Attached files
| file | filename |
|---|---|
| EX-31.1 - EXHIBIT 31.1 - Anika Therapeutics, Inc. | a6645325ex31_1.htm |
| EX-21.1 - EXHIBIT 21.1 - Anika Therapeutics, Inc. | a6645325ex21_1.htm |
| EX-23.1 - EXHIBIT 23.1 - Anika Therapeutics, Inc. | a6645325ex23_1.htm |
| EX-32.1 - EXHIBIT 32.1 - Anika Therapeutics, Inc. | a6645325ex32_1.htm |
| EX-31.2 - EXHIBIT 31.2 - Anika Therapeutics, Inc. | a6645325ex31_2.htm |
| EX-10.34 - EXHIBIT 10.34 - Anika Therapeutics, Inc. | a6645325ex10_34.htm |
| EX-10.36 - EXHIBIT 10.36 - Anika Therapeutics, Inc. | a6645325ex10_36.htm |
| EX-10.33 - EXHIBIT 10.33 - Anika Therapeutics, Inc. | a6645325ex10_33.htm |
| EX-10.35 - EXHIBIT 10.35 - Anika Therapeutics, Inc. | a6645325ex10_35.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
|
(Mark One)
|
||
|
x
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE
ACT OF 1934
|
|
|
For the fiscal year ended December 31, 2010
|
||
| o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15 (d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
|
For the transition period from to
|
||
Commission File Number 000-21326
Anika Therapeutics, Inc.
(Exact Name of Registrant as Specified in Its Charter)
|
Massachusetts
(State or Other Jurisdiction of Incorporation or Organization)
|
04-3145961
(IRS Employer Identification No.)
|
32 Wiggins Avenue, Bedford, Massachusetts 01730
(Address of Principal Executive Offices) (Zip Code)
(781) 457-9000
(Registrant’s Telephone Number, Including Area Code)
Securities registered pursuant to Section 12(b) of the Act: Common Stock, par value $.01 per share
Preferred Stock Purchase Rights
Name of Each Exchange on Which Registered: NASDAQ Global Select Market
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15 (d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one)
|
Large accelerated filer o
|
Accelerated filer x
|
Non-accelerated filer o
(Do not check if a smaller
reporting company)
|
Smaller reporting company o
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No x
The aggregate market value of voting and non-voting equity held by non-affiliates of the Registrant as of June 30, 2010, the last day of the Registrant’s most recently completed second fiscal quarter, was $79,383,470 based on the close price per share of Common Stock of $5.89 as of such date as reported on the NASDAQ Global Select Market. Shares of our Common Stock held by each executive officer, director and each person or entity known to the
registrant to be an affiliate have been excluded in that such persons may be deemed to be affiliates; such exclusion shall not be deemed to constitute an admission that any such person is an “affiliate” of the registrant. At March 14, 2011, there were issued and outstanding 13,458,168 shares of Common Stock, par value $.01 per share.
Documents Incorporated By Reference
The registrant intends to file a proxy statement pursuant to Regulation 14A within 120 days of the end of the fiscal year ended December 31, 2010. Portions of such proxy statement are incorporated by reference into Part III of this Annual Report on Form 10-K.
ANIKA THERAPEUTICS, INC.
TABLE OF CONTENTS
|
Page
|
||
|
6
|
||
|
14
|
||
|
25
|
||
|
25
|
||
|
26
|
||
|
26
|
||
| Purchases of Equity Securities |
27
|
|
|
28
|
||
|
|
||
| Operations |
29
|
|
|
44
|
||
|
45
|
||
|
|
||
| Disclosure |
72
|
|
|
72
|
||
|
72
|
||
|
73
|
||
|
73
|
||
|
|
||
| Stockholder Matters |
73
|
|
|
73
|
||
|
73
|
||
|
73
|
||
|
78
|
||
FORM 10-K
ANIKA THERAPEUTICS, INC.
For Fiscal Year Ended December 31, 2010
This Annual Report on Form 10-K, including the documents incorporated by reference into this Annual Report on Form 10-K, contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Securities Exchange Act of 1934, including, without limitation, statements regarding:
|
|
·
|
Our future sales and product revenue, including geographic expansions, possible retroactive price adjustments, and expectations of unit volumes or other offsets to price reductions;
|
|
|
·
|
Our manufacturing capacity and efficiency gains and work-in-process manufacturing operations;
|
|
|
·
|
The timing, scope and rate of patient enrollment for clinical trials;
|
|
|
·
|
The development of possible new products;
|
|
|
·
|
Our ability to achieve or maintain compliance with laws and regulations;
|
|
|
·
|
The timing of and/or receipt of the Food and Drug Administration (“FDA”), foreign or other regulatory approvals, clearances, and/or reimbursement approvals of current, new or potential products, and any limitations on such approvals;
|
|
|
·
|
Our intention to seek patent protection for our products and processes, and protect our intellectual property;
|
|
|
·
|
Our ability to effectively compete against current and future competitors;
|
|
|
·
|
Negotiations with potential and existing partners, including our performance under any of our existing and future distribution or supply agreements or our expectations with respect to sales and sales threshold milestones pursuant to such agreements;
|
|
|
·
|
The level of our revenue or sales in particular geographic areas and/or for particular products, and the market share for any of our products;
|
|
|
·
|
Our current strategy, including our corporate objectives and research and development and collaboration opportunities;
|
|
|
·
|
Our and Bausch & Lomb’s performance under the non-exclusive, two-year extension of the supply agreement for AMVISC and AMVISC Plus ophthalmic viscoelastic products, and our expectations regarding revenue from ophthalmic products;
|
|
|
·
|
Our ability, and the ability of our distribution partner, to market our aesthetic dermatology product;
|
|
|
·
|
Our ability to commercialize AnikaVisc and our expectations regarding such commercialization and the potential profits generated thereby;
|
|
|
·
|
Our expectations regarding our joint health products, including expectations regarding new products, expanded uses of existing products, new distribution and revenue growth;
|
|
|
·
|
Our intention to increase market share for joint health products in international and domestic markets or otherwise penetrate growing markets for osteoarthritis of the knee and other joints;
|
|
|
·
|
our expectations regarding next generation osteoarthritis/joint health product developments, clinical trials, regulatory approvals and commercial launches;
|
|
|
·
|
Our expectations regarding HYVISC sales;
|
-3-
|
|
·
|
Our ability to identify a new distribution partner for HYDRELLE™ in the United States and our ability to directly distribute HYDRELLE™ in the interim period and the impact such plan may have on future sales of this product;
|
|
|
·
|
Our ability to license our aesthetics product to new distribution partners outside of the United States; our ability, and the ability of our distribution partners, to market our aesthetic dermatology product; and our expectations regarding the distribution and sales of our ELEVESS product and the timing thereof;
|
|
|
·
|
Our expectations regarding product gross margin;
|
|
|
·
|
Our expectations regarding our U.S. MONOVISC trials and the results of the related premarket approval (“PMA”) filing with the FDA, including the requested Advisory Panel review and the likelihood of our obtaining such approval and/or the anticipated timing thereof;
|
|
|
·
|
Our expectations regarding the commencement of a clinical trial for CINGAL and our ability to obtain regulatory approvals for CINGAL;
|
|
|
·
|
Our expectations regarding our existing aesthetics product’s line extensions;
|
|
|
·
|
Our expectation for increases in operating expenses, including research and development and selling, general and administrative expenses;
|
|
|
·
|
The rate at which we use cash, the amounts used and generated by operations, and our expectation regarding the adequacy of such cash;
|
|
|
·
|
Our expectation for capital expenditures spending and future amounts of interest income and expense;
|
|
|
·
|
Possible negotiations or re-negotiations with existing or new distribution or collaboration partners;
|
|
|
·
|
Our expectations regarding our existing manufacturing facility and the Bedford, MA facility; our expectations related to costs, including financing costs, to build-out and occupy the new facility, the timing of construction, and our ability to obtain FDA licensure for the facility; and our expectation regarding the impact of our Bedford, MA facility on our business and the amount of the annual depreciation expense associated therewith;
|
|
|
·
|
Our abilities to comply with debt covenants;
|
|
|
·
|
Our ability to obtain additional funds through equity or debt financings, strategic alliances with corporate partners and other sources, to the extent our current sources of funds are insufficient;
|
|
|
·
|
Our abilities to successfully integrate Fidia Advanced Biopolymers S.r.1. (“FAB”), our recently acquired subsidiary, into the Company and manage the operation from one with losses, into a company generating profits;
|
|
|
·
|
Our abilities to integrate our research and development activity with those of FAB and effectively prioritize the many projects underway at both companies;
|
|
|
·
|
Our ability to obtain U.S. approval for the orthopedic and other products of FAB, including the timing and potential success of such efforts, and to expand sales of these products in the U.S., including the impact such efforts may have on our revenue;
|
|
|
·
|
Our ability to commercialize MONOVISC and the FAB products directly to customers, and the potential increase in expenses associated therewith; and
|
|
|
·
|
Our ability to successfully defend the Company against lawsuits and claims, including the Genzyme lawsuit, and the uncertain financial impact such lawsuits and claims and related defense costs may have on the Company.
|
Furthermore, additional statements identified by words such as “will,” “likely,” “may,” “believe,” “expect,” “anticipate,” “intend,” “seek,” “designed,” “develop,” “would,” “future,” “can,” “could” and other expressions that are predictions of or indicate future events and trends and which do not relate to historical matters, also identify forward-looking statements.
-4-
You should not rely on forward-looking statements because they involve known and unknown risks, uncertainties and other factors, some of which are beyond our control, including those factors described in the section titled “Risk Factors” in this Annual Report on Form 10-K or elsewhere in this report. These risks, uncertainties and other factors may cause our actual results, performance or achievement to be materially different from the anticipated future results, performance or achievement, expressed or implied by the forward-looking statements. These forward-looking statements are based upon the current assumptions of our management and are only expectations of future results. You should carefully review all of these factors, and you should be aware that there may be other factors that could cause these differences, including those factors discussed in the sections titled “Business” and “Management’s Discussions and Analysis of Financial Condition and Results of Operations” elsewhere in this Annual Report on Form 10-K. We undertake no obligation to publicly update or revise any forward-looking statement to reflect changes in underlying assumptions or factors, new information, future events or other changes.
-5-
ITEM 1. BUSINESS
Overview
Anika Therapeutics, Inc. (“Anika,” and together with its subsidiaries, the “Company,” “we,” “us,” or “our”) was incorporated in 1992 as a Massachusetts company. Anika develops, manufactures and commercializes therapeutic products for tissue protection, healing and repair. These products are based on hyaluronic acid (“HA”), a naturally occurring, biocompatible polymer found throughout the body. Due to its unique biophysical and biochemical properties, HA plays an important role in a number of physiological functions such as the protection and lubrication of soft tissues and joints, the maintenance of the structural integrity of tissues, and the transport of molecules to and within cells.
Anika acquired 100% of the issued and outstanding stock of FAB on December 30, 2009 from Fidia Farmaceutici S.p.A., a privately held Italian corporation, for a purchase price consisting of $17.0 million in cash and 1,981,192 shares of the Company’s common stock valued at $16.8 million based on the closing stock price of $8.49 per share. See Item 8: Financial Statements, Note 16, for additional information regarding this transaction. In December of 2010, FAB’s name was changed to Anika Therapeutics S.r.l., but to avoid confusion, we will continue to refer to it as “FAB” in the rest of this document.
FAB has over 20 products currently commercialized, primarily in Europe. These products are also all made from hyaluronic acid, and based on two technologies “HYAFF”, which is a solid form of HA, and ACP gel, an autocross-linked polymer of HA. Both technologies are protected by an extensive portfolio of owned and licensed patents. With the acquisition of FAB, the Company now offers therapeutic products in the following areas:
|
Anika
|
FAB
|
|||||||
|
Orthobiologics
|
X | X | ||||||
|
Dermal
|
||||||||
| Advanced wound care | X | |||||||
|
Aesthetic dermatology
|
X | |||||||
|
Ophthalmic
|
X | |||||||
|
Surgical
|
||||||||
| Anti-adhesion | X |
X
|
||||||
|
Ear, nose and throat care (“ENT”)
|
X
|
|||||||
|
Veterinary
|
X | |||||||
The Company plans to commercialize MONOVISC and certain FAB products in the U.S. once we receive FDA approval to market. In 2011, upon FDA approval, we will begin adding resources and materials to implement this commercialization strategy.
The following sections provide more specific information on our products and related activities:
Orthobiologics
The Company’s orthobiologics products are used in a wide range of treatments from providing relief from the pain of osteoarthritis, to regenerating damaged tissue such as cartilage defects. Osteoarthritis is a debilitating disease causing pain, swelling and restricted movement in joints. It occurs when the cartilage in a joint gradually deteriorates due to the effects of mechanical stress, which can be caused by a variety of factors including the normal aging process. In an osteoarthritic joint, particular regions of articulating surfaces are exposed to irregular forces, which result in the remodeling of tissue surfaces that disrupt the normal equilibrium or mechanical function. As osteoarthritis advances, the joint gradually loses its ability to regenerate cartilage tissue and the cartilage layer attached to the bone deteriorates to the point where eventually the bone becomes exposed. Advanced osteoarthritis often requires surgery and the possible implantation of artificial joints. The current treatment options for osteoarthritis before joint replacement surgery include viscosupplementation, analgesics, non-steroidal anti-inflammatory drugs and steroid injections.
-6-
Our joint health products include ORTHOVISC, ORTHOVISC mini, and MONOVISC. ORTHOVISC is available in the U.S., Canada, Turkey and other international markets for the treatment of osteoarthritis of the knee, and in Europe for the treatment of osteoarthritis in all joints. ORTHOVISC mini is available in Europe, and is designed for the treatment of osteoarthritis in small joints. MONOVISC is our single injection osteoarthritis treatment indicated for all joints in Europe, and for the knee in Turkey and Canada. ORTHOVISC mini and MONOVISC are our two newest joint health products and became available in certain international markets during the second quarter of 2008.
In the U.S., ORTHOVISC is indicated for the treatment of pain caused by osteoarthritis of the knee in patients who have failed to respond adequately to conservative non-pharmacologic therapy and to simple analgesics, such as acetaminophen. It is a sterile, clear, viscoelastic solution of hyaluronan dissolved in physiological saline, and dispensed in a single-use syringe. A complex sugar of the glycosaminoglycan family, hyaluronan is a high molecular weight polysaccharide composed of repeating disaccharide units of sodium glucuronate and N-acetylglucosamine. ORTHOVISC is injected into joints in a series of three intra-articular injections one week apart. ORTHOVISC became available for sale in the U.S. on March 1, 2004, and is marketed by DePuy Mitek, under the terms of a ten-year licensing, distribution, supply and marketing agreement which was entered into in December 2003 (the “JNJ Agreement”).
We have a number of distribution relationships servicing international markets including Canada, Europe, Turkey, the Middle East, Latin America, and Asia. We will continue to seek to establish distribution relationships in other regions. See the sections captioned “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Management Overview” and “Risk Factors”.
With the acquisition of FAB, we now offer several additional products used in connection with orthopedic regenerative medicine. The products currently available in Europe include Hyalograft C Autograft for cartilage regeneration; Hyalofast, a biodegradable support for human bone marrow mesenchymal stem cells; Hyalonect, a woven gauze used as a graft wrap; and Hyaloss, HYAFF fibers used to mix blood/bone grafts to form a paste for bone regeneration. FAB also offers Hyaloglide, an ACP gel used in tenolysis treatment, but with potential for flexor tendon adhesion prevention, and in the shoulder for adhesive capsulitis with additional clinical data. FAB’s products are commercialized directly in Italy, and through a network of distributors, primarily in Europe, the Middle East, Argentina, and Korea. One of Anika’s areas of focus is to seek U.S. approval of a number of these products, as Anika believes it has the opportunity to expand its sales of these products in the U.S. In this regard, in October 2010, Anika filed 510(k) applications with the FDA to gain marketing clearance for three FAB products: Hyalofast®, Hyaloglide®, and Hyalonect®, but is currently unable to predict the timing of receipt of such clearance.
Dermal
Our aesthetic dermatology business is designed as a family of products for facial wrinkles and scar remediation, and is intended to compete with collagen-based and other HA-based products currently on the market. Our initial aesthetic dermatology product is a dermal filler based on our proprietary chemically modified, cross-linked HA, and is approved in Europe, Canada, the U.S. and certain countries in South America. Internationally, this product is marketed under the ELEVESS name, and in the U.S. under the name HYDRELLE. Coapt Systems, Inc. (“Coapt”) began selling HYDRELLE in the third quarter of 2009. In July 2010, Coapt made a general assignment for the benefit of creditors and an assignee began the liquidation of Coapt’s assets. The Company’s Distribution Agreement with Coapt has been terminated, and the Company has directly sold HYDRELLE in the interim while it reviews its franchise strategy and opportunities for new distribution partners.
With the acquisition of FAB, the Company entered the field of advanced wound care products. FAB offers over seven products for the treatment of skin wounds ranging from burns to diabetic ulcers. The products cover a variety of wound treatment solutions including debridement agents, advanced therapies and skin substitutes. Leading products include Hyalograft 3D, for the regeneration of skin; and Hyalomatrix, for treatment of burns and ulcers and the only product not contra-indicated for 3 rd degree burns. FAB’s products are commercialized directly in Italy, and through a network of distributors, primarily in Europe, the Middle East, Argentina, and Korea. Several of the products are also approved for sale in the United States, including Hyalomatrix and Hyalofill, and the Company is exploring domestic distribution opportunities.
Ophthalmic
Our ophthalmic business includes HA viscoelastic products used in ophthalmic surgery. The ophthalmic products we manufacture include the AMVISC and AMVISC Plus product line, STAARVISC-II, Optivisc (formerly ShellGel), and recently FDA-approved AnikaVisc. They are injectable, high molecular weight HA products used as viscoelastic agents in ophthalmic surgical procedures such as cataract extraction and intraocular lens implantation. These products coat, lubricate and protect sensitive tissue such as the endothelium, and maintain the shape of the eye, thereby facilitating ophthalmic surgical procedures.
-7-
Anika previously manufactured the AMVISC product line for Bausch & Lomb (“B&L”) under the terms of a supply agreement that expired on December 31, 2010 (the “2004 B&L Agreement”) for viscoelastic products used in ophthalmic surgery. Effective January 1, 2011 we entered into a non-exclusive, two year contract with B&L intended to transition the manufacture of AMVISC and AMVISC Plus to an alternative, recently acquired low-cost supplier to B&L. Under the 2004 B&L Agreement, the Company was restricted in its ability to commercialize viscoelastic products to only existing customers (STAAR Surgical Company and Hoya Surgical Optics, Inc.). That restriction has now expired, and the Company is free to market its own viscoelastic product AnikaVisc. B&L accounted for 21% of product revenue for the year ended 2010, and product revenue is expected to be significantly lower in 2011 under the new transition contract. Operating margins under the 2004 B&L Agreement were low, and the Company expects to see margin improvement through commercialization of its new AnikaVisc product. There can be no assurance that AnikaVisc will be successfully sold or that it will generate any profit for the Company. See also Item 1A. “Risk Factors.”
Surgical
INCERT, approved for sale in Europe and Turkey, is designed as a family of HA based products, with chemically modified, cross-linked HA, for prevention of post-surgical adhesions. Surgical adhesions occur when fibrous bands of tissues form between adjacent tissue layers during the wound healing process. Although surgeons attempt to minimize the formation of adhesions, they nevertheless occur quite frequently after surgery. Adhesions in the abdominal and pelvic cavity can cause particularly serious problems such as intestinal blockage following abdominal surgery, and infertility following pelvic surgery. Fibrosis following spinal surgery can complicate re-operation and may cause pain. INCERT is currently marketed in four countries. We see potential for expanded indications for the use of INCERT, but have made this a secondary goal to the successful launch and expanded distribution of our joint health and dermatology products. There are currently no plans at this time to distribute INCERT in the U.S. Anika co-owns issued U.S. patents covering the use of INCERT for adhesion prevention. See the section captioned “Patent and Proprietary Rights.”
Hyalobarrier and Hyalobarrier Endo are a clinically proven post operative adhesion barrier approved for abdominal indications. The products are currently commercialized by FAB in Europe, the Middle East and certain Asian countries through a distribution network, but are not approved in the U.S.
FAB offers several products used in connection with the treatment of ENT disorders. The lead product is Merogel, a thick, viscous hydrogel composed of cross-linked hyaluronic acid—a biocompatible agent that creates a moist wound-healing environment. FAB is partnered with Medtronic for worldwide distribution.
Veterinary
HYVISC is a high molecular weight injectable HA product for the treatment of joint dysfunction in horses due to non-infectious synovitis associated with equine osteoarthritis. HYVISC has viscoelastic properties that lubricate and protect the tissues in horse joints. HYVISC is distributed by Boehringer Ingelheim Vetmedica, Inc. in the United States.
See Note 13 to our Consolidated Financial Statements, “Revenue by Product Group, by Significant Customer and by Geographic Region,” for a discussion regarding our segments and geographic sales.
Research and Development of Potential Products
Anika’s research and development efforts primarily consist of the development of new medical applications for our HA-based technology, the management of clinical trials for certain product candidates, the preparation and processing of applications for regulatory approvals or clearances at all relevant stages of product development, and process development and scale-up manufacturing activities relative to our existing and new products. Our development focus includes chemically modified formulations of HA designed for longer residence time in the body. For the years ended December 31, 2010, 2009 and 2008, these expenses were $6.9 million, $8.2 million, and $7.4 million, respectively. We anticipate that we will continue to commit significant resources to research and development, including clinical trials, in the future.
-8-
With the acquisition of FAB, we have enhanced both our research and development capabilities and our pipeline of candidate products. FAB has research and development programs for new products including Hyalobone, a bone tissue filler; Hyalospine, an adhesion prevention gel for use after spinal surgery; and Hyalofast, to repair cartilage defects.
In addition to the FAB products in the preceding paragraph, additional products in development include MONOVISC for U.S. marketing approval, and additional next generation joint health products. Our first next generation osteoarthritis product is MONOVISC, a single-injection treatment product that uses a non-animal source HA. MONOVISC is also our first osteoarthritis product based on our proprietary crosslinked HA-technology. We received Conformité Européene (“CE”) Mark approval for the MONOVISC product in October 2007, and began sales in Europe during the second quarter of 2008, following a small, post-marketing clinical study. In the U.S., we filed an investigational device exemption, or an IDE application, with the FDA, and completed the clinical segment of the U.S. MONOVISC pivotal trial in June 2009, and a follow-on retreatment study in September 2009. We filed the final module of our MONOVISC PMA containing the clinical data in December 2009. We were informed that there were deficiencies in our submissions through a deficiency/non-approvable letter, which is the FDA's mechanism for informing companies of deficiencies. We submitted additional data and analyses throughout 2010, and have been informed by FDA that deficiencies remain. Acting on an option presented by the FDA to resolve the remaining open issues, Anika requested a review by the Orthopedic Advisory Panel. The Company has not yet received a date for an Advisory Panel meeting. We continue to believe that Monovisc should receive FDA approval. Our second single-injection osteoarthritis product under development is CINGAL™, which is based on the same technology platform used in MONOVISC, but with an added active therapeutic molecule to provide broad pain relief for a long period of time.
During the past year, we have integrated the research and development efforts of Anika and FAB and prioritized our new product development activities. There is a risk that our efforts will not be successful in (1) developing our existing product candidates, (2) expanding the therapeutic applications of our existing products, or (3) resulting in new applications for our HA technology. There is also a risk that we may choose not to pursue development of potential product candidates. We may not be able to obtain regulatory approval for any new applications we develop. Furthermore, even if all regulatory approvals are obtained, there can be no assurances that we will achieve meaningful sales of such products or applications. See Item 1A. “Risk Factors.”
Patent and Proprietary Rights
Our products and trademarks, including our Company name, product names and logos, are proprietary. We rely on a combination of patent protection, trade secrets and trademark laws, license agreements, confidentiality and other contractual provisions to protect our proprietary information.
We have a policy of seeking patent protection for patentable aspects of our proprietary technology. Our issued patents expire between 2011 and 2023. Anika co-owns certain U.S. patents and a patent application with claims relating to the chemical modification of HA and certain adhesion prevention uses and certain drug delivery uses of HA. Anika also solely owns patents covering composition of matter and certain manufacturing processes. FAB’s issued patents expire between 2011 and 2026. The FAB patent estate is extensive and intertwined with its former parent company, Fidia Farmaceutici S.p.A, through a cross-licensing agreement which provides both companies with access to each others patents to the extent required to support their own products. We intend to seek patent protection for products and processes developed in the course of our activities when we believe such protection is in our best interest and when the cost of seeking such protection is not inordinate relative to the potential benefits. See also the section captioned “Risk Factors—We may be unable to adequately protect our intellectual property rights.”
Other entities have filed patent applications for or have been issued patents concerning various aspects of HA-related products or processes. In addition, the products or processes we develop may infringe the patent rights of others in the future. Any such infringement may have a material adverse effect on our business, financial condition, and results of operations. See also the section captioned “Risk Factors—We may be unable to adequately protect our intellectual property rights. ”
We also rely upon trade secrets and proprietary know-how for certain non-patented aspects of our technology. To protect such information, we require certain customers and vendors, and all employees, consultants and licensees to enter into confidentiality agreements limiting the disclosure and use of such information. These agreements, however, may not provide adequate protection. See also the section captioned “Risk Factors—We may be unable to adequately protect our intellectual property rights.”
We have granted Depuy Mitek an exclusive, non-transferable royalty bearing license to use and sell ORTHOVISC (and other products developed pursuant to the JNJ Agreement) in the U.S., as well as a license to manufacture and have manufactured such products in the event that we are unable to supply them with products in accordance with the terms of the JNJ Agreement.
-9-
Government Regulation
United States Regulation
Our research (including clinical research), development, manufacture, and marketing of products are subject to regulation by numerous governmental authorities in the U.S. and other countries. Medical devices and pharmaceuticals are subject to extensive and rigorous regulation by the FDA and by other federal, state and local authorities. The Federal Food, Drug and Cosmetic Act (“FDC Act”) and respective regulations govern the conditions of safety, efficacy, clearance, approval, manufacture, quality system requirements, labeling, packaging, distribution, storage, record keeping, reporting, marketing, advertising, and promotion of our products. Noncompliance with applicable requirements can result in, among other things, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, failure of the government to grant premarket clearance or approval of products, withdrawal of clearances and approvals, and criminal prosecution.
Medical products regulated by the FDA are generally classified as drugs, biologics, and/or medical devices. Medical devices intended for human use are classified into three categories (Class I, II or III), on the basis of the controls deemed reasonably necessary by the FDA to assure their safety and efficacy. Class I devices are subject to general controls, for example, labeling and adherence to the FDA’s Good Manufacturing Practices/Quality System Regulation (“GMP/QSR”). Most Class I devices are exempt from the FDA review process and some are exempt from Good Manufacturing Practice. Class II devices are subject to general and special controls (for example, performance standards, postmarket surveillance, and patient registries). Most Class II devices are subject to premarket notification and may be subject to clinical testing for purposes of premarket notification and clearance for marketing. Class III is the most stringent regulatory category for medical devices. Most Class III devices require premarket approval (“PMA”) from the FDA.
AMVISC, AMVISC Plus, ShellGel/Optivisc, STAARVISC, and AnikaVisc are approved as Class III medical devices in the U.S. for intraocular ophthalmic surgical procedures in intraocular use in humans. ORTHOVISC is approved as a Class III medical device in the U.S. for treatment of pain resulting from osteoarthritis of the knee in humans. HYDRELLE™ is approved as a Class III medical device in the U.S. for treatment of facial wrinkles and folds, such as nasolabial folds. HYVISC is approved as an animal drug for intra-articular injection in horse joints to treat degenerative joint disease associated with synovitis. Most HA products for human use are regulated as medical devices. We believe that our INCERT product, should we decide to seek U.S. approval to market, will have to meet the regulatory requirements for Class III devices and will require clinical trials and a PMA submission. Our subsidiary, FAB, has three advanced wound care products approved in the U.S. as Class II devices through premarket notification (510(k))--Hyalomatrix, Hyalofill-R, and Hyalofill-F. All of FAB’s ENT products are 510(k) cleared by Medtronic as Class II devices. The FDA’s 510(k) clearance process is under review and changes to the process may have an impact on current or future product approvals. Three products were submitted for 510(k) clearance in 2010: Hyaloglide, Hyalofast and Hyalonect. There has been delay in the FDA’s review process and the Company is unable to predict the timing of receipt of these clearances. There is no guarantee that the clearance process for these products will be successful or that additional data will not be required to support clearance.
Unless a new device is exempted from premarket notification, its manufacturer must obtain marketing clearance from the FDA through premarket notification (510(k)) or approval through PMA before the device can be introduced to the market. Product development and approval within the FDA regulatory framework takes a number of years and involves the expenditure of substantial resources. This regulatory framework may change or additional regulations may arise at any stage of our product development process and may affect approval of, or delay an application related to, a product, or require additional expenditures by us. There can be no assurance that the FDA review of marketing applications will result in product approval on a timely basis, if at all. The PMA approval process is lengthy, expensive, and typically requires, among other things, valid scientific evidence which generally includes extensive data such as pre-clinical and clinical trial data to demonstrate a reasonable assurance of safety and effectiveness.
Human clinical trials in the U.S. for significant risk devices must be conducted under Good Clinical Practice (“GCP”) regulations through Investigational Device Exemption (“IDE”), which must be submitted to the FDA and either be approved or be allowed to become effective before the trials may commence. There can be no assurance that submission of an IDE will result in the ability to commence clinical trials. In addition, the IDE approval process could result in significant delays. Even if the FDA approves an IDE or allows an IDE for a clinical investigation to become effective, clinical trials may be suspended at any time for a number of reasons. Among others, these reasons may include: a) failure to comply with applicable requirements; b) inadequacy of informed consent; and c) the data generated suggests that: the risks to clinical subjects are not outweighed by the anticipated benefits to clinical subjects and the importance of the knowledge to be gained, the investigation is scientifically unsound, or there is reason to believe that the device, as used, is ineffective. A trial may be terminated if serious unanticipated adverse events present an unreasonable risk to subjects. If clinical studies are suspended or terminated, we may be unable to continue the development of the investigational products affected.
-10-
Upon completion of required clinical trials, for Class III medical devices, results might be presented to the FDA in a PMA application. In addition to the results of clinical investigations, the New Drug Application (“NDA”) applicant must submit other information relevant to the safety and efficacy of the device, including, among other things, the results of non-clinical tests and clinical trials; a full description of the device and its components; a full description of the methods, facilities and controls used for manufacturing; and proposed labeling. The FDA also conducts an on-site inspection to determine whether an applicant conforms to the FDA’s current Quality System Regulation (“QSR”), formerly known as GMP. FDA review of the PMA may not result in timely, or any, PMA approval, and there may be significant conditions on approval, including limitations on labeling and advertising claims and the imposition of post-market testing, tracking, or surveillance requirements. We have completed the clinical trail and PMA submissions for our MONOVISC product, which is currently under review by FDA. We have requested review of MONOVISC by the Orthopedic Advisory Panel, but we have not yet received a date for a panel meeting.
Upon completion of required clinical trials for pharmaceuticals, results might be presented to the FDA in a NDA or New Animal Drug Application (“NADA”). In addition to the results of clinical investigations, the NDA or NADA applicant must submit other information relevant to the safety and efficacy of the product, including, among other things, the results of non-clinical tests and clinical trials; a full description of the product formulation; a full description of the methods, facilities and controls used for manufacturing; and proposed labeling. The FDA also conducts an on-site inspection to determine whether an applicant conforms with the FDA’s current cGMP related to pharmaceuticals. FDA review of the NDA or NADA may not result in timely, or any, FDA approval, and there may be significant conditions on approval, including limitations on labeling and advertising claims and the imposition of post-market testing, tracking, or surveillance requirements.
Post-approval product or manufacturing changes where such change affects the safety and efficacy of the medical products as well as the use of a different facility for manufacturing, could necessitate additional review and approval by the FDA. Post-approval changes in labeling, packaging or promotional materials may also necessitate further review and approval by the FDA.
Legally marketed products are subject to continuing requirements by the FDA relating to design control, manufacturing, quality control and quality assurance, maintenance of records and documentation, reporting of adverse events, and labeling and promotion. The FDC Act requires medical product manufacturers to comply with QSR for medical devices and cGMP regulations related to pharmaceuticals. The FDA enforces these requirements through periodic inspections of manufacturing facilities. To ensure full compliance with requirements set forth in the GMP/QSR regulations, manufacturers must continue to expend time, money and effort in the area of production and quality control to ensure full technical compliance. Other federal, state, and local agencies may inspect manufacturing establishments as well.
A set of regulations known as the Medical Device Reporting and Drug Adverse Events Reporting System regulations obligates manufacturers to inform the FDA whenever information reasonably suggests that one of their medical products may have caused or contributed to a death or serious injury, or when one of their devices malfunctions and if the malfunction were to recur, the device or a similar device would be likely to cause or contribute to a death or serious injury.
The process of obtaining approvals from the FDA and foreign regulatory authorities can be costly, time consuming, and subject to unanticipated delays. Approvals of our products, processes or facilities may not be granted on a timely basis or at all, and we may not have available resources or be able to obtain the financing needed to develop certain of such products. Any failure or delay in obtaining such approvals could adversely affect our ability to market our products in the U.S. and in other countries.
In addition to regulations enforced by the FDA, we are subject to regulation under the Occupational Safety and Health Act, the Environmental Protection Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act and other existing and future federal, state and local laws and regulations as well as those of foreign governments. Federal, state and foreign regulations regarding the manufacture and sale of medical products are subject to change. We cannot predict what impact, if any, such changes might have on our business.
-11-
FDA Warning Letter
In July 2008, we received a Warning Letter (the “Warning Letter”) from the FDA in response to an earlier FDA Form 483 Notice of Observations issued to us following an inspection at our manufacturing facility in Woburn, Massachusetts. The Company submitted corrective action plans, which have been accepted by the FDA and resulted in the clearance of the Warning Letter in September 2010.
Foreign Regulation
In addition to regulations enforced by the FDA, we and our products are subject to certain foreign regulations. International regulatory bodies often establish regulations governing product standards, packing requirements, labeling requirements, import restrictions, tariff regulations, duties, and tax requirements ORTHOVISC is approved for sale and is marketed in Canada, Europe, Turkey, and parts of the Middle East. In the European Union (“EU”), ORTHOVISC is sold under the CE mark authorization, a certification required under European Union medical device regulations.
The CE mark, achieved in 1996, allows ORTHOVISC to be marketed without further approvals in most of the EU nations as well as other countries that recognize EU device regulations. ORTHOVISC ® mini, a treatment for osteoarthritis targeting small joints, is available in Europe under CE mark authorization received in 2008. In August 2004, we received an EC Design Examination Certificate which entitled us to affix a CE mark to INCERT-S as a barrier to adhesion formation following surgery. AMVISC ® and AMVISC ® Plus are CE marked, and in May 2005, we received an EC Design Examination Certificate which entitled us to affix a CE mark to ShellGel™/OptiVisc as an ophthalmic viscoelastic surgical device. Staarvisc, an ophthalmic viscoelastic surgical device, is licensed in Canada from May 2002. We received EU CE Mark approval for ELEVESS during the second quarter of 2007. MONOVISC, a medical device for treatment of pain associated with osteoarthritis, was approved in the EU in October 2007 and in Canada in August 2009. In addition, Anika received approval for several of its products in Latin America, Korea, Turkey, Middle East, UAE, Saudi Arabia, and other international markets.
Almost all of FAB’s products are CE marked for European sale. In addition, FAB has received approval for several of its products in Argentina, Egypt, Hong Kong, Iran, Israel, Korea, Malaysia, Singapore, Mexico, Cyprus, Saudi Arabia, Taiwan, Turkey, and the United Arab Emirates. FAB’s tissue engineered products Hyalograft C Autograft, Hyalograft 3D Autograft and Laserskin Autograft are currently marketed in Europe. However, the regulations for marketing of these products in Europe have been changed. Effective January 1, 2013, new regulations mandate these products to be approved by the European Medicines Agency (“EMA”) as Advanced Therapeutics Medical Products (“ATMP”) in order to remain on the EU market. FAB continues to be in discussion with the EMA and is implementing a plan to qualify for the new status while continuing to sell these products in the EU. There can be no assurance that required approvals will be obtained in a timely fashion. We may not be able to achieve and/or maintain the compliance required for CE marking or other foreign regulatory approvals for any or all of our products. The requirements relating to the conduct of clinical trials, product licensing, marketing, pricing, advertising, promotion and reimbursement also vary widely from country to country.
Competition
We compete with many companies, including, among others, large pharmaceutical firms and specialized medical products companies across all of our product lines. Many of these companies have substantially greater financial resources, larger research and development staffs, more extensive marketing and manufacturing organizations and more experience in the regulatory process than us. We also compete with academic institutions, governmental agencies and other research organizations, which may be involved in research, development and commercialization of products. Many of our competitors also compete against us in securing relationships with collaborators for their research and development and commercialization programs.
Competition in our industry is based primarily on product efficacy, safety, timing and the scope of regulatory approvals, availability of supply, marketing and sales capability, reimbursement coverage, product pricing and patent protection.
-12-
Some of the principal factors that may affect our ability to compete in our HA development and commercialization markets include:
|
|
·
|
The quality and breadth of our technology and technological advances;
|
|
|
·
|
Our ability to complete successful clinical studies and obtain FDA marketing and foreign regulatory approvals prior to our competitors;
|
|
|
·
|
Our ability to recruit and retain skilled employees; and
|
|
|
·
|
The availability of substantial capital resources to fund discovery, development and commercialization activities or the ability to defray such costs through securing relationships with collaborators for our research and development and commercialization programs.
|
We are aware of several companies that are developing and/or marketing products utilizing HA for a variety of human applications. In some cases, competitors have already obtained product approvals, submitted applications for approval or have commenced human clinical studies, either in the U.S. or in certain foreign countries. All of the Company’s products face substantial competition. There exist major worldwide competing products, made from HA and other materials, for use in ophthalmic surgery, orthopedics, surgical adhesion prevention, advanced wound care, ENT and cosmetic dermal fillers. There is a risk that we will be unable to compete effectively against our current or future competitors.
Employees
As of December 31, 2010, we had 114 employees, 42 of whom are located outside the U.S. and were added as a result of the FAB acquisition. We consider our relations with our employees to be good. None of our U.S. employees are represented by labor unions, and most of the employees based in Italy are represented by unions adding complexity and additional risks to the wage and employment decision process.
Environmental Laws
We believe that we are in compliance with all federal, state and local environmental regulations with respect to our manufacturing facilities and that the cost of ongoing compliance with such regulations does not have a material effect on our operations. Our leased Woburn manufacturing facility is located within the Wells G&H Superfund site in Woburn, Massachusetts. We have not been named and are not a party to any legal proceedings regarding the Wells G&H Superfund site.
Product Liability
The testing, marketing and sale of human health care products entail an inherent risk of allegations of product liability, and we cannot assure you that substantial product liability claims will not be asserted against us. Although we have not received any material product liability claims to date and have coverage under our insurance policy of $5,000,000 per occurrence and $5,000,000 in the aggregate, we cannot assure you that if material claims arise in the future, our insurance will be adequate to cover all situations. Moreover, we cannot assure you that such insurance, or additional insurance, if required, will be available in the future or, if available, will be available on commercially reasonable terms. Any product liability claim, if successful, could have a material adverse effect on our business, financial condition, and results of operation.
Available Information
Our Annual Reports on Form 10-K, including our consolidated financial statements, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and other information, including amendments and exhibits to such reports, filed or furnished pursuant to the Securities Exchange Act of 1934, as amended, are available free of charge in the “SEC Filings” section of our website located at http://www.anikatherapeutics.com, as soon as reasonably practicable after the reports are filed with or furnished to the Securities and Exchange Commission (“SEC”). The information on our website is not part of this Annual Report on Form 10-K. Reports filed with the SEC may be viewed at www.sec.gov or obtained at the SEC Public Reference Room at 100F Street NE, Washington, D.C. Information regarding the operation of the Public Reference Room may be obtained by calling the SEC at 1-800-SEC-0330.
-13-
ITEM 1A. RISK FACTORS
Our operating results and financial condition have varied in the past and could in the future vary significantly depending on a number of factors. From time to time, information provided by us, or statements made by our employees, contain “forward-looking” information that involves risks and uncertainties. In particular, statements contained in this Annual Report on Form 10-K, and in the documents incorporated by reference into this Annual Report on Form 10-K, that are not historical facts, including, but not limited to statements concerning new products, product development, regulatory approval, and offerings, product and price competition, competition and strategy, customer diversification, product price and inventory, contingent consideration payments, deferred revenues, economic and market conditions, potential government regulation, seasonal factors, international expansion, revenue recognition, profits, growth of revenues, composition of revenues, cost of revenues, operating expenses, sales, marketing and support expenses, general and administrative expenses, product gross profit, interest income, interest expense, anticipated operating and capital expenditure requirements, cash inflows, contractual obligations, tax rates, stock-based compensation, leasing and subleasing activities, acquisitions, liquidity, litigation matters, intellectual property matters, distribution channels, stock price, third party licenses and potential debt or equity financings constitute forward-looking statements and are made under the safe harbor provisions of Section 27 of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. These statements are neither promises nor guarantees. Our actual results of operations and financial condition have varied and could in the future vary significantly from those stated in any forward-looking statements. The following factors, among others, could cause actual results to differ materially from those contained in forward-looking statements made in this Form 10-K, in the documents incorporated by reference into this Form 10-K or presented elsewhere by our management from time to time. Such factors, among others, could have a material adverse effect upon our business, results of operations and financial condition.
Our business is subject to comprehensive and varied government regulation and, as a result, failure to obtain FDA or other U.S. and foreign governmental approvals for our products may materially adversely affect our business, results of operations and financial condition.
Product development and approval within the FDA framework takes a number of years and involves the expenditure of substantial resources. There can be no assurance that the FDA will grant approval for our new products on a timely basis if at all, or that FDA review will not involve delays that will adversely affect our ability to commercialize additional products or expand permitted uses of existing products, or that the regulatory framework will not change, or that additional regulation will not arise at any stage of our product development process which may adversely affect approval of or delay an application or require additional expenditures by us. In the event our future products are regulated as human drugs or biologics, the FDA’s review process of such products typically would be substantially longer and more expensive than the review process to which they are currently subject as devices.
Products in development include a next generation HYDRELLE/ELEVESS™ line extension, and joint health related products. Our first next generation osteoarthritis product is MONOVISC, a single-injection treatment product that uses a non-animal source HA. MONOVISC is also our first osteoarthritis product based on our proprietary crosslinked HA- technology. We received CE Mark approval for MONOVISC in October 2007. We have completed a pivotal trial in the U.S., and submitted the results for a PMA application in December 2009. We were informed that there were deficiencies in our submissions through a deficiency/non-approvable letter, which is the FDA's mechanism for informing companies of deficiencies. We submitted additional data and analyses throughout 2010, and have been informed by FDA that deficiencies remain. Acting on an option presented by the FDA to resolve the remaining open issues, Anika requested a review by the Orthopedic Advisory Panel. The Company has not yet received a date for an Advisory Panel meeting. There can be no assurance that the FDA will grant the Company's request for a orthopedic advisory panel meeting. Even if such request is granted, the panel may not find favorably for Anika, or the FDA may not accept the panel's findings even if such findings are favorable to Anika.
Our second single-injection osteoarthritis product is Cingal, which is based on the technology platform used in MONOVISC, with an added active therapeutic molecule to provide broad pain relief for a long period of time. In addition, in October 2010, Anika filed 510(k) applications with the FDA to gain marketing clearance for three FAB products: Hyalofast®, Hyaloglide®, and Hyalonect®. There has been delay in the FDA’s review process and the Company is unable to predict the timing of receipt of these clearances. There can be no guarantee that the clearance process for these product will be successful or that additional data will not be required to support clearance.
FAB’s tissue engineered products Hyalograft C Autograft, Hyalograft 3D Autograft and Laserskin Autograft are currently marketed in Europe. However, the regulations for marketing of these products in Europe have changed. Effective January 1, 2013, new regulations mandate these products to be approved by the European Medicines Agency (“EMA”) in order to remain on the EU market. FAB continues to be in discussion with the EMA and is implementing a plan to qualify for the new status. There can be no assurance that approval will be timely obtained.
-14-
In addition, we cannot assure you that:
|
|
·
|
We will begin or successfully complete U.S. clinical trials for next generation products;
|
|
|
·
|
The clinical data will support the efficacy of these products;
|
|
|
·
|
We will be able to successfully complete the FDA or foreign regulatory approval or clearance process, where required;
|
|
|
·
|
Additional clinical trials will support a PMA application and/or FDA approval or other foreign regulatory approvals, where required, in a timely manner or at all; or
|
|
|
·
|
European and other regulations may not change for the marketing of cell based products and thus impact our ability to continue commercialization of these products.
|
We also cannot assure you that any delay in receiving FDA approvals will not adversely affect our competitive position. Furthermore, even if we do receive FDA approval or clearance:
|
|
·
|
The approval may include significant limitations on the indications and other claims sought for use for which the products may be marketed;
|
|
|
·
|
The approval may include other significant conditions of approval such as post-market testing, tracking, or surveillance requirements; and
|
|
|
·
|
Meaningful sales may never be achieved.
|
Once obtained, marketing approval can be withdrawn by the FDA for a number of reasons, including, among others, the failure to comply with regulatory requirements, or the occurrence of unforeseen problems following initial approval. We may be required to make further filings with the FDA under certain circumstances. The FDA’s regulations require a PMA supplement for certain changes if they affect the safety and effectiveness of an approved device, including, but not limited to, new indications for use, labeling changes, process or manufacturing changes, the use of a different facility to manufacture, process or package the device, and changes in performance or design specifications. Our failure to receive approval of a PMA supplement regarding the use of a different manufacturing facility or any other change affecting the safety or effectiveness of an approved device on a timely basis, or at all, may have a material adverse effect on our business, financial condition, and results of operations. The FDA could also limit or prevent the manufacture or distribution of our products and has the power to require the recall of such products. It also might be necessary for us, in applicable circumstances, to initiate a voluntary recall per FDA regulations of one or several of our products. Significant delay or cost in obtaining, or failure to obtain FDA approval to market products, any FDA limitations on the use of our products, or any withdrawal or suspension of approval or rescission of approval by the FDA could have a material adverse effect on our business, financial condition, and results of operations.
In addition, all FDA approved or cleared products manufactured by us must be manufactured in compliance with the FDA’s Good Manufacturing Practices (“GMP”) regulations and, for medical devices, the FDA’s Quality System Regulations (“QSR”). Ongoing compliance with QSR and other applicable regulatory requirements is enforced through periodic inspection by state and federal agencies, including the FDA. The FDA may inspect our facilities, from time to time, to determine whether we are in compliance with regulations relating to medical device and pharmaceutical companies, including regulations concerning manufacturing, testing, quality control and product labeling practices. We cannot assure you that we will be able to comply with current or future FDA requirements applicable to the manufacture of our products.
FDA regulations depend heavily on administrative interpretation and we cannot assure you that the future interpretations made by the FDA or other regulatory bodies, with possible retroactive effect, will not adversely affect us. In addition, changes in the existing regulations or adoption of new governmental regulations or policies could prevent or delay regulatory approval of our products.
Failure to comply with applicable regulatory requirements could result in, among other things, warning letters, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, refusal of the FDA to grant pre-market clearance or pre-market approval for devices or drugs, withdrawal of approvals and criminal prosecution.
-15-
In July 2008, we received a Warning Letter (the “Warning Letter”) from the FDA in response to an earlier FDA Form 483 Notice of Observations issued to us following an inspection at our manufacturing facility in Woburn, Massachusetts. The Company submitted corrective action plans, which have been accepted by the FDA and resulted in the clearance of the Warning Letter in September 2010.
In addition to regulations enforced by the FDA, we are subject to other existing and future federal, state, local and foreign regulations. International regulatory bodies often establish regulations governing product standards, packing requirements, labeling requirements, quality system and manufacturing requirements, import restrictions, tariff regulations, duties and tax requirements. We cannot assure you that we will be able to achieve and/or maintain compliance required for CE marking or other foreign regulatory approvals for any or all of our products or that we will be able to produce our products in a timely and profitable manner while complying with applicable requirements. Federal, state, local and foreign regulations regarding the manufacture and sale of medical products are subject to change. We cannot predict what impact, if any, such changes might have on our business.
The process of obtaining approvals from the FDA and other regulatory authorities can be costly, time consuming, and subject to unanticipated delays. We cannot assure you that approvals or clearances of our products will be granted or that we will have the necessary funds to develop certain of our products. Any failure to obtain, or delay in obtaining such approvals or clearances, could adversely affect our ability to market our products.
Uncertain economic conditions, including the credit crisis affecting the financial markets and global recession, could adversely affect our business, results of operations and financial condition.
The worldwide financial markets have experienced turmoil, characterized by volatility in security prices, rating downgrades of investments and reductions in available credit. These events materially and adversely impacted the availability of financing to a wide variety of businesses, and the resulting uncertainty led to reductions in capital investments, overall spending levels, future product plans, and sales projections across industries and markets.
The financial markets remain uncertain and renewed turmoil could have a material adverse impact on our business, our ability to achieve planned results of operations and our financial condition by:
|
|
·
|
Reducing demand for our products;
|
|
|
·
|
Increasing risk of order cancellations or delays;
|
|
|
·
|
Increasing pressure on the prices for our products;
|
|
|
·
|
Creating greater difficulty in collecting accounts receivable; and
|
|
|
·
|
Increasing the risks to our liquidity, including the possibility that we might not have sufficient access to cash when needed.
|
We are unable to predict the likelihood of renewed disruption in financial markets and adverse economic conditions in the U.S. and other countries.
Substantial competition could materially affect our financial performance.
We compete with many companies, including, among others, large pharmaceutical companies, specialized medical products companies and healthcare companies. Many of these companies have substantially greater financial resources, larger research and development staffs, more extensive marketing and manufacturing organizations and more experience in the regulatory process than us. We also compete with academic institutions, governmental agencies and other research organizations that may be involved in research, development and commercialization of products. Because a number of companies are developing or have developed HA products for similar applications and have received FDA approval, the successful commercialization of a particular product will depend in part upon our ability to complete clinical studies and obtain FDA marketing and foreign regulatory approvals prior to our competitors, or, if regulatory approval is not obtained prior to our competitors, to identify markets for our products that may be sufficient to permit meaningful sales of our products. For example, we are aware of several companies that are developing and/or marketing products utilizing HA for a variety of human applications. In some cases, competitors have already obtained product approvals, submitted applications for approval or have commenced human clinical studies, either in the U.S. or in certain foreign countries. There exist major competing products for the use of HA in ophthalmic surgery. In addition, certain HA products made by our competitors for the treatment of osteoarthritis in the knee have received FDA approval before ours and have been marketed in the U.S. since 1997, as well as select markets in Canada, Europe and other countries. To date, the FDA approved nine HA products for the treatment of facial wrinkles which have been marketed internationally for a number of years. There can be no assurance that we will be able to compete against current or future competitors or that competition will not have a material adverse effect on our business, financial condition and results of operations.
-16-
We are uncertain regarding the success of our clinical trials.
Several of our products do require clinical trials to determine their safety and efficacy for U.S. and international marketing approval by regulatory bodies, including the FDA. There can be no assurance that we will be able to successfully complete the U.S. or international regulatory approval process for products in development. In addition, there can be no assurance that we will not encounter additional problems that will cause us to delay, suspend or terminate our clinical trials. In addition, we cannot make any assurance that clinical trials will be deemed sufficient in size and scope to satisfy regulatory approval requirements, or, if completed, will ultimately demonstrate these products to be safe and efficacious. We completed a pivotal clinical trial on MONOVISC and submitted the data as part of our PMA filing in December 2009. We were informed that there were deficiencies in our submissions through a deficiency/non-approvable letter, which is the FDA's mechanism for informing companies of deficiencies. We submitted additional data and analyses throughout 2010, and have been informed by FDA that deficiencies remain. Acting on an option presented by the FDA to resolve the remaining open issues, Anika requested a review by the Orthopedic Advisory Panel. The Company has not yet received a date for an Advisory Panel meeting.
We are dependent upon marketing and distribution partners and the failure to maintain strategic alliances on acceptable terms will have a material adverse effect on our business, financial condition and results of operations.
Our success will be dependent, in part, upon the efforts of our marketing and distribution partners and the terms and conditions of our relationships with such partners. We cannot assure you that such partners will not seek to renegotiate their current agreements on terms less favorable to us or terminate such agreements. We are continuing to seek to establish long-term distribution relationships in regions not covered by existing agreements, but can make no assurances that we will be successful in doing so. There can be no assurance that we will be able to identify or engage appropriate distribution or collaboration partners or effectively transition to any such partners. There can be no assurance that we will obtain European or other reimbursement approvals or, if such approvals are obtained, they will be obtained on a timely basis or at a satisfactory level of reimbursement.
We may need to obtain the assistance of additional marketing partners to bring new and existing products to market and to replace certain marketing partners. The failure to establish strategic partnerships for the marketing and distribution of our products on acceptable terms will have a material adverse effect on our business, financial condition, and results of operations.
Anika has never directly commercialized products on our own before.
We have announced our desire to pursue the option of directly commercializing MONOVISC and certain FAB orthopedic products in the United States. Historically Anika has sold its products through a network of distributors, and there can be no assurance that we will successfully find and hire the appropriate people to succeed in a direct commercialization effort. We will be competing against larger companies with greater resources and portfolios of products for access to the customer. In addition, we will have limited resources for advertising and promotion of the products.
Our future success depends upon market acceptance of our existing and future products.
Our success will depend in part upon the acceptance of our existing and future products by the medical community, hospitals and physicians and other health care providers, third-party payers, and end-users. Such acceptance may depend upon the extent to which the medical community and end-users perceive our products as safer, more effective or cost-competitive than other similar products. Ultimately, for our new products to gain general market acceptance, it may also be necessary for us to develop marketing partners for the distribution of our products. There can be no assurance that our new products will achieve significant market acceptance on a timely basis, or at all. Failure of some or all of our future products to achieve significant market acceptance could have a material adverse effect on our business, financial condition, and results of operations.
-17-
We may be unable to adequately protect our intellectual property rights.
Our efforts to enforce our intellectual property rights may not be successful. We rely on a combination of copyright, trademark, patent and trade secret laws, confidentiality procedures and contractual provisions to protect our proprietary rights. Our success will depend, in part, on our ability to obtain and enforce patents, protect trade secrets, obtain licenses to technology owned by third parties when necessary, and conduct our business without infringing on the proprietary rights of others. The patent positions of pharmaceutical, medical products and biotechnology firms, including ours, can be uncertain and involve complex legal and factual questions. There can be no assurance that any patent applications will result in the issuance of patents or, if any patents are issued, whether they will provide significant proprietary protection or commercial advantage, or will not be circumvented by others. In the event a third party has also filed one or more patent applications for any of its inventions, we may have to participate in interference proceedings declared by the United States Patent and Trademark Office (“PTO”) to determine priority of invention, which could result in failure to obtain, or the loss of, patent protection for the inventions and the loss of any right to use the inventions. Even if the eventual outcome is favorable to us, such interference proceedings could result in substantial cost to us, and diversion of management’s attention away from our operations. Filing and prosecution of patent applications, litigation to establish the validity and scope of patents, assertion of patent infringement claims against others and the defense of patent infringement claims by others can be expensive and time consuming. There can be no assurance that in the event that any claims with respect to any of our patents, if issued, are challenged by one or more third parties, that any court or patent authority ruling on such challenge will determine that such patent claims are valid and enforceable. An adverse outcome in such litigation could cause us to lose exclusivity covered by the disputed rights. If a third party is found to have rights covering products or processes used by us, we could be forced to cease using the technologies or marketing the products covered by such rights, could be subject to significant liabilities to such third party, and could be required to license technologies from such third party. Furthermore, even if our patents are determined to be valid, enforceable, and broad in scope, there can be no assurance that competitors will not be able to design around such patents and compete with us using the resulting alternative technology. We have a policy of seeking patent protection for patentable aspects of our proprietary technology. We intend to seek patent protection with respect to products and processes developed in the course of our activities when we believe such protection is in our best interest and when the cost of seeking such protection is not inordinate. However, no assurance can be given that any patent application will be filed, that any filed applications will result in issued patents or that any issued patents will provide us with a competitive advantage or will not be successfully challenged by third parties. The protections afforded by patents will depend upon their scope and validity, and others may be able to design around our patents.
Other entities have filed patent applications for or have been issued patents concerning various aspects of HA-related products or processes. There can be no assurance that the products or processes developed by us will not infringe on the patent rights of others in the future. Any such infringement may have a material adverse effect on our business, financial condition, and results of operations.
We also rely upon trade secrets and proprietary know-how for certain non-patented aspects of our technology. To protect such information, we require all employees, consultants and licensees to enter into confidentiality agreements limiting the disclosure and use of such information. There can be no assurance that these agreements provide meaningful protection or that they will not be breached, that we would have adequate remedies for any such breach, or that our trade secrets, proprietary know-how, and our technological advances will not otherwise become known to others. In addition, there can be no assurance that, despite precautions taken by us, others have not and will not obtain access to our proprietary technology. Further, there can be no assurance that third parties will not independently develop substantially equivalent or better technology.
Pursuant to the 2004 B&L Agreement, we have agreed to transfer to Bausch & Lomb, upon expiration of the term of the 2004 B&L Agreement on December 31, 2010, our manufacturing process, know-how and technical information, which relate only to AMVISC products.
Effective January 1, 2011, we entered into a non-exclusive, two year contract with B&L intended to transition the manufacture of AMVISC and AMVISC Plus to an alternative, recently acquired low-cost supplier to B&L. Under the 2004 B&L Agreement, the Company was restricted in its ability to commercialize viscoelastic products to only existing customers (STAAR Surgical Company and Hoya Surgical Optics, Inc.). That restriction has now expired and we are free to market our own viscoelastic product AnikaVisc.
-18-
B&L accounted for 21% of product revenue for the year ended 2010 and product revenue is expected to be significantly lower in 2011 under the new transition contract. Operating margins under the 2004 B&L Agreement were low, and the Company expects to see margin improvement through commercialization of its new AnikaVisc product. There can be no assurance that AnikaVisc will be successfully sold or that it will generate any profit for the Company.
Our manufacturing processes involve inherent risks and disruption could materially adversely affect our business, financial condition and results of operations.
The operation of biomedical manufacturing plants involves many risks, including the risks of breakdown, failure or substandard performance of equipment, the occurrence of natural and other disasters, and the need to comply with the requirements of directives of government agencies, including the FDA. In addition, we rely on a single supplier for certain key raw materials and a small number of suppliers for a number of other materials required for the manufacturing and delivery of our HA products. Although we believe that alternative sources for many of these and other components and raw materials that we use in our manufacturing processes are available, any supply interruption could harm our ability to manufacture our products until a new source of supply is identified and qualified. We may not be able to find a sufficient alternative supplier in a reasonable time period, or on commercially reasonable terms, if at all, and our ability to produce and supply our products could be impaired.
Furthermore, our manufacturing processes and research and development efforts involve animals and products derived from animals. We procure our animal-derived raw materials from qualified vendors, control for contamination and have processes that effectively inactivate infectious agents; however, we cannot assure you that we can completely eliminate the risk of transmission of infectious agents. Furthermore, regulatory authorities could in the future impose restrictions on the use of animal-derived raw materials that could impact our business.
The utilization of animals in research and development and product commercialization is subject to increasing focus by animal rights activists. The activities of animal rights groups and other organizations that have protested animal based research and development programs or boycotted the products resulting from such programs could cause an interruption in our manufacturing processes and research and development efforts. The occurrence of material operational problems, including but not limited to the events described above, could have a material adverse effect on our business, financial condition, and results of operations during the period of such operational difficulties.
Our new facility construction and validation processes could materially adversely affect our operations.
We entered into a new lease on January 4, 2007, for a new headquarters facility consisting of approximately 134,000 square feet of general office, research and development and manufacturing space located in Bedford, Massachusetts. The lease has an initial term of ten and a half years, and commenced on approximately May 1, 2007 when certain agreed upon landlord improvements were completed. We commenced the build-out of the new facility during the second quarter of 2007. Our administrative, marketing, regulatory, and research and development personnel moved into the Bedford facility in November 2007. The remaining build-out was completed in mid-2008 and validation and approval for operation in the new manufacturing space is ongoing.
We received FDA approval to manufacture our terminally sterilized product, ELEVESS™, in our Bedford facility in November 2010. We believed that we had an agreement with the FDA to temporarily move certain critical equipment used to manufacture our ophthalmic and orthopedic products to Bedford, validate its use at that facility and then briefly return it to service in Woburn. We understood such validation data and reports would be utilized as part of a final inspection of the Bedford facility scheduled for December 2010. That final inspection did not occur and will not occur now until the equipment is permanently installed in Bedford. In order to fill product orders and build sufficient safety stock to accommodate any further approval delays, we currently expect manufacturing of the ophthalmic and orthopedic products to continue in Woburn into June 2011.
We provide no assurance that the installation of such equipment or the validation and approval processes for our Bedford facility will be completed in a timely fashion, if at all. Furthermore, we cannot assure you that the transition from the existing facilities to the new facility will be efficient and successful. In the event the validation or approval is delayed or the move transition is unsuccessful, it may result in business interruptions. We have incurred additional expense as a result of maintaining two facilities, and will continue to incur additional expenses if we have to maintain both facilities, for a prolonged period.
-19-
Our financial performance depends on the continued growth and demand for our products and we may not be able to successfully manage the expansion of our operations.
Our future success depends on substantial growth in product sales. There can be no assurance that such growth can be achieved or, if achieved, can be sustained. There can be no assurance that even if substantial growth in product sales and the demand for our products is achieved, we will be able to:
|
|
·
|
Develop the necessary manufacturing capabilities;
|
|
|
·
|
Obtain the assistance of additional marketing partners;
|
|
|
·
|
Attract, retain and integrate the required key personnel; and
|
|
|
·
|
Implement the financial, accounting and management systems needed to manage growing demand for our products.
|
Our failure to successfully manage future growth could have a material adverse effect on our business, financial condition, and results of operations.
We engage in acquisitions as a part of our growth strategy in which we will incur a variety of costs and may never realize the anticipated benefits of such acquisitions.
Our business strategy includes the acquisition of businesses, technologies, services or products that we believe are a strategic fit with our business. Such acquisitions could reduce stockholders’ ownership, cause us to incur debt, expose us to liabilities and result in amortization expenses related to intangible assets with definite lives. In addition, acquisitions involve other risks, including diversion of management resources otherwise available for ongoing development of our business and risks associated with entering new markets with which we have limited experience or where distribution alliances with experienced distributors are not available. Our future profitability may depend in part upon our ability to develop further our resources to adapt to these new products or business areas and to identify and enter into satisfactory distribution networks. Moreover, we may fail to realize the anticipated benefits of any acquisition as rapidly as expected or at all, or the acquired business may not perform in accordance with our expectations. We may also incur significant expenditures in anticipation of an acquisition that is never realized.
We may not realize the expected benefits from acquisitions due to difficulties integrating the businesses, operations and product lines.
Our ability to achieve the benefits of acquisitions depends in part on the integration and leveraging of technology, products, operations, sales and marketing channels and personnel. If we undertake any acquisition, the process of integrating an acquired business may result in unforeseen operating difficulties and expenditures and may absorb significant management attention that would otherwise be available for ongoing development of our business even if completed in a timely and efficient manner.
We may have difficulty successfully integrating acquired businesses, the domestic and foreign operations or the product lines, and as a result, we may not realize any of the anticipated benefits of the acquisitions. Moreover, we may lose key clients or employees of acquired businesses as a result of the change in ownership to us. Additionally, we cannot assure that our growth rate will equal the growth rates that have been experienced by us and the acquired companies, respectively, operating as separate companies in the past.
Customer, vendor and employee uncertainty about the effects of any acquisitions could harm us.
We and the customers of any companies we acquire may, in response to the consummation of any acquisitions, delay or defer purchasing decisions. Any delay or deferral in purchasing decisions by customers could adversely affect our business. Similarly, employees of acquired companies may experience uncertainty about their future role until or after we execute our strategies with regard to employees of acquired companies. This may adversely affect our ability to attract and retain key management, sales, marketing and technical personnel following an acquisition.
-20-
The acquisitions we have made or may make in the future may make us the subject of lawsuits from either an acquired company’s stockholders, an acquired company’s previous stockholders or our current stockholders.
We may be the subject of lawsuits from either an acquired company’s stockholders, an acquired company’s previous stockholders or our current stockholders. These lawsuits could result from the actions of the acquisition target prior to the date of the acquisition, from the acquisition transaction itself or from actions after the acquisition. Defending potential lawsuits could cost us significant expense and detract management’s attention from the operation of the business. Additionally, these lawsuits could result in the cancellation of or the inability to renew, certain insurance coverage that would be necessary to protect our assets.
Attractive acquisition opportunities may not be available to us in the future.
We will consider the acquisition of other businesses. However, we may not have the opportunity to make suitable acquisitions on favorable terms in the future, which could negatively impact the growth of our business. In order to pursue such opportunities, we may require significant additional financing, which may not be available to us on favorable terms, if at all. The availability of such financing is limited by the recent tightening of the global credit markets. We expect that our competitors, many of which have significantly greater resources than we do, will compete with us to acquire compatible businesses. This competition could increase prices for acquisitions that we would likely pursue.
Sales of our products are largely dependent upon third party reimbursement and our performance may be harmed by health care cost containment initiatives.
In the U.S. and other markets, health care providers, such as hospitals and physicians, that purchase health care products, such as our products, generally rely on third party payers, including Medicare, Medicaid and other health insurance and managed care plans, to reimburse all or part of the cost of the health care product. We depend upon the distributors for our products to secure reimbursement and reimbursement approvals. Reimbursement by third party payers may depend on a number of factors, including the payer’s determination that the use of our products is clinically useful and cost-effective, medically necessary and not experimental or investigational. Since reimbursement approval is required from each payer individually, seeking such approvals can be a time consuming and costly process which, in the future, could require us or our marketing partners to provide supporting scientific, clinical and cost-effectiveness data for the use of our products to each payer separately. Significant uncertainty exists as to the reimbursement status of newly approved health care products, and any failure or delay in obtaining reimbursement approvals can negatively impact sales of our new products. In addition, third party payers are increasingly attempting to contain the costs of health care products and services by limiting both coverage and the level of reimbursement for new therapeutic products and by refusing, in some cases, to provide coverage for uses of approved products for disease indications for which the FDA has not granted marketing approval. Also, Congress and certain state legislatures have considered reforms that may affect current reimbursement practices, including controls on health care spending through limitations on the growth of Medicare and Medicaid spending. There can be no assurance that third party reimbursement coverage will be available or adequate for any products or services developed by us. Outside the U.S., the success of our products is also dependent in part upon the availability of reimbursement and health care payment systems. Domestic and international reimbursement laws and regulations may change from time to time. Lack of adequate coverage and reimbursement provided by governments and other third party payers for our products and services, including change of classification by CMS for ORTHOVISC under a unique Q-code for Medicare/Medicaid reimbursement, could have a material adverse effect on our business, financial condition, and results of operations.
We may seek financing in the future, which could be difficult to obtain and which could dilute your ownership interest or the value of your shares.
We had cash and cash equivalents of approximately $28.2 million at December 31, 2010. Our future capital requirements and the adequacy of available funds will depend, however, on numerous factors, including:
|
|
·
|
Market acceptance of our existing and future products;
|
|
|
·
|
The success and sales of our products under various distributor agreements;
|
|
|
·
|
The successful commercialization of products in development;
|
|
|
·
|
Progress in our product development efforts;
|
-21-
|
|
·
|
The magnitude and scope of such product development efforts;
|
|
|
·
|
Any potential acquisitions of products, technologies or businesses;
|
|
|
·
|
Progress with preclinical studies, clinical trials and product clearances by the FDA and other agencies;
|
|
|
·
|
The cost and timing of our efforts to manage our manufacturing capabilities and related costs;
|
|
|
·
|
The cost and timing of validation and approval processes for our new manufacturing space;
|
|
|
·
|
The cost of filing, prosecuting, defending and enforcing patent claims and other intellectual property rights and the cost of defending any other legal proceeding;
|
|
|
·
|
Competing technological and market developments;
|
|
|
·
|
The development of strategic alliances for the marketing of certain of our products;
|
|
|
·
|
The terms of such strategic alliances, including provisions (and our ability to satisfy such provisions) that provide upfront and/or milestone payments to us;
|
|
|
·
|
Our abilities to meet debt covenant and repayment requirements; and
|
|
|
·
|
The cost of maintaining adequate inventory levels to meet current and future product demands.
|
To the extent funds generated from our operations, together with our existing capital resources are insufficient to meet future requirements, we will be required to obtain additional funds through equity or debt financings, strategic alliances with corporate partners and others, or through other sources. The terms of any future equity financings may be dilutive to you and the terms of any debt financings may contain restrictive covenants, which limit our ability to pursue certain courses of action. Our ability to obtain financing is dependent on the status of our future business prospects as well as conditions prevailing in the relevant capital markets. No assurance can be given that any additional financing will be made available to us or will be available on acceptable terms should such a need arise.
We are subject to debt covenants and any failure to comply with these could materially adversely affect our business, financial condition and results of operations.
On January 31, 2008, we entered into a Credit Agreement (the “Credit Agreement”). Under the Credit Agreement, our lender made periodic loans to us through December 31, 2008. We borrowed $16,000,000 in 2008, the maximum allowed amount under the Credit Agreement. At December 31, 2008, the borrowings were converted into a 7-year term loan. On December 30, 2009, the Credit Agreement was amended as part of the FAB acquisition. The Credit Agreement was entered into in order to finance the construction and validation of our Bedford facility. Construction of the new facility commenced in the spring of 2007 and was substantially completed in mid-2008. Validation of our new manufacturing facility will continue into 2011. See Note 15 to our Consolidated Financial Statements for additional information relative to this debt facility.
There can be no assurance that we will be successful in qualifying the new facility under the FDA and European Union regulations. The Credit Agreement contains certain debt covenants, representations and warranties that we must comply with. If we do not comply with the specified covenants and restrictions, we could be in default under our Credit Agreement. Our ability to comply with the provisions of our Credit Agreement governing our other indebtedness may be affected by changes in the economic or business conditions or other events beyond our control.
We could become subject to product liability claims, which, if successful, could materially adversely affect our business, financial condition and results of operations.
The testing, marketing and sale of human health care products entail an inherent risk of allegations of product liability, and there can be no assurance that substantial product liability claims will not be asserted against us. Although we have not received any material product liability claims to date and have an insurance policy of $5,000,000 per occurrence and $5,000,000 in the aggregate to cover such claims should they arise, there can be no assurance that material claims will not arise in the future or that our insurance will be adequate to cover all situations. Moreover, there can be no assurance that such insurance, or additional insurance, if required, will be available in the future or, if available, will be available on commercially reasonable terms. Any product liability claim, if successful, could have a material adverse effect on our business, financial condition and results of operations.
-22-
Our business is dependent upon hiring and retaining qualified management and technical personnel.
We are highly dependent on the members of our management and technical staff, the loss of one or more of whom could have a material adverse effect on us. We have experienced a number of management changes in recent years. There can be no assurances that such management changes will not adversely affect our business. We believe that our future success will depend in large part upon our ability to attract and retain highly skilled, technical, managerial and manufacturing personnel. We face significant competition for such personnel from other companies, research and academic institutions, government entities and other organizations. There can be no assurance that we will be successful in hiring or retaining the personnel we require. The failure to hire and retain such personnel could have a material adverse effect on our business, financial condition and results of operations.
We are subject to environmental regulations and any failure to comply with applicable laws could subject us to significant liabilities and harm our business.
We are subject to a variety of local, state and federal government regulations relating to the storage, discharge, handling, emission, generation, manufacture and disposal of toxic, or other hazardous substances used in the manufacture of our products. Any failure by us to control the use, disposal, removal or storage of hazardous chemicals or toxic substances could subject us to significant liabilities, which could have a material adverse effect on our business, financial condition, and results of operations.
As our international sales and operations grow, including through our recent acquisition of FAB, we could become increasingly subject to additional economic, political and other risks that could harm our business.
Since we manufacture and sell our products worldwide, our business is subject to risks associated with doing business internationally. During the years ended December 31, 2010 and 2009, approximately, 31% and 25%, respectively, of our product sales were to international distributors. However, as a result of our acquisition of FAB, we anticipate the percentage of our product sales resulting from international operations to increase in fiscal year 2011 offset by lower B&L revenue. As a result of this international growth, we have become increasingly subject to a variety of risks, which could cause fluctuations in the results of our international and domestic operations. These risks include:
|
|
·
|
The impact of recessions and other economic conditions in economies, including Europe in particular, outside the United States;
|
|
|
·
|
Instability of foreign economic, political and labor conditions;
|
|
|
·
|
Unfavorable labor regulations applicable to European operations, such as severance and the unenforceability of non-competition agreements in the European Union;
|
|
|
·
|
The impact of strikes, work stoppages, work slowdowns, grievances, complaints, claims of unfair labor practices or other collective bargaining disputes;
|
|
|
·
|
Difficulties in complying with restrictions imposed by regulatory or market requirements, tariffs or other trade barriers or by U.S. export laws;
|
|
|
·
|
Imposition of governmental controls limiting the volume of international sales;
|
|
|
·
|
Longer accounts receivable payment cycles;
|
|
|
·
|
Potentially adverse tax consequences, including, if required, difficulties transferring funds generated in non-U.S. jurisdictions to the U.S. in a tax efficient manner;
|
|
|
·
|
Difficulties in protecting intellectual property;
|
-23-
|
|
·
|
Difficulties in managing international operations; and
|
|
|
·
|
Burdens of complying with a wide variety of foreign laws.
|
Our success depends, in part, on our ability to anticipate and address these risks. We cannot guarantee that these or other factors will not adversely affect our business or operating results.
Currency exchange rate fluctuations may have a negative impact on our reported earnings.
Approximately 15% of our business from continuing operations during fiscal year 2010 was conducted in functional currencies other than the U.S. dollar, which is our reporting currency. As a result of our acquisition of FAB, we anticipate this percentage to increase to approximately 20% during fiscal year 2011. Thus, currency fluctuations among the U.S. dollar and the other currencies in which we do business have caused and will continue to cause foreign currency transaction gains and losses. Currently, we attempt to manage foreign currency risk through the matching of assets and liabilities. In the future, we may undertake to manage foreign currency risk through additional hedging methods. We recognize foreign currency gains or losses arising from our operations in the period incurred. We cannot guarantee that we will be successful in managing foreign currency risk or in predicting the effects of exchange rate fluctuations upon our future operating results because of the variability of currency exposure and the potential volatility of currency exchange rates.
Our stock price has been and may remain highly volatile, and we cannot assure you that market making in our common stock will continue.
The market price of shares of our common stock may be highly volatile. Factors such as announcements of new commercial products or technological innovations by us or our competitors, disclosure of results of clinical testing or regulatory proceedings, governmental regulation and approvals, developments in patent or other proprietary rights, public concern as to the safety of products developed by us and general market conditions may have a significant effect on the market price of our common stock. The trading price of our common stock could be subject to wide fluctuations in response to quarter-to-quarter variations in our operating results, material announcements by us or our competitors, governmental regulatory action, conditions in the health care industry generally or in the medical products industry specifically, or other events or factors, many of which are beyond our control. In addition, the stock market has experienced extreme price and volume fluctuations which have particularly affected the market prices of many medical products companies and which often have been unrelated to the operating performance of such companies. Our operating results in future quarters may be below the expectations of equity research analysts and investors. In such event, the price of our common stock would likely decline, perhaps substantially.
No person is under any obligation to make a market in the common stock or to publish research reports on us, and any person making a market in the common stock or publishing research reports on us may discontinue market making or publishing such reports at any time without notice. There can be no assurance that an active public market in our common stock will be sustained.
Our charter documents contain anti-takeover provisions that may prevent or delay an acquisition of us.
Certain provisions of our Restated Articles of Organization and Amended and Restated By-laws could have the effect of discouraging a third party from pursuing a non-negotiated takeover of us and preventing certain changes in control. These provisions include a classified Board of Directors, advance notice to the Board of Directors of stockholder proposals, limitations on the ability of stockholders to remove directors and to call stockholder meetings, the provision that vacancies on the Board of Directors be filled by vote of a majority of the remaining directors. In addition, the Board of Directors renewed a Shareholders Rights Plan in April 2008. We are also subject to Chapter 110F of the Massachusetts General Laws which, subject to certain exceptions, prohibits a Massachusetts corporation from engaging in any of a broad range of business combinations with any “interested stockholder” for a period of three years following the date that such stockholder became an interested stockholder. These provisions could discourage a third party from pursuing a takeover of us at a price considered attractive by many stockholders, since such provisions could have the effect of preventing or delaying a potential acquirer from acquiring control of us and our Board of Directors.
-24-
Our revenues are derived from a small number of customers, the loss of which could materially adversely affect our business, financial condition and results of operations.
We have historically derived the majority of our revenues from a small number of customers, most of whom resell our products to end-users and most of whom are significantly larger companies than us. For the year ended December 31, 2010, five customers accounted for 82% of product revenue. We expect to continue to be dependent on a small number of large customers for the majority of our revenues. Our failure to generate as much revenue as expected from these customers or the failure of these customers to purchase our products would seriously harm our business. In addition, if present and future customers terminate their purchasing arrangements with us, significantly reduce or delay their orders, or seek to renegotiate their agreements on terms less favorable to us, our business, financial condition, and results of operations will be adversely affected. If we accept terms less favorable than the terms of the current agreement, such renegotiations may have a material adverse effect on our business, financial condition, and/or results of operations. Furthermore, in any future negotiations we may be subject to the perceived or actual leverage that these customers may have given their relative size and importance to us. Any termination, change, reduction or delay in orders could seriously harm our business, financial condition, and results of operations. Accordingly, unless and until we diversify and expand our customer base, our future success will significantly depend upon the timing and size of future purchases by our largest customers and the financial and operational success of these customers. The loss of any one of our major customers or the delay of significant orders from such customers, even if only temporary, could reduce or delay our recognition of revenues, harm our reputation in the industry, and reduce our ability to accurately predict cash flow, and, as a consequence, could seriously harm our business, financial condition, and results of operations.
We may not fully realize the benefits of our acquisitions or strategic alliances.
In December 2009, we acquired FAB which we accounted for as a business combination. We may not be able to realize the expected synergies and cost savings from the integration with our existing operations or technologies that we may acquire. In addition, the integration process for our acquisitions may be complex, costly, and time consuming and include unanticipated issues, expenses and liabilities. We may have difficulty in developing, manufacturing and marketing the products of a newly acquired company in a manner that enhances the performance of our combined businesses or product lines and allows us to realize value from expected synergies. Following an acquisition, we may not achieve the revenue or net income levels that justify the acquisition. Acquisitions may also result in one-time charges, such as write-offs or restructuring charges, or in the future, impairment of goodwill or acquired IPR&D, which adversely affect our operating results. Additionally, we may fund acquisitions of new businesses, strategic alliances or joint ventures by utilizing our cash, incurring debt, issuing shares of our common stock, or by other means.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
Our corporate headquarters is located in Bedford, Massachusetts, where we lease approximately 134,000 square feet of administrative, research and development and manufacturing space. We entered into this lease on January 4, 2007, and the lease commenced on May 1, 2007 for an initial term of ten and a half years. We have an option under the Lease to extend its terms for up to four periods beyond the original expiration date subject to the condition that we notify the landlord that we are exercising each option at least one year prior to the expiration of the original or current term thereof. The first three renewal options each extend the term an additional five years with the final renewal option extending the term six years. Our administrative, marketing, regulatory, and research and development personnel moved into the Bedford facility in November of 2007. The remaining build-out at the Bedford facility was completed in mid-2008. We received FDA approval to manufacture our terminally sterilized product, ELEVESS™, in our Bedford facility in November 2010 and are waiting to schedule the final FDA inspection for the manufacture of the Company’s ophthalmic and orthopedic products. As a result, the validation for this manufacturing space will continue into 2011.
Our prior corporate headquarters was located in Woburn, Massachusetts and the lease for that facility ended on December 31, 2007. We also lease approximately 37,000 square feet of space at a separate location in Woburn, Massachusetts, which currently houses our manufacturing facility and warehouse for several major products. This facility has received all FDA, state and European regulatory approvals to operate as a sterile device and drug manufacturer. We extended our lease for this facility to June 30, 2011. As part of the acquisition of FAB, we now lease approximately 26,000 square feet of laboratory, warehouse and office space in Abano Terme, Italy. The lease commenced on December 30, 2009 for an initial term of six (6) years. For the year ended December 31, 2010, we had aggregate facility lease expenses of approximately $2,888,277.
-25-
Our aggregate expenditures to build out the Bedford facility, which will serve as our corporate headquarters and manufacturing facility for the foreseeable future, were approximately $34.6 million through December 31, 2010. We have borrowed $16 million under our Credit Agreement which we entered into on January 31, 2008. There can be no assurance that we will be successful in re-qualifying the new facility under the FDA and European Union regulations, in which case we may need to further extend our Woburn lease.
ITEM 3. LEGAL PROCEEDINGS
On July 7, 2010, Genzyme Corporation filed a complaint against the Company in the United States District Court for the District of Massachusetts seeking unspecified damages and equitable relief. The Complaint alleges that the Company has infringed U.S. Patent No. 5,143,724 by manufacturing MONOVISC in the United States for sale outside the United States and will infringe U.S. Patent Nos. 5,143,724 and 5,399,351 if the Company begins manufacture and sale of MONOVISC in the United States. On August 30, 2010, the Company filed an answer denying liability. The Company believes that neither MONOVISC, nor its manufacture, does or will infringe any valid and enforceable claim of the asserted patents. Management has assessed and determined that contingent losses related to this matter are remote and inestimable. Therefore, pursuant to Accounting Standards Codification (“ASC”) 450, Contingencies, an accrual has not been recorded for this loss contingency.
Artes Medical, Inc. (“Artes”), the former U.S. distributor of HYDRELLE, filed a liquidating bankruptcy case under Chapter 7 of the United States Bankruptcy Code. Artes’ Trustee in Bankruptcy demanded the Company pay $359,768 to the Trustee, representing the total amount of three payments received by the Company from Artes within the 90 days prior to the filing of Artes' liquidating bankruptcy. The Trustee asserts that the payments are recoverable as preferences under the Bankruptcy Code.
The Company believes that the payments it received either do not meet the legal requirements of avoidable preferences or are subject to one or more exceptions to the Trustee's powers to recover preferences and has recently so advised the Trustee. Management has assessed and determined that contingent losses related to this matter are remote. Therefore, pursuant to ASC 450, an accrual has not been recorded for this loss contingency.
We are also involved in various other legal proceedings arising in the normal course of business. Although the outcomes of these other legal proceedings are inherently difficult to predict, we do not expect the resolution of these other legal proceedings to have a material adverse effect on our financial position, results of operations or cash flow.
ITEM 4. (Removed and Reserved).
-26-
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
COMMON STOCK INFORMATION
Our common stock has traded on the NASDAQ Global Select Market since November 25, 1997, under the symbol “ANIK.” The following table sets forth, for the periods indicated, the high and low sales prices of our common stock on the NASDAQ Global Select Market. These prices represent prices between dealers and do not include retail mark-ups, markdowns, or commissions and may not necessarily represent actual transactions.
|
Year Ended December 31, 2010
|
High
|
Low
|
||||||
|
First Quarter
|
$ | 7.97 | $ | 6.04 | ||||
|
Second Quarter
|
7.40 | 5.83 | ||||||
|
Third Quarter
|
6.48 | 4.83 | ||||||
|
Fourth Quarter
|
6.98 | 5.30 | ||||||
|
Year Ended December 31, 2009
|
High
|
Low
|
||||||
|
First Quarter
|
$ | 5.01 | $ | 3.05 | ||||
|
Second Quarter
|
5.80 | 4.51 | ||||||
|
Third Quarter
|
7.15 | 4.81 | ||||||
|
Fourth Quarter
|
9.05 | 6.11 | ||||||
At December 31, 2010, the closing price per share of our common stock was $6.67 as reported on the NASDAQ Global Select Market and there were approximately 232 holders of record. We believe that the number of beneficial owners of our common stock at that date was substantially greater.
We have never declared or paid any cash dividends on our common stock. We currently intend to retain earnings, if any, for use in our business and do not anticipate paying cash dividends on our common stock in the foreseeable future. Payment of future dividends, if any, on our common stock will be at the discretion of our Board of Directors after taking into account various factors, including our financial condition, operating results, anticipated cash needs, and plans for expansion.
Performance Graph (Unaudited)
Set forth below is a graph comparing the total returns of the Company, the NASDAQ Composite Index and the NASDAQ Biotechnology Index. The graph assumes $100 is invested on December 31, 2005 in the Company's Common Stock and each of the indices.
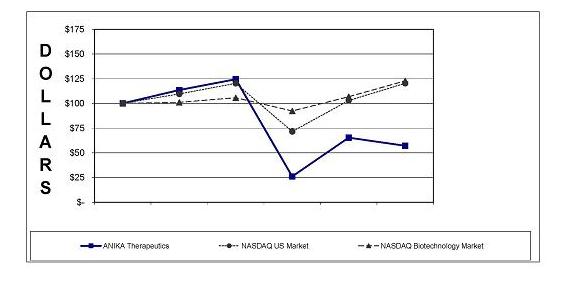
|
Dec-05
|
Dec-06
|
Dec-07
|
Dec-08
|
Dec-09
|
Dec-10
|
|||||||||||||||||||
|
Anika Therapeutics Inc
|
$ | 100.00 | $ | 113.52 | $ | 124.47 | $ | 26.01 | $ | 65.27 | $ | 57.06 | ||||||||||||
|
NASDAQ Composite Index
|
100.00 | 109.52 | 120.27 | 71.51 | 102.89 | 120.29 | ||||||||||||||||||
|
NASDAQ Biotechnology Index
|
100.00 | 101.02 | 105.65 | 92.31 | 106.74 | 122.76 | ||||||||||||||||||
-27-
ITEM 6. SELECTED FINANCIAL DATA
The following selected consolidated financial data should be read in conjunction with the Consolidated Financial Statements and the Notes thereto and Management’s Discussion and Analysis of Financial Condition and Results of Operations included elsewhere in this Annual Report on Form 10-K. The Balance Sheet Data at December 31, 2010 and 2009 and the Statement of Operations Data for each of the three years ended December 31, 2010, 2009 and 2008 have been derived from the audited Consolidated Financial Statements for such years, included elsewhere in this Annual Report on Form 10-K. The Balance Sheet Data at December 31, 2008, 2007 and 2006, and the Statement of Operations Data for each of the two years in the period ended December 31, 2007 have been derived from the audited Consolidated Financial Statements for such years not included in this Annual Report on Form 10-K.
|
Statement of Operations Data
|
||||||||||||||||||||
|
(In thousands, except per share data)
|
||||||||||||||||||||
|
Years ended December 31,
|
||||||||||||||||||||
|
2010
|
2009
|
2008
|
2007
|
2006
|
||||||||||||||||
|
Product Revenue
|
$ | 52,736 | $ | 37,321 | $ | 33,055 | $ | 26,905 | $ | 23,953 | ||||||||||
|
Licensing, milestone and contract revenue
|
2,821 | 2,815 | 2,725 | 3,925 | 2,887 | |||||||||||||||
|
Total revenue
|
55,557 | 40,136 | 35,780 | 30,830 | 26,840 | |||||||||||||||
|
Cost of product revenue
|
23,827 | 13,670 | 13,189 | 11,881 | 11,118 | |||||||||||||||
|
Product gross profit
|
28,909 | 23,651 | 19,866 | 15,024 | 12,835 | |||||||||||||||
|
Product gross margin
|
55 | % | 63 | % | 60 | % | 56 | % | 54 | % | ||||||||||
|
Total operating expenses
|
48,019 | 34,549 | 31,533 | 24,242 | 21,413 | |||||||||||||||
|
Net Income
|
4,316 | 3,688 | 3,629 | 6,035 | 4,604 | |||||||||||||||
|
Diluted net income per common share
|
0.32 | 0.32 | 0.32 | 0.53 | 0.41 | |||||||||||||||
|
Diluted common shares outstanding
|
13,647 | 11,562 | 11,461 | 11,454 | 11,155 | |||||||||||||||
|
Balance Sheet Data
|
||||||||||||||||||||
|
(In thousands)
|
||||||||||||||||||||
|
Years ended December 31,
|
||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
|
Cash, cash equivalents and short-term investments
|
$ | 28,202 | $ | 24,427 | $ | 43,194 | $ | 39,406 | $ | 47,167 | ||||||||||
|
Working capital
|
36,952 | 33,307 | 46,798 | 41,805 | 52,145 | |||||||||||||||
|
Total assets
|
128,937 | 129,431 | 95,821 | 79,497 | 68,114 | |||||||||||||||
|
Retained earnings
|
25,786 | 21,470 | 17,782 | 14,153 | 8,118 | |||||||||||||||
|
Stockholder's equity
|
85,190 | 82,144 | 60,757 | 54,961 | 45,488 | |||||||||||||||
Anika acquired 100% of the issued and outstanding stock of FAB on December 30, 2009 from Fidia Farmaceutici S.p.A., a privately held Italian corporation, for a purchase price consisting of $17.0 million in cash and 1,981,192 shares of the Company’s common stock valued at $16.8 million based on the closing stock price of $8.49 per share. See Note 16 to our Consolidated Financial Statements for additional information regarding the acquisition.
-28-
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following section of this Annual Report on Form 10-K titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” contains statements that are not statements of historical fact and are forward-looking statements within the meaning of the federal securities laws. These statements involve known and unknown risks, uncertainties, and other factors that may cause our actual results, performance, or achievement to differ materially from anticipated results, performance, or achievement, expressed or implied in such forward-looking statements. These statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties. We discuss many of these risks and uncertainties at the beginning of this Annual Report on Form 10-K and under Item 1 “Business” and Item 1A “Risk Factors.” The following discussion should also be read in conjunction with the Consolidated Financial Statements of Anika Therapeutics, Inc. and the Notes thereto appearing elsewhere in this report.
Management Overview
Anika Therapeutics, Inc. (“Anika,” and together, with its subsidiaries, the “Company”) develops, manufactures and commercializes therapeutic products for tissue protection, healing, and repair. These products are based on hyaluronic acid (“HA”), a naturally occurring, biocompatible polymer found throughout the body. Due to its unique biophysical and biochemical properties, HA plays an important role in a number of physiological functions such as the protection and lubrication of soft tissues and joints, the maintenance of the structural integrity of tissues, and the transport of molecules to and within cells.
Anika acquired 100% of the issued and outstanding stock of FAB on December 30, 2009 from Fidia Farmaceutici S.p.A. (“Fidia”), a privately held Italian corporation, for a purchase price consisting of $17.0 million in cash and 1,981,192 shares of the Company’s common stock valued at $16.8 million based on the closing stock price of $8.49 per share. See Note 16 to our Consolidated Financial Statements for additional information regarding the acquisition.
FAB has over 20 products currently commercialized, primarily in Europe. These products are also all made from hyaluronic acid, and based on two technologies “HYAFF”, which is a solid form of HA, and ACP gel, an autocross-linked polymer of HA. Both technologies are protected by an extensive portfolio of owned and licensed patents. With the acquisition of FAB, the Company now offers therapeutic products in the following areas:
|
Anika
|
FAB
|
|||||||
|
Orthobiologics
|
X | X | ||||||
|
Dermal
|
||||||||
| Advanced wound care | X | |||||||
|
Aesthetic dermatology
|
X | |||||||
|
Ophthalmic
|
X | |||||||
|
Surgical
|
||||||||
| Anti-adhesion | X |
X
|
||||||
|
Ear, nose and throat care (“ENT”)
|
X
|
|||||||
|
Veterinary
|
X | |||||||
Orthobiologics
Anika’s orthobiologics business contributed 58% to our product revenue in the year ended December 31, 2010. This includes FAB’s products which added $1,923,256 to orthobiologics revenue in 2010. Our orthobiologics products consist of joint health and orthopedic products. Joint health products include ORTHOVISC, ORTHOVISC mini, and MONOVISC. ORTHOVISC is available in the U.S., Canada, and some international markets for the treatment of osteoarthritis of the knee, and in Europe for the treatment of osteoarthritis in all joints. ORTHOVISC mini is available in Europe and is designed for the treatment of osteoarthritis in small joints. MONOVISC is our single injection osteoarthritis treatment indicated for all joints in Europe, and for the knee in Turkey and Canada. ORTHOVISC mini, and MONOVISC are our two newest joint health products and became available in certain international markets during the second quarter of 2008.
Anika has marketed ORTHOVISC, our product for the treatment of osteoarthritis of the knee, internationally since 1996 through various distribution agreements. International sales of ORTHOVISC contributed 9% of product revenue for the year ended December 31, 2010. Our strategy is to continue to add new products, to expand the indications for usage of these products, and to add additional countries to our distribution network. The joint health area has been the fastest growing area for the Company, growing from 27% of our product revenue in 2006 to 58% of our product revenue in 2010. We continue to seek new distribution partnerships around the world and we expect total joint health product sales to increase in 2011 compared to 2010.
-29-
With the acquisition of FAB, we now offer several orthopedic products used in connection with regenerative medicine. The products currently available in Europe, include Hyalograft C Autograft for cartilage regeneration; Hyalofast, a biodegradable support for human bone marrow mesenchymal stem cells; Hyalonect, a woven gauze used as a graft wrap; and Hyaloss, HYAFF fibers used to mix blood/bone grafts to form a paste for bone regeneration. We also offer Hyaloglide, an ACP gel used in tenolysis treatment, but with potential for flexor tendon adhesion prevention, and in the shoulder for adhesive capsulitis with additional clinical data. These products are commercialized directly in Italy, and through a network of distributors, primarily in Europe, the Middle East, Argentina, and Korea. Anika believes that the U.S. market offers excellent expansion potential to increase revenue, and this will continue to be a major focus area for the Company.
Dermal
Our dermal products consist of advanced wound care products, a field new to the Company with the acquisition of FAB, and aesthetic dermal fillers. Altogether, our dermal products contributed 7% of our product revenue for the year ended December 31, 2010. We offer over seven products for treatment of skin wounds ranging from burns to diabetic ulcers. The products cover a variety of wound treatment solutions including debridement agents, advanced therapies and skin substitutes. Leading products include Hyalograft 3D, for the regeneration of skin; and Hyalomatrix, for treatment of burns and ulcers and the only product not contra-indicated for 3 rd degree burns. These products are commercialized directly in Italy, and through a network of distributors, primarily in Europe, the Middle East, Argentina, and Korea. Several of the products are also approved for sale in the United States, and the Company is exploring distribution opportunities.
Our aesthetic dermatology business is designed as a family of products for facial wrinkles and scar remediation, and is intended to supplant collagen-based products and to compete with other HA-based products currently on the market. Our initial aesthetic dermatology product is a dermal filler based on our proprietary chemically modified, cross-linked HA, and is approved in Europe, Canada, the U.S., Korea, and certain countries in South America. Internationally, this product is marketed under the ELEVESS name. This product has been marketed in the U.S. under the name of HYDRELLETM.
Coapt Systems, Inc. (“Coapt”) began selling HYDRELLETM in the third quarter of 2009 under a distribution agreement granting Coapt an exclusive and non-transferable right to market HYDRELLE™ in the United States. On July 2, 2010 we were notified by Coapt that it had filed for an Assignment for the Benefit of Creditors under the laws of the State of California. The Company’s Distribution Agreement with Coapt was subsequently terminated. The Company plans to directly distribute HYDRELLETM in the interim while it reviews its franchise strategy. We recorded $500,064 and $1,471,165 of aesthetic dermatology revenue in 2010 and 2009, respectively, with revenue in 2009 primarily derived from sales to Coapt.
Ophthalmic
Our ophthalmic business includes HA viscoelastic products used in ophthalmic surgery. For the year ended December 31, 2010, sales of ophthalmic products contributed 23% of our product revenue, 94% of which represents sales to Bausch & Lomb. Anika manufactures the AMVISC product line for Bausch & Lomb under the terms of a supply agreement that expired on December 31, 2010 (the “2004 B&L Agreement”) for viscoelastic products used in ophthalmic surgery. Effective January 1, 2011, the parties entered into a non-exclusive, two year contract intended to transition the manufacture of AMVISC and AMVISC Plus to an alternative, recently acquired low-cost supplier to B&L. Under the 2004 B&L Agreement, the Company was restricted in its ability to commercialize viscoelastic products to only existing customers (STAAR Surgical Company and Hoya Surgical Optics, Inc.). That restriction has now expired and the Company is free to market its own viscoelastic product called AnikaVisc. B&L accounted for 21% of product revenue for the year ended 2010, but is expected to be significantly lower in 2011 under the new transition contract. Operating margins under the 2004 B&L Agreement were low, and the Company expects to see margin improvement through commercialization of its new AnikaVisc product. There can be no assurance that AnikaVisc will be successfully sold or that it will generate any profit for the Company. See Item 1A. “Risk Factors.”
-30-
Surgical
Our surgical group consists of products used to prevent surgical adhesions, and to treat ENT disorders. For the year ended December 31, 2010, sales of surgical products contributed 7% of our product revenue, INCERT, approved for sale in Europe and Turkey is a chemically modified, cross-linked HA barrier gel, for prevention of post-surgical adhesions in connection with spinal surgeries such as discectomies with a laminectomy or laminotomy. INCERT is currently marketed in four countries. We see potential for expanded indications for the use of INCERT, but have made this a secondary goal to the successful launch and expanded distribution of our joint health and advanced wound care products. There are currently no plans to distribute INCERT in the U.S.
Hyalobarrier and Hyalobarrier Endo are also clinically proven post operative adhesion barrier products, approved for abdominal indications. The products are currently commercialized by FAB in Europe, the Middle East and certain Asian countries through a distribution network but are not approved in the U.S.
FAB also offers several products used in connection with the treatment of ENT disorders. The lead product is Merogel, a thick, viscous hydrogel composed of cross-linked hyaluronic acid, a biocompatible agent that creates a moist wound-healing environment. FAB is partnered with Medtronic for worldwide distribution or Merogel.
Veterinary
U.S. sales of HYVISC, our product for the treatment of equine osteoarthritis, contributed 5% to product revenue for the year ended December 31, 2010. We continue to look at other veterinary applications and opportunities to expand geographic territories.
Research and Development
Anika’s research and development efforts primarily consist of the development of new medical applications for our HA-based technology, the management of clinical trials for certain product candidates, the preparation and processing of applications for regulatory approvals or clearances at all relevant stages of product development, and process development and scale-up manufacturing activities relative to our existing and new products. Our development focus includes chemically modified formulations of HA designed for longer residence time in the body. Our investment in R&D has been important over the years, and varies considerably depending on the number and size of clinical trials and studies underway. For the years 2010 and 2009, these expenses were $6.9 million and $8.2 million, respectively. We anticipate that we will continue to commit significant resources to research and development, including clinical trials, in the future.
With the acquisition of FAB, we have enhanced both our research and development capabilities and our pipeline of candidate products. FAB has research and development programs for new products including Hyalobone, a bone tissue filler; Hyalospine, an adhesion prevention gel for use after spinal surgery; and Hyalofast, to repair cartilage defects. Other key projects include obtaining FDA approval to market FAB’s suite of orthopedic products in the U.S. These products consist of Hyalofast, Hyaloglide, and Hyalonect.
In addition to the FAB products in the preceding paragraph, additional products in development include MONOVISC for U.S. marketing approval, and our first next generation osteoarthritis product. MONOVISC is a single-injection treatment product that uses a non-animal source HA. MONOVISC is also our first osteoarthritis product based on our proprietary crosslinked HA-technology. We received CE Mark approval for the MONOVISC product in October 2007, and began sales in Europe during the second quarter of 2008, following a small, post-marketing clinical study. In the U.S., we filed an investigational device exemption, or an IDE application, with the FDA, and completed the clinical segment of the U.S. MONOVISC pivotal trial in June 2009, and a follow-on retreatment study in September 2009. We filed the final module of our MONOVISC PMA containing the clinical data in December 2009. We were informed that there were deficiencies in our submissions through a deficiency/non-approvable letter, which is the FDA's mechanism for informing companies of deficiencies. We submitted additional data and analyses throughout 2010, and have been informed by FDA that deficiencies remain. Acting on an option presented by the FDA to resolve the remaining open issues, Anika requested a review by the Orthopedic Advisory Panel. The Company has not yet received a date for an Advisory Panel meeting. We continue to believe that Monovisc should receive FDA approval. Our second single-injection osteoarthritis product under development is CINGAL™, which is based on the same technology platform used in MONOVISC, with an added active therapeutic molecule to provide broad pain relief for a long period of time. During the past year, we have integrated the research and development efforts of Anika and FAB, and prioritized our new product development activities.
-31-
Summary of Critical Accounting Policies; Significant Judgments and Estimates
Our discussion and analysis of our financial condition and results of operations are based upon our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities. We monitor our estimates on an on-going basis for changes in facts and circumstances, and material changes in these estimates could occur in the future. Changes in estimates are recorded in the period in which they become known. We base our estimates on historical experience and other assumptions that we believe to be reasonable under the circumstances. Actual results may differ from our estimates if past experience or other assumptions do not turn out to be substantially accurate.
We have identified the policies below as critical to our business operations and the understanding of our results of operations. The impact and any associated risks related to these policies on our business operations is discussed throughout “Management’s Discussion and Analysis of Financial Condition and Results of Operations” where such policies affect our reported and expected financial results. For a detailed discussion on the application of these and other accounting policies, see Note 2 in the Notes to the Consolidated Financial Statements of this Annual Report on Form 10-K for the year ended December 31, 2010.
Foreign Currency Translation
The functional currency of our foreign subsidiary is the Euro. Assets and liabilities of the foreign subsidiary are translated using the exchange rate existing on each respective balance sheet date. Revenues and expenses are translated using the monthly average exchange rates prevailing throughout the year. The translation adjustments resulting from this process are included as a component of accumulated other comprehensive income (loss).
Fair Value Measurements
Fair value is defined as the price that would be received from selling an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. When determining the fair value measurements for assets and liabilities required to be recorded at fair value, we consider the principal or most advantageous market in which we would transact and consider assumptions that market participants would use when pricing the asset or liability, such as inherent risk, transfer restrictions, and risk of nonperformance. The accounting standard establishes a fair value hierarchy that requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value.
A financial instrument’s categorization within the fair value hierarchy is based upon the lowest level of input that is significant to the fair value measurement. Three levels of inputs that may be used to measure fair value:
|
|
•
|
Level 1 – Valuation is based upon quoted prices for identical instruments traded in active markets. Level 1 instruments include securities traded on active exchange markets, such as the New York Stock Exchange.
|
|
|
•
|
Level 2 – Valuation is based upon quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active and model-based valuation techniques for which all significant assumptions are observable in the market.
|
|
|
•
|
Level 3 – Valuation is generated from model-based techniques that use significant assumptions not observable in the market. These unobservable assumptions reflect our own estimates of assumptions market participants would use in pricing the asset or liability.
|
Allowance for Doubtful Accounts
We maintain allowances for doubtful accounts for estimated losses resulting from the inability of our customers to make required payments. In determining the adequacy of the allowance for doubtful accounts, management specifically analyzes individual accounts receivable, historical bad debts, customer concentrations, customer credit-worthiness, current economic conditions, accounts receivable aging trends and changes in our customer payment terms.
-32-
Inventories
Inventories are stated at the lower of cost or market, with cost being determined using the first-in, first-out (“FIFO”) method. Work-in-process and finished goods inventories include materials, labor, and manufacturing overhead.
The Company’s policy is to write-down inventory when conditions exist that suggest inventory may be in excess of anticipated demand or is obsolete based upon assumptions about future demand for the Company’s products and market conditions. The Company regularly evaluates the ability to realize the value of inventory based on a combination of factors including, but not limited to: historical usage rates, forecasted sales or usage, product end of life dates, and estimated current or future market values. Purchasing requirements and alternative usage avenues are explored within these processes to mitigate inventory exposure.
Revenue Recognition - General
We recognize revenue from product sales when all of the following criteria are met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the seller's price to the buyer is fixed or determinable; and collection from the customer is reasonably assured.
Product Revenue
Revenues from product sales are recognized when title and risk of loss have passed to the customer, which is typically upon shipment to the customer. Amounts billed or collected prior to recognition of revenue are classified as deferred revenue. When determining whether risk of loss has transferred to customers on product sales, or if the sales price is fixed or determinable, the Company evaluates both the contractual terms and conditions of its distribution and supply agreements as well as its business practices.
Product revenue also includes royalties. Royalty revenue is based on our distributors’ sales and recognized in the same period our distributors record their sale of products manufactured by us. On a quarterly basis we record royalty revenue based upon sales projections provided to us by our distributor customers. If necessary we adjust our estimates based upon final sales data received prior to issuing our annual audited financial statements.
Licensing, Milestone and Contract Revenue
Licensing, milestone, and contract revenue consists of revenue recognized on initial and milestone payments, as well as contractual amounts received from partners. The Company’s business strategy includes entering into collaborative license, development and/or supply agreements with partners for the development and commercialization of the Company’s products.
The terms of the agreements typically include non-refundable license fees, funding of research and development, payments based upon achievement of certain milestones, and royalties on product sales. The Company evaluates each agreement, and elements within each agreement, in accordance with ASC Subtopic 605-25, Multiple Element Arrangements (“ASC 605-25”). Under ASC 605-25, in order to account for an element as a separate unit of accounting, the element must have had stand-alone value and there must have been objective and reliable evidence of fair value of the undelivered elements. In general, non-refundable upfront fees and milestone payments were recognized as revenue over the term of the arrangement as the Company completes its performance obligations.
Property and Equipment
Property and equipment are recorded at cost and depreciated using the straight-line method over their estimated useful lives. Computer hardware and software are typically amortized over three to five years, and furniture and fixtures over three to eight years. Leasehold improvements are amortized over the shorter of their useful lives or the remaining terms of the related leases. Property and equipment under capital leases are amortized over the lesser of the lease terms or their estimated useful lives. Maintenance and repairs are charged to expense when incurred; additions and improvements are capitalized. When an item is sold or retired, the cost and related accumulated depreciation is relieved, and the resulting gain or loss, if any, is recognized in income.
-33-
Goodwill and Acquired Intangible Assets
Goodwill is the amount by which the purchase price of acquired net assets in a business combination exceeded the fair values of net identifiable assets on the date of acquisition. Acquired In-Process Research and Development (“IPR&D”) represents the fair value assigned to research and development assets that we acquire that have not been completed at the date of acquisition or are pending regulatory approval in certain jurisdictions. The value assigned to acquired IPR&D is determined by estimating the costs to develop the acquired technology into commercially viable products, estimating the resulting revenue from the projects, and discounting the net cash flows to present value.
Goodwill and IPR&D, are evaluated for impairment annually or more frequently if events or changes in circumstances indicate that the asset might be impaired. Factors we consider important, on an overall company basis, that could trigger an impairment review include significant underperformance relative to historical or projected future operating results, significant changes in our use of the acquired assets or the strategy for our overall business, significant negative industry or economic trends, a significant decline in our stock price for a sustained period, or a reduction of our market capitalization relative to net book value.
To conduct impairment tests of goodwill, the fair value of the reporting unit is compared to its carrying value. If the reporting unit’s carrying value exceeds its fair value, we record an impairment loss to the extent that the carrying value of goodwill exceeds its implied fair value. We estimate the fair value for reporting units using discounted cash flow valuation models which require the use of significant estimates and assumptions including but not limited to: risk free rate of return on an investment, weighted average cost of capital, future revenue, operating margin, working capital and capital expenditure needs. Our annual assessment for impairment of goodwill as of November 30, 2010 indicated that the fair value of our reporting units exceeded the carrying value of the reporting units. There can be no assurance that, at the time future impairment tests are completed, a material impairment charge will not be recorded.
To conduct impairment tests of IPR&D, the fair value of the IPR&D project is compared to its carrying value. If the carrying value exceeds its fair value, we record an impairment loss to the extent that the carrying value of the IPR&D project exceeds its fair value. We estimate the fair values for IPR&D projects using discounted cash flow valuation models which require the use of significant estimates and assumptions including but not limited to: estimating the timing of and expected costs to complete the in process projects, projecting regulatory approvals, estimating future cash flows from product sales resulting from completed projects and in process projects, and developing appropriate discount rates. Our annual assessment for impairment of IPR&D indicated that the fair value of our IPR&D as of November 30, 2010 exceeded their respective carrying values. There can be no assurance that, at the time future impairment tests are completed, a material impairment charge will not be recorded.
Long-Lived Assets
Long-lived assets primarily include property and equipment and intangible assets with finite lives (including purchased software and trade names). Purchased software is amortized over 2 to 10 years and trade names are amortized over 10 years. We review long-lived assets for impairment when events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable or that the useful lives of those assets are no longer appropriate. Each impairment test is based on a comparison of the undiscounted cash flows to the recorded value of the asset. If impairment is indicated, the asset is written down to its estimated fair value based on a discounted cash flow analysis.
Stock-Based Compensation
We measure the compensation cost of employee services received in exchange for an award of equity instruments based on the grant-date fair value of the underlying award. That cost is recognized over the period during which an employee is required to provide service in exchange for the award. See Note 10 for a description of the types of stock-based awards granted, the compensation expense related to such awards, and detail of equity-based awards outstanding. See Note 14 of the accompanying Consolidated Financial Statements for details relative to the tax benefit recognized in the consolidated statement of operations for stock-based compensation.
Income Taxes
Our income tax expense includes U.S. and international income taxes. Certain items of income and expense are not reported in tax returns and financial statements in the same year. The tax effects of these differences are reported as deferred tax assets and liabilities. Deferred tax assets are recognized for the estimated future tax effects of deductible temporary differences and tax operating loss and credit carryforwards. Changes in deferred tax assets and liabilities are recorded in the provision for income taxes. We assess the likelihood that our deferred tax assets will be recovered from future taxable income and, to the extent we believe that it is more likely than not that all or a portion of deferred tax assets will not be realized, we establish a valuation allowance. To the extent we establish a valuation allowance or increase this allowance in a period, we include an expense within the tax provision in the consolidated statement of operations.
-34-
Comprehensive Income
Comprehensive income consists of net income and other comprehensive income (loss), which includes foreign currency translation adjustments. For the purposes of comprehensive income disclosures, we do not record tax provisions or benefits for the net changes in the foreign currency translation adjustment, as we intend to reinvest permanently undistributed earnings of our foreign subsidiary. Accumulated other comprehensive income (loss) is reported as a component of stockholders' equity and, as of December 31, 2010 and 2009, was comprised solely of cumulative translation adjustment gains.
Segment Information
Operating segments, as defined under U.S. GAAP, are components of an enterprise about which separate financial information is available that is evaluated regularly by the chief operating decision maker, or decision-making group, in deciding how to allocate resources and in assessing performance. The Company’s chief operating decision maker is its Chief Executive Officer. Based on the criteria established by ASC 280, Segment Reporting, the Company has one reportable operating segment the results of which are disclosed in the accompanying Consolidated Financial Statements.
Results of Operations
Year ended December 31, 2010 compared to year ended December 31, 2009
The historical results of operations discussion below for 2009 pertains only to Anika Therapeutics, Inc. and does not include a discussion of FAB’s operation or its impact on Anika’s historical results.
|
Statement of Operations Detail
|
||||||||||||||||
|
Year Ended December 31,
|
||||||||||||||||
|
2010
|
2009
|
Inc/(Dec)
|
Inc/(Dec)
|
|||||||||||||
|
Product revenue
|
$ | 52,735,730 | $ | 37,320,906 | 15,414,824 | 41 | % | |||||||||
|
Licensing, milestone and contract revenue
|
2,820,864 | 2,814,798 | 6,066 | 0 | % | |||||||||||
|
Total revenue
|
55,556,594 | 40,135,704 | 15,420,890 | 38 | % | |||||||||||
|
Operating expenses:
|
||||||||||||||||
|
Cost of product revenue
|
23,826,604 | 13,670,228 | 10,156,376 | 74 | % | |||||||||||
|
Research & development
|
6,874,633 | 8,181,532 | (1,306,899 | ) | -16 | % | ||||||||||
|
Selling, general & administrative
|
17,317,671 | 10,545,351 | 6,772,320 | 64 | % | |||||||||||
|
Acquisition-related expenses
|
- | 2,151,854 | (2,151,854 | ) | -100 | % | ||||||||||
|
Total operating expenses
|
48,018,908 | 34,548,965 | 13,469,943 | 39 | % | |||||||||||
|
Income from operations
|
7,537,686 | 5,586,739 | 1,950,947 | 35 | % | |||||||||||
|
Interest income (expense), net
|
(194,620 | ) | (74,480 | ) | (120,140 | ) | 161 | % | ||||||||
|
Income before income taxes
|
7,343,066 | 5,512,259 | 1,830,807 | 33 | % | |||||||||||
|
Provision for income taxes
|
3,027,071 | 1,824,692 | 1,202,379 | 66 | % | |||||||||||
|
Net income
|
$ | 4,315,995 | $ | 3,687,567 | 628,428 | 17 | % | |||||||||
|
Product gross profit
|
28,909,126 | 23,650,678 | 5,258,448 | 22 | % | |||||||||||
|
Product gross margin
|
55 | % | 63 | % | -9 | % | -13 | % | ||||||||
-35-
Total Revenue. Total revenue for the year ended December 31, 2010 increased by $15,420,890 to $55,556,594. The increase in total revenue was primarily due to the addition of FAB results, and increased Joint Health product revenue in 2010.
Product revenue by product line. Product revenue for the year ended December 31, 2010 was $52,735,730, an increase of $15,414,824, or 41%, compared to the prior year. Excluding the contributions of FAB, Anika’s product revenue grew 18% for the year compared to the prior year.
|
Year Ended December 31,
|
||||||||||||||||
|
2010
|
2009
|
Inc/(Dec)
|
Inc/(Dec)
|
|||||||||||||
|
Orthobiologics
|
$ | 30,741,305 | $ | 22,879,899 | $ | 7,861,406 | 34 | % | ||||||||
|
Dermal
|
3,564,616 | 1,471,165 | 2,093,451 | 142 | % | |||||||||||
|
Ophthalmic surgery
|
11,971,787 | 10,573,915 | 1,397,872 | 13 | % | |||||||||||
|
Surgical
|
3,883,444 | 121,445 | 3,761,999 |
NM
|
||||||||||||
|
Veterinary
|
2,574,578 | 2,274,482 | 300,096 | 13 | % | |||||||||||
| $ | 52,735,730 | $ | 37,320,906 | $ | 15,414,824 | 41 | % | |||||||||
|
NM = Not Meaningful
|
||||||||||||||||
Revenue from orthobiologics increased $7,861,406, or 34%, in 2010. The improvement in orthobiologics product revenue was due to increases in domestic ORTHOVISC revenue, as well as increased sales of MONOVISC in Europe, Turkey and Canada in 2010 compared with 2009. Our U.S. joint health product revenue for 2010 totaled $22,353,753, compared to $16,930,419 in 2009, an increase of 32%. This increase reflects DePuy Mitek’s continued market penetration to an estimated market share of 13% in 2010 versus 11% share in 2009. International joint health product revenue in 2010 increased 9% to $6,469,110, from $5,949,479, in 2009. The increase in international revenue was driven by higher product shipments to new and existing customers in Eastern Europe and the Middle East, partially offset by continued weakness in sales in Southern Europe. FAB’s orthopedic product revenue was $1,918,442 for the year ended December 31, 2010, or approximately 4% of our product revenue. Although FAB’s orthopedic revenue was not included in the same period in 2009, it did achieve significant increases in sales of Hyalograft C Autograft and Hyalofast as compared to 2009. We expect orthobiologics revenue to increase in 2011 compared to 2010, both domestically and internationally.
Dermal revenue increased $2,093,451, or 142%, to $3,564,616 in 2010 from $1,471,165 in 2009. The increase was primarily due to the addition of FAB’s advanced wound care products revenue for which totaled $3,064,552 in 2010. Aesthetic dermatology revenue was $500,064 for the year ended December 31, 2010, versus $1,471,165 for the prior year. In July 2010, our former U.S. distributor, Coapt, filed for protection from creditors and we terminated our agreement with them. Coapt contributed approximately $47,000 and $1,132,000 of aesthetic dermatology revenue in 2010 and 2009, respectively. The Company has directly distributed HYDRELLETM in the interim to existing customers while it reviews its franchise strategy and opportunities for new distribution partners. We continue to seek additional marketing and distribution partners to commercialize our aesthetic products outside the U.S. The aesthetics’ market is crowded with many large companies, and our sales expectations in this area are modest.
Revenue from ophthalmic products in 2010 increased $1,397,872, or 13%, to $11,971,787 compared to revenue for these products in 2009. The increase was primarily attributable to order timing and inventory management by our partners. As previously disclosed, Anika has been a contract manufacturer for Bausch & Lomb for over 20 years, and the current Supply Agreement with B&L expired on December 31, 2010. Effective January 1, 2011, we entered into a non-exclusive, two year contract with B&L intended to transition the manufacture of AMVISC and AMVISC Plus to an alternative, recently acquired low-cost supplier to B&L. B&L accounted for 21% of product revenue for the year ended 2010, but is expected to be significantly lower in 2011 under the new transition contract. Operating margins under the 2004 B&L Agreement were low, and the Company expects to see margin improvement through commercialization of its new AnikaVisc product. We are unable to quantify the amount of revenue we will derive from B&L and AnikaVisc in 2011, but expect our overall ophthalmic revenue to decline significantly in 2011 compared to 2010.
Sales of our surgical products were $3,883,444 and $121,445 for the years ended December 31, 2010 and 2009, respectively. This product group consists primarily of FAB’s anti-adhesion and ENT products acquired in December 2009 and, as a result, revenue from this product group were significantly higher in 2010 as compared to 2009. Our anti-adhesion products include INCERT, Hyalobarrier and Hyalobarrier Endo. Our leading ear, nose and throat care product is Merogel. FAB is partnered with Medtronic for worldwide distribution (except for Italy) of its ENT products. We expect surgical product revenue to be relatively flat in 2011 compared to 2010.
-36-
Veterinary revenue increased $300,096, or 13%, to $2,574,578 in 2010 as compared with $2,274,482 in 2009. We believe the increase for the period was primarily due to inventory management by our partner, Boehringer Ingelheim Vetmedica. Sales of HYVISC are made to a single customer under an exclusive agreement which was extended in December 2010 to December 31, 2014. We expect HYVISC revenue to be relatively flat in 2011 compared to 2010.
Licensing, milestone and contract revenue. Licensing, milestone and contract revenue for the year ended December 31, 2010 was $2,820,864, compared to $2,814,798 for 2009. The increase was due to licensing and grant revenue earned by our FAB subsidiary, partially offset by a decrease in maintenance and contract revenue from Anika. Licensing and milestone revenue includes the ratable recognition of the $27,000,000 in up-front and milestone payments related to the JNJ Agreement. These amounts are being recognized in income ratably over the ten-year expected life of the agreement, or $2,700,000 per year.
Product gross profit and margin. Product gross profit for the year ended December 31, 2010 was $28,909,126, or 55% of product revenue, compared with $23,650,678, or 63% of product revenue, for the year ended December 31, 2009. The increase in product gross profit was primarily due to increased sales including the additional revenue from FAB. The decrease in the product gross margin was primarily due to the addition of lower margin FAB products into Anika’s overall mix, and was also negatively affected by inventory reserves and duplicate manufacturing expenditures during the continued transition from Woburn to the Company’s Bedford, Massachusetts facility. FAB currently operates at a lower volume and outsources most manufacturing to its former parent company, Fidia, contributing to its current lower gross margins. The Company plans to transfer a significant portion of the FAB product manufacturing to its location in Bedford, MA over the next two years.
The Company wrote down inventory by approximately $480,000 during 2010 related to aesthetic and joint health products due to the Coapt bankruptcy and the delay in FDA approval of MONOVISC. Looking forward, we expect a continued impact of duplicate facilities on our results during 2011 as we complete the transition of operations from our Woburn, MA facility to our Bedford, MA facility. As of December 31, 2010, non-U.S. MONOVISC and aesthetic product manufacturing has been moved to the Bedford facility. Transition of the remaining products will take place by product line and result in manufacturing activities occurring in both our Woburn and Bedford in 2011. Commencing in 2011, the Bedford, MA facility is expected to add in excess of $2.2 million to annual depreciation expense once completely on-line.
Research and development. Research and development (“R&D”)expenses for the year ended December 31, 2010 decreased by $1,306,899, or 16%, to $6,874,633 from $8,181,532 for the prior year. The decrease in research and development expenses was primarily due to the higher costs incurred in 2009 in connection with the Company’s U.S.-based clinical trials for MONOVISC, and the post-marketing aesthetic dermatology “people of color” study during the year ended December 31, 2009. The MONOVISC clinical trial was completed in late 2009. This decrease was partially offset by the addition of R&D costs at FAB. Research and development during 2010 was primarily for manufacturing validation activities at our Bedford facility, as well as other continuing new product development projects in Italy and the U.S. We expect research and development expenses will increase significantly in the future with the additional pipeline of new products.
Selling, general and administrative. Selling, general and administrative expenses for the year ended December 31, 2010, increased by $4,620,466, or 36%, to $17,317,671 from $12,697,205 in the prior year, which also includes $2.2 million of acquisition-related non-recurring expenses in connection with our acquisition of FAB The increase was primarily due to the addition of FAB to the Company, including integration and infrastructure development costs, as well as costs related to the creation of MONOVISC marketing materials and reimbursement strategy consulting. We expect general and administrative expenses for 2011 will increase significantly as we prepare to commercialize MONOVISC in the U.S.
Interest income, net. Net interest expense was $194,620 for the year ended December 31, 2010, compared to a net interest expense of $74,480 in 2009. Interest expense incurred was capitalized during the construction and validation of our Bedford, MA facility prior to July 1, 2009, and is the primary reason for the increased expense in 2010 versus 2009.
Income taxes. Provisions for income taxes were $3,027,071 and $1,824,692 for the years ended December 31, 2010 and 2009, respectively. The increase in effective tax rate in 2010 of 8.1%, and difference from the U.S. federal statutory rate, is primarily due to three factors: the foreign rate differential associated with the net operating loss incurred by FAB, lower R&D spending in 2010 resulting in a decrease in the associated R&D credit earned, and continued lower capital spending on the Bedford facility resulting in lower state investment tax credits in 2010.
-37-
A reconciliation of the U.S. federal statutory tax rate to the effective tax rate for the periods ending December 31 is as follows:
| Years ended December 31, | ||||||||
|
2010
|
2009
|
|||||||
|
Statutory federal income tax rate
|
34.0 | % | 34.0 | % | ||||
|
State tax expense, net of federal benefit
|
7.8 | % | 6.2 | % | ||||
|
State deferred tax assets, net of federal benefit, due to rate change
|
0.5 | % | (0.8 | )% | ||||
|
Permanent items, including nondeductible expenses
|
(1.8 | )% | 8.8 | % | ||||
|
State investment tax credit
|
(0.8 | )% | (5.6 | )% | ||||
|
Federal and state research and development credits
|
(2.2 | )% | (8.4 | )% | ||||
|
Foreign rate differential
|
3.1 | % | 0.0 | % | ||||
|
Other
|
0.6 | % | (1.1 | )% | ||||
|
Tax expense
|
41.2 | % | 33.1 | % | ||||
During 2010, the Company concluded its audit by the Massachusetts Department of Revenue (“DOR”) for its 2006 and 2007 tax returns and the statue of limitations expired on certain other previously-reserved positions. The combination of these items resulted in a reduction to our reserve for uncertain tax positions, and a related income tax benefit, of approximately $41,000. In 2010, the Company recorded an additional reserve of approximately $37,000 as the IRS is currently auditing the Company’s taxes for the years ended December 31, 2008. See Note 14 to our Consolidated Financial Statements for additional information.
Net income. For the year ended December 31, 2010 net income was $4,315,995 or $0.32 per diluted share compared to $3,687,567 or $0.32 per diluted share for the same period last year. The primary driver for this increase in net income was an increase in product sales with a more favorable product mix, and lower clinical spending. Earnings per diluted share did not increase from 2009 due to the 1,981,192 increase is outstanding shares resulting from the acquisition of FAB on December 30, 2009. See Note 16 to our Consolidated Financial Statements for additional information.
Year ended December 31, 2009 compared to year ended December 31, 2008
The historical results of operations discussion below pertains only to Anika Therapeutics, Inc. and does not include a discussion of FAB’s operation or its impact on Anika’s historical results.
|
Statement of Operations Detail
|
||||||||||||||||
|
Year Ended December 31,
|
||||||||||||||||
|
2009
|
2008
|
Inc/(Dec)
|
Inc/(Dec)
|
|||||||||||||
|
Product revenue
|
$ | 37,320,906 | $ | 33,054,787 | 4,266,119 | 13 | % | |||||||||
|
Licensing, milestone and contract revenue
|
2,814,798 | 2,725,000 | 89,798 | 3 | % | |||||||||||
|
Total revenue
|
40,135,704 | 35,779,787 | 4,355,917 | 12 | % | |||||||||||
|
Operating expenses:
|
||||||||||||||||
|
Cost of product revenue
|
13,670,228 | 13,188,516 | 481,712 | 4 | % | |||||||||||
|
Research & development
|
8,181,532 | 7,399,049 | 782,483 | 11 | % | |||||||||||
|
Selling, general & administrative
|
10,545,351 | 10,965,493 | -420,142 | (4 | )% | |||||||||||
|
Acquisition-related expenses
|
2,151,854 | - | 2,151,854 | 0 | % | |||||||||||
|
Total operating expenses
|
34,548,965 | 31,553,058 | 2,995,907 | 9 | % | |||||||||||
|
Income from operations
|
5,586,739 | 4,226,729 | 1,360,010 | 32 | % | |||||||||||
|
Interest income (expense), net
|
(74,480 | ) | 498,512 | -572,992 | (115 | )% | ||||||||||
|
Income before income taxes
|
5,512,259 | 4,725,241 | 787,018 | 17 | % | |||||||||||
|
Provision for income taxes
|
1,824,692 | 1,096,046 | 728,646 | 66 | % | |||||||||||
|
Net income
|
$ | 3,687,567 | $ | 3,629,195 | 58,372 | 2 | % | |||||||||
|
Product gross profit
|
23,650,678 | 19,866,271 | 3,784,407 | 19 | % | |||||||||||
|
Product gross margin
|
63 | % | 60 | % | 5 | % | ||||||||||
-38-
Total Revenue. Total revenue for the year ended December 31, 2009 increased by $4,355,917 to $40,135,704. The increase in total revenue was primarily due to increased Joint Health and Aesthetic Dermatology product revenue in 2009.
Product revenue by product line. Product revenue for the year ended December 31, 2009 was $37,320,906, an increase of $4,266,119, or 13%, compared to the prior year.
|
Year Ended December 31,
|
||||||||||||||||
|
2009
|
2008 |
Inc/(Dec)
|
Inc/(Dec)
|
|||||||||||||
|
Orthobiologics
|
$ | 22,879,899 | $ | 18,707,669 | $ | 4,172,230 | 22 | % | ||||||||
|
Dermal
|
1,471,165 | 505,273 | 965,892 | 191 | % | |||||||||||
|
Ophthalmic surgery
|
10,573,915 | 10,678,615 | (104,700 | ) | (1 | )% | ||||||||||
|
Surgical
|
121,445 | 134,780 | (13,335 | ) | (10 | )% | ||||||||||
|
Veterinary
|
2,274,482 | 3,028,450 | (753,968 | ) | (25 | )% | ||||||||||
| $ | 37,320,906 | $ | 33,054,787 | $ | 4,266,119 | 13 | % | |||||||||
Our orthobiologics or joint health products consist of ORTHOVISC, ORTHOVISC mini and MONOVISC, the latter two of which are currently only available outside the United States. Revenue from joint health products increased $4,172,230, or 22%, in 2009. The improvement in joint health product revenue was due to increases in both international and domestic ORTHOVISC revenue, as well as increased sales of MONOVISC and ORTHOVISC mini in Europe, Turkey and Canada in 2009 compared with only a partial period during 2008. Our U.S. joint health product revenue for 2009 totaled $16,930,420, compared to $13,222,454 in 2008, an increase of 28%. This increase reflects DePuy Mitek’s underlying volume driven sales increases to end-users as a result of their continued marketing efforts. International joint health product revenue in 2009 increased 8% to $5,949,479, from $5,485,215, in 2008. The increase in international revenue was due to increased product shipments to Canada, France, Egypt, Hungary and Austria.
Ophthalmic products sales decreased $104,700, or 1%, to $10,573,915. The decrease was primarily attributable to order timing and inventory management by our partners.
Veterinary revenue decreased $753,968, or 25%, to $2,274,482 in 2009 as compared with $3,028,450 in 2008. We believe the decrease for the period was primarily due to inventory management by our partner, Boehringer Ingelheim Vetmedica. Sales of HYVISC are made to a single customer under an exclusive agreement which was extended in April 2006 to December 31, 2010.
Dermal revenue increased $965,892, or 191%, to $1,471,165 in 2009 from $505,273 in 2008. The increase was primarily due to the commencement of sales in the third quarter of 2009 by our new United States distributor, Coapt Systems, Inc. Coapt is marketing the product in the U.S. under the brand name HYDRELLE™. Aesthetics revenue in 2008 was primarily from Artes Medical, Inc., our former U.S. ELEVESS distributor. Our distribution agreement with Artes Medical, Inc. was terminated in the fourth quarter of 2008 as a result of Artes’ Chapter 7 bankruptcy filing. We added several additional international distributors in the second half of 2009, and we continue to seek additional marketing and distribution partners to commercialize our aesthetic products outside the U.S. The aesthetics’ market is crowded with many large companies, and our growth expectations in this area are modest.
Licensing, milestone and contract revenue. Licensing, milestone and contract revenue for the year ended December 31, 2009 was $2,814,798, compared to $2,725,000 for 2008. The increase was due to a short term product development contract with an existing partner. Licensing and milestone revenue includes the ratable recognition of the $27,000,000 in up-front and milestone payments related to the JNJ agreement. These amounts are being recognized in income ratably over the ten-year expected life of the agreement, or $2,700,000 per year.
Product gross profit and margin. Product gross profit for the year ended December 31, 2009 was $23,650,678, or 63% of product revenue, compared with $19,866,271, or 60% of product revenue, for the year ended December 31, 2008. The increase in product gross profit dollars and margin was primarily due to increased sales of our more profitable joint health products resulting in a more favorable product mix compared to 2008, and increased manufacturing activity in our Woburn facility to build inventory in preparation for moving our operations to the Bedford facility.
Research and development. Research and development expenses for the year ended December 31, 2009 increased by $782,483, or 11%, to $8,181,532 from $7,399,049 for the prior year. The increase in research and development expenses was primarily related to our ongoing U.S.-based clinical trials for MONOVISC, the post-marketing aesthetics dermatology “people of color” study, manufacturing validation activities at our Bedford facility, as well as other continuing new product development projects.
-39-
Selling, general and administrative. Selling, general and administrative expenses for the year ended December 31, 2009, excluding acquisition-related expense, decreased by $420,142 or 4%, to $10,545,351 from $10,965,493 in the prior year. The decrease was primarily due to a decrease in marketing expenses. The spending in 2008 included additional marketing expenses as a result of the MONOVISC and ORTHOVISC mini product launches in Europe.
Acquisition-related Expenses. We incurred $2.2 million of acquisition-related non-recurring expenses in 2009 in connection with our acquisition of FAB. We did not have corresponding acquisition-related expenses in 2008.
Interest income, net. Net interest expense was $74,480 for the year ended December 31, 2009, compared to a net interest income of $498,512 in 2008. The decrease was primarily attributable to lower interest rates as a result of the current rate environment, and lower available cash and invested balances in 2009 compared to 2008. The net interest expense for 2009 represents interest expense for facility related asset retirement obligations, and the interest expense on our outstanding debt balance, which was capitalized during the construction and validation of our Bedford, MA facility prior to July 1, 2009.
Income taxes. Provisions for income taxes were $1,824,692 and $1,096,046 for 2009 and 2008, respectively. The increase in effective tax rate in 2009 of 9.9% and difference from the U.S. federal statutory rate is primarily due to two factors: approximately $1.3 million of the FAB acquisition expenses are non-deductible for income tax purposes, and lower capital spending on the Bedford facility as the project spending peaked in 2008, resulting in lower state investment tax credits in 2009. Partially offsetting this increase was greater state and federal research and development spending resulting in higher credits in 2009 compared to 2008.
A reconciliation of the U.S. federal statutory tax rate to the effective tax rate for the periods ending December 31 is as follows:
| Years ended December 31, | ||||||||
|
2009
|
2008
|
|||||||
|
Computed expected tax expense
|
34.0 | % | 34.0 | % | ||||
|
State tax expense (net of federal benefit)
|
6.2 | % | 4.6 | % | ||||
|
State deferred tax assets rate change
|
(0.8 | )% | 2.6 | % | ||||
|
Permanent items, including nondeductible expenses
|
8.8 | % | 0.6 | % | ||||
|
State investment tax credit
|
(5.6 | )% | (11.1 | )% | ||||
|
Federal and state research and development credits
|
(8.4 | )% | (5.8 | )% | ||||
|
Other
|
(1.1 | )% | (1.7 | )% | ||||
|
Tax expense
|
33.1 | % | 23.2 | % | ||||
During the third quarter of 2008, the Company concluded its audit by the Massachusetts DOR for its 2004 and 2005 tax returns, which resulted in a reduction to its FIN 48 tax reserves and a related income tax benefit of approximately $100,000. In 2008, the Company recorded additional provision of approximately $121,000 related to the reduction of its deferred tax assets as a result of newly enacted changes in the Commonwealth of Massachusetts to gradually reduce future corporate income tax rates.
Net income. For the year ended December 31, 2009, net income was $3,687,567 or $0.32 per diluted share compared to $3,629,195, or $0.32 per diluted share, for the same period last year. The primary driver for the increase in net income was an increase in product sales with a more favorable product mix.
Liquidity and Capital Resources
We require cash to fund our operating expenses and to make capital expenditures. We expect that our requirements for cash to fund these uses will increase as the scope of our operations expand, particularly as a result of our acquisition of FAB. Historically we have funded our cash requirements from available cash and investments on hand. In 2008, we borrowed $16.0 million from Bank of America to partially fund our Bedford, Massachusetts facility capital project, and have spent approximately $34.0 million to date on this project to expand our operations and capabilities. In addition, in 2009 we spent approximately $16.2 million in cash, net of cash acquired, in connection with the FAB acquisition. Cash and cash equivalents totaled $28.2 million compared to $24.4 million, and working capital totaled approximately $37.0 million and $33.3 million, at December 31, 2010 and December 31, 2009, respectively. The Company believes it has adequate financial resources to support its business for the foreseeable future.
-40-
Cash provided by operating activities was $7,853,461, $3,094,705 and $3,407,231 for 2010, 2009, and 2008 respectively. Cash provided by operating activities increased by $4,758,756 in 2010 from 2009. This increase in operating cash was primarily due to higher net income, partially offset by an increase in net working capital requirements of approximately $3,326,000.
Cash used in investing activities was $2,679,677, $20,217,869 and $12,804,552 in 2010, 2009 and 2008 respectively. Cash used in investing activities in 2010 was primarily the result of capital expenditures related to the build-out of our Bedford, MA facility, as well as expenditure at FAB to establish computer network and financial systems. Cash used for investing activities in 2009 was primarily due to the acquisition of FAB, as well as additional capital expenditures related to the build-out of our new facility. We expect the new facility capital project to cost approximately $34 million in total (including interior construction, equipment, furniture and fixtures). Through December 31, 2010, nearly all such funds have been spent in connection with the build-out. Build-out at the new facility commenced in May 2007 and validation of the facility is expected to be completed in 2011. There can also be no assurance that we will be successful in qualifying the Bedford, MA facility under the FDA and European Union regulations.
Cash used in financing activities was $1,337,320 for 2010 and $1,643,501 for 2009, compared to cash provided by financing activities of $16,687,407 for 2008. Cash used in financing activities in 2010 and 2009 was primarily due to our principal payments on long-term debt in the amount of $1.6 million. On January 31, 2008, the Company entered into an unsecured credit facility and during 2008 borrowed $16 million to finance its new facility project. The credit facility was converted to a 7 year term loan on December 31, 2008. Also reflected in the cash provided by financing activities for all three years were proceeds from the exercise of stock options, including any associated tax benefits, as well as debt issuance costs related to our loan amendment with Bank of America.
Off Balance Sheet Arrangements
We do not use special purpose entities or other off-balance sheet financing techniques except for operating leases as disclosed in the contractual obligations table below that we believe have or are reasonably likely to have a current or future material effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity or capital resources.
Recent Accounting Pronouncements
In July 2010, the Financial Accounting Standards Board (“FASB”) issued Accounting Standard Update (“ASU”) No. 2010-20, Disclosures about the Credit Quality of Financing Receivables and the Allowance for Credit Losses. ASU No. 2010-20 requires disclosure about the credit quality of accounts receivables with terms exceeding one year and any related allowance for credit losses. These new disclosures are required for interim and fiscal periods ending after December 15, 2010 (our fourth quarter of fiscal 2010). Adoption of this standard did not have a material impact on our consolidated financial position, results of operations, or cash flows.
In April 2010, the FASB issued ASU No. 2010-017, Revenue Recognition — Milestone Method, (“ASU 2010-017”). ASU 2010-017 provides guidance in applying the milestone method of revenue recognition to research or development arrangements. Under this guidance management may recognize revenue contingent upon the achievement of a milestone in its entirety, in the period in which the milestone is achieved, only if the milestone meets all the criteria within the guidance to be considered substantive. This ASU is effective on a prospective basis for research and development milestones achieved in fiscal years beginning on or after June 15, 2010. Early adoption is permitted; however, we have elected to implement ASU 2010-17 prospectively, and as a result, the effect of this guidance will be limited to future transactions. We do not expect adoption of this standard to have a material impact on our consolidated financial position, results of operations, or cash flows.
In December 2010, the FASB issued ASU No. 2010-027, Fees Paid to the Federal Government by Pharmaceutical Manufacturers, (“ASU 2010-027”). ASU 2010-027 provides guidance concerning the recognition and classification of the new annual fee payable by branded prescription drug manufactures and importers on branded prescription drugs which was mandated under the health care reform legislation enacted in the U.S. in March 2010. Under this new accounting standard, the annual fee would be presented as a component of operating expenses and recognized over the calendar year such fees are payable using a straight-line method of allocation unless another method better allocates the fee over the calendar year. This ASU is effective for calendar years beginning on or after December 31, 2010, when the fee initially becomes effective. We do not expect adoption of this standard to have a material impact on our consolidated financial position, results of operations, or cash flows.
-41-
Contractual Obligations and Other Commercial Commitments
To-date, we have limited commitments for purchases of inventories. We have incurred significant capital investments related to the build-out of our new facility in Bedford, Massachusetts, as well as the FAB acquisition. Our future capital requirements and the adequacy of available funds will depend, on numerous factors, including:
|
|
·
|
Market acceptance of our existing and future products;
|
|
|
·
|
The success and sales of our products under current and future distribution agreements;
|
|
|
·
|
The successful commercialization of products in development;
|
|
|
·
|
Progress in our product development efforts;
|
|
|
·
|
The magnitude and scope of such efforts;
|
|
|
·
|
Any potential acquisitions of products, technologies or businesses;
|
|
|
·
|
Progress with pre-clinical studies, clinical trials and product clearances by the FDA and other agencies;
|
|
|
·
|
The cost of maintaining adequate manufacturing capabilities;
|
|
|
·
|
The cost of filing, prosecuting, defending and enforcing patent claims and other intellectual property rights;
|
|
|
·
|
Competing technological and market developments;
|
|
|
·
|
The development of strategic alliances for the marketing of certain of our products;
|
|
|
·
|
The terms of such strategic alliances, including provisions (and our ability to satisfy such provisions) that provide upfront and/or milestone payments to us;
|
|
|
·
|
The cost of maintaining adequate inventory levels to meet current and future product demands;
|
|
|
·
|
The contractual obligation to make principal and interest debt payments;
|
|
|
·
|
Our success with respect to our recently announced plan to utilize a direct sales force; and
|
|
|
·
|
The successful integration of our subsidiary FAB
|
We cannot assure you that we will record profits in future periods. To the extent that funds generated from our operations, together with our existing capital resources are insufficient to meet future requirements, we will be required to obtain additional funds through equity or debt financings, strategic alliances with corporate partners, or through other sources. No assurance can be given that any additional financing will be made available to us or will be available on acceptable terms should such a need arise. However, we believe that our existing cash and cash equivalents and future cash provided by operating activities will be sufficient to meet our working capital and capital expenditure needs over the next 12 months. See Item 1A. “Risk Factors.”
The terms of any future equity financings may be dilutive to our stockholders and the terms of any debt financings may contain restrictive covenants, which could limit our ability to pursue certain courses of action. Our ability to obtain financing is dependent on the status of our future business prospects as well as conditions prevailing in the relevant capital markets. No assurance can be given that any additional financing may be made available to us or may be available on acceptable terms should such a need arise.
-42-
The table below summarizes our non-cancelable operating leases and contractual obligations at December 31, 2010:
|
Payments due by period
|
||||||||||||||||||||
|
Total
|
Less than 1
year
|
2-3 years
|
4-5 years
|
More than 5
years
|
||||||||||||||||
|
Operating Leases (1)
|
$ | 12,340,208 | $ | 2,610,613 | $ | 3,321,729 | $ | 3,284,892 | $ | 3,122,975 | ||||||||||
|
Purchase Commitments
|
2,239,265 | 2,239,265 | - | - | - | |||||||||||||||
|
Long Term Debt (2)
|
13,476,913 | 1,783,519 | 3,494,833 | 8,198,561 | - | |||||||||||||||
| $ | 28,056,385 | $ | 6,633,396 | $ | 6,816,562 | $ | 11,483,453 | $ | 3,122,975 | |||||||||||
|
(1)
|
Included in this line is a lease we entered into on January 4, 2007, pursuant to which we lease a corporate headquarters facility, consisting of approximately 134,000 square feet of general office, research and development and manufacturing space located in Bedford, Massachusetts. The Lease has an initial term of ten and a half years, and commenced on May 1, 2007. We have an option under the Lease to extend its terms for up to four periods beyond the original expiration date subject to the condition that we notify the landlord that we are exercising each option at least one year prior to the expiration of the original or current term thereof. The first three renewal options each extend the term an additional five years with the final renewal option extending the term six years. The lease covering the Company’s existing manufacturing facility located in Woburn, Massachusetts is also included in the table above. Our administrative, research and development personnel began occupying the Bedford facility in November of 2007, and the build-out and validation for the new manufacturing space is expected to be completed in 2011. Also included in the table above is the lease entered into in Italy related to FAB. The lease commenced on December 30, 2009 and is for the next 6 years.
|
|
(2)
|
On January 31, 2008, the Company entered into an unsecured Credit Agreement (the “Agreement”) with Bank of America. Pursuant to the terms of the Agreement, our lender has agreed to provide the Company with an unsecured revolving credit facility through December 31, 2008 of up to a maximum principal amount at any time outstanding of $16,000,000. The Company borrowed the maximum amount as of December 31, 2008. On December 31, 2008, all outstanding revolving credit loans were converted into a term loan with quarterly principal payments of $400,000 and a final installment of $5,200,000 due on the maturity date of December 31, 2015. In connection with the acquisition of FAB, the Company entered into a Consent and First Amendment to our original loan with Bank of America. As part of this amendment, the interest rate for Eurodollar based loans was increased and is payable at a rate based upon (at the Company’s election) either Bank of America’s prime rate or LIBOR plus 125 basis points. This increased from the original loan amount of prime rate or LIBOR plus 75 basis points. In addition, the Company has pledged to the lender sixty-five percent (65%) of the stock of FAB. The Agreement contains customary representations and warranties of the Company, affirmative and negative covenants regarding the Company’s operations, financial covenants regarding the maintenance by the Company of a specified quick ratio and consolidated fixed charge coverage ratio, and events of default. The table includes expected principal and interest payments. For the purpose of this calculation, interest payments are based on the carrying rate of the debt at December 31, 2010, throughout the life of the obligation.
|
-43-
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
As of December 31, 2010, we did not utilize any derivative financial instruments, market risk sensitive instruments or other financial and commodity instruments for which fair value disclosure would be required under ASC 825, Financial Instruments. Our investments consist of money market funds primarily invested in U.S. Treasury obligations and repurchase agreements secured by U.S. Treasury obligations, and municipal bonds that are carried on our books at amortized cost, which approximates fair market value.
Primary Market Risk Exposures
Our primary market risk exposures are in the areas of interest rate risk and currency rate risk. We have two supplier contracts denominated in foreign currencies. Unfavorable fluctuations in exchange rates would have a negative impact on our financial statements. The impact of changes in currency exchange rates for the two contracts on our financial statements was immaterial in 2010. Our investment portfolio of cash equivalents and long-term debt are subject to interest rate fluctuations. As of December 31, 2010, the Company is subject to interest rate risk on $12.8 million of variable rate debt. The interest payable on our debt is determined based (at the Company’s election) on either an interest rate based on LIBOR plus 1.25% or the lender’s prime rate and, therefore, is affected by changes in market interest rates. Based on the outstanding debt amount as of December 31, 2010, we would have a decrease (increase) in future annual cash flows of approximately $128,000 for every 1% increase (decrease) in the interest rate.
A significant portion of FAB’s revenue, and all operating expenses, are denominated in Euros which leaves the Company vulnerable to foreign exchange risk.
-44-
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
ANIKA THERAPEUTICS, INC. AND SUBSIDIARIES
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| 46 | ||||
| 47 | ||||
| 2008 | 48 | |||
| 2009 and 2008 | 49 | |||
| 2008 | 50 | |||
| 51 |
-45-
To Board of Directors and Stockholders of Anika Therapeutics, Inc.
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations, stockholders' equity and cash flows present fairly, in all material respects, the financial position of Anika Therapeutics, Inc. and its subsidiaries at December 31, 2010 and 2009, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2010 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2010 based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company's management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management's Report on Internal Control over Financial Reporting appearing under Item 9A. Our responsibility is to express opinions on these financial statements and on the Company's internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
As discussed in Note 16 to the consolidated financial statements the Company changed the manner in which it accounts for business combinations effective January 1, 2009.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
PricewaterhouseCoopers LLP
Boston, Massachusetts
March 16, 2011
-46-
|
Anika Therapeutics, Inc. and Subsidiaries
|
||||||||
|
December 31,
|
||||||||
|
ASSETS
|
2010
|
2009
|
||||||
|
Current assets:
|
||||||||
|
Cash and cash equivalents
|
$ | 28,201,932 | $ | 24,426,990 | ||||
| Accounts receivable, net of reserves of $30,000 and $29,261 at December | ||||||||
|
31, 2010 and 2009, respectively
|
14,819,868 | 11,778,743 | ||||||
|
Inventories
|
8,949,745 | 8,547,339 | ||||||
|
Current portion deferred income taxes
|
1,990,609 | 2,183,827 | ||||||
|
Prepaid expenses and other
|
2,360,182 | 2,892,858 | ||||||
|
Total current assets
|
56,322,336 | 49,829,757 | ||||||
|
Property and equipment, at cost
|
49,696,989 | 47,172,403 | ||||||
|
Less: accumulated depreciation
|
(12,715,595 | ) | (11,424,788 | ) | ||||
| 36,981,394 | 35,747,615 | |||||||
|
Long-term deposits and other
|
384,988 | 413,228 | ||||||
|
Intangible assets, net
|
25,764,185 | 29,975,451 | ||||||
|
Deferred income taxes
|
392,005 | 3,506,362 | ||||||
|
Goodwill
|
9,091,960 | 9,959,008 | ||||||
|
Total Assets
|
$ | 128,936,868 | $ | 129,431,421 | ||||
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
||||||||
|
Current liabilities:
|
||||||||
|
Accounts payable
|
$ | 9,694,355 | $ | 6,354,761 | ||||
|
Accrued expenses
|
5,375,585 | 5,816,170 | ||||||
|
Deferred revenue
|
2,700,000 | 2,751,467 | ||||||
|
Current portion of long-term debt
|
1,600,000 | 1,600,000 | ||||||
|
Total current liabilities
|
19,369,940 | 16,522,398 | ||||||
|
Other long-term liabilities
|
1,560,205 | 1,775,386 | ||||||
|
Long-term deferred revenue
|
5,399,995 | 8,099,996 | ||||||
|
Deferred tax liability
|
6,216,582 | 8,090,139 | ||||||
|
Long-term debt
|
11,200,000 | 12,800,000 | ||||||
|
Commitments and contingencies (Note 9)
|
||||||||
|
Stockholders’ equity:
|
||||||||
| Preferred stock, $.01 par value; 1,250,000 shares authorized, no shares | ||||||||
|
issued and outstanding at December 31, 2010 and 2009, respectively
|
- | - | ||||||
| Common stock, $.01 par value; 30,000,000 shares authorized, 13,482,384 | ||||||||
| and 13,418,772 shares issued and outstanding at December 31, 2010 | ||||||||
|
and 2009, respectively
|
134,823 | 134,188 | ||||||
|
Additional paid-in-capital
|
61,817,558 | 60,539,768 | ||||||
|
Accumulated currency translation adjustment
|
(2,547,776 | ) | - | |||||
|
Retained earnings
|
25,785,541 | 21,469,546 | ||||||
|
Total stockholders’ equity
|
85,190,146 | 82,143,502 | ||||||
|
Total Liabilities and Stockholders’ Equity
|
$ | 128,936,868 | $ | 129,431,421 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
-47-
|
Anika Therapeutics, Inc. and Subsidiaries
|
||||||||||||
|
For the Years Ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Product revenue
|
$ | 52,735,730 | $ | 37,320,906 | 33,054,787 | |||||||
|
Licensing, milestone and contract revenue
|
2,820,864 | 2,814,798 | 2,725,000 | |||||||||
|
Total revenue
|
55,556,594 | 40,135,704 | 35,779,787 | |||||||||
|
Operating expenses:
|
||||||||||||
|
Cost of product revenue
|
23,826,604 | 13,670,228 | 13,188,516 | |||||||||
|
Research & development
|
6,874,633 | 8,181,532 | 7,399,049 | |||||||||
|
Selling, general & administrative
|
17,317,671 | 10,545,351 | 10,965,493 | |||||||||
|
Acquisition-related expenses
|
- | 2,151,854 | - | |||||||||
|
Total operating expenses
|
48,018,908 | 34,548,965 | 31,553,058 | |||||||||
|
Income from operations
|
7,537,686 | 5,586,739 | 4,226,729 | |||||||||
|
Interest income (expense), net
|
(194,620 | ) | (74,480 | ) | 498,512 | |||||||
|
Income before income taxes
|
7,343,066 | 5,512,259 | 4,725,241 | |||||||||
|
Provision for income taxes
|
3,027,071 | 1,824,692 | 1,096,046 | |||||||||
|
Net income
|
$ | 4,315,995 | $ | 3,687,567 | $ | 3,629,195 | ||||||
|
Basic net income per share:
|
||||||||||||
|
Net income
|
$ | 0.34 | $ | 0.32 | $ | 0.32 | ||||||
|
Basic weighted average common shares outstanding
|
12,624,495 | 11,386,989 | 11,308,124 | |||||||||
|
Diluted net income per share:
|
||||||||||||
|
Net income
|
$ | 0.32 | $ | 0.32 | $ | 0.32 | ||||||
|
Diluted weighted average common shares outstanding
|
13,646,533 | 11,562,304 | 11,460,801 | |||||||||
|
Net income
|
$ | 4,315,995 | $ | 3,687,567 | $ | 3,629,195 | ||||||
|
Other comprehensive income (loss)
|
||||||||||||
|
Foreign currency translation adjustment
|
(2,547,776 | ) | - | - | ||||||||
|
Comprehensive income
|
$ | 1,768,219 | $ | 3,687,567 | $ | 3,629,195 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
-48-
|
Anika Therapeutics, Inc. and Subsidiaries
|
||||||||||||||||||||||||
|
Accumulated
|
||||||||||||||||||||||||
|
Common Stock
|
Other
|
Total
|
||||||||||||||||||||||
|
Number of
|
$.01 Par
|
Additional Paid
|
Retained
|
Comprehensive
|
Stockholders'
|
|||||||||||||||||||
|
Shares
|
Value
|
in Capital
|
Earnings
|
Income (Loss)
|
Equity
|
|||||||||||||||||||
|
Balance, December 31, 2007
|
11,223,273 | $ | 112,233 | $ | 40,695,940 | $ | 14,152,784 | $ | $ | 54,960,957 | ||||||||||||||
| Issuance of common stock for | ||||||||||||||||||||||||
|
employee equity awards
|
154,350 | 1,543 | 515,439 | 516,982 | ||||||||||||||||||||
| Tax benefit related to stock based | ||||||||||||||||||||||||
|
compensation
|
258,146 | 258,146 | ||||||||||||||||||||||
|
Stock based compensation expense
|
1,391,704 | 1,391,704 | ||||||||||||||||||||||
|
Net income
|
- | 3,629,195 | 3,629,195 | |||||||||||||||||||||
|
Balance, December 31, 2008
|
11,377,623 | 113,776 | 42,861,229 | 17,781,979 | 60,756,984 | |||||||||||||||||||
| Issuance of common stock for | ||||||||||||||||||||||||
|
employee equity awards
|
59,957 | 600 | 2,550 | 3,150 | ||||||||||||||||||||
| Acquisition of Fidia Advanced | ||||||||||||||||||||||||
|
Biopolymers S.r.l.
|
1,981,192 | 19,812 | 16,800,508 | 16,820,320 | ||||||||||||||||||||
| Tax shortfall related to stock based | ||||||||||||||||||||||||
|
compensation
|
(82,544 | ) | (82,544 | ) | ||||||||||||||||||||
|
Stock based compensation expense
|
958,025 | 958,025 | ||||||||||||||||||||||
|
Net income
|
3,687,567 | 3,687,567 | ||||||||||||||||||||||
|
Balance, December 31, 2009
|
13,418,772 | 134,188 | 60,539,768 | 21,469,546 | 82,143,502 | |||||||||||||||||||
| Issuance of common stock for | ||||||||||||||||||||||||
|
employee equity awards
|
63,612 | 635 | 196,609 | 197,244 | ||||||||||||||||||||
| Tax benefit related to stock based | ||||||||||||||||||||||||
|
compensation
|
(21,188 | ) | (21,188 | ) | ||||||||||||||||||||
|
Stock based compensation expense
|
1,102,369 | 1,102,369 | ||||||||||||||||||||||
|
Net income
|
4,315,995 | (2,547,776 | ) | 1,768,219 | ||||||||||||||||||||
|
Balance, December 31, 2010
|
13,482,384 | $ | 134,823 | $ | 61,817,558 | $ | 25,785,541 | $ | (2,547,776 | ) | $ | 85,190,146 | ||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
-49-
|
Anika Therapeutics, Inc. and Subsidiaries
|
||||||||||||
|
For the year ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Cash flows from operating activities:
|
||||||||||||
|
Net income
|
$ | 4,315,995 | $ | 3,687,567 | $ | 3,629,195 | ||||||
| Adjustments to reconcile net income to net cash provided | ||||||||||||
|
by operating activities:
|
||||||||||||
|
Depreciation and amortization
|
3,320,352 | 1,293,468 | 1,433,012 | |||||||||
|
Amortization of premium on short-term investment
|
- | - | 1,974 | |||||||||
|
Stock-based compensation expense
|
1,102,617 | 958,025 | 1,391,704 | |||||||||
|
Deferred income taxes
|
1,953,946 | 1,735,947 | 377,045 | |||||||||
|
Provision for doubtful accounts
|
302,723 | - | - | |||||||||
|
Provision for inventory
|
699,057 | 350,220 | 138,290 | |||||||||
|
Tax benefit from exercise of stock options
|
(65,434 | ) | (27,349 | ) | (258,146 | ) | ||||||
|
Changes in operating assets and liabilities, net of effect of acquisition:
|
||||||||||||
|
Accounts receivable
|
(3,716,478 | ) | (1,697,673 | ) | 377,552 | |||||||
|
Inventories
|
(1,220,359 | ) | (1,871,545 | ) | (1,267,926 | ) | ||||||
|
Prepaid expenses and other current assets
|
445,653 | (774,764 | ) | 876,576 | ||||||||
|
Long-term deposits and other
|
28,239 | 93,559 | (73,706 | ) | ||||||||
|
Accounts payable
|
5,784,731 | 141,083 | (129,662 | ) | ||||||||
|
Accrued expenses
|
(2,188,082 | ) | 1,718,307 | (733,070 | ) | |||||||
|
Deferred revenue
|
(2,751,468 | ) | (2,680,831 | ) | (2,774,485 | ) | ||||||
|
Other long-term liabilities
|
(158,028 | ) | 168,691 | 364,686 | ||||||||
|
Net cash provided by operating activities
|
7,853,461 | 3,094,705 | 3,407,231 | |||||||||
|
Cash flows from investing activities:
|
||||||||||||
|
Proceeds from maturity of short-term investment
|
- | - | 3,500,000 | |||||||||
|
Purchase of property and equipment, net
|
(2,784,977 | ) | (3,962,232 | ) | (16,246,494 | ) | ||||||
|
Payment for the acquisition of FAB, net of cash acquired
|
- | (16,255,637 | ) | - | ||||||||
|
Reduction in purchase price of acquisition
|
105,300 | - | - | |||||||||
|
Other assets
|
- | - | (58,058 | ) | ||||||||
|
Net cash used in investing activities
|
(2,679,677 | ) | (20,217,869 | ) | (12,804,552 | ) | ||||||
|
Cash flows from financing activities:
|
||||||||||||
|
Principal payments on debt
|
(1,600,000 | ) | (1,600,000 | ) | - | |||||||
|
Proceeds from long-term debt
|
- | - | 16,000,000 | |||||||||
|
Debt Issuance Costs
|
- | (74,000 | ) | (87,721 | ) | |||||||
|
Proceeds from exercise of stock options
|
197,246 | 3,150 | 516,982 | |||||||||
|
Tax benefit from exercise of stock options
|
65,434 | 27,349 | 258,146 | |||||||||
|
Net cash (used in) provided by financing activities
|
(1,337,320 | ) | (1,643,501 | ) | 16,687,407 | |||||||
|
Exchange rate impact on cash
|
(61,522 | ) | - | - | ||||||||
|
Increase (decrease) in cash and cash equivalents
|
3,774,942 | (18,766,665 | ) | 7,290,086 | ||||||||
|
Cash and cash equivalents at beginning of period
|
24,426,990 | 43,193,655 | 35,903,569 | |||||||||
|
Cash and cash equivalents at end of period
|
$ | 28,201,932 | $ | 24,426,990 | $ | 43,193,655 | ||||||
|
Supplemental disclosure of cash flow information:
|
||||||||||||
|
Cash paid for income taxes
|
$ | 360,000 | $ | 1,210,000 | $ | 10,000 | ||||||
|
Cash paid for interest
|
$ | 222,919 | $ | 208,053 | $ | 191,137 | ||||||
|
Supplemental disclosure of cash flow information:
|
||||||||||||
|
Fair value of assets of FAB and product lines
|
$ | - | $ | 50,539,846 | $ | - | ||||||
|
Cash paid for FAB and product lines
|
$ | - | $ | 17,055,000 | $ | - | ||||||
|
Fair value of common stock issued to acquire FAB
|
$ | - | $ | 16,820,320 | $ | - | ||||||
|
Liabilities assumed of acquired businesses and product lines
|
$ | - | $ | 16,664,611 | $ | - | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
-50-
Anika Therapeutics, Inc. and Subsidiaries
1. Nature of Business
Anika Therapeutics, Inc. (“Anika,” the “Company,” “we,” “us,” or “our”) develops, manufactures and commercializes therapeutic products for tissue protection, healing and repair. These products are based on hyaluronic acid (“HA”), a naturally occurring, biocompatible polymer found throughout the body. Due to its unique biophysical and biochemical properties, HA plays an important role in a number of physiological functions such as the protection and lubrication of soft tissues and joints, the maintenance of the structural integrity of tissues, and the transport of molecules to and within cells.
On December 30, 2009, Anika Therapeutics, Inc. entered into a Sale and Purchase Agreement (the “Purchase Agreement”) with Fidia Farmaceutici S.p.A., a privately held Italian corporation, pursuant to which the Company acquired 100% of the issued and outstanding stock of Fidia Advanced Biopolymers S.r.l. (“FAB”), a privately held Italian corporation, for a purchase price consisting of $17.1 million in cash and 1,981,192 shares of the Company’s common stock valued at $16.8 million based on the closing stock price of $8.49 per share.
The Company is subject to risks common to companies in the biotechnology and medical device industries including, but not limited to, development by the Company or its competitors of new technological innovations, dependence on key personnel, protection of proprietary technology, commercialization of existing and new products, and compliance with FDA government regulations and approval requirements as well as the ability to grow the Company’s business.
2. Summary of Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Principles of Consolidation
The accompanying consolidated financial statements include the accounts of Anika Therapeutics, Inc. and its wholly owned subsidiaries, Anika Securities, Inc. (a Massachusetts Securities Corporation), and Anika Therapeutics S.r.l. All intercompany balances and transactions have been eliminated in consolidation.
Foreign Currency Translation
The functional currency of our foreign subsidiary is the euro. Assets and liabilities of the foreign subsidiary are translated using the exchange rate existing on each respective balance sheet date. Revenues and expenses are translated using the monthly average exchange rates prevailing throughout the year. The translation adjustments resulting from this process are included as a component of accumulated other comprehensive income (loss).
Fair Value Measurements
Fair value is defined as the price that would be received from selling an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. When determining the fair value measurements for assets and liabilities required to be recorded at fair value, we consider the principal or most advantageous market in which we would transact and consider assumptions that market participants would use when pricing the asset or liability, such as inherent risk, transfer restrictions, and risk of nonperformance. The accounting standard establishes a fair value hierarchy that requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value.
-51-
A financial instrument’s categorization within the fair value hierarchy is based upon the lowest level of input that is significant to the fair value measurement. Three levels of inputs that may be used to measure fair value:
|
|
•
|
Level 1 – Valuation is based upon quoted prices for identical instruments traded in active markets. Level 1 instruments include securities traded on active exchange markets, such as the New York Stock Exchange.
|
|
|
•
|
Level 2 – Valuation is based upon quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active and model-based valuation techniques for which all significant assumptions are observable in the market.
|
|
|
•
|
Level 3 – Valuation is generated from model-based techniques that use significant assumptions not observable in the market. These unobservable assumptions reflect our own estimates of assumptions market participants would use in pricing the asset or liability.
|
Our significant financial assets and liabilities measured at fair value on a recurring basis as of December 31, 2010 and 2009 were as follows:
| December 31, 2010 | |||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Cash equivalents - money market transations | $ | 20,244, 955 | $ | - | $ | - | $ | 20,244,955 | |||||||
| December 31, 2009 | |||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Cash equivalents - money market accounts | $ | 20,212,991 | $ | - | $ | - | $ | 20,212,991 | |||||||
The carrying value of our debt instrument was $12,800,000 and $14,400,000 at December 31, 2010 and 2009, respectively. The estimated fair value of our debt instrument approximated book value at both dates using market observable inputs and interest rate measurements.
Allowance for Doubtful Accounts
We maintain allowances for doubtful accounts for estimated losses resulting from the inability of our customers to make required payments. In determining the adequacy of the allowance for doubtful accounts, management specifically analyzes individual accounts receivable, historical bad debts, customer concentrations, customer credit-worthiness, current economic conditions, accounts receivable aging trends and changes in our customer payment terms. Our allowance for doubtful accounts on trade accounts receivable was $30,000 and $29,261 at December 31, 2010 and 2009, respectively. There was no activity relative to the allowance for doubtful accounts during 2008.
Uncollectible trade accounts receivable written-off were $301,984 and $93,484 in 2010 and 2009, respectively. Provisions for bad debt expense were $302,723 and $62,745 in 2010 and 2009, respectively, and are included in general and administrative expenses in the accompanying consolidated statements of operations.
Revenue Recognition - General
We recognize revenue from product sales when all of the following criteria are met: persuasive evidence of an arrangement exists; delivery has occurred or services have been rendered; the seller's price to the buyer is fixed or determinable; and collection from the customer is reasonably assured.
Product Revenue
Revenues from product sales are recognized when title and risk of loss have passed to the customer, which is typically upon shipment to the customer. Amounts billed or collected prior to recognition of revenue are classified as deferred revenue. When determining whether risk of loss has transferred to customers on product sales, or if the sales price is fixed or determinable, the Company evaluates both the contractual terms and conditions of its distribution and supply agreements as well as its business practices.
Product revenue also includes royalties. Royalty revenue is based on our distributors’ sales and recognized in the same period our distributors record their sale of products manufactured by us. On a quarterly basis we record royalty revenue based upon sales projections provided to us by our distributor customers. If necessary we adjust our estimates based upon final sales data received prior to issuing our annual audited financial statements.
-52-
Licensing, Milestone and Contract Revenue
Licensing, milestone, and contract revenue consist of revenue recognized on initial and milestone payments, as well as contractual amounts received from partners. The Company’s business strategy includes entering into collaborative license, development and/or supply agreements with partners for the development and commercialization of the Company’s products.
The terms of the agreements typically include non-refundable license fees, funding of research and development, payments based upon achievement of certain milestones, and royalties on product sales. The Company evaluates each agreement, and elements within each agreement, in accordance with ASC Subtopic 605-25, Multiple Element Arrangements (“ASC 605-25”). Under ASC 605-25, in order to account for an element as a separate unit of accounting, the element must have had stand-alone value and there must have been objective and reliable evidence of fair value of the undelivered elements. In general, non-refundable upfront fees and milestone payments were recognized as revenue over the term of the arrangement as the Company completes its performance obligations.
Grant Research Revenue
With the FAB acquisition, the Company assumed two grant contracts with the European Community related to cell-based tissue engineered products and disc regeneration research. FAB coordinates the fiscal activities for a group of participating companies and universities, and accounts for these contracts by recording an accounts receivable for the reimbursable expenses incurred under the contract, and records a liability for any amounts due to the other participants. Expenses are recorded as incurred.
Cash, Cash Equivalents and Marketable Investments
We consider only those investments which are highly liquid, readily convertible to cash, and that mature within three months from date of purchase to be cash equivalents. Marketable investments are those with original maturities in excess of three months.
At December 31, 2010 and 2009, cash equivalents were comprised of money market funds secured by U.S. Treasury obligations, which approximates fair market value. We had no marketable investments at December 31, 2010 and 2009, respectively.
Concentration of Credit Risk and Significant Customers
The Company has no significant off-balance sheet risks related to foreign exchange contracts, option contracts or other foreign hedging arrangements. The Company currently maintains its cash equivalent balance with one major national financial institution.
The Company, by policy, routinely assesses the financial strength of its customers. As a result, the Company believes that its accounts receivable credit risk exposure is limited and generally has not experienced significant write-downs in its accounts receivable balances with the single exception of Coapt Systems, Inc. (“Coapt”).
On May 15, 2009, the Company entered into a Distribution Agreement with Coapt, granting Coapt an exclusive and non-transferable right to market HYDRELLE™ in the United States. The Company was to receive payments for the supply of HYDRELLE™ to Coapt and royalties on future Coapt net product sales to its customers. On July 2, 2010 we were notified by Coapt that it had filed for an Assignment for the Benefit of Creditors under the laws of the State of California. As a result of Coapt entering receivership during 2010 we reserved for, and subsequently wrote-off, approximately $270,000 associated with invoices for product and royalties left unpaid at the time of Coapt’s filing.
As of December 31, 2010, Johnson and Johnson , Bausch and Lomb, Medtronics Xomed, Azienda USL Roma, and Biomeks, combined, represented 55% of the Company’s accounts receivable balance. As of December 31, 2009, Bausch & Lomb, Johnson and Johnson, Boehringer Ingelheim Vetmedica, Coapt, and Rivex, combined, represented 53% of the Company’s accounts receivable balance.
-53-
Inventories
Inventories are stated at the lower of cost or market, with cost being determined using the first-in, first-out (FIFO) method. Work-in-process and finished goods inventories include materials, labor, and manufacturing overhead.
The Company’s policy is to write-down inventory when conditions exist that suggest inventory may be in excess of anticipated demand or is obsolete based upon assumptions about future demand for the Company’s products and market conditions. The Company regularly evaluates the ability to realize the value of inventory based on a combination of factors including, but not limited to: historical usage rates, forecasted sales or usage, product end of life dates, and estimated current or future market values. Purchasing requirements and alternative usage avenues are explored within these processes to mitigate inventory exposure.
When recorded, inventory write-downs are intended to reduce the carrying value of inventory to its net realizable value. Inventory of $8,949,745 and $8,547,339 as of December 31, 2010 and 2009 is stated net of inventory write-downs of $631,028 and $150,000, respectively. If actual demand for the Company’s products deteriorates, or market conditions are less favorable than those projected, additional inventory write-downs may be required.
Property and Equipment
Property and equipment are recorded at cost and depreciated using the straight-line method over their estimated useful lives. Computer hardware and software are typically amortized over three to five years, and furniture and fixtures over three to eight years. Leasehold improvements are amortized over the shorter of their useful lives or the remaining terms of the related leases which range from six months to 22 years at December 31, 2010. Property and equipment under capital leases are amortized over the lesser of the lease terms or their estimated useful lives. Maintenance and repairs are charged to expense when incurred; additions and improvements are capitalized. When an item is sold or retired, the cost and related accumulated depreciation is relieved, and the resulting gain or loss, if any, is recognized in income.
Goodwill and Acquired Intangible Assets
Goodwill is the amount by which the purchase price of acquired net assets in a business combination exceeded the fair values of net identifiable assets on the date of acquisition. Acquired In-Process Research and Development (“IPR&D”) represents the fair value assigned to research and development assets that we acquire that have not been completed at the date of acquisition or are pending regulatory approval in certain jurisdictions. The value assigned to acquired IPR&D is determined by estimating the costs to develop the acquired technology into commercially viable products, estimating the resulting revenue from the projects, and discounting the net cash flows to present value.
Goodwill and IPR&D, are evaluated for impairment annually or more frequently if events or changes in circumstances indicate that the asset might be impaired. Factors we consider important, on an overall company basis, that could trigger an impairment review include significant underperformance relative to historical or projected future operating results, significant changes in our use of the acquired assets or the strategy for our overall business, significant negative industry or economic trends, a significant decline in our stock price for a sustained period, or a reduction of our market capitalization relative to net book value.
To conduct impairment tests of goodwill, the fair value of the reporting unit is compared to its carrying value. If the reporting unit’s carrying value exceeds its fair value, we record an impairment loss to the extent that the carrying value of goodwill exceeds its implied fair value. We estimate the fair value for reporting units using discounted cash flow valuation models which require the use of significant estimates and assumptions including but not limited to: risk free rate of return on an investment, weighted average cost of capital, future revenue, operating margin, working capital and capital expenditure needs. Our annual assessment for impairment of goodwill as of November 30, 2010 indicated that the fair value of our reporting units exceeded the carrying value of the reporting units. There can be no assurance that, at the time future impairment tests are completed, a material impairment charge will not be recorded.
To conduct impairment tests of IPR&D, the fair value of the IPR&D project is compared to its carrying value. If the carrying value exceeds its fair value, we record an impairment loss to the extent that the carrying value of the IPR&D project exceeds its fair value. We estimate the fair values for IPR&D projects using discounted cash flow valuation models which require the use of significant estimates and assumptions including but not limited to: estimating the timing of and expected costs to complete the in process projects, projecting regulatory approvals, estimating future cash flows from product sales resulting from completed projects and in process projects, and developing appropriate discount rates. Our annual assessment for impairment of IPR&D indicated that the fair value of our IPR&D as of November 30, 2010 exceeded their respective carrying values. There can be no assurance that, at the time future impairment tests are completed, a material impairment charge will not be recorded.
During the years ended December 31, 2010, 2009, and 2008, the Company did not record any impairment losses.
Long-Lived Assets
Long-lived assets primarily include property and equipment and intangible assets with finite lives (including purchased software and trade names). Purchased software is amortized over 2 to 10 years and trade names are amortized over 10 years. We review long-lived assets for impairment when events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable or that the useful lives of those assets are no longer appropriate. Each impairment test is based on a comparison of the undiscounted cash flows to the recorded value of the asset. If impairment is indicated, the asset is written down to its estimated fair value based on a discounted cash flow analysis.
-54-
Research and Development
Research and development costs consist primarily of salaries and related expenses for personnel and fees paid to outside consultants and outside service providers, including costs associated with licensing, milestone and contract revenue. Research and development costs are expensed as incurred.
Advertising Expenses
Promotional costs associated with new product introduction initiatives are deferred and charged to operations the first time the programs are publicly launched. All other advertising costs are expensed as incurred. Advertising expenses were negligible for all periods presented.
Income Taxes
Our income tax expense includes U.S. and international income taxes. Certain items of income and expense are not reported in tax returns and financial statements in the same year. The tax effects of these differences are reported as deferred tax assets and liabilities. Deferred tax assets are recognized for the estimated future tax effects of deductible temporary differences, tax operating losses, and credit carryforwards (including investment tax credits). Changes in deferred tax assets and liabilities are recorded in the provision for income taxes. We assess the likelihood that our deferred tax assets will be recovered from future taxable income and, to the extent we believe that it is more likely than not that all or a portion of deferred tax assets will not be realized, we establish a valuation allowance. To the extent we establish a valuation allowance or increase this allowance in a period, we include an expense within the tax provision in the consolidated statement of operations.
Stock-Based Compensation
We measure the compensation cost of employee services received in exchange for an award of equity instruments based on the grant-date fair value of the underlying award. That cost is recognized over the period during which an employee is required to provide service in exchange for the award. See Note 10 for a description of the types of stock-based awards granted, the compensation expense related to such awards, and detail of equity-based awards outstanding. See Note 14 for detail of the tax benefit recognized in the consolidated statement of operations related to stock-based compensation.
Comprehensive Income
Comprehensive income consists of net income and other comprehensive income (loss), which includes foreign currency translation adjustments. For the purposes of comprehensive income disclosures, we do not record tax provisions or benefits for the net changes in the foreign currency translation adjustment, as we intend to reinvest permanently undistributed earnings of our foreign subsidiary. Accumulated other comprehensive income (loss) is reported as a component of stockholders' equity and, as of December 31, 2010 and 2009, was comprised solely of cumulative translation adjustment gains.
Segment Information
Operating segments, as defined under U.S. GAAP, are components of an enterprise about which separate financial information is available that is evaluated regularly by the chief operating decision maker, or decision-making group, in deciding how to allocate resources and in assessing performance. The Company’s chief operating decision maker is its Chief Executive Officer. Based on the criteria established by ASC 280, Segment Reporting, the Company has one reportable operating segment the results of which are disclosed in the accompanying consolidated financial statements.
Recent Accounting Pronouncements
In July 2010, the FASB issued Accounting Standard Update (“ASU”) No. 2010-20, Disclosures about the Credit Quality of Financing Receivables and the Allowance for Credit Losses. ASU No. 2010-20 requires disclosure about the credit quality of accounts receivables with terms exceeding one year and any related allowance for credit losses. These new disclosures are required for interim and fiscal periods ending after December 15, 2010 (our fourth quarter of fiscal 2010). Adoption of this standard did not have a material impact on our consolidated financial position, results of operations, or cash flows.
-55-
In April 2010, the FASB issued ASU No. 2010-017, Revenue Recognition — Milestone Method, (“ASU 2010-017”). ASU 2010-017 provides guidance in applying the milestone method of revenue recognition to research or development arrangements. Under this guidance management may recognize revenue contingent upon the achievement of a milestone in its entirety, in the period in which the milestone is achieved, only if the milestone meets all the criteria within the guidance to be considered substantive. This ASU is effective on a prospective basis for research and development milestones achieved in fiscal years, beginning on or after June 15, 2010. Early adoption is permitted; however, we have elected to implement ASU 2010-17 prospectively, and as a result, the effect of this guidance will be limited to future transactions. We do not expect adoption of this standard to have a material impact on our consolidated financial position, results of operations, or cash flows.
In December 2010, the FASB issued ASU No. 2010-027, Fees Paid to the Federal Government by Pharmaceutical Manufacturers, (“ASU 2010-027”). ASU 2010-027 provides guidance concerning the recognition and classification of the new annual fee payable by branded prescription drug manufactures and importers on branded prescription drugs which was mandated under the health care reform legislation enacted in the U.S. in March 2010. Under this new accounting standard, the annual fee would be presented as a component of operating expenses and recognized over the calendar year such fees are payable using a straight-line method of allocation unless another method better allocates the fee over the calendar year. This ASU is effective for calendar years beginning on or after December 31, 2010, when the fee initially becomes effective. We do not expect adoption of this standard to have a material impact on our consolidated financial position, results of operations, or cash flows.
3. Earnings per Share (“EPS”)
Basic EPS is calculated by dividing net income by the weighted average number of shares outstanding during the period. Unvested restricted shares, although legally issued and outstanding, are not considered outstanding for purposes of calculating basic earnings per share. Diluted EPS is calculated by dividing net income by the weighted average number of shares outstanding plus the dilutive effect, if any, of outstanding stock options, stock appreciation rights (“SAR’s”), restricted shares and restricted stock units (collectively “RSA’s”) using the treasury stock method.
The following table provides share information used in the calculation of the Company's basic and diluted earnings per share:
|
Year ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Shares used in the calculation of
|
||||||||||||
|
Basic earnings per share
|
12,624,495 | 11,386,989 | 11,308,124 | |||||||||
|
Effect of dilutive securities:
|
||||||||||||
|
Stock options, SAR's, and RSA's
|
1,022,038 | 175,315 | 152,677 | |||||||||
|
Diluted shares used in the calculation of
|
||||||||||||
|
earnings per share
|
13,646,533 | 11,562,304 | 11,460,801 | |||||||||
Stock options to purchase 1,210,970, 924,007 and 757,153 shares for 2010, 2009 and 2008, respectively, were excluded from the computation of diluted EPS as their effect would have been anti-dilutive.
At December 31, 2010, 2009 and 2008, 20,630, 46,965 and 83,395 shares of issued and outstanding unvested restricted stock were excluded from the basic earnings per share calculation in accordance with ASC 260.
4. Inventories
Inventories consist of the following:
| December 31, | ||||||||
|
2010
|
2009
|
|||||||
|
Raw materials
|
$ | 2,882,944 | $ | 2,535,496 | ||||
|
Work-in-process
|
1,787,473 | 3,188,241 | ||||||
|
Finished goods
|
4,279,328 | 2,823,602 | ||||||
|
Total
|
$ | 8,949,745 | $ | 8,547,339 | ||||
-56-
5. Property and Equipment
Property and equipment is stated at cost and consists of the following:
| December 31, | ||||||||
|
2010
|
2009
|
|||||||
|
Machinery and equipment
|
$ | 9,972,821 | $ | 9,859,867 | ||||
|
Furniture and fixtures
|
640,931 | 608,361 | ||||||
|
Leasehold improvements
|
12,074,288 | 12,117,091 | ||||||
|
Construction in progress
|
27,008,950 | 24,587,084 | ||||||
|
Subtotal
|
49,696,989 | 47,172,403 | ||||||
|
Less accumulated depreciation
|
(12,715,595 | ) | (11,424,788 | ) | ||||
|
Total
|
$ | 36,981,394 | $ | 35,747,615 | ||||
Depreciation expense was $1,308,713, $1,234,644 and $1,374,189 for the years ended December 31, 2010, 2009 and 2008, respectively. The above amounts, at December 31, 2010, include $1,025,350 of machinery and equipment, $15,688 of furniture and fixtures, and $522,197 of leasehold improvements from the FAB acquisition.
6. Acquired Intangible Assets, Net
In November 2007, in connection with the termination of the agreement with Galderma which originally granted to Galderma the worldwide rights to commercialization, distribution, and marketing of ELEVESS products, the Company reacquired the worldwide rights and control of the future development and marketing of ELEVESS. The intangible asset realized during this process was the ELEVESS trade name.
On December 30, 2009, in connection with the acquisition of FAB, the Company purchased various intangible assets. The Company finalized the purchase price allocation relative to this acquisition during the fourth quarter of 2010. See Note 16 for additional disclosure relative to this subject.
We completed our annual impairment review as of November 30, 2010 and concluded that no impairment charge was required as of that date with respect to both goodwill and acquired intangible assets. Through December 31, 2010 there have not been any events or changes in circumstances that indicate that the carrying value of goodwill or acquired intangible assets may not be recoverable. There can be no assurance that, at the time future impairment tests are completed, a material impairment charge will not be recorded.
Amortization expense was $2,011,639, $58,823, and $58,823 for the years ended December 31, 2010, 2009 and 2008, respectively. As of December 31, 2010, amortization expense on intangible assets for the next five years is expected to be approximately $2.0 million annually.
Intangible assets, stated cost, consist of the following:
|
December 31, 2010
|
December 31, 2009 | |||||||||||||||||||||||||||
|
Gross Value
|
Currency
Translation Adjustment
|
Completed
Projects
|
Accumulated Amortization
|
Net Book
Value
|
Net Book
Value
|
Useful Life
|
||||||||||||||||||||||
|
Developed technology
|
$ | 15,700,000 | $ | (1,182,194 | ) | $ | 1,000,000 | $ | (967,854 | ) | $ | 14,549,952 | $ | 15,700,000 | 15 | |||||||||||||
|
In-process research & development
|
7,698,000 | (579,652 | ) | (1,000,000 | ) | - | $ | 6,118,348 | 7,698,000 |
Indefinite
|
||||||||||||||||||
|
Distributor relationships
|
4,700,000 | (353,905 | ) | (869,219 | ) | $ | 3,476,876 | 4,700,000 | 5 | |||||||||||||||||||
|
Patents
|
1,000,000 | (75,299 | ) | (57,794 | ) | $ | 866,907 | 1,000,000 | 16 | |||||||||||||||||||
|
Elevess trade name
|
1,000,000 | - | (247,899 | ) | $ | 752,101 | 877,451 | 9 | ||||||||||||||||||||
|
Total
|
$ | 30,098,000 | $ | (2,191,050 | ) | $ | - | $ | (2,142,765 | ) | $ | 25,764,185 | $ | 29,975,451 | ||||||||||||||
-57-
7. Accrued Expenses
Accrued expenses consist of the following:
|
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
Payroll and benefits
|
$ | 1,895,393 | $ | 2,137,067 | ||||
|
Professional fees
|
417,751 | 1,470,007 | ||||||
|
Clinical trial costs
|
149,319 | 129,509 | ||||||
|
FAB research grants
|
2,021,003 | 1,625,044 | ||||||
|
Other
|
892,119 | 454,543 | ||||||
|
Total
|
$ | 5,375,585 | $ | 5,816,170 | ||||
8. Deferred Revenue
In December 2003, the Company entered into a ten-year licensing and supply agreement (the “JNJ Agreement”) with Ortho Biotech Products, L.P., a member of the Johnson & Johnson family of companies, to market ORTHOVISC in the U.S. In mid-2005, the agreement was assigned to DePuy Mitek, Inc., a subsidiary of Johnson & Johnson. Under the JNJ Agreement, DePuy Mitek performs sales, marketing and distribution functions and licensed the right to further develop and commercialize ORTHOVISC as well as other new products for the treatment of pain associated with osteoarthritis based on the Company’s viscosupplementation technology. In support of the license, the JNJ Agreement provides that DePuy Mitek will fund post-marketing clinical trials for new indications of ORTHOVISC.
The Company received an initial payment of $2,000,000 upon entering into the JNJ Agreement, a milestone payment of $20,000,000 in February 2004, as a result of obtaining FDA approval of ORTHOVISC and a milestone payment of $5,000,000 in December 2004 for planned upgrades to our manufacturing operations. The Company evaluated the terms of the JNJ Agreement and determined that the upfront fee and milestone payments did not meet the conditions to be recognized separately from the supply agreement, therefore, the Company has deferred non-refundable payments received of $27,000,000 which we are recognizing ratably over the expected ten year term of the JNJ Agreement. Current and long-term deferred revenue related to the JNJ Agreement were $8,099,995 and $10,799,996 at December 31, 2010 and 2009, respectively.
9. Commitments and Contingencies
Leasing Arrangements
The Company’s headquarters facility is located in Bedford, Massachusetts, where the Company leases approximately 134,000 square feet of administrative, manufacturing, and research and development (“R&D”) space. This lease was entered into on January 4, 2007, and the lease commenced on May 1, 2007 for an initial term of ten and one-half years. The Company has an option under the lease to extend its terms for up to four periods beyond the original expiration date subject to the condition that we notify the landlord that we are exercising each option at least one year prior to the expiration of the original or current term thereof. The first three renewal options each extend the term an additional five years with the final renewal option extending the term six years.
The Company’s administrative and R&D personnel moved into the Bedford facility in November of 2007. The build-out of the Bedford facility and required validation process for the manufacturing space will be completed during 2011. We also lease approximately 37,000 square feet of space at a separate location in Woburn, Massachusetts, for our current manufacturing facility and warehouse. The Woburn manufacturing lease was extended in February of 2011 and is scheduled to end on June 30, 2011. See Note 18 for additional information.
As part of the acquisition of FAB, we now lease approximately 26,000 square feet of laboratory, warehouse and office space in Abano Terme, Italy. The lease commenced on December 30, 2009 for an initial term of six (6) years.
Rental expense in connection with the various facility leases totaled $2,888,277, $1,651,713 and $1,486,472, for the years ended December 31, 2010, 2009, and 2008, respectively.
-58-
The Company’s future lease commitments as of December 31, 2010 are as follows:
|
2011
|
$ | 2,610,613 | ||
|
2012
|
1,638,531 | |||
|
2013
|
1,683,198 | |||
|
2014
|
1,642,446 | |||
|
2015 and thereafter
|
4,765,421 | |||
| $ | 12,340,208 |
Warranty and Guarantor Arrangements
In certain of our contracts, the Company warrants to its customers that the products it manufactures conform to the product specifications as in effect at the time of delivery of the specific product. The Company may also warrant that the products it manufactures do not infringe, violate, or breach any U.S. patent or intellectual property rights, trade secret, or other proprietary information of any third party. On occasion, the Company contractually indemnifies its customers against any and all losses arising out of, or in any way connected with, any claim or claims of breach of its warranties or any actual or alleged defect in any product caused by the negligence or acts or omissions of the Company. The Company maintains a products liability insurance policy that limits its exposure to these risks. Based on the Company’s historical activity, in combination with its liability insurance coverage, the Company believes the estimated fair value of these indemnification agreements is immaterial. The Company has no accrued warranties at December 31, 2010 and 2009, respectively, and has no history of claims paid.
Legal Proceedings
On July 7, 2010, Genzyme Corporation filed a complaint against the Company in the United States District Court for the District of Massachusetts seeking unspecified damages and equitable relief. The Complaint alleges that the Company has infringed U.S. Patent No. 5,143,724 by manufacturing MONOVISC in the United States for sale outside the United States and will infringe U.S. Patent Nos. 5,143,724 and 5,399,351 if the Company begins manufacture and sale of MONOVISC in the United States. On August 30, 2010, the Company filed an answer denying liability. The Company believes that neither MONOVISC, nor its manufacture, does or will infringe any valid and enforceable claim of the asserted patents. Management has assessed and determined that contingent losses related to this matter are remote and inestimable. Therefore, pursuant to Accounting Standards Codification (“ASC”) 450, Contingencies, an accrual has not been recorded for this loss contingency.
Artes Medical, Inc. (“Artes”), the former U.S. distributor of HYDRELLE, filed a liquidating bankruptcy case under Chapter 7 of the United States Bankruptcy Code. Artes’ Trustee in Bankruptcy asked the Company to pay $359,768 to the Trustee, representing the total amount of three payments received by the Company from Artes within the 90 days prior to the filing of Artes' liquidating bankruptcy. The Trustee asserts that the payments are recoverable as preferences under the Bankruptcy Code.
The Company believes that the payments it received either do not meet the legal requirements of avoidable preferences or are subject to one or more exceptions to the Trustee's powers to recover preferences and has recently so advised the Trustee. Management has assessed and determined that contingent losses related to this matter are remote. Therefore, pursuant to ASC 450, an accrual has not been recorded for this loss contingency.
We are also involved in various other legal proceedings arising in the normal course of business. Although the outcomes of these other legal proceedings are inherently difficult to predict, we do not expect the resolution of these other legal proceedings to have a material adverse effect on our financial position, results of operations or cash flow.
10. Equity Incentive Plan
The Anika Therapeutics, Inc. Stock Option and Incentive Plan, as amended, (the “2003 Plan”) provides for grants of nonqualified and incentive stock options, common stock, restricted stock, restricted stock units, and stock appreciation rights (“SARs”) to employees, directors, officers and consultants. The 2003 Plan was originally approved by the Board of Directors on April 4, 2003, approved by the Company’s shareholders on June 4, 2003, and reserved 1,500,000 shares of common stock for grant pursuant to its terms.
-59-
On May 29, 2009, the Board of Directors approved changes to the 2003 Plan and adopted the Amended and Restated 2003 Stock Option and Incentive Plan (the “Amended 2003 Plan”), to increase the number of shares available to grant by 850,000. The Amended 2003 Plan was approved by the Company’s shareholders on June 5, 2009, and resulted in a total of 2,350,000 shares of common stock being reserved for issuance under the Amended 2003 Plan.
The Company may satisfy the awards upon exercise, or upon fulfillment of the vesting requirements for other equity-based awards, with either newly-issued shares or shares reacquired by the Company. Stock-based awards are granted with an exercise price equal to the market price of the Company’s stock on the date of grant. Awards contain service or performance conditions and generally become exercisable ratably over one to four years.
The 2003 Plan succeeds the Anika Therapeutics, Inc. 1993 Stock Option Plan (“1993 Plan”) which expired according to its terms in 2003. As of December 31, 2010, there were 225,376 shares still outstanding under the 1993 Plan included in the total outstanding options of 1,625,253. There are 524,929 options available for future grant at December 31, 2010.
The Company estimates the fair value of stock options and SARs using the Black-Scholes valuation model. Fair value of restricted stock is measured by the grant-date price of the Company’s shares. Key input assumptions used to estimate the fair value of stock options and SARs include the exercise price of the award, the expected award term, the expected volatility of the Company’s stock over the option’s expected term, the risk-free interest rate over the award’s expected term, and the Company’s expected annual dividend yield.
The Company uses historical data on exercise of stock options and other factors to estimate the expected term of share-based awards. The Company also evaluates forfeitures periodically and adjusts accordingly. The expected volatility assumption is based on the historical volatility of the Company’s common stock. The risk-free interest rate assumption is based on U.S. Treasury interest rates at the time of grant.
The fair value of each stock option and SAR award during 2010, 2009, and 2008 was estimated on the grant date using the Black-Scholes option-pricing model with the following assumptions:
|
December 31,
|
|||||||||
|
2010
|
2009
|
2008
|
|||||||
|
Risk free interest rate
|
1.11% to 1.88%
|
1.54% to 1.89%
|
1.44% to 2.82%
|
||||||
|
Expected volatility
|
57.60% |
59.35% to 61.03%
|
58.15% to 63.37%
|
||||||
|
Expected lives (years)
|
4 | 4 | 4 | ||||||
|
Expected dividend yield
|
0.00% | 0.00% | 0.00% | ||||||
The Company recorded $1,102,617, $958,025 and $1,391,704 of share-based compensation expense for the years ended December 31, 2010, 2009 and 2008, respectively, for stock options, SARs and restricted stock awards. The Company presents the expenses related to stock-based compensation awards in the same expense line items as cash compensation paid to each of its employees.
Combined stock options and SARs activity under our plans is summarized as follows for the years ended December 31, 2010, 2009, and 2008:
|
2010
|
2009
|
2008
|
||||||||||||||||||||||
|
Weighted
|
Weighted
|
Weighted
|
||||||||||||||||||||||
|
Exercise
|
Exercise
|
Exercise
|
||||||||||||||||||||||
|
Number of
|
Price Per
|
Number of
|
Price Per
|
Number of
|
Price Per
|
|||||||||||||||||||
|
Shares
|
Share
|
Shares
|
Share
|
Shares
|
Share
|
|||||||||||||||||||
|
Options and SAR's outstanding at
|
||||||||||||||||||||||||
|
beginning of year
|
1,372,933 | $ | 7.13 | 1,094,683 | $ | 9.00 | 1,093,479 | $ | 7.93 | |||||||||||||||
|
Granted
|
450,750 | $ | 6.35 | 365,000 | $ | 4.33 | 179,130 | $ | 10.50 | |||||||||||||||
|
Cancelled
|
(69,333 | ) | $ | 7.02 | (76,750 | ) | $ | 20.93 | (29,126 | ) | $ | 6.43 | ||||||||||||
|
Expired
|
(71,547 | ) | $ | 9.55 | (7,000 | ) | $ | 4.80 | - | $ | - | |||||||||||||
|
Exercised
|
(57,550 | ) | $ | 3.96 | (3,000 | ) | $ | 1.05 | (148,800 | ) | $ | 3.47 | ||||||||||||
|
Options and SAR's outstanding at
|
||||||||||||||||||||||||
|
end of year
|
1,625,253 | $ | 6.92 | 1,372,933 | $ | 7.13 | 1,094,683 | $ | 9.00 | |||||||||||||||
Of the 1,625,253 options and SARs outstanding at December 31, 2010, approximately 1,575,000 are vested or are expected to vest with a weighted-average exercise price of approximately $6.95 and an aggregate intrinsic value of approximately $1,425,000. The weighted average remaining contractual term of the vested and expected to vest options and SARs was 6.11 years as of December 31, 2010.
-60-
As of December 31, 2010, total unrecognized compensation costs related to non-vested options and SARs was approximately $1,500,000 and is expected to be recognized over a weighted average period of 2.8 years.
There were 551,343 options exercisable at December 31, 2010 with a weighted-average exercise price of $6.29 and a weighted-average remaining contractual term of 2.60 years.
There were 332,782 SARs exercisable at December 31, 2010 with a weighted-average exercise price of $9.49 and a weighted-average remaining contractual term of 6.69 years.
The aggregate intrinsic value of stock options and SARs fully vested at December 31, 2010, 2009 and 2008 were $1,434,768, $1,646,441, and $482,853, respectively. The aggregate intrinsic value of stock options and SARs outstanding at December 31, 2010, 2009 and 2008, were $2,135,991, $2,880,716 and $482,853, respectively.
The total intrinsic value of options and SARs exercised were $181,290, $12,770 and $729,313 for the years ended December 31, 2010, 2009 and 2008, respectively.
The total fair value of options and SARs vested during the years ended December 31, 2010, 2009, and 2008 were $761,906, $812,732, and $727,765 respectively.
The Company received $197,245, $3,150 and $516,982 for exercises of stock options during the years ended December 31, 2010, 2009 and 2008, respectively.
The restricted stock activity for the years ended December 31, 2010, 2009 and 2008 are as follows:
|
2010
|
2009
|
2008
|
||||||||||||||||||||||
|
Weighted
|
Weighted
|
Weighted
|
||||||||||||||||||||||
|
Number of
|
Grantd Date
|
Number of
|
Grantd Date
|
Number of
|
Grantd Date
|
|||||||||||||||||||
|
Shares
|
Fair Value
|
Shares
|
Fair Value
|
Shares
|
Fair Value
|
|||||||||||||||||||
|
Nonvested at Beginning of year
|
94,977 | $ | 6.94 | 83,395 | $ | 10.71 | 17,225 | $ | 11.82 | |||||||||||||||
|
Granted
|
23,580 | $ | 6.36 | 47,600 | $ | 3.05 | 77,170 | $ | 10.58 | |||||||||||||||
|
Cancelled
|
(4,750 | ) | $ | 6.13 | (7,082 | ) | $ | 9.62 | (5,850 | ) | $ | 11.39 | ||||||||||||
|
Expired
|
- | $ | - | - | $ | - | - | $ | - | |||||||||||||||
|
Vested/Released
|
(36,722 | ) | $ | 6.98 | (28,936 | ) | $ | 10.74 | (5,150 | ) | $ | 11.76 | ||||||||||||
|
Nonvested at end of year
|
77,085 | $ | 5.48 | 94,977 | $ | 6.94 | 83,395 | $ | 10.71 | |||||||||||||||
The total fair value of restricted stock and restricted stock units vested during the year ended December 31, 2010 was $229,780.
11. Shareholder Rights Plan
On April 4, 2008 the Board of Directors of the Company adopted a Shareholder Rights Plan (“2008 Plan”) that replaced the Company’s former Shareholder Rights Plan. Under the 2008 Plan, the Rights generally become exercisable if:
|
(1)
|
A person becomes an “Acquiring Person” by acquiring 15% or more of the Company’s Common Stock, or
|
|
(2)
|
A person commences a tender offer that would result in that person owning 15% or more of the Company’s Common Stock.
|
In the event that a person becomes an “Acquiring Person,” each holder of a Right (other than the Acquiring Person) would be entitled to acquire such number of shares of preferred stock which are equivalent to shares of the Company’s Common Stock having a value of twice the exercise price of the Right. If, after any such event, the Company enters into a merger or other business combination transaction with another entity, each holder of a Right would then be entitled to purchase, at the then-current exercise price, shares of the acquiring company’s common stock having a value of twice the exercise price of the Right.
-61-
The current exercise price per Right is $75.00. The Rights may be redeemed in whole, but not in part, at a price of $0.01 per Right (payable in cash, shares of the Company’s Common Stock, or other consideration deemed appropriate by the Board of Directors) by the Board of Directors only until the earlier of :
(1) The time at which any person becomes an “Acquiring Person”, or
(2) The Expiration Date.
At any time after any person becomes an “Acquiring Person”, the Board of Directors may, at its option, exchange all or any part of the then outstanding and exercisable Rights for shares of the Company’s Common Stock at an exchange ratio specified in the Rights Plan. Notwithstanding the foregoing, the Board of Directors generally will not be empowered to affect such exchange at any time after any person becomes the beneficial owner of 50% or more of the Company’s Common Stock.
In connection with the establishment of the Rights Plan, the Board of Directors approved the creation of Preferred Stock of the Company designated as Series B Junior Participating Cumulative Preferred Stock with a par value of $0.01 per share. The Board also reserved 175,000 shares of preferred stock for issuance upon exercise of the Rights. Until a Right is exercised, the holder will have no rights as a stockholder of the Company, beyond those as an existing stockholder, including the right to vote or to receive dividends.
12. Employee Benefit Plan
U.S. employees are eligible to participate in the Company’s 401(k) savings plan. Employees may elect to contribute a percentage of their compensation to the Plan, and the Company will make matching contributions up to a limit of 5% of an employee’s compensation. In addition, the Company may make annual discretionary contributions. For the years ended December 31, 2010, 2009, and 2008, the Company made matching contributions of $291,107, $323,876 and $301,155 respectively.
13. Revenue by Product Group, by Significant Customer and by Geographic Region; Geographic Information
Product revenue by product group is as follows:
|
Year Ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Orthobiologics
|
$ | 30,741,305 | $ | 22,879,899 | $ | 18,707,669 | ||||||
|
Dermal
|
3,564,616 | 1,471,165 | 505,273 | |||||||||
|
Ophthalmic surgery
|
11,971,787 | 10,573,915 | 10,678,615 | |||||||||
|
Surgical
|
3,883,444 | 121,445 | 134,780 | |||||||||
|
Veterinary
|
2,574,578 | 2,274,482 | 3,028,450 | |||||||||
| $ | 52,735,730 | $ | 37,320,906 | $ | 33,054,787 | |||||||
Product revenue by significant customers as a percent of product revenues is as follows:
|
Percent of Product Revenue
|
||||||||||||
|
Year Ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Depuy Mitek
|
42.7 | % | 45.4 | % | 40.0 | % | ||||||
|
Bausch & Lomb Incorporated
|
21.2 | % | 26.6 | % | 29.8 | % | ||||||
|
Medtronic
|
10.2 | % | 0.0 | % | 0.0 | % | ||||||
|
Boehringer
|
4.9 | % | 6.1 | % | 9.2 | % | ||||||
|
Biomeks
|
3.3 | % | 4.4 | % | 5.7 | % | ||||||
| 82.3 | % | 82.5 | % | 84.7 | % | |||||||
-62-
Revenues by geographic location in total and as a percentage of total revenues are as follows:
|
Year Ended December 31,
|
||||||||||||||||||||||||
|
2010
|
2009
|
2008
|
||||||||||||||||||||||
|
Percentage of
|
Percentage of
|
Percentage of
|
||||||||||||||||||||||
|
Revenue
|
Revenue
|
Revenue
|
Revenue
|
Revenue
|
Revenue
|
|||||||||||||||||||
|
Geographic Location:
|
||||||||||||||||||||||||
|
United States
|
$ | 38,313,594 | 69.0 | % | $ | 30,196,213 | 75.2 | % | $ | 26,789,515 | 74.9 | % | ||||||||||||
|
Europe
|
12,976,985 | 23.4 | % | 6,536,835 | 16.3 | % | 5,667,215 | 15.8 | % | |||||||||||||||
|
Other
|
4,266,015 | 7.7 | % | 3,402,656 | 8.5 | % | 3,323,057 | 9.3 | % | |||||||||||||||
|
Total
|
$ | 55,556,594 | 100 | % | $ | 40,135,704 | 100.0 | % | $ | 35,779,787 | 100.0 | % | ||||||||||||
The Company recorded licensing, milestone and contract revenue of $2,820,864, $2,814,798 and $2,725,000 for the year ended December 31, 2010, 2009, and 2008, respectively. Substantially all licensing, milestone and contract revenue was derived in the United States for all years presented.
Net long-lived assets, consisting of net property and equipment, are subject to geographic risks because they are generally difficult to move and to effectively utilize in another geographic area in a reasonable time period and because they are relatively illiquid.
Net long-lived assets by principal geographic areas were as follows:
|
Year Ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
United States
|
$ | 34,826,815 | $ | 34,056,615 | $ | 32,246,683 | ||||||
|
Italy
|
2,154,579 | 1,691,000 | - | |||||||||
|
Total
|
$ | 36,981,394 | $ | 35,747,615 | $ | 32,246,683 | ||||||
-63-
14. Income Taxes
Income Tax Expense
The components of the Company’s income before income taxes and our provision for (benefit from) income taxes consist of the following:
|
Year ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Income (loss) before income taxes
|
||||||||||||
|
Domestic
|
$ | 11,944,795 | $ | 5,512,259 | $ | 4,725,241 | ||||||
|
Foreign
|
(4,601,729 | ) | - | - | ||||||||
| $ | 7,343,066 | $ | 5,512,259 | $ | 4,725,241 | |||||||
|
Year ended December 31,
|
||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
|
Provision for (benefit from) income taxes:
|
||||||||||||
|
Current provision:
|
||||||||||||
|
Federal
|
$ | 1,063,841 | $ | (2,908 | ) | $ | 765,578 | |||||
|
State
|
(6,920 | ) | (18,237 | ) | (46,577 | ) | ||||||
|
Foreign
|
- | - | - | |||||||||
| 1,056,921 | (21,145 | ) | 719,001 | |||||||||
|
Deferred provision:
|
||||||||||||
|
Federal
|
2,828,029 | 2,010,097 | 693,732 | |||||||||
|
State
|
479,529 | (164,260 | ) | (316,687 | ) | |||||||
|
Foreign
|
(1,337,408 | ) | ||||||||||
| 1,970,150 | 1,845,837 | 377,045 | ||||||||||
|
Total expense
|
$ | 3,027,071 | $ | 1,824,692 | $ | 1,096,046 | ||||||
Deferred Tax Assets and Liabilities
Significant components of the Company’s deferred tax assets and liabilities consist of the following:
|
December 31,
|
||||||||
|
2010
|
2009
|
|||||||
|
Deferred tax assets:
|
||||||||
|
Deferred revenue
|
$ | 3,078,098 | $ | 4,161,014 | ||||
|
Stock-based compensation expense
|
1,347,412 | 1,116,599 | ||||||
|
Tax credit carry forward
|
1,072,993 | 1,646,935 | ||||||
|
Net operating loss carryforward, foreign
|
2,063,055 | 1,545,434 | ||||||
|
Accrued expenses and other
|
565,503 | 640,254 | ||||||
|
Inventory reserve
|
170,240 | 57,519 | ||||||
|
Deferred tax asset
|
$ | 8,297,301 | $ | 9,167,755 | ||||
|
December 31,
|
||||||||
| 2010 | 2009 | |||||||
|
Deferred tax liabilities:
|
||||||||
|
Intangibles related to FAB acquisition
|
$ | (8,279,637 | ) | $ | (9,635,573 | ) | ||
|
Depreciation
|
(3,851,614 | ) | (1,932,133 | ) | ||||
|
Deferred tax liability
|
$ | (12,131,251 | ) | $ | (11,567,706 | ) | ||
-64-
Tax Rate
The reconciliation between the U.S. federal statutory rate and our effective rate is summarized as follows:
|
Year ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Statutory federal income tax rate
|
34.0 | % | 34.0 | % | 34.0 | % | ||||||
|
State tax expense, net of federal tax benefit
|
7.8 | % | 6.2 | % | 4.6 | % | ||||||
|
State deferred tax expense, net of federal benefit,
|
||||||||||||
|
due to rate change
|
0.5 | % | (0.8 | )% | 2.6 | % | ||||||
|
Permanent items, including nondeductible expenses
|
(1.8 | )% | 8.8 | % | 0.6 | % | ||||||
|
State investment tax credit
|
(0.8 | )% | (5.6 | )% | (11.1 | )% | ||||||
|
Federal and state research and development credits
|
(2.2 | )% | (8.4 | )% | (5.8 | )% | ||||||
|
Foreign rate differential
|
3.1 | % | 0.0 | % | 0.0 | % | ||||||
|
Other
|
0.6 | % | (1.1 | )% | (1.7 | )% | ||||||
|
Tax expense
|
41.2 | % | 33.1 | % | 23.2 | % | ||||||
As of December 31, 2010, the Company had net operating losses (“NOL”) for federal income tax purposes in Italy of $7,148,138. For Massachusetts state income tax purposes the Company also had R&D tax credit carryforwards of $158,153 and investment tax credit carryforwards of $1,314,571.
In connection with the preparation of the financial statements, the Company performed an analysis to ascertain if it was more likely than not that it would be able to utilize, in future periods, the net deferred tax assets associated with its NOL carryforward, R&D tax credit carryforward, and its investment tax credit carryforward. We have concluded that the positive evidence outweighs the negative evidence and, thus, that those deferred tax assets not otherwise subject to a valuation allowance are realizable on a “more likely than not” basis. As such, we have not recorded a valuation allowance at December 31, 2010, and 2009, respectively.
-65-
Accounting for Uncertainty in Income Taxes
A reconciliation of the beginning and ending amount of our unrecognized tax benefits is summarized as follows:
|
Year ended December 31,
|
||||||||||||
|
2010
|
2009
|
2008
|
||||||||||
|
Unrecognized tax benefit, beginning of year
|
$ | 40,900 | $ | 40,900 | $ | 203,954 | ||||||
|
Tax positions related to current year
|
- | - | 6,249 | |||||||||
|
Tax positions related to prior years
|
37,427 | - | 8,443 | |||||||||
|
Settlements
|
(3,089 | ) | - | (68,221 | ) | |||||||
|
Statute expirations
|
(37,810 | ) | - | (109,525 | ) | |||||||
|
Unrecognized tax benefit, end of year
|
$ | 37,428 | $ | 40,900 | $ | 40,900 | ||||||
In the normal course of business, Anika and its subsidiaries may be periodically examined by various taxing authorities. We file income tax returns in the U.S. federal jurisdiction, in certain U.S. states, and in Italy. The associated tax filings remain subject to examination by applicable tax authorities for a certain length of time following the tax year to which those filings relate. The 2007 through 2009 tax years remain subject to examination by the IRS for U.S. federal and for state tax purposes. The 2006 through 2009 tax years remain subject to examination by the appropriate governmental authorities for Italy.
We do not anticipate experiencing any significant increases or decreases in our unrecognized tax benefits within the twelve months following December 31, 2010.
We incurred expenses related to stock-based compensation in 2010, 2009 and 2008 of $1,102,617, $958,025, and $1,391,704, respectively. Accounting for the tax effects of stock-based awards requires that we establish a deferred tax asset as the compensation is recognized for financial reporting prior to recognizing the tax deductions. The tax benefit recognized in the consolidated statement of operations related to stock-based compensation totaled $244,746, $230,812, and $463,212 in 2010, 2009 and 2008, respectively.
Upon the settlement of the stock-based awards (i.e., exercise, vesting, forfeiture or cancellation), the actual tax deduction is compared with the cumulative financial reporting compensation cost and any excess tax deduction is considered a windfall tax benefit, and is tracked in a "windfall tax benefit pool" to offset any future tax deduction shortfalls and will be recorded as increases to additional paid-in capital in the period when the tax deduction reduces income taxes payable. We follow the with-and-without approach for the direct effects of windfall/shortfall items and to determine the timing of the recognition any related benefits. We recorded net shortfalls of approximately $21,000 and $83,000 in 2010 and 2009, respectively.
15. Long-term Debt
On January 31, 2008, the Company entered into an unsecured Credit Agreement (the “Agreement”) with Bank of America, under which the Company was provided with a revolving credit line through December 31, 2008 of up to a maximum principal amount outstanding of $16,000,000. The Company borrowed the maximum amount of $16,000,000 in 2008 to finance its new facility construction and capital project validation. On December 31, 2008, the outstanding revolving credit loans were converted into a term loan with quarterly principal payments of $400,000 and a final installment of $5,200,000 due on the maturity date of December 31, 2015. Interest on the term loan was originally payable at a rate based upon, at the Company’s election, either Bank of America’s prime rate or LIBOR plus 75 basis points. The Company recorded approximately $171,000 as deferred issuance costs which continue to be amortized over the life of the debt facility.
In connection with the acquisition of FAB, the Company entered into a Consent and First Amendment to the original loan facility with Bank of America. As part of this amendment, the interest rate for Eurodollar based loans was increased and is payable at a rate based upon, at the Company’s election, either Bank of America’s prime rate or LIBOR plus 125 basis points. In addition, the Company pledged to the lender sixty-five percent (65%) of the stock of Anika Therapeutics S.r.l. We also incurred $74,000 of fees charged by Bank of America which were capitalized in accordance with ASC Subtopic 470-50, Debt – Modifications and Extinguishments, as the Consent and Amendment represents a debt modification. The fees are being amortized over the remaining life of the debt facility.
-66-
The Agreement contains customary representations and warranties of the Company, affirmative and negative covenants regarding the Company’s operations, financial covenants regarding maintenance by the Company of a specified quick ratio and consolidated fixed charge coverage ratio, and events of default. We are in compliance with all covenants specified in the debt agreement.
As of December 31, 2010 and 2009, the Company had a total outstanding debt balance of $12,800,000 and $14.400,000, respectively, of which $1,600,000 was recorded as current at each date.
For the year ended December 31, 2009, the Company capitalized interest expense of $98,183 as part of construction in progress related to the Company’s new facility build-out. Interest capitalization was recorded in accordance with ASC Subtopic 835-20, Capitalization of Interest Costs. The Company began expensing all interest costs incurred beginning after July 1, 2009.
Long-term debt principal payments are $1,600,000 for each of the next four years with the remaining principal of $6,400,000 due in the fifth and final year. The estimated fair value of our debt instrument approximated book value at December 31, 2010.
16. Acquisition
On December 30, 2009, we completed our acquisition of Fidia Advanced Biopolymers S.r.l., a privately held Italian corporation (“FAB”) for a purchase price consisting of $17.0 million in cash and 1,981,192 shares of the Company’s common stock (the “Acquisition”). FAB’s operating results and cash flow changes were immaterial for the one day of post-acquisition activity. FAB has over 20 products currently on the market, all manufactured from hyaluronic acid, the same basic material used by Anika. The acquisition complements our portfolio of products, broadens its pipeline of potential new products, and advances our vision to offer orthopedic doctors protective and regenerative products.
The 1,981,192 shares of the Company’s stock issued includes 800,000 shares to be held in escrow for a period of up to two years in order to satisfy any future indemnification claims under the Purchase Agreement. The issued shares are also subject to a one year holding period. The Purchase Agreement was based on an estimated closing balance sheet with a minimum working capital amount to be delivered.
Effective January 1, 2009, the Company implemented the newly-issued accounting standard for business combinations. The transaction was accounted for under the acquisition method of accounting in accordance with ASC 805, Business Combinations. Under ASC 805, all of the assets acquired and liabilities assumed in the transaction are recognized at their acquisition-date fair values, while transaction costs and restructuring costs associated with the transaction are expensed as incurred.
FAB’s results of operations have been included in our consolidated financial statements beginning January 1, 2010. Our results of operations prior to this acquisition, presented on a pro forma basis, are found further below.
Purchase Price
The $33.9 million purchase price for FAB is based on the acquisition-date fair value of the consideration transferred, which included cash and the issuance of shares of Anika stock, which was calculated based on the closing price of the Company's common stock of $8.49 per share on December 30, 2009.
The acquisition-date fair value of the consideration consisted of the following:
|
Fair Value of
Consideration
|
||||
|
Cash
|
$ | 17,055,000 | ||
|
Common stock
|
16,820,320 | |||
| $ | 33,875,320 | |||
During the second quarter of 2010 Anika and Fidia Farmaceutici S.p.A. (“Fidia”) agreed to a final working capital settlement whereby Fidia paid us $105,300. This settlement is not reflected in the above table.
-67-
Allocations of Assets and Liabilities
The Company allocated the purchase price for FAB, based on the acquired fair value of the net tangible assets and intangible assets, goodwill and a deferred tax liability. The difference between the aggregate purchase price and the fair value of assets acquired and liabilities assumed, after consideration of deferred taxes, was allocated to goodwill.
During the fourth quarter of 2010 we completed the purchase price allocation for the acquisition of FAB. During this process, which occurred within the measurement period, new information about facts which existed at the date of acquisition came to our attention. As a result, some of the amounts previously estimated have changed. The changes in estimates resulting from the finalization of this process resulted in an increase of inventory of approximately $106,000, an increase in other net assets of approximately $18,000, a decrease in IPR&D of approximately $3,602,000, and a decrease in deferred tax liabilities of approximately $1,171,000. As a result of finalization of the valuation there was a net increase to goodwill of approximately $2,307,000.
The measurement period adjustments represent updates made to the preliminary purchase price allocation based upon the finalization of the valuation estimates occurring during the measurement period subsequent to the acquisition and preliminary accounting date. These measurement period adjustments have been retrospectively applied to the December 31, 2009 balance sheet in accordance with ASC 805, Business Combinations. There is no significant impact to the Company’s Consolidated Statement of Operations for any periods prior to the year ended December 31, 2010.
The following table summarizes the fair values of the assets acquired and liabilities assumed at the acquisition date based upon the completed valuation and resulting measurement period adjustments:
|
Inventory
|
$ | 1,506,260 | ||
|
Other assets and liabilities, net
|
(244,346 | ) | ||
|
Property and equipment
|
1,691,000 | |||
|
Acquired intangible assets
|
29,098,000 | |||
|
Goodwill
|
9,959,008 | |||
|
Deferred tax liability
|
(8,134,603 | ) | ||
|
Total purchase price
|
$ | 33,875,320 |
Changes to the carrying amount of goodwill for the fiscal year ended 2010 were as follows:
|
Balance at December 31, 2009
|
$ | 9,959,008 | ||
|
Reduction in purchase price of subsidiary
|
(105,300 | ) | ||
|
Effect of foreign currency adjustments
|
(761,751 | ) | ||
|
Balance at December 31, 2010
|
$ | 9,091,957 |
The intangible assets identified in the purchase price allocation represent primarily developed technology, acquired in-process research and development, patents, and distributor relationship assets. Under the acquisition method of ASC 805, $21.4 million of these assets are recorded at their fair value and amortized over their estimated lives. The remaining amount represents IPR&D, which is accounted for as an indefinite-lived intangible asset. The goodwill recognized is largely attributable to establishing a deferred tax liability for the acquired intangible assets, which are not deductible for income tax purposes.
All intangible assets are tested for impairment on an annual basis, or earlier if impairment indicators are present. See Note 2 for additional disclosure regarding our accounting policies relative to this and other subjects.
IPR&D primarily revolves around obtaining U.S. approval for several of FAB’s orthopedic products to gain access to this important market. Costs to complete the projects are estimated at $7 million to $13 million spread over the next six years, and involve primarily clinical studies and regulatory costs, which are deemed to be of moderate difficulty. IPR&D value was estimated using a multi-period excess earnings approach. The primary risks associated with the projects include generating sufficient data to support efficacy, and thereby gaining regulatory approval. There can be no assurance that the Company will be successful in completing development or obtaining regulatory approval; and if successful, that meaningful sales will occur.
-68-
Acquisition-related Expenses
In connection with the acquisition of FAB, the Company incurred approximately $2.2 million in expenses, which are reflected as acquisition-related expenses on the consolidated statements of operations in 2009. These costs include costs to investigate, document, close, and complete regulatory compliance requirements.
The FAB balance sheet amounts that have been included in the 2009 consolidated balance sheet consist of the following:
|
December 31,
2009
|
||||
|
ASSETS
|
||||
|
Cash and cash equivalents
|
$ | 799,363 | ||
|
Accounts receivable
|
4,662,649 | |||
|
Inventories
|
1,506,260 | |||
|
Prepaid expenses and other
|
1,553,461 | |||
|
Property and equipment
|
1,691,000 | |||
|
Intangible assets
|
29,098,000 | |||
|
Goodwill
|
9,959,008 | |||
|
Total Assets
|
$ | 49,269,741 | ||
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
||||
|
Accounts Payable
|
$ | 4,080,413 | ||
|
Accrued expenses
|
2,448,225 | |||
|
Other long-term liabilities
|
775,644 | |||
|
Deferred tax liability
|
8,090,139 | |||
|
Stockholders’ equity
|
33,875,320 | |||
|
Total Liabilities and Stockholders’ Equity
|
$ | 49,269,741 | ||
The unaudited financial information in the table below summarizes the combined results of operations of Anika and FAB, on a pro forma basis, as though the companies had been combined as of the beginning of each period presented. The pro forma financial information is presented for comparative purposes only and is not necessarily indicative of the results of operations that actually would have been achieved if the acquisition had taken place at the beginning of the respective periods. The pro forma financial information is based on Anika's results of operations for each period presented, combined with FAB’s results of operations for 2009 and 2008, respectively.
The pro forma financial information includes the amortization charges from acquired intangible assets, acquisition-related expenses, and the related tax effects.
The Pro Forma (unaudited) combined Statement of Operations for the years ended December 31, 2009 and 2008, had FAB been included are as follows:
|
Year ended December 31,
|
||||||||
|
2009
|
2008
|
|||||||
|
(unaudited)
|
(unaudited)
|
|||||||
|
Total revenue
|
$ | 52,570,705 | $ | 50,196,000 | ||||
|
Net income
|
$ | (1,350,081 | ) | $ | (317,000 | ) | ||
|
Diluted net income per share:
|
||||||||
|
Net income
|
(0.10 | ) | (0.02 | ) | ||||
|
Diluted weighted average common shares outstanding
|
13,532,640 | 13,442,000 | ||||||
17. Related Party
In connection with the acquisition of FAB by Anika on December 30, 2009, Fidia Farmaceutici S.p.A ("Fidia") acquired ownership of 1,981,192 shares of the Company's common stock, or approximately 14.7% of the outstanding shares of the Company as of December 31, 2010, thus becoming a "related party" under the Securities and Exchange Commission regulations. See Note 16 to the consolidated financial statements for further description of the acquisition.
-69-
As part of the acquisition, the Company, primarily through FAB, entered into a series of operating agreements with Fidia as follows:
| Agreement Type | Description | Term in Years | ||
| Lease | Rent of space in Abano Terme, Italy | Six | ||
| Finished goods supply | Manufacture and supply of goods | Three | ||
| Raw material supply | Hyaluronic acid power | Five | ||
| Accounts receivable | Collection of trade receivables outstanding as of | Two | ||
| management | December 30, 2009. | |||
| Marketing and Promotion | Promote FAB products in Italy through | Three | ||
| Fidia sales force |
Historically FAB has relied on Fidia, its former parent company, for several functional activities. In connection with the purchase of FAB, the Company has negotiated a lease for approximately 26,000 square feet of office, laboratory and warehouse space in Abano Terme, Italy, and a finished goods supply agreement. In addition, accounting and purchasing was performed by Fidia on behalf of FAB during 2010 under a services agreement. Finally, Fidia has agreed to promote FAB's products in Italy through its existing 140 person sales force. At December 31, 2010, FAB had a net payable to Fidia for past products and services of approximately $6.4 million.
18. Subsequent Events
Anika manufactures the AMVISC product line for Bausch & Lomb (“B&L”) under the terms of a supply agreement that expired on December 31, 2010 (the “2004 B&L Agreement”) for viscoelastic products used in ophthalmic surgery. Effective January 1, 2011, the parties entered into a non-exclusive, two year contract intended to transition the manufacture of AMVISC and AMVISC Plus to an alternative, recently affiliated low-cost supplier to B&L. Under the 2004 B&L Agreement, the Company was restricted in its ability to commercialize viscoelastic products to only existing customers (STAAR Surgical Company and Hoya Surgical Optics, Inc.). That restriction has now expired and the Company is free to market its own viscoelastic product - AnikaVisc.
B&L accounted for 21% of product revenue for the year ended 2010, but is expected to be significantly lower in 2011 under the new transition contract. Margins under the 2004 B&L Agreement were very low, and the Company expects to see margin improvement through commercialization of its new AnikaVisc product. There can be no assurance that AnikaVisc will be successfully sold or that it will generate any profit for the Company.
In February 2011 we extended the lease on our Woburn, Massachusetts facility which currently houses our manufacturing and warehousing operations for several major products. The lease is for $176,902 per month and expires on June 30, 2011.
-70-
19. Quarterly Financial Data (Unaudited)
|
Quarter ended
|
Quarter ended
|
Quarter ended
|
Quarter ended
|
|||||||||||||
|
Year 2010
|
December 31,
|
September 30,
|
June 30,
|
March 31,
|
||||||||||||
|
Product revenue
|
$ | 14,193,352 | $ | 13,179,399 | $ | 13,720,929 | $ | 11,642,050 | ||||||||
|
Total revenue
|
14,721,493 | 13,869,214 | 14,499,800 | 12,466,087 | ||||||||||||
|
Cost of product revenue
|
6,702,674 | 6,108,502 | 5,891,752 | 5,123,675 | ||||||||||||
|
Gross profit on product revenue
|
7,490,678 | 7,070,897 | 7,829,177 | 6,518,375 | ||||||||||||
|
Net income
|
$ | 1,350,701 | $ | 1,184,265 | $ | 1,066,752 | $ | 714,280 | ||||||||
|
Per common share information:
|
||||||||||||||||
|
Basic net income per share
|
$ | 0.11 | $ | 0.09 | $ | 0.08 | $ | 0.06 | ||||||||
|
Basic common shares outstanding
|
12,641,394 | 12,633,405 | 12,645,889 | 12,614,808 | ||||||||||||
|
Diluted net income per share
|
$ | 0.10 | $ | 0.09 | $ | 0.08 | $ | 0.05 | ||||||||
|
Diluted common shares outstanding
|
13,672,245 | 13,622,603 | 13,642,322 | 13,628,376 | ||||||||||||
|
Quarter ended
|
Quarter ended
|
Quarter ended
|
Quarter ended
|
|||||||||||||
|
Year 2009
|
December 31,
|
September 30,
|
June 30,
|
March 31,
|
||||||||||||
|
Product revenue
|
$ | 9,943,940 | $ | 10,087,130 | $ | 8,770,763 | $ | 8,519,073 | ||||||||
|
Total revenue
|
10,618,940 | 10,792,764 | 9,523,676 | 9,200,324 | ||||||||||||
|
Cost of product revenue
|
3,613,028 | 3,551,374 | 3,294,160 | 3,211,666 | ||||||||||||
|
Gross profit on product revenue
|
6,330,912 | 6,535,756 | 5,476,603 | 5,307,407 | ||||||||||||
|
Net income
|
$ | 697,148 | $ | 1,511,925 | $ | 955,774 | $ | 522,720 | ||||||||
|
Per common share information:
|
||||||||||||||||
|
Basic net income per share
|
$ | 0.06 | $ | 0.13 | $ | 0.08 | $ | 0.05 | ||||||||
|
Basic common shares outstanding
|
11,408,790 | 11,385,679 | 11,384,949 | 11,366,545 | ||||||||||||
|
Diluted net income per share
|
$ | 0.06 | $ | 0.13 | $ | 0.08 | $ | 0.05 | ||||||||
|
Diluted common shares outstanding
|
11,653,048 | 11,575,907 | 11,548,079 | 11,496,518 | ||||||||||||
|
Quarter ended
|
Quarter ended
|
Quarter ended
|
Quarter ended
|
|||||||||||||
|
Year 2008
|
December 31,
|
September 30,
|
June 30,
|
March 31,
|
||||||||||||
|
Product revenue
|
$ | 8,284,557 | $ | 8,523,765 | $ | 8,378,936 | $ | 7,867,529 | ||||||||
|
Total revenue
|
8,965,804 | 9,205,015 | 9,060,189 | 8,548,779 | ||||||||||||
|
Cost of product revenue
|
2,822,930 | 3,504,986 | 3,644,530 | 3,216,070 | ||||||||||||
|
Gross profit on product revenue
|
5,461,627 | 5,018,779 | 4,734,406 | 4,651,459 | ||||||||||||
|
Net income
|
$ | 1,094,505 | $ | 1,104,203 | $ | 812,929 | $ | 617,558 | ||||||||
|
Per common share information:
|
||||||||||||||||
|
Basic net income per share
|
$ | 0.10 | $ | 0.10 | $ | 0.07 | $ | 0.06 | ||||||||
|
Basic common shares outstanding
|
11,352,383 | 11,329,422 | 11,327,457 | 11,525,282 | ||||||||||||
|
Diluted net income per share
|
$ | 0.10 | $ | 0.10 | $ | 0.07 | $ | 0.05 | ||||||||
|
Diluted common shares outstanding
|
11,456,691 | 11,485,989 | 11,516,177 | 11,612,720 | ||||||||||||
-71-
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
|
(a)
|
Evaluation of disclosure controls and procedures.
|
As required by Rule 13a-15 under the Securities Exchange Act of 1934 (“Exchange Act”), we carried out an evaluation under the supervision and with the participation of our management, including our chief executive officer and chief financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the period covered by this report. Based upon that evaluation, the chief executive officer and chief financial officer have concluded that our disclosure controls and procedures are effective to ensure that information required to be disclosed by us in reports we file or submit under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in Securities and Exchange Commission rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by the Company in the reports it files or submits under the Exchange Act is accumulated and communicated to the Company’s management, including our chief executive officer and chief financial officer, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure. On an on-going basis, we review and document our disclosure controls and procedures, and our internal control over financial reporting, and may from time to time make changes aimed at enhancing their effectiveness and to ensure that our systems evolve with our business.
|
(b)
|
Changes in internal controls over financial reporting.
|
There were no changes in our internal control over financial reporting during the fourth quarter of fiscal year 2010 that have materially affected, or that are reasonably likely to materially affect, our internal controls over financial reporting.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Because of its inherent limitations, internal control over financial reporting can provide only reasonable assurance and may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2010. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) in Internal Control—Integrated Framework.
Based on our assessment and those criteria, our management believes that the Company maintained effective internal control over financial reporting as of December 31, 2010.
The effectiveness of our internal control over financial reporting as of December 31, 2010 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm which has audited the consolidated financial statements contained in this Form 10-K, as stated in their report which is included herein.
ITEM 9B. OTHER INFORMATION
None.
-72-
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required under this item is incorporated herein by reference to the Company’s definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the Securities and Exchange Commission not later than 120 days after the close of the Company’s fiscal year ended December 31, 2010.
ITEM 11. EXECUTIVE COMPENSATION
The information required under this item is incorporated herein by reference to the Company’s definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the Securities and Exchange Commission not later than 120 days after the close of the Company’s fiscal year ended December 31, 2010.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required under this item and Item 5 of this Annual Report on Form 10-K under the heading “Equity Compensation Plan Information” is incorporated herein by reference to the Company’s definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the Securities and Exchange Commission not later than 120 days after the close of the Company’s fiscal year ended December 31, 2010.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required under this item is incorporated herein by reference to the Company’s definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the Securities and Exchange Commission not later than 120 days after the close of the Company’s fiscal year ended December 31, 2010.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required under this item is incorporated herein by reference to the Company’s definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the Securities and Exchange Commission not later than 120 days after the close of the Company’s fiscal year ended December 31, 2010.
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
|
(a)
|
Documents filed as part of Form 10-K.
|
|
(1)
|
Financial Statements
|
|
Report of Independent Registered Public Accounting Firm
|
[46]
|
|
|
Consolidated Balance Sheets
|
[47]
|
|
|
Consolidated Statements of Operations
|
[48]
|
|
|
Consolidated Statements of Stockholder’s Equity
|
[49]
|
|
|
Consolidated Statements of Cash Flows
|
[50]
|
|
|
Notes to Consolidated Financial Statements
|
[51-65]
|
|
(2)
|
Schedules
|
Schedules have been omitted as all required information has been disclosed in the financial statements and related footnotes.
|
(3)
|
Exhibits
|
-73-
The list of Exhibits filed as a part of this Annual Report on Form 10-K are set forth in the Exhibit Index (b) below.
|
(b) Exhibit No.
|
Description
|
|
|
(2) Plan of Acquisition, Reorganization, Arrangement, Liquidation or Succession:
|
||
|
2.1
|
Sale and Purchase Agreement, dated December 30, 2009, by and between Fidia Farmaceutici S.p.A., as Seller, and the Company, as Buyer, incorporated herein by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on January 6, 2010.
|
|
|
(3) Articles of Incorporation and Bylaws:
|
||
|
3.1
|
Restated Articles of Organization of the Company, incorporated herein by reference to Exhibit 3.1 to the Company’s Registration Statement on Form 10 (File no. 000-21326), filed with the Securities and Exchange Commission on March 5, 1993.
|
|
|
3.2
|
Certificate of Vote of Directors Establishing a Series of Convertible Preferred Stock, incorporated herein by reference to the Exhibits to the Company’s Registration Statement on Form 10 (File no. 000-21326), filed with the Securities and Exchange Commission on March 5, 1993.
|
|
|
3.3
|
Amendment to the Restated Articles of Organization of the Company, incorporated herein by reference to Exhibit 3.1 to the Company’s Quarterly Report on Form 10-QSB for the quarterly period ended November 30, 1996 (File no. 000-21326), filed with the Securities and Exchange Commission on January 14, 1997.
|
|
|
3.4
|
Amendment to the Restated Articles of Organization of the Company, incorporated herein by reference to Exhibit 3.1 to the Company’s Quarterly Report on Form 10-QSB for the quarterly period ended June 30, 1998 (File no. 001-14027), filed with the Securities and Exchange Commission on August 14, 1998.
|
|
|
3.5
|
Amendment to the Restated Articles of Organization of the Company, incorporated herein by reference to Exhibit 3.3 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2002 (File no. 001-14027), filed with the Securities and Exchange Commission on August 14, 2002.
|
|
|
3.6
|
Amended and Restated Certificate of Vote of Directors Establishing a Series of Preferred Stock of the Company classifying and designating the Series B Junior Participating Cumulative Preferred Stock, incorporated herein by reference to Exhibit 3.1 to the Company’s Registration Statement on Form 8-A12B (File no. 001-14027), filed with the Securities and Exchange Commission on April 7, 2008.
|
|
|
3.7
|
Amendment to the Restated Articles of Organization of the Company, incorporated herein by reference to Exhibit 3.7 to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2008 (File no. 001-14027), filed with the Securities and Exchange Commission on March 9, 2009.
|
|
|
3.8
|
Amended and Restated Bylaws of the Company, incorporated herein by reference to Exhibit 3.6 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2002 (File no. 001-14027), filed with the Securities and Exchange Commission on August 14, 2002.
|
|
|
(4) Instruments Defining the Rights of Security Holders
|
||
|
4.1
|
Shareholder Rights Agreement, dated as of April 7, 2008, between the Company and American Stock Transfer & Trust Company, incorporated herein by reference to Exhibit 4.1 to the Company’s Registration Statement on Form 8-A12B (File no. 001-14027), filed with the Securities and Exchange Commission on April 7, 2008.
|
|
|
(10) Material Contracts
|
||
|
10.1
|
Commercial Lease, dated March 10, 1995, between the Company and Cummings Properties Management, Inc., incorporated herein by reference to Exhibit 10.8 to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2000 (File no. 001-14027), filed with the Securities and Exchange Commission on April 2, 2001.
|
|
|
10.2
|
Amendment to Lease #1, dated December 11, 1997, between the Company and Cummings Properties Management, Inc., incorporated herein by reference to Exhibit 10.9 to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2000 (File no. 001-14027), filed with the Securities and Exchange Commission on April 2, 2001.
|
|
-74-
|
10.3
|
Lease Extension, dated March 23, 1998, between the Company and Cummings Properties Management, Inc., incorporated herein by reference to Exhibit 10.10 to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2000 (File no. 001-14027), filed with the Securities and Exchange Commission on April 2, 2001.
|
|
|
10.4
|
Amendment to Lease #2, dated September 27, 1999, between the Company and Cummings Properties LLC, incorporated herein by reference to Exhibit 10.11 to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2000 (File no. 001-14027), filed with the Securities and Exchange Commission on April 2, 2001.
|
|
|
10.5
|
Commercial Lease, dated July 9, 1999, between the Company and Cummings Properties LLC, incorporated herein by reference to Exhibit 10.12 to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2000 (File no. 001-14027), filed with the Securities and Exchange Commission on April 2, 2001.
|
|
|
10.6
|
Stipulation and Agreement of Compromise, Settlement and Release, dated May 25, 2001, in connection with In Re Anika Therapeutics, Inc. Securities Litigation, incorporated herein by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2001 (File no. 001-14027), filed with the Securities and Exchange Commission on August 14, 2001.
|
|
|
10.7
|
Amendment to Lease #3, dated November 1, 2001, between the Company and Cummings Properties, LLC, incorporated herein by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2001 (File no. 001-14027), filed with the Securities and Exchange Commission on November 14, 2001.
|
|
|
10.8
|
Lease Extension, dated October 8, 2003, between the Company and Cummings Properties, LLC, incorporated herein by reference to Exhibit 10.36 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2003 (File no. 001-14027), filed with the Securities and Exchange Commission on November 14, 2003.
|
|
|
**10.9
|
License Agreement, dated as of December 20, 2003, by and between the Company and Ortho Biotech Products, L.P., incorporated herein by reference to Exhibit 10.38 to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2003 (File no. 001-14027), filed with the Securities and Exchange Commission on March 30, 2004.
|
|
|
**10.10
|
Supply Agreement, dated as of December 15, 2004, by and between the Company and Bausch & Lomb Incorporated, incorporated herein by reference to Exhibit 10.43 to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2004 (File no. 001-14027), filed with the Securities and Exchange Commission on March 16, 2005.
|
|
|
†10.11
|
Form of Incentive Stock Option Agreement under the Company’s Amended and Restated 2003 Stock Option and Incentive Plan, incorporated herein by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on October 5, 2004.
|
|
|
†10.12
|
Form of Non-Qualified Stock Option Agreement for Non-Employee Directors under the Company’s Amended and Restated 2003 Stock Option and Incentive Plan, incorporated herein by reference to Exhibit 10.4 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on October 5, 2004.
|
|
|
†10.13
|
Form of Stock Appreciation Right Agreement for Employees under the Company’s Amended and Restated 2003 Stock Option and Incentive Plan, incorporated herein by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended March 31, 2006 (File no. 001-14027), filed with the Securities and Exchange Commission on May 9, 2006.
|
|
|
†10.14
|
Form of Stock Appreciation Right Agreement for Non-Employee Directors under the Company’s Amended and Restated 2003 Stock Option and Incentive Plan, incorporated herein by reference to Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended March 31, 2006 (File no. 001-14027), filed with the Securities and Exchange Commission on May 9, 2006.
|
-75-
|
10.15
|
Lease, dated January 3, 2007, between the Company and Farley White Wiggins, LLC, incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on January 10, 2007.
|
|
|
10.16
|
Credit Agreement, dated as of January 31, 2008, among the Company, Anika Securities, Inc., Bank of America, N.A., and the other lenders party thereto (the “Credit Agreement”), incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on February 6, 2008.
|
|
|
†10.17
|
Anika Therapeutics, Inc. Senior Executive Incentive Compensation Plan, incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on February 6, 2008.
|
|
|
†10.18
|
Form of Performance Share Award Agreement under the Company’s Amended and Restated 2003 Stock Option and Incentive Plan, incorporated herein by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on February 6, 2008.
|
|
|
†10.19
|
Employment Agreement, dated October 17, 2008, between the Company and Charles H. Sherwood, Ph.D., incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on October 22, 2008.
|
|
|
†10.20
|
Employment Agreement, dated October 17, 2008, between the Company and Kevin Quinlan, incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on October 22, 2008.
|
|
|
†10.21
|
Form of Restricted Stock Award Agreement for Employees under the Company’s Amended and Restated 2003 Stock Option and Incentive Plan, incorporated herein by reference to Exhibit 10.27 to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2007 (File no. 001-14027), filed with the Securities and Exchange Commission on March 12, 2008.
|
|
|
†10.22
|
Anika Therapeutics, Inc. Non-Employee Director Compensation Policy, incorporated herein by reference to Exhibit 10.28 to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2007 (File no. 001-14027), filed with the Securities and Exchange Commission on March 12, 2008.
|
|
|
†10.23
|
Form of Restricted Deferred Stock Unit Award Agreement for Non-Employee Directors under the Company’s Amended and Restated 2003 Stock Option and Incentive Plan, incorporated herein by reference to Exhibit 10.25 to the Company’s Annual Report on Form 10-K for the fiscal year ended December 31, 2008 (File no. 001-14027), filed with the Securities and Exchange Commission on March 9, 2009.
|
|
|
†10.24
|
Letter Agreement, dated April 27, 2009, by and between the Company and Frank J. Luppino, incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on May 29, 2009.
|
|
|
†10.25
|
Amended and Restated 2003 Stock Option and Incentive Plan, incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on June 11, 2009.
|
|
|
†10.26
|
Employment Agreement, dated September 10, 2009, between the Company and Frank J. Luppino, incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on September 14, 2009.
|
|
|
†10.27
|
Employment Agreement, dated September 10, 2009, between the Company and William J. Mrachek, incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on September 14, 2009.
|
|
|
10.28
|
Registration Rights Agreement, dated December 30, 2009, between the Company and Fidia Farmaceutici S.p.A., incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on January 6, 2010.
|
-76-
|
10.29
|
Lease Agreement, dated December 30, 2009, between Fidia Farmaceutici S.p.A. and Fidia Advanced Biopolymers S.r.l., incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on January 6, 2010.
|
|
|
10.30
|
Tolling Agreement, dated December 30, 2009, between Fidia Farmaceutici S.p.A. and Fidia Advanced Biopolymers S.r.l., incorporated herein by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on January 6, 2010.
|
|
|
10.31
|
Consent and First Amendment to the Credit Agreement, dated as of December 30, 2009, by and among the Company, Anika Securities, Inc., Bank of America, N.A. and each lender signatory thereto, incorporated herein by reference to Exhibit 10.4 to the Company’s Current Report on Form 8-K (File no. 001-14027), filed with the Securities and Exchange Commission on January 6, 2010.
|
|
|
10.32
|
Pledge Agreement on a Quota of Fidia Advanced Biopolymers S.r.l., dated March 12, 2010, dated March 12, 2010, by the Company in favor of Bank of America, N.A., incorporated herein by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q (File no. 001-14027), filed with the Securities and Exchange Commission on May 10, 2010.
|
|
|
*†10.33
|
Amendment No. 1 to Employment Agreement by and between the Company and Charles H. Sherwood, Ph.D, dated December 8, 2010.
|
|
|
*†10.34
|
Amendment No. 1 to Employment Agreement by and between the Company and Kevin W. Quinlan, dated December 8, 2010.
|
|
|
*†10.35
|
Amendment No. 1 to Employment Agreement by and between the Company and Frank J. Luppino, dated December 8, 2010.
|
|
|
*†10.36
|
Amendment No. 1 to Employment Agreement by and between the Company and William J. Mrachek, dated December 8, 2010.
|
|
| †10.37 | 1993 Stock Option Plan, as amended, incorporated herein by reference to the Company's Proxy Statement (File no. 001-14027), filed with the Securities and Exchange Commission on April 28, 2000. | |
|
(11) Statement Regarding the Computation of Per Share Earnings
|
||
|
11.1
|
See Note 3 to the Financial Statements included herewith.
|
|
|
(21) Subsidiaries of the Registrant
|
||
|
*21.1
|
List of Subsidiaries of the Registrant.
|
|
|
(23) Consent of Experts
|
||
|
*23.1
|
Consent of PricewaterhouseCoopers LLP, an independent registered public accounting firm.
|
|
|
(31) Rule 13a-14(a) / 15d-14(a) Certifications
|
||
|
*31.1
|
Certification of Charles H. Sherwood, Ph.D. pursuant to Rule 13a-14(a) or 15d-14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
*31.2
|
Certification of Kevin W. Quinlan pursuant to Rule 13a-14(a) or 15d-14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
(32) Section 1350 Certification
|
||
|
***32.1
|
Certification of Charles H. Sherwood, Ph.D. and Kevin W. Quinlan, pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002.
|
|
|
*
|
Filed herewith.
|
|
**
|
Certain portions of this document have been omitted pursuant to a confidential treatment request filed with the Commission. The omitted portions have been filed separately with the Commission.
|
|
***
|
Furnished herewith.
|
|
†
|
Denotes compensatory plan or arrangement.
|
-77-
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
ANIKA THERAPEUTICS, INC.
|
|||
|
Date: March 16, 2011
|
By:
|
/s/ CHARLES H. SHERWOOD, PH.D.
|
|
|
Charles H. Sherwood, Ph.D.
|
|||
| Chief Executive Officer | |||
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
||
|
/s/ CHARLES H. SHERWOOD, PH.D.
|
Chief Executive Officer and Director
|
March 16, 2011
|
||
|
Charles H. Sherwood, Ph.D.
|
(Principal Executive Officer)
|
|
||
|
/s/ KEVIN W. QUINLAN
|
Chief Financial Officer
|
March 16, 2011
|
||
|
Kevin W. Quinlan
|
(Principal Accounting Officer)
|
|
||
|
/s/ JOSEPH L. BOWER
|
Director
|
March 16, 2011
|
||
|
Joseph L. Bower
|
|
|||
|
/s/ EUGENE A. DAVIDSON, PH.D.
|
Director
|
March 16, 2011
|
||
|
Eugene A. Davidson, Ph.D.
|
|
|||
|
/s/ RAYMOND J. LAND
|
Director
|
March 16, 2011
|
||
|
Raymond J. Land
|
|
|||
|
/s/ JOHN C. MORAN
|
Director
|
March 16, 2011
|
||
|
John C. Moran
|
|
|||
|
/s/ JEFFERY S. THOMPSON
|
Director
|
March 16, 2011
|
||
|
Jeffery S. Thompson
|
|
|||
|
/s/ STEVEN E. WHEELER
|
Director
|
March 16, 2011
|
||
|
Steven E. Wheeler
|
-78-