Attached files
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2010.
Or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission File Number 000-51171
ZALICUS INC.
(Exact name of registrant as specified in its charter)
| Delaware | 04-3514457 | |
| (State or other jurisdiction of incorporation or organization) |
(IRS Employer Identification Number) | |
| 245 First Street Third Floor Cambridge, Massachusetts |
02142 | |
| (Address of Principal Executive Offices) | (Zip Code) | |
Registrant’s telephone number, including area code: (617) 301-7000
Securities registered pursuant to Section 12(b) of the Exchange Act:
| Title of Each Class |
Name of Exchange on Which Registered | |
| Common Stock, par value $0.001 | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Exchange Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. Check One:
| Large Accelerated Filer: ¨ | Accelerated Filer: x | |
| Non-Accelerated Filer: ¨ | Smaller Reporting Company: ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No x
The aggregate market value of voting common equity of the registrant held by non-affiliates of the registrant was approximately $95,771,609 on June 30, 2010. For purposes of the foregoing sentence, the term “affiliate” includes each director and executive officer of the registrant and affiliates of such persons. The computation of the aggregate market value is based upon the closing price of the common stock as reported on the NASDAQ Global Market on June 30, 2010.
As of March 8, 2011, the registrant had 94,566,880 shares of common stock, par value $0.001 per share, outstanding.
Specified portions of the registrant’s definitive Proxy Statement relating to the registrant’s Annual Meeting of Stockholders, which is expected to be filed pursuant to Regulation 14A within 120 days after the end of the registrant’s fiscal year ended December 31, 2010 are incorporated by reference in Part III of this Annual Report on Form 10-K.
ZALICUS INC.
ANNUAL REPORT
ON FORM 10-K
| PAGE | ||||||
| PART I |
||||||
| Item 1. |
3 | |||||
| Item 1A. |
28 | |||||
| Item 1B. |
46 | |||||
| Item 2. |
46 | |||||
| Item 3. |
46 | |||||
| Item 4. |
46 | |||||
| PART II |
||||||
| Item 5. |
47 | |||||
| Item 6. |
50 | |||||
| Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
52 | ||||
| Item 7A. |
64 | |||||
| Item 8. |
64 | |||||
| Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
64 | ||||
| Item 9A. |
65 | |||||
| Item 9B. |
67 | |||||
| PART III |
||||||
| Item 10. |
67 | |||||
| Item 11. |
67 | |||||
| Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
67 | ||||
| Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
67 | ||||
| Item 14. |
68 | |||||
| PART IV |
||||||
| Item 15. |
68 | |||||
| 69 | ||||||
2
PART I
FORWARD-LOOKING STATEMENTS
This annual report on Form 10-K includes statements with respect to Zalicus Inc. (“Zalicus”) and its subsidiaries (“we”, “our” or “us”), which constitute “forward-looking statements” within the meaning of the safe harbor provisions of the United States Private Securities Litigation Reform Act of 1995. Words such as “believe,” “anticipate,” “expect,” “estimate,” “intend,” “plan,” “project,” “will be,” will continue,” “will result,” “seek,” “could,” “may,” “might,” or any variations of such words or other words with similar meanings are intended to identify such forward-looking statements. Forward-looking statements in this annual report on Form 10-K include, without limitation, statements regarding our future expectations; any projections of financing needs, revenue, expenses, earnings or losses from operations, or other financial items; any statements of the plans, strategies and objectives of management for future operations; any statements concerning product candidate research, development and commercialization plans and timelines; any statements regarding safety and efficacy of product candidates; any statements regarding plans for outlicensing of our clinical or pre-clinical product candidates and seeking collaborations; any statements regarding timing of initiating and completing clinical and pre-clinical trials and studies; any statements of expectation or belief; and any statements regarding other matters that involve known and unknown risks, uncertainties and other factors that may cause actual results, levels of activity, performance or achievements to differ materially from results expressed in or implied by this annual report on Form 10-K.
The risks, uncertainties and assumptions referred to above include risks that are described in this annual report on Form 10-K in the section entitled “Risk Factors” and elsewhere and that are otherwise described from time to time in our Securities and Exchange Commission reports filed after this report. You are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date of this annual report on Form 10-K. We specifically disclaim any obligation to update these forward-looking statements in the future, except as required by law.
| Item 1. | Business |
Overview
We are a biopharmaceutical company that discovers and develops novel treatments for patients suffering from pain and immuno-inflammatory diseases. We have entered into multiple revenue-generating collaborations with large pharmaceutical companies, including a commercial collaboration for Exalgo™, and we have a portfolio of clinical-stage product candidates targeting pain and immuno-inflammatory diseases. We also apply our selective ion channel modulation platform and combination high throughput screening technology to discover new product candidates for our portfolio and our collaborators in the areas of pain, inflammation, oncology and infectious disease. In December of 2009, we completed a merger, which we refer to as the Neuromed Merger, with Neuromed Pharmaceuticals Inc., or Neuromed, pursuant to which Neuromed Pharmaceuticals Ltd. became a wholly-owned subsidiary of Zalicus. In September of 2010, we changed our name from CombinatoRx, Incorporated to Zalicus Inc. We also changed the name of our subsidiaries, including Neuromed Pharmaceuticals Ltd., which is now named Zalicus Pharmaceuticals Ltd, and which we refer to herein as Zalicus Canada.
On March 1, 2010, the United States Food and Drug Administration, or the FDA, approved the New Drug Application for Exalgo (hydromorphone HCl) extended-release tablets, for the management of moderate to severe pain in opioid tolerant patients requiring continuous, around-the-clock opioid analgesia for an extended period of time. Exalgo is an extended release formulation of hydromorphone, an opioid analgesic that has been used in an immediate release formulation to treat pain for many years. Exalgo employs the OROS® PUSH-PULL™ osmotic delivery system designed to release hydromorphone at a controlled rate over an extended period of time, allowing for once-daily administration. Zalicus Canada acquired the rights to Exalgo in 2007 and advanced it successfully through Phase 3 clinical development, the submission of a new drug application in May
3
2009 and its approval by the FDA in March 2010. The United States commercial rights to Exalgo were acquired in June 2009 by Mallinckrodt, Inc., a subsidiary of Covidien, plc, or Covidien. Covidien is responsible for all commercialization activities for Exalgo in the United States, including marketing and sales, and for all post-approval regulatory activities. We received a $40.0 million milestone payment following FDA approval of Exalgo, and we receive tiered royalties quarterly on net sales of Exalgo by Covidien following its commercial launch in April 2010.
We have been advancing a portfolio of product candidates into or through clinical research and development, and we have a number of product candidates that are in late-stage preclinical development. Our clinical and preclinical product candidates targeting pain and inflammatory diseases include:
| • | Synavive™, a low-dose glucocorticoid product candidate, with amplified immuno-inflammatory benefits, being developed to treat immuno-inflammatory disorders, initially rheumatoid arthritis; |
| • | Z160, an N-type calcium channel blocker product candidate we have selected for clinical development in chronic pain; |
| • | Z944, a T-type calcium channel blocker product candidate we have selected for clinical development in acute or chronic inflammatory pain; and |
| • | Additional preclinical N-type and T-type calcium channel blockers and sodium channel blockers being evaluated as potential candidates for development in chronic pain. |
Our ion channel programs have the potential to produce a new class of analgesics for the treatment of both acute and chronic pain, with the potential for safety and efficacy advantages over existing analgesics. We believe our N-type calcium channel blockers and sodium channel blockers have the potential to be efficacious across a wide variety of chronic pain states and that our T-type calcium channel blockers show promise in acute and chronic pain.
Combination approaches are an emerging clinical standard in the treatment of cancer, infectious diseases, inflammatory diseases and pain. We believe that our combination high throughput screening technology platform, or cHTS™, has been validated clinically by the identification of unique synergies underlying our most advanced clinical product candidate, Synavive, and through our research collaborations with Novartis Institutes of Biomedical Research, Inc., or Novartis, and Amgen Inc., or Amgen, who have adopted cHTS as an important addition to their oncology discovery efforts. Our cHTS platform technology profiles the activity of individual agents and combinations in cell-based phenotypic assays at high throughput to discover synergistic multi-target drug candidates that may generate novel intellectual property and expand development pipelines. Our proprietary software solution, Chalice™, works with the cHTS technology platform to extract valuable insights, discoveries, and data trends from high-throughput combination activity profiling experiments. We seek strategic partnerships to leverage the power of the cHTS technology platform in the areas of cancer, inflammation and infectious disease.
Exalgo
On March 1, 2010, the FDA approved the New Drug Application for Exalgo (hydromorphone HCl) extended-release tablets, for the management of moderate to severe pain in opioid tolerant patients requiring continuous, around-the-clock opioid analgesia for an extended period of time. Exalgo is an extended release formulation of hydromorphone, an opioid analgesic that is five to eight times more potent than morphine and has a long history of use for the control of pain. Other oral hydromorphone products currently available in the United States are immediate release formulations, requiring dosing several times per day. Exalgo employs the OROS® PUSH-PULL™ osmotic delivery system designed to release hydromorphone at a controlled rate over an extended period of time allowing for once-daily administration. The same OROS® hydromorphone formulation as Exalgo is currently marketed for severe pain by Janssen-Cilag, a wholly-owned subsidiary of Johnson & Johnson, in several European countries under the brand name JURNISTA™. The OROS PUSH-PULL formulation technology utilized for Exalgo has been used clinically for more than two decades and in more than a dozen marketed products including Procardia XL®, DITROPAN XL® and CONCERTA®.
4
Our subsidiary, Zalicus Canada, acquired the United States rights to Exalgo in 2007 and advanced it successfully through Phase 3 clinical development, the submission of a new drug application in May 2009 and its approval by the FDA in March 2010. The United States commercial rights to Exalgo were acquired in June 2009 by Covidien and Covidien is responsible for all commercialization activities for Exalgo in the United States, including marketing and sales, and for all post-approval regulatory activities. We received a $40.0 million milestone payment following FDA approval of Exalgo. Exalgo was commercially launched in the United States by Covidien on April 26, 2010, and Covidien has steadily increased Exalgo managed care access, formulary listings and pharmacy shelf space allocations since the launch. We received tiered royalties on net sales of Exalgo by Covidien on a quarterly basis since its commercial launch. We have recognized $1.6 million in royalty revenue from Exalgo through December 31, 2010.
Background
Hydromorphone, a widely prescribed opioid, is sold under the brand name Dilaudid® and in generic formulations. Hydromorphone is a mature generic product generally not supported by significant sales force coverage and was, until the FDA approval of Exalgo, available only in immediate release formulations in the United States. Other strong oral opioids such as morphine, fentanyl, oxymorphone and oxycodone are available in both immediate release as well as extended release formulations and have been supported by significant sales force coverage within the last five years. Despite these factors, in the five year period from 2004 to 2008, on a percentage basis, the volume of prescriptions of hydromorphone grew more rapidly than these other strong opioid products. For example, during this five year period, immediate release hydromorphone prescription volume, on a percentage basis, outgrew fentanyl (all forms combined) which is available in a long acting transdermal patch, DURAGESIC®, and fast-acting buccal formulations, Actiq® and FENTORA®.
OROS ® PUSH-PULL™ Technology
ALZA Corporation’s, or ALZA’s, OROS® PUSH-PULLTM technology delivery system is an oral osmotic technology that has been used for more than two decades and has been successful in the development of once-daily dosage options for several short-acting drugs. Examples of these formulations include Procardia XL®, DITROPAN XL® and CONCERTA®. Formulations that use OROS® PUSH-PULL™ technology vary greatly as the design of each system is unique, formulated to provide a specific delivery pattern. OROS® hydromorphone as utilized for Exalgo uses an osmotic pump to control the release of hydromorphone over an extended period. The tablet core is made up of two layers: a “drug” layer containing hydromorphone and non-medicinal ingredients and a “push” layer containing osmotic agents. These layers are surrounded by a hard, non-dissoluble outer shell that has a precision laser- drilled hole through it. Water from the gastrointestinal tract enters the tablet through this semi-permeable outer shell, causing the push layer to expand, which expels the drug into the gastrointestinal tract at a controlled rate. The outer shell itself does not dissolve and the residual tablet is excreted.
Market for Opioids to Treat Moderate to Severe Chronic Pain
The United States market for long-acting strong opioids includes extended release formulations of morphine, oxycodone, oxymorphone and fentanyl. In the United States, prior to the FDA approval of Exalgo, there was no long-acting version of hydromorphone. Exalgo was developed to be an extended release formulation of hydromorphone for the management of moderate to severe pain in opioid tolerant patients requiring continuous, around-the-clock opioid analgesia for an extended period of time.
For the past decade in the United States, market sales data indicates that, in general, more potent extended release formulations of long-acting opioids, such as fentanyl in the DURAGESIC® patch and oxycodone in OxyContin®, have generated significantly higher sales revenue compared to less potent morphine products, such as AVINZA® and KADIAN®.
5
Hydromorphone, a widely prescribed opioid, is intermediate in potency between oxycodone and fentanyl. Fentanyl is available for chronic use only as a patch and, while patches may be suitable for many patients, prescription data indicates that the majority of patients on long-term opioid therapy are treated with oral opioids. Orally-administered oxymorphone, a relatively new long-acting strong opioid, is more potent than oxycodone but is less potent than hydromorphone and has a shorter history with prescribing physicians than hydromorphone.
Benefits of Exalgo
We believe that Exalgo provides the benefit of a convenient once-daily formulation of hydromorphone, the most potent oral, long-acting opioid in the United States, for the management of moderate to severe pain in opioid tolerant patients requiring continuous, around-the-clock opioid analgesia for an extended period of time. For these patients, frequent administration of immediate release strong opioids may be inconvenient, particularly if they need to take medication during the night. Patient convenience and potentially patient compliance may be improved with the less frequent dosing intervals offered by products such as once-daily Exalgo. Exalgo also has the potential to combine the benefits of oral therapies, such as ease of titration and intake, with the benefits of transdermal therapies, such as stable blood drug levels and an extended duration of action.
Our Product Candidates
All of our product candidates are focused on diseases with continuing medical needs and potentially large commercial markets. Our principal drug development programs are in the areas of pain and inflammation.
Synavive
We currently have one clinical stage product candidate, Synavive, targeting immuno-inflammatory diseases. Immuno-inflammatory diseases include rheumatoid arthritis, osteoarthritis, lupus (SLE), polymyalgia rheumatic, ulcerative colitis and Crohn’s disease.
Rheumatoid Arthritis Background. Rheumatoid arthritis is a chronic disease, mainly characterized by inflammation of the lining, or synovium, of the joints. According to the Arthritis Foundation, rheumatoid arthritis affects approximately one percent of the United States population, or approximately 2.1 million people. Rheumatoid arthritis can lead to long-term joint damage, resulting in chronic pain, loss of function and disability. Because it is a chronic disease, rheumatoid arthritis continues indefinitely, and frequent flares in disease activity can occur. Rheumatoid arthritis is also a systemic disease, which means it can affect other organs in the body. Studies have shown that early aggressive treatment of rheumatoid arthritis can limit joint damage, which in turn limits loss of movement, increases ability to work, reduces medical costs and delays or prevents the need for surgery.
Osteoarthritis Background. Osteoarthritis is one of the most common degenerative joint diseases and a frequent cause of physical disability among older adults. In the United States more than 21 million people suffer from osteoarthritis. Osteoarthritis affects the hands, lower back, neck, and weight-bearing joints such as the knees, hips, and feet. Symptoms of osteoarthritis range from stiffness and intermittent mild pain to severe joint pain and impaired biomechanical function. Although there is no cure for most forms of osteoarthritis, various therapies can help patients manage symptoms such as non-steroidal anti-inflammatory drugs (NSAIDS), COX-2 inhibitors, local analgesics, opioids, intra-articular corticosteroid injection and surgery.
Synavive Background
Synavive is designed to enhance the anti-inflammatory benefits of glucocorticoids, without associated dose-dependent side effects. It is being developed in a uniquely engineered formulation and contains the cardiovascular agent, dipyridamole, and a very low dose of the glucocorticoid, prednisolone. In proof-of-concept clinical trials, Synavive demonstrated an anti-inflammatory effect in subjects with rheumatoid arthritis and osteoarthritis and was generally well-tolerated. We believe Synavive works through a novel mechanism of action in which dipyridamole selectively amplifies prednisolone’s anti-inflammatory activities without increases in
6
adverse effects typically associated with higher doses of glucocorticoids. After consultation with the FDA and other regulatory authorities in 2011, we are planning to initiate a Phase 2b clinical trial in subjects with rheumatoid arthritis in the second quarter of 2011. The planned Phase 2b clinical trial, titled SYNERGY, is a 12-week, five-arm, global, double-blind, placebo-controlled study to evaluate the safety and efficacy of Synavive in approximately 250 subjects with moderate to severe rheumatoid arthritis. Subjects who complete the core SYNERGY study will be eligible to participate in a one-year extension study designed to investigate the long-term safety and durability of response for Synavive.
Rheumatoid Arthritis Clinical Results. We studied Synavive in a multi-center randomized, blinded, placebo-controlled Phase 2a clinical trial of 59 subjects with rheumatoid arthritis. Subjects were enrolled in this study with established rheumatoid arthritis and moderate disease activity as determined by DAS28 scores of greater than 4.5 and CRP levels of greater than 2.2 mg/L. DAS28 is a composite disease activity score using 28 joint counts that is used to monitor disease activity in rheumatoid arthritis patients and CRP is the inflammatory biomarker C-reactive protein. Subjects were required to be on a disease-modifying anti-rheumatic drug, or DMARD (such as methotrexate or sulfasalazine) for at least three months and on a stable dose of DMARD therapy for a minimum of 28 days prior to enrollment. Synavive was dosed in this trial using 3 mg of prednisolone plus 200 mg dipyridamole for the first week of treatment and 3 mg prednisolone plus 400 mg of dipyridamole for the following five weeks of treatment. The clinical trial compared Synavive plus a DMARD to placebo plus DMARD in subjects with rheumatoid arthritis.
In this trial, Synavive demonstrated a statistically significant improvement on the primary endpoint of reduction of CRP, with a 50% median reduction from baseline to day 42 compared to a 19% reduction with control (p=0.024). Importantly, Synavive demonstrated statistically significant improvements in two clinically meaningful measures of efficacy: (i) ACR20, a standard measure developed by the American College of Rheumatology to rate rheumatoid arthritis disease improvement and (ii) DAS28. In this study Synavive demonstrated a statistically significant 63% ACR-20 response at day 42 compared to a 30% response with placebo (p=0.025) and a statistically significant DAS28 score, with a -1.6 mean change from baseline to day 42 compared to -0.7 with control (p=0.016). Patients are classified as ACR20 responders if they demonstrate at least a 20% improvement from baseline in tender and swollen joint count and at least 3 of 5 other symptom-related criteria. Data provided for the rheumatoid arthritis trial of Synavive are for the per protocol population; statistical significance remained consistent in the intent-to-treat population. In this rheumatoid arthritis trial, Synavive was generally well tolerated, and there were no serious adverse events reported for subjects treated with Synavive. The most common adverse events observed with Synavive that occurred with a frequency of greater than 5% were headache, gastro-intestinal symptoms and dizziness, known side effects of dipyridamole.
Knee Osteoarthritis Clinical Results. We studied Synavive in a multi-center, randomized, double-blind, placebo-controlled Phase 2 clinical trial of 279 subjects with knee osteoarthritis, the COMET-1 study. The COMET-1 study was a standard flare design where subjects with active disease needed to demonstrate an increase in knee pain as determined by the Western Ontario and McMaster University Osteoarthritis, or WOMAC, question #1 (related to pain while walking on a flat surface) upon withdrawal of their NSAID or COX-2 inhibitor drug therapy to be eligible. In the study, subjects were randomized to three different doses of Synavive (2.7mg prednisolone and 360mg, 180mg or 90mg of dipyridamole), 2.7mg of prednisolone alone or placebo. Subjects were dosed for a total of 14 weeks (98 days) including an initial two-week dipyridamole titration phase. The primary endpoint of this study, to assess the efficacy of Synavive compared to placebo, was the change in WOMAC question #1 calculated from baseline to day 98. Secondary and ancillary endpoints included the full WOMAC pain, stiffness, physical function sub-scales and patient global assessment scores and hand pain. Subjects who completed the 14-week core study were eligible to participate in a one-year, open-label extension study designed to investigate the long-term safety and durability of response for Synavive.
The COMET-1 study was completed in September 2008, and the preliminary analysis of the primary and certain secondary endpoints from the trial were disclosed on October 6, 2008. While there was a numerical trend in favor of Synavive and an observed dose-response relationship, Synavive did not demonstrate a statistically
7
significant increased WOMAC response compared to placebo for the primary endpoint, nor when compared to prednisolone alone, both analyzing the intent-to-treat, or ITT, population from the study. We also conducted pre-specified analyses of the data from the COMET-1 study using a modified ITT, or mITT, analysis, which accounts for subjects who violated the protocol by commencing use of a prohibited medication (such as an NSAID or COX-2 inhibitor) prior to their end-of-study visit. For the mITT analysis, the last observation prior to this protocol violation was carried forward for all efficacy measures. In the mITT analysis, the trends favoring Synavive over placebo were enhanced for WOMAC, and were statistically significant for high-dose Synavive (2.7 mg prednisolone/360 mg dipyridamole) compared to placebo in the more comprehensive assessments of improvement as measured by the WOMAC pain, stiffness, and physical function subscales. These effects are comparable to current osteoarthritis therapies, including NSAIDs and COX-2 inhibitors, based on reviews of published data. In addition, in a pre-specified analysis, a beneficial effect of Synavive was observed for those subjects in the study who also experienced significant hand pain, a 45% reduction versus 23% observed with placebo, and 26% observed with prednisolone alone, thus corroborating activity observed in an earlier Phase 2a proof-of-concept clinical study of Synavive in subjects with hand osteoarthritis.
In the COMET-1 study, Synavive was generally well tolerated, and no serious adverse events were reported relating to Synavive. The most commonly reported adverse event was headache. The rate of drop out from headache of 4% was evenly distributed across all active arms, including prednisolone. In addition, there was no evidence of increased hemoglobin A1c, fasting plasma glucose or triglycerides in the Synavive arms as compared to placebo. Mean systolic blood pressure at the end of 14 weeks of treatment was unchanged in the prednisolone alone arm (a known side effect of glucocorticoids), while the high-dose Synavive combination slightly reduced this measure of blood pressure. Of the 279 subjects enrolled, 191 (68%) completed the study. Primary reasons for discontinuation included adverse event (11%), subject request (8%) and disease progression/lack of efficacy (6%). A total of 141 subjects who completed the 14-week duration of the COMET-1 study enrolled in an open-label extension study designed to investigate the long-term safety and durability of response for Synavive. The COMET-1 open-label extension study of Synavive completed dosing in June 2009 and no serious adverse events related to Synavive have been reported for subjects in the COMET-1 extension study.
Hand Osteoarthritis Clinical Results. Prior to initiating the COMET-1 Phase 2 clinical trial of Synavive in knee osteoarthritis, we studied Synavive in a randomized, blinded, placebo-controlled Phase 2a clinical trial in 83 subjects with moderate to severe osteoarthritis of the hand. The study met its primary endpoint of improvement in joint pain using the Australian Canadian Osteoarthritis, or AUSCAN, index. The AUSCAN Osteoarthritis Index is a composite patient-reported outcome tool developed specifically for hand osteoarthritis, similar to the WOMAC index which is used for knee and hip osteoarthritis. Analysis of the primary endpoint shows mean change from baseline in the Synavive group of a 31% improvement in pain, compared to mean change from baseline in the placebo group of a 7% improvement in pain (p=0.007). Synavive demonstrated improvements in clinical secondary endpoints including stiffness, the AUSCAN physical function subscale, joint pain and patient global assessment scores.
| Clinical Measure |
Synavive Mean Improvement |
Placebo Mean Improvement |
Synavive P Value |
Synavive Mean Baseline |
Synavive Improvement from Baseline*** |
|||||||||||||||
| Pain* |
102.4 mm | 31 mm | 0.006 | ** | 309.3 mm | 33 | % | |||||||||||||
| Stiffness |
20.3 mm | 8.3 mm | 0.023 | ** | 62.9 mm | 32 | % | |||||||||||||
| Physical Function |
115.8 mm | 53.1 mm | 0.081 | 584.2 mm | 20 | % | ||||||||||||||
| Joint Pain |
23.5 mm | 6.3 mm | 0.002 | ** | 59.8 mm | 39 | % | |||||||||||||
| Patient Global |
23.4 mm | 4.6 mm | <0.001 | ** | 61.5 mm | 38 | % | |||||||||||||
Notes:
| * | Primary endpoint |
| ** | Statistically significant |
| *** | Calculation (mean improvement/mean baseline x 100) |
8
Subjects enrolled in the clinical trial had moderate to severe hand osteoarthritis as determined by American College of Rheumatology criteria and a score on the pain dimension of the AUSCAN scale above a pre-specified minimum. Synavive was generally well tolerated, and there were no serious adverse events reported from subjects taking Synavive. The most common adverse events observed with Synavive were headache and nausea, known side effects of dipyridamole.
Our Ion Channel Product Candidates for Pain
One of our key platform discovery technologies is our advanced drug design and electrophysiological screening process to discover new compounds that selectively target neuronal calcium and sodium channels. The current treatment paradigms for pain rely on three major classes of medicines: opiates, non-steroidal anti-inflammatories and gabapentanoids, a class of anti-convulsants with analgesic properties, all of which have significant safety, tolerability and dependence issues. We are using our experience with ion channel research and development in our efforts to discover and develop novel proprietary compounds to potentially treat pain by selectively blocking N-type and T-type calcium channels or selected sodium channels. We believe our ion channel modulator programs have the potential to produce a new class of analgesics for the treatment of both acute and chronic pain, with the potential for safety and efficacy advantages over existing analgesics.
Background of Clinical Pain. Pain results from sensory nerve stimulation often associated with actual or potential tissue damage. Specific nerve fibers carry the pain signal across the nervous system to the brain, where it is recognized as pain. Pain is generally characterized on two dimensions, intensity and duration. Pain intensity is typically expressed as mild, moderate or severe. Mild pain results from relatively common conditions such as headaches, sprains or strains. Moderate pain results from conditions such as surgery, severe strains or sprains. Severe pain results from serious underlying illnesses such as cancer, AIDS, osteoarthritis, lower back pain and diabetic neuropathy. Pain duration is expressed as acute or chronic. Acute pain often subsides in a short period of time and is typically associated with tissue injury such as surgery, a cut, a joint dislocation, or pressure on a nerve. Chronic pain persists for long periods of time and may involve underlying changes in the nervous system producing unusual sensitivity to touch, shooting pains, aching and other often disabling pain symptoms.
Background on Pain Treatment Market. NSAIDs, including COX-2 inhibitors, used to treat mild to moderate pain, are widely prescribed within the pain pharmaceutical market. NSAIDs are drugs with analgesic, fever-reducing and anti-inflammatory effects. As a class, NSAIDs are usually prescribed as first-line treatment; however, their relatively low potency may result in insufficient pain control for the patient. The long-term use of NSAIDs may result in side effects such as gastrointestinal bleeding, liver and kidney damage and cardiovascular-related complications.
Opioids have long been prescribed to treat moderate to severe pain and are regarded as the most potent class of analgesics. When used for extended periods, however, opioids can lead to side-effects, the development of tolerance, dependence and addiction. Tolerance means that increasing doses of opioids are required to maintain effective pain relief. Dependence means reliance on the drug and the existence of significant withdrawal symptoms upon cessation of drug administration. Addiction refers to drug-seeking behaviors characterized by a continued craving for the opioid and the need to use it for effects other than pain relief. As a consequence of these and other serious side effects, opioids are usually prescribed when other treatments for chronic pain have failed. In general, the more severe or chronic the pain, the more likely an opioid will be prescribed.
Despite the availability of many drugs to treat chronic pain, the results of a 2006 survey of chronic pain sufferers conducted by the American Pain Foundation, found that approximately 51% of the respondents felt that they had little or no control over their pain. We believe that this lack of adequate pain control, particularly in patients with moderate to severe chronic pain, represents a significant therapeutic gap in current pain management.
9
Our Ion Channel Programs
N-type and T-type calcium channels and sodium channels in cells are important to the regulation of the body’s nervous and cardiovascular systems. We are developing novel proprietary compounds to potentially treat pain and other diseases by selectively blocking ion channels in the neuronal cells responsible for pain signal transmission. We believe that our ion channel programs have the potential to produce a new class of analgesics for the treatment of both acute and chronic pain, with potential safety and efficacy advantages over existing analgesics.
Overview of Calcium Channel Biology
Calcium ions play critical roles in the biochemistry, physiology and anatomy of cells and organisms. The rapid entry of calcium ions into cells is mediated by a class of proteins called voltage-gated calcium channels. These channels respond to electrical signals by the opening of a calcium-selective pore in the cell membrane. Calcium channels are involved in a large number of normal physiological processes including muscle contraction, hormone secretion, gene expression, and electrical signaling in the nervous system.
In order to carry out the multiple physiological functions that calcium channels help regulate, the human genome encodes ten distinct types of calcium channels. These ten different calcium channels have been traditionally classified into five designations; L-type (four gene subtypes), T-type (three gene subtypes), and one gene subtype for each of the R-type, P/Q-type and N-type. Of particular relevance for pharmaceutical development, each of the different types of calcium channels is known to perform distinct physiological functions and offers the opportunity to target specific drugs to specific calcium channels and human disease indications.
For example, L-type calcium channels are responsible for triggering contraction of both heart muscle and blood vessel smooth muscle. As such, L-type channels have been the selective target for drugs treating cardiovascular disease including hypertension and cardiac arrhythmias. These well-known calcium channel blockers have been on the market for over 40 years and provide proof-of-concept that calcium channels represent a valid target for therapeutic intervention and may address large commercial opportunities. Our Chief Scientific Officer, Dr. Terrance Snutch, was the first to describe that the various calcium channels in the nervous system are encoded by a family of distinct genes. In addition to being the first to clone these important clinical targets, Dr. Snutch, recognizing the potential pharmaceutical significance of the N-type and T-type calcium channels, devised our innovative screening platforms and invented our initial proprietary calcium channel blocker product candidates. Building on this work, we are currently pursuing two calcium channel programs: one targeting the N-type calcium channels, and a second focused on targeting the T-type calcium channel gene subtypes. We also believe that there may also be an opportunity for development of mixed N/T-type calcium channel product candidates.
N-type Calcium Channel Program and Z160
Our N-type calcium channel program is focused on developing drug candidates to treat chronic pain and our orally-administered N-type calcium channel blockers, including Z160 (formerly referred to as NMED-160, have shown efficacy in multiple animal models of pain, and in the case of Z160, have been well tolerated in Phase 1 and Phase 2a clinical trials. Our N-type calcium channel programs, including Z160, were previously licensed to Merck & Co. Inc., or Merck, as part of a research collaboration and license agreement which terminated in September 2009. Upon termination of the agreement with Merck, Merck’s exclusive license to our N-type calcium channel intellectual property terminated, and we have an exclusive license to all jointly owned patents created in collaboration with Merck relating to the N-type calcium channel compounds Merck was advancing.
Z160. Z160 is an N-type calcium channel blocker that has shown efficacy in multiple animal models of neuropathic and inflammatory pain, and has been well tolerated in Phase 1 and Phase 2a clinical trials in approximately 200 subjects. After the termination of the Merck collaboration, we have developed proprietary formulations of Z160 to increase its bioavailability and improve its pharmacokinetic and pharmacodynamic properties. Based on these improved formulations of Z160, we are planning to advance Z160 into Phase 1 clinical development by the end of 2011.
10
N-type Background. N-type calcium channels are expressed exclusively in the nervous system and are highly concentrated in the dorsal horn of the spinal cord where incoming sensory pain information is processed and then relayed to the brain. The changes in calcium concentration in certain spinal cord nerve cells due to calcium entering through N-type calcium channels are directly linked to pain signal transmission, especially as it relates to chronic inflammatory and neuropathic pain conditions. Our focus is on controlling the flow of calcium into spinal nerve cells through the N-type calcium channel and thus on reducing pain signaling as it relates to chronic inflammatory, neuropathic, and other complex pain conditions.
A number of pharmaceutical companies have attempted to develop orally and intravenously active small molecules that block the N-type calcium channel. We believe that, by uniquely combining rational drug design with innovative biological assays, we can produce orally available N-type calcium channel blockers like Z160.
Current Market Opportunity for N-type Product Candidates. We believe that oral N-type calcium channel blockers have the potential to become a new class of oral analgesics. Efficacy of N-type calcium channel blockers was first demonstrated by Prialt®, a small protein N-type calcium channel blocker. Prialt® has been shown to be efficacious in various pain states such as cancer pain, lower back pain, osteoarthritis, herpes zoster neuropathy and AIDS neuropathy. Prialt®, currently marketed in several countries including the United States, U.K. and Germany, is reported to not cause some of the common opioid side effects such as tolerance, withdrawal, nausea, vomiting or respiratory depression; however, its use is limited by its own side effects and the requirement that it be administered with an intrathecal pump directly into a patient’s spinal column. Our orally-administered small molecule N-type calcium channel blockers, including Z160, show efficacy in a variety of animal models of chronic pain resulting from inflammation or nerve injury.
Potential Benefits of Oral N-type Calcium Channel Blockers. Limitations with existing pain therapies may be addressed by our N-type calcium channel blocker product candidates. As a small molecule, orally available N-type calcium channel blockers have the potential to improve:
| • | Efficacy. Despite the availability of many drugs to treat chronic pain, results of a 2006 survey by the American Pain Foundation of chronic pain sufferers found that 51% of the respondents felt that they had little or no control over their pain. For some patients, pain may not be under control due to ineffective medications or the inability to tolerate effective doses of currently available analgesics. Deletion of the N-type channel gene in animals, which has the effect of eliminating N-type calcium channel activity, results in the animals retaining normal acute sensation, yet with limited pain response in reaction to inflammatory or nerve damage. Consistent with this observation, the existing N-type calcium channel blocker, Prialt®, has demonstrated efficacy across a range of severe chronic pain states and has been approved for use in patients whose pain is not relieved by morphine. |
| • | Safety Profile. NSAIDs and opioid analgesics used over an extended period of time may lead to significant side effects, providing the opportunity for the introduction of a new class of analgesics with a more favorable safety profile. We have tested Z160 in human clinical trials in approximately 200 subjects and found it to be well tolerated and to exhibit a good safety profile with no serious drug-related adverse effects. |
| • | Tolerance and Addiction. Patients often require increasing doses of opioids to maintain effective pain relief, commonly referred to as tolerance. Patients who abruptly stop using opioids commonly experience withdrawal symptoms. Some patients using opioids experience euphoria and can progress to addictive behaviors and abuse. Unlike the currently marketed strong opioids, the N-type blocker Prialt® is not a controlled substance in the United States. |
| • | Drug Delivery. As a small protein, the N-type calcium channel blocker Prialt®, must be administered using a pump implanted in the patient’s spine. Our N-type calcium channel blockers are all small organic molecules that are expected to be orally administered using tablet and capsule formulations. |
11
We believe that oral N-type calcium channel blockers such as Z160 have the potential to become a new class of analgesics.
T-type Calcium Channel Program and Z944
Our T-type calcium channel program is focused on developing drug candidates to treat a variety of chronic pain conditions. Our orally-administered T-type calcium channel blockers have shown efficacy in animal models of inflammatory pain. Z944, a novel oral T-type calcium channel blocker that has demonstrated preclinical efficacy in multiple inflammatory pain models, has been selected to move into IND-enabling toxicology studies in 2011 with the goal of initiating Phase 1 clinical development in 2011.
T-type Background. The wide distribution of T-type calcium channels found in brain, heart, endocrine cells and other tissues provides for the possibility of developing therapeutics for multiple indications, including treatment of pain or epilepsy. We have identified and cloned three proprietary gene targets for T-type calcium channels that we believe have therapeutic potential for pain. Blocking T-type calcium channels in animal models has been shown to produce relief of acute and chronic pain from mild to moderate intensities. Our research indicates that blocking a specific T-type calcium channel subtype represents an attractive potential therapeutic target for pain intervention. We have discovered proprietary pre-clinical T-type calcium channel blockers such as Z944 that show efficacy in animal models of inflammatory pain.
Potential Benefits of Oral T-type Calcium Channel Blockers. Limitations with existing pain therapies may be addressed by our T-type calcium channel blocker product candidates. The efficacy, safety, tolerance and addiction benefits of T-type calcium channel blockers are expected to be similar to those of the N-type calcium channel blockers described above. Our T-type calcium channel blockers also have the potential to improve:
| • | Efficacy. The inflammatory pain market currently served by NSAIDs and COX2 inhibitors approaches approximately $4.0 billion for mild to moderate pain; however, NSAIDs are often insufficient for acute and chronic pain and are associated with gastro-intestinal and cardiovascular side effects. The COX2 market has contracted due to the cardiovascular safety concerns that lead to the market withdrawal of Vioxx™. Our T-type calcium channel blockers, such as Z944, have shown efficacy in chronic inflammatory pain models providing a novel approach for the treatment of inflammatory pain as an alternative to the NSAIDs. In addition, T-type calcium channels may have utility in other therapeutic indications such as epilepsy. |
| • | Safety Profile. Our T-type calcium channel blockers have been profiled for target and off-target affinities. We believe that the state-dependent selectivity toward the T-type channel is advantageous, providing the opportunity for the introduction of a new class of analgesics with a more favorable safety profile. |
| • | Drug Delivery. Our T-type calcium channel blockers are all small organic molecules that are expected to be orally administered using tablet and capsule formulations. |
Overview of Sodium Channel Biology
In addition to the N-type and T-type calcium channels being targeted for pain, we have been actively pursuing another class of ion channel blockers for pain drug discovery and therapeutic intervention. Sodium channels regulate pain signaling by mediating sodium ion currents that contribute to the excitability of both peripheral pain-sensing neurons and also neurons within the spinal cord that relay pain signals to the brain. Several distinct types of sodium channels are important for setting the threshold and influencing the frequency, sustainability and intensity of pain signaling.
Of the ten sodium channel genes found in humans, the Nav1.7 and Nav1.8 types are of particular interest related to multiple chronic pain conditions as they are validated in humans. For example, naturally occurring
12
loss-of-function genetic mutations in the Nav1.7 channel lead to the complete absence of pain sensation while other gain-of-function Nav1.7 mutations cause severe chronic pain syndromes. Further, in animal models it has been shown that the suppression of either Nav1.7 or Nav1.8 channels reduces various kinds of acute and neuropathic pain.
Sodium Channel Program
Addressing these attractive targets for pain intervention, we have utilized our considerable expertise in ion channels to develop a research and development program to design, characterize and develop novel, orally available agents that block the functioning of sodium channels. The goal is to specifically target the increased firing and hypersensitivity in peripheral and spinal cord neurons that express Nav1.7 and Nav1.8 and that are associated with chronic inflammatory and neuropathic pain.
We have generated a pipeline of preclinical agents shown to affect sodium channels. A number of these new compounds have been found to both reduce the excitability of neurons and to reverse pain hypersensitivity in animal models of acute and neuropathic pain. The Nav1.7 and Nav1.8 blocking compound Z212 uniquely acts by preferentially attenuating hyperexcitable neurons while largely sparing normally firing neurons. We hope that targeting sodium channels in the peripheral and central nociceptive signaling pathways through a unique mechanism of action has the potential to lead to novel classes of safe and effective pain therapeutics. We plan to advance Z212 into further preclinical and toxicology studies and to evaluate our other sodium channel compounds for potential clinical candidacy.
We believe that the targeting of selective sodium channels both complements the existing N-type and T-type calcium channel pain programs and by further integrating many preclinical aspects of the programs, provides for cost-effective, value-added synergies to our drug discovery platform.
Our cHTS Drug Discovery Technology
Combination approaches are an emerging clinical standard in the treatment of cancer, infectious diseases, inflammatory diseases and pain. We believe that our combination high throughput screening technology platform, or cHTS™, has been validated clinically by the identification of unique synergies underlying our most advanced clinical product candidate, Synavive, and through our research collaborations with Novartis and Amgen, who have adopted cHTS as an important addition to their oncology discovery efforts.
The cHTS platform technology profiles the activity of individual agents and combinations in cell-based phenotypic assays at high throughput to discover synergistic multi-target drug candidates. This technology generates novel intellectual property and expands development pipelines. We seek strategic partnerships to leverage the power of the cHTS technology platform in the areas of cancer, inflammation and infectious disease.
Our proprietary cHTS technology platform provides a solution to quantitatively assess how drugs or other probes of biological targets interact with diverse genetic backgrounds using cell-based phenotypic screening assays. This technology offers collaboration partners a high-throughput capability to identify synergistic interactions between small-molecule drugs, therapeutic antibodies and proteins, or siRNAs that can be developed as therapeutic candidates. Combinations are now emerging as the standard of care in indications such as cancer, inflammation, diabetes, and infectious disease. Unfortunately, the standard approach of developing combination therapies at the clinical stage misses the opportunity to identify novel multi-target mechanisms that better address the systems biology of disease and the emergence of resistance. In addition to the ability to identify synergistic multi-target drug candidates that generate novel intellectual property and expand development pipelines, our cHTS technology can also be deployed to define genetic determinants of chemical and multi-target drug sensitivity to identify likely responder populations and companion diagnostics. This approach provides collaboration partners with an opportunity to optimize the value of their existing product candidates, and improve
13
their return on investment in less ideal candidates, by identifying synergistic combinations that amplify drug activity and potency within an indication, expand therapeutic utility into new indications, and extend patent estates through new compositions and use.
We also receive interest from collaboration partners to profile how their drugs or clinical candidates interact with standard-of-care agents in particular indications. Most recently we have engaged with multiple large pharmaceutical companies to characterize the activity of very large sets of combinations across multiple cancer and tissue types to better understand how various drugs and combinations of interest interact with diverse cellular networks. We expect this type of collaboration leveraging the power of the cHTS technology to produce future value for us and our strategic partners.
Our proprietary software solution, Chalice™, was developed by us to enable the visualization and analysis of combination activity profiling data. This software provides a unique and generalized capability to assemble combination test data into matrices for visualization and analysis, and has utility across diverse therapeutic indications. The platform requires only test article identification, concentration, and data measurable information to produce a rich set of combination results suitable for the analysis of a wide variety of data types including: biochemical assays (enzyme inhibition, gene expression, etc.), cell-based assays (proliferation, apoptosis, etc.), and in vivo biomarker or activity assays. In addition, test article and experimental data attributes can be utilized to visualize combination activity profiles to facilitate meta-analyses and drive hypothesis generation. For example, visualizing combination synergy measures with the oncogenic status of cell-lines tested may suggest possible predictors of response that could then be validated. The Chalice software works with the cHTS technology platform to extract valuable insights, discoveries, and data trends from high-throughput combination activity profiling experiments.
Collaborations
We intend to continue to seek collaborations with pharmaceutical and biotechnology companies to support the development and commercialization of our product candidates and to obtain access to additional development, commercial or financial resources. We also plan to continue to engage in selected discovery research collaborations to utilize our proprietary discovery research platforms to discover new product candidates for ourselves or our collaborators and to generate ongoing revenue.
Active Collaborations
Mallinckrodt Inc., a subsidiary of Covidien plc
In June 2009, prior to the merger with us, Neuromed entered into an asset purchase agreement with Mallinckrodt, a subsidiary of Covidien, to sell all of the tangible and intangible assets associated with Exalgo, including the rights to develop and commercialize the product candidate in the United States. As part of the agreement, Neuromed received upfront and initial milestone payments of $15.0 million. Upon approval of Exalgo by the FDA on March 1, 2010, we received an additional milestone payment of $40.0 million, and we are eligible to receive tiered royalties on Covidien’s net sales of Exalgo. Covidien will continue to pay these royalties on net sales for as long as it is selling Exalgo, although the royalty rate will be reduced upon the earlier to occur of generic competition or June 11, 2024.
We have also entered into a development and transition services agreement with Covidien, pursuant to which we have performed certain clinical development and regulatory activities relating to the FDA approval of Exalgo. These activities are at Covidien’s cost and expense, capped at $16.0 million. Through December 31, 2010, we have received $8.3 million in funding and expense reimbursement under this agreement. The development and transition services agreement terminates when the tasks required to be completed by us under the approved development plan are complete. The development and transition services agreement may be terminated by either party after thirty days’ notice or upon an unremedied material breach.
14
Novartis
In May 2009, we entered into a research collaboration and license agreement with Novartis focused on the discovery of novel anti-cancer combinations. Through the collaboration, we are using our proprietary cHTS platform to screen a unique library of molecules, including Novartis compounds, in multiple cell lines representing a broad spectrum of cancers to potentially discover novel single agent and combination therapies to treat various cancers.
Under the terms of the collaboration agreement, we received an initial payment of $4.0 million and will receive annual research support payments of up to $3.0 million, plus certain expenses. In addition, the collaboration agreement may provide us with up to $58.0 million for each combination product candidate advanced by Novartis upon achievement of certain clinical, regulatory and commercial milestones. The research program had an initial two-year term that could be extended by Novartis for three additional one-year periods. In January 2011, Novartis elected to extend the research program for an additional year, through May 2012. We also entered into a software license agreement with Novartis, where we provided Novartis with a non-exclusive license to use our proprietary Chalice analyzer software in connection with the collaboration and other Novartis research programs for approximately five years.
The library to be screened under the collaboration consists of certain Novartis oncology compounds and compounds from our library of approved drugs and other molecules. Novartis will own and have an exclusive license to intellectual property generated under the collaboration to research, develop and commercialize their approved or active development-stage compounds. We will own and have an exclusive license to intellectual property generated under the collaboration to research, develop and commercialize compounds from our library. Intellectual property generated under the collaboration using certain compounds from the Novartis library will be jointly owned by Novartis and us and non-exclusively licensed to allow each party to research, develop and commercialize product candidates. Under the collaboration agreement, Novartis retains an option, exercisable once per year of the research collaboration, to exclusively license a portion of this jointly owned intellectual property if certain conditions are met. Novartis also has a right of first negotiation to exclusively license the intellectual property owned by us that was discovered as a part of the collaboration, under terms to be negotiated by the parties at such time.
The collaboration agreement may be terminated by either party after ninety days’ notice upon an unremedied material breach and upon thirty days’ notice in the event of bankruptcy of the other party. Novartis may terminate the collaboration agreement after sixty days’ notice in the event of a change in control or liquidation of us, as defined in the collaboration agreement.
Fovea Pharmaceuticals SA, a subsidiary of Sanofi Aventis
On January 30, 2006, we entered into a research and license agreement with Fovea Pharmaceuticals SA, or Fovea. Under the terms of the agreement, Fovea agreed to conduct, at its own expense, preclinical and clinical development of combination drug candidates it selected from our portfolio of product candidates for certain ophthalmic indications, including creating ophthalmic formulations for these selected drug candidates. Fovea was acquired by Sanofi Aventis in October 2009.
On July 22, 2009, we amended and restated the agreement with Fovea. Under the amended and restated agreement, we have granted Fovea an exclusive worldwide license to certain drug combinations to treat allergic and inflammatory diseases of the front of the eye. Fovea has advanced one such combination, Prednisporin (FOV1101), through Phase 2b clinical development for allergic conjunctivitis. Prednisporin (FOV1101) is a topical ocular drug candidate, containing low doses of the glucocorticoid, prednisolone acetate and the immunosuppressant cyclosporine A. Fovea has developed a proprietary co-formulation of Prednisporin and is seeking to develop Prednisporin to treat inflammatory ocular diseases such as allergic conjunctivitis. Under our agreement with Fovea, they control all aspects of the clinical development of Prednisporin. For Prednisporin, we
15
have received payments totaling $1.5 million, and are eligible to receive up to $24.0 million in additional development and regulatory milestone payments for each combination successfully developed by Fovea, an additional $15.0 million milestone payment for the approval of a combination in a specified additional indication. We are also eligible to receive royalties for each product commercialized by Fovea in connection with the agreement.
The agreement has no definite term; however, Fovea’s royalty payment obligations terminate on the later of 15 years from the date of the first commercial sale of an exclusively licensed combination and the expiration of all patents covering a royalty bearing product under the license agreement, each on a country-by-country basis. The agreement may be terminated by either party upon an unremedied material breach. In addition, if Fovea fails to develop a product candidate it selects pursuant to specified diligence milestones, after discussions between the parties, the agreement may be terminated by us. We may terminate the agreement if Fovea fails to make required undisputed payments and either party may terminate the agreement upon the insolvency of the other party.
Amgen Inc.
In December 2009, we entered into a Research Collaboration Agreement with Amgen, focused on identifying synergistic combinations for two oncology targets of interest to Amgen. Under the Pilot Agreement, we received a $750,000 payment in January 2010 to fund the initial research plan, and Amgen also agreed to reimburse us for laboratory supplies consumed. The initial research plan ended in September 2010, and Amgen elected for us to do follow-up research at an annual rate of $300,000 per full-time employee equivalent, plus the reimbursement of laboratory supplies. Amgen will also pay us a $1.0 million milestone payment for each Investigational New Drug application filing by Amgen for a product candidate with new intellectual property generated by the collaboration. The agreement also provides Amgen with a non-exclusive license to use our Chalice analyzer software in connection with the research collaboration. Through December 31, 2010, we have received $1.0 million in funding and expense reimbursement under this agreement.
USAMRIID
In December 2008, we entered into a cooperative research and development agreement with the United States Army Medical Research Institute for Infectious Diseases, or USAMRIID, focused on discovering agents to prevent or treat Ebola, Marburg and Lassa virus infections. Under the agreement, which expires in September 2011, we and USAMRIID are undertaking a joint research project, and we are eligible to receive up to approximately $1.4 million in funding. In May 2010, Zalicus entered into a cooperative research and development agreement with USAMRIID focused on discovering agents to prevent or treat Alphavirus infections, which are mosquito borne viruses whose infection can cause severe and fatal encephalitis in humans. Under the agreement, which expires in June 2012, we and USAMRIID are undertaking a joint research project, and we are eligible to receive up to approximately $1.1 million in funding. Through December 31, 2010, we have received approximately $1.2 million in funding from these agreements with USAMRIID.
Previous Collaborations
Merck & Co., Inc.
In March 2006, Neuromed entered into a research collaboration and license agreement with Merck for the research, development and commercialization of N-type calcium channel blockers. Under the terms of the agreement, Merck made a $25.0 million initial cash payment to Neuromed, and Neuromed also received research funding and milestones under the agreement. The Merck agreement was terminated in September 2009, and the rights to the previously licensed intellectual property and product candidates were returned to Neuromed prior to the merger with us.
16
Angiotech Pharmaceuticals, Inc.
In October 2005, we entered into a research and license agreement with Angiotech Pharmaceuticals, Inc., or Angiotech, under which we granted Angiotech an exclusive, royalty-bearing license to up to ten compounds to be selected by Angiotech from our portfolio of clinical and preclinical product candidates or Chalice database. This license was for Angiotech’s research, development and potential commercialization of the licensed compounds as drug components to be used with medical devices or interventional medicine products to treat conditions in specific areas of the human body. In addition, we agreed to use our cHTS technology in a joint research project with Angiotech to screen different disease-specific assay combinations of compounds to be developed and commercialized by Angiotech for use in combination with medical devices or with interventional medicine products in Angiotech’s field.
Under the research and license agreement, Angiotech paid us a $27.0 million up-front license execution fee, and on June 8, 2007, Angiotech agreed to extend the research project beyond the original 30-month term to a total term of five years and paid an additional license execution fee of $7.0 million. On November 10, 2009, we and Angiotech mutually agreed to terminate the research and license agreement.
CHDI, Inc.
In August 2005, we entered into a research agreement with CHDI, Inc., or CHDI, a foundation aimed at preventing and treating Huntington’s disease, to perform joint research and development to discover and perform preclinical development of product candidates for the treatment of Huntington’s disease. Under the terms of the research agreement, we received approximately $5.4 million of research and development funding from CHDI. We and CHDI jointly owned the intellectual property covering product candidates discovered in the collaboration. Joint research and development activities under the research agreement were completed in December 2008, and we assigned our joint ownership interest in the intellectual property from the research project to CHDI.
Cystic Fibrosis Foundation Therapeutics
On May 31, 2006, we entered into a research, development and commercialization agreement with Cystic Fibrosis Foundation Therapeutics Incorporated, or CFFT, the nonprofit drug discovery and development affiliate of the Cystic Fibrosis Foundation, to discover and develop novel therapeutics built from synergistic drug combinations to treat cystic fibrosis. Under the terms of the agreement, CFFT awarded us approximately $7.4 million in research funding and expenses during the term of the research and development project. On May 14, 2009, we and CFFT mutually agreed to end the cystic fibrosis research program being conducted under the research, development and commercialization agreement, effective August 15, 2009.
The DMD Foundations
In November 2007, we entered into a sponsored research collaboration agreement with an entity formed by Charley’s Fund and the Nash Avery Foundation, two nonprofit organizations founded to support Duchenne Muscular Dystrophy, or DMD, research. In October 2008, GMT Charitable Research, LLC, an affiliate of a charitable organization focused on finding therapies for DMD joined the sponsored research agreement. Under the agreement with these DMD foundations, we were seeking to identify novel disease-modifying multi-targeted treatments for DMD, the most common childhood form of muscular dystrophy. Under the terms of the agreement, we received approximately $3.45 million in research funding and reimbursement of additional expenses during the term of the DMD research and development project. The research and development collaboration for DMD expired on December 31, 2009. The DMD Foundations have exercised their rights to retain the intellectual property developed under the collaboration in the field of DMD.
17
PGxHealth, a subsidiary of Clinical Data, Inc.
In August 2009, we and PGxHealth, LLC, or PGx, a subsidiary of Clinical Data, Inc., entered into a collaboration agreement relating to the potential development of ATL313, an adenosine A2A receptor agonist compound owned by PGx, as a combination therapy in the cancer field. We had used our cHTS technology to discover that adenosine A2A agonists synergize with existing and emerging standard-of-care drugs for the treatment of multiple myeloma and certain other B-cell malignancies, which supports the rationale for employing a systematic combination screening approach to oncology drug discovery. Under the terms of the collaboration agreement, which included cross licenses to the other party’s intellectual property, we funded and advanced the preclinical development of ATL313 as a combination therapy in the cancer field. As part of our focus on developing product candidates for pain and inflammation, on March 1, 2011, we provided notice of termination of the collaboration agreement and our associated license to ATL313.
NIAID
In April 2005, we were awarded an approximately $4.4 million research grant from the National Institutes of Allergy and Infectious Diseases, or NIAID, which was payable over five years to perform research and preclinical development in the area of bioterrorism defense. Through December 31, 2010, we have received approximately $3.5 million in funding under this grant.
Patents and Other Proprietary Rights
Our success depends in part on our ability and the ability of our collaborators and licensors to obtain and maintain intellectual property protection for their drug candidates, technology and know-how, to operate without infringing the proprietary rights of others and to prevent others from infringing on our proprietary rights. Our policy is to seek to protect our proprietary chemical compounds, combination drug candidates and technologies by, among other methods, filing United States and foreign patent applications related to our proprietary technology, inventions and improvements that are important to the development of our business. We also rely on trade secrets, know-how, continuing technological innovation and in-licensing opportunities to develop and maintain our proprietary position. We file patent applications for the proprietary technology that we consider important to our business, directed primarily to compounds, compositions and our methods of use as drug candidates.
As of March 4, 2011, our patent estate, on a worldwide basis, includes 115 issued patents and approximately 173 pending patent applications, with claims covering all of our current clinical stage product candidates and select preclinical and research programs. Of the 115 issued patents, 36 are issued in the United States. Of the 173 pending patent applications, 28 are United States non-provisional applications and 7 are United States provisional applications.
Exalgo is covered by United States patents related to formulations, including oral osmotic formulations, of hydromorphone, all of which will expire on July 7, 2014. On October 29, 2010, Covidien received a Paragraph IV Certification Notice Letter from Watson Laboratories, Inc.—Florida, a subsidiary of Watson Pharmaceuticals, Inc., notifying them that it had submitted an Abbreviated New Drug Application, or ANDA, to the FDA for approval to market a generic version of hydromorphone, the active pharmaceutical ingredient in Exalgo, prior to the expiration of U.S. Patent No. 5,914,131, which expires on July 7, 2014, and is listed in the Orange Book. The notice letter further stated that Watson is asserting that claims in U.S. Patent No. 5,914,131 are not infringed and/or are invalid. On December 10, 2010, Covidien’s Mallinckrodt Inc. subsidiary filed a patent infringement lawsuit against Watson Laboratories, Inc.—Florida, Watson Pharma, Inc. and Watson Pharmaceuticals, Inc., in response to the ANDA filing, which we refer to as the Watson litigation. The complaint, which was filed in the United States District Court for the District of New Jersey, alleges infringement of the referenced patent. Under current United States law, the filing of the lawsuit automatically prevents the FDA from approving the ANDA for 30 months from Covidien’s receipt of Watson’s Paragraph IV notification letter on October 29, 2010, or until April 29, 2013, unless the court enters judgment in favor of Watson in less than
18
30 months or finds that a party has failed to cooperate reasonably to expedite the lawsuit. In the lawsuit, the court may find the patents that are the subject of the notice letter invalid, not infringed and/or unenforceable. This could lead to the approval of a generic version of Exalgo prior to the expiration of the 5,914,131 patent on July 7, 2014, which could result in reduced sales of Exalgo and a reduction in the royalties payable to us by Covidien on sales of Exalgo.
One issued United States patent, which expires in October 2022, covers the method of use of Synavive to treat certain immuno-inflammatory diseases, including rheumatoid arthritis. Another patent, issued in multiple countries in Europe which expires in October 2022, covers the pharmaceutical composition and method of use of Synavive to treat certain immuno-inflammatory diseases, including rheumatoid arthritis. We have also received a notice of allowance for a United States patent application covering the pharmaceutical composition of Synavive, which, if issued, would expire no earlier than October 2022. The actual expiration date of this patent would be determined by the United States Patent and Trademark Office at the time of issuance in accordance with the patent term extension rules. We also have pending United States patent applications relating to Synavive which, if issued as patents, would be expected to expire between 2024 and 2029. These applications include claims covering the pharmaceutical composition, other methods of use and formulation of Synavive.
Two pending United States patent applications that relate to Prednisporin are licensed to Fovea, and if these applications result in patents, they would be expected to expire in 2026. These applications include claims covering the pharmaceutical composition and methods of use of Prednisporin in certain ophthalmic diseases.
For our combination product candidates such as Synavive or Prednisporin, it is our current practice to seek the issuance of extensive claims in our patent applications directed to the following:
| • | pharmaceutical compositions comprising the active pharmaceutical ingredients in the combination; |
| • | pharmaceutical compositions comprising structural, functional, or mechanistic analogs of the active pharmaceutical ingredients in the combination; |
| • | methods of treating diseases by administering the active pharmaceutical ingredients in the combination or their analogs; |
| • | pharmaceutical compositions or kits or packages, including the active pharmaceutical ingredients in the combination or their analogs and instructions for the treatment of diseases; and |
| • | compositions and methods of use for formulations, preferred routes of administration, dosages and other properties for our more advanced product candidates. |
To maximize the protection and potential value of our intellectual property relating to ion channels, we are building a portfolio of issued and pending patents, which currently include 164 issued and pending patents, that cover broad and specific claims for new chemical entities as well as research tools and targets. Issued and pending patents cover the following areas: (a) composition of matter utilizing the core chemistry backbone as well as the specific chemistry of many compounds; (b) use of such compounds in certain ion channel related disorders such as pain; (c) novel gene sequences; and (d) assays and research techniques.
Seven issued United States patents, which expire in 2018, cover the method of use and pharmaceutical composition of our N-type calcium channel blocker Z160, and its chemical analogs. We also have pending United States patent applications relating to Z160 which, if issued as patents, would be expected to expire between 2018 and 2032. These applications include claims covering the methods of use and formulation of Z160.
Four additional United States patents, which expire between 2018 and 2024, cover the methods of use and pharmaceutical composition of our N-type calcium channel blockers. We also have pending four United States patent applications relating to our N-type calcium channel blockers which, if issued as patents, would be expected to expire between 2027 and 2031. These applications include claims covering the pharmaceutical
19
composition and methods of use for our N-type calcium channel blockers. Five issued United States patents, which expire between 2018 and 2024, cover the methods of use and pharmaceutical composition of our T-type calcium channel blockers. We also have pending six United States patent applications relating to our T-type calcium channel blockers which, if issued as patents, would be expected to expire between 2028 and 2031. These applications include claims covering the pharmaceutical composition and methods of use for our T-type calcium channel blockers.
Eight additional issued United States patents, which expire between 2018 and 2026, cover the methods of use and pharmaceutical composition of our mixed N and T-type calcium channel blockers. We also have pending six United States patent applications relating to our mixed N and T-type calcium channel blockers which, if issued as patents, would be expected to expire between 2026 and 2031. These applications include claims covering the pharmaceutical composition and methods of use for our mixed N and T-type calcium channel blockers.
Four pending United States patent applications relate to our sodium channel blockers which, if issued as patents, would be expected to expire between 2030 and 2032. These applications include claims covering the pharmaceutical composition and methods of use for our sodium channel blockers.
In addition to seeking patent protection in the United States, we generally file patent applications in European countries, Canada, Japan and additional foreign countries on a selective basis in order to further protect the inventions that we or our collaboration partners consider important to the development of our potential foreign business. As we develop novel formulations of our product candidates and learn more about the most promising dose ratios, pharmacokinetic and pharmacodynamic parameters and mechanism of action information for our drug candidates, we intend to file additional patent applications to augment the core composition of matter and method of use patents we have been issued and are currently seeking.
In all of our activities, we rely on proprietary materials and information, trade secrets, and know-how to conduct research and development activities and to attract and retain collaborative partners, licensees, and customers. our attempts to protect our trade secrets by entering into confidentiality agreements with third parties, employees, and consultants. Our employees and consultants are also asked to sign agreements requiring that they assign to us their interests in patents and other intellectual property arising from their work for us. We also require all employees to sign an agreement not to engage in any conflicting employment or activity during their employment with us, and not to disclose or misuse our confidential information.
In certain instances, a patent term can be extended to recapture a portion of the term effectively lost as a result of the FDA regulatory review period. The duration of foreign patents varies in accordance with provisions of applicable local law, but typically is 20 years from the earliest effective filing date. Our patent estate, based on patents existing now and patents we expect will be issued based on pending applications, will expire on dates ranging from 2018 to 2032.
The actual protection afforded our product candidates by a patent varies from country to country and depends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patent.
Government Regulation
The FDA and comparable regulatory authorities in other countries impose substantial requirements upon companies involved in the clinical development, manufacture, marketing and distribution of drugs. These agencies and other federal, state, provincial and local entities regulate research and development activities and the testing, manufacturing, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion, and export and import of pharmaceutical products such as those that we are developing. In addition, companies developing or commercializing Controlled Substances such as Exalgo face regulations above and beyond other pharmaceutical companies.
20
United States Food and Drug Administration Approval Process
United States Government Regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, and the agency’s implementing regulations. If we fail to comply with the applicable United States requirements at any time during the product development process, clinical testing, and the approval process or after approval, we, our product candidates or, once approved, our products may become subject to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, license suspension or revocation, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, civil penalties or criminal prosecution. Any FDA enforcement action could have a material adverse effect on us.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| • | completion of extensive pre-clinical laboratory tests, pre-clinical animal studies and formulation studies all performed in accordance with the FDA’s good laboratory practice regulations; |
| • | submission to the FDA’s Center for Drug Evaluation and Research, or CDER, of an investigational new drug application, or IND, which must become effective before human clinical trials may begin. The IND contains the plan for the clinical trial. CDER specialists carefully review the IND to determine whether there are any flaws in the initial studies and whether the overall development plan is feasible and reasonably designed to minimize health risks to the participants; |
| • | performance of adequate and well-controlled clinical trials to establish the safety and efficacy of the product candidate for each proposed indication; |
| • | submission to the FDA of a new drug application, or NDA; |
| • | for Controlled Substances such as Exalgo, a risk management plan describing the sponsor’s plan for minimizing the risk of abuse of its product is required; |
| • | the FDA may require compliance with risk evaluation and mitigation strategies, or REMS; |
| • | the FDA may request feedback by an Advisory Committee on the application and ask for their advice regarding approval, risk/benefit, risk management, and/or specific labeling claims. FDA is not obligated to follow recommendations of an Advisory Committee, but may consider such advice prior to completion of its own internal decision making; |
| • | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product candidate is produced to assess compliance with current good manufacturing practice regulations; and |
| • | FDA review and approval of the NDA before any commercial marketing, sale or shipment of the product. |
In addition, the Drug Enforcement Administration, or DEA, plays an important role in the scheduling of medications and will be particularly involved in the commercialization of opioid products such as Exalgo. The testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals of any product candidate we may develop will be granted by the FDA or the DEA on a timely basis, if at all.
Pre-clinical Tests
Pre-clinical tests include laboratory evaluations of product chemistry, formulation and stability, as well as animal studies to evaluate toxicity. The results of the pre-clinical tests, together with manufacturing information
21
and analytical data, are submitted to the FDA as part of an IND. The IND automatically becomes effective 30 days after receipt by the FDA unless the FDA, within the 30-day time period, raises concerns or questions about the conduct of the clinical trial, such as subjecting human research subjects to unreasonable health risks. In that case, the IND sponsor and the FDA must resolve any outstanding FDA concerns before the clinical trials can begin. Submission of an IND may result in the FDA not allowing the trials to commence or not allowing the trial to commence on the terms originally specified in the IND. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development, and the FDA must grant permission, either explicitly or implicitly (by not objecting), before each clinical trial can begin.
In March 2006, the FDA released Guidance for Industry: Nonclinical Safety Evaluation of Drug Combinations. The guidance discusses what preclinical studies are appropriate to support the clinical study and approval of new combination products and therapies. In the case of new products composed of previously marketed drugs, the guidance states that generally the FDA believes sufficient clinical and preclinical data will exist for each drug component separately. Therefore, in such a case, the issues to be resolved before the new product is tested in humans generally relate to possible interactions between the components of the proposed product. The guidance identifies specific potential interaction issues to be considered and suggests the type of testing that may be appropriate to resolve any issues that require such testing.
Clinical Trials
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing, among other things, the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND. An independent Institutional Review Board, or IRB, for each medical center proposing to conduct a clinical trial must also review and approve a plan for any clinical trial before it can begin at that center and the IRB must monitor the trial until it is completed. The FDA, the IRB or the sponsor may suspend or discontinue a clinical trial at any time for various reasons, including a finding that the subjects are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive good clinical practice requirements and the requirements for informed consent.
During the conduct of a clinical trial, a company is required to monitor the investigators’ compliance with the clinical study protocol and other FDA requirements, including the requirements to submit reports to the sponsor, the IRB, and the FDA, and to keep detailed records regarding study findings and use and disposition of the study drug. Although monitoring can help reduce the risk of inadequate compliance by study investigators, it cannot eliminate this risk entirely. Inadvertent regulatory noncompliance by the investigator, or intentional investigator misconduct, can jeopardize the usefulness of study results and, in rare circumstances, require a company to repeat a study. A company must report to the FDA any adverse event that is both unexpected and serious and for which there is a reasonable possibility that the event may have been caused by the investigational drug. In addition, a company must within seven days report to the FDA any unexpected fatal or life-threatening event that may have been caused by the drug. The FDA may stop the trials by placing a “clinical hold” on such trials because of concerns about the safety of the product being tested. Such holds can cause substantial delay and in some cases may require abandonment of a product candidate.
22
For the purposes of an NDA submission and approval, clinical trials are typically conducted in the following four sequential phases, which may overlap:
| Phase 1 | Trials are initially conducted with relatively few subjects to test the drug candidate for safety, dosage tolerance, absorption, metabolism, distribution and excretion in healthy humans, or, on occasion, in patients with the disease or condition under trial to gain an early indication of its effectiveness or tolerance in a patient population. | |
| Phase 2 | Trials are generally conducted with a relatively small number of subjects to: | |
| – evaluate dosage tolerance and appropriate dosage; | ||
| – identify possible adverse effects and safety risks; and | ||
| – evaluate the efficacy of the drug for specific indications in patients with the disease or condition under trial. | ||
| Phase 3 | Trials, commonly referred to as “pivotal” or “registration” trials, are typically conducted when phase 2 clinical trials demonstrate that a dose range of the drug candidate is effective and has an acceptable safety profile. Phase 3 clinical trials are undertaken with large numbers of patients (several hundred to several thousand) to further evaluate dosage, to provide substantial evidence of clinical efficacy and to further test for safety in an expanded and diverse patient population at multiple, geographically dispersed clinical trial sites. | |
| Phase 4 | Post-approval trials, to further assess the drug’s safety and effectiveness, are sometimes required by the FDA as a condition of approval. | |
When two or more drugs are combined in a single dosage form, as our product candidate Synavive is, the data submitted to the FDA must ordinarily show that each component makes a contribution to the claimed effects and that the dosage of each component (amount, frequency, duration) is such that the combination is safe and effective for a significant patient population requiring such concurrent therapy as defined in the labeling for the drug. This FDA policy may necessitate more elaborate and expensive clinical trials for combination candidates than would be required for a single-agent pharmaceutical because the trials may need to be designed to study the combined agent, each drug as a single agent and a placebo.
When FDA approval is sought for a new use of a previously approved drug, the sponsor must demonstrate that the drug is safe and effective for the proposed use. However, because pre-existing information on the drug’s safety is available, the safety data required for FDA approval of a previously approved drug is ordinarily less than the safety data required to support approval of a new drug. The FDA may not require us to submit some types of safety data, such as data from certain types of animal and human pharmacokinetic studies for our product candidates that are combinations of previously approved products. The FDA’s specific requirements will be determined on a case-by-case basis for each product candidate. It is possible that our combination product candidates could present new safety issues because the previously approved drugs are being used in combinations or because the proposed combination products are being used under different circumstances than the components are used as single agents. For example, the combination might be proposed for long-term use for a chronic condition while the single agents are used short-term for acute conditions. In such a case, the FDA may require additional animal or human studies to address any safety issues.
Special Protocol Assessment Process
In the United States, certain protocols for clinical trials can be submitted to the FDA for special protocol assessment, or SPA. Under a SPA, the applicant and the FDA reach an agreement on the design and size of the clinical trial. This agreement can be in writing and cannot be changed after the clinical trial begins except: (a) with written agreement of the applicant and the FDA; or (b) if the director of the FDA reviewing division determines that “a substantial scientific issue essential to determining the safety or effectiveness of the drug” was identified after testing began.
23
New Drug Applications
The results of the pre-clinical testing and of the clinical trials, together with other detailed information, including extensive manufacturing information and information on the composition of the product, are submitted to the FDA in the form of an NDA requesting approval to market the product for one or more specified indications. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use.
Submission of a NDA does not assure FDA approval for marketing. After the application is submitted, the FDA initially determines whether all pertinent data and information have been submitted before accepting the application for filing. After the application is accepted for filing, the FDA begins its substantive review. The FDA typically will request a review of the data in the NDA and recommendation regarding approval by an advisory committee consisting of outside experts. The FDA may accept or reject the advisory committee’s recommendations, or accept them with modifications. The application review process generally takes a year or longer to complete, although reviews of drugs that meet a medical need for serious or life-threatening diseases may be accelerated or prioritized for a six-month review. The FDA may deny approval of an application. Any such denial may require extensive additional testing, which could take years to complete, in order to make the application approvable, or the denial may be based on considerations that cannot be favorably resolved through additional testing. In some circumstances, the FDA may approve an application even though some unanswered questions remain about the product, if the applicant agrees to conduct post-marketing studies. The FDA may impose other conditions of approval as well. Expedited or accelerated approvals may require additional larger confirmatory clinical studies to be conducted following approval.
Product approval may be withdrawn if compliance with regulatory requirements is not maintained or if post-marketing adverse events associated with the product are reported that cannot be addressed satisfactorily through changes to the product’s labeling or warnings to healthcare professionals. The FDA requires reporting of certain safety and other information that becomes known to a manufacturer of an approved product. A company may become aware of such information from reports of adverse events suspected to be related to the product, voluntarily provided to the company and/or to the FDA by physicians and other healthcare professionals, or from published scientific data. In some circumstances, the FDA may require the company to make changes to its approved product labeling or to issue safety warnings to healthcare professionals or the public, which may have a negative impact on product sales. In addition, the Amendments Act of 2007 provides the FDA with expanded authority over drug products after approval, including the authority to require post-approval studies and clinical trials, labeling changes based on new safety information, and compliance with risk evaluation and mitigation strategies, or REMS, approved by the FDA. The FDA’s exercise of this authority could result in delays or increased costs during the period of product candidate development, clinical trials and regulatory review and approval, increased costs to assure compliance with new post-approval regulatory requirements, and potential restrictions on the sale of approved products, which could lead to lower product revenues to us or our collaborators. Manufacturing and sales may also be disrupted or delayed in the event of failure to comply with all required current good manufacturing practice, or cGMP, as determined by FDA investigators in periodic inspections of manufacturing facilities. Upon approval, a drug or biological product may only be marketed for the approved indications, in the approved dosage forms, and at the approved dosage. The nature of marketing claims that we will be permitted to make in the labeling and advertising of our products will be limited to those specified in an FDA approval.
Other United States Regulatory Requirements
Any end products manufactured or distributed by us or our collaborators and licensors pursuant to FDA approvals are subject to continuing regulation by the FDA, including recordkeeping and reporting requirements. Adverse event experience with the product must be reported to the FDA in a timely fashion and the FDA may mandate a pro-active search for these adverse effects. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced
24
inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including cGMPs, which impose certain procedural and documentation requirements upon them and their third-party manufacturers. Failure to comply with the statutory and regulatory requirements can subject a manufacturer to possible legal or regulatory action.
In addition to regulations enforced by the FDA, we are also subject to regulation under the Occupational Safety and Health Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act, and other present and potential future federal, state, provincial and local statutes and regulations. Our research and development involves the controlled use of hazardous materials, chemicals, and various radioactive compounds. Although we believe that our safety procedures for storing, handling, using, and disposing of such materials comply with the standards prescribed by applicable regulations, the risk of accidental contaminations or injury from these materials cannot be completely eliminated. In the event of such an accident, we could be held liable for any damages that result, and any such liability could materially affect our ongoing business.
International Regulation
Outside the United States, our ability to market a product is contingent upon receiving marketing authorization from the appropriate regulatory authorities. The requirements governing the conduct of clinical trials, marketing authorization, pricing, and reimbursement vary widely from country to country. At present, foreign marketing authorizations are applied for at a national level, although within the European Union, centralized procedures are available to companies wishing to market a product in more than one European Union member state. If the regulatory authorities are satisfied that adequate evidence of safety, quality, and efficacy has been presented, a marketing authorization will be granted. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
Third Party Reimbursement and Pricing Controls
Sales of pharmaceutical products depend in significant part on the availability of coverage and adequate reimbursement from government and other third-party payers, including the United States Medicare and Medicaid programs. Third-party payers are increasingly challenging the pricing of pharmaceutical products and may not consider our product candidates or those of its collaborators cost-effective or may not provide coverage of, and adequate reimbursement for, any future product candidates, in whole or in part.
Competition
The development and commercialization of pharmaceutical products is highly competitive. We compete against a wide range of pharmaceutical, biotechnology and life science companies that have greater resources than us, including existing research and development programs in the markets we plan to target. We must compete with these companies both in regard to the discovery technology we use to identify potential product candidates and in regard to the development and commercialization of our product candidates themselves.
Exalgo and our other product candidates for pain and inflammation, if approved, will compete with existing proprietary and generic opioid and non-opioid analgesics as well as new molecules, formulations and technologies that may be developed or commercialized in the future for the treatment of pain or inflammatory diseases. Any of these drugs and drug delivery technologies may receive government approval or gain market acceptance more rapidly than our product candidates, may offer therapeutic, safety or cost advantages over Exalgo or our other product candidates or may cure their targeted diseases or their underlying causes completely. As a result, Exalgo or our product candidates, even if they are approved, may become non-competitive or obsolete. In October 2010, Watson Laboratories, Inc.—Florida notified Covidien that it had submitted an ANDA to the FDA for approval to market a generic version of hydromorphone, the active pharmaceutical ingredient in Exalgo, prior to the expiration of United States Patent No. 5,914,131, which expires on July 7, 2014. If Watson
25
Laboratories, Inc.—Florida, or other third parties, are successful in gaining approval to market a generic version of Exalgo prior to the expiration of the 5,914,131 patent on July 7, 2014, Exalgo may face considerable competition which could result in reduced sales of Exalgo and a reduction in the royalties payable to us by Covidien on sales of Exalgo. For a detailed discussion of the Watson litigation, please see “Business—Patents and Other Proprietary Rights.”
In regard to our product candidates, we file patent applications on the composition and use of the drug combinations we discover. If we obtain the patent protection we are seeking for our product candidates, we believe that this will give us the exclusive rights to market products covered by our patents. We also believe that, if obtained, we should be able to use our patents to prevent the makers of either of the drugs included in our combination products from marketing their drug for use together with the other drug that comprises the product. We are also developing or plan to develop, for each of our combination product candidates, a customized formulation that will be selected for its pharmacology, dosage strength, and route of delivery based on the activity and pharmacology of the drugs when delivered together in combination. Where appropriate, we will seek to protect these formulations by patent applications or as trade secrets. We intend to seek regulatory approval for our product candidates as new drugs, and the expense and time involved in seeking regulatory approval for a new drug may deter potential competitors.
Our ability to commercialize our product candidates will be limited to the extent that we are unable to obtain patent protection for our product candidates or patent or trade secret protection for our formulations. Competitors may also be able to use similar component drugs or different combinations of our component drugs to develop combination products that are not covered by our patents. In addition, the approved drugs that are combined to produce our product candidates are likely to be commercially available at lower prices, so physicians may be able to prescribe the individual drugs already approved and marketed by other companies instead of our combination products, and it would be difficult or impossible for us to enforce our patents, if obtained, to prevent this practice.
In addition to potential competition from other combination drugs, all of our product candidates will face competition from single agent pharmaceuticals. The target markets for our product candidates and those of our collaborators, are all very competitive, with existing approved products holding substantial market share and other product candidates being developed by other pharmaceutical or biotechnology companies.
Principal competitive factors impacting drug development and commercialization include:
| • | improved patient outcomes; |
| • | demonstrable safety of product candidates; |
| • | acceptance of products by physicians and other healthcare providers; |
| • | research and drug development capabilities; |
| • | intellectual property positions; |
| • | sales and marketing capabilities; and |
| • | availability of capital resources to fund research, development and commercialization activities. |
In regard to our ion channel discovery platform, we protect our trade secrets around our assays and experimental techniques in order to give us a competitive advantage in discovering compounds that modulate ion channels. Many large and small companies have sought to discover and develop ion channel modulators for the treatment of pain and other diseases, and may be more successful in discovering ion channel modulators using similar or different techniques.
In regard to our cHTS discovery technology and Chalice analyzer software, we protect our trade secrets in order to give us the exclusive right to use our technologies. Many companies have already developed and employ high throughput screening technologies. Should these companies seek to apply these technologies to the discovery of combination drugs, our drug discovery technology may be rendered obsolete or noncompetitive.
26
Many of the companies competing against us have financial and other resources substantially greater than our own. In addition, many of our competitors have significantly greater experience in clinical testing, obtaining FDA and other regulatory approvals and in the manufacture and commercialization of products.
Manufacturing
Covidien is responsible for arranging for the manufacturing of Exalgo. We have no manufacturing capabilities. We rely and plan to continue to rely on third parties to manufacture our compounds and product candidates for research, development, preclinical and clinical trials. We believe that there are several manufacturing sources available to us on commercially reasonable terms to meet our preclinical and clinical requirements.
We plan to rely on third parties to manufacture commercial quantities of products we successfully develop, if any. Among the conditions for FDA or other regulatory approval of a pharmaceutical product is the requirement that the manufacturer’s quality control and manufacturing procedures conform to cGMP, which must be followed at all times. The FDA typically inspects manufacturing facilities every two years, and other regulators inspect manufacturing facilities as well. In complying with cGMP regulations, pharmaceutical manufacturers must expend resources and time to ensure compliance with product specifications as well as production, record keeping, quality control, reporting, and other requirements. We plan to seek suitable third-party manufacturing arrangements for the commercial production of a product candidate.
Employees
As of March 8, 2011, we employed 58 persons. A total of 21 of our employees hold Ph.D. or M.D. degrees. Approximately 44 employees are engaged in research and development, and 14 employees are engaged in business development, intellectual property, finance, human resources, legal, and other administrative functions. Our workforce is non-unionized, and we believe that our relations with employees are good.
Segment and Geographic Information
For additional segment and geographic information, please See “Management’s Discussion and Analysis” under the heading “Overview” and Note 13, “Segment and Geographic Information”, to our consolidated financial statements for information about our operating segments and financial information about geographic areas.
Corporate and Available Information
We were incorporated in Delaware in 2000. Our principal executive offices are located at 245 First Street, Third Floor, Cambridge, Massachusetts 02142. We have two subsidiaries: Zalicus Pharmaceuticals Ltd. and Zalicus Securities Corp. Our Internet website is www.zalicus.com. We make available free of charge through our website our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Securities Exchange Act of 1934, as amended. We have made these reports available through our website at the same time that they become available on the Securities and Exchange Commission’s website. The public may read and copy any materials that we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an Internet site (http://www.sec.gov) that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC.
Our code of conduct and ethics, corporate governance guidelines, and the charters of the Audit Committee, Compensation Committee and Nominating and Corporate Governance Committee are all available on the corporate governance section of our website at www.zalicus.com/investors. Stockholders may request a free copy of any of these documents by writing to Zalicus Inc., 245 First Street, Third Floor, Cambridge, Massachusetts 02142, Attn: General Counsel.
27
“Zalicus”, “cHTS”, “Chalice” and “Synavive” are trademarks of Zalicus or its subsidiaries. Other trademarks, service marks and trade names referred to in this Form 10-K are the property of their respective owners.
| Item 1A. | Risk Factors |
Risks Related to Discovery, Development and Commercialization of Drug Products
Our ability to generate ongoing revenue from Exalgo sales depends on Covidien.
We cannot assure you of the level of sales of Exalgo that Covidien will generate. Covidien has historically marketed generic drugs and Exalgo is one of the few branded drugs it markets. Covidien markets other opioids which may compete with Exalgo. We cannot control whether Covidien will devote sufficient resources to maximize sales of Exalgo and we have no control over the size of the sales force that it will assemble. Similarly, we also cannot control Covidien’s decisions regarding the commercialization of Exalgo, including the launch of Exalgo, subsequent strategic sales initiatives, any decision to scale back its sales initiatives or withdraw Exalgo from the market for any reason, including because Covidien wants to increase sales of a competitive product at Exalgo’s expense. If sales of Exalgo by Covidien are low, the royalties we expect to receive on sales of Exalgo will be low, which, in turn, could have a material adverse effect on our business, financial position and results of operations.
Our royalty revenue from Covidien relating to the net sales of Exalgo could vary significantly in future periods and royalty revenue from Exalgo sales during quarterly periods may not be predictive of future royalty revenue to us from sales of Exalgo.
Our revenue from net sales of Exalgo by Covidien may fluctuate from period to period due to:
| • | the rate of market acceptance of Exalgo as a new therapy by physicians and payors; |
| • | fluctuations in future sales of Exalgo due to competition from other products, including other extended-release opioids; |
| • | reimbursement and pricing for Exalgo under commercial or government plans; |
| • | permitted deductions from gross sales of Exalgo relating to estimates of sales returns, credits and allowances, normal trade and cash discounts, managed care sales rebates and other allocated costs; |
| • | inventory levels of Exalgo with wholesalers not matching actual demand for Exalgo; |
| • | the duration of market exclusivity of Exalgo; |
| • | the timing of approvals, if any, for other competitive products or the future availability of generic versions of branded long-acting opioids; |
| • | manufacturing difficulties; or |
| • | other factors that affect the sales of a pharmaceutical product. |
All of these factors are outside our control, and any of these events may cause our revenues to fluctuate significantly from quarter to quarter, and in some cases may cause our operating results for a particular quarterly or annual period to vary in a material manner.
Covidien may not be able to maintain or protect the proprietary rights necessary for the long-term commercialization of Exalgo, particularly given the Watson litigation described below, which could result in reduced royalty revenue to us from sales of Exalgo by Covidien.
Covidien has obtained rights to a limited number of patents directed toward the OROS formulation of hydromorphone that is used in Exalgo. Third parties may seek to market generic versions of Exalgo by filing an ANDA with the FDA, in which they claim that patents protecting Exalgo that are held by Covidien and listed
28
with the FDA in the Orange Book, are invalid, unenforceable and/or not infringed. This type of ANDA is referred to as a Paragraph IV filing. If an ANDA or 505(b)(2) application containing a Paragraph IV certification is submitted to the FDA and accepted as a reviewable filing by the agency, the ANDA or 505(b)(2) applicant must then, within 20 days, provide notice to the NDA holder and patent owner stating that the application has been submitted and providing the factual and legal basis for the applicant’s opinion that the patent is invalid or not infringed.
On October 29, 2010, Covidien received a Paragraph IV Certification Notice Letter from Watson Laboratories, Inc. – Florida, a subsidiary of Watson Pharmaceuticals, Inc., notifying them that it had submitted an ANDA to the FDA for approval to market a generic version of hydromorphone, the active pharmaceutical ingredient in Exalgo, prior to the expiration of United States Patent No. 5,914,131, which expires on July 7, 2014, and is listed in the Orange Book. The notice letter further stated that Watson is asserting that claims in United States Patent No. 5,914,131 are not infringed and/or are invalid. On December 10, 2010, Covidien’s Mallinckrodt Inc. subsidiary filed a patent infringement lawsuit against Watson Laboratories, Inc.—Florida, Watson Pharma, Inc. and Watson Pharmaceuticals, Inc., in response to the ANDA filing, which we refer to as the Watson litigation. The complaint, which was filed in the United States District Court for the District of New Jersey, alleges infringement of the referenced patent. Under current United States law, the filing of the lawsuit automatically prevents the FDA from approving the ANDA for 30 months from Covidien’s receipt of Watson’s Paragraph IV notification letter on October 29, 2010, or until April 29, 2013, unless the court enters judgment in favor of Watson in less than 30 months or finds that a party has failed to cooperate reasonably to expedite the lawsuit. In the Watson litigation, the court may find the patents that are the subject of the notice letter invalid, not infringed and/or unenforceable. This could lead to the approval of a generic version of Exalgo prior to the expiration of the 5,914,131 patent on July 7, 2014, which could result in reduced sales of Exalgo and a reduction in the royalties payable to us by Covidien on sales of Exalgo. The conduct of the Watson litigation and any decisions regarding litigation strategy or potential settlement are in the complete control of Covidien, and Zalicus has no rights to participate in the Watson litigation or otherwise direct its prosecution or settlement. During the period in which the Watson litigation is pending, the uncertainty of its outcome may cause our stock price to decline. In addition, an adverse result in the Watson litigation, whether appealable or not, will likely cause our stock price to decline and may have a material adverse effect on our results of operations and financial condition. Any final, unappealable, adverse result in the Watson litigation will likely have a material adverse effect on our results of operations and financial condition and cause our stock price to decline. Our ability to continue to generate significant royalty revenues from Exalgo is dependent upon Covidien’s ability to prevail in the Watson litigation or to otherwise resolve the Watson litigation on favorable terms.
Zalicus’s approach to the discovery and development of drugs is unproven and may never lead to commercially viable products.
Zalicus’s various approaches to drug discovery and development using our ion channel technology platform or our cHTS technology platform are complex and unproven. Previously unrecognized or unexpected defects in or limitations to Zalicus’s drug discovery technologies or drug development strategies may emerge, which we may also be unable to overcome or mitigate. None of the product candidates identified or developed to date, through the application of Zalicus’s business model and drug discovery technologies, has been approved by any regulatory agency for commercial sale or been commercialized.
We have committed resources to the development of our ion channel drug discovery technology, and our future success depends in part on the successful development of this technology and product candidates identified by it. The scientific evidence to support the feasibility of developing drugs that modulate ion channels is limited, and many companies with more resources than us have not been able to successfully develop drugs that modulate ion channels.
29
Zalicus’s current business model also involves the ability of Zalicus’s proprietary high throughput discovery technology to identify additional promising product candidates. Combination high throughput screening involves testing large numbers of compounds in cell-based assays using automated systems that measure the biological activity of the compounds and provide detailed data regarding the results. Because Zalicus’s combination high throughput discovery technology is unproven in identifying drugs that can be approved, we cannot be certain that we will be able to discover additional drug combinations that show promising effects in Zalicus’s cell-based disease models and pre-clinical studies, which we can advance into clinical trials. As a result, we may not be able to identify additional product candidates. Many other companies with substantially greater resources than ours use high throughput screening for drug discovery. These or other companies may have developed or could in the future develop combination screening technology equal or superior to Zalicus’s technology. In addition, regulatory approval for a combination drug generally requires clinical trials to compare the activity of each component drug with the combination. As a result, it may be more difficult and costly to obtain regulatory approval of a combination drug than of a new drug containing only a single active pharmaceutical ingredient. In some instances, we may choose to advance product candidates where one or more of the compounds of the combination are not approved drugs, which may lead to longer clinical development timelines for these types of product candidates. For these and other reasons, Zalicus’s approach to drug discovery and development may not be successful and Zalicus’s current business model may not generate viable products or revenue. Even if Zalicus’s approach is theoretically viable, we may not complete the significant research and development or obtain the financial resources and personnel required to further develop and apply Zalicus’s discovery technology, advance promising product candidates to and through clinical trials, and obtain required regulatory approvals.
Our results to date provide only a limited basis for predicting whether any of our product candidates will be safe or effective, or receive regulatory approval.
All of our product candidates are in an early stage of development and their risk of failure is high. The data supporting Zalicus’s drug discovery and development programs is derived from either laboratory and pre-clinical studies and limited early stage clinical trials that were not all designed to be statistically significant or proof-of-concept or Phase 2 clinical trials, some of which are exploratory in nature. We cannot predict when or if any one of Zalicus’s product candidates will prove effective or safe in humans or will receive regulatory approval. If we are unable to discover or successfully develop drugs that are effective and safe in humans, we will not have a viable business.
Exalgo is a member of a class of drugs that may cause undesirable side effects that could limit their marketability.
Opioids, including hydromorphone, the active ingredient in Exalgo, are known to have serious side effects, including dependence and addiction. These or other undesirable side effects could negatively affect sales of Exalgo. In addition, if we or others later identify additional undesirable side effects caused by Exalgo, one or more of the following could occur:
| • | regulatory authorities may require the addition of labeling statements, such as a contraindication or a “black box” warning that the drug carries significant risks of serious or life-threatening adverse effects; |
| • | regulatory authorities may withdraw their approval of Exalgo; |
| • | the FDA may require that the way Exalgo is administered be changed, that additional clinical trials be conducted or that the labeling of the product be changed; and |
| • | our reputation may suffer. |
Any of these events could prevent Exalgo from achieving or maintaining market acceptance or could substantially increase the costs and expenses of commercializing Exalgo, which in turn could decrease the royalties we expect to receive from sales of Exalgo.
30
Exalgo could be tampered with for the purpose of drug abuse. Misuse or abuse of Exalgo may result in adverse regulatory or other actions, including withdrawals of regulatory approvals and litigation.
Misuse or abuse of drugs, including opioids such as hydromorphone, the active ingredient in Exalgo, could result in serious, even fatal, consequences. Abusers of pharmaceutical drugs may misuse or abuse Exalgo, to, among other things, accelerate the release of opioids. If effective methods to misuse or abuse Exalgo are employed, adverse action from regulatory authorities may result, including:
| • | withdrawal of regulatory approvals; |
| • | delays or interruption in commercialization; |
| • | product recalls or seizures; |
| • | suspension of manufacturing; |
| • | withdrawals of previously approved marketing indications; |
| • | injunctions, suspensions, or revocations of marketing licenses; and |
| • | product liability or class action litigation. |
If any of these events were to occur, Covidien may not be able to continue to sell Exalgo. In these circumstances, we could suffer reduced revenues. Further, in response to these circumstances, regulatory authorities may impose new regulations concerning the manufacture and sale of drugs such as Exalgo. Such regulations may include new labeling requirements, additional risk management plan requirements to further minimize the risk of abuse, restrictions on the prescription and sale of Exalgo and mandatory reformulation of products in order to make abuse more difficult. Any such new regulations may be difficult and expensive for Covidien to comply with, may adversely affect Covidien’s sales and may have a material adverse effect on our business and cash flows.
FDA approval of our drug candidates, subjects Zalicus and our collaborators to ongoing FDA obligations and continued regulatory review, such as continued safety reporting requirements, and we and our collaborators may also be subject to additional FDA post-marketing obligations or new regulations, all of which may result in significant expense and limit our ability to commercialize our drug candidates or Covidien’s ability to commercialize Exalgo.
Any regulatory approvals that we receive for our drug candidates, may be subject to limitations on the indicated uses for which the drug may be marketed or contain requirements for potentially costly post-marketing follow-up studies. In addition, the labeling, packaging, adverse event reporting, storage, advertising, promotion and record keeping for Exalgo, or any other drug candidate the FDA may approve, is subject to extensive regulatory requirements. The subsequent discovery of previously unknown problems with a drug, including but not limited to adverse events of unanticipated severity or frequency, or the discovery that adverse events previously observed in pre-clinical research or clinical trials that were believed to be minor actually constitute much more serious problems, may result in restrictions on the marketing of the drug, and could include withdrawal of the drug from the market.
The FDA’s policies may change, and additional government regulations may be enacted that could prevent or delay regulatory approval of our drug candidates or may adversely impact the commercialization of Exalgo. For example, the FDA has commenced the process of developing a common “Risk Evaluation and Mitigation Strategy,” or REMS, for all opioid drugs. According to FDA’s announcement, opioid drugs affected by this announcement include both brand name and generic products that contain various drug components, including hydromorphone, the active ingredient in Exalgo. Exalgo is subject to its own unique REMS program, and may ultimately be subject to the common REMS program for approved opioid drugs. If we are not, or, in the case of Exalgo, Covidien is not, able to maintain regulatory compliance, including compliance with a required REMS program, we or Covidien may be subject to fines, suspension or withdrawal of regulatory approvals, product
31
recalls, seizure of products, operating restrictions and criminal prosecution. Any of these events could prevent us from marketing our drug candidates or prevent Covidien from marketing Exalgo and our business could suffer accordingly. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action, either in the United States or abroad.
The approved drugs included in Zalicus’s combination product candidates, including Synavive, may have their own adverse or unacceptable side effects or may produce adverse or unacceptable side effects when delivered in combination, and we may not be able to achieve an acceptable level of side effect risks, compared to the potential therapeutic benefits, for Zalicus’s combination product candidates.
The approved drugs included in Zalicus’s combination product candidates, including Synavive, have known adverse side effects. These side effects may be acceptable when an ingredient is used in its approved dosage to achieve a therapeutic benefit for its currently approved indications, but the side effect risk compared to the therapeutic benefit may not be acceptable when used for the intended indications for the product candidate. These side effects may also make it difficult to obtain regulatory or other approval to initiate clinical trials of Zalicus’s combination product candidates. In addition, the therapeutic effect of an approved drug in its currently approved indications may be inappropriate or undesirable in the intended indication for Zalicus’s combination product candidate. Also, Zalicus’s discovery technology is not designed to and does not detect adverse side effects that may result from the combination of the two drugs. The adverse side effects of an approved drug may be enhanced when it is combined with the other approved drug in the product candidate or other drugs patients are taking, or the combined drugs in a product candidate may produce additional side effects. Adverse side effects could, in any of these situations, require pre-clinical and Phase 1 studies testing for combination side effects or prevent successful development and commercialization of some or all of Zalicus’s combination product candidates, because the risks may outweigh the therapeutic benefit of the combination.
The development of a product candidate could be adversely affected by safety or efficacy issues that subsequently arise or become the focus of increased attention or publicity regarding use of either of the approved drugs which comprise the product candidate or similar drugs. We could be forced to abandon a product candidate or an approved product due to adverse side effects from long-term or other use of one of the active pharmaceutical ingredients in the product candidate or product, even if such long-term or other use is not contemplated for such product candidate or product.
Zalicus’s combination product candidate Synavive, seeks to increase the therapeutic effect of a reduced-dose of the oral glucocorticoid prednisolone by the combination with dipyridamole that serves as an enhancer agent. The adverse side effects of prednisolone and dipyridamole are significant and generally increase as their dosage and/or duration of therapy increases. As a result, the success of Synavive depends upon the ability of an acceptable dose of dipyridamole to selectively amplify the therapeutic effect of a reduced-dose of prednisolone, without causing unacceptable expected or unexpected adverse side effects. As a result, Zalicus’s combination product candidates could have greater adverse side effects than anticipated and could fail to achieve risk-to-benefit profiles that would justify their continued development.
Significant adverse side effects of the components of Synavive include, headache, nausea, dizziness, diarrhea, muscle and bone loss, diabetes, dyslipidemia, osteoporosis, fractures, weakness, adrenal suppression, infections, abdominal distress, peptic ulceration, arrhythmias, cataracts, glaucoma and myopathy. These side effects are not the only side effects of the components of Synavive. The occurrence of these or other significant adverse side effects could make it inadvisable to continue development of Synavive or lead to difficulty in obtaining regulatory or other approval for clinical trials of Synavive, the termination of clinical trials or could result in product liability claims.
Synavive is a combination of approved drugs that are commercially available and marketed by other companies. As a result, Synavive, if approved, may be subject to substitution and competition.
The approved drugs that are combined in Synavive are commercially available at prices lower than the prices at which we would seek to market Synavive, if it is approved. Even with its new formulation, we cannot be
32
sure that physicians will view Synavive, if approved, as sufficiently superior to a treatment regimen of the individual active pharmaceutical ingredients as to justify the significantly higher cost we expect to seek for Synavive, and they may prescribe the individual drugs already approved and marketed by other companies instead of Synavive. Even if we are issued patents covering the composition of matter, method of use or formulations of Synavive, those patents may be ineffective as a practical matter to protect against physicians prescribing the individual generic drugs instead of Synavive. To the extent that the price of Synavive is significantly higher than the prices of the individual components, physicians may have a greater incentive to write prescriptions for the individual components instead of for Synavive, and this may limit how we price Synavive. Similar concerns could also limit the reimbursement amounts private health insurers or government agencies in the United States are prepared to pay for Synavive, which could also limit market and patient acceptance of Synavive, and could negatively impact our revenues and net income, if any. Physicians might also prescribe the individual components of Synavive prior to its approval, which could adversely affect our development of Synavive due to our lack of control over the administration to patients of the combination of active pharmaceutical ingredients in Synavive, the occurrence of adverse effects, and other reasons. Such pre-approval use could also adversely affect our ability to market and commercialize Synavive if it is approved.
In many jurisdictions where we plan to market Synavive, including Europe, Japan and Canada, the pricing of prescription drugs is controlled by the government or regulatory agencies. Regulatory agencies in these countries could determine that the pricing for Synavive should be based on prices for their active pharmaceutical ingredients when sold separately, rather than allowing us to market Synavive at a premium as a new drug.
We may not be able to create commercially viable pharmaceutical formulations of Synavive or our ion channel product candidates.
The success of Synavive will depend on our ability to develop a formulation that is superior to a treatment regimen of prednisolone and dipyridamole taken separately. We have developed or are developing proprietary formulations of Synavive. In addition, we have developed or will need to develop proprietary formulations of ion channel product candidates, including Z160. Developing such proprietary formulations is costly and difficult, and we have limited experience in developing formulations ourselves. We are relying on and expect to rely on third-party suppliers to develop the pharmaceutical formulations, delivery methods or packaging for Zalicus’s product candidates and they may not be successful in doing so or may experience delays in doing so that could delay clinical trials, and ultimately our ability to obtain approval of Zalicus’s product candidates, including Synavive. Defects in the formulation, delivery method or packaging of any of Zalicus’s product candidates could delay our ability to conduct clinical trials or require us to repeat clinical trials using a revised formulation, delivery method or packaging. If we are unsuccessful in creating commercially viable formulations, delivery methods or packaging, we may never generate product revenue or be profitable.
We may not be able to initiate and complete clinical trials for our product candidates.
Conducting clinical studies for any of our product candidates requires finding appropriate clinical sites and clinical investigators, securing approvals for such studies from the independent review board at each such site and local regulatory authorities and enrolling sufficient numbers of patients. We may not be able to arrange for appropriate clinical trials for our product candidates, secure the necessary approvals or enroll the necessary number of participants. Zalicus plans to initiate a Phase 2b clinical trial of Synavive in rheumatoid arthritis during 2011. In 2008, Zalicus terminated enrollment in a Phase 2b clinical trial of Synavive in rheumatoid arthritis due to delays in the enrollment associated with competing therapies otherwise available to the relevant patient population, enrollment criteria that required the discontinuance of glucocorticoid use by subjects and issues with third-party contract research organizations and third-party suppliers of clinical trial material. Zalicus cannot be certain that similar challenges will not arise with respect to the currently planned Phase 2b study of Synavive. In addition, we cannot guarantee that outside clinical investigators conducting clinical trials will conduct them in compliance with applicable United States or foreign regulations. Clinical sites may fail the FDA’s or other regulatory agencies’ inspections or reviews, and our trials could be halted for these or other
33
reasons. Zalicus plans to contract with third-party clinical research organizations and other parties to conduct virtually all aspects of Zalicus’s Phase 2b and other Phase 1 clinical trials for Zalicus’s product candidates. These organizations may not adequately or completely perform their contractual obligations regarding the trials, or may not diligently or completely perform their tasks with respect to clinical trials under their supervision. As a result of these risks, our clinical trials may be extended, delayed or terminated, which could delay the receipt of clinical results for our product candidates, which could delay, impede or stop the development, regulatory approval or successful commercialization of our product candidates.
We may be unable to find safe and effective doses or dose ratios for Zalicus’s product candidates without extensive clinical trials and substantial additional costs, if at all.
We must select the doses, including the amount, frequency and duration, of each of the active pharmaceutical ingredients included in Zalicus’s product candidates, and the relative amounts, or dose ratios, of these doses, including the doses of prednisolone and dipyridamole contained in our product candidate Synavive. Our clinical trials in humans may show that the doses or dose ratios we select based on Zalicus’s in vitro screening, animal testing or early clinical trials do not achieve the desired therapeutic effect in humans, or achieve this effect only in a small part of the population. Even if the doses or dose ratios we select show efficacy in humans, the resulting doses or dose ratios of active pharmaceutical ingredients may not have acceptable safety profiles for targeted indications. Furthermore, even if we believe that pre-clinical and clinical studies adequately demonstrate that the doses or dose ratios we select for Zalicus’s product candidates are safe and effective in humans, the FDA or other regulatory agencies in foreign jurisdictions may conclude that the clinical trials do not support this conclusion. We may be required to conduct additional long-term clinical studies and provide more evidence substantiating the safety and effectiveness of the doses or selected dose ratios in a significant patient population. If we need to adjust the doses or dose ratios, we may need to conduct new clinical trials. We may also be required to make different doses or dose ratios available for different types of patients. All of this may result in significant delays and additional costs or prevent commercialization of Zalicus’s product candidates.
We may fail to select or capitalize on the most scientifically, clinically or commercially promising or profitable product candidates.
We may make incorrect determinations as to which product candidates should proceed to initial clinical trials, later stage clinical development and potential commercialization. Our decisions to allocate finite research, management and financial resources toward particular product candidates or therapeutic areas may not lead to the development of viable commercial products and may divert resources from better opportunities. Similarly, our decisions to delay or terminate drug development programs may also be incorrect and could cause us to miss valuable opportunities.
A material component of our business strategy is to establish and maintain collaborative relationships to fund research and possible development and commercialization of product candidates, by us or by our collaborators. If we or any collaborator terminates or fails to perform any obligations under our collaboration agreements, the development and commercialization of product candidates under these agreements could be delayed or terminated.
A material component of our business strategy is to establish and maintain collaborative arrangements with pharmaceutical and biotechnology companies, and to seek grants from agencies of the United States government, to fund research and possible development and commercialization of drug products for the treatment of diseases. We have established collaborative royalty and milestone-based agreements with Covidien for Exalgo and with Sanofi-Aventis/Fovea for Prednisporin and funded discovery research agreements with Novartis, USAMRIID and others, if any of our product candidates continue to advance through preclinical or clinical development, we intend to continue to seek collaborative relationships to obtain discovery or clinical development funding and expertise, as well as domestic or international sales, marketing and distribution capabilities.
34
The process of establishing collaborative relationships is difficult, time-consuming and involves significant uncertainty. Moreover, it may be difficult to maintain or perform under collaboration arrangements, as our funding resources may be limited or our collaborators may seek to renegotiate or terminate their relationships due to unsatisfactory research or clinical results, a change in business strategy, a change of control or other reasons.
See “—Our ability to generate ongoing revenue from Exalgo depends on Covidien.” If we or any collaborator fails to fulfill any responsibilities in a timely manner, or at all, our research, clinical development or commercialization efforts related to that collaboration could be delayed or terminated. Additionally it may become necessary for us to assume responsibility for activities that would otherwise have been the responsibility of our collaborator. Further, if we are unable to establish and maintain collaborative relationships on acceptable terms, we may have to delay or discontinue further development of one or more of our product candidates, undertake development and commercialization activities at our own expense or find alternative sources of funding.
Zalicus’s collaborations are generally new, and we have only a short history of working together with our collaborators and cannot predict the success of any of these collaborations. Our collaborations typically involve a complex allocation of responsibilities, costs and benefits and provide for milestone payments to us upon the achievement of specified clinical and regulatory milestones. Our collaborations also may provide us with royalty-based revenue if product candidates are successfully commercialized. Under the Covidien, Novartis, Sanofi-Aventis/Fovea and other collaborations, we will rely on our collaborators to provide resources to develop new product candidates and to potentially achieve these milestones and commercialize any new products. We may not be able to achieve any of the milestones provided in the Novartis, Sanofi-Aventis/Fovea or other collaboration agreements or derive any license or royalty revenue with respect to our collaborations.
We have no sales or distribution capabilities and may not obtain the collaboration, development, commercialization, manufacturing or other third-party relationships required to develop, commercialize and manufacture some or all of our product candidates.
We have no sales or distribution capabilities and lack many of the resources, capabilities and experience necessary to clinically develop, formulate, manufacture, test, market and sell pharmaceuticals. As a result, to succeed in our business plan, we will be dependent on the efforts of third parties. We depend on collaborators, licensees, clinical research organizations and other third parties to formulate product candidates and to conduct clinical trials for some or all of our product candidates. We also rely on third-party manufacturers to manufacture all clinical trial supplies of our product candidates and they or we will rely on obtaining sufficient quantities of the approved drugs in Synavive from sources acceptable to the FDA and other regulators for early stage clinical trials.
Our third-party manufacturers may encounter difficulties performing their obligations in a timely manner and in accordance with applicable governmental regulations, including problems involving: inconsistent production yields; poor quality control and assurance or inadequate process controls; and lack of compliance with regulations set forth by the FDA or other foreign regulatory agencies. We typically engage only a single contract manufacturer to make any product candidate which can exacerbate the impact of any such difficulties. Under our agreements with Covidien, we have no responsibility for manufacturing Exalgo. However, any manufacturing difficulties or related regulatory issues could impact Covidien’s sales of Exalgo, if any, which would reduce the significant revenue we expect to receive from royalties on net sales of Exalgo.
We expect to be able to develop and commercialize many of our product candidates only with the participation of pharmaceutical or biotechnology company collaborators or by out-licensing rights to the product candidates. Pharmaceutical and biotechnology companies and others may be reluctant to collaborate with Zalicus or to license rights to Zalicus’s product candidates due to the unproven nature of Zalicus’s drug discovery and development approach, the fact that the active pharmaceutical ingredients in Synavive are approved generic drugs, the risk that healthcare providers may substitute the component active pharmaceutical ingredients for Synavive, concerns regarding the pricing of and reimbursement for Zalicus’s product candidates if they are successfully developed, or other factors.
35
We cannot guarantee that we will be able to successfully negotiate agreements for relationships with collaborators, partners, licensees, clinical investigators, manufacturers and other third parties on favorable terms, if at all. If we are unable to obtain these agreements, we may not be able to clinically develop, formulate, manufacture, test, obtain regulatory approvals for or commercialize our product candidates. We expect to expend substantial funds and management time and effort to enter into relationships with these third parties and, if we enter successfully into such relationships, to manage these relationships. However, we cannot control the amount or timing of resources our contract partners will devote to our research and development programs, product candidates or potential product candidates, and we cannot guarantee that these parties will succeed in a timely fashion, if at all. Due to the recent volatility in the financial markets and tightening of the credit markets, there may be a disruption or delay in the performance or satisfaction of commitments to us by third party providers which could have a material adverse effect on our business.
We may not be able to gain market acceptance of our product candidates, which would prevent us from becoming profitable.
We cannot be certain that any of our product candidates, if approved, will gain market acceptance among physicians, patients, healthcare payors, pharmaceutical companies or others. Demonstrating the safety and efficacy of our product candidates and obtaining regulatory approvals will not guarantee future revenue. Sales of medical products largely depend on the reimbursement of patients’ medical expenses by government healthcare programs and private health insurers. Governments and private insurers closely examine medical products to determine whether they should be covered by reimbursement and if so, the level of reimbursement that will apply. We cannot be certain that third-party payors will sufficiently reimburse sales of our products, or enable us to sell our products, if approved, at profitable prices. Sales of medical products also depend on physicians’ willingness to prescribe the treatment, which is likely to be based on a determination by these physicians that the products are safe, therapeutically effective and cost-effective. We cannot predict whether physicians, other healthcare providers, government agencies or private insurers will determine our products are safe, therapeutically effective and cost effective relative to competing treatments, including a treatment regimen of the individual approved drugs included in Zalicus’s combination products.
Disputes under key agreements could delay or prevent development or commercialization of our product candidates.
Any agreements we have or may enter into with third parties, such as collaboration, license, formulation supplier, manufacturing, testing, clinical research organization or clinical trial agreements, may give rise to disputes regarding the rights and obligations of the parties to such agreements. Disagreements could develop over rights to ownership or use of intellectual property, the scope and direction of research and development, the approach for regulatory approvals or commercialization strategy. We intend to conduct research programs in a range of therapeutic areas, but our pursuit of these opportunities could result in conflicts with the other parties to these agreements who may be developing or selling pharmaceuticals or conducting other activities in these same therapeutic areas. Any disputes or commercial conflicts could lead to the termination of these agreements, delay progress of our product development programs, compromise our ability to renew agreements or obtain future agreements, lead to the loss of intellectual property rights or result in costly litigation.
Risks Related to Financial Results and Need for Additional Financing
We may be unable to raise the substantial additional capital that it will need to sustain our operations.
We will need substantial additional funds to support our planned operations. Based on current operating plans, we expect our resources to be sufficient to fund our operations into 2014. We may, however, need to raise additional funds before that time if our research and development expenses exceed current expectations or our collaboration funding and Exalgo royalties are less than current assumptions or expectations. This could occur for many reasons, including:
| • | we determine to acquire or license rights to additional product candidates or new technologies; |
36
| • | some or all of our product candidates fail in clinical or pre-clinical studies or prove to be less commercially promising than we expect or we are forced to seek additional product candidates; |
| • | our product candidates require more extensive clinical or pre-clinical testing or clinical trials take longer to complete than we currently expect; |
| • | we advance more of our product candidates than expected into costly later stage clinical trials; |
| • | we advance more of our pre-clinical product candidates than expected into early stage clinical trials; |
| • | our revenue generating collaboration agreements are terminated; |
| • | we determine or are required to conduct more discovery research than expected to develop additional product candidates; |
| • | we are required, or consider it advisable, to acquire or license rights from one or more third parties. |
While we expect to seek additional funding through public or private financings, we may not be able to obtain financing on acceptable terms, or at all. In addition, the terms of any financings may be dilutive to, or otherwise adversely affect, holders of Zalicus common stock. We may also seek additional funds through arrangements with collaborators or others. These arrangements would generally require us to relinquish rights to some of our technologies, product candidates or products, and we may not be able to enter into such agreements on acceptable terms, if at all. The arrangements also may include issuances of equity, which may also be dilutive to, or otherwise adversely affect, holders of Zalicus common stock. Many people believe that participants in financial markets in the United States are increasingly less willing to fund drug discovery companies like Zalicus. There can be no assurance that we will be able to access equity or credit markets in order to finance our operations or expand development programs for any of our product candidates, or that there will not be a further deterioration in financial markets and confidence in economies. We may also have to scale back or further restructure our operations. If we are unable to obtain additional funding on a timely basis, we may be required to curtail or terminate some or all of our research or development programs.
Zalicus has a history of operating losses. We expect to incur significant operating losses and may never be profitable. Zalicus common stock is a highly speculative investment.
Zalicus commenced operations in March 2000 and has no approved products of its own and has generated no direct product revenue. Zalicus has incurred operating losses since Zalicus’s inception in 2000. As of December 31, 2010, Zalicus had an accumulated deficit of $254.7 million. We have spent, and expect to continue to spend, significant resources to fund research and development of our product candidates and to enhance our drug discovery technologies. We expect to incur substantial operating losses over the next several years due to our ongoing research, development, pre-clinical testing, and potential clinical trial activities. As a result, our accumulated deficit will continue to increase.
Our product candidates are in the early stages of development and may never result in any revenue. We will not be able to generate product revenue unless and until one of our product candidates successfully completes clinical trials and receives regulatory approval. We may seek to obtain revenue from collaboration or licensing agreements with third parties. Other than our agreement with Covidien, our current collaboration and license agreements may not provide us with material, sustainable ongoing future revenue, and we may not be able to enter into additional collaboration agreements. Even if we eventually generate product revenues, we may never be profitable, and if we ever achieve profitability, we may not be able to sustain it.
Risks Related to Regulatory Approvals
The regulatory approval process is costly and lengthy and we may not be able to successfully obtain all required regulatory approvals.
The pre-clinical development, clinical trials, manufacturing, marketing, testing and labeling of pharmaceuticals and medical devices are all subject to extensive regulation by numerous governmental authorities and agencies in
37
the United States and other countries. We or our collaborators must obtain regulatory approval for product candidates before marketing or selling any of them. The approval process is typically lengthy and expensive, and approval is never certain. It is not possible to predict how long the approval processes of the FDA or any other applicable federal or foreign regulatory authority or agency for any of our products will take or whether any such approvals ultimately will be granted. The FDA and foreign regulatory agencies have substantial discretion in the drug and medical device approval process, and positive results in pre-clinical testing or early phases of clinical studies offer no assurance of success in later phases of the approval process. Generally, pre-clinical and clinical testing of products and medical devices can take many years and require the expenditure of substantial resources, and the data obtained from these tests and trials can be susceptible to varying interpretations that could delay, limit or prevent regulatory approval. Any delay in obtaining, or failure to obtain, approvals could prevent or adversely affect the marketing of our products or our collaborator’s products and our ability to generate product revenue. The risks associated with the approval process include delays or rejections in the regulatory approval process based on the failure of clinical or other data to meet expectations, or the failure of the product or medical device to meet a regulatory agency’s requirements for safety, efficacy and quality. In addition, regulatory approval, if obtained, may significantly limit the indicated uses for which a product may be marketed.
We or our collaborators may delay, suspend or terminate clinical trials to obtain marketing authorization of any of our product candidates or their associated medical devices or products at any time for reasons including:
| • | ongoing discussions with the FDA or comparable foreign authorities regarding the scope or design of clinical trials; |
| • | delays or the inability to obtain required approvals from institutional review boards or other governing entities at clinical sites selected for participation in our clinical trials; |
| • | delays in enrolling patients and volunteers into clinical trials; |
| • | lower than anticipated retention rates of patients and volunteers in clinical trials; |
| • | the need to repeat clinical trials as a result of inconclusive or negative results or poorly executed testing; |
| • | lack of effectiveness of a product candidate in other clinical trials; |
| • | lack of sufficient funds for further clinical development; |
| • | insufficient supply or deficient quality of product candidate materials or other materials necessary to conduct clinical trials; |
| • | unfavorable regulatory inspection of a manufacturing, testing, labeling or packaging facility for drug substance or drug product; |
| • | unfavorable regulatory inspection and review of a clinical or pre-clinical trial site or records of any clinical or pre-clinical investigation; |
| • | serious and unexpected drug-related side effects or serious adverse safety events experienced by participants in clinical trials or by patients following commercialization; or |
| • | the placement of a clinical hold on a product candidate in an ongoing clinical trial. |
Positive or timely results from pre-clinical studies and early clinical trials do not ensure positive or timely results in late stage clinical trials or product approval by the FDA or any other regulatory authority. Product candidates that show positive pre-clinical or early clinical results often fail in later stage clinical trials. Data obtained from pre-clinical and clinical activities is susceptible to varying interpretations, which could delay, limit, or prevent regulatory approvals.
We may not be able to conduct clinical trials at preferred sites, enlist clinical investigators, enroll sufficient numbers of patients, or begin or successfully complete clinical trials in a timely fashion, if at all. Any failure to
38
perform may delay or terminate the trials. Our current clinical trials may be insufficient to demonstrate that our potential products are active, safe, or effective and as a result we may decide to abandon further development of such product candidates. Additional clinical trials may be required if clinical trial results are negative or inconclusive, which will require us to incur additional costs and significant delays. If we do not receive the necessary regulatory approvals, we will not be able to generate product revenues and will not become profitable. We may encounter significant delays in the regulatory process that could result in excessive costs that may prevent us from continuing to develop our product candidates. In addition, the failure to comply with applicable regulatory requirements may result in criminal prosecution, civil penalties, product recalls, withdrawal of product approval, mandatory restrictions or other actions that could impair our ability to conduct our business.
The FDA and other regulatory agencies may require more extensive or expensive trials for Synavive than may be required for single agent pharmaceuticals.
To obtain regulatory approval for Synavive, we expect to be required to show that each active pharmaceutical ingredient in Synavive makes a contribution to the combined product candidate’s claimed effects and that the dosage of each component, including amount, frequency and duration, is such that the combination is safe and effective for a significant patient population requiring such concurrent therapy. As a result, we are typically required to conduct clinical trials comparing each component drug with the combination. This could require us to conduct more extensive and more expensive clinical trials with Synavive than would be the case for many single agent pharmaceuticals. The need to conduct such trials could make it more difficult and costly to obtain regulatory approval of Synavive compared to a new drug containing only a single active pharmaceutical ingredient.
Even if we receive regulatory approvals for marketing Zalicus’s product candidates, if we fail to comply with continuing regulatory requirements, we could lose regulatory approvals, and our business would be adversely affected.
The FDA and other regulatory authorities continue to review therapeutic products and medical devices even after they receive initial approval. If we receive approval to commercialize any product candidates, the manufacturing, testing, marketing, sale and distribution of these drugs and medical devices will be subject to continuing regulation, including compliance with quality systems regulations, good manufacturing practices, adverse event reporting requirements and prohibitions on promoting a product for unapproved uses. Furthermore, heightened Congressional scrutiny on the adequacy of the FDA’s drug approval process and the agency’s efforts to assure the safety of marketed drugs has resulted in the enactment of legislation, the FDA Amendments Act of 2007, addressing, among other things, drug safety issues. This law provides the FDA with expanded authority over drug products after approval, including the authority to require post-approval studies and clinical trials, labeling changes based on new safety information, and compliance with REMS approved by the FDA. The FDA’s exercise of this authority could result in delays or increased costs during the period of product candidate development, clinical trials and regulatory review and approval, increased costs to assure compliance with new post-approval regulatory requirements, and potential restrictions on the sale of approved products, which could lead to lower product revenues to us or our collaborators. Enforcement actions resulting from failure to comply with government requirements could result in fines, suspension of approvals, withdrawal of approvals, recalls of products, product seizures, operating restrictions, and civil or criminal penalties. These enforcement actions could affect the manufacturing, testing, marketing, sale and distribution of our products.
Legislative or regulatory reform of the health care system in the United States and foreign jurisdictions may affect our ability to profitably sell our products, if approved.
Our and our collaborators’ ability to commercialize our future products successfully will depend in part on the extent to which reimbursement for the products will be available from government and health administration authorities, private health insurers and other third-party payors. The continuing efforts of the U.S. and foreign governments, insurance companies, managed care organizations and other payors of health care services to contain or reduce health care costs may adversely affect our ability to set prices for our products which we believe are fair, and our ability to generate revenues and achieve and maintain profitability.
39
Specifically, in both the United States and some foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the health care system in ways that could affect our ability to sell our products profitably. Congress has passed America’s Affordable Health Choices Act of 2009 and is considering a number of proposals that are intended to reduce or limit the growth of health care costs and which could significantly transform the market for pharmaceuticals products. We expect further federal and state proposals and health care reforms to continue to be proposed by legislators, which could limit the prices that can be charged for the products we develop and may limit our commercial opportunity. In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, also called the Medicare Modernization Act, or MMA, changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for drugs. In addition, this legislation provided authority for limiting the number of drugs that will be covered in any therapeutic class. As a result of this legislation and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. These cost reduction initiatives and other provisions of this legislation could decrease the coverage and price that we receive for any approved products and could seriously harm our business. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates, and any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private payors.
The continuing efforts of government and other third-party payors to contain or reduce the costs of health care through various means may limit our commercial opportunity. It will be time consuming and expensive for us or our partners to go through the process of seeking reimbursement from Medicare and private payors. Our products, including Exalgo, may not be considered cost effective, and government and third-party private health insurance coverage and reimbursement may not be available to patients for any of our future products or sufficient to allow us or our collaborators, including Covidien, to sell our products on a competitive and profitable basis. Our results of operations could be adversely affected by the MMA and additional prescription drug coverage legislation, by the possible effect of this legislation on amounts that private insurers will pay and by other health care reforms that may be enacted or adopted in the future. In addition, increasing emphasis on managed care in the United States will continue to put pressure on the pricing of pharmaceutical products. Cost control initiatives could decrease the price that we or any potential collaborators could receive for any of our future products and could adversely affect our profitability.
In some foreign countries, including major markets in the European Union and Japan, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take 6 to 12 months or longer after the receipt of regulatory marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we or our collaborators may be required to conduct a clinical study that compares the cost-effectiveness of our product candidates to other available therapies. Such pharmacoeconomic studies can be costly and the results uncertain. Our business could be harmed if reimbursement of our or our collaborators’ products are unavailable or limited in scope or amount or if pricing is set at unsatisfactory levels.
Federal laws or regulations on drug importation could make lower cost versions of our future products available, which could adversely affect our revenues, if any.
The prices of some drugs are lower in other countries than in the United States because of government regulation and market conditions. Under current law, importation of drugs into the United States is generally not permitted unless the drugs are approved in the United States and the entity that holds that approval consents to the importation. Various proposals have been advanced to permit the importation of drugs from other countries to provide lower cost alternatives to the products available in the United States. In addition, the MMA requires the Secretary of Health and Human Services to promulgate regulations for drug reimportation from Canada into the United States under some circumstances, including when the drugs are sold at a lower price in Canada than in the United States.
40
If the laws or regulations are changed to permit the importation of drugs into the United States in circumstances not now permitted, such a change could have an adverse effect on our business by making available lower priced alternatives to our future products.
Failure to obtain regulatory and pricing approvals in foreign jurisdictions could delay or prevent commercialization of our products abroad.
If we succeed in developing any products (other than Exalgo), we intend to market them in the European Union and other foreign jurisdictions. In order to do so, we must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval abroad may differ from that required to obtain FDA approval. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval and additional risks particularly because there may be additional variations between how our combination products, including Synavive, and single agent drugs are treated in foreign jurisdictions. We may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. We and our collaborators may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market outside the United States. The failure to obtain these approvals could materially adversely affect our business, financial condition and results of operations. Even if we are successful at obtaining these approvals, regulatory agencies in foreign countries where the pricing of prescription drugs or medical devices is controlled by the government, could determine that pricing for Zalicus’s combination products should be based on prices for the existing drugs that comprise the active pharmaceutical ingredients in Zalicus’s combination products instead of allowing us to price Zalicus’s combination products at a premium as novel medicines.
Risks Related to Intellectual Property
Our success depends upon our ability to obtain and maintain intellectual property protection for our products and technologies.
Our success will depend on our ability to obtain and maintain adequate protection of our intellectual property, including our proprietary drug discovery technologies and any products or product candidates we plan to develop. We intend to apply for patents with claims covering our technologies, processes, products and product candidates when and where we deem it appropriate to do so and plan to take other steps to protect our intellectual property. We have applied for patent protection covering our clinical and pre-clinical product candidates in the United States, and some, but not all, foreign countries. In countries where we have not and do not seek patent protection, third parties may be able to manufacture and sell our products without our permission, and we may be unable to stop them from doing so. The patents covering Exalgo expire in July 2014, or they may be successfully challenged prior to expiration, after which Covidien may face generic competition for the specific formulation of Exalgo. One patent covering Exalgo is the subject of ongoing patent litigation with Watson Pharmaceuticals.
Similar to other biotechnology companies, our patent position is generally uncertain and involves complex legal and factual questions. In addition, the laws of some countries do not protect proprietary rights to the same extent as the laws of the United States, and other biotechnology companies have encountered significant problems in protecting and defending their proprietary rights in non-US jurisdictions. Whether filed in the United States or abroad, our patent applications may be challenged or may fail to result in issued patents. In addition, our existing patents and any future patents may not be sufficiently broad to prevent others from practicing our technologies or from developing or commercializing competing products. Furthermore, others may independently develop or commercialize similar or alternative technologies or drugs, or design around our patents. Our patents may be challenged, invalidated or fail to provide any competitive advantages.
The United States Patent and Trademark Office and similar agencies in foreign jurisdictions may not agree with our view that our combination product candidates are patentable or novel and non-obvious, and on this basis
41
may deny patent protection. Even if we receive patent protection, others, including those who own patent or trade secret rights associated with the approved drugs that are active pharmaceutical ingredients of these product candidates, may attempt to invalidate our patent or trade secret rights. Even if our patent or trade secret rights are not directly challenged, disputes among third parties could lead to the weakening or invalidation of intellectual property rights.
If we do not obtain or are unable to maintain adequate patent or trade secret protection for products in the United States, competitors could duplicate them without repeating the extensive testing that we will be required to undertake to obtain approval of the products by the FDA and other regulatory authorities. Regardless of any patent protection, under the current statutory framework the FDA is prohibited by law from approving any generic version of any of Zalicus’s combination products for three years after it has approved Zalicus’s combination product and for five years in the case of other product candidates. Upon the expiration of that period, or if that time period is altered, the FDA could approve a generic version of Zalicus’s product unless we have patent protection sufficient to enforce our rights. Without sufficient patent protection, the applicant for a generic version of Zalicus’s product would be required only to conduct a relatively inexpensive study to show that its product is bioequivalent to Zalicus’s product and would not have to repeat the studies that we conducted to demonstrate that the product is safe and effective. In the absence of adequate patent protection in other countries, competitors may similarly be able to obtain regulatory approval of products that duplicate Zalicus’s products.
We may not be able to develop or commercialize our product candidates due to intellectual property rights held by third parties.
If a third party holds a patent to a composition or method of use of an approved drug that is a component of one or more of Zalicus’s combination product candidates or a formulation technology related to Zalicus’s planned formulation of a combination product candidate, we may not be able to develop or commercialize such product candidates without first obtaining a license to such patent, or waiting for the patent to expire. Our business will be harmed if we are unable to use the optimal formulation or methods of use of the component drugs that comprise our product candidates. This may occur because the formulations or methods of use are covered by one or more third-party patents, and a license to such patents is unavailable or is available on terms that are unacceptable.
We may be unable to in-license intellectual property rights or technology necessary to develop and commercialize Zalicus’s products.
Depending on its ultimate formulation and method of use, before we can develop, make, use, or sell a particular product candidate, we may need to obtain a license from one or more third parties who have patent or other intellectual property rights covering components of Zalicus’s product candidate or its method of use. There can be no assurance that such licenses will be available on commercially reasonable terms, or at all. Because Zalicus’s combination product candidates are based on combinations of existing drugs, there may be a significant number of patents covering both the active pharmaceutical ingredients in Zalicus’s combination product candidates and their method of use. If a third party does not offer us a necessary license or offers a license only on terms that are unattractive or unacceptable, we might be unable to develop and commercialize one or more of Zalicus’s combination product candidates.
Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information.
In our activities, we rely substantially upon proprietary materials, information, trade secrets and know-how to conduct our research and development activities, and to attract and retain collaborators, licensees and customers. We take steps to protect our proprietary rights and information, including the use of confidentiality and other agreements with our employees and consultants and in our academic and commercial relationships.
42
However, these steps may be inadequate, agreements may be violated, or there may be no adequate remedy available for a violation of an agreement. Our proprietary information may be inadvertently disclosed or we may lose the protection of our trade secrets. Our competitors may independently develop substantially equivalent proprietary information or may otherwise gain access to our trade secrets, which could adversely affect our ability to compete in the market.
Litigation or third-party claims of intellectual property infringement could require substantial time and money to resolve. Unfavorable outcomes in these proceedings could limit our intellectual property rights and our activities.
We may need to resort to litigation to enforce or defend our intellectual property rights, including any patents issued to us. If a competitor or collaborator files a patent application claiming technology also invented by us, in order to protect our rights, we may have to participate in an expensive and time consuming interference proceeding before the United States Patent and Trademark Office. We cannot guarantee that our product candidates will be free of claims by third parties alleging that we have infringed their intellectual property rights. Third parties may assert that we are employing their proprietary technologies without authorization and they may resort to litigation to attempt to enforce their rights. Third parties may have or obtain patents in the future and claim that the use of our technology or any of our product candidates infringes their patents. We may not be able to develop or commercialize product candidates because of patent protection others have. Our business will be harmed if we cannot obtain a necessary or desirable license, can obtain such a license only on terms we consider to be unattractive or unacceptable, or if we are unable to redesign our product candidates or processes to avoid actual or potential patent or other intellectual property infringement.
Our efforts to obtain, protect and defend our patent and other intellectual property rights, whether we are successful or not, may require us to incur substantial costs, including the diversion of management and technical personnel. An unfavorable ruling in patent or intellectual property litigation could subject us to significant liabilities to third parties, require us to cease developing, manufacturing or selling the affected products or using the affected processes, require us to license the disputed rights from third parties, or result in awards of substantial damages against us. In addition, defending patent or other intellectual property litigation, whether we are successful or not, can be very expensive and may require us to incur substantial costs, including the diversion of management and technical personnel. During the course of any patent litigation, there may be public announcements of the results of hearings, motions, and other interim proceedings or developments in the litigation. If securities analysts or investors regard these announcements as negative, the market price of Zalicus common stock may decline. General proclamations or statements by key public figures may also have a negative impact on the perceived value of our intellectual property.
There can be no assurance that we would prevail in any intellectual property infringement action, will be able to obtain a license to any third-party intellectual property on commercially reasonable terms, successfully develop non-infringing alternatives on a timely basis, or license non-infringing alternatives, if any exist, on commercially reasonable terms. Any significant intellectual property impediment to our ability to develop and commercialize our products could seriously harm our business and prospects.
Risks Related to the Biotechnology and Pharmaceutical Industry
Our industry is highly competitive and our competitors and potential competitors may develop products and technologies that make ours less attractive or obsolete.
The development and commercialization of pharmaceutical products is highly competitive. Many companies, universities, and research organizations developing competing product candidates have greater resources and significantly greater experience in research and development, manufacturing, marketing, sales, distribution, financial and technical regulatory matters than we have. In addition, many competitors have greater name recognition and more extensive collaborative relationships. Our competitors could commence and
43
complete clinical testing of their product candidates, obtain regulatory approvals, and begin commercial-scale manufacturing of their products faster than we are able to for our products. They could develop drug discovery technology or products that would render Zalicus’s drug discovery technologies and our product candidates, and those of our collaborators, obsolete and noncompetitive.
Our competitors already have high throughput screening technologies and if they employ these technologies to discover combination drugs, they may render Zalicus’s technologies or Zalicus’s approach to combination drug discovery and development obsolete or noncompetitive. Even when we successfully identify a new combination product candidate, our ability to commercialize any combination product candidate may be limited to the extent that we are unable to obtain patent protection for such a product candidate or patent or trade secret protection for its newly identified formulation. Our competitors may also be able to use similar component drugs or different combinations of component drugs to develop combination products that are not covered by our patents. Our competitors’ ability to market competitive drugs would adversely impact our ability to generate revenues. In addition, the approved drugs that are combined to produce our combination product candidates are likely to be commercially available at lower prices, so physicians may be able to prescribe the individual drugs already approved and marketed by other companies instead of prescribing our combination products. This practice could adversely impact our ability to generate revenues and would be difficult or impossible for us to prevent by enforcing our combination patents, if obtained.
Zalicus’s drug discovery technologies compete against well-established techniques to discover new drugs. If we are unable to compete effectively against these existing techniques and the companies that support them, then we may not be able to commercialize our product candidates or achieve a competitive position in the market. In addition, any product candidates that we do discover will face competition from existing pharmaceuticals. Our competitors’ success is discovering new drugs and marketing existing drugs which compete with our product candidates would adversely affect our ability to generate revenues.
We may have significant product liability exposure which may harm our business and our reputation.
We face exposure to product liability and other claims if our product candidates, products or processes are alleged to have caused harm. These risks are inherent in the testing, manufacturing, and marketing of human therapeutic products and medical devices. We maintain product liability insurance covering our clinical trials of our product candidates. We may not have sufficient insurance coverage, and we may not be able to obtain sufficient coverage at a reasonable cost, if at all. Our inability to obtain product liability insurance at an acceptable cost or to otherwise protect against potential product liability claims could prevent or inhibit the commercialization of any products or product candidates that we develop. If we are sued for any injury caused by our products, product candidates or processes, our liability could exceed our product liability insurance coverage and our total assets. Claims against us, regardless of their merit or potential outcome, may also divert significant management time and resources, generate negative publicity or hurt our ability to obtain physician endorsement of our products or expand our business.
We use and generate materials that may expose us to expensive and time-consuming legal claims.
Our development programs involve the use of hazardous materials, chemicals, and biological materials. We are subject to foreign, federal, state and local laws and regulations governing the use, manufacture, storage, and disposal of materials and waste products. We believe that our safety procedures for handling these materials comply with the standards prescribed by laws and regulations. However, we may incur significant costs to comply with current or future environmental laws and regulations. In addition, we cannot completely eliminate the risk of contamination or injury from hazardous materials. In the event of an accident, an injured party may seek to hold us liable for any damages that result. Any liability could exceed the limits or fall outside the coverage of our insurance, and we may not be able to maintain insurance on acceptable terms, if at all.
44
Risks Related to an Investment in Our Common Stock
Future sales of common stock by former Neuromed stockholders may cause the price of Zalicus common stock to fall.
After the FDA approved Exalgo, shares issued in connection with the Neuromed Merger were released from an escrow arrangement such that pre-merger Zalicus stockholders only owned approximately 40% of the then outstanding shares of Zalicus common stock. Before this merger, the former Neuromed stockholders held shares of a private company that were difficult to sell. If the former Neuromed stockholders seek to sell substantial amounts of our common stock in the public market to monetize their merger proceeds, particularly if these sales are in a rapid or disorderly manner, or investors perceive that these sales could occur, the market price of our common stock could decrease significantly. These sales might also make it more difficult for us to sell equity securities at an appropriate time and price.
Our common stock has a volatile public trading price.
The market price for our common stock has been volatile, and market prices for securities of companies comparable to us have been highly volatile. In addition, the stock market as a whole and biotechnology and other life science stocks in particular have experienced significant recent price declines and volatility. Like our common stock, these stocks have experienced significant price and volume fluctuations for reasons unrelated to the operating performance of the individual companies. Factors giving rise to this volatility may include:
| • | disclosure of the sales performance of Exalgo by Covidien, or matters related to the Watson litigation; |
| • | disclosure by our collaboration partners, including Sanofi-Aventis/Fovea and Novartis regarding our collaborations and the related product candidates, including Prednisporin; |
| • | disclosure of actual or potential preclinical or clinical results with respect to product candidates we are developing; |
| • | regulatory developments in both the United States and abroad; |
| • | developments concerning proprietary rights, including patents and litigation matters; |
| • | public concern about the safety or efficacy of our product candidates or technology, their components, or related technology or new technologies generally; |
| • | public announcements by our competitors or others regarding new products or new product candidates; and |
| • | general market conditions and comments by securities analysts and investors. |
Fluctuations in our operating losses could adversely affect the price of our common stock.
Our operating losses may fluctuate significantly on a quarterly basis. Some of the factors that may cause our operating losses to fluctuate on a period-to-period basis include royalties from sales of Exalgo, the status of our clinical and pre-clinical development programs, level of expenses incurred in connection with our clinical and pre-clinical development programs, restructuring costs, implementation or termination of collaboration, licensing, manufacturing or other material agreements with third parties, non-recurring revenue or expenses under any such agreement, and compliance with regulatory requirements. Period-to-period comparisons of our historical and future financial results may not be meaningful, and investors should not rely on them as an indication of future performance. Our fluctuating losses may fail to meet the expectations of securities analysts or investors. Our failure to meet these expectations may cause the price of our common stock to decline.
Anti-takeover provisions in our charter documents and provisions of Delaware law may make an acquisition more difficult and could result in the entrenchment of management.
We are incorporated in Delaware. Anti-takeover provisions of Delaware law and our charter documents may make a change in control or efforts to remove management more difficult. Also, under Delaware law, our board
45
of directors may adopt additional anti-takeover measures. The existence of the following provisions of Delaware law and our sixth amended and restated charter, as amended, or our amended and restated bylaws could limit the price that investors might be willing to pay in the future for shares of our common stock.
Our charter authorizes our board of directors to issue up to 5,000,000 shares of preferred stock and to determine the terms of those shares of stock without any further action by our stockholders. If the board of directors exercises this power to issue preferred stock, it could be more difficult for a third party to acquire a majority of our outstanding voting stock and vote the stock they acquire to remove management or directors.
Our charter also provides staggered terms for the members of our board of directors. Under Section 141 of the Delaware General Corporation Law and our charter, our directors may be removed by stockholders only for cause and only by vote of the holders of 75% of voting shares then outstanding. These provisions may prevent stockholders from replacing the entire board in a single proxy contest, making it more difficult for a third party to acquire control without the consent of our board of directors. These provisions could also delay the removal of management by the board of directors with or without cause. In addition our amended and restated bylaws limit the ability our stockholders to call special meetings of stockholders.
Our equity incentive plans generally permit our board of directors to provide for acceleration of vesting of options granted under these plans in the event of certain transactions that result in a change of control. If our board of directors uses its authority to accelerate vesting of options, this action could make an acquisition more costly, and it could prevent an acquisition from going forward.
Under Section 203 of the Delaware General Corporation Law, a corporation may not engage in a business combination with any holder of 15% or more of its capital stock until the holder has held the stock for three years unless, among other possibilities, the board of directors approves the transaction in advance.
| Item 1B. | Unresolved Staff Comments |
None.
| Item 2. | Properties |
We currently lease approximately 23,000 square feet of laboratory and office space in Cambridge, Massachusetts. Our subsidiary, Zalicus Canada, currently leases approximately 12,000 square feet of office and laboratory space in Vancouver, British Columbia, Canada. We believe that our current facilities are sufficient for our current operations.
| Item 3. | Legal Proceedings |
None.
| Item 4. | (Removed and Reserved) |
46
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Price of and Dividends on Our Common Stock and Related Stockholder Matters.
Our common stock is listed for quotation on the NASDAQ Global Market under the symbol “ZLCS.” Trading of our common stock commenced following our initial public offering on November 9, 2005. The following table sets forth the high and low sale prices per share of our common stock as reported on the NASDAQ Global Market for the periods indicated.
| Common Stock Price | ||||||||
| High | Low | |||||||
| Fiscal year ended December 31, 2010 |
||||||||
| First quarter |
$ | 1.72 | $ | 0.76 | ||||
| Second quarter |
$ | 2.02 | $ | 1.17 | ||||
| Third quarter |
$ | 1.69 | $ | 1.17 | ||||
| Fourth quarter |
$ | 1.72 | $ | 1.01 | ||||
| Fiscal year ended December 31, 2009 |
||||||||
| First quarter |
$ | 0.99 | $ | 0.25 | ||||
| Second quarter |
$ | 1.34 | $ | 0.49 | ||||
| Third quarter |
$ | 2.60 | $ | 0.70 | ||||
| Fourth quarter |
$ | 1.86 | $ | 0.81 | ||||
On March 8, 2011, the reported last sale price of our common stock on the NASDAQ Global Market was $2.13 per share. As of March 8, 2011, there were approximately 85 holders of record of our common stock.
We have never paid cash dividends on our common stock. We currently do not anticipate paying cash dividends on our common stock in the foreseeable future. We currently intend to retain earnings, if any, to finance the growth and development of our business. Payment of future dividends, if any, will be at the discretion of our board of directors and will depend on our financial condition, results of operations, capital requirements, restrictions contained in current or future financing instruments and other factors that our board of directors deems relevant.
In connection with the loan and security agreement with Oxford entered into on December 22, 2010, and as previously disclosed on our Current Report on Form 8-K filed with the Securities and Exchange Commission on December 22, 2010, we issued to Oxford a warrant to purchase 64,748 shares of our common stock. The warrant is exercisable, in whole or in part, immediately, and has a per share exercise price of $1.39 and may be exercised on a cashless basis. The warrant will terminate on the earlier of December 22, 2017 and the closing of a merger or consolidation transaction in which Zalicus is not the surviving entity. In reliance on Rule 506 of Regulation D, the “safe harbor” for the private offering exemption of Section 4(2) of the Securities Act and because the offering satisfied all of the requirements thereunder, this warrant was not registered under the Securities Act.
47
Securities Authorized For Issuance Under Equity Compensation Plans
| Plan Category |
Number of Securities to be Issued upon Exercise of Outstanding Options, Warrants or Rights(1) |
Weighted Average Exercise Price of Outstanding Options, Warrants or Rights(2) |
Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plan (Excluding Securities Reflected in Column(a))(1)(3) |
|||||||||
| (a) | (b) | (c) | ||||||||||
| Equity compensation plans approved by security holders |
7,860,447 | $ | 2.03 | 11,738,775 | ||||||||
| Equity compensation plans not approved by security holders |
— | — | — | |||||||||
| Total |
7,860,447 | $ | 2.03 | 11,738,775 | ||||||||
| (1) | As of December 31, 2010. |
| (2) | For outstanding restricted stock units, the exercise price was deemed to be $0. |
| (3) | Our Amended and Restated 2004 Incentive Plan (the “2004 Plan”) contains an “evergreen provision” that allows for an annual increase in the number of shares of common stock available for issuance under the 2004 Plan, which annual increase is and will be added on the first day of each fiscal year from 2011 through 2015, inclusive, and will be equal to the least of: (i) 4,000,000 shares of common stock, (ii) 4% of the outstanding shares on that date or (iii) such lesser amount determined by the board of directors. The Compensation Committee of the board of directors elected not to increase the number of shares of common stock available for issuance under the 2004 Plan for 2011. |
48
Comparative Stock Performance Graph
The information contained in the performance graph shall not be deemed to be “soliciting material” or to be “filed” with the Securities and Exchange Commission, and such information shall not be incorporated by reference into any future filing under the Securities Act or Exchange Act, except to the extent that Zalicus specifically incorporates it by reference into such filing.
The comparative stock performance graph below compares the cumulative total stockholder return (assuming reinvestment of dividends, if any) from investing $100 on December 31, 2005, and plotted at the close of the last trading day of the fiscal year ended December 31, 2010, in each of (i) Zalicus common stock, (ii) the Nasdaq Global Stock Market Index, which is referred to as the Nasdaq Stock Market Index, and (iii) the Nasdaq Global Stock Market Biotechnology Index, which is referred to as the Nasdaq Biotechnology Index; except, in the case of the Nasdaq Stock Market Index and the Nasdaq Biotechnology Index, the stock performance graph below reflects an investment date of December 31, 2005.
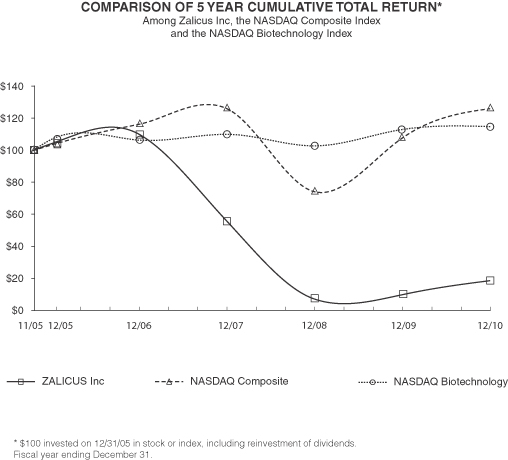
| Cumulative Total Return | ||||||||||||||||||||||||
| 12/05 | 12/06 | 12/07 | 12/08 | 12/09 | 12/10 | |||||||||||||||||||
| ZALICUS INC. |
104.20 | 110.32 | 56.56 | 7.90 | 10.57 | 19.32 | ||||||||||||||||||
| NASDAQ COMPOSITE |
103.41 | 116.35 | 126.25 | 74.61 | 107.78 | 125.93 | ||||||||||||||||||
| NASDAQ BIOTECHNOLOGY |
106.69 | 106.54 | 110.17 | 102.94 | 113.09 | 114.80 | ||||||||||||||||||
49
| Item 6. | Selected Financial Data |
The historical financial data set forth below should be read in conjunction with our “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and the related notes thereto appearing elsewhere in this Annual Report on Form 10-K. The selected financial data in this section are not intended to replace the financial statements. We have derived the statement of operations data for the years ended December 31, 2010, 2009 and 2008 and the balance sheet data as of December 31, 2010 and 2009 from our consolidated financial statements included elsewhere in this annual report, which have been audited by Ernst & Young LLP, our independent registered public accounting firm. We derived the statement of operations data for the year ended December 31, 2007 and 2006 and the balance sheet data as of December 31, 2008, 2007 and 2006 from our audited consolidated financial statements, as adjusted for the divestiture of our Singapore subsidiary which was accounted for as a discontinued operation, which are not included herein. See the notes to the financial statements for an explanation of the method used to determine the number of shares used in determining basic and diluted net loss per common share.
| Year Ended December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| (in thousands, except share and per share amounts) | ||||||||||||||||||||
| Statement of Operations Data: |
||||||||||||||||||||
| Revenue: |
||||||||||||||||||||
| Collaborations |
$ | 45,969 | $ | 16,320 | $ | 11,462 | $ | 11,728 | $ | 11,725 | ||||||||||
| Government contracts and grants |
772 | 953 | 842 | 2,069 | 1,122 | |||||||||||||||
| Total revenue |
46,741 | 17,273 | 12,304 | 13,797 | 12,847 | |||||||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
23,011 | 21,244 | 55,296 | 51,412 | 32,428 | |||||||||||||||
| General and administrative |
12,115 | 17,081 | 14,469 | 16,879 | 18,008 | |||||||||||||||
| Amortization of intangible asset |
18,736 | 520 | — | — | — | |||||||||||||||
| Gain on legal settlement |
— | (3,700 | ) | — | — | — | ||||||||||||||
| Restructuring |
— | 2,736 | 4,637 | — | — | |||||||||||||||
| Total operating expenses |
53,862 | 37,881 | 74,402 | 68,291 | 50,436 | |||||||||||||||
| Loss from operations |
(7,121 | ) | (20,608 | ) | (62,098 | ) | (54,494 | ) | (37,589 | ) | ||||||||||
| Interest income |
132 | 257 | 2,264 | 5,029 | 5,686 | |||||||||||||||
| Interest expense |
(12 | ) | (28 | ) | (651 | ) | (734 | ) | (283 | ) | ||||||||||
| (Loss) gain on revaluation of contingent consideration |
(29,286 | ) | 12,068 | — | — | — | ||||||||||||||
| Other income (expense) |
32 | (281 | ) | (4 | ) | — | 8 | |||||||||||||
| Loss on early extinguishment of debt |
— | — | (195 | ) | — | — | ||||||||||||||
| Gain on bargain purchase |
— | 9,809 | — | — | — | |||||||||||||||
| Net (loss) income before provision for income taxes |
(36,255 | ) | 1,217 | (60,684 | ) | (50,199 | ) | (32,178 | ) | |||||||||||
| Benefit (provision) for income taxes |
1,210 | 67 | 108 | (46 | ) | (51 | ) | |||||||||||||
| Net (loss) income from continuing operations |
(35,045 | ) | 1,284 | (60,576 | ) | (50,245 | ) | (32,229 | ) | |||||||||||
| Discontinued operations: |
||||||||||||||||||||
| Loss from operations of discontinued subsidiary |
— | (1,536 | ) | (4,557 | ) | (3,098 | ) | (2,059 | ) | |||||||||||
| Gain on disposal of discontinued operations |
— | 15,640 | — | — | — | |||||||||||||||
| Gain (loss) on discontinued operations |
— | 14,104 | (4,557 | ) | (3,098 | ) | (2,059 | ) | ||||||||||||
| Net (loss) income |
$ | (35,045 | ) | $ | 15,388 | $ | (65,133 | ) | $ | (53,343 | ) | $ | (34,288 | ) | ||||||
| Net (loss) income per share—basic and diluted: |
||||||||||||||||||||
| From continuing operations |
$ | (0.42 | ) | $ | 0.03 | $ | (1.74 | ) | $ | (1.68 | ) | $ | (1.18 | ) | ||||||
| From discontinued operations |
— | 0.38 | (0.13 | ) | (0.10 | ) | (0.08 | ) | ||||||||||||
| Net (loss) income per share—basic and diluted |
$ | (0.42 | ) | $ | 0.41 | $ | (1.87 | ) | $ | (1.78 | ) | $ | (1.26 | ) | ||||||
| Weighted-average number of common shares used in net (loss) income per share calculation: |
||||||||||||||||||||
| Basic |
82,663,645 | 37,338,042 | 34,848,701 | 30,025,830 | 27,223,319 | |||||||||||||||
| Diluted |
82,663,645 | 37,491,237 | 34,848,701 | 30,025,830 | 27,223,319 | |||||||||||||||
50
| Year Ended December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 As adjusted |
||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents and short-term investments |
$ | 44,619 | $ | 23,330 | $ | 39,653 | $ | 100,026 | $ | 109,563 | ||||||||||
| Working capital |
38,803 | 18,350 | 27,572 | 88,393 | 94,275 | |||||||||||||||
| Total assets |
82,669 | 88,152 | 66,497 | 132,243 | 138,335 | |||||||||||||||
| Long-term debt, less current portion |
2,523 | — | — | 4,534 | 2,527 | |||||||||||||||
| Accumulated deficit |
(254,653 | ) | (219,608 | ) | (251,471 | ) | (186,213 | ) | (132,748 | ) | ||||||||||
| Total stockholders’ equity |
62,997 | 52,913 | 15,875 | 75,235 | 87,050 | |||||||||||||||
51
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
You should read the following discussion and analysis of our financial condition and results of operations in conjunction with our financial statements and related notes appearing elsewhere in this annual report. The following discussion contains forward-looking statements that involve risks and uncertainties. Our actual results and the timing of certain events could differ materially from those anticipated in these forward-looking statements as a result of certain factors, including those discussed below and elsewhere in this annual report, particularly under the heading “Risk Factors.”
Overview
We are a biopharmaceutical company that discovers and develops novel treatments for patients suffering from pain and immuno-inflammatory diseases. We have entered into multiple revenue-generating collaborations with large pharmaceutical companies and have a portfolio of clinical-stage product candidates targeting pain and immuno-inflammatory diseases. We also apply our selective ion-channel modulation platform and combination high throughput screening technology to discover new product candidates for our portfolio or for our collaborators in the areas of pain, inflammation, oncology and infectious disease.
On December 21, 2009, we completed our merger, which we refer to as the Neuromed Merger, with Neuromed Pharmaceuticals Inc., or Neuromed, pursuant to which Neuromed Pharmaceuticals Ltd. became a wholly-owned subsidiary of Zalicus. Upon the consummation of the Neuromed Merger, former Neuromed stockholders were issued an aggregate of 14,937,654 shares of our common stock and 67,826,875 shares of our common stock were issued into escrow. When the FDA did not approve Exalgo™ prior to January 1, 2010, 29,943,752 shares of our common stock held in escrow were cancelled. On March 1, 2010, the FDA approved Exalgo for the management of moderate to severe pain in opioid tolerant patients requiring continuous, around-the-clock opioid analgesia for an extended period of time, resulting in the release to former Neuromed stockholders of the remaining 37,883,123 shares of our common stock held in escrow.
On September 8, 2010, we changed our name from CombinatoRx, Incorporated to Zalicus Inc. We also changed the name of our subsidiaries, including Neuromed Pharmaceuticals Ltd., which is now named Zalicus Pharmaceuticals Ltd, and which we refer to herein as Zalicus Canada. In conjunction with the name change, our trading symbol for our common stock on the Nasdaq Global Market was changed from “CRXX” to “ZLCS”.
The United States commercial rights to Exalgo were acquired by Covidien from Neuromed in June 2009. Under the agreement, Covidien is responsible for all commercialization activities for Exalgo in the United States, including marketing and sales, and for all post-approval regulatory activities. We received a $40.0 million milestone payment following FDA approval of Exalgo, and are eligible to receive tiered royalties on net sales of Exalgo by Covidien following its commercial launch in April 2010. We have recognized $1.6 million in revenue related to these royalties through December 31, 2010.
Our most advanced product candidate is Synavive, a product candidate we have been developing to treat immuno-inflammatory disorders. We have studied Synavive in a Phase 2a clinical trial in rheumatoid arthritis. During 2008 we studied Synavive in a multi-center Phase 2 clinical trial of 279 subjects with knee osteoarthritis, the COMET-1 study. The COMET-1 study was completed in September 2008, and the results of the study were disclosed in October 2008. Subjects who completed the 14-week duration of the COMET-1 study were eligible to participate in an extension study designed to investigate the long-term safety and durability of response for Synavive. The COMET-1 extension study of Synavive was completed in June 2009. We are currently planning to advance Synavive into a Phase 2b clinical trial in rheumatoid arthritis. We have also been performing research and development on our proprietary ion channel modulators which have been advancing through preclinical research and development. We are currently planning to advance one or more ion channel modulators into Phase 1 clinical development in 2011. We have also been using our combination high throughput screening technology platform, or cHTS, to perform our obligations with our collaboration partners. In addition to Synavive, during 2009, we had been advancing three other product candidates through clinical research and development: CRx-401 for Type 2 diabetes, CRx-191 for psoriasis and CRx-197 for psoriasis. We currently are not planning to advance the development of these product candidates.
52
As of December 31, 2010, we had an accumulated deficit of $254.7 million. We had net loss from continuing operations of $35.0 million for the year ended December 31, 2010 and net income from continuing operations of $1.3 million for the year ended December 31, 2009.
Our management currently uses consolidated financial information in determining how to allocate resources and assess performance. We have determined that we conduct operations in one business segment. For the years ended December 31, 2010, 2009 and 2008, revenues from customers located outside the United States were $0.5 million, $11.4 million and $5.5 million, respectively. As of December 31, 2010, $0.8 million and $6.1 million of our long-lived assets were located in Canada and the United States, respectively.
Critical Accounting Policies
Our discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with United States generally accepted accounting principles, or GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments, including those related to business combinations, intangibles and other long-lived assets, revenue recognition, stock-based compensation, accrued expenses and income taxes. We base our estimates on historical experience, known trends and events and various other factors that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
We believe that several accounting policies are important to understanding our historical and future performance. We refer to these policies as “critical” because these specific areas generally require us to make judgments and estimates about matters that are uncertain at the time we make the estimate, and different estimates—which also would have been reasonable—could have been used, which would have resulted in different financial results. We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our financial statements. It is important that the discussion of our operating results that follows be read in conjunction with the critical accounting policies discussed below.
Business Combinations
We assign the value of the consideration transferred to acquire or merge with a business to the tangible assets and identifiable intangible assets acquired and liabilities assumed on the basis of their fair values at the date of acquisition. The consideration transferred in the Neuromed Merger included shares of our common stock and the fair value of contingent consideration. The fair value of contingent consideration was determined based upon a probability-weighted estimate of the incremental number of shares of our common stock to be issued to former Neuromed stockholders at our stock price at merger closing. The probability weighted estimate reflected our assessment of the likelihood that Exalgo would be approved by the FDA during each of the four time periods included in the merger agreement and the corresponding pre-determined amount of escrow shares that would be released to the former Neuromed stockholders as a result thereof. The probabilities assigned to each Exalgo FDA approval outcome ranged from 10% to 40% and were determined based upon a review of four other product candidates for the treatment of pain that were in or had recently completed the FDA review process that were not yet approved or had long approval delays. We classified the fair value of contingent consideration as a long-term liability in the balance sheet and reassessed the fair value of the contingent consideration at December 31, 2009. In connection therewith, we recorded a gain of $12.1 million primarily as a result of a decrease in our stock price from the date of merger closing through December 31, 2009 and the FDA approval of Exalgo not occurring before January 1, 2010 impacting our probability assessment of the amount of shares expected to be released from escrow. We reassessed the fair value of the contingent consideration through March 1, 2010, the date that the FDA approved Exalgo. We recorded other expense of $29.3 million in the first quarter of 2010 associated with the remeasurement of the contingent consideration to fair value upon release of the incremental shares from escrow to the former Neuromed shareholders.
53
We assess the fair value of assets acquired, including intangible assets, using a variety of methods including present-value models. Each asset is measured at fair value from the perspective of a market participant. Transaction costs and restructuring costs associated with the transaction are expensed as incurred. In the event the fair value of the assets acquired less the fair value of the liabilities assumed exceeds the value of the consideration transferred, as was the case with the Neuromed Merger, a bargain purchase would be deemed to have occurred and a gain would be recorded in our statement of operations.
The determination and allocation of the consideration transferred requires management to make significant estimates and assumptions, especially at the acquisition date with respect to the fair value of the contingent consideration and intangible assets acquired.
Intangible Assets
We have a significant intangible asset in our rights to Exalgo milestones and royalties as a result of the Neuromed Merger. Our intangible asset has a finite life and is being amortized based upon the pattern in which we expect to utilize the economic benefits. Determining the economic lives of acquired finite-lived intangible assets requires us to make significant judgments and estimates, and changes in those estimates could materially impact our results of operations.
The value of the intangible asset was initially determined by a risk-adjusted, discounted cash flow approach. We assess the potential impairment of the intangible asset whenever events or changes in circumstances indicate that the carrying value may not be recoverable. In connection with this assessment, we consider the cash flow forecast for projected royalties and any other factors that we are aware of that could impact the amount of royalties we might receive in future periods. Future adverse changes or other unforeseeable factors could result in an impairment charge that would materially impact our future results of operations and financial position in the relevant reporting period.
Revenue Recognition
We have entered into collaborative research and development agreements with other pharmaceutical and biotechnology companies, government agencies and charitable foundations. These agreements are generally in the form of research and development and license agreements. The agreements are for Exalgo and for development of early-stage compounds and are generally focused on specific disease areas. The agreements provide for nonrefundable up-front payments, milestone payments upon achieving significant milestone events and in some cases ongoing research funding. The agreements also contemplate royalty payments on sales if and when the product receives marketing approval by the FDA or other regulatory agency.
We evaluate our agreements with software license components in order to determine whether the arrangements should be accounted for under revenue recognition guidance for software or if other applicable revenue recognition guidance should be applied. Revenue arrangements with multiple deliverables are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered elements. The consideration received is allocated among separate elements based on their respective fair values. Revenue is recognized when there is persuasive evidence of an arrangement, delivery has occurred, the price is fixed or determinable and collection is reasonably assured. License fees or other amounts received in advance of performance obligations, or in cases where we have a continuing obligation to perform services, are deferred and recognized over the performance period. Revenues from milestone payments that are deemed to be substantive and represent the culmination of a separate earnings process are recorded when the milestone is achieved. Contract revenues are recorded as the services are performed. Royalty revenue is recognized based upon net sales of licensed products as provided by the licensee and is recognized in the period the sales occur. The periods over which revenue should be recognized are subject to estimates by management and may change over the course of the collaborative agreement. Such a change could have a material impact on the amount of revenue we record in future periods.
54
Revenue under government grants or cost reimbursement contracts is recognized as we perform the underlying research and development activities.
Stock-Based Compensation
We estimate the value of our stock options using the Black-Scholes model which requires us to make assumptions regarding stock price volatility and expected term. Historically we analyzed the volatilities and expected terms of a peer group of companies and utilized a stock price volatility in the Black-Scholes model that reflects the average volatility of this peer group and our own historical volatility. We continued to increase the weight applied to our own historical volatility over time. In 2009, we determined that we had sufficient trading history as a public company and began to use our own historical volatility to estimate stock price volatility. We utilize the expected terms from this analysis of peer companies to support the expected term used in the Black-Scholes model, as we do not believe we have sufficient historical data to support this assumption.
We estimate the level of stock-based award forfeitures expected to occur and record compensation cost only for those awards that are ultimately expected to vest. We believe that using an average of our own historical data for percentage of option cancellations and actual employee turnover rates derived from periods of business activity most likely to be representative of future periods to be the most appropriate measure of forfeitures. Changes in estimated forfeitures are recognized through a cumulative catch-up adjustment in the period of change and will also impact the amount of compensation expense to be recognized in future periods. We have determined that the reductions in work force that occurred in the fourth quarter of 2008 and July 2009 and the divestiture of CombinatoRx Singapore in the second quarter of 2009 and discussed below under “Results of Operations—Gain from Discontinued Operations” were not likely to recur and, therefore, excluded these employees and their stock-based awards from the forfeiture rate calculation.
As of December 31, 2010, there was approximately $2.6 million of total stock-based compensation expense not yet recognized relating to non-vested awards granted under our stock option plans, restricted stock awards and restricted stock units. This expense is net of estimated forfeitures and is expected to be recognized over a weighted-average period of approximately 2.6 years. The amount of stock-based compensation expense to be recorded in any future period cannot be accurately predicted due to the uncertainty of future grant levels and actual forfeitures to be recorded. Additionally, changes to the assumptions used in the Black-Scholes model could cause a material change in the amount of stock-based compensation expense to be recorded in future reporting periods.
Accrued Expenses
As part of the process of preparing our consolidated financial statements, we are required to estimate certain accrued expenses. This process involves identifying services that third parties have performed on our behalf and estimating the amount of service performed and the associated cost incurred for these services as of the balance sheet date in our consolidated financial statements. Examples of estimated accrued expenses for our business are professional service fees, such as attorneys and accountants, contract service fees, such as amounts due to clinical research organizations who are supporting clinical trials for our product candidates preclinical and toxicology research services providers and formulation development providers. In connection with these service fees, our estimates are most affected by our understanding of the status and timing of services provided relative to the actual level of services incurred by the service providers. In the event that we do not identify certain costs that have been incurred or we under- or over-estimates the level of services or the costs of such services, our reported expenses for a reporting period could be understated or overstated.
Income Taxes
In preparing our consolidated financial statements, we estimate our income tax liability in each of the jurisdictions in which we operate by estimating our actual current tax expense together with assessing temporary differences resulting from differing treatment of items for tax and financial reporting purposes. These differences
55
result in deferred tax assets and liabilities, which are included in our consolidated balance sheets. Significant management judgment is required in assessing the realizability of our deferred tax assets. In performing this assessment, we consider whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. In making this determination, under the applicable financial accounting standards, we are allowed to consider the scheduled reversal of deferred tax liabilities, projected future taxable income, and the effects of tax planning strategies. Our estimates of future taxable income include, among other items, our estimates of future income tax deductions related to the exercise of stock options. In the event that actual results differ from our estimates, we adjust our estimates in future periods and we may need to establish a valuation allowance, which could materially impact our financial position and results of operations.
We account for uncertain tax positions using a “more-likely-than-not” threshold for recognizing and resolving uncertain tax positions. The evaluation of uncertain tax positions is based on factors that include, but are not limited to, changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, the effective settlement of matters subject to audit, new audit activity and changes in facts or circumstances related to a tax position. We evaluate uncertain tax positions on a quarterly basis and adjust the level of the liability to reflect any subsequent changes in the relevant facts surrounding the uncertain positions. Our liabilities for uncertain tax positions can be relieved only if the contingency becomes legally extinguished through either payment to the taxing authority or the expiration of the statute of limitations, the recognition of the benefits associated with the position meet the “more-likely-than-not” threshold or the liability becomes effectively settled through the examination process. We consider matters to be effectively settled once the taxing authority has completed all of its required or expected examination procedures, including all appeals and administrative reviews; we have no plans to appeal or litigate any aspect of the tax position; and we believe that it is highly unlikely that the taxing authority would examine or re-examine the related tax position. We also accrue for potential interest and penalties, related to unrecognized tax benefits in general and administrative expense.
Results of Operations
Years Ended December 31, 2010 and 2009
Revenue. For the year ended December 31, 2010, we recorded $46.7 million of revenue primarily from our collaboration agreements with Covidien, Novartis, Amgen, Fovea, and from government contracts and grants from NIAID and USAMRIID. We received a $40.0 million milestone payment from Covidien following the FDA approval of Exalgo on March 1, 2010. Covidien launched the commercial sale of Exalgo in the second quarter of 2010, and we began to receive royalty payments from Covidien in the second quarter of 2010 based upon a percentage of Covidien’s net sales of Exalgo. During the year ended December 31, 2010, we recognized $1.6 million in revenue related to royalties from Covidien based on sales of Exalgo. For the year ended December 31, 2009, we recorded $17.3 million of revenue from our collaboration agreements with Novartis, Angiotech, CFFT, CHDI, Fovea, the DMD Foundations, and from government contracts and grants from NIAID and USAMRIID. We expect revenue for the year ending December 31, 2011 to be significantly lower than revenue for the year ended December 31, 2010 due to the milestone earned from Covidien being a one-time payment upon the approval of Exalgo.
Research and Development Expense. Research and development expense for the year ended December 31, 2010 was $23.0 million compared to $21.2 million for the year ended December 31, 2009. The $1.8 million increase was primarily due to an increase of $2.9 million in salary and benefit expenses related to the addition of Zalicus Canada personnel, a $3.5 million increase in consulting expense, preclinical expenses, formulations and lab supplies and a $0.2 million increase in stock-based compensation expense related to the approval of Exalgo satisfying certain performance based vesting criteria for stock-based awards offset by a decrease of $2.0 million in external clinical expenses and a $2.8 million decrease in facilities, depreciation and other overhead costs associated with the 2009 restructuring. We expect our research and development expense for the year ended December 31, 2011 to increase as we plan to advance Synavive into a Phase 2b clinical trial and advance one or more ion channel product candidates into Phase 1 clinical trials.
56
The table below summarizes our allocation of research and development expenses to Exalgo, our clinical programs, including Synavive, and our preclinical and discovery programs for the years ended December 31, 2010 and 2009. Our internal project costing methodology, which was implemented in 2007, does not allocate all of the personnel and other indirect costs from all of our research and development departments to specific clinical and preclinical programs, and such unallocated costs are further summarized in the table below. Other clinical program costs consist primarily of the personnel and other expenses for our clinical operations department, the majority of which supported the development of Exalgo and Synavive. Collaboration discovery and preclinical program costs consist of the direct costs allocated to all of our cHTS research collaborations, as well as personnel and other expenses allocated to our internally funded preclinical programs, other than our ion channel program. Other clinical and preclinical program costs consist primarily of the personnel and other expenses for our formulations, pharmacology, regulatory and discovery departments, the majority of which supported the development of our clinical product candidates, including Exalgo and Synavive, as well as our preclinical product candidates, including candidates from our ion channel program. Infrastructure and support costs consist of facility costs, depreciation and amortization and costs for research and development support personnel such as our informatics and facilities departments. Due to the uncertainty in drug development and the stage of development of our pre-clinical and clinical programs, we are unable to predict the nature, specific timing and estimated costs to complete the development of our product candidates or the timing of when material cash inflows may commence.
| Year Ended December 31, |
||||||||
| 2010 | 2009 | |||||||
| (in thousands) | ||||||||
| Exalgo |
$ | 659 | $ | — | ||||
| Synavive |
2,346 | 3,173 | ||||||
| Other clinical program costs |
1,605 | 1,527 | ||||||
| Total clinical program costs |
4,610 | 4,700 | ||||||
| Collaboration discovery and preclinical program costs |
4,199 | 5,256 | ||||||
| Ion channel program costs |
4,747 | — | ||||||
| Unallocated clinical and preclinical program costs |
2,023 | 1,681 | ||||||
| Infrastructure and support costs |
5,899 | 8,303 | ||||||
| Noncash employee and non-employee stock-based compensation expense |
1,533 | 1,304 | ||||||
| Total research and development expenses |
$ | 23,011 | $ | 21,244 | ||||
General and Administrative Expense. General and administrative expense for the year ended December 31, 2010 was $12.1 million compared to $17.1 million for the year ended December 31, 2009. The $5.0 million decrease was primarily due to the $3.6 million decrease in legal and consulting fees related to the Neuromed Merger and a decrease of $1.2 million in stock-based compensation expense. We expect our general and administrative expenses for the year ending December 31, 2011 to be consistent with 2010.
Amortization of Intangible Asset. For the years ended December 31, 2010 and 2009, we recorded $18.7 million and $0.5 million, respectively, of amortization expense related to the Exalgo intangible asset acquired in the Neuromed Merger. The increase is due to amortization being recorded for a full year in 2010. The intangible asset is being amortized in a manner which reflects our estimate of the future undiscounted cash flows we expect to receive over the estimated useful life of Exalgo. We expect to record amortization expense of $5.1 million in the year ending December 31, 2011.
Interest Income. Interest income decreased to $0.1 million for the year ended December 31, 2010 from $0.3 million for the year ended December 31, 2009. The decrease in interest income was primarily caused by significantly lower average interest rates for the securities held in our investment portfolio.
Interest Expense. Interest expense, for the years ended December 31, 2010 and 2009, was less than $0.1 million. We expect interest expense to increase in the year ending December 31, 2011 as a result of the term loan payable being outstanding for an entire year and the possibility of additional borrowings in 2011.
57
Loss on Revaluation of Contingent Consideration. In the year ended December 31, 2010, we recognized a loss in the amount of $29.3 million on the revaluation of contingent consideration in connection with the Neuromed Merger compared to a gain of $12.1 million for the year ended December 31, 2009. This loss represents the change in fair value of the contingent consideration at December 31, 2009 and March 1, 2010 when the contingent consideration was settled. On March 1, 2010, 37,883,123 escrow shares were released to former Neuromed stockholders as settlement of the contingent consideration. The change in value is due to the fair value of shares released from escrow being greater than our probability weighted estimate as of December 31, 2009.
Benefit for Income Taxes. In the years ended December 31, 2010 and 2009, we recognized a tax benefit of $1.2 million and $0.1 million, respectively. The income tax benefit for the year ended December 31, 2010 was primarily related to the reversal of an unrecognized tax benefit for an uncertain tax position due to the expiration of the relevant statute of limitations.
Gain from Discontinued Operations. On June 2, 2009, we divested our 51% equity ownership interest in CombinatoRx Singapore, by selling our 2,602,041 ordinary shares of CombinatoRx Singapore to the other shareholder of CombinatoRx Singapore, BioMedical Sciences, for nominal consideration. In connection with the divestiture, we entered into a termination agreement with CombinatoRx Singapore and BioMedical Sciences, pursuant to which the parties agreed to terminate all of the prior agreements among us, CombinatoRx Singapore and BioMedical Sciences relating to the joint funding and operations of CombinatoRx Singapore. As a result of the divestiture and the termination of the prior agreements, CombinatoRx Singapore is no longer affiliated with us, and the issued and outstanding preferred shares and convertible promissory notes issued by CombinatoRx Singapore and held by BioMedical Sciences and are no longer convertible into shares of our common stock. We recorded a gain from discontinued operations of $14.1 million during the year ended December 31, 2009.
Years Ended December 31, 2009 and 2008
Revenue. For the year ended December 31, 2009, we recorded $17.3 million of revenue from our collaboration agreements with Novartis, Angiotech, CFFT, CHDI, Fovea and the DMD Foundations and from government contracts and grants from NIAID and USAMRIID. For the year ended December 31, 2008, we recorded $12.3 million of revenue from our research and collaboration agreements with Angiotech, CFFT, CHDI, HenKan and the DMD Foundations and from government contracts and grants from NIAID and USAMRIID. The $5.0 million increase in revenue is primarily due to $7.0 million of accelerated revenue recognition due to the early termination of our agreement with Angiotech.
Research and Development Expense. Research and development expense for the year ended December 31, 2009 was $21.2 million compared to $55.3 million for the year ended December 31, 2008. The $34.1 million decrease from the 2008 period to the 2009 period was primarily due to a decrease of $16.7 million in preclinical and external clinical expenses, a $10.8 million decrease in compensation and benefit expenses, associated with reduced headcount attributable to the 2008 and 2009 restructurings, a $5.6 million decrease in consulting, lab supplies, facilities, depreciation and other overhead costs related to our 2008 and 2009 restructurings, as well as a $1.0 million decrease in non-cash stock-based compensation expense.
The table below summarizes our allocation of research and development expenses to our clinical programs, including Synavive, CRx-401, CRx-191 and CRx-197 for the years ended December 31, 2009 and 2008. Our internal project costing methodology does not allocate all of the personnel and other indirect costs from all of our research and development departments to specific clinical and preclinical programs, and such unallocated costs are further summarized in the table below. Unallocated clinical program costs consist primarily of the personnel and other expenses for our clinical operations, medical affairs, biostatistics, data systems, medical writing and clinical program leadership departments, all of which support the development of the clinical product candidates Synavive, CRx-401, CRx-191 and CRx-197. Preclinical program costs consist of the personnel and other expenses allocated to our internally funded preclinical programs and the direct costs allocated to all of our research collaborations,
58
including the personnel costs of our alliance management department. Other clinical and preclinical program costs consist primarily of the personnel and other expenses for our formulations, pharmacology, regulatory, quality, new products and discovery departments, all of which supported the development of both our clinical product candidates Synavive, CRx-401, CRx-191 and CRx-197, as well as our preclinical product candidates. Infrastructure and support costs consist of facility costs, depreciation and amortization and costs for research and development support personnel such as our informatics and facilities departments.
| Year Ended December 31, |
||||||||
| 2009 | 2008 | |||||||
| (in thousands) | ||||||||
| Synavive |
$ | 3,173 | 21,021 | |||||
| CRx-401 |
1,171 | 3,491 | ||||||
| CRx-197 |
— | 2,448 | ||||||
| CRx-191 |
(34 | ) | 1,196 | |||||
| Other clinical program costs |
390 | 2,286 | ||||||
| Total clinical program costs |
4,700 | 30,442 | ||||||
| Preclinical program costs |
5,256 | 9,906 | ||||||
| Unallocated clinical and preclinical program costs |
1,681 | 5,414 | ||||||
| Infrastructure and support costs |
8,303 | 7,236 | ||||||
| Noncash employee and non-employee stock-based compensation expense |
1,304 | 2,298 | ||||||
| Total research and development expenses |
$ | 21,244 | $ | 55,296 | ||||
General and Administrative Expense. General and administrative expense for the year ended December 31, 2009 was $17.1 million compared to $14.5 million for the year ended December 31, 2008. The $2.6 million increase was primarily due to the $4.4 million increase in professional and consulting fees associated with our strategic realignment and the Neuromed Merger offset by a $0.8 million decrease in non-cash stock-based compensation and a $1.1 million decrease in compensation and benefits expense associated with reduced headcount attributable to the 2008 and 2009 restructurings.
Restructuring. For the year ended December 31, 2009, we recorded a net restructuring charge of $2.7 million primarily related to the third quarter 2009 restructuring related to the continued implementation of our plan to focus our efforts on our funded drug discovery and conserving capital in connection with the Neuromed Merger. The charge consisted of $2.7 million for severance and related benefits for approximately 23 employees, $1.8 million of accelerated stock-based compensation and $1.0 million of lease termination costs, partially offset by $2.8 million of non-cash credits related to the write-off of deferred rent and lease incentive obligations associated with the facilities that we exited.
Amortization of Intangible Asset. We recorded $0.5 million of amortization related to the Exalgo intangible asset acquired in the Neuromed Merger. The intangible asset is being amortized in a manner which reflects our estimate of the future undiscounted cash flows we expect to receive over the estimated useful life of Exalgo.
Gain on Legal Settlement. On April 30, 2009, we filed a lawsuit against Aptuit, Inc., or Aptuit, in the Supreme Court for the State of New York, New York County, Commercial Division. We asserted claims against Aptuit for fraudulent inducement, breach of contract, breach of the implied covenant of good faith and fair dealing and unjust enrichment arising out of Aptuit’s manufacture and distribution of our product candidate Synavive for a worldwide Phase 2b clinical trial targeting subjects with rheumatoid arthritis. On December 29, 2009, we entered into a settlement agreement with Aptuit to settle this lawsuit. Pursuant to the terms of the settlement agreement, Aptuit agreed to pay us $3.7 million, and the parties agreed to a mutual release of claims and causes of action that were asserted or that could have been asserted in the action. We received the $3.7 million payment in January 2010.
59
Interest Income. Interest income decreased to $0.3 million for the year ended December 31, 2009 from $2.3 million for the year ended December 31, 2008. The decrease in interest income was primarily caused by decreases in our average cash and short-term investments balances and significantly lower average interest rates for the securities held in our investment portfolio.
Interest Expense. Interest expense for the year ended December 31, 2009 was less than $0.1 million compared to $0.7 million for the year ended December 31, 2008. This decrease was due to the repayment of equipment lines of credit in December 2008.
Gain on Bargain Purchase. In the year ended December 31, 2009, we recognized a gain on bargain purchase in the amount of $9.8 million related to the acquisition of Neuromed. The gain on bargain purchase represents the excess fair value of the net assets acquired less the total consideration transferred to the former Neuromed stockholders.
Gain on Revaluation of Contingent Consideration. In the year ended December 31, 2009, we recognized a gain in the amount of $12.1 million on the revaluation of contingent consideration in connection with the Neuromed Merger. This gain represents the change in fair value of the contingent consideration between the merger date and December 31, 2009. The change in value is due to FDA approval not occurring before January 1, 2010 that impacted our probability assessment and a decline in the closing price of our common stock between the merger date and December 31, 2009.
Liquidity and Capital Resources
Since our inception in March 2000, we have funded our operations principally through private and public offerings of our equity securities and, to a lesser extent, from debt financing, payments from our collaboration partners and proceeds from litigation. As of December 31, 2010, we had cash, cash equivalents and short-term investments of approximately $46.5 million, which includes $1.9 million of restricted cash. Our funds are primarily invested in short-term, government agency securities, United States Treasury money market funds and short-term corporate debt securities, and as such, we do not believe there is significant risk in our investment portfolio as of December 31, 2010.
We expect our resources to be sufficient to fund our planned operations into 2014. However, we may require significant additional funds earlier than we currently expect if our research and development expenses exceed our current expectations or Exalgo royalties or our collaboration funding is less than our current expectations. We may seek additional funding through collaboration agreements and public or private financings of debt or equity capital. However, funding may not be available to us on acceptable terms or at all. If we are unable to obtain funding on a timely basis, we may be required to significantly curtail one or more of our research or development programs or our operations. We also could be required to seek funds through arrangements with collaborators or others that may require us to relinquish rights to some of our technologies or product candidates which we would otherwise pursue on our own.
Operating Activities. Our operating activities provided cash of $18.7 million for the year ended December 31, 2010 and used cash of $25.0 million and $55.6 million for the years ended December 31, 2009 and 2008, respectively. The $43.7 million increase in net cash flows from operating activities is primarily attributed to the $40.0 million milestone received from Covidien related to the FDA approval of Exalgo and a reduction in merger related operating expenses during the year ended December 31, 2010. This increase is also attributed to working capital adjustments in accounts receivable, accounts payable, accrued expenses and accrued restructuring expense.
The decrease in our net use of cash in operating activities in the year ended December 31, 2009 compared to the year ended December 31, 2008 is primarily attributed to the increase in income from continuing operations of $61.9 million as well as adjustments for non-cash gains and losses and adjustments for changes in working capital during the year.
60
Investing Activities. Our investing activities used cash of $27.6 million for the year ended December 31, 2010 and provided cash of $23.4 million and $57.8 million in the years ended December 31, 2009 and 2008, respectively. The cash used in investing activities for the year ended December 31, 2010 was primarily due to net purchases of short-term investments of $27.3 million and purchases of fixed assets of $1.1 million. The cash provided by investing activities in the years ended December 31, 2009 and 2008, respectively, was primarily due to sales and maturities of short-term investments. Purchases of property and equipment decreased from $2.3 million for the year ended December 31, 2008 to $0.6 million for the year ended December 31, 2009. For the year ended December 31, 2009, our cash provided by investing activities included $6.2 million of cash acquired in the Neuromed Merger. In addition, in 2009, our restricted cash balance decreased by $1.5 million, related to the amendment to the lease for our Cambridge, Massachusetts facility.
Financing Activities. Our financing activities generated cash proceeds of $2.9 during the year ended December 31, 2010. These proceeds relate primarily to the $2.9 million of net proceeds from Term Loan A under the loan and security agreement entered into on December 22, 2010. Our financing activities were not a significant source or use of cash during the year ended December 31, 2009. Our financing activities used $3.4 million of cash proceeds as of December 31, 2008. The financing activities in the year ended December 31, 2008 consisted of the issuance of $5.0 million of convertible promissory notes by CombinatoRx Singapore to BioMedical Sciences in August 2008 included in financing activities from discontinued operations, offset by the repayment of indebtedness to General Electric Capital Corporation, or GECC, in the amount of $8.5 million.
Oxford Loan and Security Agreement
On December 22, 2010, we entered into a loan and security agreement with Oxford Finance Corporation, or Oxford, pursuant to which Oxford agreed to lend us up to $20.0 million. Upon entering into the loan and security agreement, we borrowed $3.0 million from Oxford as Term Loan A. Under the terms of the loan and security agreement, we may, in our sole discretion, borrow from Oxford up to an additional $8.5 million, at any time on or before July 15, 2011, called Term Loan B, and up to an additional $8.5 million, at any time on or before January 15, 2012, called Term Loan C, collectively with Term Loan A and Term Loan B, called the Term Loans. Our wholly owned subsidiary, Zalicus Canada, is also a party to the loan and security agreement as a co-borrower. Our obligations under the loan and security agreement are secured by a first priority security interest in substantially all of our assets, including those of Zalicus Canada, other than intellectual property.
We are required to pay interest on Term Loan A on a monthly basis through and including July 1, 2011. Beginning August 1, 2011 through the maturity of Term Loan A on July 1, 2014, we will be required to make payments of outstanding principal and interest on Term Loan A in 36 equal monthly installments. Interest is payable on Term Loan A at an annual interest rate of 10.26%. If we enter into Term Loan B or Term Loan C, interest on each term loan will accrue at an annual fixed rate equal to greater of (i) 10.25% or (ii) the sum of (a) the 90-day U.S. Dollar LIBOR rate prior to the funding of such term loan, plus (b) 9.96%. Payments due under Term Loan B or Term Loan C, if borrowed, are interest only, payable monthly, in arrears, for six months following the funding of each term loan, and will consist of 36 payments of principal and interest which are payable monthly, in arrears, and all unpaid principal and accrued and unpaid interest on Term Loan B or Term Loan C would be due and payable 42 months after the funding of any each term loan.
Upon the last payment date of the amounts borrowed under the loan and security agreement, whether on the maturity date of one of the Term Loans, on the date of any prepayment or on the date of acceleration in the event of a default, we will be required to pay Oxford a final payment fee equal to 1.5% of any of the Term Loans borrowed. In addition, if we repay all or a portion of the Term Loans prior to maturity, we will pay Oxford a prepayment fee of four percent of the total amount prepaid if the prepayment occurs prior to the first anniversary of the funding of the relevant Term Loan, two percent of the total amount prepaid if the prepayment occurs between the first and second anniversary of the funding of the relevant Term Loan, and one percent of the total amount prepaid if the prepayment occurs on or after the second anniversary of the funding of the relevant Term Loan.
Upon the occurrence of an event of default, as defined in the loan and security agreement, including payment defaults, breaches of covenants, a material adverse change in the collateral, our business, operations or condition
61
(financial or otherwise) and certain levies, attachments and other restraints on our business, the interest rate will be increased by five percentage points and all outstanding obligations will become immediately due and payable.
In connection with the loan and security agreement, we issued to Oxford a warrant to purchase 64,748 shares of our common stock. The warrant is exercisable, in whole or in part, immediately, and has a per share exercise price of $1.39 and may be exercised on a cashless basis. The warrant will terminate on the earlier of December 22, 2017 and the closing of a merger or consolidation transaction in which Zalicus is not the surviving entity.
If we borrow Term Loan B or Term Loan C, upon the funding of each Term Loan, the warrant will become exercisable for an additional number of shares of our common stock determined by dividing 3.0% of the amount of each such Term Loan by the volume-weighted average price of our common stock on the Nasdaq Global Market for the ten trading days prior to the funding of such Term Loan. The exercise price for any additional shares of our common stock that may be purchased by the Lender pursuant to the Warrant following our borrowing Term Loan B or Term Loan C will be the volume-weighted average price of our common stock on Nasdaq for the ten trading days prior to the funding of such Term Loan.
Shelf Registration Statement and Equity Offering
On April 16, 2010, we filed a shelf registration statement on Form S-3 under the Securities Act of 1933, as amended (SEC File No. 333-166116), with the SEC covering the registration of senior debt securities, subordinated debt securities, preferred stock, common stock, warrants, units or any combination of the foregoing securities, in an aggregate amount of $50.0 million, which was declared effective by the SEC on April 29, 2010. These securities may be offered from time to time in amounts, at prices and on terms to be determined at the time of sale. We believe the shelf registration statement provides additional financing flexibility to meet potential future funding requirements and the ability to take advantage of potentially attractive capital market conditions.
On February 9, 2011, we entered into an equity distribution agreement with Wedbush Securities Inc., or Wedbush, under which we may, from time to time, utilize our effective shelf registration statement to offer and sell our common stock having aggregate sales proceeds of up to $20.0 million through Wedbush, or to Wedbush, for resale. Sales of our common stock through Wedbush will be made by means of ordinary brokers’ transactions on the Nasdaq Global Market or otherwise at market prices prevailing at the time of sale, in block transactions, or as otherwise agreed upon by us and Wedbush. Wedbush will use commercially reasonable efforts to sell our common stock from time to time, based upon instructions from us (including any price, time or size limits or other customary parameters or conditions we may impose). We will pay Wedbush a commission, or allow a discount, as the case may be, in each case equal to 3.0% of the gross offering proceeds of any common stock sold through Wedbush as agent. We have agreed to reimburse Wedbush for certain expenses incurred by them in connection with the offering, up to an aggregate of $80,000, plus up to an additional $20,000 per calendar quarter related to ongoing maintenance, due diligence expenses and other expenses; provided, however, that in no event will we be liable for any such quarterly expenses in excess of $150,000 in the aggregate. We also may sell our common stock to Wedbush, as principal for its own account, at a price to be agreed upon at the time of sale.
As of March 8, 2011, we have sold an aggregate of 4,813,600 shares of common stock under the equity distribution agreement at an average price of approximately $2.21 per share for gross proceeds of approximately $10.7 million. Net proceeds were approximately $10.2 million after deducting Wedbush’s commissions and expenses to establish the agreement.
Discontinued Operations. On June 2, 2009, we divested our 51% equity ownership interest in CombinatoRx Singapore, by selling our 2,602,041 ordinary shares of CombinatoRx Singapore to the other shareholder of CombinatoRx Singapore, BioMedical Sciences, for nominal consideration. In connection with the divestiture, we entered into a termination agreement with CombinatoRx Singapore and BioMedical Sciences, pursuant to which the parties agreed to terminate all of the prior agreements among us, CombinatoRx Singapore and BioMedical Sciences relating to the joint funding and operations of CombinatoRx Singapore. As a result of the divestiture
62
and the termination of the prior agreements, CombinatoRx Singapore is no longer affiliated with us, and the issued and outstanding preferred shares and convertible promissory notes issued by CombinatoRx Singapore and held by BioMedical Sciences and are no longer convertible into shares of our common stock.
We also entered into a share purchase agreement with CombinatoRx Singapore and BioMedical Sciences and an intellectual property assignment agreement with CombinatoRx Singapore. Under the intellectual property assignment agreement, CombinatoRx Singapore has been assigned and retains all infectious disease intellectual property developed by CombinatoRx Singapore with our assistance since the formation of CombinatoRx Singapore. Under the share purchase agreement, we agreed not to compete with CombinatoRx Singapore in the discovery and development of product candidates to treat certain infectious diseases in substantially all markets until June 2, 2010. The results of operations and the assets and the liabilities related to the divestiture of our Singapore subsidiary in June 2009 have been accounted for as discontinued operations. Accordingly, the results of operations from prior periods have been reclassified to discontinued operations as a result of the divestiture.
Contractual Obligations and Commitments
The following table summarizes our contractual obligations at December 31, 2010 and the effects such obligations are expected to have on our liquidity and cash flows in future periods.
| Contractual Obligations |
Total | 2011 | 2012 through 2013 |
2014 through 2015 |
After 2016 |
|||||||||||||||
| Short and long-term debt: |
||||||||||||||||||||
| Notes payable issued to Oxford(1) |
$ | 3,697 | $ | 640 | $ | 2,332 | $ | 725 | $ | — | ||||||||||
| Operating lease obligations: |
||||||||||||||||||||
| Cambridge facility(2) |
7,354 | 1,114 | 2,449 | 2,459 | 1,332 | |||||||||||||||
| Vancouver facility(3) |
246 | 246 | — | — | — | |||||||||||||||
| Total contractual obligations |
$ | 11,297 | $ | 2,000 | $ | 4,781 | $ | 3,184 | $ | 1,332 | ||||||||||
| (1) | On December 22, 2010, we borrowed $3.0 million under Term Loan A of the loan and security agreement with Oxford. This term loan bears interest at 10.25%. We are required to make monthly payments of interest only through July 1, 2011 and then will be required to make 36 equal monthly payments of principal and interest. See Note 9 of our audited consolidated financial statements for additional information relating to the agreement with Oxford. |
| (2) | On October 18, 2005, we entered into a lease agreement for approximately 40,000 square feet of office and laboratory space in Cambridge, Massachusetts. On March 9, 2006, we entered into an amendment to the lease agreement to lease an additional approximately 23,000 square feet of laboratory space. On August 3, 2009, we entered into a second amended lease agreement to reduce our leased space to approximately 23,000 square feet of office and laboratory space. The lease term, as amended, extends through January 2017. Our rent obligations under the lease, as amended, are reflected as operating lease obligations in the table above. The amounts in the table above do not include management fees payable to our landlord for our leased space, which are meant to cover our allocable share of property taxes, utilities and other common area costs. Our payment obligations under the amended lease are supported by a standby letter of credit totaling $1.8 million. |
| (3) | In connection with the Neuromed merger, we assumed a sublease renewal and amendment agreement between Zalicus Canada and Discovery Parks Incorporated, dated November 2, 2009, relating to our office and laboratory facility in Vancouver, British Columbia, Canada. Under the terms of this sublease we subleased approximately 24,600 square feet of office and laboratory space, which was reduced to approximately 12,000 square feet in 2010. The lease will expire on December 31, 2011. We have the option to extend the lease for an additional year. Our obligations under the lease are reflected as operating lease obligations in the table above. |
63
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements or relationships with unconsolidated entities of financial partnerships, such as entities often referred to as structured finance or special purpose entities.
Climate Change
Recent scientific studies have suggested that emissions of certain gases, commonly referred to as “greenhouse gases,” may be contributing to warming of the earth’s atmosphere. As a result, there have been a variety of regulatory developments, proposals or requirements and legislative initiatives that have been introduced in the United States (as well as other parts of the world) that are focused on restricting the emission of carbon dioxide, methane and other greenhouse gases. Based upon our existing and currently expected future operations, we do not believe that climate change itself, or any current or future legislation aimed at preventing its advance, will have a significant effect on our operations.
| Item 7A. | Quantitative and Qualitative Disclosure about Market Risk |
We are exposed to market risk related to changes in interest rates and changes in the exchange rate of the United States dollar to the Canadian dollar. As of December 31, 2010, we had unrestricted cash, cash equivalents and marketable securities of $44.6 million consisting of cash and highly liquid short-term investments. Our cash is deposited in and invested through highly rated financial institutions in North America and Canada. Our marketable securities are subject to interest rate risk and will decrease in value if market interest rates increase. If market interest rates were to increase immediately and uniformly by 10% from levels at December 31, 2010, we estimate that the fair value of our investments will decline by an immaterial amount, and therefore, our exposure to interest rate changes is immaterial.
The amounts outstanding under Term Loan A from Oxford are fixed at an annual interest rate of 10.25%. The loan and security agreement entered into with Oxford in December of 2010 allows for additional borrowings in the form of two additional term loans. In the event, we enter into the additional term loans, the interest rate will be the greater of (i) 10.25% or (ii) the 90-day LIBOR U.S. dollar rate prior to the funding of the term loan plus an additional 9.96%. In the event we make additional borrowings under the loan and security agreement, changes in LIBOR interest rates may increase the interest rates we would pay on such term loans and increase our cost of capital which would have a significant impact to our financial condition.
Transactions by our subsidiary, Zalicus Canada, may be denominated in Canadian dollars, however, the entity’s functional currency is the United States dollar. Exchange gains or losses resulting from the translation between the currency in which a transaction is denominated and functional currency of Zalicus Canada are included in net loss for our consolidated financial statements. Fluctuations in exchange rates, primarily between the United States dollar and the Canadian dollar, may adversely affect our results of operations, financial position and cash flows. We do not hedge this exposure.
| Item 8. | Financial Statements and Supplementary Data |
The information called for by this item is indexed on page F-1 of this Annual Report on Form 10-K and is contained on pages F-2 through F-35.
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
64
| Item 9A. | Controls and Procedures |
| (a) | Evaluation of Disclosure Controls and Procedures |
As required by Rule 13a-15(b) of the Securities Exchange Act of 1934, as amended (the “1934 Act”), the company’s management, including the Chief Executive Officer and the Chief Financial Officer, conducted an evaluation as of the end of the period covered by this Annual Report on Form 10-K of the effectiveness of the design and operation of the company’s disclosure controls and procedures. Based on that evaluation, the company’s Chief Executive Officer and Chief Financial Officer concluded that the company’s disclosure controls and procedures are effective at the reasonable assurance level in ensuring that information required to be disclosed by the company in the reports that it files or submits under the 1934 Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms.
| (b) | Management’s Report on Internal Control Over Financial Reporting |
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the 1934 Act as a process designed by, or under the supervision of, the issuer’s principal executive and principal financial officers, or persons performing similar functions, and effected by the issuer’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
| (1) | Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the issuer; |
| (2) | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the issuer are being made only in accordance with authorizations of management and directors of the issuer; and |
| (3) | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the issuer’s assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2010. In making this assessment, the company’s management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework. Based on this assessment, our management concluded that, as of December 31, 2010, our internal control over financial reporting is effective based on those criteria.
Our independent registered accounting firm has issued an audit report on our internal control over financial reporting. The report appears below:
65
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Zalicus Inc.
We have audited Zalicus Inc’s. internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Zalicus Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion Zalicus Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets as of December 31, 2010 and 2009, and the related consolidated statements of operations, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2010 of Zalicus Inc. and our report dated March 10, 2011 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Boston, Massachusetts
March 10, 2011
66
Changes in Internal Control
There has been no change to the company’s internal control over financial reporting during the last quarter of the fiscal year covered by this Annual Report on Form 10-K that has materially affected, or is reasonably likely to materially affect, the company’s internal control over financial reporting.
| Item 9B. | Other Information |
None.
PART III
| Item 10. | Directors and Executive Officers |
Information concerning our directors and executive officers will appear in our Proxy Statement for the 2011 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before May 2, 2011, under the captions “Election of Directors” and “Executive Officers.” Such information is incorporated herein by reference.
Information concerning compliance with Section 16(a) of the Act of 1934 will appear in the company’s Proxy Statement for the 2011 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before May 2, 2011, under the caption “Section 16(a) Beneficial Ownership Reporting Compliance.” Such information is incorporated herein by reference.
Information about our Audit Committee, including the members of the Committee, and our Audit Committee financial experts, will appear in our Proxy Statement for the 2011 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before May 2, 2011, under the captions “The Audit Committee” and “Audit Committee Financial Experts.” Such information is incorporated herein by reference.
Information concerning our Code of Ethics and Conduct will appear in our Proxy Statement for the 2011 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before May 2, 2011, under the caption “Code of Ethics and Conduct.” Such information is incorporated herein by reference.
| Item 11. | Executive Compensation |
Information in response to this item will appear in our Proxy Statement for the 2011 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before May 2, 2011, under the captions “Executive Compensation,” “Director Compensation” and “Report of the Compensation Committee on Executive Compensation.” Such information is incorporated herein by reference.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
Information concerning security ownership of certain beneficial owners and management will appear in our Proxy Statement for the 2011 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before May 2, 2011, under the caption “Security Ownership of Certain Beneficial Owners and Management.” Such information is incorporated herein by reference.
| Item 13. | Certain Relationships and Related Transactions |
Information concerning certain relationships and related transactions will appear in the company’s Proxy Statement for the 2011 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before May 2, 2011, under the caption “Certain Relationships and Related Transactions.” Such information is incorporated herein by reference.
67
| Item 14. | Principal Accountant Fees and Services |
Information concerning principal accounting fees and services will appear in our Proxy Statement for the 2011 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before May 2, 2011, under the caption “Independent Public Accountants.” Such information is incorporated herein by reference.
PART IV
| Item 15. | Exhibits, Financial Statement Schedules |
(a)(1) Financial Statements.
The consolidated financial statements filed as part of this Annual Report on Form 10-K are listed and indexed at page F-1.
(a)(2) Financial Statement Schedules.
Certain schedules are omitted because they are not applicable, or not required, or because the required information is included in the consolidated financial statements or notes thereto.
(a)(3) Exhibits.
The Exhibits listed in the Exhibit Index immediately preceding the Exhibits are filed as a part of this Annual Report on Form 10-K.
68
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| ZALICUS INC. | ||
| By: | /s/ MARK CORRIGAN | |
| Mark Corrigan President and Chief Executive Officer | ||
Date: March 10, 2011
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the date indicated.
| Signature |
Title |
Date | ||
| /s/ MARK CORRIGAN Mark Corrigan |
President and Chief Executive Officer (Principal Executive Officer) | March 10, 2011 | ||
| /s/ JUSTIN A. RENZ Justin A. Renz |
Senior Vice President and Chief Financial Officer (Principal Financial and Accounting Officer) | March 10, 2011 | ||
| /s/ SALLY CRAWFORD Sally Crawford |
Director | March 10, 2011 | ||
| /s/ TODD FOLEY Todd Foley |
Director | March 10, 2011 | ||
| /s/ FRANK HAYDU Frank Haydu |
Director | March 10, 2011 | ||
| /s/ WILLIAM HUNTER William Hunter |
Director | March 10, 2011 | ||
| /s/ MICHAEL KAUFFMAN Michael Kauffman |
Director | March 10, 2011 | ||
| /s/ W. JAMES O’SHEA W. James O’Shea |
Director | March 10, 2011 | ||
| /s/ HARTLEY RICHARDSON Hartley Richardson |
Director | March 10, 2011 | ||
69
Zalicus Inc.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| Page | ||||
| F-2 | ||||
| Consolidated Balance Sheets as of December 31, 2010 and 2009 |
F-3 | |||
| F-4 | ||||
| F-5 | ||||
| F-7 | ||||
| F-9 | ||||
F-1
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Zalicus Inc.
We have audited the accompanying consolidated balance sheets of Zalicus Inc. (the “Company”) as of December 31, 2010 and 2009, and the related consolidated statements of operations, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2010. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Zalicus Inc. at December 31, 2010 and 2009, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2010, in conformity with U.S. generally accepted accounting principles.
We have also audited, in accordance with standards of the Public Company Accounting Oversight Board (United States), the Company’s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 10, 2011, expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Boston, Massachusetts
March 10, 2011
F-2
Consolidated Balance Sheets
(in thousands except per share data)
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 2,820 | $ | 8,779 | ||||
| Restricted cash |
650 | 750 | ||||||
| Short-term investments |
41,799 | 14,551 | ||||||
| Accounts receivable |
1,605 | 2,927 | ||||||
| Prepaid expenses and other current assets |
965 | 5,415 | ||||||
| Total current assets |
47,839 | 32,422 | ||||||
| Property and equipment, net |
6,898 | 8,380 | ||||||
| Intangible asset, net |
26,687 | 45,423 | ||||||
| Restricted cash and other assets |
1,245 | 1,927 | ||||||
| Total assets |
$ | 82,669 | $ | 88,152 | ||||
| Liabilities and stockholders’ equity |
||||||||
| Current liabilities: |
||||||||
| Current portion of term loan payable |
$ | 273 | $ | — | ||||
| Accounts payable |
1,840 | 4,269 | ||||||
| Accrued expenses |
4,269 | 5,495 | ||||||
| Deferred revenue |
2,370 | 2,750 | ||||||
| Current portion of lease incentive obligation |
284 | 284 | ||||||
| Accrued restructuring |
— | 1,274 | ||||||
| Total current liabilities |
9,036 | 14,072 | ||||||
| Term loan payable, net of current portion |
2,523 | — | ||||||
| Deferred revenue, net of current portion |
3,667 | 2,667 | ||||||
| Deferred rent, net of current portion |
743 | 775 | ||||||
| Lease incentive obligation, net of current portion |
1,442 | 1,726 | ||||||
| Other long-term liabilities |
2,261 | 3,235 | ||||||
| Contingent consideration |
— | 12,764 | ||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.001 par value; 5,000 shares authorized; no shares issued and outstanding |
— | — | ||||||
| Common stock, $0.001 par value; 200,000 shares authorized; 89,113 and 117,828 shares issued and outstanding at December 31, 2010 and 2009, respectively |
89 | 118 | ||||||
| Additional paid-in capital |
317,581 | 272,405 | ||||||
| Accumulated other comprehensive loss |
(20 | ) | (2 | ) | ||||
| Accumulated deficit |
(254,653 | ) | (219,608 | ) | ||||
| Stockholders’ equity |
62,997 | 52,913 | ||||||
| Total liabilities and stockholders’ equity |
$ | 82,669 | $ | 88,152 | ||||
See accompanying notes.
F-3
Consolidated Statements of Operations
(in thousands, except share and per share amounts)
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Revenue: |
||||||||||||
| Collaborations |
$ | 45,969 | $ | 16,320 | $ | 11,462 | ||||||
| Government contracts and grants |
772 | 953 | 842 | |||||||||
| Total revenue |
46,741 | 17,273 | 12,304 | |||||||||
| Operating expenses: |
||||||||||||
| Research and development |
23,011 | 21,244 | 55,296 | |||||||||
| General and administrative |
12,115 | 17,081 | 14,469 | |||||||||
| Amortization of intangible asset |
18,736 | 520 | — | |||||||||
| Gain on legal settlement |
— | (3,700 | ) | — | ||||||||
| Restructuring |
— | 2,736 | 4,637 | |||||||||
| Total operating expenses |
53,862 | 37,881 | 74,402 | |||||||||
| Loss from operations |
(7,121 | ) | (20,608 | ) | (62,098 | ) | ||||||
| Interest income |
132 | 257 | 2,264 | |||||||||
| Interest expense |
(12 | ) | (28 | ) | (651 | ) | ||||||
| (Loss) gain on revaluation of contingent consideration |
(29,286 | ) | 12,068 | — | ||||||||
| Other income (expense) |
32 | (281 | ) | (4 | ) | |||||||
| Loss on early extinguishment of debt |
— | — | (195 | ) | ||||||||
| Gain on bargain purchase |
— | 9,809 | — | |||||||||
| Net (loss) income before provision for income taxes |
(36,255 | ) | 1,217 | (60,684 | ) | |||||||
| Benefit for income taxes |
1,210 | 67 | 108 | |||||||||
| Net (loss) income from continuing operations |
(35,045 | ) | 1,284 | (60,576 | ) | |||||||
| Discontinued operations: |
||||||||||||
| Loss from operations of discontinued subsidiary |
— | (1,536 | ) | (4,557 | ) | |||||||
| Gain on disposal of discontinued operations |
— | 15,640 | — | |||||||||
| Gain (loss) on discontinued operations |
— | 14,104 | (4,557 | ) | ||||||||
| Net (loss) income |
$ | (35,045 | ) | $ | 15,388 | $ | (65,133 | ) | ||||
| Net (loss) income per share—basic and diluted: |
||||||||||||
| From continuing operations |
$ | (0.42 | ) | $ | 0.03 | $ | (1.74 | ) | ||||
| From discontinued operations |
— | 0.38 | (0.13 | ) | ||||||||
| Net (loss) income per share—basic and diluted |
$ | (0.42 | ) | $ | 0.41 | $ | (1.87 | ) | ||||
| Weighted average number of common shares used in net (loss) income per share calculation: |
||||||||||||
| Basic |
82,663,645 | 37,338,042 | 34,848,701 | |||||||||
| Diluted |
82,663,645 | 37,491,237 | 34,848,701 | |||||||||
See accompanying notes.
F-4
Consolidated Statements of Stockholders’ Equity
(in thousands, except share amounts)
| Common Stock | Additional Paid-in Capital |
Accumulated Other Comprehensive Income (Loss) |
Accumulated Deficit |
Total | ||||||||||||||||||||
| Shares | Par Value |
|||||||||||||||||||||||
| Balance at December 31, 2007 |
34,822,170 | 35 | 261,187 | 226 | (186,213 | ) | 75,235 | |||||||||||||||||
| Net loss |
— | — | — | — | (65,133 | ) | (65,133 | ) | ||||||||||||||||
| Unrealized loss on investments |
— | — | — | (153 | ) | — | (153 | ) | ||||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (65,286 | ) | |||||||||||||||||
| Exercise of stock options |
264,706 | — | 273 | — | — | 273 | ||||||||||||||||||
| Issuance of restricted stock to employees |
45,000 | — | — | — | — | — | ||||||||||||||||||
| Cancellation of restricted stock |
(42,039 | ) | — | (131 | ) | — | — | (131 | ) | |||||||||||||||
| Accretion of dividends on redeemable convertible preferred stock |
— | — | — | — | (125 | ) | (125 | ) | ||||||||||||||||
| Stock-based compensation expense |
— | — | 5,872 | — | — | 5,872 | ||||||||||||||||||
| Stock-based compensation expense related to non-employees |
— | — | 37 | — | — | 37 | ||||||||||||||||||
| Balance at December 31, 2008 |
35,089,837 | $ | 35 | $ | 267,238 | $ | 73 | $ | (251,471 | ) | $ | 15,875 | ||||||||||||
| Net income |
— | — | — | — | 15,388 | 15,388 | ||||||||||||||||||
| Unrealized loss on investments |
— | — | — | (75 | ) | — | (75 | ) | ||||||||||||||||
| Comprehensive income |
— | — | — | — | — | 15,313 | ||||||||||||||||||
| Exercise of stock options |
11,857 | — | 2 | — | — | 2 | ||||||||||||||||||
| Cancellation of restricted stock |
(37,813 | ) | — | — | — | — | — | |||||||||||||||||
| Accretion of dividends on redeemable convertible preferred stock |
— | — | — | — | (53 | ) | (53 | ) | ||||||||||||||||
| Stock-based compensation expense |
— | — | 5,891 | — | — | 5,891 | ||||||||||||||||||
| Stock-based compensation expense related to non-employees |
— | — | 2 | — | — | 2 | ||||||||||||||||||
| Singapore divestiture |
— | — | (16,528 | ) | — | 16,528 | ||||||||||||||||||
| Issuance of shares of common stock in connection with Neuromed merger |
14,937,591 | 15 | 15,868 | — | — | 15,883 | ||||||||||||||||||
| Issuance of shares into escrow in connection with Neuromed merger |
67,826,875 | 68 | (68 | ) | — | — | — | |||||||||||||||||
| Balance at December 31, 2009 |
117,828,347 | 118 | $ | 272,405 | $ | (2 | ) | $ | (219,608 | ) | $ | 52,913 | ||||||||||||
See accompanying notes.
F-5
Zalicus Inc.
Consolidated Statements of Stockholders’ Equity (Continued)
(in thousands, except share amounts)
| Common Stock | Additional Paid-in Capital |
Accumulated Other Comprehensive Income (Loss) |
Accumulated Deficit |
Total | ||||||||||||||||||||
| Shares | Par Value |
|||||||||||||||||||||||
| Balance at December 31, 2009 |
117,828,347 | 118 | $ | 272,405 | $ | (2 | ) | $ | (219,608 | ) | $ | 52,913 | ||||||||||||
| Net loss |
— | — | — | — | (35,045 | ) | (35,045 | ) | ||||||||||||||||
| Unrealized loss on investments |
— | — | — | (18 | ) | — | (18 | ) | ||||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (35,063 | ) | |||||||||||||||||
| Exercise of stock options |
502,179 | — | 484 | — | — | 484 | ||||||||||||||||||
| Issuance of restricted stock to employees |
726,045 | 1 | (391 | ) | — | — | (390 | ) | ||||||||||||||||
| Stock-based compensation expense |
— | — | 2,928 | — | — | 2,928 | ||||||||||||||||||
| Cancellation of escrow shares in connection with Neuromed merger |
(29,943,752 | ) | (30 | ) | 30 | — | — | — | ||||||||||||||||
| Release of escrow shares in connection with Neuromed merger |
— | — | 42,050 | — | — | 42,050 | ||||||||||||||||||
| Issuance of warrants in connection with term loan |
— | — | 75 | — | — | 75 | ||||||||||||||||||
| Balance at December 31, 2010 |
89,112,819 | $ | 89 | $ | 317,581 | $ | (20 | ) | $ | (254,653 | ) | $ | 62,997 | |||||||||||
See accompanying notes.
F-6
Consolidated Statements of Cash Flows
(in thousands)
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Operating activities |
||||||||||||
| Net (loss) income |
$ | (35,045 | ) | $ | 15,388 | $ | (65,133 | ) | ||||
| Less: Net loss from discontinued operations |
— | (1,536 | ) | (4,557 | ) | |||||||
| Gain on disposal of subsidiary |
— | 15,640 | — | |||||||||
| (Loss) income from continuing operations |
(35,045 | ) | 1,284 | (60,576 | ) | |||||||
| Adjustments to reconcile (loss) income from continuing operations to net cash provided by (used in) operating activities from continuing operations: |
||||||||||||
| Depreciation and amortization |
21,163 | 5,601 | 3,212 | |||||||||
| Noncash restructuring charge and benefit |
— | (977 | ) | 289 | ||||||||
| Noncash interest expense |
3 | 28 | 64 | |||||||||
| Noncash rent expense |
(284 | ) | (490 | ) | (643 | ) | ||||||
| Stock-based compensation expense |
2,928 | 3,909 | 5,692 | |||||||||
| Gain on bargain purchase |
— | (9,809 | ) | — | ||||||||
| Loss (gain) on revaluation of contingent consideration |
29,286 | (12,068 | ) | — | ||||||||
| Loss on sale of fixed assets |
52 | 355 | — | |||||||||
| Loss on foreign exchange |
175 | 34 | — | |||||||||
| Decrease in deferred rent |
(32 | ) | (78 | ) | (81 | ) | ||||||
| Changes in assets and liabilities: |
||||||||||||
| Decrease in accounts receivable |
1,322 | 1,309 | 519 | |||||||||
| Decrease (increase) in prepaid expenses and other assets |
4,559 | (2,967 | ) | 1,286 | ||||||||
| (Decrease) increase in accounts payable |
(2,429 | ) | (1,308 | ) | 957 | |||||||
| (Decrease) increase in accrued restructuring |
(1,274 | ) | (1,596 | ) | 2,870 | |||||||
| Decrease in accrued expenses and other long-term liabilities |
(2,366 | ) | (841 | ) | (162 | ) | ||||||
| Increase (decrease) in deferred revenue |
620 | (6,292 | ) | (5,549 | ) | |||||||
| Net cash provided by (used in) operating activities from continuing operations |
18,678 | (23,906 | ) | (52,122 | ) | |||||||
| Net cash used in operating activities from discontinued operations |
— | (1,088 | ) | (3,486 | ) | |||||||
| Net cash provided by (used in) operating activities |
18,678 | (24,994 | ) | (55,608 | ) | |||||||
| Investing activities |
||||||||||||
| Cash acquired in connection with Neuromed merger |
— | 6,156 | — | |||||||||
| Purchases of property and equipment |
(1,094 | ) | (613 | ) | (2,283 | ) | ||||||
| Proceeds from sale of property and equipment |
97 | 588 | — | |||||||||
| Loss on sale of equity interest in subsidiary |
— | (6,240 | ) | — | ||||||||
| Purchases of short-term investments |
(369,849 | ) | (89,288 | ) | (275,532 | ) | ||||||
| Sales and maturities of short-term investments |
342,583 | 111,286 | 335,764 | |||||||||
| Decrease in restricted cash |
700 | 1,500 | — | |||||||||
| Net cash (used in) provided by investing activities from continuing operations |
(27,563 | ) | 23,389 | 57,949 | ||||||||
| Net cash used in investing activities from discontinued operations |
— | (16 | ) | (144 | ) | |||||||
| Net cash (used in) provided by investing activities |
(27,563 | ) | 23,373 | 57,805 | ||||||||
F-7
Zalicus Inc.
Consolidated Statements of Cash Flows (Continued)
(in thousands)
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Financing activities |
||||||||||||
| Proceeds from exercise of stock options |
484 | 2 | 273 | |||||||||
| Repurchases of common stock |
(391 | ) | — | (131 | ) | |||||||
| Net proceeds from term loan |
2,868 | — | — | |||||||||
| Payment of deferred financing costs |
(26 | ) | — | — | ||||||||
| Repayment of notes payable |
— | — | (7,258 | ) | ||||||||
| Net cash provided by (used in) financing activities from continuing operations |
2,935 | 2 | (7,116 | ) | ||||||||
| Net cash provided by financing activities from discontinued operations |
— | — | 3,714 | |||||||||
| Net cash provided by (used in) financing activities |
2,935 | 2 | (3,402 | ) | ||||||||
| Effect of exchange rate changes on cash and cash equivalents |
(9 | ) | 18 | — | ||||||||
| Net decrease in cash and cash equivalents |
(5,959 | ) | (1,601 | ) | (1,205 | ) | ||||||
| Cash and cash equivalents at beginning of the period(1) |
8,779 | 10,380 | 11,585 | |||||||||
| Cash and cash equivalents at end of the period(1) |
$ | 2,820 | $ | 8,779 | $ | 10,380 | ||||||
| Supplemental disclosure of cash flow information |
||||||||||||
| Cash paid for interest |
$ | — | $ | — | $ | 705 | ||||||
| Cash paid for income taxes |
$ | — | $ | — | $ | 20 | ||||||
| Supplemental disclosure of noncash investing and financing activities |
||||||||||||
| Consideration transferred in merger with Neuromed |
$ | — | $ | 40,715 | $ | — | ||||||
| Fair value of warrant issued in connection with issuance of term loan |
$ | 75 | $ | — | $ | — | ||||||
| (1) | Cash and cash equivalents as of December 31, 2008 includes cash and cash equivalents of the Company’s Singapore subsidiary of $7,341. |
See accompanying notes.
F-8
Notes to Consolidated Financial Statements
(in thousands, except share and per share amounts)
| 1. | Nature of the Business |
On September 8, 2010, CombinatoRx, Incorporated changed its name to Zalicus Inc. In conjunction with the name change, the trading symbol for its common stock on the Nasdaq Global Market changed from “CRXX” to “ZLCS”. Zalicus Inc. was formed as a Delaware corporation on March 28, 2000. Zalicus Inc., and its subsidiaries (the “Company”), is a biopharmaceutical company developing drug candidates with a focus on the treatment of pain and inflammation. To date, the Company has devoted substantially all of its resources to the development of its drug discovery technologies and the research and development of its drug candidates, including conducting preclinical and clinical trials and seeking intellectual property protection for its technology and product candidates.
The Company is subject to risks common to companies in the life science industry. All of its current product candidates are in preclinical or clinical development. If it does not successfully commercialize any of its product candidates, it will be unable to generate product revenue or achieve profitability.
The Company has a limited operating history and has incurred losses from operations since inception, resulting in an accumulated deficit of $254,653 at December 31, 2010. The Company may seek additional funding through public or private equity or debt financings and collaboration agreements. Additional funding, if needed, may not be available to the Company on acceptable terms or at all. Any additional equity financing would be dilutive to existing stockholders, and any debt financing, if available, may involve restrictive covenants that could adversely impact how the Company conducts its business. If the Company is unable to obtain funding on a timely basis, it may be required to significantly curtail its business or one or more of its research or development programs. The Company also could be required to seek funds through arrangements with collaborators or others that may require the Company to relinquish rights to some of its technologies or product candidates which the Company would otherwise develop and pursue on its own.
| 2. | Summary of Significant Accounting Policies |
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly-owned, controlled subsidiaries. All intercompany transactions have been eliminated in consolidation.
On December 21, 2009, the Company merged with Neuromed Pharmaceuticals, Inc. and its subsidiaries (“Neuromed”) (See Note 3.) The merger was accounted for under the acquisition method, and accordingly, the results of operations of Neuromed have been included in the consolidated results of operations since the merger date.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Actual results could differ from those estimates.
Foreign Currency Transactions
The functional currency of the Company’s foreign subsidiaries is the United States dollar. Foreign currency transaction gains and losses are recorded in the consolidated statement of operations. Net Foreign exchange losses of $175 and $33 were recorded in other expense in 2010 and 2009, respectively.
F-9
Research and Development
Research and development costs include all direct costs, including cash compensation, stock-based compensation and benefits for research and development personnel, supplies and materials, direct external costs including costs of clinical trials, formulation manufacturing, preclinical programs, collaboration expenses, external consultants, other outside costs and infrastructure and overhead related to the development of drug candidates. These costs have been charged to research and development expense as incurred.
Comprehensive (Loss) Income
Comprehensive (loss) income is the change in equity of a company during a period from transactions and other events and circumstances, excluding transactions resulting from investments by owners and distributions to owners. Comprehensive (loss) income includes net (loss) income and unrealized loss on investments for all periods presented.
The Company’s total comprehensive (loss) income consists of the following:
| Year Ended December 31, | ||||||||
| 2010 | 2009 | |||||||
| Net (loss) income |
$ | (35,045 | ) | $ | 15,388 | |||
| Other comprehensive loss: |
||||||||
| Unrealized loss on investments |
(18 | ) | (75 | ) | ||||
| Comprehensive (loss) income |
$ | (35,063 | ) | $ | 15,313 | |||
Revenue Recognition
Collaborations
The Company has entered into collaborative research and development agreements with other pharmaceutical and biotechnology companies, government agencies and charitable foundations. These agreements are generally in the form of research and development and license agreements. The agreements are primarily for early-stage compounds and are generally focused on specific disease areas. The agreements provide for nonrefundable up-front payments, milestone payments upon achieving significant milestone events and in some cases ongoing research funding. The agreements also contemplate royalty payments on sales if and when the product receives marketing approval by the United States Food and Drug Administration (“FDA”) or other regulatory agency.
The Company evaluates the arrangements with software license components in order to determine whether the arrangement should be accounted for under revenue recognition guidance for software or if other applicable revenue guidance should be applied. Revenue arrangements with multiple deliverables are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered elements. The consideration received is allocated among separate elements based on their respective fair values. Revenue is recognized when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed or determinable, and collection is reasonably assured. License fees or other amounts received in advance of performance obligations, or in cases where the Company has a continuing obligation to perform services, are deferred and recognized over the performance period. Revenues from milestone payments that are deemed to be substantive and represent the combination of a separate earnings process are recorded when the milestone is achieved. Contract revenues are recorded as services are performed. Royalty revenue is recognized based upon net sales of licensed products as provided by the relevant license and is recognized in the period the sales occur. The periods over which revenue is recognized are subject to estimates by management and may change over the course of a collaborative agreement.
F-10
Government Contracts and Grants
Revenue under government grants or cost reimbursement contracts is recognized as the Company performs the underlying research and development activities.
Concentrations of Credit Risk
Financial instruments that potentially expose the Company to concentrations of credit risk consist of cash, cash equivalents, short-term investments and accounts receivable. Short-term investments consist of corporate debt securities, government agency securities and Treasury money market funds. The Company maintains its cash, cash equivalents and marketable securities at high-quality financial institutions. The Company limits the amount of investment in any one type of investment, thereby reducing credit risk concentrations. The Company does not believe there is significant concentration of credit risk related to accounts receivable since its customers are primarily large well-capitalized pharmaceutical companies, foundations or government agencies.
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original or remaining maturity of three months or less at the date of purchase to be cash equivalents, except for those funds managed by the Company’s investment manager, which are classified as short-term investments. Cash equivalents consist primarily of money market instruments.
Fair Value Disclosure
The Company has certain financial assets and liabilities recorded at fair value which have been classified as Level 1, 2 or 3 within the fair value hierarchy as described in the accounting standards for fair value measurements.
| • | Level 1 – Quoted market prices in active markets for identical assets or liabilities. Assets utilizing Level 1 inputs include money market funds, U.S. government securities and bank deposits; |
| • | Level 2 – Inputs other than Level 1 inputs that are either directly or indirectly observable, such as quoted market prices, interest rates and yield curves. Assets utilizing Level 2 inputs include U.S. agency securities, including direct issuance bonds and corporate bonds; and |
| • | Level 3 – Unobservable inputs developed using estimates and assumptions developed by the Company, which reflect those that a market participant would use. |
The following tables summarize the financial instruments measured at fair value on a recurring basis in the accompanying consolidated balance sheet as of December 31, 2010 and 2009:
| Fair Value Measurement as of December 31, 2010 | Total | |||||||||||||||
| Level 1 | Level 2 | Level 3 | ||||||||||||||
| Assets: |
||||||||||||||||
| Short-term investments |
$ | 16,993 | $ | 24,806 | $ | — | $ | 41,799 | ||||||||
| Fair Value Measurement as of December 31, 2009 | Total | |||||||||||||||
| Level 1 | Level 2 | Level 3 | ||||||||||||||
| Assets: |
||||||||||||||||
| Short-term investments |
$ | 8,781 | $ | 5,770 | $ | — | $ | 14,551 | ||||||||
| Liabilities: |
||||||||||||||||
| Contingent consideration |
$ | — | $ | — | $ | 12,764 | $ | 12,764 | ||||||||
F-11
The Company’s Level 2 securities are valued using third-party pricing sources. These sources generally use interest rates and yield curves observable at commonly quoted intervals of similar assets as observable inputs for pricing.
As discussed in Note 3, on December 21, 2009, the Company acquired intangible assets as a result of the merger with Neuromed. As of December 31, 2009, the Company’s liability for contingent consideration was valued using management’s estimates of the number of shares that would be released to the former stockholders of Neuromed upon the outcome and related timing of FDA approval of Exalgo™ (see Note 3). The valuation was dependent on the Company’s stock price and an assessment of the final number of Escrow Shares (as defined below in Note 3) that were to be issued to former Neuromed stockholders. As such, this valuation was determined to be a Level 3 valuation, as the primary inputs were unobservable. On March 1, 2010, the shares held in escrow were released to the former Neuromed stockholders upon the FDA’s approval of Exalgo. The Company valued the liability prior to settlement using the closing stock price on March 1, 2010. This valuation was a Level 1 valuation, as the Company’s stock price is an observable input. The Company determines when transfers between levels are recognized based on the actual date of the circumstance that caused the transfer.
The following table provides a roll forward of the fair value of the contingent consideration, where fair value is determined by Level 3 inputs:
| Balance at January 1, 2010 |
$ | 12,764 | ||
| Additions |
— | |||
| Change in fair value |
29,286 | |||
| Release of escrow shares |
(42,050 | ) | ||
| Balance at December 31, 2010 |
$ | — | ||
As discussed in Note 9, the proceeds from the Oxford financing were allocated between Term Loan A and the warrant issued in connection with the financing using the relative fair value method. The fair value of the warrant was determined using the Black-Scholes method which uses the Company’s stock price, the historical volatility of the price of the Company’s common stock and published U.S. Treasury rates. This valuation was determined to be a Level 2 valuation. The fair value of the loan was determined by calculating the present value of future cash outflows by the Company under the terms of the loan discounted by an estimated market rate for a loan with similar terms issued without warrants. This valuation was determined to be a Level 2 valuation.
Fair Value of Financial Instruments
The carrying values of the Company’s financial instruments, which include cash equivalents, short-term investments, accounts payable, accrued expenses and term loan payable, approximate their fair values due to their short maturities.
Property and Equipment
Property and equipment are recorded at cost and depreciated over their estimated useful lives using the straight-line method. Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation are removed from the accounts, and any resulting gain or loss is credited or charged to the statement of operations. Repairs and maintenance costs are expensed as incurred.
Discontinued Operations
On June 2, 2009, the Company divested its 51% equity ownership interest in CombinatoRx Singapore, by selling its 2,602,041 ordinary shares of CombinatoRx Singapore to the other shareholder of CombinatoRx Singapore, BioMedical Sciences, for nominal consideration.
F-12
In connection with the divestiture, the Company entered into a termination agreement with CombinatoRx Singapore and BioMedical, pursuant to which the parties agreed to terminate all of the prior agreements among the Company, CombinatoRx Singapore and BioMedical Sciences relating to the joint funding and operations of CombinatoRx Singapore. As a result of the divestiture and the termination of the prior agreements, CombinatoRx Singapore is no longer affiliated with the Company, and the issued and outstanding preferred shares and convertible promissory notes issued by CombinatoRx Singapore and held by BioMedical Sciences are no longer convertible into shares of its common stock.
The Company also entered into a share purchase agreement with CombinatoRx Singapore and BioMedical Sciences and an intellectual property assignment agreement with CombinatoRx Singapore. Under the intellectual property assignment agreement, CombinatoRx Singapore was assigned and retains all infectious disease intellectual property developed by CombinatoRx Singapore with the Company’s assistance since the formation of CombinatoRx Singapore. Under the share purchase agreement, the Company agreed not to compete with CombinatoRx Singapore in the discovery and development of product candidates to treat certain infectious diseases in substantially all markets until June 2, 2010. The results of operations and the assets and the liabilities related to the divestiture of the Company’s Singapore subsidiary in June 2009 have been accounted for as discontinued operations. The Company recorded a $15,640 gain on the divestiture of CombinatoRx Singapore in June 2009. The gain was calculated as the difference between (1) the consideration received and the carrying value of the noncontrolling equity interest and (2) the carrying value of the assets and liabilities of CombinatoRx Singapore. Accordingly, the results of operations from prior periods have been reclassified to discontinued operations as a result of the divestiture.
Business Combinations
The Company assigns the value of the consideration transferred to acquire or merge with a business to the tangible assets and identifiable intangible assets acquired and liabilities assumed on the basis of their fair values at the date of acquisition. The Company assesses the fair value of assets, including intangible assets, using a variety of methods including present-value models. Each asset is measured at fair value from the perspective of a market participant. Transaction costs and restructuring costs associated with the transaction are expensed as incurred. Consideration transferred is measured on the date of the transaction. The consideration transferred in excess of the fair value of the assets acquired less the fair value of the liabilities assumed, if any, is recorded as goodwill on the Company’s balance sheet. In the event the fair value of the assets acquired less the fair value of the liabilities assumed exceeds the value of the consideration transferred, a bargain purchase would be deemed to have occurred and a gain would be recorded on the Company’s statement of operations.
Impairment of Intangibles and Long-Lived Assets
The Company continually monitors whether events or circumstances have occurred that indicate that the estimated remaining useful life of its intangible assets or long-lived assets may warrant revision or that the carrying value of these assets may be impaired. The carrying value for intangible and long-lived assets with finite lives is reviewed for impairment when events or changes in circumstances indicate the carrying value of the assets may not be recoverable. An impairment loss would be recognized when estimated undiscounted future cash flows from the use of the asset and its eventual disposition is less than its carrying value. Any write-downs are treated as permanent reductions in the carrying value of the assets. As of December 31, 2010 and 2009, the Company concluded that none of the Company’s intangible and long-lived assets that were held and used were impaired.
Capitalized Software
The Company capitalizes certain internal and external costs incurred to develop internal use software. Capitalized software development costs are included in property and equipment and are depreciated over estimated useful lives (five years) when development is complete. The net book value of the Company’s capitalized software was $122 and $170 at December 31, 2010 and 2009, respectively.
F-13
Accounting for Stock-Based Compensation
The Company recognizes, as expense, the estimated fair value of all share-based payments to employees. The Company accounts for transactions in which services are received from non-employees in exchange for equity instruments based on the fair value of such services received or of the equity instruments issued, whichever is more reliably measured.
Income Taxes
Deferred taxes are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect in the years in which the differences are expected to reverse. Valuation allowances are provided, if, based upon the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized. The Company accounts for uncertain tax positions using a “more-likely-than-not” threshold for recognizing and resolving uncertain tax positions. The evaluation of uncertain tax positions is based on factors including, but not limited to, changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, the effective settlement of matters subject to audit, new audit activity and changes in facts or circumstances related to a tax position. The Company evaluates this tax position on a quarterly basis. The Company also accrues for potential interest and penalties, related to unrecognized tax benefits in general and administrative expense. The Company’s evaluation was performed for the tax years ended December 31, 2004, 2005, 2006, 2007, 2008, 2009 and 2010, the tax years which remain subject to examination by major tax jurisdictions as of December 31, 2010.
The Company may from time to time be assessed interest or penalties by major tax jurisdictions, although any such assessments historically have not impacted the financial results of the Company. In the event the Company would receive an assessment for interest and/or penalties, it would be classified in the consolidated financial statements as general and administrative expense.
Net Income (Loss) per Share
Net income (loss) per share is calculated using the two-class method, which is an earnings allocation formula that determines net income (loss) per share for the holders of the Company’s common shares and Escrow Shares issued in connection with the Neuromed merger. (See Note 3.) The Escrow Shares contained participation rights in any dividend paid by the Company while the shares were held in escrow. Net income available to common shareholders and participating Escrow Shares is allocated to each share equally as if all of the earnings for the period had been distributed. The escrow shares did not include a contractual obligation to share in losses of the Company and were not included in the calculation of net loss per share in the periods that have a net loss. Diluted net income (loss) per share was calculated using the treasury stock method for all dilutive outstanding warrants, stock options, restricted stock awards and restricted stock units.
Recently Issued Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the FASB or other standard setting bodies that are adopted by the Company as of the specified effective date. Unless otherwise discussed, the Company believes that the impact of recently issued standards that are not yet effective will not have a material impact on its financial position or results of operations upon adoption.
In October 2009, the Financial Accounting Standards Board, or FASB, issued Accounting Standards Update (ASU) No. 2009-13, Multiple-Deliverable Revenue Arrangements (ASU No. 2009-13). ASU No. 2009-13, which amends existing revenue recognition accounting pronouncements and provides accounting principles and application guidance on whether multiple deliverables exist, how the arrangement should be separated, and the consideration allocated. This guidance eliminates the requirement to establish the fair value of undelivered products and services and instead provides for separate revenue recognition based upon management’s estimate
F-14
of the selling price for an undelivered item when there is no other means to determine the fair value of that undelivered item. Previous accounting principles required that the fair value of the undelivered item be the price of the item either sold in a separate transaction between unrelated third parties or the price charged for each item when the item is sold separately by the vendor. This was difficult to determine when the product was not individually sold because of its unique features. If the fair value of all of the elements in the arrangement was not determinable, then revenue was deferred until all of the items were delivered or fair value was determined. This new approach is effective prospectively for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010, which for the Company means no later than January 1, 2011. While the Company does not expect the adoption of this standard to have a material impact on its financial position and results of operations, this standard may impact the Company in the event it completes future transactions or modifies existing collaborative relationships.
In October 2009, FASB updated its guidance on software revenue recognition rules. According to this update, tangible products containing software components and non-software components that function together to deliver the tangible product’s essential functionality, are no longer within the scope of the software revenue guidance. This update requires that hardware components of a tangible product containing software components always be excluded from the software revenue guidance. This update provides additional guidance on how to determine which software, if any, relating to the tangible product should be excluded from the scope of the software revenue guidance. This update will be effective prospectively for revenue arrangements entered into or materially modified in fiscal years beginning on or after June 15, 2010, but early adoption is permitted. This update must be adopted in the same period using the same transition method as indicated above in the update to revenue arrangements with multiple deliverables. The Company does not expect the adoption of this standard to have a material impact on its financial position and results of operations; however, this standard may impact the Company in the event it completes future transactions.
In January 2010, the FASB issued updated guidance to amend the disclosure requirements related to recurring and nonrecurring fair value measurements. This update requires new disclosures on significant transfers of assets and liabilities between Level 1 and Level 2 of the fair value hierarchy (including the reasons for these transfers) and the reasons for any transfers in or out of Level 3. This update also requires a reconciliation of recurring Level 3 measurements about purchases, sales, issuances and settlements on a gross basis. In addition to these new disclosure requirements, this update clarifies certain existing disclosure requirements. For example, this update clarifies that reporting entities are required to provide fair value measurement disclosures for each class of assets and liabilities rather than each major category of assets and liabilities. This update also clarifies the requirement for entities to disclose information about both the valuation techniques and inputs used in estimating Level 2 and Level 3 fair value measurements. This update became effective for the Company with the interim and annual reporting period beginning January 1, 2010, except for the requirement to provide the Level 3 activity of purchases, sales, issuances, and settlements on a gross basis, which is effective for the Company beginning January 1, 2011. Other than requiring additional disclosures, adoption of this update did not have a material effect on the Company’s consolidated financial statements.
| 3. | Merger with Neuromed Pharmaceuticals, Inc. |
On December 21, 2009, the Company completed the merger with Neuromed Pharmaceuticals, Inc. (“Neuromed”). Under the terms of the merger agreement and a related escrow agreement (the “Escrow Agreement”), at closing the Company issued 14,937,654 new shares of its common stock (the “Firm Shares”) to Neuromed stockholders and placed 67,826,875 new shares in escrow for the benefit of Neuromed stockholders (the “Escrow Shares”). Of the Escrow Shares subject to the Escrow Agreement, an aggregate of 19,916,872 shares (the “Holdback Shares”) were placed into escrow and would or would not be released to Neuromed stockholders depending upon the timing of the FDA’s approval of Exalgo and an aggregate of 47,910,003 shares (the “Milestone Shares”) were placed into escrow and would or would not be released to Neuromed stockholders depending upon the timing of the FDA’s approval of Exalgo. Former Neuromed stockholders had voting and other ownership rights with respect to the Holdback Shares but no voting rights with respect to the Milestone
F-15
Shares. As a result, at the closing of the merger with Neuromed, current Zalicus stockholders effectively retained approximately 50% of the outstanding voting shares of common stock of Zalicus immediately after the merger, former Neuromed stockholders effectively owned or controlled approximately 48.5% of the outstanding voting shares of common stock of Zalicus immediately after the merger (a portion of which was subject to the terms of the Escrow Agreement), and certain Neuromed directors, officers and other employees effectively held approximately 1.5% of the outstanding shares of common stock of Zalicus immediately after the merger in the form of shares underlying restricted stock unit awards granted under the Neuromed special equity incentive plan. The release of the Escrow Shares, and any resulting adjustment of the relative ownership percentage of the then outstanding shares of common stock of the combined company, was based upon the timing of the FDA’s approval of Exalgo and, subject to the terms and conditions of the Escrow Agreement:
| • | If FDA approval of Exalgo were received on or before December 31, 2009, all of the Escrow Shares would be released to the former Neuromed stockholders, resulting in the pre-merger Zalicus stockholders owning approximately 30% of the then outstanding shares of common stock of the combined company. |
| • | If FDA approval of Exalgo were received on or after January 1, 2010 and on or before September 30, 2010, a portion of the Escrow Shares would be released to the former Neuromed stockholders and a portion would be cancelled, resulting in the pre-merger Zalicus stockholders owning approximately 40% of the then outstanding shares of common stock of the combined company. |
| • | If FDA approval of Exalgo were received on or after October 1, 2010 and on or before December 31, 2010, a portion of the Escrow Shares would be released to the former Neuromed stockholders and a portion would be cancelled, resulting in the pre-merger Zalicus stockholders owning approximately 60% of the then outstanding shares of common stock of the combined company. |
| • | If FDA approval of Exalgo were not received on or before December 31, 2010, all of the remaining Escrow Shares would be cancelled, resulting in the pre-merger Zalicus stockholders owning approximately 70% of the then outstanding shares of common stock of the combined company. |
The Company evaluated the considerations in ASC sections 805 Business Combinations and 810 Consolidations and concluded that it was the accounting acquirer in the merger. As such, it recorded the assets acquired and liabilities assumed from Neuromed at their estimated fair values as of the acquisition date of December 21, 2009.
Valuation of Consideration Transferred
The consideration transferred to effect the acquisition of Neuromed consisted of the following:
| Fair value of Zalicus shares issued in the merger |
$ | 15,883 | ||
| Estimated fair value of contingent consideration |
24,832 | |||
| Total consideration transferred |
$ | 40,715 | ||
The value of the shares issued in the merger was based upon the closing price of the Company’s common stock of $1.07 on December 21, 2009, the date of the closing of the merger with Neuromed.
The fair value of contingent consideration at the merger date was determined based upon a probability-weighted estimate of the incremental number of shares of the Company’s common stock to be issued to former Neuromed stockholders at the stock price on the date the merger closed. The probability-weighted estimate reflected the Company’s assessment of the likelihood that Exalgo would be approved by the FDA during each of the four time periods included in the merger agreement and the corresponding pre-determined amount of Escrow Shares that would be released to the former Neuromed stockholders as a result thereof. The probabilities assigned to each Exalgo FDA approval outcome ranged from 10% to 40% and were determined based upon a review of
F-16
four other product candidates for the treatment of pain that were in or had recently completed the FDA review process that were not yet approved or had long approval delays. The overall probability-weighted estimated outcome of the Exalgo approval scenarios on December 21, 2009 resulted in the Neuromed stockholders and pre-merger Zalicus stockholders owning 47.5% and 52.5%, respectively, of the combined company. This fair value measurement was based on significant inputs not observable in the market and thus represents a Level 3 measurement within the fair value hierarchy.
The Company reassessed the fair value of the contingent consideration at December 31, 2009 and classified the fair value of contingent consideration of $12,764 as a long-term liability in the consolidated balance sheet at December 31, 2009. In connection therewith, the Company recorded a gain of $12,068 primarily as a result of the FDA approval of Exalgo not occurring before January 1, 2010 impacting the Company’s assessment of the amount of shares expected to be released from escrow and a decrease in the Company’s stock price from the date of merger closing through December 31, 2009.
On March 1, 2010, the FDA approved the New Drug Application for Exalgo, and the contingency was resolved. As a result, 37,883,123 Escrow Shares were released to former Neuromed stockholders. The fair value of the contingent consideration liability was $42,050 as of March 1, 2010, and the Company recorded a loss of $29,286 in the statement of operations in the year ended December 31, 2010. The fair value of the contingent consideration was recorded as additional paid-in capital upon the release of the Escrow Shares.
Allocation of Assets and Liabilities Acquired
In accordance with accounting standards, any excess of fair value of acquired net assets over the consideration transferred in the acquisition results in a gain on bargain purchase. Prior to recording a gain, the acquiring entity must reassess whether all acquired assets and assumed liabilities have been identified and recognized and perform re-measurements to verify that the consideration paid, assets acquired, and liabilities assumed have been properly valued. The Company underwent such a reassessment, and as a result, recorded a gain on bargain purchase of approximately $9,809 in the year ended December 31, 2009.
The bargain purchase was primarily a result of the contingent consideration being valued using probability weighted assumptions to estimate the number of shares that would ultimately be released to the Neuromed shareholders at the stock price at the merger closing. The actual value of the consideration ultimately received by the Neuromed shareholders was dependent on the amount of shares released and the closing price of the Company’s common stock on the date the shares were released from escrow.
The allocation of the purchase price was based upon a valuation of certain assets and liabilities acquired. The purchase price allocation was as follows (in thousands):
| Cash and equivalents |
$ | 6,156 | ||
| Short-term investments |
10 | |||
| Accounts receivable |
3,799 | |||
| Prepaid expense and other current assets |
1,434 | |||
| Property and equipment |
1,622 | |||
| Intangible assets |
45,943 | |||
| Total assets acquired |
58,964 | |||
| Accounts payable |
2,734 | |||
| Accrued expenses |
5,706 | |||
| Total liabilities assumed |
8,440 | |||
| Total net assets acquired |
50,524 | |||
| Total consideration transferred |
40,715 | |||
| Gain on bargain purchase |
$ | 9,809 | ||
F-17
The above estimated fair values of assets acquired and liabilities were based on the information that was available as of the acquisition date to estimate the fair value of assets acquired and liabilities assumed. The Company believes that information provided a reasonable basis for estimating the fair values of assets acquired and liabilities.
The estimated fair value of the intangible asset of $45,943 relates to projected future cash flows associated with the commercial rights to Exalgo that were sold to Covidien in June 2009. The intangible asset is being amortized in a manner which reflects estimates of future undiscounted cash flows expected to be generated from Exalgo over an estimated useful life of five years, representing the estimated remaining patent life of Exalgo from June 2009.
| 4. | Discontinued Operations |
On June 2, 2009, the Company divested its 51% equity ownership interest in CombinatoRx (Singapore) Pte. Ltd. (“CombinatoRx Singapore”), by selling its 2,602,041 ordinary shares of CombinatoRx Singapore to the other shareholder of CombinatoRx Singapore, BioMedical Sciences Investment Fund Pte. Ltd. (“BioMedical Sciences”), for nominal consideration. In connection with the divestiture, the Company, CombinatoRx Singapore and BioMedical Sciences entered into a termination agreement pursuant to which the parties agreed to terminate all of the prior agreements among the Company, CombinatoRx Singapore and BioMedical Sciences relating to the joint funding and operations of CombinatoRx Singapore. As a result of the Divestiture and the termination of the prior agreements, CombinatoRx Singapore is no longer affiliated with the Company, and the issued and outstanding preferred shares and convertible promissory notes issued by CombinatoRx Singapore and held by BioMedical Sciences are no longer convertible into shares of the Company’s common stock. The Company also entered into a share purchase agreement with CombinatoRx Singapore and BioMedical Sciences and an intellectual property assignment agreement with CombinatoRx Singapore. Under the intellectual property assignment agreement, CombinatoRx Singapore has been assigned and retains all infectious disease intellectual property developed by CombinatoRx Singapore with the assistance of the Company since the formation of CombinatoRx Singapore. Under the share purchase agreement, the Company agreed not to compete with CombinatoRx Singapore in the discovery and development of product candidates to treat certain infectious diseases in substantially all markets until June 2, 2010.
The Company recorded a $15,640 gain on the divestiture of CombinatoRx Singapore in June 2009. The gain was calculated as the difference between the consideration received and the carrying value of the noncontrolling equity interest and the carrying value of the assets and liabilities of CombinatoRx Singapore. The results of operations related to the divestiture of the Company’s Singapore subsidiary in June 2009 were accounted for as discontinued operations. Accordingly, the accompanying consolidated financial statements and notes have been retrospectively adjusted to reclassify discontinued operations for all periods presented.
F-18
| 5. | Short-Term Investments |
Short-term investments consist primarily of investments with original maturities greater than ninety days and less than one year when purchased. The Company classifies these investments as available-for-sale. Unrealized gains and losses are included in other comprehensive income (loss).
Available-for-sale securities at December 31, 2010 and 2009 consist of the following:
| Amortized Cost |
Unrealized Gains |
Unrealized Losses |
Fair Value |
|||||||||||||
| December 31, 2010— |
||||||||||||||||
| Corporate debt securities |
$ | 24,826 | $ | 3 | $ | (23 | ) | $ | 24,806 | |||||||
| Treasury money market funds |
16,993 | — | — | 16,993 | ||||||||||||
| $ | 41,819 | $ | 3 | $ | (23 | ) | $ | 41,799 | ||||||||
| December 31, 2009— |
||||||||||||||||
| Government agency securities |
$ | 7,845 | $ | — | $ | (2 | ) | $ | 7,843 | |||||||
| Treasury money market funds |
6,708 | — | — | 6,708 | ||||||||||||
| $ | 14,553 | $ | — | $ | (2 | ) | $ | 14,551 | ||||||||
The amortized cost and estimated fair value of investments in debt securities, which excludes money market funds, at December 31, 2010 and 2009, by contractual maturity, were as follows:
| December 31, 2010 | December 31, 2009 | |||||||||||||||
| Cost | Estimated Fair Value |
Cost | Estimated Fair Value |
|||||||||||||
| Maturing in one year or less |
$ | 21,898 | $ | 21,888 | $ | 7,845 | $ | 7,843 | ||||||||
| Maturing in more than one year |
$ | 2,928 | $ | 2,918 | $ | — | $ | — | ||||||||
The cost of securities sold is determined based on the specific identification method for purposes of recording realized gains and losses. Gross realized gains and losses on the sales of investments have not been material to the Company’s results of operations for all periods presented. As a matter of investment policy, the Company does not invest in auction rate securities.
| 6. | Property and Equipment |
Property and equipment consist of the following:
|
Estimated Useful |
December 31, | |||||||||
| 2010 | 2009 | |||||||||
| Leasehold improvements |
Lesser of useful life or life of lease | $ | 5,434 | $ | 4,783 | |||||
| Laboratory equipment |
5 | 7,500 | 7,166 | |||||||
| Computer equipment |
3 | 1,067 | 1,072 | |||||||
| Construction in progress |
— | 356 | 527 | |||||||
| Capitalized software |
5 | 815 | 815 | |||||||
| Furniture and fixtures |
3 | 410 | 409 | |||||||
| 15,582 | 14,772 | |||||||||
| Less: accumulated depreciation |
(8,684 | ) | (6,392 | ) | ||||||
| $ | 6,898 | $ | 8,380 | |||||||
Depreciation expense for the years ended December 31, 2010, 2009 and 2008 was approximately $2,427, $5,081, and $3,212, respectively.
F-19
| 7. | Intangible Assets |
The intangible asset relates to rights to receive milestone payments and royalties from Covidien for the commercial rights to Exalgo that were acquired as part of the merger with Neuromed. The intangible asset was initially recorded at a value of $45,943 with a useful life of five years, representing the remaining patent life of Exalgo. The intangible asset is being amortized in a manner which reflects estimates of future undiscounted cash flows expected to be generated from Exalgo. As of December 31, 2010, the remaining useful life of the intangible asset is three and a half years. The Company recorded amortization expense of $18,736 and $520 for the years ended December 31, 2010 and 2009, respectively.
The Company estimates that the remaining amortization for the intangible asset will be as follows:
| 2011 |
$ | 5,141 | ||
| 2012 |
8,505 | |||
| 2013 |
8,588 | |||
| 2014 |
4,453 | |||
| Total |
$ | 26,687 | ||
| 8. | Restructuring |
In the fourth quarter of 2008, the Company implemented a strategic realignment to focus the business on identifying novel product candidates based on unexpected biological synergies. The strategic realignment included two reductions in force eliminating, in total, approximately 77 employees, or approximately 65% of the Company’s workforce at the time. The Company incurred total restructuring charges of $5,095, of which $4,637 was incurred in the fourth quarter of 2008. These restructuring charges included $3,257 of severance payments and related benefits, $1,704 of facility exit costs, of which $1,314 pertained to the impairment of leasehold improvements and other fixed assets and $390 pertained to the net lease obligation, and $134 of other associated costs. In the first quarter of 2009, the Company recorded the $458 balance of the restructuring charges, which represented severance payments and related benefits for employees who provided service beyond the minimum retention period, or 60 days. In the second quarter of 2009, the Company recorded a restructuring credit of $442 related to a change in estimate of facility exit costs that were originally recorded in the fourth quarter of 2008.
On July 1, 2009, in connection with the entry into the merger agreement on June 30, 2009 with Neuromed (as discussed in Note 3), the Company’s Board of Directors committed to a restructuring plan that resulted in a workforce reduction of 20 employees, or approximately 36% of the Company’s workforce at the time. The restructuring was a result of a continued strategic realignment of the Company to focus its efforts on its funded drug discovery and on conserving capital in connection with the Neuromed merger.
As a result of the July 1, 2009 restructuring plan, the Company recorded a restructuring charge of $2,597 in the third quarter of 2009, consisting of termination benefits and facility exit costs. The restructuring charge included termination benefits of $4,400, which consisted of $2,574 of cash severance and related benefits and $1,826 of accelerated stock-based compensation. The Company began paying severance and related benefits in the third quarter of 2009 and continued making payments into the first quarter of 2010.
As a result of the Company vacating certain lab premises in the third quarter of 2009, the Company recorded a restructuring credit of $1,803 in the third quarter of 2009 associated with facility exit costs. The credit to restructuring expense was comprised of lease termination payments of $1,000 under an amendment to the lease (as discussed in Note 17), which were offset by the reduction of deferred rent and lease incentive obligations totaling $2,803. In connection with the Company’s decision to vacate a portion of its laboratory and office premises in the second quarter of 2009, the Company recorded $1,151 and $1,383 within research and development and general and administrative expenses in the second quarter and third quarter of 2009, respectively, of accelerated amortization of leasehold improvements associated with these premises to reflect a remaining useful life commensurate with the estimated date that the Company would vacate the relevant premises.
F-20
The following tables summarize the activity in accrued restructuring through December 31, 2010 related to the Company’s 2008 and 2009 strategic alignments.
| Balance at December 31, 2008 |
Charges | Change in Estimate |
Non-Cash Items |
Payments | Balance at December 31, 2009 |
|||||||||||||||||||
| Termination benefits |
$ | 1,503 | $ | 4,981 | $ | — | $ | (1,826 | ) | $ | (4,384 | ) | $ | 274 | ||||||||||
| Facilities |
1,367 | (1,803 | ) | (442 | ) | 2,803 | (925 | ) | 1,000 | |||||||||||||||
| Total |
$ | 2,870 | $ | 3,178 | $ | (442 | ) | $ | 977 | $ | (5,309 | ) | $ | 1,274 | ||||||||||
| Balance at December 31, 2009 |
Payments | Balance at December 31, 2010 |
||||||||||
| Termination benefits |
$ | 274 | $ | (274 | ) | $ | — | |||||
| Facilities |
1,000 | (1,000 | ) | — | ||||||||
| Total |
$ | 1,274 | $ | (1,274 | ) | $ | — | |||||
As discussed in Note 17, the Company made payments on the facility component of accrued restructuring through July 1, 2010.
| 9. | Notes Payable |
On December 22, 2010, the Company entered into a loan and security agreement (the “Loan and Security Agreement”) with Oxford Finance Corporation (the “Lender”) pursuant to which the Lender agreed to lend the Company up to $20,000. Upon entering into the Loan and Security Agreement, the Company borrowed $3,000 from the Lender (“Term Loan A”). Under the terms of the Loan and Security Agreement, the Company may, in its sole discretion, borrow from the Lender up to an additional $8,500, at any time on or before July 15, 2011 (“Term Loan B”) and up to an additional $8,500, at any time on or before January 15, 2012 (“Term Loan C”, collectively with Term Loan A and Term Loan B, the “Term Loans”). The Company’s wholly owned subsidiary, Zalicus Pharmaceuticals Ltd. (the “Subsidiary”), is also a party to the Loan and Security Agreement as a co-borrower. The Company’s obligations under the Loan and Security Agreement are secured by a first priority security interest in substantially all of the assets of the Company and the Subsidiary, other than intellectual property.
The Company is required to pay interest on Term Loan A on a monthly basis through and including July 1, 2011. Beginning August 1, 2011 through the maturity of Term Loan A on July 1, 2014, the Company will be required to make payments of outstanding principal and interest on Term Loan A in 36 equal monthly installments. Interest is payable on Term Loan A at an annual interest rate of 10.26%. If the Company enters into Term Loan B or Term Loan C, interest on each term loan will accrue at an annual fixed rate equal to greater of (i) 10.25% or (ii) the sum of (a) the 90-day U.S. Dollar LIBOR rate prior to the funding of such term loan, plus (b) 9.96%. Payments due under Term Loan B or Term Loan C, if borrowed, are interest only, payable monthly, in arrears, for six months following the funding of each term loan, and will consist of 36 payments of principal and interest which are payable monthly, in arrears, and all unpaid principal and accrued and unpaid interest on Term Loan B or Term Loan C would be due and payable 42 months after the funding of any each term loan.
Upon the last payment date of the amounts borrowed under the Loan and Security Agreement, whether on the maturity date of one of the Term Loans, on the date of any prepayment or on the date of acceleration in the event of a default, the Company will be required to pay the Lender a final payment fee equal to 1.5% of any of the Term Loans borrowed. In addition, if the Company repays all or a portion of the Term Loans prior to maturity, it will pay the Lender a prepayment fee of four percent of the total amount prepaid if the prepayment occurs prior to the first anniversary of the funding of the relevant Term Loan, two percent of the total amount prepaid if the prepayment occurs between the first and second anniversary of the funding of the relevant Term Loan, and one percent of the total amount prepaid if the prepayment occurs on or after the second anniversary of the funding of the relevant Term Loan.
F-21
Upon the occurrence of an event of default, including payment defaults, breaches of covenants, a material adverse change in the collateral, the Company’s business, operations or condition (financial or otherwise) and certain levies, attachments and other restraints on the Company’s business, the interest rate will be increased by five percentage points and all outstanding obligations will become immediately due and payable. The Loan and Security Agreement also contains a subjective acceleration clause, which provides the Lender the ability to demand repayment of the loan early upon a material adverse change, as defined. The portion of the Term Loan A that is not due within 12 months of December 31, 2010 has been classified as long-term, as the Company believes a material adverse change is remote.
In connection with the Loan and Security Agreement, the Company issued to the Lender a warrant to purchase 64,748 shares of the Company’s common stock (the “Warrant”). The Warrant is exercisable, in whole or in part, immediately, and has a per share exercise price of $1.39 and may be exercised on a cashless basis. The Warrant will terminate on the earlier of December 22, 2017 and the closing of a merger or consolidation transaction in which Zalicus is not the surviving entity.
If the Company borrows Term Loan B or Term Loan C, upon the funding of each Term Loan, the Warrant will become exercisable for an additional number of shares of the Company’s common stock determined by dividing 3.0% of the amount of each such Term Loan by the volume-weighted average price of the Company’s common stock on Nasdaq for the ten trading days prior to the funding of such Term Loan. The exercise price for any additional shares of the Company’s common stock that may be purchased by the Lender pursuant to the Warrant following the Company borrowing Term Loan B or Term Loan C will be the volume-weighted average price of the Company’s common stock on Nasdaq for the ten trading days prior to the funding of such Term Loan.
The fair value of the Warrant issued in connection with Term Loan A was $81 and was recorded as a discount to Term Loan A. The Company also paid the Lender a facility fee of $100 and reimbursed the Lender certain costs associated with the Loan and Security Agreement of $31 both of which were also recorded as a discount to Term Loan A. The discount is being amortized to interest expense over the 42 month period that Term Loan A is outstanding using the effective interest method.
Future principal payments under the Loan and Security Agreement at December 31, 2010, are as follows:
| 2011 |
$ | 364 | ||
| 2012 |
939 | |||
| 2013 |
1,040 | |||
| 2014 |
657 | |||
| Total |
$ | 3,000 | ||
| 10. | Research and Development Agreements |
Mallinckrodt Inc., a subsidiary of Covidien plc
In June 2009, Neuromed entered into an asset purchase agreement with Mallinckrodt, a subsidiary of Covidien (“Covidien”), to sell all of the tangible and intangible assets associated with Exalgo, including the rights to develop and commercialize the product candidate in the United States. As part of the agreement, Neuromed received upfront payments of $15,000. The Company received a milestone payment of $40,000 following FDA approval of Exalgo in March 2010 and is eligible for tiered royalties on Covidien’s net sales of Exalgo. Covidien will continue to pay these royalties on net sales for as long as it is selling Exalgo although the royalty rate will be reduced upon the earlier to occur of generic competition or June 11, 2024. For the year ending December 31, 2010, total revenue recognized from Covidien represents 90.6% of total revenue.
Covidien launched the commercial sale of Exalgo in the second quarter of 2010. The Company recognized $1,615 of revenue related to royalties from Covidien’s sale of Exalgo during the year ended December 31, 2010. The Company received payments related to Exalgo royalties totaling $1,238 during the year ended December 31, 2010.
F-22
On October 29, 2010, Covidien received a Paragraph IV Certification Notice Letter from Watson Laboratories, Inc.—Florida, a subsidiary of Watson Pharmaceuticals, Inc., notifying them that it had submitted an Abbreviated New Drug Application (“ANDA”) to the FDA for approval to market a generic version of hydromorphone, the active pharmaceutical ingredient in Exalgo, prior to the expiration of U.S. Patent No. 5,914,131, which expires on July 7, 2014, and is listed in the Orange Book. The notice letter further stated that Watson is asserting that claims in U.S. Patent No. 5,914,131 are not infringed and/or are invalid. On December 10, 2010, Covidien’s Mallinckrodt Inc. subsidiary filed a patent infringement lawsuit against Watson Laboratories, Inc.–Florida, Watson Pharma, Inc. and Watson Pharmaceuticals, Inc., in response to the ANDA filing, which the Company refers to as the Watson litigation. The complaint, which was filed in the U.S. District Court for the District of New Jersey, alleges infringement of the referenced patent. Under current U.S. law, the filing of the lawsuit automatically prevents the FDA from approving the ANDA for 30 months from Covidien’s receipt of Watson’s Paragraph IV notification letter on October 29, 2010, or until April 29, 2013, unless the court enters judgment in favor of Watson in less than 30 months or finds that a party has failed to cooperate reasonably to expedite the lawsuit. In the Watson litigation, the court may find the patents that are the subject of the notice letter invalid, not infringed and/or unenforceable. This could lead to the approval of a generic version of Exalgo prior to the expiration of the 5,914,131 patent on July 7, 2014, which could result in reduced sales of Exalgo and a reduction in the royalties payable to us by Covidien on sales of Exalgo. The conduct of the Watson litigation and any decisions regarding litigation strategy or potential settlement are in the complete control of Covidien, and Zalicus has no rights to participate in the Watson litigation or otherwise direct its prosecution or settlement.
Neuromed also entered into a development and transition services agreement with Covidien, pursuant to which the Company will perform certain clinical development and regulatory activities relating to the FDA approval of Exalgo. These activities are at Covidien’s cost and expense, capped at $16,000. Through December 31, 2010, $8,292 has been billed and received related to the development and transition services agreement. The Company recorded $678 of revenue and received payments related to development and transition services totaling $1,959 during the year ended December 31, 2010. For the year ended December 31, 2010, the Company recorded unbilled receivables for this agreement totaling $47.
Novartis
In May 2009, Zalicus entered into a research collaboration and license agreement with Novartis Institutes for BioMedical Research, Inc. (“Novartis”), focused on the discovery of novel anti-cancer combinations. Through the collaboration, the Company will use its proprietary cHTS platform to screen a unique library of molecules, including Novartis compounds, in multiple cell lines representing a broad spectrum of cancers to potentially discover novel single agent and combination therapies to treat various cancers.
Under the terms of the collaboration agreement, the Company received an initial payment of $4,000 and will receive annual research support payments of up to $3,000, plus certain expenses. In addition, the collaboration agreement may provide the Company with up to $58,000 for each combination product candidate advanced by Novartis upon achievement of certain clinical, regulatory and commercial milestones. The research program has an initial two-year term that may be extended by Novartis for three additional one-year periods. In January 2011, Novartis elected to extend the research program for an additional contract year. The Company also entered into a software license agreement with Novartis, where the Company provided Novartis with a non-exclusive license to use its proprietary Chalice™ analyzer software in connection with the collaboration and other Novartis research programs for approximately five years.
The library to be screened under the collaboration will consist of certain Novartis oncology compounds and compounds from the Company’s library of approved drugs and other molecules. Novartis will own and have an exclusive license to intellectual property generated under the collaboration to research, develop and commercialize their approved or active development-stage compounds. The Company will own and have an exclusive license to intellectual property generated under the collaboration to research, develop and commercialize compounds from the Company’s library. Intellectual property generated under the collaboration
F-23
using certain compounds from the Novartis library will be jointly owned by Novartis and the Company and non-exclusively licensed to allow each party to research, develop and commercialize product candidates. Under the collaboration agreement, Novartis retains an option, exercisable once per year of the research collaboration, to exclusively license a portion of this jointly owned intellectual property if certain conditions are met. Novartis also has a right of first negotiation to exclusively license the intellectual property owned by the Company that was discovered as a part of the collaboration, under terms to be negotiated by the parties at such time.
The collaboration agreement may be terminated by either party after ninety days’ notice upon an unremedied material breach and upon thirty days’ notice in the event of bankruptcy of the other party. Novartis may terminate the collaboration agreement after sixty days’ notice in the event of a change in control or liquidation of us, as defined in the collaboration agreement. The Company is recognizing the total consideration under the agreement of $13,000 ratably over the five-year software license term. The Company recorded $2,000 and $1,333 of revenue related to the research and license agreement for the years ended December 31, 2010 and 2009, respectively. The Company received payments related to the research and license agreement totaling $3,000 and $5,250 during the years ended December 31, 2010 and 2009, respectively. For the years ended December 31, 2010 and 2009, respectively, the Company recorded unbilled receivables for this agreement totaling $750 and $0.
Amgen Inc.
In December 2009, Zalicus entered into a Research Collaboration Agreement (the “Pilot Agreement”) with Amgen Inc. (“Amgen”) focused on identifying synergistic combinations for two oncology targets of interest to Amgen. Under the Pilot Agreement, Zalicus received a $750 payment in January 2010 to fund the initial research plan, and Amgen also agreed to reimburse Zalicus for laboratory supplies consumed. Amgen will also pay Zalicus a $1,000 milestone payment for each Investigational New Drug application filing by Amgen for a product candidate with new intellectual property generated by the collaboration. The Pilot Agreement also provides Amgen with a non-exclusive license to use the Zalicus Chalice analyzer software in connection with the research collaboration. The initial research plan ended in September 2010, and Amgen elected for Zalicus to do follow-up research at a per annum rate of $300 per full-time employee equivalent , plus the reimbursement of laboratory supplies. The Company recorded $976 of revenue related to the Pilot Agreement for the year ended December 31, 2010. The Company received payments related to the Pilot Agreement totaling $975 during the year ended December 31, 2010.
Fovea Pharmaceuticals SA
On January 30, 2006, the Company entered into a research and license agreement (the “Original Fovea Agreement”) with Fovea Pharmaceuticals SA (“Fovea”). Under the terms of the Original Fovea Agreement, Fovea agreed to fund and conduct preclinical and clinical development in ophthalmology of combination drug candidates it selects from the Company’s portfolio, including creating ophthalmic formulations. Under the Original Fovea Agreement, the Company also granted to Fovea an exclusive worldwide license to certain preclinical drug combinations to treat specified diseases of the front of the eye. In consideration for the license of these combinations to treat specified diseases of the front of the eye, the Company is entitled to receive license execution fees, development milestones and royalties from Fovea if certain conditions within the Original Fovea Agreement are satisfied.
On June 12, 2007, the Company and Fovea amended and restated the Original Fovea Agreement (the “Amended Fovea Agreement”). Under the Amended Fovea Agreement, Fovea will continue to conduct, at its own expense, preclinical and clinical development for certain ophthalmic indications of combination drug candidates it has selected from the Company’s portfolio of product candidates. Fovea is obligated to develop selected combination candidates pursuant to specified development criteria through the end of Phase 2b clinical trials.
F-24
On July 22, 2009, the Company and Fovea amended and restated their Amended and Restated Research and License Agreement, dated as of June 12, 2007 (the “Amended and Restated Agreement—Amended”). Under the Amended and Restated Agreement—Amended, in exchange for Fovea’s development investment, the Company has granted Fovea an exclusive worldwide license to certain drug combinations to treat allergic and inflammatory diseases of the front of the eye. Fovea has advanced one such combination, Prednisporin (FOV-1101), through a Phase 2b clinical trial for allergic conjunctivitis. For these licensed combinations, the Company has received payments totaling $1,500, and is eligible to receive up to an additional $24,000 in development and regulatory milestone payments for each combination successfully developed by Fovea, an additional $15,000 milestone payment for the approval of a combination in a specified additional indication and in the event these license combinations are licensed by Fovea to a third party, commercialization milestones of up to an additional $25,000. The Company is also eligible to receive royalties for each product commercialized by Fovea in connection with the agreement.
Under the Amended Fovea Agreement, the Company may also receive up to approximately $20,000 in development milestone payments for each licensed combination successfully developed and approved by the regulatory authorities in the European Union, United States and Japan. The Company could receive an additional milestone payment of $10,000 for the approval by any regulatory authority of a licensed combination developed to treat a specifically identified indication within the agreement. The Company is also eligible to receive royalties for product(s) commercialized by Fovea. On December 30, 2006, Fovea selected the licensed combination compounds as provided in the Original Fovea Agreement. In June 2009, the Company received and recognized as revenue $250 upon Fovea’s successful filing of an IND with the FDA. In June 2010, the Company received and recognized as revenue $500 upon Fovea’s initiation of Phase 2b clinical testing.
Angiotech Pharmaceuticals, Inc.
In October 2005, the Company entered into a research and license agreement (the “R&L Agreement”) with Angiotech Pharmaceuticals, Inc. (“Angiotech”), under which the Company granted Angiotech a royalty-bearing license for up to ten compounds to be selected by Angiotech from the Company’s portfolio of clinical and preclinical product candidates or Chalice database, as well as an option to purchase the same rights to an additional five compounds. This license was for Angiotech’s research, development and potential commercialization of the licensed compounds as drug components to be used in Angiotech’s field with medical devices or interventional medicine products to treat conditions in specific areas of the human body. In addition, the Company agreed to use its combination high throughput screening technology in a joint research project to screen combinations of compounds that may be developed and commercialized by Angiotech for use in combination with medical devices or with interventional medicine products in Angiotech’s field. The Company received a $27,000 up-front license fee upon execution of the R&L Agreement in 2005. As contemplated by the original agreement, on June 8, 2007, Angiotech agreed to extend the research project beyond the original 30-month term to a total term of five years for an additional license execution fee of $7,000. The original three-year research project performance period included a six-month period beyond the 30 months where the Company was required to provide Angiotech with all reasonable assistance required in order to transfer the licensed information to Angiotech. The Company recognized $11,152 and $4,957 of revenue under this agreement in 2009 and 2008, respectively, which represented 64.6% and 35.1% of the Company’s total revenue in 2009 and 2008, respectively. In November 2009, the Company and Angiotech agreed to terminate the R&L Agreement. Under the terms of the termination, the Company is not required to refund to Angiotech any funds paid to it under the Agreement and has no significant continuing obligations under the agreement; and therefore, recognized the remaining deferred revenue balance of $7,022 in the year ended December 31, 2009.
Cystic Fibrosis Foundation Therapeutics
On May 31, 2006, the Company entered into a research, development and commercialization agreement (the “CF Agreement”) with Cystic Fibrosis Foundation Therapeutics Incorporated (“CFFT”). Under the terms of the CF Agreement, the Company was awarded up to $13,825 in research funding and expenses.
F-25
During the year ended December 31, 2009, the Company received payments of $1,249 and recognized $1,783 of revenue under the CF Agreement, which represented 10.3% of the Company’s total revenue in 2009. During the year ended December 31, 2008, the Company received payments of $3,235 and recognized $3,209 of revenue under the CF Agreement, which represented 22.7% of the Company’s total revenue in 2008. On May 14, 2009, the Company and CFFT mutually agreed to end the cystic fibrosis research program being conducted under the Agreement. The research program ended on August 15, 2009, and the CF Agreement survived as modified by the Company and CFFT on May 14, 2009.
The DMD Foundations
On November 7, 2007, the Company entered into a sponsored research collaboration agreement with an entity formed by Charley’s Fund and the Nash Avery Foundation (the “DMD Foundations”), two nonprofit organizations founded to support Duchenne muscular dystrophy, or DMD research. In October 2008, GMT Charitable Research, LLC, an affiliate of a charitable organization focused on finding therapies for DMD joined the sponsored research collaboration agreement. Under the agreement, the Company was seeking to identify novel disease-modifying multi-targeted treatments for DMD, the most common childhood form of muscular dystrophy. Under the terms of the agreement, the Company was eligible to receive up to $3,450 in research funding and reimbursement of additional expenses during the term of the DMD research and development project. The research and development collaboration for the DMD Foundations expired on December 31, 2009. The DMD Foundations have exercised their rights to an exclusive, fully-paid and sublicensable license to the intellectual property developed under the collaboration in the field of DMD. The Company recorded revenue of $1,800 and $1,643 in 2009 and 2008, respectively.
CHDI, Inc.
In August 2005, the Company entered into a research agreement with CHDI, Inc. to perform joint research and development to discover and perform preclinical development of product candidates for the treatment of Huntington’s disease. Under the terms of this agreement as amended and restated in February 2007, subject to satisfaction of conditions, the Company could receive up to $6,695 in research funding over a four-year period and was eligible to receive milestone and revenue sharing payments under certain circumstances if a product candidate is commercialized. Joint research and development activities under the research agreement were completed in December 2008, and Zalicus has assigned its joint ownership interest in the intellectual property from the research project to CHDI. The Company recorded revenue of $2 and $1,154 in 2009 and 2008, respectively, based upon research services performed and costs incurred.
PGx Health, a Subsidiary of Clinical Data, Inc.
In August 2009, the Company and PGxHealth, LLC, or PGx, a subsidiary of Clinical Data, Inc., entered into a collaboration agreement relating to the potential development of ATL313, an adenosine A2A receptor agonist compound owned by PGx, as a combination therapy in the cancer field. The Company has previously discovered that adenosine A2A agonists synergize with existing and emerging standard-of-care drugs for the treatment of multiple myeloma and certain other B-cell malignancies.
Under the terms of the collaboration agreement, the Company would fund and advance the preclinical and clinical development of ATL313 as a combination therapy in the cancer field. On March 1, 2011, the Company provided notice of termination of the collaboration agreement.
NIAID
In April 2005, the Company received a grant from the National Institutes of Allergy and Infectious Diseases (“NIAID”) to perform research and preclinical development in the area of bioterror defense. The Company recorded $188, $432 and $580 of revenue related to the research and preclinical development agreement for the
F-26
years ended December 31, 2010, 2009 and 2008, respectively. The Company received payments related to the research and preclinical development agreement totaling $250, $384 and $595 during the years ended December 31, 2010, 2009 and 2008, respectively.
USAMRIID
In March 2008, the Company entered into a cooperative research and development agreement with the United States Army Medical Research Institute for Infectious Diseases, or USAMRIID, focused on discovering agents to prevent or treat Ebola virus infections, which was extended in October 2010. Under the agreement, which expires in September 2011, the Company and USAMRIID will undertake a joint research project, and the Company is eligible to receive up to $1,387 in funding. Through December 31, 2010, the Company has received approximately $1,166 in funding from this agreement.
The Company recorded $425, $520 and $262 of revenue related to the cooperative research and development agreement for the years ended December 31, 2010, 2009 and 2008, respectively. The Company received payments related to the research and development agreement totaling $456, $605 and $105 during the years ended December 31, 2010, 2009 and 2008, respectively.
In May 2010, Zalicus entered into a cooperative research and development agreement with USAMRIID focused on discovering agents to prevent or treat Alphavirus infections. Under the agreement, which expires in June 2012, the Company and USAMRIID are undertaking a joint research project, and the Company is eligible to receive up to approximately $1,056 in funding. Through December 31, 2010, the Company has received approximately $74 in funding from this agreement. The Company recorded $159 of revenue related to the research and development agreement for the year ended December 31, 2010.
Other Pilot Research Agreements
The Company recorded $200 of revenue related to other pilot research agreements for the year ended December 31, 2010. The Company received payments related to these agreements totaling $295 during the year ended December 31, 2010.
| 11. | Common Stock |
Each share of common stock is entitled to one vote. The holders of common stock are also entitled to receive dividends whenever funds are legally available and when declared by the Board of Directors, subject to the prior rights of holders of all classes of stock outstanding.
On December 21, 2009, the Company issued 14,937,654 shares of common stock to the former Neuromed stockholders and placed 67,826,875 shares into escrow as consideration for the merger with Neuromed. (See Note 3.). The shares held in escrow would either be released from escrow or returned to the Company and cancelled based upon the timing of the FDA approval of Exalgo.
Because the FDA did not approve Exalgo prior to January 1, 2010, 29,943,752 Escrow Shares were returned to the Company on January 1, 2010 and were subsequently cancelled.
On March 1, 2010, the FDA approved Exalgo, and 37,883,123 shares held in escrow were released to former Neuromed stockholders.
The Company has reserved a total of 8,021,447 shares of common stock for the exercise of stock options and warrants at December 31, 2010. The Company has also issued a warrant to purchase 10,019 shares of common stock to Silicon Valley Bank with an exercise price of $7.88 per share and a term that expires on April 25, 2011; warrants to purchase 51,870 shares of common stock to entities affiliated with Lighthouse
F-27
Capital Partners with an exercise price of $6.75 per share that expires on September 7, 2011; warrants to purchase 9,363 shares of common stock to General Electric Capital Corporation with an exercise price of $6.75 per share that expire on September 15, 2014 and June 28, 2015; and a warrant to purchase 64,748 shares of common stock to Oxford at an exercise price of $1.39 per share that expires on December 21, 2017.
| 12. | Stock Compensation Plans |
In 2000, the Company adopted the 2000 Stock Plan (“2000 Plan”), as amended, under which 3,028,571 shares of the Company’s common stock were reserved for issuance to employees, officers, directors, advisors and consultants. Options granted under the 2000 Plan may be incentive stock options or non-statutory stock options. As of December 31, 2010, there were no options available to grant under the 2000 Plan.
In December 2004, the Board of Directors and stockholders adopted the 2004 Incentive Plan, which was effective upon the Company’s initial public offering on November 9, 2005. The 2004 Plan includes an “evergreen provision” that allows for an annual increase in the number of shares of common stock available for issuance under the 2004 Plan, which annual increase will be added on the first day of each fiscal year from 2010 through 2019, inclusive, and will be equal to the least of (i) 4,000,000 shares of common stock, (ii) 4% of the outstanding shares on that date or (iii) such lesser amount determined by the Board of Directors. On December 21, 2009, the Compensation Committee of the Board of Directors, in conjunction with the Company’s Annual Meeting, authorized an increase in the number of shares of common stock reserved for issuance. As of December 21, 2009, 20,000,000 shares of common stock were reserved for issuance under the 2004 Plan. The Compensation Committee of the Board of Directors elected not to increase the number of shares of common stock available for issue under the 2004 Plan for 2010. The 2004 Plan provides for the grant of incentive stock options, non-statutory stock options, restricted stock and unrestricted stock awards, stock appreciation rights, cash awards, performance awards and restricted stock units. Awards under the 2004 Plan may be granted to employees, directors, consultants and advisors. As of December 31, 2010, there were 11,738,775 shares available for future issuance under the 2004 plan.
On December 21, 2009, the shareholders approved an exchange program that permitted eligible employees to exchange outstanding options with exercise prices greater than or equal to $1.31 per share, for a lesser number of options with an exercise price equal to the closing price on the day the exchange program closed. On December 21, 2009, the Company granted an aggregate of 941,065 new stock options in exchange for the eligible 1,490,750 stock options surrendered. The exercise price of the new stock options was $1.07 per share, which was the closing price of the Company’s common stock on December 21, 2009. The new stock options were granted under the 2004 Plan. No incremental stock-based compensation expense was recognized for the exchange because the fair value of the new options approximated the fair value of the surrendered options.
The Board of Directors, or the Compensation Committee of the Board of Directors, administers the 2000 Plan and the 2004 Plan and has sole discretion to grant options to purchase shares of the Company’s common stock and other stock-based awards or to delegate to certain officers of the Company the ability to make specified grants. The Compensation Committee or the respective officers of the Company determine the exercise price and the period over which options become exercisable. However, incentive stock options may not be granted at less than 100% of the fair market value of the Company’s common stock as determined by the Compensation Committee at the time of grant, or for a term in excess of ten years. For holders of more than 10% of the Company’s total combined voting power of all classes of stock, incentive stock options may not be granted at less than 110% of the fair market value of the Company’s common stock at the date of grant, and for a term not to exceed five years.
F-28
A summary of the status of the Company’s stock option plans at December 31, 2010 and changes during the year then ended are presented in the table and narrative below:
| Options | Weighted- Average Exercise Price |
Weighted Average Remaining Contractual Term |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding at December 31, 2009 |
4,108,663 | $ | 3.50 | |||||||||||||
| Granted |
3,872,000 | 0.98 | ||||||||||||||
| Exercised |
(502,179 | ) | 0.96 | |||||||||||||
| Cancelled |
(368,037 | ) | 4.32 | |||||||||||||
| Outstanding at December 31, 2010 |
7,110,447 | $ | 2.26 | 6.20 | $ | 3,225 | ||||||||||
| Vested or expected to vest at December 31, 2010 |
6,558,244 | $ | 2.37 | 5.97 | $ | 2,888 | ||||||||||
| Exercisable at December 31, 2010 |
3,108,041 | $ | 3.92 | 2.70 | $ | 785 | ||||||||||
The aggregate intrinsic value in the table above represents the value (the difference between the Company’s closing common stock price on the last trading day of the year ended December 31, 2010 and the exercise price of the options, multiplied by the number of in-the-money options) that would have been received by the option holders had all option holders exercised their options on December 31, 2010. As of December 31, 2010, there was $2,069 of total unrecognized stock-based compensation expense related to stock options granted under the plans. The expense is expected to be recognized over a weighted-average period of 2.5 years. The weighted-average grant date fair value of options for the years ended December 31, 2010, 2009 and 2008 was $0.80, $0.10 and $4.17, respectively. The intrinsic value of stock options exercised for the years ended December 31, 2010, 2009 and 2008 was $230, $6 and $733, respectively, and represents the difference between the exercise price of the option and the market price of the Company’s common stock on the dates exercised.
The Company valued stock options using a Black-Scholes method of valuation and has applied the weighted-average assumptions set forth in the following table. The resulting fair value is recorded as compensation cost on a straight line basis over the requisite service period, which generally equals the option vesting period. Since the Company completed its initial public offering in November 2005, it did not have sufficient history as a publicly traded company to evaluate its volatility factor and expected term. As such, the Company analyzed the volatilities and expected terms of several peer companies to support the assumptions used in its calculations. The Company averaged the volatilities and expected terms of these peer companies with sufficient trading history and similar vesting terms to generate the assumptions detailed below. The Company also began to include its own historical volatility in the expected volatility calculation once it was available and has continued to increase the weight applied to its own historical volatility over time. In 2010, the Company determined it had sufficient trading history as a publicly traded company to utilize the historical volatility of its common stock to calculate an annual expected volatility input. The risk-free interest rates used are based on the United States Treasury yield curve in effect for periods corresponding with the expected life of the stock option. The Company has estimated forfeitures based upon an average of its historical data of option cancellations and employee turnover rates. Changes in estimated forfeitures are recognized through a cumulative true-up adjustment in the period of change. The Company determined that the reduction in work force that occurred in the fourth quarter of 2008 and continued into 2009 is a one-time occurrence and, therefore, excluded these stock awards for these terminated employees from the forfeiture rate calculation.
F-29
During the years ended December 31, 2010, 2009 and 2008, respectively, the weighted-average assumptions used in the Black-Scholes model were as follows:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Volatility factor |
108.69 | % | 108.96 | % | 56.86 | % | ||||||
| Risk-free interest rate |
2.47 | % | 2.69 | % | 2.68 | % | ||||||
| Dividend yield |
— | % | — | % | — | % | ||||||
| Expected term (in years) |
5.8 | 5.8 | 5.7 | |||||||||
Restricted Stock
A summary of the status of non-vested restricted stock awards as of December 31, 2010 is as follows:
| Restricted Stock Awards |
Weighted- Average Grant Date Fair Value |
|||||||
| Non-vested at December 31, 2009 |
18,750 | $ | 4.57 | |||||
| Granted |
— | — | ||||||
| Vested |
(6,250 | ) | $ | 4.57 | ||||
| Canceled |
— | — | ||||||
| Non-vested at December 31, 2010 |
12,500 | $ | 4.57 | |||||
As of December 31, 2010, there was $27 of total unrecognized stock-based compensation expense related to non-vested restricted stock awards granted under the 2004 Plan. The expense is expected to be recognized over a weighted-average period of 1.1 years. The total fair value of shares vested for the years ended December 31, 2010, 2009 and 2008 was $29, $446 and $939, respectively.
The Company issued performance-based Restricted Stock Units (“RSUs”) in 2009 and 2010 to certain employees and directors. If the performance measure is not achieved, a portion of the vesting of the RSU grant is time based contingent upon the grantee’s continued employment or board membership with the Company. The fair value of RSUs is based on the closing price of the Company’s common stock on the award date. Expense for performance-based RSUs is recognized when it is probable the performance goal will be achieved. On March 1, 2010, the performance goal for the December 21, 2009 RSU grants was achieved, and all corresponding expense, or $1,150, was recognized immediately as the awards vested in full. On March 1, 2010, the performance goal was achieved for the RSUs granted on January 15, 2010 such that all of the awards are expected to ultimately vest. The expense for this grant will be recognized on a straight-line basis over the four year vesting period. Prior to 2009, the Company had not granted awards of RSUs. A summary of the status of non-vested RSUs as of December 31, 2010 is as follows:
| Restricted Stock Units |
Weighted- Average Grant Date Fair Value |
Weighted Average Remaining Contractual Term |
Aggregate Intrinsic Value |
|||||||||||||
| Non-vested at December 31, 2009 |
1,077,976 | $ | 1.07 | |||||||||||||
| Granted |
750,000 | 0.95 | ||||||||||||||
| Vested |
(1,077,976 | ) | 1.07 | |||||||||||||
| Cancelled |
— | — | ||||||||||||||
| Non-Vested at December 31, 2010 |
750,000 | $ | 0.95 | 3.04 | $ | 1,185 | ||||||||||
As of December 31, 2010, there was $455 of total unrecognized stock-based compensation expense related to non-vested restricted stock units granted under the 2004 Plan. The expense is expected to be recognized over a weighted-average period of 3 years.
F-30
| 13. | Segment and Geographic Information |
Operating segments are defined as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker, or decision-making group, in deciding how to allocate resources and in assessing performance. The Company views its operations and manages its business in one operating segment. The Company operates in two geographic segments: the United States and Canada. As of December 31, 2010, $6,098 and $800 of the Company’s long-lived assets were located in the United States and Canada, respectively. As of December 31, 2009, $6,839 and $1,541 of the Company’s long-lived assets were located in the United States and Canada, respectively. Revenues from customers located outside the United States for the years ended December 31, 2010, 2009 and 2008 were $500, $11,402 and $5,457, respectively.
| 14. | Income Taxes |
The benefit for income taxes for the years ended December 31, 2010, 2009 and 2008 are as follows, in thousands:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| U.S.: |
||||||||||||
| Current |
$ | 300 | $ | 67 | $ | 108 | ||||||
| Deferred |
— | — | — | |||||||||
| Total U.S. |
300 | 67 | 108 | |||||||||
| Foreign: |
||||||||||||
| Current |
910 | — | — | |||||||||
| Deferred |
— | — | — | |||||||||
| Total foreign |
910 | — | — | |||||||||
| Benefit for income taxes |
$ | 1,210 | $ | 67 | $ | 108 | ||||||
A reconciliation of the expected income tax benefit (expense) computed using the federal statutory income tax rate to the Company’s effective income tax rate is as follows for the years ended December 31, 2010, 2009 and 2008:
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Income tax computed at federal statutory tax rate |
34.0 | % | 34.0 | % | 34.0 | % | ||||||
| State taxes, net of federal benefit |
3.0 | % | (5.5 | )% | 0.2 | % | ||||||
| Impact of state rate change |
(0.5 | )% | 5.2 | % | — | % | ||||||
| Gain on revaluation of contingent consideration |
(27.5 | )% | (26.8 | )% | — | % | ||||||
| Gain on bargain purchase |
— | % | (21.8 | )% | — | % | ||||||
| Merger costs |
— | % | 11.5 | % | — | % | ||||||
| Divestiture of subsidiary |
— | % | (34.7 | )% | — | % | ||||||
| Expiration of net operating losses |
— | % | (5.2 | )% | — | % | ||||||
| Change in valuation allowance |
(11.7 | )% | 48.9 | % | (35.6 | )% | ||||||
| Other |
3.3 | % | 0.1 | % | (1.1 | )% | ||||||
| Stock-based compensation |
(0.8 | )% | 0.3 | % | (1.4 | )% | ||||||
| Research and development credits |
3.0 | % | (6.3 | )% | 4.2 | % | ||||||
| Permanent differences |
0.5 | % | (0.2 | )% | (0.1 | )% | ||||||
| Total |
3.3 | % | (0.5 | )% | 0.2 | % | ||||||
F-31
The Company has incurred net operating losses since inception. At December 31, 2010, the Company had domestic federal and state net operating loss carryforwards of approximately $42,113 and $17,641, respectively, available to reduce future taxable income, which expire at various dates beginning in 2009 through 2029. At December 31, 2010, the Company also had federal and state research and development tax credit carryforwards of approximately $125 and $123, respectively, available to reduce future tax liabilities and which expire in 2030 and 2015, respectively. The Company also had foreign net operating loss carryforwards of approximately $40,964, which expire at various dates through 2029. The Company also has foreign research and development tax credit carryforwards of $7,180, which never expire. The net operating loss carryforwards included $20 of federal and state net operating losses that are attributable to stock option exercises which will be recorded as an increase in additional paid-in-capital once they are “realized”. Utilization of the net operating loss and research and development credit carryforwards may be subject to a substantial annual limitation under Internal Revenue Code Section 382 of the Internal Revenue Code of 1986 due to ownership change limitations that could occur in the future.
Deferred taxes consist of the following:
| As of December 31, | ||||||||
| 2010 | 2009 | |||||||
| Net operating loss carryforwards . |
$ | 25,527 | $ | 67,863 | ||||
| Research and development credits |
7,411 | 18,419 | ||||||
| Capitalized research and development costs |
6,492 | 27,620 | ||||||
| Stock-based compensation |
5,474 | 5,222 | ||||||
| Depreciation and amortization |
1,704 | 1,473 | ||||||
| Accrued expenses |
45 | 473 | ||||||
| Capitalized financing costs |
124 | 637 | ||||||
| Deferred revenue |
1,042 | — | ||||||
| Other |
(389 | ) | (759 | ) | ||||
| Deferred tax asset |
47,430 | 120,948 | ||||||
| Deferred tax asset valuation allowance |
(47,430 | ) | (120,948 | ) | ||||
| Net deferred tax asset |
$ | — | $ | — | ||||
As required by ASC 740, management of the Company has evaluated the positive and negative evidence bearing upon the realizability of its deferred tax assets. Management has determined at this time that it is more likely than not that the Company will not recognize the benefits of its federal and state deferred tax assets and, as a result, a valuation allowance of $47,430 and $120,948 has been established at December 31, 2010 and 2009, respectively. The decrease in the valuation allowance was $75,518 for the year ended December 31, 2010, and the increase in the valuation allowance was $29,407 and $20,871 for the years ended December 31, 2009 and 2008, respectively.
As a result of the acquisition of Neuromed, the Company recorded a liability for unrecognized tax benefits of $2,685 and interest and penalties of $560 in other long-term liabilities in accordance with ASC 740-10. During the year ended December 31, 2010, the Company reversed $910 of unrecognized tax benefits and $306 of related accrued interest and penalties due to the expiration of the statute of limitations for certain tax years. The Company expects to reverse $1,438 of unrecognized tax benefits during the year ending December 31, 2011 due to the expiration of statute of limitations for certain years. The Company has not, as yet, conducted a study of its research and development credit carryforwards. This study may result in an increase or decrease to the Company’s research and development credit carryforwards; however, until a study is completed and any adjustment is known, no amounts are being presented as an uncertain tax position. A full valuation allowance has been provided against the Company’s research and development credits, and if an adjustment is required, this adjustment would be offset by an adjustment to the valuation allowance. As a result, there would be no impact to the consolidated balance sheet, statement of operations or cash flows if an adjustment were required.
F-32
The following is a reconciliation of the Company’s gross uncertain tax positions at December 31, 2010 and 2009 (in thousands):
| December 31, 2008 |
$ | — | ||
| Decrease related to positions taken in prior years |
— | |||
| Increases related to positions taken by acquired subsidiary |
2,685 | |||
| December 31, 2009 |
2,685 | |||
| Decrease related to positions taken in prior years |
— | |||
| Expiration of statute of limitations |
(910 | ) | ||
| Foreign exchange revaluation |
103 | |||
| December 31, 2010 |
$ | 1,878 | ||
Of this amount of unrecognized tax benefits, approximately $1,878 and $2,685, if recognized, would result in a reduction of the Company’s effective tax rate for the years ended December 31, 2010 and 2009, respectively. The tax years 2000 through 2009 remain open to examination by major taxing jurisdictions to which the Company is subject, which are primarily in the United States and Canada, as carryforward attributes generated in years past may still be adjusted upon examination by the Internal Revenue Service, Revenue Canada or state or provincial tax authorities if they have or will be used in a future period. The Company is currently not under examination by the Internal Revenue Service or any other jurisdictions for any tax years. For the year ended December 31, 2010, the Company recognized $108 of interest expense related to uncertain tax positions. As of December 31, 2010 and 2009, the Company has accrued $383 and $560 of interest related to uncertain tax positions, respectively.
| 15. | Accrued Expenses |
Accrued expenses consisted of the following:
| As of December 31, | ||||||||
| 2010 | 2009 | |||||||
| Accrued payroll and related benefits |
$ | 1,721 | $ | 2,340 | ||||
| Accrued clinical trial costs |
994 | 36 | ||||||
| Accrued professional fees |
464 | 1,394 | ||||||
| Accrued other expenses |
1,090 | 1,725 | ||||||
| $ | 4,269 | $ | 5,495 | |||||
| 16. | Gain on Legal Settlement |
On April 30, 2009, the Company filed a lawsuit against Aptuit, Inc. (“Aptuit”), in the Supreme Court for the State of New York, New York County, Commercial Division. In the action, the Company asserted claims against Aptuit for fraudulent inducement, breach of contract, breach of the implied covenant of good faith and fair dealing, and unjust enrichment arising out of Aptuit’s manufacture and distribution of the Company’s product candidate Synavive for a worldwide Phase 2b clinical trial targeting subjects with rheumatoid arthritis. On December 29, 2009, the Company entered into a settlement agreement with Aptuit to settle this lawsuit. Pursuant to the terms of the settlement agreement, Aptuit agreed to pay $3,700 to the Company, and the parties agreed to a mutual release of claims and causes of action that were asserted, or that could have been asserted in the action. The amount was included in prepaid expenses and other current assets at December 31, 2009 and the Company received the $3,700 payment in January 2010.
F-33
| 17. | Commitments |
On October 18, 2005, the Company entered into a lease agreement for approximately 40,000 square feet of office and laboratory space located in Cambridge, Massachusetts. The initial term of the lease commenced on September 1, 2006 for the office space and December 1, 2006 for the laboratory space and was to extend until November 30, 2016 with two five-year renewal options. The Company had the right to use and controlled physical access to the leased premises beginning on December 6, 2005. Thus, the effective lease term began on that date. In March 2006, the Company amended (the “First Amendment”) the October 18, 2005 operating lease agreement. The First Amendment provided for 23,199 square feet of additional laboratory space. The Company has committed to lease this additional laboratory space through January 2017. In addition, the First Amendment extended the original lease term of the existing space an additional two months though January 2017.
Additionally, the lease, as amended, contains rent escalation, rent holiday, and leasehold improvement incentives. Rent escalation and rent holiday are being accounted for as rent expense under the straight-line method. In connection with the lease, the Company received approximately $6,900 in leasehold improvement incentives from the landlord. These leasehold improvement incentives are being accounted for as a reduction in rent expense ratably over the lease term. The balance from these leasehold improvement incentives is included in current portion of lease incentive obligation and lease incentive obligation, net of current portion in the balance sheets at December 31, 2010 and 2009. Leasehold improvements are amortized using the straight-line method over the estimated useful lives of the assets or the term of the lease, whichever is shorter.
On August 3, 2009, the Company entered into a Second Amendment (the “Amendment”) to the Office and Laboratory Lease Agreement (the “Lease”). Prior to the Amendment of the Lease, which, as amended, expires in January 2017, the Company leased approximately 63,000 square feet of office and laboratory space.
In accordance with the terms of the Amendment, the Company and the landlord agreed that the Company’s occupancy of approximately 18,000 square feet of leased premises (the “Office Premises”) would cease as of June 16, 2009, and that the Company would be liable for rent payments and occupancy costs on the Office Premises through September 30, 2009.
In addition, the Company and the Landlord agreed that the Company’s occupancy and liability for rent payments and occupancy costs of approximately 22,000 square feet of leased premises (the “Lab Premises”) would cease as of September 15, 2009.
Under the terms of the Lease, as amended by the Amendment, the Company will continue to lease and occupy approximately 23,000 square feet of office and laboratory space at the Facility with a lease term until January 2017.
Under the Amendment, as consideration for the right to cease occupancy of the Office Premises, the Company paid the Landlord $500 on October 1, 2009. In addition, as consideration for the right to cease occupancy of the Lab Premises, the Company would pay the Landlord $500 on or before January 1, 2010 and $500 on or before July 1, 2010. The Company made both payments in the year ended December 31, 2010.
In connection with the Amendment, the Company’s Letter of Credit for the benefit of the Landlord was reduced from $4,000 to $2,500, effective December 1, 2009; was reduced to: $1,800 effective as of December 1, 2010; and will be reduced to $1,200 effective as of December 1, 2011; and $800 effective as of December 1, 2012. The certificate of deposit that secures the letter of credit is included in restricted cash on the consolidated balance sheet in the amount of $1,800 and $2,500 as of December 31, 2010 and 2009, respectively.
In connection with the Neuromed merger (as discussed in Note 3), the Company assumed a sublease renewal and amendment agreement between Zalicus Pharmaceuticals Ltd. and Discovery Parks Incorporated, dated November 2, 2009, relating to the Company’s office and laboratory facility in Vancouver, British
F-34
Columbia, Canada. Under the terms of this lease the Company leased approximately 24,600 square feet of office and laboratory space, which was reduced to approximately 12,000 square feet. The lease will expire on December 31, 2011. The Company has the option to extend the lease for one additional year.
The Company also leases certain office equipment under various operating leases. Total rent expense was $1,676, $1,860 and $2,750, of which $0, $58 and $148 was classified as discontinued operations, for the years ended December 31, 2010, 2009 and 2008, respectively.
Future minimum lease payments under noncancelable operating leases at December 31, 2010, are as follows:
| Year ending December 31, |
||||
| 2011 |
$ | 1,359 | ||
| 2012 |
1,220 | |||
| 2013 |
1,229 | |||
| 2014 |
1,230 | |||
| 2015 |
1,230 | |||
| Thereafter |
1,332 | |||
| $ | 7,600 | |||
| 18. | Net Income (Loss) Per Share |
The Company presents basic net income (loss) per share and diluted net income (loss) per share. Basic income (loss) per share is based on the weighted average number of shares outstanding during the period. Diluted income (loss) per share reflects the per share effect of dilutive common stock equivalents using the treasury stock method for all outstanding warrants, stock options, restricted stock awards and restricted stock units.
For the period that the Escrow Shares issued in the Neuromed merger were held in Escrow, the Company calculated the net income (loss) per share using the two-class method, which is an earnings allocation formula that determines net income (loss) per share for the holders of the Company’s common shares and Escrow Shares issued in connection with the Neuromed merger. (See Note 3.) The Escrow Shares contained participation rights in any dividend paid by the Company while the shares were held in escrow. Net income available to common shareholders and participating Escrow Shares was allocated to each share equally as if all of the earnings for the period had been distributed. Prior to being released from escrow, during the period of January 1, 2010 to March 1, 2010, the Escrow Shares were not considered in the calculation of basic net loss per share for the year ended December 31, 2010, since they did not include a contractual obligation to share in the losses of the Company.
F-35
The following table sets forth the computation of basic and diluted earnings per share (in thousands, except per share data):
| Year Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Net (loss) income from continuing operations |
$ | (35,045 | ) | $ | 1,284 | $ | (60,576 | ) | ||||
| Discontinued operations: |
||||||||||||
| Gain (loss) from operations of discontinued subsidiary |
— | 14,104 | (4,557 | ) | ||||||||
| Net (loss) income |
$ | (35,045 | ) | $ | 15,388 | (65,133 | ) | |||||
| Weighted-average common shares outstanding |
82,663,645 | 35,479,771 | 34,848,701 | |||||||||
| Weighted-average participating escrow shares |
— | 1,858,271 | — | |||||||||
| Total shares for basic earnings per share |
82,663,645 | 37,338,042 | 34,848,701 | |||||||||
| From continuing operations |
$ | (0.42 | ) | $ | 0.03 | $ | (1.74 | ) | ||||
| From discontinued operations |
— | 0.38 | (0.13 | ) | ||||||||
| Net (loss) income per share—basic |
$ | (0.42 | ) | $ | 0.41 | $ | (1.87 | ) | ||||
| Net (loss) income per share—diluted information: |
||||||||||||
| Weighted-average common shares outstanding |
82,663,645 | 37,338,042 | 34,848,701 | |||||||||
| Weighted-average diluted common share equivalents |
— | 153,195 | — | |||||||||
| Weighted-average common shares and diluted common share equivalents |
82,663,645 | 37,491,237 | 34,848,701 | |||||||||
| From continuing operations |
$ | (0.42 | ) | $ | 0.03 | $ | (1.74 | ) | ||||
| From discontinued operations |
— | 0.38 | (0.13 | ) | ||||||||
| Net income (loss) per share—diluted |
$ | (0.42 | ) | $ | 0.41 | $ | (1.87 | ) | ||||
The following potentially dilutive securities outstanding prior to the use of the treasury stock method have been excluded from the computation of diluted weighted-average shares outstanding for the years ended December 31, 2010, 2009 and 2008, as they would be anti-dilutive.
| As of December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Options outstanding |
7,110,447 | 3,287,042 | 5,966,316 | |||||||||
| Unvested restricted stock awards and units |
762,500 | 467,905 | 103,438 | |||||||||
| Warrants outstanding |
136,000 | 96,252 | 96,252 | |||||||||
| 19. | Employee Benefit Plans |
In May 2001, the Company adopted the Zalicus Inc. 401(k) Plan (“401(k) Plan”). The 401(k) Plan allows employees to make pre-tax contributions up to the maximum allowable amount set by the IRS. Under the 401(k) Plan, the Company may make discretionary contributions as approved by the Board of Directors. During 2010, 2009 and 2008, the Company made contributions of $273, $295 and $601, respectively.
Effective December 1, 2007, the Company approved the Zalicus Nonqualified Deferred Compensation Plan (the “NQ Plan”), a non-qualified tax-deferred compensation plan in which certain senior managers and officers of the Company may participate. The NQ Plan provides a tax-favorable vehicle for deferring cash compensation, including base salary and bonus awards. Under the NQ Plan, each year a participant may defer up to 25% of his or her base salary and up to 100% of his or her annual cash bonus pay. The participant will at all times be vested in the portion of his or her account attributable to the compensation the participant has elected to defer under the
F-36
NQ Plan. The Company has established a special account for each participant, however, the Company’s obligation to pay the balance credited to such account will at all times be an unfunded and unsecured obligation of the Company and rank on parity with other unsecured and unsubordinated indebtedness of the Company from time to time outstanding.
The Company may also credit to the account of each eligible participant who makes deferrals a matching contribution in an amount equal to 100% of the deferrals contributed by the participant for such plan year, up to a maximum amount equal to: (i) four percent (4%) of each such participant’s cash compensation for such year, less (ii) the amount of matching contributions made to the Company’s qualified 401(k) Plan for such year on behalf of such participant. In order to be eligible for a matching contribution for a given year, a participant must be employed by the Company on the date the matching contribution is credited to the NQ Plan, which is currently planned to be the January following a participant’s election, and have deferred the maximum amount permitted under the Company’s tax-qualified Section 401(k) Plan for such year. A participant will become 100% vested in any employer contributions credited to his or her account upon the participant’s death, disability or a change in control (as defined in the NQ Plan). Deferred balances are credited to each participant’s account under the NQ Plan and will be credited, at periodic intervals, with earnings that track the actual rate of return for such period realized by the investment fund or funds or index or indices selected by such participant from the range of investment vehicles offered under the NQ Plan. Deferred amounts are paid, at the participant’s option, either in a lump sum or in annual installments over a period of up to ten years upon separation from service or up to five years for scheduled in-service withdrawals. The Company contributed approximately $60 to the NQ Plan as of December 31, 2010.
| 20. | Quarterly Financial Information (unaudited) |
| First Quarter Ended March 31, 2010 |
Second Quarter Ended June 30, 2010 |
Third Quarter Ended September 30, 2010 |
Fourth Quarter Ended December 31, 2010 |
|||||||||||||
| Revenue |
$ | 41,330 | $ | 2,928 | $ | 1,166 | $ | 1,317 | ||||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
7,381 | 4,617 | 5,062 | 5,951 | ||||||||||||
| General and administrative |
3,862 | 2,569 | 2,828 | 2,856 | ||||||||||||
| Amortization of intangible asset |
4,684 | 4,685 | 4,683 | 4,684 | ||||||||||||
| Total operating expenses |
15,927 | 11,871 | 12,573 | 13,491 | ||||||||||||
| Income (loss) from operations |
25,403 | (8,943 | ) | (11,407 | ) | (12,174 | ) | |||||||||
| Interest income |
7 | 30 | 55 | 40 | ||||||||||||
| Interest expense |
— | — | — | (12 | ) | |||||||||||
| Loss on revaluation of contingent consideration |
(29,286 | ) | — | — | — | |||||||||||
| Other (expense) income |
(142 | ) | 15 | (18 | ) | 177 | ||||||||||
| Net loss before benefit for income taxes |
(4,018 | ) | (8,898 | ) | (11,370 | ) | (11,969 | ) | ||||||||
| Benefit for income taxes |
910 | 300 | — | — | ||||||||||||
| Net loss |
$ | (3,108 | ) | $ | (8,598 | ) | $ | (11,370 | ) | $ | (11,969 | ) | ||||
| Net loss per share—basic and diluted |
$ | (0.05 | ) | $ | (0.10 | ) | $ | (0.13 | ) | $ | (0.13 | ) | ||||
| Weighted-average number of common shares used in net loss per share calculation |
63,310,675 | 88,946,220 | 89,014,241 | 89,031,018 | ||||||||||||
F-37
| First Quarter Ended March 31, 2009 |
Second Quarter Ended June 30, 2009 |
Third Quarter Ended September 30, 2009 |
Fourth Quarter Ended December 31, 2009 |
|||||||||||||
| Revenue |
$ | 2,633 | $ | 3,318 | $ | 2,742 | $ | 8,580 | ||||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
7,137 | 6,106 | 4,881 | 3,120 | ||||||||||||
| General and administrative |
3,647 | 4,824 | 3,554 | 5,056 | ||||||||||||
| Restructuring |
441 | (425 | ) | 2,597 | 123 | |||||||||||
| Amortization of intangible asset |
— | — | — | 520 | ||||||||||||
| Gain on legal settlement |
— | — | — | (3,700 | ) | |||||||||||
| Total operating expenses |
11,225 | 10,505 | 11,032 | 5,119 | ||||||||||||
| (Loss) income from operations |
(8,592 | ) | (7,187 | ) | (8,290 | ) | 3,461 | |||||||||
| Interest income |
128 | 60 | 54 | 15 | ||||||||||||
| Interest expense |
(16 | ) | (11 | ) | (1 | ) | — | |||||||||
| Gain on revaluation of contingent consideration |
— | — | — | 12,068 | ||||||||||||
| Gain on bargain purchase |
— | — | — | 9,809 | ||||||||||||
| Other income (expense) |
4 | (11 | ) | (25 | ) | (249 | ) | |||||||||
| Net (loss) income before benefit for income taxes |
(8,476 | ) | (7,149 | ) | (8,262 | ) | 25,104 | |||||||||
| Benefit for income taxes |
— | — | — | 67 | ||||||||||||
| Net (loss) income from continuing operations |
(8,476 | ) | (7,149 | ) | (8,262 | ) | 25,171 | |||||||||
| Discontinued operations: |
||||||||||||||||
| Loss from operations of discontinued subsidiary |
(1,007 | ) | (529 | ) | — | — | ||||||||||
| Gain on disposal of subsidiary |
— | 15,640 | — | — | ||||||||||||
| (Loss) gain on discontinued operations |
(1,007 | ) | 15,111 | — | — | |||||||||||
| Net (loss) income |
$ | (9,483 | ) | $ | 7,962 | $ | (8,262 | ) | $ | 25,171 | ||||||
| Net (loss) income per share—basic and diluted: |
||||||||||||||||
| From continuing operations |
$ | (0.24 | ) | $ | (0.20 | ) | $ | (0.24 | ) | $ | 0.57 | |||||
| From discontinued operations |
(0.03 | ) | 0.43 | — | — | |||||||||||
| Net (loss) income per share—basic and diluted |
$ | (0.27 | ) | $ | 0.23 | $ | (0.24 | ) | $ | 0.57 | ||||||
| Weighted-average number of common shares used in net (loss) income per share calculation: |
||||||||||||||||
| Basic |
35,013,381 | 35,026,106 | 35,038,881 | 44,198,131 | ||||||||||||
| Diluted |
35,013,381 | 35,026,106 | 35,038,881 | 44,503,062 | ||||||||||||
| 21. | Subsequent Events |
On February 9, 2011, the Company entered into an equity distribution agreement with Wedbush Securities Inc., or Wedbush, pursuant to which the Company could issue and sell shares of its common stock having an aggregate offering price of up to $20,000 from time to time through Wedbush acting as agent and/or principal. The Company agreed to pay Wedbush a commission, or allow a discount, of 3.0% of the gross proceeds from each sale. As of March 8, 2011, the Company had sold an aggregate of 4,813,600 shares of common stock at an average price of approximately $2.21 per share for gross proceeds of approximately $10,656. Net proceeds were approximately $10,160 after deducting Wedbush’s commission and expenses to establish the agreement.
F-38
EXHIBIT INDEX
| Exhibit No. |
Description |
Incorporated by Reference to: | ||||||||||||||||
| Form or Schedule |
Exhibit No. |
Filing Date with SEC |
SEC File Number |
|||||||||||||||
| 2.1 | Agreement and Plan of Merger, dated as of June 30, 2009, by and among the Registrant, PawSox, Inc., Neuromed Pharmaceuticals Inc., Neuromed Pharmaceuticals Ltd. and the Stockholder Representative named therein. | 8-K | 2.1 | 07/01/2009 | 000-51171 | |||||||||||||
| 2.2 | Escrow Agreement, dated as of June 30, 2009, by and among the Registrant, Computershare Trust Company, N.A. and the Stockholder Representative. | 8-K | 2.2 | 07/01/2009 | 000-51171 | |||||||||||||
| 3.1 | Sixth Amended and Restated Certificate of Incorporation of the Registrant. | S-1/A | 3.2 | 11/04/2005 | 333-121173 | |||||||||||||
| 3.2 | Certificate of Amendment to the Registrant’s Sixth Amended and Restated Certificate of Incorporation. | 8-K | 3.1 | 12/21/2009 | 000-51171 | |||||||||||||
| 3.3 | Certificate of Amendment to the Registrant’s Sixth Amended and Restated Certificate of Incorporation. | 8-K | 3.1 | 09/09/2010 | 000-51171 | |||||||||||||
| 3.4 | Amended and Restated By-Laws of the Registrant. | 8-K | 3.2 | 09/09/2010 | 000-51171 | |||||||||||||
| 4.1 | Specimen Common Stock Certificate of the Registrant. | 10-Q | 4.1 | 11/10/2010 | 000-51171 | |||||||||||||
| 4.2 | Warrant issued to General Electric Capital Corporation on September 15, 2004, to purchase up to 8,892 shares of the Registrant’s Common Stock. | S-1 | 10.7 | 12/10/2004 | 333-121173 | |||||||||||||
| 4.3 | Warrant issued to General Electric Capital Corporation on June 28, 2005, to purchase 471 shares of the Registrant’s Common Stock. | 10-K | 4.5 | 03/20/2006 | 000-51171 | |||||||||||||
| 4.4 | Warrant issued to Silicon Valley Bank on April 25, 2001, to purchase up to 10,019 shares of the Registrant’s Common Stock. | S-1 | 10.9 | 12/10/2004 | 333-121173 | |||||||||||||
| 4.5 | Registration Rights Agreement dated as of April 25, 2001, by and between Silicon Valley Bank and the Registrant. | S-1 | 10.10 | 12/10/2004 | 333-121173 | |||||||||||||
| 4.6 | Form of Warrant to purchase shares of the Registrant’s Stock, together with a schedule of warrant holders. | S-1 | 10.11 | 12/10/2004 | 333-121173 | |||||||||||||
| 4.7 | Registration Rights Agreement, dated as of June 30, 2009, among the Registrant and the investors set forth therein. | S-4 | 4.11 | 08/07/2009 | 333-161146 | |||||||||||||
| 4.8 | Form of Warrant issued to Oxford Finance Corporation on December 22, 2010. | 8-K | 4.1 | 12/22/2010 | 000-51171 | |||||||||||||
| #10.1 | 2000 Stock Option Plan, as amended. | S-1 | 10.1 | 12/10/2004 | 333-121173 | |||||||||||||
| #10.2 | Amended and Restated 2004 Incentive Plan. | S-8 | 4.1 | 04/16/2010 | 333-166118 | |||||||||||||
| #10.3 | Form Incentive Stock Option Agreement under the 2004 Incentive Plan. | 10-K | 10.3 | 03/20/2006 | 000-51171 | |||||||||||||
| Exhibit No. |
Description |
Incorporated by Reference to: | ||||||||||||||||
| Form or Schedule |
Exhibit No. |
Filing Date with SEC |
SEC File Number |
|||||||||||||||
| #10.4 | Form Non-Qualified Option Agreement under the 2004 Incentive Plan. | 10-K | 10.4 | 03/20/2006 | 000-51171 | |||||||||||||
| #10.5 | Nonqualified Deferred Compensation Plan, effective December 1, 2007. | S-8 | 4.1 | 11/30/2007 | 333-147745 | |||||||||||||
| #10.6 | Employment Letter Agreement with Jason F. Cole, dated as of January 23, 2006. | 10-K | 10.15 | 03/20/2006 | 000-51171 | |||||||||||||
| #10.7 | Amendment to Employment Agreement with Jason Cole, dated December 15, 2008. | 10-K | 10.13 | 03/16/2009 | 000-51171 | |||||||||||||
| #10.8 | Letter Agreement Re: Retention Bonus with Jason Cole, dated December 16, 2008. | 10-K | 10.14 | 03/16/2009 | 000-51171 | |||||||||||||
| #10.9 | Amended and Restated Restricted Stock Award Agreement, dated September 5, 2008, by and between Jason Cole and the Registrant. | 10-Q | 10.25 | 11/10/2008 | 000-51171 | |||||||||||||
| #10.10 | Employment Letter Agreement with Justin Renz, dated as of August 31, 2006. | S-4 | 10.14 | 08/07/2009 | 333-161146 | |||||||||||||
| #10.11 | Letter Agreement Re: Retention Bonus with Justin Renz, dated December 12, 2008. | S-4 | 10.15 | 08/07/2009 | 333-161146 | |||||||||||||
| #10.12 | Letter Agreement Re: Severance Arrangements with Justin Renz, dated December 12, 2008. | S-4 | 10.16 | 08/07/2009 | 333-161146 | |||||||||||||
| #10.13 | Separation Agreement, dated as of January 14, 2010 between the Registrant and Robert Forrester. | 10-Q | 10.13 | 05/12/2010 | 000-51171 | |||||||||||||
| #10.14 | Employment Agreement with Mark H. N. Corrigan, dated as of March 8, 2010. | 8-K | 10.1 | 03/08/2010 | 000-51171 | |||||||||||||
| #10.15 | Form Incentive Stock Option Agreement with Mark H. N. Corrigan, dated as of January 15, 2010. | 8-K | 10.1 | 01/20/2010 | 000-51171 | |||||||||||||
| #10.16 | Form Nonstatutory Stock Option Agreement with Mark H. N. Corrigan, dated as of January 15, 2010. | 8-K | 10.2 | 01/20/2010 | 000-51171 | |||||||||||||
| #10.17 | Form Restricted Stock Unit Agreement between the Registrant and Mark Corrigan, dated as of January 15, 2010. | 8-K | 10.3 | 01/20/2010 | 000-51171 | |||||||||||||
| #10.18 | Revised Offer Letter from Neuromed Pharmaceuticals accepted by Christopher C. Gallen, dated May 27, 2004. | S-4 | 10.45 | 08/07/2009 | 333-161146 | |||||||||||||
| #10.19 | Confidentiality and Assignment of Proprietary Developments Agreement between Neuromed Technologies Inc. and Christopher C. Gallen, dated April 2005. | S-4 | 10.46 | 08/07/2009 | 333-161146 | |||||||||||||
| #10.20 | Award Letter from Neuromed Pharmaceuticals to Christopher C. Gallen, accepted and agreed to as of June 19, 2009. | S-4 | 10.43 | 08/07/2009 | 333-161146 | |||||||||||||
| #10.21 | Neuromed Pharmaceuticals Inc. Special Equity Incentive Plan. | S-4/A | 10.51 | 09/16/2009 | 333-161146 | |||||||||||||
| Exhibit No. |
Description |
Incorporated by Reference to: | ||||||||||||||||
| Form or Schedule |
Exhibit No. |
Filing Date with SEC |
SEC File Number |
|||||||||||||||
| #10.22 | The Registrant’s Form of Restricted Stock Unit Agreement for awards granted under the Neuromed Pharmaceuticals Inc. Special Equity Incentive Plan (Directors). | S-4/A | 10.52 | 09/16/2009 | 333-161146 | |||||||||||||
| #10.23 | The Registrant’s Form of Restricted Stock Unit Agreement for awards granted under the Neuromed Pharmaceuticals Inc. Special Equity Incentive Plan (Executives). | S-4/A | 10.53 | 09/16/2009 | 333-161146 | |||||||||||||
| 10.24 | Form of Indemnification Agreement for Directors. | 8-K | 10.1 | 12/21/2009 | 000-51171 | |||||||||||||
| 10.25 | Research Project Cooperative Agreement, dated April 10, 2005, between the National Institutes of Health and the Registrant. | S-1/A | 10.37 | 08/19/2005 | 333-121173 | |||||||||||||
| 10.26 | Termination Agreement, dated as of June 2, 2009, by and among CombinatoRx (Singapore) Pte Ltd, Biomedical Sciences Investment Fund Pte Ltd and the Registrant. | 8-K | 10.1 | 06/02/2009 | 000-51171 | |||||||||||||
| 10.27 | Share Purchase Agreement, dated as of June 2, 2009, by and among CombinatoRx (Singapore) Pte Ltd, Biomedical Sciences Investment Fund Pte Ltd and the Registrant. | 8-K | 10.2 | 06/02/2009 | 000-51171 | |||||||||||||
| 10.28 | Intellectual Property Assignment Agreement, dated as of June 2, 2009, by and between CombinatoRx (Singapore) Pte Ltd and the Registrant. | 8-K | 10.3 | 06/02/2009 | 000-51171 | |||||||||||||
| 10.29 | Transition Services Agreement, dated as of June 2, 2009, by and between CombinatoRx (Singapore) Pte Ltd and the Registrant. | 8-K | 10.4 | 06/02/2009 | 000-51171 | |||||||||||||
| †10.30 | Research and License Agreement, dated as of October 3, 2005, between Angiotech Pharmaceuticals, Inc. and the Registrant. | S-1/A | 10.46 | 10/05/2005 | 333-121173 | |||||||||||||
| 10.31 | Second Amended and Restated Research and License Agreement, dated July 22, 2009, between Fovea Pharmaceuticals SA and the Registrant. | 8-K | 10.1 | 07/23/2009 | 000-51171 | |||||||||||||
| 10.32 | Office and Laboratory Lease Agreement, dated as of October 18, 2005, by and between MA-Riverview, 245 First Street, L.L.C. and the Registrant. | S-1/A | 10.48 | 10/24/2005 | 333-121173 | |||||||||||||
| 10.33 | First Amendment to Office and Laboratory Lease Agreement, dated as of March 9, 2006, by and between MA-Riverview, 245 First Street, L.L.C. and the Registrant. | 10-K | 10.44 | 03/20/2006 | 000-51171 | |||||||||||||
| 10.34 | Second Amendment to Office and Laboratory Lease Agreement, dated as of August 3, 2009, by and between MA-Riverview, 245 First Street, L.L.C. and the Registrant. | 8-K | 10.1 | 08/04/2009 | 000-51171 | |||||||||||||
| Exhibit No. |
Description |
Incorporated by Reference to: | ||||||||||||||||
| Form or Schedule |
Exhibit No. |
Filing Date with SEC |
SEC File Number |
|||||||||||||||
| 10.35 | Sponsored Research Collaboration Agreement, dated November 7, 2007, between Dart Therapeutics, LLC and the Registrant. | 8-K | 10.1 | 11/08/2007 | 000-51171 | |||||||||||||
| 10.36 | First Amendment to Sponsored Research Collaboration Agreement, dated as of October 20, 2008, between the Registrant, DART Therapeutics, LLC and GMT Charitable Research, LLC. | 8-K | 10.1 | 10/22/2008 | 000-51171 | |||||||||||||
| †10.37 | Research Collaboration and License Agreement, dated as of May 1, 2009, by and between the Registrant and Novartis Institutes for BioMedical Research, Inc. | 10-Q | 10.43 | 05/11/2009 | 000-51171 | |||||||||||||
| 10.38 | Software License Agreement, dated as of May 1, 2009, by and between the Registrant and Novartis Institutes for BioMedical Research, Inc. | 10-Q | 10.44 | 05/11/2009 | 000-51171 | |||||||||||||
| 10.39 | Collaboration Agreement, dated August 11, 2009, by and between PGxHealth, LLC and the Registrant. | 8-K | 10.1 | 08/13/2009 | 000-51171 | |||||||||||||
| †10.40 | Asset Purchase Agreement, dated as of June 11, 2009, between Neuromed Development Inc. and Mallinckrodt Inc. and amendments thereto. | S-4/A | 10.35 | 10/13/2009 | 333-161146 | |||||||||||||
| †10.41 | Development and Transition Services Agreement, dated as of June 11, 2009, by and among Neuromed Development Inc., Neuromed Pharmaceuticals Ltd. and Mallinckrodt Inc. | S-4/A | 10.40 | 10/13/2009 | 333-161146 | |||||||||||||
| 10.42 | Agreement by and among Mallinckrodt Inc., a Covidien company, ALZA Corporation and Neuromed Pharmaceuticals Ltd. dated February 11, 2010. | 10-Q | 10.42 | 05/12/2010 | 000-51171 | |||||||||||||
| 10.43 | Sub-Lease Agreement between Neuromed Pharmaceuticals Inc. and Discovery Parks, Inc. | S-4/A | 10.56 | 10/09/2009 | 333-161146 | |||||||||||||
| 10.44 | Sub-Lease Renewal and Amendment Agreement between Neuromed Pharmaceuticals Ltd. and Discovery Parks, Inc., dated as of November 2, 2009. | 10-K | 10.54 | 03/26/2010 | 000-51171 | |||||||||||||
| 10.45 | Sub-Lease Renewal Agreement between Neuromed Pharmaceuticals Ltd. and Discovery Parks, Inc., dated as of September 16, 2010. | 10-Q | 10.45 | 11/10/2010 | 000-51171 | |||||||||||||
| 10.46 | Lease Agreement between Neuromed Pharmaceuticals Inc. and Six Tower Bridge Associates. | S-4/A | 10.57 | 10/09/2009 | 333-161146 | |||||||||||||
| 10.47 | Second Amendment to Lease Agreement between Neuromed Pharmaceuticals Inc. and Six Tower Bridge Associates, dated as of November 17, 2009. | 10-K | 10.56 | 03/26/2010 | 000-51171 | |||||||||||||
| 10.48 | Termination Agreement between the Registrant and Angiotech Pharmaceuticals, Inc., dated as of November 10, 2009. | 8-K | 10.1 | 11/10/2009 | 000-51171 | |||||||||||||
| Exhibit No. |
Description |
Incorporated by Reference to: | ||||||||||||||||
| Form or Schedule |
Exhibit No. |
Filing Date with SEC |
SEC File Number |
|||||||||||||||
| 10.49 | Termination Agreement between the Registrant and Cystic Fibrosis Foundation Therapeutics, dated as of May 14, 2009. | 8-K | 10.1 | 05/20/2009 | 000-51171 | |||||||||||||
| 10.50 | Loan and Security Agreement dated as of December 22, 2010, by and among the Registrant and Oxford Finance Corporation. | 8-K | 10.1 | 12/22/2010 | 000-51171 | |||||||||||||
| 10.51 | Equity Distribution Agreement dated February 9, 2011, between the Registrant and Wedbush Securities Inc. | 8-K | 10.1 | 02/09/2011 | 000-51171 | |||||||||||||
| *21.1 | List of subsidiaries. | |||||||||||||||||
| *23.1 | Consent of Ernst & Young LLP. | |||||||||||||||||
| *31.1 | Certification of Chief Executive Officer pursuant to Exchange Act rules 13a-14 or 15d-14. | |||||||||||||||||
| *31.2 | Certification of Chief Financial Officer pursuant to Exchange Act rules 13a-14 or 15d-14. | |||||||||||||||||
| *32.1 | Certifications of Chief Executive Officer and Chief Financial Officer pursuant to Exchange Act rules 13a-14(b) or 15d-14(b) and 18 U.S.C. Section 1350. | |||||||||||||||||
| * | Filed herewith. |
| † | Confidential treatment requested as to certain portions, which portions are omitted and filed separately with the Securities and Exchange Commission. |
| # | Management contract or compensatory plan or arrangement filed as an Exhibit to this report pursuant to Items 15(a) and 15(c) of Form 10-K. |