufacturers to
pay certain rebates or otherwise provide discounts to secure coverage of drug products. To control healthcare expenditures generally, in the United States, the EU and other potentially significant markets for our product candidates, government authorities and third party payors are increasingly attempting to limit or regulate the price of medical products and services, particularly for new and innovative products and therapies. The measures taken often have resulted in lower average selling prices. Further, the increased emphasis on managed healthcare in the United States and on country and regional pricing and reimbursement controls in the EU places additional pressure on product pricing, reimbursement and usage, which may adversely affect our future product sales and results of operations. These pressures can arise from rules and practices of managed care groups, judicial decisions and governmental laws and regulations related to Medicare, Medicaid and healthcare reform, as well as drug coverage and reimbursement policies and pricing in general.
Some of the additional requirements and restrictions on coverage and reimbursement levels imposed by third-party payors influence the purchase of healthcare services and products. For example, there may be limited coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA-approved drug products for a particular indication. There may also be formulary placements that result in lower reimbursement levels and higher cost-sharing borne by patients. Further, third-party payors are increasingly examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to the costs required to obtain the FDA approvals. Our products may not be considered medically necessary or cost-effective. Even if a third-party payor determines to provide coverage for a drug product, adequate reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in drug development.
Healthcare legislative proposals to reform healthcare or reduce costs under government insurance programs may also result in lower reimbursement for our drugs and drug candidates or exclusion of our drugs and drug candidates from coverage altogether. The cost containment measures that healthcare payors and providers are instituting and any healthcare reform could significantly reduce our revenues from the sale of any of our drug candidates, if approved. We cannot provide any assurances that we will be able to obtain and maintain third party coverage or adequate reimbursement for any of our approved drug candidates in whole or in part.
Healthcare Reform
With respect to legislative reform, in the United States, there have been and continue to be a number of significant legislative initiatives to contain healthcare costs. For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Affordable Care Act, was passed, which substantially changes the way health care is financed by both governmental and private insurers, and significantly impacts the U.S. pharmaceutical industry. The Affordable Care Act, among other things, subjects biologic products to potential competition by lower-cost biosimilars, addresses a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, increases the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extends the rebate program to individuals enrolled in Medicaid managed care organizations, establishes annual fees and taxes on manufacturers of certain branded prescription drugs, and a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D.
In addition, other legislative changes have been proposed and adopted in the United States since the Affordable Care Act was enacted. On August 2, 2011, the Budget Control Act of 2011 among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect in April 2013, and will remain in effect through 2024 unless additional Congressional action is taken.
We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our product candidates or additional pricing pressures.
Other Healthcare Laws
We may also be subject to healthcare regulation and enforcement by the federal government and the states and foreign governments in which we conduct our business. These laws include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, privacy and security and physician sunshine laws and regulations.
The federal Anti-Kickback Statute prohibits, among other things, any person from knowingly and willfully offering, soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs. The Anti-Kickback Statute is subject to evolving interpretations. In the past, the government has enforced the Anti-Kickback Statute to reach large settlements with healthcare companies based on sham consulting and other financial arrangements with physicians. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act. The majority of states also have anti-kickback laws which establish similar prohibitions and in some cases may apply to items or services reimbursed by any third-party payor, including commercial insurers.
Additionally, the civil False Claims Act prohibits knowingly presenting or causing the presentation of a false, fictitious or fraudulent claim for payment to the U.S. government. Actions under the False Claims Act may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Violations of the False Claims Act can result in very significant monetary penalties and treble damages. The federal government is using the False Claims Act, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical and biotechnology companies throughout the U.S., for example, in connection with the promotion of products for unapproved uses and other sales and marketing practices. The government has obtained multi-million and multi-billion dollar settlements under the False Claims Act in addition to individual criminal convictions under applicable criminal statutes. Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, also created new federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying,
concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
There has also been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers. The Affordable Care Act, among other things, imposes new reporting requirements on drug manufacturers for payments made by them to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests that are not timely, accurately and completely reported in an annual submission. Drug manufacturers were required to begin collecting data on August 1, 2013 and submit reports to the government by March 31, 2014 and June 30, 2014, and the 90th day of each subsequent calendar year. Certain states also mandate implementation of compliance programs, impose restrictions on drug manufacturer marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians.
We may also be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH, and their respective implementing regulations, including the final omnibus rule published on January 25, 2013, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates,” defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways, thus complicating compliance efforts.
The Foreign Corrupt Practices Act
We are subject to the U.S. Foreign Corrupt Practices Act, or FCPA, which generally prohibits certain individuals and entities subject to its jurisdiction from paying, offering, or authorizing payment of anything of value, directly or indirectly to a “foreign official” with the corrupt intent of improperly influencing any act or decision of the official in order to assist the individual or business in obtaining or retaining business or any improper business advantage. The activities of our business partners and intermediaries could also subject us to potential liability under the FCPA, as we could be held responsible for their improper conduct while acting on our behalf. In addition, the FCPA requires companies whose securities are publicly listed in the United States to comply with accounting provisions requiring the company to maintain books and records that accurately and fairly reflect transactions of the corporation and to devise and maintain an adequate system of internal accounting controls for its operations. Activities that violate the FCPA, even if they occur outside of the United States, can result in criminal and civil fines, imprisonment, disgorgement, mandatory compliance monitor oversight, debarment from government contracts, and reputational damage.
We are also subject to similar anti-bribery laws in other jurisdictions in which we conduct or expect to conduct business, including the United Kingdom’s Bribery Act of 2010, which also prohibits commercial bribery. These laws are complex and far-reaching in nature, and may require us in the future to alter one or more of our practices to be in compliance with these laws or any changes in
these laws or the interpretation thereof. Any violations of the FCPA or other anti-corruption laws, or allegations of such violations, could disrupt our operations, involve significant management distraction, involve significant costs and expenses, including legal fees, and could result in a material adverse effect on our business, prospects, financial condition, or results of operations.
Europe
The European Medicines Agency, or EMA, is responsible for the evaluation and approval of marketing authorization applications for human and veterinary medicines in the European Union, or EU, under the “centralized procedure” set forth in Regulation (EC) No. 726/2004. The centralized procedure consists of “a single application, a single evaluation and a single authorisation” that allows applicants to obtain a marketing authorization that is valid throughout the entire European Economic Area, which is comprised of the 28 Member States of the EU plus Iceland, Norway, and Liechtenstein. In addition, each EU Member State also has its own procedures for authorizing, within its territory, medicines that fall outside of the scope of the centralized procedure.
Use of the centralized procedure is mandatory for biosimilars that are developed using one of the biotechnological processes enumerated in the Annex of Regulation (EC) No 726/2004.284. These include recombinant DNA technology, hybridoma and monoclonal antibody methods, and “controlled expression of genes coding for biologically active proteins in prokaryotes and eukaryotes including transformed mammalian cells.” Use of the centralized procedure is permitted, but not mandatory, where the reference medicinal product was authorized via the centralized procedure. It may also be used in cases where the reference medicinal product was not authorized via the centralized procedure, if the biosimilar product applicant demonstrates that either: (1) its product constitutes a significant therapeutic, scientific, or technical innovation; or (2) granting a single European Community marketing authorization is in the interest of patients in the European Community as a whole. Other biosimilar products may be authorized by individual member countries on a national level.
The legal basis for authorization of biosimilars in the EU is set forth in Article 10(4) of Directive 2001/83/EC, as amended by Directive 2004/27/EC,292 and Article 6 of Regulation (EC) No. 726/2004. The primary objective of the evaluation of an application pursuant to Article 10(4) is to determine the similarity of a proposed biosimilar to a reference product. The regulation does not, however, establish an explicit standard for determining biosimilarity. Rather, “[w]hether a medicinal product would be acceptable using the ‘similar biological medicinal product’ approach depends on the state of the art of analytical procedures, the manufacturing processes employed, as well as clinical and regulatory experiences.” In addition, comparability studies are needed to generate evidence substantiating the similar nature, in terms of quality, safety and efficacy, of the new similar biological medicinal product and the chosen reference medicinal product authorized in the Community.
Article 10(4) of Directive 2001/83/EC establishes the pre-clinical and clinical testing requirements to support applications for biosimilars that do not meet the definition of generic medicinal products due to differences in raw materials or manufacturing processes. Annex I of the Directive sets forth the type and quantity of supplementary data that must be provided. To demonstrate biosimilarity between a proposed biosimilar medicinal product and a reference medicinal product, the EMA expects a robust head-to-head comparison that assesses quality, safety and efficacy. To demonstrate similar quality, the EMA requires studies comparing the structure and biological activity of the active ingredients in the proposed biosimilar and the reference medicinal product. Comparisons of safety and effectiveness must demonstrate the absence of significant differences between the proposed and reference products in terms of benefits and risks, including the risks of adverse immune reactions. Similar biological medicinal products are also subject to post-marketing monitoring requirements consistent with those applicable to the reference product. The EMA requires clinical safety of biosimilars to be monitored closely on an ongoing basis during the post-approval phase including continued benefit-risk assessment.
The EMA has issued a number of general and product-specific guidance documents clarifying its regulation of biosimilars. These guidance documents include guidance for industry on the non-clinical and clinical aspects of the development of biosimilars, as well as product-class-specific guidelines for the development of biosimilar epoetins, filgrastims, insulins, growth hormones, alfa interferons, monoclonal antibodies, beta interferons, follitropins, and low-molecular-weight heparins. Although the EMA recognizes that the diversity of biological medicinal products requires a case-by-case determination of the amount of data required for a particular application, the guidelines issued to date indicate that the demonstration of comparability requires, among other things, an assessment of purity and impurity profiles of the active substance and medicinal product, characterization of the physicochemical properties including composition and primary and higher order structures, and an assessment of biological activity with “biological assays using different approaches to measure the biological activity,” as appropriate.
The EU regulatory framework does not provide authority for the EMA to determine whether the biosimilar may be used interchangeably with the reference product. Instead, the EMA advises patients to speak with their doctors and pharmacists about the possibility of switching between a reference product and a biosimilar product. In addition, individual Member States have the authority to determine whether or not a biosimilar is interchangeable with, or should automatically be substituted for, the reference product. Throughout the EU, all similar biological medicinal products must include the following statement in their Summary of Product Characteristics, or SmPC: “[Invented Name] is a Biosimilar medicinal product. Detailed information is available on the website of the European Medicines Agency http://www.ema.europa.eu.”
A reference biological product receives a period of eight years of data exclusivity during which time applications for biosimilars may not be filed, starting from the date of the reference product’s initial authorization. In addition, the EMA may not issue a marketing authorization for a similar biological medicinal product application for a total of 10 years after the reference product’s approval, providing the reference product with an additional two years of market exclusivity after the initial eight years of data exclusivity. Market exclusivity may be extended for an additional year if the reference product sponsor obtains approval for a second significant new indication during the data exclusivity period. This framework is known as the “8 + 2 (+ 1)” exclusivity period. In addition, the patent protections available for reference products may further restrict or delay the development and approval of biosimilars. However, Article 11 of Directive 2001/83/EC and Article 3.3(b) of Regulation No. 726/2004 allow applicants and marketing authorization holders to exclude from their proposed product information those parts of the reference product’s SmPC that refer to indications or dosage forms that are covered by unexpired patents. Where patent protection of the reference product differs across Member States, the EMA allows the submission of duplicate applications for the biosimilar product to the extent that the reference product is protected by patents for certain therapeutic indications or pharmaceutical forms. Therefore, the duplicate application may contain more or fewer indications or pharmaceutical forms than the original application, as necessary to market the product in Member States where specific indications or pharmaceutical forms are protected by patents. An applicant that submits such applications must commit to extend the indications or pharmaceutical forms of the duplicate marketing authorization, or to withdraw the duplicate marketing authorization, as soon as the patents expire to ensure harmonization of the SmPCs across the EU.
Brazil
The regulatory body for approval of pharmaceuticals in Brazil is the Brazilian Health Surveillance Agency (Agência Nacional de Vigilância Sanitária, ANVISA). The rules concerning the registration of biosimilars are provided by ANVISA’s Resolution RDC n. 55/2010, which was drafted based on different international regulations and guidelines such as Health Canada (Canada), EMA (Europa),
CECMED (Cuba), KFDA (Korea) and the World Health Organization’s Similar Biological Product Guidelines.
According to RDC n. 55/2010, there are two categories of biological and biotechnological products in Brazil: (i) reference biological products are denominated “new biological products” and (ii) biosimilars are treated simply as “biological products.” While new biological products are registered with ANVISA by means of filing a dossier containing all production, quality control and non-clinical and clinical data (Phase 1, 2 and 3 trials), there are two possible alternatives for registering a biosimilar: (i) the Development by Comparability and (ii) Individual Development pathways.
Through the Development by Comparability pathway, a biosimilar’s quality, efficacy and safety are compared to a new biological product already registered with ANVISA (the reference biologic or comparator), including its cellular origin, manufacturing process, quality attributes and clinical and non-clinical studies. Based on the comparability data presented, the clinical and non-clinical development requirements can be simplified. Development by Comparability also enables the extrapolation of efficacy and safety data to other indications.
The Individual Development pathway is recommended for non-innovative products for which the comparability exercise is not possible. In this case, the manufacturer may apply for registration with production, quality control and clinical (Phase 1 and 2) and non-clinical non-comparative data. The extension of the Phase 1 and 2 clinical trials may be reduced according to the molecule complexity and specific attributes. However, this pathway requires a comparative Phase 3 clinical trial (non-inferiority, clinical equivalence or superiority), except for hemoderivatives, vaccines and oncological products. This pathway does not allow for extrapolation of efficacy and safety data to other indications.
In both scenarios, RDC n. 55/2010 still requires that the manufacturer present a list of documents related to efficacy, quality and safety of the product for ANVISA’s evaluation. In the case of a product manufactured abroad, ANVISA also requires that such product be already registered in the country of origin. GMP Certificates issued by both ANVISA and by the country of origin are also mandatory.
China
China’s State Food and Drug Administration, or SFDA, has established a number of regulatory requirements for the registration, manufacture, quality standards, marketing, distribution, importation, pricing control, advertising and labeling of pharmaceutical products, including biosimilars, in China.
Pursuant to the Administrative Measures Governing Registration of Medicines issued by SFDA on July 10, 2007 and effective on October 1, 2007, a pharmaceutical product, whether manufactured domestically or imported from outside of China, must be registered and approved by SFDA or its local counterparts before it may be manufactured in or imported into China. Different registration requirements and formalities are applicable to the registration of new pharmaceutical products, generic pharmaceutical products and imported pharmaceutical products respectively. Biosimilar products manufactured within China and imported into China are treated as new pharmaceutical products and imported pharmaceutical products, respectively, in respect of registration requirements and formalities. A new pharmaceutical product must be registered and approved by SFDA before it can be manufactured. Once on the market, any change in dosage form or route of administration of the approved new pharmaceutical product, or any claim of a new indication for such product, shall be subject to the same scrutiny and procedure as if such change or new indication were a registration of a new pharmaceutical product. As a part of the registration process, the manufacturer of a new pharmaceutical product is also required to conduct pre-clinical trials, apply to SFDA for permission to conduct clinical trials, and, after clinical trials are completed, file clinical data with the SFDA for approval. Once a pharmaceutical product is approved by SFDA as a new pharmaceutical product and its manufacturer has met the requisite manufacturing requirements and is in possession of a manufacturing permit, SFDA will issue a New Medicine Certificate together with an approval number
to the manufacturer and may, at its discretion, impose a monitoring period of not more than five years. During the monitoring period, SFDA will monitor the safety of the new pharmaceutical product, and will refrain from both (i) registering an identical pharmaceutical product manufactured or imported by another pharmaceutical company, and (ii) approving applications made by another pharmaceutical company for changes to the ingredients of its registered product which will render it identical to the new product. On January 7, 2009, SFDA promulgated the Administrative Measures Governing the Special Examination and Approval of New Medicine Registration, under which manufacturers of certain types of new pharmaceutical products may apply to follow the special examination and approval procedure with respect to clinical trial applications or production applications as the case may be. Under such special examination and approval procedure, SFDA will strengthen communications with the applicant and the application will be reviewed on an accelerated basis. On October 29, 2014, the Center for Drug Evaluation of the SFDA issued a draft for public comment called “Technical Guidelines for Biosimilar R&D and Evaluation.” If and when the draft is finalized and implemented into law, whether in the currently published form or otherwise, the requirements for registration of biosimilars in China may change in ways that we cannot predict.
A manufacturer is required to obtain a manufacturing permit from the relevant provincial counterpart of SFDA before it may carry out manufacturing operations. The issuance of such permit is conditional upon satisfactory inspection of the applicant’s manufacturing facilities and the applicant’s satisfaction of certain personnel qualifications, sanitary conditions, quality assurance systems, management structure and equipment standards requirements. Pursuant to the Regulations on the Implementation of the Law of the People’s Republic of China on the Administration of Medicines, which took effect on September 15, 2002, and the Measures on the Supervision and Administration of the Manufacture of Medicines, which took effect on August 5, 2004, a manufacturing permit is valid for five years and application for its renewal should be made to the relevant provincial counterpart of SFDA at least six months prior to its expiry. Manufacturers must also obtain GMP certification for production of pharmaceutical products in China. The Good Manufacturing Practice for Medicines (2010 revised edition) promulgated by the Ministry of Health and effective on March 1, 2011 is made up of a set of detailed guidelines on practices governing different aspects of the production of pharmaceutical products, including institutional qualifications, staff qualifications, production premises and facilities, equipment, hygiene, production management, quality control and assurance, product distribution and recall, documentation and raw material management. A GMP certificate is valid for a term of 5 years and application for its renewal should be made to the provincial counterpart of SFDA at least six months prior to its expiry.
India
In India, biosimilars, (which are more commonly known as “similar biologics”) are regulated pursuant to (i) the Drugs and Cosmetics Act, 1940; (ii) the Drugs and Cosmetics Rules, 1945 as amended from time to time; (iii) Rules for the Manufacture, Use, Import, Export and Storage of Hazardous Micro Organisms, Genetically Engineered Organisms or Cells, 1986 notified under the Environment Protection Act, 1986; (iv) Recombinant DNA Safety Guidelines & Regulations, 1990; (v) Guidelines for generating Pre-Clinical & Clinical Data for R-DNA vaccines, diagnostics & other Biologicals, 1999; (vi) Good Environmental Practice guidelines issued by the Ministry of Environment and Forests, including Good Practices in Environmental Regulation and Good Practices in Animal Experimentation; (vii) CDSCO Guidance for Industry, 2008 (read with clarificatory Circular dated 5th August, 2010), which covers: (a) Submission of Clinical Trial Application for Evaluating Safety and Efficacy; (b) Requirements for permission of New Drugs Approval; (c) Post approval changes in biological products: Quality, Safety and Efficacy documents; and (d) Preparation of the Quality Information for Drug Submission for New Drug Approval: Biotechnological/Biological Products. issued by the CDSCO with respect to the requirements for post-approval changes; (viii) Guidelines and Handbook for Institutional Biosafety Committees, 2011; and (ix) Guidelines on Recall & Rapid Alert
System for Drugs (Including Biologicals & Vaccines) (effective from 23rd November 2012). In addition, the Central Drugs Standard Control Organization, or CDSCO, established by the Ministry of Health and Welfare, Government of India, which is the overarching regulatory body in the Ministry for the regulation of drugs, issues additional requirements for approval of drugs or clarification on the requirements from time to time.
In June 2012, the CDSCO and the Department of Biotechnology issued a guidance document titled “Guidelines on Similar Biologics.” The Guidelines lay down the regulatory pathway for a biologic claiming to be similar to an already authorized reference biologic. Pursuant to the Guidelines, to obtain approval for a biosimilar, the sponsor must establish that the proposed biosimilar is similar to the reference biologic, which is authorized for marketing in India. The similarity of the proposed biosimilar to the reference product is demonstrated through quality characterization studies that compare the molecular and quality attributes of the proposed biosimilar with those of the reference biologic. The testing of the proposed biosimilar should also be sufficient to ensure that the proposed biosimilar meets acceptable levels of safety, quality and efficacy. Biosimilars are subject to a number of premarket regulatory requirements, including comparability exercises for quality, pre-clinical and clinical studies, along with post-market regulatory obligations, including post-approval testing. The Guidelines are applicable both to similar biologics developed in India and similar biologics imported into India.
The authorities involved in the approval process of biosimilars include the Review Committee on Genetic Manipulation, or RCGM, set up in the Department of Biotechnology Ministry of Science and Technology, Government of India; the Genetic Engineering Appraisal Committee, or GEAC, in the Ministry of Environment and Forests; and CDSCO, in the Ministry of Health and Welfare. The RCGM is responsible for authorizing import and export for research and development and review of data up to preclinical evaluation. The GEAC is a statutory body established for review and approval of activities involving large-scale use of genetically engineered organisms (also referred as living modified organisms) and products thereof in research and development, industrial production, environmental release and field applications. The CDSCO is the apex regulatory body for the regulation of drugs, including biosimilars. It is responsible for the grant of import and export licenses, clinical trial approvals and authorizations for marketing and manufacturing. The State Food & Drug Administration works with CDSCO in each state within India and is responsible for the issuance of licenses to manufacture similar biologics in India. Other bodies involved in the approval process for biosimilars are the Recombinant DNA Advisory Committee, the Institutional Biosafety Committees, the State Biosafety Co-ordination Committees, the District Level Committees, and the State Licensing Authority.
Facilities
Our headquarters is located in Boston, MA, where we occupy approximately 8,000 square feet of office space. The term of our lease expires in 2016. The premises are anticipated to be sufficient for current and future operations and we expect to be able to renew the lease for these premises upon its expiration.
Employees
As of January 2015, we had 33 full-time employees. None of our employees is represented by a labor union and we consider our employee relations to be good.