Attached files
| file | filename |
|---|---|
| EX-4.5 - EXHIBIT 4.5 - HUMANIGEN, INC | ex4_5.htm |
| EX-32.2 - EXHIBIT 32.2 - HUMANIGEN, INC | ex32_2.htm |
| EX-32.1 - EXHIBIT 32.1 - HUMANIGEN, INC | ex32_1.htm |
| EX-31.2 - EXHIBIT 31.2 - HUMANIGEN, INC | ex31_2.htm |
| EX-31.1 - EXHIBIT 31.1 - HUMANIGEN, INC | ex31_1.htm |
| EX-23.1 - EXHIBIT 23.1 - HUMANIGEN, INC | ex23_1.htm |
| EX-21.1 - EXHIBIT 21.1 - HUMANIGEN, INC | ex21_1.htm |
| EX-10.20 - EXHIBIT 10.20 - HUMANIGEN, INC | ex10_20.htm |
| EX-10.19 - EXHIBIT 10.19 - HUMANIGEN, INC | ex10_19.htm |
| EX-10.18 - EXHIBIT 10.18 - HUMANIGEN, INC | ex10_18.htm |
| EX-10.17 - EXHIBIT 10.17 - HUMANIGEN, INC | ex10_17.htm |
| EX-10.16 - EXHIBIT 10.16 - HUMANIGEN, INC | ex10_16.htm |
| EX-10.15 - EXHIBIT 10.15 - HUMANIGEN, INC | ex10_15.htm |
| EX-10.14 - EXHIBIT 10.14 - HUMANIGEN, INC | ex10_14.htm |
| EX-10.5 - EXHIBIT 10.5 - HUMANIGEN, INC | ex10_5.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| (Mark One) | |
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| For the Fiscal Year Ended December 31, 2020 | |
| Or | |
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number: 001-35798
HUMANIGEN, INC.
(Exact name of registrant as specified in its charter)
| Delaware (State or other jurisdiction of incorporation or organization) |
77-0557236 (I.R.S. Employer Identification No.) |
533 Airport Boulevard, Ste. 400
Burlingame, CA 94005
(Address of Principal Executive Offices) (Zip Code)
(650) 243-3100
(Registrant’s Telephone Number, Including Area Code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
| Common Stock | HGEN | The Nasdaq Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes x No ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company" in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ¨ | Accelerated filer ¨ | Non-accelerated filer ☒ | Smaller reporting company x | Emerging growth company ¨ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. Yes ¨ No x
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No x
The aggregate market value of the registrant’s voting stock held by non-affiliates as of June 30, 2020, was approximately $370,406,889 based on the closing price of $24.55 of the Common Stock of the registrant as reported on the OTCQB Venture Market operated by OTC Markets Group, Inc. on such date. As of March 5, 2021, there were 53,482,364 shares of the registrant’s Common Stock, par value $0.001 per share, outstanding.
Indicate by check mark whether the registrant has filed all documents and reports required to be filed by Section 12, 13 or 15(d) of the Securities Exchange Act of 1934 subsequent to the distribution of securities under a plan confirmed by a court. Yes x No ¨
DOCUMENTS INCORPORATED BY REFERENCE
The definitive proxy statement relating to the registrant’s Annual Meeting of Stockholders to be held on June 17, 2021, is incorporated by reference in Part III to the extent described therein.
Humanigen, Inc.
Form 10-K
Index
| 2 |
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains statements that discuss future events or expectations, projections of results of operations or financial condition, trends in our business, business prospects and strategies and other “forward-looking” information. In some cases, you can identify “forward-looking statements” by words like “may,” “will,” “should,” “expects,” “plans,” “anticipates,” “believes,” “estimates,” “predicts,” “intends,” “potential” or “continue” or the negative of those words and other comparable words. These statements may relate to, among other things, our expectations regarding the scope, progress, expansion, and costs of researching, developing and commercializing our product candidates; our opportunity to benefit from various regulatory incentives; expectations for our financial results, revenue, operating expenses and other financial measures in future periods; and the adequacy of our sources of liquidity to satisfy our working capital needs, capital expenditures, and other liquidity requirements. Actual events or results may differ materially due to known and unknown risks, uncertainties and other factors such as:
| · | the evolution of scientific discovery around the coronavirus, COVID-19 and the lung and other organ and systems dysfunction resulting in some patients may indicate that cytokine storm is caused by or results from something other than elevated GM-CSF levels; |
| · | the results of our Phase 3 trial of lenzilumab for the prevention and treatment of cytokine storm in patients hospitalized with COVID-19; |
| · | the enrollment, timing and results of the ACTIV-5/BET study sponsored by NIH for the use of lenzilumab and remdesivir in patients hospitalized with COVID-19; |
| · | the possibility that FDA might not grant an EUA for lenzilumab or, if one were granted, that its duration might be shorter than anticipated or the EUA might be conditioned to a greater degree than anticipated; |
| · | the duration and impact of the COVID-19 pandemic; |
| · | our ability to timely source adequate supply of bulk drug substance for our development and if approved, commercial products from third-party manufacturers on which we depend; |
| · | the negative impact that may result from manufacturing delays due to displacement of scheduled production at certain of our Contract Manufacturing Organizations (CMO) as mandated from Rated Orders issued under the Defense Production Act (DPA) or the shortage of raw materials and critical components we are experiencing and expect to continue to experience relating to Rated Orders and increased demand for production at our CMOs; |
| · | if an EUA is granted for lenzilumab, our ability to accurately forecast and predict future revenues in the U.S. and outside the U.S. coupled with our ability to produce sufficient quantities on a timely basis to meet demand; |
| · | our ability to research, develop and commercialize our product candidates, including our ability to do so before our competitors develop and commercialize competing products or alternative therapies or vaccines that may reduce the demand for our product candidates; |
| · | our ability to execute our strategy and business plan focused on developing our proprietary monoclonal antibody portfolio and our GM-CSF knockout gene-editing CAR-T platform; |
| · | our ability to enter into partnerships with potential collaborators with development, regulatory and commercialization expertise to enable us to pursue the other initiatives in our development pipeline; |
| · | our ability to successfully pursue the Kite collaboration and the enrollment, timing and results of pending and planned clinical trials contemplated by the collaboration; |
| · | the initiation and successful completion of the acute GvHD study in the UK with the IMPACT Partnership; |
| · | our ability to attain the additional financing we will need to pursue our development initiatives, manufacturing requirements and commercialize our product candidates on favorable terms or at all; |
| · | our ability to successfully maintain the listing of our common stock on the Nasdaq Capital Market; |
| · | the timing of the initiation, enrollment and completion and results of planned clinical trials; |
| 3 |
| · | the potential, if any, for future development of any of our present or future products; |
| · | increasing levels of market acceptance of CAR-T therapies and stem cell transplants and the development of a market for lenzilumab in these therapies; |
| · | our ability to successfully progress, partner or complete further development of our programs; |
| · | the potential timing and outcomes of development, preclinical and clinical studies of lenzilumab, ifabotuzumab, HGEN005, any of our CAR-T projects and the uncertainties inherent in development, preclinical and clinical testing; |
| · | our ability to identify and develop additional uses for our products; |
| · | our ability to attain market exclusivity and/or to obtain, maintain, protect and enforce our intellectual property and to operate our business without infringing, misappropriating or otherwise violating, the intellectual property rights of others; |
| · | the outcome of pending, threatened or future litigation; |
| · | acquisitions or in-licensing or out-licensing transactions that we may pursue may fail to perform as expected; |
| · | our ability to obtain and maintain regulatory approval of our product candidates, and any related restrictions; |
| · | limitations and/or warnings in the label of an approved product candidate or one that is granted an EUA; |
| · | changes in the regulatory landscape that may prevent us from pursuing or realizing any of the expected benefits from the various regulatory incentives, or the imposition of regulations that affect our products; |
| · | the success, progress, timing and costs of our efforts to evaluate or consummate various strategic alternatives if in the best interests of our stockholders; and |
| · | the accuracy of our estimates regarding expenses, future revenues, capital requirements and needs for additional financing. |
These are only some of the factors that may affect the forward-looking statements contained in this annual report. For a discussion identifying additional important factors that could cause actual results to vary materially from those anticipated in the forward-looking statements, see “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Risk Factors” in this Annual Report on Form 10-K. You should review these risk factors for a more complete understanding of the risks associated with an investment in our securities. However, we operate in a competitive and rapidly changing environment and new risks and uncertainties emerge, are identified or become apparent from time to time. It is not possible for us to predict all risks and uncertainties that could have an impact on the forward-looking statements contained in this annual report. You should be aware that the forward-looking statements contained in this annual report are based on our current views and assumptions. We undertake no obligation to revise or update any forward-looking statements made in this annual report to reflect events or circumstances after the date hereof or to reflect new information or the occurrence of unanticipated events, except as required by law.
| 4 |
PART I
Overview
We are a clinical stage biopharmaceutical company, developing our portfolio of immuno-oncology and immunology monoclonal antibodies. We are focusing our efforts on the development of our lead product candidate, lenzilumab™, our proprietary Humaneered® (“Humaneered”) anti-human granulocyte-macrophage colony-stimulating factor (“GM-CSF”) monoclonal antibody. Lenzilumab is a monoclonal antibody that has been demonstrated in animal models to neutralize GM-CSF, a cytokine that we believe is of critical importance in the hyperinflammatory cascade, sometimes referred to as cytokine release syndrome (“CRS”) or cytokine storm, associated with COVID-19, chimeric antigen receptor T-cell (“CAR-T”) therapy and acute Graft versus Host Disease (“GvHD”) associated with bone marrow transplants. GM-CSF neutralization with lenzilumab has been shown to reduce downstream inflammatory cytokines, prevent CRS and reduce its associated neurologic toxicities in-vivo in validated preclinical human xenograft models (Blood. 2019 Feb 14;133(7):697-709).
Lenzilumab has completed full enrollment in a Phase 3, multi-center, double-blind, placebo-controlled potential registrational trial for hospitalized, hypoxic patients with COVID-19 pneumonia. If the data from the trial are favorable, based on our discussions with the U.S. Food and Drug Administration (“FDA”), including a Type B meeting, and in consultation with the regulatory experts at Operation Warp Speed, we plan on filing an Emergency Use Authorization (“EUA”) application in the second quarter of 2021. If the EUA is granted, we could begin to commercialize lenzilumab for the treatment of hospitalized COVID-19 pneumonia patients. We also intend to file a Biologics License Application (“BLA”) in 2021, again assuming the data from the Phase 3 trial are favorable and support such an application. Based on discussions with FDA, we understand that a BLA will require us to generate and present more clinical and manufacturing data than would be required to support an EUA. If the ACTIV-5/BET study described below is successful and completed on a timely basis, we plan to include the results in our BLA filing. COVID-19 infections are widespread and as of February 26, 2021 in the United States, there were over 28 million cases, nearly 1.8 million hospitalizations and over 510 thousand deaths. Several vaccines have been developed which show efficacy in excess of 90%, however the slow rollout of inoculations and the emergence of numerous COVID-19 variants lead us to believe the need for therapeutics to treat COVID-19 currently exists and will continue to exist.
Lenzilumab has been selected to be part of the ACTIV-5 “Big Effect Trial” (ACTIV-5/BET NCT). This study is sponsored by the National Institutes of Health (“NIH”). ACTIV-5/BET is designed to determine whether certain approved therapies or investigational drugs in late-stage clinical development show promise against COVID-19 and, therefore, merit advancement into larger clinical trials. ACTIV-5/BET, currently has 17 active U.S. sites enrolling and is expected to enroll in up to 40 U.S. sites, will evaluate lenzilumab with remdesivir, compared to placebo and remdesivir, in hospitalized COVID-19 patients, with approximately 100 patients expected to be assigned to each study arm. We are providing lenzilumab for the study, which is fully funded by NIH.
Lenzilumab is also being studied in a multicenter phase 1b/2 potential registrational trial as a sequenced therapy with Yescarta® (axicabtagene ciloleucel) to reduce CRS and neurotoxicity in patients with relapsed or refractory diffuse large B-cell lymphoma (“DLBCL”) (NCT04314843). This trial currently has 10 active sites and is being conducted in partnership with Kite Pharmaceuticals, Inc., a Gilead company (“Kite”), which markets Yescarta. We are also in the final planning stages for a Phase 2/3 trial for lenzilumab to treat patients who have undergone allogeneic hematopoietic stem cell therapy (“HSCT”) who are at high and intermediate risk for acute GvHD. The trial is expected to be conducted by the IMPACT Partnership, a collection of 22 stem cell transplant centers located in the United Kingdom.
Our proprietary, patented Humaneered technology platform is a method for converting existing antibodies (typically murine) into engineered, high-affinity human antibodies designed for therapeutic use, particularly with acute and chronic conditions. We have developed or in-licensed targets or research antibodies, typically from academic institutions, and then applied our Humaneered technology to produce them. Lenzilumab and our other two product candidates, ifabotuzumab and HGEN005, are Humaneered monoclonal antibodies. Our Humaneered antibodies are closer to human antibodies than chimeric or conventionally humanized antibodies and have a high affinity for their target but low immunogenicity. We believe our Humaneered antibodies offer additional advantages, such as high potency, a slow off-rate and a lower likelihood to induce an inappropriate immune response or infusion related reactions.
| 5 |
Scientific Rationale and Background on Lenzilumab
Lenzilumab neutralizes GM-CSF. The coronavirus pandemic, which has been triggered by the SARS-CoV-2 virus and leads to the condition referred to as COVID-19, is characterized in the later and sometimes fatal stages by lung dysfunction and, in many patients, multi-organ impairment, which is triggered by Cytokine Release Syndrome (“CRS”), or cytokine storm. Publications have pointed to GM-CSF as being a signature cytokine in this process, with elevated GM-CSF levels correlated to poorer outcomes and sometimes ventilator use and Intensive Care Unit (“ICU”) admission.
COVID-19 has three distinct phases: early infection, pulmonary/inflammatory and hyperinflammatory. As shown on the diagram below, lenzilumab is being studied for use in patients that are in the pulmonary/inflammatory phase who are hospitalized and hypoxic.

The severe clinical features associated with some COVID-19 infections result from an inflammation-induced lung injury which may require supplemental oxygen through a nasal cannula, non-invasive or invasive mechanical ventilation or Extra Corporeal Mechanical Oxygenation (ECMO) and sometimes ICU care. This lung injury is a result of a hyperinflammatory dysregulation of the immune system and associated with cytokine storm. The lung injury that leads to death is not directly related to the virus but appears to be a result of a hyper-reactive immune response to the virus triggering a cytokine storm that can continue even after viral titers remain stable or even begin to fall.
CRS is characterized by an elevation of inflammatory cytokines resulting in fever, hypotension, capillary leak syndrome, pulmonary edema, disseminated intravascular coagulation, respiratory failure, and Acute Respiratory Distress Syndrome (“ARDS”). The development of CRS as a direct result of immune hyper-stimulation has been previously described in patients with autoimmune and lymphoproliferative diseases, as well as in patients with B-cell malignancies receiving CAR-T therapy. Over the last five years, preclinical studies and correlative science from clinical trials in CAR-T therapy have shed light on the pathophysiology, development, characterization, and management of CRS.
| 6 |
CRS is characterized by activation of myeloid cells and release of inflammatory cytokines, including GM-CSF, monocyte chemoattractant protein -1 (MCP-1), macrophage inflammatory protein 1α (MIP-1α), Interferon gamma-induced protein 10 (IP-10), interleukin-6 (IL-6), and interleukin-1 (IL-1). The cascade, once initiated, can quickly evolve into a cytokine storm, resulting in further activation, expansion and trafficking of myeloid cells, leading to abnormal endothelial activation, increased vascular permeability, and disseminated intravascular coagulation.
Data from National Scientific Review (2020, Vol. 7, No. 6) titled “Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients”, supports the hypothesis that GM-CSF induced cytokine storm immune mechanisms have contributed to patient mortality with the current pandemic strain and, we believe, in multiple variants, of coronavirus, and there is increasing acceptance that this pathophysiology may be responsible for worsening of clinical status and poor outcomes. The authors noted that steroid treatment in such cases has been disappointing in terms of outcome but suggested that a monoclonal antibody that targets GM-CSF may prevent or curb the hyper-active immune response caused by COVID-19 in this setting. Three recent publications point to GM-CSF as a so-called ‘signature cytokine’ including the largest inflammatory marker study in over 600 patients from a multicenter study in the UK. (Uncontrolled Innate and Impaired Adaptive Immune Responses in Patients with COVID-19 ARDS, deProst et al, AJRCCM Articles in Press., American Thoracic Society, August 31, 2020, 10.1164/rccm.202005-1885OC) (The dysregulated innate immune response in severe COVID-19 pneumonia that could drive poorer outcome, Blot et al. J Transl Med (2020) 18:457) (Elevated antiviral, myeloid and endothelial inflammatory markers in severe COVID-19, Openshaw et al, MedRxIV, doi).
While early studies demonstrated elevated GM-CSF levels in both ICU and non-ICU treated COVID-19 patients, one of these three more recent studies showed a positive association with disease severity and outcome, in agreement with reports of elevated frequencies of GM-CSF+ Th1 cells in patients with COVID-19 requiring ICU treatment. The authors analyzed serial plasma samples from patients hospitalized with COVID-19 recruited through the prospective multicenter ISARIC clinical characterization protocol in the UK. Elevated levels of numerous mediators including angiopoietin-2, IP-10, and GM-CSF were seen at recruitment in patients who later died. This analysis shows that, at the time of enrollment, different COVID-19 outcome groups were already identifiable and associated with distinct patterns of inflammatory mediators and that markers such as GM-CSF were particularly associated with enhanced disease severity and strikingly elevated in fatal cases of COVID-19. An influx of monocytes/macrophages has been described in the lung parenchyma in fatal COVID-19, combined with a mononuclear cell pulmonary artery vasculitis, and presence of pro-inflammatory monocyte-derived macrophages in bronchoalveolar lavage fluid from patients with severe COVID-19. The elevation of GM-CSF in severe disease could contribute to monocyte recruitment and activation leading to this vasculitis, alongside the role of GM-CSF in the recruitment of neutrophils to the pulmonary vasculature.
A second independent study reported serum concentrations of GM-CSF were significantly higher in COVID-19 patients. In this study, increased serum concentrations of IL-8, IL-10 and GM-CSF were associated with disease severity. Strikingly, in this study the serum concentrations of IL-6, IL-1Ra and IL-8 not only showed no, or only marginally, significant differences between COVID-19 and non-COVID-19 ARDS patients but were also not associated with day-28 mortality in COVID-19 patients. Further, the serum concentrations of IL-6 and IL-1Ra correlated with the SOFA score, indicating that these cytokines rather behave as biomarkers of organ-failure-associated hyper-inflammation, consistent with their previously reported association with patient severity and outcome in cohort studies merging patients with mild to severe disease. Interestingly, the concentrations of CCL19/MIP/3b, CCL20/MIP-3a and CCL5/RANTES, which recruit monocytes and T cells, remained stable over time. These chemokines are secreted by CD14+CD16+ inflammatory monocytes, which are enriched in the blood of COVID-19 patients with severe disease. In line with this observation, the serum concentrations of GM-CSF were significantly higher in COVID-19 patients.
A third independent study reported a significant correlation between the duration of mechanical ventilation (“MV”) and GM-CSF (p < 0.0001), IL-10 (p < 0.0001), IP-10 (p < 0.0001), MCP-1 (p = 0.001), CX3CL1 (p = 0.0233), and Granzyme B (p = 0.0143). Based on these results, they performed a multivariate linear regression to identify factors associated with the duration of MV in the first 30 days, using two models (i.e., SOFA score (model 1) or PaO2: FiO2 ratio (model 2) as the variable of adjustment to account for severity). Only GM-CSF was independently associated with a longer duration of MV in both models. The authors estimated an excess of 22.11 ± 8.36 min of MV per increase of 1 pg/mL of GM-CSF (p = 0.0105, model 1; and p < 0.0001, model 2).
| 7 |
Similar to patients receiving CAR-T therapy, the development of CRS in patients with COVID-19 has been associated with elevation of CRP, ferritin, MCP-1, MIP-1 alpha, INF-gamma, TNF-alpha, and IL-6, as well as correlating with respiratory failure, ARDS, and adverse clinical outcomes. Most significantly, high levels of GM-CSF-secreting Th17 T-cells have been associated with disease severity, myeloid cell trafficking to the lungs, and ICU admission. This indicates that post-COVID-19 CRS is caused by a similar mechanism, induced by activation of myeloid cells and their trafficking to the lung, resulting in lung injury and ARDS. Tissue CD14+ myeloid cells produce GM-CSF and IL-6, further triggering a cytokine storm cascade. Single-cell RNA sequencing of bronchoalveolar lavage samples from COVID-19 patients with severe ARDS demonstrated an overwhelming infiltration of newly-arrived inflammatory myeloid cells compared to mild COVID-19 disease and healthy controls, consistent with a hyperinflammatory CRS-mediated pathology. We believe that these new data suggest that GM-CSF may be a critical triggering cytokine in the increased mortality in COVID-19.
Lenzilumab has been shown to prevent cytokine storm in animal models and this work has been published in peer reviewed journals. These data demonstrate that GM-CSF neutralization results in a reduction in IL-6, MCP-1, MIP-1α, IP-10, vascular endothelial growth factor (VEGF), and tumor necrosis factor-α (TNFα) levels, demonstrating that GM-CSF is an upstream regulator of many inflammatory cytokines that are important in the pathophysiology of CRS. GM-CSF depletion results in modulation of myeloid cell behavior, a specific decrease in their inflammatory cytokines, and a reduction in tissue trafficking, while enhancing T-cell expansion and effector functions.
The Phase 3 program in COVID-19 is complementary to the programs in CAR-T and GvHD, which are also focused on preventing or reducing cytokine storm in those disease states. Positive results from the Phase 3 trial in COVID-19 may be predictive of results in these other settings, which are also characterized by cytokine storm.
As a leader in GM-CSF pathway science, we believe that we have the ability to transform prevention and treatment of CRS in SARS-CoV-2 infection. SARS-Cov-2, is one of a group of several betacoronaviruses, which includes the viruses responsible for Severe Acute Respiratory Syndrome (SARS-CoV) and Middle East Respiratory Syndrome (MERS-CoV). These viruses infect predominantly the lower lung and cause fatal pneumonia. Other coronaviruses infect the upper respiratory tract and cause some cases of the common cold. Initially, the clinical course of COVID-19 could be mistaken for influenza infection – patients in both cases often suffer from aches and pains throughout the body, fever, cough and general malaise. However, rapid nasal or throat swab tests for SARS-CoV-2 infection have been developed and are in widespread use. Exposure to people who are known to have suffered from the condition or carriers of SARS-CoV-2 also increases the clinical suspicion of possible infection. Data generated during the SARS and MERS outbreaks point to cytokine storm as a phase of the illness which is characterized by an immune hyperactive phase, which can progress to lung dysfunction and death. In patients who clinically deteriorate, there can be multi-system effects, including hematologic and coagulation disorders, renal, cardiovascular and neurologic impairment, some of which may further complicate respiratory compromised patients. The natural history of SARS infection shows viral load decreases as patients enter the second phase of the illness, which is often characterized by cytokine storm and the elevation of certain biomarkers.
We also believe we have the ability to transform CAR-T therapy and a broad range of other T-cell engaging therapies, including both autologous and allogeneic cell transplantation. There is a direct correlation between the efficacy of CAR-T therapy and the incidence of life-threatening toxicities (referred to as the efficacy/toxicity linkage). We have begun enrolling patients in ZUMA-19, in collaboration with our partner Kite, to assess effect of the sequential therapy of lenzilumab with Yescarta, the leading CAR-T, in reducing or minimizing cytokine storm and neurotoxicity in patients receiving CAR-T, and potentially building further on Yescarta’s current category-leading efficacy. Depending on the final number of patients enrolled, this may serve as a Phase 1b/2 potential registration study.
We believe that our GM-CSF neutralization and gene-editing CAR-T platform technologies have the potential to reduce the inflammatory cascade associated with serious and potentially life-threatening CAR-T therapy-related side-effects while preserving and potentially improving the efficacy of the CAR-T therapy itself, thereby breaking the efficacy/toxicity linkage. Clinical correlative analysis and preclinical in vivo evidence points to GM-CSF as the key initiator of the inflammatory cascade resulting in CAR-T therapy’s side-effects, including CRS and NT. GM-CSF has also been linked to the suppressive myeloid cell axis through recruitment of myeloid-derived suppressor cells (“MDSCs”) that reduce CAR-T cell expansion and hamper CAR-T cell efficacy. Our strategy is to continue to pioneer the use of GM-CSF neutralization and GM-CSF gene knockout technologies to improve efficacy and prevent or significantly reduce the serious side-effects associated with CAR-T therapy.
| 8 |
We believe that our GM-CSF pathway science, assets and expertise create two technology platforms to assist in the development of next-generation CAR-T therapies:
| · | Lenzilumab has the potential to be used in combination with any FDA-approved or development stage T-cell therapy, including CAR-T therapy, as well as in combination with other cell therapies such as allogeneic HSCT such as bone marrow transplants, to make these treatments safer and more effective |
| · | In addition, our GM-CSF knockout gene-editing CAR-T platform has the potential to create next-generation CAR-T therapies that may inherently avoid any efficacy/toxicity linkage, thereby potentially preserving the benefits of the CAR-T therapy while reducing or altogether avoiding its serious and potentially life-threatening side-effects. |
We have utilized a precision medicine approach and personalized the development of lenzilumab based on specific genetic mutations or biomarkers at baseline. We are collaborating with IMPACT, a clinical trial partnership of 22 transplant centers in the United Kingdom, in planning a randomized, placebo controlled, Phase 2/3 study focused on early intervention with lenzilumab in patients at high risk or intermediate risk for steroid-refractory acute GvHD based on specific biomarkers. The goal of the trial, as it is currently contemplated, would be to determine the efficacy and safety of lenzilumab in reducing non-relapse mortality at six months.
We have completed enrollment and reported on a Phase 1 study of lenzilumab as monotherapy in refractory chronic myelomonocytic leukemia (CMML) and are planning a Phase 2 study of lenzilumab in combination with azacitidine (current standard therapy) in newly-diagnosed CMML patients with certain genetic mutations. The Phase 1 clinical trial in patients with CMML identified the MTD or recommended Phase 2 dose of lenzilumab, and assessed lenzilumab’s safety, pharmacokinetics, and clinical activity. The results were reported at the 2019 American Society of Hematology (ASH) conference.
We have also reported on a Phase 2 study with lenzilumab in severe asthma in patients uncontrolled on high-dose steroid therapy.
Our Pipeline
Our lenzilumab-based clinical-stage pipeline comprises a Phase 3 potential registration study in COVID-19 which has fully enrolled, the NIH-sponsored and funded ACTIV-5/BET program in COVID-19, a currently enrolling Phase 1b/2 study (ZUMA-19) as sequenced therapy of lenzilumab and Yescarta, a planned Phase 2/3 study in acute GvHD, and a planned Phase 2 study in CMML, the latter two of which we expect will be majority funded by partners. While the majority of our clinical programs involve lenzilumab, we have also fully enrolled an ifabotuzumab Phase 1 study in GBM.
| 9 |
Except for the potential of lenzilumab for COVID-19 under an EUA, our product candidates are in the clinical stage of development and will require substantial time, resources, research and development, and regulatory approval prior to commercialization. Furthermore, it may be years before any of our products are approved for use, if at all. Our current pipeline is depicted below:
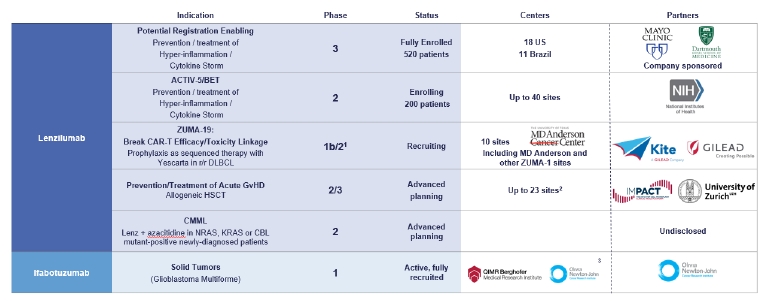
1 Phase 3 may not be necessary for approval in ZUMA-19; precedent CAR-Ts to date have been approved on Phase 2 data
2 UK
3 US, EU, Australia
Lenzilumab
Lenzilumab neutralizes human GM-CSF and has the potential to prevent or reduce poor outcomes associated with COVID-19. We have completed enrollment in a Phase 3 multi-center, randomized, placebo-controlled, double-blinded, clinical trial in the setting of COVID-19. There were 18 clinical sites across the US and 11 sites in Brazil involved in the study. This trial will assess the safety and efficacy of lenzilumab in improving ventilator-free survival in hospitalized hypoxic adult patients with confirmed COVID-19 pneumonia.
There are currently no products approved by the FDA for the prevention of CRS/cytokine storm associated with COVID-19. There are numerous products currently in development for COVID-19 which can be broadly categorized as direct acting antivirals, immunomodulators, anticoagulants and other preventative strategies such as vaccines and neutralizing antibodies. Remdesivir (a direct acting antiviral) was approved by FDA for COVID-19. There is no evidence that remdesivir offers a mortality benefit and the largest remdesivir-based global study conducted by the World Health Organization failed to show benefit from remdesivir treatment. Other direct acting antiviral agents such as lopinavir/ritonavir and hydroxychloroquine (with or without a macrolide) have not demonstrated efficacy in randomized controlled trials. Other treatments available under EUA include baricitinib (Olumiant) in combination with remdesivir (Veklury), bamlanivimab alone and with Etesevimab, and REGEN-COV (casirivimab and imdevimab). Published data to date from randomized controlled clinical trials in the setting of COVID-19 and the two leading IL-6 inhibitors, Actemra (tocilizumab) and Kevzara (sarilumab) has been mixed. Recently published results from the The Randomised Evaluation of COVID-19 Therapy (RECOVERY) trial has demonstrated that an anti-inflammatory treatment, tocilizumab, reduces the risk of death when given to hospitalized patients with severe COVID-19.
This is an open label study. The study also showed that tocilizumab shortens the time until patients are successfully discharged from hospital and reduces the need for a mechanical ventilator. The relative reduction in mortality was 14% and the relative reduction in progressing to IMV or death was 15%. A total of 2,022 patients were randomly allocated to receive tocilizumab by intravenous infusion and were compared with 2,094 patients randomly allocated to usual care alone. 82% of patients were taking a systemic steroid such as dexamethasone. Treatment with tocilizumab significantly reduced deaths: 596 (29%) of the patients in the tocilizumab group died within 28 days compared with 694 (33%) patients in the usual care group (rate ratio 0·86; [95% confidence interval [CI] 0·77 to 0·96]; p=0·007), a 14% relative risk reduction and an absolute difference of 4%. Tocilizumab also increased the probability of discharge alive within 28 days from 47% to 54% (rate ratio 1·23, [95% CI 1·12 to 1·34], p<0·0001). These benefits were seen in all patient subgroups, including those requiring oxygen via a simple face mask through to those requiring mechanical ventilators in an intensive care unit. Among patients not on invasive mechanical ventilation when entered into the trial, tocilizumab significantly reduced the chance of progressing to invasive mechanical ventilation or death from 38% to 33% (risk ratio 0·85, [95% CI 0·78 to 0·93], p=0·0005) a 15% relative risk reduction. However, there was no evidence that tocilizumab had any effect on the chance of successful cessation of invasive mechanical ventilation. In June 2020, the RECOVERY trial found that the inexpensive and widely available steroid dexamethasone reduces death for patients with severe COVID-19. Dexamethasone became part of standard-of-care given to all such patients. In this trial, the benefits of tocilizumab were seen to be in addition to those of steroids.
| 10 |
In addition, IL-1 and IL-1 receptor inhibitors, respectively Ilaris (canakinumab) and Kineret (anakinra) have both failed to demonstrate benefit in Phase 3 studies, as has the oral JAK inhibitor Jakafi (ruxolitinib) and the BTK inhibitor Calquence (acalabrutinib). The anti-GM-CSF agent, otilimab failed in an 806-patient randomized controlled study sponsored by GlaxoSmithKline (“GSK”). In a post-hoc analysis of a subgroup of patients older than 70, there was an apparent benefit in the proportion of patients alive and free of respiratory failure at day 28 and mortality at day 60. A 350-patient follow-on Phase 3 study is planned to determine if the benefit in the patients older than 70 can be confirmed. It is unclear whether the anti-GM-CSF agents, gimsilumab and namilumab are continuing development for COVID-19 and the primary endpoint for mavrilimumab for COVID-19 in a 40-patient study was recently missed, though there were some promising data in this small Phase 2 study.
As an upstream regulator of cytokine storm, GM-CSF neutralization with lenzilumab may offer advantages over other immunomodulator strategies that either target other downstream cytokines such as IL-1, IL-6, CCR5 or MIP-1a or are broadly immunosuppressive and target cytokine signaling pathways non-selectively through JAK inhibition. In addition, lenzilumab is the only immunomodulator that was in an active clinical trial to prevent cytokine storm prior to COVID-19 and is currently the only agent in an active Phase 3 trial targeting GM-CSF, with other such agents in earlier stage studies.
During 2020, we achieved several significant milestones relating to lenzilumab. In April 2020 lenzilumab was granted emergency single use IND authorization from the FDA (often referred to as compassionate use) to treat patients with COVID-19. On June 15, 2020, we announced that Mayo Clinic published data derived from the compassionate use of lenzilumab in the treatment of 12 patients hospitalized in the Mayo Clinic system. Under applicable FDA rules, a patient cannot receive a compassionate use drug unless FDA has issued an individual patient emergency IND authorization, which the Mayo Clinic requested and received from FDA prior to each individual patient dosing of lenzilumab. Accordingly, there was no randomized control group in the Mayo Clinic program. We did not pre-select patients to receive lenzilumab through the compassionate use program and did not deny any requests for compassionate use. Mayo Clinic clinicians solely determined which patients for which they would request emergency IND authorization from the FDA. As discussed below the results of the compassionate use lenzilumab compared to the contemporaneous control group were recently published in the Mayo Clinic Proceedings.
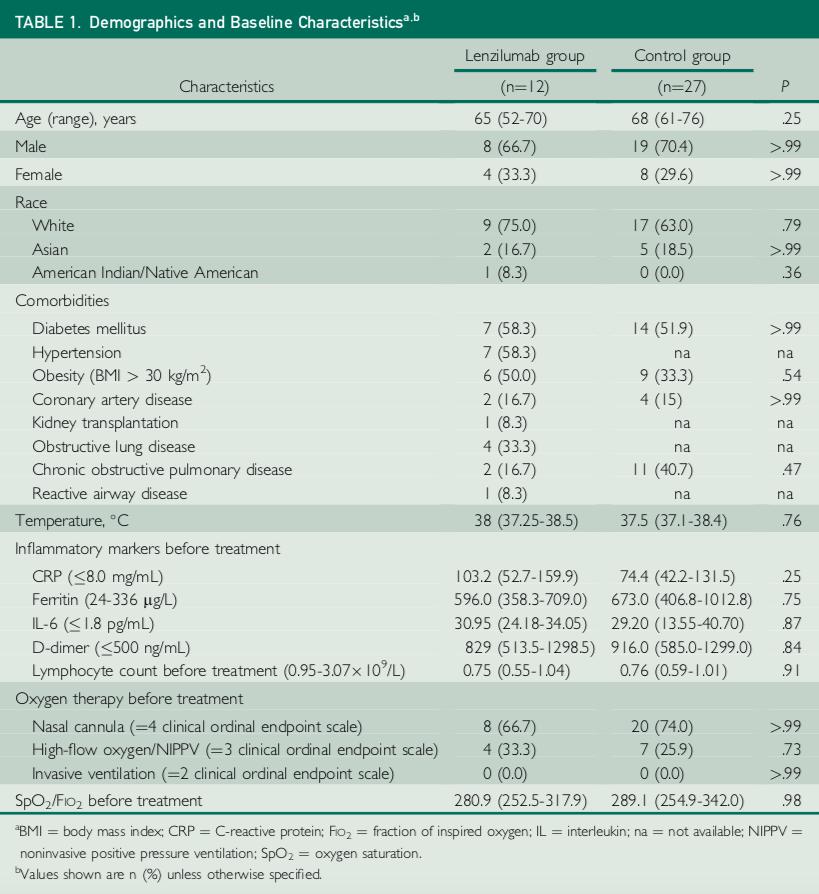
The patients receiving lenzilumab were hospitalized, required oxygen supplementation (hypoxic) as a result of COVID-19 but did not include any patients on invasive mechanical ventilation. They were also viewed as being at high risk of further disease progression. All patients had elevation in at least one inflammatory biomarker prior to receiving lenzilumab. All patients had at least one co-morbidity associated with poor outcomes in COVID-19 and several patients had multiple co-morbidities.
On September 1, 2020, we announced that Mayo Clinic Proceedings, a premier peer-reviewed journal, had published a manuscript reporting the first case-cohort data of lenzilumab in COVID-19 patients. Control patients were identified from an electronic registry of COVID-19 patients in the same centers as cases and matched for age, sex, disease severity, and baseline oxygen requirements. At the time of selection, the clinical outcomes for the matched control patients were not known.
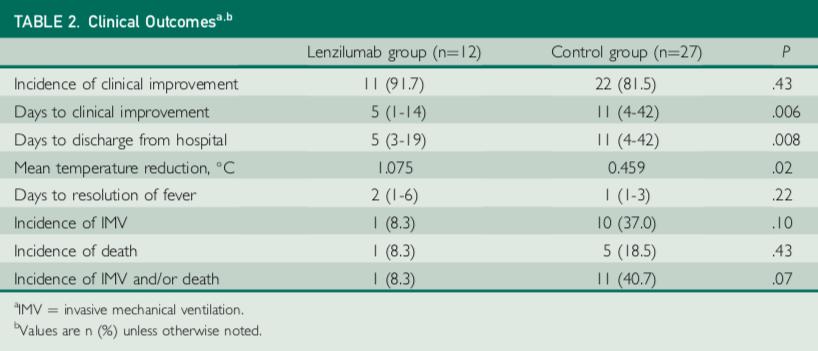
| 11 |
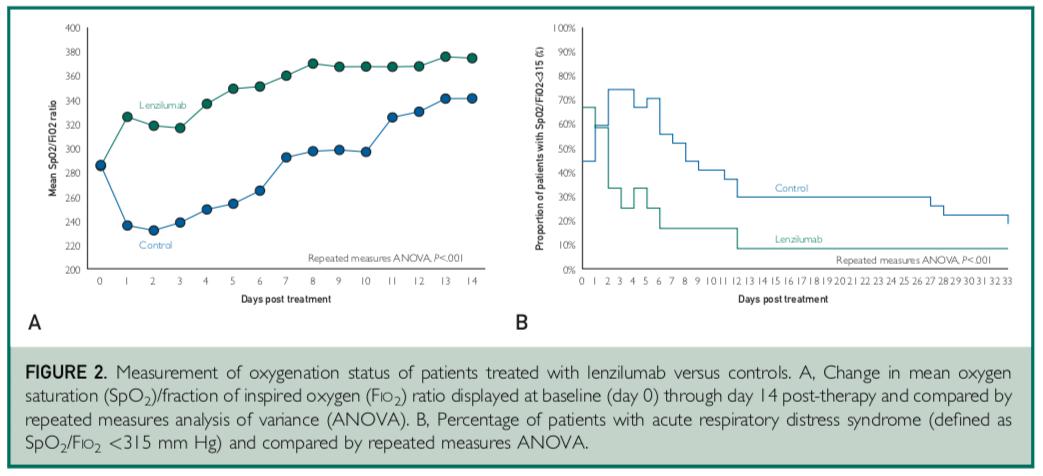
The study involved a total of 39 patients, 12 treated with lenzilumab, and 27 contemporaneous matched control patients who received standard of care treatment. Lenzilumab treatment was associated with an 80% relative reduction in risk of progression to IMV and/or death compared to matched controls (8% vs. 41%, p=0.07). Median time to a 2-point clinical improvement on the 8-point hospital ordinal scale was five days versus 11 days in the control arm (p=0.006). Ventilator-free survival favored lenzilumab versus controls (p=0.06). Median time to resolution of acute respiratory distress syndrome (ARDS) was one day in the lenzilumab treatment arm versus eight days in the control group (p<0.001). Mean SpO2/FiO2 ratios post-therapy were significantly improved in the lenzilumab patients versus controls (p<0.001). Patients treated with lenzilumab were discharged in a median of five days versus 11 days in the control arm (p=0.008).
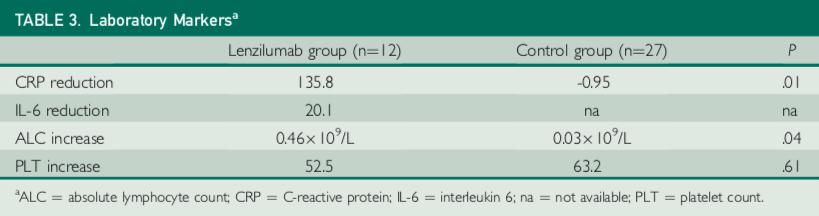
Lenzilumab treatment was also associated with a significant reduction in the inflammatory marker CRP relative to the control group (p=0.01) and an improvement in lymphocyte counts relative to the control group (p=0.04). There were no treatment-emergent adverse events attributable to lenzilumab.
The data from the case-control study suggest that GM-CSF neutralization with lenzilumab may have an effect through a dual mechanism of action to restore balance to dysregulated immune response induced by SARS-CoV-2 by suppressing myeloid inflammatory response and improving T-cell counts thought to be responsible for viral clearance.
| 12 |
| 13 |

| 14 |
On October 29, 2020, we announced that National Institute of Allergy and Infectious Diseases (“NIAID”), a part of National Institutes of Health (“NIH”), which is part of the U.S. Department of Health and Human Services (“HHS”) as represented by the Division of Microbiology and Infectious Diseases (“DMID”), and Humanigen enrolled the first patient in the NIAID-sponsored ACTIV-5/Big Effect Trial (“BET”) in hospitalized patients with COVID-19. BET will help advance NIAID’s strategic plan for COVID-19 research, which includes conducting studies to advance high-priority therapeutic candidates. Identification of agents with novel mechanisms of action for therapy is a strategic priority.
ACTIV-5 will evaluate the combination of lenzilumab and remdesivir on treatment outcomes versus placebo and remdesivir in hospitalized COVID-19 patients. The trial is expected to enroll 100 patients in each arm of the study with an interim analysis for efficacy after 50 patients have been enrolled in each arm. With data from ACTIV-5 and our ongoing Phase 3 study, we expect to have data from more than 700 hospitalized COVID-19 patients, in the aggregate.
Lenzilumab also has the potential to prevent or reduce serious and sometime fatal side-effects associated with CAR-T therapy (CRS and neurotoxicity), a cancer therapy; See “--CAR-T Overview” later in this Item 1 for a fuller description of the therapy. Preclinical data generated in collaboration with the Mayo Clinic (the “Mayo Clinic”), which was published in ‘blood®’, a premier journal in hematology, indicates that the use of lenzilumab in combination with CAR-T therapy may also enhance the proliferation and improve the efficacy of CAR-T therapy. This may also result in durable, or longer term, responses in CAR-T therapies. There are currently no products approved by the FDA for the prevention of CAR-T therapy-related side effects, nor are there any approved therapies for the treatment of CAR-T therapy related NT.
We are advancing the development of lenzilumab in combination with CAR-T therapy through a non-exclusive clinical collaboration with Kite, pursuant to which we are conducting a multi-center potential registration Phase 1b/2 study of lenzilumab with Kite’s Yescarta in patients with relapsed or refractory B-cell lymphoma, including DLBCL (the “Study”). The Study has been designated the nomenclature ‘ZUMA-19’, consistent with the other Kite CAR-T studies, which also receive a ‘ZUMA’ designation, including ZUMA-1, the study that served as the registrational study for Yescarta. The primary objective of ZUMA-19 is to determine the effect of lenzilumab on the safety and efficacy of Yescarta. Kite’s Yescarta is one of four CAR-T therapies that have been approved by the FDA and is the CAR-T therapy market leader. Our collaboration with Kite is currently the only clinical collaboration which is now enrolling patients with the potential to improve both the safety and efficacy of CAR-T therapy, including in 5 centers that were participants in ZUMA-1. We also plan to measure other potentially beneficial effects on efficacy and healthcare resource utilization. In addition, lenzilumab’s success in preventing serious and potentially life-threatening side-effects could offer economic benefits to medical system payers by making the CAR-T therapy capable of being administered, and follow-up care subsequently monitored and managed, potentially on an out-patient basis in certain patients and circumstances. In turn, we believe that delivering such provider and payer benefits might accelerate the use of the CAR-T therapy itself, and thereby permit us to generate further revenues from sales of lenzilumab.
| 15 |
In addition to COVID-19 and CAR-T therapy, we are committed to advancing our diverse platform for GM-CSF axis suppression for a broad range of other T-cell engaging therapies, including both autologous and allogeneic next generation CAR-T therapies, bi-specific antibody therapies, as well as other cell-based immunotherapies in development, including allogeneic HSCT, with our current and future partners.
In July 2019, we entered into an exclusive worldwide license agreement (the “Zurich Agreement”) with UZH. Under the Zurich Agreement, we have in-licensed certain technologies that we believe may be used to prevent or treat GvHD a serious and potentially fatal condition associated with transplantation, thereby expanding our development platform to include improving the safety and effectiveness of allogeneic HSCT, a potentially curative therapy for patients with hematological cancers. We believe that cytokine storm may be responsible, at least in part, for the emergence of GvHD in this setting. There are currently no FDA-approved agents for the prevention of GvHD nor treatment of GvHD in patients identified as high risk by certain biomarkers. We believe that GM-CSF neutralization with lenzilumab has the potential to prevent or treat GvHD without compromising, and potentially improving, the beneficial GvL effect in patients undergoing allogeneic HSCT, thereby making allogeneic HSCT safer. Several recent papers have been published which support this approach, including in Science Translational Medicine in November 2018 and in ‘blood advances’ in October 2019.
We aim to position lenzilumab as a necessary companion product to any allogeneic HSCT and as a part of the standard pre-conditioning that all patients receiving allogeneic HSCT should receive or as an early treatment option in patients identified as high risk for GvHD.
Given our interest in developing lenzilumab to prevent CRS/cytokine storm in COVID-19 as well as in the treatment of rare cancers and other orphan conditions such as GvHD, we believe that we have the opportunity to benefit from various regulatory incentives, such as coronavirus treatment acceleration program (CTAP), orphan drug exclusivity, breakthrough therapy designation, fast track designation, priority review and accelerated approval.
GM-CSF Gene Knockout
We are advancing our GM-CSF knockout gene-editing CAR-T platform through an exclusive worldwide license agreement (the “Mayo Agreement”) that we entered into in June 2019 with the Mayo Foundation for Medical Education and Research (the “Mayo Foundation”). Under the Mayo Agreement, we have in-licensed certain technologies that we believe may be used to create CAR-T cells lacking GM-CSF expression through various gene-editing tools, including CRISPR-Cas9. We believe that our GM-CSF knockout gene-editing CAR-T platform has the potential to create next-generation CAR-T therapies that improve the efficacy and safety profile of CAR-T therapy. In addition, we have and continue to file intellectual property encompassing a broad range of gene-editing approaches related to GM-CSF knockout.
Preclinical data indicates that GM-CSF gene knockout CAR-T cells show improved overall survival in animals compared to wild-type CAR-T cells in addition to the expected benefits of reduced serious side-effects associated with CAR-T therapy. We are establishing a platform of next-generation combinatorial gene knockout CAR-T cells that have potential to be applied across both autologous and allogeneic approaches and we are also investigating multiple CAR-T cell designs using precise dual and triple gene editing to significantly enhance the anti-tumor activity while simultaneously preventing CAR-T therapy induced toxicities. Through targeted gene expression and modulating cytokine activation signaling, we may be able to increase the proportion of fitter T-cells produced during expansion, increase their proliferative potential, and inhibit activation-induced cell death, thereby improving the cancer killing activity of our engineered CAR-T cells thereby making them more effective and safer in the treatment of cancers. Initial data were published in an abstract that was presented at the December 2019 American Society of Hematology (ASH) meeting and also won an ASH Abstract Achievement award.
| 16 |
We plan to continue development of this technology in combination approaches that could add to the observed efficacy benefits of current generation CAR-T products. In addition, we anticipate that our GM-CSF knockout gene-editing CAR-T platform may be a future backbone for controlling the serious side-effects that hamper CAR-T therapy that lead to serious and sometimes fatal outcomes for patients as a result of the CAR-T therapy itself.
CAR-T Overview
As of the date of this filing, four CAR-T therapies that have been approved by the FDA, Gilead/Kite’s Yescarta, and Tecartus, Novartis’s Kymriah and Bristol Myers Squibb’s Breyanzi (lisocabtagene maraleucel; liso-cel), seek to treat forms of B-cell cancers such as various types of Non-Hodgkin Lymphoma (“NHL”), including DLBCL and acute lymphoblastic leukemia (“ALL”) and Mantle Cell Lymphoma (“MCL”) that are refractory or in second or later stage relapse. Although patients suffering from these aggressive cancers frequently undergo multiple treatments, including chemotherapy, radiation and targeted therapy including stem cell transplants, the five-year survival rate has been severely limited and patients who do not respond to, or have relapsed following at least two courses of standard treatment, have no other treatment options and a very poor outcome. According to the Surveillance, Epidemiology, and End Results (“SEER”) program of the National Cancer Institute, which is a source of epidemiologic information on the incidence and survival rates of cancer in the U.S., it is estimated that up to 10,000 patients per year in the U.S. with relapsed or refractory (r/r) B-cell NHL and ALL who have failed at least two prior systemic therapies may be eligible for CAR-T therapy. In addition, if CAR-T therapy is approved as an earlier second line option versus stem cell transplantation, an additional 10,000 to 12,000 patients may be eligible for treatment. However, this is predicated on improving the benefit-to-risk profile of CAR-T therapy, addressing the severe life-threatening adverse events currently associated with these agents and breaking the efficacy/toxicity linkage.
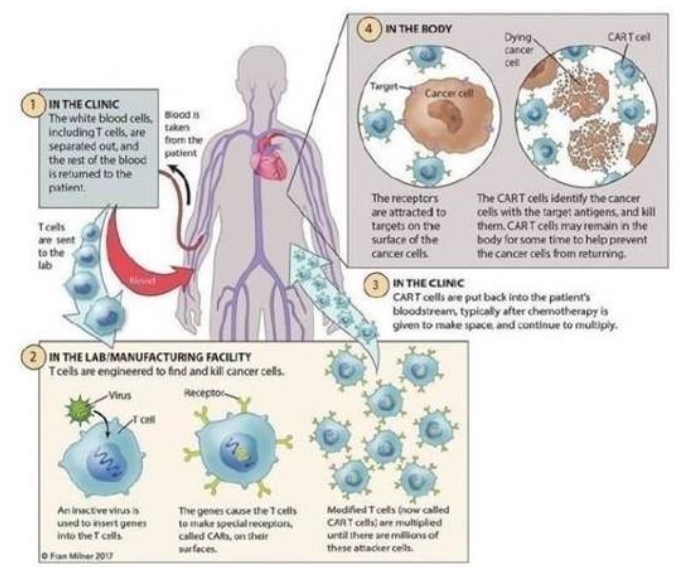
The FDA-approved CAR-T therapies have demonstrated the effectiveness of using targeted immuno-cellular engineering to cause a patient’s own T-cells to fight certain cancers that have not responded to standard therapies. T-cells are often called the “workhorses” of the immune system because of their role in coordinating the immune response and killing cells infected by pathogens and cancer cells. As depicted below, each of the FDA-approved CAR-T therapies is currently a one-time treatment that involves multiple steps:
| · | Harvesting white blood cells from the patient’s blood, also known as apheresis; |
| · | Engineering T-cells within this population to express cancer-specific receptors; |
| · | Increasing and purifying the number of genetically re-engineered T-cells; and |
| · | Infusing the functional cancer-specific T-cells back into the patient to allow for expansion and targeting the cancer cells. |
| 17 |
Kymriah, Yescarta and Tecartus each received FDA approval for adults with r/r DLBCL on the basis of one pivotal, single-arm Phase 2 study (ZUMA-1, in the case of Yescarta, and ZUMA-2, in the case of Tecartus) which served as the pivotal registration trial for each product in this indication, a markedly accelerated process that indicates the FDA’s view of the strong potential of these novel CAR-T therapy treatments to address an unmet need and improve patient outcomes. The number of evaluable patients in the studies that led to FDA approval for Kymriah, Yescarta and Tecartus in large B-cell lymphoma was 68, 101 and 68 respectively. Moreover, Kymriah also received FDA approval for the treatment of pediatrics and adolescents with relapsed refractory (“r/r”) acute lymphoblastic leukemia (“ALL”) based on a single phase 2 study. In the Kymriah registration study in the DLBCL population, 160 patients were enrolled and 68 were evaluable.
ZUMA-1, the single Phase 2 study that led to the FDA approval of Yescarta in r/r DLBCL, showed similarly positive results. The study enrolled 111 patients (101 were evaluable) with large B-cell lymphoma at advanced stages despite having undergone at least two previous treatments, with approximately 20% of patients already having undergone a stem cell transplant. The CR rate within three months of CAR-T treatment, given as a single infusion, was 58%, which dropped to 46% after six months. The CR rate after two years of CAR-T treatment, given as a single infusion, has been reported as 37%.
ZUMA-2, a single-arm, open-label Phase 2 study that led to the FDA approval of Tecartus for treatment of adult patients with relapsed or refractory mantle cell lymphoma (MCL), enrolled a total of 74 patients (68 were evaluable) that had received BTK inhibitor therapy previously. In an intention-to-treat analysis of all 74 patients enrolled, 85% of patients were reported to have had an objective response, and 59% had a complete response (https://www.nejm.org/doi/10.1056/NEJMoa1914347)
| 18 |
The FDA approval of Breyanzi is based on data from the TRANSCEND NHL 001 (017001) trial in which 268 patients with R/R LBCL received Breyanzi, the largest pivotal trial in third-line plus R/R LBCL that included patients with a broad range of histologies and high-risk disease. In the trial, Breyanzi was administered in the inpatient and outpatient settings.
Encouraged by the success of the Phase 2 studies, since the initial FDA approvals were granted to Novartis for Kymriah, the CAR-T therapy market has seen rapid expansion, with Gilead/Kite and Novartis and scores of other biotechnology companies actively working to progress CAR-T therapies as potential treatments for numerous blood and solid tumor cancers. The fourth entrant to the US market, Brevanzi from BMS has been recently approved.
Kymriah, Yescarta, Tecartus and Brevanzi are autologous individualized CD19 targeted CAR-T therapies. Development is also ongoing to move each agent to earlier lines of therapy for DLBCL (rather than as salvage therapy for patients who have exhausted other options), in other types of B-cell NHL and for the treatment of chronic lymphocytic leukemia (“CLL”). According to SEER, as well as the American Cancer Society's Cancer Statistics Center and World Health Organization Union for International Cancer Control, it is estimated that up to 10,000 patients with r/r B-cell hematologic malignancies (including DLBCL, ALL, CLL) per year may potentially benefit from CD19 targeted CAR-T therapies. In addition, if CAR-T therapy is approved as an earlier second-line option versus stem cell transplantation, an additional 10,000 to 12,000 patients may be eligible for treatment. Moreover, there are two B-cell maturation antigen (“BCMA”) targeted CAR-T therapies in phase 2 development for relapsed or refractory multiple myeloma and several other novel CAR-T therapies targeting various antigens and neo-antigens in development for a number of hematologic and solid cancers. While there may be individual differences between CAR-T therapy products, the overall toxicity profile is generally expected to be generally consistent with that reported for Yescarta, Tescarta and for Kymriah and it is known that various development-stage BCMA and other CAR-T therapies are hampered by the emergence of cytokine storm and other serious and potentially fatal side-effects.
CAR-T Therapy
The four FDA-approved CAR-T therapies are not without significant limitations. Despite the exciting prospects for treating patients with limited options, significant and potentially life-threatening side-effects associated with CAR-T therapy, including NT and CRS, remain a significant unmet need that must be addressed. Because NT and CRS can be life-threatening and have proven fatal in many instances, and because each product bears a “Boxed” warning from the FDA (the strictest FDA warning label intended to alert patients and providers about serious and life-threatening risks associated with a particular drug), patients seeking to benefit from Yescarta, Tescartus, Kymriah or Brevanzi generally may only do so if the treatment center is in compliance with the Risk Evaluation and Mitigation Strategy (“REMS”) program required by FDA.
REMS is a drug safety program that the FDA can require for certain medications with serious safety concerns to help ensure the benefits of the medication outweigh its risks and are intended to assist and train certified treatment centers on the management of these serious side-effects. For example, each hospital and its associated clinics have a minimum of two doses of tocilizumab available on-site for each patient for the potential treatment of moderate to severe cases of CRS. We believe the REMS requirement may have adversely impacted both market uptake and usage to date. Both CRS and NT are caused by a large-scale release of pro-inflammatory cytokines and chemokines induced by the CAR-T therapy, sometimes referred to as a “cytokine storm”. The first placebo-controlled, randomized study evaluating tocilizumab for the treatment of cytokine storm, in this case in COVID-19 patients with cytokine storm, failed to demonstrate an effect. However, the Recovery study, an open-label, randomized trial conducted in the UK demonstrated an improvement in mortality (HR=.86, 14% relative risk reduction) and an improvement in progression to IMV and/or death (HR=.85, 15% relative risk reduction).
According to the package inserts for Yescarta and Kymriah, up to 94% of patients treated with Yescarta or Kymriah in the clinical trial setting experienced CRS (with up to 49% of cases being severe or grade >3 in nature) and up to 87% experienced NT (with up to 31% of cases being severe or grade >3 in nature) despite the availability and utilization of tocilizumab. Moreover, based on feedback from leading treatment centers in the US, approximately 30 to 60% of patients receiving CAR-T therapy require admission to the ICU and in some cases require an extended stay, with multiple interventions, including ventilator support and other supportive measures, to be urgently administered to manage these side-effects. Some patients can suffer seizures, coma, brain swelling, heart arrhythmias, organ failure and serious and life-threatening clotting disorders, not only causing more complex and potentially fatal medical consequences, but significantly adding to cost of patient care. These can be particularly challenging and concerning issues, especially in younger and pediatric patients.
| 19 |
It is important to note that patients suffering from severe or critical COVID-19 pneumonia can often suffer similar medical conditions and extended hospitalization and ICU care.
Researchers who evaluated 1,254 patients who underwent CAR-T therapy at 86 hospitals over the past two years reported that the median ICU stay was 15 to 19 days with a median overall cost ranging from $85,726 to $242,730, not including the cost of the CAR-T therapy itself (Harris, et al. TCT 2019 Abstracts 500, 501). In addition, there have also been deaths reported as a result of these serious side-effects. A publication assessing 636 patients who had received either of the two FDA-approved CAR-T therapies (348 patients on Yescarta and 288 on Kymriah) authored by Anand and Burns, et al. in the Journal of Clinical Oncology (37, 2019 (suppl; abstract 2540)) reported that 15% of CAR-T treated patients (10% receiving Yescarta and 21% receiving Kymriah) died from factors not associated with disease progression (i.e., non-relapse mortality) and the primary driver of non-relapse mortality was NT and/or CRS. Therefore, these serious side-effects are associated with significant mortality rates, despite the availability of approved supportive care measures, even as CAR-T therapies are administered only in trained and certified treatment centers staffed by experts in the field. If such side-effects can be ameliorated or eradicated, and adequate data is submitted to FDA, the “Black Box” warning and REMS program could potentially be scaled back or removed. Further, this may allow a larger proportion of patients to receive CAR-T administration in the out-patient setting. This could convey significant medical and financial benefits and also change the reimbursement dynamic in favor of providers, while opening up a larger cohort of potentially suitable patients.
There are currently no FDA-approved products for the prevention or treatment of NT or for the prevention of CRS associated with CAR-T therapy. Medicines used to manage NT and CRS, such as tocilizumab and corticosteroids, have not adequately controlled the side-effects, and steroids may have a detrimental impact on the efficacy of the CAR-T therapy itself while tocilizumab may increase the risk of CAR-T therapy induced NT and is correlated with an increased risk of infections, including severe infections. Further, these medicines have not undergone prospective clinical trials for use in this patient population. Tocilizumab is only approved for the treatment of severe cases of CRS, but is not approved for prevention of CRS, nor is it approved for either prevention or treatment of NT.
The approval of tocilizumab in CRS was granted as a result of case studies and not as a result of a planned, prospective clinical study in this patient population, as would be typical. Studies testing tocilizumab for the prevention of CRS have shown tocilizumab to significantly worsen the rate of NT across all grades as well as the more serious grades 3 and above, as compared to the rate in patients who did not receive tocilizumab prophylactically. In addition, recent publications question the efficacy of tocilizumab in CRS. For example, studies testing tocilizumab as a prophylactic therapy for CRS have shown the rates of overall CRS remained unaltered as compared to the rate in patients who did not receive tocilizumab prophylactically (Locke et al. American Society of Hematology (“ASH”) 2017, Abstract 1547). Further, a publication authored by Le, et al. in The Oncologist (2018, 28(8); 943-947) assessing 60 patients who had received either Yescarta or Kymriah and had suffered from CRS having received tocilizumab and/or steroids after the onset of CRS, reported that only approximately half of the patients responded at day 11.
These data, along with the Anand/Burns data and the Locke data discussed above, demonstrate that improvements in the ability to prevent or mitigate NT and CRS are needed. Such improvements would help remove these major impediments to uptake and utility of CAR-T therapies, improve healthcare utilization and improve overall patient outcomes. Managing patients with these side-effects can consume a significant amount of in-hospital resources, including extended stays in the ICU. The primary driver of non-drug related costs associated with CAR-T therapy is the length of stay in the hospital, particularly if this includes ICU admission. Non-drug related costs for patients who develop CRS and/or NT are approximately double that of patients who do not develop these serious toxicities. Further, as the potential benefit of CAR-T therapies are explored in earlier lines of hematologic cancers (rather than as salvage therapy for patients who have exhausted other options), as well as moving use of CAR-T therapies into solid tumors, the need to address serious side-effects becomes paramount.
| 20 |
In addition to improving patient outcomes, the ability to significantly reduce the incidence and severity of NT and prevent CRS associated with CAR-T therapy may offer significant benefits in making these treatments more cost-effective. Hospital reimbursement for patients who are treated only as an out-patient is profoundly different from, and more favorable to the hospital than, the reimbursement afforded to treatments for patients who are admitted or re-admitted to the hospital within a 72-hour period. Unfortunately, at present, the need to identify, treat and manage NT and CRS generally has prevented CAR-T therapies from being administered, and follow-up care monitored and managed, on an out-patient basis. Again, 30-60% of patients receiving CAR-T therapy require admission to the ICU, in some cases requiring an extended stay, with multiple interventions, investigations and treatments needing to be urgently administered. As a result, in some institutions, the treating physician may require the hospital to reserve a bed in the ICU as a prerequisite to administering the CAR-T therapy in case the patient needs to be hospitalized in an attempt to manage the adverse effects from NT and CRS. This is especially challenging in the context of the COVID-19 pandemic where many ICU units are already overwhelmed with COVID-19 patients. At other institutions, the patient is admitted as an in-patient and is required to remain in the hospital for at least a week, with discharge being subject to satisfactory short-term outcomes and no emergence of complications. Even in institutions where the CAR-T therapy is initially administered in an out-patient setting, the patient is closely monitored daily for several weeks and is required to stay within a short distance from the hospital in case the patient needs to be admitted to the hospital on an emergency basis, requiring additional lodging, food and other costs to be incurred by the patient, the payer, or both. In some situations, these patients are re-admitted to the hospital on an emergency basis as an in-patient if complications ensue. If a patient is admitted or re-admitted to the hospital as an in-patient, the hospital reimbursement dynamics may change in a manner which is negative for the hospital, the payer and the patient. This dynamic also changes typical hospital reimbursement, depending on when in the treatment cycle the patient is admitted or re-admitted. In addition, certain treatment centers do not accept patients who are not potentially able to be treated as an out-patient and refer such patients to other centers who may be willing to treat them as in-patients, primarily as a result of the reimbursement handicap that would accrue as a result of in-patient coding, billing and reimbursement, which generally leads to the hospital system losing money because of the in-patient care reimbursement. Further, the COVID-19 pandemic has placed further stress on the hospital, ICU and broader healthcare systems of almost all countries affected by the outbreak.
While both Kymriah and Yescarta have been approved by European regulators for market authorization, prescriptions have been limited as Kite and Novartis work to establish reimbursement arrangements intended to facilitate access to the treatments on a discounted basis consistent with the governmental mandates to curb healthcare spending. These dynamics, and the additional complexity of treating patients with serious and potentially life-threatening side-effects in the hospital and/or ICU, mean that enabling true out-patient administration and follow-up would confer significant benefits to patients, payers and the hospital system. Lenzilumab, if proven to be able to abrogate these serious side-effects as well as improve efficacy, may offer a solution.
Other T-cell Engaging Therapies
In addition to CAR-T therapy, we are committed to advancing our diverse platform for GM-CSF axis suppression for a broad range of other T-cell engaging therapies, including both autologous and allogeneic next generation CAR-T therapies, bi-specific antibody therapies, as well as other cell-based immunotherapies in development, to break the efficacy/toxicity linkage, including for the prevention and/or treatment of GvHD in patients undergoing allogeneic HSCT. Many of these treatment options may lead to serious side-effects and have ample room for improved efficacy.
We believe that GM-CSF neutralization with lenzilumab has the potential to prevent or reduce GvHD without compromising, and potentially improving, the beneficial GvL effect in patients undergoing allogeneic HSCT, thereby making allogeneic HSCT safer. Allogeneic HSCT is a potentially curative therapy for patients with hematological cancers. Although a potentially life-saving treatment for patients suffering from hematological cancers, between 40-60% of patients receiving HSCT treatments experience acute or chronic GvHD, which together carries a 50% mortality rate. After being transplanted into the patient, donor-derived T cells are responsible for mediating the beneficial GvL effect. In many cases, however, donor-derived T cells that remain within the graft itself have also been linked to destruction of healthy tissue in the patient (the host), with particular risk of destroying cells in the patient’s skin, gut, and liver, resulting in GvHD. Although depleting donor grafts of T cells can prevent or reduce the risk of GvHD, this results in a reduced GvL effect, thereby having a detrimental impact on the efficacy of the allogeneic HSCT treatment itself and leading to increased relapse rates. We expect that the use of allogeneic HSCT may be hampered by GvHD complications. A recent study published in ‘blood advances’ an official journal of the American Society of Hematology, suggests that neutralizing or blocking GM-CSF may limit or prevent GvHD in the gastrointestinal tract (Gartlan, K., et al, October 8, 2019, vol. 3, no.19).
| 21 |
There are currently no FDA-approved agents for the prevention of GvHD, and there is a significant unmet medical need for an agent that can uncouple the beneficial GvL effect from harmful GvHD. At this time, pre-conditioning regimens for HSCT treatments vary significantly by treatment centers, including by unapproved, or “off-label”, use of agents that have been approved by the FDA for other uses only. We believe there to be a significant unmet medical need and lenzilumab, if proven to be able to prevent GvHD in allogeneic HSCTs, may offer a solution.
Allogeneic HSCT Overview
Allogeneic HSCT, which involves transferring stem cells from a healthy donor to the patient, has demonstrated effectiveness in treating hematological cancers. As depicted below, allogeneic HSCT involves multiple steps:
| · | Collecting blood from a healthy donor; |
| · | Processing the donor’s blood to remove the stem cells before returning the rest of the donor’s blood back to the donor; |
| · | Pre-conditioning the patient with high-dose chemotherapy and/or radiation; and |
| · | Infusing the donor’s stem cells into the patient to allow for the production of new blood cells. |
| 22 |
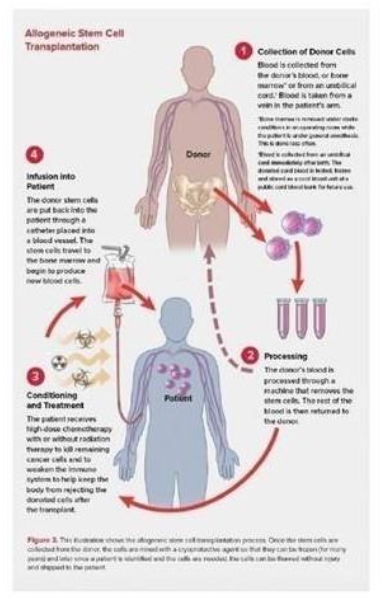
The overall number of allogeneic HSCT treatments continues to increase annually in the US and abroad. In 2019, approximately 10,000 allogeneic HSCT treatments are expected to be performed in the US, with similar trends expected in Europe.
Combining CAR-T Therapies with Lenzilumab
We believe lenzilumab has the potential to improve the efficacy and safety of CAR-T therapy and that the use of lenzilumab may minimize or eradicate the incidence, frequency, duration and/or severity of NT and/or CRS that frequently appear in CAR-T patients. Further, GM-CSF neutralization may enhance CAR-T proliferation and effector functions and potentially confer additional benefits in terms of durable efficacy and healthcare resource utilization.
We also believe lenzilumab may improve the value proposition of CAR-T and allogeneic HSCT therapies and facilitate their use and acceptance throughout the healthcare systems in the US and abroad.
Our current clinical and regulatory development plan in marketed CAR-T therapies is focused on a collaboration agreement we executed with Kite in May 2019 (the “Kite Agreement”), with Kite marketing two of only four approved CAR-T therapies (Yescarta) is the market leader by a large margin. Pursuant to the Kite Agreement, the parties have agreed to conduct a multi-center Phase 1b/2 study (ZUMA-19) of lenzilumab with Kite’s Yescarta in patients with relapsed or refractory B-cell lymphoma. The primary objective of ZUMA-19 is to determine the effect of lenzilumab on the safety of Yescarta. In addition, efficacy and healthcare resource utilization will be assessed. The Kite Agreement is non-exclusive. Depending upon FDA feedback, we believe ZUMA-19 may serve as the basis for registration for lenzilumab. On June 30, 2020, we announced that the first patient had been infused in the ZUMA-19 study.
| 23 |
Combining Allogeneic HSCT with Lenzilumab
In addition to CAR-T therapy, we are committed to advancing our diverse platform for GM-CSF axis suppression for a broad range of other T-cell engaging therapies, including both autologous and allogeneic next generation CAR-T therapies, bi-specific antibody therapies as well as other cell-based immunotherapies in development, with our current and future partners.
We believe that GM-CSF neutralization using lenzilumab has the potential to make allogeneic HSCT safer and more effective. Similar to GM-CSF neutralization with lenzilumab breaking the efficacy/toxicity linkage with CAR-T therapy, GM-CSF neutralization has demonstrated potential to attenuate GvHD while maintaining the beneficial GvL effect in patients undergoing allogeneic HSCT.
In July 2019, we entered into the Zurich Agreement with UZH. Under the Zurich Agreement, we have in-licensed certain technologies that we believe may be used to prevent or treat GvHD, thereby expanding our development platform to include improving the safety and effectiveness of allogeneic HSCT, a potentially curative therapy for patients with hematological cancers. The technology was recently featured in a November 2018 research article published in Science Translational Medicine, where the authors demonstrated in a murine model of GvHD, that donor T cell-derived GM-CSF drives GvHD through activation, expansion, and trafficking of myeloid cells but has no effect on the GvL response. Neutralization of GM-CSF (either using a neutralizing antibody or through GM-CSF gene knock-out) was able to uncouple the myeloid-mediated immunopathology resulting in GvHD from the T cell-mediated control of leukemic cells. This discovery provides a clear mechanistic proof-of-concept for neutralizing GM-CSF to prevent GvHD without compromising, and potentially improving, the GvL effect in patients undergoing allogeneic HSCT. Corroborating data related to the critical effect GM-CSF has on GvHD development in HSCT was published recently by Gartlan et al.
The strong link between T cell-mediated efficacy and myeloid cell mediated toxicity mirrors the findings that have been reported with CAR-T therapies where T cell-produced GM-CSF has emerged as a key driver of the myeloid inflammatory cascade resulting in NT and CRS and potentially impairing improved CAR-T therapy efficacy through effects on myeloid-derived suppressor cells. GM-CSF neutralization has the potential to eliminate or reduce the off-target inflammatory cascade while preserving the on-target efficacy of T cell therapies, thereby breaking the efficacy/toxicity linkage.
We believe that GM-CSF neutralization with lenzilumab has the potential to prevent or treat GvHD without compromising, and potentially improving, the beneficial GvL effect in patients undergoing allogeneic HSCT, thereby making allogeneic HSCT safer. Accordingly, we aim to position lenzilumab as a “must have” companion product to any allogeneic HSCT and as a part of the standard pre-conditioning that all patients receiving allogeneic HSCT should receive or as an early treatment option in patients identified as high risk for GvHD.
Lenzilumab in CMML
We believe that lenzilumab also holds promise in CMML, a rare form of hematologic cancer with no FDA-approved treatment options and a three-year overall survival rate of 20% and median overall survival of 20 months, and potentially in JMML, a rare pediatric form of leukemia. CMML is a clonal stem cell disorder of which monocytosis is a key feature. Approximately 40% of CMML patients carry NRAS/KRAS/CBL mutations which are associated with GM-CSF hypersensitivity. CMML has features of myelodysplastic syndrome (“MDS”), including abnormal, dysplastic bone marrow cells; cytopenia; transfusion dependence; and of myeloproliferative neoplasms, including overproduction of white blood cells, organomegaly (e.g., splenomegaly and hepatomegaly) and extramedullary disease. About 15 to 20% of CMML cases progress to acute myeloid leukemia, or AML. According to the American Cancer Society, approximately 1,100 individuals in the United States are newly diagnosed annually with CMML, with the majority of these new patients being age 60 or older. These patients are typically unsuitable for stem cell transplants.
| 24 |
In a Phase 1 study, 3 of 6 patients with NRAS/KRAS/CBL mutations demonstrated a clinical response by the MDS/MPN International Working Group criteria. Final results of this study were presented at the 2019 ASH annual meeting and published in ‘blood’. Building on this successful Phase 1 study in CMML with lenzilumab, we are planning a Phase 2 study in combination with azacitidine in newly-diagnosed CMML patients who express NRAS/KRAS/CBL mutations which are known to be hypersensitive to GM-CSF and therefore may lend themselves to responsiveness to lenzilumab treatment. The Phase 2 study will be conducted by Dr Daniel Thomas, MBBS PhD FRACP FRCPA B Med Sci (Hons) Associate Professor, South Australia Health and Medical Research Institute (SAHMRI), University of Adelaide in Australia and supported with a grant from the Medical Research Future Fund (MRFF).
Gene-edited CAR-T Therapies using GM-CSF Gene Knockout
We believe that our GM-CSF knockout gene-editing CAR-T platform has the potential to create next-generation CAR-T therapies that improve the efficacy and safety profile of CAR-T therapy via gene-edited CAR-T cells which can be engineered to lack the ability to produce GM-CSF, thereby avoiding any efficacy/toxicity linkage and potentially preserving and improving upon the benefits of the CAR-T therapy while altogether avoiding its serious and potentially life-threatening side-effects.
We are advancing our GM-CSF knockout gene-editing CAR-T platform through the Mayo Agreement that we entered into in June 2019 with the Mayo Foundation. Under the Mayo Agreement, we have in-licensed certain technologies that we believe may be used to create CAR-T cells lacking GM-CSF expression through various gene-editing tools including CRISPR-Cas9. The Mayo Agreement broadened our leadership position in the GM-CSF neutralization space and expanded our discovery platform aimed at improving CAR-T therapy to include gene-edited CAR-T cells.
Preclinical data indicates that GM-CSF gene knockout CAR-T cells show improved overall survival compared to GM-CSF-expressing CAR-T cells in addition to the expected benefits of reduced serious side-effects associated with CAR-T therapy. We are establishing a platform of next-generation combinatorial gene knockout CAR-T cells that have potential to be applied across both autologous and allogeneic approaches and we are also investigating multiple CAR-T cell designs using precise dual and triple gene editing to significantly enhance the anti-tumor activity while simultaneously preventing CAR-T therapy induced toxicities. Through targeted gene expression and modulating cytokine activation signaling, we may be able to increase the proportion of fitter T-cells produced during expansion, increase the proliferative potential, and inhibit activation induced cell death, thereby improving the cancer killing activity of our engineered CAR-T cells thereby making them more effective and safer in the treatment of cancers. Preclinical data indicates that CAR-T cells express GM-CSF and signal through GM-CSF receptors upon activation in an autocrine fashion. GM-CSF knockout CAR-T cells are not able to signal through this pathway which results in a gene expression profile distinct from GM-CSF-expressing CAR-T cells after in vitro expansion. This includes lower levels of Fas expression which may indicate a less differentiated state of the CAR-T cells. These data were presented at the 2019 ASH annual meeting and the abstract was the recipient of an Abstract Achievement Award. We continue to explore the phenotypic pattern of GM-CSF knockout CAR-T cells relative to GM-CSF-expressing CAR-T cells and possible implications for improved safety and efficacy. We plan to continue development of this technology in combination approaches that could add to the observed efficacy benefits of current generation CAR-T products.
Kite Collaboration
Our current clinical and regulatory development plan is focused on the Kite Agreement we executed in May 2019. Pursuant to the Kite Agreement, the parties have agreed to conduct a multi-center potential registration Phase 1b/2 study (ZUMA-19) of lenzilumab with Kite’s Yescarta in patients with relapsed or refractory B-cell lymphoma, including DLBCL. The primary objective of ZUMA-19 is to determine the effect of lenzilumab on the safety of Yescarta. In addition, efficacy and healthcare resource utilization will be assessed. Kite is the sponsor of ZUMA-19 and responsible for its conduct. On June 30, 2020, we announced that the first patient had been infused in the ZUMA-19 study.
| 25 |
The Kite Agreement provides that we and Kite will split only the out-of-pocket costs incurred in conducting the ZUMA-19 study, including third-party expenses incurred in accordance with a mutually agreed budget. We currently project we will be responsible for an aggregate of up to approximately $8 million in out-of-pocket costs, assuming up to a total of 72 patients are recruited for a multi-center study. Each party will otherwise be responsible for its own internal costs, including internal personnel costs, incurred in connection with the Study.
In addition, the parties have agreed to enter into certain additional agreements in connection with ZUMA-19, including a quality and a supply agreement that will obligate us, at our expense, to supply certain quantities of lenzilumab in final form for administration to subjects in ZUMA-19. Kite is responsible, at its expense, to supply Yescarta. Kite will be responsible for other costs related to ZUMA-19.
The parties have formed a Joint Development Committee (“JDC”) to oversee ZUMA-19, its progress and administration, and other matters between the parties and their obligations set forth in the Kite Agreement. The JDC comprises representatives of each of Humanigen and Kite. Kite’s JDC designees will have decision-making authority with respect to (1) certain operational matters in conducting ZUMA-19, such as selection of participating sites and engagement of third party service providers; and (2) amendments to the ZUMA-19 protocol established by the JDC that do not directly relate to our investigational product or the part of the ZUMA-19 Study combination that consists solely of our investigational product; but only, in each case, to the extent that such decisions by the Kite JDC designees do not result in an increase in the mutually agreed-upon budget by fifteen percent or more. Neither Kite designees nor our designees will have final decision-making authority on matters directly related to the investigational product of the other party, including the other party’s investigational product that is used in or a part of ZUMA-19.
We and Kite will jointly own all ZUMA-19 data and sample data, including case report forms, findings, conclusions and other results from ZUMA-19 that relates to each party’s investigational product that is used in combination or sequence in ZUMA-19, and not solely to either party’s investigational product. ZUMA-19 data and sample data that relates solely to a party’s own investigational product will be owned solely by that party. We and Kite will own our respective background intellectual property and neither party has been granted a commercial license to use the respective background intellectual property of the other party. Each party will own any inventions that relate to its own investigational product used in ZUMA-19 or sample data, however, any new inventions related to, or covering, the combination of each party’s investigational product used in ZUMA-19 will be jointly owned by the parties. Kite will control any preparation, filing, prosecution and maintenance of any patent covering any new inventions related to, or covering, the combination of each party’s investigational product used in ZUMA-19 and the parties will equally share costs for all such patents. We will continue to own all world-wide rights to lenzilumab and the intellectual property related to lenzilumab, including use of lenzilumab with CAR-T therapy.
Unless previously terminated, the Kite Agreement will continue until the first anniversary of the date Kite provides the final ZUMA-19 study report to us or the termination of the study. Kite may elect to terminate or suspend the study at any time. Each party may terminate the Kite Agreement under certain circumstances, including (1) for an uncured breach by the other party; (2) if a party determines that the study may unreasonably affect patient safety; (3) upon certain actions by regulatory authorities that are adverse to the study; or (4) if a party determines to discontinue the development of its investigational product.
The Kite Agreement imposes additional obligations on the parties, such as confidentiality obligations, obligations to comply with applicable law related to patient privacy and data protection, and potential indemnification obligations. The Kite Agreement is non-exclusive.
Worldwide License for the Prevention of GvHD through GM-CSF Neutralization from UZH
In July 2019, we entered into the Zurich Agreement with UZH. Under the Zurich Agreement, we have in-licensed certain technologies that we believe may be used to prevent GvHD through GM-CSF neutralization. The Zurich Agreement covers various patent applications filed by UZH which complement and broaden our position in the application of GM-CSF neutralization and expands our development platform to include improving allogeneic HSCT.
| 26 |
Worldwide License to Gene-Editing Technology from the Mayo Foundation
In June 2019, we entered into an exclusive worldwide license with the Mayo Foundation. Under the Mayo Agreement, we have in-licensed certain technologies that we believe may be used to create CAR-T cells lacking GM-CSF expression through various gene-editing tools including CRISPR-Cas9. The license covers various patent applications and know-how developed by the Mayo Foundation in collaboration with us. These licensed technologies complement and broaden our leadership position in the CAR-T/GM-CSF neutralization space and expand our discovery platform aimed at improving CAR-T therapy to include gene-edited CAR-T cells. With this license agreement, we significantly expanded our intellectual property portfolio to include gene-edited CAR-T cells which can be engineered to lack the ability to produce GM-CSF which may improve the efficacy and safety profile of CAR-T therapy. In addition, we have and continue to file intellectual property encompassing a broad range of gene-editing approaches related to GM-CSF knockout.
Lenzilumab
Overview and Mechanism of Action
Lenzilumab is a novel monoclonal antibody designed to target and neutralize human GM-CSF, which could also be described as a ‘myeloid inflammatory factor’. We used our proprietary and patent protected Humaneered antibody development platform to develop lenzilumab.
There is extensive evidence linking GM-CSF expression to serious and potentially life-threatening outcomes in other respiratory conditions such as COVID-19 pneumonia. GM-CSF and other downstream cytokines that are triggered by GM-CSF are elevated in patients who suffer poorer outcomes, including ICU admission and invasive mechanical ventilation. Neutralizing GM-CSF with lenzilumab may result in fewer days to symptom resolution, fewer days in hospital, minimization, or absence of admission to the ICU and use of invasive mechanical ventilation.
In addition, there is extensive published scientific evidence linking GM-CSF expression to serious and potentially life-threatening side-effects associated with CAR-T therapy and reduced efficacy through recruitment of MDSCs. Our focus for lenzilumab development is investigating its potential to improve efficacy of CAR-T therapy and to prevent or ameliorate CAR-T therapy-related NT and CRS. Following CAR-T therapy administration, GM-CSF produced by CAR-T cells initiates a signaling cascade of inflammation that results in the trafficking and recruitment of myeloid cells to the tumor site. These myeloid cells produce key cytokines known to be associated with the development of NT and CRS thereby perpetuating the inflammatory cascade. Peer-reviewed publications in leading journals by well-recognized experts have reported that GM-CSF blood levels are elevated early after CAR-T cell administration and reach significant peak levels in patients who suffer serious NT as a side-effect of CAR-T therapy.
GM-CSF seems to be critical for the initiation of CRS, NT and the inflammatory cascade in COVID-19 and following CAR-T administration. GM-CSF acts as an upstream ‘initiator’ and the precursor to other cytokines involved in the cascade. GM-CSF gene knock-out (k/o) mice, or animals that lack a functional myeloid compartment, do not develop CRS and have normal levels of downstream cytokines, including IL-6, IL-1 and MCP-1/CCL2. Animals that are k/o for IL-1, INF-gamma and IL-6 still develop CRS in models which recapitulate this syndrome. This is very telling, given that some investigators believe IL-6 to be causative of CRS. In addition, in pivotal clinical studies, IL-6 has not been shown to correlate with severe NT or CRS emergence. In the COVID-19 setting, multiple late-stage clinical trials have failed to demonstrate a benefit in patients hospitalized with COVID-19 pneumonia when dosed with IL-6 inhibitors. Recently published results from the RECOVERY trial have demonstrated that an anti-inflammatory treatment, tocilizumab, reduces the risk of death when given to hospitalized patients with severe COVID-19. Furthermore, IL-6 inhibitors have Black Box Warnings in the product label and package inserts related to an increase of potentially fatal infections, including respiratory and an increase in viral reactivation. In the COVID-19 setting, these may be particularly concerning, given the viral and respiratory issues at work. The benefit:risk to patients who are not end-stage and with co-morbidities where the additional risk posed by these warnings may need to be carefully assessed before using IL-6 inhibitors off-label and in studies in COVID-19 patients.
| 27 |
The lack of GM-CSF does not affect T-cell mediated cancer-killing, or cytotoxicity, as GM-CSF k/o animals had equivalent effector to target cell (E:T) ratios and cytotoxic activity against tumor cells in our published studies. GM-CSF is required for CCR2+ monocytes to initiate and sustain neuro-inflammation. It is postulated that GM-CSF induces CCR2+ inflammatory myeloid derived cells to infiltrate into the CNS, activating microglial cells; the activated microglial cells then increase their expression of CCL2/MCP-1 to further recruit inflammatory myeloid cells in a self-perpetuating manner, forming a positive feedback loop. Research from CAR-T clinical trials demonstrated that fever and elevated MCP-1 levels 36 hours post CAR-T treatment were most predictive of severe CRS and NT across patients with NHL, ALL and CLL, providing further support for the mechanism by which GM-CSF may contribute to these toxicities.
The field of COVID-19 is extremely dynamic and fast-moving, with multiple publications and other information appearing at a very rapid pace, as clinical practice endeavors to optimize care for patients.
There are many other publications that point to the pivotal role of GM-CSF in CRS and NT in CAR-T therapy, as well as potentially hampering efficacy. Based on these publications and extensive discussions with leading key opinion leaders, we believe that lenzilumab, used as alongside CAR-T therapy offers several potential benefits, including:
| · | Lower rates of severe/grades >3 CRS and all grades of CRS; |
| · | Lower rates of severe/grades >3 NT and all grades of NT; |
| · | Lower rates of ICU admissions and duration of hospitalization; |
| · | Improved anti-tumor response (e.g. ORR, CR) and overall patient outcomes; |
| · | Improved duration of response and reduced relapse rates; |
| · | Improved cost effectiveness and reduced direct/indirect costs; |
| · | Improved reimbursement, and preferential formulary placement for CAR-T; |
| · | Expansion of CAR-T beyond the relapse/refractory setting to second-line and potential first-line use due to improved benefit-risk profile, increasing utilization to a significantly larger pool of patients; |
| · | Expansion of CAR-T into solid tumor treatments; and |
| · | Scaling back or removal of current CAR-T required REMS programs and “Black Box” warnings due to improved benefit-risk profile of CAR-T. |
Preclinical studies in mice conducted at the Mayo Clinic using human ALL blasts, human CD19 CAR-T, and human peripheral blood mononuclear cells (PBMCs), demonstrated that blockade of GM-CSF with lenzilumab prevented the onset of CRS, reduced neuro-inflammation by 75% (as assessed by quantitative MRI) and maintained the integrity of the blood-brain barrier (BBB) compared to CAR-T plus control antibody treatment, where CRS, neuro-inflammation and BBB disruption can be profoundly affected. The administration of lenzilumab in combination with CAR-T therapy led to a significant (5-fold) increase in proliferation of CAR-T cells and improved CAR-T effector function, presumably due to a decrease in MDSC expansion and trafficking which is known to be promulgated by GM-CSF. GM-CSF neutralization in combination with CAR-T therapy reduced relapse, enhanced anti-tumor response and improved overall survival compared to CAR-T therapy alone and these data were published in ‘blood’. Moreover, the combination of lenzilumab and CAR-T reduced myeloid cell infiltration into the CNS and resulted in significantly better leukemic control as quantified by flow cytometry compared to CAR-T and control antibody. Human data from CAR-T clinical trials suggests that the only cytokines associated with grade > 3 NT are GM-CSF, IL-2 and IL-15. Moreover, in patients who developed severe NT, there was a 17-fold increase in myeloid cell trafficking into the CNS further establishing the role of GM-CSF in the expansion and trafficking of myeloid cells in the toxicities associated with CAR-T therapy.
We are also developing lenzilumab alongside allogeneic HSCT for patients with hematological cancers. Accordingly, we are assessing plans to investigate use of lenzilumab as a necessary companion product to any allogeneic HSCT and as a part of the standard pre-conditioning that all patients receiving allogeneic HSCT should receive or as an early treatment option for patients identified as high risk by certain biomarkers.
| 28 |
Clinical data also shows the potential for lenzilumab as a treatment for certain autoimmune and other inflammatory conditions, including eosinophilic asthma, rheumatoid arthritis (RA), ankylosing spondylitis (“AS”), psoriatic arthritis (“PsA”), inflammatory bowel disease (“IBD”), juvenile idiopathic arthritis (“JIA”), giant cell arteritis (“GCA”), atopic dermatitis (“AD”) and systemic lupus erythematosus (“SLE”). There is potential for a range of other oncology, immunology and autoimmune conditions and we are investigating partnering lifecycle management opportunities for lenzilumab in high value markets with strong unmet medical needs.
Development Program
As a Treatment and/or Prevention of Cytokine Storm Associated with COVID-19
We have completed enrollment in a Phase 3 multi-center, randomized, placebo-controlled, double-blinded, clinical trial in the setting of COVID-19. There were 18 clinical sites recruiting patients into this study in the US and 11 sites in Brazil. The Phase 3 trial is assessing the safety and efficacy of lenzilumab in reducing severe outcomes in hospitalized adult patients with confirmed COVID-19 pneumonia.
There are currently no products approved by the FDA for the prevention of CRS/cytokine storm associated with COVID-19. There are numerous products currently in development for COVID-19 which can be broadly categorized as direct acting antivirals, immunomodulators, and other preventative strategies such as vaccines or neutralizing antibodies., Remdesivir (a direct acting antiviral) was approved by FDA for COVID-19. There is no evidence that remdesivir offers a mortality benefit and the largest remdesivir-based global study conducted by the World Health Organization failed to show benefit from remdesivir treatment. Other direct acting antiviral agents such as lopinavir/ritonavir and hydroxychloroquine (with or without a macrolide) have not demonstrated efficacy in randomized controlled trials.
Other than the recently published results from the RECOVERY trial which demonstrated that an anti-inflammatory treatment, tocilizumab, in addition to steroids, reduces the risk of death when given to hospitalized patients with severe COVID-19, no immunomodulator therapy has proven efficacy in a randomized controlled clinical trial in the setting of COVID-19. Other studies involving tocilizumab have failed to demonstrate benefit. In addition, IL-1 and IL-1 receptor inhibitors, respectively Ilaris (canakinumab) and Kineret (anakinra) have both failed to demonstrate benefit in Phase 3 studies, as has the oral JAK inhibitor Jakafi (ruxolitinib) and the BTK inhibitor Calquence (acalabrutinib). Baricitinib a JAK inhibitor is authorized for use in combination with remdesivir for COVID-19. The anti-GM-CSF agent, otilimab failed in an 806-patient randomized controlled study sponsored by GlaxoSmithKline (“GSK”). In a post-hoc analysis of a subgroup of patients older than 70, there was an apparent benefit in the proportion of patients alive and free of respiratory failure at day 28 and mortality at day 60. A 350-patient follow-on Phase 3 study is planned to determine if the benefit in patients older than 70 can be confirmed. It is unclear whether the anti-GM-CSF agents gimsilumab and namilumab are continuing development for COVID-19 and the primary endpoint for mavrilimumab for COVID-19 in a 40 patient study was recently missed, though there were some promising data in this Phase 2 study.
As an upstream regulator of cytokine storm, GM-CSF neutralization with lenzilumab may offer advantages over other immunomodulator strategies that either target other downstream cytokines such as IL-1, IL-6, or MIP-1 alpha or are broadly immunosuppressive and target cytokine signaling pathways non-selectively through JAK inhibition. In addition, lenzilumab is the only immunomodulator that was in an active clinical trial to prevent cytokine storm prior to COVID-19 and is currently the only agent in an active Phase 3 trial targeting GM-CSF.
Lenzilumab was granted emergency single use IND authorization from the FDA (often referred to as compassionate use) to treat patients with COVID-19. On June 15, 2020, we announced that Mayo Clinic published data derived from the compassionate use of lenzilumab in the treatment of 12 patients hospitalized in the Mayo Clinic system. Under applicable FDA rules, a patient cannot receive a compassionate use drug unless FDA has issued an individual patient emergency IND authorization, which the Mayo Clinic requested from FDA prior to each individual patient dosing of lenzilumab. Accordingly, there was no randomized control group in the Mayo Clinic program. We did not pre-select patients to receive lenzilumab through the compassionate use program and did not deny any requests for compassionate use. Mayo Clinic clinicians solely determined which patients for which they would request emergency IND authorization from the FDA.
| 29 |
The patients receiving lenzilumab were hospitalized and required supplemental oxygen as a result of COVID-19 pneumonia. They were also viewed as being at high risk of further disease progression. All patients had elevation in at least one inflammatory biomarker prior to receiving lenzilumab. All patients had at least one co-morbidity associated with poor outcomes in COVID-19 and several patients had multiple co-morbidities: 58% had diabetes mellitus, 58% had hypertension, 58% had underlying lung diseases, 50% were obese (defined as a BMI greater than 30), 17% had chronic kidney disease and 17% had coronary artery disease. The median age was 65 years.
Patients receiving lenzilumab showed rapid clinical improvement with an 80% relative reduction in the risk of progressing to IMV and/or death, a median time to median time to discharge of five days versus 11 days for controls. Patients also demonstrated rapid improvement in oxygenation and inflammatory cytokines consistent with the improved clinical outcomes.
As a Sequenced Therapy in Combination with CD19 Targeted CAR-T Therapies
Our current clinical and regulatory development plan is focused on the Kite Agreement we executed in May 2019. Pursuant to the Kite Agreement, the parties have agreed to conduct a multi-center Phase 1b/2 study (ZUMA-19) of lenzilumab with Kite’s Yescarta in patients with relapsed or refractory B-cell lymphoma, including DLBCL which is currently enrolling. Kite is the sponsor of ZUMA-19 and is responsible for its conduct. The primary objective of ZUMA-19 is to determine the effect of lenzilumab on the safety of Yescarta. In addition, efficacy and healthcare resource utilization will be assessed. On June 30, 2020, we announced that the first patient had been infused in the ZUMA-19 study.
Kite’s Yescarta is one of four CAR-T therapies that have been approved by FDA and is the leading CAR-T by revenue. Our collaboration with Kite is the only current clinical collaboration that is enrolling patients with the potential to improve both the safety and efficacy of CAR-T therapy. The Kite Agreement is non-exclusive. Depending upon FDA feedback, and the number of patients enrolled, we believe ZUMA-19 may serve as the basis for registration for lenzilumab.
As a Companion to Allogeneic HSCT
We believe lenzilumab has potential to prevent or reduce GvHD in allogeneic HSCT, thereby making allogeneic HSCT safer as a potentially curative therapy for patients with hematological cancers. In July 2019, we entered into an exclusive worldwide license agreement with UZH. Under the Zurich Agreement, we have in-licensed certain technologies that we believe may be used to prevent GvHD through GM-CSF neutralization, thereby expanding our development platform to include improving the safety and effectiveness of allogeneic HSCT. A study published in ‘blood advances’, an official journal of the American Society of Hematology, suggests that neutralizing or blocking GM-CSF may limit or prevent GvHD in the gastrointestinal tract (Gartlan, K., et al, October 8, 2019, vol. 3, no.19).
We plan to study lenzilumab as a companion to allogeneic HSCT for patients with hematological cancers. We are collaborating with IMPACT, a clinical trial partnership of 22 transplant centers in the United Kingdom, in planning a potential randomized, placebo controlled, Phase 2/3 study focused on early intervention with lenzilumab in patients at high risk or intermediate risk for steroid refractory acute GvHD based on specific biomarkers. The goal of the trial, as it is currently contemplated, would be to determine the efficacy and safety of lenzilumab in reducing non-relapse mortality at six months.
Other Inflammatory Conditions
Previous clinical studies of lenzilumab include a repeat-dose, Phase 2 clinical trial of lenzilumab in rheumatoid arthritis (“RA”) with the inclusion of a run-in portion. On completing the run-in portion of this trial, we reassessed the increasingly competitive RA market and chose to redirect our study of lenzilumab to other areas given the competitive intensity and diminishing levels of unmet need in RA relative to some other medical areas. In addition to the 520 patients enrolled in the COVID-19 study and an expected 200 patients in the ACTIV-5 study, we have generated tolerability data in 125 patients in various clinical studies, including in asthma and CMML.
| 30 |
Gene-edited CAR-T Therapies using GM-CSF Gene Knockout
We believe that our GM-CSF knockout gene-editing CAR-T platform has the potential to create next-generation CAR-T therapies that improve the efficacy and safety profile of CAR-T therapy via gene-edited CAR-T cells which can be engineered to lack the ability to produce GM-CSF, thereby avoiding any efficacy/toxicity linkage and potentially preserving and improving upon the benefits of the CAR-T therapy while altogether avoiding its serious and potentially life-threatening side-effects.
We are advancing our GM-CSF knockout gene-editing CAR-T platform through the Mayo Agreement that we entered into in June 2019 with the Mayo Foundation. Under the Mayo Agreement, we have in-licensed certain technologies that we believe may be used to create CAR-T cells lacking GM-CSF expression through various gene-editing tools including CRISPR-Cas9. The Mayo Agreement broadened our leadership position in the GM-CSF neutralization space and expanded our discovery platform aimed at improving CAR-T therapy to include gene-edited CAR-T cells.
Preclinical data indicates that GM-CSF knockout CAR-T cells show improved overall survival compared to GM-CSF-expressing CAR-T cells, in addition to the expected benefits of reduced serious side-effects associated with CAR-T therapy. We are establishing a platform of next-generation combinatorial gene knockout CAR-T cells that have potential to be applied across both autologous and allogeneic approaches and we are also investigating multiple CAR-T cell designs using precise dual and triple gene editing to significantly enhance the anti-tumor activity while simultaneously preventing CAR-T therapy induced toxicities. Through targeted gene expression and modulating cytokine activation signaling, we may be able to increase the proportion of fitter T-cells produced during expansion, increase the proliferative potential, and inhibit activation-induced cell death, thereby improving the cancer killing activity of our engineered CAR-T cells thereby making them more effective and safer in the treatment of cancers. Preclinical data indicates that CAR-T cells express GM-CSF and signal through GM-CSF receptors upon activation in an autocrine fashion. GM-CSF knockout CAR-T cells are not able to signal through this pathway which results in a gene expression profile distinct from GM-CSF-expressing CAR-T cells after in vitro expansion. This includes lower levels of Fas expression which may indicate a less exhausted state of the CAR-T cells. These data were presented at the 2019 ASH annual meeting.
We continue to explore exhaustion markers of GM-CSF knockout CAR-T cells relative to GM-CSF-expressing CAR-T cells and possible implications for improved safety and efficacy. We plan to continue development of this technology in combination approaches that could add to the observed efficacy benefits of current generation CAR-T products.
Ifabotuzumab
Ifabotuzumab is a Humaneered monoclonal antibody, formerly referred to as KB004, which targets EphA3, and in which the antibody carbohydrate chains lack fucose, thereby enhancing the targeted cell-killing activity of the antibody. We believe that ifabotuzumab has the potential for treating solid tumors, hematologic malignancies and serious pulmonary conditions. In 2006, we entered into a license agreement with Ludwig Institute of Cancer Research (“LICR”) pursuant to which LICR granted certain exclusive rights to the ifabotuzumab prototype (referred to as IIIA4) as well as EphA3 intellectual property.
Ifabotuzumab binds to EphA3, which plays an important role in cell positioning and tissue organization during fetal development but is not thought to be expressed nor play a significant role in healthy adults. EphA3 is a tyrosine kinase receptor, aberrantly expressed on the tumor vasculature and tumor stroma in several solid tumors including melanoma, breast cancer, small and non-small cell lung cancer (“SCLC” and “NSCLC”), colorectal cancer, gastric cancer, renal cancer, GBM, and prostate cancer, making it an attractive target for a range of cancers. Publications related to certain cancers have indicated that EphA3 tumor cell expression correlates with cancer growth and a poor prognosis. EphA3 is overexpressed in GBM and, in particular, in the most aggressive mesenchymal subtype. Importantly, EphA3 is highly expressed on the tumor-initiating cell population in glioma and appears to be critically involved in maintaining tumor cells in a less differentiated state by modulating mitogen-activated protein kinase signaling. EphA3 knockdown or depletion of EphA3-positive tumor cells may reduce tumorigenic potential to a degree comparable to treatment with a therapeutic radiolabeled EphA3-specific monoclonal antibody. We believe EphA3 is a functional, targetable receptor in GBM and other solid tumors. A study published in December 2018 in ‘Cancers’ showed that an antibody drug conjugate (“ADC”) comprising IIIA4 (a predecessor monoclonal antibody and prototype for ifabotuzumab) showed significant survival benefit in mice with GBM.
| 31 |
Anti-EphA3 treatment has shown encouraging preclinical results in multiple experiment types, including patient primary tumor cell assays, colony forming assays, and xenograft mouse models. Upon binding to EphA3, ifabotuzumab causes cell killing to occur either through antibody-dependent, cell-mediated cytotoxicity or through direct apoptosis, and in the case of tumor neovasculature, through cell rounding and blood vessel disruption. Given the expression pattern of EphA3 in multiple tumor types, ifabotuzumab may have the potential to kill cancer cells and the tumor stem cell microenvironment, providing for long-term responses while sparing normal cells.
We completed the Phase 1 dose escalation portion of a Phase 1/2 clinical trial of ifabotuzumab in multiple hematologic malignancies for which the preliminary results were published in the journal Leukemia Research in 2016. We have conducted a Phase 1/2 trial for ifabotuzumab in multiple hematologic malignancies. The most common adverse event attributed to ifabotuzumab in this trial was infusion reactions (chills, fever, nausea, hypertension, and rapid heart rate) which is an expected safety finding based on the mechanism of action. The majority of infusion reactions were mild-to-moderate in severity and resolved with temporary stoppage of infusion and/or use of medications to treat symptoms. In 2014, we completed the Phase 1 dose escalation portion of our study, primarily treating patients with AML as well as patients with MDS and myelofibrosis (“MF”).
A Phase 1 safety and imaging trial of radio-labeled ifabotuzumab in recurrent glioblastoma multiforme, a particularly aggressive and deadly form of brain cancer, has fully enrolled at two centers in Australia, the Olivia Newton-John Cancer Research Institute (“ONJCRI”) in Melbourne and the Queensland Institute for Medical Research in Brisbane and interim data have been presented at both American Association for Cancer Research (“AACR”) and Society for Neuro-Oncology (“SNO”) in 2019, with final data expected to be presented in 2021. The lead investigators at the ONJCRI, are also evaluating an antibody-drug conjugate (“ADC” or “ADCs”) based on ifabotuzumab in solid tumor models. The Phase 1 clinical trial has enrolled all 12 patients. Preliminary imaging data reported at AACR and at SNO demonstrated that administration of ifabotuzumab resulted in rapid, specific targeting of GBM tumors in all patients. Whole body bio-distribution imaging demonstrated no normal tissue uptake of the antibody. Post-treatment MRI scans showed predominant T2/Flair changes which were consistent with treatment effect on tumor vasculature. We continue to explore partnering opportunities and seek governmental and institutional grants to facilitate the further development of ifabotuzumab in a range of cancer types.
We are in discussions with separate and various parties and may initiate partnerships to pursue some of the following activities:
| · | Complete the on-going clinical study and preclinical studies with various ADCs that are based on ifabotuzumab (in partnership with leading centers in Australia); and |
| · | Develop bi-specific antibodies based on ifabotuzumab. |
Centers in Australia have worked independently on IIIA4, the murine antibody parent of ifabotuzumab, as an ADC in mice and a December 2018 publication in the journal ‘Cancers’ showed that in mice engrafted with GBM, treatment with an ADC based on IIIA4 showed significantly improved survival.
| 32 |
HGEN005
HGEN005 is a Humaneered monoclonal antibody, formerly referred to as KB005, which targets the EMR1, and in which the antibody carbohydrate chains lack fucose, thereby enhancing the targeted cell-killing activity of the antibody.
A major limitation of current eosinophil-targeted therapies is incomplete depletion of tissue eosinophils and/or lack of cell selectivity. Eosinophils are a type of white blood cell. If too many eosinophils are produced in the body, chronic inflammation and tissue and organ damage may result. The origin and development of eosinophilic disorders is mostly due to eosinophils infiltrating tissue. EMR1 is expressed exclusively on eosinophils, making it an ideal target for the treatment of eosinophilic disorders. Regardless of the eosinophilic disorder, mature eosinophils express EMR1 in tissue, blood and bone marrow in patients with eosinophilia. We believe that because of its high selectivity, HGEN005 has significant potential to treat serious eosinophil diseases.
In preclinical work, HGEN005’s anti-EMR1 activity resulted in dramatically enhanced NK killing of eosinophils from normal and eosinophilic donors and also induced a rapid and sustained depletion of eosinophils in a non-human primate model without any clinically significant adverse events. We have engaged with NIH to discuss expanding the initial work they have conducted utilizing HGEN005 and discussions are underway with a leading center in the US to perform the IND-enabling work.
Our Humaneered Technology
Our proprietary and patented Humaneered technology platform is a method for converting existing antibodies (typically murine) into engineered, high-affinity human antibodies designed for therapeutic use, particularly for chronic conditions. We have developed or in-licensed targets or research (mouse) antibodies, typically from academic institutions, and then applied our Humaneered technology to them. Lenzilumab, ifabotuzumab and HGEN005 are Humaneered antibodies. In aggregate, our Humaneered antibodies have been tested clinically in more than 200 patients with no evidence of serious immunogenicity. Our Humaneered antibodies are closer to human antibodies than chimeric or conventionally humanized antibodies and have a high affinity for their target but low immunogenicity. Specifically, our Humaneered technology generates an antibody from an existing antibody with the required specificity as a starting point and, we believe, provides the following additional advantages:
| · | high potency; |
| · | slow off-rate; |
| · | high solubility; |
| · | retention of identical target epitope specificity and generation of higher affinity antibodies; |
| · | high antibody expression yields; |
| · | attractive cost-of-goods; |
| · | physiochemical properties that facilitate process development and formulation; |
| · | lack of aggregation at high concentration; |
| · | very-near-to-human germ line sequence (which may mean they are less likely to induce an inappropriate immune response when used chronically, which has proven to be the case in clinical studies); and |
| · | an optimized antibody processing time of three to six months. |
As we are focused on progressing our current portfolio of antibodies through clinical development and out-licensing, we are not currently dedicating additional resources to the research or development of additional Humaneered antibodies other than our existing portfolio of lenzilumab, ifabotuzumab and HGEN005.
Intellectual Property
Intellectual property (IP) is an important part of our strategy and has been a key focus area for the Company. We have and continue to file aggressively on our own inventions and in-license intellectual property and technology as it relates to our therapeutic interests. We believe that we are a leader in the field of GM-CSF neutralization to ameliorate cytokine storm as it relates to COVID-19, CAR-T and GvHD.
| 33 |
Licensing and Collaborations
CRADA
On November 5, 2020, we and the Department of Defense (DoD) Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense (JPEO-CBRND or JPEO) entered into a Cooperative Research and Development Agreement (“CRADA”) in collaboration with the Biomedical Advanced Research and Development Authority (BARDA), part of the Office of the Assistant Secretary for Preparedness and Response (ASPR) at the U.S. Department of Health and Human Services (HHS), in support of Operation Warp Speed (“OWS”), to assist in the development of our lead product candidate, lenzilumab, in advance of a potential EUA for COVID-19.
On January 22, 2021, we announced an expansion of the CRADA that the company had previously entered with JPEO on November 5, 2020, was subsequently co-signed by BARDA. This provides Humanigen with access to manufacturing capacity reserved by BARDA for fill-finish product to accelerate the drug product manufacturing of lenzilumab.
Pursuant to the CRADA, we have been provided access to a full-scale, integrated team of OWS manufacturing, and regulatory subject matter experts, leading decision makers and statistical support in anticipation of applying for EUA and subsequently a BLA for lenzilumab as a potential treatment for COVID-19. The CRADA also provides that OWS regulatory experts will work with us on FDA communications, meetings and regulatory filings. The CRADA aims to support the lenzilumab Phase 3 clinical trial output, focusing on efficiently generating an EUA and BLA submission. In addition to providing access under EUA, a goal of the CRADA is to ensure lenzilumab receives the benefits provided by Public Law 115-92.
Clinical Trial Agreement with the National Institute of Allergy and Infectious Diseases
On July 24, 2020, we entered into a clinical trial agreement (the “Clinical Trial Agreement”) with NIAID, part of the National Institutes of Health, which is part of the United States Government Department of Health and Human Services, as represented by the Division of Microbiology and Infectious Diseases. Pursuant to the Clinical Trial Agreement, lenzilumab will be an agent to be evaluated in the NIAID-sponsored ACTIV-5/Big Effect Trial (BET) in hospitalized patients with COVID-19.
ACTIV-5 will evaluate the combination of lenzilumab and Gilead’s investigational antiviral, remdesivir, on treatment outcomes versus placebo and remdesivir in hospitalized COVID-19 patients. The trial is expected to enroll 100 patients in each arm of the study with an interim analysis for efficacy after 50 patients have been enrolled in each arm.
Pursuant to the Clinical Trial Agreement, NIAID will serve as sponsor and will be responsible for supervising and overseeing ACTIV-5. The Company will be responsible for providing lenzilumab to NIAID without charge and in quantities to ensure a sufficient supply of lenzilumab.
The University of Zurich
On July 19, 2019, we entered into the Zurich Agreement with UZH. Under the Zurich Agreement, we have in-licensed certain technologies that we believe may be used to prevent GvHD through GM-CSF neutralization. The Zurich Agreement covers various patent applications filed by UZH which complement and broaden our position in the application of GM-CSF neutralization and expands our development platform to include improving allogeneic HSCT.
The Zurich Agreement required an initial one-time payment of $100,000, which we paid to UZH on July 29, 2019. The Zurich Agreement also requires the payment of annual license maintenance fees, as well as milestones and royalties upon the achievement of certain regulatory and commercialization milestones.
| 34 |
The term of the Zurich Agreement runs from July 19, 2019 until the expiration date of the longest-lived patent right, subject to termination rights specified in the agreement.
The Mayo Foundation for Medical Education and Research
On June 19, 2019, we entered into the Mayo Agreement with the Mayo Foundation. Under the Mayo Agreement, we have in-licensed certain technologies that we believe may be used to create CAR-T cells lacking GM-CSF expression through various gene-editing tools including CRISPR-Cas9. The license covers various patent applications and know-how developed by Mayo Foundation in collaboration with us. These licensed technologies complement and broaden our position in the GM-CSF neutralization space and expand our discovery platform aimed at improving CAR-T to include gene-edited CAR-T cells. The Mayo Agreement requires the payment of milestones and royalties upon the achievement of certain regulatory and commercialization milestones.
The term of the Mayo Agreement runs from May 29, 2019 until the later of: (i) the expiration date of the last to expire of the patent rights granted; or (ii) the conclusion of a period of ten (10) years from the date of the first commercial sale, subject to termination rights specified in the agreement.
Kite
On May 30, 2019, we entered into the Kite Agreement. Pursuant to the Kite Agreement, the parties agreed to conduct a multi-center Phase 1b/2 study (ZUMA-19) of lenzilumab with Kite’s Yescarta in patients with relapsed or refractory B-cell lymphoma. The primary objective of ZUMA-19 is to determine the effect of lenzilumab on the safety of Yescarta. Various other important parameters, including efficacy and healthcare resource utilization, will also be measured. Kite is the sponsor of ZUMA-19 and responsible for its conduct. On June 30, 2020, we announced that the first patient had been infused in the ZUMA-19 study.
The Kite Agreement provides that we and Kite will split only the out-of-pocket costs incurred in conducting ZUMA-19, including third-party expenses incurred in accordance with a mutually agreed budget. We currently project we will be responsible for an aggregate of up to approximately $8 million in out-of-pocket costs, assuming up to a total of 72 patients are recruited for ZUMA-19 as a multi-center Study. Each party will otherwise be responsible for its own internal costs, including internal personnel costs, incurred in connection with ZUMA-19.
For more information about the Kite Agreement, see “Business—Kite Collaboration.”
The Ludwig Institute for Cancer Research
In 2004, we entered into a license agreement with the LICR pursuant to which LICR granted to us an exclusive license for intellectual property rights and materials related to chimeric anti-GM-CSF antibodies that formed the basis for lenzilumab. Under the agreement, we were granted an exclusive license to develop antibodies related to LICR’s antibodies against GM-CSF. We are responsible for using commercially reasonable efforts to research, develop, and sell lenzilumab. We pay LICR a quarterly license fee and are obligated to pay to LICR a royalty from 1.5% to 3% of net sales of licensed products, subject to certain potential offsets and deductions. Our royalty obligation applies on a country-by-country and licensed product-by-licensed product basis, and will begin on the first commercial sale of a licensed product in a given country and end on the later of the expiration of the last to expire patent covering a licensed product in a given country (which in the US is currently expected in 2029 for the composition of matter and 2038 for methods of use in CAR-T) or 10 years from first commercial sale of such licensed product in the country. We must also pay to LICR a certain percentage of sublicensing revenue received by us. Payments made to LICR under this license for the twelve months ended December 31, 2020 and 2019 were $0.1 million and $0.1 million, respectively.
| 35 |
Other License Agreements
LICR and ifabotuzumab
In 2006, we entered into a license agreement with LICR pursuant to which LICR granted to us certain exclusive rights to the ifabotuzumab prototype (IIIA4) which targets EphA3 and EphA3-related intellectual property. Under the agreement, we obtained rights to develop and commercialize products made through use of licensed patents and any improvements thereto, including human or Humaneered antibodies that bind to or modulate EphA3. We paid LICR an upfront option fee of $0.05 million and a further $0.05 million upon our exercise of the option for the exclusive license outlined above. We are responsible for contingent milestone payments of less than $2.5 million and royalties of 3% of net sales subject to certain potential offsets and deductions. In addition, we are obligated to pay to LICR a percentage of certain payments we receive from any sublicensee in consideration for a sublicense. Our royalty obligation exists on a country-by-country and licensed product-by-licensed product basis, which will begin on the first commercial sale and end on the later of the expiration of the last to expire patent covering such licensed product in such country, which in the US is currently expected in 2031, or 10 years from first commercial sale of such licensed product in such country.
BioWa and Lonza
In 2010, we entered into a license agreement with BioWa, Inc. (“BioWa”), and Lonza Sales AG (“Lonza”) pursuant to which BioWa and Lonza granted us a non-exclusive, royalty-bearing, sub-licensable license under certain know-how and patents related to antibody expression and antibody-dependent cellular cytotoxicity enhancing technology using BioWa and Lonza’s Potelligent® CHOK1SV technology. This technology is used to enhance the cell killing capabilities of antibodies and is currently used by us in connection with our development of ifabotuzumab. Under this agreement, we owe annual license fees, milestone payments in connection with certain regulatory and sales milestones and royalties in the low single digits on net sales of products developed under the agreement. The agreement expires upon the expiration of royalty payment obligations under the agreement, is terminable at will by us upon written notice, is terminable by BioWa and Lonza if we challenge or otherwise oppose any licensed patents under the agreement and is terminable by either party upon the occurrence of an uncured material breach or insolvency.
Patents and Trade Secrets
We use a combination of patent, trade secret and other intellectual property protections to protect our product candidates and platforms. We will be able to protect our product candidates from unauthorized use by third parties only to the extent they are covered by valid and enforceable patents or to the extent our technology is effectively maintained as trade secrets. Intellectual property is an important part of our strategy. We have and continue to file aggressively on our own inventions and in-license intellectual property and technology as it relates to our therapeutic interests. Our success will depend in part on our ability to obtain, maintain, defend and enforce patent rights for and to extend the life of patents covering lenzilumab, ifabotuzumab, HGEN005, our Humaneered technology, and our GM-CSF gene-editing CAR-T platform technologies, to preserve trade secrets and proprietary know how, and to operate without infringing the patents and proprietary rights of third parties. We actively seek patent protection, if available, in the US and select foreign countries for the technology we develop. We have 111 registered patents, including 16 registered in the US and 95 registered in foreign countries. Of the 111 registered patents, 87 are owned by us, 9 are owned jointly with a third party and 15 are exclusively licensed from a third party. We also have 53 patent applications pending globally, of which 21 are owned by us, 7 are owned jointly with a third party and 25 are exclusively licensed from a third party.
Using our Humaneered technology, we have developed and own two composition of matter US patents covering lenzilumab and related anti-GM-CSF antibodies that provide patent protection through April 2029, a granted composition of matter patent in 15 European countries and certain foreign countries, and have two U.S. patents and six additional pending patent applications in the US and one PCT international patent application covering various methods of treatment, including in the CAR-T space covering a broad and comprehensive range of approaches to neutralizing GM-CSF, including the use of GM-CSF k/o CAR-T cells, which, if granted, are expected to confer protection to at least October 2038. We also have two currently pending foreign patent applications that are jointly owned with a third party for anti-EphA3 antibodies and their use, and we developed and own an issued US composition of matter patent covering ifabotuzumab and related anti-EphA3 antibodies, which is currently expected to expire in 2031, and own 22 foreign patents to anti-EphA3 antibodies and therapeutic uses, in addition to owning four US patents to therapeutic methods using anti-EphA3 antibodies and seven foreign patents owned jointly with a third party covering methods of treatment with anti-EphA3 antibodies. The two US and 26 foreign patents to our Humaneered technology cover methods of producing human antibodies that are very specific for target antigens using only a small region from mouse antibodies.
| 36 |
We cannot be certain that any of our pending patent applications, or those of our licensors, will result in issued patents. In addition, because the patent positions of biopharmaceutical companies are highly uncertain and involve complex legal and factual questions, the patents we own and license, or any further patents we may own or license, may not prevent other companies from developing similar or therapeutically equivalent products, even though we may be able to prevent their commercial use without our permission if our intellectual property allows for such limitations. Patents also will not protect our products if competitors devise ways of making or using these products without legally infringing our patents. We cannot be assured that our patents will not be challenged by third parties or that we will be successful in any defense we undertake.
In addition, changes in patent laws, rules or regulations or in their interpretations by the courts may materially diminish the value of our intellectual property or narrow the scope of our patent protection, which could have a material adverse effect on our business and financial condition. However, prospective partners may have to license or otherwise come to an agreement with us if they wish to use our products and those products and methods of use of such products have issued patents in those territories.
We also rely on trade secrets, technical know-how and continuing innovation to develop and maintain our competitive position. We seek to protect our proprietary information by requiring our employees, consultants, contractors, outside scientific collaborators, and other advisors to execute non-disclosure and confidentiality agreements and our employees to execute assignment of invention agreements to us on commencement of their employment. Agreements with our employees also prevent them from bringing any proprietary rights of third parties to us. We also require confidentiality or material transfer agreements from third parties that receive our confidential data or materials.
Manufacturing and Raw Materials
We outsource all development activities, including the development of formulation prototypes. We do not have, nor do we plan to have, any manufacturing facilities. Instead, we have adopted a manufacturing strategy of contracting with third parties for the manufacture of drug substance and product. Additional contract manufacturers are used to fill, label, package, and distribute investigational drug products. The use of contract manufacturers can be expensive, complicated and time consuming and could delay clinical trials, drug approval and potential product launch. We believe our outsourcing approach allows us to maintain a leaner staffing approach and a more flexible infrastructure, allowing us to focus our expertise on developing our products. It does however mean that we have to carefully plan the availability of manufacturing ‘slots’ and the availability of drug for investigation in preclinical and clinical trials. We have been proactive and aggressive in pursuing manufacturing slots for lenzilumab in connection with our clinical trial programs and expect to commit the necessary resources to ensure that our manufacturing capabilities do not hinder our ability to attain regulatory approvals for its use. During the latter half of 2020, and continuing into 2021, it has become more challenging and expensive to obtain these slots due to increased demand for production at our CMOs, many of which also manufacture vaccines and other therapeutics. Even after being reserved, the manufacturing slots for which we contract are subject to risk of displacement as mandated from Rated Orders issued under the Defense Production Act (DPA). This displacement risk has caused us to expend significant efforts to manage our supply chain. Despite those efforts, we have experienced and expect to continue to experience shortages of raw materials and critical components necessary for our manufacturing processes.
| 37 |
During the year ended December 31, 2020, we entered into agreements with several contract manufacturing organizations (“CMOs”) for the manufacture of bulk drug substance (“BDS”) for our lenzilumab clinical trial activities in COVID-19 as well as for a potential launch of lenzilumab in anticipation of an EUA in 2021. We have also entered into agreements for filling, finishing and packaging of the drug. These agreements represent large financial commitments, including upfront amounts prior to commencement of manufacturing and progress payments through the course of the manufacturing process and include payments for the initial technology transfer. A summary of the various CMOs with whom we have contracted for services is as follows:
| Contract Manufacturer | Services |
| Catalent Pharma Solutions, LLC | Bulk drug substance manufacture, fill and finish, clinical packaging |
| Lonza Sales AG | Bulk drug substance manufacture |
| Patheon Biologics LLC | Bulk drug substance manufacture |
| Avid BioServices, Inc. | Bulk drug substance manufacture |
| Emergent BioSolutions, Inc. | Fill and finish |
| Ajinomoto Bio-Pharma Services | Fill and finish |
| Bryllan LLC | Fill and finish |
| PCI Pharma Services | Commercial packaging |
Sales and Marketing
We outsource our sales and marketing infrastructure necessary to market and sell our products, if authorized or approved. The establishment of a sales and marketing operation can be expensive, complicated and time consuming and could delay any potential product launch. As our drug candidates progress, we may also seek strategic alliances and partnerships with third parties including those with existing infrastructure and with government bodies. The following summarizes our important sales and marketing arrangements.
Eversana Agreement
On January 10, 2021, we announced that we had entered into a master services agreement (the “Eversana Agreement”) with Eversana Life Science Services, LLC (“Eversana”) pursuant to which Eversana will provide us multiple services from its integrated commercial platform in preparation for the potential commercialization of lenzilumab, for treatment of hospitalized and hypoxic COVID-19 patients.
Under the Eversana Agreement, Eversana will serve as our end-to-end commercial partner, providing us with a full suite of services in connection with the potential launch of lenzilumab. Eversana services initially will comprise marketing, market access and field solution services, and may expand to other areas including patient services, compliance services, professional services, data and analytics, and health economics outcome research, as may be negotiated by the parties and set forth in statements of work delivered in accordance with the Eversana Agreement.
We will pay Eversana fees and reimburse it for its expenses in performing the services as established in applicable statements of work. Eversana has agreed to defer its fees pending our receipt of an EUA from the FDA for lenzilumab for hospitalized and hypoxic COVID-19 patients. If we have not received an EUA by April 30, 2021 or such later date as to which the parties otherwise may agree, either party may terminate the Agreement immediately; Eversana will not receive the deferred fees upon such termination.
The Eversana Agreement provides for a one-year term and will renew for subsequent one-year terms unless either party provides a notice of non-renewal. After the first year, we may terminate the Eversana Agreement upon advance written notice to Eversana. The Eversana Agreement contains customary provisions allowing either party to terminate the Eversana Agreement as a result of certain changes in law and material breaches and certain insolvency events by or relating to the other party.
The Eversana Agreement imposes customary mutual obligations on the parties to protect and not disclose the confidential information and intellectual property of the other, and contains insurance, non-solicitation, indemnification and limitation of liability provisions customary for service contracts of this type.
Cardinal Agreement
On September 9, 2020 we entered into an agreement with Cardinal Health (“Cardinal”) to provide shipping, receiving, returns, warehousing, invoicing, collections, data collection and exchange, as well as other third-party logistics services for lenzilumab in preparation for receipt of an EUA for COVID-19. The agreement is for a three-year term and is subject to annual renewal thereafter. It is anticipated that initially Cardinal will take title of the product while we finalize US state licensing. Cardinal will receive monthly fees for services as well as a percentage of the Wholesaler Acquisition Cost (WAC) while operating in the title model.
| 38 |
Competition
We compete in an industry characterized by rapidly advancing technologies, intense competition, a changing regulatory and legislative landscape and a strong emphasis on the benefits of intellectual property protection and regulatory exclusivities. Our competitors include pharmaceutical companies, other biotechnology companies, academic institutions, government agencies and other private and public research organizations. We compete with these parties to develop potential biologic therapies to make CAR-T therapy and allogeneic HSCT safer and more effective and to develop a potential treatment for hematologic cancers, in addition to recruiting highly qualified personnel. Our product candidates, if successfully developed and approved, may compete with established therapies, with new treatments that may be introduced by our competitors, including competitors relying to a large extent on our drug approvals or on our biologics approvals, or with generic copies of our product approved by FDA, as bio-similars, referencing our drug products. Many of our potential competitors have substantially greater scientific, research, and product development capabilities, as well as greater financial, marketing, sales and human resources capabilities than we do.
In addition, many specialized biotechnology firms have formed collaborations with large, established companies to support the research, development and commercialization of products that may be competitive with ours. Accordingly, our competitors may be more successful with respect to their products than we may be in developing, commercializing, and achieving widespread market acceptance for our products. In addition, our competitors’ products may be more effective or more effectively marketed and sold than any treatment we or our development partners may commercialize and may render our product candidates obsolete or non-competitive before we can recover the expenses related to developing and supporting the commercialization of any of our product candidates. Developments by competitors may render our product candidates obsolete or noncompetitive. After one of our product candidates is approved, FDA may also approve a generic version with the same or similar dosage form, safety, strength, route of administration, quality, performance characteristics and intended use as our product. These bio-similar equivalents would be less costly to bring to market and could generally be offered at lower prices, thereby limiting our ability to gain or retain market share. However, our product candidates are all biologics and, as such, would benefit from 12 years market exclusivity from first licensure.
The acquisition or licensing of pharmaceutical products is also very competitive, and a number of more established companies, which have acknowledged strategies to in-license or acquire products, may have competitive advantages as may other emerging companies taking similar or different approaches to product acquisitions. The more established companies may have a competitive advantage over us due to their size, cash flows, institutional experience, and historical corporate reputation.
Lenzilumab and COVID-19 competition
There are currently no products approved by the FDA for the prevention of CRS/cytokine storm associated with COVID-19. There are numerous products currently in development for COVID-19 which can be broadly categorized as direct acting antivirals, immunomodulators, anticoagulants and other preventative strategies such as vaccines and neutralizing antibodies. Remdesivir (a direct acting antiviral) was approved by FDA for COVID-19. There is no evidence that remdesivir offers a mortality benefit and the largest remdesivir-based global study conducted by the World Health Organization failed to show benefit from remdesivir treatment. Other direct acting antiviral agents such as lopinavir/ritonavir and hydroxychloroquine (with or without a macrolide) have not demonstrated efficacy in randomized controlled trials. Other treatments available under EUA include baricitinib (Olumiant) in combination with remdesivir (Veklury), bamlanivimab alone and with Etesevimab, and REGEN-COV (casirivimab and imdevimab). Published data to date from randomized controlled clinical trials in the setting of COVID-19 and the two leading IL-6 inhibitors, Actemra (tocilizumab) and Kevzara (sarilumab) has been mixed. Recently published results from The Randomised Evaluation of COVID-19 Therapy (RECOVERY) trial have demonstrated that an anti-inflammatory treatment, tocilizumab, reduces the risk of death when given to hospitalized patients with severe COVID-19. The study also showed that tocilizumab shortens the time until patients are successfully discharged from hospital and reduces the need for a mechanical ventilator. The relative reduction in mortality was 14% and the relative reduction in progressing to IMV or death was 15%. A total of 2,022 patients were randomly allocated to receive tocilizumab by intravenous infusion and were compared with 2,094 patients randomly allocated to usual care alone. 82% of patients were taking a systemic steroid such as dexamethasone. Treatment with tocilizumab significantly reduced deaths: 596 (29%) of the patients in the tocilizumab group died within 28 days compared with 694 (33%) patients in the usual care group (rate ratio 0·86; [95% confidence interval [CI] 0·77 to 0·96]; p=0·007), an absolute difference of 4%. Tocilizumab also increased the probability of discharge alive within 28 days from 47% to 54% (rate ratio 1·23, [95% CI 1·12 to 1·34], p<0·0001). These benefits were seen in all patient subgroups, including those requiring oxygen via a simple face mask through to those requiring mechanical ventilators in an intensive care unit. Among patients not on invasive mechanical ventilation when entered into the trial, tocilizumab significantly reduced the chance of progressing to invasive mechanical ventilation or death from 38% to 33% (risk ratio 0·85, [95% CI 0·78 to 0·93], p=0·0005). However, there was no evidence that tocilizumab had any effect on the chance of successful cessation of invasive mechanical ventilation. In June 2020, the RECOVERY trial found that the inexpensive and widely available steroid dexamethasone reduces death for patients with severe COVID-19. Dexamethasone became part of standard-of-care given to all such patients. In this trial, the benefits of tocilizumab were seen to be in addition to those of steroids.
| 39 |
In addition, IL-1 and IL-1 receptor inhibitors, respectively Ilaris (canakinumab) and Kineret (anakinra) have both failed to demonstrate benefit in Phase 3 studies, as has the oral JAK inhibitor Jakafi (ruxolitinib) and the BTK inhibitor Calquence (acalabrutinib). The anti-GM-CSF agent, otilimab failed in an 806-patient randomized controlled study sponsored by GlaxoSmithKline (“GSK”). In a post-hoc analysis of a subgroup of patients older than 70, there was an apparent benefit in the proportion of patients alive and free of respiratory failure at day 28 and mortality at day 60. A 350-patient follow-on Phase 3 study is planned to determine if the benefit in the patients older than 70 can be confirmed. It is unclear whether the anti-GM-CSF agents gimsilumab and namilumab are continuing development for COVID-19 and the primary endpoint for mavrilimumab for COVID-19 in a 40 patient study was recently missed, though there were some promising data in this small Phase 2 study.
Lenzilumab and CAR-T-related toxicities competition
Significant ongoing concerns for clinicians, caregivers, patients and FDA regarding CAR-T therapy include, durability of response, long-term outcomes, manufacturing process, time to delivery of active CAR-T product, serious and potentially life-threatening side-effects, namely NT and CRS, frequency and duration of hospitalization and ICU admission, health resource utilization, cost effectiveness and reimbursement. Kymriah, Yescarta, Tecartus and Brevanzi carry “Black Box” warnings in their labels for NT and CRS and are subject to a REMS program, such that on-going data has to be provided to FDA and CAR-T therapy can only be administered in strictly controlled environments at trained centers.
FDA approval of tocilizumab (anti-IL-6 receptor blocker, Genetech’s Actemra®) with or without high-dose corticosteroids, for the management of severe cases of CRS, was announced in conjunction with approval of Kymriah solely as part of its REMS program based on a retrospective analysis of 45 patients who received tocilizumab. Approval was subsequently also granted for tocilizumab as a treatment (not prevention) of moderate to severe CRS, despite the lack of an IND application, NDA or conduct of a prospective trial of tocilizumab in this setting. Only 20% of Kymriah or Yescarta patients treated with tocilizumab had resolution of signs and symptoms 6 days after onset of CRS and ~50% patients responded 11 days after onset of CRS. Tocilizumab is not approved for the prevention of CRS or for the prevention or treatment of NT. Tocilizumab is also not approved for the treatment of mild cases of CRS.
| 40 |
There are no FDA-approved therapies for the prevention of CAR-T therapy induced NT and CRS or for the treatment of CAR-T therapy induced NT. The CAR-T therapy-associated TOXicity Working Group currently recommends intensive monitoring, accurate grading and aggressive supportive care with the anti-IL-6 receptor blocker tocilizumab and/or high-dose corticosteroids. These agents are reserved only for the treatment of severe cases of CRS and are not approved for prevention. Since corticosteroids have been reported to suppress T-cell function and/or induce T-cell apoptosis, they may limit the effectiveness of CAR-T therapy and use has been generally limited to treatment of severe cases of CRS refractory to tocilizumab and severe cases of NT. Sometimes high-dose corticosteroids are used alongside tocilizumab. Tocilizumab has not been found to be effective in the prevention or management of CAR-T therapy induced NT. In fact, the prophylactic use of tocilizumab has been shown to increase both overall rates of NT and rates of severe NT with overall rates of CRS remaining unchanged in an expanded safety cohort from a CAR-T trial. As tocilizumab is an anti-IL-6 receptor blocker, serum IL-6 levels have been shown to increase shortly after administration of tocilizumab which may increase passive diffusion of IL-6 into the CNS and increase the risk of NT. In patients who received prophylactic tocilizumab, a 17-fold increase in CD14+ myeloid cells was seen in the CSF of patients who developed severe NT vs those who did not. A similar dynamic may occur with other cytokine receptor blocking monoclonal antibodies that are also being explored in this setting (e.g. anti-IL-1Ra, anti-GM-CSFRa). As the antibody directly binds the cytokine receptor, the receptor may get saturated causing the cytokine to get dislodged which leads to an initial increase in the level of the circulating cytokine. In this setting, elevated levels of pro-inflammatory cytokines can further propagate the inflammatory cascade and result in higher levels of NT and CRS when the receptor blocker is administered prophylactically. In addition, IL-1 levels have not been shown to correlate to CRS and NT in CAR-T clinical trials and there is no evidence in the clinical or preclinical setting available to support the use of GM-CSF receptor alpha blockade with CAR-T cell therapy. There are data that suggest that the mechanism of GM-CSF receptor alpha blockade may be interfere with CAR-T expansion and potentially efficacy. Our approach with lenzilumab is a different mechanism of action entirely and we have published data which shows expansion of CAR-T cells in animal models.
Other experimental approaches being explored include development of next-generation, CAR-T constructs, including introducing suicide genes into CAR-T cells using herpes simplex virus thymidine kinase (HSV-TK) or inducible caspase-9 (iCasp9) genes with “on / off” switches, incorporating a co-stimulatory molecule into T-cells, using RNA-guided DNA targeting technology or other epitope-based / gene-editing technology. While it is possible that some of these approaches could result in lower rates of NT and/or CRS, preliminary data suggests that improvements in the safety profile are associated with lower rates of efficacy and durable response. This is not surprising given the linkage that has been shown to exist between CAR-T cell expansion, efficacy and toxicity. In addition, an agent comprised of a mixture of single-stranded oligonucleotides that is purified from the intestinal mucosa of pigs, defibrotide (Jazz Pharmaceuticals, Defitelio®) is being evaluated to reduce NT although there is no preclinical data supporting its use in this indication and the mechanisms of action is uncertain. Defibrotide is approved for the treatment of severe veno-occlusive disease (VOD) in adults and children who have undergone chemotherapy and a stem-cell transplant and is associated with a 37% rate of hypotension, or low blood pressure. Hypotension is a hallmark of CRS, which occurs very frequently with patients receiving CAR-T therapy. There are also several other anti-GM-CSF compounds that are in various stages of development, however the focus of these compounds appears to be in chronic autoimmune disorders such as rheumatoid arthritis, ankylosing spondylitis, giant cell arteritis and related disorders.
Government Regulation
Drug Development and Approval in the U.S.
As a biopharmaceutical company operating in the U.S., we are subject to extensive regulation by FDA and by other federal, state, and local regulatory agencies. FDA regulates biological products such as our product candidates under the U.S. Federal Food and Cosmetic Act (FDCA), the Public Health Service Act (PHSA) and their implementing regulations. Under the PHSA, an FDA-approved biologics license application (BLA) is required to market a biological product, or biologic, in the U.S. These laws and regulations set forth, among other things, requirements for preclinical and clinical testing, development, approval, labeling, manufacture, storage, record keeping, reporting, distribution, import, export, advertising, and promotion of our products and product candidates. Biologics receive 12 years market exclusivity from first licensure.
Applications Relying on the Applicant’s Clinical Data
The approval process for a BLA under the PHSA requires the conduct of extensive studies and the submission of large amounts of data by the applicant. The biologic development process for these applications will generally include the following phases:
| 41 |
Preclinical Testing. Before testing any new biologic in human subjects in the U.S., a company must generate extensive preclinical data. Preclinical testing generally includes laboratory evaluation of product chemistry and formulation, as well as toxicological and pharmacological studies in several animal species to assess the quality and safety of the product. Animal studies must be performed in compliance with FDA’s Good Laboratory Practice (GLP) regulations and the U.S. Department of Agriculture’s Animal Welfare Act.
IND Application. Human clinical trials in the U.S. cannot commence until an IND application is submitted and becomes effective. A company must submit preclinical testing results to FDA as part of the IND application, and FDA must evaluate whether there is an adequate basis for testing the product in initial clinical studies in human volunteers. Unless FDA raises concerns, the IND application becomes effective 30 days following its receipt by FDA. Once human clinical trials have commenced, FDA may stop the clinical trials by placing them on “clinical hold” because of concerns about the safety of the product being tested, or for other reasons.
Clinical Trials. Clinical trials involve the administration of the product to healthy human volunteers or to patients, under the supervision of a qualified investigator. The conduct of clinical trials is subject to extensive regulation, including compliance with FDA’s bioresearch monitoring regulations and Good Clinical Practice (“GCP”) requirements, which establish standards for conducting, recording data from, and reporting the results of clinical trials. GCP requirements are intended to assure that the data and reported results are credible and accurate, and that the rights, safety, and well-being of study participants are protected.
Clinical trials must be conducted under protocols that detail the study objectives, parameters for monitoring safety, and the efficacy criteria, if any, to be evaluated. Each protocol is submitted to FDA as part of the IND application. In addition, each clinical trial must be reviewed, approved, and conducted under the auspices of an Institutional Review Board (IRB), at the institution conducting the clinical trial. Companies sponsoring the clinical trials, investigators, and IRBs also must comply with regulations and guidelines for obtaining informed consent from the study subjects, complying with the protocol and investigational plan, adequately monitoring the clinical trial, and timely reporting of adverse events. Foreign studies conducted under an IND application must meet the same requirements that apply to studies being conducted in the U.S. Data from a foreign study not conducted under an IND application may be submitted in support of a BLA if the study was conducted in accordance with GCP and FDA is able to validate the data. A study sponsor is required to publicly post certain details about active clinical trials and clinical trial results on the government website clinicaltrials.gov.
Human clinical trials are typically conducted in three sequential phases, although the phases may overlap with one another and, notably, in the CAR-T setting, FDA has granted approval to both currently marketed CAR-T therapies (Kite’s Yescarta, Tescartus and Novartis’s Kymriah) based on Phase 2 data and to tocilizumab without any prospective data in the CAR-T setting:
| · | Phase 1 clinical trials include the initial administration of the investigational product to humans, typically to a small group of healthy human subjects, but occasionally to a group of patients with the targeted disease or disorder. Phase I clinical trials generally are intended to determine the metabolism and pharmacologic actions of the product, the side-effects associated with increasing doses, and, if possible, to gain early evidence of effectiveness. |
| · | Phase 2 clinical trials generally are controlled studies that involve a relatively small sample of the intended patient population and are designed to develop data regarding the product’s effectiveness, to determine dose response and the optimal dose range, and to gather additional information relating to safety and potential adverse effects. |
| · | Phase 3 clinical trials are conducted after preliminary evidence of effectiveness has been obtained and are intended to gather additional information about safety and effectiveness necessary to evaluate the product’s overall risk-benefit profile, and to provide a basis for physician labeling. Generally, Phase 3 clinical development programs consist of expanded, large-scale studies of patients with the target disease or disorder to obtain statistical evidence of the efficacy and safety of the drug at the proposed dosing regimen, or with the safety, purity, and potency of a biological product. |
| 42 |
| · | The FDA does not always require every approved therapy to complete Phase 1 through 3 studies to secure approval. Approval through expedited routes is at the discretion of FDA. |
The sponsoring company, FDA, or the IRB may suspend or terminate a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk. Further, success in early-stage clinical trials does not assure success in later-stage clinical trials. Data obtained from clinical activities are not always conclusive and may be subject to alternative interpretations that could delay, limit, or prevent regulatory approval.
BLA Submission and Review
After completing clinical testing of an investigational biologic product, a sponsor must prepare and submit a BLA for review and approval by FDA. A BLA is a comprehensive, multi-volume application that must include, among other things, sufficient data establishing the safety, purity and potency of the proposed biological product for its intended indication. The application includes all relevant data available from pertinent preclinical and clinical trials, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls and proposed labeling. When a BLA is submitted, FDA conducts a preliminary review to determine whether the application is sufficiently complete to be accepted for filing. If it is not, FDA may refuse to accept the application for filing and may request additional information, in which case the application must be resubmitted with the supplemental information and review of the application is delayed.
FDA performance goals, which are target dates and other aspirational measures of agency performance to which the agency, Congressional representatives, and industry agree through negotiations that occur every five years, generally provide for action BLA applications within 10 months of submission or 10 months from acceptance for filing for an original BLA. FDA is not expected to meet those target dates for all applications, however, and the deadline may be extended in certain circumstances, such as when the applicant submits new data late in the review period. In practice, the review process is often significantly extended by FDA requests for additional information or clarification. In some circumstances, FDA can expedite the review of new biologics deemed to qualify for priority review, such as those intended to treat serious or life-threatening conditions that demonstrate the potential to address unmet medical needs. In those cases, the targeted action date is six months from submission, or for biologics constituting original biological products, six months from the date that FDA accepts the application for filing.
As part of its review, FDA may refer a BLA to an advisory committee for evaluation and a recommendation as to whether the application should be approved. Although FDA is not bound by the recommendation of an advisory committee, the agency usually has followed such recommendations. FDA may also determine that a REMS program is necessary to ensure that the benefits of a new product outweigh its risks, and that the product can therefore be approved. A REMS program may include various elements, ranging from a medication guide or patient package insert to limitations on who may prescribe or dispense the product, depending on what FDA considers necessary for the safe use of the product. Under the Pediatric Research Equity Act, a BLA must include an assessment, generally based on clinical study data, of the safety and effectiveness of the subject drug or biological product in relevant pediatric populations, unless the requirement is waived or deferred. Receiving orphan drug designation from FDA is one situation where such a requirement may be waived.
After review of a BLA, FDA may determine that the product cannot be approved, or may determine that it can only be approved if the applicant cures deficiencies in the application, in which case the agency endeavors to provide the applicant with a complete list of the deficiencies in correspondence known as a Complete Response Letter (“CRL”). A CRL may request additional information, including additional preclinical or clinical data. Even if such additional information and data are submitted, FDA may decide that the BLA still does not meet the standards for approval. Data from clinical trials are not always conclusive and FDA may interpret data differently than the sponsor interprets them. Additionally, as a condition of approval, FDA may impose restrictions that could affect the commercial success of a drug or require post-approval commitments, including the completion within a specified time period of additional clinical studies, which often are referred to as “Phase IV” studies or “post-marketing requirements.” Obtaining regulatory approval often takes several years, involves the expenditure of substantial resources, and depends on a number of factors, including the severity of the disease in question, the availability of alternative treatments, and the risks and benefits demonstrated in clinical trials.
| 43 |
Post-approval modifications to the drug or biologic product, such as changes in indications, labeling, or manufacturing processes or facilities, may require a sponsor to develop additional data or conduct additional preclinical or clinical trials. The proposed changes would need to be submitted in a new or supplemental BLA, which would then require FDA approval.
Regulatory Exclusivities
Biologics Price Competition and Innovation Act
In 2010, the Biologics Price Competition and Innovation Act (BPCIA) was enacted, creating an abbreviated approval pathway for biologic products that are biosimilar to, and possibly interchangeable with, reference biological products licensed under a BLA. The BPCIA also provides innovator manufacturers of original reference biological products 12 years of exclusive use from first licensure before biosimilar versions can be licensed in the US. This means that FDA may not approve an application for a biosimilar version of a reference biological product until 12 years after the date of first licensure of the reference biological product (with a potential six-month extension of exclusivity if certain pediatric studies are conducted and the results reported to FDA), although a biosimilar application may be submitted four years after the date of first licensure of the reference biological product. Additionally, the BPCIA establishes procedures by which the biosimilar applicant must provide information about its application and product to the reference product sponsor, and by which information about potentially relevant patents is shared and litigation over patents may proceed in advance of approval of the biosimilar product, although the interpretation of those procedures has been subject to litigation and appears to continue to evolve. The BPCIA also provides a period of exclusivity for the first biosimilar to be determined by FDA to be interchangeable with the reference product.
FDA approved the first biosimilar product under the BPCIA in 2015, and the agency continues to refine the procedures and standards it will apply in implementing this approval pathway. FDA has released guidance documents interpreting specific aspects of the BPCIA in the years since. We would expect lenzilumab, ifabotuzumab and HGEN005, as biologics, to each receive at least 12 years exclusivity from first licensure if they are approved.
Pediatric Studies and Exclusivity
Under the Pediatric Research Equity Act, a BLA must contain data adequate to assess the safety and effectiveness of the product for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. Sponsors must also submit pediatric study plans prior to the assessment data. Those plans must contain an outline of the proposed pediatric study or studies the applicant plans to conduct, including study objectives and design, any deferral or waiver requests and other information required by regulation. The applicant, the FDA, and the FDA’s internal review committee must then review the information submitted, consult with each other and agree upon a final plan. The FDA or the applicant may request an amendment to the plan at any time.
For products intended to treat a serious or life-threatening disease or condition, the FDA must, upon the request of an applicant, meet to discuss preparation of the initial pediatric study plan or to discuss deferral or waiver of pediatric assessments. In addition, FDA will meet early in the development process to discuss pediatric study plans with sponsors and FDA must meet with sponsors by no later than the end-of-Phase I meeting for serious or life-threatening diseases and by no later than ninety (90) days after FDA’s receipt of the study plan.
The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after licensing of the product for use in adults, or full or partial waivers from the pediatric data requirements. Additional requirements and procedures relating to deferral requests and requests for extension of deferrals are contained in Food and Drug Administration Safety and Innovation Act (FDASIA). Unless otherwise required by regulation, the pediatric data requirements do not apply to products with orphan designation.
| 44 |
The FDA Reauthorization Act of 2017 established new requirements to govern certain molecularly targeted cancer indications. Any company that submits a BLA three years after the date of enactment of that statute must submit pediatric assessments with the BLA if the biologic is intended for the treatment of an adult cancer and is directed at a molecular target that FDA determines to be substantially relevant to the growth or progression of a pediatric cancer. The investigation must be designed to yield clinically meaningful pediatric study data regarding the dosing, safety and preliminary potency to inform pediatric labeling for the product. Deferrals and waivers as described above are also available.
Pediatric exclusivity is another type of exclusivity in the U.S. and, if granted, provides for the attachment of an additional six months of marketing protection to the term of any existing regulatory exclusivity, including the non-patent and orphan exclusivity. This six-month exclusivity may be granted if a BLA sponsor submits pediatric data that fairly respond to a written request from the FDA for such data. The data do not need to show the product to be effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the FDA’s request, the additional protection is granted. If reports of requested pediatric studies are submitted to and accepted by the FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity or patent protection cover the product are extended by a further six months. This is not a patent term extension, but it effectively extends the regulatory period during which the FDA cannot license another application.
Orphan Drug Designation
The Orphan Drug Act provides incentives for the development of therapeutic products intended to treat rare diseases or conditions. Rare diseases and conditions generally are those affecting less than 200,000 individuals in the U.S., but also include diseases or conditions affecting more than 200,000 individuals in the U.S. if there is no reasonable expectation that the cost of developing and making available in the U.S. a drug for such disease or condition will be recovered from sales in the U.S. of such product.
If a sponsor demonstrates that a therapeutic product, including a biological product, is intended to treat a rare disease or condition, and meets certain other criteria, FDA grants orphan drug designation to the product for that use. FDA may grant multiple orphan designations to different companies developing the same product for the same indication, until the one company is the first to be able to secure successful approval for that product. The first product approved with an orphan drug designated indication is granted seven years of orphan drug exclusivity from the date of approval for that indication. During that period, FDA generally may not approve any other application for the same product for the same indication, although there are exceptions, most notably when the later product is shown to be clinically superior to the product with exclusivity. FDA can also revoke a product’s orphan drug exclusivity under certain circumstances, including when the holder of the approved orphan drug application is unable to assure the availability of sufficient quantities of the product to meet patient needs.
A sponsor of a product application that has received an orphan drug designation is also granted tax incentives for clinical research undertaken to support the application. In addition, FDA will typically coordinate with the sponsor on research study design for an orphan drug and may exercise its discretion to grant marketing approval based on more limited product safety and efficacy data than would ordinarily be required, based on the limited size of the applicable patient population.
Expedited Programs for Serious Conditions
FDA has implemented a number of expedited programs to help ensure that therapies for serious or life-threatening conditions, and for which there is unmet medical need, are approved and available to patients as soon as it can be concluded that the therapies’ benefits justify their risks. Among these programs are the following:
| 45 |
Fast Track Designation
FDA may designate a product for fast-track review if it is intended, whether alone or in combination with one or more other products, for the treatment of a serious or life-threatening disease or condition and where non-clinical or clinical data demonstrates the potential to address unmet medical need for such a disease or condition. A product can also receive fast track review if it receives breakthrough therapy designation.
For fast-track products, sponsors may have greater interactions with FDA and FDA may initiate review of sections of a fast-track product’s application before the application is complete, also referred to as a ‘rolling review’. This rolling review may be available if FDA determines, after preliminary evaluation of clinical data submitted by the sponsor, that a fast-track product may be effective. The sponsor must also provide, and FDA must approve, a schedule for the submission of the remaining information and the sponsor must pay applicable user fees. Furthermore, FDA’s time period goal for reviewing a fast-track application does not begin until the last section of the complete application is submitted. Finally, the fast-track designation may be withdrawn by FDA if FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
Breakthrough Therapy Designation
A product may be designated as a breakthrough therapy if it is intended, either alone or in combination with one or more other products, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The designation includes all of the features of fast-track designation, as well as more intensive FDA interaction and guidance. FDA may take certain actions with respect to breakthrough therapies, including holding meetings with the sponsor throughout the development process; providing timely advice to the product sponsor regarding development and approval; involving more senior staff in the review process; assigning a cross-disciplinary project lead for the review team; and taking other steps to design efficient clinical trials.
Accelerated Approval
Under the accelerated approval pathway, FDA may approve a drug or biologic based on a surrogate endpoint that is reasonably likely to predict clinical benefit; qualifying products must target a serious or life-threatening illness and provide meaningful therapeutic benefit to patients over existing treatments. In clinical trials, a surrogate endpoint is a measurement of laboratory or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient feels, functions, or survives. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A product candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or to confirm a clinical benefit during post-marketing studies, would allow FDA to withdraw the product from the market on an expedited basis. All promotional materials for product candidates approved under accelerated regulations are subject to prior review by FDA.
Priority Review
FDA may designate a product for priority review if it is a product that treats a serious condition and, if approved, would provide a significant improvement in safety or effectiveness. FDA generally determines, on a case-by-case basis, whether the proposed product represents a significant improvement in safety and effectiveness when compared with other available therapies. Significant improvement may be illustrated by evidence of increased effectiveness in the treatment of a condition, elimination or substantial reduction of a treatment-limiting product reaction, documented enhancement of patient compliance that may lead to improvement in serious outcomes, and evidence of safety and effectiveness in a new subpopulation. A priority designation is intended to direct overall attention and resources to the evaluation of such applications and will shorten FDA’s goal for acting on a marketing application from the standard targeted ten months to a target of six months review.
| 46 |
Created in 2012 under the FDASIA and extended with the 21st Century Cures Act in 2016, FDA is authorized under section 529 of the FDCA to grant a PRV to BLA sponsors receiving FDA approval for a product to treat a rare pediatric disease, defined as a disease that affects fewer than 200,000 individuals in the U.S., and where more than 50% of the patients affected are aged from birth to 18 years. We believe that our product candidates which may assist with the treatment of rare pediatric cancers or other rare pediatric diseases may qualify for a PRV under this program, depending on the indication.
The PRV program allows the voucher holder to obtain priority review for a product application that would otherwise not receive priority review, shortening FDA’s target review period to a targeted six months following acceptance of filing of an NDA or BLA, or four months shorter than the standard review period. The voucher may be used by the sponsor who receives it, or it may be sold to another sponsor for use in that sponsor’s own marketing application. The sponsor who uses the voucher is required to pay additional user fees on top of the standard user fee for reviewing an NDA or BLA.
We anticipate submitting applications for one or more of these expedited programs and the targeted therapeutic indications.
Regenerative Medicine Advanced Therapy Designation
Recently, through the 21st Century Cures Act, or Cures Act, Congress also established another expedited program, called a RMAT designation. The Cures Act directs the FDA to facilitate an efficient development program for and expedite review of RMATs. To qualify for this program, the product must be a cell therapy, therapeutic tissue engineering product, human cell and tissue product, or a combination of such products, and not a product solely regulated as a human cell and tissue product. The product must be intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition, and preliminary clinical evidence must indicate that the product has the potential to address an unmet need for such disease or condition. Advantages of the RMAT designation include all the benefits of the fast track and breakthrough therapy designation programs, including early interactions with the FDA. These early interactions may be used to discuss potential surrogate or intermediate endpoints to support accelerated approval.
Post-Licensing Regulation
Once a BLA is approved and a product marketed, a sponsor will be required to comply with all regular post-licensing regulatory requirements as well as any post-licensing requirements that the FDA may have imposed as part of the licensing process. The sponsor will be required to report, among other things, certain adverse reactions and manufacturing problems to the FDA, provide updated safety and potency or efficacy information and comply with requirements concerning advertising and promotional labeling requirements. Manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including cGMP regulations, which impose certain procedural and documentation requirements upon manufacturers. Changes to the manufacturing processes are strictly regulated and often require prior FDA approval before being implemented. Accordingly, the sponsor and its third-party manufacturers must continue to expend time, money, and effort in the areas of production and quality control to maintain compliance with cGMP regulations and other regulatory requirements.
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act (PDMA) and its implementing regulations, as well as the Drug Supply Chain Security Act (DSCA), which regulate the distribution and tracing of prescription drug samples at the federal level and set minimum standards for the regulation of distributors by the states. The PDMA, its implementing regulations and state laws limit the distribution of prescription pharmaceutical product samples, and the DSCA imposes requirements to ensure accountability in distribution and to identify and remove counterfeit and other illegitimate products from the market.
| 47 |
Employees
We currently have ten full-time employees and no part-time employees. We contract with several part-time independent consultants performing manufacturing, regulatory and clinical development, intellectual property, public relations, investor relations, commercial, and finance and accounting functions. None of our employees are represented by labor unions or covered by collective bargaining agreements.
Information about our Executive Officers
The following sets forth the names, ages and current positions of our executive officers as of March 1, 2021.
Cameron Durrant, M.D., MBA, age 60, has served as our Chairman of our Board since January 2016, and as our Chief Executive Officer since March 2016. In addition, Dr. Durrant served as our Interim Chief Financial Officer from July 1, 2019 to July 31, 2020. From May 2014 to January 2016, Dr. Durrant served as Founder and Director of Taran Pharma Limited, a private semi-virtual specialty pharma company developing and registering treatments in Europe for orphan conditions. Dr. Durrant served as President and Chief Executive Officer of ECR Pharmaceuticals Co., Inc., a subsidiary of Hi-Tech Pharmacal Co., Inc., from September 2012 to April 2014 until its acquisition by Akorn. He previously has been a senior executive at Johnson and Johnson, Pharmacia Corporation, GSK and Merck. Dr. Durrant was a director of Immune Pharmaceuticals Inc. from July 2014 to September 2018 and serves on the boards of directors of two privately held healthcare companies. Dr. Durrant earned his medical degree from the Welsh National School of Medicine, Cardiff, UK, his DRCOG from the Royal College of Obstetricians and Gynecologists, London, UK, his MRCGP from the Royal College of General Practitioners, London, UK, and his MBA from Henley Management College, Oxford, UK.
Timothy Morris, CPA, age 59, joined the Company as Chief Operating and Financial Officer on August 1, 2020. Prior to assuming his position as a Company officer, Mr. Morris served as a member of our Board since June 2016. Mr. Morris served as the Chief Financial Officer of Iovance Biotherapeutics, Inc., a biopharmaceutical company, from August 2017 until June 2020. From March 2014 to June 2017, Mr. Morris served as Chief Financial Officer and Head of Business Development of AcelRx Pharmaceuticals, Inc., a specialty pharmaceutical company. From November 2004 to December 2013, Mr. Morris served as Senior Vice President Finance and Global Corporate Development, Chief Financial Officer of VIVUS, Inc. a biopharmaceutical company. Mr. Morris received his BS in Business with an emphasis in Accounting from California State University, Chico, and is a Certified Public Accountant (Inactive).
David Tousley, MBA, CPA, age 65, was appointed as our Chief Accounting and Administrative Officer, Corporate Secretary and Treasurer on July 6, 2020 and served in that capacity until his resignation effective March 5, 2021. From January 2007 to July 6, 2020, Mr. Tousley served as Principal of Stratium Consulting Services (“Stratium Consulting”), assisting private and public companies with strategic and financial planning and management. Previously, Mr. Tousley served as the Chief Financial Officer for DARA Biosciences, Inc., a publicly traded specialty pharmaceutical company focused on oncology treatment and oncology supportive care. Mr. Tousley has over 35 years of business experience including biotech, specialty pharmaceuticals and full phase pharmaceutical companies. He has held President, Chief Operating Officer and Chief Financial Officer roles in companies such as Pasteur Merieux Connaught (known today as Sanofi Pasteur), AVAX Technologies, Inc., airPharma, LLC, and PediaMed Pharmaceuticals, Inc., and has led companies in all aspects of operations, including pharmaceutical development. He has managed in both the private and public company environment and has led business development activities, including joint ventures, partnerships, acquisitions and divestitures in the U.S., Europe and Australia. Mr. Tousley is a Certified Public Accountant, a member of the American Institute of Certified Public Accountants, holds an undergraduate degree from Rutgers College and earned his MBA in accounting from Rutgers Graduate School of Business. Mr. Tousley previously served as our interim Chief Financial Officer from October 2016 until his resignation in August 2017.
Dale Chappell, M.D., MBA, age 50, was appointed as our Chief Scientific Officer on July 6, 2020. Dr. Chappell is the managing member of BH Management, a private investment manager that specializes in biopharmaceuticals and a significant stockholder, a position he has held since 2002. Since April 2015, Dr. Chappell has served as CEO, President and CFO of Cheval US Holdings, Inc., a private investment company with holdings in the hospitality industry. Previously, Dr. Chappell was an associate with Chilton Investment Company, covering healthcare, and an analyst at W.P. Carey & Company. Dr. Chappell, who received his MD from Dartmouth Medical School and his MBA from Harvard Business School, began his career as a Howard Hughes Medical Institute fellow at the National Cancer Institute where he studied tumor immunology, worked as a researcher in the labs of Dr. Steven A. Rosenberg (widely thought of as one of the pioneers in CAR-T therapy) and Dr. Nicholas P. Restifo (a leading researcher in the field of immunology) and is published in the field of GM-CSF. Dr. Chappell served as a member of the Board from June 2016 to November 2017. Prior to joining the Company in a full-time role as our Chief Scientific Officer, Dr. Chappell advised and consulted with management as our ex-officio chief scientific officer.
| 48 |
Edward P. Jordan, MBA, age 53, was appointed as our Chief Commercial Officer on August 24, 2020. Mr. Jordan has more than two decades of commercial operations experience at leading biotechnology and pharmaceutical companies, having launched over a dozen products and developed new therapeutic markets in the U.S. and abroad. From 2016 until joining the Company, Mr. Jordan served as Senior Vice President of DBV Technologies, where he built the North American commercial organization in preparation for the launch of a lifesaving pediatric biologic. Prior to DBV Technologies, from January 2014 to July 2015, Mr. Jordan was the Senior Vice President, Hematology and Oncology Sales and Marketing at AMAG Pharmaceuticals. Prior to AMAG Pharmaceuticals, Mr. Jordan served in executive roles at Teva Pharmaceuticals. Mr. Jordan began his career at Schering-Plough, prior to the acquisition by Merck, and spent 18 years in sales and marketing leadership positions. Mr. Jordan received dual undergraduate degrees from The University of Rhode Island and an MBA from Southern New Hampshire University.
Available Information
We were incorporated on March 15, 2000 in California and reincorporated as a Delaware corporation in September 2001. Effective August 7, 2017, we changed our legal name to Humanigen, Inc. Our principal offices are located at 533 Airport Boulevard, Suite 400, Burlingame, CA 94010, and our telephone number is (650) 243-3100. Our website address is www.humanigen.com. Our common stock is currently listed on the Nasdaq Capital Market. We operate in a single segment.
Our website and the information contained on, or that can be accessed through, the website will not be deemed to be incorporated by reference in, and are not considered part of, this Annual Report on Form 10-K. Our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and amendments to reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Securities Exchange Act of 1934, as amended, are available free of charge on the Investor Relations portion of our website as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. In addition, the SEC maintains an internet site that contains the reports, proxy and information statements, and other information we electronically file with or furnish to the SEC, located at www.sec.gov.
| 49 |
Risk Factor Summary
The following summary highlights the material risks that may affect our business, operating results, financial condition and prospects, as more fully described in the pages that follow this summary.
| · | The COVID-19 pandemic may adversely affect our business. |
| · | Vaccines and other treatments authorized or approved for COVID-19 may decrease or eliminate hospitalizations for COVID-19 patients which would negatively impact the need for lenzilumab. |
| · | If our competitors receive FDA approval for treatments or vaccines for COVID-19, our commercial opportunity may be reduced or eliminated. |
| · | We have a history of operating losses and we may never become profitable. |
| · | We are substantially dependent on third parties to plan and conduct the referenced studies and clinical trials in our development pipeline, to manufacture and to commercialize and distribute our product candidate. |
| · | If we are unable to obtain additional financing to fund our operations, we may be unable to complete the development and commercialization of our product candidates. |
| · | The scientific rationale behind the hypothesis that GM-CSF is a cause of the cytokine storm that leads to adverse results in COVID-19 patients is still being tested and may not prove accurate. |
| · | We may not be able to develop, obtain regulatory approval for, or successfully commercialize, any of our product candidates, and our product candidates are at an early stage of development. |
| · | The adoption of CAR-T therapies as the potential standard of care for treatment of certain cancers is uncertain and, if not adopted as anticipated, the market for lenzilumab may not develop. |
| · | We may not ultimately qualify for or benefit from various regulatory incentives. |
| · | If our existing or future collaborators cease development efforts under our collaboration agreements (including our agreement with Kite and the IMPACT partnership), or if any of those agreements are terminated or are ultimately not successful, our business could be adversely affected. |
| · | We have relied and may in the future rely on third parties to conduct investigator-sponsored trials (“ISTs”) of our products, which affords the investigators the ability to retain significant control over the design and conduct of the trials, as well as the use of the data generated from their efforts. |
| · | Our ability to pursue our clinical development programs could be delayed or unsuccessful and we may not be able to obtain regulatory approval for or commercialize our product candidates when expected or at all. |
| · | We may experience delays in commencing or conducting our clinical trials, in receiving data from third parties or in the continuation or completion of clinical testing. |
| · | Our product candidates are subject to extensive regulation, compliance with which is costly and time consuming, may cause unanticipated delays, or may prevent the receipt of the required approvals to commercialize our product candidates. |
| · | The results of preclinical studies and early clinical trials are not always predictive of future results. |
| · | We face risks related to the development, manufacturing and distribution of our product candidates, which may delay or impair our ability to obtain an EUA or BLA or to successfully commercialize any FDA-approved product candidate. These risks include manufacturing delays due to displacement of scheduled production at certain of our Contract Manufacturing Organizations (CMO) as mandated from Rated Orders issued under the Defense Production Act (DPA) or the shortage of raw materials and critical components we are experiencing and expect to continue to experience relating to Rated Orders and increased demand for production capability from others at our CMOs. |
| 50 |
| · | Interim, “top-line” or preliminary data from our clinical trials may not be favorable and may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data. |
| · | We may be unable to accurately predict demand for lenzilumab and to accurately forecast any sales. |
| · | The regulatory pathway for lenzilumab is highly dynamic and continues to evolve and may result in unexpected or unforeseen challenges. |
| · | Even if EUA or regulatory approval is received for our product candidates, the later discovery of previously unknown problems associated with the use of any product candidate may adversely affect our business. |
| · | We face risks associated with clinical operations abroad. |
| · | If we fail to attract and retain key management and clinical development personnel, or if the attention of such personnel is diverted, we may be unable to successfully manage our business. |
| · | If our competitors develop treatments for the target indications of our product candidates that are approved more quickly, marketed more successfully or are demonstrated to be safer or more effective than our product candidates, or if FDA approves generic or biosimilar competitors to our products post-approval, our commercial opportunity will be reduced or eliminated. |
| · | Any FDA approved product candidate may not achieve broad market acceptance among physicians, healthcare payers and the medical community. |
| · | Reimbursement may be limited or unavailable in certain market segments for our product candidates, and governments may impose price controls. |
| · | We outsource our sales and marketing capabilities and rely on agreements with third parties to market and sell any product candidates we may successfully develop. |
| · | We face potential product liability exposure and, if successful claims are brought against us, we may incur substantial liability for a product candidate and may have to limit its commercialization. |
| · | Our employees and consultants may engage in misconduct or other improper activities, including noncompliance with regulatory standards. |
| · | We may encounter difficulties in managing our growth and expanding our operations successfully. |
| · | Even if we are able to obtain regulatory approval for our product candidates, we will continue to be subject to ongoing and extensive regulatory requirements. |
| · | Our ability to utilize our net operating loss carryforwards may be subject to certain limitations. |
| · | Currently pending, threatened or future litigation or governmental proceedings or inquiries could result in material adverse consequences, including judgments or settlements. |
| · | We may not be able to identify or complete various strategic alternatives, and future acquisitions of and investments in new businesses could impact our business and financial condition. |
| · | If we fail to adequately protect or enforce our intellectual property rights or secure rights to patents of others, the value of our intellectual property rights would diminish. |
| · | If we infringe on the rights of third parties, we could be prevented from selling products and be forced to defend against litigation and pay damages. |
| · | A significant portion of our total outstanding shares are eligible to be sold into the market, which may affect the market price of our common stock. |
| · | Our executive officers, directors and principal stockholders, if they choose to act together, will continue to have the ability to significantly influence all matters submitted to stockholders for approval. |
| · | Our common stock may be delisted if we fail to comply with ongoing Nasdaq Capital Market standards. |
| · | Raising additional funds by issuing securities or through licensing or lending arrangements may cause dilution to our existing stockholders or restrict or otherwise affect our operations. |
| · | Our stock price is volatile, and purchasers of our common stock could incur substantial losses. |
| 51 |
In addition to the other information set forth in this Annual Report on Form 10-K and other filings we have made and will make in the future with the SEC, you should carefully consider the following risk factors and uncertainties, which could materially affect our business, financial condition or results of operations in future periods. Additional risks not currently known to us or that we currently deem to be immaterial also may materially adversely affect our business, financial condition or results of operations in future periods.
Risks Related to Our Business and Industry
Risks Related to COVID-19 and Lenzilumab
The fluid and unpredictable nature of the COVID-19 pandemic, which began in late 2019 and has spread worldwide, may affect our ability to conduct the clinical trial with Kite and the IMPACT partnership, delay the initiation of planned and future clinical trials, disrupt regulatory activities, or have other adverse effects on our business and operations. In addition, this pandemic has caused substantial volatility in the financial markets and may adversely impact economies worldwide, both of which could result in adverse effects on our business and operations.
In December 2019, an outbreak of the respiratory illness COVID-19 caused by a strain of novel coronavirus, SARS-Cov-2, was first reported in China. The COVID-19 outbreak has spread worldwide, causing many governments to implement measures to slow the spread of the outbreak through quarantines, strict travel restrictions, heightened border scrutiny, and other measures. COVID-19 infections are widespread and as of February 26, 2021 in the United States, there were over 28 million cases, nearly 1.8 million hospitalizations and over 510 thousand deaths. Several vaccines have been developed which show efficacy in excess of 90%, however the slow rollout of inoculations and the emergence of numerous COVID-19 variants lead us to believe the need for therapeutics to treat COVID-19 currently exists and will continue to exist for the next several years. The outbreak and government measures taken in response have had a significant impact, both direct and indirect, on businesses and commerce, as worker shortages have occurred; supply chains have been disrupted; facilities and production have been suspended; and demand for certain goods and services, such as medical services and supplies, has spiked, while demand for other goods and services, such as travel, has fallen. The future progression of the outbreak and its effects on our business and operations are uncertain.
We and our third-party contract research organizations (“CROs”) clinical sites and CMOs may experience disruptions in supply of product candidates and/or procuring items that are essential for our research and development activities, including raw materials used in the manufacturing of our product candidates, medical and laboratory supplies used in our clinical trials or preclinical studies or animals that are used for preclinical testing, in each case, for which there may be shortages because of ongoing efforts to address the outbreak. These delays have impacted our overall manufacturing supply chain operations to date, and we continue to explore back up or alternative sources of supply, any future disruption in the supply chain from the COVID-19 outbreak, or any continued outbreak could have a material adverse impact on our clinical trial plans and business operations.
Additionally, we have enrolled, and will seek to enroll, patients in our clinical trials at sites located in many areas affected by COVID-19 and, as a result, our trials may be impacted. In addition, even if sites are actively recruiting, we may face difficulties recruiting or retaining patients in our ongoing and planned clinical trials if patients are affected by the virus or are fearful of visiting or traveling to our clinical trial sites because of the outbreak. Prolonged delays or closure to enrollment in our trials or patient discontinuations could have a material adverse impact on our clinical trial plans and timelines.
In addition, our ability to collect all data requested of patients enrolled in our clinical trials during this pandemic is being impacted to varying degrees by COVID-19. Clinical trial data collection generally continues for each of our clinical trials, but at a slower pace, and in some instances, we encounter disruption of collection of complete study data. This could have a material adverse impact on our data analysis.
| 52 |
The response to the COVID-19 pandemic may redirect resources with respect to regulatory and intellectual property matters in a way that would adversely impact our ability to progress regulatory approvals and protect our intellectual property. In addition, we may face impediments to regulatory meetings and approvals due to measures intended to limit in-person interactions.
Any negative impact that the COVID-19 outbreak has on the ability of our suppliers to provide materials for our product candidates or on recruiting or retaining patients in our clinical trials or our ability to collect patient data could cause costly delays to clinical trial activities, which could adversely affect our ability to obtain regulatory approval for and to commercialize our product candidates, increase our operating expenses, and have a material adverse effect on our financial results. Furthermore, any negative impact that the outbreak has on the ability of our CROs to deliver data sets and execute on experimentation could cause substantial delays for our discovery activities and materially impact our ability to fuel our pipeline with new product candidates.
Any negative impact that the COVID-19 pandemic has on recruiting or retaining patients in our clinical trials, obtaining complete clinical trial data or on the ability of our suppliers to provide materials for our product candidates could cause additional delays to clinical trial and developmental activities, which could materially and adversely affect our ability to obtain regulatory approval for and to commercialize our product candidates, increase our operating expenses, affect our ability to raise additional capital, and have a material adverse effect on our financial results.
The COVID-19 pandemic has already caused significant volatility in the financial markets, and may continue to cause such disruptions, which could impact our ability to raise additional funds through public offerings and may also impact the volatility of our stock price and trading in our stock. Moreover, the pandemic has also significantly impacted economies worldwide, which could result in adverse effects on our business and operations. We cannot be certain what the overall impact of the COVID-19 pandemic will be on our business. It has the potential to adversely affect our business, financial condition, results of operations, and prospects.
If our competitors develop and receive FDA approval for treatments or vaccines for COVID-19, our commercial opportunity may be reduced or eliminated.
On October 22, 2020, FDA approved remdesivir for use in adult and certain pediatric patients for the treatment of COVID-19 requiring hospitalization. Baricitinib has also been approved for use with remdesivir for the treatment of COVID-19. Several neutralizing antibodies have been approved to prevent progression in the early stages of infection. Three vaccines have been authorized by the FDA with efficacy rates ranging from 66% to 90%. There are numerous other companies working on therapies to treat COVID-19 and/or vaccines to prevent or treat COVID-19. If additional competing therapies are approved, and/or a safe, efficacious, accessible preventative vaccine is approved, such approval could have a material adverse impact on our ability to commercialize lenzilumab as a therapy for COVID-19. The speed at which all parties are acting to create and test many treatments and vaccines for COVID-19 is unusual and evolving or changing plans or priorities within the FDA and other branches of the U.S. government involved in the fight against the pandemic, including changes based on new knowledge of COVID-19, new COVID-19 variants and how the disease affects the human body, may significantly affect the regulatory timeline for lenzilumab. In addition, there are numerous clinical trial programs in progress for COVID-19 therapies which are all competing for largely the same patient population for enrollment. This problem is exacerbated by changing rates of infection and spread, which make it challenging to ensure there are sufficient centers for the ACTIV-5 trial in areas with sufficient potential patient populations. Results from clinical testing may raise new questions and require us to redesign proposed clinical trials. Many of our competitors and potential competitors have substantially greater scientific, research, and product development capabilities, as well as greater financial, marketing, sales and human resources capabilities than we do. In addition, many specialized biotechnology firms have formed collaborations with large, established companies to support the research, development and commercialization of products that may be competitive with ours.
| 53 |
The scientific rationale behind the hypothesis that GM-CSF is a cause of the cytokine storm that leads to adverse results in COVID-19 patients is still being tested and may not prove accurate.
The hypothesis that elevated GM-CSF levels may contribute to cytokine storm-induced immune mechanisms that places patients at greater risk of ICU admission and mortality with the current pandemic strain of coronavirus is unproven. Certain data are the subject of pre-publication papers that have not been peer-reviewed and may not be substantiated. If this hypothesis is not ultimately proven through clinical trials which are underway, the potential for lenzilumab to play a meaningful role in a COVID-19 therapy likely would decrease or be eliminated. We cannot assure you that our exploratory efforts in this respect will be fruitful.
We face risks related to the development, manufacturing and distribution of lenzilumab as a treatment for COVID-19, which has not been granted an EUA or approved by FDA and has not been proven through a randomized double-blind placebo clinical trial to be safe or effective for any use.
We have completed enrollment in a Phase 3 multi-center, randomized, placebo-controlled, double-blinded, clinical trial of lenzilumab as a potential treatment for COVID-19 pneumonia. However, there is no assurance of favorable results from our clinical trial, or future clinical trials including the NIH sponsored ACTIV-5/BET, or that our Phase 3 clinical trial will be finalized in the currently anticipated timeline or at all.
It is also possible that FDA and other regulatory authorities may not grant an EUA or subsequently approve lenzilumab for the treatment of COVID-19 pneumonia, or that any such EUA or approval, if granted, may have significant limitations on its use. Three COVID-19 vaccines have been authorized by the FDA with efficacy over 90%. The inoculation rollout has been slow but widespread uptake of a safe, effective, scalable and affordable vaccine also will have a negative impact on the demand for lenzilumab over the long term, even if an EUA or approval were issued. As a result, we may never successfully commercialize lenzilumab in COVID-19 or realize a return on our significant investment in the development, supply, and commercialization of lenzilumab for this purpose.
Furthermore, even if an EUA were granted to permit lenzilumab to be commercialized for use in the treatment of COVID-19 pneumonia, the authorization would be temporary and might be expressly conditioned or limited by FDA. An EUA does not take the place of the formal BLA submission, review and approval process. While there are ongoing clinical trials to evaluate the safety clinical profile and the efficacy of lenzilumab, there is no assurance of favorable results from any ongoing or future clinical trials, or that one or more of such trials will be completed in the currently anticipated timelines or at all. It is also possible that FDA and other regulatory authorities may not approve lenzilumab for the treatment of COVID-19, or that any marketing approvals, if granted, may have significant limitations on its use. Further, we may make a strategic decision to discontinue development of lenzilumab in this indication if other parties are successful in developing a more effective treatment for COVID-19. As a result, we may never successfully commercialize lenzilumab for use in COVID-19 patients.
Manufacturing problems at our third-party manufacturers or their requirements to displace our reserved manufacturing slots to comply with government orders could cause inventory shortages and delay or impair our ability to obtain an EUA or BLA or other regulatory approval or delay shipments of lenzilumab for commercial use, which may adversely affect our results of operations.
We believe that our ability to obtain an EUA and, ultimately, a BLA to permit lenzilumab to be used commercially for patients with COVID-19 pneumonia depends at least in part on our ability to demonstrate to FDA that we will be able to scale the manufacturing to produce a sufficient quantity of dosages to begin to address the potential demand for the product. We have contracted and expect to continue to contract with third-party manufacturers to produce lenzilumab. We depend on these third parties to perform the manufacturing of lenzilumab effectively, timely, and in compliance with Good Manufacturing Practices (“GMP”), which are extensive regulations governing manufacturing processes, stability testing, record-keeping and quality standards as defined by FDA. Similar regulations are in effect in other jurisdictions.
| 54 |
Our complete reliance on third-party manufacturers to produce lenzilumab subjects us to additional risks, including the possible breach of the manufacturing agreement by the third party, the possible cancellation, delay or modification of contracted manufacturing slots by the third party, or the termination or nonrenewal of the agreement by the third party at a time that is costly or inconvenient for us. In addition, in an effort to increase the availability of needed medical products, vaccinations or related therapies in response to the COVID-19 pandemic, governments have required and may continue require our third-party manufacturers or our suppliers to allocate manufacturing capacity (for example pursuant to the U.S Defense Production Act) in a way that adversely affects our ability to obtain regulatory approval for and to commercialize our product candidates. During the latter half of 2020, and continuing into 2021, it has become more challenging and expensive to obtain these slots due to increased demand for production at our CMOs, many of which also manufacture vaccines and other therapeutics. Even after being reserved, the manufacturing slots for which we contract are subject to risk of displacement as mandated from Rated Orders issued under the Defense Production Act (DPA). This displacement risk has caused us to expend significant efforts to manage our supply chain. Despite those efforts, we have experienced and expect to continue to experience shortages of raw materials and critical components we necessary for our manufacturing processes. Accordingly, the inability to manage our manufacturing capacity could delay our ability to generate revenues from sales of lenzilumab.
Our third-party manufacturers are independent entities subject to their own unique operational and financial risks that are out of our control. If we or any of these third-party manufacturers fail to perform as required, this could cause delays in our clinical trials and applications for regulatory approval. Further, we may have to pay the costs of manufacturing any batch that fails to pass quality inspection or meet regulatory approval. In addition, we, our third-party manufacturers may only be able to produce some of our products at one or a limited number of facilities and, therefore, we have limited manufacturing capacity for certain products, and we may not be able to locate additional or replacement facilities on a reasonable basis or at all. Our sales of such products could also be adversely impacted by our reliance on such limited number of facilities. To the extent these risks materialize and affect their performance obligations to us, our financial results may be adversely affected.
In addition, the process of manufacturing biologics is extremely susceptible to product loss due to contamination, equipment failure or improper installation or operation of equipment, or vendor or operator error. Even minor deviations from normal manufacturing processes could result in reduced production yields, product defects and other supply disruptions. If microbial, viral or other contaminations are discovered in our products or in the manufacturing facilities in which our products are made, such manufacturing facilities may need to be closed for an extended period of time to investigate and remedy the contamination. Lenzilumab produced in these facilities may be quarantined or recalled and not available for clinical or commercial use. As we encounter any of these difficulties, our ability to manufacture and sell any products that may be authorized or approved for commercial use could be impaired, which could have an adverse effect on our business.
We may not be able to obtain materials or supplies necessary to conduct clinical trials or, following requisite regulatory authorizations or approvals, to manufacture and sell our products, which could limit our ability to generate revenues.
We need access to certain supplies and products to conduct our clinical trials and, if an EUA or BLA or other approval were to be received, to manufacture and sell our products. If we are unable to purchase sufficient quantities of these materials or find suitable alternative materials in a timely manner, our development efforts for our product candidates may be delayed or our ability to manufacture our products could be limited, which could limit our ability to generate revenues.
Suppliers of key components and materials must be named in the EUA, BLA or other marketing authorization application filed with the regulatory authority for any product candidate for which we are seeking marketing approval, and significant delays can occur if the qualification of a new supplier is required. Even after a manufacturer is qualified by the regulatory authority, the manufacturer must continue to expend time, money and effort in the area of production and quality control to ensure full compliance with GMP. Manufacturers are subject to regular periodic inspections by regulatory authorities following initial approval. If, as a result of these inspections, a regulatory authority determines that the equipment, facilities, laboratories or processes do not comply with applicable regulations and conditions of product approval, the regulatory authority may suspend the manufacturing operations. If the manufacturing operations of any of the single suppliers for our products are suspended, we may be unable to generate sufficient quantities of commercial or clinical supplies of product to meet market demand, which could in turn decrease our revenues and harm our business. In addition, if deliveries of materials from our suppliers were interrupted for any reason, we may be unable to supply our product candidates in development for clinical trials or ship them to customers, if authorized or approved for commercial use. In addition, some of our products and the materials that we utilize in our operations are manufactured at only one facility or from a single vendor, which we may not be able to replace in a timely manner and on commercially reasonable terms, or at all. Problems with any of the single suppliers we depend on, including in the event of a disaster, such as an earthquake, equipment failure or other difficulty, may negatively impact our development and commercialization efforts.
| 55 |
A significant portion of the raw materials and intermediates used to manufacture our product candidates are supplied by third-party manufacturers and corporate partners outside of the United States. As a result, any political or economic factors in a specific country or region, including any changes in or interpretations of trade regulations, compliance requirements or tax legislation, that would limit or prevent third parties outside of the United States from supplying these materials could adversely affect our ability conduct our pending or contemplated clinical trials.
If we were to encounter any of these difficulties, our ability to conduct clinical trials on product candidates and to manufacture and sell any products that may be authorized or approved for commercial use could be impaired, which could have an adverse effect on our business.
Interim, “top-line” or preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose preliminary or “top-line” data from our clinical trials, which is based on a preliminary analysis of then-available top-line data, and the results and related findings and conclusions are subject to change following a full analysis of all data related to the particular trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the top-line results that we report may differ from future results of the same trials, or different conclusions or considerations may qualify such results once additional data have been received and fully evaluated. Top-line data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, top-line data should be viewed with caution until the final data are available. We may also disclose interim data from our clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Adverse differences between preliminary or interim data and final data could significantly harm our business prospects. Further, disclosure of preliminary or interim data by us or by our competitors could result in volatility in the price of shares of our common stock.
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our business in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure, and any information we determine not to disclose may ultimately be deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding a particular drug, product candidate or our business. If the top-line data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for and commercialize in our current or any our future product candidate, our business, operating results, prospects or financial condition may be harmed.
Our inability to accurately predict demand for lenzilumab makes it difficult for us to accurately forecast any sales and may cause our forecasts to fluctuate and could adversely affect our stock price.
We may be unable to accurately predict demand for lenzilumab, or other approved or unapproved products for COVID-19, as demand depends on a number of factors, including rollout and effectiveness of approved vaccines, competing drug products and other treatments that may be developed for COVID-19 and other illnesses and conditions and federal and state social measures design to slow the spread of COVID-19. If lenzilumab is commercialized, we may be unsuccessful in estimating user demand and may not be effective in matching inventory levels and locations to actual end user demand, in particular if the COVID-19 outbreak is effectively contained or the risk of coronavirus infection is significantly diminished. In addition, adverse changes in economic conditions, increased competition, including through approved vaccines or other therapeutic, or other factors may cause hospitals or other users or distributors of lenzilumab to reduce inventories of our products, which would reduce their orders for lenzilumab, even if end user demand has not changed. As a result, even if lenzilumab receives an EUA or BLA approval for use in COVID-19 patients, our revenues may be difficult to predict and vulnerable to extreme fluctuations.
| 56 |
There can be no assurance that lenzilumab, even if approved, would ever become profitable, due to government interest and public perception regarding a vaccine and treatments for COVID-19 related complications.
As a result of the emergency situations in many countries, there is a heightened risk that a COVID-19 therapy or other treatments for COVID-19 pneumonia and symptoms may be subject to adverse governmental actions in certain countries, including intellectual property expropriation, compulsory licenses, strict price controls or other actions. Additionally, we may need to, or we may be required by governmental or non-governmental authorities to, set aside specific quantities of doses of lenzilumab for designated purposes or geographic areas. We may face challenges related to the allocation of supply of lenzilumab, particularly with respect to geographic distribution. Thus, even if lenzilumab is approved, such governmental actions may limit our ability to recoup our current and future expenses incurred to develop and commercialize lenzilumab.
Furthermore, public sentiment regarding commercialization of a COVID-19 therapy or other treatment may limit or negate our ability to generate revenues from sales of lenzilumab. Given that COVID-19 has been designated as a pandemic and represents an urgent public health crisis, we are likely to face significant public attention and scrutiny over any future business models and pricing decisions with respect to lenzilumab. If we are unable to successfully manage these risks, we could face significant reputational harm, which could negatively affect the price of our common stock.
The regulatory pathway for lenzilumab is highly dynamic and continues to evolve and may result in unexpected or unforeseen challenges.
To date, lenzilumab has moved rapidly through the regulatory review process of FDA. The speed at which all parties are acting to create and test many therapeutics and vaccines for COVID-19 is unusual and evolving or changing plans or priorities within FDA and foreign regulatory authorities, including changes based on new knowledge of COVID-19 and how the disease affects the human body, may significantly affect the regulatory timeline for lenzilumab. Results from clinical testing may raise new questions and require us to redesign proposed clinical trials, including revising proposed endpoints or adding new clinical trial sites or cohorts of subjects.
For example, FDA on June 30, 2020 adopted guidance outlining the FDA’s current recommendations regarding the data needed to facilitate clinical development and licensure of vaccines to prevent COVID-19. Although we intend to design any future clinical trials for lenzilumab in accordance with FDA and other applicable regulatory guidance, we cannot be certain that, as the regulatory pathway continues to evolve, we will be able to complete a clinical trial in accordance with FDA’s guidance and regulations then in effect. A failure to complete a clinical trial in accordance with guidance and regulations then in effect could impair our ability to obtain approval for lenzilumab, which may adversely affect our operating results, reputation and ability to raise capital and enter into or maintain collaborations to advance lenzilumab.
Additionally, FDA has the authority to grant an EUA to allow unapproved medical products to be used in an emergency to diagnose, treat, or prevent serious or life-threatening diseases or conditions when there are no adequate, approved, and available alternatives. If we are granted an EUA for lenzilumab, we would be able to commercialize lenzilumab prior to FDA approval. However, FDA may revoke an EUA where it is determined that the underlying health emergency no longer exists or warrants such authorization, and we cannot predict how long, if ever, an EUA would remain in place. Such revocation could adversely impact our business in a variety of ways, including if lenzilumab is not yet approved by FDA and if we and our manufacturing partners have invested in the supply chain to provide lenzilumab under an EUA.
| 57 |
Even if Emergency Use Authorization or regulatory approval is received for lenzilumab, the later discovery of previously unknown problems associated with the use of lenzilumab may result in restrictions, including withdrawal of the product from the market, and lead to significant liabilities and reputational damage.
Because the path to marketing approval of any vaccine for COVID-19, or drug used to treat COVID-19 pneumonia or other symptoms is unclear, lenzilumab may be widely used and in circulation in the United States or another country prior to our receipt of marketing approval. Unexpected safety issues, including any that we have not yet observed in our Phase 3 clinical trial for lenzilumab, could lead to significant reputational damage to us and our platforms going forward and other issues, including delays in our other programs, the need for re-design of our clinical trial and the need for significant additional financial resources.
We also may be restricted or prohibited from marketing or manufacturing lenzilumab, even after obtaining product approval, if previously unknown problems with lenzilumab or its manufacture are subsequently discovered. We cannot provide assurance that newly discovered or developed safety issues will not arise following regulatory approval. With the use of any drug product or treatment by a wide patient population, serious adverse events may occur from time to time that did not arise in the clinical trials of the product or that initially appeared to be unrelated to the vaccine itself and only with the collection of subsequent information were found to be causally related to the product. Any such safety issues could cause us to suspend or cease marketing of our approved products, possibly subject us to substantial liabilities, and adversely affect our ability to generate revenue and our financial condition.
We currently have no internal sales and marketing capabilities and will rely on partners such as Eversana and other third parties to market and sell lenzilumab if we attain an EUA or other regulatory approval for its commercialization, and any product candidates we may successfully develop. We or they may not be able to effectively market and sell any such product candidates.
While we have entered into several agreements with commercial partners who will provide sales and marketing support as well as logistic and distribution services, we currently do not have the internal sales and marketing infrastructure in place that would be necessary to sell and market products. We have engaged Eversana to serve as our end-to-end commercial partner in marketing and selling lenzilumab, if approved, and have entered into other arrangements with distribution partners as described under “Item 1. Business—Sales and Marketing.” There can be no assurance that our partners will be effective in marketing and selling lenzilumab.
If we or Eversana fail to hire, train, retain and manage qualified sales personnel, market our product successfully or on a cost-effective basis or otherwise terminate our agreement, our ability to generate revenue will be limited and we will need to identify and retain an alternative third-party, or develop our own sales and marketing capability. The establishment of a sales and marketing operation can be expensive and time consuming and could delay any product candidate launch.
We may experience significant volatility in the market price of our common stock following announcements and releases regarding our ongoing development of lenzilumab as a potential COVID-19 therapy.
Biopharmaceutical companies that are developing potential therapeutics and vaccines to combat COVID-19, including our company, have experienced significant volatility in the price of their securities upon publication of preclinical and clinical data as well as news about their development programs. Since our initial announcement of our plans to develop lenzilumab as a therapy for COVID-19 patients, our common stock has experienced wide variations in daily trading volume and price movements. We expect that over the coming months we may make public several additional data and clinical updates with respect to lenzilumab and our commercialization efforts for lenzilumab. We cannot predict public reaction or the impact on the market price of our common stock once further announcements regarding lenzilumab or developments from our on-going clinical trial are announced. Given the attention being paid to the COVID-19 pandemic and the public scrutiny of COVID-19 development announcements and data releases to date, we expect that any public announcements we make in the coming months regarding the ongoing development and commercialization of lenzilumab will attract significant attention and scrutiny and that, as a result, the price of our common stock may be particularly volatile during this time.
| 58 |
Other Risks Related to Our Business and Industry
We have a history of operating losses, we expect to continue to incur losses, and we may never become profitable.
We have incurred net losses in nearly every year since our inception. For the fiscal year ended December 31, 2020 we incurred a net loss of $89.5 million, and we have an accumulated deficit of $374.4 million as of December 31, 2020. Since inception, we have recognized a nominal amount of revenue from payments for funded research and development and for license or collaboration fees, $0.3 million of which was recognized in 2020. We expect to make substantial expenditures and incur additional operating losses in the future to further develop and commercialize our product candidates. Our accumulated deficit is expected to increase significantly as we continue our development and clinical trial efforts. Our ability to achieve and sustain profitability depends on obtaining regulatory approvals for and successfully commercializing our product candidates, either alone or with third parties. We do not currently have the required approvals to market any of our product candidates and we may never receive them. We may not be profitable even if we or any future development partners succeed in commercializing any of our product candidates. Because of the numerous risks and uncertainties associated with developing and commercializing our product candidates, we are unable to predict the extent of any future losses or when we will become profitable, if at all.
Our ability to execute on all of the initiatives in our development pipeline is substantially dependent on third parties to plan and conduct the referenced studies and clinical trials.
Our success depends on our ability to negotiate agreements with third parties with resources to plan and conduct the initiatives, studies and clinical trials we are pursuing. If we are not able to reach agreements with current or future partners for it, we will not be able to execute on each of these particular initiatives, studies and trials. Our inability to identify and complete negotiations with any such third party therefore could have a material and adverse impact on our ability to pursue our business plan in respect of the applicable element of our pipeline, which in turn could have a material adverse effect on our business.
We will need to obtain additional financing to fund our operations and, if we are unable to obtain such financing, we may be unable to complete the development and commercialization of our product candidates.
We believe that the Company’s cash and cash equivalents will be sufficient to fund our planned operations and capital expenditure requirements for at least 12 months. This evaluation is based on relevant conditions and events that are currently known or reasonably knowable. As a result, we could deplete our available capital resources sooner than we currently expect, and a delay in obtaining or failure to obtain an EUA could further constrain our cash resources. We have based these estimates on assumptions that may prove to be wrong, and our operating projections, including our projected net revenue following the potential receipt of an EUA for lenzilumab in COVID-19 patients, may change as a result of many factors currently unknown to us.
We expect we will need to obtain additional financing to fund our future operations, including for the development, manufacturing, distribution and commercialization of lenzilumab for patients with COVID-19 and other indications including CRS and GvHD and our other product candidates. We may need to obtain additional financing to conduct additional trials for the approval of our product candidates if requested by regulatory authorities in the US and other countries, and to complete the development of any additional product candidates we own or might acquire. Moreover, our fixed expenses such as salaries, committed payments to our contract manufacturers, and other contractual commitments, are substantial and are expected to increase in the future. Our need to raise funds will depend on a number of factors, including our ability to establish additional relationships for the manufacture of lenzilumab and our ability to commence commercialization and begin generating revenues from product sales if lenzilumab were to be issued an EUA and eventually approved under a BLA.
| 59 |
Until we can generate a sufficient amount of revenue, we may finance future cash needs through public or private equity offerings, license agreements, grant financing and support from governmental agencies, convertible debt, other debt financings, collaborations, strategic alliances and marketing, supply, distribution or licensing arrangements. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available, we may be required to delay or reduce the scope of or eliminate one or more of our research or development programs, our commercialization efforts or our manufacturing commitments and capacity. We may seek to access the public or private capital markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time. In addition, if we raise additional funds through collaborations, strategic alliances or marketing, supply, distribution, or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams or product candidates or to grant licenses on terms that may not be favorable to us.
Our business depends on the success of our current product candidates. We cannot be certain that we will be able to obtain regulatory approval for, or successfully commercialize, any of our product candidates.
We have a limited pipeline of product candidates and we do not plan to conduct active research at this time for discovery of new molecules or antibodies. We depend on the successful continued development and regulatory approval of our current product candidates for our future business success. Since the fall of 2017, our primary focus has been the development of lenzilumab. We are also working to create next-generation gene-edited CAR-T therapies using GM-CSF gene knockout technologies, as well as working to develop ifabotuzumab and related products. We will need to successfully enroll and complete clinical trials of lenzilumab and ifabotuzumab, and potentially obtain regulatory approval to market these products. The future clinical, regulatory and commercial success of our product candidates is subject to a number of risks, including the following:
| · | we may not be able to enroll adequate numbers of eligible patients in the clinical trials we propose to conduct, whether alone or through collaborations, including the collaboration with Kite and the IMPACT partnership; |
| · | we may not have sufficient financial and other resources to fund our clinical trials or collaborations; |
| · | we may not be able to provide acceptable evidence of safety and efficacy for our product candidates; |
| · | the results of our clinical trials or collaborations may not meet the level of statistical or clinical significance, or product safety, required to move to the next stage of development or, ultimately, obtain marketing approval from the FDA; |
| · | we may not be able to obtain, maintain and enforce our patents and other intellectual property rights; and |
| · | we may not be able to obtain and maintain commercial manufacturing arrangements with third-party manufacturers or establish commercial-scale manufacturing capabilities. |
Furthermore, even if we do receive regulatory approval to market any of our product candidates, any such approval may be subject to limitations on the indicated uses for which we may market the product. If any of our product candidates are unsuccessful, that could have a substantial negative impact on our business.
Accordingly, we cannot assure you that our product candidates will be successfully developed or commercialized. If we or any future development partners are unable to develop, or obtain regulatory approval for or, if approved, successfully commercialize, one or more of our product candidates, we may not be able to generate sufficient revenue to continue our business.
Our product candidates are at an early stage of development and may not be successfully developed or commercialized.
Our product candidates are in the early stages of development and will require substantial clinical development, testing, and regulatory approval prior to commercialization. Of the large number of drugs in development, only a small percentage successfully completes the FDA regulatory approval process and are commercialized. Accordingly, we cannot assure you that our product candidates will be successfully developed or commercialized. If we or any future development partners are unable to develop, or obtain regulatory approval for or, if approved, successfully commercialize, one or more of our product candidates, we may not be able to generate sufficient revenue to continue our business.
| 60 |
The adoption of CAR-T therapies as the potential standard of care for treatment of certain cancers is uncertain, and dependent on the efforts of a limited number of market entrants, and if not adopted as anticipated, the market for lenzilumab or next-generation gene-edited CAR-T therapies may be limited or not develop.
We are seeking to advance the development of lenzilumab to address, among other things, the serious and potentially fatal side effects associated with CAR-T therapies and to improve the efficacy of these treatments. We are also working to create next-generation gene-edited CAR-T therapies using GM-CSF gene knockout technologies. Although two CAR-T therapies have been approved by FDA to date, the use of engineered T cells as a potential cancer treatment is a recent development and may not be broadly accepted by physicians, patients, hospitals, cancer treatment centers, payers and others in the medical community. The degree of market acceptance of any approved product candidates will depend on a number of factors, including:
| · | the efficacy and safety as demonstrated in clinical trials; |
| · | the clinical indications for which the product candidate is approved; |
| · | acceptance by physicians, major operators of hospitals and clinics, and patients of the product candidate as a safe and effective treatment; |
| · | the potential and perceived advantages of product candidates over alternative treatments; |
| · | the safety of product candidates seen in a broader patient group, including its use outside the approved indications; |
| · | competitive approaches to tackle similar issues; |
| · | the cost of treatment in relation to alternative treatments; |
| · | the availability of adequate reimbursement and pricing by payers; |
| · | relative convenience and ease of administration; |
| · | the prevalence and severity of adverse events; |
| · | the effectiveness of sales and marketing efforts; and |
| · | the ability to manage any unfavorable publicity relating to the product candidate. |
If the medical and payer community is not sufficiently persuaded of the safety, efficacy and cost-effectiveness of CAR-T therapy and the potential advantages of using lenzilumab compared to existing and future therapeutics, and there is not significant market acceptance of CAR-T therapy as the standard of care for treatment of certain cancers, the market for lenzilumab or next-generation gene-edited CAR-T therapies may be limited or not develop, and our stock price could be adversely affected.
CAR-T therapies currently in early development purport to incorporate technology that may minimize or eliminate the adverse side-effects that we believe have impaired the uptake of the approved CAR-T therapies. If these developing therapies are proven equally efficacious in their proposed indications and approved for use by FDA and other regulatory agencies, the market growth for the currently approved CAR-T therapies may be limited, impairing demand for lenzilumab.
In recent months, several biotechnology companies describing business plans focusing on development of CAR-T therapies have completed or announced they are pursuing initial public offerings, or “IPOs”. Several of these companies have described their belief that their therapies will not result in the same level of CRS or NT as has been experienced in use of previously FDA-approved CAR-T therapies. While these products are in early-stage development, the data is limited and these products have not yet been approved for use by FDA, if any such product were also proven equally efficacious and subsequently approved, the market for lenzilumab may not develop or grow as anticipated. Recently the FDA approved Breyanzi), a new CAR-T cell therapy for adults with relapsed or refractory Large B-cell Lymphoma. Grade ≥3 cytokine release syndrome and Grade ≥3 neurologic toxicities following Breyanzi treatment occurred in 4% and 12% of patients, respectively. If new CAR-T therapies with lower occurrences of CRS and NT are approved, the market for lenzilumab in conjunction with CAR-T therapy may be diminished. Any such failure of a market for lenzilumab to develop could adversely affect our stock price.
| 61 |
Our business could target benefits from various regulatory incentives, such as Emergency Use Authorization, orphan drug exclusivity, breakthrough therapy designation, fast track designation, and priority review, but we may not ultimately qualify for or benefit from these arrangements.
We may seek various regulatory incentives, such as Emergency Use Authorization, orphan drug exclusivity, breakthrough therapy designation, fast track designation, accelerated approval, priority review and Priority Review Vouchers (“PRVs”), where available, that provide for certain periods of exclusivity, expedited review and/or other benefits, and we may also seek similar designations elsewhere in the world. Often, regulatory agencies have broad discretion in determining whether or not products qualify for such regulatory incentives and benefits. We cannot guarantee that we will be able to receive orphan drug status from FDA or equivalent regulatory designations elsewhere. We also cannot guarantee that we will obtain breakthrough therapy or fast track designation, which may provide certain potential benefits such as more frequent meetings with FDA to discuss the development plan, intensive guidance on an efficient drug development program, and potential eligibility for rolling review or priority review. Legislative developments in the U.S., including recent proposed legislation that would restrict eligibility for PRVs, may affect our ability to qualify for these programs in the future.
Even if we are successful in obtaining beneficial regulatory designations by FDA or other regulatory agency for our product candidates, such designations may not lead to faster development or regulatory review or approval, and it does not increase the likelihood that our product candidates will receive marketing approval. We may not be able to obtain or maintain such designations for our product candidates, and our competitors may obtain these designations for their product candidates, which could impact our ability to develop and commercialize our product candidates or compete with such competitors, which would adversely impact our business, financial condition or results of operations.
There is a limited amount of information about us upon which investors can evaluate our product candidates and business prospects, including because we have a limited operating history developing product candidates, have not yet successfully commercialized any products, have changed our strategy and have a small management team.
Our primary focus is developing our proprietary monoclonal antibody portfolio, which comprises lenzilumab, ifabotuzumab and HGEN005. We are also currently developing our GM-CSF knockout gene-editing CAR-T platform to create next-generation CAR-T therapies that preserve the benefits of CAR-T therapy while altogether avoiding its serious and potentially life-threatening side-effects. Our limited operating history developing clinical-stage product candidates may make it more difficult for us to succeed or for investors to be able to evaluate our business and prospects. In addition, as an early-stage clinical development company, we have limited experience in development activities, including conducting clinical trials, or seeking and obtaining regulatory approvals, even though our executives have had relevant experience at other companies. We currently have ten full-time employees and therefore are heavily dependent on external consultants and expert vendors for scientific, clinical manufacturing and regulatory expertise. We have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in the biopharmaceutical area. To execute our business plan we will need to successfully:
| · | execute our product candidate development activities, including successfully completing our clinical trial programs, including our collaborations with Kite and the IMPACT partnership; |
| · | obtain required regulatory approvals or authoriztions for the development and commercialization of our product candidates; |
| · | manage our costs and expenses related to clinical trials, regulatory approvals, manufacturing and commercialization; |
| · | secure substantial additional funding; |
| · | develop and maintain successful strategic relationships; |
| · | build and maintain a strong intellectual property portfolio; |
| · | build and maintain appropriate clinical, sales, manufacturing, distribution, and marketing capabilities on our own or through third parties; and |
| 62 |
| · | gain market acceptance and favorable reimbursement status for our product candidates. |
If we are unsuccessful in accomplishing these objectives, we may not be able to develop product candidates, raise capital, expand our business, or continue our operations.
Our collaboration with Kite is critically important to our business. If we are unable to maintain this collaboration, or if this collaboration is not successful, our business could be adversely affected.
In May 2019, we entered into the Kite Agreement to conduct a multi-center Phase 1b/2 study of lenzilumab with Kite’s Yescarta in patients with relapsed or refractory B-cell lymphoma. See "Business — Kite Collaboration."
Pursuant to the terms of the Kite Agreement, Kite may elect to terminate or suspend the Study at any time. Because we currently rely on Kite for a substantial portion of our discovery capabilities, if Kite delays or fails to perform its obligations under the Kite Agreement, disagrees with our interpretation of the terms of the collaboration or our discovery plan or terminates the Kite Agreement, our pipeline of product candidates would be adversely affected. Kite may also fail to properly maintain or defend the intellectual property we have licensed from them, or even infringe upon, our intellectual property rights, leading to the potential invalidation of our intellectual property or subjecting us to litigation or arbitration, any of which would be time-consuming and expensive. Additionally, either party has the right to terminate the Kite Agreement under certain circumstances. If our collaboration with Kite is terminated, the development of lenzilumab would be materially delayed or harmed.
In addition to our collaboration with Kite and the IMPACT partnership, we may, in the future, seek to enter into collaborations with other third parties for the discovery, development and commercialization of our product candidates. If our collaborators cease development efforts under our collaboration agreements, or if any of those agreements are terminated, these collaborations may fail to lead to commercial products and we may never receive milestone payments or future royalties under these agreements.
We may in the future seek to enter into agreements with other third-party collaborators for research, development and commercialization of other therapeutic technologies or product candidates. Biopharmaceutical companies are our likely future collaborators for any marketing, distribution, development, licensing, or broader collaboration arrangements. If we fail to enter into future collaborations on commercially reasonable terms, or at all, or such collaborations are not successful, we may not be able to execute our strategy to develop our product candidates or therapies that we believe could benefit from the resources of either larger biopharmaceutical companies or those specialized in a particular area of relevance.
With respect to our existing Kite Agreement and with any future collaboration agreements, we have limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of our product candidates. Moreover, our ability to generate revenues from these arrangements will depend on our collaborators' abilities to successfully perform the functions assigned to them in these arrangements.
Collaborations involving our product candidates pose the following risks to us:
| · | collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations; |
| · | collaborators may not pursue development and commercialization of our product candidates or may elect not to continue or renew development or commercialization programs based on preclinical studies or clinical trial results, changes in the collaborators' strategic focus or available funding, or external factors such as an acquisition that diverts resources or creates competing priorities; |
| · | collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing; |
| · | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidates if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours; |
| 63 |
| · | collaborators with marketing and distribution rights to one or more products may not commit sufficient resources to the marketing and distribution of such product or products; |
| · | collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to litigation or potential liability; |
| · | collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability; |
| · | disputes may arise between the collaborators and us that result in the delay or termination of the research, development or commercialization of our product candidates or that result in costly litigation or arbitration that diverts management attention and resources; and |
| · | collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates. |
As a result of the foregoing, our current and any future collaboration agreements may not lead to development or commercialization of our product candidates in the most efficient manner or at all. If a collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development or commercialization program could be delayed, diminished or terminated. Any failure to successfully develop or commercialize our product candidates pursuant to our current or any future collaboration agreements could have a material and adverse effect on our business, financial condition, results of operations and prospects.
Moreover, to the extent that any of our existing or future collaborators were to terminate a collaboration agreement, we may be forced to independently develop these product candidates, including funding preclinical studies or clinical trials, assuming marketing and distribution costs and defending intellectual property rights, or, in certain instances, abandon product candidates altogether, any of which could result in a change to our business plan and have a material adverse effect on our business, financial condition, results of operations and prospects.
We have relied and may in the future rely on third parties to conduct investigator-sponsored trials (“ISTs”) of our products, which is cost-effective for us but affords the investigators the ability to retain significant control over the design and conduct of the trials, as well as the use of the data generated from their efforts.
We have relied and may in the future rely on third parties to conduct and sponsor clinical trials relating to lenzilumab, our GM-CSF gene knockout platform and our other immunotherapies, ifabotuzumab and HGEN005. Such ISTs may provide us with valuable clinical data that can inform our future development strategy in a cost-efficient manner, but we do not control the design or conduct of the ISTs, and it is possible that the FDA or non-U.S. regulatory authorities will not view these ISTs as providing adequate support for future clinical trials, whether controlled by us or third parties, for any one or more reasons, including elements of the design or execution of the trials or safety concerns or other trial results.
These arrangements provide us limited information rights with respect to the ISTs, including access to and the ability to use and reference the data, including for our own regulatory filings, resulting from the ISTs. However, we would not have control over the timing and reporting of the data from ISTs, nor would we own the data from the ISTs. If we are unable to confirm or replicate the results from the ISTs or if negative results are obtained, we would likely be further delayed or prevented from advancing further clinical development. Further, if investigators or institutions breach their obligations with respect to the clinical development of our product candidates, or if the data proves to be inadequate compared to the first-hand knowledge, we might have gained had the ISTs been sponsored and conducted by us, then our ability to design and conduct any future clinical trials ourselves may be adversely affected.
| 64 |
If the third parties conducting our clinical trials do not conduct the trials in accordance with our agreements with them, our ability to pursue our clinical development programs could be delayed or unsuccessful and we may not be able to obtain regulatory approval for or commercialize our product candidates when expected or at all.
We do not have the ability to conduct all aspects of our preclinical testing or clinical trials ourselves. Therefore, the timing of the initiation and completion of these trials is uncertain and may occur on substantially different timing from our estimates. We also use CROs to conduct our clinical trials and rely on medical institutions, clinical investigators, CROs, and consultants to conduct our trials in accordance with our clinical protocols and regulatory requirements. Our CROs, investigators, and other third parties play a significant role in the conduct of these trials and subsequent collection and analysis of data.
There is no guarantee that any CROs, investigators, or other third parties on which we rely for administration and conduct of our clinical trials will devote adequate time and resources to such trials or perform as contractually required. If any of these third parties fails to meet expected deadlines, fails to adhere to our clinical protocols, or otherwise performs in a substandard manner, our clinical trials may be extended, delayed, or terminated. If any of our clinical trial sites terminates for any reason, we may experience the loss of follow-up information on subjects enrolled in our ongoing clinical trials unless we are able to transfer those subjects to another qualified clinical trial site. In addition, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and may receive cash or equity compensation in connection with such services. If these relationships and any related compensation result in perceived or actual conflicts of interest, the integrity of the data generated at the applicable clinical trial site may be jeopardized.
We may experience delays in commencing or conducting our clinical trials, in receiving data from third parties or in the continuation or completion of clinical testing, which could result in increased costs to us, delay our ability to generate product candidate revenue or, ultimately, render us unable to complete the development and commercialization of our product candidates.
We have product candidates in clinical development and preclinical development. The risk of failure for each of our product candidates is high. It is impossible to predict when or if any of our product candidates will prove effective or safe in humans or will receive regulatory approval. Before obtaining marketing approval from regulatory authorities for the sale of any product candidate, we must complete preclinical development and then conduct extensive clinical trials to demonstrate the safety and efficacy of our product candidates in humans.
Before we can initiate clinical trials in the United States for any new product candidates, we are required to submit the results of preclinical testing to FDA as part of an IND application, along with other information including information about product candidate chemistry, manufacturing, and controls and our proposed clinical trial protocol. For our programs already underway, we are required to report or provide information to appropriate regulatory authorities in order to continue with our testing programs. If we are unable to make timely regulatory submissions for any of our programs, it will delay our plans for our clinical trials. If those third parties do not make the required data available to us, we will likely have to identify and contract with another third party, and/or develop all necessary preclinical and clinical data on our own, which will lead to significant delays and increase development costs of the product candidate. In addition, FDA may require us to conduct additional preclinical testing for any product candidate before it allows us to initiate clinical testing under any IND application, which may lead to additional delays and increase the costs of our preclinical development. Moreover, despite the presence of an active IND application for a product candidate, clinical trials can be delayed for a variety of reasons, including delays in:
| · | identifying, recruiting, and enrolling qualified subjects to participate in a clinical trial; |
| · | identifying, recruiting, and training suitable clinical investigators; |
| · | reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and trial sites, the terms of which can be subject to extensive negotiation, may be subject to modification from time to time, and may vary significantly among different CROs and trial sites; |
| · | obtaining and maintaining sufficient quantities of a product candidate for use in clinical trials, either as a result of transferring the manufacturing of a product candidate to another site or manufacturer, deferring ordering or production of product in order to conserve resources or mitigate risk, having product in inventory become no longer suitable for use in humans, or other reasons that reduce or delay availability of drug supply; |
| 65 |
| · | obtaining and maintaining Institutional Review Board (“IRB”) or ethics committee approval to conduct a clinical trial at an existing or prospective site; |
| · | retaining or replacing participants who have initiated a clinical trial but may withdraw due to adverse events from the therapy, insufficient efficacy, fatigue with the clinical trial process, or personal issues; |
| · | developing any companion diagnostic necessary to ensure the study enrolls the target population; |
| · | being required by the FDA to add more patients or sites or to conduct additional trials; or |
| · | the FDA placing a clinical trial on hold. |
Once a clinical trial has begun, recruitment and enrollment of subjects may be slower than we anticipate. Numerous companies and institutions are conducting clinical studies in similar patient populations which can result in competition for qualified patients. In addition, clinical trials will take longer than we anticipate if we are required, or believe it is necessary, to enroll additional subjects than originally planned. Clinical trials may also be delayed as a result of ambiguous or negative interim results. Further, a clinical trial may be suspended or terminated by us, an IRB, an ethics committee, or a data safety monitoring committee overseeing the clinical trial, any of our clinical trial sites with respect to that site, or FDA or other regulatory authorities, due to several factors, including:
| · | failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
| · | inspection of the clinical trial operations or clinical trial site by the FDA or other regulatory authorities; |
| · | inability to provide timely supply of drug product due to shortages or other issues with the drug product; |
| · | unforeseen safety issues, known safety issues that occur at a greater frequency or severity than we anticipate, or any determination that the clinical trial presents unacceptable health risks; or |
| · | lack of adequate funding to continue the clinical trial or unforeseen significant incremental costs related to the trial. |
Additionally, if any future development partners do not develop the licensed product candidates in the time and manner that we expect, or at all, the clinical development efforts related to these licensed product candidates could be delayed or terminated. In addition, our ability to enforce our partners’ obligations under any future collaboration efforts may be limited due to time and resource constraints, competing corporate priorities of our future partners, and other factors.
Any delays in the commencement of our clinical trials may delay or preclude our ability to further develop or pursue regulatory approval for our product candidates. Changes in U.S. and foreign regulatory requirements and guidance also may occur, and we may need to amend clinical trial protocols to reflect these changes. Amendments may require us to resubmit our clinical trial protocols to IRBs for re-examination, which may affect the costs, timing, and likelihood of a successful completion of a clinical trial.
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, if we are unable to successfully complete clinical trials of our product candidates or other testing, if the results of these trials or tests are not positive or are only modestly positive or if there are safety concerns, we may:
| · | be delayed in obtaining marketing approval for our product candidates; |
| · | not obtain marketing approval or EUA at all; |
| · | obtain approval for indications or patient populations that are not as broad as intended or desired; |
| · | obtain approval with labeling that includes significant use or distribution restrictions or safety warnings; |
| · | be subject to additional post-marketing testing requirements; or |
| · | have the product removed from the market after obtaining marketing approval. |
| 66 |
Our product development costs will also increase if we experience delays in testing or in obtaining marketing approvals. We do not know whether any of our preclinical studies or clinical trials will begin as planned, will need to be restructured or will be completed on schedule, or at all. We may also determine to change the design or protocol of one or more of our clinical trials, including to add additional arms or patient populations, which could result in increased costs and expenses and/or delays. If we or any future development partners experience delays in the completion of, or if we or any future development partners must terminate, any clinical trial of any product candidate our ability to obtain regulatory approval for that product candidate will be delayed and the commercial prospects, if any, for the product candidate may suffer as a result. Significant preclinical study or clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our product candidates and may harm our business and results of operations. In addition, many of these factors may also ultimately lead to the denial of regulatory approval of a product candidate.
Our product candidates are subject to extensive regulation, compliance with which is costly and time consuming, may cause unanticipated delays, or may prevent the receipt of the required approvals to commercialize our product candidates.
The clinical development, approval, manufacturing, labeling, storage, record-keeping, advertising, promotion, import, export, marketing, and distribution of our product candidates are subject to extensive regulation by FDA in the United States and by comparable authorities in foreign markets. In the United States, we are not permitted to market our product candidates until we receive regulatory approval from FDA. The process of obtaining regulatory approval is expensive, often takes many years, and can vary substantially based upon the type, complexity, and novelty of the products involved, as well as the target indications. Approval policies or regulations may change, and FDA has substantial discretion in the drug approval process, including the ability to delay, limit, or deny approval of a product candidate for many reasons. Despite the time and expense invested in clinical development of product candidates, regulatory approval is never guaranteed. FDA or other comparable foreign regulatory authorities can delay, limit, or deny approval of a product candidate for many reasons, including:
| · | such authorities may disagree with the design or implementation of our or any future development partners’ clinical trials; |
| · | such authorities may not accept clinical data from trials that are conducted at clinical facilities or in countries where the standard of care is potentially different from the United States; |
| · | the results of clinical trials may not demonstrate the safety or efficacy required by such authorities for approval; |
| · | we or any future development partners may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; |
| · | such authorities may disagree with our interpretation of data from preclinical studies or clinical; |
| · | such authorities may find deficiencies in the manufacturing processes or facilities of third-party manufacturers with which we or any future development partners contract for clinical and commercial supplies; or |
| · | the approval policies or regulations of such authorities may significantly change in a manner rendering our or any future development partners’ clinical data insufficient for approval. |
With respect to foreign markets, approval procedures vary widely among countries and, in addition to the aforementioned risks, can involve additional product testing, administrative review periods, and agreements with pricing authorities. In addition, events raising questions about the safety of certain marketed pharmaceuticals may result in increased caution by FDA and comparable foreign regulatory authorities in reviewing new drugs based on safety, efficacy or other regulatory considerations and may result in significant delays in obtaining regulatory approvals. Any delay in obtaining, or inability to obtain, applicable regulatory approvals may delay or prevent us or any future development partners from commercializing our product candidates.
| 67 |
The results of preclinical studies and early clinical trials are not always predictive of future results. Any product candidate we or any future development partners advance into clinical trials may not have favorable results in later clinical trials, if any, or receive regulatory approval.
Drug development has substantial inherent risk. We or any future development partners will be required to demonstrate through adequate and well-controlled clinical trials that our product candidates are effective, with a favorable benefit-risk profile, for use in their target populations for their intended indications before we can seek regulatory approvals for their commercial sale. Drug development is a long, expensive and uncertain process, and delay or failure can occur at any stage of development, including after commencement of any of our clinical trials. Success in early clinical trials does not mean that later clinical trials will be successful because product candidates in later-stage clinical trials may fail to demonstrate sufficient safety or efficacy despite having progressed through initial clinical testing. Furthermore, our future trials will need to demonstrate sufficient safety and efficacy for approval by regulatory authorities in larger patient populations. Companies frequently suffer significant setbacks in advanced clinical trials, even after earlier clinical trials have shown promising results. In addition, only a small percentage of drugs under development result in the submission of an NDA or BLA to FDA and even fewer are approved for commercialization.
In addition, serious adverse or undesirable side effects may emerge or be identified during later stages of development that were not observed in earlier stages. If our product candidates, either alone or in combination with other therapeutics, are associated with serious adverse events or undesirable side effects or unacceptable drug-drug interactions in clinical trials or have characteristics that are unexpected in clinical trials or preclinical testing, we may need to abandon their development or limit development to more narrow uses or subpopulations in which the serious adverse events, undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective. In pharmaceutical development, many compounds that initially show promise in early-stage or clinical testing are later found to cause side effects that prevent further development of the compound. In addition, if third parties manufacture or use our product candidates without our permission, and generate adverse events or unacceptable side effects, this could also have an adverse impact on our development efforts.
Unacceptable adverse events caused by any of our product candidates that we advance into clinical trials could cause us or regulatory authorities to interrupt, delay, or halt clinical trials and could result in the denial of regulatory approval by FDA or other regulatory authorities for any or all targeted indications and markets. This in turn could prevent us from completing development or commercializing the affected product candidate and generating revenue from its sale. We have not yet successfully completed testing of any of our product candidates for the treatment of the indications for which we intend to seek approval in humans, and we currently do not know the extent of adverse events, if any, that will be observed in individuals who receive any of our product candidates. If any of our product candidates cause unacceptable adverse events in clinical trials, we may not be able to obtain regulatory approval or commercialize such product candidates.
We face risks associated with clinical operations abroad, which may adversely affect our financial condition and results of operations.
The Brazilian Health Regulatory Agency, Anvisa, has granted us permission to expand our Phase 3 study of lenzilumab in patients with COVID-19 in Brazil and we enrolled patients in that country. In addition, the Mexican regulatory agency, COFEPRIS, has granted us permission to expand the Phase 3 study of lenzilumab in patients with COVID-19 in Mexico. We did not open any sites in Mexico or enroll any patients in that country; however, we may do so in the future for a different study. Partners also are conducting or planning to conduct clinical trials involving lenzilumab in Australia, Korea and the United Kingdom and potentially other countries in patients with COVID-19 and other indications. We have not received any authorization or approval to sell lenzilumab in any country but would expect to commence commercial operations, through a partner or on our own, once any such authorization or approval were received. Operations abroad are accompanied by certain financial, political, economic and other risks, including those listed below:
| · | Foreign Currency Exchange: Operations internationally may subject us to risks related to foreign currency exchange risks as we make payments, or incur obligations, denominated in foreign currencies. We cannot predict future fluctuations in the foreign currency exchange rates of the U.S. dollar. If the U.S. dollar appreciates significantly against certain currencies and our practices do not sufficiently offset the effects of such appreciation, our results of operations would be adversely affected, and our stock price may decline. |
| · | Anti-Bribery: We are subject to the U.S. Foreign Corrupt Practices Act and similar worldwide anti-bribery laws that will govern our international operations with respect to payments to government officials. Our international operations would be heavily regulated and require significant interaction with foreign officials. In certain circumstances, strict compliance with anti-bribery laws may conflict with local customs and practices or may require us to interact with doctors and hospitals, some of which may be state controlled, in a manner that is different than local custom. In addition, despite our efforts, our policies and procedures may not protect us from reckless or criminal acts committed by persons who act on our behalf. Enforcement activities under anti-bribery laws could subject us to administrative and legal proceedings and actions, which could result in civil and criminal sanctions, including monetary penalties and exclusion from health care programs. |
| 68 |
| · | Other risks inherent in conducting foreign operations include: |
| o | International operations, including any use of third-party manufacturers, distributors, CROs and collaboration arrangements outside the United States, expose us to increased risk of theft of our intellectual property and other proprietary technology, particularly in jurisdictions with less robust intellectual property protections than the United States, as well as restrictive government actions against our intellectual property and other foreign assets such as nationalization, expropriation or the imposition of compulsory licenses. |
| o | We may be subject to protective economic policies taken by foreign governments, such as trade protection measures and import and export licensing requirements, which may result in the imposition of trade sanctions or similar restrictions by the United States or other governments. |
| o | Our foreign operations, third-party manufacturers, CROs or strategic partners could be subject to business interruptions for which we or they may be uninsured or inadequately insured. |
| o | Our operations may also be adversely affected if there is political instability or disruption in any other geographic region where we may have operations, which could impact our ability to do business in those areas. |
If we were to encounter any of these risks, our foreign operations may be adversely affected, which could have an adverse effect on our overall business and results of operations.
If we fail to attract and retain key management and clinical development personnel, or if the attention of such personnel is diverted, we may be unable to successfully manage our business and develop or commercialize our product candidates.
We will need to effectively manage our managerial, operational, financial, and other resources in order to successfully pursue our clinical development and commercialization efforts. As a company with a limited number of personnel, we are heavily affected by turnover and highly dependent on the expertise of the members of our senior management, in particular our Chief Executive Officer, Dr. Cameron Durrant, and Dr. Dale Chappell, the controlling owner of the Black Horse Entities (as defined below), our current Chief Scientific Officer and a director. Furthermore, we rely on third party consultants for a variety of services. We cannot predict the impact of the loss of such individuals or the loss of services of any of our other senior management, should they occur, or the difficulty in replacing such individuals. Such losses could delay or prevent the further development and potential commercialization of our product candidates and, if we are not successful in finding suitable replacements, could harm our business.
If our competitors develop treatments for the target indications of our product candidates that are approved more quickly, marketed more successfully or are demonstrated to be safer or more effective than our product candidates, or if FDA approves generic or biosimilar competitors to our products post-approval, our commercial opportunity will be reduced or eliminated.
We compete in an industry characterized by rapidly advancing technologies, intense competition, a changing regulatory and legislative landscape and a strong emphasis on the benefits of intellectual property protection and regulatory exclusivities. Our competitors include pharmaceutical companies, other biotechnology companies, academic institutions, government agencies and other private and public research organizations. We compete with these parties in immunotherapy and oncology treatments and in recruiting highly qualified personnel. Our product candidates, if successfully developed and approved, may compete with established therapies, with new treatments that may be introduced by our competitors, including competitors relying on our biologics approvals under section 351(k) of the Public Health Service Act, or with generic copies of our products approved by FDA under an abbreviated new drug application (“ANDA”), referencing our drug products. We believe that competitors are actively developing competing products to our product candidates. See “Competition” in the “Business” section of this prospectus for a discussion of competition with respect to our current product candidates.
| 69 |
Many of our competitors and potential competitors have substantially greater scientific, research, and product development capabilities, as well as greater financial, marketing, sales and human resources capabilities than we do. In addition, many specialized biotechnology firms have formed collaborations with large, established companies to support the research, development and commercialization of products that may be competitive with ours. Accordingly, our competitors may be more successful with respect to their products than we may be in developing, commercializing, and achieving widespread market acceptance for our products. If a competitor obtains approval for an orphan drug that is the same drug or the same biologic as one of our candidates before we do, we will be blocked from obtaining FDA approval for seven years from the date of the competitor’s product, unless we can establish that our product is clinically superior to the previously-approved competitor’s product or we can meet another exception, such as by showing that the competitor has failed to provide an adequate supply of its product to patients after approval. In addition, our competitors’ products may be more effective or more effectively marketed and sold than any treatment we or our development partners may commercialize and may render our product candidates obsolete or non-competitive before we can recover the expenses related to developing and supporting the commercialization of any of our product candidates. Developments by competitors may render our product candidates obsolete or noncompetitive. After one of our product candidates is approved, FDA may also approve a generic version with the same dosage form, safety, strength, route of administration, quality, performance characteristics and intended use as our product. These generic equivalents would be less costly to bring to market and could generally be offered at lower prices, thereby limiting our ability to gain or retain market share.
The acquisition or licensing of pharmaceutical products is also very competitive, and a number of more established companies, which have acknowledged strategies to in-license or acquire products, may have competitive advantages as may other emerging companies taking similar or different approaches to product acquisitions. The more established companies may have a competitive advantage over us due to their size, cash flows, institutional experience and historical corporate reputation.
We are subject to a multitude of manufacturing risks, any of which could substantially increase our costs and limit supply of our products.
We are wholly dependent on third party contract manufacturers for the timely supply of adequate quantities of our products which meet or exceed requisite quality and production standards for use in clinical and nonclinical studies. Given the extensive risks, scope, complexity, cost, regulatory requirements, and commitment of resources associated with developing the capabilities to manufacture one or more of our products, we have no present plan or intention of developing in-house manufacturing capabilities for nonclinical, clinical or commercial scale production, beyond our current supervision and management of our third-party contract manufacturers. In addition, in order to balance risk and conserve financial and human resources, we have and may continue from time to time to defer commitment to production of product, which could result in delays to the continued progress of our clinical and nonclinical testing.
In addition to the foregoing, the process of manufacturing our products is complex, highly regulated, and subject to several risks, including but not limited to the following:
| · | We, and our contract manufacturers, must comply with FDA’s current Good Manufacturing Practice, (“cGMP”), regulations and guidance. We, and our contract manufacturers, may encounter difficulties in achieving quality control and quality assurance and may experience shortages in qualified personnel. We, and our contract manufacturers, are subject to inspections by FDA and comparable agencies in other jurisdictions to confirm compliance with applicable regulatory requirements. Any failure to follow cGMP or other regulatory requirements or any delay, interruption or other issues that arise in the manufacture, fill-finish, packaging, or storage of our products as a result of a failure of our facilities or the facilities or operations of third parties to comply with regulatory requirements, or a failure to pass any regulatory authority inspection, could significantly impair our ability to develop and commercialize our products, including leading to significant delays in the availability of products for our clinical studies or the termination or hold on a clinical study, or the delay or prevention of a filing or approval of marketing applications for our product candidates. Significant noncompliance could also result in the imposition of sanctions, including injunctions, civil penalties, failure of regulatory authorities to grant marketing approvals for our product candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of products, operating restrictions, adverse publicity, and criminal prosecutions, any of which could damage our reputation. If we are not able to maintain regulatory compliance, we may not be permitted to market our products and/or may be subject to product recalls, seizures, injunctions, or criminal prosecution. Any adverse developments affecting manufacturing operations for our products may result in shipment delays, inventory shortages, lot failures, product withdrawals or recalls, or other interruptions in the supply of our products. Once our product candidates are approved, we may also have to take inventory write-offs and incur other charges and expenses for products that fail to meet specifications, undertake costly remediation efforts or seek more costly manufacturing alternatives. |
| 70 |
| · | The manufacturing facilities in which our products are made could be adversely affected by equipment failures, plant closures, capacity constraints, competing customer priorities or changes in corporate strategy or priorities, process changes or failures, changes in business models or operations, materials or labor shortages, natural disasters, power failures and numerous other factors. |
| · | We are wholly dependent upon third party CMOs for the timely supply of adequate quantities of requisite quality product for our nonclinical, clinical and, if approved by regulatory authorities, commercial scale production. |
If any product candidate that we successfully develop does not achieve broad market acceptance among physicians, patients, healthcare payers and the medical community, the revenue that it generates may be limited.
Even if our product candidates receive regulatory approval, they may not gain market acceptance among physicians, patients, healthcare payers, and the medical community. Coverage and reimbursement of our product candidates by third-party payers, including government payers, generally is also necessary for commercial success. The degree of market acceptance of any approved product candidates will depend on several factors, including:
| · | the efficacy and safety as demonstrated in clinical trials; |
| · | the clinical indications for which the product candidate is approved; |
| · | acceptance by physicians, major operators of hospitals and clinics, and patients of the product candidate as a safe and effective treatment; |
| · | the potential and perceived advantages of product candidates over alternative treatments; |
| · | the safety of product candidates seen in a broader patient group, including its use outside the approved indications; |
| · | the cost of treatment in relation to alternative treatments; |
| · | the availability of adequate reimbursement and pricing by payers; |
| · | relative convenience and ease of administration; |
| · | the prevalence and severity of adverse events; |
| · | the effectiveness of our sales and marketing efforts; and |
| · | the ability to manage any unfavorable publicity relating to the product candidate. |
If any product candidate is approved but does not achieve an adequate level of acceptance by physicians, hospitals, healthcare payers, and patients, we may not generate sufficient revenue from that product candidate and may not become or remain commercially attractive as a standalone indication for that product.
| 71 |
Reimbursement may be limited or unavailable in certain market segments for our product candidates, which could make it difficult for us to sell our product candidates profitably.
Market acceptance and sales of our product candidates will depend significantly on the availability of adequate insurance coverage and reimbursement from third-party payers for any of our product candidates and may be affected by existing and future health care reform measures. Government authorities and third-party payers, such as private health insurers and health maintenance organizations, decide which drugs they will pay for and establish reimbursement levels. Reimbursement by a third-party payer may depend upon a number of factors including the third-party payer’s determination that use of a product candidate is:
| · | a covered benefit under its health plan; |
| · | safe, effective, and medically necessary; |
| · | appropriate for the specific patient; |
| · | cost-effective; and |
| · | neither experimental nor investigational. |
Obtaining coverage and reimbursement approval for a product candidate from a government or other third-party payer is a time-consuming and costly process that could require us to provide supporting scientific, clinical, and cost effectiveness data for the use of our product candidates to the payer. We may not be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement. We cannot be sure that coverage or adequate reimbursement will be available for any of our product candidates. Also, we cannot be sure that reimbursement amounts will not reduce the demand for, or the price of, our product candidates. If reimbursement is not available or is available only to limited levels or with restrictions, we may not be able to commercialize certain of our product candidates profitably, or at all, even if approved.
In the United States and in certain foreign jurisdictions, there have been a number of legislative and regulatory changes to the health care system that could affect our ability to sell our product candidates profitably. In particular, the Medicare Modernization Act of 2003 revised the payment methods for many product candidates under Medicare. This has resulted in lower rates of reimbursement. There have been numerous other federal and state initiatives designed to reduce payment for pharmaceuticals.
As a result of legislative proposals and the trend toward managed health care in the United States, third-party payers are increasingly attempting to contain health care costs by limiting both coverage and the level of reimbursement of new drugs. They may also refuse to provide coverage of approved product candidates for medical indications other than those for which FDA has granted market approvals. As a result, significant uncertainty exists as to whether and how much third-party payers will reimburse patients for their use of newly approved drugs, which in turn will put pressure on the pricing of drugs. We could be subject to pricing pressures in connection with the sale of our product candidates due to the trend toward managed health care, the increasing influence of health maintenance organizations, and additional legislative proposals as well as country, regional, or local healthcare budget limitations.
Similar concerns about the costs of treatment have been raised in Europe and the United Kingdom, where the cost effectiveness of CAR-T therapies have been an impediment to utilization of Kymriah and Yescarta. If CAR-T companies are not able to convince regulators and payers in national healthcare systems that the benefits of a CAR-T therapy outweigh its costs, the market for lenzilumab might not develop.
Governments may impose price controls, which may adversely affect our future profitability.
We intend to seek approval to market our future product candidates in the United States and potentially in foreign jurisdictions. If we obtain approval in one or more foreign jurisdictions, we will be subject to rules and regulations in those jurisdictions relating to our product candidates. In some foreign countries, particularly in the European Union and the United Kingdom, the pricing of prescription pharmaceuticals and biologics is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product candidate. If reimbursement of our future products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, we may be unable to achieve or sustain profitability.
| 72 |
We face potential product liability exposure and, if successful claims are brought against us, we may incur substantial liability for a product candidate and may have to limit its commercialization.
The use of our product candidates in clinical trials and the sale of any product candidates for which we may obtain marketing approval expose us to the risk of product liability claims. Product liability claims may be brought against us or any future development partners by participants enrolled in our clinical trials, patients, health care providers, or others using, administering, or selling our product candidates. If we cannot successfully defend ourselves against any such claims, or have insufficient insurance protection, we would incur substantial liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
| · | withdrawal of clinical trial participants; |
| · | termination of clinical trial sites or entire trial programs; |
| · | costs of related litigation; |
| · | substantial monetary awards to trial participants or other claimants; |
| · | decreased demand for our product candidates and loss of revenue; |
| · | impairment of our business reputation; |
| · | diversion of management and scientific resources from our business operations; and |
| · | the inability to commercialize our product candidates. |
We have obtained limited product liability insurance coverage for our clinical trials domestically and in selected foreign countries where we are conducting clinical trials. As such, our insurance coverage may not reimburse us or may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive and in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to product liability. We intend to expand our insurance coverage for product candidates to include the sale of commercial products if we obtain marketing approval for our product candidates in development; however, we may be unable to obtain commercially reasonable product liability insurance for any product candidates approved for marketing. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us, particularly if judgments exceed our insurance coverage, could decrease our working capital and adversely affect our business.
Our employees and consultants may engage in misconduct or other improper activities, including noncompliance with regulatory standards, which could have a material adverse effect on our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees or consultants could include intentional failures to comply with FDA regulations or similar regulations of comparable foreign regulatory authorities, failure to provide accurate information to FDA or comparable foreign regulatory authorities, failure to comply with manufacturing standards, failure to comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable foreign regulatory authorities, failure to report financial information or data accurately, violations of anti-bribery laws, or failure to disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee or consultant misconduct could also involve the improper use of confidential information obtained in the course of our business, which could result in civil or criminal legal actions, regulatory sanctions, or serious harm to our reputation. We have adopted a Code of Business Conduct and Ethics and other corporate policies, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant fines or other sanctions.
| 73 |
We may encounter difficulties in managing our growth and expanding our operations successfully.
As we seek to advance our product candidates through clinical trials, we will need to expand our development, regulatory, manufacturing, marketing, and sales capabilities, and contract with third parties to provide these capabilities for us. As our operations expand, we expect that we will need to manage additional relationships with various development partners, suppliers, and other third parties. Future growth will impose significant added responsibilities on members of management. Our future financial performance and our ability to commercialize our product candidates and to compete effectively will depend in part on our ability to manage any future growth effectively. To that end, we must be able to manage our development efforts and clinical trials effectively. We may not be able to accomplish these tasks and our failure to accomplish any of them could prevent us from successfully growing our company.
We and any future development partners, third-party manufacturers and suppliers use hazardous materials, and any claims relating to improper handling, storage, or disposal of these materials could be time consuming or costly.
We and any future development partners, third-party manufacturers and suppliers may use hazardous materials, including chemicals and biological agents and compounds that could be dangerous to human health and safety or the environment. Our operations and the operations of our development partner, third-party manufacturers and suppliers also produce hazardous waste products. Federal, state, and local laws and regulations govern the use, generation, manufacture, storage, handling, and disposal of these materials and wastes. Compliance with applicable environmental laws and regulations may be expensive and current or future environmental laws and regulations may impair our product development efforts. In addition, we cannot eliminate the risk of accidental injury or contamination from these materials or wastes. We do not carry specific biological or hazardous waste insurance coverage and our property, casualty, and general liability insurance policies specifically exclude coverage for damages and fines arising from biological or hazardous waste exposure or contamination. Accordingly, in the event of contamination or injury we could be held liable for damages or be penalized with fines in an amount exceeding our resources, and our clinical trials or regulatory approvals could be suspended.
Healthcare reform measures, when implemented, could hinder or prevent our commercial success.
There have been, and likely will continue to be, legislative and regulatory proposals at the federal and state levels directed at broadening the availability of health care and containing or lowering the cost of health care. We cannot predict the initiatives that may be adopted in the future. The continuing efforts of the government, insurance companies, managed care organizations, and other payers of healthcare services to contain or reduce costs of health care may adversely affect:
| · | the demand for any drug products for which we may obtain regulatory approval; |
| · | our ability to set a price that we believe is fair for our product candidates; |
| · | our ability to generate revenue and achieve or maintain profitability; |
| · | the level of taxes that we are required to pay; and |
| · | the availability of capital. |
We and any of our future development partners will be required to report to regulatory authorities if any of our approved products cause or contribute to adverse medical events, and any failure to do so would result in sanctions that would materially harm our business.
If we and any future development partners are successful in commercializing our products, FDA and foreign regulatory authorities would require that we and any future development partners report certain information about adverse medical events if those products may have caused or contributed to those adverse events. The timing of our obligation to report would be triggered by the date we become aware of the adverse event as well as the nature of the event. We and any future development partners may fail to report adverse events we become aware of within the prescribed timeframe. We and any future development partners may also fail to appreciate that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we and any future development partners fail to comply with our reporting obligations, FDA or a foreign regulatory authority could take action including criminal prosecution, the imposition of civil monetary penalties, seizure of our products, or delay in approval or clearance of future products.
| 74 |
Our product candidates for which we intend to seek approval as biologic products may face competition sooner than anticipated.
With the enactment of the Biologics Price Competition and Innovation Act of 2009, or the BPCIA, as part of the Affordable Care Act, an abbreviated pathway for the approval of biosimilar and interchangeable biological products was created. The abbreviated regulatory pathway establishes legal authority for FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as ‘‘interchangeable’’ based on its similarity to an existing brand product. Under the BPCIA, an application for a biosimilar product cannot be approved by FDA until 12 years after the original branded product was approved under a BLA. The law is complex and is still being interpreted and implemented by FDA. As a result, its ultimate impact, implementation, and meaning are subject to uncertainty. While it is uncertain when such processes intended to implement BPCIA may be fully adopted by FDA, any such processes could have a material adverse effect on the future commercial prospects for our biological products.
We believe that any of our product candidates approved as biological products under a BLA should qualify for the 12-year period of exclusivity. However, there is a risk that FDA will not consider our product candidates to be reference products for competing products, potentially creating the opportunity for biosimilar competition sooner than anticipated. Moreover, the extent to which a biosimilar, once approved, will be substituted for any one of our reference products in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing. Finally, there is a risk that the 12-year exclusivity period could be reduced which could negatively affect our products.
In addition, foreign regulatory authorities may also provide for exclusivity periods for approved biological products. For example, biological products in Europe may be eligible for a 10-year period of exclusivity. However, biosimilar products have been approved under a sub-pathway of the centralized procedure since 2006. The pathway allows sponsors of a biosimilar product to seek and obtain regulatory approval based in part on the clinical trial data of an originator product to which the biosimilar product has been demonstrated to be ‘‘similar.’’ In many cases, this allows biosimilar products to be brought to market without conducting the full suite of clinical trials typically required of originators. It is unclear whether we and our development partner would face competition to our products in European markets sooner than anticipated.
We may in the future be subject to various U.S. federal and state laws pertaining to health care fraud and abuse, including anti-kickback, self-referral, false claims and fraud laws, and any violations by us of such laws could result in fines or other penalties.
If one or more of our product candidates is approved, we will likely be subject to the various U.S. federal and state laws intended to prevent health care fraud and abuse. The federal anti-kickback statute prohibits the offer, receipt, or payment of remuneration in exchange for or to induce the referral of patients or the use of products or services that would be paid for in whole or part by Medicare, Medicaid, or other federal health care programs. Remuneration has been broadly defined to include anything of value, including cash, improper discounts, and free or reduced-price items and services. Many states have similar laws that apply to their state health care programs as well as private payers. Violations of the anti-kickback laws can result in exclusion from federal health care programs and substantial civil and criminal penalties.
The False Claims Act imposes liability on persons who, among other things, present or cause to be presented false or fraudulent claims for payment by a federal health care program. The False Claims Act has been used to prosecute persons submitting claims for payment that are inaccurate or fraudulent, that are for services not provided as claimed, or for services that are not medically necessary. The False Claims Act includes a whistleblower provision that allows individuals to bring actions on behalf of the federal government and share a portion of the recovery of successful claims. If our marketing or other arrangements were determined to violate the False Claims Act or anti-kickback or related laws, then our revenue could be adversely affected, which would likely harm our business, financial condition, and results of operations.
| 75 |
State and federal authorities have aggressively targeted medical technology companies for alleged violations of these anti-fraud statutes, based on improper research or consulting contracts with doctors, certain marketing arrangements that rely on volume-based pricing, off-label marketing schemes, and other improper promotional practices. Companies targeted in such prosecutions have paid substantial fines in the hundreds of millions of dollars or more, have been forced to implement extensive corrective action plans or corporate integrity agreements, and have often become subject to consent decrees severely restricting the manner in which they conduct their business. If we become the target of such an investigation or prosecution based on our contractual relationships with providers or institutions, or our marketing and promotional practices, we could face similar sanctions, which would materially harm our business.
Also, the Foreign Corrupt Practices Act and similar worldwide anti-bribery laws generally prohibit companies and their intermediaries from making improper payments to non-U.S. officials for the purpose of obtaining or retaining business. We cannot assure you that our internal control policies and procedures will protect us from reckless or negligent acts committed by our employees, future distributors, partners, collaborators, or agents. Violations of these laws, or allegations of such violations, could result in fines, penalties, or prosecution and have a negative impact on our business, results of operations and reputation.
Legislative or regulatory healthcare reforms in the United States may make it more difficult and costly for us to obtain regulatory approval of our product candidates and to produce, market, and distribute our products after approval is obtained.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the regulatory approval, manufacture, and marketing of regulated products or the reimbursement thereof. In addition, FDA regulations and guidance are often revised or reinterpreted by FDA in ways that may significantly affect our business and our products. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of our current product candidates or any future product candidates. We cannot determine what effect changes in regulations, statutes, legal interpretation, or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require:
| · | changes to manufacturing methods; |
| · | additional studies, including clinical studies; |
| · | recall, replacement, or discontinuance of one or more of our products; and |
| · | additional record-keeping. |
Each of these would likely entail substantial time and cost and could materially harm our business and our financial results. In addition, delays in receipt of or failure to receive regulatory approvals for any future products would harm our business, financial condition, and results of operations.
Even if we are able to obtain regulatory approval for our product candidates, we will continue to be subject to ongoing and extensive regulatory requirements, and our failure to comply with these requirements could substantially harm our business.
If we receive regulatory approval for our product candidates, we will be subject to ongoing FDA obligations and continued regulatory oversight and review, such as continued safety reporting requirements, and we may also be subject to additional FDA post-marketing obligations. If we are not able to maintain regulatory compliance, we may not be permitted to market our product candidates and/or may be subject to product recalls or seizures.
| 76 |
If the FDA approves any of our product candidates, the labeling, manufacturing, packaging, storage, distribution, export, adverse event reporting, advertising, promotion and record-keeping for our products will be subject to extensive regulatory requirements. Violations of these regulatory requirements or the subsequent discovery of previously unknown problems with the products, including adverse events of unanticipated severity or frequency, may result in:
| · | the issuance of warning or untitled letters; |
| · | requirements to conduct post-marking clinical trials; |
| · | restrictions on the marketing and distribution of the product, including potential withdrawal of the product from the market; |
| · | suspension of ongoing clinical trials; |
| · | the issuance of product recalls, import and export restrictions, seizures, and detentions; and |
| · | the issuance of injunctions, or imposition of other civil and/or criminal penalties. |
Our ability to utilize our net operating loss carryforwards and certain other tax attributes may be subject to certain limitations.
We have incurred substantial losses during our history and do not expect to become profitable in the foreseeable future and may never achieve profitability. To the extent that we continue to generate taxable losses, unused losses will carry forward to offset future taxable income, if any, until such unused losses expire. We may be unable to use these losses to offset income before such unused losses expire. The Tax Cuts and Jobs Act, enacted in 2017, limited the use of net operating loss carryforwards for periods beginning after 2017 to eighty percent of taxable income in the period to which the losses were carried. However, this limitation on the use of the carryforwards was eliminated by the Coronavirus Aid, Relief and Economic Security Act (the “CARES” Act) for tax years beginning before January 1, 2021. In addition, Section 382 of the Internal Revenue Code of 1986, as amended, may limit the utilization of net operating loss carryforwards. Under Section 382, if a corporation undergoes an ‘‘ownership change’’ (generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period), the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes to offset its post-change income may be limited. We have recently and in the past experienced ownership changes that have resulted in limitations on the use of a portion of our net operating loss carryforwards. If we experience further ownership changes our ability to utilize our net operating loss carryforwards could be further limited.
We rely completely on third parties, most of which are sole source suppliers, to supply drug substance and manufacture drug product for our clinical trials and preclinical studies and intend to rely on other third parties to produce commercial supplies of product candidates, and our dependence on third parties could adversely impact our business.
We are completely dependent on third-party suppliers, most of which are sole source suppliers of the drug substance and drug product for our product candidates. We regularly evaluate potential alternate sources of supply but there can be no assurance that any such suppliers would be available, acceptable, or successful. The costs of manufacturing our drug candidates are high, and we will require additional capital to ensure that we can maintain an adequate supply to conduct our contemplated development programs.
If our third-party suppliers do not supply sufficient quantities for product candidates to us on a timely basis and in accordance with applicable specifications and other regulatory requirements, there could be a significant interruption of our supplies, which would adversely affect clinical development of the product candidate, including affecting our ability to enroll in and timely progress clinical trials. Furthermore, if any of our contract manufacturers cannot successfully manufacture material that conforms to our specifications and with regulatory requirements, we will not be able to secure and/or maintain regulatory approval, if any, for our product candidates.
We will also rely on our contract manufacturers to purchase from third-party suppliers the materials necessary to produce our product candidates for our anticipated clinical trials. There are a small number of suppliers for certain capital equipment and raw materials used to manufacture our product candidates. We do not have any control over the process or timing of the acquisition of these raw materials by our contract manufacturers. Moreover, we currently do not have agreements in place for the commercial production of these raw materials. Any significant delay in the supply of a product candidate or the raw material components thereof for an ongoing clinical trial could considerably delay completion of that clinical trial, product candidate testing, and potential regulatory approval of that product candidate.
| 77 |
We do not expect to have the resources or capacity to commercially manufacture any of our proposed product candidates, if approved, and will likely continue to be dependent on third-party manufacturers. Our dependence on third parties to manufacture and supply us with clinical trial materials and any approved product candidates may adversely affect our ability to develop and commercialize our product candidates on a timely basis.
We may not be successful in establishing and maintaining development partnerships and licensing agreements, which could adversely affect our ability to develop and commercialize product candidates.
Part of our strategy is to enter into development partnerships and licensing agreements. We face significant competition in seeking appropriate partners and the negotiation process is time consuming and complex. Even if we are successful in securing a development partnership, we may not be able to continue it. Moreover, we may not be successful in our efforts to establish a development partnership or other alternative arrangements for any of our other existing or future product candidates and programs because, among other reasons, our research and development pipeline may be insufficient, our product candidates and programs may be deemed to be at too early a stage of development for collaborative effort and/or third parties may not view our product candidates and programs as having the requisite potential to demonstrate safety and efficacy. Even if we are successful in our efforts to establish new development partnerships, the terms that we agree upon may not be favorable to us and we may not be able to maintain such development partnerships if, for example, development or approval of a product candidate is delayed or sales of an approved product candidate are disappointing. Any delay in entering into new development partnership agreements related to our product candidates could delay the development and commercialization of our product candidates and reduce their competitiveness if they reach the market.
Moreover, if we fail to establish and maintain additional development partnerships related to our product candidates:
| · | the development of our current or future product candidates may be terminated or delayed; |
| · | our cash expenditures related to development of certain of our current or future product candidates would increase significantly and we may need to seek additional financing; |
| · | we may be required to hire additional employees or otherwise develop expertise, such as sales and marketing expertise, for which we have not budgeted; and |
| · | we will bear all of the risk related to the development of any such product candidates. |
Our or any new partner’s failure to develop, manufacture or effectively commercialize our product would result in a material adverse effect on our business and results of operations and would likely cause our stock price to decline.
Currently pending, threatened or future litigation or governmental proceedings or inquiries could result in material adverse consequences, including judgments or settlements.
We are, or may from time to time become, involved in lawsuits and other legal or governmental proceedings. See Note 12 to the Consolidated Financial Statements included in this Annual Report on Form 10-K for information regarding currently pending litigation that could have a material impact on the Company. Many of these matters raise complicated factual and legal issues and are subject to uncertainties and complexities, all of which make the matters costly to address. The timing of the final resolutions to any such lawsuits, inquiries, and other legal proceedings is uncertain.
| 78 |
Additionally, the possible outcomes or resolutions to these matters could include adverse judgments or settlements, either of which could require substantial payments, adversely affecting our consolidated financial condition, results of operations and cash flows. Any judgment against us, the entry into any settlement agreement, or the imposition of any fine could have a material adverse effect on our consolidated financial condition, results of operations and cash flows.
Future acquisitions of and investments in new businesses could impact our business and financial condition.
From time to time, we may acquire or invest in businesses or partnerships that we believe could complement our business and drug candidates. The pursuit of such acquisitions or investments may divert the attention of management and cause us to incur various expenses, regardless of whether the acquisition or investment is ultimately completed. In addition, acquisitions and investments may not perform as expected and we may be unable to realize the expected benefits, synergies, or developments that we may initially anticipate. Further, if we are able to successfully identify and acquire additional businesses, we may not be able to successfully integrate the acquired personnel or operations, or effectively manage the combined business following the acquisition, any of which could harm our business and financial condition.
In addition, to the extent we finance any acquisition or investment in cash, it would reduce our cash reserves, and to the extent the purchase price is paid with shares of our common or preferred stock, it could be dilutive to our current stockholders. To the extent we finance any acquisition or investment with the proceeds from the incurrence of debt, this would increase our level of indebtedness and could negatively affect our liquidity, credit rating and restrict our operations. Moreover, we may face contingent liabilities in connection with any acquisitions or investments.
Risks Related to Intellectual Property
If we fail to adequately protect or enforce our intellectual property rights or secure rights to patents of others, the value of our intellectual property rights would diminish, and our business and competitive position would suffer.
Our success, competitive position and future revenues will depend in part on our ability and the abilities of our licensors and licensees to obtain and maintain patent protection for our products, methods, processes and other technologies, to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of third parties. We have an active patent protection program that includes filing patent applications on new compounds, formulations, delivery systems and methods of making and using products and prosecuting these patent applications in the United States and abroad. As patents issue, we also file continuation applications as appropriate. Although we have taken steps to build what we believe to be a strong patent portfolio, we cannot predict:
| · | the degree and range of protection any patents will afford us against competitors, including whether third parties find ways to invalidate or otherwise circumvent our licensed patents; |
| · | if and when patents will issue in the United States or any other country; |
| · | whether or not others will obtain patents claiming aspects similar to those covered by our licensed patents and patent applications; |
| · | whether we will need to initiate litigation or administrative proceedings to protect our intellectual property rights, which may be costly whether we win or lose; |
| · | whether any of our patents will be challenged by our competitors alleging invalidity or unenforceability and, if opposed or litigated, the outcome of any administrative or court action as to patent validity, enforceability or scope; |
| · | whether a competitor will develop a similar compound that is outside the scope of protection afforded by a patent or whether the patent scope is inherent in the claims modified due to interpretation of claim scope by a court; |
| · | whether there were activities previously undertaken by a licensor that could limit the scope, validity or enforceability of licensed patents and intellectual property; or |
| 79 |
| · | whether a competitor will assert infringement of its patents or intellectual property, whether or not meritorious, and what the outcome of any related litigation or challenge may be. |
Our success also depends upon the skills, knowledge and experience of our scientific and technical personnel, our consultants and advisors as well as our licensors, sublicensees and contractors. To help protect our proprietary know-how and our inventions for which patents may be unobtainable or difficult to obtain, we rely on trade secret protection and confidentiality agreements. To this end, we require all employees, consultants and board members to enter into agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business. These agreements may not provide adequate protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure or the lawful development by others of such information. If any of our trade secrets, know-how or other proprietary information is disclosed, the value of our trade secrets, know-how and other proprietary rights would be significantly impaired, and our business and competitive position would suffer.
Due to legal and factual uncertainties regarding the scope and protection afforded by patents and other proprietary rights, we may not have meaningful protection from competition.
Our long-term success will substantially depend upon our ability to protect our proprietary technologies from infringement, misappropriation, discovery and duplication and avoid infringing the proprietary rights of others. Our patent rights, and the patent rights of biopharmaceutical companies in general, are highly uncertain and include complex legal and factual issues. These uncertainties also mean that any patents that we own or may obtain in the future could be subject to challenge, and even if not challenged, may not provide us with meaningful protection from competition. Patents already issued to us or our pending applications may become subject to dispute, and any dispute could be resolved against us.
If some or all of our or any licensor’s patents expire or are invalidated or are found to be unenforceable, or if some or all of our patent applications do not result in issued patents or result in patents with narrow, overbroad, or unenforceable claims, or claims that are not supported in regard to written description or enablement by the specification, or if we are prevented from asserting that the claims of an issued patent cover a product of a third party, we may be subject to competition from third parties with products in the same class of products as our product candidates or products with the same active pharmaceutical ingredients as our product candidates, including in those jurisdictions in which we have no patent protection.
Our commercial success will depend in part on obtaining and maintaining patent and trade secret protection for our product candidates, as well as the methods for treating patients in the product indications using these product candidates. We will be able to protect our product candidates and the methods for treating patients in the applicable product indications using these product candidates from unauthorized use by third parties only to the extent that we or our exclusive licensor owns or controls such valid and enforceable patents or trade secrets.
Even if our product candidates and the methods for treating patients for prescribed indications using these product candidates are covered by valid and enforceable patents and have claims with sufficient scope, disclosure and support in the specification, the patents will provide protection only for a limited amount of time. Our and any licensor’s ability to obtain patents can be highly uncertain and involve complex and in some cases unsettled legal issues and factual questions. Furthermore, different countries have different procedures for obtaining patents, and patents issued in different countries provide different degrees of protection against the use of a patented invention by others. Therefore, if the issuance to us or any licensor, in a given country, of a patent covering an invention is not followed by the issuance, in other countries, of patents covering the same invention, or if any judicial interpretation of the validity, enforceability, or scope of the claims in, or the utility, written description or enablement in, a patent issued in one country is not similar to the interpretation given to the corresponding patent issued in another country, our ability to protect our intellectual property in those countries may be limited. Changes in either patent laws or in interpretations of patent laws in the United States and other countries may materially diminish the value of our intellectual property or narrow the scope of our patent protection.
| 80 |
We may be subject to competition from third parties with products in the same class of products as our product candidates, or products with the same active pharmaceutical ingredients as our product candidates in those jurisdictions in which we have no patent protection. Even if patents are issued to us or any licensor regarding our product or methods of using them, those patents can be challenged by our competitors who can argue such patents are invalid or unenforceable on a variety of grounds, including lack of utility, lack sufficient written description or enablement, utility, or that the claims of the issued patents should be limited or narrowly construed. Patents also will not protect our product candidates if competitors devise ways of making or using these products without legally infringing our patents. The current U.S. regulatory environment may have the effect of encouraging companies to challenge branded drug patents or to create non-infringing versions of a patented product in order to facilitate the approval of ANDAs for generic substitutes. These same types of incentives encourage competitors to submit NDAs that rely on literature and clinical data not prepared for or by the drug sponsor, providing another less burdensome pathway to approval.
If we infringe the rights of third parties, we could be prevented from selling products and be forced to defend against litigation and pay damages.
There is a risk that we are infringing the proprietary rights of third parties because numerous United States and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields that are the focus of our development and manufacturing efforts. Others might have been the first to make the inventions covered by each of our or any licensor’s pending patent applications and issued patents and/or might have been the first to file patent applications for these inventions. In addition, because patent applications take many months to publish and patent applications can take many years to issue, there may be currently pending applications, unknown to us or any licensor, which may later result in issued patents that cover the production, manufacture, synthesis, commercialization, formulation or use of our product candidates. In addition, the production, manufacture, synthesis, commercialization, formulation or use of our product candidates may infringe existing patents of which we are not aware. Defending ourselves against third-party claims, including litigation in particular, would be costly and time consuming and would divert management’s attention from our business, which could lead to delays in our development or commercialization efforts. If third parties are successful in their claims, we might have to pay substantial damages or take other actions that are adverse to our business.
If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and may have to:
| · | obtain licenses, which may not be available on commercially reasonable terms, if at all; |
| · | redesign our products or processes to avoid infringement, which may not be possible or could require substantial funds and time; |
| · | stop using the subject matter claimed in patents held by others, which could cause us to lose the use of one or more of our drug candidates; |
| · | pay damages royalties, or other amounts; or |
| · | grant a cross license to our patents to another patent holder. |
We expect that, as our drug candidates move further into clinical trials and commercialization and our public profile is raised, we will be more likely to be subject to such claims.
We may fail to comply with any of our obligations under existing agreements pursuant to which we license or have otherwise acquired rights or technology, which could result in the loss of rights or technology that are material to our business.
We are a party to technology licenses and have acquired certain assets and rights that are important to our business and we may enter into additional licenses or acquire additional assets and rights in the future. We currently hold licenses from Ludwig Institute for Cancer Research (“LICR”), BioWa, Inc. (“BioWa”), Lonza Sales AG (“Lonza”) Mayo Foundation (“Mayo”) and the University of Zurich (“UZH”). These licenses impose various commercial, contingent payments, royalty, insurance, indemnification, and other obligations on us. If we fail to comply with these obligations, the licensor may have the right to terminate the license or take back rights or assets, in which event we would lose valuable rights under our collaboration agreements, potential claims and our ability to develop product candidates.
| 81 |
We may be subject to claims that our consultants or independent contractors have wrongfully used or disclosed alleged trade secrets of their other clients or former employers to us.
As is common in the biotechnology and pharmaceutical industry, we engage the services of consultants to assist us in the development of our product candidates. Many of these consultants were previously employed at or may have previously or may be currently providing consulting services to, other biotechnology or pharmaceutical companies including our competitors or potential competitors. We may become subject to claims that our company or a consultant inadvertently or otherwise used or disclosed trade secrets or other information proprietary to their former employers or their former or current clients. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to our management team.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on product candidates in all countries throughout the world would be prohibitively expensive, and we intend to seek patent protection only in selected countries. Our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our product candidates and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
| 82 |
Risks Related to Our Common Stock
A significant portion of our total outstanding shares are eligible to be sold into the market in the near future, which could cause the market price of our common stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market or the perception in the market that the holders of a large number of shares intend to sell shares, could depress the market price of our common stock and could impair our future ability to obtain capital, especially through an offering of equity securities. In addition, holders of a substantial number of shares of our common stock have rights, subject to certain conditions, to require us to file registration statements covering their shares or to include their shares in registration statements that we may file for ourselves or other stockholders.
We also have registered all shares of common stock that we may issue under our equity compensation plans or that are issuable upon exercise of outstanding options. These shares can be freely sold in the public market upon issuance and once vested, subject to volume limitations applicable to affiliates. If any of these additional shares are sold, or if it is perceived that they will be sold, in the public market, the market price of our common stock could decline.
We have also entered into a Controlled Equity OfferingSM Sales Agreement (the “Sales Agreement”) with Cantor Fitzgerald & Co., under which we may issue and sell from time to time shares of common stock having an aggregate gross sales price of up to $100,000,000 through Cantor Fitzgerald, as sales agent. Sales of a substantial number of shares under the Sales Agreement, or the perception that those sales may occur, could cause the market price of our common stock to decline.
Further, certain shares of our common stock that are currently outstanding but have not been registered for resale may currently be sold under Rule 144 under the Securities Act or the Securities Act. Sales of a substantial number of these shares in the public market, or the perception that those sales may occur, could cause the market price of our common stock to decline.
Our executive officers, directors and principal stockholders, if they choose to act together, will continue to have the ability to significantly influence all matters submitted to stockholders for approval, and this concentration of ownership may contribute to volatility in our stock price.
We have a relatively small public float due to the ownership percentage of our executive officers and directors, and greater than 5% stockholders. Our directors, executive officers, and the other holders of more than 5% of our common stock together with their affiliates beneficially owned approximately 50.7% of our common stock as of March 5, 2021.
Some of these persons or entities may have interests that are different from our other stockholders, which could prevent or discourage unsolicited acquisition proposals or offers for our common stock that may be in the best interest of our other stockholders. This may also adversely affect the trading price of our common stock because investors may perceive disadvantages in owning stock in companies with a significant concentration of ownership.
As a result of our small public float, our common stock may be less liquid, experience reduced daily trading volume and have greater stock price volatility than the common stock of companies with broader public ownership. In addition, the trading of a relatively small volume of shares of our common stock may result in significant volatility in our stock price. If and to the extent ownership of our common stock becomes more concentrated, whether due to increased ownership by our directors and executive officers or other principal stockholders, or other factors, our public float would further decrease, which in turn would likely result in increased stock price volatility.
Additionally, because a large amount of our stock is closely held, we may experience low trading volume or large fluctuations in share price and volume due to sales by our principal stockholders. If our existing stockholders, particularly our directors, executive officers and the holders of more than 5% of our common stock, or their affiliates or associates, sell substantial amounts of our common stock in the public market, or are perceived by the public market as intending to sell substantial amounts of our common stock, the trading price of our common stock could decline significantly.
| 83 |
Despite our listing on the Nasdaq Capital Market, there can be no assurance that an active trading market for our common stock will develop or be sustained, and the Nasdaq Capital Market may subsequently delist our common stock if we fail to comply with ongoing listing standards.
On September 18, 2020, our common stock commenced trading on the Nasdaq Capital Market under the symbol “HGEN.” The Nasdaq Capital Market’s rules for listed companies will require us to meet certain financial, public float, bid price and liquidity standards on an ongoing basis in order to continue the listing of our common stock. To satisfy the initial listing standards for the Nasdaq Capital Market, we have had to execute a reverse stock split. In addition to specific listing and maintenance standards, the Nasdaq Capital Market has broad discretionary authority over the continued listing of securities, which it could exercise with respect to the listing of our common stock.
As a listed company, we are required to meet the continued listing requirements applicable to all Nasdaq Capital Market companies. If we fail to meet those standards, as applied by Nasdaq in its discretion, our common stock may be subject to delisting. We intend to take all commercially reasonable actions to maintain our Nasdaq listing. If our common stock is delisted in the future, it is not likely that we will be able to list our common stock on another national securities exchange and, as a result, we expect our securities would be quoted on an over-the-counter market; however, if this were to occur, our stockholders could face significant material adverse consequences, including limited availability of market quotations for our common stock and reduced liquidity for the trading of our securities. In addition, in the event of such delisting, we could experience a decreased ability to issue additional securities and obtain additional financing in the future.
The National Securities Markets Improvement Act of 1996, which is a federal statute, prevents or preempts the states from regulating the sale of certain securities, which are referred to as “covered securities.” Because our common stock is listed on Nasdaq, shares of our common stock qualify as covered securities under the statute. Although the states are preempted from regulating the sale of our securities, the federal statute does allow the states to investigate companies if there is a suspicion of fraud, and, if there is a finding of fraudulent activity, then the states can regulate or bar the sale of covered securities in a particular case. Further, if we were no longer listed on Nasdaq, our securities would not qualify as covered securities under the statute, and we would be subject to regulation in each state in which we offer our securities.
Further, there can be no assurance that an active trading market for our common stock will be sustained despite our listing on the Nasdaq Capital Market.
Raising additional funds by issuing securities or through licensing or lending arrangements may cause dilution to our existing stockholders, restrict our operations or require us to relinquish proprietary rights.
To the extent that we raise additional capital by issuing equity securities, the share ownership of existing stockholders will be diluted. To the extent that additional capital is raised through the sale of equity or convertible debt securities, the issuance could result in further dilution to our stockholders by causing a reduction in their proportionate ownership and voting power.
Any future debt financing may involve covenants that restrict our operations, including, among other restrictions, limitations on our ability to incur liens or additional debt, pay dividends, redeem our stock, make certain investments, and engage in certain merger, consolidation, or asset sale transactions. In addition, if we raise additional funds through licensing arrangements, it may be necessary to grant potentially valuable rights to our product candidates or grant licenses on terms that are not favorable to us.
| 84 |
Any material weaknesses in our internal control over financing reporting that we may identify in the future could adversely affect investor confidence, impair the value of our common stock and increase our cost of raising capital.
If we were to identify any material weaknesses or significant deficiencies in our internal controls over financial reporting in the future, our operating results might be harmed, we may fail to meet our reporting obligations or fail to prevent or detect material misstatements in our financial statements. Any such failure could, in turn, affect the future ability of our management to certify that internal control over our financial reporting is effective. Inferior internal control over financial reporting could also subject us to the scrutiny of the SEC and other regulatory bodies which could cause investors to lose confidence in our reported financial information and could subject us to civil or criminal penalties or stockholder litigation, which could have an adverse effect on our results of operations and the market price of our common stock.
In addition, if we or our independent registered public accounting firm identify deficiencies in our internal control over financial reporting, the disclosure of that fact, even if quickly remedied, could reduce the market’s confidence in our financial statements and harm our share price. Furthermore, deficiencies could result in future non-compliance with Section 404 of the Sarbanes-Oxley Act of 2002. Such non-compliance could subject us to a variety of administrative sanctions, including review by the SEC or other regulatory authorities.
Our stock price is volatile and purchasers of our common stock could incur substantial losses.
The market price of our common stock may fluctuate significantly in response to a number of factors. These factors include those discussed in this “Risk Factors” section of this prospectus and others such as:
| · | delay or failure in initiating or completing preclinical studies or clinical trials, or unsatisfactory results of these trials and the resulting impact on ongoing product development; |
| · | the success, progress, timing and costs of our efforts to evaluate or consummate various strategic alternatives if in the best interests of our stockholders; |
| · | our ability to maintain the listing of our common stock on the Nasdaq Capital Market; |
| · | announcements regarding equity or debt financing transactions; |
| · | sales or potential sales of substantial amounts of our common stock or securities convertible into our common stock; |
| · | announcements or social media comments about us or about our competitors including clinical trial results, regulatory approvals, or new product candidate introductions; |
| · | developments concerning our development partner, licensors or product candidate manufacturers; |
| · | litigation and other developments relating to our patents or other proprietary rights or those of our competitors; |
| · | conditions in the pharmaceutical or biotechnology industries and the economy as a whole; |
| · | governmental regulation and legislation; |
| · | recruitment or departure of members of our board of directors, management team or other key personnel; |
| · | changes in our operating results; |
| · | any financial projections we may provide to the public, any changes in these projections, our failure to meet these projections, or changes in recommendations by any securities analysts that elect to follow our common stock; |
| · | change in securities analysts’ estimates of our performance, or our failure to meet analysts’ expectations; and |
| · | price and volume fluctuations in the overall stock market or resulting from inconsistent trading volume levels of our shares. |
In recent years, the stock market in general, and the market for pharmaceutical and biotechnological companies in particular, has experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to changes in the operating performance of the companies whose stock is experiencing those price and volume fluctuations. Broad market and industry factors may seriously affect the market price of our common stock, regardless of our actual operating performance.
| 85 |
In addition, public statements by us, government agencies, traditional and social media or others relating to the COVID-19 pandemic (including currently authorized vaccines and therapeutics and efforts to develop additional COVID-19 vaccines, treatments or therapies) have in the past resulted, and may in the future result, in significant fluctuations in our stock price. Given the global focus on the COVID-19 pandemic, any information in the public arena on this topic, whether or not accurate, could have an outsized impact (either positive or negative) on our stock price. Information related to our Phase 3 trial of lenzilumab, the ACTIV-5 trial or our development, manufacturing and distribution efforts with respect to lenzilumab, or information regarding such efforts by competitors, may also impact our stock price.
We have never paid and do not intend to pay cash dividends and, consequently, the ability to achieve a return on any investment in our common stock will depend on appreciation in the price of our common stock.
We have never paid cash dividends on any of our capital stock, and we currently intend to retain future earnings, if any, to fund the development and growth of our business. Therefore, a holder of our stock is not likely to receive any dividends on our common stock for the foreseeable future. Since we do not intend to pay dividends, the ability to receive a return on an investment in our common stock will depend on any future appreciation in the market value of our common stock. There is no guarantee that our common stock will appreciate or even maintain the price at which it was purchased.
Anti-takeover provisions in our charter documents and Delaware law, could discourage, delay, or prevent a change in control of our company and may affect the trading price of our common stock.
We are a Delaware corporation and the anti-takeover provisions of the Delaware General Corporation Law may discourage, delay, or prevent a change in control by prohibiting us from engaging in a business combination with an interested stockholder for a period of three years after the person becomes an interested stockholder, even if a change in control would be beneficial to our existing stockholders.
Our Amended and Restated Certificate of Incorporation, as amended (the “Charter”), and our Second Amended and Restated Bylaws (the “Bylaws”) may discourage, delay, or prevent a change in our management or control over us that stockholders may consider favorable. Our Charter and Bylaws:
| · | provide that vacancies on our board of directors, including newly created directorships, may be filled only by a majority vote of directors then in office; |
| · | do not provide stockholders with the ability to cumulate their votes; and |
| · | require advance notification of stockholder nominations and proposals. |
In addition, our Charter permits the Board to issue up to 25 million shares of preferred stock with such powers, rights, terms and conditions as may be designated by the Board upon the issuance of shares of preferred stock at one or more times in the future. Specifically, the Charter permits the Board to approve the future issuance of all or any shares of the preferred stock in one or more series, to determine the number of shares constituting any series and to determine any voting powers, conversion rights, dividend rights, and other designations, preferences, limitations, restrictions and rights relating to such shares without any further authorization by our stockholders. The Board’s power to issue preferred stock could have the effect of delaying, deterring or preventing a transaction or a change in control of our company that might otherwise be in the best interest of our stockholders.
General Risk Factors
Our internal computer systems, or those of our third-party vendors, collaborators or other contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs, compromise sensitive information related to our business or prevent us from accessing critical information, potentially exposing us to liability or otherwise adversely affecting our business.
| 86 |
Our internal computer systems and those of our current and any future third-party vendors, collaborators and other contractors or consultants are vulnerable to damage or interruption from computer viruses, computer hackers, malicious code, employee theft or misuse, denial-of-service attacks, sophisticated nation-state and nation-state-supported actors, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we seek to protect our information technology systems from system failure, accident and security breach, if such an event were to occur and cause interruptions in our operations, it could result in a disruption of our development programs and our business operations, whether due to a loss of our trade secrets or other proprietary information or other disruptions. For example, the loss of clinical trial data from clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. If we were to experience a significant cybersecurity breach of our information systems or data, the costs associated with the investigation, remediation and potential notification of the breach to counterparties and data subjects could be material. In addition, our remediation efforts may not be successful. If we do not allocate and effectively manage the resources necessary to build and sustain the proper technology and cybersecurity infrastructure, we could suffer significant business disruption, including transaction errors, supply chain or manufacturing interruptions, processing inefficiencies, data loss or the loss of or damage to intellectual property or other proprietary information.
To the extent that any disruption or security breach were to result in a loss of, or damage to, our or our third-party vendors’, collaborators’ or other contractors’ or consultants’ data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability including litigation exposure, penalties and fines, we could become the subject of regulatory action or investigation, our competitive position could be harmed and the further development and commercialization of our product candidates could be delayed. Any of the above could have a material adverse effect on our business, financial condition, results of operations or prospects.
Our insurance policies are expensive and protect us only from some business risks, which leaves us exposed to significant uninsured liabilities.
We do not carry insurance for all categories of risk that our business may encounter. Some of the policies we currently maintain include general liability, employment practices liability, property, inventory and cargo, auto, workers’ compensation, products liability, and directors’ and officers’ insurance. We do not know, however, if we will be able to maintain existing insurance with adequate levels of coverage. Any significant, uninsured liability may require us to pay substantial amounts, which would adversely affect our working capital and results of operations.
If securities analysts do not publish research or publish unfavorable research about our business, our stock price and trading volume could decline.
The trading market for a company’s common stock often is based in part on the research and reports that securities and industry analysts publish about the company. If additional analyst coverage does develop, and an analyst downgrades our stock or publishes unfavorable research about our business, or if our clinical trials or operating results fail to meet the analysts’ expectations, our stock price would likely decline.
Changes in laws or regulations relating to data privacy and security, or any actual or perceived failure by us to comply with such laws and regulations, or contractual or other obligations relating to data privacy and security, could have a material adverse effect on our reputation, results of operations, financial condition and cash flows.
We are, and may increasingly become, subject to various laws and regulations, as well as contractual obligations, relating to data privacy and security in the jurisdictions in which we operate. The regulatory environment related to data privacy and security is increasingly rigorous, with new and constantly changing requirements applicable to our business, and enforcement practices are likely to remain uncertain for the foreseeable future. These laws and regulations may be interpreted and applied differently over time and from jurisdiction to jurisdiction, and it is possible that they will be interpreted and applied in ways that may have a material adverse effect on our business, financial condition, results of operations or prospects.
| 87 |
In the United States, various federal and state regulators, including governmental agencies like the Consumer Financial Protection Bureau and the Federal Trade Commission, have adopted, or are considering adopting, laws and regulations concerning personal information and data security. In particular, regulations promulgated pursuant to the Health Insurance Portability and accountability Act of 1996 (“HIPAA”) establish privacy and security standards that limit the use and disclosure of protected health information and require the implementation of safeguards to protect the privacy, integrity and availability of protected health information. Determining whether protected health information has been handled in compliance with applicable privacy standards and our contractual obligations can be complex and may be subject to changing interpretation. If we fail to comply with applicable HIPAA privacy and security standards, we could face civil and criminal penalties. In addition, state attorneys general are authorized to bring civil actions seeking either injunctions or damages in response to violations that threaten the privacy of state residents. We cannot be sure how these regulations will be interpreted, enforced or applied to our operations.
Certain state laws may be more stringent or broader in scope, or offer greater individual rights, with respect to personal information than federal, international, or other state laws, and such laws may differ from each other, all of which may complicate compliance efforts. For example, the California Consumer Privacy Act (“CCPA”), which increases privacy rights for California residents and imposes obligations on companies that process their personal information, came into effect on January 1, 2020. Among other things, the CCPA requires covered companies to provide new disclosures to California consumers and provide such consumers new data protection and privacy rights, including the ability to opt-out of certain sales of personal information. The CCPA provides for civil penalties for violations, as well as a private right of action for certain data breaches that result in the loss of personal information. This private right of action may increase the likelihood of, and risks associated with, data breach litigation.
Internationally, laws, regulations and standards in many jurisdictions apply broadly to the collection, use, retention, security, disclosure, transfer and other processing of personal information. For example, the E.U. General Data Protection Regulation (“GDPR”), which became effective in May 2018, greatly increased the European Commission’s jurisdictional reach of its laws and adds a broad array of requirements for handling personal data. EU member states are tasked under the GDPR to enact, and have enacted, certain implementing legislation that adds to and/or further interprets the GDPR requirements and potentially extends our obligations and potential liability for failing to meet such obligations. The GDPR, together with national legislation, regulations and guidelines of the EU member states governing the processing of personal data, impose strict obligations and restrictions on the ability to collect, use, retain, protect, disclose, transfer and otherwise process personal data. In particular, the GDPR includes obligations and restrictions concerning the consent and rights of individuals to whom the personal data relates, the transfer of personal data out of the European Economic Area, security breach notifications and the security and confidentiality of personal data. The GDPR authorizes fines for certain violations of up to 4% of global annual revenue or €20 million, whichever is greater.
All of these evolving compliance and operational requirements impose significant costs, such as costs related to organizational changes, implementing additional protection technologies, training associates and engaging consultants, which are likely to increase over time. In addition, such requirements may require us to modify our data processing practices and policies, distract management or divert resources from other initiatives and projects, all of which could have a material adverse effect on our results of operations, financial condition and cash flows. Any failure or perceived failure by us to comply with any applicable federal, state or similar foreign laws and regulations relating to data privacy and security could result in damage to our reputation and our relationship with our customers, as well as proceedings or litigation by governmental agencies or customers, including class action privacy litigation in certain jurisdictions, which would subject us to significant fines, sanctions, awards, penalties or judgments, all of which could have a material adverse effect on our business, financial condition, results of operations or prospects.
We review and explore strategic alternatives on an on-going basis, but there can be no assurance that we will be successful in identifying or completing any strategic alternative or that any such strategic alternative will yield additional value for our stockholders.
We regularly review strategic alternatives to ensure our current structure optimizes our ability to execute our strategic plan and to maximize stockholder value. The review of strategic alternatives could result in, among other things, a sale, merger, consolidation or business combination, asset divestiture, partnering, licensing or other collaboration agreements, or potential acquisitions or recapitalizations, in one or more transactions, or continuing to operate with our current business plan and strategy. There can be no assurance that the exploration of strategic alternatives will result in the identification or consummation of any transaction.
| 88 |
In addition, we may incur substantial expenses associated with identifying and evaluating potential strategic alternatives. The process of exploring strategic alternatives may be time consuming and disruptive to our business operations and if we are unable to effectively manage the process, our business, financial condition and results of operations could be adversely affected. We also cannot assure that any potential transaction or other strategic alternative, if identified, evaluated and consummated, will provide greater value to our stockholders than that reflected in our current stock price. Any potential transaction would be dependent upon a number of factors that may be beyond our control, including, among other factors, market conditions, industry trends, the interest of third parties in our business or product candidates and the availability of financing to potential buyers on reasonable terms.
| 89 |
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
We have leased an office in Burlingame, California, which lease will expire on August 31, 2021.
Please see Note 12 to the Condensed Consolidated Financial Statements for a summary of material pending legal proceedings.
ITEM 4. MINE SAFETY DISCLOSURES
None.
| 90 |
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock is currently listed on the Nasdaq Capital Market under the symbol “HGEN”. From June 26, 2017 until September 17, 2020, our common stock was quoted on the OTCQB Venture Market operated by OTC Markets Group, Inc. under the symbol “HGEN”.
Reverse Stock Split
Effective as of 4:30 p.m. Eastern Time on September 11, 2020 (the “Effective Time”), we amended our charter to effect a reverse stock split at a ratio of 1-for-5. No fractional shares were issued in connection with the reverse stock split. Stockholders of record otherwise entitled to receive fractional shares of common stock received cash (without interest or deduction) in lieu of such fractional share interests.
The reverse stock split reduced the total number of shares of our common stock outstanding as of the Effective Time from approximately 210.9 million shares to approximately 42.2 million shares. The par value per share and other terms of our common stock were not affected by the reverse stock split, and the number of authorized shares of the Company’s common stock remains at 225,000,000.
The reverse stock split was accounted for retroactively and is reflected in our common stock, stock option and warrant activity as of and during the years ended December 31, 2020 and 2019. Unless stated otherwise, all share data in this Annual Report on Form 10-K have been adjusted, as appropriate, to reflect the reverse stock split.
Holders of Common Stock
As of March 5, 2021, we had 53,482,364 shares of common stock outstanding held by approximately 40 stockholders of record. The actual number of stockholders is greater than this number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers and other nominees.
ITEM 6. SELECTED FINANCIAL DATA
Information requested by this Item is not applicable as we are electing scaled disclosure requirements available to Smaller Reporting Companies with respect to this Item.
| 91 |
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis together with our Consolidated Financial Statements and the notes thereto included elsewhere in this Annual Report on Form 10-K. This discussion contains forward-looking statements that involve risks and uncertainties. For additional discussion, see “SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS” above.
Overview
We are a clinical stage biopharmaceutical company, developing our portfolio of immuno-oncology and immunology monoclonal antibodies. We are focusing our efforts on the development of our lead product candidate, lenzilumab™, our proprietary Humaneered® (“Humaneered”) anti-human granulocyte-macrophage colony-stimulating factor (“GM-CSF”) monoclonal antibody. Lenzilumab is a monoclonal antibody that has been demonstrated in animal models to neutralize GM-CSF, a cytokine that we believe is of critical importance in the hyperinflammatory cascade, sometimes referred to as cytokine release syndrome (“CRS”) or cytokine storm, associated with COVID-19, chimeric antigen receptor T-cell (“CAR-T”) therapy and acute Graft versus Host Disease (“GvHD”) associated with bone marrow transplants. GM-CSF neutralization with lenzilumab has been shown to reduce downstream inflammatory cytokines, prevent CRS and reduce its associated neurologic toxicities in-vivo in validated preclinical human xenograft models (Blood. 2019 Feb 14;133(7):697-709).
Lenzilumab has completed full enrollment in a Phase 3, multi-center, double-blind, placebo-controlled potential registrational trial for hospitalized, hypoxic patients with COVID-19 pneumonia. If the data from the trial are favorable, based on our discussions with the U.S. Food and Drug Administration (“FDA”), including a Type B meeting, and in consultation with the regulatory experts at Operation Warp Speed, we plan on filing an Emergency Use Authorization (“EUA”) application in the second quarter of 2021. If the EUA is granted, we could begin to commercialize lenzilumab for the treatment of hospitalized COVID-19 pneumonia patients. We also intend to file a Biologics License Application (“BLA”) in 2021, again assuming the data from the Phase 3 trial are favorable and support such an application. Based on discussions with FDA, we understand that a BLA will require us to generate and present more clinical and manufacturing data than would be required to support an EUA. If the ACTIV-5/BET study described below is successful and completed on a timely basis, we plan to include the results in our BLA filing. COVID-19 infections are widespread and as of February 26, 2021 in the United States, there were over 28 million cases, nearly 1.8 million hospitalizations and over 510 thousand deaths. Several vaccines have been developed which show efficacy in excess of 90%, however the slow rollout of inoculations and the emergence of numerous COVID-19 variants lead us to believe the need for therapeutics to treat COVID-19 currently exists and will continue to exist.
Lenzilumab has been selected to be part of the ACTIV-5 “Big Effect Trial” (ACTIV-5/BET NCT). This study is sponsored by the National Institutes of Health (“NIH”). ACTIV-5/BET is designed to determine whether certain approved therapies or investigational drugs in late-stage clinical development show promise against COVID-19 and, therefore, merit advancement into larger clinical trials. ACTIV-5/BET, currently has 17 active sites enrolling and is expected to enroll in up to 40 U.S. sites, will evaluate lenzilumab with remdesivir, compared to placebo and remdesivir, in hospitalized COVID-19 patients, with approximately 100 patients expected to be assigned to each study arm. We are providing lenzilumab for the study, which is fully funded by NIH.
Lenzilumab is also being studied in a multicenter phase 1b/2 potential registrational trial as a sequenced therapy with Yescarta® (axicabtagene ciloleucel) to reduce CRS and neurotoxicity in patients with relapsed or refractory diffuse large B-cell lymphoma (“DLBCL”) (NCT04314843). This trial currently has 10 active sites and is being conducted in partnership with Kite Pharmaceuticals, Inc., a Gilead company (“Kite”), which markets Yescarta. We are also in the final planning stages for a Phase 2/3 trial for lenzilumab to treat patients who have undergone allogeneic hematopoietic stem cell therapy (“HSCT”) who are at high and intermediate risk for acute GvHD. The trial is expected to be conducted by the IMPACT Partnership, a collection of 22 stem cell transplant centers located in the United Kingdom.
| 92 |
Our proprietary, patented Humaneered technology platform is a method for converting existing antibodies (typically murine) into engineered, high-affinity human antibodies designed for therapeutic use, particularly with acute and chronic conditions. We have developed or in-licensed targets or research antibodies, typically from academic institutions, and then applied our Humaneered technology to produce them. Lenzilumab and our other two product candidates, ifabotuzumab and HGEN005, are Humaneered monoclonal antibodies. Our Humaneered antibodies are closer to human antibodies than chimeric or conventionally humanized antibodies and have a high affinity for their target but low immunogenicity. We believe our Humaneered antibodies offer additional advantages, such as high potency, a slow off-rate and a lower likelihood to induce an inappropriate immune response or infusion related reactions.
Our Pipeline
Our lenzilumab-based clinical-stage pipeline comprises a Phase 3 potential registration study in COVID-19 which has fully enrolled, the NIH-sponsored and funded ACTIV-5/BET program in COVID-19, a currently enrolling Phase 1b/2 study (ZUMA-19) as sequenced therapy of lenzilumab and Yescarta, a planned Phase 2/3 study in acute GvHD, and a planned Phase 2 study in CMML, the latter two of which we expect will be majority funded by partners. While the majority of our clinical programs involve lenzilumab, we have also fully enrolled an ifabotuzumab Phase 1 study in GBM.
Except for the potential of lenzilumab for COVID-19 under an EUA, our product candidates are in the clinical stage of development and will require substantial time, resources, research and development, and regulatory approval prior to commercialization. Furthermore, it may be years before any of our products are approved for use, if at all. Our current pipeline is depicted below:
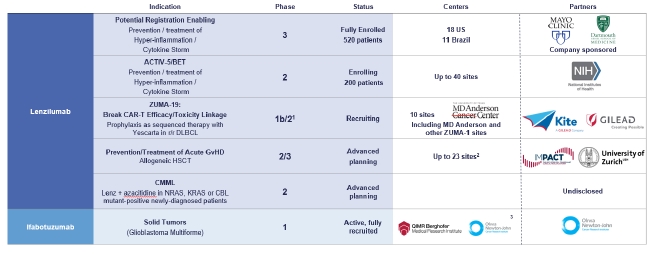
1 Phase 3 may not be necessary for approval in ZUMA-19; precedent is CAR-Ts to date have been approved on Phase 2 data
2 UK
3 US, EU, Australia
Results of Operations
General
At December 31, 2020, we had an accumulated deficit of $374.4 million, primarily as a result of research and development and general and administrative expenses as well as costs incurred in reorganization. While we may in the future generate revenue from a variety of sources, including license fees, milestone payments, and research and development payments in connection with strategic partnerships, our product candidates are at an early stage of development and may never be successfully developed or commercialized. Accordingly, we expect to continue to incur substantial losses from operations for the foreseeable future, and there can be no assurance that we will ever generate significant revenue or profits.
| 93 |
Research and Development Expenses
Conducting research and development is central to our business model. We expense both internal and external research and development costs as incurred. We track external research and development costs incurred by project for each of our clinical programs. Our external research and development costs consist primarily of:
| · | expenses incurred under agreements with contract research organizations, investigative sites, and consultants that conduct our clinical trials and a substantial portion of our pre-clinical activities; |
| · | the cost of acquiring and manufacturing clinical trial and EUA materials prior to receiving an EUA; and |
| · | other costs associated with development activities, including additional studies. |
Other research and development costs consist primarily of internal research and development costs such as salaries and related fringe benefit costs for our employees (such as workers compensation and health insurance premiums), stock-based compensation charges, and external costs not allocated to one of our clinical programs. Internal research and development costs generally benefit multiple projects and are not separately tracked per project.
The following table shows our total research and development expenses for the years ended December 31, 2020 and 2019:
| Year Ended December 31, | ||||||||
| (in thousands) | 2020 | 2019 | ||||||
| External Costs | ||||||||
| Lenzilumab | $ | 71,341 | $ | 2,046 | ||||
| Ifabotuzumab | 225 | 104 | ||||||
| Internal costs | 1,147 | 466 | ||||||
| Total research and development | $ | 72,713 | $ | 2,616 | ||||
Selling, General and Administrative Expenses
Selling, general and administrative expenses consist principally of personnel-related costs, costs related to our preparation for commercialization of lenzilumab, professional fees for legal and patent expenses, consulting, audit and tax services, investor and public relations costs, rent and other general operating expenses not otherwise included in research and development.
For the years ended December 31, 2020 and 2019, Selling, general and administrative expenses were $15.8 million and $6.3 million, respectively.
| 94 |
Comparison of Years Ended December 31, 2020 and 2019 ($000’s)
| Year Ended December 31, | Increase/ (Decrease) | |||||||||||||||
| (in thousands) | 2020 | 2019 | Amount | % | ||||||||||||
| Revenue: | ||||||||||||||||
| License revenue | $ | 312 | $ | - | $ | 312 | 100 | |||||||||
| Total revenue | 312 | - | 312 | 100 | ||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | 72,713 | 2,616 | 70,097 | 2,680 | ||||||||||||
| Selling, general and administrative | 15,797 | 6,328 | 9,469 | 150 | ||||||||||||
| Total operating expenses | 88,510 | 8,944 | 79,566 | 890 | ||||||||||||
| Loss from operations | (88,198 | ) | (8,944 | ) | 79,254 | 886 | ||||||||||
| Interest expense | (1,336 | ) | (1,349 | ) | (13 | ) | (1 | ) | ||||||||
| Other expense, net | (1 | ) | (1 | ) | - | - | ||||||||||
| Net loss | $ | (89,535 | ) | $ | (10,294 | ) | $ | 79,241 | 770 | |||||||
License revenue increased $0.3 million in 2020 from $0 for the year ended December 31, 2019 to $0.3 million for the year ended December 31, 2020. The increase is due to the signing of the South Korea Agreement, described in more detail in Note 3 to the Consolidated Financial Statements included in this Annual Report on Form 10-K.
Research and development expenses increased $70.1 million in 2020 from $2.6 million for the year ended December 31, 2019 to $72.7 million for the year ended December 31, 2020. The increase is primarily due to increased clinical trial and clinical material manufacturing costs related to the COVID-19 clinical trials. We expect our development costs to decrease in 2021 as a result of the completion of the COVID-19 clinical trials and related manufacturing costs which comprised a significant portion of our development expense in 2020 upon grant of EUA will be classified as Cost of Goods Sold in 2021.
Selling, general and administrative expenses increased $9.5 million in 2020 from $6.3 million for the year ended December 31, 2019 to $15.8 million for the year ended December 31, 2020. The increase is primarily due to increased compensation costs of $2.0 million, related to the hiring of a new Chief Operating Officer and Chief Financial Officer and a new Chief Accounting and Administrative Officer, as well as an increase in bonus costs for our Chief Executive Officer, an increase in public and investor relations expense of $4.9 million, which amount includes the issuance of warrants with a fair value of $2.0 million to certain third party consultants, and an increase in legal fees of $0.8 million incurred in connection with financing preparations and other corporate actions taken. We have also incurred $1.6 million in commercial operations expense as we prepare for a potential commercial launch under an EUA in COVID-19 including the hiring of a Chief Commercial Officer and the engagement of professional services organizations. We expect that we will continue to scale our support operations in connection with the filing for approval of lenzilumab in COVID-19 in the US and in Europe and will increase our commercial operations spending in preparation for potential commercialization under an EUA.
Interest expense and Other expense, net was essentially flat on a year-over-year basis.
| 95 |
Income Taxes
As of December 31, 2020, we had net operating loss carryforwards of approximately $166.2 million to offset future federal income taxes which expire in the years 2021 through 2037, and approximately $261.4 million that may offset future state income taxes which expire in the years 2028 through 2040. We also have federal net operating loss carryforwards generated in 2018, 2019 and 2020 of $104.9 million that have no expiration date as a result of the tax law changes signed into law on December 22, 2017. Current federal and state tax laws include substantial restrictions on the utilization of net operating losses and tax credits in the event of an ownership change. Even if the carryforwards are available, they may be subject to annual limitations, lack of future taxable income, or future ownership changes that could result in the expiration of the carryforwards before they are utilized. At December 31, 2020, we recorded a 100% valuation allowance against our deferred tax assets of approximately $82.3 million, as at that time our management believed it was uncertain that they would be fully realized. If we determine in the future that we will be able to realize all or a portion of our deferred tax assets, an adjustment to our valuation allowance would increase net income in the period in which we make such a determination.
Liquidity and Capital Resources
Since our inception, we have financed our operations primarily through proceeds from the public offerings and private placements of our common and preferred stock, debt financings, interest income earned on cash, and cash equivalents, and marketable securities, borrowings against lines of credit, and receipts from prior collaboration agreements. At December 31, 2020, we had cash and cash equivalents of $67.7 million. As of March 5,2021, we had cash and cash equivalents of $74.3 million.
Primary Sources of and Uses of Cash
The following table sets forth the primary sources and uses of cash and cash equivalents for each of the periods presented below ($000’s):
| Twelve Months Ended December 31, | ||||||||
| (In thousands) | 2020 | 2019 | ||||||
| Net cash (used in) provided by: | ||||||||
| Operating activities | $ | (69,853 | ) | $ | (4,001 | ) | ||
| Investing activities | (20 | ) | - | |||||
| Financing activities | 137,466 | 3,330 | ||||||
| Net increase (decrease) in cash, cash equivalents and restricted cash | $ | 67,593 | $ | (671 | ) | |||
Net cash used in operating activities was $69.9 million and $4.0 million for the years ended December 31, 2020 and 2019, respectively. Cash used in operating activities in 2019 primarily related to our net loss of $10.3 million, adjusted for non-cash items, such as $2.0 million in stock-based compensation, changes in operating assets and liabilities of $4.1 million and other non-cash items of $0.2 million. Cash used in operating activities in 2020 primarily related to our net loss of $89.0 million, adjusted for non-cash items, such as $2.1 million in stock-based compensation, changes in operating assets and liabilities of $16.6 million and other non-cash items of $0.4 million.
Net cash used in investing activities was $20,000 for the year ended December 31, 2020. This relates to the acquisition of intangible assets. There was no such investment for the year ended December 31, 2019.
Net cash provided by financing activities was $137.5 million for the year ended December 31, 2020 and consists primarily of $67.0 million received from the issuance of common stock in the 2020 Private Placement (as defined below) in June 2020, $72.7 million received from the issuance of common stock in the 2020 Underwritten Offering (as defined below) in September 2020, $0.5 million received from the issuance of the 2020 Convertible Notes, $0.6 million received from the exercise of stock options, $0.3 million received from the issuance of the 2020 Bridge Notes and $0.1 million received from the issuance of common stock under the equity line of credit with Lincoln Park Capital Fund, LLC (“Lincoln Park”), offset by $0.5 million for the payoff of the 2020 Convertible Notes, $2.4 million for the payoff of the 2019 and 2020 Bridge Notes and $0.8 million for the payoff of the Notes payable to vendors.
| 96 |
Net cash provided by financing activities was $3.3 million for the year ended December 31, 2019. This amount consists primarily of $2.0 million received from the issuance of the 2019 Bridge Notes (as defined below) entered into in June and November 2019, $1.3 million from the issuance of the 2019 Notes (as defined below) entered into in April 2019, $0.3 million received from the exercise of stock options, $0.2 million received from the issuance of common stock under the equity line of credit with Lincoln Park, offset by $0.5 million in payments made against the notes payable to vendors.
Recent Financings
2020 Private Placement
On June 1, 2020, we entered into a securities purchase agreement (the “Purchase Agreement”) with certain accredited investors (the “Investors”) to complete a private placement of our common stock (the “Private Placement”). The closing of the Private Placement occurred on June 2, 2020 (the “Closing Date”). At the closing, we issued and sold 16,505,743 shares of our common stock (the “Shares”) at a purchase price of $4.35 per share, for aggregate gross proceeds of approximately $71.8 million. We used a portion of the proceeds to retire the following indebtedness:
| · | the outstanding principal amount and accrued and unpaid interest on the Company’s convertible promissory notes issued in March 2020, which approximated $0.5 million, were repaid in full, and the notes were extinguished; |
| · | the outstanding principal and accrued and unpaid interest, amounting to approximately $2.4 million, on short-term, secured bridge loans made to the Company in 2019 and 2020 were repaid in full and the related liens were released; and |
| · | the remaining outstanding principal and accrued and unpaid interest, amounting to approximately $0.8 million, on certain notes payable to vendors in accordance with the bankruptcy plan. |
We used the remaining proceeds from the Private Placement to fund our Phase 3 study of lenzilumab in COVID-19, to secure manufacturing capacity, to progress Chemistry, Manufacturing and Controls (“CMC”) work, to prepare for commercialization in the event of approval of lenzilumab for use in COVID-19 patients, our collaboration with Kite and other development programs, as well as for working capital and other general corporate purposes. See Note 12 to the Consolidated Financial Statements included in this Annual Report on Form 10-K for material information regarding two complaints filed against us in connection with the Private Placement.
2020 Underwritten Public Offering
On September 17, 2020, we entered into an underwriting agreement (the “Underwriting Agreement”) with J.P. Morgan Securities LLC and Jefferies LLC, as representatives of the several underwriters, in connection with the public offering of 8,000,000 of the Company’s shares of common stock. Pursuant to the Underwriting Agreement, we granted the underwriters a 30-day option to purchase an additional 1,200,000 shares of common stock, which option was exercised in full by the underwriters on September 18, 2020.
| 97 |
As a result of the pricing of the public offering, our common stock commenced trading on the Nasdaq Capital Market under the symbol “HGEN.”
The aggregate gross proceeds from the sale of the full 9,200,000 shares in the offering were approximately $78.2 million. We expect to use the proceeds from the offering to support our manufacturing, production and commercial preparation activities relating to lenzilumab as a potential therapy for COVID-19 patients and for working capital and other general corporate purposes.
Controlled Equity Offering
On December 31, 2020, we entered into a Controlled Equity OfferingSM Sales Agreement (the “Sales Agreement”) with Cantor Fitzgerald & Co., under which we may issue and sell from time to time shares of the Company’s common stock, having an aggregate gross sales price of up to $100,000,000 through Cantor Fitzgerald, as sales agent. As of December 31, 2020, no sales were made under the Sales Agreement. During the period from January 1, 2020 through March 5, 2021, we sold 1,796,858 shares of our common stock under the Sales Agreement for net proceeds of $36.2 million.
Secured Term Loan Facility
On March 10, 2021, we executed a Loan and Security Agreement with Hercules Capital, Inc., as agent for its affiliates serving as lenders thereunder, pursuant to which we established a term loan facility in aggregate principal amount of up to $80 million (the “Term Loan”). The Term Loan provides us with the ability to draw an initial amount of $25.0 million, with up to an additional $55.0 million available for future draws subject to achievement of future milestones and satisfaction of other conditions. See Part II, Item 9B “Other Information” for additional information regarding the Term Loan.
Liquidity
We believe that our cash and cash equivalents will be sufficient to fund our planned operations and capital expenditure requirements for at least 12 months. This evaluation is based on relevant conditions and events that are currently known or reasonably knowable. As a result, we could deplete our available capital resources sooner than we currently expect, and a delay in obtaining or failure to obtain an EUA could further constrain our cash resources. We have based these estimates on assumptions that may prove to be wrong, and our operating projections, including our projected net revenue following the potential receipt of an EUA for lenzilumab in COVID-19 patients, may change as a result of many factors currently unknown to us. If we are unable to raise additional capital when needed or on acceptable terms, we would be forced to delay, reduce, or eliminate our research and development programs and commercialization efforts.
We expect we will need to obtain additional financing to fund our future operations, including for the development, manufacturing, distribution and commercialization of lenzilumab for patients with COVID-19 pneumonia and other indications including CRS and GvHD and our other product candidates. We may need to obtain additional financing to conduct additional trials for the approval of our product candidates if requested by regulatory authorities in the US and other countries, and to complete the development of any additional product candidates we own or might acquire. Moreover, our fixed expenses such as salaries, committed payments to our contract manufacturers, and other contractual commitments, including those for our other research and clinical collaborations, are substantial and are expected to increase in the future. Our need to raise funds will depend on a number of factors, including our ability to establish additional relationships for the manufacture of lenzilumab and our ability to commence commercialization and begin generating revenues from product sales if lenzilumab were to be issued an EUA and eventually approval under a BLA.
Until we can generate a sufficient amount of revenue, we may finance future cash needs through public or private equity offerings, license agreements, grant financing and support from governmental agencies, convertible debt, borrowings under our Term Loan with Hercules Capital and other debt financings, collaborations, strategic alliances and marketing, supply, distribution, or licensing arrangements. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available, we may be required to delay or reduce the scope of or eliminate one or more of our research or development programs, our commercialization efforts or our manufacturing commitments and capacity. We may seek to access the public or private capital markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time. In addition, if we raise additional funds through collaborations, strategic alliances or marketing, supply, distribution, or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams or product candidates or to grant licenses on terms that may not be favorable to us.
| 98 |
Capital Commitments and Capital Resources
We currently anticipate that top-line data from our Phase 3 trial of lenzilumab in hospitalized and hypoxic patients with COVID-19 will be available before the end of the first quarter of 2021. We have been preparing to submit an application for EUA for lenzilumab in the event data from the Phase 3 clinical trial are favorable. Even if the data are favorable, we cannot provide any assurance that we will receive an EUA from FDA. If EUA is received, we currently anticipate potential product launch, subject to the terms and conditions of the EUA, could occur as early as the second quarter of 2021.
To support our application for EUA and the potential launch of lenzilumab under an EUA, we have entered into agreements with several organizations for clinical trial services, commercialization services and contract manufacturing services as described in more detail elsewhere in this Annual Report. Our agreements with contract manufacturing organizations, more fully described under “Item 1. Business—Manufacturing and Raw Materials,” generally require us to commit to non-refundable up-front costs to secure manufacturing slots. Firm commitments under these agreements for 2021 are estimated to be $53 million, and we have committed to pay approximately $16 million in 2021 for other services related to potential product launch, We expect to be able to fund these firm commitments from our cash and cash equivalents on hand, which approximated $74.3 million as of March 5, 2021.
Our agreements for contract manufacturing also provide for our payment against performance by the manufacturers of their obligations under the contracts. We estimate our total commitments in respect of our current contractual arrangements to be approximately $244 million in 2021, of which approximately $210 million is for manufacturing of lenzilumab. If the data from the COVID-19 Phase 3 clinical trial are favorable and an EUA is granted in the second quarter of 2021, we expect to be able to satisfy the bulk of the cash requirements associated with our manufacturing commitments from revenues from the commercial sale of lenzilumab, supplemented as necessary with proceeds from the sale of our equity securities; the incurrence of debt; upfront and milestone payments from licensees; and government funding or financial support, if offered.
While our clinical manufacturing agreements specify maximum payment terms, they also generally contain standard terms that provide us the right to cancel pending orders prior to the initiation of manufacturing and, if the manufacturer is able to fill the slots with other products, to do so with limited further payment obligations to the manufacturers. Accordingly, in the event we do not receive an EUA, we would seek to terminate these agreements and related commitments. Given our sense of the demand for biologic manufacturing, sterile fill/finish capacity and the materials and components required, in the event of cancellation of production orders, we believe, the spending under these agreements would be limited.
See “Contracts” below for additional information regarding our agreements with Eversana, our contract manufacturers and our CRADA with DOD.
Operating Leases
We lease a small office space in Burlingame, California. Such lease will expire on August 31, 2021.
Other Financings
Notes Payable to Vendors
On June 30, 2016, we issued promissory notes in an aggregate principal amount of approximately $1.2 million to certain claimants in accordance with the bankruptcy plan. The notes were unsecured, accrued interest at 10% per annum and became due and payable in full, including principal and accrued interest on June 30, 2019. In July and August, 2019, following the receipt of proceeds from the 2019 Bridge Notes, we used approximately $0.5 million of the proceeds to retire a portion of these notes, including accrued interest. After giving effect to these payments, the aggregate principal amount and accrued but unpaid interest on these notes approximated $1.1 million as of December 31, 2019 and we had accrued $0.3 million in interest expense. In June and July 2020, we used the proceeds from the Private Placement to repay the remaining outstanding principal including accrued and unpaid interest on these notes and the notes were extinguished. As of December 31, 2020, all the notes have been repaid.
| 99 |
Advance Notes
In June, July and August 2018, we received an aggregate of $0.9 million of proceeds from advances made to us (the “Advance Notes”) by four different lenders including Dr. Cameron Durrant, the Company’s Chairman and Chief Executive Officer; Cheval, an affiliate of BHC, the Company’s controlling stockholder at the time; and Ronald Barliant, a director of the Company (collectively the “Advance Note Lenders”). The Advance Notes accrued interest at a rate of 7% per year, compounded annually.
In accordance with their terms, on May 30, 2019, in connection with our announcement of the Kite Agreement, the Advance Note Lenders converted the amounts due under the Advance Notes into common stock at the conversion price of $2.25 per share. We issued a total of 435,924 shares of common stock in connection with the conversion.
2018 Convertible Notes
Commencing September 19, 2018, we delivered a series of convertible promissory notes (the “2018 Notes”) evidencing an aggregate of $2.5 million of loans made to us by six different lenders, including an affiliate of BHC, our controlling stockholder at the time. The 2018 Notes accrued interest at a rate of 7% per annum.
The 2018 Notes were convertible into our equity securities in three different scenarios, including if we sold our equity securities on or before the date of repayment of the 2018 Notes in any financing transaction that resulted in gross proceeds to us of less than $10 million (a “Non-Qualified Financing”). Our sales of shares pursuant to the purchase agreement with Lincoln Park constituted a Non-Qualified Financing. Commencing on April 2, 2020, the holders of the 2018 Notes notified the Company of their exercise of their conversion rights under the 2018 Notes. See “2019 Convertible Notes” for additional information regarding the conversion of 2018 Notes by the holders.
As of December 31, 2019, the Company had accrued $0.2 million in interest related to these promissory notes. Interest expense recorded during the year ended December 31, 2020 was approximately $817,000.
2019 Convertible Notes
Commencing on April 23, 2019, we delivered a series of convertible promissory notes (the “2019 Notes” and together with the 2018 Notes, the “Convertible Notes”) evidencing an aggregate of $1.3 million of loans made to us. The 2019 Notes accrued interest at a rate of 7.5% per annum.
The 2019 Notes were convertible into our equity securities in four different scenarios, including if the Company sold its equity securities on or before the date of repayment of the 2019 Notes in any financing transaction that resulted in gross proceeds to us of less than $10 million (a “Non-Qualified Financing”). Our sales of shares pursuant to the purchase agreement with LPC constituted a Non-Qualified Financing. Commencing on April 2, 2020, holders of the Convertible Notes, including Cheval, an affiliate of BHC, the Company’s controlling stockholder at the time, notified us of their exercise of their conversion rights under the Convertible Notes. Pursuant to the exemption from registration afforded by Section 3(a)(9) under the Securities Act, we issued an aggregate of 2,397,915 shares of our common stock upon the conversion of $4.3 million in aggregate principal and interest on the Convertible Notes that were converted, which obligations were retired. Of these, we issued 316,666 shares to Cheval. Dr. Dale Chappell, who was serving as our ex-officio chief scientific officer at the time and currently serves as our Chief Scientific Officer, controls BHC and reports beneficial ownership of all shares held by it and its affiliates, including Cheval. After giving effect to the shares issued upon such conversions, no convertible notes issued in 2018 or 2019 were outstanding as of December 31, 2020.
As of December 31, 2019, we had accrued $0.1 million in interest related to the 2019 Notes. Interest expense related to the 2019 Notes, recorded during the year ended December 31, 2020, was approximately $219,000.
| 100 |
2019 Bridge Notes
On June 28, 2019, we issued three short-term, secured bridge notes (the “June Bridge Notes”) evidencing an aggregate of $1.7 million of loans made to us by three parties: Cheval, an affiliate of BHC, our controlling stockholder at the time, lent $750,000; Nomis Bay, our second largest stockholder at the time, lent $750,000; and Dr. Durrant, our Chief Executive Officer and Chairman of our Board of Directors, lent $200,000. The $1.7 million in proceeds was recorded as Advance notes in the Consolidated Balance Sheet as of December 31, 2019. As previously disclosed, the June Bridge Notes were repaid in June 2020 with proceeds from the Private Placement, and the June Bridge Notes were extinguished.
On November 12, 2019, we issued two short-term, secured bridge notes (the “November Bridge Notes” and together with the June Bridge Notes, the “2019 Bridge Notes”) evidencing an aggregate of $350,000 of loans made us by two parties: Cheval, an affiliate of BHC, our controlling stockholder at the time, lent $250,000; and Cameron Durrant, M.D., MBA, our Chief Executive Officer and Chairman of our Board of Directors, lent $100,000. As previously disclosed, the November Bridge Notes were repaid in June 2020 with proceeds from the Private Placement.
In April 2020, we issued two short-term, secured bridge notes (the “April Bridge Notes” and together with the June Bridge Notes and the November Bridge Notes, the “Bridge Notes”) evidencing an aggregate of $350,000 of loans made to us: Cheval, an affiliate of BHC, our controlling stockholder at the time, loaned $100,000, and Nomis Bay, our second largest stockholder, loaned $250,000. The proceeds from the April Bridge Notes were used for working capital and general corporate purposes. As previously disclosed, these April Bridge Notes were repaid in June 2020 with proceeds from the Private Placement, and these bridge notes were extinguished. The Bridge Notes were secured by a lien on substantially all the Company’s assets, which liens have been released.
The Bridge Notes accrued interest at a rate of 7.0% per annum. Interest expense related to the Bridge Notes, recorded during the year ended December 31, 2020, was approximately $66,000.
2020 Convertible Redeemable Notes
In March 2020, we delivered two convertible redeemable promissory notes (the “2020 Notes”) evidencing loans with an aggregate principal amount of $518,333 made to us. The 2020 Notes accrued interest at a rate of 7.0% per annum and contained an original issue discount of $33,000 and $18,833, respectively. We used the proceeds from the 2020 Notes for working capital. As previously disclosed, the notes were repaid in June 2020 with proceeds from the Private Placement, and the notes were extinguished.
Interest expense related to the 2020 Notes, recorded during the year ended December 31, 2020, was approximately $165,000. Interest expense includes the original issue discount amortization of approximately $52,000 for the year ended December 31, 2020.
Equity Line of Credit
On November 8, 2019, we entered into a purchase agreement and a registration rights agreement with Lincoln Park, pursuant to which we had the right to sell to Lincoln Park up to $20,000,000 in shares of our common stock, subject to certain limitations and conditions set forth in the purchase agreement.
In connection with the signing of the ELOC Purchase Agreement on November 8, 2019, we issued 141,318 shares of its common stock to LPC. The issuance of the shares was recorded as debt issuance costs in Common stock and Additional paid-in capital with no net effect on Stockholders’ equity (deficit). During the months of December 2019 and January 2020, we issued a total of 140,000 shares for aggregate proceeds of $0.3 million under the ELOC Purchase Agreement.
On June 2, 2020, following completion of the Private Placement, we notified LPC of our decision to terminate the ELOC Purchase Agreement. The termination of the ELOC Purchase Agreement became effective on June 3, 2020.
| 101 |
Contracts
Eversana Agreement
On January 10, 2021, we announced that we had entered into a master services agreement (the “Eversana Agreement”) with Eveersana Life Science Services, LLC (“Eversana”) pursuant to which Eversana will serve as our end-to-end commercial partner, providing us with a full suite of services in connection with the potential launch of lenzilumab.
We will pay Eversana fees and reimburse it for its expenses in performing the services as established in applicable statements of work. Eversana has agreed to defer its fees pending our receipt of an EUA from the U.S. Food and Drug Administration for lenzilumab for hospitalized and hypoxic COVID-19 patients. If we have not received an EUA by April 30, 2021 or such later date as to which the parties otherwise may agree, either party may terminate the Agreement immediately; Eversana will not receive the deferred fees upon such termination.
We estimate that with approval of an EUA and commencement of commercial activities that our first-year commitment to Eversana will be up to $20 million.
Manufacturing Agreements
During the year ended December 31, 2020 we have entered into agreements with several contract manufacturing organizations (“CMOs”) to have them manufacture bulk drug substance (“BDS”) for our lenzilumab clinical trial activities in COVID-19 as well as to manufacture BDS for a potential launch of lenzilumab in anticipation of an EUA in 2021. We have also entered into agreements for filling, finishing and packaging of the drug. These agreements represent large commitments, including upfront amounts prior to commencement of manufacturing and progress payments through the course of the manufacturing process and include payments for technology transfer. As of December 31, 2020, the Company estimates that its commitments to be incurred during the first quarter of 2021 will amount to approximately $48 million. The agreements contain customary cancellation clauses.
License Agreements
We are obligated to make future payments to third parties under in-license agreements, including sublicense fees, royalties, and payments that become due and payable on the achievement of certain development and commercialization milestones.
We record upfront and milestone payments made to third parties under licensing arrangements as an expense. Upfront payments are recorded when incurred and milestone payments are recorded when the specific milestone has been achieved.
License with the University of Zurich
On July 19, 2019, we entered into the Zurich Agreement with UZH. Under the Zurich Agreement, we have in-licensed certain technologies that we believe may be used to prevent GvHD through GM-CSF neutralization. The Zurich Agreement required an initial one-time payment of $100,000, which we paid to UZH on July 29, 2019. The Zurich Agreement also requires the payment of annual license maintenance fees, as well as milestones and royalties upon the achievement of certain regulatory and commercialization milestones.
License with the Mayo Foundation for Medical Education and Research
On June 19, 2019, we entered into the Mayo Agreement with the Mayo Foundation. Under the Mayo Agreement, we have in-licensed certain technologies that we believe may be used to create CAR-T cells lacking GM-CSF expression through various gene-editing tools including CRISPR-Cas9. Pursuant to the Mayo Agreement, we were required to pay $200,000 to the Mayo Foundation within six months of the effective date of the Mayo Agreement, or upon completion of a qualified financing, whichever is earlier. We paid the initial payment following completion of the Private Placement. The Mayo Agreement also requires the payment of milestones and royalties upon the achievement of certain regulatory and commercialization milestones.
| 102 |
Collaboration Agreement with Kite
On May 30, 2019, we entered into the Kite Agreement. The Kite Agreement provides that we and Kite will split only the out-of-pocket costs actually incurred in conducting the Study, including all third-party expenses incurred in accordance with a mutually agreed budget. We currently project we will be responsible for an aggregate of approximately $4 million in out-of-pocket costs, assuming up to a total of 36 patients are recruited for a multi-center Study. Each party will otherwise be responsible for its own internal costs, including internal personnel costs, incurred in connection with the Study.
Outlicensing Agreements
The South Korea Agreement
On November 3, 2020, we entered into a License Agreement (the “South Korea Agreement”) with KPM Tech Co., Ltd. (“KPM”) and its affiliate, Telcon RF Pharmaceutical, Inc. (together with KPM, the “Licensee”). Pursuant to the South Korea Agreement, among other things, we granted the Licensee a license under certain patents and other intellectual property to develop and commercialize our lead product candidate, lenzilumab (the “Product”), for treatment of COVID-19 pneumonia, in South Korea and the Philippines (the “Territory”), subject to certain reservations and limitations. The Licensee will be responsible for gaining regulatory approval for, and subsequent commercialization of, lenzilumab in those territories.
As consideration for the license, the Licensee has agreed to pay us (i) an up-front license fee of $6.0 million, payable promptly following the execution of the License Agreement, (ii) up to an aggregate of $14.0 million in two payments based on achievement by the Company of two specified milestones in the US, and (iii) subsequent to the receipt by the Licensee of the requisite regulatory approvals, double-digit royalties on the net sales of lenzilumab in South Korea and the Philippines. The Licensee has agreed to certain development and commercial performance obligations. It is expected that we will supply lenzilumab to the Licensee for a minimum of 7.5 years at a cost plus basis from an existing or future manufacturer. The Licensee has agreed to certain minimum purchases of lenzilumab on an annual basis.
Indemnification
In the normal course of business, we enter into contracts and agreements that contain a variety of representations and warranties and provide for general indemnifications. Our exposure under these agreements is unknown because it involves claims that may be made against us in the future but have not yet been made. To date, we have not paid any claims or been required to defend any action related to our indemnification obligations. However, we may record charges in the future as a result of these indemnification obligations.
Off-Balance Sheet Arrangements
We currently have no off-balance sheet arrangements, such as structured finance, special purpose entities, or variable interest entities.
| 103 |
Critical Accounting Policies and Critical Accounting Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our Consolidated Financial Statements, which have been prepared in accordance with accounting principles generally accepted in the U.S., or GAAP. The preparation of our financial statements in conformity with GAAP requires our management to make estimates and assumptions that affect the amounts and disclosures reported in the Consolidated Financial Statements and accompanying notes. Actual results could differ materially from those estimates. Our management believes judgment is involved in determining revenue recognition, the fair value-based measurement of stock-based compensation and accruals. Our management evaluates estimates and assumptions as facts and circumstances dictate. As future events and their effects cannot be determined with precision, actual results could differ from these estimates and assumptions, and those differences could be material to the Consolidated Financial Statements. If our assumptions change, we may need to revise our estimates, or take other corrective actions, either of which may also have a material adverse effect on our statements of operations, liquidity and financial condition.
While our significant accounting policies are described in more detail in Note 2 to our Consolidated Financial Statements included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report on Form 10-K, we believe the following accounting policies to be critical to the judgments and estimates used in the preparation of our financial statements.
Accrued Research and Development Expenses
As part of the process of preparing our Consolidated Financial Statements, we are required to estimate our accrued research and development expenses. This process involves reviewing contracts and purchase orders, reviewing the terms of our license agreements, communicating with our applicable personnel to identify services that have been performed on our behalf, and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual cost. Some of our service providers invoice us monthly in arrears for services performed. We make estimates of our accrued expenses as of each balance sheet date based on facts and circumstances known to us at that time. Examples of estimated accrued research and development expenses include fees to:
| · | contract research organizations and other service providers in connection with clinical studies; |
| · | contract manufacturers in connection with the production of clinical trial materials; and |
| · | vendors in connection with preclinical development activities. |
We base our expenses related to clinical studies on our estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and contract research organizations that conduct and manage clinical studies on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract, and may result in uneven payment flows and expense recognition. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing these costs, we estimate the time period over which services will be performed for which we have not been invoiced and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we adjust the accrual accordingly. Our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in our reporting changes in estimates in any particular period.
Stock-Based Compensation
Our stock-based compensation expense for stock options is estimated at the grant date based on the award’s fair value as calculated by the Black-Scholes option pricing model and is recognized as expense over the requisite service period. The Black-Scholes option pricing model requires various highly judgmental assumptions including expected volatility and expected term. The expected volatility is based on the historical stock volatilities of several of our publicly listed peers over a period equal to the expected terms of the options as we do not have a sufficient trading history to use the volatility of our own common stock. To estimate the expected term, we have opted to use the simplified method, which is the use of the midpoint of the vesting term and the contractual term. If any of the assumptions used in the Black-Scholes option pricing model changes significantly, stock-based compensation expense may differ materially in the future from that recorded in the current period. In addition, we are required to estimate the expected forfeiture rate and only recognize expense for those shares expected to vest. We estimate the forfeiture rate based on historical experience and our expectations regarding future pre-vesting termination behavior of employees. To the extent our actual forfeiture rate is different from our estimate, stock-based compensation expense is adjusted accordingly.
| 104 |
Revenue Recognition
Our revenue to date has been generated primarily through license agreements and research and development collaboration agreements. We had no revenues for the year ending December 31, 2019 and have recorded revenue from licensing of $0.3 million for the year ending December 31, 2020. Commencing January 1, 2018, we recognize revenue in accordance with Accounting Standards Update (“ASU”) 2014-09, “Revenue from Contracts with Customers (Topic 606)” (“ASC 606”). The core principle of ASC 606 is that an entity should recognize revenue to depict the transfer of promised goods and/or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods and/or services. To determine the appropriate amount of revenue to be recognized for arrangements that we determine are within the scope of ASC 606, the Company performs the following steps: (i) identify the contract(s) with the customer, (ii) identify the performance obligations in the contract, (iii) determine the transaction price, (iv) allocate the transaction price to the performance obligations in the contract and (v) recognize revenue when (or as) each performance obligation is satisfied.
Revenue under technology licenses and collaborative agreements typically consists of nonrefundable and/or guaranteed license fees, collaborative research funding, and various milestone and future product royalty or profit-sharing payments.
The fair value of deliverables under the arrangement may be derived using a best estimate of selling price if vendor specific objective evidence and third-party evidence is not available. Deliverables under the arrangement will be separate units of accounting if a delivered item has value to the customer on a standalone basis and if the arrangement includes a general right of return for the delivered item, delivery or performance of the undelivered item is considered probable and substantially in our control.
We recognize upfront license payments as revenue upon delivery of the license only if the license has standalone value from any undelivered performance obligations and that value can be determined. The undelivered performance obligations typically include manufacturing or development services or research and/or steering committee services. If the fair value of the undelivered performance obligations can be determined, then these obligations would be accounted for separately. If the license is not considered to have standalone value, then the license and other undelivered performance obligations would be accounted for as a single unit of accounting. In this case, the license payments and payments for performance obligations are recognized as revenue over the estimated period of when the performance obligations are performed or deferred indefinitely until the undelivered performance obligation is determined.
Whenever we determine that an arrangement should be accounted for as a single unit of accounting, we determine the period over which the performance obligations will be performed, and revenue will be recognized. Revenue is recognized using a proportional performance or straight-line method. The proportional performance method is used when the level of effort required to complete performance obligations under an arrangement can be reasonably estimated. The amount of revenue recognized under the proportional performance method is determined by multiplying the total payments under the contract, excluding royalties and payments contingent upon achievement of milestones, by the ratio of the level of effort performed to date to the estimated total level of effort required to complete performance obligations under the arrangement. If we cannot reasonably estimate the level of effort to complete performance obligations under an arrangement, we recognize revenue under the arrangement on a straight-line basis over the period we are expected to complete our performance obligations. Significant management judgment is required in determining the level of effort required under an arrangement and the period over which we are expected to complete our performance obligations under an arrangement.
| 105 |
Our collaboration agreements typically entitle us to additional payments upon the achievement of development, regulatory and sales performance-based milestones. If the achievement of a milestone is considered probable at the inception of the collaboration, the related milestone payment is included with other collaboration consideration, such as upfront fees and research funding, in our revenue calculation. Typically, these milestones are not considered probable at the inception of the collaboration. As such, milestones will typically be recognized in one of two ways depending on the timing of when the milestone is achieved. If the milestone is achieved during the performance period, then we will only recognize revenue to the extent of the proportional performance achieved at that date, or the proportion of the straight-line basis achieved at that date, and the remainder will be recorded as deferred revenue to be amortized over the remaining performance period. If the milestone is achieved after the performance period has completed and all performance obligations have been delivered, then we will recognize the milestone payment as revenue in its entirety in the period the milestone was achieved.
Recently Issued Accounting Pronouncements
In June 2018, the FASB issued ASU 2018-07, “Compensation – Stock Compensation (Topic 718), Improvements to Nonemployee Share-Based Payment Accounting”. This ASU expands the scope of Topic 718 to include share-based payment transactions for acquiring goods and services from nonemployees and is effective for fiscal years beginning after December 15, 2018, including interim periods within that fiscal year. Early adoption is permitted. We adopted the standard effective January 1, 2019. The adoption of this standard did not have a material impact on our Consolidated Financial Statements and related disclosures.
In November 2018, the FASB issued ASU No. 2018-18, “Collaborative Arrangements (Topic 808)—Clarifying the Interaction between Topic 808 and Topic 606”. ASU 2018-18 makes targeted improvements for collaborative arrangements by clarifying that certain transactions between collaborative arrangement participants should be accounted for as revenue under Topic 606 when the collaborative arrangement participant is a customer in the context of a unit of account. In those situations, all the guidance in Topic 606 should be applied, including recognition, measurement, presentation, and disclosure requirements. In addition, unit-of-account guidance in Topic 808 was aligned with the guidance in Topic 606 (that is, a distinct good or service) when an entity is assessing whether the collaborative arrangement or a part of the arrangement is within the scope of Topic 606. ASU 2018-18 is effective for fiscal years beginning after December 15, 2019, and interim periods within those fiscal years. Early adoption is permitted, including adoption in any interim period. The amendments in this Update should be applied retrospectively to the date of initial application of Topic 606. We adopted the standard effective January 1, 2020. The adoption of this standard did not have a material impact on our Consolidated Financial Statements and related disclosures.
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board (“FASB”) or other standard setting bodies that we adopt as of the specified effective date. Unless otherwise discussed, the Company does not believe that the impact of recently issued standards that are not yet effective will have a material impact on its financial position or results of operations upon adoption.
| 106 |
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Information requested by this Item is not applicable as we are electing scaled disclosure requirements available to Smaller Reporting Companies with respect to this Item.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Our Consolidated Financial Statements and The Report of Independent Registered Public Accounting Firm are included in this Annual Report on Form 10-K on pages F-1 through F-33.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our Chief Executive Officer and our Chief Financial Officer evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2020. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms.
Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is accumulated and communicated to our management, including our Chief Executive Officer and our Chief Financial Officer, to allow timely decisions regarding required disclosure. Based on this evaluation, our Chief Executive Officer and our Chief Financial Officer concluded that our disclosure controls and procedures were effective as of December 31, 2020.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) and 15d-15(f) under the Exchange Act). Our Chief Executive Officer and our Chief Financial Officer assessed the effectiveness of our internal control over financial reporting as of December 31, 2020. In making this assessment, our Chief Executive Officer and our Chief Financial Officer used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO, in Internal Control—Integrated Framework. Based on that assessment and using the COSO criteria, our Chief Executive Officer and our Chief Financial Officer concluded that, as of December 31, 2020, our internal control over financial reporting was effective.
In our 2019 Annual Report on Form 10-K, our Chief Executive Officer who was also our Interim Chief Financial Officer concluded that, as of December 31, 2019, our internal control over financial reporting was not effective because of the material weaknesses described below.
A material weakness is defined as “a deficiency, or a combination of deficiencies in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the company’s annual or interim financial statements will not be prevented or detected on a timely basis.”
The ineffectiveness of our internal control over financial reporting at December 31, 2019 was due to the following material weaknesses which each reflected the limited number of accounting and financial reporting personnel as of December 31, 2019: (i) an inability to complete our financial statement close process in a timely and accurate manner; (ii) an insufficient degree of segregation of duties amongst our accounting and financial reporting personnel; and (iii) a lack of technical competency in review and approval of financial reporting processes.
| 107 |
During 2020, our management remediated the material weaknesses identified above by, among other things, (i) adding highly qualified accounting and financial reporting personnel and consultants, (ii) modifying our internal processes to ensure a proper segregation of duties and oversight, and (iii) establishing a formal disclosure committee comprised of managers from all functional areas of the Company who are now closely engaged in our quarterly and annual reporting and filing process.
As we are a non-accelerated filer, our independent registered public accounting firm is not required to issue an attestation report on our internal control over financial reporting.
Changes in Internal Control Over Financial Reporting
Other than as described above, there has been no change in our internal control over financial reporting during the year ended December 31, 2020, that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Inherent Limitations of Controls
Management does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent or detect all errors and all fraud. Controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within the Company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the controls. The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Over time, controls may become inadequate because of changes in conditions, or deterioration in the degree of compliance with the policies or procedures. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
On March 10, 2021, we executed a Loan and Security Agreement with Hercules Capital, Inc., or Hercules, as agent for its affiliates serving as lenders thereunder. The agreement provides a loan in the aggregate principal amount of $80 million (the “Term Loan”). The Term Loan provides us debt with the ability to draw an initial amount of $25.0 million, and the potential for two additional term loans: a second tranche in the amount of $35.0 million or $25.0 million, which we may become entitled to draw through September 15, 2021 subject to our receipt of emergency use authorization for lenzilumab for the treatment of hospitalized patients with COVID-19 pneumonia; and a third tranche, a $20.0 million term loan which we may become entitled to draw through June 15, 2022 in the discretion of Hercules if we have drawn the second term loan and request additional funding in support of our strategic initiatives. We expect to draw the initial $25.0 million term loan in March 2021, at which time we will pay Hercules customary fees and reimburse it for certain of its expenses associated with the establishment of the Term Loan.
| 108 |
We will be required to repay all amounts borrowed on March 1, 2025, subject to a one-year extension option that we may exercise if we have received FDA approval of a biologics license application (“BLA”) for the use of lenzilumab for the treatment of hospitalized patients with COVID-19 pneumonia, and the FDA-authorized label for lenzilumab is generally consistent with that sought in our BLA filing, and paid Hercules certain fees and expenses associated with the extension.
Amounts drawn will bear interest at a floating rate equal to the greater of either (i) 8.75% plus the prime rate as reported in The Wall Street Journal minus 3.25%, and (ii) 8.75% (such greater amount, the “Base Interest Rate”). Subject to there not having occurred any default or event of default under the loan agreement, the Base Interest Rate will be reduced by 25 basis points upon the occurrence of each of the first three of the four following events to occur:
| · | if we achieve the protocol-specified primary efficacy endpoint for the pivotal Phase 3 study of lenzilumab for COVID-19, (clinicaltrials.gov identifier NCT04351152), and receive EUA for the use of lenzilumab for the treatment of hospitalized patients with COVID-19 pneumonia; |
| · | if we achieve product revenue from lenzilumab that is invoiced and/or recognized as revenue (as determined in accordance with GAAP) solely from the sale of lenzilumab (“Net Lenzilumab Product Revenue”) of at least $100.0 million; |
| · | if we achieve Net Lenzilumab Product Revenue of at least $250.0 million; and |
| · | if we achieve Net Lenzilumab Product Revenue of at least $350.0 million. |
No principal payments will be due during an interest-only period, commencing on the initial borrowing date and continuing to April 1, 2023, subject to extension to April 1, 2024 and potentially October 1, 2024 under certain conditions. Following the interest-only period, the outstanding balance of the loan will be required to be repaid monthly, continuing through the maturity date.
We may prepay amounts drawn under the agreement in full prior to the maturity date then in effect, subject to payment of prepayment charges equal to:
| · | 2.0% of the amounts borrowed, if the prepayment occurs on or prior to March 10, 2022; |
| · | 1.5% of the amounts borrowed, if the prepayment occurs after March 10, 2022 and before March 10, 2023; and |
| · | 1.0% of the amounts borrowed, if the prepayment occurs after March 10, 2023 and before March 10, 2024. |
In addition, on the earliest to occur of (i) the maturity date, (ii) the date we prepay the outstanding principal amount of the term loans, or (iii) the date the outstanding principal amount of the term loans otherwise becomes due, we will owe Hercules an end of term charge equal to 6.75% of the aggregate amount of term loans funded by Hercules.
| 109 |
As a condition to obtaining the term loan facility, we granted Hercules a security interest in substantially all of our assets and personal property not otherwise subject to existing or permitted liens. In addition, the loan agreement contains customary representations and warranties and events of default for a term loan facility of this size and type. Under the loan agreement, we also agreed to comply with certain customary affirmative and negative covenants that will become effective upon our initial draw of the first term loan, including covenants to:
| · | maintain $10.0 million in unrestricted cash, which amount may be increased to $20.0 million if we borrow the second tranche of the term loan; |
| · | comply with certain requirements to provide Hercules with financial information and other rights to inspect our books and records and the collateral for the term loans; |
| · | refrain from incurring debt that is not expressly subordinated to the term loans; “Rule 144A-style” convertible notes in aggregate principal amount up to $250.0 million; and other permitted indebtedness; |
| · | refrain from granting (or permitting to exist) liens on our assets and properties, other than certain permitted liens, including in respect of our patents and other intellectual property; |
| · | refrain from making certain investments, other than permitted acquisitions and certain other permitted investments; |
| · | refrain from repurchasing our stock or paying dividends, subject to limited exceptions; |
| · | refrain from transferring any material portion of our assets; and |
| · | refrain from entering into any merger or consolidation in which we are not the surviving entity. |
All of these covenants will not apply upon repayment of any borrowings under the Term Loan.
Certain of the baskets for permitted investments and other exceptions to the covenants described above will increase in the event we raise more than $100.0 million in unrestricted net cash proceeds from one or more bona fide equity financings prior to March 31, 2022. Further, the covenants in the loan agreement will not prohibit us from pursuing our strategy of entering into out-bound license agreements for lenzilumab that may be exclusive as to specific geographic regions outside the United States, nor will the covenants prohibit us from entering into co-development or co-promotion or commercialization agreements relating to lenzilumab, so long as such agreements generally are negotiated on arms’-length and commercially reasonable terms.
While the term loans are outstanding, the lenders will have the right to convert a portion of the principal amount outstanding under the term loan (ranging from $5.0 million to $10.0 million in the aggregate) into shares of our common stock at a conversion price equal to $19.57 per share, subject to customary anti-dilution adjustments.
| 110 |
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information contained in our definitive proxy statement for our 2021 annual meeting of stockholders under the captions “ELECTION OF DIRECTORS”, “INFORMATION ABOUT OUR EXECUTIVE OFFICERS” and “INFORMATION REGARDING THE BOARD AND CORPORATE GOVERNANCE” is hereby incorporated by reference. Certain other information relating to our Executive Officers appears in Part I of this Annual Report on Form 10-K under the heading “Information about our Executive Officers.”
ITEM 11. EXECUTIVE COMPENSATION
The information contained in our definitive proxy statement for our 2021 annual meeting of stockholders under the captions “EXECUTIVE COMPENSATION” is hereby incorporated by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information contained in our definitive proxy statement for our 2021 annual meeting of stockholders under the captions “SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT” is hereby incorporated by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information contained in our definitive proxy statement for our 2021 annual meeting of stockholders under the captions “CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS” and “DIRECTOR INDEPENDENCE” is hereby incorporated by reference.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information contained in our definitive proxy statement for our 2021 annual meeting of stockholders under the captions “RATIFICATION OF HORNE LLP AS OUR INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM” is hereby incorporated by reference.
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a) The following documents are filed as part of this report:
| (1) | Financial Statements—See Index to Consolidated Financial Statements at Part I, Item 8 on page F-1 of this Annual Report on Form 10-K. |
| (2) | All financial statement schedules have been omitted because they are not applicable or not required or because the information is included elsewhere in the financial statements or the Notes thereto. |
| (3) | See the accompanying Index to Exhibits filed as a part of this Annual Report, which list is incorporated by reference in this Item. |
(b) See the accompanying Index to Exhibits filed as a part of this Annual Report.
| 111 |
(c) Other schedules are not applicable.
None.
| 112 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized on March 10, 2021.
| Humanigen, Inc. | |||
|
By: |
/s/ Cameron Durrant, M.D., MBA |
||
|
Cameron Durrant, M.D., MBA Chief Executive Officer and Chairman of the Board of Directors |
|||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Cameron Durrant | ||||
| Cameron Durrant, M.D., MBA | Chairman of the Board and Chief Executive Officer (Principal Executive Officer) |
March 10, 2021 | ||
| /s/ Timothy Morris | ||||
| Timothy Morris, CPA | Chief Operating Officer and Chief Financial Officer (Principal Financial and Accounting Officer) |
March 10, 2021 | ||
| /s/ Ronald Barliant | ||||
| Ronald Barliant, JD | Director | March 10, 2021 | ||
| /s/ Rainer Boehm | ||||
| Rainer Boehm, M.D. | Director | March 10, 2021 | ||
| /s/ Cheryl Buxton | ||||
| Cheryl Buxton | Director | March 10, 2021 | ||
| /s/ Dale Chappell | ||||
| Dale Chappell, M.D., MBA | Director | March 10, 2021 |
| 113 |
Index to Consolidated Financial Statements
Contents
|
Report of Independent Registered Public Accounting Firm |
F-2 | |
| Consolidated Balance Sheets | F-4 | |
| Consolidated Statements of Operations | F-5 | |
| Consolidated Statements of Stockholders’ Equity (Deficit) | F-6 | |
| Consolidated Statements of Cash Flows | F-7 | |
| Notes to Consolidated Financial Statements | F-8 |
| F-1 |
Report of Independent Registered Public Accounting Firm
To Shareholders and the Board of Directors of Humanigen, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Humanigen, Inc. and subsidiary (the Company) as of December 31, 2020 and 2019, the related consolidated statements of operations and comprehensive loss, stockholders’ equity (deficit) and cash flows for the years then ended, and the related notes to the consolidated financial statements (collectively, the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2020 and 2019, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of a critical audit matter does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing separate opinions on the critical audit matter or on the accounts or disclosures to which they relate.
Revenue Recognition – Estimated Variable Consideration Associated Milestones and Related Payments Under Exclusive License Agreements
Milestone payments are considered variable consideration, which requires management to make estimates of when achievement of a particular milestone becomes probable. Milestone payments are included in the transaction price when it becomes probable that such inclusion would not result in a significant revenue reversal. As discussed in Note 2 to the consolidated financial statements, the Company entered into an exclusive license agreement with KPM Tech Co., LTD (KPM) and Telecon RF Pharmaceutical, Inc.(Telecon) (and collectively, the “Licensee”) in November 2020. Under the agreement, the Licensee made an upfront payment of $6.0 million to the Company. The upfront payment is being recognized as revenue ratably over a 29-month period through March of 2023, the expected period over which the Company expects the services to be performed. The Company will receive an aggregate of $14.0 million in milestone payments upon the achievement of two specified milestones. Milestone payments will be recognized as revenue when it becomes probable of achievement. Subsequent to the receipt of requisite regulatory approvals, the Licensee will make royalty payments to the Company on a quarterly basis over the term of the agreement.
| F-2 |
We identified the assessment of Licensee revenue recognition as a critical audit matter. The determination of the nature of the performance obligation and when it is satisfied was complex and involved subjective auditor judgment.
The primary procedures we performed to address this critical audit matter included the following. We examined the license agreements between the Company and the Licensee. We evaluated management’s assessment of the accounting treatment of this transaction, including the determination of the Company’s performance obligations. These procedures included testing the effectiveness of controls relating to the revenue recognition process, including the controls over the milestone probability assessment and the amortization period of upfront payment performed by management. These procedures also included, among others, reading the license agreement and testing management’s process for determining regulatory approval in South Korea and the Philippines was probable.
/s/ HORNE LLP
We have served as the Company's auditor since 2016.
Ridgeland, Mississippi
March 10, 2021
| F-3 |
Humanigen, Inc.
Consolidated Balance Sheets
(in thousands, except share and per share data)
| December 31, 2020 | December 31, 2019 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 67,737 | $ | 143 | ||||
| Prepaid expenses and other current assets | 475 | 309 | ||||||
| Total current assets | 68,212 | 452 | ||||||
| Intangible assets | 20 | - | ||||||
| Restricted cash | 70 | 71 | ||||||
| Total assets | $ | 68,302 | $ | 523 | ||||
| Liabilities and stockholders’ equity (deficit) | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 13,078 | $ | 5,046 | ||||
| Accrued expenses | 5,463 | 3,308 | ||||||
| Deferred revenue - current | 1,874 | - | ||||||
| Bridge notes | - | 2,113 | ||||||
| Convertible notes - current | - | 2,033 | ||||||
| Notes payable to vendors | - | 1,094 | ||||||
| Total current liabilities | 20,415 | 13,594 | ||||||
| Non-current liabilities: | ||||||||
| Deferred revenue - non-current | 2,342 | - | ||||||
| Convertible notes - non current | - | 1,247 | ||||||
| Total non-current liabilities: | 2,342 | 1,247 | ||||||
| Total liabilities | 22,757 | 14,841 | ||||||
| Stockholders’ equity (deficit): | ||||||||
| Common stock, $0.001 par value: 225,000,000 shares authorized at | ||||||||
| December 31, 2020 and December 31, 2019; 51,626,508 and 22,806,890 shares issued and outstanding at December 31, 2020 and December 31, 2019, respectively | 52 | 22 | ||||||
| Additional paid-in capital | 419,923 | 270,555 | ||||||
| Accumulated deficit | (374,430 | ) | (284,895 | ) | ||||
| Total stockholders’ equity (deficit) | 45,545 | (14,318 | ) | |||||
| Total liabilities and stockholders’ equity (deficit) | $ | 68,302 | $ | 523 | ||||
See accompanying notes.
| F-4 |
Humanigen, Inc.
Consolidated Statements of Operations
(in thousands, except share and per share data)
| Twelve Months Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Revenue: | ||||||||
| License revenue | $ | 312 | $ | - | ||||
| Total revenue | 312 | - | ||||||
| Operating expenses: | ||||||||
| Research and development | 72,713 | 2,616 | ||||||
| Selling, general and administrative | 15,797 | 6,328 | ||||||
| Total operating expenses | 88,510 | 8,944 | ||||||
| Loss from operations | (88,198 | ) | (8,944 | ) | ||||
| Other expense: | ||||||||
| Interest expense | (1,336 | ) | (1,349 | ) | ||||
| Other expense, net | (1 | ) | (1 | ) | ||||
| Net loss | $ | (89,535 | ) | $ | (10,294 | ) | ||
| Basic and diluted net loss per common share | $ | (2.42 | ) | $ | (0.46 | ) | ||
| Weighted average common shares outstanding used to | ||||||||
| calculate basic and diluted net loss per common share | 36,963,030 | 22,361,250 | ||||||
See accompanying notes.
| F-5 |
Humanigen, Inc.
Consolidated Statements of Stockholders’ Equity (Deficit)
(in thousands, except share and per share data)
| Total | ||||||||||||||||||||
| Additional | Stockholders’ | |||||||||||||||||||
| Common Stock | Paid-In | Accumulated | Equity | |||||||||||||||||
| Shares | Amount | Capital | Deficit | (Deficit) | ||||||||||||||||
| Balances at January 1, 2019 | 21,979,506 | $ | 22 | $ | 266,469 | $ | (274,601 | ) | $ | (8,110 | ) | |||||||||
| Issuance of common stock for payment of compensation | 30,445 | - | 137 | - | 137 | |||||||||||||||
| Issuance of stock options for payment of compensation | - | - | 207 | - | 207 | |||||||||||||||
| Issuance of common stock in exchange for services | 21,972 | - | 83 | - | 83 | |||||||||||||||
| Issuance of common stock upon option exercise | 97,725 | - | 325 | - | 325 | |||||||||||||||
| Convertible note beneficial conversion feature | - | - | 143 | - | 143 | |||||||||||||||
| Conversion of advance notes | 435,924 | - | 981 | - | 981 | |||||||||||||||
| Issuance of common stock in connection with equity line of credit | 241,318 | - | 185 | - | 185 | |||||||||||||||
| Stock-based compensation expense | - | - | 2,025 | - | 2,025 | |||||||||||||||
| Net loss | - | - | - | (10,294 | ) | (10,294 | ) | |||||||||||||
| Balances at December 31, 2019 | 22,806,890 | $ | 22 | $ | 270,555 | $ | (284,895 | ) | $ | (14,318 | ) | |||||||||
| Issuance of common stock, net of expenses | 25,745,744 | 27 | 139,733 | - | 139,760 | |||||||||||||||
| Issuance of common stock for conversion of debt | 2,397,916 | 3 | 4,313 | - | 4,316 | |||||||||||||||
| Issuance of common stock in exchange for services | 45,064 | - | 302 | - | 302 | |||||||||||||||
| Issuance of stock options for payment of compensation | - | - | 180 | - | 180 | |||||||||||||||
| Issuance of common stock for payment of compensation | 17,317 | - | 78 | - | 78 | |||||||||||||||
| Issuance of warrant for services | - | - | 2,070 | - | 2,070 | |||||||||||||||
| Issuance of common stock upon option exercise | 390,668 | - | 572 | - | 572 | |||||||||||||||
| Issance of common stock upon warrant exercise | 222,909 | - | 10 | - | 10 | |||||||||||||||
| Stock-based compensation expense | - | - | 2,110 | - | 2,110 | |||||||||||||||
| Net loss | - | - | - | (89,535 | ) | (89,535 | ) | |||||||||||||
| Balances at December 31, 2020 | 51,626,508 | $ | 52 | $ | 419,923 | $ | (374,430 | ) | $ | 45,545 | ||||||||||
See accompanying notes.
| F-6 |
Humanigen, Inc.
Consolidated Statements of Cash Flows
(in thousands)
| Twelve Months Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Operating activities: | ||||||||
| Net loss | $ | (89,535 | ) | $ | (10,294 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock based compensation expense | 2,110 | 2,025 | ||||||
| Issuance of common stock for payment of compensation | 78 | 137 | ||||||
| Issuance of common stock in exchange for services | 302 | 83 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other assets | (166 | ) | 176 | |||||
| Accounts payable | 8,032 | 2,190 | ||||||
| Accrued expenses | 9,326 | 1,682 | ||||||
| Net cash used in operating activities | (69,853 | ) | (4,001 | ) | ||||
| Investing activities: | ||||||||
| Purchase of intangible assets | (20 | ) | - | |||||
| Net cash used in investing activities | (20 | ) | - | |||||
| Financing activities: | ||||||||
| Net proceeds from issuance of common stock | 139,760 | 185 | ||||||
| Proceeds from exercise of stock options | 572 | 325 | ||||||
| Proceeds from exercise of warrants | 10 | - | ||||||
| Net proceeds from issuance of convertible notes | 467 | 1,275 | ||||||
| Net proceeds from issuance of PPP loan | 83 | - | ||||||
| Net proceeds from issuance of bridge notes | 350 | 2,050 | ||||||
| Payments on PPP loan | (83 | ) | - | |||||
| Payments on bridge notes | (2,400 | ) | - | |||||
| Payments on convertible notes | (518 | ) | - | |||||
| Payments on notes payable to vendors | (775 | ) | (505 | ) | ||||
| Net cash provided by financing activities | 137,466 | 3,330 | ||||||
| Net increase (decrease) in cash, cash equivalents and restricted cash | 67,593 | (671 | ) | |||||
| Cash, cash equivalents and restricted cash, beginning of period | 214 | 885 | ||||||
| Cash, cash equivalents and restricted cash, end of period | $ | 67,807 | $ | 214 | ||||
| Supplemental cash flow disclosure: | ||||||||
| Cash paid for interest | $ | 672 | $ | 13 | ||||
| Supplemental disclosure of non-cash investing and financing activities: | ||||||||
| Conversion of notes payable and related accrued interest and fees to common stock | $ | 4,316 | $ | 981 | ||||
| Beneficial conversion feature of convertible notes | $ | - | $ | 143 | ||||
| Issuance of stock options in lieu of cash compensation | $ | 180 | $ | 207 | ||||
| Issuance of warrants for services | $ | 2,070 | $ | - | ||||
See accompanying notes.
| F-7 |
Notes to Consolidated Financial Statements
(in thousands unless otherwise indicated, except share and per share data)
1. Organization and Description of Business
Description of the Business
Humanigen, Inc. (the “Company”) was incorporated on March 15, 2000 in California and reincorporated as a Delaware corporation in September 2001 under the name KaloBios Pharmaceuticals, Inc. Effective August 7, 2017, the Company changed its legal name to Humanigen, Inc.
The Company is a clinical stage biopharmaceutical company, developing its portfolio of immuno-oncology and immunology monoclonal antibodies. The Company is focusing its efforts on the development of its lead product candidate, lenzilumab™, its proprietary Humaneered® (“Humaneered”) anti-human granulocyte-macrophage colony-stimulating factor (“GM-CSF”) monoclonal antibody. Lenzilumab is a monoclonal antibody that has been demonstrated in animal models to neutralize GM-CSF, a cytokine that we believe is of critical importance in the hyperinflammatory cascade, sometimes referred to as cytokine release syndrome (“CRS”) or cytokine storm, associated with COVID-19, chimeric antigen receptor T-cell (“CAR-T”) therapy and acute Graft versus Host Disease (“GvHD”) associated with bone marrow transplants. GM-CSF neutralization with lenzilumab has been shown to reduce downstream inflammatory cytokines, prevent CRS and reduce its associated neurologic toxicities in-vivo in validated preclinical human xenograft models (Blood. 2019 Feb 14;133(7):697-709).
Lenzilumab has completed full enrollment in a Phase 3, multi-center, double-blind, placebo-controlled potential registrational trial for hospitalized, hypoxic patients with COVID-19 pneumonia. If the data from the trial are favorable, based on the Company’s discussions with the U.S. Food and Drug Administration (“FDA”), including a Type B meeting, and in consultation with the regulatory experts at Operation Warp Speed, the Company plans on filing an Emergency Use Authorization (“EUA”) application in the second quarter of 2021. If the EUA is granted, the Company could begin to commercialize lenzilumab for the treatment of hospitalized COVID-19 pneumonia patients. The Company also intends to file a Biologics License Application (“BLA”) in 2021, again assuming the data from the Phase 3 trial are favorable and support such an application. Based on discussions with FDA, the Company understands that a BLA will require it to generate and present more clinical and manufacturing data than would be required to support an EUA. If the ACTIV-5/BET study described below is successful and completed on a timely basis, the Company plans to include the results in its BLA filing. COVID-19 infections are widespread and as of February 26, 2021 in the United States, there were over 28 million cases, nearly 1.8 million hospitalizations and over 510 thousand deaths.. Several vaccines have been developed which show efficacy in excess of 90%, however the slow rollout of inoculations and the emergence of numerous COVID-19 variants lead us to believe the need for therapeutics to treat COVID-19 currently exists and will continue to exist.
Lenzilumab has been selected to be part of the ACTIV-5 “Big Effect Trial” (ACTIV-5/BET NCT 04583969). This study is sponsored by the National Institutes of Health (“NIH”). ACTIV-5/BET is designed to determine whether certain approved therapies or investigational drugs in late-stage clinical development show promise against COVID-19 and, therefore, merit advancement into larger clinical trials. ACTIV-5/BET, currently has 17 active U.S. sites enrolling and is expected to enroll in up to 40 U.S. sites, will evaluate lenzilumab with remdesivir, compared to placebo and remdesivir, in hospitalized COVID-19 patients, with approximately 100 patients expected to be assigned to each study arm. The Company is providing lenzilumab for the study, which is fully funded by NIH.
Lenzilumab is also being studied in a multicenter phase 1b/2 potential registrational trial as a sequenced therapy with Yescarta® (axicabtagene ciloleucel) to reduce CRS and neurotoxicity in patients with relapsed or refractory diffuse large B-cell lymphoma (“DLBCL”) (NCT 04314843). This trial currently has 10 active sites and is being conducted in partnership with Kite Pharmaceuticals, Inc., a Gilead company (“Kite”), which markets Yescarta. The Company is also in the final planning stages for a Phase 2/3 trial for lenzilumab to treat patients who have undergone allogeneic hematopoietic stem cell therapy (“HSCT”) who are at high and intermediate risk for acute GvHD. The trial is expected to be conducted by the IMPACT Partnership, a collection of 22 stem cell transplant centers located in the United Kingdom.
| F-8 |
The Company’s proprietary, patented Humaneered technology platform is a method for converting existing antibodies (typically murine) into engineered, high-affinity human antibodies designed for therapeutic use, particularly with acute and chronic conditions. The Company has developed or in-licensed targets or research antibodies, typically from academic institutions, and then applied our Humaneered technology to produce them. Lenzilumab and the Company’s other two product candidates, ifabotuzumab and HGEN005, are Humaneered monoclonal antibodies. The Company’s Humaneered antibodies are closer to human antibodies than chimeric or conventionally humanized antibodies and have a high affinity for their target but low immunogenicity. The Company believes its Humaneered antibodies offer additional advantages, such as high potency, a slow off-rate and a lower likelihood to induce an inappropriate immune response or infusion related reactions.
Our Pipeline
The Company’s lenzilumab-based clinical-stage pipeline comprises a Phase 3 potential registration study in COVID-19 which has fully enrolled, the NIH-sponsored and funded ACTIV-5/BET program in COVID-19, a currently enrolling Phase 1b/2 study (ZUMA-19) as sequenced therapy of lenzilumab and Yescarta, a planned Phase 2/3 study in acute GvHD, and a planned Phase 2 study in CMML, the latter two of which the Company expects will be majority funded by partners. While the majority of the Company’s clinical programs involve lenzilumab, it has also fully enrolled an ifabotuzumab Phase 1 study in GBM.
Except for the potential of lenzilumab for COVID-19 under an EUA, the Company’s product candidates are in the clinical stage of development and will require substantial time, resources, research and development, and regulatory approval prior to commercialization. Furthermore, it may be years before any of its products are approved for use, if at all. The Company’s current pipeline is depicted below:
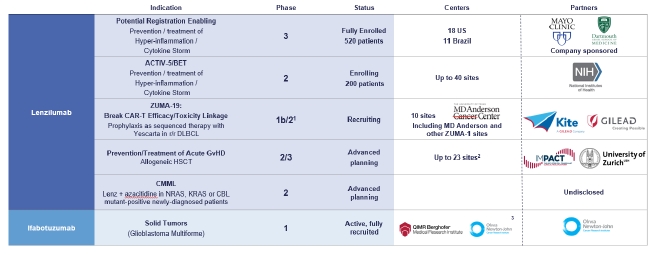
1 Phase 3 may not be necessary for approval in ZUMA-19; precedent is CAR-Ts to date have been approved on Phase 2 data
2 UK
3 US, EU, Australia
Liquidity and Going Concern
The Consolidated Financial Statements for the years ended December 31, 2020 and 2019 were prepared on the basis of a going concern, which contemplates that the Company will be able to realize assets and discharge liabilities in the normal course of business. However, the Company has incurred net losses since its inception and has negative operating cash flows. These conditions raised substantial doubt about the Company’s ability to continue as a going concern as described in the Company’s historical Condensed Consolidated Financial Statements included in its Annual Report on Form 10-K for the fiscal year ended December 31, 2019 and in its Quarterly Reports on Form 10-Q for the quarters ended March 31, 2020 and June 30, 2020.
| F-9 |
The Company has been continuing to advance its efforts to submit an application for EUA for lenzilumab, with potential product launch subject to EUA being granted as early as the second quarter of 2021, in the event data from the Phase 3 clinical trial are favorable. The Company currently anticipates that top-line data from the trial will be available in the first quarter of 2021. To support the potential launch of lenzilumab under an EUA, the Company has entered into agreements with several organizations for clinical trial services, contract manufacturing services and commercialization services, as more fully described under “Item 1. Business—Manufacturing and Raw Materials.” The agreements currently in effect commit the Company to spend approximately $58 million during the first quarter of 2021 in support of these efforts. The Company expects to be able to fund these cash commitments from its cash and cash equivalents on hand, which approximated $67.7 million as of December 31, 2020, although it may pursue other funding opportunities on an opportunistic basis to support our liquidity.
The Company is working with its existing contract manufacturing organizations and with additional contract manufacturing organizations to bolster its ability to supply lenzilumab in the event of receipt of EUA. Most of these manufacturing agreements, like the ones currently in place, are expected to require payment of upfront fees upon execution and further payments against performance of the manufacturing services to be provided, often over a lengthy performance period. Given the competitive environment, it is not possible for the Company to estimate the aggregate amount of these potential future payments or the timing in which they may be made. If the COVID-19 clinical study is successful and an EUA is granted in the second quarter of 2021, the Company expects to be able to satisfy the bulk of the cash requirements associated with its manufacturing commitments from revenues from the commercial sale of lenzilumab, supplemented as necessary with proceeds from the sale of its equity securities; the incurrence of debt; upfront and milestone payments from licensees; and government funding or financial support, if offered.
Following completion of the 2020 Private Placement in June 2020 and the 2020 Underwritten Public Offering in September 2020, as well as net proceeds received from sales of its common stock under the Controlled Equity Offering Sales Agreement of $36.2 million (unaudited), as of March 5, 2021, the Company had cash and cash equivalents of $74.3 million (unaudited). Accordingly, the Company re-evaluated its potential going concern disclosure requirements in accordance with ASC 205-40-50 as of the date of filing. Upon completion of this evaluation, the Company has concluded that its existing cash and cash equivalents have alleviated that prior substantial doubt about the Company’s ability to continue as a going concern.
The Company believes that its cash and cash equivalents will be sufficient to fund its planned operations and capital expenditure requirements for at least 12 months. This evaluation is based on relevant conditions and events that are currently known or reasonably knowable. As a result, the Company could deplete its available capital resources sooner than it currently expects, and a delay in obtaining or failure to obtain an EUA could further constrain its cash resources. The Company has based these estimates on assumptions that may prove to be wrong, and its operating projections, including its projected net revenue following the potential receipt of an EUA for lenzilumab in COVID-19 patients, may change as a result of many factors currently unknown to it. If the Company is unable to raise additional capital when needed or on acceptable terms, it would be forced to delay, reduce, or eliminate its research and development programs and commercialization efforts. Alternatively, the Company might raise funds through strategic collaborations, public or private financings or other arrangements. Such funding, if needed, may not be available on favorable terms, or at all. The financial statements included herein do not include any adjustments that might result from the outcome of these uncertainties.
2. Summary of Significant Accounting Policies
Basis of Presentation and Use of Estimates
The accompanying Consolidated Financial Statements have been prepared in accordance with U.S. generally accepted accounting principles (GAAP) and include all adjustments necessary for the presentation of the Company’s consolidated financial position, results of operations and cash flows for the periods presented. The Consolidated Financial Statements include the accounts of the Company and its wholly owned subsidiaries. These financial statements have been prepared on a basis that assumes that the Company will continue as a going concern, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business.
| F-10 |
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts and disclosures reported in the Consolidated Financial Statements and accompanying notes. Actual results could differ materially from those estimates. The Company believes judgment is involved in accounting for the fair value-based measurement of stock-based compensation, accruals, convertible notes and warrants. The Company evaluates its estimates and assumptions as facts and circumstances dictate. As future events and their effects cannot be determined with precision, actual results could differ from these estimates and assumptions, and those differences could be material to the Consolidated Financial Statements.
Concentration of Credit Risk
Cash, cash equivalents, and marketable securities consist of financial instruments that potentially subject the Company to a concentration of credit risk in the event of a default by the related financial institution holding the securities, to the extent of the value recorded in the balance sheet. The Company invests cash that is not required for immediate operating needs primarily in highly liquid instruments with lower credit risk. The Company has established guidelines relating to the quality, diversification, and maturities of securities to enable the Company to manage its credit risk.
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original maturity of 90 days or less at the time of purchase to be cash equivalents. Cash and cash equivalents consist of deposits with commercial banks in checking, interest-bearing and demand money market accounts.
Restricted Cash
Restricted cash at December 31, 2020 and 2019 of $70 thousand and $71 thousand, respectively, relates to a standby letter of credit in the amount of approximately $50 thousand issued in connection with certain insurance policy coverage maintained by the Company and restricted cash related to a credit card facility in the amount of approximately $20 thousand.
Debt Issue Costs
Debt issuance costs related to a recognized debt liability are presented on the balance sheet as a direct deduction from the carrying amount of that debt liability, consistent with debt discounts and are amortized to interest expense over the term of the related debt using the effective interest method.
Research and Development Expenses
Development costs incurred in the research and development of new product candidates are expensed as incurred, including expenses that may or may not be reimbursed under research and development collaboration arrangements. Research and development costs include, but are not limited to, salaries, benefits, stock-based compensation, laboratory supplies, allocated overhead, fees for professional service providers and costs associated with product development efforts, including preclinical studies and clinical trials.
The Company estimates preclinical study and clinical trial expenses based on the services performed, pursuant to contracts with research institutions and clinical research organizations that conduct and manage preclinical studies and clinical trials on its behalf. In accruing service fees, the Company estimates the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from the estimate, the Company will adjust the accrual accordingly. Payments made to third parties under these arrangements in advance of the receipt of the related services are recorded as prepaid expenses until the services are rendered.
The Company records upfront and milestone payments made to third parties under licensing arrangements as an expense. Upfront payments are recorded when incurred and milestone payments are recorded when the specific milestone has been achieved.
| F-11 |
Revenue Recognition
The Company’s revenue to date has been generated primarily through license agreements and research and development collaboration agreements. The Company recorded $0.3 million for the year ending December 31, 2020 related to a licensing agreement dated November 3, 2020 (See Note 3 – License Revenue) and had no revenues for the year ending December 31, 2019. Commencing January 1, 2018, the Company recognizes revenue in accordance with FASB Accounting Standards Codification (“ASC”) Topic 606 – Revenue from Contracts with Customers. The core principle of ASC 606 is that an entity should recognize revenue to depict the transfer of promised goods and/or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods and/or services. To determine the appropriate amount of revenue to be recognized for arrangements that the Company determines are within the scope of ASC 606, the Company performs the following steps: (i) identify the contract(s) with the customer, (ii) identify the performance obligations in the contract, (iii) determine the transaction price, (iv) allocate the transaction price to the performance obligations in the contract and (v) recognize revenue when (or as) each performance obligation is satisfied.
Revenue under technology licenses and collaborative agreements typically consists of nonrefundable and/or guaranteed license fees, collaborative research funding, and various milestone and future product royalty or profit-sharing payments.
The fair value of deliverables under the arrangement may be derived using a best estimate of selling price if vendor specific objective evidence and third-party evidence is not available. Deliverables under the arrangement will be separate units of accounting if a delivered item has value to the customer on a standalone basis and if the arrangement includes a general right of return for the delivered item, delivery or performance of the undelivered item is considered probable and substantially in the Company’s control.
The Company recognizes upfront license payments as revenue upon delivery of the license only if the license has standalone value from any undelivered performance obligations and that value can be determined. The undelivered performance obligations typically include manufacturing or development services or research and/or steering committee services. If the fair value of the undelivered performance obligations can be determined, then these obligations would be accounted for separately. If the license is not considered to have standalone value, then the license and other undelivered performance obligations would be accounted for as a single unit of accounting. In this case, the license payments and payments for performance obligations are recognized as revenue over the estimated period of when the performance obligations are performed or deferred indefinitely until the undelivered performance obligation is determined.
Whenever the Company determines that an arrangement should be accounted for as a single unit of accounting, the Company determines the period over which the performance obligations will be performed, and revenue will be recognized. Revenue is recognized using a proportional performance or straight-line method. The proportional performance method is used when the level of effort required to complete performance obligations under an arrangement can be reasonably estimated. The amount of revenue recognized under the proportional performance method is determined by multiplying the total payments under the contract, excluding royalties and payments contingent upon achievement of milestones, by the ratio of the level of effort performed to date to the estimated total level of effort required to complete performance obligations under the arrangement. If the Company cannot reasonably estimate the level of effort to complete performance obligations under an arrangement, the Company recognizes revenue under the arrangement on a straight-line basis over the period the Company is expected to complete its performance obligations. Significant management judgment is required in determining the level of effort required under an arrangement and the period over which the Company is expected to complete its performance obligations under an arrangement.
The Company’s collaboration agreements typically entitle the Company to additional payments upon the achievement of development, regulatory and sales performance-based milestones. If the achievement of a milestone is considered probable at the inception of the collaboration, the related milestone payment is included with other collaboration consideration, such as upfront fees and research funding, in the Company’s revenue calculation. Typically, these milestones are not considered probable at the inception of the collaboration. As such, milestones will typically be recognized in one of two ways depending on the timing of when the milestone is achieved. If the milestone is achieved during the performance period, then the Company will only recognize revenue to the extent of the proportional performance achieved at that date, or the proportion of the straight-line basis achieved at that date, and the remainder will be recorded as deferred revenue to be amortized over the remaining performance period. If the milestone is achieved after the performance period has completed and all performance obligations have been delivered, then the Company will recognize the milestone payment as revenue in its entirety in the period the milestone was achieved. See Note 3 for information on the 2020 Korea License Agreement.
| F-12 |
Leases
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842) (“ASU 2016-02”), which sets out the principles for the recognition, measurement, presentation and disclosure of leases for both lessees and lessors. The FASB subsequently issued ASU No. 2018-10 and 2018-11 in July 2018, which provide clarifications and improvements to ASU 2016-02 (collectively, the “new lease standard”).
ASU No. 2018-11 provides the optional transition method which allows companies to apply the new lease standard at the adoption date instead of at the earliest comparative period presented and continue to apply the provisions of the previous lease standard in its annual disclosures for the comparative periods. The new lease standard requires lessees to present a right-of-use asset and a corresponding lease liability on the balance sheet. Additional footnote disclosures related to leases are also required.
On January 1, 2019, the Company adopted the new lease standard using the optional transition method and certain other practical expedients. Under the practical expedient package elected, the Company is not required to reassess whether expired or existing contracts are or contain a lease; and is not required to reassess the lease classifications or reassess the initial direct costs associated with expired or existing leases.
The new lease standard also provides practical expedients for an entity’s ongoing accounting. The Company elected the short-term lease recognition exemption for all leases that qualify. This means, for those leases that qualify, we will not recognize right of use assets or lease liabilities, and this includes not recognizing right of use assets or lease liabilities for existing short-term leases of those assets in transition. The Company elected the practical expedient to not separate lease and non-lease components for certain classes of assets.
During 2019 and a portion of 2020, the Company sub-leased office space under a short-term lease in Burlingame, California. On September 1, 2020 we entered into a one-year lease for a small office in the same building in Burlingame, California for $1,200 per month. Management has determined the lease term to be less than 12 months, including renewals, and therefore has not recorded a right-of-use asset and corresponding liability under the short-term lease recognition exemption. Lease costs for the years ended December 31, 2020 and 2019 totaled approximately $6,100 and $10,700, respectively and are included in the Consolidated Statements of Operations.
Because the Company has elected to adopt the transitional practical expedients, Management was not required to reassess whether any existing or expired contracts contained embedded leases. The Company has not entered into any contracts during the 2020 fiscal year that contain an embedded lease.
Stock-Based Compensation Expense
The Company measures stock-based compensation expense for stock awards at the grant date, based on the fair value-based measurement of the award, and the expense is recorded over the related service period, generally the vesting period, net of estimated forfeitures. The Company calculates the fair value-based measurement of stock options using the Black-Scholes valuation model and the single-option method and recognizes expense using the straight-line attribution approach.
Income Taxes
The Company accounts for income taxes under an asset-and-liability approach. Deferred income taxes reflect the impact of temporary differences between assets and liabilities recognized for tax and financial reporting purposes measured by applying enacted tax rates and laws that will be in effect when the differences are expected to reverse, net operating loss carryforwards and tax credits. Valuation allowances are provided when necessary to reduce net deferred tax assets to an amount that is more likely than not to be realized. The Company’s policy is to include interest and penalties related to unrecognized tax benefits within the Company’s provision for income taxes.
| F-13 |
Net Loss Per Common Share
Basic net loss per common share is calculated by dividing the net loss attributable to common stockholders by the weighted-average number of common shares outstanding during the period, without consideration for potentially dilutive securities. Diluted net loss per share is computed by dividing the net loss attributable to common stockholders by the weighted-average number of common shares and potentially dilutive securities outstanding for the period determined using the treasury-stock and if-converted methods. For purposes of the diluted net loss per share calculation, stock options, restricted stock units and common stock warrants are considered to be potentially dilutive securities but are excluded from the calculation of diluted net loss per share because their effect would be anti-dilutive and therefore, basic and diluted net loss per share were the same for all periods presented.
The Company’s potential dilutive securities, which include stock options, restricted stock units and warrants have been excluded from the computation of diluted net loss per share as the effect would be to reduce the net loss per common share and be antidilutive. Therefore, the denominator used to calculate both basic and diluted net loss per common share is the same in all periods presented.
The following shares subject to outstanding potentially dilutive securities have been excluded from the computations of diluted net loss per common share as the effect of including such securities would be antidilutive:
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Options to purchase common stock | 3,728,149 | 3,176,336 | ||||||
| Warrants to purchase common stock | 51,238 | 66,238 | ||||||
| 3,779,387 | 3,242,574 | |||||||
Segment Reporting
The Company determines its segment reporting based upon the way the business is organized for making operating decisions and assessing performance. The Company operates in only one segment, which is related to the development of pharmaceutical products.
3. License Revenue
On November 3, 2020, the Company entered into a License Agreement (the “South Korea Agreement”) with KPM Tech Co., Ltd. (“KPM”) and its affiliate, Telcon RF Pharmaceutical, Inc. (together with KPM, the “Licensee”). Pursuant to the South Korea Agreement, among other things, the Company granted the Licensee a license under certain patents and other intellectual property to develop and commercialize the Company’s lead product candidate, lenzilumab (the “Product”), for treatment of COVID-19 pneumonia, in South Korea and the Philippines (the “Territory”), subject to certain reservations and limitations. The Licensee will be responsible for gaining regulatory approval for, and subsequent commercialization of, lenzilumab in the Territory.
As consideration for the license, the Licensee has agreed to pay the Company (i) an up-front license fee of $6.0 million, payable promptly following the execution of the License Agreement, (ii) up to an aggregate of $14.0 million in two payments based on achievement by the Company of two specified milestones in the US, and (iii) subsequent to the receipt by the Licensee of the requisite regulatory approvals, double-digit royalties on the net sales of lenzilumab in South Korea and the Philippines. The Licensee has agreed to certain development and commercial performance obligations. It is expected that the Company will supply lenzilumab to the Licensee for a minimum of 7.5 years at a cost-plus basis from an existing or future manufacturer. The Licensee has agreed to certain minimum purchases of lenzilumab on an annual basis.
The Company assessed the South Korea Agreement in accordance with ASC 606 and ASC 808 – Collaborative Arrangements and determined that its performance obligations under the South Korea agreement include (i) the exclusive, royalty-bearing, sublicensable license to lezilumab, (ii) the manufacturing supply services to be provided by the Company, (iii) cooperation and assistance to be provided by the Company to the Licensee with regulatory authorities in the Territory and (iv) its obligation to serve on a joint steering committee (Items iii and iv above collectively, the “Research and Development Services” or the “Services”). The Company concluded that in the initial period leading up to regulatory approval in the Territory (the “Initial Period”), the license was not distinct since it was of no benefit to Licensee without the aforementioned Services and that, as such, the license and the Services should be bundled as a single performance obligation.
| F-14 |
The Company has concluded that the nature of its promise is to stand ready to provide Research and Development Services as needed during the Performance Period (as defined below). The Company has further concluded that for all of the increments of time during the Performance Period its promise of standing ready to provide the Services is substantially the same. While the specific tasks performed during each increment of time will vary, the nature of the overall promise to provide the Services remains the same throughout the Performance Period.
Since the provision of the license and the Services are considered a single performance obligation, the $4.5 million upfront payment ($6.0 million net of withholding taxes and other fees and royalties) is being recognized as revenue ratably over the 29-month period through March of 2023 (the “Performance Period”), the expected period over which the Company conservatively expects the Services to be performed with approval in the Territory expected by the end of March 2023. Therefore, in November and December 2020, the Company has recognized license revenue totaling approximately $0.3 million.
Licensee’s purchases of lezilumab for development purposes or for commercial requirements, represent options under the agreement and revenues will therefore be recognized when control of the product is transferred to Licensee.
4. Savant Arrangements
On February 29, 2016, the Company entered into a binding letter of intent (the “LOI”) with Savant Neglected Diseases, LLC (“Savant”). The LOI provided that the Company would acquire certain worldwide rights relating to benznidazole from Savant. On June 30, 2016, the Company and Savant entered into an Agreement for the Manufacture, Development and Commercialization of Benznidazole for Human Use (the “MDC Agreement”), pursuant to which the Company acquired certain worldwide rights relating to benznidazole. The MDC Agreement consummates the transactions contemplated by the LOI.
In addition, on June 30, 2016, the Company and Savant also entered into a Security Agreement (the “Security Agreement”), pursuant to which the Company granted Savant a continuing senior security interest in the assets and rights acquired by the Company pursuant to the MDC Agreement and certain future assets developed from those acquired assets.
On June 30, 2016, in connection with the MDC Agreement, the Company issued to Savant a five-year warrant to purchase 200,000 shares of the Company’s Common Stock, at an exercise price of $2.25 per share, subject to adjustment. See Note 7.
On May 26, 2017, the Company submitted its benznidazole Investigational New Drug Application (“IND”) to the Food and Drug Administration (“FDA”) which became effective on June 26, 2017. The Company recorded expense of $1.0 million during the year ended December 31, 2017 as Research and development expense related to the milestone achievement associated with the IND being declared effective.
On July 10, 2017, FDA notified the Company that it granted Orphan Drug Designation to benznidazole for the treatment of Chagas disease. The Company recorded expense of $1.0 million during the year ended December 31, 2017 as Research and development expense related to the milestone achievement associated with Orphan Drug Designation.
The $2.0 million in milestone payments due Savant are included in Accrued expenses in the accompanying Consolidated Balance Sheets as of December 31, 2020 and 2019.
In July 2017, the Company commenced litigation against Savant alleging that Savant breached the MDC Agreement and is seeking a declaratory judgement. Savant has asserted counterclaims for breaches of contract under the MDC Agreement and the Security Agreement. See Note 12 below for more information regarding the Savant litigation.
| F-15 |
5. Debt
Notes Payable to Vendors
On June 30, 2016, the Company issued promissory notes in an aggregate principal amount of approximately $1.2 million to certain claimants in accordance with the Company’s Plan of Reorganization (the “Plan”) filed with the United States Bankruptcy Court for the District of Delaware (the “Bankruptcy Court”) (Case No. 15-12628 (LSS) (the “Bankruptcy Case”) which became effective June 30, 2016, at which time the Company emerged from its Chapter 11 bankruptcy proceedings. The notes were unsecured, accrued interest at 10% per annum and became due and payable in full, including principal and accrued interest on June 30, 2019. In July and August, 2019, following the receipt of proceeds from the 2019 Bridge Notes, the Company used approximately $0.5 million of the proceeds to retire a portion of these notes, including accrued interest. After giving effect to these payments, the aggregate principal amount and accrued but unpaid interest on these notes approximated $1.1 million as of December 31, 2019 and the Company had accrued $0.3 million in interest expense. In June and July 2020, the Company used the proceeds from the Private Placement (as defined below) to repay the remaining outstanding principal including accrued and unpaid interest on these notes and the notes were extinguished. As of December 31, 2020, all the notes have been repaid.
Advance Notes
In June, July and August, 2018 the Company received an aggregate of $0.9 million of proceeds from advances made to the Company (the “Advance Notes”) by four different lenders including Dr. Cameron Durrant, the Company’s Chairman and Chief Executive Officer; Cheval Holdings, Ltd. (“Cheval”), an affiliate of Black Horse Capital, L.P. (“BHC”), the Company’s controlling stockholder at the time; and Ronald Barliant, a director of the Company. The Advance Notes accrued interest at a rate of 7% per year, compounded annually.
In accordance with their terms, on May 30, 2019, in connection with the Company’s announcement of the Collaboration Agreement with Kite, the lenders converted the amounts due under the Advance Notes into the Company’s common stock at the conversion price of $2.25 per share. The Company issued a total of 435,924 shares of common stock in connection with the conversion.
Convertible Notes
2018 Convertible Notes
Commencing September 19, 2018, the Company delivered a series of convertible promissory notes (the “2018 Notes”) evidencing an aggregate of $2.5 million of loans made to the Company by six different lenders, including an affiliate of BHC, the Company’s controlling stockholder at the time. The 2018 Notes accrued interest at a rate of 7% per annum and, in general, were set to mature twenty-four months from the date the 2018 Notes were signed. The Company used the proceeds from the 2018 Notes for working capital.
The 2018 Notes were convertible into equity securities of the Company in three different scenarios, including if the Company sold its equity securities on or before the date of repayment of the 2018 Notes in any financing transaction that resulted in gross proceeds to the Company of less than $10 million (a “Non-Qualified Financing”). In connection with a Non-Qualified Financing, the noteholders were able to convert their remaining 2018 Notes into either (i) such equity securities as the noteholder would acquire if the principal and accrued but unpaid interest thereon (the “Conversion Amount”) were invested directly in the financing on the same terms and conditions as given to the financing investors in the Non-Qualified Financing, or (ii) common stock at a conversion price equal to $2.25 per share (subject to ratable adjustment for any stock split, stock dividend, stock combination or other recapitalization occurring subsequent to the date of the Notes).
The Company’s sales of shares pursuant to the ELOC Purchase Agreement with Lincoln Park constituted a Non-Qualified Financing. Commencing on April 2, 2020, the holders of the 2018 Notes notified the Company of their exercise of their conversion rights under the 2018 Notes. See “2019 Convertible Notes” for additional information regarding the conversion of 2018 Notes by the holders.
| F-16 |
As of December 31, 2019, the Company had accrued $0.2 million in interest related to these promissory notes. Interest expense recorded during the year ended December 31, 2020 was approximately $817 thousand.
2019 Convertible Notes
Commencing on April 23, 2019, the Company delivered a series of convertible promissory notes (the “2019 Notes” and together with the 2018 Notes, the “Convertible Notes”) evidencing an aggregate of $1.3 million of loans made to the Company. The 2019 Notes accrued interest at a rate of 7.5% per annum and, in general, were set to mature twenty-four months from the date the 2019 Notes were signed. The Company used the proceeds from the 2019 Notes for working capital.
The 2019 Notes were convertible into equity securities of the Company in four different scenarios, including if the Company sold its equity securities on or before the date of repayment of the 2019 Notes in any financing transaction that resulted in gross proceeds to the Company of less than $10 million (a “Non-Qualified Financing”). In connection with a Non-Qualified Financing, the noteholders were able to convert their remaining 2019 Notes into either (i) such equity securities as the noteholder would acquire if the Conversion Amount were invested directly in the financing on the same terms and conditions as given to the financing investors in the Non-Qualified Financing, or (ii) common stock at a conversion price equal to $6.25 per share (subject to ratable adjustment for any stock split, stock dividend, stock combination or other recapitalization occurring subsequent to the date of the 2019 Notes).
The Company’s sales of shares pursuant to the ELOC Purchase Agreement with LPC constituted a Non-Qualified Financing. Commencing on April 2, 2020, holders of the Convertible Notes, including Cheval, an affiliate of BHC, the Company’s controlling stockholder at the time, notified the Company of their exercise of their conversion rights under the Convertible Notes. Pursuant to the exemption from registration afforded by Section 3(a)(9) under the Securities Act, the Company issued an aggregate of 2,397,915 shares of its common stock upon the conversion of $4.3 million in aggregate principal and interest on the Convertible Notes that were converted, which obligations were retired. Of these, the Company issued 316,666 shares to Cheval. Dr. Dale Chappell, who was serving as the Company’s ex-officio chief scientific officer at the time and currently serves as its Chief Scientific Officer, controls BHC and reports beneficial ownership of all shares held by it and its affiliates, including Cheval. After giving effect to the shares issued upon such conversions, no convertible notes issued in 2018 or 2019 were outstanding as of December 31, 2020.
As of December 31, 2019, the Company had accrued $0.1 million in interest related to the 2019 Notes. Interest expense related to the 2019 Notes, recorded during the year ended December 31, 2020, was approximately $219 thousand.
The Advance Notes, the 2018 Notes and the 2019 Notes had an optional voluntary conversion feature in which the holder could convert the notes in the Company’s common stock at maturity at a conversion rate of $2.25 per share for the Advance Notes and the 2018 Notes and at a conversion rate of $6.25 for the 2019 Notes. The intrinsic value of this beneficial conversion feature was $1.8 million upon the issuance of the Advance Notes, the 2018 Notes and the 2019 Notes and was recorded as additional paid-in capital and as a debt discount which was accreted to interest expense over the term of the Advance Notes and Notes. Interest expense includes debt discount amortization of $785 thousand for the year ended December 31, 2020.
The Company evaluated the embedded features within the Advance Notes, the 2018 Notes and the 2019 Notes to determine if the embedded features are required to be bifurcated and recognized as derivative instruments. The Company determined that the Advance Notes, the 2018 Notes and the 2019 Notes contain contingent beneficial conversion features (“CBCF”) that allow or require the holder to convert the Advance Notes, the 2018 Notes and the 2019 Notes, as applicable, to Company common stock at a conversion rate of $2.25 per share for the Advance Notes and the 2018 Notes and $6.25 for the 2019 Notes, but did not contain embedded features requiring bifurcation and recognition as derivative instruments. Upon the occurrence of a CBCF that results in conversion of the Advance Notes, the 2018 Notes or the 2019 Notes to Company common stock, the remaining unamortized discount will be charged to interest expense. Upon conversion of the Advance Notes on May 30, 2019, the remaining unamortized discount was charged to interest expense. Upon the conversion of the Convertible Notes in April 2020, the remaining related unamortized discount was charged to interest expense.
| F-17 |
2020 Convertible Redeemable Notes
On March 13, 2020 and March 19, 2020 (the “Issuance Dates”), the Company delivered two convertible redeemable promissory notes (the “2020 Notes”) evidencing loans with an aggregate principal amount of $518,333 made to the Company.
The 2020 Notes accrued interest at a rate of 7.0% per annum and were set to mature on March 13, 2021 and March 19, 2021, respectively. The 2020 Notes contained an original issue discount of $33,000 and $18,833, respectively. The Company used the proceeds from the 2020 Notes for working capital.
The notes could be redeemed by the Company at any time before the 270th day following issuance, at a redemption price equal to the principal and accrued but unpaid interest on the notes to the date of redemption, plus a premium that increases on day 61 and day 121 from the issuance date. Accordingly, the notes were repaid in June 2020 with proceeds from the Private Placement, and the notes were extinguished.
The Company evaluated the embedded features within the 2020 Notes and determined that the embedded features are required to be bifurcated and recognized as stand-alone derivative instruments. The variable-share settlement features within the 2020 Notes qualify as redemption features and meet the net settlement criterion for qualification as a stand-alone derivative. In determining the fair value of the bifurcated derivative, the Company evaluated the likelihood of conversion of the 2020 Notes to Company stock. As the Company believed it would have adequate funding prior to the six-month anniversary of the 2020 Notes, the first conversion option for the holders of the 2020 Notes, and it had the intent to either begin making amortizing payments or to pay off the 2020 Notes in their entirety prior to that date, the fair value was determined to be $0. The original issue discount was accreted to interest expense and the remaining balance was charged to interest expense upon payoff.
Interest expense related to the 2020 Notes, recorded during the year ended December 31, 2020, was approximately $165 thousand.
Interest expense includes the original issue discount amortization of approximately $52 thousand for the year ended December 31, 2020.
Bridge Notes
On June 28, 2019, the Company issued three short-term, secured bridge notes (the “June Bridge Notes”) evidencing an aggregate of $1.7 million of loans made to the Company by three parties: Cheval, an affiliate of BHC, the Company’s controlling stockholder at the time, lent $750,000; Nomis Bay LTD, the Company’s second largest stockholder, lent $750,000; and Dr. Cameron Durrant, the Company’s Chief Executive Officer and Chairman of the Board of Directors (the “Board”), lent $200,000. The proceeds from the June Bridge Notes were used to satisfy a portion of the unsecured obligations incurred in connection with the Company’s emergence from bankruptcy in 2016 and for working capital and general corporate purposes. Of the $1.7 million in proceeds received, $950,000 was received on June 28, 2019 and was recorded as Advance notes in the Condensed Consolidated Balance Sheet as of June 30, 2019. The remaining proceeds of $750,000 were received July 1, 2019 and recorded accordingly.
The June Bridge Notes accrued interest at a rate of 7.0% per annum and after giving effect to multiple extensions, were set to mature on December 31, 2020. The June Bridge Notes could become due and payable at such earlier time as the Company raised more than $3,000,000 in a bona fide financing transaction or upon a change in control. Accordingly, the June Bridge Notes were repaid in June 2020 with proceeds from the Private Placement, and the June Bridge Notes were extinguished.
On November 12, 2019, the Company issued two short-term, secured bridge notes (the “November Bridge Notes” and together with the June Bridge Notes, the “2019 Bridge Notes”) evidencing an aggregate of $350,000 of loans made to the Company by two parties: Cheval, an affiliate of BHC, our controlling stockholder at the time, lent $250,000; and Dr. Cameron Durrant, our Chief Executive Officer and Chairman of our Board, lent $100,000. The proceeds from the November Bridge Notes were used for working capital and general corporate purposes.
The November Bridge Notes ranked on par with the June Bridge Notes and possessed other terms and conditions substantially consistent with those notes. The November Bridge Notes accrued interest at a rate of 7.0% per annum and after giving effect to multiple extensions, were set to mature on December 31, 2020. The November Bridge Notes could become due and payable at such earlier time as the Company raised more than $3,000,000 in a bona fide financing transaction or upon a change in control. Accordingly, the November Bridge Notes were repaid in June 2020 with proceeds from the Private Placement.
| F-18 |
In April 2020, the Company issued two short-term, secured bridge notes (the “April Bridge Notes” and together with the June Bridge Notes and the November Bridge Notes, the “Bridge Notes”) evidencing an aggregate of $350,000 of loans made to the Company: Cheval, an affiliate of BHC, the Company’s controlling stockholder at the time, loaned $100,000, and Nomis Bay, the Company’s second largest stockholder, loaned $250,000. The proceeds from the April Bridge Notes were used for working capital and general corporate purposes.
The April Bridge Notes ranked on par with the June Bridge Notes and the November Bridge Notes and possessed other terms and conditions substantially consistent with them. The notes accrued interest at a rate of 7.0% per annum and were set to mature on December 31, 2020. The April Bridge Notes could become due and payable at such earlier time as the Company raised more than $10,000,000 in a bona fide financing transaction or upon a change in control. Accordingly, these April Bridge Notes were repaid in June 2020 with proceeds from the Private Placement, and these bridge notes were extinguished.
The Bridge Notes were secured by a lien on substantially all the Company’s assets, which liens have been released.
Interest expense related to the Bridge Notes, recorded during the year ended December 31, 2020, was approximately $66 thousand.
6. Warrants to Purchase Common Stock
On June 19, 2013, the Company issued a warrant to purchase up to an aggregate of 1,238 shares of common stock at an exercise price of $484.80 per share. The warrant expires on the tenth anniversary of its issuance date. As of December 31, 2020, these warrants were fully vested.
On December 4, 2015, the Company issued a warrant to purchase up to an aggregate of 25,000 shares of common stock at an exercise price of $146.60 per share. The warrant expired on December 4, 2020.
On June 30, 2016, in connection with the MDC Agreement described in Note 5, the Company issued to Savant a five-year warrant (the “Savant Warrant”) to purchase 40,000 shares of the Company’s Common Stock, at an exercise price of $11.25 per share, subject to adjustment. The Savant Warrant was exercisable for 25% of the shares immediately and exercisable for the remaining shares upon reaching certain regulatory related milestones. In addition, pursuant to the MDC Agreement, the Company had granted Savant certain “piggyback” registration rights for the shares issuable under the Savant Warrant.
The Company determined the initial fair value of the Savant Warrant to be approximately $0.7 million as of June 30, 2016. The Company reevaluated the performance conditions and expected vesting of the Warrant quarterly during 2017 and 2018 and recorded a reduction of expense of approximately $0.1 million during the year ended December 31, 2017. The expense reduction was due to a decline in the fair value, which reduction was included in Research and development expenses. Specifically, as a result of the FDA granting accelerated and conditional approval of a benznidazole therapy manufactured by the Chemo Group (“Chemo”) for the treatment of Chagas disease and awarding Chemo a neglected tropical disease PRV, the Company re-evaluated the final two vesting milestones and concluded that the probability of achievement of these milestones had decreased to 0%.
As of December 31, 2019, 20,000 of these warrants were fully vested and 20,000 were not vested.
On June 30, 2020, Savant exercised 20,000 warrants in a cashless exercise resulting in 10,909 shares being issued to Savant in July 2020. The remaining unvested warrant for an aggregate of up to 20,000 shares will expire on June 30, 2021.
The Company will continue to reevaluate the performance conditions and expected vesting of the Savant Warrant on a quarterly basis until all performance conditions have been met or the warrants expire.
| F-19 |
On April 22, 2020, in connection with investor relation services, the Company issued two warrants to purchase up to an aggregate of 8,000 shares of common stock at an exercise price of $0.05 per share. The warrants were to vest upon either a change of control, as defined in the warrant agreement, an uplisting to a national securities exchange, or eight years from the issuance date and were to expire two years after full vesting. On September 18, 2020, the Company’s common stock commenced trading on the Nasdaq Capital Market and on the same day the warrants became vested and were exercised for proceeds of $400.
On May 20, 2020, in connection with investor relation services, the Company issued a warrant to purchase up to an aggregate of 4,000 shares of common stock at an exercise price of $0.05 per share. The warrants were to vest upon either a change of control, as defined in the warrant agreement, an uplisting to a national securities exchange, or eight years from the issuance date and were to expire two years after full vesting. On September 18, 2020, the Company’s common stock commenced trading on the Nasdaq Capital Market and on the same day the warrants became vested and were exercised for proceeds of $200.
On May 20, 2020, in connection with manufacturing consulting services, the Company issued a warrant to purchase up to an aggregate of 30,000 shares of common stock at an exercise price of $4.30 per share. The warrants were fully vested on the date of issue and expire ten years from the issuance date.
On September 14, 2020, in connection with investor relation services, the Company issued three warrants to purchase up to an aggregate of 200,000 shares of common stock at an exercise price of $0.05 per share. The warrants were to vest upon either a change of control, as defined in the warrant agreement, an uplisting to a national securities exchange, or eight years from the issuance date and were to expire two years after full vesting. On September 18, 2020, the Company’s common stock commenced trading on the Nasdaq Capital Market and on the same day the warrants became vested and were exercised for proceeds of $10,000.
7. Commitments and Contingencies
Operating Leases
The Company leased office space in Brisbane, California, which lease was month-to-month and commenced in April 2018. The Company terminated the lease on November 19, 2019 and entered into a sub-lease agreement for office space in Burlingame, California. The sub-lease initial term expired on March 31, 2020 and was renewed until September 1, 2020 at which point the Company entered into a new lease agreement in the same building which lease will expire on August 31, 2021.
As of December 31, 2020, the Company had future minimum lease payments of approximately $10 thousand.
Rent expense was approximately $3 thousand and $10 thousand for the years ended December 31, 2020 and December 31, 2019, respectively.
Indemnification
The Company has certain agreements with service providers with which it does business that contain indemnification provisions pursuant to which the Company typically agrees to indemnify the party against certain types of third-party claims. The Company accrues for known indemnification issues when a loss is probable and can be reasonably estimated. The Company would also accrue for estimated incurred but unidentified indemnification issues based on historical activity. As the Company has not incurred any indemnification losses to date, there were no accruals for or expenses related to indemnification issues for any period presented.
8. Stockholders’ Equity
Reverse Stock Split
Effective as of 4:30 p.m. Eastern Time on September 11, 2020 (the “Effective Time”), the Company amended its charter to effect a reverse stock split at a ratio of 1-for-5 (the “Split Ratio”). No fractional shares were issued in connection with the reverse stock split. Stockholders of record otherwise entitled to receive fractional shares of common stock received cash (without interest or deduction) in lieu of such fractional share interests.
| F-20 |
The reverse stock split reduced the total number of shares of the Company’s common stock outstanding as of the Effective Time from approximately 210.9 million shares to approximately 42.2 million shares. The par value per share and other terms of the Company’s common stock were not affected by the reverse stock split, and the number of authorized shares of the Company’s common stock remains at 225,000,000.
The reverse stock split resulted in a proportionate adjustment in the number of shares reserved for issuance under the 2020 Equity Plan, such that a total of 7,000,000 shares of the Company’s common stock were reserved for issuance under the 2020 Equity Plan following the Effective Time. In addition, proportionate adjustments were made to the number of shares covered by, and the exercise price applicable to, each outstanding stock option award under the 2012 Equity Plan and outstanding warrants issued by the Company, in each case to give effect to the Split Ratio and the reverse stock split.
The reverse stock split was accounted for retroactively and is reflected in the Company’s common stock, stock option and warrant activity as of and during the years ended December 31, 2020 and 2019. Unless stated otherwise, all share data in this Annual Report on Form 10-K have been adjusted, as appropriate, to reflect the reverse stock split.
Lincoln Park Capital Purchase Agreement
On November 8, 2019, the Company entered into a purchase agreement (the “ELOC Purchase Agreement”) and a registration rights agreement with LPC, pursuant to which the Company had the right to sell to LPC up to $20,000,000 in shares of the Company’s common stock, $0.001 par value per share (the “Common Stock”), subject to certain limitations and conditions set forth in the ELOC Purchase Agreement.
In connection with the signing of the ELOC Purchase Agreement on November 8, 2019, the Company issued 141,318 shares of its common stock to LPC. The issuance of the shares was recorded as debt issuance costs in Common stock and Additional paid-in capital with no net effect on Stockholders’ equity (deficit).
During the months of December 2019 and January 2020, the Company issued a total of 140,000 shares for aggregate proceeds of $0.3 million under the ELOC Purchase Agreement.
On June 2, 2020, following completion of the Private Placement described below, the Company notified LPC of its decision to terminate the ELOC Purchase Agreement. The termination of the ELOC Purchase Agreement became effective on June 3, 2020.
2020 Private Placement
On June 1, 2020, the Company entered into a securities purchase agreement (the “Purchase Agreement”) with certain accredited investors (the “Investors”) to complete a private placement of our common stock (the “Private Placement”). The closing of the Private Placement occurred on June 2, 2020 (the “Closing Date”). At the closing, the Company issued and sold 16,505,743 shares of its common stock (the “Shares”) at a purchase price of $4.35 per share, for aggregate gross proceeds of approximately $71.8 million. The Company used a portion of the proceeds to retire certain indebtedness, as further described in Note 6 - Debt. See Note 12 for information regarding two complaints filed against us in connection with the Private Placement.
On the Closing Date, the Company and the Investors also entered into a registration rights agreement (the “Registration Rights Agreement”) pursuant to which the Company agreed to prepare and file a registration statement (the “Resale Registration Statement”) for the resale of the Shares with the Securities and Exchange Commission.
Subject to certain limitations and an overall cap, the Company may be required to pay liquidated damages to the investors at a rate of 2% of the invested capital for each occurrence (and continuation for 30 consecutive days thereafter) of a breach by the Company of certain of its obligations under the Registration Rights Agreement.
| F-21 |
The Purchase Agreement also required that the Company use its commercially reasonable efforts to achieve a listing of the Common Stock on a national securities exchange, subject to certain limitations set forth in the Purchase Agreement. On July 6, 2020, the Company applied to have its common stock approved for listing on the Nasdaq Capital Market. On September 18, 2020, the Company’s common stock commenced trading on the Nasdaq Capital Market under the symbol “HGEN.”
2020 Underwritten Public Offering
On September 17, 2020, the Company entered into an underwriting agreement (the “Underwriting Agreement”) with J.P. Morgan Securities LLC and Jefferies LLC, as representatives of the several underwriters, in connection with the public offering of 8,000,000 of the Company’s shares of common stock. Pursuant to the Underwriting Agreement, the Company granted the underwriters a 30-day option to purchase an additional 1,200,000 shares of common stock, which option was exercised in full by the underwriters on September 18, 2020.
As a result of the pricing of the public offering, the Company’s common stock commenced trading on the Nasdaq Capital Market under the symbol “HGEN.”
The aggregate gross proceeds from the sale of the full 9,200,000 shares in the offering were approximately $78.2 million. The Company expects to use the proceeds from the offering to support its manufacturing, production and commercial preparation activities relating to lenzilumab as a potential therapy for COVID-19 patients and for working capital and other general corporate purposes.
The Company has reserved the following shares of common stock for issuance as of December 31, 2020:
| Warrants to purchase common stock | 51,238 | |||
| Options: | ||||
| Outstanding under the 2020 Equity Incentive Plan | 637,286 | |||
| Outstanding under the 2012 Equity Incentive Plan | 3,090,821 | |||
| Outstanding under the 2001 Equity Incentive Plan | 42 | |||
| Available for future grants under the 2020 Equity Incentive Plan | 6,349,911 | |||
| 10,129,298 |
Controlled Equity Offering
On December 31, 2020, the Company entered into a Controlled Equity OfferingSM Sales Agreement (the “Sales Agreement”) with Cantor Fitzgerald & Co., under which the Company may issue and sell from time to time shares of the Company’s common stock, $0.001 par value per share, having an aggregate gross sales price of up to $100,000,000 through Cantor Fitzgerald, as sales agent. As of December 31, 2020 no sales were made under the Sales Agreement.
2020 Equity Plan
On July 27, 2020, the Board unanimously approved, and recommended that the Company’s stockholders approve, the 2020 Equity Plan, to ensure that the Board and its compensation committee (the “Compensation Committee”) will be able to make the types of awards, and covering the number of shares, as necessary to meet the Company’s compensatory needs. On July 29, 2020, the 2020 Equity Plan was approved by the holders of approximately 63% of the Company’s outstanding shares of common stock on that date. The 2020 Equity Plan became effective on September 11, 2020 following the Effective Time of the reverse stock split.
Immediately following the Effective Time, a total of 7,000,000 shares of the Company’s common stock were reserved for issuance under the 2020 Equity Plan. The Board or Compensation Committee may grant the following types of awards under the 2020 Equity Plan: stock options, stock appreciation rights, restricted stock, stock awards, restricted stock units, performance shares, performance units, cash-based awards and substitute awards. The 2020 Equity Plan will remain in effect until the tenth anniversary of its effective date, unless terminated earlier by the Board.
| F-22 |
As of December 31, 2020, there were 6,349,911 shares available for grant under the 2020 Equity Incentive Plan.
2012 Equity Plan
The 2020 Equity Plan replaced the 2012 Equity Plan, under which no further grants will be made. However, any outstanding awards under the 2012 Equity Plan will continue in accordance with the terms of the 2012 Equity Plan and any award agreement executed in connection with such outstanding awards. At the Effective Time of the reverse stock split, proportionate adjustments were made to the number of shares covered by, and the exercise price applicable to, each outstanding stock option award under the 2012 Equity Plan to give effect to the Split Ratio and the reverse stock split.
Under the 2012 Equity Plan, the Company could grant shares, stock units, stock appreciation rights, performance cash awards and/or options to employees, directors, consultants, and other service providers. For options, the per share exercise price could not be less than the fair market value of a Company common share on the date of grant. Awards generally vest and become exercisable over three to four years and expire 10 years from the date of grant. Options generally become exercisable as they vest following the date of grant.
As of December 31, 2020, there were no shares available for grant under the 2012 Equity Incentive Plan.
2001 Equity Incentive Plan
Under the Company’s 2001 Stock Plan (the “2001 Plan”), the Company was able to grant shares and/or options to purchase up to 426,030 shares of common stock to employees, directors, consultants, and other service providers. In connection with the 2012 Plan taking effect, the 2001 Plan was terminated in August 2012. However, the awards under the 2001 Plan outstanding as of the termination of the 2001 Plan continued to be governed by their existing terms.
Stock Option Activity
The following table summarizes stock option activity for the years ended December 31, 2020 and 2019:
| Number of Shares | Weighted Average Price (per | Weighted- Average Remaining Contractual Term (in years) | Aggregate Intrinsic Value ($000's) (2) | |||||||||||||
| Outstanding at January 1, 2019 | 3,081,863 | $ | 4.75 | |||||||||||||
| Granted | 294,191 | 3.90 | ||||||||||||||
| Exercised | (97,725 | ) | 3.35 | |||||||||||||
| Cancelled (forfeited) | (101,984 | ) | 3.10 | |||||||||||||
| Cancelled (expired) | (9 | ) | 48.40 | |||||||||||||
| Outstanding at December 31, 2019 | 3,176,336 | 4.75 | ||||||||||||||
| Granted | 981,164 | 7.35 | ||||||||||||||
| Exercised | (429,330 | ) | 3.55 | |||||||||||||
| Cancelled (expired) | (21 | ) | 58.40 | |||||||||||||
| Outstanding at December 31, 2020 | 3,728,149 | $ | 5.57 | 7.6 | $ | 44,479 | ||||||||||
| Options vested and expected to vest | 3,715,473 | $ | 5.57 | 7.6 | $ | 44,328 | ||||||||||
| Exercisable | 2,999,502 | $ | 5.07 | 7.2 | $ | 10 | ||||||||||
______________________
| (1) | The weighted average price per share is determined using exercise price per share for stock options. |
| (2) | The aggregate intrinsic value is calculated as the difference between the exercise price of the option and the fair value of the Company’s common stock for in-the-money options at December 31, 2020. |
| F-23 |
The stock options outstanding and exercisable by exercise price at December 31, 2020 are as follows:
| Stock Options Outstanding | Stock Options Exercisable | |||||||||||||||||||
| Range of Exercise Prices | Number of Shares | Weighted- Average Remaining Contractual Life In Years | Weighted- Average Exercise Price Per Share | Number of Shares | Weighted- Average Exercise Price Per Share | |||||||||||||||
| $1.65 - $3.33 | 2,534,965 | 7.30 | $ | 3.14 | 2,372,837 | $ | 3.18 | |||||||||||||
| $4.20 - $9.05 | 343,065 | 8.95 | $ | 6.30 | 176,190 | $ | 4.92 | |||||||||||||
| $9.65 - $16.50 | 587,286 | 9.31 | $ | 10.57 | 187,642 | $ | 12.53 | |||||||||||||
| $16.90 - $16.90 | 252,604 | 5.62 | $ | 16.90 | 252,604 | $ | 16.90 | |||||||||||||
| $17.00 - $18.00 | 10,125 | 5.72 | $ | 17.01 | 10,125 | $ | 17.01 | |||||||||||||
| $58.40 - $86.80 | 42 | 0.09 | $ | 67.87 | 42 | $ | 67.87 | |||||||||||||
| $216.40 - $240.00 | 62 | 0.12 | $ | 225.92 | 62 | $ | 225.92 | |||||||||||||
| 3,728,149 | 7.65 | $ | 5.57 | 2,999,502 | $ | 5.07 | ||||||||||||||
The total fair value of options vested for the years ended December 31, 2020 and 2019 was $2.2 million and $2.2 million, respectively.
Stock-Based Compensation
The Company’s stock-based compensation expense for stock options is estimated at the grant date based on the award’s fair value as calculated by the Black-Scholes option pricing model and is recognized as expense over the requisite service period. The Black-Scholes option pricing model requires various highly judgmental assumptions including expected volatility and expected term. The expected volatility is based on the historical stock volatilities of several of the Company’s publicly listed peers over a period equal to the expected terms of the options as the Company does not have a sufficient trading history to use the volatility of its own common stock. To estimate the expected term, the Company has opted to use the simplified method, which is the use of the midpoint of the vesting term and the contractual term. If any of the assumptions used in the Black-Scholes option pricing model changes significantly, stock-based compensation expense may differ materially in the future from that recorded in the current period. In addition, the Company is required to estimate the expected forfeiture rate and only recognize expense for those shares expected to vest. The Company estimates the forfeiture rate based on historical experience and its expectations regarding future pre-vesting termination behavior of employees. The Company reviews its estimate of the expected forfeiture rate annually, and stock-based compensation expense is adjusted accordingly.
The weighted-average fair value-based measurement of stock options granted under the Company’s stock plans in the years ended December 31, 2020 and 2019 was $7.35 and $3.95 per share, respectively. The fair value- based measurement of stock options granted under the Company’s stock plans was estimated at the date of grant using the Black-Scholes model with the following assumptions:
| F-24 |
| Year Ended December 31, | ||||
| 2020 | 2019 | |||
| Expected term | 5 - 6 years | 5 - 6 years | ||
| Expected volatility | 95% - 111% | 96% - 99% | ||
| Risk-free interest rate | 0.28% - 1.57% | 1.74% - 2.59% | ||
| Expected dividend yield | 0% | 0% | ||
Total expense for stock option grants recognized was as follows:
| Year ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Selling,general and administrative | $ | 1,773 | $ | 1,928 | ||||
| Reseach and development | 337 | 97 | ||||||
| Total stock-based compensation | $ | 2,110 | $ | 2,025 | ||||
At December 31, 2020, the Company had $4.1 million of total unrecognized compensation expense, net of estimated forfeitures, related to outstanding stock options that will be recognized over a weighted-average period of 2.3 years.
| F-25 |
9. Income Taxes
No provision for federal income taxes has been recorded for the years ended December 31, 2020 and 2019 due to net losses and the valuation allowance established.
Deferred tax assets and liabilities reflect the net tax effects of net operating loss and tax credit carryovers and the temporary differences between the carrying amounts of assets and liabilities for financial reporting and the amounts used for income tax purposes. Significant components of the Company's deferred tax assets are as follows:
| As of December 31, | ||||||||
| 2020 | 2019 | |||||||
| Deferred tax assets: | ||||||||
| Net operating losses | $ | 75,149 | $ | 50,144 | ||||
| Research and other credits | 2,178 | 2,178 | ||||||
| Stock based compensation | 3,256 | 3,001 | ||||||
| In-Process research and development | 1,193 | 1,253 | ||||||
| Other | 665 | 854 | ||||||
| Total deferred tax assets | 82,441 | 57,430 | ||||||
| Valuation allowance | (82,441 | ) | (57,430 | ) | ||||
| Net deferred tax assets | $ | - | $ | - | ||||
A reconciliation of the statutory tax rates and the effective tax rates for the years ended December 2020 and 2019 is as follows:
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Statutory rate | 21.0 | % | 21.0 | % | ||||
| Valuation allowance | (27.9 | )% | (26.0 | )% | ||||
| Nondeductible stock compensation | - | % | (0.3 | )% | ||||
| Other | 6.9 | % | 5.3 | % | ||||
| Effective tax rate | - | % | - | % | ||||
Realization of deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Accordingly, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance increased by $25.0 million during 2020 and increased by $2.7 million during 2019.
At December 31, 2020, the Company had federal net operating loss carryforwards of approximately $166.2 million, which expire in the years 2021 through 2037, and state net operating loss carryforwards of approximately $261.4 million, which expire in the years 2028 through 2040. The Company also has federal net operating loss carryforwards generated in 2018, 2019 and 2020 of $104.9 million that have no expiration date as a result of the December 22, 2017 Tax Cuts and Jobs Act tax reform legislation.
At December 31, 2020, the Company had federal research and development credit carryforwards of approximately $1.3 million, which expire in the years 2022 through 2035 and state research and development credit carryforwards of approximately $2.2 million. The state research and development credit carryforwards can be carried forward indefinitely.
During 2013, the Company completed a Section 382 study in accordance with the Internal Revenue Code of 1986, as amended, and similar state provisions. The study concluded that the Company has experienced several ownership changes since inception. This causes the Company's utilization of its net operating loss and tax credit carryforwards to be subject to substantial annual limitations. These results are reflected in the above carryforward amounts and deferred tax assets. The Company's ability to utilize its net operating loss and tax credit carryforwards are further limited as a result of subsequent ownership changes. All such limitations could result in the expiration of carryforwards before they are utilized. An ownership change may have occurred during 2015, 2016, 2017, 2018, 2019 and 2020, or all five years and in connection with the Restructuring Transactions that occurred in 2018. As a result, tax attributes such as net operating losses and research and development credits may be subject to further limitation.
| F-26 |
ASC 740 requires that the Company recognize the financial statement effects of a tax position when it is more likely than not, based on the technical merits, that the position will be sustained upon examination.
A reconciliation of the beginning and ending amount of unrecognized tax benefits is as follows:
| Balance at December 31, 2018 | $ | 1,060 | ||
| Additions based on tax positions related to prior year | - | |||
| Additions based on tax positions related to current year | - | |||
| Balance at December 31, 2019 | 1,060 | |||
| Additions based on tax positions related to prior year | - | |||
| Additions based on tax positions related to current year | - | |||
| Balance at December 31, 2020 | $ | 1,060 |
There were no interest or penalties related to unrecognized tax benefits. Substantially all of the unrecognized tax benefit, if recognized to offset future taxable income would affect the Company’s tax rate. The Company does not anticipate that the amount of existing unrecognized tax benefits will significantly increase or decrease within the next 12 months. Because of net operating loss carryforwards, substantially all of the Company’s tax years remain open to federal tax and state tax examination.
The Company files income tax returns in the U.S. federal jurisdiction, California and Florida. Federal and California corporation income tax returns beginning with the 2001 tax year remain subject to examination by the Internal Revenue Service and the California Franchise Tax Board, respectively.
10. Employee Benefit Plan
The Company has established a 401(k) tax-deferred savings plan (the “401(k) Plan”), which permits participants to make contributions by salary deduction pursuant to Section 401(k) of the Internal Revenue Code. The Company is responsible for administrative costs of the 401(k) Plan. The Company may, at its discretion, make matching contributions to the 401(k) Plan. No employer contributions have been made to the plan for the years ended December 31, 2020 and 2019.
11. Litigation
Savant Litigation
On July 10, 2017, the Company filed a complaint against Savant in the Superior Court for the State of Delaware, New Castle County (the “Delaware Court”). KaloBios Pharmaceuticals, Inc. v. Savant Neglected Diseases, LLC, No. N17C-07-068 PRW-CCLD. The Company asserted breach of contract and declaratory judgment claims against Savant arising under the MDC Agreement. See Note 8 - “Savant Arrangements” for more information about the MDC Agreement. The Company alleges that Savant has breached its MDC Agreement obligations to pay cost overages that exceed a budgetary threshold as well as other related MDC Agreement representations and obligations. In the litigation, the Company has alleged that as of June 30, 2017, Savant was responsible for aggregate cost overages of approximately $3.4 million, net of a $0.5 million deductible under the MDC. The Company asserts that it is entitled to offset $2.0 million in milestone payments due Savant against the cost overages.
On July 12, 2017, Savant removed the case to the Bankruptcy Court, claiming that the action is related to or arises under the Bankruptcy Case from which we emerged in July 2016. In re KaloBios Pharmaceuticals, Inc., No. 15-12628 (LSS) (Bankr. D. Del.). On July 27, 2017, Savant filed an Answer and Counterclaims. Savant’s filing alleges breaches of contracts under the MDC Agreement and the Security Agreement, claiming that the Company breached its obligations to pay the milestone payments and other related representations and obligations. On August 1, 2017, the Company moved to remand the case back to the Delaware Court (the “Motion to Remand”).
| F-27 |
On August 2, 2017, Savant sent a foreclosure notice to the Company, demanding that it provide the Collateral as defined in the Security Agreement for inspection and possession on August 9, 2017, with a public sale to be held on September 1, 2017. The Company moved for a Temporary Restraining Order (the “TRO”) and Preliminary Injunction in the Bankruptcy Court on August 4, 2017. Savant responded on August 7, 2017. On August 7, 2017, the Bankruptcy Court granted the Company’s motion for a TRO, entering an order prohibiting Savant from collecting on or selling the Collateral, entering our premises, issuing any default notices to us, or attempting to exercise any other remedies under the MDC Agreement or the Security Agreement. On August 9, 2017, the parties have stipulated to continue the provisions of the TRO in full force and effect until further order of the appropriate court, which the Bankruptcy Court signed that same day (the “Stipulated Order”).
On January 22, 2018, Savant wrote to the Bankruptcy Court requesting dissolution of the TRO and the Stipulated Order. On January 29, 2018, the Bankruptcy Court granted the Motion to Remand and denied Savant’s request to dissolve the TRO and Stipulated Order, ordering that any request to dissolve the TRO and Stipulated Order be made to the Delaware Court.
On February 13, 2018 Savant made a letter request to the Delaware Court to dissolve the TRO and Stipulated Order. Also, on February 13, 2018, the Company filed its Answer and Affirmative defenses to Savant’s Counterclaims. On February 15, 2018 the Company filed a letter opposition to Savant’s request to dissolve the TRO and Stipulated Order and requesting a status conference. A hearing on Savant’s request to dissolve the TRO and Stipulated Order was held before the Delaware Court on March 19, 2018. The Delaware Court denied Savant’s request to dissolve the TRO and Stipulated order, which remain in effect.
On April 11, 2018, the Company advised the Delaware Court that it would meet and confer with Savant regarding a proposed case management order and date for trial. On April 26, 2018 the Delaware Court so-ordered a proposed case management order submitted by the Company and Savant. The schedule in the case management order was modified by stipulation on August 24, 2018.
On April 8, 2019, the Company moved to compel Savant to produce documents in response to the Company’s document requests. The parties thereafter agreed to a discovery schedule through June 30, 2019, which the Superior Court so ordered, and the parties produced documents to each other.
On June 4, 2019, Savant filed a complaint against the Company and Madison in the Delaware Court of Chancery (the “Chancery Action”) seeking to “recover as damages that amounts owed to it under the MDC Agreement, and to reclaim Savant’s intellectual property,” among other things. Savant also requested leave to move to dismiss the Company’s complaint on the grounds that the Company’s transfer of assets to Madison was champertous. On June 10, 2019, the Company requested by letter that the Superior Court hold a contempt hearing because the Chancery Action violated the TRO entered by the Bankruptcy Court, the terms of which have been extended by stipulation of the parties. On June 18, 2019, the Superior Court held a telephonic status conference. The parties agreed that the Chancery Action should be consolidated with the Superior Court action, after which the Superior Court would address the parties’ motions.
On July 22, 2019, the Company moved for contempt against Savant. Savant filed its opposition on July 29, 2019. On August 12, 2019, the Superior Court denied the Company’s motion for contempt.
On July 23, 2019, Savant moved for summary judgment on the issue of champerty. The Company filed its response and cross-motion for summary judgment on August 27, 2019. Savant filed its reply on September 10, 2019 and the Company filed its cross-reply on September 20, 2019. The motion is fully briefed and was argued at a hearing on February 3, 2020. On August 17, 2020 the Superior Court issued a memorandum opinion denying Savant’s motion for summary judgment on the issue of champerty.
On July 26, 2019, the Company moved to modify the previously agreed-upon discovery schedule to extend discovery through December 31, 2019, which the Superior Court granted. In subsequent orders, the discovery schedule was further extended until the end of June 2020.
On July 30, 2019, the Company filed a motion to dismiss Savant’s Chancery Action. Savant filed an amended complaint on September 4, 2019, and the Company filed its opening brief in support of its motion to dismiss on October 11, 2019. That motion is fully briefed and was argued at a hearing on February 3, 2020. On August 17, 2020 the Superior Court issued a memorandum opinion denying the Company’s motion to dismiss Savant’s Chancery Action.
| F-28 |
On August 19, 2019, Savant moved to dismiss the Company’s amended Superior Court complaint. On September 27, 2019, the Company filed an opposition to Savant’s motion and, in the alternative, requested leave to file a second amended complaint against Savant. Savant consented to the filing of the second amended complaint and withdrew their motion to dismiss. Savant filed a partial motion to dismiss against a co-defendant on October 30, 2019. That motion is fully briefed and was argued at a hearing on February 3, 2020. At the February 3, 2020 hearing, the Court reserved judgment on the parties’ reciprocal motions. On August 17, 2020, the Superior Court issued a memorandum opinion granting in part and denying in part the Savant’s motion to dismiss the Company’s amended Superior Court complaint. The Superior Court found the Company lacked standing because it assigned its interest to its co-plaintiff Madison Joint Venture LLC (“Madison”). However, the Superior Court found that Madison had standing to proceed. Although the Company was dismissed from all counts of the Superior Court Action it continues to possess an interest in the Superior Court Action because of its ownership of Madison and remains a litigant because of Savant’s Chancery Action. Savant’s motion to dismiss the Company’s amended Superior Court complaint was denied in all other respects.
On May 22, 2020, upon the request of the parties, the Superior Court stayed both Delaware actions until July 29, 2020.
On June 30, 2020, Savant exercised 100,000 warrants in a cashless exercise resulting in 54,545 shares being issued to Savant.
On July 24, 2020, the parties submitted a joint status report in the Delaware actions. The parties also requested a status conference with the Court to discuss moving the trial from October 2020 to some later time.
On August 20, 2020, the Court held the requested status conference and ordered that a consolidated trial for the Superior Court Action and Chancery Action would be held in April 2021. The parties subsequently agreed to a five-day trial starting on April 12, 2021.
On September 29, 2020, upon the request of the parties, the Superior Court revised the existing discovery schedule. Under the revised discovery schedule fact discovery closed on November 9, 2020 and expert discovery closed on December 18, 2020.
On October 7, 2020, Savant moved for leave to amend its complaint in the Chancery Action to add a claim for breach of contract related to its exercise of 100,000 warrants on June 30, 2020. Savant claims that the Company’s issuance of 54,545 shares was insufficient under the warrant. On November 23, 2020, the Superior Court granted Savant’s motion for leave to amend its complaint in the Chancery Action. The Company answered the second amended complaint in the Chancery Action on December 11, 2020.
On November 30, 2020, the Superior Court revised the discovery schedule to permit the Company to take additional discovery regarding Savant’s second amended complaint in the Chancery Action. Under this revised schedule, the Company had until December 18, 2020 to take additional discovery. Further, the deadline to complete expert discovery was moved to January 5, 2021. A consolidated trial for the Superior Court Action and Chancery Action remains scheduled for April 2021.
On December 18, 2020, fact discovery closed.
On January 5, 2021, the Company moved for leave to amend the Superior Court complaint to add Scott Freeman as a defendant. Savant opposed the motion for leave to amend on February 5, 2021. Briefing was completed on March 5, 2021. Further, the deadline for expert discovery closed.
On January 19, 2021, the Company and Savant filed competing motions for summary judgment on their respective claims in the Superior Court Action and Chancery Action. Oppositions to the motions for summary judgment were filed on February 5, 2021 and replies were filed on February 24, 2021. Briefing is now complete and a hearing is scheduled for March 18, 2021.
| F-29 |
Private Placement Litigation
On June 15, 2020, a complaint was filed against the Company and Dr. Durrant in the Commercial Division of the Supreme Court of the State of New York. The case caption is Alliance Texas Holdings, LLC et al. v. Humanigen, Inc. et al., Index No. 652490/2020 (“Alliance Texas Holdings Case”). The plaintiffs in the Alliance Texas Holdings Case comprise a group of 17 prospective investors introduced to Humanigen by Noble Capital Markets, Inc. (“Noble”), which had been engaged as a non-exclusive placement agent in connection with the Private Placement. The plaintiffs had indicated interest in purchasing shares of common stock in the Private Placement but, due to the strength of demand for shares from other prospective investors introduced to the company by J.P. Morgan Securities LLC, the lead placement agent for the Private Placement, the plaintiffs were not allocated any investment amount. The plaintiffs allege that the Company and Dr. Durrant breached a contractual obligation to deliver shares of common stock to the plaintiffs. The plaintiffs seek to recover for losses due to alleged fraudulent misstatements and the Company’s failure to deliver shares to them and seek equitable relief in the form of specific performance. The Defendants filed a Motion to Dismiss the Complaint in the Alliance Texas Holdings Case on July 24, 2020. The Parties completed briefing on the Motion to Dismiss on September 4, 2020. The Motion to Dismiss is currently being considered by the court.
On June 19, 2020, Noble filed a separate complaint against the Company in the Circuit Court of the Fifteenth Judicial Circuit in and for Palm Beach County, Florida, also arising from the Private Placement. On July 13, 2020, the Defendants removed Noble’s complaint to United States District Court for the Southern District of Florida. The case caption is Noble Capital Markets, Inc. v. Humanigen, Inc., Case No. 9:20-CV-81131-WPD (the “Noble Case”). Noble’s complaint alleges that Humanigen breached the terms of its engagement letters with Noble by refusing to pay it the sales commissions it would have earned had its prospective investors received the entire allocation of shares sought in the Private Placement, as opposed to the $4 million of shares actually allocated to Noble and its clients. Noble is seeking payment in full of the commission, damages for Humanigen’s alleged tortious interference with Noble’s business relationship with the investors it introduced to Humanigen, but which were not allocated shares in the Private Placement, and attorneys’ fees. The Company filed its answer and affirmative defenses in response to Noble’s complaint on February 2, 2021. The parties are currently engaged in discovery.
The Company believes that the claims made in each complaint are without merit, and it is prepared to defend itself vigorously.
12. License and Collaboration Agreements
Kite Agreement
On May 30, 2019, the Company entered into a collaboration agreement (the “Kite Agreement”) with Kite Pharmaceuticals, Inc., pursuant to which the Company and Kite are conducting a multi-center Phase 1b/2 study of lenzilumab with Kite’s Yescarta in patients with relapsed or refractory B-cell lymphoma, including diffuse large B-cell lymphoma (“DLBCL”). The primary objective of the study is to determine the effect of lenzilumab on the safety of Yescarta.
Pursuant to the Kite Agreement, the Company shall supply lenzilumab to the collaboration for use in the study and will contribute up to approximately $8.0 million towards the out-of-pocket costs of the study, depending on the number of patients enrolled into the study. During the month of September 2020, the Company paid $2.0 million to Kite towards its contribution for the study, which payment was recorded as Research and development expense in the year ended December 31, 2020.
Mayo Agreement
On June 19, 2019 the Company entered into an exclusive worldwide license agreement (the “Mayo Agreement”) with the Mayo Foundation for Medical Education and Research (“Mayo”) for certain technologies used to create CAR-T cells lacking GM-CSF expression through various gene-editing tools including CRISPR-Cas9 (GM-CSF knock-out). The license covers various patent applications and know-how developed by Mayo in collaboration with the Company. These licensed technologies complement and broaden the Company’s position in the GM-CSF neutralization space and expand the Company’s discovery platform aimed at improving CAR-T to include gene-edited CAR-T cells.
| F-30 |
Pursuant to the Mayo Agreement, the Company agreed to pay $200,000 to Mayo within six months of the effective date, or upon completion of a qualified financing, whichever is earlier. The Mayo Agreement also requires the payment of milestones and royalties upon the achievement of certain regulatory and commercialization milestones. The initial payment was recorded as Research and development expense in June 2019. The Company paid the initial payment in June 2020 subsequent to the closing of the Private Placement.
Zurich Agreement
On July 19, 2019 the Company entered into an exclusive worldwide license agreement (the “Zurich Agreement”) with the University of Zurich (“UZH”) for technology used to prevent or treat Graft versus Host Disease (“GvHD”) through GM-CSF neutralization. The Zurich Agreement covers various patent applications filed by UZH which complement and broaden the Company’s position in the application of GM-CSF and expands the Company’s development platform to include improving allogeneic Hematopoietic Stem Cell Transplantation (“HSCT”).
Pursuant to the Zurich Agreement, the Company paid $100,000 to UZH in July 2019. The Zurich Agreement also requires the payment of annual maintenance fees and milestones and royalties upon the achievement of certain regulatory and commercialization milestones. The license payment of $100,000 was recorded as Research and development expense in July 2019.
CRADA
On November 5, 2020, the Company and the Department of Defense (DoD) Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense (JPEO-CBRND or JPEO) entered into a Cooperative Research and Development Agreement (“CRADA”) in collaboration with the Biomedical Advanced Research and Development Authority (BARDA), part of the Office of the Assistant Secretary for Preparedness and Response (ASPR) at the U.S. Department of Health and Human Services (HHS), in support of OWS, to assist in the development of the Company’s lead product candidate, lenzilumab, in advance of a potential EUA for COVID-19.
On January 22, 2021, the Company announced an expansion of the CRADA that it had previously entered with JPEO on November 5, 2020, was subsequently co-signed by BARDA. This provides the Company with access to manufacturing capacity reserved by BARDA for fill-finish product to accelerate the drug product manufacturing of lenzilumab.
Pursuant to the CRADA, the Company has been provided access to a full-scale, integrated team of OWS manufacturing, and regulatory subject matter experts, leading decision makers and statistical support in anticipation of applying for EUA and subsequently a BLA for lenzilumab as a potential treatment for COVID-19. The CRADA also provides that OWS regulatory experts will work with the Company on FDA communications, meetings and regulatory filings. The CRADA aims to support the lenzilumab Phase 3 clinical trial output, focusing on efficiently generating an EUA and BLA submission. In addition to providing access under EUA, a goal of the CRADA is to ensure lenzilumab receives the benefits provided by Public Law 115-92.
Manufacturing Agreements
During the year ended December 31, 2020 the Company has entered into agreements with several contract manufacturing organizations (“CMOs”) to have them manufacture bulk drug substance (“BDS”) for our lenzilumab clinical trial activities in COVID-19 as well as to manufacture BDS for a potential launch of lenzilumab in anticipation of an EUA in 2021. We have also entered into agreements for filling, finishing and packaging of the drug. These agreements represent large commitments, including upfront amounts prior to commencement of manufacturing and progress payments through the course of the manufacturing process and include payments for technology transfer. As of December 31, 2020, the Company estimates that its commitments to be incurred during the first quarter of 2021 will amount to approximately $48 million. The agreements contain customary cancellation clauses.
| F-31 |
13. Related Party Transactions
Commencing September 19, 2018, the Company delivered a series of convertible promissory notes evidencing an aggregate of $2.5 million of loans made to the Company by six different lenders, including an affiliate of BHC, the Company’s controlling stockholder. See Note 6 for a further discussion of the 2018 Notes.
On June 28, 2019, the Company issued three short-term, secured bridge notes evidencing an aggregate of $1.7 million of loans made to the Company by three parties including Dr. Cameron Durrant, our Chairman and Chief Executive Officer and Cheval Holdings, Ltd., an affiliate of Black Horse Capital, L.P., our controlling stockholder. See Note 6 for a further discussion of the 2019 Bridge Notes.
On November 12, 2019, the Company issued two short-term, secured bridge notes (the “November Bridge Notes” and together with the June Bridge Notes, the “2019 Bridge Notes”) evidencing an aggregate of $350,000 of loans made to the Company by two parties, including Dr. Cameron Durrant, our Chairman and Chief Executive Officer and Cheval Holdings, Ltd., an affiliate of Black Horse Capital, L.P., our controlling stockholder. See Note 6 for a further discussion of the 2019 Bridge Notes.
14. Subsequent Events
Eversana Agreement
On January 10, 2021, the Company announced that it had entered into a master services agreement (the “Eversana Agreement”) with Eversana Life Science Services, LLC (“Eversana”) pursuant to which Eversana will provide the Company multiple services from its integrated commercial platform in preparation for the potential commercialization of lenzilumab, the Company’s lead product candidate, for treatment of hospitalized and hypoxic COVID-19 patients.
Under the Eversana Agreement, Eversana will serve as the Company’s end-to-end commercial partner, providing the Company with a full suite of services in connection with the potential launch of lenzilumab. Eversana services initially will comprise marketing, market access and field solution services, and may expand to other areas including patient services, compliance services, professional services, data and analytics, and health economics outcome research, as may be negotiated by the parties and set forth in statements of work delivered in accordance with the Eversana Agreement.
The Company will pay Eversana fees and reimburse it for its expenses in performing the services as established in applicable statements of work. Eversana has agreed to defer its fees pending the Company’s receipt of an EUA from the U.S. Food and Drug Administration for lenzilumab for hospitalized and hypoxic COVID-19 patients. If the Company has not received an EUA by April 30, 2021 or such later date as to which the parties otherwise may agree, either party may terminate the Agreement immediately; Eversana will not receive the deferred fees upon such termination.
The Eversana Agreement provides for a one-year term and will renew for subsequent one-year terms unless either party provides a notice of non-renewal. After the first year, the Company may terminate the Eversana Agreement upon advance written notice to Eversana. The Eversana Agreement contains customary provisions allowing either party to terminate the Eversana Agreement as a result of certain changes in law and material breaches and certain insolvency events by or relating to the other party.
The Eversana Agreement imposes customary mutual obligations on the parties to protect and not disclose the confidential information and intellectual property of the other, and contains insurance, non-solicitation, indemnification and limitation of liability provisions customary for service contracts of this type.
CRADA Amendment
On January 22, 2021, the Company announced an expansion to the Cooperative Research and Development Agreement (as amended and restated, the “CRADA”) that the Company had previously entered into with the Department of Defense Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense (JPEO-CBRND), to gain access to manufacturing capacity reserved by the Biomedical Advanced Research and Development Authority (BARDA), part of the Office of the Assistant Secretary for Preparedness and Response (ASPR) at the U.S. Department of Health and Human Services. The CRADA supports development of the Company’s lead product candidate, lenzilumab, in advance of a potential Emergency Use Authorization (“EUA”) for COVID-19.
| F-32 |
The CRADA, now co-signed by BARDA, provides the Company with access to manufacturing capacity reserved by BARDA for fill-finish product to accelerate the drug product manufacturing of lenzilumab. The initial agreement, originally signed in November 2020 and amended and restated on January 21, 2021, complements the Company’s development efforts for lenzilumab by providing access to a full-scale, integrated team of manufacturing and regulatory subject matter experts and statistical support in anticipation of applying for EUA and subsequently a Biologics License Application (“BLA”) for lenzilumab as a potential treatment for COVID-19.
Enrollment Completion
On January 29, 2021, the Company announced that it had completed enrollment for its pivotal Phase 3 study of lenzilumabTM for COVID-19.
Controlled Equity Offering Sales
During the period from January 1, 2021 through March 5, 2021, the Company sold 1,796,858 shares of its common stock under the Sales Agreement for net proceeds of $36.2 million.
Secured Term Loan Facility
On March 10, 2021, we executed a Loan and Security Agreement with Hercules Capital, Inc., as agent for its affiliates serving as lenders thereunder, pursuant to which we established a term loan facility in aggregate principal amount of up to $80 million (the “Term Loan”). The Term Loan provides us with the ability to draw an initial amount of $25.0 million, with up to an additional $55.0 million available for future draws subject to achievement of future milestones and satisfaction of other conditions. See Part II, Item 9B “Other Information” for additional information regarding the Term Loan.
| F-33 |
EXHIBIT INDEX
| 101.INS | XBRL Instance Document | |
| 101.SCH | XBRL Taxonomy Extension Schema | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | |
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document | |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document |
**Indicates management contract or compensatory plan.
***The certifications attached as Exhibits 32.1 and 32.2 that accompanies this Annual Report on Form 10-K are not deemed filed with the Securities and Exchange Commission and are not to be incorporated by reference into any filing of Registrant under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date of this Annual Report on Form 10-K, irrespective of any general incorporation language contained in such filing.
†Confidential treatment has been granted with respect to certain portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission.
††Certain portions of this exhibit have been omitted pursuant to Item 601(b)(10)(iv) of Regulation S-K. The Company will furnish supplementally an unredacted copy of such exhibit to the Securities and Exchange Commission or its staff upon request.