Attached files
| file | filename |
|---|---|
| EX-32.2 - EXHIBIT 32.2 - Marker Therapeutics, Inc. | tv499451_ex32-2.htm |
| EX-32.1 - EXHIBIT 32.1 - Marker Therapeutics, Inc. | tv499451_ex32-1.htm |
| EX-31.2 - EXHIBIT 31.2 - Marker Therapeutics, Inc. | tv499451_ex31-2.htm |
| EX-31.1 - EXHIBIT 31.1 - Marker Therapeutics, Inc. | tv499451_ex31-1.htm |
UNITED STATES SECURITIES AND EXCHANGE
COMMISSION
Washington, D.C. 20549
FORM 10-Q
| x | Quarterly Report Under Section 13 or 15(d) of the Securities Exchange Act of 1934 for the quarterly period ended June 30, 2018 |
| ¨ | Transition Report Under Section 13 or 15(d) of the Securities Exchange Act of 1934 for the transition period from _____ to _____. |
Commission File Number: 001-37939

TAPIMMUNE INC.
(Name of registrant in its charter)
| NEVADA | 45-4497941 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
|
5 West Forsyth Street, Suite 200 Jacksonville, FL |
32202 | |
| (Address of principal executive offices) | (Zip Code) | |
| 904-516-5436 | ||
| (Issuer's telephone number) |
Indicate by check mark whether the registrant (1) filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act during the past 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, non-accelerated filer, a smaller reporting company, or an emerging growth company. See definition of “accelerated filer”, “large accelerated filer”, “smaller reporting company”, and “emerging growth company” in Rule 12b-2 of the Exchange Act (check one):
| ¨ | Large accelerated filer | ¨ | Accelerated filer |
| ¨ | Non-accelerated filer (Do not check | x | Smaller reporting company |
| if smaller reporting company) | ¨ | Emerging growth company | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
As of August 2, 2018, the Company had 13,710,544 shares of common stock issued and outstanding.
| PART I. | FINANCIAL INFORMATION |
| Item 1. | Financial Statements |
CONDENSED CONSOLIDATED BALANCE SHEETS
(UNAUDITED)
| June 30, | December 31, | |||||||
| 2018 | 2017 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash | $ | 7,782,962 | $ | 5,129,289 | ||||
| Prepaid expenses and deposits | 108,716 | 51,150 | ||||||
| Total current assets | 7,891,678 | 5,180,439 | ||||||
| Total assets | $ | 7,891,678 | $ | 5,180,439 | ||||
| LIABILITIES AND STOCKHOLDERS' EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable and accrued liabilities | $ | 3,595,152 | $ | 1,508,312 | ||||
| Warrant liability | 147,000 | 9,000 | ||||||
| Promissory note | 5,000 | 5,000 | ||||||
| Total current liabilities | 3,747,152 | 1,522,312 | ||||||
| Total liabilities | 3,747,152 | 1,522,312 | ||||||
| COMMITMENTS AND CONTINGENCIES | ||||||||
| Stockholders' equity: | ||||||||
| Preferred stock - $0.001 par value, 5 million shares authorized at June 30, 2018 and December 31, 2017, respectively | ||||||||
| Series A, $0.001 par value, 1.25 million shares designated, 0 shares issued and outstanding as of June 30, 2018 and December 31, 2017, respectively | - | - | ||||||
| Series B, $0.001 par value, 1.5 million shares designated, 0 shares issued and outstanding as of June 30, 2018 and December 31, 2017, respectively | - | - | ||||||
| Common stock, $0.001 par value, 41.7 million shares authorized, 13.6 million and 10.6 million shares issued and outstanding as of June 30, 2018 and December 31, 2017, respectively | 13,624 | 10,616 | ||||||
| Additional paid-in capital | 170,287,725 | 161,067,538 | ||||||
| Accumulated deficit | (166,156,823 | ) | (157,420,027 | ) | ||||
| Total stockholders' equity | 4,144,526 | 3,658,127 | ||||||
| Total liabilities and stockholders' equity | $ | 7,891,678 | $ | 5,180,439 | ||||
See accompanying notes to these unaudited condensed consolidated financial statements.
| 1 |
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS
(UNAUDITED)
| For the three months ended | For the six months ended | |||||||||||||||
| June 30, | June 30, | |||||||||||||||
| 2018 | 2017 | 2018 | 2017 | |||||||||||||
| Revenues: | ||||||||||||||||
| Grant income | $ | 205,994 | $ | - | $ | 205,994 | $ | - | ||||||||
| Total revenues | 205,994 | - | 205,994 | - | ||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 1,826,837 | $ | 1,202,725 | $ | 3,426,387 | $ | 2,191,817 | ||||||||
| General and administrative | 3,052,954 | 1,190,517 | 4,650,890 | 2,618,310 | ||||||||||||
| Total operating expenses | 4,879,791 | 2,393,242 | 8,077,277 | 4,810,127 | ||||||||||||
| Loss from operations | (4,673,797 | ) | (2,393,242 | ) | (7,871,283 | ) | (4,810,127 | ) | ||||||||
| Other income (expense): | ||||||||||||||||
| Change in fair value of warrant liabilities | (139,000 | ) | 7,500 | (138,000 | ) | 4,500 | ||||||||||
| Debt extinguishment gain | - | 492,365 | - | 492,365 | ||||||||||||
| Net loss | $ | (4,812,797 | ) | $ | (1,893,377 | ) | $ | (8,009,283 | ) | $ | (4,313,262 | ) | ||||
| Net loss per share, Basic and Diluted | $ | (0.41 | ) | $ | (0.22 | ) | $ | (0.71 | ) | $ | (0.51 | ) | ||||
| Weighted average number of common shares outstanding | 11,838,371 | 8,576,634 | 11,233,755 | 8,503,521 | ||||||||||||
See accompanying notes to these unaudited condensed consolidated financial statements.
| 2 |
CONDENSED CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY
(UNAUDITED)
| Common Stock | Additional Paid- | Accumulated | Total Stockholders' | |||||||||||||||||
| Shares | Par value | in Capital | Deficit | Equity | ||||||||||||||||
| Balance at January 1, 2018 | 10,615,724 | $ | 10,616 | $ | 161,067,538 | $ | (157,420,027 | ) | $ | 3,658,127 | ||||||||||
| Issuance of common stock in private placement | 1,300,000 | 1,300 | 3,118,700 | - | 3,120,000 | |||||||||||||||
| Stock options exercised for cash | 10,416 | 10 | 18,115 | - | 18,125 | |||||||||||||||
| Stock warrants exercised for cash | 1,446,881 | 1,447 | 4,259,638 | - | 4,261,085 | |||||||||||||||
| Stock warrants cashless exercised | 118,425 | 118 | (118 | ) | - | - | ||||||||||||||
| Stock-based compensation | 132,825 | 133 | 1,096,339 | - | 1,096,472 | |||||||||||||||
| Fair value of repriced warrants as inducement | - | - | 727,513 | (727,513 | ) | - | ||||||||||||||
| Net loss | - | - | - | (8,009,283 | ) | (8,009,283 | ) | |||||||||||||
| Balance, June 30, 2018 | 13,624,271 | $ | 13,624 | $ | 170,287,725 | $ | (166,156,823 | ) | $ | 4,144,526 | ||||||||||
See accompanying notes to these unaudited condensed consolidated financial statements.
| 3 |
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(UNAUDITED)
| For the six months ended | ||||||||
| June 30, | ||||||||
| 2018 | 2017 | |||||||
| Cash Flows from Operating Activities: | ||||||||
| Net loss | $ | (8,009,283 | ) | $ | (4,313,262 | ) | ||
| Reconciliation of net loss to net cash used in operating activities: | ||||||||
| Changes in fair value of warrant liabilities | 138,000 | (4,500 | ) | |||||
| Stock-based compensation | 1,096,472 | 647,387 | ||||||
| Debt extinguishment gain | - | (492,365 | ) | |||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and deposits | (57,566 | ) | (109,032 | ) | ||||
| Accounts payable and accrued expenses | 2,086,840 | 336,135 | ||||||
| Net cash used in operating activities | (4,745,537 | ) | (3,935,637 | ) | ||||
| Cash Flows from Financing Activities: | ||||||||
| Proceeds from issuance of common stock and warrants in private placement, net of offering costs | 3,120,000 | 5,408,343 | ||||||
| Proceeds from exercise of stock warrants, net of offering costs | 4,261,085 | 638,666 | ||||||
| Proceeds from exercise of stock options | 18,125 | - | ||||||
| Net cash provided by financing activities | 7,399,210 | 6,047,009 | ||||||
| Net increase in cash | 2,653,673 | 2,111,372 | ||||||
| Cash at beginning of period | 5,129,289 | 7,851,243 | ||||||
| Cash at end of period | $ | 7,782,962 | $ | 9,962,615 | ||||
| For the six months ended | ||||||||
| June 30, | ||||||||
| 2018 | 2017 | |||||||
Supplemental schedule of non-cash financing activities: | ||||||||
Fair value of repriced warrants as inducement | $ | 727,513 | $ | 622,042 | ||||
Stock warrants cashless exercised | $ | 118 | $ | - | ||||
See accompanying notes to these unaudited condensed consolidated financial statements.
| 4 |
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
June 30, 2018
(Unaudited)
Note 1: Nature of Operations
TapImmune Inc. (the “Company” or “we”), a Nevada corporation incorporated in 1991, is a biotechnology company focusing on immunotherapy specializing in the development of innovative peptide and gene-based immunotherapeutics and vaccines for the treatment of oncology. Unlike other vaccine technologies that narrowly address the initiation of an immune response, TapImmune's approach broadly stimulates the cellular immune system by enhancing the function of killer T-cells and T-helper cells and by restoring antigen presentation in tumor cells allowing their recognition by the immune system.
NOTE 2: Basis of Presentation
The accompanying unaudited condensed consolidated financial statements have been prepared in accordance with the accounting principles generally accepted in the United States of America (“U.S. GAAP”) for interim financial information and pursuant to the instructions to Form 10-Q and Article 8 of Regulation S-X of the Securities and Exchange Commission (“SEC”) and on the same basis as the Company prepares its annual audited consolidated financial statements. In the opinion of management, the accompanying unaudited condensed consolidated financial statements reflect all adjustments, consisting of normal recurring adjustments, considered necessary for a fair presentation of such interim results.
The results for the condensed consolidated statement of operations are not necessarily indicative of results to be expected for the year ending December 31, 2018 or for any future interim period. The condensed consolidated balance sheet at June 30, 2018 has been derived from unaudited financial statements; however, it does not include all of the information and notes required by U.S. GAAP for complete financial statements. The accompanying condensed consolidated financial statements should be read in conjunction with the consolidated financial statements for the year ended December 31, 2017, and notes thereto included in the Company’s annual report on Form 10-K filed on March 23, 2018.
NOTE 3: LIQUIDITY AND FINANCIAL CONDITION
The Company’s activities since inception have consisted principally of acquiring product and technology rights, raising capital, and performing research and development. Successful completion of the Company’s development programs and, ultimately, the attainment of profitable operations are dependent on future events, including, among other things, its ability to access potential markets; secure financing, develop a customer base; attract, retain and motivate qualified personnel; and develop strategic alliances and collaborations. From inception, the Company has been funded by a combination of equity and debt financings.
The Company expects to continue to incur substantial losses over the next several years during its development phase. To fully execute its business plan, the Company will need to complete certain research and development activities and clinical studies. Further, the Company’s product candidates will require regulatory approval prior to commercialization. These activities may span many years and require substantial expenditures to complete and may ultimately be unsuccessful. Any delays in completing these activities could adversely impact the Company. The Company plans to meet its capital requirements primarily through issuances of debt and equity securities and, in the longer term, revenue from product sales.
As of June 30, 2018, the Company had cash of approximately $7.8 million. Historically, the Company had net losses and negative cash flows from operations. Management intends to continue its research efforts and to finance operations of the Company through debt and/or equity financings. Management plans to seek additional debt and/or equity financing through private or public offerings. There can be no assurance that the Company will be successful in obtaining additional financing on favorable terms, or at all. The Company has no sources of revenue to provide incoming cash flows to sustain its future operations. The Company’s ability to pursue its planned business activities is dependent upon successful efforts to raise additional capital. These factors raise substantial doubt regarding the Company’s ability to continue as a going concern. The financial statements do not include any adjustments that might result from the outcome of these uncertainties.
| 5 |
Note 4: SIGNIFICANT ACCOUNTING POLICIES
There have been no material changes in the Company’s significant accounting policies to those previously disclosed in the Company’s annual report on Form 10-K, which was filed with the SEC on March 23, 2018.
New Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board (“FASB”) or other standard setting bodies that we adopt as of the specified effective date. Unless otherwise discussed, we do not believe that the impact of recently issued standards that are not yet effective will have a material impact on our financial position or results of operations upon adoption.
Recent Accounting Pronouncements Adopted in the Year
Compensation-Stock Compensation
In May 2017, the FASB issued Accounting Standards Update (“ASU”) No. 2017-09, Compensation—Stock Compensation (Topic 718): Scope of Modification Accounting, which clarifies when to account for a change to the terms or conditions of a share-based payment award as a modification. Under the new guidance, modification accounting is required only if the fair value, the vesting conditions, or the classification of the award (as equity or liability) changes as a result of the change in terms or conditions. It is effective prospectively for the annual period beginning after December 15, 2017 and interim periods within that annual period. Early adoption is permitted. The Company adopted ASU 2017-09 on January 1, 2018; the adoption of ASU 2017-09 did not have a material impact on its financial condition or results of operations, as the Company has not had any modifications to share-based payment awards. However, if the Company does have a modification to an award in the future, it will follow the guidance in ASU 2017-09.
Revenue from Contracts with Customers
In May 2014, the FASB issued ASU No. 2014-09, “Revenue from Contracts with Customers (Topic 606)” (ASU 2014-09) as modified by ASU No. 2015-14, “Revenue from Contracts with Customers (Topic 606): Deferral of the Effective Date,” ASU 2016-08, “Revenue from Contracts with Customers (Topic 606): Principal versus Agent Considerations (Reporting Revenue Gross versus Net),” ASU No. 2016-10, “Revenue from Contracts with Customers (Topic 606): Identifying Performance Obligations and Licensing,” and ASU No. 2016-12, “Revenue from Contracts with Customers (Topic 606): Narrow-Scope Improvements and Practical Expedients.” The revenue recognition principle in ASU 2014-09 is that an entity should recognize revenue to depict the transfer of goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. In addition, new and enhanced disclosures will be required. Companies may adopt the new standard either using the full retrospective approach, a modified retrospective approach with practical expedients, or a cumulative effect upon adoption approach. The Company adopted the new standard effective January 1, 2018, using the modified retrospective approach. The only impact of the adoption of ASU 2014-09 was to reclassify the Company's grant income as revenue.
Recent Accounting Pronouncements Not Yet Adopted
Accounting for Certain Financial Instruments with Down Round Features
On July 13, 2017, the FASB has issued a two-part ASU, No. 2017-11, (i). Accounting for Certain Financial Instruments with Down Round Features and (ii) Replacement of the Indefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Noncontrolling Interests With a Scope Exception.
The ASU is effective for public business entities for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2018 and the interim periods within that annual period. Early adoption is permitted. The Company will be evaluating the impact of adopting this standard on the consolidated financial statements and disclosures.
Improvements to Nonemployee Share-Based Payment Accounting
In June 2018, the FASB issued ASU 2018-07 “Improvements to Nonemployee Share-Based Payment Accounting”, which simplifies the accounting for share-based payments granted to nonemployees for goods and services. Under the ASU, most of the guidance on such payments to nonemployees would be aligned with the requirements for share-based payments granted to employees. The amendments are effective for fiscal years beginning after December 15, 2019, and interim periods within fiscal years beginning after December 15, 2020. Early adoption is permitted, but no earlier than an entity’s adoption date of Topic 606. The Company is currently evaluating the impact of the new standard on its consolidated financial statements.
| 6 |
Note 5: NET LOSS PER SHARE APPLICABLE TO COMMON SHAREHOLDER
Basic loss per common share is computed by dividing net loss by the weighted average number of common shares outstanding during the reporting period. Diluted loss per common share is computed similarly to basic loss per common share except that it reflects the potential dilution that could occur if dilutive securities or other obligations to issue common stock were exercised or converted into common stock.
The following table sets forth the computation of net loss per share:
| Six Months Ended | Six Months Ended | |||||||||||||||
| June 30, | June 30, | |||||||||||||||
| 2018 | 2017 | 2018 | 2017 | |||||||||||||
| Numerator: | ||||||||||||||||
| Net loss | $ | (4,812,797 | ) | $ | (1,893,377 | ) | $ | (8,009,283 | ) | $ | (4,313,262 | ) | ||||
| Denominator: | ||||||||||||||||
| Weighted average common shares outstanding | 11,838,371 | 8,576,634 | 11,233,755 | 8,503,521 | ||||||||||||
| Net loss per share data: | ||||||||||||||||
| Basic and Diluted | $ | (0.41 | ) | $ | (0.22 | ) | $ | (0.71 | ) | $ | (0.51 | ) | ||||
The following securities, rounded to the thousand, were not included in the diluted net loss per share calculation because their effect was anti-dilutive for the periods presented:
| Six Months Ended | ||||||||
| June 30, | ||||||||
| 2018 | 2017 | |||||||
| Common stock options | 439,000 | 455,000 | ||||||
| Common stock purchase warrants | 4,871,000 | 6,544,000 | ||||||
| Potentially dilutive securities | 5,310,000 | 6,999,000 | ||||||
Note 6: WARRANT LIABILITY AND FAIR VALUE MEASUREMENTS
A summary of quantitative information with respect to valuation methodology and significant unobservable inputs used for the Company’s common stock purchase warrants that are categorized within Level 3 of the fair value hierarchy for the six months ended June 30, 2018 and 2017 is as follows:
| June 30, | June 30, | |||||||
| 2018 | 2017 | |||||||
| Stock price | $ | 9.43 | $ | 3.88 | ||||
| Exercise price | $ | 8.67 | $ | 1.20 | ||||
| Contractual term (years) | 1.32 | 1.03 | ||||||
| Volatility (annual) | 83 | % | 78 | % | ||||
| Risk-free rate | 1 | % | 1 | % | ||||
| Dividend yield (per share) | 0 | % | 0 | % | ||||
The foregoing assumptions are reviewed quarterly and are subject to change based primarily on management’s assessment of the probability of the events described occurring. Accordingly, changes to these assessments could materially affect the valuations.
| 7 |
Financial Liabilities Measured at Fair Value on a Recurring Basis
Financial liabilities measured at fair value on a recurring basis are summarized below and disclosed on the balance sheet under Warrant liability:
| Fair value measured at June 30, 2018 | |||||||||||||||
| Quoted prices in active | Significant other | Significant | |||||||||||||
| markets | observable inputs | unobservable inputs | Fair value at | ||||||||||||
| (Level 1) | (Level 2) | (Level 3) | June 30, 2018 | ||||||||||||
| Warrant liability | $ | - | $ | - | $ | 147,000 | $ | 147,000 | |||||||
| Fair value measured at December 31, 2017 | |||||||||||||||
| Quoted prices in active | Significant other | Significant | |||||||||||||
| markets | observable inputs | unobservable inputs | Fair value at | ||||||||||||
| (Level 1) | (Level 2) | (Level 3) | December 31, 2017 | ||||||||||||
| Warrant liability | $ | - | $ | - | $ | 9,000 | $ | 9,000 | |||||||
The fair value accounting standards define fair value as the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is determined based upon assumptions that market participants would use in pricing an asset or liability. Fair value measurements are rated on a three-tier hierarchy as follows:
| · | Level 1 inputs: Quoted prices (unadjusted) for identical assets or liabilities in active markets; |
| · | Level 2 inputs: Inputs, other than quoted prices included in Level 1, that are observable either directly or indirectly; and |
| · | Level 3 inputs: Unobservable inputs for which there is little or no market data, which require the reporting entity to develop its own assumptions. |
There were no transfers between Level 1, 2 or 3 during the six months ended June 30, 2018.
The following table presents changes in Level 3 liabilities measured at fair value for the six months ended June 30, 2018:
| Warrant | ||||
| Liability | ||||
| Balance - December 31, 2017 | $ | 9,000 | ||
| Change in fair value of warrant liability | 138,000 | |||
| Balance – June 30, 2018 | $ | 147,000 | ||
Note 7: STOCKHOLDERS’ EQUITY
2018 Common Stock Transactions
Common Stock Purchase Agreement
On May 14, 2018, the Company’s largest stockholder Eastern Capital Limited entered into a Common Stock Purchase Agreement with the Company pursuant to which it purchased 1,300,000 shares of common stock at a price per share of $2.40 providing gross proceeds to the Company of $3.12 million.
Exercise and Repricing of Warrants Held by Existing Institutional Investors
On May 14, 2018, certain institutional holders of outstanding warrants entered into Warrant Exercise Agreements with the Company that provide for an amendment to the exercise price of the warrants being exercised at $2.50 per share. Upon closing of the Warrant Exercise Agreements, such institutional holders immediately exercised warrants for 782,505 shares of common stock providing aggregate proceeds to the Company of approximately $2.0 million.
The fair value relating to the modification of exercise prices on the repriced and exercised warrants was treated as deemed dividend on the statement of stockholders’ equity of $728,000.
A weighted average summary of quantitative information with respect to valuation methodology and significant unobservable inputs used for the Company’s common stock purchase warrants that are included in the modification is as follows:
| 8 |
| Weighted Average Inputs | ||||||||
| Before | After | |||||||
| Modification | Modification | |||||||
| Exercise price | $ | 9.93 | $ | 2.50 | ||||
| Contractual term (years) | 2.37 | 2.37 | ||||||
| Volatility (annual) | 79 | % | 79 | % | ||||
| Risk-free rate | 1.5 | % | 1.5 | % | ||||
| Dividend yield (per share) | 0 | % | 0 | % | ||||
Exercise of Stock Warrants
During June 2018, shareholders exercised 782,800 shares of common stock pursuant to stock warrants providing aggregate proceeds to the Company of approximately $2.3 million. 118,425 of the stock warrants exercised were exercised on a cashless basis, resulting in a cancellation of 83,130 stock warrants.
Exercise of Stock Options
In January 2018, 10,416 shares of common stock were issued pursuant to stock option exercises at an exercise price equal to $1.74 per share.
Consulting Arrangements
During the six months ended June 30, 2018, the Company issued 132,825 shares of common stock as part of consulting agreements. The fair value of the common stock of approximately $644,000 was recognized as stock-based compensation, $563,000 in general and administrative expenses and $81,000 in research and development expenses.
Share Purchase Warrants
A summary of the Company’s share purchase warrants as of June 30, 2018 and changes during the period is presented below:
| Weighted Average | ||||||||||||||||
| Number of | Weighted Average | Remaining Contractual | Total Intrinsic | |||||||||||||
| Warrants | Exercise Price | Life (in years) | Value | |||||||||||||
| Balance - January 1, 2018 | 6,520,000 | $ | 6.11 | 3.16 | $ | 1,739,000 | ||||||||||
| Issued | - | - | - | - | ||||||||||||
| Cashless exercised | (118,000 | ) | 4.01 | - | - | |||||||||||
| Exercised for cash | (1,447,000 | ) | 2.95 | - | - | |||||||||||
| Expired or Cancelled | (84,000 | ) | 4.01 | - | - | |||||||||||
| Balance - June 30, 2018 | 4,871,000 | $ | 5.94 | 2.76 | $ | 20,417,000 | ||||||||||
Note 8: STOCK-BASED COMPENSATION
The Company recorded approximately $960,000 and $271,000 of stock-based compensation expense for the three months ended June 30, 2018 and 2017, respectively. The Company recorded approximately $1,096,000 and $647,000 of stock-based compensation expense for the six months ended June 30, 2018 and 2017, respectively.
At June 30, 2018, the total stock-based compensation cost related to unvested awards not yet recognized was $159,000. The expected weighted average period compensation costs to be recognized was 0.48 years. Future option grants will impact the compensation expense recognized.
$596,000 and $364,000 of stock-based compensation expenses for the three months ended June 30, 2018 were included in general and administrative expenses and research and development expenses, respectively, on the condensed consolidated statements of operations.
$629,000 and $467,000 of stock-based compensation expenses for the six months ended June 30, 2018 were included in general and administrative expenses and research and development expenses, respectively, on the condensed consolidated statements of operations.
| 9 |
Note 9: grant income
During the six months ended June 30, 2018, the Company received $0.2 million of a grant awarded to Mayo Foundation from the U.S. Department of Defense for the Phase II Clinical Trial of TPIV200. The grant compensated the Company for clinical supplies manufactured and provided by the Company for the clinical study. In accordance with Accounting Standards Update No. 2014-09, “Revenue from Contracts with Customers (Topic 606)” issued by the Financial Accounting Standards Board, the Company recorded the $0.2 million of grant income as revenue.
| 10 |
| Item 2. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
This quarterly report on Form 10-Q contains forward-looking statements within the meaning of Section 21E of the Securities and Exchange Act of 1934, as amended, that involve risks and uncertainties. All statements other than statements relating to historical matters including statements to the effect that we “believe”, “expect”, “anticipate”, “plan”, “target”, “intend” and similar expressions should be considered forward-looking statements. Our actual results could differ materially from those discussed in the forward-looking statements as a result of a number of important factors, including factors discussed in this section and elsewhere in this quarterly report on Form 10-Q, and the risks discussed in our other filings with the Securities and Exchange Commission. Readers are cautioned not to place undue reliance on these forward-looking statements, which reflect management’s analysis, judgment, belief or expectation only as the date hereof. We assume no obligation to update these forward-looking statements to reflect events or circumstance that arise after the date hereof.
As used in this quarterly report: (i) the terms “we”, “us”, “our”, “TapImmune” and the “Company” mean TapImmune Inc. and its wholly owned subsidiary, GeneMax Pharmaceuticals Inc. which wholly owns GeneMax Pharmaceuticals Canada Inc., unless the context otherwise requires; (ii) “SEC” refers to the Securities and Exchange Commission; (iii) “Securities Act” refers to the Securities Act of 1933, as amended; (iv) “Exchange Act” refers to the Securities Exchange Act of 1934, as amended; and (v) all dollar amounts refer to United States dollars unless otherwise indicated.
The following should be read in conjunction with our unaudited condensed consolidated interim financial statements and related notes for the three and six months ended June 30, 2018 included in this quarterly report, as well as our Annual Report on Form 10-K for the year ended December 31, 2017 filed on March 23, 2018.
Company Overview
We are a clinical-stage immuno-oncology company specializing in the development of innovative peptide and gene-based immunotherapeutics and vaccines for the treatment of cancer and metastatic disease. We are actively advancing our clinical programs by expanding our Folate Receptor Alpha program (TPIV200) for breast and ovarian cancers and our HER2/neu peptide antigen program (TPIV110) in Phase II clinical trials. In parallel, we are developing a proprietary DNA expression technology named PolyStart™ to improve the ability of the cellular immune system to recognize and destroy diseased cells. We plan to complete the pre-clinical development of our PolyStart™ vaccine and move it into the clinic as an integral component of a prime-boost vaccine methodology.
We are a leader in the development of immunotherapies for women's cancers, with multiple Phase 2 and Phase 1b/2 clinical studies for the treatment of ovarian and breast cancer. The company's peptide or nucleic acid-based immunotherapeutic products comprise one or multiple naturally processed epitopes (NPEs) designed to comprehensively stimulate a patient's killer T-cells and helper T-cells, and to restore or further augment antigen presentation by using proprietary nucleic acid-based expression systems. Our technologies may be used as stand-alone medications or in combination with current treatment modalities.
Immuno-oncology has become the most rapidly growing sector in the pharmaceutical and biotech industry. The approval and success of checkpoint inhibitors, including ipilimumab and nivolumab (Yervoy® and Opdivo®, respectively, Bristol Myers Squibb), pembrolizumab (Keytruda®, Merck & Co.), avelumab (Bavencio®, EMD Serono), durvalumab (Imfinzi™, AstraZeneca), and atezolizumab (Tecentriq®, Genentech), together with the development and approval of CAR T-cell therapies sponsored by Novartis, Juno Therapeutics, and Kite Pharma, has provided much momentum in this sector. In addition, new evidence points to the increasing use of combination immunotherapies for the treatment of cancer. This has provided greater justification and opportunities for the successful development of T-cell vaccines in combination with other approaches.
On May 23, 2017, the U.S. Food and Drug Administration (“FDA”) approved expanded use of Keytruda for immunotherapy. The FDA granted accelerated approval to a treatment for patients whose cancers have a specific genetic feature (biomarker). This is the first time the FDA has approved a cancer treatment based on a common biomarker rather than the location in the body where the tumor originated.
We believe the strength of our science and development approaches is becoming more widely appreciated, particularly as our clinical program has now generated positive Phase I data using our two products in clinical programs in breast and ovarian cancers.
| 11 |
We continue to focus primarily on our Phase II triple-negative breast cancer trials using TPIV200 (which has achieved Fast Track and Orphan Drug Status), and are planning for the next Phase II HER2/neu breast cancer trial.
We expect to continue to prosecute our PolyStart™ patent filings and develop new PolyStart™ constructs to facilitate collaborative efforts in our current clinical indications. We will also evaluate those indications where others have already indicated interest in combination therapies.
We believe that these fundamental programs and corporate activities have positioned our company to capitalize on the acceptance of immunotherapy as a leading therapeutic strategy in cancer and infectious diseases.
We are continuously working on improving our product formulation and supply. TPIV200 and TPIV110 are both off-the-shelf, lyophilized products that only require reconstitution and mixing with GM-CSF at the clinical site before injection. We believe our off-the-shelf product may provide a significant competitive advantage over autologous products that require preparation for each patient. We also believe the investments we have made in the formulation work for both very stable products will result in commercially viable products consistent with typically high pharmaceutical profit margins.
The Phase I data produced for both TPIV200 and TPIV100 in collaboration with the Mayo Clinic are the driving force behind the high-value collaborations we have established and maintained with organizations such as Mayo Clinic, AstraZeneca, Memorial Sloan Kettering, and the U.S. Department of Defense. As we move forward into advanced Phase II studies, some of which incorporate collaborations with prestigious third-party organizations, we believe they will represent further independent validation of the potential of our technology.
Recent Developments
Merger Agreement
On May 15, 2018, we and our wholly owned subsidiary, (formed for purposes of the Merger) Timberwolf Merger Sub, Inc., a Delaware corporation (“Merger Sub”), and Marker Therapeutics, Inc., a Delaware corporation (“Marker”), entered into an Agreement and Plan of Merger and Reorganization (the “Merger Agreement”). Subject to the terms and conditions set forth in the Merger Agreement, MergerSub will merge with and into Marker (the “Merger”), with Marker surviving the Merger as a wholly owned subsidiary of TapImmune (the “Surviving Corporation”).
At the effective time of the Merger (the “Effective Time”), each outstanding share of Marker’s common stock will be converted into the right to receive (i) shares of TapImmune’s common stock, par value $0.001 per share (“TapImmune Common Stock”), in an amount equal to the exchange ratio calculated pursuant to the Merger Agreement (the “Stock Exchange Ratio”), and (ii) warrants to purchase TapImmune Common Stock, in an amount equal to the exchange ratio calculated pursuant to the Merger Agreement (the “Warrant Exchange Ratio”).
The Merger Agreement contains customary representations, warranties and covenants made by us and Marker, including covenants relating to obtaining the requisite approvals of the stockholders of TapImmune and Marker, indemnification of directors and officers, and TapImmune’s and Marker’s conduct of their respective businesses between the date of signing of the Merger Agreement and the closing of the Merger.
The issuance of TapImmune Common Stock and other transactions contemplated by the Merger Agreement are subject to approval by TapImmune’s stockholders. The Merger is subject to other customary closing conditions, including, among other things, the accuracy of the representations and warranties, subject generally to an overall material adverse effect qualification, compliance by the parties with their respective covenants and no existence of any law or order preventing the Merger and related transactions.
| 12 |
The Merger Agreement contains certain termination rights for both us and Marker and provides for the payment of a termination fee of $1,500,000 by us to Marker upon termination of the Merger Agreement under specified circumstances. In connection with a termination of the Merger Agreement under specified circumstances involving competing transactions, a willful, intentional and material breach of the non-solicitation obligations by us, a change in our board of directors’ recommendation of the Merger to the stockholders or other triggering events, we may be required to pay Marker reimbursement for certain fees and expenses up to $500,000. In connection with a termination of the Merger Agreement under specified circumstances involving the failure of Marker stockholders to approve the Merger Agreement within 24 hours of signing the Merger Agreement, intentional and material breach of the non-solicitation obligations by Marker or other triggering events, Marker may be required to pay our reimbursement for certain fees and expenses up to $500,000. The Merger Agreement may also be terminated by either us or Marker if the merger has not been consummated by September 15, 2018, subject to an extension of an additional 60 days if our proxy statement is being reviewed or commented upon by the SEC.
Following the Merger, the board of directors of the Company will consist of six directors and will be comprised of (i) three members designated by Marker, and (ii) three members designated by us.
Common Stock Purchase Agreement
On May 18, 2018, we closed on the sale of 1,300,000 shares of common stock for $2.40 per share pursuant to a Common Stock Purchase Agreement with an existing accredited investor in a private placement under Rule 506 of Regulation D. Aggregate gross proceeds were approximately $3.1 million.
Exercise of Warrants Held by Existing Institutional Investors
Also on May 18, 2018, we and certain existing institutional investors, who are holders of various warrants to purchase shares of TapImmune common stock, closed on Warrant Exercise Agreements in which TapImmune agreed to reduce the exercise price for a portion of the investors’ previously purchased Series C, Series D, Series E and Series F warrants from $6.00, $9.00, $15.00 and $7.20, respectively per share to $2.50 per share, provided that the investors exercise such warrants for cash immediately, which they did, for 782,506 shares and aggregate proceeds of approximately $2.0 million. The shares of common stock underlying the exercised warrants are registered for resale under the Form S-3 Registration Statement (File no. 333-220538) declared effective by the SEC on December 29, 2017.
Private Placement
On June 8, 2018, in connection with, and in furtherance of, the Merger Agreement, we entered into Securities Purchase Agreements for a private placement with a select group of institutional and accredited investors (the “Purchasers”). Pursuant to the Securities Purchase Agreements, the Purchasers have agreed to purchase 17,500,000 shares of the Company’s common stock, par value $0.001, at $4.00 per share, for gross offering proceeds of $70 million. Each share of common stock will be issued with a warrant to purchase 0.75 additional shares of the Company’s common stock at an exercise price of $5.00 per share for an aggregate of 13,125,000 Warrants. In accordance with NASDAQ Stock Market Rule 5635, the completion of the issuance and sale of the common stock and Warrants pursuant to the Securities Purchase Agreements is subject to the approval of the private placement by the Company’s stockholders. The Warrants will be immediately exercisable upon issuance at closing and will have a term of five years. Subject to obtaining shareholder approval of the private placement, the issuance and sale of the common stock and Warrants pursuant to the Securities Purchase Agreements is expected to close concurrently with our merger with Marker.
Intellectual Property Strategies
A key component to success is having a comprehensive patent strategy that continually updates and extends patent coverage for key products. It is highly unlikely that early patents will extend through ultimate product marketing, so extending patent life is an important strategy for ensuring product protection.
We have three active patent families that we are supporting:
1. Filed patents on the PolyStart™ expression vector (owned by TapImmune and filed in 2014: this IP covers the use with TAP). We announced the allowance of this patent in February 2016.
2. Filed patents on HER2/neu Class II and Class I antigens: exclusive license from Mayo Foundation; and
3. Filed patents on Folate Receptor Alpha antigens: exclusive license from Mayo Foundation.
While doing the studies on the path to successful product development takes time, we believe we have put together a team that can deliver the highest quality data in the least amount of time. The strength of our product pipeline and access to leading scientists and institutions gives us a unique opportunity to make a major contribution to global health care.
| 13 |
With respect to the broader market, a major driver and positive influence on our activities has been the emergence and general acceptance of the potential of a new generation of immunotherapies that promise to change the standard of care for cancer. The immunotherapy sector has been greatly stimulated by the approval of Provenge® for prostate cancer and Yervoy™ for metastatic melanoma, progression of the areas of immune checkpoint inhibitors and adoptive T-cell therapy, as well as multiple other approaches reaching Phase II and Phase III status.
We believe that through our combination of technologies, we are well positioned to be a leading player in this emerging market. It is important to note that many of the late-stage immunotherapies currently in development do not represent competition to our programs, but instead offer synergistic opportunities to partner our antigen-based immunotherapeutics and the PolyStart™ expression system. Thus, the use of naturally processed T-cell antigens discovered using samples derived from cancer patients, plus our PolyStart™ expression technology to improve antigen presentation to T-cells, could not only produce an effective cancer vaccine in its own right, but could also enhance the efficacy of other immunotherapy approaches such as CAR-T and checkpoint inhibitors.
| 14 |
Products and Technology in Development-Clinical
TPIV200
Phase I Human Clinical Trials – Folate Alpha Breast and Ovarian Cancers – Mayo Clinic
Folate Receptor Alpha (“FRa”) is overexpressed in over 80% of breast cancers and in addition, over 90% of ovarian cancers, for which the only treatment options are surgery, radiation therapy and chemotherapy, leaving a very important and urgent clinical need for a new therapeutic. Time to recurrence is relatively short for ovarian cancer and survival prognosis is extremely poor after recurrence. In the United States alone, there are approximately 30,000 ovarian cancer patients and 40,000 triple-negative breast cancer patients newly diagnosed every year.
We have completed a 21-patient Phase I clinical trial for the FRa vaccine. Twenty-one patients with breast or ovarian cancer, who had undergone standard surgery and adjuvant treatment, were treated with one cycle of cyclophosphamide. Following this, patients were vaccinated intradermally with a mixture of the five FRa peptides adjuvanted with GM-CSF (now called TPIV200) on day one of a 28-day cycle for a maximum of six vaccination cycles. The vaccine was well-tolerated and safe and 20 out of 21 evaluable patients showed positive immune responses, providing a strong rationale for progressing to Phase II trials. Further, the data showed that 16 out of 16 patients in the observation stage still showed immune responses (Source: published online 15Mar2018; DOI: 10.1158/1078-0432.CCR-17-2499). We have developed a commercial quality lyophilized formulation of the peptides in a single vial for reconstitution and injection. Good Manufacturing Practice (“GMP”) manufacturing for the Phase II trials has been completed.
On July 27, 2015, we exercised our option agreement with Mayo Foundation with the signing of a worldwide exclusive license agreement to commercialize a proprietary Folate Receptor Alpha vaccine technology for all cancer indications. As part of this agreement, the IND from the Folate Receptor Alpha Phase I Trial was transferred from Mayo Foundation to us for amendment for Phase II Clinical Trials on our lead product.
On September 15, 2015, we announced that our collaborators at the Mayo Foundation had been awarded a grant of $13.3 million from the U.S. Department of Defense. This grant, commencing September 15, 2015, covers the costs for a 280-patient Phase II Clinical Trial of Folate Receptor Alpha Vaccine in patients with triple-negative breast cancer. We are working closely with Mayo Foundation on this clinical trial by providing clinical and manufacturing expertise, as well as providing GMP vaccine formulations. These vaccine formulations are being developed for multiple Phase II clinical programs in triple-negative breast and ovarian cancer in combination with other immunotherapeutics. This Phase II study of TPIV200 in the treatment of triple-negative breast cancer began enrolling patients in late 2017.
On December 9, 2015, we announced that we received Orphan Drug Designation from the U.S. Food & Drug Administration’s Office of Orphan Products Development (“OOPD”) for our cancer vaccine TPIV200 in the treatment of ovarian cancer. The TPIV200 ovarian cancer clinical program will now receive benefits including tax credits on clinical research and seven-year market exclusivity upon receiving marketing approval. TPIV200 is a multi-epitope peptide vaccine that targets Folate Receptor Alpha which is overexpressed in multiple cancers including over 90% of ovarian cancer cells.
On February 3, 2016, we announced that the U.S. FDA designated the investigation of multiple-epitope FRa Vaccine (TPIV200) for maintenance therapy in subjects with platinum-sensitive advanced ovarian cancer who achieved stable disease or partial response following completion of standard-of-care chemotherapy, as a Fast Track Development Program. We began enrolling a Phase II study in this indication in 2017.
We have opened multiple clinical sites and have completed enrollment of patients in a Phase II trial of our Folate Receptor Alpha cancer vaccine, TPIV200, in the treatment of triple-negative breast cancer, one of the most difficult-to-treat cancers representing a clear unmet medical need. The open-label, 80-patient clinical trial is designed to evaluate dosing regimens, efficacy, and immune responses in women with triple-negative breast cancer and is fully enrolled. Key data from the trial is expected to be included in a future New Drug Application submission to the FDA for marketing clearance. This trial is sponsored and conducted by TapImmune.
| 15 |
On April 21, 2016, we announced our participation in an ovarian cancer study sponsored by Memorial Sloan Kettering Cancer Center in New York City in collaboration with AstraZeneca Pharmaceuticals in ovarian cancer patients who are not responsive to platinum, a commonly used chemotherapy for ovarian cancer. This study, a Phase II study of TPIV200 is currently enrolling ovarian cancer patients and is designed to look at the effects of combination therapy with AstraZeneca’s checkpoint inhibitor durvalumab. The study will enroll 40 patients and is open-label. Because they are unresponsive to platinum, these patients have no real options left. If the combination therapy proves effective, we believe it would address a critical unmet need. TPIV200 has received Orphan Drug designation for use in the treatment of ovarian cancer. Although we have no business relationship with AstraZeneca, we are paying for one-half of the costs of the clinical study, in addition to providing our TPIV200 for the study.
A Company-sponsored Phase II study in platinum-sensitive ovarian cancer patients was initiated in 2017. This study is designed to evaluate TPIV200 with GM-CSF in a randomized, placebo-controlled fashion during the first maintenance period after primary surgery and chemotherapy. Patients at this stage of their treatment have the highest potential for an immunotherapeutic effect and no other approved treatment options. The study will enroll up to 120 patients over the next year and a half, with an interim analysis planned in the first half of 2019.
TPIV 100/110
Phase I Human Clinical Trials – HER2/neu+ Breast Cancer – Mayo Clinic
A Phase I study using TPIV 100 (four HER2/neu peptides adjuvanted with GM-CSF) was completed in 2015. Final safety analysis on all the patients treated is complete and shown to be safe. In addition, 19 out of 20 evaluable patients showed robust T-cell immune responses to the antigens in the vaccine composition providing a solid case for advancement to Phase II in 2017. An additional secondary endpoint incorporated into this Phase I Trial will be a two-year follow on recording time to disease recurrence in the participating breast cancer patients.
For Phase I(b)/II studies, we plan to add a Class I peptide, licensed from the Mayo Clinic (April 16, 2012), to the four Class II peptides, producing TPIV 110 (five peptide product). Management believes that the combination of Class I and Class II HER2/neu antigens, gives us the leading HER2/neu vaccine platform. We are amending the IND to incorporate the fifth peptide in the Phase I(b)/II study. Discussions with the FDA have resulted in a pre-clinical development project that should allow us to file the amended IND in mid-2018.
Products and Technology-Pre-clinical
Polystart
On February 7, 2017, we announced that we received a Notice of Allowance from the U.S. Patent and Trademark Office of our patent application titled, “Chimeric nucleic acid molecules with non-AUG initiation sequences and uses thereof,” which represents our first patent on our Polystart program. We anticipate additional patent filings in connection with our research and development in this area. We plan to develop Polystart as both a stand-alone therapy and as a ‘boost strategy’ to be used synergistically with our peptide-based vaccines for breast and ovarian cancers.
| 16 |
TapImmune’s Clinical Program Pipeline
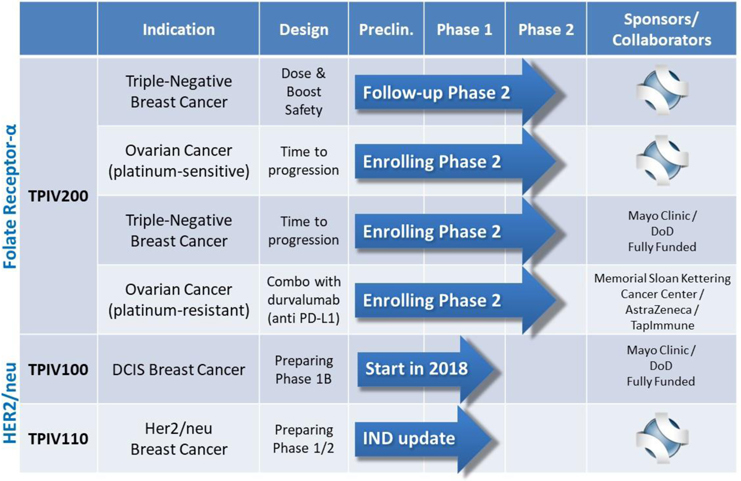
Refer to the “Clinical Program Pipeline Status Updates” section below for latest updates on above clinical pipeline chart.
In addition to the exciting clinical developments, our peptide vaccine technology may be coupled with our recently developed in-house PolyStart™ nucleic acid-based technology, which is designed to make vaccines significantly more effective by producing four times the required peptides for the immune systems to recognize and act on.
Clinical Program Pipeline Status Updates
Completed GMP Manufacturing Scale Up and Second Clinical Lot of TPIV200; to Supply Additional Phase II Clinical Trials
We successfully completed a multi-gram production scale-up as well as GMP manufacturing of a second clinical lot of TPIV200. The vaccine supply will be used in the company’s ongoing Phase II study in platinum-sensitive ovarian cancer, as well as the planned 280-patient Phase II study sponsored by the Mayo Clinic and funded by the U.S. Department of Defense for treating triple-negative breast cancer. We also made various improvements to the vaccine manufacturing process, resulting in, what we believe to be, a superior formulation of the vaccine that is more amenable to large-scale manufacturing and commercialization.
Announcement of Publication of Clinical Trial Results for the TPIV200 Cancer Vaccine in Clinical Cancer Research
On March 15, 2018, we announced the publication of clinical data from a Phase I trial of TPIV200, our multi-epitope T-cell vaccine targeting Folate Receptor Alpha (“FRa”) in patients with ovarian and breast cancer. The results show that TPIV200 vaccination was well tolerated by all patients and over 90% developed robust and durable antigen-specific immune responses against FRa without regard for HLA type, which aligns with the intended mechanism of action of the vaccine.
| 17 |
Enrollment Completed: Phase II TPIV200 Trial in Triple-Negative Breast Cancer
We have completed enrollment and are now treating and following the patients in a Phase II trial of our Folate Receptor Alpha cancer vaccine, TPIV200, in the treatment of triple-negative breast cancer, one of the most difficult cancers to treat, representing a clear unmet medical need. The open-label, 80-patient clinical trial is designed to evaluate dosing regimens, pre-treatment, efficacy, and immune responses in women with triple-negative breast cancer. Key data from the trial is expected to be included in a future Biologics License Application submission to the FDA for marketing clearance. This trial is sponsored and conducted by TapImmune.
An independent Data Safety Monitoring Board (DSMB) reviews the safety every quarter in this ongoing Phase II study enrolling women with stage I-III triple-negative breast cancer who have completed initial surgery and chemo/radiation therapy. The randomized four-arm study is evaluating two doses of TPIV200 (a high dose and a low dose), each of which will be tested both with and without immune priming with cyclophosphamide prior to vaccination. Safety reviews are conducted quarterly and have shown no safety issues. The study completed enrollment at the end of 2017, with interim data expected in mid-2018. Details regarding this trial can be found at www.clinicaltrials.gov under identifier numbers NCT02593227 and FRV-002.
Enrolling Patients: Phase II TPIV200 Trial in Platinum-Sensitive Ovarian Cancer
We have opened multiple clinical sites and have enrolled half of the patients in a Phase II trial of TPIV200 for a 120-patient study on ovarian cancer patients who are responsive to platinum. We have received the FDA’s Fast Track designation to develop TPIV200 as a maintenance in women with Stage III and IV ovarian cancer who are in remission following their first round of successful platinum-based chemotherapy. This multi-center, double-blind efficacy study is sponsored and conducted by TapImmune. We expect to complete enrollment mid-2019. An interim analysis is planned based upon 50% patient progression, which we anticipate completing in the first half of 2019. Details regarding this trial can be found at www.clinicaltrials.gov under identifier numbers NCT02978222 and FRV-004. TPIV200 has also received Orphan Drug designation for use in the treatment of ovarian cancer.
Enrolling Patients: Phase II Mayo Clinic-U.S. DOD Trial of TPIV200 in Triple-Negative Breast Cancer
Patients are being enrolled in this Phase II study of TPIV200 in the treatment of triple-negative breast cancer, conducted by the Mayo Clinic and sponsored by the U.S. DOD. The 280-patient study is led by Dr. Keith Knutson of the Mayo Clinic in Jacksonville, Florida. Dr. Knutson is the inventor of the technology and a member of the Scientific Advisory Board at TapImmune. While we are supplying doses of TPIV200 for the trial and being reimbursed for the costs associated with manufacturing, the costs associated with conducting this study are being funded by a $13.3 million grant made by the DOD to the Mayo Clinic.
Enrolling Patients: Phase II Trial at Memorial Sloan Kettering of TPIV200 in Platinum-Resistant Ovarian Cancer
A Phase II study of TPIV200 in ovarian cancer patients who are not responsive to platinum, a commonly used chemotherapy for ovarian cancer, being sponsored by Memorial Sloan Kettering Cancer Center (“MSKCC”) in collaboration with AstraZeneca and TapImmune, has begun enrollment for a 40-patient study. The open-label study is designed to evaluate a combination therapy which includes our TPIV200 T-cell vaccine and AstraZeneca’s checkpoint inhibitor, durvalumab. Because they are unresponsive to platinum, these patients have no real remaining options. If the combination therapy proves effective, we believe it would address a critical unmet need. We successfully completed enrollment of the first safety cohort. This may enable MSKCC to increase the number of patients that can be enrolled and will subsequently increase the study’s enrollment rate. Currently more than 50% of patients have been enrolled. An interim analysis is planned in the second half of 2018. Details regarding this trial can be found at www.clinicaltrials.gov under identifier numbers NCT02764333.
Open IND with FDA for TPIV110 in 2018: Phase II Protocol Now in Preparation
We have enhanced the formulation of our second cancer vaccine product, TPIV110 (the five-peptide product), following very strong safety and immune responses from a Phase I Mayo Clinic study using TPIV100 (the four-peptide product). TPIV110 targets HER2/neu+, which makes it applicable to breast, ovarian, and colorectal cancers. The enhanced TPIV product adds a fifth antigen that should produce an even more robust immune response activating both CD4+ (helper) and CD8+ (killer) T-cells. We have participated in a pre-Investigational New Drug (“pre-IND”) meeting with the FDA and will file the amended IND containing the fifth peptide in mid-2018.
Mayo Clinic to Vaccinate Women With Ductal Carcinoma In Situ (DCIS) Using TapImmune TPIV100 HER2-targeted T-Cell Vaccine
On March 14, 2017, we announced that our partners at the Mayo Clinic received a grant from the U.S. Department of Defense to conduct a Phase IB study of our HER2-targeted vaccine candidate TPIV100 in an early form of breast cancer called DCIS. This is the second TapImmune vaccine to be tested in a fully funded study sponsored by the Mayo Clinic. Our collaborators at Mayo Clinic announced a $3.8 million grant which we believe would fully fund this trial. If the study is successful, our vaccine may eventually augment or even replace standard surgery and chemotherapy, and potentially could become part of a routine immunization schedule for preventing breast cancer in healthy women. The study is expected to enroll 40-45 women with DCIS and begin to commence such enrollment in mid-2018.
| 18 |
Results of Operations
In this discussion of the Company’s results of operations and financial condition, amounts, other than per-share amounts, have been rounded to the nearest thousand dollars.
Three Months Ended June 30, 2018 Compared to Three Months Ended June 30, 2017
We recorded a net loss of $4.8 million or ($0.41) basic and diluted per share during the three months ended June 30, 2018 compared to a net loss of $1.9 million or ($0.22) basic and diluted per share during the three months ended June 30, 2017. The change in net loss period over period was due to the following changes:
Revenue
Grant income
During the three months ended June 30, 2018, we received $206,000 of a grant awarded to Mayo Foundation from the US Department of Defense for the Phase II Clinical Trial of TPIV200. The grant compensated us for clinical supplies manufactured by us and provided for the clinical study.
Operating Expenses
Operating expenses incurred during the three months ended June 30, 2018 were $4.9 million compared to $2.4 million in the prior period. Significant changes in operating expenses are outlined as follows:
| · | Research and development costs during the three months ended June 30, 2018 were $1.8 million compared to $1.2 million during the prior year period. The three months ended June 30, 2018 had increased expenses from the prior period relating to our clinical trials. |
| · | General and administrative expenses increased to $3.1 million during the three months ended June 30, 2018 from $1.2 million during the prior year period. This was due to increased expenses relating to: |
| o | stock-based compensation for employees and outside consultants, |
| o | compensation expenses resulting from increased headcount, |
| o | expenses relating to the announced and proposed merger agreement, |
| o | investor relations expenses, and |
| o | increased legal, audit and other professional fees. |
Other Expense
Change in fair value of warrant liabilities
The change in fair value of warrant liabilities for the three months ended June 30, 2018 was $139,000 as compared to ($8,000) for the three months ended June 30, 2017. This increase by $139,000 for the three months ended June 30, 2018 is reflected by a corresponding expense in the condensed consolidated statement of operations.
Debt extinguishment gain
In 2003 we entered into a license agreement with a foreign based third-party for certain adenovirus technology. The license agreement was amended several times between inception and 2008 at which time it was amended and restated and had a fixed three-year term expiring in 2011. During such time, we did not pursue the technology and have not undertaken further work in the area covered by the technology license. Neither we nor the third-party took further actions under or pursuant to the license agreement. We carried a historical accrual of approximately $0.5 million under the amended license agreement related to certain obligations provided for in the license agreement. The license agreement was governed by the laws of a foreign jurisdiction. We sought and obtained legal advice related to such accrued obligations under the expired license agreement. We relied upon a judicial conclusion, as opined upon by outside legal counsel in the applicable foreign jurisdiction, that a court in such foreign jurisdiction would grant relief releasing us from liability under the license agreement, and in accordance with Accounting Standards Codification 405 “Extinguishment of Liabilities”, we recorded a debt extinguishment gain of $0.5 million and reduced the liability amount owed to $0 during the three months ended June 30, 2017.
| 19 |
Six Months Ended June 30, 2018 Compared to Six Months Ended June 30, 2017
We recorded a net loss of $8.0 million or ($0.71) basic and diluted per share during the six months ended June 30, 2018 compared to a net loss of $4.3 million or ($0.51) basic and diluted per share during the six months ended June 30, 2017. The change in net loss period over period was due to the following:
Revenue
Grant income
During the six months ended June 30, 2018, we received $206,000 of a grant awarded to Mayo Foundation from the US Department of Defense for the Phase II Clinical Trial of TPIV200. The grant compensated us for clinical supplies manufactured by us and provided for the clinical study.
Operating Expenses
Operating expenses incurred during the six months ended June 30, 2018 were $8.1 million compared to $4.8 million in the prior period. Significant changes in operating expenses are outlined as follows:
| · | Research and development costs during the six months ended June 30, 2018 were $3.4 million compared to $2.2 million during the prior year period. The six months ended June 30, 2018 had increased expenses from the prior period relating to our clinical trials. |
| · | General and administrative expenses increased to $4.7 million during the six months ended June 30, 2018 from $2.6 million during the prior year period. This was due to increased expenses relating to: |
| o | stock-based compensation for employees and outside consultants, |
| o | compensation expenses resulting from increased headcount, |
| o | expenses relating to the announced and proposed merger agreement, |
| o | investor relations expenses, and |
| o | increased legal, audit and other professional fees. |
Other Expense
Change in fair value of warrant liabilities
The change in fair value of warrant liabilities for the six months ended June 30, 2018 was $138,000 as compared to ($5,000) for the six months ended June 30, 2017. This increase by $138,000 for the six months ended June 30, 2018 is reflected by a corresponding expense in the condensed consolidated statement of operations.
Debt extinguishment gain
In 2003 we entered into a license agreement with a foreign based third-party for certain adenovirus technology. The license agreement was amended several times between inception and 2008 at which time it was amended and restated and had a fixed three-year term expiring in 2011. During such time, we did not pursue the technology and have not undertaken further work in the area covered by the technology license. Neither we nor the third-party took further actions under or pursuant to the license agreement. We carried a historical accrual of approximately $0.5 million under the amended license agreement related to certain obligations provided for in the license agreement. The license agreement was governed by the laws of a foreign jurisdiction. We sought and obtained legal advice related to such accrued obligations under the expired license agreement. We relied upon a judicial conclusion, as opined upon by outside legal counsel in the applicable foreign jurisdiction, that a court in such foreign jurisdiction would grant relief releasing us from liability under the license agreement, and in accordance with Accounting Standards Codification 405 “Extinguishment of Liabilities”, we recorded a debt extinguishment gain of $0.5 million and reduced the liability amount owed to $0 during the six months ended June 30, 2017.
| 20 |
Liquidity and Capital Resources
We have not generated any revenues since inception. We have financed our operations primarily through public and private offerings of our stock and debt including warrants and the exercises thereof.
The following table sets forth our cash and working capital as of June 30, 2018 and December 31, 2017:
| June 30, | December 31, | |||||||
| 2018 | 2017 | |||||||
| Cash | $ | 7,782,000 | $ | 5,129,000 | ||||
| Working Capital | $ | 4,144,000 | $ | 3,658,000 | ||||
Cash Flows
The following table summarizes our cash flows for the six months ended June 30, 2018 and 2017:
| Six Months Ended | ||||||||
| June 30, | ||||||||
| 2018 | 2017 | |||||||
| Net Cash provided by (used in): | ||||||||
| Operating activities | $ | (4,746,000 | ) | $ | (3,936,000 | ) | ||
| Financing activities | $ | 7,399,000 | $ | 6,047,000 | ||||
| Net increase in cash | $ | 2,653,000 | $ | 2,111,000 | ||||
| 21 |
Financings
Our financing activities during the six months ended June 30, 2018 were as follows:
Common Stock Purchase Agreement
On May 14, 2018, the Company’s largest stockholder Eastern Capital Limited entered into a Common Stock Purchase Agreement with the Company pursuant to which it purchased 1,300,000 shares of common stock at a price per share of $2.40 providing gross proceeds to the Company of $3.12 million.
Exercise and Repricing of Warrants Held by Existing Institutional Investors
On May 14, 2018, certain institutional holders of outstanding warrants entered into Warrant Exercise Agreements with the Company that provide for an amendment to the exercise price of the warrants being exercised at $2.50 per share. Upon closing of the Warrant Exercise Agreements, such institutional holders immediately exercised warrants for 782,505 shares of common stock providing aggregate proceeds to the Company of approximately $2.0 million.
The fair value relating to the modification of exercise prices on the repriced and exercised warrants was treated as deemed dividend on the statement of stockholders’ equity of $728,000.
Exercise of Stock Warrants
During June 2018, shareholders exercised warrants and acquired 782,800 shares of common stock providing aggregate proceeds to the Company of approximately $2.3 million. 118,425 of the stock warrants exercised were exercised on a cashless basis, resulting in a cancellation of 83,130 stock warrants.
Exercise of Stock Options
In January 2018, a former officer exercised 10,416 shares of common stock pursuant to stock options providing proceeds of $18,000.
Future Capital Requirements
As of June 30, 2018, we had working capital of $4.1 million, compared to working capital of $3.7 million as of December 31, 2017.
The discussion below excludes the closing of the proposed merger with Marker Therapeutics and the concurrent closing of the $70 million private placement.
We expect our expenses to continue at a similar pace through 2018 primarily to continue funding our in-process Phase II clinical trials. Two of our clinical studies are expected to be funded by a total of $17.1 million of grants made by the DOD to the Mayo Clinic. Our collaborators at Mayo Clinic announced a $3.8 million grant which we expect would fully fund a Phase II clinical trial in DCIS that we had planned for our HER2/neu+ vaccine.
Our capital requirements for 2018 and beyond will depend on numerous factors, including the success of our research and development, the resources we devote to develop and support our technologies and our success in pursuing strategic licensing and funded product development collaborations with external partners as well as other strategic initiatives we may determine to pursue. Subject to our ability to raise additional capital, we expect to incur substantial expenditures to further develop our technologies including continued increases in costs related to research, nonclinical testing and clinical studies and trials, as well as costs associated with our capital raising efforts and being a public company.
We believe our existing cash will fund our operations into the first quarter of fiscal 2019. We will require substantial additional capital to conduct research and development, to fund nonclinical testing and Phase II clinical trials of our licensed, patented technologies, and to begin cultivating collaborative relationships for the Phase II and future Phase III clinical testing. Our plans could include seeking both equity and debt financing, alliances or other partnership agreements with entities interested in our technologies, or other business transactions that could generate sufficient resources to ensure continuation of our operations and research and development programs.
We expect to continue to seek additional funding for our operations. Any such required additional capital may not be available on reasonable terms, if at all. If we were unable to obtain additional financing, we may be required to reduce the scope of, delay or eliminate some or all of our planned clinical testing and research and development activities, which could harm our business. The sale of additional equity or debt securities may result in additional dilution to our shareholders. If we raise additional funds through the issuance of debt securities or preferred stock, these securities could have rights senior to those holders of our common stock and could contain covenants that could restrict our operations. We also will require additional capital beyond our currently forecasted amounts.
| 22 |
Because of the numerous risks and uncertainties associated with research, development and commercialization of our product candidates, we are unable to estimate the exact amounts of our future working capital requirements. Our future funding requirements will depend on many factors, including, but not limited to:
| · | the number and characteristics of the product candidates we pursue; |
| · | the scope, progress, results and costs of researching and developing our product candidates, and conducting pre-clinical and clinical trials including the research and development expenditures we expect to make in connection with our license agreements with Mayo Foundation; |
| · | the amount and timing of transaction expenses we incur in connection with the pending Marker merger agreement; |
| · | strategic transactions we may undertake; |
| · | the timing of, and the costs involved in, obtaining regulatory approvals for our product candidates; |
| · | our ability to maintain current research and development licensing agreements and to establish new strategic partnerships and collaborations, licensing or other arrangements and the financial terms of such agreements; |
| · | our ability to achieve our milestones under our licensing arrangements and the payment obligations we may have under such agreements; |
| · | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims, including litigation costs and the outcome of such litigation; and |
| · | the timing, receipt and amount of sales of, or royalties on, our future products, if any. |
We have based our estimates on assumptions that may prove to be wrong. We may need to obtain additional funds sooner or in greater amounts than we currently anticipate.
Various conditions outside of our control may detract from our ability to raise additional capital needed to execute our plan of operations, including overall market conditions in the international and local economies. We recognize that the United States economy has suffered through a period of uncertainty during which the capital markets have been impacted, and that there is no certainty that these levels will stabilize or reverse despite the optics of an improving economy. Any of these factors could have a material impact upon our ability to raise financing and, as a result, upon our short-term or long-term liquidity.
Critical Accounting Policies
The condensed consolidated financial statements are prepared in conformity with U.S. GAAP, which require the use of estimates, judgments and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent liabilities at the date of the financial statements, and the reported amounts of expenses in the periods presented. We believe that the accounting estimates employed are appropriate and resulting balances are reasonable; however, due to inherent uncertainties in making estimates, actual results could differ from the original estimates, requiring adjustments to these balances in future periods. The critical accounting estimates that affect the consolidated financial statements and the judgments and assumptions used are consistent with those described under Part II, Item 7 of our Annual Report on Form 10-K for the year ended December 31, 2017.
Going Concern
The below excludes the closing of the proposed merger with Marker Therapeutics and the concurrent closing of the $70 million private placement.
We have no sources of revenue to provide incoming cash flows to sustain our future operations. As outlined above, our ability to pursue our planned business activities is dependent upon our successful efforts to raise additional capital.
These factors raise substantial doubt regarding our ability to continue as a going concern. Our condensed consolidated financial statements have been prepared on a going concern basis, which implies that we will continue to realize our assets and discharge our liabilities in the normal course of business. Our financial statements do not include any adjustments to the recoverability and classification of recorded asset amounts and classification of liabilities that might be necessary should we be unable to continue as a going concern.
| 23 |
Off-Balance Sheet Arrangements
We have not entered into any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes of financial condition, revenues, expenses, results of operations, liquidity, capital expenditures or capital resources that are material to investors.
| Item 3. | Quantitative and Qualitative Disclosures About Market Risk |
We are a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and are not required to provide the information required under this item.
| Item 4. | Controls and Procedures |
| (a) | Evaluation of Disclosure Controls and Procedures |
Our management, with the participation of our Chief Executive Officer and our Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures (as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of the end of the period covered by this report. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed in the reports we file or submit under the Exchange Act is accumulated and communicated to management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. Based on such evaluation, our Chief Executive Officer and our Chief Financial Officer have concluded that, as of the end of the period covered by this report, our disclosure controls and procedures are not effective in recording, processing, summarizing and reporting, on a timely basis, information required to be disclosed by us in the reports that we file or submit under the Exchange Act.
It should be noted that any system of controls is based in part upon certain assumptions designed to obtain reasonable (and not absolute) assurance as to its effectiveness, and there can be no assurance that any design will succeed in achieving its stated goals.
| (b) | Changes in Internal Control Over Financial Reporting |
There have been no changes in our internal control over financial reporting during the three months ended June 30, 2018 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| Item 1. | Legal Proceedings |
Management is not aware of any material legal proceedings and there are no pending material procedures that would affect the property of the Company. Management is not aware of any legal proceedings contemplated by any government authority or any other party involving the Company. As of the date of this Quarterly Report, no director, officer or affiliate is (i) a party adverse to us in any legal proceeding, or (ii) has an adverse interest to us in any legal proceeding.
| Item 1A. | Risk Factors |
In addition to the risk factors set forth in Part I — Item 1A — “Risk Factors” of our Annual Report on Form 10-K for the fiscal year ended December 31, 2017 (the “10-K”) and Part II — Item 1A — “Risk Factors” of our Quarterly Report on Form 10-Q for the quarterly period ended March 31, 2018 (the “10-Q”), investors should consider the following risk factors arising from our intention to combine with Marker Therapeutics, Inc. (“Marker”) through a “merger of equals” business combination (the “merger”, and the Company and Marker following completion of the merger, the “Combined Company”) as well as the other risk factors set forth in the preliminary proxy statement we filed with the SEC on July 13, 2018 in connection with the proposed merger, which also include risks related to Marker, Marker’s product candidates, governmental regulation of Marker and Marker’s intellectual property which may affect the Combined Company upon completion of the Merger. On May 15, 2018, we entered into a definitive merger agreement (the “merger agreement”) with Marker. Pursuant to the terms of the merger agreement, Marker will be merged with and into a wholly-owned acquisition subsidiary of the Company formed by the Company in connection with the merger. Upon completion of the merger, the separate existence of the acquisition subsidiary will cease and Marker will be a wholly owned subsidiary of the Company and we will continue as the Combined Company in the merger and renamed Marker Therapeutics, Inc. Following a vote of the Company’s Stockholders, the merger is expected to close in the third or fourth quarter of 2018, although we cannot assure you that the transaction will close during that time or at all. The risk factors below related to the proposed merger with Marker and the Combined Company upon completion of the merger should be read in conjunction with the risk factors set forth in the 10-K and 10-Q and the other information contained in this report as our business, financial condition or results of operations could be adversely affected if any of these risks actually occur.
| 24 |
RISKS RELATED TO THE PROPOSED MERGER WITH MARKER
If the proposed merger with Marker is not consummated, TapImmune’s business could suffer materially and TapImmune’s stock price could decline.
The consummation of the proposed merger with Marker is subject to a number of closing conditions, including the approval of the stock issuance pursuant to the merger agreement by TapImmune stockholders, and other customary closing conditions.
If the proposed merger is not consummated, TapImmune may be subject to a number of material risks, and its business and stock price could be adversely affected, as follows:
· TapImmune has incurred, and expects to continue to incur, significant expenses related to the proposed merger with Marker even if the merger is not consummated.
· The market price of TapImmune common stock may decline to the extent that the current market price reflects a market assumption that the proposed merger will be completed.
· The merger agreement contains covenants relating to TapImmune’s solicitation of competing acquisition proposals and the conduct of TapImmune’s business between the date of signing the merger agreement and the closing of the merger. As a result, significant business decisions and transactions before the closing of the merger require the consent of Marker. Accordingly, TapImmune may be unable to pursue business opportunities that would otherwise be in its best interest as a standalone company. If the merger agreement is terminated after TapImmune has invested significant time and resources in the merger process, TapImmune will have a limited ability to obtain additional financing to fund its operations on a standalone basis.
· TapImmune could be obligated to pay Marker a $1.5 million termination fee in connection with the termination of the merger agreement, depending on the reason for the termination. Additionally, in connection with the termination of the merger agreement, depending on the reason for the termination, TapImmune may be obligated to pay up to $500,000 of out-of-pocket costs incurred by Marker in connection with the transactions and any legal fees incurred by Marker in connection with preparation of the proxy statement.
· TapImmune would need to raise additional capital independently of the proposed merger to continue to operate its business on a stand-alone basis and this capital might not be available on acceptable terms, if at all.
· The merger agreement places certain restrictions on the conduct of our business, which may have delayed or prevented us from undertaking business opportunities that, absent the merger agreement, we may have pursued.
· Litigation related to any failure to complete the merger or related to any enforcement proceeding commenced against us to perform our obligations under the merger agreement.
· TapImmune’s prospective customers, collaborators and other business partners and investors in general may view the failure to consummate the merger as a poor reflection on TapImmune’s business or prospects.
In addition, if the merger agreement is terminated and TapImmune’s board of directors determines to seek another business combination, it may not be able to find a third party willing to provide equivalent or more attractive consideration than the consideration to be provided by each party in the merger. In such circumstances, TapImmune’s board of directors may elect to, among other things, divest all or a portion of TapImmune’s business, or take the steps necessary to liquidate all of TapImmune’s business and assets, and in either such case, the consideration that TapImmune receives may be less attractive than the consideration to be received by TapImmune pursuant to the merger agreement.
Any delay in completing the merger may reduce or eliminate the benefits expected to be achieved thereunder.
The merger is subject to a number of conditions beyond our control that may prevent, delay or otherwise materially adversely affect its completion. We cannot predict whether and when these conditions will be satisfied. Any delay in completing the merger could cause the Combined Company not to realize some or all of the operational and other benefits that we expect to achieve if the merger is successfully completed within its expected time frame.
| 25 |
Completion of the merger is subject to a number of conditions, which, if not satisfied or waived, may result in termination of the merger agreement.
The merger agreement contains a number of conditions to completion of the merger, including, among others:
· receipt of our requisite shareholder approval;
· the absence of any temporary restraining order, preliminary or permanent injunction or other judgment, order or decree issued by any court of competent jurisdiction or other legal restraint that prohibits or makes illegal the completion of the merger;
· the absence of a stop order or proceedings threatened or initiated by the SEC for that purpose;
· the accuracy of the representations and warranties made in the merger agreement by us, subject to certain materiality thresholds, and each party having performed, in all material respects, all obligations required to be performed by it under the merger agreement at or prior to the effective time of the merger; and
· the non-occurrence of any fact, circumstance, development, event, change, occurrence or effect that, individually or in the aggregate, has had or would reasonably be expected to have a material adverse effect on either party.
Many of the conditions to completion of the merger are not within our control, and we cannot predict when or if these conditions will be satisfied. If any of these conditions are not satisfied or waived prior to the outside deadline for consummating the merger, it is possible that the merger agreement may be terminated. The original deadline for consummating the merger is September 15, 2018, but it will be extended for 60 days to November 14, 2018 since the Company has received notice that the proxy statement is being reviewed by the SEC. Although we have agreed in the merger agreement to use reasonable best efforts, subject to certain limitations, to complete the merger in the most expeditious manner practicable, these and other conditions to completion of the merger may fail to be satisfied.
The merger will cause dilution to the Combined Company, which may negatively affect the market price of common stock of the Combined Company.
In connection with the completion of the merger, we expect to issue approximately 13.7 million shares of our common stock exclusive of any warrants that are expected to be issued and we expect to issue 17.5 million shares in connection with the concurrent private placement, exclusive of any warrants expected to be issued in connection therewith. The issuance of these new shares of our common stock could have the effect of depressing the market price of common stock of the Combined Company.
The announcement and pendency of the proposed merger with Marker could adversely affect TapImmune’s business.
The announcement and pendency of the proposed merger could adversely affect TapImmune’s business for a number of different reasons, many of which are not within TapImmune’s control, including as follows:
· Some of TapImmune’s suppliers, distributors, collaborators, and other business partners may seek to change or terminate their relationships with TapImmune as a result of the proposed merger;
· As a result of the proposed merger, current and prospective employees could experience uncertainty about their future roles within the Combined Company. This uncertainty may adversely affect TapImmune’s ability to retain its key employees, who may seek other employment opportunities; and
· TapImmune’s management team may be distracted from day-to-day operations as a result of the proposed merger.
Some of TapImmune’s and Marker’s officers and directors have conflicts of interest that may influence them to support or approve the merger.
Certain officers and directors of TapImmune and Marker participate in arrangements that provide them with interests in the merger that are different from yours, including, among others, to the extent applicable, their continued service as an officer or director of the Combined Company, severance benefits, the acceleration of restricted stock and stock option vesting and continued indemnification. These interests, among others, may influence such officers and directors of TapImmune and Marker to support or approve the merger.
The merger may be completed even though material adverse changes may result from the announcement of the merger, industry-wide changes and other causes.
In general, either party can refuse to complete the merger if there is a material adverse change affecting the other party between May 15, 2018, the date of the merger agreement, and the closing. However, some types of changes do not permit either party to refuse to complete the merger, even if such changes would have a material adverse effect on TapImmune or Marker, to the extent they resulted from the following (unless, in some cases, they have a materially disproportionate effect on TapImmune or Marker, as the case may be):
| 26 |
· any rejection by a governmental body of a registration or filing by TapImmune or Marker relating to TapImmune or Marker’s intellectual property rights;
· any change in the cash position of TapImmune or Marker that results from operations in the ordinary course of business;
· conditions generally affecting the industries in which TapImmune or Marker and its subsidiaries participate or the U.S. or global economy or capital markets as a whole, to the extent that such conditions do not have a disproportionate impact on Marker and its subsidiaries, taken as a whole;
· any failure by TapImmune or Marker or any of its subsidiaries to meet internal projections or forecasts on or after the date of the merger agreement, provided that any such effect, change, event, circumstance, or development causing or contributing to any such failure to meet projections or forecasts may constitute a material adverse effect of TapImmune or Marker and may be taken into account in determining whether a material adverse effect has occurred;
· the execution, delivery, announcement, or performance of obligations under the merger agreement or the announcement, pendency or anticipated consummation of the merger;
· any natural disaster or any acts of terrorism, sabotage, military action, or war or any escalation or worsening thereof; or
· any changes after the date of the merger agreement in U.S. GAAP or applicable laws.
If adverse changes occur but TapImmune and Marker must still complete the merger, the Combined Company’s stock price may suffer.
During the pendency of the merger, TapImmune may not be able to enter into a business combination with another party because of restrictions in the merger agreement.
Covenants in the merger agreement impede the ability of TapImmune or Marker to make acquisitions or complete other transactions that are not in the ordinary course of business pending completion of the merger. As a result, if the merger is not completed, the parties may be at a disadvantage to their competitors. In addition, while the merger agreement is in effect and subject to limited exceptions, each party is prohibited from soliciting, initiating, encouraging or taking actions designed to facilitate any inquiries or the making of any proposal or offer that could lead to the entering into certain extraordinary transactions with any third party, such as a sale of assets, an acquisition of TapImmune common stock, a tender offer for TapImmune common stock or a merger or other business combination outside the ordinary course of business, which transactions could be favorable to such party’s stockholders.
The market price of the Combined Company’s common stock may decline as a result of the merger.
The market price of the Combined Company’s common stock may decline as a result of the merger for a number of reasons including if:
· the Combined Company does not achieve the perceived benefits of the merger as rapidly or to the extent anticipated by financial or industry analysts;
· the effect of the merger on the Combined Company’s business and prospects is not consistent with the expectations of financial or industry analysts; or
· investors react negatively to the effect on the Combined Company’s business and prospects from the merger.
TapImmune stockholders may not realize a benefit from the merger commensurate with the ownership dilution they will experience in connection with the merger.
If the Combined Company is unable to realize the strategic and financial benefits currently anticipated from the merger, TapImmune stockholders will have experienced substantial dilution of their ownership interest without receiving any commensurate benefit. Significant management attention and resources will be required to integrate and operate the Combined Company. Delays in this process could adversely affect the Combined Company’s business, financial results, financial condition and stock price following the merger. Even if the Combined Company is able to integrate the business operations successfully, there can be no assurance that this integration will result in the realization of the full benefits of synergies, innovation, and operational efficiencies that may be possible from this integration and that these benefits will be achieved within a reasonable period of time.
| 27 |
Because the lack of a public market for the Marker shares makes it difficult to value Marker, TapImmune may pay consideration in the merger that is greater than the fair market value of the Marker shares.
The outstanding capital stock of Marker is privately held and is not traded in any public market. The lack of a public market makes it extremely difficult to determine the fair market value of Marker. Since the percentage of TapImmune’s equity to be issued to Marker stockholders was determined based on negotiations between the parties, it is possible that the value of the TapImmune common stock to be issued in connection with the merger will be greater than the fair market value of Marker.
The Combined Company will incur significant transaction costs as a result of the merger, including investment banking, legal, and accounting fees. In addition, the Combined Company will incur significant consolidation and integration expenses which cannot be accurately estimated at this time. These costs could include the planned relocation of certain operations from Jacksonville, Florida to Houston, Texas as well as other transition and start-up costs associated with the clinical programs to be conducted by the Combined Company after the merger. Actual transaction costs may substantially exceed TapImmune’s estimates and may have an adverse effect on the Combined Company’s financial condition and operating results.
Marker’s principal stockholders, executive officers, and directors will own a significant percentage of TapImmune common stock and will be able to exert significant control over matters submitted to the stockholders for approval.
Immediately following the effective time of the merger between Marker and TapImmune, and after taking into account the issuance of shares in the private placement transaction occurring concurrently with the merger, Marker’s stockholders are expected to own, on a fully-diluted basis (assuming the exercise of all outstanding warrants and options), approximately 27.5%, and TapImmune’s current stockholders are expected to own approximately 27.5%, of TapImmune common stock.
After the merger with TapImmune, Marker’s stockholders will beneficially own a significant percentage of TapImmune common stock. This significant concentration of share ownership may adversely affect the trading price for TapImmune common stock because investors often perceive disadvantages in owning stock in companies with large stockholders. These stockholders, if they acted together, could significantly influence all matters requiring approval by the stockholders following the merger, including the election of directors and the approval of mergers or other business combination transactions. The interests of these stockholders may not always coincide with the interests of other stockholders.
The merger may limit the use of the NOL carryforwards and other tax attributes of both TapImmune and Marker to offset future taxable income of the Combined Company.
Under Section 382 of the Internal Revenue Code of 1986, as amended, or the Code, if a corporation undergoes an “ownership change” (generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period), the corporation’s ability to use its pre-change net operating loss, which is referred to as NOL, carryforwards, and other pre-change tax attributes (such as research tax credits) to offset its post-change income may be limited.
As of December 31, 2017, TapImmune had federal NOL carryforwards of approximately $41.7 million and state NOL carryforwards of approximately $21.9 million. The merger may result in an ownership change for TapImmune under Section 382 of the Code and may limit the use of the NOL carryforwards and other tax attributes of TapImmune to offset future taxable income of the Combined Company for both federal and state income tax purposes. These tax attributes are subject to expiration at various times in the future to the extent that they have not been applied to offset the taxable income of the Combined Company. These limitations may affect the Combined Company’s effective tax rate in the future.
RISKS RELATED TO THE COMBINED COMPANY UPON COMPLETION OF MERGER
Risks Related to the Combined Company’s Business and Product Candidates
The Combined Company’s future success will be highly dependent upon its key personnel, and its ability to attract, retain, and motivate additional qualified personnel.
The Combined Company’s ability to compete in the highly competitive biotechnology and pharmaceutical industries depends upon its ability to attract and retain highly qualified managerial, scientific, and medical personnel. The Combined Company will be highly dependent on its management, scientific, and medical personnel, including Peter Hoang, its President and Chief Executive Officer, Ann Leen, Ph.D., who is expected to be its Chief Scientific Officer following completion of the merger, and Juan Vera, M.D., who is expected to be its Chief Development Officer following completion of the merger. The loss of the services of any of the Combined Company’s executive officers, other key employees, and other scientific and medical advisors, and the Combined Company’s inability to find suitable replacements could result in delays in product development and harm to the Combined Company’s business. In particular, Dr. Leen is the key person who has produced Marker’s MultiTAA T cell therapy-based product. A priority of the Combined Company will be to quickly train additional qualified scientific and medical personnel in the Combined Company to ensure the ability to maintain business continuity. Any delays in training such personnel could delay the development, manufacture, and clinical trials of the Combined Company’s product candidates.
| 28 |
The Combined Company also anticipates hiring additional scientific and medical personnel to grow its business. The Combined Company will conduct operations in Houston, Texas. This region is headquarters to many other biopharmaceutical companies and many academic and research institutions. Competition for skilled personnel in the combined companies market is intense and may limit its ability to hire and retain highly qualified personnel on acceptable terms or at all. If the Combined Company is not successful in attracting and retaining highly qualified personnel, it may not be able to successfully implement its business strategy.
The Combined Company’s strategic relationship with Baylor College of Medicine, or BCM, is dependent, in part, upon its relationship with key medical and scientific personnel and advisors.
Marker’s therapy has been developed through its collaboration with the Center for Cell and Gene Therapy at BCM, founded by Malcom K. Brenner, M.D., Ph.D., a recognized pioneer in immuno-oncology. In addition to Dr. Brenner, Marker’s founders include, Ann Leen, Ph.D., Juan Vera, M.D., Helen Heslop, M.D., DSc (Hon) and Cliona Rooney, Ph.D., who have significant experience in this field and are all affiliated with the Center for Cell and Gene Therapy at BCM. Dr. Leen and Dr. Vera are expected to serve as the Combined Company’s Chief Scientific Officer and Chief Development Officer, respectively, following completion of the merger. In addition, Dr. Brenner, Dr. Heslop and Dr. Rooney have agreed to join the Combined Company’s newly formed Scientific Advisory Board that will become effective in conjunction with the merger.
The Combined Company’s strategic relationship with BCM will be dependent, in part, on its relationship with these key employees and advisors, and in particular Dr. Leen and Dr. Vera, who are also employed with the Center for Cell and Gene Therapy at BCM. If the Combined Company loses Dr. Leen or Dr. Vera, or if either leaves their position at BCM, the Combined Company’s relationship with BCM may deteriorate, and its business could be harmed.
The Combined Company, and certain of its key medical and scientific personnel, will need additional agreements in place with BCM to expand its development, manufacture, and clinical trial efforts.
Although the Combined Company will have an exclusive license agreement with BCM under which Marker received a worldwide, exclusive license to BCM’s rights in and to three patent families to develop and commercialize the MultiTAA product candidates, the Combined Company will need to enter into additional agreements with BCM with respect to (i) a strategic alliance to advance pre-clinical research, early stage clinical trials, and Phase II clinical trials with respect to the Combined Company’s product candidates, as well as continued access to its clinical data, (ii) sponsored research for investigators within the Center for Cell and Gene Therapy at BCM, and (iii) product manufacturing and support, including personnel and space at the institution for the foreseeable future. Any delays in entering into new strategic agreements with BCM related to the Combined Company’s product candidates could delay the development, manufacture, and clinical trials of its product candidates.
The multiple roles of certain of the Combined Company’s officers and directors could limit their time and availability to the Combined Company, and create, or appear to create, conflicts of interest.
After completion of the merger, Dr. Leen and Dr. Vera will continue to be employees of BCM, and will be contractually obligated to spend a significant portion of their time for BCM. In addition, Dr. Leen and Dr. Vera are co-founders and members of ViraCyte, and perform services from time to time for ViraCyte LLC, or ViraCyte. ViraCyte is owned by the same principal stockholder group as Marker and has technology which is being developed under a license agreement with BCM by the same research group at BCM. More specifically, ViraCyte is a clinical stage biopharmaceutical company, which is investigating and developing virus-specific T cell therapy technology for the prevention and/or treatment of viral infections. Accordingly, Dr. Leen and Dr. Vera may have other commitments that would, at times, limit their availability to the Combined Company, and other research being conducted by Dr. Leen and Dr. Vera may, at times, receive higher priority than research on the Combined Company’s programs, which may, in turn, delay the development or commercialization of the Combined Company’s product candidates.
| 29 |
In addition, John Wilson is a member, director and officer of ViraCyte and will be a director of the Combined Company after the consummation of the merger. Dr. Leen and Dr. Vera are also co-founders and members of ViraCyte, and perform services for ViraCyte from time to time, and Dr. Vera will be a director of the Combined Company after the consummation of the merger. All of these individuals will have certain fiduciary or other obligations to the Combined Company after the consummation of the merger and certain fiduciary or other obligations to ViraCyte and, in the case of Dr. Leen and Dr. Vera, to BCM. Such multiple obligations may in the future result in a conflict of interest with respect to presenting other potential business opportunities to the Combined Company or to ViraCyte. A conflict of interest also may arise concerning the timing of the parties’ planned and ongoing clinical trials, investigational new drug application filings and the parties’ opportunities for marketing their respective product candidates. In addition, they may be faced with decisions that could have different implications for the Combined Company than for ViraCyte. Consequently, there is no assurance that these members of the Combined Company’s board and management would always act in the Combined Company’s best interests in all situations should a conflict arise.
Product development involves a lengthy and expensive process with an uncertain outcome, and results of earlier pre-clinical and clinical trials may not be predictive of future clinical trial results.
Clinical testing is expensive and generally takes many years to complete, and the outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of pre-clinical testing and early clinical trials of the Combined Company’s product candidates may not be predictive of the results of larger, later-stage controlled clinical trials. Product candidates that have shown promising results in early-stage clinical trials may still suffer significant setbacks in subsequent clinical trials. Marker’s clinical trials to date have been conducted on a small number of patients in a single clinical site for a limited number of indications. The Combined Company will have to conduct larger, well-controlled trials in its proposed indications at multiple sites to verify the results obtained to date and to support any regulatory submissions for further clinical development of Marker’s product candidates. TapImmune’s and Marker’s assumptions related to Marker’s products, such as with respect to lack of toxicity and manufacturing cost estimates, are based on early limited clinical trials and current manufacturing process at BCM and may prove to be incorrect. In addition, the initial estimates of the clinical cost of development may prove to be inadequate, particularly if clinical trial timing or outcome is different than predicted or regulatory agencies require further testing before approval. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles despite promising results in earlier, smaller clinical trials. Moreover, clinical data are often susceptible to varying interpretations and analyses. The Combined Company does not know whether any Phase II, Phase III, or other clinical trials it may conduct will demonstrate consistent or adequate efficacy and safety with respect to the proposed indication for use sufficient to receive regulatory approval or market its product candidates.
The Combined Company may not be able to expand its manufacturing processes to other third-party manufacturing facilities or successfully create its own manufacturing infrastructure for supply of its requirements of product candidates for use in clinical trials and for commercial sale.
The Combined Company will not own any facility that may be used as its clinical-scale manufacturing and processing facility following the merger. The Combined Company anticipates it will initially rely solely on the cGMP manufacturing facility within BCM for the manufacturing of its product candidates. If the cGMP manufacturing facility of BCM, which does manufacturing for itself and other parties, experiences capacity constraints, disruptions, or delays in manufacturing the Combined Company’s products, the Combined Company’s planned clinical trials and necessary manufacturing capabilities will be disrupted or delayed, which will adversely affect the Combined Company’s ability to conduct and further develop its business as currently planned. Further, the cGMP manufacturing facility is most likely too small to conduct the pivotal clinical studies being planned by the Combined Company, so the Combined Company will need to develop its own cGMP manufacturing capacity that will be adequate for such clinical trials.
In 2019, the Combined Company currently intends to begin developing additional cGMP manufacturing capacity of its own that would be capable of supporting its manufacturing needs with respect to its clinical trials, particularly with respect to pivotal studies. TapImmune and Marker expect that the Combined Company’s manufacturing strategy will involve the use of one or more Contract Manufacturing Organizations, or CMOs, or the Combined Company will establish its own capabilities and infrastructure, including a manufacturing facility. Establishment of the Combined Company’s own manufacturing facility is subject to many risks. For example, the establishment of a cell-therapy manufacturing facility is a complex endeavor requiring knowledgeable individuals. Creating an internal manufacturing infrastructure will rely upon building out a complex facility and finding personnel with an appropriate background and training to staff and operate the facility. Should it be unable to find these individuals, the Combined Company may need to rely on external contractors or train additional personnel to fill needed roles. There are a small number of individuals with experience in cell therapy, and the competition for these individuals is high.
The Combined Company would expect that development of its own manufacturing facility could provide it with enhanced control of material supply for both clinical trials and the commercial market, enable the more rapid implementation of process changes, and allow for better long-term margins. However, neither TapImmune nor Marker has any experience as a company in developing a manufacturing facility and may never be successful in developing the Combined Company’s own manufacturing facility or capability. The Combined Company may establish multiple manufacturing facilities as it expands its commercial footprint to multiple geographies, which may lead to regulatory delays or prove costly. Even if the Combined Company is successful, its manufacturing capabilities could be affected by cost-overruns, unexpected delays, equipment failures, labor shortages, natural disasters, power failures, transportation difficulties and numerous other factors that could prevent the Combined Company from realizing the intended benefits of its manufacturing strategy and have a material adverse effect on the Combined Company’s clinical development and/or commercialization plans.
| 30 |
In addition, the manufacturing process for any products that the Combined Company may develop is subject to the U.S. Food and Drug Administration, or FDA, and foreign regulatory authority approval process, and the Combined Company will need to contract with manufacturers who can meet all applicable FDA and foreign regulatory authority requirements on an ongoing basis. If the Combined Company or its CMOs are unable to reliably produce products to specifications acceptable to the FDA, or other regulatory authorities, the Combined Company may not obtain or maintain the approvals it needs to commercialize such products. Even if the Combined Company obtains regulatory approval for any of its product candidates, there is no assurance that either the Combined Company or its CMOs will be able to manufacture the approved product to specifications acceptable to the FDA or other regulatory authorities, to produce it in sufficient quantities to meet the requirements for the potential launch of the product, or to meet potential future demand. Any of these challenges could delay completion of clinical trials, require bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay approval of the Combined Company’s product candidate, impair commercialization efforts, increase its cost of goods, and have an adverse effect on its clinical development and/or commercialization plans.
Regardless of whether the Combined Company engages additional CMOs to manufacture its products or establishes its own manufacturing facility, in order to transfer the Combined Company’s manufacturing from or expand its manufacturing capabilities beyond BCM pursuant to its development plans, whether through additional third parties or by developing its own manufacturing capabilities, the Combined Company will need access to the Standard Operating Procedures and the specific Batch Production Records that are used to manufacture the product candidates. If BCM fails to transfer Marker’s manufacturing processes, or impedes the Combined Company’s ability to transfer the manufacturing processes of its products to the Combined Company or third-party manufacturers, the Combined Company’s planned clinical trials and additional necessary manufacturing capabilities will be delayed, which will adversely affect the Combined Company’s ability to conduct and further develop its business as currently planned.
The Combined Company will be dependent on third-party vendors to design, build, maintain and support its manufacturing and cell processing facilities.
As a result of the Combined Company’s strategy to outsource its manufacturing, it will rely very heavily on BCM and other third-party manufacturers to perform the Combined Company’s manufacturing of Marker’s products for its clinical trials. Marker also licenses a significant portion of its technology from others and, at this time, does not own any intellectual properties or technologies. The Combined Company intends to rely on its contract manufacturers to produce large quantities of materials needed for clinical trials and potential product commercialization. Third-party manufacturers may not be able to meet the Combined Company’s needs concerning timing, quantity, or quality. If the Combined Company is unable to contract for a sufficient supply of needed materials on acceptable terms, or if it should encounter delays or difficulties in its relationships with manufacturers, its clinical testing may be delayed, thereby delaying the submission of products for regulatory approval or the market introduction and subsequent sales of its products. Any such delay may lower the Combined Company’s revenues and potential profitability.
If any third party breaches or terminates its agreement with the Combined Company,or fails to conduct its activities in a timely manner, the commercialization of the Combined Company’s products under development could be slowed down or blocked completely. It is possible that third parties relied upon by the Combined Company will change their strategic focus, pursue alternative technologies, or develop alternative products, either on their own or in collaboration with others, as a means for developing treatments for the diseases targeted by the Combined Company’s collaborative programs, or for other reasons. The effectiveness of these third parties in marketing their own products may also affect the revenues and earnings of the Combined Company.
The Combined Company intends to continue to enter into additional third-party agreements in the future. However, the Combined Company may not be able to negotiate any additional agreements successfully. Even if established, these relationships may not be scientifically or commercially successful.
The Combined Company’s manufacturing process is reliant upon the specialized equipment, and other specialty materials, which may not be available to the Combined Company on acceptable terms or at all. For some of this equipment and materials, the Combined Company relies or may rely on sole source vendors or a limited number of vendors, which could impair its ability to manufacture and supply its products.
The Combined Company will depend on a limited number of vendors for supply of certain materials and equipment used in the manufacture of its product candidates. For example, the Combined Company will purchase equipment and reagents critical for the manufacture of its product candidates from Wilson Wolf Manufacturing Corporation (a company controlled by a Marker stockholder, John Wilson, who will become a director of the Combined Company), JPT Peptide Technologies and other suppliers. Some of the Combined Company’s suppliers may not have the capacity to support commercial products manufactured under cGMP by biopharmaceutical firms or may otherwise be ill-equipped to support the Combined Company’s needs. The Combined Company also may not have supply contracts with many of these suppliers,and may not be able to obtain supply contracts with them on acceptable terms or at all. Accordingly, the Combined Company may not be able to obtain key materials and equipment to support clinical or commercial manufacturing.
| 31 |
For some of this equipment and materials, the Combined Company will rely, and may in the future rely, on sole-source vendors or a limited number of vendors. An inability to continue to source product from any of these suppliers, which could be due to regulatory actions or requirements affecting the supplier, adverse financial, or other strategic developments experienced by a supplier, labor disputes or shortages, unexpected demands, or quality issues, could adversely affect the Combined Company’s ability to satisfy demand for its product candidates, which could adversely and materially affect the Combined Company’s operating results or its ability to conduct clinical trials, either of which could significantly harm its business.
As the Combined Company continues to develop and scale its manufacturing process, it may need to obtain rights to and supplies of specific materials and equipment to be used as part of that process. For example, Marker’s manufacturing process is based, in part, upon the G-Rex® cell culture device manufactured by Wilson Wolf Manufacturing Corporation, which is used by many cell therapy developers, both in commercial and academic settings. The Combined Company will not own any exclusive rights to the G-Rex® that could be used to prevent third parties from developing similar and competing processes. The Combined Company may not be able to obtain rights to such materials and equipment on commercially reasonable terms, or at all, and if the Combined Company is unable to alter its process in a commercially viable manner to avoid the use of such materials or find a suitable substitute, it would have a material adverse effect on its business.
The Combined Company may enter into one or more transactions with entities controlled by one of its directors, which could pose a conflict of interest.
John Wilson, currently a significant stockholder in Marker and who will be a director of the Combined Company, is also CEO and co-founder of Wilson Wolf Manufacturing Corporation, which is the sole source vendor that provides Marker with the G-Rex® cell culture device for the large-scale production of T cells used in Marker’s manufacturing process. Marker does not currently have a supply contract with Wilson Wolf Manufacturing for the G-Rex®. The Combined Company plans to negotiate a supply contract with Wilson Wolf Manufacturing for the purchase of G-Rex® devices. The Combined Company also plans to engage Wilson Wolf Manufacturing in discussions to customize the G-Rex® further to optimally match the Combined Company’s manufacturing requirements, as well as to develop a scalability plan to drive efficiencies for a commercial product. There may be conflicts of interest between the Combined Company and Wilson Wolf Manufacturing. There can be no assurance that Wilson Wolf Manufacturing will agree to enter into any contract with the Combined Company, or that the terms of any such agreements will be in the best interests of the Combined Company, or will have terms no less favorable to the Combined Company than could have been obtained from unaffiliated third parties.
The future results of the Combined Company will suffer if the Combined Company does not effectively manage its expanded operations following the completion of the merger.
Following the completion of the merger, the size of the business of the Combined Company will increase significantly beyond the current size of either us or Marker. The Combined Company’s future success depends, in part, upon its ability to manage this expanded business, which will pose substantial challenges for management, including challenges related to the management and monitoring of new operations and associated increased costs, complexity and allocation of financial resources. If the Combined Company is unsuccessful in managing its integrated operations, or if it does not realize the expected operating efficiencies, cost savings and other benefits currently anticipated from the merger, the operations and financial condition of the Combined Company could be adversely affected and the Combined Company may not be able to take advantage of business development opportunities.
The Combined Company may be unable to fully realize the competitive synergies that are projected to be achieved through the combination of our services and Marker’s offerings.
Part of the strategic rationale for the merger is the opportunity for the Combined Company to potentially drive additional value through the utilization by us of Marker’s capabilities. However, the utilization of Marker’s offerings is still evolving and subject to a number of risks and uncertainties, including the following:
· government regulatory agencies and legislative bodies, including agencies and legislatures regulating the use of clinical trials, may impose new conditions or restrictions which affect the Combined Company’s use of Marker’s data;
· implementation of any operational plans to develop new cancer treatments from the Marker offerings will likely be complex and challenging to achieve, and may be subject to delays and cost overruns and there is no assurance that the research and development can be carried out effectively;
| 32 |
· clinical research is a complex and evolving area, and creating effective approaches to drive more effective and efficient research outcomes is difficult and challenging; and
· third parties outside of our control (including suppliers and regulators) may impose restrictions or conditions which affect the projected timing and successful achievement of our benefits from the transaction.
The Combined Company is unable to predict the extent to which these factors will inhibit its business plans and any one of them could result in decreased or delays in post-closing performance by the Combined Company.
We may fail to realize all of the anticipated benefits of the merger or those benefits may take longer to realize than expected. The Combined Company may also encounter significant difficulties in integrating the two businesses.
Our ability to realize the anticipated benefits of the transaction will depend, to a large extent, on the Combined Company’s ability to integrate the two businesses. The combination of two independent businesses is a complex, costly and time-consuming process. As a result, we and Marker will be required to devote significant management attention and resources to integrating the business practices and operations. The integration process may disrupt the businesses and, if implemented ineffectively, would restrict the realization of the full-expected benefits. The failure to meet the challenges involved in integrating the two businesses and to realize the anticipated benefits of the transaction could cause an interruption of or a loss of momentum in, the activities of the Combined Company and could adversely affect the results of operations of the Combined Company.
In addition, the overall integration of the businesses may result in material unanticipated problems, expenses, liabilities, competitive responses, and diversion of management’s attention. The difficulties of combining the operations of the companies include, among others:
· difficulties in achieving successful development of Marker’s offerings, business opportunities and growth prospects from the combination;
· difficulties in the integration of the companies’ businesses;
· difficulties in managing the expanded operations of a significantly larger and more complex company;
· challenges in attracting and retaining key personnel; and
· potential unknown liabilities and unforeseen increased expenses or delays associated with the merger.
Many of these factors will be outside of the control of the Combined Company and any one of them could result in increased costs, and diversion of management’s time and energy, which could materially impact the business, financial condition and results of operations of the Combined Company. In addition, even if the operations of the businesses are integrated successfully, the full benefits of the transaction may not be realized, including growth opportunities that are expected. These benefits may not be achieved within the anticipated time frame, or at all. Further, additional unanticipated costs may be incurred in the integration of the businesses of ours and Marker. All of these factors could negatively impact or decrease or delay the expected benefits of the transaction and negatively impact the price of the Combined Company’s shares. As a result, there is no assurance that the combination of us and Marker will result in the realization of the full benefits anticipated from the merger.
Risks Related to Combined Company’s Financial Condition and Need for Additional Capital
Management will have broad discretion as to the use of the proceeds from the private placement transaction, and the Combined Company may not use the proceeds effectively.
The Combined Company’s management will have broad discretion as to the application of the net proceeds from the private placement transaction for general corporate purposes and working capital to advance the development of the Combined Company’s product candidates. Management may spend the proceeds in ways that do not necessarily improve its operating results or enhance the value of its common stock.
The Combined Company will require additional financing before it can generate any revenue from operations.
After consummation of the merger and the private placement transaction, the Combined Company anticipates having sufficient cash on hand to fund its operations for at least the next thirty months. The product candidates of the Combined Company, however, remain in the early stages of development and the Combined Company anticipates it will be years before it is able to generate any revenue from operations. Accordingly, the Combined Company will need additional debt or equity financing in the future to execute its business plan, complete its future clinical trials, and to add manufacturing, sales, marketing, and customer support personnel in the future to advance the commercialization of its products. The Combined Company will operate in a market that makes its prospects difficult to evaluate, and achievement of positive cash flow from operations will depend upon revenue resulting from the successful development of its product candidates, which depend upon regulatory clearance.
| 33 |
In the future, if the Combined Company fails to satisfy the continued listing standards of NASDAQ, it may not be able to sell shares of its common stock to raise additional capital. In addition, future market conditions may limit the ability of the Combined Company to raise capital on favorable terms, or at all, and the terms of any public or private offerings of debt or equity securities likely would be significantly dilutive to existing stockholders at such time. There is no guarantee that the Combined Company will be able to obtain any of the additional debt or equity financing that will be required after completion of the merger and the private placement transaction on commercially reasonable terms or at all. If the Combined Company fails to obtain the necessary debt or equity financing when needed, it may not be able to execute its planned development and commercialization efforts, which would have a material adverse effect on the Combined Company’s growth strategy, the results of its operations and financial condition and stock price. If the Combined Company is unable to generate sufficient capital from operations or raise additional funds, it may need to consider other alternative actions, including one or more of the following:
· delay, scale-back, or eliminate research and development of some or all of the Combined Company’s product candidates;
· license third parties to develop and commercialize products or technologies that TapImmune would otherwise seek to develop and commercialize ourselves;
· attempt to sell the company;
· cease operations; or
· declare bankruptcy.
The occurrence of any of the foregoing events would have a material adverse effect on the Combined Company’s growth strategy, the results of its operations and financial condition, and stock price, and there can be no assurance that it would be able to continue as a going concern.
The issuance of additional equity securities may negatively impact the trading price of the Combined Company’s common stock.
TapImmune has issued equity securities in the past, will issue equity securities in the merger and private placement transaction, and expects to continue to issue equity securities to finance the activities of the Combined Company in the future. In addition, outstanding options and warrants to purchase its common stock may be exercised, and additional options and warrants may be issued, resulting in the issuance of additional shares of common stock. The issuance by the Combined Company of additional equity securities, including the shares of common stock issuable upon exercise of the warrants issued by TapImmune in the private placement transaction, would result in dilution to the Combined Company’s stockholders, and even the perception that such an issuance may occur could have a negative impact on the trading price of the Combined Company’s common stock.
The Combined Company will have a significant number of outstanding warrants and options, and future sales of the shares obtained upon exercise of these options or warrants could adversely affect the market price of the Combined Company’s common stock.
Upon completion of the merger and private placement transaction, the Combined Company will have outstanding warrants to purchase up to 23,657,372 shares of its common stock at a weighted average exercise price of $4.74 per share, and options exercisable for an aggregate of 439,467 shares of common stock at a weighted average exercise price of $6.77 per share, in each case calculated as if the merger had been consummated as of June 29, 2018. TapImmune has committed to register the resale of all the shares issuable upon exercise of these warrants, and they will be freely tradable by the exercising party upon issuance. Upon such registration, the holders may sell these shares in the public markets from time to time, without limitations on the timing, amount, or method of sale. If the Combined Company’s stock price rises, the holders may exercise their warrants and options and sell a large number of shares. This could cause the market price of the Combined Company’s common stock to decline and cause existing stockholders to experience significant further dilution.
| Item 2. | Unregistered Sales of Equity Securities and Use of Proceeds |
| (a) | We issued the following unrestricted securities during the period covered by this report to the named individual pursuant to exemptions under the Securities Act of 1933 including Section 4(2): |
On April 10, 2018, we issued 15,000 shares of common stock to Omnicor Media, LLC pursuant to a vendor agreement.
| 34 |
On April 13, 2018, we issued 33,334 shares of common stock to Collision Capital, LLC pursuant to a vendor agreement.
On May 18, 2018, we issued 12,849 shares of common stock to Richard Kenney, pursuant to a consulting services agreement.
On May 31, 2018, we issued 50,000 shares of common stock to Caro Partners, LLC pursuant to a vendor agreement.
On June 18, 2018, we issued 11,600 shares of common stock to Corporate Profile pursuant to a vendor agreement.
| Item 3. | Defaults Upon Senior Securities |
None.
| Item 4. | Mine Safety Disclosure |
Not applicable.
| Item 5. | Other Information |
Not applicable.
| Item 6. | Exhibits |
The following exhibits are included with this Quarterly Report on Form 10-Q:
Exhibit 101
101.INS - XBRL Instance Document
101.SCH - XBRL Taxonomy Extension Schema Document
101.CAL - XBRL Taxonomy Extension Calculation Linkbase Document
101.DEF - XBRL Taxonomy Extension Definition Linkbase Document
101.LAB - XBRL Taxonomy Extension Label Linkbase Document
101.PRE - XBRL Taxonomy Extension Presentation Linkbase Document
| 35 |
In accordance with the requirements of the Exchange Act, the registrant caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Date: August 9, 2018
TAPIMMUNE INC.
| /s/ Peter L. Hoang | ||
| Peter L. Hoang President and Chief Executive Officer and Principal Executive Officer | ||
| /s/ Michael J. Loiacono | ||
| Michael J. Loiacono Chief Financial Officer and Principal Accounting Officer | ||
| 36 |