Attached files
| file | filename |
|---|---|
| EX-32 - EXHIBIT 32 - Accelerate Diagnostics, Inc | axdx-12312017xexh32.htm |
| EX-31.2 - EXHIBIT 31.2 - Accelerate Diagnostics, Inc | axdx-12312017xexh312.htm |
| EX-31.1 - EXHIBIT 31.1 - Accelerate Diagnostics, Inc | axdx-12312017xexh311.htm |
| EX-23.1 - EXHIBIT 23.1 - Accelerate Diagnostics, Inc | axdx-12312017xexh231.htm |
| EX-21 - EXHIBIT 21 - Accelerate Diagnostics, Inc | axdx-12312017xexh21.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
þ ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2017
o TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
Commission file number: 001-31822
ACCELERATE DIAGNOSTICS, INC.
(Exact name of registrant as specified in its charter)
Delaware | 84-1072256 |
(State or other jurisdiction | (I.R.S. Employer Identification No.) |
3950 South Country Club, Suite 470 | |
Tucson, Arizona | 85714 |
(Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code:
(520) 365-3100
Securities registered pursuant to Section 12(b) of the Act:
Title of each class | Name of each exchange on which registered |
Common Stock, $0.001 par value per share | The NASDAQ Stock Market LLC (NASDAQ Capital Market) |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. o Yes þ No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. o Yes þ No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. þ Yes o No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§229.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). þ Yes o No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer | þ | |
Accelerated filer | o | |
Non-accelerated file | o (Do not check if a smaller reporting company) | |
Smaller reporting company | o | |
Emerging growth company | o | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. o
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). o Yes þ No
The aggregate market value of the shares of the registrant’s common stock held by non-affiliates on June 30, 2017, the last day of the registrant’s most recently completed second fiscal quarter, was approximately $841.8 million based on the closing price quoted on the NASDAQ Stock Market.
There were 55,766,359 shares of common stock of the registrant outstanding as of February 23, 2018.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive proxy statement relating to the registrant’s 2018 Annual Meeting of Stockholders are incorporated by reference in Part III of this Form 10-K.
TABLE OF CONTENTS
2
Introductory Note
Except as otherwise indicated by the context, references in this Annual Report on Form 10-K (this “Form 10-K“) to the “Company,” “Accelerate,” “we,” “us” or “our” are references to the combined business of Accelerate Diagnostics, Inc.
The Accelerate Pheno™ system is also generically referred to herein as the “ID/AST System” or “Accelerate ID/AST System.”
Forward-Looking Statements
This Annual Report on Form 10-K contains certain forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”) and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and the Company intends that such forward-looking statements be subject to the safe harbors created thereby. These forward-looking statements, which can be identified by the use of words such as “may,” “will,” “expect,” “anticipate,” “estimate,” or “continue,” or variations thereon or comparable terminology, include the plans and objectives of management for future operations, including plans and objectives relating to the products and future economic performance of the Company. In addition, all statements other than statements of historical facts that address activities, events, or developments the Company expects, believes, or anticipates will or may occur in the future, and other such matters, are forward-looking statements.
The forward-looking statements included herein are based on current expectations that involve a number of risks and uncertainties. These forward-looking statements are based on assumptions that the Company will retain key management personnel, the Company will be able to protect its intellectual property, the Company’s ability to respond to technological change, that the Company will accurately anticipate market demand for the Company’s products and that there will be no material adverse change in the Company’s operations or business. Assumptions relating to the foregoing involve judgments with respect to, among other things, future economic, competitive and market conditions and future business decisions, all of which are difficult or impossible to predict accurately and many of which are beyond the control of the Company. Although the Company believes that the assumptions underlying the forward-looking statements are reasonable, any of the assumptions could prove inaccurate and, therefore, there can be no assurance that the results contemplated in forward-looking statements will be realized. We undertake no obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events or otherwise.
The following discussion should be read in conjunction with the Company’s audited financial statements and related notes included elsewhere herein. The Company’s future operating results may be affected by various trends and factors which are beyond the Company's control. These include, among other factors, general public perception of issues and solutions, and other uncertain business conditions that may affect the Company’s business. The Company cautions the reader that a number of important factors discussed herein, and in other reports, filed with the Securities and Exchange Commission, including but not limited to the risks in the section entitled “Risk Factors” in this Form 10-K, could affect the Company’s actual results and cause actual results to differ materially from those discussed in forward-looking statements.
Industry and other data
We obtained the industry, statistical and market data from our own internal estimates and research as well as from industry and general publications and research, surveys and studies conducted by third parties. Industry publications, studies and surveys generally state that they have been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. While we believe that each of these studies and publications is reliable, we have not independently verified statistical, market and industry data from third-party sources. While we believe our internal Company research is reliable and the market definitions are appropriate, neither such research nor these definitions have been verified by any independent source.
3
PART I
Item 1. Business
Overview
Accelerate Diagnostics, Inc. (“Accelerate”) is an in vitro diagnostics company dedicated to providing solutions that improve patient outcomes and lower healthcare costs through the rapid diagnosis of serious infections. Microbiology laboratories are in need of new tools to address what the U.S. Centers for Disease Control and Prevention calls one of the most serious healthcare threats of our time, antibiotic resistance. A significant contributing factor to the rise of resistance is the overuse and misuse of antibiotics, which is exacerbated by a lack of timely diagnostic results. The delay of identification and antibiotic susceptibility results is often due to the reliance by microbiology laboratories on traditional culture-based tests that often take two to three days to complete. Our technology platform is built to address these challenges by delivering significantly faster and more accurate testing of infectious pathogens in various patient sample types.
Since 2004, we have focused our efforts on the research, development, and commercialization of an innovative rapid diagnostic platform, the Accelerate Pheno™ system. The Accelerate Pheno™ system utilizes genotypic technology to identify (ID) infectious pathogens and phenotypic technology to conduct antibiotic susceptibility testing, (AST) which determines whether live bacterial and fungal cells are resistant or susceptible to a particular antibiotic. The Accelerate PhenoTest™ BC Kit provides ID and AST results for patients suspected of bacteremia or fungemia, both life-threatening conditions with high morbidity and mortality risk. The Accelerate PhenoTest™ BC Kit is a highly multiplexed panel targeting over 80% of the routine and significant pathogens causing blood stream infections and over 90% of the antibiotics useful in treating those pathogens.
On June 30, 2015, we declared our conformity to the European In Vitro Diagnostic Directive 98/79 EC and applied a CE Mark to the Accelerate Pheno™ system and the Accelerate PhenoTest™ BC Kit for in vitro diagnostic use. On February 23, 2017, the U.S. Food and Drug Administration (“FDA”) granted our de novo request to market our Accelerate Pheno™ system and Accelerate PhenoTest™ BC Kit.
In 2017, we began selling the Accelerate Pheno™ system in hospitals in the United States, Europe, and the Middle East. Consistent with the Company's business model revenues were generated from the sale of the instrument and the sale of single use consumable test kits.
History
We were incorporated in 1982 in Colorado under the name Sage Resources Corp., and through a series of subsequent transactions, we became Accelerate Diagnostics, Inc., a Delaware corporation, in December 2012.
From 2001 to 2012, we focused primarily upon furthering the research and development of the OpTest portfolio of technologies (“OpTest”) that we acquired from DDx, Inc. in 2001 and the development of revenue producing products related to that technology. The purchase of OpTest provided us with a proprietary surface chemistry formulation, which led to our OptiChem and other surface chemistry products, and quantitative bio-analytical measurement instruments.
In 2012, our Board of Directors and management team established a new strategic direction for the Company, which was (1) to focus on the internal development, manufacture, and commercialization of the Accelerate Pheno™ system and (2) to discontinue efforts to develop and actively market OptiChem and our other surface chemistry products. Our Board of Directors and management team decided to pursue this new strategic direction based on the belief that we could internally develop and commercialize the Accelerate Pheno™ system, formerly called the BacCel System.
Since the adoption of the new strategic direction in 2012, we have made significant investments in research and development personnel, facilities, equipment, and consumables to support the internal development of the Accelerate Pheno™ system. The Company has also invested in the hiring of regulatory, manufacturing, quality, sales, and marketing personnel experienced in the manufacture and commercialization of medical devices.
This strategic direction required the Company to raise additional capital. In June 2012, the Company raised
4
$14.4 million through the sale of common stock to Abeja Ventures, LLC. In March 2013, the Company obtained additional capital through the exercise of warrants issued to Abeja Ventures, LLC in the aggregate amount of $20.1 million. In August 2013, the Company completed a rights offering that raised gross proceeds of $20.0 million. In April 2014, the Company completed a rights offering that raised gross proceeds of $45.0 million. In December 2015, the Company completed a publicly marketed common stock offering that raised gross proceeds of $109.3 million. In May 2017, the Company completed another publicly marketed common stock offering that raised additional gross proceeds of $89.0 million.
Clinical Need
Antibiotic resistance has a significant healthcare impact, costing the U.S. an estimated $55 billion per year in healthcare and productivity costs. This estimate includes $20 billion in direct costs and $35 billion in indirect costs, such as lost productivity and sick days. Increasing infection rates and misuse of antibiotics results in serious treatment complications. Recent studies have shown that the number of hospital-acquired infections in the United States ranges from 214,700 to 1.4 million per year, contributing to an estimated 75,000 deaths per year. According to the CDC, there are approximately 2 million illnesses per year attributable to antibiotic resistance. Moreover, inappropriate antibiotic use is widespread. Of the approximately 35 million patients admitted to U.S. hospitals each year, 56% are put on empiric antibiotic therapy, of which more than half are on inappropriate or unnecessary antibiotics.
AST is designed to address these challenges by delivering significantly faster and more accurate testing of infectious pathogens in various patient sample types. Studies have shown that even a modest decrease in the time it takes to deliver an AST result correlates to reduced length and cost of hospital stay per patient. One such study showed that a five hour reduction in the time to receive an AST result delivered a two-day reduction in length of stay and a reduction in patient treatment costs of $1,750 per patient. Based on our analysis, we estimate that the Accelerate Pheno™ system is capable of delivering clinically-actionable results in approximately 19 hours from the time a blood sample is received by the laboratory, while current solutions often require 2-3 days to deliver these results. Recent studies have established that the Accelerate Pheno™ system is between 24 and 36 hours faster to identification results and 36 to 54 hours faster to AST results.
Market Opportunity
Recent reports have indicated estimated compound annual growth rates in the Global Hospital Acquired Infections Testing Market, which we believe includes the market segment in which we operate, of 19.1% over the next decade. We believe this growth is driven by the entrance of new technologies coupled with higher volumes due to enhanced screening, immune-compromised patients and increasing challenges with multidrug-resistant organisms. Across North America, Europe and Asia Pacific geographies, we estimate there are over 16 million high-acuity tests completed annually across various sample types including blood, respiratory, urine, intra-abdominal and other sterile body fluids. We estimate there are over 5 million high-acuity tests annually for blood culture samples globally, including over 4 million in North America, Europe and various countries in the Asia Pacific region.
In addition, based on information compiled from various competitor annual reports and other publicly available information, as well as our own estimates, we believe there are nearly 20,000 global instrument placements currently, consisting of approximately 10,000 bioMerieux Vitek 2® automated instrument installations, 6,000 Danaher Microscan® Systems installations and 4,000 instruments from other companies. We believe these placements reflect the number of potential placements for the Accelerate Pheno™ system.
Certain recent government initiatives are complementary to the Accelerate Pheno™ system. Centers for Medicare and Medicaid Services (“CMS”) programs designed to decrease hospital-acquired infections directly impact hospital budgets via reimbursement cuts, incentivizing providers to enhance infection-management protocols. These programs include the Medicare Hospital-Acquired Condition Reduction Program and the Hospital Readmissions Reduction Program. Similarly, on March 27, 2015, the White House released the National Action Plan for Combating Antibiotic-Resistant Bacteria, which directly and indirectly promotes rapid susceptibility testing. The plan identifies several milestones to accomplish this goal, such as calling on the National Institutes of Health to fund new projects and provide prizes aimed at the development of rapid diagnostic tests that characterize antibiotic susceptibility and improve antibiotic stewardship; mandating implementation of antibiotic stewardship programs by all hospitals participating in Medicare and Medicaid; and calling on the FDA and CMS evaluate new regulatory pathways to promote development and adoption of innovative infectious disease diagnostics. The antibiotic stewardship programs are being implemented by many hospitals in advance of the 2020 implementation deadline.
5
Products
The Accelerate Pheno™ system is the Company’s first in vitro diagnostic platform and is intended for the identification and antibiotic susceptibility testing of pathogens most commonly associated with serious or health care-associated infections, including Gram-positive and Gram-negative organisms as well as yeast. The system leverages long-accepted bacteriological testing principles enhanced by proprietary technology and automation enabling the analysis of live microbial cells. It detects and identifies pathogens directly from a single patient sample followed by antimicrobial susceptibility testing based on the identification results. Antimicrobial susceptibility is determined by morphokinetic cellular analysis (“MCA”), a process that evaluates the change of individual cells and microcolonies in response to a range of antibiotics over time. The system’s combined technologies and automation dramatically reduce the need for time-consuming traditional bacterial culturing, thus eliminating the major source of delay with current testing methods. Identification results are typically available within 90 minutes of presenting the patient sample to the system, and susceptibility results, including minimum inhibitory concentrations (“MIC”), are available about 5 five hours after identification results. In the case of our first test kit for positive blood culture samples, a blood culture screening step is required, which we estimate takes an average of approximately twelve hours to complete before the sample is introduced to the Accelerate Pheno™ system. This combined turnaround time is a significant improvement over the multiple days currently required to obtain AST results, with MIC details, using conventional testing methods.
The Accelerate Pheno™ system features walk-away automation and consists of a fixed instrument and proprietary single-use test kit. The instrument consists of module(s) connected to a single analysis computer, which allows hospitals to acquire various numbers of modules to address their particular test volume. In order to run a patient sample on the Accelerate Pheno™ system a laboratory technician would pipette the patient sample into our system, insert the Accelerate PhenoTest™ BC Kit, and initiate the run. In the case of our initial test, a positive blood culture sample is introduced to the system through pipetting directly from the blood culture bottle into our Accelerate PhenoTest™ BC Kit.
The Accelerate Pheno™ system is the result of over a decade of technology development and several years of instrument design and engineering. The system comprises of custom-engineered functional components, including a robotic pipettor for fluidic manipulation, an optical system with both dark-field and fluorescent illumination, and an imaging system. These sensor components, among others, are used in the four processes that follow, each of which is a crucial component in delivering the rapid ID and AST results.
These processes include:
• | Automated specimen preparation. The initial step in the process is the automated purification of samples through an on-board and proprietary process to separate live organisms from sample debris. |
• | Live-cell immobilization. Following preparation, the purified sample is moved to the imaging cassette where pathogens are immobilized onto the cassette surface such that they can be imaged and analyzed in a stationary position during the identification and antibiotic susceptibility testing. |
• | Identification testing via fluorescent in situ hybridization (FISH). The now immobilized cells are tested with our proprietary FISH probes to enable identification. Because the genetic sequences of bacteria are distinctive, the binding of fluorescently labeled probes indicates the presence of a specific target sequence of RNA associated with a single or group of bacterial species or yeasts. When the probe finds a targeted sequence, it binds to it—generating a fluorescent signal—which is visible by the imaging system on the Accelerate Pheno™ system. Positive fluorescent signals from more than one target probe indicate polymicrobial samples and a universal bacterial stain discriminates target from non-target bacteria or fungi. The identification result is presented on the Accelerate Pheno™ system's graphic user interface in approximately 90 minutes from the introduction of the sample into the Accelerate Pheno™ system. |
• | Susceptibility testing via live-cell optical analysis. With the identification of the pathogen known, the system’s software determines the antibiotic panel to be used for susceptibility testing. These antibiotics, growth media, and additional patient sample are introduced to additional channels on the optical cassette. Finally, our proprietary imaging platform and algorithms determine the minimum inhibitory concentration of the bacteria by observing which antibiotics arrested live cell growth and led to cell death and which antibiotics were ineffective in ceasing live cell growth. The susceptibility test result is presented approximately five hours after the conclusion of the identification test. |
6
The Accelerate Pheno™ system has been the subject of dozens of scientific posters and studies. Recent studies and associated publications have covered subjects including time savings, performance, and opportunity rates for clinical interventions. Published study abstracts and links to full papers are available on our website at http://acceleratediagnostics.com/updates/#publications.
Research and Development
The Company plans to continue making significant investments in the research and development of new applications for existing technologies and in the research and development of new complementary technologies.
Since the completion and launch of the Accelerate Pheno™ system and Accelerate PhenoTest™ BC Kit, the Company has focused on product improvements and the development of additional test kits to address opportunities in sample types including but not limited to respiratory, urine, intra-abdominal and other sterile body fluids. Similar to the Accelerate PhenoTest™ BC Kit, the objective is to develop test kits that work seamlessly on the Accelerate Pheno™ system and deliver substantial benefits to the microbiology laboratory and to physicians in the treatment of serious infections. Proof of concept research has already been completed on some of these sample types, which are in various stages of research and development.
We anticipate seeking separate regulatory approval for each additional test kit. If and when we determine that we will pursue regulatory approvals for those applications, we would likely include the identification of the most prevalent infectious pathogens found in each specimen type and the applicable antimicrobial agents for treatment.
Our research activity is also focused on shortening the time required to distinguish between positive and negative whole blood and potentially urine samples prior to using the Accelerate Pheno™ system.
The Company's research and development expense for the years ended December 31, 2017, 2016 and 2015, is included in the consolidated statement of operations and comprehensive loss.
Intellectual Property
We rely on a combination of patent, copyright, trademark and trade secret laws, employee and third-party non-disclosure agreements, license agreements, and other intellectual property protection methods to protect our proprietary rights. We intend to continue developing intellectual property, and we intend to aggressively protect our position in key technologies. Our patented technology covers key components of the Accelerate Pheno™ system and is, thus, very important to the Company. Our patents are focused on several key technologies, including our automated process for sample preparation, automated immobilization process, and methods for imaging and analysis of individual pathogen cells. The Company’s first patent on the Accelerate Pheno™ system technology, U.S. Patent No. 7,341,841 titled “Rapid Microbial Detection and Antimicrobial Susceptibility Testing” was issued on March 11, 2008. The patent specification covers methods used to derive identification and antibiotic susceptibility from tests on individual immobilized bacterial cells. As of December 31, 2017, we had 36 issued patents worldwide, including 17 patents issued in the United States and 19 issued outside the United States. Our patents are set to expire on various dates in 2022 through 2032. Additionally, as of December 31, 2017, we had 24 patent applications pending worldwide, including 11 U.S. applications and 13 applications outside the United States. The Company believes that its patent suite would make it difficult for any other company to conduct rapid antibiotic susceptibility testing of individual pathogens utilizing our technology. From a trademark perspective, we had 20 pending and 15 registered marks protecting our brand and prospective products both domestically and internationally.
Sales, Marketing, and Distribution
The target customer for our products is hospital microbiology laboratories that perform identification and antibiotic susceptibility testing. Globally, the vast majority of hospitals with an intensive care unit have such a laboratory. In general, we utilize our own sales force to market the Accelerate Pheno™ system directly to our targeted customers. However, in select geographies, we plan to use third-party distributors to market the product.
The business while not seasonal is influenced by the timing of hospital budget and tender approval cycles which vary by geography. Due to the relatively long sales cycles back-logs are not typical and inventory is managed based on an estimation of demand forecasts.
7
Competition
To the best of our knowledge, no other company has a single product with capabilities matching those of the Accelerate Pheno™ system. The leading companies with automated microbiological testing products include Becton, Dickinson and Company (“BD”), bioMerieux, Danaher Corporation (“Danaher”), Bruker Corporation, Luminex Corporation, T2 Biosystems, Abbott and Thermo Fisher Scientific’s subsidiary TREK Diagnostics Systems, Inc. (“TREK”). These companies provide products for the broad-based culturing and analysis of a wide variety of bacteria. However, only BD, bioMerieux, Danaher and TREK market products that perform AST, and we believe that none of our competitors’ products is able to provide AST results as quickly as the Accelerate Pheno™ system.
Our competitors’ AST products require purified bacterial strains or “isolates” for analysis, which require at least overnight culturing of a sample to produce enough organisms to test. We believe these standard culturing methods, including enrichment growth and colony isolation, cannot achieve the speed that the Accelerate Pheno™ system provides.
Potential competitors for rapid AST have recently made announcements at various trade shows, including - but not limited to - Quantamatrix and Lifescale. While we do not have visibility into all of these companies’ respective stages of development, we believe these are early stage and will require years to achieve FDA approval.
Industry Developments
The clinical microbiology industry is subject to rapid technological changes, and new products are frequently introduced for rapid bacterial identification using genes or other molecular markers (“molecular diagnostics”). Numerous acquisitions, licenses, and distribution arrangements have been announced over the last few years for such products. However, we do not believe that any of these technologies offers the advantages afforded by the Accelerate Pheno™ system. For example, gene detection can be highly sensitive and specific for the identification of pathogens, but very few antibiotic resistance mechanisms are simple enough to allow accurate guidance for drug selection. Even in those rare instances that have a direct relationship between a gene and effective resistance, such as particular Methicillin-Resistant Staphylococcus Aureus (MRSA) strains, leading literature has reported novel mutations that escape detection by recently commercialized tests.
Fundamental biological limitations arise from the complexity of the majority of drug resistance expression mechanisms. This complexity precludes direct interpretation of molecular marker presence or absence and extrapolating to prescription guidance. Many new diagnostic technologies also require prior isolation of cultured colonies in order to assure accuracy. The time required to obtain such isolates, with a minimum of overnight turnaround, prevents these technologies from serving as rapid diagnostics for treatment decision support.
Another new technology receiving wide attention is mass spectrometry, and particularly the matrix-assisted laser desorption ionization time of flight version (“MALDI-TOF”), such as the Biotyper® system from Bruker Corporation. Bruker Corporation has agreements with a number of companies for distribution, including BD, TREK, and Siemens. bioMerieux has a similar system for distribution with Shimadzu Corporation. These systems build an empiric database from protein spectra acquired from many thousands of purified bacterial and fungal strains. They require a pure strain isolate for analysis and enrichment culturing to produce enough material to analyze. Some research papers on these systems report attempts to directly analyze isolate or blood culture smears, but results are not as reliable as those from samples prepared using a cleanup process to produce crude protein extracts.
MALDI-TOF systems have a major advantage over other molecular methods in identifying a very broad range of organisms. Cost of ownership is also substantially below that of older molecular methods. But the requirement for extensive organism enrichment and purification, as well as the inability to quantify live organisms or distinguish samples derived from viable organisms, substantially limits this technology from time-critical decision support. In this respect, the Accelerate Pheno™ system provides a substantial advantage for more rapid test results. Finally, as with the older molecular methods, MALDI-TOF systems cannot identify major drug resistance expression and faces the same fundamental biological barriers as gene detection.
Government Regulation
Our products under development and our operations are subject to significant government regulation. In the United States, our products are regulated as medical devices by the FDA and other federal, state, and local regulatory authorities.
8
FDA Regulation of Medical Devices
The FDA and other U.S. and foreign governmental agencies regulate, with respect to medical devices:
• | design, development, manufacturing, and storage; |
• | testing, content, and language of instructions for use and storage; |
• | labeling; |
• | pre-clinical testing and clinical trials; |
• | product safety; |
• | advertising, promotion, marketing, sales, and distribution; |
• | pre-market clearance and approval; |
• | record-keeping procedures; |
• | advertising and promotion; |
• | recalls and corrective field actions; |
• | post-market reporting, including reporting of deaths, serious injuries, and malfunctions that, if they were to recur, could lead to death or serious injury; |
• | post-market studies and surveillance; and |
• | product import and export. |
In the United States, numerous laws and regulations govern all the processes by which medical devices are brought to market and marketed. These include the Federal Food, Drug and Cosmetic Act (the “FDCA”) and the FDA's regulations implementing the law codifying the FDCA.
FDA Pre-market Clearance and Approval Requirements
Each medical device we seek to commercially distribute in the United States must first receive 510(k) clearance, approval of a reclassification petition or de novo classification request, or pre-market approval from the FDA, unless specifically exempted by the FDA. The FDA categorizes medical devices into one of three classes - Class I, II, or III - based on their risks and the regulatory controls necessary to provide a reasonable assurance of safety and effectiveness. Class I devices generally pose the lowest risk to the patient and/or user and Class III devices pose the highest risk. Regulatory control increases from Class I to Class III. The device classification regulation defines the regulatory requirements for a general device type. Generally, in order to market or commercially distribute a Class I, II, and III device intended for human use in the United States, for which a Premarket Approval application (PMA) is not required, one must submit a 510(k) to FDA unless, as noted, the device is exempt from the 510(k) pre-market notification requirements of the FDCA. Per the FDA, generally, most Class I devices are exempt from Premarket Notification 510(k); most Class II devices require Premarket Notification 510(k); and most Class III devices require a PMA.
510(k) Clearance Process
To obtain 510(k) clearance, we must submit a pre-market notification to the FDA demonstrating that the proposed device is substantially equivalent to a device that has previously obtained 510(k) clearance, a device that has been classified into Class I or II, or a device that was legally marketed before May 28, 1976 and that is not yet subject to an FDA order requiring pre-market approval. In rare cases, Class III devices may be cleared through the 510(k) process. The FDA has committed to review most 510(k) decisions within 90 days, but the review clock may be stopped due to requests for additional information. A decision may take significantly longer, and clearance is never
9
assured. Although many 510(k) pre-market notifications are cleared without clinical data, in some cases, the FDA requires significant clinical data to support substantial equivalence. In reviewing a pre-market notification submission, the FDA may request additional information, including clinical data, which may significantly prolong the review process.
After a device receives 510(k) clearance, any subsequent modification of the device that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, requires a new 510(k) clearance or, in some cases, approval of a PMA. The FDA requires each manufacturer to make this determination initially, but the FDA may review any such decision and may disagree with a manufacturer's determination. If the FDA disagrees with a manufacturer's determination, the FDA may require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or approval of a PMA is obtained. Under these circumstances, the FDA may also subject a manufacturer to enforcement action and sanctions, including those described below. In addition, the FDA is currently evaluating the 510(k) process and may make substantial changes to regulatory requirements, including changes that could affect which devices are eligible for 510(k) clearance, the FDA’s ability to rescind 510(k) clearances, and additional requirements that may significantly impact the 510(k) review process.
Pre-market Approval Process
A PMA generally must be submitted if the medical device is in Class III or cannot be cleared through the 510(k) process. A PMA must be supported by extensive technical, preclinical, clinical, manufacturing, and labeling data to demonstrate to the FDA's satisfaction the safety and effectiveness of the device.
After a PMA is submitted and filed, the FDA begins an in-depth review of the submitted information. During this review, the FDA may request additional information or clarification of information already provided. Also during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. In addition, the FDA will conduct a pre-approval inspection of the manufacturing facility to ensure compliance with Quality System Regulation (“QSR”), which imposes elaborate development, testing, control, documentation and other quality assurance requirements on the design and manufacturing process. The FDA has committed to review most PMAs within 180 days where an advisory panel is not required and within 320 days where an advisory panel is required, but the review clock may be stopped due to requests for additional information. A decision may take significantly longer, and approval is never assured. The FDA may approve a PMA with post-approval conditions intended to ensure the safety and effectiveness of the device including restrictions on labeling, promotion, sale, and distribution and collection of safety data. Failure to comply with the conditions of approval can result in enforcement action and sanctions, including those described below. New PMAs or PMA supplements are required for significant modifications to the manufacturing process, labeling of the product, or design of a device that is approved through the PMA process. PMA supplements often require submission of the same type of information as an original PMA, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA and may not require as extensive clinical data or the convening of an advisory panel.
De novo Classification Process
Medical device types that the FDA has not previously classified as Class I, II, or III are automatically classified into Class III regardless of the level of risk they pose. The Food and Drug Administration Modernization Act of 1997 established a new route to market for low-to-moderate risk medical devices that are automatically placed into Class III due to the absence of a predicate device, called the “Request for Evaluation of Automatic Class III Designation,” or the de novo classification procedure. This procedure allows a manufacturer whose novel device is automatically classified into Class III to request down-classification of its medical device into Class I or Class II on the basis that the device presents low or moderate risk, rather than requiring the submission and approval of a PMA. Prior to the enactment of the Food and Drug Administration Safety and Innovation Act (“FDASIA”) in July 2012, a medical device could only be eligible for de novo classification if the manufacturer first submitted a 510(k) pre-market notification and received a determination from the FDA that the device was not substantially equivalent to a predicate device. FDASIA streamlined the de novo classification pathway by permitting manufacturers to also request de novo classification directly without first submitting a 510(k) pre-market notification to the FDA and receiving a not substantially equivalent determination. Under FDASIA, the FDA is required to classify the device within 120 days following receipt of such a direct de novo request; however this time period can be extended if questions and/or requests for additional information are asked of the applicant. If the manufacturer seeks classification into Class II, the manufacturer should include a draft proposal for special controls that are necessary to provide a reasonable assurance of the safety and effectiveness of the medical device. In addition, the FDA may reject a de novo request if the FDA identifies a legally marketed predicate device that would be appropriate for a 510(k), determines that the device is not low-to-moderate risk, or
10
determines that general controls would be inadequate to control the risks and special controls cannot be developed.
In July of 2016, we submitted a de novo request for evaluation of automatic Class III Designation to the FDA for Accelerate Pheno™ system and Accelerate PhenoTest™ BC Kit, and on February 23, 2017, the FDA granted our de novo request to market the Accelerate Pheno™ system and Accelerate PhenoTest™ BC Kit.
Clinical Trials
Clinical trial data is typically required to support a PMA and is sometimes required for a 510(k) pre-market notification. Initiation of a clinical trial generally requires submission of an application for an Investigational Device Exemption (an “IDE”) to the FDA. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the investigational protocol is scientifically sound. The IDE application must be approved in advance by the FDA for a specified number of patients, unless the product is deemed a non-significant risk device and eligible for abbreviated IDE requirements. Clinical trials for a significant risk device may begin once the IDE application is approved by the FDA as well as the appropriate institutional review boards at the clinical trial sites and the informed consent of the patients participating in the clinical trial is obtained. After a trial begins, the FDA may place it on hold or terminate if it concludes that the clinical subjects are exposed to unacceptable risks. Any trials we conduct must be conducted in accordance with FDA regulations as well as other federal regulations and state laws concerning human subject protection and privacy. Moreover, the results of a clinical trial may not be sufficient to obtain clearance or approval of the product.
Clinical trial sponsors may also be subject to the Medicare Secondary Payer laws, which prohibit Medicare from making a payment if payment has been made or can reasonably be expected to be made by other plans, such as liability insurance plans (including self-insurance). Section 111 of the Medicare, Medicaid, and SCHIP Extension Act of 2007 (“MMSEA”) established mandatory reporting requirements with respect to Medicare beneficiaries who receive settlements, judgments, awards, or other payment from liability insurance (including self-insurance) plans. When payments are made by sponsors of clinical trials for complications or injuries arising out of the trials, such payments are considered to be payments by liability insurance (including self-insurance) and must be reported. Section III of the MMSEA includes authority for CMS to impose civil monetary penalties against liability insurance (including self-insurance) plans that are determined to be non-compliant with the applicable reporting requirements.
Pervasive and Continuing Regulation
After a medical device is placed on the market, numerous FDA regulatory requirements apply, including the following:
• | the QSR, which imposes elaborate development, testing, control, documentation, and other quality assurance requirements on the design and manufacturing process; |
• | establishment registration, which requires establishments involved in the production and distribution of medical devices, intended for commercial distribution in the United States, to register with the FDA; |
• | medical device listing, which requires manufacturers to list the devices they have in commercial distribution with the FDA; |
• | labeling regulations and various statutory provisions, which prohibit false or misleading labeling, as well as the promotion of products for unapproved or “off-label” uses, and impose other restrictions on labeling; and |
• | post-market reporting requirements, which require that manufacturers report to the FDA deaths, serious injuries, and malfunctions that, if they were to recur, could lead to death or serious injury, recalls, and corrective field actions. |
In certain cases, advertising is also subject to scrutiny by the Federal Trade Commission in addition to the FDA. The FDA and other agencies actively enforce these and other applicable laws and regulations, accordingly. Failure to comply with applicable requirements may result in enforcement action by the FDA and/or the U.S. Department of Justice, which may include one or more of the following administrative or judicial sanctions:
• | untitled letters or warning letters; |
11
• | fines, injunctions, and civil penalties; |
• | mandatory recall or seizure of our products; |
• | administrative detention or banning of our products; |
• | operating restrictions, partial suspension, or total shutdown of production; |
• | import holds; |
• | refusing to approve pending 510(k) notifications or PMAs; |
• | revocation of 510(k) clearance or pre-market approvals previously granted; and |
• | criminal prosecution and penalties. |
International Regulation
Sales of medical devices outside the United States are subject to foreign government regulations, which vary substantially from country to country. In order to market our products in other countries, we must obtain regulatory approvals and comply with extensive safety and quality regulations in other countries. The time required to obtain approval by a foreign country may be longer or shorter than that required for FDA clearance or approval, and the requirements may differ significantly.
In the European Economic Area, or EEA, which comprises the 28 Member States of the EU plus Liechtenstein, Norway and Iceland, in vitro medical devices are required to conform with the essential requirements of the EU Directive on in vitro diagnostic medical devices (Directive 98/79/EC, as amended). To demonstrate compliance with the essential requirements, the manufacturer must undergo a conformity assessment procedure. The conformity assessment varies according to the type of medical device and its classification. For low-risk devices, the conformity assessment can be carried out internally, but for higher risk devices (self-test devices and those included in List A and B of Annex II of Directive 98/79/EC) it requires the intervention of an accredited EEA Notified Body. If successful, the conformity assessment concludes with the drawing up by the manufacturer of an EC Declaration of Conformity entitling the manufacturer to affix the CE mark to its products and to sell them throughout the EEA. The EC Declaration of Conformity was received by the Company in 2015.
Other Healthcare Laws
With the FDA’s granting of our de novo request to market the Accelerate Pheno™ system and Accelerate PhenoTest™ BC Kit, we are actively commercializing the Accelerate Pheno™ system. Such business activities, including the activities of any third-party distributors that we retain, will be subject to additional healthcare laws and regulations and related enforcement by the federal government as well as the governments of states and foreign jurisdictions where we conduct our business. These laws and regulations include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, privacy and security, and physician payment transparency laws and regulations. Violations of these laws or regulations can result in criminal or civil sanctions, including substantial fines and, in some cases, exclusion from participation in federal healthcare programs, such as Medicare and Medicaid. The following discussion describes certain federal and state healthcare laws and regulations that may impact our operations and the operations of our customers, but is not intended to be an exhaustive discussion of all potentially applicable federal and state health laws and regulations.
The U.S. federal Anti-Kickback Statute prohibits any person from knowingly and willfully offering, soliciting, receiving, or providing remuneration, directly or indirectly, overtly or covertly, in cash or in kind, in exchange for or to induce either the referral of an individual for an item or service, or the purchasing, leasing, ordering, or arranging for or recommending the purchase, lease, or order of any good, facility, item, or service for which payment may be made, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. A person need not have actual knowledge of the Anti-Kickback Statute or specific intent in order to commit a violation, and several courts have interpreted the intent requirement of the Anti-Kickback Statute to mean that if any one purpose of an arrangement is to induce referrals or purchases of federal healthcare program business, the Anti-Kickback Statute has been violated. In addition to criminal fines and penalties set forth under the Anti-Kickback Statute, violations of
12
the Anti-Kickback Statute can result in exclusion or debarment from participation in the federal healthcare programs, as well as substantial penalties under the Civil Monetary Penalties Statute, which imposes penalties against any person or entity that is determined to have presented or caused to be presented a claim to a federal healthcare program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. A violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act, which, as discussed below, imposes liability on any person or entity that knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal healthcare program. Several states also have anti-kickback laws which establish similar prohibitions and, in some cases, may apply to items or services reimbursed by any third-party payer, including commercial insurers.
The federal False Claims Act imposes liability on any person or entity that knowingly presents or causes to be presented a false or fraudulent claim for payment to, or approval by, the U.S. government. Liability under the False Claims Act can give rise to treble damages and civil penalties of up to $21,916 per claim. In addition to actions initiated by the government itself, the qui tam provisions of the False Claims Act authorize private individuals to bring False Claims Act actions on behalf of the federal government alleging that the defendant has submitted a false claim to the federal government, and to share in a percentage of the recovery. In recent years, the government and qui tam relators have initiated suits resulting in multi-million and multi-billion dollar settlements under the False Claims Act in addition to criminal convictions under applicable criminal statutes. Given the significant size of actual and potential settlements, it is expected that the government and qui tam relators will continue to devote substantial resources and use the False Claims Act to investigate and prosecute healthcare companies’ compliance with applicable fraud and abuse laws.
The federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) created federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme or artifice to defraud any healthcare benefit program, including private third-party payers or to obtain—by means of false or fraudulent pretenses, representations, or promises—any of the money or property owned by or under the custody or control of any healthcare benefit program; and knowingly and willfully falsifying, concealing, or covering up a material fact or making any materially false, fictitious, or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. The Affordable Care Act amended certain sections of the HIPAA criminal statutes such that a person need not have actual knowledge of the applicable statute or specific intent in order to have committed a healthcare fraud violation.
As stated above, many states and foreign countries have adopted similar fraud and abuse laws that may be broader in scope and may apply regardless of payer. Violations of any of these laws can lead to additional risk such as risk of plaintiff class actions, state attorney general actions, and investigation by agencies such as the Department of Justice (“DOJ”) or the Federal Trade Commission (“FTC”).
The Physician Payment Sunshine Act, implemented by Section 6002 of the Affordable Care Act, imposes transparency requirements on certain manufacturers, referred to as “applicable manufacturers,” of drugs, devices, biological, or medical supplies for which payment is available under Medicare, Medicaid, the Children’s Health Insurance Program (“CHIP”), or a waiver of a plan offered under CHIP. Applicable manufacturers must track and report to the CMS certain payments or “transfers of value” provided to U.S. licensed physicians and teaching hospitals during the preceding calendar year, as well as certain ownership and investment interests held by U.S. licensed physicians and their immediate family members. CMS releases the reported data on a public website on an annual basis. Failure to report as required under the Physician Payment Sunshine Act could subject applicable manufacturers to significant financial penalties, while tracking and reporting the required payments and transfers of value may result in considerable administrative expense. Several states currently have similar laws, and more states may enact similar legislation, some of which may be broader in scope. For example, certain states require the implementation of compliance programs, compliance with industry ethics codes, implementation of gift bans, and spending limits, and/or reporting of gifts, compensation, and other remuneration to healthcare professionals.
We also may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act (“HITECH”), and their respective implementing regulations, including the final omnibus rule published by the Department of Health and Human Services Office for Civil Rights (“OCR”) in January 2013, restrict the use and disclosure of patient-identifiable health information, mandate the adoption of standards relating to the privacy and security of patient-identifiable health information, and require us to report certain security breaches to healthcare provider customers with respect to such information where we are acting as a HIPAA business associate, as that term is defined, to that customer. In addition to HIPAA criminal penalties, HITECH created four new tiers of civil and monetary penalties and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts
13
to enforce the federal HIPAA privacy and security laws and seek attorneys' fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances and impose reporting requirements for data breaches, many of which differ from each other and HIPAA in significant ways and may not have the same effect, thus complicating compliance efforts.
The use of certain diagnostic products by our potential customers is affected by the Clinical Laboratory Improvement Amendments (“CLIA”) and related federal and state regulations that provide for regulation of laboratory testing. CLIA is intended to ensure the quality and reliability of clinical laboratories in the United States by mandating specific standards in the areas of personnel qualifications, administration, and participation in proficiency testing, patient test management, quality assurance, quality control, and inspections. Current or future CLIA requirements or the promulgation of additional regulations affecting laboratory testing may prevent some laboratories, hospitals, providers, or other customers with laboratories from using some or all of our diagnostic products.
Healthcare Reform
In the United States and several foreign jurisdictions, there have been, and we expect there may continue to be, a number of legislative and regulatory changes to the healthcare system seeking to reduce healthcare costs that could affect our future results of operations as we begin to commercialize our products.
In the United States, the Affordable Care Act (“ACA”), enacted in March 2010, made changes that are expected to have a continued and significant impact on the medical device industry and clinical laboratories, including the way healthcare is delivered and financed by governmental and private insurers. For example, the legislation provided for reductions in the Medicare clinical laboratory fee schedule and, since 2013 (with exception of a moratorium period from January 1, 2016 through December 31, 2019), has required that certain medical device manufacturers pay an excise tax in an amount equal to 2.3% of the price for which such manufacturer sells its medical devices.
In addition, frequently in recent years, other legislative, regulatory, and political changes aimed at regulating healthcare delivery in general and clinical laboratories in particular have been proposed and adopted in the United States. Payment and reimbursement for the laboratory industry and hospital and other healthcare provider services have been under significant pressure. Most recently, in January 2015, the Department of Health and Human Services (“HHS”) announced a plan to shift the Medicare program and the healthcare system at large toward paying providers based on quality, rather than the quantity of care provided to patients.
Reimbursement
We do not believe that hospitals will specifically seek reimbursement from the government or private insurance companies for their purchase of the Accelerate Pheno™ system or the Accelerate PhenoTest™ BC Kit. Instead, we believe that hospitals will recoup such costs by obtaining reimbursement from the government or private insurance companies for in-bed occupancies, which traditionally includes all testing required for admitted patients.
Hospitals, clinical laboratories, and other healthcare provider customers that may purchase our products, if approved, generally bill various third-party payers to cover all or a portion of the costs and fees associated with diagnostic tests, including the cost of the purchase of our products. We currently expect that all of our diagnostic tests will be performed in a hospital inpatient setting, where governmental payers, such as Medicare, generally reimburse hospitals a single bundled payment that is based on the patient’s diagnosis under a classification system known as the Medicare severity diagnosis-related groups (“MS-DRGs”) classification for all items and services provided to the patient during a single hospitalization, regardless of whether our diagnostic tests are performed during such hospitalization.
Operations
In January 2013, we relocated our headquarters from Denver, Colorado, to Tucson, Arizona, where we currently lease approximately 52,092 square feet of office, manufacturing and laboratory space. Further information regarding our Tucson facility is included in Item 2. Properties included elsewhere in this report, and details regarding our lease arrangement are included in Item 8, Note 17, Commitments and Contingencies to the audited consolidated financial statements included elsewhere in this report.
We assemble the Accelerate Pheno™ system instrument and Accelerate PhenoTest™ BC Kit in our facilities in Tucson, Arizona. The Accelerate Pheno™ system requires certain components that are custom-fabricated to our
14
specifications. Such components include injection-molded plastic components, die-cut laminates, and machined mechanical components. We own the necessary production tooling and believe that we will be able to qualify secondary sources as needed to support future demand for the Accelerate Pheno™ system.
Raw Materials
We purchase many different types of raw materials, including plastics, glass, metals, electronic and mechanical sub-assemblies and various biological and chemical products. We seek to ensure continuity of raw material supply by securing multiple options for sourcing. However, our components are custom-made by only a few outside suppliers. In certain instances, we have a sole source supply for key product components of the Accelerate Pheno™ system. We have entered into supply agreements with most of our suppliers to help ensure component availability and flexible purchasing terms with respect to the purchase of such components.
Employees
We have 239 employees as of December 31, 2017. We have not entered into any collective bargaining agreements and consider our labor practices and employee relations to be good.
Available Information
We regularly file reports with the SEC, including Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and any other filings required by the SEC. We make these reports available free of charge in the investor relations section of our corporate website (http://ir.axdx.com/) as soon as reasonably practicable after such material is electronically filed with or furnished to the SEC. References to our corporate website address in this report are intended to be inactive textual references only, and none of the information contained on our website is part of this report or incorporated in this report by reference.
The public may inspect and copy materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Room 1580, Washington, DC 20549. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. You may also access these materials, and other information regarding issuers like us that file information electronically with the SEC, from the SEC’s internet website at http://www.sec.gov/.
Item 1A. Risk Factors
Investing in our securities involves a high degree of risk. You should carefully consider the risks described below, in addition to the other information included or incorporated by reference in this Annual Report on Form 10-K, including our financial statements and the related notes. If any of the following risks materializes, our business, financial condition, results of operations or growth prospects could be materially adversely affected, and the value of an investment in our common stock may decline significantly.
Risks Related to Our Business and Strategy
Our future profitability and continued existence is dependent in large part upon the successful commercialization of the Accelerate Pheno™ system and further development and commercialization of complimentary products.
Our principal business strategy involves the successful commercialization of the Accelerate Pheno™ system, development of associated test kits and the future development and commercialization of complimentary products. On June 30, 2015, we declared our conformity to the European In Vitro Diagnostic Directive 98/79 EC and CE Mark of the Accelerate Pheno™ system and Accelerate PhenoTest™ BC Kit for in vitro diagnostic use and on February 23, 2017, received FDA clearance of the Accelerate Pheno™ system and Accelerate PhenoTest™ BC Kit. We have and will continue to dedicated a significant amount of resources to market and sell the Accelerate Pheno™ system. Likewise we plan to continue our investment in additional test kit development and commercialization of the Accelerate Pheno™ system in the United States and other jurisdictions in which we intend to pursue marketing authorization. There can be no assurance that we will successfully commercialize the Accelerate Pheno™ system, associated Accelerate PhenoTest™ BC Kit, or further develop and commercialize complimentary products. We may be required to expend significantly more resources than planned in this process, and as a result we may have to cease investing in the Accelerate Pheno™ system or developing other products.
15
If we are not successful in the development of product improvements and additional test kits and commercialization of the Accelerate Pheno™ system, such failure could lead to impairment of certain of our intellectual property and may result in our ceasing operations.
Further, if we are not successful in conveying to hospitals that the Accelerate Pheno™ system provides equivalent or superior diagnostic information in a shorter period of time compared to existing technologies, or that the Accelerate Pheno™ system improves patient outcomes or decreases healthcare costs, we may experience reluctance from hospitals to order our product. If we fail to successfully commercialize the Accelerate Pheno™ system, we may never receive a return on the significant investments in product development, sales and marketing, regulatory compliance, manufacturing and quality assurance we have made and further investments we intend to make, and may fail to generate revenue and gain economies of scale from such investments.
Our future product candidates have not obtained marketing authorization from the FDA, and they may never obtain such marketing authorization or other regulatory clearance.
Our success in part depends on our ability to obtain additional product marketing authorizations from the FDA for product candidates in our pipeline. If our attempts to obtain marketing authorization or other regulatory clearance are unsuccessful, we may be unable to generate sufficient revenue to sustain and grow our business. Our future product candidates may not be sufficiently sensitive or specific to obtain, or may prove to have other characteristics that preclude our obtaining, marketing authorization from the FDA or regulatory clearance. The process of obtaining regulatory clearance is expensive and time-consuming and can vary substantially based upon, among other things, the type, complexity and novelty of our product candidates. Changes in regulatory policy, changes in or the enactment of additional statutes or regulations or changes in regulatory review for each submitted product application may cause delays in the clearance of, or receipt of marketing authorization from the FDA for, a product candidate or rejection of a regulatory application altogether. The FDA has substantial discretion in the de novo review and clearance processes and may refuse to accept any application or may decide that our data are insufficient for clearance and require additional pre-clinical, clinical or other studies. In addition, varying interpretations of the data obtained from pre-clinical and clinical testing could delay, limit or prevent marketing authorization from the FDA or regulatory clearance of a product candidate. Any marketing authorization from the FDA or regulatory clearance we ultimately obtain may be limited or subject to restrictions or post-market commitments that render the product candidate not commercially viable.
If we do not achieve our projected development goals in the time frames we announce and expect, the commercialization of our products may be delayed and, as a result, our stock price may decline.
From time to time, we estimate the timing of the accomplishment of various scientific, clinical, regulatory and other product development goals. These goals may include the commencement or completion of clinical trials and the submission of regulatory filings. From time to time, we may publicly announce the expected timing of some of these goals. All of these goals are, and will be, based on a variety of assumptions. The actual timing of these goals can vary significantly compared to our estimates, in some cases for reasons beyond our control. We may experience numerous unforeseen events that could delay or prevent our ability to receive marketing approval or commercialize our product candidates, including the uncertainties and risks set forth in these risk factors. If we do not meet our goals as publicly announced, the commercialization of our product candidates may be delayed and, as a result, our stock price may decline.
We may not be able to enhance the capabilities of our current and new products to keep pace with our industry’s rapidly changing technology and customer requirements.
Our industry is characterized by rapid technological changes, frequent new product introductions and enhancements and evolving industry standards. Our future success will depend significantly on our ability to enhance our current products and develop or acquire and market new products that keep pace with technological developments and evolving industry standards as well as respond to changes in customer needs. New technologies, techniques or products could emerge that might offer better combinations of price and performance than the products and systems that we plan to sell. It is critical to our success that we anticipate changes in technology and customer requirements and physician, hospital and healthcare provider practices and successfully introduce new, enhanced and competitive technologies to meet our prospective customers’ needs on a timely and cost-effective basis. At the same time, however, we must carefully manage our introduction of new products. If potential customers believe that such products will offer enhanced features or be sold for a more attractive price, they may delay purchases until such products are available.
16
We are developing additional uses for the Accelerate Pheno™ system. Any failure or delay in launching new applications may compromise our ability to achieve our growth objectives.
We are developing additional uses for the Accelerate Pheno™ system, including the ability to test on additional specimen types (e.g., respiratory, urine, and intra-abdominal samples). We may have problems applying our technologies to additional specimen types, and our new applications may not be as effective in detection as our initial applications. We may also encounter difficulties obtaining regulatory approval for additional uses of the Accelerate Pheno™ system. Any failure or delay in launching new applications may compromise our ability to achieve our growth objectives.
There can be no assurance that we will be successful in developing or acquiring product enhancements or new products to address changing technologies and customer requirements adequately, that we can introduce such products on a timely basis or that any such products or enhancements will be successful in the marketplace. If we are unable to successfully develop or acquire new products or if the market does not accept our products, or if we experience difficulties or delays in the final development and commercialization of our products, including the Accelerate Pheno™ system, we may be unable to attract additional customers for our products or license our products to other strategic partners.
The failure of the Accelerate Pheno™ system or any future diagnostic products to perform as expected could significantly impair our reputation and the public image of our products, and we may be subject to legal claims arising from any defects or errors.
Our success will depend on the market’s confidence that our technologies can provide reliable, high-quality diagnostic results. We believe that our customers are likely to be particularly sensitive to any defects or errors in the Accelerate Pheno™ system. If we experience disruptions or other performance problems with the Accelerate Pheno™ system or any future diagnostic product, we could face warranty and liability claims against us or our reputation could suffer as a result of such failures. We cannot assure you that our product liability insurance would adequately protect our assets from the financial impact of defending a product liability claim. Any product liability claim brought against us, with or without merit, could increase our product liability insurance rates or prevent us from securing insurance coverage in the future. In addition, the FDA and similar foreign governmental authorities have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture of a product or in the event that a product poses an unacceptable risk to health. A recall, material liability claim or other occurrence that harms our reputation or decreases market acceptance of our products could cause us to incur significant costs, divert the attention of our key personnel or cause other significant customer relations problems.
We have limited revenues from our products and no assurance of future revenues.
We have received limited revenue from sales of the Accelerate Pheno™ system, Accelerate PhenoTest™ BC Kit, and products using our OptiChem technology, and have discontinued efforts to develop and actively market OptiChem and our other surface chemistry products. During the years ended December 31, 2017, 2016 and 2015, we experienced losses from operations. Our future revenues are dependent on the successful commercialization of the Accelerate Pheno™ system, and there can be no assurance that we will be successful. If we are unsuccessful in generating revenues from such product, we will likely continue to experience losses from operations and negative cash flow as we have in the past.
Until we received FDA approval to market the Accelerate Pheno™ system, we were a development-stage company and therefore incurred significant losses in recent years. While we are in the early years of commercializing the Accelerate Pheno™ system, we may continue to incur losses. We cannot be certain that we will achieve or sustain profitability.
We have incurred significant costs in connection with the development and commercialization of our technology, and there is no assurance that we will achieve sufficient revenues to offset anticipated operating costs. We have incurred significant losses in recent years and may incur losses in the future. Although we can now begin to commercialize our technology, we expect that our selling, general and administrative expenses will continue to increase due to the additional costs associated with establishing and expanding a dedicated sales force and other marketing efforts for the Accelerate Pheno™ system. Our ability to achieve or sustain profitability depends on numerous factors including the market acceptance of our product, future product development and our market penetration and margins. If we are unsuccessful in generating revenues from the Accelerate Pheno™ system, we will likely continue to experience
17
losses from operations and negative cash flow. Although we anticipate deriving revenues from the sale of our products, no assurance can be given that these products can be sold on a net profit basis. If we achieve profitability, we cannot give any assurance that we will be able to sustain or increase profitability on a quarterly or annual basis in the future.
We have limited experience in marketing and selling the Accelerate Pheno™ system.
We have limited experience marketing and selling the Accelerate Pheno™ system. We have established an initial sales force to market the Accelerate Pheno™ system directly to our target customers, and we plan to continue to build this sales force. In select geographies outside of the United States and Europe, we may also use third-party distributors to market our product.
Our future sales will depend in large part on our ability to successfully establish an effective sales force. Because we have no experience in marketing and selling the Accelerate Pheno™ system, our ability to forecast demand, the infrastructure required to support such demand and the sales cycle of our potential customers is unproven.
Moreover, we do use third-party distribution partners for certain geographic areas outside of the United States and Europe, and there is no guarantee that we will be able to enter into such arrangements on favorable terms. Distributors may not commit the necessary resources to market and sell the Accelerate Pheno™ system effectively or may choose to favor marketing the products of our competitors. If distributors do not perform adequately, or if we are unable to enter into effective arrangements with distributors in particular geographic areas, we may not realize our full potential for sales and growth in these areas.
If treatment guidelines for bacterial infections change, or the standard of care evolves, we may need to redesign and seek new marketing authorization from the FDA for our product candidates.
If treatment guidelines for bacterial infections change, or the standard of care evolves, we may need to redesign and seek new marketing authorization from the FDA or other regulatory clearance for our product candidates. If treatment guidelines change so that different treatments become desirable, the Accelerate Pheno™ system may no longer provide the information sought by physicians, and we could be required to seek marketing authorization from the FDA or other regulatory clearance for a revised product.
We may not be able to correctly estimate or control our future operating expenses, which could lead to cash shortfalls.
Our operating expenses may fluctuate significantly in the future as a result of a variety of factors, many of which may be outside of our control. These factors include, but are not limited to:
• | the expenses we incur for research and development required to maintain and improve our technology, including the final development of the Accelerate Pheno™ system; |
• | the expenses we incur in connection with the development, marketing authorization and regulatory clearance of the use of the Accelerate Pheno™ system to test on additional specimen types; |
• | the costs of preparing, filing, prosecuting, defending and enforcing patent claims and other intellectual property related costs, including litigation costs and the results of such litigation; |
• | the expenses we incur in connection with commercialization activities, including product marketing, sales and distribution expenses; |
• | the costs incurred to build manufacturing capabilities; |
• | the expenses to implement our sales strategy; |
• | the costs to attract and retain personnel with the skills required for effective operations; and |
• | the costs associated with being a public company. |
Our budgeted expense levels are based in part on our expectations concerning future revenues from sales of the Accelerate Pheno™ system, as well as our assessment of the future investments needed to expand our
18
commercial organization and support research and development activities in connection with the Accelerate Pheno™ system. We may be unable to reduce our expenditures in a timely manner to compensate for any unexpected events or a shortfall in revenue. Accordingly, a shortfall in demand for our products or other unexpected events could have an immediate and material impact on our cash levels.
Breaches of our information technology systems could have a material adverse effect on our operations and potentially result in liability, depending on the type of breach and information compromised.
We rely on information technology systems to process, transmit and store electronic information, which may include protected health information, in our day-to-day operations. In addition, our research and development operations are highly dependent on our information technology and storage. Our information technology systems have been subjected to computer viruses or other malicious codes and phishing attacks, and we expect to be subject to similar viruses and codes in the future. These attacks could result in our intellectual property, unsecured protected health information, and other confidential information being lost or stolen, including the disclosure of our trade secrets, disruption of our operations, loss of valuable research and development data, the need to notify individuals whose information was disclosed, increased costs for security measures or remediation costs and diversion of management attention and other negative consequences. While we will continue to implement protective measures to reduce the risk of and detect future cyber incidents, cyber-attacks are becoming more sophisticated and frequent, and the techniques used in such attacks change rapidly. There can be no assurance that our protective measures will prevent future attacks that could have a significant impact on our business.
We are dependent on our key employees. If we are unable to recruit, train and retain qualified personnel, we may not achieve our goals.
Because of the complex and technical nature of our products and the dynamic market in which we compete, our future success depends on our ability to recruit, train and retain key personnel, including our senior management, research and development, science and engineering, manufacturing and sales and marketing personnel. In particular, we are highly dependent on the management and business expertise of Lawrence Mehren, our President and Chief Executive Officer. We do not maintain key person life insurance for Mr. Mehren or any of our employees. Our industry is very competitive for qualified personnel. To the extent that the services of Mr. Mehren would be unavailable to us, we may be unable to employ another qualified person with the appropriate background and expertise to replace Mr. Mehren on terms suitable to us. Our growth depends, in particular, on attracting, retaining and motivating highly trained sales personnel with the necessary scientific background and ability to understand our systems and pathogens at a technical level. In addition, we may need additional employees at our manufacturing facilities to meet demand for our products as we scale up our sales and marketing operations.
We face competition from industry participants who may have greater resources than we do.
The industry in which we compete is subject to rapid technological changes, and we face and expect to continue to face competition for our products. We may also face competition from non-medical device companies. Many of our competitors and potential competitors may have substantially greater research and development, financial, manufacturing, customer support, sales and marketing resources, larger customer bases, longer operating histories, greater name recognition and more established relationships in the industry than we do. In addition, some of our competitors may, individually or together with companies affiliated with them, have greater human and scientific resources than we do. Our competitors could develop technologies and methods that are more effective than our current and proposed technologies, including but not limited to the Accelerate Pheno™ system.
We cannot assure you that we will effectively compete or that we will be successful in the face of increasing competition from new products and technologies introduced by existing or new competitors. In addition, we cannot assure you that our future competitors do not have or will not develop products or technologies that enable them to produce competitive products with greater capabilities or at lower costs than our products. Any failure to compete effectively could materially and adversely affect our business, financial condition and operating results.
We generate a portion of our future revenue internationally and are subject to various risks relating to our international activities which could adversely affect our operating results.
We market and sell the Accelerate Pheno™ system in other countries outside of the United States. In order to market our products in certain foreign jurisdictions, we, or our distributors or partners, must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements regarding safety and efficacy and governing,
19
among other things, clinical studies and commercial sales and distribution of our products. The approval procedure varies among countries and can involve additional testing. In addition, in many countries outside the United States, a product must be approved for reimbursement before the product can be approved for sale in that country. We may not obtain approvals from regulatory authorities outside the United States on a timely basis, if at all, which could harm our ability to expand into markets outside the United States. In addition, engaging in international business involves a number of other difficulties and risks, including:
• | required compliance with existing and changing foreign healthcare and other regulatory requirements and laws, such as those relating to patient privacy or handling of bio-hazardous waste; |
• | required compliance with anti-bribery laws, such as the U.S. Foreign Corrupt Practices Act and the U.K. Bribery Act, data privacy requirements, labor laws and anti-competition regulations; |
• | export and import restrictions; |
• | various reimbursement and insurance regimes; |
• | laws and business practices favoring local companies; |
• | longer payment cycles and difficulties in enforcing agreements and collecting receivables through certain foreign legal systems; |
• | political and economic instability; |
• | potentially adverse tax consequences, tariffs, customs charges, bureaucratic requirements and other trade barriers; |
• | foreign exchange controls; |
• | fluctuations due to changes in foreign currency exchange rates; |
• | difficulties and costs of staffing and managing foreign operations; and |
• | impediments with protecting or procuring intellectual property rights. |
Our employees, independent contractors, principal investigators, consultants, commercial partners, vendors and other agents may engage in misconduct or other improper activities, including non-compliance with legal standards and requirements.
We are exposed to the risk of fraud or other misconduct by our employees, independent contractors, principal investigators, consultants, commercial partners, vendors and other agents. Misconduct by these parties could include intentional, reckless or negligent failures to: (i) comply with the laws and regulations of the FDA, CMS, the HHS Office of Inspector General, Office for Civil Rights and other similar foreign regulatory bodies; (ii) provide true, complete and accurate information to the FDA and other similar regulatory bodies; (iii) comply with manufacturing requirements of the FDA and other similar regulatory bodies and manufacturing standards we have established; (iv) comply with healthcare fraud and abuse laws and regulations in the United States and similar foreign fraudulent misconduct laws; or (v) report financial information or data accurately, or disclose unauthorized activities to us. These laws may impact, among other things, our activities with principal investigators and research subjects, as well as our sales, marketing and education programs. In particular, the promotion, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing, unauthorized use of protected health information and data breaches, and other abusive practices. These laws may restrict or prohibit a wide range of activities related to pricing, discounting, marketing and promotion, patient support, royalty, consulting, research and other business arrangements, as well as the improper use of patient information obtained in the course of clinical studies. We currently have a code of conduct applicable to all of our employees, but it is not always possible to identify and deter employee misconduct, and our code of conduct and the other precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses, or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition
20
of civil, criminal and administrative penalties, damages, monetary fines, disgorgement, individual imprisonment, corporate integrity agreements, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of our operations. Any of these actions or investigations could result in substantial costs to us, including legal fees, and divert the attention of management from operating our business.
We may be unable to successfully manage our growth.
We expect to continually expand our operations to support the commercialization of the Accelerate Pheno™ system and future products. We intend to continue to develop a targeted sales force in connection with our commercialization efforts in the United States and in other countries. Our growth has placed and will continue to place a significant strain on our management, operating and financial systems and our sales, marketing and administrative resources. As a result of our growth, operating costs may escalate faster than planned, and some of our internal systems and processes, including those relating to manufacturing our products, may need to be enhanced, updated or replaced.
We also plan to introduce additional test kits for use on the Accelerate Pheno™ system to enable its use with other sample types (e.g., blood, respiratory, urine, and intra-abdominal samples), and plan to invest in the development of additional instruments, tests and other microbiology solutions. If we cannot effectively manage our expanding operations, manufacturing capacity and costs, including scaling to meet increased demand, we may not be able to continue to grow or we may grow at a slower pace than expected.
We may in the future be subject to shareholder lawsuits, including purported class actions, which is expensive and could divert the attention of management away from our business. In addition, any adverse result of such litigation could negatively impact our financial condition or results of operations.
In the past, companies such as Accelerate that have experienced volatility in the market price of their stock have been subject to an increased incidence of securities class action litigation and other shareholder lawsuits. We may in the future be the target of this type of litigation. Shareholder lawsuits against us, our officers or directors could result in substantial costs and divert the attention of management away from operating our business and other concerns, which could harm our business.
Current macroeconomic conditions and the uncertain economic outlook may remain challenging for the foreseeable future.
Global economic conditions may remain challenging and uncertain for the foreseeable future. These conditions not only limit our access to capital but also make it difficult for our customers, our vendors and us to accurately forecast and plan future business activities, and they could cause U.S. and foreign hospitals and other customers to slow spending on our products, which would delay and lengthen sales cycles. Some of our customers rely on government research grants to fund technology purchases. If negative trends in the economy affect the government’s allocation of funds to research, there may be less grant funding available for certain of our customers to purchase technologies from us. Certain of our customers may face challenges gaining timely access to sufficient credit or may otherwise be faced with budget constraints, which could result in decreased purchases of our products or in an impairment of their ability to make timely payments to us. If our customers do not make timely payments to us, we may be required to assume greater credit risk relating to those customers and increase our allowance for doubtful accounts, and our days sales outstanding would be negatively impacted. Although we maintain allowances for doubtful accounts for estimated losses resulting from the inability of our customers to make required payments, we may not continue to experience the same loss rates that we have in the past. Additionally, challenging macroeconomic conditions and market turbulence may also impact our suppliers, causing them to be unable to supply in a timely manner sufficient quantities of customized components, thereby impairing our ability to manufacture on schedule and at commercially reasonable costs.
Compliance with public company corporate governance and reporting is complex and expensive.
We are subject to laws and regulations affecting our domestic and international operations in a number of areas. Many laws and regulations, notably those adopted in connection with the Sarbanes-Oxley Act of 2002 by the Securities and Exchange Commission (the “SEC”), the Dodd-Frank Wall Street Reform and Consumer Protection Act and The NASDAQ Stock Market, impose obligations on public companies, such as ours, which have increased the scope, complexity and cost of corporate governance, reporting and disclosure practices. Compliance with these laws, regulations and similar requirements may be onerous, requires substantial management time and oversight and
21
requires us to incur significant additional accounting, legal and compliance costs. Any such costs, which may rise in the future as a result of changes in these laws and regulations or in their interpretation could individually or in the aggregate make our products and services more expensive, delay the introduction of new products in one or more regions, or cause us to change or limit our business practices. In addition, our larger competitors may be in a better position to absorb the costs of being a public company. We have implemented policies and procedures designed to ensure compliance with applicable laws and regulations, but there can be no assurance that our employees, contractors or agents will not violate such laws and regulations or our policies and procedures.
Additionally, changes to existing accounting rules and standards and the implementation of new accounting rules or standards, such as tax accounting or revenue recognition rules, may adversely impact our reported financial results and business, and may further require us to incur greater accounting fees.
Our estimates of market opportunity and forecasts of market growth may prove to be inaccurate, and even if the market in which we compete achieves the forecasted growth, our business could fail to grow at similar rates, if at all.
Market opportunity estimates and growth forecasts are subject to significant uncertainty and are based on assumptions and estimates that may not prove to be accurate. The estimates and forecasts in this 10-K relating to the size and expected growth of our market, total available market, estimated test and placement volume and estimated pricing, may prove to be inaccurate, which may have negative consequences, such as us overestimating our potential market opportunity. Even if the market in which we compete meets our size estimates and forecasted growth, our business could fail to grow at similar rates, if at all.
Our reputation, ability to do business and financial results may be impaired by improper conduct by any of our employees, agents or any business partners.
We cannot provide assurance that our internal controls and compliance systems will always protect us from acts committed by employees, agents or business partners that would violate U.S. and/or non-U.S. laws, including the laws governing payments to government officials, bribery, fraud, kickbacks and false claims, pricing, sales and marketing practices, conflicts of interest, competition, export and import compliance, insider trading, money laundering and data privacy. In particular, the U.S. Foreign Corrupt Practices Act, the U.K. Bribery Act and similar anti-bribery laws in other jurisdictions generally prohibit companies and their intermediaries from making improper payments to government officials for the purpose of obtaining or retaining business. Any such improper actions or allegations of such acts could damage our reputation and subject us to civil or criminal investigations in the U.S. and in other jurisdictions and related shareholder lawsuits, could lead to substantial civil and criminal, monetary and non-monetary penalties and could cause us to incur significant legal and investigatory fees.
Risks related to our intellectual property
If we are unable to effectively protect our intellectual property, our business would be harmed.
In addition to patent protection, we rely on trademark, copyright, trade secret protection and confidentiality agreements to protect intellectual property rights related to our proprietary technologies, both in the United States and in other countries. If we fail to protect our intellectual property, third parties may be able to compete more effectively against us and we may incur substantial litigation costs in our attempts to recover or restrict use of our intellectual property. We own or exclusively license 17 issued U.S. patents and 11 pending U.S. patent applications, including provisional and non-provisional filings. We also own 32 pending or granted non-U.S. counterpart patent applications. We own 35 pending and registered marks in the United States and foreign countries. In addition to our patents and trademarks, we possess an array of unpatented proprietary technology and know-how, and we license intellectual property rights to and from third parties. The strength of patents in our field involves complex legal and scientific questions. In addition, patent law continuously evolves and might change the legal framework under which our patent claims would be interpreted and adjudicated in the future. Uncertainty created by these questions and potential legal changes means that our patents may provide only limited protection and may not adequately protect our rights or permit us to gain or keep any competitive advantage. In addition, competitors could purchase our products and attempt by reverse engineering to replicate some or all of the competitive advantages we derive from our development efforts, willfully infringe our intellectual property rights, design around our protected technology or develop their own competitive technologies that fall outside of the protections provided by our intellectual property rights. If our intellectual property,
22
including licensed intellectual property, does not adequately protect our market position against competitors’ products and methods, our competitive position could be adversely affected, as could our business.
Further, if we are unable to prevent unauthorized disclosure of our non-patented intellectual property, and there is no guarantee that we will have any such enforceable trade secret protection, we may not be able to establish or maintain a competitive advantage. In addition, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad.
We may not be successful in our currently pending or future patent applications, and even if such applications are successful, we cannot guarantee that the resulting patents will sufficiently protect our products and proprietary technology.
We cannot assure you that any of our currently pending or future patent applications will result in issued patents with claims that adequately cover our products and technologies in the United States or in other foreign countries, and we cannot predict how long it will take for such patents to be issued. Further, issuance of a patent is not conclusive as to its inventorship or scope, and there is no guarantee that our issued patents will include claims that are sufficiently broad to cover our technologies or to provide meaningful protection from our competitors. Further, we cannot be certain that all relevant prior art relating to our patents and patent applications has been identified. Accordingly, there may be prior art that can invalidate our issued patents or prevent a patent from issuing from a pending patent application, or will preclude our ability to obtain patent claims that have a scope broad enough to provide meaningful protection from our competitors.
Even if patents do successfully issue and even if such patents cover our products and technologies, we cannot assure you that other parties will not challenge the validity, enforceability or scope of such issued patents in the United States and in foreign countries, including by proceedings such as reexamination, inter-partes review, interference, opposition, or other patent office or court proceedings. The strength of patents in our field involves complex legal and scientific questions. Moreover, we cannot assure you that if such patents were challenged in court or before a regulatory agency that the patent claims will be held valid, enforceable, to be sufficiently broad to cover our technologies or to provide meaningful protection from our competitors. Nor can we assure you that the court or agency will uphold our ownership rights in such patents. Accordingly, we cannot guarantee that we will be successful in defending challenges made against our patents and patent applications. Any successful third-party challenge to our patents could result in the unenforceability or invalidity of such patents, or narrowing of claim scope, such that we could be deprived of patent protection necessary for the successful commercialization of our products and technologies, which could adversely affect our business. To this end, we note that one of our issued European Patents, EP No. 1831692, is the subject of an Opposition proceeding within the European Patent Office. The Opposition, filed by our competitor bioMerieux, alleges that, inter alia, this issued patent claims subject matter that lacks novelty and inventive step in view of the state of the art at the time of filing. We disagree with bioMerieux’s contentions, and intend to vigorously defend our patent as properly issued by the European Patent Office. Our initial filing in response to bioMerieux’s allegations is due by early May 2018. After any additional briefing by both Parties to the Opposition, an oral hearing before the European Patent Office may be scheduled, which we do not anticipate arising until some point in 2019. Should we fail in our defense against bioMerieux’s allegations, the opposed patent potentially could be revoked or the claims may be amended such that they no longer cover aspects of our commercialized products.
Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our inventions, provide exclusivity for our products and technologies or prevent others from designing around our claims. Others may independently develop similar or alternative products and technologies or duplicate any of our products and technologies. These products and technologies may not be covered by claims of issued patents for which we are the right holder. Any of these outcomes could impair our ability to prevent competition from third parties, which may have an adverse impact on our business.
Further, if we encounter delays in regulatory approvals, the period of time during which we could market a product under patent protection could be reduced. Since patent applications in the United States and most other countries are confidential for a period of time after filing, and some remain so until issued, we cannot be certain that we were the first to make the inventions covered by our pending patent applications, or that we were the first to file any patent application related to a product candidate. Furthermore, if third parties have filed such patent applications, an interference proceeding in the United States can be initiated by a third party to determine who was the first to invent any of the subject matter covered by the patent claims of our applications. In addition, patents have a limited lifespan.
23
In the United States, the natural expiration of a patent is generally 20 years after it is filed. Various extensions may be available; however the life of a patent, and the protection it affords, is limited.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive and time consuming.
Third parties may infringe or misappropriate our intellectual property, including our existing patents and patent claims that may be allowed in the future. As a result, we may be required to file infringement claims to stop third-party infringement or unauthorized use. Further, we may not be able to prevent misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
If we file an infringement action against a third party, that party may challenge the scope, validity or enforceability of our patents, requiring us to engage in complex, lengthy and costly litigation or other proceedings. Such litigation and administrative proceedings could result in revocation of our patents or amendment of our patent claims such that they no longer cover our product candidates. They may also put our pending patent applications at risk of not issuing or issuing with limited and potentially inadequate scope to cover our product candidates. The outcome following legal assertions of invalidity and unenforceability is unpredictable.
Enforcing our intellectual property rights through litigation is very expensive and time-consuming. Some of our competitors may be able to sustain the costs of litigation more effectively than we can because of greater financial resources. Patent litigation and other proceedings may also absorb significant management time and reduce employee productivity. Furthermore, because of the substantial amount of discovery required in connection with U.S. intellectual property litigation or administrative proceedings, there is a risk that some of our confidential information could be compromised by disclosure. The occurrence of any of the foregoing could have a material adverse effect on our business, financial condition or results of operations.
We could face claims that our proprietary technologies infringe on the intellectual property rights of others.
Due to the significant number of U.S. and foreign patents issued to, and other intellectual property rights owned by, entities operating in the industry in which we operate, we believe that there is a risk of litigation arising from infringement of these patents and other rights. Third parties may assert infringement or other intellectual property claims against us or our licensees.
In addition, patent applications in the United States and many foreign jurisdictions are typically not published until 18 months after the earliest filing date for which a benefit is claimed. For this reason, and because publications in the scientific literature often lag behind actual discoveries, despite our best efforts we cannot be certain that others have not filed patent applications for technology covered by our issued patents or our pending applications or that we were the first to invent the technology. Another party may have filed or may in the future file patent applications covering our products or technology similar to ours. Under the “first to invent” rules applicable to patents filed before March 2013, any such patent application may have priority over our patent applications or patents, which could further require us to obtain rights to issued patents covering such technologies. If another party has filed a U.S. patent application on inventions similar to ours, we may have to participate in an interference proceeding declared by the U.S. Patent and Trademark Office to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful if the other party had independently arrived at the same or similar invention prior to our own invention, resulting in a loss of our U.S. patent position with respect to such inventions.
We may have to pay substantial damages, including treble damages, for past infringement if it is ultimately determined that our products infringe on a third party’s proprietary rights. In addition, even if such claims are without merit, defending a lawsuit may result in substantial expense to us and divert the efforts of our technical and management personnel. We may also be subject to significant damages or injunctions against development and sale of some of our products. Furthermore, claims of intellectual property infringement may require us to enter into royalty or license agreements with third parties, and we may be unable to obtain royalty or license agreements on commercially acceptable terms, if at all.
We may be subject to claims by third parties asserting that our employees or we have misappropriated their intellectual property, or claiming ownership of what we regard as our own intellectual property.
24
Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that these employees or we have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such employee’s former employer. Litigation may be necessary to defend against these claims.
In addition, while it is our policy to require our employees and contractors who may be involved in the development of intellectual property in the performance of their work to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing an enforceable agreement with each party who in fact develops intellectual property that we regard as our own. Relevant assignment agreements may not be self-executing or may be breached, and we may be forced to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of what we regard as our intellectual property.
If we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in prosecuting or defending against such claims, litigation could result in substantial costs and be a distraction to management.
Risks related to our research and development activities
We have a single research and development facility and we may be unable to continue to conduct our research and development activities if we lose this facility. If our facility or our equipment were damaged or destroyed, or if we experience a significant disruption in our operations for any reason, our ability to continue to operate our business could be materially harmed.
We currently conduct all of our research and development and product development activities in our existing facility in Tucson, Arizona. If this facility were to be damaged, destroyed or otherwise unable to operate, whether due to fire, floods, storms, tornadoes, other natural disasters, employee malfeasance, terrorist acts, power outages or otherwise, or if our business is disrupted for any other reason, we may not be able to continue the development of future products or test our products as promptly as our potential customers expect, or possibly not at all, and we would have no other means of conducting such activities until we were able to restore such capabilities at the current facility or develop an alternative facility. Further, in such an event, we may lose revenue and significant time during which we might otherwise have conducted research and development and product development activities. Further, we may not be able to maintain our relationships with our licensees or customers.
The manufacture of components of the Accelerate Pheno™ system involves complex processes, sophisticated equipment and strict adherence to specifications and quality systems procedures. Any unforeseen manufacturing problems, such as contamination of our facility, equipment malfunction or failure to strictly follow procedures or meet specifications, could result in delays or shortfalls in production of our products. Identifying and resolving the cause of any manufacturing issues could require substantial time and resources. If we are unable to keep up with future demand for our products by successfully manufacturing and shipping our products in a timely manner, our revenue growth could be impaired and market acceptance of our product candidates could be adversely affected.
While we carry a nominal amount of business interruption insurance to cover lost revenue and profits, this insurance does not cover all possible situations. If we have underestimated our insurance needs with respect to an interruption, or if an interruption is not subject to coverage under our insurance policies, we may not be able to cover our losses. In addition, our business interruption insurance would not compensate us for the loss of opportunity and potential adverse impact on relations with our licensees or customers.
We use hazardous materials in some of our research, development and manufacturing processes and face the accompanying risks and regulations governing environmental safety.
Our operations are subject to complex and stringent environmental, health, safety and other governmental laws and regulations that both public officials and private individuals may seek to enforce. In particular, our research activities sometimes involve the controlled use of various hazardous materials. Although we believe that our safety procedures for handling and disposing of such materials comply with the standards prescribed by state and federal regulations, the risk of accidental contamination or injury from these materials cannot be completely eliminated, and we may not be in compliance with these regulations. In addition, existing laws and regulations may also be revised or reinterpreted, or new laws and regulations may become applicable to us, whether retroactively or prospectively,
25
causing us to incur additional compliance costs and/or change the manner in which we operate. We could be held liable for any damages that might result from any accident or release involving hazardous materials.
Disruptions in the supply of raw materials, consumable goods or other key product components, or issues associated with their quality from our single source suppliers, could result in a significant disruption in sales and profitability.
We must manufacture or engage third parties to manufacture components of our products in sufficient quantities and on a timely basis, while maintaining product quality, acceptable manufacturing costs and complying with regulatory requirements. Our components are custom-made by only a few outside suppliers. In certain instances, we have a sole source supply for key product components of the Accelerate Pheno™ system. We may be unable to satisfy our forecasted demand from existing suppliers for our products, or we may be unable to find alternative suppliers for key product components or ancillary items at reasonably comparable prices. If this occurs, we may be unable to manufacture our products and/or meet our customers’ needs in a timely manner or at all.
Additionally, we have entered into supply agreements with most of our suppliers to help ensure component availability and flexible purchasing terms with respect to the purchase of such components. If our suppliers discontinue production of a key component for one or more of our products, we may be unable to identify or secure a viable alternative on reasonable terms, or at all, which could limit our ability to manufacture our products. While we may be able to modify our product candidates to utilize a new source of components, we may need to secure marketing authorization from the FDA or other regulatory clearance for the modified product, and it could take considerable time and expense to perform the requisite tasks prior to seeking such authorization.
In determining the required quantities of our products and our manufacturing schedule, we will need to make significant judgments and estimates regarding factors such as market trends and any seasonality with respect to our sales. Because of the inherent nature of estimates and our lack of experience marketing the Accelerate Pheno™ system, there could be significant differences between our estimates and the actual amounts of products that we require. This can result in shortages if we fail to anticipate demand, or excess inventory and write-offs if we order more than we need.
Reliance on third-party manufacturers entails risk to which we would not be subject if we manufactured these components ourselves, including:
• | reliance on third parties for regulatory compliance and quality assurance; |
• | possible breaches of manufacturing agreements by the third parties because of factors beyond our control; |
• | possible regulatory violations or manufacturing problems experienced by our suppliers; |
• | possible termination or non-renewal of agreements by third parties, based on their own business priorities, at times that are costly or inconvenient for us; |
• | the potential obsolescence and/or inability of our suppliers to obtain required components; |
• | the potential delays and expenses of seeking alternate sources of supply or manufacturing services; |
• | the inability to qualify alternate sources without impacting performance claims of our products; |
• | reduced control over pricing, quality and timely delivery due to the difficulties in switching to alternate suppliers or assemblers; and |
• | increases in prices of raw materials and key components. |
The manufacturing operations for the Accelerate Pheno™ system use highly technical processes involving unique, proprietary techniques. In addition, the manufacturing equipment we use would be costly to repair or replace and could require substantial lead time to repair or replace. Any interruption in our operations or decrease in the production capacity of our manufacturing facility or the facilities of any of our suppliers because of equipment failure, natural disasters such as earthquakes, tornadoes and fires, or otherwise, would limit our ability to meet customer demand for our products. In the event of a disruption, we may lose customers and we may be unable to regain those
26
customers thereafter. Our insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all.
We have made and intend to make significant additional investments in research and development, but there is no guarantee that any of these investments will ultimately result in a commercial product that will generate revenues.
The Accelerate Pheno™ system integrates several of our component products, systems and processes. We have dedicated significant resources on research and development activities, and we intend to spend significantly more on research and development activities. Notwithstanding these investments, we anticipate that we will have to spend additional funds in the research and development of the Accelerate Pheno™ system, particularly with respect to its use for additional specimen types. There can be no assurance that we will be able to obtain marketing authorization from the FDA of the Accelerate Pheno™ system for its use with additional specimen types. There can also be no assurance that we will be able to develop additional types of tests and instruments in the future.
Risks related to government regulation
Legislative and Administrative Action May Have an Adverse Effect on Our Company
Political, economic and regulatory influences are subjecting the health care industry in the U.S. to fundamental change. We cannot predict what other legislation relating to our business or to the health care industry may be enacted, including legislation relating to third-party reimbursement, or what effect such legislation may have on our business, prospects, operating results and financial condition. We expect federal and state legislators to continue to review and assess alternative health care delivery and payment systems, and possibly adopt legislation affecting further changes in the health care delivery system. Such laws may contain provisions that may change the operating environment for hospitals and managed care organizations. Health care industry participants may react to such legislation by curtailing or deferring expenditures and initiatives, including those relating to our products. Future legislation could result in modifications to the existing public and private health care insurance systems that would have a material adverse effect on the reimbursement policies discussed above. With growing pressures on government budgets due to the current economic downturn, government efforts to contain or reduce health care spending are likely to gain increasing emphasis. It is uncertain what impact the presidency of Donald Trump will have on health care spending in light of his promise to repeal the Affordable Care Act. If enacted and implemented, any measures to restrict health care spending could result in decreased revenue from our products and decrease potential returns from our research and development initiatives. Furthermore, we may not be able to successfully neutralize any lobbying efforts against any initiatives we may have with governmental agencies.
We and our suppliers, contract manufacturers and customers are subject to various governmental laws and regulations, and we may incur significant expenses to comply with, and experience delays in our product commercialization as a result of, these laws and regulations.
Our operations are affected by various state, federal, and international healthcare, environmental, anti-corruption, fraud and abuse (including anti-kickback and false claims laws), privacy, and employment laws as well as international political sanctions. Violations of these laws and and sanctions can result in criminal or civil penalties, including substantial fines and, in some cases, exclusion from participation in federal health care programs such as Medicare and Medicaid and individual liability and imprisonment.
We are also subject to extensive regulation by the FDA pursuant to the Federal Food, Drug, and Cosmetic Act, by comparable agencies in foreign countries and by other regulatory agencies and governing bodies. Following the introduction of a product, these and other government agencies will periodically review our manufacturing processes, product performance and compliance with applicable requirements.
We are also subject to various U.S. healthcare related laws regulating sales, contracting, marketing, and other business arrangements and the use and disclosure of individually identifiable health information. These include but are not limited to:
• | The federal Anti-Kickback Statute, which prohibits persons from knowingly and willfully offering, providing, soliciting, or receiving any remuneration, directly or indirectly, in exchange for or to induce the referral of an individual, or the purchasing, leasing, ordering, recommending, furnishing or arranging for a good or service, for which payment may be made under a federal health care program, such as Medicare or |
27
Medicaid.
• | The Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which prohibits knowingly and willfully (i) executing a scheme to defraud any health care benefit program, including private payers, or (ii) falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for items or services under a health care benefit program. |
• | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, which also restricts the use and disclosure of protected health information, mandates the adoption of standards relating to the privacy and security of protected health information, and requires us to report certain security breaches to health care provider customers with respect to such information where we are acting as a HIPAA business associate to that customer. |
• | The federal Physician Payment Sunshine Act, which requires manufacturers of certain medical devices to track payments or other transfers of value given to U.S. licensed physicians or teaching hospitals and to report this data to CMS annually for subsequent public disclosure. |
• | The federal False Claims Act, which imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal health care program. The qui tam provisions of the False Claims Act allow a private individual to bring actions on behalf of the federal government alleging that the defendant has submitted a false claim to the federal government and to share in any monetary recovery. |
Similar requirements have been adopted by many states and foreign countries. Violations of any of these laws can lead to additional legal risk such as risk of plaintiff class actions, state Attorney General actions, and investigations by the Federal Trade Commission, among others.
Failure to comply with applicable requirements, or later discovery of previously unknown problems with our products or manufacturing processes, including our failure or the failure of one of our contract manufacturers to take satisfactory corrective action in response to an adverse inspection, can result in, among other things:
• | administrative or judicially imposed sanctions; |
• | injunctions or the imposition of civil penalties; |
• | recall or seizure of our products; |
• | total or partial suspension of production or distribution; |
• | withdrawal or suspension of marketing clearances or approvals; |
• | clinical holds; |
• | warning letters; |
• | refusal to permit the import or export of our products; |
• | criminal prosecution; and |
• | exclusion or debarment from participation in federal health care programs such as Medicare and Medicaid. |
Any of these actions, in combination or alone, could prevent us from marketing, distributing and selling our products.
In addition, we have developed, configured and we intend to market our products to meet customer needs created by these various regulations. Any significant change in these regulations could reduce demand for our products. Governmental agencies may also impose new requirements regarding registration, labeling or prohibited materials
28
that may require us to modify or re-register products already on the market or otherwise adversely impact our ability to market our products.
In addition, a product defect or regulatory violation could lead to a government-mandated or voluntary recall by us. We believe that the FDA would request that we initiate a voluntary recall if a product was defective or presented a risk of injury or gross deception. Regulatory agencies in other countries have similar authority to recall devices because of material deficiencies or defects in design or manufacture that could endanger health. Any recall would divert management attention and financial resources, could cause the price of our shares of common stock to decline, expose us to product liability or other claims (including contractual claims from parties to whom we sold products) and harm our reputation with customers.
The use of our diagnostic products by our customers is also affected by the Clinical Laboratory Improvement Amendments (“CLIA”) and related federal and state regulations that provide for regulation of laboratory testing. CLIA is intended to ensure the quality and reliability of clinical laboratories in the United States by mandating specific standards in the areas of personnel qualifications, administration, participation in proficiency testing, patient test management, quality assurance, quality control and inspections. Current or future CLIA requirements or the promulgation of additional regulations affecting laboratory testing may prevent some laboratories, hospitals, providers or other customers with laboratories from using some or all of our diagnostic products.
Maintaining adequate sales of our product may depend on the availability of adequate reimbursement to our customers from third-party payers, including government programs such as Medicare and Medicaid, private insurance plans, and managed care programs.
Maintaining and growing sales of our product, if approved, may depend in part on the availability of adequate reimbursement to our customers from third-party payers, including government programs such as Medicare and Medicaid, private insurance plans and managed care programs. Hospitals, clinical laboratories and other healthcare provider customers that may purchase our products generally bill various third-party payers to cover all or a portion of the costs and fees associated with diagnostic tests, including the cost of the purchase of our products. We currently expect that all of our diagnostic tests will be performed in a hospital inpatient setting, where governmental payers, such as Medicare, generally reimburse hospitals a single bundled payment that is based on the patient’s diagnosis under a classification system known as the Medicare severity diagnosis-related groups (MS-DRGs) classification for all items and services provided to the patient during a single hospitalization, regardless of whether our diagnostic tests are performed during such hospitalization. As a result, our customers’ access to adequate payment by government and private insurance plans is central to the acceptance of our products. We may be unable to sell our products, if approved, on a profitable basis if third-party payers reduce their current levels of payment or if our costs of production increase faster than increases in reimbursement levels.
Additionally, third-party payers are increasingly reducing reimbursement for medical products and services. In addition, the U.S. government, state legislatures, and foreign governments have and may continue to implement cost-containment measures and more restrictive policies, including price controls and restrictions on reimbursement. For example, the Budget Control Act of 2011 (the “Budget Control Act”) established a process to reduce federal budget deficits through an automatic “sequestration” process if deficit reductions targets are not otherwise reached. Under the terms of the Budget Control Act, sequestration imposes cuts to a wide range of federal programs, including Medicare, which is subject to a two percent cut. The Bipartisan Budget Act of 2013 extended the two percent sequestration cut for Medicare through fiscal year 2023, and a bill signed by President Obama on February 15, 2014 further extended this cut for an additional year, through fiscal year 2024. For fiscal year 2024, however, Medicare sequestration amounts will be realigned such that there will be a four percent sequester for the first six months and no sequester for the second six months, under the Protecting Access to Medicare Act of 2014.
While we cannot predict whether third-party reimbursement to our customers will be adequate, cost-containment measures and similar efforts by third-party payers, including government programs such as Medicare and Medicaid, could substantially impact the sales of our products and potentially limit our net revenue and results.
We may be adversely affected by healthcare policy changes, including additional healthcare reform and changes in managed healthcare.
Healthcare reform and the growth of managed care organizations have been considerable forces in the medical diagnostics industry and in recent political discussions. These forces have placed, and are expected to continue to place, constraints on the levels of overall pricing for healthcare products and services as well as the coverage available
29
by public and private insurance and thus, could have a material adverse effect on the future profit margins of our products or the amounts that we are able to receive from third parties for the licensing of our products. Changes in the United States healthcare market could also force us to alter our approach to selling, marketing, distributing and servicing our products and customer base. In and outside the United States, changes to government reimbursement policies could reduce the funding that healthcare service providers have available for diagnostic product expenditures, which could have a material adverse impact on the use of the products we are developing and our future sales, license and royalty fees and profit margin.
For example, the Affordable Care Act (the “ACA”), enacted in March 2010, made changes that have significantly impacted the medical device industry and other healthcare providers. The legislation requires, among other mandates, that certain medical device manufacturers pay an excise tax in an amount equal to 2.3% of the price for which such manufacturer sells its medical devices (with exception of a moratorium period from January 1, 2016 through December 31, 2019). The ACA also requires CMS to reduce payments to hospitals reimbursed under Medicare’s Inpatient Prospective Payment System (“IPPS”) that have excess readmissions. These and other applicable requirements set forth under the ACA and its current and future implementing regulations may significantly increase our costs, and/or reduce our customer’s ability to obtain adequate reimbursement for tests performed with our products, which could adversely affect our business and financial condition.
In recent years, other legislative, regulatory, and political changes aimed at regulating healthcare delivery in general and clinical laboratory tests in particular have been proposed and adopted in the United States. Reimbursement for the laboratory industry is under significant pressure. In January 2015, HHS announced a plan to shift the Medicare program and the healthcare system at large, toward paying providers based on quality, rather than the quantity of care provided to patients. On July 31, 2015, CMS issued a final rule addressing the IPPS for fiscal year 2016, which included penalties for readmissions and for hospitals in the worst performing quartile of the Hospital Acquired Condition Reduction Program. With respect to clinical laboratory tests, on April 1, 2014, President Obama signed into law the Protecting Access to Medicare Act of 2014 (“PAMA”), revamping Medicare’s clinical laboratory reimbursement system to tie Medicare payment rates to private market rates beginning in 2017. CMS then released a proposed rule on September 25, 2015, to implement such provisions of PAMA, requiring applicable clinical laboratories to report private payer reimbursement rates and volume data for tests on the clinical laboratory fee schedule. These measures can result in reduced prices, added costs, and decreased test utilization for our customers, although the full impact on our business of the ACA, changes to the IPPS, PAMA, and other applicable laws, regulations, and policies is uncertain.
We cannot predict whether future healthcare initiatives will be implemented at the federal or state level or in countries outside of the United States in which we may do business, or the effect of any future legislation or regulation will have on our industry generally, our ability to successfully commercialize the Accelerate Pheno™ system, and our overall business operations. Changes in healthcare policy could substantially impact the sales of our tests, increase costs and divert management’s attention from our business. For example, expansion in the government’s regulation of the United States healthcare system may result in decreased profits to us, lower reimbursements to our customers for laboratory testing or reduced medical procedure volumes.
The regulatory processes applicable to our products and operations are expensive, time-consuming, and uncertain and may prevent us from obtaining required approvals for the commercialization of our products.
Our products, including the Accelerate Pheno™ system, are regulated as medical device products by the FDA and comparable agencies of other countries. In particular, FDA regulations govern activities such as product development, product testing, product labeling, product storage, premarket clearance or approval, manufacturing, advertising, promotion, product sales, reporting of certain product failures and distribution. Some of our products, depending on their intended use, will require approval of a premarket approval application (“PMA”) or clearance of a 510(k) notification from the FDA prior to marketing. The FDA has committed to review most 510(k) decisions within 90 days, but the review may be delayed due to requests for additional information. A decision may take significantly longer, and clearance is never assured. The PMA process is much more costly, lengthy and uncertain. The FDA has committed to review most PMAs within 180 days where an advisory panel is not required and within 320 days where an advisory panel is required, but the review may be delayed due to requests for additional information. A decision may take significantly longer, and approval is never assured. In the 510(k) clearance process, the FDA must determine that a proposed device is “substantially equivalent” to a device legally on the market, known as a “predicate” device, with respect to intended use, technology and safety and effectiveness, in order to clear the proposed device for marketing. Clinical data is sometimes required to support substantial equivalence. The PMA pathway requires an applicant to demonstrate the safety and effectiveness of the device based, in part, on extensive data, including technical, preclinical, clinical trial, manufacturing and labeling data. The PMA process is typically required for devices that are
30
deemed to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices. However, some devices are automatically subject to the PMA pathway regardless of the level of risk they pose, because they have not previously been classified into a lower risk class by the FDA. Manufacturers of these devices may request that the FDA review such devices in accordance with the de novo classification procedure, which allows a manufacturer whose novel device would otherwise require the submission and approval of a PMA prior to marketing to request down-classification of the device on the basis that the device presents low or moderate risk. If the FDA agrees with the down-classification, the applicant will then receive authorization to market the device. This device type can then be used as a predicate device for future 510(k) submissions.
The FDA can delay, limit or deny clearance or approval of a device for many reasons, including:
• | we may not be able to demonstrate to the FDA’s satisfaction that our product candidates are safe and effective, sensitive and specific diagnostic tests, for their intended users; |
• | the data from our pre-clinical studies and clinical trials may be insufficient to support clearance or approval, where required; and |
• | the manufacturing process or facilities we or our contract manufacturers use may not meet applicable requirements. |
With respect to those future products where a PMA is not required, we cannot assure you that we will be able to obtain 510(k) clearances with respect to those products. The process of obtaining regulatory clearances or approvals, or completing the de novo classification process, to market a medical device can be costly and time consuming, and we may not be able to successfully obtain pre-market reviews on a timely basis, if at all. Further, even if we were to obtain regulatory clearance, it may not be for the uses we believe are important or commercially attractive, in which case we would not be permitted to market our product for those uses.
Clinical trial data is typically required to support a PMA and is sometimes required for a 510(k) pre-market notification. Although many 510(k) pre-market notifications are cleared without clinical data, in some cases, the FDA requires significant clinical data to support substantial equivalence. Clinical trials are expensive and time-consuming. In addition, the commencement or completion of any clinical trials may be delayed or halted for any number of reasons, including product performance, changes in intended use, changes in medical practice and the opinion of evaluator Institutional Review Boards.
Additionally, since 2009, the FDA has significantly increased the scrutiny applied to its oversight of companies subject to its regulations by hiring new investigators and increasing inspections of manufacturing facilities. The FDA has also undertaken initiatives related to enhancement of the 510(k) review process and has proposed significant changes to the regulation of laboratory developed tests (“LDTs”). We continue to monitor these developments and analyze how they will impact the approval of our products. These and other actions proposed by the FDA’s Center for Devices and Radiological Health could result in significant changes to the 510(k) process, which could complicate the product approval process, although we cannot predict the effect of such changes and cannot ascertain if such changes will have a substantive impact on the approval of our products. If we fail to adequately respond to the increased scrutiny and streamlined 510(k) submission process, our business may be adversely impacted.
Failure to comply with the applicable requirements can result in, among other things, warning letters, administrative or judicially imposed sanctions such as injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, refusal to grant premarket clearance or PMA for devices, withdrawal of marketing clearances or approvals, or criminal prosecution. With regard to products for which we seek 510(k) clearance or PMA approval from the FDA, any failure or material delay to obtain such clearance or approval could harm our business. If the FDA were to disagree with our regulatory assessment and conclude that approval or clearance is necessary to market the products, we could be forced to cease marketing the products and seek approval or clearance. Once clearance or approval has been obtained for a product, there is an obligation to ensure that all applicable FDA and other regulatory requirements continue to be met.
In addition, it is possible that the current regulatory framework could change or additional regulations could arise at any stage during our product development or marketing, which may adversely affect our ability to obtain or maintain approval of our products. For example, in response to industry and healthcare provider concerns regarding the predictability, consistency and rigor of the 510(k) regulatory pathway, the FDA initiated an evaluation of the program, and in January 2011, announced several proposed actions intended to reform the review process governing the
31
clearance of medical devices. The FDA undertook these reform actions to improve the efficiency and transparency of the clearance process, as well as bolster patient safety. In addition, as part of the Food and Drug Administration Safety and Innovation Act, Congress reauthorized the Medical Device User Fee Amendments with various FDA performance goal commitments and enacted several “Medical Device Regulatory Improvements” and miscellaneous reforms that are further intended to clarify and improve medical device regulation both pre- and post-approval. Any delay in, or failure to receive or maintain, clearance or approval for our product candidates could prevent us from generating revenue from these product candidates. Additionally, the FDA and other regulatory authorities have broad enforcement powers. Regulatory enforcement or inquiries, or other increased scrutiny on us, could affect the perceived safety and efficacy of our product candidates and dissuade our customers from using our product candidates, if and when they are authorized for marketing.
Our manufacturing facility located in Tucson, Arizona, where we assemble and produce the Accelerate Pheno™ system, is subject to periodic regulatory inspections by the FDA and other federal and state and foreign regulatory agencies. For example, this facility is subject to Quality System Regulations (“QSR”) of the FDA and is subject to annual inspection and licensing by the State of Arizona. If we fail to maintain this facility in accordance with the QSR requirements, international quality standards or other regulatory requirements, our manufacturing process could be suspended or terminated, which would prevent us from being able to provide products to our customers in a timely fashion.
Sales of our diagnostic product candidates outside the United States are subject to foreign regulatory requirements governing clinical studies, vigilance reporting, marketing approval, manufacturing, product licensing, pricing and reimbursement. These regulatory requirements vary greatly from country to country. As a result, the time required to obtain approvals outside the United States may differ from that required to obtain FDA marketing authorization from the FDA, and we may not be able to obtain foreign regulatory approvals on a timely basis or at all. Marketing authorization from the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure clearance or approval by regulatory authorities in other countries or by the FDA. Foreign regulatory authorities could require additional testing. Failure to comply with these regulatory requirements, or to obtain required clearances or approvals, could impair our ability to commercialize our diagnostic product candidates outside of the United States.
Modifications to our products, if cleared or approved, may require new 510(k) clearances or pre-market approvals, or may require us to cease marketing or recall the modified products until clearances are obtained.
Any modification to a device authorized for marketing that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, design or manufacture, requires a new 510(k) clearance or, possibly, approval of a PMA supplement or new PMA. The FDA requires every manufacturer to make this determination in the first instance, but the FDA may review any manufacturer’s decision. The FDA may not agree with our decisions regarding whether new clearances or approvals are necessary. If the FDA disagrees with our determination and requires us to submit new 510(k) notifications, PMA supplements or PMAs for modifications to previously cleared or approved products for which we conclude that new clearances or approvals are unnecessary, we may be required to cease marketing or to recall the modified product until we obtain clearance or approval, and we may be subject to significant regulatory fines or penalties.
Furthermore, the FDA’s ongoing review of the 510(k) program may make it more difficult for us to make modifications to any products for which we obtain clearance, either by imposing more strict requirements on when a manufacturer must submit a new 510(k) for a modification to a previously cleared product, or by applying more onerous review criteria to such submissions. The practical impact of the FDA’s continuing scrutiny of the 510(k) program remains unclear.
We rely on third parties to conduct studies of our products that may be required by the FDA or other regulatory authorities, and those third parties may not perform satisfactorily.
We rely on third parties, including medical investigators, to conduct studies on our products. Our reliance on these third parties for clinical development activities will reduce our control over these activities. These third parties may not complete activities on schedule or conduct studies in accordance with regulatory requirements or our study design. If applicable, our reliance on third parties that we do not control will not relieve us of any applicable requirement to prepare, and ensure compliance with, various procedures required under good clinical practices. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if the third parties need to be replaced or if the quality or accuracy of the data they obtain is compromised due to their
32
failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our studies may be extended, delayed, suspended or terminated, and we may not be able to obtain marketing authorization from the FDA or regulatory clearance for our products.
If we obtain marketing authorization from the FDA, a recall of our products, either voluntarily or at the direction of the FDA, or the discovery of serious safety issues with our products that leads to corrective actions, could have a significant adverse impact on us.
The FDA and similar foreign governmental authorities have the authority to require the recall of commercialized products. Manufacturers may, under their own initiative, recall a product if any material deficiency in a device is found. A government-mandated or voluntary recall by us or one of our distributors could occur as a result of an unacceptable risk to health, component failures, manufacturing errors, design or labeling defects or other deficiencies and issues. Under the FDA’s medical device reporting regulations, we are required to report to the FDA any incident in which our product may have caused or contributed to a death or serious injury or in which our product malfunctioned and, if the malfunction were to recur, would likely cause or contribute to death or serious injury. Repeated product malfunctions may result in a voluntary or involuntary product recall. Recalls of any of our products would divert managerial and financial resources, have an adverse effect on our reputation, and may impair our ability to produce our products in a cost-effective and timely manner in order to meet our customers’ demands. Depending on the corrective action we take to redress a product’s deficiencies or defects, the FDA may require, or we may decide that we will need to obtain, new approvals or clearances for the device before we may market or distribute the corrected device. Seeking such approvals or clearances may delay our ability to replace the recalled devices in a timely manner. Moreover, if we do not adequately address problems associated with our devices, we may face additional regulatory enforcement action, including FDA warning letters, product seizure, injunctions, administrative penalties, or civil or criminal fines. We may also be required to bear other costs or take other actions that may have a negative impact on our sales as well as face significant adverse publicity or regulatory consequences, which could harm our ability to market our products in the future.
Any adverse event involving our products could result in future voluntary corrective actions, such as recalls or customer notifications, or agency action, such as inspection, mandatory recall or other enforcement action. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, would require the dedication of our time and capital, distract management from operating our business and may harm our reputation.
Risks related to our common stock
Our stock price has been volatile and may continue to be volatile and traded on low volumes.
The trading price of our common stock has been, and is likely to continue to be, highly volatile. One factor contributing to volatility in the price of our common stock is the low trading volume currently prevailing in the market for our shares. Further, due to the concentration of the stock the sale of one individual shareholder may have a material impact on the price of the stock. The market value of your investment in our common stock may rise or fall sharply at any time because of this volatility and also because of significant short positions that may be taken by investors from time to time in our common stock. During the year ended December 31, 2017, the sale price for our common stock ranged from $16.75 to $30.45 per share, and during the year ended December 31, 2016, the sale price for our common stock ranged from $10.29 to $28.5 per share. The market prices for securities of medical technology companies like us historically have been highly volatile, and the market has experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies.
The short interest in our common stock is high.
As of December 31, 2017, the number of shares shorted was high as compared to the number of shares in the public float. A high short interest can be a contributing factor to a highly volatile stock price.
The ownership of our common stock is highly concentrated.
As of December 31, 2017, our directors and executive officers, together with members of their immediate families, as a group, will beneficially own, in the aggregate, approximately 49% of our outstanding capital stock, including 29% held by our director, Jack Schuler. As a result, these stockholders will be able to affect the outcome of, or exert significant influence over, all matters requiring stockholder approval, including the election and removal of directors and any change in control. In particular, this concentration of ownership of our common stock could have the effect
33
of delaying or preventing a change in control of us or otherwise discouraging or preventing a potential acquirer from attempting to obtain control of us. This, in turn, could have a negative effect on the market price of our common stock. It could also prevent our stockholders from realizing a premium over the market prices for their shares of common stock. Moreover, the interests of this concentration of ownership may not always coincide with our interests or the interests of other stockholders. The concentration of ownership also contributes to the low trading volume and volatility of our common stock.
Future sales of shares of our common stock may depress the price of our shares and be dilutive to our existing stockholders.
We cannot predict whether future issuances of shares of our common stock or the availability of shares for resale in the open market will decrease the market price per share of our common stock. Any sales by us or by our existing stockholders of a substantial number of shares of our common stock in the public market, or the perception that such sales might occur, may cause the market price of our shares to decline. The exercise of any options or warrants, the issuance of our common stock in connection with acquisitions and other issuances of our common stock could have an adverse effect on the market price of the shares of our common stock.
To the extent that we raise additional funds through the sale of equity or convertible debt securities, the issuance of such securities will result in dilution to our stockholders. Investors purchasing shares or other securities in the future could have rights superior to existing stockholders. In addition, we have a significant number of options and warrants outstanding. If the holders of these options or warrants exercise such securities, you may incur further dilution.
We may require additional capital in the future, and you may incur dilution to your stock holdings.
We have primarily relied upon capital from the sale of our securities to fund our operations. Although we are beginning to commercialize the Accelerate Pheno™ system, there can be no assurance that our commercialization efforts will be successful or that we will not continue to incur operating losses. If capital requirements vary materially from those currently forecast by management, we may require additional capital sooner than expected. We may also require additional capital in the future to expand our product offerings, expand our sales and marketing infrastructure, increase our manufacturing capacity, fund our operations, and continue our research and development activities. Our future funding requirements will depend on many factors, including:
• | our ability to obtain marketing authorization from the FDA or clearance from the FDA to market our product candidates; |
• | market acceptance of our product candidates, if cleared; |
• | the cost and timing of establishing sales, marketing and distribution capabilities; |
• | the cost of our research and development activities; |
• | the ability of healthcare providers to obtain coverage and adequate reimbursement by third-party payers for procedures using our products; |
• | the cost and timing of marketing authorization or regulatory clearances; |
• | the cost of goods associated with our product candidates; |
• | the cost of customer disruptions due to supply disruptions; |
• | the effect of competing technological and market developments; and |
• | the extent to which we acquire or invest in businesses, products and technologies, including entering into licensing or collaboration arrangements for product candidates, although we currently have no commitments or agreements to complete any such transactions. |
If we require additional capital, we may attempt to raise it through a variety of strategies, including the issuance and sale of additional shares of our common stock. Issuances of additional shares of our common stock or preferred
34
stock in the future, whether in connection with a rights offering, follow-on offering or otherwise, would dilute existing stockholders and may adversely affect the market price of our common stock.
We cannot assure you that we will be able to obtain additional funds on acceptable terms, or at all. Debt financing, if available, may involve covenants restricting our operations or our ability to incur additional debt. Any debt or additional equity financing that we raise may contain terms that are not favorable to us or our stockholders. If we raise additional funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish some rights to our technologies or our products, or grant licenses on terms that are not favorable to us. If we are unable to raise adequate funds, we may have to liquidate some or all of our assets or delay, reduce the scope of or eliminate some or all of our product development.
If we do not have, or are not able to obtain, sufficient funds, we may be required to delay additional product development or license to third parties the rights to commercialize our products or technologies that we would otherwise seek to commercialize ourselves. We also may have to reduce marketing, customer support or other resources devoted to our product candidates or cease operations. Any of these factors could harm our operating results.
Negative reports issued by securities analysts, and the election by securities analysts not to cover us, may have a negative impact on the market price of our common stock.
The trading market for our common stock may be affected in part by the research and reports that industry or financial analysts publish about us or our business, and our failure to achieve analyst earnings estimates. It may be difficult for companies such as ours, with smaller market capitalizations, to attract securities analysts that will cover our common stock. The lack of research coverage may adversely affect the market price of our common stock. If one or more of the analysts who elects to cover us downgrades our stock, our stock price may decline rapidly. If one or more of these analysts ceases coverage of our Company, we could lose visibility in the market, which in turn may cause our stock price to decline.
We do not anticipate paying any cash dividends on our capital stock in the foreseeable future.
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all of our future earnings, if any, to finance the growth and development of our business. Any future debt agreements may also preclude us from paying dividends. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future.
Provisions in our certificate of incorporation and bylaws and Delaware law may delay or prevent acquisition of our Company, which could adversely affect the value of our common stock.
Provisions contained in our certificate of incorporation and bylaws, as well as provisions of the Delaware General Corporation Law, could delay or make it more difficult to remove incumbent directors or for a third party to acquire us, even if a takeover would benefit our stockholders. For example, our board of directors may fill any vacancy on the board of directors, whether such vacancy occurs as a result of an increase in the number of directors or otherwise. Stockholders may only take action by written consent if acting unanimously. Special meetings of the stockholders may be called only by the President, a Vice President, our board of directors or the holders of not less than one-tenth of all the shares entitled to vote at the meeting. Additionally, our board of directors has the authority to cause us to issue, without any further vote or action by the stockholders, up to 5.0 million shares of preferred stock, par value $0.001 per share, in one or more series, to fix the number of shares constituting such series and the designation of such series, the voting powers, if any, of the shares of such series, and the preferences and relative, participating, optional or other special rights, if any, and any qualifications, limitations or restrictions thereof, of the shares of such series. The issuance of shares of preferred stock may have the effect of delaying, deferring or preventing a change in control of our Company without further action by the stockholders, even where stockholders are offered a premium for their shares. Moreover, we are subject to the provisions of Section 203 of the General Corporation Law of the State of Delaware, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
Item 1B. Unresolved Staff Comments
Not applicable.
35
Item 2. Properties
Our headquarters is located in Tucson, Arizona, and we have other offices in Europe. As of December 31, 2017 and 2016, we leased approximately 53,833 and 53,448 square feet of office, laboratory and manufacturing space, respectively. We believe that our currently leased facilities are adequate to meet our needs for the foreseeable future. The leases have a remaining term through 2022. See Item 8, Note 17, Commitments and Contingencies for additional details regarding the leases.
Item 3. Legal Proceedings
We are from time to time subject to various claims and legal actions in the ordinary course of our business. Other than the patent Opposition proceeding discussed under the heading ”Risks related to our intellectual property,” in Item 1A, Risk Factors of this Annual Report on Form 10-K, which is incorporated herein by reference, we believe that there are currently no claims or legal actions that would reasonably be expected to have a material adverse effect on our results of operations or financial condition.
Item 4. Mine Safety Disclosures
Not applicable.
PART II
Item 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issue Purchases of Equity Securities
Market Information
The information in the following table sets forth the quarterly high and low sales price per share of our Common Stock, as reported by the NASDAQ Capital market, for the period from January 1, 2016 through December 31, 2017.
Common Stock Closing Price | ||
Quarter Ended | High | Low |
March 31, 2016 | $22.97 | $10.29 |
June 30, 2016 | 17.17 | 10.87 |
September 30, 2016 | 28.50 | 14.39 |
December 31, 2016 | 27.95 | 19.50 |
March 31, 2017 | 29.40 | 19.20 |
June 30, 2017 | 30.45 | 22.65 |
September 30, 2017 | 29.50 | 19.65 |
December 31, 2017 | 30.10 | 16.75 |
Performance Graph
The following Performance Graph compares the cumulative 5-year total stockholder return on our common stock relative to the cumulative total returns of the NASDAQ Composite index (XCMP) and the NASDAQ Biotechnology index (XNBI). An investment of $100 (with reinvestment of all dividends) is assumed to have been made in our common stock and in each of the indexes on December 31, 2012 and its relative performance is tracked through December 31, 2017. The Performance Graph and related information shall not be deemed to be “soliciting material” or to be “filed” with the Securities and Exchange Commission, nor shall such information be incorporated by reference into any filing under the Securities Act of 1933 or Securities Exchange Act of 1934, each as amended, except to the extent that we specifically incorporate it by reference into such filing.
36
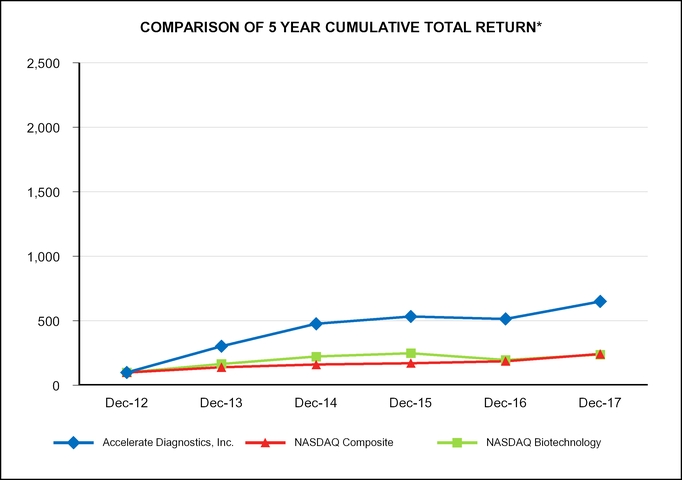
Dec-12 | Dec-13 | Dec-14 | Dec-15 | Dec-16 | Dec-17 | |
Accelerate Diagnostics, Inc. | 100.00 | 302.73 | 476.18 | 533.25 | 514.89 | 650.12 |
NASDAQ Composite | 100.00 | 140.12 | 160.78 | 171.97 | 187.22 | 242.71 |
NASDAQ Biotechnology | 100.00 | 165.97 | 223.07 | 249.32 | 196.09 | 238.51 |
* $100 invested on 12/31/2012 in stock or index, including reinvestment of dividends.
Holders
As of February 23, 2018, we had approximately 125 record owners of our Common Stock.
Dividends Paid and Dividend Policy
Holders of Common Stock are entitled to receive dividends as may be declared by the Board of Directors out of funds legally available. To date, no dividends have been declared by the Board of Directors. We currently intend to retain all available funds and any future earnings for use in the operation of our business and do not anticipate paying any cash dividends on our Common Stock for the foreseeable future.
Future cash dividends, if any, will be at the discretion of our Board of Directors and will depend upon our future operations and earnings, capital requirements and surplus, general financial condition, contractual restrictions and other factors as our Board of Directors may deem relevant. We do not intend to pay any cash dividends on our Common Stock in the foreseeable future.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
None.
37
Equity Compensation Plan Information
The table set forth below presents the securities authorized for issuance with respect to compensation plans under which equity securities are authorized for issuance as of December 31, 2017:
Equity Compensation Plan | |||||||
Plan category | Number of securities to be issued upon exercise of outstanding options, restricted stock, warrants and rights | Weighted average exercise price of available outstanding options, restricted stock, warrants and rights | Number of securities remaining for future issuance under equity compensation plans (excluding securities reflected in the 1st column) | ||||
Equity compensation plans approved by security holders | 7,352,281 | $ | 10.13 | 3,429,049 | |||
Equity compensation plans not approved by security holders | — | — | — | ||||
Total | 7,352,281 | $ | 10.13 | 3,429,049 | |||
Item 6. Selected Financial Data
The following selected consolidated financial data has been derived from our audited consolidated financial statements for the years ended December 31, 2013, through 2017. The information below is not necessarily indicative of the results of future operations, and should be read in conjunction with Item 7, Management's Discussion and Analysis of Financial Condition and Results of Operations and the consolidated financial statements and related notes thereto included in Item 8, Financial Statements and Supplementary Data in order to fully understand factors that may affect the comparability of the information presented below.
Selected Consolidated Financial Data (in thousands except per share data) | |||||||||||||||
12/31/2017 | 12/31/2016 | 12/31/2015 | 12/31/2014 | 12/31/2013 | |||||||||||
Net sales | $ | 4,177 | $ | 246 | $ | 147 | $ | 122 | $ | 48 | |||||
Net loss | (64,028 | ) | (66,374 | ) | (45,498 | ) | (30,933 | ) | (15,282 | ) | |||||
Basic and diluted loss per share (1) | (1.18 | ) | (1.29 | ) | (1.01 | ) | (0.71 | ) | (0.41 | ) | |||||
Cash dividends | — | — | — | — | — | ||||||||||
12/31/2017 | 12/31/2016 | 12/31/2015 | 12/31/2014 | 12/31/2013 | |||||||||||
Total assets | $ | 125,512 | $ | 82,852 | $ | 139,324 | $ | 69,801 | $ | 43,431 | |||||
Long-term obligations | 21 | — | — | 13 | — | ||||||||||
(1) Loss per share has been adjusted for the effects of the April 2014 rights offering.
38
Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations (“MD&A”) summarizes our change in fiscal year financial condition, results of operations, recent developments, the significant factors affecting our results of operations, capital resources and liquidity, off-balance sheet arrangements, contractual obligations, as well as discusses recent accounting pronouncements and our critical accounting policies and estimates. You should read the following discussion and analysis together with our financial statements, including the related notes, which are included in this Annual Report on Form 10-K. Certain information contained in the discussion and analysis set forth below and elsewhere in this report, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. See Item 1A, Risk Factors of this Annual Report on Form 10-K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements in this report.
39
Changes in Results of Operations: Comparison of fiscal years ended December 31, 2017, December 31, 2016 and December 31, 2015
December 31, | December 31, | ||||||||||||||||||||||
(in thousands) | (in thousands) | ||||||||||||||||||||||
2017 | 2016 | $ Change | % Change | 2016 | 2015 | $ Change | % Change | ||||||||||||||||
Net sales | $ | 4,177 | $ | 246 | $ | 3,931 | 1,598 | % | $ | 246 | $ | 147 | $ | 99 | 67 | % | |||||||
During the year ended December 31, 2017, total revenues increased compared to the year ended December 31, 2016, due to sales of Accelerate Pheno™ systems and Accelerate PhenoTest™ BC kits. The increase was in connection with the FDA granting Accelerate’s de novo request to market the Accelerate Pheno™ system and Accelerate PhenoTest™ BC kit.
During the year ended December 31, 2016, total revenues increased compared to the year ended December 31, 2015, due to sales of Research Use Only Accelerate Pheno™ systems.
December 31, | December 31, | ||||||||||||||||||||||
(in thousands) | (in thousands) | ||||||||||||||||||||||
2017 | 2016 | $ Change | % Change | 2016 | 2015 | $ Change | % Change | ||||||||||||||||
Cost of sales | $ | 1,002 | $ | — | $ | 1,002 | 100 | % | $ | — | $ | — | $ | — | — | % | |||||||
Gross profit | $ | 3,175 | $ | 246 | $ | 2,929 | 1,191 | % | $ | 246 | $ | 147 | $ | 99 | 67 | % | |||||||
During the year ended December 31, 2017, cost of sales and gross profit increased compared to the year ended December 31, 2016, as a result of higher sales. Inventory without a cost basis was sold to customers during the year ended December 31, 2017. This inventory comprised pre-launch inventory previously not capitalized and expensed in a previous period. Cost of sales associated with this inventory during the year ended December 31, 2017, would have been an additional $868,000.
December 31, | December 31, | ||||||||||||||||||||||
(in thousands) | (in thousands) | ||||||||||||||||||||||
2017 | 2016 | $ Change | % Change | 2016 | 2015 | $ Change | % Change | ||||||||||||||||
Research and development | $ | 22,301 | $ | 29,564 | $ | (7,263 | ) | (25 | )% | $ | 29,564 | $ | 27,142 | $ | 2,422 | 9 | % | ||||||
Research and development expenses for the year ended December 31, 2017, decreased as compared to the year ended December 31, 2016. The decrease was primarily the result of clinical trial expenses not recurring in the current period. Additionally, on January 1, 2017, the regulatory review process had progressed to a point that objective and persuasive evidence of approval was sufficiently probable, and a future economic benefit existed for the Accelerate Pheno™ system and Accelerate PhenoTest™ BC kit. As a result, the Company started capitalizing pre-launch inventory for the Accelerate Pheno™ system and Accelerate PhenoTest™ BC kit on January 1, 2017. Prior to January 1, 2017, all pre-launch inventory was expensed, because a future economic benefit could not be asserted.
Research and development expenses for the year ended December 31, 2016, increased as compared to the year ended December 31, 2015. The increase was primarily the result of increasing employee headcount, clinical trial fees, and increased purchases of laboratory and instrument engineering supplies to support research and development.
Pre-launch inventory not capitalized in accordance with U.S. GAAP, which included instruments and consumables charged to research and development were $411,000, $4.6 million and $2.7 million for the years ended December 31, 2017, 2016 and 2015, respectively.
Research and development expenses include non-cash equity-based compensation of $3.7 million, $1.6 million and $2.5 million for the years ended December 31, 2017, 2016 and 2015, respectively. The increase in non-cash equity-based compensation was primarily driven by an increase in the number of employees and stock option grants.
40
December 31, | December 31, | ||||||||||||||||||||||
(in thousands) | (in thousands) | ||||||||||||||||||||||
2017 | 2016 | $ Change | % Change | 2016 | 2015 | $ Change | % Change | ||||||||||||||||
Sales, general and administrative | $ | 45,058 | $ | 37,183 | $ | 7,875 | 21 | % | $ | 37,183 | $ | 18,554 | $ | 18,629 | 100 | % | |||||||
Sales, general and administrative expenses for the years ended December 31, 2017, 2016 and 2015, increased year over year due to an increase in salaries and related expenses as we ramp up our sales and marketing operations globally.
Pre-launch inventory not capitalized in accordance with U.S. GAAP, which included instruments and consumables charged to sales, general and administrative expenses, were $50,000, $4.1 million and $804,000 for the years ended December 31, 2017, 2016 and 2015, respectively.
Sales, general and administrative expenses include non-cash equity-based compensation of $10.1 million, $7.2 million and $5.9 million for the years ended December 31, 2017, 2016 and 2015, respectively. The increase in non-cash equity-based compensation was primarily driven by an increase in the number of employees and stock option grants.
December 31, | December 31, | ||||||||||||||||||||||
(in thousands) | (in thousands) | ||||||||||||||||||||||
2017 | 2016 | $ Change | % Change | 2016 | 2015 | $ Change | % Change | ||||||||||||||||
Loss from operations | $ | (64,184 | ) | $ | (66,501 | ) | $ | 2,317 | (3 | )% | $ | (66,501 | ) | $ | (45,549 | ) | $ | (20,952 | ) | 46 | % | ||
During the year ended December 31, 2017, loss from operations decreased compared to the year ended December 31, 2016. The decrease is the result of higher sales and the Company capitalizing inventory in connection with the FDA granting Accelerate’s de novo request to market the Accelerate Pheno™ system and Accelerate PhenoTest™ BC kit.
During the year ended December 31, 2016, loss from operations increased compared to the year ended December 31, 2015. The increase was the result of our continued investments in research and development, expanded laboratory and operational space and increased employee headcount, along with other factors.
Loss from operations includes non-cash equity-based compensation of $13.9 million, $8.8 million and $8.4 million for the years ended December 31, 2017, 2016 and 2015. Further losses are anticipated as we continue to investments in research and development, expanded laboratory and operational space, increased employee headcount and other factors as we continue to develop and commercialize products.
December 31, | December 31, | ||||||||||||||||||||||
(in thousands) | (in thousands) | ||||||||||||||||||||||
2017 | 2016 | $ Change | % Change | 2016 | 2015 | $ Change | % Change | ||||||||||||||||
Total other income, net | $ | 649 | $ | 394 | $ | 255 | 65 | % | $ | 394 | $ | 51 | $ | 343 | 673 | % | |||||||
Other income during the year ended December 31, 2017, increased compared to the year ended December 31, 2016, primarily due to an increase in interest and dividends as a result of higher investment balances in 2017. The interest and dividends were offset in part by other expenses.
Other income during the year ended December 31, 2016, increased compared to the year ended December 31, 2015, primarily due to an increase in interest and dividends as a result of higher investment balances in 2016. The interest and dividends were offset in part by other expenses.
41
December 31, | December 31, | ||||||||||||||||||||||
(in thousands) | (in thousands) | ||||||||||||||||||||||
2017 | 2016 | $ Change | % Change | 2016 | 2015 | $ Change | % Change | ||||||||||||||||
Provision for income taxes | $ | (493 | ) | $ | (267 | ) | $ | (226 | ) | 85 | % | $ | (267 | ) | $ | — | $ | (267 | ) | 100 | % | ||
Due to net losses incurred, we have only recorded tax provisions related to tax liabilities generated by our foreign subsidiaries for international income taxes.
Capital Resources and Liquidity
Our primary source of liquidity has been from sales of shares of common stock. As of December 31, 2017, the Company had $109.2 million in cash and cash equivalents and available-for-sale securities, an increase of $31.4 million from $77.8 million at December 31, 2016. The primary reason for the change in these assets was a public offering that occurred during the year ended December 31, 2017.
The Company is subject to Lease Agreements. The future minimum lease payments under the Lease Agreements are included in Item 8, Note 17, Commitments and Contingencies.
As of December 31, 2017, management believes that current cash balances will be more than sufficient to fund our capital and liquidity needs for the next twelve months.
The following summarizes selected items in the Company’s consolidated statements of cash flows for years ended December 31, 2017, 2016 and 2015 (in thousands):
Cash Flow Summary (in thousands) | |||||||||
12/31/2017 | 12/31/2016 | 12/31/2015 | |||||||
Net cash used in operating activities | $ | (55,746 | ) | $ | (53,408 | ) | $ | (35,126 | ) |
Net cash used in investing activities | (25,728 | ) | (49,568 | ) | (2,675 | ) | |||
Net cash provided by financing activities | 90,427 | 1,762 | 104,823 | ||||||
The net cash used in operating activities was $55.7 million, $53.4 million and $35.1 million during the years ended December 31, 2017, 2016 and 2015, respectively. These losses are the result of our continued investments in research and development, expanded laboratory and operational space, increased employee headcount and other factors as we develop and begin to commercialize the Company’s products.
The net cash used in investing activities was $25.7 million, $49.6 million and $2.7 million during the years ended December 31, 2017, 2016 and 2015, respectively, and was primarily comprised of purchases of available-for-sale investments, offset by sales and maturities of available-for-sale investments.
The net cash provided by financing activities was $90.4 million during the year ended December 31, 2017. This is primarily from proceeds received from a public offering. The net cash provided by financing activities was $1.8 million during the year ended December 31, 2016. This is primarily from proceeds received from the recovery of related party short swing profits and exercised options and warrants; which were partially offset by cash paid for common stock issuance expenses. The net cash provided by financing activities was $104.8 million during the year ended December 31, 2015. This is primarily from proceeds received from a public offering. Further information regarding the 2017 and 2015 public offerings is described in Item 8, Note 12, Public Offerings.
Our primary use of capital has been for the continued development and investment in commercialization readiness of the Accelerate Pheno™ system. We believe our capital requirements will continue to be met with our existing cash balance and those provided under exercises of stock options and/or additional issuance of equity or debt securities. However, if capital requirements vary materially from those currently planned, we may require additional capital sooner than expected. There can be no assurance that such capital will be available in sufficient amounts or on terms acceptable to us, if at all. Additional issuances of equity or convertible debt securities will result in dilution to our current common stockholders.
42
Off-Balance Sheet Arrangements
We did not have any off-balance sheet arrangements as of December 31, 2017.
Contractual Obligations
The Company has certain contractual obligations and commercial commitments as disclosed in Item 8, Note 17, Commitments and Contingencies that do not meet the definition of long term debt obligations, capital leases, operating leases or purchase obligations. The Company has entered into Lease Agreements as described in Item 2, Properties and Item 8, Note 17, Commitments and Contingencies. The operating lease obligations associated with Lease Agreements over the next five years are (in thousands):
Payments due by Period (in thousands) | ||||||||||||||||||
Contractual Obligations | Total | 2018 | 2019 | 2020 | 2021 | 2022 | ||||||||||||
Operating Lease Obligations | $ | 1,454 | $ | 1,119 | $ | 200 | $ | 84 | $ | 47 | $ | 4 | ||||||
Total | $ | 1,454 | $ | 1,119 | $ | 200 | $ | 84 | $ | 47 | $ | 4 | ||||||
Recent Accounting Pronouncements
A discussion relating to recent accounting pronouncements can be found in Item 8, Note 2, Summary of Significant Accounting Policies.
Critical Accounting Policies and Estimates
We consider our accounting policies related to deferred tax assets, equity-based compensation, inventory and revenue to be critical accounting policies. A number of significant estimates, assumptions, and judgments are inherent in our calculations, which are based on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. Actual results could differ materially from these estimates.
Inventory
Inventory is stated at the lesser of cost or net realizable value, with cost determined on the first-in-first-out method. The allocation of production overhead to inventory costs is based on normal production capacity. Abnormal amounts of idle facility expense and spoilage are expensed as incurred and not included in overhead subject to capitalization. The Company maintains provisions for excess and obsolete inventory based on management’s estimates of forecasted demand and, where applicable, product expiration. The Company adopted Accounting Standards Update (“ASU”) 2015-11, Simplifying the Measurement of Inventory (Topic 310) Inventory on January 1, 2017. This ASU simplifies the subsequent measurement of inventory by using only the lower of cost or net realizable value. The adoption did not have an effect on the Company’s consolidated financial statements.
Revenue
The Company recognizes revenue in accordance with ASC 605, Revenue Recognition, when persuasive evidence of an arrangement exists, the price is fixed or determinable, collection is reasonably assured and delivery of products has occurred or services have been rendered. Additional considerations include whether the applicable fee arrangement contains future delivery or performance obligations that should be divided into separate accounting units, whether the arrangement requires the Company to retain risks consistent with a collaborative arrangement, and/or whether any of the fees are contingent on the achievement of future milestones.
Product revenue is derived from the sale or rental of our instruments and sales of related consumable products. When an instrument is sold, revenue is generally recognized upon installation of the unit consistent with contract terms, which do not include a right of return. When a consumable product is sold, revenue is generally recognized upon shipment. Revenue is recognized net of reserves, which includes rebates.
43
We also provide instruments to customers under bundled rental agreements. Under these agreements, we install the instrument in the customer’s facility and provide service. The customer agrees to purchase consumable products at a stated price over the term of the agreement, which is typically five years or less. Contracts sometimes have renewal clauses but such clauses do not provide for a bargain renewal option or penalize the customer if they do not renew. The instrument remains the Company’s property throughout the term of the agreement and there is no transfer of title upon expiration. Revenue is recognized as consumable products are shipped or delivered, depending on contract terms.
For multiple element arrangements, the total consideration for an arrangement is allocated among the separate elements in the arrangement based on a selling price hierarchy. The selling price hierarchy for a deliverable is based on: (1) vendor specific objective evidence (“VSOE”), if available; (2) third party evidence of selling price if VSOE is not available; or (3) an estimated selling price, if neither VSOE nor third party evidence is available. Estimated selling price is our best estimate of the selling price of an element in a transaction. The Company limits the amount of revenue recognized for delivered elements to the amount that is not contingent on the future delivery of products or services or other future performance obligations.
Deferred revenue represents amounts received but not yet earned under existing agreements.
Effective January 1, 2018, the Company will adopt changes to its revenue recognition policy. Refer to Item 8, Note 2, Summary of Significant Accounting Policies for further details.
Equity-Based Compensation
The Company awards stock options and other equity-based instruments to its employees, directors and consultants. Compensation cost related to equity-based instruments is based on the fair value of the instrument on the grant date, and is recognized over the requisite service period on a straight-line basis over the vesting period for each tranche (an accelerated attribution method).
The Company estimates the fair value of stock option awards, including modifications of stock option awards, using the Black-Scholes option pricing model. This model derives the fair value of stock options based on certain assumptions related to expected stock price volatility, expected option life, risk-free interest rate and dividend yield.
• | Volatility: The expected volatility is based on the historical volatility of the Company's stock price over the most recent period commensurate with the expected term of the stock option award. |
• | Expected term: The estimated expected term for employee awards is based on the calculation published by the SEC in SAB110 for use when there is not a sufficient history of employee exercise patterns. For consultant awards, the estimated expected term is the same as the life of the award. |
• | Risk-free interest rate: The risk-free interest rate is based on published U.S. Treasury rates for a term commensurate with the expected term. |
• | Dividend yield: The dividend yield is estimated as zero as the Company has not paid dividends in the past and does not have any plans to pay any dividends in the foreseeable future. |
The Company records the fair value of Restricted Stock Units (“RSU's”) or Stock Grants (“SG's”) based on published closing market price on the day before the grant date.
In January 2017, the Company implemented ASU 2016-09, Compensation-Stock Compensation (Topic 718) Improvements to Employee Share-Based Payment Accounting. Pursuant to this guidance, we made a policy election to account for forfeitures as they occur rather than on an estimated basis. For periods prior to the adoption of this ASU, the Company estimated the forfeiture rate of unvested awards based on the forfeitures in the previous twelve-month period. The rate was calculated separately for awards to the board of directors/executives and all other awards. Further information regarding this change is included in Note 15, Employee and Consultant Equity-Based Compensation.
The Company also has an employee stock purchase program whereby eligible employees can elect payroll deductions that are subsequently used to purchase common stock at a discounted price. There is no compensation recorded for this program as (i) the purchase discount does not exceed the issuance costs that would have been
44
incurred to raise a significant amount of capital by a public offering, (ii) substantially all employees that meet limited employment qualifications may participate on an equitable basis, and (iii) the plan doesn't incorporate option features that would require compensation to be recorded.
See Note 15, Employee and Consultant Equity-Based Compensation for further information.
Income Taxes and Deferred Tax Assets
Deferred tax assets and liabilities are recorded for the estimated future tax effects of temporary differences between the tax basis of assets and liabilities and amounts reported in the accompanying balance sheets. The change in deferred tax assets and liabilities for the period represents the deferred tax provision or benefit for the period. Effects of changes in enacted tax laws in deferred tax assets and liabilities are reflected as an adjustment to the tax provision or benefit in the period of enactment.
We regularly review our deferred tax assets for recoverability and establish a valuation allowance based on historical taxable income, projected future taxable income, and the expected timing of the reversals of existing temporary differences. As of December 31, 2017, and 2016, we have established a valuation allowance equal to our net deferred tax asset, as we have not been able to determine that we will generate sufficient future taxable income to allow us to realize the deferred tax assets. See Item 8, Note 16, Income Taxes for additional information.
Tax Cuts and Jobs Act
On December 22, 2017, President Trump signed into law the Tax Cuts and Jobs Act (the Tax Act”). The Tax Act reduced the corporate tax rate from the maximum federal statutory rate of 35% to 21%. The Tax Act states that the 21% corporate tax rate is effective for tax years beginning on or after January 1, 2018. However, existing tax law, which was not amended under the Tax Act, governs when a change in tax rate is effective. Existing tax law provides that if the taxable year includes the effective date of any rate change (unless the change is the first date of the taxable year), taxes should be calculated by applying a blended rate to the taxable income for the year. For the Company, the net deferred tax assets for US purposes have been revalued to 21% as of December 31, 2017 with a corresponding adjustment to the valuation allowance. Therefore, the reduction in the US corporate tax rate had no impact to the Company's earnings.
Due to uncertainties which currently exist in the interpretation of the provisions of the Tax Cuts and Jobs Act of 2017 regarding Internal Revenue Code Section 162(m), the Company has not evaluated the potential impacts of IRC Section 162(m) as amended by the Tax Cuts and Jobs Act of 2017 on its financial statements.
In conjunction with the Tax Act, the SEC staff issued Staff Accounting Bulletin No. 118 (“SAB 118”) to address the application of U.S. GAAP in situations when a registrant does not have the necessary information available, prepared, or analyzed (including computations) in reasonable detail to complete the accounting for certain income tax effects of the Tax Act. The Company has recognized the provisional tax impacts related to deemed repatriated earnings and the revaluation of deferred tax assets and liabilities and included these amounts in its consolidated financial statements for the year ended December 31, 2017. The ultimate impact may differ from these provisional amounts, possibly materially, due to, among other things, additional analysis, changes in interpretations and assumptions the Company has made, additional regulatory guidance that may be issued, and actions the Company may take as a result of the Tax Act.
Item 7A. Quantitative and Qualitative Disclosures
Interest Rate Risk
Our investment portfolio is exposed to market risk from changes in interest rates. The fair market value of fixed rate securities may be adversely impacted by fluctuations in interest rates while income earned on floating rate securities may decline as a result of decreases in interest rates. We have historically maintained a relatively short average maturity for our investment portfolio, and we believe a hypothetical 100 basis point adverse move in interest rates along the entire interest rate yield curve would change the fair value of our interest sensitive financial instruments by approximately $555,000 for the year ended December 31, 2017 and $398,000 for the year ended December 31, 2016. The increase in interest rate risk is due to the increase in available-for-sale investments carried as of December 31, 2017.
45
Under our current policies, we do not use interest rate derivative instruments to manage exposure to interest rate changes. We attempt to ensure the safety and preservation of our invested principal funds by limiting default risk, market risk and reinvestment risk. The goals of our investment policy are preservation of capital, fulfillment of liquidity needs and fiduciary control of cash and investments. We also seek to maximize income from our investments without assuming significant risk. To achieve our goals, we maintain a portfolio of cash equivalents and investments in a variety of securities that management believes to be of high credit quality. Further information regarding our investments is included in Item 8, Note 6, Investments.
Item 8. Financial Statements and Supplementary Data
Financial Statements of Accelerate Diagnostics, Inc.
Report of Independent Registered Public Accounting Firm
Consolidated Balance Sheets as of December 31, 2017, and 2016
Consolidated Statements of Operations and Comprehensive Loss for the years ended December 31, 2017, 2016 and 2015
Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2017, 2016 and 2015
Consolidated Statements of Cash Flow for the years ended December 31, 2017, 2016 and 2015
Notes to Consolidated Financial Statements
46
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of Accelerate Diagnostics, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Accelerate Diagnostics, Inc. (the Company) as of December 31, 2017 and 2016, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2017, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated February 28, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2013.
Phoenix, Arizona
February 28, 2018
47
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of Accelerate Diagnostics, Inc.
Opinion on Internal Control over Financial Reporting
We have audited Accelerate Diagnostics, Inc.’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Accelerate Diagnostics, Inc. (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of December 31, 2017 and 2016, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2017, and the related notes of the Company and our report dated February 28, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Phoenix, Arizona
February 28, 2018
48
ACCELERATE DIAGNOSTICS, INC.
CONSOLIDATED
BALANCE SHEETS
(in thousands, except share data)
December 31, | ||||||
2017 | 2016 | |||||
ASSETS | ||||||
Current assets: | ||||||
Cash and cash equivalents | $ | 28,513 | $ | 19,244 | ||
Investments | 80,648 | 58,519 | ||||
Trade accounts receivable | 1,946 | 34 | ||||
Inventory | 8,063 | — | ||||
Prepaid expenses | 850 | 468 | ||||
Other current assets | 468 | 183 | ||||
Total current assets | 120,488 | 78,448 | ||||
Property and equipment, net | 4,890 | 4,258 | ||||
Intellectual property, net | 134 | 146 | ||||
Total assets | $ | 125,512 | $ | 82,852 | ||
LIABILITIES AND STOCKHOLDERS' EQUITY | ||||||
Current liabilities: | ||||||
Accounts payable | $ | 2,080 | $ | 992 | ||
Accrued liabilities | 3,636 | 3,009 | ||||
Deferred revenue and income | 1,071 | 35 | ||||
Total current liabilities | 6,787 | 4,036 | ||||
Long-term deferred income | — | 1,000 | ||||
Other long term liabilities | 21 | — | ||||
Total liabilities | 6,808 | 5,036 | ||||
Commitments and contingencies | ||||||
Stockholders' equity: | ||||||
Preferred shares, $0.001 par value; | ||||||
5,000,000 preferred shares authorized and none outstanding as of December 31, 2017 and December 31, 2016 | — | — | ||||
Common stock, $0.001 par value; | ||||||
75,000,000 common shares authorized with 55,673,810 shares issued and outstanding on December 31, 2017 and 75,000,000 authorized with 51,516,309 shares issued and outstanding on December 31, 2016 | 56 | 52 | ||||
Contributed capital | 360,620 | 255,257 | ||||
Accumulated deficit | (241,972 | ) | (177,289 | ) | ||
Accumulated other comprehensive income (loss) | — | (204 | ) | |||
Total stockholders' equity | 118,704 | 77,816 | ||||
Total liabilities and stockholders' equity | $ | 125,512 | $ | 82,852 | ||
See accompanying notes to consolidated financial statements.
49
ACCELERATE DIAGNOSTICS, INC.
CONSOLIDATED
STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(in thousands, except per share data)
Years Ended December 31, | |||||||||
2017 | 2016 | 2015 | |||||||
Net sales | $ | 4,177 | $ | 246 | $ | 147 | |||
Cost of sales | 1,002 | — | — | ||||||
Gross profit | 3,175 | 246 | 147 | ||||||
Costs and expenses: | |||||||||
Research and development | 22,301 | 29,564 | 27,142 | ||||||
Sales, general and administrative | 45,058 | 37,183 | 18,554 | ||||||
Total costs and expenses | 67,359 | 66,747 | 45,696 | ||||||
Loss from operations | (64,184 | ) | (66,501 | ) | (45,549 | ) | |||
Other income (expense): | |||||||||
Interest expense | — | — | (4 | ) | |||||
Foreign currency exchange loss | (75 | ) | (77 | ) | (19 | ) | |||
Interest and dividend income | 908 | 494 | 74 | ||||||
Other expense, net | (184 | ) | (23 | ) | — | ||||
Total other income, net | 649 | 394 | 51 | ||||||
Net loss before income taxes | (63,535 | ) | (66,107 | ) | (45,498 | ) | |||
Provision for income taxes | (493 | ) | (267 | ) | — | ||||
Net loss | $ | (64,028 | ) | $ | (66,374 | ) | $ | (45,498 | ) |
Basic and diluted net loss per share | $ | (1.18 | ) | $ | (1.29 | ) | $ | (1.01 | ) |
Weighted average shares outstanding | 54,073 | 51,276 | 44,998 | ||||||
Other comprehensive loss: | |||||||||
Net loss | $ | (64,028 | ) | $ | (66,374 | ) | $ | (45,498 | ) |
Net unrealized loss on available-for-sale investments | (117 | ) | (64 | ) | (20 | ) | |||
Foreign currency translation adjustment | 321 | (128 | ) | 1 | |||||
Comprehensive loss | $ | (63,824 | ) | $ | (66,566 | ) | $ | (45,517 | ) |
See accompanying notes to consolidated financial statements.
50
ACCELERATE DIAGNOSTICS, INC.
CONSOLIDATED
STATEMENTS OF STOCKHOLDERS’ EQUITY
(in thousands)
Shares | Common Stock Amount | Contributed Capital | Accumulated Deficit | Accumulated Other Comprehensive Income (Loss) | Total Stockholders’ Equity | ||||||||||||
Balances, January 1, 2015 | 44,640 | $ | 45 | $ | 131,356 | $ | (65,417 | ) | $ | 7 | $ | 65,991 | |||||
Net loss | — | — | — | (45,498 | ) | — | (45,498 | ) | |||||||||
Issuance of common stock | 6,426 | 6 | 103,345 | — | — | 103,351 | |||||||||||
Exercise of options | 125 | — | 805 | — | — | 805 | |||||||||||
Unrealized loss on available-for-sale securities | — | — | — | — | (20 | ) | (20 | ) | |||||||||
Foreign currency translation adjustment | — | — | — | — | 1 | 1 | |||||||||||
Equity-based compensation | — | — | 8,388 | — | — | 8,388 | |||||||||||
Balances, December 31, 2015 | 51,191 | 51 | 243,894 | (110,915 | ) | (12 | ) | 133,018 | |||||||||
Net loss | — | — | — | (66,374 | ) | — | (66,374 | ) | |||||||||
Exercise of options and warrants | 314 | 1 | 1,496 | — | — | 1,497 | |||||||||||
Issuance of common stock under employee purchase plan | 11 | — | 226 | — | — | 226 | |||||||||||
Short swing profits (net of costs) | — | — | 866 | — | — | 866 | |||||||||||
Unrealized loss on available-for-sale securities | — | — | — | — | (64 | ) | (64 | ) | |||||||||
Foreign currency translation adjustment | — | — | — | — | (128 | ) | (128 | ) | |||||||||
Equity-based compensation | — | — | 8,775 | — | — | 8,775 | |||||||||||
Balances, December 31, 2016 | 51,516 | 52 | 255,257 | (177,289 | ) | (204 | ) | 77,816 | |||||||||
Net loss | — | — | — | (64,028 | ) | — | (64,028 | ) | |||||||||
Issuance of common stock | 3,085 | 3 | 83,221 | — | — | 83,224 | |||||||||||
Exercise of options and warrants | 1,045 | 1 | 6,605 | — | — | 6,606 | |||||||||||
Issuance of common stock under employee purchase plan | 28 | — | 597 | — | — | 597 | |||||||||||
Unrealized loss on available-for-sale securities | — | — | — | — | (117 | ) | (117 | ) | |||||||||
Foreign currency translation adjustment | — | — | — | — | 321 | 321 | |||||||||||
Cumulative impact of accounting change for forfeitures | — | — | — | (655 | ) | — | (655 | ) | |||||||||
Equity-based compensation | — | — | 14,940 | — | — | 14,940 | |||||||||||
Balances, December 31, 2017 | 55,674 | $ | 56 | $ | 360,620 | $ | (241,972 | ) | $ | — | $ | 118,704 | |||||
See accompanying notes to consolidated financial statements.
51
ACCELERATE DIAGNOSTICS, INC.
CONSOLIDATED
STATEMENT OF CASH FLOWS
(in thousands)
Years Ended December 31, | |||||||||
2017 | 2016 | 2015 | |||||||
Cash flows from operating activities: | |||||||||
Net loss | $ | (64,028 | ) | $ | (66,374 | ) | $ | (45,498 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | |||||||||
Depreciation | 2,184 | 2,340 | 1,782 | ||||||
Amortization of intangible assets | 12 | 11 | 10 | ||||||
Amortization of investment discount | 326 | 374 | 188 | ||||||
Equity-based compensation | 13,933 | 8,775 | 8,388 | ||||||
Realized gain on available-for-sale securities | — | (6 | ) | — | |||||
Loss on disposal of property and equipment | 240 | 23 | — | ||||||
(Increase) decrease in assets: | |||||||||
Accounts receivable | (1,912 | ) | 43 | 1 | |||||
Inventory | (7,759 | ) | — | — | |||||
Prepaid expense and other | (174 | ) | 1,292 | (1,241 | ) | ||||
Other current assets | (285 | ) | (171 | ) | (12 | ) | |||
Increase (decrease) in liabilities: | |||||||||
Accounts payable | 1,064 | (1,242 | ) | 329 | |||||
Accrued liabilities | 596 | 1,619 | 827 | ||||||
Deferred revenue and income | 36 | (92 | ) | 100 | |||||
Deferred compensation | 21 | — | — | ||||||
Net cash used in operating activities | (55,746 | ) | (53,408 | ) | (35,126 | ) | |||
Cash flows from investing activities: | |||||||||
Purchases of equipment | (2,966 | ) | (2,409 | ) | (3,656 | ) | |||
Purchase of available-for-sale securities | (82,333 | ) | (74,075 | ) | (12,418 | ) | |||
Sales of available-for-sale securities | 11,522 | 9,716 | 141 | ||||||
Maturity of available-for-sale securities | 48,049 | 17,200 | 13,258 | ||||||
Net cash used in investing activities | (25,728 | ) | (49,568 | ) | (2,675 | ) | |||
Cash flows from financing activities: | |||||||||
Exercise of warrants and options | 6,606 | 1,497 | 805 | ||||||
Common stock issuance cost | — | (814 | ) | — | |||||
Issuance of common stock and warrants | 83,821 | 226 | 104,165 | ||||||
Recovery of related party short-swing profits | — | 866 | — | ||||||
Payments on capital lease obligations | — | (13 | ) | (147 | ) | ||||
Net cash provided by financing activities | 90,427 | 1,762 | 104,823 | ||||||
Effect of exchange rate on cash | 316 | (127 | ) | — | |||||
Increase (decrease) in cash and cash equivalents | 9,269 | (101,341 | ) | 67,022 | |||||
Cash and cash equivalents, beginning of period | 19,244 | 120,585 | 53,563 | ||||||
Cash and cash equivalents, end of period | $ | 28,513 | $ | 19,244 | $ | 120,585 | |||
See accompanying notes to consolidated financial statements.
52
ACCELERATE DIAGNOSTICS, INC.
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1. ORGANIZATION AND NATURE OF BUSINESS; BASIS OF PRESENTATION; PRINCIPLES OF CONSOLIDATION
Accelerate Diagnostics, Inc. (“we” or “us” or “our” or “Accelerate” or “the Company”) is an in vitro diagnostics company dedicated to providing solutions which improve patient outcomes and lower healthcare costs through the rapid diagnosis of serious infections.
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles, (“U.S. GAAP”), and applicable rules and regulations of the United States Securities and Exchange Commission (“SEC”), regarding annual financial reporting.
All amounts are rounded to the nearest thousand dollars unless otherwise indicated.
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries after elimination of intercompany transactions and balances.
Reclassification
Certain prior year amounts have been reclassified for consistency with the current year presentation and had no effect on our net income, stockholders’ equity or cash flows. In the current period presentation and the revised prior period presentation, depreciation and amortization expenses are reported as a component of the individual costs and expenses as part of the statements of operations and comprehensive loss. The amount of depreciation and amortization expenses now reported as a component of research and development costs for the years ended December 31, 2017, 2016 and 2015, were $1.1 million, $1.4 million and $1.1 million, respectively. The amount of depreciation and amortization expenses now reported as a component of sales, general and administrative costs for the years ended December 31, 2017, 2016 and 2015, were $632,000, $983,000 and $672,000, respectively.
NOTE 2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Use of Estimates
The Company’s consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles (“GAAP”). The preparation of the Company’s consolidated financial statements requires the Company to make estimates and assumptions that affect the reported amounts of assets and liabilities and the related disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. The more significant areas requiring the use of management estimates and assumptions relate to accounts receivable, inventories, property and equipment, intangible assets, accruals, warranty liabilities, tax valuation accounts and stock-based compensation. Actual result could differ materially from those estimates.
Estimated Fair Value of Financial Instruments
The Company follows ASC Topic 820, Fair Value Measurements and Disclosures which has defined fair value and requires the Company to establish a framework for measuring fair value and disclose fair value measurements. The framework requires the valuation of assets and liabilities subject to fair value measurements using a three tiered approach and fair value measurement be classified and disclosed in one of the following three categories:
• | Level 1: Unadjusted quoted prices in active markets that are accessible at the measurement date for identical, unrestricted assets or liabilities; |
• | Level 2: Quoted prices for similar assets and liabilities in active markets, quoted prices in markets that are not active, or inputs that are observable, either directly or indirectly, for substantially the full term of |
53
the asset or liability;
• | Level 3: Prices or valuation techniques that require inputs that are both significant to the fair value measurement and unobservable (i.e. supported by little or no market activity). |
The carrying amounts of financial instruments such as cash and cash equivalents, trade accounts receivable, prepaid expenses, accounts payable, accrued liabilities, and other current liabilities approximate the related fair values due to the short-term maturities of these instruments.
See Note 5, Fair Value of Financial Instruments for further information and related disclosures regarding the Company’s fair value measurements.
Cash and Cash Equivalents
All highly liquid investments with an original maturity of three months or less at time of purchase are considered to be cash equivalents. Cash and cash equivalents include overnight repurchase agreement accounts and other investments. As part of our cash management process, excess operating cash is invested in overnight repurchase agreements with our bank. Repurchase agreements and other investments classified as cash and cash equivalents are not deposits and are not insured by the U.S. Government, the FDIC or any other government agency and involve investment risk including possible loss of principal. We believe however, that the market risk arising from holding these financial instruments is minimal.
Investments
The Company invests excess funds in various investments which are primarily held in the custody of major financial institutions. Investments consist of debt securities in U.S. government and agency securities, corporate debt securities and certificates of deposit. Management classifies its investments as available-for-sale investments and records these investments in the condensed consolidated balance sheet at fair value. The Company considers all available-for-sale securities, including those with maturity dates beyond 12 months, as available to support current operational liquidity needs. Unrealized gains or losses for available-for-sale securities are included in accumulated other comprehensive income (loss), a component of stockholders’ equity. The Company classifies its investments as current based on the nature of the investments and their availability for use in current operations.
The Company assesses whether an other-than-temporary impairment loss has occurred due to declines in fair value or other market conditions when an investment’s fair value remains less than its cost for more than twelve months. This assessment includes a determination of whether the investment is expected to recover in value and whether the Company has the intent and ability to hold the investment until the anticipated recovery in value occurs. When an investment is identified as having an other-than-temporary impairment loss, we adjust the cost basis of the investment down to fair value resulting in a realized loss. The new cost basis is not changed for subsequent recoveries in fair value and temporary future increases or decreases in fair value are included in other comprehensive income (loss).
Inventory
Inventory is stated at the lesser of cost or net realizable value, with cost determined on the first-in-first-out method. The allocation of production overhead to inventory costs is based on normal production capacity. Abnormal amounts of idle facility expense and spoilage are expensed as incurred and not included in overhead subject to capitalization. The Company maintains provisions for excess and obsolete inventory based on management’s estimates of forecasted demand and, where applicable, product expiration. The Company adopted Accounting Standards Update (“ASU”) 2015-11, Simplifying the Measurement of Inventory (Topic 310) Inventory on January 1, 2017. This ASU simplifies the subsequent measurement of inventory by using only the lower of cost or net realizable value. The adoption did not have an effect on the Company’s consolidated financial statements.
Accounts Receivable
Accounts receivable consist of amounts due to the Company for sales to customers and are recorded net of an allowance for doubtful accounts. Allowances on accounts receivable are recorded when circumstances indicate collection is doubtful for a particular accounts receivable. Receivables are written off if reasonable collection efforts prove unsuccessful. The Company provides for allowances on a specific account basis.
54
The Company has not included an allowance for doubtful accounts as of December 31, 2017 and 2016, due to no circumstances arising that would indicate collection of any particular or grouping of customer accounts receivable is doubtful.
Property and Equipment
Property and equipment are recorded at cost. Maintenance and repairs are charged to expense as incurred and expenditures for major improvements are capitalized. Gains and losses from retirement or replacement are included in costs and expenses. Depreciation of property and equipment is computed using the straight-line method over the estimated useful life of the assets, ranging from one to seven years. Leasehold improvements are depreciated over the remaining life of the lease or the life of the asset, whichever is less. Property and equipment includes diagnostic instruments used for sales demonstrations and instruments under rental agreements. The Company retains title to the instruments under these arrangements. See Note 8, Property and Equipment for further information and related disclosures.
Long-lived Assets
Long-lived assets and certain identifiable intangibles to be held and used by the Company are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. The Company continuously evaluates the recoverability of its long-lived assets based on estimated future cash flows from and the estimated fair value of such long-lived assets, and provides for impairment if such undiscounted cash flows or the estimated fair value are insufficient to recover the carrying amount of the long-lived asset.
Warranty Reserve
Instruments are typically sold with a one year limited warranty, while kits and accessories are typically sold with a sixty days limited warranty. Accordingly, a provision for the estimated cost of the limited warranty repair is recorded at the time revenue is recognized. Our estimated warranty provision is based on our estimate of future repair events and the related estimated cost of repairs. The Company periodically assesses the adequacy of the warranty reserve and adjusts the amount as necessary. The expense incurred for these provisions is included in cost of sales on the consolidated statements of operations and comprehensive loss.
Product warranty reserve activity for the years ended December 31, 2017 and 2016 is as follows (in thousands):
2017 | 2016 | |||||
Beginning balance | $ | 1 | $ | — | ||
Provisions | 331 | 18 | ||||
Warranty expenses incurred | (140 | ) | (17 | ) | ||
Ending balance | $ | 192 | $ | 1 | ||
We had no warranty reserve in 2015 as there were no commercial sales in 2015 that included warranty coverage.
Revenue Recognition
The Company recognizes revenue in accordance with ASC 605, Revenue Recognition, when persuasive evidence of an arrangement exists, the price is fixed or determinable, collection is reasonably assured and delivery of products has occurred or services have been rendered. Additional considerations include whether the applicable fee arrangement contains future delivery or performance obligations that should be divided into separate accounting units, whether the arrangement requires the Company to retain risks consistent with a collaborative arrangement, and/or whether any of the fees are contingent on the achievement of future milestones.
Product revenue is derived from the sale or rental of our instruments and sales of related consumable products. When an instrument is sold, revenue is generally recognized upon installation of the unit consistent with contract terms,
55
which do not include a right of return. When a consumable product is sold, revenue is generally recognized upon shipment. Revenue is recognized net of reserves, which includes rebates.
We also provide instruments to customers under bundled rental agreements. Under these agreements, we install the instrument in the customer’s facility and provide service. The customer agrees to purchase consumable products at a stated price over the term of the agreement which is typically five years or less. Contracts sometimes have renewal clauses but such clauses do not provide for a bargain renewal option or penalize the customer if they do not renew. The instrument remains the Company’s property throughout the term of the agreement and there is no transfer of title upon expiration. Revenue is recognized as consumable products are shipped or delivered, depending on contract terms.
For multiple element arrangements, the total consideration for an arrangement is allocated among the separate elements in the arrangement based on a selling price hierarchy. The selling price hierarchy for a deliverable is based on: (1) vendor specific objective evidence (“VSOE”), if available; (2) third party evidence of selling price if VSOE is not available; or (3) an estimated selling price, if neither VSOE nor third party evidence is available. Estimated selling price is our best estimate of the selling price of an element in a transaction. The Company limits the amount of revenue recognized for delivered elements to the amount that is not contingent on the future delivery of products or services or other future performance obligations.
Deferred revenue represents amounts received but not yet earned under existing agreements.
Effective January 1, 2018, the Company will adopt changes to its revenue recognition policy. Refer to Recent Accounting Pronouncements for further details.
Cost of Sales
Cost of sales consists of raw materials, depreciation, direct labor and stock-based compensation expense, manufacturing overhead, facility costs and warranty costs.
Shipping and Handling
Shipping and handling costs billed to customers are included as a component of revenue. The corresponding expense incurred with third party carriers is included as a component of sales, general and administrative costs on the consolidated statements of operations and comprehensive loss.
Leases
The Company accounts for leases in accordance with ASC 840, Leases, which requires leases to be classified as either operating or capital leases. In general, the Company classifies leases as capital leases when there is either a transfer of ownership at the end of the lease term, the lease contains a bargain purchase option, the lease term is seventy-five percent or more of the estimated economic life of the leased property or the minimum lease payments are ninety percent or more of the fair value at lease inception. Other leases are classified as operating leases.
Operating lease rent is recorded as an operating expense monthly. For capital leases, both an asset and liability are recorded at the inception of the lease based on the present value of lease payments. The asset is included with property and equipment on the balance sheet and amortization is recorded on a straight-line basis over the term of the lease with the amortization expense included with depreciation on the statements of operations and comprehensive loss. For the liability, the amount due within the next year is recorded as capital lease obligations and the amount due in more than a year is recorded as long-term capital lease obligation on the balance sheet. Interest expense is recorded based on the implicit or explicit interest rate used in the lease and is included as non-operating interest expense on the statements of operations and comprehensive loss.
Equity-Based Compensation
The Company awards stock options and other equity-based instruments to its employees, directors and consultants. Compensation cost related to equity-based instruments is based on the fair value of the instrument on the grant date, and is recognized over the requisite service period on a straight-line basis over the vesting period for each tranche (an accelerated attribution method).
56
The Company estimates the fair value of stock option awards, including modifications of stock option awards, using the Black-Scholes option pricing model. This model derives the fair value of stock options based on certain assumptions related to expected stock price volatility, expected option life, risk-free interest rate and dividend yield.
• | Volatility: The expected volatility is based on the historical volatility of the Company's stock price over the most recent period commensurate with the expected term of the stock option award. |
• | Expected term: The estimated expected term for employee awards is based on the calculation published by the SEC in SAB110 for use when there is not a sufficient history of employee exercise patterns. For consultant awards, the estimated expected term is the same as the life of the award. |
• | Risk-free interest rate: The risk-free interest rate is based on published U.S. Treasury rates for a term commensurate with the expected term. |
• | Dividend yield: The dividend yield is estimated as zero as the Company has not paid dividends in the past and does not have any plans to pay any dividends in the foreseeable future. |
The Company records the fair value of Restricted Stock Units (“RSU's”) or Stock Grants (“SG's”) based on published closing market price on the day before the grant date.
In January 2017, the Company implemented ASU 2016-09, Compensation-Stock Compensation (Topic 718) Improvements to Employee Share-Based Payment Accounting. Pursuant to this guidance, we made a policy election to account for forfeitures as they occur rather than on an estimated basis. For periods prior to the adoption of this ASU, the Company estimated the forfeiture rate of unvested awards based on the forfeitures in the previous twelve-month period. The rate was calculated separately for awards to the board of directors/executives and all other awards. Further information regarding this change is included in Note 15, Employee and Consultant Equity-Based Compensation.
See Note 15, Employee and Consultant Equity-Based Compensation for further information.
Income Taxes and Deferred Tax Assets
Deferred tax assets and liabilities are recorded for the estimated future tax effects of temporary differences between the tax basis of assets and liabilities and amounts reported in the accompanying balance sheets. The change in deferred tax assets and liabilities for the period represents the deferred tax provision or benefit for the period. Effects of changes in enacted tax laws in deferred tax assets and liabilities are reflected as an adjustment to the tax provision or benefit in the period of enactment.
The Company follows the provisions of ASC 740, Income Taxes, to account for any uncertainty in income taxes with respect to the accounting for all tax positions taken (or expected to be taken) on any income tax return. This guidance applies to all open tax periods in all tax jurisdictions in which the Company is required to file an income tax return. Under U.S. GAAP, in order to recognize an uncertain tax benefit the taxpayer must be more likely than not certain of sustaining the position, and the measurement of the benefit is calculated as the largest amount that is more likely than not to be realized upon resolution of the position. Interest and penalties, if any, would be recorded within tax expense.
Foreign Currency Translation and Foreign Currency Transactions
The Company follows ASC 830 Foreign Currency Matters, which provides guidance on foreign currency transactions and translation of financial statements. Adjustments resulting from translating foreign functional currency financial statements into U.S. Dollars are included in the foreign currency translation adjustment, a component of accumulated other comprehensive income (loss) in the consolidated statements of stockholder's equity.
The Company has assets and liabilities, including receivables and payables, which are denominated in currencies other than their functional currency. These balance sheet items are subject to re-measurement, the impact of which is recorded in foreign currency exchange gain and loss, within the consolidated statement of operations and comprehensive loss.
Earnings Per Share
57
The Company follows ASC 260, Earnings Per Share, which requires companies to present basic earnings per share and diluted earnings per share. Basic earnings (loss) per share includes no dilution and is computed by dividing income (loss) available to common stockholders by the weighted average number of common shares outstanding for the period. Diluted earnings per share are computed similarly to basic earnings (loss) per share except the denominator includes additional common shares that would have been outstanding if warrants and share-based payments had been issued. Diluted earnings are not presented when the effect of adding such additional common shares is antidilutive.
Earnings per share are restated when certain transactions or events, including rights offerings determined to have bonus elements have occurred.
See Note 14, Earnings Per Share for further information.
Comprehensive Loss
The Company follows ASC 220, Reporting Comprehensive Income, which establishes standards for reporting and displaying comprehensive income (loss) and its components (revenues, expenses, gains and losses) in a full set of general-purpose financial statements. The Company holds investments classified as available-for-sale securities and records the change in fair market value as a component of comprehensive income (loss). The Company also has adjustments resulting from translating foreign functional currency financial statements into U.S. Dollars which is included as a component of comprehensive income (loss).
Recent Accounting Pronouncements
In May 2017, the Financial Accounting Standards Board (“FASB”) issued ASU 2017-09, Compensation—Stock Compensation (Topic 718) Scope of Modification Accounting. This amendment clarifies when to account for a change to the terms or conditions of a share-based payment award as a modification. Under the new guidance, modification accounting is required only if the fair value, the vesting conditions, or the classification of the award (as equity or liability) changes as a result of the change in terms or conditions. It is effective prospectively for the annual period ending December 31, 2018 and interim periods within that annual period. Early adoption is permitted. Historically, modifications to our share-based awards is rare. As such, we do not expect the application of this standard to have a significant impact on our consolidated financial statements.
In March 2017, the FASB issued ASU 2017-08, Receivable-Nonrefundable Fees and Other Costs (Topic 310-20) Premium Amortization on Purchased Callable Debt Securities. This amendment shortens the amortization period for certain callable debt securities held at a premium. Specifically, the amendment requires premiums to be amortized to the earliest call date. The amendments do not require an accounting change for securities held at a discount; the discount continues to be amortized to maturity. The guidance is effective for public business entities for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2018. Early adoption is permitted, including adoption in an interim period. If an entity early adopts in an interim period, any adjustments should be reflected as of the beginning of the fiscal year that includes that interim period. The amendments should be applied on a modified retrospective basis, with a cumulative-effect adjustment directly to retained earnings as of the beginning of the period of adoption. We are currently assessing the impact this will have on our consolidated financial statements and the timing of adoption.
In June 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2016-13, Financial Instruments-Credit Losses (Topic 326) Measurement of Credit Losses on Financial Instruments, which amends the guidance on measuring credit losses on financial assets (including trade accounts receivable and available for sale debt securities) held at amortized cost. Currently, an “incurred loss” methodology is used for recognizing credit losses which delays recognition until it is probable a loss has been incurred. The amendment requires assets valued at amortized cost to be presented at the net amount expected to be collected using an allowance for credit losses. Reversal of credit losses on available for sale debt securities will be recorded in the current period net income. The amendment will be effective for us on January 1, 2020, with early adoption permitted. We do not anticipate this guidance will have a significant impact on our financial statements and plan to adopt on the effective date.
In February 2016, the FASB issued ASU 2016-02, Leases (Topic 842). This replaces the existing standards relating to leases for both lessees and lessors. For lessees, the new standard requires most leases to be recorded on the balance sheet with expenses recognized much like the existing standard. For lessors, the new standard modifies
58
the classification criteria and accounting for sales-type and direct financing leases and eliminates leveraged leases. For both lessees and lessors, the standard eliminates real estate-specific provisions, changes some of the presentation and disclosure requirements, and changes sale and leaseback criteria. The ASU is required for us on January 1, 2019, with early adoption permitted. We are currently assessing the impact this will have on our consolidated financial statements and the timing of adoption.
In January, 2016, the FASB issued ASU 2016-01, Financial Instruments - Overall. This standard requires equity investments, with some exceptions, be measured at fair value with valuation changes recognized in net income, simplifies the impairment assessment of some equity investments, eliminates the requirement to disclose the methods and significant assumptions used to estimate the fair value for financial instruments measured at amortized cost, requires the use of the exit price notion when measuring the fair value of financial instruments, requires separate presentation of some changes in other comprehensive income, requires separate presentation of financial assets and financial liabilities by measurement category and form of financial assets, and clarifies the need for a valuation allowance on some deferred tax assets. The ASU is required for us on January 1, 2018. We do not expect the adoption of ASU 2016-01 to have a significant impact on our consolidated financial statements.
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2014-09, Revenue from Contracts with Customers (“Topic 606”). Topic 606 supersedes the revenue recognition requirements in ASU Topic 605, Revenue Recognition (“Topic 605”), and requires the recognition of revenue when promised goods or services are transferred to customers in an amount that reflects the considerations to which the entity expects to be entitled to in exchange for those goods or services. Topic 606 also includes Subtopic 340-40, Other Assets and Deferred Costs - Contracts with Customers, which discusses the deferral of incremental costs of obtaining a contract with a customer, including the period of amortization of such costs. Collectively, we refer to Topic 606 and Subtopic 340-40 as the “new standard”. The two permitted transition methods under the new standard are the full retrospective method, in which the new standard would be applied to each prior reporting period presented and the cumulative effect of applying the new standard would be recognized at the earliest period shown, or the modified retrospective method, in which the cumulative effect of applying the new standard would be recognized at the date of initial application. In the fourth quarter of 2017, we substantially completed our assessment of the new standard, including completing our historical contract reviews and our evaluation of incremental costs of obtaining a contract. We will adopt the new standard on January 1, 2018 under the modified retrospective method. We expect an immaterial transition adjustment and an immaterial impact to our net earnings on an ongoing basis.
NOTE 3. CONCENTRATION OF CREDIT RISK
Financial instruments that potentially subject the Company to concentrations of credit risk consist primarily of cash equivalents, short-term investments and accounts receivable, including receivables from major customers.
The Company’s main financial institution for banking operations held 82% and 57% of the Company’s cash and cash equivalents as of December 31, 2017 and December 31, 2016, respectively.
The Company grants credit to domestic and international clients in various industries. Exposure to losses on accounts receivable is principally dependent on each client's financial position. At December 31, 2017 and 2016, 24% and 29%, respectively, of the outstanding receivable balance was with one customer.
NOTE 4. FDA CLEARANCE
On February 23, 2017, the U.S. Food and Drug Administration (“FDA”) granted Accelerate’s de novo request to market the Accelerate Pheno™ system and Accelerate PhenoTest™ BC kit for identification and antibiotic susceptibility testing of pathogens directly from positive blood culture samples.
Due to various factors, the Company manufactured inventory in advance of regulatory approval (pre-launch inventory).
On January 1, 2017, the regulatory review process had progressed to a point that objective and persuasive evidence of approval was sufficiently probable, and a future economic benefit existed. On January 1, 2017, the Company started capitalizing pre-launch inventory. Additional information regarding inventory is included in Item 8, Note 7, Inventory.
Prior to January 1, 2017, all pre-launch inventory was expensed because a future economic benefit could not
59
be asserted. Costs associated with the Company’s purchase of inventory were reported as research and development costs, or if the inventory was used in marketing evaluations, as sales, general and administrative costs on the consolidated statements of operations and comprehensive loss.
NOTE 5. FAIR VALUE OF FINANCIAL INSTRUMENTS
The following tables represent the financial instruments measured at fair value on a recurring basis on the financial statements of the Company and the valuation approach applied to each class of financial instruments at December 31, 2017 and 2016 (see Note 2, Summary of Significant Accounting Policies for further information):
December 31, 2017 | ||||||||||||
(in thousands) | ||||||||||||
Quoted Prices in Active Markets for Identical Assets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | Total | |||||||||
Assets: | ||||||||||||
Cash and cash equivalents: | ||||||||||||
Money market funds | $ | 7,832 | $ | — | $ | — | $ | 7,832 | ||||
Investments: | ||||||||||||
Certificates of deposit | — | 13,441 | — | 13,441 | ||||||||
US Treasury securities | 14,716 | — | — | 14,716 | ||||||||
US Agency securities | — | 8,459 | — | 8,459 | ||||||||
Commercial paper | — | 9,171 | — | 9,171 | ||||||||
Asset-backed securities | — | 3,025 | — | 3,025 | ||||||||
Corporate notes and bonds | — | 31,836 | — | 31,836 | ||||||||
Total investments | 14,716 | 65,932 | — | 80,648 | ||||||||
Total assets measured at fair value | $ | 22,548 | $ | 65,932 | $ | — | $ | 88,480 | ||||
December 31, 2016 | ||||||||||||
(in thousands) | ||||||||||||
Quoted Prices in Active Markets for Identical Assets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | Total | |||||||||
Assets: | ||||||||||||
Cash and cash equivalents: | ||||||||||||
Money market funds | $ | 10,970 | $ | — | $ | — | $ | 10,970 | ||||
Investments: | ||||||||||||
Certificates of deposit | — | 7,257 | — | 7,257 | ||||||||
US Treasury securities | 8,544 | — | — | 8,544 | ||||||||
US Agency securities | — | 4,501 | — | 4,501 | ||||||||
Asset-backed securities | — | 5,557 | — | 5,557 | ||||||||
Corporate notes and bonds | — | 32,660 | — | 32,660 | ||||||||
Total investments | 8,544 | 49,975 | — | 58,519 | ||||||||
Total assets measured at fair value | $ | 19,514 | $ | 49,975 | $ | — | $ | 69,489 | ||||
Money market funds are included in cash and cash equivalents on the consolidated balance sheet.
Level 1 assets are priced using quoted prices in active markets for identical assets which include money market
60
funds and U.S. Treasury securities as these specific assets are liquid.
Level 2 available-for-sale securities are priced using quoted market prices for similar instruments or nonbinding market prices that are corroborated by observable market data. The Company uses inputs such as actual trade data, benchmark yields, broker/dealer quotes, and other similar data, which are obtained from quoted market prices, independent pricing vendors, or other sources, to determine the ultimate fair value of these assets and liabilities. The Company uses such pricing data as the primary input to make its assessments and determinations as to the ultimate valuation of its investment portfolio and has not made, during the periods presented, any material adjustments to such inputs. There were no transfers between levels during the year ended December 31, 2017.
NOTE 6. INVESTMENTS
The following tables summarize the Company’s available-for-sale investments at December 31, 2017 and 2016 (in thousands):
AVAILABLE-FOR-SALE INVESTMENTS | ||||||||||||
December 31, 2017 | ||||||||||||
(in thousands) | ||||||||||||
Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Fair Value | |||||||||
Certificates of deposit | $ | 13,441 | $ | — | $ | — | $ | 13,441 | ||||
US Treasury securities | 14,787 | — | (71 | ) | 14,716 | |||||||
US Agency securities | 8,510 | — | (51 | ) | 8,459 | |||||||
Commercial paper | 9,171 | — | — | 9,171 | ||||||||
Asset-backed securities | 3,026 | — | (1 | ) | 3,025 | |||||||
Corporate notes and bonds | 31,906 | — | (70 | ) | 31,836 | |||||||
Total | $ | 80,841 | $ | — | $ | (193 | ) | $ | 80,648 | |||
AVAILABLE-FOR-SALE INVESTMENTS | ||||||||||||
December 31, 2016 | ||||||||||||
(in thousands) | ||||||||||||
Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Fair Value | |||||||||
Certificates of deposit | $ | 7,257 | $ | — | $ | — | $ | 7,257 | ||||
US Treasury securities | 8,553 | 1 | (10 | ) | 8,544 | |||||||
US Agency securities | 4,514 | — | (13 | ) | 4,501 | |||||||
Asset-backed securities | 5,554 | 3 | — | 5,557 | ||||||||
Corporate notes and bonds | 32,717 | 3 | (60 | ) | 32,660 | |||||||
Total | $ | 58,595 | $ | 7 | $ | (83 | ) | $ | 58,519 | |||
The following table summarizes the maturities of the Company’s available-for-sale securities at December 31, 2017, and 2016 (in thousands):
61
AVAILABLE-FOR-SALE INVESTMENT MATURITIES | ||||||||||||
(in thousands) | ||||||||||||
At December 31, 2017 | At December 31, 2016 | |||||||||||
Amortized Cost | Fair Value | Amortized Cost | Fair Value | |||||||||
Due in less than 1 year | $ | 55,801 | $ | 55,735 | $ | 45,391 | $ | 45,344 | ||||
Due in 1-5 years | 25,040 | 24,913 | 13,204 | 13,175 | ||||||||
Total | $ | 80,841 | $ | 80,648 | $ | 58,595 | $ | 58,519 | ||||
Proceeds from sales of marketable securities (including principal paydowns) for the years ended December 31, 2017, and 2016, were $11.5 million and $9.7 million, respectively. The Company determines gains and losses of marketable securities based on specific identification of the securities sold. There were no realized gains from sales of marketable securities the year ended December 31, 2017, and $6,000 gross realized gains from sales of marketable securities for year ended December 31, 2016. The gross proceeds associated with the realized gains for the year ended December 31, 2016 were $7.2 million. No material balances were reclassified out of accumulated other comprehensive income (loss) for the years ended December 31, 2017, 2016 and 2015.
No other-than-temporary impairments are recorded as no investments had a fair value that remained less than its cost for more than twelve months as of December 31, 2017, and 2016. The Company does not intend to sell investments and it is more likely than not that we will not be required to sell investments before recovering the amortized cost.
Additional information regarding the fair value of our financial instruments is included in Item 8, Note 5, Fair Value of Financial Instruments.
NOTE 7. INVENTORY
Inventory is stated at the lesser of cost or net realizable value, with cost determined on the first-in-first-out method. The allocation of production overhead to inventory costs is based on normal production capacity. Abnormal amounts of idle facility expense and spoilage are expensed as incurred, and not included in overhead subject to capitalization. The Company maintains provisions for excess and obsolete inventory based on management’s estimates of forecasted demand and, where applicable, product expiration. Inventories consisted of the following at December 31, (in thousands):
2017 | 2016 | |||||
Raw materials | $ | 4,220 | $ | — | ||
Work in process | 377 | — | ||||
Finished goods | 3,466 | — | ||||
Inventory, net | $ | 8,063 | $ | — | ||
See Note 4, FDA Clearance for further information related to 2016 inventory balances.
NOTE 8. PROPERTY AND EQUIPMENT
Property and equipment are recorded at cost and consisted of the following at December 31, (in thousands).
62
PROPERTY AND EQUIPMENT | ||||||
(in thousands) | ||||||
2017 | 2016 | |||||
Computer equipment | $ | 2,756 | $ | 2,270 | ||
Technical equipment | 3,348 | 2,427 | ||||
Facilities | 3,621 | 3,387 | ||||
Instruments | 1,400 | — | ||||
Capital projects in progress | 349 | 1,010 | ||||
Total property and equipment | $ | 11,474 | $ | 9,094 | ||
Accumulated depreciation - other | (6,584 | ) | (4,836 | ) | ||
Net property and equipment | $ | 4,890 | $ | 4,258 | ||
Depreciation expense (which includes amortization of capital lease assets) for the years ended December 31, 2017, 2016 and 2015, was $2.2 million, $2.3 million and $1.8 million, respectively.
NOTE 9. LICENSE AGREEMENTS AND GRANTS
National Institute of Health Grant
In February 2015, the National Institute of Health awarded Denver Health and the Company a five-year, $5.0 million grant to develop a fast and reliable identification and categorical susceptibility test for carbepenem-resistant Enterobacteriaceae directly from whole blood. The cumulative award amount under these subawards is $885,000. The amounts invoiced for the years ended December 31, 2017, 2016 and 2015 was $240,000, $74,000 and $483,000, respectively.
Arizona Commerce Authority
In August 2012, the Company entered into a Grant Agreement (the “Grant Agreement”) with the Arizona Commerce Authority, an agency of the State of Arizona (the “Authority”), pursuant to which the Authority provided certain state and county sponsored incentives for the Company to relocate its corporate headquarters to, and expand its business within, the State of Arizona (the “Project”). Pursuant to the Grant Agreement, the Authority agreed to provide a total grant in the amount of $1.0 million (the “Grant”) for the use by the Company in the advancement of the Project. The Grant is payable out of an escrow account in four installments, upon the achievement of the following milestones:
• | Milestone 1 – Relocation of Company’s operations and corporate headquarters to Arizona and creation of 15 Qualified Jobs (as defined below). |
• | Milestone 2 – Creation of 30 Qualified Jobs (including Qualified Jobs under Milestone 1). |
• | Milestone 3 – Creation of 40 Qualified Jobs (including Qualified Jobs under Milestones 1 and 2). |
• | Milestone 4 – Creation of 65 Qualified Jobs (including Qualified Jobs under Milestones 1, 2 and 3) and capital investment of at least $4.5 million. |
For purposes of the Grant Agreement, a “Qualified Job” is a job that is permanent, full-time, new to Arizona, and for which the Company pays average (across all Qualified Jobs identified by the Company in its discretion) annual wages of at least $63,000 and offers health insurance benefits and pays at least 65% of the premiums associated with such benefits. The amount of each installment payment will be determined in accordance with a formula specified in the Grant Agreement. The Grant Agreement also contains other customary provisions, including representations, warranties and covenants of both parties. As of December 31, 2017, the Company has collected all of the $1.0 million in milestones. The full amount is recorded in deferred revenue and income until the economic development provisions of the grant have been satisfied in full, as there are “claw-back” provisions which would require repayment of certain amounts received if employment levels are not sustained during the term of the arrangement. Once the “claw-back” provisions expire in January 2018, we will recognize the grant as an offset to expense. Further details are included in Note 10, Deferred Revenue and Income.
63
In January 2018, the “claw-back” provisions expired and the $1.0 million was recognized as an offset to expense.
Arizona R&D Refundable Tax Credit Program
The Company has applied for and met the program requirements to receive a “Certificate of Qualification” from the Arizona Commerce Authority (“Authority”) which allows the Company to be eligible for a partial refund of research and development investments (“Arizona R&D Refundable Tax Credit Program”). The amounts collected under this program are recorded as an offset to research and development expenses, and for the years ended December 31, 2017, 2016 and 2015, were $0, $1.2 million and $647,000, respectively. If the amount received for this program is later determined to be incorrect or invalid, the excess may need to be repaid.
NOTE 10. DEFERRED REVENUE AND INCOME
Deferred revenue consists of amounts received for products or services not yet delivered or earned. Deferred income consists of amounts received for commitments not yet fulfilled. If we anticipate that the revenue or income will not be earned within the following twelve months, the amount is reported as long-term deferred income. A summary of the balances as of December 31, follows (in thousands):
2017 | 2016 | |||||
Products and services not yet delivered | 71 | 35 | ||||
Arizona Commerce Authority grant | 1,000 | — | ||||
Total current deferred revenue and income | $ | 1,071 | $ | 35 | ||
Arizona Commerce Authority grant | $ | — | $ | 1,000 | ||
Total long-term deferred income | $ | — | $ | 1,000 | ||
Through December 31, 2017, we received $1.0 million in milestone payments from the Arizona Commerce Authority under the Grant Agreement described in Note 9, License Agreements and Grants. As of December 31, 2017, no such payments have been recognized in income, and we do not anticipate recognizing such payments as income until the “claw-back” provisions under the Grant Agreement expire in 2018.
In January 2018, the “claw-back” provisions expired and the $1.0 million was recognized as an offset to expense.
Deferred revenue of $114,000 was recognized and recorded as a component of net sales on the consolidated statements of operations and comprehensive loss for the year ended December 31, 2016. No material deferred revenue was recognized and recorded as a component of net sales during the year ended December 31, 2017.
NOTE 11. STOCK PURCHASE
In 2012, we entered into a Securities Purchase Agreement with Abeja Ventures, LLC (“Abeja”), pursuant to which the Company agreed to sell and issue to Abeja at a purchase price of $1.03 per share for an aggregate purchase price of $14.4 million; (i) 14.0 million shares of the Company’s common stock (“Common Stock”); (ii) a warrant to purchase 7.0 million shares of Common Stock at an exercise price of $1.03 per share (the “$1.03 Warrant”); and (iii) another warrant to purchase 7.0 million shares of Common Stock at an exercise price of $2.00 per share (the “$2.00 Warrant”), with each warrant exercisable prior to the fifth anniversary of the closing of the transactions contemplated by the Securities Purchase Agreement (collectively, the “Investment”). The purchase of Common Stock and warrants pursuant to the Investment, which was consummated in June 2012, qualified for equity treatment under U.S. GAAP. The respective values of the warrants and Common Stock were calculated using their relative fair values and both are classified under Contributed Capital. The value therefore recorded for the warrants was $5.9 million and for the Common Stock was $8.5 million.
Both warrants were exercisable until June 26, 2017, which was the fifth anniversary of the date on which the warrants were issued.
64
In 2013, Abeja exercised in full its warrant to purchase 7.0 million shares of Common Stock at an exercise price of $1.03 per share. On the same date, Abeja also exercised the 92% of its warrant to purchase an additional 7.0 million shares of Common Stock at an exercise price of $2.00 per share (Abeja exercised such warrant for 6.4 million shares, leaving 571,160 shares unexercised on that date). The Company received aggregate funds of $20.1 million in connection with such exercises. Shares issued by the Company in connection with the warrant exercises were issued directly to the members of Abeja on a pro rata basis in accordance with their membership interests and written exercise instructions provided to the Company by Abeja. Immediately after giving effect to the warrant exercises, Abeja also distributed in kind to its members (on a pro rata basis in accordance with their membership interests) the remaining shares of Common Stock held by that entity.
In 2016, warrants to purchase 155,289 shares were exercised at an exercise price of $2.00 leaving 415,871 warrants for shares unexercised at December 31, 2016. The Company received funds totaling $311,000 in connection with these exercises which are recorded as common stock and contributed capital in the consolidated balance sheet.
In 2017, warrants to purchase 370,307 shares were exercised at an exercise price of $2.00 leaving 45,564 warrants for shares unexercised. The Company received funds totaling $741,000 in connection with these exercises which are recorded as common stock and contributed capital in the Consolidated Balance Sheet. The remaining 45,564 warrants for shares expired on June 26, 2017 and went unexercised. At December 31, 2017 the Company did not carry any warrants for shares due to all warrants for shares being exercised or expired.
NOTE 12. PUBLIC OFFERINGS
On December 9, 2015, the Company published a prospectus supplement underwritten by J.P. Morgan and Piper Jaffray (“2015 Underwriters”) offering 5.6 million shares of common stock with an option for the 2015 Underwriters to purchase up to 838,000 additional shares of common stock for a total of 6.4 million shares. The public offering price was $17.00 per share and underwriting discounts and commissions were $1.19 per share.
Affiliates, including entities affiliated with one of Company's directors, Jack Schuler, and which together are our largest stockholders, purchased 2.9 million of the offered shares at the public offering price of $17.00 per share. This includes various types of sales or offers to sell. The 2015 Underwriters did not receive any underwriting discount or commissions on the sale of 2.4 million shares to such affiliates.
The public offering was finalized and 6.4 million shares of common stock were delivered to the purchasers on or around December 15, 2015. Proceeds from the sale totaled $109.3 million less underwriting and other expenses of $5.9 million for net proceeds of $103.4 million.
On May 9, 2017, the Company published a prospectus supplement underwritten by J.P. Morgan Securities LLC, William Blair & Company, L.L.C., Piper Jaffray & Co. and BTIG, LLC (“2017 Underwriters”) offering 2.8 million shares of common stock with an option for the 2017 Underwriters to purchase up to 413,000 additional shares of common stock for a total of 3.2 million shares. The public offering price was $28.85 per share and underwriting discounts and commissions were $1.73.
The public offering was finalized and 2.8 million shares of common stock were delivered to the purchasers on or around May 15, 2017. The 2017 Underwriters partially exercised their option to purchase an additional 335,000 shares, with the sale closing on June 14, 2017, and the option as to the remaining shares expired June 15, 2017. Proceeds from the sales totaled $89.0 million less underwriting discounts, commissions and other costs of $5.8 million for net proceeds of $83.2 million.
The net proceeds will be used for general corporate purposes and to fund our commercialization efforts. We may also use a portion of the net proceeds to acquire or invest in complementary businesses, technologies, product candidates or other intellectual property, although we have no present commitments or agreements to do so. Accordingly, we will retain broad discretion over the use of these proceeds.
NOTE 13. RELATED PARTY TRANSACTION
In June 2016, the Company recorded a net amount of $866,000 related to the recovery of short-swing profits under Section 16(b) of the Securities Exchange Act of 1934, as amended. The Company recognized these related
65
party proceeds as an increase to contributed capital on the consolidated balance sheet.
NOTE 14. EARNINGS PER SHARE
The financial statements show basic earnings (loss) per share.
The Company’s net loss for the periods presented caused the inclusion of all outstanding warrants, restricted stocks and options to purchase our Common Stock to be antidilutive. As of December 31, 2017, 2016 and 2015, there were Common Stock options, restricted stocks and warrants exercisable for 7,352,281, 7,313,245 and 6,778,580 shares of Common Stock, respectively, which were not included in diluted loss per share as the effect was antidilutive.
NOTE 15. EMPLOYEE AND CONSULTANT EQUITY-BASED COMPENSATION
The Company has three equity based compensation plans, which are discussed below:
Non-Qualified Stock Option Plan
The Non-Qualified Stock Option Plan (the “Non-Qualified Plan”) was a stockholder-approved plan that provided for stock option grants to independent contractors, technical advisors and directors of the Company. The exercise price of each option, which has a maximum 10 year life, was established by the Company's Compensation Committee on the date of grant.
As of December 31, 2017, there were 280,000 options exercised under the Non-Qualified Plan and 0 that remain outstanding. The Non-Qualified Plan has been replaced by the 2012 Omnibus Equity Incentive Plan, so no further options are available for grant.
2004 Omnibus Stock Option Plan
In December 2004, the Company’s stockholders approved the Omnibus Stock Option Plan and reserved 500,000 shares of its authorized but unissued Common Stock for stock options to be granted to employees, independent contractors, technical advisors and directors of the Company. The authorized shares in this plan were increased by 5,000,000 shares to an aggregate amount of 5,500,000 upon stockholder approval during the fiscal year ended July 31, 2012.
As of December 31, 2017, there were 658,994 options exercised under the 2004 Omnibus Stock Option Plan and 3,281,006 that remain outstanding. The 2004 Omnibus Stock Option Plan has been replaced by the 2012 Omnibus Equity Incentive Plan, so no further options are available for grant.
2012 Omnibus Equity Incentive Plan
In December 2012, the Company’s stockholders approved the Company’s 2012 Omnibus Equity Incentive Plan to replace all prior plans (“Prior Plans”). The Prior Plans remain in effect until all awards granted under those plans have been exercised, forfeited, canceled, expired or otherwise terminated.
In connection with the approval of such plan, all stock options, totaling 1,677,500 formerly available for new awards under the Prior Plans were transferred to the 2012 Omnibus Equity Incentive Plan.
During the Company's Annual Meeting of Stockholders, stockholders approved amendments to the Company's 2012 Omnibus Equity Incentive Plan increasing the number of stock options of Common Stock reserved and available for grant by 4,000,000 in May 2014 and 2,000,000 in May 2017 resulting in a total of 7,677,500 reserved shares.
Stock options granted under this plan typically vest either (i) one year after grant date, (ii) monthly over a one year period, (iii) annually over a five year period, or (iv) 40% two years after grant date and the remaining 60% monthly over the next three years. The maximum term is ten years.
Restricted Stock Units (“RSU's”) granted under this plan typically vest either (i) annually over a five year period, or (ii) immediately.
Stock Grants (“SG's”) granted under this plan vest immediately.
66
As of December 31, 2017, there were 621,216 options exercised as well as 18,011 RSU's and SG's released under the 2012 Omnibus Equity Incentive Plan. There were 4,071,275 shares remaining outstanding, leaving 2,966,998 available for grant.
In December 2015, the Board of Directors voted to restrict new grants under the 2012 Omnibus Equity Incentive Plan to 200,000 shares until such time as the Company’s total available shares are increased. The Company's total available shares were increased in March 2016 terminating the restriction of new grants.
Combined Stock Option Plans
The following table summarizes option activity under all plans during the years ending December 31, 2017, and December 31, 2016 and shows the exercisable shares as of December 31, 2017:
Stock Option Activity | |||||
Number of Shares | Weighted Average Exercise Price per Share | ||||
Options Outstanding January 1, 2016 | 6,167,170 | $ | 6.91 | ||
Granted | 1,024,050 | 14.05 | |||
Forfeited | (147,663 | ) | 18.43 | ||
Exercised | (158,743 | ) | 7.47 | ||
Expired | (27,690 | ) | 4.87 | ||
Options Outstanding December 31, 2016 | 6,857,124 | 7.72 | |||
Granted | 1,208,542 | 23.84 | |||
Forfeited | (79,962 | ) | 17.82 | ||
Exercised | (656,846 | ) | 8.93 | ||
Expired | (727 | ) | 24.45 | ||
Options Outstanding December 31, 2017 | 7,328,131 | 10.16 | |||
Exercisable December 31, 2017 | 5,163,899 | 6.38 | |||
The cash received from the exercise of options during the year ending December 31, 2017, was $5.9 million and the tax benefit realized was $0 for the same period. Upon exercise, shares are issued from shares authorized and held in reserve. The intrinsic value of options exercised was $12.1 million, $2.2 million and $2.0 million for the years ending December 31, 2017, 2016 and 2015, respectively.
The total fair value of options vesting during the period was $12.0 million, $6.6 million, and $7.3 million for the years ending December 31, 2017, 2016 and 2015, respectively.
The Company accounts for all option grants using the Black-Scholes option pricing model in accordance with ASC 718 for options granted or extended. The table below summarizes the inputs used to calculate the estimated fair value of options awarded during the respective periods:
67
Black-Scholes Assumptions for Options Granted | |||||||||
Years ended December 31, | |||||||||
2017 | 2016 | 2015 | |||||||
Expected term (in years) | 6.23 | 6.43 | 6.26 | ||||||
Volatility | 77 | % | 86 | % | 91 | % | |||
Expected dividends | — | — | — | ||||||
Risk free interest rates | 2.1 | % | 1.6 | % | 1.7 | % | |||
Estimated forfeitures | — | % | 8.5 | % | 5.6 | % | |||
Weighted average fair value | $ | 16.24 | $ | 10.35 | $ | 16.69 | |||
In general, option awards have a requisite service period and unvested options are forfeited upon employee or consultant termination. In years prior to 2016, the Company estimated a forfeiture rate which was applied to outstanding options to determine options expected to be forfeited with the remaining outstanding options being those expected to vest. Effective January 2017, the Company implemented ASU 2016-09, Compensation-Stock Compensation (Topic 718) Improvements to Employee Share-Based Payment Accounting and made a policy election to account for forfeitures as they occur. Further information regarding this implementation is included below in this note.
The following table shows summary information for outstanding options and options that are exercisable (vested) as of December 31, 2017:
Stock Option Supplemental Information | ||||||
Options Outstanding | Options Exercisable | |||||
Number of options | 7,328,131 | 5,163,899 | ||||
Weighted average remaining contractual term (in years) | 6.16 | 5.19 | ||||
Weighted average exercise price | $ | 10.16 | $ | 6.38 | ||
Weighted average fair value | $ | 7.48 | $ | 4.74 | ||
Aggregate intrinsic value (in millions) | $ | 117.7 | $ | 102.4 | ||
The aggregate intrinsic value in the table above represents the total pretax intrinsic value that would have been received by the option holders had all option holders exercised their options on that date. It is calculated as the difference between the Company’s closing stock price of $26.20 on the last trading day of 2017 and the exercise price multiplied by the number of shares for options where the exercise price is below the closing stock price. This amount changes based on the fair market value of the Company’s stock.
In December 2015, in connection with the Company's public offering, we modified the stock options of three of our executive officers to restrict the ability to exercise until such time as additional available unissued shares have been authorized. Such authorization was obtained from a majority of our shareholders as of December 31, 2015, and was filed with the State of Delaware in March 2016. There was an immaterial amount of incremental compensation associated with this modification.
The following table summarizes RSU and SG activity during the years ending December 31, 2017, and December 31, 2016:
68
RSU and SG Activity | ||||
Number of Shares | Weighted Average Grant Date Fair Value per Share | |||
RSU's & SG Outstanding January 1, 2016 | 40,250 | 20.91 | ||
Granted | — | — | ||
Forfeited | — | — | ||
Vested/released | — | — | ||
RSU's & SG's outstanding December 31, 2016 | 40,250 | 20.91 | ||
Granted | 1,911 | 22.40 | ||
Forfeited | — | — | ||
Vested/released | (18,011 | ) | 21.07 | |
RSU's & SG's outstanding December 31, 2017 | 24,150 | 20.91 | ||
The total fair value of RSU's and SG's vested and released during the period was $379,000, $0, and $0 for the years ending December 31, 2017, 2016 and 2015, respectively.
The Company records compensation cost based on the fair value of the award. The table below summarizes the weighted average fair value of RSU's and SG's awarded for the years ending December 31,:
RSU and SG Grants | ||||||
2017 | 2016 | 2015 | ||||
Weighted average fair value | 22.40 | — | 20.91 | |||
The expense and tax benefits recognized on the Company’s consolidated statements of operations and comprehensive loss related to options for the years ending December 31, is summarized below (in thousands):
Equity-Based Compensation Expenses and Tax Benefit (in thousands) | |||||||||
2017 | 2016 | 2015 | |||||||
Cost of Sales | $ | 99 | $ | — | $ | — | |||
Research and development | $ | 3,738 | $ | 1,585 | $ | 2,479 | |||
Sales, general and administrative | 10,096 | 7,190 | 5,909 | ||||||
Total equity-based compensation expense | 13,933 | 8,775 | 8,388 | ||||||
Recognized tax benefit | $ | — | $ | — | $ | — | |||
As of December 31, 2017, $304,000 and $48,000 of equity-based compensation expense was a component of capitalized inventory and property and equipment, respectively.
As of December 31, 2017, unrecognized equity-based compensation cost related to unvested stock options, and unvested RSU's was $15.9 million and $174,000, respectively. This is expected to be recognized over the years 2018 through 2022.
In January 2017, we implemented ASU 2016-09, Compensation-Stock Compensation (Topic 718) Improvements to Employee Share-Based Payment Accounting. Pursuant to this guidance, we made a policy election to account for forfeitures as they occur rather than on an estimated basis and, therefore, equity based compensation expense for the year ended December 31, 2017 has been calculated based on actual forfeitures in our condensed consolidated statements of operations and comprehensive loss, rather than our previous approach which was net of estimated forfeitures. Share-based compensation expense for the years ending December 31, 2016 and December 31, 2015 is recorded net of estimated forfeitures, which were based on historical forfeitures and adjusted to reflect changes in facts and circumstances, if any. This change was accounted for using the modified retrospective transition method.
69
This election resulted in a cumulative-effect adjustment which increased our accumulated deficit and additional paid-in capital by $655,000 for all outstanding awards as of January 1, 2017. We believe this election simplifies several aspects of the accounting for share-based payment transactions.
This new guidance requires that we record excess tax benefits and tax deficiencies related to the settlement of employee stock-based compensation to the income tax expense line item on our condensed consolidated statements of operations and comprehensive loss. The new guidance also states that previously unrecognized excess tax benefits should be recognized on a modified retrospective basis as of the beginning of the annual period of adoption. At January 1, 2017, we recorded approximately $1.5 million of additional deferred tax assets, which are fully offset by a valuation allowance. Accordingly, the adoption of ASU 2016-09 did not result in an adjustment to retained earnings for the cumulative effect of the tax benefit of the stock compensation.
The new guidance also requires excess tax benefits to be classified as an operating activity in the statement of cash flows rather than as a financing activity. Additionally, ASU 2016-09 requires that the minimum tax withholding paid on behalf of employees for share-based awards be classified as a financing activity in the statement of cash flows. Adoption of ASU 2016-09 did not result in any adjustments to prior period disclosures on the Company's consolidated statement of cash flows.
NOTE 16. INCOME TAXES
The components of the pretax loss from operations for the years ended December 31, are as follows (in thousands):
2017 | 2016 | 2015 | |||||||
U.S. Domestic | $ | (46,849 | ) | $ | (48,539 | ) | $ | (44,415 | ) |
Foreign | (16,686 | ) | (17,568 | ) | (1,083 | ) | |||
Pretax loss from operations | $ | (63,535 | ) | $ | (66,107 | ) | $ | (45,498 | ) |
The components of the provision for income taxes for the years ended December 31, is presented in the following table:
2017 | 2016 | 2015 | |||||||
Current: | |||||||||
Federal | $ | — | $ | — | $ | — | |||
State | — | — | — | ||||||
Foreign | 493 | 267 | — | ||||||
Total current provision | 493 | 267 | — | ||||||
Deferred: | |||||||||
Federal | — | — | — | ||||||
State | — | — | — | ||||||
Foreign | — | — | — | ||||||
Total deferred provision | — | — | — | ||||||
Total provision | $ | 493 | $ | 267 | $ | — | |||
On December 22, 2017, President Trump signed into law the Tax Cuts and Jobs Act (the Tax Act”). The Tax Act reduced the corporate tax rate from the maximum federal statutory rate of 35% to 21%. The Tax Act states that the 21% corporate tax rate is effective for tax years beginning on or after January 1, 2018. However, existing tax law, which was not amended under the Tax Act, governs when a change in tax rate is effective. Existing tax law provides that if the taxable year includes the effective date of any rate change (unless the change is the first date of the taxable year), taxes should be calculated by applying a blended rate to the taxable income for the year. For the Company, the net deferred tax assets for US purposes have been revalued to 21% as of December 31, 2017 with a corresponding adjustment to the valuation allowance. Therefore, the reduction in the US corporate tax rate had no impact to the Company's earnings.
70
Due to uncertainties which currently exist in the interpretation of the provisions of the Tax Cuts and Jobs Act of 2017 regarding Internal Revenue Code Section 162(m), the Company has not evaluated the potential impacts of IRC Section 162(m) as amended by the Tax Cuts and Jobs Act of 2017 on its financial statements.
In conjunction with the Tax Act, the SEC staff issued Staff Accounting Bulletin No. 118 (“SAB 118”) to address the application of U.S. GAAP in situations when a registrant does not have the necessary information available, prepared, or analyzed (including computations) in reasonable detail to complete the accounting for certain income tax effects of the Tax Act. The Company has recognized the provisional tax impacts related to deemed repatriated earnings and the revaluation of deferred tax assets and liabilities and included these amounts in its consolidated financial statements for the year ended December 31, 2017. The ultimate impact may differ from these provisional amounts, possibly materially, due to, among other things, additional analysis, changes in interpretations and assumptions the Company has made, additional regulatory guidance that may be issued, and actions the Company may take as a result of the Tax Act.
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s net deferred income taxes for the years ended December 31, are as follows (in thousands):
2017 | 2016 | |||||
Deferred tax assets: | ||||||
Net operating loss carryforward | $ | 41,086 | $ | 42,013 | ||
Property & equipment | 524 | 690 | ||||
Inventory | 58 | 1,735 | ||||
Stock options | 8,195 | 7,208 | ||||
Intangible assets, definite-lived | 42 | 220 | ||||
General business credit | 6,651 | 3,984 | ||||
Deferred revenue | 253 | 370 | ||||
Other | 45 | 121 | ||||
Valuation allowance | (56,854 | ) | (56,341 | ) | ||
Deferred tax assets | $ | — | $ | — | ||
As of December 31, 2017, the Company generated regular tax federal net operating losses of approximately $173.4 million. The Company's ability to realize tax benefit from the net operating loss is subject to annual limitation under Internal Revenue Code Section 382. The Company will never get the benefit of $4.2 million of the net operating losses generated prior to June 26, 2012. The deferred tax asset has been adjusted to reflect the Section 382 limitation. The net operating losses available for future use are approximately $169.2 million. As a result of The Act, for U.S. income tax purposes, net operating losses can be carried forward for up to 20 years. The Company’s federal net operating losses will begin to expire in 2023.
As of December 31, 2017, the Company has generated Arizona net operating losses of approximately $142.8 million. The Company's Arizona net operating losses will begin to expire in 2033.
The net deferred tax asset valuation allowance is $56.9 million as of December 31, 2017, compared to $56.3 million as of December 31, 2016. The valuation allowance is based on management’s assessment that it is more likely than not that the Company will not have taxable income in the foreseeable future. Due to the Company's consolidated loss position, the Company maintains a valuation allowance against its deferred tax assets.
The Company began commercialization of its products in Europe in 2016 and has subsidiaries in the Netherlands, France, Germany, Italy, Spain and the United Kingdom. The Company intends to treat earnings from its foreign subsidiaries as permanently reinvested.
The difference between the U.S. federal statutory income tax rate and the Company’s effective tax rate for years ending December 31, is as follows:
71
2017 | 2016 | 2015 | ||||
U.S. federal statutory income tax rate | (34.00 | )% | (34.00 | )% | (34.00 | )% |
State taxes, net of federal tax benefit | (2.62 | ) | (1.69 | ) | (2.93 | ) |
Permanent and other differences | (2.31 | ) | (0.17 | ) | 0.11 | |
Change in tax rates | (1.02 | ) | 0.67 | — | ||
Tax rate differential | 8.99 | 8.62 | 0.42 | |||
Tax cuts and jobs act | 38.46 | — | — | |||
Unrecognized tax benefits | 1.20 | 1.09 | 0.40 | |||
Nondeductible equity and other compensation | (4.31 | ) | 1.17 | 2.86 | ||
Limitation on net operating losses due to §382 | — | — | — | |||
Credit for increased research activities | (4.42 | ) | (3.31 | ) | (2.67 | ) |
Change in Valuation allowance | 0.81 | 28.02 | 35.81 | |||
0.78 | % | 0.40 | % | — | % | |
The Company's uncertain tax positions at December 31, as follows (in thousands):
2017 | 2016 | 2015 | |||||||
Balance at beginning of year | $ | 1,101 | $ | 343 | $ | 161 | |||
Increases for prior positions | 97 | 37 | — | ||||||
Increases for current year positions | 943 | 721 | 182 | ||||||
Other increases | — | — | — | ||||||
Decreases due to settlements | — | — | — | ||||||
Expiration of the statute of limitations for the assessment of taxes | — | — | — | ||||||
Other decreases | — | — | — | ||||||
Balance at end of year | $ | 2,141 | $ | 1,101 | $ | 343 | |||
These uncertain positions are not expected to change within the next twelve months. Of the $2.1 million of uncertain tax positions, $65,000 would impact the effective tax rate, if reversed. The Company accounts for interest on uncertain tax positions within tax expense. The Company's foreign subsidiaries are subject to applicable jurisdiction examination for all years of operations. The Company did not accrue interest or penalties for these uncertain tax positions as of December 31, 2017.
The Company has incurred federal net operating losses dating to the tax year ended July 31, 2004. As such, all loss carryovers are subject to adjustment under IRS and state examination, depending on the jurisdiction in which they were incurred. In 2016, the IRS began an examination of the Company’s 2014 income tax return. The Company completed the IRS audit of the 2014 income tax return with no change.
NOTE 17. COMMITMENTS AND CONTINGENCIES
General
Estimated losses from contingencies are accrued by a charge to income when information available prior to issuance of the financial statements indicates that it is probable that a liability could be incurred and the amount of the loss can be reasonably estimated. Legal expenses associated with the contingency are expensed as incurred. If a loss contingency is not probable or reasonably estimable, disclosure of the contingency and estimated range of loss, if determinable, is made in the financial statements when it is at least reasonably possible that a material loss could be incurred.
Leases
The Company has entered into lease agreements, lease amendments, and lease extensions (“Lease Agreements”) for office, laboratory and manufacturing space located in Tucson, Arizona and Europe, the last of which expires in 2018. Total rent expense including common area charges was $1.3 million, $1.1 million and $685,000 for
72
the years ended December 31, 2017, 2016 and 2015, respectively. Future minimum lease payments under these agreements are as follows (in thousands):
2018 | $ | 1,119 | |
2019 | 200 | ||
2020 | 84 | ||
2021 | 47 | ||
Thereafter | 4 | ||
Total | $ | 1,454 | |
Clinical Trial Agreements
The Company has entered into master agreements with clinical trial sites in which we typically pay a set amount for start-up costs and then pay for work performed. These agreements typically indemnify the clinical trial sites from any and all losses arising from third party claims as a result of the Company's negligence, willful misconduct or misrepresentation. Clinical trial master agreement expense was $27,000, $2.1 million and $1.6 million and for years ended December 31, 2017, 2016 and 2015, respectively. The expense incurred as part of the clinical trial is included in research and development on the Company's consolidated statements of operations and comprehensive loss.
Marketing Study and Contracted Research
The Company has entered into marketing study agreements and contracted research agreements with research institutions and hospitals in which we typically pay a set amount for start-up costs and then pay for work performed. These agreements typically indemnify the sites from any and all losses arising from third party claims as a result of the Company's negligence, willful misconduct or misrepresentation.
NOTE 18. INDUSTRY AND GEOGRAPHIC INFORMATION
The Company operates as one operating segment. Sales to customers outside the U.S. represented 28%, 3% and 0% of total revenue for the years ended December 31, 2017, 2016 and 2015, respectively. As of December 31, 2017, 2016, balances due from foreign customers, in U.S. dollars, were $1.0 million and $3,000, respectively.
For the year ended December 31, 2017 the Company had sales to one customer in excess of 10%; this customers balance represented 18% of total net sales.
The following presents long-lived assets (excluding intangible assets) and total net revenue by geographic territory (in thousands):
Long lived assets as of | Total revenue | ||||||||||||||
December31, | for the years ended December 31, | ||||||||||||||
2017 | 2016 | 2017 | 2016 | 2015 | |||||||||||
Domestic | $ | 3,779 | $ | 4,150 | $ | 3,016 | $ | 238 | $ | 147 | |||||
Foreign | 1,111 | 108 | 1,161 | 8 | — | ||||||||||
$ | 4,890 | $ | 4,258 | $ | 4,177 | $ | 246 | $ | 147 | ||||||
73
NOTE 19. SUPPLEMENTAL DATA (UNAUDITED)
The following is a summary of unaudited selected quarterly financial information for the three months ended (in thousands, except per share data):
December 31, | September 30, | June 30, | March 31, | |||||||||
2017 | 2017 | 2017 | 2017 | |||||||||
Revenue | $ | 2,120 | $ | 828 | $ | 699 | $ | 530 | ||||
Gross profit | $ | 1,470 | $ | 637 | $ | 564 | $ | 504 | ||||
Loss from operations | $ | (16,136 | ) | $ | (17,315 | ) | $ | (16,423 | ) | $ | (14,310 | ) |
Net loss | $ | (16,296 | ) | $ | (17,075 | ) | $ | (16,457 | ) | $ | (14,200 | ) |
Basic and diluted net loss per share | $ | (0.29 | ) | $ | (0.31 | ) | $ | (0.31 | ) | $ | (0.27 | ) |
December 31, | September 30, | June 30, | March 31, | |||||||||
2016 | 2016 | 2016 | 2016 | |||||||||
Revenue | $ | 39 | $ | 24 | $ | 20 | $ | 163 | ||||
Gross profit | $ | 39 | $ | 24 | $ | 20 | $ | 163 | ||||
Loss from operations | $ | (16,024 | ) | $ | (17,416 | ) | $ | (17,889 | ) | $ | (15,172 | ) |
Net loss | $ | (16,135 | ) | $ | (17,299 | ) | $ | (17,866 | ) | $ | (15,074 | ) |
Basic and diluted net loss per share | $ | (0.31 | ) | $ | (0.34 | ) | $ | (0.35 | ) | $ | (0.29 | ) |
74
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
Based on an evaluation under the supervision and with the participation of the Company’s management, the Company’s Principal Executive Officer and Principal Financial Officer have concluded that the Company’s disclosure controls and procedures as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act were effective as of December 31, 2017, to ensure that information required to be disclosed by the Company in reports that it files or submits under the Exchange Act is (i) recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms and (ii) accumulated and communicated to the Company’s management, including its Principal Executive Officer and Principal Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of our financial statements for external purposes in accordance with U.S. generally accepted accounting principles. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. Under the supervision and with the participation of our management, including our CEO and CFO, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control-Integrated Framework (2013), issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under the framework in Internal Control-Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2017.
The effectiveness of our internal control over financial reporting as of December 31, 2017 has been audited by Ernst & Young LLP, our independent registered public accounting firm, as stated in their report which is included in Item 8. Financial Statements and Supplementary Data.
Attestation Report of Independent Registered Public Accounting Firm
The attestation report required under this Item 9A is contained in PART II, Item 8, Report of Independent Public Accounting Firm of this Annual Report on Form 10-K.
Changes in Internal Control Over Financial Reporting
During the year ended December 31, 2017, in connection with the Company’s preparations to commercialize the Accelerate Pheno™ system and Accelerate PhenoTest™ BC Kit the Company implemented additional internal controls related to revenue recognition and inventory.
Item 9B. Other Information
Not Applicable.
PART III
Certain information required by Part III is omitted from this Annual Report on Form 10-K because the required
75
information will be incorporated by reference to our definitive proxy statement for our 2018 Annual Meeting of Stockholders, to be filed with the SEC pursuant to Regulation 14A of the Exchange Act (the “Proxy Statement”) not later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K.
Item 10. Directors, Executive Officers and Corporate Governance
The information required by this Item will be disclosed in the Proxy Statement and is incorporated by reference to the Proxy Statement.
Item 11. Executive Compensation
The information required by this Item will be disclosed in the Proxy Statement and is incorporated by reference to the Proxy Statement.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this Item will be disclosed in the Proxy Statement and is incorporated by reference to the Proxy Statement.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this Item will be disclosed in the Proxy Statement and is incorporated by reference to the Proxy Statement.
Item 14. Principal Accounting Fees and Services
The information required by this Item will be disclosed in the Proxy Statement and is incorporated by reference to the Proxy Statement.
Item 15. Exhibits, Financial Statement Schedules
a) Documents filed as part of this report
1) All financial statements
Page | |
2) Financial Statement Schedules
All financial statement schedules have been omitted, since the required information is not applicable or because the information required is included in the financial statements and notes thereto.
b) Exhibits required by Item 601 of Registration S-K
76
The information required by this Item is set forth on the exhibit index that follows the signature page of this report.
77
Item 16. Form 10-K Summary
None.
EXHIBIT INDEX
Exhibit No. | Description | Filing Information | |
Incorporated by reference to Appendix B of the Registrant’s Definitive Proxy Statement on Schedule 14A filed on November 13, 2012 | |||
Incorporated by reference to Exhibit A to the Registrant’s Definitive Information Statement on Schedule 14C filed on July 12, 2013 | |||
Incorporated by reference to Exhibit 3.1 to the Registrant’s Current Report on Form 8-K filed on March 15, 2016 | |||
Incorporated by reference to Exhibit 3.2 filed with the Registrant’s Annual Report on Form 10-K for the fiscal year ended July 31, 2012. | |||
Incorporated by reference to Exhibit 4.2 filed with the Registrant’s Annual Report on Form 10-K for the fiscal year ended July 31, 2012 | |||
Incorporated by reference to Appendix A of the Registrant’s Definitive Proxy Statement on Schedule 14A filed on November 15, 2004 | |||
Incorporated by reference to Annex C of the Registrant’s Definitive Proxy Statement on Schedule 14A filed on May 17, 2012 | |||
Incorporated by reference to Exhibit 4.4 filed with the Registrant’s Form S-8 Registration Statement (No. 333-182930) on July 30, 2012 | |||
Incorporated by reference to Exhibit 10.5 filed with the Registrant’s Annual Report on Form 10-K for the fiscal year ended July 31, 2012 | |||
Incorporated by reference to Exhibit 10.9 filed with the Registrant’s Annual Report on Form 10-K for the fiscal year ended July 31, 2012 | |||
Incorporated by reference to Exhibit 10.10 filed with the Registrant’s Annual Report on Form 10-K for the fiscal year ended July 31, 2012 | |||
Incorporated by reference to Exhibit 10.11 filed with the Registrant’s Annual Report on Form 10-K for the fiscal year ended July 31, 2012 | |||
Incorporated by reference to Exhibit 10.12 filed with the Registrant’s Annual Report on Form 10-K for the fiscal year ended July 31, 2012 | |||
78
Incorporated by reference to Appendix C of the Registrant’s Definitive Proxy Statement on Schedule 14A filed on November 13, 2012 | |||
Incorporated by reference to Exhibit 99.2 to the Form S-8 Registration Statement (No. 333-187439) filed by the Registrant on March 22, 2013 | |||
Incorporated by reference to Exhibit 99.3 to the Form S-8 Registration Statement (No. 333-187439) filed by the Registrant on March 22, 2013 | |||
Incorporated by reference to Exhibit 99.4 to the Form S-8 Registration Statement (No. 333-187439) filed by the Registrant on March 22, 2013 | |||
Incorporated by reference to Appendix A of the Registrant’s Definitive Proxy Statement on Schedule 14A filed on April 15, 2014 | |||
Filed herewith | |||
Filed herewith | |||
Filed herewith | |||
Filed herewith | |||
Filed herewith | |||
101 | XBRL Instance Document | ||
101 | XBRL Taxonomy Extension Schema Document | ||
101 | XBRL Taxonomy Calculation Linkbase Document | ||
101 | XBRL Taxonomy Extension Definition Linkbase Document | ||
101 | XBRL Taxonomy Label Linkbase Document | ||
101 | XBRL Taxonomy Presentation Linkbase Document | ||
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
ACCELERATE DIAGNOSTICS, INC. | ||
February 28, 2018 | By: /s/ Lawrence Mehren | |
Lawrence Mehren President and Chief Executive Officer | ||
Power of Attorney
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Lawrence Mehren, as his attorney-in-fact, with the power of substitution, for him in any and all capacities, to sign any amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that said attorney-in-fact, or his substitute or substitutes, may do or cause to be done by virtue hereof.
79
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the registrant and in the capacities and on the date indicated.
Signature | Title | Date | ||
/s/ Lawrence Mehren Lawrence Mehren | President, Chief Executive Officer and Director | February 28, 2018 | ||
/s/ Steve Reichling Steve Reichling | Corporate Secretary, Chief Financial Officer and Chief Accounting Officer | February 28, 2018 | ||
/s/ John Patience John Patience | Chairman of the Board of Directors | February 28, 2018 | ||
/s/ Jack Schuler Jack Schuler | Director | February 28, 2018 | ||
/s/ Matthew W. Strobeck, Ph.D. Matthew W. Strobeck, Ph.D. | Director | February 28, 2018 | ||
/s/ Frank ten Brink Frank ten Brink | Director | February 28, 2018 | ||
/s/ Mark Miller Mark Miller | Director | February 28, 2018 | ||
/s/ Charles Watts, M.D. Charles Watts | Director | February 28, 2018 | ||
/s/ Tom Brown Tom Brown | Director | February 28, 2018 | ||
80