Attached files
| file | filename |
|---|---|
| EX-3.8 - EX-3.8 - MABVAX THERAPEUTICS HOLDINGS, INC. | d846215dex38.htm |
| EX-4.12 - EX-4.12 - MABVAX THERAPEUTICS HOLDINGS, INC. | d846215dex412.htm |
| EX-23.1 - EX-23.1 - MABVAX THERAPEUTICS HOLDINGS, INC. | d846215dex231.htm |
| EX-4.11 - EX-4.11 - MABVAX THERAPEUTICS HOLDINGS, INC. | d846215dex411.htm |
| EX-4.13 - EX-4.13 - MABVAX THERAPEUTICS HOLDINGS, INC. | d846215dex413.htm |
| EX-10.15 - EX-10.15 - MABVAX THERAPEUTICS HOLDINGS, INC. | d846215dex1015.htm |
| EX-10.30 - EX-10.30 - MABVAX THERAPEUTICS HOLDINGS, INC. | d846215dex1030.htm |
| EX-10.29 - EX-10.29 - MABVAX THERAPEUTICS HOLDINGS, INC. | d846215dex1029.htm |
| EX-32.1 - EX-32.1 - MABVAX THERAPEUTICS HOLDINGS, INC. | d846215dex321.htm |
| EX-99.1 - EX-99.1 - MABVAX THERAPEUTICS HOLDINGS, INC. | d846215dex991.htm |
| EX-32.2 - EX-32.2 - MABVAX THERAPEUTICS HOLDINGS, INC. | d846215dex322.htm |
| EX-31.2 - EX-31.2 - MABVAX THERAPEUTICS HOLDINGS, INC. | d846215dex312.htm |
| EX-31.1 - EX-31.1 - MABVAX THERAPEUTICS HOLDINGS, INC. | d846215dex311.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended December 31, 2014
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Transition period from to .
Commission file number: 0-31265
MABVAX THERAPEUTICS HOLDINGS, INC.
(Exact name of Registrant as specified in its charter)
| Delaware | 93-0987903 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) | |
| 11588 Sorrento Valley Rd., Suite 20, San Diego, CA | 92121 | |
| (Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number, including area code: (858) 259-9405
Securities registered pursuant to Section 12(b) of the Act: None
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| None |
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, $0.01 par value per share
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES ¨ NO x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. YES ¨ NO x
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. YES x NO ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). YES x NO ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this Chapter) is not contained herein, and will not be contained to the best of Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | ¨ | Accelerated filer | ¨ | |||
| Non-accelerated filer | ¨ | Smaller reporting company | x | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act.). YES ¨ NO x
The aggregate market value of the voting common stock held by non-affiliates of the Registrant was approximately $6,627,000 as of June 30, 2014, based upon the closing sale price on the OTCQB Market of $11.60 per share reported on such date.
As of March 31, 2015, there were 9,615,035 shares of the registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Certain information required by Part III Of this Annual Report on Form 10-K is incorporated by reference from the registrant’s definitive proxy statement for the annual meeting of stockholders, which will be filed with the Securities and exchange Commission within 120 days after the close of the registrant’s fiscal year ended December 31, 2014.
Table of Contents
MABVAX THERAPEUTICS HOLDINGS, INC.
2014 ANNUAL REPORT ON FORM 10-K
TABLE OF CONTENTS
| Page | ||||||
| PART I | ||||||
| Item 1. |
4 | |||||
| Item 1A. |
15 | |||||
| Item 1B. |
26 | |||||
| Item 2. |
26 | |||||
| Item 3. |
26 | |||||
| Item 4. |
27 | |||||
| PART II | ||||||
| Item 5. |
27 | |||||
| Item 6. |
28 | |||||
| Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
28 | ||||
| Item 7A. |
36 | |||||
| Item 8. |
36 | |||||
| Item 9. |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
36 | ||||
| Item 9A. |
37 | |||||
| Item 9B. |
37 | |||||
| PART III | ||||||
| Item 10. |
37 | |||||
| Item 11. |
37 | |||||
| Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
37 | ||||
| Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
38 | ||||
| Item 14. |
38 | |||||
| PART IV | ||||||
| Item 15. |
38 | |||||
Table of Contents
Disclosure Regarding Forward-Looking Statements
This Annual Report on Form 10-K, including the documents that we incorporate by reference, contains statements indicating expectations about future performance and other forward-looking statements. Forward-looking statements relate to future events or our future financial performance. We generally identify forward-looking statements by terminology such as “may,” “will,” “should,” “expects,” “plans,” “anticipates,” “could,” “intends,” “target,” “projects,” “contemplates,” “believes,” “estimates,” “predicts,” “potential” or “continue” or the negative of these terms or other similar words, although not all forward-looking statements contain these words. Forward-looking statements include, but are not limited to, statements regarding our or our management’s expectations, hopes, beliefs, intentions or strategies regarding the future, such as our estimates regarding anticipated operating losses, future performance, future revenues and projected expenses; our liquidity and our expectations regarding our needs for and ability to raise additional capital; our ability to manage our expenses effectively and raise the funds needed to continue our business; our ability to retain the services of our current executive officers, directors and principal consultants; our ability to obtain and maintain regulatory approval of our existing products and any future products we may develop; the initiation, timing, progress and results of our preclinical and clinical trials, research and development programs; regulatory and legislative developments in the United States and foreign countries; the timing, costs and other limitations involved in obtaining regulatory approval for any product; the further preclinical or clinical development and commercialization of our product candidates; the potential benefits of our product candidates over other therapies; our ability to enter into any collaboration with respect to product candidates; the performance of our third-party manufacturers; our ability to obtain and maintain intellectual property protection for our products and operate our business without infringing upon the intellectual property rights of others; the successful development of our sales and marketing capabilities; the size and growth of the potential markets for our products and our ability to serve those markets; the rate and degree of market acceptance of any future products; our reliance on key scientific management or personnel; the payment and reimbursement methods used by private or governmental third-party payers; and other factors discussed elsewhere in this report or any document incorporated by reference herein or therein.
The words “believe,” “may,” “will,” “estimate,” “continue,” “anticipate,” “intend,” “expect,” “plan” and similar expressions may identify forward-looking statements, but the absence of these words does not mean that a statement is not forward-looking. The forward-looking statements contained in this report are based on our current expectations and beliefs concerning future developments and their potential effects on us. There can be no assurance that future developments affecting us will be those that we have anticipated. These forward-looking statements involve a number of risks, uncertainties (many of which are beyond our control) or other assumptions that may cause actual results or performance to be materially different from those expressed or implied by these forward-looking statements. These risks and uncertainties include, but are not limited to, those factors described in the section titled “Risk Factors.” Should one or more of these risks or uncertainties materialize, or should any of our assumptions prove incorrect, actual results may vary from those projected in these forward-looking statements. We undertake no obligation to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, except as may be required under applicable securities laws. The section entitled “Risk Factors,” as well as other sections in this report or incorporated by reference into this report, discuss some of the factors that could contribute to these differences.
The forward-looking statements made in this report relate only to events as of the date on which the statements are made. We undertake no obligation to update any forward-looking statement to reflect events or circumstances after the date on which the statement is made or to reflect the occurrence of unanticipated events.
This report also contains market data related to our business and industry. These market data include projections that are based on a number of assumptions. While we believe these assumptions to be reasonable and sound as of the date of this report, if these assumptions turn out to be incorrect, actual results may differ from the projections based on these assumptions. As a result, our markets may not grow at the rates projected by these data, or at all. The failure of these markets to grow at these projected rates may have a material adverse effect on our business, results of operations, financial condition and the market price of our common stock.
MabVax®, MabVax Therapeutics® and our corporate logo are trademarks or registered trademarks of MabVax Therapeutics Holdings, Inc. All other brand names or trademarks appearing in this Annual Report are the property of their respective holders.
3
Table of Contents
| Item 1. | Business. |
Company Background
We are a Delaware corporation, originally incorporated in 1988 under the name Terrapin Diagnostics, Inc. in the state of Delaware, and subsequently renamed “Telik, Inc.” in 1998, and MabVax Therapeutics Holdings, Inc. in September 2014. Our principal corporate office in the United States is at 11588 Sorrento Valley Road, Suite 20, San Diego, CA 92121 telephone: (858) 259-9405. Our internet address is www.mabvax.com. On July 8, 2014, we consummated a merger with MabVax Therapeutics, pursuant to which our subsidiary Tacoma Acquisition Corp. merged with and into MabVax Therapeutics, with MabVax Therapeutics surviving as our wholly owned subsidiary. This transaction is referred to as the Merger. Immediately following the Merger, the former holders of the issued and outstanding securities of MabVax Therapeutics held approximately 85% of our issued and outstanding securities.
On September 8, 2014, we filed an amended and restated certificate of incorporation to increase the authorized number of shares of our common stock to a new total of 150,000,000 shares, increase the number of shares of our preferred stock to a new total of 15,000,000 shares, and change our name from “Telik, Inc.” to “MabVax Therapeutics Holdings, Inc.”
Overview
We are a clinical-stage biopharmaceutical company focused on discovering and developing innovative vaccine and monoclonal (produced from a single DNA sequence encoded into multiple cells that all produce the same single antibody) antibody-based therapeutics for the diagnosis and treatment of cancer. We generate our pipeline of antibody-based product candidates from patients who have been vaccinated with propriety vaccines licensed from Memorial Sloan Kettering Cancer Center (“MSKCC”). Our approach of surveying the protective immune response from many patients to identify the ideal monoclonal antibody candidate against a specific target on the surface of a cancer cell is a novel next-generation human antibody technology platform. We believe our approach to antibody discovery identifies the antibody candidates with superior performance characteristics while minimizing many of the toxicity and off target binding drawbacks (phenomenon occurring when antibodies bind to non-cancer cells) of other discovery technologies. Our lead antibody candidates have been recovered from patients who had substantially better treatment outcomes than almost all other patients in clinical trials conducted by us and our partners.
Our therapeutic vaccines were developed at MSKCC and are exclusively licensed to us pursuant to agreements entered into in 2008. These vaccines are administered in the adjuvant (period following conventional treatment consisting of watchful waiting) setting and have shown in clinical studies to elicit a protective antibody response. The antibodies are intended to seek out circulating tumor cells and micrometastases (small clusters of cancer cells) to kill them before they can cause cancer recurrence. Our lead cancer vaccines targeting recurrent sarcoma (soft tissue cancer) and ovarian cancer are currently in proof of concept Phase II multi-center clinical trials. Both trials have received substantial federal grant monies to support their development. A vaccine to address the orphan disease neuroblastoma (cancer starting in the adrenal gland of children which may spread to nervous tissue and bone marrow) has completed an initial Phase I trial at MSKCC yielding encouraging results. This vaccine product candidate is expected be ready for a Phase II trial in 2015. We and MSKCC have completed additional Phase I vaccine clinical trials in melanoma, ovarian cancer, breast cancer, and small cell lung cancer over the last three years.
Market Opportunity and Competition
Antibodies as targeted therapeutic treatments for cancer have become a significant market accounting for more than $20 billion in revenue according to Global Data’s commercial database Pharma eTrack. Eight of the leading twenty therapeutic products for the treatment of cancer are antibodies according to Global Data’s commercial database Pharma eTrack. There are more than 150 monoclonal antibodies in development for cancer alone according to Global Data’s commercial database Pharma eTrack. The focus on the development of new monoclonal antibody based drugs is expected to continue for multiple reasons. Targeted therapies can attack cancer cells while minimizing damage to normal cells in the patient. Antibodies are complex molecules and are difficult and expensive to duplicate with biosimilars and therefore have a potentially longer commercial life. Currently monoclonal antibodies receive very favorable reimbursement levels from federal, state, and private insurance providers. This favorable treatment is expected to continue.
We have focused our initial antibody development efforts on matching the early antibody discoveries against diseases for which there remains a significant unmet medical need. Our lead antibody candidate targets an antigen over expressed on metastatic pancreatic, colon, breast, and small cell lung cancers. Patients who develop metastatic disease with these cancers have a significantly poorer prognosis. In the case of pancreatic and small cell lung cancer we believe that there are no treatments currently available that focus on metastatic disease. We are developing this lead antibody as a stand-alone therapeutic agent as well as
4
Table of Contents
combining it with a radiolabel for use as a novel PET imaging agent. According to the National Cancer Institute’s SEER database, there are approximately 100,000 patients annually who develop metastatic disease from these cancers who could potentially benefit from this antibody if it is successfully developed. This product has completed its preclinical development and GMP manufacturing has been initiated to produce Phase I clinical material by mid-2015. We expect to conduct a Phase I in the second half of 2015.
We have initiated development efforts on a second antibody that targets an antigen over expressed on the surface of sarcoma, melanoma, and neuroblastoma. The use of a targeted antibody therapy in neuroblastoma in a study sponsored by the National Cancer Institute and published in the New England Journal of Medicine has demonstrated that antibodies targeting this antigen can be effective. We are focused on developing an antibody-based drug for the treatment of sarcoma. While there are newer agents being developed to treat sarcoma, it is a difficult to treat cancer with unacceptably high recurrence rates. We believe that there are approximately 30,000 patients who could potentially benefit from this antibody if it is successfully developed. This product is in preclinical development and is expected to move to GMP manufacturing in late 2016.
Recent Developments
In the past year, full enrollment has been achieved in both the sarcoma and ovarian cancer vaccine Phase II clinical trials. We expect to analyze the endpoint of progression free survival, or PFS, and both clinical programs are following patients to the OS endpoint that is expected to be reached in 2016. Our discussions with the FDA at the time we developed the protocols for our vaccine trials indicated that the OS endpoint was the primary evaluation criteria for their evaluation of the efficacy and safety of the vaccines. The results of the Phase I trial with our neuroblastoma vaccine were published recently in Clinical Cancer Research. The study report describes an encouraging study result with a small cohort of difficult to treat patients who have repeatedly relapsed.
As noted in our Management’s Discussion and Analysis of Financial Results, our lead antibody candidate, 5B1, is being developed for the treatment of pancreatic and colon cancer.
Product Candidates
5B1 Antibody Program
Of the many tumor restricted monoclonal antibodies resulting from immunization of mice with human cancer cells, the majority have been directed against carbohydrate antigens (structures consistently expressed and are targets for therapeutic intervention) expressed at the cell surface. The carbohydrate antigen sialyl Lewisa (sLea) is the antigen recognized by monoclonal antibody CA19.9 and it is widely expressed on tumors of the gastrointestinal tract. These tumor types are generally classified as epithelial tumors (a broad classification of tumor types) and include pancreatic, colon, stomach, ovarian, breast, and small cell lung cancers. A CA19.9 serum test is the only FDA validated marker for pancreatic cancer and is a commercial test that is readily available and used frequently to aid in the diagnosis of pancreatic cancer. Circulating epithelial cancer cells over-express sLea (abbreviation for the antigen sialyl Lewis A) and as a ligand (binding partner) for E selectin (a structure in the inner lining of blood vessels) this antigen facilitates tumor—tissue interactions that are key events for tumor metastasis. Patient outcomes appear to be worse in patients with metastatic tumors expressing higher levels of sLea according to articles published by T. Ben-David and colleagues in Immunology Letters in 2008 and YI Kawamura in Cancer Research in 2005. Because these tumor types express very high numbers of the sLea antigen on their cell surface, it makes the antigen an attractive target for therapeutic intervention.
We have created a series of fully human monoclonal antibodies against sLea. One antibody in particular, 5B1, has demonstrated exceptionally high affinity and specificity for sLea, and has very good efficacy in multiple tumor xenograft models (human cancer cells engrafted into mice) in studies conducted by MSKCC. 5B1 is a fully human full-length monoclonal antibody we discovered by capturing a portion of the immune response from seven stage IV breast cancer patients who were being vaccinated in a Phase I trial in 2008 at MSKCC with one of our licensed vaccines. Six of the original seven patients who participated in the trial are alive today and the patient from whom we recovered the 5B1 antibody is disease free five years post vaccination.
We have conducted tissue microarray work (normal and cancer tissue samples placed on slides treated with the antibody to determine if an antibody binds to such tissue samples) with commercially available tissue samples of both normal and cancer tissues. The results of this work indicated that the 5B1 antibody bound to multiple types of epithelial tumors, including pancreatic, colon, bladder, ovarian, breast, and small cell lung cancer tissues. The antibody did not bind to normal tissues except for the exocrine cells at the ductal border of secretory cells (primarily cells in the gastrointestinal tract that face into the digestive system and not inward to the body) in epithelial tissues; those sites are less accessible to the immune system. Both characteristics combined with the significant cytotoxicity demonstrated in in vitro testing led us to move to xenographic animal model testing. 5B1 has demonstrated in our pre-clinical studies good anti-tumor activity in a variety of animal models with multiple tumor types. Specifically, we have obtained positive results from animal models examining human pancreatic, colon, and small cell lung cancers.
5
Table of Contents
We have entered into a manufacturing agreement with Patheon (f.k.a. Gallus BioPharmaceuticals) to manufacture clinical supplies of the antibody, and plan to enter a Phase I clinical trial in 2015 to determine the safety and pharmacokinetics (assessment of the distribution and metabolism of a drug) of the 5B1antibody in patients of different cancer types. We expect that the clinical trial will help us determine if the antibody demonstrates anti-tumor activity over a range of doses tested. We expect the preliminary results of our Phase I studies to be initially available in 2015 with full Phase I results in 2016.
5B1 Imaging Program
Circulating biomarkers (substances released by certain cells that can be measured to assess if a patient has or is likely to have a particular type of cancer) such as CA19.9 are important clinical tools for early detection and diagnosis as well as monitoring of therapeutic progress, and detection of tumor recurrence in oncology. However, false positive readings due to biomarker production from benign disorders in unrelated host tissues are a significant problem. We believe that probing the site(s) of biomarker secretion with an imaging tool could be a broadly useful strategy to enhance the fidelity of diagnosis and staging of cancers such as pancreatic ductal adenocarcinoma, or PDAC, a notoriously occult (difficult to diagnose and treat) cancer. Moreover, such a tool could guide patient stratification for directed therapeutic intervention, surgical planning and aide in the evaluation of tumor response to chemotherapy and radiation therapy. To address this opportunity clinically, together with Dr. Jason Lewis’ radiochemistry laboratory at MSKCC, we developed 89Zr-5B1, a fully human, antibody-based radiotracer (combined antibody and radiolabel) targeting tumor-associated CA19.9. In preclinical studies,89Zr-5B1 localized to tumors in multiple models representing diseases with both undetectable and clinical relevant circulating CA19.9 serum levels. Among these, 89Zr-5B1 detected tumor in an orthotopic model (xenograph model where human cancer cells are surgically implanted in the corresponding organ of the mouse) of PDAC, an elusive cancer for which the serum assay (actual test for a biomarker) is measured in a human, but with limited specificity in part because of the frequency of CA19.9 secretion from benign hepatic pathologies (certain non-cancer conditions such as inflammation causing the production of the target biomarkers resulting in a false reading). Of further note, in preliminary experiments 89Zr-5B1 showed better tumor specificity (targeting only a specific type of cancer cell) compared to the commonly used 2-deoxy-2-(18F)-fluoro-D-glucose, or FDG, imaging agent, which relies on increased tumor metabolism relative to nonmalignant cells, and is known to lack sensitivity and specificity in pancreas cancers and other slow growing cancers.
To facilitate the development of the 5B1based antibody conjugated to a radiolabel as a novel PET imaging agent for pancreatic cancer, we applied for and received a development contract from NIH pursuant to which NIH may provide up to $1.75 million in non-dilutive funding for this project. We will be reach the clinic in 2015 with this novel product. We expect the preliminary results of our Phase I study to be initially available in 2015 with full Phase I results in 2016
5B1 Antibody Drug Conjugate
We observed in our clinical studies that certain types of cancer cells internalized the 5B1 antibody. These were primarily pancreatic cancer tumor types. We believe that this characteristic of the 5B1 antibody could be highly useful in constructing an antibody-drug conjugate. The development of antibody-drug conjugates is an area of intense competitive development with few companies capable of producing viable linker and toxin technologies that can be coupled with an antibody which, in this case, serves as the targeting mechanism. According to an analysis conducted utilizing the online database Global Data, over the last few years more than 21 technology access licensing deals worth more than $6 billion have been completed by biopharmaceutical companies to gain access to these technologies. We were able to identify and form an early collaboration with Heidelberg Pharm who has developed its own proprietary linker and toxin technology. For the collaboration, we were able to supply the 5B1 antibody and Heidelberg has conducted both in vitro and in vivo experiments demonstrating the significant potential utility of this combination. We are hoping to expand this collaboration and continue a joint development program to bring this new product to the clinic.
Follow-on Antibody Products From Our Discovery Library
1B7 and 31F9 Antibody Program
We have discovered multiple fully-human antibodies to the antigen GD2, which is significantly over expressed on sarcoma, melanoma, and neuroblastoma. These are three related cancers classified as neuroectodermal cancers (sarcoma, melanoma, neuroblastoma). We discovered these antibodies by examining the immune response from more than 60 patients who participated in our sarcoma vaccine trial over the last three years. According to an article published in the New England Journal of Medicine by Yu and colleagues in 2010, antibodies against GD2 have already been validated as effective therapeutic agents in a well-controlled Phase III clinical trial that produced statistically significant improvement in time to progression of disease in children suffering
6
Table of Contents
from neuroblastoma. Each of the two potential development candidates have specificity and affinity for the GD2 target and demonstrate significant ability to kill cancer cells that express GD2. Each antibody has a unique set of characteristics and, as part of the preclinical evaluation program, researchers at MSKCC will provide further in vitro and in vivo testing in multiple models of disease to help us decide which candidate to move forward toward clinical testing. We are currently targeting sarcoma as the primary indication for which we plan to develop an anti-GD2 antibody product.
Antibody Discovery
We have discovered and recombinantly expressed more than 100 fully human antibodies to eleven separate targets over expressed on multiple types of cancer. The antigenic targets are incorporated in various combinations making up the eight vaccines that were licensed from MSKCC and have been in at least Phase I clinical trials. To date, six separate vaccines have entered early stage clinical programs and we have received antibody discovery material from all patients who participated in five of the trials. Exclusive access to these patient samples is covered under separate licenses with MSKCC. We have discovered multiple antibodies to important cancer targets such as MUC1, GD3, GM2, Fucosyl-GM1, Globo-H, Tn, sTn, and TF. As we continue to raise additional capital and add capacity, it is our intention to begin moving these early product development candidates forward toward clinical evaluation.
Current Approach to Treatment of Pancreatic Cancer
According to an article in the Journal of Advanced Practitioner in Oncology, or Advanced Practitioner, by Sheena Daniels, DNP, ARNP, FNP-BC and colleagues in 2011, for the more than 80% of patients diagnosed with pancreatic cancer who will not be candidates for resection and a large number (80%) of patients who were able to have resection but will develop metastases within 2 to 3 years, the drug gemcitabine has been established as the standard of care because of its documented advantage in OS and more favorable side-effect profile. In patients who are not resectable (able to remove through surgery), combination therapy with a gemcitabine-based systemic regimen followed by consolidation with chemoradiation has produced some favorable median survival durations.
Newer Therapeutic Agents
According to Daniels’ article in the Advanced Practitioner, in spite of aggressive treatment, patients with unresectable pancreateic cancer have a median survival of 10 to 14 months and an estimated 1 in 4 patients who undergo pancreatic resection do not recover sufficiently from surgery to allow administration of systemic chemotherapy. Newer targeted agents have been studied in combination with gemcitabine but did not correlate with improved OS. However, according to Daniel, the combination of gemcitabine and erlotinib did improve overall survival from 5.91 months to 6.24 months and 1-year survival improved to 23% vs 17%. These results were not duplicated using multi-targeted kinase inhibitors. There are several ongoing trials combining additional chemotherapeutic agents with gemcitabine and early results show improvements in PFS and OS but come at a significant cost of increased adverse events. Nab-paclitaxel combined with gemcitabine has produced positive results in an early Phase II trial and those results were confirmed in a subsequent Phase III trial. Nab-paclitaxel is now approved for treatment of pancreatic cancer and the combination of gemcitabine and Nab-paclitaxel is considered first line therapy in many institutions.
We believe there is a significant unmet medical need for newer agents that can treat metastatic disease. Pancreatic cancer is an area of intense research with much of the late stage clinical development efforts targeted toward advanced and metastatic disease. According to the online database BCIQ from BioCentury, to date there are 92 products in all three stages of clinical development from a total of 86 companies and there are 14 products in Phase III or pivotal trials at this time.
Potential Market Opportunity for the Full Length Therapeutic Antibody 5B1
There is a critical unmet medical need for new and better treatment for metastatic pancreatic and colon cancer. According to the National Cancer Institutes SEER database, the five-year survival rate for patients with metastatic pancreatic cancer is just 1.8%. There are 43,000 new pancreatic cancer patients per year and more than half of them present at initial diagnosis with metastatic disease. According to the SEER database, the five-year survival rate for patients with metastatic colon cancer is only slightly better at 12%. According to the SEER database, while most of the 141,000 new patients can be treated successfully initially, almost half of all colon cancer patients will develop metastatic disease. Thus, adding the number of metastatic disease patients for these two cancers alone represent 96,000 new metastatic cancer patients per year. Using the cost of current antibody therapies as a baseline, the market potential for an annual patient population of 96,000 is in excess of $1 billion per year.
Pancreatic Cancer Imaging and Diagnosis
We believe that the radiolabeled 5B1 antibody represents the only human derived agent in development specifically aimed at improving imaging in pancreatic cancer. Since the antigen targeted by the 5B1 antibody is significantly and preferentially over
7
Table of Contents
expressed on metastatic pancreatic cancer, this development effort represents a potentially important step forward in the diagnosis, staging, and assessment of the majority of pancreatic cancer patients. We believe that the market opportunity for a 5B1antibody-based radiopharmaceutical is significant in multiple ways. The ability of physicians to accurately diagnose, stage, and assess treatment outcomes in pancreatic cancer would be very important. Accurate determinations on the extent of disease and resectability are essential to improve outcomes in this cancer. Potentially almost all patients diagnosed with pancreatic cancer could benefit from 89Zr-hu5B1 scan. Accordingly, improvements in the sensitivity and specificity of one of the primary diagnostic tools could have a significant impact on clinical outcome.
Currently, according to Daniels’ article in the Advanced Practitioner, all screening methods for pancreatic cancer have limitations. The symptoms associated with the disease are often vague and attributed to other more benign etiologies. Diagnosing pancreatic cancer is often challenging, as the presenting symptoms of pancreatic, hepatobiliary (portion of the liver, pancreas, and biliary tract), and upper gastrointestinal cancers are similar. Screening for pancreatic cancer, according to Daniel, is difficult primarily because there are no tumor markers that can be screened at an early stage of disease. Therefore, according to Daniel, the majority of pancreatic cancers are diagnosed at a late stage of disease hampering efforts to provide curative therapy. Pancreatic cancer is typically diagnosed by a combination of history and physical examination, coupled with CT, ultrasound, and PET imaging. In patients deemed to be at high risk for metastasis or for whom the staging is indeterminate, the diagnosis is confirmed by fine needle aspiration or laparoscopy along with FDG-PET. Accurate staging, particularly identification of distant metastases, is of paramount importance in order to properly select patients who are the most likely to benefit from surgery. Differential diagnosis between pancreatic cancer and pancreatitis (non-cancerous inflammation of the liver) is a common problem with imaging modalities.
Recent Developments in ImmunoPET Imaging
2-deoxy-2-(18F)-fluoro-D-glucose, or FDG, combined with PET imaging alone, or FDG-PET, combined with CT scanning, or FDG-PET/CT, is commonly used to augment the diagnosis and staging of pancreatic cancer. According to an article in The American Journal of Surgery by Lan and colleagues in 2012, FDG-PET is the principal imaging agent available today for enhanced PET imaging for pancreatic cancer. FDG is a radiolabeled form of glucose and because cancer cells normally have elevated metabolic rates, highly active cells will incorporate this glucose marker and as a result can potentially help pinpoint pancreatic cancer and potential metastases.
To date, there have been 17 studies examining the accuracy of FDG-PET and the conclusions are mixed. While studies are heterogeneous (not of a similar type; not uniform), the utility of FDG-PET was reasonably established for the primary diagnosis purposes only, utility of FDG-PET/CT for determining recurrence and staging was not confirmed. Additional studies have evaluated the diagnostic thinking impact of FDG-PET with regards to patient management and diagnostic work-up of pancreatic cancer. Findings from FDG-PET lead to changes in the pre-treatment staging as well as the decisions regarding treatment management because of changes in resectability status. The majority of findings demonstrated previously unsuspected metastases and resulted in cancellation of previously planned surgical resection. Roughly half the time the newly identified metastases had not been detected by initial CT scans.
The cost-effectiveness of adding FDG-PET to the routine diagnostic procedures to determining staging and eligibility for surgery among patients with presumed resectable pancreatic cancer has been examined in a limited number of studies. Cost savings were identified primarily by identifying patients who were initially staged for surgery and later deemed ineligible because of detected metastases.
Although FDG is used extensively and successfully in many cancers, because of the targeting characteristics of this compound as a marker of glucose metabolism, the sensitivity and specificity of FDG are not optimal in all cancer types. The shortfalls of imaging with FDG, such as inadequate differentiations between post-therapy inflammation and tumor, poor imaging in slow-growing tumors, and high uptake in normal cells such as brain and gut, have remained as the justification for the development of newer PET tracers.
FDG-PET Reimbursement
The Department of Health and Human Services has completed a Technology Assessment for FDG-PET in pancreatic cancer. The Centers for Medicare and Medicaid Services, or the Center, utilized the Technology Assessment and has issued a National Coverage Determination for FDG for oncologic conditions. The Center has elected to cover the expense of using FDG-PET for the diagnosis and staging of pancreatic cancer. However, the Technology Assessment found insufficient support for fully covering FDG-PET for restaging and monitoring response to treatment. Private insurance carriers follow the Center’s recommendation and cover FDG-PET for diagnosis and staging.
8
Table of Contents
Potential Market Opportunity
To our knowledge, the radiolabeled 5B1 antibody represents one of the only agents in development specifically aimed at improving imaging in pancreatic cancer. Since the antigen targeted by the 5B1 antibody is significantly and preferentially over expressed on metastatic pancreatic cancer, this development effort represents a potentially important step forward in the diagnosis, staging, and assessment of the majority of pancreatic cancer patients. Using an imaging agent that is specific for a pancreatic cancer antigen would be greatly preferable to an indirect marker that can produce false positive results in high metabolic rate tissues (tissues where there is an elevated metabolism of sugar due to a variety of causes including cancer) or a false negative in slow growing cancers. We believe that the market opportunity for a 5B1 antibody-based radiopharmaceutical is significant in multiple ways. First the ability of physicians to accurately diagnose, stage, and assess treatment outcomes in pancreatic cancer would be very important. Accurate determinations on extent of disease and resectability are essential to improving outcomes in this cancer. We believe improvements in the sensitivity and specificity of one of the primary diagnostic tools would be useful and that potentially almost all patients diagnosed with pancreatic cancer could benefit from a 89Zr-hu5B1 antibody scan. Using existing utilization and reimbursement rates for the current standard of care product, FDG-PET, annual revenues for a 5B1 antibody-based radiopharmaceutical could exceed $100 million per year. Since the regulatory pathway is significantly less expensive and time consuming than a therapeutic agent, we believe that this companion diagnostic product opportunity will complement the full-length therapeutic antibody and antibody-drug conjugate products.
This project is also important to us because of the potential enablement of the full-length therapeutic antibody. The 89Zr-hu5B1 antibody will be essential in the clinical development of the 5B1 antibody. It would help improve our understanding of the antibody’s in vivo behavior including the interaction with its critical disease target, mechanism of action, distribution, and potential toxicities. Just as important, we believe the 5B1antibody-based radiopharmaceutical would enable physicians to identify patients with the greatest chance of benefiting from treatment with the antibody and that the 5B1Db-based radiopharmaceutical would enable accurate diagnosis, staging, and assessment of treatment outcome of a new antibody treatment for advanced pancreatic cancer.
License Agreement with MSKCC
We have licensed from MSKCC the exclusive world-wide developmental and commercial rights to all antibodies we discover using lymphocytes (immune system cells from with antibodies are secreted) from vaccinated clinical trial participants enrolled in any of the clinical trials involving the vaccines licensed to us. MSKCC has issued patents or has patents pending on all underlying vaccines. This patent portfolio includes 35 issued patents in the US and rest of world along with 7 patent applications. We own all monoclonal antibodies produced by the antibody discovery program and these antibodies are generally patented once their potential therapeutic utility has been sufficiently demonstrated in animal models. A provisional patent application for the anti-sLea antibodies described in this application has been filed.
We are unique in that only a very small number of oncology focused companies are vaccinating patients with carbohydrate-based vaccines intended to elicit an antibody response and, to the best of our knowledge, we are the only company deriving antibodies from the lymphocytes of vaccinated patients. Since these carbohydrates are very difficult to make immunogenic and the carbohydrates cannot be easily manufactured, it is very difficult if not impossible to find a source for human antibodies to these antigens beyond that utilized by us.
Vaccine Program
We have licensed exclusive rights from MSKCC to exploit key aspects of the work of Dr. Livingston (who is also a member of our board of directors) and colleagues, who over the last 30 years have developed a series of monovalent (targeting a single tumor cell surface antigen) cancer vaccines against cancers of neuroectodermal and epithelial (breast, ovarian colon, pancreatic) origin as well as small cell lung cancer, or SCLC. These target molecules on malignant cells, known as carbohydrate antigens, are the most extensively expressed antigenic targets on the cell surface of these types of cancers and play a key role in tumor progression and metastasis. We expect to benefit from the years of work and significant expense already invested in the development and testing of the vaccines incorporating these antigens. Researchers at MSKCC have progressively developed highly immunogenic monovalent vaccines to each of the 11 validated target antigens that comprise the licensed vaccines. These monovalent vaccines or the combination of the monovalent forms into polyvalent vaccines (targeting multiple antigens) have been tested and refined not only in animal models but also in multiple clinical trials establishing immunogenicity, tolerability, and therapeutic utility. Our license agreement with MSKCC calls for MSKCC to complete all preclinical and Phase I clinical trial work at MSKCC’s expense at which point the Investigational New Drug Application, or IND, would be transferred to us for continued development.
Our lead cancer vaccines targeting recurrent sarcoma and ovarian cancer are currently in proof of concept Phase II multi-center clinical trials. Both trials are fully enrolled, and have received substantial federal grant monies to support their development.
9
Table of Contents
A vaccine to address the orphan disease neuroblastoma has completed an initial Phase I trial at MSKCC yielding encouraging results. We expect the vaccine product to be ready for a Phase II trial by early 2015. MSKCC and MabVax Holdings have completed additional Phase I vaccine clinical trials in melanoma, ovarian cancer, and small cell lung cancer over the last three years.
Our Vaccines Intend to Address Recurrent Cancer, An Unmet Medical Need
Despite undergoing potentially curative surgical resection or combination therapy, a significant number of patients with cancer will have their cancer recur. Patients with recurrent cancer have a significantly lower survival rate and incur much higher medical costs compared to those whose disease does not recur. Multiple clinical studies have demonstrated that additional courses of chemotherapy or radiation in the adjuvant setting do not or only minimally improve outcomes for these patient groups. Thus, in the majority of cases, the current standard of care following treatment of metastatic disease and the achievement of disease-free status is watchful waiting. With recurrence rates of 95% in stage IV sarcoma and 70% in stage III and IV ovarian cancer respectively, there is an unmet medical need for new treatments for recurrent disease.
Medical Solution and Rationale
According to an article in Human Vaccines by Livingston and colleagues published in 2006, the adjuvant setting is the ideal time for immune intervention and in particular for administration of monoclonal antibodies or cancer vaccines aimed at instructing the immune system to identify and kill the few remaining circulating cancer cells. We believe that passively administered or vaccine induced antibodies against selected cell surface antigens are ideally suited for eradication of free tumor cells and micrometastases. This is the role of antibodies against most infectious diseases, which has been accomplished against cancer cells in a variety of pre-clinical models and recent clinical trials. We also believe that if antibodies of sufficient titer can be administered or induced against tumor antigens to eliminate tumor cells from the blood and lymphatic systems and to eradicate micrometastases, this could dramatically change the approach to treating the cancer patient. If successful, establishment of new metastasis would no longer be possible, so aggressive local therapies including surgery or radiation therapy, and intralesional (injection of cancer treatment directly into a tumor) treatments, combined with our immunotherapeutic agents could result in long term control of metastatic cancers.
Vaccine Purpose Determines Vaccine Design
According to Livingston’s article in Human Vaccines, the majority of carbohydrate antigens are recognized exclusively by B-lymphocytes and antibodies and the optimal approach for augmenting antibody responses against defined antigens involves conjugation of the antigens to a highly immunogenic carrier protein. This is the approach currently used in a variety of vaccines against bacterial pathogens and is the approach we believe to be optimal for cancer associated carbohydrate antigens (antigens/targets made of carbohydrates; also known as sugar structures) as well.
We have explored a variety of methods for augmenting the antibody response to defined antigens including vaccine production with autologous (derived from a subject’s own cells) or allogeneic (derived from another subject’s cells) tumor cell lines or lysates, the use of multiple different immunological adjuvants, chemical modification of antigens to render them more immunogenic, conjugation to different immunological carrier proteins and adherence of antigens to a variety of vehicles including liposomes (artificial structures that can carry active agents into the body), polystyrene beads (small synthetic beads), BCG and Salmonella. The conclusions from these studies are that the use of an immunogenic carrier protein plus a potent immunological adjuvant is the optimal approach. The studies established that the optimal carrier protein was KLH, while the optimal adjuvants were saponins (class of adjuvants) such as QS-21 or OPT-821obtained from the bark of Quillaia saponaria (type of tree indigenous to South America). Both conjugation to KLH and use of a saponin (substance derived from Quillaia saponaria) adjuvant are required for an optimal response. The role of the carrier protein in these conjugate vaccines was induction of a potent T-cell response against KLH, resulting in a cascade of cytokines (immune system cells essential to immune response) which then provide help for the antibody response against both KLH and more weakly immunogenic molecules attached to KLH. The primary role of the saponin adjuvants appeared to be augmentation of the immune response against KLH.
Previous Human Clinical Experience
There are an increasing number of clinical trials showing that passively administered monoclonal antibodies, or mAbs, against cell surface antigens such as HER2/neu, EGF receptor, VEGF, CD20, CD33 and CD52 have demonstrated clinical efficacy against human cancers and leukemias. Specifically, according to an article in Clinical Breast Cancer by Jahanzeb there is evidence from a series of recently described clinical trials with Trastuzumab (Herceptin®, against HER2/neu) used in breast cancer patients in the adjuvant setting confirming a striking recurrence free and overall survival advantage. This is a more dramatic response than seen with the same mAb used in the more advanced disease setting. Finally, naturally acquired or vaccine induced antibodies
10
Table of Contents
against cancer cell surface antigens such as GM2 and sTn have correlated with improved prognosis in several different clinical settings. Murine (mouse origin) monoclonal antibodies 3F8 against GD2 and R24 against GD3 have each induced clinical responses in a significant proportion of melanoma patients in the advanced disease setting. 3F8 and R24 are murine monoclonal antibodies and so can only be administered briefly before human anti-mouse antibodies, or HAMA, induction leads to decreased clinical availability.
There has been a significant amount of clinical work in the development of the monovalent and polyvalent versions of these cancer vaccines that stretches over two decades. In the Investigator’s Brochure portion of the INDs submitted to FDA on behalf of the sarcoma and ovarian cancer vaccine trials, we list the 30 Phase I clinical trials testing immunogenicity and tolerability of each of the monovalent vaccines to date. Refinements in antigen configuration, selection of carrier molecules, selection of adjuvant, vaccination schedules, and dose ranging have all lead to the optimal configuration of the current vaccines. According to Livingston’s article in Human Vaccines, the current monovalent vaccines all induce an immune response of IgM and IgG antibodies capable of killing targeted cancer cells.
Concern for Autoimmune Disease
Antigens on cancer cells are generally either autoantigens or slightly modified autoantigens (antigens or targets that do not trigger an immune response) so autoimmunity is a concern with any cancer vaccine. This concern is either as a consequence of cross reactivity of the specific immune response generated against cancer antigens (also present on normal organs) or as a consequence of immune modulation resulting from the immunological adjuvant or other components of the vaccine that may generate non-specific immune modulation. These concerns are largely mitigated, however, by the extensive experience and low toxicity profile consistently observed with the individual components of this vaccine in the clinic either alone or paired up with other components of this vaccine.
The Basis for Polyvalent Immunotherapy
There are at least three reasons for the expectation that carbohydrate vaccines against cancer should contain multiple antigens (be polyvalent). First, heterogeneity is an essential feature of malignancy with patient-to-patient tumor variability and even cell-to-cell heterogeneity in the same tumor is the norm. Second, heterogeneity also characterizes the immune responsiveness of immunized hosts as a consequence of a variety of known and unknown factors including previous immunologic experience and genetic background. The final basis for polyvalent vaccines is a consequence of the previous two reasons. Anti-tumor effector mechanisms are proportional to the amount of antibody bound to the cell surface, which will in turn be proportional to the number of different antigens in the vaccine.
Addressing the issue of tumor heterogeneity will also inform our decisions regarding which antibody candidates to develop. We expect that our current work aimed at improving the quality and control of manufacturing monoclonal antibodies will eventually allow us to explore the development of “cocktails” of mAbs which in turn would allow us to continue to improve the utility of antibodies against various cancers.
Sarcoma Vaccine
Background
Sarcomas are rare neoplasms (tumor that has caused a lump) that arise from the mesenchymal (connective tissue such as bone or cartilage) tissues of the body. According to the NCI’s website, in the United States, there are approximately 13,000 cases diagnosed each year, representing less than 1% of all new cancers. Of these sarcomas, roughly 80% originate from soft tissue, with the remainder originating from bone. Prognosis remains poor, with more than 5,000 patients in the US dying of disease each year. The overall prevalence of sarcoma patients in the US is thought to be approximately 100,000.
As in other malignancies, disease recurrence and metastasis are common in sarcoma. Metastases may involve any organ of the body, but, according to the NCI’s website approximately 20% of adult patients with extremity sarcomas will have isolated lung metastasis at some point during their disease course, with some amenable to complete surgical resection. Additional patients will have solitary or oligometastatic (cancer that has spread to multiple locations throughout the body) disease affecting other sites of the body that will be amenable to complete resection. Favorable prognostic indicators in recurrent sarcoma include a long disease-free interval from the time of primary resection, the number and location of metastatic lesions, and a long tumor doubling time.
Osteosarcomas (bone based sarcomas), rhabdomyosarcomas (sarcomas arising from muscle tissue), and other non-rhabdomyosarcomas such as Ewing sarcoma (tumor arising in bone or soft tissue and primarily occurring in teenagers and young adults) are high-risk sarcomas that occur most commonly in teens and young adults. According to the NCI’s website, approximately 30% of patients will present with metastases or recur following initial therapy for metastatic disease. These recurrences are generally treated with a combination of chemotherapy, surgery, and/or radiotherapy, with more than 90% of these patients achieving a complete response to therapy according to the NCI’s website.
11
Table of Contents
Despite undergoing potentially curative surgical resection or combination therapy, according to an article by DA Potter and colleagues published in the Journal of Clinical Oncology in 1985, the majority of recurrent sarcoma patients die as a result of further recurrences. The overwhelming majority of pediatric patients ultimately succumb to their disease following the development of recurrent, chemoresistant disease, and their prognosis remains unacceptably poor despite aggressive multimodality treatment. The addition of chemotherapy to surgical resection has not been shown to improve outcome in adult sarcomas. Thus, in the majority of cases, the current standard of care following treatment of metastatic disease and the achievement of disease-free status is expectant management.
Sarcoma Vaccine Clinical Program
We initiated a randomized, multicenter, double-blind Phase II clinical trial in July of 2010. A total of 136 patients were enrolled. Patients who entered the study had stage IV metastatic disease and were cleared by surgery. Patients were vaccinated 10 times over 84 weeks and monitored throughout the study period. The study was powered to show a statistical improvement in both progression free survival (measured at the mid-point of the study) and overall survival. In October of 2013, MabVax Holdings presented the mid-point results to the independent Drug Safety Monitoring Board, or DSMB. The DSMB concluded that there were no unanticipated or clinically worrisome safety concerns. Injection site reactions were the most common adverse events followed by fatigue, fever, and flu-like symptoms. In addition, the DSMB recommended that investigators and patients should remain blinded as to treatment assignment and the patients should continue to be followed to assess overall survival. In addition, given the acceptable safety profile observed in both arms of the study, the DSMB recommended that after investigators and patients are informed of the (blinded) results of this analysis and with their consent, the last of the patients still receiving vaccinations should be allowed to continue treatment.
In this study, the sarcoma vaccine elicited an antibody response intended to kill circulating tumor cells and micrometastases in all but one of the vaccinated patients. However, the DSMB concluded that the study did not reach statistical significance for its primary efficacy endpoint of a 50% improvement in time to recurrence. The study has not yet accumulated a sufficient number of events to evaluate the secondary endpoint of overall survival. Based on our discussions with the FDA prior to the initiation of the study, the overall survival endpoint will be considered the primary endpoint for the measurement of efficacy. As such we plan to follow all patients in the study until sufficient numbers of events (deaths) have occurred to allow analysis of this endpoint. We expect that the event threshold will be reached in 2016.
Potential Commercial Opportunity
Sarcomas are a diverse group of malignant tumors that develop from fat, muscles, nerves, joints, blood vessels, bones, and deep skin tissues. Soft tissue sarcomas are more deadly in part due to the lack of detectable symptoms at early disease stages and prognosis remains poor, with more than 5,000 patients in the US dying of disease each year according to the National Cancer Institute, or NCI. The NCI estimates 5-year survival rates of 60%. Additionally, according to the NCI, an article in CA: A Cancer Journal for Clinicians by A. Jemal published in 2006, and articles published by the American Cancer Society, the overall prevalence of sarcoma patients in the US is thought to be approximately 100,000, recurrence rates vary depending on the particular subtype of sarcoma but generally range from 30% to 50% and patients whose sarcoma recurs have a significantly poorer prognosis which declines even further as the number of recurrences increase over the course of the disease.
The current standard of care for patients who have been successfully treated for their cancer and rendered free of detectable disease is watchful waiting. According to an article in The Lancet Oncology in 2012, additional treatments of chemotherapy or radiation have not been proven to prevent or prolong the time to onset of cancer recurrence. Consequently, we anticipate that the sarcoma vaccine will be added as an additional treatment to the current treatment paradigm and not displace an existing treatment. We expect that patients who have experienced one or more recurrences will be the initial candidates for vaccine therapy. This would be consistent with the early clinical trials. Over time, as the product demonstrates utility, we anticipate that usage will migrate toward earlier and earlier treatment to include patients who have been diagnosed and treated for sarcoma but not yet experienced a recurrence in an effort to block the progress of the cancer at an earlier and more likely productive time period.
We have estimated the ex-manufacturer price for this therapy at $30,000 to $50,000 per course of treatment, and an average estimated cost of cancer therapy of $32,000. Treatment is defined as a course of 10 vaccinations over 84 weeks as per the Phase II clinical trial protocol. Standard of care calls for patient check-ups quarterly after successful initial treatment to achieve the minimal disease status. The first 5 vaccinations require visits to the physician more frequently than quarterly but beyond that initial treatment sequence, the remaining vaccinations are delivered at the time of the standard check-up. Treatment can be given in the physician office setting with no special administration equipment required. The cost of the proposed vaccine therapy can be favorably compared to cycle costs for targeted vaccine and monoclonal antibody products ranging from $20,000 to $100,000 per
12
Table of Contents
treatment cycle. According to the NCI, the average monthly direct cost of all cancer treatments across all cancers studied was $2,647 per month and the total cost for treatment was more than $32,600. There are substantial indirect costs associated with hospitalizations, short-term disability, and absenteeism resulting from cancer recurrence that can drive treatment costs higher. We project that annual revenue from a sarcoma vaccine could range from $150 million to $300 million annually based on internal projections considering the number of patients, the percentage of patients with metastatic disease, the cost of treatment, and market penetration rates.
The ultimate success of the sarcoma vaccine will depend on several factors, including, but not limited to the following: (i) The percentage of patients whose cancer does not recur or for whom recurrence is delayed, (ii) In those patients who respond to the vaccine, whether the prevention of recurrence or the delay of recurrence is for a clinically meaningful period of time, and (iii) The willingness of third-party payers and the government to pay for the addition of the vaccine therapy to the existing treatment paradigm. The clinical development program should answer the first two questions positively. As with almost all new therapies in cancer, the last question will require a substantial amount of work with these important participants in the healthcare system. If we can demonstrate reduced recurrence rates in sarcoma, we believe we can also demonstrate the financial benefits of reduced or unnecessary further treatments when recurrent sarcoma is prevented or delayed.
Ovarian Vaccine Program
Background
According to the NCI, ovarian cancer is the most lethal gynecologic cancer. According to materials available on the NCI’s website there are more than 21,800 new cases each year with almost 14,000 deaths per year. It is estimated that there are 174,000 surviving ovarian cancer patients. Recurrence rates are extremely high at 70% and 5-year survival is still very poor at just over 40%. The current standard treatment for patients with advanced ovarian cancer consists of aggressive surgical cytoreduction (resection of a tumor to the extent possible followed by radiation treatment) followed by taxane (chemical anti-cancer drug) and platinum-based chemotherapy. While the median overall survival for optimally debulked patients has increased to 65.6 months, less than 30% of patients will remain free of disease. Many patients will have chemotherapy-sensitive disease initially at recurrence, and can reenter successive remissions with additional treatment. Subsequent remissions are of progressively shorter duration until chemotherapy resistance uniformly develops. We believe that immune directed therapy is ideally suited for patients who are in clinical remission when the disease burden is lowest, and evaluating treatments designed to prolong the duration of such remissions remains a high priority. We also believe that antibodies are well suited for eradicating tumor cells from the bloodstream and eliminating early tissue invasion. Preclinical models have demonstrated the clearance of circulating tumor cells and the elimination of systemic micrometastasis through the use of both passively administered and vaccine induced antibodies.
Ovarian cancers express a rich array of cell-surface antigens. These include carbohydrate epitopes such as GM2, Globo-H, Lewisy, sialyl Tn, or STn, Tn, Thompson Friedreich antigen, or TF, and mucin 1, or MUC1. According to an article in Cancer Immunology Immunotherapy written by Livingston that was published in 1997, for the production of antibodies against defined cell-surface antigens such as these, the best approach has been described to include chemical conjugation of the antigen to a highly immunogenic carrier protein plus the use of a potent immunological adjuvant. The best carrier protein in our experience has been keyhole limpet hemocyanin (a sea creature such as a limpet or snail from which copper-based highly immunogenic blood is extracted), or KLH, and the best immunological adjuvant has been a saponin such as QS-21 or OPT-821. Pre-clinical data supports the hypothesis that polyvalent vaccines will likely be required due to tumor cell heterogeneity, heterogeneity of the human immune response, and the correlation between overall antibody titer against tumor cells and antibody effector mechanisms.
Ovarian Vaccine Clinical Program
A randomized, multicenter, double-blind Phase II clinical trial in ovarian cancer with a pentavalent (a vaccine that has multiple antigens) vaccine was initiated in July of 2010. While this vaccine was included in the group of vaccines exclusively licensed to us in 2008, a NIH grant award co-authored by Dr. Philip Livingston was made which fully funded the planned Phase II clinical trial. Management of the trial was assigned to the Gynecologic Oncology Group, or GOG. We contributed to the development of the IND and provided financial support for the manufacture of the clinical material. A total of 164 patients were enrolled. Patients who entered the study had metastatic ovarian cancer and have been treated with cytoreductive surgery and chemotherapy. They were in complete clinical remission as defined by CA-125 levels within normal, negative physical examination and no evidence of disease by CT scan. Patients were vaccinated 10 times over 84 weeks and monitored throughout the study period. The study was powered to show a statistical improvement in both progression free survival (measured at the mid-point of the study) and overall survival. The study has not achieved a sufficient number of events to trigger the mid-point analysis. Based on discussions with the principal investigator, the GOG plans to recommend that investigators and patients remain blinded as to treatment assignment and the patients should continue to be followed to assess overall survival. MabVax Holdings anticipates that results from the overall survival endpoint will be announced in 2016.
13
Table of Contents
Potential Commercial Opportunity
We have estimated that the ex-manufacturer price for this therapy to be $40,000 per course of treatment. Treatment is defined as a course of 10 vaccinations over 84 weeks as per the Phase II clinical trial protocol. Standard of care calls for patient check-ups quarterly after successful initial treatment to achieve the minimal disease status. The first 5 vaccinations require visits to the physician more frequently than quarterly but beyond that initial treatment sequence, the remaining vaccinations are delivered at the time of the standard check-up. Treatment can be given in the physician office setting with no special administration equipment required. The cost of the proposed vaccine therapy can be favorably compared to cycle costs for targeted vaccine and monoclonal antibody products ranging from $20,000 to $100,000 per treatment cycle. The average monthly direct cost of all cancer treatments across all cancers studied was $2,647 per month and the total cost for treatment was $32,629. There are substantial indirect costs associated with hospitalizations, short-term disability, and absenteeism resulting from cancer recurrence that can drive treatment costs higher. We project that annual revenue from an ovarian vaccine could range from $200 million to $400 million annually.
The ultimate success of the ovarian cancer vaccine will depend on several factors including, but not limited to: (i) The percentage of patients whose cancer does not recur or for whom recurrence is delayed, (ii) In those patients who respond to the vaccine, whether the prevention of recurrence or the delay of recurrence is for a clinically meaningful period of time, and (iii) The willingness of third-party payers and the government to pay for the addition of the vaccine therapy to the existing treatment paradigm. The clinical development program should answer the first two questions positively. As with almost all new therapies in cancer, the last question will require a substantial amount of work with these important participants in the healthcare system. If we can demonstrate reduced recurrence rates in sarcoma, we believe we can also demonstrate the financial benefits of reduced or unnecessary further treatments when recurrent sarcoma is prevented or delayed.
Neuroblastoma Vaccine
Introduction and Overview
Neuroblastoma is the most common extra-cranial solid tumor in children. According to the NCI and an article in the Annals of Pharmacology by Parsons and colleagues in 2013, there are approximately 700 new cases per year in the United States so it is certainly an orphan disease. According to the NCI SEER data, patients with high risk features, defined by clinical and tumor biologic parameters at diagnosis have an expected survival of only 45% despite intensive induction chemotherapy (very high dose chemotherapy), surgical resection, myeloablative consolidation chemotherapy with stem cell support, radiation,(radiation doses intended to eliminate the immune system of the patient followed by stem cell reconstitution of the system) focal radiation (precise pin-point radiation doses) and post-consolidation treatment (chemotherapy treatment post resection), with 13-cis-retinoic acid, or cisRA, and anti-GD2 mAb ch14.18 immunotherapy. MabVax Holdings believes that these results, plus the potentially severe toxicities of chemotherapy and radiotherapy, are compelling reasons for pursuing novel therapeutic approaches.
In particular we believe that, there is a need for therapies that selectively eliminate neuroblastoma cells in the setting of minimal or limited amounts of residual disease following intensive induction therapy. According to an article in Clinical Cancer Research by Matthay and Yu in 2012, neuroblastoma is unique in its abundant expression of the gangliosides GD2 and GD3. Each of these antigenic targets is an outstanding target for immune attack against neuroblastoma cells. In experimental animal studies conducted by Livingston and Ragupathi, administered or vaccine-induced antibodies against GD2, GD3 and other cell surface antigens were able to eliminate micrometastasis in settings similar to the treatment of patients in complete remission but with a high likelihood of relapse. The basis for emphasis on a bivalent (having two antigens raising an immune response to two targets) vaccine containing each of these agents are tumor cell heterogeneity, heterogeneity of the human immune response and the correlation between overall antibody titer against the tumor cell surface and effector mechanisms such as opsonization, complement-dependent cytotoxicity (how antibodies marshal other immune system cells to kill the cells to which they have attached, or CDC, or antibody dependent cellular cytotoxicity, or ADCC.
Vaccines based on self-antigens such as these three must be potently immunogenic to overcome immune tolerance and tumor-mediated immune suppression. Based on the results of studies discussed by Livingston in an article published in 2006 in Human Vaccines, the most effective vaccine for breaking tolerance in man appears to be composed of the tumor-associated self-antigen conjugated to a highly immunogenic carrier protein such KLH combined with a potent saponin adjuvant. Monovalent KLH conjugate vaccines against GD2, and GD3 (plus saponin adjuvant) have been constructed, tested and optimized in patients with melanoma or small cell lung cancer. Each induces high titers of antibodies against the immunizing antigen and against tumor cells expressing these antigens in most patients and these antibodies mediate effector mechanisms such as CDC and ADCC. The minimal optimal dose of each has been determined and safety confirmed through multiple Phase I studies. In addition, a Phase I trial of a bivalent vaccine containing GD2-KLH, GD3-KLH and escalating doses of saponin adjuvant co-administered with oral ß-glucan in children with neuroblastoma has been completed (Kushner, et. al.) Safety, immunogenicity and the optimal saponin adjuvant dose were determined and the vaccinated subjects in this study were noted to have an unexpectedly favorable outcome. Finally, a Phase II randomized, multicenter, double blind trial of a trivalent ganglioside vaccine plus saponin adjuvant is currently underway in subjects with sarcoma who are disease-free after resection of metastatic disease. We believe that this collective experience provides the rationale for undertaking the current study in high-risk neuroblastoma subjects who are in second or subsequent complete remission or have only limited residual disease.
14
Table of Contents
Results of a Recent Phase I trial of Bivalent Vaccine in Combination with Oral ß-glucan
MSKCC carried out a Phase I trial in subjects with high-risk neuroblastoma to assess the toxicity of escalating doses of the immunological adjuvant OPT-821 in a bivalent vaccine containing fixed doses of GD2L and GD3L, each covalently attached to the immunological carrier protein KLH (Kushner, 2012). The study subjects were in second (or later) complete, very good partial, or partial remission. Subjects received seven subcutaneous injections over 52 weeks,
Thirteen of the 15 subjects received the entire 12 months of protocol treatment. No dose limiting toxicity was noted in any of the subjects. Most subjects had detectable anti-GD2 or anti-GD3 antibodies. Twelve remain relapse-free at 21+-36+ (median 29+) months. One had a focal relapse (recurrence of the cancer at a specific spot) (supraclavicular node) at 21 months. Two other subjects had early focal relapses (2.3 and 4.6 months). PFS was 87+9% at 12 months and 78+11% at 24 months. Overall survival is 100% (follow-up >22months). These results are in marked contrast to the expected relapse rate at 12 months of 50% to 60%.
Clinical Plan
We have received the Phase I portion of an SBIR grant for support in production of the clinical trial material required for a planned Phase II clinical trial. The Company has also established a relationship with a consortium of thirteen academic hospital based neuroblastoma treatment programs called New Advances in Neuroblastoma Therapy, or NANT. We entered into a Letter of Intent with NANT and the NANT Scientific Review Committee has reviewed our jointly developed Phase II clinical protocol which was approved by this committee with only minor modifications. Because the market opportunity for a vaccine to treat recurrent neuroblastoma is very small, we have worked to bring in additional non-dilutive financing. We hope to expand the SBIR award to the Phase II portion of the grant which could offset $1 million in Phase II clinical expenses. We are currently conducting standard release and stability testing on the recently completed clinical material. Test results will be available in the second quarter of 2015. We are planning to obtain the orally bioavailable immune system stimulant product that was used effectively in the Phase I trial at MSKCC. That product will be available later in the year. Once that last adjunctive treatment is secured, we will be able to seek an IND to initiate the clinical trial.
Patents
As of March 12, 2015, we were the exclusive licensee, sole assignee or co-assignee of 20 granted United States patents, 16 pending United States patent applications and 41 international patents through MabVax Therapeutics covering the monovalent vaccines that make up the polyvalent vaccine products. The major claims of these patents cover composition of the vaccine, methods of treatment, chemical modification of antigens, and synthesis. We also own or have rights to 2 patent applications in the United States and 6 in other geographic areas currently pending, covering aspects of ovarian cancer and breast cancer. We also have 21 granted United States patents, 12 pending United States patent applications and 99 international patents obtained, and 88 international patent applications that are also being maintained by the Company from prior to the Merger while we continue to evaluate the usefulness of these patents and patent applications to the Company.
The Intellectual Property Rights of the Company used prior to the Merger are not used in the company’s current operations. The Company may enter into one or more agreements relating to the sale or license of such Intellectual Property Rights, but is not otherwise actively maintaining any registrations or making filings required in connection with any such Intellectual Property Rights, and certain of such Intellectual Property Rights may already have expired or been deemed abandoned.
| Item 1A. | Risk Factors. |
Our business faces significant risks, some of which are set forth below to enable readers to assess, and be appropriately apprised of, many of the risks and uncertainties applicable to the forward-looking statements made in this Annual Report. You should carefully consider these risk factors as each of these risks could adversely affect our business, operating results and financial condition. If any of the events or circumstances described in the following risks actually occurs, our business may suffer, the trading price of our common stock could decline and our financial condition or results of operations could be harmed. Given these risks and uncertainties, you are cautioned not to place undue reliance on forward-looking statements. These risks should be read in conjunction with the other information set forth in this Annual Report. There may be additional risks faced by our business, though we do believe that the risks set forth below reflect the more important ones.
15
Table of Contents
We will be required to raise additional funds to finance our operations and remain a going concern; we may not be able to do so when necessary, and/or the terms of any financings may not be advantageous to us.
Our operations to date have consumed substantial amounts of cash. Negative cash flows from our operations are expected to continue over at least the next several years. Our cash utilization amount is highly dependent on the progress of our product development programs, particularly, the results of our preclinical and clinical studies and those of our partners, the cost, timing and outcomes of regulatory approval for our product candidates, and the rate of recruitment of patients in our human clinical trials. In addition, the further development of our ongoing clinical trials will depend on upcoming analysis and results of those studies and our financial resources at that time.
We are aggressively pursuing forms of capital infusion including public or private financing, strategic partnerships or other arrangements with organizations that have capabilities and/or products that are complementary to our own capabilities and/or products, in order to continue the development of our product candidates. However, there can be no assurances that we will complete any financings, strategic alliances or collaborative development agreements, and the terms of such arrangements may not be advantageous to us.
Our ongoing capital requirements will depend on numerous factors, including: the progress and results of preclinical testing and clinical trials of our product candidates under development; the costs of complying with the FDA and other domestic and foreign regulatory agency requirements, the progress of our research and development programs and those of our partners; the time and costs expended and required to obtain any necessary or desired regulatory approvals; the resources that we devote to manufacturing expenditures; our ability to enter into licensing arrangements, including any unanticipated licensing arrangements that may be necessary to enable us to continue our development and clinical trial programs; the costs and expenses of filing, prosecuting and, if necessary, enforcing our patent claims, or defending against possible claims of infringement by third-party patent or other technology rights; the cost of commercialization activities and arrangements, if any, that we undertake; and, if and when approved, the demand for our products, which demand depends in turn on circumstances and uncertainties that cannot be fully known, understood or quantified unless and until the time of approval, including the range of indications for which any product is granted approval.
We anticipate that we will need to raise additional funds through public or private financing, strategic partnerships or other arrangements. As further discussed below with respect to risks related to ownership of our capital stock, any additional equity financing will be dilutive to our current stockholders and debt financing, if available, may involve restrictive covenants. If we raise funds through collaborative or licensing arrangements, we may be required to relinquish, on terms that are not favorable to us, rights to some of our technologies or product candidates that we would otherwise seek to develop or commercialize. Our failure to raise capital when needed could materially harm our business, financial condition and results of operations.
We have a history of losses, and we anticipate that we will continue to incur losses in the future; our auditors have included in their audit report an explanatory paragraph as to substantial doubt as to our ability to continue as a going concern.
We have experienced net losses every year since our inception and, as of December 31, 2014, had an accumulated deficit of $24,550,308. Our auditors have included in their audit report a “going concern” explanatory paragraph as to substantial doubt as to our ability to continue as a going concern that assumes the realization of our assets and the satisfaction of our liabilities and commitments in the normal course of business. We anticipate continuing to incur substantial additional losses over at least the next several years due to, among other factors, expenses related to the following: the GMP manufacture of our 5B1 antibody to create clinical trial supplies, conducting Phase I clinical trials with the 5B1 antibody, preclinical testing of follow-on antibody candidates, investor and public relations, SEC compliance efforts, anticipated research and development activities and the general and administrative expenses associated with each of these activities. We have not yet commercialized any product candidates. Our ability to attain profitability will depend upon our ability to develop and commercialize products that are effective and commercially viable, to obtain regulatory approval for the manufacture and sale of our products and to license or otherwise market our products successfully. We may never achieve profitability, and even if we do, we may not be able to sustain being profitable.
If we are unable to obtain required regulatory approvals, we will be unable to market and sell our product candidates.
Our product candidates are subject to extensive governmental regulations relating to development, clinical trials, manufacturing, oversight of clinical investigators, recordkeeping and commercialization. Rigorous preclinical testing and clinical trials and an extensive regulatory review and approval process are required to be successfully completed in the United States and in each foreign jurisdiction in which we offer our products before a new drug or other product can be sold in such jurisdictions. Satisfaction of these and other regulatory requirements is costly, time consuming, uncertain, and subject to unanticipated delays. The time required to obtain approval by the FDA, or the regulatory authority in such other jurisdictions is unpredictable and often exceeds five years following the commencement of clinical trials, depending upon the complexity of the product candidate and the requirements of the applicable regulatory agency.
In connection with the clinical development of our product candidates, we face risks that:
| • | the product candidate may not prove to be safe and efficacious; |
16
Table of Contents
| • | patients may die or suffer serious adverse effects for reasons that may or may not be related to the product candidate being tested; |
| • | we may fail to maintain adequate records of observations and data from our clinical trials, to establish and maintain sufficient procedures to oversee, collect data from, and manage clinical trials, or to monitor clinical trial sites and investigators to the satisfaction of the FDA or other regulatory agencies; |
| • | the results of later-phase clinical trials may not confirm the results of earlier clinical trials; and |
| • | the results from clinical trials may not meet the level of statistical significance or clinical benefit-to-risk ratio required by the FDA or other regulatory agencies for marketing approval. |
Only a small percentage of product candidates for which clinical trials are initiated receive approval for commercialization. Furthermore, even if we do receive regulatory approval to market a product candidate, any such approval may be subject to limitations such as those on the indicated uses for which we may market a particular product candidate.
Our product candidates have not completed clinical trials, and may never demonstrate sufficient safety and efficacy in order to do so.
Our product candidates are in the clinical and pre-clinical stages of development. In order to achieve profitable operations, we alone, or in collaboration with others, must successfully develop, manufacture, introduce and market our products. The time frame necessary to achieve market success for any individual product is long and uncertain. The products we are currently developing will require significant additional research, development and preclinical and clinical testing prior to application for commercial use or sale. A number of companies in the biotechnology and pharmaceutical industries have suffered significant setbacks in clinical trials, even after showing promising results in early or later-stage studies or clinical trials. Although we have obtained some favorable results to-date in preclinical studies and clinical trials of certain of our potential products, such results may not be indicative of results that will ultimately be obtained in or throughout such clinical trials, and clinical trials may not show any of our products to be safe or capable of producing a desired result. Additionally, we may encounter problems in our clinical trials that may cause us to delay, suspend or terminate those clinical trials.
Further, our research or product development efforts may not be successfully completed, any compounds we currently have under development may not be successfully developed into drugs, may not receive regulatory approval on a timely basis, if at all, and competitors may develop and bring to market products or technologies that render our potential products obsolete. If any of these events occur, our business would be materially and adversely affected.
If clinical trials or regulatory approval processes for our product candidates are prolonged, delayed or suspended, we may be unable to commercialize our product candidates on a timely basis, which would require us to incur additional costs and delay our receipt of any revenue from potential product sales.
We cannot predict whether we will encounter problems with any of our completed, ongoing or planned clinical trials that will cause us or any regulatory authority to delay or suspend those clinical trials or delay the analysis of data derived from them. A number of events, including any of the following, could delay the completion of our ongoing and planned clinical trials and negatively impact our ability to obtain regulatory approval for, and to market and sell, a particular product candidate:
| • | conditions imposed on us by the FDA or another foreign regulatory authority regarding the scope or design of our clinical trials; |
| • | delays in obtaining, or our inability to obtain, required approvals from institutional review boards or other reviewing entities at clinical sites selected for participation in our clinical trials; |
| • | insufficient supply of our product candidates or other materials necessary to conduct and complete our clinical trials; |
| • | slow enrollment and retention rate of subjects in our clinical trials; |
| • | serious and unexpected drug-related side effects related to the product candidate being tested; and |
| • | delays in meeting manufacturing and testing standards required for production of clinical trial supplies. |
Commercialization of our product candidates may be delayed by the imposition of additional conditions on our clinical trials by the FDA or any other applicable foreign regulatory authority or the requirement of additional supportive studies by the FDA or such foreign regulatory authority. In addition, clinical trials require sufficient patient enrollment, which is a function of many factors, including the size of the patient population, the nature of the trial protocol, the proximity of patients to clinical sites, the
17
Table of Contents
availability of effective treatments for the relevant disease, the conduct of other clinical trials that compete for the same patients as our clinical trials, and the eligibility criteria for our clinical trials. Our failure to enroll patients in our clinical trials could delay the completion of the clinical trial beyond its expectations. In addition, the FDA could require us to conduct clinical trials with a larger number of subjects than we may have projected for any of our product candidates. We may not be able to enroll a sufficient number of patients in a timely or cost-effective manner. Furthermore, enrolled patients may drop out of our clinical trials, which could impair the validity or statistical significance of the clinical trials.
We do not know whether our clinical trials will begin as planned, will need to be restructured, or will be completed on schedule, if at all. Delays in our clinical trials will result in increased development costs for our product candidates, and our financial resources may be insufficient to fund any incremental costs. In addition, if our clinical trials are delayed, our competitors may be able to bring products to market before we do and the commercial viability of our product candidates could be limited.
Our product candidates will remain subject to ongoing regulatory review even if they receive marketing approval, and if we fail to comply with continuing regulations, we could lose these approvals and the sale of any of our approved commercial products could be suspended.
Even if we receive regulatory approval to market a particular product candidate, the manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion, and record keeping related to the product will remain subject to extensive regulatory requirements. If we fail to comply with the regulatory requirements of the FDA and other applicable domestic and foreign regulatory authorities or discover any previously unknown problems with any approved product, manufacturer, or manufacturing process, we could be subject to administrative or judicially imposed sanctions, including:
| • | restrictions on the products, manufacturers, or manufacturing processes; |
| • | warning letters; |
| • | civil or criminal penalties; |
| • | fines; |
| • | injunctions; |
| • | product seizures or detentions; |
| • | pressure to initiate voluntary product recalls; |
| • | suspension or withdrawal of regulatory approvals; and |
| • | refusal to approve pending applications for marketing approval of new products or supplements to approved applications. |
Our industry is highly competitive, and our product candidates may become obsolete.
We are engaged in a rapidly evolving field. Competition from other pharmaceutical companies, biotechnology companies and research and academic institutions is intense and likely to increase. Many of those companies and institutions have substantially greater financial, technical and human resources than we do. Those companies and institutions also have substantially greater experience in developing products, conducting clinical trials, obtaining regulatory approval and in manufacturing and marketing pharmaceutical products. Our competitors may succeed in obtaining regulatory approval for their products more rapidly than we do. Competitors have developed or are in the process of developing technologies that are, or in the future may be, the basis for competitive products. We are aware of potential competitors developing products similar to our sarcoma vaccine, ovarian cancer vaccine and pancreatic cancer antibodies product candidates. Our competitors may succeed in developing products that are more effective and/or cost competitive than those we are developing, or that would render our product candidates less competitive or even obsolete. In addition, one or more of our competitors may achieve product commercialization or patent protection earlier than we do, which could materially adversely affect our business.
If physicians and patients do not accept our future products or if the market for indications for which any product candidate is approved is smaller than expected, we may be unable to generate significant revenue, if any.
Even if any of our product candidates obtain regulatory approval, they may not gain market acceptance among physicians, patients, and third-party payers. Physicians may decide not to recommend our treatments for a variety of reasons including:
| • | timing of market introduction of competitive products; |
| • | demonstration of clinical safety and efficacy compared to other products; |
18
Table of Contents
| • | cost-effectiveness; |
| • | limited or no coverage by third-party payers; |
| • | convenience and ease of administration; |
| • | prevalence and severity of adverse side effects; |
| • | restrictions in the label of the drug; |
| • | other potential advantages of alternative treatment methods; and |
| • | ineffective marketing and distribution support of its products. |
If any of our product candidates are approved, but fail to achieve market acceptance or such market is smaller than anticipated, we may not be able to generate significant revenue and our business would suffer.
As we evolve from a company that is primarily involved in clinical development to a company that is also involved in commercialization, we may encounter difficulties in expanding our operations successfully.
As we advance our product candidates through clinical trials, we will need to expand our development, regulatory, manufacturing, marketing and sales capabilities and may need to further contract with third parties to provide these capabilities. As our operations expand, we likely will need to manage additional relationships with such third parties, as well as additional collaborators, distributors, marketers and suppliers.
Maintaining third party relationships for these purposes will impose significant added responsibilities on members of our management and other personnel. We must be able to: manage our development efforts effectively; recruit and train sales and marketing personnel; manage our participation in the clinical trials in which our product candidates are involved effectively; and improve our managerial, development, operational and finance systems, all of which may impose a strain on our administrative and operational infrastructure.
If we enter into arrangements with third parties to perform sales, marketing or distribution services, any product revenues that we receive, or the profitability of these product revenues to us, are likely to be lower than if we were to market and sell any products that we develop without the involvement of these third parties. In addition, we may not be successful in entering into arrangements with third parties to sell and market our products or in doing so on terms that are favorable to us. We likely will have little control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively. If we do not establish sales and marketing capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing our products.
The uncertainty associated with pharmaceutical reimbursement and related matters may adversely affect our business.
Market acceptance and sales of any one or more of our product candidates will depend on reimbursement policies and may be affected by future healthcare reform measures in the United States and in foreign jurisdictions. Government authorities and third-party payers, such as private health insurers and health maintenance organizations, decide which drugs they will cover and establish payment levels. We cannot be certain that reimbursement will be available for any of our product candidates. Also, we cannot be certain that reimbursement policies will not reduce the demand for, or the price paid for, our products. If reimbursement is not available or is available on a limited basis, we may not be able to successfully commercialize any product candidates that we develop.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, also called the Medicare Modernization Act, or MMA, changed the way Medicare covers and pays for pharmaceutical products. The legislation established Medicare Part D, which expanded Medicare coverage for outpatient prescription drug purchases by the elderly but provided authority for limiting the number of drugs that will be covered in any therapeutic class. The MMA also introduced a new reimbursement methodology based on average sales prices for physician-administered drugs.
The United States and several foreign jurisdictions are considering, or have already enacted, a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. Among policy makers and payers in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access to healthcare. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives. We expect to experience pricing pressures in connection with the sale of any products that it develops due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative proposals.
19
Table of Contents
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively, ACA, became law in the U.S. The goal of ACA is to reduce the cost of health care and substantially change the way health care is financed by both government and private insurers. While we cannot predict what impact on federal reimbursement policies this legislation will have in general or on our business specifically, the ACA may result in downward pressure on pharmaceutical reimbursement, which could negatively affect market acceptance of, and the price we charge for, any products we develop that receive regulatory approval. We also cannot predict the impact of ACA on our business, as many of the ACA reforms require the promulgation of detailed regulations implementing the statutory provisions, which have not yet been fully promulgated and implemented.
We only have a limited number of employees to manage and operate our business.
As of March 31, 2015, we had a total of 12 full-time employees and 2 part-time employees. Our focus on limiting cash utilization requires us to manage and operate our business in a highly efficient manner. We cannot assure you that we will be able to retain adequate staffing levels to run our operations and/or to accomplish all of the objectives that we otherwise would seek to accomplish.
We depend heavily on our executive officers, directors, and principal consultants and the loss of their services would materially harm our business.
We believe that our success depends, and will likely continue to depend, upon our ability to retain the services of our current executive officers, directors, principal consultants and others. In addition, we have established relationships with universities, hospitals and research institutions, which have historically provided, and continue to provide, us with access to research laboratories, clinical trials, facilities and patients. The loss of the services of any of these individuals or institutions would have a material adverse effect on our business.
Due in part to our limited financial resources, we may fail to select or capitalize on the most scientifically, clinically or commercially promising or profitable indications or therapeutic areas for our product candidates or those that are in-licensed, and/or we may be unable to pursue the clinical trials that we would like to pursue.
We have limited technical, managerial and financial resources to determine the indications on which we should focus the development efforts related to our product candidates. Due to our limited available financial resources, we may have curtailed clinical development programs and activities that might otherwise have led to more rapid progress of our product candidates through the regulatory and development processes.
We may make incorrect determinations with regard to the indications and clinical trials on which to focus the available resources that we do have. Furthermore, we cannot assure you that we will be able to retain adequate staffing levels to run our operations and/or to accomplish all of the objectives that we otherwise would seek to accomplish. Our decisions to allocate our research, management and financial resources toward particular indications or therapeutic areas for our product candidates may not lead to the development of viable commercial products and may divert resources from better opportunities. Similarly, our decisions to delay or terminate drug development programs may also cause us to miss valuable opportunities.
If the third parties on which we rely for the conduct of our clinical trials and results do not perform our clinical trial activities in accordance with good clinical practices and related regulatory requirements, we may be unable to obtain regulatory approval for or commercialize our product candidates.
We use independent clinical investigators and other third-party service providers to conduct and/or oversee the clinical trials of our product candidates and expect to continue to do so for the foreseeable future. We rely heavily on these parties for successful execution of our clinical trials. Nonetheless, we are responsible for confirming that each of our clinical trials is conducted in accordance with the FDA’s requirements and our general investigational plan and protocol.
The FDA requires us and our clinical investigators to comply with regulations and standards, commonly referred to as good clinical practices, for conducting and recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the trial participants are adequately protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements. Third parties may not complete activities on schedule or may not conduct our clinical trials in accordance with regulatory requirements or the respective trial plans and protocols. The failure of these third parties to carry out their obligations could delay or prevent the development, approval and commercialization of our product candidates or result in enforcement action against us.
20
Table of Contents
We have limited manufacturing capacity and have relied on, and expect to continue to rely on, third-party manufacturers to produce our product candidates.
We do not own or operate manufacturing facilities for the production of clinical or commercial quantities of our product candidates, and we lack the resources and the capabilities to do so. As a result, we currently rely, and expect to rely for the foreseeable future, on third-party manufacturers to supply our product candidates. Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured our product candidates or products ourselves, including:
| • | reliance on third-parties for manufacturing process development, regulatory compliance and quality assurance; |
| • | limitations on supply availability resulting from capacity and scheduling constraints of third-parties; |
| • | the possible breach of manufacturing agreements by third-parties because of factors beyond our control; and |
| • | the possible termination or non-renewal of the manufacturing agreements by the third-party, at a time that is costly or inconvenient to us. |
If we do not maintain our key manufacturing relationships, we may fail to find replacement manufacturers or develop our own manufacturing capabilities, which could delay or impair our ability to obtain regulatory approval for our products and substantially increases our costs or deplete profit margins, if any. If we do find replacement manufacturers, we may not be able to enter into agreements with them on terms and conditions favorable to us and there could be a substantial delay before new facilities could be qualified and registered with the FDA and other foreign regulatory authorities.
The FDA and other foreign regulatory authorities require manufacturers to register manufacturing facilities. The FDA and corresponding foreign regulators also inspect these facilities to confirm compliance with current cGMPs. Contract manufacturers may face manufacturing or quality control problems causing drug substance production and shipment delays or a situation where the contractor may not be able to maintain compliance with the applicable cGMP requirements. Any failure to comply with cGMP requirements or other FDA, EMA and comparable foreign regulatory requirements could adversely affect our clinical research activities and our ability to develop our product candidates and market our products following approval.
Our current and anticipated future dependence upon others for the manufacture of our product candidates may adversely affect our future profit margins and our ability to develop our product candidates and commercialize any products that receive regulatory approval on a timely basis.
We depend extensively on our patents and proprietary technology and the patents and proprietary technology we license from others, and we must protect those assets in order to preserve our business.
Although we expect to seek patent protection for any compounds we discover and/or for any specific use we discover for new or previously known compounds, any or all of such compounds or new uses may not be subject to effective patent protection. Further, the development of regimens for the administration of our vaccines, which involve specifications for the frequency, timing and amount of dosages, has been, and we believe may continue to be, important to our efforts, although those processes, as such, may not be patentable. In addition, our issued patents may be declared invalid or our competitors may find ways to avoid the claims in the patents.
Our success will depend, in part, on our ability to obtain patents, protect our trade secrets and operate without infringing on the proprietary rights of others. As of February 6, 2015, we were the exclusive licensee, sole assignee or co-assignee of 20 granted United States patents, 16 pending United States patent applications and 41 international patents through MabVax Therapeutics, and 21granted United States patents, 12 pending United States patent applications and 99 international patents obtained and 88 international patent applications are being maintained by the Company from prior to the Merger while we continue to evaluate the usefulness to the Company. The patent position of pharmaceutical and biotechnology firms like us are generally highly uncertain and involves complex legal and factual questions, resulting in both an apparent inconsistency regarding the breadth of claims allowed in United States patents and general uncertainty as to their legal interpretation and enforceability. Accordingly, patent applications assigned or exclusively licensed to us may not result in patents being issued, any issued patents assigned or exclusively licensed to us may not provide us with competitive protection or may be challenged by others, and the current or future granted patents of others may have an adverse effect on our ability to do business and achieve profitability. Moreover, because some of the basic research relating to one or more of our patent applications and/or patents were performed at various universities and/or funded by grants, one or more universities, employees of such universities and/or grantors could assert that they have certain rights in such research and any resulting products. Further, others may independently develop similar products, may
21
Table of Contents
duplicate our products, or may design around our patent rights. In addition, as a result of the assertion of rights by a third-party or otherwise, we may be required to obtain licenses to patents or other proprietary rights of others in or outside of the United States. Any licenses required under any such patents or proprietary rights may not be made available on terms acceptable to us, if at all. If we do not obtain such licenses, we could encounter delays in product market introductions during our attempts to design around such patents or could find that the development, manufacture or sale of products requiring such licenses is foreclosed. In addition, we could incur substantial costs in defending suits brought against us or in connection with patents to which we hold licenses or in bringing suit to protect our own patents against infringement.
We require employees and the institutions that perform our preclinical and clinical trials to enter into confidentiality agreements with us. Those agreements provide that all confidential information developed or made known to a party to any such agreement during the course of the relationship with us be kept confidential and not be disclosed to third-parties, except in specific circumstances. Any such agreement may not provide meaningful protection for our trade secrets or other confidential information in the event of unauthorized use or disclosure of such information.
With respect to our vaccine programs we have licensed in rights to antibody programs and other programs from third parties. If these license agreements terminate or expire, we may lose the licensed rights to some or all of our product candidates. We may not be able to continue to develop them or, if they are approved, market or commercialize them.
We depend on license agreements with third-parties for certain intellectual property rights relating to our product candidates, including, but not limited to, the license of certain intellectual property rights from MSKCC. In general, our license agreements require us to make payments and satisfy performance obligations in order to keep these agreements in effect and retain our rights under them. These payment obligations can include upfront fees, maintenance fees, milestones, royalties, patent prosecution expenses, and other fees. These performance obligations typically include diligence obligations. If we fail to pay, be diligent or otherwise perform as required under our license agreements, we could lose the rights under the patents and other intellectual property rights covered by these agreements. If disputes arise under any of our in-licenses, including our in-licenses from MSKCC, we could lose our rights under these agreements. Any such dispute may not be resolvable on favorable terms, or at all. Whether or not any disputes of this kind are favorably resolved, our management’s time and attention and our other resources could be consumed by the need to attend to these disputes and our business could be harmed by the emergence of such a dispute.
If we lose our rights under these agreements, we might not be able to develop any related product candidates further, or following regulatory approval, if any, we might be prohibited from marketing or commercializing these product candidates. In particular, patents previously licensed to us might, after termination of an agreement, be used to stop us from conducting these activities.
If product liability lawsuits are successfully brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates and any products that we may develop.
The testing and marketing of medical products entail an inherent risk of product liability. Although we are not aware of any historical or anticipated product liability claims or specific causes for concern, if we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates and any products that we may develop. In addition, product liability claims may also result in withdrawal of clinical trial volunteers, injury to our reputation and decreased demand for any products that we may commercialize. We currently carry product liability insurance that covers our clinical trials up to a $10.0 million annual aggregate limit. We will need to increase the amount of coverage if and when we have a product that is commercially available. If we are unable to obtain sufficient product liability insurance at an acceptable cost, potential product liability claims could prevent or inhibit the commercialization of any products that we may develop, alone or with corporate partners.
Our restated certificate of incorporation, our amended and restated by-laws and Delaware law could deter a change of our management which could discourage or delay offers to acquire us.
Certain provisions of Delaware law and of our restated certificate of incorporation, as amended, and amended and restated by-laws, could discourage or make it more difficult to accomplish a proxy contest or other change in our management or the acquisition of control by a holder of a substantial amount of our voting stock. It is possible that these provisions could make it more difficult to accomplish, or could deter, transactions that stockholders may otherwise consider to be in their best interests or in our best interests. These provisions include:
| • | establishing a classified board of directors requiring that members of the board be elected in different years, which lengthens the time needed to elect a new majority of the board; |
| • | authorizing the issuance of “blank check” preferred stock that could be issued by our board of directors to increase the number of outstanding shares or change the balance of voting control and thwart a takeover attempt; |
22
Table of Contents
| • | prohibiting cumulative voting in the election of directors, which would otherwise allow for less than a majority of stockholders to elect director candidates; |
| • | limiting the ability of stockholders to call special meetings of the stockholders; |
| • | prohibiting stockholder action by written consent and requiring all stockholder actions to be taken at a meeting of our stockholders; and |
| • | establishing 90 to 120 day advance notice requirements for nominations for election to the board of directors and for proposing matters that can be acted upon by stockholders at stockholder meetings. |
The price of our common stock is volatile, and is likely to continue to fluctuate due to reasons beyond our control.
The market price of our common stock has been, and likely will continue to be, highly volatile. Factors, including our financial results or our competitors’ financial results, clinical trial and research development announcements and government regulatory action affecting our potential products in both the United States and foreign countries, have had, and may continue to have, a significant effect on our results of operations and on the market price of our common stock. We cannot assure you that any investment in our common stock will not fluctuate significantly. One or more of these factors could significantly harm our business and cause a decline in the price of our common stock in the public market. Sales of shares of common stock registered for resale or eligible for resale pursuant to Rule 144 under the Securities Act as amended, as well as future sales of our common stock by existing stockholders, or the perception that sales may occur at any time, could adversely affect the market price of our common stock.
Substantial future sales of our common stock by us or by our existing stockholders could cause our stock price to fall.
Additional equity financings or other share issuances by us, including shares issued in connection with strategic alliances and corporate partnering transactions, could adversely affect the market price of our common stock. Sales by existing stockholders of a large number of shares of our common stock in the public market or the perception that additional sales could occur could cause the market price of our common stock to drop. Substantially all of our outstanding shares of common stock were freely tradable and, in limited cases, subject to certain volume, notice and manner of sale restrictions under Rule 144 of the Securities Act of 1933.
If we do not progress in our programs as anticipated, our stock price could decrease.
For planning purposes, we estimate the timing of a variety of clinical, regulatory and other milestones, such as when a certain product candidate will enter clinical development, when a clinical trial will be completed or when an application for regulatory approval will be filed. Our estimates are based on present facts and a variety of assumptions. Many of the underlying assumptions are outside of our control. If milestones are not achieved when we estimated that they would be, investors could be disappointed, and our stock price may decrease.
Our stock price may be volatile, you may not be able to resell your shares at or above your purchase price.
Our stock prices and the market prices for securities of biotechnology companies in general have been highly volatile, with recent significant price and volume fluctuations, and may continue to be highly volatile in the future. For example, during the year ended December 31, 2014, our common stock traded between $1.51 per share and $16.48 per share. The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of our common stock, some of which are beyond our control:
| • | developments regarding, or the results of, our clinical trials; |
| • | announcements of technological innovations or new commercial products by our competitors or us; |
| • | our issuance of equity or debt securities, or disclosure or announcements relating thereto; |
| • | developments concerning proprietary rights, including patents; |
| • | developments concerning our collaborations; |
| • | publicity regarding actual or potential medical results relating to products under development by our competitors or us; |
| • | regulatory developments in the United States and foreign countries; |
23
Table of Contents
| • | litigation; |
| • | economic and other external factors or other disaster or crisis; or |
| • | period-to-period fluctuations in our financial results. |
We have been, and in the future may be, subject to securities class action lawsuits and shareholder derivative actions. These, and potential similar or related litigation, could result in substantial damages and may divert management’s time and attention from our business.
We have been, and may in the future be, the target of securities class actions or shareholder derivative claims. Any such actions or claims could result in substantial damages and may divert management’s time and attention from our business.
The rights of our common stockholders are limited by and subordinate to the rights of the holders of Series A-1 Preferred Stock and Series B Preferred Stock; these rights may have a negative effect on the value of shares of our common stock.
The holders of the Series A-1 Preferred Stock and Series B Preferred Stock have rights and preferences generally superior to those of the holders of common stock. The existence of these superior rights and preferences may have a negative effect on the value of shares of our common stock. These rights are more fully set forth in the Series A-1 certificate of designations and Series B certificate of designations, respectively, and include, but are not limited to:
| • | the right to receive a liquidation preference, prior to any distribution of our assets to the holders of our common stock, in an amount equal to $ 1.676708000 per share for the Series A-1 Preferred Stock and $2.00 per share for the Series B Preferred Stock, as of December 31, 2014, subject to adjustments, and all accrued and unpaid dividends; |
| • | the right to convert into shares of our common stock at the conversion price set forth in the Series A-1 certificate of designations and Series B certificate of designations, respectively, which may be adjusted as set forth therein; and |
| • | the right to receive dividends in arrears at a rate of 8% per annum in preference to the holders of our common stock and to receive dividends made to holders of our common stock on an as converted basis. |
The holders of Series A-1 Preferred Stock and Series B Preferred Stock have the right to block certain fundamental transactions as further described in the Series A-1 certificate of designations and Series B certificate of designations, respectively; these holders may use this right to negotiate terms more favorable to the holders of the Series A-1 Preferred Stock and Series B Preferred Stock to the detriment of the holders of other classes of our capital stock or may prevent us from completing transactions favorable to us and the holders of other classes of our capital stock.
As further set forth in the Series A-1 certificate of designations, the holders of a majority of the issued and outstanding Series A-1 convertible preferred stock, Series B convertible preferred stock or Hudson Bay Opportunities Fund LP and/or its affiliates, or Hudson Bay, have the right to approve or block certain transactions, including transactions that:
| • | create or issue additional or other capital stock or securities exchangeable for or convertible or exercisable into capital stock pari passu with or senior to the Series A-1 convertible preferred stock or Series B convertible preferred stock; |
| • | reclassify, alter or amend any of our existing securities that are pari passu with the Series A-1 Preferred Stock or Series B Preferred Stock; |
| • | change the authorized number of shares of our capital stock; |
| • | create or issue debt securities; |
| • | authorize or effect payment of dividends or distributions on our capital stock; |
| • | authorize or effect change of control, dissolution or liquidation events; |
| • | amend or repeal our certificate of incorporation or bylaws; |
| • | amend, alter or repeal preferences, special rights or other powers of the Series A-1 convertible preferred stock or Series B convertible preferred stock; |
| • | avoid the observance or performance of the terms of the Series A-1 or Series B certificates of designations; and |
| • | effect any change in our principal business. |
24
Table of Contents
Hudson Bay and/or the Series A-1 convertible preferred stockholders and/or the Series B convertible preferred stockholders may have interests differing from or detrimental to other holders of other classes of our capital stock with respect to these fundamental transactions. Obtaining the consent of these holders may delay or limit our ability to enter into transactions that may be beneficial to the holders of other classes of our capital stock.
A limited public trading market may cause volatility in the price of our common stock.
Our common stock is currently quoted on the OTCQB marketplace. The quotation of our common stock on the OTCQB marketplace does not assure that a meaningful, consistent and liquid trading market currently exists, and in recent years such market has experienced extreme price and volume fluctuations that have particularly affected the market prices of many smaller companies like us. Our common stock is subject to this volatility. Sales of substantial amounts of common stock, or the perception that such sales might occur, could adversely affect prevailing market prices of our common stock and our stock price may decline substantially in a short time and our stockholders could suffer losses or be unable to liquidate their holdings. Because our common stock does not trade on a national securities exchange, our common stock is subject to the securities laws of the various states and jurisdictions of the United States in addition to federal securities law. While we may register our common stock or qualify for exemptions for our common stock in one of more states, if we fail to do so the investors in those states where we have not taken such steps may not be allowed to purchase our stock or those who presently hold our stock may not be able to resell their shares without substantial effort and expense. These restrictions and potential costs could be significant burdens on our stockholders.
The floating conversion price for our Series B Preferred Stock may lead to significant shareholder dilution and a corresponding drop in the market price of our common stock.
As described above and in the Series B certificate of designations, our Series B Preferred Stock is convertible into our common stock at a “floating” conversion price. Following the Adjustment Date (as defined in the Series B certificate of designations), this conversion price adjusts according to the market price of our common stock. If the market price of our common stock declines, shares of Series B Preferred Stock will be convertible into a greater number of shares of common stock, which could have the effect of diluting the ownership interest of all other holders of our common stock. If the market price of our common stock were to decline significantly, this dilution could be substantial. Furthermore, any such dilution may cause the market price of the common stock to decline further, resulting in additional dilution and a continued potential adverse effect on the common stock price thereafter and could result in an imbalance of supply and demand for our common stock and reduce its price. This series of events could lead to a repetitive cycle, further and successively driving the market price of our common stock downward. The further the market price of our common stock declines, the further the floating conversion price will fall and the greater the number of shares we will have to issue upon conversion. In addition to affecting the market price of our common stock and diluting the value of each share of common stock for existing shareholders, the floating conversion price and the effects that it may have may also make it more difficult for us to raise capital in the future, which could adversely affect our ability to operate or grow our business.
We may not be able to achieve secondary trading of our stock in certain states because our common stock is no longer nationally traded, which could subject our stockholders to significant restrictions and costs.
Our common stock is not currently eligible for trading on the NASDAQ Capital Market or on a national securities exchange. Therefore, our common stock is subject to the securities laws of the various states and jurisdictions of the United States in addition to federal securities law. While we may register our common stock or qualify for exemptions for our common stock in one of more states, if we fail to do so the investors in those states where we have not taken such steps may not be allowed to purchase our stock or those who presently hold our stock may not be able to resell their shares without substantial effort and expense. These restrictions and potential costs could be significant burdens on our stockholders.
Our management and our independent registered public accounting firm identified certain material weaknesses in our internal controls over financial reporting upon completion of our audit in May of 2014 that were not fully remediated as of December 31, 2014. While we have implemented certain internal controls to eliminate material weaknesses, if we are unable to maintain effective internal controls, we may not be able to produce timely and accurate financial statements, and our independent registered public accounting firm could conclude that our internal controls over financial reporting are not effective, which could adversely impact investor confidence and our stock price.
In connection with the audit of MabVax Therapeutics’ financial statements as of and for the year ended December 31, 2013, MabVax Therapeutics management and its independent registered public accounting firm identified material weaknesses in its internal control over financial reporting relating to the reporting of non-routine complex transactions and the lack of segregation of
25
Table of Contents
duties. These material weaknesses were primarily the result of a limited number of employees in the accounting department at MabVax Therapeutics. In June 2014, MabVax Therapeutics added an assistant controller, a person dedicated solely to processing accounts payable, and another person dedicated to reviewing and reporting on clinical trials progress and expenses. These persons have continued to work in these capacities following the Merger. On March 25, 2015 we filed with the SEC an amended quarterly report for the quarter ended September 30, 2014, to correct certain errors in our financial statements for that period, namely an error in accounting for employee severance expense relative to the July 2014 Merger. Additional errors may occur if we are not able to eliminate the material weaknesses in our internal controls over financial reporting. Our management is responsible for maintaining, implementing and testing our internal controls over financial reporting. These efforts are intended to maintain an effective control environment but may not be sufficient to remediate any material weaknesses identified by our independent registered accounting firm and may not prevent significant deficiencies from occurring.
A material weakness is a deficiency, or combination of deficiencies, such that there is reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis by our employees. A control system, no matter how well designed and operated, can provide only reasonable assurance that the control system’s objectives will be met. There are inherent limitations in all control systems and no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and all instances of fraud will be detected. If management identifies future material weaknesses in our internal controls over financial reporting, if we are unable to comply with the requirements of Section 404 of the Sarbanes-Oxley Act in a timely manner, if we are unable to assert that our internal controls over financial reporting are effective, or if our independent registered public accounting firm is unable to express an opinion as to the effectiveness of our internal control over financial reporting, if and when required, investors may lose confidence in the accuracy and completeness of our financial reports and the value of our capital stock could be negatively affected, and our could become subject to criminal and civil investigations by the stock exchange or marketplace on which our securities are then listed, the SEC or other regulatory authorities.
| Item 1B. | Unresolved Staff Comments. |
None
| Item 2. | Properties. |
MabVax Therapeutics entered into a lease agreement in August 2012, with a lease term ending on July 31, 2015, for 5,955 square feet of office space at 11588 Sorrento Valley Road in San Diego, California. Following the Merger, we consolidated all corporate offices to this San Diego location. We believe that our existing facilities are adequate for our current and projected needs in the future.
| Item 3. | Legal Proceedings. |
On May 30, 2014, a putative class action complaint, or the complaint, was filed in Santa Clara County Superior Court, State of California, captioned Cadillac Partners, on Behalf of Itself and All Others Similarly Situated, v. Michael M. Wick, et al., or the litigation. The suit alleged the defendants breached certain fiduciary duties, or aided and abetted a breach of fiduciary duties, in connection with the Company’s Merger with MabVax Therapeutics. The complaint asserts claims concerning the private placement of our Series B Preferred Stock and transactions contemplated by the Merger Agreement, each of which were entered into on May 12, 2014, and is brought against MabVax Holdings, MabVax Therapeutics, past and current members of our board, and the investors participating in the private placement transaction.
On July 16, 2014, MabVax Holdings and all other parties to the litigation entered into an agreement which, if consummated, will settle the litigation, or the proposed settlement. Among many other terms, under the proposed settlement we agreed to provide the supplemental disclosure filed as definitive additional materials to the definitive proxy on June 30, 2014, and MabVax Holdings, MabVax Therapeutics and all defendants will receive a broad release of any and all claims pertaining to the Series B Preferred Stock private placement, the Merger, the prior disclosure and a wide variety of other matters. The proposed settlement also calls for the parties to ask the court to, among other things, enter orders enjoining other shareholders from bringing similar actions, certifying the putative settlement class, and approving the proposed settlement as a fair, final, and binding resolution of the litigation. Under the proposed settlement, MabVax Holdings, MabVax Therapeutics and the other defendants have expressly denied the allegations of the complaint and denied engaging in any other misconduct, nor will any of them make any payment or in any respect amend the negotiated terms of the since-consummated Series B Preferred Stock private placement and Merger. Finally, under the proposed settlement, MabVax Holdings, MabVax Therapeutics and the other defendants have not agreed to pay any legal fees, or reimburse any expenses, allegedly incurred by the plaintiffs who filed the complaint; instead, MabVax Holdings and MabVax Therapeutics expect that counsel for those plaintiffs will present any such disputed claim for legal fees and expenses to the court for resolution.
The proposed settlement remains contingent upon a number of future events, including, without limitation, court certification of the putative class and entry of a final, non-appealable order and final judgment approving the settlement (including the broad releases and other terms set forth therein).
26
Table of Contents
| Item 4. | Mine Safety Disclosures. |
Not applicable.
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. |
Our common stock trades on the OTCQB under the symbol “MBVX”. The following table sets forth the high and low sales prices for our common stock for each quarterly period within the two most recent fiscal years. All stock prices included in the following table are adjusted for the 1 for 8 reverse stock split on September 8, 2014.
| High | Low | |||||||
| 2014 |
||||||||
| Quarter ended March 31, 2014 |
$ | 15.20 | $ | 9.52 | ||||
| Quarter ended June 30, 2014 |
$ | 16.48 | $ | 9.68 | ||||
| Quarter ended September 30, 2014 |
$ | 15.00 | $ | 5.00 | ||||
| Quarter ended December 31, 2014 |
$ | 6.70 | $ | 1.51 | ||||
| 2013 |
||||||||
| Quarter ended March 31, 2013 |
$ | 24.40 | $ | 10.48 | ||||
| Quarter ended June 30, 2013 |
$ | 14.00 | $ | 9.36 | ||||
| Quarter ended September 30, 2013 |
$ | 13.04 | $ | 8.40 | ||||
| Quarter ended December 31, 2013 |
$ | 17.20 | $ | 9.36 | ||||
Holders
As of February 24, 2015, there were 88 stockholders of record of our common stock, one of which is Cede & Co., a nominee for Depository Trust Company, or DTC. Shares of common stock that are held by financial institutions as nominees for beneficial owners are deposited into participant accounts at DTC, and are considered to be held of record by Cede & Co. as one stockholder.
Dividends
We have never paid our stockholders cash dividends, and we do not anticipate paying any cash dividends in the foreseeable future as we intend to retain any earnings for use in our business. Any future determination to pay dividends will be at the discretion of our board of directors.
Securities Authorized for Issuance under Equity Compensation Plans
Information relating to compensation plans under which our equity securities are authorized for issuance is presented in Part III, Item 12 of this Form 10-K.
27
Table of Contents
Performance Graph
The following graph compares our cumulative total stockholder return for the past five years to two indices: the Nasdaq U.S. Index and the Nasdaq Pharmaceutical Stocks Index. This graph assumes the investment of $100 on December 31, 2009 in our common stock, the Nasdaq U.S. Index; and the Nasdaq Pharmaceutical Stocks Index. All values assume reinvestment of the full amount of all dividends and are calculated as of the last stock trading day of each year:
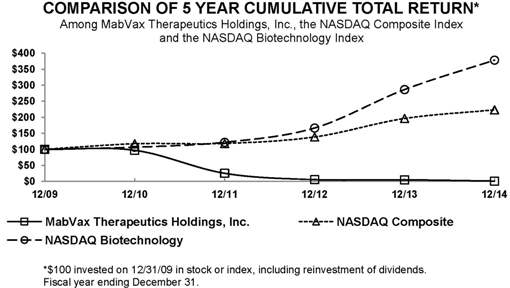
Source: Research Data Group. The information under “Performance Graph” is not deemed to be “soliciting material” or “filed” with the Securities and Exchange Commission or subject to Regulation 14A or 14C, or to the liabilities of Section 18 of the Exchange Act of 1934, as amended and is not to be incorporated by reference in any filing of MabVax under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date of this Annual Report on Form 10-K and irrespective of any general incorporation language in those filings.
| Item 6. | Selected Financial Data. |
The information under this Item is not required to be provided by smaller reporting companies.
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations. |
You should read the following discussion and analysis in conjunction with “Item 8. Financial Statements and Supplementary Data” included below in this Annual Report on Form 10-K, or Annual Report. Operating results are not necessarily indicative of results that may occur in future periods.
This discussion and analysis contains forward-looking statements that involve a number of risks, uncertainties and assumptions. Actual events or results may differ materially from our expectations. Important factors that could cause actual results to differ materially from those stated or implied by our forward-looking statements include, but are not limited to, those set forth in “Item 1A. Risk Factors” in this Annual Report. All forward-looking statements included in this Annual Report are based on information available to us as of the time we file this Annual Report and, except as required by law, we undertake no obligation to update publicly or revise any forward-looking statements.
Overview
We have been engaged in the discovery, development and commercialization of proprietary human monoclonal antibody products and vaccines for the diagnosis and treatment of a variety of cancers. We have discovered a pipeline of human monoclonal antibody products based on the protective immune responses generated by patients who have been immunized against targeted cancers. Therapeutic vaccines under development were discovered at Memorial Sloan Kettering Cancer Center, or MSKCC, and are exclusively licensed to MabVax Therapeutics. We operate in only one business segment.
We have incurred net losses since inception, and we expect to incur substantial losses for the foreseeable future as we continue our research and development activities. To date, we have funded operations primarily through government grants, the sale of preferred stock, equity securities, non-equity payments from collaborators and interest income. The process of developing our products will require significant additional research and development, preclinical testing and clinical trials, as well as regulatory approval. We expect these activities, together with general and administrative expenses, to result in substantial operating losses for the foreseeable future. We will not receive product revenue unless we, or our collaborative partners, complete clinical trials, obtain regulatory approval and successfully commercialize one or more of our products.
28
Table of Contents
During the year ended December 31, 2014, our loss from operations was $8,392,896 and our net loss was $7,917,853. Net cash used in operating activities for the year ended December 31, 2014 was $7,662,019 and cash and cash equivalents at December 31, 2014 were $1,477,143. As of December 31, 2014, we had an accumulated deficit of $24,550,308.
We are subject to risks common to biopharmaceutical companies, including the need for capital, risks inherent in our research, development and commercialization efforts, preclinical testing, clinical trials, uncertainty of regulatory and marketing approvals, enforcement of patent and proprietary rights, potential competition and retention of key employees. In order for a product to be commercialized, it will be necessary for us to conduct preclinical tests and clinical trials, demonstrate efficacy and safety of our product candidates to the satisfaction of regulatory authorities, obtain marketing approval, enter into manufacturing, distribution and marketing arrangements, obtain market acceptance and, in many cases, obtain adequate reimbursement from government and private insurers. We cannot provide assurance that we will ever generate revenues or achieve and sustain profitability in the future or obtain the necessary working capital for our operations.
Pre-Merger Private Financings
From February 13, 2014 through July 7, 2014, MabVax Therapeutics completed a series of financing transactions and exercise of warrants totaling approximately $7.3 million net of approximately $300,000 in issuance costs.
Merger Agreement
Upon the terms and subject to the satisfaction of the conditions described in the Merger Agreement, Tacoma Corp. was merged with and into MabVax Therapeutics on July 8, 2014, with MabVax Therapeutics surviving the Merger as a wholly-owned subsidiary of MabVax Therapeutics Holdings. The Merger was intended to qualify as a tax-free reorganization for U.S. Federal income tax purposes.
On July 7, 2014, our stockholders approved the Merger, and the Merger closed and became effective on July 8, 2014. All shares of MabVax Therapeutics Series A preferred stock and MabVax Therapeutics Series B preferred stock were automatically converted into shares of MabVax Therapeutics Holdings common stock immediately prior to the Merger. Upon the effective date of the Merger (a) all outstanding shares of MabVax Therapeutics common stock were converted into and exchanged for shares of our common stock at an exchange rate calculated in accordance with the methodology set forth in the Merger Agreement, which resulted in the issuance of 2.223284 shares of our common stock for every share of MabVax Therapeutics common stock, (b) all outstanding shares of MabVax Therapeutics Series C-1 preferred stock were converted into and exchanged for shares of our Series A-1 preferred stock at a rate of two shares of MabVax Therapeutics Series C-1 preferred stock per each share of our Series A-1 preferred stock, (c) each outstanding MabVax Therapeutics option and warrant to purchase MabVax Therapeutics common stock became options and warrants to purchase our common stock (and the number of such shares and exercise price was adjusted as calculated in accordance with the methodology set forth in the Merger Agreement), and (d) each outstanding MabVax Therapeutics warrant to purchase MabVax Therapeutics preferred stock was cancelled for no consideration.
As a result of the consummation and upon the closing of the Merger, the former stockholders, option holders and warrant holders of MabVax Therapeutics were issued, based on the methodology set forth in the Merger Agreement (which excluded certain out of the money convertible securities and calculated others on a net-exercise or cashless basis under the terms of the convertible securities), approximately 85% of the outstanding shares of our common stock on a fully diluted basis and our stockholders, option holders and warrant holders immediately prior to the Merger owned approximately 15% of the outstanding shares of our common stock on a fully diluted basis (such percentages calculated based on the methodology set forth in the Merger Agreement). As a result of the Merger, a change of control of MabVax Therapeutics Holdings occurred.
The total consideration for the transaction was approximately $6,416,000, based on the market price of MabVax Therapeutics Holdings common stock, since management has determined that this was the most reliable measure of fair value.
The following intangible assets were investigated by the Company:
Patents.The Company determined the patents were “ancillary” patents around molecules developed by MabVax Therapeutics Holdings and did not see much benefit that may be derived from the patents. Further, based on efforts by an investment banking firm to market this asset group without success, the Company determined the patents do not have any value from a market participant perspective. The Company will continue to assess the patent portfolio for possible uses in the product development programs that were underway at the Company prior to the Merger.
29
Table of Contents
In Process Research and Development (“IPR&D”). The Company did not see much benefit from MabVax Therapeutics Holdings’ IPR&D projects and does not plan to continue funding such projects. Management has also retained outside industry experts to determine if there is any future benefit to pursue the IPR&D projects. Feedback from these industry experts confirmed little to no prospects for the acquired IPR&D projects and suggested not to spend another $20 million attempting to get the assets through the remaining approval process. Further, based on an investment banking firm’s marketing of these IPR&D projects, industry expert feedback and management’s perspective, the IPR&D projects do not have any value, from a market participant perspective, without further investment by the Company.
Trade Names and Trademarks. Management believes there is not much benefit that may be derived from MabVax Therapeutics Holdings’ trade names and trademarks around the name “Telik”. Management has asked its legal counsel to discontinue registration of such trade names and trademarks. Further, based on an investment banking firm’s marketing of the trade name portfolio and the Company’s perspective, the trade names and trademarks of MabVax Therapeutics Holdings do not have any value, from a market participant perspective.
Non-compete Agreements and Contingent Consideration. No non-compete agreements and no contingent consideration were included as part of the transaction.
For accounting purposes, because a change in control took place with a business, the Merger is treated as a “reverse acquisition” and MabVax Therapeutics is considered the accounting acquirer. As a result, the historical financial statements of the private company MabVax Therapeutics constitute the historical financial statements of the merged companies. The transaction is considered a business combination as the accounting acquirer, MabVax Therapeutics Holdings, is considered an operating entity. For accounting purposes, the private company MabVax Therapeutics is treated as the continuing reporting entity.
The issuance of shares of our common stock and preferred stock in the Merger were approved by our stockholders in the stockholders’ meeting held on July 7, 2014. The amendments to our amended and restated certificate of incorporation related to an increase in the authorized number of shares of our common and preferred stock and a potential reverse stock split to meet the initial NASDAQ listing standards required as a result of the Merger and other transactions contemplated by the Merger Agreement were not approved at such meeting.
Reverse Stock Split, Name Change and Increase in Authorized Shares
In a subsequent special stockholders meeting held on September 8, 2014 our stockholders approved, among other items, authorization for our Board of Directors to effectuate a reverse split in the ratio range of 5:1 to 15:1. Following the stockholders meeting, our Board of Directors approved a reverse split of 8:1, or the Reverse Split, on the Effective Date. The stockholders also approved and our Board of Directors implemented an amendment to our amended and restated certificate of incorporation to change our name from “Telik, Inc.” to “MabVax Therapeutics Holdings, Inc.” and to increase the authorized number of our common shares to a new total of 150,000,000 and our authorized number of preferred shares to a new total of 15,000,000.
On the Effective Date, immediately and without further action by the Company’s stockholders, every 8 shares of the Company’s common stock, issued and outstanding immediately prior to the Effective Time, were automatically converted into 1 share of the Company’s common stock. As a result of the Reverse Split and calculated as of the Record Date, the number of outstanding shares of our common stock was reduced to 1,741,617, excluding outstanding and unexercised share options and warrants and subject to adjustment for fractional shares. No fractional shares were issued as a result of the Reverse Split and, in lieu of these fractional shares, any holder of less than 1 share of the Company’s common stock was entitled to receive cash for such holder’s fractional share equal to the product of such fraction multiplied by the average of the last reported bid and ask prices of the Company’s common stock at 4:00 p.m., Eastern time, end of regular trading hours on OTCQB marketplace, during the 10 consecutive trading days ending on the last trading day prior to the Effective Date. Further, any options, warrants and contractual rights outstanding as of the Effective Date that were subject to adjustment were adjusted in accordance with their terms. These adjustments included, without limitation, changes to the number of shares of the Company’s common stock that may be obtained upon exercise or conversion of these securities, and changes to the applicable exercise or purchase price of such securities.
Shares of our common stock began to trade on the OTCQB marketplace on a post-split basis under the name “MabVax Therapeutics Holdings, Inc.” on September 10, 2014 under the new CUSIP number 55414P108. Commencing on October 10, 2014, shares of our common stock begin trading on the OTCQB marketplace under the trading symbol “MBVX.”
30
Table of Contents
Clinical Product Development
Our therapeutic vaccines were developed at MSKCC and are exclusively licensed to MabVax Therapeutics pursuant to agreements entered into by and between MabVax Therapeutics and MSKCC in 2008. These vaccines are administered in the adjuvant setting and have been shown to elicit a protective antibody response in clinical studies. The antibodies are intended to seek out circulating tumor cells and micrometastases to kill them before they can cause cancer recurrence. Our lead cancer vaccines targeting recurrent sarcoma and ovarian cancer are currently in proof of concept Phase II multi-center clinical trials. Both trials have received substantial federal grant monies to support their development. A vaccine to address the orphan disease neuroblastoma has completed an initial Phase I trial at MSKCC yielding encouraging results. The neuroblastoma vaccine product is expected be ready for a Phase II trial by early 2015. MSKCC and MabVax Therapeutics have completed additional Phase I vaccine clinical trials in melanoma, ovarian cancer, and small cell lung cancer over the last three years.
Preclinical Drug Product Development
Our lead antibody candidate, 5B1, is being developed as a therapeutic product and as a diagnostic imaging product. The antibody targets carbohydrate antigen sialyl Lewis, which is widely expressed on tumors of the gastrointestinal tract, including pancreatic, colon and stomach cancers, as well as ovarian, breast, and small cell lung cancers. We are also developing the 5B1 antibody conjugated to a radiolabel as a novel PET imaging agent to assist in the diagnosis of pancreatic cancer. The advanced preclinical study results of our work in tumor imaging using our 5B1 antibody conjugated to a radiolabel were published in the Journal of Nuclear Medicine. We subsequently applied for and received a contract from the National Institutes of Health (the “NIH”) for the development of the 5B1 based PET imaging agent. We also discovered and are developing multiple fully-human antibodies to the antigen GD2.
Nasdaq Listing Compliance
As disclosed on our Current Report on Form 8-K filed with the SEC on November 14, 2013, MabVax Therapeutics Holdings received a notice letter from The Nasdaq Stock Market, LLC (“NASDAQ”) indicating that based on stockholders’ equity of $2,441,000 disclosed in our Form 10-Q for the period ended September 30, 2013, the Company did not comply with the minimum $2,500,000 stockholders’ equity requirement for continued listing on the Nasdaq Capital Market set forth in Listing Rule 5550(b)(1). MabVax Therapeutics Holdings also failed to meet the stockholders’ equity requirement set forth in 5550(b)(1) prior to the Merger.
On July 9, 2014, following the Merger, we received a notice from the NASDAQ Hearings Panel (the “Panel”) informing us of the Panel’s decision to delist our common shares from the NASDAQ Capital Market effective as of the open of business on Friday, July 11, 2014. We were before the Panel for the failure to maintain the minimum $2.5 million shareholders’ equity requirement as required by NASDAQ Listing Rule 5550(b)(1).
We then submitted an application to OTC Marketplace LLC to list shares of our common stock on the OTCQB marketplace while we pursue an appeals process with NASDAQ. Shares of our common stock began trading on the OTCQB marketplace on Friday, July 11, 2014 under the trading symbol “TELK” and currently trade under the symbol “MBVX”.
In connection with the Merger, we filed an Initial Listing Application for the post-Merger combined entity pursuant to NASDAQ Listing Rule 5110(a) and as disclosed in our Proxy Statement filed with the SEC on June 3, 2014, as supplemented and amended. We also solicited the approval of the holders of a majority of our issued and outstanding stock for certain amendments to our charter documents to, among other things, effect a reverse split, in order to meet the minimum mid-price of $4.00 a share, which is also an Initial Listing Standard as part of the merged company.
We failed to obtain stockholder approval for this reverse split; however, we met the minimum stockholders’ equity requirement set forth in 5550(b)(1) immediately following the Merger. However, partially as a result of our failure to obtain approval for a reverse stock split, the bid price of our common stock traded on the NASDAQ Capital Market on July 9, 2014 failed to meet the NASDAQ Initial Listing Standards required for NASDAQ’s approval of our Initial Listing Application.
31
Table of Contents
On July 9, 2014, we appealed the Panel’s decision to the NASDAQ Listing and Hearing Review Council (the “Council”) requesting an expedited review of the Panel’s delisting decision regarding our request for an opportunity to demonstrate our compliance with the NASDAQ Initial Listing Standards. On December 8, 2014, we received notice that the Council denied our appeal, and the NASDAQ subsequently filed a Form 25 with the Securities and Exchange Commission to finalize removal of the Common Stock from listing on NASDAQ. Our Common Stock has continued to trade on the OTCQB since July 11, 2014.
Results of Operations
Revenues
Revenues for the years ended December 31, 2014 and 2013 were $314,175 and $366,368, respectively, primarily from grant revenues. Future revenues will depend upon the extent to which we obtain approval of new grants or enter into new collaborative research agreements and the amounts of payments relating to such agreements.
| Years Ended December 31, | % change | |||||||||||
| 2014 | 2013 | 2013 to 2014 | ||||||||||
| Revenues |
$ | 314,175 | $ | 366,368 | -14 | % | ||||||
For the year ended December 31, 2014, MabVax Therapeutics recognized revenues of $314,175, as compared to $366,368 for the same period in the prior year. This decrease was primarily due to less work performed on grant contracts in 2014 as compared to work performed on grants in 2013. Revenues earned in 2014 were from the NIH Imaging Contract, which began on September 20, 2013 and continued in 2014 with a Phase II portion of the SBIR contract from NCI being awarded for $1.5 million. Revenues for 2013 represent both the work performed under the NIH imaging contract starting in September 2013, as well as work performed in connection with the remainder of a Phase II NIH grant to support the NCI sarcoma vaccine trial for a vaccine intended to prevent the recurrence of sarcoma, or the NCI Sarcoma Vaccine Grant.
Research and Development Expenses
Research and development expenses for the years ended December 31, 2014 and 2013 were $3,502,730 and $2,967,278, respectively. Our research and development costs consist primarily of clinical trial site costs, clinical data management and statistical analysis support, drug storage and distribution, regulatory services and other outside services related to drug development.
| Years Ended December 31, | % change | |||||||||||
| 2014 | 2013 | 2013 to 2014 | ||||||||||
| Research and development |
$ | 3,502,730 | $ | 2,967,278 | 18 | % | ||||||
Total research and development expenses for the year ended December 31, 2014 increased by 18%, or $535,452, compared to the same period in 2013 primarily due to initiating GMP manufacturing development of our lead antibody candidate 5B1 at Patheon (f.k.a. Gallus BioPharmaceuticals) and increased staffing to support in-house management of patient monitoring for the sarcoma clinical trial. Expenses in the same period a year ago were primarily for direct labor, supplies and third party costs in connection with the sarcoma vaccine trial as well as the initial contract expenses under the NIH Imaging Contract.
Stock-based compensation expense included in research and development expenses for the years ended December 31, 2014 and 2013 was $163,019 and $166,796, respectively.
We expect our total research and development expenditures in the next twelve months to increase as we continue GMP manufacturing of 5B1 and producing finished clinical drug product. We are seeking additional capital to fund the initial clinical study of 5B1 in humans intended to start later in 2015. In the event we are unable to obtain sufficient funding for clinical development of 5B1, we will need to defer the start of clinical trials until funding is in place. If we are unable to obtain additional funding for 5B1, our total research and development expenditures will decrease substantially until the additional funding is raised.
The process of conducting the clinical research necessary to obtain FDA approval is costly and time consuming. Current FDA requirements for a new human drug to be marketed in the United States include:
| • | the successful conclusion of preclinical laboratory and animal tests, if appropriate, to gain preliminary information on the product’s safety; |
32
Table of Contents
| • | filing with the FDA of an IND, to conduct initial human clinical trials for drug candidates; |
| • | the successful completion of adequate and well-controlled human clinical trials to establish the safety and efficacy of the product candidate; and |
| • | filing by the company and acceptance and approval by the FDA of an NDA for a product candidate to allow commercial distribution of the drug. |
We consider the active management and development of our clinical pipeline to be crucial to our long-term success. The actual probability of success for each product candidate and clinical program may be impacted by a variety of factors, including, among others, the quality of the candidate, the validity of the target and disease indication, early clinical data, investment in the program, competition, manufacturing capability and commercial viability. Due to these and other factors, it is difficult to give accurate guidance on the anticipated proportion of our research and development investments or the future cash inflows from these programs.
General and Administrative Expenses
General and administrative expenses for the years ended December 31, 2014 and 2013 were $5,204,341 and $1,442,483, respectively.
| Years Ended December 31, | % change | |||||||||||
| 2014 | 2013 | 2013 to 2014 | ||||||||||
| General and administrative |
$ | 5,204,341 | $ | 1,442,483 | 261 | % | ||||||
The increase in general and administrative expenses of 261%, or $3,761,858 in 2014, compared to the same period in 2013, was primarily due to increases of approximately $1,840,000 related to legal work, mostly in connection with the Merger, $684,000 in investor relations and other public company expenses, $300,000 in professional fees related to accounting and auditing, $282,000 in stock based compensation expense related to the Board of Directors’ grants, $194,000 related to insurance, and additional headcount primarily in the finance, accounting and general administration areas.
Stock-based compensation expense included in general and administrative expenses for the years ended December 31, 2014 and 2013 was $441,957 and $159,848, respectively.
We expect future general and administrative expenses to increase in 2015 as we continue to operate as a public company and complete additional regulatory filings. We also plan to complete capital restructurings of the Company as we expect it will be needed in 2015 to allow for additional financing initiatives.
Interest Income and Interest Expense
| Years Ended December 31, | % change | |||||||||||
| 2014 | 2013 | 2013 to 2014 | ||||||||||
| Interest and other income (expense), net |
$ | (379 | ) | $ | (1,578 | ) | -76 | % | ||||
Interest and other income and expense, net was $379 and $1,578 for the years ended December 31, 2014 and 2013, respectively.
Warrant Liability
Change in fair value of warrant liability for the year ended December 31, 2014 was $475,422. The decrease was mainly due to the decline in Company’s stock price. We calculate the value of our warrant liability on a quarterly basis, or when other events and circumstances occur, using the Black Scholes valuation model.
33
Table of Contents
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with GAAP. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements as well as the reported revenues and expenses during the reporting periods. On an on-going basis, we evaluate our estimates and judgments related to our operating costs. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ significantly from these estimates under different assumptions or conditions.
Our critical accounting policies include:
Revenue recognition. Revenue from grants is based upon internal and subcontractor costs incurred that are specifically covered by the grant, including a facilities and administrative rate that provides funding for overhead expenses. NIH grants are recognized when MabVax Therapeutics incurs internal expenses that are specifically related to each grant, in clinical trials at the clinical trial sites, by subcontractors who manage the clinical trials, and provided the grant has been approved for payment. U.S. grant awards are based upon internal research and development costs incurred that are specifically covered by the grant, and revenues are recognized when MabVax Therapeutics incurs internal expenses that are related to the approved grant.
Any amounts received by MabVax Therapeutics pursuant to the NIH grants prior to satisfying our revenue recognition criteria are recorded as deferred revenue.
Clinical trial expenses. We accrue clinical trial expenses based on work performed. In determining the amount to accrue, we rely on estimates of total costs incurred based on the enrollment of subjects, the completion of trials and other events defined in contracts. We follow this method because we believe reasonably dependable estimates of the costs applicable to various stages of a clinical trial can be made. However, the actual costs and timing of clinical trials are highly uncertain, subject to risks, and may change depending on a number of factors. Differences between the actual clinical trial costs and the estimated clinical trial costs that we have accrued in any prior period are recognized in the subsequent period in which the actual costs become known. Historically, these differences have not been material; however, material differences could occur in the future.
Stock-based compensation. Our stock-based compensation programs include grants of stock options to employees, non-employee directors and non-employee consultants. Stock-based compensation cost is measured at the grant date, based on the calculated fair value of the award, and is recognized as an expense, under the straight-line method, over the employee’s requisite service period (generally the vesting period of the equity grant).
We account for equity instruments, including stock options, issued to employees and non-employees in accordance with authoritative guidance for equity based payments. Stock options issued are accounted for at their estimated fair value determined using the Black Scholes option-pricing model. The fair value of options granted to non-employees is re-measured as they vest, and the resulting increase in value, if any, is recognized as expense during the period the related services are rendered.
Warrant liability. We calculate the value of our warrant liability on a quarterly basis, or when other events and circumstances occur, using as a first step the Black Scholes valuation model, taking into consideration the warrant exercise price, the probability of certain exercise price re-pricing scenarios, the market price for the common stock on the date of measurement, the risk-free interest rate, the dividend yield, the volatility of a comparable period in which the warrant may be exercised, and the remaining life of the warrant, and then as a second step we test our valuation for reasonableness based on settlement offers we have received from the holder of the warrant. If the settlement offer is within a reasonable period of time from when we do our calculation, and is not materially different from the value we recorded using the Black Scholes model, then we retain the value established with our model. If the settlement offer were to reflect a materially different amount near the date of our calculation, then we would record the settlement offer.
Income taxes. Significant judgment is required by management to determine our provision for income taxes, our deferred tax assets and liabilities, and the valuation allowance to record against our net deferred tax assets, which are based on complex and evolving tax regulations throughout the world. Our tax calculation is impacted by tax rates in the jurisdictions in which we are subject to tax and the relative amount of income earned in each jurisdiction. Our deferred tax assets and liabilities are determined using the enacted tax rates expected to be in effect for the years in which those tax assets are expected to be realized.
The effect of an uncertain income tax position is recognized as the largest amount that is “more-likely-than-not” to be sustained under audit by the taxing authority. An uncertain income tax position will not be recognized if it has less than a 50% likelihood of being sustained.
34
Table of Contents
The realization of our deferred tax assets is dependent upon our ability to generate sufficient future taxable income. We establish a valuation allowance when it is more-likely-than-not that the future realization of all or some of the deferred tax assets will not be achieved. The evaluation of the need for a valuation allowance is performed on a jurisdiction-by-jurisdiction basis, and includes a review of all available evidence, both positive and negative. As of December 31, 2014, MabVax Therapeutics concluded that it was more-likely-than-not that its deferred tax assets would not be realized.
Liquidity and Capital Resources
To date, we have financed our operations principally through net proceeds received from private equity and preferred stock financings, and grants through the NIH and SBIR programs. We have experienced negative cash flow from operations each year since our inception. As of December 31, 2014, we had an accumulated deficit of $24,550,308. We expect to continue to incur increased expenses, resulting in losses, over at least the next several years due to, among other factors, our continuing and planned clinical trials and anticipated research and development activities. We had cash of $1,477,143 as of December 31, 2014.
| 2014 | 2013 | |||||||
| December 31: |
||||||||
| Cash and cash equivalents |
$ | 1,477,143 | $ | 354,254 | ||||
| Working capital deficit |
$ | (1,055,335 | ) | $ | (875,924 | ) | ||
| Current ratio |
0.64:1 | 0.31:1 | ||||||
| December 31: |
||||||||
| Cash provided by (used in): |
||||||||
| Operating activities |
$ | (7,662,019 | ) | $ | (2,851,218 | ) | ||
| Investing activities |
$ | 1,452,476 | $ | (8,718 | ) | |||
| Financing activities |
$ | 7,332,432 | $ | 2,792,993 | ||||
Sources and Uses of Cash
Due to the significant research and development expenditures and the lack of any approved products to generate revenue, we have not been profitable and have generated operating losses since we incorporated in 1988. As such, we have funded our research and development operations through government grants and contracts, sales of equity, collaborative arrangements with corporate partners, and interest earned on investments. At December 31, 2014, we had available cash and cash equivalents of $1,477,143. Our cash and cash equivalents balances are held primarily in checking accounts. Cash in excess of immediate requirements is invested with regard to liquidity and capital preservation. Wherever possible, we seek to minimize the potential effects of concentration and degrees of risk.
Cash Flows from Operating Activities. Cash used in operating activities for 2014 was $7,662,019 compared to $2,851,218 for the same period in 2013. Net loss of $7,917,853 in 2014 included non-cash charges of $604,976 for stock-based compensation and $12,241 in depreciation, partially offset by a $475,422 reduction in fair value of the Series B warrants. The Company was able to reduce cash used in operating activities by slowing of payments on invoices for services primarily related to legal expense, partially offset by increased payments to clinical trial sites engaged in our two vaccine clinical trials and increase grant receivable for funds to be received related to work performed on our grants contracts. Cash used in 2013 resulted from net loss of $4,044,971 and included non-cash charges of $326,644 for stock-based compensation and $35,366 for depreciation. Cash used in operating activities in 2013 was reduced by a $539,633 reduction in prepaid expenses.
Cash Flows from Investing Activities. Cash provided by investing activities for 2014 was $1,452,476 compared to $8,718 in cash used in investing activities for 2013. Cash provided in 2014 was primarily from $1,497,283 in cash received in the Merger, offset by $44,807 used to purchase property and equipment. Cash used in 2013 was primarily for property and equipment.
Cash Flows from Financing Activities. Cash provided by financing activities for 2014 was $7,332,432 compared to $2,792,993 provided in 2013. Cash provided by financing activities in 2014 included $2,884,333 from sales of our common stock, $2,973,655 from sales of preferred stock, and $1,472,502 from exercises of Series C-1 warrants. Cash provided by financing activities in 2013 was due to net proceeds received from issuances of preferred stock.
Working Capital Deficit. Working capital deficit increased to $1,055,335 at December 31, 2014 compared with $875,924 at December 31, 2013. The increase in deficit was primarily due to the completion of the merger and costs associated with becoming a public company and was partially offset by sales of our common stock and preferred stock.
35
Table of Contents
We believe our cash and cash equivalents as of December 31, 2014 will be sufficient to fund our projected operating requirements through approximately October 2015. In order to continue our current and future operations and continue our clinical product development programs through 2016, we will depend on our ability to obtain additional funding in a timely manner. We are uncertain about our ability to raise sufficient funds to continue our existing operations beyond 2016. We continue to explore alternatives that could include partnerships involving one or more of our product candidates, licensing arrangements with one or more of our product development candidates, merger with or acquisition by another company, or some other arrangement through which the value of our assets to stockholders could be enhanced. We may raise funds through arrangements with collaborators or others that may require us to relinquish rights to certain product candidates that we might otherwise seek to develop or commercialize independently. Our failure to raise capital when needed could materially harm our business, financial condition and results of operations. See risk factors.
Our future capital uses and requirements depend on numerous factors, including the following:
| • | the progress and success of preclinical studies and clinical trials of our product candidates; |
| • | the progress and number of research programs in development; |
| • | the costs associated with conducting Phase I and II clinical trials; |
| • | the costs and timing of obtaining regulatory approvals; |
| • | our ability to establish, and the scope of, any new collaborations; |
| • | our ability to meet the milestones identified in our collaborative agreements that trigger payments; |
| • | the costs and timing of obtaining, enforcing and defending our patent and intellectual property rights; and |
| • | competing technological and market developments. |
Future Contractual Obligations.
MabVax Therapeutics currently has rental payment obligations under a non-cancelable operating lease at 11588 Sorrento Valley Road that expires on July 31, 2015. Future lease obligations for the next seven months as of July 31, 2015 amount to $77,117. Our master lease and sublease of our facility located at 3165 Porter Drive in Palo Alto, California (the “Porter Drive Facility”) were terminated on February 28, 2013 and we entered into a termination agreement with ARE on February 19, 2013 to voluntarily surrender its premises. As a result of the termination agreement, we were relieved of further obligations under the master lease and further rights to rental income under the sublease and paid a termination fee of approximately $700,000. In addition to the termination fee, if we receive $15 million or more in additional financing in the aggregate, an additional termination fee of $590,504 will be due to ARE, but will otherwise be forgiven.
In connection with the Merger, we signed separation agreements in May 2014 with nine MabVax employees and agreed to pay severances and health benefits upon closing of the Merger subject to certain provisions in the agreement. The total in severance and benefits costs to be paid out subsequent to the Merger is approximately $748,000, of which approximately $6,000 remains as of December 31, 2014.
Off-Balance Sheet Arrangements
We have no material off-balance sheet arrangements as defined in Regulation S-K 303(a)(4)(ii).
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk. |
We do not hold any derivative financial instruments, commodity-based instruments or other long-term debt obligations.
| Item 8. | Financial Statements and Supplementary Data. |
All information required by this item is included in Item 15 of Part IV of this Annual Report and is incorporated into this item by reference.
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure. |
None
36
Table of Contents
| Item 9A. | Controls and Procedures. |
(I) Evaluation of Disclosure Controls and Procedures and Changes in Internal Control over Financial Reporting
Based on their evaluation as of December 31, 2014, our Chief Executive Officer and Chief Financial Officer have concluded that our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended) were not effective.
We made changes in our internal control over financial reporting during the year ended December 31, 2014, that are intended to eliminate a material weakness over the financial reporting on non-routine complex accounting matters such that they are completed in a timely fashion. We also added to the staff three people to help with segregation of duties. Effective December 2014, for any significant material, complex, non-routine event or transaction, the Company retains a third party expert to review the technical memo documenting the accounting treatment for the transaction. Regarding segregation of duties, in June 2014, we added an assistant controller, a person dedicated solely to processing accounts payable, and another person dedicated to reviewing and reporting on clinical trials progress and expenses. These persons have continued to work in these capacities following the Merger; however, not all remediation was in place by December 31, 2014, to eliminate our material weakness.
(II) Management’s Report on Internal Control over Financial Reporting
Our management, including our Chief Executive Officer and Chief Financial Officer, does not expect that our procedures or our internal controls will prevent or detect all error and all fraud. An internal control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations in all control systems, no evaluation of our controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2014. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in its Internal Control — Integrated Framework (2013). Based on our assessment, our management has concluded that, as of December 31, 2014, our internal control over financial reporting was not effective due to material weakness noted above.
This annual report does not include an attestation report of the company’s independent registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by the company’s independent registered public accounting firm pursuant to rules of the Securities and Exchange Commission that permit the company to provide only management’s report in this annual report.
| Item 9B. | Other Information. |
None.
| Item 10. | Directors, Executive Officers and Corporate Governance. |
Information responsive to this Item will be included in our definitive proxy statement relating to our 2015 annual meeting of stockholders to be filed by us with the Securities and Exchange Commission no later than 120 days after the close of our fiscal year ended December 31, 2014 (the “Proxy Statement”) and is incorporated herein by reference.
| Item 11. | Executive Compensation. |
Information responsive to this Item will be included in our definitive proxy statement relating to our 2015 annual meeting of stockholders to be filed by us with the Securities and Exchange Commission no later than 120 days after the close of our fiscal year ended December 31, 2014 (the “Proxy Statement”) and is incorporated herein by reference.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
Information responsive to this Item will be included in our definitive proxy statement relating to our 2015 annual meeting of stockholders to be filed by us with the Securities and Exchange Commission no later than 120 days after the close of our fiscal year ended December 31, 2014 (the “Proxy Statement”) and is incorporated herein by reference.
37
Table of Contents
| Item 13. | Certain Relationships and Related Transactions, and Director Independence. |
Information responsive to this Item will be included in our definitive proxy statement relating to our 2015 annual meeting of stockholders to be filed by us with the Securities and Exchange Commission no later than 120 days after the close of our fiscal year ended December 31, 2014 (the “Proxy Statement”) and is incorporated herein by reference.
| Item 14. | Principal Accounting Fees and Services |
Information responsive to this Item will be included in our definitive proxy statement relating to our 2015 annual meeting of stockholders to be filed by us with the Securities and Exchange Commission no later than 120 days after the close of our fiscal year ended December 31, 2014 (the “Proxy Statement”) and is incorporated herein by reference.
| Item 15. | Exhibits and Financial Statement Schedules. |
(a) The following documents are filed as part of this Annual Report:
| 1. | Financial Statements. Our financial statements and the Report of Independent Registered Public Accounting Firm, are included in Part IV of this Report on the pages indicated: |
MABVAX THERAPEUTICS HOLDINGS, INC.
INDEX TO FINANCIAL STATEMENTS
| F-1 | ||||
| F-2 | ||||
| F-3 | ||||
| F-4 | ||||
| F-7 | ||||
| F-8 |
| 2. | Financial Statement Schedules. All schedules are omitted because they are not applicable or the required information is shown in the financial statements or the notes thereto. |
| 3. | Exhibits: |
| Exhibit No. |
Description |
Form |
Filing Date/Period |
Exhibit |
||||||
| 2.1 |
Agreement and Plan of Merger and Reorganization, dated May 12, 2014, between the Company, Tacoma Acquisition Corp., Inc. and MabVax Therapeutics, Inc. | 8-K | 5/12/2014 | 2.1 | ||||||
| 2.2 |
Amendment No. 1, dated as of June 30, 2014, by and between the Company and MabVax Therapeutics, Inc. | 8-K | 7/1/2014 | 2.1 | ||||||
| 2.3 |
Amendment No. 2 to the Agreement and Plan of Merger, dated July 7, 2014, by and among the Company, Tacoma Acquisition Corp. and MabVax Therapeutics, Inc. | 8-K | 7/9/2014 | 2.1 | ||||||
| 3.1 |
Certificate of Designations, Preferences and Rights of Series A-1 Convertible Preferred Stock | 8-K | 7/9/2014 | 3.1 | ||||||
| 3.2 |
Amended and Restated Certificate of Designations, Preferences and Rights of Series B Convertible Preferred Stock | 8-K | 7/9/2014 | 3.2 | ||||||
| 3.3 |
Certificate of Designations, Preferences and Rights of Series C Convertible Preferred Stock | 8-K | 9/3/2014 | 3.1 | ||||||
| 3.4 |
Amended and Restated Certificate of Incorporation | 8-K | 9/9/2014 | 3.1 | ||||||
| 3.5 |
Certificate of Amendment of Amended and Restated Certificate of Incorporation | 8-K | 9/9/2014 | 3.2 | ||||||
| 3.6 |
Amended and Restated Bylaws | 8-K | 12/14/2007 | 3.2 | ||||||
38
Table of Contents
| 3.7 |
Certificate of Designations, Preferences and Rights of Series D Convertible Preferred Stock | 8-K | 3/26/2015 | 3.1 | ||||||
| 3.8** |
Certificate of Designations, Preferences and Rights of Series E Convertible Preferred Stock | |||||||||
| 4.1 |
Securities Purchase Agreement, dated May 12, 2014, between the Company and the investors identified on the Schedule of Buyers therein and the Form of Registration Rights Agreement, attached thereto as Exhibit C | 8-K | 5/12/2014 | 10.1 | ||||||
| 4.2 |
Securities Purchase Agreement, dated as of February 12, 2014, between MabVax Therapeutics, Inc. and the purchasers set forth on the signature pages thereto including that certain Amendment No. 1 to Securities Purchase Agreement, dated as of May 12, 2014, between MabVax Therapeutics, Inc. and the persons and entities identified on the signature pages thereto | 8-K | 5/12/2014 | 10.3 | ||||||
| 4.3 |
Registration Rights Agreement, dated as of February 12, 2014, between MabVax Therapeutics, Inc. and the persons and entities identified on the signature pages thereto | 8-K | 5/12/2014 | 10.2 | ||||||
| 4.4 |
Omnibus Amendment and Stockholder Consent, dated July 7, 2014, by and among the Company and the Purchasers | 8-K | 7/9/2014 | 10.1 | ||||||
| 4.5 |
Form of Parent Common Stock Warrant | 8-K | 7/9/2014 | 4.1 | ||||||
| 4.6 |
Form of Warrant to Purchase Common Stock | 8-K | 7/9/2014 | 4.2 | ||||||
| 4.7 |
Form of Exchange Agreement | 8-K | 9/3/2014 | 10.1 | ||||||
| 4.8 |
Form of Waiver Letter | 8-K | 9/3/2014 | 10.2 | ||||||
| 4.9 |
Form of Common Stock Certificate | S-1 | 9/29/2014 | 4.1 | ||||||
| 4.10 |
Form of Waiver Extension Letter | 8-K | 9/30/2014 | 10.1 | ||||||
| 4.11** |
Form of Subscription Agreement, dated March 31, 2015, between the Company and the subscribers set forth on the signature pages thereto | |||||||||
| 4.12** |
Form of Common Stock Purchase Warrant | |||||||||
| 4.13** |
Form of Registration Rights Agreement, dated March 31, 2015, between the Company and the persons and entities identified on the signature pages thereto | |||||||||
| 10.1 |
Separation Agreement and Release, dated May 12, 2014, between Michael M. Wick and the Company | 8-K | 5/12/2014 | 10.4 | ||||||
| 10.2 |
Separation Agreement and Release, dated May 12, 2014, between William P. Kaplan and the Company | 8-K | 5/12/2014 | 10.5 | ||||||
| 10.3 |
Separation Agreement and Release, dated May 12, 2014, between Steven R. Schow and the Company | 8-K | 5/12/2014 | 10.6 | ||||||
| 10.4 |
Separation Agreement and Release, dated May 12, 2014, between Wendy K. Wee and the Company | 8-K | 5/12/2014 | 10.7 | ||||||
| 10.5 |
Michael Wick Resignation Letter, dated July 7, 2014 | 8-K | 7/9/2014 | 99.1 | ||||||
| 10.6 |
Edward W. Cantrall Resignation Letter, dated July 7, 2014 | 8-K | 7/9/2014 | 99.2 | ||||||
| 10.7 |
Steven R. Goldring Resignation Letter, dated July 7, 2014 | 8-K | 7/9/2014 | 99.3 | ||||||
| 10.9 |
Richard B. Newman Resignation Letter, dated July 7, 2014 | 8-K | 7/9/2014 | 99.4 | ||||||
| 10.10 |
Employment Agreement, dated July 8, 2014, by and between MabVax Therapeutics, Inc. and J. David Hansen | 10-Q | 8/8/2014 | 10.9 | ||||||
| 10.11 |
Employment Agreement, dated July 8, 2014, by and between MabVax Therapeutics, Inc. and Gregory P. Hanson | 10-Q | 8/8/2014 | 10.10 | ||||||
| 10.12 |
Employment Agreement, dated July 8, 2014, by and between MabVax Therapeutics, Inc. and Wolfgang W. Scholz, Ph.D. | 10-Q | 8/8/2014 | 10.11 | ||||||
| 10.13 |
Securities Purchase Agreement, dated July 8, 2014, by and between MabVax Therapeutics, Inc. and certain institutional investors set forth therein | 10-Q | 8/8/2014 | 10.12 | ||||||
| 10.14 |
Form of Indemnification Agreement | 8-K | 9/9/2014 | 10.1 | ||||||
39
Table of Contents
| 10.15** |
Second Amended and Restated MabVax Therapeutics Holdings, Inc. 2014 Employee, Director and Consultant Equity Incentive Plan | |||||||||
| 10.16 |
Non-Employee Director Compensation Policy | S-1 | 9/29/2014 | 10.22 | ||||||
| 10.17 |
Standard Industrial Net Lease, dated as of May 23, 2008, by and between MabVax Therapeutics, Inc. and Sorrento Square | S-1 | 9/29/2014 | 10.23 | ||||||
| 10.18 |
First Amendment to that Standard Industrial Net Lease, dated May 6, 2010, by and between MabVax Therapeutics, Inc. and Sorrento Square | S-1 | 9/29/2014 | 10.24 | ||||||
| 10.19 |
Second Amendment to that Standard Industrial Net Lease, dated August 1, 2012, by and between the Company and Sorrento Square | S-1 | 9/29/2014 | 10.25 | ||||||
| 10.20 |
Employment Agreement, dated July 21, 2014, 2014, by and between MabVax Therapeutics, Inc. and Paul Maffuid, Ph.D. | S-1 | 9/29/2014 | 10.31 | ||||||
| 10.21 |
Development and Manufacturing Services Agreement, dated April 15, 2014, by and between MabVax Therapeutics, Inc. and Gallus BioPharmaceuticals NJ, LLC | S-1/A | 10/14/2014 | 10.26 | ||||||
| 10.22 |
Exclusive License Agreement for “Polyvalent Conjugate Vaccines for Cancer” (SK#14491), dated as of June 30, 2008, by and between MabVax Therapeutics, Inc. and Sloan-Kettering Institute for Cancer Research | S-1/A | 10/14/2014 | 10.27 | ||||||
| 10.23 |
Research and License Agreement, dated as of April 7, 2008, by and between MabVax Therapeutics, Inc. and Sloan-Kettering Institute for Cancer Research | S-1/A | 10/14/2014 | 10.28 | ||||||
| 10.24 |
Exclusive License to Unimolecular Antibodies, dated October 13, 2011, by and between MabVax Therapeutics, Inc. and Sloan-Kettering Institute for Cancer Research | S-1/A | 10/14/2014 | 10.29 | ||||||
| 10.25 |
Option Agreement, dated August 29, 2014, by and between MabVax Therapeutics, Inc. and Juno Therapeutics, Inc. | S-1/A | 10/14/2014 | 10.30 | ||||||
| 10.26 |
SBIR Contract from National Cancer Institute | S-1/A | 10/14/2014 | 10.34 | ||||||
| 10.27 |
Form of Exchange Agreement (Series A-1 Preferred Stock and Series A-1 Warrants). | 8-K | 3/26/2015 | 10.1 | ||||||
| 10.28 |
Form of Exchange Agreement (Series B Preferred Stock and Series B Warrants). | 8-K | 3/26/2015 | 10.2 | ||||||
| 10.29** |
2008 Equity Incentive Plan | |||||||||
| 10.30** |
Form of Option Agreement, 2008 Equity Incentive Plan | |||||||||
| 11.1 |
Statement of per share earnings | S-1 | 9/29/2014 | 11.1 | ||||||
| 14.1 |
Code of Ethics | 10-K | 3/5/2004 | 14.1 | ||||||
| 21.1 |
Subsidiaries of the Registrant | S-1 | 9/29/2014 | 21.1 | ||||||
| 23.1** |
Consent of Independent Registered Public Accounting Firm | |||||||||
| 31.1** |
Certification of Principal Executive Officer required by Section 302 of the Sarbanes-Oxley Act of 2002 | |||||||||
| 31.2** |
Certification of Principal Financial Officer required by Section 302 of the Sarbanes-Oxley Act of 2002 | |||||||||
| 32.1*** |
Certification of Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |||||||||
| 32.2 *** |
Certification of Chief Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |||||||||
| 99.1** |
Temporary Hardship Exemption per Regulation S-T | |||||||||
| 101**** |
Interactive data file | |||||||||
| ** | Filed herewith |
| *** | Furnished herewith |
| **** | To be filed by amendment per Temporary Hardship Exemption under Regulation S-T. |
40
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Date: March 31, 2015
| MABVAX THERAPEUTICS HOLDINGS, INC | ||
| By: | /s/ J. David Hansen | |
| J. David Hansen | ||
| President and Chief Executive Officer | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ J. David Hansen J. David Hansen |
Chairman of the Board, President and Chief Executive Officer (Principal executive officer) |
March 31, 2015 | ||
| /s/ Gregory P. Hanson Gregory P. Hanson |
Chief Financial Officer (Principal financial and accounting officer) |
March 31, 2015 | ||
| /s/ Kenneth M. Cohen Kenneth M. Cohen |
Director | March 31, 2015 | ||
| /s/ Robert E. Hoffman Robert E. Hoffman |
Director | March 31, 2015 | ||
| /s/ Philip O. Livingston Philip O. Livingston, M.D. |
Director | March 31, 2015 | ||
| /s/ Paul V. Maier Paul V. Maier |
Director | March 31, 2015 | ||
| /s/ Jeffrey V. Ravetch Jeffrey V. Ravetch, M.D., Ph.D. |
Director | March 31, 2015 | ||
| /s/ Michael M. Wick Michael M. Wick, M.D., Ph.D. |
Director | March 31, 2015 | ||
41
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
MabVax Therapeutics Holdings, Inc.
We have audited the accompanying consolidated balance sheets of MabVax Therapeutics Holdings, Inc. (the “Company”) as of December 31, 2014 and 2013, and the related consolidated statements of operations, redeemable convertible preferred stock, convertible preferred stock and stockholders’ equity (deficit), and cash flows for the years then ended. MabVax Therapeutics Holdings, Inc.’s management is responsible for these consolidated financial statements. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of MabVax Therapeutics Holdings, Inc. as of December 31, 2014 and 2013, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has incurred recurring operating losses and is dependent on additional financing to fund operations. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are described in Note 1 to the consolidated financial statements. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ CohnReznick LLP
San Diego, California
March 31, 2015
F-1
Table of Contents
MABVAX THERAPEUTICS HOLDINGS, INC.
Consolidated Balance Sheets
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 1,477,143 | $ | 354,254 | ||||
| Grants receivable |
84,344 | — | ||||||
| Prepaid expenses - clinical operations |
— | — | ||||||
| Prepaid expenses |
334,629 | 44,408 | ||||||
| Other current assets |
14,675 | — | ||||||
|
|
|
|
|
|||||
| Total current assets |
1,910,791 | 398,662 | ||||||
| Property and equipment, net |
57,053 | 24,487 | ||||||
| Goodwill |
6,826,003 | — | ||||||
| Other long term assets |
11,017 | 14,285 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 8,804,864 | $ | 437,434 | ||||
|
|
|
|
|
|||||
| Liabilities, Redeemable Convertible Preferred Stock and Stockholders’ Equity (Deficit) |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 1,313,247 | $ | 66,977 | ||||
| Accrued compensation |
230,381 | 169,123 | ||||||
| Accrued clinical operations and site costs |
494,110 | 773,523 | ||||||
| Related party liabilities |
— | 240,000 | ||||||
| Accrued lease contingency fee |
590,504 | — | ||||||
| Other accrued expenses |
245,421 | 24,963 | ||||||
| Warrant liability |
92,463 | — | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
2,966,126 | 1,274,586 | ||||||
|
|
|
|
|
|||||
| Commitments and contingencies: |
||||||||
| Redeemable convertible preferred stock: |
||||||||
| MabVax Series A redeemable convertible preferred stock, 956,240 shares authorized, 956,240 shares issued and outstanding as of December 31, 2013 with a liquidation preference of $8,013,996 as of December 31, 2013 |
— | 5,787,906 | ||||||
| MabVax Series B redeemable convertible preferred stock, 2,000,000 shares authorized, 891,485 shares issued and outstanding as of December 31, 2013 with a liquidation preference of $6,509,866 as of December 31, 2013 |
— | 6,737,276 | ||||||
| MabVax Therapeutics Holdings Series B redeemable convertible preferred stock, 1,250,000 shares authorized, issued and outstanding as of December 31, 2014 with a liquidation preference of $2,627,123 as of December 31, 2014 |
1,838,025 | — | ||||||
|
|
|
|
|
|||||
| Total redeemable convertible preferred stock |
1,838,025 | 12,525,182 | ||||||
|
|
|
|
|
|||||
| Stockholders’ equity (deficit): |
||||||||
| Series A-1 convertible preferred stock, 2,763,000 shares authorized, 1,593,389 shares issued and outstanding as of December 31, 2014, with a liquidation preference of $2,860,233 as of December 31, 2014 |
4,029,576 | — | ||||||
| Series C convertible preferred stock, 200,000 shares authorized, 96,571 shares issued and outstanding as of December 31, 2014 with no liquidation preference |
966 | — | ||||||
| Common stock, $0.01 par value; 150,000,000 shares authorized as of December 31, 2014, 2,802,867 and 230,503 shares issued and outstanding as of December 31, 2014 and December 31, 2013, respectively |
28,029 | 2,305 | ||||||
| Additional paid-in capital |
24,492,450 | 607,913 | ||||||
| Accumulated deficit |
(24,550,308 | ) | (13,972,552 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity (deficit) |
4,000,713 | (13,362,334 | ) | |||||
|
|
|
|
|
|||||
| Total liabilities, redeemable convertible preferred stock and stockholders’ equity (deficit) |
$ | 8,804,864 | $ | 437,434 | ||||
|
|
|
|
|
|||||
See Accompanying Notes to Consolidated Financial Statements.
F-2
Table of Contents
MABVAX THERAPEUTICS HOLDINGS, INC.
Consolidated Statements of Operations
| For the Years Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| Revenues: |
||||||||
| Grants |
$ | 304,175 | $ | 366,368 | ||||
| Other |
10,000 | — | ||||||
|
|
|
|
|
|||||
| Total revenues |
314,175 | 366,368 | ||||||
|
|
|
|
|
|||||
| Operating costs and expenses: |
||||||||
| Research and development |
3,502,730 | 2,967,278 | ||||||
| General and administrative |
5,204,341 | 1,442,483 | ||||||
|
|
|
|
|
|||||
| Total operating costs and expenses |
8,707,071 | 4,409,761 | ||||||
|
|
|
|
|
|||||
| Loss from operations |
(8,392,896 | ) | (4,043,393 | ) | ||||
| Interest and other income (expense) |
(379 | ) | (1,578 | ) | ||||
| Change in fair value of warrant liability |
475,422 | — | ||||||
|
|
|
|
|
|||||
| Net loss |
(7,917,853 | ) | (4,044,971 | ) | ||||
| Deemed dividend on Series A-1 preferred stock |
(2,214,911 | ) | (691,812 | ) | ||||
| Accretion of preferred stock dividends |
(444,992 | ) | — | |||||
|
|
|
|
|
|||||
| Net loss available to common stockholders |
$ | (10,577,756 | ) | $ | (4,736,783 | ) | ||
|
|
|
|
|
|||||
| Basic and diluted net loss per share |
$ | (9.51 | ) | $ | (20.55 | ) | ||
|
|
|
|
|
|||||
| Shares used to calculate basic and diluted net loss per share |
1,112,481 | 230,503 | ||||||
|
|
|
|
|
|||||
See Accompanying Notes to Consolidated Financial Statements.
F-3
Table of Contents
MABVAX THERAPEUTICS HOLDINGS, INC.
Consolidated Statements of Redeemable Convertible Preferred Stock, Convertible Preferred Stock and Stockholders’ Equity (Deficit)
| Redeemable Convertible Preferred Stock | ||||||||||||||||||||||||||||||||||||
| MabVax Series A | MabVax Series B | MabVax Series C-1 | Series B | |||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | Total | ||||||||||||||||||||||||||||
| Balance, December 31, 2012 |
956,240 | $ | 5,787,906 | 480,928 | $ | 3,252,471 | — | — | — | — | $ | 9,040,377 | ||||||||||||||||||||||||
| Issuance of Series B preferred stock from February 1 to December 18 at $6.82 per share, net of issuance costs of $7,007 |
— | — | 410,557 | 2,792,993 | — | — | — | — | 2,792,993 | |||||||||||||||||||||||||||
| Deemed dividend related to beneficial conversion feature of series B preferred |
— | — | — | 691,812 | — | — | — | — | 691,812 | |||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2013 |
956,240 | 5,787,906 | 891,485 | 6,737,276 | — | — | — | — | 12,525,182 | |||||||||||||||||||||||||||
| Exercise of Series B warrant in January at $0.01 per share |
— | — | 194,281 | 1,942 | — | — | — | — | 1,942 | |||||||||||||||||||||||||||
| Conversion of $240,000 in accounts payable into 44,466 shares of common stock on February 12, 2014 |
— | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Issuance of MabVax Series C-1 preferred stock in February at $0.84 per share, net of issuance costs of $126,345 |
— | — | — | — | 3,697,702 | 2,973,655 | — | — | 2,973,655 | |||||||||||||||||||||||||||
| Deemed dividend related to beneficial conversion feature of MabVax Series C-1 preferred |
— | — | — | — | — | 2,214,911 | — | — | 2,214,911 | |||||||||||||||||||||||||||
| Issuance of common stock at $9.32 per share, net of issuance costs of $156,303 in June and July |
— | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Reclassification of Series A and Series B to equity in June |
(956,240 | ) | (5,787,906 | ) | (1,085,766 | ) | (6,739,218 | ) | — | — | — | — | (12,527,124 | ) | ||||||||||||||||||||||
| Conversion of Series A to common stock on July 8, 2014 |
— | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Conversion of Series B to common stock on July 8, 2014 |
— | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Accretion of redemption value for Series C-1 to July 8, 2014 |
— | — | — | — | — | 99,200 | — | — | 99,200 | |||||||||||||||||||||||||||
| Exercise of Series C-1 warrant on July 7, 2014 |
— | — | — | — | 1,827,979 | 1,472,502 | — | — | 1,472,502 | |||||||||||||||||||||||||||
| Accretion of redemption value for Series C-1 warrant to July 8, 2014 |
— | — | — | — | — | 47,120 | — | — | 47,120 | |||||||||||||||||||||||||||
| Conversion of Series C-1 into Series A-1 on July 8, 2014 |
— | — | — | — | (5,525,681 | ) | (6,807,388 | ) | — | — | (6,807,388 | ) | ||||||||||||||||||||||||
| Accretion of redemption value for Series A-1 from July 8 to Dec 31, 2014 |
— | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Acquisition of MabVax Therapeutics Holdings (f.k.a. Telik, Inc.) at exchange ratio of 2.223284 shares of MabVax Therapeutics Holdings for every share of MabVax, including 4,205,411 common and 1,250,000 Series B preferred stock outstanding in July |
— | — | — | — | — | — | 1,250,000 | 1,710,902 | 1,710,902 | |||||||||||||||||||||||||||
| Accretion of redemption value for Series B from May 12, 2014 |
— | — | — | — | — | — | — | 127,123 | 127,123 | |||||||||||||||||||||||||||
| Exchange of common stock for Series C on September 3, 2014 |
— | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Elimination of fractional shares resulting from Reverse Split on September 8, 2014 |
— | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Shares issued in connection with exercise of warrants on a cashless basis in September and October |
— | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Conversion of Series A-1 into Common stock from November 13 to Dec 31, 2014 |
— | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Conversion of Series C into Common stock from October to December, 2014 |
— | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Balance at December 31, 2014 |
— | $ | — | — | $ | — | — | $ | — | 1,250,000 | $ | 1,838,025 | $ | 1,838,025 | ||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
See Accompanying Notes to Consolidated Financial Statements.
F-4
Table of Contents
MABVAX THERAPEUTICS HOLDINGS, INC.
Consolidated Statements of Redeemable Convertible Preferred Stock, Convertible Preferred Stock and Stockholders’ Equity (Deficit)
| Convertible Preferred Stock | ||||||||||||||||||||||||||||||||
| MabVax Series A | MabVax Series B | Series A-1 | Series C | |||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | |||||||||||||||||||||||||
| Balance, December 31, 2012 |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Issuance of Series B preferred stock from February 1 to December 18 at $6.82 per share, net of issuance costs of $7,007 |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Deemed dividend related to beneficial conversion feature of series B preferred |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 31, 2013 |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Exercise of Series B warrant in January at $0.01 per share |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Conversion of $240,000 in accounts payable into 44,466 shares of common stock on February 12, 2014 |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Issuance of MabVax Series C-1 preferred stock in February at $0.84 per share, net of issuance costs of $126,345 |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Deemed dividend related to beneficial conversion feature of MabVax Series C-1 preferred |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Issuance of common stock at $9.32 per share, net of issuance costs of $156,303 in June and July |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Reclassification of Series A and Series B to equity in June |
956,240 | 5,787,906 | 1,085,766 | 6,739,218 | — | — | — | — | ||||||||||||||||||||||||
| Conversion of Series A to common stock on July 8, 2014 |
(956,240 | ) | (5,787,906 | ) | — | — | — | — | — | — | ||||||||||||||||||||||
| Conversion of Series B to common stock on July 8, 2014 |
— | — | (1,085,766 | ) | (6,739,218 | ) | — | — | — | — | ||||||||||||||||||||||
| Accretion of redemption value for Series C-1 to July 8, 2014 |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Exercise of Series C-1 warrant on July 7, 2014 |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Accretion of redemption value for Series C-1 warrant to July 8, 2014 |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Conversion of Series C-1 into Series A-1 on July 8, 2014 |
— | — | — | — | 2,762,841 | 6,807,388 | — | — | ||||||||||||||||||||||||
| Accretion of redemption value for Series A-1 from July 8 to Dec 31, 2014 |
— | — | — | — | — | 171,549 | — | — | ||||||||||||||||||||||||
| Acquisition of MabVax Therapeutics Holdings (f.k.a. Telik, Inc.) at exchange ratio of 2.223284 shares of MabVax Therapeutics Holdings for every share of MabVax, including 4,205,411 common and 1,250,000 Series B preferred stock outstanding in July |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Accretion of redemption value for Series B from May 12, 2014 |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Exchange of common stock for Series C on September 3, 2014 |
— | — | — | — | — | — | 118,970 | 1,190 | ||||||||||||||||||||||||
| Elimination of fractional shares resulting from Reverse Split on September 8, 2014 |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Shares issued in connection with exercise of warrants on a cashless basis in September and October |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Conversion of Series A-1 into Common stock from November 13 to Dec 31, 2014 |
— | — | — | — | (1,169,452 | ) | (2,949,361 | ) | — | — | ||||||||||||||||||||||
| Conversion of Series C into Common stock from October to December, 2014 |
— | — | — | — | — | — | (22,399 | ) | (224 | ) | ||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Balance at December 31, 2014 |
— | $ | — | — | $ | — | 1,593,389 | $ | 4,029,576 | 96,571 | $ | 966 | ||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
See Accompanying Notes to Consolidated Financial Statements.
F-5
Table of Contents
MABVAX THERAPEUTICS HOLDINGS, INC.
Consolidated Statements of Redeemable Convertible Preferred Stock, Convertible Preferred Stock and Stockholders’ Equity (Deficit)
| Additional Paid-in Capital |
Total Stockholders’ Equity (Deficit) |
|||||||||||||||||||
| Common Stock | Accumulated Deficit |
|||||||||||||||||||
| Shares | Amount | |||||||||||||||||||
| Balance, December 31, 2012 |
230,503 | $ | 2,305 | $ | 281,269 | $ | (9,235,769 | ) | $ | (8,952,195 | ) | |||||||||
| Issuance of Series B preferred stock from February 1 to December 18 at $6.82 per share, net of issuance costs of $7,007 |
— | — | — | — | — | |||||||||||||||
| Deemed dividend related to beneficial conversion feature of series B preferred |
— | — | — | (691,812 | ) | (691,812 | ) | |||||||||||||
| Stock-based compensation |
— | — | 326,644 | — | 326,644 | |||||||||||||||
| Net loss |
— | — | — | (4,044,971 | ) | (4,044,971 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance at December 31, 2013 |
230,503 | 2,305 | 607,913 | (13,972,552 | ) | (13,362,334 | ) | |||||||||||||
| Exercise of Series B warrant in January at $0.01 per share |
— | — | — | — | — | |||||||||||||||
| Conversion of $240,000 in accounts payable into 44,466 shares of common stock on February 12, 2014 |
44,466 | 445 | 239,555 | — | 240,000 | |||||||||||||||
| Issuance of MabVax Series C-1 preferred stock in February at $0.84 per share, net of issuance costs of $126,345 |
— | — | — | — | — | |||||||||||||||
| Deemed dividend related to beneficial conversion feature of MabVax Series C-1 preferred |
— | — | — | (2,214,911 | ) | (2,214,911 | ) | |||||||||||||
| Issuance of common stock at $9.32 per share, net of issuance costs of $156,303 in June and July |
326,264 | 3,263 | 2,881,070 | — | 2,884,333 | |||||||||||||||
| Reclassification of Series A and Series B to equity in June |
— | — | — | — | 12,527,124 | |||||||||||||||
| Conversion of Series A to common stock on July 8, 2014 |
265,749 | 2,657 | 5,785,249 | — | — | |||||||||||||||
| Conversion of Series B to common stock on July 8, 2014 |
301,746 | 3,017 | 6,736,201 | — | — | |||||||||||||||
| Accretion of redemption value for Series C-1 to July 8, 2014 |
— | — | — | (99,200 | ) | (99,200 | ) | |||||||||||||
| Exercise of Series C-1 warrant on July 7, 2014 |
— | — | — | — | — | |||||||||||||||
| Accretion of redemption value for Series C-1 warrant to July 8, 2014 |
— | — | — | (47,120 | ) | (47,120 | ) | |||||||||||||
| Conversion of Series C-1 into Series A-1 on July 8, 2014 |
— | — | — | — | 6,807,388 | |||||||||||||||
| Accretion of redemption value for Series A-1 from July 8 to Dec 31, 2014 |
— | — | — | (171,549 | ) | — | ||||||||||||||
| Acquisition of MabVax Therapeutics Holdings (f.k.a. Telik, Inc.) at exchange ratio of 2.223284 shares of MabVax Therapeutics Holdings for every share of MabVax, including 4,205,411 common and 1,250,000 Series B preferred stock outstanding in July |
572,858 | 5,729 | 4,699,997 | — | 4,705,726 | |||||||||||||||
| Accretion of redemption value for Series B from May 12, 2014 |
— | — | — | (127,123 | ) | (127,123 | ) | |||||||||||||
| Exchange of common stock for Series C on September 3, 2014 |
(148,713 | ) | (1,487 | ) | 297 | — | — | |||||||||||||
| Elimination of fractional shares resulting from Reverse Split on September 8, 2014 |
— | — | (293 | ) | — | (293 | ) | |||||||||||||
| Shares issued in connection with exercise of warrants on a cashless basis in September and October |
488,659 | 4,887 | (4,887 | ) | — | — | ||||||||||||||
| Conversion of Series A-1 into Common stock from November 13 to Dec 31, 2014 |
693,335 | 6,933 | 2,942,428 | — | — | |||||||||||||||
| Conversion of Series C into Common stock from October to December, 2014 |
28,000 | 280 | (56 | ) | — | — | ||||||||||||||
| Stock-based compensation |
— | — | 604,976 | — | 604,976 | |||||||||||||||
| Net loss |
— | — | — | (7,917,853 | ) | (7,917,853 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance at December 31, 2014 |
2,802,867 | $ | 28,029 | $ | 24,492,450 | $ | (24,550,308 | ) | $ | 4,000,713 | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
See Accompanying Notes to Consolidated Financial Statements.
F-6
Table of Contents
MABVAX THERAPEUTICS HOLDINGS, INC.
Consolidated Statements of Cash Flows
| For the Years Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| Operating activities |
||||||||
| Net loss |
$ | (7,917,853 | ) | $ | (4,044,971 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
| Depreciation and amortization |
12,241 | 35,366 | ||||||
| Stock-based compensation |
604,976 | 326,644 | ||||||
| Change in fair value of warrants |
(475,422 | ) | — | |||||
| Increase (decrease) in operating assets and liabilities excluding effects of the Merger: |
||||||||
| Grants receivable |
(84,344 | ) | 19,845 | |||||
| Other receivables |
28,316 | — | ||||||
| Prepaid expenses - clinical operations |
— | 539,633 | ||||||
| Prepaid expenses and other |
(117,004 | ) | 13,061 | |||||
| Accounts payable |
1,246,270 | 21,946 | ||||||
| Accrued clinical operations and site costs |
(279,413 | ) | 128,485 | |||||
| Accrued compensation |
(789,014 | ) | 80,138 | |||||
| Related party liabilities |
— | 45,000 | ||||||
| Other accrued expenses |
109,228 | (16,365 | ) | |||||
|
|
|
|
|
|||||
| Net cash used in operating activities |
(7,662,019 | ) | (2,851,218 | ) | ||||
|
|
|
|
|
|||||
| Investing activities |
||||||||
| Purchases of property and equipment |
(44,807 | ) | (8,718 | ) | ||||
| Proceeds from acquisition of Telik, Inc. |
1,497,283 | — | ||||||
|
|
|
|
|
|||||
| Net cash provided by (used in) investing activities |
1,452,476 | (8,718 | ) | |||||
|
|
|
|
|
|||||
| Financing activities |
||||||||
| Issuances of preferred stock, net of issuance costs |
2,973,655 | 2,792,993 | ||||||
| Proceeds from exercise of MabVax Series B warrant |
1,942 | — | ||||||
| Proceeds from exercise of MabVax Series C-1 warrants |
1,472,502 | — | ||||||
| Proceeds from issuance of common stock, net of issuance costs |
2,884,333 | — | ||||||
|
|
|
|
|
|||||
| Net cash provided by financing activities |
7,332,432 | 2,792,993 | ||||||
|
|
|
|
|
|||||
| Net change in cash and cash equivalents |
1,122,889 | (66,943 | ) | |||||
| Cash and cash equivalents at beginning of year |
354,254 | 421,197 | ||||||
|
|
|
|
|
|||||
| Cash and cash equivalents at end of year |
$ | 1,477,143 | $ | 354,254 | ||||
|
|
|
|
|
|||||
| Supplemental disclosures of cash flow information: |
||||||||
| Cash paid during the period for income taxes |
$ | 800 | $ | 1,526 | ||||
|
|
|
|
|
|||||
| Supplemental disclosures of non-cash investing and financing information: |
||||||||
| Deemed dividend on beneficial conversion feature for preferred stock |
$ | 2,214,911 | $ | 691,812 | ||||
|
|
|
|
|
|||||
| Goodwill on acquisition of Telik, Inc. |
$ | 6,826,003 | $ | — | ||||
|
|
|
|
|
|||||
| Warrant liability upon acquisition of Telik, Inc. |
$ | 567,885 | $ | — | ||||
|
|
|
|
|
|||||
| Accretion of redemption value for Series A-1 and B preferred stock |
$ | 444,992 | $ | — | ||||
|
|
|
|
|
|||||
| Issuance of common stock for accounts payable |
$ | 240,000 | $ | — | ||||
|
|
|
|
|
|||||
| Conversion of Series A and Series B redeemable preferred stock into common stock |
$ | 12,527,124 | $ | — | ||||
|
|
|
|
|
|||||
| Conversion of Series C-1 redeemable preferred stock into Series A-1 preferred stock |
$ | 6,807,388 | $ | — | ||||
|
|
|
|
|
|||||
| Acquisition of MabVax Therapeutics Holdings in relation to the merger |
$ | 4,705,726 | $ | — | ||||
|
|
|
|
|
|||||
| Conversion of Series A-1 preferred stock to common stock |
$ | 2,949,361 | $ | — | ||||
|
|
|
|
|
|||||
| Warrants exercised to purchase common stock on a cashless basis to purchase 488,659 shares of common stock. See Note 7. |
$ | 4,887 | $ | — | ||||
|
|
|
|
|
|||||
| Conversion of common stock to Series C preferred stock |
$ | 1,190 | $ | — | ||||
|
|
|
|
|
|||||
| Conversion of Series C preferred stock to common stock |
$ | 224 | $ | — | ||||
|
|
|
|
|
|||||
See Accompanying Notes to Consolidated Financial Statements.
F-7
Table of Contents
MABVAX THERAPEUTICS HOLDINGS, INC.
Notes to Consolidated Financial Statements
1. Nature of Operations and Basis of Presentation
MabVax Therapeutics Holdings, Inc. (f.k.a. Telik, Inc. and referred to herein as “MabVax Therapeutics Holdings” or the “Company”) (OTCQB: MBVX) was incorporated in the state of Delaware on October 20, 1988. On July 8, 2014, Tacoma Acquisition Corp., a Delaware corporation and wholly owned subsidiary of MabVax Therapeutics Holdings (“Tacoma Corp.”) merged with MabVax Therapeutics, Inc., a Delaware corporation (“MabVax Therapeutics”) pursuant to an Agreement and Plan of Merger, dated May 12, 2014, by and among MabVax Therapeutics Holdings, Tacoma Corp. and MabVax Therapeutics, as amended by that certain Amendment No. 1 to the Merger Agreement, dated June 30, 2014, by and among the parties thereto and by that certain Amendment No. 2 to the Merger Agreement, dated July 7, 2014, by and among the parties thereto (such agreement as amended, the “Merger Agreement”; such Merger, the “Merger”). Unless the context otherwise requires, references to “we,” “our,” “us,” or the “Company” in this Annual Report mean MabVax Therapeutics Holdings on a consolidated financial statement basis with our wholly-owned subsidiary following the Merger, MabVax Therapeutics, as applicable.
Par value and additional paid-in capital for December 31, 2013 has been restated to reflect the par value for shares post-merger and the September 8, 2014, 8-for-1 Reverse Split (as defined in note 5).
We are a clinical stage biopharmaceutical company engaged in the discovery, development and commercialization of proprietary human monoclonal antibody products and vaccines for the treatment of a variety of cancers. We have discovered a pipeline of human monoclonal antibody products based on the protective immune responses generated by patients who have been immunized against targeted cancers. Therapeutic vaccines under development were discovered at Memorial Sloan Kettering Cancer Center (“MSKCC”), and are exclusively licensed to MabVax Therapeutics. We operate in only one business segment.
We are continuing to evaluate the technology and development programs that were under way at the Company prior to the Merger and plan to continue developing MabVax Therapeutics’ pre-Merger pipeline.
We have incurred net losses since inception and expect to incur substantial losses for the foreseeable future as the Company continues research and development activities. To date, we have funded operations primarily through government grants, the sale of preferred stock, equity securities, non-equity payments from collaborators and interest income. The process of developing the Company’s products will require significant additional research and development, preclinical testing and clinical trials, as well as regulatory approval. We expect these activities, together with general and administrative expenses, to result in substantial operating losses for the foreseeable future. We will not receive revenue unless the Company or its collaborative partners complete clinical trials, obtain regulatory approval and successfully commercialize one or more products; or the Company licenses its technology after achieving one or more milestones of interest to a potential partner.
Liquidity and Going Concern
The accompanying consolidated financial statements have been prepared on the going concern basis, which assumes that the Company will continue to operate as a going concern and which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. As reflected in the accompanying consolidated financial statements, the Company had a net loss of $7,917,853, net cash used in operating activities of $7,662,019 and net cash provided by investing activities of $1,452,476, for the year ended December 31, 2014. As of December 31, 2014, the Company had $1,477,143 in cash and cash equivalents and an accumulated deficit of $24,550,308.
From February 13, 2014 through July 7, 2014, MabVax Therapeutics Holdings completed a series of financing transactions totaling approximately $7.3 million net of approximately $300,000 in issuance costs, through the sale of MabVax Therapeutics Holdings preferred stock, MabVax Therapeutics Holdings common stock and exercise of MabVax Therapeutics Holdings warrants.
The Company anticipates that it will continue to incur net losses into the foreseeable future as it: (i) continues to identify and advance a number of potential drug candidates into clinical and preclinical development activities, (ii) initiates manufacturing of its lead antibody candidate 5B1 and continues to fund its operations, and (iii) expands its corporate infrastructure, including the costs associated with being a public company. Without additional funding, management believes that the Company will not have sufficient funds to meet its obligations beyond October 2015, unless the Company is able to raise additional capital. These conditions give rise to substantial doubt as to the Company’s ability to continue as a going concern. The accompanying consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
F-8
Table of Contents
The Company plans to continue to fund its losses from operations and capital funding needs through equity or debt financings, strategic collaborations, licensing arrangements, asset sales, government grants or other arrangements. However, the Company cannot be sure that such additional funds will be available on reasonable terms, or at all. If the Company is unable to secure adequate additional funding, the Company may be forced to make reductions in spending, extend payment terms with suppliers, liquidate assets where possible, and/or suspend or curtail planned programs. In addition, if the Company does not meet its payment obligations to third parties as they come due, it may be subject to litigation claims. Even if the Company is successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. Any of these actions could materially harm the Company’s business, results of operations, and future prospects.
If the Company raises additional funds by issuing equity securities, substantial dilution to existing stockholders would result. If the Company raises additional funds by incurring debt financing, the terms of the debt may involve significant cash payment obligations as well as covenants and specific financial ratios that may restrict the Company’s ability to operate its business.
2. Summary of Significant Accounting Policies
Basis of Presentation
The accompanying consolidated financial statements reflect all of our activities, including those of our wholly owned subsidiaries. All material intercompany accounts and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America (“GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Management believes that these estimates are reasonable; however, actual results may differ from these estimates.
Cash and Cash Equivalents
The Company considers all highly liquid investments purchased with original maturities of three months or less to be cash equivalents. The Company minimizes its credit risk associated with cash and cash equivalents by periodically evaluating the credit quality of its primary financial institution. The balance at times may exceed Federally insured limits. The Company has not experienced any losses on such accounts.
Fair Value of Financial Instruments
The Company’s financial instruments consist of cash and cash equivalents, grants receivable, other receivable, prepaid expenses and other assets, accounts payable, related party payables and warrant liabilities, all of which are generally considered to be representative of their respective fair values because of the short-term nature of those instruments.
Grants Receivable
Grants receivable at December 31, 2014 represent amounts due under the NIH Imaging Contract Phase II with the National Cancer Institute (the “NCI”), a division of the National Institutes of Health, or NIH (collectively, the “NIH Grants”). The Company considers the grants receivable to be fully collectible; accordingly, no allowance for doubtful accounts has been established. If amounts become uncollectible, they are charged to operations.
Property and Equipment
Property and equipment are carried at cost less accumulated depreciation. Depreciation of property and equipment is computed using the straight-line method over the estimated useful lives of the assets, which are generally three to five years. Leasehold improvements are amortized over the lesser of the life of the lease or the life of the asset.
F-9
Table of Contents
Impairment of Long-lived Assets
The Company evaluates its long-lived assets with definite lives, such as property and equipment, for impairment. The Company records impairment losses on long-lived assets used for operations when indicators of impairment are present and the undiscounted cash flows estimated to be generated by those assets are less than the carrying value of the assets. There have not been any impairment losses of long-lived assets for the years ended December 31, 2014 and 2013.
Impairment of Goodwill
The Company applies the GAAP principles related to Intangibles – Goodwill and Other to test for goodwill impairment annually. During the fourth quarter, there was a triggering event that occurred as a result of the decline in the Company’s market capitalization. As a result, the Company went to a step 1 analysis utilizing an external valuation firm to value the Company. Based upon the analysis performed no impairment was noted, therefore step 2 was not required. The Company has concluded that no impairment of Goodwill has taken place for the year ended December 31, 2014.
Revenue Recognition
Revenue from grants are based upon internal and subcontractor costs incurred that are specifically covered by the grant, including a facilities and administrative rate that provides funding for overhead expenses. NIH Grants are recognized when the Company incurs internal expenses that are specifically related to each grant, in clinical trials at the clinical trial sites, by subcontractors who manage the clinical trials, and provided the grant has been approved for payment. U.S. Treasury grant awards are based upon internal research and development costs incurred that are specifically covered by the grant, and revenues are recognized when the Company incurs internal expenses that are related to the approved grant. The Company records revenue associated with the NIH Grants as the related costs and expenses are incurred. Any amounts received by the Company pursuant to the NIH Grants prior to satisfying the Company’s revenue recognition criteria are recorded as deferred revenue.
Research and Development Costs
Research and development expenses, which consist primarily of salaries and other personnel costs, clinical trial costs and preclinical study fees, manufacturing costs for non-commercial products, and the development of earlier-stage programs and technologies, are expensed as incurred when these expenditures have no alternative future uses. A significant portion of the development activities are outsourced to third parties, including contract research organizations. In such cases, the Company may be required to estimate related service fees incurred.
Stock-based Compensation
The Company’s stock-based compensation programs include grants of stock options to employees, non-employee directors and non-employee consultants. Stock-based compensation cost is measured at the grant date, based on the calculated fair value of the award, and is recognized as an expense, under the straight-line method, over the employee’s requisite service period (generally the vesting period of the equity grant).
The Company accounts for equity instruments, including stock options, issued to non-employees in accordance with authoritative guidance for equity based payments to non-employees. Stock options issued to non-employees are accounted for at their estimated fair value determined using the Black Scholes option-pricing model. The fair value of options granted to non-employees is re-measured as they vest, and the resulting increase in value, if any, is recognized as expense during the period the related services are rendered.
Income Taxes
The Company uses the asset and liability method of accounting for income taxes. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to basis differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized. As of December 31, 2014 and 2013, all deferred tax assets were fully offset by a valuation allowance.
F-10
Table of Contents
The Company accrues interest and penalties, if any, on underpayment of income taxes related to unrecognized tax benefits as a component of income tax expense in its consolidated statements of operations.
Fair Value Measurements
Level 1 fair value inputs are quoted prices for identical items in active, liquid and visible markets such as stock exchanges. Level 2 fair value inputs are observable information for similar items in active or inactive markets, and appropriately consider counterparty creditworthiness in the valuations. Level 3 fair value inputs reflect our best estimate of inputs and assumptions market participants would use in pricing an asset or liability at the measurement date. The inputs are unobservable in the market and significant to the valuation estimate.
3. Recent Accounting Pronouncements
The Company has historically reported as a development stage company. In the period ended June 30, 2014, the Company elected to early adopt FASB Accounting Standards Update (“ASU”) No. 2014-10, “Development Stage Entities (Topic 915): Elimination of Certain Financial Reporting Requirements.” The adoption of this ASU allows the Company to remove the inception to date information and all references to development stage.
In May 2014, the FASB issued ASU No. 2014-09, “Revenue from Contracts with Customers” (Topic 606). ASU No. 2014-09 supersedes the revenue recognition requirements in Topic 605, “Revenue Recognition,” and most industry-specific revenue recognition guidance throughout the Industry Topics of the Accounting Standards Codification. Additionally, this update supersedes some cost guidance included in Subtopic 605-35, “Revenue Recognition-Construction-Type and Production-Type Contracts.” The core principle of the guidance is that an entity should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. It is effective for the first interim period within annual reporting periods beginning after December 15, 2016, and early adoption is not permitted. Entities may choose from two adoption methods, with certain practical expedients. We are currently reviewing this standard to assess the impact on the Company’s future financial statements and evaluating the available adoption methods.
In June 2014, the FASB issued ASU No. 2014-12, “Compensation—Stock Compensation” (Topic 718): “Accounting for Share-Based Payments When the Terms of an Award Provide That a Performance Target Could Be Achieved after the Requisite Service Period,” which requires that a performance target that affects vesting, and that could be achieved after the requisite service period, be treated as a performance condition. As such, the performance target should not be reflected in estimating the grant date fair value of the award. ASU No. 2014-12 is effective for annual reporting periods beginning after December 15, 2015, including interim periods within that reporting period, although early adoption is permitted. We are currently reviewing this standard to assess the impact on the Company’s future financial statements.
In August 2014, the FASB issued ASU No. 2014-15, (“ASU 2014-15”), “Disclosure of Uncertainties About an Entity’s Ability to Continue as a Going Concern”. ASU 2014-15 requires management to perform interim and annual assessments of an entity’s ability to continue as a going concern within one year of the date the financial statements are issued and provides guidance on determining when and how to disclose going concern uncertainties in the financial statements. Certain disclosures will be required if conditions give rise to substantial doubt about an entity’s ability to continue as a going concern. ASU 2014-15 applies to all entities and is effective for annual and interim reporting periods ending after December 15, 2016, with early adoption permitted. Management is currently evaluating the impact of the adoption of the updated standard on the financial statements and disclosures.
Management does not believe that any other recently issued, but not yet effective, accounting standards if currently adopted would have a material effect on the accompanying consolidated financial statements.
F-11
Table of Contents
4. Property and Equipment, Net
Property and equipment consisted of the following as of December 31, 2014 and 2013:
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Furniture and fixtures |
$ | 8,979 | $ | 8,979 | ||||
| Office equipment |
31,170 | 21,850 | ||||||
| Lab equipment |
321,884 | 286,397 | ||||||
|
|
|
|
|
|||||
| 362,033 | 317,226 | |||||||
| Less accumulated depreciation and amortization |
(304,980 | ) | (292,739 | ) | ||||
|
|
|
|
|
|||||
| Totals |
$ | 57,053 | $ | 24,487 | ||||
|
|
|
|
|
|||||
Depreciation expense for the years ended December 31, 2014 and 2013 was $12,241 and $35,366, respectively.
5. Reverse Stock Split, Name Change and Increase in Authorized Shares
On September 8, 2014, MabVax Therapeutics Holdings filed an amended and restated certificate of incorporation to increase the authorized number of shares of our common stock to a new total of 150,000,000 shares, increase the number of shares of our preferred stock to a new total of 15,000,000 shares, and change the name of the Company from “Telik, Inc.” to “MabVax Therapeutics Holdings, Inc.” The amendment and restatement of the certificate of incorporation effectuating the name change and above authorized share increases were approved by our stockholders at the special stockholder meeting on September 8, 2014 and by our Board of Directors at a meeting of the Board held on September 8, 2014.
On September 8, 2014, following the filing of the amended and restated certificate disclosed above, MabVax Therapeutics Holdings filed a certificate of amendment to the amended and restated certificate of incorporation to effect an 8-for-1 reverse stock split on common stock (the “Reverse Split”), effective as of 4:01 p.m. Eastern Time (the “Effective Time”) on September 8, 2014 (the “Effective Date”). The Reverse Split was approved by our stockholders at the special stockholder meeting held on September 8, 2014 and by the Board of Directors at a meeting of the Board held on September 8, 2014.
On the Effective Date, immediately and without further action by our stockholders, every 8 shares of our common stock, issued and outstanding immediately prior to the Effective Time, were automatically converted into 1 share of our common stock. As a result of the Reverse Split and calculated as of the Record Date, the number of outstanding shares of our common stock was reduced from 13,932,937 to 1,741,617, excluding outstanding and unexercised share options and warrants and subject to adjustment for fractional shares. No fractional shares were issued as a result of the Reverse Split and, in lieu of these fractional shares, any holder of less than 1 share of our common stock was entitled to receive cash for such holder’s fractional share equal to the product of such fraction multiplied by the average of the last reported bid and ask prices of our common stock at 4:00 p.m., Eastern time, end of regular trading hours on OTCQB marketplace, during the 10 consecutive trading days ending on the last trading day prior to the Effective Date. Further, any options, warrants and contractual rights outstanding as of the Effective Date that were subject to adjustment were adjusted in accordance with their terms. These adjustments included, without limitation, changes to the number of shares of our common stock that may be obtained upon exercise or conversion of these securities, and changes to the applicable exercise or purchase price of such securities.
Shares of our common stock began to trade on the OTCQB marketplace on a post-split basis under the name MabVax Therapeutics Holdings, Inc. on September 10, 2014 under the new CUSIP number 55414P108. MabVax Therapeutics Holdings retained the same CUSIP number when its common stock began trading on the OTCQB marketplace under the trading symbol MBVX on October 10, 2014.
All prior periods in these consolidated financial statements have been adjusted to reflect the effects of the Merger and the Reverse Split, unless otherwise indicated.
6. Merger with MabVax Therapeutics, Inc.
On May 12, 2014, the Company entered into a Merger Agreement. Upon the terms and subject to the satisfaction of the conditions described in the Merger Agreement, Tacoma Corp. was merged with and into private company MabVax Therapeutics on July 8, 2014, with MabVax Therapeutics surviving the Merger as a wholly-owned subsidiary of MabVax Therapeutics Holdings. The Merger is intended to qualify as a tax-free reorganization for U.S. Federal income tax purposes.
On July 7, 2014, the stockholders of MabVax Therapeutics Holdings approved the Merger, and the Merger closed and became effective on July 8, 2014. At the effective date of the Merger: (a) all shares of MabVax Therapeutics Series A
F-12
Table of Contents
preferred stock and all shares of MabVax Therapeutics Series B preferred stock were automatically converted into shares of MabVax Therapeutics Holdings common stock, (b) all outstanding shares of MabVax Therapeutics common stock were converted into and exchanged for shares of MabVax Therapeutics Holdings common stock at an exchange rate calculated in accordance with the methodology set forth in the Merger Agreement, which resulted in 2.223284 shares of MabVax Therapeutics Holdings common stock for every share of MabVax Therapeutics common stock, (c) all outstanding shares of MabVax Therapeutics Series C-1 preferred stock were converted into and exchanged for shares of MabVax Therapeutics Holdings Series A-1 preferred stock at a rate of two shares of MabVax Therapeutics Series C-1 per each share of MabVax Therapeutics Holdings Series A-1 preferred stock, (d) each outstanding MabVax Therapeutics option and warrant to purchase MabVax Therapeutics common stock became options and warrants to purchase shares of MabVax Therapeutics Holdings common stock (and the number of such shares and exercise price was adjusted as calculated in accordance with the methodology set forth in the Merger Agreement), and (e) each outstanding MabVax Therapeutics warrant to purchase MabVax Therapeutics preferred stock was cancelled for no consideration.
As a result of the consummation of the Merger, as of the closing date, the former stockholders, option holders and warrant holders of MabVax Therapeutics were issued, based on the methodology set forth in the Merger Agreement (which excluded certain out of the money convertible securities and calculated others on a net-exercise or cashless basis under the terms of the convertible securities), approximately 85% of the outstanding shares of MabVax Therapeutics Holdings common stock on a fully diluted basis and the stockholders, option holders and warrant holders of MabVax Therapeutics Holdings prior to the Merger owned approximately 15% of the outstanding shares of MabVax Therapeutics Holdings common stock on a fully diluted basis (such percentages calculated based on the methodology set forth in the Merger Agreement). As a result of the Merger, a change of control of MabVax Therapeutics Holdings occurred.
For accounting purposes, the Merger is treated as a “reverse acquisition”. The private company MabVax Therapeutics is considered the accounting acquirer, and the public company MabVax Therapeutics Holdings is considered the legal acquirer and accounting acquiree. The private company MabVax Therapeutics is the accounting acquirer because it owns a majority of the merged company (approximately 85%). As a result, the historical financial statements of the private company MabVax Therapeutics constitute the historical financial statements of the merged companies. The transaction is considered a business combination as MabVax Therapeutics Holdings is considered an operating entity. For accounting purposes, MabVax Therapeutics is treated as the continuing reporting entity.
The issuance of shares of our common stock and preferred stock in the Merger was approved by our stockholders in the annual stockholder meeting held on July 7, 2014. Amendments to our amended and restated certificate of incorporation related to an increase in the authorized number of shares of our common stock and preferred stock and a proposed reverse stock split to maintain Nasdaq listing maintenance standards and other transactions contemplated by the Merger Agreement were not approved at this meeting. As a result of our not getting stockholder approval of a proposed reverse stock split at the July 7, 2014 annual stockholders’ meeting, we were unable to meet all of the listing requirements for the Nasdaq Exchange and our common stock began trading on the OTCQB market under the stock symbol MBVX. There is no impact on accounting for the Merger on July 8, 2014, as a result of not getting stockholder approval on all matters presented at the July 7, 2014 annual meeting.
The purchase price is based upon the fair value of MabVax Therapeutics Holdings (f.k.a. Telik, Inc.) common stock outstanding of 572,887 shares as of July 8, 2014, multiplied by the stock closing price at July 8, 2014 of $11.20, or approximately $6,416,000. The consideration transferred is based on the market price of MabVax Therapeutics Holdings since management has determined that this was the most reliable measure of fair value, taking into consideration a third party valuation we received for financial reporting purposes as outlined under the Financial Accounting Standards Board Accounting Standards Codification Topic 805: Business Combination in connection with the Merger.
F-13
Table of Contents
The total estimated purchase price of the acquisition as of July 8, 2014 is as follows:
Purchase Consideration:
| (In thousands) | ||||||||
| Purchase Consideration |
$ | 6,416 | ||||||
| Telik Assets: |
||||||||
| Cash and Cash Equivalents |
$ | 1,497 | ||||||
| Accounts Receivable |
31 | |||||||
| Prepaids and Other Current Assets |
182 | |||||||
|
|
|
|||||||
| (1,710 | ) | |||||||
| Telik Liabilities: |
||||||||
| Accrued Compensation |
$ | 850 | ||||||
| Accrued Liabilities |
111 | |||||||
| Accrued Contingent Termination Fee |
591 | |||||||
| Warrant Liability |
568 | |||||||
|
|
|
|||||||
| 2,120 | ||||||||
|
|
|
|||||||
| Goodwill |
$ | 6,826 | ||||||
|
|
|
|||||||
The Company noted a triggering event relating to its goodwill due to the decrease in public market cap during the year ended December 31, 2014. Factors contributing to a low implied value of the Company on the stock exchange included thinly traded stock and significant stock sales from a significant investor, as well as lack of visibility on public exchanges of potentially dilutive securities that are disclosed in the Company’s public filings, that if converted would show substantially more shares outstanding than reported on public stock exchanges. Therefore, the Company performed a step 1 analysis using an independent valuation firm to determine if there was in fact an impairment of goodwill that needed to be recorded. The valuation took into consideration a recent re-capitalization and financing for the Company as a basis for determining the valuation of the Company and the Company concluded that no impairment had taken place. Goodwill is not deductible for tax purposes.
7. Redeemable Convertible Preferred Stock, Convertible Preferred Stock, Common Stock and Warrants
MabVax Therapeutics Series A and MabVax Therapeutics Series B preferred stock (Pre-Merger MabVax Therapeutics Issuances)
During February 2013 through December 2013, the Company sold an additional 410,557 shares of MabVax Therapeutics Series B redeemable convertible preferred stock in exchange for $2,792,993 in funds, net of issuance costs of $7,007. The Company also issued warrants to purchase an additional 194,281 shares of MabVax Therapeutics Series B redeemable convertible preferred stock at an exercise price of $0.01 per share (the “Series B Warrant”). The Series B Warrant is exercisable immediately and has a term of five years. Because the Series B Warrant is immediately convertible at the option of the holder, the Company recorded a deemed dividend of $691,812 from the beneficial conversion feature associated with the issuance of the MabVax Therapeutics Series B redeemable convertible preferred stock and the Series B Warrant.
The Company valued the warrants at fair value at the date the warrants were issued, using the Black Scholes valuation model with the following assumptions; contractual term of five years, volatility of 86%, no dividend yield and a risk-free interest rate of 0.28%.
As of December 31, 2013, the holders of shares of MabVax Therapeutics Series A redeemable convertible preferred stock and MabVax Therapeutics Series B redeemable convertible preferred stock were entitled to cumulative cash dividends of 8% per annum, when and if declared by the MabVax Therapeutics Board of Directors. Such dividends would have been in preference to and prior to any payment of any dividend on shares of MabVax Therapeutics common stock. Cumulative preferred stock dividends, when and if declared, for the MabVax Therapeutics Series A redeemable convertible preferred stock totaled $2,114,818 and the MabVax Therapeutics Series B redeemable convertible preferred stock totaled $430,944, as of December 31, 2013, and were reduced to zero in February 2014 as a result of the MabVax Therapeutics Series C-1 Preferred Stock Financing.
In January 2014, holders of warrants to purchase shares of MabVax Therapeutics Series B redeemable convertible preferred stock exercised their rights to purchase 194,281 shares of MabVax Therapeutics Series B redeemable convertible preferred stock for proceeds of $1,942.
F-14
Table of Contents
In February 2014, the holders of MabVax Therapeutics Series A redeemable convertible preferred stock and MabVax Therapeutics Series B redeemable convertible preferred stock waived any rights to all prior accrued dividends they may have had a right to receive and amended the MabVax Therapeutics certificate of incorporation to eliminate their right to accrue dividends in the future as an inducement to buyers in the MabVax Therapeutics Series C-1 Preferred Stock Financing. The effect of this change reduced the liquidation preference for the MabVax Therapeutics Series A redeemable convertible preferred stock by $2,187,762 and the MabVax Therapeutics Series B redeemable convertible preferred stock by $486,938 as of February 12, 2014.
No dividends were ever declared by the MabVax Therapeutics Board of Directors since MabVax Therapeutics’ inception on either of the MabVax Therapeutics Series A redeemable convertible preferred stock or the MabVax Therapeutics Series B redeemable convertible preferred stock.
Removal of Redemption Rights – As of December 31, 2013, the holders of a majority interest of the MabVax Therapeutics Series A redeemable convertible preferred stock and MabVax Therapeutics Series B redeemable convertible preferred stock held a right to redeem (the “MabVax Therapeutics Redemption Right”), at any time on or after the fifth anniversary of the issuance date, upon request of at least 60% of the holders thereof, all of their preferred stock at a redemption price of $6.17 and $6.82 per share of MabVax Therapeutics Series A redeemable convertible preferred stock and MabVax Therapeutics Series B redeemable convertible preferred stock, respectively, exclusive of dividends. Due to these terms, MabVax Therapeutics classified all of the MabVax Therapeutics preferred stock as mezzanine equity (outside of permanent equity) as of December 31, 2013. In March 2014, the majority of holders, or more than 60%, of the MabVax Therapeutics Series A redeemable convertible preferred stock and MabVax Therapeutics Series B redeemable convertible preferred stock agreed by letter commitment to MabVax Therapeutics to relinquish the MabVax Therapeutics Redemption Right, and MabVax Therapeutics reclassified the presentation on the consolidated balance sheets as permanent equity following the agreement.
Liquidation preference – As of December 31, 2013, in the event of any voluntary or involuntary liquidation, dissolution or winding up of MabVax Therapeutics, the MabVax Therapeutics Series A redeemable convertible preferred stockholders and MabVax Therapeutics Series B redeemable convertible preferred stockholders were entitled to be paid an amount equal to $6.17 and $6.82 per share, respectively, plus all declared and unpaid dividends, as adjusted to reflect any stock splits, stock dividends or other recapitalization. In addition, after setting apart or paying in full the MabVax Therapeutics Series A redeemable convertible preferred stock and MabVax Therapeutics Series B redeemable convertible preferred stock liquidation preference, any remaining assets of MabVax Therapeutics available for distribution to its stockholders would have been distributed to all stockholders of MabVax Therapeutics with holders of MabVax Therapeutics preferred stock participating on an as converted basis without actually converting their MabVax Therapeutics preferred stock into shares of MabVax Therapeutics common stock. In the event that upon liquidation or dissolution, the assets and funds of MabVax Therapeutics would have been insufficient to permit the payment to MabVax Therapeutics preferred stockholders of the full preferential amounts, then the entire assets and funds of MabVax Therapeutics legally available for distribution were to be distributed ratably first to the holders of MabVax Therapeutics Series B preferred stock, second to the holders of MabVax Therapeutics Series A preferred stock and third on a pro rata basis to all stockholders of MabVax Therapeutics on an as-converted basis.
Series C-1 preferred stock purchase agreement
On February 12, 2014, MabVax Therapeutics entered into a Securities Purchase Agreement (the “MabVax Therapeutics Securities Purchase Agreement”) and issued 3,697,702 shares of MabVax Therapeutics Series C-1 preferred stock, warrants to purchase 2,055,260 shares of MabVax Therapeutics common stock at $3.62 a share (the “MabVax Therapeutics Series C Common Warrants”) and warrants to purchase 1,848,851 shares of MabVax Therapeutics Series C-1 preferred stock at $0.84 a share (the “MabVax Therapeutics Series C Preferred Warrants”), respectively, for aggregate gross proceeds of $3,100,000, less issuance costs of $126,345 (the “MabVax Therapeutics Series C-1 Financing”). The MabVax Therapeutics Series C Common Warrants and Preferred Warrants were exercisable immediately. The MabVax Series C Common Warrants would have expired on February 13, 2022, and the MabVax Therapeutics Series C Preferred Warrants would have expired upon registration of the shares of MabVax Therapeutics common stock (or a successor entity) under the Securities Act. Because the warrants are immediately convertible at the option of the holder, MabVax Therapeutics recorded a deemed dividend of $2,214,911 from the beneficial conversion feature associated with the issuance of the MabVax Series C-1 preferred stock and the MabVax Therapeutics Series C Common Stock Warrants and the MabVax Therapeutics Series C Preferred Stock Warrants.
In connection with the MabVax Therapeutics Series C-1 Financing, MabVax Therapeutics agreed to use its reasonable best efforts to raise at least an additional $3,000,000 through the sale and issuance of shares of MabVax Therapeutics common stock initially intended to be at $15.08 per share (the “Subsequent Capital Raise”). Substantially all of the investors
F-15
Table of Contents
in the MabVax Therapeutics Series C-1 Financing executed a financing commitment letter (such letters, the “Financing Commitment Letters”) to purchase a pro rata number of shares of MabVax Therapeutics common stock at the purchase price of $15.08 per share, representing in the aggregate at least $750,000, subject to certain terms and conditions, including a condition that MabVax Therapeutics raise at least $3,000,000 from new investors in the Subsequent Capital Raise. In addition, each such commitment letter provided that, in the event that less than $3,000,000 was raised from new investors in the Subsequent Capital Raise and subject to certain terms and conditions, each investor party to such letter was required to purchase shares of MabVax Therapeutics preferred stock to be designated as MabVax Therapeutics Series C-2 convertible preferred stock at $15.08 per share and in the aggregate amount of up to $3,000,000 (the “Backstop Capital Raise”).
On May 12, 2014, MabVax Therapeutics and certain investors amended the MabVax Therapeutics Securities Purchase Agreement to, among other things, (i) lower the price per share of the Subsequent Capital Raise from $15.08 to $9.93 per share, and (ii) provide that the price per share payable by investors as set forth in the Financing Commitment Letters would henceforth be the lower of (A) $15.08 a share and (B) the lowest price paid in the Subsequent Capital Raise. The price per share of the Backstop Capital Raise was not changed as a result of the amendment. On July 7, 2014, prior to the Merger, MabVax Therapeutics raised over $3.0 million from the sale of common stock and the Backstop Capital Raise was no longer in effect.
The MabVax Therapeutics Series C-1 preferred stock allowed the holders to require that MabVax Therapeutics redeem their shares of MabVax Therapeutics Series C-1 preferred stock, including any accrued but unpaid dividends, upon the occurrence of any of the following events (each, a “Triggering Event”): (i) the suspension of trading of common stock following registration of such shares, (ii) the failure to issue shares of MabVax Therapeutics common stock upon conversion of any MabVax Therapeutics Series C-1 preferred stock, (iii) the failure to authorize sufficient shares of MabVax Therapeutics common stock to permit the conversion of all outstanding shares of MabVax Therapeutics Series C-1 preferred stock and exercise of all MabVax Therapeutics Series C Common Warrants and MabVax Therapeutics Series C Warrants, (iv) failure to make certain required payments to the holders in excess of $25,000, (v) a default on indebtedness in the aggregate amount of $100,000, (vi) bankruptcy events, (vii) judgments requiring payments in excess of $100,000, (viii) consummation of a change of control with an entity which did not have a class of securities registered for trading, (ix) failure of MabVax Therapeutics to initiate the process of becoming publicly traded (either through a merger into a public company or the filing of a registration statement) within 4 months of the closing of the MabVax Therapeutics Series C-1 Financing, (x) failure to complete such Merger within one year or such registration within 4 months of the closing of the MabVax Therapeutics Series C-1 Financing, (xi) issuance of common stock in violation of certain restrictions relating to employee equity, (xii) issuance of debt in violation of any agreement relating to the MabVax Therapeutics Series C-1 Financing, (xiii) failure to convert MabVax Therapeutics Series A preferred stock or MabVax Therapeutics Series B preferred stock on or prior to the date shares of MabVax Therapeutics common stock became publicly tradable, (xiv) any deviation of 20% or more from the annual budget approved by such holders, (xv) any deviation of 5% or more with respect to auditing and investors’ relations expenses, (xvi) failure to deliver the 2013 audited financials within 45 days of the closing of the MabVax Therapeutics Series C-1 Financing, (xvii) any deviation of any line item of the 2013 audited financials from those set forth in the 2013 unaudited financials delivered in connection with the MabVax Therapeutics Series C-1 Financing or (xviii) a breach of any representation, warranty, covenant or other term or condition of any agreement relating to the MabVax Therapeutics Series C-1 Financing. Certain Triggering Events had occurred as of May 9, 2014, but were subsequently waived by the holders of the MabVax Therapeutics Series C-1 preferred stock.
On July 8, 2014, the date of the Merger, all MabVax Therapeutics Series C-1 preferred stock was converted into shares of MabVax Therapeutics Holdings Series A-1 preferred stock, and the Triggering Events were removed. Because of the removal of the Triggering Events as of the Merger date, the MabVax Therapeutics Holdings Series A-1 convertible preferred stock is presented on the consolidated balance sheet as permanent equity as of December 31, 2014.
Conversion
After giving effect to the Merger and Reverse Split, the holders of our Series A-1 preferred stock may at any time voluntarily convert each share into a number of fully paid shares of our common stock determined by dividing the liquidation preference (described below) by the initial conversion price of $1.6767 per share. Conversion is subject to (a) proportional adjustment for certain dilutive issuances, splits, combinations and other recapitalizations or reorganizations and (b) a full ratchet anti-dilution adjustment upon issuance of shares of common stock (or securities convertible into shares of common stock) at a price per share (or with a conversion or exercise price per share) less than the applicable conversion price, and subject to customary carve outs and exclusions.
Under the terms described for a mandatory conversion, all outstanding shares of our Series A-1 preferred stock shall be automatically converted into shares of our common stock upon the affirmative election of the holders of a majority of the
F-16
Table of Contents
issued and outstanding shares of our Series A-1 preferred stock. In the event that the Company does not issue the shares of its common stock upon conversion of any shares of its Series A-1 preferred stock, certain penalties, which may be paid in the form of cash or additional shares of its common stock, will accrue. The number of shares of our common stock issuable upon conversion of our Series A-1 preferred stock held by any particular holder, together with all affiliates of such holder, is capped at 4.99% of the issued and outstanding shares of common stock of the Company. Any shares in excess of such amount will be held in abeyance until such time as the issuance of such shares of common stock would not put such holder, together will all affiliates of such holder, above 4.99%. An individual holder may elect to increase this limit to up to 9.99% effective 61 days after providing notice to the Company.
Dividends
The Company’s Series A-1 stockholders are entitled to cumulative dividends on each share held at a rate of 8% per annum on the Stated Value (as defined in the Series A-1 certificate of designations) from and after the first date of issuance of any Series A-1 whether or not declared by the Board and whether or not there are funds legally available for the payment of dividends. Such dividends are in preference to and prior to any payment of any dividend on shares of our Series B preferred stock, our Series C preferred stock or our common stock. If any dividend is declared and paid on any shares of our common stock, Series B preferred stock or Series C preferred stock, a dividend shall be declared and paid on shares of our Series A-1 preferred stock on an “as converted” basis. The Company is accreting the dividends in accordance with the agreement.
Liquidation preference
In the event of any voluntary or involuntary liquidation, dissolution or winding up of the Company, our Series A-1 preferred stockholders shall be paid an amount equal to $1.6767 per share, plus all accrued dividends, as adjusted to reflect any stock splits, stock dividends or other recapitalization. In addition, after setting apart or paying in full the Series A-1 preferred stock, and Series B preferred stock liquidation preference, any remaining assets of the Company available for distribution to stockholders, if any, shall be distributed to all stockholders of the Company with holders of our preferred stock participating on an as converted basis without actually converting their preferred stock into common stock.
In the event that upon liquidation or dissolution, the assets and funds of the Company are insufficient to permit the payment to its preferred stockholders of the full preferential amounts, then the entire assets and funds of the Company legally available for distribution are to be distributed ratably first to the holders of shares of our Series A-1 preferred stock, second to holders of our Series B preferred stock and third on a pro rata basis to all stockholders of the Company on an as-converted basis.
Voting rights
Each holder of our Series A-1 preferred stock is entitled to the number of votes equal to the number of shares of our common stock into which such holder’s shares are convertible. In addition, the consent of the Required Holders (as defined in the Series A-1 preferred stock certificate of designations) is required in certain circumstances.
Registration of Common Stock Issuable upon Conversion of Series A-1 Preferred Stock, and Conversions
On October 14, 2014, the Company filed an Amendment No. 1 to a Registration Statement on Form S-1 (the “Form S-1”) that was initially filed on September 29, 2014, for the purpose of registering additional shares of MabVax Therapeutics Holdings common stock issuable upon conversion of outstanding shares of MabVax Therapeutics Holdings Series A-1 preferred stock. The Form S-1, as amended, to register 1,615,070 shares of common stock, was declared effective by the SEC at 4:00 p.m. Eastern Standard Time on November 12, 2014.
From November 13, 2014, to December 31, 2014, holders of Series A-1 preferred stock converted 1,169,452 shares into 693,335 shares of common stock.
Exercise of MabVax Therapeutics Series C Preferred Warrants
On July 7, 2014, MabVax Therapeutics received $1.5 million in exchange for the exercise by holders of the MabVax Therapeutics Series C Preferred warrants to purchase 1,827,979 shares of MabVax Therapeutics Series C-1 preferred stock.
F-17
Table of Contents
MabVax Therapeutics Holdings Series B Redeemable Convertible Preferred Stock
On May 12, 2014 (the “Closing Date”), MabVax Therapeutics Holdings entered into a securities purchase agreement (the “Series B Purchase Agreement”) with certain purchasers the “Purchasers” pursuant to which MabVax Therapeutics Holdings agreed to issue and sell to the Purchasers, subject to customary closing conditions, an aggregate of 1,250,000 shares of MabVax Therapeutics Series B redeemable convertible preferred stock and warrants (the “Series B Common Warrants”) to purchase up to an additional 78,125 shares of MabVax Therapeutics Holdings common stock, with an aggregate purchase price of $2,500,000, or $2.00 for each share of our Series B redeemable convertible preferred stock and related Series B Common Warrant (such transaction collectively, the “Series B Private Placement”). The closing of the Series B Private Placement took place on the Closing Date.
On May 8, 2014, MabVax Therapeutics Holdings filed a certificate of designation for the MabVax Therapeutics Holdings Series B preferred stock with the Secretary of State of the State of Delaware. The certificate of designations authorized 1,250,000 shares of Series B preferred stock. Holders of MabVax Therapeutics Series B redeemable convertible preferred stock (the “Holders”) are entitled to cumulative dividends on each share held at a rate of 8% per annum on the Stated Value (as defined in the certificate of designations). Upon a liquidation event, the Holders are entitled to a liquidation preference per share, prior to any distribution of the Company’s assets to the holders of its common stock, in an amount equal to the Stated Value plus accrued and unpaid dividends. After payment to the Holders of the full preferential amount, the Holders will, on a pari passu basis with the holders of the Company’s common stock, participate in the distribution of any remaining assets of the Company, subject to certain limitations. Each Holder may elect to convert their Series B preferred stock into shares of the Company’s common stock at the applicable conversion rate in effect at the time of such conversion. However, the Company shall not effect conversion of the Series B redeemable convertible preferred stock to the extent such conversion would result in the beneficial owner acquiring beneficial ownership of more than 4.99% of the Company’s outstanding common stock post-conversion, including any shares of its common stock issuable upon exercise or conversion of other convertible securities held by such beneficial owner. The Company obtained stockholder approval for the securities being issued in the Series B Private Placement at the annual stockholder meeting held on July 7, 2014. The conversion rate is subject to full ratchet anti-dilution protection upon certain dilutive issuances of our common stock or convertible securities of the Company. Such conversion price will be subject to adjustment from and after the earlier of: (i) the date that some or all of the Registerable Securities (as defined below) have become registered pursuant to an effective registration statement and (ii) six months after the Closing Date at which time the conversion price of the Series B preferred stock shall equal the lower of (a) the initial conversion price and (b) 90% of the average of the 10 lowest weighted average prices of the Company’s common stock during the 20 trading days immediately preceding applicable date of the conversion, of which the latter condition was reached on November 14, 2014. The Holders may also require the Company to redeem their shares of Series B redeemable convertible preferred stock prior to a change of control, as set forth in the certificate of designations. The certificate of designations further provides that the Holders are entitled to certain participation rights on issuances by the Company to holders of common stock in order to maintain their proportionate ownership, subject to certain customary exclusions, such as issuances pursuant to Company option plans, and in connection with the Merger.
The Series B Common Warrants became exercisable six months from the Closing Date, or November 12, 2014, expire five years from the Closing Date and may be exercised for cash or otherwise may be net-exercised. The Series B Common Warrants initially had a per share exercise price of $26.64. On the 60th day following the earlier of (i) the date all of the shares underlying the Warrants become registered pursuant to an effective registration statement and (ii) six months following the Closing Date (in each case, the “Reset Date”), the exercise price shall be reset to equal the lower of (i) the current exercise price and (ii) 90% of the average of the 10 lowest weighted average prices of Common Stock during the 20 trading days immediately preceding the Reset Date. The price was reset to $1.57 on January 11, 2015. The exercise price is subject to full ratchet anti-dilution adjustment for any issuances of common stock and convertible securities for common stock below the current conversion price, consistent with the terms of the Series B preferred stock.
In connection with the Series B Private Placement, the Company also entered into a Registration Rights Agreement with the Purchasers (the “Series B Registration Rights Agreement”). Pursuant to the Series B Registration Rights Agreement, the Company agreed to file a registration statement with the SEC covering resales of the Warrant Shares and the shares issuable upon conversion of the Series B preferred stock (together, the “Series B Registerable Securities”) by the Purchasers no later than 60 days following the Closing Date, and to use its commercially reasonable best efforts to have such registration statement declared effective as soon as practicable. The Company bears all expenses of such registration of the resale of the Registerable Securities. On September 3, 2014, the Required Holders (as defined in the Series B preferred stock certificate of designations) temporarily waived the 60 day registration deadline for a five day period.
As a result of the Series B Warrants’ anti-dilution provision, the Series B Warrants are recorded as a current liability on our consolidated balance sheet. The outstanding warrant was valued at $92,463 and $567,885 as of December 31, 2014, and July 8, 2014 or the acquisition date, respectively. Our outstanding warrants are revalued on each balance sheet date, with changes in the fair value between reporting periods recorded in the consolidated statements of operations.
F-18
Table of Contents
Warrants are valued using the Black-Scholes-Merton model. The warrant has only partial down round protection, as it has a price reset only on a down round financing, and not an increase in number of shares convertible with the warrant. The Company concluded that using the Black-Scholes-Merton model for the valuation as of December 31, 2014, is fairly accurate compared to a recent buyout offer. The fair value of warrants is estimated using the following assumptions, which, except for risk-free interest rate, are Level 3 inputs:
Warrant liability valuation assumptions
| As of December 31, 2014 |
As of July 8, 2014 |
|||||||
| Risk-free interest rate |
1.75 | % | 1.60 | % | ||||
| Dividend yield |
— | % | — | % | ||||
| Expected volatility |
86.67 | % | 101.60 | % | ||||
| Expected life of options, in years |
4.36 | 4.90 | ||||||
| Market price for common stock |
$ | 1.82 | $ | 11.60 | ||||
| Warrant exercise price, adjusted |
$ | 1.80 | $ | 26.64 | ||||
The following table presents information about our financial instruments that are measured at fair value on a recurring basis as of December 31, 2014 and indicates the fair value hierarchy of the valuation techniques utilized to determine such fair value:
| Basis of Fair Value Measurement at December 31, 2014 | ||||||||||||||||
| December 31, 2014 | Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
|||||||||||||
| Financial liabilities: |
||||||||||||||||
| Warrants |
$ | 92,463 | $ | — | $ | — | $ | 92,463 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total financial liabilities |
$ | 92,463 | $ | — | $ | — | $ | 92,463 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The changes in the value of the warrant liability during the year ended December 31, 2014 were as follows:
| Fair value - beginning of year |
$ | — | ||
| Fair value on acquisition |
567,885 | |||
| Change in fair value |
(475,422 | ) | ||
|
|
|
|||
| Fair value - end of year |
$ | 92,463 | ||
|
|
|
There were no transfers between Level 1 and Level 2 measurements for the years ended December 31, 2014 and no required disclosure as of December 31, 2013.
Exchange Agreement and Series C Preferred Stock
On September 3, 2014, MabVax Therapeutics Holdings and certain holders of its issued and outstanding common stock entered into an Exchange Agreement (the “Exchange Agreement”) pursuant to which such holders agreed to exchange 148,713 shares of MabVax Therapeutics Holdings common stock for an aggregate of 118,970 shares of newly designated MabVax Therapeutics Holdings Series C preferred stock. From October to December 2014, holders converted 22,399 shares of Series C preferred stock into 28,000 shares of common stock.
F-19
Table of Contents
As contemplated by the Exchange Agreement and as approved by the Board of Directors, the Company filed with the Secretary of State of the State of Delaware a certificate of designations for the Series C preferred stock, on September 3, 2014. Holders of the Series C preferred stock are entitled to vote on an as converted basis on matters presented to the Company’s stockholders and, upon liquidation, share in distributions on a pari passu basis with the holders of the Company’s common stock in amounts available for distribution following payments required to be made to the holders of the Series A-1 preferred stock and Series B preferred stock. Each share of Series C preferred stock is convertible into 1.25 shares of our common stock subject to adjustment and the conversion limitations set forth in the Series C certificate of designations. When and as declared by the Board of Directors, the holders of the Series C preferred stock shall be entitled to receive dividends on an as converted basis (without regard to any limitations on conversion) with the holders of the Company’s common stock.
The terms of the Exchange Agreement and Series C Certificate of Designations were determined by arms-length negotiation between the parties. The shares of common stock issuable pursuant to the Exchange Agreement have been, or will be, upon settlement, issued in reliance on the exemption from registration contained in Section 3(a)(9) of the Securities Act for securities exchanged by an issuer and an existing security holder where no commission or other remuneration is paid or given directly or indirectly by the issuer for soliciting such exchange.
MabVax Common Stock Financing
From June 27 to July 7, 2014, MabVax Therapeutics Holdings issued approximately 326,000 shares of common stock for aggregate proceeds of approximately $2,884,000, net of issuance costs of approximately $156,000, in a private placement transaction (the “MabVax Common Stock Private Placement”), pursuant to Common Stock Purchase Agreements by and among MabVax Therapeutics and certain institutional investors party thereto (the “MabVax Purchase Agreements”). Pursuant to the MabVax Purchase Agreements, MabVax Therapeutics agreed to issue the purchasers participating in closings held under the MabVax Common Stock Private Placement prior to the closing of the Merger additional “anti-dilution” shares of MabVax Therapeutics common stock, for no additional consideration should MabVax Therapeutics sell shares of its common stock in the future (subject to certain customary exceptions, such as upon the conversion or exercise of then outstanding convertible securities, the securities issued in the Merger and issuances under the MabVax Therapeutics option plan) at a price lower than $9.14 per share prior to the first to occur of (x) December 31, 2015 and (y) the date on which MabVax Therapeutics raises an aggregate of $10,000,000. The number of additional shares would be calculated on a weighted average based on the price per share of equity securities sold by MabVax Therapeutics following the initial closing of the MabVax Common Stock Private Placement and in no event would a purchaser be issued a number of additional shares of MabVax Therapeutics common stock in excess of 33% of the number of shares initially purchased by such purchaser and held as of the date of any anti-dilution adjustment. These shares of MabVax Therapeutics common stock issued in the MabVax Common Stock Private Placement were converted into shares of MabVax Therapeutics Holdings common stock in connection with the Merger. MabVax Therapeutics’ obligations with respect to the anti-dilution provisions in the Merger were assumed by MabVax Therapeutics Holdings, and these provisions now apply to sales of MabVax Therapeutics Holdings common stock. As of December 31, 2014, no sales of common stock had taken place since the MabVax Common Stock Private Placement that would have caused the issuance of anti-dilution shares.
Temporary Waiver of Warrant Exercise Period
On the effective date of the Merger and pursuant to the Merger Agreement, MabVax Therapeutics Holdings issued as part of its securities to the holders of MabVax Therapeutics in exchange for securities owned by MabVax Therapeutics’ security holders, warrants to purchase up to an aggregate of 2,055,268 shares of MabVax Therapeutics Holdings common stock, with an exercise price of $3.62 per share and expiring on July 10, 2023 (the “Merger Warrants”).
The preamble of the Merger Warrants contains limitations prohibiting the Merger Warrant holders from exercising the Merger Warrants prior to the one year anniversary of the effective date of the Merger, or July 8, 2015.
On September 3, 2014, the Company sent a letter to the holders of the issued and outstanding Merger Warrants (the “Waiver Letter”), waiving, on a limited basis from September 3 through September 12, 2014, the requirement set forth in the preamble of the Merger Warrants that the Merger Warrants may not be exercised until July 8, 2015, and permitting the Merger Warrants to be exercised, either through payment of the exercise price or on a net “cashless” basis, at any time during the period commencing on the date of the letter and ending on and including September 12, 2014 (the “Waiver Period”). The Waiver Letter also provides that, with respect to exercises pursuant to the Waiver Letter during the Waiver Period, the number of shares of common stock issuable upon cashless exercise shall be determined in accordance with the formula set forth in the Waiver Letter rather than the formula set forth in Section 1(d) of the Merger Warrant.
F-20
Table of Contents
On October 3, 2014, following the Company’s delivery on September 30, 2014, of a second letter to the holders of the issued and outstanding Merger Warrants (the “Waiver Extension Letter”), waiving, on a limited basis for a four day period, the requirement set forth in the preamble of the Merger Warrants that the Merger Warrants may not be exercised until July 8, 2015, and permitting the Merger Warrants to be exercised, either through payment of the exercise price or on a net “cashless” basis, at any time during the period commencing on the date of the letter and ending on and including October 3, 2014 (the “Waiver Extension Period”). The Waiver Extension Letter also provides that, with respect to exercises pursuant to the Waiver Extension Letter during the Waiver Extension Period, the number of shares of the Company’s common stock issuable upon cashless exercise shall be determined in accordance with the formula set forth in the Waiver Extension Letter rather than the formula set forth in Section 1(d) of the Merger Warrant.
The Company’s management issued the temporary waiver of the warrant exercise period with the intention of gradually increasing the number of its publicly held shares in furtherance of the Company’s continued efforts to satisfy NASDAQ’s Initial Listing Standards and regain trading eligibility for shares of its common stock on the NASDAQ Capital Market. Shares of the Company’s common stock issued upon exercise of the Merger Warrants will not be registered for resale during the Waiver Extension Period and will be subject to resale restrictions per Rule 144 as promulgated by the Securities Act.
For the year ended December 31, 2014, 488,659 additional shares of the Company’s common stock had been issued pursuant to the exercise and delivery of 775,219 Merger Warrants in accordance with the terms of the Waiver Letter and the Waiver Extension Letter. As of December 31, 2014, the number of warrants outstanding was 1,280,049 shares and 78,125 shares of the Merger Warrants exercisable into common stock and the Series B Common Warrants, respectively.
8. Related Party Transactions
The Company incurred consulting fees of $240,000 with a former board member and another founder of the Company during the year ended December 31, 2013. The Company recorded a $240,000 related party liability as of December 31, 2013.
In February 2014, MabVax Therapeutics issued approximately 44,000 shares of common stock to related parties in settlement of $240,000 in related party liabilities for consulting services.
In connection with the Merger, MabVax Therapeutics Holdings (f.k.a. Telik, Inc.) signed separation agreements in May 2014 with nine employees and agreed to pay severances and health benefits upon closing of the Merger subject to certain provisions in the agreement. The total in severance and benefits costs to be paid out subsequent to the Merger is approximately $748,000. At December 31, 2014, the accrued severance and benefits costs are approximately $6,000.
9. Stock-based Activity
Stock Incentive Plan
In September 2008, the Company’s stockholders approved the 2008 Stock Incentive Plan (the “2008 Plan”) which became effective in September 2008 and under which 65,507 shares of the Company’s common stock were initially reserved for issuance to employees, non-employee directors and consultants of the Company. In November 2012, the Company increased the authorized shares under the plan to 155,893. On February 14, 2013, the 2008 Plan terminated and no further grants of equity may be made thereunder.
In June 2014, MabVax Therapeutics Inc.’s stockholders approved the amended 2014 Stock Incentive Plan (the “2014 Plan”) which became effective and was adopted by the Company in the Merger in July 2014. The 2014 Plan authorized the issuance of up to 351,443 shares, 152,017 of which are contingent upon the forfeiture, expiration or cancellation of the 2008 Reserved Shares.
The 2014 Plan provides for the grant of incentive stock options, non-incentive stock options, stock appreciation rights, restricted stock awards, and restricted stock unit awards to eligible recipients. The maximum term of options granted under the Stock Plan is ten years. Employee option grants will generally vest 25% on the first anniversary of the original vesting date, and the balance vests monthly over the next three years. The vesting schedules for grants to non-employee directors and consultants will be determined by the Company’s Compensation Committee. Stock options are generally not exercisable prior to the applicable vesting date, unless otherwise accelerated under the terms of the applicable stock plan agreement.
F-21
Table of Contents
Stock-based Compensation
Total estimated stock-based compensation expense, related to all of the Company’s stock-based payment awards recognized under ASC 718, “Compensation—Stock Compensation” was comprised of the following:
| Years Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| Research and development |
$ | 163,019 | $ | 166,796 | ||||
| General and administrative |
441,957 | 159,848 | ||||||
|
|
|
|
|
|||||
| Total share-based compensation expense |
$ | 604,976 | $ | 326,644 | ||||
|
|
|
|
|
|||||
Stock-based Award Activity
The following table summarizes the Company’s stock option activity for the years ended December 31, 2014 and 2013:
| Options Outstanding |
Weighted- Average Exercise Price |
|||||||
| Outstanding at December 31, 2012 |
58,639 | $ | 0.83 | |||||
| Granted |
93,378 | 1.44 | ||||||
| Exercised |
— | — | ||||||
| Forfeited/cancelled/expired |
— | — | ||||||
|
|
|
|||||||
| Outstanding at December 31, 2013 |
152,017 | $ | 1.19 | |||||
| Granted |
90,876 | 8.47 | ||||||
| Exercised |
— | — | ||||||
| Forfeited/cancelled/expired |
— | — | ||||||
|
|
|
|||||||
| Outstanding and expected to vest at December 31, 2014 |
242,893 | $ | 3.92 | |||||
|
|
|
|||||||
| Vested and exercisable at December 31, 2014 |
154,877 | $ | 3.77 | |||||
|
|
|
|||||||
The total unrecognized compensation cost related to unvested stock option grants as of December 31, 2014 was $750,405 and the weighted average period over which these grants are expected to vest is 2.5 years. The Company has assumed a forfeiture rate of zero. The weighted average remaining contractual life of stock options outstanding at December 31, 2014 is 7.9 years.
None of the stock options granted to employees during the year ended December 31, 2014 were vested at December 31, 2014, as they generally vest over a four year period and vesting does not start until the one-year anniversary of the grant date. During the year ended December 31, 2014, the Company granted five new board members appointed in connection with the Merger an aggregate of 55,580 in stock options, which were immediately vested on the grant date.
Valuation Assumptions
The Company used the Black-Scholes-Merton option valuation model, or the Black Scholes model, to determine the stock-based compensation expense recognized under ASC 718. The Company’s expected stock-price volatility assumption was based solely on the weighted average of the historical and implied volatility of comparable companies whose share prices are publicly available. The expected term of stock options granted was based on the simplified method in accordance with Staff Accounting Bulletin No. 110, or SAB 110, as the Company’s historical share option exercise experience did not provide a reasonable basis for estimation. The risk-free interest rate was based on the U.S. Treasury yield for a period consistent with the expected term of the stock award in effect at the time of the grant.
| Years Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| Risk-free interest rate |
0.1 to 2 | % | 0.6 | % | ||||
| Dividend yield |
— | % | — | % | ||||
| Expected volatility |
84 to 100 | % | 86 | % | ||||
| Expected life of options, in years |
5 and 6.25 | 5 | ||||||
| Weighted-average grant date fair value |
$ | 4.73 | $ | 11.84 | ||||
F-22
Table of Contents
Because the Company had a net operating loss carryforward as of December 31, 2014, no tax benefits for the tax deductions related to stock-based compensation expense were recognized in the Company’s Consolidated Statements of Operations. Additionally, no stock options were exercised in the years ended December 31, 2014 and 2013.
Common stock reserved for future issuance
Common stock reserved for future issuance consists of the following at December 31, 2014:
| Common stock reserved for conversion of preferred stock and warrants |
2,591,256 | |||
| Common stock options outstanding |
242,893 | |||
| Authorized for future grant or issuance under the Stock Plan |
326,431 | |||
|
|
|
|||
| Total |
3,160,580 | |||
|
|
|
10. Net Loss per Share
The Company calculates basic and diluted net loss per share using the weighted-average number of shares of common stock outstanding during the period.
When the Company is in a net loss position, it excludes from the calculation of diluted net loss per share all potentially dilutive stock options, preferred stock and warrants, and the diluted net loss per share is the same as the basic net loss per share for such periods. If the Company was to be in a net income position, the weighted-average number of shares used to calculate the diluted net income per share would include the potential dilutive effect of in-the-money securities, as determined using the treasury stock method.
The table below presents the potentially dilutive securities that would have been included in the calculation of diluted net loss per share if they were not antidilutive for the periods presented.
| Years Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| Stock options |
44,615 | 103,417 | ||||||
| MabVax Series A redeemable convertible preferred stock |
137,607 | 265,749 | ||||||
| MabVax Series B redeemable convertible preferred stock |
156,247 | 189,020 | ||||||
| MabVax Series C-1 redeemable convertible preferred stock |
412,444 | — | ||||||
| Series B redeemable convertible preferred stock |
102,895 | — | ||||||
| Series A-1 preferred stock |
742,658 | — | ||||||
| Series C preferred stock |
47,023 | — | ||||||
|
|
|
|
|
|||||
| Total |
1,643,489 | 558,186 | ||||||
|
|
|
|
|
|||||
F-23
Table of Contents
11. Contracts and Agreements
NCI Sarcoma Vaccine Grant
In July 2010, the National Cancer Institute (“NCI”) awarded the Company a Small Business Innovation Research (“SBIR”) Program grant to support the Company’s program to conduct a Phase II clinical trial for a vaccine intended to prevent the recurrence of sarcoma (the “NCI Sarcoma Vaccine Grant”). The Company received the Phase II portion of the grant, which amounted to approximately $1,829,000 and covered the period from April 2011 to January 2013. The Company records revenue associated with the NIH Grants as the related costs and expenses are incurred. For the year ended December 31, 2013, the Company recorded $201,355 of revenue associated with the NCI Sarcoma Vaccine Grant.
NCI Neuroblastoma Vaccine Grant
In July 2012, the NCI awarded the Company a SBIR Program grant to support the Company’s program to manufacture the clinical material and develop an Investigational New Drug Application for a vaccine to prevent the recurrence of Neuroblastoma (the “NCI Neuroblastoma Vaccine Grant”). The project period for Phase I of the grant ended in December 2012 and the Company received a one-year extension on the project. The Company records revenue associated with the NIH Grants as the related costs and expenses are incurred. For the years ended December 31, 2014 and 2013, the Company recorded $32,355 and $102,521 of revenue associated with the NCI Neuroblastoma Vaccine Grant, respectively.
NCI PET Imaging Agent Grant
In September 2013, the NCI awarded the Company a SBIR Program Contract to support the Company’s program to develop a PET imaging agent for pancreatic cancer using a fragment of the Company’s 5B1 antibody (the “NCI PET Imaging Agent Grant”). The project period for Phase I of the grant award of approximately $250,000 covered a nine-month period which commenced in September 2013 and ended in June 2014.
On August 25, 2014, the Company was awarded a $1.5 million contract for the Phase II portion of the NCI PET Imaging Agent Grant. The contract is intended to support a major portion of the preclinical work being conducted by the Company, together with its collaboration partner, MSKCC, to develop a novel Positron Emission Tomography (“PET”) imaging agent for detection and assessment of pancreatic cancer. The total contract amount for Phase I and Phase II of approximately $1,749,000 supports research work through June 2016.
The Company records revenue associated with the NCI PET Imaging Agent Grant as the related costs and expenses are incurred. For the years ended December 31, 2014 and 2013, the Company recorded $271,820 and $62,492 of revenue associated with the NCI PET Imaging Agent Grant, respectively.
Juno Therapeutics Option Agreement
On August 29, 2014, MabVax Therapeutics entered into an Option Agreement (the “Option Agreement”) with Juno Therapeutics, Inc. (“Juno”). Pursuant to the Option Agreement, MabVax Therapeutics granted Juno the option to obtain an exclusive, world-wide, royalty-bearing license (the “License”) authorizing Juno to develop, make, have made, use, import, have imported, sell, have sold, offer for sale and otherwise exploit certain patents MabVax Therapeutics developed with respect to fully human antibodies with binding specificity against human GD2 or sialyl Lewis A antigens (the “Patents”) and certain MabVax Therapeutics controlled biologic materials. Juno may exercise its option to purchase the License until the earlier of June 30, 2016 or 90 days from the date MSKCC completes its research with respect to the Patents in accordance with the terms of agreements by and between MSKCC and MabVax Therapeutics.
The Option Agreement may be terminated by either party (i) upon material breach of the other party if the breach is not cured within 30 days, or (ii) with 60 days’ prior written notice in the event the other party becomes the subject of a voluntary or involuntary petition in bankruptcy. Juno may terminate the Option Agreement at any time upon 30 days’ prior written notice. MabVax Therapeutics may terminate the Option Agreement if Juno, or any Juno employee or affiliate, is a party to any action or proceeding in which Juno, or any Juno employee or affiliate, opposes the Patents or otherwise seeks a determination that any of the Patents are invalid or unenforceable if Juno, or as applicable, its employee and/or affiliate, fails to discontinue its involvement in such an action within 10 days of receiving notice from MabVax Therapeutics.
As consideration for the grant of the exclusive option to purchase the License, Juno has agreed to pay MabVax Therapeutics a one-time up-front option fee in the low five figures. Should the option be exercised, MabVax Therapeutics would expect to negotiate with Juno to pay amounts that include MabVax Therapeutics license fees, milestone payments, and royalty-based compensation in connection with entering into a License. The terms of the License including the financial terms are expected to be agreed upon at a future date.
F-24
Table of Contents
12. Commitments and contingencies
Litigation
On May 30, 2014, a class action lawsuit was commenced in Santa Clara County Superior Court, State of California, on behalf of Cadillac Partners and others similarly situated, naming as defendants, MabVax Therapeutics, the Company and the Company’s directors, Hudson Bay Capital Management LP, Bio IP Ventures LLC, Hudson Bay Master Fund Ltd., and Hudson Bay IP Opportunities Master Fund LP. The suit alleged the defendants breached certain fiduciary duties, or aided and abetted a breach of fiduciary duties, in connection with the Company’s Merger with MabVax Therapeutics. In support of their purported claims, the plaintiff alleged, among other things, that the Company’s board has historically failed to fulfill its fiduciary duty to its stockholders, and claiming with respect to the Series B Private Placement and the Merger, the such transactions involved an inadequate sales process and included preclusive deal protection devices, and that the Company’s board of directors would receive personal benefits not available to its public stockholders as a result of the Merger. The plaintiff sought to enjoin the Merger and obtain damages as well as attorneys’ and expert fees and costs.
On June 29, 2014, the parties entered into a Stipulation and Settlement (the “Settlement”), pursuant to which the Company agreed to file with the SEC certain supplemental disclosures in connection with the Merger. The Settlement is subject to certain confirmatory discovery to be undertaken by the plaintiff and to the parties’ agreement on the payment of the plaintiff’s attorneys’ fees and expenses.
On July 16, 2014, the Company and all other parties to the litigation entered into an agreement which, if consummated, will settle the litigation (the “Proposed Settlement”). Among many other terms, under the Proposed Settlement the Company and all defendants will receive a broad release of any and all claims pertaining to the Series B Private Placement, the Merger, the prior disclosure and a wide variety of other matters. The Proposed Settlement also calls for the parties to ask the court to, among other things, enter orders enjoining other stockholders from bringing similar actions, certifying the putative settlement class, and approving the Proposed Settlement as a fair, final, and binding resolution of the litigation. Under the Proposed Settlement, the Company and the other defendants have expressly denied the allegations of the complaint and denied engaging in any other misconduct, nor will any of them make any payment or in any respect amend the negotiated terms of the since-consummated Series B Private Placement and Merger. Finally, under the Proposed Settlement, the Company and the other defendants have not agreed to pay any legal fees, or reimburse any expenses, allegedly incurred by the plaintiffs who filed the complaint; instead, the Company expects that counsel for those plaintiffs will present any such disputed claim for legal fees and expenses to the court for resolution.
Operating Leases
In connection with the Merger, the Company recorded a $590,504 contingent lease termination fee, related to the termination of the master lease and sublease of the Porter Drive Facility by MabVax Therapeutics Holdings (f.k.a. Telik, Inc.), which is payable to ARE-San Francisco No. 24 (“ARE”) if the Company receives $15 million or more in additional financing in the aggregate, but otherwise forgiven.
The Company leases its corporate office and laboratory space under an operating lease that, as amended on August 1, 2010, expires on July 31, 2015. The lease contains an option to cancel at various dates prior to the termination date by paying a cancellation penalty. The Company has provided a refundable security deposit of $11,017 to secure its obligations under the lease, which has been included in other long-term assets in the accompanying consolidated financial statements. We recognize rent expense on a straight-line basis over the term the lease. Rent expense of $115,118 and $138,783 was recognized in the years ended December 31, 2014 and 2013, respectively.
Minimum future annual operating lease obligations are as follows as of December 31, 2014:
| 2015 |
$ | 77,117 | ||
|
|
|
|||
| Total |
$ | 77,117 | ||
|
|
|
F-25
Table of Contents
Restructuring Plan upon Closing of the Merger
In connection with the Merger, the Company signed separation agreements in May 2014 with nine employees and agreed to pay severances and health benefits upon closing of the Merger subject to certain provisions in the agreements. Approximately $6,000 in severance and benefits costs remain as of December 31, 2014.
13. Income taxes
The components of the provision for income taxes for the years ended December 31, 2014 and 2013 is as follows:
| 2014 | 2013 | |||||||
| Current: |
||||||||
| Federal |
$ | — | $ | — | ||||
| State |
— | — | ||||||
|
|
|
|
|
|||||
| — | — | |||||||
|
|
|
|
|
|||||
| Deferred: |
||||||||
| Federal |
$ | — | $ | — | ||||
| State |
— | — | ||||||
|
|
|
|
|
|||||
| — | — | |||||||
|
|
|
|
|
|||||
| Income tax expense |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s net deferred tax assets are as follows as of December 31, 2014 and 2013:
| 2014 | 2013 | |||||||
| Deferred tax assets: |
||||||||
| Net operating loss carryforwards |
$ | 9,478,000 | $ | 4,932,000 | ||||
| Tax credits |
4,128,000 | 90,000 | ||||||
| Accrued expenses and other |
225,000 | 35,500 | ||||||
|
|
|
|
|
|||||
| Total deferred tax assets |
13,831,000 | 5,057,500 | ||||||
| Less valuation allowance |
(13,831,000 | ) | (5,057,500 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax assets |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
The Company has evaluated the available evidence supporting the realization of its gross deferred tax assets, including the amount and timing of future taxable income, and has determined that it is more likely than not that the deferred tax assets will not be realized. Due to such uncertainties surrounding the realization of the Company’s deferred tax assets, the Company maintains a valuation allowance of $13,831,000 against its deferred tax assets as of December 31, 2014. Realization of the deferred tax assets will be primarily dependent upon the Company’s ability to generate sufficient taxable income prior to the expiration of its net operating losses.
During the year, MabVax Therapeutics, Inc. merged with Telik, Inc. in a tax-free reorganization. As a result of the merger, all components of Telik’s deferred tax assets are now included as deferred tax assets of MabVax Therapeutics, Inc. These pre-merger deferred tax assets are net operating loss carryforwards of $1,672,000, research and development credit carryforwards of $3,903,000, as well as other deferred tax asset items of $53,000, in total equaling $5,628,000. The current year change in these assets has been reflected in the provision for income taxes.
As of December 31, 2014, the Company had net operating loss carryforwards of approximately $23,909,000 and $23,773,000 for federal and state income tax purposes, respectively. These may be used to offset future taxable income and will begin to expire in varying amounts in 2028 to 2034. The Company also has research and development credits of approximately $194,000 and $5,960,000 for federal and state income tax purposes, respectively. The federal credits may be used to offset future taxable income and will begin to expire at various dates beginning in 2030 through 2034. The state credits may be used to offset future taxable income, and such credits carryforward indefinitely.
F-26
Table of Contents
The Company is subject to taxation in the U.S. and California jurisdictions. Currently, no historical years are under examination. The Company’s tax years ending December 31, 2014 and 2013 are subject to examination by the U.S. and state taxing authorities due to the carryforward of unutilized net operating losses and research and development credits.
Utilization of the Company’s net operating loss carryforwards and research and development credit carryforwards may be subject to a substantial annual limitation due to an “ownership change” that may have occurred, or that could occur in the future, as defined and required by Section 382 of the Internal Revenue Code of 1986, as amended (the “Code”), as well as similar state provisions. These ownership changes may limit the amount of net operating loss carryforwards and research and development credit carryforwards, and other tax attributes that can be utilized annually to offset future taxable income and tax, respectively. Any limitation may result in the expiration of a portion of the net operating loss carryforwards or research and development credit carryforwards before utilization. The net operating loss carryforwards and research and development credit carryforwards inherited as a result of the merger with Telik, Inc. have been severely limited under these rules and will likely not be realized.
In general, an “ownership change” results from a transaction or series of transactions over a three-year period resulting in an ownership change of more than 50% of the outstanding stock of a company by certain stockholders or public groups. The Company intends to complete a study in the future to assess whether an ownership change has occurred or whether there have been multiple ownership changes since the Company’s formation, and will complete such study before the use of any of the aforementioned attributes.
The provision for income taxes differs from the amount computed by applying the U.S. federal statutory tax rate (34% in 2014 and 2013) to income taxes as follows:
| 2014 | 2013 | |||||||
| Tax benefit computed at 34% |
$ | (2,692,100 | ) | $ | (1,375,300 | ) | ||
| State tax provision, net of federal tax benefit |
(462,800 | ) | (227,400 | ) | ||||
| Change in valuation allowance |
3,146,000 | 1,542,600 | ||||||
| Other |
8,900 | 60,100 | ||||||
|
|
|
|
|
|||||
| Tax provision (benefit) |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
The Company has adopted ASC 740-10-25. This interpretation clarifies the criteria for recognizing income tax benefits under ASC 740, “Accounting for Income Taxes”, and requires additional disclosures about uncertain tax positions. Under ASC 740-10-25 the financial statement recognition of the benefit for a tax position is dependent upon the benefit being more likely than not to be sustainable upon audit by the applicable taxing authority. If this threshold is met, the tax benefit is then measured and recognized at the largest amount that is greater than 50 percent likely of being realized upon ultimate settlement.
F-27
Table of Contents
14. Subsequent Events
On January 11, 2015, the Series B Common Warrants reached the Reset Date, in accordance with the original terms of the agreement, and the warrant exercise price was reset to $1.57.
On January 14, 2015, holders of the Series C preferred stock converted 96,571 shares into 120,714 shares of common stock.
Between January 10, 2015 and February 25, 2015, holders of the Series A-1 preferred stock converted 64,019 shares into 38,456 shares of common stock.
Between March 3, 2015 and March 20, 2015, holders of the Company’s Series B Preferred Stock converted a total of 106,437 of those shares into 276,883 shares of common stock.
Exchange of Preferred Stock and Warrants
On March 25, 2015, the Company entered into separate exchange agreements (the “Exchange Agreements”) with certain holders (each an “Exchange Holder”; collectively the “Exchange Holders”) of the Company’s Series A-1 preferred stock and Merger Warrants (the “Series A-1 Exchange Securities”) and holders of the Company’s Series B preferred stock and Series B Warrants (the “Series B Exchange Securities” and, collectively with the Series A-1 Exchange Securities, the “Exchange Securities”), all previously issued by the Company. Pursuant to the Exchange Agreements, the Exchange Holders exchanged the Exchange Securities and relinquished any and all other rights they may have had pursuant to the Exchange Securities, their respective governing agreements and certificates of designation, including any related registration rights, in exchange for an aggregate of 2,588,407 shares of the Company’s common stock and an aggregate of 237,647 shares of the Company’s newly designated Series D Convertible preferred stock (the “Series D preferred stock” and together with the common stock issuable pursuant to the Exchange Agreements and the common stock issuable upon conversion of the Series D preferred stock, the “Securities”).
Additionally, for as long as a certain principal holder of Exchange Securities holds Securities issued pursuant to the Exchange Agreements, subject to certain exceptions, the Company is restricted from issuing any shares of common stock or securities convertible into common stock, enter into any equity line of credit or issue any floating or variable priced equity linked instrument.
No commission or other payment was received by the Company in connection with the Exchange Agreements.
Series D Preferred Stock
As contemplated by the Exchange Agreements and as approved by the Company’s Board of Directors, the Company filed with the Secretary of State of the State of Delaware a Certificate of Designation of Preferences, Rights and Limitations of Series D Convertible Preferred Stock (the “Series D Certificate of Designations”), on March 25, 2015. Pursuant to the Series D Certificate of Designations, the Company designated 1,000,000 shares of its blank check preferred stock as Series D preferred stock. Each share of Series D preferred stock has a stated value of $0.01 per share. In the event of a liquidation, dissolution or winding up of the Company, each share of Series D preferred stock will be entitled to a per share preferential payment equal to the stated value. Each share of Series D preferred stock is convertible into 100 shares of common stock. The conversion ratio is subject to adjustment in the event of stock splits, stock dividends, combination of shares and similar recapitalization transactions. The Company is prohibited from effecting the conversion of the Series D preferred stock to the extent that, as a result of such conversion, the holder beneficially owns more than 4.99% (provided that certain investors elected to block their beneficial ownership initially at 2.49% in the Exchange Agreements), in the aggregate, of the issued and outstanding shares of the Company’s common stock calculated immediately after giving effect to the issuance of shares of common stock upon the conversion of the Series D preferred stock (the “Beneficial Ownership Limitation”). Each share of Series D preferred stock entitles the holder to vote on all matters voted on by holders of common stock. With respect to any such vote, each share of Series D preferred stock entitles the holder to cast such number of votes equal to the number of shares of common stock such shares of Series D preferred stock are convertible into at such time, but not in excess of the Beneficial Ownership Limitation.
After giving effect to the transactions contemplated by the Exchange Agreements, and prior to Private Placement Financing noted in our Subsequent Events the Company had 5,827,327 shares of common stock issued and outstanding and 237,647 shares of Series D preferred stock outstanding convertible into an aggregate of 23,764,700 shares of common stock, without giving effect to any Beneficial Ownership Limitation.
As of March 25, 2015, pursuant to the terms of the Exchange Agreements, the MabVax Therapeutics Securities Purchase Agreement, Series A-1 Registration Rights Agreement, the Series B Purchase Agreement and the Series B Registration Rights Agreement were terminated, and all rights covenants, agreements and obligations contained therein, are of no further force or effect.
F-28
Table of Contents
Private Placement Transaction
On March 31, 2015 the Company accepted subscription agreements (the “Subscription Agreements”) in a private placement issuance of 6,661,000 Units, as described below, and received proceeds of $4,662,957, net of $332,793 in issuance costs. The Company also agreed to issue and sell, subject to customary closing conditions, additional Units for an aggregate private placement of up to 21,333,333 shares of the Company’s common stock (or, for purchasers who would hold 5% or more of the Company’s common stock, shares of the Company’s Series E Convertible preferred stock, par value $0.01 per share (the “Series E preferred stock”) convertible into an equivalent number of shares of such common stock) (such shares of common stock and Series E preferred stock, the “PIPE Shares”) and, for each share of common stock so purchased (or issuable upon conversion of each share of Series E preferred stock so purchased) warrants to purchase one-half of one share of common stock (collectively, the “Private Placement” and the “PIPE Warrants” and, together with the PIPE Shares, the “Units”). Upon closing, the Company will sell Units with an aggregate purchase price of up to $16,000,000 (or $0.75 for each Unit). The Series E preferred stock is described below.
The PIPE Warrants are exercisable upon issuance at the Closing Date (as defined in the Subscription Agreement), expire 30 months from the Closing Date and may be exercised for cash or on a cashless basis. The PIPE Warrants will initially have a per share exercise price of $1.50, subject to certain adjustments. The Company is prohibited from effecting the exercise of the PIPE Warrants to the extent that, as a result of such exercise, the holder beneficially owns more than 4.99% in the aggregate, of the issued and outstanding shares of the Company’s common stock calculated immediately after giving effect to the issuance of shares of common stock upon the exercise of the PIPE Warrants.
In connection with the Private Placement, the Company also entered into a Registration Rights Agreement with the PIPE Purchasers (the “PIPE Registration Rights Agreement”). Pursuant to the PIPE Registration Rights Agreement, the Company has agreed to file a registration statement with the SEC covering resales of up to 25% of common stock issued under the Subscription Agreements and shares issuable upon conversion of the Series E preferred stock (together, the “Registrable Securities”) by the PIPE Purchasers no later than 60 days following the Closing Date, and to use its commercially reasonable best efforts to have such registration statement declared effective with 120 days after filing. The Company will bear all expenses of such registration of the resale of the Registrable Securities. PIPE Purchasers also may be required under certain circumstances to agree to refrain from resales of a percentage of their securities upon request of an underwriter or placement agent in a future offering.
Series E Preferred Stock
As approved by the Company’ Board of Directors, the Company filed with the Secretary of State of the State of Delaware a Certificate of Designation of Preferences, Rights and Limitations of Series E Convertible preferred stock (the “Series E Certificate of Designations”), on March 31, 2015. Pursuant to the Series E Certificate of Designations, the Company designated 100,000 shares of its blank check preferred stock as Series E preferred stock. Each share of Series E preferred stock has a stated value of $75.00 per share. In the event of a liquidation, dissolution or winding up of the Company, each share of Series E preferred stock will be entitled to a per share preferential payment equal to $0.01 per share. Each share of Series E preferred stock is convertible into 100 shares of common stock. The conversion ratio is subject to adjustment in the event of stock splits, stock dividends, combination of shares and similar recapitalization transactions. In addition, until the earlier of (i) twenty-four (24) months from the Final Closing Date (as defined in the Subscription Agreement), (ii) the date the Company consummates a financing (excluding proceeds from the sale of the Series E preferred stock) in which the Company receives gross proceeds of at least Ten Million Dollars ($10,000,000) and (iii) the date the Company’s common stock is listed for trading on a national securities exchange, if the Company issues or sells any shares of common stock at a price less than $0.75 (a “New Issuance”), the Conversion Price of the Series E preferred stock is automatically adjusted to the New Issuance price. The Company is prohibited from effecting the conversion of the Series E preferred stock to the extent that, as a result of such conversion, the holder beneficially owns more than 4.99%, in the aggregate, of the issued and outstanding shares of the Company’s common stock calculated immediately after giving effect to the issuance of shares of common stock upon the conversion of the Series E preferred stock (the “Series E Beneficial Ownership Limitation”). Each share of Series E preferred stock entitles the holder to vote on all matters voted on by holders of common stock. With respect to any such vote, each share of Series E preferred stock entitles the holder to cast such number of votes equal to the number of shares of common stock such shares of Series E preferred stock are convertible into at such time, but not in excess of the Series E Beneficial Ownership Limitation. All, none or a portion of the Series E preferred stock may be issued in connection with the Subscription Agreements including with respect to any subscriptions that may be accepted in the discretion of the Company in connection with any closings which the Company may elect to accept following the date of this report.
F-29
Table of Contents
Issuance of Common Stock under Common Stock Purchase Agreement
In connection with the July 2014 Private Placement Transaction, or July 2014 Financing, the Company assumed certain obligations to issue additional shares to investors in the July 2014 Financing if a subsequent financing was at a price per share lower than the price per share in the July 2014 Financing. The Company therefore issued an aggregate of 88,093 shares of common stock that were required to be issued in connection with the Private Placement.
Amendment of Equity Incentive Plan
On March 31, 2015 the Company approved a Second Amended and Restated 2014 Employee, Director and Consultant Equity Incentive Plan (the “Plan”), effective as of and contingent upon the consummation of the initial closing of the sale of Units pursuant to the Subscription Agreement, to increase the number of shares reserved for issuance under the Plan from 158,073 to 8,360,789 shares of common stock. Additional changes to the Plan include:
| • | An “evergreen” provision to reserve additional shares for issuance under the Plan on an annual basis commencing on the first day of fiscal 2016 and ending on the second day of fiscal 2024, such that the number of shares that may be issued under the Plan shall be increased by an amount equal to the lesser of: (i) 8,000,000 or the equivalent of such number of shares after the administrator, in its sole discretion, has interpreted the effect of any stock split, stock dividend, combination, recapitalization or similar transaction in accordance with the Plan; (ii) the number of shares necessary such that the total shares reserved under the Plan equals (x) 15% of the number of outstanding shares of common stock on such date (assuming the conversion of all outstanding shares of Preferred Stock (as defined in the Plan) and other outstanding convertible securities and exercise of all outstanding warrants to purchase common stock) plus (y) 229,000; and (iii) an amount determined by the Board; |
| • | Provide that no more than 3,000,000 shares may be granted to any participant in any fiscal year. |
| • | Provisions to allow for performance based equity awards to be issued by the Company in accordance with Section 162(m) of the Internal Revenue Code. |
F-30