Attached files
| file | filename |
|---|---|
| EX-10.5D - EX-10.5D - REVA Medical, Inc. | rva-ex105d_479.htm |
| EX-32.1 - EX-32.1 - REVA Medical, Inc. | rva-ex321_7.htm |
| EX-31.2 - EX-31.2 - REVA Medical, Inc. | rva-ex312_6.htm |
| EX-31.1 - EX-31.1 - REVA Medical, Inc. | rva-ex311_9.htm |
| EX-23.1 - EX-23.1 - REVA Medical, Inc. | rva-ex231_8.htm |
| EX-10.20 - EX-10.20 - REVA Medical, Inc. | rva-ex1020_481.htm |
| EX-10.19 - EX-10.19 - REVA Medical, Inc. | rva-ex1019_480.htm |
| EX-10.18 - EX-10.18 - REVA Medical, Inc. | rva-ex1018_549.htm |
| EX-10.5C - EX-10.5C - REVA Medical, Inc. | rva-ex105c_478.htm |
| EX-3.2 - EX-3.2 - REVA Medical, Inc. | rva-ex32_482.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
|
☑ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
or
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission file number: 000-54192
REVA MEDICAL, INC.
(Exact name of registrant as specified in its charter)
|
Delaware |
|
33-0810505 |
|
(State or other jurisdiction of incorporation or organization) |
|
(I.R.S. Employer Identification No.) |
|
|
|
|
|
5751 Copley Drive, San Diego, CA 92111 |
|
(858) 966-3000 |
|
(Address of principal executive offices including zip code) |
|
(Registrant’s telephone number, including area code)
|
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act: Common Stock, $0.0001 par value per share
|
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. |
|
Yes ☐ No ☑ |
|||
|
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. |
|
Yes ☐ No ☑ |
|||
|
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. |
|
Yes ☑ No ☐ |
|||
|
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or such shorter period that the registrant was required to submit and post such files). |
|
Yes ☑ No ☐ |
|||
|
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. |
|
☑ |
|||
|
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or emerging growth company. See the definitions of "large accelerated filer," "accelerated filer", "smaller reporting company" and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one): |
|||||
|
Large accelerated filer ☐ |
Accelerated filer ☑ |
||||
|
Non-accelerated filer ☐ (Do not check if smaller reporting company) |
Smaller reporting company ☐ |
||||
|
|
Emerging growth company ☐ |
||||
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. |
|
☐ |
|||
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). |
|
Yes ☐ No ☑ |
|||
The aggregate market value of the common equity held by non-affiliates of the registrant as of June 30, 2017 totaled approximately $180,077,000 based on the closing price for the registrant's common stock trading in the form of CHESS Depositary Interests, or CDIs, as reported by the Australian Securities Exchange and based on the closing currency exchange rate in effect that day. Such value excludes common stock and CDIs held by directors, executive officers, and 10% or greater stockholders as of June 30, 2017. The identification of 10% or greater stockholders as of June 30, 2017 is based on Schedule 13G and amended Schedule 13G reports publicly filed before June 30, 2017. This calculation does not reflect a determination that such parties are affiliates for any other purposes.
As of February 23, 2018, there were 41,245,820 shares of the registrant's common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
|
Document Description |
|
Portions of registrant's definitive proxy statement for its 2018 annual meeting of stockholders, to be filed pursuant to Regulation 14A within 120 days after its December 31, 2017 fiscal year end, are incorporated by reference into Part III (items 10, 11, 12, 13, and 14) of this report.
|
FORM 10-K — ANNUAL REPORT
For the Fiscal Year Ended December 31, 2017
TABLE OF CONTENTS
Forward-Looking Statements
This Annual Report on Form 10-K for the year ended December 31, 2017, or “Form 10-K,” contains forward-looking statements concerning our business, operations, and financial performance and condition, as well as our plans, objectives, and expectations for business operations and financial performance and condition. Any statements contained herein other than statements of historical facts may be deemed to be forward-looking statements. You can identify these statements by words such as “aim,” “anticipate,” “assume,” “believe,” “could,” “due,” “estimate,” “expect,” “forecast,” “goal,” “intend,” “may,” “objective,” “plan,” “predict,” “project,” “potential,” “positioned,” “should,” “target,” “will,” “would,” and other similar expressions that are predictions of or indicate future events and future trends. These forward-looking statements are based on current expectations, estimates, forecasts, and projections about our business and the industry in which we operate and management’s beliefs and assumptions and are not guarantees of future performance or developments and involve known and unknown risks, uncertainties, and other factors that are in some cases beyond our control. We caution readers that forward-looking statements are not guarantees of future performance and our actual results may differ materially from those anticipated, projected, or assumed in the forward-looking statements in this Form 10-K. Factors that can cause our actual results to differ materially from those anticipated in the forward-looking statements include, but are not limited to, the risks described under “Risk Factors,” including:
|
|
• |
failure of our Fantom scaffold, or any future product, to meet our required clinical specifications; |
|
|
• |
our inability to obtain regulatory clearance or approval for any of our products; |
|
|
• |
failure of our products to gain market acceptance domestically or internationally; |
|
|
• |
less than anticipated growth in the market for bioresorbable scaffolds generally; |
|
|
• |
changes in the regulatory environment which may adversely impact the commercialization of our products and result in significant additional capital expenditures; |
|
|
• |
refusal of third-party payors to reimburse our customers for use of our products; |
|
|
• |
our history of net losses and our expectation of operating losses for the foreseeable future; |
|
|
• |
increases in our projected expenditures on research and development and administrative activities; |
|
|
• |
our inability to attract or retain skilled personnel for our product development and commercialization efforts; |
|
|
• |
our inability to protect our intellectual property and operate our business without infringing upon the intellectual rights of others, which could result in litigation and significant expenditures; |
|
|
• |
our inability to repay our convertible notes when, and if, required or otherwise comply with their requirements; |
|
|
• |
changes in the fair value of our convertible notes and the gains or losses that may arise upon such changes; |
|
|
• |
failure to complete financings to fund our operations when needed or on terms favorable to us; and, |
|
|
• |
our ability to continue as a going concern; |
Stockholders, potential investors, and other readers are urged to consider these factors carefully in evaluating forward-looking statements and are cautioned not to place undue reliance on forward-looking statements. Forward-looking statements speak only as of the date of this Form 10-K. Unless required by law, we do not intend to publicly update or revise any forward-looking statements to reflect new information or future events or otherwise. You should, however, review the factors and risks we describe in the reports we will file from time to time with the Securities and Exchange Commission after the date of this Form 10-K.
General Information
Unless the context implies otherwise, references in this report and the information incorporated herein by reference to “REVA Medical,” “REVA,” the “Company,” “we,” “us,” and “our” refer to REVA Medical, Inc. and its consolidated subsidiary.
Our product name Fantom® is a registered trademark in the United States, Australia and the European Union. We have pending applications to register the mark in Brazil, China and Japan. All other trademarks, trade names, and service marks appearing in this report are the property of their respective owners. Use or display by us of other parties’
- 2 -
trademarks, trade dress, or products is not intended to and does not imply a relationship with, or endorsement or sponsorship of, us by the trademark or trade dress owner.
Unless indicated otherwise in this Form 10-K, all references to “$” or “dollars” refer to United States dollars, the lawful currency of the United States of America. References to “A$” refer to Australian dollars, the lawful currency of the Commonwealth of Australia.
Overview
We are a medical device company focused on the development and commercialization of polymer-based bioresorbable products for vascular applications. On April 3, 2017, our second generation and first commercial product, Fantom, was approved for sale under a CE Mark, which allows us to commercialize in Europe and other jurisdictions that recognize the CE Mark. Fantom is a sirolimus-eluting bioresorbable scaffold designed specifically for coronary vascular applications. We believe Fantom is uniquely positioned to meet a market opportunity because of its distinctive features. Fantom is the only bioresorbable scaffold made from Tyrocore™, our proprietary tyrosine-derived polymer designed specifically for vascular scaffold applications. Tyrocore is inherently x-ray visible, making Fantom the first and only bioresorbable scaffold that is fully visible under x-ray. Fantom is designed with thin struts while maintaining strength and with distinct ease-of-use features such as expansion with one continuous inflation and no requirement for refrigerated storage.
In February 2018, we received CE approval for our third generation product, Fantom Encore, in the 2.5 millimeter diameter size. Fantom Encore has thinner struts than Fantom (95 microns versus 125 microns in the 2.5 millimeter diameter size) and comparable strength and visibility. Fantom Encore has the thinnest struts of any commercially available bioresorbable scaffold. We believe that thin struts are associated with better healing and clinical outcomes. Reductions in strut thickness are considered one of the most important improvements for bioresorbable scaffolds.
We began our commercial launch of Fantom late in the second quarter of 2017 and shipped our first product in the third quarter of 2017. Our sales strategy involves a phased approach beginning first with direct sales in Germany, Switzerland and Austria. We will expand geographically as we move into additional phases of our launch throughout 2018 and beyond. We anticipate commercializing Fantom Encore later in 2018. We expect that the Fantom Encore family will ultimately replace the Fantom family.
Although we initiated commercial sales of Fantom in the third quarter of 2017, we are still very early in the commercialization stage. The withdrawal of Absorb, a competitor’s product, in 2017, and the negative publicity related to Absorb’s safety have severely impacted the market for bioresorbable scaffolds, and companies with bioresorbable scaffolds that were made from the same polylactic acid polymer, or PLLA, as Absorb have reduced scale and appear to have abandoned their efforts to commercialize such scaffolds. Because Fantom is not made with the same polymer as Absorb, we continue to believe that we can commercialize Fantom despite the impact that the withdrawal of Absorb has had on the market and demonstrate the benefits of bioresorbable technology. That said, we must now rebuild the market for bioresorbable scaffolds, which can be more challenging than selling into an existing, healthy market. Our rebuilding activities include educating physicians regarding the unique features of Fantom and Fantom Encore, continuing to publish results from our pivotal clinical trial (FANTOM II) and conducting and initiating additional clinical trials to build the clinical evidence needed to support market adoption.
In 2018, we plan to expand our product portfolio into peripheral arterial disease by applying for CE Mark with our bioresorbable scaffold technology for below-the-knee revascularization. The majority of the arteries below the knee have similar sizes and dimensions to coronary arteries, making this a viable treatment option. Resorbable technology is attractive in this application because of the frequent need for retreatment in this patient population. If and when we receive CE Mark, we plan to conduct a pilot trial in a small number of centers to assess product performance, inform product development activities and determine commercial strategy.
As of December 31, 2017, our cash, cash equivalents and investment securities totaled $20.0 million, which, based on our current operating plans and projections, we believe will be sufficient to fund our operating and capital needs through the first quarter of 2019. Our projections are predicated on us achieving certain minimum levels of sales of Fantom and Fantom Encore. If we are unable to achieve these levels of sales, we may be compelled to reduce operating and capital expenditures or sell certain assets. For information regarding our liquidity and capital resources,
- 3 -
see “Item 1A. Risk Factors— Risks Related to Our Business” and "Part II, Item 7—Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources," below.
Our company was founded in California in June 1998 as MD3, Inc. We changed our name to REVA Medical, Inc. in March 2002. We reincorporated from the State of California to the State of Delaware in October 2010; as a result, the rights of our stockholders are governed by the Delaware General Corporation Law. We formed a wholly owned subsidiary in Germany in 2007 to facilitate our clinical trials and our planned commercialization of products; we have not used this subsidiary yet for any operating activities. Our principal executive offices are located at 5751 Copley Drive, San Diego, California 92111, and our telephone number is (858) 966-3000.
Market Opportunity
Coronary Artery Disease
Cardiovascular disease, or CVD, is a term used to describe all diseases and conditions that relate to the heart and blood vessels in the body. Coronary arteries, which supply blood to the heart, are susceptible to the buildup of plaque and the formation of lesions, which can inhibit or block blood flow, a condition known as coronary artery disease. If arteries become too narrow as a result of plaque buildup, the heart (“cardiac”) muscle may become starved of nutrients and oxygen, resulting in chest pain known as angina. As artery narrowing becomes more severe, death of cardiac muscle downstream from the blockage can occur due to a lack of oxygen. The sudden death of cardiac muscle can result in a heart attack, or “myocardial infarction.”
CVD is a leading cause of death. A January 2015 report published by the World Health Organization cited CVD as the number one cause of death globally, with an estimated 17.5 million deaths in 2012, representing 31 percent of all global deaths. According to the 2017 Update of Heart Disease and Stroke Statistics published by the American Heart Association, or AHA, coronary heart disease accounts for more than 360,000 deaths in the United States, or approximately one in every seven deaths, and coronary artery disease costs an estimated $199.6 billion in direct and indirect costs. According to the AHA, each year about 790,000 people in the United States will have a heart attack.
The European Heart Network reported in early 2017 that ischemic (coronary) heart disease is the single leading cause of death in Europe, accounting for approximately 1.7 million deaths per year, or 14 percent of all male and 12 percent of all female deaths. In addition, the Australia Institute of Health and Welfare reported that coronary artery disease kills more Australians than any other disease, accounting for 21,500 deaths in 2011, or 15 percent of all deaths in Australia. In 2011, an estimated 69,900 people in Australia over the age of 25 had a heart attack.
Current Interventional Treatments for Coronary Artery Disease
There are various methods to prevent, slow progression, reduce symptoms, and reverse (“treat”) coronary artery disease. Lifestyle factors contribute to the development of coronary artery disease and lifestyle interventions such as eating healthy and being physically active are used for prevention and treatment. Evidence shows that healthy lifestyle alternatives are not being universally adopted. As an added therapy, medications such as beta blockers, diuretics, aspirin, nitroglycerin, and calcium channel blockers are used to reduce blood pressure and cholesterol levels and/or aid in the treatment of coronary artery disease. Although lifestyle changes and drug therapy can improve quality of life and prolong survival, genetic factors must also be considered, and many patients will require an invasive surgery or a minimally invasive treatment such as stenting, to improve cardiac health.
Surgical or minimally invasive procedures, developed and used over the past four decades, quickly and safely restore blood flow by either surgically rerouting the flow around a plaque buildup or by reopening the artery with an interventional procedure. As technology has advanced, procedure-related complications have decreased, costs have been reduced, and procedure and recovery times have been shortened. Physicians have rapidly adopted each new advancement. The main treatment options used by physicians and available to patients are:
|
|
• |
Coronary Artery Bypass Surgery: An extremely invasive procedure requiring open heart surgery. The bypass is achieved by removing a vein or artery from somewhere else in a patient’s body and connecting it to the blocked artery, bypassing the blockage. This allows oxygen-rich blood to reach the heart muscle. |
- 4 -
|
|
bypass surgery, the long-term effectiveness of balloon angioplasty is limited by restenosis, a re-narrowing of the artery caused by the elastic recoil of the artery wall or formation of arterial scar tissue. Restenosis typically requires another angioplasty procedure or bypass surgery. Also, some patients experience abrupt vessel closure after angioplasty, leading to complications such as heart attack, emergency bypass surgery, or death. |
|
|
• |
Bare Metal Stents: A minimally invasive therapy introduced in the 1990s to address the issues of abrupt vessel closure and restenosis following balloon angioplasty. Stents are small permanently implanted tube-like devices that are inserted into an artery to prop it open and facilitate blood flow. While bare metal stents minimized the issues and complications of abrupt vessel closure, restenosis continued to be a significant problem. |
|
|
• |
Drug-Eluting Metal Stents: A metal stent that additionally delivers a therapeutic drug to help minimize buildup of scar tissue during healing. After bare metal stents were introduced, physicians determined that restenosis resulted from the trauma of the procedure and stent, rather than from the underlying coronary artery disease. To overcome restenosis, delivery of pharmacological agents from stents was developed; the drugs used range from cytotoxic types (for example, paclitaxel) to immunosuppressants (sirolimus, zotarolimus, and everolimus). Patients usually also undergo treatment with aspirin and anti-clotting or anti-platelet drugs, such as clopidogrel (sold as Plavix) or ticlopidine (sold as Ticlid) after stenting, to reduce the incidence of blood clots, or “thrombosis.” |
In coronary stenting, we believe the key measures of success or failure of the therapy are:
|
|
• |
Target Lesion Revascularization, or “TLR,” which measures the incidence of required re-stenting or bypass surgery due to a failure of the initial coronary angioplasty and stenting; and |
|
|
• |
Major Adverse Cardiac Events, or “MACE,” which are events of cardiac death, ischemia (“heart attack”), or TLR. |
|
|
• |
Bioresorbable Scaffolds: A stent therapy that achieves the benefits of metal stents with the added advantage that the stent dissolves after the artery has healed. After drug-eluting metal stents were introduced and studies showed they successfully lowered the rates of restenosis, safety concerns arose when long-term studies suggested occurrence of late stent thrombosis and TLR. Drug-eluting bioresorbable scaffolds were devised to achieve the proven benefits of drug-eluting metal stents but eliminate the long-term risks of a permanent implant and allow an artery to return to its natural function. While numerous coronary bioresorbable scaffolds have been under development, due to the many technical challenges of the technology, we believe that only three are currently available for sale, all of which are available for sale only under CE Mark. |
Coronary Stent Market
In 2017, annual worldwide sales of coronary stent sales approximated $3.8 billion according to JP Morgan Equity Research Interventional Cardiology Market Model. According to analyst reports, approximate 2017 annual coronary stent sales were:
|
|
• |
$1.4 billion in the United States from approximately 1.2 million stent implants; |
|
|
• |
$2.0 billion in Europe and Asia (excluding Japan) from approximately 4.3 million stent implants; and, |
|
|
• |
$0.4 billion in Japan from approximately 230,000 stent implants. |
Sales of bioresorbable scaffolds began in 2012 in locations outside the United States following CE Mark of Abbott’s Absorb scaffold. Two other companies obtained a CE Mark for their bioresorbable scaffolds in subsequent years and Absorb became commercially available in more than 100 countries, including the United States. Of the worldwide stent sales, bioresorbable scaffold sales were estimated to be $86.0 million in 2013, with an increase to an estimated $127.0 million in 2016. In 2017, Abbott withdrew Absorb from the worldwide market due to low commercial sales. Additionally, Abbott reported high rates of adverse events in its clinical studies. Absorb was made with PLLA. Our bioresorbable scaffold, Fantom, is made from a proprietary material called Tyrocore. The withdrawal of Absorb will have a negative impact on the market for the next several years. We believe that Fantom will be able to rebuild the confidence in and potential for bioresorbable scaffolds as we build clinical and commercial data. See “—Competition” for additional information.
Our Technology
REVA is a leader in bioresorbable polymer technologies for vascular applications. Our bioresorbable polymers are derived from the naturally occurring tyrosine amino acid. The molecular structure of tyrosine includes a phenyl ring. The phenyl ring structure is the backbone for polycarbonate polymers. We have differentiated our polycarbonate
- 5 -
polymers by binding iodine to the phenyl ring. The resulting iodinated polycarbonate polymer can be leveraged in many vascular applications such as coronary artery disease, peripheral artery disease, and embolic particles.
REVA has developed four polymer families based on the iodinated polycarbonate structure. We have also developed expertise and capabilities to develop, manufacture, and test these polymers. We have the ability to tailor the polymer properties to the clinical application by modifying characteristics such as strength, flexibility, degradation and resorption time, and the capability to delivery drugs. The first polymer that we have developed into a commercial product is called Tyrocore. Tyrocore is the material used to make the Fantom bioresorbable scaffold.
We believe Tyrocore offers the following advantages as compared to other polymer-based scaffold materials:
|
|
• |
Strength: Tyrocore is strong owing to the polycarbonate structure that forms the polymer backbone. In addition, the manufacturing process contributes to the strength of the final material. Tyrocore’s strength confers advantageous properties to the final scaffold such as a thinner profile, low recoil, and greater over-expansion range. |
|
|
• |
Visibility: The use of covalently-bound iodine in Tyrocore enables our entire scaffold to be visible under x-ray, including standard fluoroscopy, providing visibility approximating that of metal stents. Other commercially available bioresorbable scaffolds utilize metal “markers” at each end of a scaffold; under x-ray, these metal markers are the only visible portion of those scaffolds and they remain in the vessel wall permanently. Our improved visibility allows interventional cardiologists to more accurately assess the implant quality and position. |
|
|
• |
Biocompatibility: Tyrocore has been tested for biocompatibility in a variety of preclinical and clinical studies. In a 12-month preclinical study of our desaminotyrosine polycarbonate polymer, no adverse biological reactions occurred during the scaffold material degradation. Additionally, we performed human clinical trials with the Fantom bioresorbable scaffold. Imaging results have been reported at the 6, 9, and 24-month time points and demonstrated excellent vessel healing. |
|
|
• |
Degradation and Resorption: Tyrocore degrades into benign metabolites (consisting of monomers, water, and carbon dioxide) that are cleared from the body. Tyrocore is designed to maintain structural integrity for a period of time and then to degrade and resorb. This resorption removes all foreign material from the body. |
We license Tyrocore and related improvements from Rutgers, The State University of New Jersey, or “Rutgers.” See “— Distribution and License Agreements” for additional information. We work in collaboration with Rutgers to continually develop and enhance Tyrocore.
Our Products
REVA’s first commercial product is Fantom, a drug-eluting bioresorbable scaffold. Bioresorbable scaffolds are an alternative to metal stents to hold an artery open during healing after a percutaneous coronary intervention procedure. Unlike metal stents, which are a permanent implant, bioresorbable scaffolds provide support to the artery during the healing process and then degrade and resorb from the artery. This resorption removes all foreign material related to the scaffold from the body.
Bare metal and drug-eluting stents are small, mesh tubes that are placed in the artery during a percutaneous coronary intervention procedure to prop and hold the artery open during healing. These stents are made from a metal alloy and drug-eluting stents are coated with a mixture of drug and polymer. After the drug-eluting stent is placed in the artery, the drug elutes into the vessel wall to reduce vessel narrowing during healing. Over time, the stent heals into the artery wall where it remains as a permanent implant and can interfere with future diagnostic or therapeutic procedures, as well as result in adverse events. In fact, per one study conducted, drug-eluting stents are associated with an annual risk of adverse events of 0.2-0.7% from 5 to 10 years after treatment1.
Bioresorbable scaffolds, on the other hand, become fully encapsulated into the artery wall, and resorb from the body after the artery has fully healed. There are three primary potential benefits of bioresorbable scaffolds compared to bare metal and drug-eluting metal stents:
|
|
1 |
Yamaji K, et al. Ten-year clinical outcomes of first-generation drug-eluting stents: the Sirolimus-Eluting vs. Paclitaxel-Eluting Stents for Coronary Revascularization (SIRTAX) VERY LATE trial. EHJ (2016)37:3386-95. |
- 6 -
|
|
are the only scaffolds that are 100% bioresorbable. All other bioresorbable scaffolds contain small permanent markers for limited x-ray visibility which remain behind once the scaffold is resorbed. |
|
|
• |
Enhanced Applications for Diagnostic Procedures: Metal stents can interfere with non-invasive imaging technologies such as magnetic resonance imaging (MRI) and computed tomography scanning (CT scanning). Bioresorbable scaffolds are made from polymers, which do not interfere with these imaging modalities. |
|
|
• |
Options for Future Medical Treatment: Coronary artery disease is typically progressive and many patients will require additional treatments. We believe that the polymer material of bioresorbable scaffolds and its bioresorption make future medical procedures easier because there is no metal to interfere. A patient may undergo re-stenting, be treated for lesions located in the same artery as the original stent or undergo surgical procedures. These treatments may be inhibited by the existence of a metal stent, whereas the resorption of a scaffold helps to ensure all treatment options remain available. |
Fantom consists of a standard deformable stent design fabricated from our proprietary polymer, Tyrocore, that we coat with sirolimus, a proven anti-proliferative drug. This drug is available from a number of different sources and has been approved by both European and U.S. regulatory bodies. We coat the outside surface of our scaffold using a polymer solution containing a target dose of sirolimus. The Tyrocore used for the coating solution is the same Tyrocore used in the scaffold structure. Through our preclinical studies, we have demonstrated a controlled release of the drug over 30 days; most of the drug is released within 90 days. We believe our release characteristics optimize the efficacy of the drug.
Fantom was designed with the following features that we believe make it commercially competitive with other bioresorbable scaffolds and drug-eluting metal stents:
|
|
• |
Strong and Resilient: Fantom is made with Tyrocore, which, in conjunction with our scaffold design and our method of processing, maintains the strength and structural integrity necessary to support an artery during the critical 90-day healing period after implant. We believe our specific polymer formulation is inherently less prone to cracking and breakage than other polymers. This strength and resiliency supports our thinner struts, a broader over-expansion range and single step inflation; three very important differentiating factors for Fantom. |
|
|
• |
Thin Profiles and Ease of Use: Due to the strength of the Tyrocore polymer, Fantom is designed with a thinner profile than other commercially available bioresorbable scaffolds. Thin profiles have been shown to improve vessel healing and reduce adverse events. Fantom is implanted using a standard balloon catheter and is compatible with a standard 6-french delivery catheter size. Our scaffolds do not require any change to traditional storage or handling or to the method of deployment. |
|
|
• |
Visible Using Standard Imaging Techniques: Fantom is the only commercially available bioresorbable scaffold that is fully visible under x-ray. X-ray visibility allows the physician to see the scaffold during implant to ensure accurate placement within the vessel, which has the potential to reduce adverse events. Competitive bioresorbable scaffolds are designed with metallic radiopaque markers on each end of the scaffold which may provide guidance on the approximate location of the scaffold but do not inform the physician with full details of scaffold deployment. |
|
|
• |
Standard Resorption Rate: Fantom loses its molecular weight within approximately one year after implant, which allows the vessel to move more naturally. It loses its mass over the next three years, with total resorption taking approximately four years. |
|
|
• |
Biocompatible and Safe: Fantom is made with Tyrocore, and has been demonstrated to be biocompatible in preclinical testing. In a 12-month study during which the scaffold was degrading, there was no indication of adverse biological reactions, consistent with the other tests of the polymer. Reported results from the FANTOM II human clinical trial has shown a low rate of Major Adverse Cardiac Events at 24 months. |
In February 2018, we received CE approval for our third generation coronary scaffold, Fantom Encore, in the 2.5 millimeter diameter size. Fantom Encore has thinner struts than Fantom (95 microns versus 125 microns in the 2.5 millimeter diameter size) and comparable strength to Fantom. We anticipate commercializing Fantom Encore later in 2018.
We anticipate allocating a small amount of resources to new product testing in 2018, primarily focused on expanding the size matrix of Fantom Encore and developing a product for a below-the-knee vascular application. The majority of the arteries below the knee have similar sizes and dimensions to coronary arteries, making this a viable treatment
- 7 -
option. Resorbable technology is attractive in this application because of the frequent need for retreatment in this patient population. If and when we receive CE Mark, we plan to conduct a pilot trial in a small number of centers to assess product performance, inform product development activities and determine commercial strategy.
Our Business Strategy
Our goal is to become a world leader in the production and sale of bioresorbable vascular products for use in humans. To achieve this goal, we are pursuing the following business strategies:
|
|
• |
Gain Commercial Acceptance and Demonstrate Clinical Safety and Efficacy of Fantom and Fantom Encore: We are commercializing Fantom in targeted countries based on our CE Mark. The safety and efficacy of Fantom is supported by data reported from the FANTOM II clinical trial which showed low rates of Major Adverse Cardiac Events of 4.2% in 240 patients at 12 months and 5.6% in the initial 125 patients who completed 24 months of follow-up by the fall of 2017. We will continue to follow and report clinical results on patients in the FANTOM II clinical trial through 5 years. In addition, we are conducting trials in more complex patients and launching a post-market trial. We believe the combined clinical and commercial evidence will support the broad adoption of Fantom and Fantom Encore. |
|
|
• |
Expand Geographically: We are executing our commercialization in phases. Initially, we have launched with a direct sales force in Germany, Switzerland and Austria. During 2018 we will evaluate and expand into additional countries that accept the CE Mark. We will go direct or with distributor partners depending on specific market dynamics. The countries we will be considering during 2018 include Italy, Spain, Turkey, Brazil, Belgium, Denmark, and The Netherlands. |
|
|
• |
Advance Fantom Product Line: We intend to continue to enhance Fantom, by refining or adding features and expanding the available sizes. We intend to commercialize Fantom Encore in 2018 and to begin to expand our product size matrix. |
|
|
• |
Build Awareness and Support among Leading Physicians: Our commercial strategies include collaboration with key opinion leaders in the field of interventional cardiology. We believe these key physicians will be advocates of our technology and important in the market adoption of our products once approved. Additionally, we will look to these physicians to generate and publish scientific data that further supports the benefits of our scaffolds. |
|
|
• |
Leverage Our Technology Platform into Other Therapeutic Areas: We believe our technology is applicable to therapies beyond coronary artery disease. For example, we intend to pursue the use of our technology to treat peripheral arterial disease, which is an expanding market. Resorbable technology is attractive for peripheral arterial disease because of the frequent need for retreatment in this patient population. We are pursuing CE Mark for a Tyrocore-based product in a below-the-knee application. Arteries below the knee have similar dimensions to coronary arteries making this a viable treatment option. Treatments for other peripheral artery applications require research and development of polymers with properties designed specifically for those applications. |
|
|
• |
Provide the Highest Quality Products for Our Customers and Patients: We have assembled a team of employees and consultants who are experienced professionals in the medical device industry and who are focused on patient safety and product quality. We incorporate these principles in every aspect of our products, including development, manufacturing, quality assurance, and clinical research. We intend to offer only the highest quality products to patients and physician customers. |
|
|
• |
Expand and Strengthen Our Intellectual Property Portfolio: We plan to continue to expand our current intellectual property portfolio. While we believe that our current portfolio will allow us to effectively market our products for the treatment of coronary artery disease, we plan to create, originate, license, and acquire additional intellectual property to enhance our existing position and enable us to more effectively expand and protect our technology. |
|
|
• |
Explore Licensing Opportunities: We intend to explore opportunities to leverage our intellectual property portfolio by out-licensing our technology, polymer and/or products to third parties or through the establishment of partnerships. For example, we may seek a partner to license our polymer for use as embolic beads. |
Preclinical Testing
We have tested our bioresorbable scaffolds during their development, a period spanning over 12 years. Our preclinical tests show the technology to be safe and effective, with over 1,000 scaffolds tested across various animal
- 8 -
models. Our bench tests confirm the intended product features, with over 30,000 scaffolds tested in various manners. Our preclinical tests generally comprise the following:
|
|
• |
Comparative Testing: We compared our scaffolds to commercially available metal stents and, to a lesser extent, bioresorbable scaffolds. Our tests show that our scaffolds maintain the openness of the artery in the 90 days following implant and allow the lumen size (the inside area of the artery) to increase during resorption. Comparatively, the lumen size of arteries implanted with metal stents was almost unchanged. |
|
|
• |
Strength and Fatigue Testing: We conducted engineering and life cycle tests with equipment that replicates both the physiological conditions in the coronary artery as well as measures the maximum stress levels that our technology can withstand. These tests demonstrated satisfactory scaffold design and polymer strength, low levels of polymer embrittlement, and resistance to fatigue failure prior to significant degradation of the scaffold. |
|
|
• |
Biocompatibility Testing: The biological response to our scaffold has been evaluated by assessing healing in animal coronary arteries using standard microscopy for stented arteries (where the vessel is examined under a microscope). These tests have demonstrated that the polymer is safe and no adverse response occurs in the artery, including while the polymer degrades. |
|
|
• |
Rate of Degradation Testing: Our degradation rate tests demonstrate that our scaffolds maintain their structural integrity and strength for at least 90 days, the healing time of an artery following the implant procedure. By design, at 12 months the scaffold no longer has significant mechanical strength and the polymer continues to resorb and be eliminated from the body for approximately four years. A study of the byproducts resulting from the resorption of our scaffold showed no accumulation in key organs or tissues of the animal’s body and that the byproducts are cleared from the body. |
|
|
• |
Toxicity Testing: Among other tests, we performed an ISO-10993-1 test for genotoxicity. Our test showed that there is no change to the DNA or chromosomes of cells tested and that our polymer is not genotoxic. We have conducted preclinical tests for several other types of toxicity that also demonstrated the polymer is safe. |
|
|
• |
Drug Testing: Implanting a stent can injure an artery and the body’s wound-healing process can cause excessive scar tissue to form inside the stent, referred to as “in-stent restenosis.” The drug sirolimus minimizes overgrowth of tissue, thereby minimizing in-stent restenosis. It has been used in drug-eluting stents, has a demonstrated safety profile, and is proven effective at reducing restenosis. We tested the effects of sirolimus, which we apply to the surface of our scaffold in a coating. Our studies demonstrated no major drug toxicity. |
This testing has shown that our technology was sufficiently safe and effective in animals to support continued product development. We used the data from our preclinical tests in our application for CE Mark of Fantom.
Clinical Studies and Regulatory Strategy
We have targeted Europe, and other countries that recognize the European CE Mark, as our initial commercial markets. For each jurisdiction in which we perform clinical studies or commercialize products, we are subject to significant regulatory requirements, with which we intend to fully comply.
The European Medical Devices Directive 93/42/EEC sets out the essential requirements for clinical studies, product approval, and CE Mark in the European Union; there are numerous other directives and standards regulating the design, manufacture, clinical trials, and labeling for medical devices. We obtained CE Mark approval for Fantom in April 2017. We intend to seek additional regulatory approvals for Fantom in Australia, Japan and China, in addition to seeking a Premarket Approval Application (PMA) in the United States.
Prior to its approval, Fantom had been implanted in 247 patients in the FANTOM I and FANTOM II clinical trials conducted in eight countries outside the United States. We used six-month clinical results from 117 patients in the FANTOM II clinical trial for our CE Mark application. We have reported additional data from the FANTOM II clinical trial which showed low rates of Major Adverse Cardiac Events of 4.2% in 240 patients at 12 months and 5.6% in the initial 125 patients who completed 24 months of follow-up by the fall of 2017. We will continue to follow and report clinical results on patients in the FANTOM II clinical trial through 5 years.
Prior to the FANTOM I and II clinical trials, we enrolled a total of 165 patients in three clinical trials between 2007 and 2014 with predecessor scaffolds, which combined our proprietary “slide and lock” designs with our proprietary polymer formulations. While these predecessor, first generation scaffolds demonstrated viability of the technology, we believed enhanced characteristics were needed and, therefore, developed our Fantom scaffold during 2014.
- 9 -
We have two additional clinical trials of Fantom ongoing evaluating use of our bioresorbable scaffold in broader patient populations and are in the process of initiating a third trial. In the first trial, we are enrolling patients in the FANTOM II Cohort C Study, which is specifically related to more complex patients with lesions longer than those treated in the first two cohorts and patients with multiple vessel disease. We anticipate enrollment of 30 patients in this trial by the end of 2018. In the second trial, we are enrolling patients in a pilot trial to specifically examine the safety and performance of Fantom in ST-elevation myocardial infarction (STEMI) indication. We anticipate enrollment of 10 to 20 patients in this trial by the end of 2018. In addition to those two indication expansion trials, we are in the process of initiating a larger scale post-market registry trial. In this trial we will be assessing the continued safety of Fantom when used in regular clinical practice in approximately 1,500 patients. We expect to begin enrollment in this trial in the second quarter of 2018 and to follow patients through 5 years of clinical follow-up.
We have partnered with a distributor who is seeking regulatory approval in Brazil. We anticipate approval and initiation of commercial sales in Brazil in 2018.
We are currently in discussions with the FDA to conduct a clinical trial in the United States, which is expected to be a randomized trial of between 1,800 and 2,200 patients. We are working towards having conditional Investigational Device Exemption (IDE) approval to conduct the clinical trial by the end of 2018. We will need to obtain additional funding to conduct a U.S. clinical trial.
Manufacturing
Manufacturing of medical devices is subject to strict quality requirements. See “—Government Regulation” for additional information. Our manufacturing operations take place at our facility in San Diego, California. The facility includes laboratories for polymer development and synthesis, chemistry, engineering, and product assembly, including clean rooms and quality control laboratories. We believe our facility has the capacity to produce the quantities of Fantom that will be needed for initial commercial sales and for ongoing clinical trials. An independent third-party has certified that our facility conforms to EN ISO 13485:2012. We may expand our manufacturing beyond our current facility at such point when sales growth forecasts suggest we will reach the capacity of our facility.
The process to manufacture Fantom involves seven main steps, most of which are conducted on-site and some of which are outsourced. Our strategy to outsource selected processes is intended to minimize capital and operating costs while maintaining required quality standards. The manufacturing steps are as follows:
|
|
• |
Polymer Manufacture: Performed at our facility. |
|
|
• |
Polymer Tube Fabrication: Performed at our facility. |
|
|
• |
Lasing of Polymer Tubes: Outsourced to a domestic third party. |
|
|
• |
Drug Coating: Drug purchased from a foreign supplier; coating prepared and applied at our facility. |
|
|
• |
Catheter System: System purchased from a domestic supplier; coating purchased from a foreign supplier. |
|
|
• |
Assembly, Mounting on the Catheter, Quality Assurance, and Packaging: Performed at our facility. |
|
|
• |
Sterilization: Outsourced to a domestic third-party. |
Certain materials used in our products are purchased from, and certain portions of our manufacturing process are completed by, third-parties. See “Item 1A. Risk Factors—Risks Related to Our Business—We are dependent on a limited number of third-party suppliers for delivery system catheters, sirolimus and other components, and the loss of any of these suppliers, or their inability to provide us with an adequate supply of materials that meet our quality and other requirements, could harm our business.”
In February 2017, we entered in an agreement with a third-party logistics provider located in The Netherlands. Under this agreement, the third party is responsible for the warehousing and distribution of our products, as well as order processing and accounts receivable, with respect to our sales in the EU. Products are stored in a temperature-controlled environment and in accordance with ISO 13485 requirements.
Competition
While physicians may recommend alternative treatments for CVD, such as drug therapy, bypass surgery, angioplasty, or bare metal stenting, we expect the primary competition for our products to be drug-eluting stents. The market leaders for metal stents (bare and drug-eluting) are Abbott, Boston Scientific Corporation and Medtronic. The
- 10 -
drug-eluting stent industry is highly competitive. Competitive conditions include technological innovation, price, convenience of use, service, product performance and design, product potential for overall cost-effectiveness, name and brand recognition, relationships with physicians and patients, product safety and the availability of supporting clinical data, sales, marketing and distribution capabilities, and intellectual property protection.
Drug-eluting stents were first available in 2002. Since then, drug-eluting stent technology experienced rapid innovation with improvements in material, design, and clinical performance. Today, product sales from three manufacturers, Abbott, Boston Scientific, and Medtronic, account for approximately 95% of the global drug-eluting stent market with minimal differences in product performance and market share. We expect that future innovation in drug-eluting stents will involve incremental performance improvements that do not translate into meaningful clinical benefit.
Bioresorbable scaffolds are the next innovation in CVD treatment that has the potential to deliver a meaningful improvement in clinical benefit for patients. Of the three major drug-eluting stent manufacturers, only Abbott has commercialized a bioresorbable scaffold. Abbott began selling its bioresorbable scaffold, named Absorb, outside of the United States in 2012 and achieved rapid adoption and early commercial success. During this time, Abbott conducted multiple large clinical studies and then secured FDA approval in July 2016. In September 2017, Abbott withdrew Absorb from the worldwide market due to low commercial sales. Additionally, Abbott reported high rates of adverse events in its clinical studies. The clinical outcomes and resulting withdrawal of Absorb has had a significant negative impact on the adoption of bioresorbable scaffolds.
Today there are three categories of bioresorbale scaffolds based on the scaffold material: polylactic acid (also known as polylactide or PLLA), magnesium alloy, and Tyrocore, which is REVA’s proprietary polymer for Fantom. PLLA is the most commonly used material for bioresorbable scaffolds. Scaffolds with CE Mark in this category include Absorb from Abbott and DESolve from Elixir Medical. Based on clinical trials of which we are aware, a number of other companies are developing PLLA bioresorbable scaffolds, including Amaranth Medical and Meril Life Sciences. The adverse events reported in the Absorb clinical studies have raised concerns about the utility of PLLA as an appropriate material for bioresorbable scaffolds. Today there is minimal commercial activity from companies with PLLA scaffolds. Abbott stated that it is developing a next generation polylactic acid bioresorbable scaffold, which it could advance into clinical trials in the future.
In June 2016, Biotronik received CE Mark for its Magmaris magnesium alloy bioresorbable scaffold based on data from 118 patients enrolled in the BIOSOLVE II clinical trial. Magmaris is a first generation scaffold with a similar strut profile to Absorb, and the magnesium alloy is not visible under x-ray. Magmaris offers a limited size matrix with only six sizes available. The ability of Biotronik to obtain reimbursement for and secure adoption of its scaffold may further define the bioresorbable scaffold market.
Fantom and Fantom Encore are made from our proprietary polymer called Tyrocore. We secured CE Mark for Fantom in April 2017 and for the 2.5 mm diameter of Fantom Encore in February 2018. Compared to first generation PLLA and magnesium alloy scaffolds, Fantom and Fantom Encore offer thinner strut profiles, improved ease-of-use, and full x-ray visibility. Fantom and Fantom Encore face competition from the negative perceptions of bioresorbable scaffolds created by the market withdrawal of Absorb and from drug-eluting stents. This competition will have a negative impact on our commercialization activities for the next several years while we generate clinical data and gain commercial experience. However, we believe that Fantom and Fantom Encore are differentiated from Absorb and other bioresorbable scaffolds based on their unique features and that we will be able to rebuild the confidence in bioresorbable scaffolds.
Products in the CVD market, including our products, can be subject to rapid product adoption or obsolescence, as well as regulatory changes. We compete with established medical device companies, as well as earlier-stage companies that often have differentiated technology and potentially superior solutions. Although we believe Fantom has technological advantages over competitive products, these advantages may be reduced or eliminated, should competitors introduce new products. We also compete to recruit and retain qualified scientific and management personnel, establish clinical trial sites and patient registrations, and acquire technologies complementary to our programs or advantageous to our business.
See “Item 1A. Risk Factors—Risks Related to Our Business—We compete against companies that have longer operating histories, more established or approved products, and greater resources, which may prevent us from achieving market penetration or improving operating results” for additional information regarding our competition and the competitive landscape.
- 11 -
Since inception, we have devoted a significant amount of resources to develop our technology. Our research and development expenses, which include the costs to conduct our human clinical trials, were $12.8 million in 2017, $18.2 million in 2016 and $16.8 million in 2015. We expect our research and development expenditures to decrease in 2018 as we dedicate a significant portion of our resources to the commercialization of Fantom and supporting registry studies. If and when we successfully establish commercial feasibility of Fantom, we expect our research and development expenses to increase and we may focus on new product development and initiation of new clinical trials of Fantom, including, if we receive FDA approval for it and if we have raised sufficient capital to support it, an FDA clinical trial of Fantom.
Sales and Marketing
We commenced commercial activities for Fantom during the second quarter of 2017 and revenues were first recorded in the third quarter of 2017. We have built a small sales force that is initially focused on a small number of targeted accounts to drive adoption and prove the commercial viability of Fantom. We plan a broader roll-out following the initial sales efforts, mostly using independent distributors.
Generally, we expect our planned targeted roll-out to occur as follows:
|
|
• |
Initial Markets: The European Union, or EU, will be our initial commercial market target since the CE Mark is our first regulatory approval. Our initial countries of focus are Germany, Switzerland and Austria. |
|
|
• |
Follow-on Markets: Other countries that accept the CE Mark or the clinical data from the FANTOM II clinical trial, including Italy, Spain, Brazil and select countries in the Middle East, will be our second commercial market targets. |
|
|
• |
Longer Term: We will target the United States, Australia, Japan and China commercial markets in the future because they require more extensive, and expensive, clinical trials. |
We have considered many aspects of commercial sales, including product pricing. In most countries, a significant portion of medical expenses is covered by third-party payors. Reimbursement in the EU varies by country and often by hospital. We believe that numerous hospitals have established budgets to purchase coronary stents and the purchase decision is often driven by the interventional cardiologists working at those hospitals. Since bioresorbable scaffolds were introduced to the market in 2012, they have traditionally been priced at a significant premium to drug-eluting stents. Fantom is priced at a premium as well, which may hinder or prohibit its broad adoption.
We will continue to monitor and, if we believe appropriate, revise our sales and marketing plans and strategy based on market changes and acceptance, competitor activities, and timing of regulatory approvals.
Third-Party Reimbursement
In most countries, a significant portion of patient medical expense is covered by third-party reimbursement, consisting of both government-funded and private insurance programs. While each payor develops and maintains its own coverage and reimbursement policies, the vast majority of payors have established policies for stents. We believe that our products generally will fall within existing reimbursement guidelines, or within new reimbursement guidelines that were established by other bioresorbable scaffold companies, although some refinement in policies may be needed for our products. Some countries may require us to gather additional clinical data before agreeing to coverage and reimbursement for our products. We intend to complete the requisite clinical studies and obtain coverage and reimbursement approval in countries where it makes economic sense to do so. To date, the customers who have purchased Fantom in the European countries in which it has been sold have been able to obtain third-party reimbursement. However, reimbursement guidelines differ from region to region. Before we can obtain reimbursement for our products in Australia, Japan, China, the United States, or any other country, we will need to obtain appropriate regulatory approvals for product sales in such countries.
Intellectual Property
We rely on a combination of patents, trade secrets, and copyrights, together with non-disclosure and confidentiality agreements, to establish and protect our proprietary rights in our technologies. Our patents and patent applications covering the fundamental technology underlying our “slide and lock” design have been developed internally, while the technology underlying our polymer has been either licensed or developed by us.
- 12 -
As of February 23, 2018, on a worldwide basis, our patent portfolio comprised approximately 281 issued and pending U.S. and foreign patents that we own directly or for which we are the licensee. Our latest patent expiration date with respect to these patents is 2036. We have been issued 52 U.S. patents and have 11 U.S. patent applications that are pending examination or have been allowed by the United States Patent and Trademark Office. For these 63 technology patents and applications, we have sought intellectual property protection outside the United States and have been granted 189 foreign patents and have 29 pending foreign applications. We do not know if any of our pending patent applications will be issued, nor do we know whether our patents, if issued, will adequately cover our technology or will be able to be successfully enforced. Even if valid and enforceable, our patents may not be sufficiently broad to prevent others from inventing a scaffold like ours, despite our patent rights. We believe that the remaining lives of our patents provide adequate time to generate revenues from sales, subject to timing of the clinical pathway and regulatory approvals.
We actively monitor our intellectual property position and review new developments to identify prudent extensions to our patent portfolio to ensure protection of our key technology, as well as to maximize our defensive strategy through the coverage of similar technology developments. We employ an in-house patent attorney and utilize external patent counsel to assist us in managing our intellectual property portfolio. The stent industry has been subject to numerous patent filing and infringement lawsuits. Whether we would, upon commercialization, infringe any patent claim will not be known with certainty unless and until a court interprets a patent claim in the context of litigation. If an infringement allegation is made against us, we may seek to invalidate the asserted patent claim and may allege non-infringement of the asserted patent claim. In order for us to invalidate a U.S. patent claim, we would need to rebut the presumption of validity afforded to patents issued in the United States with clear and convincing evidence of invalidity, which is a high burden of proof. To date, none of our patents or patent applications has been subject to reexamination, interference, or other legal challenge.
We require all employees to sign confidentiality and invention assignment agreements under which they are bound to assign to us inventions made during the term of their employment. These agreements prohibit our employees from using, disclosing, or bringing onto the premises any proprietary information belonging to a third party. In addition, our consultants are required to sign agreements under which they must assign to us any inventions that relate to our business. These agreements also prohibit our consultants from incorporating into any inventions the proprietary rights of third parties without informing us. It is our policy to require all employees to document potential inventions and other intellectual property in laboratory notebooks and to disclose inventions to patent counsel in written form.
We also rely on confidentiality restrictions and trade secrets to protect our technology. We generally require our consultants and other parties who may be exposed to our proprietary technology to sign non-disclosure agreements which prohibit such parties from disclosing or using our proprietary information except as may be authorized by us.
Distribution and License Agreements
Boston Scientific Corporation Agreement
In 2007, we entered into a distribution option agreement with Boston Scientific Corporation, or BSC, in which we granted BSC an option to negotiate country-by-country or worldwide exclusive rights to sell, market, and distribute our scaffolds. BSC’s election window to exercise the option was triggered in 2017 when we delivered an extensive set of positive clinical data supporting Fantom’s performance. BSC did not exercise the option and it expired in August 2017. With expiration of the option, all aspects of the distribution option agreement are now concluded.
Rutgers License
In July 2010, we entered into an exclusive license agreement with Rutgers, The State University of New Jersey (“Rutgers”) that superseded the exclusive license agreement we entered into with Rutgers in 2004. Under the 2010 license agreement, we have an exclusive, worldwide right, including sublicensing rights, to develop and commercialize products that utilize certain polymers in the vascular field. Terms of this agreement require us to pay annual license fees to Rutgers until a product is commercially sold in a major market. Also, to maintain our rights under this agreement, we must achieve certain development and commercialization milestones. The term of the this agreement continues until the expiration of the last to expire of the patents licensed to us, which we believe is 2036. Rutgers may sublicense certain technology that Rutgers invented, we jointly invented with Rutgers, or that we solely invented, outside our field of use. If Rutgers sublicenses inventions and improvements solely owned by us, Rutgers will pay us a percentage of all income and consideration Rutgers receives from such sublicenses.
- 13 -
The royalties due under the 2010 license agreement vary depending upon type of product, use of product, stage of product, location of sale, and ultimate sales volume and price. We believe the royalties will range from a minimum of approximately $15 to a maximum of approximately $50 per product sale, with license provisions for escalating minimum royalties that could be as high as $2.2 million per year. Additionally, in the event we receive certain milestone payments related to this technology, the agreement requires that up to 40 percent of the milestone amount be paid to the licensors. The agreement required annual licensing payments of $175,000 until the underlying technology was commercialized in 2017. The agreement also requires other payments to occur during commercialization that could total $950,000, payment of $350,000 upon a change in control of our ownership, payments of up to $300,000 annually to extend regulatory filing periods related to certain technology (of which, payments totaling up to $250,000 per year during the years 2016, 2017, and 2018 may be deferred to January 1, 2019), and payment of patent filing, maintenance, and defense fees.
Government Regulation
United States
We do not currently have any products approved for sale in the United States. We are in discussions with the FDA regarding our preclinical data and our clinical trial strategy for Fantom. We believe that these discussions will continue throughout 2018. In order to commence a clinical trial in the United States, we will need to obtain additional financing.
Fantom (and Fantom Encore) is considered a combination product because it combines two regulated components in a single product: a drug and a medical device. In the United States, the FDA assigns the review of a combination product, based on the product’s “primary mode of action,” to one of its centers, such as the Center for Drug Evaluation and Research (“CDER”) or the Center for Devices and Radiological Health, or CDRH. The center to which the product is assigned will have primary jurisdiction over the PMA of the product.
Because the primary mode of action for Fantom is that of a medical device, when, and if, we apply for approval in the United States, Fantom will be reviewed by the FDA under the Federal Food, Drug, and Cosmetic Act with CDRH having primary responsibility for review and regulation of our products. As a result, we expect our clinical trial of drug-eluting scaffolds to be conducted under an IDE application in accordance with 21 CFR Part 812. However, it is possible the FDA may assign our products to CDER. Based on FDA precedent and jurisdictional statements to date, we believe that the drug component of our products will not require separate FDA approval and that it will be reviewed in the context of our PMA, with CDRH consulting with CDER as needed. Even if the FDA assigns our products to be regulated by CDER, the drug component of the product will likely not require separate CDER approval but will be evaluated in the context of our PMA as a whole, with application of drug standards as deemed appropriate by FDA based on the circumstances.
FDA regulations govern the following activities that we and our suppliers, licensors, and partners perform and will continue to perform to ensure that the products we distribute domestically or export internationally are safe and effective for their intended uses:
|
|
• |
product design, development, and testing; |
|
|
• |
product manufacturing and production; |
|
|
• |
product safety; |
|
|
• |
product labeling and storage; |
|
|
• |
record keeping; |
|
|
• |
premarket approval; |
|
|
• |
advertising and promotion; |
|
|
• |
product sales and distribution; and, |
|
|
• |
post-marketing requirements including monitoring for and reporting of adverse events and malfunctions. |
Clinical Trials: Clinical trial data is almost always required to support a PMA application. Clinical trials of our scaffolds in the United States will require submission of an IDE application, supported with appropriate data, and approvals by the FDA and institutional review boards. Clinical trials must be conducted in accordance with applicable regulations and must adhere to extensive record keeping and reporting requirements. We, the FDA, or the institutional review board at a clinical site may suspend a clinical trial at any time for any reason, including a belief that the risks
- 14 -
to the patients in a trial outweigh the anticipated benefits. U.S. clinical trials of the scope we anticipate for our products can typically take years to complete and may encounter challenges at any stage that may require a trial to be halted.
Premarket Clearance and Approval Requirements: The FDA classifies medical devices into one of three classes. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting, or implantable devices or devices not substantially equivalent to a previously cleared 510(k) device, are placed in Class III, requiring PMA. Our scaffolds are Class III devices and will require FDA approval. A PMA must be supported by extensive data, including but not limited to, technical, preclinical, clinical, manufacturing, and labeling to demonstrate to the FDA’s satisfaction the safety and efficacy of the device. A PMA must also contain a full description of the device and its components and a full description of the methods, facilities, and controls used for manufacturing of the device.
Product Modifications: New PMAs or PMA supplements are required for all significant modifications to a manufacturing process, labeling, use, or design of a device that is approved through the PMA process. PMA supplements often require submission of the same type of information as an initial application, except the supplement is limited to information needed to support the device changes. Certain modifications may not require as extensive clinical data or the convening of an advisory panel.
Pervasive and Continuing Regulation: When a device is approved for sale, numerous regulatory requirements apply to the commercial product. These include:
|
|
• |
Good Manufacturing Practices, or GMP, and Quality System Regulations, or QSR, that require manufacturers, including third-party suppliers, to follow stringent design, testing, control, documentation, and other quality assurance procedures during all aspects of the manufacturing process; |
|
|
• |
labeling and promotion regulations, which limit the manner in which companies can market their products and impose requirements for content and format of labeling and promotional materials, and FDA prohibitions against promotion of products for unapproved or “off-label” uses; |
|
|
• |
medical device reporting regulations, which require manufacturers to report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur; |
|
|
• |
post-market surveillance regulations, which will apply when necessary to protect the public health or to provide additional safety and efficacy data for the device; and, |
|
|
• |
specific conditions of approval that may be imposed on a specific PMA. |
The FDA has broad post-market and regulatory enforcement powers. When, and if, we are approved to sell in the United States, we will be subject to unannounced inspections by the FDA and the Food and Drug Branch of the California Department of Health Services to determine our compliance with QSR and other regulations. The manufacturing facilities of our suppliers may also be inspected by the FDA or other regulatory authorities to determine their compliance with GMP regulations. The FDA monitors marketing and promotional activities for matters of concern and may receive complaints from competitors or other third parties regarding our products.
In addition, discovery of previously unknown problems with a medical device, manufacturer, or facility may result in restrictions on the manufacturing or marketing of an approved device, including costly recalls or withdrawal of the device from the market. The FDA also has the authority to require repair, replacement, or refund of any medical device that has been manufactured or distributed. Failure to comply with applicable regulatory requirements may result in enforcement action being taken by the FDA, which may include any of the following sanctions:
|
|
• |
inspectional observations or warning letters, identifying concerns that must be corrected; |
|
|
• |
fines, injunctions, consent decrees, and civil penalties; |
|
|
• |
recall or seizure of our products; |
|
|
• |
operating restrictions, partial suspension, or total shutdown of production; |
|
|
• |
refusing our requests for PMA or new intended uses; |
|
|
• |
withdrawing PMA approval; and/or, |
|
|
• |
criminal prosecution. |
- 15 -
Fraud and Abuse: We are directly, or indirectly through our business associates, subject to various state and federal fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute and the False Claims Act. These laws may impact, among other things, our proposed sales and marketing programs. The federal Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering, receiving, or providing payments, directly or indirectly, in exchange for, or to induce, either the referral of an individual, or the furnishing or arranging for a good or service, for which payment is made under a federal program such as Medicare or Medicaid. This statute is broad and prohibits many arrangements and practices that are lawful outside the health care industry. Recognizing that this statute is broad and may technically prohibit many innocuous or beneficial arrangements, Congress authorized a series of safe harbor regulations. The safe harbors set forth provisions that give some assurance to health care providers and others that they will not be prosecuted. The failure of a transaction or arrangement to fit precisely within a safe harbor does not necessarily mean that it is illegal or that prosecution will be pursued; however, conduct and arrangements that do not fully satisfy each applicable safe harbor may result in increased scrutiny by enforcement authorities. All parties to a prohibited transaction may be prosecuted, whether any party sought or received payment from any federally funded program. Penalties for violations of the Anti-Kickback Statute include criminal and civil sanctions such as fines, imprisonment, and possible exclusion from Medicare, Medicaid, and other federal health care programs. Many states have adopted laws similar to the federal statute.
The U.S. False Claims Act prohibits persons from knowingly filing, or causing to be filed, a false claim or using false statements to obtain payment from the federal government. Suits filed under the False Claims Act, known as “qui tam” actions, can be brought by any individual on behalf of the government and such individuals, commonly referred to as “whistleblowers,” may share in any amounts paid to the government in fines or settlement. The frequency of filing of qui tam actions has increased significantly in recent years, causing more health care companies to defend False Claims. When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties ranging from $5,500 to $11,000 for each separate False Claim. Various states have also enacted laws modeled after the federal False Claims Act. Similarly, the federal Civil Monetary Penalty statute imposes penalties of up to $50,000 per violation for filing certain types of proscribed claims or engaging in prohibited acts.
In addition to the laws described above, the Health Insurance Portability and Accountability Act of 1996 created two new federal crimes: health care fraud and false statements relating to health care matters. The health care fraud statute prohibits knowingly and willfully executing a scheme to defraud any health care benefit program, including those of private payors. A violation of this statute is a felony and may result in fines, imprisonment, or exclusion from government sponsored programs. The false statements statute prohibits knowingly and willfully falsifying, concealing, or covering up a material fact or making any materially false, fictitious, or fraudulent statement in connection with the delivery of, or payment for, health care benefits, items, or services. A violation of this statute is a felony and may result in fines or imprisonment.
The penalties for violating any of the laws described above or other applicable state and federal fraud and abuse laws, include civil and criminal penalties, damages, fines, exclusion from government health care programs, and the operating sanctions.
Patient Protection and Affordable Care Act: Our operations may be impacted by the federal Patient Protection and Affordable Care Act of 2010, as modified by the Health Care and Education Reconciliation Act of 2010, which is referred to as the Affordable Care Act, or ACA. Among other things, the ACA imposed a 2.3 percent excise tax on sales of medical devices sold in the United States and intended for human use; such excise tax is suspended through 2019 and we are unable to predict whether the suspension will be continued beyond 2019. There is no exemption for small companies. If not permanently eliminated, we believe the tax will apply to our scaffolds when we begin commercial sales of our products in the U.S. The ACA also requires (under what are referred to as “Sunshine” or “Open Payments” requirements) manufacturers of covered devices to report details regarding certain payments and other financial arrangements with physicians and teaching hospitals. These reporting provisions preempt state laws that require reporting of the same information, but not those that require reports of different or additional information. Failure to comply results in significant civil monetary penalties. We expect compliance with the ACA to impose significant administrative and financial burdens on us.
Environmental Regulation: We are subject to numerous federal, state, and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control, and disposal of hazardous substances. Some of these laws require us to obtain licenses or permits to conduct our operations. We have numerous policies and procedures in place to ensure compliance with these laws and to minimize the risk of occupational exposure to hazardous materials. We do not expect our operations to produce quantities of hazardous or toxic waste or radiation that would require the use of extraordinary disposal practices. Although the costs to comply
- 16 -
with these laws and regulations have not been material, we cannot predict the impact of new or amended laws or regulations or any changes in the way existing and future laws and regulations are interpreted or enforced, nor can we ensure we will be able to obtain or maintain any required licenses or permits.
International
International sales of medical devices are subject to foreign governmental regulations, which vary substantially from country to country. The time required to obtain clearance or approval by a particular country may be longer or shorter than that required for FDA clearance or approval, and the requirements may be different.
The primary regulatory environment in Europe is the EU, which consists of 28 countries. Three members of the European Free Trade Association, Iceland, Norway, and Liechtenstein have voluntarily adopted medical device laws and regulations that mirror those of the EU. Other countries, such as Switzerland, have entered into Mutual Recognition Agreements, or MRA, and allow the sale of medical devices that meet EU requirements.
The EU has three core directives concerning medical devices: Medical Devices Directive, or MDD, In-Vitro Diagnostic Medical Devices Directive, and Active Implantable Medical Devices Directive. Also, the European Committees for Standardization have set forth voluntary standards regulating the design, manufacture, clinical trials, labeling, and adverse event reporting for medical devices. Prior to marketing or using a medical device in the EU, it must undergo a conformity assessment process as set forth in the relevant medical devices directives (Conformité Européenne, or CE). Once a medical device is approved for CE Mark, it can be commercially distributed in the EU, the member states of the European Free Trade Association, and countries with MRAs. The method of assessing conformity varies depending on the type and class of product, but normally involves a self-assessment by the manufacturer and an assessment by a third-party notified body, an independent and neutral institution appointed in an EU country. The assessment may also include an audit of the manufacturer’s quality system and specific testing of the device for compliance with ISO 13485, which are voluntary harmonized standards. Each EU member country implements the MDD into national laws that are enforced by a competent authority in that country. For example, the authority in the United Kingdom is the Medicines and Healthcare Products Regulatory Agency. In addition to obtaining CE Mark, many EU countries require completion of a formal registration process before products can be commercially sold. This in-country process may delay our ability to commercialize.
Before any medical device can be supplied within Australia, it must be included on the Australian Register of Therapeutic Goods and comply with the provisions of the Australian Therapeutic Goods Act. While much of the documentation produced for obtaining the CE Mark in Europe can be used to obtain registration in Australia and the regulatory requirements are similar to European regulations, compliance generally requires the following:
|
|
• |
full technical documentation demonstrating compliance to all relevant standards and regulations; |
|
|
• |
full quality assurance certification to the key international standard; and, |
|
|
• |
the ability of the manufacturer to undertake post market surveillance processes. |
Employees
As of December 31, 2017, we had 51 employees, 50 of whom were full-time. A total of 18 were in research and development, 19 were in manufacturing and 14 were in selling, general and administrative functions. We have never had a work stoppage and none of our employees are covered by collective bargaining agreements or are represented by a labor union.
Available Information
We make available on our website, free of charge, our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and any amendments to those reports, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Australian Securities Exchange, or the ASX, and the U.S. Securities and Exchange Commission, or the SEC. Our website address is www.revamedical.com. Our SEC reports can be accessed in the Investor Relations section of our website. The public may read and copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Room 1580, Washington, DC 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an internet site that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC at www.sec.gov. The information contained on the websites referenced in this Form 10-K is not part of this or any other report we file with or furnish to the SEC.
- 17 -
Our executive officers and their ages and backgrounds as of February 23, 2018, are as follows:
Regina E. Groves, age 59, was appointed as our Chief Executive Officer in September 2015. Her background encompasses over 30 years in medical devices, executive leadership, and financial management. Prior to joining REVA, from 2008, Ms. Groves served as Vice President and General Manager of AF Solutions, Cardiac Rhythm and Heart Failure division of Medtronic, a leading global medical technology company. Previously she held other senior positions at Medtronic, McKinsey & Company, Inc., and several health care companies. Ms. Groves received her M.B.A. from Harvard Graduate School of Business Administration and her B.S. in Pharmacy from the University of Florida.
Brandi L. Roberts, age 44, has served as our Chief Financial Officer and Corporate Secretary since August 2017. Her experience encompasses over 20 years of public accounting and finance experience, including 18 years at publicly traded pharmaceutical, medical technology, and life science companies. Ms. Roberts most recently served as Chief Financial Officer at Mast Therapeutics, Inc., a publicly traded US-based biopharmaceutical company. Previously she held senior positions at Alphatec Spine, Artes Medical, Stratagene and Pfizer. She is a certified public accountant and received her B.S. in Business Administration from the University of Arizona and her M.B.A. from the University of San Diego.
Jeffrey A. Anderson, age 51, has served as our Senior Vice President of Clinical and Regulatory affairs since December 2013 and as our Vice President of Clinical and Regulatory affairs since February 2011, a position he previously held at REVA from 2004 to 2008. He has over 25 years of experience in the medical device industry, including his positions of Vice President of Clinical & Regulatory Affairs and Vice President of Research & Development for Neomend, a biomedical device company engaged in the development and commercialization of surgical wound healing products, where he served from October 2008 through February 2011. Additionally, Mr. Anderson has held senior positions at Abbott Vascular, Jomed, CRS Clinical Research, and Medtronic. He received his B.S. in Physics from California State University at Fullerton.
Richard M. Kimes, age 56, has served as our Senior Vice President of Operations since January 2016. His background comprises over 25 years of medical device operations. Prior to joining REVA, Mr. Kimes was President of Advantage Consulting, a firm specializing in operations management, since December 2013. From May 2013 to December 2013, he was Executive Vice President of Operations for Elixir Medical Corporation, a stent company. Prior to that, from 2009 through May 2013, he was Senior Vice President of Operations for Volcano Corporation, a medical imaging equipment company. He has also held senior positions with mNemoscience GmbH, Guidant Corporation, IMED Corp., and Becton Dickinson Corporation, all medical device companies. Mr. Kimes received his B.S. in Mechanical Engineering from the University of Utah.
You should carefully consider the risks described below and all of the other information set forth elsewhere in this Form 10-K, including our consolidated financial statements and the related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” in evaluating our business, our prospects, and our securities. If any of the risks, events or developments described below occurs, our business, financial condition, results of operations, and the price of our CDIs and common stock could be materially and adversely affected. Additional risks and uncertainties not presently known to us, or risks we currently consider immaterial, could also affect our business, financial condition, results of operations, and the price of our CDIs and common stock.
Risks Related to Our Business
Our available cash is limited and we will need additional funding to pursue our long-term business strategy; there can be no assurance that we will be able to obtain such funding on a timely basis or on commercially reasonable terms, if at all. If we are unable to raise additional funding, there would be a material adverse effect on our business, including our ability to remain in business.
As of December 31, 2017, we had cash, cash equivalents and investment securities of approximately $20.0 million, which we believe will be sufficient to fund our operating and capital needs through the first quarter of 2019. As of December 31, 2017, we had current liabilities and long-term liabilities of approximately $2.7 million and $112.8 million, respectively, including convertible notes with an aggregate face value of $72.1 million that mature or that we
- 18 -
may be required to redeem in November 2019. See “We have a significant amount of indebtedness that we may not be able to repay in accordance with its terms,” below. To pursue our long-term business strategy, we will need additional capital. We have a plan to address our capital needs, which includes pursuing sales expansion and executing business development and strategic opportunities. We are also evaluating public or private sales of our equity or debt securities. However, we may not be able to successfully execute our plan or obtain sufficient additional funding through any of those alternatives on satisfactory terms, if at all. If we are unable to execute our plan or secure additional funding when needed, including if needed to repay our outstanding convertible notes, we may need to reduce operating activities and personnel, sell assets, such as our intellectual property, and/or declare bankruptcy, and we may not be able to remain in business.
We have a significant amount of indebtedness that we may not be able to repay in accordance with its terms.
The convertible notes we issued in 2014, which have a face value of $25.0 million and accrue interest at the rate of 7.54 percent per annum, compounded annually, mature in November 2019. The convertible notes we issued in 2017, which have a face value of $47.1 million and accrue interest at the rate of 8.00 percent per annum, compounded annually, mature in May and June 2022. Each holder of the 2017 convertible notes has a one-time right to require us to redeem such holder’s note (face value plus accrued interest) in November 2019. Accordingly, we may be required to repay an aggregate of $72.1 million plus accrued interest in November 2019. If the noteholders collectively, or individually, call for redemption of the 2017 convertible notes, or if we are unable to convert or extend the maturity date of the 2014 convertible notes, we most likely would not have the cash to repay the notes, and the noteholders could commence legal action against us and/or we may need to reduce operating activities and personnel, sell assets, such as our intellectual property, and/or declare bankruptcy, and we may not be able to remain in business.
In addition, the convertible notes include certain events of default, including without limitation failure to make a payment obligation and failure to observe other covenants. In the event of default, the noteholders have the right to call for the immediate redemption of their notes.
We have a history of net losses and negative cash flows and we may never achieve or maintain profitability.
We are in the very early stages of commercialization. We have incurred net operating losses since our inception, including net operating losses of approximately $21.3 million, $26.8 million and $24.0 million for the fiscal years ended December 31, 2017, 2016 and 2015, respectively. As of December 31, 2017, our accumulated deficit was approximately $382.2 million. Although we initiated commercialization activities in 2017, based on our current expectations, it will take a significant amount of time to generate sufficient revenues to cover anticipated costs, and we may never achieve or maintain profitability.
Unless we are able to significantly accelerate our sales, we expect to continue to incur significant operating losses and cash outflows through 2018 and 2019 as we incur costs associated with, among other matters:
|
|
• |
seeking regulatory approvals in the EU, Australia, Japan, China and United States for Fantom and/or Fantom Encore; |
|
|
• |
expanding our manufacturing capabilities, broadening our infrastructure, and initiating and growing sales and marketing capabilities to commercialize our products; and |
We cannot predict the extent of our future operating losses and accumulated deficit, we may never generate sufficient revenues or positive cash flow to achieve or sustain profitability, and we may be unable to repay our convertible notes when required to do so, either at maturity or earlier. To become and remain profitable, we must succeed in commercializing products with significant market potential. This will require us to succeed in a range of challenging activities, including those listed above. We may not succeed in these activities and we may be unsuccessful in developing alternatives; therefore, we may not ever achieve profitability. If we do achieve profitability, we may not be able to sustain it. Our failure to achieve or sustain profitability could negatively affect the value of our securities
- 19 -
and our ability to attract and retain personnel, raise capital, execute our long-term business strategy or continue operations.
In addition to our current capital needs, we may need additional funding in the future to continue to meet our operating, capital, and debt service needs, and we may be unable to raise capital when needed or on acceptable terms.
Our future operating and capital requirements will depend on many factors, including the timing and achievement of regulatory approval of our products, the growth of revenue, the amount of intellectual property and technology expenditures, the number and size of our clinical trials, the extent of new product development, and the timing of repayment of our convertible notes, should they become due and payable. Until we generate a level of revenue to support our cost structure, we expect to continue to incur substantial net cash outflows and we may need to raise additional capital in the future to continue to meet our operating, capital, and debt service needs. We may not be able to obtain sufficient additional funding on satisfactory terms, if at all. Additionally, we may be limited under the terms of our convertible notes as to the type, quantity, timing, or other aspects of a financing, unless the noteholders agree. See also, “Raising additional capital may cause dilution to our existing stockholders, require us to relinquish proprietary rights or restrict our operations,” below.
Because our need for capital arises as a result of significant past cash outflows and losses, the continuing occurrence of losses and cash outflows may make it difficult for us to raise necessary capital when needed, which would force us to delay, reduce, or eliminate our product development programs or commercialization efforts. In addition, we may incur substantial costs in connection with evaluating and negotiating future capital-raising transactions, the effect of which may be to shorten the period through which our current operating funds will sustain us. Even if we incur costs in pursuing, evaluating and negotiating particular capital-raising transactions, our efforts may not prove successful.
Our ability to generate revenue depends upon receipt of regulatory approval and successfully commercializing our scaffolds, which we may be unable to accomplish.
On April 3, 2017, our first product, Fantom, was approved for sale under a CE Mark, which allows us to commercialize in Europe and other jurisdictions that recognize the CE Mark. On February 21, 2018, our second product, Fantom Encore in the 2.5 millimeter diameter size, was approved for sale under a CE Mark. To successfully execute our long-term business strategy, we believe will need to obtain regulatory approval for other markets, including the United States. Once approved, if approved, our products will also require significant marketing and distribution efforts before they can generate any revenue. Our efforts to generate revenue may not succeed for many reasons including:
|
|
• |
we may not receive regulatory approvals in the markets we seek; |
|
|
• |
our products may not be accepted in the marketplace by physicians and patients; |
|
|
• |
by offering only one type of product, we would not have the ability to bundle products to drive sales; |
|
|
• |
physicians may not receive adequate coverage and reimbursement for procedures using our products; |
|
|
• |
we may not be able to manufacture or distribute our products in commercial quantities at an acceptable cost; |
|
|
• |
new product introductions by our competitors or any rapid technological change may make our technology and product candidates obsolete; |
|
|
• |
our Fantom scaffold may not continue to demonstrate the same safety and efficacy results in the long-term that we have seen in the short-term and, therefore, may not be commercially supported; and |
|
|
• |
we may be sued for infringement of intellectual property rights which could prevent us from manufacturing or selling our products. |
We cannot market our products in the United States until we receive a premarket approval, or PMA, from the FDA. Our long-term business strategy is based in part on our expectations regarding the timing for receipt of regulatory approvals and if we experience delays in the approval process, or ultimately do not receive approval, we may be unable to reduce our expenditures in a timely manner to compensate for such delay or denial, and we may not have adequate financial or other resources to complete the approval process or continue in business. Accordingly, a significant delay in the regulatory approval process, or a denial of approval, would have a material adverse effect on our ability to successfully sell our products and on our financial condition.
- 20 -
We depend on our ability to successfully commercialize our Fantom scaffold, and factors that negatively impact its commercial potential, including failures by our competitors, will adversely affect our business, financial condition, and results of operations.
Fantom is our first commercial product and our ability to successfully generate revenues and to consider additional products for commercialization depends on our ability to successfully market and sell Fantom. The degree of market acceptance for this scaffold will depend on many factors, including:
|
|
• |
its perceived advantages and disadvantages compared to existing stents and other treatments and technologies; |
|
|
• |
the success of our sales and marketing initiatives; |
|
|
• |
the selling price of Fantom and the third-party coverage and reimbursement for procedures using Fantom. |
If our Fantom scaffold does not achieve an adequate level of acceptance by physicians, patients, and health care payors, or if competing bioresorbable scaffolds being marketed ultimately prove to be unsuccessful or cause continuing negative sentiments about bioresorbables, we may not be able to successfully commercialize Fantom, generate sufficient revenues, or generate or maintain positive gross margins. Abbott, a competitor with significantly greater resources than what we have, withdrew its first generation bioresorbable product, Absorb, from the worldwide market in September 2017 due to low commercial sales. Additionally, Abbott reported high rates of adverse events in its clinical studies. Even if Fantom does achieve market acceptance, we may not be able to sustain it or otherwise achieve it to a degree that would support the ongoing viability of our operations.
If we fail to obtain and maintain adequate reimbursement for our products by third-party payors, there may be no commercially viable markets for our products or the markets may be much smaller than expected.
The markets for our products depend on the availability and levels of reimbursement by governmental and other health care payment systems including private insurance, which vary significantly by country. Government and other third-party payors continually attempt to contain or reduce health care costs by challenging prices charged for products and services and they may attempt to limit coverage and level of reimbursement of new products, such as ours. To obtain reimbursement or pricing approval in some countries, we may be required to produce additional clinical data, which may involve one or more clinical trials, that compares the cost-effectiveness of our products to other available therapies. In addition, the efficacy, safety, performance, and cost-effectiveness of our products in comparison to any competing products may determine the availability and level of reimbursement for our products.
We believe that future reimbursement may be subject to increased restrictions both in the United States and in international markets. Future legislation, regulation, or reimbursement policies of third-party payors may adversely affect the demand for our products and limit our ability to sell our products on a profitable basis. We cannot predict how pending or future legislative and regulatory proposals will influence the manner in which medical devices, including ours, are purchased or covered and reimbursed. If reimbursement for our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, sales of our products would be impaired, and our future revenues would be materially adversely affected.
To be commercially successful, we must effectively demonstrate to physicians the merits of our products compared to those of our competitors.
Physicians play a significant role in determining the course of treatment and, ultimately, the product used to treat a patient. As a result, our success depends, in large part, on demonstrating to physicians the value of our products in the treatment of their patients. To do so requires that we demonstrate the merits of our products and underlying technology compared to those of our competitors. Physicians who do not use our products may be hesitant to do so for the following or other reasons:
- 21 -
|
|
• |
the time required for physicians and medical staff education and training on new products, techniques and equipment; |
|
|
• |
lack or perceived lack of clinical evidence supporting patient benefit relative to competing products; |
|
|
• |
our products not being included on hospital formularies or integrated delivery network or group purchasing organization preferred vendor lists; |
|
|
• |
less attractive coverage and/or reimbursement within healthcare payment systems for our products and procedures compared to other products and procedures; |
|
|
• |
other costs associated with the introduction of new products and the equipment necessary to use new products; and |
In addition, we believe recommendations and support of our products by influential physicians are essential for market acceptance and adoption. If we do not receive support from such physicians or long-term data does not show the benefits of using our products, physicians may not use our products.
If we are not successful in convincing physicians of the merits of our products, we may be unable to maintain or grow our sales or achieve or sustain profitability.
We must successfully educate and train physicians and their staff on the proper use of Fantom.
Although most physicians may have adequate knowledge on how to use Fantom based on their clinical training and experience, we believe that the most effective way to introduce and build market demand for our products is by directly training physicians in the use of Fantom. Convincing physicians to dedicate the time and energy necessary for adequate training is challenging, and we cannot assure that we will be successful in these efforts. If physicians are not properly trained, they may not use Fantom, and, as a result, we may be unable to maintain or grow our sales or achieve or sustain profitability. If physicians are not properly trained they may also misuse or ineffectively use Fantom, which may result in unsatisfactory patient outcomes, patient injury, negative publicity or lawsuits against us, any of which could have a significant adverse effect on our business, financial condition and results of operations.
Although we believe our training methods for physicians are conducted in compliance with applicable regulations, if a regulatory agency determines that our training constitutes promotion of an unapproved use or promotion of an intended purpose not covered by the current CE mark affixed to Fantom, they could request that we modify our training or subject us to regulatory enforcement actions.
We compete against companies that have longer operating histories, more established or approved products, and greater resources, which may prevent us from achieving market penetration or improving operating results.
Competition in the stent industry is intense. Fantom competes with, and to the extent we can commercialize other products, such other products will compete with, products offered by established medical device companies, as well as earlier-stage companies that often have differentiated technology and potentially superior solutions. Global stent sales are dominated by Abbott, Boston Scientific Corporation, and Medtronic, who together recorded an estimated 94 percent of the $3.8 billion worldwide stent sales in 2017, according to JP Morgan Equity Research Interventional Cardiology Market Model. All three of those companies, as well as many of our other competitors, have access to greater financial, technical, research and development, marketing, manufacturing, sales, distribution, administrative, consulting and other resources than we do. Our competitors may be more effective at developing products, at differentiating their products from our and other competitor products and at designing, executing, analyzing the results of and publishing data from clinical studies. Our competitors may also have: stronger intellectual property portfolios; broader product offerings and products supported by more extensive clinical data; more established distribution networks; entrenched relationships with physicians; significantly greater name recognition as well as more recognizable trademarks for products similar to the products that we may seek to sell; more established relationships with healthcare providers and payors; greater experience in obtaining and maintaining FDA and other regulatory clearances or approvals for products and product enhancement; and greater experience in launching, marketing and selling products than we do. The frequent introduction by competitors of products that are, or claim to be, superior to our products, or that are alternatives to our existing or planned products may also create market confusion that may make it difficult to differentiate the benefits of our products over competing products. In addition, the entry of multiple new products and competitors may lead some of our competitors to employ pricing strategies that could adversely affect the pricing of our products and pricing in the stent market generally.
- 22 -
Our ability to compete effectively depends upon our ability to distinguish our company and our products from our competitors and their products. We believe the factors affecting our competitive position include:
|
|
• |
name and brand recognition; |
|
|
• |
relationships with physicians and patients; |
|
|
• |
the availability of other products and procedures, including bundled product offerings; |
|
|
• |
product performance and design; |
|
|
• |
product safety and the availability of supporting clinical data; |
|
|
• |
sales, marketing and distribution capabilities; |
|
|
• |
success and timing of new product development and introductions; and |
|
|
• |
intellectual property protection. |
The stent industry has a history of rapid and significant technological change and competition intensifies as technical advances are made. Our competitors may develop and commercialize stents or other medical device or pharmaceutical products that are safer or more effective, have fewer side effects, or are less expensive than products we may develop. For example, development of less-invasive technologies for treating cardiovascular disease could limit the market potential for our scaffolds. We also compete to recruit and retain qualified scientific and management personnel, establish clinical trial sites and patient registrations, and acquire technologies complementary to our programs or advantageous to our business. For all these reasons, we may not be able to compete successfully against current and future competitors.
Product liability claims could damage our reputation or adversely affect our business.
The design, manufacture, and sale of medical devices for human use, particularly implantable life-sustaining devices like our scaffolds, carry inherent risks of product liability and other damage claims. In addition, if physicians are not sufficiently trained in the use of our products, they may misuse or ineffectively use our products, which may result in unsatisfactory patient outcomes or patient injury. We could become the subject of product liability lawsuits alleging that component failures, malfunctions, manufacturing flaws, design defects or inadequate disclosure of product-related risks or product-related information resulted in an unsafe condition or injury to patients.
Product liability or other claims against us would be expensive to defend and would divert our management’s attention, and, if we are not successful in defending the claim, can result in substantial monetary awards against us or costly settlements. Further, successful product liability claims made against one or more of our competitors could cause claims to be made against us or expose us to a perception that we are vulnerable to similar claims. Any product liability claim brought against us, with or without merit and regardless of the outcome or whether it is fully pursued, may result in: decreased demand for our products; injury to our reputation; significant litigation costs; product recalls; loss of revenue; the inability to commercialize new products; and adverse publicity regarding our products. Any of these may have a material and adverse effect on our reputation with existing and potential customers and on our business, financial condition and results of operations
Our insurance policies are expensive and protect us only from some risks, which will leave us exposed to significant uninsured liabilities.
We do not carry insurance for all categories of risk that our business may encounter. Some of the policies we currently maintain include clinical trial and limited product liability insurance. We cannot be certain that such insurance will be sufficient to cover all claims that may be made against us. Our insurance policies generally renew on an annual basis and we may not be able to maintain or increase such insurance on acceptable terms or at reasonable costs. A successful claim brought against us in excess, or outside, of our insurance coverage could seriously harm our business, financial condition, results of operations, and prospects, and could also materially and adversely damage our reputation and affect our ability to attract and retain customers, whether or not such claim had merit.
We have limited manufacturing capabilities and personnel, and if we are unable to provide an adequate supply of our scaffolds, we may not be able to meet our commercial demands.
We currently have limited resources and have very limited experience in manufacturing commercial products. We believe we can manufacture quantities of Fantom to support initial sales volumes, but to achieve larger volumes or to
- 23 -
manufacture additional products, we will need to enhance our production and manufacturing operations. The significant technical and regulatory challenges to increasing manufacturing capacity and efficiency will require additional capital investment and the addition of experienced personnel. We may not successfully enhance our manufacturing capabilities in a timely or economical manner, or at all. In addition, we may not receive or continue the necessary regulatory approvals for our manufacturing facilities. If we are unable to manufacture a sufficient or consistent supply, or if our manufacturing processes yield substandard product or do not conform to regulatory standards, our revenues, business, and financial prospects would be adversely affected.
If our manufacturing facility is damaged or our manufacturing processes are interrupted, we could experience supply disruptions, lost revenues and our business could be seriously harmed.
We currently manufacture our scaffolds at our facility in San Diego, California. If, for any reason, that facility is damaged or we encounter a disruption to our manufacturing processes, including due to a natural disaster (such as earthquake, wildfires or other fires), power loss, communications failure, unauthorized entry or other events, we would have no means to manufacture until we were able to restore our facility or procure alternative facilities. In addition, our facility could be difficult to replace and would require substantial lead time to repair or replace. The property damage and business interruption insurance coverage on this facility that we maintain might not cover all losses under such circumstances, and we may not be able to renew or obtain such insurance in the future on acceptable terms with adequate coverage or at reasonable costs.
We are dependent on a limited number of third-party suppliers for delivery system catheters, sirolimus and other components, and the loss of any of these suppliers, or their inability to provide us with an adequate supply of materials that meet our quality and other requirements, could harm our business.
Outside suppliers, some of whom are sole-source suppliers, provide us with delivery system catheters, sirolimus and other components used in the manufacture of our Fantom scaffold. We have not entered into agreements with such suppliers that guarantees the supply of such components. We strive to maintain sufficient inventory of delivery system catheters, sirolimus and other components so that our production will not be significantly disrupted if a particular item is not available to us for a period of time. Any such disruption in our production could harm our reputation, business, financial condition and results of operations.
Although we believe there are alternative supply sources, replacing our current suppliers may be impractical or difficult in many instances. For example, we could have difficulty obtaining similar products from other suppliers that are acceptable to regulatory authorities. In addition, if we are required to transition to new suppliers for delivery system catheters, sirolimus and other components, the use of items furnished by these alternative suppliers could require us to alter our operations, and if we are required to change the manufacturer of a critical aspect of our products, we will be required to verify that the new manufacturer maintains facilities, procedures and operations that comply with our quality and applicable regulatory requirements, which could further impede our ability to manufacture our products in a timely manner. Transitioning to a new supplier could be time-consuming and expensive, may result in interruptions in our operations and product delivery, could affect the performance specifications of our products or could require that we modify the design of those systems.
If we are unable to obtain sufficient quantities of delivery system catheters, sirolimus and other components that meet our quality and other requirements on a timely basis for any reason, we may not be able to produce sufficient quantities of our products to meet market demand until a new or alternative supply source is identified and qualified and, as a result, we could lose customers, our reputation could be harmed and our business could suffer. Furthermore, an uncorrected defect or supplier’s variation in delivery system catheters, sirolimus and other components that are incompatible with our manufacturing, unknown to us, could harm our ability to manufacture products.
We rely on a single third-party logistics provider for distribution of our products in the European Union, and its failure to distribute our products would adversely affect sales.
We rely on a single third-party logistics provider for warehousing and distribution of our commercial supply of our products in the European Union. Although this third-party only stores a portion of our commercial supply of our products at its warehouse, the use of a single third-party increases the risk that a fire or damage from another type of disaster at its warehouse may result in a disruption of our commercialization efforts.
If our third-party logistics provider does not fulfill its contractual obligations to us, or refuses or fails to adequately distribute our products, or the agreement is terminated without adequate notice, shipments of our
- 24 -
products, and associated revenues, would be adversely affected. In addition, we expect that it may take a significant amount of time if we were required to change our third-party logistics provider.
If we are unable to retain or hire key personnel, we may not be able to sustain or grow our business.
Our ability to operate successfully and manage our potential future growth depends upon our ability to attract, retain, and motivate highly skilled and qualified research, technical, clinical, regulatory, sales, marketing, manufacturing, managerial, and financial personnel. We compete for talent with numerous companies, as well as universities and non-profit research organizations. Competition, particularly for key positions, is intense among companies in our industry, particularly in the San Diego, California area, and many of the organizations against which we compete for qualified personnel have greater financial and other resources and different risk profiles than our company, which may make them more attractive employers.
Except with respect to our employment agreements with our Chief Executive Officer, Chief Financial Officer, and Senior Vice President of Clinical and Regulatory Affairs, we have not entered into any employment agreements with our employees. We do not maintain key person life insurance on any of the members of our senior management team. All of our employees, including the members of our senior management team, may terminate their employment with us at any time without notice. The loss of key personnel for any reason or our inability to hire, retain, and motivate additional qualified personnel in the future could prevent us from sustaining or growing our business.
We are commercializing outside the U.S. and are subject to the risks of operating in foreign markets.
We have obtained CE Mark approval of Fantom and intend to seek additional regulatory approvals for Fantom in Australia, Japan, and China, in addition to seeking a PMA in the United States. We may also seek regulatory approvals outside the U.S. for future products. We are subject to a variety of risks as a result of having international operations. If and when we expand into other foreign markets, our exposure to these risks will increase and we may be subject to new risks. The risks to which we are currently subject and the new risks to which we may be subject include the following, any one of which may adversely impact our business, financial condition, and results of operations:
|
|
• |
failure to fulfill foreign regulatory requirements on a timely basis, or at all, to market our products; |
|
|
• |
availability of, and changes in, reimbursement within prevailing foreign health care payment systems; |
|
|
• |
differing laws and regulations, business and clinical practices, and patient preferences in foreign countries; |
|
|
• |
difficulties managing foreign relationships and operations, including relationships with foreign partners, sales or marketing agents, or distributors, and the costs of enforcing contractual obligations in foreign jurisdictions; |
|
|
• |
limited protection for intellectual property rights in some countries; |
|
|
• |
difficulty in collecting accounts receivable and longer collection periods; |
|
|
• |
recessions, political instability, and changes in diplomatic and trade relationships in foreign countries; |
|
|
• |
currency exchange rate fluctuations; and |
|
|
• |
potentially adverse tax consequences. |
In addition, expansion into foreign markets imposes additional burdens on our executive and administrative personnel, research and sales departments, and general managerial resources. Our efforts to introduce our current or future products into foreign markets may not be successful, in which case we may have expended significant resources without realizing the expected benefit. Ultimately, the investment required for expansion into foreign markets could exceed the results of operations generated from this expansion.
Our results may be impacted by changes in foreign currency exchange rates.
Currently, we are allowed to market and sell our Fantom scaffold only in Europe and other jurisdictions that recognize the CE Mark. As a result, to the extent we generate revenues, it is in various foreign currencies, currently euros. We also incur operating expenses in euros. We cannot predict accurately the consolidated effects of exchange rate fluctuations upon our future operating results because of the variability of currency exposure in our revenues and operating expenses and the potential volatility of currency exchange rates. Although we address currency risk management through regular operating and financing activities, those actions may not prove to be fully effective. In addition, for those foreign customers who purchase our products in U.S. dollars, currency exchange rate fluctuations between the U.S. dollar and the currencies in which those customers do business may have a negative effect on the
- 25 -
demand for our products in foreign countries where the U.S. dollar has increased in value compared to the local currency. Converting our earnings from international operations to U.S. dollars for use in the U.S. can also raise challenges, including problems moving funds out of the countries in which the funds were earned and difficulties in collecting accounts receivable in foreign countries where the usual accounts receivable payment cycle is longer. To date, we have not used risk management techniques to hedge the risks associated with foreign currency exchange rate fluctuations. Even if we were to implement hedging strategies, not every exposure can be hedged and, where hedges are put in place based on expected foreign currency exchange exposure, they are based on forecasts that may vary or that may later prove to have been inaccurate. As a result, fluctuations in foreign currency exchange rates or our failure to successfully hedge against these fluctuations could have a material adverse effect on our operating results and financial condition.
We are dependent on information technology and if our information technology fails to operate adequately or fails to properly maintain the integrity of our data, our business could be materially and adversely affected.
We depend significantly on sophisticated information technology, or IT, for our infrastructure and to support business decisions. Our IT needs require an ongoing commitment of significant resources to maintain, protect and enhance existing systems and develop new systems to keep pace with new technology, evolving regulatory standards, the increasing need to protect patient and customer information and changing customer patterns. While we have disaster recovery plans in place, there can be no such guarantee that such plans will be completely effective. Any significant breakdown, intrusion, interruption, corruption or destruction of these systems could have a material and adverse effect on our business, financial condition and results of operations.
Security breaches, loss of data and other disruptions could compromise sensitive information related to our business, prevent us from accessing critical information or expose us to liability, which could adversely affect our business and our reputation.
In the ordinary course of our business, we collect and store sensitive data, including legally protected patient health information, personally identifiable information about our employees, intellectual property, and proprietary business information. We manage and maintain our applications and data utilizing on-site systems. These applications and data encompass a wide variety of business-critical information including research and development information, commercial information and business and financial information.
Although our computer and information systems are protected through physical and software safeguards, they are still vulnerable to system malfunction, computer viruses, cyber-attacks, breaches or interruptions due to employee error or malfeasance, terrorist attacks, earthquakes, fire, flood, other natural disasters, power loss, computer systems failure, data network failure, Internet failure, or lapses in compliance with privacy and security mandates. Any such attack, virus, breach or interruption could compromise our networks and the information stored there could be accessed by unauthorized parties, publicly disclosed, lost or stolen. These events could lead to the unauthorized access and result in the misappropriation or unauthorized disclosure of confidential information belonging to us or to our employees, partners, customers or suppliers. We have measures in place that are designed to detect and respond to such security incidents and breaches of privacy and security mandates. The techniques used by criminal elements to attack computer systems are sophisticated, change frequently and may originate from less regulated and remote areas of the world. As a result, we may not be able to address these techniques proactively or implement adequate preventative measures. If our IT systems are compromised, due to a data breach or otherwise, we could be subject to legal claims or proceedings, liability under laws that protect the privacy of personal information, government enforcement actions and regulatory penalties, fines, damages, enforcement actions and we could lose trade secrets or other confidential information, the occurrence of which could harm our business and could have a material and adverse effect on our business, financial condition and results of operations. Unauthorized access, loss or dissemination could also interrupt our operations, including our ability to bill our customers, provide customer support services, conduct research and development activities, process and prepare company financial information, manage various general and administrative aspects of our business and damage our reputation, any of which could adversely affect our business.
The recently passed comprehensive tax reform bill could adversely affect our business and financial condition.
On December 22, 2017, President Trump signed into law new legislation that significantly revises the Internal Revenue Code of 1986, as amended. The newly enacted federal income tax law, among other things, contains significant changes to corporate taxation, including reduction of the corporate tax rate from a top marginal rate of 35% to a flat rate of 21%, limitation of the tax deduction for interest expense to 30% of adjusted earnings (except for certain small businesses), effective for net operating losses incurred in taxable years beginning after December 31, 2017, limitation of the deduction for net operating losses to 80% of current year taxable income and elimination of net
- 26 -
operating loss carrybacks, one time taxation of offshore earnings at reduced rates regardless of whether they are repatriated, elimination of U.S. tax on foreign earnings (subject to certain important exceptions), immediate deductions for certain new investments instead of deductions for depreciation expense over time, and modifying or repealing many business deductions and credits. Notwithstanding the reduction in the corporate income tax rate, the overall impact of the new federal tax law is uncertain and our business and financial condition could be adversely affected. In addition, it is uncertain how various states will respond to the newly enacted federal tax law. The impact of this tax reform on holders of our securities is also uncertain and could be adverse. Investors should consult with their legal and tax advisors with respect to this legislation and the potential tax consequences of investing in or holding our securities.
Risk Factors Related to Regulation
We cannot predict if there will be long-term issues from use of our products in patients. If our products cause adverse or unexpected results, we will be significantly negatively impacted.
Even though the results from our clinical trials have been favorable, the long-term use of our products could produce unexpected results. If our Fantom scaffold, or any future product, should demonstrate adverse issues such as restenosis, stroke, thrombosis, and/or death, we may need to suspend or terminate clinical trials or cease commercial sales until, and unless, we can address the issues. There is no guarantee that we will be able to successfully address and overcome any adverse issues arising from our products, which may significantly impair the product value, as well as the underlying technology value.
Regulatory or other factors could impact completion of current clinical trials or initiation of future trials. Any delay in completing clinical trials could have an adverse effect in bringing products to market.
Our existing and anticipated clinical trials could be substantially delayed or prevented by several factors, including the failure of patients to complete the clinical trial or our inability to monitor patients adequately after implant and other factors such as:
|
|
• |
governmental and regulatory delays or changes in regulatory requirements, policies, or guidelines; |
|
|
• |
varying interpretation of data by regulatory agencies; and |
|
|
• |
perceived lack of product efficacy during clinical trials. |
Any delay in initiating clinical trials, or in obtaining data from the trials, could have an adverse impact on our commercialization timelines, which would have an adverse impact on our ability to generate revenue, which will adversely affect our business, financial condition, and results of operations.
Even with successful data from clinical trials, we may not receive regulatory approval to market and sell our products, which could negatively impact our future prospects.
The process of obtaining marketing approval or clearance from regulatory authorities to market and sell our Fantom scaffold or any future products or enhancements or modifications to any products, could:
|
|
• |
take a significant period of time or more time than we expect; |
|
|
• |
require the expenditure of substantial resources; |
|
|
• |
involve rigorous preclinical and clinical testing; and |
|
|
• |
require changes to our products and/or result in limitations on the indicated uses of the products. |
There can be no assurance that we will receive the required approvals from the regulatory authorities or, if we do receive the required approvals, that we will receive them on a timely basis or that we otherwise will be able to satisfy the conditions of such approval, if any. The failure to receive product approvals by the regulatory authorities will have a material adverse effect on our business, financial condition, and results of operations.
We plan to operate in multiple regulatory environments that require costly and time consuming approvals.
We will need to obtain regulatory approval in each jurisdiction in which we intend to commercialize our products. The regulatory requirements will vary from country to country. In addition, the laws and regulations regarding the manufacture and sale of our products will be subject to future changes, as are administrative interpretations and policies of regulatory agencies. If we fail to comply with applicable laws or regulations, we could be subject to
- 27 -
enforcement actions. Enforcement actions could include product seizures, recalls, withdrawal of clearances or approvals, and civil and criminal penalties, which, in each case, would harm our business.
We may not meet regulatory quality standards applicable to our manufacturing and quality processes, which could have an adverse effect on our business, financial condition, or results of operations.
Even after products receive marketing approval, they can be withdrawn due to failure to comply with regulatory standards or the occurrence of problems following initial approval. As a device manufacturer, we will be required to demonstrate and maintain compliance with a variety of regulatory requirements. In the EU, we are required to maintain certain ISO certifications in order to sell our products and we undergo periodic inspections by notified bodies to obtain and maintain these certifications. Additionally, suppliers of components and products that we use in the manufacture of our products must also comply with applicable regulatory requirements.
We have received a Certificate of Registration certifying that our Quality Management System complies with the requirements of ISO 13485:2012 and we have undertaken to ensure our suppliers comply with applicable regulatory requirements. In the future, if we or our suppliers fail to comply with a regulation, the relevant regulatory authority may withdraw our approval to market, require a product recall, or take other enforcement action. Compliance is subject to continual review and is rigorously monitored through periodic inspections. If we fail to take satisfactory corrective action in response to an adverse inspection, we could be subject to enforcement actions, including a public warning letter, a shutdown of or restrictions on our manufacturing, delays in approval of a product, refusal to permit the import or export of our products, a recall or seizure of our products, fines, injunctions, civil or criminal penalties, or other sanctions, any of which could cause our business and operating results to materially suffer.
Our operations involve hazardous materials and we must comply with environmental laws and regulations, which can be expensive.
Our research, development, and manufacturing activities involve the controlled use of hazardous chemicals. Our operations also produce hazardous waste products. We are subject to a variety of federal, state, and local regulations relating to the use, handling, storage, and disposal of these materials. We generally contract with third parties for the disposal of such substances. We cannot eliminate the risk of accidental contamination or injury from these materials. We may be required to incur substantial costs to comply with current or future environmental and safety regulations. If an accident or contamination occurred, we would likely incur significant costs to remedy the situation and may be subject to civil penalties or criminal fines. Current or future environmental regulation may impair our research, development, or production efforts.
Health care reform legislation could adversely affect our future revenue and financial condition.
In recent years in the United States and other countries, there have been numerous initiatives for reforms affecting the availability of, and reimbursement for, health care services. These initiatives have ranged from proposals that would fundamentally change health care reimbursement programs, to minor modifications of existing programs, to requirements for comparative effectiveness analysis. The ultimate content or timing of any future health care reform legislation, and its impact on medical device companies such as ours, is impossible to predict. If significant reforms are made to the United States or other health care systems, they may have a material adverse effect on our financial condition and results of operations.
Our future operations may also be impacted by the ACA, if it remains in effect, or any replacement health care legislation. Among other things, we believe the 2.3 percent excise tax on sales of medical devices intended for use by humans, which is currently suspended, would apply to our scaffolds if and when we sell in the U.S. We are unable to predict whether the suspension will be continued beyond 2019 or eliminated entirely.
The ACA also requires (under what are referred to as “Sunshine” or “Open Payments” requirements) manufacturers of covered devices to report details regarding certain payments and other financial arrangements with physicians and teaching hospitals. These reporting provisions preempt state laws that require reporting of the same information, but not those that require reports of different or additional information. Failure to comply subjects the manufacturer to significant civil monetary penalties. Congress and the Trump administration have proposed various steps to revise, repeal, or delay implementation of various aspects of the ACA. If the ACA is significantly changed, or repealed, or if any replacement health care legislation is enacted, our business, including our financial results could be negatively impacted. We expect compliance with the ACA, or any future healthcare regulations, to impose significant administrative and financial burdens on us.
- 28 -
We are subject to various federal and state laws pertaining to health care fraud and abuse. Any violations of such laws could result in fines, penalties, or other criminal prosecution. In addition, compliance with these laws may result in significant additional expense to us and limit our ability to commercialize our products.
Our commercial, research, and other financial relationships with health care providers and institutions are subject to various federal and state laws intended to prevent health care fraud and abuse. We are also subject to regulation by other regional, national, state, and local agencies, including the Department of Justice, the Office of Inspector General of the U.S. Department of Health and Human Services and other regulatory bodies. Violations of any of these laws and regulations could result in penalties or fines being assessed against us, significant additional compliance expense, or even a limitation on our ability to commercialize our products.
The federal Anti-Kickback Statute prohibits the knowing offer, receipt, or payment of remuneration in exchange for or to induce the referral of patients or the use of products or services that would be paid for in whole or part by Medicare, Medicaid, or other federal health care programs. Remuneration has been broadly defined to include anything of value, including cash, improper discounts, and free or reduced price items and services. Many states have similar laws that apply to their state health care programs as well as private payors. Violations of the anti-kickback laws can result in exclusion from federal health care programs and substantial civil and criminal penalties.
The U.S. False Claims Act, or FCA, imposes liability on persons who, among other things, present false or fraudulent claims for payment by a federal health care program. The FCA has been used to prosecute persons submitting claims for payment that are inaccurate or fraudulent, including services that are not medically necessary. The FCA includes a whistleblower provision that allows individuals to bring actions on behalf of the federal government and share in a portion of the recovery of successful claims. If our marketing or other arrangements were determined to violate anti-kickback or related laws, including the FCA, then our revenues could be adversely affected, which would likely have a material adverse effect on our business, financial conditions, and results of operations. Similarly, the federal Civil Monetary Penalty statute imposes significant penalties for filing certain types of improper claims or engaging in prohibited acts related to federal program integrity.
State and federal authorities have aggressively targeted medical device companies for alleged violations including improper research or consulting contracts with doctors, certain marketing arrangements that rely on volume-based pricing, off-label marketing schemes, and other improper promotional practices. Compliance with the federal and state laws is difficult and time consuming and companies that violate them may face substantial penalties. Because of the breadth of these laws and the lack of extensive legal guidance in the form of regulations or court decisions, it is possible that some of our business activities or those of our commercial partners could be subject to challenge under one or more of these laws, which could have a material adverse effect on our business and financial condition and growth prospects. Companies targeted in prosecutions have paid fines in the hundreds of millions of dollars or more, have been forced to implement extensive corrective action plans, and have often been subject to consent decrees severely restricting the manner in which they conduct business. If we become the target of such an investigation or prosecution based on our contractual relationships with providers or institutions, or our marketing and promotional practices, we could face similar sanctions which would materially negatively affect our business.
If we are found to have violated laws protecting the privacy or security of patient information, we could be subject to civil suits and civil or criminal penalties, which could result in liability and harm our reputation or business.
We are subject to privacy laws in the countries in which we do business. We have in place a specific Australian Privacy Policy and plan to expand our privacy policies to cover the privacy laws we are, or will be, subject to. These laws, including the federal and state privacy laws in the United States, are designed to protect the privacy and security of personally identifiable information, including patient health information and patient records, by, among other things, limiting its use and disclosure, establishing patient rights, requiring security safeguards, and mandating notice to the government and individuals if information is compromised (i.e., a breach).
Many local jurisdictions also have similar laws protecting the privacy and security of personally identifiable information, including breach notification requirements. If we violate applicable privacy laws, we could be subject to civil lawsuits based on state law or tort (including class actions) and civil or criminal penalties, which could increase our liabilities, harm our reputation, and have a material adverse effect on our business, financial condition, and results of operations.
- 29 -
Risk Factors Related to Intellectual Property
We rely on certain licenses for patents and other technology related to our products. The termination of these license agreements could delay or prevent us from being able to commercialize our products.
We have licensed certain patent rights and other technology that we use for our scaffolds. For example, we license a majority of the polymer technology that we use in our scaffolds from Rutgers University. In order to maintain our rights under the license agreement we entered into with Rutgers for this technology, we must satisfy certain development and commercialization obligations. If we fail to satisfy these obligations, Rutgers might license some or all this technology to one or more parties, any of which could be one of our competitors, and our ability to compete may be substantially and adversely diminished. Furthermore, if we fail to comply with material obligations under the license agreement or if it were terminated for any reason, we could lose rights to this technology that are important to our business. The license agreement expires on the expiration date of the last patent to expire under the agreement, which we believe is currently approximately 2036; if we need to renew the license, there is no guarantee we will be able to renew it on commercially reasonable terms, if at all.
If we are unable to obtain, maintain, and enforce intellectual property protection covering our products, others may be able to make, use, or sell products similar to ours, which could materially and adversely affect our ability to compete.
Our success depends in part on obtaining, maintaining, and enforcing the intellectual property rights, including patents, that cover our scaffolds and future products. If we are unable to protect our intellectual property, others may make, use, or sell products that are substantially the same as ours without incurring the sizeable costs, including development and licensing costs, we have incurred, which would materially and adversely affect our ability to compete in the market.
Currently, our patent portfolio comprises approximately 281 issued and pending U.S. and foreign patents that we own directly or license and that expire between 2018 and 2036. Pending patent applications could further extend our patent portfolio life. However, we might not receive approval of pending applications or future patent applications, and issued patents may be found by a court to be invalid or otherwise unenforceable. Even if our patents are determined to be valid and enforceable, they may not be sufficiently broad to prevent others from marketing products similar to ours, or designing around our patents despite our patent rights, nor do they provide us with freedom to operate unimpeded by the patent rights of others.
As we have licensed certain intellectual property from third parties, we rely on them to file and prosecute patent applications, maintain patents, and otherwise protect that intellectual property. We cannot be certain that such third parties have or will comply with applicable laws and regulations or that their activities will result in valid and enforceable patents and other intellectual property rights. In addition, we cannot be certain that our licensors will allocate sufficient resources to enforce or defend their patents to sufficiently protect our interests.
The patent positions of medical device companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in patents in these fields has emerged to date in the United States or in many foreign jurisdictions.
Both the U.S. Supreme Court and the Court of Appeals for the Federal Circuit have made, and will likely continue to make, changes in how the U.S. patent laws are interpreted. In addition, Congress is currently considering legislation that would change provisions of the patent law. We cannot predict future changes in the interpretation of patent laws or changes to patent laws that might be enacted into law. Those changes may materially affect our patents, our ability to obtain patents, or the patents and applications of our collaborators and licensors. The patent situation in the medical device and disease diagnostic fields outside the United States is even more uncertain.
We have numerous foreign patents and applications.
The laws of some foreign jurisdictions do not protect intellectual property rights to the same extent as laws in the United States and many companies have encountered significant difficulties in obtaining, protecting, and defending such rights in foreign jurisdictions. If we encounter such difficulties or we are otherwise precluded from effectively protecting our intellectual property rights in foreign jurisdictions, our business prospects could be substantially harmed.
- 30 -
We rely on trade-secret protection for certain processes for which patents are difficult to obtain or enforce. We may not be able to protect our trade secrets adequately as we have limited control over our licensors, collaborators, and suppliers.
Although we use reasonable efforts to protect our trade secrets, our employees, consultants, suppliers, scientific collaborators, and other advisors may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third party illegally obtained and used any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets. We rely, in part, on non-disclosure and confidentiality agreements with our employees, consultants, and other parties to protect our trade secrets and other proprietary technology. These agreements may be breached and we may not have adequate remedies for any breach. Moreover, others may independently develop equivalent proprietary information or third parties may otherwise gain access to our trade secrets and proprietary knowledge. Any disclosure of confidential data into the public domain or to third parties could allow our competitors to use the information against us.
Claims that our current or future products infringe or misappropriate the proprietary rights of others could materially and adversely affect our ability to sell those products and cause us to incur additional costs.
Intellectual property rights play a critical role in stents and stent delivery systems. We face significant risks relating to our patents and to patents held by others. If any intellectual property claim against us is successful, we could be prevented from commercializing our scaffolds or other future product candidates. There are numerous U.S. and foreign-issued patents and pending patent applications owned by third parties with patent claims in areas that relate to our scaffolds. Also, because patent applications can take many years to be issued, there may be other pending applications, unknown to us, that may result in future patents that pose a material risk to us. We are aware of patents owned by others, to which we do not have licenses, that relate to, among other things:
|
|
• |
stent structures, materials, and designs; |
|
|
• |
catheters used to deliver stents; and |
|
|
• |
polymer and stent manufacturing and coating processes. |
We expect that we could be increasingly subject to third-party infringement claims as we receive regulatory approval to sell products, our revenues increase, we are faced with more competitors, or the functionality of products and technology in different industry segments overlaps. Third parties may currently have, or may eventually be issued, patents on which our current or future products or technologies may infringe. Any of these third parties might make a claim of infringement against us.
All the major companies in the stent and related markets, including Boston Scientific Corporation, Abbott, and Medtronic have been involved in patent litigation relating to stents since at least 1997. The stent and related markets have experienced rapid technological change and obsolescence in the past, and our competitors have strong incentives to stop or delay the introduction of new products and technologies by their competitors. We may pose a competitive threat to many companies in the stent and related markets. Accordingly, many of these companies will have a strong incentive to take steps, through patent litigation or otherwise, to prevent us from commercializing our products.
Any litigation, regardless of its outcome, would likely result in significant expenses and the diversion of resources and management time. In addition, litigation in which we are accused of infringement may cause negative publicity, adversely impact prospective customers, cause product shipment delays, prohibit us from manufacturing, marketing, or selling our products, require us to develop non-infringing technology, make substantial payments to third parties, or enter into royalty or license agreements, which may not be available on acceptable terms, or at all.
If a successful claim of infringement were made against us and we could not develop non-infringing technology, invalidate the claim, or license the infringed or similar technology on a timely and cost-effective basis, our revenues may decrease substantially and we could be exposed to significant liability. A court could enter orders that temporarily, preliminarily, or permanently prevent us or our customers from making, using, selling, offering to sell, or importing our current or future products, or could enter an order mandating that we undertake certain remedial activities. Claims that we have misappropriated the confidential information or trade secrets of third parties can have a similar negative impact on our reputation, business, financial condition, or results of operations.
- 31 -
We may need to initiate lawsuits to protect our patents or other intellectual property rights, which, regardless of outcome, could be expensive and which, if lost, could result in loss of intellectual property rights, which would harm our business.
We rely on patents to protect a portion of our intellectual property and competitive position. Patent law relating to the technology fields in which we operate is still evolving and, consequently, patent positions in the medical device industry are generally uncertain. In order to protect or enforce our patent rights, we may initiate patent litigation against third parties, such as infringement suits or interference proceedings. Litigation may be necessary to:
|
|
• |
assert claims of infringement; |
|
|
• |
enforce our patents; |
|
|
• |
protect our trade secrets or know-how; or |
|
|
• |
determine the enforceability, scope, and validity of the proprietary rights of others. |
Any lawsuits that we initiate could be expensive, take significant time, and divert management’s attention from other business concerns. Litigation also puts our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing. Additionally, we may provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially valuable. The occurrence of any of these events may have a material adverse effect on our business, financial condition, and results of operations.
Risks Related to Our CDIs and Common Stock
The market price of our CDIs and common stock may be volatile and fluctuate significantly, which could result in substantial losses for investors.
Our securities are listed for sale only on the Australian Securities Exchange, or ASX, in the form of CHESS Depositary Interests, or CDIs. We are very early in the commercialization stage. Until we achieve substantial commercialization, start generating significant revenues and cash receipts, are able to service our convertible notes, demonstrate regular measurable performance, and list our securities for sale on an additional stock exchange, the market for our CDIs may continue to be illiquid and the market price of our CDIs may continue to be volatile. In addition to the matters described in this “Risk Factors” section, the market price of our CDIs may fluctuate due to other risks and factors, such as changes in general economic, industry, and market conditions and other factors including:
|
|
• |
our development progress, including delays or advancements in our timelines; |
|
|
• |
changes to the regulatory status of our scaffolds and future product candidates; |
|
|
• |
any reported adverse events in our human clinical trials; |
|
|
• |
technology innovations, new products, contracts, acquisitions, or strategic alliances by our competitors or us; |
|
|
• |
changes in the estimates of the future size and growth rate of our markets; |
|
|
• |
changes in market valuations or earnings of our competitors; |
|
|
• |
changes in legislation or regulatory policies, practices, or actions; |
|
|
• |
the commencement or outcome of litigation involving our company, our general industry, or both; |
|
|
• |
recruitment or departure of one or more members of our executive management team or board of directors; |
|
|
• |
changes in our capital structure, such as future issuances of securities or the incurrence of additional debt; |
|
|
• |
actual or expected sales of our CDIs or common stock by existing holders; |
|
|
• |
the overall trading volume of our CDIs; and |
|
|
• |
failure to service our debt or complete additional financings to fund our operations when needed or on terms favorable to us or on terms that are not overly dilutive to our current securityholders. |
Stock markets in general, and submarkets for medical technology companies in particular, have experienced volatility that has often been unrelated to the operating performance of companies. These broad market and industry factors may materially affect the market price of our CDIs. Litigation has often been brought against companies whose securities have experienced volatility in market price. Class-action litigation, even if unsuccessful, could be costly to
- 32 -
defend and divert management’s attention and resources, which could materially harm our financial condition and results of operations.
Investors may experience difficulty in trading our CDIs due to their relatively limited liquidity on the ASX.
Although our CDIs are listed on the ASX, there can be no guarantee of a readily liquid market for them, particularly since a small number of securityholders own a majority of our outstanding shares. It may be more difficult for an investor to realize an investment on the ASX than it would be to realize an investment in a company whose shares or other securities are quoted on NYSE, NASDAQ, or any other stock exchange.
We may not retain our ASX listing and we may not qualify for listing on another securities exchange.
We cannot assure investors that we will always retain a listing on ASX and our common stock is not currently listed for trading on a U.S. or any other securities exchange. In connection with the issuance of our 2014 convertible notes, we agreed to use reasonable efforts to list our common stock on NASDAQ, however, at this time, we are still evaluating our listing alternatives. If we fail to retain our ASX listing or if we do not list our common stock on another securities exchange, certain investors may decide to sell their securities and/or there may not be a market for the securities, which could have an adverse impact on the price of the securities. There is no assurance that we can qualify in the future for listing any of our securities on the New York Stock Exchange, the NASDAQ Stock Market, or any other exchange.
Some of our existing stockholders can exert control over us and may not make decisions that are in the best interests of all stockholders.
As of February 23, 2018, officers, directors, and stockholders holding more than five percent of our outstanding shares collectively controlled approximately 64 percent of our outstanding common stock. As a result, these stockholders, if they act together, would be able to exert a significant degree of influence over our management and affairs and over matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. Accordingly, this concentration of ownership may harm the market price of our shares by delaying or preventing a change in control, even if a change is in the best interests of our other stockholders. In addition, the interests of this concentration of ownership may not always coincide with the interests of other stockholders and, accordingly, they could cause us to enter into transactions or agreements that we would not otherwise consider.
Future sales of our common stock may depress the market price of our CDIs.
The holders of an aggregate of approximately 17.6 million shares of our outstanding common stock, as well as the holders of our convertible notes, if such notes are converted into common stock, have the right to cause us to file a registration statement that would register the resale of such shares on their behalf and to include their shares in registration statements that we may file on behalf of other stockholders. In addition, shares of common stock reserved for issuance under our 2010 Equity Incentive Plan, as amended, have been registered under the Securities Act of 1933, as amended, and, accordingly, any shares of stock issued in accordance with such plan may be freely sold under the federal securities laws and may be tradable under state securities laws if the holder satisfies such laws or is exempt from them. Additionally, our 2010 Equity Incentive Plan provides for annual increases in the number of shares available for issuance under the plan, which we intend to register annually. From time to time, we also may sell additional shares of common stock, or securities convertible, exercisable or exchangeable for shares of our common stock, in subsequent public offerings or private placements. Sales of a substantial number of shares of our common stock or such securities or CDIs in the public market, whether by us or by our stockholders, or the perception that these sales may occur, could cause the market price of our CDIs to decline and make it more difficult for holders to sell our CDIs or shares of common stock.
Raising additional capital may cause dilution to our existing stockholders, require us to relinquish proprietary rights or restrict our operations.
We may raise additional capital at any time and may do so through one or more financing alternatives, including public or private sales of our equity securities, debt financings, collaborations, licensing arrangements or other strategic transactions. Each of these financing alternatives carries certain risks. Raising capital through the issuance of common stock, or securities convertible, exercisable or exchangeable for shares of our common stock, may depress the market price of our stock and may substantially dilute our existing stockholders. In addition, our currently outstanding convertible notes are convertible into shares of our common stock at any time at the option of the holder. The conversion rate of our 2014 convertible notes of $2.17275 per share is favorable for the noteholders, as it is below
- 33 -
the $3.52 market price of our common stock as of February 23, 2018. If we instead seek to raise capital through strategic transactions, such as licensing arrangements, we may be required to relinquish valuable rights. Debt financings could involve covenants that restrict our operations. These restrictive covenants may include limitations on additional borrowing and specific restrictions on the use of our assets, as well as prohibitions on our ability to create liens or make investments and may, among other things, preclude us from making distributions to stockholders (either by paying dividends or redeeming stock) and taking other actions beneficial to our stockholders. In addition, investors could impose more one-sided investment terms and conditions on companies that have or are perceived to have limited remaining funds or limited ability to raise additional funds. The lower our cash balance, the more difficult it is likely to be for us to raise additional capital on commercially reasonable terms, or at all.
We have broad discretion in the use of our assets and our investment of these assets may not yield a favorable return, which could harm our business and depress the market price of our securities.
Our management has discretion in the application of our assets and other resources and may use them for a broad range of purposes. Accordingly, securityholders will have to rely upon our management’s judgment with respect to the use of our assets. Management may spend a portion or all our assets in ways that holders of our securities may not desire or that may not yield a significant return, or any return at all. The failure by our management to apply these funds effectively could harm our business and depress the market price of our securities. Pending their use, we may also invest our assets in a manner that does not produce income or that loses value.
We do not currently intend to pay dividends on our CDIs or common stock; consequently, the return on an investment in our securities will depend on appreciation in the market price of our CDIs.
We currently intend to invest our future earnings, if any, to fund the development and growth of our business. The payment of dividends will be at the discretion of our board of directors and will depend on our operating results, capital needs, financial condition, future prospects, debt covenants, contractual arrangements, restrictions imposed by applicable law, and other factors our board of directors may deem relevant. The terms of our convertible notes limit our ability to pay dividends. If we do not pay dividends, the return on an investment in our CDIs will depend on any future appreciation in the market price of our CDIs. There is no guarantee that our CDIs will appreciate or even maintain the price at which they were purchased.
We incur exchange rate risks relating to our listing on the ASX.
Our securities, in the form of CDIs, are listed on the ASX and priced in Australian dollars. However, we report in U.S. dollars. As a result, movements in foreign exchange rates may cause the price of our CDIs to fluctuate for reasons unrelated to our financial condition or performance and may result in a discrepancy between our actual results of operations and investors’ expectations of returns on our CDIs expressed in Australian dollars.
Failure to comply with U.S. public company laws and regulations as well as the listing requirements of the ASX could cause investors to lose confidence in us and could have a material adverse effect on our business and on the market price of our CDIs.
As a company with securities registered with the SEC and with securities listed on the ASX, we incur substantial legal, accounting, and other stockholder and reporting compliance expenses. In addition, changing laws, regulations, and standards relating to corporate governance and public disclosure, including SEC regulations, may increase legal and financial compliance costs and make some corporate activities more time consuming. Since our securities are traded on the ASX, we must comply with ASX Listing Rules. We believe our policies and procedures are designed to provide reasonable assurance of ASX Listing Rules compliance; however, if we do not follow those procedures and policies, or they are not sufficient to prevent non-compliance, we could be subject to delisting, liability, fines, and lawsuits. These laws, regulations, and standards are subject to varying interpretations and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. We expend significant management resources to comply with securities regulations, which may divert attention from revenue-generating activities. If our efforts to comply with new laws, regulations, and standards are unsuccessful, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
We are required, pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, or Section 404, to maintain internal control over financial reporting and to assess and furnish reports as to the effectiveness of our internal control over financial reporting. Additionally, our independent auditors are required to report on, among other things, the effectiveness of our internal control over financial reporting. Our internal control over financial reporting process is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Although we
- 34 -
believe we have developed and maintain effective internal control over financial reporting, they may become inadequate because of changes in conditions or our degree of compliance, or weaknesses may be discovered. If we, or our auditors, are unable to state that our internal control over financial reporting is effective and in compliance with Section 404, or we are unable to produce timely or accurate financial reports, we may be subject to sanctions or investigations, and investors may lose confidence in our financial reports, which would have a material adverse effect on our business, the market price of our CDIs, and our ability to access the capital markets.
Failure to comply with the SEC and ASX rules and regulations might also make it more difficult for us to obtain certain types of insurance, including director and officer liability insurance, and we might be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, on committees of our board of directors, or as members of senior management.
As a Delaware corporation, an acquisition of us, which may be beneficial to our stockholders, and attempts by our stockholders to replace or remove the current members of our board and management may be more difficult.
We are a Delaware corporation, subject to the provisions of Delaware General Corporation Law. Those laws, in addition to certain provisions of our certificate of incorporation and our bylaws, could discourage, delay, or prevent a merger, acquisition, or other change of control that stockholders may consider favorable. Furthermore, these provisions could prevent or frustrate attempts by our stockholders to replace or remove members of our board of directors. These provisions also could limit the price that investors might be willing to pay in the future for our CDIs, thereby depressing the market price of our CDIs. These provisions:
|
|
• |
allow the authorized number of directors to be changed only by resolution of our board of directors and provide that our stockholders may only remove our directors for cause; |
|
|
• |
establish a classified board of directors so that not all directors may be elected at one time; |
|
|
• |
authorize our board of directors to issue, without stockholder approval but subject to ASX Listing Rules, up to 100,000,000 shares of common stock or up to 5,000,000 shares of preferred stock, that, if issued, would dilute ownership and operate as a “poison pill” to help prevent an acquisition that is not approved by our board of directors; |
|
|
• |
require that stockholder actions occur at a duly called stockholder meeting or by unanimous written consent; |
|
|
• |
establish advance notice requirements for stockholder nominations to our board of directors or for stockholder proposals that can be voted at stockholder meetings; |
|
|
• |
limit who may call stockholder meetings to the chairman of our board of directors, our board of directors (pursuant to a resolution adopted by a majority of the total number of directors that we would have if there were no vacancies) or our chief executive officer; and |
|
|
• |
require approval from no less than 80 percent of the outstanding shares of our capital stock to amend certain provisions of our certificate of incorporation and bylaws. |
In addition, provisions of Section 203 of the Delaware General Corporation Law may, unless certain criteria are met, prohibit large stockholders, in particular those owning 15 percent or more of the voting rights on our common stock, from merging or combining with us for a prescribed period of time.
Item 1B. Unresolved Staff Comments
We do not have any unresolved staff comments relating to our periodic or current reports.
Our headquarters is located at 5751 Copley Drive, San Diego, California, where we lease and occupy approximately 37,000 square feet of research, lab, manufacturing and office space. The lease expires in May 2025.
We do not own any real property. We believe that our leased facility, which is in good operating condition, is suitable to meet our current needs, as well as our needs through our first few years of commercial sales of Fantom.
- 35 -
We may consider additional or different facilities and locations for manufacturing as needed to meet our manufacturing needs.
We may from time to time become subject to ordinary routine litigation incidental to our business. As of the date of filing this report, there is no material pending legal proceedings to which we are a party or to which any of our property is subject.
Item 4. Mine Safety Disclosures
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters, and Issuer Purchases of Equity Securities
Market Information
Shares of our common stock began trading in the form of CHESS Depositary Interests, or CDIs, on the Australian Securities Exchange, or ASX, under the symbol “RVA” on December 23, 2010. Prior to such time, there was no public market for our securities. Each CDI represents one-tenth of a share of our common stock.
The high and low sales prices for our CDIs during each quarter during the two most recent years, and on an equivalent basis as converted to common stock and U.S. dollars, are set forth below. The dollar amounts are after giving effect to the ten-for-one CDI-to-common stock exchange ratio and after converting to U.S. dollars using the closing exchange rate applicable on the relevant date as reported by the Reserve Bank of Australia.
|
|
|
CDI Price Range |
|
|
Stock Price Range |
|
||||||||||
|
|
|
Low |
|
|
High |
|
|
Low |
|
|
High |
|
||||
|
Year Ended December 31, 2017: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
First quarter |
|
AUD 0.87 |
|
|
|
AUD 1.10 |
|
|
$ |
6.59 |
|
|
$ |
8.10 |
|
|
|
Second quarter |
|
AUD 0.80 |
|
|
|
AUD 1.11 |
|
|
$ |
6.05 |
|
|
$ |
8.35 |
|
|
|
Third quarter |
|
AUD 0.65 |
|
|
|
AUD 0.86 |
|
|
$ |
5.13 |
|
|
$ |
6.60 |
|
|
|
Fourth quarter |
|
AUD 0.56 |
|
|
|
AUD 0.74 |
|
|
$ |
4.24 |
|
|
$ |
5.76 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2016: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
First quarter |
|
AUD 0.81 |
|
|
|
AUD 1.22 |
|
|
$ |
5.89 |
|
|
$ |
8.65 |
|
|
|
Second quarter |
|
AUD 0.95 |
|
|
|
AUD 1.28 |
|
|
$ |
7.02 |
|
|
$ |
9.32 |
|
|
|
Third quarter |
|
AUD 1.04 |
|
|
|
AUD 1.35 |
|
|
$ |
7.73 |
|
|
$ |
10.34 |
|
|
|
Fourth quarter |
|
AUD 0.95 |
|
|
|
AUD 1.32 |
|
|
$ |
7.05 |
|
|
$ |
10.06 |
|
As of February 23, 2018, we had 41,245,820 shares of common stock issued and outstanding with 800 holders of record. The holders included CHESS Depositary Nominee Pty Limited, which held 26,866,792 shares of our common stock, or approximately 65% of the outstanding shares, in the form of CDIs on behalf of the CDI holders. As of February 23, 2018, there were 741 registered owners of our CDIs.
Stock Price Performance Graph
The following graph compares the total return on our common stock (after giving effect to the ten-for-one CDI-to-common stock ratio and after converting to U.S. dollars using closing exchange rate applicable on the relevant date as reported by the Reserve Bank of Australia) to (i) the NASDAQ Composite Index and (ii) the NASDAQ Biotechnology Index for the five years ending December 31, 2017:
- 36 -
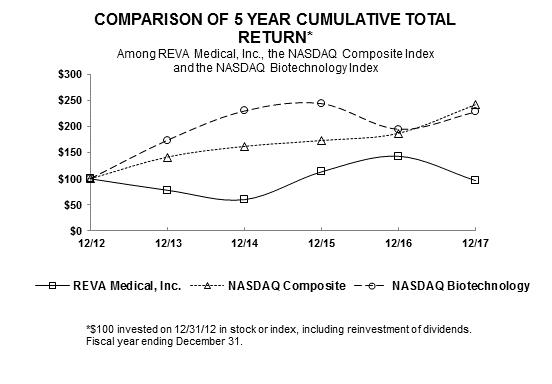
The performance graph assumes $100 was invested in each of our common stock, the NASDAQ Composite Index and the NASDAQ Biotechnology Index as of the market close on January 1, 2012. The closing price of a share of our common stock on such date was $6.30. The comparisons in the table are disclosures in accordance with SEC requirements and are not intended to forecast or be indicative of possible future performance of our common stock. The graph shall not be deemed “soliciting material” or to be “filed” for purposes of Section 18 of the Exchange Act or otherwise subject to the liabilities under that Section, and shall not be deemed to be incorporated by reference into any of our filings under the Securities Act or the Exchange Act.
Dividend Policy
We have never declared or paid any cash dividends on our common stock. We currently intend to retain all available funds and any future earnings to support operations and finance the growth and development of our business and do not intend to pay cash dividends on our common stock or CDIs for the foreseeable future. In addition, our outstanding convertible notes limit our ability to pay dividends. Any future determination related to our dividend policy will be made at the discretion of our board of directors.
Recent Sales of Unregistered Securities
None.
Item 6. Selected Financial Data
The following tables present selected financial data for the five-year period ended December 31, 2017 (in thousands, except per share data). The selected financial data should be read together with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and related notes included elsewhere in this Form 10-K. We derived our statements of operations data for the years ended December 31, 2017, 2016, and 2015 and our balance sheet data as of December 31, 2017 and 2016 from our audited financial statements appearing elsewhere in this Form 10-K. We derived our statements of operations data for the years ended December 31, 2014 and 2013 and our balance sheet data as of December 31, 2015, 2014, and 2013 from
- 37 -
our audited financial statements that are not included in this Form 10-K. Our financial information is prepared and presented in accordance with generally accepted accounting principles in the United States, or U.S. GAAP.
|
|
Year Ended December 31, |
|
|||||||||||||||||
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||||
|
Consolidated Statement of Operations Data: |
|
|
|||||||||||||||||
|
Revenue |
$ |
45 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
Cost of revenue |
|
42 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Gross profit |
|
3 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating Expense: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
12,760 |
|
|
|
18,171 |
|
|
|
16,760 |
|
|
|
14,318 |
|
|
|
19,212 |
|
|
Selling, general and administrative |
|
8,572 |
|
|
|
8,609 |
|
|
|
7,210 |
|
|
|
7,645 |
|
|
|
8,731 |
|
|
Loss from operations |
|
(21,329 |
) |
|
|
(26,780 |
) |
|
|
(23,970 |
) |
|
|
(21,963 |
) |
|
|
(27,943 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other Income (Expense): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
57 |
|
|
|
3 |
|
|
|
9 |
|
|
|
8 |
|
|
|
30 |
|
|
Interest expense |
|
(6,690 |
) |
|
|
(2,053 |
) |
|
|
(1,904 |
) |
|
|
(986 |
) |
|
|
— |
|
|
Loss on issuance of convertible notes and warrants to purchase common stock |
|
(520 |
) |
|
|
— |
|
|
|
— |
|
|
|
(15,627 |
) |
|
|
— |
|
|
Gain (loss) on change in fair value of convertible notes and warrant liability |
|
35,731 |
|
|
|
(25,247 |
) |
|
|
(56,788 |
) |
|
|
(12,542 |
) |
|
|
— |
|
|
Other income (expense) |
|
(115 |
) |
|
|
(21 |
) |
|
|
59 |
|
|
|
73 |
|
|
|
(9 |
) |
|
Net Income (Loss) |
$ |
7,134 |
|
|
$ |
(54,098 |
) |
|
$ |
(82,594 |
) |
|
$ |
(51,037 |
) |
|
$ |
(27,922 |
) |
|
Net Income (Loss) Per Share: (1) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income (loss) per share - basic |
$ |
0.17 |
|
|
$ |
(1.28 |
) |
|
$ |
(2.38 |
) |
|
$ |
(1.53 |
) |
|
$ |
(0.84 |
) |
|
Shares used to compute net loss per share - basic |
|
41,811,326 |
|
|
|
42,120,545 |
|
|
|
34,680,634 |
|
|
|
33,382,381 |
|
|
|
33,124,655 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per share - diluted |
$ |
(0.40 |
) |
|
$ |
(1.28 |
) |
|
$ |
(2.38 |
) |
|
$ |
(1.53 |
) |
|
$ |
(0.84 |
) |
|
Shares used to compute net loss per share - diluted |
|
53,317,482 |
|
|
|
42,120,545 |
|
|
|
34,680,634 |
|
|
|
33,382,381 |
|
|
|
33,124,655 |
|
___________
|
(1) |
See Note 13 to our consolidated financial statements for an explanation of the method used to compute the diluted net loss per share and the number of shares used in the computation of the diluted per share amounts. |
|
|
Year Ended December 31, |
|
|||||||||||||||||
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||||
|
Balance Sheet Data: |
|
|
|||||||||||||||||
|
Cash and cash equivalents |
$ |
18,544 |
|
|
$ |
6,674 |
|
|
$ |
16,895 |
|
|
$ |
25,814 |
|
|
$ |
19,229 |
|
|
Investment securities |
|
1,470 |
|
|
|
— |
|
|
|
— |
|
|
|
995 |
|
|
|
1,492 |
|
|
Working capital |
|
18,491 |
|
|
|
(91,664 |
) |
|
|
13,996 |
|
|
|
24,351 |
|
|
|
17,656 |
|
|
Total assets |
|
22,661 |
|
|
|
9,483 |
|
|
|
20,071 |
|
|
|
30,195 |
|
|
|
24,785 |
|
|
Convertible notes payable |
|
99,368 |
|
|
|
91,655 |
|
|
|
75,365 |
|
|
|
37,780 |
|
|
|
— |
|
|
Common stock warrant liability |
|
4,176 |
|
|
|
— |
|
|
|
19,622 |
|
|
|
15,389 |
|
|
|
— |
|
|
Total liabilities |
|
115,474 |
|
|
|
99,076 |
|
|
|
100,635 |
|
|
|
56,644 |
|
|
|
3,960 |
|
|
Accumulated deficit |
|
(382,157 |
) |
|
|
(389,238 |
) |
|
|
(335,140 |
) |
|
|
(252,546 |
) |
|
|
(201,509 |
) |
|
Total stockholders' equity (deficit) |
|
(92,813 |
) |
|
|
(89,593 |
) |
|
|
(80,564 |
) |
|
|
(26,449 |
) |
|
|
20,825 |
|
- 38 -
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of operations in conjunction with the “Selected Financial Data” and our consolidated financial statements and related notes thereto included elsewhere in this Form 10-K. In addition to historical information, the following discussion and analysis includes forward-looking statements that involve risks, uncertainties, and assumptions. Actual results and the timing of events could differ materially from those anticipated by these forward-looking statements as a result of many factors, including those discussed under “Risk Factors” included elsewhere in this Form 10-K. Also see “Forward-Looking Statements” included elsewhere in this Form 10-K.
Overview
We are a medical device company focused on the development and commercialization of polymer-based bioresorbable products for vascular applications. On April 3, 2017, our first product, Fantom, was approved for sale under a CE Mark, which allows us to commercialize in Europe and other jurisdictions that recognize the CE Mark. Fantom is a sirolimus-eluting bioresorbable scaffold designed specifically for coronary vascular applications.
In February 2018, we received CE approval for our third generation product, Fantom Encore, in the 2.5 millimeter diameter size. Fantom Encore has thinner struts than Fantom (95 microns versus 125 microns in the 2.5 millimeter diameter size) and comparable strength and visibility. Fantom Encore has the thinnest struts of any commercially available bioresorbable scaffold. We believe that thin struts are associated with better healing and clinical outcomes. Reductions in strut thickness are considered by physicians to be one of the most important improvements for bioresorbable scaffolds.
We began our commercial launch of Fantom late in the second quarter of 2017 and shipped our first product in the third quarter of 2017. We are launching Fantom in a phased approach beginning first with direct sales in Germany, Switzerland and Austria. We will continue to expand geographically as we move into additional phases of our launch.
Although we initiated commercial sales of Fantom in the third quarter of 2017, we are still very early in the commercialization stage. The withdrawal of Absorb, a competitor’s product, in 2017, and the negative publicity related to Absorb’s safety have severely impacted the market for bioresorbable scaffolds, and companies with bioresorbable scaffolds that were made from the same polylactic acid polymer, or PLLA, as Absorb have reduced scale and abandoned their efforts to commercialize such scaffolds. Because Fantom is not made with the same PLLA polymer as Absorb, we continue to believe that we can commercialize Fantom despite the impact that the withdrawal of Absorb had on the market and capitalize on the benefits of bioresorbable technology. That said, we must now rebuild the market for bioresorbable scaffolds, which can be more challenging than selling into an existing, healthy market. Our rebuilding activities include educating physicians regarding the unique features of Fantom and Fantom Encore, continuing to publish results from our pivotal clinical trial (FANTOM II) and conducting and initiating additional clinical studies to build the clinical evidence needed to support market adoption.
In 2018, we plan to expand our product portfolio into peripheral arterial disease by applying for CE Mark with our bioresorbable scaffold technology for below-the-knee revascularization. The majority of the arteries below the knee have similar sizes and dimensions to coronary arteries, making this a viable treatment option. Resorbable technology is attractive in this application because of the frequent need for retreatment in this patient population. If and when we receive CE Mark, we plan to conduct a pilot trial in a small number of centers to assess product performance, inform product development activities and determine commercial strategy.
As of December 31, 2017, our cash, cash equivalents and investment securities totaled $20.0 million, which, based on our current operating plans and projections, we believe will be sufficient to fund our operating and capital needs through the first quarter of 2019. We have incurred substantial losses since our inception; as of December 31, 2017, we had accumulated a deficit of approximately $382.2 million. See “—Liquidity and Capital Resources,” below.
Highlights of our Results of Operations
We began commercialization of our first product, Fantom, in 2017 and first recorded revenue and cost of goods sold in the third quarter of 2017. We also began recording sales and marketing expenses in 2017 as we built our sales force and marketing team. During 2017, we continued to work on product development activities and clinical studies. As commercialization did not commence in earnest until the second half of 2017, research and development expenses were our most significant expense for 2017. Our general and administrative expenses have remained consistent from 2016 to 2017. We also incur other non-operating expenses that primarily arise from the convertible notes and warrants that we issued in 2014 and 2017.
- 39 -
Research and Development Expenses: Our research and development, or R&D, expenses arise from a combination of internal and external costs. Our internal costs primarily consist of employee salaries and benefits, facility and other overhead expenses, and engineering and other supplies that we use in our labs for prototyping, testing, and other development activities. Our external costs primarily consist of contract research, engineering consulting, polymer consulting and certain production costs, polymer lasing costs, catheter system and anti-restenotic drug purchases, preclinical and clinical trial expenses, regulatory consulting, and license fees paid for the technology underlying our polymer materials. All R&D costs are expensed when incurred.
Our R&D expenses were 60 percent and 68 percent of total operating expenses for the years ended December 31, 2017 and 2016, respectively. These percentages have decreased from our historical averages of 70 to 75 percent of total operating expenses as we transitioned to commercialization of Fantom. While we expect our R&D expenses to decrease in 2018 as we dedicate a significant portion of our resources to the commercialization of Fantom and supporting registry studies, we believe R&D expenses will still be a significant portion of our operating expenses as we continue to research, prove feasibility, and develop additional products.
Selling, General and Administrative Expenses: Our selling, general and administrative, or SG&A, expenses consist primarily of salaries and benefits for our executive officers, administrative and marketing staff and sales force, corporate office and other overhead expenses, legal expenses including costs, audit and tax fees, sales and marketing expenses, investor relations and other public company costs, and travel expenses.
Our SG&A expenses were 40 percent and 32 percent of total operating expenses for the years ended December 31, 2017 and 2016, respectively. These percentages have increased from our historical averages of 25 to 30 percent of total operating expenses as we expanded our corporate infrastructure to support the commercialization of Fantom and the ongoing needs of being a public company.
Other Income (Expense): Following our issuance of convertible notes and warrants in 2014 and 2017, the components of other income and expense primarily comprise interest expense on the convertible notes and gains or losses related to the changes in fair values of the convertible notes and warrants. We account for the convertible notes and warrants (until they are exercised) at fair value, which means we remeasure their fair values at each reporting date and, if those fair values change, record a corresponding gain (upon a decrease in fair value) or loss (upon an increase in fair value) in our statement of operations. During 2017, due primarily to a decrease in the trading price of our common stock of approximately 33 percent, the value of the convertible notes and warrants decreased and we recorded a $35.7 million gain on the change in value.
Until the convertible notes are either repaid or converted into common stock, we expect our other income and expense to fluctuate, and possibly by a significant amount, by future gains or losses on the changes in their fair value. Also, we will continue to accrue and record interest expense on the notes at the rate of 7.54 percent per annum for the convertible notes we issued in 2014 and 8.0 percent per annum for the convertible notes we issued in 2017 until they are either converted or repaid.
Income Taxes: We have reported operating losses for all periods through December 31, 2017; therefore no provision for income taxes has been recorded. While we had deferred tax assets of approximately $76.0 million as of December 31, 2017, we have established a valuation allowance against the entire balance of deferred tax assets due to the uncertainty surrounding our ability to generate future taxable income to be able to realize those tax assets.
Critical Accounting Policies and Significant Estimates
Our consolidated financial statements are prepared in accordance with US GAAP. Their preparation requires us to make and use estimates, assumptions, and judgments that affect the reported amounts of assets, liabilities, stockholders’ equity, expenses, and the presentation and disclosures related to those items. We base our estimates and assumptions on historical experience and other factors that we believe to be reasonable under the circumstances. We evaluate our estimates and assumptions on an ongoing basis; changes in our estimates and assumptions are reasonably likely to occur from period to period. Additionally, actual results could differ significantly from the estimates we make. To the extent there are material changes in our estimates or material differences between our estimates and our actual results, our future financial statement presentation, financial condition, results of operations, and cash flows will be affected.
While our significant accounting policies are described in more detail in Note 3 to our consolidated financial statements included elsewhere in this Form 10-K, we believe the following accounting policies involve a greater
- 40 -
degree of judgment and complexity than our other accounting policies and, therefore, are the most critical to understanding and evaluating our consolidated financial condition and results of operations.
Revenue: We received our first order for Fantom in June 2017. We sell Fantom to hospitals, and title and risk of loss transfer upon delivery to these hospitals. We recognize revenue when all of the following four criteria are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred; (3) the fee is fixed or determinable; and (4) collectability is reasonably assured. We also consider any return or exchange rights. We analyze product reorder rates to evaluate and determine whether exchange rights exist and are likely to be exercised. If the revenue recognition criteria are not met, we defer the recognition of revenue by recording deferred revenue until such time that all criteria are met.
We recognized $45,000 of revenue during the year ended December 31, 2017. Total billings for shipped product for this period were $203,000; the amount by which total billings for shipped product exceeded recognized revenue was recorded as deferred revenue.
Accounts receivable consist of trade receivables recorded upon shipment of product reduced by reserves when necessary for estimated bad debts. Accounts receivable are recorded at the invoiced amount and do not bear interest. Credit is extended based on an evaluation of the customer’s financial condition. The allowance for doubtful accounts is determined based on current customer information and other relevant factors, including specific identification of past due accounts. Once a receivable is deemed to be uncollectible, such balance is charged against the allowance. As of December 31, 2017, our allowance for doubtful accounts was $0.
R&D Costs: R&D costs are expensed as incurred. These costs include salaries, employee benefits, laboratory supplies, consulting services, manufacturing products and services, preclinical and clinical costs, technology license fees, laboratory equipment depreciation, facility costs, and certain indirect costs.
Stock-Based Compensation: Stock-based compensation expense is recorded in connection with stock options, restricted stock awards, and restricted stock unit awards, or RSUs, to employees, directors, and consultants. We have granted stock options, restricted stock, and RSUs that vest based on the passage of time (time-based vesting awards) as well as stock options and RSUs that vest based on achievement of performance milestones (performance-based vesting awards).
For time-based vesting stock options granted to employees and directors, we determine compensation expense based on estimated grant date fair values utilizing the Black-Scholes option valuation model. The Black-Scholes model requires the input of assumptions, including volatility, the expected term, and the fair value of the underlying common stock on the date of grant, among other inputs. For time-based vesting restricted stock awards and RSUs, the grant date fair value is equal to the closing market price of our common stock on the date of award. We use the straight-line method to allocate compensation expense to reporting periods over each recipient’s requisite service period, which is generally from one to four years. All stock-based compensation expense is recorded as either research and development or selling, general and administrative expense based on a recipient’s work classification.
For performance-based vesting stock options and RSUs, we record compensation expense for only the performance milestones that are probable of being achieved, with such expense recorded on a straight-line basis over the expected vesting period. We reassess our performance-based estimates each reporting period and, if the estimated service period changes, we recognize all remaining compensation expense over the remaining service period and, if the probability of achievement changes to or from “probable,” we recognize the cumulative effect. Whenever an award recipient terminates service prior to achievement of a performance milestone, the recipient’s unvested awards are cancelled and the related compensation expense previously recorded is reversed.
For stock options granted to consultants, all of which are time-based vesting, we estimate fair values at the date of grant and at each subsequent reporting period and record compensation expense during the consultant’s service period. We estimate the fair value utilizing the Black-Scholes option valuation model with the same approach to inputs and assumptions as we use to estimate the fair value of employee options, except we use the remaining term as the expected life of the option.
Inventory: We received CE Mark approval of our Fantom scaffold on April 3, 2017, at which time we began capitalizing raw material purchases and commercial scaffold production costs to inventory. Inventory is stated at the lower of cost or net realizable value based on the first-in, first-out cost method. Our policy is to record an estimated allowance against inventory for unsalable, obsolete, or impaired inventory, with a corresponding increase to cost of
- 41 -
revenue. We record the cost of products to be used in research and development or clinical trials as research and development expense when they are identified as such.
Convertible Notes: Convertible notes are analyzed at issue date to determine balance sheet classification, issue discounts or premiums, and embedded or derivative features. Embedded or derivative features are evaluated in accordance with accounting guidance for derivative securities and, if the features give rise to separate accounting, we make an election to account for the notes at cost or at fair value. If fair value accounting is elected on the issue date, we record the difference between the issue price of the notes and their fair value as a gain or loss in our consolidated statement of operations. We remeasure the fair value at each reporting date and record a gain (upon a decrease in fair value) or loss (upon an increase in fair value), as a component of other income (expense) in our consolidated statement of operations. Inputs to the models include the market value of the underlying stock, a life equal to the contractual life of the notes, incremental borrowing rates that correspond to debt with similar credit worthiness, and estimated volatility based on the historical prices of our trading securities. For each periodic valuation, we also make assumptions as to our abilities to test and commercialize our product, to obtain future financings when and if needed, and to comply with the terms and conditions of any outstanding convertible notes.
Following an analysis of their embedded and derivative features, we elected to utilize fair value accounting for all issues of convertible notes as management believes the convertible notes will be converted into common stock, rather than repaid, and the fair value method of accounting provides a more appropriate value of these liabilities than would be provided under the cost method.
Common Stock Warrants: The fair value of warrants issued for the purchase of common stock is recorded as a liability whenever warrants call for issuance of registered shares upon exercise, a condition that we may not be able to satisfy at the time of exercise, and which, if not so satisfied, will result in a net settlement of warrants. Until the time warrants are exercised or expire, the fair value is assessed at each reporting date. Any change in value is recorded as a gain or loss component of other income (expense) in our consolidated statement of operations. Inputs to the valuation models are of the same nature as those used to value our convertible notes.
Results of Operations
During 2017, our operating activities focused on finalizing processes for commercial operations which commenced in the third quarter of 2017. We also continued follow-up assessments of patients in our FANTOM II clinical trial. We started two additional clinical trials of Fantom evaluating use of our bioresorbable scaffold in broader patient populations. Additionally, we completed a financing in which we issued convertible notes and warrants in May 2017 and June 2017, receiving net cash proceeds of approximately $32.5 million.
During 2016, our operating activities consisted of clinical trial enrollments, which were completed in March 2016 with a total of 240 patients enrolled, performing follow-up assessments of the patients, collecting the related clinical data to support the CE Mark submission that was completed in August 2016, and refining our manufacturing processes in preparation for the commercialization of Fantom.
Comparison of the Years Ended December 31, 2017 and 2016 (dollars in thousands):
|
|
Year Ended |
|
|
|
|
|||||||||
|
|
December 31, |
|
|
Change |
|
|||||||||
|
|
2017 |
|
|
2016 |
|
|
$ |
|
|
% |
|
|||
|
Revenue |
$ |
45 |
|
|
$ |
— |
|
|
$ |
45 |
|
|
100% |
|
|
R&D expenses |
|
12,760 |
|
|
|
18,171 |
|
|
|
(5,411 |
) |
|
-30% |
|
|
SG&A expenses |
|
8,572 |
|
|
|
8,609 |
|
|
|
(37 |
) |
|
0% |
|
|
Interest expense |
|
6,690 |
|
|
|
2,053 |
|
|
|
4,637 |
|
|
226% |
|
|
Loss on issuance of convertible notes and warrants |
|
520 |
|
|
|
— |
|
|
|
520 |
|
|
-100% |
|
|
Gain/(loss) on change in fair values of convertible notes and warrant liability |
|
35,731 |
|
|
|
(25,247 |
) |
|
|
60,978 |
|
|
242% |
|
|
Interest income and other (expense)/income |
|
(58 |
) |
|
|
(18 |
) |
|
|
(40 |
) |
|
-222% |
|
We recognized $45,000 of revenue for the year ended December 31, 2017 compared to no revenue for the year ended December 31, 2016.
R&D expenses decreased $5.4 million, or 30%, to $12.8 million for the year ended December 31, 2017, compared to $18.2 million for the year ended December 31, 2016. The decrease in R&D expenses in 2017 compared to 2016
- 42 -
was primarily due to a $1.7 million decrease in R&D materials, $1.3 million decrease in stock-based compensation, $1.2 million decrease in clinical costs, $0.6 million decrease in personnel costs and a $0.5 million decrease in testing and validation costs.
The $1.7 million decrease in R&D materials and $0.5 million decrease in testing and validation costs was related to reduced activities in these areas as we transitioned from R&D to commercialization. The $1.3 million decrease in stock-based compensation and $0.6 million decrease in personnel costs was related to the reduction in force that occurred in the third quarter of 2017. The $1.2 million decrease in clinical costs was related to decreased activity in 2017 as enrollment of patients in the FANTOM II clinical trial enrolled was completed in March 2016.
SG&A expenses were $8.6 million for the year ended December 31, 2017, consistent with SG&A expenses for the year ended December 31, 2016. Although SG&A expenses in 2017 and 2016 were consistent, stock-based compensation decreased by $1.4 million in 2017, which was offset by increases of $0.5 million in personnel expenses, $0.4 million in sales and marketing expenses, $0.4 million in audit and tax expenses and $0.1 million in consulting expenses.
The $1.4 million decrease in stock-based compensation and $0.5 million increase in personnel expenses was primarily related to executive retirements that occurred in the third quarter of 2017. The $0.4 million increase in sales and marketing expenses was related to the launch of Fantom in the second quarter of 2017. The $0.4 million increase in audit and tax expenses and $0.1 million in consulting expenses was related to the accounting for the convertible notes issued in 2017 and accounting for items related to commercialization (i.e. revenue, cost of goods sold, accounts receivable, inventory).
Our other non-operating expenses for the year ended December 31, 2017 were primarily comprised of interest expense, the loss on the issuance of convertible notes and warrants in 2017 and the gain on the change in fair value of convertible notes and warrant liability.
Interest expense increased $4.6 million, or 226%, to $6.7 million for the year ended December 31, 2017, compared to $2.1 million for the year ended December 31, 2016. The $4.6 million increase in interest expense was related to the convertible notes issued in 2017 and continuing compounding interest on the convertible notes issued in 2014.
Gain on change in fair value of convertible notes and warrant liability was $35.7 million for the year ended December 31, 2017 compared to a loss of $25.2 million for the year ended December 31, 2016. The gain in 2017 was related to a $30.7 million gain on the change in fair values of the convertible notes issued in 2014, a $2.5 million gain on the change in fair values of the convertible notes issued in 2017 and a $2.5 million gain on the change in fair values on the warrants issued in connection with the convertible notes issued in 2017. The loss in 2016 was related to a $16.3 million loss on the change in fair values of the convertible notes issued in 2014 and a $9.0 million loss on the change in fair values on the warrants issued in connection with such convertible notes between January 1, 2016 and the exercise date of February 12, 2016.
Comparison of the Years Ended December 31, 2016 and 2015 (dollars in thousands):
|
|
Year Ended |
|
|
|
|
|||||||||
|
|
December 31, |
|
|
Change |
|
|||||||||
|
|
2016 |
|
|
2015 |
|
|
$ |
|
|
% |
|
|||
|
R&D expenses |
$ |
18,171 |
|
|
$ |
16,760 |
|
|
$ |
1,411 |
|
|
8% |
|
|
SG&A expenses |
|
8,609 |
|
|
|
7,210 |
|
|
|
1,399 |
|
|
19% |
|
|
Interest expense |
|
2,053 |
|
|
|
1,904 |
|
|
|
149 |
|
|
8% |
|
|
Loss on change in fair values of convertible notes and warrant liability |
|
(25,247 |
) |
|
|
(56,788 |
) |
|
|
31,541 |
|
|
56% |
|
|
Interest income and other (expense)/income |
|
(18 |
) |
|
|
68 |
|
|
|
(86 |
) |
|
-126% |
|
R&D expenses increased $1.4 million, or 8%, to $18.2 million for the year ended December 31, 2016, compared to $16.8 million for the year ended December 31, 2015. The increase in R&D expenses in 2016 compared to 2015 was primarily due to a $1.0 million increase in R&D materials, $0.6 million increase in personnel costs and a $0.4 million increase in testing and validation costs, offset by a $0.6 million decrease in preclinical costs and $0.2 million decrease in clinical costs.
The $1.0 million increase in R&D materials, $0.6 million increase in personnel costs and $0.4 million increase in testing and validation costs was related to increased activity as we prepared for commercialization. The $0.6 million decrease in preclinical costs was related to the timing of studies and related analyses. The $0.2 million decrease in
- 43 -
clinical costs was related to decreased activity in 2016 as enrollment of patients in the FANTOM II clinical trial was completed in March 2016.
SG&A expenses increased $1.4 million, or 19%, to $8.6 million for the year ended December 31, 2016, compared to $7.2 million for the year ended December 31, 2015. The increase in SG&A expenses in 2016 compared to 2015 was primarily due to a $1.5 million increase in stock-based compensation, offset by a $0.2 million increase in travel costs.
The $1.5 million increase in stock-based compensation was related to equity grants to our Chief Executive Officer and members of our Board of Directors in late 2015 and 2016. The $0.2 million decrease in travel costs was related to less clinical support travel following the completion of enrollment in the FANTOM II clinical trial.
Our other non-operating expenses for the year ended December 31, 2016 were primarily comprised of interest expense and the loss on the change in fair value of convertible notes and warrant liability.
Interest expense increased $0.2 million, or 8%, to $2.1 million for the year ended December 31, 2016, compared to $1.9 million for the year ended December 31, 2015. The $0.2 million increase in interest expense was related to compounding interest on the convertible notes issued in 2014.
Loss on change in fair value of convertible notes and warrant liability was $25.2 million for the year ended December 31, 2016 compared to $56.8 million for the year ended December 31, 2015. The loss in 2016 was related to a $16.3 million loss on the change in fair values of the convertible notes issued in 2014 and a $9.0 million loss on the change in fair values on the warrants issued in connection with such convertible notes between January 1, 2016 and the exercise date of February 12, 2016. The loss in 2015 was related to a $37.6 million loss on the change in fair values of the convertible notes issued in 2014 and a $19.2 million loss on the change in fair values on the warrants issued in connection with such convertible notes.
Liquidity and Capital Resources
Sources of Liquidity
As of December 31, 2017, we had a cash, cash equivalents and investment securities balance of $20.0 million, which we believe is sufficient to fund our operating and capital needs through the first quarter of 2019. See “—Operating Capital and Capital Expenditure Requirements,” below.
During the second quarter of 2017, we completed a two-stage financing. In May 2017, we issued 338 convertible notes and in June 2017 we issued 133 convertible notes, each with a face value of $100,000, for total gross cash proceeds of $47.1 million. We used a portion of the proceeds from this financing to repurchase 1,732,260 shares of our common stock from one of the investors in the financing at $7.212 per share, for a total repurchase price of $12.5 million, and incurred transaction costs of $2.1 million, resulting in net proceeds from this financing of $32.5 million. As part of the financing, we issued warrants to purchase 2,119,500 shares of our common stock at an initial exercise price of $5.00 per share.
The convertible notes we issued in 2014 mature in November 2019 and the convertible notes we issued in May and June 2017 mature in May and June 2022, respectively. No payments of interest or principal are required on any of the notes until maturity. However, each holder of the notes we issued in 2017 has a right to request that we redeem the notes (face value plus accrued interest) on November 4, 2019. Accordingly, we may be required to repay an aggregate of $72.1 million plus accrued interest in November 2019. If the noteholders collectively, or individually, call for redemption of the 2017 convertible notes, or if we are unable to convert or extend the maturity date of the 2014 convertible notes, we most likely would not have the cash to repay the convertible notes, and the noteholders could commence legal action against us and/or we may need to reduce operating activities and personnel, sell assets, such as our intellectual property, and/or declare bankruptcy, and we may not be able to remain in business. Management believes that it is more likely that all the convertible notes will be converted into shares of our common stock, rather than redeemed, if the value of the underlying equity continues to increase and, therefore, management has no plans to redeem any of the convertible notes.
While the warrants we issued in connection with the convertible notes we issued in May and June 2017 are immediately exercisable, have a five-year life, and only allow for cash exercise, management does not look to the warrants as a source of funding for operating or capital needs because exercise is at the holders’ option.
- 44 -
Below is a summary of our cash flows for the periods indicated.
|
|
Year Ended December 31, |
|
|||||||||
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Net cash used for operating activities |
$ |
(18,858 |
) |
|
$ |
(21,259 |
) |
|
$ |
(19,082 |
) |
|
Net cash provided by (used for) investing activities |
$ |
(1,856 |
) |
|
$ |
(729 |
) |
|
$ |
138 |
|
|
Net cash provided by financing activities |
$ |
32,584 |
|
|
$ |
11,767 |
|
|
$ |
10,025 |
|
|
Net increase (decrease) in cash and cash equivalents |
$ |
11,870 |
|
|
$ |
(10,221 |
) |
|
$ |
(8,919 |
) |
Net Cash Flow Used for Operating Activities
Net cash used for operating activities during 2017 primarily reflects the loss from operations of $21.3 million and $0.6 million used for changes in operating assets and liabilities, offset by non-cash expenses of $2.0 million for stock-based compensation and $1.1 million of depreciation and amortization. The gain from the change in fair value of convertible notes and warrant liability, the loss on issuance of convertible notes and warrants to purchase common stock and the interest on convertible notes recorded during 2017 were non-cash items that had no effect on cash flows.
Net cash used for operating activities during 2016 primarily reflects the loss from operations of $26.8 million and $0.3 million used for changes in operating assets and liabilities, offset by non-cash expenses of $4.7 million for stock-based compensation, $1.1 million of depreciation and amortization, and $21,000 of other non-cash expense. The loss from the change in fair value of convertible notes and warrant liability and the interest on convertible notes recorded during 2016 were non-cash items that had no effect on cash flows.
Net cash used for operating activities during 2015 primarily reflects the loss from operations of $24.0 million, offset by non-cash expenses of $3.4 million for stock-based compensation, $1.1 million of depreciation and amortization, $0.2 million from changes in operating assets and liabilities, and $46,000 of other non-cash expense. The loss from the change in fair value of convertible notes and warrant liability and the interest on convertible notes recorded during 2015 were non-cash items that had no effect on cash flows.
Net Cash Flow Provided by (Used for) Investing Activities
Net cash used for investing activities during 2017 consisted of investment purchases of $1.5 million and property and equipment purchases of $0.4 million.
Net cash used for investing activities during 2016 consisted of property and equipment purchases of $0.7 million.
Net cash provided by investing activities during 2015 consisted of $1.0 million in net maturities of investments offset by $0.9 million in purchases of property and equipment.
Net Cash Flow Provided by Financing Activities
Net cash provided by financing activities in 2017 consisted of $47.1 million in proceeds from the issuance of convertible notes and warrants and $0.1 million in proceeds from the issuance of common stock upon the exercise of stock options, offset by $12.5 million to repurchase 1,732,260 shares of our common stock and $2.1 million for financing transaction costs.
Net cash provided by financing activities in 2016 consisted of $11.4 million in proceeds from the issuance of common stock upon the exercise of 4,375,000 warrants originally issued in 2014 and $0.4 million in proceeds from the issuance of common stock upon exercise of stock options.
Net cash provided by financing activities in 2015 consisted of $9.5 million in proceeds from the issuance of common stock upon the exercise of 4,375,000 warrants originally issued in 2014 and $0.6 million in proceeds from the issuance of common stock upon exercise of employee stock options. These receipts were offset by a $50,000 payment in 2015 for issuance costs incurred in 2014 in connection with the issuance of convertible notes.
- 45 -
Operating Capital and Capital Expenditure Requirements
On April 3, 2017, our first product, Fantom, was approved for sale under a CE Mark, which allows us to commercialize in Europe and other jurisdictions that recognize the CE Mark. We initiated commercial sales in July 2017. Fantom is our first commercial product; prior to 2017, we have not commercialized any products or generated any revenue since our inception in June 1998. Although we initiated commercial sales of Fantom in the third quarter of 2017, we are still very early in the commercialization stage. We have been negatively impacted by the withdrawal of Absorb, a competitor’s product, in 2017 and the negative publicity around Absorb’s safety. We are focused on rebuilding the market for bioresorbable scaffolds and educating physicians regarding the unique features of Fantom versus Absorb. We are conducting and initiating additional clinical studies to build the clinical evidence needed to support market adoption. During 2018, we will be expanding our commercial and clinical efforts to additional markets that accept the CE Mark or allow registration based on our existing clinical evidence.
We have incurred substantial losses since our inception. As of December 31, 2017, we had accumulated a deficit of $382.2 million. Until we generate revenue at a level to support our cost structure, we expect to continue to incur substantial operating losses and net cash outflows. We may never become profitable and even if we do attain profitability, we may not be able to sustain profitability or positive cash flows on a recurring basis. Until we generate positive cash flows from operations on a sustainable basis, we plan to continue to fund our operating and capital needs from our current cash resources and proceeds from future capital raising efforts. As of December 31, 2017, we had a cash, cash equivalents and investment securities balance of $20.0 million, which we believe is sufficient to fund our operating and capital needs through the first quarter of 2019. See “—Sources of Liquidity,” above.
Until we are able to significantly accelerate our sales, we do not anticipate generating positive cash flows in 2018 or 2019, and therefore, will need to raise further capital to support our operations and our ongoing costs, and to conduct a U.S. clinical trial, if we determine to do so. We have a plan to address our capital needs, which includes accelerating our revenue by pursuing sales expansion and executing business development and strategic opportunities. We are also evaluating public or private sales of our equity or debt securities. In addition, the convertible notes we issued in 2014 mature in November 2019 and each holder of the convertible notes we issued in 2017 has a redemption right that it may exercise in November 2019. The aggregate face value of all such convertible notes is $72.1 million and the aggregate accrued interest on all such notes was $8.8 million as of December 31, 2017. If the holders of the 2017 convertible notes collectively, or individually, call for redemption, or if we are unable to convert or extend the maturity date of the 2014 convertible notes, we most likely will not have the cash to repay the notes.
There can be no assurance that we will be successful in accelerating our revenue or raising additional capital. Additionally, we may be limited under the terms of our convertible notes as to the type, quantity, timing, or other aspects of a financing, unless the noteholders agree. If we are unable to significantly increase revenue or raise additional capital when needed or on acceptable terms, we would need to consider a delay, reduction or cessation of our research and development programs and our commercialization efforts. There can be no assurance that our efforts will result in the resolution of our liquidity needs. If we are not able to continue as a going concern, holders of our common stock could lose their investment. The accompanying consolidated financial statements do not include any adjustments that might result should we be unable to continue as a going concern.
Because of the numerous risks and uncertainties associated with developing, testing, and commercializing medical devices such as our bioresorbable scaffolds, our estimates as to the amounts and timing of capital outlays and operating expenditures are subject to change. Our ongoing funding requirements will depend on many factors, including, but not limited to:
|
|
• |
the success of our sales and marketing initiatives; |
|
|
• |
the success, or failure, of our competitors who marketed bioresorbable scaffolds before us, including their ability to identify and remedy the causes of very late stent thrombosis reported from their products; |
|
|
• |
our ability to provide additional clinical data regarding Fantom’s potential long-term benefits; |
|
|
• |
the time and effort it will take to successfully complete our clinical trials and analyze patient data; |
|
|
• |
the requirements, cost, and timing of regulatory approvals; |
|
|
• |
the time and effort required to refine and scale-up manufacturing processes and the cost of establishing commercial supplies of our products; |
- 46 -
|
|
• |
the scope of research and development for any of our other product opportunities and the terms and timing of any collaborative, licensing, or other arrangements that we may establish; and |
|
|
• |
the cost of filing and prosecuting patentable technologies and defending and enforcing our patent and other intellectual property rights and the effect of competing technological and market developments. |
Our ongoing capital requirements will also depend on the extent to which we acquire or invest in businesses, products, and technologies; we currently have no commitments or agreements relating to any of these types of transactions. We believe our current San Diego facility has the capacity to produce the quantities of Fantom that will be needed for our initial commercial sales and, therefore, do not have any plans for facility expansion at this time.
Contractual Obligations, Commitments, and Contingencies
The following table summarizes our outstanding contractual obligations as of December 31, 2017 (dollars in thousands):
|
|
Payments Due by Period |
|
|||||||||||||||||
|
|
< 1 Year |
|
|
1 to 3 Years |
|
|
3 to 5 Years |
|
|
Thereafter |
|
|
Total |
|
|||||
|
Operating lease obligations |
$ |
716 |
|
|
$ |
1,504 |
|
|
$ |
1,670 |
|
|
$ |
2,248 |
|
|
$ |
6,138 |
|
|
Deferred technology license fees |
|
— |
|
|
|
500 |
|
|
|
— |
|
|
|
— |
|
|
|
500 |
|
|
Purchase obligations |
|
610 |
|
|
|
24 |
|
|
|
— |
|
|
|
— |
|
|
|
634 |
|
|
|
$ |
1,326 |
|
|
$ |
2,028 |
|
|
$ |
1,670 |
|
|
$ |
2,248 |
|
|
$ |
7,272 |
|
Our convertible notes are not included in the table above as we believe they will be converted into common stock rather than repaid. Our operating lease obligations represent the contractual rental payments due under the lease for our headquarters, as amended in October 2017, which matures in May 2025.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements.
Recently Adopted and Issued Accounting Pronouncements
We have set forth the applicable recent adopted and issued accounting pronouncements in Note 3 to our consolidated financial statements included elsewhere in this Form 10-K. We adopted ASU 2016-09, Stock Compensation: Improvements, in 2017 and, accordingly, provide the disclosures required by that pronouncement. We do not believe adoption of other applicable pronouncements will result in a material effect on our financial statements or related disclosures.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
Interest Rate Sensitivity
As of December 31, 2017, we had cash and cash equivalents and investment securities totaling $20.0 million which are comprised of cash in checking and savings accounts, money market funds and FDIC insured certificates of deposit. The primary objective of our investment activities is to preserve principal and liquidity while maximizing income without significantly increasing risk. We do not enter into investments for trading or speculative purposes. Our convertible notes bear interest at a fixed rate and are not subject to interest rate fluctuations. We do not believe that an immediate 10% increase in interest rates would have a material effect on our operating results or cash flows.
We are subject to foreign currency exchange risk because we conduct our clinical trials primarily in the EU and Australia. The effect of foreign currency rate fluctuations with respect to the amounts we incur to hospitals, physicians, and other suppliers in connection with such clinical trials, which are denominated primarily in the currencies of the EU and Australia, has been immaterial through December 31, 2017.
We are also subject to foreign currency exchange risk related to our commercial sales activities in Europe, which we initiated during the third quarter of 2017. We denominate these sales in the currencies of the EU and are subject to
- 47 -
foreign currency exchange risk on these sales until we receive payment. The effect of foreign currency rate fluctuations with respect to such sales has been immaterial through December 31, 2017.
We do not enter into foreign currency hedging transactions. At our current levels of clinical trial activity and international sales, we believe our exposure to foreign currency rate fluctuations is minor. Our German subsidiary is non-operational; until such time as it conducts operations, the effects of exchange rate fluctuations on its net assets are immaterial to our financial statements.
Inflation
Inflation generally affects us by increasing our cost of labor and clinical trial costs. We do not believe that inflation has had a material effect on our business, financial condition or results of operations during the periods presented.
Item 8. Financial Statements and Supplementary Data
Our consolidated financial statements, and accompanying notes, and the Report of Grant Thornton LLP, our Independent Registered Public Accounting Firm, are included in this Annual Report on Form 10-K on pages F-1 through F-25.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
Item 9A. Controls and Procedures
Management’s Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) under the Securities Exchange Act of 1934 as a process designed by, or under the supervision of, the Company’s principal executive and principal financial officers, or persons performing similar functions, and effected by the Company’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
|
|
• |
pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the Company’s assets; |
|
|
• |
provide reasonable assurance that transactions are recorded as necessary to permit the preparation of financial statements in accordance with generally accepted accounting principles, and that the receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and, |
|
|
• |
provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the Company’s assets that could have a material effect on the financial statements. |
Our internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. GAAP. Management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our internal controls will prevent or detect all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of internal controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
- 48 -
Our management, including our Chief Executive Officer and Chief Financial Officer, conducted an assessment of the effectiveness of our internal control over financial reporting as of the end of the fiscal year covered by this Form 10-K. In making this assessment, our management used the criteria set forth in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 Framework). Based on their assessment, management has concluded that, as of December 31, 2017, our internal control over financial reporting was effective based on such criteria.
Our independent registered public accounting firm has issued an audit report on our internal control over financial reporting, which appears on page F-2 of this Form 10-K.
Evaluation of Disclosure Controls and Procedures
Based on an evaluation under the supervision and with the participation of our management, our Chief Executive Officer and Chief Financial Officer have concluded that our disclosure controls and procedures as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act were effective as of December 31, 2017 to provide reasonable assurance that information required to be disclosed by us in reports that we file or submit under the Exchange Act is (i) recorded, processed, summarized and reported within the time periods specified in the SEC rules and forms and (ii) accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure.
Changes in Internal Control Over Financial Reporting
There has not been any change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934) that occurred during the quarterly period ended December 31, 2017 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
None.
- 49 -
Item 10. Directors, Executive Officers, and Corporate Governance
The information required by this item related to our directors is incorporated by reference to our Definitive Proxy Statement for our 2018 Annual Meeting of Stockholders, to be filed with the SEC within 120 days of December 31, 2017, or the 2018 Proxy Statement, under the heading “Election of Directors.”
The information required by this item related to our executive officers is set forth under “Executive Officers” in Item 1 of Part I of this Form 10-K and is incorporated herein by reference.
Code of Business Conduct and Ethics
We have adopted a Code of Business Conduct and Ethics that applies to all of our officers, directors, and employees. We have posted a copy of our Code of Business Conduct and Ethics, and intend to post updates to this code, or any waivers of its requirements, in the Corporate Governance section on our website at www.revamedical.com. If we make any substantive amendments to the Code of Business Conduct and Ethics or grant any waiver from any of its provisions to any executive officer or director, we will disclose the nature of the amendment or waiver on our website within four business days.
Australian Disclosure Requirements
Because we are listed on the ASX, we are required to comply with various disclosure requirements as set out in the ASX Listing Rules. The following information is provided to comply with the ASX Listing Rules and is not intended to fulfill SEC information required by Part III of this Annual Report on Form 10-K.
Substantial Holders at February 23, 2018
The number of our equity securities held by our substantial security holders (i.e., those holders, who together with their affiliates, have an interest in at least five percent of our voting securities), assuming the conversion of common stock held by those security holders into CHESS Depositary Interests, or “CDIs” (ten CDIs are equivalent to one share of common stock), based on our review of our shareholder registers and available public filings, as of February 23, 2018 are as follows:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total Holdings |
|
||||
|
Security Holder |
|
Number of Common Shares Held |
|
% of Total Common Shares Outstanding |
|
|
Number of CDIs Held |
|
% of Total CDIs Outstanding |
|
|
Equivalent Number of CDIs Held |
|
% of Total Securities Outstanding |
|
||||||
|
Brookside Capital and affiliates |
|
|
— |
|
— |
|
|
|
29,650,222 |
|
11.0% |
|
|
|
29,650,222 |
|
7.2% |
|
|||
|
Cerberus and affiliates |
|
|
— |
|
— |
|
|
|
28,844,260 |
|
10.7% |
|
|
|
28,844,260 |
|
7.0% |
|
|||
|
Citicorp Nominees Pty Limited |
|
|
— |
|
— |
|
|
|
37,643,372 |
|
14.0% |
|
|
|
37,643,372 |
|
9.1% |
|
|||
|
Domain Partners and affiliates |
|
|
3,703,688 |
|
25.8% |
|
|
|
— |
|
— |
|
|
|
37,036,880 |
|
9.0% |
|
|||
|
Elliott Associates, L.P. |
|
|
3,227,031 |
|
22.4% |
|
|
|
— |
|
— |
|
|
|
32,270,310 |
|
7.8% |
|
|||
|
Goldman Sachs International |
|
|
4,375,000 |
|
30.4% |
|
|
|
— |
|
— |
|
|
|
43,750,000 |
|
10.6% |
|
|||
|
Group Outcome Investors/Robert B. Stockman |
|
|
1,761,506 |
|
12.3% |
|
|
|
1,199,264 |
|
0.5% |
|
|
|
18,814,324 |
|
4.5% |
|
|||
|
Merrill Lynch (Australia) Nominees Pty Limited |
|
|
— |
|
— |
|
|
|
26,287,301 |
|
9.8% |
|
|
|
26,287,301 |
|
6.4% |
|
|||
|
Kenneth Rainin Trust and affiliates |
|
|
— |
|
— |
|
|
|
13,470,695 |
|
5.0% |
|
|
|
13,470,695 |
|
3.3% |
|
|||
|
Senrigan Capital and affiliates |
|
|
— |
|
— |
|
|
|
62,192,706 |
|
23.2% |
|
|
|
62,192,706 |
|
15.1% |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total securities held by > 5% holders |
|
|
13,067,225 |
|
90.9% |
|
|
|
199,287,820 |
|
74.2% |
|
|
|
329,960,070 |
|
80.0% |
|
|||
|
Total securities held by all other holders |
|
|
1,311,803 |
|
9.1% |
|
|
|
69,380,100 |
|
25.8% |
|
|
|
82,498,130 |
|
20.0% |
|
|||
- 50 -
Distribution of Security Holders as of February 23, 2018
As of February 23, 2018, we had a total of 41,245,820 shares of common stock issued and outstanding, a portion of which were held as CDIs (ten CDIs are equivalent to one share of common stock). The table below presents the number of shares of common stock (including restricted stock) and CDIs held, as well as the number of shares underlying outstanding stock options to purchase common stock, convertible notes, warrants and restricted stock units.
|
|
Common Stock |
|
|
CDIs |
|
|
Options (unlisted) |
|
|
Convertible Notes (unlisted) |
|
|
Warrants (unlisted) |
|
|
Restricted Stock Units (unlisted) |
|
||||||||||||||||||||||||||||||
|
|
# of Holders |
|
|
# of Shares |
|
|
# of Holders |
|
|
# of CDIs |
|
|
# of Holders |
|
|
# of Shares |
|
|
# of Holders |
|
|
# of Shares |
|
|
# of Holders |
|
|
# of Shares |
|
|
# of Holders |
|
|
# of Shares |
|
||||||||||||
|
1 – 1,000 |
|
11 |
|
|
|
4,114 |
|
|
|
129 |
|
|
|
58,660 |
|
|
|
1 |
|
|
|
1,000 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
||||||
|
1,001 – 5,000 |
|
10 |
|
|
|
26,795 |
|
|
|
195 |
|
|
|
595,989 |
|
|
|
3 |
|
|
|
9,000 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
||||||
|
5,001 – 10,000 |
|
7 |
|
|
|
47,793 |
|
|
|
123 |
|
|
|
1,030,853 |
|
|
|
1 |
|
|
|
10,000 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
|
5 |
|
|
|
30,000 |
|
||||
|
10,001 – 100,000 |
|
21 |
|
|
|
665,574 |
|
|
|
237 |
|
|
|
7,985,896 |
|
|
|
15 |
|
|
|
605,620 |
|
|
|
2 |
|
|
|
115,540 |
|
|
|
5 |
|
|
|
283,500 |
|
|
|
10 |
|
|
|
374,000 |
|
|
100,001 and over |
|
10 |
|
|
|
13,634,752 |
|
|
|
57 |
|
|
|
258,996,522 |
|
|
|
13 |
|
|
|
5,920,871 |
|
|
|
10 |
|
|
|
16,832,557 |
|
|
|
5 |
|
|
|
1,836,000 |
|
|
|
2 |
|
|
|
287,500 |
|
|
Total holders and securities |
|
59 |
|
|
|
14,379,028 |
|
|
|
741 |
|
|
|
268,667,920 |
|
|
|
33 |
|
|
|
6,546,491 |
|
|
|
12 |
|
|
|
16,948,097 |
|
|
|
10 |
|
|
|
2,119,500 |
|
|
|
17 |
|
|
|
691,500 |
|
The number of shareholders holding less than a marketable parcel of CDIs (being a parcel of securities not less than A$500) as of February 23, 2018 was 135.
Top 20 CDI Holders as of February 23, 2018
Following are the top 20 holders of our CDIs on February 23, 2018 (does not include holdings in common stock):
|
|
Holder |
|
|
|
Number of CDIs Held |
|
|
|
|
% of CDIs Outstanding |
|
||
|
1 |
Citicorp Nominees Pty Limited |
|
|
|
|
68,305,814 |
|
|
|
|
|
25.4 |
% |
|
2 |
JP Morgan Nominees Australia Limited |
|
|
|
|
51,554,519 |
|
|
|
|
|
19.2 |
% |
|
3 |
HSBC Custody Nominees (Australia) Limited <No 2 A/C> |
|
|
|
|
44,334,296 |
|
|
|
|
|
16.5 |
% |
|
4 |
HSBC Custody Nominees (Australia) Limited – GSCO ECA |
|
|
|
|
38,937,333 |
|
|
|
|
|
14.5 |
% |
|
5 |
Merrill Lynch (Australia) Nominees Pty Limited |
|
|
|
|
26,287,301 |
|
|
|
|
|
9.8 |
% |
|
6 |
Frederic H Moll |
|
|
|
|
3,345,610 |
|
|
|
|
|
1.2 |
% |
|
7 |
Trienos Group LLC |
|
|
|
|
3,000,000 |
|
|
|
|
|
1.1 |
% |
|
8 |
HSBC Custody Nominees (Australia) Limited |
|
|
|
|
2,730,607 |
|
|
|
|
|
1.0 |
% |
|
9 |
UBS Nominees Pty Ltd |
|
|
|
|
2,000,000 |
|
|
|
|
|
0.7 |
% |
|
10 |
Warman Investments Pty Ltd |
|
|
|
|
1,451,771 |
|
|
|
|
|
0.5 |
% |
|
11 |
Lightstorm Pty Ltd <Hotspice A/C> |
|
|
|
|
1,240,840 |
|
|
|
|
|
0.5 |
% |
|
12 |
Viking Management Services Pty Ltd <VHK Superannuation Fund A/C> |
|
|
|
|
1,159,121 |
|
|
|
|
|
0.4 |
% |
|
13 |
Moore Family Nominee Pty Ltd <Moore Family Super Fund A/C> |
|
|
|
|
1,100,000 |
|
|
|
|
|
0.4 |
% |
|
14 |
Mr Robert Thomas + Mrs Kyrenia Thomas <Rob Thomas Super Fund A/C> |
|
|
|
|
1,100,000 |
|
|
|
|
|
0.4 |
% |
|
15 |
Mrs Danielle Susan Borgas |
|
|
|
|
1,006,000 |
|
|
|
|
|
0.4 |
% |
|
16 |
Mr Antony Richard Kerr + Mr Peter Michael Clerk <AR Kerr Family A/C> |
|
|
|
|
900,000 |
|
|
|
|
|
0.3 |
% |
|
17 |
Dr. Philip James Currie + Mrs. Anne Jennifer Currie <Currie Family Superfund A/C> |
|
|
|
|
662,521 |
|
|
|
|
|
0.2 |
% |
|
18 |
BT Portfolio Services Limited <Wade Family Super Fund A/C> |
|
|
|
|
646,394 |
|
|
|
|
|
0.2 |
% |
|
19 |
Mr. Robert Arthur Schneider |
|
|
|
|
525,000 |
|
|
|
|
|
0.2 |
% |
|
20 |
Unibram Pty Ltd <Michael Hoy Superfund A/C> |
|
|
|
|
525,000 |
|
|
|
|
|
0.2 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total CDIs held by top 20 CDI holders |
|
|
|
|
250,812,127 |
|
|
|
|
|
93.1 |
% |
|
|
Total CDIs held by all other CDI holders |
|
|
|
|
17,855,793 |
|
|
|
|
|
6.9 |
% |
|
|
Total CDIs outstanding |
|
|
|
|
268,667,920 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
- 51 -
The table below provides a list of the top 20 holders of our securities as of February 23, 2018, taking into account securities held in the form of both common stock and CDIs and prepared on the assumption that all CDIs are held as common stock. Related but separate legal entities are not aggregated for the purposes of the table below.
|
|
Security Holder |
|
Shares of Common Stock Held |
|
|
CDIs Held (common stock equivalent) |
|
|
Total Number of Securities Held |
|
|
% of Outstanding Capital |
|
||||
|
1 |
Senrigan Capital |
|
|
— |
|
|
|
6,219,271 |
|
|
|
6,219,271 |
|
|
15.1% |
|
|
|
2 |
Goldman Sachs International |
|
|
4,375,000 |
|
|
|
— |
|
|
|
4,375,000 |
|
|
10.6% |
|
|
|
3 |
Citicorp Nominees Pty Limited |
|
|
— |
|
|
|
3,764,337 |
|
|
|
3,764,337 |
|
|
9.1% |
|
|
|
4 |
Domain Partners V, L.P. |
|
|
3,703,688 |
|
|
|
— |
|
|
|
3,703,688 |
|
|
9.0% |
|
|
|
5 |
Elliott Associates, L.P. |
|
|
3,227,031 |
|
|
|
— |
|
|
|
3,227,031 |
|
|
7.8% |
|
|
|
6 |
Brookside Capital Partners Fund, LP |
|
|
— |
|
|
|
2,965,022 |
|
|
|
2,965,022 |
|
|
7.2% |
|
|
|
7 |
Merrill Lynch (Australia) Nominees Pty Limited |
|
|
— |
|
|
|
2,628,730 |
|
|
|
2,628,730 |
|
|
6.4% |
|
|
|
8 |
HSCB Custody Nominees (Australia) Limited No. 2 |
|
|
— |
|
|
|
1,650,226 |
|
|
|
1,650,226 |
|
|
4.0% |
|
|
|
9 |
Kenneth Rainin Trust and affiliates |
|
|
|
|
|
|
1,347,070 |
|
|
|
1,347,070 |
|
|
3.3% |
|
|
|
10 |
Group Outcome Investors I, LLC |
|
|
1,341,175 |
|
|
|
— |
|
|
|
1,341,175 |
|
|
3.2% |
|
|
|
11 |
Cerberus Series Four Holdings, LLC |
|
|
— |
|
|
|
1,046,486 |
|
|
|
1,046,486 |
|
|
2.5% |
|
|
|
12 |
Cerberus International, Ltd |
|
|
— |
|
|
|
995,553 |
|
|
|
995,553 |
|
|
2.4% |
|
|
|
13 |
JP Morgan Nominees Australia Limited |
|
|
— |
|
|
|
959,585 |
|
|
|
959,585 |
|
|
2.3% |
|
|
|
14 |
Gordon E. Nye |
|
|
6,300 |
|
|
|
821,485 |
|
|
|
827,785 |
|
|
2.0% |
|
|
|
15 |
Bob and Lisa Stockman |
|
|
420,331 |
|
|
|
119,926 |
|
|
|
540,257 |
|
|
1.3% |
|
|
|
16 |
Cerberus Partners, L.P. |
|
|
— |
|
|
|
520,641 |
|
|
|
520,641 |
|
|
1.3% |
|
|
|
17 |
HSBC Custody Nominees (Australia) Limited GSCO |
|
|
— |
|
|
|
393,733 |
|
|
|
393,733 |
|
|
1.0% |
|
|
|
18 |
C. Raymond Larkin Jr. |
|
|
351,749 |
|
|
|
— |
|
|
|
351,749 |
|
|
0.9% |
|
|
|
19 |
Frederic H. Moll |
|
|
— |
|
|
|
334,561 |
|
|
|
334,561 |
|
|
0.8% |
|
|
|
20 |
Trienos Group LLC |
|
|
— |
|
|
|
300,000 |
|
|
|
300,000 |
|
|
0.7% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total securities held by top 20 holders (stated as common stock) |
|
|
13,425,274 |
|
|
|
24,066,626 |
|
|
|
37,491,900 |
|
|
90.9% |
|
|
|
|
Total securities held by all other holders (stated as common stock) |
|
|
953,754 |
|
|
|
2,800,166 |
|
|
|
3,753,920 |
|
|
9.1% |
|
|
Unlisted Options, Unlisted Convertible Notes Payable, Unlisted Warrants and Unlisted Restricted Stock Units
As of February 23, 2018, we had 6,546,491 unlisted options to purchase shares of common stock on issue under the 2010 Equity Incentive Plan and the 2001 Stock Option/Stock Issuance Plan. These options are held by 33 individuals. With the exception of our Chief Executive Officer, Regina E. Groves, who holds 2,234,000 options representing 34.1% of the outstanding options, no other single person holds 20% or more of the outstanding options.
As of February 23, 2018, we had issued 721 unlisted convertible notes, each with a face value of $100,000. 250 unlisted convertible notes are related to a transaction that occurred in November 2014; each of which is convertible into 46,024.62 shares of common stock. These convertible notes are held equally by two entities, Goldman Sachs International and Senrigan Master Fund. 471 unlisted convertible notes are related to a two-part transaction that occurred in May and June 2017; each of which is convertible into 11,554.01 shares of common stock. These convertible notes are held by ten entities. With the exception of Medtronic, who holds 2,021,953 of these convertible notes, if converted, which represents 37.2% of the 2017 convertible notes, if converted. No other entity holds 20% or more of these convertible notes, if converted.
As of February 23, 2018, we had 691,500 unlisted restricted stock units on issue; each restricted stock unit entitles the holder to one share of common stock upon vesting. These restricted stock units are held by 17 individuals. The only individuals holding 20 percent or more of the RSUs are Jeffrey A. Anderson, who holds 170,500 RSUs, and Regina E. Groves, who holds 117,000 RSUs.
- 52 -
Our amended and restated certificate of incorporation and by-laws provide that each stockholder has one vote for every share of common stock entitled to vote and held by such stockholder on a record date. In addition, although holders of restricted stock are subject to restrictions on transfer until vesting, holders of restricted stock have the same voting rights as holders of shares of common stock.
If holders of CDIs wish to attend our general meetings, they will be able to do so. Under the ASX Listing Rules, REVA Medical, Inc., as an issuer of CDIs, must allow CDI holders to attend any meeting of the holders of the underlying securities unless relevant U.S. law at the time of the meeting prevents CDI holders from attending those meetings. In order to vote at such meetings, CDI holders have the following options:
|
|
• |
instructing CHESS Depositary Nominee or “CDN,” as the legal owner, to vote the shares of REVA Medical common stock underlying their CDIs in a particular manner. The instruction form must be completed and returned to our share registry prior to the meeting; |
|
|
• |
informing REVA Medical that they wish to nominate themselves or another person to be appointed as CDN’s proxy for the purposes of attending and voting at the general meeting; and, |
|
|
• |
converting their CDIs into a holding of shares of REVA Medical common stock and voting these at the meeting (however, if thereafter the former CDI holder wishes to sell their investment on ASX, it would be necessary to convert shares of common stock back to CDIs). This must be done prior to the record date for the meeting. |
Because holders of CDIs do not appear on REVA Medical’s share register as the legal holders of the common stock, they will not be entitled to vote at our stockholder meetings unless one of the above steps is undertaken. Proxy forms and details of these alternatives will be included in each notice of meeting sent to CDI holders by REVA Medical.
Holders of options and warrants to purchase stock, convertible notes, and restricted stock units are not entitled to vote.
Required Statements
REVA Medical makes the following disclosures:
|
|
• |
There is no current on-market buy-back of the Company’s securities. |
|
|
• |
REVA Medical, Inc. is incorporated in the state of Delaware in the United States of America. |
|
|
• |
REVA Medical, Inc. is not subject to Chapters 6, 6A, 6B, or 6C of the Corporations Act dealing with the acquisitions of shares (including substantial shareholdings and takeovers). |
|
|
• |
Under the Delaware General Corporation Law, shares are generally freely transferable subject to restrictions imposed by U.S. federal or state securities laws, by our certificate of incorporation or bylaws, or by an agreement signed with the holders of the shares at issue. Our amended and restated certificate of incorporation and bylaws do not impose any specific restrictions on transfer. Delaware General Corporation Law prohibits a publicly held Delaware Corporation from engaging in a “business combination” with an “interested shareholder” for a period of three years following the time the person became an interested shareholder, unless the business combination or acquisition of shares that resulted in a shareholder’s becoming an interested shareholder is approved in a prescribed manner. A “business combination” can include a merger, asset or share sale, or other transaction resulting in a financial benefit to an interested shareholder. Generally, an interested shareholder is a person who, together with its affiliates and associates, owns (or within three years prior to the determination of interested shareholder status did own) 15 percent or more of a corporation’s voting shares. The existence of this provision would be expected to have an anti-takeover effect with respect to transactions not approved in advance by the Board, including discouraging attempts that might result in a premium over the market price for the shares of common stock held by shareholders. |
|
|
• |
REVA Medical, Inc. has not listed its securities for quote on any exchange other than the ASX. |
General Information
The name of the Company Secretary is Brandi L. Roberts. The address of our office in the United States, which is our principal administrative office, is REVA Medical, Inc., 5751 Copley Dr., San Diego, California 92111, telephone +1 (858) 966-3000.
- 53 -
The address of our registered office in Australia is c/o Buchan Pty Ltd, Suite 4, Level 14, 6 O’Connell Street, Sydney NSW 2000, telephone +61 2 9237 2800.
Registers of CDI securities are held at Computershare Investor Services Pty Limited, Level 3, 60 Carrington Street, Sydney NSW 2000, Australia, Investor Enquiries 1300 855 080. Registers of common stock securities are held at Computershare Trust Company, N.A., 250 Royall Street, Canton, MA 02021, USA, Investor Inquiries (800) 962-4284.
Quotation has been granted for CDIs (and the underlying shares of common stock) on the ASX Limited.
Australian Corporate Governance Statement
Our Board of Directors (the “Board”) is committed to promoting and strengthening good corporate governance practices and a culture of good corporate governance and ethical conduct throughout the Company. The Board has evaluated the Company’s corporate governance policies and practices in light of the ASX Corporate Governance Council’s Corporate Governance Principles and Recommendations 3rd Edition (“ASX Recommendations”) for the Company’s financial year ended December 31, 2017 and confirms that the Company’s corporate governance framework is generally consistent with the ASX Recommendations, other than as set forth below. Following is a summary of the approach adopted and used by the Company during the year ended December 31, 2017, using the same numbering sequence as contained in the ASX Recommendations.
Principle 1 — Lay solid foundations for management and oversight
Recommendation 1.1 — Establish the roles and responsibilities of the board and management; disclose those matters expressly reserved to the board and those delegated to senior executives
The roles and responsibilities of the Board and of management have been established. The Board’s responsibilities are defined by the Company’s Corporate Governance Guidelines, a copy of which is available in the Corporate Governance section on the Company’s website at www.revamedical.com. Management is responsible for implementing the strategic objectives set by the Board, to carry out the day-to-day operations of the Company, and to make accurate, timely, and clear reports to the Board. There is a clear delineation between the Board’s responsibility for the Company’s strategy and activities and management’s responsibilities for the day-to-day management of operations.
Recommendation 1.2 — Undertake appropriate assessments prior to appointing a board member; provide all relevant material information to stockholders regarding a candidate proposed for election to the board
The Company’s Corporate Governance Guidelines provide general criteria for Board member qualification. Additionally, the Nominating and Corporate Governance Committee of the Board is responsible for assessing the qualifications and background of Board candidates and appointees. Prior to recommending a new Board candidate, the Committee undertakes to check a candidate’s independence, experience, education, and general character. Annually and prior to recommending re-election of a director, the Committee assesses performance, interests and independence, outside commitments, and availability for Board responsibilities. Only qualified candidates are recommended for appointment or candidacy. The backgrounds and qualifications of directors who are recommended for election or re-election are provided to stockholders in the Company’s proxy statement. A partial list of the evaluations to be made of Board candidates is contained in the Nominating and Corporate Governance Committee’s charter, which is available on the Company’s website at www.revamedical.com in the Corporate Governance section Each Board member’s background is also available on the Company’s website in the Corporate Governance section.
The skills, experience, expertise, diversity, independence, and related information for each of our directors holding office as of February 23, 2018 are set forth below:
C. Raymond Larkin, Jr., age 70, has served as a director since July 2017 and chairman since September 2017. He currently serves as a principal of Group Outcome L.L.C., a merchant banking firm concentrating on medical technologies. Prior to Group Outcome, Mr. Larkin was Chairman and CEO at Eunoe Inc., an investigative device for slowing and stopping progression of Alzheimer's disease. From 2001 to 2007, he served as a part-time Venture Partner at Cutlass Capital, a venture capital firm. Prior to that, he was President and Chief Executive Officer of Nellcor Puritan Bennett, Inc., a respiratory products company. Mr. Larkin also held positions of increasing responsibility at Bentley Laboratories/American Hospital Supply from 1976 to 1983. Mr. Larkin currently serves as board chairman of Align Technology, and also served as board chairman of Heartware, Inc. prior to its acquisition by Medtronic, plc. He received his B.S. degree in Industrial Management from LaSalle University and is a former captain in the United
- 54 -
States Marine Corps. Mr. Larkin is qualified to sit on our Board due to his experience serving on multiple other boards of directors, his experience in executive positions with life science companies and his experience at venture capital firms.
Ross Breckenridge, MD, MRCP, PhD, age 48, has served as a director since January 2015. Dr. Breckenridge is the Chief Executive Officer of Silver Creek Pharmaceuticals, Inc., since September 2016. Prior to that, he was a senior clinical lecturer and Programme Director for the Masters Programme in Clinical and Experimental Medicine at University College London since 2006. He was a Fellow of the Royal College of Physicians (London) and a Consultant Physician at University College London Hospital from 2006 until September 2016. Dr. Breckenridge has provided consultation services to investors in the biotech and healthcare sector since 1998. He is a current board member of the Cornelia de Lange Society of Great Britain and has sat on numerous other medical and corporate boards. He obtained his medical degree from Oxford University, followed by his PhD in Developmental Biology at the University of Cambridge. He then completed his training in Clinical Pharmacology at University College London. Dr. Breckenridge is qualified to sit on our Board due to his extensive medical background, particularly as it relates to research of cardiac disease, his experience serving on multiple other boards of directors, and his general business proficiency.
Brian H. Dovey, age 76, has served as a director since June 2001 and as Chairman of the Board from March 2016 through September 2017. Since 1988, Mr. Dovey has been a partner of Domain Associates, LLC, a private venture capital management firm focused on life sciences, where he has led innovative investments not only in life science companies, but also has established and directed new initiatives such as the collaboration between Domain and Rusnano. Since joining Domain, he has served on the board of directors of over 35 private and public companies and has been chairman of six, including REVA. Mr. Dovey currently sits on the board of three public companies: REVA, Orexigen Therapeutics, Inc., and Miramar Labs, Inc. (since May 2016). Prior to joining Domain, Mr. Dovey spent six years at Rorer Group, Inc. (now part of Sanofi-Aventis), a pharmaceutical and medical device company listed on the NYSE. As president of Rorer from 1986 to 1988, he was the primary architect of the company’s strategic shift to pharmaceuticals. Previous to that, he was President of Survival Technology, Inc., a start-up medical products company. Mr. Dovey serves on the board of directors and is also chairman at the Center for Venture Education (Kauffman Fellows Program) and serves on the La Jolla Playhouse board of trustees. He was the former chair and currently serves on the board of trustees of the Wistar Institute, a leader in preclinical biomedical research in the non-profit sector. Mr. Dovey has served as both president and chairman of the National Venture Capital Association. He is a former board member of the industry association representing the medical device industry, as well as the association representing consumer pharmaceuticals. He is a trustee emeritus of Germantown Academy and is a former trustee of the University of Pennsylvania School of Nursing and the Sanford-Burnham Institute for Medical Research. Mr. Dovey received his B.A. in mathematics from Colgate University and his MBA from the Harvard Business School. Mr. Dovey is qualified to sit on our Board due to his extensive financial background, his experience in corporate governance and risk management, his service as a director on over 35 private and public companies, his broad executive experience with medical device companies, and his extensive experience at a health care venture capital firm.
Regina E. Groves, age 59, joined REVA as Chief Executive Officer in September 2015 and was appointed as a director in June 2017. Her background encompasses more than 30 years’ experience in medical devices, executive leadership, and financial management. Prior to joining REVA, Ms. Groves served as Vice President and General Manager of the AF Solutions, Cardiac Rhythm and Heart Failure division of Medtronic, a leading global medical technology company. Previously she held positions at Medtronic of Vice President of Quality and Regulatory for the Cardiac Rhythm Disease Management (“CRDM”) business and Vice President and General Manager for Patient Management CRDM. Her experience also includes positions with McKinsey & Company, Inc., several health care companies, including direct providers, and involvement with start-up companies. Ms. Groves received her MBA from Harvard Graduate School of Business Administration and her BS in Pharmacy from the University of Florida. She currently serves on the board of three private companies. Ms. Groves is qualified to sit on our Board due to her significant experience in executive positions with medical device companies and her broad business background.
Scott Huennekens, age 53, has served as a director since March 2015. Since December 2015 he is President and Chief Executive Officer of Verb Surgical, Inc., a collaboration between Alphabet, Inc. (formerly Google) and Johnson & Johnson, focused on developing a comprehensive robotic surgical solutions platform. Previously, from April 2002 to February 2015, Mr. Huennekens was President and Chief Executive Officer of Volcano Corporation, a manufacturer of intravascular imaging equipment for coronary and peripheral applications. Prior to 2002, he served as President and Chief Executive Officer of Digirad Corporation, a diagnostic imaging solutions provider, and also held senior positions at Baxter International, Inc. in the Edwards Cardiovascular Division and the Novacor division. Mr. Huennekens currently serves on the Medical Device Manufacturers Association (“MDMA”) board and he served on the board of EndoChoice until November 2016. He received his B.S. in Business Administration from the
- 55 -
University of Southern California and an MBA from Harvard Business School. Mr. Huennekens is qualified to sit on our Board due to his vast experience in executive positions with medical equipment manufacturers, his broad business background, his experience serving on multiple other boards of directors, and his strong financial background, including his work early in his career at Deloitte, a provider of tax, audit, and advisory services.
Stephen N. Oesterle, MD, age 67, was appointed as a director in February 2018. He is currently a venture partner at New Enterprise Associates and serves as a Senior Advisor to EQT Partners of Sweden and Temasek Holdings in Singapore. He is also a member of the Board of Directors at Baxter International. Dr. Oesterle was formerly Senior Vice President for Medicine and Technology at Medtronic, Inc. and a member of its Executive Operating Committee for 14 years. He oversaw long term internal technology investments while participating in strategic corporate investments in emerging private companies. Additionally, he served as a member of Medtronic’s Business Development and Strategy Committee that approved all corporate acquisitions. Dr. Oesterle also served as a director of Heartware, Inc. prior to its acquisition by Medtronic, plc. Dr. Oesterle graduated summa cum laude from Harvard and received his M.D. from Yale Medical School; he completed his residency at Massachusetts General Hospital. Following medical school, he completed a fellowship in Interventional Cardiology at Stanford University Hospital. Dr. Oesterle then served on the faculties at Harvard and Stanford medical schools where he directed Invasive Cardiology Services at each hospital. Dr. Oesterle is qualified to sit on our Board due to his extensive medical background, particularly as it relates to cardiology, his experience at health care venture capital firms and his general business proficiency.
Robert B. Stockman, age 64, our co-founder and a member of our Board, served as our Board Chairman from 1999 until March 2016; he was our Chief Executive Officer from August 2010 to September 2015. He served as a director of HeartWare Limited/HeartWare International, Inc., a NASDAQ-listed medical device company (formerly also ASX-listed), between December 2006 and August 2016 when they were acquired by Medtronic, Inc. He previously served on the board of ZELTIQ Aesthetics, Inc., a medical technology company listed on NASDAQ, from July 2010 until April 2012. Since 1999, Mr. Stockman has been the President and Chief Executive Officer of Group Outcome LLC, a U.S.-based merchant banking firm that deploys its capital and that of its financial partners in private equity and venture capital investments in medical technology companies. Mr. Stockman also co-founded Centrimed, Inc., an internet-based software company, that was acquired by the Global Healthcare Exchange, LLC, and led the buyouts of Ioptex, an intraocular lens manufacturer, and two Johnson & Johnson divestitures, “A” Company Orthodontics, Inc. and Critikon Company, LLC, each of which was subsequently acquired. Prior to establishing Group Outcome LLC, Mr. Stockman spent 18 years with Johnston Associates, Inc. and Narragansett Capital Corporation, where he focused on venture capital investments and merger advisory work in health care. Mr. Stockman holds a Bachelor’s Degree from Harvard College and an MBA from The Tuck School at Dartmouth College, where he serves on Tuck’s Board of Overseers. Mr. Stockman is qualified to sit on our Board due to his extensive experience as an entrepreneur driving the growth of five medical products companies, his experience as an executive of several medical device companies, and his experience as an executive in the investment banking industry, particularly in private equity and venture capital investments in medical technology. Mr. Stockman’s qualifications also include his strong financial background, including his work early in his career at Price Waterhouse, a provider of tax, audit, and advisory services, and his ability to provide financial expertise to the Board, including an understanding of financial statements, corporate finance, accounting, and capital markets.
Robert Thomas, age 72, has served as a director since July 2010. He was a director and non-executive Chairman of the Board of HeartWare Limited/HeartWare International, Inc., a NASDAQ-listed medical device company (formerly also ASX-listed), between November 2004 and August 2016 when they were acquired by Medtronic, Inc. He is currently a director of several Australian public companies, including Starpharma Limited (Chairman) and Biotron Limited. Mr. Thomas was a director of Virgin Australia Limited from 2007 through February 28, 2018. Between October 2004 and September 2008, Mr. Thomas was a consultant to Citigroup Corporate and Investment Bank. Between March 2003 and September 2004, he was Chairman of Global Corporate and Investment Bank, Citigroup Global Markets, Australia and New Zealand. Prior to that time, Mr. Thomas was Chief Executive Officer of Citigroup’s Corporate and Investment Bank (formerly known as Salomon Smith Barney), Australia and New Zealand from October 1999 until February 2003. Mr. Thomas is Chairman of Aus Bio Limited, a director of O’Connell Street Associates, and Chairman of Gragher Retail Securities. Mr. Thomas holds a Bachelor of Economics from Monash University, Australia. He is a member of the Stockbrokers Association of Australia and is a Master Stockbroker. Mr. Thomas is also a Fellow of the Financial Services Institute of Australia and the Australian Institute of Company Directors. He is on the board of the NSW State Library Foundation. Mr. Thomas is qualified to sit on our Board due to his extensive investment banking experience, including his leadership of finance and strategic transactions, his involvement with medical device companies, and his experience in governance and risk management across a wide range of industries. Mr. Thomas also brings capital market and economics expertise to the Board from his years of service as a securities analyst and experience as a director of ASX-listed companies.
- 56 -
Recommendation 1.3 — Provide a written agreement with each director and senior executive setting out the terms of his or her appointment
The terms of Board membership are set forth in the Company’s Corporate Governance Guidelines and remuneration to Board members is provided in accordance with stockholder approvals following the Compensation Committee’s recommendation. While the Company does not have a separate written agreement with each of its Board members, it believes these guidelines are adequate to provide a clear understanding of the roles and responsibilities of Board members. In the case of senior executives, the Company has provided a letter of employment to each executive detailing the terms of employment and has developed job descriptions setting forth the position, duties, and reporting structure. Where there are any agreed entitlements upon termination, such agreed items are set forth in the employment letters. For the year ended December 31, 2017, there were no material variations to any of the Company’s employment letters.
Recommendation 1.4 — The Company secretary is accountable directly to the board on all matters to do with the proper functioning of the board
The role and responsibilities of the Company’s secretary are set forth in the Company’s bylaws. The Board is responsible for electing or appointing the secretary and for prescribing the duties and powers of the secretary. The secretary is responsible for preparing and maintaining the appropriate corporate records, including such items as meeting notices, meeting minutes, and stock ledgers, and to provide such records to the Board as requested or required. The secretary is accountable to the Board on all matters to do with the proper functioning of the Board. Each director is able to communicate freely and directly with the secretary and vice versa.
Recommendation 1.5 — Establish and maintain a gender-based diversity policy and provide an annual report of the Company’s measurable objectives for achieving gender diversity
The Company has adopted a Diversity Policy, which includes measurable objectives for achieving gender diversity and provisions for the Board to annually assess both the objectives and the Company’s progress in achieving them. A copy of the Diversity Policy is available in the Corporate Governance section on the Company’s website at www.revamedical.com.
The Board continued to evaluate the gender diversity of the Company’s employees, its senior management, and its Board during 2017 and determined that the gender diversity continued at levels generally consistent with the prior year and in line with expectations. The Board endorsed the Company’s objective for diversity to remain at the same relative proportions, if not higher, of females in each category measured. The base level expectations for females are a minimum 15 percent of Board members, 30 percent of senior management, and 40 percent of employees. As of December 31, 2017, the Company reports that women represented 14 percent of its Board members, 63 percent of its senior executives (those positions of director and higher), and 49 percent of its entire workforce, which was aligned with its Diversity Policy.
Recommendation 1.6 — Establish and maintain a process to periodically evaluate the board, the board committees, and individual directors and provide an annual report of the undertaking of such process
The Company’s Corporate Governance Guidelines provide for annual assessments of the performance of the Board and each committee of the Board, to be provided to the Nominating and Corporate Governance Committee. The performance assessments include evaluations of numerous items, including each Board and committee member’s independence and skill levels, process and effectiveness in addressing Company, Board, and committee matters, interactions with management and outside service providers, meeting attendance, and governance items, including annual charter reviews. The assessments are to be completed by individual Board members, aggregated by the Nominating and Corporate Governance Committee, and evaluated and discussed by the Board and the individual committees of the Board. Such Board and committee assessments were performed and evaluated for the year ended December 31, 2017 in accordance with the guidelines.
The Company’s Corporate Governance Guidelines do not call for evaluation of each individual director. The size of the Board and each committee is relatively small, Board and committee meetings are held frequently throughout the year, and the process to assess the Board and each committee considers the involvement and effectiveness of the individual directors. These factors allow for continuous self-assessment, as well as Board level assessments and feedback, of individual performance and contribution.
- 57 -
Recommendation 1.7 — Establish and disclose the process to evaluate the performance of senior executives and provide an annual report of the undertaking of such process
The Company’s employment and personnel policies provide for annual performance evaluations and goal setting for all employees, including senior executives. The Compensation Committee of the Board, in accordance with its charter, annually reviews the performance of each senior executive and reviews and approves each personal performance goal, then subsequently measures attainment of the goals. The assessments made by the Compensation Committee are reported to the Board. In accordance with the established processes, the performance of the senior executives of the Company was evaluated by the Company’s Compensation Committee and Board for the year ended December 31, 2017.
Principle 2 — Structure the board to add value
Recommendation 2.1 — Establish a nomination committee and disclose its charter and membership
The Board has established a Nominating and Corporate Governance Committee to oversee the selection and appointment practices of the Company. Prior to September 18, 2017, Anne Keating (Chair), Dr. Ross Breckenridge and Gordon Nye were on the Nominating and Corporate Governance Committee. Ms. Keating and Mr. Nye resigned from the Board effective June 1, 2017 and July 13, 2017, respectively. On September 18, 2017, the Board appointed the following three members to the Compensation Committee: C. Raymond Larkin Jr. (Chair), Brian Dovey and Scott Huennekens. All members of the Committee are non-executives and are considered independent directors for both ASX and SEC purposes. The Committee held three meetings during the year ended December 31, 2017; all members attended all meetings. A copy of the Nominating and Corporate Governance Committee Charter is available in the Corporate Governance section on the Company’s website at www.revamedical.com.
Recommendation 2.2 — Establish and disclose a board skills matrix setting out the mix of skills and diversity the board currently has or is looking to achieve
The Nominating and Corporate Governance Committee of the Board is responsible for developing and recommending the mix of skills and diversity for Board and committee members. The committee continually assesses the needs of the Company and the current mix of skills provided by Board members. As the Company transitioned to a commercial company in 2017, the skill mix of the Board has been updated to ensure appropriate representation.
Recommendation 2.3 — Disclose director independence and length of service
The Company considers a director to be independent when that director is free from any interest and any business or other relationship that could, or could reasonably be perceived to, materially interfere with the director’s decisions relating to the Company or with the director’s ability to act in the best interests of the Company. In accordance with the Corporate Governance Guidelines, a director will not be considered to be independent until the Board affirmatively determines that such director meets all applicable standards. Annually, the Board will review its determinations based on recommendations from the Nominating and Corporate Governance Committee.
At the Company’s expense, the Board collectively or the directors acting as individuals are entitled to seek advice from independent external advisors in relation to any matter that is considered necessary to fulfill their relevant duties and responsibilities. Individual directors seeking such advice must obtain approval of the Chairman (which may not be unreasonably withheld). Any advice so obtained will be made available to all Board members.
The composition and tenure of the members of the Board as of December 31, 2017, as well as each member’s independence status during 2017, was as follows:
|
|
|
|
|
|
|
|
|
Committees |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
Nominating |
|
|
|
|
|
|
|
|
|
|
|
|
|
and Corp. |
|
Director |
|
Director Position |
|
Year Appointed |
|
Independent |
|
Audit |
|
Compensation |
|
Governance |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C. Raymond Larkin, Jr. |
|
Chairman |
|
2017 |
|
Yes |
|
− |
|
− |
|
Chair |
|
Dr. Ross A. Breckenridge |
|
Non-Executive |
|
2015 |
|
Yes |
|
X |
|
X |
|
− |
|
Brian H. Dovey |
|
Non-Executive |
|
2001 |
|
(1) |
|
− |
|
Chair |
|
X |
|
Regina E. Groves |
|
Executive |
|
2017 |
|
No (2) |
|
− |
|
− |
|
− |
|
R. Scott Huennekens |
|
Non-Executive |
|
2015 |
|
Yes |
|
Chair |
|
− |
|
X |
|
Robert B. Stockman |
|
Non-Executive |
|
1999 |
|
No (2) |
|
− |
|
− |
|
− |
|
Robert B. Thomas |
|
Non-Executive |
|
2010 |
|
Yes |
|
X |
|
X |
|
− |
- 58 -
|
(1) |
Mr. Dovey is an Independent Director under the rules of NASDAQ and the SEC, but not considered independent under the ASX. |
|
(2) |
Mr. Stockman was employed as our Chief Executive Officer until September 18, 2015. Ms. Groves is currently employed as our Chief Executive Officer. Under ASX, NASDAQ, and SEC rules, a director employed by the Company is not independent until three years after such employment terminates. |
Recommendation 2.4 — A majority of board members should be independent
A majority of the Company’s Board was independent during the year ended December 31, 2017. Mr. Dovey is a principal in a firm that has invested in our Company and for ASX purposes he is not considered to be independent. Ms. Groves is our Chief Executive Officer and is not considered an independent director. Mr. Stockman was Chief Executive Officer until September 2015 and, as a result, will not be considered an independent director until September 2018. All other members of our Board are considered to be independent.
Recommendation 2.5 — The Chair of the board should be an independent director and should not be the chief executive officer
Mr. Larkin was appointed Chairman of our Board in September 2017. Mr. Larkin is considered an independent director under ASX Listing Rules.
Recommendation 2.6 — Establish a program for inducting new directors and ongoing development opportunities for directors
The Nominating and Corporate Governance Committee of the Board continually assesses the needs of the Company and the skills and knowledge required of its Board members. On appointment, new directors are provided with induction information that generally includes historical information about the Company and its operations, details of the Company's directors’ and officers’ insurance, the Company's Corporate Governance Guidelines, and other Company governance policies. The induction process also involves one-on-one discussions with the Chairman and other directors and briefings from senior management to help new directors participate actively in Board decision making at the earliest opportunity. When it is necessary, resources are provided for the Board as a whole, and for individual Board members as needed, to supplement their skills and knowledge and fill any identified gaps. Any outside expertise guidance or training undertaken by Board members in furtherance of their responsibilities to the Company is generally paid by the Company.
Principle 3 — Act ethically and responsibly
Recommendation 3.1 — Establish a code of conduct and disclose a summary of the code
The Company has adopted a Code of Business Conduct and Ethics, an Insider Trading Policy, and a Related Party Transaction Policy. A copy of each policy is available in the Corporate Governance section on the Company’s website at www.revamedical.com. These Company codes apply to all directors, senior executives, and employees and, in general, call for personal integrity, ethical conduct, and balanced business approaches and dealings. The policies are reinforced on a regular basis and provide for disciplinary action for any violations.
Principle 4 — Safeguard integrity in corporate reporting
Recommendation 4.1 — Establish an audit committee
The Board has established an Audit Committee to oversee the management of the Company’s financial and internal risks and reporting. The Audit Committee has adopted and is governed by a formal charter, a copy of which is available in the Corporate Governance section on the Company’s website at www.revamedical.com. The Audit Committee regularly reports to the Board about Committee activities, issues, and related recommendations.
The Audit Committee comprises three members, who are Mr. Huennekens (Chair), Dr. Breckenridge and Mr. Thomas. All Committee members are non-executive directors and independent. The Committee held five meetings during 2017; all members attended all meetings.
All members of the Audit Committee are considered to be financially literate and familiar with financial and accounting matters and qualified to adequately understand the financial and accounting matters that relate to the Company. Mr. Huennekens is considered to be a financial professional with appropriate financial and accounting expertise.
- 59 -
Recommendation 4.2 — Receive declarations from the Chief Executive Officer and Chief Financial Officer prior to approving financial statements regarding compliance with accounting standards, accuracy and fairness of disclosures, and the systems of internal controls and risk management underlying the financial statements
The Company is a U.S. SEC registrant and, as such, complies with SEC requirements in addition to the ASX Listing rules. In accordance with SEC requirements, the Company’s Chief Executive Officer and its Chief Financial Officer review and assess the financial statements and related disclosures and the underlying financial records, internal controls, and policies and procedures. At each financial reporting date, the Audit Committee of the Board is provided certifications, which are filed with the SEC and lodged with the ASX, by the Chief Executive Officer and the Chief Financial Officer regarding their assessments of the financial statements and underlying internal controls. During the year ended December 31, 2017, such certifications were filed with the SEC and lodged with the ASX.
Recommendation 4.3 — The Company’s external auditor should attend its AGM
The Company’s policy is to ensure its external auditor attends the Annual General Meeting of stockholders, in person, to have an opportunity to make a statement, if desired, and to respond to appropriate questions from security holders regarding the audit. The Company’s auditor for the year ended December 31, 2017 was Grant Thornton LLP, who attended the AGM.
Principle 5 — Make timely and balanced disclosure
Recommendation 5.1 — Establish a policy for continuous disclosure and disclose that policy
The Company is committed to providing timely and balanced disclosure to the market in accordance with its continuous disclosure obligations, as required under ASX Listing Rules. In accordance with its commitment to fully comply with these obligations and to ensure accountability at a senior management level for that compliance, the Company has adopted a Continuous Disclosure Policy, together with other internal mechanisms and reporting requirements. A copy of the Company’s Continuous Disclosure Policy is available on its website at www.revamedical.com in the Corporate Governance section. In addition, copies of all the Company’s ASX announcements, financial reports, and related public information are also available on the Company’s website.
Principle 6 — Respect the rights of security holders
Recommendation 6.1 — Provide information about the Company and its governance via a website
The Company is committed to providing ready access to information about the Company, its approach and policies regarding governance, and its reports. Accordingly, the Company hosts and maintains a website at www.revamedical.com that includes information about the Company, its products, methods of contact, answers to frequently asked questions, and a separate section with information for Investors. In addition to providing links to the Company’s ASX trading information, news releases, and ASX and SEC filings, the Investor section includes information about the Company’s directors and senior management, committee composition and charters, and its corporate governance policies.
Recommendation 6.2 — Design and implement a program to facilitate effective two-way communication with investors
The Company has adopted a Shareholder Communication Policy that supports effective two-way communication with its shareholders. The Shareholder Communication Policy is included in the Company’s Corporate Governance Guidelines, a copy of which is available in the Corporate Governance section on the Company’s website at www.revamedical.com. The Company seeks to utilize numerous modes of communication, including electronic communication, to ensure that its communication with Shareholders is frequent, clear, and accessible. Additionally, the Company announces briefing calls in advance of such calls, provides relevant information on its website, and maintains internal records of matters discussed with shareholders. Shareholders are entitled to and encouraged to participate in briefing calls and/or contact the Company directly with questions or concerns. Contact information in both Australia and the U.S. is provided in each communication with shareholders, as well as on the Company’s website.
- 60 -
Recommendation 6.3 — Facilitate and encourage shareholder participation at meetings
All shareholders are invited to attend the Company’s annual meeting either in person or by proxy. To facilitate attendance, the Company arranges the annual meeting to be held in an easily accessed and well-known public location in Sydney or San Diego and announces the date and location of the meeting in advance of the meeting. Notices of the meeting are mailed to all securityholders. The Board regards the annual meeting as an excellent forum in which to discuss issues relevant to the Company and accordingly encourages full participation by shareholders. Shareholders have an opportunity to submit questions to the Board and auditors. The meeting may also be audio cast and/or webcast to provide access to those shareholders who are unable to attend the annual general meeting in person.
Recommendation 6.4 — Provide security holders the option to receive communications electronically
The Company’s share registry is managed by Computershare Investor Services, who provides security holders the option to receive and send communications from and to the share registry electronically. Additionally, the share registry facilitates electronic distribution of Company materials. In addition, the Company provides ongoing electronic notices and reports to shareholders and other third parties who have provided their electronic contact details to the Company and have requested to receive such notices and reports electronically. The Company provides an e-mail alert subscription form on its website at www.revamedical.com under the Investor section that allows the subscriber to select which information to receive about the Company. The selections include press releases, ASX announcements, SEC filings, and webcasts and events.
Principle 7 — Recognize and manage risk
Recommendation 7.1 — Establish a committee to oversee risk
While the Company has adopted a Risk Management Policy that sets forth the process to identify, assess, and manage risk in the Company’s business operations, it has not established a formal Risk Committee. A copy of the Risk Management Policy is available in the Corporate Governance section on the Company’s website at www.revamedical.com. In addition to following its Risk Management Policy, the Board and its committees have developed its charters and policies with a focus on risk identification and management. The Board’s role in risk oversight includes receiving reports from external auditors, internal auditors, other independent parties, and from members of management on a regular basis regarding material risks faced by the Company and applicable mitigation strategies and activities. The reports from management are provided at least quarterly. The reports cover the critical areas of operations, research and development, regulatory and quality affairs, intellectual property, clinical developments, and legal and financial affairs, as well as management’s assessment of risks facing the Company. The Board and its committees consider these reports, discuss matters with management and independent parties, and identify and evaluate any potential strategic or operational risks and appropriate activity to address those risks, thereby ensuring effectiveness in identifying and managing material business risks.
Recommendation 7.2 — Review the risk management framework and disclose the results of such review
While the Board does not currently conduct a formal annual review of the material risks to the Company and the methods used to identify and communicate those risks, the Board continually assesses these matters and believes this current approach is effective. Additionally, the Audit Committee reviews an annual fraud risk assessment. As the Company moves forward with commercialization and anticipates related additional business risks in 2018, it intends to develop a formal review process of the Company’s risk identification and management processes.
Recommendation 7.3 — Disclose the structure and role of internal audit
The Company engages a third party independent firm for its internal audit function. This independent internal audit firm reports directly to the Audit Committee and is responsible for developing independent risk-based reviews and testing of the Company’s system of internal controls over financial reporting. The independent internal audit firm shares its results and reports with management and the Company’s external auditors and provides recommendations for improvements if necessary.
- 61 -
Recommendation 7.4 — Disclose material exposures to economic, environmental, and social sustainability risks
The Company provides a complete assessment of risks to the business in the “Risk Factors” section of this Annual Report on Form 10-K. Considering its pre-revenue stage, location of facilities, and intended products and markets, the Company does not believe it has exposure to material economic, environmental, or social sustainability risks beyond those discussed in the “Risk Factors” section.
Principle 8 — Remunerate fairly and responsibly
Recommendation 8.1 — Establish a remuneration committee
The Board has established a Compensation Committee to review and assess executive and director compensation. The Compensation Committee has adopted and is governed by a formal charter, a copy of which is available in the Corporate Governance section on the Company’s website at www.revamedical.com. The Compensation Committee regularly reports to the Board about Committee activities, issues, and related recommendations.
Prior to September 18, 2017, Gordon Nye (Chair), Brian Dovey and Robert Thomas were on the Compensation Committee. Mr. Nye resigned from the Board effective July 13, 2017. On September 18, 2017, the Board appointed the following three members to the Compensation Committee: C. Raymond Larkin Jr. (Chair), Brian Dovey and Scott HuennekensThe Committee comprises three members, who are Mr. Dovey (Chair), Dr. Breckenridge and Mr. Thomas. Dr. Breckenridge and Mr. Thomas are both considered to be independent for ASX purposes; however, Mr. Dovey is not considered to be independent for ASX purposes but is considered to be independent under the SEC rules. The Compensation Committee, therefore, consists of a majority of independent directors. The Committee held two meetings during 2017. All Committee members attended all meetings.
Recommendation 8.2 — Disclose the policies and practices regarding remuneration of directors and senior executives
In accordance with its charter, the Compensation Committee is responsible for ensuring that the policies and practices regarding compensation for directors and senior executives are defined and disclosed. The Company has adopted a non-executive director compensation policy pursuant to which directors are compensated for their services to the Board. Non-executive director compensation comprises a base fee, committee membership fees, chair fees, and the ability to receive annual equity grants at the Board’s discretion (subject to shareholders’ approval being obtained as required under the ASX Listing Rules). The Company has adopted a separate executive compensation program that consists of base salary, equity-based incentives, performance-based cash bonuses, severance benefits, and other customary benefits such as health insurance on the same basis as provided to all other employees. None of the Company’s non-executive directors are entitled to any retirement benefits.
The Company discloses compensation details, including philosophy, policy, and compensation payments for each director and each executive officer in its annual Proxy Statement as lodged with the ASX, filed with the SEC, and provided to shareholders ahead of the Annual General Meeting. A copy of the prior proxy statements can be found in the Investors section of the Company’s website at www.revamedical.com.
While the Compensation Committee reviews and reports compensation items to the Board for both non-executive directors and executive management, including each individual’s skills, knowledge, and contributions to the Company, the Committee does not provide a separate report of compensation by gender.
Further information regarding the Compensation Committee, as required by Item 10 of this Annual Report on Form 10-K, will be contained in our 2018 Proxy Statement. Such information is incorporated herein by reference.
Recommendation 8.3 — Disclose the policy regarding permitted equity-based transactions
The Company provides compensation in the form of equity-based awards to non-executive directors (upon approval by shareholders), senior executives, and employees of the Company. Awards are made under the Company’s 2010 Equity Incentive Plan, as amended, which has been approved by shareholders. The Company’s Insider Trading Policy, a copy of which is available in the Corporate Governance section on the Company’s website at www.revamedical.com, sets out the Company’s policy that prohibits certain transactions involving REVA’s securities, including short-term or speculative transactions and publicly traded options, short sales, puts and calls, hedging, and other transactions.
- 62 -
Item 11. Executive Compensation
The information required by this item is incorporated by reference to our 2018 Proxy Statement under the headings “Non-Employee Director Compensation” and “Executive Compensation.”
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this item is incorporated by reference to our 2018 Proxy Statement under the heading “Security Ownership of Certain Beneficial Owners and Management.”
The following table sets forth information regarding outstanding options and shares reserved for future issuance as of December 31, 2017 under equity compensation plans approved by our stockholders. We do not have any equity compensation plans that have not been approved by stockholders.
|
Plan Category |
|
Number of Shares to be Issued on Vesting or Exercise of Outstanding Awards |
|
|
Weighted Average Exercise Price of Outstanding Stock Options |
|
|
Number of Shares Remaining Available for Future Issuance (1) |
|
|||
|
Equity compensation plans approved by stockholders (2) |
|
|
6,697,991 |
|
|
|
$6.65 |
|
|
|
9,144,512 |
|
__________
|
|
(1) |
Our 2010 Equity Incentive Plan, as amended, contains a provision for an automatic increase each January 1st of the number of shares available for grant. The automatic increase shall be the lesser of (i) 3% of the number of shares of our common stock issued and outstanding on January 1st or (ii) a number of shares set by our Board of Directors. |
|
|
(2) |
Consists of grants and awards from our 2001 Stock Option/Stock Issuance Plan and our 2010 Equity Incentive Plan, as amended, including 6,120,491 outstanding options to purchase common stock and 577,500 restricted stock units that each entitles the holder to one share of our common stock upon vesting. |
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this item is incorporated by reference to our 2018 Proxy Statement under the heading “Related Party Transactions.”
Item 14. Principal Accounting Fees and Services
The information required by this item is incorporated by reference to our 2018 Proxy Statement under the heading “Audit and Non-Audit Fees.”
- 63 -
Item 15. Exhibits and Financial Statement Schedules
The following documents are filed as part of this Annual Report on Form 10-K:
|
1. |
Financial Statements — The following financial statements are included in this report: |
Reports of Independent Registered Public Accounting Firm
Consolidated Balance Sheets
Consolidated Statements of Operations and Comprehensive Income (Loss)
Consolidated Statements of Cash Flows
Consolidated Statements of Stockholders’ Equity (Deficit)
Notes to Consolidated Financial Statements
|
2. |
List of Financial Statement Schedules — All schedules are omitted because they are not applicable or the required information is shown in the financial statements or notes thereto. |
|
3. |
Exhibits. |
|
|
|
|
|
Filed |
|
|
|
|
|
|
|
Exhibit |
|
|
|
with this |
|
Incorporated by Reference |
||||
|
Number |
|
Description of Exhibits |
|
10-K |
|
Form |
|
File No. |
|
Date Filed |
|
3.1 |
|
|
|
|
S-1/A |
|
333-168852 |
|
10/22/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3.2 |
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.1 |
|
|
|
|
S-1/A |
|
333-168852 |
|
11/12/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.2(a) |
|
|
|
|
DEF14A |
|
000-54192 |
|
10/14/2014 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.2(b) |
|
First Amendment to Amended and Restated Investors’ Rights Agreement dated September 24, 2014 |
|
|
|
DEF14A |
|
000-54192 |
|
5/15/2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.1(a) |
|
|
|
|
S-1 |
|
333-168852 |
|
8/13/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.1(b) |
|
|
|
|
S-1 |
|
333-168852 |
|
8/13/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.1(c) |
|
|
|
|
S-1 |
|
333-168852 |
|
8/13/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.1(d) |
|
|
|
|
S-1 |
|
333-168852 |
|
8/13/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.1(e) |
|
|
|
|
S-1 |
|
333-168852 |
|
8/13/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.1(f) |
|
|
|
|
8-K |
|
000-54192 |
|
11/23/2011 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.1(g) |
|
|
|
|
10-Q |
|
000-54192 |
|
11/7/2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
- 64 -
|
|
|
|
|
Filed |
|
|
|
|
|
|
|
Exhibit |
|
|
|
with this |
|
Incorporated by Reference |
||||
|
Number |
|
Description of Exhibits |
|
10-K |
|
Form |
|
File No. |
|
Date Filed |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.2(a) |
|
|
|
|
10-Q |
|
000-54192 |
|
11/9/2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.2(b) |
|
|
|
|
10-Q |
|
000-54192 |
|
11/6/2014 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.2(c) |
|
|
|
|
10-Q |
|
000-54192 |
|
11/9/2016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.3 |
|
|
|
|
S-1/A |
|
333-168852 |
|
9/21/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.4(a) |
|
|
|
|
S-1 |
|
333-168852 |
|
8/13/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.4(b) |
|
|
|
|
S-1 |
|
333-168852 |
|
8/13/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.4(c) |
|
Form of Addendum to Stock Option Agreement (2001 Stock Option Plan)* |
|
|
|
S-1 |
|
333-168852 |
|
8/13/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.5(a) |
|
2010 Equity Incentive Plan* (as amended and restated as May 13, 2014) |
|
|
|
DEF14A |
|
000-54192 |
|
4/2/2014 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.5(b) |
|
Form of Stock Option Agreement (2010 Equity Incentive Plan)* |
|
|
|
S-1/A |
|
333-168852 |
|
11/12/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.5(c) |
|
Form of Restricted Stock Unit Agreement (2010 Equity Incentive Plan)* |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.5(d) |
|
Form of Restricted Stock Agreement (2010 Equity Incentive Plan)* |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.6 |
|
Form of Stock Option Agreement entered into with Robert Thomas and Anne Keating* |
|
|
|
S-1/A |
|
333-168852 |
|
11/12/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.7 |
|
|
|
|
S-1 |
|
333-168852 |
|
8/13/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.8 |
|
|
|
|
10-K |
|
000-54192 |
|
3/17/2014 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.9 |
|
Employment Agreement, dated October 21, 2010, by and between REVA Medical, Inc. and Robert Schultz* |
|
|
|
S-1/A |
|
333-168852 |
|
11/12/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.10 |
|
|
|
|
S-1/A |
|
333-168852 |
|
11/12/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.11 |
|
|
|
|
10-K |
|
000-54192 |
|
3/17/2014 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.12 |
|
Employment Agreement, dated September 21, 2015, by and between REVA Medical, Inc. and Regina E. Groves* |
|
|
|
8-K |
|
000-54192 |
|
8/21/2015 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.13 |
|
Employment Agreement, dated January 18, 2016 by and between REVA Medical, Inc. and Richard M. Kimes* |
|
|
|
10-K |
|
000-54192 |
|
3/10/2016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
10.14 |
|
|
|
|
10-Q |
|
000-54192 |
|
11/7/2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.15(a) |
|
|
|
|
DEF14A |
|
000-54192 |
|
10/14/2014 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.15(b) |
|
|
|
|
DEF14A |
|
000-54192 |
|
3/9/2016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.15(c) |
|
|
|
|
8-K |
|
000-54192 |
|
4/26/2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
- 65 -
|
|
|
|
|
Filed |
|
|
|
|
|
|
|
Exhibit |
|
|
|
with this |
|
Incorporated by Reference |
||||
|
Number |
|
Description of Exhibits |
|
10-K |
|
Form |
|
File No. |
|
Date Filed |
|
|
|
|
|
8-K |
|
000-54192 |
|
4/26/2017 |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
10.17 |
|
|
|
|
8-K |
|
000-54192 |
|
4/26/2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.18 |
|
Consulting Agreement, dated July 5, 2017, by and between REVA Medical, Inc. and Brandi L. Roberts |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.19 |
|
Consulting Agreement, dated July 13, 2017, by and between REVA Medical, Inc. and Robert K. Schultz |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.20 |
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
21.1 |
|
|
|
|
S-1 |
|
333-168852 |
|
8/13/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
23.1 |
|
Consent of Grant Thornton LLP, Independent Registered Public Accounting Firm |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
24.1 |
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.1 |
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.2 |
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
32.1 (1) |
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
99.1 |
|
|
|
|
S-1/A |
|
333-168852 |
|
10/22/2010 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.INS |
|
XBRL Instance Document |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.SCH |
|
XBRL Taxonomy Extension Schema Document |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.CAL |
|
XBRL Calculation Linkbase Document |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.DEF |
|
XBRL Taxonomy Definition Linkbase Document |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.LAB |
|
XBRL Taxonomy Label Linkbase Document |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.PRE |
|
XBRL Taxonomy Presentation Linkbase Document |
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
_______________________
|
|
* |
Management Compensatory Plan or Arrangement |
|
|
** |
Confidential treatment has been granted with respect to certain portions of this exhibit. |
|
|
(1) |
These certifications are being furnished solely to accompany this annual report pursuant to 18 U.S.C. Section 1350, and are not being filed for purposes of Section 18 of the Securities Exchange Act of 1934 and are not to be incorporated by reference into any filing of REVA Medical, Inc., whether made before or after the date hereof, regardless of any general incorporation language in such filing. |
None.
- 66 -
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
REVA Medical, Inc. |
|
|
|
|
|
|
Dated: March 7, 2018 |
By: |
/s/ Regina E. Groves |
|
|
Name: |
Regina E. Groves |
|
|
Title: |
Chief Executive Officer |
|
|
|
(principal executive officer) |
Power of Attorney
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Regina E. Groves and Brandi L. Roberts, jointly and severally, his or her attorneys-in-fact, each with the power of substitution, for him or her in any and all capacities, to sign any amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact, or his substitute or substitutes, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Signature |
|
Title |
|
Date |
|
|
|
|
|
|
|
/s/ Regina E. Groves |
|
Chief Executive Officer and Director |
|
March 7. 2018 |
|
Regina E. Groves |
|
(principal executive officer) |
|
|
|
|
|
|
|
|
|
/s/ Brandi L. Roberts |
|
Chief Financial Officer |
|
March 7. 2018 |
|
Brandi L. Roberts |
|
(principal financial and accounting officer) |
|
|
|
|
|
|
|
|
|
/s/ C. Raymond Larkin Jr. |
|
Chairman of the Board |
|
March 7. 2018 |
|
C. Raymond Larkin Jr. |
|
|
|
|
|
|
|
|
|
|
|
/s/ Ross A. Breckenridge |
|
Director |
|
March 7. 2018 |
|
Dr. Ross A. Breckenridge |
|
|
|
|
|
|
|
|
|
|
|
/s/ Brian H. Dovey |
|
Director |
|
March 7. 2018 |
|
Brian H. Dovey |
|
|
|
|
|
|
|
|
|
|
|
/s/ R. Scott Huennekens |
|
Director |
|
March 7. 2018 |
|
R. Scott Huennekens |
|
|
|
|
|
|
|
|
|
|
|
/s/ Stephen N. Oesterle |
|
Director |
|
March 7. 2018 |
|
Stephen N. Oesterle |
|
|
|
|
|
|
|
|
|
|
|
/s/ Robert B. Stockman |
|
Director |
|
March 7. 2018 |
|
Robert B. Stockman |
|
|
|
|
|
|
|
|
|
|
|
/s/ Robert B. Thomas |
|
Director |
|
March 7. 2018 |
|
Robert B. Thomas |
|
|
|
|
- 67 -
|
|
PAGE |
|
|
|
|
F-2 |
|
|
F-4 |
|
|
Consolidated Statements of Operations and Comprehensive Income (Loss) |
F-5 |
|
F-6 |
|
|
F-7 |
|
|
F-8 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
REVA Medical, Inc.
Opinion on internal control over financial reporting
We have audited the internal control over financial reporting of REVA Medical, Inc. (a Delaware corporation) and subsidiary (the “Company”) as of December 31, 2017, based on criteria established in the 2013 Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on criteria established in the 2013 Internal Control—Integrated Framework issued by COSO.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the consolidated financial statements of the Company as of and for the year ended December 31, 2017, and our report dated March 7, 2018 expressed an unqualified opinion on those financial statements.
Basis for opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and limitations of internal control over financial reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ GRANT THORNTON LLP
San Diego, California
March 7, 2018
F-2
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders
REVA Medical, Inc.
Opinion on the financial statements
We have audited the accompanying consolidated balance sheets of REVA Medical, Inc. (a Delaware corporation) and subsidiary (the “Company”) as of December 31, 2017 and 2016, the related consolidated statements of operations and comprehensive income (loss), cash flows, and stockholders’ equity (deficit) for each of the three years in the period ended December 31, 2017, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the Company’s internal control over financial reporting as of December 31, 2017, based on criteria established in the 2013 Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”), and our report dated March 7, 2018 expressed an unqualified opinion thereon.
Basis for opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ GRANT THORNTON LLP
We have served as the Company’s auditor since 2014.
San Diego, California
March 7, 2018
F-3
(in thousands, except share and per share amounts)
|
|
December 31, |
|
|||||
|
|
2017 |
|
|
2016 |
|
||
|
Assets |
|
|
|
|
|
|
|
|
Current Assets: |
|
|
|
|
|
|
|
|
Cash and cash equivalents |
$ |
18,544 |
|
|
$ |
6,674 |
|
|
Investment securities |
|
1,470 |
|
|
|
— |
|
|
Accounts receivable |
|
63 |
|
|
|
— |
|
|
Inventory |
|
627 |
|
|
|
— |
|
|
Prepaid expenses and other current assets |
|
438 |
|
|
|
472 |
|
|
Total current assets |
|
21,142 |
|
|
|
7,146 |
|
|
Non-Current Assets: |
|
|
|
|
|
|
|
|
Property and equipment, net |
|
1,492 |
|
|
|
2,277 |
|
|
Other non-current assets |
|
27 |
|
|
|
60 |
|
|
Total non-current assets |
|
1,519 |
|
|
|
2,337 |
|
|
Total Assets |
$ |
22,661 |
|
|
$ |
9,483 |
|
|
|
|
|
|
|
|
|
|
|
Liabilities and Stockholders' Deficit |
|
||||||
|
Current Liabilities: |
|
|
|
|
|
|
|
|
Accounts payable |
$ |
756 |
|
|
$ |
778 |
|
|
Accrued expenses and other current liabilities |
|
1,737 |
|
|
|
2,173 |
|
|
Deferred revenue |
|
158 |
|
|
|
— |
|
|
Convertible notes |
|
— |
|
|
|
91,655 |
|
|
Accrued interest on convertible notes |
|
— |
|
|
|
4,204 |
|
|
Total current liabilities |
|
2,651 |
|
|
|
98,810 |
|
|
Long-Term Liabilities: |
|
|
|
|
|
|
|
|
Convertible notes |
|
99,368 |
|
|
|
— |
|
|
Accrued interest on convertible notes |
|
8,779 |
|
|
|
— |
|
|
Common stock warrant liability |
|
4,176 |
|
|
|
— |
|
|
Other long-term liabilities |
|
500 |
|
|
|
266 |
|
|
Total long-term liabilities |
|
112,823 |
|
|
|
266 |
|
|
Total Liabilities |
|
115,474 |
|
|
|
99,076 |
|
|
|
|
|
|
|
|
|
|
|
Commitments and contingencies (Note 9) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stockholders' Deficit: |
|
|
|
|
|
|
|
|
Common stock ― $0.0001 par value; 100,000,000 shares authorized; 41,245,820 and 42,851,477 shares issued and outstanding at December 31, 2017 and December 31, 2016, respectively |
|
4 |
|
|
|
4 |
|
|
Additional paid-in capital |
|
289,342 |
|
|
|
299,643 |
|
|
Accumulated other comprehensive loss |
|
(2 |
) |
|
|
(2 |
) |
|
Accumulated deficit |
|
(382,157 |
) |
|
|
(389,238 |
) |
|
Total Stockholders' Deficit |
|
(92,813 |
) |
|
|
(89,593 |
) |
|
Total Liabilities and Stockholders' Deficit |
$ |
22,661 |
|
|
$ |
9,483 |
|
The accompanying notes are an integral part of these financial statements.
F-4
Consolidated Statements of Operations and Comprehensive Income (Loss)
(in thousands, except share and per share amounts)
|
|
|
Year Ended December 31, |
|
|||||||||||
|
|
|
2017 |
|
|
|
2016 |
|
|
|
2015 |
|
|||
|
Revenue |
$ |
|
45 |
|
|
$ |
|
— |
|
|
$ |
|
— |
|
|
Cost of revenue |
|
|
42 |
|
|
|
|
— |
|
|
|
|
— |
|
|
Gross profit |
|
|
3 |
|
|
|
|
— |
|
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating Expense: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
12,760 |
|
|
|
|
18,171 |
|
|
|
|
16,760 |
|
|
Selling, general and administrative |
|
|
8,572 |
|
|
|
|
8,609 |
|
|
|
|
7,210 |
|
|
Loss from operations |
|
|
(21,329 |
) |
|
|
|
(26,780 |
) |
|
|
|
(23,970 |
) |
|
Other Income (Expense): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
57 |
|
|
|
|
3 |
|
|
|
|
9 |
|
|
Interest expense |
|
|
(6,690 |
) |
|
|
|
(2,053 |
) |
|
|
|
(1,904 |
) |
|
Loss on issuance of convertible notes and warrants to purchase common stock |
|
|
(520 |
) |
|
|
|
— |
|
|
|
|
— |
|
|
Gain (loss) on change in fair value of convertible notes and warrant liability |
|
|
35,731 |
|
|
|
|
(25,247 |
) |
|
|
|
(56,788 |
) |
|
Other (expense) income |
|
|
(115 |
) |
|
|
|
(21 |
) |
|
|
|
59 |
|
|
Total other income (expense) |
|
|
28,463 |
|
|
|
|
(27,318 |
) |
|
|
|
(58,624 |
) |
|
Net Income (Loss) |
$ |
|
7,134 |
|
|
$ |
|
(54,098 |
) |
|
$ |
|
(82,594 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income (loss) per share - basic |
$ |
|
0.17 |
|
|
$ |
|
(1.28 |
) |
|
$ |
|
(2.38 |
) |
|
Weighted average shares outstanding - basic |
|
|
41,811,326 |
|
|
|
|
42,120,545 |
|
|
|
|
34,680,634 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss per share - diluted |
$ |
|
(0.40 |
) |
|
$ |
|
(1.28 |
) |
|
$ |
|
(2.38 |
) |
|
Weighted average shares outstanding - diluted |
|
|
53,317,482 |
|
|
|
|
42,120,545 |
|
|
|
|
34,680,634 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive Income (Loss): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income (loss) |
$ |
|
7,134 |
|
|
$ |
|
(54,098 |
) |
|
$ |
|
(82,594 |
) |
|
Other comprehensive income (loss) |
|
0 |
|
|
|
0 |
|
|
|
(1 |
) |
|||
|
Comprehensive income (loss) |
$ |
|
7,134 |
|
|
$ |
|
(54,098 |
) |
|
$ |
|
(82,595 |
) |
The accompanying notes are an integral part of these financial statements.
F-5
Consolidated Statements of Cash Flows
(in thousands)
|
|
Year Ended December 31, |
|
|||||||||
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Cash Flows from Operating Activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Net income (loss) |
$ |
7,134 |
|
|
$ |
(54,098 |
) |
|
$ |
(82,594 |
) |
|
Non-cash adjustments to reconcile net income (loss) to net cash used for operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
1,062 |
|
|
|
1,139 |
|
|
|
1,096 |
|
|
Loss on sale of property and equipment |
|
52 |
|
|
|
— |
|
|
|
— |
|
|
Stock-based compensation |
|
2,047 |
|
|
|
4,723 |
|
|
|
3,434 |
|
|
Interest on convertible notes |
|
6,690 |
|
|
|
2,053 |
|
|
|
1,904 |
|
|
Loss on issuance of convertible notes and warrants to purchase common stock |
|
520 |
|
|
|
— |
|
|
|
— |
|
|
(Gain) loss on change in fair value of convertible notes and warrant liability |
|
(35,731 |
) |
|
|
25,247 |
|
|
|
56,788 |
|
|
Other non-cash expenses |
|
— |
|
|
|
21 |
|
|
|
46 |
|
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
Accounts receivable |
|
(63 |
) |
|
|
— |
|
|
|
— |
|
|
Inventory |
|
(570 |
) |
|
|
— |
|
|
|
— |
|
|
Prepaid expenses and other current assets |
|
84 |
|
|
|
(75 |
) |
|
|
9 |
|
|
Other non-current assets |
|
33 |
|
|
|
— |
|
|
|
— |
|
|
Accounts payable |
|
(72 |
) |
|
|
(244 |
) |
|
|
365 |
|
|
Accrued expenses and other current liabilities |
|
(436 |
) |
|
|
(90 |
) |
|
|
33 |
|
|
Deferred revenue |
|
158 |
|
|
|
— |
|
|
|
— |
|
|
Other long-term liabilities |
|
234 |
|
|
|
65 |
|
|
|
(163 |
) |
|
Net cash used for operating activities |
|
(18,858 |
) |
|
|
(21,259 |
) |
|
|
(19,082 |
) |
|
Cash Flows from Investing Activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Purchases of property and equipment |
|
(386 |
) |
|
|
(729 |
) |
|
|
(857 |
) |
|
Purchases of investments |
|
(1,470 |
) |
|
|
— |
|
|
|
— |
|
|
Maturities of investments |
|
— |
|
|
|
— |
|
|
|
995 |
|
|
Net cash (used for) provided by investing activities |
|
(1,856 |
) |
|
|
(729 |
) |
|
|
138 |
|
|
Cash Flows from Financing Activities: |
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from issuances of common stock |
|
92 |
|
|
|
11,767 |
|
|
|
10,075 |
|
|
Repurchase of common stock |
|
(12,493 |
) |
|
|
— |
|
|
|
— |
|
|
Proceeds from (costs of) issuance of convertible notes payable and warrants, net |
|
44,985 |
|
|
|
— |
|
|
|
(50 |
) |
|
Net cash provided by financing activities |
|
32,584 |
|
|
|
11,767 |
|
|
|
10,025 |
|
|
Net increase (decrease) in cash and cash equivalents |
|
11,870 |
|
|
|
(10,221 |
) |
|
|
(8,919 |
) |
|
Cash and cash equivalents at beginning of period |
|
6,674 |
|
|
|
16,895 |
|
|
|
25,814 |
|
|
Cash and Cash Equivalents at End of Period |
$ |
18,544 |
|
|
$ |
6,674 |
|
|
$ |
16,895 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental Non-Cash Information: |
|
|
|
|
|
|
|
|
|
|
|
|
Property and equipment in accounts payable |
$ |
11 |
|
|
$ |
18 |
|
|
$ |
50 |
|
|
Adjustment to beginning accumulated deficit upon adoption of ASU 2016-09 |
$ |
53 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Warrant liability transferred to equity upon exercise |
$ |
— |
|
|
$ |
28,579 |
|
|
$ |
14,970 |
|
The accompanying notes are an integral part of these financial statements.
F-6
Consolidated Statements of Stockholders’ Equity (Deficit)
(in thousands, except share and per share amounts)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other |
|
|
|
|
|
|
Total |
|
||
|
|
Common Stock |
|
|
Additional |
|
|
Comprehensive |
|
|
Accumulated |
|
|
Stockholders' |
|
|||||||||
|
|
Shares |
|
|
Amount |
|
|
Paid-In Capital |
|
|
Income/(Loss) |
|
|
Deficit |
|
|
Deficit |
|
||||||
|
Balance at December 31, 2014 |
|
33,529,778 |
|
|
$ |
3 |
|
|
$ |
226,095 |
|
|
$ |
(1 |
) |
|
$ |
(252,546 |
) |
|
$ |
(26,449 |
) |
|
Net loss and comprehensive loss |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(82,594 |
) |
|
|
(82,594 |
) |
|
Common stock issued upon exercise of stock options for cash at $1.25 to $5.60 per share |
|
251,208 |
|
|
|
0 |
|
|
|
570 |
|
|
|
— |
|
|
|
— |
|
|
|
570 |
|
|
Common stock issued upon exercise of warrants for cash at $2.17275 per share |
|
4,375,000 |
|
|
|
1 |
|
|
|
9,505 |
|
|
|
— |
|
|
|
— |
|
|
|
9,506 |
|
|
Fair value of warrant liability transferred to equity upon warrant exercise |
|
— |
|
|
|
— |
|
|
|
14,970 |
|
|
|
— |
|
|
|
— |
|
|
|
14,970 |
|
|
Stock-based compensation expense |
|
— |
|
|
|
— |
|
|
|
3,434 |
|
|
|
— |
|
|
|
— |
|
|
|
3,434 |
|
|
Other comprehensive loss |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(1 |
) |
|
|
— |
|
|
|
(1 |
) |
|
Balance at December 31, 2015 |
|
38,155,986 |
|
|
$ |
4 |
|
|
$ |
254,574 |
|
|
$ |
(2 |
) |
|
$ |
(335,140 |
) |
|
$ |
(80,564 |
) |
|
Net loss and comprehensive loss |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(54,098 |
) |
|
|
(54,098 |
) |
|
Common stock issued upon exercise of stock options for cash at $1.40 to $4.00 per share |
|
132,916 |
|
|
0 |
|
|
|
360 |
|
|
|
— |
|
|
|
— |
|
|
|
360 |
|
|
|
Common stock issued upon exercise of warrants for cash at $2.6073 per share |
|
4,375,000 |
|
|
0 |
|
|
|
11,407 |
|
|
|
— |
|
|
|
— |
|
|
|
11,407 |
|
|
|
Common stock issued upon vesting of restricted stock units |
|
160,000 |
|
|
0 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
0 |
|
|
|
Common stock issued upon net exercise of stock options |
|
27,575 |
|
|
0 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
0 |
|
|
|
Fair value of warrant liability transferred to equity upon warrant exercise |
|
— |
|
|
|
— |
|
|
|
28,579 |
|
|
|
— |
|
|
|
— |
|
|
|
28,579 |
|
|
Stock-based compensation expense |
|
— |
|
|
|
— |
|
|
|
4,723 |
|
|
|
— |
|
|
|
— |
|
|
|
4,723 |
|
|
Other comprehensive loss |
|
— |
|
|
|
— |
|
|
|
— |
|
|
0 |
|
|
|
— |
|
|
|
0 |
|
|
|
Balance at December 31, 2016 |
|
42,851,477 |
|
|
$ |
4 |
|
|
$ |
299,643 |
|
|
$ |
(2 |
) |
|
$ |
(389,238 |
) |
|
$ |
(89,593 |
) |
|
Net income and comprehensive income |
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
7,134 |
|
|
|
7,134 |
|
|
Reversal of forfeiture estimates |
|
— |
|
|
|
— |
|
|
|
53 |
|
|
|
— |
|
|
|
(53 |
) |
|
|
— |
|
|
Common stock issued upon vesting of restricted stock units |
|
47,800 |
|
|
0 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
0 |
|
|
|
Common stock issued upon exercise of stock options for cash at $1.40 to $1.50 per share |
|
65,000 |
|
|
0 |
|
|
|
92 |
|
|
|
— |
|
|
|
— |
|
|
|
92 |
|
|
|
Common stock issued upon net exercise of stock options |
|
13,803 |
|
|
0 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
0 |
|
|
|
Stock repurchase associated with convertible debt financing |
|
(1,732,260 |
) |
|
0 |
|
|
|
(12,493 |
) |
|
|
|
|
|
|
|
|
|
|
(12,493 |
) |
|
|
Stock-based compensation expense |
|
— |
|
|
|
— |
|
|
|
2,047 |
|
|
|
— |
|
|
|
— |
|
|
|
2,047 |
|
|
Other comprehensive income |
|
— |
|
|
|
— |
|
|
|
— |
|
|
0 |
|
|
|
— |
|
|
|
0 |
|
|
|
Balance at December 31, 2017 |
|
41,245,820 |
|
|
$ |
4 |
|
|
$ |
289,342 |
|
|
$ |
(2 |
) |
|
$ |
(382,157 |
) |
|
$ |
(92,813 |
) |
The accompanying notes are an integral part of these financial statements.
F-7
REVA Medical, Inc.
Notes to Consolidated Financial Statements
REVA Medical, Inc. was incorporated in California in 1998 under the name MD3, Inc. In March 2002, we changed our name to REVA Medical, Inc. In October 2010 we reincorporated in Delaware. We established a non-operating wholly owned subsidiary, REVA Germany GmbH, in 2007. In these Notes to Consolidated Financial Statements, the terms “REVA,” the “Company,” “us,” “we,” or “our” refer to REVA and its consolidated subsidiary unless context dictates otherwise.
We are a medical device company focused on developing and commercializing products for use in humans, utilizing our proprietary bioresorbable polymer technologies. On April 3, 2017, we received approval for the marketing and sale of our first product, the Fantom scaffold, in Europe and other jurisdictions that recognize the CE Mark. Our Fantom scaffold is a sirolimus-eluting bioresorbable scaffold used to treat coronary artery disease in humans. We received our first customer order late in the second quarter of 2017 and we recorded our first order shipments and revenues in the third quarter of 2017. Prior to CE Mark, Fantom had been implanted in 247 patients in the FANTOM I and FANTOM II clinical trials conducted in eight countries outside the United States. We used the six-month clinical results from 117 patients in the FANTOM II clinical trial for CE Mark application, which we submitted in 2016.
In December 2010, we completed an initial public offering (the “IPO”) of our common stock in Australia and registered with the U.S. Securities and Exchange Commission (“SEC”) and, consequently, became an SEC reporting company. Our common stock is traded in the form of CHESS Depositary Interests (“CDIs”) on the Australian Securities Exchange (“ASX”); each share of our common stock is equivalent to ten CDIs. Our trading symbol is “RVA.AX.” We may pursue a listing of our common stock on a U.S. stock exchange, at which time we would become dual-listed, if we maintain our listing on the ASX.
2. Capital Resources and Basis of Presentation
Capital Resources: The accompanying consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. We have incurred significant operating losses since inception and have relied on our ability to fund our operations primarily through equity and debt financings. At December 31, 2017, we had an accumulated deficit of $382.2 million and our cash, cash equivalents and investment securities totaled $20.0 million. Based on our current operating plans and projections, we believe our cash, cash equivalents and investment securities of $20.0 million will be sufficient to fund our operations through the first quarter of 2019. Our projections are predicated on us achieving certain minimum levels of sales of our Fantom scaffold. If we are unable to achieve these levels of sales, we may be compelled to reduce operating and capital expenditures or sell certain assets.
Although we initiated commercial sales of Fantom in the third quarter of 2017, we are still very early in the commercialization stage. The withdrawal of Absorb, a competitor’s product, in 2017, and the negative publicity related to Absorb’s safety have severely impacted the market for bioresorbable scaffolds, and companies with bioresorbable scaffolds that were made from the same polylactic acid polymer, or PLLA, as Absorb have reduced scale and abandoned their efforts to commercialize such scaffolds. Because Fantom is not made with the same polymer as Absorb, we continue to believe that we can commercialize Fantom despite the impact that the withdrawal of Absorb has had on the market and demonstrate the benefits of bioresorbable technology. That said, we must now rebuild the market for bioresorbable scaffolds, which can be more challenging than selling into an existing, healthy market. Our rebuilding activities include educating physicians regarding the unique features of Fantom and Fantom Encore, continuing to publish results from our pivotal clinical trial (FANTOM II) and conducting and initiating additional clinical studies to build the clinical evidence needed to support market adoption
Until we generate revenue at a level to support our cost structure, we expect to continue to incur substantial operating losses and net cash outflows. We may never become profitable and even if we do attain profitability, we may not be able to sustain profitability or positive cash flows on a recurring basis. Unless we are able to significantly accelerate our sales, we do not anticipate generating positive cash flows in 2018 or 2019, and therefore, will need to raise further capital to support our operations and our ongoing costs, and to conduct a U.S. clinical trial, if we determine to do so. We have a plan to address our capital needs, which includes accelerating our revenue by pursuing sales expansion and executing business development and strategic opportunities. We are also evaluating public or private sales of our equity or debt securities. In addition, the convertible notes we issued in 2014 mature in November 2019 and each holder of the convertible notes we issued in 2017 has a redemption right that it may exercise in November 2019. The
F-8
REVA Medical, Inc.
Notes to Consolidated Financial Statements
aggregate face value of all such convertible notes and accrued interest is $72.1 million and $8.8 million, respectively, as of December 31, 2017. See Note 7 Convertible Notes and Warrants to Purchase Common Stock for additional information. If we are unable to significantly increase revenue or raise additional capital when needed or on acceptable terms, we would need to consider a delay, reduction or cessation of our research and development programs and our commercialization efforts. There can be no assurance that our efforts will result in the resolution of our liquidity needs. If we are not able to continue as a going concern, holders of our common stock and our convertible notes could lose their investment. The accompanying consolidated financial statements do not include any adjustments that might result should we be unable to continue as a going concern.
Basis of Presentation: We have prepared the accompanying consolidated financial statements in accordance with accounting principles generally accepted in the United States (“U.S. GAAP”). The consolidated financial statements include the accounts of REVA and our wholly owned subsidiary, REVA Germany GmbH. All intercompany transactions and balances, if any, have been eliminated in consolidation.
Use of Estimates: The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from our estimates.
3. Significant Accounting Policies
Cash Equivalents: We consider all highly liquid investments with original maturities of three months or less at the date of purchase to be cash equivalents. Cash equivalents are carried at cost, which we believe approximates fair value due to the short-term maturities of these investments.
Investment Securities: Investment securities are marketable equity or debt securities. All of our investment securities are “available-for-sale” securities and carried at fair value. Fair value for securities with short maturities and infrequent secondary market trades typically is determined by using a curve-based evaluation model that utilizes quoted prices for similar securities. The evaluation model takes into consideration the days to maturity, coupon rate and settlement date convention. Net unrealized gains or losses on these securities are included in accumulated other comprehensive loss, which is a separate component of stockholders’ deficit. Realized gains and realized losses are included in other income (expense) while amortization of premiums and accretion of discounts are included in interest income. Interest and dividends on available-for-sale securities are included in interest income. We periodically evaluate our investment securities for impairment. If we determine that a decline in fair value of any investment security is other than temporary, then the cost basis would be written down to fair value and the decline in value would be charged to other expense at that time.
Our investment securities are under the custodianship of a major financial institution and consist of certificates of deposit that are insured by the Federal Deposit Insurance Corporation. We have classified all of our available-for-sale investment securities as current assets on our consolidated balance sheets because we consider them to be highly liquid and available for use, if needed, in current operations. As of December 31, 2017, none of our $1.5 million of investment securities had contractual maturity dates of more than one year.
Inventory: We received CE Mark approval of our Fantom scaffold on April 3, 2017, at which time we began capitalizing raw material purchases and commercial scaffold production costs to inventory. Inventory is stated at the lower of cost or net realizable value based on the first-in, first-out cost method (“FIFO”). Our policy is to record an estimated allowance against inventory for unsalable, obsolete, or impaired inventory, with a corresponding increase to cost of revenue. We record the cost of products to be used in research and development or clinical trials as research and development expense when inventory is requisitioned for such use.
Impairment of Long-Lived Assets: We review our long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable and exceeds its undiscounted future cash flows. The amount of impairment, if any, is determined by comparing an asset’s estimated fair value to the asset’s respective carrying amount. During the years ended December 31, 2017, 2016, and 2015 we determined there were no indications of long-lived asset impairment.
F-9
REVA Medical, Inc.
Notes to Consolidated Financial Statements
Convertible Notes: Convertible notes are analyzed at issue date to determine balance sheet classification, issue discounts or premiums, and embedded or derivative features. Embedded or derivative features are evaluated in accordance with accounting guidance for derivative securities and, if the features give rise to separate accounting, we make an election to account for the notes at cost or at fair value. If fair value accounting is elected on the issue date, we record the difference between the issue price of the notes and their fair value as a gain or loss in our consolidated statement of operations. We remeasure the fair value at each reporting date and record a gain (upon a decrease in fair value) or loss (upon an increase in fair value), as a component of other income (expense) in our consolidated statement of operations. Inputs to the models include the market value of the underlying stock, a life equal to the contractual life of the notes, incremental borrowing rates that correspond to debt with similar credit worthiness, and estimated volatility based on the historical prices of our trading securities. For each periodic valuation, we also make assumptions as to our abilities to test and commercialize our product, to obtain future financings when and if needed, and to comply with the terms and conditions of any outstanding notes payable.
Following an analysis of their embedded and derivative features, we elected to utilize fair value accounting for all issues of convertible notes as management believes the convertible notes will be converted into common stock, rather than repaid, and the fair value method of accounting provides a more appropriate value of these liabilities than would be provided under the cost method.
Common Stock Warrants: The fair value of warrants issued for the purchase of common stock is recorded as a liability whenever warrants call for issuance of registered shares upon exercise, a condition that we may not be able to satisfy at the time of exercise, and which, if not so satisfied, will result in a net settlement of warrants. Until the time warrants are exercised or expire, the fair value is assessed at each reporting date. Any change in value is recorded as a gain or loss component of other income (expense) in our consolidated statement of operations. Inputs to the valuation models are of the same nature as those used to value our convertible notes.
Revenue: We received our first order for Fantom in June 2017. We sell Fantom to hospitals; and title and risk of loss transfer upon delivery to these hospitals. We recognize revenue when all of the following four criteria are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred; (3) the fee is fixed or determinable; and (4) collectability is reasonably assured. We also consider any return or exchange rights. We analyze product reorder rates to evaluate and determine whether return or exchange rights exist and are likely to be exercised. If the revenue recognition criteria are not met, we defer the recognition of revenue by recording deferred revenue until such time that all criteria are met.
We recognized $45,000 of revenue during the year ended December 31, 2017. Total billings for shipped product for this period were $203,000; the amount by which total billings for shipped product exceeded recognized revenue was recorded as deferred revenue.
Accounts receivable consist of trade receivables recorded upon shipment of product reduced by reserves when necessary for estimated bad debts. Accounts receivable are recorded at the invoiced amount and do not bear interest. Credit is extended based on an evaluation of the customer’s financial condition. The allowance for doubtful accounts is determined based on current customer information and other relevant factors, including specific identification of past due accounts. Once a receivable is deemed to be uncollectible, such balance is charged against the allowance. As of December 31, 2017, our allowance for doubtful accounts was $0.
Research and Development: Research and development costs are expensed as incurred. These costs include salaries, employee benefits, laboratory supplies, consulting services, manufacturing products and services, preclinical and clinical costs, technology license fees, laboratory equipment depreciation, facility costs, and certain indirect costs.
Income Taxes: We account for income taxes using the asset and liability method, under which the current income tax expense or benefit is the amount of income tax expected to be payable or refundable in the current year. Deferred tax assets and liabilities are recorded for the estimated future tax consequences of temporary differences between the financial statement carrying amounts of assets and liabilities and their respective tax bases, and for operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the year in which the temporary differences are expected to be recovered or settled.
F-10
REVA Medical, Inc.
Notes to Consolidated Financial Statements
We evaluate the realizability of our deferred tax assets and establish a valuation allowance when it is more likely than not that all or a portion of our deferred tax assets will not be realized. In making such a determination, we consider all available positive and negative evidence, including future reversals of existing taxable temporary differences, projected future taxable income, tax planning strategies, and results of recent operations. If we determine that we would be able to realize our deferred tax assets in the future in excess of their net recorded amount, we would make an adjustment to the deferred tax asset valuation allowance, which would reduce the provision for income taxes.
We account for the uncertainty in income tax components based on tax positions taken or expected to be taken in a tax return. To recognize a benefit, a tax position must be more likely than not to be sustained upon examination by taxing authorities. We do not recognize tax benefits that have a less than 50 percent likelihood of being sustained. Our policy is to recognize interest and tax penalties related to unrecognized tax benefits in income tax expense; no interest or tax penalties on uncertain tax benefits have been recorded through December 31, 2017.
Stock-Based Compensation: Stock-based compensation expense is recorded in connection with stock options, restricted stock awards, and restricted stock unit awards (“RSUs”) to employees, directors, and consultants. We have granted stock options, restricted stock, and RSUs that vest based on the passage of time (time-based vesting awards) as well as stock options and RSUs that vest based on achievement of performance milestones (performance-based vesting awards).
For time-based vesting stock options granted to employees and directors, we determine compensation expense based on estimated grant date fair values utilizing the Black-Scholes option valuation model. The Black-Scholes model requires the input of assumptions, including volatility, the expected term, and the fair value of the underlying common stock on the date of grant, among other inputs. For time-based vesting restricted stock awards and RSUs, the grant date fair value is equal to the closing market price of our common stock on the date of award. We use the straight-line method to allocate compensation expense to reporting periods over each recipient’s requisite service period, which is generally from one to four years. All stock-based compensation expense is recorded as either research and development or selling, general and administrative expense based on a recipient’s work classification.
For performance-based vesting stock options and RSUs, we record compensation expense for only the performance milestones that are probable of being achieved, with such expense recorded on a straight-line basis over the expected vesting period. We reassess our performance-based estimates each reporting period and, if the estimated service period changes, we recognize all remaining compensation expense over the remaining service period and, if the probability of achievement changes to or from “probable,” we recognize the cumulative effect. Whenever an award recipient terminates service prior to achievement of a performance milestone, the recipient’s unvested awards are cancelled and the related compensation expense previously recorded is reversed.
For stock options granted to consultants, all of which are time-based vesting, we estimate fair values at the date of grant and at each subsequent reporting period and record compensation expense during the consultant’s service period. We estimate the fair value utilizing the Black-Scholes option valuation model with the same approach to inputs and assumptions as we use to estimate the fair value of employee options, except we use the remaining term as the expected life of the option.
Foreign Currency: The functional currency of our subsidiary REVA Germany GmbH is the Euro. Balance sheet accounts of our subsidiary are translated into United States dollars using the exchange rate in effect at the balance sheet date while expenses are translated using the average exchange rate in effect during the period. Gains and losses arising from translation of our subsidiary’s financial statements are recorded to other comprehensive income (loss). These gains and losses, in the aggregate, were insignificant through December 31, 2017.
Concentrations: Financial instruments that potentially subject us to concentrations of credit risk are primarily cash, cash equivalents and investment securities. Investment securities are invested in accordance with our investment policy. Our audit committee approved an investment policy that sets our investment parameters and limitations with objectives of preserving principal and liquidity. Periodically, we maintain deposits at financial institutions in excess of government insured limits. We invest our cash balances in major financial institutions that we believe have high credit quality and have not experienced any losses on such accounts. We do not believe we are exposed to significant credit risk.
F-11
REVA Medical, Inc.
Notes to Consolidated Financial Statements
We require customized components that currently are available from a limited number of sources. We source certain components included in our products from single vendors.
As we recently commenced commercial operations, our revenue in 2017 is more concentrated than we expect it to be after we expand commercially. Our top customer represented 26% of our total shipments in 2017. All of our shipments as of December 31, 2017 were made to customers outside of the United States as we only have approval to sell in countries that recognize CE Mark.
We maintain inventory at our third-party logistics provider in The Netherlands. As of December 31, 2017, $127,000 of our inventory was held at this location.
Segment Information: We operate in one business segment, which is the development and commercialization of medical devices.
Recently Adopted Accounting Pronouncements: We adopted ASU 2016-09, Stock Compensation: Improvements to Employee Share-Based Payment Accounting, effective January 1, 2017. ASU 2016-09 simplifies certain aspects of accounting for stock-based compensation, including the accounting for income taxes, the option to recognize forfeiture credits as they occur rather than as an estimate of future activity, and classifications in the statement of cash flows. Upon the adoption, we recorded a cumulative effect adjustment to increase our accumulated deficit by approximately $53,000, with a corresponding increase to additional paid-in capital, to reverse our forfeiture estimate for unvested awards. All forfeitures occurring after adoption are being recognized in the consolidated statement of operations in the reporting period in which they occur. We had $1.8 million of forfeitures during the year ended December 31, 2017 related to a reduction in force that occurred in July 2017. See Note 11 Stock-Based Compensation.
Recently Issued Accounting Pronouncements: In May 2014, the Financial Accounting Standards Board, or FASB, issued ASU 2014-09, Revenue from Contracts with Customers, which introduced Accounting Standards Codification 606, Revenue from Contracts with Customers (“ASC 606”), an updated standard on revenue recognition. The standard outlines a single comprehensive model to use in accounting for revenue arising from contracts with customers and supersedes most current revenue recognition guidance. Revenue recognized under ASC 606 will represent the consideration an entity expects to be entitled to in exchange for the transfer of goods or services to a customer; it also requires additional disclosures about the nature, amount, timing, and uncertainty of revenue and cash flows arising from customer contracts. The standard permits two methods of adoption: retrospectively to each prior reporting period (full retrospective method), or retrospectively with the cumulative effect of initially applying the guidance recognized at the date of initial application (modified retrospective method). We adopted ASC 606 effective January 1, 2018 and will utilize the modified retrospective method for adoption. The adoption of the standard will not result in a material transition adjustment. Total billings for shipped product for the year ended December 31, 2017 were $203,000. We are in the process of finalizing the new required disclosures.
In January 2016, the FASB issued ASU No. 2016-01, “Financial Instruments – Overall (Subtopic 825-10): Recognition and Measurement of Financial Assets and Financial Liabilities” (“ASU 2016-01”). The amendments in this update require an entity to present separately in other comprehensive income the portion of the total change in the fair value of a liability resulting from a change in the instrument-specific credit risk when the entity has elected to measure the liability at fair value in accordance with the fair value option for financial instruments. In addition, the amendments in this update eliminate the requirement to disclose the method(s) and significant assumptions used to estimate the fair value that is required to be disclosed for financial instruments measured at amortized cost on the balance sheet for public entities. For public business entities, the amendments in ASU 2016-01 are effective for fiscal years beginning after December 15, 2017, including interim periods within those fiscal years. Except for the early application guidance discussed in ASU 2016-01, early adoption of the amendments in this update is not permitted. We adopted ASU 2016-01 effective January 1, 2018. We are in the process of analyzing the impact of this guidance on our consolidated financial statements and do not yet know if the changes will be material.
F-12
REVA Medical, Inc.
Notes to Consolidated Financial Statements
In February 2016, ASU 2016-02, Leases (Topic 842), was issued. The amendments in this update require a lessee to recognize on the balance sheet a liability to make lease payments (the lease liability) and a right-of-use asset representing its right to use the underlying asset for the lease term for all leases with terms greater than twelve months. For leases less than twelve months, an entity is permitted to make an accounting policy election by class of underlying asset not to recognize lease assets and lease liabilities. If a lessee makes this election, it should recognize lease expense for such leases generally on a straight-line basis over the lease term. The amendments in this update are effective for fiscal years beginning with fiscal year 2019, including interim periods within those years, with early adoption permitted. We are currently in the process of evaluating the impact of adoption of the amendments in this update on our consolidated balance sheet and results of operations; however, adoption of the amendments in this update is expected to have a material impact on our consolidated balance sheet, including an increase in assets and liabilities representing the present value of our future lease payments.
In July 2017, ASU 2017-11, Earnings Per Share, Distinguishing Liabilities from Equity, Derivatives and Hedging, was issued. ASU 2017-11 changes the accounting treatment and the earnings per share calculation for certain instruments with down round features. The amendments in this update should be applied using a cumulative-effect adjustment as of the beginning of the fiscal year of adoption or retrospective adjustment to each period presented. This update is effective for annual periods beginning after December 15, 2018, and interim periods within those periods. We are in the process of determining the impact the adoption will have on our Consolidated Financial Statements as well as whether to early adopt the new guidance.
4. Inventory
The Company began capitalizing inventory upon CE Mark approval in the second quarter of 2017. Inventory consisted of the following at December 31, 2017 (in thousands):
|
|
December 31, |
|
|
|
|
2017 |
|
|
|
Raw materials |
$ |
255 |
|
|
Work in process |
|
61 |
|
|
Finished goods |
|
329 |
|
|
Excess and obsolete reserve |
|
(18 |
) |
|
|
$ |
627 |
|
5. Property and Equipment
Property and equipment are stated at cost, less accumulated depreciation. Property and equipment are depreciated using the straight-line method over the estimated useful lives of the assets, which is generally three to five years. Leasehold improvements are amortized over the economic life of the asset or the lease term, whichever is shorter. Upon disposition or retirement of an asset, its cost and related accumulated depreciation are written off and any gain or loss is recognized in the consolidated statement of operations
Property and equipment at December 31, 2017 and 2016 are as follows (in thousands):
|
|
December 31, |
|
|||||
|
|
2017 |
|
|
2016 |
|
||
|
Furniture, office equipment, and software |
$ |
601 |
|
|
$ |
655 |
|
|
Laboratory equipment |
|
5,705 |
|
|
|
6,604 |
|
|
Leasehold improvements |
|
2,422 |
|
|
|
2,412 |
|
|
|
|
8,728 |
|
|
|
9,671 |
|
|
Accumulated depreciation and amortization |
|
(7,236 |
) |
|
|
(7,394 |
) |
|
|
$ |
1,492 |
|
|
$ |
2,277 |
|
F-13
REVA Medical, Inc.
Notes to Consolidated Financial Statements
6. Accrued Expenses and Other Current Liabilities
Accrued expenses and other current liabilities at December 31, 2017 and 2016 are as follows (in thousands):
|
|
December 31, |
|
|||||
|
|
2017 |
|
|
2016 |
|
||
|
Accrued salaries and other employee costs |
$ |
1,296 |
|
|
$ |
1,456 |
|
|
Accrued operating expenses |
|
404 |
|
|
|
519 |
|
|
Accrued use taxes and other |
|
37 |
|
|
|
198 |
|
|
|
$ |
1,737 |
|
|
$ |
2,173 |
|
7. Convertible Notes and Warrants to Purchase Common Stock
In May 2017, we issued 338 convertible notes and in June 2017 we issued 133 convertible notes (collectively, the “2017 Notes”), each with a face value of $100,000, for total gross cash proceeds of $47.1 million. We used a portion of the proceeds from this financing to repurchase 1,732,260 shares of our common stock from one of the investors in the 2017 Notes at $7.212 per share, for a total repurchase price of $12.5 million, and incurred transaction costs of $2.1 million, resulting in net proceeds from this financing of $32.5 million. The 2017 Notes are convertible at any time at the holders’ election; the conversion rate as of December 31, 2017 was $8.655 per share, which, if converted at that conversion rate, would result in issuing 5,441,941 shares of common stock upon conversion. The conversion rate may decrease depending on the price at which we issue securities in future financings, if any, to a minimum of $7.212 per share. The 2017 Notes mature five years from issue date, if not converted or redeemed earlier. Interest accrues at the rate of 8.0 percent per annum, compounded annually, and is payable upon redemption or maturity; accrued interest is not payable or convertible upon conversion of the notes. Each holder of the 2017 Notes has a right to request that we redeem the notes (face value plus accrued interest) on November 4, 2019, if they have not been previously converted or redeemed, if the holders have provided at least 30 days’ written notice to elect such a redemption.
On their issue dates, we evaluated the 2017 Notes and, following an analysis of the embedded and derivative features, made an irrevocable election to account for the notes at fair value. The fair value on December 31, 2017 was estimated to be $38.4 million, $8.7 million below the $47.1 million face value of the 2017 Notes.
In November 2014, we issued 250 convertible notes (the “2014 Notes”), each with a face value of $100,000, for total gross cash proceeds of $25.0 million. The 2014 Notes are convertible at any time at the holders’ election into a total of 11,506,156 shares of common stock, which reflects a conversion rate of $2.17275 per share. The 2014 Notes mature on November 14, 2019, if not converted or redeemed earlier. Interest accrues at the rate of 7.54 percent per annum, compounded annually, and is payable upon redemption or maturity; accrued interest is not payable or convertible upon conversion of the 2014 Notes. Effective June 1, 2017, the terms of the 2014 Notes that provided the holders with a one-time option to require us to redeem the notes on June 30, 2017 and that provided for an automatic conversion of the 2014 Notes were eliminated, and the 2014 Notes were modified to be subordinate to the 2017 Notes. Our stockholders approved the foregoing modifications to the terms of the 2014 Notes.
On their issue date, we evaluated the 2014 Notes and, following an analysis of the embedded and derivative features, we made an irrevocable election to account for the notes at fair value. Following the modifications to the notes that were effective on June 1, 2017, we continued to account for the 2014 Notes under the fair value method. The fair values of the 2014 Notes as of December 31, 2017 and 2016 were calculated to be $61.0 million and $91.7 million, respectively. The fair value as of December 31, 2017 was $36.0 million higher than the $25.0 million face value of the 2014 Notes.
Changes in the fair value of the 2014 Notes and 2017 Notes, which collectively we refer to as convertible notes, are recorded as gains or losses in the other income (expense) portion of our consolidated statement of operations. During the years ended December 31, 2017, 2016 and 2015, we accrued $4.6 million, $2.1 million and $1.9 million in interest expense on the convertible notes, respectively. Accrued interest on the 2014 Notes and 2017 Notes as of December 31, 2017 and 2016, in the aggregate, was $8.8 million and $4.2 million, respectively. An additional $2.1 million of transaction costs related to the issuance of the 2017 Notes was recorded as interest expense during the year ended December 31, 2017.
F-14
REVA Medical, Inc.
Notes to Consolidated Financial Statements
In connection with issuing the 2017 Notes, in May 2017 and June 2017 we issued warrants to purchase up to 2,119,500 shares of our common stock to the purchasers of the 2017 Notes. The warrants are immediately exercisable and expire five years from issue date. The exercise price of each warrant is $5.00 per share, which may increase depending on the price at which we issue securities in future financings, if any, to a maximum of $7.212 per share. The fair value of the warrants on December 31, 2017 was estimated to be $4.2 million. Changes in the fair value of the warrants are recorded as gains or losses in the other income (expense) portion of our consolidated statement of operations.
The aggregate fair value of the 2017 Notes and the warrants on their issue dates was estimated to be $47.6 million, which was $0.5 million higher than the $47.1 million issue price; we recorded this difference as a loss on issuance in our consolidated statement of operations.
The warrants we issued in November 2014 in connection with issuance of the 2014 Notes were exercised in full on or before February 12, 2016. Prior to their exercise, we recorded their change in fair value in our consolidated statement of operations. The loss on the change in fair value from January 1, 2016 to February 12, 2016 was $9.0 million.
As previously discussed, the 2014 Notes mature in November 2019 and each holder of the 2017 Notes has a redemption right that it may exercise in November 2019. The aggregate face value of all such convertible notes is $72.1 million and the aggregate accrued interest on all such notes was $8.8 million as of December 31, 2017. If the holders of the 2017 Notes collectively, or individually, call for redemption, or if we are unable to convert or extend the maturity date of the 2014 Notes, we most likely will not have the cash to repay the notes.
8. Fair Value Measurements
Our cash equivalents, investment securities, convertible notes and common stock warrant liability are carried at fair value. The fair value of financial assets and liabilities is measured under a framework that establishes “levels” which are defined as follows: (i) Level 1 fair value is determined from observable, quoted prices for identical assets or liabilities; (ii) Level 2 fair value is determined from quoted prices for similar items in active markets or quoted prices for identical or similar items in markets that are not active, and (iii) Level 3 fair value is determined using the entity’s own assumptions about the inputs that market participants would use in pricing an asset or liability.
The fair values of our cash equivalents, investment securities, convertible notes and common stock warrant liability are summarized in the following tables (in thousands):
|
|
December 31, 2017 |
|
|||||||||||||
|
|
Total |
|
|
Fair Value Determined Under: |
|
||||||||||
|
|
Fair Value |
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
||||
|
Assets: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash equivalents |
$ |
4,388 |
|
|
$ |
4,388 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Investment securities |
$ |
1,470 |
|
|
$ |
— |
|
|
$ |
1,470 |
|
|
$ |
— |
|
|
Liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Convertible notes payable |
$ |
99,368 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
99,368 |
|
|
Common stock warrant liability |
$ |
4,176 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
4,176 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2016 |
|
|||||||||||||
|
|
Total |
|
|
Fair Value Determined Under: |
|
||||||||||
|
|
Fair Value |
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
||||
|
Assets: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash equivalents |
$ |
6,655 |
|
|
$ |
6,655 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Convertible notes payable |
$ |
91,655 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
91,655 |
|
The fair values of our 2017 Notes as of December 31, 2017 and the fair values of our 2014 Notes as of December 31, 2016 were determined utilizing a Least Squares Monte Carlo simulation model; the fair value of our warrants to
F-15
REVA Medical, Inc.
Notes to Consolidated Financial Statements
purchase common stock was determined using either a Least Squares Monte Carlo simulation model or a Black-Scholes valuation model, depending on their exercise price and other features. These models require use of unobservable inputs that are determined by management, with the assistance of independent experts. These inputs represent our best estimates, but involve certain inherent uncertainties. We use the market value of the underlying stock, a life equal to the contractual life of the financial instrument, incremental borrowing rates and bond yields that correspond to instruments of similar credit worthiness and the instrument’s remaining life, an estimate of volatility based on the historical prices of our trading securities, and we make assumptions as to our abilities to test and commercialize our product(s), to obtain future financings when and if needed, to comply with the terms and conditions of our convertible notes, and the probability of a change in control event.
A summary of the weighted average assumptions used to value these Level 3 liabilities is as follows:
|
|
Year Ended December 31, |
|
|||||
|
|
2017 |
|
|
2016 |
|
||
|
Market price per share of common stock |
$ |
5.31 |
|
|
$ |
7.90 |
|
|
Risk-free interest rate |
2.1% |
|
|
2.0% |
|
||
|
Expected volatility of common stock |
45.0% |
|
|
79.7% |
|
||
|
Expected life (in years) |
|
4.37 |
|
|
|
2.90 |
|
|
Bond yield of equivalent securities |
26.5% |
|
|
27.0% |
|
||
A significant change in the market price per share, expected volatility, or bond yield of equivalent securities, in isolation, would result in significantly higher or lower fair value measurements. In combination, changes in these inputs could result in a significantly higher or lower fair value measurement if the input changes were to be aligned, or could result in a minimally higher or lower fair value measurement if the input changes were of a compensating nature.
As the 2014 Notes were significantly in the money and no longer had complex features as of December 31, 2017, we used an “as-converted” method for calculating the fair value of such notes. This involved multiplying the number of shares into which the 2014 Notes convert (11,506,156 shares) by the Company’s stock price as of December 29, 2017 (the last trading day of the year). We performed an evaluation as to whether the as-converted method would yield a materially different result from the Least Squares Monte Carlo simulation model used in previous quarters and determined that it would not.
A reconciliation of the convertible notes and common stock warrant liability that are measured and recorded at fair value on a recurring basis using significant unobservable inputs (Level 3) is as follows:
|
|
Convertible |
|
|
Common Stock |
|
||
|
|
Notes Payable |
|
|
Warrant Liability |
|
||
|
Balance at December 31, 2014 |
$ |
37,780 |
|
|
$ |
15,389 |
|
|
Total unrealized losses on change in fair value |
|
37,585 |
|
|
|
19,203 |
|
|
Net settlements upon exercise of warrants |
|
— |
|
|
|
(14,970 |
) |
|
Balance at December 31, 2015 |
$ |
75,365 |
|
|
$ |
19,622 |
|
|
Total unrealized losses on change in fair value |
|
16,290 |
|
|
|
8,957 |
|
|
Net settlements upon exercise of warrants |
|
— |
|
|
|
(28,579 |
) |
|
Balance at December 31, 2016 |
$ |
91,655 |
|
|
$ |
— |
|
|
Net issuances |
|
40,954 |
|
|
|
6,666 |
|
|
Total unrealized gains on change in fair value |
|
(33,241 |
) |
|
|
(2,490 |
) |
|
Balance at December 31, 2017 |
$ |
99,368 |
|
|
$ |
4,176 |
|
9. Commitments and Contingencies
We license certain patents and other intellectual property rights related to the composition and coating of our bioresorbable scaffold and our other biomaterial products. Terms of these licenses include provisions for royalty payments on future sales of products, if any, utilizing this technology, with provisions for minimum royalties once product sales begin. The amount of royalties varies depending upon type of product, use of product, stage of product, location of sale, and ultimate sales volume, and ranges from a minimum of approximately $15 per unit sold to a maximum of approximately $50 per unit sold, with license provisions for escalating minimum royalties that could be
F-16
REVA Medical, Inc.
Notes to Consolidated Financial Statements
as high as $2.2 million per year. Additionally, in the event we sublicense the technology and receive certain milestone payments, the licenses require that up to 40 percent of the milestone amount be paid to the licensors.
Additional terms of the technology licenses include annual license fees of $175,000 until the underlying technology has been commercialized. Because we began commercial sales of our Fantom scaffold in July 2017, these annual license fees will not continue after 2017. Terms of the licenses also include other payments to occur during commercialization that could total $950,000, payment of $350,000 upon a change in control of ownership of the Company, payments of up to $300,000 annually to extend filing periods related to certain technology (of which, payments totaling up to $250,000 per year during the years 2016, 2017, and 2018 are being deferred to January 1, 2019; accordingly, $500,000 was accrued as a long-term liability at December 31, 2017), and payment of patent filing, maintenance, and defense fees. The license terms remain in effect until the last patent expires.
In connection with our operating and business activities, we periodically enter into contracts with consultants and suppliers. These contracts are generally cancelable by either party with 30 days’ prior written notice. As of December 31, 2017, the minimum future payments on these contracts totaled approximately $634,000.
We lease approximately 37,000 square feet of office and lab space for our corporate headquarters in San Diego, California. In October 2017, we amended this lease to extend the expiration date by 88 months from January 2018 to May 2025. Effective February 1, 2018, our monthly rent became $66,000 and it will increase every February by three percent. The amended lease also contains a leasehold improvement allowance of $787,000 and rent abatements of $274,000.
We record rent expense on a straight-line basis over the life of the lease; the difference between average rent expense and cash payments for rent is recorded as a deferred liability. As of December 31, 2017, our deferred rent totaled $17,000, which was classified as a current liability. We recorded rent expense of $758,000, $770,000, and $794,000 for the years ended December 31, 2017, 2016, and 2015, respectively.
Future minimum payments under the lease are as follows (in thousands):
|
|
Minimum |
|
|
|
Year Ending December 31, |
Payments |
|
|
|
2018 |
$ |
716 |
|
|
2019 |
|
741 |
|
|
2020 |
|
763 |
|
|
2021 |
|
786 |
|
|
2022 |
|
884 |
|
|
Therafter |
|
2,248 |
|
|
|
$ |
6,138 |
|
10. Capital Stock
Our certificate of incorporation, as amended, authorizes us to issue 100,000,000 shares of common stock, par value $0.0001 per share, 25,000,000 shares of Class B common stock, par value $0.0001 per share and 5,000,000 shares of undesignated preferred stock, par value $0.0001 per share. As of December 31, 2017 and 2016, 41,245,820 and 42,851,477, respectively, shares of common stock were outstanding and no shares of Class B common stock or undesignated preferred stock were outstanding.
Certain shareholders, as well as the holders of our convertible notes, if such convertible notes are converted into common stock, have the right to cause us to file a registration statement that would register the resale of such shares on their behalf and to include their shares in registration statements that we may file on behalf of other stockholders.
11. Stock-Based Compensation
The Plan: Our 2010 Equity Incentive Plan, as amended (the “Plan”), provides for grants of incentive and non-qualified stock options for purchase of our common stock at a price per share equal to the closing market price on the
F-17
REVA Medical, Inc.
Notes to Consolidated Financial Statements
date of grant, and for awards of restricted stock units (“RSUs”) and restricted stock, for which there is no consideration payable by a recipient. An RSU entitles the recipient to one share of our common stock upon vesting. All stock issuances under the Plan are made with new shares from our authorized but unissued common stock. The number of shares reserved under the Plan may be increased annually by up to three percent of our outstanding stock. On January 1, 2017, an additional 1,285,544 shares were added to the Plan, resulting in a total of 9,144,512 shares reserved for issuance under the Plan as of December 31, 2017. Option activity under the Plan is as follows:
|
|
Options Outstanding |
|
|
Weighted Average Exercise Price |
|
|
Weighted Average Remaining Contractual Term (Years) |
|
|
Aggregate Intrinsic Value |
|
||||
|
Balance at December 31, 2014 |
|
4,243,425 |
|
|
$ |
7.01 |
|
|
|
|
|
|
|
|
|
|
Granted |
|
2,152,500 |
|
|
$ |
4.50 |
|
|
|
|
|
|
|
|
|
|
Cancelled |
|
(232,292 |
) |
|
$ |
2.85 |
|
|
|
|
|
|
|
|
|
|
Exercised |
|
(251,208 |
) |
|
$ |
2.27 |
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2015 |
|
5,912,425 |
|
|
$ |
6.46 |
|
|
|
6.50 |
|
|
$ |
7,873,000 |
|
|
Granted |
|
570,100 |
|
|
$ |
8.22 |
|
|
|
|
|
|
|
|
|
|
Cancelled |
|
(106,834 |
) |
|
$ |
10.81 |
|
|
|
|
|
|
|
|
|
|
Exercised |
|
(247,499 |
) |
|
$ |
4.04 |
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2016 |
|
6,128,192 |
|
|
$ |
6.65 |
|
|
|
5.94 |
|
|
$ |
13,857,000 |
|
|
Granted |
|
897,100 |
|
|
$ |
6.80 |
|
|
|
|
|
|
|
|
|
|
Cancelled |
|
(783,123 |
) |
|
$ |
7.42 |
|
|
|
|
|
|
|
|
|
|
Exercised |
|
(121,678 |
) |
|
$ |
2.81 |
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2017 |
|
6,120,491 |
|
|
$ |
6.65 |
|
|
|
6.00 |
|
|
$ |
3,571,000 |
|
|
Exercisable at December 31, 2017 |
|
5,153,771 |
|
|
$ |
7.03 |
|
|
|
5.68 |
|
|
$ |
2,899,000 |
|
|
Vested at December 31, 2017 |
|
3,973,578 |
|
|
$ |
7.10 |
|
|
|
4.72 |
|
|
$ |
2,750,000 |
|
Employees, non-employee directors, and consultants are eligible to participate in the Plan. For purposes of determining stock-based compensation expense, we include non-employee directors with employees; we account for consultant compensation expense separately.
The term of awards granted under the Plan may not exceed ten years. Vesting periods of awards are determined by our board of directors.
A majority of the vesting periods of outstanding stock options is four years, with 25 percent vesting on the one-year anniversary of the vesting commencement date and 75 percent vesting in equal monthly installments thereafter. A majority of the options are exercisable at any time but, if exercised prior to vesting, are subject to a lapsing right of repurchase by us at the exercise price until fully vested. As of December 31, 2017 and 2016, no unvested options had been exercised and, therefore, no shares were subject to repurchase.
During March 2015, we granted a total of 316,000 options that vest based on achievement of certain performance milestones. We estimated the vesting term for each performance milestone on the date of grant, and on each reporting date thereafter, based on our internal timelines and operating projections. Our estimates of vesting ranged from approximately nine to 30 months at the grant date in March 2015; we estimated the weighted average remaining vesting term to be 12 months as of both December 31, 2017 and 2016. A total of 65 percent of these options had vested as of December 31, 2017. During the years ended December 31, 2017 and 2016, 63,000 and 12,250 unvested options were cancelled, respectively.
During 2013, we awarded 87,500 shares of restricted stock; 25 percent of each award vests on each annual anniversary date of the award. As of December 31, 2017, all of these awards had vested and none had been cancelled.
F-18
REVA Medical, Inc.
Notes to Consolidated Financial Statements
RSU activity under the Plan is as follows:
|
|
RSUs |
|
|
Performance |
|
|
Time |
|
|||
|
|
Outstanding |
|
|
Based |
|
|
Based |
|
|||
|
Balance at December 31, 2014 |
|
— |
|
|
|
— |
|
|
|
— |
|
|
Granted |
|
984,200 |
|
|
|
824,200 |
|
|
|
160,000 |
|
|
Cancelled |
|
— |
|
|
|
— |
|
|
|
— |
|
|
Vested |
|
— |
|
|
|
— |
|
|
|
— |
|
|
Balance at December 31, 2015 |
|
984,200 |
|
|
|
824,200 |
|
|
|
160,000 |
|
|
Granted |
|
47,800 |
|
|
|
— |
|
|
|
47,800 |
|
|
Cancelled |
|
(118,000 |
) |
|
|
(118,000 |
) |
|
|
— |
|
|
Vested |
|
(160,000 |
) |
|
|
— |
|
|
|
(160,000 |
) |
|
Balance at December 31, 2016 |
|
754,000 |
|
|
|
706,200 |
|
|
|
47,800 |
|
|
Granted |
|
397,300 |
|
|
|
162,500 |
|
|
|
234,800 |
|
|
Cancelled |
|
(526,000 |
) |
|
|
(479,200 |
) |
|
|
(46,800 |
) |
|
Vested |
|
(47,800 |
) |
|
|
— |
|
|
|
(47,800 |
) |
|
Balance at December 31, 2017 |
|
577,500 |
|
|
|
389,500 |
|
|
|
188,000 |
|
We estimated the vesting term for each performance-based RSU on the award date, and on each reporting date thereafter, based on our internal timelines and operating projections. As of December 31, 2017, we estimated the remaining weighted average vesting term to be 8.1 months for the RSUs granted in 2015 and 6 months for the RSUs granted in 2017.
Time-based RSUs generally vest over one year for non-employee directors and ratably over three years for employees.
No tax benefits arising from stock-based compensation have been recognized in our consolidated statements of operations through December 31, 2017.
Grants and Awards to Employees: We account for option grants, restricted stock awards, and RSUs to employees based on their estimated fair values on the date of grant or award, with the resulting stock-based compensation recorded over the requisite service period on a straight-line basis. The fair value of restricted stock and RSUs is equal to the closing market price of our common stock on the date of award. The fair value of option grants was estimated on the date of grant using the following weighted-average assumptions:
|
|
Year Ended December 31, |
|
|||||||||||
|
|
2017 |
|
|
|
|
2016 |
|
|
2015 |
|
|||
|
Risk-free interest rate |
2.2% |
|
|
|
1.6% |
|
|
1.8% |
|
||||
|
Expected volatility of common stock |
65.4% |
|
|
|
|
57.6% |
|
|
55.6% |
|
|||
|
Expected life in years |
|
6.21 |
|
|
|
|
|
6.13 |
|
|
|
6.16 |
|
|
Dividend yield |
0.0% |
|
|
|
|
0.0% |
|
|
0.0% |
|
|||
The assumed risk-free interest rate was based on the implied yield on a U.S. Treasury zero-coupon issue with a remaining term equal to the expected life of the option. The assumed volatility was calculated from the historical market prices of a selected group of publicly traded companies considered to be our peers; we use peer group data because we have limited historical trading data for our common stock, but adjusted the 2016 volatility upward by approximately ten percent to allow us to move toward using historical trading data for our common stock, which is more volatile than our peer group. In 2017, we began to use our historical trading price of our common stock; our common stock began trading on our IPO date of December 23, 2010, which provides approximately 7 years of history as December 31, 2017. For options that have time-based vesting, the expected option life was calculated using the simplified method under the accounting standard for stock compensation and a ten-year option expiration; we use the simplified method because we do not yet have adequate history as a public company traded on a U.S. stock exchange to establish a reasonable expected life. For options that have performance-based vesting, the expected life was calculated based on our internal timelines and operating projections. The expected dividend yield of zero reflects that we have not paid cash dividends since inception and do not intend to pay cash dividends in the foreseeable future.
F-19
REVA Medical, Inc.
Notes to Consolidated Financial Statements
The options granted during the years ended December 31, 2017, 2016 and 2015 had a weighted average grant date fair value of $4.15, $4.48 and $2.40, respectively.
The aggregate intrinsic value of options exercised during the years ended December 31, 2017, 2016 and 2015 was $362,000, $976,000 and $511,000, respectively.
For the options and RSUs that vest upon achievement of performance milestones, we record compensation expense for only those milestones that are probable of being achieved.
The vest-date fair value of RSUs that vested during the year ended December 31, 2017 and 2016 was $320,000 and $1.3 million, respectively. No RSUs vested in the year ended December 31, 2015.
Stock-based compensation arising from employee options and awards under the Plan is as follows (in thousands):
|
|
Year Ended December 31, |
|
|||||||||
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Research and development expense |
$ |
(109 |
) |
|
$ |
1,260 |
|
|
$ |
1,502 |
|
|
Selling, general and administrative expense |
|
2,156 |
|
|
|
3,423 |
|
|
|
1,905 |
|
|
|
$ |
2,047 |
|
|
$ |
4,683 |
|
|
$ |
3,407 |
|
In the years ended December 31, 2017 and 2016, we reversed stock-based compensation of $1.8 million and $163,000, respectively, related to unvested awards for employees that were terminated.
As of December 31, 2017, we had approximately $7.3 million of total unrecognized compensation costs related to unvested employee awards that are expected to be recognized over a weighted average period of 1.7 years.
Stock Options to Consultants: We account for stock options granted to consultants at their fair value. Under this method, the fair value is estimated at each reporting date during the vesting period using the Black-Scholes option valuation model. The resulting stock-based compensation expense, or income if the fair value declines in a reporting period, is recorded over the consultant’s service period.
Fully vested options to purchase 7,500 shares of common stock were granted to consultants during the year ended December 31, 2016. No options were granted to consultants during the years ended December 31, 2017 or 2015.
Consultant stock-based compensation expense is recorded to the financial statement line item for which the consultant’s services are rendered. Stock-based compensation expense arising from consultant options is as follows (in thousands):
|
|
Year Ended December 31, |
|
|||||||||
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Research and development expense |
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
Selling, general and administrative expense |
|
— |
|
|
|
40 |
|
|
|
27 |
|
|
|
$ |
— |
|
|
$ |
40 |
|
|
$ |
27 |
|
F-20
REVA Medical, Inc.
Notes to Consolidated Financial Statements
A reconciliation of the tax provision to the amount computed by applying the statutory federal rate to the net income/(loss) is summarized as follows (in thousands):
|
|
Year Ended December 31, |
|
|||||||||
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Federal income taxes at 34% |
$ |
2,426 |
|
|
$ |
(18,393 |
) |
|
$ |
(28,082 |
) |
|
State income taxes, net of federal benefit |
|
(226 |
) |
|
|
(1,484 |
) |
|
|
(1,513 |
) |
|
Research and development tax credits |
|
(556 |
) |
|
|
(889 |
) |
|
|
(650 |
) |
|
Changes in fair value of convertible notes and common stock warrant liability |
|
(11,972 |
) |
|
|
8,584 |
|
|
|
19,308 |
|
|
Increase in valuation allowance |
|
(24,704 |
) |
|
|
10,583 |
|
|
|
8,789 |
|
|
Accrued interest on convertible notes |
|
1,555 |
|
|
|
698 |
|
|
|
944 |
|
|
Expiration of state net operating losses |
|
— |
|
|
|
641 |
|
|
|
692 |
|
|
State rate adjustment |
|
1,411 |
|
|
|
— |
|
|
|
— |
|
|
Tax Cuts and Jobs Act of 2017 |
|
31,541 |
|
|
|
— |
|
|
|
— |
|
|
Stock-based compensation expense |
|
815 |
|
|
|
223 |
|
|
|
287 |
|
|
Other |
|
(290 |
) |
|
|
37 |
|
|
|
225 |
|
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
Our deferred income taxes reflect the net tax effects of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of our deferred tax assets and liabilities are as follows:
|
|
December 31, |
|
|||||
|
|
2017 |
|
|
2016 |
|
||
|
Net operating loss carryforwards |
$ |
57,278 |
|
|
$ |
80,382 |
|
|
Research and development credits |
|
9,636 |
|
|
|
8,422 |
|
|
Amortization |
|
4,422 |
|
|
|
4,302 |
|
|
Stock-based compensation expense |
|
3,723 |
|
|
|
6,766 |
|
|
Depreciation |
|
265 |
|
|
|
416 |
|
|
Accrued operating expenses |
|
11 |
|
|
|
22 |
|
|
Debt issuance costs |
|
479 |
|
|
|
142 |
|
|
Other |
|
196 |
|
|
|
262 |
|
|
|
|
76,010 |
|
|
|
100,714 |
|
|
Valuation allowance |
|
(76,010 |
) |
|
|
(100,714 |
) |
|
|
$ |
— |
|
|
$ |
— |
|
As of December 31, 2017, we had aggregate federal and California state net operating loss carryforwards of approximately $221.3 million and $154.6 million, respectively, which may be available to offset future taxable income for income tax purposes. The federal and California net operating loss carryforwards begin to expire in 2019 and 2027, respectively.
F-21
REVA Medical, Inc.
Notes to Consolidated Financial Statements
As of December 31, 2017, we also had federal and California state research tax credit carryforwards of approximately $7.5 million and $6.8 million, respectively. The federal research tax credit carryforwards begin to expire in 2020. The California state research tax carryforwards have no expiration.
Under Internal Revenue Code Sections 382 and 383, annual use of our net operating loss and research tax credit carryforwards to offset taxable income may be limited based on cumulative changes in ownership. An analysis of the impact of this provision from December 1, 1999 through December 31, 2017 has been performed and it was determined that, although ownership changes have occurred, the carryovers should be available for use by the Company before they expire, provided we generate sufficient future taxable income. Future ownership changes could result in limitations and may impact the realizability of these loss and credit carryforwards in future periods.
On December 22, 2017, new tax reform legislation in the U.S., known as the Tax Cuts and Jobs Act of 2017 (the “Act”) was signed into law. At December 31, 2017, the Company has not yet completed its accounting assessment for the tax effects of the enactment of the Act; however, as described below, the Company has made a reasonable estimate of the effects on the existing deferred tax balances.
As a result of the lower enacted corporate tax rate, the Company has remeasured certain deferred tax assets and liabilities based on the rates at which they are expected to reverse in the future, which is generally 21%. The provisional amount recorded related to the remeasurement of our deferred tax balance was $31.5 million that is fully offset by a corresponding decrease to our valuation allowance.
Staff Accounting Bulletin No. 118 ("SAB 118") was issued to address the application of US GAAP in situations when a registrant does not have the necessary information available, prepared, or analyzed (including computations) in reasonable detail to complete the accounting for certain income tax effects of the Act. In accordance with SAB 118, the Company has provisionally determined that there is no deferred tax benefit or expense with respect to the remeasurement of certain deferred tax assets and liabilities due to the full valuation allowance against net deferred tax assets. The Company is still analyzing certain aspects of the Act and refining its calculations, which could potentially affect the measurement of these balances or potentially give rise to new deferred tax amounts. Additional analysis of the law and the impact to the Company will be performed and any impact will be recorded in the respective quarter in 2018.
As of December 31, 2017, we had deferred tax assets of $76.0 million and have established a valuation allowance against those deferred tax assets due to the uncertainty surrounding our ability to generate future taxable income to realize those assets. The change in the valuation allowance for the years ended December 31, 2017 and 2016 was ($24.7 million) and $10.6 million, respectively.
We recognize a tax benefit from an uncertain tax position when it is more likely than not that the position will be sustained upon examination, including resolutions of any related appeals or litigation processes, based on the technical merits. Income tax positions must meet a more-likely-than-not recognition at the effective date to be recognized. As of December 31, 2017, the unrecognized tax benefits recorded were approximately $3.6 million. We do not anticipate a significant change in the unrecognized tax benefits within the next 12 months.
A reconciliation of the beginning and ending amount of gross unrecognized tax benefits for 2017 and 2016, excluding interest and penalties, is as follows:
|
|
December 31, |
|
|||||
|
|
2017 |
|
|
2016 |
|
||
|
Balance at beginning of year |
$ |
3,345 |
|
|
$ |
4,298 |
|
|
Additions (reductions) for prior year tax positions |
|
— |
|
|
|
(1,297 |
) |
|
Additions for current year tax positions |
|
222 |
|
|
|
344 |
|
|
|
$ |
3,567 |
|
|
$ |
3,345 |
|
Due to our valuation allowance position, none of the unrecognized tax benefits, if recognized, will impact our effective tax rate. Our policy is to record interest and penalties within tax expense. As of December 31, 2017 and 2016, we had no accrued interest or penalties related to uncertain tax positions.
F-22
REVA Medical, Inc.
Notes to Consolidated Financial Statements
The Company is subject to taxation in the U.S. federal and state jurisdictions. As of December 31, 2017, the Company is no longer subject to U.S. federal and state examinations by tax authorities for years before 2012 and 2011, respectively. However, to the extent allowed by law, the tax authorities may have the right to examine prior periods where net operating losses were generated and carried forward, and make adjustments up to the amount of the net operating loss carryforward amount. The Company is not currently under IRS, state or local tax examination.
13. Net Income (Loss) Per Common Share
Basic net income (loss) per common share is calculated by dividing the net income (loss) attributable to common stockholders by the weighted average number of common shares outstanding for the period, without consideration for common stock equivalents. Diluted net loss per share is computed by dividing the net loss attributable to common stockholders by the weighted average number of common share equivalents outstanding for the period determined using the treasury-stock method and the if-converted method, as applicable. For this calculation, common stock options and restricted stock subject to forfeiture are considered to be common stock equivalents; common stock equivalents are used in the calculation of diluted net loss per share only when their effect is dilutive.
Basic net income (loss) per share reconciles to fully diluted net loss per share as follows (dollars in thousands):
|
|
Year Ended December 31, |
|
|||||||||||
|
|
2017 |
|
|
2016 |
|
|
|
|
2015 |
|
|||
|
Diluted Net Loss: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income (loss) used for basic net income (loss) per share |
$ |
7,134 |
|
|
$ |
(54,098 |
) |
|
|
|
$ |
(82,594 |
) |
|
Interest expense on 2014 convertible notes |
|
2,202 |
|
|
|
— |
|
|
|
|
|
— |
|
|
Gain on change in fair value of 2014 convertible notes |
|
(30,672 |
) |
|
|
— |
|
|
|
|
|
— |
|
|
|
$ |
(21,336 |
) |
|
$ |
(54,098 |
) |
|
|
|
$ |
(82,594 |
) |
|
Weighted Average Shares Used to Compute Diluted Net Loss per Share: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Shares used for basic net income (loss) per share |
|
41,811,326 |
|
|
|
42,120,545 |
|
|
|
|
|
34,680,634 |
|
|
Common share equivalents – assumed conversion of 2014 convertible notes (see Note 7) |
|
11,506,156 |
|
|
|
— |
|
|
|
|
|
— |
|
|
|
|
53,317,482 |
|
|
|
42,120,545 |
|
|
|
|
|
34,680,634 |
|
The following weighted average shares were excluded from the computations of diluted net loss per share because including them would have been antidilutive.
|
|
Year Ended December 31, |
|
|||||||||
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Weighted Average Shares Excluded: |
|
|
|
|
|
|
|
|
|
|
|
|
Options to purchase common stock |
|
6,352,118 |
|
|
|
6,355,093 |
|
|
|
4,812,372 |
|
|
Unvested restricted stock |
|
5,171 |
|
|
|
31,528 |
|
|
|
61,623 |
|
|
Restricted stock units |
|
726,379 |
|
|
|
882,779 |
|
|
|
768,908 |
|
|
Warrants to purchase common stock |
|
1,334,749 |
|
|
|
502,049 |
|
|
|
7,647,260 |
|
|
Common share equivalents of convertible notes |
|
3,427,047 |
|
|
|
11,506,156 |
|
|
|
11,506,156 |
|
|
|
|
11,845,464 |
|
|
|
19,277,605 |
|
|
|
24,796,319 |
|
14. Retirement Plan
In 2003 we adopted a qualified 401(k) profit sharing plan (the “401(k) Plan”) for the benefit of our employees. Employees are eligible to participate in the 401(k) Plan the month following hire and may defer up to the maximum
F-23
REVA Medical, Inc.
Notes to Consolidated Financial Statements
allowed under IRS regulations, on an annual basis. We match 25 percent of an employee’s deferral amount, up to a maximum of four percent of qualified compensation. We may, at our discretion, make additional contributions. Employees are immediately vested in the employer matching contributions. Our contributions to the 401(k) Plan were $45,000, $49,000, and $42,000 for the years ended December 31, 2017, 2016, and 2015, respectively.
15. Related Parties
Our related parties include the members of our board of directors, investors with five percent or more of our outstanding shares of common stock, and holders of our convertible notes. Other than approved board compensation, the amendment to the 2014 Notes, issuance of the 2017 Notes and warrants and exercise of warrants to purchase common stock (all discussed above), we had no related party transactions during the years ended December 31, 2017 and 2016. See Note 7 Convertible Notes Payable and Warrants to Purchase Common Stock.
16. Selected Quarterly Financial Information
The following table presents selected quarterly financial information that has been derived from our unaudited quarterly consolidated financial statements, which, in the opinion of management, include all adjustments (consisting only of normal recurring items) necessary for a fair presentation. The quarterly per share data presented below was calculated separately and may not sum to the annual figures presented in the consolidated financial statements. These operating results are also not necessarily indicative of results for any future period.
|
|
Quarter Ended |
|
|
Year Ended |
|
||||||||||||||
|
|
March 31, |
|
|
June 30, |
|
|
September 30, |
|
|
December 31, |
|
|
December 31, |
|
|||||
|
2017 (unaudited) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue |
$ |
— |
|
|
$ |
— |
|
|
$ |
17 |
|
|
$ |
28 |
|
|
$ |
45 |
|
|
Loss from operations |
|
(6,066 |
) |
|
|
(5,060 |
) |
|
|
(4,769 |
) |
|
|
(5,434 |
) |
|
|
(21,329 |
) |
|
Gain on change in fair values |
|
8,138 |
|
|
|
8,178 |
|
|
|
12,304 |
|
|
|
7,111 |
|
|
|
35,731 |
|
|
Net income (loss) |
|
1,424 |
|
|
|
(503 |
) |
|
|
6,053 |
|
|
|
160 |
|
|
|
7,134 |
|
|
Basic net income (loss) per common share |
$ |
0.03 |
|
|
$ |
(0.01 |
) |
|
$ |
0.15 |
|
|
$ |
0.00 |
|
|
$ |
0.17 |
|
|
Diluted net loss per common share |
$ |
(0.11 |
) |
|
$ |
(0.01 |
) |
|
$ |
(0.04 |
) |
|
$ |
(0.10 |
) |
|
$ |
(0.40 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2016 (unaudited) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from operations |
$ |
(7,481 |
) |
|
$ |
(7,031 |
) |
|
$ |
(6,149 |
) |
|
$ |
(6,119 |
) |
|
$ |
(26,780 |
) |
|
Gain (loss) on change in fair values |
|
(32,764 |
) |
|
|
2,966 |
|
|
|
(17,269 |
) |
|
|
21,820 |
|
|
|
(25,247 |
) |
|
Net income (loss) |
|
(40,798 |
) |
|
|
(4,555 |
) |
|
|
(23,943 |
) |
|
|
15,198 |
|
|
|
(54,098 |
) |
|
Basic net income (loss) per common share |
$ |
(1.01 |
) |
|
$ |
(0.11 |
) |
|
$ |
(0.56 |
) |
|
$ |
0.36 |
|
|
$ |
(1.28 |
) |
|
Diluted net loss per common share |
$ |
(1.01 |
) |
|
$ |
(0.11 |
) |
|
$ |
(0.56 |
) |
|
$ |
(0.11 |
) |
|
$ |
(1.28 |
) |
For the quarterly periods provided above, when the Company recognized net income, diluted net loss per share is computed by dividing the net loss attributable to common stockholders by the weighted average number of common share equivalents outstanding for the period determined using the treasury-stock method and the if-converted method, as applicable. For purpose of this calculation, common stock options and restricted stock subject to forfeiture are considered to be common stock equivalents; common share equivalents are included in the calculation of diluted net loss per share only when their effect is dilutive.
F-24
REVA Medical, Inc.
Notes to Consolidated Financial Statements
Basic net income per share reconciles to fully diluted net loss per share as follows (dollars in thousands):
|
|
Quarter Ended |
|
|||||||||||||||
|
|
March 31, 2017 |
|
|
September 30, 2017 |
|
|
December 31, 2017 |
|
|
December 31, 2016 |
|
||||||
|
Diluted Net Loss: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
Net income used for basic net income per share |
$ |
1,424 |
|
|
$ |
6,053 |
|
|
$ |
160 |
|
|
$ |
15,198 |
|
||
|
Interest expense on 2014 convertible notes payable |
|
538 |
|
|
|
550 |
|
|
|
571 |
|
|
|
531 |
|
||
|
Gain on change in fair value of 2014 convertible notes payable |
|
(8,138 |
) |
|
|
(8,741 |
) |
|
|
(6,213 |
) |
|
|
(21,820 |
) |
||
|
|
$ |
(6,176 |
) |
|
$ |
(2,138 |
) |
|
$ |
(5,482 |
) |
|
$ |
(6,091 |
) |
||
|
Weighted Average Shares Used to Compute Diluted Net Loss per Share: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
Shares used for basic net income per share |
|
42,838,158 |
|
|
|
41,197,348 |
|
|
|
41,245,820 |
|
|
|
42,747,769 |
|
||
|
Common share equivalents |
|
11,506,156 |
|
|
|
11,506,156 |
|
|
|
11,506,156 |
|
|
|
11,506,156 |
|
||
|
|
|
54,344,314 |
|
|
|
52,703,504 |
|
|
|
52,751,976 |
|
|
|
54,253,925 |
|
||
F-25