Attached files
| file | filename |
|---|---|
| EX-32.1 - EXHIBIT 32.1 - Anika Therapeutics, Inc. | exh_321.htm |
| EX-31.2 - EXHIBIT 31.2 - Anika Therapeutics, Inc. | exh_312.htm |
| EX-31.1 - EXHIBIT 31.1 - Anika Therapeutics, Inc. | exh_311.htm |
| EX-23.2 - EXHIBIT 23.2 - Anika Therapeutics, Inc. | exh_232.htm |
| EX-23.1 - EXHIBIT 23.1 - Anika Therapeutics, Inc. | exh_231.htm |
| EX-21.1 - EXHIBIT 21.1 - Anika Therapeutics, Inc. | exh_211.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| (Mark One) | |
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
For the fiscal year ended December 31, 2017
| |
| o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15 (d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to | |
Commission File Number 000-21326
Anika Therapeutics, Inc.
(Exact Name of Registrant as Specified in Its Charter)
Massachusetts (State or Other Jurisdiction of Incorporation or Organization) |
04-3145961 (IRS Employer Identification No.) |
32 Wiggins Avenue, Bedford, Massachusetts 01730
(Address of Principal Executive Offices) (Zip Code)
(781) 457-9000
(Registrant’s Telephone Number, Including Area Code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Name of Each Exchange on Which Registered |
| Common Stock, par value $0.01 per share | NASDAQ Global Select Market |
| Preferred Stock Purchase Rights | NASDAQ Global Select Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15 (d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer x | Accelerated filer o | Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller reporting company o | Emerging growth company o |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No x
The aggregate market value of voting and non-voting equity held by non-affiliates of the Registrant (without admitting that any person whose shares are not included in such calculation is an affiliate) as of June 30, 2017, the last day of the Registrant’s most recently completed second fiscal quarter, was $702,114,713 based on the close price per share of common stock of $49.34 as of such date as reported on the NASDAQ Global Select Market.
At February 9, 2018, there were issued and outstanding 14,693,928 shares of common stock, par value $0.01 per share.
Documents Incorporated By Reference
The registrant intends to file a proxy statement pursuant to Regulation 14A within 120 days of the end of the fiscal year ended December 31, 2017. Portions of such proxy statement are incorporated by reference into Part III of this Annual Report on Form 10-K.
| 2 |
ANIKA THERAPEUTICS, INC.
TABLE OF CONTENTS
References in this Annual Report on Form 10-K to “we,” “us,” “our,” “our company,” and other similar references refer to Anika Therapeutics, Inc. and its subsidiaries unless the context otherwise indicates.
ANIKA, ANIKA THERAPEUTICS, ANIKAVISC, CINGAL, HYAFF, HYDRELLE, HYVISC, INCERT, MONOVISC, and ORTHOVISC are our registered trademarks, and HYALOSS, ELEVESS, OPTIVISC, and SHELLGEL are our trademarks. This Annual Report on Form 10-K also contains registered marks, trademarks, and trade names that are the property of other companies and licensed to us.
| 3 |
FORM 10-K
ANIKA THERAPEUTICS, INC.
For Fiscal Year Ended December 31, 2017
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains "forward-looking statements" within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934 concerning our business, consolidated financial condition, and results of operations. The Securities and Exchange Commission ("SEC") encourages companies to disclose forward-looking statements so that investors can better understand a company’s future prospects and make informed investment decisions. Forward-looking statements are subject to risks and uncertainties, many of which are outside our control, which could cause actual results to differ materially from these statements. Therefore, you should not rely on any of these forward-looking statements. Forward-looking statements can be identified by such words as "will," "likely," "may," "believe," "expect," "anticipate," "intend," "seek," "designed," "develop," "would," "future," "can," "could," and other expressions that are predictions of or indicate future events and trends and that do not relate to historical matters. All statements other than statements of historical facts included in this Annual Report regarding our strategies, prospects, financial condition, operations, costs, plans, and objectives are forward-looking statements. Examples of forward-looking statements include, among others, statements regarding expected future operating results, expectations regarding the timing and receipt of regulatory results, anticipated levels of capital expenditures, and expectations of the effect on our financial condition of claims, litigation, and governmental and regulatory proceedings.
Please refer to "Risk Factors" for important factors that we believe could cause actual results to differ materially from those in our forward-looking statements. Any forward-looking statement made by us in this Annual Report on Form 10-K is based only on information currently available to us and speaks only as of the date on which it is made. We undertake no obligation to publicly update any forward-looking statement, whether written or oral, that may be made from time to time, whether as a result of new information, future developments, or otherwise.
| 4 |
Overview
We are a global, integrated orthopedic medicines company committed to improving the lives of patients with degenerative orthopedic diseases and traumatic conditions with clinically meaningful therapies along the continuum of care, from palliative pain management to regenerative tissue repair. We have over two decades of global expertise developing, manufacturing, and commercializing products based on our proprietary hyaluronic acid (“HA”) technology. Our orthopedic medicine portfolio includes ORTHOVISC, MONOVISC, and CINGAL, which alleviate pain and restore joint function by replenishing depleted HA, and HYALOFAST, a solid HA-based scaffold to aid cartilage repair and regeneration.
Our therapeutic offerings consist of products in the following areas: Orthobiologics, Dermal, Surgical, and Other, which includes our ophthalmic and veterinary products. All of our products are based on HA, a naturally occurring, biocompatible polymer found throughout the body. Due to its unique biophysical and biochemical properties, HA plays an important role in a number of physiological functions such as the protection and lubrication of soft tissues and joints, the maintenance of the structural integrity of tissues, and the transport of molecules to and within cells.
Our proprietary technologies for modifying the HA molecule allow product properties to be tailored specifically to therapeutic use. Our patented technology chemically modifies HA to allow for longer residence time in the body. We also offer products made from HA based on two other technologies: HYAFF, which is a solid form of HA, and ACP gel, an autocross-linked polymer of HA. Our technologies are protected by an extensive portfolio of owned and licensed patents.
Since our inception as a Massachusetts corporation in 1992, we have utilized a commercial partnership model for the distribution of our products to end users. Our strong, worldwide network of distributors has historically provided, and continues to provide, a solid foundation for our revenue growth and territorial expansion. In 2015, we made the strategic decision to commercialize our next generation viscosupplementation product, CINGAL, in the United States by utilizing a direct sales model, initially through the engagement of a contract sales organization. Ultimately, we intend to transition the direct sales function into our company as part of a broader buildout of our commercial capabilities. We believe that the combination of the direct and distribution commercial models will maximize the revenue and profitability potential from our current and future product portfolio.
In the fourth quarter of 2017, we completed all planned activities related to the strategic project we began in 2015 to insource the manufacturing of our HYAFF-based products to our Bedford, Massachusetts global headquarters facility. Final costs totaled $23.0 million for this project. These products were previously manufactured by a third-party contract manufacturer in Italy. Our main purposes behind this strategic initiative are to enhance our research and development capabilities with the aim of accelerating future product development and to improve the efficiency of our manufacturing processes.
The following sections provide more specific information about our products and related activities:
Orthobiologics
Our Orthobiologics products primarily consist of viscosupplementation and regenerative orthopedic products. These products are used in a wide range of treatments, from providing pain relief from osteoarthritis to regenerating damaged tissue such as cartilage. Osteoarthritis is a debilitating disease causing pain, swelling, and restricted movement in joints. It occurs when the cartilage in a joint gradually deteriorates due to the effects of mechanical stress, which can be caused by a variety of factors, including the normal aging process. In an osteoarthritic joint, particular regions of articulating surfaces are exposed to irregular forces, which results in the remodeling of tissue surfaces that disrupt the normal equilibrium or mechanical function. As osteoarthritis advances, the joint gradually loses its ability to regenerate cartilage tissue, and the cartilage layer attached to the bone eventually deteriorates to the point that the bone becomes exposed. Advanced osteoarthritis often requires surgery and the possible implantation of artificial joints. The current treatment options for osteoarthritis, prior to joint replacement surgery, include viscosupplementation, analgesics, non-steroidal anti-inflammatory drugs, and steroid injections.
| 5 |
Our viscosupplementation franchise includes ORTHOVISC, ORTHOVISC mini, MONOVISC, and CINGAL. ORTHOVISC is available in the United States, Canada, and other international markets for the treatment of osteoarthritis of the knee, and in Europe and certain international markets for the treatment of osteoarthritis in all synovial joints. ORTHOVISC mini is available in Europe, and it is designed for the treatment of osteoarthritis in small joints. MONOVISC is our single injection osteoarthritis treatment indicated for all synovial joints in Europe and certain international markets, including India and Australia, and for the knee in the United States, Turkey, and Canada. ORTHOVISC has been marketed by us internationally since 1996, and it was approved by the FDA for sale in the United States in 2004. ORTHOVISC mini and MONOVISC became available in certain international markets in the second quarter of 2008. MONOVISC was approved by the FDA for sale in the United States in February 2014, and the related U.S. commercial introduction of the product occurred in April 2014. In the United States, our viscosupplementation franchise, consisting of our ORTHOVISC and MONOVISC products, continues to maintain a market leadership position. CINGAL, our second single-injection osteoarthritis product, received regulatory approval from Health Canada in November 2015 for the treatment of pain associated with osteoarthritis of the knee. In March 2016, we received CE Mark approval of CINGAL as a viscoelastic supplement or as a replacement for synovial fluid in human joints. We successfully achieved commercial launch of the product in Canada during May 2016 and in the European Union during June 2016. CINGAL is also distributed in certain countries in Asia. Upon achievement, if any, of regulatory approval in the United States, we plan to commercialize the product through a direct sales model, initially through the engagement of a contract sales organization, with the ultimate goal of transitioning the direct sales function into our company as part of a broader buildout of our commercial capabilities. For additional information about CINGAL in the United States, see the section captioned “Business—Research and Development of Potential Products.”
In the United States, ORTHOVISC is indicated for the treatment of pain caused by osteoarthritis of the knee in patients who have failed to respond adequately to conservative, non-pharmacologic therapy and to simple analgesics, such as acetaminophen. ORTHOVISC is a sterile, clear, viscous solution of hyaluronan dissolved in physiological saline and dispensed in a single-use syringe. A complex sugar of the glycosaminoglycan family, hyaluronan is a high-molecular-weight polysaccharide composed of repeating disaccharide units of sodium glucuronate and N-acetyl glucosamine. ORTHOVISC is injected into joints in a series of three intra-articular injections one week apart. ORTHOVISC became available for sale in the United States on March 1, 2004, and it is marketed by DePuy Synthes Mitek Sports Medicine, a division of DePuy Orthopaedics, Inc. (“Mitek”) under the terms of an initial ten-year licensing, distribution, supply, and marketing agreement which was entered into in December 2003 (the “Mitek ORTHOVISC Agreement”). The Mitek ORTHOVISC Agreement has been extended for two additional five-year terms, and it will now expire on December 20, 2023, unless it is further extended by Mitek. Outside of the U.S., we have a number of distribution relationships servicing international markets including Canada, Europe, the Middle East, Latin America, and Asia. We will continue to seek to establish distribution relationships in other key markets. See the sections captioned “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Management Overview” and “Risk Factors.”
In the United States, MONOVISC is also indicated for the treatment of pain caused by osteoarthritis of the knee in patients who have failed to respond adequately to conservative, non-pharmacologic therapy and to simple analgesics, such as acetaminophen. MONOVISC is a sterile, clear, viscous solution of partially cross-linked sodium hyaluronate in a phosphate buffered saline solution. A treatment of MONOVISC is comprised of one injection of the product delivered directly into the affected joint. MONOVISC became available for sale in the United States in April 2014, and it is also marketed by Mitek under the terms of a fifteen-year licensing, distribution, supply, and marketing agreement, which was entered into on December 21, 2011 (the “Mitek MONOVISC Agreement”). Outside of the United States, we have a number of distribution relationships servicing international markets including Canada, Europe, the Middle East, Latin America, Asia, Australia, and certain other international countries. We continue to seek to establish distribution relationships in other key markets. See the sections captioned “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Management Overview” and “Risk Factors.”
| 6 |
In addition to the four viscosupplementation products discussed above, we also offer several additional products used in connection with orthopedic regenerative medicine. These products are based on the HYAFF technology and are currently available in Europe, South America, Asia, and certain other international markets. They include HYALOFAST, a biodegradable support for human bone marrow mesenchymal stem cells used for cartilage regeneration and as an adjunct for microfracture surgery; HYALONECT, a resorbable knitted fabric mesh for use in orthopedic and trauma reconstructive procedures to maintain the relative position of engrafted bone tissue or bone fragments from comminuted fractures; and HYALOSS MATRIX, HYAFF fibers used to mix blood/bone grafts to form a paste for bone regeneration. We also offer HYALOGLIDE, an ACP gel used in tenolysis treatment, with the potential for use in flexor tendon adhesion prevention and for use in the shoulder for prevention of adhesive capsulitis with additional clinical data. This product is commercialized through a network of distributors, primarily in Europe and the Middle East. We also received CE Mark approval in December 2016 for a product which utilizes our proprietary HA technology to treat pain associated with lateral epicondylitis, better known as tennis elbow. Outside of the United States, this product is marketed under the trade name ORTHOVISC-T. Additionally, in the second quarter of 2016, we submitted an Investigational Device Exemption (“IDE”) to the FDA to conduct a Phase III clinical trial for this treatment, and the IDE was approved by the FDA in June 2016. In addition to these products, we also received 510(k) clearance for an injectable HA-based bone repair treatment in December 2017. In total, Orthobiologics products accounted for 87%, 87%, and 84% of our product revenue in 2017, 2016, and 2015 respectively.
Dermal
Our dermal products consist of advanced wound care products, based on the HYAFF technology, and an aesthetic dermal filler, based on our proprietary chemically modified, cross-linked HA technology. Products utilizing our HYAFF technology are used for the treatment of skin wounds, ranging from burns to diabetic ulcers. The products cover a variety of wound treatment solutions including debridement agents, advanced therapies to aid healing, and scaffolds used as skin substitutes. Leading products include HYALOMATRIX and HYALOFILL, for the treatment of complex wounds such as burns and ulcers. The dermal products are commercialized through a network of distributors, primarily in the United States, Europe, Latin America, and the Middle East. Products cleared for sale in the United States include HYALOMATRIX, HYALOFILL, HYALOGRAN, HYALOSAFE, and HYALOMATRIX 3D. In 2014, we entered into an agreement with Medline Industries, Inc. with a current expiration date of December 31, 2022 to commercialize HYALOMATRIX in the United States.
Our aesthetic dermatology product is a dermal filler based on our proprietary, chemically modified, cross-linked HA, and it is commercially available in select countries in the Middle East. Internationally, this product is marketed under the ELEVESS name. In the United States, the trade name is HYDRELLE, although the product is not currently marketed in the United States.
Surgical
Our surgical business consists of products used to prevent post-surgical adhesions after abdominal-pelvic, spinal, and ear, nose, and throat (“ENT”) surgeries. HYALOBARRIER is a clinically proven, post-operative adhesion barrier for use in the abdominopelvic area. The product is currently commercialized in Europe, the Middle East, and certain African and Asian countries through a distribution network, but it is not approved for sale in the United States. INCERT, approved for sale and commercialized through a network of distributors in Europe and Malaysia, is a chemically modified, cross-linked HA product, for the prevention of spinal post-surgical adhesions. There are currently no plans at this time to distribute INCERT in the United States.
Surgical adhesions occur when fibrous bands of tissues form between adjacent tissue layers during the wound healing process. Although surgeons attempt to minimize the formation of adhesions, they nevertheless occur quite frequently after surgery. Adhesions in the abdominal and pelvic cavity can cause particularly serious problems such as intestinal blockage following abdominal surgery and infertility following pelvic surgery. Fibrosis following spinal surgery can complicate re-operation and may cause pain.
We also offer several products used in connection with the treatment of ENT disorders. The lead products are MEROGEL, a woven fleece nasal packing, and MEROGEL INJECTABLE, a thick, viscous hydrogel composed of cross-linked hyaluronic acid—a biocompatible agent that creates a moist wound-healing environment. We have partnered with Medtronic XoMed, Inc. (“Medtronic”) for worldwide distribution of these ENT products.
| 7 |
Other
Our other products include our ophthalmic and veterinary products, which are legacy products and not a part of our core business. Our ophthalmic business includes injectable, high molecular weight HA products used as viscoelastic agents in ophthalmic surgical procedures such as cataract extraction and intraocular lens implantation. These products coat, lubricate, and protect sensitive tissue such as the endothelium, and they function to maintain the shape of the eye, thereby facilitating ophthalmic surgical procedures. Our veterinary product, HYVISC, is a high molecular weight injectable HA product for the treatment of joint dysfunction in horses due to non-infectious synovitis associated with equine osteoarthritis. HYVISC has viscoelastic properties that lubricate and protect the tissues in horse joints. HYVISC is distributed by Boehringer Ingelheim Vetmedica, Inc. (“Boehringer”) in the United States and in selected countries in the Middle East.
See Note 15 “Revenue by Product Group, by Significant Customer and by Geographic Location; Geographic Information” to our consolidated financial statements included elsewhere in this Annual Report on Form 10-K for a discussion regarding our segments and geographic sales.
See also the section captioned “Risk Factors—Risks Related to Our Business and Industry—We experience quarterly sales volume variation, which makes our future results difficult to predict and makes period-to-period comparisons potentially not meaningful” for a discussion regarding the effect that quarterly sales volume variation could have on our business and financial performance.
See also the section captioned “Risk Factors —Risks Related to Our Business and Industry—A significant portion of our revenues are derived from a small number of customers, the loss of which could materially adversely affect our business, financial condition and results of operations” for a discussion regarding our dependence on large-volume customers and the effects that the loss of any such customer could have on our business and financial performance.
See also the section captioned “Risk Factors—Risks Related to Our Business and Industry—Our manufacturing processes involve inherent risks, and disruption could materially adversely affect our business, financial condition and results of operations” for a discussion of the sources and availability of raw materials related to the manufacture of our products.
Research and Development of Potential Products
Our research and development efforts primarily consist of the development of new medical applications for our HA-based technology, the management of clinical trials for certain product candidates, the preparation and processing of applications for regulatory approvals or clearances at all relevant stages of product development, and process development and scale-up manufacturing activities for our existing and new products. Our development focus includes products for tissue protection, repair, and regeneration. For the years ended December 31, 2017, 2016 and 2015, these expenses were $18.8 million, $10.7 million, and $9.0 million, respectively. We anticipate that we will continue to commit significant resources to, and increase our aggregate spending on, research and development activities, including in relation to preclinical activities and clinical trials, in the future.
Our second single-injection osteoarthritis product under development in the United States is CINGAL, which is composed of our proprietary cross-linked HA material combined with an approved steroid, and it is designed to provide both short- and long-term pain relief to patients. We completed an initial CINGAL phase III clinical trial, including the associated statistical analysis for 368 enrolled patients, during the fourth quarter of 2014 with data indicating that the product met all primary and secondary endpoints relative to placebo set forth for the trial. During the first half of 2015, we completed a CINGAL retreatment study with 242 patients who had participated in the phase III clinical trial and reported safety data related to the retreatment study. This initial phase III clinical trial and the associated retreatment study supported the Health Canada and CE Mark approvals of the product. The commercial launch of the product in both Canada and the European Union occurred in the second quarter of 2016. In the United States, after discussions with the U.S. Food and Drug Administration (“FDA”) related to the regulatory pathway for CINGAL, we conducted a formal meeting with the FDA’s Office of Combination Products (“OCP”) to present and discuss our data in September 2015, and we submitted a formal request for designation with OCP a month later. In its response to our formal request for designation, OCP assigned the product to the FDA’s Center for Drug Evaluation and Research (“CDER”) as the lead agency center for premarket review and regulation. We held a meeting with CDER in September 2016 to align on an approval framework and on submission requirements for a New Drug Application (“NDA”) for CINGAL, including the execution of an additional Phase III clinical trial to supplement our existing CINGAL pivotal study data. We submitted an Investigational New Drug Application (“IND”) in late 2016, and discussions with CDER indicated no objections to our clinical protocol design. As a result, we commenced work on this second Phase III clinical trial in the first quarter of 2017, and the first patient was treated in the second quarter of 2017. Enrollment of 576 patients in this second Phase III clinical trial was completed during October 2017. We expect to complete the six-month follow-up in this Phase III clinical trial during the second quarter of 2018 and to submit our NDA to FDA as expeditiously as possible thereafter. We have also initiated an additional three-month extended follow-up study in conjunction with the second Phase III clinical trial to investigate the efficacy of CINGAL over this longer period, and the first patients were enrolled in this follow-up study in the fourth quarter of 2017. This extended follow-up study will not impact the timeline for submission of the NDA for CINGAL following the completion of the second Phase III clinical trial.
| 8 |
We have several research and development programs underway for new products, including for HYALOFAST (in the United States), an innovative product for cartilage tissue repair, and other early stage regenerative medicine development programs. HYALOFAST received CE Mark approval in September 2009, and it is commercially available in Europe and certain international countries. During the first quarter of 2015, we submitted an Investigational Device Exemption (“IDE”) for HYALOFAST to the FDA, which was approved in July 2015. We commenced patient enrollment in a clinical trial in December 2015, and we are advancing site initiations and patient enrollment activities. In the second quarter of 2016, a supplement to the HYALOFAST IDE was approved to expand the inclusion criteria for the clinical study. The purpose of this supplement is to allow us to increase enrollment rates with the ultimate goal of decreasing the time needed to complete the clinical trial. In addition, we are currently proceeding with other research and development programs, one of which utilizes our proprietary HA technology to treat pain associated with common repetitive overuse injuries, such as lateral epicondylitis, also known as tennis elbow. We submitted a CE Mark application for this treatment during the first quarter of 2016 and received a CE Mark for the treatment of pain associated with tennis elbow in December 2016. We expect to begin enrolling patients in a post-market clinical study in relation to the CE Mark for this product before the end of the second quarter of 2018. Outside of the United States, this product will be marketed under the trade name ORTHOVISC-T. In the second quarter of 2016, we submitted an IDE to the FDA to conduct a phase III clinical trial for this treatment, which was approved by the FDA in June 2016. We also have other research and development programs underway focused on expanding the indications of our current products, including one program being conducted and funded by our U.S. MONOVISC distribution partner, Mitek, seeking to expand MONOVISC’s indication to include the treatment of pain associated with osteoarthritis of the hip. In the third quarter of 2017, we also submitted an application to the FDA for 510(k) clearance of an injectable HA-based bone repair treatment. The 510(k) clearance was received from FDA in December 2017. In addition to other early stage research and developments initiatives we are currently undertaking, we are working to expand our regenerative medicine pipeline with a new product candidate in the form of an implant for rotator cuff repair utilizing our proprietary solid HA.
In June 2015, we entered into an agreement with the Institute for Applied Life Sciences at the University of Massachusetts Amherst to collaborate on research to develop a novel modality for the treatment of rheumatoid arthritis. The agreement with the University of Massachusetts Amherst was extended in January 2018, and the next phase of the research will focus on optimizing the drug delivery system with the goal of advancing a novel therapeutic candidate into clinical trials to support regulatory submission. We also recently entered into an agreement with the University of Liverpool to develop an injectable mesenchymal stem cell therapy for the treatment of age-related osteoarthritis with the goal of bringing a therapeutics candidate through clinical trials to market to meet an unmet therapeutic need.
Our research and development efforts may not be successful in (1) developing our existing product candidates, (2) expanding the therapeutic applications of our existing products, or (3) resulting in new applications for our HA technology. There is also a risk that we may choose not to pursue development of potential product candidates. We may not be able to obtain regulatory approval for any new applications we develop. Furthermore, even if all regulatory approvals are obtained, there can be no assurances that we will achieve meaningful sales of such products or applications.
| 9 |
See also the section captioned “Risk Factors—Risks Related to Our Business and Industry—Failure to obtain, or any delay in obtaining, FDA or other U.S. and foreign governmental approvals for our products may have a material adverse effect on our business, financial condition and results of operations.” for a discussion regarding the impact of government regulations on our product development activities.
Patent and Proprietary Rights
Our products and trademarks, including our corporate name, product names, and logos, are proprietary. We rely on a combination of patent protection, trade secrets and trademark laws, license agreements, and confidentiality and other contractual provisions to protect our proprietary information.
We have a policy of seeking patent protection for patentable aspects of our proprietary technology. In the United States, we own 20 patents, 1 of which is co-owned with other parties, license 12 patents, and have 4 patent applications currently pending. These U.S. patents have expiration dates through 2030. Internationally, we own 167 patents, 7 of which are co-owned with other parties, license 79 patents, and have 12 patent applications currently pending. Outside of the United States, we own, co-own, license, or have filed for patents in 34 jurisdictions. Our international patents have expiration dates through 2032. In 2017, we were granted 18 new patents in Austria, Belgium, Brazil, Czech Republic, Denmark, France, Germany, Great Britain, Hungary, Ireland, Italy, Luxembourg, the Netherlands, Norway, Poland, Spain, Sweden, and Switzerland. Many of these patents, including all licensed patents, belong to the Anika S.r.l. patent estate, which is extensive and partly intertwined with its former parent company Fidia Farmaceutici S.p.A. (“Fidia”) through a patent licensing agreement that provides Anika S.r.l. with access to certain of Fidia’s patents to the extent required to support Anika S.r.l.’s products. In 2017, 2 of the patents belonging to the Anika S.r.l. patent estate expired in the United States, and 4 expired internationally. We intend to seek patent protection for products and processes developed in the course of our activities when we believe such protection is in our best interests and when the cost of seeking such protection is not inordinate relative to the potential benefits.
Other entities have filed patent applications for, or have been issued patents concerning, various aspects of HA-related products or processes. In addition, the products or processes we develop may infringe the patent rights of others in the future. Any such infringement may have a material adverse effect on our business, financial condition, and results of operations.
We rely upon trade secrets and proprietary know-how for certain non-patented aspects of our technology. To protect such information, we require certain customers and vendors, and all employees, consultants, and licensees to enter into confidentiality agreements limiting the disclosure and use of such information. These agreements, however, may not provide adequate protection.
See also the section captioned “Risk Factors—Risks Related to Our Intellectual Property—We may be unable to adequately protect our intellectual property rights, which could have a material impact on our business and future financial results” for a discussion of the risks we face with respect to protecting intellectual property we develop.
We have granted Mitek an exclusive and non-transferable, royalty-bearing license to develop, commercialize, and sell ORTHOVISC and MONOVISC in the United States pursuant to the Mitek ORTHOVISC Agreement and the Mitek MONOVISC Agreement. These agreements include a license to manufacture and have manufactured such products in the event that we are unable to supply Mitek with ORTHOVISC or MONOVISC in accordance with the terms of the relevant agreement. We have also granted Mitek the exclusive, royalty free right to use the trademarks ORTHOVISC and MONOVISC in connection with the marketing, distribution, and sale of the licensed products within the United States.
Government Regulation
The clinical development, manufacturing, and marketing of our products are subject to governmental regulation in the United States, the European Union, and other territories worldwide. Various statutes, regulations, directives, and guidelines, including the Food, Drug, and Cosmetic Act in the United States, govern the development, design, non-clinical and clinical research, testing, manufacture, safety, efficacy, labeling, packaging, storage, record keeping, premarket clearance or approval, adverse event reporting, advertising, and promotion of our products. Product development and approval within these various regulatory frameworks takes a number of years and involves the expenditure of substantial resources. Pharmaceutical and medical device manufacturers are also inspected regularly by the FDA and other applicable regulatory bodies.
| 10 |
Medical products regulated by the FDA are generally classified as drugs, biologics, or medical devices. Drugs and biologic products undergo rigorous preclinical testing prior to beginning clinical trials. Clinical trials for new drugs or biologic products include Phase I trials in healthy volunteers to understand safety, dosage tolerance, and pharmacokinetics, Phase II trials in a limited patient population to identify initial efficacy and side effects, and Phase III pivotal trials to statistically evaluate the safety and efficacy of the product. Medical devices intended for human use are classified into three categories (Class I, II or III) on the basis of the controls deemed reasonably necessary by the FDA to assure their safety and effectiveness. Class II devices are cleared for marketing under the premarket notification 510(k) regulatory pathway, which may include clinical testing. Class III devices require pre-market approval based on valid scientific evidence of safety and effectiveness, including evidence elicited through appropriate clinical testing. The failure to adequately demonstrate the quality, safety, and efficacy of a product under development can delay or prevent regulatory approval of the product. In order to gain marketing approval, we must submit to the relevant regulatory authority for review information on the quality aspects of the product as well as the non-clinical and clinical data. The FDA undertakes this review in the United States.
In the European Union, medical devices must be CE Marked in order to be marketed. CE marking a device involves working with a Notified Body, and in some cases a Competent Authority, to demonstrate that the device meets all applicable requirements of the Medical Devices Directive and that our Quality Management System is compliant. Drug approval in the European Union follows one of several possible processes: (i) a centralized procedure involving members of the European Medicines Agency’s Committee for Medicinal Products for Human Use; (ii) a “mutual recognition procedure” in which an individual country's regulatory agency approves the product followed by “mutual recognition” of this approval by regulatory agencies of other countries; or (iii) a decentralized procedure in which the approval is sought through the regulatory agencies of multiple countries at the same time.
Approval timelines can range from several months to several years, or applications can be denied entirely. The approval process can be affected by a number of factors. For example, additional studies or clinical trials may be requested during the review, which may delay marketing approval and involve unbudgeted costs. As a condition of approval, the regulatory agency may require post-marketing surveillance to monitor for adverse effects, and may require other additional studies, as it deems appropriate. After approval for an initial indication, further clinical studies are generally necessary to gain approval for any additional indications. The terms of any approval, including labeling content, may be more restrictive than expected and could affect the marketability of a product.
As a condition of approval, the relevant regulatory agency requires that the product continues to meet applicable regulatory requirements related to quality, safety, and efficacy, and it requires strict procedures to monitor and report any adverse effects. Where adverse effects occur or may occur, the regulatory agency may require additional studies or changes to the labeling. Compelling new “adverse” data may result in a product approval being withdrawn at any stage following review by an agency and discussion with the product manufacturer.
The branch of the FDA responsible for product marketing oversight routinely reviews company marketing practices and also may impose pre-clearance requirements on materials intended for use in marketing of approved drug products. We are also subject to various U.S. federal and state laws pertaining to healthcare fraud and abuse, including anti-kickback and false claims laws. Similar review and regulation of advertising and marketing practices exists in the other geographic areas where we operate.
The FDA has broad regulatory compliance and enforcement powers. If the FDA determines that we failed to comply with applicable regulatory requirements, it can take a variety of compliance or enforcement actions, including, without limitation, issuing an FDA Form 483 notice of inspectional observations or a warning letter, imposing civil money penalties, suspending or delaying issuance of approvals, requiring product recall, imposing a total or partial shutdown of production, withdrawal of approvals or clearances already granted, pursuing product seizures, consent decrees or other injunctive relief, or criminal prosecution through the Department of Justice. The FDA can also require us to repair, replace, or refund the cost of products that we manufactured or distributed. Outside the United States, regulatory agencies may exert a range of similar powers.
| 11 |
See also the sections captioned “Risk Factors—Risks Related to Our Business and Industry—Failure to obtain, or any delay in obtaining, FDA or other U.S. and foreign governmental approvals for our products may have a material adverse effect on our business, financial condition and results of operations,” “Risk Factors—Risks Related to Our Business and Industry—Once obtained, we cannot guarantee that FDA or international product approvals will not be withdrawn or that relevant agencies will not require other corrective action, and any withdrawal or corrective action could materially affect our business and financial results,” “Risk Factors—Risks Related to Our Business and Industry—Our operations and products are subject to extensive regulation, compliance with which is costly and time consuming, and our failure to comply may result in substantial penalties, including recalls of our products,” and “Risk Factors—Risks Related to Our Business and Industry—Any changes in FDA or international regulations related to product approval, including those that apply retroactively, could adversely affect our competitive position and materially affect our business and financial results” for a discussion regarding the potential impact of government regulations on our business and financial results.
Competition
We compete with many companies including large pharmaceutical firms and specialized medical products companies across all of our product lines. Many of these companies have substantially greater financial resources, larger research and development staffs, more extensive marketing and manufacturing organizations, and more experience in the regulatory processes than we have. We also compete with academic institutions, government agencies, and other research organizations, which may be involved in the research and development and commercialization of products. Many of our competitors also compete against us in securing relationships with collaborators for their research and development and commercialization programs.
We compete with other market participants primarily on the efficacy of our products, our products’ reputation for safety, our focus on HA-based products, and the breadth of our HA-based product portfolio. Other factors that impact competition in our industry are the timing and scope of regulatory approvals, the availability of raw material and finished product supply, marketing and sales capability, reimbursement coverage, product pricing, and patent protection. Some of the principal factors that may affect our ability to compete in the HA development and commercialization markets include:
| · | The quality and breadth of our continued development of our technology portfolio; |
| · | Our ability to complete successful clinical studies and obtain FDA marketing and foreign regulatory approvals prior to our competitors; |
| · | The successful execution of our commercial strategies, including our direct commercialization initiative for CINGAL; |
| · | Our ability to recruit and retain skilled employees; and |
| · | The availability of capital resources to fund strategic activities related to the significant expansion of our business or product portfolio. |
We are aware of several companies that are developing and/or marketing products utilizing HA for a variety of human applications. In some cases, competitors have already obtained product approvals, submitted applications for approval, or commenced human clinical studies, either in the United States or in certain foreign countries. All of our products face substantial competition. There exist major worldwide competing products, made from HA and other materials, for use in orthopedics, surgical adhesion prevention, advanced wound care, ENT, cosmetic dermatology, ophthalmic surgery, and the treatment of equine osteoarthritis. There is a risk that we will be unable to compete effectively against our current or future competitors. Additionally, legislation and regulation aimed at curbing rising healthcare costs has resulted in a consolidation trend in the healthcare industry to create larger companies, including hospitals, with greater market power. In turn, this has led to greater and more intense competition in the provision of products and services to market participants. Important market makers, like group purchasing organizations, have increased their negotiating leverage, and if these market makers demand significant price concessions or if we are excluded as a supplier by these market makers, our product revenue could be adversely impacted.
| 12 |
See also the sections captioned “Risk Factors—Risks Related to Our Business and Industry—Substantial competition could materially affect our financial performance” and “Risk Factors—Risks Related to Our Business and Industry—Our business may be adversely affected if consolidation in the healthcare industry leads to demand for price concessions or if we are excluded from being a supplier by a group purchasing organization or similar entity” for additional discussion of the impact competition could have on our business and financial results.
Employees
As of December 31, 2017, we had 123 employees, 21 of whom were located outside the United States. We consider our relations with our employees to be good. None of our U.S. employees are represented by labor unions, but certain employees based in Italy are represented by unions, adding complexity and additional risks to the wage and employment decision processes.
Environmental Laws
We believe that we are in compliance with all foreign, federal, state, and local environmental regulations with respect to our manufacturing facilities and that the cost of ongoing compliance with such regulations does not have a material effect on our operations.
Product Liability
The testing, marketing, and sale of human health care products entails an inherent risk of allegations of product liability, and we cannot assure that substantial product liability claims will not be asserted against us. Although we have not received any material product liability claims to date and have coverage under our insurance policy of $5.0 million per occurrence and $5.0 million in the aggregate, we cannot assure that if material claims arise in the future, our insurance will be adequate to cover all situations. Moreover, we cannot assure that such insurance, or additional insurance, if required, will be available in the future or, if available, will be available on commercially reasonable terms. Any product liability claim, if successful, could have a material adverse effect on our business, financial condition, and results of operation.
Available Information
Our Annual Reports on Form 10-K, including our consolidated financial statements, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and other information, including amendments and exhibits to such reports, filed or furnished pursuant to the Securities Exchange Act of 1934, as amended, are available free of charge in the “SEC Filings” section of our website located at http://www.anikatherapeutics.com, as soon as reasonably practicable after the reports are filed with or furnished to the SEC. The information on our website is not part of this Annual Report on Form 10-K. Reports filed with the SEC may be viewed at www.sec.gov or obtained at the SEC Public Reference Room at 100 F Street NE, Washington, D.C. 20549. Information regarding the operation of the Public Reference Room may be obtained by calling the SEC at 1-800-SEC-0330.
Our operating results and financial condition have varied in the past and could vary significantly in the future depending on a number of factors. You should consider carefully the risks and uncertainties described below, in addition to the other information contained in this Annual Report on Form 10-K, before deciding whether to purchase our common stock. If any of the following risks actually occurs, our business, financial condition, results of operations, and future prospects could be materially and adversely affected. In that event, the trading price of our common stock could decline, and you could lose part or all of your investment.
| 13 |
Risks Related to Our Business and Industry
Failure to obtain, or any delay in obtaining, FDA or other U.S. and foreign governmental approvals for our products may have a material adverse effect on our business, financial condition and results of operations.
Several of our current products, and any future products we may develop, will require clinical trials to determine their safety and efficacy for United States and international marketing approval by regulatory bodies, including the FDA. Product development and approval within the FDA framework takes a number of years and involves the expenditure of substantial resources. There can be no assurance that the FDA will accept submissions related to our new products or the expansion of the indications of our current products, and, even if submissions are accepted, there can be no guarantee that the FDA will grant approval for our new products, including CINGAL, HYALOFAST, or other line extensions of our current products, or for the expansion of indications of our current products on a timely basis, if at all. In addition to regulations enforced by the FDA, we are subject to other existing and future federal, state, local, and foreign regulations applicable to product approval, which may vary significantly across jurisdictions. Additional approval of existing products may be required when changes to such products may affect the safety and effectiveness, including for new indications for use, labeling changes, process or manufacturing changes, the use of a different facility to manufacture, process or package the device, and changes in performance or design specifications. Failure to obtain regulatory approvals of our products, including any changes to existing products, could have an adverse material impact on our business, financial condition, and results of operations.
Even if ultimately granted, FDA and international regulatory approvals may be subject to significant, unanticipated delays throughout the regulatory approval process. Internally, we make assumptions regarding product approval timelines, both in the United States and internationally, in our business planning, and any delay in approval could materially affect our competitive position in the relevant product market and our projections related to future business results.
We cannot be certain that product approvals, both in the United States and internationally, will not include significant limitations on the product indications, and other claims sought for use, under which the products may be marketed. The relevant approval or clearance may also include other significant conditions of approval such as post-market testing, tracking, or surveillance requirements. Any of these factors could significantly impact our competitive position in relation to such products and could have a negative impact on the sales of such products.
Once obtained, we cannot guarantee that FDA or international product approvals will not be withdrawn or that relevant agencies will not require other corrective action, and any withdrawal or corrective action could materially affect our business and financial results.
Once obtained, marketing approval can be withdrawn by the FDA or comparable foreign regulatory agencies for a number of reasons, including the failure to comply with ongoing regulatory requirements or the occurrence of unforeseen problems following initial approval. Regulatory authorities could also limit or prevent the manufacture or distribution of our products. Any regulatory limitations on the use of our products or any withdrawal or suspension of approval or rescission of approval by the FDA or a comparable foreign regulatory agency could have a material adverse effect on our business, financial condition, and results of operations.
Our operations and products are subject to extensive regulation, compliance with which is costly and time consuming, and our failure to comply may result in substantial penalties, including recalls of our products.
The FDA and foreign regulatory bodies impose extensive regulations applicable to our operations and products, including regulations governing product standards, packing requirements, labeling requirements, quality system and manufacturing requirements, import restrictions, tariff regulations, duties, and tax requirements. We cannot assure you that we will be able to achieve and maintain compliance required for FDA, CE marking, or other foreign regulatory approvals for any or all of our operations and products or that we will be able to produce our products in a timely and profitable manner while complying with applicable requirements.
Failure to comply with applicable regulatory requirements could result in substantial penalties, including warning letters, fines, injunctions, civil penalties, seizure of products, total or partial suspension of production, refusal to grant pre-market clearance or pre-market approval for devices or drugs, withdrawal of approvals, and criminal prosecution. Additionally, regulatory authorities have the power to require the recall of our products. It also might be necessary for us, in applicable circumstances, to initiate a voluntary recall per regulatory requirements of one or several of our products. The imposition of any of the foregoing penalties, whether voluntarily or involuntary, could have a material negative impact on our business, financial condition, and results of operations.
| 14 |
Any changes in FDA or international regulations related to product approval, including those that apply retroactively, could adversely affect our competitive position and materially affect our business and financial results.
FDA and foreign regulations depend heavily on administrative interpretation, and we cannot assure you that future interpretations made by the FDA or other regulatory bodies, with possible retroactive effect, will not adversely affect us. Additionally, any changes, whether in interpretation or substance, in existing regulations or policies, or any future adoption of new regulations or policies by relevant regulatory bodies, could prevent or delay approval of our products. In the event our future, or current, products, including HA generally, are classified, or re-classified, as human drugs, combination products, or biologics by the FDA or an applicable international regulatory body, the applicable review process related to such products is typically substantially longer and substantially more expensive than the review process to which they are currently subject as medical devices, which could materially impact our competitive position, business, and financial results.
We are implementing a direct sales model to commercialize our CINGAL product, as well as certain other future products, in the United States and we may face unforeseen difficulties and delays in implementing this new model, which could affect our business and financial results.
For the first time, we are implementing a direct sales model to market and promote one of our products, CINGAL, in the United States, initially through a contract sales organization, with the ultimate goal of transitioning the direct sales function into our company as part of a broader buildout of our commercial capabilities. We may also use this direct model to commercialize other of our products in the United States in the future. Our success in utilizing this sales model will initially depend in part on our ability to successfully develop and implement the necessary internal and external resources to manage the contract sales organization and the sales of the product. Our longer term success will depend on our ability to transition the direct sales function into our company and to manage all resources associated with this function. We cannot assure you that there will not be unforeseen roadblocks or delays in finalizing the contracts related to, and implementing, the relationship with the contract sales organization, nor we can we assure you that we will not face setbacks in transitioning the direct sales function into our organization. The initial implementation timeline of this direct sales model is also dependent on CINGAL obtaining FDA approval in a timely manner, of which there is no guarantee. Failure to implement our direct sales model in a timely fashion or to successfully manage the implementation or transition process could materially impact our competitive position, business, and financial results.
Substantial competition could materially affect our financial performance.
We compete with many companies, including large pharmaceutical companies, specialized medical products companies, and healthcare companies. Many of these companies have substantially greater financial resources, larger research and development staffs, more extensive marketing and manufacturing organizations, and more experience in the regulatory process than us. We also compete with academic institutions, government agencies, and other research organizations that may be involved in research, development, and commercialization of products similar to our own. Because a number of companies are developing or have developed HA products for similar applications and have received FDA approval, the successful commercialization of a particular product will depend in part upon our ability to complete clinical studies and obtain FDA marketing and foreign regulatory approvals prior to our competitors, or, if regulatory approval is not obtained prior to our competitors, to identify markets for our products that may be sufficient to permit meaningful sales of our products. For example, we are aware of several companies that are developing and/or marketing products utilizing HA for a variety of human applications. In some cases, competitors have already obtained product approvals, submitted applications for approval, or have commenced human clinical studies, either in the United States or in certain foreign countries. There exist major competing products for the use of HA in ophthalmic surgery. In addition, certain HA products made by our competitors for the treatment of osteoarthritis in the knee received FDA approval before ours and have been marketed in the United States since 1997, as well as select markets in Canada, Europe, and other countries. There can be no assurance that we will be able to compete against current or future competitors or that competition will not have a material adverse effect on our business, financial condition, and results of operations.
| 15 |
We may rely on third parties to support certain aspects of our clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval or commercialize our products, and our business could be substantially harmed.
We have hired experienced clinical development and regulatory staff, and we have also retained the services of knowledgeable external service providers, including consultants and clinical research organizations, to develop and supervise our clinical trials and regulatory processes. Despite our internal investment in staffing, we will remain dependent upon these third party contract research organizations to carry out portions of our clinical and preclinical research studies for the foreseeable future. As a result, we have had and will have less control over the conduct of the clinical trials, the timing and completion of the trials, the required reporting of adverse events, and the management of data developed through the trials than would be the case if we were relying entirely on our own staff. Outside parties may have staffing difficulties, may undergo changes in priorities or may become financially distressed, adversely affecting their willingness or ability to conduct our trials. Failure by these third parties to comply with regulatory requirements or to meet timing expectations may require us to repeat clinical or preclinical trials, which would delay the regulatory approval process, or require substantial unexpected expenditures.
We are dependent upon marketing and distribution partners and the failure to maintain strategic alliances on acceptable terms will have a material adverse effect on our business, financial condition, and results of operations.
Our success will be dependent, in part, upon the efforts of our marketing and distribution partners and the terms and conditions of our relationships with such partners. One partner, Mitek accounted for 73% of our product revenue in fiscal year 2017. We cannot assure you that our partners, including Mitek, will not seek to renegotiate their current agreements on terms less favorable to us or terminate such agreements. A failure to renew these partnerships on terms satisfactory to us, or at all, could result in a material adverse effect on our operating results.
We continue to seek to establish long-term distribution relationships in regions and countries not covered by existing agreements, and we may need to obtain the assistance of additional marketing partners to bring new and existing products to market and to replace certain marketing partners. There can be no assurance that we will be able to identify or engage appropriate distribution or collaboration partners or effectively transition to any such partners. The failure to establish strategic partnerships for the marketing and distribution of our products on acceptable terms and within our planned timeframes could have a material adverse effect on our business, financial condition, and results of operations.
We must achieve market acceptance of our products in order to be successful in the future.
Our success will depend in part upon the acceptance of our existing and future products by the medical community, hospitals, physicians, other health care providers, third-party payers, and end-users. Such acceptance may depend upon the extent to which the medical community and end-users perceive our products as safer, more effective, or more cost-competitive than other similar products. Ultimately, for our new products to gain general market acceptance, it may also be necessary for us to develop marketing partners or viable commercial strategies for the distribution of our products. There can be no assurance that our new products will achieve significant market acceptance on a timely basis, or at all. Failure of some or all of our future products to achieve significant market acceptance could have a material adverse effect on our business, financial condition, and results of operations.
Our manufacturing processes involve inherent risks, and disruption could materially adversely affect our business, financial condition, and results of operations.
The operation of biomedical manufacturing plants involves many risks, including the risks of breakdown, failure, or substandard performance of equipment, the occurrence of natural and other disasters, and the need to comply with the requirements of directives of government agencies, including the FDA. In addition, we rely on a single supplier for certain key raw materials and a small number of suppliers for a number of other materials required for the manufacturing and delivery of our HA products. Although we believe that alternative sources for many of these and other components and raw materials that we use in our manufacturing processes are available, we cannot be certain that the supply of key raw materials, specifically HA, will continue be available at current levels or will be sufficient to meet our future needs. Any supply interruption could harm our ability to manufacture our products until a new source of supply is identified and qualified. We may not be able to find sufficient alternative suppliers in a reasonable time period, or on commercially reasonable terms, if at all, and our ability to produce and supply our products could be impaired.
| 16 |
We use raw materials derived from animal sources to produce certain of our products, and there is no guarantee that we will be able to continue to utilize this source of material in the future.
Our manufacturing processes and research and development efforts for some of our ophthalmic and veterinary products involve products derived from animals. We procure our animal-derived raw materials from a qualified vendor, who controls for contamination and has processes that effectively inactivate infectious agents; however, we cannot assure you that we can completely eliminate the risk of transmission of infectious agents. Furthermore, regulatory authorities could in the future impose restrictions on the use of animal-derived raw materials that could impact our business.
The utilization of animals in research and development and product commercialization is subject to increasing focus by animal rights activists. The activities of animal rights groups and other organizations that have protested animal based research and development programs or boycotted the products resulting from such programs could cause an interruption in our manufacturing processes and research and development efforts. The occurrence of material operational problems, including but not limited to the events described above, could have a material adverse effect on our business, financial condition, and results of operations during the period of such operational difficulties and beyond.
We lease properties in the United States and Italy, and there is no guarantee that these leaseholds will be without issue or sufficient to support future growth.
We lease approximately 134,000 square feet of administrative, research and development, and manufacturing space in Bedford, MA and approximately 33,000 square feet of office, research and development, training, and warehousing space in Padova, Italy. The current term of the Bedford lease extends to 2022, and the current term of the Padova lease extends to 2032, each with several options for renewal. Please see Item 2 – Properties for additional information on our current leases. The nature of these leaseholds presents certain risks. We must maintain a positive working relationship with the respective owners as a dispute with either owner over payment, maintenance, or any other matter could be disruptive to our business. Additionally, there is a possibility that changes to our business or the geographic location of the facilities could make either location less suitable to our operations. Any renegotiation or termination of either lease could result in substantial cost or business interruption to our operations. Additionally, there is no guarantee that our current space will be sufficient to support our future growth or that any future relocation or expansion of our operations would be completed smoothly or in a timely manner due to, among other things, unexpected construction delays or unexpected difficulties related to the achievement of necessary permitting. Any business disruption as a result of any of these factors could have a material impact on our business, financial condition, and results of operations.
Our financial performance depends on the continued sales growth and increasing demand for our products and we may not be able to successfully manage the expansion of our operations.
Our future success depends on substantial growth in product sales. There can be no assurance that such growth can be achieved or, if achieved, sustained. There can be no assurance that, even if substantial growth in product sales and the demand for our products is achieved, we will be able to:
| · | Develop and maintain the necessary manufacturing capabilities; |
| · | Obtain the assistance of additional marketing partners or develop appropriate alternative sales strategies; |
| 17 |
| · | Attract, retain, and integrate required key personnel; and |
| · | Implement the financial, accounting, and management systems needed to manage growing demand for our products. |
Our failure to successfully manage future growth could have a material adverse effect on our business, financial condition, and results of operations.
We may face circumstances in the future that will result in impairment charges, including, but not limited to, goodwill impairment and In-Process Research and Development (“IPR&D”) charges.
As of December 31, 2017, we had long-lived assets, including goodwill and IPR&D, of $76.3 million. If the fair value of any of our long-lived assets decreases as a result of an economic slowdown, a downturn in the markets where we sell products and services, or a downturn in our financial performance or future outlook, we may be required to record an impairment charge on such assets.
We are required to test intangible assets with indefinite life periods for potential impairment annually and on an interim basis if there are indicators of a potential impairment. We also are required to evaluate amortizable intangible assets and fixed assets for impairment if there are indicators of a possible impairment. Impairment charges could have a negative impact on our results of operations and financial position, as well as on the market price of our common stock.
Customer, vendor, and employee uncertainty about the effects of any acquisitions could harm us.
We and the customers of any companies we acquire may, in response to the consummation of any acquisitions, delay or defer purchasing decisions. Any delay or deferral in purchasing decisions by customers could adversely affect our business. Similarly, employees of acquired companies may experience uncertainty about their future role until or after we execute our strategies with regard to employees of acquired companies. This may adversely affect our ability to attract and retain key management, sales, marketing, and technical personnel following an acquisition.
We may engage in acquisitions as a part of our future growth strategy, which exposes us to a variety of risks that could adversely affect our business operations.
Our business strategy includes the acquisition of businesses, technologies, services, or products that we believe are a strategic fit with our business. We may fund these acquisitions by utilizing our cash, incurring debt, issuing additional shares of our common stock, or by other means. Completed acquisitions may expose us to a number of risks and expenses, including unanticipated liabilities, amortization expenses related to intangible assets with definite lives, or risks associated with entering new markets with which we have limited experience or where commercial alliances with experienced partners or existing sales channels are not available. Whether or not completed, acquisitions may result in diversion of management resources otherwise available for ongoing development of our business and significant expenditures.
We may not be able to realize the expected benefits of any completed acquisitions, including growth synergies and cost savings from the integration of acquired businesses or assets with our existing operations and technologies, as rapidly as expected, or at all. In addition, the integration and reorganization processes for our acquisitions may be complex, costly, and time consuming and include unanticipated issues, expenses, and liabilities. We may have difficulty in developing, manufacturing, and marketing the products of a newly acquired company in a manner that enhances the performance of our combined businesses or product lines and allows us to realize value from expected synergies. Moreover, we may lose key clients or employees of acquired businesses as a result of the change in ownership to us. Following an acquisition, we may not achieve the revenue or net income levels that justify the acquisition. Acquisitions may also result in one-time charges, such as write-offs or restructuring charges, impairment of goodwill or acquired In-Process Research and Development, which could adversely affect our operating results. The failure to achieve the expected benefits of any acquisition may harm our business, financial condition, and results of operations.
| 18 |
The acquisitions we have made or may make in the future may make us the subject of lawsuits from either an acquired company’s stockholders, an acquired company’s previous stockholders, or our current stockholders.
We may be the subject of lawsuits from either an acquired company’s stockholders, an acquired company’s previous stockholders, or our current stockholders. These lawsuits could result from the actions of the acquisition target prior to the date of the acquisition, from the acquisition transaction itself, or from actions after the acquisition. Defending potential lawsuits could cost us significant expense and distract management’s attention from the operation of the business. Additionally, these lawsuits could result in the cancellation of, or the inability to renew, certain insurance coverage that would be necessary to protect our assets.
Attractive acquisition opportunities may not be available to us in the future.
We may consider the acquisition of other businesses. However, we may not locate suitable acquisition targets or have the opportunity to make acquisitions of such targets on favorable terms in the future, which could negatively impact the growth of our business. In order to pursue such opportunities, we may require significant additional financing, which may not be available to us on favorable terms, if at all. The availability of such financing is limited by the continued tightening of the global credit markets. We expect that our competitors, many of which have significantly greater resources than we do, will compete with us to acquire compatible businesses. This competition could increase prices for acquisitions that we would likely pursue.
Sales of our products are largely dependent upon third party reimbursement and our performance may be harmed by health care cost containment initiatives.
In the United States and other foreign markets, health care providers, such as hospitals and physicians, that purchase health care products, such as our products, generally rely on third party payers, including Medicare, Medicaid, and other health insurance and managed care plans, to reimburse all or part of the cost of the health care product. We generally depend upon the distributors of our products to secure reimbursement and reimbursement approvals. Reimbursement by third party payers, both in the United States and internationally, may depend on a number of factors, including the payer’s determination that the use of our products is clinically useful and cost-effective, medically necessary, and not experimental or investigational. Since reimbursement approval is required from each payer individually, seeking such approvals can be a time consuming and costly process which, in the future, could require us or our marketing partners to provide supporting scientific, clinical, and cost-effectiveness data for the use of our products to each payer separately. Significant uncertainty exists as to the reimbursement status of newly approved health care products, and any failure or delay in obtaining reimbursement approvals can negatively impact sales of our new products.
In addition, third party payers are increasingly attempting to contain the costs of health care products and services by limiting both coverage and the level of reimbursement for new therapeutic products and by refusing, in some cases, to provide coverage for uses of approved products for disease indications for which the FDA, or the applicable foreign regulatory agency, has granted marketing approval. Also, the U.S. Congress, certain state legislatures, and certain foreign governments and regulatory agencies have considered reforms, including, among other items, the potential repeal of the Affordable Care Act in the United States, which may affect current reimbursement practices and create additional uncertainty about the pricing of our products, including the potential implementation of controls on health care spending through limitations on the growth of Medicare and Medicaid spending. There can be no assurance that third party reimbursement coverage will be available or adequate for any products or services developed by us. Outside the United States, the success of our products is also dependent in part upon the availability of reimbursement and health care payment systems. Domestic and international reimbursement laws and regulations may change from time to time. Lack of adequate coverage and reimbursement provided by governments and other third party payers for our products and services, including continuing coverage for MONOVISC and ORTHOVISC in the United States, and any change of classification by the Centers for Medicare and Medicaid Services for ORTHOVISC and MONOVISC, could have a material adverse effect on our business, financial condition, and results of operations.
| 19 |
We may seek additional financing in the future, which could be difficult to obtain and which could dilute your ownership interest or the value of your shares.
We had cash, cash equivalents, and investments of $157.3 million at December 31, 2017. In addition, and subject to certain constraints, we have $50.0 million of credit available to us under our Senior Revolving Credit Facility as of December 31, 2017. Our future capital requirements and the adequacy of available funds will depend, however, on numerous factors, including:
| · | Market acceptance of our existing and future products; |
| · | The success and sales of our products under various distributor agreements and other appropriate commercial strategies, including the ability of our partners to achieve third party reimbursement for our products; |
| · | The successful commercialization of products in development; |
| · | Progress in our product development efforts; |
| · | The magnitude and scope of such product development efforts; |
| · | Any potential acquisitions of products, technologies, or businesses; |
| · | Progress with preclinical studies, clinical trials, and product approvals and clearances by the FDA and other agencies; |
| · | The cost and timing of our efforts to manage our manufacturing capabilities and related costs; |
| · | The cost of filing, prosecuting, defending, and enforcing patent claims and other intellectual property rights and the cost of defending any other legal proceeding; |
| · | Competing technological and market developments; |
| · | The development of strategic alliances for the marketing of certain of our products; |
| · | The terms of such strategic alliances, including provisions (and our ability to satisfy such provisions) that provide upfront and/or milestone payments to us; and |
| · | The cost of maintaining adequate inventory levels to meet current and future product demand. |
To the extent funds generated from our operations, together with our existing capital resources, are insufficient to meet future requirements, we will be required to obtain additional funds through equity or debt financings, through strategic alliances with corporate partners and others, or through other sources. The terms of any future equity financings may be dilutive to our investors and the terms of any debt financings may contain restrictive covenants, which limit our ability to pursue certain courses of action. Our ability to obtain financing is dependent on the status of our future business prospects as well as conditions prevailing in the relevant capital markets at the time we seek financing. No assurance can be given that any additional financing will be made available to us or will be available on acceptable terms should such a need arise.
We could become subject to product liability claims, which, if successful, could materially adversely affect our business, financial condition, and results of operations.
The testing, marketing, and sale of human health care products entail an inherent risk of allegations of product liability, and there can be no assurance that substantial product liability claims will not be asserted against us. Although we have not received any material product liability claims to date and have an insurance policy of $5.0 million per occurrence and $5.0 million in the aggregate to cover such product liability claims should they arise, there can be no assurance that material claims will not arise in the future or that our insurance will be adequate to cover all situations. Moreover, there can be no assurance that such insurance, or additional insurance, if required, will be available in the future or, if available, will be available on commercially reasonable terms. Any product liability claim, if successful, could have a material adverse effect on our business, financial condition, and results of operations.
| 20 |
Our business is dependent upon hiring and retaining qualified management and technical personnel.
We are highly dependent on the members of our management and technical staff, the loss of one or more of whom could have a material adverse effect on us. We have experienced a number of management changes in recent years, and there can be no assurances that any future management changes will not adversely affect our business. We believe that our future success will depend in large part upon our ability to attract and retain technical and highly skilled executive, managerial, professional, and technical personnel. We face significant competition for such personnel from competitive companies, research and academic institutions, government entities, and other organizations. There can be no assurance that we will be successful in hiring or retaining the personnel we require. The failure to hire and retain such personnel could have a material adverse effect on our business, financial condition, and results of operations.
We are subject to environmental regulations and any failure to comply with applicable laws could subject us to significant liabilities and harm our business.
We are subject to a variety of local, state, federal, and foreign government regulations relating to the storage, discharge, handling, emission, generation, manufacture, and disposal of toxic or other hazardous substances used in the manufacture of our products. Any failure by us to control the use, disposal, removal, or storage of hazardous chemicals or toxic substances could subject us to significant liabilities, which could have a material adverse effect on our business, financial condition, and results of operations.
As our international sales and operations grow, we could become increasingly subject to additional economic, political, and other risks that could harm our business.
Since we manufacture and sell our products worldwide, our business is subject to risks associated with doing business internationally. During the years ended December 31, 2017, 2016, and 2015, 20%, 19%, and 18%, respectively, of our product sales were to international distributors. We continue to be subject to a variety of risks, which could cause fluctuations in the results of our international and domestic operations. These risks include:
| · | The impact of recessions and other economic conditions in economies, including Europe in particular, outside the United States; |
| · | Instability of foreign economic, political, and labor conditions; |
| · | Unfavorable labor regulations applicable to our European operations, such as severance and the unenforceability of non-competition agreements in the European Union; |
| · | The impact of strikes, work stoppages, work slowdowns, grievances, complaints, claims of unfair labor practices, or other collective bargaining disputes; |
| · | Difficulties in complying with restrictions imposed by regulatory or market requirements, tariffs, or other trade barriers or by U.S. export laws; |
| · | Imposition of government controls limiting the volume of international sales; |
| · | Longer accounts receivable payment cycles; |
| · | Potentially adverse tax consequences, including, if required or applicable, difficulties transferring funds generated in non-U.S. jurisdictions to the United States in a tax efficient manner; |
| · | Difficulties in protecting intellectual property, especially in international jurisdictions; |
| 21 |
| · | Difficulties in managing international operations; and |
| · | Burdens of complying with a wide variety of foreign laws. |
Our success depends, in part, on our ability to anticipate and address these risks. We cannot guarantee that these or other factors will not adversely affect our business or operating results.
Currency exchange rate fluctuations may have a negative impact on our reported earnings.
Approximately 5% of our business during 2017 was conducted in functional currencies other than the U.S. dollar, which is our reporting currency. Thus, currency fluctuations among the U.S. dollar and the other currencies in which we do business have caused and will continue to cause foreign currency transaction gains and losses. Currently, we attempt to manage foreign currency risk through the matching of assets and liabilities. In the future, we may undertake to manage foreign currency risk through additional hedging methods. We recognize foreign currency gains or losses arising from our operations in the period incurred. We cannot guarantee that we will be successful in managing foreign currency risk or in predicting the effects of exchange rate fluctuations upon our future operating results because of the variability of currency exposure and the potential volatility of currency exchange rates.
A significant portion of our revenues are derived from a small number of customers, the loss of which could materially adversely affect our business, financial condition and results of operations.
We have historically derived the majority of our revenues from a small number of customers who resell our products to end-users, and most of these customers are significantly larger companies than us. For the year ended December 31, 2017, five customers accounted for 83% of product revenue, with Mitek alone accounting for 73% of product revenue. We expect to continue to be dependent on a small number of large customers, especially Mitek, for the majority of our revenues for the foreseeable future. The failure of these customers to purchase our products in the amounts they historically have or in amounts that we expect would seriously harm our business.
In addition, if present and future customers terminate their purchasing arrangements with us, significantly reduce or delay their orders, or seek to renegotiate their agreements on terms less favorable to us, our business, financial condition, and results of operations will be adversely affected. If we accept terms less favorable than the terms of the current agreements, such renegotiations may have a material adverse effect on our business, financial condition, and/or results of operations. Furthermore, in any future negotiations we may be subject to the perceived or actual leverage that these customers may have given their relative size and importance to us. Any termination, change, reduction, or delay in orders could seriously harm our business, financial condition, and results of operations. Accordingly, unless and until we diversify and expand our customer base, or develop alternative commercial strategies, our future success will significantly depend upon the timing and size of future purchases by our largest customers, and the financial and operational success of these customers. The loss of any one of our major customers or the delay of significant orders from such customers, even if only temporary, could reduce or delay our recognition of revenues, harm our reputation in the industry, and reduce our ability to accurately predict cash flow, and, as a consequence, it could seriously harm our business, financial condition, and results of operations.
Information security breaches or business system disruptions, including our ongoing phased implementation of our enterprise resource planning (ERP) system, may adversely affect our business.
We rely on our information technology infrastructure and management information systems to effectively run our business. While we have not previously experienced a material information security breach caused by illegal hacking, computer viruses, or acts of vandalism or terrorism, we may in the future be subject to such a breach. Our security measures or those of our third-party service providers may not detect or prevent such breaches. Any such compromise to our information security could result in an interruption in our operations, the unauthorized publication of our confidential business or proprietary information, the unauthorized release of customer, vendor, or employee data, the violation of privacy, or other laws and exposure to litigation, any of which could harm our business and operating results. In addition, there may be other challenges and risks as we upgrade and standardize our business systems, including with respect to our newly implemented ERP system and the planned system enhancements thereto, which could adversely affect our business, financial condition, or results of operations.
| 22 |
Our business may be adversely affected if consolidation in the healthcare industry leads to demand for price concessions or if we are excluded from being a supplier by a group purchasing organization or similar entity.
Because healthcare costs have risen significantly over the past decade, numerous initiatives and reforms have been launched by legislators, regulators, and third-party payers to curb these costs. As a result, there has been a consolidation trend in the healthcare industry to create larger companies, including hospitals, with greater market power. As the healthcare industry consolidates, competition to provide products and services to industry participants has become and may continue to become more intense. This may result in greater pricing pressures and the exclusion of certain suppliers from important markets as group purchasing organizations, independent delivery networks, and large single accounts continue to use their market power to consolidate purchasing decisions. If a group purchasing organization excludes us from being one of their suppliers, our net sales could be adversely impacted. We expect that market demand, government regulation, third-party reimbursement policies, and societal pressures will continue to change the worldwide healthcare industry, which may exert further downward pressure on the prices of our products.
We experience quarterly sales volume variation, which makes our future results difficult to predict and makes period-to-period comparisons potentially not meaningful.
We experience quarterly fluctuations in our products sales as a result of multiple factors, many of which are outside of our control. These quarterly fluctuations create uncertainty as to the volume of sales that we may achieve in a given period. As a result, comparing our operating results on a period-to-period basis might not be meaningful. You should not rely on our past results as an indication of our future performance. Our operating results could be disproportionately affected by a reduction in revenue because a proportionately smaller amount of our expenses varies with our revenue. As a result, our quarterly operating results are difficult to predict, even in the near term.
Risks Related to Our Intellectual Property
We may be unable to adequately protect our intellectual property rights, which could have a material impact on our business and future financial results.
Our efforts to enforce our intellectual property rights may not be successful. We rely on a combination of copyright, trademark, patent, and trade secret laws, confidentiality procedures, and contractual provisions to protect our proprietary rights. Our success will depend, in part, on our ability to obtain and enforce patents and trademarks, to protect trade secrets, to obtain licenses to technology owned by third parties when necessary, and to conduct our business without infringing on the proprietary rights of others. The patent positions of pharmaceutical, medical product, and biotechnology firms, including ours, can be uncertain and involve complex legal and factual questions. There can be no assurance that any patent applications will result in the issuance of patents or, if any patents are issued, that they will provide significant proprietary protection or commercial advantage or will not be circumvented by others. Filing and prosecution of patent applications, litigation to establish the validity and scope of patents, assertion of patent infringement claims against others, and the defense of patent infringement claims by others can be expensive and time consuming. There can be no assurance that, in the event that any claims with respect to any of our patents, if issued, are challenged by one or more third parties, any court or patent authority ruling on such challenge will determine that such patent claims are valid and enforceable. An adverse outcome in such litigation or patent review process could cause us to lose exclusivity covered by the disputed rights. If a third party is found to have rights covering products or processes used by us, we could be forced to cease using the technologies or marketing the products covered by such rights, we could be subject to significant liabilities to such third party, and we could be required to license technologies from such third party in order to continue production of the products. Furthermore, even if our patents are determined to be valid, enforceable, and broad in scope, there can be no assurance that competitors will not be able to design around such patents and compete with us using the resulting alternative technology. We have a policy of seeking patent protection for patentable aspects of our proprietary technology. We intend to seek patent protection with respect to products and processes developed in the course of our activities when we believe such protection is in our best interest and when the cost of seeking such protection is not inordinate. However, no assurance can be given that any patent application will be filed, that any filed applications will result in issued patents, or that any issued patents will provide us with a competitive advantage or will not be successfully challenged by third parties. The protections afforded by patents will depend upon their scope and validity, and others may be able to design around our patents.
| 23 |
We also rely upon trade secrets and proprietary know-how for certain non-patented aspects of our technology. To protect such information, we require all employees, consultants, and licensees to enter into confidentiality agreements limiting the disclosure and use of such information. There can be no assurance that these agreements provide meaningful protection or that they will not be breached, that we would have adequate remedies for any such breach, or that our trade secrets, proprietary know-how, and our technological advances will not otherwise become known to others. In addition, there can be no assurance that, despite precautions taken by us, others have not and will not obtain access to our proprietary technology. Further, there can be no assurance that third parties will not independently develop substantially equivalent or better technology.
There can be no assurance that we will not infringe upon the intellectual property rights of others, which could have a significant impact on our business and financial results.
Other entities have filed patent applications for, or have been issued patents concerning, various aspects of HA-related products or processes. There can be no assurance that the products or processes developed by us will not infringe on the patent rights of others in the future. The cost of defending infringement suits is typically large, and there is no guarantee that any future defense would be successful. In addition, infringement could lead to substantial damages payouts or our inability to produce or market certain of our current or future products. As a result, any such infringement may have a material adverse effect on our business, financial condition, and results of operations.
Risks Related to Ownership of Our Common Stock
Our stock price may be highly volatile, and we cannot assure you that market making in our common stock will continue.
The market price of shares of our common stock may be highly volatile. Factors such as announcements of new commercial products or technological innovations by us or our competitors, disclosure of results of clinical testing or regulatory proceedings, government regulation and approvals, developments in patent or other proprietary rights, public concern as to the safety of products developed by us, and general market conditions may have a significant effect on the market price of our common stock. The trading price of our common stock could be subject to wide fluctuations in response to quarter-to-quarter variations in our operating results, material announcements by us or our competitors, governmental regulatory action, conditions in the health care industry generally or in the medical products industry specifically, or other events or factors, many of which are beyond our control. In addition, the stock market has experienced extreme price and volume fluctuations, which have particularly affected the market prices of many medical products companies and which often have been unrelated to the operating performance of such companies. Our operating results in future quarters may be below the expectations of equity research analysts and investors. In such an event, the price of our common stock would likely decline, perhaps substantially.
If securities or industry analysts do not publish or cease publishing research or reports about us, our business, or our market, or if they adversely change their recommendations regarding our stock, our stock price and trading volume could decline.
The trading market for our common stock is influenced by the research and reports that securities or industry analysts may publish about us, our business, our market, or our competitors. No person is under any obligation to publish research or reports on us, and any person publishing research or reports on us may discontinue doing so at any time without notice. If adequate research coverage is not maintained on our company or if any of the analysts who cover us downgrade our stock or publish inaccurate or unfavorable research about our business or provide relatively more favorable recommendations about our competitors, our stock price would likely decline. If any analysts who cover us were to cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline.
| 24 |
We do not intend to pay dividends on our common stock in the foreseeable future.
We have never declared or paid any cash dividends on our common stock. We currently intend to retain earnings, if any, for use in our business and do not anticipate paying cash dividends on our common stock in the foreseeable future. Accordingly, investors are not likely to receive any dividends on their common stock in the foreseeable future, and their ability to achieve a return on their investment will therefore depend on appreciation in the price of our common stock.
Our charter documents contain anti-takeover provisions that may prevent or delay an acquisition of our company.
Certain provisions of our Restated Articles of Organization and Amended and Restated By-laws could have the effect of discouraging a third party from pursuing a non-negotiated takeover of us and preventing certain changes in control. These provisions include a classified Board of Directors, advance notice to the Board of Directors of stockholder proposals, limitations on the ability of stockholders to remove directors and to call stockholder meetings, and the provision that vacancies on the Board of Directors be filled by vote of a majority of the remaining directors. In addition, the Board of Directors adopted a ten-year Shareholders Rights Plan in April 2008. We are also subject to Chapter 110F of the Massachusetts General Laws which, subject to certain exceptions, prohibits a Massachusetts corporation from engaging in any of a broad range of business combinations with any “interested stockholder” for a period of three years following the date that such stockholder becomes an interested stockholder. All of these provisions, policies, and plans are reviewed periodically by our Board of Directors. These provisions could discourage a third party from pursuing a takeover of us at a price considered attractive by many stockholders, since such provisions could have the effect of preventing or delaying a potential acquirer from acquiring control of us and our Board of Directors.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
Our corporate headquarters is located in Bedford, Massachusetts, where we lease approximately 134,000 square feet of administrative, research and development, and manufacturing space. We entered into this lease in January 2007, and the lease commenced in May 2007 for an initial term of ten and a half years. In October 2016, we exercised the first option under the lease to extend its term for five years. There are three additional renewal periods, each of which is subject to the condition that we notify the landlord of our exercise of such option at least one year prior to the expiration of the then current term. Two additional renewal options each extend the term an additional five years, and the final renewal option extends the term an additional six years.
In October 2015, Anika S.r.l. entered into a build-to-suit lease agreement for a new European headquarters facility consisting of approximately 33,000 square feet of general office, research and development, training, and warehousing space located in Padova, Italy. This lease, which has an initial term of fifteen years, commenced in February 2017 in accordance with the lease agreement, as amended in February 2017. The lease will automatically renew for up to three additional six-year terms, subject to certain terms and conditions. Anika S.r.l. may elect to early withdraw from this lease subject to certain financial penalties after six years and with no penalties after the ninth year. The lease provides for an initial yearly rent of approximately $0.3 million.
Prior to April 2017, Anika S.r.l. leased approximately 28,000 square feet of laboratory, warehouse, and office space in Abano Terme, Italy from Fidia. The lease commenced in December 2009. In December 2016, following discussions between Anika S.r.l. and Fidia, Anika S.r.l. notified Fidia of its intention to terminate this lease agreement as of March 2017, in accordance with the terms of the lease.
In 2017, we had aggregate facility lease expenses of approximately $1.8 million. We believe that the capacity of our Bedford, Massachusetts corporate headquarters is sufficient to satisfy our needs for the immediately foreseeable future. We also believe that Anika S.r.l.’s leased facility in Padova, Italy will be sufficient to satisfy its needs for the foreseeable future.
| 25 |
We are involved from time-to-time in various legal proceedings arising in the normal course of business. Although the outcomes of these legal proceedings are inherently difficult to predict, we do not expect the resolution of these proceedings to have a material adverse effect on our financial position, results of operations, or cash flow.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Common Stock Information
Our common stock has traded on the NASDAQ Global Select Market since November 25, 1997, under the symbol “ANIK.” The following table sets forth, for the periods indicated, the high and low sales prices of our common stock on the NASDAQ Global Select Market. These prices represent prices between dealers and do not include retail mark-ups, markdowns, or commissions, and they may not necessarily represent actual transactions.
| Year Ended December 31, 2017 | High | Low | ||||||
| First Quarter | $ | 52.23 | $ | 41.72 | ||||
| Second Quarter | 49.68 | 43.04 | ||||||
| Third Quarter | 58.21 | 45.71 | ||||||
| Fourth Quarter | 59.94 | 52.14 | ||||||
| Year Ended December 31, 2016 | High | Low | ||||||
| First Quarter | $ | 47.24 | $ | 35.07 | ||||
| Second Quarter | 53.68 | 42.36 | ||||||
| Third Quarter | 54.96 | 45.52 | ||||||
| Fourth Quarter | 50.19 | 41.38 | ||||||
At December 31, 2017, the closing price per share of our common stock was $53.91 as reported on the NASDAQ Global Select Market, and there were 135 holders of record. We believe that the number of beneficial owners of our common stock at that date was substantially greater, due to shares being held by intermediaries.
We have never declared or paid any cash dividends on our common stock. We currently intend to retain earnings, if any, for use in our business and do not anticipate paying cash dividends on our common stock in the foreseeable future. Payment of future dividends, if any, on our common stock will be at the discretion of our Board of Directors after taking into account various factors, including our financial condition, operating results, anticipated cash needs, and plans for expansion.
Accelerated Share Repurchase Program
On February 26, 2016, we entered into an accelerated stock repurchase agreement with Morgan Stanley & Co. LLC (“Morgan Stanley”) pursuant to a Fixed Dollar Accelerated Share Repurchase Transaction (“ASR Agreement") to purchase $25.0 million of shares of its common stock. Pursuant to the terms of the ASR Agreement, we paid Morgan Stanley $25.0 million in cash and received an initial delivery of 0.4 million shares of our common stock on February 29, 2016 based on a closing market price of $46.40 per share and the applicable contractual discount.
| 26 |
On August 26, 2016, we settled the approximately $7.5 million remaining under the ASR Agreement, which was recorded as an equity forward sale contract and was included in additional paid-in capital in stockholders' equity in the consolidated balance sheet as it met the criteria for equity accounting. Pursuant to the terms of the ASR Agreement, the final number of shares and the average purchase price was determined at the end of the applicable purchase period, which was August 26, 2016. Based on the volume-weighted average price since the effective date of the ASR Agreement less the applicable contractual discount, Morgan Stanley delivered 0.1 million additional shares to us on August 31, 2016. In total, 0.5 million shares were repurchased under the ASR Agreement at an average repurchase price of $47.08 per share. These shares are held by us as authorized but unissued shares pursuant to Massachusetts law. The initial and final delivery of shares resulted in immediate reductions of the outstanding shares used to calculate the weighted-average common shares outstanding for basic and diluted net income per share.
Performance Graph
Set forth below is a graph comparing the total returns of our company, the NASDAQ Composite Index, and the NASDAQ Biotechnology Index. The graph assumes $100 is invested on December 31, 2012 in our common stock and each of the indices. Past performance is not indicative of future results.
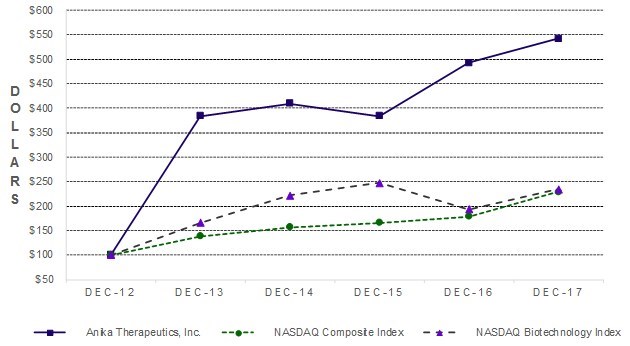
| Dec-12 | Dec-13 | Dec-14 | Dec-15 | Dec-16 | Dec-17 | |||||||||||||||||||
| Anika Therapeutics, Inc. | $ | 100.00 | $ | 383.90 | $ | 409.86 | $ | 383.90 | $ | 492.56 | $ | 542.35 | ||||||||||||
| NASDAQ Composite Index | $ | 100.00 | $ | 138.32 | $ | 156.85 | $ | 165.84 | $ | 178.28 | $ | 228.63 | ||||||||||||
| NASDAQ Biotechnology Index | $ | 100.00 | $ | 165.61 | $ | 222.08 | $ | 247.44 | $ | 193.79 | $ | 234.60 | ||||||||||||
Securities Authorized for Issuance Under Equity Compensation Plans
For information regarding securities authorized for issuance under our employee stock-based compensation plans, see Part III, Item 12, Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters, included elsewhere in this Annual Report on Form 10-K.
ITEM 6. SELECTED FINANCIAL DATA
The following selected consolidated financial data should be read in conjunction with the Consolidated Financial Statements and the Notes thereto and the section captioned “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this Annual Report on Form 10-K. The Balance Sheet Data at December 31, 2017 and 2016 and the Statement of Operations Data for each of the three years ended December 31, 2017, 2016, and 2015 have been derived from the audited Consolidated Financial Statements for such years, included elsewhere in this Annual Report on Form 10-K. The Balance Sheet Data at December 31, 2015, 2014, and 2013, and the Statement of Operations Data for each of the two years in the period ended December 31, 2014 and 2013 have been derived from audited consolidated financial statements for such years not included in this Annual Report on Form 10-K.
| 27 |
| Years ended December 31, | ||||||||||||||||||||
| 2017 | 2016 | 2015 | 2014 | 2013 | ||||||||||||||||
| Statements of Operations Data: | (in thousands, except per share data) | |||||||||||||||||||
| Product revenue | $ | 107,783 | $ | 102,932 | $ | 87,696 | $ | 75,474 | $ | 71,774 | ||||||||||
| Licensing, milestone and contract revenue | 5,637 | 447 | 5,303 | 30,121 | 3,307 | |||||||||||||||
| Total revenue | 113,420 | 103,379 | 92,999 | 105,595 | 75,081 | |||||||||||||||
| Cost of product revenue | 27,364 | 24,027 | 21,053 | 20,930 | 22,765 | |||||||||||||||
| Product gross profit | 80,419 | 78,905 | 66,643 | 54,544 | 49,009 | |||||||||||||||
| Product gross margin | 75 | % | 77 | % | 76 | % | 72 | % | 68 | % | ||||||||||
| Total operating expenses | 67,691 | 52,772 | 44,865 | 44,148 | 42,474 | |||||||||||||||
| Net income | 31,816 | 32,547 | 30,758 | 38,319 | 20,575 | |||||||||||||||
| Diluted net income per common share | $ | 2.11 | $ | 2.15 | $ | 2.01 | $ | 2.51 | $ | 1.39 | ||||||||||
| Diluted common shares outstanding | 15,068 | 15,116 | 15,321 | 15,269 | 14,826 | |||||||||||||||
| Years ended December 31, | ||||||||||||||||||||
| 2017 | 2016 | 2015 | 2014 | 2013 | ||||||||||||||||
| Balance Sheet Data: | (in thousands) | |||||||||||||||||||
| Cash, cash equivalents and investments | $ | 157,256 | $ | 124,761 | $ | 138,458 | $ | 106,906 | $ | 63,333 | ||||||||||
| Working capital | 193,254 | 161,641 | 159,155 | 131,863 | 84,650 | |||||||||||||||
| Total assets | 282,617 | 240,246 | 235,748 | 192,808 | 156,042 | |||||||||||||||
| Long-term liabilities | 6,054 | 8,674 | 7,622 | 8,737 | 11,125 | |||||||||||||||
| Retained earnings | 199,511 | 168,209 | 135,662 | 104,904 | 66,584 | |||||||||||||||
| Stockholders' equity | 263,491 | 222,773 | 210,848 | 178,098 | 135,634 | |||||||||||||||
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following section contains statements that are not statements of historical fact and are forward-looking statements within the meaning of the federal securities laws. These statements involve known and unknown risks, uncertainties, and other factors that may cause our actual results, performance, or achievement to differ materially from anticipated results, performance, or achievement, expressed or implied in such forward-looking statements. These statements reflect our current views with respect to future events, are based on assumptions, and are subject to risks and uncertainties. We discuss many of these risks and uncertainties at the beginning of this Annual Report on Form 10-K and under the sections captioned “Business” and “Risk Factors.” The following discussion should also be read in conjunction with the consolidated financial statements and the Notes thereto appearing elsewhere in this Annual Report on Form 10-K.
Management Overview
We are a global, integrated orthopedic medicines company committed to improving the lives of patients with degenerative orthopedic diseases and traumatic conditions with clinically meaningful therapies along the continuum of care, from palliative pain management to regenerative tissue repair. We have over two decades of global expertise developing, manufacturing, and commercializing products based on our proprietary HA technology. Our orthopedic medicine portfolio includes ORTHOVISC, MONOVISC, and CINGAL, which alleviate pain and restore joint function by replenishing depleted HA, and HYALOFAST, a solid HA-based scaffold to aid cartilage repair and regeneration.
| 28 |
Our therapeutic offerings consist of products in the following areas: Orthobiologics, Dermal, Surgical, and Other, which includes our ophthalmic and veterinary products. All of our products are based on HA, a naturally occurring, biocompatible polymer found throughout the body. Due to its unique biophysical and biochemical properties, HA plays an important role in a number of physiological functions such as the protection and lubrication of soft tissues and joints, the maintenance of the structural integrity of tissues, and the transport of molecules to and within cells.
Our proprietary technologies for modifying the HA molecule allow product properties to be tailored specifically to therapeutic use. Our patented technology chemically modifies HA to allow for longer residence time in the body. We also offer products made from HA based on two other technologies: HYAFF, which is a solid form of HA, and ACP gel, an autocross-linked polymer of HA. Our technologies are protected by an extensive portfolio of owned and licensed patents.
Since our inception in 1992, we have utilized a commercial partnership model for the distribution of our products to end users. Our strong, worldwide network of distributors has historically provided, and continues to provide, a solid foundation for our revenue growth and territorial expansion. In 2015, we made the strategic decision to commercialize our next generation viscosupplementation product, CINGAL, in the United States by utilizing a direct sales model, initially through the engagement of a contract sales organization. Ultimately, we intend to transition the direct sales function into our company as part of a broader buildout of our commercial capabilities. We believe that the combination of the direct and distribution commercial models will maximize the revenue and profitability potential from our current and future product portfolio.
In the fourth quarter of 2017, we completed all planned activities related to the strategic project we began in 2015 to insource the manufacturing of our HYAFF-based products to our Bedford, Massachusetts facility at a total cost of $23.0 million. These products were previously manufactured by a third-party contract manufacturer in Italy. Our main purposes behind this strategic move are to enhance our research and development capabilities with the aim of accelerating future product development and to improve the efficiency of our manufacturing processes.
The following sections provide more information about our products:
Orthobiologics
Our orthobiologics business contributed 87% of our product revenue for the year ended December 31, 2017. Our orthobiologics products primarily consist of viscosupplementation and regenerative orthopedic products. Our viscosupplementation products include ORTHOVISC, ORTHOVISC mini, and MONOVISC, each of which is commercialized in various territories worldwide, and CINGAL, which we launched internationally in Canada and the European Union in the second quarter of 2016 after receiving Health Canada and CE Mark approval. ORTHOVISC is available in the United States, Canada, and some international markets for the treatment of osteoarthritis of the knee, and in Europe and other international markets for the treatment of osteoarthritis in all synovial joints. It has been marketed by us in the United States since 2004 and internationally since 1996 through various distribution agreements. ORTHOVISC mini is available in Europe and is designed for the treatment of osteoarthritis in small joints. MONOVISC is our first single injection osteoarthritis treatment indicated for all synovial joints in Europe and certain international markets, and for the knee in the United States, Turkey, and Canada. ORTHOVISC mini and MONOVISC both became available in certain international markets through our network of distributors during the second quarter of 2008, and the commercial introduction of MONOVISC in the United States occurred in April 2014. We are currently seeking regulatory approval for CINGAL, our second single-injection osteoarthritis product, in the United States.
| 29 |
We currently offer several orthopedic products used in connection with regenerative medicine. The products currently available in Europe and certain international markets include HYALOFAST, a biodegradable support for human bone marrow mesenchymal stem cells used for cartilage regeneration and as an adjunct for microfracture surgery; HYALONECT, a resorbable knitted fabric mesh for use in orthopedic and trauma reconstructive procedures to maintain the relative position of engrafted bone tissue or bone fragments from comminuted fractures; and HYALOSS, HYAFF fibers used to mix blood/bone grafts to form a paste for bone regeneration. We also offer HYALOGLIDE, an ACP gel used in tenolysis treatment that, with additional clinical data, may demonstrate potential for flexor tendon adhesion prevention and for the treatment of adhesive capsulitis prevention in the shoulder. This product is commercialized through a network of distributors, primarily in Europe and the Middle East. We believe that the U.S. market offers excellent expansion potential to increase revenue for these products, and this will continue to be a focus area for us moving forward.
In addition to the products discussed above, we received CE Mark approval in December 2016 for a product which utilizes our proprietary HA technology to treat pain associated with lateral epicondylitis, better known as tennis elbow. Outside of the United States, this product is marketed under the trade name ORTHOVISC-T. Additionally, in the second quarter of 2016, we submitted an IDE to the FDA to conduct a Phase III clinical trial for this treatment, and the IDE was approved by the FDA in June 2016. We also received 510(k) clearance for an injectable HA-based bone repair treatment in December 2017.
Our strategy is to continue to add new products, to expand the indications for use of both our current and any new products, and to expand our commercial reach. The orthobiologics area has been our fastest growing area, generating 88% growth from 2012 to 2017. We continue to seek new distribution partnerships around the world, in concert with entering new markets with other appropriate sales strategies, and we expect total orthobiologics product sales to increase in 2018 compared to 2017 based mainly on increased sales to existing and new partners, as well as additional MONOVISC and CINGAL product launches in certain international countries. Additionally, if we achieve FDA approval of CINGAL, we plan to utilize a direct sales model to commercialize the product in the United States initially through the engagement of a contract sales organization with the ultimate goal of transitioning the direct sales function to our company as part of a broader buildout of our commercial capabilities.
Dermal
Our dermal products contributed 3% to our product revenue for the year ended December 31, 2017 and consist of advanced wound care products, which are based on the HYAFF technology, and an aesthetic dermal filler. We offer products for the treatment of skin wounds ranging from burns to diabetic ulcers. The products cover a variety of wound treatment solutions including debridement agents, advanced therapies, and scaffolds used as skin substitutes. Leading products include HYALOMATRIX and HYALOFILL for the treatment of complex wounds, such as burns and ulcers, and for use in connection with the regeneration of skin. Our dermal products are commercialized through a network of distributors, primarily in the United States, Europe, Latin America, and the Middle East. Products cleared for sale in the United States include HYALOMATRIX, HYALOFILL, HYALOGRAN, HYALOSAFE, and HYALOMATRIX 3D. We have a commercial partnership agreement with Medline Industries, Inc. to commercialize HYALOMATRIX in the United States on an exclusive basis through 2022.
Our aesthetic dermatology product is a dermal filler based on our proprietary, chemically modified, cross-linked HA, and it is primarily commercialized in certain countries in the Middle East. Internationally, this product is marketed under the ELEVESS trade name. In the United States, the trade name is HYDRELLE, although the product is not currently marketed in the United States.
Surgical
Our surgical group consists of products used to prevent surgical adhesions and to treat ENT disorders. For the year ended December 31, 2017, sales of surgical products contributed 5% of our product revenue. HYALOBARRIER is a clinically proven post-operative adhesion barrier for use in the abdomino-pelvic area. The product is currently commercialized in Europe, the Middle East, and certain African and Asian countries through a distribution network, but it is not approved in the United States. INCERT, approved for sale in Europe, Turkey, and Malaysia, is a chemically modified, cross-linked HA product used for the prevention of post-surgical spinal adhesions. There are no plans at this time to distribute INCERT in the United States.
We also offer several products used in connection with the treatment of ENT disorders. The lead products are MEROGEL, a woven fleece nasal packing, and MEROGEL INJECTABLE, a thick, viscous hydrogel composed of cross-linked HA, a biocompatible agent that creates a moist wound-healing environment. We partner with Medtronic for the worldwide distribution of these products.
| 30 |
Other
Our other products include our ophthalmic and veterinary products, which constituted 5% of our product revenue for the year ended December 31, 2017. These legacy products are not a part of our core business. Our ophthalmic business includes HA viscoelastic products used in ophthalmic surgery. Sales of ophthalmic products contributed 1% of our product revenue and sales of HYVISC, our veterinary product used for the treatment of equine osteoarthritis, contributed 4% of our product revenue for the year ended December 31, 2017.
Research and Development
Our research and development efforts primarily consist of the development of new medical applications for our HA-based or other technologies, the management of clinical trials for certain product candidates, the preparation and processing of applications for regulatory approvals or clearances at all relevant stages of product development, and process development and scale-up manufacturing activities for our existing and new products. Our development focus includes products for tissue protection, repair, and regeneration. For the years ended December 31, 2017, 2016, and 2015, these expenses were $18.8 million, $10.7 million, and $9.0 million, respectively. We anticipate that we will continue to commit significant resources in the near future to research and development activities, including in relation to preclinical activities and clinical trials. These activities are aimed at the delivery of a steady cascade of new product development and launches over the next several years.
Our second single-injection osteoarthritis product under development in the United States is CINGAL, which is composed of our proprietary cross-linked HA material combined with an approved steroid and is designed to provide both short- and long-term pain relief to patients. We completed an initial CINGAL phase III clinical trial, including the associated statistical analysis for 368 enrolled patients, during the fourth quarter of 2014 with data indicating that the product met all primary and secondary endpoints set forth for the trial. During the first half of 2015, we completed a CINGAL retreatment study with 242 patients who had participated in the phase III clinical trial and reported safety data related to the retreatment study. This initial phase III clinical trial and the associated retreatment study supported the Health Canada and CE Mark approval of the product, and the commercial launch of the product in both Canada and the European Union occurred in the second quarter of 2016. In the United States, after discussions with the FDA related to the regulatory pathway for CINGAL, we conducted a formal meeting with OCP to present and discuss our data in September 2015, and we submitted a formal request for designation with OCP a month later. In its response to our formal request for designation, OCP assigned the product to CDER as the lead agency center for premarket review and regulation. We held a meeting with CDER at the end of September 2016 to align on an approval framework and on submission requirements for an NDA for CINGAL, including the execution of an additional Phase III clinical trial to supplement our strong, existing CINGAL pivotal study data. We submitted an IND in late 2016, and discussions with CDER to this point indicate that they do not have objections to our clinical protocol design. As a result, we commenced work on this second Phase III clinical trial in the first quarter of 2017, and the first patient was treated in the second quarter of 2017. Enrollment of the 576 patients in this second Phase III clinical trial was completed during October 2017. We expect to complete the six-month follow-up for this Phase III clinical trial during the second quarter of 2018 and to submit our NDA to FDA as expeditiously as possible thereafter. We have also initiated an additional three-month extended follow-up study in conjunction with the second Phase III clinical trial to investigate the efficacy of CINGAL over this longer period, and the first patients were enrolled in this follow-up study in the fourth quarter of 2017. This extended follow-up study will not impact the timeline for submission of the NDA for CINGAL following the completion of the second Phase III clinical trial.
We have several research and development programs underway for new products, including for HYALOFAST (in the United States), an innovative product for cartilage tissue repair, and other early stage regenerative medicine development programs. HYALOFAST received CE Mark approval in September 2009, and it is commercially available in Europe and certain international countries. During the first quarter of 2015, we submitted an IDE for HYALOFAST to the FDA, which was approved in July 2015. We commenced patient enrollment in a clinical trial in December 2015, and we are advancing site initiations and patient enrollment activities. In the second quarter of 2016, a supplement to the HYALOFAST IDE was approved to expand the inclusion criteria for the clinical study. The purpose of this supplement is to allow us to increase enrollment rates with the ultimate goal of decreasing the time needed to complete the clinical trial. We are also currently proceeding with other research and development programs, one of which utilizes our proprietary HA technology to treat pain associated with common repetitive overuse injuries, such as lateral epicondylitis, also known as tennis elbow. We submitted a CE Mark application for this treatment during the first quarter of 2016 and received a CE Mark for the treatment of pain associated with tennis elbow in December 2016. We expect to begin enrolling patients in a post-market clinical study in relation to the CE Mark for this product before the end of the second quarter of 2018. Outside of the United States, this product will be marketed under the trade name ORTHOVISC-T. In the second quarter of 2016, we submitted an IDE to the FDA to conduct a phase III clinical trial for this treatment, which was approved by the FDA in June 2016. We also have other research and development programs underway focused on expanding the indications of our current products, including one program being conducted and funded by our U.S. MONOVISC distribution partner, Mitek, seeking to expand MONOVISC’s indication to include the treatment of pain associated with osteoarthritis of the hip. In third quarter of 2017, we also submitted an application to the FDA for 510(k) clearance of an injectable HA-based bone repair treatment. The 510(k) clearance was received from the FDA in December 2017. In addition to other early stage research and developments initiatives we are currently undertaking, we are working to expand our regenerative medicine pipeline with a new product candidate in the form of an implant for rotator cuff repair utilizing our proprietary solid HA.
| 31 |
In June 2015, we entered into an agreement with the Institute for Applied Life Sciences at the University of Massachusetts Amherst to collaborate on research to develop a therapy for rheumatoid arthritis. The purpose of this research is to develop a novel modality for the treatment of rheumatoid arthritis. The agreement with the University of Massachusetts Amherst was extended in January 2018, and the next phase of the research will focus on optimizing the drug delivery system with the goal of advancing a novel therapeutic candidate into clinical trials to support regulatory submission. We also recently entered into an agreement with the University of Liverpool to develop an injectable mesenchymal stem cell therapy for the treatment of age-related osteoarthritis with the goal of bringing a therapeutics candidate through clinical trials to market to meet an unmet therapeutic need.
Summary of Critical Accounting Policies; Significant Judgments and Estimates
Our discussion and analysis of our financial condition and results of operations are based upon our consolidated financial statements included elsewhere in this Annual Report on Form 10-K, which consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and the related disclosure of contingent assets and liabilities. We monitor our estimates on an ongoing basis for changes in facts and circumstances, and material changes in these estimates could occur in the future. Changes in estimates are recorded in the period in which they become known. We base our estimates on historical experience and other assumptions that we believe to be reasonable under the circumstances. Actual results may differ from our estimates if past experience or other assumptions do not turn out to be substantially accurate.
We have identified the policies below as critical to our business operations and the understanding of our results of operations. The impact and any associated risks related to these policies on our business operations are discussed throughout this section captioned “Management’s Discussion and Analysis of Financial Condition and Results of Operations” where such policies affect our reported and expected financial results. For a detailed discussion on the application of these and other accounting policies, see Note 2 to the consolidated financial statements included elsewhere in this Annual Report on Form 10-K.
Revenue Recognition - General
We recognize revenue from product sales when all of the following criteria are met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the seller's price to the buyer is fixed or determinable; and collection from the customer is reasonably assured.
Product Revenue
Revenue from product sales is recognized when title and risk of loss have passed to the customer, which is typically upon shipment to the customer. Amounts billed or collected prior to recognition of revenue are classified as deferred revenue. When determining whether risk of loss has transferred to customers on product sales, or if the sales price is fixed or determinable, we evaluate both the contractual terms and conditions of our distribution and supply agreements as well, as our business practices.
| 32 |
Product revenue also includes royalties. Royalty revenue is based on our distributors’ sales and is recognized in the same period our distributors record their sale of products manufactured by us. On a quarterly basis we record royalty revenue based upon estimated or reported sales results provided to us by our distributor customers.
Licensing, Milestone and Contract Revenue
Licensing, milestone and contract revenue consists of revenue recognized on initial and milestone payments, as well as contractual amounts received from partners. Our business strategy includes entering into collaborative license, development, and/or supply agreements with partners for the development and commercialization of our products.
The terms of the agreements typically include non-refundable license fees, funding of research and development, and payments based upon achievement of certain milestones. Under ASC 605-25, Multiple Element Arrangements, in order to account for an element as a separate unit of accounting, the element must have objective and reliable evidence of selling price of the undelivered elements. In general, non-refundable upfront fees and milestone payments that do not relate to other elements are recognized as revenue over the term of the arrangement as we complete our performance obligations.
Inventories
Inventories are stated at the lower of standard cost and net realizable value, with approximate cost determined using the first-in, first-out method. Work-in-process and finished goods inventories include materials, labor, and manufacturing overhead. Inventory costs associated with product candidates that have not yet received regulatory approval are capitalized if we believe there is probable future commercial use and future economic benefit.
Our policy is to write-down inventory when conditions exist that suggest inventory may be in excess of anticipated demand or is obsolete based upon assumptions about future demand for our products and market conditions. We regularly evaluate our ability to realize the value of inventory based on a combination of factors including, but not limited to, historical usage rates, forecasted sales or usage, product end of life dates, and estimated current or future market values. Purchasing requirements and alternative usage avenues are explored within these processes to mitigate inventory exposure.
Goodwill and Acquired In-Process Research and Development
Goodwill is the amount by which the purchase price of acquired net assets in a business combination exceeded the fair values of net identifiable assets on the date of acquisition. Acquired IPR&D represents the fair value assigned to research and development assets that we acquire that have not been completed at the date of acquisition or are pending regulatory approval in certain jurisdictions. The value assigned to the acquired IPR&D is determined by estimating the costs to develop the acquired technology into commercially viable products, estimating the resulting revenue from the projects, and discounting the net cash flows to present value.
Goodwill and IPR&D are evaluated for impairment annually, or more frequently if events or changes in circumstances indicate that the asset might be impaired. Factors we consider important, on an overall company basis, that could trigger an impairment review include significant underperformance relative to historical or projected future operating results, significant changes in our use of the acquired assets or the strategy for our overall business, significant negative industry or economic trends, a significant decline in our stock price for a sustained period, or a reduction of our market capitalization relative to net book value.
To conduct impairment tests of goodwill, the fair value of the reporting unit is compared to its carrying value. If the reporting unit’s carrying value exceeds its fair value, we record an impairment loss to the extent that the carrying value of goodwill exceeds its implied fair value. Our annual assessment for impairment of goodwill as of November 30, 2017 indicated that the fair value of our reporting unit exceeded the carrying value of the reporting unit.
| 33 |
To conduct impairment tests of IPR&D, the fair value of the IPR&D project is compared to its carrying value. If the carrying value exceeds its fair value, we record an impairment loss to the extent that the carrying value of the IPR&D project exceeds its fair value. We estimate the fair value for IPR&D projects using discounted cash flow valuation models, which require the use of significant estimates and assumptions, including, but not limited to, estimating the timing of and expected costs to complete the in-process projects, projecting regulatory approvals, estimating future cash flows from product sales resulting from completed projects and in-process projects, and developing appropriate discount rates. During the fourth quarter of 2015, we performed an impairment review of our IPR&D projects as we reassessed our research and development strategy. We recorded an impairment charge of $0.7 million due to the decision to discontinue further development efforts needed to commercialize our Hemostatic Patch in-process development project. Our annual assessment for impairment of IPR&D indicated that the fair value of our other IPR&D assets as of November 30, 2017 exceeded their respective carrying values.
Through December 31, 2017, there have not been any events or changes in circumstances that indicate that the carrying value of goodwill or acquired intangible assets may not be recoverable. We continue to monitor and evaluate the financial performance of our business, including the impact of general economic conditions, to assess the potential for the fair value of the reporting unit to decline below its book value. There can be no assurance that, at the time future impairment tests are completed, a material impairment charge will not be recorded.
Long-Lived Assets
Long-lived assets primarily include property and equipment and intangible assets with finite lives. Our intangible assets are comprised of purchased developed technologies, distributor relationships, patents, and a trade name. The distributor relationships and trade name were fully amortized as of December 31, 2017. These intangible assets are carried at cost, net of accumulated amortization. Amortization is recorded on a straight-line basis over the intangible assets' useful lives, which range from 5 to 16 years. We review long-lived assets for impairment when events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable or that the useful lives of those assets are no longer appropriate. Each impairment test is based on a comparison of the undiscounted cash flows to the recorded value of the asset. If impairment is indicated, the asset is written down to its estimated fair value based on a discounted cash flow analysis.
Stock-Based Compensation
We measure the compensation cost of award recipients’ services received in exchange for an award of equity instruments based on the grant-date fair value of the underlying award. That cost is recognized over the period during which an employee is required to provide service in exchange for the award. For performance based awards with financial achievement targets, we recognize expense using the graded vesting methodology based on the number of shares expected to vest. Compensation cost associated with performance grants is estimated using the Black-Scholes valuation method multiplied by the expected number of shares to be issued, which is adjusted based on the estimated probabilities of achieving the performance goals. Changes to the probability assessment and the estimated shares expected to vest will result in adjustments to the related share-based compensation expense that will be recorded in the period of the change. If the performance targets are not achieved, no compensation cost is recognized and any previously recognized compensation cost is reversed. See Note 12, Equity Incentive Plan, to the consolidated financial statements included elsewhere in this Annual Report on Form 10-K for a description of the types of stock-based awards granted, the compensation expense related to such awards, and detail of equity-based awards outstanding. See Note 16, Income Taxes, to the consolidated financial statements included elsewhere in this Annual Report on Form 10-K for details related to the tax benefit recognized in the consolidated statement of operations for stock-based compensation.
Income Taxes
Our income tax expense includes U.S. and international income taxes. Certain items of income and expense are not reported in tax returns and financial statements in the same year. The tax effects of these differences are reported as deferred tax assets and liabilities. Deferred tax assets are recognized for the estimated future tax effects of deductible temporary differences and tax operating loss and credit carry-forwards. Changes in deferred tax assets and liabilities are recorded in the provision for income taxes. We assess the likelihood that our deferred tax assets will be recovered from future taxable income, and to the extent we believe that it is more likely than not that all or a portion of deferred tax assets will not be realized, we establish a valuation allowance. To the extent we establish a valuation allowance or increase this allowance in a period, we include an expense within the tax provision in the consolidated statement of operations.
| 34 |
Results of Operations
Year ended December 31, 2017 compared to year ended December 31, 2016
Statement of Operations Detail
| Years Ended December 31, | ||||||||||||||||
| 2017 | 2016 | $ Inc/(Dec) | % Inc/(Dec) | |||||||||||||
| (in thousands, except percentages) | ||||||||||||||||
| Product revenue | $ | 107,783 | $ | 102,932 | $ | 4,851 | 5 | % | ||||||||
| Licensing, milestone and contract revenue | 5,637 | 447 | 5,190 | 1,161 | % | |||||||||||
| Total revenue | 113,420 | 103,379 | 10,041 | 10 | % | |||||||||||
| Operating expenses: | ||||||||||||||||
| Cost of product revenue | 27,364 | 24,027 | 3,337 | 14 | % | |||||||||||
| Research & development | 18,787 | 10,732 | 8,055 | 75 | % | |||||||||||
| Selling, general & administrative | 21,540 | 18,013 | 3,527 | 20 | % | |||||||||||
| Total operating expenses | 67,691 | 52,772 | 14,919 | 28 | % | |||||||||||
| Income from operations | 45,729 | 50,607 | (4,878 | ) | (10 | %) | ||||||||||
| Interest income, net | 473 | 263 | 210 | 80 | % | |||||||||||
| Income before income taxes | 46,202 | 50,870 | (4,668 | ) | (9 | %) | ||||||||||
| Provision for income taxes | 14,386 | 18,323 | (3,937 | ) | (21 | %) | ||||||||||
| Net income | $ | 31,816 | $ | 32,547 | $ | (731 | ) | (2 | %) | |||||||
| Product gross profit | $ | 80,419 | $ | 78,905 | $ | 1,514 | 2 | % | ||||||||
| Product gross margin | 75 | % | 77 | % | ||||||||||||
Total revenue
Total revenue for the year ended December 31, 2017 increased by $10.0 million, as compared to the prior year, to $113.4 million. This increase was primarily due to the growth of our orthobiologics franchise, specifically an increase in global MONOVISC revenue and our achievement of $5.0 million of milestone revenue in 2017 for reaching a target MONOVISC U.S. end-user sales threshold set forth in the Mitek MONOVISC Agreement.
Product revenue
Product revenue for the year ended December 31, 2017 was $107.8 million, an increase of $4.9 million, or 5.0%, compared to the prior year. Product revenue increases in our Orthobiologics and Other franchises were partially offset by a moderate decrease in our Surgical product revenue. The following table presents comparative product revenue analysis by product franchise:
| Years Ended December 31, | ||||||||||||||||
| 2017 | 2016 | $ Inc/(Dec) | % Inc/(Dec) | |||||||||||||
| Orthobiologics | $ | 93,816 | $ | 89,695 | $ | 4,121 | 5 | % | ||||||||
| Dermal | 2,755 | 2,759 | (4 | ) | (0 | %) | ||||||||||
| Surgical | 5,262 | 5,427 | (165 | ) | (3 | %) | ||||||||||
| Other | 5,950 | 5,051 | 899 | 18 | % | |||||||||||
| $ | 107,783 | $ | 102,932 | $ | 4,851 | 5 | % | |||||||||
| 35 |
Orthobiologics
Our orthobiologics franchise consists of our joint health and orthopedic products. Overall, revenue from our orthobiologics franchises increased $4.1 million, or 5%, in 2017 as compared to 2016. The growth in 2017 reflected growing end-user demand, continued market penetration, and increased revenue from worldwide MONOVISC and CINGAL sales. The increase in viscosupplementation revenue in 2017 was driven primarily by increased sales of MONOVISC resulting from a robust and growing end-user demand. We expect orthobiologics revenue to continue to grow in 2018, led by MONOVISC revenue in domestic and international markets, including revenue from sales in India, Australia, New Zealand, and Taiwan, as well as increased revenue from CINGAL internationally.
Dermal
Our dermal franchise consists of advanced wound care products, which are based on our HYAFF technology, and aesthetic dermal fillers. Our advanced wound care products treat complex skin wounds ranging from burns to diabetic ulcers, with HYALOMATRIX and HYALOFILL as the lead products. Dermal revenue had no significant change in 2017 as compared to 2016. The revenue, in part, is derived from the agreement we entered into with Medline Industries, Inc. to commercialize HYALOMATRIX in the United States on an exclusive basis through 2022. We expect dermal revenue to increase modestly in 2018 as compared to 2017.
Surgical
Our surgical franchise consists of products used to prevent surgical adhesions and to treat ear, nose, and throat (“ENT”) disorders. Sales of our surgical products decreased $0.2 million, or 3%, in 2017 as compared to 2016. The decrease of surgical product revenue was primarily due to a decrease in sales generated by our ENT products. Our surgical franchise consists primarily of our anti-adhesion products, including INCERT and HYALOBARRIER, and our ENT offerings, of which MEROGEL is the leading product. We are partnered with Medtronic for the worldwide distribution of our ENT products. We expect surgical product revenue to increase modestly in 2018 as compared to 2017 primarily due to increased worldwide sales of our surgical anti-adhesions products.
Other
Other product revenue includes revenues from ophthalmic and veterinary products. The other product revenue increased in 2017 from 2016 due to a recovery from weak 2016 sales volume for these franchises. We expect other revenue to increase in 2018 as compared to 2017, primarily driven by continued increases in ophthalmic revenue.
Licensing, milestone and contract revenue
Licensing, milestone and contract revenue for the year ended December 31, 2017 was $5.6 million, compared to $0.4 million for 2016. The year-over-year increase was primarily the result of the recognition of milestone revenue during the year ended December 31, 2017. During the second quarter of 2017, we fully recognized revenue for a milestone payment of $5.0 million under Mitek MONOVISC Agreement as a result of U.S. MONOVISC 12-month end-user sales exceeding $100 million. We expect that our licensing, milestone and contract revenue in 2018 will be approximately equal to such revenue received in 2017.
Product gross profit and margin
Product gross profit for the year ended December 31, 2017 was $80.4 million, or 75% of product revenue, as compared with $78.9 million, or 77% of product revenue, for the year ended December 31, 2016. The increase in product gross profit was primarily due to the increased volume compared to the prior year, while the decrease in product gross margin was due to inventory write-offs in 2017, as well as initial start-up costs associated with our insourcing of the manufacturing of HYAFF-based products to the Company’s Bedford facility. We expect gross margin to remain at similar levels in 2018 with potential opportunities for improvement due to increased sales volume and manufacturing process improvements.
| 36 |
Research and development
Research and development expenses for the year ended December 31, 2017 increased by $8.1 million, or 75%, as compared to the prior year, mainly due to an increase in expenses for our HYALOFAST and CINGAL phase III clinical trials. We also increased our pre-clinical product development activities, including with respect to achieving a 510(k) clearance of an HA-based, injectable, calcium phosphate bone graft substitute material. Research and development expense as a percentage of total revenue was 17% in 2017 and 10% in 2016. Research and development spending is expected to increase in 2018 compared to 2017 as we further develop new products and line extensions and initiate new clinical trials based on our existing technology assets, including CINGAL and HYALOFAST, as well as increase early-stage activities for other products and line extensions in the pipeline, such as our research collaborations with the University of Massachusetts Amherst and the University of Liverpool.
Selling, general and administrative
Selling, general and administrative expenses for the year ended December 31, 2017 increased by $3.5 million, or 20%, as compared to 2016. The increase was primarily as a result of increased personnel related costs, external professional fees, and additions to our allowance for doubtful accounts. We expect selling, general and administrative expenses for 2018 will increase to reflect the support, including CINGAL pre-launch expenses and the implementation of improved operational and financial technology platforms, required to grow our business both domestically and internationally.
Income taxes
Provisions for income taxes were $14.4 million and $18.3 million for the years ended December 31, 2017 and 2016, respectively. The decrease in the effective tax rate in 2017 of 4.9%, as compared to 2016, is primarily due to the revaluation of the deferred tax liability as a result of the Tax Cuts and Jobs Act tax reform legislation and an increased benefit from research and development activities. In accordance with Staff Accounting Bulletin No. 118, which provides guidance on accounting for the tax effects of the 2017 Tax Act, the Company has recorded a reasonable estimate of the impact on the consolidated financial statements. The provisional amounts incorporate assumptions made based upon the Company’s current interpretation and implementation guidance of the 2017 Tax Act.
A reconciliation of the U.S. federal statutory tax rate to the effective tax rate for the periods ending December 31 is as follows:
| Years ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Statutory federal income tax rate | 35.0 | % | 35.0 | % | ||||
| State tax expense, net of federal benefit | 4.8 | % | 4.5 | % | ||||
| Impact of rate change on deferred taxes | (4.9 | )% | 0.0 | % | ||||
| Permanent items, including nondeductible expenses | 0.6 | % | 0.5 | % | ||||
| State investment tax credit | (0.7 | )% | (0.1 | )% | ||||
| Federal, state and foreign research and development credits | (1.4 | )% | (0.9 | )% | ||||
| Foreign rate differential | 0.5 | % | (0.1 | )% | ||||
| Domestic production deduction | (2.8 | )% | (2.9 | )% | ||||
| Effective income tax rate | 31.1 | % | 36.0 | % | ||||
As of December 31, 2017, we had gross net operating losses (“NOL”) for income tax purposes in Italy of $4.0 million with no expiration date. In connection with the preparation of the financial statements, we performed an analysis to ascertain if it was more likely than not that we would be able to utilize, in future periods, the net deferred tax assets associated with our NOL carry-forward. We have concluded that the positive evidence outweighs the negative evidence and, thus, that the deferred tax assets not otherwise subject to a valuation allowance are realizable on a “more likely than not” basis. As such, we have not recorded a valuation allowance at December 31, 2017 or 2016.
| 37 |
In the normal course of business, Anika and its subsidiaries may be periodically examined by various taxing authorities. We file income tax returns in the U.S. federal jurisdiction, in certain U.S. states, and in Italy. The associated tax filings remain subject to examination by applicable tax authorities for a certain length of time following the tax year to which those filings relate. The 2014 through 2016 tax years remain subject to examination by the IRS and other taxing authorities for U.S. federal and state tax purposes. The 2011 through 2016 tax years remain subject to examination by the appropriate governmental authorities for Italy.
Net income
For the year ended December 31, 2017, net income was $31.8 million, or $2.11 per diluted share, compared to $32.5 million, or $2.15 per diluted share, for the same period in the prior year. The decrease in net income and diluted earnings per share was primarily a result of increased expenses for our HYALOFAST and CINGAL phase III clinical trials and increases in personnel related costs, external professional fees, and additions to our allowance for doubtful accounts. These increased expenses are offset by increased total revenue and a decreased effective federal income tax rate as a result of the 2017 Income Tax Reform Legislation.
Year ended December 31, 2016 compared to year ended December 31, 2015
Statement of Operations Detail
| Years Ended December 31, | ||||||||||||||||
| 2016 | 2015 | $ Inc/(Dec) | %Inc/(Dec) | |||||||||||||
| (in thousands, except percentages) | ||||||||||||||||
| Product revenue | $ | 102,932 | $ | 87,696 | $ | 15,236 | 17 | % | ||||||||
| Licensing, milestone and contract revenue | 447 | 5,303 | (4,856 | ) | (92 | %) | ||||||||||
| Total revenue | 103,379 | 92,999 | 10,380 | 11 | % | |||||||||||
| Operating expenses: | ||||||||||||||||
| Cost of product revenue | 24,027 | 21,053 | 2,974 | 14 | % | |||||||||||
| Research & development | 10,732 | 8,987 | 1,745 | 19 | % | |||||||||||
| Selling, general & administrative | 18,013 | 14,825 | 3,188 | 22 | % | |||||||||||
| Total operating expenses | 52,772 | 44,865 | 7,907 | 18 | % | |||||||||||
| Income from operations | 50,607 | 48,134 | 2,473 | 5 | % | |||||||||||
| Interest income (expense), net | 263 | 120 | 143 | 119 | % | |||||||||||
| Income before income taxes | 50,870 | 48,254 | 2,616 | 5 | % | |||||||||||
| Provision for income taxes | 18,323 | 17,496 | 827 | 5 | % | |||||||||||
| Net income | $ | 32,547 | $ | 30,758 | $ | 1,789 | 6 | % | ||||||||
| Product gross profit | $ | 78,905 | $ | 66,643 | $ | 12,262 | 18 | % | ||||||||
| Product gross margin | 77 | % | 76 | % | ||||||||||||
Total revenue
Total revenue for the year ended December 31, 2016 increased by $10.4 million to $103.4 million compared to the prior year. This increase was primarily due to the growth of our orthobiologics franchise, specifically an increase in global MONOVISC revenue, which was partially offset by our receipt of $5 million of milestone revenue in 2015 for the achievement of a target MONOVISC U.S. end user sales threshold.
| 38 |
Product revenue
Product revenue for the year ended December 31, 2016 was $102.9 million, an increase of $15.2 million, or 17%, compared to the prior year. Product revenue increases in our Orthobiologics and Dermal franchises were partially offset by moderate decreases in product revenue in our Surgical and Other franchises. Included in product revenue for the year ended December 31, 2015 was approximately $1.8 million and $0.5 millions of non-recurring revenue recorded in the second and third quarter of 2015, respectively, related to a high end-user average selling price for MONOVISC products sold to our U.S. partner, Mitek, prior to the fourth quarter of 2014. Products sold to Mitek after the third quarter of 2014 are not impacted by this arrangement, which will not result in additional related revenue.
| Years Ended December 31, | ||||||||||||||||
| 2016 | 2015 | $ Inc/(Dec) | % Inc/(Dec) | |||||||||||||
| (in thousands, except percentages) | ||||||||||||||||
| Orthobiologics | $ | 89,695 | $ | 73,247 | $ | 16,448 | 22 | % | ||||||||
| Dermal | 2,759 | 2,266 | 493 | 22 | % | |||||||||||
| Surgical | 5,427 | 5,812 | (385 | ) | (7 | %) | ||||||||||
| Other | 5,051 | 6,371 | (1,320 | ) | (21 | %) | ||||||||||
| $ | 102,932 | $ | 87,696 | $ | 15,236 | 17 | % | |||||||||
Orthobiologics
Revenue from our orthobiologics franchises increased $16.4 million, or 22%, in 2016 as compared to 2015. The growth in 2016 reflected a growing end-user demand, continued market penetration, increased revenue from worldwide MONOVISC sales, and CINGAL revenue associated with the product’s commercial launch in Canada and Europe. ORTHOVISC and MONOVISC revenue in the U.S. also increased 22% in 2016 as compared to 2015, while international viscosupplementation product revenue in 2016 increased 23% year-over-year. The increase in international viscosupplementation revenue in 2016 was driven primarily by increased sales of MONOVISC resulting from a robust and growing end-user demand.
Dermal
Dermal revenue increased $0.5 million, or 22%, in 2016 as compared to 2015. The increase primarily reflects revenue from the agreement we entered into with Medline Industries, Inc. to commercialize HYALOMATRIX in the United States on an exclusive basis through 2022.
Surgical
Sales of our surgical products decreased slightly in 2016 as compared to 2015. The decrease of surgical product revenue was primarily due to a decrease in sales generated by our ENT products and unfavorable impact from foreign currency exchange rate fluctuations compared with the same periods in the prior year. Our surgical franchise consists primarily of our anti-adhesion products, including INCERT and HYALOBARRIER, and our ENT offerings, of which MEROGEL is the leading product. We are partnered with Medtronic for the worldwide distribution of our ENT products.
Other
Other product revenue includes revenues from ophthalmic and veterinary products. The other product revenue decreased in 2016 from 2015 due to a decrease in sales generated by our veterinary and ophthalmic franchises.
Licensing, milestone and contract revenue
Licensing, milestone and contract revenue for the year ended December 31, 2016 was $0.4 million, compared to $5.3 million for 2015. The year over year decrease was primarily the result of the recognition of licensing and milestone revenue for the year ended December 31, 2015 of $5.0 million for the achievement of a milestone payment under the Mitek MONOVISC Agreement. During the fourth quarter of 2015, we collected and fully recognized revenue for a milestone payment of $5.0 million as a result of U.S. MONOVISC 12-month end-user sales exceeding $50 million.
| 39 |
Product gross profit and margin
Product gross profit for the year ended December 31, 2016 was $78.9 million, or 77% of product revenue, as compared with $66.6 million, or 76% of product revenue, for the year ended December 31, 2015. The increase in product gross profit was primarily due to improvements in the overall revenue mix compared to the prior year, with increased sales of our higher-margin products as a percentage of our total product sales.
Research and development
Research and development expenses for the year ended December 31, 2016 increased by $1.7 million, or 19%, as compared to the prior year, mainly due to an increase in expenses for our HYALOFAST phase III clinical trial. Included in our 2015 results was a $0.7 million expense resulting from an impairment charge related to IPR&D that was recorded in connection with our acquisition of Anika S.r.l. The charge resulted from a decision to discontinue development of the acquired Hemostatic Patch in-process development project. Research and development expense as a percentage of total revenue was 10% for the years ended 2016 and 2015.
Selling, general and administrative
Selling, general and administrative expenses for the year ended December 31, 2016 increased by $3.2 million, or 22%, as compared to 2015. The increase was primarily a result of increased headcount and external professional fees.
Income taxes
Provisions for income taxes were $18.3 million and $17.5 million for the years ended December 31, 2016 and 2015, respectively. The decrease in the effective tax rate in 2016 of 0.3%, as compared to 2015, is primarily due to an increased benefit from research and development credits.
A reconciliation of the U.S. federal statutory tax rate to the effective tax rate for the periods ending December 31 is as follows:
| Years ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| Statutory federal income tax rate | 35.0 | % | 35.0 | % | ||||
| State tax expense, net of federal benefit | 4.5 | % | 4.8 | % | ||||
| Impact of rate change on deferred taxes | 0.0 | % | 0.0 | % | ||||
| Permanent items, including nondeductible expenses | 0.5 | % | (0.3 | %) | ||||
| State investment tax credit | (0.1 | %) | 0.0 | % | ||||
| Federal, state and foreign research and development credits | (0.9 | %) | (0.4 | %) | ||||
| Foreign rate differential | (0.1 | %) | 0.1 | % | ||||
| Domestic production deduction | (2.9 | %) | (2.9 | %) | ||||
| Effective income tax rate | 36.0 | % | 36.3 | % | ||||
The decrease in permanent items, including nondeductible expenses, was mainly due to the impact on Anika S.r.l.’s long-term deferred tax assets for the decrease in Italy’s tax rate, effective January 1, 2017. The increase in the federal, state, and foreign research and development credit was mainly due to increased qualified research and development expenses.
As of December 31, 2016, we had gross net operating losses (“NOL”) for income tax purposes in Italy of $5.2 million with no expiration date. In connection with the preparation of the financial statements, we performed an analysis to ascertain if it was more likely than not that we would be able to utilize, in future periods, the net deferred tax assets associated with our NOL carry-forward. We have concluded that the positive evidence outweighs the negative evidence and, thus, that the deferred tax assets not otherwise subject to a valuation allowance are realizable on a “more likely than not” basis. As such, we have not recorded a valuation allowance at December 31, 2016 or 2015.
| 40 |
Net income
For the year ended December 31, 2016, net income was $32.5 million, or $2.15 per diluted share, compared to $30.8 million, or $2.01 per diluted share, for the same period in the prior year. The increase in net income and diluted earnings per share was primarily a result of increased worldwide product revenue and improved operating profit.
Concentration of Risk
We have historically derived the majority of our revenues from a small number of customers, most of whom resell our products to end-users and most of whom are significantly larger companies than us. For the year ended December 31, 2017, five customers accounted for 83% of product revenue, with Mitek alone accounting for 73% of product revenue. We expect to continue to be dependent on a small number of large customers, especially Mitek, for the majority of our revenues for the foreseeable future, even with our implementation of a direct sales model for CINGAL in the United States. The failure of these customers to purchase our products in the amounts they historically have or in amounts that we expect would seriously harm our business.
In addition, if present and future customers terminate their purchasing arrangements with us, significantly reduce or delay their orders, or seek to renegotiate their agreements on terms less favorable to us, our business, financial condition, and results of operations will be adversely affected. If we accept terms less favorable than the terms of the current agreements, such renegotiations may have a material adverse effect on our business, financial condition, and/or results of operations. Furthermore, in any future negotiations we may be subject to the perceived or actual leverage that these customers may have given their relative size and importance to us. Any termination, change, reduction, or delay in orders could seriously harm our business, financial condition, and results of operations. Accordingly, unless and until we diversify and expand our customer base, our future success will significantly depend upon the timing and size of future purchases by our largest customers and the financial and operational success of these customers. The loss of any one of our major customers or the delay of significant orders from such customers, even if only temporary, could reduce or delay our recognition of revenues, harm our reputation in the industry, and reduce our ability to accurately predict cash flow, and, as a consequence, it could seriously harm our business, financial condition, and results of operations.
See Note 15, Revenue by Product Group, by Significant Customer and by Geographic Location; Geographic Information, to the consolidated financial statements included elsewhere in this Annual Report on Form 10-K for information regarding significant customers.
Liquidity and Capital Resources
We require cash to fund our operating expenses and to make capital expenditures. We expect that our requirements for cash to fund these uses will increase as our operations expand. Historically we have generated positive cash flow from operations, which, together with our available cash, investments, and debt, have met our cash requirements. Cash, cash equivalents, and investments totaled $157.3 million and $124.8 million, and working capital totaled $193.3 million and $161.6 million, at December 31, 2017 and December 31, 2016, respectively. In addition, we have $50.0 million of available credit under our Senior Revolving Credit Facility as of December 31, 2017. We believe that we have adequate financial resources to support our business for at least the twelve months from the issuance date of our financial statements.
Cash provided by operating activities was $40.8 million, $24.4 million, and $39.9 million for 2017, 2016, and 2015, respectively. The increase in cash provided by operations was due primarily to increases in accounts payable and decreases in accounts receivable offset in part by increases in inventory.
Cash used in investing activities was $12.5 million, $6.8 million, and $30.2 million for 2017, 2016, and 2015, respectively. The increase in cash used in investing activities in 2017 as compared to 2016 was mainly the result of increased purchase of investments in 2017. In the fourth quarter of 2017, we completed all planned activities related to the strategic project at a cost of $23.0 million that we began in 2015 to insource the manufacturing of our HYAFF-based products to our Bedford, Massachusetts facility. Cash used in property and equipment investing activities decreased year-over-year in 2017 as the largest portion of the investment in this strategic project was made in 2016.
| 41 |
Cash provided (used) by financing activities was $0.3 million, ($24.0) million, and $1.1 million for 2017, 2016, and 2015, respectively. Cash provided by financing activities in 2017 was attributable to proceeds from the exercise of stock options. The increase in cash used in financing activities in 2016 as compared to 2015 was primarily attributable to the $25.0 million accelerated share repurchase program initiated in February 2016 and concluded in August 2016. For a description of the accelerated share repurchase program, see “Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities Accelerated Share Repurchase Program.”
Contractual Obligations and Other Commercial Commitments
The table below summarizes our non-cancelable operating leases, purchase commitments, and contractual obligations related to future periods which are not reflected in our consolidated balance sheet at December 31, 2017. Purchase commitments relate primarily to non-cancellable inventory commitments and capital expenditures entered in the normal course of business:
| Payments due by period (in thousands) | ||||||||||||||||||||
| Less than | More than | |||||||||||||||||||
| Total | 1 year | 1 - 3 years | 3 - 5 years | 5 years | ||||||||||||||||
| Operating Leases (1) | $ | 10,583 | $ | 1,879 | $ | 3,797 | $ | 3,596 | $ | 1,311 | ||||||||||
| Purchase Commitments (2) | 22,053 | 19,286 | 2,767 | - | - | |||||||||||||||
| Year Ended December 31, 2017 | $ | 32,636 | $ | 21,165 | $ | 6,564 | $ | 3,596 | $ | 1,311 | ||||||||||
| (1) | Includes a lease we entered into in January 2007, pursuant to which we lease our corporate headquarters facility, which consists of approximately 134,000 square feet of general office, research and development, and manufacturing space located in Bedford, Massachusetts. The lease has an initial term of ten and one-half years, and commenced in May 2007. In February 2017, we finalized the exercise of its first option under the lease to extend the terms from November 1, 2017 through October 31, 2022, including the determination of a new annual base rent of $1.5 million which is included in the disclosure above. No other terms of this lease were altered. We have an option under this lease to extend its lease-term for up to three additional periods subject to the condition that the Company notify the landlord that we are exercising each option at least one year prior to the expiration of the original or then-current term. The next two renewal options each extend the term an additional five years, while the final renewal option extends the term by six years. This schedule does not include the amounts that would be due if the company exercised the renewal options. Also includes a lease entered into pursuant to which Anika S.r.l. leases its Italian facility. In October 2015, Anika S.r.l, entered into a build-to-suit lease agreement for a new European headquarters facility consisting of approximately 33,000 square feet of general office, research and development, training, and warehousing space located in Padova, Italy. This lease has an initial term of fifteen years which commenced in February 2017. The lease will automatically renew for up to three additional six-year terms, subject to certain terms and conditions. We have the ability to withdraw from this lease subject to certain financial penalties after six years and with no penalties after the ninth year. As such, lease commitments through the ninth year are included in the table above. The lease provides for an initial yearly rent of approximately $0.3 million. See the section captioned “Item 2—Properties” in this Annual Report on Form 10-K for additional discussion regarding these leases. |
| (2) | Includes purchase commitments for materials, clinical trials, and other day to day business requirements. |
Accounting for Off-Balance Sheet Arrangements
We do not use special purpose entities or other off-balance sheet financing techniques, except for operating leases as disclosed in the contractual obligations table above, that we believe have or are reasonably likely to have a current or future material effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, or capital resources.
| 42 |
Recent Accounting Pronouncements
A discussion of recent accounting pronouncements is included in Note 2 to the consolidated financial statements in this Annual Report on Form 10-K.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Primary Market Risk Exposures
We manage our investment portfolio in accordance with our investment policy. The primary objectives of our investment policy are to preserve principal, maintain a high degree of liquidity to meet operating and other needs, and obtain competitive returns subject to prevailing market conditions without significantly increasing risk. To achieve this objective, we maintain our portfolio of cash equivalents and investments in a variety of high quality securities, including money market funds and bank certificates of deposits. The investments are classified as available-for-sale and consequently are recorded at fair value with unrealized gains or losses reported as a separate component of accumulated other comprehensive income. Our portfolio of cash equivalents and investments is subject to interest rate fluctuations, changes in credit quality of the issuer, and other factors.
Foreign Exchange Risk
Our primary market risk exposures are in the area of currency exchange rate risk. A significant portion of Anika S.r.l.’s revenue and operating expenses are denominated in Euros. We are utilizing clinical vendors which are located in various countries outside of the United States and invoice us in their local currency. We do not engage in foreign currency hedging arrangements for our accounts payable, and, consequently, foreign currency fluctuations may adversely affect our earnings. In addition, we have one major supplier contract denominated in a foreign currency. Gains and losses arising from transactions denominated in foreign currencies are primarily related to intercompany accounts that have been determined to be temporary in nature and cash, accounts payable, and accounts receivable denominated in non-functional currencies. Unfavorable fluctuations in exchange rates would have a negative impact on our financial statements. The impact of currency exchange rate fluctuations for the contract on our financial statements was immaterial in 2017. In the future, we may undertake to manage foreign currency risk through additional hedging methods. We recognize foreign currency gains or losses arising from our operations in the period incurred.
| 43 |
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
ANIKA THERAPEUTICS, INC. AND SUBSIDIARIES
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| 44 |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Anika Therapeutics, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Anika Therapeutics, Inc. and subsidiaries (the "Company") as of December 31, 2017, the related consolidated statements of operations, cash flows, and stockholders’ equity for the year ended December 31, 2017, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2017, and the results of its operations and its cash flows for the year ended December 31, 2017, in conformity with the accounting principles generally accepted in the United States of America (GAAP).
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated February 26, 2018, expressed an unqualified opinion on the Company's internal control over financial reporting.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provides a reasonable basis for our opinion.
/s/ Deloitte & Touche LLP
Boston, Massachusetts
February 26, 2018
We have served as the Company’s auditor since 2017.
| 45 |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Anika Therapeutics, Inc.
In our opinion, the consolidated balance sheet as of December 31, 2016 and the related consolidated statements of operations and comprehensive income, of stockholders' equity, and of cash flows for each of the two years in the period ended December 31, 2016 present fairly, in all material respects, the financial position of Anika Therapeutics, Inc. and its subsidiaries as of December 31, 2016, and the results of their operations and their cash flows for each of the two years in the period ended December 31, 2016, in conformity with accounting principles generally accepted in the United States of America. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits. We conducted our audits of these financial statements in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
/s/ PricewaterhouseCoopers LLP
Boston, Massachusetts
February 24, 2017
| 46 |
Anika Therapeutics, Inc. and Subsidiaries
(in thousands, except per share data)
| December 31, | ||||||||
| ASSETS | 2017 | 2016 | ||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 133,256 | $ | 104,261 | ||||
| Investments | 24,000 | 20,500 | ||||||
| Accounts receivable, net of reserves of $1,914 and $194 at December 31, 2017 and December 31, 2016, respectively | 23,825 | 27,598 | ||||||
| Inventories, net | 22,035 | 15,983 | ||||||
| Prepaid expenses and other current assets | 3,211 | 2,098 | ||||||
| Total current assets | 206,327 | 170,440 | ||||||
| Property and equipment, net | 56,183 | 52,296 | ||||||
| Other long-term assets | 1,254 | 69 | ||||||
| Intangible assets, net | 10,635 | 10,227 | ||||||
| Goodwill | 8,218 | 7,214 | ||||||
| Total assets | $ | 282,617 | $ | 240,246 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 6,747 | $ | 2,303 | ||||
| Accrued expenses and other current liabilities | 6,326 | 6,496 | ||||||
| Total current liabilities | 13,073 | 8,799 | ||||||
| Other long-term liabilities | 660 | 2,126 | ||||||
| Deferred tax liability | 5,393 | 6,548 | ||||||
| Commitments and contingencies (Note 11) | ||||||||
| Stockholders’ equity: | ||||||||
| Preferred stock, $0.01 par value; 1,250 shares authorized, no shares issued and outstanding at December 31, 2017 and December 31, 2016, respectively | - | - | ||||||
| Common stock, $0.01 par value; 60,000 shares authorized, 14,688 and 14,627 shares issued and outstanding at December 31, 2017 and December 31, 2016, respectively | 147 | 146 | ||||||
| Additional paid-in-capital | 68,617 | 61,735 | ||||||
| Accumulated other comprehensive loss | (4,784 | ) | (7,317 | ) | ||||
| Retained earnings | 199,511 | 168,209 | ||||||
| Total stockholders’ equity | 263,491 | 222,773 | ||||||
| Total liabilities and stockholders’ equity | $ | 282,617 | $ | 240,246 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
| 47 |
Anika Therapeutics, Inc. and Subsidiaries
Consolidated Statements of Operations and Comprehensive Income
(in thousands, except per share data)
| For the Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Product Revenue | $ | 107,783 | $ | 102,932 | $ | 87,696 | ||||||
| Licensing, milestone and contract revenue | 5,637 | 447 | 5,303 | |||||||||
| Total revenue | 113,420 | 103,379 | 92,999 | |||||||||
| Operating expenses: | ||||||||||||
| Cost of product revenue | 27,364 | 24,027 | 21,053 | |||||||||
| Research & development | 18,787 | 10,732 | 8,987 | |||||||||
| Selling, general & administrative | 21,540 | 18,013 | 14,825 | |||||||||
| Total operating expenses | 67,691 | 52,772 | 44,865 | |||||||||
| Income from operations | 45,729 | 50,607 | 48,134 | |||||||||
| Interest income, net | 473 | 263 | 120 | |||||||||
| Income before income taxes | 46,202 | 50,870 | 48,254 | |||||||||
| Provision for income taxes | 14,386 | 18,323 | 17,496 | |||||||||
| Net income | $ | 31,816 | $ | 32,547 | $ | 30,758 | ||||||
| Basic net income per share: | ||||||||||||
| Net income | $ | 2.18 | $ | 2.22 | $ | 2.06 | ||||||
| Basic weighted average common shares outstanding | 14,575 | 14,682 | 14,934 | |||||||||
| Diluted net income per share: | ||||||||||||
| Net income | $ | 2.11 | $ | 2.15 | $ | 2.01 | ||||||
| Diluted weighted average common shares outstanding | 15,068 | 15,116 | 15,321 | |||||||||
| Net income | $ | 31,816 | $ | 32,547 | $ | 30,758 | ||||||
| Foreign currency translation adjustment | 2,533 | (668 | ) | (2,154 | ) | |||||||
| Comprehensive income | $ | 34,349 | $ | 31,879 | $ | 28,604 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| 48 |
Anika Therapeutics, Inc. and Subsidiaries
Consolidated Statements of Stockholders' Equity
(in thousands)
| Accumulated | ||||||||||||||||||||||||
| Common Stock | Other | Total | ||||||||||||||||||||||
| Number of | $.01 Par | Additional Paid | Retained | Comprehensive | Stockholders' | |||||||||||||||||||
| Shares | Value | in Capital | Earnings | Loss | Equity | |||||||||||||||||||
| Balance, December 31, 2014 | 14,852 | $ | 149 | $ | 77,540 | $ | 104,904 | $ | (4,495 | ) | $ | 178,098 | ||||||||||||
| Issuance of common stock for equity awards | 185 | 1 | 1,073 | - | - | 1,074 | ||||||||||||||||||
| Tax benefit related to equity awards | - | - | 847 | - | - | 847 | ||||||||||||||||||
| Stock-based compensation expense | - | - | 2,225 | - | - | 2,225 | ||||||||||||||||||
| Net income | - | - | - | 30,758 | - | 30,758 | ||||||||||||||||||
| Other comprehensive loss | - | - | - | - | (2,154 | ) | (2,154 | ) | ||||||||||||||||
| Balance, December 31, 2015 | 15,037 | $ | 150 | $ | 81,685 | $ | 135,662 | $ | (6,649 | ) | $ | 210,848 | ||||||||||||
| Issuance of common stock for equity awards | 121 | 1 | 1,006 | - | - | 1,007 | ||||||||||||||||||
| Tax benefit related to equity awards | - | - | 647 | - | - | 647 | ||||||||||||||||||
| Stock-based compensation expense | - | - | 3,392 | - | - | 3,392 | ||||||||||||||||||
| Repurchase of common stock | (531 | ) | (5 | ) | (24,995 | ) | - | - | (25,000 | ) | ||||||||||||||
| Net income | - | - | - | 32,547 | - | 32,547 | ||||||||||||||||||
| Other comprehensive loss | - | - | - | - | (668 | ) | (668 | ) | ||||||||||||||||
| Balance, December 31, 2016 | 14,627 | $ | 146 | $ | 61,735 | $ | 168,209 | $ | (7,317 | ) | $ | 222,773 | ||||||||||||
| Issuance of common stock for equity awards | 61 | 1 | 313 | - | - | 314 | ||||||||||||||||||
| Stock-based compensation expense | - | - | 5,807 | 5,807 | ||||||||||||||||||||
| Cumulative effect of change in accounting for stock-based compensation | - | - | 762 | (514 | ) | - | 248 | |||||||||||||||||
| Net income | - | - | - | 31,816 | - | 31,816 | ||||||||||||||||||
| Other comprehensive income | - | - | - | - | 2,533 | 2,533 | ||||||||||||||||||
| Balance, December 31, 2017 | 14,688 | $ | 147 | $ | 68,617 | $ | 199,511 | $ | (4,784 | ) | $ | 263,491 | ||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| 49 |
Anika Therapeutics, Inc. and Subsidiaries
Consolidated Statements of Cash Flows
(in thousands)
| For the years ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Cash flows from operating activities: | ||||||||||||
| Net income | $ | 31,816 | $ | 32,547 | $ | 30,758 | ||||||
| Adjustments to reconcile net income to net cash provided by operating activities: | ||||||||||||
| Depreciation and amortization | 4,290 | 3,734 | 3,775 | |||||||||
| Loss on disposal of fixed assets | 150 | - | - | |||||||||
| Stock-based compensation expense | 5,807 | 3,392 | 2,225 | |||||||||
| Deferred income taxes | (1,198 | ) | (65 | ) | (747 | ) | ||||||
| Provision for doubtful accounts | 1,609 | 52 | 38 | |||||||||
| Provision for inventory | 695 | 654 | 210 | |||||||||
| Non-cash impairment charges for IPR&D | - | - | 697 | |||||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Accounts receivable | 2,674 | (6,201 | ) | (4,996 | ) | |||||||
| Inventories | (6,521 | ) | (1,738 | ) | (2,939 | ) | ||||||
| Prepaid expenses, other current and long-term assets | (1,454 | ) | (898 | ) | 89 | |||||||
| Accounts payable | 3,890 | (5,059 | ) | 5,625 | ||||||||
| Accrued expenses and other current liabilities | (605 | ) | 1,566 | (199 | ) | |||||||
| Income taxes payable | 367 | (3,552 | ) | 5,484 | ||||||||
| Other long-term liabilities | (708 | ) | 16 | (109 | ) | |||||||
| Net cash provided by operating activities | 40,812 | 24,448 | 39,911 | |||||||||
| Cash flows from investing activities: | ||||||||||||
| Proceeds from maturity of investments | 41,500 | 46,500 | 24,250 | |||||||||
| Purchase of investments | (45,000 | ) | (39,249 | ) | (45,251 | ) | ||||||
| Purchase of property and equipment, net | (8,980 | ) | (14,014 | ) | (9,225 | ) | ||||||
| Net cash used in investing activities | (12,480 | ) | (6,763 | ) | (30,226 | ) | ||||||
| Cash flows from financing activities: | ||||||||||||
| Repurchase of common stock | - | (25,000 | ) | - | ||||||||
| Proceeds from exercise of equity awards | 314 | 1,007 | 1,074 | |||||||||
| Net cash (used in) provided by financing activities | 314 | (23,993 | ) | 1,074 | ||||||||
| Exchange rate impact on cash | 349 | (138 | ) | (208 | ) | |||||||
| Increase (Decrease) in cash and cash equivalents | 28,995 | (6,446 | ) | 10,551 | ||||||||
| Cash and cash equivalents at beginning of period | 104,261 | 110,707 | 100,156 | |||||||||
| Cash and cash equivalents at end of period | $ | 133,256 | $ | 104,261 | $ | 110,707 | ||||||
| Supplemental disclosure of cash flow information: | ||||||||||||
| Cash paid for income taxes | $ | 15,088 | $ | 22,826 | $ | 12,724 | ||||||
| Non-cash Investing Activities: | ||||||||||||
| Purchases of property and equipment included in accounts payable and accrued expenses | $ | 1,891 | $ | 1,257 | $ | 1,949 | ||||||
| Build-to-suit lease agreement | $ | - | $ | 1,723 | $ | 30 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| 50 |
Anika Therapeutics, Inc. and Subsidiaries
Notes to Consolidated Financial Statements
(amounts in thousands, except share and per share amounts or as otherwise noted)
1. Nature of Business
Anika Therapeutics, Inc. (the “Company”) is a global, integrated orthopedic medicines company committed to improving the lives of patients with degenerative orthopedic diseases and traumatic conditions with clinically meaningful therapies along the continuum of care, from palliative pain management to regenerative tissue repair. The Company has over two decades of global expertise developing, manufacturing, and commercializing products based on the Company’s proprietary Hyaluronic Acid (“HA”) technology. The Company’s orthopedic medicine portfolio includes ORTHOVISC, MONOVISC, and CINGAL, which alleviate pain and restore joint function by replenishing depleted HA, and HYALOFAST, a solid HA-based scaffold to aid cartilage repair and regeneration.
The Company is subject to risks common to companies in the biotechnology and medical device industries including, but not limited to, development by the Company or its competitors of new technological innovations, dependence on key personnel, protection of proprietary technology, commercialization of existing and new products, and compliance with U.S. Food and Drug Administration (“FDA”) and foreign regulations and approval requirements, as well as the ability to grow the Company’s business through appropriate commercial strategies.
2. Summary of Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of Anika Therapeutics, Inc. and its wholly owned subsidiaries, Anika Securities, Inc. (a Massachusetts Securities Corporation), and Anika Therapeutics S.r.l. All intercompany balances and transactions have been eliminated in consolidation.
Foreign Currency Translation
The functional currency of the Company’s foreign subsidiary is the Euro. Assets and liabilities of the foreign subsidiary are translated using the exchange rate existing on each respective balance sheet date. Revenues and expenses are translated using the average exchange rates for the period. The translation adjustments resulting from this process are included in stockholders’ equity as a component of accumulated other comprehensive loss which resulted in a gain (loss) from foreign currency translation of $2.5 million, ($0.7) million, and ($2.2) million for the years ended December 31, 2017, 2016, and 2015, respectively.
The Company recognized a gain (loss) from foreign currency transactions of $0.7 million, ($0.3) million, and ($0.4) million during the years ended December 31, 2017, 2016, and 2015, respectively.
Fair Value Measurements
Fair value is defined as the price that would be received from selling an asset, or paid to transfer a liability, in an orderly transaction between market participants at the measurement date. When determining the fair value measurements for assets and liabilities required to be recorded at fair value, the Company considers the principal or most advantageous market in which it would transact and considers assumptions that market participants would use when pricing the asset or liability, such as inherent risk, transfer restrictions, and risk of non-performance. The accounting standard establishes a fair value hierarchy that requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value.
| 51 |
A financial instrument’s categorization within the fair value hierarchy is based upon the lowest level of input that is significant to the fair value measurement. Three levels of inputs that may be used to measure fair value are:
| • | Level 1 – Valuation is based upon quoted prices for identical instruments traded in active markets. Level 1 instruments include securities traded on active exchange markets, such as the New York Stock Exchange. |
| • | Level 2 – Valuation is based upon quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active and model-based valuation techniques for which all significant assumptions are directly observable in the market. |
| • | Level 3 – Valuation is generated from model-based techniques that use significant assumptions not observable in the market. These unobservable assumptions reflect the Company’s own estimates of assumptions market participants would use in pricing the instrument. |
The Company’s financial assets have been classified as Levels 1 and 2. The Company’s financial assets (which include cash equivalents and investments) have been initially valued at the transaction price and subsequently valued, at the end of each reporting period, utilizing third party pricing services or other market observable data.
Allowance for Doubtful Accounts
The Company maintains an allowance for doubtful accounts for estimated losses resulting from the inability of its customers to make required payments, which is included in selling, general and administrative expenses in the accompanying consolidated statements of operations. In determining the adequacy of the allowance for doubtful accounts, management specifically analyzes individual accounts receivable, historical bad debts, customer concentrations, customer credit-worthiness, current economic conditions, accounts receivable aging trends, and changes in the Company’s customer payment terms. A summary of activity in the allowance for doubtful accounts is as follows:
| December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Balance, beginning of the year | $ | 194 | $ | 167 | $ | 147 | ||||||
| Amounts provided | 1,609 | 52 | 38 | |||||||||
| Amounts written off | (6 | ) | (16 | ) | (3 | ) | ||||||
| Translation adjustments | 117 | (9 | ) | (15 | ) | |||||||
| Balance, end of the year | $ | 1,914 | $ | 194 | $ | 167 | ||||||
Revenue Recognition - General
The Company recognizes revenue when all of the following criteria are met: persuasive evidence of an arrangement exists, risk of loss has passed or services have been rendered, the seller's price to the buyer is fixed or determinable, and collection from the customer is reasonably assured.
Product Revenue
Revenues from product sales are recognized when title and risk of loss have passed to the customer, which is typically upon shipment to the customer. Amounts billed or collected prior to recognition of revenue are classified as deferred revenue. When determining whether risk of loss has transferred to customers on product sales, or if the sales price is fixed or determinable, the Company evaluates both the contractual terms and conditions of its distribution and supply agreements as well as its business practices.
| 52 |
Product revenue also includes royalties. Royalty revenue is based on distributors’ sales and is recognized in the same period distributors record their sale of products manufactured by the Company. On a quarterly basis the Company records royalty revenue based upon sales provided to it by its distributor customers.
Pursuant to the Health Care and Education Reconciliation Act of 2010, in conjunction with the Patient Protection and Affordable Care Act, a medical device excise tax (“MDET”) became effective on January 1, 2013 for sales of certain medical devices. Some of the Company’s product sales are subject to the provisions of the MDET. The Company elected to recognize any amounts related to the MDET under the gross method as allowed under ASC 605-45. Amounts included in revenues and costs of goods sold for the MDET in 2015 were immaterial. There were no amounts reported for 2016 and 2017 as the 2.3% MDET has been suspended by Congress from January 1, 2016 through 2020.
Licensing, Milestone and Contract Revenue
Licensing, milestone and contract revenue consists of revenue recognized on initial and milestone payments, as well as contractual amounts received from partners. The Company’s business strategy includes entering into collaborative license, development, and/or supply agreements with partners for the development and commercialization of the Company’s products. Under the milestone method, the Company recognizes a consideration that is contingent upon the achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone is substantive in its entirety. A milestone is considered substantive when it meets all of the following criteria:
| 1. | The consideration is commensurate with either the entity’s performance to achieve the milestone or the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity’s performance to achieve the milestone; |
| 2. | The consideration relates solely to past performance; and |
| 3. | The consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. |
A milestone is defined as an event (i) that can only be achieved based in whole or in part on either the entity’s performance or on the occurrence of a specific outcome resulting from the entity’s performance, (ii) for which there is substantive uncertainty at the date the arrangement is entered into that the event will be achieved, and (iii) that would result in additional payments being due to the Company. Non substantive milestones are recognized when there are no further obligations by the Company.
The terms of the agreements typically include non-refundable license fees, funding of research and development, and payments based upon achievement of certain milestones. The Company adopted ASU 2009-13, Revenue Recognition in January 2011, which amended ASC Subtopic 605-25, Multiple Element Arrangements (“ASC 605-25”) to require the establishment of a selling price hierarchy for determining the allocable selling price of an item. Under ASC 605-25, as amended by ASU 2009-13, in order to account for an element as a separate unit of accounting, the element must have objective and reliable evidence of selling price of the undelivered elements. In general, non-refundable up-front fees and milestone payments that do not relate to other elements are recognized as revenue over the term of the arrangement as the Company completes its performance obligations.
Cash and Cash Equivalents
The Company considers only those investments which are highly liquid, readily convertible to cash, and that mature within three months from date of purchase to be cash equivalents. The Company’s cash equivalents consist of money market funds, mutual funds, and bank certificates of deposit with an original maturity of less than 90 days.
| 53 |
Investments
The Company’s investments consist of bank certificates of deposit with an original maturity of more than 90 days. The Company has designated all investments as available-for-sale, and therefore such investments are reported at fair value, with unrealized gains and losses recorded in accumulated other comprehensive income (loss). For securities sold prior to maturity, the cost of securities sold is based on the specific identification method. Realized gains and losses on the sale of investments are recorded in interest income, net. Interest is recorded when earned. Investments with original maturities greater than approximately three months and remaining maturities less than one year are classified as short-term investments. Investments with remaining maturities greater than one year are classified as long-term investments. The Company considers securities with maturities of three months or less from the purchase date to be cash equivalents.
All of the Company’s investments are subject to a periodic impairment review. The Company recognizes an impairment charge when a decline in the fair value of its investments below the cost basis is judged to be other-than-temporary. Factors considered in determining whether a loss is temporary include the extent and length of time the investment's fair value has been lower than its cost basis, the financial condition and near-term prospects of the investee, extent of the loss related to credit of the issuer, the expected cash flows from the security, the Company’s intent to sell the security, and whether or not the Company will be required to sell the security prior the expected recovery of the investment's amortized cost basis. During the years ended December 31, 2017 and 2016, the Company did not record any other-than-temporary impairment charges on its available-for-sale securities because the Company does not intend to sell the securities and it is not more likely than not that the Company will be required to sell these securities before the recovery of their cost basis.
Concentration of Credit Risk and Significant Customers
The Company has no significant off-balance sheet risks related to foreign exchange contracts, option contracts, or other foreign hedging arrangements. The Company’s cash equivalents and investments are held with two major international financial institutions.
The Company, by policy, routinely assesses the financial strength of its customers. As a result, the Company believes that its accounts receivable credit risk exposure is limited.
As of December 31, 2017 and 2016, DePuy Synthes Mitek Sports Medicine, a division of DePuy Orthopaedics, Inc. (“Mitek”), represented 68% and 66%, respectively, of the Company’s accounts receivable balance, no other single customer accounted for more than 10% of accounts receivable in either period.
Inventories
Inventories are stated at the lower of standard cost and net realizable value, with approximate cost determined using the first-in, first-out method. Work-in-process and finished goods inventories include materials, labor, and manufacturing overhead. Inventory costs associated with product candidates that have not yet received regulatory approval are capitalized if the Company believes there is probable future commercial use and future economic benefit.
The Company’s policy is to write-down inventory when conditions exist that suggest inventory may be in excess of anticipated demand or is obsolete based upon assumptions about future demand for the Company’s products and market conditions. The Company regularly evaluates the ability to realize the value of inventory based on a combination of factors including, but not limited to, historical usage rates, forecasted sales or usage, product end of life dates, and estimated current or future market values. Purchasing requirements and alternative usage avenues are explored within these processes to mitigate inventory exposure.
When recorded, inventory write-downs are intended to reduce the carrying value of inventory to its net realizable value. Inventory of $22.0 million and $16.0 million as of December 31, 2017 and 2016, respectively, is stated net of inventory reserves of approximately $1.7 million and $0.9 million, respectively. If actual demand for the Company’s products deteriorates, or if market conditions are less favorable than those projected, additional inventory write-downs may be required.
| 54 |
Property and Equipment
Property and equipment are recorded at cost and depreciated using the straight-line method over their estimated useful lives, which are typically:
| Asset | Estimated useful life (in years) | |||
| Computer equipment and software | 3 - 5 years | |||
| Furniture and fixtures | 5 - 7 years | |||
| Equipment | 5-15 years | |||
| Leasehold improvements | Shorter of useful life or term of lease | |||
Maintenance and repairs are charged to expense when incurred; additions and improvements are capitalized. Fully depreciated assets are retained in the accounts until they are no longer used and no further charge for depreciation is made in respect of these assets. When an item is sold, retired or removed from service, the cost and related accumulated depreciation is relieved, and the resulting gain or loss, if any, is recognized in income.
Construction-in-process is stated at cost, which includes the cost of construction and other direct costs attributable to the construction. Construction-in-process is not depreciated until such time as the relevant assets are completed and put into use.
Goodwill and Acquired Intangible Assets
Goodwill is the amount by which the purchase price of acquired net assets in a business combination exceeded the fair values of net identifiable assets on the date of acquisition. Acquired IPR&D represents the fair value assigned to research and development assets that the Company acquires that have not been completed at the date of acquisition or are pending regulatory approval in certain jurisdictions. The value assigned to the acquired IPR&D is determined by estimating the costs to develop the acquired technology into commercially viable products, estimating the resulting revenue from the projects, and discounting the net cash flows to present value.
Goodwill and IPR&D are evaluated for impairment annually or more frequently if events or changes in circumstances indicate that the asset might be impaired. Factors the Company considers important, on an overall company basis, that could trigger an impairment review include significant underperformance relative to historical or projected future operating results, significant changes in the Company’s use of the acquired assets or the strategy for its overall business, significant negative industry or economic trends, a significant decline in the Company’s stock price for a sustained period, or a reduction of its market capitalization relative to net book value.
To conduct impairment tests of goodwill, the fair value of the reporting unit is compared to its carrying value. If the reporting unit’s carrying value exceeds its fair value, the Company records an impairment loss to the extent that the carrying value of goodwill exceeds its implied fair value. The Company’s annual assessment for impairment of goodwill as of November 30, 2017 indicated that the fair value of its reporting unit exceeded the carrying value of the reporting unit.
To conduct impairment tests of IPR&D, the fair value of the IPR&D project is compared to its carrying value. If the carrying value exceeds its fair value, the Company records an impairment loss to the extent that the carrying value of the IPR&D project exceeds its fair value. The Company estimates the fair value for IPR&D projects using discounted cash flow valuation models, which require the use of significant estimates and assumptions, including but not limited to, estimating the timing of and expected costs to complete the in-process projects, projecting regulatory approvals, estimating future cash flows from product sales resulting from completed projects and in-process projects, and developing appropriate discount rates. During the fourth quarter of 2015, the Company performed an impairment review of its IPR&D projects as it reassessed its research and development strategy. In 2015, the Company recorded an impairment charge of $0.7 million due to the decision to discontinue further development efforts needed to commercialize the Hemostatic Patch in-process development project. The Company’s annual assessment for impairment of IPR&D indicated that the fair value of its other IPR&D assets as of November 30, 2017 and 2016 exceeded their respective carrying values.
| 55 |
Long-Lived Assets
Long-lived assets primarily include property and equipment and intangible assets with finite lives. The Company’s intangible assets are comprised of purchased developed technologies, patents, and trade names. These intangible assets are carried at cost, net of accumulated amortization. Amortization is recorded on a straight-line basis over the intangible assets' useful lives, which range from approximately five to sixteen years. The Company reviews long-lived assets for impairment when events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable or that the useful lives of those assets are no longer appropriate. Each impairment test is based on a comparison of the undiscounted cash flows to the recorded value of the asset. If impairment is indicated, the asset is written down to its estimated fair value based on a discounted cash flow analysis.
Research and Development
Research and development costs consist primarily of clinical trials, salaries and related expenses for personnel, and fees paid to outside consultants and outside service providers, including costs associated with licensing, milestone and contract revenue. Research and development costs are expensed as incurred.
Stock-Based Compensation
The Company has stock-based compensation plans under which it grants various types of equity-based awards, including restricted stock units (“RSUs”), restricted stock awards (“RSAs”), performance options, and stock options. The Company measures the compensation cost of award recipients’ services received in exchange for an award of equity instruments based on the grant date fair value of the underlying award. That cost is recognized over the period during which an employee is required to provide service in exchange for the award.
For performance-based options with financial and business milestone achievement targets, the Company recognizes expense using the graded vesting methodology over the service period. Compensation cost associated with performance-based options is based on the probable outcome of the performance conditions. Changes to the probability assessment and the estimated shares expected to vest will result in adjustments to the related stock-based compensation expense that will be recorded in the period of the change. If the performance targets are not achieved, no compensation cost is recognized, and any previously recognized compensation cost is reversed. The Company recorded $0.8 million, $0.3 million, and $0.4 million related to performance-based options in 2017, 2016, and 2015, respectively.
See Note 12, Equity Incentive Plan, to the consolidated financial statements included elsewhere in this Annual Report on Form 10-K for a description of the types of stock-based awards granted, the compensation expense related to such awards, and detail of equity-based awards outstanding.
Income Taxes
The Company’s income tax expense includes U.S. and international income taxes. Certain items of income and expense are not reported in tax returns and financial statements in the same year. The tax effects of these timing differences are reported as deferred tax assets and liabilities. Deferred tax assets are recognized for the estimated future tax effects of deductible temporary differences, tax operating losses, and tax credit carry-forwards (including investment tax credits). Changes in deferred tax assets and liabilities are recorded in the provision for income taxes. The Company assesses the likelihood that its deferred tax assets will be recovered from future taxable income and, to the extent it believes that it is more likely than not that all or a portion of deferred tax assets will not be realized, the Company establishes a valuation allowance to reduce the deferred tax assets to the appropriate valuation. To the extent the Company establishes a valuation allowance or increases or decreases this allowance in a given period, it includes the related tax expense or tax benefit within the tax provision in the consolidated statement of operations in that period.
Comprehensive Income
Comprehensive income consists of net income and other comprehensive income (loss), which includes foreign currency translation adjustments. For the purposes of comprehensive income disclosures, the Company does not record tax provisions or benefits for the net changes in the foreign currency translation adjustment, as it intends to indefinitely reinvest undistributed earnings of its foreign subsidiary. Accumulated other comprehensive loss is reported as a component of stockholders' equity.
| 56 |
Segment Information
Operating segments are components of an enterprise about which separate financial information is available that is evaluated regularly by the chief operating decision maker, or decision-making group, in deciding how to allocate resources and in assessing performance. The Company’s chief operating decision maker is its Chief Executive Officer. Based on the criteria established by ASC 280, Segment Reporting, the Company has one operating and reportable segment.
Contingencies
In the normal course of business, the Company is involved from time-to-time in various legal proceedings and other matters such as contractual disputes, which are complex in nature and have outcomes that are difficult to predict. The Company records accruals for loss contingencies to the extent that it concludes that it is probable that a liability has been incurred and the amount of the related loss can be reasonably estimated. The Company considers all relevant factors when making assessments regarding these contingencies. Although the outcomes of any potential legal proceedings are inherently difficult to predict, the Company does not expect the resolution of any potential legal proceedings to have a material adverse effect on its financial position, results of operations, or cash flow.
Subsequent Events
Events occurring subsequent to December 31, 2017 have been evaluated for potential recognition or disclosure in the consolidated financial statements. As a result of the evaluation, no subsequent events were required to be recognized or disclosed.
Recent Accounting Pronouncements
Recently Issued
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2014-09, Revenue from Contracts with Customers. ASU 2014-09 supersedes the revenue recognition requirements in “Topic 605, Revenue Recognition” and requires entities to recognize revenue in a way that depicts the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. In July 2015, the FASB issued a one-year deferral making it effective for annual reporting periods by public business entities beginning on or after December 15, 2017 while also providing for early adoption not to occur before the original effective date. The Company adopted the new standard on a modified retrospective basis on January 1, 2018.
The Company developed an implementation plan to assess the impact of the new guidance on its operations, financial results, and related disclosures. To date, the Company has substantially completed its assessment of the potential areas of the balance sheet and financial statement components impacted. The Company has prepared its accounting policy memorandum and assessment of the quantitative impact of adoption, including the impact of the new guidance on its results of operations and internal controls. Based on procedures performed to date, the Company has concluded that the adoption of the new standard will not have a material impact on its annual revenues. The Company does not anticipate a material adjustment to beginning retained earnings as of the adoption on January 1, 2018.
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842). ASU 2016-02 amends existing leasing accounting requirements. The most significant change will result in the recognition of lease assets and lease liabilities by lessees for virtually all leases. The new guidance will also require significant additional disclosures about the amount, timing, and uncertainty of cash flows from leases. ASU 2016-02 is effective for fiscal years and interim periods beginning after December 15, 2018. Upon adoption, entities are required to recognize and measure leases at the beginning of the earliest period presented using a modified retrospective approach. Early adoption is permitted, and a number of optional practical expedients may be elected to simplify the impact of adoption. The Company is assessing ASU 2016-02 and the impact that adopting this new accounting standard will have on its consolidated financial statements and footnote disclosures.
| 57 |
In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments (Topic 326) Credit Losses. ASU 2016-13 changes the impairment model for most financial assets and certain other instruments. Under the new standard, entities holding financial assets and net investment in leases that are not accounted for at fair value through net income are to be presented at the net amount expected to be collected. An allowance for credit losses will be a valuation account that will be deducted from the amortized cost basis of the financial asset to present the net carrying value at the amount expected to be collected on the financial asset. ASU 2016-13 is effective as of January 1, 2020. Early adoption is permitted. The adoption of this standard is not expected to have a material impact on the Company’s consolidated financial statements or footnote disclosures.
Recently Adopted
In March 2016, the FASB issued ASU No. 2016-09, Compensation (Topic 718) Stock Compensation. The ASU identifies areas for simplification involving several aspects of accounting for share-based payment transactions, including the income tax consequences, classification of awards as equity or liabilities, an option to recognize gross stock compensation expense with actual forfeitures recognized as they occur, and certain classifications on the statement of cash flows. ASU 2016-09 is effective as of January 1, 2017. Since January 1, 2017, the Company has recognized excess tax benefits and tax deficiencies related to share-based payments in the Consolidated Statements of Operations and Comprehensive Income as a component of the provision for income taxes on a prospective basis. Such excess tax benefits and tax deficiencies were previously recorded in equity. The Company also began presenting tax-related cash flows resulting from share-based payments as operating activities in the Consolidated Statements of Cash Flows and retrospectively revised prior periods to reflect this provision. Accordingly, the Consolidated Statement of Cash Flows for the years ended December 31, 2016 and 2015, was revised by increasing net cash provided by operating activities by $0.6 million and $0.8 million and by decreasing net cash used in financing activities by $0.6 million and $0.8 million, respectively. Lastly, as of January 1, 2017, the Company elected to recognize forfeitures as they occur rather than estimate forfeitures each period on a modified retrospective basis. Accordingly, the Company recognized a cumulative $0.5 million reduction to retained earnings at the beginning of 2017. Previously, the Company used historical data on the exercise of stock options and other factors to evaluate and estimate the expected term of share-based awards to evaluate actual forfeiture rates periodically and adjusted the expected forfeiture rate assumption within the model. See Note 16, Income Taxes, to the consolidated financial statements included elsewhere in this Annual Report on Form 10-K for additional information regarding the impacts on the consolidated financial statements.
3. Investments
All of the Company’s investments are classified as available-for-sale and are carried at fair value with unrealized gains and losses recorded as a component of accumulated other comprehensive income, net of related income taxes. The Company held bank certificates of deposits of $24.0 million and $20.5 million at December 31, 2017 and 2016, respectively. There were no unrealized gains or losses on the Company’s available-for-sale securities at December 31, 2017 or 2016.
4. Fair Value Measurements
The Company’s investments are all classified within Levels 1 and 2 of the fair value hierarchy. The Company’s investments classified within Level 1 of the fair value hierarchy are valued based quoted prices in active markets. Level 2 are based on matrix pricing compiled by third party pricing vendors, using observable market inputs such as interest rates, yield curves, and credit risk. For cash and cash equivalents, current receivables, accounts payable, interest accrual and short-term debts, the carrying amounts approximate fair value, because of the short maturity of these instruments, and therefore fair value information is not included in the table below.
| 58 |
The fair value hierarchy of the Company’s cash equivalents and investments at fair value is as follows:
| Fair Value Measurements at Reporting Date Using | ||||||||||||||||
| December 31, 2017 | Quoted Prices in Active Markets for Identical Assets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | |||||||||||||
| Cash equivalents: | ||||||||||||||||
| Money market fund | $ | 5,893 | $ | 5,893 | $ | - | $ | - | ||||||||
| Bank certificates of deposit | 500 | - | 500 | - | ||||||||||||
| Total cash equivalents | $ | 6,393 | $ | 5,893 | $ | 500 | $ | - | ||||||||
| Investments: | ||||||||||||||||
| Bank certificates of deposit | $ | 24,000 | $ | - | $ | 24,000 | $ | - | ||||||||
| Fair Value Measurements at Reporting Date Using | ||||||||||||||||
| December 31, 2016 | Quoted Prices in Active Markets for Identical Assets (Level 1) | Significant Other Observable Inputs (Level 2) | (Level 3) | |||||||||||||
| Cash equivalents: | ||||||||||||||||
| Money market funds | $ | 68,352 | $ | - | $ | 68,352 | $ | - | ||||||||
| Bank certificates of deposit | 750 | - | 750 | - | ||||||||||||
| Total cash equivalents | $ | 69,102 | $ | - | $ | 69,102 | $ | - | ||||||||
| Investments: | ||||||||||||||||
| Bank certificates of deposit | 20,500 | - | 20,500 | - | ||||||||||||
| Total investments | $ | 20,500 | $ | - | $ | 20,500 | $ | - | ||||||||
The Company did not have transfers in or out of Level 3 of the fair value hierarchy during the years ended December 31, 2017 and 2016. As of December 31, 2017, the Company’s exchange traded money market fund is reported as a Level 1 cash equivalent, previously it was reported as Level 2 cash equivalent.
5. Earnings per Share (“EPS”)
Basic EPS is calculated by dividing net income by the weighted average number of shares outstanding during the period. Unvested RSA’s, although legally issued and outstanding, are not considered outstanding for purposes of calculating basic earnings per share. Diluted EPS is calculated by dividing net income by the weighted average number of shares outstanding plus the dilutive effect, if any, of outstanding stock options, stock appreciation rights (“SAR’s”), RSA’s, and RSU’s using the treasury stock method.
The following table provides share information used in the calculation of the Company's basic and diluted earnings per share:
| Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Shares used in the calculation of basic earnings per share | 14,575 | 14,682 | 14,934 | |||||||||
| Effect of dilutive securities: | ||||||||||||
| Stock options, SAR's, RSA's and RSU's | 493 | 434 | 387 | |||||||||
| Diluted shares used in the calculation of earnings per share | 15,068 | 15,116 | 15,321 | |||||||||
Stock options to purchase 0.5 million shares, 0.4 million shares, and 0.2 million shares for the years ended December 31, 2017, 2016, and 2015, respectively, were excluded from the computation of diluted EPS as their effect would have been anti-dilutive. The anti-dilutive restricted shares for the years 2017, 2016 and 2015 were not significant.
| 59 |
At December 31, 2017, 2016, and 2015, 0.1 million shares of issued and outstanding unvested RSA’s were excluded from the basic earnings per share.
On February 26, 2016, the Company entered into an accelerated stock repurchase agreement with Morgan Stanley & Co. LLC (“Morgan Stanley”) pursuant to a Fixed Dollar Accelerated Share Repurchase Transaction (“ASR Agreement") to purchase $25.0 million of shares of its common stock. Pursuant to the terms of the ASR Agreement, the Company paid Morgan Stanley $25.0 million in cash and received an initial delivery of 0.4 million shares of the Company’s common stock on February 29, 2016 based on a closing market price of $46.40 per share and the applicable contractual discount.
On August 26, 2016, the Company settled the approximately $7.5 million remaining under the ASR Agreement, which was recorded as an equity forward sale contract and was included in additional paid-in capital in stockholders' equity in the consolidated balance sheet as it met the criteria for equity accounting. Pursuant to the terms of the ASR Agreement, the final number of shares and the average purchase price was determined at the end of the applicable purchase period, which was August 26, 2016. Based on the volume-weighted average price since the effective date of the ASR Agreement less the applicable contractual discount, Morgan Stanley delivered 0.1 million additional shares to the Company on August 31, 2016. In total, 0.5 million shares were repurchased under the ASR Agreement at an average repurchase price of $47.08 per share. These shares are held by the Company as authorized but unissued shares pursuant to Massachusetts law. The initial and final delivery of shares resulted in immediate reductions of the outstanding shares used to calculate the weighted-average common shares outstanding for basic and diluted net income per share.
6. Inventories
Inventories consist of the following:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Raw materials | $ | 11,296 | $ | 5,884 | ||||
| Work-in-process | 6,062 | 5,559 | ||||||
| Finished goods | 4,677 | 4,540 | ||||||
| Total | $ | 22,035 | $ | 15,983 | ||||
7. Property and Equipment
Property and equipment is stated at cost and consists of the following:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Equipment and software | $ | 37,137 | $ | 27,456 | ||||
| Furniture and fixtures | 1,947 | 1,126 | ||||||
| Leasehold improvements | 31,459 | 27,796 | ||||||
| Construction in progress | 5,830 | 22,695 | ||||||
| Subtotal | 76,373 | 79,073 | ||||||
| Less accumulated depreciation | (20,190 | ) | (26,777 | ) | ||||
| Total | $ | 56,183 | $ | 52,296 | ||||
Construction-in-progress at December 31, 2017, primarily represents the costs incurred for the implementation of a new ERP that was placed in service in January 2018. Construction-in-progress at December 31, 2016 primarily represented the costs being incurred in our strategic project that we began in 2015 to insource the manufacturing of our HYAFF-based products to our Bedford, Massachusetts facility, which was placed in service in December 2017. In 2017 we retired $9.8 million of fully depreciated assets that were no longer in use.
| 60 |
Depreciation expense was $3.3 million, $2.7 million, and $2.7 million for the years ended December 31, 2017, 2016, and 2015, respectively.
8. Acquired Intangible Assets, Net
Intangible assets consist of the following:
| December 31, 2017 | December 31, 2016 | |||||||||||||||||||||||||||||||
| Gross Value | Accumulated Currency Translation Adjustment | Accumulated Amortization | Net Book Value | Accumulated Currency Translation Adjustment | Accumulated Amortization | Net Book Value | Useful Life | |||||||||||||||||||||||||
| Developed technology | $ | 17,100 | $ | (2,550 | ) | $ | (7,723 | ) | $ | 6,827 | $ | (3,442 | ) | $ | (6,816 | ) | $ | 6,842 | 15 | |||||||||||||
| In-process research & development | 4,406 | (1,015 | ) | 3,391 | (1,433 | ) | - | 2,973 | Indefinite | |||||||||||||||||||||||
| Distributor relationships | 4,700 | (415 | ) | (4,285 | ) | - | (415 | ) | (4,285 | ) | - | 5 | ||||||||||||||||||||
| Patents | 1,000 | (152 | ) | (431 | ) | 417 | (207 | ) | (381 | ) | 412 | 16 | ||||||||||||||||||||
| Elevess trade name | 1,000 | - | (1,000 | ) | - | - | (1,000 | ) | - | 9 | ||||||||||||||||||||||
| Total | $ | 28,206 | $ | (4,132 | ) | $ | (13,439 | ) | $ | 10,635 | $ | (5,497 | ) | $ | (12,482 | ) | $ | 10,227 | ||||||||||||||
On December 30, 2009, in connection with the acquisition of Anika S.r.l., the Company purchased various intangible assets.
In 2015, the Company recorded an impairment charge totaling $0.7 million to write-off in-process research and development that was recorded in connection with its acquisition of Anika S.r.l. Subsequent to an evaluation in the fourth quarter of the ongoing research and development efforts surrounding the Hemostatic Patch IPR&D project, the Company determined it would discontinue further development efforts needed to commercialize this technology. As a result of this decision, an impairment charge was recorded. These amounts are included in research and development expenses on the Company’s consolidated statements of operations.
The Company performed an annual assessment of IPR&D intangible assets as of November 30, 2017. Based upon that assessment, for the fiscal year 2017 there were no events or changes in circumstances that would result in a change in the carrying value of IPR&D.
Total amortization expense was $1.0 million, $1.1 million, and $1.1 million for the years ended December 31, 2017, 2016, and 2015, respectively. Amortization expense on intangible assets is expected to be approximately $1.0 million in 2018, $1.0 million annually through 2021, and approximately $3.1 million in aggregate thereafter.
9. Goodwill
The Company completed its annual impairment review as of November 30, 2017 and concluded that no impairment in the carrying value exists as of that date with respect to goodwill. Through December 31, 2017, there have not been any events or changes in circumstances that indicate that the carrying value of goodwill may not be recoverable. Changes in the carrying value of goodwill were as follows:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Balance, beginning | $ | 7,214 | $ | 7,482 | ||||
| Effect of foreign currency adjustments | 1,004 | (268 | ) | |||||
| Balance, ending | $ | 8,218 | $ | 7,214 | ||||
| 61 |
10. Accrued Expenses
Accrued expenses consist of the following:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Compensation and related expenses | $ | 2,893 | $ | 3,089 | ||||
| Facility construction costs | - | 804 | ||||||
| Research grants | 419 | 463 | ||||||
| Clinical trial costs | 2,318 | 227 | ||||||
| Professional fees | 448 | 802 | ||||||
| Deferred rent | - | 231 | ||||||
| Other | 248 | 880 | ||||||
| Total | $ | 6,326 | $ | 6,496 | ||||
11. Commitments and Contingencies
Leasing Arrangements
On October 9, 2015, our Italian subsidiary, Anika Therapeutics S.r.l. (“Anika S.r.l.”) entered into a build-to-suit lease agreement with Consorzio Zona Industriale E Porto Fluviale di Padova (“ZIP”) as landlord, pursuant to which Anika S.r.l. leases a new European headquarters facility, consisting of approximately 33,000 square feet of general office, research and development, training, and warehousing space located in Padova, Italy. The lease has an initial term of fifteen years, which commenced on March 1, 2017. The lease will automatically renew for up to three additional six-year terms, subject to certain terms and conditions. The Company has the ability to withdraw from this lease subject to certain financial penalties after six years and with no penalties after the ninth year. Beginning on the commencement date, the lease provides for an initial yearly rent of approximately $0.3 million.
Construction of the new facility commenced during the first quarter of 2016. During the period of construction, the Company was the deemed owner of the facility. Accordingly, the landlord's costs of constructing the facility were capitalized, as a non-cash transaction, offset by a corresponding facility lease obligation in the Company’s consolidated balance sheet. When the construction concluded on March 1, 2017, the Company removed the construction-in-process asset of $3.1 million and related liability from its consolidated balance sheet. The Company commissioned ZIP for additional tenant improvements of $0.8 million, which are recorded within Other long-term assets. The lease is accounted for as an operating lease based on the Company’s assessment of the applicable accounting principles.
Prior to April 2017, Anika S.r.l. leased approximately 28,000 square feet of laboratory, warehouse, and office space in Abano Terme, Italy that served as headquarters for Anika S.r.l. On December 29, 2016 Anika S.r.l. notified the landlord of its intention to terminate the lease agreement as of March 31, 2017.
Rental expense in connection with the various facility leases totaled $1.8 million, $1.3 million, and $1.3 million for the years ended December 31, 2017, 2016, and 2015, respectively. The increased expense in 2017 is primarily a result of finalizing the exercise of our first option under the lease to extend the terms from November 1, 2017 through October 31, 2022, including the determination of a new annual base rent for the Company’s headquarters facility in Bedford, Massachusetts.
| 62 |
The Company’s future lease commitments as of December 31, 2017 are as follows:
| 2018 | $ | 1,879 | ||
| 2019 | 1,880 | |||
| 2020 | 1,916 | |||
| 2021 | 1,924 | |||
| 2022 | 1,673 | |||
| 2023 and thereafter | 1,311 | |||
| Total | $ | 10,583 |
Warranty and Guarantor Arrangements
In certain of its contracts, the Company warrants to its customers that the products it manufactures conform to the product specifications as in effect at the time of delivery of the specific product. The Company may also warrant that the products it manufactures do not infringe, violate or breach any U.S. patent or intellectual property rights, trade secret, or other proprietary information of any third party. On occasion, the Company contractually indemnifies its customers against any and all losses arising out of, or in any way connected with, any claim or claims of breach of its warranties or any actual or alleged defect in any product caused by the negligence or acts or omissions of the Company. The Company maintains a products liability insurance policy that limits its exposure to these risks. Based on the Company’s historical activity, in combination with its liability insurance coverage, the Company believes the estimated fair value of these indemnification agreements is immaterial. The Company has no accrued warranties at December 31, 2017 or 2016, respectively, and has no history of claims paid.
Legal Proceedings
The Company is involved from time-to-time in various legal proceedings arising in the normal course of business. Although the outcomes of potential legal proceedings are inherently difficult to predict, the Company does not expect the resolution of potential legal proceedings to have a material adverse effect on its financial position, results of operations, or cash flow.
12. Equity Incentive Plan
Equity Incentive Plan
The Anika Therapeutics, Inc. Stock Option and Incentive Plan, as amended, (the “2003 Plan”) provides for grants of nonqualified and incentive stock options, common stock, RSA’s, RSU’s, and SAR’s to employees, directors, officers, and consultants. The 2003 Plan was originally approved by the Board of Directors on April 4, 2003, approved by the Company’s shareholders on June 4, 2003, and reserved 1,500,000 shares of common stock for grant pursuant to its terms.
On May 29, 2009, the Board of Directors approved changes to the 2003 Plan and adopted the Amended and Restated 2003 Stock Option and Incentive Plan (the “Amended 2003 Plan”) to increase the number of shares available to grant by 850,000. The Amended 2003 Plan was approved by the Company’s shareholders on June 5, 2009, and it resulted in a total of 2,350,000 shares of common stock being reserved for issuance under the Amended 2003 Plan.
At the 2011 Annual Meeting of Stockholders on June 7, 2011, the shareholders of the Company approved the Anika Therapeutics, Inc. Second Amended and Restated Stock Option and Incentive Plan (the “2003 Plan”), which, among other things, increased the number of shares reserved for issuance under the Company’s predecessor stock option and incentive plan by 800,000 to 3,150,000 shares. Pursuant to this amendment and restatement to the 2003 Plan approved by the Company’s shareholders, each share award issued after June 7, 2011 other than stock options or SAR’s will reduce the number of total shares available for grant by 1.9 shares.
At the 2013 Annual Meeting of Stockholders on June 18, 2013, the shareholders of the Company approved an additional amendment to the Amended 2003 Plan, which, among other things, increased the number of shares reserved for issuance under the Company’s stock option and incentive plan by 650,000 to 3,800,000 shares. Pursuant to this amendment and restatement to the 2003 Plan approved by the Company’s shareholders, each share award issued after June 18, 2013 other than stock options or SAR’s will reduce the number of total shares available for grant by 1.5 shares.
| 63 |
On June 13, 2017, the Company’s shareholders approved the Anika Therapeutics, Inc. 2017 Omnibus Incentive Plan (the “2017 Plan”). The 2017 Plan replaced the 2003 Plan, as the plan under which future grants to employees, directors, officers, and consultants will be made. The 2017 Plan was originally approved by the Company’s Board of Directors on March 31, 2017. The terms of the 2017 Plan provide for the grant of incentive stock options, nonqualified stock options, SAR’s, RSA’s, RSU’s, and performance options that may be settled in cash, stock, or other property. In accordance with the 2017 Plan approved by the Company’s shareholders, each share award other than stock options or SAR’s will reduce the number of total shares available for grant by 2.0 shares. Subject to adjustment for specified types of changes in our capitalization, no more than 1.2 million shares of common stock may be issued under the 2017 Plan. There are 1.1 million shares available for future grant at December 31, 2017.
The Company may satisfy the awards upon exercise, or upon fulfillment of the vesting requirements for other equity-based awards, with either newly-issued shares or shares reacquired by the Company. Stock-based awards are granted with an exercise price equal to the market price of the Company’s stock on the date of grant. Awards contain service conditions or service and performance conditions, and they generally become exercisable ratably over one to four years.
The Company estimates the fair value of stock options and SAR’s using the Black-Scholes valuation model. Fair value of restricted stock is measured by the grant-date price of the Company’s shares. Key input assumptions used to estimate the fair value of stock options and SAR’s include the exercise price of the award, the expected award term, the expected volatility of the Company’s stock over the option’s expected term, the risk-free interest rate over the award’s expected term, and the Company’s expected annual dividend yield.
The expected volatility assumption is evaluated against the historical volatility of the Company’s common stock over a four-year average, and it is adjusted if there are material changes in historical volatility. The risk-free interest rate assumption is based on U.S. Treasury interest rates at the time of grant.
The fair value of each stock option during 2017, 2016, and 2015 was estimated on the grant date using the Black-Scholes option-pricing model with the following assumptions:
| 2017 | 2016 | 2015 | ||||||||||
| Risk free interest rate | 1.60% | - | 1.86% | 0.94% | - | 1.55% | 1.15% | - | 1.46% | |||
| Expected volatility | 38.74% | - | 44.31% | 47.33% | - | 51.61% | 53.15% | - | 54.65% | |||
| Expected life (years) | 4.0 | 4.5 | 4.5 | |||||||||
| Expected dividend yield | 0.00% | 0.00% | 0.00% | |||||||||
Stock Options and Restricted Stock
During the year ended December 31, 2017, a total of 85,109 stock options and 26,306 RSA’s were granted under the 2017 Plan, and a total of 407,635 stock options were granted under the 2003 Plan. The stock options granted to employees become exercisable or vest ratably over a three-year period. In January 2017, the Company executed its annual grant under the 2003 Plan of 9,970 RSU’s to non-employee directors; these RSU’s vest over a one-year period.
| 64 |
The Company recorded $5.8 million, $3.4 million, and $2.2 million of stock-based compensation expense for the years ended December 31, 2017, 2016, and 2015, respectively, for stock options, SAR’s, RSA’s and RSU’s. The Company presents the expenses related to stock-based compensation awards in the same expense line items as cash compensation paid to each of its employees as follows:
| 2017 | 2016 | 2015 | ||||||||||
| Cost of product revenue | $ | 439 | $ | 148 | $ | 42 | ||||||
| Research & development | 564 | 467 | 269 | |||||||||
| Selling, general & administrative | 4,804 | 2,777 | 1,914 | |||||||||
| Total stock-based compensation expense | $ | 5,807 | $ | 3,392 | $ | 2,225 | ||||||
Combined stock options and SAR’s activity under the Company’s plans is summarized as follows for the years ended December 31, 2017 and 2016, respectively:
| 2017 | 2016 | |||||||||||||||
| Weighted | Weighted | |||||||||||||||
| Average | Average | |||||||||||||||
| Exercise | Exercise | |||||||||||||||
| Number of | Price Per | Number of | Price Per | |||||||||||||
| Shares | Share | Shares | Share | |||||||||||||
| Options and SAR's outstanding at beginning of year | 979,569 | $ | 26.15 | 762,260 | $ | 18.75 | ||||||||||
| Granted | 440,688 | $ | 50.22 | 354,275 | $ | 40.77 | ||||||||||
| Cancelled | (74,527 | ) | $ | 45.56 | (58,841 | ) | $ | 30.05 | ||||||||
| Expired | (589 | ) | $ | 32.86 | (3,310 | ) | $ | 11.37 | ||||||||
| Exercised | (17,941 | ) | $ | 20.56 | (74,815 | ) | $ | 15.46 | ||||||||
| Options and SAR's outstanding at end of year | 1,327,200 | $ | 33.70 | 979,569 | $ | 26.15 | ||||||||||
All the 1,327,200 stock options and SAR’s outstanding at December 31, 2017 are vested or are expected to vest, with a weighted-average exercise price of $33.70 as well as an aggregate intrinsic value of $27.6 million related to these awards. The weighted average remaining contractual term of the vested and expected to vest stock options and SAR’s was 7.0 years as of December 31, 2017.
As of December 31, 2017, total unrecognized compensation costs related to non-vested stock options and SAR’s was approximately $7.6 million and is expected to be recognized over a weighted average period of 2.1 years.
The exercisable options and SAR’s at December 31, 2017 are as follows:
| Outstanding | Weighted Average Exercise Price | Weighted Average Remaining Term (in years) | ||||||||||
| Incentive stock options | 182,472 | $ | 14.12 | 4.3 | ||||||||
| Nonqualified stock options | 374,211 | $ | 20.18 | 5.3 | ||||||||
| Performance options | 32,598 | $ | 38.79 | 7.7 | ||||||||
| SAR's | 35,250 | $ | 6.36 | 2.1 | ||||||||
The aggregate intrinsic value of stock options and SAR’s fully vested at December 31, 2017 and 2016 was $22.0 million and $16.7 million, respectively. The aggregate intrinsic value of stock options and SAR’s outstanding at December 31, 2017 and 2016 was $27.6 million and $22.3 million, respectively.
| 65 |
The total intrinsic value of stock options and SAR’s exercised was $0.5 million and $2.1 million for the years ended December 31, 2017 and 2016, respectively.
The total fair value of stock options and SAR’s vested during the years ended December 31, 2017 and 2016 was approximately $2.1 and $1.3 million, respectively.
The Company received $0.3 million and $1.0 million for exercises of stock options during the years ended December 31, 2017 and 2016, respectively.
The RSA and RSU activity for the years ended December 31, 2017 and 2016 is as follows:
| 2017 | 2016 | |||||||||||||||
| Weighted | Weighted | |||||||||||||||
| Average | Average | |||||||||||||||
| Number of | Grant Date | Number of | Grant Date | |||||||||||||
| Shares | Fair Value | Shares | Fair Value | |||||||||||||
| Unvested at Beginning of year | 207,077 | $ | 36.44 | 150,384 | $ | 34.29 | ||||||||||
| Granted | 67,567 | $ | 52.03 | 87,158 | $ | 38.11 | ||||||||||
| Cancelled | - | $ | - | (4,950 | ) | $ | 36.20 | |||||||||
| Expired | - | $ | - | - | $ | - | ||||||||||
| Vested/Released | (45,418 | ) | $ | 35.32 | (25,515 | ) | $ | 33.35 | ||||||||
| Unvested at end of year | 229,226 | $ | 42.47 | 207,077 | $ | 36.44 | ||||||||||
The total fair value of RSA’s and RSU’s vested during the years ended December 31, 2017 and 2016 was $2.3 million and $1.0 million.
13. Employee Benefit Plan
The Company’s U.S. employees are eligible to participate in the Company’s 401(k) savings plan. Employees may elect to contribute a percentage of their compensation to the plan, and the Company will make 140% matching contributions up to a limit of 5% of an employee’s eligible compensation. In addition, the Company may make annual discretionary contributions. The Company made matching contributions of $0.6 million, $0.6 million, and $0.4 million for the years ended December 31, 2017, 2016, and 2015, respectively.
14. Shareholder Rights Plan
On April 4, 2008, the Board of Directors of the Company adopted a Shareholder Rights Plan (the “2008 Plan”) that replaced the Company’s former Shareholder Rights Plan. Under the 2008 Plan, the Rights generally become exercisable if:
(1) A person becomes an “Acquiring Person” by acquiring 15% or more of the Company’s common stock, or
(2) A person commences a tender offer that would result in that person owning 15% or more of the Company’s common stock.
In the event that a person becomes an “Acquiring Person,” each holder of a Right (other than the Acquiring Person) would be entitled to acquire a number of shares of preferred stock equivalent to shares of the Company’s common stock having a value of twice the exercise price of the Right. If, after any such event, the Company enters into a merger or other business combination transaction with another entity, each holder of a Right would then be entitled to purchase, at the then-current exercise price, shares of the acquiring company’s common stock having a value of twice the exercise price of the Right.
The current exercise price per Right is $75.00. The Rights may be redeemed in whole, but not in part, at a price of $0.01 per Right (payable in cash, shares of the Company’s common stock, or other consideration deemed appropriate by the Board of Directors) by the Board of Directors only until the earlier of:
| 66 |
(1) The time at which any person becomes an “Acquiring Person,” or
(2) The Expiration Date.
At any time after any person becomes an “Acquiring Person,” the Board of Directors may, at its option, exchange all or any part of the then outstanding and exercisable Rights for shares of the Company’s common stock at an exchange ratio specified in the 2008 Plan. Notwithstanding the foregoing, the Board of Directors generally will not be empowered to affect such exchange at any time after any person becomes the beneficial owner of 50% or more of the Company’s common stock.
In connection with the establishment of the 2008 Plan, the Board of Directors approved the creation of Preferred Stock of the Company designated as Series B Junior Participating Cumulative Preferred Stock with a par value of $0.01 per share. The Board also reserved 175,000 shares of preferred stock for issuance upon exercise of the Rights. Until a Right is exercised, the holder will have no rights as a stockholder of the Company, beyond those as an existing stockholder, including the right to vote or to receive dividends.
15. Revenue by Product Group, by Significant Customer and by Geographic Location; Geographic Information
Product revenue by product group is as follows:
| Years Ended December 31, | ||||||||||||||||||||||||
| 2017 | 2016 | 2015 | ||||||||||||||||||||||
| Revenue | Percentage of Product Revenue | Revenue | Percentage of Product Revenue | Revenue | Percentage of Product Revenue | |||||||||||||||||||
| Orthobiologics | $ | 93,816 | 87 | % | $ | 89,695 | 87 | % | $ | 73,247 | 84 | % | ||||||||||||
| Dermal | 2,755 | 3 | % | 2,759 | 3 | % | 2,266 | 2 | % | |||||||||||||||
| Surgical | 5,262 | 5 | % | 5,427 | 5 | % | 5,812 | 7 | % | |||||||||||||||
| Other | 5,950 | 5 | % | 5,051 | 5 | % | 6,371 | 7 | % | |||||||||||||||
| $ | 107,783 | 100 | % | $ | 102,932 | 100 | % | $ | 87,696 | 100 | % | |||||||||||||
Product revenue from our sole significant customer, Mitek, as a percentage of our total product revenue was 73%, 75%, and 72% for the years ended December 31, 2017, 2016, and 2015, respectively.
ORTHOVISC became available for sale in the United States on March 1, 2004, and it is marketed exclusively by Mitek under the terms of an initial ten-year licensing, distribution, supply, and marketing agreement entered into in December 2003. The agreement was extended by Mitek for additional five-year terms in 2012 and in 2017, with the current agreement to expire on December 20, 2023.
In December 2011, the Company entered into a fifteen-year licensing agreement with Mitek to exclusively market MONOVISC in the United States. The agreement provides certain milestone payments to the Company when rolling end-user sales of U.S. MONOVISC exceed certain target sales goals. For the years ended December 31, 2017, 2016, and 2015, the Company recognized milestone revenue of $5.0 million, $0.0 million, and $5.0 million, respectively, as a result of MONOVISC achieving end-user sales in 2015 of $50 million within a consecutive 12-month period, and end-user sales in 2017 of $100 million within a consecutive 12-month period. Under the terms of the agreement, there are additional milestone revenue that may be achieved in future years.
| 67 |
Total revenue by geographic location based on the location of the customer in total and as a percentage of total revenue are as follows:
| Years Ended December 31, | ||||||||||||||||||||||||
| 2017 | 2016 | 2015 | ||||||||||||||||||||||
| Total | Percentage of | Total | Percentage of | Total | Percentage of | |||||||||||||||||||
| Revenue | Revenue | Revenue | Revenue | Revenue | Revenue | |||||||||||||||||||
| Geographic Location: | ||||||||||||||||||||||||
| United States | $ | 92,905 | 82 | % | $ | 83,972 | 81 | % | $ | 76,621 | 82 | % | ||||||||||||
| Europe | 12,435 | 11 | % | 10,953 | 11 | % | 8,756 | 9 | % | |||||||||||||||
| Other | 8,080 | 7 | % | 8,454 | 8 | % | 7,622 | 9 | % | |||||||||||||||
| Total | $ | 113,420 | 100 | % | $ | 103,379 | 100 | % | $ | 92,999 | 100 | % | ||||||||||||
The Company recorded licensing, milestone, and contract revenue of $5.6 million, $0.4 million, and $5.3 million for the years ended December 31, 2017, 2016, and 2015, respectively; substantially all was derived in the United States with the exception of 2016 which was derived in the Middle East and Latin America.
Net long-lived assets, consisting of net property and equipment, are subject to geographic risks because they are generally difficult to move and to effectively utilize in another geographic area in a reasonable time period and because they are relatively illiquid. Net tangible long-lived assets by principal geographic areas are as follows:
| Years Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| United States | $ | 52,828 | $ | 49,140 | ||||
| Italy | 3,355 | 3,156 | ||||||
| Total | $ | 56,183 | $ | 52,296 | ||||
16. Income Taxes
New Tax Legislation
On December 22, 2017, the President of the United States signed into law the Tax Cuts and Jobs Act (the “2017 Tax Act”) tax reform legislation. This legislation makes significant changes to the U.S. tax law, including a reduction in the corporate tax rate from the current rate of 35% to 21% starting in 2018. As a result of the enacted law, the Company was required to revalue deferred tax assets and liabilities at the future rate. This revaluation resulted in a $2.3 million income tax benefit in continuing operations and a corresponding reduction in the deferred tax liability. The other provisions of the 2017 Tax Act did not have a material impact on the 2017 consolidated financial statements.
In accordance with Staff Accounting Bulletin No. 118, which provides guidance on accounting for the tax effects of the 2017 Tax Act, the Company has recorded a reasonable estimate of the impact on the consolidated financial statements. The provisional amounts incorporate assumptions made based upon the Company’s current interpretation and implementation guidance of the 2017 Tax Act. The Company does not expect a significant adjustment to the recorded amounts.
| 68 |
Income Tax Expense
The components of the Company’s income before income taxes and its provision for (benefit from) income taxes consist of the following:
| Years ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Income before income taxes | ||||||||||||
| Domestic | $ | 48,446 | $ | 50,181 | $ | 48,608 | ||||||
| Foreign | (2,244 | ) | 689 | (354 | ) | |||||||
| $ | 46,202 | $ | 50,870 | $ | 48,254 | |||||||
| Years ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Provision for (benefit from) income taxes: | ||||||||||||
| Current provision: | ||||||||||||
| Federal | $ | 12,608 | $ | 14,982 | $ | 14,572 | ||||||
| State | 2,737 | 3,265 | 3,635 | |||||||||
| Foreign | 31 | 302 | 249 | |||||||||
| 15,376 | 18,549 | 18,456 | ||||||||||
| Deferred provision: | ||||||||||||
| Federal | (426 | ) | (70 | ) | (370 | ) | ||||||
| State | (68 | ) | (84 | ) | (33 | ) | ||||||
| Foreign | (496 | ) | (72 | ) | (557 | ) | ||||||
| (990 | ) | (226 | ) | (960 | ) | |||||||
| Total provision | $ | 14,386 | $ | 18,323 | $ | 17,496 | ||||||
Deferred Tax Assets and Liabilities
Significant components of the Company’s deferred tax assets and liabilities consist of the following:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carry forward, foreign | $ | 959 | $ | 1,253 | ||||
| Stock-based compensation expense | 2,309 | 1,882 | ||||||
| Foreign currency exchange | 265 | 677 | ||||||
| Accrued expenses and other | 496 | 308 | ||||||
| Inventory reserve | 740 | 640 | ||||||
| Deferred tax assets | $ | 4,769 | $ | 4,760 | ||||
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Deferred tax liabilities: | ||||||||
| Acquisition-related Intangibles | $ | (2,743 | ) | $ | (2,932 | ) | ||
| Depreciation | (7,419 | ) | (8,376 | ) | ||||
| Deferred tax liabilities | $ | (10,162 | ) | $ | (11,308 | ) | ||
| Net deferred tax liabilities | $ | (5,393 | ) | $ | (6,548 | ) | ||
| 69 |
Tax Rate
The reconciliation between the U.S. federal statutory rate and the Company’s effective rate is summarized as follows:
| Years ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Statutory federal income tax rate | 35.0 | % | 35.0 | % | 35.0 | % | ||||||
| State tax expense, net of federal benefit | 4.8 | % | 4.5 | % | 4.8 | % | ||||||
| Impact of rate change on deferred taxes | (4.9 | %) | 0.0 | % | 0.0 | % | ||||||
| Permanent items, including nondeductible expenses | 0.6 | % | 0.5 | % | (0.3 | %) | ||||||
| State investment tax credit | (0.7 | %) | (0.1 | %) | 0.0 | % | ||||||
| Federal, state and foreign research and development credits | (1.4 | %) | (0.9 | %) | (0.4 | %) | ||||||
| Foreign rate differential | 0.5 | % | (0.1 | %) | 0.1 | % | ||||||
| Domestic production deduction | (2.8 | %) | (2.9 | %) | (2.9 | %) | ||||||
| Effective income tax rate | 31.1 | % | 36.0 | % | 36.3 | % | ||||||
As of December 31, 2017, the Company had NOL’s for income tax purposes in Italy of $4.0 million that do not expire.
Accounting for Uncertainty in Income Taxes
The Company had no unrecognized tax benefits for the years ended December 31, 2017 and 2016, respectively. The Company does not anticipate experiencing any significant increases or decreases in its unrecognized tax benefits within the twelve months following December 31, 2017.
In the normal course of business, Anika and its subsidiaries may be periodically examined by various taxing authorities. We file income tax returns in the U.S. federal jurisdiction, in certain U.S. states, and in Italy. The associated tax filings remain subject to examination by applicable tax authorities for a certain length of time following the tax year to which those filings relate. The 2014 through 2016 tax years remain subject to examination by the IRS and other taxing authorities for U.S. federal and state tax purposes. The 2011 through 2016 tax years remain subject to examination by the appropriate governmental authorities for Italy.
Upon the settlement of certain stock-based awards (i.e., exercise, vesting, forfeiture, or cancellation), the actual tax deduction is compared with cumulative financial reporting compensation cost, and any excess tax deduction related to these awards is considered a windfall tax benefit. With the adoption of ASU 2016-09 in 2017, the Company records windfall tax benefits to income tax expense. Prior to the adoption of ASU 2016-09, such benefits were tracked in a “windfall tax benefit pool” as part of additional paid-in capital. The Company follows the with-and-without approach for the direct effects of windfall/shortfall items and to determine the timing of the recognition of any related benefits. The Company recorded a windfall tax benefit in income tax expense of $0.4 million in 2017. The amount of excess tax benefits previously recognized through additional paid-in capital was $0.6 million and $0.9 million in 2016 and 2015, respectively.
17. Revolving Credit Agreement
On October 24, 2017, the Company, as borrower, entered into a new five-year agreement with Bank of America, N.A., as administrative agent, swingline lender and issuer of letters of credit, for a $50.0 million senior revolving line of credit (the “Credit Agreement”). Subject to certain conditions, the Company may request up to an additional $50.0 million in commitments for a maximum aggregate commitment of $100.0 million, which requests must be approved by the Revolving Lenders (as defined in the Credit Agreement). Loans under the Credit Agreement generally bear interest equal to, at the Company’s option, either: (i) LIBOR plus the Applicable Margin, as defined below, or the (ii) Base Rate, defined as the highest of: (a) the Federal Funds Rate plus 0.50%, (b) Bank of America, N.A.’s prime rate and (c) the one month LIBOR adjusted daily plus 1.0%, plus the Applicable Margin. The Applicable Margin ranges from 0.25% to 1.75% based on the Company’s consolidated leverage ratios at the time of the borrowings under the Credit Agreement. The Company has agreed to pay a commitment fee in an amount that is equal to 0.25% per annum on the actual daily unused amount of the credit facility and that is due and payable quarterly in arrears. Loan origination costs are included in Other long-term assets and are being amortized over the five-year term of the Credit Agreement. As of December 31, 2017, there are no outstanding borrowings under the Credit Agreement and the Company is in compliance with the terms of the Credit Agreement.
| 70 |
The Credit Agreement contains customary representations, warranties, affirmative and negative covenants, including financial covenants, events of default and indemnification provisions in favor of the Lenders (as defined in the Credit Agreement). The covenants include restrictions governing the Company’s leverage ratio and interest coverage ratio, its incurrence of liens and indebtedness, and its entry into certain merger and acquisition transactions or dispositions and other matters, all subject to certain exceptions. The financial covenants require the Company not to exceed certain maximum leverage and interest coverage ratios. The Lenders have been granted a first priority lien and security interest in substantially all of the Company’s assets, except for certain intangible assets.
18. Quarterly Financial Data (Unaudited)
| Quarter ended | Quarter ended | Quarter ended | Quarter ended | |||||||||||||
| Year 2017 | December 31 | September 30 | June 30 | March 31 | ||||||||||||
| Product revenue | $ | 28,884 | $ | 27,178 | $ | 28,340 | $ | 23,381 | ||||||||
| Total revenue | 29,388 | 27,184 | 33,462 | 23,386 | ||||||||||||
| Cost of product revenue | 8,716 | 6,250 | 6,315 | 6,083 | ||||||||||||
| Gross profit on product revenue | 20,168 | 20,928 | 22,025 | 17,298 | ||||||||||||
| Net income | $ | 8,067 | $ | 6,887 | $ | 11,369 | $ | 5,493 | ||||||||
| Per common share information: | ||||||||||||||||
| Basic net income per share | $ | 0.55 | $ | 0.47 | $ | 0.78 | $ | 0.38 | ||||||||
| Basic common shares outstanding | 14,596 | 14,579 | 14,588 | 14,576 | ||||||||||||
| Diluted net income per share | $ | 0.53 | $ | 0.46 | $ | 0.76 | $ | 0.37 | ||||||||
| Diluted common shares outstanding | 15,141 | 15,115 | 15,044 | 15,043 | ||||||||||||
| Quarter ended | Quarter ended | Quarter ended | Quarter ended | |||||||||||||
| Year 2016 | December 31 | September 30 | June 30 | March 31 | ||||||||||||
| Product revenue | $ | 28,296 | $ | 25,783 | $ | 26,575 | $ | 22,278 | ||||||||
| Total revenue | 28,726 | 25,789 | 26,581 | 22,283 | ||||||||||||
| Cost of product revenue | 7,539 | 4,998 | 6,065 | 5,425 | ||||||||||||
| Gross profit on product revenue | 20,757 | 20,785 | 20,510 | 16,853 | ||||||||||||
| Net income | $ | 8,085 | $ | 8,952 | $ | 8,615 | $ | 6,895 | ||||||||
| Per common share information: | ||||||||||||||||
| Basic net income per share | $ | 0.56 | $ | 0.61 | $ | 0.59 | $ | 0.46 | ||||||||
| Basic common shares outstanding | 14,538 | 14,625 | 14,679 | 14,875 | ||||||||||||
| Diluted net income per share | $ | 0.54 | $ | 0.59 | $ | 0.57 | $ | 0.45 | ||||||||
| Diluted common shares outstanding | 14,979 | 15,077 | 15,111 | 15,307 | ||||||||||||
| 71 |
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
| (a) | Evaluation of disclosure controls and procedures. |
As required by Rule 13a-15 under the Securities Exchange Act of 1934 (“Exchange Act”), we carried out an evaluation under the supervision and with the participation of our management, including our chief executive officer and chief financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the period covered by this report. Based upon that evaluation, the chief executive officer and chief financial officer have concluded that our disclosure controls and procedures are effective as of December 31, 2017 to ensure that information required to be disclosed by us in reports we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in SEC rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by our company in the reports we file or submit under the Exchange Act is accumulated and communicated to our management, including our chief executive officer and chief financial officer, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure. On an on-going basis, we review and document our disclosure controls and procedures, and our internal control over financial reporting, and we may from time to time make changes aimed at enhancing their effectiveness and ensuring that our systems evolve with our business.
| (b) | Changes in internal controls over financial reporting. |
There were no changes in our internal control over financial reporting during the fourth quarter of fiscal year 2017 that have materially affected, or that are reasonably likely to materially affect, our internal controls over financial reporting.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles in the United States.
Because of its inherent limitations, internal control over financial reporting can provide only reasonable assurance, and it may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2017. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in its 2013 Internal Control—Integrated Framework.
Based on its assessment and those criteria, our management believes that our company maintained effective internal control over financial reporting as of December 31, 2017.
The effectiveness of our internal control over financial reporting as of December 31, 2017 has been audited by Deloitte & Touche LLP an independent registered public accounting firm, as stated in their report which is included below in this Item 9A of this annual report on Form 10-K.
| 72 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of Anika Therapeutics, Inc.
Opinion on Internal Control over Financial Reporting
We have audited the internal control over financial reporting of Anika Therapeutics, Inc. and subsidiaries (the “Company”) as of December 31, 2017, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control — Integrated Framework (2013) issued by COSO.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated financial statements as of and for the year ended December 31, 2017, of the Company and our report dated February 26, 2018, expressed an unqualified opinion on those financial statements.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management's Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Deloitte & Touche LLP
Boston, Massachusetts
February 26, 2018
| 73 |
None.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the SEC not later than 120 days after the close of our fiscal year ended December 31, 2017.
ITEM 11. EXECUTIVE COMPENSATION
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the SEC not later than 120 days after the close of our fiscal year ended December 31, 2017.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required under this item and Item 5 of this Annual Report on Form 10-K under the heading “Equity Compensation Plan Information” is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the SEC not later than 120 days after the close of our fiscal year ended December 31, 2017.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the SEC not later than 120 days after the close of our fiscal year ended December 31, 2017.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the SEC not later than 120 days after the close of our fiscal year ended December 31, 2017.
| 74 |
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
| (a) | Documents filed as part of Form 10-K. | |
| (1) Financial Statements | ||
| Reports of Independent Registered Public Accounting Firms | 45 | |
| Consolidated Balance Sheets | 47 | |
| Consolidated Statements of Operations and Comprehensive Income | 48 | |
| Consolidated Statements of Stockholder’s Equity | 49 | |
| Consolidated Statements of Cash Flows | 50 | |
| Notes to Consolidated Financial Statements | 51-71 | |
| (2) Schedules |
Schedules have been omitted as all required information has been disclosed in the financial statements and related footnotes.
| (3) Exhibits |
The list of Exhibits filed as a part of this Annual Report on Form 10-K is set forth in the Exhibit Index (b) below.
| 75 |
| 76 |
| 77 |
| Filed | Incorporated by Reference | |||||||||
| Exhibit Number |
Description | with this Form 10-K |
Form | Filing Date with SEC |
Exhibit Number | |||||
| 21.1 | List of Subsidiaries of Anika Therapeutics, Inc. | X | ||||||||
| 23.1 | Consent of Deloitte & Touche LLP | X | ||||||||
| 23.2 | Consent of PricewaterhouseCoopers LLP | X | ||||||||
| 31.1 | Certification of Principal Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | X | ||||||||
| 31.2 | Certification of Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | X | ||||||||
| **32.1 | Certification pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | X | ||||||||
| ***101 | The following materials from the Annual Report on Form 10-K of Anika Therapeutics, Inc. for the fiscal year ended December 31, 2017, formatted in xBRL: (i) Consolidated Balance Sheets as of December 31, 2017 and December 31, 2016; (ii) Consolidated Statements of Operations for the Years Ended December 31, 2017, December 31, 2016, and December 31, 2015; (iii) Consolidated Statements of Stockholders’ Equity for the Years Ended December 31, 2017, December 31, 2016, and December 31, 2015; (iv) Consolidated Statements of Cash Flows for the Years Ended December 31, 2017, December 31, 2016, and December 31, 2015; and (v) Notes to Consolidated Financial Statements | X | ||||||||
| † | Management contract or compensatory plan or arrangement. |
| * | Certain portions of this document have been omitted pursuant to a confidential treatment request filed with the Securities and Exchange Commission. The omitted portions have been filed separately with the Commission. |
| ** | The certification attached as Exhibit 32.1 that accompanies this Form 10-K is not deemed filed with the SEC and is not to be incorporated by reference into any filing of Anika Therapeutics, Inc. under the Securities Act of 1933 or the Securities Exchange Act of 1934, whether made before or after the date of this Form 10-K, irrespective of any general incorporation language contained in such filing. |
| *** | Pursuant to Rule 406T of Regulation S-T, XBRL (Extensible Business Reporting Language) information is deemed not filed or a part of a registration statement or prospectus for purposes of sections 11 or 12 of the Securities Act of 1933, is deemed not filed for purposes of section 18 of the Securities Exchange Act of 1934 and otherwise is not subject to liability under these sections. |
None.
| 78 |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| ANIKA THERAPEUTICS, INC. | |||
| Date: February 26, 2018 | By: | /s/ CHARLES H. SHERWOOD, PH.D. | |
| Charles H. Sherwood, Ph.D. | |||
| Chief Executive Officer | |||
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ CHARLES H. SHERWOOD, PH.D. | Chief Executive Officer, Director | |||
| Charles H. Sherwood, Ph.D. | (Principal Executive Officer) | February 26, 2018 | ||
| /s/ SYLVIA CHEUNG | Chief Financial Officer | |||
| Sylvia Cheung | (Principal Accounting Officer and Principal Financial Officer) | February 26, 2018 | ||
| /s/ JOSEPH L. BOWER | Director | |||
| Joseph L. Bower | February 26, 2018 | |||
| /s/ RAYMOND J. LAND | Director | |||
| Raymond J. Land | February 26, 2018 | |||
| /s/ GLENN R. LARSEN, PH.D. | Director | |||
| Glenn R. Larsen, Ph.D. | February 26, 2018 | |||
| /s/ JEFFERY S. THOMPSON | Director | |||
| Jeffery S. Thompson | February 26, 2018 | |||
| /s/ STEVEN E. WHEELER | Director | |||
| Steven E. Wheeler | February 26, 2018 |
79