Attached files
| file | filename |
|---|---|
| EX-32.2 - EX-32.2 - AVEO PHARMACEUTICALS, INC. | d846897dex322.htm |
| EX-23.1 - EX-23.1 - AVEO PHARMACEUTICALS, INC. | d846897dex231.htm |
| EX-32.1 - EX-32.1 - AVEO PHARMACEUTICALS, INC. | d846897dex321.htm |
| EX-21.1 - EX-21.1 - AVEO PHARMACEUTICALS, INC. | d846897dex211.htm |
| EX-31.1 - EX-31.1 - AVEO PHARMACEUTICALS, INC. | d846897dex311.htm |
| EX-31.2 - EX-31.2 - AVEO PHARMACEUTICALS, INC. | d846897dex312.htm |
| EX-10.27 - EX-10.27 - AVEO PHARMACEUTICALS, INC. | d846897dex1027.htm |
| EXCEL - IDEA: XBRL DOCUMENT - AVEO PHARMACEUTICALS, INC. | Financial_Report.xls |
| EX-10.30 - EX-10.30 - AVEO PHARMACEUTICALS, INC. | d846897dex1030.htm |
| EX-10.28 - EX-10.28 - AVEO PHARMACEUTICALS, INC. | d846897dex1028.htm |
| EX-10.35 - EX-10.35 - AVEO PHARMACEUTICALS, INC. | d846897dex1035.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended: December 31, 2014
Or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number: 001-34655
AVEO PHARMACEUTICALS, INC.
(Exact Name of Registrant as Specified in Its Charter)
| Delaware | 04-3581650 | |
| (State or Other Jurisdiction of Incorporation or Organization) |
(I.R.S. Employer Identification No.) |
650 East Kendall Street
Cambridge, Massachusetts 02142
(Address of Principal Executive Offices) (zip code)
Registrant’s telephone number, including area code: (617) 299-5000
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class |
Name of each exchange on which registered | |
| Common Stock, $.001 par value | NASDAQ Global Market |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | ¨ | Accelerated filer | x | |||
| Non-accelerated filer | ¨ (Do not check if a smaller reporting company) | Smaller reporting company | ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of the registrant’s common stock, $0.001 par value per share (“Common Stock”), held by non-affiliates of the registrant, based on the last reported sale price of the Common Stock on the NASDAQ Global Market at the close of business on June 30, 2014, was $88,784,981.
The number of shares outstanding of the registrant’s Common Stock as of February 28, 2015: 52,138,394.
Documents incorporated by reference:
Portions of our definitive proxy statement for our 2015 annual meeting of stockholders are incorporated by reference into Part III of this Annual Report on Form 10-K.
Table of Contents
AVEO PHARMACEUTICALS, INC.
| Page No. | ||||||
| 2 | ||||||
| Item 1. |
2 | |||||
| Item 1A. |
30 | |||||
| Item 1B. |
54 | |||||
| Item 2. |
54 | |||||
| Item 3. |
55 | |||||
| Item 4. |
55 | |||||
| 56 | ||||||
| Item 5. |
56 | |||||
| Item 6. |
58 | |||||
| Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
59 | ||||
| Item 7A. |
83 | |||||
| Item 8. |
85 | |||||
| Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
122 | ||||
| Item 9A. |
122 | |||||
| Item 9B. |
125 | |||||
| 126 | ||||||
| Item 10. |
126 | |||||
| Item 11. |
126 | |||||
| Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
126 | ||||
| Item 13. |
Certain Relationships and Related Person Transactions, and Director Independence |
126 | ||||
| Item 14. |
126 | |||||
| 127 | ||||||
| Item 15. |
127 | |||||
| 128 | ||||||
Table of Contents
References to AVEO
Throughout this Form 10-K, the words “we,” “us,” “our” and “AVEO”, except where the context requires otherwise, refer to AVEO Pharmaceuticals, Inc. and its consolidated subsidiaries, and “our board of directors” refers to the board of directors of AVEO Pharmaceuticals, Inc.
Forward-Looking Information
This report contains forward-looking statements regarding, among other things, our future discovery and development efforts, our collaborations, our future operating results and financial position, our business strategy, and other objectives for our operations. You can identify these forward-looking statements by their use of words such as “anticipate,” “believe,” “estimate,” “expect,” “forecast,” “intend,” “plan,” “project,” “target,” “will” and other words and terms of similar meaning. You also can identify them by the fact that they do not relate strictly to historical or current facts. There are a number of important risks and uncertainties that could cause our actual results to differ materially from those indicated by forward-looking statements. These risks and uncertainties include those inherent in pharmaceutical research and development, such as adverse results in our drug discovery, preclinical trials and clinical development activities, our dependence on our existing and future strategic partners, our ability to obtain any necessary financing to conduct our planned activities, decisions made by the U.S. Food and Drug Administration and other regulatory authorities with respect to the development and commercialization of our drug candidates, our ability to obtain, maintain and enforce intellectual property rights for our drug candidates and other risk factors. Please refer to the section entitled “Risk Factors” in Part I—Item 1A of this report for a description of these risks and uncertainties. Unless required by law, we do not undertake any obligation to publicly update any forward-looking statements.
1
Table of Contents
| ITEM 1. | Business |
Overview
We are a biopharmaceutical company committed to developing targeted therapies through biomarker-driven insights to provide substantial improvements in patient outcomes where significant unmet medical needs exist. Our proprietary platform has delivered unique insights into cancer and related disease. Our strategy is to leverage these biomarker insights and partner resources to advance the development of our clinical pipeline. As further described below, we currently are exploring partnership opportunities to fund the further development of three of our four development programs, including our lead program for tivozanib.
Our development programs, which seek to advance our clinical stage assets, are as follows:
| • | Tivozanib: In 2006, we acquired exclusive rights to develop and commercialize tivozanib, outside of Asia pursuant to a license agreement we entered into with Kirin Brewery Co. Ltd. (now Kyowa Hakko Kirin), or KHK. Tivozanib is a potent, selective long half-life vascular endothelial growth factor tyrosine kinase inhibitor of all three vascular endothelial growth factors, or VEGF TKI. In a global, phase 3 clinical trial comparing the efficacy and safety of tivozanib with Nexavar® (sorafenib), an approved therapy, for first-line treatment of renal cell carcinoma, or RCC, the trial met its primary endpoint for progression-free survival, or PFS, but showed a non-statistically significant trend favoring the sorafenib arm in overall survival, or OS. Based on a review of our application for approval of the use of tivozanib for the treatment of first line advanced RCC, in June 2013, the U.S. Food and Drug Administration, or FDA, issued a Complete Response Letter informing us that they would not approve tivozanib at this time based on these study data. |
In August 2014, as discussed below under the heading “Strategic Partnerships,” our strategic collaboration with Astellas for the development of tivozanib terminated. Upon reversion back to AVEO of rights previously granted Astellas, we reevaluated our tivozanib regulatory and development strategy, as well as partnering opportunities. In November 2014, we filed a letter of intent with the European Medicines Agency, allowing us to evaluate the opportunity for submitting a Marketing Authorization Application for tivozanib for the treatment of RCC based on clinical trials conducted to date. Moreover, we are evaluating the opportunity to conduct an additional adequate and well-controlled randomized trial of tivozanib in the refractory RCC setting using PFS as the primary endpoint and OS as a secondary endpoint to address the OS concerns raised by the FDA from our tivozanib study. We plan to have discussion with the FDA to evaluate if such a study design coupled with the prior study findings would allow for an advanced RCC label for tivozanib and after such discussion, we will explore the opportunity for future clinical development in conjunction with our funding resources.
In November 2014, we entered into a research and exclusive option agreement with Ophthotech Corporation, or Ophthotech, under which we granted Ophthotech an option to develop and commercialize tivozanib for the potential diagnosis, prevention and treatment of non-oncologic diseases or conditions of the eye in humans.
Additionally, we recently announced results from a predefined biomarker analysis of our BATON-CRC study, a randomized phase 2 clinical trial of modified FOLFOX6, a commonly used chemotherapy, combined with tivozanib or Avastin® (bevacizumab), which both target angiogenesis signaling pathways, in first line treatment of metastatic colorectal cancer, or CRC. In this study, among prospectively defined biomarkers, patients with low neuropilin-1, a cell surface protein that modulates blood vessel development (which we refer to a NRP-1), showed an improved PFS versus patients with high NRP-1 in both treatment arms, supporting the value of NRP-1 as a potential prognostic marker for angiogenesis inhibitors. Further, patients with low serum NRP-1 demonstrated longer PFS when treated with tivozanib compared to bevacizumab, which suggests that first line colorectal cancer patients with low NRP-1 levels may benefit from treatment with tivozanib over bevacizumab, a standard of care in this disease.
2
Table of Contents
Based on the NRP-1 findings, we plan to discuss potential registration studies with the FDA and are currently exploring additional partnership opportunities to further the development of tivozanib in CRC. We are also evaluating the opportunity to pursue the registration of tivozanib for RCC in the European Union based on prior study findings.
We are currently exploring additional partnership opportunities to further the development of tivozanib.
| • | Ficlatuzumab: Ficlatuzumab is a potent Hepatocyte Growth Factor, or HGF, inhibitory antibody. HGF is the sole known ligand of the c-Met receptor which is believed to trigger many activities that are involved in cancer development and metastasis. We have completed two phase 1 clinical studies of ficlatuzumab administered as a single agent and in combination with erlotinib, a tyrosine kinase inhibitor, or TKI, of the epidermal growth factor receptor, or EGFR, and a phase 2 clinical study evaluating ficlatuzumab in combination with gefitinib, an EGFR TKI, in first line non-small cell lung cancer, or NSCLC. The phase 2 trial failed to demonstrate a statistically significant benefit in the intent-to-treat population. However, an exploratory analysis using a serum-based molecular diagnostic test, known as VeriStrat®, identified a sub-population of patients who experienced a progression free survival and overall survival benefit from the addition of ficlatuzumab to gefitinib. VeriStrat is commercially available to help physicians guide treatment decisions for patients with second line advanced NSCLC. Data from the exploratory analyses with VeriStrat prompted the development of a separate investigational companion diagnostic device called BDX004. Based upon the exploratory analyses, BDX004 may be indicative of a predictive biomarker for the combination of ficlatuzumab and EGFR TKI over EGFR TKI alone in the first line EGFR mutation patients who have been previously identified to not respond well to the current standard of care. |
In April 2014, we entered into a worldwide agreement with Biodesix, Inc. to develop and commercialize ficlatuzumab with BDX004, a serum based diagnostic test which has been derived from the VeriStrat test, employing the same methodology and data processing algorithms as VeriStrat, for use in a confirmatory clinical trial. Pursuant to the agreement, we are conducting a phase 2 proof of concept study of ficlatuzumab, which we refer to as the FOCAL study, in combination with erlotinib in first line advanced NSCLC patients who have an EGFR mutation selected using the BDX004 test to identify the patient subset that is most likely to benefit from the addition of ficlatuzumab to the EGFR TKI. Biodesix will fund up to $15 million of the cost of this study, as well as all of the costs associated with development and registration of BDX004, and any additional development, regulatory and commercial costs for ficlatuzumab will be shared equally. Subject to regulatory approval, AVEO will lead worldwide commercialization of ficlatuzumab.
| • | AV-203: AV-203 is a potent anti-ErbB3 specific monoclonal antibody with high ErbB3 affinity. We have observed potent anti-tumor activity in mouse models. AV-203 selectively inhibits the activity of the ErbB3 receptor and our preclinical studies suggest that neuregulin-1, or NRG1 (also known as heregulin), levels predict AV-203 anti-tumor activity in preclinical models. We have completed a phase 1 dose escalation study of AV-203, which established a recommended phase 2 dose of AV-203 at 20mg/kg intravenously every 2 weeks, demonstrated good tolerability and promising early signs of activity, and reached the maximum planned dose of AV-203 monotherapy. No anti-drug antibodies were detected, and pharmacokinetic results indicated a dose-proportional increase in levels of AV-203. The expansion cohort of this study among patients with a specific biomarker has been discontinued. We are seeking to pursue further clinical development of AV-203 with a strategic partner. |
| • | AV-380 Program: AV-380 is a potent humanized IgG1 inhibitory monoclonal antibody targeting growth differentiating factor-15, or GDF15, a divergent member of the TGF-ß family, for the potential treatment or prevention of cachexia. Cachexia is defined as a multi-factorial syndrome of involuntary weight loss characterized by an ongoing loss of skeletal muscle mass (with or without loss of fat mass) that cannot be fully reversed by conventional nutritional support and leads to progressive functional impairment. Cachexia is associated with various cancers as well as diseases outside of cancer including |
3
Table of Contents
| chronic kidney disease, or CKD, congestive heart failure or CHF, and chronic obstructive pulmonary disease, or COPD. We believe that AV-380 represents a unique approach to treating cachexia because it addresses key underlying mechanisms of the syndrome and focuses a significant area of patient need. It is estimated that approximately 30% of all cancer patients die due to cachexia and over half of cancer patients who die do so with cachexia present. (J Cachexia Sarcopenia Muscle 2010). In the United States alone, the estimated prevalence of cancer cachexia alone is over 400,000 patients (Am J Clin Nutr 2006). |
In September 2014, we presented the results from four preclinical studies of AV-380 in various in vivo cachexia models and in vitro assays at the 2nd Cancer Cachexia Conference held in Montreal Canada. Our research was also selected for presentation in an oral session at the conference. We believe that our research sets forth our proof of concept for GDF15, by demonstrating that GDF15 is elevated in cachectic animal models and patients versus non-cachectic, administration of GDF15 induces cachexia and inhibition of GDF15 reverses cachexia.
In connection with the AV-380 program, we have in-licensed certain patents and patent applications from St. Vincent’s Hospital in Sydney, Australia. We have demonstrated preclinical proof-of-concept for AV-380 in multiple cancer cachexia models and have completed cell line development and manufacturing of our first cGMP batch, in preparation for potential future clinical development. We are evaluating partnership opportunities to continue the development of AV-380.
Product Pipeline
We were founded with the goal of developing a fundamentally new kind of pre-clinical cancer model designed to overcome many of the limitations of traditional xenograft models, and thereby improve the probability of success in developing new cancer drugs. We utilized these novel models to identify and validate target genes that drive tumor growth, to identify drugs that can block the function of these targets, and to identify patients who are most likely to respond favorably to treatment with such drugs. Our cancer models, together with the various techniques we have developed to use these models to aid in the discovery and development of new cancer drugs, are collectively referred to as our Human Response Platform.
Tivozanib: Inhibitor of VEGF Receptors 1, 2 & 3
Tivozanib is a potent, selective long half-life inhibitor of all three VEGF receptors that is designed to optimize VEGF blockade while minimizing off-target toxicities. The demonstrated clinical results for tivozanib are supported by its core biochemical properties of potency, selectivity and long half-life inhibition of all three VEGF receptors. The potency of tivozanib across VEGF receptors 1, 2 and 3 provides a comprehensive blockade of the VEGF pathway. Its high level of selectivity for all three VEGF receptors is designed to minimize unintended side effects, such as fatigue, diarrhea and hand-foot syndrome, which are often associated with the currently approved therapies. Hypertension and dysphonia were the most commonly reported side effects in patients treated with tivozanib.
In 2012, we announced detailed data from our global, phase 3 clinical trial comparing the efficacy and safety of tivozanib with Nexavar® (sorafenib), an approved therapy, for first-line treatment of RCC. This phase 3 trial met its primary endpoint PFS but showed a non-statistically significant trend favoring the sorafenib arm in overall survival. Based on a review of our application for approval of the use of tivozanib for the treatment of first line advanced RCC, in June 2013, the U.S. Food and Drug Administration issued a Complete Response Letter informing us that they would not approve tivozanib at this time based on these study data.
In August 2014, our collaboration and license agreement with Astellas terminated, at which time all rights for the development and commercialization of tivozanib reverted to AVEO. We had entered into the collaboration and license agreement with Astellas in February 2011, pursuant to which we and Astellas shared responsibility for tivozanib, including expenses for continued development and any future commercialization of tivozanib, in North America and Europe. Upon reversion back to AVEO of rights previously granted Astellas, we reevaluated our tivozanib regulatory and development strategy, as well as partnering opportunities.
4
Table of Contents
We are currently evaluating the opportunity for submitting a Marketing Authorization Application for tivozanib with the European Medicines Agency for the treatment of RCC based on the trials conducted to date. In January 2015, we announced our receipt of confirmation from the European Medicine Agency that tivozanib is eligible for submission of an application for a European Union Marketing Authorization under the Agency’s centralized procedure for the treatment of RCC. Confirmation of eligibility for submission is not predictive of the European Medicines Agency’s approval of a Marketing Authorization Application. Tivozanib has previously been granted orphan drug designation in Europe for the treatment of RCC.
We also have evaluated tivozanib in additional clinical programs including our BATON (Biomarker Assessment of Tivozanib in ONcology) program, assessing biomarkers in solid tumors that may be predictive of clinical response to tivozanib in patients with metastatic colorectal cancer, and other clinical trials assessing locally recurrent or metastatic triple negative breast cancer.
The BATON-BC study in patients with breast cancer, led by AVEO, initiated patient enrollment in December 2012 in a randomized, double-blind, multicenter phase 2 clinical trial, evaluating the efficacy of tivozanib in combination with paclitaxel compared to placebo in combination with paclitaxel in patients with locally recurrent or metastatic triple negative breast cancer who have received no more than one systemic therapy for advanced or metastatic breast cancer. On January 30, 2014, we announced that we and Astellas jointly decided to discontinue the BATON-BC clinical trial, due to insufficient enrollment.
The BATON-CRC study, led by Astellas, which enrolled a total of 265 patients randomized 2 to 1, was an open-label, phase 2 study with a primary endpoint evaluating the superiority of tivozanib in combination with modified FOLFOX6, a standard chemotherapy, compared to bevacizumab in combination with modified FOLFOX6 as first-line treatment in patients with advanced metastatic colorectal cancer. On December 13, 2013, we announced that the study was unlikely to meet the primary endpoint in the intent-to-treat population and on February 14, 2014, we announced that we and Astellas agreed to discontinue this study. The data from the preplanned interim analysis of this study was presented at the European Society for Medical Oncology, or ESMO, on September 29, 2014. The final data through February 28, 2014, including predefined biomarker data from the study, will be presented at the American Association for Cancer Research, or AACR Tumor Angiogenesis and Vascular Normalization Conference on March 6, 2015.
An objective of the BATON-CRC study was the assessment of prospectively defined biomarkers that may be predictive of response in selected patient subpopulations. Among these, patients with low neuropilin-1 (NRP-1) showed an improved PFS versus patients with high NRP-1 in both treatment arms, supporting the value of NRP-1 as a potential prognostic marker for angiogenesis inhibitors. Further, patients with low serum NRP-1 demonstrated longer PFS when treated with tivozanib (17.9 months, n=52), compared to bevacizumab (11.2 months, n=28) (HR=0.380, p=0.0075). Patients with high NRP-1 had inferior PFS outcomes regardless of treatment assignment, with progression free survival of 7.3 months and 7.5 months for the tivozanib and bevacizumab arms, respectively. As soluble NRP-1 is known to bind to VEGF and is believed to inhibit VEGF binding to VEGF Receptor 2, we hypothesize that VEGF inhibitors may only be effective in patients with low serum NRP-1 levels, and that in patients with low serum NRP-1, a more complete blockade of VEGF pathway inhibition may be beneficial. Of note, exploratory biomarker analyses from two prior studies with tivozanib in RCC presented at the 17th Annual Symposium on Anti-Angiogenesis and Immune Therapies in February 2015 indicated that NRP-1 is a possible biomarker of tivozanib efficacy in patients with RCC. Based on the scientific rationale and biomarker findings, we are currently evaluating opportunities for further development of tivozanib in CRC patients with low NRP-1 levels.
We are evaluating the opportunity to conduct an additional adequate and well-controlled randomized trial of tivozanib in the refractory RCC setting using PFS as the primary endpoint and OS as a secondary endpoint to address the OS concerns raised by the FDA from our TIVO-1 study. We plan to have discussion with the FDA to evaluate if such a study design coupled with the prior study findings would allow for an advanced RCC label for tivozanib. Based on the NRP-1 findings, we plan to discuss potential registration studies with the FDA and are
5
Table of Contents
currently exploring additional partnership opportunities to further the development of tivozanib in CRC. We are also evaluating the opportunity to pursue the registration of tivozanib for RCC in the European Union based on prior study findings.
In November 2014, we entered into a Research and Exclusive Option Agreement with Ophthotech Corporation, pursuant to which we provided Ophthotech an exclusive option to enter into a definitive license agreement under which we would grant Ophthotech the right to develop and commercialize tivozanib outside of Asia for the potential diagnosis, prevention and treatment of non-oncologic diseases or conditions of the eye in humans. Pursuant to this option agreement, we granted to Ophthotech an exclusive, royalty free license or sublicense, as applicable, under our intellectual property rights solely to perform the research and development activities related to the use of tivozanib as set forth in the development plan during the option period described below. These activities include formulation work for ocular administration, preclinical research and the conduct of a phase 1/2a, proof of concept clinical trial of a product containing tivozanib in patients with wet age-related macular degeneration. Ophthotech may exercise its option at any time until the latest to occur of: (i) twelve (12) months after the achievement of a certain clinical efficacy milestones, (ii) ninety (90) days after the date Ophthotech is required to make certain clinical efficacy milestone payments, and (iii) thirty (30) days after AVEO and Ophthotech agree as to the definitive form of license agreement.
Ficlatuzumab: Hepatocyte Growth Factor (HGF) Inhibitory Antibody
Through the use of our Human Response Platform, our scientists identified the HGF/c-Met pathway as a significant driver of tumor growth. HGF is a protein that circulates in the blood and binds to and activates a receptor called c-Met. HGF is the sole known ligand of c-Met receptor, which is believed to trigger many activities that are involved in cancer development and metastasis. Altered HGF/c-Met signaling is observed in many tumors including lung, head and neck, gastric, bladder, breast, ovarian, prostate and colorectal cancers, certain sarcomas and in multiple myeloma and leukemias. There are no approved therapies that specifically target the HGF/c-Met pathway.
In September 2012, we presented results of the phase 2 portion of a phase 1b/2 clinical trial, which we refer to as the ficlatuzumab phase 2 trial (6162), testing a combination of ficlatuzumab with gefitinib, an epidermal growth factor receptor, or EGFR, tyrosine kinase inhibitor, randomized 1:1 versus gefitinib alone in patients with previously untreated locally advanced or metastatic non-small cell lung cancer, or NSCLC. Patients who demonstrated disease progression during treatment with gefitinib alone had the opportunity to be treated with ficlatuzumab in combination with gefitinib provided that safety was maintained and the patient continued to meet trial eligibility criteria. This randomized clinical trial, which was conducted in Asia, studied response rate and progression-free survival, or PFS, in distinct patient subsets: those with activating EGFR mutations and those with wild-type EGFR. In addition, we are evaluating patient outcome based on c-Met levels expressed in their tumors. The primary endpoint of the study was overall response rate, referred to as ORR; secondary endpoints included PFS, overall survival, or OS, and correlation of biomarkers with clinical activity. In the intent to treat, referred to as ITT, population, the addition of ficlatuzumab to gefitinib did not result in statistically significant improved ORR or PFS in Asian treatment-naïve NSCLC patients. The OS hazard ratio in the ITT population for ficlatuzumab plus gefitinib versus gefitinib monotherapy was 0.98 (95% CI 0.66, 1.46). The combination was well-tolerated, with no clinically meaningful differences in adverse event rates observed between the two arms.
In September 2014, at the 2014 Congress of the European Society for Medical Oncology, or ESMO, we presented the results of our exploratory analysis using a serum-based molecular diagnostic test to identify a patient sub-population that experienced a progression free survival and overall survival benefit on the combination therapy in the ficlatuzumab phase 2 trial. The results suggest that VeriStrat, a serum protein test that is commercially available to help physicians guide treatment decisions for patients with NSCLC, may be selective of positive clinical response for ficlatuzumab plus gefitinib over gefitinib alone. For this retrospective exploratory analysis, 180 pre-treatment serum samples analyzed with VeriStrat and were assigned a label of either “VeriStrat Good” (VSG) or “VeriStrat Poor” (VSP) (VSG=145, VSP=35). While the study failed to
6
Table of Contents
demonstrate improved OS or PFS over gefitinib alone in the intent-to-treat population, the addition of ficlatuzumab to gefitinib provided significant clinical benefit to the VSP subgroup.
Based on this data, in April 2014, we entered into a worldwide agreement with Biodesix, Inc. to develop and commercialize HGF inhibitory antibody ficlatuzumab, with Biodesix’s proprietary companion diagnostic test, BDX004, a serum protein test derived from Veristrat. Pursuant to this agreement, we are conducting the FOCAL study, a phase 2, global, randomized, double-blind, placebo controlled clinical study, evaluating ficlatuzumab, our HGF inhibitory antibody, in combination with erlotinib (Tarceva®), an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR TKI) in first line EGFR-mutated NSCLC patients. BDX004 will be used to select patients for entry into the trial.
In November 2011, we entered into an agreement with Boehringer Ingelheim International GmbH, or Boehringer Ingelheim, for large-scale process development and clinical manufacturing of ficlatuzumab. In connection with the agreement, Boehringer Ingelheim is producing ficlatuzumab at its biopharmaceutical sites in Fremont, California (drug substance) and Beberach, Germany (drug product). We have retained all rights to the development and commercialization of ficlatuzumab under our agreement with Boehringer Ingelheim.
AV-203: Anti-ErbB3 Antibody
Through the use of our Human Response Platform, we identified the importance of the ErbB3 receptor in tumor growth. ErbB3 belongs to a family of proteins that also includes epidermal growth factor receptor, or EGFR, and HER2, all of which have been implicated in promoting the growth of significant numbers of tumor types.
ErbB3 is believed to be an important receptor regulating cancer cell growth and survival, and high ErbB3 levels have been shown to correlate with poor prognoses in several tumor types. It has also been implicated in resistance to certain drugs which target EGFR in lung cancer and with resistance to radiotherapy. AV-203 inhibits the activity of the ErbB3 receptor and our preclinical studies suggest that neuregulin-1, or NRG1, levels predict AV-203 anti-tumor activity and we have filed a U.S. patent application relating to a method of predicting tumor response to ErbB3 inhibitors based on NRG1 levels.
In March 2014, we amended our option and license agreement with Biogen Idec MA Inc., or Biogen, regarding the development and commercialization of our ErbB3-targeted antibodies for the potential treatment and diagnosis of cancer and other diseases in humans. Pursuant to the amendment, Biogen agreed to terminate its rights and obligations under our agreement, including Biogen’s option to (i) obtain a co-exclusive (with AVEO) license to develop and manufacture ErbB3 targeted antibodies and (ii) obtain exclusive commercialization rights to ErbB3 products in countries in the world other than North America. Pursuant to the amendment, we are obligated to pay Biogen a specified percentage of milestone payments received by us from future partnerships after March 28, 2016 and single digit royalty payments on net sales related to the sale of ErbB3 products, up to cumulative maximum amount of $50 million. As a result, we retain worldwide rights to AV-203, a clinical stage ErbB3-targeted antibody and are obligated to in good faith use reasonable efforts to seek a collaboration partner for the purpose of funding further development and commercialization of ErbB3 targeted antibodies. We are currently exploring partnership opportunities to advance the clinical development of AV-203.
AV-380 Program in Cachexia
In 2012, we initiated a program focusing on cachexia, which we now refer to as our AV-380 program. AV-380 is a potent humanized IgG1 inhibitory monoclonal antibody targeting growth differentiating factor-15, or GDF15, a divergent member of the TGF-ß family, for the potential treatment or prevention of cachexia. Cachexia is a serious and common complication of advanced cancer and a number of chronic diseases. It is defined as a multi-factorial syndrome of involuntary weight loss characterized by an ongoing loss of skeletal muscle mass (with or without loss of fat mass) that cannot be fully reversed by conventional nutritional support and leads to progressive functional impairment. The pathophysiology is characterized by a negative protein and energy
7
Table of Contents
balance driven by a variable combination of reduced food intake and abnormal metabolism. Other symptoms or conditions associated with cachexia include anemia, breathing difficulties, edema, insulin resistance, muscle weakness/asthenia, and fatigue.
In September 2014, we presented the results from four preclinical studies of AV-380 in various in vivo cachexia models and in vitro assays at the 2nd Cancer Cachexia Conference held in Montreal Canada. Our research was also selected for presentation in an oral session at the conference. We believe our research set forth our proof of concept for GDF15, by demonstrating that GDF15 is elevated in cachectic animal models and patients versus non-cachectic, administration of GDF15 induces cachexia and inhibition of GDF15 reverses cachexia.
In connection with the AV-380 program, we have in-licensed certain patents and patent applications from St. Vincent’s Hospital in Sydney, Australia. We have demonstrated preclinical proof-of-concept for AV-380 in multiple cancer cachexia models and have completed cell line development and manufacturing of our first cGMP batch, in preparation for potential future clinical development.
We believe that cachexia represents a significant area of patient need, particularly in cancer patients. Weight loss during cancer treatment is associated with more chemotherapy-related side effects, fewer completed cycles of chemotherapy, a reduction in response to therapy and decreased survival rates (J Gastroenterol 2013; Eur J Cancer 1998; Br J Cancer 2004). In a cohort of over 3,000 patients in the U.S. studied by the Eastern Cooperative Oncology Group, or ECOG, the prevalence of weight loss even before starting chemotherapy was observed to be substantial across several cancers: over 80% in pancreatic and gastric cancers and over 50% in prostate, colorectal and lung cancers (Am Med Journal 1980). It is estimated that more than 30% of all cancer patients die due to cachexia and over half of cancer patients who die do so with cachexia present. (J Cachexia Sarcopenia Muscle 2010). In the United States alone, the estimated prevalence of cancer cachexia is over 400,000 patients (Am J Clin Nutr 2006).
We are evaluating opportunities for partnerships to continue the development of AV-380.
Competition
The biotechnology and pharmaceutical industries are highly competitive. There are many pharmaceutical companies, biotechnology companies, public and private universities and research organizations actively engaged in the research and development of products that may be similar to our products. A number of multinational pharmaceutical companies, as well as large biotechnology companies, including Roche Laboratories, Inc., or Roche, Pfizer Inc., or Pfizer, Bayer HealthCare AG, or Bayer, Sanofi-Aventis, US, LLC, Amgen, Inc., Eli Lilly and Company, or Lilly, GlaxoSmithKline plc, or GSK, GTx, Inc., Helsinn and XBiotech are pursuing the development or are currently marketing pharmaceuticals that target VEGF, HGF, ErbB3, and cachexia, or other oncology pathways on which we are focusing. It is probable that the number of companies seeking to develop products and therapies for the treatment of unmet needs in the lives of people with cancer will increase.
Many of our competitors, either alone or with their strategic partners, have greater financial, technical and human resources than we do and greater experience in the discovery and development of product candidates, obtaining FDA and other regulatory approvals of products, and the commercialization of those products. Accordingly, our competitors may be more successful than we may be in obtaining approval for drugs and achieving widespread market acceptance. Our competitors’ drugs may be safer and more effective, or more effectively marketed and sold, than any drug we may commercialize and may render our product candidates obsolete or non-competitive before we can recover the expenses of developing and commercializing any of our product candidates. We anticipate that we will face intense and increasing competition as new drugs enter the market and advanced technologies become available.
8
Table of Contents
Tivozanib Competition
There are currently ten FDA-approved drugs in oncology which target the VEGF receptors. Seven of the FDA-approved VEGF pathway inhibitors are oral small molecule receptor tyrosine kinase inhibitors, or TKIs. Nexavar (sorafenib) and Stivarga (regorafenib) are marketed by Bayer and Onyx, a subsidiary of Amgen, Sutent (sunitinib) and Inlyta (axitinib) are marketed by Pfizer, and Votrient (pazopanib) is marketed by GSK. Most of these approved VEGFR TKIs are not specific to the VEGF 1, 2 and 3 receptors. Nexavar is approved for advanced RCC and unresectable hepatocellular cancer. Stivarga is approved for refractory metastatic colorectal cancer, or mCRC, and refractory gastrointestinal stromal tumors, or GIST. Sutent is approved for advanced RCC, GIST, and progressive, well-differentiated pancreatic neuroendocrine tumors. Inlyta is approved for advanced RCC after failure of one prior systemic therapy. Votrient is approved for advanced RCC and advanced soft tissue sarcoma after prior chemotherapy. Caprelsa (vandetanib), marketed by AstraZeneca, and Cometriq (cabozantinib), marketed by Exelixis, are approved for medullary thyroid carcinoma.
Avastin (bevacizumab), marketed by Roche/Genentech, is an infused monoclonal antibody approved in combination with other anti-cancer agents for the treatment of mCRC, non-squamous non-small cell lung cancer, and metastatic RCC. It is also approved as a monotherapy for the treatment of glioblastoma in patients with progressive disease following prior therapy. Zaltrap (zif-aflibercept), marketed by Sanofi and Regeneron, is a VEGF-trap molecule that binds to multiple circulating VEGF factors, and is approved in combination with standard chemotherapy agents for treatment of second line mestatatic CRC. Cyramza (ramucirumab), marketed by Lilly, is an antibody that binds to the VEGFR-2 receptor that is approved for the treatment of advanced gastric or gastro-esophageal junction adenocarcinoma and in combination with docetaxel for the treatment of NSCLC.
Many of the approved VEGF pathway inhibitor agents are in ongoing development in additional cancer indications. Additionally, we are aware of a number of companies that have ongoing programs to develop both small molecules and biologics that target the VEGF pathway.
Ficlatuzumab Competition
We believe the products that are considered competitive with ficlatuzumab include those agents targeting the HGF/c-Met pathway. The agents exclusively targeting this pathway consist of the only other HGF-targeted antibody, Amgen’s AMG-102 (rilotumumab), initiated in a phase 3 clinical trial (which has been discontinued), as well as Lilly’s c-Met receptor antibody LY-2875358, currently in multiple phase 2 trials. In addition, Roche has conducted multiple phase 3 trials for a c-Met receptor antibody onartuzumab (MetMAb/ 5D5 Fab). Roche announced that an independent data monitoring committee recommended that its phase 3 trial of onartuzumab in second and third line NSCLC be stopped due to lack of efficacy.
Other marketed or late clinical-stage drugs which target the HGF/c-Met pathway, though not exclusively, include Pfizer’s PF-2341066 (Xalkori, crizotinib), Exelixis Inc.’s XL-184 (Cometriq, cabozantinib), ArQule, Inc.’s/ Daiichi Sankyo, Inc.’s ARQ-197 (tivantanib), Mirati Therapeutics’ (formerly MethylGene) MGCD-265, Eisai Co. Ltd.’s E-7050 (golvatinib), Exelixis Inc.’s and GSK’s XL-880 (foretinib), Incyte Corp.’s and Novartis’s INCB-028060 and Sanofi-Aventis’s SAR-125844, EMD Serono’s MSC2156119J, Amgen Astellas BioPharma’s AMG 337, and Bristol-Myers Squibb Company’s and Aslan Pharmaceuticals’ BMS-777607.
AV-203 Program Competition
We believe the most direct competitors to our AV-203 program are monoclonal antibodies that specifically target the ErbB3 receptor, including Merrimack Pharmaceuticals, Inc.’s MM-121, which is currently in phase 2 clinical development, and Daiichi Sankyo, Inc.’s and Amgen, Inc.’s patritumab (AMG-888), which recently entered phase 3 clinical development for non-small cell lung cancer. Other clinical-stage ErbB3-specific competitors include Roche’s RG-7116, Novartis’s LJM716, Regeneron’s REGN1400, GSK’s GSK-2849330 and Kolltan’s KTN-3379. Clinical stage competitor’s targeting ErbB3 in addition to other targets include Roche’s MEHD7945A, and Merrimack Pharmaceuticals, Inc.’s MM-111 and MM-141.
9
Table of Contents
AV-380 Program in Cachexia Competition
Only a limited number of agents have been approved for the treatment or prevention of cachexia caused by any disease. In the United States, Megace is the only approved agent for the treatment of cachexia (in patients with the diagnosis of AIDS). Megace and medroxyprogesterone are approved for cancer cachexia in Europe. Three agents have recently completed or are currently being studied in phase 3 trials. One agent, GTx, Inc.’s selective androgen receptor modulator, or SARM, called enobosarm (GT-024) recently completed two phase 3 trials for the prevention and treatment of muscle wasting in newly diagnosed locally advanced or metastatic non-small cell lung cancer patients. Another agent in phase 3 trials is Helsinn’s anamorelin, which has been studied in newly diagnosed locally advanced non-small cell lung cancer patients who have cachexia, but overall survival data has not yet been released. A third agent, XBiotech’s xilonix (MABp1), is in a phase 3 trial for metastatic colorectal cancer patients who are cachectic and refractory to standard therapies.
A number of agents with different mechanisms of action have completed or are currently being studied in phase 2 trials in cachexia or muscle wasting. Agents targeting the muscle regulatory molecule myostatin include Lilly’s LY2495655, Regeneron’s REGN-1033, and Atara Biosciences’ PINTA 745. Novartis is currently studying bimagrumab (BYM-338), an agent targeting the activin receptor. Drugs with other mechanisms currently in or recently completing phase 2 clinical trials include Alder Biosciences’ clazakizumab (ALD-518, targeting IL-6), PsiOxus’ MT-102 (dual acting catabolic/anabolic transforming agent), Acacia’s APD-209 (progestin/ß2 antagonist) and Ohr Pharmaceuticals’ OHR118 (cytoprotectant/immunomodulator).
Strategic Partnerships
We are party to the following collaboration and license agreements:
Ophthotech Corporation
In November 2014 we entered into a Research and Exclusive Option Agreement, or Option Agreement, with Ophthotech Corporation pursuant to which we provided Ophthotech an exclusive option to enter into a definitive license agreement whereby we would grant Ophthotech the right to develop and commercialize our VEGF factor tyrosine kinase inhibitor, tivozanib, outside of Asia for the potential diagnosis, prevention and treatment of non-oncologic diseases or conditions of the eye in humans.
Pursuant to this Option Agreement, we granted to Ophthotech an exclusive, royalty free license or sublicense, as applicable, under intellectual property rights controlled by us solely to perform the research and development activities related to the use of tivozanib for the specific purposes outlined in the agreement during the option period (as defined below). These activities include formulation work for ocular administration, preclinical research and the conduct of a phase 1/2a, proof of concept clinical trial of a product containing tivozanib in patients with wet age-related macular degeneration, or the POC Study.
Ophthotech paid us $500,000 in consideration for the grant of the option. Such amount is non-refundable and not creditable against any other amounts due under the agreement. We are obligated to make available to Ophthotech, at no cost to Ophthotech, certain quantities of tivozanib hydrochloride solely for conducting its option period research including manufacturing additional quantities of tivozanib in the event stability data indicates that the current supply will expire prior to the end of February 2017.
During the option period, if Ophthotech elects to continue the development of tivozanib for non-oncologic diseases of the eye, we are entitled to receive a one-time milestone payment of $2.0 million upon acceptance of the first Investigational New Drug application for the purpose of conducting a human clinical study of tivozanib in ocular diseases, which we refer to as the IND Submission Milestone Payment. We are also entitled to receive a one-time milestone payment of $6.0 million, which we refer to as the Clinical Efficacy Milestone Payment, on the earlier of (a) December 31, 2016 and (b) the later to occur of: (i) the achievement of a clinical milestone in the POC Study, or the Clinical Efficacy Milestone and (ii) the earlier of (A) the date twelve (12) months after our
10
Table of Contents
and Ophthotech’s agreement as to the form and substance of the KHK Amendment (as defined below) or (B) the date ninety (90) days after the entry into the KHK Amendment, subject to our right to terminate the Option Agreement on 90 days’ written notice (the date on which such payment is due, referred to as the Clinical Efficacy Milestone Payment Trigger Date).
Ophthotech may exercise the option at any time until the latest to occur of: (i) twelve (12) months after the achievement of the Clinical Efficacy Milestone, (ii) ninety (90) days after the Clinical Efficacy Milestone Payment Trigger Date, and (iii) thirty (30) days after we and Ophthotech agree as to the definitive form of license agreement, which we refer to as the Option Period.
During the Option Period, we will not grant a license to any third party that would preclude us from being able to grant to Ophthotech the rights and licenses that are contemplated by the definitive license agreement, and we will not engage in any research, development or commercialization of tivozanib in the field covered by the contemplated definitive license agreement, except as specified in the Option Agreement.
The terms of the Option Agreement are subject to our obligations to Kirin Brewery Co. Ltd. (now Kyowa Hakko Kirin), or KHK, under a license agreement entered into by us with KHK in 2006, pursuant to which we acquired exclusive rights to develop and commercialize tivozanib for all human diseases outside of Asia, referred to as the KHK License Agreement. A percentage of all payments received by us under the Option Agreement and any definitive license agreement must be paid to KHK. We are required to maintain the KHK Agreement in effect, and not enter into any amendment or termination thereof that would adversely affect our rights, during the option period.
During the option period, we and Ophthotech are obligated to negotiate in good faith the form and substance of a definitive license agreement, as well as the form and substance of an amendment to our license agreement with KHK (such amendment referred to as the KHK Amendment) to modify certain rights and obligations of the parties and sublicensees thereunder, particularly with respect to rights to improvements that are not specifically related to tivozanib, and regulatory affairs matters.
Upon exercise of the option, Ophthotech is required to pay us a one-time option exercise fee of $2.0 million in addition to the IND Submission Milestone Payment if such payment has not then been previously paid. If upon exercise of the option, the Clinical Efficacy Milestone Payment Trigger Date has not yet occurred, we shall be entitled to the Clinical Efficacy Milestone Payment at such time that the Clinical Efficacy Milestone Payment Date does occur if the license agreement remains in effect as of such date. The license agreement, if entered into upon Ophthotech’s exercise of the Option, will provide for us to be entitled to receive (i) $10.0 million upon meeting certain efficacy and safety endpoints in phase 2 clinical trials that would enable the commencement of a phase 3 clinical trial, (ii) $20.0 million upon marketing approval in the United States, (iii) $20.0 million upon marketing approval in the UK, Germany, Spain, Italy and France and (iv) up to $45.0 million in sales-based milestone payments. Ophthotech would also be required to pay tiered, double digit royalties, up to a mid-teen percentage, on net sales of tivozanib or products containing tivozanib.
Either party may terminate the Option Agreement in the event of an uncured material breach of the Option Agreement by the other party which remains uncured for a period of ninety (90) days (or thirty (30) days for a breach relating to non-payment), or upon bankruptcy or like proceedings relating to the other party. Ophthotech may terminate the Option Agreement at any time upon ninety (90) days’ prior written notice to us. In addition, we may terminate the Option Agreement upon thirty (30) days’ prior written notice to Ophthotech if Ophthotech challenges certain patents controlled by us related to tivozanib. Unless terminated as provided above, the Option Agreement will expire upon the expiration of the option or the entry into the definitive license agreement.
Biodesix
In April 2014, we entered into a worldwide agreement with Biodesix to develop and commercialize our HGF inhibitory antibody ficlatuzumab, with BDX004, a proprietary companion diagnostic test developed by
11
Table of Contents
Biodesix and based upon the exploratory analyses with VeriStrat®, a serum protein test that is commercially available to help physicians guide treatment decisions for patients with advanced NSCLC.
Under the agreement, we granted Biodesix perpetual, non-exclusive rights to certain intellectual property, including all clinical and biomarker data related to ficlatuzumab, to develop and commercialize BDX004. Biodesix granted us perpetual, non-exclusive rights to certain intellectual property, including diagnostic data related to BDX004, with respect to the development and commercialization of ficlatuzumab; each license includes the right to sublicense, subject to certain exceptions. Pursuant to a joint development plan, as monitored by a joint steering committee, we retain primary responsibility for clinical development of ficlatuzumab in a phase 2 proof of concept, or POC, clinical study of ficlatuzumab for non-small cell lung cancer, in which BDX004, a diagnostic test derived from VeriStrat will be used to select clinical trial subjects, referred to as the FOCAL study. The FOCAL study will be fully funded by Biodesix up to a maximum of $15 million, referred to as the Cap. Biodesix will also be responsible for all of the costs associated with development and registration of BDX004. After the Cap is reached, we and Biodesix will share equally in the costs of the FOCAL study, and we and Biodesix will each be responsible for 50% of development and regulatory costs associated with all future ficlatuzuamab clinical development trials agreed-upon by Biodesix and us, including all milestone payments and royalties payable to third parties, if any.
Pending marketing approval of ficlatuzumab and subject to a commercialization agreement to be entered into after receipt of results from the FOCAL study, each party would share equally in commercialization profits and losses, subject to our right to be the lead commercialization party.
Biodesix is solely responsible for the BDX004 development costs, as well as BDX004 sales and marketing costs. Subject to and following the approval of the BDX004 test as a companion diagnostic for ficlatuzumab, Biodesix has agreed to make the BDX004 test available and use commercially reasonable efforts to seek reimbursement in all geographies where ficlatuzumab is approved. We have agreed to reimburse Biodesix a pre-specified amount, under certain circumstances for VeriStrat tests performed.
Prior to the first commercial sale of ficlatuzumab and after the earlier of (i) the Cap being reached or (ii) the completion of the FOCAL study, each party has the right to elect to discontinue participating in further development or commercialization efforts with respect to ficlatuzumab, which is referred to as an Opt-Out. If either we or Biodesix elects to Opt-Out, with such party referred to as the Opting-Out Party, then the Opting-Out Party shall not be responsible for any future costs associated with developing and commercializing ficlatuzumab other than any ongoing clinical trials. After election of an Opt-Out, the non-opting out party shall have sole decision-making authority with respect to further development and commercialization of ficlatuzumab. Additionally, the Opting-Out Party shall be entitled to receive, if ficlatuzumab is successfully developed and commercialized, a royalty equal to 10% of net sales of ficlatuzumab throughout the world, if any, subject to offsets under certain circumstances.
If Biodesix elects to Opt-Out, it will continue to be responsible for its development and commercialization obligations with respect to BDX004. If we elect to Opt-Out, we will continue to make the existing supply of ficlatuzumab available to Biodesix for the purposes of enabling Biodesix to complete the development of ficlatuzumab, and Biodesix will have the right to commercialize ficlatuzumab.
Prior to any Opt-Out, the parties shall share equally in any payments received from a third party licensee; provided, however, after any Opt-Out, the Opting-Out Party shall be entitled to receive only a reduced portion of such third party payments. The agreement will remain in effect until the expiration of all payment obligations between the parties related to development and commercialization of ficlatuzumab, unless earlier terminated.
St. Vincent’s Hospital
In July 2012, we entered into a license agreement with St. Vincent’s Hospital Sydney Limited, which we refer to as St. Vincent’s, under which we obtained an exclusive, worldwide license to research, develop,
12
Table of Contents
manufacture and commercialize products for therapeutic applications that benefit from inhibition or decreased expression or activity of MIC-1, which is also known as GDF15. We believe GDF15 is a novel target for cachexia and we are using this license in our AV-380 program for cachexia. Under the agreement, we have the right to grant sublicenses subject to certain restrictions. We have a right of first negotiation to obtain an exclusive license to certain improvements that St. Vincent’s or third parties may make to licensed therapeutic products. Under the license agreement, St. Vincent’s also granted us non-exclusive rights for certain related diagnostic products and research tools.
Under the license agreement, we are obligated to use diligent efforts to conduct research and clinical development and commercially launch at least one licensed therapeutic product, and to maximize profits from licensed therapeutic products for the benefit of us and St. Vincent’s. Subject to certain conditions, we have also agreed to achieve specified research, development and regulatory milestones by specified dates. If we do not achieve a given milestone by the agreed date, we have the option of paying the amount we would have been obligated to pay had we timely achieved the milestone, and, if we do so, St. Vincent’s will not have the right to terminate the license agreement based on our failure to timely achieve such milestone.
We have also agreed that, for as long as there is a valid claim in the licensed patents, we will not, and we will ensure that our affiliates and our sublicensees do not, develop or commercialize any product, other than a licensed therapeutic product, for the treatment, prevention or prophylaxis of cachexia, decreased appetite or body weight, that binds to GDF15 or the GDF15 receptor and that is a GDF15 antagonist, and will not license or induce any other person to do the same.
In connection with entering into the license agreement with St. Vincent’s, we paid St. Vincent’s an upfront license fee of $0.7 million and a low five-figure amount to reimburse St. Vincent’s for patent-related expenses it incurred with respect to a specified licensed patent.
Under our license agreement with St. Vincent’s, we may be required to:
| • | make milestone payments, up to an aggregate total of $9.2 million, upon achievement of specified research, development and regulatory milestones for the first three indications for licensed therapeutic products, some of which payments may be increased by a mid to high double-digit percentage rate for milestones payments made after we grant any sublicense under the license agreement, depending on the sublicensed territory or territories; |
| • | pay tiered royalty payments equal to a low-single-digit percentage of any net sales we or our sublicensees make of licensed therapeutic products. The royalty rate escalates within the low-single-digit range during each calendar year based on increasing licensed therapeutic product sales during such calendar year. Our royalty payment obligations for a licensed therapeutic product in a particular country end on the later of 10 years after the date of first commercial sale of such licensed therapeutic product in such country or expiration of the last-to-expire valid claim of the licensed patents covering such licensed therapeutic product in such country, and are subject to offsets under certain circumstances; |
| • | pay St. Vincent’s sublicensing fees of up to an aggregate amount in the low-to-mid six-digits, depending on the sublicensed territory or territories, at the time we grant any sublicense; and |
| • | reimburse St. Vincent’s for some or all of the reasonable costs and expenses it incurs in patent management, filing, prosecuting and maintaining the licensed patents. |
The license agreement will remain in effect until the later of 10 years after the date of first commercial sale of licensed therapeutic products in the last country in which a commercial sale is made, or expiration of the last-to-expire valid claim of the licensed patents, unless we elect, or St. Vincent’s elects, to terminate the license agreement earlier.
St. Vincent’s has the right to terminate the agreement due to any patent-related challenge by us, our affiliates or any sublicensee, or if we or our affiliates or any sublicensee cause or induce any other person to make a patent-related challenge, and such challenge continues after a specified cure period.
13
Table of Contents
We have the right to terminate the agreement on six months’ notice if we terminate our GDF15 research and development programs as a result of the failure of a licensed therapeutic product in pre-clinical or clinical development, or if we form the reasonable view that further GDF15 research and development is not commercially viable, and we are not then in breach of any of our obligations under the agreement. If we form the reasonable view that further GDF15 research and development is not commercially viable and terminate the agreement before we start a phase 1 clinical trial on a licensed therapeutic product, we will be required to pay St. Vincent’s a low-to-mid six-figure termination payment.
Any termination of the agreement, in whole or in part, will result in a loss of our rights to the relevant licensed patents and know-how. If St. Vincent’s terminates the agreement in its entirety due to our breach, insolvency or a patent-related challenge, or we terminate the agreement due to a development failure or lack of commercial viability, as described above, St. Vincent’s will have a non-exclusive license from us to certain intellectual property rights and know-how relating to the licensed therapeutic products, and we must transfer to St. Vincent’s certain then-existing regulatory approvals and related documents for the licensed therapeutic products.
Astellas Pharma
In February 2011, we entered into a collaboration and license agreement with Astellas and certain of its indirect wholly-owned subsidiaries pursuant to which we and Astellas made plans to develop and seek to commercialize tivozanib for the treatment of a broad range of cancers. On February 12, 2014, Astellas exercised its right to terminate the agreement. The termination of the agreement became effective August 11, 2014, at which time tivozanib rights returned to us. In accordance with the collaboration and license agreement, we and Astellas agreed to equally share committed development costs, including the costs of completing certain tivozanib clinical development activities that were initiated as part of our partnership with Astellas.
Biogen Idec
In March 2009, we entered into an exclusive option and license agreement with Biogen Idec International GmbH, or Biogen Idec, regarding the development and commercialization of our discovery-stage ErbB3-targeted antibodies for the potential treatment and diagnosis of cancer and other diseases in humans outside of North America. Under the agreement, we were responsible for developing ErbB3 antibodies through completion of the first phase 2 clinical trial designed in a manner that, if successful, would generate data sufficient to support advancement to a phase 3 clinical trial. In March 2014, we amended our agreement with Biogen Idec, whereby Biogen Idec agreed to the termination of its rights and obligations under the agreement, including Biogen Idec’s option to (i) obtain a co-exclusive (with us) license to develop and manufacture ErbB3 targeted antibodies and (ii) obtain exclusive commercialization rights to ErbB3 products in countries in the world other than North America. As a result, we retain worldwide rights to AV-203, a clinical stage ErbB3-targeted antibody. Pursuant to the amendment, we are obligated to in good faith use reasonable efforts to seek a collaboration partner for the purpose of funding further development and commercialization of ErbB3-targeted antibodies. Pursuant to the amendment, we are obligated to pay Biogen Idec a percentage of milestone payments received by us from future partnerships after March 28, 2016 and single digit royalty payments on net sales related to the sale of ErbB3 products, up to cumulative maximum amount of $50.0 million.
Kyowa Hakko Kirin
In December 2006, we entered into a license agreement with Kirin Brewery Co. Ltd. (now Kyowa Hakko Kirin), which we sometimes refer to as KHK, under which we obtained an exclusive license, with the right to grant sublicenses subject to certain restrictions, to research, develop, manufacture and commercialize tivozanib, pharmaceutical compositions thereof and associated biomarkers. Our exclusive license covers all territories in the world, except for Asia. KHK has retained rights to tivozanib in Asia. Under the license agreement, we obtained exclusive rights in our territory under certain KHK patents, patent applications and know-how related to
14
Table of Contents
tivozanib, to research, develop, make, have made, use, import, offer for sale, and sell tivozanib for the diagnosis, prevention and treatment of any and all human diseases and conditions. We and KHK each have access to and can benefit from the other party’s clinical data and regulatory filings with respect to tivozanib and biomarkers identified in the conduct of activities under the license agreement.
Under the license agreement, we are obligated to use commercially reasonable efforts to develop and commercialize tivozanib in our territory, including meeting certain specified diligence goals. Prior to the first anniversary of the first post-marketing approval sale of tivozanib in our territory, neither we nor any of our subsidiaries has the right to conduct certain clinical trials of, seek marketing approval for or commercialize any other cancer product that also works by inhibiting the activity of the VEGF receptor.
Upon entering into the license agreement with KHK, we made a one-time cash payment in the amount of $5.0 million. In March 2010, we made a $10.0 million milestone payment to KHK in connection with the dosing of the first patient in our phase 3 clinical trial of tivozanib. In December 2012, we made a $12.0 million milestone payment to KHK in connection with the acceptance by the FDA of our NDA filing for tivozanib. The total maximum payments for clinical and regulatory milestones under our license agreement with KHK are $60.0 million, in the aggregate.
We also made a $22.5 million payment to KHK during the year ended December 31, 2011 related to the up-front license payment received under the collaboration and license agreement with Astellas which we entered into in February 2011. We are also required to pay tiered royalty payments on net sales we make of tivozanib in our territory, which range from the low to mid teens as a percentage of net sales. The royalty rate escalates within this range based on increasing tivozanib sales. Our royalty payment obligations in a particular country in our territory begin on the date of the first commercial sale of tivozanib in that country, and end on the later of 12 years after the date of first commercial sale of tivozanib in that country or the date of the last to expire of the patents covering tivozanib that have been issued in that country. In the event we sublicense the rights licensed to us under the license agreement with KHK, we are required to pay KHK a specified percentage of any amounts we receive from any third party sublicensees, other than amounts we receive in respect of research and development funding or equity investments, subject to certain limitations.
The license agreement will remain in effect until the expiration of all of our royalty and sublicense revenue obligations to Kyowa Hakko Kirin, determined on a product-by-product and country-by-country basis, unless we elect to terminate the license agreement earlier. If we fail to meet our obligations under the agreement and are unable to cure such failure within specified time periods, Kyowa Hakko Kirin can terminate the agreement, resulting in a loss of our rights to tivozanib and an obligation to assign or license to Kyowa Hakko Kirin any intellectual property or other rights we may have in tivozanib, including our regulatory filings, regulatory approvals, patents and trademarks for tivozanib.
Intellectual Property Rights
Patent Rights
We have built a strong intellectual property portfolio, and, whenever possible, we have multiple tiers of patent protection for our product candidates. With respect to tivozanib, we have exclusively licensed patents that cover the molecule and its therapeutic use (patent expiration 2022, with the possibility of patent term extension to 2027 in the United States), a key step in manufacturing the molecule, and a crystal form of the molecule, i.e., a polymorph with low hygroscopicity used in the clinical formulation. With respect to tivozanib, we have:
| • | U.S. patents: 3 issued; none pending; expirations ranging from 2018 to 2023 |
| • | European patents: 3 granted; none pending; expirations ranging from 2018 to 2023 |
| • | Canadian patents: 1 granted; none pending; expiration 2022 |
| • | Australian patents: 1 granted; none pending; expiration 2022 |
15
Table of Contents
Complementing these in-licensed patents relating to tivozanib are two of our own issued U.S. patents that cover different biomarker tests for identifying human patients likely to respond to treatment with tivozanib, and an issued U.S. patent on a method of using tivozanib in combination with temsirolimus. We also have two U.S. patent applications relating to the use of certain biomarkers, including Neuropilin-1, for identifying patients, including patients with colorectal cancer, likely to respond to treatment with tivozanib. With respect to tivozanib related technologies, we have:
| • | U.S. patents: 3 issued; 2 pending; expirations ranging from 2029 to 2036 |
| • | Canadian patents: none granted; 1 pending; expiration 2030 |
| • | Australian patents: none granted; 1 pending; expiration 2030 |
| • | International applications: 1 pending |
With respect to our anti-HGF antibodies, including ficlatuzumab, we have eight U.S. patents covering our anti-HGF antibodies, nucleic acids and expression vectors encoding the antibodies, host cells, methods of making the antibodies, and methods of treatment using the antibodies. With respect to our anti-HGF antibody program we have:
| • | U.S. patents: 8 granted; 2 pending; expirations ranging from 2027 to 2028 |
| • | European patents: 2 granted; none pending; expirations 2027 |
| • | Japanese patents: 3 granted; 1 pending; expirations 2027 |
| • | Canadian patents: 0 granted; 2 pending; expirations 2027 |
| • | Australian patents: 1 granted; none pending; expiration 2027 |
With respect to our anti-ErbB3 antibodies, including AV-203, we have a U.S. patent covering AV-203 and a pending U.S. application covering nucleic acids and expression vectors encoding the antibodies, host cells, and methods of making the antibodies, and a U.S. patent application relating to a method of predicting tumor response to our anti-ErbB3 antibody. With respect to our anti- ErbB3 antibody program we have:
| • | U.S. patents: 1 granted; 2 pending; expirations ranging from 2031 to 2032 |
| • | European patents: none granted; 2 pending; expirations ranging from 2031 to 2032 |
| • | Japanese patents: none granted; 2 pending; expirations ranging from 2031 to 2032 |
| • | Canadian patents: none granted; 2 pending; expirations ranging from 2031 to 2032 |
| • | Australian patents: none granted; 2 pending; expirations ranging from 2031 to 2032 |
With respect to our anti-GDF15 antibodies, we have exclusively licensed certain patent rights from Saint Vincent’s Hospital in the field of GDF15 inhibition for therapeutic, preventative and palliative applications, including increasing appetite and/or body weight in subjects where decreased appetite and/or body weight loss due to elevated expression or amounts of GDF15. A U.S. Patent covering method of increasing appetite and /or body weight administering an effective amount of an anti-GDF15 antibody is expected to expire in 2029, which includes approximately 4 years of patent term adjustment granted by the U.S. Patent and Trademark Office. We also have rights in a granted European patent in the field of GDF15 inhibition for decreased appetite and/or body weight due to elevated expression or amounts of GDF15 in patients with cancer, and plan to pursue broader claims in a divisional patent application. The granted European patents will expire in 2025.
With respect to the licensed technologies, we have:
| • | U.S. patents: 1 issued; 2 pending; expirations ranging from 2025 to 2029 |
| • | European patents: 2 granted; 3 pending; expirations ranging from 2016 to 2028 |
| • | Japanese patents: 1 allowed; 2 pending; expirations ranging from 2025 to 2028. |
16
Table of Contents
| • | Canadian patents: 1 granted; 2 pending; expiration 2016 to 2028 |
| • | Australian patents: 3 granted; none pending; expiration 2016 to 2028 |
Complementing these in-licensed patents relating to GDF15 inhibition, we have filed our own U.S. and international patent applications that cover our GDF15 antibodies. The patent, if issued, is expected to expire in 2033. We have also filed four U.S. provisional patent applications covering the use of our inhibitory GDF15 antibodies in improving cardiac, renal, and pulmonary function in patients with congestive heart failure, chronic kidney disease, and chronic obstructive pulmonary disease, respectively, as well as use in conjunction with chemotherapeutic agents to increase survival in a cancer cachexia patient.
In addition to patents relating to tivozanib and our ficlatuzumab, AV-203, and anti-GDF15 antibody programs, our patent portfolio contains a number of other patents and patent applications relevant to our business. We own a granted U.S. patent and issued foreign counterparts covering a method of making a chimeric mouse cancer model. We also own a granted U.S. patent and issued foreign counterparts covering a method of producing primary tumor material via directed complementation. We also own a granted U.S. patent and pending U.S. patent application covering a mouse model that contains a human breast tumor. We own pending patent applications that cover a general method for identifying new, multi-gene biomarkers for predicting response to an anti-cancer drug of interest, as well as specific multi-gene biomarkers identified by using the same method. With respect to our technology platforms, we have:
| • | U.S. patents: 3 issued; 2 pending; expirations ranging from 2024 to 2032 |
| • | European patents: 2 granted; 1 pending; expirations ranging from 2024 to 2032 |
| • | Japanese patents: 2 granted; 1 pending; expirations ranging from 2024 to 2032 |
| • | Canadian patents: 1 granted; 1 pending; expirations ranging from 2026 to 2032 |
| • | Australian patents: 2 granted; 1 pending; expirations ranging from 2024 to 2032 |
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the earliest date of filing a non-provisional patent application. In the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office in granting a patent. A U.S. patent term may be shortened, if a patent is terminally disclaimed by its owner, over another patent.
The patent term of a patent that covers an FDA-approved drug may also be eligible for patent term extension, which permits patent term restoration as compensation for the patent term lost during the FDA regulatory review process. The Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Act, permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the drug is under regulatory review. Patent extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, and only one patent applicable to an approved drug may be extended. Similar provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved drug. In the future, if and when our pharmaceutical products receive FDA approval, we expect to apply for patent term extensions on patents covering those products.
Many pharmaceutical companies, biotechnology companies and academic institutions are competing with us in the field of oncology and filing patent applications potentially relevant to our business. With regard to tivozanib, we are aware of a third party United States patent, and corresponding foreign counterparts, that contain broad claims related to the use of an organic compound that, among other things, inhibits tyrosine phosphorylation of a VEGF receptor caused by VEGF binding to such VEGF receptor. We are also aware of third party United States patents that contain broad claims related to the use of a tyrosine kinase inhibitor in
17
Table of Contents
combination with a DNA damaging agent such as chemotherapy or radiation and we have received written notice from the owners of such patents indicating that they believe we may need a license from them in order to avoid infringing their patents. With regard to ficlatuzumab, we are aware of two separate families of United States patents, United States patent applications and foreign counterparts, with each of the two families being owned by a different third party, that contain broad claims related to anti-HGF antibodies having certain binding properties and their use. With regard to AV-203, we are aware of a third party United States patent that contains broad claims relating to anti-ErbB3 antibodies. In the event that an owner of one or more of these patents were to bring an infringement action against us, we may have to argue that our product, its manufacture or use does not infringe a valid claim of the patent in question. Furthermore, if we were to challenge the validity of any issued United States patent in court, we would need to overcome a statutory presumption of validity that attaches to every United States patent. This means that in order to prevail, we would have to present clear and convincing evidence as to the invalidity of the patent’s claims. There is no assurance that a court would find in our favor on questions of infringement or validity.
Over the years, we have attempted to identify potential third party intellectual property issues during the early stages of research of our research programs, in order to minimize the cost and disruption of resolving such issues. From time to time, we have found it necessary or prudent to obtain licenses from third party intellectual property holders. Where licenses are readily available at reasonable cost, such licenses are considered a normal cost of doing business. In other instances, however, we may have used the results of freedom-to-operate studies to guide our research away from areas where we believed we were likely to encounter obstacles in the form of third party intellectual property. For example, where a third party holds relevant intellectual property and is a direct competitor, a license might not be available on commercially reasonable terms or available at all.
In spite of our efforts to avoid obstacles and disruptions arising from third party intellectual property, it is impossible to establish with certainty that our technology platform or our product programs will be free of claims by third party intellectual property holders. Even with modern databases and on-line search engines, literature searches are imperfect and may fail to identify relevant patents and published applications. Even when a third party patent is identified, we may conclude upon a thorough analysis, that we do not infringe the patent or that the patent is invalid. If the third party patent owner disagrees with our conclusion and we continue with the business activity in question, patent litigation may be initiated against us. Alternatively, we might decide to initiate litigation in an attempt to have a court declare the third party patent invalid or non-infringed by our activity. In either scenario, patent litigation typically is costly and time-consuming, and the outcome is uncertain. The outcome of patent litigation is subject to uncertainties that cannot be quantified in advance, for example, the credibility of expert witnesses who may disagree on technical interpretation of scientific data. Ultimately, in the case of an adverse outcome in litigation, we could be prevented from commercializing a product or using certain aspects of our technology platform as a result of patent infringement claims asserted against us. This could have a material adverse effect on our business.
To protect our competitive position, it may be necessary to enforce our patent rights through litigation against infringing third parties. Litigation to enforce our own patent rights is subject to the same uncertainties discussed above. In addition, however, litigation involving our patents carries the risk that one or more of our patents will be held invalid (in whole or in part, on a claim-by-claim basis) or held unenforceable. Such an adverse court ruling could allow third parties to commercialize our products or our platform technology, and then compete directly with us, without making any payments to us.
Trade Secrets
For some aspects of our proprietary technology, trade secret protection is more appropriate than patent protection. For example, our proprietary bioinformatics software tools and databases are protected as trade secrets. Our bioinformatics tools and databases give us the means to store, analyze, interpret and integrate the large volume of data generated from our various tumor models and from analysis of human clinical samples from clinical trials. We continually make incremental improvements in our proprietary software tools, as we tailor
18
Table of Contents
them to the changing needs of our research and development programs. In general, trade secret protection can accommodate this continuing evolution of our bioinformatics system better than other forms of intellectual property protection.
Trademarks
We seek trademark protection in the U.S. and foreign jurisdictions where available and when appropriate. We have filed to register several trademarks intended for potential use in the marketing of tivozanib. We own a U.S. trademark that we use in connection with our research and development (Human Response Platform). We also own a U.S. trademark (The Human Response™) and a U.S. trademark application (AVEO Oncology The Human Response™) that we use in connection with our business, in general.
Manufacturing
We currently contract with third parties, to the extent we require, for the manufacture of our product candidates and intend to do so in the future for both clinical and potential commercial needs. We do not own or operate manufacturing facilities for the production of clinical or commercial quantities of our product candidates. We currently have no plans to build our own clinical or commercial scale manufacturing capabilities. Although we rely on contract manufacturers, we have personnel with extensive manufacturing experience to oversee the relationships with our contract manufacturers.
One of our contract manufacturers has manufactured what we believe to be sufficient quantities of tivozanib’s drug substance to support our ongoing clinical trials. In addition, we currently engage a separate contract manufacturer to manufacture, package, label and distribute clinical supplies of tivozanib on an as–needed basis.
We are responsible for all process development and all manufacturing of ficlatuzumab for future development and commercialization and have an agreement with Boehringer Ingelheim for large-scale process development and clinical manufacturing of ficlatuzumab. In connection with the agreement, Boehringer Ingelheim has produced ficlatuzumab at its biopharmaceutical sites in Fremont, California (drug substance) and Biberach, Germany (drug product).
In August 2010, we entered into an agreement with Gallus BioPharmaceuticals, LLC (previously known as Laureate Pharma, Inc. and now part of Patheon), or Gallus, for the clinical manufacture of AV-203 drug substance. Gallus has produced two batches of AV-203 drug substance for future clinical trials at its site in Princeton, New Jersey. AV-203 drug product was produced at Microtest Laboratories, Inc. in Agawam, Massachusetts. Microtest Laboratories was purchased by ATS Labs and the combined company will operate under the name Accuratus Lab Services.
On March 9, 2014, we entered into a manufacturing agreement with AbbVie Inc. for the process, development and manufacture of clinical drug substance of AV-380. Abbvie has manufactured two lots of AV-380 drug substance in Worchester, Massachusetts for future clinical trials.
To date, our third-party manufacturers have met our manufacturing requirements. We believe that there are alternate sources of supply that can satisfy our current clinical requirements, although we cannot be certain that identifying and establishing relationships with such sources, if necessary, would not result in significant delay or material additional costs.
Government Regulation
Government authorities in the United States (including federal, state and local authorities) and in other countries, extensively regulate, among other things, the manufacturing, research and clinical development,
19
Table of Contents
marketing, labeling and packaging, distribution, post-approval monitoring and reporting, advertising and promotion, and export and import of pharmaceutical products, such as those we are developing. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
United States Government Regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and related regulations. Drugs are also subject to other federal, state and local statutes and regulations. Biological products are subject to regulation by the FDA under the FDCA, the Public Health Service Act, and related regulations, and other federal, state and local statutes and regulations. Failure to comply with the applicable U.S. regulatory requirements at any time during the product development process, approval process or after approval, may be the basis for administrative or judicial sanctions. These sanctions could include the imposition by the FDA or an Institutional Review Board, or IRB, of a clinical hold on trials, the FDA’s refusal to approve pending applications or supplements, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us.
The Investigational New Drug Process
An Investigational New Drug application, or an IND, is a request for authorization from the FDA to administer an investigational drug or biological product to humans. Such authorization must be secured prior to interstate shipment (usually to clinical investigators) and administration of any new drug or biological product to humans that is not the subject of an approved New Drug Application or Biologics License Application, except under limited circumstances.
To conduct a clinical investigation with an investigational new drug or biological product, we are required to file an IND with the FDA in compliance with Title 21 of the Code of Federal Regulations (CFR), Part 312. These regulations contain the general principles underlying the IND submission and the general requirements for an IND’s content and format.
The central focus of the initial IND submission is on the general investigational plan and the protocol(s) for human studies. The IND also includes results of animal studies or other human studies, as appropriate, as well as manufacturing information, analytical data and any available clinical data or literature to support the use of the investigational new drug or biological product. An IND must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to the proposed clinical trials as outlined in the IND. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before clinical trials can begin. Accordingly, submission of an IND may or may not result in the FDA allowing clinical trials to commence.
Clinical trials involve the administration of the investigational drug or biological product to patients under the supervision of qualified investigators in accordance with Good Clinical Practices, or GCPs. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety, and the efficacy criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. Additionally, approval must also be obtained from each clinical site’s independent IRB before the trials may be initiated. All participants in our clinical trials must provide their informed consent in writing in compliance with GCPs and the ethical principles that have their origin in the Declaration of Helsinki.
20
Table of Contents
The clinical investigation of an investigational drug or biological product is generally divided into three phases. Although the phases are usually conducted sequentially, they may overlap or be combined. The three phases of an investigation are as follows:
| • | Phase 1. Phase 1 includes the initial introduction of an investigational new drug or biological product into humans. Phase 1 clinical trials are typically closely monitored and may be conducted in patients with the target disease or condition or healthy volunteers. These studies are designed to evaluate the safety, dosage tolerance, metabolism and pharmacologic actions of the investigational drug or biological product in humans, the side effects associated with increasing doses, and if possible, to gain early evidence on effectiveness. During phase 1 clinical trials, sufficient information about the investigational drug’s or biological product’s pharmacokinetics and pharmacological effects may be obtained to permit the design of well-controlled and scientifically valid phase 2 clinical trials. The total number of participants included in phase 1 clinical trials varies, but is generally in the range of 20 to 80. |
| • | Phase 2. Phase 2 includes the controlled clinical trials conducted to preliminarily or further evaluate the effectiveness of the investigational drug or biological product for a particular indication(s) in patients with the disease or condition under study, to determine dosage tolerance and optimal dosage, and to identify possible adverse side effects and safety risks associated with the drug or biological product. Phase 2 clinical trials are typically well-controlled, closely monitored, and conducted in a limited patient population, usually involving no more than several hundred participants. |
| • | Phase 3. Phase 3 clinical trials are generally controlled clinical trials conducted in an expanded patient population generally at geographically dispersed clinical trial sites. They are performed after preliminary evidence suggesting effectiveness of the drug or biological product has been obtained, and are intended to further evaluate dosage, clinical effectiveness and safety, to establish the overall benefit-risk relationship of the investigational drug or biological product, and to provide an adequate basis for product approval. Phase 3 clinical trials usually involve several hundred to several thousand participants. |
The FDA’s primary objectives in reviewing an IND are to assure the safety and rights of patients and to help assure that the quality of the investigation will be adequate to permit an evaluation of the drug’s effectiveness and safety and of the biological product’s safety, purity and potency. The decision to terminate development of an investigational drug or biological product may be made by either a health authority body such as the FDA, an IRB or ethics committee, or by us for various reasons. Additionally, some trials are overseen by an independent group of qualified experts organized by the trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether or not a trial may move forward at designated check points based on access that only the group maintains to available data from the study. Suspension or termination of development during any phase of clinical trials can occur if it is determined that the participants or patients are being exposed to an unacceptable health risk. Other reasons for suspension or termination may be made by us based on evolving business objectives and/or competitive climate.
In addition, there are requirements and industry guidelines that require the posting of ongoing clinical trials on public registries, and the disclosure of designated clinical trial results.
The NDA/BLA Approval Process
In order to obtain approval to market a drug or biological product in the United States, a marketing application must be submitted to the FDA that provides data establishing the safety and effectiveness of the drug product for the proposed indication, and the safety, purity and potency of the biological product for its intended indication. The application includes all relevant data available from pertinent preclinical and clinical trials, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls and proposed labeling, among other things. Data can come
21
Table of Contents
from company-sponsored clinical trials intended to test the safety and effectiveness of a use of a product, or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and effectiveness of the investigational drug product and the safety, purity and potency of the biological product to the satisfaction of the FDA.
The steps required before an investigational drug or biological product may be marketed in the United States generally include:
| • | Completion of preclinical laboratory tests, animal studies and formulation studies in compliance with the FDA’s Good Laboratory Practices, or GLP, regulations; |
| • | Submission to the FDA of an IND to support human clinical testing; |
| • | Approval by an IRB at each clinical site before each trial may be initiated; |
| • | Performance of adequate and well-controlled clinical trials in accordance with GCP to establish the safety and efficacy of the investigational drug product for each targeted indication or the safety, purity and potency of the biological product for its intended indication; |
| • | Submission of an NDA or Biologics License Application, or BLA, to the FDA; |
| • | Satisfactory completion of an FDA Advisory Committee review, if applicable; |
| • | Satisfactory completion of an FDA inspection of the manufacturing facilities at which the investigational drug or biological product is produced to assess compliance with current good manufacturing practices, or cGMP, and to assure that the facilities, methods and controls are adequate to preserve the product’s identity, strength, quality and purity; and |
| • | FDA review and approval of the NDA or BLA. |
In most cases, the NDA or BLA must be accompanied by a substantial user fee; there may be some instances in which the user fee is waived.
The FDA will initially review the NDA or BLA for completeness before it accepts the NDA or BLA for filing. The FDA has 60 days from its receipt of an NDA or BLA to determine whether the application will be accepted for filing based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. After the NDA submission is accepted for filing, the FDA reviews the NDA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. After the BLA submission is accepted for filing, the FDA reviews the BLA to determine, among other things, whether the product is safe, pure and potent and the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, purity and potency. The FDA may refer applications for novel drug or biological products or drug or biological products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes outside clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA or BLA, the FDA will carefully review and typically require changes to the proposed product labeling. The FDA will also inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA or BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. If the FDA determines the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will
22
Table of Contents
request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete. Data obtained from clinical activities are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to develop our product candidates and secure necessary governmental approvals, which could delay or preclude us from marketing our products. Even if the FDA approves a product, it may limit the approved indications for use, impose prominent warnings, or place other conditions on any approvals that could restrict the commercial application of the products such as a requirement that we implement special risk management measures through a Risk Evaluation and Mitigation Strategy. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Post-Approval Regulation
After regulatory approval of a drug or biological product is obtained, we are required to comply with a number of post-approval requirements. For example, as a condition of approval of an NDA or BLA, the FDA may require post-marketing testing, including phase 4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization. Regulatory approval of oncology products often requires that patients in clinical trials be followed for long periods to determine the overall survival benefit of the drug or biologic. In addition, as a holder of an approved NDA or BLA, we would be required to report, among other things, certain adverse events and production problems to the FDA, to provide updated safety and efficacy information, and to comply with requirements concerning advertising and promotional labeling for any of our products. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval to assure and preserve the long term stability of the drug or biological product. The FDA periodically inspects manufacturing facilities to assess compliance with cGMP, which imposes extensive procedural, substantive and record keeping requirements. In addition, changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon us and any third party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our product candidates. Future FDA, foreign regulatory authorities, and state inspections may identify compliance issues at our facilities or at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of previously unknown problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved NDA or BLA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action that could delay or prohibit further marketing. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures.
Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development and/or could significantly impact the requirements imposed on us after approval.
23
Table of Contents
Europe / Rest of World Government Regulation
In addition to regulations in the United States, we will be subject to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a clinical trial application much like the IND prior to the commencement of human clinical trials. In the European Union, for example, a clinical trial application, or CTA, must be submitted to each country’s national health authority and an independent ethics committee, much like the FDA and IRB, respectively. Once the CTA is approved in accordance with a country’s requirements, and relevant ethics committees have issued positive opinions, the clinical trial covered by the CTA may proceed. In all cases, the clinical trials must be conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
To obtain regulatory approval of an investigational drug or biological medicinal product under European Union regulatory systems, we must submit a marketing authorization application. The application used to file the NDA or BLA in the United States is similar to that required in Europe, with the exception of, among other things, country-specific documentation requirements. The requirements and process governing pricing and reimbursement in the European Union vary from country to country.
For other countries outside of the European Union, such as countries in Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, any clinical trials that we sponsor must be conducted in accordance with GCP and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Compliance
During all phases of development (pre- and post-marketing), failure to comply with the applicable regulatory requirements may result in administrative or judicial sanctions. These sanctions could include the FDA’s imposition of a clinical hold on trials or the suspension of clinical trials by other regulatory authorities, refusal to approve pending applications, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, product detention or refusal to permit the import or export of products, injunctions, fines, civil penalties or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us.
Available Special Regulatory Procedures
Formal Meetings
We are encouraged to engage and seek guidance from health authorities relating to the development and review of investigational drugs and biologics, as well as marketing applications. In the United States, there are different types of official meetings that may occur between us and the FDA. Each meeting type is subject to different procedures. Conclusions and agreements from each of these meetings are captured in the official final meeting minutes issued by the FDA.
The EMA and national medicines regulators within the EU also provide the opportunity for dialogue with us. At the EMA level, this is usually done in the form of Scientific Advice, which is given by the Scientific
24
Table of Contents
Advice Working Party of the Committee for Medicinal Products for Human Use, or CHMP. A fee is incurred with each Scientific Advice procedure.
Advice from either the FDA or EMA is typically provided based on questions concerning, for example, quality (chemistry, manufacturing and controls testing), nonclinical testing and clinical studies, and pharmacovigilance plans and risk-management programs. Advice is not legally binding with regard to any future marketing authorization application of the product concerned. To obtain binding commitments from the FDA on the design and size of clinical trials intended to form the primary basis of an effectiveness claim, Special Protocol Assessment procedures are available. Where the FDA agrees to a Special Protocol Assessment, or SPA, the agreement may not be changed by either the sponsor or the FDA except if the sponsor and the FDA agree to a change, or a senior FDA official determines that a substantial scientific issue essential to determining the safety or effectiveness of the product was identified after the testing began. An SPA is not binding if new circumstances arise, and there is no guarantee that a study will ultimately be adequate to support an approval even if the study is conducted according to the terms of an SPA.
Orphan Drug Designation
The FDA may grant orphan drug designation to drugs and biological products intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the United States, or if it affects more than 200,000 individuals in the United States and there is no reasonable expectation that the cost of developing and making the drug or biological product for this type of disease or condition will be recovered from sales in the United States. In the European Union, the EMA’s Committee for Orphan Medicinal Products, or COMP, may recommend orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than 5 in 10,000 persons in the European Union. Additionally, designation is granted for products intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the drug in the European Union would be sufficient to justify the necessary investment in developing the drug or biological product. In addition, the COMP may only recommend orphan drug designation when the product in question offers a significant clinical benefit over existing approved products for the relevant indication. Following a positive opinion by the COMP, the European Commission adopts a decision granting orphan status. The COMP will reassess orphan status in parallel with EMA review of a marketing authorization application and orphan status may be withdrawn at that stage if it no longer fulfills the orphan criteria.
In the United States, orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages and user-fee waivers. In addition, if a product receives the first FDA approval for the indication for which it has orphan designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other application to market the same drug or biological product for the same indication for a period of 7 years, except in limited circumstances, such as a showing of clinical superiority over the product with orphan exclusivity or if the product with orphan exclusivity experiences a shortage.
In the European Union, orphan drug designation also entitles a party to financial incentives such as reduction of fees or fee waivers and 10 years of market exclusivity is granted following drug or biological product approval. During this period, regulators may not accept an application for or approve any similar medicinal product, unless there are supply shortages or the similar product is safer, more effective or otherwise clinically superior than the approved orphan drug. This period may be reduced to 6 years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
25
Table of Contents
Pediatric Development
In the United States, Section 505A of the Federal Food, Drug, and Cosmetic Act (21 U.S.C. 355a, Pediatric Studies of Drugs) provides for an additional 6 months of marketing exclusivity for a drug if reports are filed of investigations studying the use of the drug product in a pediatric population in response to a written request from the FDA. Separate from this potential exclusivity benefit, NDAs and BLAs must contain data (or a proposal for post-marketing activity) to assess the safety and effectiveness of an investigational drug or biological product for the claimed indications in all relevant pediatric populations in order to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults or full or partial waivers if certain criteria are met. Discussions about pediatric development plans can be discussed with the FDA at any time, but usually occur any time between the end-of-phase 2 meeting and submission of the NDA or BLA.
For the EMA, a Pediatric Investigation Plan, and/or a request for waiver or deferral, is required for submission prior to submitting a marketing authorization application.
Authorization Procedures in the European Union
There are two types of marketing authorization procedures for medicinal products in the European Union; the centralized authorization procedure and national authorization procedures.
| • | Centralized procedure. The centralized procedure gives rise to marketing authorizations that are valid throughout the European Union and, by extension, in three European Economic Area, or EEA member states, Norway, Iceland and Liechtenstein. Applicants file marketing authorizations with the EMA, where they are reviewed by a relevant scientific committee, which is most likely the Committee for Medicinal Products for Human Use, or CHMP. The EMA forwards CHMP positive opinions to the European Commission, which uses them as the basis for a decision granting a marketing authorization. The centralized procedure is compulsory for human medicines that are: derived from biotechnology processes, such as recombinant DNA technology, controlled expression of genes in prokaryotes and eukaryotes and hybridoma and monoclonal antibody methods. It is also mandatory for products containing a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders, viral diseases or autoimmune diseases and other immune dysfunctions, and officially designated orphan medicines. For medicines that do not fall within these categories, an applicant has the option of submitting an application for a centralized marketing authorization to the EMA, as long as the CHMP accepts that the medicine concerned is a significant therapeutic, scientific or technical innovation, or if its authorization would be in the interest of public health. |
| • | National authorization procedures. There are also two other possible routes to authorize medicinal products in more than one EU or EEA country, which are available for investigational drug products that fall outside the scope of the centralized procedure: |
| • | Decentralized procedure. Using the decentralized procedure, an applicant may apply for simultaneous authorization in more than one EEA country of medicinal products that have not yet been authorized in any EEA country and that do not fall within the mandatory scope of the centralized procedure. The applicant selects a so-called reference member state, or RMS, to take the lead in the review of the application. Other member states are expected to recognize the RMS decision, unless they identify a serious risk to public health. If the member states cannot resolve any such concerns between themselves, the matter is referred to the CHMP for an opinion and ultimately a binding European Commission decision. |
| • | Mutual recognition procedure. In the mutual recognition procedure, a medicine is first authorized in one EEA RMS, in accordance with the national procedures of that country. Following this, |
26
Table of Contents
| further marketing authorizations can be sought from other EEA countries in a procedure whereby the countries concerned agree to recognize the validity of the original, national marketing authorization. As in the decentralized procedure, these concerned member states must recognize the RMS approval unless they identify a serious risk to the public health. If the member states cannot reach a consensus between themselves, the matter can be referred to the CHMP. |
Priority Review / Standard Review (United States) and Accelerated Review (European Union)
Based on results of phase 3 clinical trials, an NDA or BLA may receive either priority or standard review from the FDA. Priority review is given where preliminary estimates indicate that a product, if approved, would provide a significant improvement in safety or effectiveness of the treatment, diagnosis or prevention of a serious condition. Under PDUFA V, effective October 1, 2012, where an application receives priority review, the target date for FDA action will be 8 months from submission in the case of an application for a new chemical entity and 6 months from submission in the case of products that do not contain a new chemical entity. Where an application receives standard review, the target date for FDA action will be 12 months from submission in the case of an application for a new chemical entity and 10 months from submission in the case of products that do not contain a new chemical entity.
Under the centralized procedure in the European Union, the maximum timeframe for the evaluation of a marketing authorization application is 210 days (excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP). Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest, defined by three cumulative criteria: the seriousness of the disease (e.g. heavy disabling or life-threatening diseases) to be treated; the absence or insufficiency of an appropriate alternative therapeutic approach; and anticipation of high therapeutic benefit. In this circumstance, EMA ensures that the opinion of the CHMP is given within 150 days.
Biosimilars
The 2010 healthcare reform legislation created an approval pathway for biosimilars (i.e., follow-on version of innovative biologics). The European regulatory bodies also have authority to approve biosimilars. Because many issues under the U.S. biosimilar legislation remain unresolved (including the scope of exclusivity for new biologics), it is difficult to predict how this legislation will affect us. Our products may face significant competition from biosimilars (as well as traditional generic drugs) in the United States and abroad.
Employees
As of December 31, 2014, we had 57 employees worldwide. In January 2015, as part of a strategic restructuring, we eliminated approximately two-thirds of our workforce, or 40 positions. We expect to have approximately 20 employees following completion of our strategic restructuring. None of our employees is represented by a labor union or is covered by a collective bargaining agreement. We consider our relationship with our employees to be good.
Research and Development Costs
Our research and development costs were $38.3 million, $68.5 million, and $91.4 million for the years ended December 31, 2014, 2013 and 2012, respectively. These costs consist of the cost of our own independent research and development efforts and the costs associated with collaborative research and development and in-licensing arrangements. Research and development costs, including upfront fees and milestones paid to collaboration partners, are expensed as incurred if the underlying products have not received regulatory approval and have no alternative future use.
27
Table of Contents
Segment and Geographic Information
We view our operations and manage our business in one operating segment. As of December 31, 2014, we operate only in the United States.
Executive Officers
The following table lists the positions, names and ages of our executive officers as of March 5, 2015:
Executive Officers
| Michael P. Bailey |
49 | Chief Executive Officer, President and Director and Acting Chief Financial Officer | ||||
| Michael N. Needle |
55 | Chief Medical Officer |
Michael P. Bailey was appointed President and Chief Executive Officer and a member of our Board of Directors effective January 6, 2015. Mr. Bailey joined our company in September 2010 as our Chief Commercial Officer and was named our Chief Business Officer in June 2013. Prior to joining our company, Mr. Bailey served as Senior Vice President, Business Development and Chief Commercial Officer at Synta Pharmaceuticals from 2008 to September 2010. From 1999 to 2008, Mr. Bailey worked at ImClone. During his nine-year tenure at ImClone, he was responsible for commercial aspects of the planning and launch of ERBITUX® (cetuximab) across multiple oncology indications, as well as new product planning for the ImClone development portfolio, which included CYRAMZA® (ramucirumab) and necitumumab. In addition, Mr. Bailey was a key member of the strategic leadership committees for ImClone and its North American and worldwide partnerships and led their commercial organization, most recently as Senior Vice President of Commercial Operations. Prior to his role at ImClone. Mr. Bailey managed the cardiovascular development portfolio at Genentech, Inc. from 1997 to 1999. Mr. Bailey started his career in the pharmaceutical industry as part of Smith-Kline Beecham’s Executive Marketing Development Program, where he held a variety of commercial roles from 1992 to 1997, including sales, strategic planning, and product management. Mr. Bailey received a B.S. in psychology from St. Lawrence University and an M.B.A. in international marketing from the Mendoza College of Business at University of Notre Dame.
Michael N. Needle, MD was appointed Chief Medical Officer on January 9, 2015. Dr. Needle has more than 15 years of pharmaceutical industry experience in drug development and regulatory affairs. This includes central roles in the development of oncology and hematology drugs, including Erbitux® (cetuximab), Revlimid® (lenalidomide) and Pomalyst® (pomolidimide). He most recently served as Chief Medical Officer for Array BioPharma from April 2013 to September 2014. Prior to Array, Dr. Needle was Chief Medical Officer of the Multiple Myeloma Research Foundation and Consortium (MMRF) from April 2012 to April 2013. Prior to MMRF, he held multiple Vice President level positions at Celgene in Clinical Research and Development in Oncology, Strategic Medical Business Development, and Pediatric Strategy from March 2004 to April 2010. Dr. Needle also served as the Vice President of Clinical Affairs at ImClone Systems Incorporated from April 2000 to February 2004. Dr. Needle received his fellowship in Pediatric Hematology/Oncology at the Children’s Hospital Medical Center, the Fred Hutchinson Cancer Research Center of the University of Washington in Seattle and the University of Texas M.D. Anderson Cancer Center in Houston. Dr. Needle has held faculty positions at the University of Pennsylvania and Columbia University. Dr. Needle graduated from Binghamton University with a Bachelor of Arts in Physics and received his medical degree from SUNY Downstate Medical Center, in Brooklyn, New York.
28
Table of Contents
Available Information
We file reports and other information with the SEC as required by the Securities Exchange Act of 1934, as amended, which we refer to as the Exchange Act. You can find, copy, and inspect information we file at the SEC’s public reference room, which is located at 100 F Street, N.E., Room 1580, Washington, DC 20549. Please call the SEC at 1-800-SEC-0330 for more information about the operation of the SEC’s public reference room. You can review our electronically filed reports and other information that we file with the SEC on the SEC’s web site at http://www.sec.gov.
We were incorporated under the laws of the State of Delaware on October 19, 2001 as GenPath Pharmaceuticals, Inc. and changed our name to AVEO Pharmaceuticals, Inc. on March 1, 2005. Our principal executive offices are located at 650 East Kendall Street, Cambridge, Massachusetts, 02142, and our telephone number is (617) 299-5000. Our Internet website is http://www.aveooncology.com. We make available free of charge through our website our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Exchange Act. We also make available, free of charge on our website, the reports filed with the SEC by our executive officers, directors and 10% stockholders pursuant to Section 16 under the Exchange Act. We make these reports available through our website as soon as reasonably practicable after we electronically file such reports with, or furnish such reports to, the SEC, or, in the case of Section 16 reports, as soon as reasonably practicable after copies of those filings are provided to us by the filing persons. In addition, we regularly use our website to post information regarding our business, product development programs and governance, and we encourage investors to use our website, particularly the information in the section entitled “For Investors” and “For Media,” as a source of information about us.
The foregoing references to our website are not intended to, nor shall they be deemed to, incorporate information on our website into this report by reference.
29
Table of Contents
Our business is subject to numerous risks. We caution you that the following important factors, among others, could cause our actual results to differ materially from those expressed in forward-looking statements made by us or on our behalf in this Annual Report on Form 10-K and other filings with the SEC, press releases, communications with investors and oral statements. Any or all of our forward-looking statements in this Annual Report on Form 10-K and in any other public statements we make may turn out to be wrong. They can be affected by inaccurate assumptions we might make or by known or unknown risks and uncertainties. Many factors mentioned in the discussion below will be important in determining future results. Consequently, no forward-looking statement can be guaranteed. Actual future results may differ materially from those anticipated in our forward-looking statements. We undertake no obligation to update any forward-looking statements, whether as a result of new information, future events or otherwise. You are advised, however, to consult any further disclosure we make in our reports filed with the SEC.
Risks Related to Our Financial Position and Capital Requirements
We may not be successful in establishing and maintaining strategic partnerships to further the development of each of our therapeutic programs. A failure to obtain such partnerships in the near future will have a material adverse effect on our operations and business.
We currently are exploring partnership opportunities to fund the further development of a majority of our development programs, including our lead program for tivozanib as well as AV-203 and AV-380. Accordingly, our success will depend in significant part on our ability to attract and maintain strategic partners and strategic relationships with major biotechnology or pharmaceutical companies to support the development and commercialization of these product candidates. In these partnerships, we would expect our strategic partner to provide substantial funding, as well as significant capabilities in research, development, marketing and sales
We face significant competition in seeking appropriate strategic partners and the negotiation process is time-consuming and complex. Moreover, we may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for any product candidates and programs because our development pipeline may be deemed insufficient, our product candidates and programs may be deemed to be at too early of a stage of development for collaborative effort or third parties may not view our product candidates and programs as having the requisite potential to demonstrate safety and efficacy.
Even if we are successful in our efforts to establish new strategic partnerships, the terms that we agree upon may not be favorable to us and we may not be able to maintain such strategic partnerships if, for example, development or approval of a product candidate is delayed or sales of an approved product are disappointing. Any delay in entering into new strategic partnership agreements related to our product candidates could have an adverse effect on our business or our operating plan, including delaying the development and commercialization of our product candidates.
Moreover, if we fail to establish and maintain additional strategic partnerships related to our product candidates:
| • | we will have limited resources with which to continue to operate our business and we may not be able to successfully complete any other strategic transactions; |
| • | the development of certain of our product candidates may be terminated or delayed; and |
| • | our cash expenditures related to development of our product candidates would increase significantly and we do not have the cash resources to develop our product candidates on our own. |
30
Table of Contents
We will require substantial additional financing, and a failure to obtain this necessary capital when needed would force us to delay, limit, reduce or terminate our research, product development or commercialization efforts.
We will require substantial funds to continue our development programs and to fulfill our planned operating goals. In particular, our currently planned operating and capital requirements include the need for substantial working capital to support our development activities for tivozanib, ficlatuzumab, AV-203 and AV-380. Moreover, we have future payment obligations and cost-sharing arrangements under certain of our collaboration and license agreements. For example, under our agreements with KHK and St. Vincent’s, we are required to make certain clinical and regulatory milestone payments, have royalty obligations with respect to product sales and are required to pay a specified percentage of sublicense revenue in certain instances. Moreover, under our agreement with Biodesix, we are obligated to share any costs for the phase 2 FOCAL study that exceed $15 million. Accordingly, we will need substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, or if we are unable to procure partnership arrangements to advance our programs, we would be forced to delay, reduce or eliminate our research and development programs and any future commercialization efforts.
We believe that our existing cash and cash equivalents will allow us to fund our operating plan into the third quarter of 2016.
Because of the numerous risks and uncertainties associated with the development and commercialization of pharmaceutical products, we are unable to estimate the exact amounts of our working capital requirements. Our future capital requirements depend on many factors, including:
| • | our ability to establish and maintain strategic partnerships, licensing or other arrangements and the financial terms of such agreements; |
| • | the number and characteristics of the product candidates we pursue; |
| • | the scope, progress, results and costs of developing our product candidates, and conducting preclinical and clinical trials; |
| • | the timing of, and the costs involved in, obtaining regulatory approvals for our product candidates; |
| • | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims, including litigation costs and the outcome of such litigation; |
| • | the absence of any breach, acceleration event or event of default under our loan agreement with Hercules or under any other agreements with third parties; |
| • | the outcome of lawsuits against us, including the current lawsuits described under “Part I, Item 3—Legal Proceedings;” |
| • | the cost of commercialization activities if any of our product candidates are approved for sale, including marketing, sales and distribution costs; |
| • | the cost of manufacturing our product candidates and any products we successfully commercialize; |
| • | our ability to secure alternative leasing or subleasing arrangements for new office space and to achieve related cost savings with respect to termination of our lease at 650 East Kendall Street, Cambridge, and |
| • | the timing, receipt and amount of sales of, or royalties on, our future products, if any. |
In addition, it is possible that Hercules Technology II, L.P. and Hercules Technology III, L.P., affiliates of Hercules Technology Growth, which we refer to collectively as Hercules, could take the position that the recent restructuring which we consummated constitutes a material adverse change under our loan and security agreements with Hercules, under which we had $21.6 million in loans outstanding as of December 31, 2014, which could trigger a repayment of all principal and interest due under the loan, unless such event of default is waived by Hercules.
31
Table of Contents
Raising additional capital may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or drug candidates.
We may seek to sell additional equity or debt securities or obtain additional credit facilities. The sale of additional equity or convertible debt securities may result in additional dilution to our stockholders. If we raise additional funds through the issuance of debt securities or preferred stock or through additional credit facilities, these securities and/or the loans under credit facilities could provide for rights senior to those of our common stock and could contain covenants that would restrict our operations. We may require additional capital beyond our currently forecasted amounts. Additional funds may not be available when we need them, on terms that are acceptable to us, or at all.
We also expect to seek additional funds through arrangements with collaborators, licensees or other third parties. These arrangements would generally require us to relinquish or encumber rights to some of our technologies or drug candidates, and we may not be able to enter into such arrangements on acceptable terms, if at all.
We anticipate that we will require additional funding. If we are unable to obtain such additional funding on a timely basis, whether through payments under existing or future collaborations or license agreement or sales of debt or equity, we may be required to delay, limit, reduce or terminate our clinical trials or development activities for one or more of our product candidates.
If we fail to regain compliance with the requirements for continued listing on the NASDAQ Global Select Market, our common stock could be delisted from trading on the NASDAQ Market, which would adversely affect the liquidity of our common stock and our ability to raise additional capital.
Our common stock is currently listed for quotation on the NASDAQ Global Select Market. We are required to meet specified financial requirements in order to maintain our listing on the NASDAQ Global Select Market. On December 29, 2014, we received a deficiency letter from the Listing Qualifications Department, or the Staff, of The NASDAQ Stock Market notifying us that, for the last 30 consecutive business days, the bid price for our common stock had closed below the minimum $1.00 per share requirement for continued inclusion on The NASDAQ Global Market. We were provided an initial period of 180 calendar days, or until June 29, 2015, to regain compliance with the listing requirements. If, at any time before June 29, 2015, the bid price for our common stock closes at $1.00 or more for a minimum of 10 consecutive business days we may be eligible to regain compliance with the minimum bid requirement. Under certain circumstances, NASDAQ could require that the minimum bid price exceed $1.00 for more than ten consecutive days before determining that we comply with NASDAQ’s continued listing standards. If in the future we fail to satisfy the NASDAQ Global Select Market’s continued listing requirements, we may transfer to the OTC Bulletin Board. Any potential delisting of our common stock from the NASDAQ Global Select Market would make it more difficult for our stockholders to sell our stock in the public market and would likely result in decreased liquidity, limited availability of market quotations for shares of our common stock, limited availability of news and analyst coverage regarding our company, a decreased ability to issue additional securities and increased volatility in the price of our common stock.
We may experience difficulties, delays or unexpected costs and not achieve anticipated benefits and savings from our recently announced restructuring plan, and our restructuring activities may adversely affect our business.
In January 2015, our Board of Directors approved a strategic restructuring that eliminated our internal research function, given that our material programs where at preclinical and clinical stages of development, to better align our resources with our future clinically focused strategic plans. This restructuring resulted in the reduction of approximately two-thirds of our workforce, leaving us with approximately 20 employees. The reduction in force resulted in the loss of numerous long-term employees, the loss of institutional knowledge and expertise and the reallocation of certain employment responsibilities, all of which could adversely affect
32
Table of Contents
operational efficiencies, employee performance and retention. Because of these reductions, we are outsourcing certain corporate functions, which make us more dependent on third parties for the performance of functions that are critical to our business and the development of our product candidates. To the extent that we are unable to effectively reallocate employee responsibilities, retain key employees, establish and maintain agreements with competent third-party contractors on terms that are acceptable to us, or effectively manage the work performed by any retained third-party contractors, our ability to advance our business or product candidates may be significantly impaired and our strategic goals and our financial results may be adversely affected.
Restructuring plans may yield unintended consequences, such as attrition beyond our intended reduction in workforce and reduced employee morale, which may cause our employees who were not affected by the reduction in workforce to seek alternate employment. Furthermore, employees whose positions will be eliminated in connection with these restructuring plans may seek future employment with our competitors. Although all our employees are required to sign a confidentiality agreement with us at the time of hire, we cannot assure you that the confidential nature of our proprietary information will be maintained in the course of such future employment. Additionally, as a result of our restructuring activities we may experience a loss of continuity, loss of accumulated knowledge and/or inefficiency during transitional periods. If we cannot successfully manage the transition of our restructured operations, we may be unsuccessful in executing our business strategy, which would have a material adverse effect on our financial condition and results of operations.
We anticipate that we will continue to incur significant operating costs for the foreseeable future. It is uncertain if we will ever attain profitability in the future, which would depress the market price of our common stock.
We have incurred net losses in all prior reporting periods, other than for the year ended December 31, 2011, including a net loss of $52.7 million during the twelve months ended December 31, 2014. As of December 31, 2014, we had an accumulated deficit of $480.0 million. To date, we have not commercialized any products or generated any revenues from the sale of products, and absent the realization of sufficient revenues from product sales, we may never attain profitability in the future. Our losses have resulted principally from costs incurred in our discovery and development activities. We anticipate that we will continue to incur significant operating costs over the next several years as we seek to develop our product candidates.
If we do not successfully develop and obtain regulatory approval for our existing and future pipeline product candidates and effectively manufacture, market and sell any product candidates that are approved, we may never generate product sales, and even if we do generate product sales, we may never achieve or sustain profitability on a quarterly or annual basis. Our failure to become and remain profitable would depress the market price of our common stock and could impair our ability to raise capital, expand our business, diversify our product offerings or continue our operations.
We and certain of our former officers and present and former directors have been named as defendants in multiple lawsuits that could result in substantial costs and divert management’s attention.
We, and certain of our present and former officers and directors, were named as defendants in a consolidated class action lawsuit initiated in 2013 that generally alleges that we and certain of our former officers and present and former directors violated Sections 10(b) and/or 20(a) of the Securities Exchange Act of 1934 and Rule 10b-5 promulgated thereunder by making allegedly false and/or misleading statements concerning the phase 3 trial design and results for our TIVO-1 study in an effort to lead investors to believe that the drug would receive approval from the FDA. The complaints seek unspecified damages, interest, attorneys’ fees, and other costs. Additionally, we received a subpoena from the SEC requesting documents and information concerning tivozanib, including related communications with the FDA, investors and others. Moreover, a plaintiff has filed a derivative complaint allegedly on our behalf, naming us, as a nominal defendant and also naming as defendants present and former members of the our board of directors, alleging breach of fiduciary duty and abuse of control
33
Table of Contents
between January 2012 and May 2013 with respect to allegedly misleading statements and omissions regarding tivozanib. The derivative complaint seeks, among other relief, unspecified damages, costs and expenses, including attorneys’ fees, an order requiring us to implement certain corporate governance reforms, restitution from the defendants and such other relief as the court might find just and proper.
We intend to engage in a vigorous defense of these lawsuits and are fully cooperating with the SEC regarding its fact-finding inquiry. However, we are unable to predict the outcome of these matters at this time. Moreover, any conclusion of these matters in a manner adverse to us would have a material adverse effect on our financial condition and business. For example, we could incur substantial costs not covered by our directors’ and officers’ liability insurance, suffer a significant adverse impact on our reputation and divert management’s attention and resources from other priorities, including the execution of business plans and strategies that are important to our ability to grow our business, any of which could have a material adverse effect on our business. In addition, any of these matters could require payments that are not covered by, or exceed the limits of, our available directors’ and officers’ liability insurance, which could have a material adverse effect on our operating results or financial condition
Our business is in early stage of development, which may make it difficult for you to evaluate the success of our business to date and to assess our future viability.
All of our product candidates are in early stages of development. We have not yet demonstrated our ability to obtain marketing approvals, manufacture a commercial scale medicine, or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful commercialization. Typically, it takes about ten to 15 years to develop one new medicine from the time it is discovered to when it is available for treating patients. Preclinical studies and clinical trials may involve highly uncertain results and a high risk of failure. Moreover, positive data from preclinical studies and clinical trials of our product candidates may not be predictive of results in ongoing or subsequent preclinical studies and clinical trials. Consequently, any predictions you make about our future success or viability may not be as accurate as they could be if we had a longer operating history.
In addition, as an early stage business, we may encounter unforeseen expenses, difficulties, complications, delays and other known and unknown factors. To be profitable, we will need to transition from a company with a research focus to a company capable of supporting commercial activities. We may not be successful in such a transition.
Risks Related to Development, Clinical Testing and Regulatory Approval of Our Drug Candidates
All of our product candidates are still in preclinical and clinical development. Preclinical testing and clinical trials of our product candidates may not be successful, or may not result in approval by the FDA. If we are unable to obtain marketing approval or commercialize our product candidates, or experience significant delays in doing so, our business will be materially harmed.
Development of our product candidates, all of which are still in preclinical and clinical development, is at an early stage and we may not successfully develop a drug candidate that becomes a commercially viable drug. Our ability to generate product revenues, which we do not expect for many years, if ever, will depend heavily on the successful development and eventual commercialization of our product candidates. This process can take many years to complete, requiring the expenditure of substantial resources with highly uncertain results and a high risk of failure. Moreover, positive data from preclinical studies and clinical trials of our product candidates may not be predictive of results in ongoing or subsequent preclinical studies and clinical trials and may not be predictive of success in gaining any regulatory or marketing approvals necessary for commercialization.
The regulatory process can vary substantially based upon a variety of factors, including the type, complexity and novelty of the product candidates involved. If any of our product candidates are not shown to be safe and
34
Table of Contents
effective in humans through clinical trials, we and/or our strategic partners will not be able to obtain regulatory approval for such product candidate, and the resulting delays in developing other product candidates and conducting related preclinical studies and clinical trials would have a material adverse effect on our business, financial condition and results of operations.
The success of our product candidates will depend on several factors, many of which are beyond our control, including the following:
| • | our ability to maintain collaborations with our strategic partners and establish new collaborations; |
| • | successful enrollment in, and completion of, clinical trials and preclinical studies; |
| • | our ability to demonstrate to the satisfaction of the FDA, and equivalent foreign regulatory agencies, the safety, efficacy and clinically meaningful benefit of our product candidates through completed, ongoing and any future clinical and non-clinical trials; |
| • | our ability to obtain additional funding when needed; |
| • | achieving and maintaining compliance with all regulatory requirements applicable to pharmaceutical products; |
| • | the prevalence and severity of adverse side effects; |
| • | the ability of our third-party manufacturers to manufacture clinical trial and commercial supplies and to develop, validate and maintain commercially viable manufacturing processes that are compliant with current Good Manufacturing Practices, or cGMP; |
| • | the availability, relative cost, safety and efficacy of alternative and competing treatments; |
| • | acceptance of the product by patients, the medical community and third-party payors; |
| • | launching commercial sales of the product, whether alone or in collaboration with others; and |
| • | our ability to avoid third-party patent interference or patent infringement claims. |
If we do not achieve one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully commercialize our product candidates, which would materially harm our business.
Any failure or delay in completing clinical trials for our product candidates, or unfavorable results from such trials, may prevent us from obtaining regulatory approval or commercializing product candidates on a timely basis, or at all, which would require us to incur additional costs and delay receipt of any product revenue.
We cannot predict whether we will encounter problems with any of our ongoing clinical trials or clinical trials we may conduct to further develop our product candidates, which could cause us or regulatory authorities to delay, suspend or terminate those clinical trials. The completion of clinical trials for product candidates may be delayed, suspended or terminated for many reasons, including:
| • | delays in patient enrollment, and variability in the number and types of patients available for clinical trials, or high drop-out rates of patients in our clinical trials; |
| • | our inability to obtain even additional funding when needed; |
| • | delays or failure in reaching agreement on acceptable clinical trial contracts or clinical trial protocols with prospective sites; |
| • | failure of our third-party contractors or our investigators to comply with regulatory requirements or otherwise meet their contractual obligations in a timely manner; |
35
Table of Contents
| • | delays or failure in obtaining the necessary approvals from regulators or institutional review boards in order to commence a clinical trial at a prospective trial site, or their suspension or termination of a clinical trial once commenced; |
| • | our inability to manufacture or obtain from third parties materials sufficient to complete our preclinical studies and clinical trials; |
| • | difficulty in maintaining contact with patients after treatment, resulting in incomplete data; |
| • | poor effectiveness of our product candidates during clinical trials, including without limitation, a failure to meet study objectives or obtain the requisite level of statistical significance imposed by the FDA or other regulatory agencies; |
| • | safety issues, including serious adverse events associated with our product candidates; |
| • | governmental or regulatory delays and changes in regulatory requirements, policy and guidelines; or |
| • | varying interpretations of data by the FDA and similar foreign regulatory agencies. |
Clinical trials often require the enrollment of large numbers of patients, and suitable patients may be difficult to identify and recruit. Our ability to enroll sufficient numbers of patients in our clinical trials depends on many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites, the eligibility criteria for the trial, competing clinical trials, the availability of approved effective drugs and the perception of the efficacy and safety of our product candidates. We may experience delays or difficulties in enrolling patients in our current and future trials. For example, in January 2014, we and Astellas decided to discontinue the BATON (Biomarker Assessment of Tivozanib in ONcology) breast cancer clinical trial, a phase 2 study in patients with locally recurrent or metastatic triple negative breast cancer, due to insufficient enrollment. If we fail to enroll and maintain the number of patients for which the clinical trial was designed, the statistical power of that clinical trial may be reduced which would make it harder to demonstrate that the product candidate being tested in such clinical trial is safe and effective. Additionally, we may not be able to enroll a sufficient number of qualified patients in a timely or cost-effective manner.
We, the FDA, other applicable regulatory authorities or institutional review boards may suspend or terminate clinical trials of a product candidate at any time if we or they believe the patients participating in such clinical trials are being exposed to unacceptable health risks or for other reasons.
Significant clinical trial delays could allow our competitors to obtain marketing approval before we do or shorten the patent protection period during which we may have the exclusive right to commercialize our product candidates. Our product development costs also will increase if we experience delays in completing clinical trials. In addition, it is impossible to predict whether legislative changes will be enacted, or whether FDA regulations, guidance or interpretations will be changed, or what the impact of such changes, if any, may be. If we experience any such problems, we may not have the financial resources to continue development of the product candidate that is affected or the development of any of our other product candidates.
If we are unable to successfully develop companion diagnostics for certain of our therapeutic product candidates, or experience significant delays in doing so, we may not realize the full commercial potential of these therapeutics.
A component of our business strategy may be to develop, in collaboration with a third party, companion diagnostics for some of our therapeutic product candidates. There has been limited success to date industry-wide in developing companion diagnostics. To be successful, our collaborator will need to address a number of scientific, technical, regulatory and logistical challenges. We have limited experience in the development of diagnostics and may not be successful in developing appropriate diagnostics to pair with any of our therapeutic product candidates that receive marketing approval. The FDA and similar regulatory authorities outside the United States are generally expected to regulate companion diagnostics as medical devices. In each case,
36
Table of Contents
companion diagnostics require separate regulatory approval prior to commercialization. For example, BDX004, our companion diagnostic test for ficlatuzumab in our FOCAL study, requires separate approval by the FDA, for which we must rely on Biodesix to obtain. Given our limited experience in developing diagnostics, we expect to rely in part on third parties for their design, development and manufacture. If we, or any third parties that we engage to assist us, are unable to successfully develop companion diagnostics for our therapeutic product candidates, or experience delays in doing so, the development of our therapeutic product candidates may be adversely affected, our therapeutic product candidates may not receive marketing approval and we may not realize the full commercial potential of any therapeutics that receive marketing approval. As a result, our business would be harmed, possibly materially.
Even if we receive regulatory approval for any of our product candidates, we will be subject to ongoing FDA requirements and continued regulatory review, which may result in significant additional expense. Additionally, our product candidates, if approved, could be subject to labeling and other restrictions, post-approval requirements and market withdrawal and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our products.
Any regulatory approvals that we or our strategic partners receive for our product candidates may also be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing requirements and testing, including post-approval clinical trials, surveillance to monitor the safety and efficacy of the product candidate, and implementation of a risk evaluation and mitigation strategy. In addition, if the FDA approves any of our product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMP and good clinical practices, or GCP, for any clinical trials that we conduct post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
| • | restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls; |
| • | fines, warning letters or holds on clinical trials; |
| • | refusal by the FDA to approve pending applications or supplements to approved applications filed by us or our strategic partners, or suspension or revocation of product license approvals; |
| • | product seizure or detention, or refusal to permit the import or export of products; and |
| • | injunctions or the imposition of civil or criminal penalties. |
The FDA’s policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would adversely affect our business.
Failure to obtain regulatory approval in jurisdictions outside the United States will prevent us from marketing our products abroad.
We intend to market our products, if approved, in international markets, which will require separate regulatory approvals and compliance with numerous and varying regulatory requirements. The approval
37
Table of Contents
procedures vary among countries and may involve requirements for additional testing, and the time required to obtain approval may differ from that required to obtain FDA approval. In addition, in many countries outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that country. In some cases, the price that we intend to charge for our product is also subject to approval. Approval by the FDA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or jurisdictions or by the FDA. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. We may not obtain foreign regulatory approvals on a timely basis, if at all. We and our strategic partners may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market.
Risks Related to Our Business and Industry
We face substantial competition, which may result in others discovering, developing or commercializing products before, or more successfully, than we do.
Our future success depends on our ability to demonstrate and maintain a competitive advantage with respect to the design, development and commercialization of product candidates. Our objective is to design, develop and commercialize new products with superior efficacy, convenience, tolerability and safety. We expect any product candidate that we commercialize with our strategic partners will compete with existing, market-leading products.
Many of our potential competitors have substantially greater financial, technical and personnel resources than we have and several are already marketing products to treat the same indications, and having the same biological targets, as the product candidates we are developing, including with respect to cachexia. In addition, many of these competitors have significantly greater commercial infrastructures than we have. We will not be able to compete successfully unless we effectively:
| • | design and develop products that are superior to other products in the market in terms of, among other things, both safety and efficacy; |
| • | obtain patent and/or other proprietary protection for our processes and product candidates; |
| • | obtain required regulatory approvals; |
| • | obtain favorable reimbursement, formulary and guideline status; and |
| • | collaborate with others in the design, development and commercialization of new products. |
Established competitors may invest heavily to quickly discover and develop novel compounds that could make our product candidates obsolete. In addition, any new product that competes with an approved product must demonstrate compelling advantages in efficacy, convenience, tolerability and safety in order to obtain approval, to overcome price competition and to be commercially successful. If we are not able to compete effectively against our current and future competitors, our business will not grow and our financial condition and operations will suffer.
We may not achieve development and commercialization goals in the time frames that we publicly estimate, which could have an adverse impact on our business and could cause our stock price to decline.
We set goals, and make public statements regarding our expected timing for certain accomplishments, such as the initiation and completion of clinical trials, filing and approval of regulatory applications for our product candidates and other developments and milestones under our research and development programs. The actual timing of these events can vary significantly due to a number of factors, including, without limitation, delays or failures in our and our current and potential future collaborators’ preclinical studies or clinical trials, the amount of time, effort and resources committed to our programs by us and our current and potential future collaborators and the uncertainties inherent in the regulatory approval process. As a result, there can be no assurance that our
38
Table of Contents
or our current and potential future collaborators’ preclinical studies and clinical trials will advance or be completed in the time frames we expect or announce, that we or our current and potential future collaborators will make regulatory submissions or receive regulatory approvals as planned or that we or our current and potential future collaborators will be able to adhere to our current schedule for the achievement of key milestones under any of our programs. If we or any collaborators fail to achieve one or more of the milestones described above as planned, our business could be materially adversely affected and the price of our common stock could decline.
Because we have limited experience in developing and commercializing pharmaceutical products, there is a limited amount of information about us upon which you can evaluate our business and prospects.
Although certain of our employees may have experience in developing and commercializing pharmaceutical products, as an organization we have limited experience in developing and commercializing pharmaceutical products and have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biopharmaceutical area. For example, to execute our business plan, we will need to successfully:
| • | execute product development activities; |
| • | obtain required regulatory approvals for the development and commercialization of our product candidates; |
| • | build and maintain a strong intellectual property portfolio; |
| • | build and maintain robust sales, distribution, reimbursement and marketing capabilities; |
| • | obtain reimbursement and gain market acceptance for our products; |
| • | develop and maintain successful strategic relationships and partnerships; and |
| • | manage our spending as costs and expenses increase due to clinical trials, regulatory approvals and commercialization. |
If we are unsuccessful in accomplishing these objectives, we may not be able to develop product candidates, raise capital, expand our business or continue our operations.
If we fail to attract and keep senior management, we may be unable to successfully develop our product candidates, conduct our clinical trials and commercialize our product candidates.
Our success depends in part on our continued ability to attract, retain and motivate highly qualified management personnel. We are highly dependent upon our senior management, as well as others on our management team. The reduction in force related to the restructuring we announced on January 7, 2015, together with the departure within the last year of several executive officers, could not only make it difficult to retain other remaining employees, but also could make it difficult to attract additional personnel, to the extent we need to replace departed employees. The loss of services of employees, and in particular, of or one or more of our members of management could delay or prevent our ability to successfully maintain or enter into new licensing arrangements or collaborations, the successful development of our product candidates, the completion of our planned clinical trials or the commercialization of our product candidates. We do not carry “key person” insurance covering any members of our senior management. Our employment arrangements with all of these individuals are “at will,” meaning they or we can terminate their service at any time.
We face intense competition for qualified individuals from numerous pharmaceutical and biotechnology companies, universities, governmental entities and other research institutions, many of which have substantially greater resources with which to reward qualified individuals than we do. We may face challenges in retaining our
39
Table of Contents
existing senior management and key employees and recruiting new employees to join our company as our business needs change. We may be unable to attract and retain suitably qualified individuals, and our failure to do so could have an adverse effect on our ability to implement our future business plans.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with FDA regulations, to provide accurate information to the FDA, to comply with manufacturing standards we have established, to comply with federal and state health-care fraud and abuse laws and regulations, to report financial information or data accurately or to disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a code of business conduct and ethics, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
In addition, during the course of our operations, our directors, executives and employees may have access to material, nonpublic information regarding our business, our results of operations or potential transactions we are considering. Despite the adoption of an Insider Trading Policy, we may not be able to prevent a director, executive or employee from trading in our common stock on the basis of, or while having access to, material, nonpublic information. If a director, executive or employee was to be investigated, or an action was to be brought against a director, executive or employee for insider trading, it could have a negative impact on our reputation and our stock price. Such a claim, with or without merit, could also result in substantial expenditures of time and money, and divert attention of our management team from other tasks important to the success of our business.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize any products. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability, and a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Even successful defense could require significant financial and management resources. Regardless of the merits or eventual outcome, product liability claims may result in:
| • | decreased demand for our product candidates or products that we may develop; |
| • | injury to our reputation; |
| • | withdrawal of clinical trial participants; |
| • | costs to defend the related litigation; |
| • | diversion of management’s time and our resources; |
40
Table of Contents
| • | substantial monetary awards to trial participants or patients; |
| • | product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| • | loss of revenue; |
| • | the inability to commercialize our product candidates; and |
| • | a decline in our stock price. |
Our inability to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of products we develop. We currently carry product liability insurance covering our clinical studies in the amount of $20 million in the aggregate. Although we maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We will have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts.
We rely significantly upon information technology and any failure, inadequacy, interruption or security lapse of that technology, including any cyber security incidents, could harm our ability to operate our business effectively.
Despite the implementation of security measures, our internal computer systems and those of third parties with which we contract are vulnerable to damage from cyber-attacks, computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. System failures, accidents or security breaches could cause interruptions in our operations, and could result in a material disruption of our clinical and commercialization activities and business operations, in addition to possibly requiring substantial expenditures of resources to remedy. The loss of clinical trial data could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate public disclosure of confidential or proprietary information, we could incur liability and our product development and commercialization efforts could be delayed.
Risks Related to Commercialization of Our Product Candidates
Our commercial success depends upon attaining significant market acceptance of our product candidates, if approved, including among physicians, patients, healthcare payors and, in the cancer market, acceptance by the major operators of cancer clinics.
Even if one of our product candidates obtains regulatory approval, the product may not gain market acceptance among physicians, healthcare payors, patients and the medical community. Market acceptance of any products for which we receive approval depends on a number of factors, including:
| • | the efficacy and safety of the product candidate, as demonstrated in clinical trials; |
| • | the clinical indications for which the drug is approved; |
| • | acceptance by physicians, major operators of cancer clinics, healthcare payors, physician networks and patients of the drug as a safe and effective treatment; |
| • | the potential and perceived advantages over alternative treatments; |
| • | the cost of treatment in relation to alternative treatments; |
| • | the availability of adequate reimbursement and pricing by third parties and government authorities; |
41
Table of Contents
| • | the continued projected growth of oncology drug markets; |
| • | relative convenience and ease of administration; |
| • | the prevalence and severity of adverse side effects; and |
| • | the effectiveness of our sales and marketing efforts. |
If our approved drugs fail to achieve market acceptance, we would not be able to generate significant revenue.
Reimbursement may be limited or unavailable in certain market segments for our product candidates, which could make it difficult for us to sell any approved products profitably.
Market acceptance and sales of our product candidates will depend significantly on the availability of adequate coverage and reimbursement from third-party payors for any of our product candidates and may be affected by existing and future healthcare reform measures. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which drugs they will pay for and establish reimbursement levels. Reimbursement by a third-party payor may depend upon a number of factors, including the third-party payor’s determination that use of a product is:
| • | a covered benefit under its health plan; |
| • | safe, effective and medically necessary; |
| • | appropriate for the specific patient; |
| • | cost-effective; and |
| • | neither experimental nor investigational. |
Obtaining coverage and reimbursement approval for a product from a government or other third-party payor is a time-consuming and costly process that could require us to provide supporting scientific, clinical and cost-effectiveness data for the use of our products to the payor. We may not be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement. We cannot be sure that coverage or adequate reimbursement will be available for any of our product candidates. Also, we cannot be sure that reimbursement amounts will not reduce the demand for, or the price of, our products. If reimbursement is not available or is available only to limited levels, we may not be able to commercialize certain of our products.
In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory changes to the healthcare system that could impact our ability to sell our products profitably. In particular, the Medicare Modernization Act of 2003 revised the payment methodology for many products under Medicare. This has resulted in lower rates of reimbursement. There have been numerous other federal and state initiatives designed to reduce payment for pharmaceuticals that may set reimbursement or pricing at unsatisfactory levels.
As a result of legislative proposals and the trend towards managed healthcare in the United States, third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement of new drugs. They may also refuse to provide any coverage of approved products for medical indications other than those for which the FDA has granted market approvals. As a result, significant uncertainty exists as to whether and how much third-party payors will reimburse patients for their use of newly approved drugs, which in turn will put pressure on the pricing of drugs. We expect to experience pricing pressures in connection with the sale of any products we may develop or commercialize due to the trend toward managed healthcare, the increasing influence of health maintenance organizations, additional legislative proposals, as well as country, regional, or local healthcare budget limitations. Any products that we may develop or commercialize may not be considered cost-effective, and coverage and reimbursement may not be available or sufficient to allow us to sell our products on a profitable basis.
42
Table of Contents
Foreign governments may impose price controls, which may adversely affect our future profitability.
We and our strategic partners intend to seek approval to market our future products in both the United States and in foreign jurisdictions. If approval is obtained in one or more foreign jurisdictions, we and our strategic partners will be subject to rules and regulations in those jurisdictions relating to our product.
In some foreign countries, particularly in countries in the European Union, the pricing of prescription pharmaceuticals and biologics is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product candidate. If reimbursement of our future products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, we may be unable to achieve or sustain profitability.
Healthcare reform measures could hinder or prevent our product candidates’ commercial success.
The U.S. government and other governments have shown significant interest in pursuing healthcare reform. Any government-adopted reform measures could adversely impact the pricing of healthcare products and services in the U.S. or internationally and the amount of reimbursement available from governmental agencies or other third-party payors. The continuing efforts of the U.S. and foreign governments, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce healthcare costs may adversely affect our ability to set prices which we believe are fair for any products we may develop and commercialize, and our ability to generate revenues and achieve and maintain profitability.
New laws, regulations and judicial decisions, or new interpretations of existing laws, regulations and decisions, that relate to healthcare availability, methods of delivery or payment for products and services, or sales, marketing or pricing, may limit our potential revenue, and we may need to revise our research and development programs. The pricing and reimbursement environment may change in the future and become more challenging due to several reasons, including policies advanced by the U.S. government, new healthcare legislation or fiscal challenges faced by government health administration authorities. Specifically, in both the U.S. and some foreign jurisdictions, there have been a number of legislative and regulatory proposals and initiatives to change the healthcare system in ways that could affect our ability to sell any products we may develop and commercialize profitably. Some of these proposed and implemented reforms could result in reduced reimbursement rates for our potential products, which would adversely affect our business strategy, operations and financial results.
For example, in March 2010, President Obama signed into law a legislative overhaul of the U.S. healthcare system, known as the Patient Protection and Affordable Care Act of 2010, as amended by the Healthcare and Education Affordability Reconciliation Act of 2010, or the PPACA, which may have far reaching consequences for life science companies like us. As a result of this legislation, substantial changes could be made to the current system for paying for healthcare in the United States, including changes made in order to extend medical benefits to those who currently lack insurance coverage. Extending coverage to a large population could substantially change the structure of the health insurance system and the methodology for reimbursing medical services, drugs and devices. These structural changes could entail modifications to the existing system of private payors and government programs, such as Medicare and Medicaid, creation of a government-sponsored healthcare insurance source, or some combination of both, as well as other changes. Restructuring the coverage of medical care in the United States could impact the reimbursement for prescribed drugs, biopharmaceuticals, medical devices, or our product candidates. If reimbursement for our approved product candidates, if any, is substantially less than we expect in the future, or rebate obligations associated with them are substantially increased, our business could be materially and adversely impacted.
Further federal and state proposals and healthcare reforms could limit the prices that can be charged for the product candidates that we develop and may further limit our commercial opportunity. Our results of operations could be materially adversely affected by the PPACA, by Medicare prescription drug coverage legislation, by the possible effect of such current or future legislation on amounts that private insurers will pay and by other healthcare reforms that may be enacted or adopted in the future.
43
Table of Contents
We have limited sales, marketing, reimbursement and distribution experience and we will have to invest significant resources to develop those capabilities.
We have limited sales, marketing, reimbursement and distribution experience. To develop these capabilities, we will have to invest significant amounts of financial and management resources, some of which will be committed prior to any confirmation that any of our product candidates will be approved for commercial sale. We could face a number of additional risks in developing our commercial infrastructure, including:
| • | we may not be able to attract and build an effective marketing or sales force; |
| • | the cost of establishing a marketing or sales force may not be justifiable in light of the revenues generated by any particular product; and |
| • | our direct sales and marketing efforts may not be successful. |
Furthermore, we may elect in the future to utilize strategic partners or contract sales forces to assist in the commercialization of other products, if approved. We may have limited or no control over the sales, marketing and distribution activities of these third parties. Our future revenues may depend heavily on the success of the efforts of these third parties.
Risks Related to Our Dependence on Third Parties
If any of our current or future strategic partners fails to perform its obligations or terminates its agreement with us, the development and commercialization of the product candidates under such agreement could be delayed or terminated and our business could be substantially harmed.
As part of our business strategy, we plan to enter into additional strategic partnerships in the future. If any current or future strategic partners do not devote sufficient time and resources to its arrangements with us, we may not realize the potential commercial benefits of the arrangement, and our results of operations may be adversely affected. In addition, if any strategic partner were to breach or terminate its arrangements with us, the development and commercialization of the affected product candidate could be delayed, curtailed or terminated because we may not have sufficient financial resources or capabilities to continue development and commercialization of the product candidate on its own, and we may find it difficult to attract a new alliance partner for such product candidate. For example, Biodesix can opt-out of its agreement with us after the completion of the proof of concept trial prior to the first commercial sale of ficlatuzumab, at which point Biodesix would not be responsible for any future costs associated with developing and commercializing ficlatuzumab other than any ongoing clinical studies.
Much of the potential revenue from any strategic partnership we may enter into in the future will likely consist of contingent payments, such as royalties payable on sales of any successfully developed drugs. Any such contingent revenue will depend upon our, and our strategic partners’, ability to successfully develop, introduce, market and sell new drugs. In some cases, we will not be involved in these processes, and we will depend entirely on our strategic partners. Any of our future strategic partners may fail to develop or effectively commercialize these drugs because it:
| • | decides not to devote the necessary resources because of internal constraints, such as limited personnel with the requisite scientific expertise, limited cash resources or specialized equipment limitations, or the belief that other product candidates may have a higher likelihood of obtaining regulatory approval or may potentially generate a greater return on investment; |
| • | does not have sufficient resources necessary to carry the product candidate through clinical development, regulatory approval and commercialization; or |
| • | cannot obtain the necessary regulatory approvals. |
If one or more of our strategic partners fails to develop or effectively commercialize product candidates for any of the foregoing reasons, we may not be able to replace the strategic partner with another partner to develop
44
Table of Contents
and commercialize a product candidate under the terms of the strategic partnership. We may also be unable to obtain, on terms acceptable to us, a license from such strategic partner to any of its intellectual property that may be necessary or useful for us to continue to develop and commercialize a product candidate. Any of these events could have a material adverse effect on our business, results of operations and our ability to achieve future profitability, and could cause our stock price to decline.
We rely on third-party manufacturers to produce our preclinical and clinical product candidate supplies and we intend to rely on third parties to produce commercial supplies of any approved product candidates. Any failure by a third-party manufacturer to produce supplies for us may delay or impair our ability to complete our clinical trials or commercialize our product candidates.
We do not possess all of the capabilities to fully commercialize any of our product candidates on our own. We have relied upon third-party manufacturers for the manufacture of our product candidates for preclinical and clinical testing purposes and intend to continue to do so in the future. If we are unable to arrange for third-party manufacturing sources, or to do so on commercially reasonable terms, we may not be able to complete development of such other product candidates or market them.
Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured product candidates ourselves, including reliance on the third party for regulatory compliance and quality assurance, the possibility of breach of the manufacturing agreement by the third party because of factors beyond our control (including a failure to synthesize and manufacture our product candidates in accordance with our product specifications), failure of the third party to accept orders for supply of drug substance or drug product and the possibility of termination or nonrenewal of the agreement by the third-party, based on its own business priorities, at a time that is costly or damaging to us. In addition, the FDA and other regulatory authorities require that our product candidates be manufactured according to cGMP and similar foreign standards. Any failure by our third-party manufacturers to comply with cGMP or failure to scale-up manufacturing processes as needed, including any failure to deliver sufficient quantities of product candidates in a timely manner, could lead to a delay in, or failure to obtain, regulatory approval of any of our product candidates. In addition, such failure could be the basis for action by the FDA to withdraw approvals for product candidates previously granted to us and for other regulatory action, including recall or seizure, fines, imposition of operating restrictions, total or partial suspension of production or injunctions.
We rely on our manufacturers to purchase from third-party suppliers the materials necessary to produce our product candidates for our clinical studies. There are a small number of suppliers for certain capital equipment and raw materials that we use to manufacture our product candidates. Such suppliers may not sell this capital equipment or these raw materials to our manufacturers at the times we need them or on commercially reasonable terms. We do not have any control over the process or timing of the acquisition of this capital equipment or these raw materials by our manufacturers. Moreover, we currently do not have any agreements for the commercial production of these raw materials. Any significant delay in the supply of a product candidate or the raw material components thereof for an ongoing clinical trial due to the need to replace a third-party manufacturer could considerably delay completion of our clinical studies, product testing and potential regulatory approval of our product candidates. If our manufacturers or we are unable to purchase these raw materials after regulatory approval has been obtained for our product candidates, the commercial launch of our product candidates would be delayed or there would be a shortage in supply, which would impair our ability to generate revenues from the sale of our product candidates.
Because of the complex nature of many of our early stage compounds and product candidates, our manufacturers may not be able to manufacture such compounds and product candidates at a cost or in quantities or in a timely manner necessary to develop and commercialize related products. If we successfully commercialize any of our drugs, we may be required to establish or access large-scale commercial manufacturing capabilities. In addition, as our drug development pipeline increases and matures, we will have a greater need for clinical trial and commercial manufacturing capacity. We do not own or operate manufacturing facilities for the
45
Table of Contents
production of clinical or commercial quantities of our product candidates and we currently have no plans to build our own clinical or commercial scale manufacturing capabilities. To meet our projected needs for commercial manufacturing in the event that one or more of our product candidates gains marketing approval, third parties with whom we currently work will need to increase their scale of production or we will need to secure alternate suppliers.
We rely on third parties, such as clinical research organizations, or CROs, to conduct clinical trials for our product candidates, and if they do not properly and successfully perform their obligations to us, we may not be able to obtain regulatory approvals for our product candidates.
We, in consultation with our strategic partners, where applicable, design the clinical trials for our product candidates, but we have relied, and will rely, on contract research organizations and other third parties to assist us in managing, monitoring and otherwise carrying out many of these trials. We compete with larger companies for the resources of these third parties.
Although we plan to continue to rely on these third parties to conduct our ongoing any future clinical trials, we are responsible for ensuring that each of our clinical trials is conducted in accordance with its general investigational plan and protocol. Moreover, the FDA and foreign regulatory agencies require us to comply with regulations and standards, commonly referred to as good clinical practices, for designing, conducting, monitoring, recording, analyzing, and reporting the results of clinical trials to assure that the data and results are credible and accurate and that the rights, integrity and confidentiality of trial participants are protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements.
The third parties on whom we rely generally may terminate their engagements with us at any time. If we are required to enter into alternative arrangements because of any such termination, the introduction of our product candidates to market could be delayed.
If these third parties do not successfully carry out their duties under their agreements with us, if the quality or accuracy of the data they obtain is compromised due to their failure to adhere to our clinical trial protocols or regulatory requirements, or if they otherwise fail to comply with clinical trial protocols or meet expected deadlines, our clinical trials may not meet regulatory requirements. If our clinical trials do not meet regulatory requirements or if these third parties need to be replaced, our preclinical development activities or clinical trials may be extended, delayed, suspended or terminated. If any of these events occur, we may not be able to obtain regulatory approval of our product candidates and our reputation could be harmed.
Risks Related to Our Intellectual Property Rights
We could be unsuccessful in obtaining adequate patent protection for one or more of our product candidates, which could result in competition and a decrease in the potential market share for our product candidates.
We cannot be certain that patents will be issued or granted with respect to applications that are currently pending, or that issued or granted patents will not later be found to be invalid and/or unenforceable. The patent position of biotechnology and pharmaceutical companies is generally uncertain because it involves complex legal and factual considerations. The standards applied by the United States Patent and Trademark Office and foreign patent offices in granting patents are not always applied uniformly or predictably. For example, there is no uniform worldwide policy regarding patentable subject matter or the scope of claims allowable in biotechnology and pharmaceutical patents. Consequently, patents may not issue from our pending patent applications. As such, we do not know the degree of future protection that we will have on our proprietary products and technology. The scope of patent protection that the U.S. Patent and Trademark Office will grant with respect to the antibodies in our antibody product pipeline is uncertain. It is possible that the U.S. Patent and Trademark Office will not allow broad antibody claims that cover closely related antibodies as well as the specific antibody. Upon receipt of FDA approval, competitors would be free to market antibodies almost identical to ours, including biosimilar antibodies, thereby decreasing our market share.
46
Table of Contents
Issued patents covering one or more of our products could be found invalid or unenforceable if challenged in court.
If we or one of our corporate partners were to initiate legal proceedings against a third-party to enforce a patent covering one of our products, the defendant could counterclaim that our patent is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet one or more statutory requirements for patentability, including, for example, lack of novelty, obviousness, lack of written description or non-enablement. In addition, patent validity challenges may, under certain circumstances, be based upon non-statutory obviousness-type double patenting, which, if successful, could result in a finding that the claims are invalid for obviousness-type double patenting or the loss of patent term, including a patent term adjustment granted by the United States Patent and Trademark Officer, if a terminal disclaimer is filed to obviate a finding of obviousness-type double patenting. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the U.S. Patent and Trademark Office, or made a misleading statement, during prosecution. Although we have conducted due diligence on patents we have exclusively in-licensed, and we believe that we have conducted our patent prosecution in accordance with the duty of candor and in good faith, the outcome following legal assertions of invalidity and unenforceability during patent litigation is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on one of our products or certain aspects of our Human Response Platform. Such a loss of patent protection could have a material adverse impact on our business.
Claims that our platform technologies, our products or the sale or use of our products infringe the patent rights of third parties could result in costly litigation or could require substantial time and money to resolve, even if litigation is avoided.
We cannot guarantee that our platform technologies, our products, or the use of our products, do not infringe third-party patents. Third parties might allege that we are infringing their patent rights or that we have misappropriated their trade secrets. Such third parties might resort to litigation against us. The basis of such litigation could be existing patents or patents that issue in the future.
It is also possible that we failed to identify relevant third-party patents or applications. For example, applications filed before November 29, 2000 and certain applications filed after that date that will not be filed outside the United States remain confidential until patents issue. Patent applications in the United States and elsewhere are published approximately 18 months after the earliest filing, which is referred to as the priority date. Therefore, patent applications covering our products or platform technology could have been filed by others without our knowledge. Additionally, pending patent applications which have been published can, subject to certain limitations, be later amended in a manner that could cover our platform technologies, our products or the use of our products.
With regard to tivozanib, we are aware of a third-party United States patent, and corresponding foreign counterparts, that contain broad claims related to use of an organic compound, that, among other things, inhibits the tyrosine phosphorylation of a VEGF receptor caused by VEGF binding to such VEGF receptor. We are also aware of third-party United States patents that contain broad claims related to the use of a tyrosine kinase inhibitor in combination with a DNA damaging agent such as chemotherapy or radiation and we have received written notice from the owners of such patents indicating that they believe we may need a license from them in order to avoid infringing their patents. With regard to ficlatuzumab, we are aware of two separate families of United States patents, United States patent applications and foreign counterparts, with each of the two families being owned by a different third-party, that contain broad claims related to anti-HGF antibodies having certain binding properties and their use. With regard to AV-203, we are aware of a third-party United States patent that contains broad claims relating to anti-ErbB3 antibodies. Based on our analyses, if any of the above third-party patents were asserted against us, we do
47
Table of Contents
not believe our proposed products or activities would be found to infringe any valid claim of these patents. If we were to challenge the validity of any issued United States patent in court, we would need to overcome a statutory presumption of validity that attaches to every United States patent. This means that in order to prevail, we would have to present clear and convincing evidence as to the invalidity of the patent’s claims. There is no assurance that a court would find in our favor on questions of infringement or validity.
In order to avoid or settle potential claims with respect to any of the patent rights described above or any other patent rights of third parties, we may choose or be required to seek a license from a third-party and be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we or our strategic partners were able to obtain a license, the rights may be non-exclusive, which could result in our competitors gaining access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations, if, as a result of actual or threatened patent infringement claims, we are unable to enter into licenses on acceptable terms. This could harm our business significantly.
Defending against claims of patent infringement or misappropriation of trade secrets could be costly and time-consuming, regardless of the outcome. Thus, even if we were to ultimately prevail, or to settle at an early stage, such litigation could burden us with substantial unanticipated costs. In addition, litigation or threatened litigation could result in significant demands on the time and attention of our management team, distracting them from the pursuit of other company business.
Unfavorable outcomes in intellectual property litigation could limit our research and development activities and/or our ability to commercialize certain products.
If third parties successfully assert intellectual property rights against us, we might be barred from using aspects of our technology platform, or barred from developing and commercializing related products. Prohibitions against using specified technologies, or prohibitions against commercializing specified products, could be imposed by a court or by a settlement agreement between us and a plaintiff. In addition, if we are unsuccessful in defending against allegations of patent infringement or misappropriation of trade secrets, we may be forced to pay substantial damage awards to the plaintiff. There is inevitable uncertainty in any litigation, including intellectual property litigation. There can be no assurance that we would prevail in any intellectual property litigation, even if the case against us is weak or flawed. If litigation leads to an outcome unfavorable to us, we may be required to obtain a license from the patent owner in order to continue our research and development programs or to market our product(s). It is possible that the necessary license will not be available to us on commercially acceptable terms, or at all. This could limit our research and development activities, our ability to commercialize specified products, or both.
Most of our competitors are larger than we are and have substantially greater resources. They are, therefore, likely to be able to sustain the costs of complex patent litigation longer than we could. In addition, the uncertainties associated with litigation could have a material adverse effect on our ability to raise the funds necessary to continue our clinical trials, continue our internal research programs, in-license needed technology, or enter into strategic partnerships that would help us bring our product candidates to market.
In addition, any future patent litigation, interference or other administrative proceedings will result in additional expense and distraction of our personnel. An adverse outcome in such litigation or proceedings may expose us or our strategic partners to loss of our proprietary position, expose us to significant liabilities, or require us to seek licenses that may not be available on commercially acceptable terms, if at all.
Intellectual property litigation may lead to unfavorable publicity that harms our reputation and causes the market price of our common stock to decline.
During the course of any patent litigation, there could be public announcements of the results of hearings, rulings on motions, and other interim proceedings in the litigation. If securities analysts or investors regard these
48
Table of Contents
announcements as negative, the perceived value of our products, programs, or intellectual property could be diminished. Accordingly, the market price of our common stock may decline.
AV-380 and tivozanib are protected by patents exclusively licensed from other companies or institutions. If the licensors terminate the licenses or fail to maintain or enforce the underlying patents, our competitive position and our market share in the markets for any of our approved products will be harmed.
We are a party to several license agreements under which certain aspects of our business depend on patents and/or patent applications owned by other companies or institutions. In particular, we hold exclusive licenses from St. Vincent’s for therapeutic applications that benefit from inhibition or decreased expression or activity of MIC-1, which we refer to as GDF15 and which we are using in our AV-380 program and from KHK for tivozanib. We may enter into additional license agreements as part of the development of our business in the future. Our licensors may not successfully prosecute certain patent applications under which we are licensed and on which our business depends. Even if patents issue from these applications, our licensors may fail to maintain these patents, may decide not to pursue litigation against third-party infringers, may fail to prove infringement, or may fail to defend against counterclaims of patent invalidity or unenforceability. In addition, in spite of our best efforts, our licensors might conclude that we have materially breached our license agreements and might therefore terminate the license agreements, thereby removing our ability to obtain regulatory approval and to market products covered by these license agreements. If these in-licenses are terminated, or if the underlying patents fail to provide the intended market exclusivity, competitors would have the freedom to seek regulatory approval of, and to market, products identical to ours. This could have a material adverse effect on our competitive business position and our business prospects.
Confidentiality agreements with employees and third parties may not prevent unauthorized disclosure of trade secrets and other proprietary information.
In addition to patents, we rely on trade secrets, technical know-how, and proprietary information concerning our business strategy in order to protect our competitive position in the field of oncology. In the course of our research, development and business activities, we often rely on confidentiality agreements to protect our proprietary information. Such confidentiality agreements are used, for example, when we talk to vendors of laboratory or clinical development services or potential strategic partners. In addition, each of our employees is required to sign a confidentiality agreement upon joining our company. We take steps to protect our proprietary information, and we seek to carefully draft our confidentiality agreements to protect our proprietary interests. Nevertheless, there can be no guarantee that an employee or an outside party will not make an unauthorized disclosure of our proprietary confidential information. This might happen intentionally or inadvertently. It is possible that a competitor will make use of such information, and that our competitive position will be compromised, in spite of any legal action we might take against persons making such unauthorized disclosures.
Trade secrets are difficult to protect. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, or outside scientific collaborators might intentionally or inadvertently disclose our trade secret information to competitors. Enforcing a claim that a third-party illegally obtained and is using any of our trade secrets is expensive and time-consuming, and the outcome is unpredictable. In addition, courts outside the United States sometimes are less willing than U.S. courts to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
Our research and development strategic partners may have rights to publish data and other information to which we have rights. In addition, we sometimes engage individuals or entities to conduct research relevant to our business. The ability of these individuals or entities to publish or otherwise publicly disclose data and other information generated during the course of their research is subject to certain contractual limitations. These contractual provisions may be insufficient or inadequate to protect our confidential information. If we do not apply for patent protection prior to such publication, or if we cannot otherwise maintain the confidentiality of our proprietary technology and other confidential information, then our ability to obtain patent protection or to protect our trade secret information may be jeopardized.
49
Table of Contents
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage. The following examples are illustrative:
| • | Others may be able to make compounds that are similar to our product candidates but that are not covered by the claims of the patents that we own or have exclusively licensed. |
| • | We or our licensors or strategic partners might not have been the first to make the inventions covered by the issued patent or pending patent application that we own or have exclusively licensed. |
| • | We or our licensors or strategic partners might not have been the first to file patent applications covering certain of our inventions. |
| • | Others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights. |
| • | It is possible that our pending patent applications will not lead to issued patents. |
| • | Issued patents that we own or have exclusively licensed may not provide us with a competitive advantage; for example, our issued patents may not be broad enough to prevent the commercialization of competitive antibodies that are biosimilar to one or more of our antibody products, or may be held invalid or unenforceable, as a result of legal challenges by our competitors. |
| • | Our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets. |
| • | We may not develop additional proprietary technologies that are patentable. |
| • | The patents of others may have an adverse effect on our business. |
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharma industry involve both technological complexity and legal complexity. Therefore, obtaining and enforcing biopharma patents is costly, time-consuming and inherently uncertain. In addition, several recent events have increased uncertainty with regard to our ability to obtain patents in the future and the value of patents once obtained. Among these, in September 2011, patent reform legislation passed by Congress was signed into law. The new patent law introduces changes including a first-to-file system for determining which inventors may be entitled to receive patents, and a new post-grant review process that allows third parties to challenge newly issued patents. It remains to be seen how the biopharma industry will be affected by such changes in the patent system. In addition, the Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in specified circumstances or weakening the rights of patent owners in specified situations. Depending on decisions by the U.S. Congress, the federal courts, and the U.S. Patent and Trademark Office, the laws and regulations governing patents could change in unpredictable ways that could weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
50
Table of Contents
Risks Related to Ownership of Our Common Stock
The market price of our common stock has been, and is likely to continue to be, highly volatile, and could fall below the price you paid. A significant decline in the value of our stock price could also result in securities class-action litigation against us.
The market price of our common stock has been, and is likely to continue to be, highly volatile and subject to wide fluctuations in price in response to various factors, many of which are beyond our control, including:
| • | new products, product candidates or new uses for existing products introduced or announced by our strategic partners, or our competitors, and the timing of these introductions or announcements; |
| • | actual or anticipated results from and any delays in our clinical trials; |
| • | results of regulatory reviews relating to the approval of our product candidates; |
| • | the results of our efforts to discover, develop, acquire or in-license additional product candidates or products; |
| • | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| • | announcements by us of material developments in our business, financial condition and/or operations; |
| • | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures and capital commitments; |
| • | additions or departures of key scientific or management personnel; |
| • | conditions or trends in the biotechnology and biopharmaceutical industries; |
| • | actual or anticipated changes in earnings estimates, development timelines or recommendations by securities analysts; |
| • | general economic and market conditions and other factors that may be unrelated to our operating performance or the operating performance of our competitors, including changes in market valuations of similar companies; and |
| • | sales of common stock by us or our stockholders in the future, as well as the overall trading volume of our common stock. |
In addition, the stock market in general and the market for biotechnology and biopharmaceutical companies in particular have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. These broad market and industry factors may seriously harm the market price of our common stock, regardless of our operating performance.
In the past, following periods of volatility in the market, such as the volatility in our stock price following our May 2, 2013 announcement regarding the vote of the Oncologic Drugs Advisory Committee of the FDA, or ODAC, securities class-action litigation has often been instituted against companies. For example, we, and certain of our executive officers, have been named as defendants in a consolidated purported class action lawsuit following our announcement of the ODAC vote. Moreover, a plaintiff has filed a derivative complaint allegedly on our behalf, naming us as a nominal defendant and also naming as defendants present and former members of our board of directors, alleging breach of fiduciary duty and abuse of control between January 2012 and May 2013 with respect to allegedly misleading statements and omissions regarding tivozanib. We and certain of our executive officers have been named as defendants in multiple lawsuits that could result in substantial costs and divert management’s attention. These proceedings and other similar litigation, if instituted against us, could result in substantial costs and diversion of management’s attention and resources, which could materially and adversely affect our business and financial condition.
51
Table of Contents
Our management has broad discretion over our use of available cash and cash equivalents and might not spend our available cash and cash equivalents in ways that increase the value of your investment.
Our management has broad discretion on where and how to use our cash and cash equivalents and you will be relying on the judgment of our management regarding the application of our available cash and cash equivalents to fund our operations. Our management might not apply our cash or cash equivalents in ways that increase the value of your investment. We expect to use a substantial portion of our cash to fund existing and future research and development of our preclinical and clinical product candidates, with the balance, if any, to be used for working capital and other general corporate purposes, which may in the future include investments in, or acquisitions of, complementary businesses, joint ventures, partnerships, services or technologies. Our management might not be able to yield a significant return, if any, on any investment of this cash. You will not have the opportunity to influence our decisions on how to use our cash reserves.
Fluctuations in our quarterly operating results could adversely affect the price of our common stock.
Our quarterly operating results may fluctuate significantly. Some of the factors that may cause our operating results to fluctuate on a period-to-period basis include:
| • | the status of our preclinical and clinical development programs; |
| • | the level of expenses incurred in connection with our preclinical and clinical development programs, including development and manufacturing costs relating to our preclinical and clinical development candidates; |
| • | any intellectual property infringement lawsuit or other litigation in which we may become involved; |
| • | the implementation of our current restructuring and cost-savings strategies; |
| • | the implementation or termination of collaboration, licensing, manufacturing or other material agreements with third parties, and non-recurring revenue or expenses under any such agreement; |
| • | costs associated with lawsuits against us, including the current purported class action and derivative lawsuits described elsewhere in this Quarterly Report under “Part I, Item 3—Legal Proceedings;” |
| • | changes in our loan agreement with Hercules, including the existence of any event of default that may accelerate payments due thereunder; and |
| • | compliance with regulatory requirements. |
Period-to-period comparisons of our historical and future financial results may not be meaningful, and investors should not rely on them as an indication of future performance. Our fluctuating results may fail to meet the expectations of securities analysts or investors. Our failure to meet these expectations may cause the price of our common stock to decline.
Unstable market and economic conditions may have serious adverse consequences on our business, financial condition and stock price.
As widely reported, global credit and financial markets have been experiencing extreme volatility, and in some cases, disruptions, over the past several years, in many cases, over extended periods. Although certain of these trends have recently showed signs of reversing, there can be no assurance that rapid or extended periods of deterioration in credit and financial markets and confidence in economic conditions will not occur. Our general business strategy may be adversely affected by external economic conditions and a volatile business environment or unpredictable and unstable market conditions. If the equity and credit markets are not favorable at any time we seek to raise capital, it may make any necessary debt or equity financing more difficult, more costly, and more dilutive. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance and stock price and could require us to delay or abandon clinical development plans. In addition, there is a risk that one or more of our current service providers, manufacturers or other partners may not survive economically turbulent times, which could directly affect our ability to attain our operating goals on schedule and on budget.
52
Table of Contents
At December 31, 2014, we had $52.3 million of cash and cash equivalents consisting of cash on deposit with banks, money market funds, U.S. government agency securities, and corporate debt securities, including commercial paper. As of the date of this report, we are not aware of any downgrades, material losses, or other significant deterioration in the fair value of our cash equivalents. However, no assurance can be given that deterioration in conditions of the global credit and financial markets would not negatively impact our current portfolio of cash equivalents or our ability to meet our financing objectives. Dislocations in the credit market may adversely impact the value and/or liquidity of cash equivalents owned by us.
There is a possibility that our stock price may decline because of volatility of the stock market and general economic conditions.
Future sales of shares of our common stock, including shares issued upon the exercise of currently outstanding options, could negatively affect our stock price.
A substantial portion of our outstanding common stock can be traded without restriction at any time. Some of these shares are currently restricted as a result of securities laws, but will be able to be sold, subject to any applicable volume limitations under federal securities laws with respect to affiliate sales, in the near future. As such, sales of a substantial number of shares of our common stock in the public market could occur at any time. These sales, or the perception in the market that the holders of a large number of shares intend to sell such shares, could reduce the market price of our common stock. In addition, we have a significant number of shares that are subject to outstanding options. The exercise of these options and the subsequent sale of the underlying common stock could cause a further decline in our stock price. These sales also might make it difficult for us to sell equity securities in the future at a time and at a price that we deem appropriate.
Provisions in our certificate of incorporation, our by-laws or Delaware law might discourage, delay or prevent a change in control of our company or changes in our management and, therefore, depress the market price of our common stock.
Provisions of our certificate of incorporation, our by-laws or Delaware law may have the effect of deterring unsolicited takeovers or delaying or preventing a change in control of our company or changes in our management, including transactions in which our stockholders might otherwise receive a premium for their shares over then current market prices. In addition, these provisions may limit the ability of stockholders to approve transactions that they may deem to be in their best interest. These provisions include:
| • | advance notice requirements for stockholder proposals and nominations; |
| • | the inability of stockholders to act by written consent or to call special meetings; |
| • | the ability of our board of directors to make, alter or repeal our by-laws; and |
| • | the ability of our board of directors to designate the terms of and issue new series of preferred stock without stockholder approval, which could be used to institute a rights plan, or a poison pill, that would work to dilute the stock ownership of a potential hostile acquirer, likely preventing acquisitions that have not been approved by our board of directors. |
In addition, Section 203 of the Delaware General Corporation Law prohibits a publicly-held Delaware corporation from engaging in a business combination with an interested stockholder, generally a person which together with its affiliates owns, or within the last three years has owned, 15% of our voting stock, for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner.
The existence of the foregoing provisions and anti-takeover measures could limit the price that investors might be willing to pay in the future for shares of our common stock. They could also deter potential acquirers of our company, thereby reducing the likelihood that a stockholder could receive a premium for shares of our common stock held by a stockholder in an acquisition.
53
Table of Contents
Our business could be negatively affected as a result of the actions of activist shareholders.
Proxy contests have been waged against companies in the biopharmaceutical industry over the last few years. If faced with a proxy contest, we may not be able to successfully respond to the contest, which would be disruptive to our business. Even if we are successful, our business could be adversely affected by a proxy contest because:
| • | responding to proxy contests and other actions by activist shareholders may be costly and time-consuming, and may disrupt our operations and divert the attention of management and our employees; |
| • | perceived uncertainties as to the potential outcome of any proxy contest may result in our inability to consummate potential acquisitions, collaborations or in-licensing opportunities and may make it more difficult to attract and retain qualified personnel and business partners; and |
| • | if individuals that have a specific agenda different from that of our management or other members of our board of directors are elected to our board as a result of any proxy contest, such an election may adversely affect our ability to effectively and timely implement our strategic plan and create additional value for our stockholders. |
Failure to maintain effective internal controls in accordance with Section 404 of the Sarbanes-Oxley Act could have a material adverse effect on our ability to produce accurate financial statements and on our stock price.
Section 404 of the Sarbanes-Oxley Act of 2002 requires us, on an annual basis, to review and evaluate our internal controls, and requires our independent registered public accounting firm to attest to the effectiveness of our internal controls. Despite our efforts, we can provide no assurance as to our, or our independent registered public accounting firm’s, conclusions with respect to the effectiveness of our internal control over financial reporting under Section 404. There is a risk that neither we nor our independent registered public accounting firm will be able to continue to conclude within the prescribed timeframe that our internal control over financial reporting is effective as required by Section 404. This could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
We do not expect to pay any cash dividends for the foreseeable future.
You should not rely on an investment in our common stock to provide dividend income. We do not anticipate that we will pay any cash dividends to holders of our common stock in the foreseeable future. Instead, we plan to retain any earnings to maintain and expand our existing operations. In addition, our ability to pay cash dividends is currently prohibited by the terms of our debt financing arrangements and any future debt financing arrangement may contain terms prohibiting or limiting the amount of dividends that may be declared or paid on our common stock. Accordingly, investors must rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize any return on their investment. As a result, investors seeking cash dividends should not purchase our common stock.
| ITEM 1B. | Unresolved Staff Comments |
None.
| ITEM 2. | Properties |
We sublease our principal facilities, which consist of approximately 49,185 square feet of office, research and laboratory space located at 650 East Kendall Street, Cambridge, Massachusetts. In accordance with the terms of our lease at 650 East Kendall Street, Cambridge, Massachusetts, we are exiting our current facility through the end of May and plan to move into a new facility to better accommodate our current needs. We are currently in negotiations with various landlords regarding such new office space.
54
Table of Contents
| ITEM 3. | Legal Proceedings |
Two class action lawsuits have been filed against us and certain of our present and former officers and members of our board of directors, (Tuan Ha-Ngoc, David N. Johnston, William Slichenmyer and Ronald DePinho), in the United States District Court for the District of Massachusetts, one captioned Paul Sanders v. Aveo Pharmaceuticals, Inc., et al., No. 1:13-cv-11157-JLT, filed on May 9, 2013, and the other captioned Christine Krause v. AVEO Pharmaceuticals, Inc., et al., No. 1:13-cv-11320-JLT, filed on May 31, 2013. On December 4, 2013, the District Court consolidated the complaints as In re AVEO Pharmaceuticals, Inc. Securities Litigation et al., No. 1:13-cv-11157-DJC, and an amended complaint was filed on February 3, 2014. The amended complaint purports to be brought on behalf of shareholders who purchased our common stock between January 3, 2012 and May 1, 2013. The amended complaint generally alleges that we and certain of our present and former officers and directors violated Sections 10(b) and/or 20(a) of the Securities Exchange Act of 1934 and Rule 10b-5 promulgated thereunder by making allegedly false and/or misleading statements concerning the phase 3 trial design and results for our TIVO-1 study in an effort to lead investors to believe that the drug would receive approval from the FDA. The amended complaint seeks unspecified damages, interest, attorneys’ fees, and other costs. On April 4, 2014, we filed a motion to dismiss the consolidated class action complaint with prejudice. Lead plaintiffs filed an opposition to the motion to dismiss on June 10, 2014, and we filed a reply to the opposition on July 10, 2014. The Court heard oral argument on our motion to dismiss on July 22, 2014. We deny any allegations of wrongdoing and intend to vigorously defend against this lawsuit. However, there is no assurance that we will be successful in our defense or that insurance will be available or adequate to fund any settlement or judgment or the litigation costs of the action. Moreover, we are unable to predict the outcome or reasonably estimate a range of possible loss at this time.
On April 4, 2014, Karen J. van Ingen, a purported purchaser of AVEO stock, filed a derivative complaint allegedly on behalf of AVEO in the United States District Court for the District of Massachusetts, Civil Action No. 1:14-cv-11672-DJC, naming AVEO, as a nominal defendant and also naming as defendants present and former members of our board of directors, including Tuan Ha-Ngoc, Henri A. Termeer, Kenneth M. Bate, Anthony B. Evnin, Robert Epstein, Raju Kucherlapati, Robert C. Young, and Kenneth E. Weg. The complaint alleges breach of fiduciary duty and abuse of control between January 2012 and May 2013 with respect to allegedly misleading statements and omissions regarding tivozanib. The complaint seeks, among other relief, unspecified damages, costs and expenses, including attorneys’ fees, an order requiring us to implement certain corporate governance reforms, restitution from the defendants and such other relief as the court might find just and proper. On July 25, 2014, defendants filed a motion to dismiss the derivative complaint with prejudice. Plaintiff filed an opposition to the motion to dismiss on September 23, 2014, and we filed a reply to the opposition on October 23, 2014. The Court heard oral argument on our motion to dismiss on January 7, 2015. We deny any allegations of wrongdoing and intend to vigorously defend this lawsuit. However, there is no assurance that we will be successful in our defense or that insurance will be available or adequate to fund any settlement or judgment or the litigation costs of this action. Moreover, we are unable to predict the outcome or reasonably estimate a range of possible loss at this time.
On July 3, 2013, we received a subpoena from the SEC, requesting documents and information concerning tivozanib, including related communications with the FDA, investors and others. We are fully cooperating with the SEC regarding this fact-finding inquiry. The SEC has informed us that this inquiry should not be construed as an indication that any violations of law have occurred or that the SEC has any negative opinion of any person, entity or security.
| ITEM 4. | Mine Safety Disclosures |
Not applicable.
55
Table of Contents
| ITEM 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
MARKET PRICE INFORMATION
Our common stock is traded on the NASDAQ Global Market under the symbol “AVEO”. The following table sets forth the high and low sale prices per share for our common stock for the periods indicated:
| High | Low | |||||||
| 2013 |
||||||||
| First Quarter |
$ | 8.94 | $ | 6.35 | ||||
| Second Quarter |
$ | 8.40 | $ | 2.25 | ||||
| Third Quarter |
$ | 2.68 | $ | 2.03 | ||||
| Fourth Quarter |
$ | 2.35 | $ | 1.54 | ||||
| High | Low | |||||||
| 2014 |
||||||||
| First Quarter |
$ | 2.07 | $ | 1.49 | ||||
| Second Quarter |
$ | 1.85 | $ | 1.00 | ||||
| Third Quarter |
$ | 1.92 | $ | 1.06 | ||||
| Fourth Quarter |
$ | 1.19 | $ | 0.61 | ||||
HOLDERS
At February 23, 2015, there were approximately 54 holders of record of our common stock. We believe that the number of beneficial owners of our common stock at that date was substantially greater.
DIVIDENDS
We have never declared or paid any cash dividends on our common stock and our ability to pay cash dividends is currently prohibited by the terms of our debt financing arrangements. We currently intend to retain earnings, if any, for use in our business and do not anticipate paying cash dividends on our common stock in the foreseeable future. Payment of future dividends, if any, on our common stock will be at the discretion of our board of directors after taking into account various factors, including our financial condition, operating results, anticipated cash needs, and plans for expansion.
SECURITIES AUTHORIZED FOR ISSUANCE UNDER EQUITY COMPENSATION PLANS
The information required by this item will be set forth in the definitive proxy statement we will file in connection with our 2015 Annual Meeting of Stockholders and is incorporated by reference herein.
PURCHASE OF EQUITY SECURITIES
We did not purchase any of our equity securities during the period covered by this Annual Report on Form 10-K.
RECENT SALES OF UNREGISTERED SECURITIES
Except as disclosed in our Current Report on Form 8-K filed with the Securities and Exchange Commission on September 26, 2014, we did not issue any unregistered securities during the period covered by this Annual Report on Form 10-K.
56
Table of Contents
Comparative Stock Performance Graph
The information included under the heading “Comparative Stock Performance Graph” in this Item 5 of Part II of this Annual Report on Form 10-K shall not be deemed to be “soliciting material” or subject to Regulation 14A or 14C, shall not be deemed “filed” for purposes of Section 18 of the Exchange Act, or otherwise subject to the liabilities of that section, nor shall it be deemed incorporated by reference in any filing under the Securities Act of 1933, as amended, or the Exchange Act.
Set forth below is a graph comparing the total cumulative returns of AVEO, the NASDAQ Composite Index and the NASDAQ Biotechnology Index. The graph assumes $100 was invested on March 12, 2010 in our common stock and each of the indices and that all dividends, if any, are reinvested.
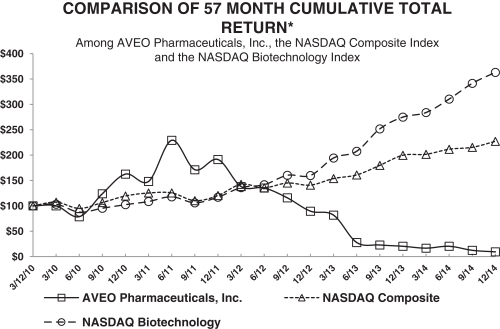
*$100 invested on 3/12/10 in stock or 2/28/10 in index, including reinvestment of dividends.
Fiscal year ending December 31.
| Company/Market/Peer Group |
3/12/2010 | 12/31/2010 | 12/31/2011 | 12/31/2012 | 12/31/2013 | 12/31/2014 | ||||||||||||||||||
| AVEO Pharmaceuticals |
$ | 100.00 | $ | 162.63 | $ | 191.32 | $ | 89.54 | $ | 20.36 | $ | 9.35 | ||||||||||||
| NASDAQ Composite Index |
$ | 100.00 | $ | 119.48 | $ | 120.34 | $ | 140.89 | $ | 199.70 | $ | 227.07 | ||||||||||||
| NASDAQ Biotechnology Index |
$ | 100.00 | $ | 102.34 | $ | 117.12 | $ | 159.87 | $ | 274.94 | $ | 363.12 | ||||||||||||
57
Table of Contents
| ITEM 6. | Selected Financial Data |
The following selected consolidated financial data should be read in conjunction with our Consolidated Financial Statements and the Accompanying Notes thereto and Management’s Discussion and Analysis of Financial Condition and Results of Operations included elsewhere in this Annual Report on Form 10-K. The Balance Sheet Data at December 31, 2014 and 2013 and the Statement of Operations Data for each of the three years in the period ended December 31, 2014 have been derived from our audited Consolidated Financial Statements for such years, included elsewhere in this Annual Report on Form 10-K. The Balance Sheet Data at December 31, 2012, 2011 and 2010, and the Statement of Operations Data for each of the two years in the period ended December 31, 2011 have been derived from the audited Consolidated Financial Statements for such years not included in this Annual Report on Form 10-K.
Our historical results for any prior period are not necessarily indicative of results to be expected in any future period.
| Years Ended December 31, |
||||||||||||||||||||
| 2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| Statement of operations data: |
||||||||||||||||||||
| Revenue |
$ | 18,123 | $ | 1,293 | $ | 19,286 | $ | 164,849 | $ | 44,682 | ||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
38,254 | 68,468 | 91,358 | 101,735 | 86,345 | |||||||||||||||
| General and administrative |
18,589 | 28,712 | 36,932 | 29,167 | 14,763 | |||||||||||||||
| Restructuring and lease exit |
11,729 | 8,017 | 2,633 | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating expenses |
68,572 | 105,197 | 130,923 | 130,902 | 101,108 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| (Loss) income from operations |
(50,449 | ) | (103,904 | ) | (111,637 | ) | 33,947 | (56,426 | ) | |||||||||||
| Other income and expense: |
||||||||||||||||||||
| Other income (expense), net |
66 | (123 | ) | 247 | 10 | 900 | ||||||||||||||
| Interest expense |
(2,388 | ) | (3,127 | ) | (3,501 | ) | (3,836 | ) | (3,389 | ) | ||||||||||
| Interest income |
32 | 125 | 497 | 527 | 126 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Other expense, net |
(2,290 | ) | (3,125 | ) | (2,757 | ) | (3,299 | ) | (2,363 | ) | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net (loss) income before benefit for income taxes |
(52,739 | ) | (107,029 | ) | (114,394 | ) | 30,648 | (58,789 | ) | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net (loss) income |
$ | (52,739 | ) | $ | (107,029 | ) | $ | (114,394 | ) | $ | 30,648 | $ | (58,789 | ) | ||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net (loss) income per share—basic |
$ | (1.01 | ) | $ | (2.10 | ) | $ | (2.64 | ) | $ | 0.77 | $ | (2.30 | ) | ||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Weighted average number of common shares used in net (loss) income per share calculation—basic |
52,289 | 50,928 | 43,374 | 39,715 | 25,582 | |||||||||||||||
| Net (loss) income per share—diluted |
$ | (1.01 | ) | $ | (2.10 | ) | $ | (2.64 | ) | $ | 0.74 | $ | (2.30 | ) | ||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Weighted average number of common shares and dilutive common share equivalents used in net (loss) income per share calculation—diluted |
52,289 | 50,928 | 43,374 | 41,473 | 25,582 | |||||||||||||||
| As of December 31, | ||||||||||||||||||||
| 2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Balance sheet data: |
||||||||||||||||||||
| Cash, cash equivalents, and marketable securities |
$ | 52,306 | $ | 118,506 | $ | 160,602 | $ | 275,440 | $ | 140,198 | ||||||||||
| Working capital |
18,773 | 97,511 | 151,551 | 199,786 | 103,360 | |||||||||||||||
| Total assets |
70,662 | 146,346 | 207,469 | 295,050 | 151,048 | |||||||||||||||
| Loans payable, including current portion, net of discount |
20,652 | 19,205 | 26,037 | 24,170 | 23,402 | |||||||||||||||
| Accumulated deficit |
(480,028 | ) | (427,289 | ) | (320,260 | ) | (205,866 | ) | (236,514 | ) | ||||||||||
| Total stockholders’ equity |
20,606 | 69,938 | 118,938 | 223,541 | 71,770 | |||||||||||||||
58
Table of Contents
| ITEM 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and the related notes appearing elsewhere in this report. Some of the information contained in this discussion and analysis or set forth elsewhere in this report, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. You should read the “Risk Factors” section in Part 1—Item 1A. of this report for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are a biopharmaceutical company committed to developing targeted therapies through biomarker-driven insights to provide substantial improvements in patient outcomes where significant unmet medical needs exist. Our proprietary platform has delivered unique insights into cancer and related disease. Our strategy is to leverage these biomarker insights and partner resources to advance the development of our clinical pipeline. As further described below, we currently are exploring partnership opportunities to fund the further development of three of our four development programs, including our lead program for tivozanib.
Our development programs, which seek to advance our clinical stage assets, are as follows:
| • | Tivozanib: In 2006, we acquired exclusive rights to develop and commercialize tivozanib, outside of Asia pursuant to a license agreement we entered into with Kirin Brewery Co. Ltd. (now Kyowa Hakko Kirin), or KHK. Tivozanib is a potent, selective long half-life vascular endothelial growth factor tyrosine kinase inhibitor of all three vascular endothelial growth factors, or VEGF TKI. In a global, phase 3 clinical trial comparing the efficacy and safety of tivozanib with Nexavar® (sorafenib), an approved therapy, for first-line treatment of renal cell carcinoma, or RCC, the trial met its primary endpoint for progression-free survival, or PFS, but showed a non-statistically significant trend favoring the sorafenib arm in overall survival, or OS. Based on a review of our application for approval of the use of tivozanib for the treatment of first line advanced RCC, in June 2013, the U.S. Food and Drug Administration, or FDA, issued a Complete Response Letter informing us that they would not approve tivozanib at this time based on these study data. |
In August 2014, as discussed below under the heading “Strategic Partnerships,” our strategic collaboration with Astellas for the development of tivozanib terminated. Upon reversion back to AVEO of rights previously granted Astellas, we reevaluated our tivozanib regulatory and development strategy, as well as partnering opportunities. In November 2014, we filed a letter of intent with the European Medicines Agency, allowing us to evaluate the opportunity for submitting a Marketing Authorization Application for tivozanib for the treatment of RCC based on clinical trials conducted to date. Moreover, we are evaluating the opportunity to conduct an additional adequate and well-controlled randomized trial of tivozanib in the refractory RCC setting using PFS as the primary endpoint and OS as a secondary endpoint to address the OS concerns raised by the FDA from our tivozanib study. We plan to have discussion with the FDA to evaluate if such a study design coupled with the prior study findings would allow for an advanced RCC label for tivozanib and after such discussion, we will explore the opportunity for future clinical development in conjunction with our funding resources.
In November 2014, we entered into a research and exclusive option agreement with Ophthotech Corporation, or Ophthotech, under which we granted Ophthotech an option to develop and commercialize tivozanib for the potential diagnosis, prevention and treatment of non-oncologic diseases or conditions of the eye in humans.
Additionally, we recently announced results from a predefined biomarker analysis of our BATON-CRC study, a randomized phase 2 clinical trial of modified FOLFOX6, a commonly used chemotherapy, combined with tivozanib or Avastin® (bevacizumab), which both target angiogenesis
59
Table of Contents
signaling pathways, in first line treatment of metastatic colorectal cancer, or CRC. In this study, among prospectively defined biomarkers, patients with low neuropilin-1, a cell surface protein that modulates blood vessel development (which we refer to a NRP-1), showed an improved PFS versus patients with high NRP-1 in both treatment arms, supporting the value of NRP-1 as a potential prognostic marker for angiogenesis inhibitors. Further, patients with low serum NRP-1 demonstrated longer PFS when treated with tivozanib compared to bevacizumab, which suggests that first line colorectal cancer patients with low NRP-1 levels may benefit from treatment with tivozanib over bevacizumab, a standard of care in this disease.
Based on the NRP-1 findings, we plan to discuss potential registration studies with the FDA and are currently exploring additional partnership opportunities to further the development of tivozanib in CRC. We are also evaluating the opportunity to pursue the registration of tivozanib for RCC in the European Union based on prior study findings.
We are currently exploring additional partnership opportunities to further the development of tivozanib.
| • | Ficlatuzumab: Ficlatuzumab is a potent Hepatocyte Growth Factor, or HGF, inhibitory antibody. HGF is the sole known ligand of the c-Met receptor which is believed to trigger many activities that are involved in cancer development and metastasis. We have completed two phase 1 clinical studies of ficlatuzumab administered as a single agent and in combination with erlotinib, a tyrosine kinase inhibitor, or TKI, of the epidermal growth factor receptor, or EGFR, and a phase 2 clinical study evaluating ficlatuzumab in combination with gefitinib, an EGFR TKI, in first line non-small cell lung cancer, or NSCLC. The phase 2 trial failed to demonstrate a statistically significant benefit in the intent-to-treat population. However, an exploratory analysis using a serum-based molecular diagnostic test, known as VeriStrat®, identified a sub-population of patients who experienced a progression free survival and overall survival benefit from the addition of ficlatuzumab to gefitinib. VeriStrat is commercially available to help physicians guide treatment decisions for patients with second line advanced NSCLC. Data from the exploratory analyses with VeriStrat prompted the development of a separate investigational companion diagnostic device called BDX004. Based upon the exploratory analyses, BDX004 may be indicative of a predictive biomarker for the combination of ficlatuzumab and EGFR TKI over EGFR TKI alone in the first line EGFR mutation patients who have been previously identified to not respond well to the current standard of care. |
In April 2014, we entered into a worldwide agreement with Biodesix, Inc. to develop and commercialize ficlatuzumab with BDX004, a serum based diagnostic test which has been derived from the VeriStrat test, employing the same methodology and data processing algorithms as VeriStrat, for use in a confirmatory clinical trial. Pursuant to the agreement, we are conducting a phase 2 proof of concept study of ficlatuzumab, which we refer to as the FOCAL study, in combination with erlotinib in first line advanced NSCLC patients who have an EGFR mutation selected using the BDX004 test to identify the patient subset that is most likely to benefit from the addition of ficlatuzumab to the EGFR TKI. Biodesix will fund up to $15 million of the cost of this study, as well as all of the costs associated with development and registration of BDX004, and any additional development, regulatory and commercial costs for ficlatuzumab will be shared equally. Subject to regulatory approval, AVEO will lead worldwide commercialization of ficlatuzumab.
| • | AV-203: AV-203 is a potent anti-ErbB3 specific monoclonal antibody with high ErbB3 affinity.We have observed potent anti-tumor activity in mouse models. AV-203 selectively inhibits the activity of the ErbB3 receptor and our preclinical studies suggest that neuregulin-1, or NRG1 (also known as heregulin), levels predict AV-203 anti-tumor activity in preclinical models. We have completed a phase 1 dose escalation study of AV-203, which established a recommended phase 2 dose of AV-203 at 20mg/kg intravenously every 2 weeks, demonstrated good tolerability and promising early signs of activity, and reached the maximum planned dose of AV-203 monotherapy. No anti-drug antibodies were detected, and pharmacokinetic results indicated a dose-proportional increase in levels of AV-203. |
60
Table of Contents
| The expansion cohort of this study among patients with a specific biomarker has been discontinued. We are seeking to pursue further clinical development of AV-203 with a strategic partner. |
| • | AV-380 Program: AV-380 is a potent humanized IgG1 inhibitory monoclonal antibody targeting growth differentiating factor-15, or GDF15, a divergent member of the TGF-ß family, for the potential treatment or prevention of cachexia. Cachexia is defined as a multi-factorial syndrome of involuntary weight loss characterized by an ongoing loss of skeletal muscle mass (with or without loss of fat mass) that cannot be fully reversed by conventional nutritional support and leads to progressive functional impairment. Cachexia is associated with various cancers as well as diseases outside of cancer including chronic kidney disease, or CKD, congestive heart failure or CHF, and chronic obstructive pulmonary disease, or COPD. We believe that AV-380 represents a unique approach to treating cachexia because it addresses key underlying mechanisms of the syndrome and focuses a significant area of patient need. It is estimated that approximately 30% of all cancer patients die due to cachexia and over half of cancer patients who die do so with cachexia present. (J Cachexia Sarcopenia Muscle 2010). In the United States alone, the estimated prevalence of cancer cachexia alone is over 400,000 patients (Am J Clin Nutr 2006). |
In September 2014, we presented the results from four preclinical studies of AV-380 in various in vivo cachexia models and in vitro assays at the 2nd Cancer Cachexia Conference held in Montreal Canada. Our research was also selected for presentation in an oral session at the conference. We believe that our research sets forth our proof of concept for GDF15, by demonstrating that GDF15 is elevated in cachectic animal models and patients versus non-cachectic, administration of GDF15 induces cachexia and inhibition of GDF15 reverses cachexia.
In connection with the AV-380 program, we have in-licensed certain patents and patent applications from St. Vincent’s Hospital in Sydney, Australia. We have demonstrated preclinical proof-of-concept for AV-380 in multiple cancer cachexia models and have completed cell line development and manufacturing of our first cGMP batch, in preparation for potential future clinical development. We are evaluating partnership opportunities to continue the development of AV-380.
We have devoted substantially all of our resources to our drug discovery efforts comprising research and development, conducting clinical trials for our product candidates, protecting our intellectual property and the general and administrative functions of these operations. We have generated no revenue from product sales through December 31, 2014, and through such date have principally funded our operations through:
| • | $396.3 million of non-dilutive capital in the form of license fees, milestone payments and research and development funding received from our strategic partners; |
| • | $169.6 million of funding from the sale of convertible preferred stock to investors prior to our initial public offering, including $77.5 million of equity sales to our strategic partners; |
| • | $89.7 million of gross proceeds from the sale of common stock in connection with the completion of our initial public offering; |
| • | $36.5 million of loan proceeds in connection with our loan agreement with Hercules Technology II, L.P. and Hercules Technology III, L.P.; |
| • | $68.3 million of gross proceeds from private placements of our common stock; and |
| • | $168.7 million of gross proceeds from the sale of common stock in connection with follow-on public offerings of our common stock in June 2011 and January 2013. |
We do not have a history of being profitable and, as of December 31, 2014, we had an accumulated deficit of $480.0 million. We anticipate that we will continue to incur significant operating costs over the next several years as we continue to execute on our clinical development strategy. We will need additional financing to support our operating activities.
61
Table of Contents
Strategic Partnerships
Ophthotech Corporation
In November 2014 we entered into a Research and Exclusive Option Agreement, or Option Agreement, with Ophthotech Corporation pursuant to which we provided Ophthotech an exclusive option to enter into a definitive license agreement whereby we would grant Ophthotech the right to develop and commercialize our VEGF factor tyrosine kinase inhibitor, tivozanib, outside of Asia for the potential diagnosis, prevention and treatment of non-oncologic diseases or conditions of the eye in humans.
Pursuant to this Option Agreement, we granted to Ophthotech an exclusive, royalty free license or sublicense, as applicable, under intellectual property rights controlled by us solely to perform the research and development activities related to the use of tivozanib for the specific purposes outlined in the agreement during the option period (as defined below). These activities include formulation work for ocular administration, preclinical research and the conduct of a Phase 1/2a, proof of concept clinical trial of a product containing tivozanib in patients with wet age-related macular degeneration, or the POC Study.
Ophthotech paid us $500,000 in consideration for the grant of the option. Such amount is non-refundable and not creditable against any other amounts due under the agreement. We are obligated to make available to Ophthotech, at no cost to Ophthotech, certain quantities of tivozanib hydrochloride solely for conducting its option period research including manufacturing additional quantities of tivozanib in the event stability data indicates that the current supply will expire prior to the end of February 2017. For additional information regarding the terms of the Option Agreement, see “Item 1, Business – Strategic Partnerships.”
Activities under the Option Agreement were evaluated under ASC 605-25 Revenue Recognition—Multiple Element Arrangements, or ASC 605-25, to determine whether such activities represented a multiple element revenue arrangement. The Option Agreement includes the following non-contingent deliverables: our obligation to grant an exclusive option to Ophthotech to enter into a License Agreement to develop and commercialize products incorporating tivozanib for treatment of AMD and other diseases of the eye outside of Asia during the Option Period; our obligation to enter into an amendment with KHK to modify the terms of the existing KHK agreement to negotiate a mutually acceptable form of license agreement; and our obligation to transfer research-grade tivozanib API for Ophthotech to conduct the Option Period research.
We determined that the delivered Option Grant Deliverable, or our obligation to grant an exclusive option to Ophthotech to enter into a License Agreement to develop and commercialize products incorporating tivozanib for treatment of AMD and other diseases of the eye outside of Asia during the Option Period, did not have standalone value from the remaining deliverables since Ophthotech could not obtain the intended benefit of the option without the remaining deliverables. Similarly, the remaining deliverables have no standalone value without the Option Grant Deliverable. We are accounting for the deliverables as one unit of accounting.
Under the Option Agreement, we received a cash payment related to up-front Grant Option fee of $0.5 million during the year ended December 31, 2014. We deferred the upfront Option Grant payment and are recording the deferred revenue over our period of performance which is estimated to be through December 2016, or the life of the agreement. We recorded approximately $38,000 of revenue associated with the Option Grant Deliverable during the year ended December 31, 2014.
Biodesix
In April 2014, we entered into a worldwide agreement with Biodesix to develop and commercialize our HGF inhibitory antibody ficlatuzumab, with BDX004, a proprietary companion diagnostic test developed by Biodesix and based upon an exploratory analyses with VeriStrat®, a serum protein test that is commercially available to help physicians guide treatment decisions for patients with advanced NSCLC. For additional information regarding the terms of this agreement, see “Part I, Item 1, Business – Strategic Partnerships.”
62
Table of Contents
Activities under the agreement with Biodesix were evaluated under ASC 605-25 Revenue Recognition—Multiple Element Arrangements, or ASC 605-25, to determine such activities represented a multiple element revenue arrangement. The agreement with Biodesix includes the following non-contingent deliverables: perpetual, non-exclusive rights to certain intellectual property including clinical and biomarker data related to ficlatuzumab for use in developing and commercializing BDX004; our obligation to deliver technology improvements and data developed during the FOCAL study to Biodesix; our obligation to participate in the joint steering committee during the FOCAL study; our obligation to perform certain development activities in associated with the FOCAL study; and our obligation to supply clinical material for use in conducting the FOCAL study; and our obligation to deliver clinical specimens and data during the FOCAL study. We concluded that any deliverables that would be delivered after the FOCAL study is complete are contingent deliverables because these services are contingent upon the results of the FOCAL study. As these deliverables are contingent, and are not at an incremental discount, they are not evaluated as deliverables at the inception of the arrangement. These contingent deliverables will be evaluated and accounted for separately as each related contingency is resolved. As of December 31, 2014, no contingent deliverables had been provided by us.
We have determined that the delivered item, or the perpetual, non-exclusive rights to certain intellectual property for use in developing and commercializing BDX004 did not have standalone value from the remaining deliverables since Biodesix could not obtain the intended benefit of the license without the remaining deliverables. Since the remaining deliverables will be performed over the same period of performance there is no difference in accounting for the deliverables as one of unit or multiple units of accounting, and therefore, we are accounting for the deliverables as one unit of accounting.
We record the consideration earned while conducting the FOCAL Study, which consists of reimbursement by Biodesix for expenses related to the trial under the Cap, as a reduction to research and development expense using the proportional performance method over the respective period of performance. As a result of the cost sharing provisions in the agreement, we reduced research and development expenses by approximately $2.7 million during the year ended December 31, 2014. The amount due to us from Biodesix pursuant to the cost-sharing provision was $1.8 million at December 31, 2014.
St. Vincent’s Hospital
In July 2012, we entered into a license agreement with St. Vincent’s Hospital Sydney Limited, which we refer to as St. Vincent’s, under which we obtained an exclusive, worldwide license to research, develop, manufacture and commercialize products for therapeutic applications that benefit from inhibition or decreased expression or activity of MIC-1, which is also known as GDF15. We believe GDF15 is a novel target for cachexia and we are exploiting this license in our AV-380 program for cachexia. Under the agreement, we have the right to grant sublicenses subject to certain restrictions. We have a right of first negotiation to obtain an exclusive license to certain improvements that St. Vincent’s or third parties may make to licensed therapeutic products. Under the license agreement, St. Vincent’s also granted us non-exclusive rights for certain related diagnostic products and research tools. For additional information regarding the terms of the agreement with St. Vincent’s, see “Part I, Item 1, Business – Strategic Partnerships.”
Biogen Idec
In March 2009, we entered into an exclusive option and license agreement with Biogen Idec International GmbH, or Biogen Idec, regarding the development and commercialization of our discovery-stage ErbB3-targeted antibodies for the potential treatment and diagnosis of cancer and other diseases in humans outside of North America. Under the agreement, we were responsible for developing ErbB3 antibodies through completion of the first phase 2 clinical trial designed in a manner that, if successful, will generate data sufficient to support advancement to a phase 3 clinical trial. In March 2014, we amended our agreement with Biogen Idec, whereby Biogen Idec agreed to the termination of its rights and obligations under the agreement, including Biogen Idec’s option to (i) obtain a co-exclusive (with us) license to develop and manufacture ErbB3 targeted antibodies and
63
Table of Contents
(ii) obtain exclusive commercialization rights to ErbB3 products in countries in the world other than North America. As a result, we retain worldwide rights to AV-203, a clinical stage ErbB3-targeted antibody. Pursuant to the amendment, we are obligated to in good faith use reasonable efforts to seek a collaboration partner for the purpose of funding further development and commercialization of ErbB3-targeted antibodies. Pursuant to the amendment, we are obligated to pay Biogen Idec a percentage of milestone payments received by us from future partnerships after March 28, 2016 and single digit royalty payments on net sales related to the sale of ErbB3 products, up to cumulative maximum amount of $50.0 million. For additional information regarding the terms of Biogen Idec agreement, see “Part I, Item 1, Business – Strategic Partnerships.”
The deliverables under the original Biogen Idec agreement included an option for a co-exclusive, worldwide license to develop and manufacture ErbB3 antibody products and an option for an exclusive license to commercialize ErbB3 antibody products in all countries in the world other than North America. We determined that these deliverables did not have standalone value due to the fact that the program was still in preclinical development and required our experience to advance development of the product candidates. As such, we determined that the agreement should be accounted for as one unit of accounting.
Under the terms of the original agreement, Biogen Idec made up-front and milestone-based cash payments totaling $20.0 million. Of the $20.0 million received, $10.0 million was associated with milestones that were considered substantive and these amounts were included in revenue when earned. The remaining $10.0 million was amortized as additional license revenue over our period of substantial involvement.
We concluded that the amendment entered into in March 2014 materially modified the terms of the agreement and, as a result, required application of the guidance included in ASC 605-25. Based upon the terms of the amended arrangement, the remaining deliverables included our obligation to seek a collaboration partner to fund further development of the program and our obligation to continue development and commercialization of the licensed products if a collaboration partner is secured. We concluded that our obligation to use best efforts to seek a collaboration partner does not have standalone value from our efforts to continue development and commercialization of the licensed products and thus the deliverables should be treated as a single unit of accounting.
Upon modifying the arrangement, we had $14.7 million of deferred revenue remaining to be amortized. We are not entitled to receive any further consideration from Biogen Idec under the amended arrangement. We allocated a portion of the remaining deferred revenue to the undelivered unit of accounting based upon our best estimate of the selling price. We determined the best estimate of the selling price to be approximately $0.6 million and recognized the remaining $14.1 million as collaboration revenue in the three months ended March 31, 2014. The deferred revenue associated with the undelivered unit of accounting is being recognized on a straight-line basis over the expected period of performance, or through December 2015, based upon our historical experience with marketing our product candidates to potential partners.
The best estimate of the selling price was based upon a cost approach pursuant to which we estimated the costs expected to be incurred in executing a partnership agreement and then applied a reasonable markup. We estimated future cash outflows for several possible outcomes, including the execution of a partnership at different times within a reasonable range and partnerships of differing complexity. We estimated our cash outflows for each scenario based upon the expected costs associated with the relevant employees and the expected level of effort to be expended to seek and execute a partnership. Our analysis also considered the legal charges we anticipate we will incur. Changes to the assumptions within the reasonable range of possible values would not have a material impact on the amounts recorded in current or future periods.
Under the agreement, we recorded revenue of $14.5 million and $0.9 million during the years ended December 31, 2014 and 2013, respectively.
64
Table of Contents
Astellas Pharma
In February 2011, we entered into a collaboration and license agreement with Astellas and certain of its indirect wholly-owned subsidiaries pursuant to which we and Astellas made plans to develop and seek to commercialize tivozanib for the treatment of a broad range of cancers. On February 12, 2014, Astellas exercised its right to terminate the agreement. The termination of the agreement became effective August 11, 2014, at which time tivozanib rights returned to us. In accordance with the collaboration and license agreement, we and Astellas agreed to equally share committed development costs, including the costs of completing certain tivozanib clinical development activities that were initiated as part of our partnership with Astellas.
In connection with the agreement, we received an initial cash payment of $125.0 million, comprised of a $75.0 million license fee and $50.0 million in research and development funding, both of which are non-creditable and non-refundable against any amounts due under the collaboration agreement. We retained net proceeds of approximately $97.6 million of the initial cash payment from Astellas, after payments to KHK and strategic, legal and financial advisors. In December 2012, we received a $15.0 million milestone payment from Astellas in connection with the acceptance by the FDA of our NDA filing for tivozanib for the treatment of patients with advanced RCC. We have accounted for the joint development and commercialization activities in North America and Europe as a joint risk-sharing collaboration in accordance with Accounting Standards Codification, or ASC, 808 Collaborative Arrangements. In addition, these joint development and commercialization activities were not deemed to be separate deliverables under the agreement with Astellas.
Payments from Astellas with respect to Astellas’ share of tivozanib development and commercialization costs incurred by us pursuant to the joint development plan are recorded as a reduction to research and development expense and general and administrative expense in the accompanying consolidated financial statements due to the joint risk-sharing nature of the activities in North America and Europe. As a result of the cost-sharing provisions in the Astellas Agreement, the Company reduced research and development expense by $3.5 million, $15.8 million and $34.1 million during the years ended December 31, 2014, 2013, and 2012, respectively. The Company also reduced general and administrative expense by $0.1 million, $2.8 million, and $3.3 million during the years ended December 31, 2014, 2013, and 2012, respectively, as a result of the cost-sharing provisions in the Astellas Agreement. The net amount due to the Company from Astellas pursuant to the cost-sharing provisions was $0.6 million at December 31, 2014.
Activities under the agreement with Astellas outside of the joint development and commercialization activities in North America and Europe were evaluated under ASC 605-25 to determine if they represented a multiple element revenue arrangement. The agreement with Astellas included the following deliverables outside of the joint development and commercialization activities in North America and Europe: a co-exclusive license to develop and commercialize tivozanib in North America and Europe; a royalty-bearing license to develop and commercialize tivozanib in the royalty territory, which includes our obligation to provide access to clinical and regulatory information resulting from the activities in North America and Europe to Astellas for its development and commercialization of tivozanib in the royalty territory; and our obligation to supply clinical material to Astellas for development of tivozanib in the royalty territory. All of these deliverables were deemed to have stand-alone value and to meet the criteria to be accounted for as separate units of accounting under ASC 605-25.
We allocated the up-front consideration of $125.0 million to the deliverables based on our best estimate of selling price of each deliverable using the relative selling price method as we did not have vendor specific objective evidence or third-party evidence for such deliverables. We allocated $120.2 million of the up-front consideration from Astellas to the co-exclusive license in North America and Europe and $4.8 million of the up-front consideration from Astellas to the combined deliverable representing a royalty-bearing license to develop and commercialize tivozanib in the royalty territory along with our obligation to provide access to clinical and regulatory information resulting from the activities in North America and Europe to Astellas for its use in the royalty territory. The relative selling price for our obligation to supply clinical material to Astellas for development in the royalty territory had de minimis value.
65
Table of Contents
We recorded the $120.2 million relative selling price of the co-exclusive license granted in North America and Europe as collaboration revenue during the three months ended March 31, 2011 upon delivery of the license, and deferred approximately $4.8 million of revenue representing the relative selling price of the royalty-bearing license to develop and commercialize tivozanib in the royalty territory along with our obligation to provide access to clinical and regulatory information resulting from the activities in North America and Europe to Astellas for its use in the royalty territory. We were recording the $4.8 million ratably over the period of our performance through April 2022, the remaining patent life of tivozanib. Upon being notified that the collaboration would be terminated effective August 2014, we reassessed the period of performance associated with the royalty territory deliverable and accelerated the recognition of the remaining deferred revenue such that the balance would be recognized through August 2014. This change in estimate resulted in the recognition of an additional $3.2 million during the year ended December 31, 2014. We recorded approximately $3.6 million, $0.4 million, and $0.4 million of revenue associated with the Royalty Territory Deliverable during each of the years ended December 31, 2014, 2013 and 2012.
Kyowa Hakko Kirin
In December 2006, we entered into a license agreement with Kirin Brewery Co. Ltd. (now Kyowa Hakko Kirin), which we sometimes refer to as KHK, under which we obtained an exclusive license, with the right to grant sublicenses subject to certain restrictions, to research, develop, manufacture and commercialize tivozanib, pharmaceutical compositions thereof and associated biomarkers. Our exclusive license covers all territories in the world, except for Asia. KHK has retained rights to tivozanib in Asia. Under the license agreement, we obtained exclusive rights in our territory under certain KHK patents, patent applications and know-how related to tivozanib, to research, develop, make, have made, use, import, offer for sale, and sell tivozanib for the diagnosis, prevention and treatment of any and all human diseases and conditions. We and KHK each have access to and can benefit from the other party’s clinical data and regulatory filings with respect to tivozanib and biomarkers identified in the conduct of activities under the license agreement.
Under the license agreement, we are obligated to use commercially reasonable efforts to develop and commercialize tivozanib in our territory, including meeting certain specified diligence goals. Prior to the first anniversary of the first post-marketing approval sale of tivozanib in our territory, neither we nor any of our subsidiaries has the right to conduct certain clinical trials of, seek marketing approval for or commercialize any other cancer product that also works by inhibiting the activity of the VEGF receptor. For additional information regarding the terms of the license agreement with KHK, see “Item 1, Business – Strategic Partnerships.”
Upon entering into the license agreement with KHK, we made a one-time cash payment in the amount of $5.0 million. In March 2010, we made a $10.0 million milestone payment to KHK in connection with the dosing of the first patient in our phase 3 clinical trial of tivozanib. In December 2012, we made a $12.0 million milestone payment to KHK in connection with the acceptance by the FDA of our NDA filing for tivozanib. The total maximum payments for clinical and regulatory milestones under our license agreement with KHK are $60.0 million, in the aggregate.
We also made a $22.5 million payment to KHK during the year ended December 31, 2011 related to the up-front license payment received under the collaboration and license agreement with Astellas which we entered into in February 2011. We are also required to pay tiered royalty payments on net sales we make of tivozanib in our territory, which range from the low to mid teens as a percentage of net sales. The royalty rate escalates within this range based on increasing tivozanib sales. Our royalty payment obligations in a particular country in our territory begin on the date of the first commercial sale of tivozanib in that country, and end on the later of 12 years after the date of first commercial sale of tivozanib in that country or the date of the last to expire of the patents covering tivozanib that have been issued in that country. In the event we sublicense the rights licensed to us under the license agreement with KHK, we are required to pay KHK a specified percentage of any amounts we receive from any third party sublicensees, other than amounts we receive in respect of research and development funding or equity investments, subject to certain limitations.
66
Table of Contents
Financial Overview
During 2014, we initiated several cost containment activities that have reduced operating expenses by approximately 35% in 2014 in comparison to year ended December 31, 2013. With these activities in place, we were able to finish 2014 with $52.3 million in cash and cash equivalents. In January 2015, our Board of Directors approved a strategic restructuring that eliminated our internal research function to better align our resources with our future clinically focused strategic plans given that our material programs were at preclinical and clinical stages of development. As part of this restructuring, we eliminated approximately two thirds of our workforce, or 40 positions, across the organization. The restructuring was substantially completed by January 15, 2015 and we expect will it be fully completed by March 31, 2015. The reduction in force is expected to reduce compensation expenses by approximately $6.0 million in 2015. We expect to incur total restructuring charges of approximately $4.5 million, consisting of severance and benefit costs.
Revenue
To date, we have not generated any revenue from product sales. All of our revenue to date has been derived from license fees, milestone payments, premium over the fair value of convertible preferred shares sold to our strategic partners, and research and development payments received from our strategic partners.
In the future, we may generate revenue from a combination of product sales, license fees, milestone payments and research and development payments in connection with strategic partnerships, and royalties resulting from the sales of products developed under licenses of our intellectual property. We expect that any revenue we generate will fluctuate from quarter to quarter as a result of the timing and amount of license fees, research and development reimbursements, milestone and other payments received under our strategic partnerships, and the amount and timing of payments that we receive upon the sale of our products, to the extent any are successfully commercialized. We do not expect to generate revenue from product sales in the near term. If we or our strategic partners fail to complete the development of our drug candidates in a timely manner or obtain regulatory approval for them, our ability to generate future revenue, and our results of operations and financial position, would be materially adversely affected.
Research and Development Expenses
Research and development expenses have historically consisted of expenses incurred in connection with the discovery and development of our product candidates. These expenses consist primarily of:
| • | employee-related expenses, which include salaries, benefits and stock-based compensation expense; |
| • | expenses incurred under agreements with contract research organizations, investigative sites and consultants that conduct our clinical trials and a substantial portion of our preclinical studies; |
| • | the cost of acquiring and manufacturing clinical trial materials, as well as commercial materials prior to our anticipated launch of tivozanib; |
| • | the cost of completing certain tivozanib clinical development activities that were initiated as part of our partnership with Astellas; |
| • | facilities, depreciation and other allocated expenses, which include direct and allocated expenses for rent and maintenance of facilities and equipment, and depreciation of fixed assets; |
| • | license fees for, and milestone payments related to, in-licensed products and technology; and |
| • | costs associated with outsourced development activities, regulatory approvals and medical affairs. |
We expense research and development costs as incurred. Nonrefundable advance payments for goods and services that will be used in future research and development activities are expensed when the activity has been performed or when the goods have been received rather than when the payment is made.
67
Table of Contents
Research and development expenses are net of amounts reimbursed under our agreements with Astellas and Biodesix for Astellas’ and Biodesix’ respective shares of development costs incurred by us under our joint development plans with each respective partner.
We track external development expenses and personnel expense on a program-by-program basis and allocate common expenses, such as scientific consultants and laboratory supplies, to each program based on the personnel resources allocated to such program. Facilities, depreciation, stock-based compensation, research and development management and research and development support services are not allocated among programs and are considered overhead. We expect our overhead expenses to decrease in future periods as a result of our January 2015 restructuring which should reduce our facilities requirement by more than 80% of our current space, including the elimination of lab and vivarium needs. Below is a summary of our research and development expenses for the years ended December 31, 2014, 2013 and 2012:
| Years Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| (in thousands) | ||||||||||||
| Tivozanib |
$ | 9,530 | $ | 25,060 | $ | 41,183 | ||||||
| AV-380 Program in Cachexia |
12,968 | 4,308 | 2,560 | |||||||||
| Ficlatuzumab |
1,579 | 12,573 | 13,097 | |||||||||
| AV-203 |
1,843 | 5,698 | 8,247 | |||||||||
| Other pipeline programs |
72 | 1,299 | 4,769 | |||||||||
| Other research and development |
67 | 376 | 1,027 | |||||||||
| Platform collaborations |
— | — | 1,340 | |||||||||
| Overhead |
12,195 | 19,154 | 19,135 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total research and development expenses |
$ | 38,254 | $ | 68,468 | $ | 91,358 | ||||||
|
|
|
|
|
|
|
|||||||
Tivozanib
On November 27, 2012, the FDA accepted for filing our NDA for tivozanib, our lead product candidate, with the proposed indication for the treatment of patients with advanced renal cell carcinoma, or RCC. On May 2, 2013, we were informed by the FDA that its Oncologic Drugs Advisory Committee believed that our NDA for investigational agent tivozanib did not demonstrate a favorable benefit-to-risk evaluation for the treatment of advanced RCC, and in June 2013, we received a Complete Response letter from the FDA informing us that the FDA will not approve in its present form our NDA for our investigational agent tivozanib for the treatment of patients with advanced RCC.
Our strategy for development of tivozanib included a focus on the exploration of various biomarkers which could provide insights into tivozanib’s potential clinical benefit. Accordingly, we conducted biomarker studies referred to as the BATON (Biomarker Assessment of Tivozanib in Oncology) trials. The first, which was a single-arm phase 2 trial of tivozanib (BATON-RCC) to evaluate various biomarkers for tivozanib activity in treatment naïve advanced RCC patients, was completed in 2014. In another, we evaluated tivozanib in colorectal cancer (BATON-CRC) through a randomized phase 2 clinical trial, to evaluate tivozanib in combination with mFOLFOX6 compared to Avastin in combination with mFOLFOX6 as first-line therapy in patients with advanced metastatic colorectal cancer, or CRC. On December 13, 2013, we announced that, based on data from a September 2013 interim analysis, the BATON-CRC trial was unlikely to meet the primary endpoint of demonstrating superiority over bevacizumab in the intent-to-treat population; and on February 14, 2014, we announced that the study would be discontinued. We recently presented the final results, including biomarker data for tivozanib in first line CRC that demonstrated that patients with low expression of Neuropilin-1, a cell surface protein which modulates blood vessel development (which we refer to a NRP-1), showed an improved PFS versus patients with high expression NRP-1, supporting the value of NRP-1 as a potential prognostic marker for angiogenesis inhibitors. Further, patients with low serum NRP-1 demonstrated longer PFS when treated with tivozanib compared to bevacizumab, which we believe suggests that first line CRC patients with low NRP-1 levels may benefit from treatment with tivozanib over bevacizumab, a standard of care for this disease.
68
Table of Contents
We entered into a collaboration and license agreement with Astellas in February 2011, pursuant to which we and Astellas shared responsibility for tivozanib, including expenses for continued development and commercialization of tivozanib, in North America and Europe. We have included $3.5 million, $15.8 million and $34.1 million in research and development cost reimbursements as a reduction in tivozanib-related expenses for the years ended December 31, 2014, 2013 and 2012, respectively. We also made a $22.5 million payment to KHK during the year ended December 31, 2011 related to the up-front license payment received under the collaboration and license agreement with Astellas which we entered into in February 2011. In December 2012, we made a $12.0 million milestone payment to KHK in connection with the acceptance of our NDA filing for tivozanib. On August 11, 2014, our collaboration and license agreement with Astellas terminated pursuant to Astellas’ election to terminate and tivozanib rights were returned to us.
We and Astellas will share the costs of completing certain tivozanib clinical development activities. We have incurred approximately $9.5 million of our share of tivozanib wind down costs during 2014.We expect our share of partnership wind down costs to be approximately $2.9 million during 2015. The actual amount that we will incur may differ from this estimate depending upon our ability to expedite the termination of our existing obligations while continuing to satisfy our patient and regulatory requirements.
In November 2014, we entered into a research and exclusive option agreement with Ophthotech Corporation, or Ophthotech, under which we granted Ophthotech an option to develop and commercialize tivozanib for the potential diagnosis, prevention and treatment of non-oncologic diseases or conditions of the eye in humans. With the termination of our partnership with Astellas and the return of our tivozanib rights, we plan on pursuing partnering options to fund further tivozanib development in appropriate clinical settings.
AV-380 Program in Cachexia
In 2012, we initiated a program focusing on cachexia, which we now refer to as our AV-380 program. Cachexia is a serious and common complication of advanced cancer and a number of chronic diseases that is characterized by symptoms of unintentional weight loss, progressive muscle wasting, and a loss of appetite. Cancer cachexia is defined as a multi-factorial syndrome of involuntary weight loss characterized by an ongoing loss of skeletal muscle mass (with or without loss of fat mass) that cannot be fully reversed by conventional nutritional support and leads to progressive functional impairment. The pathophysiology is characterized by a negative protein and energy balance driven by a variable combination of reduced food intake and abnormal metabolism. Other symptoms of cachexia include anemia, breathing difficulties, edema, insulin resistance, muscle weakness/asthenia, and fatigue. Our primary research focus is in the area of cancer cachexia which we believe represents a significant are of patient need. In addition, cachexia is also associated with diseases outside of cancer including CKD, CHF, and COPD. In connection with our cachexia program, we have in-licensed certain patents and patent applications from St. Vincent’s. Appropriate IND-enabling efforts, including cell line development and manufacturing of our first cGMP batch, have been completed in preparation for potential future clinical development. We plan to pursue opportunities for partnerships to continue the development of AV-380.
In September 2014, we presented the results from four preclinical studies of AV-380 in various in vivo cachexia models and in vitro assays at the 2nd Cancer Cachexia Conference held in Montreal Canada. Our research was also selected for presentation in an oral session at the conference. We believe our research sets forth our proof of concept for GDF15, by demonstrating that GDF15 is elevated in cachectic animal models and patients versus non-cachectic, administration of GDF15 induces cachexia and inhibition of GDF15 reverses cachexia.
Following our restructuring in January 2015 which eliminated our internal research function, we expect our costs associated with this program to decrease in 2015 in comparison to 2014. We are exploring partnerships and collaborations to further the development of AV-380. Due to the unpredictable nature of preclinical and clinical development and given the early stage of this program, we are unable to estimate with any certainty the costs we will incur in the future development of AV-380.
69
Table of Contents
Ficlatuzumab
In April 2014, we entered into a worldwide agreement with Biodesix to develop and commercialize AVEO’s potent HGF inhibitory antibody ficlatuzumab, with BDX004, a proprietary companion diagnostic test, developed by Biodesix and based upon an exploratory analyses with VeriStrat®, a serum protein test that is commercially available to help physicians guide treatment decisions for patients with advanced NSCLC. In September 2014, at the 2014 Congress of the European Society for Medical Oncology, we presented detailed data from our phase 2 clinical trial comparing the combination of ficlatuzumab and gefitinib to gefitinib monotherapy in previously untreated Asian subjects with 1st line non-small cell lung cancer. In the intent-to-treat population, the addition of ficlatuzumab to gefitinib did not result in statistically significant improved overall response rate. However, an exploratory analysis in the phase 2 using a serum-based molecular diagnostic test, known as VeriStrat®, identified a sub-population of patients who showed a progression free survival and overall survival benefit from the addition of ficlatuzumab to the EGFR TKI. Pursuant to the agreement with Biodesix, Biodesix will provide up to $15 million for a phase 2 trial of ficlatuzumab in combination with erlotinib in first line advanced NSCLC patients selected using BDX004, a diagnostic test derived from VeriStrat, and fund the further development and registration of BDX004 as a companion diagnostic. After the completion of the phase 2 trial, any additional development, regulatory or commercial expenses for ficlatuzumab will be equally shared, as well as profits, if any.
In November 2011, we entered into an agreement with Boehringer Ingelheim for large-scale process development and clinical manufacturing of ficlatuzumab, and Boehringer Ingelheim has produced ficlatuzumab at its biopharmaceutical site in Fremont, California. Due to the unpredictable nature of clinical development, we are unable to estimate with any certainty the costs we will incur in the future development of ficlatuzumab.
AV-203
Through the use of our Human Response Platform, we have identified antibodies that have been shown to be potent and selective inhibitors of ErbB3 in preclinical studies. In preclinical testing, these antibodies have significantly inhibited the growth of a number of different tumors, including in breast, prostate and pancreatic cancers. We granted Biogen Idec an exclusive option to co-develop (with us) and commercialize our ErbB3-targeted antibodies for the potential treatment and diagnosis of cancer and other diseases outside of North America. Upon the selection of AV-203 as a development candidate in the first quarter of 2010, we earned a $5.0 million milestone payment from Biogen Idec, and we earned an additional $5.0 million milestone payment in June 2011 based on initiation of a GLP toxicology study. In May 2012, we announced the initiation of a phase 1 clinical trial examining the safety, tolerability and preliminary efficacy of AV-203 along with exploratory biomarkers in patients with metastatic or advanced solid tumors. In May 2014, we presented the results of our first-in-human study of AV-203 at the 2014 American Society of Clinical Oncology Annual Meeting. Among the results, the study established a recommended phase 2 dose of AV-203, demonstrated good tolerability and promising early signs of activity, and reached the maximum planned dose of AV-203 monotherapy.
In March 2014, we regained our worldwide rights from Biogen Idec to develop, manufacture and commercialize AV-203, and we are actively pursuing partnerships or collaborations to further advance the development of AV-203. Because obtaining a partnership and collaborations may be complex and unpredictable in timing and nature of terms, we are unable to estimate with any certainty the costs we will incur in the future development of AV-203.
Uncertainties of Estimates Related to Research and Development Expenses
The process of conducting preclinical studies and clinical trials necessary to obtain FDA approval for each of our product candidates is costly and time-consuming. The probability of success for each product candidate and clinical trial may be affected by a variety of factors, including, among others, the quality of the product candidate’s early clinical data, investment in the program, competition, manufacturing capabilities and commercial viability.
70
Table of Contents
At this time, we cannot reasonably estimate or know the nature, specific timing and estimated costs of the efforts that will be necessary to complete the development of our product candidates, or the period, if any, in which material net cash inflows may commence from sales of any approved products. This uncertainty is due to the numerous risks and uncertainties associated with developing drugs, including the uncertainty of:
| • | our ability to establish and maintain strategic partnerships, the terms of those strategic partnerships and the success of those strategic partnerships, if any, including the timing and amount of payments that we might receive from strategic partners; |
| • | the scope, progress, results and costs of preclinical development, laboratory testing and clinical trials for any product candidate; |
| • | the progress and results of our clinical trials; |
| • | the costs, timing and outcome of regulatory review of our product candidates; |
| • | the emergence of competing technologies and products and other adverse market developments; and |
| • | the costs of preparing, filing and prosecuting patent applications and maintaining, enforcing and defending intellectual property-related claims. |
As a result of the uncertainties associated with developing drugs, including those discussed above, we are unable to determine the duration and completion costs of current or future clinical stages of our product candidates, or when, or to what extent, we will generate revenues from the commercialization and sale of any of our product candidates. Development timelines, probability of success and development costs vary widely. We anticipate that we will make determinations as to which additional programs to pursue and how much funding to direct to each program on an ongoing basis in response to the scientific and clinical success, if any, of each product candidate, as well as ongoing assessment of each product candidate’s commercial potential. We will need to raise substantial additional capital in the future in order to fund the development of our preclinical and clinical product candidates.
General and Administrative Expenses
General and administrative expenses consist principally of salaries and related costs for personnel in executive, finance, corporate development, marketing, information technology, legal and human resource functions. Also included in general and administrative expenses are facility costs not otherwise included in research and development expenses, patent filing, prosecution and defense costs and professional fees for legal, consulting, pre-commercialization activities, auditing and tax services.
We anticipate that our general and administrative expenses will decrease due to the January 2015 restructuring. This decrease may be partially offset by an increase in legal costs associated with the ongoing shareholder litigation and SEC investigation described in this report under the heading “Legal Proceedings” above in Part I—Item 3.
Interest Income and Interest Expense
Interest income consists of interest earned on our cash, cash equivalents and marketable securities. The primary objective of our investment policy is capital preservation.
Interest expense consists of interest, amortization of debt discount, and amortization of deferred financing costs associated with our loans payable.
71
Table of Contents
Income Taxes
We calculate our provision for income taxes on ordinary income based on our projected annual tax rate for the year. We recorded a loss for the years ended December 31, 2014, 2013, and 2012, and since we maintain a full valuation allowance on all of our deferred tax assets, we have recorded no income tax provision or benefit during the years ended December 31, 2014, 2013, and 2012.
Critical Accounting Policies and Significant Judgments and Estimates
Our discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses and the disclosure of contingent assets and liabilities in our financial statements. On an ongoing basis, we evaluate our estimates and judgments, including those related to revenue recognition, accrued clinical expenses, and stock-based compensation. We base our estimates on historical experience, known trends and events and various other factors that we and our management believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Our significant accounting policies are described in more detail in the notes to our consolidated financial statements appearing elsewhere in this report. We believe the following accounting policies to be most critical to the judgments and estimates used in the preparation of our consolidated financial statements.
Revenue Recognition
Our revenues have historically been generated primarily through collaborative research, development and commercialization agreements. The terms of these agreements generally contain multiple elements, or deliverables, which may include (i) licenses, or options to obtain licenses, to our technology, (ii) research and development activities to be performed on behalf of the collaborative partner, and (iii) in certain cases, services in connection with the manufacturing of pre-clinical and clinical material. Payments to us under these arrangements typically include one or more of the following: non-refundable, up-front license fees; option exercise fees; funding of research and/or development efforts; milestone payments; and royalties on future product sales.
When evaluating multiple element arrangements, we consider whether the deliverables under the arrangement represent separate units of accounting. This evaluation requires subjective determinations and requires management to make judgments about the individual deliverables and whether such deliverables are separable from the other aspects of the contractual relationship. In determining the units of accounting, management evaluates certain criteria, including whether the deliverables have standalone value, based on the consideration of the relevant facts and circumstances for each arrangement. The consideration received is allocated among the separate units of accounting using the relative selling price method, and the applicable revenue recognition criteria are applied to each of the separate units.
We determine the estimated selling price for deliverables within each agreement using vendor-specific objective evidence, or VSOE, of selling price, if available, third-party evidence, or TPE, of selling price if VSOE is not available, or best estimate of selling price if neither VSOE nor TPE is available. Determining the best estimate of selling price for a deliverable requires significant judgment. We typically use best estimate of selling price to estimate the selling price for licenses to our proprietary technology, since we often do not have VSOE or TPE of selling price for these deliverables. In those circumstances where we utilize best estimate of selling price to determine the estimated selling price of a license to our proprietary technology, we consider market conditions as well as entity-specific factors, including those factors contemplated in negotiating the agreements as well as internally developed models that include assumptions related to the market opportunity, estimated development
72
Table of Contents
costs, probability of success and the time needed to commercialize a product candidate pursuant to the applicable license. In validating our best estimate of selling price, we evaluate whether changes in the key assumptions used to determine the best estimate of selling price will have a significant effect on the allocation of arrangement consideration between multiple deliverables.
We typically receive up-front, non-refundable payments when licensing our intellectual property in conjunction with a research and development agreement. When management believes the license to our intellectual property does not have stand-alone value from the other deliverables to be provided in the arrangement, we generally recognize revenue attributed to the license on a straight-line basis over our contractual or estimated performance period, which is typically the term of our research and development obligations. If management cannot reasonably estimate when our performance obligation ends, then revenue is deferred until management can reasonably estimate when the performance obligation ends. When management believes the license to our intellectual property has stand-alone value, we generally recognize revenue attributed to the license upon delivery. The periods over which revenue should be recognized are subject to estimates by management and may change over the course of the research and development agreement. Such a change could have a material impact on the amount of revenue we record in future periods.
Payments or reimbursements resulting from our research and development efforts for those arrangements where such efforts are considered as deliverables are recognized as the services are performed and are presented on a gross basis so long as there is persuasive evidence of an arrangement, the fee is fixed or determinable, and collection of the related receivable is reasonably assured. Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue in the accompanying balance sheets.
At the inception of each agreement that includes milestone payments, we evaluate whether each milestone is substantive and at risk to both parties on the basis of the contingent nature of the milestone. This evaluation includes an assessment of whether (a) the consideration is commensurate with either (1) the entity’s performance to achieve the milestone, or (2) the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity’s performance to achieve the milestone, (b) the consideration relates solely to past performance, and (c) the consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. We evaluate factors such as the scientific, regulatory, commercial and other risks that must be overcome to achieve the respective milestone, the level of effort and investment required to achieve the respective milestone and whether the milestone consideration is reasonable relative to all deliverables and payment terms in the arrangement in making this assessment. The conclusion as to whether milestone payments are substantive involves management judgment regarding the factors noted above.
We classify each of our milestones into one of four categories: (i) clinical and development milestones, (ii) regulatory milestones, (iii) commercial milestones, and (iv) patent-related milestones. Clinical and development milestones are typically achieved when a product candidate advances into a defined phase of clinical research or completes such phase. For example, a milestone payment may be due to us upon the initiation of a phase 3 clinical trial for a new indication, which is the last phase of clinical development and could eventually contribute to marketing approval by the FDA or other regulatory authorities. Regulatory milestones are typically achieved upon acceptance of the submission for marketing approval of a product candidate or upon approval to market the product candidate by the FDA or other regulatory authorities. For example, a milestone payment may be due to us upon the FDA’s acceptance of an NDA. Commercial milestones are typically achieved when an approved pharmaceutical product reaches certain defined levels of net sales by the licensee, such as when a product first achieves global sales or annual sales of a specified amount. Patent-related milestones are typically achieved when a patent application is filed or a patent is issued with respect to certain intellectual property related to the applicable collaboration.
Revenues from clinical and development, regulatory and patent-related milestone payments, if the milestones are deemed substantive and the milestone payments are nonrefundable, are recognized upon successful accomplishment of the milestones. We have concluded that the clinical and development, regulatory
73
Table of Contents
and patent-related milestones pursuant to our current research and development arrangements are substantive. Milestones that are not considered substantive are accounted for as license payments and recognized on a straight-line basis over the remaining period of performance. Revenues from commercial milestone payments are accounted for as royalties and are recorded as revenue upon achievement of the milestone, assuming all other revenue recognition criteria are met.
Accrued Clinical Expenses
As part of the process of preparing our financial statements, we are required to record an estimate of our accrued expenses. This process involves reviewing open contracts and purchase orders, and communicating with our applicable personnel to identify services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for such services when we have not yet been invoiced or otherwise notified of the actual costs. The majority of our service providers invoice us monthly in arrears for services performed. We make estimates of our accrued expenses as of each balance sheet date in our financial statements based on facts and circumstances known to us at that time. We periodically confirm the accuracy of our estimates with the service providers and make adjustments if necessary. Examples of estimated accrued clinical expenses include:
| • | fees paid to contract research organizations in connection with clinical studies; |
| • | fees paid to investigative sites in connection with clinical studies; |
| • | fees paid to contract manufacturers in connection with the production of clinical trial materials; and |
| • | fees paid to vendors in connection with preclinical development activities. |
We determine our expenses related to clinical studies based on our estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and contract research organizations that conduct and manage clinical studies on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we adjust the accrual accordingly. Although we do not expect our estimates to be materially different from amounts actually incurred, and our estimates have not historically been materially different, our estimates of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in us reporting amounts that are too high or too low in any particular period. Based on our level of clinical trial expenses as of December 31, 2014, if our previous estimates are 5% too high or too low, this may result in an adjustment to our accrued clinical trial expenses in future periods of approximately $0.1 million.
Stock-Based Compensation
Under our stock-based compensation programs, we periodically grant stock options and restricted stock to employees, directors and nonemployee consultants. We also issue shares under an employee stock purchase plan. The fair value of all awards is recognized in our statements of operations over the requisite service period for each award. Awards that vest as the recipient provides service are expensed on a straight-line basis over the requisite service period. Other awards, such as performance-based awards that vest upon the achievement of specified goals, are expensed using the accelerated attribution method if achievement of the specified goals is considered probable. We have also granted awards that vest upon the achievement of market conditions. Per ASC 718 Share-Based Payments, market conditions must be considered in determining the estimated grant-date fair value of share-based payments and the market conditions must be considered in determining the requisite service period over which compensation cost is recognized. We estimate the fair value of the awards with market conditions using a Monte Carlo simulation, which utilizes several assumptions including the risk-free interest
74
Table of Contents
rate, the volatility of our stock and the exercise behavior of award recipients. The grant-date fair value of the awards is then recognized over the requisite service period, which represents the derived service period for the awards as determined by the Monte Carlo simulation. Determining the amount of stock-based compensation to be recorded requires us to develop estimates of fair values of stock options as of the grant date using highly subjective assumptions.
We use the Black-Scholes option pricing model to value our stock option awards, which requires us to make certain assumptions regarding the expected volatility of our common stock price, the expected term of the option grants, the risk-free interest rate and the dividend yield with respect to our common stock. Our expected stock price volatility is based on an average of our own historical volatility and that of several peer companies. We utilized a weighted average method using our own volatility data for the time that we have been public, along with similar data for peer companies that are publicly traded. For purposes of identifying peer companies, we considered characteristics such as industry, length of trading history, similar vesting terms and in-the-money option status. Due to the lack of available quarterly data for these peer companies and a lack of our own historical data, we elected to use the “simplified” method for “plain vanilla” options to estimate the expected term of our stock option grants. Under this approach, the weighted-average expected life is presumed to be the average of the vesting term and the contractual term of the option. The risk-free interest rate used for each grant is based on the U.S. Treasury yield curve in effect at the time of grant for instruments with a similar expected life. We utilize a dividend yield of zero based on the fact that we have never paid cash dividends and have no present intention to pay cash dividends.
During the years ended December 31, 2014, 2013 and 2012, respectively, the assumptions used in the Black-Scholes pricing model for new grants were as follows:
| Years Ended December 31, | ||||||
| 2014 | 2013 | 2012 | ||||
| Volatility |
69.38%-77.92% | 64.22%-72.65% | 64.30%-66.05% | |||
| Expected Term (in years) |
5.50-6.25 | 5.50-6.25 | 5.50-6.25 | |||
| Risk-Free Interest Rates |
1.81%-2.02% | 1.01%-2.10% | 0.83%-1.33% | |||
| Dividend Yield |
— | — | — | |||
We recognized stock-based compensation expense of approximately $2.8 million, $3.9 million and $8.0 million for the years ended December 31, 2014, 2013, and 2012, respectively. During the years ending December 31, 2014, 2013 and 2012, we estimated our expected forfeiture rates to be 62%, 49% and 18%, respectively. As of December 31, 2014, we had approximately $1.9 million of total unrecognized stock-based compensation expense for stock options, which we expect to recognize over a weighted-average period of approximately 3.2 years.
As of December 31, 2014, we had $0.1 million of total unrecognized stock-based compensation expense related to restricted stock awards granted under our 2010 Stock Incentive Plan. We expect to recognize the expense over a weighted-average period of 0.9 years.
We record compensation expense only for those awards that we ultimately expect will vest. We have performed an historical analysis of option awards that were forfeited prior to vesting and recorded total stock option expense that reflected this estimated forfeiture rate. We cannot currently predict the total amount of stock-based compensation expense to be recognized in any future period because such amounts will depend on levels of stock-based payments granted in the future as well as the portion of the awards that actually vest. Forfeitures are estimated each period and adjusted if actual forfeitures differ from those estimates. Actual forfeitures may differ from our estimates as a result of significant changes in our operations, such as those stemming from our October 2012 and June 2013 restructurings.
We have historically granted stock options at exercise prices that are not less than the fair market value of our common stock.
75
Table of Contents
Results of Operations
Comparison of Years Ended December 31, 2014 and 2013
The following tables summarize the results of our operations for each of the years ended December 31, 2014 and 2013, together with the changes in those items in dollars and as a percentage:
| Years Ended December 31, |
Increase/ (decrease) |
% | ||||||||||||||
| 2014 | 2013 | |||||||||||||||
| (in thousands) | ||||||||||||||||
| Revenue |
$ | 18,123 | $ | 1,293 | $ | 16,830 | 1,302 | % | ||||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
38,254 | 68,468 | (30,214 | ) | (44 | )% | ||||||||||
| General and administrative |
18,589 | 28,712 | (10,123 | ) | (35 | )% | ||||||||||
| Restructuring and lease exit |
11,729 | 8,017 | 3,712 | 46 | % | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total operating expenses |
68,572 | 105,197 | (36,625 | ) | (35 | )% | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Loss from operations |
(50,449 | ) | (103,904 | ) | 53,455 | (51 | )% | |||||||||
| Other income (expense), net |
66 | (123 | ) | 189 | (154 | )% | ||||||||||
| Interest expense |
(2,388 | ) | (3,127 | ) | 739 | (24 | )% | |||||||||
| Interest income |
32 | 125 | (93 | ) | (74 | )% | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss |
$ | (52,739 | ) | $ | (107,029 | ) | $ | 54,290 | (51 | )% | ||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Years Ended December 31, |
Increase/ (decrease) |
% | ||||||||||||||
| Revenue |
2014 | 2013 | ||||||||||||||
| (in thousands) | ||||||||||||||||
| Strategic Partner: |
||||||||||||||||
| Astellas |
$ | 3,564 | $ | 430 | $ | 3,134 | 729 | % | ||||||||
| Ophthotech |
39 | — | 39 | 100 | % | |||||||||||
| Biogen Idec |
14,520 | 863 | 13,657 | 1,583 | % | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 18,123 | $ | 1,293 | $ | 16,830 | 1,302 | % | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
Revenue. Revenue for the year ended December 31, 2014 was 18.1 million compared to $1.3 million for the year ended December 31, 2013, an increase of approximately $16.8 million, or 1,302%. The increase was primarily due to the recognition of an additional $13.7 million of previously deferred revenue as a result of the amendment to our arrangement with Biogen. Pursuant to the amendment, Biogen agreed to the termination of its rights and obligations under the previous arrangement. As a result, we recognized as revenue all previously deferred amounts in excess of the estimated selling price of the remaining deliverables under the modified arrangement. In addition, we recognized an additional $3.1 million in connection with the change in the estimated period of performance associated with our collaboration with Astellas as a result of the termination of the agreement in August 2014.
Research and development. Research and development expenses for the year ended December 31, 2014 were $38.3 million compared to $68.5 million for the year ended December 31, 2013, a decrease of $30.2 million, or 44%. The decrease is primarily attributable to a $14.0 million decrease in employee compensation, benefits, and contract labor as well as a decrease of $6.2 million in facilities, IT, and other costs following our June 2013 restructuring; a decrease of $11.9 million in outsourced services costs primarily related to the completion of the manufacture of ficlatuzumab material in 2013; and a decrease of $11.7 million in external clinical trial, research, and medical affairs costs associated with decreased tivozanib clinical development activity. The decrease for 2014 was partially offset by a decrease of $9.5 million in reimbursements to us by Astellas for tivozanib development costs due to the corresponding decrease in tivozanib expenses, which we
76
Table of Contents
recorded as a reduction in R&D expense in the prior year period, and an increase of $4.1 million in manufacturing costs related primarily to the completion of the manufacture of AV-380 material in 2014.
Included in research and development expenses were stock-based compensation expenses of approximately $0.9 million and $2.0 million for the years ended December 31, 2014 and 2013, respectively.
General and administrative. General and administrative expenses for the year ended December 31, 2014 were $18.6 million compared to $28.7 million for the year ended December 31, 2013, a decrease of $10.1 million, or 35%. The decrease is primarily the result of a $6.0 million decrease in salaries and benefits as well as a decrease of $2.5 million in facilities and IT costs following our June 2013 restructuring, and a $6.7 million decrease in marketing and consulting costs due to termination of tivozanib pre-commercialization activities. These amounts were partially offset by a $1.6 million increase in external legal costs associated with various ongoing legal matters, an increase in depreciation expense of $0.9 million due to the acceleration of depreciation following the termination of our lease agreement of 650 East Kendall Street in September 2014, and by a $2.6 million decrease in reimbursements to us from Astellas for shared tivozanib general and administrative costs, which we recorded as a reduction in general and administrative expense in the prior year period.
Included in general and administrative expenses were stock-based compensation expenses of approximately $1.9 million and $1.8 million for the years ended December 31, 2014 and 2013, respectively.
Restructuring and lease exit. Restructuring and lease exit expense for the year ended December 31, 2014 was $11.7 million, compared to $8.0 million for the year ended December 31, 2013. The expenses incurred during 2014 relate to costs associated with partially vacating and subsequently terminating the agreement for our leased space at 650 E Kendall Street, which occurred in September 2014. The expenses incurred during 2013 relate to severance and employee benefits incurred as part of the June 2013 strategic restructuring. We expect to incur additional restructuring charges in 2015 as a result of the elimination of our internal research function as part of our strategic restructuring, which is further discussed in Footnote 17 of the Notes to the Consolidated Financial Statements appearing elsewhere in this Annual Report on Form 10-K.
Other income (expense), net. Other income (expense), net for the year ended December 31, 2014 was $0.1 million compared to $(0.1) million for the year ended December 31, 2013, an increase of $0.2 million or 154%. Other income for 2014 is primarily due to proceeds from the sale of lab equipment, while expense for 2013 is primarily due to losses on foreign exchange rates and fixed asset disposals.
Interest expense. Interest expense for the year ended December 31, 2014 was $2.4 million compared to $3.1 million for the year ended December 31, 2013, a decrease of $0.7 million, or 24%. The decrease is primarily attributable to the declining outstanding balance on our loan with Hercules Technology Growth during the first three quarters of 2014.
Interest income. Interest income for the year ended December 31, 2014 was $32,000 compared to $125,000 for the year ended December 31, 2013, a decrease of $93,000, or 74%. The decrease in interest income is primarily due to overall lower average cash, cash equivalent and marketable securities balances during the year ended December 31, 2014 compared to the year ended December 31, 2013.
77
Table of Contents
Comparison of Years Ended December 31, 2013 and 2012
The following tables summarize the results of our operations for each of the years ended December 31, 2013 and 2012, together with the changes in those items in dollars and as a percentage:
| Years Ended December 31, |
Increase/ (decrease) |
% | ||||||||||||||
| 2013 | 2012 | |||||||||||||||
| (in thousands) | ||||||||||||||||
| Revenue |
$ | 1,293 | $ | 19,286 | $ | (17,993 | ) | (93 | )% | |||||||
| Operating expenses: |
||||||||||||||||
| Research and development |
68,468 | 91,358 | (22,890 | ) | (25 | )% | ||||||||||
| General and administrative |
28,712 | 36,932 | (8,220 | ) | (22 | )% | ||||||||||
| Restructuring |
8,017 | 2,633 | 5,384 | 204 | % | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total operating expenses |
105,197 | 130,923 | (25,726 | ) | (20 | )% | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Loss from operations |
(103,904 | ) | (111,637 | ) | 7,733 | (7 | )% | |||||||||
| Other (expense) income, net |
(123 | ) | 247 | (370 | ) | (150 | )% | |||||||||
| Interest expense |
(3,127 | ) | (3,501 | ) | 374 | (11 | )% | |||||||||
| Interest income |
125 | 497 | (372 | ) | (75 | )% | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss |
$ | (107,029 | ) | $ | (114,394 | ) | $ | 7,365 | (6 | )% | ||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Years Ended December 31, |
Increase/ (decrease) |
% | ||||||||||||||
| Revenue |
2013 | 2012 | ||||||||||||||
| (in thousands) | ||||||||||||||||
| Strategic Partner: |
||||||||||||||||
| Astellas |
$ | 430 | $ | 15,430 | $ | (15,000 | ) | (97 | )% | |||||||
| OSI |
— | 1,000 | (1,000 | ) | (100 | )% | ||||||||||
| Centocor |
— | 1,973 | (1,973 | ) | (100 | )% | ||||||||||
| Biogen Idec |
863 | 863 | — | — | ||||||||||||
| Other |
— | 20 | (20 | ) | (100 | )% | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 1,293 | $ | 19,286 | $ | (17,993 | ) | (93 | )% | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
Revenue. Revenue for the year ended December 31, 2013 was $1.3 million compared to $19.3 million for the year ended December 31, 2012, a decrease of approximately $18.0 million, or 93%. The decrease was primarily due to revenue recognized during 2012 that did not recur during 2013, including a $15.0 million milestone payment earned under our collaboration agreement with Astellas related to the FDA’s acceptance of our NDA filing for tivozanib; $1.0 million in patent-related and clinical and development milestone payments earned under our agreement with OSI Pharmaceuticals, Inc., or OSI, which granted to OSI a certain licenses to use our proprietary bioinformatics platform, bioinformatics data and a proprietary gene index related to a specific target pathway ; and research funding under our collaboration agreement with Centocor Ortho Biotech Inc., relating to the worldwide development and commercialization of antibodies, including our internally-discovered antibodies targeting the Recepteur d’Origine Nantais, which agreement terminated in September 2012. Revenue during 2013 related to amortization of previously deferred revenue associated with our collaboration agreements with Astellas and Biogen Idec.
Research and development. Research and development expenses for the year ended December 31, 2013 were $68.5 million compared to $91.4 million for the year ended December 31, 2012, a decrease of $22.9 million, or 25%. The decrease is primarily attributable to a net decrease in licensing costs of $6.8 million due primarily to a milestone payment to KHK made upon the acceptance for filing by the FDA of our NDA for tivozanib during 2012; a decrease of $1.3 million in manufacturing costs related primarily to the reduction in
78
Table of Contents
scope of tivozanib packaging and distribution activities; a decrease in clinical trial costs of $13.3 million primarily due to the ongoing wind-down of tivozanib trials; a decrease of $16.5 million in salaries, benefits and contract labor following our October 2012 and June 2013 restructurings; and a decrease of $2.8 million in external research cost in line with the decrease in overall research activity. The decrease for 2013 was partially offset by a $2.2 million increase in facility and information technology costs due to additional leased space at 650 East Kendall Street; a $2.9 million increase in outsourced services costs primarily related to the completion of the manufacture of ficlatuzumab material that began in 2012; a decrease of $12.3 million in reimbursements to us by Astellas for tivozanib development costs due to the overall decrease in tivozanib expenses; and an increase in depreciation expense of $1.0 million following the completion of a portion of the build-out of our facility at 650 East Kendall Street.
Included in research and development expenses were stock-based compensation expenses of approximately $2.0 million and $3.6 million for the years ended December 31, 2013 and 2012, respectively.
General and administrative. General and administrative expenses for the year ended December 31, 2013 were $28.7 million compared to $36.9 million for the year ended December 31, 2012, a decrease of $8.2 million, or 22%. The decrease is primarily the result of a $6.5 million decrease in salaries, benefits and other hiring costs following our June 2013 restructuring and a $2.6 million decrease in marketing and consulting costs due to termination of work related to tivozanib pre-commercialization activities. These amounts were partially offset by a $0.9 million increase in facility and information technology costs due to additional leased space at 650 East Kendall Street.
Included in general and administrative expenses were stock-based compensation expenses of approximately $1.8 million and $4.4 million for the years ended December 31, 2013 and 2012, respectively.
Restructuring. Restructuring expense for the year ended December 31, 2013 was $8.0 million, compared to $2.6 million for the year ended December 31, 2012, an increase of $5.4 million, or 204%. The increase is primarily the result of the additional costs incurred in connection with our June 2013 strategic restructuring, which was announced in connection with the receipt of a Complete Response Letter from the FDA informing us that the FDA would not approve our NDA for tivozanib for the treatment of patients with advanced RCC. We did not incur any additional restructuring costs or charges with respect to our lease commitments for our headquarters and laboratory space in Cambridge, Massachusetts. The restructuring refocused our efforts on the then on-going clinical development of tivozanib in colorectal and breast cancer and on the advancement of key pipeline and preclinical assets.
Other (expense) income, net. Other (expense) income, net for the year ended December 31, 2013 was $(0.1) million compared to $0.2 million for the year ended December 31, 2012. The decrease in other (expense) income is due to increased losses on foreign exchange rates and fixed asset disposals.
Interest expense. Interest expense for the year ended December 31, 2013 was $3.1 million compared to $3.5 million for the year ended December 31, 2012, a decrease of $0.4 million, or 11%. The decrease in interest expense is due to lower average principal balances on our loan with Hercules Technology Growth.
Interest income. Interest income for the year ended December 31, 2013 was $0.1 million compared to $0.5 million for the year ended December 31, 2012, a decrease of $0.4 million, or 75%. The decrease in interest income is primarily due to overall lower average cash, cash equivalent and marketable securities balances during the year ended December 31, 2013 compared to the year ended December 31, 2012.
79
Table of Contents
Contractual Obligations and Commitments
The following table summarizes our non-cancellable contractual obligations at December 31, 2014:
| Contractual Obligations |
Payment due by period | |||||||||||||||||||
| Total | Less than 1 Year |
1 to 3 Years |
3 to 5 Years |
More than 5 Years |
||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Long-term debt (including interest) |
$ | 25,550 | $ | 14,166 | $ | 11,384 | — | — | ||||||||||||
| Operating lease obligations(1) |
6,880 | 6,880 | — | — | — | |||||||||||||||
| License agreements(2)(3) |
200 | 175 | 25 | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total contractual cash obligations |
$ | 32,630 | $ | 21,221 | $ | 11,409 | — | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| (1) | As discussed in Note 14 to our consolidated financial statements, in September 2014 we amended our lease pursuant to which we immediately surrendered a portion of our leased space at 650 E. Kendall Street in Cambridge, MA. In connection with the lease amendment, we agreed to pay our landlord a termination fee totaling $15.6 million to be settled as follows: fifty percent of the fee, or $7.8 million, to be credited against amounts due us for tenant improvements, and the remaining fifty percent to be paid in nine equal monthly installments of $0.9 million commencing on October 1, 2014. The initial $7.8 million fee was credited by our landlord against our tenant improvement allowance of $14.7 million, resulting in a net payment to us of $6.8 million which was received in September 2014. Pursuant to the lease amendment, we also agreed to surrender the remaining portion of the leased space upon ninety (90) days written notice on a date that is no earlier than March 24, 2015 and no later than September 24, 2015. We provided notice in February 2015 of our election to surrender the space on May 29, 2015. Our remaining contractual obligations at December 31, 2014 consist of the remaining portion of the fifty percent of the termination fee, or $5.2 million, payable in six equal monthly installments, and remaining non-cancellable rental payments totaling $1.7 million. |
| (2) | Under our license agreement with Kyowa Hakko Kirin, we are required to make certain milestone payments upon the achievement of specified regulatory milestones. In November 2014, we received $0.5 million of upfront payment from our collaboration agreement with Ophthotech. We are required to make a $0.2 million royalty payment to Kyowa Hakko Kirin related to the upfront payment by March 2015 and have included this amount in the table above. At this time, we cannot reasonably estimate when or if we may be required to make other additional payments to Kyowa Hakko Kirin and have not included any additional amounts in the table above. |
| (3) | As discussed in Note 7 to our consolidated financial statements, we have executed license agreements for patented technology and other technology related to research projects, including technology to humanize ficlatuzumab and other antibody product candidates. The license agreements required us to pay non-refundable license fees upon execution, and in certain cases, require milestone payments upon the achievement of defined development goals. We have not included any additional milestone payments in the table above as we are not able to make a reasonable estimate of the probability and timing of such payments, if any. In addition to the amounts in the table above, these four agreements include sales and development milestones of up to $22.5 million, $5.5 million, $9.6 million and $4.2 million per product, respectively, and single digit royalties as a percentage of sales. |
Liquidity and Capital Resources
We have funded our operations principally through the sale of equity securities sold in private placements and underwritten public offerings of equity securities, revenue and expense reimbursements from strategic partnerships, debt financing and interest income. Through December 31, 2014, we have received gross proceeds of $89.7 million from the sale of common stock in our initial public offering, $68.3 million from private placements of shares of our common stock to institutional and accredited investors, $168.7 million from a follow-on public offering of shares of our common stock, and $169.6 million from the sale of convertible preferred stock prior to becoming a public company. As of December 31, 2014, we have received an aggregate of
80
Table of Contents
$396.3 million in cash from our agreements with strategic partners, and $36.5 million in funding from our debt financing with Hercules Technology Growth and certain of its affiliates. As of December 31, 2014, we had cash and cash equivalents of approximately $52.3 million. Currently, our funds are invested in money market funds, U.S. government agency securities, and corporate debt securities, including commercial paper. The following table sets forth the primary sources and uses of cash for each of the periods set forth below:
| Years Ended December 31, |
||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| (in thousands) | ||||||||||||
| Net cash used in operating activities |
$ | (54,248 | ) | $ | (84,402 | ) | $ | (105,729 | ) | |||
| Net cash provided by investing activities |
54,710 | 12,070 | 135,247 | |||||||||
| Net cash provided by financing activities |
1,018 | 46,998 | 3,136 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net increase (decrease) in cash and cash equivalents |
$ | 1,480 | $ | (25,334 | ) | $ | 32,654 | |||||
|
|
|
|
|
|
|
|||||||
During the years ended December 31, 2014, 2013 and 2012, our operating activities used cash of $54.2 million, $84.4 million and $105.7 million, respectively. Cash used by operations for the year ended December 31, 2014 was due primarily to our net loss adjusted for non-cash items, including a $7.6 million impairment of property due to the lease exit costs incurred upon us meeting the cease use criteria for certain of our facilities during 2014, an $18.0 million recognition of deferred revenue related to the termination of our collaboration with Astellas and Biogen during the same period, and working capital changes. Cash used by operations for the years ended December 31, 2013 and 2012, respectively, was due primarily to our net losses adjusted for non-cash items.
During the years ended December 31, 2014, 2013 and 2012, our investing activities provided cash of $54.7 million, $12.1 million and $135.2 million, respectively. The cash provided by investing activities for the years ended December 31, 2014, 2013, and 2012 was the result of fewer purchases of marketable securities than the proceeds from maturities and sales of marketable securities in order to fund our ongoing operations, partially offset by purchases of property and equipment of $12.9 million, $3.7 million and $9.9 million during the years ended December 31, 2014, 2013, and 2012, respectively.
During the years ended December 31, 2014, 2013 and 2012, our financing activities provided $1.0 million, $47.0 million and $3.1 million, respectively. The cash provided by financing activities in 2014 was due to net proceeds of $8.6 million from the amendment of our loan agreement entered into with affiliates of Hercules Technology Growth, offset partially by principal payments on loans payable in the amount of $7.8 million. The cash provided by financing activities in 2013 was primarily due to the net proceeds of $53.6 million from our public offering of stock, offset by $7.1 million in principal payments on our loan from Hercules Technology Growth. The cash provided by financing activities in 2012 was due to stock option exercises of $1.6 million, as well as net proceeds of $3.7 million from the refinancing of loans payable from our loan agreement entered into with affiliates of Hercules Technology Growth, offset partially by principal payments on loans payable in the amount of $2.2 million.
Credit Facilities. On September 24, 2014, we amended our loan and security agreement, which we refer to as the amended loan agreement, with Hercules Technology II, L.P. and Hercules Technology III, L.P., affiliates of Hercules Technology Growth, which we originally entered into on May 28, 2010 and amended on December 21, 2011 and March 31, 2012. Pursuant to the amended loan agreement, we received a new loan in an aggregate principal amount of $10.0 million and amended the terms of the original loan with an outstanding principal balance of $11.6 million. We are not required to pay principal on the new loan of $10.0 million until November 1, 2015, which date may be extended if we achieve certain performance milestones, and after which time we are required to make thirty (30) principal and interest payments with the entire loan due and payable on January 1, 2018. With respect to the original loan, we are not required to pay interest until January 1, 2015, at which time we are required to commence making twelve (12) principal and interest payments. The original loan
81
Table of Contents
agreement also included an obligation to pay a deferred financing charge of $1.2 million which we paid on June 1, 2014, and which has been recorded as a loan discount and is being amortized to interest expense over the term of the original loan using the effective interest rate method. The amended loan agreement has an end-of-term payment of approximately $0.5 million due on January 1, 2018 or on such earlier date as the new loan is prepaid. The amended loan agreement also has a financial covenant with respect to the new loan, whereby we have agreed to maintain a liquidity ratio equal to or greater than 1.25 to 1.00 or the equivalent of $12.5 million in unrestricted and unencumbered cash and cash equivalents at December 31, 2014. This financial covenant shall not apply after such time as we receive favorable data both with respect to our phase 2 clinical trial of ficlatuzumab and a phase 1 clinical trial of AV-380. We must make interest payments on both loans each month the loans remain outstanding. Per annum interest is payable on both loans at the greater of 11.9% and an amount equal to 11.9% plus the prime rate of interest minus 4.75%, provided however, that the per annum interest shall not exceed 15.0%.
While we do not believe that there has been a material adverse change as defined in our loan agreement, it is possible that Hercules Technology Growth could take the position that the recent restructuring we consummated in January 2015 constitutes a material adverse change under our loan and security agreements, which could trigger a repayment of all principal and interest due under the loan, unless such event of default is waived by the lender. As of December 31, 2014 and through the date of this filing, the lenders have not asserted any events of default under the loan.
The loans are secured by a lien on all of our personal property (other than intellectual property), whether owned as of, or acquired after, the date of the amended loan agreement. As of December 31, 2014, the principal balance outstanding was $21.6 million.
Operating Capital Requirements. We anticipate that we will continue to incur significant operating losses for the next several years as we incur expenses to continue to execute on our clinical development strategy to advance our clinical stage assets.
Based on our current operating plan, we expect our $52.3 million in cash resources as of December 31, 2014 will be sufficient to fund operations into the third quarter of 2016.
Because of the numerous risks and uncertainties associated with research, development and commercialization of pharmaceutical products, we are unable to estimate the exact amounts of our working capital requirements. Our future funding requirements will depend on many factors, including, but not limited to:
| • | our ability to establish and maintain strategic partnerships, licensing or other arrangements and the financial terms of such agreements; |
| • | the number and characteristics of the product candidates we pursue; |
| • | the scope, progress, results and costs of researching and developing our product candidates, and conducting preclinical and clinical trials; |
| • | the timing of, and the costs involved in, obtaining regulatory approvals for our product candidates; |
| • | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims, including litigation costs and the outcome of such litigation; |
| • | the absence of any breach, acceleration event or event of default under our loan agreement with Hercules or under any other agreements with third parties; |
| • | the outcome of lawsuits against us, including the current lawsuits described below under “Part I, Item 3—Legal Proceedings;” |
| • | the cost of commercialization activities if any of our product candidates are approved for sale, including marketing, sales and distribution costs; |
82
Table of Contents
| • | the cost of manufacturing our product candidates and any products we successfully commercialize; |
| • | our ability to secure alternative leasing or subleasing arrangements for new office space and to achieve related cost savings with respect to termination of our lease at 650 East Kendall Street, Cambridge, and |
| • | the timing, receipt and amount of sales of, or royalties on, our future products, if any. |
We may seek to sell additional equity or debt securities or obtain additional credit facilities. The sale of additional equity or convertible debt securities may result in additional dilution to our stockholders. If we raise additional funds through the issuance of debt securities or preferred stock or through additional credit facilities, these securities and/or the loans under credit facilities could provide for rights senior to those of our common stock and could contain covenants that would restrict our operations. We may require additional capital beyond our currently forecasted amounts. Additional funds may not be available when we need them, on terms that are acceptable to us, or at all.
We also expect to seek additional funds through arrangements with collaborators, licensees and other third parties. These arrangements would generally require us to relinquish or encumber rights to some of our technologies or drug candidates, and we may not be able to enter into such arrangements on acceptable terms, if at all.
We anticipate that we will require additional funding. If we are unable to obtain such additional funding on a timely basis, whether through payments under existing or future collaborations or license agreement or sales of debt equity, we may be required to delay, limit, reduce or terminate our clinical trials or other development activities for one or more of our product candidates.
Off-Balance Sheet Arrangements
We did not have, during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined under applicable SEC rules.
| ITEM 7A. | Quantitative and Qualitative Disclosures About Market Risk |
We are exposed to market risk related to changes in interest rates. As of December 31, 2014, we had cash and cash equivalents of $52.3 million, consisting of cash on deposit with banks, money market funds, U.S. government agency securities, and corporate debt, including commercial paper. Our primary exposure to market risk is interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates, particularly because our investments are in short-term cash equivalents. Our cash equivalents are subject to interest rate risk and could fall in value if market interest rates increase. Due to the short-term duration of our investment portfolio and the low risk profile of our investments, an immediate 10% change in interest rates would not have a material effect on the fair market value of our portfolio. We have the ability to hold our cash equivalents until maturity, and therefore we would not expect our operating results or cash flows to be affected to any significant degree by the effect of a change in market interest rates on our investments. We do not currently have any auction rate securities.
Our long-term debt bears interest at variable rates. In May 2010, we entered into a loan agreement with affiliates of Hercules Technology Growth Capital pursuant to which we received a loan in the aggregate principal amount of $25.0 million. In March 2012, we entered into an amendment to the loan agreement, pursuant to which we increased the principal amount to $26.5 million. In September 2014, we entered into a further amendment to the loan agreement, pursuant to which we borrowed a new loan of $10.0 million, which is in addition to the existing loan which had an outstanding principal balance of $11.6 million. As of December 31, 2014, our aggregate principal balance outstanding on our loans was $21.6 million. Per annum interest is payable at the greater of 11.9% and 11.9% plus the prime rate of interest minus 4.75%, not to exceed 15%. As a result of the 15% maximum per annum interest rate under the amended loan agreement, we have limited exposure to changes
83
Table of Contents
in interest rates on borrowings under this loan agreement. For every 1% increase in the prime rate over 4.75%, given the amount of debt outstanding under the loan agreement as of December 31, 2014, and expected loan payments during 2015, we would have a decrease in future annual cash flows of approximately $0.2 million over the next twelve month period as a result of such 1% increase.
We are also exposed to market risk related to change in foreign currency exchange rates. We contract with contract research organizations and investigational sites that are located around the world. We are subject to fluctuations in foreign currency rates in connection with these agreements. We do not currently hedge our foreign currency exchange rate risk.
84
Table of Contents
| ITEM 8. | Financial Statements and Supplementary Data |
AVEO PHARMACEUTICALS, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
85
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of
AVEO Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of AVEO Pharmaceuticals, Inc. as of December 31, 2014 and 2013, and the related consolidated statements of operations, comprehensive (loss) income, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2014. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of AVEO Pharmaceuticals, Inc. at December 31, 2014 and 2013, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2014, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), AVEO Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), and our report dated March 6, 2015 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Boston, Massachusetts
March 6, 2015
86
Table of Contents
Consolidated Balance Sheets
(In thousands, except par value amounts)
| 2014 | 2013 | |||||||
| Assets | ||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 52,306 | $ | 50,826 | ||||
| Marketable securities |
— | 67,680 | ||||||
| Accounts receivable |
2,341 | 984 | ||||||
| Tenant improvement allowance receivable |
— | 5,833 | ||||||
| Restricted cash |
2,997 | 598 | ||||||
| Prepaid expenses and other current assets |
1,484 | 2,998 | ||||||
|
|
|
|
|
|||||
| Total current assets |
59,128 | 128,919 | ||||||
| Property and equipment, net |
11,295 | 14,140 | ||||||
| Other assets |
239 | 290 | ||||||
| Restricted cash |
— | 2,997 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 70,662 | $ | 146,346 | ||||
|
|
|
|
|
|||||
| Liabilities and stockholders’ equity | ||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 3,245 | $ | 4,238 | ||||
| Accrued expenses |
9,301 | 13,263 | ||||||
| Lease exit obligation |
4,981 | — | ||||||
| Loans payable, net of discount |
11,722 | 10,383 | ||||||
| Deferred revenue |
537 | 1,294 | ||||||
| Other liabilities |
— | 1,238 | ||||||
| Deferred rent |
10,569 | 992 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
40,355 | 31,408 | ||||||
| Loans payable, net of current portion and discount |
8,930 | 8,822 | ||||||
| Deferred revenue, net of current portion |
231 | 17,098 | ||||||
| Deferred rent, net of current portion |
— | 19,080 | ||||||
| Other liabilities |
540 | — | ||||||
| Commitments and contingencies (Note 8) |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $.001 par value: 5,000 shares authorized; no shares issued and outstanding |
— | — | ||||||
| Common stock, $.001 par value: 100,000 shares authorized; 52,289 and 51,809 shares issued and outstanding at December 31, 2014 and 2013, respectively |
52 | 52 | ||||||
| Additional paid-in capital |
500,582 | 497,177 | ||||||
| Accumulated other comprehensive loss |
— | (2 | ) | |||||
| Accumulated deficit |
(480,028 | ) | (427,289 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
20,606 | 69,938 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 70,662 | $ | 146,346 | ||||
|
|
|
|
|
|||||
See accompanying notes
87
Table of Contents
Consolidated Statements of Operations
(In thousands, except per share amounts)
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Collaboration revenue |
$ | 18,123 | $ | 1,293 | $ | 19,286 | ||||||
| Operating expenses: |
||||||||||||
| Research and development |
38,254 | 68,468 | 91,358 | |||||||||
| General and administrative |
18,589 | 28,712 | 36,932 | |||||||||
| Restructuring and lease exit |
11,729 | 8,017 | 2,633 | |||||||||
|
|
|
|
|
|
|
|||||||
| 68,572 | 105,197 | 130,923 | ||||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(50,449 | ) | (103,904 | ) | (111,637 | ) | ||||||
| Other income and expense: |
||||||||||||
| Other (expense) income, net |
66 | (123 | ) | 247 | ||||||||
| Interest expense |
(2,388 | ) | (3,127 | ) | (3,501 | ) | ||||||
| Interest income |
32 | 125 | 497 | |||||||||
|
|
|
|
|
|
|
|||||||
| Other expense, net |
(2,290 | ) | (3,125 | ) | (2,757 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (52,739 | ) | $ | (107,029 | ) | $ | (114,394 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per share: |
||||||||||||
| Net loss per share |
$ | (1.01 | ) | $ | (2.10 | ) | $ | (2.64 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted average number of common shares outstanding |
52,289 | 50,928 | 43,374 | |||||||||
|
|
|
|
|
|
|
|||||||
See accompanying notes
88
Table of Contents
Consolidated Statements of Comprehensive Loss
(In thousands)
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Net loss |
$ | (52,739 | ) | $ | (107,029 | ) | $ | (114,394 | ) | |||
| Other comprehensive (loss) income: |
||||||||||||
| Unrealized (losses) gains on available-for-sale securities |
2 | (9 | ) | 174 | ||||||||
| Foreign currency translation adjustment |
— | 26 | (26 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Comprehensive loss |
$ | (52,737 | ) | $ | (107,012 | ) | $ | (114,246 | ) | |||
|
|
|
|
|
|
|
|||||||
See accompanying notes
89
Table of Contents
Consolidated Statements of Stockholders’ Equity
(In thousands)
| Common Shares |
Additional Paid-in Capital |
Accumulated Other Comprehensive Loss |
Accumulated Deficit |
Total Stockholders’ Equity |
||||||||||||||||||||
| Transaction |
Shares | Par Value | ||||||||||||||||||||||
| Balance at December 31, 2011 |
43,254 | $ | 43 | $ | 429,531 | $ | (167 | ) | $ | (205,866 | ) | $ | 223,541 | |||||||||||
| Exercise of stock options |
220 | 1 | 825 | — | — | 826 | ||||||||||||||||||
| Stock-based compensation expense |
— | — | 8,007 | — | — | 8,007 | ||||||||||||||||||
| Issuance of common stock under employee stock purchase plan |
95 | — | 810 | — | — | 810 | ||||||||||||||||||
| Issuance of restricted stock awards, net of forfeitures |
211 | — | — | — | — | — | ||||||||||||||||||
| Change in unrealized gain/loss on investments |
— | — | — | 174 | — | 174 | ||||||||||||||||||
| Cumulative translation adjustment |
— | — | — | (26 | ) | — | (26 | ) | ||||||||||||||||
| Net loss |
— | — | — | — | (114,394 | ) | (114,394 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2012 |
43,780 | $ | 44 | $ | 439,173 | $ | (19 | ) | $ | (320,260 | ) | $ | 118,938 | |||||||||||
| Exercise of stock options |
185 | 1 | 271 | — | — | 272 | ||||||||||||||||||
| Stock-based compensation expense related to equity-classified awards |
— | — | 3,791 | — | — | 3,791 | ||||||||||||||||||
| Issuance of common stock to settle liability-classified share awards granted to directors |
39 | — | 119 | — | — | 119 | ||||||||||||||||||
| Issuance of common stock under employee stock purchase plan |
110 | — | 193 | — | — | 193 | ||||||||||||||||||
| Issuance of common stock from follow-on stock offering (net of issuance cost of $3,865) |
7,667 | 7 | 53,630 | — | — | 53,637 | ||||||||||||||||||
| Issuance of restricted stock awards, net of forfeitures |
28 | — | — | — | — | — | ||||||||||||||||||
| Change in unrealized gain/loss on investments |
— | — | — | (9 | ) | — | (9 | ) | ||||||||||||||||
| Cumulative translation adjustment |
— | — | — | 26 | — | 26 | ||||||||||||||||||
| Net loss |
— | — | — | — | (107,029 | ) | (107,029 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2013 |
51,809 | $ | 52 | $ | 497,177 | $ | (2 | ) | $ | (427,289 | ) | $ | 69,938 | |||||||||||
| Stock-based compensation expense related to equity-classified awards |
— | — | 2,750 | — | — | 2,750 | ||||||||||||||||||
| Issuance of common stock to settle liability-classified share awards granted to directors |
31 | — | 51 | — | — | 51 | ||||||||||||||||||
| Issuance of common stock under employee stock purchase plan |
139 | — | 191 | — | — | 191 | ||||||||||||||||||
| Issuance of warrants in connection with loans payable |
— | — | 413 | — | — | 413 | ||||||||||||||||||
| Issuance of restricted stock awards, net of forfeitures |
310 | — | — | — | — | — | ||||||||||||||||||
| Change in unrealized gain/loss on investments |
— | — | — | 2 | — | 2 | ||||||||||||||||||
| Net loss |
— | — | — | — | (52,739 | ) | (52,739 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2014 |
52,289 | $ | 52 | $ | 500,582 | $ | — | $ | (480,028 | ) | $ | 20,606 | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
See accompanying notes
90
Table of Contents
Consolidated Statements of Cash Flows
(in thousands)
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Operating activities |
||||||||||||
| Net loss |
$ | (52,739 | ) | $ | (107,029 | ) | $ | (114,394 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation and amortization |
6,219 | 3,775 | 2,510 | |||||||||
| Net (gain) loss on disposal of property and equipment |
(127 | ) | 83 | 42 | ||||||||
| Impairment of property and equipment |
7,600 | 65 | — | |||||||||
| Stock-based compensation |
2,808 | 3,940 | 8,007 | |||||||||
| Non-cash interest expense |
347 | 285 | 380 | |||||||||
| Amortization of premiums and discounts on investments |
221 | 1,041 | 2,446 | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Accounts receivable |
(1,357 | ) | 19,665 | (13,439 | ) | |||||||
| Tenant improvement allowance receivable |
5,833 | (2,593 | ) | (3,180 | ) | |||||||
| Prepaid expenses and other current assets |
1,503 | 3,179 | (207 | ) | ||||||||
| Other noncurrent assets |
50 | 31 | (200 | ) | ||||||||
| Restricted cash |
598 | 5 | (2,849 | ) | ||||||||
| Accounts payable |
(993 | ) | (6,390 | ) | 1,724 | |||||||
| Accrued expenses |
(2,064 | ) | (7,838 | ) | 5,254 | |||||||
| Lease exit obligation |
4,981 | — | — | |||||||||
| Deferred revenue |
(17,623 | ) | (1,293 | ) | (1,293 | ) | ||||||
| Other liabilities |
— | — | (1,249 | ) | ||||||||
| Deferred rent |
(9,505 | ) | 8,672 | 10,719 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(54,248 | ) | (84,402 | ) | (105,729 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Investing activities |
||||||||||||
| Purchases of property and equipment |
(12,942 | ) | (3,668 | ) | (9,948 | ) | ||||||
| Purchases of marketable securities |
(42,306 | ) | (175,391 | ) | (194,584 | ) | ||||||
| Proceeds from maturities and sales of marketable securities |
109,767 | 191,129 | 339,779 | |||||||||
| Proceeds from sale of property and equipment |
191 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by investing activities |
54,710 | 12,070 | 135,247 | |||||||||
|
|
|
|
|
|
|
|||||||
| Financing activities |
||||||||||||
| Proceeds from issuance of common stock, net of issuance costs |
— | 53,637 | — | |||||||||
| Proceeds from issuance of stock for stock-based compensation arrangements |
191 | 465 | 1,636 | |||||||||
| Proceeds from refinancing of loans payable |
— | — | 3,672 | |||||||||
| Proceeds from issuance of long-term debt |
10,000 | — | — | |||||||||
| Payments of debt issuance cost |
(1,388 | ) | — | — | ||||||||
| Principal payments on loans payable |
(7,785 | ) | (7,104 | ) | (2,172 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
1,018 | 46,998 | 3,136 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net increase (decrease) in cash and cash equivalents |
1,480 | (25,334 | ) | 32,654 | ||||||||
| Effect of exchange rate changes on cash and cash equivalents |
— | 26 | (26 | ) | ||||||||
| Cash and cash equivalents at beginning of period |
50,826 | 76,134 | 43,506 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents at end of period |
$ | 52,306 | $ | 50,826 | $ | 76,134 | ||||||
|
|
|
|
|
|
|
|||||||
| Supplemental cash flow and noncash investing and financing activities |
||||||||||||
| Cash paid for interest |
$ | 2,018 | $ | 2,916 | $ | 3,104 | ||||||
| Non-cash financing activity |
||||||||||||
| Fair value of warrants issued in connection with long-term debt |
$ | 413 | — | — | ||||||||
See accompanying notes
91
Table of Contents
Notes to Consolidated Financial Statements
December 31, 2014
| 1. | Nature of Business and Organization |
AVEO Pharmaceuticals, Inc. (the “Company”) is a biopharmaceutical company committed to developing targeted therapies through biomarker insights to provide substantial improvements in patient outcomes where significant unmet medical needs exist. AVEO’s proprietary platform has delivered unique insights into cancer and related disease. AVEO’s strategy for building value is to leverage these biomarker insights and partner resources to advance the development of its clinical pipeline.
The Company’s pipeline of product candidates includes Tivozanib, a potent, selective long half-life vascular endothelial growth factor tyrosine kinase inhibitor of all three vascular endothelial growth factors, or VEGF TKI, for which the Company previously demonstrated tivozanib’s safety and efficacy in first and second line RCC. However, the U.S. Food and Drug Administration issued a Complete Response letter denying AVEO’s application for approval of the use of tivozanib in first line advanced RCC. We are currently evaluating the opportunity for submitting a Marketing Authorization Application for tivozanib with the European Medicines Agency for the treatment of RCC based on the trials conducted to date.
The Company also has a pipeline of monoclonal antibodies, including:
| (i) | Ficlatuzumab, a potent anti-HFG antibody that inhibits the activity of the HGF/c-Met pathway for which the Company has completed a phase 2 clinical trial, and has entered into a partnership with Biodesix, Inc. (“Biodesix”) to advance clinical development, |
| (ii) | AV-203, a potent, high affinity inhibitor of ErbB3 function that has demonstrated anti-tumor activity in multiple preclinical models for which the Company has completed a phase 1 dose escalation trial, |
| (iii) | AV-380, a potent humanized IgG1 inhibitory monoclonal antibody targeting growth differentiating factor-15, or GDF15, a divergent member of the TGF-ß family, for the potential treatment or prevention of cachexia. In 2012, the Company initiated a program focusing on cachexia, a serious and common complication of advanced cancer and a number of chronic diseases including chronic kidney disease, congestive heart failure and chronic obstructive pulmonary disease. |
As used throughout these consolidated financial statements, the terms “AVEO,” and the “Company”, refer to the business of AVEO Pharmaceuticals, Inc. and its subsidiaries, AVEO Pharma Limited and AVEO Securities Corporation, both of which are wholly-owned.
The Company has an accumulated deficit as of December 31, 2014 of approximately $480.0 million, and will require substantial additional capital for research and product development. The Company believes that its existing cash and cash equivalents are sufficient to fund its operations through at least twelve months from the balance sheet date.
| 2. | Significant Accounting Policies |
Revenue Recognition
The Company’s revenues have been generated primarily through collaborative research, development and commercialization agreements. The terms of these agreements generally contain multiple elements, or deliverables, which may include (i) licenses, or options to obtain licenses, to the Company’s technology, (ii) research and development activities to be performed on behalf of the collaborative partner, and (iii) in certain cases, services in connection with the manufacturing of pre-clinical and clinical material. Payments to the
92
Table of Contents
Company under these arrangements typically include one or more of the following: non-refundable, up-front license fees; option exercise fees; funding of research and/or development efforts; milestone payments; and royalties on future product sales.
When evaluating multiple element arrangements, the Company considers whether the deliverables under the arrangement represent separate units of accounting. This evaluation requires subjective determinations and requires management to make judgments about the individual deliverables and whether such deliverables are separable from the other aspects of the contractual relationship. In determining the units of accounting, management evaluates certain criteria, including whether the deliverables have standalone value, based on the relevant facts and circumstances for each arrangement. The consideration received is allocated among the separate units of accounting using the relative selling price method, and the applicable revenue recognition criteria are applied to each of the separate units.
The Company determines the estimated selling price for deliverables within each agreement using vendor-specific objective evidence (“VSOE”) of selling price, if available, third-party evidence (“TPE”) of selling price if VSOE is not available, or best estimate of selling price if neither VSOE nor TPE is available. Determining the best estimate of selling price for a deliverable requires significant judgment. The Company typically uses best estimate of selling price to estimate the selling price for licenses to the Company’s proprietary technology, since the Company often does not have VSOE or TPE of selling price for these deliverables. In those circumstances where the Company utilizes best estimate of selling price to determine the estimated selling price of a license to the Company’s proprietary technology, the Company considers market conditions as well as entity-specific factors, including those factors contemplated in negotiating the agreements and internally developed models that include assumptions related to the market opportunity, estimated development costs, probability of success and the time needed to commercialize a product candidate pursuant to the license. In validating the Company’s best estimate of selling price, the Company evaluates whether changes in the key assumptions used to determine the best estimate of selling price will have a significant effect on the allocation of arrangement consideration among multiple deliverables.
The Company typically receives non-refundable, up-front payments when licensing its intellectual property in conjunction with a research and development agreement. When management believes the license to its intellectual property does not have stand-alone value from the other deliverables to be provided in the arrangement, the Company generally recognizes revenue attributed to the license on a straight-line basis over the Company’s contractual or estimated performance period, which is typically the term of the Company’s research and development obligations. If management cannot reasonably estimate when the Company’s performance obligation ends, then revenue is deferred until management can reasonably estimate when the performance obligation ends. When management believes the license to its intellectual property has stand-alone value, the Company generally recognizes revenue attributed to the license upon delivery. The periods over which revenue should be recognized are subject to estimates by management and may change over the course of the research and development agreement. Such a change could have a material impact on the amount of revenue the Company records in future periods.
Payments or reimbursements resulting from the Company’s research and development efforts for those arrangements where such efforts are considered as deliverables are recognized as the services are performed and are presented on a gross basis so long as there is persuasive evidence of an arrangement, the fee is fixed or determinable, and collection of the related receivable is reasonably assured. Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue in the accompanying balance sheets.
At the inception of each agreement that includes milestone payments, the Company evaluates whether each milestone is substantive and at risk to both parties on the basis of the contingent nature of the milestone. This evaluation includes an assessment of whether (a) the consideration is commensurate with either (1) the entity’s performance to achieve the milestone, or (2) the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity’s performance to achieve the milestone, (b) the consideration relates
93
Table of Contents
solely to past performance, and (c) the consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. The Company evaluates factors such as the scientific, regulatory, commercial and other risks that must be overcome to achieve the respective milestone, the level of effort and investment required to achieve the respective milestone and whether the milestone consideration is reasonable relative to all deliverables and payment terms in the arrangement in making this assessment.
The Company aggregates its milestones into four categories: (i) clinical and development milestones, (ii) regulatory milestones, (iii) commercial milestones, and (iv) patent-related milestones. Clinical and development milestones are typically achieved when a product candidate advances into a defined phase of clinical research or completes such phase. For example, a milestone payment may be due to the Company upon the initiation of a phase 3 clinical trial for a new indication, which is the last phase of clinical development and could eventually contribute to marketing approval by the U.S. Food and Drug Administration (“FDA”) or other global regulatory authorities. Regulatory milestones are typically achieved upon acceptance of the submission for marketing approval of a product candidate or upon approval to market the product candidate by the FDA or other global regulatory authorities. For example, a milestone payment may be due to the Company upon the FDA’s acceptance of a New Drug Application (“NDA”). Commercial milestones are typically achieved when an approved pharmaceutical product reaches certain defined levels of net sales by the licensee, such as when a product first achieves global sales or annual sales of a specified amount. Patent-related milestones are typically achieved when a patent application is filed or a patent is issued with respect to certain intellectual property related to the applicable collaboration.
Revenues from clinical and development, regulatory, and patent-related milestone payments, if the milestones are deemed substantive and the milestone payments are nonrefundable, are recognized upon successful accomplishment of the milestones. The Company has concluded that the clinical and development, regulatory and patent-related milestones pursuant to its current research and development arrangements are substantive. Milestones that are not considered substantive are accounted for as license payments and recognized on a straight-line basis over the remaining period of performance. Revenues from commercial milestone payments are accounted for as royalties and are recorded as revenue upon achievement of the milestone, assuming all other revenue recognition criteria are met.
Principles of Consolidation
The Company’s consolidated financial statements include the Company’s accounts and the accounts of the Company’s wholly-owned subsidiaries, AVEO Pharma Limited and AVEO Securities Corporation. All intercompany transactions have been eliminated.
Research and Development Expenses
Research and development expenses are charged to expense as incurred. Research and development expenses consist of costs incurred in performing research and development activities, including personnel-related costs such as salaries and stock-based compensation, facilities, research-related overhead, clinical trial costs, manufacturing costs and costs of other contracted services, license fees, and other external costs. Nonrefundable advance payments for goods and services that will be used in future research and development activities are expensed when the activity has been performed or when the goods have been received.
Cash and Cash Equivalents
The Company considers highly liquid investments with a maturity of three months or less when purchased to be cash equivalents. Cash equivalents at December 31, 2014 consisted of money market funds, U.S. government agency securities, and corporate debt securities, including commercial paper, maintained by an investment manager totaling $36.6 million. Cash equivalents at December 31, 2013 consisted of money market
94
Table of Contents
funds and corporate debt securities, including commercial paper, maintained by an investment manager totaling $45.8 million. The carrying values of our cash equivalent securities approximate fair value due to their short term maturities.
Marketable Securities
There were no marketable securities held by the Company at December 31, 2014. Marketable securities at December 31, 2013 consisted of municipal bonds, asset-backed securities, and corporate debt securities, including commercial paper, maintained by an investment manager. Credit risk is reduced as a result of the Company’s policy to limit the amount invested in any one issue. Marketable securities consist primarily of investments which have expected average maturity dates in excess of three months, but not longer than 24 months. The Company classifies these investments as available-for-sale. Unrealized gains and losses are included in other comprehensive (loss) income until realized. The cost of securities sold is based on the specific identification method. There were no realized gains or losses recognized on the sale or maturity of securities during the years ended December 31, 2014 and 2013.
Available-for-sale securities at December 31, 2013 consisted of the following:
| Amortized Cost |
Unrealized Gains |
Unrealized Losses |
Fair Value |
|||||||||||||
| (in thousands) | ||||||||||||||||
| Corporate debt securities (Due within 1 year) |
$ | 52,156 | $ | 4 | $ | (4 | ) | $ | 52,156 | |||||||
| Government agency securities (Due within 1 year) |
7,519 | — | — | 7,519 | ||||||||||||
| Asset-backed securities (Due within 1 year) |
8,007 | — | (2 | ) | 8,005 | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 67,682 | $ | 4 | $ | (6 | ) | $ | 67,680 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The aggregate fair value of securities in an unrealized loss position for less than 12 months at December 31, 2013 was $45.6 million, representing 18 securities. There were no securities in an unrealized loss position for greater than 12 months at December 31, 2013. The unrealized loss was caused by a temporary change in the market for these securities primarily caused by changes in markets interest rates. There was no change in the credit risk of the securities. To determine whether an other-than-temporary impairment exists, the Company performs an analysis to assess whether it intends to sell, or whether it would more likely than not be required to sell, the security before the expected recovery of the amortized cost basis. Where the Company intends to sell a security, or may be required to do so, the security’s decline in fair value is deemed to be other-than-temporary and the full amount of the unrealized loss is recorded in the statement of operations as an other-than-temporary impairment charge. When this is not the case, the Company performs additional analyses on all securities with unrealized losses to evaluate losses associated with the creditworthiness of the security. Credit losses are identified where the Company does not expect to receive cash flows, based on using a single best estimate, sufficient to recover the amortized cost basis of a security and these are recognized in other income (expense), net.
Marketable securities in an unrealized loss position at December 31, 2013 consisted of the following:
| Aggregate Fair Value |
Unrealized Losses |
|||||||
| (in thousands) | ||||||||
| Corporate debt securities (Due within 1 year) |
$ | 30,106 | $ | (4 | ) | |||
| Government agency securities (Due within 1 year) |
7,519 | — | ||||||
| Asset-backed securities (Due within 1 year) |
8,005 | (2 | ) | |||||
|
|
|
|
|
|||||
| $ | 45,630 | $ | (6 | ) | ||||
|
|
|
|
|
|||||
95
Table of Contents
Concentrations of Credit Risk
Financial instruments that potentially subject the Company to credit risk primarily consist of cash, cash equivalents and available-for-sale marketable securities. The Company maintains deposits in federally insured financial institutions in excess of federally insured limits.
Management believes that the Company is not exposed to significant credit risk due to the financial position of the depository institutions in which those deposits are held.
The Company’s credit risk related to marketable securities is reduced as a result of the Company’s policy to limit the amount invested in any one issue.
Fair Value Measurements
The Company records cash equivalents and marketable securities at fair value. The accounting standards for fair value measurements establish a hierarchy that distinguishes between fair value measurements based on market data (observable inputs) and those based on the Company’s own assumptions (unobservable inputs). The hierarchy consists of three levels:
| • | Level 1—Quoted market prices in active markets for identical assets or liabilities. Assets that are valued utilizing only Level 1 inputs include money market funds. |
| • | Level 2—Inputs other than Level 1 inputs that are either directly or indirectly observable, such as quoted market prices, interest rates and yield curves. Assets that are valued utilizing Level 2 inputs include U.S. government agency securities, asset-backed securities, and corporate bonds, including commercial paper. These investments have been initially valued at the transaction price and are subsequently valued, at the end of each reporting period, utilizing third party pricing services or other observable market data. The pricing services utilize industry standard valuation models, including both income and market based approaches and observable market inputs to determine value. These observable market inputs include reportable trades, benchmark yields, credit spreads, broker/dealer quotes, bids, offers, current spot rates, and other industry and economic events. The Company validates the prices provided by third party pricing services by reviewing their pricing methods and matrices, obtaining market values from other pricing sources, analyzing pricing data in certain instances and confirming that the relevant markets are active. After completing its validation procedures, the Company did not adjust or override any fair value measurements provided by pricing services as of December 31, 2014 or December 31, 2013. |
| • | Level 3—Unobservable inputs developed using estimates and assumptions developed by the Company, which reflect those that a market participant would use. The Company currently has no assets or liabilities measured at fair value on a recurring basis that utilize Level 3 inputs. |
The following tables summarize the cash equivalents and marketable securities measured at fair value on a recurring basis in the accompanying consolidated balance sheets as of December 31, 2014 and 2013.
| Fair Value Measurements of Cash Equivalents as of December 31, 2014 |
||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| (in thousands) | ||||||||||||||||
| Cash equivalents |
$ | 28,777 | $ | 7,834 | $ | — | $ | 36,611 | ||||||||
| Fair Value Measurements of Cash Equivalents and Marketable Securities as of December 31, 2013 |
||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| (in thousands) | ||||||||||||||||
| Cash equivalents |
$ | 29,865 | $ | 15,958 | $ | — | $ | 45,823 | ||||||||
| Marketable securities |
— | 67,680 | — | 67,680 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 29,865 | $ | 83,638 | $ | — | $ | 113,503 | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
96
Table of Contents
The Company recorded a liability totaling $7.3 million associated with the exit of a portion of its leased facilities. The Company measured the fair value of the liability based on the present value of the remaining termination payments. The net cash outflows were discounted using a credit-risk adjusted rate. The Company has classified this lease liability as a Level 3 fair value measurement.
The fair value of the Company’s loans payable at December 31, 2014 and 2013, computed pursuant to a discounted cash flow technique using the effective interest rate under the loan, was $21.3 million and $20.1 million, respectively. These fair values are considered a level 3 fair value measurement. The effective interest rate considers the fair value of the warrant issued in connection with the loan, loan issuance costs and a deferred charge, which approximates a market interest rate.
Tenant Improvement Allowance Receivable
Under the terms of its lease agreement, the Company was entitled to be reimbursed by the Company’s landlord for certain expenditures associated with improvements made to its leased facility at 650 East Kendall Street in Cambridge, Massachusetts. Pursuant to the lease agreement, the total amount of the improvement allowance allocated to the premises was $14.9 million. Through December 2014, the Company incurred $14.9 million of such expenditures which, were partially offset against amounts due by the Company for the lease termination fee discussed in Note 8. The net proceeds were reimbursed to the Company by its landlord on September 29, 2014.
Property and Equipment
Property and equipment are stated at cost and are depreciated using the straight-line method over the estimated useful lives of the respective assets. Maintenance and repair costs are charged to expense as incurred.
Long-lived Assets
The Company reviews long-lived assets, including property and equipment, for impairment whenever changes in business circumstances indicate that the carrying amount of the asset may not be fully recoverable. The Company recognized $7.6 million of impairment losses for the year ended December 31, 2014 related to leasehold improvements. During the year ended December 31, 2013, the Company recognized $0.1 million of impairment losses.
Stock-Based Compensation
Under the Company’s stock-based compensation programs, the Company periodically grants stock options and restricted stock to employees, directors and nonemployee consultants. The Company also issues shares under an employee stock purchase plan. The fair value of all awards is recognized in the Company’s statements of operations over the requisite service period for each award. Awards that vest as the recipient provides service are expensed on a straight-line basis over the requisite service period. Other awards, such as performance-based awards that vest upon the achievement of specified goals, are expensed using the accelerated attribution method if achievement of the specified goals is considered probable. The Company has also granted awards that vest upon the achievement of market conditions. Per ASC 718 Share-Based Payments, market conditions must be considered in determining the estimated grant-date fair value of share-based payments and the market conditions must be considered in determining the requisite service period over which compensation cost is recognized. The Company estimates the fair value of the awards with market conditions using a Monte Carlo simulation, which utilizes several assumptions including the risk-free interest rate, the volatility of the Company’s stock and the exercise behavior of award recipients. The grant-date fair value of the awards is then recognized over the requisite service period, which represents the derived service period for the awards as determined by the Monte Carlo simulation.
97
Table of Contents
The fair value of equity-classified awards to employees and directors are measured at fair value on the date the awards are granted. Awards to nonemployee consultants are recorded at their fair values and are re-measured as of each balance sheet date until the recipient’s services are complete. During the years ended December 31, 2014 and 2013, the Company recorded the following stock-based compensation expense:
| Years Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| (in thousands) | ||||||||||||
| Research and development |
$ | 899 | $ | 1,991 | $ | 3,566 | ||||||
| General and administrative |
1,902 | 1,800 | 4,380 | |||||||||
| Restructuring |
— | — | 61 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total stock-based compensation expense |
$ | 2,801 | $ | 3,791 | $ | 8,007 | ||||||
|
|
|
|
|
|
|
|||||||
Stock-based compensation expense is allocated to research and development and general and administrative expense based upon the department of the employee to whom each award was granted. No related tax benefits of the stock-based compensation expense have been recognized.
Income Taxes
The Company provides for income taxes using the asset-liability method. Under this method, deferred tax assets and liabilities are recognized based on differences between financial reporting and tax bases of assets and liabilities, and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. Uncertain tax positions are recognized if the position is more-likely-than-not to be sustained upon examination by a tax authority. Unrecognized tax benefits represent tax positions for which reserves have been established. The Company maintains a full valuation allowance on all deferred tax assets.
Segment and Geographic Information
Operating segments are defined as components of an enterprise engaging in business activities for which discrete financial information is available and regularly reviewed by the chief operating decision maker in deciding how to allocate resources and in assessing performance. The Company views its operations and manages its business in one operating segment principally in the United States. As of December 31, 2014, the Company has $1.0 million of net assets located in the United Kingdom.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States (“GAAP”) requires the Company’s management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
Recent Accounting Pronouncements
In May 2014, the FASB issued ASU No. 2014-09, Revenue from Contracts with Customers, a comprehensive new revenue recognition standard that will supersede nearly all existing revenue recognition guidance under US GAAP. The standard is effective for public entities for annual and interim periods beginning after December 15, 2016. Early adoption is not permitted under US GAAP. The Company is currently evaluating what effect, if any, this standard will have on its revenue recognition policies and its financial statements, including how the standard will be adopted.
In August 2014, the FASB issued ASU No. 2014-15, Presentation of Financial Statements—Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern. This
98
Table of Contents
ASU is intended to define management’s responsibility to evaluate whether there is substantial doubt about an organization’s ability to continue as a going concern within one year of the date of issuance of the entity’s financial statements and to provide related footnote disclosures. This guidance is effective for fiscal years beginning after December 15, 2016, with early application permitted. The Company is currently evaluating what effect, if any, the adoption of this guidance will have on the disclosures included in its consolidated financial statements.
| 3. | Loss Per Common Share |
Basic loss per share is computed by dividing net loss by the weighted-average number of common shares outstanding during the period. Diluted loss per share is computed by dividing net loss by the weighted-average number of common shares and dilutive common share equivalents then outstanding which exclude unvested restricted stock. Potential common share equivalents consist of restricted stock awards and the incremental common shares issuable upon the exercise of stock options and warrants. Since the Company had a net loss for all periods presented, the effect of all potentially dilutive securities is anti-dilutive. Accordingly, basic and diluted net loss per common share is the same.
The following table sets forth for the periods presented the potential common shares (prior to consideration of the treasury stock method) excluded from the calculation of net loss per common share because their inclusion would have been anti-dilutive:
| Outstanding
at Years Ended December 31, |
||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| (in thousands) | ||||||||||||
| Options outstanding |
5,817 | 4,297 | 4,638 | |||||||||
| Warrants outstanding |
609 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| 6,426 | 4,297 | 4,638 | ||||||||||
|
|
|
|
|
|
|
|||||||
| 4. | Property and Equipment |
Property and equipment consists of the following:
| Estimated Useful Life |
December 31, 2014 |
December 31, 2013 |
||||||||
| (in thousands) | ||||||||||
| Laboratory equipment |
5 years | $ | 7,671 | $ | 8,660 | |||||
| Computer equipment and software |
3 years | 3,913 | 4,398 | |||||||
| Office furniture |
5 years | 893 | 930 | |||||||
| Leasehold improvements |
Shorter of asset’s useful life or remaining term of lease |
14,433 | 11,959 | |||||||
| Construction in process |
— | — | 3,193 | |||||||
|
|
|
|
|
|||||||
| 26,910 | 29,140 | |||||||||
| Less accumulated depreciation and amortization |
(15,615 | ) | (15,000 | ) | ||||||
|
|
|
|
|
|||||||
| Property and equipment, net |
$ | 11,295 | $ | 14,140 | ||||||
|
|
|
|
|
|||||||
Depreciation expense for the years ended December 31, 2014, 2013 and 2012 was $6.2 million, $3.8 million and $2.5 million, respectively. In September 2014, the Company entered into a Lease Termination Agreement, pursuant to which the Company agreed to surrender the remaining 49,185 square feet of leased space no later than September 24, 2015. As a result, the Company revised the estimated useful life of its leasehold improvements related to this space, resulting in an increase in depreciation expense of approximately $2.4 million during the year ended December 31, 2014.
99
Table of Contents
| 5. | Accrued Expenses |
Accrued expenses consisted of the following:
| December 31, 2014 |
December 31, 2013 |
|||||||
| (in thousands) | ||||||||
| Manufacturing and distribution |
$ | 3,216 | $ | 1,362 | ||||
| Clinical expenses |
2,312 | 5,319 | ||||||
| Salaries and benefits |
1,744 | 2,027 | ||||||
| Professional fees |
685 | 811 | ||||||
| Restructuring |
— | 587 | ||||||
| Property and equipment |
— | 1,905 | ||||||
| Other |
1,344 | 1,252 | ||||||
|
|
|
|
|
|||||
| $ | 9,301 | $ | 13,263 | |||||
|
|
|
|
|
|||||
| 6. | Loans Payable |
On May 28, 2010, the Company entered into a loan and security agreement (the “Loan Agreement”) with Hercules Technology II, L.P. and Hercules Technology III, L.P., affiliates of Hercules Technology Growth (collectively, “Hercules”), pursuant to which the Company received a loan in the aggregate principal amount of $25.0 million. The Company was required to repay the aggregate principal balance under the Loan Agreement in 30 equal monthly installments of principal starting on January 1, 2012. On March 31, 2012, the Company entered into an amendment to the Loan Agreement, pursuant to which the Company increased the principal amount under the Loan Agreement to $26.5 million. Under the amendment to the Loan Agreement, the date on which the Company was required to begin repaying the aggregate principal balance was extended to April 1, 2013, at which point the Company began repaying such balance in 30 equal monthly installments.
On September 24, 2014, the Company further amended the Loan Agreement with Hercules (the “Amended Loan Agreement”). Pursuant to the Amended Loan Agreement, the Company received a new loan in the aggregate principal amount of $10.0 million and amended the terms of the Original Loan Agreement with an outstanding principal balance of $11.6 million.
Pursuant to the Amended Loan Agreement, the Company is not required to make principal payments on the new $10.0 million loan until November 1, 2015; provided, that such date may be extended if the Company achieves certain performance milestones, after which time, the Company is required to make 30 principal and interest payments with the entire loan due and payable on January 1, 2018. With respect to the original loan, the Company is only required to pay interest until January 1, 2015, at which time the Company is required to repay the loan in 12 monthly installments of principal and interest. The Amended Loan Agreement has an end-of-term payment of approximately $0.5 million due on January 1, 2018 or on such earlier date as the new loan is prepaid. This end-of-term payment has been recorded as a loan discount and is being amortized to interest expense over the term of the loan agreement using the effective interest rate method. The Company accounted for the Amended Loan Agreement as a loan modification in accordance with ASC 470-50, Debt—Modifications and Extinguishments.
Per annum interest is payable on principal balance of both loans at the greater of 11.9% and an amount equal to 11.9% plus the prime rate of interest minus 4.75%, provided however, that the per annum interest shall not exceed 15.0%. With respect to the new loan of $10.0 million, the unpaid principal balance and all accrued but unpaid interest will be due and payable on January 1, 2018, and with respect to the existing loan with a principal balance of $11.6 million, the unpaid principal balance and all accrued but unpaid interest will be due and payable on January 1, 2016.
In addition to the obligations and covenants currently existing under the Loan Agreement, the Amended Loan Agreement contains a financial covenant, whereby the Company has agreed to maintain, with respect to the
100
Table of Contents
new loan of $10.0 million, a liquidity ratio equal to or greater than 1.25 to 1.00 of the then outstanding loan balance or the equivalent of $12.5 million in unrestricted and unencumbered cash and cash equivalents assuming a principal balance of $10.0 million. The financial covenant shall not apply after such time that the Company receives favorable data both with respect to its phase 2 clinical trial of ficlatuzumab and a phase 1 clinical trial of AV-380.
The Loan Agreement required a deferred financing charge of $1.3 million which was paid in May 2012 related to the amendment of the Loan Agreement. The Loan Agreement also included an additional deferred financing charge of $1.2 million which was paid in June 2014. These amounts were recorded as a loan discount and are being amortized to interest expense over the term of the loan borrowed under the Loan Agreement using the effective interest rate method. The Company recorded a liability for the full amount of the charge since the payment of such amount was not contingent on any future event. The Company paid approximately $0.2 million in loan issuance costs directly to Hercules under the Loan Agreement, which were offset against the loan proceeds and are accounted for as a loan discount.
As part of the Loan Agreement, on June 2, 2010, the Company issued warrants to the lenders to purchase up to 156,641 shares of the Company’s common stock at an exercise price equal to $7.98 per share to Hercules. The Company recorded the relative fair value of the warrants of approximately $0.8 million as stockholders’ equity and as a discount to the related loan outstanding and is amortizing the value of the discount to interest expense over the term of the original loan using the effective interest method. On July 21, 2011, Hercules exercised these warrants and they are no longer outstanding.
As part of the Amended Loan Agreement, on September 24, 2014, the Company issued warrants to the lenders to purchase up to 608,696 shares of the Company’s common stock at an exercise price equal to $1.15 per share to Hercules. The Company recorded the relative fair value of the warrants of approximately $0.4 million as stockholders’ equity and as a discount to the related loan outstanding and is amortizing the value of the discount to interest expense over the term of the loan using the effective interest method. The relative fair value of the warrants was calculated using the Black-Scholes option-pricing model with the following assumptions: volatility of 71.81%, an expected term equal to the contractual life of the warrants (five years), a risk-free interest rate of 1.82% and no dividend yield. The resulting effective interest rate for the loans outstanding under the Amended Loan Agreement is approximately 16.14%.
As part of the Loan Agreement, Hercules also received an option, subject to the Company’s written consent, not to be unreasonably withheld, to purchase, either with cash or through conversion of outstanding principal under the loan, up to $2.0 million of equity of the Company sold in any sale by the Company to third parties of equity securities resulting in at least $10.0 million in net cash proceeds to the Company, subject to certain exceptions. The Company has evaluated the embedded conversion option, and has concluded that it does not need to be bifurcated and separately accounted for. No amount will be recognized for the conversion feature until such time as the conversion feature is exercised and it can be determined whether a beneficial conversion feature exists. As of December 31, 2014, the outstanding aggregate principal balance under both loans was $21.6 million.
The loans are secured by a lien on all of our personal property (other than intellectual property), whether owned as of, or acquired after, the date of the amended loan agreement. The Amended Loan Agreement defines events of default, including the occurrence of an event that results in a material adverse effect upon the Company’s business operations, properties, assets or condition (financial or otherwise), its ability to perform its obligations under and in accordance with the terms of the Amended Loan Agreement, or upon the ability of the lenders to enforce any of their rights or remedies with respect to such obligations, or upon the collateral under the Loan Agreement, the related liens or the priority thereof. As of December 31, 2014, Hercules has not asserted any events of default and the Company does not believe that there has been a material adverse change as defined in the loan agreement. The Company has classified the principal amount of the loans as current and non-current on its consolidated balance sheet based upon the expected timing of the remaining payments.
101
Table of Contents
Future minimum payments under the loans payable outstanding as of December 31, 2014 are as follows (amounts in thousands):
| Years Ending December 31: |
||||
| 2015 |
$ | 14,166 | ||
| 2016 |
4,644 | |||
| 2017 |
4,644 | |||
| 2018 |
2,096 | |||
|
|
|
|||
| 25,550 | ||||
| Less amount representing interest |
(3,398 | ) | ||
| Less discount |
(960 | ) | ||
| Less deferred charges |
(540 | ) | ||
| Less current portion |
(11,722 | ) | ||
|
|
|
|||
| Loans payable, net of current portion and discount |
$ | 8,930 | ||
|
|
|
|||
| 7. | Collaboration and License Agreements |
(a) Out-License Agreements
Ophthotech Corporation
In November 2014 the Company entered into a Research and Exclusive Option Agreement, or Option Agreement, with Ophthotech Corporation pursuant to which the Company provided Ophthotech an exclusive option to enter into a definitive license agreement whereby the Company would grant Ophthotech the right to develop and commercialize the Company’s VEGF factor tyrosine kinase inhibitor, tivozanib, outside of Asia for the potential diagnosis, prevention and treatment of non-oncologic diseases or conditions of the eye in humans.
Pursuant to this Option Agreement, the Company granted to Ophthotech an exclusive, royalty free license or sublicense, as applicable, under intellectual property rights controlled by the Company solely to perform the research and development activities related to the use of tivozanib for the specific purposes outlined in the agreement during the option period (as defined below). These activities include formulation work for ocular administration, preclinical research and the conduct of a phase 1/2a, proof of concept clinical trial of a product containing tivozanib in patients with wet age-related macular degeneration, (the “POC Study”).
Ophthotech paid the Company $500,000 in consideration for the grant of the option. Such amount is non-refundable and not creditable against any other amounts due under the agreement. The Company is obligated to make available to Ophthotech, at no cost to Ophthotech, certain quantities of tivozanib hydrochloride solely for conducting its option period research including manufacturing additional quantities of tivozanib in the event stability data indicates that the current supply will expire prior to the end of February 2017.
During the option period, if Ophthotech elects to continue the development of tivozanib for non-oncologic diseases of the eye, the Company is entitled to receive a one-time milestone payment of $2.0 million upon acceptance of the first Investigational New Drug application for the purpose of conducting a human clinical study of tivozanib in ocular diseases (the “IND Submission Milestone Payment”). The Company is also entitled to receive a one-time milestone payment of $6.0 million (the “Clinical Efficacy Milestone Payment”), on the earlier of (a) December 31, 2016 and (b) the later to occur of: (i) the achievement of a clinical milestone in the POC Study (the “Clinical Efficacy Milestone”) and (ii) the earlier of (A) the date twelve (12) months after our and Ophthotech’s agreement as to the form and substance of the KHK Amendment (as defined below) or (B) the date ninety (90) days after the entry into the KHK Amendment, subject to the Company’s right to terminate the Option Agreement on 90 days’ written notice (the date on which such payment is due, referred to as the “Clinical Efficacy Milestone Payment Trigger Date”).
102
Table of Contents
Ophthotech may exercise the option at any time until the latest to occur of: (i) twelve (12) months after the achievement of the Clinical Efficacy Milestone, (ii) ninety (90) days after the Clinical Efficacy Milestone Payment Trigger Date, and (iii) thirty (30) days after the Company and Ophthotech agree as to the definitive form of license agreement, which the Company refers to as the option period.
During the Option Period, the Company will not grant a license to any third party that would preclude the Company from being able to grant to Ophthotech the rights and licenses that are contemplated by the definitive license agreement, and the Company will not engage in any research, development or commercialization of tivozanib in the field covered by the contemplated definitive license agreement, except as specified in the Option Agreement.
The terms of the Option Agreement are subject to the Company’s obligations to Kyowa Hakko Kirin (“KHK”), under a license agreement entered into by the Company with KHK in 2006, pursuant to which the Company acquired exclusive rights to develop and commercialize tivozanib for all human diseases outside of Asia (the “KHK License Agreement”). A percentage of all payments received by the Company under the Option Agreement and any definitive license agreement must be paid to KHK. The Company is required to maintain the KHK Agreement in effect, and not enter into any amendment or termination thereof that would adversely affect the Company’s rights, during the option period.
During the option period, the Company and Ophthotech are obligated to negotiate in good faith the form and substance of a definitive license agreement, as well as the form and substance of an amendment to the Company’s license agreement with KHK (the “KHK Amendment”) to modify certain rights and obligations of the parties and sublicensees thereunder, particularly with respect to rights to improvements that are not specifically related to tivozanib, and regulatory affairs matters.
Upon exercise of the option, Ophthotech is required to pay the Company a one-time option exercise fee of $2.0 million in addition to the IND Submission Milestone Payment if such payment has not then been previously paid. If upon exercise of the option, the Clinical Efficacy Milestone Payment Trigger Date has not yet occurred, the Company shall be entitled to the Clinical Efficacy Milestone Payment at such time that the Clinical Efficacy Milestone Payment Date does occur if the license agreement remains in effect as of such date. The license agreement, if entered into upon Ophthotech’s exercise of the Option, will provide for the Company to be entitled to receive (i) $10.0 million upon meeting certain efficacy and safety endpoints in phase 2 clinical trials that would enable the commencement of a phase 3 clinical trial, (ii) $20.0 million upon marketing approval in the United States, (iii) $20.0 million upon marketing approval in the UK, Germany, Spain, Italy and France and (iv) up to $45.0 million in sales-based milestone payments. Ophthotech would also be required to pay tiered, double digit royalties, up to the mid-teens, on net sales of tivozanib or products containing tivozanib.
Either party may terminate the Option Agreement in the event of an uncured material breach of the Option Agreement by the other party which remains uncured for a period of ninety (90) days (or thirty (30) days for a breach relating to non-payment), or upon bankruptcy or like proceedings relating to the other party. Ophthotech may terminate the Option Agreement at any time upon ninety (90) days’ prior written notice to us. In addition, the Company may terminate the Option Agreement upon thirty (30) days’ prior written notice to Ophthotech if Ophthotech challenges certain patents controlled by the Company related to tivozanib. Unless terminated as provided above, the Option Agreement will expire upon the expiration of the option or the entry into the definitive license agreement.
Activities under the agreement with Ophthotech were evaluated under ASC 605-25 Revenue Recognition—Multiple Element Arrangements, or ASC 605-25, to determine whether such activities represented a multiple element revenue arrangement. The agreement with Ophthotech includes the following non-contingent deliverables: the Company’s obligation to grant an exclusive option to Ophthotech to enter into a License Agreement to develop and commercialize products incorporating tivozanib for treatment of AMD and other diseases of the eye outside of Asia during the Option Period; the Company’s obligation to enter into an
103
Table of Contents
amendment with KHK to modify the terms of the existing KHK agreement to negotiate a mutually acceptable form of license agreement; and the Company’s obligation to transfer research-grade tivozanib API for Ophthotech to conduct the Option Period research.
The Company determined that the delivered Option Grant Deliverable, or the Company’s obligation to grant an exclusive option to Ophthotech to enter into a License Agreement to develop and commercialize products incorporating tivozanib for treatment of AMD and other diseases of the eye outside of Asia during the Option Period, did not have standalone value from the remaining deliverables since Ophthotech could not obtain the intended benefit of the option without the remaining deliverables. Similarly, the remaining deliverables have no standalone value without the Option Grant Deliverable. The Company is accounting for the deliverables as one unit of accounting.
Under the agreement, the Company received a cash payment of $0.5 million during the year ended December 31, 2014. The Company deferred the payment and is recording the deferred revenue over the Company’s period of performance, which is estimated to be through December 2016. The Company recorded approximately $38,000 of revenue during the year ended December 31, 2014.
Biodesix
In April 2014, the Company entered into a worldwide agreement with Biodesix to develop and commercialize its hepatocyte growth factor (“HGF”) inhibitory antibody ficlatuzumab, with BDX004, a proprietary companion diagnostic test developed by Biodesix and derived from VeriStrat®, a serum protein test that is commercially available to help physicians guide treatment decisions for patients with advanced non-small cell lung cancer (“NSCLC”). Under the agreement, the Company granted Biodesix perpetual, non-exclusive rights to certain intellectual property, including all clinical and biomarker data related to ficlatuzumab, to develop and commercialize BDX004. Biodesix granted the Company perpetual, non-exclusive rights to certain intellectual property, including diagnostic data related to BDX004, with respect to the development and commercialization of ficlatuzumab; each license includes the right to sublicense, subject to certain exceptions. Pursuant to a joint development plan to be agreed upon by a joint steering committee, the Company retains primary responsibility for clinical development of ficlatuzumab in a proof of concept (“POC”) clinical study of ficlatuzumab for NSCLC, in which VeriStrat will be used to select clinical trial subjects, referred to as the NSCLC POC Trial. The NSCLC POC Trial will be fully funded by Biodesix up to a maximum of $15.0 million, referred to as the “Cap”. After the Cap is reached, the Company and Biodesix will share equally in the costs of the NSCLC trial, and the Company and Biodesix will each be responsible for 50% of development and regulatory costs associated with all future clinical trials agreed-upon by Biodesix and the Company, including all milestone payments and royalties payable to third parties, if any.
Pending marketing approval of ficlatuzumab and subject to a commercialization agreement to be entered into after receipt of results from the NSCLC POC Trial, each party would share equally in commercialization profits and losses, subject to the Company’s right to be the lead commercialization party.
Biodesix is solely responsible for the BDX004 development costs, as well as BDX004 sales and marketing costs. Subject to and following the approval of the BDX004 test as a companion diagnostic for ficlatuzumab, Biodesix has agreed to make the BDX004 test available and use commercially reasonable efforts to seek reimbursement in all geographies where ficlatuzumab is approved. The Company has agreed to reimburse Biodesix a pre-specified amount, under certain circumstances for BDX004 tests performed.
Prior to the first commercial sale of ficlatuzumab and after the earlier of (i) the Cap being reached or (ii) the completion of the NSCLC POC Trial, each party has the right to elect to discontinue participating in further development or commercialization efforts with respect to ficlatuzumab, which is referred to as an “Opt-Out”. If either AVEO or Biodesix elects to Opt-Out, with such party referred to as the “Opting -Out Party”, then the
104
Table of Contents
Opting-Out Party shall not be responsible for any future costs associated with developing and commercializing ficlatuzumab other than any ongoing clinical trials. After election of an Opt-Out, the non-opting out party shall have sole decision-making authority with respect to further development and commercialization of ficlatuzumab. Additionally, the Opting-Out Party shall be entitled to receive, if ficlatuzumab is successfully developed and commercialized, a royalty equal to 10% of net sales of ficlatuzumab throughout the world, if any, subject to offsets under certain circumstances.
If Biodesix elects to Opt-Out, it will continue to be responsible for its development and commercialization obligations with respect to BDX004. If AVEO elects to Opt-Out, it will continue to make the existing supply of ficlatuzumab available to Biodesix for the purposes of enabling Biodesix to complete the development of ficlatuzumab, and Biodesix will have the right to commercialize ficlatuzumab.
Prior to any Opt-Out, the parties shall share equally in any payments received from a third party licensee; provided, however, after any Opt-Out, the Opting-Out Party shall be entitled to receive only a reduced portion of such third party payments. The agreement will remain in effect until the expiration of all payment obligations between the parties related to development and commercialization of ficlatuzumab, unless earlier terminated.
Activities under the agreement with Biodesix were evaluated under ASC 605-25 Revenue Recognition—Multiple Element Arrangements, or ASC 605-25, to determine whether such activities represented a multiple element revenue arrangement. The agreement with Biodesix includes the following non-contingent deliverables: perpetual, non-exclusive rights to certain intellectual property including clinical and biomarker data related to ficlatuzumab for use in developing and commercializing BDX004; the Company’s obligation to deliver technology improvements and data developed during the NSCLC POC Trial to Biodesix; the Company’s obligation to participate in the joint steering committee during the NSCLC POC Trial; the Company’s obligation to perform certain development activities associated with the NSCLC POC Trial; and the Company’s obligation to supply clinical material for use in conducting the NSCLC POC Trial; and the Company’s obligation to deliver clinical specimens and data during the NSCLC POC Trial. The Company concluded that any deliverables that would be delivered after the NSCLC POC Trial is complete are contingent deliverables because these services are contingent upon the results of the NSCLC POC Trial. As these deliverables are contingent, and are not at an incremental discount, they are not evaluated as deliverables at the inception of the arrangement. These contingent deliverables will be evaluated and accounted for separately as each related contingency is resolved. As of December 31, 2014, no contingent deliverables had been provided by the Company.
The Company determined that the delivered item, or the perpetual, non-exclusive rights to certain intellectual property for use in developing and commercializing BDX004 did not have standalone value from the remaining deliverables since Biodesix could not obtain the intended benefit of the license without the remaining deliverables. Since the remaining deliverables will be performed over the same period of performance there is no difference in accounting for the deliverables as one unit or multiple units of accounting, and therefore, the Company is accounting for the deliverables as one unit of accounting.
The Company records the consideration earned while conducting the NSCLC POC Trial, which consists of reimbursements from Biodesix for expenses related to the trial under the Cap, as a reduction to research and development expense using the proportional performance method over the respective period of performance. As a result of the cost sharing provisions in the agreement, the Company reduced research and development expenses by approximately $2.7 million during the year ended December 31, 2014. The amount due to the Company from Biodesix pursuant to the cost-sharing provision was $1.8 million at December 31, 2014.
Biogen Idec International GmbH
In March 2009, the Company entered into an exclusive option and license agreement with Biogen Idec International GmbH, a subsidiary of Biogen Idec Inc., (collectively “Biogen Idec”) regarding the development and commercialization of the Company’s discovery-stage ErbB3-targeted antibodies for the potential treatment
105
Table of Contents
and diagnosis of cancer and other diseases outside of North America. Under the agreement, the Company is responsible for developing ErbB3 antibodies through completion of the first phase 2 clinical trial designed in a manner that, if successful, will generate data sufficient to support advancement to a phase 3 clinical trial.
In March 2014, the Company and Biogen Idec amended the exclusive option and license agreement (the “Amendment”). Pursuant to the Amendment, Biogen agreed to the termination of its rights and obligations under the agreement, including Biogen’s option to (i) obtain a co-exclusive (with AVEO) worldwide license to develop and manufacture ErbB3 targeted antibodies and (ii) obtain exclusive commercialization rights to ErbB3 products in countries in the world other than North America. As a result, AVEO has worldwide rights to AV-203. Pursuant to the Amendment, AVEO is obligated to use reasonable efforts to seek a collaboration partner for the purpose of funding further development and commercialization of ErbB3 targeted antibodies. AVEO is also obligated to pay Biogen a percentage of milestone payments received by AVEO from future partnerships after March 28, 2016 and single digit royalty payments on net sales related to the sale of ErbB3 products, if any, up to cumulative maximum amount of $50 million.
The deliverables under the original arrangement included an option for a co-exclusive, worldwide license to develop and manufacture ErbB3 antibody products and an option for an exclusive license to commercialize ErbB3 antibody products in all countries in the world other than North America. The Company determined that these deliverables did not have standalone value due to the fact that the program was still in preclinical development and required the Company’s experience to advance development of the product candidates. As such, the Company determined that the original agreement should be accounted for as one unit of accounting.
Under the terms of the original agreement, Biogen Idec made up-front and milestone-based cash payments totaling $20.0 million. Of the $20.0 million received, $10.0 million was associated with milestones that were considered substantive and these amounts were included in revenue when they were earned. The remaining $10.0 million was amortized as additional license revenue over the Company’s period of substantial involvement.
The Company concluded that the Amendment materially modified the terms of the agreement and, as a result, required the application of ASC 605-25. Based upon the terms of the Amendment, the remaining deliverables included the Company’s obligation to seek a collaboration partner to fund further development of the program and the Company’s obligation to continue development and commercialization of the licensed products if a collaboration partner is secured (“Development Deliverable”). The Company concluded that its obligation to use best efforts to seek a collaboration partner does not have standalone value from the Development Deliverable upon delivery and thus the deliverables should be treated as a single unit of accounting.
Upon modifying the arrangement, the Company had $14.7 million of deferred revenue remaining to be amortized. The Company is not entitled to receive any further consideration from Biogen Idec under the amended arrangement. The Company allocated a portion of the remaining deferred revenue to the undelivered unit of accounting based upon the Company’s best estimate of the selling price, as the Company determined that neither VSOE or TPE were available. The Company determined the best estimate of selling price to be approximately $0.6 million and recognized the remaining $14.1 million as collaboration revenue in March 2014. The deferred revenue associated with the undelivered unit of accounting is being recognized on a straight-line basis over the expected period of performance, or through December 2015, based upon the Company’s historical experience with marketing its product candidates to potential partners.
The best estimate of selling price was based upon a cost approach pursuant to which the Company estimated the costs expected to be incurred in executing a partnership agreement and then applied a reasonable markup. The Company estimated future cash outflows for several possible outcomes, including the execution of a partnership at different times within a reasonable range and partnerships of differing complexity. The Company estimated its cash outflows for each scenario based upon the expected costs associated with the relevant employees and the expected level of effort to be expended to seek and execute a partnership. The Company’s analysis also considered the legal charges that it anticipates it will incur. Changes to the Company’s assumptions within the reasonable range of possible values would not have a material impact on the amounts recorded in current or future periods.
106
Table of Contents
Under the agreement, the Company recorded revenue of $14.5 million, $0.9 million and $0.9 million during the years ended December 31, 2014, 2013 and 2012, respectively.
Astellas Pharma Inc.
In February 2011, the Company, together with its wholly-owned subsidiary AVEO Pharma Limited, entered into a Collaboration and License Agreement with Astellas (the “Astellas Agreement”), pursuant to which the Company and Astellas intended to develop and commercialize tivozanib for the treatment of a broad range of cancers. Under the terms of the Astellas Agreement, the Company and Astellas shared responsibility for continued development and commercialization of tivozanib in North America and in Europe under a joint development plan and a joint commercialization plan, respectively. Throughout the rest of the world (the “Royalty Territory”), excluding Asia, where KHK has retained all development and commercialization rights, Astellas had an exclusive, royalty-bearing license to develop and commercialize tivozanib. The terms of the Astellas Agreement were subject to the Company’s obligations to KHK under a license agreement entered into with KHK in 2006 pursuant to which the Company acquired exclusive rights to develop and commercialize tivozanib worldwide outside of Asia.
In June 2013, the Company received a complete response letter from the U.S. Department of Food and Drug Administration, or FDA, informing the Company that the FDA will not approve in its present form the Company’s New Drug Application, or NDA, for tivozanib for the treatment of patients with advanced renal cell carcinoma, or RCC. In January 2014, AVEO and Astellas jointly decided to discontinue a phase 2 breast cancer clinical trial due to insufficient enrollment. Further, Astellas elected in February 2014 to terminate the Astellas Agreement as a result of the limited scope of development for tivozanib moving forward. This termination became effective on August 11, 2014, at which time the tivozanib rights returned to the Company. In accordance with the Astellas Agreement, committed development costs, including the costs of completing certain tivozanib clinical development activities, will be shared equally. There are no refund provisions in the Astellas Agreement.
Under the Astellas Agreement, the Company received an initial cash payment of $125.0 million, comprised of a $75.0 million license fee and $50.0 million in research and development funding. The Company retained net proceeds of approximately $97.6 million of the initial cash payment from Astellas, after payments to KHK and strategic, legal and financial advisors. In December 2012, the Company received a $15.0 million milestone payment from Astellas in connection with the acceptance by the FDA of the NDA filing for tivozanib. The milestone was considered substantive and revenue was recognized upon achievement of the milestone.
The Company accounted for the joint development and commercialization activities in North America and Europe as a joint risk-sharing collaboration in accordance with ASC 808, Collaborative Arrangements. In addition, these activities were not deemed to be separate deliverables under the Astellas Agreement.
Payments from Astellas with respect to Astellas’ share of tivozanib development and commercialization costs incurred by the Company pursuant to the joint development plan were recorded as a reduction to research and development expense and general and administrative expense in the accompanying consolidated financial statements due to the joint risk-sharing nature of the activities in North America and Europe. As a result of the cost-sharing provisions in the Astellas Agreement, the Company reduced research and development expense by $3.5 million, $15.8 million and $34.1 million during the years ended December 31, 2014, 2013, and 2012, respectively. The Company also reduced general and administrative expense by $0.1 million, $2.8 million, and $3.3 million during the years ended December 31, 2014, 2013 and 2012, respectively, as a result of the cost-sharing provisions in the Astellas Agreement. The net amount due to the Company from Astellas pursuant to the cost-sharing provisions was $0.6 million at December 31, 2014.
Activities under the Astellas Agreement outside of the joint development and commercialization activities in North America and Europe, including the co-exclusive license to develop and commercialize tivozanib in North America and Europe that was delivered prior to the initiation of the collaborative activities in North
107
Table of Contents
America and Europe, were evaluated under ASC 605-25 to determine if they represented a multiple element revenue arrangement. The Astellas Agreement included the following deliverables: (1) a co-exclusive license to develop and commercialize tivozanib in North America and Europe (the “License Deliverable”); (2) a combined deliverable comprised of an exclusive royalty-bearing license to develop and commercialize tivozanib in the Royalty Territory and the Company’s obligation to provide access to clinical and regulatory information resulting from the activities in North America and Europe to Astellas for its development and commercialization of tivozanib in the Royalty Territory (the “Royalty Territory Deliverable”); and (3) the Company’s obligation to supply clinical material to Astellas for development of tivozanib in the Royalty Territory (the “Clinical Material Deliverable”). All of these deliverables were deemed to have stand-alone value and to meet the criteria to be accounted for as separate units of accounting under ASC 605-25. Factors considered in this determination included, among other things, the subject of the licenses and the research and development and commercial capabilities of Astellas.
The Company allocated the up-front consideration of $125.0 million to the deliverables based on management’s best estimate of selling price of each deliverable using the relative selling price method as the Company did not have VSOE or TPE of selling price for such deliverables. The Company’s best estimate of selling price considered discounted cash flow models, the key assumptions of which included the market opportunity for commercialization of tivozanib in North America and Europe and the Royalty Territory, the probability of successfully developing and commercializing tivozanib, the remaining development costs for tivozanib, and the estimated time to commercialization of tivozanib. The Company’s analysis included the following market conditions and entity-specific factors: (a) the specific rights provided under the license to develop and commercialize tivozanib in North America and Europe and the Royalty Territory, (b) the potential indications for tivozanib pursuant to the licenses, (c) the relevant territories for the respective licenses, (d) the stage of development of tivozanib by potential indication and estimated remaining development timelines and costs for each indication, (e) the development risk by indication, (f) the market size by indication, (g) the expected product life of tivozanib assuming commercialization and (h) the competitive environment. The Company utilized a discount rate of 15% in its analysis, representing the weighted average cost of capital derived from returns on equity for comparable companies.
The Company concluded that a change in the key assumptions used to determine best estimate of selling price for each license deliverable would not have a significant effect on the allocation of arrangement consideration.
The Company allocated up-front consideration of $120.2 million to the License Deliverable and up-front consideration of $4.8 million to the Royalty Territory Deliverable. The relative selling price of the Company’s obligation under the Clinical Material Deliverable had de minimis value.
The Company recorded the $120.2 million relative selling price of the License Deliverable as collaboration revenue during the three months ended March 31, 2011 upon delivery of the license, and deferred approximately $4.8 million of revenue representing the relative selling price of the Royalty Territory Deliverable. The Company was recording the $4.8 million of revenue attributed to the Royalty Territory Deliverable ratably over the Company’s period of performance through April 2022, the remaining patent life of tivozanib. Upon being notified that the collaboration would be terminated effective August 2014, the Company reassessed the period of performance associated with the Royalty Territory Deliverable and accelerated the recognition of the remaining deferred revenue such that the balance would be recognized through August 2014. This change in estimate resulted in the recognition of an additional $3.1 million during the year ended December 31, 2014. The Company recorded approximately $3.6 million, $0.4 million, and $0.4 million of revenue associated with the Royalty Territory Deliverable during the years ended December 31, 2014, 2013, and 2012, respectively.
Under the agreement, the Company received cash payments related to reimbursable payments and milestone payments of $4.1 million, $40.1 million, and $39.8 million during the years ended December 31, 2014, 2013 and 2012, respectively, and recorded revenue of $3.6 million, $0.4 million and $15.4 million during the years ended December 31, 2014, 2013 and 2012, respectively.
108
Table of Contents
(b) In-license Agreements
Kirin Brewery Co. Ltd. (KHK)
In December 2006, the Company entered into an exclusive license agreement, with the right to grant sublicenses, subject to certain restrictions, with Kirin Brewery Co. Ltd. (now Kyowa Hakko Kirin) (“KHK”) to research, develop, manufacture and commercialize tivozanib, pharmaceutical compositions thereof and associated biomarkers in all territories in the world except for Asia (the “KHK Agreement”). Upon entering into the KHK Agreement, the Company made a cash payment in the amount of $5.0 million.
In March 2010, the Company made a $10.0 million milestone payment to KHK in connection with the dosing of the first patient in the Company’s phase 3 clinical trial of tivozanib. The Company recorded $22.5 million of research and development expense during the year ended December 31, 2011 associated with a payment made to KHK related to the up-front license payment received under the Astellas Agreement. In December 2012, the Company made a $12.0 million milestone payment to KHK in connection with the acceptance by the FDA of the Company’s NDA filing for tivozanib, all of which was expensed as research and development expense during the year ended December 31, 2012. In connection with this payment, $6.0 million was reimbursed from Astellas and recorded as a reduction of research and development expense.
Under the KHK Agreement, the Company may be required to (i) make future milestone payments upon the achievement of specified regulatory milestones in the United States, including a possible milestone payment of $18.0 million to KHK in connection with the FDA granting marketing approval in the United States, (ii) pay tiered royalty payments on net sales it makes of tivozanib in its territory ranging from the low to mid-teens as a percentage of the Company’s net sales of tivozanib, and (iii) pay 30% of certain amounts the Company receives under the Astellas Agreement in connection with Astellas’ development and commercialization activities in Europe and the Royalty Territory related to tivozanib, including up-front license fees, milestone payments and royalties the Company may receive from Astellas (including a potential $4.5 million milestone payable to KHK in connection with the acceptance by the EMA of the filing of a Marketing Authorization Application and $9.0 million to KHK in connection with the EMA granting marketing approval in Europe), other than amounts the Company receives in respect of research and development funding or equity investments, subject to certain limitations.
The license agreement will remain in effect until the expiration of all of the Company’s royalty and sublicense revenue obligations to KHK unless either party elects to terminate the license agreement earlier. If the Company fails to meet its obligations under the agreements and is unable to cure such failure within specified time periods, KHK can terminate the agreement, resulting in a loss of the Company’s rights to tivozanib and an obligation to assign or license to KHK any intellectual property rights the Company may have in tivozanib.
St. Vincent’s Hospital
In July 2012, the Company entered into a license agreement with St Vincent’s Hospital Sydney Limited, which the Company refers to as St. Vincent’s, under which the Company obtained an exclusive, worldwide license, with the right to grant sublicenses subject to certain restrictions, under specified patent rights and related know-how, to research, develop, manufacture and commercialize products for human therapeutic, preventative and palliative applications that benefit from inhibition or decreased expression or activity of MIC-1, which the Company refers to as GDF15. The Company is exploiting this license in its AV-380 program for cachexia. The Company has a right of first negotiation to obtain an exclusive license to certain improvements that St. Vincent’s or third parties may make to licensed therapeutic products. Under the license agreement, St. Vincent’s also granted us non-exclusive rights for certain related diagnostic products and research tools.
Under the license agreement, the Company is obligated to use diligent efforts to conduct research and clinical development and commercially launch at least one licensed therapeutic product, and to maximize profits from licensed therapeutic products for the benefit of us and St. Vincent’s. Subject to certain conditions, the
109
Table of Contents
Company has also agreed to achieve specified research, development and regulatory milestones by specified dates. If the Company does not achieve a given milestone by the agreed date, the Company has the option of paying the amount the Company would have been obligated to pay had the Company timely achieved the milestone, and, if the Company does so, St. Vincent’s will not have the right to terminate the license agreement based on its failure to timely achieve such milestone.
The Company has also agreed that, for as long as there is a valid claim in the licensed patents, the Company will not, and the Company will ensure that its affiliates and its sublicensees do not, develop or commercialize any product, other than a licensed therapeutic product, for the treatment, prevention or prophylaxis of cachexia, decreased appetite or body weight, that binds to GDF15 or the GDF15 receptor and that is a GDF15 antagonist, and will not license or induce any other person to do the same.
In connection with entering into the license agreement with St. Vincent’s, the Company paid St. Vincent’s an upfront license fee of $0.7 million and a low five-figure amount to reimburse St. Vincent’s for patent-related expenses it incurred with respect to a specified licensed patent.
Under the Company’s license agreement with St. Vincent’s, the Company may be required to:
| • | make milestone payments, up to an aggregate total of $9.2 million, upon achievement of specified research, development and regulatory milestones for the first three indications for licensed therapeutic products, some of which payments may be increased by a mid to high double-digit percentage rate for milestones payments made after the Company grants any sublicense under the license agreement, depending on the sublicensed territory or territories; |
| • | pay tiered royalty payments equal to a low-single-digit percentage of any net sales the Company or its sublicensees make of licensed therapeutic products. The royalty rate escalates within the low-single-digit range during each calendar year based on increasing licensed therapeutic product sales during such calendar year. The Company’s royalty payment obligations for a licensed therapeutic product in a particular country end on the later of 10 years after the date of first commercial sale of such licensed therapeutic product in such country or expiration of the last-to-expire valid claim of the licensed patents covering such licensed therapeutic product in such country, and are subject to offsets under certain circumstances; |
| • | pay St. Vincent’s sublicensing fees of up to an aggregate amount in the low-to-mid six-digits, depending on the sublicensed territory or territories, at the time the Company grants any sublicense; and |
| • | reimburse St. Vincent’s for some or all of the reasonable costs and expenses it incurs in patent management, filing, prosecuting and maintaining the licensed patents. |
The license agreement will remain in effect until the later of 10 years after the date of first commercial sale of licensed therapeutic products in the last country in which a commercial sale is made, or expiration of the last-to-expire valid claim of the licensed patents, unless the Company elects, or St. Vincent’s elects, to terminate the license agreement earlier.
Either party has the right to terminate the agreement in connection with a material breach of the agreement by the other party that remains uncured for a specified cure period, or in connection with events relating to the other party’s insolvency or bankruptcy, or if a force majeure event continues for more than 4 months.
St. Vincent’s has the right to terminate the agreement due to any patent-related challenge by the Company, its affiliates or any sublicensee, or if the Company or its affiliates or any sublicensee cause or induce any other person to make a patent-related challenge, and such challenge continues after a specified cure period.
The Company has the right to terminate the agreement on 6 months’ notice if the Company terminates its GDF15 research and development programs as a result of the failure of a licensed therapeutic product in pre-clinical or clinical development, or if the Company forms the reasonable view that further GDF15 research
110
Table of Contents
and development is not commercially viable, and the Company is not then in breach of any of its obligations under the agreement. If the Company forms the reasonable view that further GDF15 research and development is not commercially viable and terminate the agreement before the Company starts a phase 1 clinical trial on a licensed therapeutic product, the Company will be required to pay St. Vincent’s a low-to-mid six-figure termination payment.
Any termination of the agreement, in whole or in part, will result in a loss of the Company’s rights to the relevant licensed patents and know-how. If St. Vincent’s terminates the agreement in its entirety due to the Company’s breach, insolvency or a patent-related challenge, or the Company terminates the agreement due to a development failure or lack of commercial viability, as described above, St. Vincent’s will have a non-exclusive license from us to certain intellectual property rights and know-how relating to the licensed therapeutic products, and the Company must transfer to St. Vincent’s certain then-existing regulatory approvals and related documents for the licensed therapeutic products.
Other License Agreements
The Company has entered into various cancelable license agreements for patented technology and other technology related to research projects, including technology to humanize ficlatuzumab, AV-203 and other antibody product candidates. The Company is obligated to pay annual maintenance payments of $25,000, which are recognized as research and development expense over the maintenance period. Under an additional agreement, if the parties agree to the use of the licensed technology in development of a product, the Company will be required to make a $1.0 million license payment per product. Three of these agreements also include development and sales-based milestones of up to $22.5 million, $5.5 million and $4.2 million per product, respectively, and single digit royalties as a percentage of sales.
Certain other research agreements require the Company to remit royalties in amounts ranging from 0.5% to 1.5% based on net sales of products utilizing the licensed technology. Total license expense incurred under these other license agreements amounted to $0.3 million during the year ended December 31, 2012. No expenses were incurred during the years ended December 31, 2014 and 2013. The Company has not paid any royalties to date.
| 8. | Commitments and Contingencies |
Operating Leases
The Company leases office and has leased lab space and equipment under various operating lease agreements. Rent expense under the operating leases amounted to $4.1 million, $9.4 million and $7.8 million for the years ended December 31, 2014, 2013 and 2012, respectively. For the year ended December 31, 2014, $3.1 million of rent expense is included within lease exit costs on the Company’s statement of operations.
On November 4, 2011, the Company entered into a lease agreement with the Massachusetts Institute of Technology, to lease an additional 11,500 square feet of office space located at 12 Emily Street in Cambridge, Massachusetts. The lease commenced on December 15, 2011 and expired on February 28, 2014.
On May 9, 2012, the Company entered into a lease agreement with BMR-650 E KENDALL B LLC (“BMR”), under which the Company agreed to lease 126,065 square feet of space located at 650 East Kendall Street, Cambridge, Massachusetts to be used for office, research and laboratory space. The initial term of the lease agreement was approximately twelve years and seven months (the “initial term”). The Company’s occupancy of the space occurred in two phases. The Company began the phase one occupancy on January 4, 2013 when the Company began to move some employees to occupy 26,734 square feet of office space (the
111
Table of Contents
“phase 1 space”). The Company began the second phase when the remainder of the Company’s employees moved into the remaining space (the “phase 2 space”) and during the second half of 2014. Rent payments with respect the phase 1 space commenced on January 1, 2013, and rent payments for the phase 2 space commenced on November 1, 2013 for the phase 2 space. The initial base rent expense for both the phase 1 space and the phase 2 space is $54.50 per rentable square foot per year, with 3% increases on each anniversary of the phase 1 space rent commencement date. In addition to the base rent, the Company is also responsible for its share of operating expenses and real estate taxes. In accordance with the terms of the lease agreement, the Company maintains a letter of credit securing its obligations under the lease agreement of approximately $2.9 million. The Company has determined that the lease should be classified as an operating lease. As the Company gained access to both the phase 1 space and the phase 2 space beginning in May 2012, the Company recognized rent expense of approximately $4.3 million, $6.7 million and $4.9 million related to the lease during the years ended December 31, 2014, 2013 and 2012, respectively.
In order to make the space usable for the Company’s operations, substantial improvements were made to the space. These improvements were planned, managed and carried out by the Company and the improvements were tailored to the Company’s needs. BMR agreed to reimburse the Company for up to $14.9 million of the improvements, and the Company bore all risks associated with any cost overruns that may be incurred. As such, the Company determined it was the owner of the improvements and, as such, the Company accounted for tenant improvement reimbursements from BMR as a lease incentive. The Company recorded a deferred lease incentive (included as a component of the deferred rent balance in the accompanying consolidated balance sheets) and the incentive is being amortized as an offset to rent expense over the term of the lease. Rent expense, inclusive of the escalating rent payments, is being recognized on a straight-line basis over the initial term of the lease agreement. Refer to Footnote 14 for further discussion regarding the termination of this lease.
Future annual minimum lease payments under all non-cancelable operating leases at December 31, 2014 is $1.7 million for the year ending December 31, 2015.
Employment Agreements
Certain key executives are covered by severance and change in control agreements. Under these agreements, if the executive’s employment is terminated without cause or if the executive terminates his employment for good reason, such executive will be entitled to receive severance equal to his base salary, benefits and prorated bonuses for a period of time equal to either 12 months or 18 months, depending on the terms of such executive’s individual agreement. In addition, in December 2007, the Company approved a key employee change in control severance benefits plan, which was amended in November 2009, and which provides for severance and other benefits under certain qualifying termination events upon a change in control for a period of time ranging from 6 months to 18 months, depending upon the position of the key employee.
| 9. | Income Taxes |
The Company accounts for income taxes under the provisions of ASC 740. For the years ended December 31, 2014 and 2013, the Company did not have any federal, state, or foreign income tax expense as it generated taxable losses in all filing jurisdictions. For the year ended December 31, 2011, the Company was able to utilize net operating loss carryforwards (NOLs) to fully offset taxable income in all filing jurisdictions.
112
Table of Contents
A reconciliation of the expected income tax benefit computed using the federal statutory income tax rate to the Company’s effective income tax rate is as follows for the years ended December 31, 2014, 2013 and 2012:
| December 31, 2014 |
December 31, 2013 |
December 31, 2012 |
||||||||||
| Income tax computed at federal statutory tax rate |
34.0 | % | 34.0 | % | 34.0 | % | ||||||
| State taxes, net of federal benefit |
5.3 | % | 5.1 | % | 5.1 | % | ||||||
| Research and development credits |
2.6 | % | 2.0 | % | 0.1 | % | ||||||
| Permanent differences |
(0.8 | )% | (0.9 | )% | (1.2 | )% | ||||||
| Foreign rate differential |
0.0 | % | (0.2 | )% | (0.4 | )% | ||||||
| Other |
(5.7 | )% | (0.5 | )% | (0.2 | )% | ||||||
| Change in valuation allowance |
(35.4 | )% | (39.5 | )% | (37.4 | )% | ||||||
|
|
|
|
|
|
|
|||||||
| Total |
0.0 | % | 0.0 | % | 0.0 | % | ||||||
|
|
|
|
|
|
|
|||||||
Prior to 2011, the Company had incurred net operating losses from inception. At December 31, 2014, the Company had domestic federal, state, and UK net operating loss carryforwards of approximately $424.0 million, $316.1 million, and $6.9 million respectively, available to reduce future taxable income, which expire at various dates. The federal net operating loss carryforwards expire beginning in 2024 through 2034 and the state loss carryforwards begin to expire in 2030 and continue through 2034. The Company also had federal and state research and development tax credit carryforwards of approximately $9.7 million and $4.5 million, respectively, available to reduce future tax liabilities and which expire at various dates. The federal credits expire beginning in 2022 through 2034 and the state credits begin to expire in 2015. The net operating loss and research and development carryforwards are subject to review and possible adjustment by the Internal Revenue Service and may be limited in the event of certain changes in the ownership interest of significant stockholders.
The Company’s net deferred tax assets as of December 31, 2014 and 2013 are as follows:
| 2014 | 2013 | |||||||
| (in thousands) | ||||||||
| NOL carryforwards |
$ | 162,248 | $ | 134,023 | ||||
| Research and development credits |
12,721 | 11,357 | ||||||
| Deferred revenue |
302 | 7,224 | ||||||
| Other temporary differences |
11,135 | 15,158 | ||||||
| Valuation allowance |
(186,406 | ) | (167,762 | ) | ||||
|
|
|
|
|
|||||
| $ | — | $ | — | |||||
|
|
|
|
|
|||||
A full valuation allowance has been recorded in the accompanying consolidated financial statements to offset these deferred tax assets because the future realizability of such assets is uncertain. This determination is based primarily on the Company’s historical losses. Accordingly, future favorable adjustments to the valuation allowance may be required, if and when circumstances change. The valuation allowance increased by $18.6 million during the year ended December 31, 2014, primarily due to the generation of net operating loss carryforwards.
As of December 31, 2014, the Company had federal and state net operating losses of approximately $4.1 million related to excess tax deductions that have been excluded from the above table. The benefit of these net operating losses will be recognized as an increase in additional paid in capital when it results in a reduction in taxable income.
The Company applies FASB Interpretation No. 48, “Accounting for Uncertainty in Income Taxes, an interpretation of FAS 109” (codified within ASC 740, Income Taxes), for the financial statement recognition, measurement, presentation and disclosure of uncertain tax positions taken or expected to be taken in income tax
113
Table of Contents
returns. Unrecognized tax benefits represent tax positions for which reserves have been established. The Company adopted this accounting guidance on January 1, 2009 and recorded $1.2 million of unrecognized tax benefits upon adoption. A full valuation allowance has been provided against the Company’s deferred tax assets, so that the effect of the unrecognized tax benefits is to reduce the gross amount of the deferred tax asset and the corresponding valuation allowance. Since the Company has incurred net operating losses since inception, it has never been subject to a revenue agent review. The Company is currently open to examination under the statute of limitations by the Internal Revenue Service and state jurisdictions for the tax years ended 2011 through 2014. Carryforward tax attributes generated in years past may still be adjusted upon future examination if they have or will be used in a future period. The Company is currently not under examination by the Internal Revenue Service or any other jurisdictions for any tax years.
The Company may from time to time be assessed interest or penalties by major tax jurisdictions. The Company recognizes interest and penalties related to uncertain tax positions in income tax expense. No interest and penalties have been recognized by the Company to date.
The Company anticipates that the amount of unrecognized tax benefits recorded will not change in the next twelve months.
The following is a reconciliation of the Company’s gross uncertain tax positions at December 31, 2014 and 2013:
| Year ended December 31, |
Year ended December 31, |
|||||||
| 2014 | 2013 | |||||||
| (in thousands) | ||||||||
| Amount established upon adoption |
$ | 1,200 | $ | 1,200 | ||||
| Additions for current year tax positions |
— | — | ||||||
| Additions for prior year tax positions |
— | — | ||||||
| Reductions of prior year tax positions |
— | — | ||||||
|
|
|
|
|
|||||
| Balance as of end of year |
$ | 1,200 | $ | 1,200 | ||||
|
|
|
|
|
|||||
| 10. | Common Stock and Warrants |
As of December 31, 2014, the Company had 100,000,000 authorized shares of common stock, $0.001 par value, of which 52,288,799 shares were issued and outstanding.
As part of the Amended Loan Agreement with Hercules Technology II, L.P. and Hercules Technology III, L.P., affiliates of Hercules Technology Growth (collectively, “Hercules”), on September 24, 2014, the Company issued warrants to the lenders to purchase up to 608,696 shares of the Company’s common stock at an exercise price equal to $1.15 per share to Hercules. All warrants issued during the year remain outstanding as of December 31, 2014.
Public Offering
In January 2013, the Company completed an underwritten public offering of its common stock. The total number of shares sold was 7,667,050 including the underwriters’ exercise of their over-allotment option, at the public offering price of $7.50 per share. Aggregate net proceeds to the Company were approximately $53.6 million, after deducting $3.9 million in offering related expenses and underwriting discounts and commissions.
114
Table of Contents
| 11. | Stock-Based Compensation |
Stock Incentive Plan—Overview
The Company maintains the 2010 Stock Incentive Plan (the “Plan”) for employees, consultants, advisors, and directors. The Plan provides for the grant of equity awards such as stock options and restricted stock. The Plan has been amended at various times since its approval. In March 2013, the Company’s board of directors amended the Plan to increase the number of shares of common stock reserved for issuance to 7,875,000 shares, plus the number of shares of common stock subject to awards granted under the Company’s 2002 Incentive Plan which expire, terminate or are otherwise surrendered, cancelled, forfeited or repurchased by us, up to a maximum of 625,000 shares. This amendment also adopted a fungible share pool whereby any award that is a full-value award (i.e. any restricted stock award, restricted stock unit award, or other stock-based award with a per share price or per unit purchase price lower than 100% of fair market value on the date of the grant) is counted against the share limits under the Plan as 1.5 shares for each one share of common stock subject to such full-value award. In June 2014, the Company’s stockholders approved an amendment to the Plan, which increased the annual per participant share limit under the Plan from 250,000 to 1,000,000 shares per fiscal year. No other amendments to the Plan were made.
The Company has reserved 9,018,151 shares of common stock under the Plan, and at December 31, 2014, the Company has 2,826,499 shares available for future issuance under the Plan. Shares issued upon exercise of options are generally issued from new shares of the Company. The Plan provides that the exercise price of incentive stock options cannot be less than 100% of the fair market value of the common stock on the date of the award for participants who own less than 10% of the total combined voting power of stock of the Company and not less than 110% for participants who own more than 10% of the total combined voting power of the stock of the Company. Options and restricted stock granted under the Plan vest over periods as determined by the Board, which generally are equal to four years. Options generally expire ten years from the date of grant.
Stock Incentive Plan—Employee Stock Options
The fair value of each stock option granted is estimated on the date of grant using the Black-Scholes option-pricing model using the assumptions noted in the following table:
| Years Ended December 31, | ||||||
| 2014 | 2013 | 2012 | ||||
| Volatility |
69.38%-77.92% | 64.22%-72.65% | 64.30%-66.05% | |||
| Expected Term (in years) |
5.50-6.25 | 5.50-6.25 | 5.50-6.25 | |||
| Risk-Free Interest Rates |
1.81%-2.02% | 1.01%-2.10% | 0.83%-1.33% | |||
| Dividend Yield |
— | — | — | |||
The risk-free interest rate is determined based upon the United States Treasury’s rates for U.S. Treasury zero-coupon bonds with maturities similar to those of the expected term of the options being valued. The Company does not expect to pay dividends in the foreseeable future.
The Company does not have sufficient history to support a calculation of volatility and expected term using only its historical data. As such, the Company has used a weighted-average volatility considering the Company’s own volatility since March 2010, and the volatilities of several peer companies. For purposes of identifying similar entities, the Company considered characteristics such as industry, length of trading history, similar vesting terms and in-the-money option status. Due to lack of available option activity data, the Company elected to use the “simplified” method for “plain vanilla” options to estimate the expected term of the stock option grants. Under this approach, the weighted-average expected life is presumed to be the average of the vesting term and the contractual term of the option. Additionally, the Company is required to include an estimate of the value of the awards that will be forfeited in calculating compensation costs, which the Company estimates based upon actual historical forfeitures. The forfeiture estimates are recognized over the requisite service period of the
115
Table of Contents
awards on a straight-line basis. During the years ending December 31, 2014, 2013 and 2012 the Company estimated its forfeiture rates to be 62%, 49% and 18%, respectively. Based upon these assumptions, the weighted-average grant date fair value of stock options granted during the years ended December 31, 2014, 2013, and 2012 was $1.15, $4.32 and $7.40 per share, respectively.
During the year ended December 31, 2014 the Company issued stock options to purchase 2,250,000 shares of common stock that contain market and performance –based vesting conditions that were not deemed probable of vesting at December 31, 2014. As of December 31, 2014, there was $1.9 million of total unrecognized stock-based compensation expense related to stock options granted under the Company’s 2002 Stock Incentive Plan and 2010 Stock Incentive Plan (collectively, the “Plans”). The expense is expected to be recognized over a weighted-average period of 3.2 years. The intrinsic value of options exercised was $0.5 million and $1.9 million for the years ended December 31, 2013 and 2012, respectively. No options were exercised during the year ended December 31, 2014.
The following table summarizes the activity of the Plans for the year ended December 31, 2014:
| Number of Options |
Weighted- Average Exercise Price |
Weighted- Average Remaining Contractual Life (in years) |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding at December 31, 2013 |
4,296,694 | $ | 7.36 | |||||||||||||
| Granted |
2,992,100 | $ | 1.59 | |||||||||||||
| Forfeited |
(544,232 | ) | $ | 4.15 | ||||||||||||
| Expired |
(927,249 | ) | $ | 8.89 | ||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2014 |
5,817,313 | $ | 4.45 | 3.52 | — | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Exercisable at December 31, 2014 |
3,461,864 | $ | 6.24 | 3.38 | — | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Vested or expected to vest at December 31, 2014 |
2,632,700 | $ | 7.09 | 3.04 | — | |||||||||||
|
|
|
|
|
|
|
|||||||||||
Stock Incentive Plan—Nonemployee Stock Options
During 2014, the Company granted nonqualified options to purchase 225,000 shares of common stock to nonemployee consultants, with an average exercise price of $1.47 per share. There were no stock options granted to nonemployee consultants during 2013 or 2012. The Company valued these options using the Black-Scholes option-pricing model and recognized expense related to these awards using the accelerated attribution method. The unvested options held by consultants have been revalued using the Company’s estimate of fair value at each reporting period over the vesting period. Stock-based compensation expense of approximately $0.1 million was recorded during the year ended December 31, 2014 relating to nonemployee stock option awards.
Stock Incentive Plan—Restricted Stock
The Company periodically grants awards of restricted stock to employees. These awards typically vest upon completion of the requisite service period or upon achievement of specified performance targets.
116
Table of Contents
The following table summarizes the restricted stock activity for the year ended December 31, 2014:
| Number of Shares | Weighted- Average Price |
|||||||
| Unvested at December 31, 2013 |
241,500 | $ | 2.50 | |||||
| Granted |
501,000 | 1.62 | ||||||
| Cancelled |
(191,620 | ) | 1.92 | |||||
| Expired |
— | — | ||||||
| Vested/Released |
(73,280 | ) | 2.50 | |||||
|
|
|
|
|
|||||
| Unvested at December 31, 2014 |
477,600 | $ | 1.81 | |||||
|
|
|
|
|
|||||
The fair value of restricted stock awards that vested was $0.2 million, $0.5 million, and $0.5 million for the years ended December 31, 2014, 2013, and 2012, respectively. As of December 31, 2014, there was $0.1 million of total unrecognized stock-based compensation expense related to restricted stock awards granted under the Plan. The expense is expected to be recognized over a weighted-average period of 0.9 years if all performance targets are met.
Employee Stock Purchase Plan
In February 2010, the Board of Directors adopted the 2010 Employee Stock Purchase Plan (the “ESPP”) pursuant to which the Company may sell up to an aggregate of 250,000 shares of Common Stock. The ESPP was approved by the Company’s stockholders in February 2010. The plan was amended in March 2013 to increase the total number of shares available under the ESPP for the Company to sell to 764,000. The ESPP allows eligible employees to purchase common stock at a price per share equal to 85% of the lower of the fair market value of the common stock at the beginning or end of each six month period during the term of the ESPP. The first offering period began on July 1, 2010.
Pursuant to the ESPP, the Company sold a total of 139,032 shares of common stock during the year ended December 31, 2014 at purchase prices of $1.53 and $0.71, respectively, which represent 85% of the closing price of the Company’s common stock on June 30, 2014 and December 31, 2014, respectively. For the year ended December 31, 2013, the Company sold a total of 109,610 shares of common stock at purchase prices of $2.13 and $1.56, respectively, which represent 85% of the closing price of the Company’s common stock on June 28, 2013 and December 31, 2013, respectively. For the year ended December 31, 2012, the Company sold a total of 94,592 shares of common stock at purchase prices of $10.34 and $6.84, respectively, which represent 85% of the closing price of the Company’s common stock on June 29, 2012 and December 31, 2012, respectively. The total stock-based compensation expense recorded as a result of the ESPP was approximately $0.2 million, $0.2 million and $0.3 million during the years ended December 31, 2014, 2013 and 2012, respectively.
| 12. | Employee Benefit Plan |
In 2002, the Company established the AVEO Pharmaceuticals, Inc. 401(k) Plan (the “401(k) Plan”) for its employees, which is designed to be qualified under Section 401(k) of the Internal Revenue Code. Eligible employees are permitted to contribute to the 401(k) Plan within statutory and 401(k) Plan limits. The Company makes matching contributions of 50% of the first 5% of employee contributions. The Company made matching contributions of $0.1 million, $0.4 million, and $0.6 million for the years ended December 31, 2014, 2013 and 2012, respectively.
| 13. | Strategic Restructuring |
In October 2012, the Company announced a strategic restructuring designed to optimize resources and reduce expenses. The Company’s restructuring and projected cost savings are being achieved through a combination of reduced spending on early stage research programs and a reduction in force of 48 positions, as well as the elimination of 30 previously open positions.
117
Table of Contents
In connection with the receipt of a Complete Response Letter from the FDA informing the Company that the FDA would not approve the Company’s NDA for tivozanib for the treatment of patients with advanced RCC, the Company announced a further strategic restructuring in June 2013 to refocus the Company’s efforts on the then on-going clinical development of tivozanib in colorectal and breast cancer and on the advancement of key pipeline and preclinical assets. As part of this restructuring, the Company decided not to pursue the development of tivozanib in RCC. This restructuring was completed as of December 31, 2013 and resulted in costs totaling $8.0 million, which includes impairment charges of $0.3 million.
The following table summarizes the components of the Company’s restructuring activity recorded in operating expenses and in accrued expenses in the accompanying consolidated balance sheet:
| Restructuring amounts accrued at December 31, 2013 |
Restructuring expense incurred during the year ended December 31, 2014 |
Restructuring amounts paid during the year ended December 31, 2014 |
Restructuring amounts accrued at December 31, 2014 |
|||||||||||||
| (in thousands) | ||||||||||||||||
| Employee severance, benefits and related costs. |
$ | 587 | — | $ | (587 | ) | — | |||||||||
| Restructuring amounts accrued at December 31, 2012 |
Restructuring expense incurred during the year ended December 31, 2013 |
Restructuring amounts paid during the year ended December 31, 2013 |
Restructuring amounts accrued at December 31, 2013 |
|||||||||||||
| (in thousands) | ||||||||||||||||
| Employee severance, benefits and related costs. |
$ | 1,653 | $ | 7,674 | $ | (8,740 | ) | $ | 587 | |||||||
| Contract termination costs |
— | 82 | (82 | ) | — | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
1,653 | 7,756 | (8,822 | ) | 587 | |||||||||||
| 14. | Facility Lease Exit |
During the year ended December 31, 2014, the Company completed the planned build-out of portions of the office space it leases at 650 E. Kendall Street in Cambridge, Massachusetts. Upon completion of these build-outs, the Company ceased use of these spaces and recorded liabilities totaling $15.2 million for lease exit costs. The fair value of these liabilities was determined using the credit-adjusted risk-free rate to discount the estimated future net cash outflows associated with the space that met the cease use criteria. The estimate of future net cash outflows included the Company’s expected minimum rental payments and incremental operating, utility and tax payments to the landlord less the amount of sublease income that the Company estimates it could reasonably expect to obtain during the remainder of the lease period.
On September 24, 2014, the Company entered into the Lease Termination Agreement pursuant to which the Company immediately surrendered an aggregate of 76,980 square feet of leased space that it had previously ceased using earlier in 2014. In connection with the Lease Termination Agreement, the Company agreed to pay the landlord a termination fee totaling $15.6 million to be settled as follows: fifty percent of the fee, or $7.8 million, was to be credited against amounts due the Company for tenant improvements, and the remaining fifty percent will be paid in nine equal monthly installments of $0.9 million commencing on October 1, 2014. Upon signing the Lease Termination Agreement, the Company recorded an additional $1.9 million of Lease Exit charges. The initial $7.8 million fee was credited by the landlord against the Company’s tenant improvement allowance of $14.7 million, resulting in a net payment to the Company of $6.8 million which was received in September 2014. The Company also agreed to surrender the remaining 49,185 square feet of leased space upon 90 days written notice upon a date that is no earlier than March 24, 2015 and no later than September 24, 2015. In February 2015, the Company provided notice that it will surrender the remaining space on May 29, 2015.
118
Table of Contents
Pursuant to the Lease Termination Agreement, the Company revised the estimated useful life of its leasehold improvements related to this office space and is amortizing such assets through September 2015. Similarly, the Company has accelerated the amortization of its deferred rent and leasehold improvement allowance associated with this office space through September 2015. Upon the surrender of the remaining space, the Company will have no further rights or obligations with respect to the lease.
In connection with the Lease Termination Agreement, the Company has recorded a liability related to the remaining half of the lease termination fee. The fair value of this liability was determined using the credit-adjusted risk-free rate to discount the payment stream related to this portion of the termination fee. The Company recorded the fair value of this liability, or $5.0 million, as lease exit liability at December 31, 2014.
The following table summarizes the components of the Company’s lease exit activity recorded in current liabilities:
| Lease Exit Expense incurred during the year ended December 31, 2014 |
Accretion Expense incurred during the year ended December 31, 2014 |
Amounts paid during the year ended December 31, 2014 |
Amounts offset against tenant receivable during the year ended December 31, 2014 |
Amounts accrued at December 31, 2014 |
||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Lease exit costs |
$ | 17,142 | $ | 974 | $ | (5,313 | ) | $ | (7,822 | ) | $ | 4,981 | ||||||||
In addition to the $17.1 million of expense included in the table above, lease exit expenses also include the write-off $14.0 million of deferred rent associated with the portions of the facility that met the cease use criteria under ASC 420-10 and leasehold improvements totaling $7.6 million during the year ended December 31, 2014 as the Company’s estimates of sublease income would not recover the value of the leasehold improvements.
| 15. | Quarterly Results (Unaudited) |
| Three Months Ended | ||||||||||||||||
| March 31, 2014 |
June 30, 2014 |
September 30, 2014 |
December 31, 2014 |
|||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| (unaudited) | ||||||||||||||||
| Collaboration revenue |
$ | 15,289 | $ | 1,846 | $ | 873 | $ | 115 | ||||||||
| Restructuring |
3,859 | 5,165 | 1,403 | 1,302 | ||||||||||||
| Operating expenses |
17,322 | 14,146 | 13,569 | 11,806 | ||||||||||||
| Loss from operations |
(5,892 | ) | (17,465 | ) | (14,099 | ) | (12,993 | ) | ||||||||
| Other expense, net |
(558 | ) | (494 | ) | (337 | ) | (901 | ) | ||||||||
| Net loss |
$ | (6,450 | ) | $ | (17,959 | ) | $ | (14,436 | ) | $ | (13,894 | ) | ||||
| Net loss per share—basic and diluted |
$ | (0.12 | ) | $ | (0.35 | ) | $ | (0.28 | ) | $ | (0.27 | ) | ||||
| Three Months Ended | ||||||||||||||||
| March 31, 2013 |
June 30, 2013 |
September 30, 2013 |
December 31, 2013 |
|||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| (unaudited) | ||||||||||||||||
| Collaboration revenue |
$ | 323 | $ | 324 | $ | 323 | $ | 323 | ||||||||
| Restructuring |
67 | 7,869 | 77 | 4 | ||||||||||||
| Operating expenses |
33,478 | 31,396 | 23,931 | 16,392 | ||||||||||||
| Loss from operations |
(33,155 | ) | (31,072 | ) | (23,608 | ) | (16,069 | ) | ||||||||
| Other expense, net |
(930 | ) | (841 | ) | (698 | ) | (656 | ) | ||||||||
| Net loss |
$ | (34,085 | ) | $ | (31,913 | ) | $ | (24,306 | ) | $ | (16,725 | ) | ||||
| Net loss per share—basic and diluted |
$ | (0.69 | ) | $ | (0.62 | ) | $ | (0.47 | ) | $ | (0.32 | ) | ||||
119
Table of Contents
| 16. | Legal Actions |
Two class action lawsuits have been filed against the Company and certain present and former officers and members of the Company’s board of directors, (Tuan Ha-Ngoc, David N. Johnston, William Slichenmyer and Ronald DePinho), in the United States District Court for the District of Massachusetts, one captioned Paul Sanders v. Aveo Pharmaceuticals, Inc., et al., No. 1:13-cv-11157-JLT, filed on May 9, 2013, and the other captioned Christine Krause v. AVEO Pharmaceuticals, Inc., et al., No. 1:13-cv-11320-JLT, filed on May 31, 2013. On December 4, 2013, the District Court consolidated the complaints as In re AVEO Pharmaceuticals, Inc. Securities Litigation et al., No. 1:13-cv-11157-DJC, and an amended complaint was filed on February 3, 2014. The amended complaint purports to be brought on behalf of shareholders who purchased the Company’s common stock between January 3, 2012 and May 1, 2013. The amended complaint generally alleges that the Company and certain of its present and former officers and directors violated Sections 10(b) and/or 20(a) of the Securities Exchange Act of 1934 and Rule 10b-5 promulgated thereunder by making allegedly false and/or misleading statements concerning the phase 3 trial design and results for our TIVO-1 study in an effort to lead investors to believe that the drug would receive approval from the FDA. The amended complaint seeks unspecified damages, interest, attorneys’ fees, and other costs. On April 4, 2014, the Company filed a motion to dismiss the consolidated class action complaint with prejudice. Lead plaintiffs filed an opposition to the motion to dismiss on June 10, 2014, and the Company filed a reply to the opposition on July 10, 2014. The Court heard oral argument on the Company’s motion to dismiss on July 22, 2014. The Company denies any allegations of wrongdoing and intends to vigorously defend against this lawsuit. However, there is no assurance that the Company will be successful in our defense or that insurance will be available or adequate to fund any settlement or judgment or the litigation costs of the action. Moreover, the Company is unable to predict the outcome or reasonably estimate a range of possible loss at this time.
On July 3, 2013, the Company received a subpoena from the SEC, requesting documents and information concerning tivozanib, including related communications with the FDA, investors and others. The Company is fully cooperating with the SEC regarding this fact-finding inquiry. The SEC has informed the Company that this inquiry should not be construed as an indication that any violations of law have occurred or that the SEC has any negative opinion of any person, entity or security.
On April 4, 2014, Karen J. van Ingen, a purported purchaser of AVEO stock, filed a derivative complaint allegedly on behalf of AVEO in the United States District Court for the District of Massachusetts, Civil Action No. 1:14-cv-11672-DJC, naming AVEO, as a nominal defendant and also naming as defendants present and former members of the Company’s board of directors, including Tuan Ha-Ngoc, Henri A. Termeer, Kenneth M. Bate, Anthony B. Evnin, Robert Epstein, Raju Kucherlapati, Robert C. Young, and Kenneth E. Weg. The complaint alleges breach of fiduciary duty and abuse of control between January 2012 and May 2013 with respect to allegedly misleading statements and omissions regarding tivozanib. The complaint seeks, among other relief, unspecified damages, costs and expenses, including attorneys’ fees, an order requiring the Company to implement certain corporate governance reforms, restitution from the defendants and such other relief as the court might find just and proper. On July 25, 2014, defendants filed a motion to dismiss the derivative complaint with prejudice. Plaintiff filed an opposition to the motion to dismiss on September 23, 2014, and the Company filed a reply to the opposition on October 23, 2014. The Court heard oral argument on the Company’s motion to dismiss on January 7, 2015. The Company denies any allegations of wrongdoing and intends to vigorously defend this lawsuit. However, there is no assurance that the Company will be successful in its defense or that insurance will be available or adequate to fund any settlement or judgment or the litigation costs of this action. Moreover, the Company is unable to predict the outcome or reasonably estimate a range of possible loss at this time.
| 17. | Subsequent Events |
On January 6, 2015, the Board of the Company approved a strategic restructuring of the Company that will eliminate the Company’s internal research function and align the Company’s resources with the Company’s future strategic plans.
120
Table of Contents
As part of this restructuring, the Company eliminated approximately two-thirds of the Company’s workforce, or 40 positions across the organization. The Company expects the restructuring to be fully completed by March 31, 2015. The Company currently expects to incur total restructuring charges of approximately $4.5 million, consisting of severance and benefit costs associated with the targeted staff reductions, which will be included in its results of operations for the first quarter of 2015. As a result of its strategic restructuring, the also Company intends to dispose of its laboratory equipment through sale during the first quarter of 2015. The Company expects to incur an impairment charge of approximately $0.3 million during the first quarter of 2015.
In February 2015, the Company entered into an at-the-market issuance sales agreement with MLV & Co. LLC (“MLV”), pursuant to which the Company may issue and sell shares of its common stock from time to time up to an aggregate amount of $17.9 million, at the Company’s option, through MLV as its sales agent. Sales of common stock through MLV may be made by any method that is deemed an “at-the-market” offering as defined in Rule 415 promulgated under the Securities Act of 1933, as amended, including by means of ordinary brokers’ transactions at market prices, in block transactions or as otherwise agreed by the Company and MLV. Subject to the terms and conditions of the sales agreement between the Company and MLV (the “Sales Agreement”), MLV will use commercially reasonable efforts to sell the common stock based upon the Company’s instructions (including any price, time or size limits or other customary parameters or conditions the Company may impose). The Company is not obligated to make any sales of its common stock under the Sales Agreement. Any shares sold will be sold pursuant to an effective shelf registration statement on Form S-3. The Company will pay MLV a commission of up to 3% of the gross proceeds. The Sales Agreement may be terminated by the Company at any time.
121
Table of Contents
| ITEM 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| ITEM 9A. | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Acting Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2014. In designing and evaluating our disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and our management necessarily applied its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on this evaluation, our Chief Executive Officer and Acting Chief Financial Officer concluded that as of December 31, 2014, our disclosure controls and procedures were (1) designed to ensure that material information relating to us is made known to our management including our principal executive officer and principal financial officer by others, particularly during the period in which this report was prepared and (2) effective, in that they provide reasonable assurance that information required to be disclosed by us in the reports the Company files or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms.
Management’s report on the Company’s internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) appears below.
Internal Control Over Financial Reporting
(a) Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rules 13a-15(f) and 15d-15(f) promulgated under the Exchange Act as a process designed by, or under the supervision of, the company’s principal executive and principal financial officers and effected by the company’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
| • | Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the company; |
| • | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and |
| • | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2014. In making this assessment, management used the criteria set forth by the Committee of
122
Table of Contents
Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control—Integrated Framework (2013 framework). Based on its assessment, management believes that, as of December 31, 2014, our internal control over financial reporting is effective based on those criteria.
Our independent registered public accounting firm has issued an attestation report of our internal control over financial reporting. This report appears below.
(b) Report of Independent Registered Public Accounting Firm on Internal Control over Financial Reporting
123
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of
AVEO Pharmaceuticals, Inc.
We have audited AVEO Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). AVEO Pharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying management’s report on internal control over financial reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, AVEO Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets as of December 31, 2014 and 2013, and the related consolidated statements of operations, comprehensive (loss) income, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2014 of AVEO Pharmaceuticals, Inc. and our report dated March 6, 2015 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Boston, Massachusetts
March 6, 2015
124
Table of Contents
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting that occurred during the quarter ended December 31, 2014 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| ITEM 9B. | Other Information |
None.
125
Table of Contents
| ITEM 10. | Directors, Executive Officers and Corporate Governance |
The information required by this Item 10 will be contained in the sections entitled “Election of Directors,” “Corporate Governance” and “Section 16(a) Beneficial Ownership Reporting Compliance” appearing in the definitive proxy statement we will file in connection with our 2015 Annual Meeting of Stockholders and is incorporated by reference herein. The information required by this item relating to executive officers may be found in Part I, Item 1 of this report under the heading “Business—Executive Officers” and is incorporated herein by reference.
| ITEM 11. | Executive Compensation |
The information required by this Item 11 will be contained in the sections entitled “Executive and Director Compensation,” “Executive and Director Compensation—Compensation Committee Interlocks and Insider Participation” and “Executive and Director Compensation—Compensation Committee Report” appearing in the definitive proxy statement we will file in connection with our 2015 Annual Meeting of Stockholders and is incorporated by reference herein.
| ITEM 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this Item 12 will be contained in the sections entitled “Ownership of Our Common Stock” and “Executive and Director Compensation—Equity Compensation Plan Information” appearing in the definitive proxy statement we will file in connection with our 2015 Annual Meeting of Stockholders and is incorporated by reference herein.
| ITEM 13. | Certain Relationships and Related Person Transactions, and Director Independence |
The information required by this Item 13 will be contained in the sections entitled “Certain Relationships and Related Person Transactions” appearing in the definitive proxy statement we will file in connection with our 2015 Annual Meeting of Stockholders and is incorporated by reference herein.
| ITEM 14. | Principal Accounting Fees and Services |
The information required by this Item 14 will be contained in the section entitled “Corporate Governance—Principal Accountant Fees and Services” appearing in the definitive proxy statement we will file in connection with our 2015 Annual Meeting of Stockholders and is incorporated by reference herein.
126
Table of Contents
| ITEM 15. | Exhibits and Financial Statement Schedules |
| (a) | Documents filed as part of Form 10-K. |
| (1) | Financial Statements |
Report of Independent Registered Public Accounting Firm
Consolidated Balance Sheets
Consolidated Statements of Operations
Consolidated Statements of Comprehensive Loss) Income
Consolidated Statements of Stockholders’ Equity
Consolidated Statements of Cash Flows
Notes to Consolidated Financial Statements
| (2) | Schedules |
Schedules have been omitted as all required information has been disclosed in the financial statements and related footnotes.
| (3) | Exhibits |
The Exhibits listed in the Exhibit Index are filed as a part of this Form 10-K.
127
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| AVEO PHARMACEUTICALS, INC. | ||||
| Date: March 6, 2015 | By: | /s/ MICHAEL BAILEY | ||
| Michael Bailey President & Chief Executive Officer (Principal Executive Officer) | ||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ MICHAEL BAILEY Michael Bailey |
President, Chief Executive Officer, Director and Acting Chief Financial Officer Principal Executive Officer and Principal Financial and Accounting Officer |
March 6, 2015 | ||
| /s/ KENNETH M. BATE Kenneth M. Bate |
Director | March 6, 2015 | ||
| /s/ ANTHONY B. EVNIN Anthony B. Evnin |
Director | March 6, 2015 | ||
| /s/ TUAN HA-NGOC Tuan Ha-Ngoc |
Director | March 6, 2015 | ||
| /s/ RAJU KUCHERLAPATI Raju Kucherlapati |
Director | March 6, 2015 | ||
| /s/ HENRI TERMEER Henri Termeer |
Director | March 6, 2015 | ||
| /s/ ROBERT C. YOUNG Robert C. Young |
Director | March 6, 2015 | ||
128
Table of Contents
Exhibit Index
| Exhibit |
Description of Exhibit |
Incorporated by Reference |
Filed | |||||||||
| Form | File Number |
Date of |
Exhibit |
|||||||||
| Articles of Incorporation and Bylaws | ||||||||||||
| 3.1 | Restated Certificate of Incorporation of the Registrant | 8-K | 001-34655 | 03/18/2010 | 3.1 | |||||||
| 3.2 | Second Amended and Restated Bylaws of the Registrant | S-1/A | 333-163778 | 02/08/2010 | 3.5 | |||||||
| Instruments Defining the Rights of Security Holders, Including Indentures | ||||||||||||
| 4.1 | Specimen Stock Certificate evidencing the shares of common stock | S-1/A | 333-163778 | 03/09/2010 | 4.1 | |||||||
| Material Contracts—Management Contracts and Compensatory Plans | ||||||||||||
| 10.1 | 2002 Stock Incentive Plan, as amended | S-1/A | 333-163778 | 02/23/2010 | 10.1 | |||||||
| 10.2 | Form of Incentive Stock Option Agreement under 2002 Stock Incentive Plan | S-1 | 333-163778 | 12/16/2009 | 10.2 | |||||||
| 10.3 | Form of Nonstatutory Stock Option Agreement under 2002 Stock Incentive Plan | S-1 | 333-163778 | 12/16/2009 | 10.3 | |||||||
| 10.4 | Form of Restricted Stock Agreement under 2002 Stock Incentive Plan | S-1 | 333-163778 | 12/16/2009 | 10.4 | |||||||
| 10.5 | Amended and Restated 2010 Stock Incentive Plan, as amended | 8-K | 001-34655 | 06/23/2014 | 99.1 | |||||||
| 10.6 | Form of Incentive Stock Option Agreement under 2010 Stock Incentive Plan | S-1/A | 333-163778 | 02/08/2010 | 10.6 | |||||||
| 10.7 | Form of Nonqualified Stock Option Agreement under 2010 Stock Incentive Plan | S-1/A | 333-163778 | 02/08/2010 | 10.7 | |||||||
| 10.8 | Form of Restricted Stock Agreement under 2010 Stock Incentive Plan | 10-K | 001-34655 | 03/30/2012 | 10.8 | |||||||
| 10.9 | Key Employee Change in Control Severance Benefits Plan | S-1 | 333-163778 | 12/16/2009 | 10.8 | |||||||
| 10.10 | Amended and Restated Employment Agreement, dated as of December 19, 2008, by and between the Registrant and Tuan Ha-Ngoc | S-1 | 333-163778 | 12/16/2009 | 10.9 | |||||||
| 10.11 | Severance and Change in Control Agreement, dated as of December 11, 2009, by and between the Registrant and Tuan Ha-Ngoc | S-1 | 333-163778 | 12/16/2009 | 10.10 | |||||||
| 10.12 | Severance and Change in Control Agreement, dated as of December 11, 2009, by and between the Registrant and Jeno Gyuris | S-1 | 333-163778 | 12/16/2009 | 10.12 | |||||||
| 10.13 | 2010 Employee Stock Purchase Plan, as amended | S-1/A | 333-163778 | 02/23/2010 | 10.17 | |||||||
| 10.14 | Amendment No. 1 to 2010 Employee Stock Purchase Plan | 8-K | 001-34655 | 06/04/2013 | 99.2 | |||||||
Table of Contents
| Exhibit |
Description of Exhibit |
Incorporated by Reference |
Filed |
|||||||||||
| Form | File Number |
Date of |
Exhibit |
|||||||||||
| 10.15 | Severance Agreement, dated September 13, 2010, by and between the Registrant and Michael Bailey | 10-Q | 001-34655 | 11/05/2010 | 10.1 | |||||||||
| 10.16 | Severance and Change in Control Agreement, dated as of January 24, 2013, by and between the Company and Joseph Vittiglio | 10-K | 001-34655 | 3/11/2013 | 10.18 | |||||||||
| 10.17 | Letter Agreement regarding Retention Bonus Award and Severance Agreement, dated February 3, 2014, by and between the Registrant and Jeno Gyuris | 10-Q | 001-34655 | 5/07/2014 | 10.3 | |||||||||
| 10.18 | Letter Agreement regarding Retention Bonus Award and Severance Agreement, dated February 3, 2014, by and between the Company and Michael Bailey | 10-K | 001-34655 | 3/13/2014 | 10.22 | |||||||||
| Material Contracts—Financing Agreements | ||||||||||||||
| 10.19 | Loan and Security Agreement dated May 28, 2010 by and among the Company, Hercules Technology II, L.P. and Hercules Technology III, L.P. | 8-K | 001-34655 | 06/04/2010 | 10.1 | |||||||||
| 10.20 | Amendment No. 1 to Loan and Security Agreement, dated December 21, 2011, by and among the Company, Hercules Technology II, L.P. and Hercules Technology III, L.P. | 10-K | 001-34655 | 03/30/2012 | 10.25 | |||||||||
| 10.21 | Amendment No. 2 to Loan and Security Agreement, dated March 31, 2012, by and among the Company, Hercules Technology II, L.P. and Hercules Technology III, L.P. | 8-K | 001-34655 | 04/04/2012 | 10.1 | |||||||||
| 10.22 | Amendment No. 3 to Loan and Security Agreement, dated September 24, 2014, by and among the Company, Hercules Technology Growth Capital, Inc., Hercules Capital Funding Trust 2012-1 and Hercules Technology III, L.P. | 10-Q | 001-34655 | 11/05/2014 | 10.1 | |||||||||
| 10.23 | Warrant dated as of September 24, 2014 issued by the Registrant to Hercules Technology II, L.P. and Hercules Technology III, L.P | 10-Q | 001-34655 | 11/05/2014 | 10.2 | |||||||||
| Material Contracts—Leases | ||||||||||||||
| 10.24 | Sublease, dated February 28, 2011, by and between the Company and Acceleron Pharma, Inc. | 10-Q | 001-34655 | 05/12/2011 | 10.4 | |||||||||
| 10.25 | First Amendment to Sublease, dated September 1, 2011, by and between the Company and Acceleron Pharma, Inc. | 10-K | 001-34655 | 03/30/2012 | 10.29 | |||||||||
| 10.26 | Lease, dated May 9, 2012, by and between the Company and BMR-650 E. Kendall B LLC | 10-Q | 001-34655 | 05/09/2012 | 10.3 | |||||||||
| 10.27 | First Amendment to Lease dated as of April 30, 2013 by and between the Registrant and BMR-650 E Kendall B LLC | X | ||||||||||||
| 10.28 | Second Amendment to Lease dated as of August 13, 2013 by and between the Registrant and BMR-650 E Kendall B LLC | X | ||||||||||||
Table of Contents
| Exhibit |
Description of Exhibit |
Incorporated by Reference |
Filed | |||||||||
| Form | File Number |
Date of |
Exhibit |
|||||||||
| 10.29 | Third Amendment to Lease and Lease Termination Agreement dated September 24, 2014 by and between the Registrant and BMR-650 E Kendall B LLC | 10-Q | 001-34655 | 11/05/2014 | 10.3 | |||||||
| 10.30 | Fourth Amendment to Lease dated December 1, 2014 by and between the Registrant and BMR-650 E Kendall B LLC | X | ||||||||||
| Material Contracts—License and Strategic Partnership Agreements | ||||||||||||
| 10.31† | License Agreement, dated as of December 21, 2006, by and between the Registrant and Kirin Brewery Co. Ltd. | S-1 | 333-163778 | 12/16/2009 | 10.22 | |||||||
| 10.32† | Option and License Agreement, dated as of March 18, 2009, by and between the Registrant and Biogen Idec International GmbH | S-1 | 333-163778 | 12/16/2009 | 10.26 | |||||||
| 10.33† | Amendment No. 1 to Option and License Agreement, dated as of March 18, 2014 by and between the Registrant and Biogen Idec MA Inc. | 10-Q | 001-34655 | 05/07/2014 | 10.1 | |||||||
| 10.34† | Co-Development and Collaboration Agreement, dated as of April 9, 2014 by and between the Registrant and Biodesix Inc. | 10-Q | 001-34655 | 05/07/2014 | 10.2 | |||||||
| 10.35* | Research and Exclusive License Agreement dated as of November 10, 2014 by and between the Registrant and Ophthotech Corporation | X | ||||||||||
| 10.36† | License Agreement, dated as of July 2, 2012, by and between the Registrant and St. Vincent’s Hospital Sydney Limited | 10-K | 001-34655 | 3/13/2014 | 10.35 | |||||||
| 10.37 | ATM Sales Agreement dated February 27, 2015 by and between the Company and MLV & Co. LLC | 8-K | 001-34655 | 2/27/2015 | 1.1 | |||||||
| Additional Exhibits | ||||||||||||
| 21.1 | Subsidiaries of the Registrant | X | ||||||||||
| 23.1 | Consent of Ernst & Young LLP | X | ||||||||||
| 31.1 | Certification of principal executive officer pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act of 1934, as amended. | X | ||||||||||
| 31.2 | Certification of principal financial officer pursuant to Rule 13a-14(a)/15d-14(a) of the Securities Exchange Act of 1934, as amended. | X | ||||||||||
| 32.1 | Certification of principal executive officer pursuant to 18 U.S.C. §1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | X | ||||||||||
| 32.2 | Certification of principal financial officer pursuant to 18 U.S.C. §1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | X | ||||||||||
Table of Contents
| Exhibit |
Description of Exhibit |
Incorporated by Reference |
Filed | |||||||||
| Form | File |
Date of |
Exhibit |
|||||||||
| 101.INS | XBRL Instance Document. | X | ||||||||||
| 101.SCH | XBRL Taxonomy Extension Schema Document. | X | ||||||||||
| 101.CAL | XBRL Taxonomy Calculation Linkbase Document. | X | ||||||||||
| 101DEF | XBRL Taxonomy Extension Definition Linkbase Document. | X | ||||||||||
| 101.LAB | XBRL Taxonomy Label Linkbase Document. | X | ||||||||||
| 101.PRE | XBRL Taxonomy Presentation Linkbase Document. | X | ||||||||||
| † | Confidential treatment has been granted as to certain portions, which portions have been omitted and separately filed with the Securities and Exchange Commission. |
| * | Confidential treatment has been requested as to certain portions, which portions have been omitted and separately filed with the Securities and Exchange Commission. |