Attached files
| file | filename |
|---|---|
| EX-31.1 - EXHIBIT - PAREXEL INTERNATIONAL CORP | exhibit311_2014.htm |
| EX-23.1 - EXHIBIT CONSENT - PAREXEL INTERNATIONAL CORP | exhibit231_eyconsentx2014.htm |
| EX-31.2 - EXHIBIT - PAREXEL INTERNATIONAL CORP | exhibit312_2014.htm |
| EX-32.2 - EXHIBIT - PAREXEL INTERNATIONAL CORP | exhibit322_2014.htm |
| EX-32.1 - EXHIBIT - PAREXEL INTERNATIONAL CORP | exhibit321_2014.htm |
| EX-21.1 - EXHIBIT LIST OF SUBSIDIARIES - PAREXEL INTERNATIONAL CORP | exhibit211_listofsubsidiar.htm |
| EXCEL - IDEA: XBRL DOCUMENT - PAREXEL INTERNATIONAL CORP | Financial_Report.xls |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
ý | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended June 30, 2014
OR
¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15 (d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number: 000-21244
PAREXEL INTERNATIONAL CORPORATION
(Exact name of registrant as specified in its charter)
Massachusetts | 04-2776269 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification Number) | |
195 West Street, Waltham, Massachusetts | 02451 | |
(Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number, including area code: (781) 487-9900
Securities Registered Pursuant to Section 12(b) of the Act:
Title of each class: | Name of each exchange on which registered: | |
Common Stock, $.01 par value per share | Nasdaq Global Select Market | |
Securities Registered Pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ý No ¨
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
Large Accelerated Filer | ý | Accelerated Filer | ¨ | |||
Non-accelerated Filer | ¨ (Do not check if smaller reporting company) | Smaller Reporting Company | ¨ | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No ý
The aggregate market value of common stock, $.01 par value per share, held by non-affiliates as of December 31, 2013 was approximately $2.5 billion based on the closing price of the registrant’s Common Stock as reported on the Nasdaq Global Select Market on December 31, 2013, the last business day of the registrant’s most recently completed second fiscal quarter. The registrant has assumed that all holders of 10% or more of its Common Stock, if any, are affiliates solely for purposes of calculating the aggregate market value of Common Stock held by non-affiliates. As of August 15, 2014 there were 54,732,592 shares of common stock, $.01 par value per share, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the Registrant’s Proxy Statement for the Annual Meeting of Stockholders to be held on December 4, 2014 are incorporated by reference into Part III of this report.
1
INDEX
i
PART I
ITEM 1. BUSINESS
GENERAL
PAREXEL International Corporation (“PAREXEL,” “the Company,” or “we”) is a leading biopharmaceutical services company, providing a broad range of expertise in clinical research, clinical logistics, medical communications, consulting, commercialization and advanced technology products and services to the worldwide pharmaceutical, biotechnology, and medical device industries. Our primary objective is to provide quality solutions for managing the biopharmaceutical product lifecycle with the goal of reducing the time, risk, and cost associated with the development and commercialization of new therapies. Since our incorporation in 1983, we have developed significant expertise in processes and technologies supporting this strategy. Our product and service offerings include: clinical trials management, observational studies and patient/disease registries, data management, biostatistical analysis, epidemiology, health economics/outcomes research, pharmacovigilance, medical communications, clinical pharmacology, patient recruitment, clinical supply and drug logistics, post-marketing surveillance, regulatory and product development and commercialization consulting, health policy and reimbursement and market access consulting, medical imaging services, regulatory information management (“RIM”) solutions, ClinPhone randomization and trial supply management services (“ClinPhone RTSM”), electronic data capture systems (“EDC”), clinical trial management systems (“CTMS”), web-based portals, systems integration, patient diary applications, and other product development tools and services. We believe that our comprehensive services, depth of therapeutic area expertise, global footprint and related access to patients, and sophisticated information technology, along with our experience in global drug development and product launch services, represent key competitive strengths.
Our services complement the research and development (“R&D”) and marketing functions of pharmaceutical, biotechnology, diagnostics, and medical device companies. Through our clinical research and product launch and commercialization services, we seek to help clients maximize the return on their significant investments in research and development by reducing the time, risk, and cost of clinical development and launch of new therapies. For large pharmaceutical and biotechnology companies, outsourcing these types of services to us provides those companies with a high-quality, variable cost alternative to the fixed costs associated with internal drug development. In addition, these large companies can benefit from our technical resource pool, broad therapeutic area expertise, other advisory services, and global infrastructure, all of which are designed to expedite parallel, multi-country clinical trials and accelerate time-to-market. For smaller bio-pharma companies, we provide access to expertise and a virtual and global network that enables them to develop their new products. Our vision is to integrate and build critical mass in the complementary businesses of clinical research, medical communications and drug development, as well as related information technology products and integration services. Our goal is to provide significant benefits to sponsor clients through this strategy, namely, a faster and less expensive development and launch process, as well as a clinical development strategy and expertise that support the marketing strategy for new medical products. We believe that the outsourcing of these services has increased in the past and should continue to increase in the future because of several factors, which are placing increased pressure on clients. These factors include the need to more tightly manage costs, capacity limitations, reductions in drug patent exclusivity periods, the desire to speed up patient recruitment and reduce development time, increased globalization of clinical trials, productivity issues, upcoming patent expirations, more stringent government regulations, and pricing pressure. With increased levels of investment continuing to be required and with development times being extended, we believe these trends will continue to create opportunities for companies like us that are focused on improving the efficiency of the bio-pharma product development process. Moreover, many of our clients are reassessing how they conduct their R&D activities and are now engaging in outsourcing at a more strategic level. One consequence of this reassessment is higher concentrations of their outsourced clinical development activities with a smaller number of providers. We have been successful in winning many strategic partnerships. We believe that our broad range of offerings, our global presence, our information technology solutions, and our expertise in clinical drug development position us well to continue to participate in these strategic partnerships.
We are one of the largest biopharmaceutical services companies in the world, based upon annual service revenue. Headquartered near Boston, Massachusetts, we operate in 80 locations and have approximately 15,560 employees throughout 51 countries around the world. We conduct business in healthcare markets around the world, including the United States (“U.S.”), Argentina, Australia, Austria, Belarus, Belgium, Bosnia, Brazil, Canada, Chile, China, Colombia, Croatia, Czech Republic, Denmark, Estonia, Finland, France, Georgia, Germany, Hungary, India, Indonesia, Ireland, Israel, Italy, Japan, Korea, Latvia, Lithuania, Malaysia, Mexico, the Netherlands, Norway, Peru, the Philippines, Poland, Romania, Russia, Serbia, Singapore, Slovakia, South Africa, Spain, Sweden, Taiwan, Thailand, Turkey, Ukraine, the United Kingdom (“U.K.”), and Vietnam. During our fiscal year ended June 30, 2014 (“Fiscal Year 2014”), we derived 47.0% of our service revenue from our U.S. operations and 53.0% from our non-U.S. operations. Breakdowns of service revenue by geographic region for previous years can be found in Note 16 to the consolidated financial statements included in Item 8 of this annual report.
1
We were incorporated in 1983 as a regulatory affairs consulting firm and are a Massachusetts corporation. Josef H. von Rickenbach, our Chairman of the Board and Chief Executive Officer, was a co-founder. Since our inception, we have executed a focused growth strategy embracing internal expansion as well as strategic acquisitions to expand or enhance our portfolio of services, geographic presence, therapeutic area knowledge, information technology capabilities, and client relationships.
We have completed two acquisitions over the past five fiscal years, including the acquisitions of HERON Group LTD (“HERON”) in April 2013, and LIQUENT Inc. (“LIQUENT”) in December 2012. The acquisition of HERON strengthened our ability to offer our clients a full spectrum of services that aid in developing products with reimbursement and market access strategies. Prior to the acquisition, HERON built one of the largest independent evidence-based consultancies and achieved a market leadership position. HERON helped to enhance the portfolio of services that we provide through our PAREXEL Consulting Services (“PC”) business, formerly known as PAREXEL Consulting and Medical Communication Services, and enhanced the expertise brought to the design and implementation of Phase III/IV studies. LIQUENT was a leading global provider of RIM solutions. By combining LIQUENT with our PAREXEL Informatics (“PI”, formerly known as Perceptive Informatics, Inc.) segment, we strengthened our capabilities by adding a regulatory information technology platform to provide our clients access to comprehensive regulatory agency submission planning, viewing, tracking, publishing, and registration management throughout the entire lifecycle of a product. The acquired expertise related solely to regulatory information management outsourcing, which has been reported within PI’s segment results, will be reported as part of our regulatory outsourcing service within the PC segment starting in July 2014.
DESCRIPTION OF BUSINESS
We provide a broad range of expertise in clinical research, medical communications, consulting, commercialization and informatics, and advanced technology services to the worldwide pharmaceutical, biotechnology, and medical device industries. We have three reporting segments: Clinical Research Services (“CRS”), PC, and PI.
CRS constitutes our core business and includes all phases of clinical research from “first-in-man” trials, where a medicinal entity is tested on human subjects for the first time, through post-marketing studies, following approval by the presiding regulatory body. CRS service offerings include clinical trials management, observational studies, patient/disease registries and post-marketing surveillance, data management and biostatistics, epidemiology and health economics/outcomes research, clinical logistics, pharmacovigilance, and clinical pharmacology, as well as related medical affairs, patient recruitment, and investigator site services.
PC provides technical expertise and advice in such areas as drug development, regulatory affairs, and good manufacturing practice (“GMP”) compliance consulting. In addition, PC provides a full spectrum of market development, product development, commercialization, and targeted communications services in support of product launch. PC consultants also identify alternatives and propose solutions to address clients’ product development, registration, and commercialization issues.
PI provides information technology solutions designed to improve the product development processes of our clients. PI’s portfolio of products and services includes ClinPhone RTSM, medical imaging services, RIM, CTMS, EDC, web-based portals, systems integration, and patient diary applications. These solutions are sold individually or in combination, as elements of an eClinical suite.
The revenue generated by each of our business segments for each of the last three fiscal years is described below under the heading for each segment. The gross profit of each segment for each of the last three fiscal years is described in Note 17 to the consolidated financial statements included in Item 8 of this annual report. We have a global infrastructure supporting our business segments and, therefore, assets are not identified by reportable segment.
CLINICAL RESEARCH SERVICES (CRS)
Our CRS business segment generated service revenue of $1,455.3 million, or 75.0%, of our consolidated service revenue in Fiscal Year 2014, $1,303.6 million, or 75.2%, of our consolidated service revenue in our fiscal year ended June 30, 2013 (“Fiscal Year 2013”), and $1,038.7 million, or 74.4%, of our consolidated service revenue in our fiscal year ended June 30, 2012 (“Fiscal Year 2012”).
CRS offers complete services for the design, initiation and management of clinical trial programs, a critical element in obtaining regulatory approval for biopharmaceutical products. We have performed services in connection with clinical trials in most therapeutic areas, including: Oncology, Cardiology, Infectious Diseases, Neurology, Allergy/Immunology, Endocrinology/Metabolism, Gastroenterology, Obstetrics/Gynecology, Orthopedics, Pediatrics, Psychiatry, Pulmonology, Rheumatology, Dermatology, Genitourinary, Ophthalmology, and Transplantation. Our multi-disciplinary clinical trials group examines a product’s existing preclinical and clinical data to design clinical trials to provide evidence of the product’s safety and efficacy.
CRS can manage many aspects of clinical trials including: study protocol design; Case Report Form (“CRF”) design; site and investigator recruitment; patient enrollment; study monitoring and data collection; data management; biostatistics and programming; report writing; medical services; project management; and clinical logistics.
2
Clinical trials and observational studies are monitored and conducted by CRS in adherence with Good Clinical Practice (“GCP”) and Good Pharmacoepidemiological Practice ("GPP"), respectively. The design of efficient CRFs, detailed operations manuals, and site monitoring by our clinical research associates seek to ensure that clinical investigators and their staff follow established study protocols. We have adopted standard operating procedures (“SOPs”) that are intended to satisfy regulatory requirements and serve as a tool for controlling and enhancing the quality of our worldwide clinical services.
Clinical trials represent one of the most expensive and time-consuming parts of the overall biopharmaceutical product development process. The information generated during these trials is critical to gaining marketing approval from the United States Food and Drug Administration (the “FDA”), the European Medicines Agency (the “EMA”), the Committee for Medicinal Products for Human Use (the “CHMP”), and other comparable regulatory agencies as well as market acceptance by clinicians, patients, and third-party payors. CRS clinical trial management services involve many phases of clinical trials, including Phases I, II, III, and IV. See “Government Regulations” below for additional information regarding processes involved in clinical trials.
Early Phase – The Early Phase business unit of CRS encompasses the early stages of clinical testing, when a product is first evaluated in humans to assess the potential safety and efficacy of the product. These tests vary from “first-in-man” to “dose-ranging” to “proof of concept” studies in Phases I and IIa of development. The Early Phase business unit of CRS offers clients a one-stop service where studies are performed in healthy volunteers as well as in patients from various disease populations. The support services include project and program management, drug development consulting, medical writing, handling of investigational products, data management, biostatistical and bioanalytical services. Our international network of Early Phase operations includes operations in Berlin, Germany; Baltimore, Maryland (U.S.); Glendale, California (U.S.); Bloemfontein, South Africa; and Harrow, U.K. A bioanalytical laboratory, which performs drug analyses in accordance with Good Laboratory Practices (“GLP”), a system of managed controls for laboratory and research organizations to ensure the consistency and reliability of results, is also located in Bloemfontein. With these locations, the Early Phase business unit offers clinical pharmacology services (including bioanalytical services) with more than 400 dedicated beds (cooperating partners not included) on three continents.
Phase II-III – The Phase II-III business unit of CRS encompasses the later stages of clinical testing. CRS assists clients with one or more of the aspects of clinical trials and observational studies described below. CRS performs both full-service and single- or multi-service projects. As a result, our involvement may range from participating in just one aspect of a clinical trial or observational study to all aspects. These services include the following, the majority of which are also provided by our Early Phase business unit:
• | Study Protocol Design – The protocol defines, among other things, the medical issues a study seeks to examine and the statistical tests that will be conducted. Accordingly, the protocol specifies the frequency and type of laboratory and clinical measures that are to be tracked and analyzed, the number of patients required to produce a statistically valid result, the period of time over which such patients must be tracked and the frequency and dosage of drug administration. |
• | CRF Design – Once the study protocol has been finalized, a paper or electronic CRF must be developed. The CRF is the critical document for collecting the necessary clinical data as dictated by the study protocol. It may change at different stages of a trial. |
• | Site and Investigator Recruitment – The product under investigation is administered to patients usually by third-party physicians, serving as independent contractors (referred to as investigators) at hospitals, clinics, or other locations, referred to as clinical sites. Medical devices are implemented or tested by investigators in similar settings. Potential investigators may be identified and solicited by the product sponsor. A significant portion of a trial’s success depends on the identification and recruitment of experienced investigators with an adequate base of patients who satisfy the requirements of the study protocol. We have access to thousands of investigators who have conducted clinical trials for us. We provide additional services at the clinical site to assist physicians and expedite the clinical research process. |
• | Patient Enrollment – The investigators, usually with our assistance, find and enroll patients suitable for the study. The speed with which trials can be completed is significantly affected by the rate at which patients are enrolled. Prospective patients are required to review information about the clinical test and the investigational product and its possible side effects, and sign an informed consent form to record their knowledge and acceptance of potential side effects. Patients also undergo a medical examination to determine whether they meet the requirements of the study protocol. Patients then receive the product under investigation or a control (for example, a placebo) and are examined by the investigator as specified by the study protocol. Investigators are responsible for administering the products to patients, as well as examining patients and conducting necessary tests. |
• | Study Monitoring and Data Collection – As patients are examined and tests are conducted in accordance with the study protocol and applicable regulatory requirements, data are recorded on CRFs, either electronically or paper-based. CRFs are transmitted electronically from study sites or collected by specially trained persons known as clinical |
3
monitors. To ensure that the CRFs are completed correctly and the study has been conducted in compliance with the protocol and regulatory requirements, we manage the sites closely over the telephone/internet and monitoring visits as needed. We offer several EDC technologies, which significantly enhance both the quality and timeliness of clinical data capture and collection while achieving significant efficiency savings. Our study monitoring and data collection services are designed to comply with the adverse events reporting guidelines and related regulatory requirements of the FDA and other relevant regulatory agencies. Approximately 90% of new trials are EDC-based.
• | Data Management – Our data management professionals provide a broad array of services to support the accurate collection, organization, validation, and analysis of clinical data. For instance, they assist in the design of CRFs and investigator training manuals to ensure that data are collected in an organized and consistent format in compliance with the study protocol and all applicable regulatory requirements. Databases are designed according to the analytical specifications of the project and the particular needs of the client. The use of scanning and imaging of the CRFs and the use of EDC technologies to gather and report clinical data expedites data exchange while minimizing data collection errors by permitting the verification of data integrity in a more timely manner. After the data is entered, the data management team performs an array of data abstraction, data review, medical coding, serious adverse event reconciliations, loading of electronic data (such as laboratory data), database verification, and editing and resolution of data problems. The data is then submitted in a format prescribed by the client. Our CRS business segment has extensive experience throughout the world in the creation of scientific databases for all phases of the drug development process, including the creation of customized databases to meet client-specific formats, integrated databases to support new drug application (“NDA”) and equivalent submissions and databases created and maintained in compliance with FDA, European, Asian and other regulatory specifications and requirements. |
• | Biostatistics and Programming – Our biostatistics professionals assist clients with all phases of drug development, including biostatistical consulting, database design, data analysis, and statistical reporting. These professionals develop and review protocols, design appropriate analysis plans, and design report formats to address the objectives of the study protocol as well as the client’s individual objectives. Working with programming staff, biostatisticians/epidemiologists perform appropriate analyses and produce tables, graphs, listings, and other applicable displays of results according to an analysis plan. Our biostatisticians/epidemiologists may also represent clients during panel hearings at the FDA and other regulatory agencies. |
• | Report Writing – A description of the study conducted, along with the statistical analysis of data collected during the trial and other clinical data are presented and summarized in a final report generated for inclusion in a regulatory document. |
• | Medical Services – Throughout the course of a development program, our physicians provide a wide range of medical research and consulting services to improve the efficiency and quality of clinical research, including medical supervision of clinical trials, medical monitoring of patient safety, review and reporting of adverse events, medical writing, and strategy and product development. Our medical services professionals also provide lifecycle drug safety services combining operational pharmacovigilance and pharmacovigilance consulting. Operational pharmacovigilance capabilities cover all phases of clinical development and drug safety for marketed products. |
• | Project Management – Throughout the entire spectrum of activities described above, our CRS segment provides project management services. These services entail providing overall leadership to our project team, acting as the main client liaison, project planning, managing progress against study goals and deliverables, budget management, progress and metrics reporting, and issue resolution. These project management services are offered on all types of studies – single-service, multi-service, or full-service. |
Peri/Post Approval Services – The Peri/Post Approval Services business unit of CRS encompasses many services listed within the Phase II-III business unit description. However, the Peri/Post Approval Services business unit also offers a range of additional capabilities to support lifecycle management activities:
• | Observational Research – Observational research encompasses several study designs in which groups of patients are observed within routine clinical practices. We help our clients define the needs of their target audience(s) and to develop the best study design to meet their research objectives; define the optimal regulatory authority and ethics committee submission strategies in each country or region; and implement a study management and resourcing model trained to collect the required data in the most efficient manner and to agreed-upon standards of quality. |
• | Patient-reported Outcomes – Patient reported outcome measures were developed to evaluate the impact of disease and interventions on emotional, social and physical functioning from the patient's perspective. We help our clients by advising them on the multiple variations of available tools and their appropriateness, acceptability, interpretability, precision, reliability, validity and responsiveness. |
4
• | Pharmacovigilance – Our patient safety services have been specifically designed to help bio/pharmaceutical companies meet increasing post-marketing pharmacovigilance obligations. We have established global infrastructure in drug safety management, extensive safety consulting expertise and leading capabilities in post marketing authorization safety studies enables us to offer leading post-marketing pharmacovigilance services. |
• | Market Access – We have the global infrastructure, multi-disciplinary expertise, and integrated technologies to help clients implement a market strategy that will quickly build brand value and establish a strong market position - all while continuing to monitor their new product’s long-term effects and manage related safety data and other regulatory information. |
Clinical Logistics Services – The Clinical Logistics Services business unit of CRS encompasses a full range of clinical logistics services associated with the clinical trials we conduct, that include coordinating investigational drug supply manufacturing, managing import/export requirements, labeling, warehousing, distribution, and inventory control (including the return and destruction of unused trial medication, laboratory services, and ancillary supplies).
PAREXEL CONSULTING SERVICES (PC)
Service revenue from the PC business segment represented $216.2 million, or 11.1% of consolidated service revenue in Fiscal Year 2014, $202.5 million, or 11.7% of consolidated service revenue in Fiscal Year 2013, and $167.1 million, or 12.0% of consolidated service revenue in Fiscal Year 2012.
We conduct our PC operations through five groups:
• | Integrated Product Development Consulting – Our Integrated Product Development (“IPD”) consulting group provides comprehensive product development and regulatory consulting services for pharmaceutical, biotechnology, and medical device companies in major jurisdictions in the U.S., Europe, Japan and emerging markets in Asia, the Middle East, North and South Africa, and Latin America. These services include drug and device development and regulatory strategy design, scientific and technical evaluation, writing and review services, expert liaison with the FDA, EMA and PMDA, and the preparation, review and submission of regulatory applications (both for clinical trials and for marketing authorizations) to regulatory authorities in more than 75 countries. Our IPD consulting group works closely with clients to design product development and regulatory strategies and comprehensive registration programs. Our product development and regulatory experts include individuals who have joined us from the biopharmaceutical industry and from regulatory agencies such as the FDA in the U.S. and agencies in the U.K., Germany, The Netherlands, Sweden, South Korea and France. Our experts review existing published literature and regulatory precedents, evaluate the client's scientific and technical data of a product (Non-Clinical, Clinical, Chemistry, Manufacturing and Controls (“CMC”) and Regulatory) based on their individual and collective expertise and experience, assess the competitive and regulatory environments in specific relation to our clients’ products and business goals, identify deficiencies in client product documentation (“gap analysis”), and define the steps necessary to obtain regulatory approvals in the most expeditious manner. Through these services, we help our clients obtain regulatory approval for particular products or product lines in markets around the world. |
• | Strategic Compliance Consulting – Our Strategic Compliance Consulting group offers a range of specialized clinical and manufacturing compliance consulting services designed to help pharmaceutical, biotechnology, and medical device companies achieve and maintain regulatory compliance, product quality, and process excellence. These services include clinical and manufacturing compliance strategy, assistance with addressing regulatory agency enforcement issues, risk management, GCP, GLP, Good Tissue Practice and current GMP audits, consent decrees, pre-approval inspection readiness, process optimization, organizational alignment, and training. Our Strategic Compliance group offers its clients experienced regulatory and industry professionals – formerly from the FDA or from the quality departments of major biotech, pharmaceutical, and medical device companies. |
• | Medical Communications Services – Our Medical Communications Services (“MedCom”) group assists biopharmaceutical clients in their efforts to achieve optimal market penetration for their products worldwide through expert medical communications and publications services. MedCom utilizes its expertise in strategic consultancy, market and competitive landscaping, publications planning, scientific writing, managed markets, and regulatory compliance to provide effective and compliant scientific communications to a diverse audience of provider, payer, and patient advocacy group stakeholders. An integrated communications plan can detail external and internal strategies, including communications objectives, target audiences, communications priorities and timing, key messages, key meetings and events, and target publications and media. MedCom supports marketing communication objectives across a broad spectrum of media from publications through interactive technologies. Other services include planning of meetings and exhibits in premier scientific conferences and symposia. |
• | Commercialization Consulting Services – Our Commercialization Consulting Services group provides commercialization strategies and deliverables that assist clients in understanding how changing marketplace dynamics |
5
may impact product development, product reimbursement, patient access and commercial success. We identify, gather, analyze, and communicate data that is critical to maximizing product value and commercial success. Our service lines include strategic market access planning, systematic reviews for evidence development, economic modeling and evaluation, pricing, reimbursement strategies, global value dossier writing, and engagement with Health Technology Assessment authorities. The acquisition of HERON has strengthened our ability to offer our clients a full spectrum of services that aid in developing products through reimbursement and market access strategies. We help our clients better prepare their products for the market, better prepare the market for their products and demonstrate product value in the marketplace.
• | Regulatory Outsourcing Services - Our Regulatory Outsourcing Services group combines enabling technology, operational expertise and global regulatory intelligence to deliver high-quality cost-effective regulatory affairs and regulatory operations solutions as a functional outsourced service. These services include both pre-approval and post-approval activities ranging from compilation, publishing and dispatching of large regulatory dossiers to the authoring of routine product variations, annual reports and other product lifecycle maintenance tasks. These services are used by our clients to provide a flexible outsourcing model that yields predictable year-over-year savings and addresses regulatory complexities, increased workloads, and limited budgets facing the industry. |
PAREXEL INFORMATICS (PI)
Service revenue from our PI business represented $267.9 million, or 13.9% of consolidated service revenue, in Fiscal Year 2014, $228.3 million, or 13.1% of consolidated service revenue, in Fiscal Year 2013, and $190.7 million, or 13.6% of consolidated service revenue in Fiscal Year 2012.
We conduct our PI operations through seven groups:
• | ClinPhone Randomization & Trial Supply Management (ClinPhone RTSM) – PI provides automated randomization and logistics management through its ClinPhone RTSM solutions. Our services include both Interactive Voice Response (“IVR”) and Interactive Web Response (“IWR”) technologies. The ClinPhone RTSM solutions are used in clinical trials to achieve treatment group balance, eliminate selection bias, and limit the predictability of treatment allocations, all of which are designed to comply with the latest regulatory requirements. ClinPhone RTSM allows effective real-time implementation of randomization algorithm modifications required for adaptive trial designs. |
• | Medical Imaging Services – PI offers products and services that allow our clients to apply and manage medical imaging in clinical trials. Clinical study sponsors increasingly rely on imaging as a surrogate endpoint in support of efficacy and safety. Our therapeutic and imaging experts provide a range of capabilities in the application of imaging techniques from early clinical development through peri-approval studies. These services include: |
◦ | Standardization of imaging and image management at investigative sites |
◦ | Image collection at a central location |
◦ | Development of independent review charters for review and approval by regulatory authorities |
◦ | Employing directly or subcontracting independent reviewers and training these reviewers on the assessment criteria and reviewer roles and responsibilities |
◦ | Management of the logistical processes involved in the independent review |
• | Platform Solutions (PS) - PS provides leading solutions consulting and services to integrate systems and processes to help companies simplify the concurrent use of the multiple technologies involved in clinical trials all with the purpose of giving clients better visibility and faster access to their data. We use a range of technologies including our Clinical Technology Integration Platform, which is a proprietary environment designed to facilitate seamless two-way exchange of data across different systems via reliable and repeatable integrations, our proprietary user environment which includes sophisticated reporting, analytics and visualizations, and our security technologies to manage user access to systems. Platform Solutions’ services are delivered by our dedicated experts who have an in-depth understanding of advanced technologies, clinical development processes and validated system integrations. |
• | Clinical Trial Management System (CTMS) – We offer CTMS solutions to assist biopharmaceutical companies with the complex process of planning and managing clinical trials. Our IMPACT® solution provides established global pharmaceutical companies and service organizations with flexible options that include hosted or on-premise solutions. |
• | Electronic Data Capture (EDC) – DataLabs® EDC is one of the industry’s first single data management systems that unifies the functionality of paper data entry (PDE) with the flexibility of electronic data capture (EDC). DataLabs EDC is able to combine data collected on paper with data collected electronically into one easy-to-use electronic clinical data management platform. The collected information feeds into a single database providing clients with fully |
6
integrated data. With DataLabs EDC, users are able to design a study, collect data using either method and then clean and manage that data using a single system.
• | Electronic Patient Reported Outcomes (ePRO) – Patient self-reported data is increasingly playing a key part in efficacy and quality of life assessment, patient recruitment, symptom and safety information and medical compliance monitoring. Our ePRO solutions provide the flexibility to choose among the most commonly used ePRO methods, IVR, Web, personal digital assistant (“PDA”), and computer tablet (“Tablet”): |
◦ | IVR (Interactive Voice Response) – Our IVR platform enables ePRO delivery using the subject’s own telephone, making it highly cost-effective and simple to deploy; |
◦ | Web/IWR (Interactive Web Response) – The Web offers all of the advantages and benefits of IVR as subjects use any desktop or laptop connected to the Internet to securely access the ePRO application; and |
◦ | PDA/Tablet – Depending on the specific characteristics of the protocol, a device-based ePRO solution may be best suited for a study. |
• | LIQUENT Regulatory Information Management –We offer software and professional service solutions designed to support the regulatory business processes of our life science clients. Our product suite, LIQUENT InSight®, is an end-to-end, integrated RIM platform. LIQUENT InSight provides our clients with regulatory submission planning, publishing, viewing and registration management capabilities necessary to get their products to market and effectively maintain them throughout their lifespan. We also provide a full complement of flexible regulatory affairs consulting and regulatory operational outsourcing services to help our clients meet the demands of a dynamic regulatory landscape. |
INFORMATION TECHNOLOGY
We are committed to investing in information technology designed to help us to provide high quality services, competitive and cost-effective client-facing solutions, and well-managed internal resources. We have built our solutions by developing proprietary technology as well as purchasing and integrating commercially available technology that address critical aspects of our business. The proprietary technology we use supports project proposals/budget generation, time information management, revenue and resource forecasting, clinical data entry and management, clinical trial management, project management, quality management, and procurement/expense processing.
We maintain an internal information technology group that is responsible for technological planning, applications development, program management, technical operations, and management of our worldwide computer infrastructure and voice and data networks. Our information systems are designed to support and reinforce all of our policies and procedures while enabling us to respond to the multiple needs of our different clients and regulatory systems. Our systems also enable us to respond quickly to client inquiries regarding progress on projects and, in some cases, to gain direct access to client data on client owned systems.
SALES AND MARKETING
Our sales and marketing personnel carry out our global business development activities. In addition to significant selling experience, most of these individuals have technical and/or scientific backgrounds. Our senior executives and project team leaders also participate in maintaining key client relationships and engaging in business development activities.
Each of our three reporting segments has a business development team that focuses on its particular market segment. In many cases, however, the reporting segment selling teams work together in order to provide clients with the most appropriate service offering to meet their needs. Moreover, we have developed strategic account management teams to provide clients with a single point of contact and to facilitate cross-selling opportunities.
Each business development employee is generally responsible for a specific client segment or group of clients and for strengthening and expanding an effective relationship with that client. Each individual is responsible for developing his or her client base on our behalf, responding to client requests for information, developing and defending proposals, and making presentations to clients.
Our business development group is supported by our marketing team. Our marketing activities consist primarily of market information development and analysis, strategic planning, competitive analysis, brand management, collateral development, participation in industry conferences, advertising, e-marketing, publications, and website development and maintenance. The marketing team focuses both on supporting the individual business development teams for their specific market segments as well as promoting an integrated marketing strategy and communications plan for PAREXEL as a whole.
CLIENTS
We have in the past derived, and may in the future derive, a significant portion of our service revenue from both an individual client and a core group of clients. Concentrations of business in the biopharmaceutical services industry are common and we
7
expect to continue to experience such concentration in future years due to our increasing number of strategic partnerships. Our five largest clients accounted for 47%, 50% and 40.5% of our consolidated service revenue in aggregate for Fiscal Year 2014, Fiscal Year 2013, and Fiscal Year 2012, respectively. For Fiscal Year 2014, two clients, Pfizer Inc. (“Pfizer”) and Merck & Co., Inc. (“Merck”), individually accounted for 16% and 11% of our consolidated service revenue, respectively. For Fiscal Year 2013, Pfizer and Merck individually accounted for 17% and 12% of our consolidated service revenue, respectively. No single client accounted for 10% or more of our consolidated service revenue in Fiscal Year 2012.
BACKLOG
Backlog represents anticipated service revenue from work not yet completed or performed under signed contracts, letters of intent, and pre-contract commitments that are supported by written communications. Once work commences, revenue is generally recognized over the life of the contract as services are provided. Backlog at June 30, 2014 was approximately $5.01 billion, compared with approximately $4.61 billion at June 30, 2013, an increase of 8.6%. Subject to the matters addressed in the following paragraph, we anticipate that approximately $2.1 billion of the backlog will be recognized as revenue in our fiscal year ending June 30, 2015 (“Fiscal Year 2015”).
We believe that our backlog as of any date is not necessarily a meaningful predictor of future results. Projects included in backlog are subject to cancellation, revision, or delay. As detailed more fully in the “Risk Factors” section of this annual report, clients terminate, delay, or change the scope of projects for a variety of reasons including, among others, the failure of products being tested to satisfy safety requirements, unexpected or undesirable clinical results of the product, client decisions to forego a particular study, insufficient patient enrollment or investigator recruitment, or production problems resulting in shortages of the drug. Additionally, we have been entering into an increasing number of strategic partnerships. As a result, any delay or cancellation related to these partnerships could significantly impact the conversion of backlog into revenue. Generally, our contracts can be terminated upon thirty to sixty days notice by the client.
COMPETITION
We compete with other biopharmaceutical outsourcing services companies and other clinical research organizations (“CROs”) that provide one or more of the services currently being offered by us. Some of the large biopharmaceutical services companies, such as Quintiles Transnational Corporation, Covance Inc., Pharmaceutical Product Development Inc., inVentiv Health, INC Research, and Icon plc, offer services that compete directly with our services at many levels.
We believe that the synergies arising from integrating the products and services offered by our different business units, coupled with our global infrastructure (and resulting rapid access to diverse patient populations and markets), technology products and services, and depth of expertise and experience differentiate us from our competitors. Although there are no guarantees that we will continue to do so, we believe that we compete favorably in all of our business areas and segments, as more fully described below.
CRS
The clinical outsourcing services industry is very fragmented, with several hundred providers offering varying levels of service, skills, and capabilities. Our CRS group primarily competes against in-house departments of pharmaceutical companies, other full service biopharmaceutical outsourcing services companies, small specialty CROs, and to a small extent, universities, teaching hospitals, and other site organizations. The primary competitors for our CRS business include Quintiles Transnational Corporation, Covance Inc., Pharmaceutical Product Development Inc., and Icon plc.
CRS generally competes on the basis of:
• | a broad international presence with strategically located facilities and access to end markets; |
• | the ability to organize and manage large-scale clinical trials on a global basis; |
• | the ability to recruit investigators and patients expeditiously; |
• | medical and scientific expertise in a specific therapeutic area; |
• | quality of services; |
• | breadth of services; |
• | the ability to integrate information technology with systems to improve the efficiency of clinical research; |
• | previous experience with a client or a specific therapeutic area; |
• | the ability to manage large and complex medical databases; |
• | the ability to provide statistical and regulatory services; |
• | financial strength and stability; and |
8
• | price. |
We believe that the key competitive strengths of our CRS business are its global footprint and related rapid access to diverse patient populations, therapeutic expertise, technological expertise and its experience in global drug development.
PC
Our PC segment competes with a large and diverse group of specialty service providers, including major consulting firms with pharmaceutical industry practices, large and small biopharmaceutical services companies, individual consultants, specialty medical communications services companies, and medical communication subsidiaries of large international advertising companies.
We believe that a central differentiator of our PC service offering is our combination of scientific, regulatory and commercialization expertise. We consider PC’s key competitive strengths to include a combination of deep global expertise in early and late stage drug development, regulatory strategy and submissions, GMP compliance, reimbursement and market access consulting, and global commercialization and communications strategies. We believe that this broad range of capabilities enables us to help our clients achieve their regulatory and marketing objectives.
PI
Our PI business competes primarily with biopharmaceutical services companies, information technology companies, and software companies. Companies in this segment compete based on the strength and usability of their technology offerings, their expertise and experience, and their understanding of the clinical development process. PI’s key competitive strength is its combination of technological expertise and knowledge of clinical development. Additionally, PI’s offerings provide substantial synergies to our customers and CRS services.
INTELLECTUAL PROPERTY
Our trademark “PAREXEL®” is of material importance to us. This and other trademarks have been registered in the U.S. and many foreign countries. The duration of trademark registrations varies from country to country. However, trademarks generally may be renewed indefinitely as long as they are in use and/or their registrations are properly maintained, and as long as they have not been found to have become generic.
EMPLOYEES
As of June 30, 2014, we had approximately 15,560 full-time equivalent employees. Approximately 27% of our employees are located in the United States and approximately 73% are located internationally. We believe that we have good relationships with our employees.
The success of our business depends upon our ability to attract and retain qualified professional, scientific, and technical staff. The level of competition among employers in the U.S. and overseas for skilled personnel, particularly those with Ph.D., M.D., or equivalent degrees, is high. We believe that our name recognition and our multinational presence, which allows for international transfers by employees, are an advantage in attracting qualified candidates. In addition, we believe that the wide range of clinical trials in which we participate allows us to offer broad experience to clinical researchers.
GOVERNMENT REGULATIONS
We provide clinical trial services and diverse consulting solutions to the pharmaceutical, biotechnology, and medical device industries worldwide. Lack of success in obtaining approval for the conduct of clinical trials in the countries in which we manage clinical trials on behalf of our clients can adversely affect us. We make no guarantees to our clients with regard to successful outcomes of the regulatory process, including the success of clinical trial applications or marketing authorization applications.
The clinical research services we provided in the U.S. are subject to established and evolving FDA regulations. We are obligated to comply with FDA requirements governing activities such as obtaining Institutional Review Board (“IRB”) approval and patient informed consents, verifying qualifications of investigators, reporting patients’ adverse reactions to products, and maintaining thorough and accurate records. We are also required to ensure that the computer systems we use to process human data from clinical trials are validated in accordance with the FDA’s electronic records regulations, 21 Code of Federal Regulations Part 11, which apply to the pharmaceutical and CRO industries when companies choose to use electronic records in lieu of paper records or electronic signatures in lieu of traditional signatures. We must maintain source documents for each study for specified periods, and such documents may be reviewed according to GCP or GPP standards by the study sponsors and the FDA and other regulatory agencies (for example the EMA and the Japanese Pharmaceutical and Medical Devices Agency) during audits and inspections. Non-compliance with GCP can result in the disqualification of data collected during a clinical study and in non-approval or non-clearance of a product application submitted to the FDA or other regulatory agencies around the world.
9
The clinical investigation of new drugs, biologics, and medical devices is highly regulated by government agencies around the world. The standard for the conduct of clinical research and development studies is embodied in GCP, a set of international standards and guidelines, which stipulate procedures designed to ensure the quality and integrity of data obtained from clinical testing, and to protect the rights and safety of clinical trial participants. The FDA and many other regulatory authorities require that study results submitted to such authorities be based on studies conducted in compliance with GCP. The European Union (“EU”) enacted the Clinical Trials Directive (the “Directive”) in 2004 in attempt to harmonize the requirements of the members of the EU regarding the conduct of clinical trials. The EU released in April 2014 a new legislation to repeal the Directive in a stepwise approach between mid-2016 and 2019 to increase the attractiveness for the conduct of clinical trials in its territory by reducing the bureaucratic burden. The regulation will not require adoption by each member country thus ensuring greater harmonization. The Directive requires sponsors of clinical trials to submit formal applications to national ethics committees and regulatory authorities prior to the initiation of clinical trials in any of the 28 Member States of the EU, and the new legislation will require submission through a portal at the EMA. The regulatory landscape in Asia and Latin America is heterogeneous, with each country independently enforcing its unique regulatory policies. This has improved with more cooperation between countries and a deeper awareness of regional and global differences in regulatory policies and practice, especially in Asia. As in the United States, clinical trials in the EU are expected to be carried out in compliance with detailed requirements for GCP. The international regulatory approval process, in the EU as well as many other countries, includes all of the risks and potential delays associated with the FDA approval process.
Because the FDA’s regulatory requirements have served as the model for much of the regulation of new drug development worldwide, regulatory requirements similar to those of the FDA exist in the other countries in which we operate. Our regulatory capabilities include knowledge of the specific regulatory requirements of numerous countries. For more than ten years, we have managed successful regulatory submissions for life science companies around the world. Beginning in 1990, the FDA and corresponding regulatory agencies of the EU and Japan commenced discussions to develop harmonized standards for preclinical and clinical studies and the format and content of applications for new drug approvals through a process known as the International Conference on Harmonisation (“ICH”) of Technical Requirements for Registration of Pharmaceuticals for Human use. Data from multinational studies adhering to GCP are now generally acceptable to the FDA and regulators in Australia, Canada, the EU, Japan and Latin American countries, although there can be no advance assurance that the submission of such data to any regulatory authority will result in regulatory approval for marketing of the product. The ICH process has sanctioned a single common format for drug and biologic marketing authorization applications, known as the Common Technical Document (“CTD”) in the U.S., Europe, Japan and Canada. On July 1, 2003 the CTD format became mandatory in Europe and Japan and highly recommended by the FDA in the U.S. and by the Canadian regulatory authorities. We have developed the expertise to prepare CTDs for our clients in both paper and electronic form.
REGULATION OF DRUGS AND BIOLOGICS
Before a new drug or biologic may be approved and marketed, the drug or biologic must undergo extensive testing and regulatory review in order to determine that the drug or biologic is safe and effective. It is not possible to estimate the time in which preclinical and Phase I, II and III studies will be completed with respect to a given product, although the time period may last many years. Using the U.S. regulatory environment as an example, the stages of this development process are generally as follows:
Preclinical Research (approximately 1 to 3.5 years) – In vitro (“test tube”) and animal studies must be conducted in accordance with GLP to establish the relative toxicity of the drug or biologic over a wide range of doses and to detect any potential to cause a variety of adverse conditions or diseases, including birth defects or cancer. If results warrant continuing development of the drug or biologic, the results of the studies are submitted to the FDA by the manufacturer as part of an Investigational New Drug Application (“IND”), which must be submitted to the FDA before proposed clinical testing can begin. An IND must include, among other things, preclinical data, chemistry, manufacturing and control information, and an investigational plan, and must become effective before such trials may begin. An IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions relating to one or more proposed clinical trials. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. In addition, clinical trials cannot begin at a particular study site until approved by the site's Institutional Review Board ("IRB"), which is an independent expert body charged with protecting patient safety and privacy. As a result, there can be no assurance that submission of an IND will result in the ability to commence clinical trials.
Clinical Trials (approximately 3.5 to 6 years)
Phase I consists of basic safety and pharmacology testing in approximately 20 to 80 human subjects, usually healthy volunteers or stable patients, and includes studies to evaluate the metabolic and pharmacologic action of the product in humans, how the drug or biologic works, how it is affected by other drugs, how it is tolerated and absorbed, where it goes in the body, how long it remains active, and how it is broken down and eliminated from the body.
10
Phase II includes basic efficacy (effectiveness) and dose-range testing in a limited patient population (usually 100 to 200 patients) afflicted with a specific disease or condition for which the product is intended for use, further safety testing, evaluation of effectiveness, and determination of optimal dose levels, dose schedules, and routes of administration. If Phase II studies yield satisfactory results and no hold is placed on further studies by the FDA, Phase III studies can commence.
Phase III includes larger scale, multi-center, comparative clinical trials conducted with patients afflicted by a target disease, in order to provide enough data for a valid statistical test of safety and efficacy required by the FDA and others, and to provide an adequate basis for product labeling.
The FDA receives reports on the progress of each phase of clinical testing and may require the modification, suspension, or termination of clinical trials if, among other things, an unreasonable risk is presented to patients or if the design of the trial is insufficient to meet its stated objective. In addition, information about certain clinical trials must be submitted to, and made available to the public on, the government website www.clinicaltrials.gov.
NDA or Biologic License Application (“BLA”) Preparation and Submission – Upon completion of Phase III trials, the sponsor assembles the statistically analyzed data from all phases of development, along with the chemistry and manufacturing and pre-clinical data and the proposed labeling, among other things, into a single large document, the NDA or BLA in CTD format as of July 1, 2003, which today comprises, on average, roughly 100,000 pages. Typically, an NDA or BLA must be accompanied by payment of a statutory fee.
FDA Review of NDA or BLA – The FDA carefully scrutinizes data from all phases of development to confirm that the manufacturer has complied with regulations and that the drug or biologic is safe and effective for the specific use (or “indication”) under study. The FDA may refuse to accept the NDA or BLA for filing and substantive review if certain administrative and content criteria are not satisfied. Even after accepting the submission for review, the FDA may also require additional testing or information before approval of an NDA or BLA. The FDA must deny approval of an NDA or BLA if applicable regulatory requirements are not satisfied.
Post-Marketing Surveillance and Phase IV Studies – Regulatory authorities require companies to collect and periodically report additional safety and, where appropriate, benefit data on the drug or biologic for as long as the manufacturer markets the product. The FDA and other major regulatory agencies require sponsor companies to prepare risk management plans as part of their approval requirements for marketed drugs and biologics, aimed at monitoring areas of drug risk and implementing plans for minimizing their occurrence. In addition, global agencies may impose additional post-marketing study requirements as a condition of a product’s approval to confirm the safety profile or verify clinical benefit in the “real-world” clinical setting. Regulated post marketing surveillance studies are imposed by a number of national agencies for all new products launched into their market and the data used to re-evaluate the continuation of the product license. Product approval may be withdrawn if compliance with regulatory requirements is not maintained or if a significant safety issue is identified that brings into question the clinical benefit relative to the emerging risks.
REGULATION OF MEDICAL DEVICES
Unless a medical device is exempted from pre-market approval or clearance requirements, which are described below, or is eligible for de novo review, FDA approval or clearance of the device is required before the product may be marketed in the United States. In order to obtain pre-market clearance for marketing, a manufacturer must demonstrate substantial equivalence to a similar legally marketed product by submitting a pre-market notification, or 510(k), to the FDA. The FDA may require preclinical and clinical data to support a substantial equivalence determination, and there can be no assurance the FDA will find a device substantially equivalent. Clinical trials can take extended periods of time to complete. In addition, if the FDA requires an approved Investigational Device Exemption (“IDE”) before clinical device trials may commence, there can be no guarantee that the agency will approve the IDE. The IDE approval process could also result in significant delays.
After submission of a pre-market notification containing, among other things, any data collected, the FDA may find the device substantially equivalent and the device may be marketed. After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, requires a new 510(k) clearance or could require approval of a pre-market approval application (“PMA”). If the FDA finds that a device is not substantially equivalent, the manufacturer may request that the FDA make a risk-based classification to place the device in Class I or Class II. However, if a timely request for risk-based classification is not made, or if the FDA determines that a Class III designation is appropriate, a PMA will be required before the device may be marketed.
If there is no legally marketed predicate device, a manufacturer can seek to have a device classified into Class I or Class II through the de novo review process. As a result of statutory revisions made in 2012, the de novo process can be used without first going through the 510(k) process.
The PMA approval process is lengthy, expensive, and typically requires, among other things, extensive data from preclinical testing and a well-controlled clinical trial or trials that demonstrate a reasonable assurance of safety and effectiveness. There can be no assurance that review will result in timely, or any, PMA approval. There may also be significant conditions associated
11
with the approval, including limitations on labeling and advertising claims and the imposition of post-market testing, tracking, or surveillance requirements. Even after approval, a new PMA or PMA supplement is required in the event of a modification to the device, its labeling or its manufacturing process. Typically, a PMA or 510(k) must be accompanied by payment of a statutory fee. In addition, information about certain clinical trials must be submitted to the government website www.clinicaltrials.gov, where it may be made publicly available.
REGULATION OF PATIENT INFORMATION
The confidentiality, security, use and disclosure of patient-specific information are subject to governmental regulation. Regulations to protect the safety and privacy of human subjects who participate in or whose data are used in clinical research generally require clinical investigators to obtain legally effective informed consent from identifiable research subjects before research is undertaken. Under the Health Insurance Portability and Accountability Act of 1996 ("HIPAA"), as amended by the Health Information Technology for Economics and Clinical Health (“HITECH”) Act of 2009, the U.S. Department of Health and Human Services has issued regulations mandating privacy and data security standards and breach notification requirements for certain types of individually identifiable health information, or protected health information, when used or disclosed by health care providers and other HIPAA-covered entities or business associates that provide services to or perform functions on behalf of these covered entities. HIPAA regulations generally require individuals’ written authorization before identifiable health information may be used for research, in addition to any required informed consent. HIPAA regulations also specify standards for de-identifying health information so that information can be handled outside of the HIPAA requirements and for creating limited data sets that can be used for research purposes under less stringent HIPAA restrictions.
Outside of the United States, many countries have enacted laws to safeguard the privacy and security of personal information, including individually identifiable health information. The member states of the European Union have adopted a rigorous system of data protection regulations, based upon a framework imposed by the 1995 European Commission Directive on Data Protection. These rules provide broad protections for personal information, including, among other things, notice requirements, limits on the scope and duration for which personal information may be maintained and processed, restrictions on disclosures of personal information, standards for providing individuals with control over the manner in which personal information is processed, and restrictions on transfers of such data to the United States and other countries that the European Union finds to lack “adequate” data protection laws of their own. Health-related information is recognized as a special, sensitive category of personal information, which may generally be processed only pursuant to the affirmative, or opt-in, consent of the individual to whom the information pertains. Violations of these data protection regulations are subject to administrative penalties, civil money penalties, and criminal prosecution, including corporate fines and personal liability.
In order to comply with these laws and regulations, we must maintain internal compliance policies and procedures, and we may need to implement new privacy and security measures, which may require us to make substantial expenditures or cause us to limit the products and services we offer. In addition, if we violate applicable laws, regulations, contractual commitments, or other duties relating to the use, privacy or security of health information, we could be subject to civil liability or criminal penalties and it may be necessary to modify our business practices.
REGULATION OF HEALTH INDUSTRY ARRANGEMENTS
Since the U.S. Medicare program will pay for certain costs of qualifying clinical drug trials, as well as certain reasonable and necessary items and services used to diagnose and treat complications arising from participation in clinical trials, the conduct of such trials may be subject to laws and regulations that are intended to prevent misuse of such government health care program funding. In the U.S., these laws include, among others, the False Claims Act, which prohibits submitting or causing the submission of false statements or improper claims for government health care program payments; the so-called Stark physician self-referral law, which prohibits physicians from referring or billing for certain designated health services performed or provided by an entity in which the physician or an immediate family member has a financial compensation, investment, or ownership interest; and the health care anti-kickback law, which prohibits paying, offering to pay, or receiving payment in exchange for the referral of services or devices that are covered under a federal health care program, and which therefore restrict the permissibility of financial and promotional arrangements with patients, physicians, investigators, and study sites, such as, for example, financial incentives for physicians to enroll study participants or for patients, investigators or study sites to participate in a trial. Violations of these restrictions are subject to potentially severe administrative, civil and criminal penalties that could have a substantial and material adverse effect on our business, our reputation, and our continued ability to offer our biopharmaceutical outsourcing services.
POTENTIAL LIABILITIES AND INSURANCE
Our clinical research services focus on the testing of experimental drugs and devices on human volunteers pursuant to study protocols and in accordance with laws and regulations which govern clinical trials. Clinical research involves a risk of liability for personal injury or death to patients due to, among other reasons, possible unforeseen adverse side effects or improper administration of the new drug or medical device. We do not generally provide health care services directly to patients. Rather,
12
our physicians or third party physician investigators are responsible for administering drugs and evaluating the study patients. Many of the patients enrolled in clinical trials are already seriously ill and are at risk of further illness or death.
We believe that the risk of liability to patients in clinical trials is mitigated by various regulatory requirements, including the role of IRBs, the need to obtain each patient’s informed consent, and the oversight by applicable regulatory authorities. The FDA, the Medicines and Healthcare products Regulatory Agency in the U.K., and regulatory authorities in other countries require each human clinical trial to be reviewed and approved by the IRB at each study site. An IRB is an independent ethics committee that includes both medical and non-medical personnel and is obligated to protect the interests of patients enrolled in the trial. The IRB approves and monitors the protocol and the measures designed to protect patients, such as the requirement to obtain informed consents.
To reduce our potential liability, we generally seek to incorporate indemnity provisions into our contracts with clients to protect us from liability for adverse reactions to the study drug as well as any negligent acts by the study sponsor and/or third party physician investigators. These indemnity provisions do not, however, protect us against certain of our own actions, such as those involving negligence. Moreover, these indemnities are contractual arrangements that are subject to negotiation with individual clients, and the terms and scope of such indemnities can vary from client to client and from study to study. Finally, the financial performance of these indemnities is not secured; therefore we bear the risk that an indemnifying party may not have the financial ability to fulfill its indemnification obligations. We could be materially and adversely affected if we were required to pay damages or incur defense costs in connection with an uninsured claim that is outside the scope of an indemnity or where an indemnification obligation, although applicable, is not performed in accordance with its terms.
We currently maintain a portfolio of insurance coverage, including a professional liability insurance policy, subject to deductibles and coverage limits. There can be no assurance that this insurance coverage will be adequate, or that insurance coverage will continue to be available on terms acceptable to PAREXEL.
AVAILABLE INFORMATION
Our Internet website is http://www.parexel.com. We make available through this website our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed pursuant to Sections 13(a) and 15(d) of the Securities Exchange Act of 1934 (the “Exchange Act”). We make these reports available free of charge through our website as soon as reasonably practicable after they have been electronically filed with, or furnished to, the Securities and Exchange Commission (“SEC”). Any materials we file with the SEC may also be read and copied at the SEC’s public reference room located at 100 F Street, N.E. Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for further information on the public reference room. Our SEC filings are also available to the public on the SEC’s website at www.sec.gov.
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K and, in particular, the description of our Business set forth in Part I, Item 1 and our Management’s Discussion and Analysis of Financial Condition and Results of Operations set forth in Part II, Item 7 (“MD&A”) contain or incorporate a number of forward-looking statements within the meaning of Section 27A of the Securities Act of 1933 and Section 21E of the Exchange Act.
Any statements contained in or incorporated by reference into this report that are not statements of historical fact should be considered forward-looking statements. You can identify these forward-looking statements by use of the words “believes,” “expects,” “anticipates,” “plans,” “may,” “will,” “would,” “intends,” “estimates”, and other similar expressions, whether in the negative or affirmative. These forward-looking statements are based on current expectations, estimates, forecasts and projections about the industry and markets in which we operate and management’s beliefs and assumptions and should be read in conjunction with our MD&A and our consolidated financial statements and notes to consolidated financial statements. We cannot guarantee that we actually will achieve the plans, intentions or expectations disclosed in the forward-looking statements made. There are a number of important risks and uncertainties that could cause our actual results to differ materially from those indicated by such forward-looking statements. These risks and uncertainties include, without limitation, those set forth below under the heading “Risk Factors” as well as risks that emerge from time to time that are not possible for us to predict. Forward-looking statements, like all statements in this report, speak only as of the date of this report (unless another date is indicated). We disclaim any obligation to update publicly any forward-looking statements whether as a result of new information, future events or otherwise.
13
ITEM 1A. RISK FACTORS
In addition to other information in this report, the following risk factors should be considered carefully in evaluating our company and our business. Additional risks not currently known to us or other factors not perceived by us to present significant risk to our business at this time also may impair our business operations.
Risks Associated with our Business and Operations
The loss, modification, or delay of large or multiple contracts may negatively impact our financial performance.
Our clients generally can terminate their contracts with us upon 30 to 60 days' notice or can delay the execution of services. The loss or delay of a large contract or the loss or delay of multiple contracts could adversely affect our operating results, possibly materially. We have in the past experienced large contract cancellations and delays, which have adversely affected our operating results. The loss of a strategic partner could potentially have a material adverse effect on our business and financial statements.
Clients may terminate or delay their contracts for a variety of reasons, including:
• | failure of products being tested to satisfy safety requirements; |
• | failure of products being tested to satisfy efficacy criteria; |
• | products having unexpected or undesired clinical results; |
• | client cost reductions as a result of budgetary limits or changing priorities; |
• | client decisions to forego a particular study, perhaps for economic reasons; |
• | merger or potential merger related activities involving the client; |
• | insufficient patient enrollment in a study; |
• | insufficient investigator recruitment; |
• | clinical drug manufacturing problems resulting in shortages of the product; |
• | product withdrawal following market launch; and |
• | shut down of manufacturing facilities. |
An unfavorable economic environment may negatively impact our financial performance as a result of client defaults and other factors.
Our ability to attract and retain clients, invest in and grow our business and meet our financial obligations depends on our operating and financial performance, which, in turn, is subject to numerous factors. In addition to factors specific to our business, prevailing economic conditions and financial, business, political and other factors beyond our control can also affect us. These factors include the recent European debt crisis, which highlighted concerns about the possibility of one or more sovereign debt defaults, significant bank failures or defaults and/or the exit of one or more countries from the European Monetary Union. There are continuing concerns as to the ultimate financial effectiveness of the assistance measures taken to date, and the extent to which the austerity measures may exacerbate high unemployment and test the social and political stability of weaker economies in Europe. The world has recently experienced a global macroeconomic downturn, and if global economic and market conditions, or economic conditions in Europe, the United States or other key markets, remain uncertain, persist, or deteriorate further, demand for our services could decline, and we may experience material adverse impacts on our business, operating results, and financial condition. We cannot anticipate all the ways in which the current economic climate and financial market conditions could adversely impact our business.
We are exposed to risks associated with reduced profitability and the potential financial instability of our clients, many of whom may be adversely affected by volatile conditions in the financial markets, the economy in general and disruptions to the demand for health care services and pharmaceuticals. These conditions could cause clients to experience reduced profitability and/or cash flow problems that could lead them to modify, delay or cancel contracts with us, including contracts included in our current backlog.
Some of our clients are not revenue-generating entities at this time and rely upon equity and debt investments and other external sources of capital to meet their cash requirements. Due to the poor condition of the current global economy and other factors outside of our control, these clients may lack the funds necessary to pay outstanding liabilities due to us, despite contractual obligations. For example, in the second quarter of our fiscal year ended June 30, 2009 (“Fiscal Year 2009”), one of our biopharma clients informed us that it had encountered funding difficulties when one of its major investors defaulted on a contractual investment commitment, and that, as a result, the client would be unable to make payments due to us in connection with an on-going service contract for a large Phase III clinical trial. Consequently, we recorded approximately $14.0 million in
14
reserves related to this late-stage trial, including $12.3 million in bad debt reserves. In Fiscal Year 2012, we recovered $2.3 million of proceeds from the final bankruptcy settlement. It is possible that similar situations could arise in the future, and such defaults could negatively affect our financial performance, possibly materially.
We face risks arising from the restructuring of our operations.
In April 2011, we adopted a plan to restructure our operations to reduce expenses, better align costs with current and future geographic sources of revenue, and improve operating efficiencies. The plan focused primarily on the Early Phase business and corporate functions and was completed in the third quarter of Fiscal Year 2012. The total cost of the plan was approximately $14.2 million and included the elimination of approximately 150 managerial and staff positions and the abandonment of certain property leases.
Although we expect that all costs associated with this and prior restructuring plans have been recorded as of June 30, 2014, if we incur additional restructuring charges, our financial condition and results of operations may be adversely impacted.
The fixed price nature of our contracts or failure to document change orders could hurt our operating results.
Approximately 90% of our contracts are fixed price. If we fail to accurately price our contracts, if we experience significant cost overruns that are not recovered from our clients, or if we do not properly document changes to work orders under existing contracts, our gross margins on the contracts would be reduced and we could lose money on contracts. In the past, we have had to commit unanticipated resources to complete projects, resulting in lower gross margins on those projects. We might experience similar situations in the future.
If we are unable to attract suitable investigators and volunteers for our clinical trials, our clinical development business might suffer.
The clinical research studies we run in our CRS segment rely upon the ready accessibility and willing participation of physician investigators and volunteer subjects. Investigators are typically located at hospitals, clinics or other sites and supervise administration of the study drug to patients during the course of a clinical trial. Volunteer subjects generally include people from the communities in which the studies are conducted, and the rate of completion of clinical trials is significantly dependent upon the rate of participant enrollment.
Our clinical research development business could be adversely affected if we were unable to attract suitable and willing investigators or volunteers on a consistent basis. If we are unable to obtain sufficient patient enrollment or investigators to conduct clinical trials as planned, we might need to expend substantial additional funds to obtain access to resources or else be compelled to delay or modify our plans significantly. These considerations might result in our inability to successfully achieve projected development timelines as agreed with sponsors. In rare cases, it potentially may even lead us to recommend that trial sponsors terminate ongoing clinical trials or development of a product for a particular indication.
We rely on third parties and the transportation industry for important services.
We depend on third parties to provide us with products and services critical to our business. The failure of any of these third parties to adequately provide the needed products and services including, without limitation, transportation services, could have a material adverse effect on our business. Our clinical logistics services and other businesses are also heavily reliant on air travel for transport of clinical trial kits and other material, research products, and people, and a significant disruption to the air travel system, or our access to it, could have a material adverse effect on our business.
If our business, including PI, is unable to maintain continuous, effective, reliable and secure operation of its computer hardware, software and internet applications and related tools and functions, its business will be harmed.
Our PI business involves collecting, managing, manipulating and analyzing large amounts of data, and communicating data via the Internet. In our PI business, we depend on the continuous, effective, reliable and secure operation of computer hardware, software, networks, telecommunication networks, Internet servers and related infrastructure. If the hardware or software malfunctions or access to data by internal research personnel or customers through the Internet is interrupted, our PI business could suffer. In addition, any sustained disruption in Internet access provided by third parties could adversely impact our PI business.
Although the computer and communications hardware used in our PI business is protected through physical and software safeguards, it is still vulnerable to fire, storm, flood, power loss, earthquakes, telecommunications failures, physical or software break-ins, and similar events. And while certain of our operations have appropriate disaster recovery plans in place, we currently do not have redundant facilities everywhere in the world to provide IT capacity in the event of a system failure. In addition, the PI software products are complex and sophisticated, and could contain data, design or software errors that could be difficult to detect and correct. If PI fails to maintain and further develop the necessary computer capacity and data to support the needs of our PI customers, it could result in a loss of or a delay in revenue and market acceptance. Additionally, significant
15
delays in the planned delivery of system enhancements or inadequate performance of the systems once they are completed could damage our reputation and harm our business.
Finally, long-term disruptions to infrastructure caused by events such as natural disasters, the outbreak of war, the escalation of hostilities, and acts of terrorism (particularly in areas where we have offices) could adversely affect our businesses. Although we carry property and business interruption insurance, our coverage may not be adequate to compensate us for all losses that may occur.
Our business is subject to international economic, political, and other risks that could negatively affect our results of operations or financial position.
We provide most of our services on a worldwide basis. Our service revenue from non-U.S. operations represented approximately 53.0% and 54.0% of total consolidated service revenue for Fiscal Year 2014 and Fiscal Year 2013, respectively. More specifically, our service revenue from operations in Europe, Middle East and Africa represented 37.0% and 36.0% of total consolidated service revenue for the corresponding periods. Our service revenue from operations in the Asia/Pacific region represented 13.0% and 14.0% of total consolidated service revenue for the respective periods. Accordingly, our business is subject to risks associated with doing business internationally, including:
• | changes in a specific country’s or region’s political or economic conditions, including Western Europe, in particular; |
• | the outbreak of war or hostilities in specific geographic regions, including the Ukraine, Russia and the Middle East; |
• | potential negative impact from changes in tax laws affecting any repatriation of profits; |
• | difficulty in staffing and managing widespread operations; |
• | unfavorable labor regulations applicable to our European or other international operations; |
• | changes in foreign currency exchange rates; and |
• | the need to ensure compliance with the numerous regulatory and legal requirements applicable to our business in each of these jurisdictions and to maintain an effective compliance program to ensure compliance. |
Our operating results are impacted by the health of the global and local economies in which we operate. Our business and financial performance may be adversely affected by current and future economic conditions that cause a decline in business and consumer spending, including a reduction in the availability of credit, rising interest rates, financial market volatility and recession.
If we cannot retain our highly qualified management and technical personnel, our business would be harmed.
We rely on the expertise of our Chairman and Chief Executive Officer, Josef H. von Rickenbach, and our President and Chief Operating Officer, Mark A. Goldberg, and it would be difficult and expensive to find qualified replacements with the level of specialized knowledge of our products and services and the biopharmaceutical outsourcing services industry. While we are a party to an employment agreement with Mr. von Rickenbach, it may be terminated by either party upon notice to the counterparty.
In addition, in order to compete effectively, we must attract and retain qualified sales, professional, scientific, and technical operating personnel. Competition for these skilled personnel, particularly those with a medical degree, a Ph.D. or equivalent degrees, or industry specific expertise, is intense. We may not be successful in attracting or retaining key personnel.
Risks Associated with our Financial Results
Our operating results have fluctuated between quarters and years and may continue to fluctuate in the future, which could affect the price of our common stock.
Our quarterly and annual operating results have varied and will continue to vary in the future as a result of a variety of factors. For example, our income from operations totaled $58.8 million for the fiscal quarter ended June 30, 2014, $52.2 million for the fiscal quarter ended March 31, 2014, $46.7 million for the fiscal quarter ended December 31, 2013, $41.9 million for the fiscal quarter ended September 30, 2013, and $38.3 million for the fiscal quarter ended June 30, 2013. Factors that cause these variations include:
• | the level of new business authorizations in particular quarters or years; |
• | the timing of the initiation, progress, or cancellation of significant projects; |
• | foreign currency exchange rate fluctuations between quarters or years; |
• | restructuring charges; |
• | the mix of services offered in a particular quarter or year; |
16
• | the timing of the opening of new offices or internal expansion; |
• | timing, costs and the related financial impact of acquisitions; |
• | the timing and amount of costs associated with integrating acquisitions; |
• | the timing and amount of startup costs incurred in connection with the introduction of new products, services or subsidiaries; |
• | the dollar amount of changes in contract scope finalized during a particular period; and |
• | the amount of any reserves we are required to record. |
Many of these factors, such as the timing of cancellations of significant projects and foreign currency exchange rate fluctuations between quarters or years, are beyond our control.
If our operating results do not match the expectations of securities analysts and investors, the trading price of our common stock will likely decrease.
Backlog may not result in revenue.
Our backlog is not necessarily a meaningful predictor of future results because backlog can be affected by a number of factors, including the size and duration of contracts, many of which are performed over several years. Additionally, as described above, contracts relating to our clinical development business are subject to early termination by the client, and clinical trials can be delayed or canceled for many reasons, including unexpected test results, safety concerns, regulatory developments or economic issues. Also, the scope of a contract can be reduced significantly during the course of a study. If the scope of a contract is revised, the adjustment to backlog occurs when the revised scope is approved by the client. For these and other reasons, we do not fully realize our entire backlog as service revenue.
Our revenue and earnings are exposed to exchange rate fluctuations, which have substantially affected our operating results.
Our financial statements are denominated in U.S. dollars. Because we conduct a significant portion of our operations in foreign countries, changes in foreign currency exchange rates could have and have had a significant effect on our operating results. For example, as a result of year-over-year foreign currency exchange rate fluctuation, service revenue for Fiscal Year 2014 was positively impacted by approximately $5.3 million as compared with the same period in the previous year. Exchange rate fluctuations between local currencies and the U.S. dollar create risk in several ways, including:
• | Foreign Currency Translation Risk. The revenue and expenses of our foreign operations are generally denominated in local currencies, primarily the pound sterling and the Euro, and are translated into U.S. dollars for financial reporting purposes. For Fiscal Year 2014 and Fiscal Year 2013, approximately 13.7% and 13.0% of consolidated service revenue, respectively, was from contracts denominated in Euros and service revenue from contracts denominated in pounds sterling was 2.9% and 2.6%, respectively. Accordingly, changes in exchange rates between foreign currencies and the U.S. dollar will affect the translation of foreign results into U.S. dollars for purposes of reporting our consolidated results. |
• | Foreign Currency Transaction Risk. We may be subjected to foreign currency transaction risk when our foreign subsidiaries enter into contracts or incur liabilities denominated in a currency other than the foreign subsidiary's functional (local) currency. We also may be subject to foreign currency transaction risk based upon our internal contracts and the extent of work performed by a particular region. To the extent that we are unable to shift the effects of currency fluctuations to our clients, foreign currency exchange rate fluctuations as a result of foreign currency exchange losses could have a material adverse effect on our results of operations. |
Although we try to limit these risks through the inclusion of exchange rate fluctuation provisions stated in our service contracts or by hedging transaction risk with foreign currency exchange contracts, we do not succeed in all cases. Even in those cases in which we are successful, we may still experience fluctuations in financial results from our operations outside of the U.S., and we may not be able to favorably reduce the currency transaction risk associated with our service contracts.
Our effective income tax rate may fluctuate from quarter to quarter, which may affect our earnings and earnings per share.
Our quarterly effective income tax rate is influenced by our annual projected profitability in the various taxing jurisdictions in which we operate. Changes in the distribution of profits and losses among taxing jurisdictions may have a significant impact on our effective income tax rate, which in turn could have a material adverse effect on our net income and earnings per share. Factors that affect the effective income tax rate include, but are not limited to:
• | the requirement to exclude from our quarterly worldwide effective income tax calculations losses in jurisdictions in which no tax benefit can be recognized; |
17
• | the repatriation of foreign earnings to the U.S.; |
• | actual and projected full-year pretax income; |
• | changes in tax laws in various taxing jurisdictions; |
• | audits by taxing authorities; and |
• | the establishment of valuation allowances against deferred tax assets if it is determined that it is more likely than not that future tax benefits will not be realized. |
These changes may cause fluctuations in our effective income tax rate that could cause fluctuation in our earnings and earnings per share, which could affect our stock price.
Our results of operations may be adversely affected by the results of regulatory tax examinations.
We are subject to value added tax, customs tax, sales and use tax, withholding tax, payroll tax, income tax, and other taxes as a result of the operations of our business. The regulators from the various jurisdictions in which we operate periodically perform audits. In the conduct of such audits, we may be required to disclose information of a sensitive nature and, in general, to modify the way we have conducted business with our vendors and customers until the present, which may affect our business in an adverse manner. We are also regularly subject to, and are currently undergoing, audits by tax authorities in the United States and foreign jurisdictions for prior tax years. Although we believe our tax estimates are reasonable and we intend to defend our positions through litigation if necessary, the final outcome of tax audits and related litigation is inherently uncertain and could be materially different than that reflected in our historical income tax provisions and accruals. Moreover, we could be subject to assessments of substantial additional taxes and/or fines or penalties relating to ongoing or future audits. The adverse resolution of any audits or litigation could have an adverse effect on our financial position and results of operations.
Our results of operations may be adversely affected if our goodwill or intangible assets are impaired.
As of June 30, 2014, our total assets included $421.4 of goodwill and net intangible assets. We assess the realizability of our indefinite-lived intangible assets and goodwill annually as well as whenever events or changes in circumstances indicate that these assets may be impaired. These events or changes in circumstances generally include operating losses or a significant decline in earnings associated with the acquired business or asset. Our ability to realize the value of the goodwill and indefinite-lived intangible assets will depend on the future cash flows of these businesses. These cash flows may be impacted by how well we have integrated these businesses. If we are not able to realize the value of the goodwill and indefinite-lived intangible assets, we may be required to incur material charges relating to the impairment of those assets.
Changes to our computer operating systems, programs or software could adversely impact our business.
We may make changes to our existing computer operating systems, programs and/or software in an effort to increase our operating efficiency and/or deliver better value to our clients. Such changes may cause disruptions to our operations and have an adverse impact on our business in the short term.
Our business has experienced substantial expansion in the past and such expansion and any future expansion could strain our resources if not properly managed.
We have expanded our business substantially in the past. Future rapid expansion could strain our operational, human and financial resources. In order to manage expansion, we must:
• | continue to improve operating, administrative, and information systems; |
• | accurately predict future personnel and resource needs to meet client contract commitments; |
• | track the progress of ongoing client projects; and |
• | attract and retain qualified management, sales, professional, scientific and technical operating personnel. |
If we do not take these actions and are not able to manage the expanded business, the expanded business may be less successful than anticipated. We may be required to allocate existing or future resources to the expanded business, that in either case, we would have otherwise allocated to another part of our business.
If we are unable to successfully execute our acquisition strategies, our business, results of operations and financial condition could be adversely impacted.
Historically our growth strategy has been based in part on our ability to acquire existing businesses, services or technologies. We do not know whether in the future we will be able to:
• | identify suitable businesses or technologies to buy; |
• | complete the purchase of those businesses on terms acceptable to us; |
18
• | successfully integrate their operations into our own; |
• | obtain financing necessary for an acquisition at all or on commercially acceptable terms; or |
• | retain key personnel and customers of acquired businesses. |
We compete with other potential buyers for the acquisition of existing businesses and technology. This competition may result in fewer opportunities to purchase companies that are for sale. It may also result in higher purchase prices for the businesses that we want to purchase. We may also spend time and money investigating and negotiating with potential acquisition targets but not complete the transaction. Any future acquisition could involve other risks, including the assumption of additional liabilities and expenses, issuances of potentially dilutive securities or interest-bearing debt, transaction costs, and diversion of management's attention from other business concerns.
If we are unable to successfully integrate an acquired company, the acquisition could lead to disruptions to our business. For example, in December 2012, we completed the acquisition of LIQUENT, a leading regulatory information management solutions organization. In April 2013, we acquired HERON, a leading market access and reimbursement consulting company. The success of our acquisition strategy will depend upon, among other things, our ability to:
• | assimilate the operations and services or products of the acquired company; |
• | integrate acquired personnel; |
• | retain and motivate key employees; |
• | retain customers; |
• | identify and manage risks facing the acquired company; and |
• | minimize the diversion of management’s attention from other business concerns. |
Acquisitions of companies outside of the United States may also involve additional risks, including assimilating differences in foreign business practices and overcoming language and cultural barriers.
In the event that the operations of an acquired business do not meet our performance expectations, we may have to restructure the acquired business or write-off the value of some or all of the assets of the acquired business.
Our failure to execute our acquisition strategies, including the identification of potential acquisitions, completing targeted acquisitions, and integrating completed acquisitions, could have a material adverse effect on our business, financial condition and results of operations.
Risks Associated with our Industry
We depend on the pharmaceutical and biotechnology industries, either or both of which may suffer in the short or long term.
Our revenues depend greatly on the expenditures made by the pharmaceutical and biotechnology industries in research and development. In some instances, companies in these industries are reliant on their ability to raise capital in order to fund their research and development projects. Accordingly, economic factors and industry trends that affect our clients in these industries also affect our business. If companies in these industries were to reduce the number of research and development projects they conduct or outsource, our business could be materially adversely affected.
In addition, we are dependent upon the ability and willingness of pharmaceutical and biotechnology companies to continue to spend on research and development and to outsource the services that we provide. We are therefore subject to risks, uncertainties and trends that affect companies in these industries. We have benefited to date from the tendency of pharmaceutical and biotechnology companies to outsource clinical research projects, but any downturn in these industries or reduction in spending or outsourcing could adversely affect our business. For example, if these companies expanded upon their in-house clinical or development capabilities, they would be less likely to utilize our services.
Pharmaceutical and biotechnology companies have been entering into strategic partnerships with clinical research organizations over the past few years. To the extent we are not selected or do not otherwise enter into a strategic partnership with a sponsor, future business with that sponsor may be limited.
Because we depend on a small number of industries, and there is a concentration of large clients in those industries, the loss of business from a significant client could harm our business, revenue and financial condition.
The loss of, or a material reduction in the business of, a significant client could cause a substantial decrease in our revenue and adversely affect our business and financial condition, possibly materially. In Fiscal Years 2014, 2013, and 2012, our five largest clients accounted for approximately 47%, 50%, and 40.5% of our consolidated service revenue, respectively. In Fiscal Year 2014, our two largest clients individually accounted for 16% and 11% of our consolidated service revenue, and in Fiscal Year
19
2013, our two largest clients individually accounted for 17% and 12% of our consolidated service revenue. In Fiscal Year 2015, we expect that a small number of clients will continue to represent a significant part of our consolidated revenue. This concentration may increase as a result of the increasing number of strategic partnerships into which we have been entering with sponsors. Our contracts with these clients generally can be terminated on short notice. We have in the past experienced contract cancellations with significant clients. If we lose clients, we may not be able to attract new ones, and if we lose individual projects, we may not be able to replace them.
In addition, the portion of our backlog that consists of large, multi-year awards from strategic partnerships has grown in recent years and this trend may continue in the future. A higher concentration of backlog from strategic partnerships may result in an imbalance across our project portfolio among projects in the start-up phase, which typically generate lower revenue, and projects in later stages, which typically generate higher revenue. This in turn may cause fluctuations in our revenue and profitability from period to period.
We face intense competition in many areas of our business; if we do not compete effectively, our business will be harmed.
The biopharmaceutical services industry is highly competitive and we face numerous competitors in many areas of our business. If we fail to compete effectively, we may lose clients, which would cause our business to suffer.
We primarily compete against in-house departments of pharmaceutical companies, other full service clinical research organizations (“CROs”), small specialty CROs, and, to a lesser extent, universities, teaching hospitals, and other site organizations. Some of the larger CROs against which we compete include Quintiles Transnational Corporation, Covance, Inc., Pharmaceutical Product Development Inc., and Icon plc. In addition, our PC business competes with a large and fragmented group of specialty service providers, including advertising/promotional companies, major consulting firms with pharmaceutical industry groups and smaller companies with pharmaceutical industry focus. PI competes primarily with CROs, information technology companies and other software companies. Some of these competitors, including the in-house departments of pharmaceutical companies, have greater capital, technical and other resources than we have. In addition, our competitors that are smaller specialized companies may compete effectively against us because of their concentrated size and focus.
In recent years, a number of the large pharmaceutical companies have established formal or informal alliances with one or more CROs relating to the provision of services for multiple trials over extended time periods. Our success depends in part on successfully establishing and maintaining these relationships. If we fail to do so, our revenue and results of operations could be adversely affected, possibly materially.
If we do not keep pace with rapid technological changes, our products and services may become less competitive or obsolete, especially in our PI business.
The biotechnology, pharmaceutical and medical device industries generally, and clinical research specifically, are subject to increasingly rapid technological changes. Our competitors or others might develop technologies, products or services that are more effective or commercially attractive than our current or future technologies, products or services, or render our technologies, products or services less competitive or obsolete. If our competitors introduce superior technologies, products or services and we cannot make enhancements to our technologies, products and services necessary to remain competitive, our competitive position would be harmed. If we are unable to compete successfully, we may lose clients or be unable to attract new clients, which could lead to a decrease in our revenue.
Risks Associated with Regulation or Legal Liabilities
If governmental regulation of the drug, medical device and biotechnology industry changes, the need for our services could decrease.
Governmental regulation of the drug, medical device and biotechnology product development process is complicated, extensive, and demanding. A large part of our business involves assisting pharmaceutical, biotechnology and medical device companies through the regulatory approval process. Changes in regulations that, for example, streamline procedures or relax approval standards, could eliminate or reduce the need for our services. If companies regulated by the United States Food and Drug Administration (the “FDA”) or similar foreign regulatory authorities needed fewer of our services, we would have fewer business opportunities and our revenues would decrease, possibly materially.
In the United States, the FDA and the Congress have attempted to streamline the regulatory process by providing for industry user fees that fund the hiring of additional reviewers and better management of the regulatory review process. In Europe, governmental authorities have approved common standards for clinical testing of new drugs throughout the European Union by adopting standards for Good Clinical Practices (“GCP”) and by making the clinical trial application and approval process more uniform across member states. The FDA has had GCP in place as a regulatory standard and requirement for new drug approval for many years, and Japan adopted GCP in 1998.
The United States, Europe and Japan have also collaborated for over 15 years on the International Conference on Harmonisation (“ICH”), the purpose of which is to eliminate duplicative or conflicting regulations in the three regions. The
20
ICH partners have agreed on a common format (the Common Technical Document) for new drug marketing applications that reduces the need to tailor the format to each region. Such efforts and similar efforts in the future that streamline the regulatory process may reduce the demand for our services.
Parts of our PC business advise clients on how to satisfy regulatory standards for manufacturing and clinical processes and on other matters related to the enforcement of government regulations by the FDA and other regulatory bodies. Any reduction in levels of review of manufacturing or clinical processes or levels of regulatory enforcement, generally, would result in fewer business opportunities for our business in this area.
If we fail to comply with existing regulations, our reputation and operating results would be harmed.
Our business is subject to numerous governmental regulations, primarily relating to worldwide pharmaceutical and medical device product development and regulatory approval, the conduct of clinical trials, and limitations on activities relating to delivery of health care items or services that are paid for with government health care program funding. In addition, we may be obligated to comply with or to assist our clients in complying with regulations that apply to our clients, including the Physician Payment Sunshine Act, which will require manufacturers and group purchasing organizations to report all payments or transfers of value to health care providers and teaching hospitals. If we fail to comply with these governmental regulations, such non-compliance could result in the termination of our ongoing research, development or sales and marketing projects, or the disqualification of data for submission to regulatory authorities. We also could be barred from providing clinical trial services in the future or could be subjected to civil monetary penalties or, in certain cases, criminal fines and penalties. Any of these consequences would harm our reputation, our prospects for future work and our operating results. In addition, we may have to repeat research or redo trials. If we are required to repeat research or redo trials, we may be contractually required to do so at no further cost to our clients, but at substantial cost to us.
We may lose business opportunities as a result of healthcare reform and the expansion of managed-care organizations.
Numerous governments, including the U.S. government, have undertaken efforts to control growing healthcare costs through legislation, regulation and voluntary agreements with medical care providers and drug companies. In March 2010, the United States Congress enacted healthcare reform legislation intended over time to expand health insurance coverage and impose health industry cost containment measures. The continuing implementation of this legislation may significantly impact the pharmaceutical industry. The U.S. Congress has also considered and may adopt legislation that could have the effect of putting downward pressure on the prices that pharmaceutical and biotechnology companies can charge for prescription drugs. In addition, various state legislatures and European and Asian governments may consider various types of healthcare reform in order to control growing healthcare costs. We are presently uncertain as to the effects of the enacted legislation on our business and are unable to predict what legislative proposals will be adopted in the future, if any.
If these efforts are successful, drug, medical device and biotechnology companies may react by spending less on research and development. If this were to occur, we would have fewer business opportunities and our revenue could decrease, possibly materially. In addition, new laws or regulations may create a risk of liability, increase our costs or limit our service offerings.
In addition to healthcare reform proposals, the expansion of managed-care organizations in the healthcare market and managed-care organizations’ efforts to cut costs by limiting expenditures on pharmaceuticals and medical devices could result in pharmaceutical, biotechnology and medical device companies spending less on research and development. If this were to occur, we would have fewer business opportunities and our revenue could decrease, possibly materially.
We may have substantial exposure to payment of personal injury claims and may not have adequate insurance to cover such claims.
Our CRS business primarily involves the testing of experimental drugs and medical devices on consenting human volunteers pursuant to a study protocol. Clinical research involves a risk of liability for a number of reasons, including, but not limited to:
• | personal injury or death to patients who participate in the study or who use a product approved by regulatory authorities after the clinical research has concluded; |
• | general risks associated with our Early Phase facilities, including professional malpractice of physicians, nurses and other medical care providers; and |
• | errors and omissions during a trial that may undermine the usefulness of a trial or data from the trial or study. |
In order to mitigate the risk of liability, we seek to include indemnification provisions in our CRS contracts with clients and with investigators. However, we are not able to include indemnification provisions in all of our contracts. In addition, even if we are able to include an indemnification provision in our contracts, the indemnification provisions may not cover our exposure if:
• | we had to pay damages or incur defense costs in connection with a claim that is outside the scope of an indemnification agreement; or |
21
• | a client failed to indemnify us in accordance with the terms of an indemnification agreement because it did not have the financial ability to fulfill its indemnification obligation or for any other reason. |
In addition, contractual indemnifications generally do not protect us against liability arising from certain of our own actions, such as negligence or misconduct.
We also carry insurance to cover our risk of liability. However, our insurance is subject to deductibles and coverage limits and may not be adequate to cover claims. In addition, liability coverage is expensive. In the future, we may not be able to maintain or obtain the same levels of coverage on reasonable terms, at a reasonable cost, or in sufficient amounts to protect us against losses due to claims.
Existing and proposed laws and regulations regarding confidentiality of patients’ and other individuals’ personal information could result in increased risks of liability or increased cost to us or could limit our product and service offerings.
The confidentiality, security, use and disclosure of patient-specific information are subject to governmental regulation. Regulations to protect the safety and privacy of human subjects who participate in or whose data are used in clinical research generally require clinical investigators to obtain legally effective informed consent from identifiable research subjects before research is undertaken. Under the Health Insurance Portability and Accountability Act of 1996 ("HIPAA"), as amended by the Health Information Technology for Economic and Clinical Health (“HITECH”) Act of 2009, the U.S. Department of Health and Human Services has issued regulations mandating privacy and data security standards and breach notification requirements for certain types of individually identifiable health information, or protected health information, when used or disclosed by health care providers and other HIPAA-covered entities or business associates that provide services to or perform functions on behalf of these covered entities. HIPAA regulations generally require individuals’ written authorization before identifiable health information may be used for research, in addition to any required informed consent. HIPAA regulations also specify standards for de-identifying health information so that information can be handled outside of the HIPAA requirements and for creating limited data sets that can be used for research purposes under less stringent HIPAA restrictions.
The European Union and its member states, as well as other countries, such as Canada, Argentina, Japan and other Asian countries, and state governments in the United States, have adopted and continue to implement new medical privacy and general data protection laws and regulations. In those countries, collecting, processing, using and transferring an individual’s personal data is subject to specific requirements, such as obtaining explicit consent, processing the information for limited purposes and restrictions with respect to cross-border transfers. Many countries and almost all states in the United States have adopted data security breach laws that require the user of such data to inform the affected individuals and, in some cases, government authorities and the general public of security breaches. In order to comply with these laws and regulations and corresponding contractual demands from our clients, we must maintain internal compliance policies and procedures, and we may need to implement new privacy and security measures, which may require us to make substantial expenditures or cause us to limit the products and services we offer. In addition, if we violate applicable laws, regulations, contractual commitments, or other duties relating to the use, privacy or security of health information, we could be subject to civil liability or criminal penalties and it may be necessary to modify our business practices.
Failure to achieve and maintain effective internal controls in accordance with Section 404 of the Sarbanes-Oxley Act of 2002, and delays in completing our internal controls and financial audits, could have a material adverse effect on our business and stock price.
If we fail to achieve and maintain effective internal controls, we will not be able to conclude that we have effective internal controls over financial reporting in accordance with Section 404 of the Sarbanes-Oxley Act of 2002. Failure to achieve and maintain an effective internal control environment, and delays in completing our internal controls and financial audits, could cause investors to lose confidence in our reported financial information and PAREXEL, which could result in a decline in the market price of our common stock, and cause us to fail to meet our reporting obligations in the future, which in turn could impact our ability to raise equity financing if needed in the future. Our Fiscal Year 2009 management assessment revealed a material weakness in our internal controls over financial reporting due to insufficient controls associated with accounting for the ClinPhone business combination, specifically the adoption by ClinPhone of an accounting policy for revenue recognition that was compliant with generally accepted accounting principles in the U.S. We have since changed our internal controls to address this material weakness and concluded in Fiscal Year 2013 that our internal controls related to our accounting for business combinations were designed and operating effectively. Nevertheless, we may identify other significant deficiencies or material weaknesses which we may not be able to remediate in a timely manner or at all.
We operate in many different jurisdictions and we could be adversely affected by violations of the U.S. Foreign Corrupt Practices Act and similar worldwide anti-corruption laws.
The U.S. Foreign Corrupt Practices Act (FCPA) and similar worldwide anti-corruption laws, including the U.K. Bribery Act of 2010, generally prohibit companies and their intermediaries from making improper payments to foreign officials for the
22
purpose of obtaining or retaining business. Our internal policies mandate compliance with these anti-corruption laws. We operate in many parts of the world that have experienced governmental corruption to some degree, and in certain circumstances, anti-corruption laws have appeared to conflict with local customs and practices. Despite our training and compliance programs, we cannot assure that our internal control policies and procedures always will protect us from reckless or criminal acts committed by persons associated with PAREXEL. Our continued global expansion, including in developing countries, could increase such risk in the future. Violations of these laws, or even allegations of such violations, could disrupt our business and result in a material adverse effect on our results of operations or financial condition.
Risks Associated with Indebtedness
Our indebtedness may limit cash flow available to invest in the ongoing needs of our business.
As of June 30, 2014, we had $350.0 million principal amount of debt outstanding and remaining borrowing availability of $237.5 under our credit arrangements. We may incur additional debt in the future. Our leverage could have significant adverse consequences, including:
• | requiring us to dedicate a substantial portion of any cash flow from operations to the payment of interest on, and principal of, our debt, which will reduce the amounts available to fund working capital and capital expenditures, and for other general corporate purposes; |
• | increasing our vulnerability to general adverse economic and industry conditions; |
• | limiting our flexibility in planning for, or reacting to, changes in our business and the industry in which we compete; and |
• | placing us at a competitive disadvantage to our competitors that have less debt. |
Under the terms of our various credit facilities, interest rates are fixed based on market indices at the time of borrowing and, depending upon the interest mechanism selected by us, may float thereafter. Some of our other smaller credit facilities also bear interest at floating rates. As a result, the amount of interest payable by us on our borrowings may increase if market interest rates change.
We may not have sufficient funds, our business may not generate sufficient cash flow from operations, or we may be unable to arrange for additional financing, to pay the amounts due under our existing or any future debt, or any other liquidity needs. In addition, a failure to comply with the covenants under our existing credit facilities could result in an event of default under those credit facilities. In the event of an acceleration of amounts due under our credit facilities as a result of an event of default, we may not have sufficient funds or may be unable to arrange for additional financing to repay our indebtedness or to make any required accelerated payments.
In addition, the terms of the 2013 Credit Agreement and the Note Purchase Agreement provide that upon the occurrence of a change in control, as defined in the 2013 Credit Agreement and the Note Purchase Agreement, all outstanding indebtedness under the credit facility and the Note Purchase Agreement would become due. This provision may delay or prevent a change in control that stockholders may consider desirable.
Our existing credit facilities contain covenants that limit our flexibility and prevent us from taking certain actions.
The agreements in connection with our 2013 Credit Agreement and in our short term debt facilities include a number of significant restrictive covenants. These covenants could adversely affect us by limiting our ability to plan for or react to market conditions, meet our capital needs and execute our business strategy. These covenants, among other things, limit our ability and the ability of our restricted subsidiaries to:
• | incur additional debt; |
• | buy back our common stock; |
• | make certain investments; |
• | enter into certain types of transactions with affiliates; |
• | make specified restricted payments; and |
• | sell certain assets or merge with or into other companies. |
These covenants may limit our operating and financial flexibility and limit our ability to respond to changes in our business or competitive activities. Our failure to comply with these covenants could result in an event of default, which, if not cured or waived, could result in our being required to repay these borrowings before their scheduled due date.
23
Risks Associated with our Common Stock
Our corporate governance structure, including provisions of our articles of organization, by-laws, as well as Massachusetts law, may delay or prevent a change in control or management that stockholders may consider desirable.
Provisions of our articles of organization, and by-laws, as well as provisions of Massachusetts law, may enable our management to resist an acquisition of us by a third party, or may discourage a third party from acquiring us. These provisions include the following:
• | we have divided our board of directors into three classes that serve staggered three-year terms; |
• | we are subject to Section 8.06 of the Massachusetts Business Corporation Law, which provides that directors may only be removed by stockholders for cause, vacancies in our board of directors may only be filled by a vote of our board of directors, and the number of directors may be fixed only by our board of directors; |
• | we are subject to Chapter 110F of the Massachusetts General Laws, which may limit the ability of some interested stockholders to engage in business combinations with us; and |
• | our stockholders are limited in their ability to call or introduce proposals at stockholder meetings. |
These provisions could have the effect of delaying, deferring, or preventing a change in control of us or a change in our management that stockholders may consider favorable or beneficial. These provisions could also discourage proxy contests and make it more difficult for stockholders to elect directors and take other corporate actions. These provisions could also limit the price that investors might be willing to pay in the future for shares of our stock.
In addition, our board of directors may issue preferred stock in the future without stockholder approval. If our board of directors issues preferred stock, the rights of the holders of common stock would be subordinate to the rights of the holders of preferred stock. Our board of directors’ ability to issue the preferred stock could make it more difficult for a third party to acquire, or discourage a third party from acquiring, a majority of our stock.
Our stock price has been, and may in the future be volatile, which could lead to losses by investors.
The market price of our common stock has fluctuated widely in the past and may continue to do so in the future. On August 15, 2014, the closing sales price of our common stock on the Nasdaq Global Select Market was $57.50 per share. During the period from June 30, 2009 to June 30, 2014, our common stock traded at prices ranging from a high of $57.25 per share to a low of $11.54 per share. Investors in our common stock must be willing to bear the risk of such fluctuations in stock price and the risk that the value of an investment in our common stock could decline.
Our stock price can be affected by quarter-to-quarter variations in a number of factors including, but not limited to:
• | operating results; |
• | earnings estimates by industry analysts; |
• | market conditions in our industry or the pharmaceutical and biotechnology industries; |
• | prospects of healthcare reform; |
• | changes in government regulations; |
• | general economic conditions, and |
• | our effective income tax rate. |
In addition, the stock market has from time to time experienced significant price and volume fluctuations that are not related to the operating performance of particular companies. These market fluctuations may adversely affect the market price of our common stock. Although our common stock has traded in the past at a relatively high price-earnings multiple, due in part to analysts’ expectations of earnings growth, the price of the common stock could quickly and substantially decline as a result of even a relatively small shortfall in earnings from, or a change in, analysts’ expectations.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
24
ITEM 2. PROPERTIES
As of June 30, 2014, we occupied approximately 2,570,000 square feet of building space, primarily office space, in 80 locations in 39 countries under various leases that expire between 2014 and 2035. Total square feet by region is summarized below:
Region | Square Feet | |
The Americas | 779,000 | |
Europe, Middle East & Africa | 1,247,000 | |
Asia/Pacific | 544,000 | |
Total | 2,570,000 | |
Our largest facilities are located in (a) the United States, where we occupy approximately 702,000 square feet, (b) Germany, where we occupy approximately 501,000 square feet, (c) the United Kingdom, where we occupy approximately 278,000 square feet, (d) India, where we occupy approximately 207,000 square feet, and (e) South Africa, where we occupy approximately 137,000 square feet. Our principal facilities are set forth below:
Facility | Sq. Ft. | Use of Facility | Lease Expirations | |||
Headquarters in Waltham, MA | 64,000 | CRS, PC and Corporate | 2019 | |||
Berlin, Germany | 452,000 | All Business Segments and General & Administrative | 2016 - 2035 | |||
Billerica, MA | 276,000 | All Business Segments and General & Administrative | 2018 - 2025 | |||
Hyderabad, India | 175,000 | All Business Segments and General & Administrative | 2016 - 2017 | |||
Uxbridge, UK | 88,000 | CRS, PC and General & Administrative | 2022 | |||
Nottingham, UK | 80,000 | PI and General & Administrative | 2014 - 2015 | |||
We believe that our facilities are adequate for our operations and that additional space will be available at satisfactory terms, if needed.
ITEM 3. LEGAL PROCEEDINGS
PAREXEL periodically becomes involved in various legal proceedings and claims that arise in the ordinary course of business. We believe that no matters currently pending would, in the event of an adverse outcome, have a material impact on our consolidated financial position, results of operations, or liquidity but there can be no assurance that such matters would not, in the event of an adverse outcome, have a material impact on our consolidated financial position, results of operations, or liquidity.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
25
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS, AND ISSUER PURCHASES OF EQUITY SECURITIES
MARKET INFORMATION AND HOLDERS
Our common stock is traded on the Nasdaq Global Select Market under the symbol “PRXL.” The table below shows the high and low sales prices of the common stock for each quarter of the Fiscal Years 2014 and 2013.
2014 | 2013 | |||||||
High | Low | High | Low | |||||
First Quarter | $53.55 | $46.02 | $31.56 | $25.95 | ||||
Second Quarter | $55.02 | $37.53 | $33.13 | $28.45 | ||||
Third Quarter | $57.25 | $44.89 | $39.75 | $30.39 | ||||
Fourth Quarter | $55.97 | $41.79 | $49.02 | $38.51 | ||||
As of August 15, 2014, there were approximately 144 stockholders of record of our common stock. The number does not include stockholders for which shares were held in a “nominee” or “street” name.
DIVIDENDS
We have never declared or paid any cash dividends on our capital stock, nor do we anticipate paying any cash dividends in the foreseeable future. We intend to retain future earnings for the development and expansion of our business.
Under the terms of the 2011 and 2013 Credit Agreements, which are described in “Credit Agreements” in Part II, Item 7 of this annual report, neither we nor any of our subsidiaries may pay any dividend or make any other distribution with respect to any shares of capital stock except that (a) we and our subsidiaries may declare and pay dividends with respect to equity interests payable solely in additional shares of common stock, (b) our subsidiaries may declare and pay dividends and other distributions ratably with respect to their equity interests, (c) we may make restricted payments pursuant to and in accordance with stock option plans or other benefit plans for management or employees of PAREXEL and our subsidiaries, and (d) we may make certain permitted stock repurchases.
STOCK REPURCHASE PROGRAM
Fiscal Year 2014 Share Repurchase
On June 2, 2014, we announced that our Board of Directors approved a share repurchase program (the “2014 Program”) authorizing the repurchase of up to $150.0 million of our common stock to be financed with cash on hand, cash generated from operations, existing credit facilities, or new financing. On June 13, 2014, we entered into an agreement (the “2014 Agreement”) to purchase shares of our common stock from Goldman Sachs & Co. (“GS”), for an aggregate purchase price of $150.0 million pursuant to an accelerated share purchase program. Pursuant to the 2014 Agreement, in June 2014, we paid $150.0 million to GS and received from GS 2,284,844 shares of common stock, representing 80 percent of the shares to be repurchased by us under the 2014 Agreement. The shares were repurchased at a price of $52.52 per share, which was the closing price of our common stock on the Nasdaq Global Select Market on June 13, 2014. These shares were canceled and restored to the status of authorized and unissued shares. As of June 30, 2014, we recorded the $150.0 million payment to GS as a decrease to equity in our consolidated balance sheet.
The final number of shares to be delivered to us by GS under the 2014 Agreement at program maturity, net of the initial delivery, will be adjusted based on an agreed upon discount to the average of the daily volume weighted average price of the common stock during the term of the 2014 Agreement. If the number of shares to be delivered to us at maturity is less than the initial delivery of shares by GS, we would be required to remit shares or cash, at our option, to GS in an amount equivalent to such shortfall. We expect the 2014 Program to be completed before December 31, 2014.
Fiscal Year 2013 Share Repurchase
In August 2012, our Board of Directors approved a share repurchase program (the “2013 Program”) authorizing the repurchase of up to $200.0 million of our common stock to be financed with cash on hand, cash generated from operations, existing credit facilities, or new financing. During Fiscal Year 2013, we repurchased $197.6 million of our common stock. We repurchased the remaining $2.4 million of our common stock in July 2013. The 2013 Program repurchases were effected pursuant to a $50.0 million accelerated share repurchase agreement and a $50.0 million open market agreement entered into in September 2012 and a $50.0 million accelerated share repurchase agreement (“March 2013 ASR Agreement”) and a $50.0 million open market agreement (“March 2013 Open Market Agreement”) entered in March 2013. Pursuant to the 2013 Program, we repurchased 5,458,285 shares of our common stock at an average price of $36.64 per share from September 2012 to July 2013.
26
The buyback activity also resulted in a reduction of our stockholders’ equity of $200.0 million for the value of shares repurchased and retired by the Company.
In July 2013, we purchased 51,071 shares under our March 2013 Open Market Agreement and received 101,247 shares representing the final settlement of our March 2013 ASR Agreement. With the completion of our March 2013 Open Market Agreement and the final settlement of the March 2013 ASR Agreement, the 2013 Program was completed.
The following table provides information about repurchases of our equity securities, including those under the 2014 Program described above, during the three months ended June 30, 2014:
Period | (a) Total Number of Shares (or Units) Purchased | (b) Average Price Paid per Share (or Unit) | (c) Total Number of Shares Purchased as Part of Publicly Announced Plans or Programs | (d) Approximate Dollar Value of Shares that May Yet Be Purchased Under the Plans or Programs | ||||||||
April 1, 2014 - April 30, 2014 | — | $ | — | — | $— | |||||||
May 1, 2014 - May 31, 2014 | — | $ | — | — | $— | |||||||
June 1, 2014 - June 30, 2014 | 2,284,844 | $ | 52.52 | 2,284,844 | $— | |||||||
Total | 2,284,844 | 2,284,844 | ||||||||||
27
COMPANY STOCK PERFORMANCE GRAPH
Our common stock is listed for trading on the Nasdaq Global Select Market under the symbol “PRXL.” The Stock Price Performance Graph set forth below compares the cumulative total stockholder return on our common stock for the period from June 30, 2009 through June 30, 2014, with the cumulative total return of the Nasdaq Composite Index and the Nasdaq Health Care Index over the same period. The comparison assumes $100 was invested on June 30, 2009 in PAREXEL’s common stock, in the Nasdaq Composite Index, and in the Nasdaq Health Care Index and assumes reinvestment of dividends, if any.
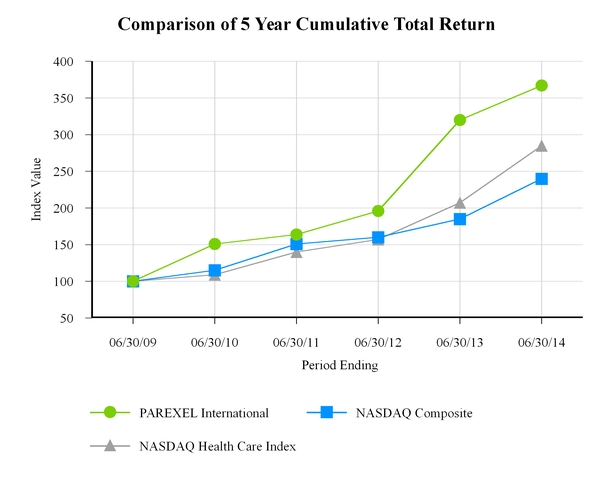
Fiscal Years Ended June 30, | ||||||||||||
Total Return Index For: | 2009 | 2010 | 2011 | 2012 | 2013 | 2014 | ||||||
PAREXEL International Stock | $100 | $151 | $164 | $196 | $320 | $367 | ||||||
Nasdaq Composite Index | $100 | $115 | $151 | $160 | $185 | $240 | ||||||
Nasdaq Health Care Index | $100 | $109 | $140 | $157 | $207 | $285 | ||||||
The stock price performance shown on the graph above is not necessarily indicative of future price performance. Information used in the graph was obtained from The Nasdaq Stock Market, a source believed to be reliable, but we are not responsible for any errors or omissions in such information.
The information included under the heading “Company Stock Performance Graph” is “furnished” and not “filed” for purposes of Section 18 of the Exchange Act, or otherwise subject to the liabilities of that section, nor shall it be deemed to be “soliciting material” subject to Regulation 14A or incorporated by reference in any filing under the Securities Act of 1933, as amended, or the Exchange Act.
28
ITEM 6. SELECTED FINANCIAL DATA
The following selected consolidated financial data of PAREXEL for the five years ended June 30, 2014 are derived from our consolidated financial statements. The information set forth below should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included as Item 7 in this annual report and the consolidated financial statements and related footnotes included as Item 8 in this annual report.
For the fiscal years ended June 30, | |||||||||||||||||||||
(in thousands, except per share data and number of employees) | 2014 | 2013 | 2012 | 2011 | 2010 | ||||||||||||||||
OPERATIONS | |||||||||||||||||||||
Service revenue | $ | 1,939,360 | $ | 1,734,442 | $ | 1,396,508 | $ | 1,212,099 | $ | 1,131,039 | |||||||||||
Income from operations | $ | 199,498 | $ | 136,123 | $ | 88,802 | $ | 81,630 | $ | 83,109 | |||||||||||
Net income | $ | 129,094 | $ | 95,972 | $ | 63,158 | $ | 48,786 | $ | 41,542 | |||||||||||
Basic earnings per share | $ | 2.28 | $ | 1.64 | $ | 1.06 | $ | 0.83 | $ | 0.72 | |||||||||||
Diluted earnings per share | $ | 2.25 | $ | 1.61 | $ | 1.05 | $ | 0.81 | $ | 0.71 | |||||||||||
FINANCIAL POSITION | |||||||||||||||||||||
Cash and marketable securities | $ | 283,812 | $ | 274,164 | $ | 213,579 | $ | 89,056 | $ | 107,413 | |||||||||||
Working capital | $ | 350,900 | $ | 403,229 | $ | 359,590 | $ | 317,298 | $ | 167,498 | |||||||||||
Total assets | $ | 1,834,000 | $ | 1,779,624 | $ | 1,532,156 | $ | 1,429,483 | $ | 1,220,710 | |||||||||||
Short-term debt | $ | 12,501 | $ | 20,399 | $ | 5,003 | $ | 5,867 | $ | 32,082 | |||||||||||
Long-term debt | $ | 337,500 | $ | 427,500 | $ | 211,784 | $ | 240,102 | $ | 183,707 | |||||||||||
Stockholders’ equity | $ | 577,681 | $ | 538,946 | $ | 609,675 | $ | 566,004 | $ | 439,555 | |||||||||||
OTHER DATA | |||||||||||||||||||||
Purchases of property and equipment | $ | 72,585 | $ | 81,089 | $ | 74,403 | $ | 60,153 | $ | 78,959 | |||||||||||
Depreciation and amortization | $ | 81,328 | $ | 73,186 | $ | 66,172 | $ | 65,480 | $ | 60,320 | |||||||||||
Number of employees | 15,560 | 14,690 | 12,695 | 10,550 | 9,720 | ||||||||||||||||
Weighted average shares | |||||||||||||||||||||
Basic | 56,504 | 58,388 | 59,464 | 58,634 | 58,062 | ||||||||||||||||
Diluted | 57,477 | 59,447 | 60,426 | 59,874 | 58,756 | ||||||||||||||||
29
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
OVERVIEW
We are a leading biopharmaceutical outsourcing services company, providing a broad range of expertise in clinical research, clinical logistics, medical communications, consulting, commercialization and advanced technology products and services to the worldwide pharmaceutical, biotechnology, and medical device industries. Our primary objective is to provide quality solutions for managing the biopharmaceutical product lifecycle with the goal of reducing the time, risk, and cost associated with the development and commercialization of new therapies. Since our incorporation in 1983, we have developed significant expertise in processes and technologies supporting this strategy. Our product and service offerings include: clinical trials management, observational studies and patient/disease registries, data management, biostatistical analysis, epidemiology, health economics / outcomes research, pharmacovigilance, medical communications, clinical pharmacology, patient recruitment, clinical supply and drug logistics, post-marketing surveillance, regulatory and product development and commercialization consulting, health policy and reimbursement and market access consulting, medical imaging services, regulatory information management ("RIM") solutions, ClinPhone randomization and trial supply management services (“RTSM”), electronic data capture systems (“EDC”), clinical trial management systems (“CTMS”), web-based portals, systems integration, patient diary applications, and other product development tools and services. We believe that our comprehensive services, depth of therapeutic area expertise, global footprint and related access to patients, and sophisticated information technology, along with our experience in global drug development and product launch services, represent key competitive strengths.
We have three reporting segments: Clinical Research Services (“CRS”), PAREXEL Consulting Services (“PC”), formerly known as PAREXEL Consulting and Medical Communication Services, and PAREXEL Informatics, Inc. (“PI”), formerly known as Perceptive Informatics, Inc.
• | CRS constitutes our core business and includes all phases of clinical research from Early Phase (encompassing the early stages of clinical testing that range from first-in-man through proof-of-concept studies) to Phase II-III and Phase IV, which we call Peri/Post-Approval Services, formerly known as Peri-Approval Clinical Excellence. Our services include clinical trials management and biostatistics, data management and clinical pharmacology, as well as related medical advisory, patient recruitment, pharmacovigilance, and investigator site services. CRS also includes our clinical supply and drug logistics business. We have aggregated Early Phase with Phase II-III and Peri/Post-Approval Services due to economic similarities in these operating segments. |
• | PC provides technical expertise and advice in such areas as drug development, regulatory affairs, product pricing and reimbursement, commercialization and strategic compliance. It also provides a full spectrum of market development, product development, and targeted communications services in support of product launch. Our PC consultants identify alternatives and propose solutions to address client issues associated with product development, registration, and commercialization. |
• | PI provides information technology solutions designed to help improve clients’ product development and regulatory submission processes. PI offers a portfolio of products and services that includes medical imaging services, ClinPhone® RTSM, IMPACT® CTMS, DataLabs® EDC, web-based portals, systems integration, electronic patient reported outcomes (“ePRO”) and LIQUENT InSight® RIM platform. These services are often bundled together and integrated with other applications to provide an eClinical solution for our clients. |
In February 2014, we announced the launch of PAREXEL Regulatory Outsourcing Services (“PROS”), which was designed to provide a focused, market-driven approach to regulatory outsourcing services in the life science industry, with a primary emphasis on post-approval regulatory activities. Effective July 1, 2014, the operating results of PROS are included in the PC segment. This service line offering was previously included within LIQUENT RIM solutions and reported within the PI segment. For interim and annual periods beginning July 1, 2014, we will disclose the reportable segment on this new basis and prior periods will be retroactively restated to reflect the change.
We conduct a significant portion of our operations in countries which are outside of the United States. Approximately 53.0% and 54.0% of our consolidated service revenue for the fiscal year 2014 and 2013 ended June 30, 2014 (“Fiscal Year 2014”) and the fiscal year ended June 30, 2013 (“Fiscal Year 2013”), respectively, were from non-U.S. operations. Because our financial statements are denominated in U.S. dollars, changes in foreign currency exchange rates can have a significant effect on our operating results. For Fiscal Year 2014, approximately 13.7% of total consolidated service revenue was from euro-denominated contracts and approximately 2.9% of total consolidated service revenue was from pound sterling-denominated contracts. For Fiscal Year 2013, approximately 13.0% of total consolidated service revenue was from euro-denominated contracts and approximately 2.6% of total consolidated service revenue was from pounds sterling-denominated contracts.
Approximately 90% of our contracts are fixed price, with some variable components, and range in duration from a few months to several years. Cash flows from these contracts typically consist of a down payment required at the time of contract execution
30
with the balance due in installments over the contract’s duration, usually on a milestone achievement basis. Revenue from these contracts is recognized generally as work is performed. As a result, the timing of client billing and cash receipts do not necessarily correspond to costs incurred and revenue recognized on contracts.
Generally, our clients can either terminate their contracts with us upon thirty to sixty days notice or delay execution of services. Clients may terminate or delay contracts for a variety of reasons, including: merger or potential merger-related activities involving the client, the failure of products being tested to satisfy safety requirements or efficacy criteria, unexpected or undesired clinical results of the product, client cost reductions as a result of budgetary limits or changing priorities, the client’s decision to forego a particular study, insufficient patient enrollment or investigator recruitment, or clinical drug manufacturing problems resulting in shortages of the product. In the cases where the contracts are canceled, services delivered through the cancellation date are due and payable by the client, including certain costs to conclude the trial or study.
CRITICAL ACCOUNTING POLICIES AND ESTIMATES
This discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the U.S. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities, revenues and expenses, and other financial information. On an ongoing basis, we evaluate our estimates and judgments. We base our estimates on historical experience and on various other factors that we believe to be reasonable under the circumstances. Actual results may differ from these estimates under different assumptions or conditions.
We regard an accounting estimate underlying our financial statements as a “critical accounting estimate” if the nature of the estimate or assumption is material due to the level of subjectivity and judgment involved, or the susceptibility of such matter to change, and if the impact of the estimate or assumption on financial condition or operating performance is material. We believe that the following accounting policies are most critical to aid in fully understanding and evaluating our reported financial results:
REVENUE RECOGNITION
We derive revenue from the delivery of services or software solutions to clients in the worldwide pharmaceutical, biotechnology, and medical device industries. We recognize revenue as services are performed when all of the following conditions are satisfied: (1) there is persuasive evidence of an arrangement; (2) the service offering has been delivered to the client; (3) the collection of fees is probable; and (4) the amount of fees to be paid by the client is fixed or determinable.
Our client arrangements in CRS generally involve multiple service deliverables, where bundled service deliverables are accounted for in accordance with Accounting Standards Codification (“ASC”) 605-25, “Multiple-Element Arrangements.” We determined that standalone value exists for each of our service deliverables and we base the selling price upon third-party evidence (“TPE”). TPE is established for each of our arrangement deliverables based on the price we charge for equivalent services when sold to other similar customers as well as our knowledge of market-pricing from the competitive bidding process for customer contracts offering similar services to comparably situated customers.
Within PI’s Clinphone® RTSM business, we offer selected software solutions through a hosted application delivered through a standard web-browser. We recognize revenue from application hosting services in accordance with ASC 985-605, “Revenue Recognition in the Software Industry” and ASC 605-25 as our customers do not have the right to take possession of the software. Revenue resulting from these hosting services is recognized over the service period.
Critical management estimates may be involved in the determination of the customer relationship period, and other revenue elements. Changes to these elements could affect the amount and timing of revenue recognition.
BILLED AND UNBILLED ACCOUNTS RECEIVABLE
Billed accounts receivable represent amounts for which invoices have been sent to clients based upon contract terms. Unbilled accounts receivable represent amounts recognized as revenue for which invoices have not yet been sent to clients due to contract terms. We maintain a provision for losses on receivables based on historical collectability and specific identification of potential problem accounts. Critical management estimates may be involved in the determination of “collectability” and the amounts required to be recorded as provisions for losses on receivables.
INCOME TAXES
Our global provision for corporate income taxes is determined in accordance with ASC 740, “Income Taxes,” which requires that deferred tax assets and liabilities be recognized for the effect of temporary differences between the book and tax basis of recorded assets and liabilities. A valuation allowance is established if it is more likely than not that future tax benefits from the deferred tax assets will not be realized. Income tax expense is based on the distribution of profit before tax among the various taxing jurisdictions in which we operate, adjusted as required by the tax laws of each taxing jurisdiction. Changes in the distribution of profits and losses among taxing jurisdictions may have a significant impact on our effective tax rate.
31
We account for uncertain tax positions in accordance with the provisions of ASC 740, which requires financial statement reporting of the expected future tax consequences of uncertain tax return reporting positions on the presumption that all relevant tax authorities possess full knowledge of those tax reporting positions, as well as all of the pertinent facts and circumstances. In addition, ASC 740 requires financial statement disclosure about uncertainty in income tax reporting positions.
We are subject to ongoing audits by federal, state and foreign tax authorities that may result in proposed assessments. Our estimate of the potential outcome for any uncertain tax issue is based on judgment. We believe we have adequately provided for any uncertain tax positions. However, future results may include favorable or unfavorable adjustments to our estimated tax liabilities in the period assessments are made or resolved or when statutes of limitation on potential assessments expire.
GOODWILL AND INDEFINITE-LIVED INTANGIBLES
Goodwill represents the excess of the cost of an acquired business over the fair value of the related net assets at the date of acquisition and is subject to annual impairment testing or more frequent testing if an event occurs or circumstances change that would more likely than not reduce the fair value below its carrying value. Our impairment testing for goodwill and an indefinite-lived intangible, the ClinPhone RTSM tradename, involves assessment of qualitative factors to determine whether it is more likely than not (a likelihood of more than 50%) that the fair value of a reporting unit or the fair value of the indefinite-lived intangibles is less than its carrying amount, including goodwill. This assessment requires management judgment on the potential impact of each qualitative factor. Based on our Fiscal Year 2014 qualitative assessment of impairment for goodwill and our ClinPhone RTSM tradename, we concluded that neither were impaired.
BUSINESS COMBINATIONS
Business combinations are accounted for under the acquisition method of accounting. Allocating the purchase price requires us to estimate the fair value of various assets acquired and liabilities assumed, including contingent consideration to be paid if specific financial targets are achieved. We are responsible for determining the appropriate valuation model and estimated fair values, and in doing so, we consider a number of factors, including information provided by an outside valuation advisor. We primarily establish fair value using the income approach based upon a discounted cash flow model. The income approach requires the use of many assumptions and estimates including future revenues and expenses, as well as discount factors and income tax rates.
Contingent consideration liabilities are remeasured to fair value each reporting period using projected financial targets, discount rates, probabilities of payment and projected payment dates. Projected contingent payment amounts are discounted back to the current period using a discounted cash flow model. Increases or decreases in projected financial targets and probabilities of payment may result in significant changes in the fair value measurements. Increases in discount rates and the time to payment may result in lower fair value measurements. Increases or decreases in any of those inputs in isolation may result in a significantly lower or higher fair value measurement.
32
RESULTS OF OPERATIONS
Note 18 to our consolidated financial statements included in this annual report provides a summary of our unaudited quarterly results of operations for Fiscal Years 2014 and 2013.
ANALYSIS BY SEGMENT
We evaluate our segment performance and allocate resources based on service revenue and gross profit (service revenue less direct costs), while other operating costs are allocated and evaluated on a geographic basis. Accordingly, we do not include the impact of selling, general, and administrative expenses, depreciation and amortization expense, interest income (expense), other income (loss), and income tax expense (benefit) in segment profitability. We attribute revenue to individual countries based upon the cost of services performed in the respective countries and inter-segment transactions are not included in service revenue. Furthermore, we have a global infrastructure supporting our business segments and therefore, assets are not identified by reportable segment. Service revenue, direct costs, and gross profit on service revenue for Fiscal Years 2014, 2013, and the fiscal year ended June 30, 2012 (“Fiscal Year 2012”) were as follows:
(in thousands) | Years Ended | Increase $ | Increase % | ||||||||||||
June 30, 2014 | June 30, 2013 | ||||||||||||||
Service revenue | |||||||||||||||
CRS | $ | 1,455,279 | $ | 1,303,569 | $ | 151,710 | 11.6 | % | |||||||
PC | 216,184 | 202,524 | 13,660 | 6.7 | % | ||||||||||
PI | 267,897 | 228,349 | 39,548 | 17.3 | % | ||||||||||
Total service revenue | $ | 1,939,360 | $ | 1,734,442 | $ | 204,918 | 11.8 | % | |||||||
Direct costs | |||||||||||||||
CRS | $ | 1,010,069 | $ | 956,513 | $ | 53,556 | 5.6 | % | |||||||
PC | 124,686 | 120,954 | 3,732 | 3.1 | % | ||||||||||
PI | 144,423 | 130,069 | 14,354 | 11.0 | % | ||||||||||
Total direct costs | $ | 1,279,178 | $ | 1,207,536 | $ | 71,642 | 5.9 | % | |||||||
Gross profit | |||||||||||||||
CRS | $ | 445,210 | $ | 347,056 | $ | 98,154 | 28.3 | % | |||||||
PC | 91,498 | 81,570 | 9,928 | 12.2 | % | ||||||||||
PI | 123,474 | 98,280 | 25,194 | 25.6 | % | ||||||||||
Total gross profit | $ | 660,182 | $ | 526,906 | $ | 133,276 | 25.3 | % | |||||||
(in thousands) | Years Ended | Increase $ | Increase % | ||||||||||||
June 30, 2013 | June 30, 2012 | ||||||||||||||
Service revenue | |||||||||||||||
CRS | $ | 1,303,569 | $ | 1,038,705 | $ | 264,864 | 25.5 | % | |||||||
PC | 202,524 | 167,125 | 35,399 | 21.2 | % | ||||||||||
PI | 228,349 | 190,678 | 37,671 | 19.8 | % | ||||||||||
Total service revenue | $ | 1,734,442 | $ | 1,396,508 | $ | 337,934 | 24.2 | % | |||||||
Direct costs | |||||||||||||||
CRS | $ | 956,513 | $ | 759,539 | $ | 196,974 | 25.9 | % | |||||||
PC | 120,954 | 97,560 | 23,394 | 24.0 | % | ||||||||||
PI | 130,069 | 114,730 | 15,339 | 13.4 | % | ||||||||||
Total direct costs | $ | 1,207,536 | $ | 971,829 | $ | 235,707 | 24.3 | % | |||||||
Gross profit | |||||||||||||||
CRS | $ | 347,056 | $ | 279,166 | $ | 67,890 | 24.3 | % | |||||||
PC | 81,570 | 69,565 | 12,005 | 17.3 | % | ||||||||||
PI | 98,280 | 75,948 | 22,332 | 29.4 | % | ||||||||||
Total gross profit | $ | 526,906 | $ | 424,679 | $ | 102,227 | 24.1 | % | |||||||
33
FISCAL YEAR ENDED JUNE 30, 2014 COMPARED WITH THE FISCAL YEAR ENDED JUNE 30, 2013
Revenue
Service revenue increased by $205.0 million, or 11.8%, to $1,939.4 million for Fiscal Year 2014 from $1,734.4 million for Fiscal Year 2013. On a geographic basis, service revenue was distributed as follows (in millions):
Fiscal Year 2014 | Fiscal Year 2013 | |||||||||||||
Region | Service Revenue | % of Total | Service Revenue | % of Total | ||||||||||
The Americas | $ | 970.9 | 50.1 | % | $ | 867.0 | 50.0 | % | ||||||
Europe, Middle East & Africa | $ | 709.2 | 36.6 | % | $ | 624.0 | 36.0 | % | ||||||
Asia/Pacific | $ | 259.3 | 13.3 | % | $ | 243.4 | 14.0 | % | ||||||
Total | $ | 1,939.4 | 100.0 | % | $ | 1,734.4 | 100.0 | % | ||||||
For Fiscal Year 2014 compared with Fiscal Year 2013, service revenue in The Americas increased by $103.9 million, or 12.0%; Europe, Middle East & Africa service revenue increased by $85.2 million, or 13.7%; and Asia/Pacific service revenue increased by $15.9 million, or 6.5%. Revenue growth in all regions was attributable to higher demand for services in all of our reporting segments, the impact of our strategic partnership wins, and additional revenue from our LIQUENT Inc.(“LIQUENT”) and HERON Group LTD (“HERON”) businesses, which we acquired in December 2012 and April 2013, respectively.
On a segment basis, CRS service revenue increased by $151.7 million, or 11.6%, to $1,455.3 million for Fiscal Year 2014 from $1,303.6 million for Fiscal Year 2013. The increase was attributable to growth in both the Phase II-III and Peri/Post Approval Service and the Early Phase businesses. Within the Phase II-III and Peri/Post Approval Service businesses, the efforts of a more productive employee base and the growth of backlog with a higher conversion rate caused revenue to increase across all of our client segments. The Early Phase business increases were due to increased demand for study services with patients.
PC service revenue increased by $13.7 million, or 6.7%, to $216.2 million for Fiscal Year 2014 from $202.5 million for Fiscal Year 2013. Higher service revenue was due primarily to $9.7 million of revenue from HERON, and the increase in consulting services associated with growth in the integrated product development consulting service line.
PI service revenue increased by $39.5 million, or 17.3%, to $267.9 million for Fiscal Year 2014 from $228.3 million for Fiscal Year 2013. The increase was primarily due to growth across all PI service lines due to higher demand for technology usage in clinical trials and the positive impact of strategic partnerships, along with $17.5 million of revenue from LIQUENT.
Reimbursement revenue consists of reimbursable out-of-pocket expenses incurred on behalf of and reimbursable by clients. Reimbursement revenue does not yield any gross profit to us, nor does it have an impact on net income.
Direct Costs
Direct costs increased by $71.6 million, or 5.9%, to $1,279.2 million for Fiscal Year 2014 from $1,207.5 million for Fiscal Year 2013. As a percentage of total service revenue, direct costs decreased to 66.0% from 69.6% for the respective periods. The gross margin improvement primarily related to the impact of various productivity and efficiency initiatives, changes in the revenue mix, and the increased sourcing of operations to low-cost countries.
On a segment basis, CRS direct costs increased by $53.6 million, or 5.6%, to $1,010.1 million for Fiscal Year 2014 from $956.5 million for Fiscal Year 2013. This increase resulted primarily from increased labor costs associated with headcount growth in CRS to match the demand of higher levels of clinical trial activity. As a percentage of service revenue, CRS direct costs decreased to 69.4% for Fiscal Year 2014 from 73.4% for Fiscal Year 2013. The decrease as a percentage of service revenue was related to the results of our operational efficiency programs, the reduction in contract staff usage, and the impact of shifting activities to low-cost countries.
PC direct costs increased by $3.7 million, or 3.1%, to $124.7 million for Fiscal Year 2014 from $121.0 million for Fiscal Year 2013. This increase was primarily due to higher headcount levels and direct costs from HERON. As a percentage of service revenue, PC direct costs decreased to 57.7% from 59.7% for the respective periods as a result of a more favorable revenue mix.
PI direct costs increased by $14.4 million, or 11.0%, to $144.4 million for Fiscal Year 2014 from $130.1 million for Fiscal Year 2013. This increase was due primarily to the inclusion of LIQUENT direct costs. As a percentage of service revenue, Perceptive direct costs decreased to 53.9% for Fiscal Year 2014 from 57.0% for Fiscal Year 2013 due to revenue growth and the impact of shifting resources to low-cost countries.
Selling, General and Administrative
Selling, general and administrative (“SG&A”) expense increased by $61.0 million, or 19.1%, to $379.8 million for Fiscal Year 2014 from $318.8 million for Fiscal Year 2013. This increase was due primarily to an increase in fixed and variable
34
compensation costs attributable to the larger employee base needed to support business growth, an increase in costs incurred to support our information technology infrastructure and facility expansion to better accommodate our growth, and an inclusion of $12.2 million in costs related to LIQUENT and HERON. As a percentage of service revenue, SG&A increased to 19.6% in Fiscal Year 2014 from 18.4% in Fiscal Year 2013.
Depreciation and Amortization
Depreciation and amortization (“D&A”) expense increased by $8.1 million, or 11.1%, to $81.3 million for Fiscal Year 2014 from $73.2 million for Fiscal Year 2013, due to higher amortization expense from the increase in intangible assets driven by the LIQUENT and HERON acquisitions and higher depreciation expense from the increasing capital expenditures from Fiscal Year 2011 to Fiscal Year 2013 to support business growth. As a percentage of service revenue, D&A was 4.2% for both Fiscal Year 2014 and Fiscal Year 2013 .
Restructuring Charge
Our restructuring plans were substantially completed by March 2012. For Fiscal Year 2014 and Fiscal Year 2013, respectively, we recorded a $0.4 million and $1.2 million net reduction in restructuring charges for adjustments to facility-related charges under our previously announced restructuring plans.
Income from Operations
Income from operations increased to $199.5 million for Fiscal Year 2014 from $136.1 million for Fiscal Year 2013. Income from operations as a percentage of service revenue ("operating margin") increased to 10.3% from 7.8% for the respective periods. This increase in operating margin was due primarily to higher gross margin, partially offset by higher SG&A and depreciation and amortization expenses.
Other Expense, Net
We recorded net other expense of $11.6 million for Fiscal Year 2014 compared with $3.0 million for Fiscal Year 2013. The $8.66 million increase was driven primarily by a $6.8 million increase in miscellaneous expenses, largely due to net foreign currency exchange losses recorded during Fiscal Year 2014 compared to the net foreign currency exchange gains recorded during Fiscal Year 2013. Additionally, the increase is attributable a $1.9 million increase in net interest expense related to a higher average debt balance in Fiscal Year 2014.
Taxes
For Fiscal Year 2014 and Fiscal Year 2013, we had effective income tax rates of 31.3% and 27.9%, respectively. The increase in Fiscal Year 2014 tax rate was primarily attributable to a shift in the geographic distribution of income which increased income subject to taxation in the United States relative to lower tax rate jurisdictions (primarily EU countries and the U.K.).
35
FISCAL YEAR ENDED JUNE 30, 2013 COMPARED WITH THE FISCAL YEAR ENDED JUNE 30, 2012
Revenue
Service revenue increased by $337.9 million, or 24.2%, to $1,734.4 million for Fiscal Year 2013 from $1,396.5 million for Fiscal Year 2012. On a geographic basis, service revenue was distributed as follows (in millions):
Fiscal Year 2013 | Fiscal Year 2012 | |||||||||||||
Region | Service Revenue | % of Total | Service Revenue | % of Total | ||||||||||
The Americas | $ | 867.0 | 50.0 | % | $ | 635.3 | 45.5 | % | ||||||
Europe, Middle East & Africa | $ | 624.0 | 36.0 | % | $ | 555.4 | 39.8 | % | ||||||
Asia/Pacific | $ | 243.4 | 14.0 | % | $ | 205.8 | 14.7 | % | ||||||
Total | $ | 1,734.4 | 100.0 | % | $ | 1,396.5 | 100.0 | % | ||||||
For Fiscal Year 2013 compared with Fiscal Year 2012, service revenue in The Americas increased by $231.7 million, or 36.5%; Europe, Middle East & Africa service revenue increased by $68.6 million, or 12.4%; and Asia/Pacific service revenue increased by $37.6 million, or 18.3%. Revenue growth in all regions was attributable to higher demand for services in all of our reporting segments and the impact of our strategic partnership wins. The conversion of backlog to revenue increased as projects matured and moved from startup phases to ongoing monitoring activities. The higher levels of service revenue growth in the Americas region was due to increased activity in the Phase II-III and Peri/Post Approval Service businesses. Service revenue growth was negatively impacted by foreign currency exchange rate fluctuations of approximately $15.1 million.
On a segment basis, CRS service revenue increased by $264.9 million, or 25.5%, to $1,303.6 million for Fiscal Year 2013 from $1,038.7 million for Fiscal Year 2012. The increase was attributable to growth in both our Phase II-III and Peri/Post Approval Service businesses and our Early Phase business. These increases were partly offset by a $12.4 million negative impact from foreign currency exchange rate movements. The Phase II-III and Peri/Post Approval Service increases were due to our success in forging strategic partnership relationships which has benefited our pipeline of work. Higher levels of new business awards won in prior periods across our entire customer base resulted in more active projects and, coupled with the efforts of a larger and more productive employee base, caused the conversion of backlog into revenue to accelerate. The revenue increase in our Early Phase business was due to improvements in our win rate among emerging clients combined with success in winning additional strategic partner relationships.
PC service revenue increased by $35.4 million, or 21.2%, to $202.5 million for Fiscal Year 2013 from $167.1 million for Fiscal Year 2012. The increase was due primarily to a $38.2 million increase in consulting services associated with growth in strategic compliance work due to higher levels of regulatory activity. These increases were partly offset by a $2.8 million decrease in our medical communications and commercialization service revenue.
PI service revenue increased by $37.7 million, or 19.8%, to $228.3 million for Fiscal Year 2013 from $190.7 million for Fiscal Year 2012. The growth was due primarily to $20.2 million in revenue from LIQUENT and a $17.5 million increase in eClinical and medical imaging services. Excluding the impact from LIQUENT, the continued growth in PI service revenue was due to higher demand for technology usage in clinical trials and the positive impact of strategic partnerships.
Reimbursement revenue consists of reimbursable out-of-pocket expenses incurred on behalf of and reimbursable by clients. Reimbursement revenue does not yield any gross profit to us, nor does it have an impact on net income.
Direct Costs
Direct costs increased by $235.7 million, or 24.3%, to $1,207.5 million for Fiscal Year 2013 from $971.8 million for Fiscal Year 2012. As a percentage of total service revenue, direct costs remained consistent at 69.6% for both periods.
On a segment basis, CRS direct costs increased by $197.0 million, or 25.9%, to $956.5 million for Fiscal Year 2013 from $759.5 million for Fiscal Year 2012. This increase resulted primarily from increased labor costs associated with headcount growth in CRS to match the demand of higher levels of clinical trial activity. Increased labor costs were also affected by upward pressure on wage rates in certain markets due to labor shortages and an increase in the number of contracted staff needed for projects in response to the higher number of studies that were initiated in Fiscal Year 2012 and the beginning of Fiscal Year 2013. The use of contracted staff declined in the latter half of Fiscal Year 2013 as full-time employees were hired to replace contract staff, and as the CRS employee base increased its productivity and efficiency. As a percentage of service revenue, CRS direct costs increased slightly to 73.4% for Fiscal Year 2013 from 73.1% for Fiscal Year 2012.
PC direct costs increased by $23.4 million, or 24.0%, to $121.0 million for Fiscal Year 2013 from $97.6 million for Fiscal Year 2012. This increase resulted primarily from higher labor costs in our consulting services unit due to increased demand related to strategic compliance work. Offsetting this increase was a $4.1 million decline in labor costs within the medical communications business. As a percentage of service revenue, PC direct costs increased to 59.7% from 58.4% for the
36
respective periods as a result of higher labor costs associated with consulting services and short-term investments directed at better positioning the business for continued growth.
PI direct costs increased by $15.3 million, or 13.4%, to $130.1 million for Fiscal Year 2013 from $114.7 million for Fiscal Year 2012. This increase was due primarily to the impact of LIQUENT, an increase in labor costs in existing businesses, and higher medical imaging "read" expenses associated with greater volume. As a percentage of service revenue, PI direct costs decreased to 57.0% for Fiscal Year 2013 from 60.2% for Fiscal Year 2012 due to the impact of shifting resources to low cost countries and a better revenue mix.
Selling, General and Administrative
SG&A expense increased by $55.3 million, or 21.0%, to $318.8 million for Fiscal Year 2013 from $263.5 million for Fiscal Year 2012. This increase was primarily due to the addition of LIQUENT and HERON SG&A costs, an increase in fixed and variable compensation costs attributable to the larger employee base needed to support business growth, and an increase in rent and other office-related expenses. As a percentage of service revenue, SG&A decreased to 18.4% in Fiscal Year 2013 from 18.9% in Fiscal Year 2012 due to leveraging of our revenue growth, effective cost management, and the benefits of past restructuring activities.
Depreciation and Amortization
D&A expense increased by $7.0 million, or 10.6%, to $73.2 million for Fiscal Year 2013 from $66.2 million for Fiscal Year 2012, primarily due to additional depreciation expense from increased capital expenditures in both Fiscal Year 2013 and Fiscal Year 2012 and higher amortization expense from the intangibles assets acquired in conjunction with the LIQUENT and HERON acquisitions. As a percentage of service revenue, D&A decreased to 4.2% for Fiscal Year 2013 from 4.7% for Fiscal Year 2012 mainly due to revenue growth.
Restructuring Charge
Our restructuring plans were substantially completed by March 2012. For Fiscal Year 2013, we recorded a $1.2 million net reduction in restructuring charges for adjustments to facility-related charges under our previously announced restructuring plans.
For Fiscal Year 2012, we recorded $6.2 million in restructuring charges under our restructuring plans, including $4.3 million in employee separation benefits and $1.9 million of facility-related costs.
Income from Operations
Income from operations increased to $136.1 million for Fiscal Year 2013 from $88.8 million for Fiscal Year 2012 due to the factors described above. Operating margin increased to 7.8% from 6.4% for the respective periods for the reasons discussed above.
Other Expense, Net
We recorded net other expense of $3.0 million for Fiscal Year 2013 compared with $9.1 million for Fiscal Year 2012. The $6.1 million decrease was due primarily to lower miscellaneous expense, partially offset by $0.3 million increase in net interest expense.
Miscellaneous income for Fiscal Year 2013 of $4.3 million was driven by $4.1 million in foreign exchange gains from certain foreign-denominated assets and liabilities.
Miscellaneous expense for Fiscal Year 2012 of $2.1 million was primarily attributable to $6.3 million of unrealized losses related to derivatives contracts and $2.4 million of losses from asset disposals and loan write-offs, partly offset by $6.6 million in foreign exchange gains from certain foreign-denominated assets and liabilities.
Taxes
For Fiscal Year 2013 and Fiscal Year 2012, we had effective income tax rates of 27.9% and 20.8%, respectively. The increase in the Fiscal Year 2013 tax rate was primarily attributable to a lower level of income tax reserve releases and a shift in the geographic distribution of income which increased income subject to taxation in the United States relative to lower tax rate jurisdictions (primarily EU countries and the UK), net of reductions in valuation allowances resulting from improved profitability in the United States. The lower tax rate for Fiscal Year 2012 was primarily the result of the release of income tax reserves and associated accruals for interest and penalties resulting from settlements with tax authorities and the expiration of statutes of limitations in Europe.
37
LIQUIDITY AND CAPITAL RESOURCES
Since our inception, we have financed our operations and growth with cash flow from operations, proceeds from the sale of equity securities, and credit facilities to fund business acquisitions and working capital. Investing activities primarily reflect capital expenditures for information systems enhancements and leasehold improvements. As of June 30, 2014, we had cash and cash equivalents of approximately $188.2 million and marketable securities of $95.6 million, of which the majority is held in countries which are outside of the U.S. since excess cash generated in the U.S. is primarily used to repay our debt obligations. Foreign cash and marketable securities balances include unremitted foreign earnings, which are invested indefinitely outside of the U.S. Our cash and cash equivalents and marketable securities are held in deposit accounts, money market funds and foreign government treasury certificates over 90 days but less than one year, which provide us with immediate and unlimited access to the funds. Repatriation of funds to the U.S. from non-U.S. entities may be subject to taxation or certain legal restrictions. Nevertheless, most of our cash resides in countries with few or no such restrictions.
DAYS SALES OUTSTANDING
Our operating cash flow is heavily influenced by changes in the levels of billed and unbilled receivables and deferred revenue. These account balances as well as days sales outstanding (“DSO”) in accounts receivable, net of deferred revenue, can vary based on contractual milestones and the timing and size of cash receipts. We calculate DSO by adding end-of-period balances for billed and unbilled account receivables, net of deferred revenue (short-term and long term) and the provision for losses on receivables, then dividing the resulting amount by the sum of total revenue plus investigator fees billed for the most recent quarter, and multiplying the resulting fraction by the number of days in the quarter. The following table presents the DSO, account receivables balances, and deferred revenue as of June 30, 2014 and June 30, 2013.
(in millions) | June 30, 2014 | June 30, 2013 | ||||||
Billed accounts receivable, net | $ | 497.1 | $ | 457.2 | ||||
Unbilled accounts receivable, net | 225.5 | 248.2 | ||||||
Total accounts receivable | 722.6 | 705.4 | ||||||
Deferred revenue | 467.0 | 408.3 | ||||||
Net receivables | $ | 255.6 | $ | 297.1 | ||||
DSO ( in days) | 32 | 42 | ||||||
The decrease in DSO for the quarter ended June 30, 2014 compared to the quarter ended June 30, 2013, was largely driven by strong collections and benefits of increased deferred revenue.
CASH FLOWS
Operating Activities
Net cash provided by operating activities was $287.2 million for Fiscal Year 2014 as compared with $183.8 million for Fiscal Year 2013. The $103.4 million increase in operating cash flows resulted primarily from $33.1 million of higher net income, a $79.2 million increase in net change of working capital and an $8.1 million increase in depreciation and amortization expenses. These increases were partially offset by a $17.5 million change in our deferred income tax provision.
Investing Activities
Net cash used for investing activities was $31.2 million for Fiscal Year 2014 as compared with $305.5 million for Fiscal Year 2013.
During Fiscal Year 2014, we made $72.6 million in capital expenditures, which was partially offset by $41.5 million in net proceeds from the sale of marketable securities.
During Fiscal Year 2013, we made net purchases of $129.6 million in marketable securities, paid $97.1 million in cash to acquire LIQUENT and HERON, and made $81.1 million in capital expenditures.
Financing Activities
Net cash used by financing activities was $224.0 million for Fiscal Year 2014 as compared with net cash provided by financing activities of $52.4 million for Fiscal Year 2013.
During Fiscal Year 2014, we paid $154.9 million for share repurchases and made net payments of $187.5 million under the 2013 Credit Agreement (as defined below), which were partially offset by $100.0 million borrowings under the Note Purchase Agreement (as defined below), $14.6 million in proceeds received related to employee stock purchases and $4.5 million in proceeds received from our receivable factoring.
38
During Fiscal Year 2013, we borrowed an additional $217.5 million under new and existing credit facilities to fund the LIQUENT and HERON acquisitions and share repurchases, received $20.9 million in proceeds from employee stock purchases, and received $10.4 million from receivable factoring. These proceeds were partially offset by $195.1 million paid for share repurchases and $1.2 million payments for debt issuance costs.
CREDIT AGREEMENTS
Note Purchase Agreement
On July 25, 2013, we issued $100.0 million principal amount of 3.11% senior notes due July 25, 2020 (the “Notes”) for aggregate gross proceeds of $100.0 million in a private placement solely to accredited investors. The Notes were issued pursuant to a Note Purchase Agreement entered into by us with certain institutional investors on June 25, 2013 (the “Note Purchase Agreement”). Proceeds from the Notes were used to pay down $100.0 million of principal borrowed under the revolving credit facility portion of the 2013 Credit Agreement. We will pay interest on the outstanding balance of the Notes at a rate of 3.11% per annum, payable semi-annually on January 25 and July 25 of each year until the principal on the Notes shall have become due and payable. We may, at our option, upon notice and subject to the terms of the Note Purchase Agreement, prepay at any time all or part of the Notes in an amount not less than 10% of the aggregate principal amount of the Notes then outstanding, plus a Make-Whole Amount (as defined in the Note Purchase Agreement). The Notes become due and payable on July 25, 2020, unless payment is required to be made earlier under the terms of the Note Purchase Agreement.
The Note Purchase Agreement includes operational and financial covenants, with which we are required to comply, including, among others, maintenance of certain financial ratios and restrictions on additional indebtedness, liens and dispositions.
In connection with the Note Purchase Agreement, certain subsidiaries of ours entered into a Subsidiary Guaranty, pursuant to which such subsidiaries guaranteed our obligations under the Notes and the Note Purchase Agreement.
As of June 30, 2014, we had $100.0 million of principal borrowed under the Note Purchase Agreement. The outstanding amounts are presented net of debt issuance cost of approximately $0.4 million in our consolidated balance sheets.
In April and May 2013, we entered into three treasury lock agreements each with a notional amount of $25.0 million in connection with the planned issuance of our Notes that were issued in July 2013. The three treasury locks were used to minimize our interest rate exposure prior to locking in the fixed interest rate on our Notes. The treasury locks matured in May 2013 when the interest rate on our Notes was fixed. The treasury locks were deemed to be fully effective in accordance with Financial Accounting Standards Board (“FASB”) ASC 815, “Derivatives and Hedging” (“ASC 815”), and as such, the unrealized gains related to these derivatives are recorded as other comprehensive income and are amortized over the life of the Notes as interest income.
Receivable Purchase Agreement
On February 19, 2013, we entered into a receivables purchase agreement (the “Receivable Agreement”) with JPMorgan Chase Bank, N.A. (“JPMorgan”). Under the Receivable Agreement, we sell to JPMorgan or other investors on an ongoing basis certain of our trade receivables, together with ancillary rights and the proceeds thereof, which arise under contracts with a client of ours, or its subsidiaries or affiliates. The Receivable Agreement includes customary representations and covenants on behalf of us, and may be terminated by either us or JPMorgan upon five business days advance notice. The Receivable Agreement provides a mechanism for accelerating the receipt of cash due on outstanding receivables. We account for the transfer of our receivables with respect to which we have satisfied the applicable revenue recognition criteria in accordance with FASB ASC 860, “Transfers and Servicing.” If we have not satisfied the applicable revenue recognition criteria for the underlying sales transaction, the transfer of the receivable is accounted for as a financing activity in accordance with FASB ASC 470, “Debt.” The accounts receivable and short-term debt balances are derecognized from our consolidated balance sheets at the earlier of the factored receivable’s due date or when all of the revenue recognition criteria are met for those billed services. For Fiscal Year 2014 and 2013, we transferred approximately $184.6 million and $36.5 million of trade receivables, respectively. As of June 30, 2013, we accounted for $10.4 million of the total transfers as financing activities which are recorded in accounts receivable and short-term debt on our consolidated balance sheets. As of June 30, 2014, no transfers were accounted for as a financing activity.
2013 Credit Agreement
On March 22, 2013, we, certain of our subsidiaries, Bank of America, N.A. (“Bank of America”), as Administrative Agent, Swingline Lender and L/C Issuer, Merrill Lynch, Pierce, Fenner & Smith Incorporated (“MLPFS”), J.P. Morgan Securities LLC (“JPM Securities”), HSBC Bank USA, National Association (“HSBC”) and U.S. Bank, National Association (“US Bank”), as Joint Lead Arrangers and Joint Book Managers, JPMorgan Chase Bank N.A. (“JPMorgan”), HSBC and US Bank, as Joint Syndication Agents, and the other lenders party thereto entered into an amended and restated agreement (the “2013 Credit Agreement”) providing for a five-year term loan of $200.0 million and a revolving credit facility in the amount of up to $300.0 million, plus additional amounts of up to $200.0 million of loans to be made available upon our request subject to specified
39
terms and conditions. A portion of the revolving credit facility is available for swingline loans of up to a sublimit of $75.0 million and for the issuance of standby letters of credit of up to a sublimit of $10.0 million.
On March 22, 2013, we drew down $107.5 million under the 2013 Credit Agreement resulting in total outstanding borrowings of $400.0 million. We used the proceeds of the borrowing (i) to repay outstanding amounts under our existing four short-term credit facilities with each of Bank of America, HSBC, TD Bank, N.A. and US Bank (collectively, the “Short Term Credit Facilities”), (ii) for stock repurchases and (iii) for other general corporate purposes.
Our obligations under the 2013 Credit Agreement are guaranteed by certain of our material domestic subsidiaries, and the obligations, if any, of any of our foreign designated borrower are guaranteed by us and certain of our material domestic subsidiaries.
Borrowings (other than swingline loans) under the 2013 Credit Agreement bear interest, at our determination, at a rate based on either (a) LIBOR plus a margin (not to exceed a per annum rate of 1.750%) based on a ratio of consolidated funded debt to consolidated earnings before interest, taxes, depreciation and amortization (“EBITDA”) (the “Leverage Ratio”) or (b) the highest of (i) prime, (ii) the federal funds rate plus 0.50%, and (iii) the one month LIBOR rate plus 1.00% (such highest rate, the “Alternate Base Rate”), plus a margin (not to exceed a per annum rate of 0.750%) based on the Leverage Ratio. Swingline loans in U.S. dollars bear interest calculated at the Alternate Base Rate plus a margin (not to exceed a per annum rate of 0.750%).
Loans outstanding under the 2013 Credit Agreement may be prepaid at any time in whole or in part without premium or penalty, other than customary breakage costs, if any, subject to the terms and conditions contained in the 2013 Credit Agreement. The 2013 Credit Agreement terminates and any outstanding loans under it mature on March 22, 2018 (the “Maturity Date”).
Repayment of the principal borrowed under the revolving credit facility (other than a swingline loan) is due on the Maturity Date. Repayment of principal borrowed under the term loan facility is as follows, with the final payment of all amounts outstanding, plus accrued interest, being due on the Maturity Date:
• | 1.25% by quarterly term loan amortization payments to be made commencing in June 2013 and made prior to June 30, 2015; |
• | 2.50% by quarterly term loan amortization payments to be made on or after June 30, 2015, but prior to June 30, 2016; |
• | 5.00% by quarterly term loan amortization payments to be made on or after June 30, 2016, but prior to June 30, 2017; |
• | 7.50% by quarterly term loan amortization payment to be made on or after June 30, 2017, but prior to the Maturity Date; and |
• | 37.50% on the Maturity Date |
Our obligations under the 2013 Credit Agreement may be accelerated upon the occurrence of an event of default, which includes customary events of default, including payment defaults, defaults in the performance of affirmative and negative covenants, the inaccuracy of representations or warranties, bankruptcy and insolvency related defaults, cross defaults to material indebtedness, defaults relating to such matters as ERISA and judgments, and a change of control default.
The 2013 Credit Agreement contains negative covenants applicable to us and our subsidiaries, including financial covenants requiring us to comply with maximum leverage ratios and minimum interest coverage ratios, as well as restrictions on liens, investments, indebtedness, fundamental changes, acquisitions, dispositions of property, making specified restricted payments (including stock repurchases exceeding an agreed to percentage of consolidated net income), and transactions with affiliates. As of June 30, 2014, we were in compliance with all covenants under the 2013 Credit Agreement.
In connection with the 2013 Credit Agreement, we agreed to pay a commitment fee on the revolving loan commitment calculated as a percentage of the unused amount of the revolving loan commitment at a per annum rate of up to 0.350% (based on the Leverage Ratio). To the extent there are letters of credit outstanding under the 2013 Credit Agreement, we will pay letter of credit fees plus a fronting fee and additional charges. We also paid various customary fees to secure this arrangement, which are being amortized using the effective interest method over the life of the debt.
As of June 30, 2014, we had $62.5 million of principal borrowed under the revolving credit facility and $187.5 million of
principal under the term loan. For Fiscal Year 2014, we made principal payments of $10.0 million on the term loan under the 2013 Credit Agreement. The outstanding amounts are presented net of debt issuance cost of approximately $2.7 million in our consolidated balance sheets. We have borrowing availability of $237.5 million under the revolving credit facility.
In September 2011, we entered into an interest rate swap agreement and an interest rate cap agreement. Prior to the execution of the 2013 Credit Agreement, the interest rate swap and cap agreements hedged principal under our 2011 Credit Agreement, as discussed below. The interest rate swap and cap agreements now hedge a portion of the principal under our 2013 Credit Agreement. Specifically, principal in the amount of $100.0 million under our 2013 Credit Agreement has been hedged with the interest rate swap agreement and carries a fixed interest rate of 1.30% plus an applicable margin. Principal in the amount of
40
$25.0 million has been hedged with the interest rate cap arrangement with an interest rate cap of 2.00% plus an applicable margin. In March 2014, the interest rate cap agreement matured and the related accumulated other comprehensive income was reclassified to net income during Fiscal Year 2014.
In May 2013, we entered into another interest rate swap agreement and hedged an additional principal amount of $100.0 million under our 2013 Credit Agreement with a fixed interest rate of 0.73% plus an applicable margin. As of June 30, 2014, our debt under the 2013 Credit Agreement, including the $200.0 million of principal hedged with both interest swap agreements, carried an average annualized interest rate of 1.60%. These interest rate hedges were deemed to be fully effective in accordance with ASC 815, “Derivatives and Hedging,” and, as such, unrealized gains and losses related to these derivatives are recorded as other comprehensive income.
2012 Term Loan and 2013 Facilities
On December 20, 2012, we entered into a new $100.0 million unsecured term loan agreement (the “2012 Term Loan”) with Bank of America, which was initially guaranteed by certain of our subsidiaries, but which guarantees were released in connection with the partial prepayment of the 2012 Term Loan in January 2013. The 2012 Term Loan was used to fund our acquisition of LIQUENT.
The 2012 Term Loan consisted of a term loan facility for $100.0 million, the full amount of which was advanced to us on December 21, 2012 and was scheduled to mature on June 30, 2013. Borrowings made under the 2012 Term Loan bore interest, at our option, at a base rate plus a margin (such margin not to exceed a per annum rate of 0.75%) based on a ratio of consolidated funded debt to EBITDA for the preceding twelve months (the “2012 Term Loan Leverage Ratio”), or at a LIBOR rate plus a margin (such margin not to exceed a per annum rate of 1.75%) based on the 2012 Term Loan Leverage Ratio. As of June 30, 2014, all outstanding amounts under the 2012 Term Loan were fully repaid with the proceeds from the 2013 Credit Agreement.
On January 22, 2013, we entered into additional short term unsecured term loan agreements with each of HSBC, TD Bank, N.A., and US Bank, each in the amount of $25.0 million (collectively, the “2013 Facilities”). The key terms of the 2013 Facilities were substantially the same as the 2012 Term Loan, including the loan maturities on June 30, 2013, except that there were no guaranties provided by any of our subsidiaries. The $75.0 million aggregate proceeds of the 2013 Facilities were used to partially pay down balances owed under the 2012 Term Loan, and in connection with such payment, Bank of America released our subsidiaries from their guaranty obligations under the 2012 Term Loan.
As of June 30, 2014, all outstanding amounts under the 2013 Facilities were fully repaid with the proceeds from the 2013 Credit Agreement.
2011 Credit Agreement
On June 30, 2011, we entered into the 2011 Credit Agreement providing for a five-year term loan of $100 million and a five-year revolving credit facility in the principal amount of up to $300 million. The borrowings all carried a variable interest rate based on LIBOR, prime, or a similar index, plus a margin (such margin not to exceed a per annum rate of 1.75%).
On March 22, 2013, the 2011 Credit Agreement was amended and restated in its entirety by the 2013 Credit Agreement. All amounts outstanding under the 2011 Credit Agreement immediately prior to the execution of the 2013 Credit Agreement were deemed to be outstanding under the terms and conditions of the 2013 Credit Agreement.
As discussed above, in September 2011, we entered into an interest rate swap and an interest rate cap agreement. Prior to the execution of the 2013 Credit Agreement, principal in the amount of $100.0 million under the 2011 Credit Agreement had been hedged with an interest rate swap agreement and carried a fixed interest rate of 1.30% plus an applicable margin. Principal in the amount of $25.0 million had been hedged with an interest rate cap arrangement with an interest rate cap of 2.00% plus an applicable margin.
Additional Lines of Credit
We have an unsecured line of credit with JP Morgan UK in the amount of $4.5 million that bears interest at an annual rate ranging between 2.00% and 4.00%. We entered into this line of credit to facilitate business transactions. At June 30, 2014, we had $4.5 million available under this line of credit.
We have a cash pool facility with RBS Nederland, NV in the amount of 5.0 million Euros that bears interest at an annual rate ranging between 2.00% and 4.00%. We entered into this line of credit to facilitate business transactions. At June 30, 2014, we had 5.0 million Euros available under this line of credit.
41
FINANCING NEEDS
Our primary cash needs are for operating expenses (such as salaries and fringe benefits, hiring and recruiting, business development and facilities), business acquisitions, stock buybacks, capital expenditures, and repayment of principal and interest on our borrowings.
Fiscal Year 2014 Share Repurchase
On June 2, 2014, we announced that our Board of Directors approved a share repurchase program (the “2014 Program”) authorizing the repurchase of up to $150.0 million of our common stock to be financed with cash on hand, cash generated from operations, existing credit facilities, or new financing. On June 13, 2014, we entered into an agreement (the “2014 Agreement”) to purchase shares of our common stock from Goldman Sachs & Co. (“GS”), for an aggregate purchase price of $150.0 million pursuant to an accelerated share purchase program. Pursuant to the 2014 Agreement, in June 2014, we paid $150.0 million to GS and received from GS 2,284,844 shares of common stock, representing 80 percent of the shares to be repurchased by us under the 2014 Agreement. The shares were repurchased at a price of $52.52 per share, which was the closing price of our common stock on the Nasdaq Global Select Market on June 13, 2014. These shares were canceled and restored to the status of authorized and unissued shares. As of June 30, 2014, we recorded the $150.0 million payment to GS as a decrease to equity in our consolidated balance sheet.
The final number of shares to be delivered to us by GS under the 2014 Agreement at program maturity, net of the initial delivery, will be adjusted based on an agreed upon discount to the average of the daily volume weighted average price of the common stock during the term of the 2014 Agreement. If the number of shares to be delivered to us at maturity is less than the initial delivery of shares by GS, we would be required to remit shares or cash, at our option, to GS in an amount equivalent to such shortfall. We expect the 2014 Program to be completed by December 31, 2014.
Credit Agreements and Note Purchase Agreement
In July 2013, we issued $100.0 million principal amount of 3.11% senior notes due July 25, 2020 for aggregate gross proceeds of $100.0 million in a private placement solely to accredited investors. We utilized the proceeds from the private placement to pay down debt outstanding under our 2013 Credit Agreement. The Note Purchase Agreement permits the proceeds from the private placement to be used for working capital purposes, stock repurchase financing, debt refinancing, and for general corporate purposes including the financing of acquisitions, subject in each case to the terms of the Note Purchase Agreement.
Our requirements for cash to pay principal and interest on our borrowings will increase significantly in future periods based on the repayment terms of our 2013 Credit Agreement and the Notes. Our primary committed external source of funds is the 2013 Credit Agreement. Our principal source of cash is from the performance of services under contracts with our clients. If we are unable to generate new contracts with existing and new clients or if the level of contract cancellations increases, our revenue and cash flow would be adversely affected (see Part II, Item 1A “Risk Factors” for further detail on these risks). Absent a material adverse change in the level of our new business bookings or contract cancellations, we believe that our existing capital resources together with cash flow from operations and borrowing capacity under existing credit facilities will be sufficient to meet our foreseeable cash needs over the next twelve months and on a longer-term basis. Depending upon our revenue and cash flow from operations, it is possible that we will require external funds to repay amounts outstanding under our 2013 Credit Agreement upon its maturity in 2018.
We expect to continue to acquire businesses that enhance our service and product offerings, expand our therapeutic expertise, and/or increase our global presence. Depending on their size, any future acquisitions may require additional external financing, and we may from time to time seek to obtain funds from public or private issuances of equity or debt securities. We may be unable to secure such financing at all or on terms acceptable to us, as a result of our outstanding borrowings, including our outstanding borrowings under the 2013 Credit Agreement.
Under the terms of the 2013 Credit Agreement, interest rates are fixed based on market indices at the time of borrowing and, depending upon the interest mechanism selected by us, may float thereafter. As a result, the amount of interest payable by us on our borrowings may increase if market interest rates change. However, we expect to mitigate the risk of increasing market interest rates with our hedging programs described below under Part II. Item 7A “Quantitative and Qualitative Disclosures About Market Risk - Foreign Currency Exchange Rates and Interest Rates.”
Capital Expenditures
We made capital expenditures of approximately $72.6 million during Fiscal Year 2014, primarily for computer software (including internally developed software), hardware, and leasehold improvements.
Fiscal Year 2013 Share Repurchase
In August 2012, our Board of Directors approved a share repurchase program (the “2013 Program”) authorizing the repurchase of up to $200.0 million of our common stock to be financed with cash on hand, cash generated from operations, existing credit
42
facilities, or new financing. During Fiscal Year 2013, we repurchased $197.6 million of our common stock. We repurchased the remaining $2.4 million of our common stock in July 2013. The 2013 Program repurchases were effected pursuant to a $50.0 million accelerated share repurchase agreement and a $50.0 million open market agreement entered into in September 2012 and a $50.0 million accelerated share repurchase agreement (“March 2013 ASR Agreement”) and a $50.0 million open market agreement (“March 2013 Open Market Agreement”) entered in March 2013. Pursuant to the 2013 Program, we repurchased 5,458,285 shares of our common stock at an average price of $36.64 per share from September 2012 to July 2013. The buyback activity also resulted in a reduction of our stockholders’ equity of $200.0 million for the value of shares repurchased and retired by the Company.
In July 2013, we purchased 51,071 shares under our March 2013 Open Market Agreement and received 101,247 shares representing the final settlement of our March 2013 ASR Agreement. With the completion of our March 2013 Open Market Agreement and the final settlement of the March 2013 ASR Agreement, the 2013 Program was completed.
DEBT, CONTRACTUAL OBLIGATIONS, CONTINGENT LIABILITIES AND GUARANTEES
The following table summarizes our contractual obligations at June 30, 2014:
(in thousands) | Less than 1 year | 2-3 years | 4-5 years | More than 5 years | Total | |||||||||||||||
Debt obligations (principal) | $ | 12,501 | $ | 70,000 | $ | 167,500 | $ | 100,000 | $ | 350,001 | ||||||||||
Operating leases | 62,103 | 91,912 | 56,384 | 99,818 | 310,217 | |||||||||||||||
Purchase obligations* | 55,036 | 23,929 | 2,608 | — | 81,573 | |||||||||||||||
Total | $ | 129,640 | $ | 185,841 | $ | 226,492 | $ | 199,818 | $ | 741,791 | ||||||||||
*includes commitments to purchase software, hardware, and services. | ||||||||||||||||||||
The above table does not include approximately $41.5 million of potential tax liabilities from unrecognized tax benefits related to uncertain tax positions. See Note 14 to our consolidated financial statements included in this annual report for more information.
We also did not include contingent payments related to the HERON acquisition, as the amounts of these payments will be determined based on the operating results of our commercialization service line for Fiscal Year 2015. The final payment related to the HERON acquisition may range from zero to $14.2 million. Any obligations will be paid during the three months ending September 30, 2015.
We have letter-of-credit agreements with banks, totaling approximately $10.5 million, guaranteeing performance under various operating leases and vendor agreements. Additionally, the borrowings under the 2013 Credit Agreement and Note Purchase Agreement are guaranteed by certain of our U.S. subsidiaries.
We periodically become involved in various claims and lawsuits that are incidental to our business. We believe, after consultation with counsel, that no matters currently pending would, in the event of an adverse outcome, either individually or in the aggregate, have a material impact on our consolidated financial position, results of operations, or liquidity.
OFF-BALANCE SHEET ARRANGEMENTS
We have no off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial position, changes in financial position, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that are material to our investors.
RESTRUCTURING PLANS
In April 2011, we adopted a plan to restructure our operations to reduce expenses, better align costs with geographic sources of revenue, and improve operating efficiencies (the “2011 Restructuring Plan”). During Fiscal Year 2011, we recorded $8.5 million in restructuring charges related to the 2011 Restructuring Plan, including approximately $1.8 million in employee separation benefits, $3.6 million in costs related to the abandonment of certain property leases, and $3.1 million in impairment charges related to exited facilities. During Fiscal Year 2012, we recorded $7.3 million in restructuring charges related to the 2011 Restructuring Plan, including $5.3 million in severance costs and $2.0 million in facility-related costs. During Fiscal Year 2013 and 2014, we recorded a $1.2 million and $0.4 million net reduction, respectively, to restructuring charges for adjustments to facility-related charges under the 2011 Restructuring Plan. The total cost of the 2011 Restructuring Plan was approximately $14.2 million and included the elimination of approximately 150 managerial and staff positions and costs related to the abandonment of certain property leases.
INFLATION
We believe the effects of inflation generally do not have a material adverse impact on our operations or financial condition.
43
RECENTLY ISSUED ACCOUNTING STANDARDS
In March 2013, the FASB issued ASU No. 2013-05, “Parent’s Accounting for the Cumulative Translation Adjustment upon Derecognition of Certain Subsidiaries or Groups of Assets within a Foreign Entity or of an Investment in a Foreign Entity.” ASU 2013-05 addresses the accounting for the cumulative translation adjustment when a parent either sells a part or all of its investment in a foreign entity or no longer holds a controlling financial interest in a subsidiary or group of assets that is a nonprofit activity or a business within a foreign entity. ASU 2013-05 is effective prospectively for fiscal years and interim periods within those fiscal years beginning after December 15, 2013 and early adoption is permitted. We do not expect the adoption of this guidance to have a material impact on our consolidated financial statements.
In May 2014, the FASB issued ASU No. 2014-09, Revenue from Contracts with Customers (“ASU 2014-09”), which stipulates that an entity should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. To achieve this core principle, an entity should apply the following steps: (1) identify the contract(s) with a customer; (2) identify the performance obligations in the contract; (3) determine the transaction price; (4) allocate the transaction price to the performance obligations in the contract; and (5) recognize revenue when (or as) the entity satisfies a performance obligation. ASU 2014-09 will be effective prospectively for fiscal years and interim periods within those fiscal years beginning after December 15, 2016, and early adoption is not permitted. The impact on our Financial Statements of adopting ASU 2014-09 is being assessed by our management.
In June 2014, the FASB issued ASU No. 2014-12, Accounting for Share-Based Payments When the Terms of an Award Provide That a Performance Target Could Be Achieved after the Requisite Service Period ("ASU 2014-12"). ASU 2014-12 requires that a performance target that affects vesting and could be achieved after the requisite service period be treated as a performance condition. A reporting entity should apply existing guidance in ASC 718, Compensation—Stock Compensation, as it relates to such awards. ASU 2014-12 is effective in the first quarter of our fiscal year ending June 30, 2017 with early adoption permitted using either of two methods: (i) prospective to all awards granted or modified after the effective date; or (ii) retrospective to all awards with performance targets that are outstanding as of the beginning of the earliest annual period presented in the financial statements and to all new or modified awards thereafter, with the cumulative effect of applying ASU 2014-12 as an adjustment to the opening retained earnings balance as of the beginning of the earliest annual period presented in the financial statements. We do not expect the adoption of this guidance to have a material impact on our consolidated financial statements.
44
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
MARKET RISK
Market risk is the potential loss arising from adverse changes in market rates and prices, such as foreign currency exchange rates, interest rates, and other relevant market rates or price changes. In the ordinary course of business, we are exposed to market risk resulting from changes in foreign currency exchange rates and interest rates, and we regularly evaluate our exposure to such changes. Our overall risk management strategy seeks to balance the magnitude of the exposure and the costs and availability of appropriate financial instruments.
FOREIGN CURRENCY EXCHANGE RATES AND INTEREST RATES
We derived approximately 53.0% of our consolidated service revenue for Fiscal Year 2014 from operations outside of the United States and 54% of our consolidated service revenue for Fiscal Year 2013 from operations outside of the United States. In addition, 13.7% of our consolidated revenue was denominated in Euros and 2.9% was denominated in pounds sterling for Fiscal Year 2014 while 13.0% of our consolidated revenue was denominated in Euros and 2.6% was denominated in pounds sterling for Fiscal Year 2013. We have no significant operations in any country in which the economy is considered to be highly inflationary. Our financial statements are denominated in U.S. dollars. Accordingly, changes in exchange rates between foreign currencies and the U.S. dollar will affect the translation of financial results into U.S. dollars for purposes of reporting our consolidated financial results.
It is our policy to mitigate the risks associated with fluctuations in foreign exchange rates and interest rates. Accordingly, we have instituted foreign currency hedging programs and an interest rate swap program. See Note 4 to our consolidated financial statements included in this annual report for more information on our hedging programs and interest rate swap program.
As of June 30, 2014, the programs with derivatives designated as hedging instruments under ASC 815 and the related notional values of the derivatives were approximately $418.1 million, including two interest rate swap agreements with a total notional value of $200 million executed to hedge our borrowings under our 2013 Credit Agreement. Under certain circumstances, such as the occurrence of significant differences between actual cash payments and forecasted cash payments, the ASC 815 programs could be deemed ineffective. In that event, the unrealized gains and losses related to these derivatives, which are currently reported in accumulated other comprehensive income, would be recognized in earnings. As of June 30, 2014, the estimated amount that could be recognized in earnings was a gain of approximately $4.2 million, net of tax.
As of June 30, 2014, the notional value of derivatives that were not designated as hedging instruments under ASC 815 was approximately $150.7 million.
During Fiscal Year 2014 and Fiscal Year 2013, we recorded foreign currency exchange losses of $3.5 million and a gain of $4.1 million, respectively. We also have exposure to additional foreign exchange rate risk as it relates to assets and liabilities that are not part of the economic hedge or designated hedging programs, but quantification of this risk is difficult to assess at any given point in time.
Our exposure to interest rate changes relates primarily to the amount of our short-term and long-term debt. Short-term debt was $12.5 million at June 30, 2014 and $20.4 million at June 30, 2013. Long-term debt was $337.5 million at June 30, 2014 and $427.5 million at June 30, 2013. Based on average short-term and long-term debt for Fiscal Year 2014, an increase in the average interest rate of 100 basis points would decrease our pre-tax earnings and cash flows by approximately $0.9 million on an annual basis.
In April and May 2013, we entered into three treasury lock agreements, each with a notional amount of $25.0 million, in connection with the planned issuance of our Notes that were issued in July 2013. The three treasury locks were used to minimize our interest rate exposure prior to locking in the fixed interest rate on our Notes. The treasury locks matured in May 2013 when the interest rates on our Notes were fixed. The treasury locks were deemed to be fully effective in accordance with ASC 815, and as such, the unrealized gains related to these derivatives are recorded as other comprehensive income and are amortized over the life of the Notes as interest income.
MARKETABLE SECURITIES
During Fiscal Year 2014, we purchased marketable securities in the form of foreign government treasury certificates that are actively traded. We expect to hold these securities to maturity. The securities are recorded at their amortized costs, which is not materially different than fair value. Securities with original maturities less than 90 days are recorded in cash and cash equivalents in our consolidated balance sheets and securities with original maturities over 90 days but less than one year are recorded as marketable securities in our consolidated balance sheets. As of June 30, 2014, all outstanding securities have original maturities less than 90 days and they are recorded at their amortized cost of $95.6 million. Since the counterparty is a stable sovereign and due to the relatively short terms of maturity (less than one year), we do not believe that these investments are at high risk of default. Nevertheless, these investments are still at risk for changes in market rates and prices.
45
Item 8. Financial Statements and Supplementary Data
PAREXEL INTERNATIONAL CORPORATION
CONSOLIDATED STATEMENTS OF INCOME AND COMPREHENSIVE INCOME
(in thousands, except per share data)
For the years ended June 30, | ||||||||||||
2014 | 2013 | 2012 | ||||||||||
Service revenue | $ | 1,939,360 | $ | 1,734,442 | $ | 1,396,508 | ||||||
Reimbursement revenue | 326,982 | 261,524 | 221,726 | |||||||||
Total revenue | 2,266,342 | 1,995,966 | 1,618,234 | |||||||||
Costs and expenses: | ||||||||||||
Direct costs | 1,279,178 | 1,207,536 | 971,829 | |||||||||
Reimbursable out-of-pocket expenses | 326,982 | 261,524 | 221,726 | |||||||||
Selling, general and administrative | 379,800 | 318,806 | 263,462 | |||||||||
Depreciation | 66,376 | 63,187 | 57,419 | |||||||||
Amortization | 14,952 | 9,999 | 8,753 | |||||||||
Restructuring (benefit) charge | (444 | ) | (1,209 | ) | 6,243 | |||||||
Total costs and expenses | 2,066,844 | 1,859,843 | 1,529,432 | |||||||||
Income from operations | 199,498 | 136,123 | 88,802 | |||||||||
Interest expense, net | (9,088 | ) | (7,238 | ) | (7,003 | ) | ||||||
Miscellaneous (expense) income, net | (2,549 | ) | 4,265 | (2,093 | ) | |||||||
Total other expense, net | (11,637 | ) | (2,973 | ) | (9,096 | ) | ||||||
Income before provision for income taxes | 187,861 | 133,150 | 79,706 | |||||||||
Provision for income taxes | 58,767 | 37,178 | 16,548 | |||||||||
Net income | $ | 129,094 | $ | 95,972 | $ | 63,158 | ||||||
Earnings per share: | ||||||||||||
Basic | $ | 2.28 | $ | 1.64 | $ | 1.06 | ||||||
Diluted | $ | 2.25 | $ | 1.61 | $ | 1.05 | ||||||
Weighted average shares: | ||||||||||||
Basic | 56,504 | 58,388 | 59,464 | |||||||||
Diluted | 57,477 | 59,447 | 60,426 | |||||||||
Comprehensive income: | ||||||||||||
Net income | $ | 129,094 | $ | 95,972 | $ | 63,158 | ||||||
Unrealized gain (loss) on derivative instruments, net of taxes | 5,078 | 624 | (1,660 | ) | ||||||||
Foreign currency translation adjustment | 27,050 | (1,523 | ) | (46,334 | ) | |||||||
Total comprehensive income | $ | 161,222 | $ | 95,073 | $ | 15,164 | ||||||
The accompanying notes are an integral part of the consolidated financial statements.
46
PAREXEL INTERNATIONAL CORPORATION
CONSOLIDATED BALANCE SHEETS
(in thousands, except share data)
June 30, 2014 | June 30, 2013 | |||||||
ASSETS | ||||||||
Current assets: | ||||||||
Cash and cash equivalents | $ | 188,171 | $ | 144,027 | ||||
Marketable securities | 95,641 | 130,137 | ||||||
Billed accounts receivable, net | 497,109 | 457,155 | ||||||
Unbilled accounts receivable, net | 225,514 | 248,219 | ||||||
Prepaid expenses | 13,641 | 19,358 | ||||||
Deferred tax assets | 54,061 | 44,236 | ||||||
Income taxes receivable | — | 4,071 | ||||||
Other current assets | 47,995 | 30,254 | ||||||
Total current assets | 1,122,132 | 1,077,457 | ||||||
Property and equipment, net | 234,164 | 224,225 | ||||||
Goodwill | 329,520 | 319,478 | ||||||
Other intangible assets, net | 91,855 | 103,514 | ||||||
Non-current deferred tax assets | 6,669 | 8,556 | ||||||
Long-term income taxes receivable | 13,406 | 11,153 | ||||||
Other assets | 36,254 | 35,241 | ||||||
Total assets | $ | 1,834,000 | $ | 1,779,624 | ||||
LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
Current liabilities: | ||||||||
Notes payable and current portion of long-term debt | $ | 12,501 | $ | 20,399 | ||||
Accounts payable | 66,483 | 54,232 | ||||||
Deferred revenue | 422,441 | 378,950 | ||||||
Accrued expenses | 52,034 | 42,211 | ||||||
Accrued employee benefits and withholdings | 175,840 | 149,290 | ||||||
Current deferred tax liabilities | 16,592 | 16,512 | ||||||
Income taxes payable | 19,384 | — | ||||||
Other current liabilities | 5,957 | 12,634 | ||||||
Total current liabilities | 771,232 | 674,228 | ||||||
Long-term debt, net of current portion | 334,443 | 424,074 | ||||||
Non-current deferred tax liabilities | 32,598 | 35,443 | ||||||
Long-term income tax liabilities | 29,525 | 46,731 | ||||||
Long-term deferred revenue | 44,523 | 29,386 | ||||||
Other liabilities | 43,998 | 30,816 | ||||||
Total liabilities | 1,256,319 | 1,240,678 | ||||||
Commitments and contingencies (Note 15) | ||||||||
Stockholders’ equity: | ||||||||
Preferred stock - $0.01 par value; 5,000,000 shares authorized, 0 shares issued and outstanding at June 30, 2014 and June 30, 2013, respectively. | — | — | ||||||
Common stock - $0.01 par value; 150,000,000 shares authorized; 54,661,877 and 56,310,582 shares issued and outstanding at June 30, 2014 and June 30, 2013, respectively. | 547 | 563 | ||||||
Additional paid-in capital | — | 113,771 | ||||||
Retained earnings | 575,044 | 454,650 | ||||||
Accumulated other comprehensive loss | 2,090 | (30,038 | ) | |||||
Total stockholders’ equity | 577,681 | 538,946 | ||||||
Total liabilities and stockholders’ equity | $ | 1,834,000 | $ | 1,779,624 | ||||
The accompanying notes are an integral part of the consolidated financial statements.
47
PAREXEL INTERNATIONAL CORPORATION
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
For the years ended June 30, | ||||||||||||
2014 | 2013 | 2012 | ||||||||||
Cash flow from operating activities: | ||||||||||||
Net income | $ | 129,094 | $ | 95,972 | $ | 63,158 | ||||||
Adjustments to reconcile net income to net cash provided by (used in) operating activities: | ||||||||||||
Depreciation and amortization | 81,328 | 73,186 | 66,172 | |||||||||
Stock-based compensation | 15,329 | 11,168 | 11,131 | |||||||||
Loss (gain) on disposal of assets | 273 | (884 | ) | 1,119 | ||||||||
Deferred income taxes | (14,704 | ) | 2,830 | (9,754 | ) | |||||||
Impairment charges | — | 1,071 | 1,150 | |||||||||
Excess tax benefit from stock-based compensation | (7,850 | ) | (5,624 | ) | (5,256 | ) | ||||||
Other non-cash items | (953 | ) | 659 | 818 | ||||||||
Changes in assets and liabilities, net of the effect from acquisitions: | ||||||||||||
Billed and unbilled accounts receivable | (23,089 | ) | (51,292 | ) | (20,083 | ) | ||||||
Prepaid expenses and other current assets | (10,803 | ) | (6,751 | ) | (1,593 | ) | ||||||
Other assets | 6,780 | (6,693 | ) | (5,460 | ) | |||||||
Accounts payable | 11,529 | 3,955 | 20,202 | |||||||||
Deferred revenue | 46,687 | 47,531 | 35,940 | |||||||||
Accrued expenses and other current liabilities | 28,861 | 18,659 | 63,303 | |||||||||
Long-term income taxes payable, net of long-term income taxes receivable | 13,119 | (1,440 | ) | 11,958 | ||||||||
Other liabilities | 11,600 | 1,468 | 1,652 | |||||||||
Net cash provided by operating activities | 287,201 | 183,815 | 234,457 | |||||||||
Cash flow from investing activities: | ||||||||||||
Purchases of marketable securities | (250,318 | ) | (312,403 | ) | (53,647 | ) | ||||||
Proceeds from sale of marketable securities | 291,824 | 182,800 | 51,529 | |||||||||
Purchases of property and equipment | (72,585 | ) | (81,089 | ) | (74,403 | ) | ||||||
Acquisition of businesses, net of cash acquired | (150 | ) | (97,099 | ) | — | |||||||
Proceeds from collection of note receivable | — | 659 | — | |||||||||
Proceeds from sale of property and equipment | — | 1,677 | — | |||||||||
Net cash used in investing activities | (31,229 | ) | (305,455 | ) | (76,521 | ) | ||||||
Cash flow from financing activities: | ||||||||||||
Proceeds from issuance of common stock, net of tax payments for cashless exercises | 6,762 | 15,271 | 12,120 | |||||||||
Payments for share repurchase | (154,906 | ) | (195,149 | ) | — | |||||||
Excess tax benefit from stock-based compensation | 7,850 | 5,624 | 5,256 | |||||||||
Borrowings under credit agreement/facility | 502,500 | 795,000 | 268,000 | |||||||||
Repayments under credit agreement/facility | (590,000 | ) | (577,500 | ) | (293,000 | ) | ||||||
Borrowings under factoring agreement | 4,497 | 10,394 | — | |||||||||
Payments for debt issuance costs | (671 | ) | (1,231 | ) | — | |||||||
Repayments under other debt | — | — | (943 | ) | ||||||||
Net cash (used in) provided by financing activities | (223,968 | ) | 52,409 | (8,567 | ) | |||||||
Effect of exchange rate changes on cash and cash equivalents | 12,140 | (321 | ) | (24,846 | ) | |||||||
Net increase (decrease) in cash and cash equivalents | 44,144 | (69,552 | ) | 124,523 | ||||||||
Cash and cash equivalents at beginning of year | 144,027 | 213,579 | 89,056 | |||||||||
Cash and cash equivalents at end of year | $ | 188,171 | $ | 144,027 | $ | 213,579 | ||||||
Supplemental disclosures of cash flow information | ||||||||||||
Non-cash debt settlement under factoring agreement | $ | 14,891 | $ | — | $ | — | ||||||
Net cash paid during year for: | ||||||||||||
Interest | $ | 13,710 | $ | 9,962 | $ | 10,802 | ||||||
Income taxes, net of refunds | $ | 61,027 | $ | 37,764 | $ | 9,709 | ||||||
The accompanying notes are an integral part of the consolidated financial statements.
48
PAREXEL INTERNATIONAL CORPORATION
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(in thousands)
Common Stock | Retained Earnings | Accumulated Other Comprehensive Income (Loss) | Total Stockholders’ Equity | ||||||||||||||||||||
Number of Shares | Par Value | Additional Paid-in Capital | |||||||||||||||||||||
Balance at June 30, 2011 | 59,004 | $ | 584 | $ | 251,045 | $ | 295,520 | $ | 18,855 | $ | 566,004 | ||||||||||||
Shares issued under stock option/restricted stock/employee stock purchase plans | 1,143 | 17 | 12,103 | 12,120 | |||||||||||||||||||
Stock-based compensation | 11,131 | 11,131 | |||||||||||||||||||||
Purchase of non-controlling interests | 5,256 | 5,256 | |||||||||||||||||||||
Unrealized loss on derivative instruments, net of taxes | (1,660 | ) | (1,660 | ) | |||||||||||||||||||
Foreign currency translation adjustment | (46,334 | ) | (46,334 | ) | |||||||||||||||||||
Net income | 63,158 | 63,158 | |||||||||||||||||||||
Balance at June 30, 2012 | 60,147 | $ | 601 | $ | 279,535 | $ | 358,678 | $ | (29,139 | ) | $ | 609,675 | |||||||||||
Shares issued under stock option/restricted stock/employee stock purchase plans, net | 1,470 | 15 | 15,256 | 15,271 | |||||||||||||||||||
Stock-based compensation | 11,168 | 11,168 | |||||||||||||||||||||
Excess tax benefit related to employee equity awards | 5,400 | 5,400 | |||||||||||||||||||||
Share repurchase | (5,306 | ) | (53 | ) | (197,588 | ) | (197,641 | ) | |||||||||||||||
Unrealized gain on derivative instruments, net of taxes | 624 | 624 | |||||||||||||||||||||
Foreign currency translation adjustment | (1,523 | ) | (1,523 | ) | |||||||||||||||||||
Net income | 95,972 | 95,972 | |||||||||||||||||||||
Balance at June 30, 2013 | 56,311 | $ | 563 | $ | 113,771 | $ | 454,650 | $ | (30,038 | ) | $ | 538,946 | |||||||||||
Shares issued under stock option/restricted stock/employee stock purchase plans, net | 788 | 8 | 6,754 | 6,762 | |||||||||||||||||||
Stock-based compensation | 15,329 | 15,329 | |||||||||||||||||||||
Excess tax benefit related to employee equity awards | 7,837 | 7,837 | |||||||||||||||||||||
Share repurchase | (2,437 | ) | (24 | ) | (143,691 | ) | (8,700 | ) | (152,415 | ) | |||||||||||||
Unrealized gain on derivative instruments, net of taxes | 5,078 | 5,078 | |||||||||||||||||||||
Foreign currency translation adjustment | 27,050 | 27,050 | |||||||||||||||||||||
Net income | 129,094 | 129,094 | |||||||||||||||||||||
Balance at June 30, 2014 | 54,662 | $ | 547 | $ | — | $ | 575,044 | $ | 2,090 | $ | 577,681 | ||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
49
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1. DESCRIPTION OF BUSINESS
PAREXEL International Corporation (“PAREXEL,” “the Company,” or “we”) is a leading biopharmaceutical outsourcing services company, providing a broad range of expertise in clinical research, clinical logistics, medical communications, consulting, commercialization and advanced technology products and services to the worldwide pharmaceutical, biotechnology, and medical device industries. Our primary objective is to provide quality solutions for managing the biopharmaceutical product lifecycle with the goal of reducing the time, risk, and cost associated with the development and commercialization of new therapies. Since our incorporation in 1983, we have developed significant expertise in processes and technologies supporting this strategy. Our product and service offerings include: clinical trials management, observational studies and patient/disease registries, data management, biostatistical analysis, epidemiology, health economics/outcomes research, pharmacovigilance, medical communications, clinical pharmacology, patient recruitment, clinical supply and drug logistics, post-marketing surveillance, regulatory and product development and commercialization consulting, health policy and reimbursement and market access consulting, medical imaging services, regulatory information management (“RIM”) solutions, randomization and trial supply management services (“RTSM”), electronic data capture systems (“EDC”), clinical trial management systems (“CTMS”), web-based portals, systems integration, patient diary applications, and other product development tools and services.
NOTE 2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation and Principles of Consolidation
The consolidated financial statements include the accounts of PAREXEL International Corporation, our wholly-owned and majority-owned subsidiaries. All inter-company accounts and transactions have been eliminated.
Net Presentation Adjustment
We have a cash pooling arrangement with RBS Nederland, NV. Pooling occurs when debit balances are offset against credit balances and the overall net position is used as a basis by the bank for calculating the overall pool interest payable or receivable amount. Each legal entity owned by us and party to this arrangement remains the owner of either a credit (deposit) or a debit (overdraft) balance. Therefore, interest income is earned by legal entities with credit balances, while interest expense is charged to legal entities with debit balances. Based on the pool’s aggregate balance, the bank then (1) recalculates the overall interest to be charged or earned, (2) compares this amount with the sum of previously charged/earned interest amounts per account and (3) additionally pays/charges the difference.
We adjusted the presentation for interest income and interest expense in our results of operation for the fiscal years ended June 30, 2013 and 2012 (“Fiscal Year 2013” and “Fiscal Year 2012”). We determined a net presentation of either interest expense or interest income better reflects the impact on our results of operations for the cash pooling arrangement compared to our prior practice of reporting interest income and expense on a gross basis. For Fiscal Years 2013 and 2012, we incurred net interest expense on our cash pooling arrangement, and therefore we netted the previously reported interest income of $3.8 million against the interest expense of $11.1 million for Fiscal Year 2013 and interest income of $5.4 million against the interest expense of $12.4 million for Fiscal Year 2012. This adjustment had no impact on our consolidated financial positions for June 30, 2013 and 2012 or our consolidated statement of cash flow for Fiscal Years 2013 and 2012.
Use of Estimates
We prepare our financial statements in conformity with U.S. generally accepted accounting principles which require us to make estimates and assumptions that affect the amounts reported in the financial statements. Estimates are used in accounting for, among other items, revenue recognition, allowance for credit losses on receivables, valuation of derivative instruments, periodic impairment reviews of goodwill and intangible assets, contingent consideration, income taxes, and the valuation of long-term assets. Our estimates are based on the facts and circumstances available at the time estimates are made, historical experience, risk of loss, general economic conditions, trends, and assessments of the probable future outcomes of these matters. Actual results could differ from those estimates. Estimates and assumptions are reviewed periodically, and the effects of changes, if any, are reflected in the statement of operations in the period in which they are determined.
Fair Values of Financial Instruments
The fair value of our cash and cash equivalents, marketable securities, accounts receivable, and accounts payable approximates the carrying value of these financial instruments because of the short-term nature of any maturities. We determine the estimated fair values of other financial instruments, using available market information and valuation methodologies, primarily discounted cash flow analysis or input from independent investment bankers.
50
Revenue Recognition
We derive revenue from the delivery of service or software solutions to clients in the worldwide pharmaceutical, biotechnology, and medical device industries. We recognize revenue when all of the following conditions are satisfied: (1) there is persuasive evidence of an arrangement; (2) the service offering has been delivered to the client; (3) the collection of fees is probable; and (4) the amount of fees to be paid by the client is fixed or determinable. Revenue recognition treatment of each business segment is described below.
Clinical Research Services (“CRS”) and PAREXEL Consulting Services (“PC”) Service Revenue
Service revenues in our CRS and PC businesses are derived principally from fee-for-service or fixed-price executory contracts, which typically involve competitive bid awards and multi-year terms. Client billing schedules and payment arrangements are prescribed under negotiated contract terms. Contract provisions do not provide for rights of return or refund, but normally include rights of cancellation with notice, in which case services delivered through the cancellation date are due and payable by the client, including certain costs to conclude the trial or study.
Our client arrangements generally involve multiple service deliverables, where bundled service deliverables are accounted for in accordance with Accounting Standards Codification (“ASC”) 605-25, “Multiple-Element Arrangements.” We determined that each of our service deliverables have standalone value. ASC 605-25 requires the allocation of contract (arrangement) value to each separate unit of accounting based on the relative selling price of the various separate units of accounting in the arrangement. ASC 605-25 requires a hierarchy of evidence be followed when determining if evidence of the selling price of an item exists such that the best evidence of selling price of a unit of accounting is vendor-specific objective evidence (“VSOE”), or the price charged when a deliverable is sold separately. When VSOE is not available to determine selling price, relevant third-party evidence (“TPE”) of selling price should be used, if available. Lastly, when neither VSOE nor TPE of selling price for similar deliverables exists, management must use its best estimate of selling price considering all relevant information that is available without undue cost and effort.
We generally are not able to establish VSOE as our deliverables are seldom sold separately. We have established TPE of selling price for each of our arrangement deliverables based on the price we charge for equivalent services when sold to other similar customers as well as our knowledge of market-pricing from the competitive bidding process for customer contracts offering similar services to comparably situated customers. Consequently, we allocate arrangement consideration at the inception of the arrangement using the relative selling prices of the deliverables within the contract based on TPE.
We recognize revenues for the separate elements of our contracts upon delivery of actual units of output and when all other revenue recognition criteria are met. Revenue from fee-for-service contracts generally is recognized as units of output are delivered. Revenue on fixed-price contracts generally is measured by applying a proportional performance model using output units, such as site or investigator recruitment, patient enrollment, data management, or other deliverables common to our CRS business. Performance-based output units are pre-defined in contracts and revenue is recognized based upon actual units of completion. Revenue related to changes in contract scope, which are subject to client approval, is recognized when realization is assured and amounts are fixed or determinable.
PAREXEL Informatics, Inc. (“PI”) Service Revenue
Service revenue is derived principally from the delivery of software solutions through our PI business segment. Software solutions include ClinPhone®RTSM, CTMS, EDC and RIM.
Within PI’s Clinphone® RTSM business, we offer selected software solutions through a hosted application delivered through a standard web-browser. We recognize revenue from application hosting services in accordance with ASC 985-605, “Revenue Recognition in the Software Industry” and ASC 605-25 as our customers do not have the right to take possession of the software. Revenue resulting from these hosting services consists of three stages: set-up (client specification and workflow), hosting and support services, and closeout reporting.
Fees charged and costs incurred in the set-up stage are deferred until the start of the hosting period and are amortized and recognized ratably over the estimated hosting period, including customary and expected extensions. Deferred costs are direct costs associated with the trial and application setup. These costs include salary and benefits associated with direct labor costs incurred during trial setup, as well as third-party subcontract fees and other contract labor costs. In the event of a contract cancellation by a client, all deferred revenue is recognized and all deferred setup costs are expensed. To the extent that termination-related fees are payable under the contract, such fees are recognized in the period of termination.
PI's Medical Imaging business provides a service allowing customers to manage the image acquisitions and the analysis and quality of data obtained during a clinical trial. Service revenue is derived from executory contracts that are tailored to meet individual client requirements. Client billing schedules and payment arrangements are prescribed under negotiated contract terms. We recognize service revenue related to our Medical Imaging business based upon a proportional performance method utilizing a unitized output method. The defined units used for revenue recognition are used to track output measures that are
51
specific to the services being provided in the contract, and may include site survey reports, project management tasks, number of reviews completed, and image receipt and processing.
Reimbursement Revenue & Investigator Fees
Reimbursable out-of-pocket expenses are reflected in our Consolidated Statements of Income under “Reimbursement revenue” and “Reimbursable out-of-pocket expenses,” as we are the primary obligor for these expenses despite being reimbursed by our clients. In addition, as is customary in our industry, we routinely subcontract on behalf of our clients with independent physician investigators in connection with clinical trials. The related investigator fees are not reflected in our Service revenue, Reimbursement revenue, Reimbursable out-of-pocket expenses, or Direct costs, because these fees are reimbursed by clients on a “pass through basis,” without risk or reward to us. The amounts of these investigator fees were $523.1 million, $421.2 million, and $250.8 million for the fiscal years ended June 30, 2014, 2013, and 2012, respectively.
Business Combinations
We account for acquisitions as business combinations in accordance with ASC Topic 805, “Business Combinations.” We allocate the amounts that we pay for each acquisition to the assets we acquire and liabilities we assume based on their fair values at the dates of acquisition, including identifiable intangible assets. We base the fair value of identifiable intangible assets acquired in a business combination on detailed valuations that use information and assumptions determined by management and which consider management's best estimates of inputs and assumptions that a market participant would use. We allocate any excess purchase price over the fair value of the net tangible and identifiable intangible assets acquired to goodwill.
Cash and Cash Equivalents
We consider all highly liquid investments purchased with original maturities of 90 days or less to be cash equivalents. We had no cash equivalents balance as of June 30, 2014 and 2013.
Marketable Securities
We account for investments in debt and equity securities in accordance with ASC 320, “Investments - Debt and Equity Securities.”
Marketable securities are held in foreign government treasury certificates that are actively traded and have original maturities over 90 days but less than one year. Our foreign government treasury certificates securities are classified as held-to-maturity based on our intent and ability to hold the securities to maturity and are recorded at amortized cost, which is not materially different than fair value. We do not intend to sell the securities and it is not more likely than not that we will be required to sell the securities before recovery of their amortized cost bases, which may be maturity. Interest and dividends related to these securities are reported as a component of interest income in our consolidated statements of income.
Concentration of Credit Risk
Financial instruments that subject us to credit risk primarily consist of cash and cash equivalents, marketable securities, derivative financial instrument contracts, and accounts receivable. We maintain our cash and cash equivalent balances with high-quality financial institutions and, consequently, we believe that such funds are subject to minimal credit risk. Our marketable securities primarily consist of foreign government treasury certificates.
We have approximately nine different counterparties in our derivative contracts, which include interest rate swaps, an interest rate cap and foreign currency hedges. Each of these counterparties is in the financial services industry and is subject to the credit risks inherent to that industry. We perform ongoing credit evaluations of these counterparties.
We perform ongoing credit evaluations related to the financial condition of our clients and, generally, do not require collateral. As of June 30, 2014, one client individually accounted for 20% of our total billed and unbilled accounts receivables. As of June 30, 2013, two clients individually accounted for 18% and 12% of our billed and unbilled accounts receivables. For the year ended June 30, 2014 (“Fiscal Year 2014”), two clients individually accounted for 16% and 11% of our consolidated service revenue. For Fiscal Year 2013, two clients individually accounted for 17% and 12% of our consolidated service revenue. No single client accounted for 10% or more of our consolidated service revenue in Fiscal Year 2012.
Billed Accounts Receivable, Unbilled Accounts Receivable and Deferred Revenue
Billed accounts receivable represent amounts for which invoices have been sent to clients based on contract terms. Unbilled accounts receivable represent amounts recognized as revenue for which invoices have not yet been sent to clients due to contract terms. Deferred revenue represents amounts billed based on contractual provisions or payments received for which revenue has not yet been earned. We maintain a provision for losses on receivables based on historical collectability and specific identification of potential problem accounts. Uncollectible invoices are written off when collection efforts have been exhausted.
52
Property and Equipment
Property and equipment is stated at cost. Depreciation is provided using the straight-line method based on estimated useful lives of 3 to 8 years for computer software and hardware, and 5 years for office furniture, fixtures and equipment. Leasehold improvements are amortized over the lesser of the estimated useful lives of the improvements or the remaining lease term, which include lease extensions when reasonably assured. Repair and maintenance costs are expensed as incurred.
Development of Software for Internal Use
PAREXEL accounts for the costs of software developed or obtained for internal use in accordance with ASC 350-40, “Internal-Use Software.” We capitalize costs of materials, consultants, payroll, and payroll-related costs for employees incurred in developing internal-use software. These costs are included in computer software in Note 5 below. Costs incurred during the preliminary project and post-implementation stages are charged to expense.
Research and Development Costs
We incur ongoing research and development costs related to core technologies used internally as well as software and technology sold externally. Unless eligible for capitalization, these costs are expensed as incurred. Research and development expense was $26.9 million, $23.8 million, and $21.0 million in Fiscal Years 2014, 2013, and 2012, respectively, and is included in selling, general and administrative expenses in the consolidated statements of income.
Goodwill
PAREXEL follows the provisions of ASC 350, “Intangibles—Goodwill and Other.” Under this statement, goodwill as well as certain other intangible assets, determined to have an indefinite life, are not amortized. Instead, these assets are evaluated for impairment at least annually or more frequently if an event occurs or circumstances change that would more likely than not reduce the fair value of the reporting unit below its carrying value. For Fiscal Years 2014 and 2013, we performed the intangibles impairment testing in accordance with the guidance of Accounting Standards Update (“ASU”) 2011-08, “Intangibles—Goodwill and Other (Topic 350): Testing Goodwill for Impairment,” and conducted an assessment of qualitative factors. We concluded that it was not more likely than not the fair value of a reporting unit was less than its carrying amount, including goodwill as of June 30, 2014 and 2013. There was no evidence of impairment of our goodwill balances as of June 30, 2014 or 2013.
Long-lived Assets and Other Intangible Assets
Long-lived assets, including fixed assets and intangible assets which have a definitive life, are reviewed for impairment when circumstances indicate that the carrying amount of assets might not be recoverable.
Indefinite-lived assets are reviewed annually or more frequently if an event occurs or circumstances change that would more likely than not reduce the fair value below the carrying value of the asset. For Fiscal Years 2014 and 2013, we performed the intangibles impairment testing in accordance with the guidance under ASU No. 2012-02, “Intangibles—Goodwill and Other (Topic 350): Testing Indefinite-Lived Intangible Assets for Impairment.” and conducted an assessment of qualitative factors and concluded that it was not more likely than not that the fair value of our indefinite-lived intangible assets was less than its carrying amount as of June 30, 2014 and 2013. There was no evidence of impairment of our indefinite-lived intangible asset balances as of June 30, 2014 or 2013.
Income Taxes
Deferred tax assets and liabilities are recorded for the expected future tax consequences of temporary differences between the carrying amounts and the tax bases of assets and liabilities. Deferred tax assets are recognized for the estimated future tax benefits of deductible temporary differences and tax operating loss and credit carryforwards and are presented net of valuation allowances. Valuation allowances are established in jurisdictions where it is more likely than not that the benefits of the associated deferred tax assets will not be realized. Deferred income tax expense represents the change in the net deferred tax asset and liability balances. Interest and penalties are recognized as a component of income tax expense
Foreign Currency
Assets and liabilities of PAREXEL’s international operations are translated into U.S. dollars at exchange rates that are in effect on the balance sheet date and equity accounts are translated at historical exchange rates. Income and expense items are translated at average exchange rates in effect during the year. Translation adjustments are accumulated in other comprehensive income (loss) as a separate component of stockholders’ equity in the consolidated balance sheet. Transaction gains and losses are included in miscellaneous expense, net in the consolidated statements of operations. Transaction gains (losses) were $(3.5) million, $4.1 million, and $0.3 million in Fiscal Years 2014, 2013, and 2012, respectively.
53
Earnings Per Share
Basic earnings per share is computed by dividing net income for the period by the weighted average number of common shares outstanding during the period. Diluted earnings per share is computed by dividing net income by the weighted average number of common shares plus the dilutive effect of outstanding stock options. We do not have any participating securities outstanding nor do we have more than one class of common stock.
Recently Implemented Accounting Standards
In December 2011, the Financial Accounting Standard Board (“FASB”) issued ASU No. 2011-11, “Balance Sheet (Topic 210): Disclosures about Offsetting Assets and Liabilities.” ASU 2011-11 requires a company to disclose information about offsetting and related arrangements to enable readers of its financial statements to understand the effects of those arrangements on its financial position. ASU 2011-11 is effective for fiscal years beginning after January 1, 2013. In January 2013, the FASB issued ASU No. 2013-01, “Clarifying the Scope of Disclosures about Offsetting Assets and Liabilities.” ASU 2013-01 was issued to limit the scope of ASU 2011-11 to derivatives (including bifurcated embedded derivatives), repurchase and reverse repurchase arrangements, and securities borrowing and lending transactions. We adopted ASU 2013-01 beginning in our fiscal quarter ended September 30, 2013. The adoption of ASU 2013-01 did not impact our consolidated financial statements.
In February 2013, the FASB issued ASU No. 2013-02, “Reporting of Amounts Reclassified Out of Accumulated Other Comprehensive Income.” ASU 2013-02 requires reporting and disclosure about changes in accumulated other comprehensive income (AOCI) balances and reclassifications out of AOCI. We adopted ASU 2013-02 beginning in our fiscal quarter ended September 30, 2013. The adoption of ASU 2013-02 did not impact our consolidated financial statements.
In July 2013, the FASB issued ASU No. 2013-11, “Income Taxes (Topic 740): Presentation of an Unrecognized Tax Benefit When a Net Operating Loss Carryforward, a Similar Tax Loss, or a Tax Credit Carryforward Exists.” ASU 2013-11 requires that entities present an unrecognized tax benefit, or a portion of an unrecognized tax benefit, as a reduction to a deferred tax asset in the financial statements for a net operating loss carryforward, a similar tax loss, or a tax credit carryforward, with certain exceptions. We adopted ASU 2013-11 beginning in our fiscal quarter ended June 30, 2014. The adoption of ASU 2013-11 did not have a material impact on our consolidated financial statements.
Recently Issued Accounting Standards
In March 2013, the FASB issued ASU No. 2013-05, “Parent’s Accounting for the Cumulative Translation Adjustment upon Derecognition of Certain Subsidiaries or Groups of Assets within a Foreign Entity or of an Investment in a Foreign Entity.” ASU 2013-05 addresses the accounting for the cumulative translation adjustment when a parent either sells a part or all of its investment in a foreign entity or no longer holds a controlling financial interest in a subsidiary or group of assets that is a nonprofit activity or a business within a foreign entity. ASU 2013-05 is effective prospectively for fiscal years and interim periods within those fiscal years beginning after December 15, 2013 and early adoption is permitted. We do not expect the adoption of this guidance to have a material impact on our consolidated financial statements.
In May 2014, the FASB issued ASU No. 2014-09, Revenue from Contracts with Customers (“ASU 2014-09”), which stipulates that an entity should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. To achieve this core principle, an entity should apply the following steps: (1) identify the contract(s) with a customer; (2) identify the performance obligations in the contract; (3) determine the transaction price; (4) allocate the transaction price to the performance obligations in the contract; and (5) recognize revenue when (or as) the entity satisfies a performance obligation. ASU 2014-09 will be effective prospectively for fiscal years and interim periods within those years beginning after December 15, 2016, early adoption is not permitted. The impact on our Financial Statements of adopting ASU 2014-09 is being assessed by our management.
In June 2014, the FASB issued ASU No. 2014-12, Accounting for Share-Based Payments When the Terms of an Award Provide That a Performance Target Could Be Achieved after the Requisite Service Period ("ASU 2014-12"). ASU 2014-12 requires that a performance target that affects vesting and could be achieved after the requisite service period be treated as a performance condition. A reporting entity should apply existing guidance in ASC 718, Compensation—Stock Compensation, as it relates to such awards. ASU 2014-12 is effective in the first quarter of our fiscal year ending June 30, 2017 with early adoption permitted using either of two methods: (i) prospective to all awards granted or modified after the effective date; or (ii) retrospective to all awards with performance targets that are outstanding as of the beginning of the earliest annual period presented in the financial statements and to all new or modified awards thereafter, with the cumulative effect of applying ASU 2014-12 as an adjustment to the opening retained earnings balance as of the beginning of the earliest annual period presented in the financial statements. We do not expect the adoption of this guidance to have a material impact on our consolidated financial statements.
54
NOTE 3 – ACQUISITIONS
LIQUENT ACQUISITION
On December 21, 2012, we acquired all of the outstanding equity securities of LIQUENT, Inc. (“LIQUENT”), a leading global provider of Regulatory Information Management (“RIM”) solutions for total cash consideration of $74.3 million. By combining LIQUENT with our PI segment, we believe that we have strengthened our regulatory capabilities by adding a regulatory information technology platform and provide our clients access to comprehensive regulatory agency submission planning, viewing, tracking, publishing, and registration management throughout the entire product lifecycle of a life sciences entity. Effective July 1, 2014, a component of LIQUENT’s RIM solutions, the regulatory outsourcing services, will be reported as part of our PC segment (see Note 17).
The acquisition was initially funded through a new $100.0 million unsecured term loan agreement (the “2012 Term Loan”) with Bank of America, N.A. (“Bank of America”) (see Note 8).
HERON ACQUISITION
On April 30, 2013, we acquired all of the outstanding equity securities of HERON, a life sciences consultancy which provides evidence-based commercialization services to support biopharmaceutical companies throughout the lifecycle of their products. The net purchase price was approximately $22.8 million, plus the potential for us to pay up to an additional $14.2 million over a twenty-six month period if specific financial targets for HERON are achieved. We determined the fair value of the contingent consideration as part of the HERON acquisition based on the probability of HERON attaining the specified financial targets and at acquisition assigned a fair value of $5.9 million to the liability. The acquisition was funded through use of existing cash. HERON's results of operations are included in our PC segment.
NOTE 4. DERIVATIVES
We are exposed to certain risks relating to our ongoing business operations. The primary risks managed by using derivative instruments are interest rate risk and foreign currency exchange rate risk. Accordingly, we have instituted interest rate and foreign currency hedging programs that are accounted for in accordance with ASC 815.
• | Our interest rate hedging program is a cash flow hedge program designed to minimize interest rate volatility. We swap the difference between fixed and variable interest amounts calculated by reference to an agreed-upon notional principal amount, at specified intervals. We also employed an interest rate cap, which matured in March 2014, that compensates us if variable interest rates rise above a pre-determined rate. Our interest rate contracts are designated as hedging instruments. |
• | Our foreign currency hedging program is a cash flow hedge program designed to mitigate foreign currency exchange rate volatility due to the foreign currency exchange exposure related to intercompany and significant external transactions. This program was expanded in the first quarter of Fiscal Year 2013 in order to reduce the impact of foreign exchange rate risk on our direct costs. In the third quarter of Fiscal Year 2014, we further expanded the program to reduce the foreign exchange rate risk on our service revenues. We primarily utilize forward currency exchange contracts and cross-currency swaps with maturities of no more than 12 months. These contracts are designated as hedging instruments. |
We also enter into other economic hedges to mitigate foreign currency exchange risk and interest rate risk related to intercompany and significant external transactions. These contracts are not designated as hedges in accordance with ASC 815.
The following table presents the notional amounts and fair values of our derivatives as of June 30, 2014 and June 30, 2013. All asset and liability amounts are reported in other current and non-current assets and other current and non-current liabilities.
55
June 30, 2014 | June 30, 2013 | ||||||||||||||
(in thousands) | Notional Amount | Asset (Liability) | Notional Amount | Asset (Liability) | |||||||||||
Derivatives designated as hedging instruments under ASC 815 | |||||||||||||||
Derivatives in an asset position: | |||||||||||||||
Interest rate contracts | $ | 100,000 | $ | 2,049 | $ | 125,000 | $ | 3,269 | |||||||
Foreign exchange contracts | 154,845 | 5,375 | 20,245 | 333 | |||||||||||
Cross-currency swap contracts | 25,560 | 528 | 27,312 | 1,032 | |||||||||||
Derivatives in a liability position: | |||||||||||||||
Interest rate contracts | 100,000 | (1,469 | ) | 100,000 | (2,208 | ) | |||||||||
Foreign exchange contracts | 37,736 | (369 | ) | 129,254 | (3,889 | ) | |||||||||
Total designated derivatives | $ | 418,141 | $ | 6,114 | $ | 401,811 | $ | (1,463 | ) | ||||||
Derivatives not designated as hedging instruments under ASC 815 | |||||||||||||||
Derivatives in an asset position: | |||||||||||||||
Foreign exchange contracts | $ | 100,849 | $ | 1,062 | $ | 20,756 | $ | 91 | |||||||
Derivatives in a liability position: | |||||||||||||||
Foreign exchange contracts | 49,863 | (133 | ) | 42,800 | (992 | ) | |||||||||
Cross-currency interest rate swap contracts | — | — | 44,580 | (1,910 | ) | ||||||||||
Total non-designated derivatives | $ | 150,712 | $ | 929 | $ | 108,136 | $ | (2,811 | ) | ||||||
Total derivatives | $ | 568,853 | $ | 7,043 | $ | 509,947 | $ | (4,274 | ) | ||||||
Under certain circumstances, such as the occurrence of significant differences between actual cash payments and forecasted cash payments, the ASC 815 programs could be deemed ineffective. We record the effective portion of any change in the fair value of derivatives designated as hedging instruments under ASC 815 to other accumulated comprehensive income (loss) in our consolidated balance sheets, net of deferred taxes, and any ineffective portion to miscellaneous (expense) income, net in our consolidated statements of income. During Fiscal Years 2014 and 2013, the amounts recorded in miscellaneous (expense) income, net in our consolidated statements of income to reflect ineffective portions of any hedges were a gain of $0.2 million and a loss of $0.8 million, respectively.
The amounts recognized for Fiscal Year 2014 and Fiscal Year 2013 in other comprehensive income (loss) are presented below:
Years Ended | ||||||||
(in thousands) | June 30, 2014 | June 30, 2013 | ||||||
Derivatives designated as hedging instruments under ASC 815 | ||||||||
Interest rate contracts, net of taxes | $ | (322 | ) | $ | 2,818 | |||
Foreign exchange contracts, net of taxes | 5,423 | (2,129 | ) | |||||
Cross-currency swap contracts, net of taxes | (23 | ) | (65 | ) | ||||
Total designated derivative unrealized gain (loss), net | $ | 5,078 | $ | 624 | ||||
The unrealized gain (loss) on derivative instruments is net of $3.2 million and $0.4 million of taxes for Fiscal Years 2014 and 2013, respectively. The estimated net amount of the existing gains that are expected to be reclassified into earnings within the next twelve months is $3.9 million.
The change in the fair value of derivatives not designated as hedging instruments under ASC 815 is recorded to miscellaneous (expense) income, net in our consolidated statements of income. The unrealized (loss) gain recognized are presented below:
Years Ended | ||||||||
(in thousands) | June 30, 2014 | June 30, 2013 | ||||||
Derivatives not designated as hedging instruments under ASC 815 | ||||||||
Cross-currency interest rate swap contracts | $ | 1,910 | $ | 2,634 | ||||
Foreign exchange contracts | 1,830 | (688 | ) | |||||
Total non-designated derivative unrealized gain (loss), net | $ | 3,740 | $ | 1,946 | ||||
56
NOTE 5. PROPERTY AND EQUIPMENT
Property and equipment at June 30, 2014 and June 30, 2013 consisted of the following:
(in thousands) | 2014 | 2013 | ||||||
Property and equipment: | ||||||||
Computer software | $ | 337,494 | $ | 282,681 | ||||
Computer hardware and office equipment | 108,717 | 94,932 | ||||||
Leasehold improvements | 97,280 | 87,982 | ||||||
Medical equipment | 17,577 | 16,337 | ||||||
Furniture and fixtures | 28,837 | 26,370 | ||||||
Office equipment and other assets | 17,930 | 19,224 | ||||||
Total | 607,835 | 527,526 | ||||||
Less: accumulated depreciation | (373,671 | ) | (303,301 | ) | ||||
Total | $ | 234,164 | $ | 224,225 | ||||
We retired $2.0 million, $13.0 million and $35.8 million of fully-depreciated assets for Fiscal Years 2014, 2013, and 2012, respectively.
NOTE 6. GOODWILL AND INTANGIBLE ASSETS
Goodwill
The changes in the carrying amount of goodwill for Fiscal Years 2014 and 2013 were as follows:
(in thousands) | Fiscal Year 2014 | Fiscal Year 2013 | ||||||
Beginning Balance | $ | 319,478 | $ | 255,455 | ||||
Goodwill arising from LIQUENT acquisition | 304 | 51,244 | ||||||
Goodwill arising from HERON acquisition | 280 | 16,631 | ||||||
Effect of changes in exchange rates used for translation | 9,458 | (3,852 | ) | |||||
Ending Balance | $ | 329,520 | $ | 319,478 | ||||
As of June 30, 2014, the carrying value of our goodwill by reportable segment was $125.5 million in CRS, $22.1 million in PC, and $181.9 million in PI.
Long-lived Assets and Other Intangible Assets
As of June 30, 2014, intangible assets consisted of the following:
(in thousands) | Weighted Average Useful Life (years) | Cost | Accumulated Amortization/ Effect of Exchange Rate Changes | Net | ||||||||||
Intangible Asset | ||||||||||||||
Customer relationships | 12.8 | $ | 104,048 | $ | (45,028 | ) | $ | 59,020 | ||||||
Technology and other intangibles | 7.9 | 35,647 | (24,191 | ) | 11,456 | |||||||||
Definite-life tradename | 7.7 | 3,900 | (673 | ) | 3,227 | |||||||||
Indefinite-life tradename * | indefinite | 22,158 | (4,006 | ) | 18,152 | |||||||||
Total intangible assets | $ | 165,753 | $ | (73,898 | ) | $ | 91,855 | |||||||
* The tradename acquired in the ClinPhone acquisition has an indefinite useful life. | ||||||||||||||
57
As of June 30, 2013, intangible assets consisted of the following:
(in thousands) | Weighted Average Useful Life (years) | Cost | Accumulated Amortization/ Effect of Exchange Rate Changes | Net | ||||||||||
Intangible Asset | ||||||||||||||
Customer relationships and backlog | 12.1 | $ | 112,274 | $ | (44,706 | ) | $ | 67,568 | ||||||
Technology and other intangibles | 7.9 | 35,647 | (20,582 | ) | 15,065 | |||||||||
Definite-life tradename | 7.7 | 3,800 | (235 | ) | 3,565 | |||||||||
Indefinite-life tradename * | indefinite | 22,158 | (4,842 | ) | 17,316 | |||||||||
Total intangible assets | $ | 173,879 | $ | (70,365 | ) | $ | 103,514 | |||||||
* The tradename acquired in the ClinPhone acquisition has an indefinite useful life. | ||||||||||||||
The changes in the carrying amounts of other intangible assets for Fiscal Years 2014 and 2013 were as follows:
(in thousands) | Fiscal Year 2014 | Fiscal Year 2013 | ||||||
Beginning Balance | $ | 103,514 | $ | 70,004 | ||||
Intangibles assets (adjusted) acquired from LIQUENT acquisition | (400 | ) | 32,600 | |||||
Intangibles assets acquired from HERON acquisition | — | 15,500 | ||||||
Amortization | (14,952 | ) | (9,999 | ) | ||||
Effect of changes in exchange rates used for translation | 3,693 | (4,591 | ) | |||||
Ending Balance | $ | 91,855 | $ | 103,514 | ||||
Estimated amortization expense for the next five years is as follows:
(in thousands) | ||||||||
FY 2015 | FY 2016 | FY 2017 | FY 2018 | FY 2019 | ||||
$13,983 | $13,050 | $10,313 | $8,861 | $7,753 | ||||
NOTE 7. RESTRUCTURING CHARGES
In April 2011, we adopted a plan to restructure our operations to reduce expenses, better align costs with current and future geographic sources of revenue, and improve operating efficiencies (the “2011 Restructuring Plan”). The 2011 Restructuring Plan focused primarily on the Early Phase business and corporate functions. The total cost of the 2011 Restructuring Plan was approximately $14.2 million and included the elimination of approximately 150 managerial and staff positions and abandonment of certain property leases.
In October 2009, we adopted a plan to restructure our operations to reduce expenses, better align costs with current and future geographic sources of revenue, and improve operating efficiencies (the “2010 Restructuring Plan”). The total cost of the 2010 Restructuring Plan was approximately $16.7 million, including the elimination of 240 managerial and staff positions and abandonment of certain property leases.
Various restructuring plans adopted by us since Fiscal Year 2005 are included in the Pre-2010 Plans.
58
Changes in the restructuring accrual during Fiscal Years 2014, 2013, and 2012 are summarized below:
Balance at | Provisions/ Adjustments | Payments/Foreign Currency Exchange | Balance at | |||||||||||||
(in thousands) | June 30, 2013 | June 30, 2014 | ||||||||||||||
2011 Restructuring Plan | ||||||||||||||||
Facilities-related | 975 | (370 | ) | (137 | ) | 468 | ||||||||||
2010 Restructuring Plan | ||||||||||||||||
Facilities-related | 468 | (74 | ) | (349 | ) | 45 | ||||||||||
Pre-2010 Plans | ||||||||||||||||
Facilities-related | 1,116 | — | (434 | ) | 682 | |||||||||||
Total | $ | 2,559 | $ | (444 | ) | $ | (920 | ) | $ | 1,195 | ||||||
Balance at | Provisions/ Adjustments | Payments/Foreign Currency Exchange | Balance at | |||||||||||||
(in thousands) | June 30, 2012 | June 30, 2013 | ||||||||||||||
2011 Restructuring Plan | ||||||||||||||||
Employee severance | $ | 1,884 | $ | (182 | ) | $ | (1,702 | ) | $ | — | ||||||
Facilities-related | 3,663 | (1,027 | ) | (1,661 | ) | 975 | ||||||||||
2010 Restructuring Plan | ||||||||||||||||
Facilities-related | 642 | — | (174 | ) | 468 | |||||||||||
Pre-2010 Plans | ||||||||||||||||
Facilities-related | 1,585 | — | (469 | ) | 1,116 | |||||||||||
Total | $ | 7,774 | $ | (1,209 | ) | $ | (4,006 | ) | $ | 2,559 | ||||||
Balance at | Provisions/ Adjustments | Payments/Foreign Currency Exchange | Balance at | |||||||||||||
(in thousands) | June 30, 2011 | June 30, 2012 | ||||||||||||||
2011 Restructuring Plan | ||||||||||||||||
Employee severance | $ | 1,622 | $ | 5,307 | $ | (5,045 | ) | $ | 1,884 | |||||||
Facilities-related | 3,720 | 2,004 | (2,061 | ) | 3,663 | |||||||||||
2010 Restructuring Plan | ||||||||||||||||
Employee severance | 1,160 | (984 | ) | (176 | ) | — | ||||||||||
Facilities-related | 855 | (84 | ) | (129 | ) | 642 | ||||||||||
Pre-2010 Plans | ||||||||||||||||
Facilities-related | 2,041 | — | (456 | ) | 1,585 | |||||||||||
Total | $ | 9,398 | $ | 6,243 | $ | (7,867 | ) | $ | 7,774 | |||||||
NOTE 8. CREDIT ARRANGEMENTS
Note Purchase Agreement
On July 25, 2013, we issued $100.0 million principal amount of 3.11% senior notes due July 25, 2020 (the “Notes”) for aggregate gross proceeds of $100.0 million in a private placement solely to accredited investors. The Notes were issued pursuant to a Note Purchase Agreement entered into by us with certain institutional investors on June 25, 2013 (the “Note Purchase Agreement”). Proceeds from the Notes were used to pay down $100.0 million of principal borrowed under the revolving credit facility portion of the 2013 Credit Agreement. We will pay interest on the outstanding balance of the Notes at a rate of 3.11% per annum, payable semi-annually on January 25 and July 25 of each year until the principal on the Notes shall have become due and payable. We may, at our option, upon notice and subject to the terms of the Note Purchase Agreement, prepay at any time all or part of the Notes in an amount not less than 10% of the aggregate principal amount of the Notes then outstanding, plus a Make-Whole Amount (as defined in the Note Purchase Agreement). The Notes become due and payable on July 25, 2020, unless payment is required to be made earlier under the terms of the Note Purchase Agreement.
The Note Purchase Agreement includes operational and financial covenants, with which we are required to comply, including, among others, maintenance of certain financial ratios and restrictions on additional indebtedness, liens and dispositions.
In connection with the Note Purchase Agreement, certain subsidiaries of ours entered into a Subsidiary Guaranty, pursuant to which such subsidiaries guaranteed our obligations under the Notes and the Note Purchase Agreement.
59
As of June 30, 2014, we had $100 million of principal borrowed under the Note Purchase Agreement. The outstanding amounts are presented net of debt issuance cost of approximately $0.4 million in our consolidated balance sheets.
In April and May 2013, we entered into three treasury lock agreements each with a notional amount of $25.0 million in connection with the planned issuance of our Notes that were issued in July 2013. The three treasury locks were used to minimize our interest rate exposure prior to locking in the fixed interest rate on our Notes. The treasury locks matured in May 2013 when the interest rate on our Notes was fixed. The treasury locks were deemed to be fully effective in accordance with ASC 815, and as such, the unrealized gains related to these derivatives are recorded as other comprehensive income and are amortized over the life of the Notes as interest income. As of June 30, 2014, our debt under the Note Purchase Agreement carried an average annualized interest rate of 3.05%.
Receivable Purchase Agreement
On February 19, 2013, we entered into a receivables purchase agreement (the “Receivable Agreement”) with JPMorgan Chase Bank, N.A. (“JPMorgan”). Under the Receivable Agreement, we sell to JPMorgan or other investors on an ongoing basis certain of our trade receivables, together with ancillary rights and the proceeds thereof, which arise under contracts with a client of ours, or its subsidiaries or affiliates. The Receivable Agreement includes customary representations and covenants on behalf of us, and may be terminated by either us or JPMorgan upon five business days advance notice. The Receivable Agreement provides a mechanism for accelerating the receipt of cash due on outstanding receivables. We account for the transfer of our receivables with respect to which we have satisfied the applicable revenue recognition criteria in accordance with FASB ASC 860, “Transfers and Servicing.” If we have not satisfied the applicable revenue recognition criteria for the underlying sales transaction, the transfer of the receivable is accounted for as a financing activity in accordance with FASB ASC 470, “Debt.” The accounts receivable and short-term debt balances are derecognized from our consolidated balance sheets at the earlier of the factored receivable’s due date or when all of the revenue recognition criteria are met for those billed services. For Fiscal Year 2014 and 2013, we transferred approximately $184.6 million and $36.5 million of trade receivables, respectively. As of June 30, 2013, we accounted for $10.4 million of the total transfers as financing activities which are recorded in accounts receivable and short-term debt on our consolidated balance sheets. As of June 30, 2014, no transfers were accounted for as a financing activity.
2013 Credit Agreement
On March 22, 2013, we, certain of our subsidiaries, Bank of America, N.A. (“Bank of America”), as Administrative Agent, Swingline Lender and L/C Issuer, Merrill Lynch, Pierce, Fenner & Smith Incorporated (“MLPFS”), J.P. Morgan Securities LLC (“JPM Securities”), HSBC Bank USA, National Association (“HSBC”) and U.S. Bank, National Association (“US Bank”), as Joint Lead Arrangers and Joint Book Managers, JPMorgan Chase Bank N.A. (“JPMorgan”), HSBC and US Bank, as Joint Syndication Agents, and the other lenders party thereto entered into an amended and restated agreement (the “2013 Credit Agreement”) providing for a five-year term loan of $200.0 million and a revolving credit facility in the amount of up to $300.0 million, plus additional amounts of up to $200.0 million of loans to be made available upon our request subject to specified terms and conditions. A portion of the revolving credit facility is available for swingline loans of up to a sublimit of $75.0 million and for the issuance of standby letters of credit of up to a sublimit of $10.0 million.
On March 22, 2013, we drew down $107.5 million under the 2013 Credit Agreement resulting in total outstanding borrowings of $400.0 million . We used the proceeds of the borrowing (i) to repay outstanding amounts under our existing four short-term credit facilities with each of Bank of America, HSBC, TD Bank, N.A. and US Bank (collectively, the “Short Term Credit Facilities”), (ii) for stock repurchases and (iii) for other general corporate purposes.
Our obligations under the 2013 Credit Agreement are guaranteed by certain of our material domestic subsidiaries, and the obligations, if any, of any of our foreign designated borrower are guaranteed by us and certain of our material domestic subsidiaries.
Borrowings (other than swingline loans) under the 2013 Credit Agreement bear interest, at our determination, at a rate based on either (a) LIBOR plus a margin (not to exceed a per annum rate of 1.750%) based on a ratio of consolidated funded debt to consolidated earnings before interest, taxes, depreciation and amortization (“EBITDA”) (the “Leverage Ratio”) or (b) the highest of (i) prime, (ii) the federal funds rate plus 0.50%, and (iii) the one month LIBOR rate plus 1.00% (such highest rate, the “Alternate Base Rate”), plus a margin (not to exceed a per annum rate of 0.750%) based on the Leverage Ratio. Swingline loans in U.S. dollars bear interest calculated at the Alternate Base Rate plus a margin (not to exceed a per annum rate of 0.750%).
Loans outstanding under the 2013 Credit Agreement may be prepaid at any time in whole or in part without premium or penalty, other than customary breakage costs, if any, subject to the terms and conditions contained in the 2013 Credit Agreement. The 2013 Credit Agreement terminates and any outstanding loans under it mature on March 22, 2018 (the “Maturity Date”).
60
Repayment of the principal borrowed under the revolving credit facility (other than a swingline loan) is due on the Maturity Date. Repayment of principal borrowed under the term loan facility is as follows, with the final payment of all amounts outstanding, plus accrued interest, being due on the Maturity Date:
• | 1.25% by quarterly term loan amortization payments to be made commencing in June 2013 and made prior to June 30, 2015; |
• | 2.50% by quarterly term loan amortization payments to be made on or after June 30, 2015, but prior to June 30, 2016; |
• | 5.00% by quarterly term loan amortization payments to be made on or after June 30, 2016, but prior to June 30, 2017; |
• | 7.50% by quarterly term loan amortization payment to be made on or after June 30, 2017, but prior to the Maturity Date; and |
• | 37.50% on the Maturity Date |
Our obligations under the 2013 Credit Agreement may be accelerated upon the occurrence of an event of default, which includes customary events of default, including payment defaults, defaults in the performance of affirmative and negative covenants, the inaccuracy of representations or warranties, bankruptcy and insolvency related defaults, cross defaults to material indebtedness, defaults relating to such matters as ERISA and judgments, and a change of control default.
The 2013 Credit Agreement contains negative covenants applicable to us and our subsidiaries, including financial covenants requiring us to comply with maximum leverage ratios and minimum interest coverage ratios, as well as restrictions on liens, investments, indebtedness, fundamental changes, acquisitions, dispositions of property, making specified restricted payments (including stock repurchases exceeding an agreed to percentage of consolidated net income), and transactions with affiliates. As of June 30, 2014, we were in compliance with all covenants under the 2013 Credit Agreement.
In connection with the 2013 Credit Agreement, we agreed to pay a commitment fee on the revolving loan commitment calculated as a percentage of the unused amount of the revolving loan commitment at a per annum rate of up to 0.350% (based on the Leverage Ratio). To the extent there are letters of credit outstanding under the 2013 Credit Agreement, we will pay letter of credit fees plus a fronting fee and additional charges. We also paid various customary fees to secure this arrangement, which are being amortized using the effective interest method over the life of the debt.
As of June 30, 2014, we had $62.5 million of principal borrowed under the revolving credit facility and $187.5 million of
principal under the term loan. For Fiscal Year 2014, we made principal payments of $10.0 million on the term loan under the 2013 Credit Agreement. The outstanding amounts are presented net of debt issuance cost of approximately $2.7 million in our consolidated balance sheets. We have borrowing availability of $237.5 million under the revolving credit facility.
In September 2011, we entered into an interest rate swap agreement and an interest rate cap agreement. Prior to the execution of the 2013 Credit Agreement, the interest rate swap and cap agreements hedged principal under our 2011 Credit Agreement, as discussed below. The interest rate swap and cap agreements now hedge a portion of the principal under our 2013 Credit Agreement. Specifically, principal in the amount of $100.0 million under our 2013 Credit Agreement has been hedged with the interest rate swap agreement and carries a fixed interest rate of 1.30% plus an applicable margin. Principal in the amount of $25.0 million has been hedged with the interest rate cap arrangement with an interest rate cap of 2.00% plus an applicable margin. In March 2014, the interest rate cap agreement matured and the related accumulated other comprehensive income was reclassified to net income during Fiscal Year 2014.
In May 2013, we entered into another interest rate swap agreement and hedged an additional principal amount of $100.0 million under our 2013 Credit Agreement with a fixed interest rate of 0.73% plus an applicable margin. As of June 30, 2014, our debt under the 2013 Credit Agreement, including the $200.0 million of principal hedged with both interest swap agreements, carried an average annualized interest rate of 1.60%. These interest rate hedges were deemed to be fully effective in accordance with ASC 815, “Derivatives and Hedging,” and, as such, unrealized gains and losses related to these derivatives are recorded as other comprehensive income.
2012 Term Loan and Facilities
On December 20, 2012, we entered into a new $100.0 million unsecured term loan agreement (the “2012 Term Loan”) with Bank of America, which was initially guaranteed by certain of our subsidiaries, but which guarantees were released in connection with the partial prepayment of the 2012 Term Loan in January 2013. The 2012 Term Loan was used to fund our acquisition of LIQUENT (see Note 3).
The 2012 Term Loan consisted of a term loan facility for $100.0 million, the full amount of which was advanced to us on December 21, 2012 and was scheduled to mature on June 30, 2013. Borrowings made under the 2012 Term Loan bore interest, at our option, at a base rate plus a margin (such margin not to exceed a per annum rate of 0.75%) based on a ratio of consolidated funded debt to EBITDA for the preceding twelve months (the “2012 Term Loan Leverage Ratio”), or at a LIBOR rate plus a margin (such margin not to exceed a per annum rate of 1.75%) based on the 2012 Term Loan Leverage Ratio. As of June 30, 2014, all outstanding amounts under the 2012 Term Loan were fully repaid with the proceeds from the 2013 Credit Agreement.
61
On January 22, 2013, we entered into additional short term unsecured term loan agreements with each of HSBC, TD Bank, N.A., and US Bank, each in the amount of $25.0 million (collectively, the “2013 Facilities”). The key terms of the 2013 Facilities were substantially the same as the 2012 Term Loan, including the loan maturities on June 30, 2013, except that there were no guaranties provided by any of our subsidiaries. The $75.0 million aggregate proceeds of the 2013 Facilities were used to partially pay down balances owed under the 2012 Term Loan, and in connection with such payment, Bank of America released our subsidiaries from their guaranty obligations under the 2012 Term Loan.
As of June 30, 2014, all outstanding amounts under the 2013 Facilities were fully repaid with the proceeds from the 2013 Credit Agreement.
2011 Credit Agreement
On June 30, 2011, we entered into the 2011 Credit Agreement providing for a five-year term loan of $100 million and a five-year revolving credit facility in the principal amount of up to $300 million. The borrowings all carried a variable interest rate based on LIBOR, prime, or a similar index, plus a margin (such margin not to exceed a per annum rate of 1.75%).
On March 22, 2013, the 2011 Credit Agreement was amended and restated in its entirety by the 2013 Credit Agreement. All amounts outstanding under the 2011 Credit Agreement immediately prior to the execution of the 2013 Credit Agreement were deemed to be outstanding under the terms and conditions of the 2013 Credit Agreement.
As discussed above, in September 2011, we entered into an interest rate swap and an interest rate cap agreement. Prior to the execution of the 2013 Credit Agreement, principal in the amount of $100.0 million under the 2011 Credit Agreement had been hedged with an interest rate swap agreement and carried a fixed interest rate of 1.30% plus an applicable margin. Principal in the amount of $25.0 million had been hedged with an interest rate cap arrangement with an interest rate cap of 2.00% plus an applicable margin. As of June 30, 2014, the interest rate swap agreements hedged a portion of the outstanding principal under our 2013 Credit Agreement.
Additional Lines of Credit
We have an unsecured line of credit with JP Morgan UK in the amount of $4.5 million that bears interest at an annual rate ranging between 2.00% and 4.00%. We entered into this line of credit to facilitate business transactions. At June 30, 2014, we had $4.5 million available under this line of credit.
We have a cash pool facility with RBS Nederland, NV in the amount of 5.0 million Euros that bears interest at an annual rate ranging between 2.00% and 4.00%. We entered into this line of credit to facilitate business transactions. At June 30, 2014, we had 5.0 million Euros available under this line of credit.
NOTE 9. STOCKHOLDERS’ EQUITY
During Fiscal Year 2014, 144,048 shares of common stock were surrendered as part of the cashless exercise of stock options and vesting of restricted stock to satisfy exercise costs and tax withholding payments.
Share Repurchase Plan
Fiscal Year 2014 Share Repurchase
On June 2, 2014, we announced that our Board of Directors approved a share repurchase program (the "2014 Program") authorizing the repurchase of up to $150.0 million of our common stock to be financed with cash on hand, cash generated from operations, existing credit facilities, or new financing. On June 13, 2014, we entered into an agreement (the “2014 Agreement”) to purchase shares of our common stock from Goldman Sachs & Co. (“GS”), for an aggregate purchase price of $150.0 million pursuant to an accelerated share purchase program. Pursuant to the 2014 Agreement, in June 2014, we paid $150.0 million to GS and received from GS 2,284,844 shares of common stock, representing 80% of the shares to be repurchased by us under the 2014 Agreement. The shares were repurchased at a price of $52.52 per share, which was the closing price of our common stock on the Nasdaq Global Select Market on June 13, 2014. These shares were canceled and restored to the status of authorized and unissued shares. As of June 30, 2014, we recorded the $150.0 million payment to GS as a decrease to equity in our consolidated balance sheet, consisting of decreases in common stock and additional paid-in capital. As additional paid-in capital was reduced to zero, the remainder was applied as a reduction in retained earnings.
The final number of shares to be delivered to us by GS under the 2014 Agreement at program maturity, net of the initial delivery, will be adjusted based on an agreed upon discount to the average of the daily volume weighted average price of the common stock during the term of the 2014 Agreement. If the number of shares to be delivered to us at maturity is less than the initial delivery of shares by GS, we would be required to remit shares or cash, at our option, to GS in an amount equivalent to such shortfall. We expect the 2014 Program to be completed by December 31, 2014.
62
Fiscal Year 2013 Share Repurchase
In August 2012, our Board of Directors approved a share repurchase program (the “2013 Program”) authorizing the repurchase of up to $200.0 million of our common stock to be financed with cash on hand, cash generated from operations, existing credit facilities, or new financing. While the 2013 Program did not obligate us to acquire any particular dollar value or number of shares of common stock, we repurchased $197.6 million of our common stock from the period September 2012 to June 2013, with the remaining $2.4 million of purchases occurring early in July 2013. This was achieved by entering into two separate $50.0 million accelerated share repurchase ("ASR") agreements and two separate $50.0 million open market agreements (the "Open Market Agreements") as described below. The Fiscal Year 2013 buyback activity also resulted in a reduction of our stockholders' equity of $197.6 million for the value of shares repurchased and retired by us.
In September 2012, we entered into an ASR agreement (the “September Agreement”) to purchase shares of our common stock from J.P. Morgan Securities LLC, as agent for JPMorgan Chase Bank, National Association, London Branch (“JPMorgan”), for an aggregate purchase price of $50.0 million. In March 2013, we finalized the settlement of the September Agreement and received an additional 234,898 shares representing the final shares delivered by JPMorgan. These shares were in addition to the initial 1,328,462 shares of our common stock delivered to us in September 2012. The total number of shares repurchased under the September Agreement was 1,563,360 at an average price per share of $31.98.
In March 2013, we entered into a second $50.0 million ASR agreement (the “March Agreement”) with JPMorgan. Pursuant to the March Agreement, JPMorgan delivered 1,044,932 shares of our common stock, representing an estimated 80% of the shares to be repurchased by us under the Agreement based on a price of $38.28 per share, which was the closing price of our common stock on March 15, 2013. In July 2013, subsequent to our fiscal year end, we finalized the settlement of the March Agreement and received an additional 101,247 shares representing the final shares delivered by JPMorgan. The total number of shares repurchased under the March Agreement was 1,146,179 at an average price per share of $43.62.
During Fiscal Year 2013, we also entered into two separate $50.0 million Open Market Agreements to buy back an additional $100.0 million of our common stock under the Program. For Fiscal Year 2013, we purchased 2,697,675 shares pursuant to the Open Market Agreements at an average price of $36.17 per share for a total of $97.6 million in common stock repurchases. As of June 30, 2013, approximately $2.4 million remained available under the 2013 Program for the purchase of additional shares. In July 2013, we purchased an additional 51,071 shares in the open market and completed our available purchases under the 2013 Program. Upon the completion of the purchases under our Open Market Agreements in July 2013, the total number of shares repurchased under the Open Market Agreements was 2,748,746 at an average price per share of $36.38.
In July 2013, with the completion of our Open Market Agreements and the final settlement of the March Agreement, the 2013 Program was completed.
NOTE 10. EARNINGS PER SHARE
The following table outlines the basic and diluted earnings per common share computations:
Years ended June 30, | ||||||||||||
(in thousands, except per share data) | 2014 | 2013 | 2012 | |||||||||
Net income attributable to common shares | $ | 129,094 | $ | 95,972 | $ | 63,158 | ||||||
Weighted average number of shares outstanding, used in computing basic earnings per share | 56,504 | 58,388 | 59,464 | |||||||||
Dilutive common stock equivalents | 973 | 1,059 | 962 | |||||||||
Weighted average shares used in computing diluted earnings per share | 57,477 | 59,447 | 60,426 | |||||||||
Basic earnings per share | $ | 2.28 | $ | 1.64 | $ | 1.06 | ||||||
Diluted earnings per share | $ | 2.25 | $ | 1.61 | $ | 1.05 | ||||||
Anti-dilutive options (excluded from the calculation of diluted earnings per share) | 536 | 503 | 2,231 | |||||||||
63
NOTE 11. ACCUMULATED OTHER COMPREHENSIVE INCOME (LOSS)
The following table reflects the activity for the components of accumulated other comprehensive income (loss), net of tax, for the years ended June 30, 2014, 2013 and 2012:
(in thousands) | Foreign Currency | Unrealized Gain/Loss on Derivatives | Total | |||||||||
Balance as of June 30, 2011 | $ | 18,719 | $ | 136 | $ | 18,855 | ||||||
Other comprehensive income before reclassifications | (46,334 | ) | (2,412 | ) | (48,746 | ) | ||||||
Loss reclassified from accumulated other comprehensive income | — | 752 | 752 | |||||||||
Net current-period other comprehensive income | $ | (46,334 | ) | $ | (1,660 | ) | $ | (47,994 | ) | |||
Balance at June 30, 2012 | $ | (27,615 | ) | $ | (1,524 | ) | $ | (29,139 | ) | |||
Other comprehensive income before reclassifications | (1,523 | ) | (1,363 | ) | (2,886 | ) | ||||||
Loss reclassified from accumulated other comprehensive income | — | 1,987 | 1,987 | |||||||||
Net current-period other comprehensive income | $ | (1,523 | ) | $ | 624 | $ | (899 | ) | ||||
Balance at June 30, 2013 | $ | (29,138 | ) | $ | (900 | ) | $ | (30,038 | ) | |||
Other comprehensive income before reclassifications | 27,050 | 6,411 | 33,461 | |||||||||
Gain reclassified from accumulated other comprehensive income | — | (1,333 | ) | (1,333 | ) | |||||||
Net current-period other comprehensive income | $ | 27,050 | $ | 5,078 | $ | 32,128 | ||||||
Balance at June 30, 2014 | $ | (2,088 | ) | $ | 4,178 | $ | 2,090 | |||||
The details regarding pre-tax gain (loss) on derivative instruments reclassified to net income from accumulated other comprehensive income are presented below:
Years Ended | Affected Line in the Consolidated Statements of Income | |||||||||||||
(in thousands) | 2014 | 2013 | 2012 | |||||||||||
Interest rate contracts | $ | (1,719 | ) | $ | (1,260 | ) | $ | (812 | ) | Interest expense, net | ||||
Foreign exchange contracts | 3,420 | (2,048 | ) | — | Direct Costs | |||||||||
Foreign exchange contracts | 281 | — | — | Service revenue | ||||||||||
Cross-currency swap contracts | 114 | 218 | (276 | ) | Miscellaneous (expense) income, net | |||||||||
Total | $ | 2,096 | $ | (3,090 | ) | $ | (1,088 | ) | ||||||
The amounts of gain (loss) reclassified from accumulated other comprehensive income into net income are net of taxes of $0.8 million, $1.1 million, and $0.3 million and for years ended June 30, 2014, 2013 and 2012, respectively.
NOTE 12. STOCK AND EMPLOYEE BENEFIT PLANS
Stock-Based Compensation
We account for stock-based compensation under ASC 718, “Compensation-Stock Compensation.” The stock option compensation cost calculated under the fair value approach is recognized over the vesting period of the stock options (generally over four years). All stock option grants are subject to graded vesting as services are rendered. The fair value for granted options is estimated at the time of the grant using the Black-Scholes option-pricing model. Expected volatilities are based on historical volatilities and we use historical data to estimate option exercise behavior. The expected term represents an estimate of the period of time we expect the options to remain outstanding based on historical exercise and post-vesting termination data. The dividend yield equals the most recent dividend payment over the market price of the stock at the beginning of the period. The risk-free interest rate is the rate at the date of grant for a zero-coupon U.S. Treasury bond with a term that approximates the expected term of the option. The following weighted average assumptions were used in the Black-Scholes
64
option-pricing model for awards issued during the respective periods:
Years ended June 30, | |||||||||
2014 | 2013 | 2012 | |||||||
Dividend yield | 0.0 | % | 0.0 | % | 0.0 | % | |||
Expected volatility | 54.1 | % | 56.4 | % | 56.9 | % | |||
Risk-free interest rate | 1.6 | % | 0.9 | % | 0.9 | % | |||
Expected term (in years) | 5.2 | 5.2 | 5.1 | ||||||
For the last three fiscal years, we recognized the following stock-based compensation expense:
Years ended June 30, | ||||||||||||
(in thousands) | 2014 | 2013 | 2012 | |||||||||
Direct costs related | $ | 3,027 | $ | 1,918 | $ | 1,815 | ||||||
Selling, general and administrative related | 12,302 | 9,250 | 9,316 | |||||||||
Total stock-based compensation | $ | 15,329 | $ | 11,168 | $ | 11,131 | ||||||
For Fiscal Years 2014, 2013, and 2012, the tax benefit related to stock compensation expense that we recognized was $4.4 million, $3.1 million, and $3.8 million, respectively. As of June 30, 2014, unearned stock-based compensation expense related to unvested awards (stock options and restricted stock) was approximately $28.6 million, which will be recognized over a weighted-average period of 2.7 years.
Stock Options
The Compensation Committee of the Board of Directors is responsible for the administration of our stock option plans and determines the term of each option, the option exercise price, the number of option shares granted, and the rate at which options become exercisable.
We adopted stock incentive plans in December 2010, December 2007, and September 2005, each of which provides for the grant of incentive stock options, non-statutory stock options, stock appreciation rights, restricted stock, restricted stock units and other stock-based award grants of up to 9,000,000 shares in aggregate to employees, officers, directors, consultants, and advisors. The granting of awards under these plans is discretionary, including the individuals who may become participants and receive awards under these plans and the number of shares they may acquire.
In September 2001, we adopted the 2001 Stock Incentive Plan, which provides for the grant of incentive and non-qualified stock options for the purchase of up to an aggregate of 2,000,000 shares of common stock to our employees, officers, directors, consultants, and advisors (and any individuals who have accepted an offer for employment).
Options under all our stock incentive plans described above expire no more than ten years from the date of grant and the expiration date and vesting period may vary at the Board of Directors’ discretion.
The following table summarizes information related to stock option activity for the respective periods:
Years ended June 30, | ||||||
(in thousands, except per share data) | 2014 | 2013 | 2012 | |||
Weighted-average fair value of options granted per share | $20.70 | $16.20 | $10.07 | |||
Intrinsic value of options exercised | $24,066 | $24,872 | $11,170 | |||
Stock option activity for the year ended June 30, 2014 was as follows:
Number of Options | Weighted-Average Exercise Price | Weighted- Average Remaining Contractual Life In Years | Aggregate Intrinsic Value (In Thousands) | ||||||||||
Balance at June 30, 2013 | 3,224,897 | $ | 23.51 | 5.5 | $ | 72,450 | |||||||
Granted | 850,455 | $ | 42.82 | ||||||||||
Exercised | (811,048 | ) | $ | 20.60 | |||||||||
Canceled | (84,125 | ) | $ | 31.14 | |||||||||
Balance at June 30, 2014 | 3,180,179 | $ | 29.21 | 5.3 | $ | 75,229 | |||||||
Exercisable at June 30, 2014 | 1,112,121 | $ | 21.37 | 3.3 | $ | 34,998 | |||||||
Expected to vest at June 30, 2014 | 1,904,051 | $ | 33.18 | 6.4 | $ | 37,519 | |||||||
65
Restricted Stock
We use restricted stock awards (“RSAs”) and restricted stock units (“RSUs”), granted under the plans described above, as a component of compensation for executive officers, non-employee members of the Board of Directors, and other employees. In general, we granted RSAs and RSUs that will vest at the end of a three-year service period for employees or one-year service period for non-employee members of the Board. The fair values of restricted stock awards and restricted stock units were based upon the closing stock prices on the day of the grants. For Fiscal Year 2014, 2013, and 2012, the fair value of restricted stock awards vested was $4.0 million, $4.6 million, and $5.2 million, respectively. Restricted stock activity for the year ended June 30, 2014 was:
Shares | Weighted-Average Grant- Date Fair Value | ||||||
Unvested Balance at June 30, 2013 | 469,266 | $ | 24.56 | ||||
Granted | 121,552 | $ | 48.90 | ||||
Vested | (167,866 | ) | $ | 23.75 | |||
Forfeited | (11,600 | ) | $ | 38.53 | |||
Unvested Balance at June 30, 2014 | 411,352 | $ | 31.69 | ||||
Employee Stock Purchase Plan
We sponsor an employee stock purchase plan (the “Purchase Plan”). The Purchase Plan allows eligible employees to purchase common stock at 95% of the fair market value of the stock on the last day of each purchase period (as defined by the Purchase Plan). The Purchase Plan also includes the automatic enrollment of contributions whereby an eligible employee’s compensation would be reduced and automatic enrollment contributions made on his/her behalf unless an affirmative election not to do so was made. The Purchase Plan is non-compensatory, and as such, no stock based compensation is recorded. An aggregate of approximately 1,800,000 shares may be issued under the Purchase Plan.
The following table summarizes the purchases under the Purchase Plan for the last three fiscal years:
Shares Purchased | Average Purchase Price | ||||||
Fiscal Year 2014 | 68,867 | $ | 43.20 | ||||
Fiscal Year 2013 | 62,777 | $ | 35.54 | ||||
Fiscal Year 2012 | 86,735 | $ | 21.81 | ||||
Savings Plan
We sponsor an employee savings plan (“the Plan”) as defined by Section 401(k) of the Internal Revenue Code of 1986, as amended. The Plan covers substantially all employees in the U.S. who elect to participate. Participants have the opportunity to invest on a pre-tax basis in a variety of mutual fund options and our stock. We match 100% of each participant’s voluntary contributions up to 3% of gross salary per payroll period subject to an annual cap of $3,000. Our contributions vest to the participants in 20% increments for each year of employment and become fully vested after five years of continuous employment. Our contributions to the Plan were approximately $9.6 million, $8.6 million, and $6.1 million for the Fiscal Years 2014, 2013, and 2012, respectively.
NOTE 13. FAIR VALUE MEASUREMENTS
We apply the provisions of ASC 820, “Fair Value Measurements and Disclosures.” ASC 820 defines fair value and provides guidance for measuring fair value and expands disclosures about fair value measurements. ASC 820 enables the reader of financial statements to assess the inputs used to develop those measurements by establishing a hierarchy for ranking the quality and reliability of the information used to determine fair values. ASC 820 requires that assets and liabilities carried at fair value be classified and disclosed in one of the following three categories:
• | Level 1 – Unadjusted quoted prices in active markets that are accessible to the reporting entity at the measurement date for identical assets and liabilities. |
• | Level 2 – Inputs other than quoted prices in active markets for identical assets and liabilities that are observable either directly or indirectly for substantially the full term of the asset or liability. Level 2 inputs include the following: |
◦ | quoted prices for similar assets and liabilities in active markets |
◦ | quoted prices for identical or similar assets or liabilities in markets that are not active |
◦ | observable inputs other than quoted prices that are used in the valuation of the asset or liabilities (e.g., interest rate and yield curve quotes at commonly quoted intervals) |
66
◦ | inputs that are derived principally from or corroborated by observable market data by correlation or other means |
• | Level 3 – Unobservable inputs for the assets or liability (i.e., supported by little or no market activity). Level 3 inputs include management’s own assumptions about the assumptions that market participants would use in pricing the asset or liability (including assumptions about risk). |
The following table sets forth by level, within the fair value hierarchy, our assets (liabilities) carried at fair value as of June 30, 2014:
(in thousands) | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
Contingent Consideration | $ | — | $ | — | $ | (5,152 | ) | $ | (5,152 | ) | ||||||
Interest Rate Derivative Instruments | — | 580 | — | 580 | ||||||||||||
Foreign Currency Exchange Contracts | — | 6,463 | — | 6,463 | ||||||||||||
Total | $ | — | $ | 7,043 | $ | (5,152 | ) | $ | 1,891 | |||||||
The following table sets forth by level, within the fair value hierarchy, our assets (liabilities) carried at fair value as of June 30, 2013:
(in thousands) | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
Contingent Consideration | $ | — | $ | — | $ | (5,934 | ) | $ | (5,934 | ) | ||||||
Interest Rate Derivative Instruments | — | (849 | ) | — | (849 | ) | ||||||||||
Foreign Currency Exchange Contracts | — | (3,425 | ) | — | (3,425 | ) | ||||||||||
Total | $ | — | $ | (4,274 | ) | $ | (5,934 | ) | $ | (10,208 | ) | |||||
Cash equivalents are measured at quoted prices in active markets. These investments are considered cash equivalents due to the short original maturity (less than 90 days) of the investments.
Marketable securities are held in foreign government treasury certificates that are actively traded and have original maturities over 90 days but less than one year. As of June 30, 2014, we had marketable securities of $95.6 million. Our marketable securities were invested in foreign government treasury certificates securities and they are classified as held-to-maturity based on our intent and ability to hold the securities to maturity and are recorded at amortized cost, which is not materially different than fair value. Interest and dividends related to these securities are reported as a component of interest income in our consolidated statements of income.
Interest rate derivative instruments are measured at fair value using a market approach valuation technique. The valuation is based on an estimate of net present value of the expected cash flows using relevant mid-market observable data inputs and based on the assumption of no unusual market conditions or forced liquidation.
Foreign currency exchange contracts are measured at fair value using a market approach valuation technique. The inputs to this technique utilize current foreign currency exchange forward market rates published by leading third-party financial news and data providers. This is observable data that represent the rates that the financial institution uses for contracts entered into at that date; however, they are not based on actual transactions so they are classified as Level 2.
Contingent consideration related to the HERON acquisition is measured at fair value using an income approach valuation technique, specifically, with probability weighted and discounted cash flow methods. Increases or decreases in the fair value of our contingent consideration liability can result from changes in discount periods and rates, as well as the likelihood of achieving financial targets. The recurring Level 3 fair value measurements of our contingent consideration liability include the following significant unobservable inputs:
Unobservable Input | Range | |
Discount rate | 18% | |
Probability of achieving financial targets | 10% to 55% | |
Projected period of payment | August 2015 | |
67
The following table provides a roll-forward of the fair value of the contingent consideration, which was determined by Level 3 inputs:
(in thousands) | Fair Value | |||
Balance at June 30, 2012 | $ | — | ||
Contingent consideration from the HERON acquisition | 5,934 | |||
Balance at June 30, 2013 | $ | 5,934 | ||
Change in fair value related to contingent consideration from HERON acquisition | (782 | ) | ||
Balance at June 30, 2014 | $ | 5,152 | ||
Contingent consideration liabilities are re-measured to fair value each reporting period using projected financial targets, discount rates, probabilities of payment and projected payment dates. Projected contingent payment amounts are discounted back to the current period using a discounted cash flow model. Projected financial targets are based on our most recent internal operational budgets and take into consideration of alternate scenarios that could result in more or less profitability for our commercialization service line. Increases or decreases in projected financial targets and probabilities of payment may result in significant changes in the fair value measurements. Increases in discount rates and the time to payment may result in lower fair value measurements. Increases or decreases in any of those inputs in isolation may result in a significantly lower or higher fair value measurement. Given the subjectivity of the inputs, the variation of estimated contingent payments related to the HERON acquisition range from zero to $14.2 million. The final obligation will be measured on June 30, 2015 based upon the operating results for the fiscal year ending June 30, 2015. For Fiscal Year 2014, the change in fair value of contingent consideration of $0.8 million was recorded in selling, general and administrative expense.
For the fiscal years ended June 30, 2014 and June 30, 2013, there were no transfers among Level 1, Level 2, or Level 3 categories. Additionally, there were no changes in the valuation techniques used to determine the fair values of our Level 2 assets or liabilities over the same periods.
The fair value of the debt under Note Purchase Agreement was estimated to be $95.4 million as of June 30, 2014, and was determined using U.S. government treasury rates and Level 3 inputs, including a credit risk adjustment.
The carrying value of our short-term and long-term debt under the 2013 Credit Agreement approximates fair value because all of the debt bears variable rate interest.
NOTE 14. INCOME TAXES
Domestic and foreign income (loss) before income taxes for the three years ended June 30 were as follows:
(in thousands) | 2014 | 2013 | 2012 | |||||||||
Domestic | $ | 98,154 | $ | 35,792 | $ | 18,347 | ||||||
Foreign | 89,707 | 97,358 | 61,359 | |||||||||
$ | 187,861 | $ | 133,150 | $ | 79,706 | |||||||
Provisions for income taxes for the three years ended June 30 were as follows:
(in thousands) | 2014 | 2013 | 2012 | |||||||||
Current: | ||||||||||||
Federal | $ | 27,264 | $ | 6,279 | $ | 8,054 | ||||||
State | 9,456 | 2,565 | 1,010 | |||||||||
Foreign | 33,242 | 26,651 | 9,608 | |||||||||
69,962 | 35,495 | 18,672 | ||||||||||
Deferred: | ||||||||||||
Federal | 4,001 | 7,650 | 2,447 | |||||||||
State | (905 | ) | (2,113 | ) | (869 | ) | ||||||
Foreign | (14,291 | ) | (3,854 | ) | (3,702 | ) | ||||||
(11,195 | ) | 1,683 | (2,124 | ) | ||||||||
$ | 58,767 | $ | 37,178 | $ | 16,548 | |||||||
68
Our consolidated effective income tax rate differed from the U.S. federal statutory income tax rate as set forth below:
(in thousands) | 2014 | % | 2013 | % | 2012 | % | |||||||||||||||
Income tax expense computed at the federal statutory rate | $ | 65,751 | 35.0 | % | $ | 46,602 | 35.0 | % | $ | 27,897 | 35.0 | % | |||||||||
State income taxes, net of federal benefit | 5,419 | 2.9 | % | 1,914 | 1.4 | % | (232 | ) | (0.3 | )% | |||||||||||
Foreign rate differential | (4,857 | ) | (2.6 | )% | (7,960 | ) | (6.0 | )% | (6,539 | ) | (8.2 | )% | |||||||||
Change in valuation allowances | (491 | ) | (0.3 | )% | (4,708 | ) | (3.5 | )% | 1,630 | 2.0 | % | ||||||||||
Change in reserves | (5,193 | ) | (2.8 | )% | 394 | 0.3 | % | (7,655 | ) | (9.6 | )% | ||||||||||
Research and development | (3,284 | ) | (1.7 | )% | (2,986 | ) | (2.2 | )% | (2,734 | ) | (3.4 | )% | |||||||||
Non-deductible losses | 69 | — | % | 440 | 0.3 | % | 678 | 0.9 | % | ||||||||||||
Other non-deductible expenses | 1,315 | 0.7 | % | 2,074 | 1.6 | % | 393 | 0.5 | % | ||||||||||||
Adjustment of net operating losses | — | — | % | 608 | 0.5 | % | 2,243 | 2.8 | % | ||||||||||||
Statutory tax rate changes | (623 | ) | (0.3 | )% | 798 | 0.5 | % | (1,047 | ) | (1.3 | )% | ||||||||||
Other, net | 661 | 0.4 | % | 2 | — | % | 1,914 | 2.4 | % | ||||||||||||
$ | 58,767 | 31.3 | % | $ | 37,178 | 27.9 | % | $ | 16,548 | 20.8 | % | ||||||||||
Provision has not been made for U.S. or additional foreign taxes on undistributed earnings of foreign subsidiaries as those earnings are indefinitely reinvested. Undistributed earnings of foreign subsidiaries that are indefinitely reinvested are approximately $423.0 million and $368.0 million at June 30, 2014 and June 30, 2013, respectively. Due to the complexities associated with this hypothetical calculation, it is not practicable to estimate the unrecognized deferred tax liability on the earnings that are indefinitely reinvested in foreign operations.
Significant components of our net deferred tax assets (liabilities) as of June 30, 2014 and June 30, 2013 were as follows:
(in thousands) | 2014 | 2013 | ||||||
Deferred tax assets: | ||||||||
U.S. loss carryforwards | $ | 1,764 | $ | 1,981 | ||||
Foreign loss carryforwards | 2,814 | 6,240 | ||||||
Accrued expenses | 38,915 | 35,340 | ||||||
Tax credit carryforwards | 5,711 | 14,559 | ||||||
Provision for losses on receivables | 920 | 1,161 | ||||||
Deferred compensation | 8,219 | 7,517 | ||||||
Deferred revenue | 12,698 | 8,949 | ||||||
Intercompany loans | 2,752 | 2,765 | ||||||
Other | 3,597 | 1,168 | ||||||
Gross deferred tax assets | 77,390 | 79,680 | ||||||
Deferred tax asset valuation allowance | (5,318 | ) | (6,023 | ) | ||||
Total deferred tax assets | 72,072 | 73,657 | ||||||
Deferred tax liabilities: | ||||||||
Property and equipment | (6,521 | ) | (5,606 | ) | ||||
Revenue recognition | (17,601 | ) | (26,537 | ) | ||||
Intangible assets | (35,372 | ) | (38,186 | ) | ||||
Other | (1,038 | ) | (2,491 | ) | ||||
Total deferred tax liabilities | (60,532 | ) | (72,820 | ) | ||||
Net deferred tax assets | $ | 11,540 | $ | 837 | ||||
69
The net deferred tax assets and liabilities included in the consolidated balance sheets as of June 30, 2014 and June 30, 2013 were as follows:
(in thousands) | 2014 | 2013 | ||||||
Current deferred tax assets | $ | 54,061 | $ | 44,236 | ||||
Non-current deferred tax assets | 6,669 | 8,556 | ||||||
Current deferred tax liabilities | (16,592 | ) | (16,512 | ) | ||||
Non-current deferred tax liabilities | (32,598 | ) | (35,443 | ) | ||||
$ | 11,540 | $ | 837 | |||||
At June 30, 2014, federal, state and foreign loss carryforwards of $2.8 million, $35.8 million and $15.8 million, respectively, were available to offset future liabilities for income taxes. The federal net operating losses expire in the years 2025 through 2034. Included in the state loss carryforwards is $3.7 million attributable to deductions from the exercise of equity awards. The benefit from these deductions will be recorded as a credit to additional paid-in capital if and when realized through a reduction of taxes paid in cash. Use of these loss carryforwards is limited based on the future income of certain subsidiaries. The state net operating losses expire in the years 2016 through 2033. Of the non-U.S. loss carryforwards, $4.8 million will expire between 2015 and 2034; the remainder does not expire. We also have U.S. foreign tax credit carryforwards of $28.0 million, which expire in the years 2018 through 2024. Included in the U.S. foreign tax credit carryforwards is $15.4 million attributable to deductions from the exercise of equity awards. The benefit from these credits will be recorded as a credit to additional paid-in capital if and when realized through a reduction of taxes paid in cash.
A valuation allowance has been established for certain future income tax benefits related to loss carryforwards and temporary tax adjustments based on an assessment that it is more likely than not that these benefits will not be realized. The decrease in the valuation allowance in Fiscal Year 2013 was principally due to improved profitability in various jurisdictions.
As of June 30, 2014, we had $41.5 million of gross unrecognized tax benefits of which $25.4 million would impact the effective tax rate if recognized. As of June 30, 2013, we had $46.6 million of gross unrecognized tax benefits of which $31.0 million would impact the effective tax rate if recognized. This reserve primarily relates to exposures for income tax matters such as changes in the jurisdiction in which income is taxable.
Unrecognized tax benefits represent favorable positions we have taken, or expect to take, on tax returns. These positions have reduced, or are expected to reduce, our income tax liability on our tax returns and financial statements. As a result of the uncertainty associated with these positions, we have established a liability that effectively reverses the previous recognition of the tax benefits, making them “unrecognized.” Our unrecognized income tax benefits, excluding accrued interest and penalties, are as follows:
(in thousands) | 2014 | 2013 | 2012 | |||||||||
Balance at beginning of year | $ | 46,591 | $ | 53,813 | $ | 62,211 | ||||||
Additions related to tax positions in prior years | 792 | — | 4,433 | |||||||||
Reductions related to tax positions in prior years | (1,115 | ) | (2,869 | ) | (1,898 | ) | ||||||
Reductions related to settlements with tax authorities | — | — | (477 | ) | ||||||||
Reductions related to the expiration of statutes | (5,630 | ) | (4,662 | ) | (6,789 | ) | ||||||
Currency translation adjustments | 833 | 309 | (3,667 | ) | ||||||||
Balance at end of year | $ | 41,471 | $ | 46,591 | $ | 53,813 | ||||||
As of June 30, 2014, we anticipate that the liability for unrecognized tax benefits for uncertain tax positions could change by up to $6.6 million in the next twelve months primarily as a result of the expiration of statutes of limitation and settlement with tax authorities. This change is composed primarily of reserves associated with the jurisdiction in which income is taxable.
We recognize interest and penalties related to income tax matters in income tax expense. As of June 30, 2014 and June 30, 2013, interest and penalties of $5.5 million and $5.0 million, respectively, were included in our liability for unrecognized tax benefits. For Fiscal Years 2014, 2013 and 2012, an expense of $0.3 million, and a benefit of $0.9 million and $2.1 million, respectively, was recorded for interest and penalties related to tax matters.
We are subject to U.S. federal income tax, as well as income tax in multiple state, local and foreign jurisdictions. All material state and local income tax matters through 2005 have been concluded. All material federal income tax matters have been concluded through 2005. Substantially all material foreign income tax matters have been concluded for all years through 2000.
70
NOTE 15. DEBT, COMMITMENTS, CONTINGENCIES AND GUARANTEES
We lease facilities under operating leases that include renewal and escalation clauses. Rent expense is recorded on a straight-line basis over the lease term. Total rent expense was $59.4 million, $54.7 million, and $52.2 million for Fiscal Years 2014, 2013, and 2012, respectively. Future minimum debt obligations, lease payments under non-cancelable leases, and purchase commitments due are as follows (excluding future potential payments in connection with the HERON acquisition - see Note 3):
(in thousands) | FY 2015 | FY 2016 | FY 2017 | FY 2018 | FY 2019 | Thereafter | Total | |||||||||||||||||||||
Debt obligations (principal) | $ | 12,501 | $ | 25,000 | $ | 45,000 | $ | 167,500 | $ | — | $ | 100,000 | $ | 350,001 | ||||||||||||||
Operating leases | 62,103 | 51,270 | 40,642 | 31,454 | 24,930 | 99,818 | 310,217 | |||||||||||||||||||||
Purchase commitments* | 55,036 | 17,783 | 6,146 | 1,588 | 1,020 | — | 81,573 | |||||||||||||||||||||
Total | $ | 129,640 | $ | 94,053 | $ | 91,788 | $ | 200,542 | $ | 25,950 | $ | 199,818 | $ | 741,791 | ||||||||||||||
*includes commitments to purchase software, hardware and services | ||||||||||||||||||||||||||||
We have letter-of-credit agreements with banks, totaling approximately $10.5 million, guaranteeing performance under various operating leases and vendor agreements. Additionally, the borrowings under the 2013 Credit Agreement and Note Purchase Agreement are guaranteed by certain of our U.S. subsidiaries.
We periodically become involved in various claims and lawsuits that are incidental to our business. We believe, after consultation with counsel, that no matters currently pending would, in the event of an adverse outcome, either individually or in the aggregate, have a material impact on our consolidated financial position, results of operations, or liquidity.
NOTE 16. GEOGRAPHIC INFORMATION
Financial information by geographic area for the three years ended June 30 was as follows:
(in thousands) | 2014 | 2013 | 2012 | |||||||||
Service revenue: | ||||||||||||
The Americas | $ | 970,894 | $ | 866,998 | $ | 635,290 | ||||||
Europe, Middle East & Africa | 709,137 | 624,010 | 555,467 | |||||||||
Asia/Pacific | 259,329 | 243,434 | 205,751 | |||||||||
Total | $ | 1,939,360 | $ | 1,734,442 | $ | 1,396,508 | ||||||
Income from operations: | ||||||||||||
The Americas | $ | 111,718 | $ | 44,743 | $ | 27,493 | ||||||
Europe, Middle East & Africa | 51,793 | 59,466 | 34,044 | |||||||||
Asia/Pacific | 35,987 | 31,914 | 27,265 | |||||||||
Total | $ | 199,498 | $ | 136,123 | $ | 88,802 | ||||||
Tangible long-lived assets: | ||||||||||||
The Americas | $ | 151,111 | $ | 146,137 | ||||||||
Europe, Middle East & Africa | 63,215 | 55,853 | ||||||||||
Asia/Pacific | 19,838 | 22,235 | ||||||||||
Total | $ | 234,164 | $ | 224,225 | ||||||||
The following countries represented greater than 10% of consolidated service revenue for the three years ended June 30:
(in thousands) | 2014 | 2013 | 2012 | |||||||||
Service revenue: | ||||||||||||
United States | $ | 902,931 | $ | 797,380 | $ | 580,340 | ||||||
United Kingdom | $ | 245,098 | $ | 227,964 | $ | 181,386 | ||||||
Germany | $ | 229,858 | $ | 187,099 | $ | 194,528 | ||||||
NOTE 17. SEGMENT INFORMATION
We have three reportable segments: CRS, PC and PI.
• | CRS constitutes our core business and includes all phases of clinical research from Early Phase (encompassing the early stages of clinical testing that range from first-in-man through proof-of-concept studies) to Phase II-III and Phase IV, which we call Peri/Post-Approval Services, formerly known as Peri-Approval Clinical Excellence. Our services |
71
include clinical trials management and biostatistics, data management and clinical pharmacology, as well as related medical advisory, patient recruitment, clinical supply and drug logistics, pharmacovigilance, and investigator site services. We aggregate Early Phase with Phase II-III and Peri/Post-Approval Services due to economic similarities in these operating segments.
• | PC provides technical expertise and advice in such areas as drug development, regulatory affairs, product pricing and reimbursement, commercialization and strategic compliance. It also provides a full spectrum of market development, product development, and targeted communications services in support of product launch. Our PC consultants identify alternatives and propose solutions to address client issues associated with product development, registration, and commercialization. HERON is included in our PC reportable segment. |
• | PI provides information technology solutions designed to help improve clients’ product development and regulatory submission processes. PI offers a portfolio of products and services that includes medical imaging services, ClinPhone® RTSM, IMPACT® CTMS, DataLabs® EDC, web-based portals, systems integration, electronic patient reported outcomes (“ePRO”), and LIQUENT InSight® RIM solutions. These services are often bundled together and integrated with other applications to provide eClinical solutions for our clients. LIQUENT is included in our PI reportable segment. |
In February 2014, we announced the launch of PAREXEL Regulatory Outsourcing Services (“PROS”), which was designed to provide a focused, market-driven approach to regulatory outsourcing services in the life science industry, with a primary emphasis on post-approval regulatory activities. Effective July 1, 2014, the operating results of PROS are included in the PC segment. This service line offering was previously included within LIQUENT RIM solutions and reported within the PI segment. For interim and annual periods beginning July 1, 2014, we will disclose the reportable segment on this new basis and prior periods will be retroactively restated to reflect the change.
We evaluate our segment performance and allocate resources based on service revenue and gross profit (service revenue less direct costs), while other operating costs are allocated and evaluated on a geographic basis. Accordingly, we do not include the impact of selling, general, and administrative expenses, depreciation and amortization expense, other income (expense), and income tax expense in segment profitability. We attribute revenue to individual countries based upon the revenue earned in the respective countries; however, inter-segment transactions are not included in service revenue. Furthermore, we have a global infrastructure supporting its business segments, and therefore, assets are not identified by reportable segment.
(in thousands) | CRS | PC | PI | TOTAL | ||||||||||||
Service revenue: | ||||||||||||||||
2014 | $ | 1,455,279 | $ | 216,184 | $ | 267,897 | $ | 1,939,360 | ||||||||
2013 | 1,303,569 | 202,524 | 228,349 | 1,734,442 | ||||||||||||
2012 | 1,038,705 | 167,125 | 190,678 | 1,396,508 | ||||||||||||
Gross profit on service revenue: | ||||||||||||||||
2014 | $ | 445,210 | $ | 91,498 | $ | 123,474 | $ | 660,182 | ||||||||
2013 | 347,056 | 81,570 | 98,280 | 526,906 | ||||||||||||
2012 | 279,166 | 69,565 | 75,948 | 424,679 | ||||||||||||
72
NOTE 18. QUARTERLY OPERATING RESULTS (UNAUDITED)
The following is a summary of unaudited quarterly results of operations for Fiscal Years 2014 and 2013:
Fiscal Year 2014 | ||||||||||||||||||||
(in thousands, except per share data) | First Quarter | Second Quarter | Third Quarter | Fourth Quarter | Total Year | |||||||||||||||
Service revenue | $ | 449,245 | $ | 487,145 | $ | 492,375 | $ | 510,595 | $ | 1,939,360 | ||||||||||
Gross profit | 146,051 | 162,638 | 169,226 | 182,267 | 660,182 | |||||||||||||||
Income from operations | 41,881 | 46,664 | 52,171 | 58,782 | 199,498 | |||||||||||||||
Net income | 25,954 | 28,329 | 34,739 | 40,072 | 129,094 | |||||||||||||||
Diluted earnings per share | $ | 0.45 | $ | 0.49 | $ | 0.60 | $ | 0.70 | $ | 2.25 | ||||||||||
Fiscal Year 2013 | ||||||||||||||||||||
(in thousands, except per share data) | First Quarter | Second Quarter | Third Quarter | Fourth Quarter | Total Year | |||||||||||||||
Service revenue | $ | 394,753 | $ | 422,068 | $ | 454,493 | $ | 463,128 | $ | 1,734,442 | ||||||||||
Gross profit | 115,349 | 120,743 | 142,576 | 148,238 | 526,906 | |||||||||||||||
Income from operations | 29,752 | 31,437 | 36,606 | 38,328 | 136,123 | |||||||||||||||
Net income | 15,064 | 21,343 | 29,524 | 30,041 | 95,972 | |||||||||||||||
Diluted earnings per share | $ | 0.25 | $ | 0.36 | $ | 0.50 | ` | $ | 0.52 | $ | 1.61 | |||||||||
73
Management’s Report on Internal Control over Financial Reporting
The management of PAREXEL International Corporation is responsible for establishing and maintaining adequate internal control over financial reporting for the Company. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Securities Exchange Act of 1934, as amended, as a process designed by, or under the supervision of, the Company’s principal executive and principal financial officers and effected by the Company’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
•Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the Company;
•Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and
•Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The Company’s management assessed the effectiveness of the Company’s internal control over financial reporting as of June 30, 2014. In making this assessment, the Company’s management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (1992 framework) (the COSO criteria) in Internal Control-Integrated Framework.
Based on the assessment, management concluded that, as of June 30, 2014, the Company’s internal control over financial reporting is effective based on those criteria.
The Company’s independent registered public accounting firm, Ernst & Young LLP, has issued an audit report on the Company’s internal control over financial reporting. This report appears on page 76.
/s/ Josef H. von Rickenbach | /s/ Ingo Bank | ||
Josef H. von Rickenbach | Ingo Bank | ||
Chairman of the Board and Chief Executive Officer | Senior Vice President and Chief Financial Officer | ||
(Principal Executive Officer) | (Principal Financial Officer) | ||
74
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of PAREXEL International Corporation
We have audited the accompanying consolidated balance sheets of PAREXEL International Corporation as of June 30, 2014 and 2013, and the related consolidated statements of income and comprehensive income, stockholders' equity and cash flows for each of the three years in the period ended June 30, 2014. Our audits also included the financial statement schedule listed in the Index at Item 15(a). These financial statements and schedule are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of PAREXEL International Corporation at June 30, 2014 and 2013, and the consolidated results of its operations and its cash flows for each of the three years in the period ended June 30, 2014, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), PAREXEL International Corporation's internal control over financial reporting as of June 30, 2014, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (1992 framework) and our report dated August 20, 2014 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Boston, Massachusetts
August 20, 2014
75
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of PAREXEL International Corporation
We have audited PAREXEL International Corporation's internal control over financial reporting as of June 30, 2014, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (1992 framework) (the COSO criteria). PAREXEL International Corporation's management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company's internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, PAREXEL International Corporation maintained, in all material aspects, effective internal control over financial reporting as of June 30, 2014, based on the COSO criteria.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of PAREXEL International Corporation as of June 30, 2014 and 2013, and the related consolidated statements of income and comprehensive income, stockholders' equity and cash flows for each of the three years in the period ended June 30, 2014 and our report dated August 20, 2014 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Boston, Massachusetts
August 20, 2014
76
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
Not applicable.
Item 9A. Controls and Procedures
(a) Evaluation of Disclosure Controls and Procedures
Management conducted an evaluation, under the supervision and with the participation of our Chief Executive Officer and Chief Financial Officer (our principal executive officer and principal financial officer, respectively), of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended) as of June 30, 2014. Based on the evaluation of our disclosure controls and procedures as of June 30, 2014, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, PAREXEL’s disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control over Financial Reporting and the independent registered public accounting firm’s attestation report on our internal control over financial reporting required under Item 308 of Regulation S-K have been included in Item 8 of this annual report.
(b) Changes in Internal Control Over Financial Reporting
No change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) occurred during the fiscal quarter ended June 30, 2014 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information
None.
77
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Information with respect to this item may be found under the captions “Elections of Directors,” “Corporate Governance,” “Executive Officers” and “Section 16(a) Beneficial Ownership Reporting Compliance” in the Proxy Statement for the Company’s 2014 Annual Meeting of Stockholders. Such information is incorporated herein by reference.
CODE OF ETHICS
PAREXEL has adopted a code of business conduct and ethics applicable to all of its employees, including our principal executive officer and principal financial officer. The code of business conduct and ethics is available on our website (www.parexel.com) under the category “Investor Relations-Corporate Governance.”
Item 11. Executive Compensation
Information with respect to this item may be found under the captions “Directors’ Compensation,” “Compensation Committee Interlocks and Insider Participation,” “Executive Compensation,” “Employment and Change of Control Agreements” and “Compensation Committee Report” in the Proxy Statement for the Company’s 2014 Annual Meeting of Stockholders. Such information is incorporated herein by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Information with respect to this item may be found under the caption “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” in the Proxy Statement for the Company’s 2014 Annual Meeting of Stockholders. Such information is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
Information with respect to this item may be found under the captions “Certain Relationships and Related Transactions” in the Proxy Statement for the Company’s 2014 Annual Meeting of Stockholders. Such information is incorporated herein by reference.
Item 14. Principal Accounting Fees and Services
Information with respect to this item may be found under the caption “Fees Paid to Independent Registered Public Accounting Firm” in the Proxy Statement for the Company’s 2014 Annual Meeting of Stockholders. Such information is incorporated herein by reference.
78
PART IV
Item 15. Exhibits and Financial Statement Schedules
(a) | 1. Financial Statements | |
The following financial statements and supplementary data are included in Item 8 of this annual report: | ||
Reports of Independent Registered Public Accounting Firm for the years ended June 30, 2014, 2013 and 2012 | ||
Consolidated Statements of Income and Comprehensive Income for each of the three years ended June 30, 2014, 2013 and 2012 | ||
Consolidated Balance Sheets at June 30, 2014 and 2013 | ||
Consolidated Statements of Cash Flows for each of the three years ended June 30, 2014, 2013 and 2012 | ||
Consolidated Statements of Stockholders’ Equity for each of the three years ended June 30, 2014, 2013 and 2012 | ||
Notes to Consolidated Financial Statements | ||
2. Financial Statement Schedules | ||
Schedule II - Valuation and Qualifying Accounts | ||
All other schedules have been omitted since the required information is not present, or not present in amounts sufficient to require submission of the schedule or because the information required is included in the consolidated financial statements or the Notes thereto. | ||
3. Exhibits | ||
Exhibits filed with this Form 10-K are included on the Exhibit Index, which is incorporated herein by reference, and are filed or furnished as part of this report or are incorporated into this report by reference. | ||
79
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
PAREXEL INTERNATIONAL CORPORATION
By: | /s/ Josef H. von Rickenbach | Dated: | August 20, 2014 | |||
Josef H. von Rickenbach Chairman of the Board and Chief Executive Officer | ||||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signatures | Title(s) | Date | ||
/s/ Josef H. von Rickenbach | August 20, 2014 | |||
Josef H. von Rickenbach | Chairman of the Board and Chief Executive Officer (Principal Executive Officer) | |||
/s/ Ingo Bank | August 20, 2014 | |||
Ingo Bank | Senior Vice President and Chief Financial Officer (Principal Financial and Accounting Officer) | |||
/s/ A. Dana Callow, Jr. | August 20, 2014 | |||
A. Dana Callow, Jr. | Director | |||
/s/ Patrick J. Fortune | August 20, 2014 | |||
Patrick J. Fortune | Director | |||
/s/ Eduard E. Holdener | August 20, 2014 | |||
Eduard E. Holdener | Director | |||
/s/ Christopher J. Lindop | August 20, 2014 | |||
Christopher J. Lindop | Director | |||
/s/ Richard L. Love | August 20, 2014 | |||
Richard L. Love | Director | |||
/s/ Ellen M. Zane | August 20, 2014 | |||
Ellen M. Zane | Director | |||
80
SCHEDULE II
PAREXEL INTERNATIONAL CORPORATION
VALUATION AND QUALIFYING ACCOUNTS
(in thousands) | Balance at beginning of year | Charged (credited) to costs and expenses | Other adjustments* | Balance at end of year | ||||||||||||
Provision for losses on receivables | ||||||||||||||||
Year ended June 30, 2012 | $ | 14,267 | $ | 818 | $ | (5,637 | ) | $ | 9,448 | |||||||
Year ended June 30, 2013 | $ | 9,448 | $ | (346 | ) | $ | (4,578 | ) | $ | 4,524 | ||||||
Year ended June 30, 2014 | $ | 4,524 | $ | (316 | ) | $ | (64 | ) | $ | 4,144 | ||||||
* Other adjustments denote the effects of foreign currency exchange, write-offs, and recoveries. | ||||||||||||||||
(in thousands) | Balance at beginning of year | Charged (credited) to income tax expense | Other adjustments* | Balance at end of year | ||||||||||||
Valuation allowance for deferred tax assets | ||||||||||||||||
Year ended June 30, 2012 | $ | 14,436 | $ | 1,179 | $ | (4,223 | ) | $ | 11,392 | |||||||
Year ended June 30, 2013 | $ | 11,392 | $ | (4,708 | ) | $ | (661 | ) | $ | 6,023 | ||||||
Year ended June 30, 2014 | $ | 6,023 | $ | (277 | ) | (428 | ) | $ | 5,318 | |||||||
* Other adjustments denote the effects of foreign currency exchange, write-offs, recoveries, acquisitions and certain reclassifications related to ASC 740. | ||||||||||||||||
81
EXHIBIT INDEX
EXHIBIT NO. | DESCRIPTION | |
3.1 | Amended and Restated Articles of Organization of the Company, as amended (filed as Exhibit 3.1 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2013 and incorporated herein by this reference). | |
3.2 | Amended and Restated By-laws of the Company, as amended (filed as Exhibit 3.2 to the Company’s Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2009 and incorporated herein by this reference). | |
4.1 | Specimen certificate representing the Common Stock of the Company (filed as Exhibit 4.1 to the Company’s Registration Statement on Form S-1 (File No. 33-97406) and incorporated herein by this reference). | |
10.1* | Second Amended and Restated 1995 Stock Plan of the Company (filed as Exhibit 10.9 to the Company’s Annual Report on Form 10-K for the year ended June 30, 1998 and incorporated herein by this reference). | |
10.2* | Form of Stock Option Agreement under the Company’s Second Amended and Restated 1995 Stock Plan (filed as Exhibit 99.1 to the Company’s current Report on Form 8-K dated September 2, 2004 and incorporated herein by this reference). | |
10.3* | Form of Stock Option Agreement for Executive Officers under the Company’s Second Amended and Restated 1995 Stock Plan (filed as Exhibit 10.3 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2004 and incorporated herein by this reference). | |
10.4* | 1998 Non-Qualified, Non-Officer Stock Option Plan, as amended (filed as Exhibit 10.16 to the Company’s Annual Report on Form 10-K for the year ended June 30, 1999 and incorporated herein by this reference). | |
10.5* | 2001 Stock Incentive Plan of the Company (filed as Exhibit 10.7.1 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2006 and incorporated herein by this reference). | |
10.6* | Amendment No. 1 to 2001 Stock Incentive Plan of the Company (filed as Exhibit 10.7.2 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2006 and incorporated herein by this reference). | |
10.7* | Form of Stock Option Agreement under the Company’s 2001 Stock Incentive Plan (filed as Exhibit 10.4 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2004 and incorporated herein by this reference). | |
10.8* | 2005 Stock Incentive Plan of the Company (filed as Exhibit 10.8 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2006 and incorporated herein by this reference). | |
10.9* | Form of Restricted Stock Agreement for non-employee directors under the Company’s 2005 Stock Incentive Plan (filed as Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended December 31, 2009 and incorporated herein by this reference). | |
10.10* | Form of Restricted Stock Agreement for executive officers under the Company’s 2005 Stock Incentive Plan (filed as Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q for the quarter ended December 31, 2009 and incorporated herein by this reference). | |
10.11* | Form of Stock Option Agreement under the Company’s 2005 Stock Incentive Plan (filed as Exhibit 10.11 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2012 and incorporated herein by this reference). | |
10.12* | 2007 Stock Incentive Plan (filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended December 31, 2007 and incorporated herein by reference). | |
10.13* | Form of Restricted Stock Unit Agreement for executive officers under the Company’s 2007 Stock Incentive Plan (filed as Exhibit 10.29 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2010 and incorporated herein by reference). | |
10.14* | Form of Restricted Stock Agreement for non-employee directors under the Company’s 2007 Stock Incentive Plan | |
10.15* | Form of Restricted Stock Agreement for executive officers under the Company’s 2007 Stock Incentive Plan | |
10.16* | Form of Stock Option Agreement under the Company’s 2007 Stock Incentive Plan | |
10.17* | 2010 Stock Incentive Plan as amended (filed as Exhibit 10.17 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2013 and incorporated herein by this reference). . | |
10.18* | Form of Restricted Stock Agreement for non-employee directors under the Company’s 2010 Stock Incentive Plan (filed as Exhibit 10.3 to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2011 and incorporated herein by reference) | |
10.19* | Form of Restricted Stock Agreement for executive officers under the Company’s 2010 Stock Incentive Plan (filed as Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2011 and incorporated herein by reference) | |
10.20* | Form of Restricted Stock Unit Agreement for executive officers under the Company’s 2010 Stock Incentive Plan (filed as Exhibit 10.4 to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2011 and incorporated herein by reference) | |
10.21* | Form of Stock Option Agreement under the Company’s 2010 Stock Incentive Plan (filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2011 and incorporated herein by reference) | |
10.22 | Lease dated June 14, 1991 between 200 West Street Limited Partnership and the Company (filed as Exhibit 10.19 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2000 and incorporated herein by this reference). | |
10.23 | First Amendment dated as of January 3, 1992 to the Lease dated June 14, 1991 between 200 West Street Limited Partnership and the Company (filed as Exhibit 10.25 to the Company’s Registration Statement on Form S-1 (File No. 33-97406) and incorporated herein by this reference). | |
10.24 | Second Amendment dated as of June 28, 1993 to the Lease dated June 14, 1991 between 200 West Street Limited Partnership and the Company (filed as Exhibit 10.28 to the Company’s Registration Statement on Form S-1 (File No. 33-97406) and incorporated herein by this reference). | |
10.25 | Third Amendment to Lease dated November 17, 1998 between Boston Properties Limited Partnership and the Company (filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended December 31, 1998 and incorporated herein by this reference). | |
10.26 | Fourth Amendment dated August 28, 2000 to the Lease dated November 17, 1998 between Boston Properties Limited Partnership and the Company (filed as Exhibit 10.22 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2000 and incorporated herein by this reference). | |
10.27 | Fifth Amendment dated as of June 29, 2007 to the lease dated June 14, 1991 by and between Boston Properties Limited Partnership and PAREXEL International, LLC (filed as Exhibit 10.12.5 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2007 and incorporated herein by this reference). | |
82
EXHIBIT NO. | DESCRIPTION | |
10.28* | Amended and Restated Employment Agreement dated April 15, 2008 between Josef H. von Rickenbach and the Company (filed as Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2008 and incorporated herein by this reference). | |
10.29* | Employment Agreement, dated February 21, 2005, between Ulf Schneider and PAREXEL GmbH (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated February 21, 2005 and incorporated herein by this reference). | |
10.30* | Amended and Restated Change of Control/Severance Agreement, dated as of October 31, 2008, by and between the Company and Mark A. Goldberg (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated October 31, 2008 and incorporated herein by reference). | |
10.31* | PAREXEL International Amended and Restated Nonqualified Deferred Compensation Plan (filed as Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2013 and incorporated herein by this reference). | |
10.32 | Lease dated April 10, 2002 between API (No. 23) Limited, Arlington Property Investments Limited, PAREXEL International Limited and the Company (filed as Exhibit 10.22 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2003 and incorporated herein by this reference). | |
10.33* | Amended and Restated Change of Control/Severance Agreement, dated as of April 15, 2008, by and between the Company and Douglas A. Batt (filed as Exhibit 10.26 to the Company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2008 and incorporated herein by this reference). | |
10.34* | Form of Key Employee Agreement for Executive Officers (US) (filed as Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2013 and incorporated herein by this reference). | |
10.35* | Form of Key Employee Agreement for Executive Officers (UK) (filed as Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q for the quarter ended March 31, 2013 and incorporated herein by this reference). | |
10.36 | Amended and Restated Credit Agreement, dated as of March 22, 2013, among PAREXEL, certain subsidiaries of PAREXEL, Bank of America, N.A., as Administrative Agent, Swingline Lender and L/C Issuer, Merill Lynch Pierce Fenner & Smith Incorporated, JPMorgan Securities LLC, HSBC Bank USA, National Association ("HSBC") and U.S. Bank National Association ("US Bank"), as Joint Lead Arrangers and Joint Book Managers, JPMorgan Chase Bank, N.A., HSBC and US Bank, as Joint Syndication Agents, and the lenders party thereto (incorporated herein by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K dated March 22, 2013 and incorporated herein by this reference). | |
10.37* | Change of Control/severance Agreement, dated as of November 5, 2009, by and between the Company and Ulf Schneider (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K dated November 5, 2009, and incorporated herein by this reference). | |
10.38* | Key Employee Agreement, dated November 1, 2012, by and between PAREXEL International Limited and Anita Cooper (filed as Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q for the quarterly period ended December 31, 2012 and incorporated herein by this reference). | |
10.39* | Key Employee Agreement, dated November 6, 2012, by and between PAREXEL International Corporation and Joseph C. Avellone (filed as Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2012 and incorporated herein by this reference). | |
10.40* | Employment Offer Letter, dated June 6, 2013, between PAREXEL International Corporation and Ingo Bank (filed as Exhibit 10.44 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2013 and incorporated herein by this reference) . | |
10.41* | Change of Control/Severance Agreement, dated as of July 1, 2013, by and between the Company and Ingo Bank (filed as Exhibit 10.45 to the Company’s Annual Report on Form 10-K for the year ended June 30, 2013 and incorporated herein by this reference). . | |
10.42 | Receivables Purchase Agreement, dated as of February 19, 2013, by and between PAREXEL International, LLC and JPMorgan Chase Bank, N.A. (filed as Exhibit 10.1 to the Company's Current Report on Form 8-K dated February 19, 2013 and incorporated herein by reference). | |
10.43 | Note Purchase Agreement, dated as of June 25, 2013, between the Company and the Purchasers listed therein (filed as Exhibit 10.1 to the Company's Current Report on Form 8-K dated June 25, 2013 and incorporated herein by reference). | |
10.44 | Letter Agreement Regarding Accelerated Share Repurchase Program by and between PAREXEL International Corporation and Goldman Sachs & Co., dated June 13, 2014 (filed as Exhibit 10.1 to the Company's Current Report on Form 8-K dated June 11, 2014 and incorporated herein by reference). | |
10.45* | Letter Agreement, dated September 30, 2013, by and between the Company and Joseph C. Avellone (filed as Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the quarterly period ended September 30, 2013 and incorporated herein by reference | |
21.1 | List of subsidiaries of the Company. | |
23.1 | Consent of Independent Registered Public Accounting Firm. | |
31.1 | CEO certification pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
31.2 | CFO certification pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
32.1 | CEO certification pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
32.2 | CFO certification pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
101.INS** | XBRL Instance Document. | |
101.SCH** | XBRL Taxonomy Extension Schema. | |
101.CAL** | XBRL Taxonomy Extension Calculation Linkbase. | |
101.DEF** | XBRL Taxonomy Extension Definition Linkbase. | |
101.LAB** | XBRL Taxonomy Extension Label Linkbase. | |
101.PRE** | XBRL Taxonomy Extension Presentation Linkbase. | |
* Denotes management contract or any compensatory plan, contract or arrangement | ||
** In accordance with Rule 406T of Regulation S-T, the information in these exhibits is furnished and deemed not filed or part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act of 1933, is deemed not filed for purposes of Section 18 of the Securities Exchange Act of 1934, and otherwise is not subject to liability under these sections | ||
83