Attached files
| file | filename |
|---|---|
| EX-32.2 - EXHIBIT 32.2 - HUMANIGEN, INC | ex32_2.htm |
| EX-32.1 - EXHIBIT 32.1 - HUMANIGEN, INC | ex32_1.htm |
| EX-31.2 - EXHIBIT 31.2 - HUMANIGEN, INC | ex31_2.htm |
| EX-31.1 - EXHIBIT 31.1 - HUMANIGEN, INC | ex31_1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-Q
(Mark One)
xQUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the quarterly period ended June 30, 2020
OR
oTRANSITION REPORT UNDER SECTION 13 OF 15(d) OF THE EXCHANGE ACT OF 1934
From the transition period from to .
Commission File Number 001-35798
Humanigen, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 77-0557236 | |
| (State or other jurisdiction of | (IRS Employer | |
| incorporation) | Identification No.) |
533 Airport Boulevard, Suite 400 Burlingame, CA 94010
(Address of principal executive offices)
(Zip Code)
Registrant’s telephone number, including area code: (650) 243-3100
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on with registered |
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes x No o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer”, “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act:
| Large accelerated filer o | Accelerated filer o | |
| Non-accelerated filer o |
Smaller reporting company x | |
| Emerging growth company o |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. o
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No x
Indicate by check mark whether the registrant has filed all documents and reports required to be filed by Section 12, 13 or 15(d) of the Securities Exchange Act of 1934 subsequent to the distribution of securities under a plan confirmed by a court. Yes x No o
As of August 13, 2020, there were 210,504,084 shares of common stock of the issuer outstanding.
HUMANIGEN, INC.
FORM 10-Q
| Item 1. | Financial Statements |
Humanigen, Inc.
Condensed Consolidated Balance Sheets
(in thousands, except share data)
(Unaudited)
| June 30, 2020 | December 31, 2019 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 41,729 | $ | 143 | ||||
| Prepaid expenses and other current assets | 770 | 309 | ||||||
| Total current assets | 42,499 | 452 | ||||||
| Restricted cash | 70 | 71 | ||||||
| Total assets | $ | 42,569 | $ | 523 | ||||
| Liabilities and stockholders’ deficit | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 2,110 | $ | 5,046 | ||||
| Accrued expenses | 8,850 | 3,308 | ||||||
| Bridge Notes | - | 2,113 | ||||||
| Convertible notes - current | - | 2,033 | ||||||
| Notes payable to vendors | 8 | 1,094 | ||||||
| Total current liabilities | 10,968 | 13,594 | ||||||
| Convertible notes - non current | - | 1,247 | ||||||
| Total liabilities | 10,968 | 14,841 | ||||||
| Stockholders’ deficit: | ||||||||
| Common stock, $0.001 par value: 225,000,000 shares authorized at | ||||||||
June 30, 2020 and December 31, 2019; 209,874,577 and 114,034,451 shares issued and outstanding at June 30, 2020 and December 31, 2019, respectively | 210 | 114 | ||||||
| Additional paid-in capital | 342,775 | 270,463 | ||||||
| Accumulated deficit | (311,384 | ) | (284,895 | ) | ||||
| Total stockholders’ deficit | 31,601 | (14,318 | ) | |||||
| Total liabilities and stockholders’ deficit | $ | 42,569 | $ | 523 | ||||
See accompanying notes.
| 3 |
Humanigen, Inc.
Condensed Consolidated Statements of Operations
(in thousands, except share and per share data)
(Unaudited)
| Three Months Ended June 30, | Six Months Ended June 30, | |||||||||||||||
| 2020 | 2019 | 2020 | 2019 | |||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 21,143 | $ | 1,234 | $ | 21,802 | $ | 1,593 | ||||||||
| General and administrative | 1,956 | 1,746 | 3,354 | 3,625 | ||||||||||||
| Total operating expenses | 23,099 | 2,980 | 25,156 | 5,218 | ||||||||||||
| Loss from operations | (23,099 | ) | (2,980 | ) | (25,156 | ) | (5,218 | ) | ||||||||
| Other expense: | ||||||||||||||||
| Interest expense | (923 | ) | (358 | ) | (1,333 | ) | (660 | ) | ||||||||
| Other income (expense), net | - | - | - | (1 | ) | |||||||||||
| Net loss | $ | (24,022 | ) | $ | (3,338 | ) | $ | (26,489 | ) | $ | (5,879 | ) | ||||
| Basic and diluted net loss per common share | $ | (0.16 | ) | $ | (0.03 | ) | $ | (0.20 | ) | $ | (0.05 | ) | ||||
| Weighted average common shares outstanding used to | ||||||||||||||||
| calculate basic and diluted net loss per common share | 153,419,159 | 111,110,926 | 132,691,688 | 110,560,662 | ||||||||||||
See accompanying notes.
| 4 |
Humanigen, Inc.
Condensed Consolidated Statements of Cash Flows
(in thousands)
(Unaudited)
| Six Months Ended | ||||||||
| June 30, | ||||||||
| 2020 | 2019 | |||||||
| Operating activities: | ||||||||
| Net loss | $ | (26,489 | ) | $ | (5,879 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock based compensation expense | 622 | 1,426 | ||||||
| Issuance of common stock for payment of compensation | 48 | 90 | ||||||
| Issuance of common stock in exchange for services | 70 | 68 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other assets | (461 | ) | 60 | |||||
| Accounts payable | (2,936 | ) | 717 | |||||
| Accrued expenses | 6,531 | 1,245 | ||||||
| Net cash used in operating activities | (22,615 | ) | (2,273 | ) | ||||
| Financing activities: | ||||||||
| Net proceeds from issuance of common stock | 67,069 | - | ||||||
| Proceeds from exercise of stock options | - | 325 | ||||||
| Net proceeds from issuance of convertible notes | 467 | 1,275 | ||||||
| Net proceeds from issuance of PPP loan | 83 | - | ||||||
| Net proceeds from issuance of bridge notes | 350 | 950 | ||||||
| Payments on PPP loan | (83 | ) | - | |||||
| Payments on bridge notes | (2,400 | ) | - | |||||
| Payments on convertible notes | (518 | ) | - | |||||
| Payments on notes payable to vendors | (768 | ) | - | |||||
| Net cash provided by financing activities | 64,200 | 2,550 | ||||||
| Net increase in cash, cash equivalents and restricted cash | 41,585 | 277 | ||||||
| Cash, cash equivalents and restricted cash, beginning of period | 214 | 885 | ||||||
| Cash, cash equivalents and restricted cash, end of period | $ | 41,799 | $ | 1,162 | ||||
| Supplemental cash flow disclosure: | ||||||||
| Cash paid for interest | $ | 668 | $ | 8 | ||||
| Supplemental disclosure of non-cash investing and financing activities: | ||||||||
| Conversion of notes payable and related accrued interest and fees to common stock | $ | 4,316 | $ | 981 | ||||
| Beneficial conversion feature of Convertible notes | $ | - | $ | 142 | ||||
| Issuance of stock options in lieu of cash compensation | $ | 165 | $ | 195 | ||||
| Issuance of warrants for services | $ | 118 | $ | - | ||||
See accompanying notes.
| 5 |
Humanigen, Inc.
Condensed Consolidated Statements of Stockholders’ Equity (Deficit)
(in thousands, except share data)
(Unaudited)
| Three and Six Months Ended June 30, 2020 | ||||||||||||||||||||
| Additional | Total | |||||||||||||||||||
| Common Stock | Paid-In | Accumulated | Stockholders’ | |||||||||||||||||
| Shares | Amount | Capital | Deficit | Equity | ||||||||||||||||
| Balances at January 1, 2020 | 114,034,451 | $ | 114 | $ | 270,463 | $ | (284,895 | ) | $ | (14,318 | ) | |||||||||
| Issuance of common stock | 200,000 | - | 65 | - | 65 | |||||||||||||||
| Issuance of common stock in exchange for services | 29,342 | - | 13 | - | 13 | |||||||||||||||
| Issuance of stock options for payment of compensation | - | - | 133 | - | 133 | |||||||||||||||
| Issuance of common stock for payment of compensation | 47,997 | - | 19 | - | 19 | |||||||||||||||
| Issuance of warrant for services | - | - | 1 | - | 1 | |||||||||||||||
| Stock-based compensation expense | - | - | 265 | - | 265 | |||||||||||||||
| Net loss | - | - | - | (2,467 | ) | (2,467 | ) | |||||||||||||
| Balances at March 31, 2020 | 114,311,790 | 114 | 270,959 | (287,362 | ) | (16,289 | ) | |||||||||||||
| Issuance of common stock for conversion of debt | 11,989,578 | 12 | 4,304 | - | 4,316 | |||||||||||||||
| Issuance of common stock, net of expenses | 82,528,718 | 83 | 66,921 | - | 67,004 | |||||||||||||||
| Issuance of common stock in exchange for services | 77,313 | - | 57 | - | 57 | |||||||||||||||
| Issuance of stock options for payment of compensation | - | - | 32 | - | 32 | |||||||||||||||
| Issuance of common stock for payment of compensation | 24,574 | - | 29 | - | 29 | |||||||||||||||
| Issuance of warrant for services | - | - | 117 | - | 117 | |||||||||||||||
| Issuance of common stock upon option exercise | 942,604 | 1 | (1 | ) | - | - | ||||||||||||||
| Stock-based compensation expense | - | - | 357 | - | 357 | |||||||||||||||
| Net loss | - | - | - | (24,022 | ) | (24,022 | ) | |||||||||||||
| Balances at June 30, 2020 | 209,874,577 | $ | 210 | $ | 342,775 | $ | (311,384 | ) | $ | 31,601 | ||||||||||
| Three and Six Months Ended June 30, 2019 | ||||||||||||||||||||
| Additional | Total | |||||||||||||||||||
| Common Stock | Paid-In | Accumulated | Stockholders’ | |||||||||||||||||
| Shares | Amount | Capital | Deficit | Deficit | ||||||||||||||||
| Balances at January 1, 2019 | 109,897,526 | $ | 110 | $ | 266,381 | $ | (274,601 | ) | $ | (8,110 | ) | |||||||||
| Issuance of common stock for payment of compensation | 93,358 | - | 90 | - | 90 | |||||||||||||||
| Issuance of stock options for payment of compensation | - | - | 195 | - | 195 | |||||||||||||||
| Issuance of common stock in exchange for services | 82,432 | - | 68 | 68 | ||||||||||||||||
| Exercise of stock options | 75,000 | - | 50 | - | 50 | |||||||||||||||
| Stock-based compensation expense | - | - | 697 | - | 697 | |||||||||||||||
| Net loss | - | - | - | (2,541 | ) | (2,541 | ) | |||||||||||||
| Balances at March 31, 2019 | 110,148,316 | 110 | 267,481 | (277,142 | ) | (9,551 | ) | |||||||||||||
| Convertible note beneficial conversion feature | - | - | 143 | 143 | ||||||||||||||||
| Conversion of advance notes | 2,179,622 | 2 | 979 | - | 981 | |||||||||||||||
| Exercise of common stock options | 413,625 | 1 | 274 | - | 275 | |||||||||||||||
| Stock-based compensation expense | - | - | 729 | - | 729 | |||||||||||||||
| Net loss | - | - | - | (3,338 | ) | (3,338 | ) | |||||||||||||
| Balances at June 30, 2019 | 112,741,563 | $ | 113 | $ | 269,606 | $ | (280,480 | ) | $ | (10,761 | ) | |||||||||
See accompanying notes.
| 6 |
Humanigen, Inc.
Notes to Condensed Consolidated Financial Statements
(Unaudited)
1. Nature of Operations
Description of the Business
The Company was incorporated on March 15, 2000 in California and reincorporated as a Delaware corporation in September 2001 under the name KaloBios Pharmaceuticals, Inc. Effective August 7, 2017, the Company changed its legal name to Humanigen, Inc. During February 2018, the Company completed the restructuring transactions announced in December 2017 and continued its transformation into a clinical-stage biopharmaceutical company by further developing its clinical stage immuno-oncology and immunology portfolio of monoclonal antibodies. The Company is currently focused on developing its novel human granulocyte-macrophage colony-stimulating factor (“GM-CSF”) neutralization and gene-knockout platforms and its portfolio of antibodies based on its proprietary Humaneered® monoclonal antibody technology and has initiated research in next-generation cell and gene therapies.
The Company’s lead product candidate is lenzilumab, a proprietary Humaneered monoclonal antibody (a biologic) that has been demonstrated to neutralize a naturally occurring inflammatory factor (GM-CSF). GM-CSF is a cytokine which acts directly on myeloid cells to cause expansion, activation, and to initiate and promote the production of other cytokines and chemokines, including IL-6, TNFa, IL-1, MCP-1, MIP-1a, and IP-10 as part of the body’s immune response. GM-CSF is thought of as a communication conduit between the innate and adaptive immune systems. Once initiated, the inflammatory cascade in certain cases may quickly evolve into a self-perpetuating “cytokine storm” as the production of cytokines and chemokines increases expansion and trafficking of myeloid cells. This, in turn, leads to abnormally high levels of inflammatory cytokines, endothelial activation, vascular permeability, disseminated intravascular coagulation, and neurologic inflammation. This cytokine storm is frequently referred to as cytokine release syndrome, or CRS. The neutralization of GM-CSF has been shown to prevent and potentially treat cytokine storm and reduce levels of inflammatory myeloid cells. Reduction of these factors demonstrates that GM-CSF is a critical upstream and early regulator of many inflammatory cytokines known to be important in the pathophysiology of CRS (Sterner RM et al. Blood 2019. 133(7): 697–709).
During 2019 and throughout the early portion of the first quarter of 2020, the Company continued to pursue its anti-GM-CSF programs to prevent or reduce the serious and potentially life-threatening side effects associated with chimeric antigen receptor T-cell (“CAR-T”) therapy and to prevent or treat graft-versus-host disease (“GvHD”) in patients undergoing allogeneic hematopoietic stem cell transplantation (“HSCT”). In collaboration with Kite Pharmaceuticals, Inc., a Gilead company (“Kite or the “Kite Collaboration”), the Company seeks to study the effect of lenzilumab on the safety of Yescarta®, axicabtagene ciloleucel (“Yescarta”), Kite’s FDA-approved CAR-T therapy. A clinical trial, ZUMA-19, is underway to measure the effect of lenzilumab in reducing CRS and neurotoxicity (NT), with a secondary endpoint of increased efficacy of Yescarta. The first patient was dosed with lenzilumab in ZUMA-19 on June 29, 2020.
The coronavirus pandemic, which is due to the SARS-CoV-2 virus and leads to the condition referred to as COVID-19, is frequently characterized in the later and sometimes fatal stages by severe and critical, progressive viral pneumonia that can progress to acute respiratory distress syndrome (“ARDS”), respiratory failure and death. Publications have indicated that ARDS in this setting is caused by the body’s autoimmune response to CRS. Published data point to GM-CSF being a key triggering cytokine, with elevated levels especially in those patients who transition to the Intensive Care Unit (“ICU”).
In response to this published data indicating that GM-CSF inhibition may play a role in treating patients with COVID-19, the Company is developing lenzilumab in patients with COVID-19 in a Phase III study. Given the severity of the pandemic and the lack of approved therapies for COVID-19, the Company believes that this single Phase III study may be suitable for registration and depending on the results of this study may file for approval with the FDA. The Company has commenced enrollment in a multicenter randomized, placebo-controlled, double-blind clinical trial to assess whether lenzilumab can reduce the time to recovery in hospitalized subjects with severe or critical COVID-19 pneumonia. The first patient was dosed with lenzilumab on May 5, 2020.
Lenzilumab was granted emergency single use Investigational New Drug Application (IND) authorization from the FDA (often referred to as compassionate use) to treat patients with COVID-19. On June 15, 2020, the Company announced that Mayo Clinic published (www.medrxiv.org/content/10.1101/2020.06.08.20125369v1) data derived from the compassionate use of lenzilumab in treatment of 12 patients hospitalized in the Mayo Clinic system. Under applicable FDA rules, a patient cannot receive a compassionate use drug unless FDA has issued an individual patient emergency IND authorization, which the Mayo Clinic requested from FDA prior to each individual patient dosing of lenzilumab. Accordingly, there was no randomized control group in the Mayo Clinic program. The Company did not pre-select patients to receive lenzilumab through the compassionate use program and did not deny any requests for compassionate use. Mayo Clinic clinicians solely determined the patients for which they would request emergency IND authorization from the FDA.
| 7 |
See Management’s Discussion and Analysis of Financial Condition and Results of Operations in Item 2 of this Quarterly Report on Form 10-Q for additional information regarding the business.
Liquidity and Going Concern
The Condensed Consolidated Financial Statements for the three and six months ended June 30, 2020 were prepared on the basis of a going concern, which contemplates that the Company will be able to realize assets and discharge liabilities in the normal course of business. However, the Company has incurred net losses since its inception and has negative operating cash flows. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. The financial statements do not include any adjustments that might be necessary if the Company is unable to continue as a going concern.
As described in more detail in Note 7, on June 1, 2020, the Company entered into a securities purchase agreement with certain accredited investors to complete a private placement of our common stock (the “Private Placement”). At the closing, the Company issued and sold 82,528,718 shares of its common stock at a purchase price of $0.87 per share, for aggregate gross proceeds of approximately $71.8 million. The Company used a portion of the proceeds to retire certain indebtedness, as further described in Note 4 - Debt. The Company expects to use the remaining proceeds from the Private Placement to fund its Phase III study of lenzilumab in COVID-19, its collaboration with Kite, initiate manufacturing activities in anticipation of potential receipt of an Emergency Use Authorization (“EUA”) from FDA and subsequent commercialization, and other development programs, as well as for working capital and other general corporate purposes.
As of June 30, 2020, the Company had cash and cash equivalents of $41.8 million. Considering the Company’s current cash resources and its current and expected levels of operating expenses, management expects to need additional capital to fund the Company’s planned operations for the next twelve months. Management may seek to raise such additional capital through equity offerings, debt financings, and/or payments under new or existing licensing or collaboration agreements. While management believes this plan to raise additional funds will alleviate the conditions that raise substantial doubt, these plans are not entirely within its control and cannot be assessed as being probable of occurring. If sufficient funds are not available on acceptable terms when needed, the Company could be required to significantly reduce its operating expenses and delay, reduce the scope of, or eliminate one or more of its development programs. If any of these events occur, the Company’s ability to achieve the development and commercialization goals would be adversely affected.
Basis of Presentation
The accompanying interim unaudited Condensed Consolidated Financial Statements have been prepared in accordance with U.S. generally accepted accounting principles (“U.S. GAAP”) for interim financial information and on a basis consistent with the annual consolidated financial statements and include all adjustments necessary for the presentation of the Company’s condensed consolidated financial position, results of operations and cash flows for the periods presented. The Condensed Consolidated Financial Statements include the accounts of the Company and its wholly owned subsidiaries. These financial statements have been prepared on a basis that assumes that the Company will continue as a going concern, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The December 31, 2019 Condensed Consolidated Balance Sheet was derived from the audited financial statements but does not include all disclosures required by U.S. GAAP. These interim financial results are not necessarily indicative of the results to be expected for the year ending December 31, 2020, or for any other future annual or interim period. The accompanying unaudited Condensed Consolidated Financial Statements should be read in conjunction with the audited consolidated financial statements and the related notes thereto included in the Company’s 2019 Form 10-K.
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts and disclosures reported in the Condensed Consolidated Financial Statements and accompanying notes. Actual results could differ materially from those estimates. The Company believes judgment is involved in determining the valuation of the fair value-based measurement of stock-based compensation and warrant valuations. The Company evaluates its estimates and assumptions as facts and circumstances dictate. As future events and their effects cannot be determined with precision, actual results could differ from these estimates and assumptions, and those differences could be material to the Condensed Consolidated Financial Statements.
| 8 |
Certain prior year amounts have been reclassified to conform to the current year presentation. Such reclassifications had no effect on prior years’ Net loss or Stockholders’ equity (deficit).
2. Summary of Significant Accounting Policies
There have been no material changes in the Company’s significant accounting policies since those previously disclosed in the 2019 Form 10-K.
Derivative Financial Instruments
The Company has equity conversion features within its 2020 Convertible Redeemable Notes that qualify as embedded derivatives under the guidance of FASB ASC Topic 815, Derivatives and Hedging. As part of that guidance, an analysis is performed on each embedded derivative to determine whether it should be bifurcated from the host instrument and recorded separately within the Consolidated Balance Sheet at its fair value. Changes in fair value are recorded in other income (expense) within the Consolidated Statement of Operations. Refer to Note 4 - Debt and Note 5 – Derivative Instruments for further discussion of the Company’s embedded derivatives.
3. Potentially Dilutive Securities
The Company’s potentially dilutive securities, which include stock options, restricted stock units and warrants, have been excluded from the computation of diluted net loss per common share as the effect of including those securities would be to reduce the net loss per common share and be antidilutive. Therefore, the denominator used to calculate both basic and diluted net loss per common share is the same in each period presented.
The following outstanding potentially dilutive securities have been excluded from the computations of diluted net loss per common share:
| As of June 30, | ||||||||
| 2020 | 2019 | |||||||
| Options to purchase common stock | 16,516,695 | 15,139,374 | ||||||
| Warrants to purchase common stock | 541,193 | 331,193 | ||||||
| 17,057,888 | 15,470,567 | |||||||
4. Debt
Notes Payable to Vendors
On June 30, 2016, the Company issued promissory notes in an aggregate principal amount of approximately $1.2 million to certain claimants in accordance with the Company’s Plan of Reorganization (the “Plan”) filed with the United States Bankruptcy Court for the District of Delaware (the “Bankruptcy Court”) (Case No. 15-12628 (LSS) (the “Bankruptcy Case”) which became effective June 30, 2016, at which time the Company emerged from its Chapter 11 bankruptcy proceedings. The notes were unsecured, accrued interest at 10% per annum and became due and payable in full, including principal and accrued interest on June 30, 2019. In July and August, 2019, following the receipt of proceeds from the 2019 Bridge Notes, the Company used approximately $0.5 million of the proceeds to retire a portion of these notes, including accrued interest. After giving effect to these payments, the aggregate principal amount and accrued but unpaid interest on these notes approximated $1.1 million as of December 31, 2019 and the Company had accrued $0.3 million in interest expense. In June and July 2020, the Company used the proceeds from the Private Placement (as defined below) to repay the remaining outstanding principal including accrued and unpaid interest on these notes and the notes were extinguished. As of June 30, 2020, one note for $8.0 thousand, including principal and accrued but unpaid interest was still outstanding; this note was repaid in July 2020.
Advance Notes
In June, July and August, 2018 the Company received an aggregate of $0.9 million of proceeds from advances made to the Company (the “Advance Notes”) by four different lenders including Dr. Cameron Durrant, the Company’s Chairman and Chief Executive Officer; Cheval Holdings, Ltd. (“Cheval”), an affiliate of Black Horse Capital, L.P. (“BHC”), the Company’s controlling stockholder at the time; and Ronald Barliant, a director of the Company. The Advance Notes accrued interest at a rate of 7% per year, compounded annually.
| 9 |
In accordance with their terms, on May 30, 2019, in connection with the Company’s announcement of the Collaboration Agreement with Kite, the lenders converted the amounts due under the Advance Notes into the Company’s common stock at the conversion price of $0.45 per share. The Company issued a total of 2,179,622 shares of common stock in connection with the conversion.
2018 Convertible Notes
Commencing September 19, 2018, the Company delivered a series of convertible promissory notes (the “2018 Notes”) evidencing an aggregate of $2.5 million of loans made to the Company by six different lenders, including an affiliate of BHC, the Company’s controlling stockholder at the time. The 2018 Notes accrued interest at a rate of 7% per annum and, in general, were set to mature twenty-four months from the date the 2018 Notes were signed. The Company used the proceeds from the 2018 Notes for working capital.
The 2018 Notes were convertible into equity securities of the Company in three different scenarios, including if the Company sold its equity securities on or before the date of repayment of the 2018 Notes in any financing transaction that resulted in gross proceeds to the Company of less than $10 million (a “Non-Qualified Financing”). In connection with a Non-Qualified Financing, the noteholders were able to convert their remaining 2018 Notes into either (i) such equity securities as the noteholder would acquire if the principal and accrued but unpaid interest thereon (the “Conversion Amount”) were invested directly in the financing on the same terms and conditions as given to the financing investors in the Non-Qualified Financing, or (ii) common stock at a conversion price equal to $0.45 per share (subject to ratable adjustment for any stock split, stock dividend, stock combination or other recapitalization occurring subsequent to the date of the Notes).
The Company’s sales of shares pursuant to the ELOC Purchase Agreement with Lincoln Park Capital Fund, LLC (“LPC”) constituted a Non-Qualified Financing. Commencing on April 2, 2020, the holders of the 2018 Notes notified the Company of their exercise of their conversion rights under the 2018 Notes. See “2019 Convertible Notes” for additional information regarding the conversion of 2018 Notes by the holders.
As of December 31, 2019, the Company had accrued $0.2 million in interest related to these promissory notes. Interest expense recorded during the three and six months ended June 30, 2020 was approximately $564 thousand and $817 thousand, respectively.
2019 Convertible Notes
Commencing on April 23, 2019, the Company delivered a series of convertible promissory notes (the “2019 Notes” and together with the 2018 Notes, the “Convertible Notes”) evidencing an aggregate of $1.3 million of loans made to the Company. The 2019 Notes accrued interest at a rate of 7.5% per annum and, in general, were set to mature twenty-four months from the date the 2019 Notes were signed. The Company used the proceeds from the 2019 Notes for working capital.
The 2019 Notes were convertible into equity securities of the Company in four different scenarios, including if the Company sold its equity securities on or before the date of repayment of the 2019 Notes in any financing transaction that resulted in gross proceeds to the Company of less than $10 million (a “Non-Qualified Financing”). In connection with a Non-Qualified Financing, the noteholders were able to convert their remaining 2019 Notes into either (i) such equity securities as the noteholder would acquire if the Conversion Amount were invested directly in the financing on the same terms and conditions as given to the financing investors in the Non-Qualified Financing, or (ii) common stock at a conversion price equal to $1.25 per share (subject to ratable adjustment for any stock split, stock dividend, stock combination or other recapitalization occurring subsequent to the date of the 2019 Notes).
The Company’s sales of shares pursuant to the ELOC Purchase Agreement with LPC constituted a Non-Qualified Financing. Commencing on April 2, 2020, holders of the Convertible Notes, including Cheval, an affiliate of BHC, the Company’s controlling stockholder at the time, notified the Company of their exercise of their conversion rights under the Convertible Notes. Pursuant to the exemption from registration afforded by Section 3(a)(9) under the Securities Act, the Company issued an aggregate of 11,989,578 shares of its common stock upon the conversion of $4.3 million in aggregate principal and interest on the Convertible Notes that were converted, which obligations were retired. Of these, the Company issued 1,583,333 shares to Cheval. Dr. Dale Chappell, who was serving as the Company’s ex-officio chief scientific officer at the time and currently serves as its Chief Scientific Officer, controls BHC and reports beneficial ownership of all shares held by it and its affiliates, including Cheval. After giving effect to the shares issued upon such conversions, no convertible notes issued in 2018 or 2019 were outstanding as of June 30, 2020.
| 10 |
As of December 31, 2019, the Company had accrued $0.1 million in interest related to the 2019 Notes. Interest expense related to the 2019 Notes, recorded during the three and six months ended June 30, 2020, was approximately $178 thousand and $219 thousand, respectively.
The Advance Notes, the 2018 Notes and the 2019 Notes had an optional voluntary conversion feature in which the holder could convert the notes in the Company’s common stock at maturity at a conversion rate of $0.45 per share for the Advance Notes and the 2018 Notes and at a conversion rate of $1.25 for the 2019 Notes. The intrinsic value of this beneficial conversion feature was $1.8 million upon the issuance of the Advance Notes, the 2018 Notes and the 2019 Notes and was recorded as additional paid-in capital and as a debt discount which is accreted to interest expense over the term of the Advance Notes and Notes. Interest expense includes debt discount amortization of $558 thousand and $785 thousand for the three and six months ended June 30, 2020, respectively.
The Company evaluated the embedded features within the Advance Notes, the 2018 Notes and the 2019 Notes to determine if the embedded features are required to be bifurcated and recognized as derivative instruments. The Company determined that the Advance Notes, the 2018 Notes and the 2019 Notes contain contingent beneficial conversion features (“CBCF”) that allow or require the holder to convert the Advance Notes, the 2018 Notes and the 2019 Notes, as applicable, to Company common stock at a conversion rate of $0.45 per share for the Advance Notes and the 2018 Notes and $1.25 for the 2019 Notes, but did not contain embedded features requiring bifurcation and recognition as derivative instruments. Upon the occurrence of a CBCF that results in conversion of the Advance Notes, the 2018 Notes or the 2019 Notes to Company common stock, the remaining unamortized discount will be charged to interest expense. Upon conversion of the Advance Notes on May 30, 2019, the remaining unamortized discount was charged to interest expense. Upon the conversion of the Convertible Notes in April 2020, the remaining related unamortized discount was charged to interest expense.
2020 Convertible Redeemable Notes
On March 13, 2020 and March 19, 2020 (the “Issuance Dates”), the Company delivered two convertible redeemable promissory notes (the “2020 Notes”) evidencing loans with an aggregate principal amount of $518,333 made to the Company.
The 2020 Notes accrued interest at a rate of 7.0% per annum and were set to mature on March 13, 2021 and March 19, 2021, respectively. The 2020 Notes contained an original issue discount of $33,000 and $18,833, respectively. The Company used the proceeds from the 2020 Notes for working capital.
The notes could be redeemed by the Company at any time before the 270th day following issuance, at a redemption price equal to the principal and accrued but unpaid interest on the notes to the date of redemption, plus a premium that increases on day 61 and day 121 from the issuance date. Accordingly, the notes were repaid in June 2020 with proceeds from the Private Placement, and the notes were extinguished.
The Company evaluated the embedded features within the 2020 Notes and determined that the embedded features are required to be bifurcated and recognized as stand-alone derivative instruments. The variable-share settlement features within the 2020 Notes qualify as redemption features and meet the net settlement criterion for qualification as a stand-alone derivative. In determining the fair value of the bifurcated derivative, the Company evaluated the likelihood of conversion of the 2020 Notes to Company stock. As the Company believed it would have adequate funding prior to the six month anniversary of the 2020 Notes, the first conversion option for the holders of the 2020 Notes, and it had the intent to either begin making amortizing payments or to pay off the 2020 Notes in their entirety prior to that date, the fair value was determined to be $0. The original issue discount was accreted to Interest expense and the remaining balance was charged to Interest expense upon payoff.
Interest expense related to the 2020 Notes, recorded during the three and six months ended June 30, 2020, was approximately $161 thousand and $165 thousand, respectively.
Interest expense includes the original issue discount amortization of approximately $50 thousand and $52 thousand for the three and six months ended June 30, 2020, respectively.
| 11 |
Bridge Notes
On June 28, 2019, the Company issued three short-term, secured bridge notes (the “June Bridge Notes”) evidencing an aggregate of $1.7 million of loans made to the Company by three parties: Cheval , an affiliate of BHC, the Company’s controlling stockholder at the time, lent $750,000; Nomis Bay LTD, the Company’s second largest stockholder, lent $750,000; and Dr. Cameron Durrant, the Company’s Chief Executive Officer and Chairman of the Board of Directors (the “Board”), lent $200,000. The proceeds from the June Bridge Notes were used to satisfy a portion of the unsecured obligations incurred in connection with the Company’s emergence from bankruptcy in 2016 and for working capital and general corporate purposes. Of the $1.7 million in proceeds received, $950,000 was received on June 28, 2019 and was recorded as Advance notes in the Condensed Consolidated Balance Sheet as of June 30, 2019. The remaining proceeds of $750,000 were received July 1, 2019 and recorded accordingly.
The June Bridge Notes accrued interest at a rate of 7.0% per annum and after giving effect to multiple extensions, were set to mature on December 31, 2020. The June Bridge Notes could become due and payable at such earlier time as the Company raised more than $3,000,000 in a bona fide financing transaction or upon a change in control. Accordingly, the June Bridge Notes were repaid in June 2020 with proceeds from the Private Placement, and the June Bridge Notes were extinguished.
On November 12, 2019, the Company issued two short-term, secured bridge notes (the “November Bridge Notes” and together with the June Bridge Notes, the “2019 Bridge Notes”) evidencing an aggregate of $350,000 of loans made to the Company by two parties: Cheval, an affiliate of BHC, our controlling stockholder at the time, lent $250,000; and Dr. Cameron Durrant, our Chief Executive Officer and Chairman of our Board, lent $100,000. The proceeds from the November Bridge Notes were used for working capital and general corporate purposes.
The November Bridge Notes ranked on par with the June Bridge Notes, and possessed other terms and conditions substantially consistent with those notes. The November Bridge Notes accrued interest at a rate of 7.0% per annum and after giving effect to multiple extensions, were set to mature on December 31, 2020. The November Bridge Notes could become due and payable at such earlier time as the Company raised more than $3,000,000 in a bona fide financing transaction or upon a change in control. Accordingly, the November Bridge Notes were repaid in June 2020 with proceeds from the Private Placement.
In April 2020, the Company issued two short-term, secured bridge notes (the “April Bridge Notes” and together with the June Bridge Notes and the November Bridge Notes, the “Bridge Notes”) evidencing an aggregate of $350,000 of loans made to the Company: Cheval, an affiliate of BHC, the Company’s controlling stockholder at the time, loaned $100,000, and Nomis Bay, the Company’s second largest stockholder, loaned $250,000. The proceeds from the April Bridge Notes were used for working capital and general corporate purposes.
The April Bridge Notes ranked on par with the June Bridge Notes and the November Bridge Notes, and possessed other terms and conditions substantially consistent with them. The notes accrued interest at a rate of 7.0% per annum and were set to mature on December 31, 2020. The April Bridge Notes could become due and payable at such earlier time as the Company raised more than $10,000,000 in a bona fide financing transaction or upon a change in control. Accordingly, these April Bridge Notes were repaid in June 2020 with proceeds from the Private Placement, and these bridge notes were extinguished.
The Bridge Notes were secured by a lien on substantially all of the Company’s assets, which liens have been released.
Interest expense related to the Bridge Notes, recorded during the three and six months ended June 30, 2020, was approximately $30 thousand and $66 thousand, respectively.
5. Derivative Instruments
The Company had certain embedded equity conversion features within its 2020 Notes that require bifurcation and recognition as stand-alone derivatives. See Note 4 – Debt for additional information and discussion on the bifurcated derivatives.
| 12 |
6. Commitments and Contingencies
Contractual Obligations and Commitments
As of June 30, 2020, other than the retirement and conversion of certain debt described in Note 4 and the license agreements described in Note 8, there were no material changes to the Company’s contractual obligations from those set forth in the 2019 Form 10-K.
Guarantees and Indemnifications
The Company has certain agreements with service providers with which it does business that contain indemnification provisions pursuant to which the Company typically agrees to indemnify the party against certain types of third-party claims. The Company accrues for known indemnification issues when a loss is probable and can be reasonably estimated. The Company would also accrue for estimated incurred but unidentified indemnification issues based on historical activity. As the Company has not incurred any indemnification losses to date, there were no accruals for or expenses related to indemnification issues for any period presented.
7. Stockholders’ Equity
Lincoln Park Capital Purchase Agreement
On November 8, 2019, the Company entered into a purchase agreement (the “ELOC Purchase Agreement”) and a registration rights agreement with LPC, pursuant to which the Company had the right to sell to LPC up to $20,000,000 in shares of the Company’s common stock, $0.001 par value per share (the “Common Stock”), subject to certain limitations and conditions set forth in the ELOC Purchase Agreement.
Under the ELOC Purchase Agreement, the Company had the right, from time to time at its sole discretion and subject to certain conditions, to direct LPC to purchase up to 100,000 shares of Common Stock, with such amounts increasing based on certain threshold prices but not to exceed $750,000 in total proceeds on any purchase date. The purchase price of shares of Common Stock pursuant to the ELOC Purchase Agreement was based on the market prices of the Common Stock at the time of such purchases as set forth in the ELOC Purchase Agreement. Such sales of Common Stock by the Company, if any, could occur from time to time, at the Company’s option, over the 36-month period expiring in December 2022.
In connection with the signing of the ELOC Purchase Agreement on November 8, 2019, the Company issued 706,592 shares of its common stock to LPC. The issuance of the shares were recorded as debt issuance costs in Common stock and Additional paid-in capital with no net effect on Stockholders’ equity (deficit).
During the months of December 2019 and January 2020, the Company issued a total of 700,000 shares for aggregate proceeds of $0.3 million under the ELOC Purchase Agreement.
On June 2, 2020, following completion of the Private Placement described below, the Company notified LPC of its decision to terminate the ELOC Purchase Agreement. The termination of the ELOC Purchase Agreement became effective on June 3, 2020.
2020 Private Placement
On June 1, 2020, the Company entered into a securities purchase agreement (the “Purchase Agreement”) with certain accredited investors (the “Investors”) to complete a private placement of our common stock (the “Private Placement”). The closing of the Private Placement occurred on June 2, 2020 (the “Closing Date”). At the closing, the Company issued and sold 82,528,718 shares of its common stock (the “Shares”) at a purchase price of $0.87 per share, for aggregate gross proceeds of approximately $71.8 million. The Company used a portion of the proceeds to retire certain indebtedness, as further described in Note 4 - Debt. The Company expects to use the remaining proceeds from the Private Placement to fund its Phase III study of lenzilumab in COVID-19, to secure manufacturing capacity, to progress Chemistry, Manufacturing and Controls (“CMC”) work, to prepare for commercialization in the event of approval of lenzilumab for use in COVID-19 patients under an EUA or otherwise, its collaboration with Kite and other development programs and for working capital and other general corporate purposes. See Note 10 for information regarding two complaints filed against us in connection with the Private Placement.
| 13 |
On the Closing Date, the Company and the Investors also entered into a registration rights agreement (the “Registration Rights Agreement”) pursuant to which the Company agreed to prepare and file a registration statement (the “Resale Registration Statement”) for the resale of the Shares with the Securities and Exchange Commission (the “SEC”),.
Subject to certain limitations and an overall cap, the Company may be required to pay liquidated damages to the investors at a rate of 2% of the invested capital for each occurrence (and continuation for 30 consecutive days thereafter) of a breach by the Company of certain of its obligations under the Registration Rights Agreement.
The Purchase Agreement also requires that the Company use its commercially reasonable efforts to achieve a listing of the Common Stock on a national securities exchange, subject to certain limitations set forth in the Purchase Agreement. On July 6, 2020, the Company applied to have its common stock approved for listing on the Nasdaq Capital Market. The Company’s ability to obtain Nasdaq’s approval of the listing application will require the Company to satisfy a number of conditions, including the Company’s ability to meet certain listing criteria including a minimum stock price and total value of public float. Accordingly, the Company can make no assurances that the application for listing will be approved.
2012 Equity Incentive Plan
Under the Company’s 2012 Equity Incentive Plan, the Company may grant shares, stock units, stock appreciation rights, performance cash awards and/or options to employees, directors, consultants, and other service providers. For options, the per share exercise price may not be less than the fair market value of a Company common share on the date of grant. Awards generally vest and become exercisable over three to four years and expire 10 years from the date of grant. Options generally become exercisable as they vest following the date of grant.
On June 1, 2020, with a limited number of shares remaining available for grant, and with the 2012 Equity Incentive Plan set to expire in 2022, the Board determined that it was appropriate to replace the 2012 Equity Incentive Plan. On July 27, 2020, the Board unanimously approved, and recommended that our stockholders approve, the Humanigen, Inc. 2020 Omnibus Incentive Compensation Plan (the “2020 Equity Plan”), to ensure that the Board and its compensation committee will be able to make the types of awards, and covering the number of shares, as necessary to meet the Company’s compensatory needs. On July 29, 2020, the 2020 Equity Plan was approved by written consent of the holders of approximately 63% of our outstanding shares of common stock on that date. Upon the “Effective Date” of the 2020 Equity Plan (as described below), an aggregate of 35,000,000 shares of our common stock will be reserved for issuance upon grants of awards made under the plan by the Board or its compensation committee. The 2020 Equity Plan will not become effective (such time, the “Effective Date”) until such time as we file an amendment to our certificate of incorporation with the Delaware Secretary of State to increase our authorized shares of common stock (described in more detail under Note 11—Subsequent Events). The 2020 Equity Plan will remain in effect until the tenth anniversary of the Effective Date, unless terminated earlier by the Board.
A summary of stock option activity for the six months ended June 30, 2020 under all of the Company’s options plans is as follows:
| Options | Weighted Average Exercise Price | |||||||
| Outstanding at January 1, 2020 | 15,881,721 | $ | 0.95 | |||||
| Granted | 1,719,395 | 0.54 | ||||||
| Exercised | (1,084,316 | ) | 0.65 | |||||
| Cancelled (forfeited) | - | - | ||||||
| Cancelled (expired) | (105 | ) | 11.68 | |||||
| Outstanding at June 30, 2020 | 16,516,695 | $ | 0.93 | |||||
| 14 |
The weighted average fair value of options granted during the six months ended June 30, 2020 was $0.40 per share.
The Company valued the options granted using the Black-Scholes options pricing model and the following weighted-average assumption terms for the six months ended June 30, 2020:
| Six months ended June 30, 2020 | |
| Exercise price | $0.38 - $1.20 |
| Market value | $0.38 - $1.20 |
| Expected term | 5 years |
| Expected volatility | 94.6% - 100.2% |
| Risk-free interest rate | 0.34% - 1.57% |
| Expected dividend yield | - % |
Stock-Based Compensation
The Company recorded stock-based compensation expense in the Condensed Consolidated Statements of Operations as follows:
| Three months ended June 30, | Six months ended June 30, | |||||||||||||||
| 2020 | 2019 | 2020 | 2019 | |||||||||||||
| General and administrative | $ | 295 | $ | 697 | $ | 506 | $ | 1,394 | ||||||||
| Research and development | 62 | $ | 32 | 116 | 32 | |||||||||||
| Total stock-based compensation | $ | 357 | $ | 729 | $ | 622 | $ | 1,426 | ||||||||
At June 30, 2020, the Company had $1.0 million of total unrecognized stock-based compensation expense, net of estimated forfeitures, related to outstanding stock options that will be recognized over a weighted-average period of 1.4 years.
8. License and Collaboration Agreements
Kite Agreement
On May 30, 2019, the Company entered into a collaboration agreement (the “Kite Agreement”) with Kite Pharmaceuticals, Inc., pursuant to which the Company and Kite are conducting a multi-center Phase 1b/2 study of lenzilumab with Kite’s Yescarta in patients with relapsed or refractory B-cell lymphoma, including diffuse large B-cell lymphoma (“DLBCL”). The primary objective of the Study is to determine the effect of lenzilumab on the safety of Yescarta.
Pursuant to the Kite Agreement, the Company shall supply lenzilumab to the collaboration for use in the study and will contribute up to approximately $8.0 million towards the out-of-pocket costs of the study, depending on the number of patients enrolled into the study.
Mayo Agreement
On June 19, 2019 the Company entered into an exclusive worldwide license agreement (the “Mayo Agreement”) with the Mayo Foundation for Medical Education and Research (“Mayo”) for certain technologies used to create CAR-T cells lacking GM-CSF expression through various gene-editing tools including CRISPR-Cas9 (GM-CSF knock-out). The license covers various patent applications and know-how developed by Mayo in collaboration with the Company. These licensed technologies complement and broaden the Company’s position in the GM-CSF neutralization space and expand the Company’s discovery platform aimed at improving CAR-T to include gene-edited CAR-T cells.
Pursuant to the Mayo Agreement, the Company agreed to pay $200,000 to Mayo within six months of the effective date, or upon completion of a qualified financing, whichever is earlier. The Mayo Agreement also requires the payment of milestones and royalties upon the achievement of certain regulatory and commercialization milestones. The initial payment was accrued as expense in Research and development in June 2019. The Company paid the initial payment in June 2020 subsequent to the closing of the Private Placement.
| 15 |
Zurich Agreement
On July 19, 2019 the Company entered into an exclusive worldwide license agreement (the “Zurich Agreement”) with the University of Zurich (“UZH”) for technology used to prevent or treat Graft versus Host Disease (“GvHD”) through GM-CSF neutralization. The Zurich Agreement covers various patent applications filed by UZH which complement and broaden the Company’s position in the application of GM-CSF and expands the Company’s development platform to include improving allogeneic Hematopoietic Stem Cell Transplantation (“HSCT”).
Pursuant to the Zurich Agreement, the Company paid $100,000 to UZH in July 2019. The Zurich Agreement also requires the payment of milestones and royalties upon the achievement of certain regulatory and commercialization milestones. The license payment of $100,000 was recorded as expense in Research and development in July 2019.
9. Savant Arrangements
On June 30, 2016 the Company and Savant Neglected Diseases, LLC (“Savant”) entered into an Agreement for the Manufacture, Development and Commercialization of Benznidazole for Human Use (the “MDC Agreement”), pursuant to which the Company acquired certain worldwide rights relating to benznidazole (the “Compound”).
In addition, on the Effective Date the Company and Savant also entered into a Security Agreement (the “Security Agreement”), pursuant to which the Company granted Savant a continuing senior security interest in the assets and rights acquired by the Company pursuant to the MDC Agreement and certain future assets developed from those acquired assets.
On the Effective Date, the Company issued to Savant a five year warrant (the “Warrant”) to purchase 200,000 shares of the Company’s Common Stock, at an exercise price of $2.25 per share, subject to adjustment. As of June 30, 2020 the Warrant was exercisable for 100,000 shares at an exercise price of $2.25 per share. On July 20, 2020, the Company issued to Savant a total of 54,545 shares of its common stock pursuant to a “net exercise” of the Warrant by Savant. The Warrant is not, and is not expected to vest or become exercisable for any additional shares of the Company’s common stock.
As a result of the FDA granting accelerated and conditional approval of a benznidazole therapy manufactured by a competitor for the treatment of Chagas disease and awarding such competitor a neglected tropical disease PRV in August 2017, the Company ceased development of benznidazole and re-evaluated the final two vesting milestones and concluded that the probability of achievement of these milestones had decreased to 0%.
In July 2017, the Company commenced litigation against Savant alleging that Savant breached the MDC Agreement and seeking a declaratory judgement. Savant has asserted counterclaims for breaches of contract under the MDC Agreement and the Security Agreement. The dispute primarily concerns the Company’s right under the MDC Agreement to offset certain costs incurred by the Company in excess of the agreed upon budget against payments due Savant. See Note 10, below, for more information regarding the Savant litigation. The aggregate cost overages as of June 30, 2017 that the Company asserts are Savant’s responsibility total approximately $3.4 million, net of a $0.5 million deductible. The Company asserts that it is entitled to offset $2.0 million in milestone payments due Savant against the cost overages, such that as of June 30, 2017, Savant owed the Company approximately $1.4 million. As of June 30, 2019, the cost overages totaled $4.1 million such that Savant owed the Company approximately $2.1 million in cost overages. Such cost overages have been charged to Research and development expense as incurred. Recovery of such cost overages, if any, will be recorded as a reduction of Research and development expense in the period received.
The $2.0 million in milestone payments due Savant are included in Accrued expenses in the accompanying Condensed Consolidated Balance Sheets as of June 30, 2020 and December 31, 2019.
10. Litigation
Savant Litigation
On July 10, 2017, the Company filed a complaint against Savant in the Superior Court for the State of Delaware, New Castle County (the “Delaware Court”). KaloBios Pharmaceuticals, Inc. v. Savant Neglected Diseases, LLC, No. N17C-07-068 PRW-CCLD. The Company asserted breach of contract and declaratory judgment claims against Savant arising under the MDC Agreement. See Note 9 - “Savant Arrangements” for more information about the MDC Agreement. The Company alleges that Savant has breached its MDC Agreement obligations to pay cost overages that exceed a budgetary threshold as well as other related MDC Agreement representations and obligations. In the litigation, the Company has alleged that as of June 30, 2017, Savant was responsible for aggregate cost overages of approximately $3.4 million, net of a $0.5 million deductible under the MDC. The Company asserts that it is entitled to offset $2.0 million in milestone payments due Savant against the cost overages, such that as of June 30, 2017 Savant owed the Company approximately $1.4 million.
| 16 |
On July 12, 2017, Savant removed the case to the Bankruptcy Court, claiming that the action is related to or arises under the Bankruptcy Case from which we emerged in July 2016. In re KaloBios Pharmaceuticals, Inc., No. 15-12628 (LSS) (Bankr. D. Del.). On July 27, 2017, Savant filed an Answer and Counterclaims. Savant’s filing alleges breaches of contracts under the MDC Agreement and the Security Agreement, claiming that the Company breached its obligations to pay the milestone payments and other related representations and obligations. On August 1, 2017, the Company moved to remand the case back to the Delaware Court (the “Motion to Remand”).
On August 2, 2017, Savant sent a foreclosure notice to the Company, demanding that it provide the Collateral as defined in the Security Agreement for inspection and possession on August 9, 2017, with a public sale to be held on September 1, 2017. The Company moved for a Temporary Restraining Order (the “TRO”) and Preliminary Injunction in the Bankruptcy Court on August 4, 2017. Savant responded on August 7, 2017. On August 7, 2017, the Bankruptcy Court granted the Company’s motion for a TRO, entering an order prohibiting Savant from collecting on or selling the Collateral, entering our premises, issuing any default notices to us, or attempting to exercise any other remedies under the MDC Agreement or the Security Agreement. On August 9, 2017, the parties have stipulated to continue the provisions of the TRO in full force and effect until further order of the appropriate court, which the Bankruptcy Court signed that same day (the “Stipulated Order”).
On January 22, 2018, Savant wrote to the Bankruptcy Court requesting dissolution of the TRO and the Stipulated Order. On January 29, 2018, the Bankruptcy Court granted the Motion to Remand and denied Savant’s request to dissolve the TRO and Stipulated Order, ordering that any request to dissolve the TRO and Stipulated Order be made to the Delaware Court.
On February 13, 2018 Savant made a letter request to the Delaware Court to dissolve the TRO and Stipulated Order. Also on February 13, 2018, the Company filed its Answer and Affirmative defenses to Savant’s Counterclaims. On February 15, 2018 the Company filed a letter opposition to Savant’s request to dissolve the TRO and Stipulated Order and requesting a status conference. A hearing on Savant’s request to dissolve the TRO and Stipulated Order was held before the Delaware Court on March 19, 2018. The Delaware Court denied Savant’s request to dissolve the TRO and Stipulated order, which remain in effect.
On April 11, 2018, the Company advised the Delaware Court that it would meet and confer with Savant regarding a proposed case management order and date for trial. On April 26, 2018 the Delaware Court so-ordered a proposed case management order submitted by the Company and Savant. The schedule in the case management order was modified by stipulation on August 24, 2018.
On April 8, 2019, the Company moved to compel Savant to produce documents in response to the Company’s document requests. The parties thereafter agreed to a discovery schedule through June 30, 2019, which the Superior Court so-ordered, and the parties produced documents to each other.
On June 4, 2019, Savant filed a complaint against the Company and Madison in the Delaware Court of Chancery (the “Chancery Action”) seeking to “recover as damages that amounts owed to it under the MDC Agreement, and to reclaim Savant’s intellectual property,” among other things. Savant also requested leave to move to dismiss the Company’s complaint on the grounds that the Company’s transfer of assets to Madison was champertous. On June 10, 2019, the Company requested by letter that the Superior Court hold a contempt hearing because the Chancery Action violated the TRO entered by the Bankruptcy Court, the terms of which have been extended by stipulation of the parties. On June 18, 2019, the Superior Court held a telephonic status conference. The parties agreed that the Chancery Action should be consolidated with the Superior Court action, after which the Superior Court would address the parties’ motions.
On July 22, 2019, the Company moved for contempt against Savant. Savant filed its opposition on July 29, 2019. On August 12, 2019, the Superior Court denied the Company’s motion for contempt.
On July 23, 2019, Savant moved for summary judgment on the issue of champerty. The Company filed its response and cross-motion for summary judgment on August 27, 2019. Savant filed its reply on September 10, 2019 and the Company filed its cross-reply on September 20, 2019. The motion is fully briefed, and was argued at a hearing on February 3, 2020. The court has not yet ruled on the motion.
| 17 |
On July 26, 2019, the Company moved to modify the previously agreed-upon discovery schedule to extend discovery through December 31, 2019, which the Superior Court granted. In subsequent orders, the discovery schedule was further extended until the end of June 2020.
On July 30, 2019, the Company filed a motion to dismiss Savant’s Chancery Action. Savant filed an amended complaint on September 4, 2019, and the Company filed its opening brief in support of its motion to dismiss on October 11, 2019. That motion is fully briefed and was argued at a hearing on February 3, 2020. The court has not yet ruled on the motion.
On August 19, 2019, Savant moved to dismiss the Company’s amended Superior Court complaint. On September 27, 2019, the Company filed an opposition to Savant’s motion and, in the alternative, requested leave to file a second amended complaint against Savant. Savant consented to the filing of the second amended complaint and withdrew their motion to dismiss. Savant filed a partial motion to dismiss against a co-defendant on October 30, 2019. That motion is fully briefed and was argued at a hearing on February 3, 2020. At the February 3, 2020 hearing, the Court reserved judgment on the parties’ reciprocal motions.
On November 18, 2019, the Court granted Savant’s Motion to Schedule a Preliminary Injunction hearing concerning the August 2017 TRO and Stipulated Order that are still in effect. On May 8, 2020, the Court granted the application for a preliminary injunction to the Company. The Stipulated Order is no longer in effect.
On May 22, 2020, upon the request of the parties, the Superior Court stayed both Delaware actions until July 29, 2020.
On July 24, 2020, the parties submitted a joint status report in the Delaware actions. The parties also requested a status conference with the Court to discuss moving the trial from October 2020 to some later time.
Private Placement Litigation
On June 15, 2020, a complaint was filed against Humanigen and Dr. Durrant in the Commercial Division of the Supreme Court of the State of New York. The plaintiffs comprise a group of 17 prospective investors introduced to Humanigen by Noble Capital Markets, Inc. (“Noble”), which had been engaged as a non-exclusive placement agent in connection with the Private Placement. The plaintiffs had indicated interest in purchasing shares of common stock in the Private Placement but, due to the strength of demand for shares from other prospective investors introduced to the company by J.P. Morgan Securities LLC, the lead placement agent for the Private Placement, the plaintiffs were not allocated any investment amount. The plaintiffs allege that the company and Dr. Durrant breached a contractual obligation to deliver shares of common stock to the plaintiffs. The plaintiffs seek to recover for losses due to alleged fraudulent misstatements and the company’s failure to deliver shares to them, and seek equitable relief in the form of specific performance.
On June 19, 2020, Noble filed a separate complaint against Humanigen in the Circuit Court of the Fifteenth Judicial Circuit in and for Palm Beach County, Florida, also arising from the Private Placement. Noble’s complaint alleges that Humanigen breached the terms of its engagement letters with Noble by refusing to pay it the sales commissions it would have earned had its prospective investors received the entire allocation of shares sought in the Private Placement, as opposed to the $4 million of shares actually allocated to Noble and its clients. Noble is seeking payment in full of the commission, damages for Humanigen’s alleged tortious interference with Noble’s business relationship with the investors it introduced to Humanigen but which were not allocated shares in the Private Placement, and attorneys’ fees.
The Company believes that the claims made in each complaint are without merit, and it is prepared to defend itself vigorously.
11. Subsequent Events
Appointment of Mr. Tousley as Chief Accounting and Administrative Officer, Corporate Secretary and Treasurer
On July 6, 2020, the Company entered into an employment agreement with David L. Tousley in connection with his appointment as the Company’s Chief Accounting and Administrative Officer, Corporate Secretary and Treasurer (the “Tousley Agreement”). The Tousley Agreement provides for an initial annual base salary for Mr. Tousley of $375,000 as well as eligibility for an annual bonus targeted at 40% of his base salary. In connection with his appointment, Mr. Tousley will be entitled to receive stock options to purchase 313,500 shares of the Company’s common stock within three business days following the Effective Date of the 2020 Equity Plan. Mr. Tousley is entitled to participate in the Company’s benefit plans available to other executives.
| 18 |
Under the Tousley Agreement, Mr. Tousley is entitled to receive certain benefits upon termination of employment under certain circumstances. If the Company terminates Mr. Tousley’s employment for any reason other than “Cause”, or if Mr. Tousley resigns for “Good Reason” (each as such term is defined in the Agreement), Mr. Tousley will receive twelve months of base salary then in effect and the amount of the actual bonus earned by Mr. Tousley under the Tousley Agreement for the year prior to the year of termination, pro-rated based on the portion of the year Mr. Tousley was employed by the Company during the year of termination, or if no bonus had been received, at minimum 50% of his target bonus.
The Tousley Agreement additionally provides that if Mr. Tousley resigns for Good Reason or the Company or its successor terminates his employment within the three month period prior to and the two year period following a change in control (as such term is defined in the Tousley Agreement), the Company must pay or cause its successor to pay Mr. Tousley a lump sum cash payment equal to one times (a) his annual salary as of the day before his resignation or termination plus (b) the aggregate bonus received by Mr. Tousley for the year immediately preceding the change in control or, if no bonus had been received, at minimum 50% of the target bonus. In addition, upon such a resignation or termination, all outstanding stock options held by Mr. Tousley will immediately vest and become exercisable.
Appointment of Dr. Chappell as Chief Scientific Officer
On July 6, 2020, the Company entered into an employment agreement with Dale Chappell in connection with his appointment as the Company’s Chief Scientific Officer (the “Chappell Agreement”). The Chappell Agreement provides for an initial annual base salary for Dr. Chappell of $410,000 as well as eligibility for an annual bonus targeted at 40% of his base salary. In connection with his appointment, Dr. Chappell will be entitled to receive stock options to purchase 668,800 shares of the Company’s common stock within three business days following the Effective Date of the 2020 Equity Plan. Dr. Chappell is entitled to participate in the Company’s benefit plans available to other executives.
Under the Chappell Agreement, Dr. Chappell is entitled to receive certain benefits upon termination of employment under certain circumstances. If the Company terminates Dr. Chappell employment for any reason other than “Cause”, or if Dr. Chappell resigns for “Good Reason” (each as such term is defined in the Chappell Agreement), Dr. Chappell will receive twelve months of base salary then in effect and the amount of the actual bonus earned by Dr. Chappell under the Chappell Agreement for the year prior to the year of termination, pro-rated based on the portion of the year Dr. Chappell was employed by the Company during the year of termination, or if no bonus had been received, at minimum 50% of his target bonus.
The Chappell Agreement additionally provides that if Dr. Chappell resigns for Good Reason or the Company or its successor terminates his employment within the three month period prior to and the two year period following a change in control (as such term is defined in the Chappell Agreement), the Company must pay or cause its successor to pay Dr. Chappell a lump sum cash payment equal to one times (a) his annual salary as of the day before his resignation or termination plus (b) the aggregate bonus received by Dr. Chappell for the year immediately preceding the change in control or, if no bonus had been received, at minimum 50% of the target bonus. In addition, upon such a resignation or termination, all outstanding stock options held by Dr. Chappell will immediately vest and become exercisable.
Appointment of Mr. Morris as Chief Financial Officer and Chief Operating Officer
On August 3, 2020, the Company announced the appointment of Timothy Morris as the Company’s Chief Operating Officer and Chief Financial Officer. In connection with his appointment as COO and CFO, Mr. Morris stepped down as a member of the Board, a position he had held since June 2016, and the size of the Board was reduced to five members.
The Company entered into an employment agreement with Mr. Morris in connection with his appointment (the “Morris Agreement”). The Morris Agreement provides for an initial annual base salary for Mr. Morris of $475,000 as well as eligibility for an annual bonus targeted at 50% of his base salary. In connection with his appointment, Mr. Morris will be entitled to receive stock options to purchase 756,580 shares of the Company’s common stock within three business days following the effective date of the Humanigen, Inc. 2020 Omnibus Incentive Compensation Plan. Mr. Morris is entitled to participate in the Company’s benefit plans available to other executives.
| 19 |
Under the Morris Agreement, Mr. Morris is entitled to receive certain benefits upon termination of employment under certain circumstances. If the Company terminates Mr. Morris’s employment for any reason other than “Cause”, or if Mr. Morris resigns for “Good Reason” (each as such term is defined in the Morris Agreement), Mr. Morris will receive his annual salary and the amount of the actual bonus earned by Mr. Morris under the Agreement for the year prior to the year of termination, pro-rated based on the portion of the year Mr. Morris was employed by the Company during the year of termination, or if no bonus had been received, 50% of his target bonus. In addition, upon such a resignation or termination, Mr. Morris will also be entitled to be reimbursed for certain monthly health plan continuation premiums for up to 12 months, and all outstanding stock options held by Mr. Morris will immediately vest and become exercisable.
The Morris Agreement additionally provides that if Mr. Morris resigns for Good Reason or the Company terminates his employment other than for Cause within the three month period prior to or the two year period following a change in control (as such term is defined in the Morris Agreement), the Company must pay or cause its successor to pay Mr. Morris a lump sum cash payment equal to one and one-half times (a) his annual salary plus (b) the aggregate bonus received by Mr. Morris for the year immediately preceding the change in control or, if no bonus had been received, 50% of the target bonus. In addition, upon such a resignation or termination, Mr. Morris will also be entitled to be reimbursed for certain monthly health plan continuation premiums for up to 18 months, and all outstanding stock options held by Mr. Morris will immediately vest and become exercisable.
Clinical Trial Agreement with the National Institute of Allergy and Infectious Diseases
On July 24, 2020, the Company entered into a clinical trial agreement (the “Clinical Trial Agreement”) with the National Institute of Allergy and Infectious Diseases (“NIAID”), part of the National Institutes of Health, which is part of the United States Government Department of Health and Human Services, as represented by the Division of Microbiology and Infectious Diseases. Pursuant to the Clinical Trial Agreement, lenzilumab will be an agent to be evaluated in the NIAID-sponsored Big Effect Trial (“BET”) in hospitalized patients with COVID-19.
BET will evaluate the combination of lenzilumab and Gilead’s investigational antiviral, remdesivir, on treatment outcomes versus placebo and remdesivir in hospitalized COVID-19 patients. The trial is expected to enroll 100 patients in each arm of the study with an interim analysis for efficacy after 50 patients have been enrolled in each arm.
Pursuant to the Clinical Trial Agreement, NIAID will serve as sponsor and will be responsible for supervising and overseeing BET. The Company will be responsible for providing lenzilumab to NIAID without charge and in quantities to ensure a sufficient supply of lenzilumab. The Clinical Trial Agreement imposes additional obligations on the Company that are reasonable and customary for clinical trial agreements of this nature, including in respect of compliance with data privacy laws and potential indemnification obligations.
Stockholder Action by Written Consent
On July 27, 2020, the Board unanimously approved and recommended, and on July 29, 2020, certain stockholders of the Company (the “Consenting Stockholders”) owning as of July 29, 2020 (the “Record Date”) approximately 63% of the Company's outstanding common stock, par value $0.001 per share (“common stock”), approved the following actions (each, an “Action” and collectively, the “Actions”) by written consent in lieu of a special meeting, in accordance with the applicable provisions of the Delaware General Corporation Law, the Company’s Amended and Restated Certificate of Incorporation, as amended (the “Charter”), and the Company’s Second Amended and Restated Bylaws:
| 1. | The approval of an amendment to Article IV of the Charter to increase the number of authorized shares of common stock from 225,000,000 to 750,000,000; |
| 2. | The approval of an amendment to Article IV of the Charter that will give the Board the discretion, until July 29, 2021, to effect a reverse stock split whereby each outstanding 2, 3, 4, 5, 6, 7, 8, 9 or 10 shares of our common stock may be combined, converted and changed into one share of common stock, with the final ratio (if any) as may be determined by and subject to final approval of the Board; and |
| 3. | The approval of the 2020 Equity Plan. |
The Company will prepare and cause to be sent or delivered to its stockholders of record as of the Record Date pursuant to Regulation 14C under the Securities Exchange Act of 1934 an information statement relating to the Actions (the “Information Statement”). In accordance with the rules and regulations of the Securities and Exchange Commission, the Actions will not become effective until at least 20 calendar days after we send the Information Statement to such stockholders. Furthermore, the Board retains sole discretion to implement or abandon a reverse stock split, based on its determination of whether effecting a reverse stock split is advisable and in the best interests of the Company and its stockholders. Therefore, a reverse stock split may not occur without further stockholder action, notwithstanding the approval provided by the Consenting Stockholders.
| 20 |
| Item 2. | Management’s Discussion and Analysis of Financial Condition and Results of Operations. |
You should read the following discussion and analysis together with our financial statements and the notes to those statements included elsewhere in this Quarterly Report on Form 10-Q and our 2019 Form 10-K for the fiscal year ended December 31, 2019. This Quarterly Report on Form 10-Q contains statements that discuss future events or expectations, projections of results of operations or financial condition, trends in our business, business prospects and strategies and other “forward-looking” information. In some cases, you can identify “forward-looking statements” by words like “may,” “will,” “should,” “expects,” “plans,” “anticipates,” “believes,” “estimates,” “predicts,” “intends,” “potential” or “continue” or the negative of those words and other comparable words. These statements may relate to, among other things, our expectations regarding the scope, progress, expansion, and costs of researching, developing and commercializing our product candidates; our opportunity to benefit from various regulatory incentives; expectations for our financial results, revenue, operating expenses and other financial measures in future periods; and the adequacy of our sources of liquidity to satisfy our working capital needs, capital expenditures, and other liquidity requirements. Actual events or results may differ materially due to known and unknown risks, uncertainties and other factors such as:
| • | the evolution of scientific discovery around the coronavirus, COVID-19 and the lung dysfunction resulting in some patients may indicate that cytokine storm is caused by or results from something other than elevated GM-CSF levels; |
| • | our ability to successfully complete our Phase III trial of lenzilumab for the prevention and treatment of cytokine storm in COVID-19 pneumonia; |
| • | the ultimate impact of the COVID-19 pandemic, or any other health epidemic, on our business, our clinical trials, our research programs, healthcare systems or the global economy as a whole; |
| • | our ability to research, develop and commercialize our product candidates, including our ability to do so before our competitors develop and receive FDA approval for treatments or vaccines for COVID-19; |
| • | our ability to execute our strategy and business plan focused on developing our proprietary monoclonal antibody portfolio and our GM-CSF knockout gene-editing CAR-T platform; |
| • | our ability to attract and retain other collaborators with development, regulatory and commercialization expertise to pursue the other initiatives in our development pipeline; |
| • | our ability to successfully pursue the Kite collaboration; |
| • | our ability to attain the additional financing we will need to pursue our development initiatives and commercialize our product candidates on favorable terms or at all; |
| • | our ability to successfully list our common stock and maintain the listing of our common stock on the Nasdaq Capital Market; |
| • | the timing of the initiation, enrollment and completion of planned clinical trials; |
| • | our ability to timely source adequate supply of our development products from third-party manufacturers on which we depend; |
| • | the potential, if any, for future development of any of our present or future products; |
| • | increasing levels of market acceptance of CAR-T therapies and the development of a market for lenzilumab in these therapies; |
| • | our ability to successfully progress, partner or complete further development of our programs; |
| • | the potential timing and outcomes of development, preclinical and clinical studies of lenzilumab, ifabotuzumab, HGEN005, any of our CAR-T projects and the uncertainties inherent in development, preclinical and clinical testing; |
| • | our ability to identify and develop additional uses for our products; |
| • | our ability to attain market exclusivity and/or to protect our intellectual property and to operate our business without infringing on the intellectual property rights of others; |
| • | the outcome of pending, threatened or future litigation; |
| • | acquisitions or in-licensing transactions that we may pursue may fail to perform as expected; |
| • | our ability to obtain and maintain regulatory approval of our product candidates, and any related restrictions; |
| • | limitations and/or warnings in the label of an approved product candidate; |
| • | changes in the regulatory landscape that may prevent us from pursuing or realizing any of the expected benefits from the various regulatory incentives, or the imposition of regulations that affect our products; |
| • | the success, progress, timing and costs of our efforts to evaluate or consummate various strategic alternatives if in the best interests of our stockholders; and |
| • | the accuracy of our estimates regarding expenses, future revenues, capital requirements and needs for additional financing. |
| 21 |
These are only some of the factors that may affect the forward-looking statements contained in this Form 10-Q. For a discussion identifying additional important factors that could cause actual results to vary materially from those anticipated in the forward-looking statements, see “Risk Factors” in Item 1A of Part I of our 2019 Form 10-K. You should review these risk factors for a more complete understanding of the risks associated with an investment in our securities. However, we operate in a competitive and rapidly changing environment and new risks and uncertainties emerge, are identified or become apparent from time to time. It is not possible for us to predict all risks and uncertainties that could have an impact on the forward-looking statements contained in this Form 10-Q. You should be aware that the forward-looking statements contained in this Form 10-Q are based on our current views and assumptions. We undertake no obligation to revise or update any forward-looking statements made in this Form 10-Q to reflect events or circumstances after the date hereof or to reflect new information or the occurrence of unanticipated events, except as required by law.
Overview
We were incorporated on March 15, 2000 in California and reincorporated as a Delaware corporation in September 2001 under the name KaloBios Pharmaceuticals, Inc. Effective August 7, 2017, we changed our legal name to Humanigen, Inc. During February 2018, we completed the financial restructuring transactions announced in December 2017 and continued our transformation into a clinical-stage biopharmaceutical company by further developing our clinical stage immuno-oncology and immunology portfolio of monoclonal antibodies.
We are a clinical stage biopharmaceutical company, developing our clinical stage COVID-19 immunology and immuno-oncology portfolio of monoclonal antibodies. We are focusing our efforts on the development of our lead product candidate, lenzilumab, our proprietary Humaneered® (“Humaneered” or “Humaneered®”) anti-human granulocyte-macrophage colony-stimulating factor (“GM-CSF”) immunotherapy.
Our proprietary and patented Humaneered technology platform is a method for converting existing antibodies (typically murine) into engineered, high-affinity human antibodies designed for therapeutic use, particularly for chronic conditions. We have developed or in-licensed targets or research (mouse) antibodies, typically from academic institutions, and then applied our Humaneered technology to them. Lenzilumab and our other two product candidates, ifabotuzumab and HGEN005, are Humaneered antibodies. Our Humaneered antibodies are closer to human antibodies than chimeric or conventionally humanized antibodies and have a high affinity for their target but low immunogenicity. Specifically, our Humaneered technology generates an antibody from an existing antibody with the required specificity as a starting point and, we believe, provides the following additional advantages:
| · | high potency; |
| · | slow off-rate; |
| · | high solubility; |
| · | retention of identical target epitope specificity and generation of higher affinity antibodies; |
| · | high antibody expression yields; |
| · | attractive cost-of-goods; |
| · | physiochemical properties that facilitate process development and formulation; |
| · | lack of aggregation at high concentration; |
| · | very-near-to-human germ line sequence (which means they are less likely to induce an inappropriate immune response when used chronically, which has proven to be the case in clinical studies); and |
| · | an optimized antibody processing time of three to six months. |
Lenzilumab is a monoclonal antibody that has been proven in animal models to neutralize GM-CSF, a cytokine that we believe is of critical importance in the inflammatory cascade, sometimes referred to as cytokine release syndrome (“CRS”) or cytokine storm, associated with COVID-19, chimeric antigen receptor T-cell (“CAR-T”) therapy and acute Graft versus Host Disease (“GvHD”) related side-effects. Lenzilumab binds to and neutralizes soluble, circulating GM-CSF.
Published scientific evidence links GM-CSF expression to serious and potentially life-threatening outcomes in respiratory conditions such as COVID-19 pneumonia. Evidence also indicates a potential causal role of GM-CSF expression in serious and potentially life-threatening side-effects associated with CAR-T therapy, and reduced efficacy in CAR-T therapies approved by the U.S. Food and Drug Administration (“FDA”). As a result, while we believe our leadership position in GM-CSF pathway science and cytokine storm presents us with a diverse set of development opportunities, we currently are focused on developing lenzilumab for three primary indications:
| · | As a therapy targeting severe outcomes in hospitalized patients with confirmed COVID-19 pneumonia; |
| 22 |
| · | As a sequenced therapy ahead of CAR-T administration in CD19 targeted CAR-T therapies; and |
| · | As an early treatment or potential prophylaxis for acute GvHD in high and intermediate risk patients. |
Our Development Program for Lenzilumab
COVID-19
We are currently enrolling patients in a Phase III multi-center, randomized, placebo-controlled, double-blinded, clinical trial in the setting of COVID-19. There are currently seventeen clinical sites across the U.S. actively recruiting patients. The Phase III trial will assess the safety and efficacy of lenzilumab in reducing severe outcomes in hospitalized adult patients with confirmed severe or critical COVID-19 pneumonia. On May 6, 2020, we announced that the first patient had been randomized in the Phase III lenzilumab COVID-19 study.
As of August 10, 2020, our Phase III study, which anticipates enrollment of a total of 300 patients, was more than 50% enrolled. We are anticipating that additional study sites will begin enrolling patients in the near term and are targeting completion of patient enrollment in the Phase III study in the third quarter of 2020.
To that end, on August 10, 2020, we announced that the Brazilian regulatory agency, Anvisa, has granted permission to commence a Phase III study of lenzilumab in patients with COVID-19 in Brazil. This study, now set to begin recruiting patients in Brazil, is expected to follow the same protocol approved by the U.S. Food and Drug Administration (FDA) in April – a multicenter, randomized, placebo-controlled, double-blinded clinical trial focused on hospitalized severe and critical adult COVID-19 patients at high risk of disease progression. We anticipate rapid enrollment in the trial in Brazil given that Brazil currently has the second highest reported rates of COVID-19 infection in the world, second only to the United States.
Based on the anticipated pace of patient enrollment, we currently estimate a primary completion date for the Phase III study to occur late in the third quarter or early in the fourth quarter of 2020. If results of the study are favorable, we would intend to file a biologics license application (BLA) for lenzilumab with FDA and, if the data submitted in connection with the BLA merit it, we might be in position to receive an emergency use authorization (EUA) from FDA and initially launch commercial activities relating to lenzilumab as early as the fourth quarter of 2020. We expect that an expanded product launch and commercialization would occur in the six months following receipt of an EUA, after which we would commence life cycle management activities for lenzilumab, which may include additional dose formulations and further international studies.
There are currently no products approved by the FDA for the prevention of CRS/cytokine storm associated with COVID-19. There are numerous products currently in development for COVID-19 which can be broadly categorized as direct-acting antivirals, immunomodulators, and other preventative strategies such as vaccines. Recently, remdesivir (a direct-acting antiviral) has been given emergency use authorization by the FDA for COVID-19 based on results from the NIAID ACTT-1 trial. In this trial, remdesivir demonstrated improvement in the primary endpoint of time to recovery reducing this measurement by four days (11 days in the remdesivir cohort vs. 15 days in the placebo cohort). There was not a statistically significant difference in mortality between the remdesivir treated cohort and the placebo cohort. Other direct acting antiviral agents such as lopinavir/ritonavir and hydroxychloroquine (with or without a macrolide) have not demonstrated efficacy in randomized controlled trials to date. In addition, no immunomodulator therapy has proven efficacy in a randomized controlled clinical trial in the setting of COVID-19 and the two leading IL-6 inhibitors, Actemra (tocilizumab) and Kevzara (sarilumab) both recently failed to demonstrate efficacy in randomized, placebo-controlled studies in COVID-19 patients.
We believe that, as an upstream regulator of cytokine storm, GM-CSF neutralization with lenzilumab may offer advantages over other immunomodulator strategies that either target other downstream cytokines such as IL-1, IL-6, CCR5 or MIP-1 alpha or are broadly immunosuppressive and target cytokine signaling pathways non-selectively through JAK inhibition. In addition, lenzilumab is the only immunomodulator that was in an active clinical trial in another indication to prevent cytokine storm prior to embarking upon the Phase III COVID-19 trial and is currently the only agent in an active Phase III trial targeting GM-CSF.
Lenzilumab was granted emergency single use Investigational New Drug Application (“IND”) authorization from the FDA (often referred to as compassionate use) to treat patients with COVID-19. On June 15, 2020, we announced that Mayo Clinic published data derived from the compassionate use of lenzilumab in treatment of 12 patients hospitalized in the Mayo Clinic system. Under applicable FDA rules, a patient cannot receive a compassionate use drug unless FDA has issued an individual patient emergency IND authorization, which the Mayo Clinic requested from FDA prior to each individual patient dosing of lenzilumab. Accordingly, there was no randomized control group in the Mayo Clinic program. We did not pre-select patients to receive lenzilumab through the compassionate use program and did not deny any requests for compassionate use. Mayo Clinic clinicians solely determined which patients for which they would request emergency IND authorization from the FDA.
The patients receiving lenzilumab had severe or critical pneumonia as a result of COVID-19. They were also viewed as being at high risk of further disease progression. All patients required oxygen supplementation and had elevation in at least one inflammatory biomarker prior to receiving lenzilumab. All patients had at least one co-morbidity associated with poor outcomes in COVID-19 and several patients had multiple co-morbidities: 58% had diabetes mellitus, 58% had hypertension, 58% had underlying lung diseases, 50% were obese (defined as a BMI greater than 30), 17% had chronic kidney disease and 17% had coronary artery disease. The median age was 65 years.
Patients receiving lenzilumab showed rapid clinical improvement with a median time to recovery of five days, median time to discharge of five days and 100% survival to the data cut-off date. Patients also demonstrated rapid improvement in oxygenation, temperature, and inflammatory cytokines consistent with the improved clinical outcomes. At the cut-off date, 11 of the 12 patients had been discharged.
| 23 |
On July 27, 2020, we announced that the National Institute of Allergy and Infectious Diseases (“NIAID”), a part of the National Institutes of Health (“NIH”), which is part of the United States Government Department of Health and Human Services (“HHS”) as represented by the Division of Microbiology and Infectious Diseases (“DMID”), and Humanigen have executed a clinical trial agreement for lenzilumab as an agent to be evaluated in the NIAID-sponsored Big Effect Trial (“BET”) in hospitalized patients with COVID-19.
BET will help advance NIAID’s strategic plan for COVID-19 research, which includes conducting studies to advance high-priority therapeutic candidates. Identification of agents with novel mechanisms of action for therapy is a strategic priority.
This trial builds on initial data from NIAID’s Adaptive COVID-19 Treatment Trial (ACTT) that demonstrated Gilead’s investigational antiviral, remdesivir, may improve time to recovery in hospitalized patients with COVID-19. BET will evaluate the combination of lenzilumab and remdesivir on treatment outcomes versus placebo and remdesivir in hospitalized COVID-19 patients. The trial is expected to enroll 100 patients in each arm of the study with an interim analysis for efficacy after 50 patients have been enrolled in each arm.
With data from the BET and our ongoing Phase III study, we expect to have data from approximately 500 hospitalized COVID-19 patients.
CAR-T Therapies
Our current clinical and regulatory development plan in the CAR-T setting is focused on a collaboration agreement we executed with Kite Pharmaceuticals, Inc., a Gilead company (“Kite”), in May 2019, which we refer to as the Kite Agreement. Pursuant to the Kite Agreement, the parties have agreed to conduct and are currently enrolling patients for a multi-center Phase 1b/2 study (“ZUMA-19”) of lenzilumab with Kite’s YESCARTA in patients with relapsed or refractory B-cell lymphoma, including diffuse large B-cell lymphoma (“DLBCL”). Kite is the sponsor of ZUMA-19 and is responsible for its conduct. The primary objective of ZUMA-19 is to determine the effect of lenzilumab on the safety of YESCARTA. In addition, efficacy and healthcare resource utilization will be assessed. On June 30, 2020, we announced that the first patient had been infused in the ZUMA-19 study. Phase Ib results are anticipated in the fourth quarter of 2020, with a Phase II interim analysis currently expected in the first half of 2021 and six-month efficacy data anticipated in the second half of 2021.
Kite’s YESCARTA is one of two CAR-T therapies that have been approved by FDA and is the leading CAR-T by revenue. We believe our collaboration with Kite is the only current clinical collaboration that is enrolling patients with the potential to improve both the safety and efficacy of CAR-T therapy. The Kite Agreement is non-exclusive. Depending upon FDA feedback, we believe ZUMA-19 may serve as the basis for registration for lenzilumab in the CAR-T setting.
GvHD
We are collaborating with IMPACT, a clinical trial partnership of 23 transplant centers in the United Kingdom, in planning a potential randomized, placebo controlled, Phase II/III study focused on early intervention with lenzilumab in patients at high risk or intermediate risk for steroid refractory acute GvHD based on specific biomarkers. The goal of the trial, as it is currently contemplated, would be to determine the efficacy and safety of lenzilumab in reducing non-relapse mortality at six months. We currently anticipate completing planning for the study in the fourth quarter of 2020, with commencement of the study in the United Kingdom occurring in the first half of 2021, and planning for the comparable study in the United States occurring in the second half of 2021.
Lenzilumab in refractory chronic myelomonocytic leukemia (CMML)
Working with partners in Australia, we are planning a Phase 2 study of lenzilumab in combination with azacitidine in newly-diagnosed CMML patients who express NRAS/KRAS/CBL mutations which are known to be hypersensitive to GM-CSF and therefore may lend themselves to responsiveness to lenzilumab treatment. CMML is a rare form of hematologic cancer with no FDA-approved treatment options and a three-year overall survival rate of 20% and median overall survival of 20 months. We currently expect to complete the planning for this study in the fourth quarter of 2020, with a goal of commencing the study in the first half of 2021.
Ifabotuzumab in solid tumors, hematologic malignancies and serious pulmonary conditions
Our clinical-stage pipeline also comprises a further Phase I study which is almost fully enrolled with ifabotuzumab in glioblastoma multiforme (GBM). We currently expect to complete the Phase I study in the fourth quarter of 2020, with results expected to be available in the first half of 2021.
Our Pipeline
In addition to these programs with lenzilumab, we are also exploring the effectiveness of our GM-CSF neutralization technologies (either through the use of lenzilumab as a neutralizing antibody, or through GM-CSF gene knockout) in combination with other CAR-T, T-cell engaging, and immunotherapy treatments to break the efficacy/toxicity linkage including the prevention and/or treatment of GvHD while preserving graft-versus-leukemia (“GvL”) benefits in patients undergoing allogeneic hematopoietic stem cell therapy (“HSCT”). In this context, GvHD is akin to cytokine storm and we believe this to be a similar mechanism and driven by elevated GM-CSF levels.
We believe that we have built a strong intellectual property position in the area of GM-CSF neutralization through multiple approaches and mechanisms, as they pertain to COVID-19, CAR-T, GvHD and multiple other oncology/transplantation, inflammation, fibrosis and autoimmune conditions which may be driven by GM-CSF.
| 24 |
Our clinical-stage pipeline also comprises a further Phase I study which is almost fully enrolled with ifabotuzumab in glioblastoma multiforme (“GBM”) and potentially other solid cancers and an additional Phase II study in CMML. We also have a focus on creating safer and more effective CAR-T therapies in hematologic malignancies and solid tumors via three key modalities:
| · | Combining FDA-approved and development stage CAR-T therapies with lenzilumab; |
| · | Creating next-generation gene-edited CAR-T therapies using GM-CSF gene knockout technologies; and |
| · | Exploring the effectiveness of our GM-CSF neutralization technologies (either through the use of lenzilumab as a neutralizing antibody or through GM-CSF gene knockout) in combination with other CAR-T, T-cell engaging, and immunotherapy treatments, including allogeneic HSCT. |
These product candidates are in the early stage of development and will require substantial time, resources, research and development, and regulatory approval prior to commercialization. Furthermore, none of these product candidates have been approved for marketing and it may be years, if this occurs at all. Our current pipeline is depicted below:
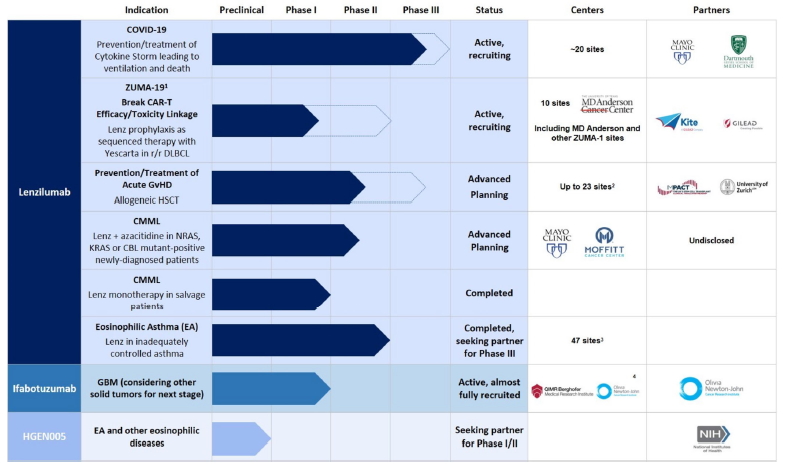
1 Phase III may not be necessary for approval in ZUMA-19; precedent is CAR-Ts to date have been approved on Phase II data
2 UK
3 US, EU, Australia
4 Australia
We will need additional capital to continue as a going concern and to support our business efforts, including obtaining regulatory approvals for our product candidates, product manufacturing and Chemistry, Manufacturing and Controls (“CMC”) work, clinical trials and other studies, and, if approved, the commercialization of our product candidates. We anticipate that we will seek additional financing from a number of sources, including, but not limited to, the sale of equity or debt securities, strategic collaborations, and licensing of our product candidates. Additional funding may not be available to us on a timely basis or at acceptable terms, if at all. Our ability to access capital when needed is not assured and, if not achieved on a timely basis, would materially harm our business, financial condition and results of operations. If adequate funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our development programs. We may also be required to sell or license to others our technologies, product candidates, or development programs that we would have preferred to develop and commercialize ourselves and on less than favorable terms, if at all. If in the best interests of our stockholders, we may also find it appropriate to enter into a strategic transaction that could result in, among other things, a sale, merger, acquisition, consolidation or business combination.
| 25 |
If management is unsuccessful in efforts to raise additional capital, based on our current levels of operating expenses, our current capital is not expected to be sufficient to fund our operations for the next twelve months. These conditions raise substantial doubt about our ability to continue as a going concern.
The condensed consolidated financial statements for the three and six months ended June 30, 2020 were prepared on the basis of a going concern, which contemplates that we will be able to realize our assets and discharge liabilities in the normal course of business. Our ability to meet our liabilities and to continue as a going concern is dependent upon the availability of future funding. The financial statements do not include any adjustments that might be necessary if we are unable to continue as a going concern.
Critical Accounting Policies and Use of Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our Condensed Consolidated Financial Statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or GAAP. The preparation of our financial statements in conformity with GAAP requires our management to make estimates and assumptions that affect the amounts and disclosures reported in the financial statements and accompanying notes. Actual results could differ materially from those estimates. Our management believes judgment is involved in determining revenue recognition, valuation of financing derivative, the fair value-based measurement of stock-based compensation, accruals and warrant valuations. Our management evaluates estimates and assumptions as facts and circumstances dictate. As future events and their effects cannot be determined with precision, actual results could differ from these estimates and assumptions, and those differences could be material to the Condensed Consolidated Financial Statements. If our assumptions change, we may need to revise our estimates, or take other corrective actions, either of which may also have a material adverse effect on our statements of operations, liquidity and financial condition.
Until December 31, 2018, we qualified as an emerging growth company (“EGC”) under the JOBS Act. Emerging growth companies can delay adopting new or revised accounting standards until such time as those standards apply to private companies. We elected to avail ourselves of this exemption from new or revised accounting standards and, therefore, we may not be subject to the same new or revised accounting standards as other smaller reporting companies that are not emerging growth companies.
We ceased to be considered as an EGC as of December 31, 2018. Accordingly, we are required to adopt new accounting standards on the same timeline as other smaller reporting companies.
There were no significant and material changes in our critical accounting policies and use of estimates during the three months ended June 30, 2020, as compared to those disclosed in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Critical Accounting Policies and Use of Estimates” in our 2019 Form 10-K, filed with the SEC on March 16, 2020.
Results of Operations
General
We have not generated net income from operations for any periods presented. At June 30, 2020, we had an accumulated deficit of $311.4 million primarily as a result of research and development and general and administrative expenses. While we may in the future generate revenue from a variety of sources, including license fees, milestone payments, and research and development payments in connection with strategic partnerships, our product candidates may never be successfully developed or commercialized and we may therefore never realize revenue from any product sales, particularly because most of our product candidates are at an early stage of development. Accordingly, we expect to continue to incur substantial losses from operations for the foreseeable future, and there can be no assurance that we will ever generate significant revenue or profits.
| 26 |
Research and Development Expenses
Conducting research and development is central to our business model. We expense both internal and external research and development costs as incurred. We track external research and development costs incurred by project for each of our clinical programs. Our external research and development costs consist primarily of:
| · | expenses incurred under agreements with contract research organizations, investigative sites, and consultants that conduct our clinical trials and a substantial portion of our pre-clinical activities; |
| · | the cost of acquiring and manufacturing clinical trial and other materials; and |
| · | other costs associated with development activities, including additional studies. |
Other research and development costs consist primarily of internal research and development costs such as salaries and related fringe benefit costs for our employees (such as workers compensation and health insurance premiums), stock-based compensation charges, travel costs, lab supplies, overhead expenses such as rent and utilities, and external costs not allocated to one of our clinical programs. Internal research and development costs generally benefit multiple projects and are not separately tracked per project.
The following table shows our total research and development expenses for the three and six months ended June 30, 2020 and 2019:
| Three Months Ended June 30, | Six Months Ended June 30, | |||||||||||||||
| (in thousands) | 2020 | 2019 | 2020 | 2019 | ||||||||||||
| External Costs | ||||||||||||||||
| Lenzilumab | $ | 20,869 | $ | 1,068 | $ | 21,352 | $ | 1,279 | ||||||||
| Ifabotuzumab | 25 | 29 | 50 | 54 | ||||||||||||
| Internal costs | 249 | 137 | 400 | 260 | ||||||||||||
| Total research and development | $ | 21,143 | $ | 1,234 | $ | 21,802 | $ | 1,593 | ||||||||
General and Administrative Expenses
General and administrative expenses consist principally of personnel-related costs, professional fees for legal, consulting, audit and tax services, rent and other general operating expenses not otherwise included in research and development.
Comparison of Three Months Ended June 30, 2020 and 2019
| Three Months Ended June 30, | Increase/ (Decrease) | |||||||||||||||
| (in thousands) | 2020 | 2019 | Amount | % | ||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 21,143 | $ | 1,234 | $ | 19,909 | 1,613 | |||||||||
| General and administrative | 1,956 | 1,746 | 210 | 12 | ||||||||||||
| Loss from operations | (23,099 | ) | (2,980 | ) | 20,119 | 675 | ||||||||||
| Interest expense | (923 | ) | (358 | ) | 565 | 158 | ||||||||||
| Net loss | $ | (24,022 | ) | $ | (3,338 | ) | $ | 20,684 | 620 | |||||||
Research and development expenses increased $19.9 million from $1.2 million for the three months ended June 30, 2019 to $21.1 million for the three months ended June 30, 2020. The increase is primarily due to increased clinical trial and clinical material manufacturing costs related to the clinical trial in COVID-19. We expect our development costs to increase significantly in the remaining six months of 2020 as a result of the recent hiring of our newly appointed Chief Scientific Officer, the securing of manufacturing capacity, progressing CMC work and the enrollment of patients in the clinical trials for COVID-19 and CAR-T therapy and our preparation for clinical trials in GvHD.
| 27 |
General and administrative expenses increased $0.2 million from $1.7 million for the three months ended June 30, 2019 to $1.9 million for the three months ended June 30, 2020. The increase is primarily due to increased legal and consulting fees incurred in connection with financing preparations and other corporate actions being planned. We expect our general and administrative costs to increase in the remaining six months of 2020 as a result of newly hired employees, including a newly appointed Chief Operating Officer and a Chief Accounting and Administrative Officer, as we scale our operations in connection with the ongoing clinical trials of lenzilumab in COVID-19 and CAR-T therapy and our preparation for clinical trials in GvHD.
Interest expense increased $0.6 million from $0.4 million recognized for the three months ended June 30, 2019 to $0.9 million for the three months ended June 30, 2020. The increase is primarily due to the payoffs of the 2020 Convertible Notes in June 2020 and the conversions of the 2018 and 2019 Convertible Notes in April 2020 and the accretion of the remaining original issue discounts and beneficial conversion factors and prepayment premiums related thereto.
Comparison of Six Months Ended June 30, 2020 and 2019
| Six Months Ended June 30, | Increase/ (Decrease) | |||||||||||||||
| (in thousands) | 2020 | 2019 | Amount | % | ||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 21,802 | $ | 1,593 | $ | 20,209 | 1,269 | |||||||||
| General and administrative | 3,354 | 3,625 | (271 | ) | (7 | ) | ||||||||||
| Loss from operations | (25,156 | ) | (5,218 | ) | 19,938 | 382 | ||||||||||
| Interest expense | (1,333 | ) | (660 | ) | 673 | 102 | ||||||||||
| Other expense, net | - | (1 | ) | (1 | ) | - | ||||||||||
| Net loss | $ | (26,489 | ) | $ | (5,879 | ) | $ | 20,610 | 351 | |||||||
Research and development expenses increased $20.2 million from $1.6 million for the six months ended June 30, 2019 to $21.8 million for the six months ended June 30, 2020. The increase is primarily due to increased clinical trial and clinical material manufacturing costs related to the clinical trial in COVID-19. We expect our development costs to increase significantly in the remaining six months of 2020 as a result of the recent hiring of our newly appointed Chief Scientific Officer, the securing of manufacturing capacity, progressing CMC work and the enrollment of patients in the clinical trials for COVID-19 and CAR-T therapy and our preparation for clinical trials in GvHD.
General and administrative expenses decreased $0.3 million from $3.6 million for the six months ended June 30, 2019 to $3.3 million for the six months ended June 30, 2020. The decrease is primarily due to decreased stock based compensation expense. We expect our general and administrative costs to increase in the remaining six months of 2020 as a result of newly hired employees, including a newly appointed Chief Operating Officer and a Chief Accounting and Administrative Officer, as we scale our operations in connection with the ongoing clinical trials of lenzilumab in COVID-19 and CAR-T therapy and our preparation for clinical trials in GvHD.
Interest expense increased $0.6 million from $0.7 million recognized for the six months ended June 30, 2019 to $1.3 million for the six months ended June 30, 2020. The increase is primarily due to the payoffs of the 2020 Convertible Notes in June 2020 and the conversions of the 2018 and 2019 Convertible Notes in April 2020 and the accretion of the remaining original issue discounts, beneficial conversion factors and prepayment premiums related thereto.
Other expense, net was essentially unchanged for the six months ended June 30, 2020 as compared to the same period in 2019.
| 28 |
Liquidity and Capital Resources
Since our inception, we have financed our operations primarily through proceeds from the public offerings of our common stock, private placements of our common and preferred stock, debt financings, interest income earned on cash, and cash equivalents, and marketable securities, and borrowings against lines of credit. At June 30, 2020, we had cash and cash equivalents of $41.8 million. As of August 13, 2020, we had cash and cash equivalents of approximately $30.8 million.
The following table sets forth the primary sources and uses of cash and cash equivalents for each of the periods presented below:
| Six Months Ended June 30, | ||||||||
| (In thousands) | 2020 | 2019 | ||||||
| Net cash (used in) provided by: | ||||||||
| Operating activities | $ | (22,615 | ) | $ | (2,273 | ) | ||
| Financing activities | 64,200 | 2,550 | ||||||
| Net increase (decrease) in cash and cash equivalents | $ | 41,585 | $ | 277 | ||||
Net cash used in operating activities was $22.6 million and $2.3 million for the six months ended June 30, 2020 and 2019, respectively. Cash used in operating activities of $22.6 million for the six months ended June 30, 2020 primarily related to our net loss of $26.5 million, adjusted for non-cash items, such as $0.6 million in stock-based compensation, an increase in accounts payable and accrued expenses of $3.6 million and net decreases in other working capital items of $0.3 million.
Net cash used in operating activities of $2.3 million for the six months ended June 30, 2019 primarily related to our net loss of $5.9 million, adjusted for non-cash items, such as $1.4 million in stock-based compensation, an increase in accounts payable and accrued expenses of $2.0 million and net increases in other working capital items of $0.2 million.
Net cash provided by financing activities was $64.2 million for the six months ended June 30, 2020 and consists primarily of $67.0 million received from the issuance of common stock in the Private Placement in June 2020, $0.5 million received from the issuance of the 2020 Convertible Notes, $0.3 million received from the issuance of the 2020 Bridge Notes and $0.1 million received from the issuance of common stock under the Purchase Agreement with LPC, offset by $0.5 million for the payoff of the 2020 Convertible Notes, $2.4 million for the payoff of the 2019 and 2020 Bridge Notes and $0.8 million for the payoff of the Notes payable to Vendors.
Equity Line of Credit
On November 8, 2019, we entered into the ELOC Purchase Agreement and a registration rights agreement with LPC, pursuant to which we had the right to sell to LPC up to $20,000,000 in shares of our common stock, subject to certain limitations and conditions set forth in the ELOC Purchase Agreement. On June 2, 2020, we notified LPC of our decision to terminate the ELOC Purchase Agreement. The termination of the ELOC Purchase Agreement became effective on June 3, 2020.
2020 Private Placement
On June 1, 2020, we entered into a securities purchase agreement (the “Purchase Agreement”) with certain accredited investors (the “Investors”) to complete a private placement of our common stock (the “Private Placement”). The closing of the Private Placement occurred on June 2, 2020 (the “Closing Date”). At the closing, we issued and sold 82,528,718 shares of our common stock (the “Shares”) at a purchase price of $0.87 per share, for aggregate gross proceeds of approximately $71.8 million. We used a portion of the proceeds to retire the following indebtedness:
| · | the outstanding principal amount and accrued and unpaid interest on the Company’s convertible promissory notes issued in March 2020, which approximated $0.6 million, were repaid in full, and the notes were extinguished; |
| · | the outstanding principal and accrued and unpaid interest, amounting to approximately $2.5 million, on short-term, secured bridge loans made to the Company in 2019 and 2020 were repaid in full and the related liens were released; and |
| 29 |
| · | the remaining outstanding principal and accrued and unpaid interest, amounting to approximately $1.1 million, on certain notes payable to vendors in accordance with the Plan. |
We expect to use the remaining proceeds from the Private Placement to fund our Phase III study of lenzilumab in COVID-19, to secure manufacturing capacity, to progress Chemistry, Manufacturing and Controls (“CMC”) work, to prepare for commercialization in the event of approval of lenzilumab for use in COVID-19 patients, our collaboration with Kite and other development programs, as well as for working capital and other general corporate purposes. See Note 10 to the Condensed Consolidated Financial Statements included in Item 1 of this Quarterly Report on Form 10-Q for material information regarding two complaints recently filed against us in connection with the Private Placement.
On the Closing Date, we also entered into a registration rights agreement with the Investors (the “Registration Rights Agreement”) pursuant to which we agreed to prepare and file a registration statement (the “Resale Registration Statement”) for the resale of the Shares with the SEC.
Subject to certain limitations and an overall cap, we may be required to pay liquidated damages to the investors at a rate of 2% of the invested capital for each occurrence (and continuation for 30 consecutive days thereafter) of a breach by the Company of certain of its obligations under the Registration Rights Agreement.
The Purchase Agreement also requires that we use our commercially reasonable efforts to achieve a listing of our common stock on a national securities exchange, subject to certain limitations set forth in the Purchase Agreement. On July 6, 2020, we filed an application to list our common stock on the Nasdaq Capital Market. Our ability to obtain Nasdaq’s approval of the listing application will require that we satisfy a number of conditions, including our ability to meet certain listing criteria including a minimum stock price and total value of public float. Accordingly, we can make no assurances that the application for listing will be approved. Unless and until our listing application is approved, our common stock will continue to trade on the OTCQB Venture Market under the symbol “HGEN.”
We will need additional capital to support our business efforts, including obtaining regulatory approvals for our product candidates, clinical trials and other studies, and, if approved, the commercialization of our product candidates. The amount of capital we will require and the timing of our need for additional capital will depend on many factors, including:
| · | the progress and timing of the Phase III COVID-19 study we are advancing and related costs we will be required to incur, including the addition of commercial manufacturing and other commercial preparation; |
| · | the progress and timing of the Study we are pursuing with Kite, and related costs we are required to incur; |
| · | the type, number, timing, progress, costs, and results of the other product candidate development programs that we are pursuing or may choose to pursue in the future; |
| · | the scope, progress, expansion, costs, and results of our other pre-clinical and clinical trials; |
| · | the timing of and costs involved in obtaining regulatory approvals; |
| · | the success, progress, timing and costs of our efforts to evaluate or consummate various strategic alternatives if in the best interests of our stockholders; |
| · | our ability to preserve our stock quotation on the OTCQB Venture Market or to list our common stock on the Nasdaq Capital Market, if our listing application is approved, or another national securities exchange; |
| · | our costs in connection with the manufacturing of drugs, whether alone or with manufacturing partners; |
| · | our ability to establish and maintain development partnering arrangements and any associated funding; |
| · | the emergence of competing products or technologies and other adverse market developments; |
| · | the costs of maintaining, expanding, and protecting our intellectual property portfolio, including potential litigation costs and liabilities; |
| · | the resources we devote to marketing, and, if approved, commercializing our product candidates; |
| · | the scope, progress, expansion and costs of manufacturing our product candidates; and |
| · | the costs associated with being a public company. |
We may pursue efforts to raise additional capital from a number of sources, including, but not limited to, the sale of equity or debt securities and strategic collaborations. Additional funding may not be available to us on a timely basis or at acceptable terms, if at all. Our ability to access capital when needed is not assured and, if not achieved on a timely basis, would materially harm our business, financial condition and results of operations. Any financing we may obtain may be dilutive to existing stockholders. If adequate funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our development programs. We may also be required to sell or license to others our technologies, product candidates, or development programs that we would have preferred to develop and commercialize ourselves and on less than favorable terms, if at all. If in the best interests of our stockholders, we may also find it appropriate to enter into a strategic transaction that could result in, among other things, a sale, merger, acquisition, consolidation or business combination.
| 30 |
If we are unsuccessful in our efforts to raise additional capital, based on our current and expected levels of operating expenses our current capital will not be sufficient to fund our operations for the next twelve months. These conditions raise substantial doubt about our ability to continue as a going concern.
Off-Balance Sheet Arrangements
We currently have no off-balance sheet arrangements, such as structured finance, special purpose entities or variable interest entities.
| Item 4. | Controls and Procedures. |
From June 2019 to July 31, 2020, Dr. Cameron Durrant, our Chief Executive Officer, acted, on an interim basis, as the Company’s principal financial officer. Subsequent to June 30, 2020, we announced the appointment of (1) Timothy Morris as our Chief Operating Officer and Chief Financial Officer, and (2) David L. Tousley as our Chief Accounting and Administrative Officer, Corporate Secretary and Treasurer.
Management’s Evaluation of our Disclosure Controls and Procedures
“Disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) promulgated under the Securities Exchange Act of 1934, as amended, or the Exchange Act, means controls and other procedures that are designed to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms.
Disclosure controls and procedures include, without limitation, those designed to ensure that this information is accumulated and communicated to our management to allow timely decisions regarding required disclosure. Management, including our Chief Executive Officer, our Chief Financial Officer, and our Chief Accounting Officer have evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this report. Based upon the evaluation, our Chief Executive Officer and our Chief Financial Officer, with the assistance of our Chief Accounting Officer, concluded that the disclosure controls and procedures were not effective as of June 30, 2020 to ensure that information required to be disclosed in the reports we file and submit under the Exchange Act is (i) recorded, processed, summarized and reported as and when required and (ii) accumulated and communicated to our management, including our Chief Executive Officer, as appropriate to allow timely discussion regarding required disclosure.
Changes in Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) and 15d-15(f) under the Exchange Act). Our Chief Executive Officer, Chief Financial Officer and Chief Accounting Officer assessed the effectiveness of our internal control over financial reporting as of June 30, 2020. In making this assessment, our Chief Executive Officer, Chief Financial Officer and our Chief Accounting Officer used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO, in Internal Control—Integrated Framework. Based on that assessment and using the COSO criteria, our Chief Executive Officer and Chief Financial Officer, with the assistance of our Chief Accounting Officer, concluded that, as of June 30, 2020, our internal control over financial reporting was not effective because of the material weaknesses described below.
A material weakness is defined as “a deficiency, or a combination of deficiencies in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the company’s annual or interim financial statements will not be prevented or detected on a timely basis.”
The ineffectiveness of our internal control over financial reporting at June 30, 2020, was due to an insufficient degree of segregation of duties among our accounting and financial reporting personnel.
During the third quarter and the remainder of 2020, we intend to work to remediate the material weaknesses identified above, which could include the addition of accounting and financial reporting personnel and/or the engagement of accounting and personnel consultants on a limited-time basis until we add a sufficient number of personnel.
| 31 |
Subsequent to June 30, 2020, we announced the appointment of (1) Timothy Morris as our Chief Operating Officer and Chief Financial Officer, effective August 1, 2020, and (2) David L. Tousley as our Chief Accounting and Administrative Officer, Corporate Secretary and Treasurer, effective July 6, 2020.
Inherent Limitations of Controls
Management does not expect that our disclosure controls and procedures or our internal control over financial reporting will prevent or detect all error and all fraud. Controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within the Company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty, and that breakdowns can occur because of a simple error or mistake. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the controls. The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions. Over time, controls may become inadequate because of changes in conditions or deterioration in the degree of compliance with the policies or procedures. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
| Item 1. | Legal Proceedings. |
Please see Note 10 to the Condensed Consolidated Financial Statements included in Item 1 of this Quarterly Report on Form 10-Q for a summary of legal proceedings and developments during the quarter ended June 30, 2020.
| Item 1A. | Risk Factors. |
The fluid and unpredictable nature of the COVID-19 pandemic, which began in late 2019 and has spread worldwide, may affect our ability to conduct our Phase III trial of lenzilumab for the prevention and treatment of cytokine storm in COVID-19 pneumonia, delay the initiation of planned and future clinical trials, disrupt regulatory activities, or have other adverse effects on our business and operations. In addition, this pandemic has caused substantial disruption in the financial markets and may adversely impact economies worldwide, both of which could result in adverse effects on our business and operations.
In December 2019, an outbreak of the respiratory illness COVID-19 caused by a strain of novel coronavirus, SARS-Cov-2, was first reported in China. The COVID-19 outbreak has spread worldwide, causing many governments to implement measures to slow the spread of the outbreak through quarantines, strict travel restrictions, heightened border scrutiny, and other measures. The outbreak and government measures taken in response have had a significant impact, both direct and indirect, on businesses and commerce, as worker shortages have occurred; supply chains have been disrupted; facilities and production have been suspended; and demand for certain goods and services, such as medical services and supplies, has spiked, while demand for other goods and services, such as travel, has fallen. The future progression of the outbreak and its effects on our business and operations are uncertain.
We and our third-party contract research organizations (“CROs”) and clinical sites may experience disruptions in supply of product candidates and/or procuring items that are essential for our research and development activities, including raw materials used in the manufacturing of our product candidates, medical and laboratory supplies used in our clinical trials or preclinical studies or animals that are used for preclinical testing, in each case, for which there may be shortages because of ongoing efforts to address the outbreak. While these delays have not materially impacted our overall manufacturing supply chain operations to date, and we continue to explore back up or alternative sources of supply, any future disruption in the supply chain from the recent COVID-19 outbreak, or any potential future outbreak could have a material adverse impact on our clinical trial plans and business operations.
Additionally, we have enrolled, and will seek to enroll, patients in our clinical trials at sites located in many areas affected by COVID-19 and, as a result, our trials may be impacted. In addition, even if sites are actively recruiting, we may face difficulties recruiting or retaining patients in our ongoing and planned clinical trials if patients are affected by the virus or are fearful of visiting or traveling to our clinical trial sites because of the outbreak. Prolonged delays or closure to enrollment in our trials or patient discontinuations could have a material adverse impact on our clinical trial plans and timelines, including our Phase III trial of lenzilumab for the prevention and treatment of cytokine storm in COVID-19 pneumonia.
| 32 |
In addition, our ability to collect all data requested of patients enrolled in our clinical trials during this pandemic is being impacted to varying degrees by COVID-19. Clinical trial data collection generally continues for each of our clinical trials, but at a slower pace, and in some instances, we encounter disruption of collection of complete study data. This could have a material adverse impact on our data analysis.
The response to the COVID-19 pandemic may redirect resources with respect to regulatory and intellectual property matters in a way that would adversely impact our ability to progress regulatory approvals and protect our intellectual property. In addition, we may face impediments to regulatory meetings and approvals due to measures intended to limit in-person interactions.
Any negative impact that the COVID-19 outbreak has on the ability of our suppliers to provide materials for our product candidates or on recruiting or retaining patients in our clinical trials or our ability to collect patient data could cause costly delays to clinical trial activities, which could adversely affect our ability to obtain regulatory approval for and to commercialize our product candidates, increase our operating expenses, and have a material adverse effect on our financial results. Furthermore, any negative impact that the outbreak has on the ability of our CROs to deliver data sets and execute on experimentation could cause substantial delays for our discovery activities and materially impact our ability to fuel our pipeline with new product candidates.
Any negative impact that the COVID-19 pandemic has on recruiting or retaining patients in our clinical trials, obtaining complete clinical trial data or on the ability of our suppliers to provide materials for our product candidates could cause additional delays to clinical trial and developmental activities, which could materially and adversely affect our ability to obtain regulatory approval for and to commercialize our product candidates, increase our operating expenses, affect our ability to raise additional capital, and have a material adverse effect on our financial results.
The COVID-19 pandemic has already caused significant disruptions in the financial markets, and may continue to cause such disruptions, which could impact our ability to raise additional funds through public offerings and may also impact the volatility of our stock price and trading in our stock. Moreover, the pandemic has also significantly impacted economies worldwide, which could result in adverse effects on our business and operations. We cannot be certain what the overall impact of the COVID-19 pandemic will be on our business. It has the potential to adversely affect our business, financial condition, results of operations, and prospects.
We need substantial additional capital to develop, manufacture and commercialize our product candidates, and our access to funding is uncertain. If we cannot obtain additional financing, we may not be able to pursue our business objectives or to remain a going concern.
We do not expect to recognize any revenues while we continue to pursue the development of lenzilumab and our other product candidates. As of June 30, 2020, the Company had cash and cash equivalents of $41.8 million. We expect our development costs to increase significantly in the remaining six months of 2020 as a result of the recent hiring of our newly appointed Chief Scientific Officer, the securing of current and future manufacturing capacity, progressing CMC work in preparation for filing for marketing approval and the enrollment of patients in the clinical trials for COVID-19 and CAR-T therapy and our preparation for clinical trials in GvHD. As a result, we expect that we will need to raise additional capital to support these business efforts.
We anticipate that we will seek additional financing from a number of sources, including, but not limited to, the sale of equity or debt securities, strategic collaborations, and licensing of our product candidates. Additional funding may not be available to us on a timely basis or on acceptable terms, if at all. Our ability to access capital when needed is not assured and, if not achieved on a timely basis, would materially harm our business, financial condition and results of operations. If adequate funds are not available, we may be required to delay, reduce the scope of, or eliminate one or more of our development programs, limit the scale and scope of our CMC work, or be unable to reserve adequate manufacturing capacity at our CMOs, which could put us at a competitive disadvantage in attaining regulatory approvals in connection with our clinical development program in COVID-19. We may also be required to sell or license to others our technologies, product candidates, or development programs that we would have preferred to develop and commercialize ourselves and on less than favorable terms, if at all. If in the best interests of our stockholders, we may also find it appropriate to enter into a strategic transaction that could result in, among other things, a sale, merger, acquisition, consolidation or business combination.
If management is unsuccessful in efforts to raise additional capital, based on our current levels of operating expenses, our current capital is not expected to be sufficient to fund our operations for the next twelve months. These conditions raise substantial doubt about our ability to continue as a going concern.
The condensed consolidated financial statements for the three and six months ended June 30, 2020 were prepared on the basis of a going concern, which contemplates that we will be able to realize our assets and discharge liabilities in the normal course of business. The financial statements do not include any adjustments that might be necessary if we are unable to continue as a going concern.
In addition, the presence of the explanatory paragraph about our ability to continue as a going concern in our financial statements, could also make it more difficult to raise the capital necessary to address our current needs.
If our competitors develop and receive FDA approval for treatments or vaccines for COVID-19, our commercial opportunity may be reduced or eliminated.
There are numerous companies working on therapies to treat COVID-19 and/or vaccines to prevent or treat COVID-19. The speed at which all parties are acting to create and test many treatments and vaccines for COVID-19 is unusual, and evolving or changing plans or priorities within the FDA, including changes based on new knowledge of COVID-19 and how the disease affects the human body, may significantly affect the regulatory timeline for lenzilumab. Results from clinical testing may raise new questions and require us to redesign proposed clinical trials. Many of our competitors and potential competitors have substantially greater scientific, research, and product development capabilities, as well as greater financial, marketing, sales and human resources capabilities than we do. In addition, many specialized biotechnology firms have formed collaborations with large, established companies to support the research, development and commercialization of products that may be competitive with ours.
Furthermore, the incidence of COVID-19 in the communities where the Phase III trial participants reside will vary across different locations. If the overall incidence of clinical deterioration of patients with COVID-19 in the lenzilumab COVID-19 Phase III trial is low, it may be difficult for this study to demonstrate differences between participants in the study who receive placebo and those who receive lenzilumab. There can be no assurance that we will receive FDA approval or Emergency Use Authorization for lenzilumab as a therapy for patients suffering from COVID-19 pneumonia, or that we would be the first to successfully develop a therapy for this indication. If we are not the first therapy approved, or if other competing therapies are approved after lenzilumab, and/or a safe, efficacious, accessible preventative vaccine is approved, such approval could have a material adverse impact on our ability to commercialize lenzilumab as a therapy for COVID-19.
| 33 |
We may experience delays in commencing or conducting our clinical trials, in receiving data from third parties or in the continuation or completion of clinical testing, which could result in increased costs to us, delay our ability to generate product candidate revenue or, ultimately, render us unable to complete the development and commercialization of our product candidates.
We have product candidates in clinical development and preclinical development. The risk of failure for each of our product candidates is high. It is impossible to predict when or if any of our product candidates will prove effective or safe in humans or will receive regulatory approval. Before obtaining marketing approval from regulatory authorities for the sale of any product candidate, we must complete preclinical development and then conduct extensive clinical trials to demonstrate the safety and efficacy of our product candidates in humans.
Before we can initiate clinical trials in the United States for any new product candidates, we are required to submit the results of preclinical testing to FDA as part of an IND application, along with other information including information about product candidate chemistry, manufacturing, and controls and our proposed clinical trial protocol. For our programs already underway, we are required to report or provide information to appropriate regulatory authorities in order to continue with our testing programs. If we are unable to make timely regulatory submissions for any of our programs, it will delay our plans for our clinical trials. If those third parties do not make the required data available to us, we will likely have to identify and contract with another third party, and/or develop all necessary preclinical and clinical data on our own, which will lead to significant delays and increase development costs of the product candidate. In addition, FDA may require us to conduct additional preclinical testing for any product candidate before it allows us to initiate clinical testing under any IND application, which may lead to additional delays and increase the costs of our preclinical development. Moreover, despite the presence of an active IND application for a product candidate, clinical trials can be delayed for a variety of reasons, including delays in:
| · | identifying, recruiting, and enrolling qualified subjects to participate in a clinical trial; |
| · | identifying, recruiting, and training suitable clinical investigators; |
| · | reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and trial sites, the terms of which can be subject to extensive negotiation, may be subject to modification from time to time, and may vary significantly among different CROs and trial sites; |
| · | obtaining and maintaining sufficient quantities of a product candidate for use in clinical trials, either as a result of transferring the manufacturing of a product candidate to another site or manufacturer, deferring ordering or production of product in order to conserve resources or mitigate risk, having product in inventory become no longer suitable for use in humans, or other reasons that reduce or delay availability of drug supply; |
| · | obtaining and maintaining Institutional Review Board (“IRB”) or ethics committee approval to conduct a clinical trial at an existing or prospective site; |
| · | retaining or replacing participants who have initiated a clinical trial but may withdraw due to adverse events from the therapy, insufficient efficacy, fatigue with the clinical trial process, or personal issues; |
| · | developing any companion diagnostic necessary to ensure the study enrolls the target population; |
| · | being required by the FDA to add more patients or sites or to conduct additional trials; or |
| · | the FDA placing a clinical trial on hold. |
Once a clinical trial has begun, recruitment and enrollment of subjects may be slower than we anticipate. Numerous companies and institutions are conducting clinical studies in similar patient populations which can result in competition for qualified patients. In addition, clinical trials will take longer than we anticipate if we are required, or believe it is necessary, to enroll additional subjects than originally planned. Clinical trials may also be delayed as a result of ambiguous or negative interim results. Further, a clinical trial may be suspended or terminated by us, an IRB, an ethics committee, or a data safety monitoring committee overseeing the clinical trial, any of our clinical trial sites with respect to that site, or FDA or other regulatory authorities, due to a number of factors, including:
| · | failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
| · | inspection of the clinical trial operations or clinical trial site by the FDA or other regulatory authorities; |
| · | inability to provide timely supply of drug product; |
| · | unforeseen safety issues, known safety issues that occur at a greater frequency or severity than we anticipate, or any determination that the clinical trial presents unacceptable health risks; or |
| · | lack of adequate funding to continue the clinical trial or unforeseen significant incremental costs related to the trial. |
| 34 |
Additionally, if any future development partners do not develop the licensed product candidates in the time and manner that we expect, or at all, the clinical development efforts related to these licensed product candidates could be delayed or terminated. In addition, our ability to enforce our partners’ obligations under any future collaboration efforts may be limited due to time and resource constraints, competing corporate priorities of our future partners, and other factors.
Any delays in the commencement of our clinical trials may delay or preclude our ability to further develop or pursue regulatory approval for our product candidates. Changes in U.S. and foreign regulatory requirements and guidance also may occur and we may need to amend clinical trial protocols to reflect these changes. Amendments may require us to resubmit our clinical trial protocols to IRBs for re-examination, which may affect the costs, timing, and likelihood of a successful completion of a clinical trial.
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, if we are unable to successfully complete clinical trials of our product candidates or other testing, if the results of these trials or tests are not positive or are only modestly positive or if there are safety concerns, we may:
| · | be delayed in obtaining marketing approval for our product candidates; |
| · | not obtain marketing approval at all; |
| · | obtain approval for indications or patient populations that are not as broad as intended or desired; |
| · | obtain approval with labeling that includes significant use or distribution restrictions or safety warnings; |
| · | be subject to additional post-marketing testing requirements; or |
| · | have the product removed from the market after obtaining marketing approval. |
Our product development costs will also increase if we experience delays in testing or in obtaining marketing approvals. We do not know whether any of our preclinical studies or clinical trials will begin as planned, will need to be restructured or will be completed on schedule, or at all. We may also determine to change the design or protocol of one or more of our clinical trials, including to add additional arms or patient populations, which could result in increased costs and expenses and/or delays. If we or any future development partners experience delays in the completion of, or if we or any future development partners must terminate, any clinical trial of any product candidate our ability to obtain regulatory approval for that product candidate will be delayed and the commercial prospects, if any, for the product candidate may suffer as a result. Significant preclinical study or clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our product candidates and may harm our business and results of operations. In addition, many of these factors may also ultimately lead to the denial of regulatory approval of a product candidate.
The results of preclinical studies and early clinical trials are not always predictive of future results. Any product candidate we or any future development partners advance into clinical trials may not have favorable results in later clinical trials, if any, or receive regulatory approval.
Drug development has substantial inherent risk. We or any future development partners will be required to demonstrate through adequate and well-controlled clinical trials that our product candidates are effective, with a favorable benefit-risk profile, for use in their target populations for their intended indications before we can seek regulatory approvals for their commercial sale. Drug development is a long, expensive and uncertain process, and delay or failure can occur at any stage of development, including after commencement of any of our clinical trials. Success in early clinical trials does not mean that later clinical trials will be successful because product candidates in later-stage clinical trials may fail to demonstrate sufficient safety or efficacy despite having progressed through initial clinical testing. Furthermore, our future trials will need to demonstrate sufficient safety and efficacy for approval by regulatory authorities in larger patient populations. Companies frequently suffer significant setbacks in advanced clinical trials, even after earlier clinical trials have shown promising results. In addition, only a small percentage of drugs under development result in the submission of a New Drug Application (“NDA”) or Biologic License Application (“BLA”) to FDA and even fewer are approved for commercialization.
| 35 |
In addition, serious adverse or undesirable side effects may emerge or be identified during later stages of development that were not observed in earlier stages. If our product candidates, either alone or in combination with other therapeutics, are associated with serious adverse events or undesirable side effects or unacceptable drug-drug interactions in clinical trials or have characteristics that are unexpected in clinical trials or preclinical testing, we may need to abandon their development or limit development to more narrow uses or subpopulations in which the serious adverse events, undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective. In pharmaceutical development, many compounds that initially show promise in early-stage or clinical testing are later found to cause side effects that prevent further development of the compound. In addition, if third parties manufacture or use our product candidates without our permission, and generate adverse events or unacceptable side effects, this could also have an adverse impact on our development efforts.
Unacceptable adverse events caused by any of our product candidates that we advance into clinical trials could cause us or regulatory authorities to interrupt, delay, or halt clinical trials and could result in the denial of regulatory approval by FDA or other regulatory authorities for any or all targeted indications and markets. This in turn could prevent us from completing development or commercializing the affected product candidate and generating revenue from its sale. We have not yet successfully completed testing of any of our product candidates for the treatment of the indications for which we intend to seek approval in humans, and we currently do not know the extent of adverse events, if any, that will be observed in individuals who receive any of our product candidates. If any of our product candidates cause unacceptable adverse events in clinical trials, we may not be able to obtain regulatory approval or commercialize such product candidates.
A significant portion of our total outstanding shares are eligible to be sold into the market in the near future, which could cause the market price of our common stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market or the perception in the market that the holders of a large number of shares intend to sell shares, could depress the market price of our common stock and could impair our future ability to obtain capital, especially through an offering of equity securities. If there are more shares of common stock offered for sale than buyers are willing to purchase, then the market price of our common stock may decline to a market price at which buyers are willing to purchase the offered shares of common stock and sellers remain willing to sell the shares.
Persons who were our stockholders prior to our issuance of common stock to the Investors pursuant to the Purchase Agreement, as further described above (see “Management’s Discussion and Analysis of Financial Condition and Results of Operations — Liquidity and Capital Resources — 2020 Private Placement”), continue to hold a substantial number of shares of our common stock. If such persons sell, or indicate an intention to sell, substantial amounts of our common stock in the public market, the trading price of our common stock could decline. In April 2018, we filed a registration statement registering additional shares of common stock that we may issue under the Humanigen, Inc. 2012 Equity Incentive Plan. These shares can be freely sold in the public market upon issuance, subject to volume limitations applicable to affiliates.
Moreover, holders of a substantial number of shares of our common stock have rights, subject to specified conditions, to require us to file a registration statement covering their shares. Registration of these shares under the Securities Act, would result in the shares becoming freely tradable without restriction under the Securities Act, except for shares held by affiliates, as defined in Rule 144 under the Securities Act. Any sales of securities by these stockholders could have a material adverse effect on the trading price of our common stock.
The concentration of our common stock owned by insiders may limit the ability of our other stockholders to influence corporate matters and may contribute to volatility in our stock price.
We have a relatively small public float due to the ownership percentage of our executive officers and directors, and greater than 5% stockholders. Our directors, executive officers, and the other holders of more than 5% of our common stock together with their affiliates beneficially owned approximately 72% of our common stock as of June 30, 2020. Some of these persons or entities may have interests that are different from our other stockholders, which could prevent or discourage unsolicited acquisition proposals or offers for our common stock that may be in the best interest of our other stockholders. This may also adversely affect the trading price of our common stock because investors may perceive disadvantages in owning stock in companies with a significant concentration of ownership.
As a result of our small public float, our common stock may be less liquid and have greater stock price volatility than the common stock of companies with broader public ownership. In addition, the trading of a relatively small volume of shares of our common stock may result in significant volatility in our stock price. If and to the extent ownership of our common stock becomes more concentrated, whether due to increased ownership by our directors and executive officers or other principal stockholders, or other factors, our public float would further decrease, which in turn would likely result in increased stock price volatility.
| 36 |
Additionally, because a large amount of our stock is closely held, we may experience low trading volume or large fluctuations in share price and volume due to large sales by our principal stockholders. If our existing stockholders, particularly our directors, executive officers and the holders of more than 5% of our common stock, or their affiliates or associates, sell substantial amounts of our common stock in the public market, or are perceived by the public market as intending to sell substantial amounts of our common stock, the trading price of our common stock could decline significantly.
In conjunction with the Purchase Agreement entered into on June 1, 2020, our directors, executive officers, and certain holders of more than 5% of our common stock together with their affiliates, which collectively beneficially owned approximately 50% of our common stock as of June 30, 2020, entered into lock-up agreements pursuant to which they have agreed to, among other things, not sell their shares of common stock or any securities convertible into or exercisable or exchangeable for common stock until 180 days after the closing of the Private Placement on June 2, 2020. Sales of a substantial number of such shares upon expiration of the lock-up agreements, the perception that such sales may occur, or early release of restrictions in the lock-up agreements, could cause the market price of our common stock to fall or make it more difficult for you to sell your common stock at a time and price that you deem appropriate.
There is a limited trading market for our securities and we do not currently have an active public market for our securities, which means you may not be able to resell shares of our common stock publicly, if at all, at times or prices you feel are fair and appropriate. An active trading market for our common stock may not develop or be sustained.
While our intention is to list our common stock on the Nasdaq Capital Market, our common stock is currently quoted on the OTCQB Venture Market, and trading in our common stock has been limited. The OTCQB Venture Market is generally understood to be a less active, and therefore less liquid, trading market than a national securities exchange. We cannot predict whether an active market for our common stock will ever develop in the future. In the absence of an active trading market:
| · | investors may have difficulty buying and selling shares of our common stock; |
| · | market visibility for shares of our common stock may be limited; |
| · | a lack of visibility for shares of our common stock may have a depressive effect on the market price for shares of our common stock; and |
| · | significant sales of our common stock, or the expectation of these sales, could materially and adversely affect the market price of our common stock. |
Our historically small trading volume in our common stock may make it difficult for our stockholders to sell their shares as and when they choose. Small trading volumes generally depress market prices. As a result, our stockholders may not be able to resell shares of our common stock publicly, if at all, at times or prices that our stockholders feel are fair or appropriate.
An inactive market may also impair our ability to raise capital and to fund operations by selling shares and may impair our ability to acquire additional intellectual property assets by using our shares as consideration.
No assurance can be given that an active market will develop for the common stock or as to the liquidity of the trading market for the common stock. The common stock may be traded only infrequently in transactions arranged through brokers or otherwise, and reliable market quotations may not be available.
We have applied to list our common stock on the Nasdaq Capital Market, but there is no assurance that our listing application will be approved by Nasdaq or that our common stock will ever be listed on the Nasdaq Capital Market or any other national securities exchange.
While we have applied to list our common stock on the Nasdaq Capital Market, we cannot ensure that we will be able to satisfy the listing standards or that our common stock will be accepted for listing on the Nasdaq Capital Market. Should we fail to satisfy the initial listing standards of the Nasdaq Capital Market, or our common stock are otherwise rejected for listing, our common stock will continue to trade on the OTCQB Venture Market, in which event the trading price of our common stock could suffer, the trading market for our common stock may be less liquid, and our common stock price may be subject to increased volatility.
| 37 |
Even if our common stock is accepted for listing on the Nasdaq Capital Market upon our satisfaction of the exchange’s initial listing criteria, there can be no assurance that an active trading market for our common stock will develop or be sustained, and the Nasdaq Capital Market may subsequently delist our common stock if we fail to comply with ongoing listing standards.
In the event we are able to list our common stock on the Nasdaq Capital Market upon our satisfaction of the exchange’s initial listing criteria, the exchange will require us to meet certain financial, public float, bid price and liquidity standards on an ongoing basis in order to continue the listing of our common stock. In addition to specific listing and maintenance standards, the Nasdaq Capital Market will have broad discretionary authority over the initial and continued listing of securities, which it could exercise with respect to the listing of our common stock.
If we fail to meet these continued listing requirements, our common stock may be subject to delisting. If our common stock is delisted from the Nasdaq Capital Market and we are not able to list our common stock on another national securities exchange, we expect our securities would be quoted on an over-the-counter market; however, if this were to occur, our stockholders could face significant material adverse consequences, including limited availability of market quotations for our common stock and reduced liquidity for the trading of our securities. In addition, in the event of such delisting, we could experience a decreased ability to issue additional securities and obtain additional financing in the future.
Further, even if our common stock is listed on the Nasdaq Capital Market, there can be no assurance that an active trading market for our common stock will develop or be sustained after our initial listing.
Future acquisitions of and investments in new businesses could impact our business and financial condition.
From time to time, we may acquire or invest in businesses or partnerships that we believe could complement our business and drug candidates. The pursuit of such acquisitions or investments may divert the attention of management and cause us to incur various expenses, regardless of whether the acquisition or investment is ultimately completed. In addition, acquisitions and investments may not perform as expected and we may be unable to realize the expected benefits, synergies or developments that we may initially anticipate. Further, if we are able to successfully identify and acquire additional businesses, we may not be able to successfully integrate the acquired personnel or operations, or effectively manage the combined business following the acquisition, any of which could harm our business and financial condition.
In addition, to the extent we finance any acquisition or investment in cash, it would reduce our cash reserves, and to the extent the purchase price is paid with shares of our common or preferred stock, it could be dilutive to our current stockholders. To the extent we finance any acquisition or investment with the proceeds from the incurrence of debt, this would increase our level of indebtedness and could negatively affect our liquidity, credit rating and restrict our operations. Moreover, we may face contingent liabilities in connection with any acquisitions or investments.
Currently pending, threatened or future litigation or governmental proceedings or inquiries could result in material adverse consequences, including judgments or settlements.
We are, or may from time to time become, involved in lawsuits, inquiries and other legal proceedings. See Note 10 to the Condensed Consolidated Financial Statements included in Item 1 of this Quarterly Report on Form 10-Q for information regarding currently pending litigation that could have a material impact on the Company. Many of these matters raise complicated factual and legal issues and are subject to uncertainties and complexities, all of which make the matters costly to address. The timing of the final resolutions to any such lawsuits, inquiries, and other legal proceedings is uncertain.
| 38 |
Additionally, the possible outcomes or resolutions to these matters could include adverse judgments or settlements, either of which could require substantial payments, adversely affecting our consolidated financial condition, results of operations and cash flows. Any judgment against us, the entry into any settlement agreement, or the imposition of any fine could have a material adverse effect on our consolidated financial condition, results of operations and cash flows.
| Item 2. | Unregistered Sales of Equity Securities and Use of Proceeds |
In the quarter ended June 30, 2020, we issued the following securities that were not registered under the Securities Act:
| · | On April 13, 2020 we issued 6,880 shares of common stock to Ness Capital and Consulting, LLC in exchange for capital markets consulting services delivered by the Consultant to us pursuant to a consulting services agreement entered into on September 13, 2019. |
| · | On May 13, 2020 we issued 8,720 shares of common stock to Ness Capital and Consulting, LLC in exchange for capital markets consulting services delivered by the Consultant to us pursuant to a consulting services agreement entered into on September 13, 2019. |
| · | In April and May 2020, we issued warrants to purchase an aggregate of 60,000 shares of common stock to Batuta Capital Advisors LLC in exchange for capital markets consulting services delivered to us pursuant to a consulting services agreement entered into in April 2020. |
| · | In May 2020, we issued an aggregate of 34,866 shares of common stock at the direction of Carter, Terry & Company, Inc. in connection with its performance of capital markets advisory services for the Company. |
| · | In May 2020, we issued warrants to purchase an aggregate of 111,000 shares of common stock to Verta Life Sciences, LLC in exchange for services delivered to us in connection with our clinical trial programs and product development activity. |
| · | In May 2020, we issued warrants to purchase an aggregate of 39,000 shares of common stock to Sage Engineering Services Ltd. in exchange for services delivered to us in connection with our clinical trial programs and product development activity. |
| · | Pursuant to the exemption from registration afforded by Section 3(a)(9) under the Securities Act, we issued an aggregate of 11,989,578 shares of our common stock upon the conversion of $4.3 million in aggregate principal and interest on convertible notes that were converted, which obligations were retired. |
| · | On May 20, 2020 we issued 26,847 shares of common stock to Ronald Barliant, a member of our Board of Directors as reimbursement for certain expenses he paid on behalf of the Company. |
| · | On June 30, 2020, Savant executed 100,000 warrant shares in a cashless exercise resulting in 54,545 shares being issued to Savant in July 2020. |
Except as otherwise described above, the sales and issuances described above were made in reliance on the exemptions from registration provided by Section 4(a)(2) of the Securities Act as transactions not involving a public offering and/or Regulation D under the Securities Act as sales to accredited investors. The purchasers in these transactions represented to us that they were accredited investors and were acquiring the shares for investment purposes and not with a view to, or for sale in connection with, any distribution thereof.
| 39 |
| Item 6. | Exhibits. |
| 40 |
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| HUMANIGEN, INC. | |||
| Date: August 14, 2020 | By: | /s/ Cameron Durrant | |
| Cameron Durrant | |||
| Chief Executive Officer | |||
| (Principal Executive Officer) | |||
| Date: August 14, 2020 | By: | /s/ David L. Tousley | |
| David L. Tousley | |||
| Chief Accounting Officer | |||
| (Principal Accounting Officer) | |||
41