Attached files
Table of Contents
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended December 31, 2011
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Transition period from to .
Commission file number: 0-31265
TELIK, INC.
(Exact name of Registrant as specified in its charter)
| Delaware | 93-0987903 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
700 Hansen Way, Palo Alto, CA 94304
(Address of principal executive offices) (Zip Code)
Registrant’s telephone number, including area code: (650) 845-7700
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common Stock, $0.01 par value per share | Nasdaq Capital Market |
Securities registered pursuant to Section 12(g) of the Act: None
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES ¨ NO x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. YES ¨ NO x
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. YES x NO ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). YES x NO ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this Chapter) is not contained herein, and will not be contained to the best of Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer ¨ Accelerated filer ¨ Non-accelerated filer ¨ Smaller reporting company x
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act.). YES ¨ NO x
The aggregate market value of the voting stock held by non-affiliates of the Registrant was approximately $22,247,357 as of June 30, 2011, based upon the closing sale price on the Nasdaq Capital Market reported on June 30, 2011. The calculation excludes approximately 25,434,924 shares held by directors, officers and stockholders whose ownership exceeded five percent of the Registrant’s outstanding Common Stock as of June 30, 2011. Exclusion of these shares should not be construed to indicate that such person controls, is controlled by or is under common control with the Registrant. The determination of affiliate status for the purposes of this calculation is not necessarily a conclusive determination for other purposes.
There were 54,456,289 shares of Registrant’s Common Stock issued and outstanding as of February 22, 2012.
DOCUMENTS INCORPORATED BY REFERENCE
Items 10, 11, 12, 13 and 14 of Part III incorporate information by reference from the definitive proxy statement to be filed on or about March 5, 2012 with the Securities and Exchange Commission pursuant to Regulation 14A for the Registrant’s Annual Meeting of Stockholders. Except with respect to the information specifically incorporated by reference in this Form 10-K, the proxy statement is not deemed to be filed as part hereof.
Table of Contents
TELIK, INC.
2011 ANNUAL REPORT ON FORM 10-K
| Page | ||||||
| PART I | ||||||
| Item 1. | 4 | |||||
| Item 1A. | 14 | |||||
| Item 1B. | 25 | |||||
| Item 2. | 25 | |||||
| Item 3. | 25 | |||||
| Item 4. | 25 | |||||
| PART II | ||||||
| Item 5. | 26 | |||||
| Item 6. | 28 | |||||
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
29 | ||||
| Item 7A. | 41 | |||||
| Item 8. | 41 | |||||
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
41 | ||||
| Item 9A. | 41 | |||||
| Item 9B | 42 | |||||
| PART III | ||||||
| Item 10. | 43 | |||||
| Item 11. | 43 | |||||
| Item 12. | Security Ownership of Certain Beneficial Owners and Management |
44 | ||||
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
44 | ||||
| Item 14. | 44 | |||||
| PART IV | ||||||
| Item 15. | 45 | |||||
| SIGNATURES |
48 | |||||
| FINANCIAL STATEMENTS |
||||||
| 49 | ||||||
| 50 | ||||||
| 51 | ||||||
| 52 | ||||||
| 53 | ||||||
| 54 | ||||||
Table of Contents
Disclosure Regarding Forward-Looking Statements
This Annual Report on Form 10-K, including the documents that we incorporate by reference, contains statements indicating expectations about future performance and other forward-looking statements that involve risks and uncertainties. We usually use words such as “may,” “will,” “should,” “expect,” “plan,” “anticipate,” “believe,” “estimate,” “predict,” “future,” “intend,” “potential,” or “continue” or the negative of these terms or similar expressions to identify forward-looking statements. These statements appear throughout this Annual Report on Form 10-K and are statements regarding our current intent, belief, or expectation, primarily with respect to our operations and related industry developments. Examples of these statements include, but are not limited to, statements regarding the following: the implications of interim or final results of our Phase 2 clinical and Phase 3 registration trials, the progress and timing of our research programs, including clinical testing, our anticipated timing for filing additional Investigational New Drug, or IND, applications with the United States Food and Drug Administration for the initiation or completion of Phase 1, Phase 2 or Phase 3 testing for any of our product candidates, the extent to which our issued and pending patents may protect our products and technology, our ability to identify new product candidates using TRAP technology (our proprietary Target-Related Affinity Profiling technology, which is discussed below), the potential of such product candidates to lead to the development of safer or more effective therapies, our ability to develop the technology derived from our collaborations and to enter into additional collaborations, our future operating expenses, our future losses, our future expenditures for research and development, our cash resources and ability to fund current and future operations. You should not place undue reliance on these forward-looking statements, which apply only as of the date of this Annual Report. Our actual results could differ materially from those anticipated in these forward-looking statements for many reasons, including the risks faced by us and described in the section of Item 1A entitled “Risk Factors,” and elsewhere in this Annual Report. Any forward-looking statement speaks only as of the date on which it is made, and we undertake no obligation to update any forward-looking statement to reflect events or circumstances after the date on which the statement is made or to reflect the occurrence of unanticipated events.
TELIK, the Telik logo, TRAP, TELCYTA and TELINTRA are trademarks or registered trademarks of Telik, Inc. All other brand names or trademarks appearing in this Annual Report are the property of their respective holders.
3
Table of Contents
PART I
| Item 1. | Business. |
Overview
Our Business and Strategy
Telik, Inc. was incorporated in Delaware in 1988 and is a clinical-stage drug development company focused on discovering and developing small molecule drugs to treat cancer. We discover our product candidates using our proprietary drug discovery technology, Target-Related Affinity Profiling, or TRAP, which we believe enables the rapid and efficient discovery of small molecule product candidates. Our business strategy is to:
| • | Advance TELINTRA through Phase 2 clinical studies, and after obtaining clinical data, enter into a partnership with a pharmaceutical or biotechnology company to assist in further development and commercialization; |
| • | Seek a partnership for TELCYTA for further development and commercialization; |
| • | Utilize our proprietary TRAP drug discovery platform to provide a pipeline of future product development candidates to address unmet needs in cancer treatment; and |
| • | Establish collaborative agreements to advance the development of these candidates. |
Clinical Product Development
TELINTRA, our lead drug product candidate in clinical development, is a small molecule glutathione analog inhibitor of the enzyme glutathione S-transferase P1-1, or GST P1-1. We are developing TELINTRA for the treatment of blood disorders associated with low blood cell levels, such as neutropenia or anemia. We completed an 86 patient Phase 2 clinical trial of TELINTRA tablets, for the treatment of patients with Myelodysplastic Syndrome, or MDS, and presented the results at the annual meeting of the American Society of Hematology, or ASH, in December 2010. In the second quarter of 2009, we initiated a Phase 2 randomized study in Severe Chronic Neutropenia, or SCN, to determine the effect of TELINTRA tablets on absolute neutrophil count in patients with this disease. Due to the scarcity of SCN patients and our focus on MDS, we plan to terminate this study once the last remaining patient completes treatment around the third quarter of 2012. In the fourth quarter of 2009, we initiated a Phase 1 dose-ranging study of TELINTRA tablets in combination with Revlimid in patients with MDS. The study was completed in the fourth quarter of 2011 and we presented the results at the annual meeting of ASH in December 2011. In June 2011, we initiated a Phase 2 clinical trial to evaluate TELINTRA tablets in patients with Revlimid refractory or resistant, deletion 5q myelodysplastic syndrome, or del 5q MDS. This multicenter trial is intended to enroll up to 117 evaluable patients. The first planned interim analysis will occur after one-third of the patients have completed therapy and depending on the results of the interim analysis, additional enrollment may continue with two additional analyses planned. The timing of the interim analyses is dependent on the availability of patients in the del 5q MDS population. In October 2011, we initiated a Phase 2b clinical trial to evaluate TELINTRA tablets in patients with transfusion dependent, non-deletion 5q MDS, who have not been treated with hypomethylating agents, or HMA. This multicenter trial is intended to enroll up to 145 evaluable patients with 2 planned interim analyses, the first of which will be performed when data from 49 evaluable patients are available and the second at 97 evaluable patients. The timing of enrollment in this study is dependent on the availability of non-deletion 5q MDS patients.
TELCYTA, our second product candidate, is a small molecule cancer drug product candidate designed to be activated in cancer cells. TELCYTA binds to GST P1-1, an enzyme that is elevated in many human cancers, such as ovarian, non-small cell lung, colorectal, and breast. GST P1-1 levels are often further elevated following treatment with many standard chemotherapy drugs and this elevation is associated with the development of resistance to these drugs.
4
Table of Contents
TELCYTA has shown clinical antitumor activity alone and in combination with standard chemotherapeutic agents in multiple Phase 2 and Phase 3 clinical trials in refractory or resistant ovarian, non-small cell lung, breast and colorectal cancer. We completed a multicenter, randomized clinical study of 125 patients of TELCYTA in combination with liposomal doxorubicin versus liposomal doxorubicin as second line therapy in platinum refractory or resistant ovarian cancer and announced results at the annual meeting of the American Society of Clinical Oncology, or ASCO, in May 2009. In May 2010, we initiated an investigator led study at a single site of TELCYTA in patients with refractory or relapsed mantle cell lymphoma, diffuse large B cell lymphoma, and multiple myeloma. Enrollment for this study is in two stages and is expected to range between 18 to 48 patients based on the number of responses observed. Based on responses observed in stage 1, we are planning to expand the study to stage 2 and add a second investigator site in the first quarter of 2012. In addition, we are currently seeking a partnership with a pharmaceutical or biotechnology company to further advance the development and commercialization of TELCYTA.
Preclinical Drug Product Development
We currently have a small molecule compound, TLK60404, in preclinical development which inhibits both Aurora kinase and VEGFR kinase. Aurora kinase is a signaling enzyme whose function is required for cancer cell division, while vascular endothelial growth factor, or VEGF, plays a key role in tumor blood vessel formation, ensuring an adequate supply of nutrients to support tumor growth. The lead compounds of our first dual inhibitor program met a development milestone in August 2008 by demonstrating anticancer activity in preclinical models of human colon cancer and human leukemia. We are conducting the required preclinical safety studies that, if successful, may support the potential filing of an IND application with the FDA.
We have also discovered TLK60357, a novel, potent small molecule inhibitor of cell division. TLK60357 inhibits the formation of microtubules that are necessary for cancer cell growth leading to persistent cancer cell block and subsequent cell death at the G2/M phase of the cell cycle. This compound demonstrates potent broad-spectrum anticancer activity against a number of human cancer cells. This compound also displays oral efficacy in multiple, standard preclinical models of cancer. Since we are currently focused on TELINTRA and TELCYTA development, no additional expenditure on this compound is expected.
In addition, we have identified TLK60596, a small-molecule dual inhibitor of VEGFR1 and VEGFR2 kinase. VEGFR1/2 kinases are known to mediate the formation of new blood vessels in human cancers allowing them to grow. In standard animal models of human colon cancer, oral administration of TLK60596 significantly reduced tumor growth. As we are currently focused on TELINTRA and TELCYTA development, no additional expenditure on this compound is expected.
Clinical Product Development Programs
Our two most advanced product candidates, TELINTRA and TELCYTA, are being developed to treat cancers for which there is significant demand for new therapies. Cancer is the second most common cause of death in the United States according to the American Cancer Society’s 2012 Cancer Facts and Figures. The five-year survival rates for patients with cancers that have spread from their original sites, although improved in recent years, are still poor. These poor survival rates reflect the limitations of current treatments and the development of resistance to available treatments. In addition, current treatments are often associated with severe toxic side effects.
TELINTRA
TELINTRA is our lead small molecule product candidate in clinical development for the treatment of blood disorders including cancer. It has a novel mechanism of action and acts by inhibiting GST P1-1, an enzyme that is involved in the control of cellular growth and differentiation. Inhibition of GST P1-1 results in the activation of the signaling molecule Jun kinase, a key regulator of the function of blood precursor cells. Preclinical tests show that TELINTRA is capable of causing the death or apoptosis of leukemic or malignant blood cells, while stimulating the growth and development of normal blood precursor cells. TELINTRA, therefore, may lead to a
5
Table of Contents
treatment for diseases that are characterized by the presence of abnormal blood cells and or low levels of normal blood cells. The combination of abnormal cells and low normal blood cell levels is found in a number of hematologic diseases, including MDS.
In addition, decreased normal blood cell levels, especially of white cells, occur as a common side effect of cancer chemotherapy and render the already weakened cancer patient susceptible to life-threatening infections. Treatment is intended to accelerate the recovery of the white blood cells levels and decrease the risk for developing an infection. TELINTRA accelerated the recovery of white blood cells (neutrophils) in several preclinical models of chemotherapy induced neutropenia. Since currently approved treatments for this complication are given by injection, the oral formulation of TELINTRA, if effective, may prove to be a convenient alternative.
TELINTRA has been studied in MDS using two formulations. A liposomal formulation was developed for intravenous administration of TELINTRA and was used in Phase 1 and Phase 2 studies in MDS patients. The results from the Phase 2 intravenous liposomal TELINTRA clinical trials demonstrated that TELINTRA treatment was associated with improvement in all three types of blood cell levels in patients with all types of MDS, including those in intermediate and high-risk groups. An oral dosage formulation (tablet) was subsequently developed and results from a Phase 1 study with TELINTRA tablets showed clinical activity and the formulation to be well tolerated. The tablet formulation of TELINTRA may offer advantages, including ease of manufacturing and oral administration and allow us to offer a product that is an alternative to the currently marketed parenterally administered drugs.
The activity and safety profile of tablet formulation allowed us to complete a Phase 2 trial of TELINTRA tablets in MDS. The results of this study were reported at the 52nd annual meeting of ASH in December 2010. The primary objective of the Phase 2 TELINTRA tablet study was to determine the efficacy of TELINTRA, defined by Hematologic Improvement, or HI, response rate according to the 2006 International Working Group criteria, or IWG 2006, as well as its safety. An additional goal of this study was to identify those patients whose MDS disease characteristics may allow us to prospectively target patients most likely to respond to TELINTRA treatment. A multivariate logistic regression analysis was conducted to identify significant MDS disease prognostic factors associated with erythroid improvement response rates, including prior MDS treatment, age, gender, the international prognostic scoring system, or IPSS, risk, Eastern Cooperative Group performance status, years from MDS diagnosis, MDS World Health Organization subtypes, anemia only versus anemia plus other cytopenias, dose schedule and starting dose. Results from this study show that:
| • | TELINTRA is the first GSTP1-1 enzyme inhibitor shown to cause clinically significant reductions in red blood cell transfusions, including transfusion independence in low to intermediate-1 risk MDS patients, as well as improvement in platelet count and white blood cell levels in certain patients. The multilineage responses and safety profile observed provides a unique clinical activity profile with attractive tolerability and safety. |
| • | TELINTRA, administered orally twice daily, appeared to be convenient and flexible for chronic treatment administration. |
| • | The hematologic improvement rates were consistent with the Phase 1 results with TELINTRA and the duration of response was enhanced using the extended dose schedules. |
| • | Prior treatment with certain agents may influence the response to subsequent TELINTRA treatment. Revlimid is currently the only drug approved for treatment of low to intermediate-1 risk MDS red blood cell transfusion dependent patients with the 5q deletion cytogenetic karyotype. TELINTRA has shown clinically significant activity in Revlimid naïve or prior Revlimid resistant patients. |
| • | Prior history of Vidaza or Dacagen treatment appears to be an important predictor of decreased TELINTRA efficacy and tolerability. These findings may assist ongoing pharmacogenomic studies to characterize the genomic profile of responders and develop a test to identify those patients that are more likely to respond to TELINTRA treatment. |
6
Table of Contents
In June 2011, we initiated a Phase 2 clinical trial to evaluate TELINTRA tablets in patients with Revlimid refractory or resistant del 5q MDS. This multicenter trial is intended to enroll up to 117 evaluable patients.
In October 2011, we initiated an additional Phase 2b clinical trial to evaluate TELINTRA tablets in patients with transfusion dependent, non-deletion 5q MDS, who have not been treated with HMA. This multicenter trial is intended to enroll up to 145 evaluable patients.
We completed a Phase 1 dose-ranging study of TELINTRA tablets in combination with Revlimid in patients with MDS to assess the potential for development of combination chemotherapy with TELINTRA and Revlimid for the treatment of MDS in the fourth quarter of 2011 and results were presented at the 53rd annual meeting of ASH in December 2011. The primary objective of the study was to establish the safety of the combination and the optimal dosing for TELINTRA in combination with the standard dose of Revlimid. The secondary objectives were to assess the efficacy as measured by rates of hematologic improvement in red blood cell, white blood cell and platelet levels, and decreases in blood transfusions. In this study, the combination of TELINTRA and Revlimid was generally well tolerated with no unexpected new toxicities and the observed toxicities were those expected from either agent alone. It also provided a unique profile of activity with platelet transfusion independence and trilineage and bilineage responses, which was also seen with single-agent TELINTRA.
We also reported the results of gene expression analyses performed on clinical samples obtained from MDS patients on our Phase 2 study at the 53rd annual meeting of ASH . The goal of these analyses is to identify patients more likely to respond to TELINTRA therapy. Pre-therapy bone marrow mononuclear cells of MDS patients treated with TELINTRA who demonstrated hematologic improvement were analyzed for gene expression by whole genome array. The top 100 differentially expressed genes of responders and non-responders were identified and revealed a number of genes and unidentified transcripts that may play a role in the MDS patient response to TELINTRA treatment. Pathway analysis of the expression data confirmed that a c-Jun N-terminal kinase, or JNK, gene set was consistently under-expressed in the pre-therapy bone marrow mononuclear cells of responders and over-expressed in non-responders. Importantly, this result was consistent with the proposed TELINTRA mechanism of action: patients whose pre-therapy marrow showed under-expression of the JNK gene set were those who benefited from TELINTRA; while those patients who over-expressed these genes were unlikely to respond to TELINTRA. In addition, it may be possible to use gene expression signatures to enable selection of MDS patients that are most likely to benefit from TELINTRA.
In addition to MDS, we are studying the use of TELINTRA tablets for the treatment of SCN, a blood disorder typified by very low neutrophil or white blood cell levels. White blood cells are important in defending the body against infections, and therefore, a patient with severely low white blood cell levels is more susceptible to life-threatening infections. A publication on November 2, 2011, in the Journal of Hematology & Oncology entitled “Oral ezatiostat HCl (Telintra®, TLK199) and Idiopathic Chronic Neutropenia (ICN): A case report of complete response of a patient with G-CSF resistant severe chronic idiopathic neutropenia following treatment with Telintra” highlights an important observation that TELINTRA produced a striking and sustained hematologic response in white blood cell levels in an ICN patient who had an inadequate response to the standard of care, granulocyte colony stimulating factors, or G-CSF. This case report describes a patient with severe ICN who experienced frequent episodes of sepsis requiring hospitalizations and prolonged courses of antibiotics for the preceding four years. She was treated with G-CSF and had delayed, variable, and transient responses. After receiving TELINTRA therapy, her white blood cell levels stabilized, temperature normalized, and chronic infections resolved for over eight months. These results may suggest a potential role for TELINTRA tablets in the treatment of patients who are not responsive to G-CSF injections. TELINTRA, a GST P1-1 inhibitor, may achieve this effect by activating JNK, promoting the growth and maturation of blood progenitor stem cells.
TELCYTA
TELCYTA is a small molecule drug product candidate we are developing for the treatment of cancer. TELCYTA binds to GST, a protein known to play an important role in the development of resistance to commonly used chemotherapeutic drugs. GST P1-1 is a type of GST that is elevated in many cancers and is often
7
Table of Contents
further elevated following treatment with standard chemotherapeutic drugs. When TELCYTA binds to GST P1-1, it releases a fragment with a proven mechanism of killing cancer cells as well as other reactive agents. In contrast to the usual situation in which GST P1-1 is involved in the destruction of chemotherapeutic drugs, GST P1-1 activates TELCYTA when TELCYTA reaches its cellular target. In this way, TELCYTA kills cancer cells by inducing cell death through a process called apoptosis.
TELCYTA has been evaluated in multiple Phase 2 and Phase 3 clinical trials, including trials using TELCYTA as monotherapy and in combination regimens in ovarian, non-small cell lung, breast and colorectal cancer. Results from these clinical trials indicate that TELCYTA monotherapy was generally well-tolerated, with mostly mild to moderate side effects, particularly when compared to the side effects and toxicities of standard chemotherapeutic drugs. When TELCYTA was evaluated in combination with standard chemotherapeutic drugs, the tolerability of the combinations was similar to that expected of each drug alone. This tolerability profile may be an important clinical advantage for TELCYTA since combination drug regimens are commonly used in cancer treatment. Clinical activity including objective tumor responses and/or disease stabilization was reported in the TELCYTA Phase 2 trials; however, TELCYTA did not meet its primary endpoints in the Phase 3 studies. Positive results from a Phase 1-2a multicenter, dose-ranging study of TELCYTA in combination with carboplatin and paclitaxel as first-line therapy for patients with non-small cell lung cancer, or NSCLC, were published in a leading peer reviewed publication. Clinical data demonstrated positive results of TELCYTA in combination with carboplatin and paclitaxel in the treatment of first-line lung cancer followed by TELCYTA maintenance therapy.
Presently we have an on-going investigator-led study at a single site of TELCYTA in patients with refractory or relapsed mantle cell lymphoma, diffuse large B cell lymphoma, and multiple myeloma. Enrollment for this study is expected to range between 18 to 48 patients based on the number of responses observed. We are currently seeking a partnership with a pharmaceutical or biotechnology company to further advance the development and commercialization of TELCYTA.
Preclinical Drug Product Development
TLK60404—Aurora Kinase/VEGFR Inhibitors
We have a small molecule compound, TLK60404, in preclinical development which inhibits both Aurora kinase and VEGFR kinase. Aurora kinase is a signaling enzyme whose function is required for cancer cell division, while VEGF plays a key role in tumor blood vessel formation, ensuring an adequate supply of nutrients to support tumor growth. The lead compounds of our first dual inhibitor program met a development milestone in August 2008 by demonstrating anticancer activity in preclinical models of human colon cancer and human leukemia. These lead compounds prevented tumor growth in preclinical models of human colon cancer and human leukemia by inhibiting Aurora kinase and VEGFR kinase. Our data support the concept that dual inhibition of Aurora kinase and VEGFR kinase represents a promising approach for anticancer therapy. A development drug product candidate, TLK60404, has been selected. We are conducting the required preclinical safety studies, which if successful may support the potential filing of an IND application with the FDA.
TLK60357—Antimitotic Agent
Using our TRAP technology, we have discovered TLK60357, a novel, potent small molecule inhibitor of cell division. TLK60357 inhibits the formation of microtubules that are necessary for cancer cell growth leading to persistent G2/M cancer cell cycle block and subsequent cell death. This compound demonstrates potent broad-spectrum anticancer activity against a number of human cancer cells. This compound also displays oral efficacy in multiple, standard preclinical models of cancer. Since we are currently focused on TELINTRA and TELCYTA development, no additional expenditure on this compound is expected.
8
Table of Contents
TLK60596—VEGFR Inhibitor
TLK60596, a potent VGFR kinase inhibitor, blocks the formation of new blood vessels in tumors. Oral administration of TLK60596 to animal models of human colon cancer significantly reduced tumor growth. As we are currently focused on TELINTRA and TELCYTA development, no additional expenditure on this compound is expected.
Research Discovery Programs
In addition to generating our current clinical product candidate portfolio, TRAP has allowed us to build our research pipeline with product candidates against targets in cancer. We have chosen to pursue those targets that have engendered a high level of interest in the drug discovery community, address important unmet clinical needs and whose modulation are expected to have a beneficial effect in treating a given disease. We are continually evaluating and prioritizing our early stage programs.
TRAP Technology
Our TRAP drug discovery technology is designed to rapidly and efficiently identify small molecule compounds that act on disease-related protein targets. TRAP technology offers solutions to the two major challenges facing drug discovery: the explosive growth in the number of new protein targets generated by the advances in genomics, and the intrinsic limitations of the Ultra High Throughput Screening, or UHTS, approach. TRAP offers several competitive advantages over UHTS, because it is able to accommodate thousands, rather than hundreds, of targets; is cost-effective to screen unproven targets for the purpose of validation; and allows the use of complex biologically relevant assays rather highly simplified assays.
We have computationally enhanced TRAP by calculating affinity fingerprints, which greatly expands the number of compounds that can be surveyed. Our small-molecule database now has over 3.5 million computed affinity fingerprints. This approach has eliminated our need to maintain a large chemical inventory, resulting in a significant cost savings. Also, since fingerprints can be computed, TRAP can guide medicinal chemistry by evaluating potential compounds before they are made, thereby reducing the time and resources needed to develop a product candidate.
Collaborative Relationships
We are seeking to enter into collaborative arrangements with third parties for clinical trials, development, manufacturing, regulatory approvals or commercialization of some of our product candidates, particularly outside North America, or in disease areas requiring larger and longer clinical trials.
We have in the past established, and we continue to seek, joint discovery programs with other pharmaceutical, biotechnology and genomics companies. These collaborations would exploit our TRAP technology platform and have the potential to identify new product development and commercialization opportunities either independently or pursuant to expanded collaborations.
Patents and Proprietary Information
Patents and other proprietary rights are very important to our business. If we have enforceable patents of sufficient scope, it can be more difficult for our competitors to use our technology to create competitive products or to obtain patents that prevent us from using technology we create. As part of our business strategy, our policy is to actively file patent applications in the United States and internationally to cover new chemical compounds, pharmaceutical compositions, methods of preparation of the compounds and compositions and therapeutic uses of the compounds and compositions, methods related to our TRAP technology, and improvements in each of these. We also rely on trade secret information, technical know-how, innovation and agreements with third parties to continuously expand and protect our competitive position.
9
Table of Contents
We have a number of patents and patent applications related to our compounds and other technology, but we cannot be certain that issued patents will be enforceable or provide adequate protection or that the pending patent applications will issue as patents. The following table shows the expiration dates in the United States and internationally for the primary patents that cover our TRAP technology and the compounds in our clinical and preclinical product candidates.
| US patent expirations |
Foreign patent expirations |
|||||||
| TRAP |
2014 | N/A | ||||||
| Product candidates |
||||||||
| TELCYTA |
2013 | 2014 | ||||||
| TELINTRA |
2014 | 2014 | ||||||
| TLK60404 |
2029 | * | 2029 | * | ||||
| TLK60357 |
2030 | * | 2030 | * | ||||
| TLK60596 |
2032 | * | 2032 | * | ||||
| * | Including pending and planned applications |
We may obtain patents for our product candidates many years before we obtain marketing approval for them. We can generally expect to obtain patent term extensions of up to five years for patents covering our product candidates in many countries when and if marketing approvals are obtained. In foreign countries, these extensions are available only if approval is obtained before the patent expires, but in the United States, extensions are available even if approval is obtained after the patent would expire normally through a system of interim extensions until approval. In addition, we are actively pursuing multiple life cycle patent applications for TELINTRA and TELCYTA, including applications related to combination therapies, polymorphs, formulations, manufacturing processes and the genomic profiles of responders.
In addition to patent coverage, we will generally be entitled to data exclusivities for our product candidates in many countries for several years after marketing approval (for example, 5 years in the United States and up to 10 years in the European Union) when and if marketing approvals are obtained.
We also rely on trade secret information, technical know-how, innovation and agreements with third parties to continuously expand and protect our proprietary position. We do not disclose our trade secrets (including significant aspects of our TRAP technology) outside Telik except where disclosure is essential to our business, and we require those individuals, companies and institutions doing business with us, including TRAP collaborators, to execute agreements to protect our trade secrets. We require our employees and consultants to execute non-disclosure and assignment of invention agreements on commencement of their employment or engagement.
Competition
Competition in the pharmaceutical and biotechnology industries is intense. The product candidates that we and our collaborative partners are developing will have to compete with existing therapies. In addition, a large number of companies are pursuing the development of pharmaceuticals that target the same diseases and conditions that we are targeting. Many pharmaceutical or biotechnology companies have products on the market and are actively engaged in the research and development of products that are competitive with our potential product candidates. Many of these companies and institutions, either alone or together with their collaborative partners, have substantially greater financial, manufacturing, sales, distribution and technical resources and more experience in research and development, clinical trials and regulatory matters, than we do. In addition, our competitors may succeed in developing technologies and drugs that are more effective, better tolerated or less costly than any which are being developed by us and our collaborative partners or which would render our technology or potential product candidates obsolete or noncompetitive.
10
Table of Contents
Regulatory Considerations
The manufacturing and marketing of our product candidates and our on-going research and development activities are subject to extensive regulation by numerous governmental authorities in the United States and other countries. In the United States, pharmaceutical products are subject to rigorous review by the FDA under the Federal Food, Drug, and Cosmetic Act, the Public Health Service Act and other federal statutes and regulations. Non-compliance with applicable requirements can result in fines, recall or seizure of products, total or partial suspension of production, refusal of the government to approve marketing applications or allow us to enter into supply contracts and criminal prosecution. The FDA also has the authority to revoke previously granted marketing authorizations.
Securing FDA approval requires the submission of extensive preclinical and clinical data and supporting information to the FDA for each indication to establish a product candidate’s safety and efficacy. The approval process may take many years, requires the expenditure of substantial resources, may involve post-marketing surveillance and may involve on-going requirements for post-marketing studies. The FDA may also require post-marketing testing and surveillance to monitor the effects of approved products or place conditions on any approvals that could restrict the commercial applications of the products. Product approvals may be withdrawn if compliance with regulatory standards is not maintained or if problems occur following initial marketing. With respect to patented products or technologies, delays imposed by the governmental approval process may materially reduce the period during which we will have exclusive rights to exploit those products or technologies.
The cost of preparing and submitting a New Drug Application, or NDA, is substantial. Under federal law, the submission of most NDAs is additionally subject to a substantial application user fee and the manufacturer and/or sponsor under an approved new drug application are also subject to annual product and establishment user fees.
Preclinical studies involve laboratory evaluation and animal studies to assess the initial efficacy and safety of a product candidate. The FDA, under its Good Laboratory Practices regulations, regulates preclinical studies. Violations of these regulations can, in some cases, lead to invalidation of the studies, requiring these studies to be replicated. The results of the preclinical studies, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND, which must be approved by the FDA before we can commence clinical trials in humans. Unless the FDA objects to an IND, the IND would become effective 30 days following its receipt by the FDA.
Clinical trials involve the administration of the investigational product candidate to humans under the supervision of a qualified principal investigator. Clinical trials in the United States must be conducted in accordance with Good Clinical Practice, or GCP, under protocols submitted to the FDA as part of the IND. In addition, each clinical trial must be approved and conducted under the auspices of an Investigational Review Board, or IRB, and with patient informed consent. The IRB will consider, among other things, ethical factors, the safety of human subjects and the possibility of liability of the institution conducting the trial.
The FDA may order the temporary or permanent discontinuation of a clinical trial at any time or impose other sanctions if it believes that the clinical trial is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
Clinical trials in the United States are conducted in three sequential phases though the phases may overlap. Phase 1 clinical trials may be performed in healthy human subjects or, depending on the disease, in patients. The goal of Phase 1 clinical trials is to establish initial data about the safety and tolerance of the product candidate in humans. In Phase 2 clinical trials, in addition to safety, the efficacy of the product candidate is evaluated in a limited number of patients with the target disease. Phase 3 clinical trials typically involve additional testing for
11
Table of Contents
safety and clinical efficacy in expanded, large-scale, multicenter studies of patients with the target disease. We have engaged contract research organizations, or CROs, to facilitate the administration of some of our clinical trials.
We and all of our contract manufacturers are required to comply with the applicable FDA current Good Manufacturing Practice, or cGMP, regulations applicable to the manufacture of the clinical and commercial supplies of our product candidates. cGMP regulations include requirements relating to quality control and quality assurance as well as the corresponding maintenance of records and documentation. Manufacturing facilities are subject to inspection by the FDA. These facilities must be approved before we can use them in the commercial manufacture of our product candidates.
Outside the United States, our ability to market a product is contingent upon receiving marketing authorization from the appropriate regulatory authorities. The requirements governing the conduct of clinical trials, marketing authorization, pricing and reimbursement vary widely from country to country. At present, foreign marketing authorizations are generally applied for and obtained at a national level, although within the European Union registration procedures are available to companies wishing to market a product in more than one European Union member state. If the foreign regulatory authority is satisfied that adequate evidence of safety, quality and efficacy has been presented, a marketing authorization will be granted for the applicable country.
Manufacturing
Isochem North America LLC, or Isochem, has in the past manufactured and supplied a limited number of batches of the active ingredient to be used in the production of TELINTRA for use in clinical trials. As such, we have qualified Isochem as a potential source of supply for clinical quantities of this ingredient. In addition, Patheon Inc., or Patheon, has in the past performed formulation development and analytical services, and produced for us limited batches of clinical trial material, relating to the tablet formation of TELINTRA. As such, we have qualified Patheon as a potential manufacturer of TELINTRA tablets. However, we currently do not have in effect any manufacturing or supply agreements with either Isochem or Patheon. Although we have not qualified any other sources for the active ingredient in TELINTRA or the manufacture of TELINTRA tablets, we have evaluated, and may consider, potential alternative sources for this material in the future.
We are currently dependent on a single source, AMRI Rensselaer, Inc., or AMRI, previously known as Organichem Corporation, for the active ingredient in TELCYTA. OSO BioPharmaceuticals Manufacturing LLC, or OSO, has in the past manufactured and supplied TELCYTA drug product for use in our clinical trials. As such, we have qualified OSO as a potential source of supply for clinical quantities of TELCYTA. However, we currently do not have in effect a manufacturing or supply agreement with OSO. Although we have not qualified any other source for the manufacturing and supply of TELCYTA drug product, we have evaluated, and may consider, potential alternative sources for TELCYTA in the future.
We intend to continue to use third-party contract manufacturers or corporate collaborators for the production of material for use in preclinical studies, clinical trials, manufacture of future products and commercialization. The manufacture of our product candidates for preclinical studies and clinical trials and commercial purposes is subject to regulations promulgated by the FDA and to other applicable domestic and foreign regulations.
Research and Development
Our goal is to develop small molecule drugs for major disease areas and this goal has been supported by our substantial research and development investments. We spent approximately $5.6 million in 2011 and $11.0 million in 2010 on research and development. We conduct research internally and also through collaborations with third parties, including universities. In 2011, approximately 75% of our research and development was conducted internally and 25% was conducted through collaborations with third parties, including consultants.
12
Table of Contents
Employees
As of February 15, 2012, our workforce consisted of one part-time and 22 full-time employees. Of these, five hold Ph.D. or M.D. degrees, or both, and one holds another advanced degree. Of our total workforce, 13 are engaged in research and development and 10 are engaged in business development, finance and administration. None of our employees are represented by a collective bargaining agreement, nor have we experienced any significant work stoppages. We believe that our relations with our employees are good.
13
Table of Contents
| Item 1A. | Risk Factors. |
Our business faces significant risks, some of which are set forth below to enable readers to assess, and be appropriately apprised of, many of the risks and uncertainties applicable to the forward-looking statements made in this Annual Report. You should carefully consider these risk factors as each of these risks could adversely affect our business, operating results and financial condition. If any of the events or circumstances described in the following risks actually occurs, our business may suffer, the trading price of our common stock could decline and our financial condition or results of operations could be harmed. Given these risks and uncertainties, you are cautioned not to place undue reliance on forward-looking statements. These risks should be read in conjunction with the other information set forth in this Annual Report. There may be additional risks faced by our business, though we do believe that the risks set forth below reflect the more important ones.
If we are unable to raise adequate funds in the future, we will not be able to continue to fund our operations, research programs, preclinical testing and clinical trials to develop and manufacture our drug product candidates; and our auditors have indicated that our recurring losses and net capital deficiency raises substantial doubt about our ability to continue as a going concern.
The process of carrying out the development of our own unpartnered product candidates to later stages of development and developing other research programs to the stage that they may be partnered to a pharmaceutical or biotechnology company will require significant additional expenditures, including the expenses associated with preclinical testing, clinical trials and obtaining regulatory approval. We believe that our existing cash and investment securities will be sufficient to support our current operating plan until the third quarter of 2012. Accordingly, our financial statements for the fiscal year ended December 31, 2011 included in this Annual Report, contain a going concern qualification from our independent registered public accounting firm. Unanticipated changes in our research and development plans or other changes affecting our operating expenses may affect actual consumption of existing cash resources. In any event, we will require substantial additional financing in the near term in order to complete the clinical trials we are currently conducting and to fund our continued operations. We do not know whether additional financing will be available when needed or that, if available, we will be able to obtain financing on terms favorable to our stockholders. In addition, the tight credit markets and concerns regarding the availability of credit, particularly in the United States, may also negatively impact our ability to raise additional capital to fund our business. If we are delisted on the Nasdaq Capital Market, our ability to raise additional capital in the public equity market will also be significantly limited. As of December 31, 2011, our accumulated deficit was $540.3 million, and we expect to incur capital outlays and operating expenditures for the next several years as we continue our clinical, research and development activities. The extent of any actual increase in operating or capital spending will depend in part on the clinical success of our product candidates. If we fail to raise adequate funds on terms acceptable to us, if at all, we will not be able to continue to fund our operations, research programs, preclinical testing, clinical trials and manufacturing efforts. Insufficient funds will affect our ability to operate as a going concern and may require us to delay, scale back, or eliminate some or all of our activities, and our continued existence would be uncertain.
Raising additional capital by issuing securities or through collaboration and licensing arrangements may cause dilution to existing stockholders or require us to relinquish rights to our technologies or product candidates.
We may seek to raise additional funds through equity or debt financings, collaborative arrangements with corporate partners or other sources. In August 2011, we filed a shelf registration statement on Form S-3 to offer and sell, from time to time, equity and debt securities in one or more offerings up to a total dollar amount of $25.0 million. On August 30, 2011 we entered into an At Market Issuance Sales Agreement, or the Sales Agreement, with McNicoll, Lewis & Vlak LLC, or MLV, pursuant to which we may issue and sell shares of our common stock having an aggregate offering price up to $7.0 million, from time to time, through MLV as our sales agent. As of December 31, 2011, we sold 484,112 shares through MLV under the Sales Agreement and received approximately $149,000 in net proceeds after deducting approximately $7,000 in commissions and other related expenses. To the extent that we raise additional capital by issuing equity securities, our stockholders will
14
Table of Contents
experience dilution. To the extent that we raise additional capital through licensing arrangements or arrangements with collaborative partners, we may be required to relinquish, on terms that are not favorable to us, rights to some of our technologies or product candidates that we would otherwise seek to develop or commercialize ourselves.
We have a history of net losses, which we expect to continue for the next several years should we have the ability to operate as a going concern. We will never be profitable unless we develop, and obtain regulatory approval and market acceptance of, our product candidates.
To date, we have not obtained regulatory approval for the commercial sale of any products, and we have not received any revenue from the commercial sale of products. Due to the significant research and development expenditures required to develop our TRAP technology and identify new product candidates, and the lack of any products to generate revenue, we have not been profitable and have incurred operating losses since we were incorporated in 1988. As of December 31, 2011, we had an accumulated deficit of $540.3 million. Should we have the ability to operate as a going concern, we expect to incur losses for the next several years as we continue our research and development activities and incur significant clinical testing and drug supply manufacturing costs. We do not anticipate that we will generate product revenue for at least several years. Our losses, among other things, have caused and will cause our stockholders’ equity and working capital to decrease. We expect that this trend will continue until we develop, and obtain regulatory approval and market acceptance of, our product candidates, if at all. We may never generate product revenue sufficient to become profitable.
Both of our most advanced drug product candidates, TELINTRA and TELCYTA, are in clinical development. If clinical trials of our product candidates are delayed or unsuccessful, or if we are unable to complete the preclinical development of our other preclinical product candidates, our business may suffer.
Preclinical testing and clinical trials are long, expensive and uncertain processes. It may take us or our collaborators several years to complete this testing, and failure can occur at any stage of the process. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and interim results of clinical trials do not necessarily predict final results. A number of companies in the pharmaceutical industry, including biotechnology companies, have suffered significant setbacks in advanced clinical trials, even after promising results in earlier clinical trials. To obtain regulatory approvals, we must, among other requirements, complete carefully controlled and well-designed clinical trials demonstrating that a particular drug is safe and effective for the applicable disease.
In 2007, we completed a Phase 1-2a clinical trial of a tablet formulation of TELINTRA in MDS, a form of pre-leukemia, to evaluate safety, pharmacokinetics, pharmacodynamics and efficacy. In May 2008, we initiated a Phase 2 clinical trial of TELINTRA tablets for the treatment of patients with MDS and announced final data at the ASH meeting in December 2010. In April 2009, we initiated a randomized Phase 2 clinical trial in SCN and in November 2009, we initiated a Phase 1 Dose-Ranging Study of TELINTRA tablets in combination with Revlimid in patients with MDS. Our success depends in part on our ability to continue clinical development of TELINTRA.
TELCYTA has been evaluated in multiple Phase 1, 2 and 3 clinical trials. Our Phase 3 trials did not achieve their primary endpoints and consequently the FDA required that we conduct additional studies of TELCYTA to complete clinical development. In May 2010, we initiated an investigator led study at a single site of TELCYTA in patients with refractory or relapsed mantle cell lymphoma, diffuse large B cell lymphoma, and multiple myeloma.
Our success depends in large part on our ability to continue clinical development of TELINTRA and TELCYTA. If we do not have sufficient capital required to conduct additional studies or if the data on future clinical trials are not positive, we may not be able to continue clinical development on TELINTRA or TELCYTA and our business will suffer.
15
Table of Contents
We rely on third-party clinical investigators to conduct our clinical trials and, as a result, we may face additional delays outside our control. We have in the past engaged contract research organizations, or CROs, to facilitate the administration of our Phase 3 clinical trials of TELCYTA. Dependence on a CRO subjects us to a number of risks. We may not be able to control the amount and timing of resources the CRO may devote to our clinical trials. Should the CRO fail to administer our Phase 3 clinical trials properly and on a timely basis, regulatory approval, development and commercialization of our product candidates will be delayed.
We do not know whether we will begin planned clinical trials on time or whether we will complete any of our on-going clinical trials on schedule, if at all. Clinical trials can be delayed for a variety of other reasons, including delays in clinical testing, obtaining regulatory approval to commence a study, delays in reaching agreement on acceptable clinical study agreement terms with prospective clinical sites, delays in obtaining institutional review board approval to conduct a study at a prospective clinical site and delays in recruiting subjects to participate in a study. Even if we are able to complete such clinical trials, we do not know whether any such trials will result in marketable products. Typically, there is a high rate of failure for product candidates in preclinical and clinical trials. We do not anticipate that any of our product candidates will reach the market for at least the next several years.
Delays in clinical testing can also materially impact our product candidates’ development costs. If we experience delays in clinical testing or approvals, our product candidates’ development costs will increase. For example, we may need to make additional payments to third-party investigators and organizations to retain their services or we may need to pay additional recruitment incentives. If the delays are significant, our financial results and the commercial prospects for our product candidates will be harmed, and our ability to become profitable will be significantly impaired or delayed.
If we do not find collaborators for our product candidates, we may have to reduce or delay our rate of product development and/or increase our expenditures.
Our strategy to develop, manufacture and commercialize our products includes entering into relationships with pharmaceutical companies to advance certain programs and reduce our expenditures with respect to such programs. Our product candidates will target highly competitive therapeutic markets in which we have limited experience and expertise. If we are unable to develop this expertise ourselves, we will need to enter into agreements with one or more biotechnology or pharmaceutical companies to provide us with the necessary resources and experience for the development and commercialization of products in these markets. In particular, we are seeking a partnership with a pharmaceutical or biotechnology company to further advance the development and commercialization of TELINTRA and TELCYTA. There are a limited number of companies with the resources necessary to develop our future products commercially, and we may be unable to attract any of these firms. The current credit and financial market conditions could also impact our ability to find a collaborator for our development programs. A company that has entered into a collaboration agreement with one of our competitors may choose not to enter into a collaboration agreement with us. We may not be able to negotiate a collaboration agreement on acceptable terms or at all. If we are not able to establish collaborative arrangements, we may have to reduce or delay further development of some of our programs and/or increase our expenditures and undertake the development activities at our own expense. If we elect to increase our expenditures to fund our development programs, we will need to obtain additional capital, which may not be available on acceptable terms or at all.
In addition, there have been a significant number of recent business combinations among biotechnology and pharmaceutical companies that have reduced the number of potential future collaborators that would be willing to enter into a collaboration agreement with us. If business combinations involving potential collaborators continue to occur, our ability to find a collaborative partner could be diminished, which could result in the termination or delay in one or more of our product candidate development programs.
16
Table of Contents
We will depend on collaborative arrangements to complete the development and commercialization of some of our product candidates. These collaborative arrangements may place the development of our product candidates outside of our control, may require us to relinquish important rights or may otherwise not be on terms favorable to us.
We expect to enter into collaborative arrangements with third parties for clinical trials, development, manufacturing, regulatory approvals or commercialization of some of our product candidates. Dependence on collaborative arrangements will subject us to a number of risks. We may not be able to control the amount and timing of resources our collaborative partners may devote to the product candidates. Our collaborative partners may experience financial difficulties. Should a collaborative partner fail to develop or commercialize a compound or product candidate to which it has rights from us, we may not receive any future milestone payments and will not receive any royalties for that compound or product candidate. Business combinations or significant changes in a collaborative partner’s business strategy may also adversely affect a partner’s willingness or ability to complete its obligations under an arrangement. If we fail to enter into additional collaborative agreements on favorable terms, our business, financial condition and results of operations could be materially adversely affected.
Some of our collaborations are for early stage programs and allow partners significant discretion in electing whether to pursue any of the planned activities. We do not anticipate significant revenues to result from these relationships until the collaborator has advanced product candidates into clinical trials, which will not occur for several years, if at all. These arrangements are subject to numerous risks, including the right of the collaboration partner to control the timing of the research and development efforts, the advancement of lead product candidates to clinical trials and the commercialization of product candidates. In addition, a collaborative partner could independently move forward with a competing lead candidate developed either independently or in collaboration with others, including our competitors.
Our ability to publicly or privately sell equity securities and the liquidity of our common stock could be adversely affected if we are delisted from the Nasdaq Capital Market and are unable to transfer our listing to another stock market.
On September 19, 2008, we received a letter from the Nasdaq Listing Qualifications Department indicating that, for 30 consecutive business days, the bid price for our common stock had closed below the minimum $1.00 per share requirement for continued inclusion on the Nasdaq Global Market under Nasdaq Marketplace Rule 5550(a)(2). The letter also stated that we were given 180 calendar days to regain compliance with this listing requirement, which could be accomplished if the bid price of our common stock closed at $1.00 per share or more for a minimum of 10 consecutive business days. Subsequently, Nasdaq implemented temporary suspensions of the minimum bid price requirement. Effective January 7, 2010, we transferred the listing of our common stock from the Nasdaq Global Market to the Nasdaq Capital Market, and we were provided an additional 180-day period to regain compliance. On April 22, 2010, we received notice from Nasdaq that we had regained compliance with the minimum bid price requirement. Subsequently, the bid price for our common stock fluctuated below the levels required by Nasdaq rules for continued listing. On July 21, 2010, we received notice from Nasdaq that the bid price for our common stock had closed below the minimum $1.00 per share requirement for continued inclusion on the Nasdaq Capital Market and that we had 180 calendar days to regain compliance. On January 19, 2011, we received a notice from Nasdaq indicating that, while we had not regained compliance with the $1.00 per share requirement, Nasdaq had determined that Telik was eligible to receive an additional 180-day period to regain compliance. On February 10, 2011, we received a notice from Nasdaq indicating that for the preceding, ten consecutive business days, the closing bid price of our common stock had been $1.00 per share or greater and, as such, we had regained compliance with the $1.00 per share minimum bid price requirement. On April 21, 2011, we received notice from Nasdaq that the bid price of our common stock had closed below the minimum $1.00 per share requirement for continued inclusion on the Nasdaq Capital Market and that we had 180 calendar days, or until October 18, 2011 to regain compliance. On October 20, 2011, we received notice from Nasdaq that we had been provided with an additional 180-day period, or until April 16, 2012, to regain compliance with the minimum $1.00 per share requirement. In order to regain compliance, at any
17
Table of Contents
time before April 16, 2012, the bid price of our common stock must close at $1.00 per share or more for a minimum of ten consecutive business days. Nonetheless, Nasdaq may require us to maintain a closing bid price of at least $1.00 per share for a longer period before determining that we have achieved compliance. If Nasdaq determines that we will not be able to cure the deficiency, or if we are otherwise ineligible, then Nasdaq will provide written notification that our common stock will be delisted, after which we may appeal the delisting determination to a Hearings Panel. In February 2012, our board of directors approved a proposal, subject to stockholder approval, to decide whether to effect a reverse stock split of all outstanding shares of our common stock and stock options at an exchange ratio of one for fifteen, one for twenty, one for twenty five or one for thirty. Our board of directors has recommended that this proposal be presented to our stockholders for approval in the 2012 Annual Meeting of Stockholders which will be held on March 28, 2012. We cannot assure you that we will be able to regain the minimum bid price, and even if so, maintain it or other Nasdaq Capital Market listing requirements.
Delisting from the Nasdaq Capital Market could adversely affect our ability to raise additional financing through the public or private sale of equity securities, would significantly affect the ability of investors to trade our securities and would negatively affect the value and liquidity of our common stock. Delisting could also have other negative results, including the potential loss of confidence by employees, the loss of institutional investor interest and fewer business development opportunities.
It may be difficult for us to retain our current employees and identify, hire and retain future employees.
Our future success depends in part upon our ability to attract and retain highly skilled personnel. Several factors could make it difficult for us to achieve this. Competition among numerous companies, academic and other research institutions for skilled personnel and experienced scientists may be intense and turnover rates high. The cost of living in the San Francisco Bay Area is high compared to other parts of the country, which could adversely affect our ability to compete for qualified personnel and increase our costs. Because of this competitive environment, we have encountered and may continue to encounter increasing difficulty attracting qualified personnel, particularly if our operations expand and the demand for these professionals increases.
In addition, we may have difficulty attracting and retaining personnel as a result of having carried out four workforce reductions since 2007, the most recent of which was completed in November 2010. We cannot assure you that future reductions or adjustments of our workforce will not be made or that issues, such as voluntary departures by some employees, associated with such reductions will not recur. These circumstances could significantly impede the achievement of our business objectives.
If our competitors develop and market products that are more effective than our product candidates or any products that we may develop, or obtain marketing approval before we do, our commercial opportunity will be reduced or eliminated.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. Some of the drugs that we are attempting to develop will have to compete with existing therapies. In addition, a large number of companies are pursuing the development of pharmaceuticals that target the same diseases and conditions that we are targeting. We face competition from pharmaceutical and biotechnology companies in the United States and abroad. Our competitors may develop new screening technologies and may utilize discovery techniques or partner with collaborators in order to develop products more rapidly or successfully than we or our collaborators are able to do.
Many of our competitors, particularly large pharmaceutical companies, have substantially greater financial, technical and human resources than we do. These organizations also compete with us to attract qualified personnel and parties for acquisitions, joint ventures, licensing arrangements or other collaborations. In addition, academic institutions, government agencies and other public and private organizations conducting research may seek patent protection with respect to potentially competing products or technologies and may establish exclusive collaborative or licensing relationships with our competitors.
18
Table of Contents
Our competitors may succeed in developing technologies and drugs that are more effective, better tolerated or less costly than any which are being developed by us or which would render our technology and potential drugs obsolete and noncompetitive. In addition, our competitors may succeed in obtaining FDA or other regulatory approvals for product candidates more rapidly than us or our collaborators. Any drugs resulting from our research and development efforts, or from our joint efforts with our existing or future collaborative partners, may not be able to compete successfully with our competitors’ existing products or product candidates under development or may not obtain regulatory approval in the United States or elsewhere.
If we do not obtain regulatory approval to market products in the United States and foreign countries, we or our collaborators will not be permitted to commercialize our product candidates.
Even if we are able to achieve success in our preclinical testing, we, or our collaborators, must provide the FDA and foreign regulatory authorities with clinical data that demonstrate the safety and efficacy of our product candidates in humans before they can be approved for commercial sale. Failure to obtain regulatory approval will prevent commercialization of our product candidates.
The pharmaceutical industry is subject to stringent regulation by a wide range of regulatory authorities. We cannot predict whether regulatory clearance will be obtained for any product candidate that we are developing or hope to develop. A pharmaceutical product cannot be marketed in the United States until it has completed rigorous preclinical testing and clinical trials and an extensive regulatory clearance process implemented by the FDA. Satisfaction of regulatory requirements typically takes many years and depends on the type, complexity and novelty of the product candidate and requires the expenditure of substantial resources. Of particular significance are the requirements covering research and development, testing, manufacturing, quality control, labeling and promotion of drugs for human use.
Before commencing clinical trials in humans, we, or our collaborators, must submit and receive approval from the FDA of an IND application. We must comply with FDA “Good Laboratory Practices” regulations in our preclinical studies. Clinical trials are subject to oversight by Institutional Review Boards, or IRBs, of participating clinical sites and the FDA and:
| • | must be conducted in conformance with the FDA regulations; |
| • | must meet requirements for IRB approval; |
| • | must meet requirements for informed consent; |
| • | must meet requirements for Good Clinical Practices; |
| • | may require large numbers of participants; and |
| • | may be suspended by us, our collaborators or the FDA at any time if it is believed that the subjects participating in these trials are being exposed to unacceptable health risks or if the FDA finds deficiencies in the IND application or the conduct of these trials. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions. |
Before receiving FDA clearance to market a product candidate, we, or our collaborators must demonstrate that the product candidate is safe and effective in the patient population that will be treated. Negative or inconclusive results or adverse medical events during a clinical trial could cause a clinical trial to be repeated, a program to be terminated and could delay approval. We typically rely on third-party clinical investigators to conduct our clinical trials and other third-party organizations to perform data collection and analysis. As a result, we may face additional delaying factors outside our control. In addition, we may encounter delays or rejections based upon additional government regulation from future legislation or administrative action or changes in FDA policy or interpretation during the period of product development, clinical trials and FDA regulatory review. Failure to comply with applicable FDA or other applicable regulatory requirements may result in criminal
19
Table of Contents
prosecution, civil penalties, recall or seizure of products, total or partial suspension of production or injunction, as well as other regulatory action. We have limited experience in conducting and managing the clinical trials necessary to obtain regulatory approval.
If regulatory clearance of a product candidate is granted, this clearance will be limited to those disease states and conditions for which the product candidate is demonstrated through clinical trials to be safe and efficacious, which could limit our market opportunity. Furthermore, product approvals, once granted, may be withdrawn if problems occur after initial marketing. We cannot ensure that any product candidate developed by us, alone or with others, will prove to be safe and efficacious in clinical trials and will meet all of the applicable regulatory requirements needed to receive marketing clearance. Regulatory clearance may also contain requirements for costly post-marketing testing and surveillance to monitor the safety and efficacy of the product candidate. If problems occur after initial marketing, such as the discovery of previously unknown problems with our product candidates, including unanticipated adverse events or adverse events of unanticipated severity or frequency, or manufacturer or manufacturing issues, marketing approval can be withdrawn.
Outside the United States, the ability to market a product depends on receiving a marketing authorization from the appropriate regulatory authorities. Most foreign regulatory approval processes include all of the risks associated with FDA clearance described above and some may include additional risks.
As our product programs advance, we may need to hire additional scientific and management personnel. Our research and development efforts will be seriously jeopardized if we are unable to attract and retain key personnel.
Our success depends in part on the continued contributions of our principal management and scientific personnel and on our ability to develop and maintain important relationships with leading academic institutions, scientists and companies in the face of intense competition for such personnel. As we plan for additional advanced clinical trials, including Phase 2 and Phase 3, we may also need to expand our clinical development personnel. If we lose the services of Dr. Wick or any of our other key personnel, our research and development efforts could be seriously and adversely affected. We have generally entered into consulting or other agreements with our scientific and clinical collaborators and advisors. We do not carry key person insurance that covers Dr. Wick or any of our other key employees.
If we or our licensees cannot obtain and defend our respective intellectual property rights, or if our product candidates, technologies or any products that we may develop are found to infringe patents of third parties, we could become involved in lengthy and costly legal proceedings that could adversely affect our business.
Our success will depend in a large part on our own and our licensees’ ability to obtain and defend patents for each party’s respective technologies and the compounds and other products, if any, resulting from the application of these technologies. The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and involve complex legal and factual questions. As a result, the degree of future protection for our proprietary rights is uncertain, and we cannot assure you that:
| • | we were the first to make the inventions covered by each of our pending patent applications; |
| • | we were the first to file patent applications for these inventions; |
| • | others will not independently develop similar or alternative technologies or duplicate any of our technologies; |
| • | any of our pending patent applications will result in issued patents; |
| • | any patents issued to us or our collaborators will provide a basis for commercially viable products, will provide us with any competitive advantages or will not be challenged by third parties; |
20
Table of Contents
| • | any of our issued patents will be valid or enforceable; or |
| • | we will develop additional proprietary technologies that are patentable. |
Accordingly, we cannot predict the breadth of claims allowed in our or other companies’ patents.
For TELINTRA, we hold compound patents in the United States and internationally that will expire in 2014. For TELCYTA, we hold compound patents in the United States and internationally that will expire in 2013 and 2014. We can generally apply for patent term extensions on the patents for TELINTRA and TELCYTA when and if marketing approvals for these compounds are obtained in the relevant countries. In foreign countries, these extensions are available only if approval is obtained before the patent expires, but in the United States, extensions are available even if approval is obtained after the patent would expire normally through a system of interim extensions until approval.
Our success will also depend, in part, on our ability to operate without infringing the intellectual property rights of others. We cannot assure you that our activities will not infringe patents owned by others. To date, we have not received any communications with the owners of related patents alleging that our activities infringe their patents. However, if our product candidates, technologies or any products that we may develop are found to infringe patents issued to third parties, the manufacture, use and sale of any products that we may develop could be enjoined, and we could be required to pay substantial damages. In addition, we may be required to obtain licenses to patents or other proprietary rights of third parties. We cannot assure you that any licenses required under any such patents or proprietary rights would be made available on terms acceptable to us, if at all. Failure to obtain such licenses could negatively affect our business.
Others may have filed and in the future may file patent applications covering small molecules or therapeutic products that are similar to ours. We cannot assure you that our patent applications will have priority over patent applications filed by others. Any legal action against us or our collaborators claiming damages and seeking to enjoin commercial activities relating to the affected products and processes could, in addition to subjecting us to potential liability for damages, require us or our collaborators to obtain a license to continue to manufacture or market the affected products and processes. We cannot predict whether we, or our collaborators, would prevail in any of these actions or that any license required under any of these patents would be made available on commercially acceptable terms, if at all. We believe that there may be significant litigation in the industry regarding patent and other intellectual property rights. If we become involved in litigation, it could consume a substantial portion of our managerial and financial resources, and we may not be successful in any such litigation.
In addition, we could incur substantial costs and use of our key employees’ time and efforts in litigation if we are required to defend against patent suits brought by third parties or if we initiate these suits, and we cannot predict whether we would be able to prevail in any of these suits.
Furthermore, some of our patents and intellectual property rights are owned jointly by us and our collaborators. While there are legal and contractual restraints on the rights of these joint owners, they may use these patents and other intellectual property in ways that may harm our business. We may not be able to prevent this type of use.
We also rely on trade secrets to protect technology, including aspects of our TRAP technology, where we believe patent protection is not appropriate or obtainable. However, trade secrets are difficult to protect. While we require employees, academic collaborators and consultants to enter into confidentiality agreements, we may not be able to adequately protect our trade secrets or other proprietary information in the event of any unauthorized use or disclosure or the lawful development by others of such information. If the identity of specific proteins or other elements of our TRAP technology become known, our competitive advantage in drug discovery could be reduced.
21
Table of Contents
Many of our collaborators and scientific advisors have publication and other rights to certain information and data gained from their collaborations and research with us. Any publication or other use could limit our ability to secure intellectual property rights or impair any competitive advantage that we may possess or realize by maintaining the confidentiality of that information or data.
If we are unable to enter into or maintain existing contracts with third parties to manufacture our product candidates or any products that we may develop in sufficient quantities and at an acceptable cost, clinical development of product candidates could be delayed and we may be unable to meet demand for any products that we may develop and lose potential revenue.
We do not currently operate manufacturing facilities for clinical or commercial production of our product candidates. We expect to continue to rely on third parties for the manufacture of our product candidates and any products that we may develop. We currently lack the resources and capability to manufacture any of our product candidates on a clinical scale or any products that we may develop on a commercial scale. As a result, we will be dependent on corporate partners, licensees or other third parties for the manufacturing of clinical and commercial scale quantities of our product candidates and any products that we may develop. Our product candidates and any products that we may develop may be in competition with other product candidates and products for access to these manufacturing facilities. For this and other reasons, our collaborators or third parties may not be able to manufacture our product candidates and products in a cost effective or timely manner. While we currently possess sufficient inventory of TELINTRA and TELCYTA that are stored in multiple locations and an additional, substantial quantity of the active ingredient in TELCYTA, if these inventories are lost or damaged, the clinical development of our product candidates or their submission for regulatory approval could be delayed, and our ability to deliver any products that we may develop on a timely basis could be impaired or precluded.
Isochem has in the past manufactured and supplied a limited number of batches of the active ingredient to be used in the production of TELINTRA for use in clinical trials. As such, we have qualified Isochem as a potential source of supply for clinical quantities of this ingredient. In addition, Patheon has in the past performed formulation development and analytical services, and produced for us limited batches of clinical trial material, relating to the tablet formation of TELINTRA. As such, we have qualified Patheon as a potential manufacturer of TELINTRA tablets. However, we currently do not have in effect any manufacturing or supply agreements with either Isochem or Patheon. Although we have not qualified any other sources for the active ingredient in TELINTRA or the manufacture of TELINTRA tablets, we have evaluated, and may consider, potential alternative sources for this material in the future.
We are currently dependent on a single source of supply of the active ingredient in TELCYTA, AMRI. OSO BioPharmaceuticals Manufacturing LLC, or OSO, has in the past manufactured and supplied TELCYTA drug product for use in our clinical trials. As such, we have qualified OSO as a potential source of supply for clinical quantities of TELCYTA. However, we currently do not have in effect a manufacturing or supply agreement with OSO. Although we have not qualified any other source for TELCYTA drug product, we have evaluated, and may consider, potential alternative sources for TELCYTA in the future.
If manufacturing is not performed in a timely manner, if our suppliers fail to perform or if our relationships with our suppliers or manufacturers should terminate, our clinical trials and commercialization of TELINTRA and TELCYTA could be delayed. We may not be able to enter into or maintain any necessary third-party manufacturing arrangements on acceptable terms, if at all. Our current dependence upon others for the manufacture of our product candidates and our anticipated dependence upon others for the manufacture of any products that we may develop may adversely affect our future profit margins and our ability to commercialize any products that we may develop on a timely and competitive basis.
22
Table of Contents
Working capital constraints may force us to delay our efforts to develop certain product candidates in favor of developing others, which may prevent us from commercializing all product candidates as quickly as possible.
Because we have limited resources, and because research and development is an expensive process, we must regularly assess the most efficient allocation of our research and development budget. As a result, we have had to prioritize development candidates and may not be able to fully realize the value of some of our product candidates in a timely manner, as they will be delayed in reaching the market, if at all.
If product liability lawsuits are successfully brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates and any products that we may develop.
The testing and marketing of medical products entail an inherent risk of product liability. Although we are not aware of any historical or anticipated product liability claims or specific causes for concern, if we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates and any products that we may develop. In addition, product liability claims may also result in withdrawal of clinical trial volunteers, injury to our reputation and decreased demand for any products that we may commercialize. We currently carry product liability insurance that covers our clinical trials up to a $10.0 million annual aggregate limit. We will need to increase the amount of coverage if and when we have a product that is commercially available. If we are unable to obtain sufficient product liability insurance at an acceptable cost, potential product liability claims could prevent or inhibit the commercialization of any products that we may develop, alone or with corporate partners.
We have implemented anti-takeover provisions which could discourage or prevent a takeover, even if an acquisition would be beneficial to our stockholders, and may prevent attempts by our stockholders to replace or remove our current management.
Provisions of our amended and restated certificate of incorporation and bylaws, as well as provisions of Delaware law, could make it more difficult for a third-party to acquire us, even if doing so would be beneficial to our stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. Because our board of directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt by our stockholders to replace current members of our management team. These provisions include:
| • | establishing a classified board of directors requiring that members of the board be elected in different years, which lengthens the time needed to elect a new majority of the board; |
| • | authorizing the issuance of “blank check” preferred stock that could be issued by our board of directors to increase the number of outstanding shares or change the balance of voting control and thwart a takeover attempt; |
| • | prohibiting cumulative voting in the election of directors, which would otherwise allow for less than a majority of stockholders to elect director candidates; |
| • | limiting the ability of stockholders to call special meetings of the stockholders; |
| • | prohibiting stockholder action by written consent and requiring all stockholder actions to be taken at a meeting of our stockholders; and |
| • | establishing 90 to 120 day advance notice requirements for nominations for election to the board of directors and for proposing matters that can be acted upon by stockholders at stockholder meetings. |
23
Table of Contents
Substantial future sales of our common stock by us or by our existing stockholders could cause our stock price to fall.
Additional equity financings or other share issuances by us, including shares issued in connection with strategic alliances and corporate partnering transactions, could adversely affect the market price of our common stock. Sales by existing stockholders of a large number of shares of our common stock in the public market or the perception that additional sales could occur could cause the market price of our common stock to drop. Substantially all of our outstanding shares of common stock were freely tradable and, in limited cases, subject to certain volume, notice and manner of sale restrictions under Rule 144 of the Securities Act of 1933.
If we do not progress in our programs as anticipated, our stock price could decrease.
For planning purposes, we estimate the timing of a variety of clinical, regulatory and other milestones, such as when a certain product candidate will enter clinical development, when a clinical trial will be completed or when an application for regulatory approval will be filed. Our estimates are based on present facts and a variety of assumptions. Many of the underlying assumptions are outside of our control. If milestones are not achieved when we estimated that they would be, investors could be disappointed, and our stock price may decrease.
Our stock price may be volatile, you may not be able to resell your shares at or above your purchase price.
Our stock prices and the market prices for securities of biotechnology companies in general have been highly volatile, with recent significant price and volume fluctuations, and may continue to be highly volatile in the future. For example, our stock price dropped by 71% on the day following the announcement in December 2006 that the preliminary top-line results of our first three Phase 3 trials did not meet primary end-points. During the twelve months ended December 31, 2011, our common stock traded between $0.16 and $1.25, and on December 31, 2011, our common stock closed at $0.20. You may not be able to sell your shares quickly or at the market price if we are delisted from the Nasdaq Capital Market or if we are unable to transfer our listing to another stock market, or if trading in our stock is not active or the volume is low. The following factors, in addition to other risk factors described in this section, may have a significant impact on the market price of our common stock, some of which are beyond our control:
| • | developments regarding, or the results of, our clinical trials; |
| • | announcements of technological innovations or new commercial products by our competitors or us; |
| • | the issuance of equity or debt securities of the Company, or disclosure or announcements relating thereto; |
| • | developments concerning proprietary rights, including patents; |
| • | developments concerning our collaborations; |
| • | publicity regarding actual or potential medical results relating to products under development by our competitors or us; |
| • | regulatory developments in the United States and foreign countries; |
| • | litigation; |
| • | economic and other external factors or other disaster or crisis; or |
| • | period-to-period fluctuations in our financial results. |
We have been, and in the future may be, subject to securities class action lawsuits and shareholder derivative actions. These, and potential similar or related litigation, could result in substantial damages and may divert management’s time and attention from our business.
We have been, in particular between 2007 and 2008, and may in the future be, the target of securities class actions or shareholder derivative claims. Any such actions or claims could result in substantial damages and may divert management’s time and attention from our business.
24
Table of Contents
| Item 1B. | Unresolved Staff Comments. |
None
| Item 2. | Properties. |
In November 2010, we entered into a 28 month lease agreement for 8,620 square feet of office space at 700 Hansen Way in Palo Alto, California and relocated our corporate offices to this facility. We also lease approximately 92,000 square feet of research and office space located at 3165 Porter Drive in Palo Alto, which we have subleased to a tenant effective November 2010 through May 2014 when our master lease expires. We believe the facility at Hansen Way is sufficient to meet our current needs.
| Item 3. | Legal Proceedings. |
None
| Item 4. | Mine Safety Disclosures. |
Not applicable.
25
Table of Contents
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. |
Market for Our Common Stock
Our common stock trades on the Nasdaq Capital Market under the symbol “TELK”. The following table sets forth the high and low sales prices for our common stock for each quarterly period within the two most recent fiscal years.
| High | Low | |||||||
| 2011 |
||||||||
| Quarter ended March 31, 2011 |
$ | 1.25 | $ | 0.75 | ||||
| Quarter ended June 30, 2011 |
$ | 1.01 | $ | 0.43 | ||||
| Quarter ended September 30, 2011 |
$ | 0.80 | $ | 0.22 | ||||
| Quarter ended December 31, 2011 |
$ | 0.37 | $ | 0.16 | ||||
| 2010 |
||||||||
| Quarter ended March 31, 2010 |
$ | 1.09 | $ | 0.72 | ||||
| Quarter ended June 30, 2010 |
$ | 1.49 | $ | 0.66 | ||||
| Quarter ended September 30, 2010 |
$ | 0.82 | $ | 0.64 | ||||
| Quarter ended December 31, 2010 |
$ | 0.95 | $ | 0.64 | ||||
Nasdaq Stock Listing Compliance
On September 19, 2008, we received notification from Nasdaq informing us that the bid price for our common stock had closed below the minimum $1.00 per share requirement for continued inclusion on the Nasdaq Global Market under Nasdaq Marketplace Rule 5550(a)(2). Effective January 7, 2010, we transferred the listing of our common stock from the Nasdaq Global Market to the Nasdaq Capital Market. On April 22, 2010, we received notification from Nasdaq that we had regained compliance with the minimum bid price requirement. Since May 20, 2010, the bid price for our common stock had again fluctuated below the levels required by Nasdaq rules for continued listing. On July 21, 2010, we received notice from Nasdaq that the bid price for our common stock had closed below the minimum $1.00 per share requirement for continued inclusion on the Nasdaq Capital Market. On February 10, 2011, we received a notification from Nasdaq that we regained compliance with the minimum bid price requirement. On April 21, 2011, we received notice from Nasdaq that the bid price of our common stock had closed below the minimum $1.00 per share requirement for continued inclusion on the Nasdaq Capital Market and that we had 180 calendar days, or until October 18, 2011 to regain compliance. On October 20, 2011, we received notice from Nasdaq that we had been provided with an additional 180-day period, or until April 16, 2012, to regain compliance with the minimum $1.00 per share requirement. In order to regain compliance, at any time before April 16, 2012, the bid price of our common stock must close at $1.00 per share or more for a minimum of ten consecutive business days. Nonetheless, Nasdaq may require us to maintain a closing bid price of at least $1.00 per share for a longer period before determining that we have achieved compliance. If Nasdaq determines that we will not be able to cure the deficiency, or if we are otherwise ineligible, then Nasdaq will provide written notification that our common stock will be delisted, after which we may appeal the delisting determination to a Hearings Panel.
As of February 22, 2012, there were 75 stockholders of record of our common stock. We have never paid our stockholders cash dividends, and we do not anticipate paying any cash dividends in the foreseeable future as we intend to retain any earnings for use in our business. Any future determination to pay dividends will be at the discretion of our board of directors.
26
Table of Contents
Performance Graph
The following graph compares our cumulative total stockholder return for the past five years to two indices: the Nasdaq U.S. Index and the Nasdaq Pharmaceutical Stocks Index. This graph assumes the investment of $100 on December 29, 2006 in our common stock, the Nasdaq U.S. Index; and the Nasdaq Pharmaceutical Stocks Index. All values assume reinvestment of the full amount of all dividends and are calculated as of the last stock trading day of each year:
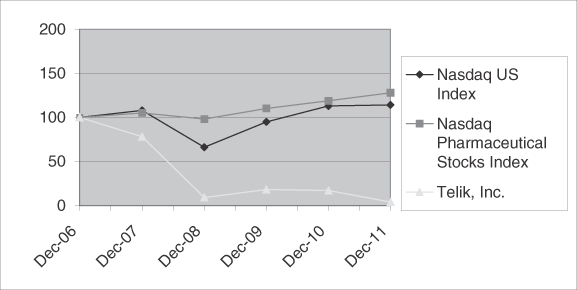
| December 29, 2006 |
December 31, 2007 |
December 31, 2008 |
December 31, 2009 |
December 31, 2010 |
December 30, 2011 |
|||||||||||||||||||
| Telik, Inc. |
$ | 100 | $ | 78 | $ | 9 | $ | 18 | $ | 17 | $ | 4 | ||||||||||||
| Nasdaq U.S. Index |
100 | 108 | 66 | 95 | 113 | 114 | ||||||||||||||||||
| Nasdaq Pharmaceutical Stocks Index |
100 | 105 | 98 | 110 | 119 | 128 | ||||||||||||||||||
Source: Nasdaq.Online. The information under “Performance Graph” is not deemed to be “soliciting material” or “filed” with the Securities and Exchange Commission or subject to Regulation 14A or 14C, or to the liabilities of Section 18 of the Exchange Act of 1934, as amended and is not to be incorporated by reference in any filing of Telik under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date of this Annual Report on Form 10-K and irrespective of any general incorporation language in those filings.
27
Table of Contents
| Item 6. | Selected Financial Data. |
The following selected historical information has been derived from the audited financial statements of Telik and should be read in conjunction with “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Item 8. Financial Statements and Supplementary Data” included elsewhere in this Annual Report on Form 10-K. The historical results are not necessarily indicative of the results of operations to be expected in the future.
| Years Ended December 31, | ||||||||||||||||||||
| 2011 | 2010 | 2009 | 2008 | 2007 | ||||||||||||||||
| (In thousands, except per share amounts) | ||||||||||||||||||||
| Statement of Operations Data: |
||||||||||||||||||||
| Operating costs and expenses: |
||||||||||||||||||||
| Research and development |
5,566 | 11,040 | 12,723 | 23,952 | 43,032 | |||||||||||||||
| General and administrative |
6,491 | 9,230 | 10,810 | 10,560 | 15,941 | |||||||||||||||
| Facility exit costs |
— | 5,360 | — | — | — | |||||||||||||||
| Restructuring costs |
— | 425 | 951 | 196 | 1,356 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating costs and expenses |
12,057 | 26,055 | 24,484 | 34,708 | 60,329 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss from operations |
(12,057 | ) | (26,055 | ) | (24,484 | ) | (34,708 | ) | (60,329 | ) | ||||||||||
| Interest income and other, net |
35 | 1,333 | 791 | 2,945 | 5,114 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss |
$ | (12,022 | ) | $ | (24,722 | ) | $ | (23,693 | ) | $ | (31,763 | ) | $ | (55,215 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Basic and diluted net loss per share |
$ | (0.22 | ) | $ | (0.46 | ) | $ | (0.44 | ) | $ | (0.60 | ) | $ | (1.05 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Shares used to calculate basic and diluted net Loss per share |
53,939 | 53,539 | 53,371 | 53,177 | 52,542 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| As of December 31, | ||||||||||||||||||||
| 2011 | 2010 | 2009 | 2008 | 2007 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents, investments and restricted Investments |
$ | 11,700 | $ | 24,064 | $ | 40,400 | $ | 63,469 | $ | 93,233 | ||||||||||
| Working capital |
9,422 | 20,736 | 39,221 | 48,778 | 69,410 | |||||||||||||||
| Total assets |
12,412 | 25,029 | 46,153 | 75,413 | 98,528 | |||||||||||||||
| Current portion of obligations and loans |
1,463 | 1,439 | 3,101 | — | — | |||||||||||||||
| Non-current portion of obligations, loans,and long-term liabilities |
1,463 | 2,923 | — | 8,000 | — | |||||||||||||||
| Accumulated deficit |
(540,329 | ) | (528,307 | ) | (503,585 | ) | (479,892 | ) | (448,129 | ) | ||||||||||
| Total stockholders’ equity |
8,299 | 18,369 | 40,934 | 62,372 | 87,319 | |||||||||||||||
28
Table of Contents
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations. |
Overview
Telik is engaged in the discovery and development of small molecule drugs. Our business strategy is to advance our drug product candidates through Phase 2 clinical studies, and to enter into a partnership with a pharmaceutical or biotechnology company to assist in further development and commercialization, license product candidates outside our therapeutic focus, and identify and develop additional drug product candidates.
We have incurred net losses since inception and expect to incur losses next year as we continue our research and development activities. During the year ended December 31, 2011, loss from operations was $12.1 million and net loss was $12.0 million. Net cash used in operations for the year ended December 31, 2011 was $12.8 million and net cash, cash equivalents, investments and restricted investments at December 31, 2011 were $11.7 million. As of December 31, 2011, we had an accumulated deficit of $540.3 million.
Our expenses consist primarily of those incurred for research and development and general and administrative costs associated with our operations. The process of carrying out the development of our product candidates to later stages of development and our research programs will require significant additional research and development expenditures, including for preclinical testing and clinical trials, as well as for manufacturing development efforts and obtaining regulatory approval. We outsource our clinical trials and our manufacturing development activities to third parties to maximize efficiency and minimize our internal overhead. To date, we have funded our operations primarily through the sale of equity securities, non-equity payments from collaborative partners and interest income.
We are subject to risks common to biopharmaceutical companies, including the need for capital, risks inherent in our research, development and commercialization efforts, preclinical testing, clinical trials, uncertainty of regulatory and marketing approvals, enforcement of patent and proprietary rights, potential competition and retention of key employees. In order for a product to be commercialized, it will be necessary for us to conduct preclinical tests and clinical trials, demonstrate efficacy and safety of our product candidates to the satisfaction of regulatory authorities, obtain marketing approval, enter into manufacturing, distribution and marketing arrangements, obtain market acceptance and, in many cases, obtain adequate reimbursement from government and private insurers. We cannot provide assurance that we will generate revenues or achieve and sustain profitability in the future.
We expect that our quarterly and annual results of operations will fluctuate for the foreseeable future due to several factors, including the timing and extent of our research and development efforts and the outcome of our clinical trial activities. The successful development of our product candidates is uncertain. As such, an accurate prediction of future operating results is difficult or impossible.
Going Concern
Our financial statements have been prepared using generally accepted accounting principles applicable to a going concern which contemplates the realization of assets and liquidation of liabilities in the normal course of business. Accordingly they do not give effect to adjustments that would be necessary should we be unable to continue as a going concern. We believe our existing cash resources will not be sufficient to fund our projected operating requirements for the next twelve months, including prosecuting our current trials, conducting research and discovery efforts towards additional product candidates and providing sufficient funds for working capital and general corporate purposes. Our independent registered public accounting firm has issued a report on our financial statements that includes an explanatory paragraph referring to our recurring losses from operations and expresses substantial doubt as to our ability to continue as a going concern without additional capital becoming available. In order to continue as a going concern, we will require substantial additional financing to fund our operations and continue our clinical product development programs, and our ability to continue as a viable entity will be dependent on our ability to obtain funding. We have been and are currently seeking collaborative arrangements with corporate partners to fund the development and commercialization of TELINTRA, our lead
29
Table of Contents
product candidate and TELCYTA, our other product candidate. We are also evaluating options to raise additional funds through equity or debt financings and sales transactions as well as other sources such as research grants from non-profit organizations. However, we cannot provide any assurances that we will be successful in obtaining additional funding. In the event we are unable to obtain additional funding, we will be required to shift our primary focus to exploring strategic alternatives available to us to create significant value for stockholders. Those alternatives include the further restructuring of our operations to conserve resources, the sale of company assets, in whole or in part, or some other arrangement through which the value of our assets to stockholders could be optimized.
Clinical Product Development
TELINTRA, our lead drug product candidate in clinical development, is a small molecule glutathione analog inhibitor of the enzyme glutathione S-transferase P1-1, or GST P1-1. We are developing TELINTRA for the treatment of blood disorders associated with low blood cell levels, such as neutropenia or anemia. We completed an 86 patient Phase 2 clinical trial of TELINTRA tablets, for the treatment of patients with Myelodysplastic Syndrome, or MDS and presented the results at the annual meeting of the American Society of Hematology, or ASH, in December 2010. In the second quarter of 2009, we initiated a Phase 2 randomized study in Severe Chronic Neutropenia, or SCN, to determine the effect of TELINTRA tablets on absolute neutrophil count in patients with this disease. Due to the scarcity of SCN patients and our focus on MDS, we plan to terminate this study once the last remaining patient completes treatment around the third quarter of 2012. In the fourth quarter of 2009, we initiated a Phase 1 dose-ranging study of TELINTRA tablets in combination with Revlimid in patients with MDS. The study was completed in the fourth quarter of 2011 and we presented the results at the annual meeting of ASH in December 2011. In June 2011, we initiated a Phase 2 clinical trial to evaluate TELINTRA tablets in patients with Revlimid refractory or resistant, deletion 5q myelodysplastic syndrome, or del 5q MDS. This multicenter trial is intended to enroll up to 117 evaluable patients. The first planned interim analysis will occur after one-third of the patients have completed therapy and depending on the results of the interim analysis, additional enrollment may continue with two additional analyses planned. The timing of the interim analyses is dependent on the availability of patients in the del 5q MDS population. In October 2011, we initiated a Phase 2b clinical trial to evaluate TELINTRA tablets in patients with transfusion dependent, non-deletion 5q MDS, who have not been treated with hypomethylating agents, or HMA. This multicenter trial is intended to enroll up to 145 evaluable patients with 2 planned interim analyses, the first of which will be performed when data from 49 evaluable patients are available and the second at 97 evaluable patients. The timing of enrollment in this study is dependent on the availability of non-deletion 5q MDS patients.
TELCYTA, our other product candidate, is a small molecule cancer drug product candidate designed to be activated in cancer cells. TELCYTA has been evaluated in multiple Phase 2 and Phase 3 clinical trials, including trials using TELCYTA as monotherapy and in combination regimens in ovarian, non-small cell lung, breast and colorectal cancer. Results from these clinical trials indicate that TELCYTA monotherapy was generally well-tolerated, with mostly mild to moderate side effects, particularly when compared to the side effects and toxicities of standard chemotherapeutic drugs. When TELCYTA was evaluated in combination with standard chemotherapeutic drugs, the tolerability of the combinations was similar to that expected of each drug alone. Clinical activity including objective tumor responses and/or disease stabilization was reported in the TELCYTA Phase 2 trials; however, TELCYTA did not meet its primary endpoints in the Phase 3 studies. In May 2010, we initiated an investigator led study at a single site of TELCYTA in patients with refractory or relapsed mantle cell lymphoma, diffuse large B cell lymphoma, and multiple myeloma. Enrollment for this study is in two stages and is expected to range between 18 to 48 patients based on the number of responses observed. Based on responses observed in stage 1, we are planning to expand study to stage 2 and adding a second investigator site in the first quarter of 2012. Completion of enrollment is expected by the second half of 2013. We are currently seeking a partnership with a pharmaceutical or biotechnology company to further advance the development and commercialization of TELCYTA.
We will need to raise additional funds in the near future in order to see our current active and planned clinical trials to completion.
30
Table of Contents
Preclinical Drug Product Development
TLK60404—Aurora Kinase/VEGFR Inhibitors
We have a small molecule compound inhibiting both Aurora kinase and VEGFR kinase. Aurora kinase is a signaling enzyme whose function is required for cancer cell division, while VEGF plays a key role in tumor blood vessel formation, ensuring an adequate supply of nutrients to support tumor growth. The lead compounds of our first dual inhibitor program met a development milestone in August 2008 by demonstrating anticancer activity in preclinical models of human colon cancer and human leukemia. These lead compounds prevented tumor growth in preclinical models of human colon cancer and human leukemia by inhibiting both Aurora kinase and VEGFR kinase. Our data support the concept that dual inhibition of Aurora kinase and VEGFR kinase represents a promising approach for anticancer therapy. A development drug product candidate, TLK60404, has been selected. We are conducting the required preclinical safety studies that, if successful, may support the potential filing of an IND application with the FDA.
TLK60357—Antimitotic Agent
Using our TRAP technology, we have discovered TLK60357, a novel, potent small molecule inhibitor of cell division. TLK60357 inhibits the formation of microtubules that are necessary for cancer cell growth leading to persistent cancer cell block and subsequent cell death at the G2/M cell cycle. This compound demonstrates potent broad-spectrum anticancer activity against a number of human cancer cells. This compound also displays oral efficacy in multiple, standard preclinical models of cancer. Since we are currently focused on TELINTRA and TELCYTA development, no additional expenditure on this compound is expected.
TLK60596—VEGFR Inhibitor
TLK60596 is a small-molecule dual inhibitor of VEGFR1 and VEGFR2 kinase. VEGFR1/2 kinases are known to mediate the formation of new blood vessels in human cancers allowing them to grow. In standard animal models of human colon cancer, oral administration of TLK60596 significantly reduced tumor growth. As we are currently focused on TELINTRA and TELCYTA development, no additional expenditure on this compound is expected.
Other
We discovered all of our drug product candidates using our proprietary technology, TRAP, which we believe enables the rapid and efficient discovery of small molecule drug product candidates. We expect to enter into collaborative arrangements with third parties, such as contract research organizations for clinical trials, development, manufacturing, regulatory approvals or commercialization of some of our products, particularly outside North America, or in disease areas requiring larger and longer clinical trials than cancer.
Nasdaq Stock Listing Compliance
On September 19, 2008, we received notification from Nasdaq informing us that the bid price for our common stock had closed below the minimum $1.00 per share requirement for continued inclusion on the Nasdaq Global Market under Nasdaq Marketplace Rule 5550(a)(2). Effective January 7, 2010, we transferred the listing of our common stock from the Nasdaq Global Market to the Nasdaq Capital Market. On April 22, 2010, we received notification from Nasdaq that we had regained compliance with the minimum bid price requirement. Since May 20, 2010, the bid price for our common stock had again fluctuated below the levels required by Nasdaq rules for continued listing. On July 21, 2010, we received notice from Nasdaq that the bid price for our common stock had closed below the minimum $1.00 per share requirement for continued inclusion on the Nasdaq Capital Market. On February 10, 2011, we received a notification from Nasdaq that we regained compliance with the minimum bid price requirement. On April 21, 2011, we received notice from Nasdaq that the bid price of our common stock had closed below the minimum $1.00 per share requirement for continued inclusion on the Nasdaq Capital Market and that we have 180 calendar days, or until October 18, 2011 to regain
31
Table of Contents
compliance. On October 20, 2011, we received notice from Nasdaq that we had been provided with an additional 180-day period, or until April 16, 2012, to regain compliance with the minimum $1.00 per share requirement. In order to regain compliance, at any time before April 16, 2012, the bid price of our common stock must close at $1.00 per share or more for a minimum of ten consecutive business days. Nonetheless, Nasdaq may require us to maintain a closing bid price of at least $1.00 per share for a longer period before determining that we have achieved compliance. In February 2012, our board of directors approved a proposal, subject to stockholder approval, to decide whether to effect a reverse stock split of all outstanding shares of our common stock and stock options to effect such reverse stock split at an exchange ratio of one for fifteen, one for twenty, one for twenty five or one for thirty. Our board of directors has recommended that this proposal be presented to our stockholders for approval in the 2012 Annual Meeting of Stockholders which will be held on March 28, 2012. If Nasdaq determines that we will not be able to cure the deficiency, or if we are otherwise ineligible, then Nasdaq will provide written notification that our common stock will be delisted, after which we may appeal the delisting determination to a Hearings Panel.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements as well as the reported revenues and expenses during the reporting periods. On an on-going basis, we evaluate our estimates and judgments related to revenue recognition and clinical development costs. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ significantly from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 2 to our financial statements appearing at the end of this Annual Report on Form 10-K, we believe the following accounting policies are critical to the process of making significant judgments and estimates in the preparation of our financial statements.
Fair Value Measurements
We invest our excess cash in money market funds, cash deposits and debt instruments of the U.S. government agency securities. In the current market environment, the assessment of the fair value of the debt securities can be difficult and subjective. Accounting Standards Codification, or ASC, 820, “Fair Value Measurements and Disclosure”, establishes three levels of inputs that may be used to measure fair value The standard describes a fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value which are the following:
| Level 1 | Quoted prices in active markets for identical assets or liabilities; |
| Level 2 | Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities; and |
| Level 3 | Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. The determination of fair value for Level 3 instruments requires the most management judgment and subjectivity. |
In the past, we used Level 3 assumptions to estimate our ARS investments. Since late 2007 the auctions for our ARS continued to fail and as a result these investments were not trading and therefore did not have a readily determinable market value. Accordingly, the estimated fair value of the ARS no longer approximated par value.
32
Table of Contents
Our ARS were held by UBS, one of our investment providers. On November 10, 2008, we accepted an offer, the “Right”, from UBS entitling us to sell our ARS originally purchased from UBS at par value at anytime during a two-year period from June 30, 2010 through July 2, 2012. We valued this put option using a discounted cash flow model based on Level 3 assumptions. The assumptions used in valuing the ARS and the put option included estimates of, based on data as of the reporting period then ended, interest rates, timing and amount of cash flows, credit and liquidity premiums, expected holding periods of the ARS, loan rates per the UBS Rights offering and bearer risk associated with UBS’s financial ability to repurchase the ARS beginning June 30, 2010. These assumptions were volatile and subject to change, and therefore could result in significant changes to the fair value of ARS. In July 2010, we exercised the Right from UBS to sell all our remaining ARS to UBS and we no longer held any ARS in our investment accounts.
Stock-based Compensation Expense
We use the fair value method under ASC 718, “Compensation—Stock Compensation” to account for share-based payment awards following the modified prospective method of adoption which provided for certain changes to the method for valuing stock-based compensation. Under ASC 718, employee stock-based compensation is estimated at the date of grant based on the employee stock award’s fair value using the Black-Scholes option-pricing model and is recognized as expense ratably over the requisite service period in a manner similar to other forms of compensation paid to employees. The Black-Scholes option-pricing model requires the use of certain subjective assumptions. The most significant of these assumptions are our estimates of the expected volatility of the market price of our stock and the expected term of the award. For the years 2009 and 2011, the expected volatilities were based solely on historical volatility data as there were insufficient traded option activities resulting from our declining stock price. We did not use any expected volatility assumptions for 2010 as there were no options granted during the year. The expected term of options granted is based on the simplified method in accordance with the SEC Staff Accounting Bulletin, or SAB, Topic 14.D.2, as our historical share option exercise experience does not provide a reasonable basis for estimation. SAB Topic 14.D.2 provides guidance to issuers on the method allowed in developing estimates of expected term of “plain vanilla” share options in accordance with ASC 718. SAB Topic 14.D.2 allows companies to continue to use the simplified method, under certain circumstances, beyond December 31, 2007. ASC 718 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. We evaluate our forfeiture rate to reflect actual historical and expected cancellations of unvested options periodically. See also Note 9, “Stockholders’ Equity,” in the Notes to Financial Statements for further information.
If factors change and we develop different assumptions in the application of ASC 718 in future periods, the compensation expense that we will then record may differ significantly from what we have recorded in the current period.
Exit and Disposal Activities
We record costs and liabilities associated with exit and disposal activities, as defined in ASC 420, “Exit or Disposal Cost Obligations”, at fair value in the period the liability is incurred. ASC 420 requires that the estimated future cash flows to be used in the fair value calculation be discounted using a credit-adjusted risk-free interest rate and that such interest rate shall have a maturity date that approximates the expected timing of future cash flows. Future cash flows related to lease obligations shall include the effect of sublease rental income and other lease operating expenses. In addition, accretion of the liability due to the passage of time is recorded as a general and administrative expense. See Note 6 “Facility Exit Costs” in the Notes to Financial Statements for further information.
Research and Development Expenses
Our research and development expenses include salaries and benefits costs, fees for contractors, consultants and third-party contract research organizations, and an allocation of facility and administrative costs. Research
33
Table of Contents
and development expenses consist of costs incurred for drug and product development, manufacturing, clinical activities, discovery research, screening and identification of product candidates, and preclinical studies. All such costs are charged to research and development expenses as incurred.
Clinical development costs are a significant component of research and development expenses. We have a history of contracting with third parties that perform various clinical trial activities on our behalf in the on-going development of our product candidates. The financial terms of these contracts are subject to negotiation and may vary from contract to contract and may result in uneven payment flows. We accrue and expense costs for clinical trial activities performed by third parties based upon estimates of the percentage of work completed during a given period of time over the life of the individual study in accordance with agreements established with third-party contract research organizations and clinical trial sites. We determine our estimates through discussion with internal clinical personnel and third-party service providers of the progress or stage of completion of trials or services and the agreed upon fee to be paid for such services in each agreement. These estimates may or may not match the actual services performed by the third-party organizations as measured by patient enrollment levels and related activities. We monitor patient enrollment levels and related activities to the extent possible; however, if we underestimate activity levels associated with various studies at a given point in time, we could record significant research and development expenses in future periods. Conversely, over estimation of activity levels could result in accrued expenses being reversed in future periods.
Use of Estimates
In preparing our financial statements to conform with generally accepted accounting principles, we make estimates and assumptions that affect the amounts reported in our financial statements and accompanying notes. Actual results may differ from these estimates.
Results of Operations
Revenues
We had no collaborative research agreements in 2011, 2010 and 2009 and currently do not expect to record any revenue in the next twelve months. Future non-product revenues, if any, will depend upon the extent to which we enter into new collaborative research agreements and the amounts of payments relating to such agreements.
Research and Development Expenses
Research and development expenses for the years ended December 31, 2011, 2010 and 2009 were $5.6 million, $11.0 million and $12.7 million. Our research and development activities consist primarily of drug development, clinical supply manufacturing, clinical activities, discovery research, screening and identification of product candidates and preclinical studies. We group these activities into two major categories: “research and preclinical” and “clinical development.”
The costs associated with research and preclinical and clinical development activities approximate the following:
| Years Ended December 31, | Annual Percent Change |
|||||||||||||||||||
| 2011 | 2010 | 2009 | 2011/2010 | 2010/2009 | ||||||||||||||||
| (in thousands, except percentages) | ||||||||||||||||||||
| Research and preclinical |
$ | 1,531 | $ | 3,000 | $ | 4,303 | (49 | )% | (30 | )% | ||||||||||
| Clinical development |
4,035 | 8,040 | 8,420 | (50 | )% | (5 | )% | |||||||||||||
|
|
|
|
|
|
|
|||||||||||||||
| Total research and development |
$ | 5,566 | $ | 11,040 | $ | 12,723 | (50 | )% | (13 | )% | ||||||||||
|
|
|
|
|
|
|
|||||||||||||||
34
Table of Contents
Total research and development expenses for the year ended December 31, 2011 decreased by 50%, or $5.5 million, compared to the same period in 2010 primarily due to the following:
| — | decreased costs of approximately $4.8 million associated with a workforce reduction as a result of our November 2010 restructuring plan and reduced facility costs as we relocated our corporate offices to a smaller facility in November 2010; |
| — | decreased clinical trial expenses of approximately $696,000 related to the completion of our Phase 2 TELINTRA tablets for MDS and $120,000 in clinical drug supply manufacturing costs; |
| — | offset by increased clinical development expenses of approximately $179,000 for our ongoing Phase 1 dose-ranging study of TELINTRA tablets in combination with lenalidomide in patients with MDS and Phase 2 TELCYTA in patients with Refractory or Relapsed Mantle Cell lymphoma, Diffuse Large B Cell Lymphoma, and Multiple Myeloma clinical studies. |
Stock-based compensation expense included in research and development expenses for the years ended December 31, 2011 and 2010 were $715,000 and $851,000.
Total research and development expenses for the year ended December 31, 2010 decreased by 13%, or $1.7 million, compared to the same period in 2009 primarily due to the following:
| — | decreased costs of approximately $1.6 million in connection with headcount reduction as a result of our restructuring in 2009 and reduced research activities; |
| — | decreased expenses of approximately $977,000 for TLK58747-Cytotoxic Small Molecule pre-clinical development program due to the reprioritization of our focus on other compounds; |
| — | decreased clinical trial expenses of approximately $765,000 related to the completion our Phase 2 TELINTRA tablets for MDS and the discontinuation of our Phase 2 TELINTRA tablets for CIN; |
| — | offset by a $531,000 increase in clinical development expenses for our ongoing Phase 1 dose-ranging study of TELINTRA tablets in combination with Lenalidomide in patients with MDS, Phase 2 TELINTRA tablets randomized study in SCN and TELCYTA in patients with Refractory or Relapsed Mantle Cell lymphoma, Diffuse Large B Cell Lymphoma, and Multiple Myeloma clinical studies; |
| — | increased stock-based compensation expense of approximately $432,000 primarily due to more stock options vested in 2010; and |
| — | in addition, the year ended December 31, 2009 included approximately a $604,000 reduction in accrued clinical trials expenses as a result of final close-out of clinical sites for completed clinical studies while there were no such adjustments in 2010. |
Stock-based compensation expense included in research and development expenses for the years ended December 31, 2010 and 2009 were $851,000 and $420,000.
We expect total research and development expenditures to increase in the next twelve months as we focus on the enrollment of our two Phase 2 clinical trials of TELINTRA in MDS and the expansion of our TELCYTA trial in multiple myeloma.
35
Table of Contents
The following table summarizes our principal drug product candidate development initiatives:
| Related R&D Expenses Years Ended December 31, |
||||||||||||
| Product |
2011 | 2010 | 2009 | |||||||||
| (in thousands) | ||||||||||||
| TELINTRA |
$ | 4,023 | $ | 7,600 | $ | 6,832 | ||||||
| TELCYTA |
1,509 | 2,107 | 923 | |||||||||
| TLK58747 |
— | 128 | 3,269 | |||||||||
| TLK60404 |
34 | 495 | 742 | |||||||||
| Other (1) |
— | 710 | 957 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total research and development expenses |
$ | 5,566 | $ | 11,040 | $ | 12,723 | ||||||
|
|
|
|
|
|
|
|||||||
| (1) | “Other” constitutes research and development activities performed by our Chemistry, Biology, Preclinical and Quality Assurance departments as these costs cannot be allocated to any individual project. |
The largest component of our total operating expenses is our on-going investment in our research and development activities and, in particular, the clinical development of our product candidate pipeline. The process of conducting the clinical research necessary to obtain FDA approval is costly and time consuming. Current FDA requirements for a new human drug to be marketed in the United States include:
| • | the successful conclusion of preclinical laboratory and animal tests, if appropriate, to gain preliminary information on the product’s safety; |
| • | filing with the FDA of an IND, to conduct initial human clinical trials for drug candidates; |
| • | the successful completion of adequate and well-controlled human clinical trials to establish the safety and efficacy of the product candidate; and |
| • | filing by the company and acceptance and approval by the FDA of a NDA for a product candidate to allow commercial distribution of the drug. |
In view of the factors mentioned above, we consider the active management and development of our clinical pipeline to be crucial to our long-term success. The actual probability of success for each product candidate and clinical program may be impacted by a variety of factors, including, among others, the quality of the candidate, the validity of the target and disease indication, early clinical data, investment in the program, competition, manufacturing capability and commercial viability. Due to these and other factors, it is difficult to give accurate guidance on the anticipated proportion of our research and development investments or the future cash inflows from these programs.
General and Administrative Expenses
| Years Ended December 31, | Annual Percent Change |
|||||||||||||||||||
| 2011 | 2010 | 2009 | 2011/2010 | 2010/2009 | ||||||||||||||||
| (in thousands, except percentages) | ||||||||||||||||||||
| General and administrative |
$ | 6,491 | $ | 9,230 | $ | 10,810 | (30 | )% | (15 | )% | ||||||||||
The decrease in general and administrative expenses of 30%, or $2.7 million, in 2011, compared to the same period in 2010, was primarily due to a decrease of $1.5 million in workforce and corporate administrative expenses as a result of the restructuring plan implemented in November 2010 and decreased facility costs of $1.9 million as we relocated our corporate offices to a smaller facility in November 2010 and were partially offset by an increase of $801,000 in legal expenses primarily relating to patent renewal activities and the filing of new patent applications. Stock-based compensation expense included in general and administrative expenses for the years ended December 31, 2011 and 2010 were $843,000 and $1.2 million.
36
Table of Contents
The decrease in general and administrative expenses of 15%, or $1.6 million, in 2010, compared to the same period in 2009, was primarily due a decrease of $618,000 in headcount and corporate administrative expenses, decreased stock-based compensation expense of approximately $571,000 as a result of higher fair value stock options fully vested in 2009 and a decrease of $393,000 in legal and professional service expenses. Stock-based compensation expense included in general and administrative expenses for the years ended December 31, 2010 and 2009 were $1.2 million and $1.8 million.
We expect future general and administrative expenses to be at the same spending level as 2011.
Facility Exit Costs
| Years Ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| (In thousands) | ||||||||||||
| Facility exit costs |
$ | — | $ | 5,360 | $ | — | ||||||
In November 2010, we ceased the use of our facility at 3165 Porter Drive in Palo Alto, California and subleased the facility to a tenant for the remaining contractual term of our master lease, which is through May 2014. We recorded a facility exit charge of $5.4 million to the statement of operations, which included $4.7 million of estimated present value of future lease-related payments through May 2014, less estimated sublease income, and an impairment charge of $1.0 million in leasehold improvements for this facility as the facility would not have any future benefits to us and their estimated fair values were determined to be zero, offset by a reduction of $335,000 in the balance of deferred rent as of November 30, 2010. At December 31, 2011, we had a remaining facility exit costs liability of $2.9 million as we paid $309,000 in 2010 and $1.5 million in 2011. See also Note 6 in the Notes to our Financial Statements for additional information.
Restructuring Costs
| Years Ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| (In thousands) | ||||||||||||
| Restructuring costs |
$ | — | $ | 425 | $ | 951 | ||||||
We have implemented workforce reductions over the past several years to reduce our operating expenses and to streamline our infrastructure based on our current preclinical and clinical trial projects. In November 2010, we recorded a restructuring charge of approximately $425,000 for severance costs, health benefits and other personnel related charges relating to a workforce reduction of eleven positions. In February 2009, we implemented a restructuring plan and reduced our workforce by 37 positions and recorded a charge of approximately $951,000. All actions associated with the 2009 and 2010 restructuring plans were completed in 2011, and we do not anticipate incurring any further costs under either plan.
Interest Income and Interest Expense
| Years Ended December 31, | Annual Percent Change |
|||||||||||||||||||
| 2011 | 2010 | 2009 | 2011/2010 | 2010/2009 | ||||||||||||||||
| (in thousands, except percentages) | ||||||||||||||||||||
| Interest and other income (expense), net |
$ | 35 | $ | 1,339 | $ | 876 | (97 | )% | 53 | % | ||||||||||
| Interest expense |
$ | — | $ | 6 | $ | 85 | (100 | )% | (93 | )% | ||||||||||
Interest and other income (expense), net were $35,000, $1.3 million and $876,000 for the years ended December 31, 2011, 2010 and 2009. The decrease of approximately $1.3 million in 2011 compared to the same period in 2010 was due primarily to a $1.2 million Qualifying Therapeutic Discovery Project grant received from the Internal Revenue Service Department of Treasury in 2010 and a decrease of $113,000 in investment income resulting from lower investment cash balances.
37
Table of Contents
The increase of approximately $463,000 in 2010 compared to the same period in 2009 was due primarily to a $1.2 million Qualifying Therapeutic Discovery Project grant received from the Internal Revenue Service Department of Treasury and was partially offset by a decrease of $344,000 in investment income resulting from lower investment cash balances and a net decrease of $374,000 due to a loss of $31,000 on the disposal of computer and laboratory equipment in 2010 compared to a gain of $343,000 recorded in 2009.
Interest expenses for the periods reported were solely for interest payments on our UBS loan. There were no interest expenses for the year ended December 31, 2011. The decrease of approximately $79,000 for the year ended December 31, 2010 compared to the same period in 2009 was due to the full payment of our UBS loan balance in February 2010.
Liquidity and Capital Resources
| 2011 | 2010 | 2009 | ||||||||||
| (In millions, except ratios) | ||||||||||||
| December 31: |
||||||||||||
| Cash, cash equivalents, investments and restricted cash |
$ | 11.7 | $ | 24.1 | $ | 40.4 | ||||||
| Working capital |
$ | 9.4 | $ | 20.7 | $ | 39.2 | ||||||
| Current ratio |
4.6 : 1 | 6.6 : 1 | 9.1 : 1 | |||||||||
| Year ended December 31: |
||||||||||||
| Cash provided by (used in): |
||||||||||||
| Operating activities |
$ | (12.8 | ) | $ | (16.7 | ) | $ | (24.1 | ) | |||
| Investing activities |
$ | 13.6 | $ | 15.3 | $ | (14.2 | ) | |||||
| Financing activities |
$ | 0.4 | $ | (3.0 | ) | $ | — | |||||
| Capital expenditures (included in investing activities above) |
$ | — | $ | — | $ | — | ||||||
Sources and Uses of Cash. Due to the significant research and development expenditures and the lack of any approved products to generate revenue, we have not been profitable and have generated operating losses since we incorporated in 1988. As such, we have funded our research and development operations through sales of equity, collaborative arrangements with corporate partners, interest earned on investments and equipment lease financings. At December 31, 2011 we had available cash, cash equivalents, investments and restricted investments of $11.7 million. Our cash and investment balances are held in a variety of interest-bearing instruments including obligations of U.S. government agencies and money market accounts. Cash in excess of immediate requirements is invested with regard to liquidity and capital preservation. Wherever possible, we seek to minimize the potential effects of concentration and degrees of risk.
Cash Flows from Operating Activities. Cash used in operations for 2011 was $12.8 million compared to $16.7 million for the same period in 2010 and $24.1 million in 2009. Net loss of $12.0 million in 2011 included non-cash charges of $1.6 million for stock based compensation and $10,000 for depreciation. Cash used in operations was further impacted by an $823,000 reduction in accounts payable primarily due to payments related to our office relocation at the end of 2010 and a $1.4 million reduction in accrued facility exit costs due to payments made on our Porter Drive facility which were partially offset by sublease payments received. Cash used in 2010 resulted from net loss of $24.7 million which included non-cash charges of $5.4 million of facility exit costs associated with the relocation of our principal executive offices, $2.1 million for stock based compensation and $343,000 for depreciation. The increase in accounts payable balance was offset by reductions in accrued expenses and did not have a significant impact on cash used in operations for 2010. Cash used in 2009 resulted from a net loss of $23.7 million which included non-cash charges of $2.2 million for stock-based compensation, $556,000 for depreciation and $1.3 million for the reduction in value of the put option associated with the Right and were partially offset by a $1.4 million increase in the fair value of marketable securities and a $342,000 gain on the disposal of property and equipment. Cash used in operations was further impacted by a $1.4 million decrease in accounts payable and a $1.3 million reduction in accrued clinical trials related primarily to final study payments as a result of the completion of our Phase 3 clinical trials.
38
Table of Contents
Cash Flows from Investing Activities. Cash provided by investing activities for 2011 was $13.6 million compared to cash provided by investing activities of $15.3 million for 2010 and cash used in investing activities in 2009 of $14.2 million. Cash provided in 2011 was primarily from $23.7 million in investment maturities and $1.7 million in investment sales partially offset by the purchase of available-for-sale investments of $11.7 million. We had no capital expenditures for the year ended December 31, 2011. Cash provided in 2010 was primarily from $35.7 million in investment maturities and $14.0 million in investment sales which included $13.8 million in sales of our ARS to UBS and was partially offset by the purchase of available-for-sale investments of $34.4 million. Cash used in 2009 was primarily for the purchase of available-for-sale investments of $42.1 million and was partially offset by $225,000 in investment sales, $27 million in investment maturities and $659,000 in proceeds from the sale of property.
Cash Flows from Financing Activities. Cash provided by financing activities for 2011 was approximately $393,000 compared to $3.0 million used in financing activities in 2010 and $47,000 provided by financing activities in 2009. Cash provided by financing activities in 2011 was primarily due to proceeds from stock sales including $149,000 in net proceeds under the At Market Issuance Sales Agreement with McNicoll, Lewis & Vlak LLC and approximately $244,000 from stock purchases under our employee stock purchase plan and stock options exercise. Cash used in financing activities for 2010 was primarily due to $3.1 million payment of our remaining UBS loan balance on February 16, 2010, offset by $64,000 in proceeds from stock purchases under our employee stock purchase plan. Financing activities for 2009 comprised of $47,000 in proceeds from stock purchases under our employee stock purchase plan.
Working Capital. Working capital decreased to $9.4 million at December 31, 2011 from $20.7 million at December 31, 2010. The decrease in working capital was primarily due to our use of cash for our clinical studies and operating expenses.
We believe our cash, cash equivalents and marketable securities as of December 31, 2011 will not be sufficient to fund our projected operating requirements for the next twelve months, including prosecuting our current trials, conducting research and discovery efforts towards additional product candidates and providing sufficient funds for working capital and general corporate purposes. In order to continue as a going concern, we will need additional capital to complete all the clinical trials we are currently conducting. We may raise funds through arrangements with collaborators or others that may require us to relinquish rights to certain product candidates that we might otherwise seek to develop or commercialize independently. Our future capital uses and requirements depend on numerous factors, including the following:
| • | the progress and success of preclinical studies and clinical trials of our product candidates; |
| • | the progress and number of research programs in development; |
| • | the costs associated with conducting Phase 2 clinical trials; |
| • | the costs and timing of obtaining regulatory approvals; |
| • | our ability to establish, and the scope of, any new collaborations; |
| • | our ability to meet the milestones identified in our collaborative agreements that trigger payments; |
| • | the costs and timing of obtaining, enforcing and defending our patent and intellectual property rights; and |
| • | competing technological and market developments. |
We need to raise additional capital or incur indebtedness in the near term to continue to fund our future operations. We may seek to raise capital through a variety of sources, including collaborative arrangements, licensing arrangements, public equity market, private equity financing, and/or public or private debt as well as other sources such as research grants from non-profit organizations. In August 2011, we filed a shelf registration statement on Form S-3 to offer and sell, from time to time, equity and debt securities in one or more offerings up to a total dollar amount of $25.0 million. On August 30, 2011 we entered into an At Market Issuance Sales
39
Table of Contents
Agreement, or the Sales Agreement, with McNicoll, Lewis & Vlak LLC, or MLV, pursuant to which we may issue and sell shares of our common stock having an aggregate offering price up to $7.0 million, from time to time, through MLV as our sales agent. In conjunction with the sales agreement, MLV would receive compensation based on an aggregate of 4% of the gross proceeds on the sale price per share of our common stock. Any sales made pursuant to the sales agreement are deemed an “at-the-market” offering and would be made pursuant to the shelf registration statement on Form S-3. For the year ended December 31, 2011 we sold 484,112 shares of our common stock and received approximately $149,000 in net proceeds from stock sales under the Sales Agreement after deducting commissions and other related expenses of approximately $7,000.
Our ability to raise additional funds will depend on clinical and regulatory events and factors related to financial, economic and market conditions, many of which are beyond our control. In addition, our ability to raise additional capital may be dependent upon our stock being quoted on the Nasdaq Capital Market. See “Nasdaq Stock Listing Compliance” above regarding our current listing status. If we are delisted, we will be substantially limited in our ability to raise capital through the sale of our securities in the public equity market. We cannot be certain that sufficient funds will be available to us when required or on satisfactory terms. If adequate funds are not available, we may be required to significantly reduce or refocus our operations or to obtain funds through arrangements that may require us to relinquish rights to certain of our products, technologies or potential markets, any of which could delay or require that we curtail our development programs or otherwise have a material adverse effect on our business, financial condition and results of operations. To the extent that additional capital is raised through the sale of equity or convertible debt securities, the issuance of such securities would result in significant ownership dilution to our existing stockholders.
If we are unable to secure additional financing on a timely basis or on terms favorable to us, we may be required to cease or reduce certain research and development projects, to sell some or all of our technology or assets or to merge all or a portion of our business with another entity. Insufficient funds may require us to delay, scale back, or eliminate some or all of our activities, and if we are unable to obtain additional funding, there will be uncertainty regarding our ability to continue as a going concern.
Our future contractual obligations at December 31, 2011 are as follows (in thousands):
| Total | 2012 | 2013-2014 | After 2014 | |||||||||||||
| Operating leases |
$ | 3,547 | $ | 1,922 | $ | 1,625 | $ | — | ||||||||
In November 2010, we entered into arrangements to sublease our facility located at 3165 Porter Drive in Palo Alto, California, which consists of approximately 92,000 square feet of research and office space. The sublease term commenced in November 2010 and will expire on May 31, 2014, the date on which the current term of our Master Lease expires. However, if the Master Lease is terminated for any reason prior to this date, the sublease will terminate concurrently.
On November 22, 2010, Telik entered into an arrangement to sublease a facility at 700 Hansen Way, Palo Alto, California in which to relocate our principal executive offices. The term of the Hansen Sublease commenced on November 29, 2010 and expires on March 31, 2013, the date on which the current term of the Hansen Master Lease expires. However, if the Hansen Master Lease is terminated for any reason prior to this date, the Hansen Sublease will terminate concurrently.
We have a contractual obligation under the terms of our manufacturing supply agreement with AMRI wherein we are obligated to purchase a majority of our United States requirements for the active ingredient in TELCYTA for a number of years. However, we currently do not have any requirements for the active ingredient. We have agreed on a pricing schedule for such supply, which will be subject to future renegotiation after a defined time period.
Off-Balance Sheet Arrangements
We have no material off-balance sheet arrangements as defined in Regulation S-K 303(a)(4)(ii).
40
Table of Contents
Recent Accounting Pronouncements
See Note 1 of Notes to Financial Statements attached to this Annual Report for a description of recent accounting pronouncements.
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk. |
The following discussion about our market risk exposure involves forward-looking statements. We do not use or hold derivative financial instruments, however we are exposed to market risk related to changes interest rates and market conditions.
Our investment policy is to manage our marketable securities portfolio to preserve principal and liquidity while maximizing the return on the investment portfolio. To minimize the exposure due to adverse shifts in interest rates we maintain investments of shorter maturities. Our marketable securities portfolio is primarily invested in U.S. government agency securities with an average maturity of under one year and a minimum investment grade rating of A or A-1 or better to minimize credit risk. Although changes in interest rates may affect the fair value of the marketable securities portfolio and cause unrealized gains or losses, such gains or losses would not be realized unless the investments were sold prior to maturity. Through our money managers, we maintain risk management control systems to monitor interest rate risk. The risk management control systems use analytical techniques, including sensitivity analysis. We have operated primarily in the United States and all funding activities with our collaborators to date have been made in U.S. dollars. Accordingly, we do not have any exposure to foreign currency rate fluctuations.
The table below presents the principal amounts and weighted-average interest rates by year of stated maturity for our investment portfolio:
| 2012 | 2013 and Beyond |
Total | Fair Value at December 31, 2011 |
|||||||||||||
| (In thousands, except percentages) | ||||||||||||||||
| Available-for-sale securities |
$ | 4,454 | — | $ | 4,454 | $ | 4,454 | |||||||||
| Average interest rate |
0.16 | % | 0.00 | % | 0.16 | % | ||||||||||
| Item 8. | Financial Statements and Supplementary Data. |
All information required by this item is included in Item 15 of Part IV of this Annual Report and is incorporated into this item by reference.
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure. |
None.
| Item 9A. | Controls and Procedures. |
(I) Evaluation of Disclosure Controls and Procedures and Changes in Internal Control over Financial Reporting
Based on their evaluation as of December 31, 2011, our Chief Executive Officer and Vice President, Finance and Controller have concluded that our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended) were effective.
There were no changes in our internal control over financial reporting during the quarter ended December 31, 2011 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
41
Table of Contents
(II) Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Our management, including our Chief Executive Officer and Vice President, Finance and Controller, does not expect that our procedures or our internal controls will prevent or detect all error and all fraud. An internal control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of the inherent limitations in all control systems, no evaluation of our controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected.
Under the supervision and with the participation of our management, including our Chief Executive Officer and Vice President, Finance and Controller, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2011. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) in Internal Control-Integrated Framework. Our management has concluded that, as of December 31, 2011, our internal control over financial reporting was effective based on these criteria.
This annual report does not include an attestation report of the company’s registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by the company’s registered public accounting firm pursuant to rules of the Securities and Exchange Commission that permit the company to provide only management’s report in this annual report.
| Item 9B. | Other Information. |
Effective as of November 14, 2011, the Rights Agreement dated November 2, 2001 between us and Wells Fargo Bank Minnesota, N.A. (as amended, the “Rights Agreement”) expired in accordance with its terms. The Rights Agreement provided our common stockholders with the right to purchase shares of Series A Junior Participating Preferred Stock (“Series A Preferred Stock”) upon the terms and subject to the conditions set forth in the Rights Agreement. As a result, the stock purchase rights under the Rights Agreement have been terminated and are no longer effective.
In connection with the expiration of the Rights Agreement, we filed a certificate of elimination with the Secretary of State of the State of Delaware on February 24, 2012 (the “Certificate of Elimination”). The Certificate of Elimination, which was effective upon filing, eliminated from our Amended and Restated Certificate of Incorporation all matters set forth in the Certificate of Designation with respect to the Series A Preferred Stock. No shares of the Series A Preferred Stock were issued or outstanding at the time of the filing of the Certificate of Elimination.
42
Table of Contents
PART III
| Item 10. | Directors, Executive Officers and Corporate Governance. |
Information regarding directors and executive officers is incorporated by reference to the information set forth under the caption “Information Regarding the Board of Directors and Corporate Governance” in our Definitive Proxy Statement pursuant to Section 14(a) of the Securities Exchange Act of 1934 for the Annual Meeting of Stockholders to be filed with the Commission on or about March 5, 2012.
We have adopted the Telik, Inc. Code of Conduct, a code of ethics with which every person who works for us is expected to comply, including without limitation our principal executive officer, principal financial officer, principal accounting officer or controller or persons performing similar functions. The Telik, Inc. Code of Conduct is filed as an exhibit to our Annual Report on Form 10-K for the period ended December 31, 2003 as filed on March 4, 2004, with the U.S. Securities and Exchange Commission, or SEC, and is incorporated herein by reference. If we make any substantive amendments to the Telik, Inc. Code of Conduct or grant to any of our directors or executive officers any waiver including any implicit waiver, from a provision of the Telik, Inc. Code of Conduct, we will disclose the nature of the waiver or amendment in a Current Report on Form 8-K.
| Item 11. | Executive Compensation. |
Information regarding executive compensation is incorporated by reference to the information set forth under the caption “Executive Compensation” in our Definitive Proxy Statement for the Annual Meeting of Stockholders to be filed with the Commission on or about March 5, 2012.
43
Table of Contents
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
Information regarding security ownership of certain beneficial owners and management is incorporated by reference to the information set forth under the caption “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” in our Definitive Proxy Statement for the Annual Meeting of Stockholders to be filed with the Commission by on or about March 5, 2012.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence. |
Information regarding certain relationships and related transactions is incorporated by reference to the information set forth under the caption “Transactions with Related Persons” in our Definitive Proxy Statement for the Annual Meeting of Stockholders to be filed with the Commission on or about March 5, 2012.
| Item 14. | Principal Accounting Fees and Services. |
Information regarding principal accountant fees and services is incorporated by reference to the information set forth under the caption “Proposal 3 – Ratification of Selection of Independent Auditors” in our Definitive Proxy Statement for the Annual Meeting of Stockholders to be filed with the Commission by on or about March 5, 2012.
44
Table of Contents
PART IV
| Item 15. | Exhibits and Financial Statement Schedules. |
(a) The following documents are filed as part of this Annual Report:
| 1. | Financial Statements. Our financial statements and the Report of Independent Registered Public Accounting Firm, are included in Part IV of this Report on the pages indicated: |
| Page | ||||
| 49 | ||||
| 50 | ||||
| 51 | ||||
| 52 | ||||
| 53 | ||||
| 54 | ||||
| 2. | Financial Statement Schedules. All schedules are omitted because they are not applicable or the required information is shown in the financial statements or the notes thereto. |
| 3. | Exhibits: |
| Exhibit |
Description | |
| 3.1 | Amended and Restated Certificate of Incorporation. (2) | |
| 3.2 | Amended and Restated Bylaws. (10) | |
| 3.3 | Registrant’s Certificate of Designation of Series A Junior Participating Preferred Stock. (5) | |
| 3.4 | Registrant’s Certificate of Elimination with respect to Series A Junior Participating Preferred Stock. | |
| 4.1 | Specimen Common Stock Certificate. (1) | |
| 10.1 | Form of Indemnity Agreement. (1) (3) | |
| 10.2 | 2011 Equity Incentive Plan and related documents (3)(14) | |
| 10.3 | 2000 Equity Incentive Plan and related documents. (3) (4) | |
| 10.4 | 2000 Employee Stock Purchase Plan and Offering. (3) (4) | |
| 10.5 | 2000 Non-Employee Directors’ Stock Option Plan and Agreement. (3) (11) | |
| 10.6 | 1996 Stock Option Plan and forms of grant thereunder. (3) (4) | |
| 10.7 | 1988 Stock Option Plan and forms of grant thereunder. (3) (4) | |
| 10.8 | Form of Non-Plan Stock Option Agreement. (3) (4) | |
| 10.9 | Telik, Inc. Executive Officer Bonus Plan. (3)(9) | |
| 10.10 | Amended and Restated Employment Agreement between Michael M. Wick, M.D., Ph.D. and Telik, dated December 17, 2008, as amended. (3) (12) | |
| 10.11 | Lease between Telik and The Board of Trustees of the Leland Stanford Junior University, dated July 25, 2002. (6) | |
| 10.12 | Lease, between Telik and The Board of Trustees of the Leland Stanford Junior University, dated November 24, 2010. (15) | |
| 10.13 | Lease, between Telik and Aricent US, Inc., dated November 22, 2010. (15) | |
| 10.14* | Manufacturing Supply Agreement dated July 1, 2004, by and between Telik and Organichem Corporation. (7) | |
45
Table of Contents
| Exhibit |
Description | |
| 10.15 | Telik, Inc. Change of Control Severance Benefit Plan, dated February 21, 2003, amended December 17, 2008. (3) (12) | |
| 10.16 | Bonuses for Fiscal Year 2009 for Named Executive Officers. (3) (9) | |
| 10.17 | Transition and Release Agreement between Telik, Inc. and Dr. Stefan Ryser, Ph.D., dated as of June 12, 2009. (3) (13) | |
| 10.18 | At Market Issuance Sales Agreement, dated August 30, 2011, by and between Telik, Inc., and McNicoll, Lewis & Vlak LLC. (16) | |
| 14.1 | Telik, Inc. Code of Conduct. (8) | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 24.1 | Power of Attorney (included on the signature pages hereto) | |
| 31.1 | Certification required by Rule 13a-14(a) or Rule 15d-14(a). | |
| 31.2 | Certification required by Rule 13a-14(a) or Rule 15d-14(a). | |
| 32.1 | Certification required by Rule 13a-14(b) or Rule 15d-14(b) and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. 1350). | |
| 101.INS** | XBRL Instance Document | |
| 101.SCH** | XBRL Taxonomy Extension Schema Document | |
| 101.CAL** | XBRL Taxonomy Extension Calculation Document | |
| 101.DEF** | XBRL Taxonomy Extension Definition Linkbase Document | |
| 101.LAB** | XBRL Taxonomy Extension Labels Linkbase Document | |
| 101.PRE** | XBRL Taxonomy Extension Presentation Link Document | |
| * | Confidential treatment has been granted for portions of this document. The information omitted pursuant to such confidential treatment order has been filed separately with the Securities and Exchange Commission. |
| ** | These interactive data files are deemed not filed or part of a registration statement or prospectus for purposes of sections 11 or 12 of the Securities Act of 1933, as amended, are deemed not filed for purposes of section 18 of the Securities Exchange Act of 1934, as amended, and otherwise are not subject to liability under these sections. |
| (1) | Incorporated by reference to exhibits to our Registration Statement on Form S-1, filed on April 3, 2000, as amended (File No. 333-33868). |
| (2) | Incorporated by reference to exhibits to our Annual Report on Form 10-K for the year ended December 31, 2001, as filed on March 27, 2002. |
| (3) | Management contract or compensatory arrangement. |
| (4) | Incorporated by reference to exhibits to our Registration Statement on Form S-8, as filed on August 30, 2000 (File No. 333-44826). |
| (5) | Incorporated by reference to exhibits to our Current Report on Form 8-K dated November 2, 2001, as filed on November 5, 2001. |
| (6) | Incorporated by reference to exhibits to our Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2002, as filed on November 13, 2002. |
| (7) | Incorporated by reference to exhibits to our Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2004, as filed on November 8, 2004. |
| (8) | Incorporated by reference to exhibits to our Annual Report on Form 10-K for the year ended December 31, 2003, as filed on March 5, 2004. |
46
Table of Contents
| (9) | Incorporated by reference to Exhibit 10.8 to our Annual Report on Form 10-K for the year ended December 31, 2006, as filed on February 28, 2007. |
| (10) | Incorporated by reference to Exhibit 3.2 to our Current Report on Form 8-K dated December 11, 2007, as filed on December 14, 2007. |
| (11) | Incorporated by reference to Exhibit 10.4 to our Annual Report on Form 10-K for the year ended December 31, 2007, as filed on March 3, 2008. |
| (12) | Incorporated by reference to exhibits to our Current Report on Form 8-K dated December 17, 2008, as filed on December 23, 2008. |
| (13) | Incorporated by reference to Exhibit 10.20 to our Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2009, as filed on August 6, 2009. |
| (14) | Incorporated by reference to Appendix E to our Proxy Statement for the Annual Meeting of Stockholders, as filed on May 16, 2011. |
| (15) | Incorporated by reference to exhibits to our Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2011, as filed on August 12, 2011. |
| (16) | Incorporated by reference to Exhibit 10.17 to our Current Report on Form 8-K dated August 30, 2011, as filed on August 31, 2011. |
47
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| TELIK, INC. | ||
| /S/ WENDY WEE | ||
| Wendy Wee Vice President, Finance and Controller (Principal Financial and Accounting Officer) | ||
Dated: February 27, 2012
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Michael M. Wick, M.D., Ph.D. and Wendy Wee, and each of them, as his true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him and in his name, place, and stead, in any and all capacities, to sign any and all amendments to this Report, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming that all said attorneys-in-fact and agents, or any of them or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated:
| Signature |
Title |
Date | ||
| /S/ MICHAEL M. WICK Michael M. Wick, M.D., Ph.D. |
Chief Executive Officer and Director (Principal Executive Officer) |
February 27, 2012 | ||
| /S/ WENDY WEE Wendy Wee |
Vice President, Finance and Controller (Principal Financial and Accounting Officer) |
February 27, 2012 | ||
| /S/ EDWARD W. CANTRALL Edward W. Cantrall, Ph.D. |
Director |
February 27, 2012 | ||
| /S/ STEVEN R. GOLDRING Steven R. Goldring, M.D. |
Director |
February 27, 2012 | ||
| /S/ RICHARD B. NEWMAN Richard B. Newman |
Director |
February 27, 2012 | ||
| /S/ HERWIG VON MORZE Herwig von Morze, Ph.D. |
Director |
February 27, 2012 | ||
48
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Telik, Inc.
We have audited the accompanying balance sheets of Telik, Inc. as of December 31, 2011 and 2010, and the related statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2011. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Telik, Inc. at December 31, 2011 and 2010, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2011, in conformity with the U.S. generally accepted accounting principles.
As discussed in Note 1 to the financial statements, Telik, Inc.’s recurring losses from operations, available cash, cash equivalents and investments and accumulated deficit raise substantial doubt about its ability to continue as a going concern. Management’s plans as to these matters also are described in Note 1.
/s/ Ernst & Young LLP
San Jose, California
February 27, 2012
49
Table of Contents
TELIK, INC.
(In thousands, except share and per share data)
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| Assets | ||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 9,046 | $ | 7,768 | ||||
| Short-term investments |
2,404 | 15,847 | ||||||
| Interest and other receivables |
25 | 214 | ||||||
| Prepaids and other current assets |
589 | 643 | ||||||
|
|
|
|
|
|||||
| Total current assets |
12,064 | 24,472 | ||||||
| Property and equipment, net |
1 | 11 | ||||||
| Restricted investments |
250 | 449 | ||||||
| Other assets |
97 | 97 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 12,412 | $ | 25,029 | ||||
|
|
|
|
|
|||||
| Liabilities and Stockholders’ Equity | ||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 109 | $ | 932 | ||||
| Accrued clinical trial costs |
153 | 226 | ||||||
| Accrued compensation |
367 | 661 | ||||||
| Accrued liabilities |
550 | 478 | ||||||
| Current portion of facility exit costs |
1,463 | 1,439 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
2,642 | 3,736 | ||||||
| Noncurrent portion of facility exit costs |
1,463 | 2,923 | ||||||
| Long-term deferred rent |
8 | 1 | ||||||
| Commitments and contingencies |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.01 par value: 5,000,000 shares authorized; none issued or outstanding |
— | — | ||||||
| Common stock, $0.01 par value: 100,000,000 shares authorized; shares issued and outstanding 54,456,289 in 2011 and 53,613,572 in 2010 |
|
545 |
|
|
536 |
| ||
| Additional paid-in capital |
548,083 | 546,141 | ||||||
| Accumulated other comprehensive loss |
— | (1 | ) | |||||
| Accumulated deficit |
(540,329 | ) | (528,307 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
8,299 | 18,369 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 12,412 | $ | 25,029 | ||||
|
|
|
|
|
|||||
See accompanying Notes to Financial Statements.
50
Table of Contents
TELIK, INC.
(In thousands, except per share amounts)
| Years Ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Operating costs and expenses: |
||||||||||||
| Research and development |
$ | 5,566 | $ | 11,040 | $ | 12,723 | ||||||
| General and administrative |
6,491 | 9,230 | 10,810 | |||||||||
| Facility exit costs |
— | 5,360 | — | |||||||||
| Restructuring costs |
— | 425 | 951 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating costs and expenses |
12,057 | 26,055 | 24,484 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(12,057 | ) | (26,055 | ) | (24,484 | ) | ||||||
| Interest and other income, net |
35 | 1,339 | 876 | |||||||||
| Interest expense |
— | (6 | ) | (85 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (12,022 | ) | $ | (24,722 | ) | $ | (23,693 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per share |
$ | (0.22 | ) | $ | (0.46 | ) | $ | (0.44 | ) | |||
|
|
|
|
|
|
|
|||||||
| Shares used to calculate basic and diluted net loss per share |
53,939 | 53,539 | 53,371 | |||||||||
|
|
|
|
|
|
|
|||||||
See accompanying Notes to Financial Statements.
51
Table of Contents
TELIK, INC.
STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands)
| Common Stock | Additional Paid-in Capital |
Accumulated Other Comprehensive Income (Loss) |
Accumulated Deficit |
Total Stockholders’ Equity |
||||||||||||||||||||
| Shares | Amount | |||||||||||||||||||||||
| Balances at December 31, 2008 |
53,290 | $ | 533 | 541,710 | $ | 21 | $ | (479,892 | ) | $ | 62,372 | |||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||
| Net loss |
— | — | — | — | (23,693 | ) | (23,693 | ) | ||||||||||||||||
| Change in unrealized gains (losses) on available-for-sale investments |
— | — | — | (23 | ) | — | (23 | ) | ||||||||||||||||
|
|
|
|||||||||||||||||||||||
| Comprehensive loss |
(23,716 | ) | ||||||||||||||||||||||
| Share-based compensation expense |
— | — | 2,231 | — | — | 2,231 | ||||||||||||||||||
| Common stock issued under stock option and purchase plans |
140 | 1 | 46 | — | — | 47 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2009 |
53,430 | 534 | 543,987 | (2 | ) | (503,585 | ) | 40,934 | ||||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||
| Net loss |
— | — | — | — | (24,722 | ) | (24,722 | ) | ||||||||||||||||
| Change in unrealized gains (losses) on available-for-sale investments |
— | — | — | 1 | — | 1 | ||||||||||||||||||
|
|
|
|||||||||||||||||||||||
| Comprehensive loss |
(24,721 | ) | ||||||||||||||||||||||
| Share-based compensation expense |
— | — | 2,092 | — | — | 2,092 | ||||||||||||||||||
| Common stock issued under stock option and purchase plans |
184 | 2 | 62 | — | — | 64 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2010 |
53,614 | 536 | 546,141 | (1 | ) | (528,307 | ) | 18,369 | ||||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||
| Net loss |
— | — | — | — | (12,022 | ) | (12,022 | ) | ||||||||||||||||
| Change in unrealized gains (losses) on available-for-sale investments |
— | — | — | 1 | — | 1 | ||||||||||||||||||
|
|
|
|||||||||||||||||||||||
| Comprehensive loss |
(12,021 | ) | ||||||||||||||||||||||
| Issuance of common stock under an At Market Issuance Sales Agreement |
484 | 5 | 144 | — | — | 149 | ||||||||||||||||||
| Share-based compensation expense |
— | — | 1,558 | — | — | 1,558 | ||||||||||||||||||
| Common stock issued under stock option and purchase plans |
358 | 4 | 240 | — | — | 244 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2011 |
54,456 | $ | 545 | $ | 548,083 | $ | — | $ | (540,329 | ) | $ | 8,299 | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
See accompanying Notes to Financial Statements.
52
Table of Contents
TELIK, INC.
(In thousands)
| Years Ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net loss |
$ | (12,022 | ) | $ | (24,722 | ) | $ | (23,693 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation |
10 | 343 | 556 | |||||||||
| (Gain) loss on the disposal of property and equipment |
— | 31 | (342 | ) | ||||||||
| Share-based compensation expense |
1,558 | 2,092 | 2,231 | |||||||||
| Facility exit costs |
— | 5,360 | — | |||||||||
| Change in value of marketable securities |
— | 111 | (1,350 | ) | ||||||||
| Change in fair value of rights to sell ARS to UBS |
— | (111 | ) | 1,309 | ||||||||
| Changes in assets and liabilities: |
||||||||||||
| Other receivables |
189 | 2 | (178 | ) | ||||||||
| Prepaid expenses and other current assets |
54 | 59 | 267 | |||||||||
| Other assets |
— | (97 | ) | — | ||||||||
| Accounts payable |
(823 | ) | 742 | (1,391 | ) | |||||||
| Accrued liabilities |
(288 | ) | (227 | ) | (1,532 | ) | ||||||
| Accrued facility exit costs |
(1,436 | ) | (308 | ) | — | |||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(12,758 | ) | (16,725 | ) | (24,123 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from investing activities: |
||||||||||||
| Purchases of investments |
(11,720 | ) | (34,411 | ) | (42,119 | ) | ||||||
| Sales of investments |
1,699 | 14,024 | 225 | |||||||||
| Maturities of investments |
23,664 | 35,660 | 27,000 | |||||||||
| Proceeds from sale of property and equipment |
— | 5 | 659 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) investing activities |
13,643 | 15,278 | (14,235 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from financing activities: |
||||||||||||
| Payments on loan provided by UBS relating to ARS |
— | (3,100 | ) | — | ||||||||
| Net proceeds from issuance of common stock |
393 | 64 | 47 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) financing activities |
393 | (3,036 | ) | 47 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net change in cash and cash equivalents |
1,278 | (4,483 | ) | (38,311 | ) | |||||||
| Cash and cash equivalents at beginning of period |
7,768 | 12,251 | 50,562 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents at end of period |
$ | 9,046 | $ | 7,768 | $ | 12,251 | ||||||
|
|
|
|
|
|
|
|||||||
| Supplemental information: |
||||||||||||
| Interest paid |
$ | — | $ | 6 | $ | 85 | ||||||
| Proceeds from sale of ARS directly applied to UBS loan relating to ARS |
— | — | $ | 4,900 | ||||||||
| Repayment of UBS loan relating to ARS using proceeds from sale of ARS |
— | — | $ | (4,900 | ) | |||||||
See accompanying Notes to Financial Statements.
53
Table of Contents
TELIK, INC.
1. Nature of Operations and Going Concern
Telik, Inc. (“Telik,” “we” or, the “Company”) was incorporated in the state of Delaware in October 1988. We are engaged in the discovery and development of small molecule therapeutics. We operate in only one business segment.
We have incurred net losses since inception and we expect to incur substantial losses for at least the next several years as we continue our research and development activities. During the year ended December 31, 2011, loss from operations was $12.1 million and net loss was $12.0 million. Net cash used in operations for the year ended December 31, 2011 was $12.8 million and aggregate cash, cash equivalents, investments and restricted investments at December 31, 2011 were $11.7 million. As of December 31, 2011, we had an accumulated deficit of $540.3 million.
To date, we have funded operations primarily through the sale of equity securities, non-equity payments from collaborators and interest income. The process of developing our products will require significant additional research and development, preclinical testing and clinical trials, as well as regulatory approval. We expect these activities, together with general and administrative expenses, to result in substantial operating losses for the foreseeable future. We will not receive product revenue unless we, or our collaborative partners, complete clinical trials, obtain regulatory approval and successfully commercialize one or more of our products.
We believe our existing cash resources will not be sufficient to fund our projected operating requirements for the next twelve months. We have been and are currently seeking collaborative arrangements with corporate partners to fund the development and commercialization of TELINTRA, our lead product candidate and TELCYTA, our other product candidate. We are also evaluating options to raise additional funds through equity or debt financings and sales transactions as well as other sources such as research grants from non-profit organizations. However, we cannot provide any assurances that we will be successful in obtaining additional funding. If we are unable to secure additional financing on a timely basis or on terms favorable to us, we may be required to cease or reduce certain research and development projects, to sell some or all of our technology or assets or to merge all or a portion of our business with another entity. Insufficient funds may require us to delay, scale back, or eliminate some or all of our activities, and if we are unable to obtain additional funding, we will continue to have uncertainty regarding our ability to continue as a going concern.
In August 2011, we filed a shelf registration statement on Form S-3 to offer and sell, from time to time, equity and debt securities in one or more offerings up to a total dollar amount of $25.0 million. On August 30, 2011, we entered into an At Market Issuance Sales Agreement, or Sales Agreement, with McNicoll, Lewis & Vlak LLC, or MLV, pursuant to which we may issue and sell shares of our common stock having an aggregate offering price of up to $7.0 million from time to time through MLV as our sales agent. For the year ended December 31, 2011, we received approximately $149,000 in net proceeds from stock sales under the Sales Agreement after deducting commissions and other related expenses. Our ability to sell shares of our common stock is subject to share volume limitations, market conditions and our continued listing on the Nasdaq Capital Market. There is no assurance that we may be able to raise any funds in the future under the Sales Agreement.
The accompanying financial statements have been prepared in accordance with U.S. generally accepted accounting principles applicable to a going concern which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. Our ability to continue as a going concern is dependent upon our ability to obtain additional equity or debt financing, attain further operating efficiencies and, ultimately, to generate revenue. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
54
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
2. Summary of Significant Accounting Policies
Use of Estimates
In preparing our financial statements to conform with U.S. generally accepted accounting principles, we make estimates and assumptions that affect the amounts reported in our financial statements and accompanying notes. Actual results may differ from these estimates.
Cash and Cash Equivalents and Short-Term Investments
We currently invest our excess cash in money market funds, cash deposits and U.S. government agency securities. Prior to July 2010, we held taxable municipal notes, some of which had an auction reset feature (auction rate securities, or ARS), and corporate notes. All investments with stated maturities of three months or less from date of purchase are classified as cash equivalents. Debt securities with original maturities greater than three months and remaining maturities less than one year are classified as short-term investments. Debt securities with remaining maturities greater than one year and which we intend to hold until maturity are classified as long-term investments. We classify all cash equivalents and non-ARS investments as available-for-sale. Available-for-sale securities are carried at estimated fair value, with unrealized gains and losses reported as a component of accumulated other comprehensive income (loss).
Realized gains or losses on the sale of investments are determined on a specific identification method, and such gains and losses are reflected as a component of interest income.
Marketable security investments are evaluated periodically for impairment. We take into account general market conditions, changes in economic environment as well as specific investment attributes, such as credit downgrade or illiquidity for each investment, the expected cash flows from the security, our intent to sell the security and whether or not we will be required to sell the security before the recovery of its amortized cost, to estimate the fair value of our investments and to determine whether impairment is other than temporary. If it is determined that a decline in fair value of any investment is other than temporary, then the unrealized loss related to credit risk would be included in interest and other income (expense), net.
Restricted Investments
Under certain operating lease agreements, we may be required from time to time to set aside cash as collateral. At December 31, 2011, we had approximately $250,000 of restricted investments and at December 31, 2010 we had approximately $449,000 related to the building lease agreement.
Fair Value of Financial Instruments
We used the provisions of ASC 820, “Fair Value Measurements and Disclosure,” to determine the fair values of our financial and nonfinancial assets and liabilities where applicable. ASC 820 defines fair value, establishes a framework for measuring fair value in generally accepted accounting principles and expands disclosure about fair value measurements. The objective of fair value measurement is to determine the price that would be received to sell the asset or paid to transfer the liability (an exit price) in an orderly transaction between market participants at the measurement date. The statement emphasizes that fair value is a market-based measurement, not an entity-specific measurement, and that market participant assumptions include assumptions about risk and effect of a restriction on the sale or use of an asset. To increase consistency and comparability in fair value measurement and related disclosures, this statement establishes a fair value hierarchy that prioritize the inputs to valuation techniques used to measure fair value into three broad levels: (1) Level 1 inputs are quoted
55
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
prices (unadjusted) in active markets for identical assets or liabilities that the reporting entity has the ability to access at the measurement date; (2) Level 2 inputs are inputs other than quoted prices included in Level 1 that are observable for the asset or liability, either directly or indirectly through corroboration with observable market data; and (3) Level 3 inputs are unobservable inputs for asset or liability that reflect the reporting entity’s own assumptions about risk and the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances.
Government agency securities are recorded at their estimated fair value. Since these government securities generally have market prices from multiple sources and it can be difficult to select the best individual price directly from the quoted prices in the active markets, therefore we use Level 2 inputs for the valuation of these securities. Using the Level 2 inputs, a “consensus price” or a weighted average price for each of these securities can be derived from a distribution-curve-based algorithm which includes market prices obtained from a variety of industrial standard data providers (e.g. Bloomberg), security master files from large financial institutions, and other third-party sources.
For our ARS and put option held prior to July 2010, we used Level 3 inputs to determine their fair values since they did not have a readily determinable market value. The assumptions used in valuing the ARS and the put option included, basing on data as of the reporting period then ended, interest rates, tax status, credit quality, expected holding periods of the ARS, insurance wraps, the portfolio composition of Federal Family Education Loan Program, or FFELP, and private loans, likelihood of redemption, loan rates per the UBS Rights offering and bearer risk associated with UBS’s financial ability to repurchase the ARS beginning June 30, 2010. These assumptions were volatile and subject to change, and therefore could have resulted in significant changes to the fair values of these financial instruments.
Property and Equipment
Property and equipment are stated at cost. We calculate depreciation using the straight-line method over the estimated useful lives of the assets, which range from three to five years. We amortize furniture and equipment leased under capital leases and leasehold improvements using the straight-line method over the estimated useful lives of the respective assets or the lease term, whichever is shorter. Amortization of assets under capital leases is included in depreciation expense.
Exit and Disposal Activities
We record costs and liabilities associated with exit and disposal activities, as defined in ASC 420, “Exit or Disposal Cost Obligations”, at fair value in the period the liability is incurred. ASC 420 requires that the estimated future cash flows to be used in the fair value calculation be discounted using a credit-adjusted risk-free interest rate and that such interest rate shall have a maturity date that approximates the expected timing of future cash flows. Future cash flows related to lease obligations shall include the effect of sublease rental income and other lease operating expenses. In addition, accretion of the liability due to the passage of time is recorded as a general and administrative expense. See Note 6 to Financial Statements for further information.
Impairment of Long-lived Assets
We regularly evaluate our long-lived assets for indicators of possible impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable, in accordance with ASC 360 and related guidance. An impairment loss would be recognized when estimated undiscounted future cash flows expected to result from the use of the asset and its eventual disposition are less
56
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
than its carrying amount. Impairment, if any, is assessed using discounted cash flows. For the year ended December 31, 2010, we recorded an impairment charge of $1.0 million against leasehold improvements. We had no impairment charge for the year ended December 31, 2011.
Research and Development
Our research and development expenses include salaries and benefits costs, fees for contractors, consultants and third party contract research organizations, and an allocation of facility and administrative costs. Research and development expenses consist of costs incurred for drug and product development, manufacturing, clinical activities, discovery research, screening and identification of product candidates, and preclinical studies. All such costs are charged to research and development expenses as incurred.
Clinical development costs are a significant component of research and development expenses. We have a history of contracting with third parties that perform various clinical trial activities on our behalf in the on-going development of our product candidates. The financial terms of these contracts are subject to negotiations and may vary from contract to contract and may result in uneven payment flows. We accrue and expense costs for clinical trial activities performed by third parties based upon estimates of the percentage of work completed over the life of the individual study in accordance to agreements established with contract research organizations and clinical trial sites. We determine our estimates through discussion with internal clinical personnel and outside service providers as to progress or stage of completion of trials or services and the agreed upon fee to be paid for such services. These estimates may or may not match the actual services performed by the organizations as determined by patient enrollment levels and related activities. We monitor patient enrollment levels and related activities to the extent possible.
Stock-based Compensation
Under the provisions of ASC 718, employee stock-based compensation is estimated using the Black-Scholes option-pricing model and is recognized as expense ratably over the requisite service period in a manner similar to other forms of compensation paid to employees. The Black-Scholes option-pricing model requires the use of certain subjective assumptions. The most significant of these assumptions are our estimates of the expected volatility of the market price of our stock and the expected term of the award. For the years 2009 and 2011, the expected volatilities were based solely on historical volatility data as there were insufficient traded option activities resulting from our declining stock price. We did not use any expected volatility assumptions for 2010 as there were no options granted during the year. The expected term of options granted is based on the simplified method in accordance with SAB Topic 14.D.2, as our historical share option exercise experience does not provide a reasonable basis for estimation. ASC 718 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. We adjust our forfeiture rate to reflect actual historical and expected cancellations of unvested options when applicable. See Note 9 to Financial Statements for additional information.
We have adopted the simplified method to calculate the beginning balance of the additional paid-in-capital ,or APIC, pool of the excess tax benefit, and to determine the subsequent impact on the APIC pool and our Statements of Cash Flows of the tax effects of employee stock-based compensation awards that were outstanding upon our adoption of ASC 718.
Comprehensive Loss
Components of other comprehensive loss, including unrealized gains and losses on available-for-sale investments, are included as part of total comprehensive loss in our statements of stockholders’ equity.
57
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
Net Loss per Share
Basic and diluted net loss per share are computed by dividing net loss by the weighted average number of common shares outstanding during the year.
The following table reflects weighted average options outstanding before application of the treasury stock method that could potentially dilute basic earnings per share in the future, but were excluded from the computation of diluted net loss per share, as their effect would have been antidilutive for the periods presented herein.
| Year Ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Weighted average outstanding options |
11,358,880 | 12,387,695 | 9,494,092 | |||||||||
Income Taxes
We apply the provisions of ASC 740, “Accounting for Income Taxes”. Under ASC 740, deferred tax liabilities or assets arise from differences between the tax basis of liabilities or assets and their basis for financial reporting, and are subject to tests of recoverability in the case of deferred tax assets. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. A valuation allowance is provided for deferred tax assets to the extent realization is not judged to be more likely than not.
ASC 740-10-25 provides detailed guidance for the financial statement recognition, measurement and disclosure of uncertain tax positions recognized in an enterprise’s financial statements in accordance with ASC 740. Income tax positions must meet a more-likely-than-not recognition threshold at the effective date to be recognized upon the adoption of Section 740-10-25 and in subsequent periods. Any potential accrued interest and penalties related to unrecognized tax benefits within operations would be recorded as income tax expense. To date, there have been no interest or penalties charged to us related to the underpayment of income taxes.
We adopted and applied ASC 740-10-25 to all income tax positions commencing from 2007. There was no impact on our financial statements upon adoption. Because of our historical significant net operating losses, we have not been subject to income tax since inception. Upon adoption of ASC 740-10-25 on January 1, 2007, we recognized a $14.2 million increase in our liability for unrecognized income tax benefits which was accounted for as a reduction to the deferred tax assets balance as of that date. At December 31, 2011, we have a liability for unrecognized tax benefits of $9.4 million, none of which, if recognized, would affect our effective tax rate. We maintain deferred tax assets that reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. These deferred tax assets include net operating loss carryforwards, research credits and capitalized research and development. The net deferred tax asset has been fully offset by a valuation allowance because of our history of losses.
Recent Accounting Pronouncements
In December 2011, the Financial Accounting Standards Board, or FASB, issued Accounting Standards Update No. 2011-12, Deferral of the Effective Date for Amendments to the Presentation of Reclassifications of Items Out of Accumulated Other Comprehensive Income in Accounting Standards Update No. 2011-05, or ASU 2011-12, to allow FASB time to redeliberate whether to present on the face of financial statements the effects of reclassifications out of accumulated other comprehensive income on the components of net income and other comprehensive income for all other periods presented. ASU 2011-12 is effective at the same time as the
58
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
amendments in ASU 2011-05 so that we will not be required to comply with these presentation requirements in ASU 2011-05. All other requirements in ASU 2011-05 are not affected by this update, including the requirement to report comprehensive income either in a single continuous financial statement or in two separate but consecutive financial statements.
In June 2011, FASB issued Accounting Standards Update No. 2011-05, Comprehensive Income (Topic 220)-Presentation of Comprehensive Income, or ASU 2011-05, to require an entity to present the total of comprehensive income, the components of net income, and the components of other comprehensive income either in a single continuous statement of comprehensive income or in two separate but consecutive statements. ASU 2011-05 eliminates the option to present the components of other comprehensive income as part of the statement of changes in stockholders’ equity. ASU 2011-05 should be applied retrospectively and is effective for fiscal years, and interim periods within those years, beginning after December 15, 2011. We are currently evaluating the potential impact of adopting ASU 2011-05 on our financial statements.
In May 2011, the FASB issued Accounting Standards Update No. 2011-04, Amendments to Achieve Common Fair Value Measurement and Disclosure Requirements in U.S. GAAP and International Financial Reporting Standards (Topic 820)-Fair Value Measurement, or ASU 2011-04, which changes the wording used to describe the requirements in U.S. GAAP for measuring fair value and for disclosing information about fair value measurements in order to improve consistency in the application and description of fair value between U.S. GAAP and International Financial Reporting Standards , or IFRS. ASU 2011-04 changes certain fair value measurement principles, clarifies the application of existing fair value measurement and enhances the disclosure requirements particularly for level 3 fair value measurements. ASU 2011-04 is effective during interim and annual periods beginning after December 15, 2011, with early adoption prohibited. The new guidance will require prospective application. We are currently evaluating the potential impact of adopting ASU 2011-04 on our financial statements.
In January 2010, the FASB issued Accounting Standards Update No. 2010-06, Fair Value Measurements and Disclosures (Topic 820)—Improving Disclosures about Fair Value Measurements, or ASU 2010-06, to improve disclosures related to fair value measurements. This guidance requires new disclosures as well as clarifies certain existing disclosure requirements. Under ASU 2010-06, companies are required to provide separate information about significant transfers in and out of Level 1 and Level 2 of the fair value hierarchy as well as the reasons for such transfers, and a reconciliation of purchases, sales, issuances, and settlements activities valued using Level 3 inputs on a gross basis. ASU 2010-06 also clarifies the requirement to determine the level of disaggregation for fair value measurement disclosures and the requirement to disclose valuation techniques and inputs used for both recurring and nonrecurring fair value measurements in either Level 2 or Level 3. ASU 2010-06 was effective for interim and annual reporting periods beginning after December 15, 2009, except for the disclosure about purchases, sales, issuances, and settlements in the rollforward of activity for Level 3 fair value measurement which was effective for fiscal years beginning after December 15, 2010, and for interim periods within those fiscal years. We have adopted ASU 2010-06 for Level 1 and 2 disclosure requirements since the quarter ended March 31, 2010 and adopted ASU 2010-06 for Level 3 requirement on January 1, 2011. The adoption of this guidance increases the level of disclosures in our financial statements related to fair value measurements.
3. Fair Value Measurements
We measure certain financial assets at fair value on a recurring basis, including cash equivalents, available-for-sale securities, trading securities and put options. The fair value of these financial assets was determined based on a three-tier fair value hierarchy as described in Note 1, which prioritizes the inputs used in measuring fair value.
59
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
The following table presents information about our financial assets that are measured at fair value on a recurring basis as of December 31, 2011 and indicates the fair value hierarchy of the valuation techniques utilized to determine such fair value:
| Fair Value Measurement at December 31, 2011 Using | ||||||||||||||||
| December 31, 2011 |
Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
|||||||||||||
| (in thousands) | ||||||||||||||||
| Available-for-sale securities: |
||||||||||||||||
| Money market funds |
$ | 5,190 | $ | 5,190 | $ | — | $ | — | ||||||||
| US government agencies |
4,454 | — | 4,454 | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
$ | 9,644 | $ | 5,190 | $ | 4,454 | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The following table presents information about our financial assets that are measured at fair value on a recurring basis as of December 31, 2010 and indicates the fair value hierarchy of the valuation techniques utilized to determine such fair value:
| Fair Value Measurement at December 31, 2010 Using | ||||||||||||||||
| December 31, 2010 |
Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
|||||||||||||
| (in thousands) | ||||||||||||||||
| Available-for-sale securities: |
||||||||||||||||
| Money market funds |
$ | 53 | $ | 53 | $ | — | $ | — | ||||||||
| US government agencies |
20,734 | — | 20,734 | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
$ | 20,787 | $ | 53 | $ | 20,734 | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
There were no transfers between Level 1 and Level 2 measurements in the years ended December 31, 2011 and 2010.
The following is a reconciliation of our investments measured at fair value on a recurring basis using significant unobservable inputs (Level 3):
| Fair Value Measurements
Using Significant Unobservable Inputs (Level 3) |
||||||||||||
| Put Option |
Auction Rate Securities |
Total | ||||||||||
| (in thousands) | ||||||||||||
| Balance at December 31, 2009 |
3,420 | 10,380 | 13,800 | |||||||||
| Impairment gain (loss) included in net loss |
111 | (111 | ) | — | ||||||||
| Sales or settlement |
(3,531 | ) | (10,269 | ) | (13,800 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Balance at December 31, 2010 |
$ | — | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
| Balance at December 31, 2011 |
$ | — | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
60
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
We estimated the fair value of our ARS and put option held prior to July 2010 using Level 3 inputs as these securities did not have a readily determinable market value. To estimate the fair value, we used a discounted cash flow approach and considered several factors including interest rates, credit quality, and expected holding period.
In 2008, we entered into an agreement with UBS whereby we received rights, or the Right, to sell all our ARS held in our UBS account at par value ($18.7 million) to UBS at any time during a two-year period beginning on June 30, 2010 and ending on July 2, 2012. On February 12, 2010, UBS repurchased $4.0 million of our ARS from the original balance of $13.8 million. On July 1, 2010, we exercised the Right and sold all our remaining ARS to UBS under the Rights Agreement at par value of $9.8 million. For the year ended December 31, 2010, we recorded (i) a reduction of approximately $3.5 million and $10.3 million in fair values of our put option and ARS respectively, as a result of certain ARS repurchased by UBS in February 2010 and the exercise of the Rights Agreement on July 1, 2010 and (ii) no gain or loss to interest and income, net due to offsetting changes in fair values of our ARS and put option. As a result, we had no ARS investments and put option balance at December 31, 2010 and at December 31, 2011. For additional information, see Note 8 to Financial Statements.
We evaluate long-lived assets for impairment on a non-recurring basis whenever events or changes in circumstances indicate that the carrying value of long-lived assets may not be recoverable. We recognize such impairment in the event the net book value of such assets exceeds the future undiscounted cash flows attributable to such assets. An impairment charge of $40,000 against laboratory equipment was recorded for the year ended December 31, 2009. For the year ended December 31, 2010, we recorded an impairment charge of $1.0 million against the leasehold improvements of the Porter Drive facility as we determined the facility would not have any future benefits to us after we relocated to a new location. For additional information, see Note 5 to Financial Statements.
4. Cash and Cash Equivalents, Investments and Restricted Investments
The following is a summary of estimated fair value of cash and cash equivalents, investments and restricted investments:
| December 31 | ||||||||
| 2011 | 2010 | |||||||
| (in thousands) | ||||||||
| Certificate of deposits |
$ | 250 | $ | 449 | ||||
| US government agencies |
4,454 | 20,734 | ||||||
| Cash and money market funds |
6,996 | 2,881 | ||||||
|
|
|
|
|
|||||
| Total |
$ | 11,700 | $ | 24,064 | ||||
|
|
|
|
|
|||||
| Reported as: |
||||||||
| Cash and cash equivalents |
$ | 9,046 | $ | 7,768 | ||||
| Short-term investments |
2,404 | 15,847 | ||||||
| Restricted investments |
250 | 449 | ||||||
|
|
|
|
|
|||||
| Total |
$ | 11,700 | $ | 24,064 | ||||
|
|
|
|
|
|||||
61
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
We had no material unrealized gains or losses for the year ended December 31, 2011. The following is a summary of amortized cost, unrealized gains and losses and estimated fair value of cash and cash equivalents and marketable debt securities held as available-for-sale for the year ended December 31, 2010.
| December 31, 2010 | ||||||||||||||||
| Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Estimated Fair Value |
|||||||||||||
| (in thousands) | ||||||||||||||||
| Certificate of deposits |
$ | 449 | $ | — | $ | — | $ | 449 | ||||||||
| US government agencies |
20,735 | 2 | (3 | ) | 20,734 | |||||||||||
| Cash and money market funds |
2,881 | — | — | 2,881 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
$ | 24,065 | $ | 2 | $ | (3 | ) | $ | 24,064 | |||||||
|
|
|
|
|
|
|
|
|
|||||||||
There were no material realized gains on sales of available-for-sale investments for the years ended December 31, 2011 and 2010. Realized gains and losses were calculated based on the specific identification method.
We had no material unrealized losses for the year ended December 31, 2011. The following tables summarize the gross unrealized losses and fair values for investments in an unrealized loss position for which other-than-temporary impairments were not recognized for the year ended December 31, 2010.
| Less Than 12 Months | 12 Months or Greater | Total | ||||||||||||||||||||||
| December 31, 2010 |
Fair Value |
Unrealized Losses |
Fair Value |
Unrealized Losses |
Fair Value |
Unrealized Losses |
||||||||||||||||||
| (in thousands) | ||||||||||||||||||||||||
| US government agencies |
$ | 8,482 | $ | (3 | ) | $ | — | $ | — | $ | 8,482 | $ | (3 | ) | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Total |
$ | 8,482 | $ | (3 | ) | $ | — | $ | — | $ | 8,482 | $ | (3 | ) | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
The following is a summary of the cost and estimated fair value of marketable debt securities, held as available-for-sale at December 31, 2011 and 2010, classified by stated maturity date of the security:
| December 31, 2011 | December 31, 2010 | |||||||||||||||
| Amortized Cost |
Fair Value |
Amortized Cost |
Fair Value |
|||||||||||||
| (in thousands) | ||||||||||||||||
| Mature in less than one year |
$ | 4,454 | $ | 4,454 | $ | 20,735 | $ | 20,734 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
$ | 4,454 | $ | 4,454 | $ | 20,735 | $ | 20,734 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
5. Property and Equipment
Property and equipment consist of the following:
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| (in thousands) | ||||||||
| Computer and lab equipment |
$ | 1,034 | $ | 1,113 | ||||
| Capitalized software |
547 | 547 | ||||||
| Office furniture and equipment |
94 | 115 | ||||||
|
|
|
|
|
|||||
| 1,675 | 1,775 | |||||||
| Less accumulated depreciation |
(1,674 | ) | (1,764 | ) | ||||
|
|
|
|
|
|||||
| Property and equipment, net |
$ | 1 | $ | 11 | ||||
|
|
|
|
|
|||||
62
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
We relocated our principal offices and ceased the use of our facility at 3165 Porter Drive, Palo Alto in November 2010. As a result, we recorded an impairment charge of $1.0 million against the leasehold improvements of the Porter Drive facility which had an original cost of $3.4 million to reflect the excess of carrying value of leasehold improvements over their estimated value which was determined to be zero as the facility would not have any future benefits to us. This impairment charge was included in our facility exit costs in the Statements of Operations. In addition, as a result of our principal offices relocation we disposed of approximately $1.1 million (at cost) of computer and laboratory equipment with a net book value of $36,000 and recorded a net loss in interest and other income (expense), net.
6. Facility Exit Costs
In November 2010, we ceased the use of our facility at 3165 Porter Drive in Palo Alto, California and subleased the facility to a tenant for the remaining contractual term of our master lease, which is through May 2014. As a result, we recorded a charge of $4.7 million which included the estimated fair value of future lease-related payments less estimated net income from sublease rental offset by a reduction of $335,000 in the balance of deferred rent related to the facility as of November 30, 2010. Future lease-related payments and rental income are scheduled to be made and received monthly until the lease and sublease expire in May 2014.
The following table summarizes the activities related to accrued facility exit costs for the year ended December 31, 2011 and 2010 (in thousands):
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| (in thousands) | ||||||||
| Beginning balance |
$ | 4,362 | $ | 4,671 | ||||
| Amounts paid during the period |
(3,822 | ) | (309 | ) | ||||
| Amounts received during the period |
2,376 | — | ||||||
| Non-cash accretion |
10 | — | ||||||
|
|
|
|
|
|||||
| Balance as of December 31, 2011 |
$ | 2,926 | $ | 4,362 | ||||
|
|
|
|
|
|||||
| Reported as current portion |
$ | 1,463 | $ | 1,439 | ||||
| Reported as noncurrent portion |
$ | 1,463 | $ | 2,923 | ||||
7. Restructuring Plans
We implemented several restructuring plans in the past years to reduce our operating expenses and to streamline our infrastructure to focus on our most advanced preclinical and clinical development programs. For the restructuring plan implemented in February 2009, we reduced our workforce by 37 positions and recorded a charge of approximately $951,000 for the year ended December 31, 2009. We paid $750,000 in the quarter ended March 31, 2009, $111,000 in the quarter ended June 30, 2009 and $90,000 in the quarter ended September 30, 2009, as severance, payroll taxes and other personnel related costs.
For the restructuring plan in November 2010, we ultimately reduced our workforce by ten positions and accrued a restructuring charge of approximately $425,000, including employee severance costs, health benefits and personnel related costs. In connection with the restructuring plan, we paid $232,000 in December 2010, $140,000 in the quarter ended March 31, 2011, $48,000 in the quarter ended June 30, 2011 and for the quarter ended September 30, 2011, we reversed $5,000 for unused health benefits.
63
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
8. Notes Payable and Commitments
Notes Payable
In connection with our acceptance of the offer to enter into the Right agreement with UBS, a “no net cost” loan in the amount of up to 75% of the market value of our ARS was made available to us. On December 31, 2008, we entered into a loan agreement with UBS and drew down $8 million with our ARS pledged as collateral. On June 10, 2009 and February 12, 2010, UBS elected to repurchase a portion of our ARS under the Rights Agreement at par value of $4.9 million and $4.0 million, respectively. Proceeds from both sales of our ARS were applied to repayment of the credit line whereby $4.9 million was paid in June 2009 leaving a balance of $3.1 million which was paid in full in February 2010. Interest paid on the loan for the year ended December 31, 2010 was approximately $6,000 which was offset entirely by interest earned on the pledged securities. We had no interest expense for the year ended December 31, 2011.
Operating Leases
In November 2010, we entered into a 28-month lease agreement for 8,620 square feet of office space at 700 Hansen Way in Palo Alto, California and relocated our corporate offices to this facility.
We also lease approximately 92,000 square feet located at 3165 Porter Drive in Palo Alto, California, which we have subleased to a tenant effective November 2010 through May 2014 when our master lease expires. Pursuant to the terms of the lease, we are required to maintain a security deposit, in the form of a letter of credit equal to approximately $250,000. This letter of credit must be secured by either a deposit account or a securities account and at December 31, 2011 the security deposit is in the form of securities that are classified in the balance sheet as restricted investments. This collateral account is managed in accordance with our investment policy, and is restricted as to withdrawal.
We also have an office equipment lease of approximately $45,000 with a remaining term of 19 months.
Future minimum rental payments under our non-cancelable operating leases as of December 31, 2011 are as follows:
| Operating Leases |
||||
| (in thousands) | ||||
| Years ending December 31, |
||||
| 2012 |
4,370 | |||
| 2013 |
3,685 | |||
| 2014 |
1,537 | |||
| 2015 |
— | |||
|
|
|
|||
| Total future minimum rental payments |
9,592 | |||
| Less aggregate future minimum rentals to be received from subleases |
(6,045 | ) | ||
|
|
|
|||
| Total |
$ | 3,547 | ||
|
|
|
|||
Rent expense under operating leases was approximately $396,000 in 2011 and $3.4 million in 2010.
64
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
9. Stockholders’ Equity
Stock Offerings
In August 2011, we filed a shelf registration statement on Form S-3 to offer and sell, from time to time, equity and debt securities in one or more offerings up to a total dollar amount of $25.0 million. On August 30, 2011 we entered into an At Market Issuance Sales Agreement with McNicoll, Lewis & Vlak LLC, or MLV, pursuant to which we may issue and sell shares of our common stock having an aggregate offering price up to $7.0 million, from time to time, through MLV as our sales agent. In conjunction with the sales agreement, MLV would receive compensation based on an aggregate of 4% of the gross proceeds on the sale price per share of our common stock. Any sales made pursuant to the sales agreement are deemed an “at-the-market” offering and would be made pursuant to the shelf registration statement on Form S-3. For the year ended December 31, 2011, we sold 484,112 shares through MLV under the sales agreement and received approximately $149,000 in net proceeds after deducting commissions and other related expenses.
Stockholder Rights Plan
In October 2001, our Board of Directors approved the adoption of a Stockholder Rights Plan, which provided for the distribution of one preferred share purchase right (a “Right”) for each outstanding share of common stock of the Company. The dividend was paid on November 14, 2001 to the stockholders of record on that date. Each Right entitles the registered holder to purchase from the Company one one-hundredth of a share of Series A Junior Participating Preferred Stock, par value $0.01 per share (the “Preferred Shares”), at a price of $90.00 per one one-hundredth of a Preferred Share (the “Purchase Price”), subject to adjustment. The Rights will be exercisable the earlier of (i) the date of a public announcement that a person, entity or group of affiliated or associated persons have acquired beneficial ownership of 20% or more of the outstanding common shares (an “Acquiring Person”), or (ii) ten business days (or such later date as may be determined by action of the Board of Directors prior to such time as any person or entity becomes an Acquiring Person) following the commencement of, or announcement of an intention to commence, a tender offer or exchange offer the consummation of which would result in any person or entity becoming an Acquiring Person. In May 2006, we amended the stockholder rights plan to exclude Eastbourne Capital Management, L.L.C., or Eastbourne, and certain related persons and entities from the definition of Acquiring Person so long as neither Eastbourne nor its affiliates or associates, either individually or in the aggregate, becomes the beneficial owner of 25% or more of the common stock then outstanding. On December 11, 2006, the plan was further amended to increase this threshold to 30%.
In the event that any person, entity or group of affiliated or associated persons become an Acquiring Person, each holder of a Right will have the right to receive, upon exercise, the number of common shares having a market value of two times the exercise price of the Right. In the event that the Company is acquired in a merger or other business combination transaction or 50% or more of its consolidated assets or earning power are sold to an Acquiring Person, its associates or affiliates or certain other persons in which such persons have an interest, each holder of a Right will have the right to receive, upon the exercise at the then-current exercise price of the Right, that number of shares of common stock of the acquiring company which at the time of such transaction will have a market value of two times the exercise price of the Right. At any time after an Acquiring Person becomes an Acquiring Person and prior to the acquisition by such Acquiring Person of 50% or more of the outstanding common shares, the Board of Directors of the Company may exchange the Rights (other than Rights owned by such person or group which have become void), in whole or in part, at an exchange ratio of one common share, or one one-hundredth of a Preferred Share, per Right (or, at the election of the Company, the Company may issue cash, debt, stock or a combination thereof in exchange for the Rights), subject to adjustment. The Rights expired on November 14, 2011. Accordingly, we filed a certificate of elimination withthe Secretary of State of the State of Delaware on February 24, 2012 (the “Certificate of Elimination”) which eliminated from
65
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
our Amended and Restated Certificate of Incorporation all matters set forth in the Certificate of Designation with respect to the Preferred Shares. No Preferred Shares were issued or outstanding at the time of the filing of the Certificate of Elimination.
2011 Equity Incentive Plan
In March 2011, we adopted the 2011 Equity Incentive Plan, or the “2011 Plan”, and reserved 3,500,000 shares of Telik common stock for issuance under the 2011 Plan. Options granted under the 2011 Plan may be either incentive stock options (“ISOs”) or nonstatutory stock options (“NSOs”). The 2011 Plan also provides for the grant of stock appreciation rights, restricted stock awards, restricted stock unit awards, performance stock awards, performance cash awards and other stock awards. For ISOs and NSOs, the option price shall be at least 100% and 85%, respectively, of the closing price of our common stock on the date of the grant. If, at any time we grant an option, and the optionee directly or by attribution owns stock possessing more than 10% of the total combined voting power of all classes of stock of Telik, the option price shall be at least 110% of the fair value and shall not be exercisable more than five years after the date of grant. Options generally vest over a period of two or four years from the date of grant. Options granted under the 2011 Plan expire no later than 10 years from the date of grant. As of December 31, 2011, there were 2,433,500 option shares outstanding and 1,066,500 shares available for future grants under the 2011 Plan.
2000 Equity Incentive Plan
In March 2000, we adopted the 2000 Equity Incentive Plan, or the “2000 Plan”, and reserved 2,000,000 shares of Telik common stock for issuance under the 2000 Plan. In addition, the 2000 Plan provides for annual increases in the number of shares available for issuance under the 2000 Plan beginning January 1, 2001. The number of additional shares to be reserved automatically will be equal to the lesser of 1,500,000 shares, 5% of the outstanding shares on the date of the annual increase or such amount as may be determined by the Board of Directors. Options granted under the 2000 Plan may be either incentive stock options (“ISOs”) or nonstatutory stock options (“NSOs”). For ISOs and NSOs, the option price shall be at least 100% and 85%, respectively, of the closing price of our common stock on the date of the grant. If, at any time we grant an option, and the optionee directly or by attribution owns stock possessing more than 10% of the total combined voting power of all classes of stock of Telik, the option price shall be at least 110% of the fair value and shall not be exercisable more than five years after the date of grant. Options generally vest over a period of two or four years from the date of grant. We have also granted performance-based options which will only vest when our Board of Directors determines we have achieved the specific performance goals. As of December 31, 2011, there were 1.2 million shares of performance-based options to be recognized when or if it is probable the specific performance goals are achieved. Options granted under the 2000 Plan expire no later than 10 years from the date of grant. The 2000 Plan expired in March 2010. There were 8.2 million stock option shares (including performance-based options) which had been granted prior to the plan’s expiration remaining outstanding as of December 31, 2011.
2000 Non-Employee Directors’ Stock Option Plan
In March 2000, we adopted the 2000 Non-Employee Directors’ Stock Option Plan, or the “Directors’ Plan”, and reserved a total of 300,000 shares of common stock for issuance thereunder. In May 2006, our stockholders approved an increase in the number of shares of common stock authorized for issuance under the Directors’ Plan by an additional 300,000 shares. Each non-employee director at the initial public offering date was granted a NSO to purchase 20,000 shares of common stock, and each non-employee director who subsequently becomes a director of Telik will be automatically granted a NSO to purchase 20,000 shares of common stock on the date on which such person first becomes a director. On February 20, 2008, our board of directors amended the Directors’ Plan such that upon the day immediately following each annual stockholder meeting each non-employee director
66
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
will automatically be granted a NSO to purchase 10,000 shares of common stock or an option to purchase an amount of shares prorated for the part of the year served as non-employee director. The exercise price of options under the Directors’ Plan will be equal to the fair market value of the common stock on the date of grant. The maximum term of the options granted under the Directors’ Plan is ten years. All grants under the Directors’ Plan will vest over a period of four years from date of grant, one fourth vesting one year after the date of the grant and thereafter the balance vesting monthly. The Directors’ Plan expired in March 2010. There were 245,000 stock option shares which had been granted prior to the plan’s expiration remaining outstanding as of December 31, 2011.
2000 Employee Stock Purchase Plan
In March 2000, we adopted the 2000 Employee Stock Purchase Plan, or the “Purchase Plan”. We reserved a total of 250,000 shares of our common stock for issuance under the Purchase Plan. In addition, the Purchase Plan provides for annual increases in the number of shares available for issuance under the Purchase Plan beginning January 1, 2001. The number of additional shares to be reserved automatically will be equal to the lesser of 150,000 shares, 1% of the outstanding shares on the date of the annual increase or such amount as may be determined by the Board of Directors. The Purchase Plan permits eligible employees to purchase common stock at a discount through payroll deductions during defined offering periods. The price at which the stock is purchased is equal to the lower of 85% of the fair market value of the common stock on the first day of the offering or 85% of the fair market value of our common stock on the purchase date. The weighted average per share fair value for stock purchase offerings under our Purchase Plan during 2011 and 2009 was $0.33 and $0.35. There were no participants enrolled in our new stock purchase offerings in 2010 under our Purchase Plan.
Reserved Shares
At December 31, 2011, shares of common stock reserved for future issuance is as follows:
| 2011 Equity incentive plan |
3,500,000 | |||
| 2000 Equity incentive plan |
8,213,449 | |||
| 2000 Non-employee directors’ stock option plan |
245,000 | |||
| 2000 Employee stock purchase plan |
584,879 | |||
|
|
|
|||
| 12,543,328 | ||||
|
|
|
67
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
Stock Option Plan Activity Summary
A summary of activity under our stock option plans is as follows:
| Shares Available for Grant |
Number of Options Outstanding |
Weighted average exercise price per share |
Weighted average remaining contractual term (in years) |
Aggregate intrinsic value (in thousands) |
||||||||||||||||
| Balance, December 31, 2009 |
2,502,136 | 12,596,938 | $ | 6.99 | ||||||||||||||||
| 1996 Plan shares expired |
(150,328 | ) | — | $ | 2.00 | |||||||||||||||
| 2000 Plan shares expired |
(4,514,257 | ) | — | (a | ) | |||||||||||||||
| 2000 Directors’ Plan shares expired |
(306,459 | ) | — | (b | ) | |||||||||||||||
| Authorized |
1,500,000 | — | — | |||||||||||||||||
| Forfeited or expired |
968,908 | (968,908 | ) | $ | 3.53 | |||||||||||||||
|
|
|
|
|
|||||||||||||||||
| Balance, December 31, 2010 |
— | 11,628,030 | $ | 7.28 | ||||||||||||||||
| 2000 Plan options expired |
— | (2,899,557 | ) | $ | 8.34 | |||||||||||||||
| Authorized |
3,500,000 | (c) | — | |||||||||||||||||
| Granted |
(2,526,000 | ) | 2,526,000 | $ | 0.69 | |||||||||||||||
| Exercised |
— | (270,024 | ) | $ | 0.80 | |||||||||||||||
| Canceled |
92,500 | (92,500 | ) | $ | 0.69 | |||||||||||||||
|
|
|
|
|
|||||||||||||||||
| Outstanding at December 31, 2011 |
1,066,500 | 10,891,949 | $ | 5.68 | 5.96 | $ | -0- | |||||||||||||
|
|
|
|
|
|||||||||||||||||
| Exercisable at December 31, 2011 |
7,885,055 | $ | 7.43 | 5.00 | $ | -0- | ||||||||||||||
|
|
|
|||||||||||||||||||
| (a) | 2000 Plan expired in March 2010. As of December 31, 2010, 4,514,257 shares expired of which 818,580 shares were from canceled stock options with a weighted average exercise price of $3.81. |
| (b) | 2000 Directors’ Plan expired in March 2010. All shares available for grant under this plan expired. |
| (c) | 2011 Equity Incentive Plan adopted in March 2011 and approved by stockholders in May 2011. |
The weighted average fair value of options granted during 2009 and 2011 was $0.62 and $0.55. There were no options granted in 2010. There were no options exercised during the year ended December 31, 2010 and 2009. The total intrinsic value of options exercised during the year ended December 31, 2011 was $29,000. The total fair value of shares vested during the years ended December 31, 2011, 2010 and 2009 was $1.5 million, $1.9 million and $3.2 million.
Stock-Based Compensation under ASC 718
Employee stock-based compensation expenses recognized in the years ended December 31, 2011, 2010 and 2009 were calculated based on awards ultimately expected to vest and have been reduced for estimated forfeitures. ASC 718 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
68
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
Total estimated stock-based compensation expense, related to all of our share-based payment awards, recognized under ASC 718 comprised of the following:
| Years Ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| (in thousands) | ||||||||||||
| Research and development |
$ | 715 | $ | 852 | $ | 420 | ||||||
| General and administrative |
843 | 1,240 | 1,811 | |||||||||
|
|
|
|
|
|
|
|||||||
| Stock-based compensation expense before taxes |
1,558 | 2,092 | 2,231 | |||||||||
| Related income tax benefits |
— | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Effect on net loss |
$ | 1,558 | $ | 2,092 | $ | 2,231 | ||||||
|
|
|
|
|
|
|
|||||||
Because we had a net operating loss carryforward as of December 31, 2011, no tax benefits for the tax deductions related to stock-based compensation expense were recognized in our Statements of Operations. Additionally, no incremental tax benefits were recognized from stock options exercised in the years ended December 31, 2011, 2010 and 2009, which would have resulted in a reclassification to reduce net cash provided by operating activities with an offsetting increase in net cash provided by financing activities. As of December 31, 2011, $854,000 of total unrecognized compensation costs, net of forfeitures, related to non-vested awards was expected to be recognized over a weighted average period of 1.29 years.
Valuation assumptions
Assumptions used in the Black-Scholes model were as follows:
| Stock Option Plans | Stock Purchase Plan | |||||||||||||||||||||||
| 2011 | 2010 | 2009 | 2011 | 2010 | 2009 | |||||||||||||||||||
| Weighted average expected stock price volatility |
104.9 | % | N/A | 99.8 | % | 88.2 | % | N/A | 140.3 | % | ||||||||||||||
| Weighted average risk-free interest rate |
2.14 | % | N/A | 2.65 | % | 0.26 | % | N/A | 0.67 | % | ||||||||||||||
| Weighted average expected life (in years) |
5.53 | N/A | 5.65 | 1.25 | N/A | 1.25 | ||||||||||||||||||
| Weighted average expected dividend yield |
— | — | — | — | — | — | ||||||||||||||||||
10. Income Taxes
We have incurred net losses since inception and, consequently, have not recorded any U.S. federal and state income taxes.
The provision for income taxes differs from the expected tax expense computed by applying the statutory federal income tax rate to loss before taxes as follows:
| Years Ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| (in thousands) | ||||||||||||
| Tax at Federal statutory rate |
$ | (4,088 | ) | $ | (8,406 | ) | $ | (8,056 | ) | |||
| State tax, net of federal income tax benefit |
(701 | ) | (1,424 | ) | (1,377 | ) | ||||||
| Research and development credit |
(51 | ) | (239 | ) | (601 | ) | ||||||
| Un-benefitted losses |
4,710 | 10,324 | 9,909 | |||||||||
| Other individually immaterial items |
130 | (255 | ) | 125 | ||||||||
|
|
|
|
|
|
|
|||||||
| Provision for taxes |
$ | — | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
69
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
Deferred income taxes reflect the net tax effects of net operating loss and tax credit carryovers and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of our deferred tax assets are as follows:
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| (in thousands) | ||||||||
| Deferred tax assets |
||||||||
| Net operating loss carryforwards |
$ | 11,476 | $ | 87,555 | ||||
| Tax credits carryforwards |
4,373 | 10,871 | ||||||
| Capitalized research expenses |
7,622 | 8,798 | ||||||
| Stock based compensation |
6,571 | 8,070 | ||||||
| Other |
1,518 | 2,198 | ||||||
|
|
|
|
|
|||||
| Total deferred tax assets |
31,560 | 117,492 | ||||||
| Valuation allowance |
(31,560 | ) | (117,492 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax assets |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
Realization of deferred tax assets is dependent upon the generation of future taxable income, the timing and amount of which are uncertain. Accordingly, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance decreased by $85.9 million at December 31, 2011 and increased by $7.1 million and $10.7 million at December 31, 2010 and 2009, respectively.
As of December 31, 2011, we had U.S. federal and state net operating losses of approximately $29.9 million and $22.4 million, respectively. If not utilized, these carryforwards will begin to expire beginning in 2012 and 2012 for federal and state purposes, respectively.
We have research credit carryforwards of approximately $6.6 million for state income tax purposes. California state research and development credits can be carried forward indefinitely.
As of December 31, 2011 the Company had net operating loss carry forwards of approximately $29.9 million and $22.4 million to offset federal and state future taxable income, respectively. These amounts have been reduced due to an ownership change which more likely than not has occurred, as defined by Sections 382 of the Internal Revenue Code, resulting in the decrease of a portion of the Company’s net operating loss carryforwards. In future periods such ownership could occur again further limiting availability of the Company’s net operating loss carryforwards.
70
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
Effective January 1, 2007, we adopted ASC 740-10-25. This interpretation clarifies the criteria for recognizing income tax benefits under ASC 740, “Accounting for Income Taxes,” and requires additional disclosures about uncertain tax positions. Under ASC 740-10-25 the financial statement recognition of the benefit for a tax position is dependent upon the benefit being more likely than not to be sustainable upon audit by the applicable taxing authority. If this threshold is met, the tax benefit is then measured and recognized at the largest amount that is greater than 50 percent likely of being realized upon ultimate settlement. Upon adoption of ASC 740-10-25 on January 1, 2007, we recognized a $14.2 million increase in our liability for unrecognized income tax benefits, and did not recognize a decrease to Retained Earnings. A reconciliation of the beginning and ending amount of the consolidated liability for unrecognized income tax benefits during the twelve-month period ended December 31, 2011 is as follows:
| 2011 | 2010 | |||||||
| (in thousands) | ||||||||
| Balance at January 1 |
$ | 9,214 | $ | 9,954 | ||||
| Additions for tax positions of prior years |
— | — | ||||||
| Additions for tax positions related to current year |
63 | 221 | ||||||
| Reductions for tax positions of prior years |
(1,132 | ) | (961 | ) | ||||
| Settlements during the current year |
NIL | NIL | ||||||
|
|
|
|
|
|||||
| Balance at December 31 |
$ | 8,145 | $ | 9,214 | ||||
Interest and penalty costs related to unrecognized tax benefits are classified as a component of “Income Tax Expense” in the accompanying statement of operations and the corresponding liability in “Income Taxes Payable” or “Prepaid Income Taxes” in the accompanying balance sheet. We, however, did not recognize any interest expense related to unrecognized tax benefits for the year ended December 31, 2011.
We file income tax returns in the U.S. federal jurisdiction and various state jurisdictions. We are subject to U.S. federal income tax examination for calendar tax years ending 2008 through 2011. Additionally, we are subject to various state income tax examinations for the 2006 through 2011 calendar tax years. The federal and U.S. state taxing authorities may choose to audit tax returns for tax years beyond the statue of limitation period due to significant tax attribute carryforwards from prior years, making adjustments only to carryforward attributes. The Company is not currently under audit in any major tax jurisdiction.
11. Subsequent Event
Reverse Stock Split
In February 2012, the Company’s board of directors approved a proposal, subject to stockholder approval, to decide whether or not to effect a reverse stock split of all outstanding shares of our common stock and stock options at an exchange ratio of one for fifteen, one for twenty, one for twenty five or one for thirty. Our board of directors has recommended that this proposal be presented to our stockholders for approval in the 2012 Annual Meeting of Stockholders which is scheduled to be held on March 28, 2012. If the reverse stock split proposal is approved by the stockholders and effected by our board of directors, our stated capital and additional paid-in-capital account on our balance sheet will be adjusted based on the exchange ratio of the reverse stock split.
71
Table of Contents
TELIK, INC.
NOTES TO FINANCIAL STATEMENTS
12. Quarterly Financial Information (unaudited)
Selected quarterly financial information is summarized below (in thousands except per share amounts):
SELECTED QUARTERLY FINANCIAL INFORMATION
| 2011 | 2010 | |||||||||||||||||||||||||||||||
| Quarter ended |
Dec. 31 | Sep. 30 | Jun. 30 | Mar. 31 | Dec. 31 | Sep. 30 | Jun. 30 | Mar. 31 | ||||||||||||||||||||||||
| Total revenues |
$ | — | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||||||||
| Operating costs and expenses: |
||||||||||||||||||||||||||||||||
| Research and development |
1,102 | 1,305 | 1,465 | 1,694 | 2,838 | 2,569 | 2,808 | 2,825 | ||||||||||||||||||||||||
| General and administrative |
1,401 | 1,550 | 1,546 | 1,994 | 2,374 | 2,101 | 2,197 | 2,558 | ||||||||||||||||||||||||
| Facility exit costs |
— | — | — | — | 5,360 | — | — | — | ||||||||||||||||||||||||
| Restructuring costs (1) |
— | — | — | — | 425 | — | — | — | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Total operating costs and expenses |
2,503 | 2,855 | 3,011 | 3,688 | 10,997 | 4,670 | 5,005 | 5,383 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Loss from operations |
(2,503 | ) | (2,855 | ) | (3,011 | ) | (3,688 | ) | (10,997 | ) | (4,670 | ) | (5,005 | ) | (5,383 | ) | ||||||||||||||||
| Interest and other income (expense), net (2) |
4 | 7 | 10 | 14 | 1,201 | 17 | 59 | 56 | ||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Net loss |
$ | (2,499 | ) | $ | (2,848 | ) | $ | (3,001 | ) | $ | (3,674 | ) | $ | (9,796 | ) | $ | (4,653 | ) | $ | (4,946 | ) | $ | (5,327 | ) | ||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Net loss per share, basic and diluted (3) |
$ | (0.05 | ) | $ | (0.05 | ) | $ | (0.06 | ) | $ | (0.07 | ) | $ | (0.18 | ) | $ | (0.09 | ) | $ | (0.09 | ) | $ | (0.10 | ) | ||||||||
| Weighted average shares used in computing net loss per share, basic and diluted |
54,142 | 53,962 | 53,956 | 53,690 | 53,614 | 53,553 | 53,522 | 53,465 | ||||||||||||||||||||||||
| (1) | Restructuring charges in 2010 related to workforce reduction by 11 positions or 28% of our workforce. |
| (2) | Interest and other income (expense), net in 2010 includes US Government Qualifying Therapeutic Discovery Project grants of $1.2 million received in the fourth quarter of 2010. |
| (3) | Net loss per share for each quarter are calculated as a discrete period; the sum of four quarters may not equal the calculated full year amount. |
72
Table of Contents
Exhibit Index
| Exhibit |
Description | |
| 3.1 | Amended and Restated Certificate of Incorporation. (2) | |
| 3.2 | Amended and Restated Bylaws. (10) | |
| 3.3 | Registrant’s Certificate of Designation of Series A Junior Participating Preferred Stock. (5) | |
| 3.4 | Registrant’s Certificate of Elimination with respect to Series A Junior Participating Preferred Stock. | |
| 4.1 | Specimen Common Stock Certificate. (1) | |
| 10.1 | Form of Indemnity Agreement. (1) (3) | |
| 10.2 | 2011 Equity Incentive Plan and related documents (3)(14) | |
| 10.3 | 2000 Equity Incentive Plan and related documents. (3) (4) | |
| 10.4 | 2000 Employee Stock Purchase Plan and Offering. (3) (4) | |
| 10.5 | 2000 Non-Employee Directors’ Stock Option Plan and Agreement. (3) (11) | |
| 10.6 | 1996 Stock Option Plan and forms of grant thereunder. (3) (4) | |
| 10.7 | 1988 Stock Option Plan and forms of grant thereunder. (3) (4) | |
| 10.8 | Form of Non-Plan Stock Option Agreement. (3) (4) | |
| 10.9 | Telik, Inc. Executive Officer Bonus Plan. (3)(9) | |
| 10.10 | Amended and Restated Employment Agreement between Michael M. Wick, M.D., Ph.D. and Telik, dated December 17, 2008, as amended. (3) (12) | |
| 10.11 | Lease between Telik and The Board of Trustees of the Leland Stanford Junior University, dated July 25, 2002. (6) | |
| 10.12 | Lease, between Telik and The Board of Trustees of the Leland Stanford Junior University, dated November 24, 2010. (15) | |
| 10.13 | Lease, between Telik and Aricent US, Inc., dated November 22, 2010. (15) | |
| 10.14* | Manufacturing Supply Agreement dated July 1, 2004, by and between Telik and Organichem Corporation. (7) | |
| 10.15 | Telik, Inc. Change of Control Severance Benefit Plan, dated February 21, 2003, amended December 17, 2008. (3) (12) | |
| 10.16 | Bonuses for Fiscal Year 2009 for Named Executive Officers. (3) (9) | |
| 10.17 | Transition and Release Agreement between Telik, Inc. and Dr. Stefan Ryser, Ph.D., dated as of June 12, 2009. (3)(13) | |
| 10.18 | At Market Issuance Sales Agreement, dated August 30, 2011, by and between Telik, Inc., and McNicoll, Lewis & Vlak LLC. (16) | |
| 14.1 | Telik, Inc. Code of Conduct. (8) | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. | |
| 24.1 | Power of Attorney (included on the signature pages hereto) | |
| 31.1 | Certification required by Rule 13a-14(a) or Rule 15d-14(a). | |
| 31.2 | Certification required by Rule 13a-14(a) or Rule 15d-14(a). | |
| 32.1 | Certification required by Rule 13a-14(b) or Rule 15d-14(b) and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. 1350). | |
| 101.INS** | XBRL Instance Document | |
| 101.SCH** | XBRL Taxonomy Extension Schema Document | |
| 101.CAL** | XBRL Taxonomy Extension Calculation Document | |
| 101.DEF** | XBRL Taxonomy Extension Definition Linkbase Document | |
| 101.LAB** | XBRL Taxonomy Extension Labels Linkbase Document | |
| 101.PRE** | XBRL Taxonomy Extension Presentation Link Document |
73
Table of Contents
| * | Confidential treatment has been granted for portions of this document. The information omitted pursuant to such confidential treatment order has been filed separately with the Securities and Exchange Commission. |
| ** | These interactive data files are deemed not filed or part of a registration statement or prospectus for purposes of sections 11 or 12 of the Securities Act of 1933, as amended, are deemed not filed for purposes of section 18 of the Securities Exchange Act of 1934, as amended, and otherwise are not subject to liability under these sections. |
| (1) | Incorporated by reference to exhibits to our Registration Statement on Form S-1, filed on April 3, 2000, as amended (File No. 333-33868). |
| (2) | Incorporated by reference to exhibits to our Annual Report on Form 10-K for the year ended December 31, 2001, as filed on March 27, 2002. |
| (3) | Management contract or compensatory arrangement. |
| (4) | Incorporated by reference to exhibits to our Registration Statement on Form S-8, as filed on August 30, 2000 (File No. 333-44826). |
| (5) | Incorporated by reference to exhibits to our Current Report on Form 8-K dated November 2, 2001, as filed on November 5, 2001. |
| (6) | Incorporated by reference to exhibits to our Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2002, as filed on November 13, 2002. |
| (7) | Incorporated by reference to exhibits to our Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2004, as filed on November 8, 2004. |
| (8) | Incorporated by reference to exhibits to our Annual Report on Form 10-K for the year ended December 31, 2003, as filed on March 5, 2004. |
| (9) | Incorporated by reference to Exhibit 10.8 to our Annual Report on Form 10-K for the year ended December 31, 2006, as filed on February 28, 2007. |
| (10) | Incorporated by reference to Exhibit 3.2 to our Current Report on Form 8-K dated December 11, 2007, as filed on December 14, 2007. |
| (11) | Incorporated by reference to Exhibit 10.4 to our Annual Report on Form 10-K for the year ended December 31, 2007, as filed on March 3, 2008. |
| (12) | Incorporated by reference to exhibits to our Current Report on Form 8-K dated December 17, 2008, as filed on December 23, 2008. |
| (13) | Incorporated by reference to Exhibit 10.20 to our Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2009, as filed on August 6, 2009. |
| (14) | Incorporated by reference to Appendix E to our Proxy Statement for the Annual Meeting of Stockholders, as filed on May 16, 2011. |
| (15) | Incorporated by reference to exhibits to our Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2011, as filed on August 12, 2011. |
| (16) | Incorporated by reference to Exhibit 10.17 to our Current Report on Form 8-K dated August 30, 2011, as filed on August 31, 2011. |
74