Attached files
| file | filename |
|---|---|
| EX-31.2 - REPROS THERAPEUTICS INC. | v215630_ex31-2.htm |
| EX-32.2 - REPROS THERAPEUTICS INC. | v215630_ex32-2.htm |
| EX-31.1 - REPROS THERAPEUTICS INC. | v215630_ex31-1.htm |
| EX-32.1 - REPROS THERAPEUTICS INC. | v215630_ex32-1.htm |
| EX-23.1 - REPROS THERAPEUTICS INC. | v215630_ex23-1.htm |
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
|
x
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
For the fiscal year ended December 31, 2010
|
or
|
¨
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
|
For the transition period from to
|
Commission File No. 001-15281
(Exact name of registrant as specified in its charter)
|
Delaware
|
76-0233274
|
|
(State or other jurisdiction of
|
(IRS Employer
|
|
incorporation or organization)
|
Identification No.)
|
|
2408 Timberloch Place, Suite B-7
|
77380
|
|
The Woodlands, Texas
|
(Zip Code)
|
|
(Address of principal executive offices)
|
(281) 719-3400
(Registrant's telephone number, including area code)
|
Title of Each Class
|
Name of Each
Exchange on Which Registered
|
|
Common Stock, $.001 par value
|
The Nasdaq Stock Market LLC
|
|
Series A Warrants
|
The Nasdaq Stock Market LLC
|
|
Series B Warrants
|
The Nasdaq Stock Market LLC
|
|
Rights to purchase Series One Junior
|
The Nasdaq Stock Market LLC
|
|
Participating Preferred Stock
|
Indicate by check mark whether the registrant is a well-known seasoned issuer (as defined in Rule 405 of the Securities Act). Yes ¨ No x
Indicate by check mark whether the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Securities Act.
Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files. Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer or a non-accelerated filer. See definition of "accelerated filer and large accelerated filer" in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer ¨
|
Accelerated filer ¨
|
|
Non-accelerated filer ¨
|
Smaller reporting company x
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Securities Exchange Act). Yes ¨ No x
The aggregate market value of the voting stock held by non-affiliates of the registrant was approximately $12,408,185 as of June 30, 2010, the last business day of the registrant's most recently completed second fiscal quarter, based on the closing sales price of the registrant's common stock on the Nasdaq Capital Market on such date of $1.44 per share. For purposes of the preceding sentence only, all directors, executive officers and beneficial owners of ten percent or more of the shares of the registrant's common stock are assumed to be affiliates.
As of March 18, 2011, there were 11,976,209 shares of the registrant's common stock outstanding.
Documents incorporated by reference: Portions of the registrant's definitive proxy statement relating to the registrant's 2011 Annual Meeting of Shareholders, which proxy statement will be filed under the Exchange Act within 120 days of the end of the registrant's fiscal year ended December 31, 2010, are incorporated by reference into Part III of this Form 10-K.
REPROS THERAPEUTICS INC
2010 FORM 10-K ANNUAL REPORT
TABLE OF CONTENTS
|
Page
|
|||
|
PART I
|
2
|
||
|
Item 1.
|
Business
|
2
|
|
|
Item 1A.
|
Risk Factors
|
11
|
|
|
Item 1B.
|
Unresolved Staff Comments
|
21
|
|
|
Item 2.
|
Properties
|
21
|
|
|
Item 3.
|
Legal Proceedings
|
22
|
|
|
Item 4.
|
(Removed and Reserved)
|
22
|
|
|
PART II
|
22
|
||
|
Item 5.
|
Market for the Registrant's Common Equity, Related Stockholder Matters & Issuer Purchases of Equity Securities
|
23
|
|
|
Item 6.
|
Selected Consolidated Financial Data
|
25
|
|
|
Item 7.
|
Management's Discussion and Analysis of Financial Condition and Results of Operations
|
26
|
|
|
Item 7A.
|
Quantitative and Qualitative Disclosures About Market Risk
|
34
|
|
|
Item 8.
|
Financial Statements and Supplementary Data
|
35
|
|
|
Item 9.
|
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
|
35
|
|
|
Item 9A.
|
Controls and Procedures
|
35
|
|
|
Item 9B.
|
Other Information
|
35
|
|
|
PART III
|
36
|
||
|
Item 10.
|
Directors, Executive Officers and Corporate Governance
|
36
|
|
|
Item 11.
|
Executive Compensation
|
36
|
|
|
Item 12.
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
|
36
|
|
|
Item 13.
|
Certain Relationship and Related Transactions, and Director Independence
|
36
|
|
|
Item 14.
|
Principal Accountant Fees and Services
|
36
|
|
|
PART IV
|
37
|
||
|
Item 15.
|
Exhibits and Financial Statement Schedules
|
37
|
This Annual Report on Form 10-K contains "forward-looking statements" within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. The words "may," "anticipate," "believe," "expect," "estimate," "project," "suggest," "intend" and similar expressions are intended to identify forward-looking statements. Such statements reflect our current views with respect to future events and financial performance and are subject to certain risks, uncertainties and assumptions, including those discussed in "Item 1. Description of Business — Business Risks." Should one or more of these risks or uncertainties materialize, or should underlying assumptions prove incorrect, actual results may vary materially from those anticipated, believed, expected, estimated, projected, suggested or intended.
PART I
|
ITEM 1.
|
BUSINESS
|
Overview
Repros Therapeutics Inc. (the “Company”, “RPRX,” “Repros,” or “we,” “us” or “our”) was organized on August 20, 1987. We are a development stage biopharmaceutical company focused on the development of new drugs to treat hormonal and reproductive system disorders.
We are developing Androxal®, an oral therapy that normalizes testicular function, for the treatment of low testosterone due to secondary hypogonadism. Secondary hypogonadism is associated with aging and we believe it is the most common cause of low testosterone in men. It is estimated that 13 million men in the U.S. experience low levels of testosterone, and the condition is becoming recognized with more frequency. In 2009, for the first time, sales of testosterone preparations for the treatment of low testosterone exceeded $1 billion worldwide and first tier pharmaceutical companies entered the low testosterone marketplace as evidenced by the acquisition of Solvay Pharmaceuticals and the subsequent active marketing of its AndroGel® product by Abbott Laboratories. Eli Lilly and Company also recently entered into a licensing agreement with a third party for a late stage topical testosterone treatment.
The Company believes Androxal® is highly differentiated from currently marketed testosterone treatments or those treatments in late stage development because it is an oral therapy and it treats the cause of secondary hypogonadism, which is inadequate pituitary hormones. We believe that by treating the cause of secondary hypogonadism it also has the potential to maintain reproductive status and potentially improve overall metabolic profiles, which we believe may improve the condition of men suffering from Type 2 diabetes. The Company held a Type B meeting with the Food and Drug Administration (“FDA”) on November 8, 2010 to discuss the FDA’s willingness to review Phase 3 protocols under a Special Protocol Assessment (“SPA”). Although the FDA advised the Company that it may proceed with Phase 3 studies, the FDA recommended that a Phase 2B study in men with secondary hypogonadism, but naïve to testosterone treatment, be conducted if the Company desired the protocols to be reviewed under an SPA. On January 3, 2011, we announced that we have received Investigational Review Board (“IRB”) approval to commence the Phase 2B study of Androxal® in men with secondary hypogonadism, and we have begun enrolling patients. Depending on the rate of subject enrollment, we hope to have the study completed by the end of 2011.
The Company is also currently conducting a Phase 2 study of the use of Androxal® in the treatment of Type 2 diabetes in hypogonadal men. Retrospective analysis of completed Androxal® studies showed that Androxal® improved fasting plasma glucose levels in men with Type 2 diabetes, an improvement not seen in similar subjects using a topical testosterone or placebo. The Company believes this effect is directly related to Androxal®’s ability to normalize the hypothalamic-pituitary-testes pathway and organ function.
We are also developing Proellex®, an orally administered selective blocker of the progesterone receptor in women, for the treatment of uterine fibroids and endometriosis. Uterine fibroids and endometriosis affect millions of women of reproductive age. We believe an effective treatment for these underserved conditions could result in sales of a safe and effective drug easily exceeding $1 billion in sales in the U.S. Proellex® had shown statistically significant results in previous Phase 2 studies for endometriosis and uterine fibroids. The Company has recently commenced a low dose escalating study as permitted by the FDA, which is intended to determine both signals of efficacy and safety for low oral doses of the drug.
Both of our product candidates have exhibited strong efficacy results in every study completed to date, and we believe the studies presently underway or scheduled to start shortly will place both programs on a clear late stage clinical development path and a solid position for licensing.
As of December 31, 2010, we had accumulated losses of $179.2 million, approximately $3.0 million in cash and cash equivalents, and our accounts payable and accrued expenses were approximately $1.3 million. On February 8, 2011, we completed a public offering of our common stock, Series A Warrants to purchase common stock and Series B Warrants to purchase common stock which resulted in approximately $11 million in gross proceeds to us, after the underwriting discount and before offering expenses. See “—Recent Developments” for a description of such offering. We believe we have sufficient funding to complete all of the Phase 2 and 2B clinical trials currently planned or underway; however, significant additional capital will be required for us to complete development of either of our product candidates. We continue to explore potential additional financing alternatives (including corporate partnering opportunities) that would provide sufficient funds to enable us to continue to develop our two product candidates through completion of the outlined clinical trials; however, there can be no assurance that we will be successful in raising any such additional funds on a timely basis or at all. The foregoing and other matters raise substantial doubt about our ability to continue as a going concern.
-2-
On October 14, 2010, the Company effected a one-for-four reverse split of its common stock. The split-adjusted shares of the Company’s common stock began trading on the Nasdaq Capital Market on October 15, 2010. The one-for-four reverse split converted all shares of the Company’s common stock issued and outstanding, plus all outstanding stock options and the number of shares of common stock available for issuance under the Company’s approved stock plans. The number of authorized shares of common stock was not affected by the reverse split. The reverse split enabled the Company to meet the continued listing rules of the Nasdaq Capital Market as evidenced by the Compliance Letter received from Nasdaq on October 29, 2010. All share and per share amounts have been retroactively adjusted to reflect the reverse stock split for all periods presented.
Androxal®
Product Overview
Our primary product candidate, Androxal®, is a single isomer of clomiphene citrate and is an orally active proprietary small molecule compound. We are developing Androxal® for men of reproductive age with low testosterone levels. Androxal® treats the underlying mechanism that causes secondary hypogonadism and restores normal testicular function. Unlike testosterone replacement which suppresses testicular function, Androxal® does not impair the reproductive status of men being treated for low testosterone. In addition, we are conducting a Phase 2 clinical trial of Androxal® as a potential treatment for Type 2 diabetes.
Testosterone is an important male hormone. Testosterone deficiency in men is linked to several negative physical and mental conditions, including loss of muscle tone, reduced sexual desire, and deterioration of memory and certain other cognitive functions. Testosterone production normally decreases as men age, sometimes leading to testosterone deficiency. The leading therapy for low testosterone is AndroGel®, a commercially available testosterone replacement cream marketed by Abbott Laboratories for the treatment of low testosterone, which we believe has had and continues to have significant sales in North America.
Based on our own clinical trial screening data, we believe over 70% of men that have low testosterone suffer from secondary hypogonadism, a pituitary defect which is characterized by suboptimal levels of LH (luteinizing hormone) and FSH (follicle stimulating hormone). LH and FSH are the pituitary hormones that stimulate testicular testosterone and sperm production, respectively. Men with secondary hypogonadism can be readily distinguished from those that have primary testicular failure via assessment of the levels of secretions of pituitary hormones, as men with primary testicular failure experience elevated secretions of pituitary hormones. In secondary hypogonadism, the low levels of LH and FSH fail to provide adequate hormone signaling to the testes, causing testosterone levels to drop to a level where we believe pituitary secretions fall under the influence of estrogen, thus further suppressing the testicular stimulation from the pituitary.
Androxal® acts centrally to restore testicular function and hence normal testosterone in the body. The administration of exogenous testosterone can restore serum testosterone levels, but does not restore testicular function and thereby generally leads to the cessation of or significant reduction in sperm production. Androxal®, by contrast, restores levels of both LH and FSH, which stimulate testicular testosterone and sperm production, respectively.
We tested Androxal® in two studies designed to show that Androxal® improved testosterone levels as well as AndroGel® in men with secondary hypogonadism. These studies indicated that Androxal® had a superior ability to improve testosterone levels when compared to AndroGel®, and that the improvement was statistically significant. In a meeting held with the FDA in the fourth quarter of 2007, however, the FDA determined that improved testosterone levels alone were not sufficient to grant approval for the drug. In the meeting held on November 8, 2010, the FDA changed its position and determined that improved testosterone levels would be sufficient to grant approval for the drug.
We also believe there may be an association between the restoration of normal pituitary function and improvement of metabolic conditions such as Type 2 diabetes. Research has been published which demonstrates that increased insulin resistance, a characteristic implicated in Type 2 diabetes, is associated with the onset of secondary hypogoandism. Based on our own clinical trial screening data, we have found hypogonadism and Type 2 diabetes to be comorbid conditions in a significant number of men. A retrospective analysis of the clinical trial data from our completed Androxal® studies showed that Androxal® improved fasting plasma glucose levels in men with Type 2 diabetes, suggesting that Androxal® modifies the endocrinologic profile in terms of both hormones and certain metabolic measures. This improvement was not seen in similar subjects using a topical testosterone or placebo. In a large trial conducted by Solvay Pharmaceuticals, AndroGel® was found to have no positive effect on glycemic control in hypogonadal men who were also Type 2 diabetic regardless of how much the exogenous testosterone concentration increased. Contrary to the results seen with exogenous testosterone, Androxal® did exhibit positive effects on glycemic control, and we believe these effects are directly related to Androxal®’s ability to normalize the hypothalamic-pituitary-testes pathway and organ function.
Androxal® will be required to undergo the full regulatory approval process, including the current Phase 2B trial, pivotal Phase 3 trial and long-term Open Label Safety Studies as well as other requirements. Androxal® is closely related chemically to the drug, Clomid®, which is approved for use in women to treat certain infertility disorders. Clomid® contains both the trans and cis isomers of clomiphene citrate; Androxal® contains only the trans isomer. The FDA has indicated that testicular tumors, gynecomastia and adverse ophthalmologic events, which have been reported in males taking Clomid®, are potential risks that should be included in informed consent forms for our Androxal® clinical trials. We do not believe that Androxal® will present with the same adverse events given its reduced half-life in the human body as compared to Clomid®. In our preclinical studies and our clinical trials to date, we have observed no evidence of any of these events except for certain ophthalmologic events in our preclinical dog study at doses significantly higher than those administered in the clinical trials.
-3-
All clinical trial results are subject to review by the FDA, and the FDA may disagree with our conclusions about safety and efficacy. We caution that the results discussed herein are based on data from non-pivotal trials and that our current Phase 2 trials, and any necessary pivotal Phase 3 and long-term Open Label Safety Trial data may not agree with these results which will be based upon significantly larger and more diverse patient populations treated for longer periods of time.
Treatment for Secondary Hypogonadism in Men Wishing to Preserve Testicular Function (Reproductive Status)
On November 8, 2010, we held a Type B meeting with the FDA to discuss whether the FDA would review our protocols for a Phase 3 trial of Androxal® in men with secondary hypogonadism under an SPA. In the meeting, the FDA recommended that a Phase 2B study in men with secondary hypogonadism but naïve to testosterone treatment be conducted if the Company desired the protocols to be reviewed under an SPA. The FDA further opined that such Phase 2B study would provide for a more solid data base for design of Phase 3 studies and eventual approval of such studies under an SPA. In our 24-patient Phase 2B proof-of-concept clinical trial which was initiated in the second quarter of 2008, we monitored the effects of Androxal® on male fertility and testicular function in patients being treated for low testosterone as compared to Testim®, a popular marketed topical testosterone medication. This trial showed that Androxal® was able to maintain sperm counts in men being treated for their low testosterone levels, whereas Testim® resulted in suppressed sperm levels.
The Company’s Phase 2B trial, which has begun enrolling patients, consists of four arms; placebo, two doses of Androxal® and topical testosterone. We hope that the study will be fully enrolled by the end of the third quarter 2011. At baseline the men should exhibit morning testosterone less than 250 ng/dl. The primary endpoint will consist of total testosterone at the end of the three month study compared to baseline. Impact on reproductive status (sperm counts) will be assessed as a safety endpoint. In a previous study, we found a statistically significant improvement in morning testosterone in a subset of men with morning testosterone less than 250 ng/dl and no deterioration of FSH in Androxal®-treated men. However, in the men on topical testosterone, 26 out of the 41 men that completed three months of dosing exhibited FSH levels below the reference limits for the hormone, with 17 below the lower limit of detection.
Unlike testosterone replacement therapies, Androxal® maintains the normal daily rhythm of testosterone peaks and valleys. We previously conducted three studies in which 24 hour testosterone levels were obtained and, unlike topical testosterone, morning testosterone was the maximum concentration observed, consistent with the normal circadian rhythm in men. We combined the three studies into one analysis, which has been submitted for FDA review. This analysis provides evidence that one assessment of testosterone between 8 a.m. and 10 a.m. correlates to the maximum value of testosterone for a given subject on a given day. We have committed to conduct one additional 24 hour study to show that Androxal®’s action in maintaining the normal rhythm is both predictable and dose-dependent.
We believe the advantages of oral delivery, maintenance of testicular function and additional metabolic benefits will be important differentiating factors for Androxal®, should it be approved. There can be no assurance, however, that we will be successful in implementing this strategy or that the FDA will approve our drug for commercial use.
Type 2 Diabetes
Our findings from a retrospective review of the clinical data from our 200 patient non-pivotal Phase 2 clinical trial showed that Androxal® therapy resulted in a significant reduction in mean fasting plasma glucose levels in men with glucose levels greater than 104 mg/dL at the outset of the trial, an outcome not seen in the placebo or AndroGel® arms of this study. Based on these results, in April 2008, we submitted a White Paper to the Division of Reproductive and Urology Products. The data demonstrated that among subjects with a serum glucose of greater than or equal to 105 mg/dL, there was a higher response rate to treatment in the Androxal® group than the placebo or AndroGel® groups, and the reduction in fasting serum glucose in this group was statistically significant. In November 2008, after the FDA reviewed this paper we received guidance from them suggesting that we open a new IND with the Division of Metabolic and Endocrine Products, or DMEP, for the investigation of Androxal® as a potential treatment for Type 2 diabetes. In December 2009, we submitted an Investigational New Drug Application (“IND”) to DMEP for the investigation of Androxal® for such purpose. On February 1, 2010, we received confirmation from DMEP that our IND was accepted and, as a result, we have initiated our Phase 2 trial. This trial will enroll 135 men with morning testosterone levels under 300 ng/dl who also have a fasting glucose level between 125 mg and 240 mg per deciliter and glycated hemoglobin, or HbA1c, levels between 7% and 9.5% - levels indicative of poor glucose control. Enrolled patients also will have been on a stable dose of an oral hypoglycemic agent for at least 2 months. We will split the men into three arms, one placebo and two doses of Androxal®, at 12.5 and 25 mg. We will look at changes in fasting glucose and HbA1C levels from baseline, along with changes in testosterone level. We hope that the study will be fully enrolled by the end of the third quarter 2011.
-4-
Proellex®
Product Overview
Proellex®, our product candidate for female reproductive health, is a new chemical entity that acts as a selective blocker of the progesterone receptor and is being developed for the treatment of symptoms associated with uterine fibroids and endometriosis. There are currently no FDA-approved orally administered drug treatments for the long-term treatment of either uterine fibroids or endometriosis. The National Uterine Fibroids Foundation estimates that 80% of all women in the U.S. have uterine fibroids, and one in four of these women have symptoms severe enough to require treatment. According to the Endometriosis Association, endometriosis affects 6.3 million women in the U.S. and Canada and millions more worldwide.
The current standards of care for uterine fibroids and endometriosis consist of surgery or short-term treatment with goanadotropin-releasing hormone (GnRH) agonists drugs, such as Lupron®. GnRH agonists induce a low estrogen, menopausal-like state and promote bone loss and are not recommended for use for more than six months.
We have conducted numerous studies with Proellex® enrolling over 750 women, roughly 700 of whom were dosed with the drug. All Proellex® studies completed to date exhibited strong efficacy signals, whether in uterine fibroids or endometriosis. In a 120 patient study of Proellex® as a treatment of uterine fibroids conducted in the United States (roughly 40 subjects per arm), both a 12.5 and 25 mg dose of Proellex® were compared to placebo. In this study each of the 12.5 and 25 mg doses achieved highly statistically significant results when compared to placebo when menstrual bleeding was assessed (p<0.0001). The two doses also achieved highly statistically significant improvement in quality of life measures using the Uterine Fibroid Symptom Quality of Life questionnaire developed and validated by Georgetown University and used in the development of device like treatments of uterine fibroids such as uterine artery embolization. There was no statistical difference in efficacy measures between the two doses. Importantly, in the Phase 2 U.S. trial a significant percentage of women stopped menstruating. Proellex® resulted in the induction of amenorrhea (cessation of menses), which we believe is a strong surrogate signal of efficacy. Over 80% of women on both the 12.5 and 25 mg doses exhibited no menses during the three month trial, whereas all women on placebo exhibited at least one menses.
Up until the summer of 2009, all side effects exhibited in the studies were considered manageable and the benefit of Proellex® far outweighed the risk. However, in Phase 3 efficacy and larger Phase 3 safety studies in diverse populations, a small number of subjects exhibited serious adverse effects associated with elevated liver enzymes. As a result of these findings, we elected to stop the trials and the FDA subsequently placed Proellex® on full clinical hold. All women that experienced elevated liver enzymes and returned for follow-up visits returned to baseline conditions with no overnight hospitalization necessary. An analysis of all the subjects that experienced such serious adverse effects showed that the effect only occurred in a small percentage of subjects that were exposed to the 50 mg dose of the drug for any period of time. Based on these findings, the Company petitioned the FDA to allow it to conduct a low dose study to demonstrate both safety and signals of efficacy in low oral doses of Proellex®, up to 12 mg administered per day. The FDA upgraded the full clinical hold to a partial hold to allow the low dose study to be conducted, which we have since commenced. In addition, the Company has undertaken two related initiatives presently at the preclinical stage. The first is the exploration of vaginal delivery as an alternative administrative route to bypass first-pass liver effects and reduce systemic exposure. The second is the screening of second generation molecules that do not possess the specific structures the Company believes induced the liver toxicity exhibited at higher doses of Proellex®.
Low Dose Study
Pursuant to the terms of the partial clinical hold currently in place as a result of the liver toxicity exhibited by Proellex®, the FDA is allowing us to run a single study to test low doses of Proellex® for signals of safety and efficacy. The new study is testing 5 different doses of Proellex® (1, 3, 6, 9 and 12 mg), with 1 mg being the first dose tested. Each dose will be compared to placebo with weekly assessments of liver function during both the placebo and drug period. Higher doses will not be studied until we are confident that it is safe to proceed to the next dose and have reported the safety findings to the FDA. Subjects will be dosed with the active drug for 10 weeks, which will allow for adequate time to determine the impact of a given dose on trends in liver function. Each dose will be tested in up to 12 different subjects and assessment of pharmacokinetic parameters will be obtained at the start of dosing and the end of the dosing period to determine overall and maximum drug exposure for a given dose. We will also monitor changes in menstrual bleeding patterns and ovulation as well as changes in endometrial thickness. The FDA required that an independent Drug Safety Monitoring Board be established and that the informed consent clearly state the liver toxicity previously experienced with Proellex®.
We believe we can complete the trial by year end 2011. As of March 18, 2011, we have begun administering the 6 mg dose. To date, no women have exhibited elevated liver enzymes indicative of an adverse event. Presuming a safe and effective dose is identified and the FDA is in agreement, we believe that we will be able to proceed with large Phase 3 efficacy trials for both uterine fibroids and endometriosis in 2012, subject to available funds, or out-license the product to a major pharmaceutical company. We believe that the evaluation of ovulation and menstrual bleeding patterns in the low dose trial will provide strong evidence for efficacy warranting further development.
Vaginal Administration
We are assessing vaginal administration of Proellex® to avoid first pass liver effects and achieve higher reproductive tract concentrations of the drug while minimizing systemic exposure. We reported results from two in vivo animal studies which confirmed reduced maximum circulating concentrations of the drug when administered vaginally, as well as efficacy signals at substantially lower doses than oral administration. Pending the outcome of dose optimization and vaginal irritation studies, we intend to open an IND for both uterine fibroids and endometriosis. We believe we will be able to leverage the experience we have gained with the oral dose in the preparation of this IND, and after a single Phase 1/2 study we will be able to test the vaginal product in a pivotal Phase 3 study. We plan on completing our preclinical proof-of-concept work by during the second quarter 2011 and will then submit a IND if warranted.
-5-
Second Generation Compound
We believe we understand the cause of the liver toxicity observed at high doses in the prior Phase 3 Proellex® studies. Our hypothesis is that liver adverse events are associated with a specific part of the chemical structure of Proellex®. To that end we have synthesized new but related molecules that are devoid of the specific toxicity-causing part of the chemical structure of Proellex® and initial preclinical screening has begun. If we are successful in identifying such a molecule, we believe we will be able to achieve high oral doses and systemic exposure, opening the path to aggressive anti progestin therapy for conditions such as breast cancer. We expect to have completed our screen of the new molecules by the end of 2011.
Other Products
We continue limited out-licensing efforts for our phentolamine-based product candidates, including VASOMAX®, which had previously been approved for marketing in several countries in Latin America for the treatment of male erectile dysfunction under the brand name, Z-Max. VASOMAX® has been on partial clinical hold in the U.S. since 1998, and no further development activities are planned.
Business Strategy
We plan to focus our clinical program on (i) the current escalating low dose study for Proellex® permitted by the FDA, (ii) the ongoing Phase 2B fertility trial for Androxal®, (iii) the ongoing Type 2 diabetes trial for Androxal®, (iv) preclinical assessment of vaginal delivery of Proellex® and (v) completing the initial identification of potential second generation Proellex® molecules. We anticipate that our current liquidity will be sufficient to complete all of these objectives; however, significant additional capital will be required for us to complete development of either of our product candidates. We will continue to explore corporate partnering opportunities for assistance in the clinical development funding and commercialization of our products, as appropriate; however, there can be no assurance that an acceptable corporate partnering opportunity will be successfully completed.
Research and Development
We have limited resources and utilize consultants and outside entities to perform clinical development and limited research activities in connection with preclinical studies and clinical trials. Our primary research and development, or R&D, expenses for 2010 were for the payment of contract research organizations and consultants in connection with our clinical trials of Proellex® and Androxal®. We believe that these expenses will continue to be our primary R&D expenses in the near future.
Proellex® License Agreement with National Institutes of Health
In 1999, we licensed rights to Proellex® from the National Institutes of Health, or NIH, under an exclusive, worldwide license in the field of treatment of human endocrinologic pathologies or conditions in steroid-sensitive tissues which expires upon the expiration of the last licensed patent. Under the terms of the agreement, we are obligated to meet certain developmental milestones as outlined in a commercial development plan, which has been amended and revised from time to time as circumstances warrant. We have recently amended the agreement to provide us with rights to certain second generation compounds under certain circumstances.
We provide annual updates to the NIH on the progress of our development of Proellex®. The NIH has the ability to terminate the agreement for lack of payment or if we are not meeting milestones as outlined in the commercial development plan and for other reasons as outlined in the agreement. Although we believe that we have a good working relationship with the NIH, there can be no assurance that all of the objectives and conditions in the commercial development plan will be met on a timely basis or at all, or that, if we fail to meet any of such objectives, the NIH will agree to amend this agreement to our satisfaction. Failure to comply with the material terms contained in the license agreement could result in termination of such agreement, which would prohibit us from further development of Proellex® and severely harm our business prospects. The NIH retains, on behalf of the government, a nonexclusive, nontransferable, worldwide license to practice the inventions licensed under the licensed patents by or on behalf of the government. For the purpose of encouraging basic research, the NIH retains the right to grant nonexclusive research licenses to third parties. Due to the work that was done on Proellex® at the NIH prior to our license agreement, the government also has certain rights to use the product in the event of a national emergency pursuant to the Patent and Trademark Laws Amendments Act of 1980, as amended.
Manufacturing
We have a five year supply agreement with Diagnostic Chemical Limited, doing business as BioVectra, for the supply of the bulk active pharmaceutical ingredient used in Androxal®. This agreement runs through July of 2012, subject to automatic one year renewals and the ability of either party to terminate upon 12 months prior notice. We have obtained all of our supply of Androxal® to date from BioVectra. We have not faced any material problems with BioVectra in supplying us with necessary quantities of Androxal® for our clinical trials and anticipate utilizing them for the remainder of our clinical supply and for commercial production if Androxal® is approved for sale. Though our relationship with BioVectra remains good, we believe that alternate manufacturers capable of manufacturing Androxal® could be identified if necessary.
-6-
Gedeon Richter was our third-party manufacturer of the active pharmaceutical ingredient for Proellex®. Due to the clinical hold, we cancelled our development and supply contract with Gedeon Richter; however, we have a large supply of Proellex® currently available for our current and planned clinical trial efforts. In the event we require an additional supply of Proellex®, we believe that we have maintained a good relationship with Gedeon Richter and that an agreement could be reached with Gedeon Richter to provide such supply when and if needed.
For the foreseeable future, we expect to continue to rely on third-party manufacturers and other third parties to produce, package and store sufficient quantities of Androxal® and Proellex®. These product candidates are complicated and expensive to manufacture. If our third-party manufacturers fail to deliver our product candidates for clinical use on a timely basis, with sufficient quality, and at commercially reasonable prices, we may be required to delay or suspend clinical trials or otherwise discontinue development and production of our product candidates. While we may be able to identify replacement third-party manufacturers or develop our own manufacturing capabilities for these product candidates, this process would likely cause a delay in the availability of our product candidates and an increase in costs. In addition, third-party manufacturers may have a limited number of facilities in which our product candidates can be produced, and any interruption of the operation of those facilities due to events such as equipment malfunction or failure or damage to the facility could result in the cancellation of shipments, loss of product in the manufacturing process or a shortfall in available product candidates.
Sales and Marketing
We have no experience in the sales, marketing and distribution of pharmaceutical products. We anticipate that we will outsource such activities to larger pharmaceutical companies, who may also conduct later stage pivotal trials of our product candidates. These companies are more capable of distributing the products to the market place. In the normal course of business we continue to explore possible partnerships with various pharmaceutical companies. If in the future we fail to reach or elect not to enter into an arrangement with a collaborative partner with respect to the sales and marketing of any of our future potential product candidates, we would need to develop a sales and marketing organization with supporting distribution capability in order to market such products directly. Significant additional expenditures would be required for us to develop such a sales and marketing organization.
Patents and Proprietary Information
Our ability to compete effectively with other companies is materially dependent on the proprietary nature of our patents and technologies. We actively seek patent protection for our proprietary technology in the United States and abroad.
Under a license agreement with the National Institutes of Health, we have exclusive rights to four issued U.S. patents, which expire in 2017, two pending U.S. patent applications, and several foreign patents and pending applications made by the NIH regarding Proellex®. We also have five pending U.S. patent applications, four foreign PCT applications and 45 foreign pending patent applications that cover various formulations of Proellex® and methods for using Proellex®.
Therapeutic uses of our Androxal® product candidate are covered in the United States by four issued U.S. patents and four pending patent applications. Foreign coverage of therapeutic uses of our Androxal® product candidate includes 42 issued foreign patents and 68 foreign pending patent applications. The issued patents and pending applications relate to methods for treating certain conditions including the treatment of testosterone deficiency in men, the treatment of metabolic syndrome and conditions associated therewith, and the treatment of infertility in hypogonadal men. Androxal® (the trans-isomer of clomiphene) is purified from clomiphene citrate. A third party individual holds two issued patents related to the use of an anti-estrogen such as clomiphene citrate and others for use in the treatment of androgen deficiency and disorders related thereto. In our prior filings with the SEC, we have described our request to the U.S. Patent and Trademark Office, or PTO, for re-examination of one of these patents based on prior art. The third party amended the claims in the re-examination proceedings, which led the PTO to determine that the amended claims are patentable in view of those publications under consideration and a re-examination certificate was issued. However, we believe that the amended claims are invalid based on additional prior art publications, and we filed a second request for re-examination by the PTO in light of a number of these additional publications and other publications cited by the PTO. The request was granted and all of the claims were finally rejected by the PTO in the re-examination. The patent holder appealed the rejections to the PTO Board of Patent Appeals and Interferences (“the Board”) which affirmed the rejection of all of the claims. The patent holder subsequently filed a request for rehearing, which led the Board to reverse the rejections of several dependent claims in view of those publications under consideration. The patent holder filed a Notice of Appeal to the Federal Circuit on September 28, 2010 contesting the rejections maintained by the Board and submitted his brief on February 8, 2011. We also believe that the second of these two patents is invalid in view of published prior art not considered by the PTO. Nevertheless, there is no assurance that either patent will ultimately be found invalid over the prior art. If such patents are not invalidated by the PTO we may be required to obtain a license from the holder of such patents in order to develop Androxal® further or attempts may be made to undertake further legal action to invalidate such patents. If such licenses were not available on acceptable terms, or at all, we may not be able to successfully commercialize or out-license Androxal®.
-7-
All of our employees and consultants have signed assignment of invention and confidentiality agreements, and each corporate partner we enter into discussions with or engage to assist in our clinical trials or manufacturing process is also required to execute appropriate confidentiality and assignment agreements protecting our intellectual property.
Competition
We are engaged in pharmaceutical product development, an industry that is characterized by extensive research efforts and rapid technological progress. Many established pharmaceutical and biotechnology companies, universities and other research institutions with financial, scientific and other resources significantly greater than ours are marketing or may develop products that directly compete with any products we may develop. These entities may succeed in developing products that are safer, more effective or less costly than products we may develop. Even if we can develop products which should prove to be more effective than those developed by other companies, other companies may be more successful than us because of greater financial resources, greater experience in conducting preclinical studies and clinical trials and in obtaining regulatory approval, stronger sales and marketing efforts, earlier receipt of approval for competing products and other factors. If we commence significant commercial sales of any products, we or our collaborators may compete in areas in which we have no experience, such as manufacturing and marketing. There can be no assurance that our products, if commercialized, will be accepted and prescribed by healthcare professionals.
Our main competitors for the treatment of testosterone deficiency are the testosterone replacement therapies currently being marketed. The current standard of care is AndroGel®, a topical gel for the replacement of testosterone. AndroGel® is marketed by Abbott Laboratories. There is another topical gel, Testim®, currently marketed by Auxilium Pharmaceuticals, and a transdermal patch, AndroDerm®, marketed by Watson Pharmaceuticals. Eli Lilly and Company also recently entered into a licensing agreement with a third party for a late stage topical testosterone treatment. In addition, other companies such as QTRX Pharmaceuticals and Clarus Therapeutics, Inc. are developing other products that would compete with Androxal®. We believe we can compete with AndroGel® and the other replacement therapies because we believe that Androxal®, besides being the only late stage oral therapy, is the only drug in development that normalizes testicular function and may provide additional metabolic benefits. Based on our clinical trial supply cost to date, we currently expect that Androxal®, if approved, can compete favorably on a cost basis with current testosterone replacement therapies.
Our main competitors for the treatment of uterine fibroids and endometriosis are GnRH agonists, especially Lupron®, the current therapeutic standard of care for uterine fibroids. Lupron® is marketed by Abbott, which has far greater resources and marketing capabilities than we have. Recently Abbott has licensed a Phase 3-ready molecule from Neurocrine Biosciences for the treatment of endometriosis. In addition, surgical treatment of both uterine fibroids and endometriosis competes with Proellex® by removing uterine fibroids and by removing misplaced tissue in women with endometriosis. We believe we can potentially compete with Lupron® and other GnRH agonists because we believe that Proellex® will not present the same side effect of a decrease in bone mineral density given its specific focus on progesterone inhibition, which differentiates it from GnRH agonists that create a low estrogen state. There are additional companies developing similar progesterone-blocking technology.
Government Regulation
Our research and development activities, preclinical studies and clinical trials, and the manufacturing, marketing and labeling of any products we may develop, are subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries. The U.S. Federal Food, Drug, and Cosmetic Act and the regulations promulgated thereunder and other federal and state statutes and regulations govern, among other things, the testing, manufacture, storage, record keeping, labeling, advertising, promotion, marketing and distribution of any products we may develop. Preclinical study and clinical trial requirements and the regulatory approval process take many years and require the expenditure of substantial resources. Additional government regulation may be established that could prevent or delay regulatory approval of our product candidates. Delays in obtaining or rejections of regulatory approvals would adversely affect our ability to commercialize any product candidate we develop and our ability to receive product revenues or to receive milestone payments or royalties from any product rights we might license to others. If regulatory approval of a product candidate is granted, the approval may include significant limitations on the indicated uses for which the product may be marketed or may be conditioned on the conduct of post-marketing surveillance studies.
The standard process required by the FDA before a pharmaceutical agent may be marketed in the United States includes: (1) preclinical tests; (2) submission to the FDA of an IND application which must become effective before human clinical trials may commence; (3) adequate and well-controlled human clinical trials to establish the safety and efficacy of the drug for its intended application; (4) submission of a new drug application, or NDA, to the FDA; and (5) FDA approval of the NDA prior to any commercial sale or shipment of the drug.
Clinical trials typically are conducted in three sequential phases, but the phases may overlap. Phase 1 typically involves the initial introduction of the drug into human subjects. In Phase 1, the drug is tested for safety and, as appropriate, for absorption, metabolism, distribution, excretion, pharmacodynamics and pharmacokinetics. Phase 2 usually involves studies in a limited patient population to evaluate preliminarily the efficacy of the drug for specific targeted indications, determine dosage tolerance and optimal dosage and identify possible adverse effects and safety risks.
-8-
Phase 3 clinical trials are undertaken to further evaluate clinical efficacy and to test further for safety within an expanded patient population at geographically dispersed clinical study sites. Phase 1, Phase 2 or Phase 3 testing may not be completed successfully within any specific time period, if at all, with respect to any products being tested by a sponsor. Furthermore, the FDA or the IRB may suspend clinical trials at any time on various grounds, including a finding that the healthy volunteers or patients are being exposed to an unacceptable health risk. This was evidenced when Proellex®, our product candidate for uterine fibroids and endometriosis, was placed on clinical hold by the FDA in summer 2009 due to liver toxicity data resulting from our clinical trials. Though the full clinical hold has been upgraded to a partial clinical hold, there can be no assurance that the partial hold will be lifted at any time.
Even if regulatory approvals for any products we may develop are obtained, we, our potential collaborators, our products, and the facilities manufacturing our products would be subject to continual review and periodic inspection. The FDA will require post-marketing reporting to monitor the safety of our products. Each drug-manufacturing establishment supplying the United States must be registered with the FDA. Manufacturing establishments are subject to periodic inspections by the FDA and must comply with the FDA’s requirements regarding current Good Manufacturing Practices, or GMP. In complying with current GMP, manufacturers must expend funds, time and effort in the area of production and quality control to ensure full technical compliance. We do not have any drug manufacturing capabilities and must rely on outside firms for this capability. The FDA stringently applies regulatory standards for manufacturing. Identification of previously unknown problems with respect to a product, manufacturer or facility may result in restrictions on the product, manufacturer or facility, including warning letters, suspensions of regulatory approvals, operating restrictions, delays in obtaining new product approvals, withdrawal of the product from the market, product recalls, fines, injunctions and criminal prosecution.
Before any products we may develop could be marketed outside of the United States, they would be subject to regulatory approval similar to FDA requirements in the United States, although the requirements governing the conduct of clinical trials, product licensing, pricing, and reimbursement vary widely from country to country. No action can be taken to market any drug product in a country until the regulatory authorities in that country have approved an appropriate application. FDA approval does not assure approval by other regulatory authorities. The current approval process varies from country to country, and the time spent in gaining approval varies from that required for FDA approval. In some countries, the sale price of a drug product must also be approved. The pricing review period often begins after market approval is granted. Even if a foreign regulatory authority approves any products we may develop, no assurance can be given that it will approve satisfactory prices for the products.
Our research and development involves the controlled use of hazardous materials and chemicals. Although we believe that our procedures for handling and disposing of those materials comply with state and federal regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. If such an accident occurs, we could be held liable for resulting damages, which could be material to our financial condition and business. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens, and the handling of biohazardous materials. Additional federal, state and local laws and regulations affecting us may be adopted in the future. Any violation of, and the cost of compliance with, these laws and regulations could materially and adversely affect us.
Third-Party Reimbursement and Pricing Controls
In the United States and elsewhere, sales of pharmaceutical products depend in significant part on the availability of reimbursement to the consumer from third-party payers, such as government and private insurance plans. Third-party payers are increasingly challenging the prices charged for medical products and services. Should any of our product candidates be approved for any commercial sales, it will be time consuming and expensive for us to go through the process of seeking reimbursement from Medicaid, Medicare and private payers.
Our products may not be considered cost effective, and coverage and reimbursement may not be available or sufficient to allow us to sell our products on a competitive and profitable basis. The passage of the Medicare Prescription Drug and Modernization Act of 2003 imposes requirements for the distribution and pricing of prescription drugs which may affect the marketing of our products.
In many foreign markets, including the countries in the European Union, pricing of pharmaceutical products is subject to governmental control. In the United States, there have been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental pricing control. While we cannot predict whether such legislative or regulatory proposals will be adopted, the adoption of such proposals could have a material adverse effect on our profitability.
The Hatch-Waxman Act
Under the U.S. Drug Price Competition and Patent Term Restoration Act of 1984, known as the Hatch-Waxman Act, newly approved drugs and indications benefit from a statutory period of non-patent marketing exclusivity. The Hatch-Waxman Act provides five year marketing exclusivity to the first applicant to gain approval of an NDA for a new chemical entity, or NCE, meaning that the FDA has not previously approved any other new drug containing the same active ingredient. Both of our current product candidates are considered NCEs. The Hatch-Waxman Act prohibits approval of an abbreviated new drug application, or ANDA, for a generic version of the drug during the five-year exclusivity period. Protection under the Hatch-Waxman Act will not prevent the filing or approval of another full NDA, however, the applicant would be required to conduct its own adequate and well-controlled clinical trials to demonstrate safety and effectiveness. The Hatch-Waxman Act also provides three years of marketing exclusivity for the approval of new NDAs with new clinical trials for previously approved drugs and supplemental NDAs, for example, for new indications, dosages, or strengths of an existing drug, if new clinical investigations are essential to the approval. This three year exclusivity covers only the new changes associated with the supplemental NDA and does not prohibit the FDA from approving ANDAs for drugs containing the original active ingredient or indications.
-9-
The Hatch-Waxman Act also permits a patent extension term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent extension cannot extend the remaining term of a patent beyond a total of 14 years. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA, plus time of active FDA review between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and it must be applied for prior to expiration of the patent and within 60 days of the approval of the NDA. The PTO, in consultation with the FDA, reviews and approves or rejects the application for patent term extension.
Litigation
See Item 3 of Part I of this Annual Report on Form 10-K for our fiscal year ended December 31, 2010.
Employees and Consultants
Employees
At December 31, 2010, we had 6 full-time employees. We also utilize consultants as well as contract research organizations and other outside specialty firms for various services such as preclinical and clinical trial support, manufacturing, regulatory approval advice and accounting and human resource management. We believe our relationship with our employees is good.
Scientific Advisors and Consultants
We benefit from consultation with prominent scientists active in fields related to our technology. For this purpose, we have part-time consulting relationships with a number of scientific advisors. At our request, these advisors review the feasibility of product development programs under consideration, provide advice about advances in areas related to our technology, and aid in recruiting personnel. All of the advisors are employed by academic institutions or other entities and may have commitments to or advisory agreements with other entities that limit their availability to us. Our advisors are required to sign an agreement providing that, if appropriate, they are to disclose and assign to us any ideas, discoveries and inventions they develop in the course of providing consulting services. We also use consultants for various administrative needs. None of our advisors are otherwise affiliated with us.
In addition to the advisors described above, we continue to engage U.S. contract research organizations to conduct our clinical trials. Under our arrangements with these contract research organizations, we design the protocols for the clinical trials and direct the contract research organizations in their efforts. We own all of the data associated with the clinical trials.
Recent Developments
On February 8, 2011, we completed an underwritten public offering of 690,000 units (including the exercise of the underwriter’s over-allotment option), consisting of an aggregate of 2,760,000 shares of our common stock, Series A Warrants to purchase 2,070,000 shares of our common stock and Series B warrants to purchase 1,690,500 shares of our common stock, at a price per unit of $17.15. Each unit consisted of four shares of our common stock, Series A Warrants exercisable for three shares of our common stock at an exercise price of $0.01 per share and Series B Warrants exercisable for 2.45 shares of our common stock at an exercise price of $2.49 per share. Gross proceeds to us, after the underwriting discount and before offering expenses, were approximately $11 million.
On March 15, 2011, we received notice from the Nasdaq Stock Market that the Company’s Series A Warrants (RPRXW) and Series B Warrants (RPRXZ) (collectively, the “Warrants”) have not had a minimum of two active and registered market makers, as required for continued inclusion by Listing Rule 5560(a) (the “Rule”). We have been provided until April 14, 2011 to regain compliance with the Rule. If, at any time before such date, the Warrants have at least two active and registered market makers for 10 consecutive trading days, we will have achieved compliance with the Rule. If compliance is not demonstrated by the Company by such date, the Warrants will be delisted from the Nasdaq Capital Market. We believe that, due to certain technical issues, relevant information related to the active and registered market makers for the Warrants was not being disseminated regarding quotes, which resulted in the issuance of such notice. We believe that such technical issues have been resolved and, therefore, we anticipate that it will demonstrate compliance with the Rule within the requisite time period.
Available Information
Our Internet site (www.reprosrx.com) makes available free of charge to all interested parties our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and all amendments to those reports, as well as all other reports and schedules filed electronically with the Securities and Exchange Commission, or SEC, as soon as reasonably practicable after such material is electronically filed with or furnished to the SEC. Interested parties may also find reports, proxy and information statements and other information on issuers that file electronically with the SEC at the SEC's Internet site (http://www.sec.gov).
-10-
|
ITEM 1A.
|
RISK FACTORS
|
You should carefully consider the risks described below before making an investment decision. You should also refer to the other information in this report, including our financial statements and the related notes incorporated by reference. The risks and uncertainties described below are not the only risks and uncertainties we face. Additional risks and uncertainties not presently known to us or that we currently deem immaterial also may impair our business operations. If any of the following risks actually occur, our business, results of operations and financial condition could suffer. In that event the trading price of our common stock could decline, and you may lose all or part of your investment in our common stock. The risks discussed below also include forward-looking statements and our actual results may differ substantially from those discussed in these forward-looking statements.
Risks Relating to Our Business
Our ability to continue as a going concern may require that we raise additional funds by the end of the second quarter of 2012, without which we may need to cease our business operations and begin liquidation proceedings.
Our ability to continue as a going concern is dependent upon our ability to obtain additional financing by the end of the second quarter of 2012 based upon our current expense and revenue assumptions. If our expenses are greater than expected or our revenues are less than expected, we may be required to raise additional funds prior to that time. We will continue to explore various financing alternatives to address our liquidity needs. No assurance can be given that we will be successful in obtaining additional financing on acceptable terms or at all. We anticipate that if we are able to secure additional financing, that such financing will result in significant dilution of the ownership interests of our stockholders and may provide certain rights to the new investors senior to the rights of our current stockholders, including but not limited to, voting rights and rights to proceeds in the event of a sale or liquidation of the Company. The current FDA partial clinical hold of our clinical trials for Proellex® will make it more difficult for us to obtain additional financing. We expect to continue to incur significant losses for the foreseeable future, and we may never achieve or sustain profitability. In the event that we are unable to obtain adequate financing to conduct operations, we may need to cease our business operations and begin liquidation proceedings. If we need to liquidate our assets, we would likely realize significantly less from them than the values at which they are carried on our financial statements. The funds resulting from the liquidation of our assets would be used first to pay off the debt owed to any creditors before any funds would be available to pay our stockholders, and any shortfall in the proceeds would directly reduce the amounts available for distribution, if any, to our creditors and to our stockholders. In the event we were required to liquidate, it is possible that stockholders would not receive any value for their shares.
If we fail to obtain the capital necessary to fund our operations, we may have to delay, reduce or eliminate our research and development programs or commercialization efforts, dispose of assets or liquidate.
We expect to make additional capital outlays and to increase operating expenditures over the next several years to support our clinical trials for Androxal® and Proellex® and related activities. Based on our current and planned clinical programs, we expect to need to raise additional capital by the end of the second quarter of 2012 or earlier if our expenses are greater than anticipated. We will continue to seek additional funding through public or private financings, including equity or debt financings, and/or through other means, including collaborations and license agreements. We do not know whether additional financing will be available when needed, or that, if available, we will obtain financing on terms favorable to our stockholders or us. If adequate funds are not available to us, we may be required to:
|
•
|
delay, reduce the scope of or eliminate one or more of our development programs;
|
|
•
|
relinquish, license or otherwise dispose of rights to technologies, product candidate or products that we would otherwise seek to develop or commercialize ourselves at an earlier stage or on terms that are less favorable than might otherwise be available; or
|
|
•
|
liquidate and dissolve our company.
|
Our future capital requirements will depend upon a number of factors, including:
|
•
|
the size, complexity, results and timing of our clinical programs;
|
|
•
|
the cost to obtain sufficient supply of the compounds necessary for our product candidates at a reasonable cost;
|
|
•
|
the time and cost involved in obtaining regulatory approvals;
|
|
•
|
the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims; and
|
|
•
|
competing technological and market developments.
|
These factors could result in variations from our currently projected operating and liquidity requirements.
-11-
Because the data from our limited clinical trials for our product candidates are not necessarily predictive of future results, we can provide no assurances that any of them will have favorable results in clinical trials or receive regulatory approval.
Before we can obtain regulatory approval for the commercial sale of any product candidate that we develop, we are required to complete preclinical development and extensive clinical trials in humans to demonstrate its safety and efficacy. To date, long-term safety and efficacy have not been demonstrated in clinical trials for any of our product candidates and in fact, our product candidate Proellex® is currently on partial clinical hold with the FDA due to safety issues experienced in our Phase 2 and Phase 3 clinical trials for endometriosis and uterine fibroids, respectively.
In addition, previous clinical trials for Androxal® have been conducted only in limited numbers of patients that may not fully represent the diversity present in larger populations. In addition, these studies have not been subjected to the exacting design requirements typically required by FDA for pivotal trials. Thus the limited data we have obtained may not predict results from studies in larger numbers of patients drawn from more diverse populations, and may not predict the ability of Androxal® to treat Type 2 diabetes or a testosterone deficiency. Furthermore, the only data that we obtained to date relating to Androxal® is to treat testosterone deficiency. We will be required to demonstrate through larger-scale clinical trials that these product candidates are safe and effective for use in a diverse population before we can seek regulatory approvals for their commercial sale.
Favorable results in our early studies or trials may not be repeated in later studies or trials, including continuing preclinical studies and large-scale clinical trials analyzed with more rigorous statistical methods, and our drug candidates in later-stage trials may fail to show desired safety and efficacy despite having progressed through earlier-stage trials. Unfavorable results from ongoing preclinical studies or clinical trials could result in delays, modifications or abandonment of ongoing or future clinical trials. Clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals. Negative or inconclusive results or adverse medical events during a clinical trial could cause a clinical trial to be delayed, repeated or terminated. In addition, we may report top-line data from time to time, which is based on a preliminary analysis of key efficacy and safety data; such data may be subject to change following a more comprehensive review of the data related to the applicable clinical trial. If Androxal®, Proellex®, or any other potential future product candidate fails to demonstrate sufficient safety and efficacy in any clinical trial, we would experience potentially significant delays in, or be required to abandon, development of that product candidate. If we delay or abandon our development efforts related to Androxal® or Proellex®, we may not be able to generate sufficient revenues to continue operations or become profitable.
We have a history of operating losses, and we expect to incur increasing net losses and may not achieve or maintain profitability for some time or at all.
We have experienced significant operating losses in each fiscal year since our inception. As of December 31, 2010, we had accumulated losses of $179.2 million, approximately $3.0 million in cash and cash equivalents, and our accounts payable and accrued expenses were approximately $1.3 million. We expect to continue incurring net losses and we may not achieve or maintain profitability for some time if at all. As we increase expenditures for the clinical development of our products, we expect our total operating losses to increase for at least the next few years. Our ability to achieve profitability will depend on, among other things, successfully completing the development of our products, obtaining regulatory approvals, establishing marketing, sales and manufacturing capabilities or collaborative arrangements with others that possess such capabilities, and raising sufficient funds to finance our activities. There can be no assurance that we will be able to achieve profitability or that profitability, if achieved, can be sustained. The uncertainties relating to the foregoing matters raise substantial doubt about our ability to continue as a going concern.
Raising additional funds by issuing securities or through collaboration and licensing arrangements may cause dilution to our stockholders, restrict our operations or require us to relinquish proprietary rights.
We may raise additional funds through public or private equity offerings, debt financings or potential corporate collaborations and licensing arrangements. We cannot be certain that additional funding will be available on acceptable terms, or at all. To the extent that we raise additional capital by issuing equity securities, our stockholders’ ownership will be diluted. Any debt financing we enter into may involve covenants that restrict our operations. These restrictive covenants may include limitations on borrowing and specific restrictions on the use of our assets, as well as prohibitions on our ability to create liens, pay dividends, redeem capital stock or make investments. In addition, if we raise additional funds through collaboration and licensing arrangements, it may be necessary to relinquish potentially valuable rights to our potential products or proprietary technologies, or grant licenses on terms that are not favorable to us. For example, we might be forced to relinquish all or a portion of our sales and marketing rights with respect to Androxal®, Proellex®, or other potential products or intellectual property.
Our stock price could decline significantly based on the results and timing of clinical trials of, and decisions affecting, our product candidates.
Results of clinical trials and preclinical studies of our current and potential product candidates may not be viewed favorably by us or third parties, including the FDA or other regulatory authorities, investors, analysts and potential collaborators. The same may be true of how we design the clinical trials of our product candidates and regulatory decisions affecting those clinical trials. Biopharmaceutical company stock prices have declined significantly when such results and decisions were unfavorable or perceived negatively or when a product candidate did not otherwise meet expectations. The final results from our clinical development programs may be negative, may not meet expectations or may be perceived negatively. The designs of our clinical trials (which may change significantly and be more expensive than currently anticipated depending on our clinical results and regulatory decisions) may also be viewed negatively by third parties. We may not be successful in completing these clinical trials on our projected timetable, if at all.
-12-
Failure to initiate additional clinical trials or delays in existing clinical trials of Androxal® and Proellex® and failure of the FDA to lift the partial clinical hold on Proellex®, or unfavorable results or decisions or negative perceptions regarding any of such clinical trials, could cause our stock price to decline significantly.
We are thinly staffed and highly dependent on a limited number of management persons and key personnel, and if we lose these members of our team or are unable to attract and retain additional qualified personnel, our future growth and ability to compete would suffer.
The competition for qualified personnel in the biopharmaceutical field is intense, and our future success depends upon our ability to attract, retain and motivate highly skilled scientific, technical and managerial employees. We have only 6 full-time employees at the present time, including Joseph S. Podolski. We are highly dependent on our professional staff for the management of our company and the development of our technologies. Mr. Podolski has an employment agreement with us. There can be no assurance that any of these employees will remain with us through development of our current product candidates. The loss of the services of any of our employees could delay or curtail our research and product development efforts.
Our plan to use collaborations to leverage our capabilities may not be successful.
As part of our business strategy, we intend to enter into collaboration arrangements with strategic partners to develop and commercialize our product candidates. For our collaboration efforts to be successful, we must identify partners whose competencies complement ours. We must also successfully enter into collaboration agreements with them on terms attractive to us and integrate and coordinate their resources and capabilities with our own. We may be unsuccessful in entering into collaboration agreements with acceptable partners or negotiating favorable terms in these agreements. In addition, we may face a disadvantage in seeking to enter into or negotiating collaborations with potential partners because other potential collaborators may have greater management and financial resources than we do. Also, we may be unsuccessful in integrating the resources or capabilities of these collaborators. In addition, our collaborators may prove difficult to work with or less skilled than we originally expected. If we are unsuccessful in our collaborative efforts, our ability to develop and market product candidates could be severely limited.
Our rights agreement and certain provisions in our charter documents and Delaware law could delay or prevent a change in management or a takeover attempt that you may consider to be in your best interest.
We have adopted certain anti-takeover provisions, including a rights agreement. The rights agreement will cause substantial dilution to any person who attempts to acquire us in a manner or on terms not approved by our board of directors.
The rights agreement and certain provisions in our certificate of incorporation and bylaws and under Delaware law could delay or prevent the removal of directors and other management and could make more difficult a merger, tender offer or proxy contest involving us that you may consider to be in your best interest. For example, these provisions:
|
•
|
allow our board of directors to issue preferred stock without stockholder approval;
|
|
•
|
limit who can call a special meeting of stockholders; and
|
|
•
|
establish advance notice requirements for nomination for election to the board of directors or for proposing matters to be acted upon at stockholder meetings.
|
Risks Relating to Our Product Development Efforts
Delays in the commencement of preclinical studies and clinical trials testing of our current and potential product candidates could result in increased costs to us and delay our ability to generate revenues.
Our product candidates will require continued preclinical studies and extensive clinical trials prior to the submission of a regulatory application for commercial sales. Because of the nature of clinical trials and our lack of sufficient capital, we do not know whether future planned clinical trials will begin on time, if at all. Delays in the commencement of preclinical studies and clinical trials could significantly increase our product development costs and delay any product commercialization. In addition, many of the factors that may cause, or lead to, a delay in the commencement of clinical trials may also ultimately lead to denial of regulatory approval of a product candidate.
The commencement of clinical trials can be delayed for a variety of reasons, including delays in:
-13-
|
•
|
demonstrating sufficient safety and efficacy in past clinical trials to obtain regulatory approval to commence a further clinical trial;
|
|
•
|
convincing the FDA that we have selected valid endpoints for use in proposed clinical trials;
|
|
•
|
reaching agreements on acceptable terms with prospective contract manufacturers for manufacturing sufficient quantities of a product candidate; and
|
|
•
|
obtaining institutional review board approval to conduct a clinical trial at a prospective site.
|
In addition, the commencement of clinical trials may be delayed due to insufficient patient enrollment, which is a function of many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites, the availability of effective treatments for the relevant disease, and the eligibility criteria for the clinical trial.
Delays in the completion of, or the termination of, clinical testing of our current and potential product candidates could result in increased costs to us, and could delay or prevent us from generating revenues.
Once a clinical trial has begun, it may be delayed, suspended or terminated by us or the FDA or other regulatory authorities due to a number of factors, including:
|
•
|
lack of adequate funding to continue clinical trials;
|
|
•
|
lack of effectiveness of any product candidate during clinical trials;
|
|
•
|
side effects experienced by trial participants or other safety issues;
|
|
•
|
slower than expected rates of patient recruitment and enrollment or lower than expected patient retention rates;
|
|
•
|
delays or inability to manufacture or obtain sufficient quantities of materials for use in clinical trials;
|
|
•
|
inadequacy of or changes in our manufacturing process or compound formulation;
|
|
•
|
delays in obtaining regulatory approvals to commence a trial, or “clinical holds” or delays requiring suspension or termination of a trial by a regulatory agency, such as the FDA, after a trial is commenced;
|
|
•
|
changes in applicable regulatory policies and regulations;
|
|
•
|
delays in identifying and reaching agreement on acceptable terms with prospective clinical trial sites;
|
|
•
|
uncertainty regarding proper dosing;
|
|
•
|
unfavorable results from on-going clinical trials and preclinical studies;
|
|
•
|
failure of our clinical research organizations to comply with all regulatory and contractual requirements or otherwise fail to perform their services in a timely or acceptable manner;
|
|
•
|
scheduling conflicts with participating clinicians and clinical institutions;
|
|
•
|
failure to construct appropriate clinical trial protocols;
|
|
•
|
insufficient data to support regulatory approval;
|
|
•
|
inability or unwillingness of medical investigators to follow our clinical protocols;
|
|
•
|
difficulty in maintaining contact with subjects during or after treatment, which may result in incomplete data;
|
|
•
|
the timing of discussions and meetings with the FDA or other regulatory authorities regarding the scope or design of our clinical trials; and
|
|
•
|
acceptability to the FDA of data obtained from clinical studies conducted in Europe or other non-United States jurisdictions.
|
Many of these factors that may lead to a delay, suspension or termination of clinical testing of a current or potential product candidate may also ultimately lead to denial of regulatory approval of a current or potential product candidate.
If we experience delays in the completion of, or termination of, clinical testing of any product candidates in the future, our financial results and the commercial prospects for our product candidates will be harmed, and our ability to generate product revenues will be delayed.
-14-
Even if we successfully complete clinical trials for Androxal® and Proellex®, there are no assurances that we will be able to submit, or obtain FDA approval of, a new drug application.
There can be no assurance that, if our clinical trials for Androxal® and Proellex® are successfully completed, we will be able to submit a new drug application, or NDA, to the FDA or that any NDA we submit will be approved by the FDA in a timely manner, if at all. After completing clinical trials for a product candidate in humans, a drug dossier is prepared and submitted to the FDA as an NDA, and includes all preclinical studies and clinical trial data relevant to the safety and effectiveness of the product at the suggested dose and duration of use for the proposed indication, in order to allow the FDA to review such drug dossier and to consider a product candidate for approval for commercialization in the United States. If we are unable to submit an NDA with respect to Androxal® or Proellex®, or if any NDA we submit is not approved by the FDA, we will be unable to commercialize that product. The FDA can and does reject NDAs and requires additional clinical trials, even when drug candidates achieve favorable results in large-scale Phase 3 clinical trials. If we fail to commercialize Androxal® or Proellex®, we may be unable to generate sufficient revenues to continue operations or attain profitability.
We rely on third parties to conduct clinical trials for our product candidates, and their failure to timely and properly perform their obligations may result in costs and delays that prevent us from obtaining regulatory approval or successfully commercializing our product candidates.
We rely on independent contractors, including researchers at clinical research organizations, or CROs, and universities, in certain areas that are particularly relevant to our research and product development plans, such as the conduct of clinical trials. The competition for these relationships is intense, and we may not be able to maintain our relationships with them on acceptable terms. Independent contractors generally may terminate their engagements at any time, subject to notice. As a result, we can control their activities only within certain limits, and they will devote only a certain amount of their time conducting research on and trials of our product candidates and assisting in developing them. If they do not successfully carry out their duties under their agreements with us, fail to inform us if these trials fail to comply with clinical trial protocols, or fail to meet expected deadlines, our clinical trials may need to be extended, delayed or terminated. We may not be able to enter into replacement arrangements without undue delays or excessive expenditures. If there are delays in testing or regulatory approvals as a result of the failure to perform by our independent contractors or other outside parties, our drug development costs will increase and we may not be able to attain regulatory approval for or successfully commercialize our product candidates.
In addition, we have no control over the financial health of our independent contractors. Several of our independent contractors are in possession of valuable and sensitive information relating to the safety and efficacy of our product candidates, and several others provide services to a significant percentage of the patients enrolled in the respective clinical trials in which such independent contractors participate. Should one or more of these independent contractors become insolvent, or otherwise are not able to continue to provide services to us, as a result of the current economic downturn or otherwise, the clinical trial in which such contractor participates could become significantly delayed and we may be adversely affected as a result of the delays and additional expenses associated with such event.
Risks Relating to Manufacturing Our Products
We currently rely on third-party manufacturers and other third parties for production of our product candidates, and our dependence on these manufacturers may impair the development of our product candidates.
Currently, we do not have the ability internally to manufacture the product candidates that we need to conduct our clinical trials. We terminated our supply agreement with Gedeon Richter for the manufacturing of Proellex® due to the clinical hold imposed by the FDA in August 2009; however, we have a large supply of Proellex® currently available for our current and planned clinical trial efforts. In the event we require an additional supply of Proellex®, we believe that we have maintained a good relationship with Gedeon Richter and that an agreement could be reached with Gedeon Richter to provide such supply when and if needed, but there is no assurance that this will be the case.
We have a five year supply agreement with Diagnostic Chemical Limited, doing business as BioVectra, for the supply of the bulk active pharmaceutical ingredient used in Androxal®. This agreement runs through July of 2012, subject to automatic one year renewals and the ability of either party to terminate upon 12 months prior notice. We have obtained all of our supply of Androxal® to date from BioVectra. We have not faced any material problems with BioVectra in supplying us with our necessary quantities of Androxal® for our clinical trials and anticipate utilizing them for commercial production if Androxal® is approved. The Company believes that should an issue with BioVectra arise an alternative supplier could be identified, but there is no assurance that this will be the case.
For the foreseeable future, we expect to continue to rely on third-party manufacturers and other third parties to produce, package and store sufficient quantities of Androxal®, Proellex®, and any future product candidates for use in our clinical trials. These product candidates are complicated and expensive to manufacture. If our third-party manufacturers fail to deliver our product candidates for clinical use on a timely basis, with sufficient quality, and at commercially reasonable prices, we may be required to delay or suspend clinical trials or otherwise discontinue development and production of our product candidates. While we may be able to identify replacement third-party manufacturers or develop our own manufacturing capabilities for these product candidates, this process would likely cause a delay in the availability of our product candidates and an increase in costs. In addition, third-party manufacturers may have a limited number of facilities in which our product candidates can be produced, and any interruption of the operation of those facilities due to events such as equipment malfunction or failure or damage to the facility by natural disasters could result in the cancellation of shipments, loss of product in the manufacturing process or a shortfall in available product candidates.
-15-
Our product candidates have only been manufactured in small quantities to date, and we may face delays or complications in manufacturing quantities of our product candidates in sufficient quantities to meet the demands of late stage clinical trials and marketing.
We cannot assure that we will be able to successfully increase the manufacturing capacity or scale-up manufacturing volume per batch, whether on our own or in reliance on third-party manufacturers, for any of our product candidates in a timely or economical manner, or at all. To date our product candidates have been manufactured exclusively by third parties in small quantities for preclinical studies and limited clinical trials. Future clinical trials of our product candidates, and future commercial sales in the event that such product candidates are approved by the FDA or foreign regulatory bodies, will require significant increased quantities of product. Significant scale-up of manufacturing requires certain additional developmental work, which the FDA must review and approve to assure product comparability. If we or our third-party manufacturers are unable to successfully increase the manufacturing capacity for a product candidate, the regulatory approval or commercial launch of that product candidate may be delayed or there may be a shortage in supply of that product candidate.
Our product candidates require precise, high-quality manufacturing which may not be available at acceptable costs.
Androxal® and Proellex® are novel compounds that have never been produced in large scale. As in the development of any new compound, there are underlying risks associated with their manufacture. These risks include, but are not limited to, cost, process scale-up, process reproducibility, construction of a suitable process plant, timely availability of raw materials, as well as regulatory issues associated with the manufacture of an active pharmaceutical agent. Any of these risks may prevent us from successfully developing Androxal® or Proellex®. Our failure, or the failure of our third-party manufacturers to achieve and maintain these high manufacturing standards, including the incidence of manufacturing errors and reliable product packaging for diverse environmental conditions, could result in patient injury or death, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could seriously hurt our business.
We may experience delays in the development of our product candidates if the third-party manufacturers of our product candidates cannot meet FDA requirements relating to Good Manufacturing Practices.
Our third-party manufacturers are required to produce our product candidates under FDA current Good Manufacturing Practices in order to meet acceptable standards for our clinical trials. If such standards change, the ability of third-party manufacturers to produce our product candidates on the schedule we require for our clinical trials may be affected. In addition, third-party manufacturers may not perform their obligations under their agreements with us or may discontinue their business before the time required by us to gain approval for or commercialize our product candidates. Any difficulties or delays in the manufacturing and supply of our product candidates could increase our costs or cause us to lose revenue or postpone or cancel clinical trials.
The FDA also requires that we demonstrate structural and functional comparability between the same drug product produced by different third-party manufacturers. Because we may use multiple sources to manufacture Androxal® and Proellex®, we may need to conduct comparability studies to assess whether manufacturing changes have affected the product safety, identity, purity or potency of any commercial product candidate compared to the product candidate used in clinical trials. If we are unable to demonstrate comparability, the FDA could require us to conduct additional clinical trials, which would be expensive and significantly delay commercialization of our product candidates.
Risks Relating to Product Commercialization
If commercialized, our product candidates may not be approved for sufficient governmental or third-party reimbursements, which would adversely affect our ability to market our product candidates.
In the United States and elsewhere, sales of pharmaceutical products depend in significant part on the availability of reimbursement to the consumer from third-party payers, such as government and private insurance plans. Third-party payers are increasingly challenging the prices charged for medical products and services. It will be time consuming and expensive for us to go through the process of seeking reimbursement from Medicaid, Medicare and private payers for Proellex® and Androxal®. Our products may not be considered cost effective, and coverage and reimbursement may not be available or sufficient to allow us to sell our products on a competitive and profitable basis. The passage of the Medicare Prescription Drug and Modernization Act of 2003 imposes requirements for the distribution and pricing of prescription drugs which may negatively affect the marketing of our potential products.
-16-
If we successfully develop products but those products do not achieve and maintain market acceptance, our business will not be profitable.
Even if our product candidates are approved for commercial sale by the FDA or other regulatory authorities, the degree of market acceptance of any approved product by physicians, healthcare professionals and third-party payers and our profitability and growth will depend on a number of factors, including:
|
•
|
relative convenience and ease of administration;
|
|
•
|
the prevalence and severity of any adverse side effects;
|
|
•
|
availability, effectiveness and cost of alternative treatments;
|
|
•
|
pricing and cost effectiveness of our drugs;
|
|
•
|
effectiveness of our or collaborators’ sales and marketing strategies; and
|
|
•
|
our ability to obtain sufficient third-party insurance coverage or reimbursement.
|
If Androxal® does not provide a treatment regime that is more beneficial than AndroGel®, the current standard of care for the treatment of testosterone deficiency, or otherwise provide patient benefit, it likely will not be accepted favorably by the market. If any products we may develop do not achieve market acceptance, then we will not generate sufficient revenue to achieve or maintain profitability.
In addition, even if our products achieve market acceptance, we may not be able to maintain that market acceptance over time if:
|
•
|
new products or technologies are introduced that are more favorably received than our products, are more cost effective or render our products obsolete;
|
|
•
|
unforeseen complications arise with respect to use of our products; or
|
|
•
|
sufficient third-party insurance coverage or reimbursement does not remain available.
|
Our liability insurance may neither provide adequate coverage nor may it always be available on favorable terms or at all.
Neither Androxal® nor Proellex® has been approved for commercial sale. However, the current and future use of our product candidates by us and potential corporate collaborators in clinical trials, and the sale of any approved products in the future, may expose us to liability claims. These claims might be made directly by consumers or healthcare providers or indirectly by pharmaceutical companies, potential corporate collaborators or others selling such products. We may experience financial losses in the future due to product liability claims. We have obtained limited general commercial liability insurance coverage for our clinical trials. We intend to expand our insurance coverage to include the sale of commercial products if we obtain marketing approval for any of our product candidates. However, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses. If a successful product liability claim or series of claims is brought against us for uninsured liabilities or for liabilities in excess of our insurance limits, our assets may not be sufficient to cover such claims and our business operations could be impaired.
We face significant competition from many companies with substantially greater resources than we have and other possible advantages.
We are engaged in biopharmaceutical product development, an industry that is characterized by extensive research efforts and rapid technological progress. The biopharmaceutical industry is also highly competitive. Our success will depend on our ability to acquire, develop and commercialize products and our ability to establish and maintain markets for any products for which we receive marketing approval. Potential competitors in North America, Europe and elsewhere include major pharmaceutical companies, specialty pharmaceutical companies and biotechnology firms, universities and other research institutions and government agencies. Many of our competitors have substantially greater research and development and regulatory capabilities and experience, and substantially greater management, manufacturing, distribution, marketing and financial resources, than we do. Accordingly, our competitors may:
|
•
|
develop or license products or other novel technologies that are more effective, safer or less costly than the product candidates that we are developing;
|
|
•
|
obtain regulatory approval for products before we do; or
|
|
•
|
commit more resources than we can to developing, marketing and selling competing products.
|
-17-
Our main competitors for the treatment of testosterone deficiency are the testosterone replacement therapies currently being marketed. The current standard of care is AndroGel®, a topical gel for the replacement of testosterone developed by Solvay Pharmaceuticals (which was acquired by Abbott Laboratories). Abbott is a much larger company than we are, with greater resources and marketing ability. Androxal® would also compete with other forms of testosterone replacement therapies such as oral treatments, patches, injectables and a tablet applied to the upper gum. There is another topical gel currently marketed by Auxilium Pharmaceuticals called Testim®, and a transdermal patch marketed by Watson Pharmaceuticals called AndroDerm®. There can be no assurance that our product candidates will be more successful than competitive products. In addition, other potential competitors may be developing testosterone therapies similar to ours.
The main therapeutic products competitive with Proellex® for the treatment of uterine fibroids and endometriosis are GnRH agonists, including Lupron® and the use of approved progestin-based contraceptives for the treatment of endometriosis. In addition, surgical treatment of both uterine fibroids and endometriosis would compete with Proellex®, if approved, by removing uterine fibroids and by removing misplaced tissue in women with endometriosis. Furthermore, Abbott has recently licensed a Phase 3-ready molecule from Neurocrine Biosciences Inc. for the treatment of endometriosis.
Risks Relating to Our Intellectual Property
There is a third party individual patent holder that claims priority over our patent application for Androxal®.
A third party individual holds two issued patents related to the use of an anti-estrogen such as clomiphene citrate and others for use in the treatment of androgen deficiency and disorders related thereto. In our prior filings with the SEC, we have described our request to the U.S. Patent and Trademark Office, or PTO, for re-examination of one of these patents based on prior art. The third party amended the claims in the re-examination proceedings, which led the PTO to determine that the amended claims are patentable in view of those publications under consideration and a re-examination certificate was issued. However, we believe that the amended claims are invalid based on additional prior art publications, and we filed a second request for re-examination by the PTO in light of a number of these additional publications and other publications cited by the PTO. The request was granted and all of the claims were finally rejected by the PTO in the re-examination. The patent holder appealed the rejections to the PTO Board of Patent Appeals and Interferences (“the Board”) which affirmed the rejection of all of the claims. The patent holder subsequently filed a request for rehearing, which led the Board to reverse the rejections of several dependent claims in view of those publications under consideration. The patent holder filed a Notice of Appeal to the Federal Circuit on September 28, 2010 contesting the rejections maintained by the Board and submitted his brief on February 8, 2011. We also believe that the second of these two patents is invalid in view of published prior art not considered by the PTO. Nevertheless, there is no assurance that either patent will ultimately be found invalid over the prior art. If such patents are not invalidated by the PTO we may be required to obtain a license from the holder of such patents in order to develop Androxal® further or attempts may be made to undertake further legal action to invalidate such patents. If such licenses were not available on acceptable terms, or at all, we may not be able to successfully commercialize or out-license Androxal®.
We licensed our rights to Proellex® from NIH and our inability to fulfill our commitments and obligations under such license may result in forfeiture of our rights.
Our rights to Proellex® are licensed exclusively to us from the NIH under a license agreement. This license agreement contains numerous detailed performance obligations, with time sensitive dates for compliance, relating to clinical development and commercialization activities required by us or our designated third-party providers, as well as additional financial milestones and royalties. Failure to achieve the benchmarks specified in the commercial development plan attached to the license agreement or meet payment obligations could result in termination of the license agreement and the loss of our rights to develop and commercialize Proellex®. We periodically update the commercial development plan as such plans evolve. There can be no assurance that we will be able to meet any or all of the performance objectives in the future on a timely basis or at all, or that, if we fail to meet any of such objectives, NIH will agree to revised objectives. NIH has the ability to terminate the agreement for an uncured material breach of the agreement, if we made a false statement or willful omission in our license application, if we do not keep Proellex® reasonably available to the public after commercial launch or if we cannot reasonably satisfy unmet health and safety needs, unless such requirement has been waived.
We cannot assure that our manufacture, use or sale of our product candidates will not infringe on the patent rights of others.
There can be no assurance that the manufacture, use or sale of any of our product candidates will not infringe the patent rights of others. We may be unable to avoid infringement of the patent rights of others and may be required to seek a license, defend an infringement action or challenge the validity of the patents in court. There can be no assurance that a license to the allegedly infringed patents will be available to us on terms and conditions acceptable to us, if at all, or that we will prevail in any patent litigation. Patent litigation is extremely costly and time-consuming, and there can be no assurance that we will have sufficient resources to defend any possible litigation related to such infringement. If we do not obtain a license on acceptable terms under such patents, or are found liable for infringement, or are not able to have such patents declared invalid, we may be liable for significant money damages, may encounter significant delays in bringing our product candidates to market, or may be precluded from participating in the manufacture, use or sale of any such product candidates, any of which would materially and adversely affect our business.
-18-
A dispute regarding the infringement or misappropriation of our proprietary rights or the proprietary rights of others could be costly and result in delays in our research and development activities.
Our commercial success depends upon our ability to develop and manufacture our product candidates and market and sell drugs, if any, and conduct our research and development activities without infringing or misappropriating the proprietary rights of others. We may be exposed to future litigation by others based on claims that our product candidates, technologies or activities infringe the intellectual property rights of others. Numerous United States and foreign issued patents and pending patent applications owned by others also exist in the therapeutic areas in, and for the therapeutic targets for, which we are developing drugs. These could materially affect our ability to develop our product candidates or sell drugs, and our activities, or those of our licensor or future collaborators, could be determined to infringe these patents. Because patent applications can take many years to issue, there may be currently pending applications, unknown to us, which may later result in issued patents that our drug candidates or technologies may infringe. There also may be existing patents, of which we are not aware, that our product candidates or technologies may infringe. Further, there may be issued patents and pending patent applications in fields relevant to our business, of which we are or may become aware, that we believe we do not infringe or that we believe are invalid or relate to immaterial portions of our overall drug discovery and development efforts. We cannot assure you that others holding any of these patents or patent applications will not assert infringement claims against us for damages or seek to enjoin our activities. We also cannot assure you that, in the event of litigation, we will be able to successfully assert any belief we may have as to non-infringement, invalidity or immateriality, or that any infringement claims will be resolved in our favor.
In addition, others may infringe or misappropriate our proprietary rights, and we may have to institute costly legal action to protect our intellectual property rights. We may not be able to afford the costs of enforcing or defending our intellectual property rights against others. There could also be significant litigation and other administrative proceedings in our industry that affect us regarding patent and other intellectual property rights. Any legal action or administrative action against us, or our collaborators, claiming damages or seeking to enjoin commercial activities relating to our drug discovery and development programs could:
|
•
|
require us, or potential collaborators, to obtain a license to continue to use, manufacture or market the affected drugs, methods or processes, which may not be available on commercially reasonable terms, if at all;
|
|
•
|
prevent us from importing, making, using, selling or offering to sell the subject matter claimed in patents held by others and subject us to potential liability for damages; or
|
|
•
|
consume a substantial portion of our managerial, scientific and financial resources; or be costly, regardless of the outcome.
|
Furthermore, because of the substantial amount of pre-trial documents and witness discovery required in connection with intellectual property litigation, there is risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, during the course of this kind of litigation, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the trading price of our common stock or warrants.
We face substantial uncertainty in our ability to protect our patents and proprietary technology.
Our ability to commercialize our products will depend, in part, on our or our licensor’s ability to obtain patents, to enforce those patents and preserve trade secrets, and to operate without infringing on the proprietary rights of others. The patent positions of biopharmaceutical companies are highly uncertain and involve complex legal and factual questions. There can be no assurance that:
|
•
|
Patent applications for and relating to our products candidates, Androxal® and Proellex®, will result in issued patents;
|
|
•
|
Patent protection will be secured for any particular technology;
|
|
•
|
Any patents that have been or may be issued to us, such as our issued patents and/or pending patent applications relating to Proellex® or Androxal®, or any patents that have been or may be issued to our licensor, such as the patent(s) and application(s) underlying our Proellex® compound, when issued, will be valid and enforceable;
|
|
•
|
any patents will provide meaningful protection to us;
|
|
•
|
others will not be able to design around the patents; or
|
|
•
|
our patents will provide a competitive advantage or have commercial application.
|
-19-
The failure to obtain and maintain adequate patent protection would have a material adverse effect on us and may adversely affect our ability to enter into, or affect the terms of, any arrangement for the marketing of any product.
We cannot assure that our patents will not be challenged by others.
There can be no assurance that patents owned by or licensed to us will not be challenged by others. We could incur substantial costs in proceedings, including interference proceedings before the PTO and comparable proceedings before similar agencies in other countries in connection with any claims that may arise in the future. These proceedings could result in adverse decisions about the patentability of our or our licensor’s inventions and products, as well as about the enforceability, validity or scope of protection afforded by the patents. Any adverse decisions about the patentability of our product candidates could cause us to either lose rights to develop and commercialize our product candidates or to license such rights at substantial cost to us. In addition, even if we were successful in such proceedings, the cost and delay of such proceedings would most likely have a material adverse effect on our business.
Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information, may not adequately protect our intellectual property, and will not prevent third parties from independently discovering technology similar to or in competition with our intellectual property.
We rely on trade secrets and other unpatented proprietary information in our product development activities. To the extent we rely on trade secrets and unpatented know-how to maintain our competitive technological position, there can be no assurance that others may not independently develop the same or similar technologies. We seek to protect trade secrets and proprietary knowledge, in part, through confidentiality agreements with our employees, consultants, advisors, collaborators and contractors. Nevertheless, these agreements may not effectively prevent disclosure of our confidential information and may not provide us with an adequate remedy in the event of unauthorized disclosure of such information. If our employees, scientific consultants, advisors, collaborators or contractors develop inventions or processes independently that may be applicable to our technologies, product candidates or products, disputes may arise about ownership of proprietary rights to those inventions and processes. Such inventions and processes will not necessarily become our property, but may remain the property of those persons or their employers. Protracted and costly litigation could be necessary to enforce and determine the scope of our proprietary rights. If we fail to obtain or maintain trade secret protection for any reason, the competition we face could increase, reducing our potential revenues and adversely affecting our ability to attain or maintain profitability.
We cannot protect our intellectual property rights throughout the world.
Filing, prosecuting, and defending patents on all of our drug discovery technologies and all of our potential drug candidates throughout the world could be prohibitively expensive. Competitors may use our technologies to develop their own drugs in jurisdictions where we have not obtained patent protection. These drugs may compete with our drugs, if any, and may not be covered by any of our patent claims or other intellectual property rights. The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States, and many companies have encountered significant problems in protecting and defending such rights in foreign jurisdictions. Many countries, including certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties (for example, the patent owner has failed to “work” the invention in that country or the third party has patented improvements). In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of the patent. Compulsory licensing of life-saving drugs is also becoming increasingly popular in developing countries either through direct legislation or international initiatives. Such compulsory licenses could be extended to include some of our drug candidates, which could limit our potential revenue opportunities. Moreover, the legal systems of certain countries, particularly certain developing countries, do not favor the aggressive enforcement of patents and other intellectual property protection, particularly those relating to biotechnology and/or pharmaceuticals, which makes it difficult for us to stop the infringement of our patents. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
Risks Related to our Common Stock and Warrants
The trading price of our common stock has been volatile and is likely to be volatile in the future.
The trading price of our common stock has been highly volatile. Since January 1, 2009 through March 18, 2011, the sale price of our stock price has fluctuated from a low of $1.11 to a high of $55.76. The market price for our common stock and warrants will be affected by a number of factors, including:
|
•
|
the denial or delay of regulatory clearances or approvals of our drug candidates or receipt of regulatory approval of competing products;
|
|
•
|
the clinical hold imposed on Proellex®, currently lifted to a partial clinical hold;
|
-20-
|
•
|
our ability to accomplish clinical, regulatory and other product development milestones;
|
|
•
|
the ability of our product candidates, if they receive regulatory approval, to achieve market success;
|
|
•
|
the performance of third-party manufacturers and suppliers;
|
|
•
|
actual or anticipated variations in our results of operations or those of our competitors;
|
|
•
|
developments with respect to patents and other intellectual property rights;
|
|
•
|
sales of common stock or other securities by us or our stockholders in the future;
|
|
•
|
additions or departures of key scientific or management personnel;
|
|
•
|
disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our products;
|
|
•
|
trading volume of our common stock and warrants;
|
|
•
|
investor perceptions about us and our industry;
|
|
•
|
public reaction to our press releases, other public announcements and SEC and other filings;
|
|
•
|
the failure of analysts to cover our common stock, or changes in analysts’ estimates or recommendations;
|
|
•
|
the failure by us to meet analysts’ projections or guidance;
|
|
•
|
general market conditions and other factors unrelated to our operating performance; and
|
|
•
|
the other factors described elsewhere in these “Risk Factors.”
|
The stock prices of many companies in the biotechnology industry have experienced wide fluctuations that have often been unrelated to the operating performance of these companies. Following periods of volatility in the market price of a company’s securities, securities class action litigation often has been initiated against a company. If any additional class action litigation is initiated against us, we may incur substantial costs and our management’s attention may be diverted from our operations, which could significantly harm our business.
Our inability to comply with the listing requirements of the Nasdaq Capital Market could result in our common stock and/or warrants being delisted, which could affect their market price and liquidity and reduce our ability to raise capital.
We are required to meet certain qualitative and financial tests (including a minimum closing bid price of $1.00 per share for our common stock) to maintain the listing of our common stock and/or warrants on the Nasdaq Capital Market. If we do not maintain compliance with the continued listing requirements for the Nasdaq Capital Market within specified periods and subject to permitted extensions, our common stock and/or warrants may be recommended for delisting (subject to any appeal we would file). If our common stock or warrants were delisted, it could be more difficult to buy or sell our common stock or warrants and to obtain accurate quotations, and the price of our common stock or warrants could suffer a material decline. Delisting would also impair our ability to raise capital.
The market price of our common stock may fall below the exercise price of our Series B Warrants.
The Series B Warrants are exercisable at any time at or prior to 5:00 p.m. Eastern time on February 8, 2016. The market price of our common stock may fall below the exercise price for such warrants prior to their expiration. Any Series B Warrants not exercised by such date of expiration will expire worthless and we will be under no further obligation to the holders of such warrants.
|
ITEM 1B.
|
UNRESOLVED STAFF COMMENTS
|
None.
|
ITEM 2.
|
PROPERTIES
|
We lease our current property under a lease agreement that expires in June 2015. This lease is for approximately 7,100 square feet of our laboratory and office space located in The Woodlands, Texas. We do not own or lease any other property and believe that our current facilities are sufficient for our needs for the foreseeable future.
-21-
|
ITEM 3.
|
LEGAL PROCEEDINGS
|
In August and September of 2009, several securities fraud class action lawsuits were filed in federal court for the Southern District of Texas against the Company and various of its current or former officers and directors. The lawsuits alleged that the defendants made certain misleading statements related to the Company's Proellex® drug. Among other claims, the lawsuits alleged that the defendants misrepresented the side effects of the drug related to liver function, and the risk that these side effects could cause a suspension of clinical trials on Proellex®. The lawsuits were consolidated under the caption In re Repros Therapeutics, Inc. Securities Litigation, Civil Action No. 09 Civ. 2530 (VDG), and the court appointed lead plaintiffs and class counsel. Lead plaintiffs filed a consolidated amended complaint making essentially the same allegations as had been made in the prior complaints. Lead plaintiffs sought to represent a class of all persons who purchased or otherwise acquired Repros common stock between July 1, 2009 and August 2, 2009, and asserted claims under the Securities Exchange Act of 1934. Defendants filed a motion to dismiss the complaint. On January 19, 2011, the court granted the defendants' motion to dismiss and entered a final judgment dismissing the case. The time for plaintiffs to file an appeal of that order expired on February 18, 2011.
On March 1, 2010, we were served with a lawsuit where we were named as a co-defendant along with one of our clinical regulatory service providers (“CRO”) relating to the Proellex® clinical trial study. The lawsuit was filed in the State of Tennessee, 30th Judicial District Chancery Court at Memphis by an investigator and claims that the CRO did not pay it amounts owing to it relating to the Proellex® study. We did not engage the investigator and under our agreement with the CRO, we believe the CRO is responsible for any such costs or damages regarding such lawsuit. Pursuant to a Settlement Agreement and Mutual Release entered into in October 2009, such CRO, on behalf of itself and its agents, released us from all claims which could be asserted by them against us. We believe such release covers the claims set forth in this lawsuit. The CRO failed to respond to the lawsuit, and a default judgment was entered against it in the amount of $172,901.29. We intend to vigorously defend any and all claims asserted by the investigator. An estimate of the possible costs or expenses to defend ourselves in this matter or risk of exposure under the litigation cannot be made at this time.See “Patents and Proprietary Information” in Item 1 for a description of judicial and regulatory proceedings involving patent matters.
|
ITEM 4.
|
(REMOVED AND RESERVED)
|
-22-
PART II
ITEM 5. MARKET FOR THE REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock is quoted on the Nasdaq Capital Market under the symbol “RPRX”. The following table shows the high and low sale prices per share of our common stock as reported by the Nasdaq Stock Market during the periods presented. Prices per share of our common stock have been adjusted to reflect the 1-for-4 reverse split of our common stock that was effected on October 14, 2010.
|
Price Range
|
||||||||
|
|
High
|
Low
|
||||||
|
2009
|
|
|
||||||
|
First Quarter
|
$
|
55.76
|
$
|
23.36
|
||||
|
Second Quarter
|
33.20
|
22.80
|
||||||
|
Third Quarter
|
24.04
|
2.60
|
||||||
|
Fourth Quarter
|
9.92
|
2.56
|
||||||
|
2010
|
|
|
||||||
|
First Quarter
|
$
|
4.88
|
$
|
2.52
|
||||
|
Second Quarter
|
4.52
|
1.44
|
||||||
|
Third Quarter
|
2.68
|
1.12
|
||||||
|
Fourth Quarter
|
4.56
|
1.11
|
||||||
|
2011
|
|
|
||||||
|
First Quarter (January 1st through March 18th)
|
$
|
6.85
|
$
|
2.37
|
||||
All of the foregoing prices reflect interdealer quotations, without retail mark-up, markdowns or commissions and may not necessarily represent actual transactions in the common stock.
On March 18, 2011, the last sale price of our common stock, as reported by the Nasdaq Capital Market, was $5.33 per share. On March 18, 2011, there were approximately 163 holders of record and approximately 3,705 beneficial holders of our common stock.
Dividend Policy
General
We have never declared or paid cash dividends on our capital stock. We currently intend to retain our future earnings, if any, for use in our business and therefore do not anticipate paying cash dividends in the foreseeable future. Payment of future dividends, if any, will be at the discretion of our board of directors after taking into account various factors, including our financial condition, operating results, current and anticipated cash needs.
Rights Plan
We are party to a rights agreement, as amended, pursuant to which a dividend consisting of one preferred stock purchase right was distributed for each share of our common stock held as of the close of business on September 13, 1999, and to each share of common stock issued thereafter until the earlier of (i) the distribution date which is defined in the rights plan, (ii) the redemption date which is defined in the rights plan or (iii) September 13, 2015. The rights plan is designed to deter coercive takeover tactics and to prevent an acquirer from gaining control of us without offering fair value to our stockholders. The rights will expire on September 13, 2015, subject to earlier redemption or exchange as provided in the rights plan. Each right entitles its holder to purchase from us one one-hundredth of a share of a new series of Series One Junior Participating Preferred Stock at a price of $20.00 per one one-hundredth of a share, subject to adjustment. The rights are generally exercisable only if a person acquires beneficial ownership of 20% or more of our outstanding common stock.
A complete description of the rights, the rights plan with Computershare Trust Company, N.A., as rights agent, and the Series One Junior Participating Preferred Stock is hereby incorporated by reference from the information appearing under the caption “Item 1. Description of the Registrant’s Securities to be Registered” contained in the Registration Statement on Form 8-A filed on September 3, 1999, and as amended by amendments to such Registration Statement on Form 8-A/A filed on September 11, 2002, October 31, 2002, June 30, 2005, January 10, 2008, October 10, 2008 and September 9, 2010.
-23-
Performance Graph
THIS INFORMATION IS REQUIRED BY ITEM 201(E) OF REGULATION S-K. SUCH INFORMATION SHALL NOT BE DEEMED TO BE “FILED” OR INCORPORATED BY REFERENCE IN FUTURE FILINGS WITH THE SEC, OR SUBJECT TO THE LIABILITIES OF SECTION 18 OF THE SECURITIES EXCHANGE ACT OF 1934, EXCEPT TO THE EXTENT THAT WE SPECIFICALLY INCORPORATE IT BY REFERENCE INTO A DOCUMENT FILED UNDER THE SECURITIES ACT OF 1933 OR THE SECURITIES EXCHANGE ACT OF 1934.
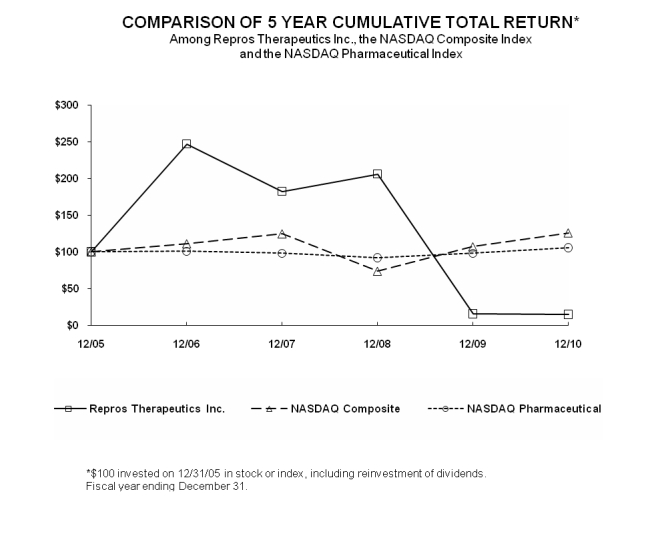
| 12/05 | 12/06 | 12/07 | 12/08 | 12/09 | 12/10 | |||||||||||||||||||
|
Repros Therapeutics Inc.
|
100.00 | 246.77 | 182.39 | 205.87 | 15.59 | 14.87 | ||||||||||||||||||
|
NASDAQ Composite
|
100.00 | 111.16 | 124.64 | 73.80 | 107.07 | 125.99 | ||||||||||||||||||
|
NASDAQ Pharmaceutical
|
100.00 | 100.74 | 97.94 | 91.84 | 98.07 | 105.79 |
-24-
ITEM 6. SELECTED CONSOLIDATED FINANCIAL DATA
The statement of operations data for the years ended December 31, 2010, 2009 and 2008, and the balance sheet data as of December 31, 2010 and 2009, have been derived from our financial statements, included elsewhere in this Annual Report on Form 10-K. The statement of operations data for the years ended December 31, 2007 and 2006, and the balance sheet data as of December 31, 2008, 2007 and 2006 have been derived from our financial statements not included in this annual report on Form 10-K. Our historical results are not necessarily indicative of results to be expected for any future period. The data presented below have been derived from financial statements that have been prepared in accordance with accounting principles generally accepted in the United States of America and should be read with our financial statements, including notes, and with "Management's Discussion and Analysis of Financial Condition and Results of Operations" included elsewhere in this annual report on Form 10-K.
STATEMENTS OF OPERATIONS DATA:
|
2010
|
2009
|
2008
|
2007
|
2006
|
||||||||||||||||
|
Revenues and Other Income:
|
||||||||||||||||||||
|
Interest income
|
$ | — | $ | 4 | $ | 433 | $ | 1,508 | $ | 596 | ||||||||||
|
Other income
|
421 | 547 | — | — | — | |||||||||||||||
|
Total revenues
|
421 | 551 | 433 | 1,508 | 596 | |||||||||||||||
|
Expenses:
|
||||||||||||||||||||
|
Research and development
|
2,904 | 23,062 | 22,575 | 12,420 | 11,912 | |||||||||||||||
|
General and administrative
|
2,285 | 4,723 | 3,060 | 2,788 | 2,879 | |||||||||||||||
|
Total expenses
|
5,189 | 27,785 | 25,635 | 15,208 | 14,791 | |||||||||||||||
|
Net loss
|
$ | (4,768 | ) | $ | (27,234 | ) | $ | (25,202 | ) | $ | (13,700 | ) | $ | (14,195 | ) | |||||
|
Net loss per share – basic and diluted (1)
|
$ | (0.59 | ) | $ | (6.28 | ) | $ | (7.54 | ) | $ | (4.38 | ) | $ | (5.60 | ) | |||||
|
Shares used in loss per share calculation
|
8,057 | 4,336 | 3,343 | 3,131 | 2,537 | |||||||||||||||
|
BALANCE SHEET DATA:
|
||||||||||||||||||||
|
Cash, cash equivalents and marketable securities
|
$ | 2,957 | $ | 1,886 | $ | 19,470 | $ | 25,903 | $ | 6,736 | ||||||||||
|
Total assets
|
4,465 | 2,960 | 22,603 | 27,599 | 7,849 | |||||||||||||||
|
Deficit accumulated during the development stage
|
(179,244 | ) | (174,476 | ) | (147,242 | ) | (122,040 | ) | (108,340 | ) | ||||||||||
|
Total stockholders' equity
|
$ | 3,167 | $ | 562 | $ | 15,614 | $ | 24,060 | $ | 3,790 | ||||||||||
|
(1)
|
See "Note 2. Summary of Significant Accounting Policies" of Notes to Consolidated Financial Statements for a description of the computation of loss per share.
|
-25-
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following management's discussion and analysis should be read in conjunction with our historical consolidated financial statements and their notes included elsewhere in this Form 10-K. This discussion contains forward-looking statements that reflect our current views with respect to future events and financial performance. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of certain factors, such as those set forth under "Risk Factors" and elsewhere in this Form 10-K.
Overview
Repros Therapeutics Inc. (the “Company”, “RPRX,” “Repros,” or “we,” “us” or “our”) was organized on August 20, 1987. We are a development stage biopharmaceutical company focused on the development of new drugs to treat hormonal and reproductive system disorders.
Our primary product candidate, Androxal®, is a single isomer of clomiphene citrate and is an orally active proprietary small molecule compound. We are developing Androxal® for men of reproductive age with low testosterone levels. Androxal® treats the underlying mechanism that causes secondary hypogonadism and restores normal testicular function. In addition, we are conducting a Phase 2 clinical trial of Androxal® as a potential treatment for Type 2 diabetes.
On November 8, 2010, we held a Type B meeting with the Food and Drug Administration (“FDA”) to discuss whether the FDA would review our protocols for a Phase 3 trial of Androxal® in men with secondary hypogonadism under a Special Protocol Assessment(“SPA”). In the meeting, the FDA recommended that a Phase 2B study in men with secondary hypogonadism but naïve to testosterone treatment be conducted if the Company desired the protocols to be reviewed under an SPA. The FDA further opined that such Phase 2B study would provide for a more solid data base for design of Phase 3 studies and eventual approval of such studies under an SPA. In our 24-patient Phase 2B proof-of-concept clinical trial which was initiated in the second quarter of 2008, we monitored the effects of Androxal® on male fertility and testicular function in patients being treated for low testosterone as compared to Testim®, a popular marketed topical testosterone medication. This trial showed that Androxal® was able to maintain sperm counts in men being treated for their low testosterone levels, whereas Testim® resulted in suppressed sperm levels. On January 3, 2011, we announced that we have received Investigation Review Board (“IRB”) approval to commence the Phase 2B study of Androxal® in men with secondary hypogonadism, and we have begun enrolling patients. Depending on the rate of subject enrollment, we hope to have the study completed by the end of 2011.
Our findings from a retrospective review of the clinical data from our 200 patient non-pivotal Phase 2 clinical trial showed that Androxal® therapy resulted in a significant reduction in mean fasting plasma glucose levels in men with glucose levels greater than 104 mg/dL at the outset of the trial, an outcome not seen in the placebo or AndroGel® arms of this study. Based on these results, in April 2008, we submitted a White Paper to the Division of Reproductive and Urology Products. The data demonstrated that among subjects with a serum glucose of greater than or equal to 105 mg/dL, there was a higher response rate to treatment in the Androxal® group than the placebo or AndroGel® groups, and the reduction in fasting serum glucose in this group was statistically significant. In November 2008, after the FDA reviewed this paper we received guidance from them suggesting that we open a new Investigational New Drug Application (“IND”) with the Division of Metabolic and Endocrine Products (“DMEP”) for the investigation of Androxal® as a potential treatment for Type 2 diabetes. In December 2009, we submitted an IND to DMEP for the investigation of Androxal® for such purpose. On February 1, 2010, we received confirmation from DMEP that our IND was accepted and, as a result, we have initiated our Phase 2 trial. We hope that the study will be fully enrolled by the end of the third quarter 2011.
Proellex®, our product candidate for female reproductive health, is a new chemical entity that acts as a selective blocker of the progesterone receptor and is being developed for the treatment of symptoms associated with uterine fibroids and endometriosis. We have conducted numerous studies with Proellex® enrolling over 750 women, roughly 700 of whom were dosed with the drug. Up until the summer of 2009, all side effects exhibited in the studies were considered manageable and the benefit of Proellex® far outweighed the risk. However, in Phase 3 efficacy and larger Phase 3 safety studies in diverse populations, a small number of subjects exhibited serious adverse effects associated with elevated liver enzymes. As a result of these findings, we elected to stop the trials and the FDA subsequently placed Proellex® on full clinical hold. All women that experienced elevated liver enzymes and returned for follow-up visits returned to baseline conditions with no overnight hospitalization necessary. An analysis of all the subjects that experienced such serious adverse effects showed that the effect only occurred in a small percentage of subjects that were exposed to the 50 mg dose of the drug for any period of time. Based on these findings, the Company petitioned the FDA to allow it to conduct a low dose study to demonstrate both safety and signals of efficacy in low oral doses of Proellex®, up to 12 mg administered per day. The FDA upgraded the full clinical hold to a partial hold to allow the low dose study to be conducted, which we have since commenced. We believe we can complete this trial by year end 2011. In addition, the Company has undertaken two related initiatives presently at the preclinical stage. The first is the exploration of vaginal delivery as an alternative administrative route to bypass first-pass liver effects and reduce systemic exposure. The second is the screening of second generation molecules that do not possess the specific structures the Company believes induced the liver toxicity exhibited at higher doses of Proellex®.
-26-
We continue limited out-licensing efforts for our phentolamine-based product candidates, including VASOMAX®, which had previously been approved for marketing in several countries in Latin America for the treatment of male erectile dysfunction under the brand name, Z-Max. VASOMAX® has been on partial clinical hold in the U.S. since 1998, and no further development activities are planned.
The clinical development of pharmaceutical products is a complex undertaking, and many products that begin the clinical development process do not obtain regulatory approval. The costs associated with our clinical trials may be impacted by a number of internal and external factors, including the number and complexity of clinical trials necessary to obtain regulatory approval, the number of eligible patients necessary to complete our clinical trials and any difficulty in enrolling these patients, and the length of time to complete our clinical trials. Given the uncertainty of these potential costs, we recognize that the total costs we will incur for the clinical development of our product candidates may exceed our current estimates
As with most biotechnology companies with drug candidates in development, the path to marketing approval by the FDA and comparable foreign agencies for each such candidate is long and uncertain. The regulatory process, both domestically and abroad, is a multi-year process with no certainty when and if a drug candidate will be approved for commercial use. The development path for a particular drug candidate typically includes a variety of clinical trials. While we have a general estimate of the timeframe for our clinical trials, the actual anticipated completion dates for each of our drug candidates are uncertain due to a wide variety of risks, including those described in the risk factors in this Annual Report on Form 10-K. The length of time for a clinical trial may vary substantially according to factors relating to the particular clinical trial, such as the type and intended use of the drug candidate, the clinical trial design and the ability to enroll suitable patients. A clinical hold,can also result in unpredictable delays and added costs. We will not receive any revenue from commercial sales unless we, or a potential partner, complete the clinical trial process, obtain regulatory approval, and successfully commercialize one or more of our product candidates. Similarly, we do not have a reasonable basis to predict when or if material net cash inflows from the commercialization and sale of our drug candidates will occur. To date, we have not commercialized any of our drug candidates to any material extent and in fact may never do so. For a discussion of the risks and uncertainties associated with the timing and costs of completing the development of the Company’s drug candidates, see the section titled “Risk Factors.”
Our results of operations may vary significantly from year to year and quarter to quarter, and depend on, among other factors, our ability to be successful in our clinical trials, the regulatory approval process in the United States and other foreign jurisdictions and the ability to complete new licenses and product development agreements. The timing of our revenues may not match the timing of our associated product development expenses. To date, research and development expenses have generally exceeded revenue in any particular period and/or fiscal year.
As of December 31, 2010, we had accumulated losses of $179.2 million, approximately $3.0 million in cash and cash equivalents, and our accounts payable and accrued expenses were approximately $1.3 million. On February 8, 2011, we completed a public offering of our common stock, Series A Warrants to purchase common stock and Series B Warrants to purchase common stock which resulted in approximately $11 million in gross proceeds to us, after the underwriting discount and before offering expenses. See “Overview—Recent Developments” for a description of such offering. We believe we have sufficient funding to complete all of the Phase 2 and 2B clinical trials currently planned or underway; however, significant additional capital will be required for us to complete development of either of our product candidates. We continue to explore potential additional financing alternatives (including corporate partnering opportunities) that would provide sufficient funds to enable us to continue to develop our two product candidates through completion of the outlined clinical trials; however, there can be no assurance that we will be successful in raising any such additional funds on a timely basis or at all. The foregoing and other matters raise substantial doubt about our ability to continue as a going concern.
On February 12, 2010, we entered into an Equity Distribution Agreement (the “Equity Distribution Agreement”) with Ladenburg Thalmann & Co. Inc. (“Ladenburg”), pursuant to which we may issue and sell from time to time through Ladenburg, as sales agent and/or principal, shares of our common stock having an aggregate offering price of up to $10 million (the “ATM Shares”). Ladenburg is not required to sell on our behalf any specific number or dollar amount of the ATM Shares, but Ladenburg, upon acceptance of written instructions from us, agreed to use its commercially reasonable efforts consistent with its customary trading and sales practices, to sell the ATM Shares up to the amount specified, and otherwise in accordance with the terms of a placement notice delivered to Ladenburg. We have no obligation to sell any ATM Shares under the Equity Distribution Agreement, and may at any time suspend sales under the Equity Distribution Agreement, provided that such suspension shall not affect either party’s obligations with respect to the ATM Shares sold prior to the receipt of notice of such suspension. Ladenburg receives a commission of 4% of the gross sales price of all ATM Shares sold through it under the Equity Distribution Agreement. The ATM Shares are issued pursuant to our shelf registration statement on Form S-3, as amended (File No. 333-163648). Cumulative through December 31, 2010, we have sold 2,448,572 ATM Shares at a weighted average share price of $2.77, for proceeds of approximately $6.4 million, net of expenses. Pursuant to General Instruction I.B.6. of Form S-3, we may not sell more than one-third of the aggregate market value of our common stock held by non-affiliates during a period of 12 calendar months immediately prior to, and including, the date of such sale of such common stock. Due to this limitation, we announced on August 3, 2010 that we had suspended this ATM offering of Company securities. Additionally, on December 30, 2010 we announced our decision to reinstate our offering of the ATM shares. As a result, we sold 286,187 ATM Shares at a weighted average share price of $2.90 in January 2011, for proceeds of approximately $831,000, net of expenses.
-27-
On October 14, 2010, the Company effected a one-for-four reverse split of its common stock. The split-adjusted shares of the Company’s common stock began trading on the Nasdaq Capital Market on October 15, 2010. The one-for-four reverse split converted all shares of the Company’s common stock issued and outstanding, plus all outstanding stock options and the number of shares of common stock available for issuance under the Company’s approved stock plans. The number of authorized shares of common stock was not affected by the reverse split. The reverse split enabled the Company to meet the continued listing rules of the Nasdaq Capital Market as evidenced by the Compliance Letter received from Nasdaq on October 29, 2010. All share and per share amounts have been retroactively adjusted to reflect the reverse stock split for all periods presented.
On February 8, 2011, we completed an underwritten public offering of 690,000 units (including the exercise of the underwriter’s over-allotment option), consisting of an aggregate of 2,760,000 shares of our common stock, Series A Warrants to purchase 2,070,000 shares of our common stock and Series B warrants to purchase 1,690,500 shares of our common stock, at a price per unit of $17.15. Each unit consisted of four shares of our common stock, Series A Warrants exercisable for three shares of our common stock at an exercise price of $0.01 per share and Series B Warrants exercisable for 2.45 shares of our common stock at an exercise price of $2.49 per share. Gross proceeds to us, after the underwriting discount and before offering expenses, were approximately $11 million.
Our common stock is traded on the Nasdaq Capital Market under our ticker symbol RPRX.
We have 6 full-time employees who utilize the services of contract research organizations, contract manufacturers and various consultants to assist us in performing clinical and regulatory services for the clinical development of our products. We are substantially dependent on our various contract groups to adequately perform the activities required to obtain regulatory approval of our products.
The value of the tax asset associated with the December 31, 2010 accumulated deficit can be substantially diminished in value to us due to various tax regulations, including change in control provisions in the tax code. For additional information relating to our net operating loss carryforward, see "Note 6. Federal Income Taxes" of the Notes to Consolidated Financial Statements. Losses have resulted principally from costs incurred in conducting clinical trials for our product candidates, in research and development activities related to efforts to develop our products and from the associated administrative costs required to support those efforts. There can be no assurance that we will be able to successfully complete the transition from a development stage company to the successful introduction of commercially viable products. Our ability to achieve profitability will depend on, among other things, successfully completing the clinical development of our products in a reasonable time frame and at a reasonable cost, obtaining regulatory approvals, establishing marketing, sales and manufacturing capabilities or collaborative arrangements with others that possess such capabilities, our and our partners' ability to realize value from our research and development programs through the commercialization of those products and raising sufficient funds to finance our activities. There can be no assurance that we will be able to achieve profitability or that profitability, if achieved, can be sustained. See "Item 1. Business — Risk Factors" and "Note 1. Organization and Operations" of Notes to Consolidated Financial Statements.
Critical Accounting Policies and the Use of Estimates
The preparation of our financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the amounts reported in our financial statements and accompanying notes. Please see Note 2, "Summary of Significant Accounting Policies", for a detailed discussion of our critical accounting policies. A brief summary of our accounting policies is provided below.
Capitalized Patent Costs
The Company capitalizes the cost associated with building its patent library for its Androxal® product. As of December 31, 2010 and 2009, other assets consist of capitalized patent costs in the amount of $1.2 million and $885,000 respectively. Patent costs, which include legal and application costs related to the patent portfolio, are being amortized over the lesser of 20 years or the estimated economic life of the patent. Amortization of patent cost expense was $76,000, $54,000 and $27,000 in 2010, 2009 and 2008, respectively. All of the $1.2 million in capitalized patent costs as of December 31, 2010 related to Androxal® patent and patent application costs.
We review capitalized patent and patent application costs for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. An impairment exists when estimated undiscounted cash flows expected to result from the patent are less than its carrying amount. The impairment loss recognized represents the excess of the patent cost as compared to its estimated fair value.
-28-
Due to the clinical hold on Proellex® and the uncertainty of future cash flows related to the Proellex® patent applications, the Company recorded an impairment charge of approximately $957,000 in 2009 related to these patent applications. Additionally, the Company concluded that it will no longer seek to protect the specific matter covered in certain Androxal® patent applications and recorded an impairment charge of approximately $318,000 in 2009 to abandon these patent applications. These charges were recorded in Research and Development expenses on the consolidated statement of operations for the year ended December 31, 2009. The remaining capitalized patent and patent application costs relating to Androxal® can continue to be used, outlicensed or sold to third parties for at least an amount management believes is sufficient to recover the carrying value of the capitalized patent costs.
Should the Company not continue development of Androxal® or should the Company not continue as a going concern, the remaining capitalized patent and patent application costs may not be recoverable, which would result in charges to operating results in future periods.
Accrued Expenses
We estimate accrued expenses as part of our process of preparing financial statements. Examples of areas in which subjective judgments may be required include costs associated with services provided by contract organizations for clinical trials, preclinical development and manufacturing of clinical materials. We accrue for costs incurred as the services are being provided by monitoring the status of the trials or services provided and the invoices received from our external service providers. In the case of clinical trials, a portion of the estimated cost normally relates to the projected cost to treat a patient in our trials, and we recognize this cost over the estimated term of the study based on the number of patients enrolled in the trial on an ongoing basis, beginning with patient enrollment. As actual costs become known to us, we adjust our accruals. To date, our estimates have not differed significantly from the actual costs incurred. However, subsequent changes in estimates may result in a material change in our accruals, which could also materially affect our balance sheet and results of operations.
R&D Expense
R&D expenses include salaries and related employee expenses, contracted regulatory affairs activities, insurance coverage for clinical trials and prior product sales, contracted research and consulting fees, facility costs and internal research and development supplies. We expense research and development costs in the period they are incurred. These costs consist of direct and indirect costs associated with specific projects as well as fees paid to various entities that perform research on our behalf.
Share-Based Compensation
We have two stock-based compensation plans at December 31, 2010, the 2000 Non-Employee Directors’ Stock Option Plan, or 2000 Director Plan and the 2004 Stock Option Plan, or 2004 Plan. Accounting standards generally require the recognition of the cost of employee services for share-based compensation based on the grant date fair value of the equity or liability instruments issued. We use the Black-Scholes option pricing model to estimate the fair value of our stock options. Expected volatility is determined using historical volatilities based on historical stock prices for a period equal to the expected term. The expected volatility assumption is adjusted if future volatility is expected to vary from historical experience. The expected term of options represents the period of time that options granted are expected to be outstanding and falls between the options’ vesting and contractual expiration dates. The risk-free interest rate is based on the yield at the date of grant of a zero-coupon U.S. Treasury bond whose maturity period equals the option’s expected term.
Income Taxes
We have had net operating losses since inception and, therefore, have not been subject to federal income taxes. We have accumulated approximately $2.4 million of research and development tax credits. As of December 31, 2010, we had approximately $153 million of net operating loss, or NOL, carryforwards for federal income tax purposes. Additionally, approximately $3.9 million of NOLs, and approximately $49,000 of research and development tax credits, expired in 2010. Accounting standards require the recognition of a deferred tax asset. However, a valuation allowance must be recorded for deferred tax assets whose recovery is deemed unlikely. As we have incurred net operating losses since inception, and there is no certainty of future revenues, our deferred tax assets have been reserved in full in the accompanying consolidated financial statements. Additionally, if the Company has an opportunity to use this NOL to off-set tax liabilities in the future, the use of this asset would be restricted based on Internal Revenue Service, state and local NOL use guidelines. The Company’s public offerings completed on February 5, 2007, October 2, 2008, September 11, 2009, October 13, 2009, February 8, 2011, the sale and issuance of the ATM Shares and the issuance of unregistered shares as part of the settlement agreements we entered into with certain of our creditors since October of 2009 may have created a change of ownership for Federal Income tax purposes. The Company has not undertaken a study to determine if this has occurred. A change in ownership for Federal Income tax purposes may result in a limitation on the use of net operating loss and tax credit carryforwards in future periods.
-29-
Results of Operations
Comparison of Years Ended December 31, 2010 and 2009
Revenues and Other Income
Total revenues and other income decreased 24% to $421,000 in 2010 as compared to $551,000 for 2009. This decrease was primarily due to a decrease of $126,000 in other income. In 2010, the Company recognized $244,000 in other income related to grant revenue received from The Department of the Treasury for investment in a qualifying therapeutic discovery project under Section 48D of the Internal Revenue Code. Additionally, we recognized approximately $177,000 in non-cash other income from settlements with certain vendors in 2010. The Company recognized $547,000 in non-cash other income related to settlements with certain vendors in the fourth quarter of 2009.
Research and Development Expenses
R&D expenses include contracted services relating to our clinical product development activities which include preclinical studies, clinical trials, regulatory affairs and bulk manufacturing scale-up activities and bulk active ingredient purchases for preclinical and clinical trials primarily relating to our two products in clinical development, which are Androxal® and Proellex®. Research and development expenses also include internal operating expenses relating to our general research and development activities. R&D expenses decreased 87% or approximately $20.2 million to $2.9 million for the year ended 2010 as compared to $23.1 million in 2009. Our primary R&D expenses for 2010 and 2009 are shown in the following table (in thousands):
|
Research and Development
|
December 31,
2010
|
December 31,
2009
|
Variance
|
Change (%)
|
||||||||||||
|
Androxal® clinical development
|
$ | 383 | $ | 786 | $ | (403 | ) | (51 | )% | |||||||
|
Proellex® clinical development
|
1,169 | 18,376 | (17,207 | ) | (94 | )% | ||||||||||
|
Payroll and benefits
|
573 | 1,384 | (811 | ) | (59 | )% | ||||||||||
|
Operating and occupancy
|
779 | 2,516 | (1,737 | ) | (69 | )% | ||||||||||
|
Total
|
$ | 2,904 | $ | 23,062 | $ | (20,158 | ) | (87 | )% | |||||||
To date through December 31, 2010 we have incurred approximately $14.7 million for the development of Androxal® and approximately $56.3 million for the development of Proellex®. These accumulated costs exclude any internal operating expenses. We are currently developing Androxal® as a treatment for men of reproductive age with low testosterone. In addition, we received confirmation on February 1, 2010 from DMEP that our IND was accepted for the investigation of Androxal® as a potential treatment for Type 2 diabetes. As a result, we have initiated a Phase 2 trial. Before the clinical hold on further Proellex® development in August 2009, we were developing Proellex® for three indications which included a pre-surgical treatment of anemia associated with uterine fibroids, a chronic treatment of symptoms associated with uterine fibroids and as a chronic treatment of symptoms associated with endometriosis. In June 2010, the FDA notified us that the full clinical hold on Proellex® had been revised to a partial clinical hold to allow us to run a single study to explore both safety and efficacy in an escalating dose fashion. As a result, we have initiated an escalating dose study and began dosing subjects in the third quarter of 2010.
The decrease in Androxal® and Proellex® clinical development expenses is primarily due to the decreased clinical development expenses related to Proellex® as a result of the discontinuation of all clinical trials in August 2009 due to the FDA’s clinical hold on Proellex®. R&D expenses were further decreased by the decreased clinical development expenses related to Androxal® due to the completion of a Phase 2B proof-of-concept clinical trial in 2009. Additionally, the new Phase 2 trial for Type 2 diabetes began dosing subjects in October 2010.
Payroll and Benefits
R&D payroll and benefit expense include salaries, non-cash stock option compensation expense and fringe benefits which decreased 59% or approximately $811,000 to $573,000 for the year ended 2010 as compared to $1.4 million in 2009. This decrease is primarily due to a decrease in headcount and the salary reduction program put in place in August 2009 and revised in May 2010. Included in payroll and benefit expense is a charge for non-cash stock option expense of $241,000 for the year ended 2010 as compared to $485,000 in the year 2009.
Operating and Occupancy
R&D operating and occupancy decreased 69% or approximately $1.7 million to approximately $779,000 for the year ended 2010 as compared to $2.5 million in 2009. Due to the clinical hold on Proellex® and the uncertainty of future cash flows related to the Proellex® patent applications, the Company recorded an impairment charge of approximately $957,000 in 2009 related to these patent applications. Additionally, the Company concluded that it will no longer seek to protect the specific matter covered in certain Androxal® patent applications and recorded an impairment charge of approximately $318,000 to abandon these patent applications in 2009. R&D operating and occupancy expenses were further decreased by approximately $128,000 in consulting fees for the year ended 2010 as compared to 2009.
-30-
General and Administrative Expenses
General and administrative expenses, or G&A, decreased 52% or approximately $2.4 million to $2.3 million for 2010 as compared to $4.7 million for 2009. Our primary G&A expenses for 2010 and 2009 are shown in the following table (in thousands):
|
General and Administrative
|
December 31,
2010
|
December 31,
2009
|
Variance
|
Change (%)
|
||||||||||||
|
Payroll and benefits
|
$ | 627 | $ | 2,039 | $ | (1,412 | ) | (69 | )% | |||||||
|
Operating and occupancy
|
1,658 | 2,684 | (1,026 | ) | (38 | )% | ||||||||||
|
Total
|
$ | 2,285 | $ | 4,723 | $ | (2,438 | ) | (52 | )% | |||||||
G&A payroll and benefit expense for both 2010 and 2009, include salaries, bonuses, non-cash stock option compensation expense and fringe benefits and decreased 69% or approximately $1.4 million to $627,000 for the year ended 2010 as compared to $2.0 million in 2009. The decrease in payroll and benefits for the year ended 2010 as compared to 2009 is primarily due to a decrease in headcount and the salary reduction program put in place in August 2009 and revised in May 2010. Included in payroll and benefit expense is a charge for non-cash stock option expense of $314,000 for the year ended 2010 as compared to $799,000 in the year 2009. Additionally, salaries for the year ended 2010 were $264,000 as compared to $949,000 for 2009.
G&A operating and occupancy expenses, which include expenses to operate as a public company, decreased 38% or approximately $1.0 million to $1.7 million in 2010 as compared to $2.7 million in 2009. The decrease is primarily due to a decrease in professional services.
Comparison of Years Ended December 31, 2009 and 2008
Revenues and Other Income
Total revenues and other income increased 27% to $551,000 in 2009 as compared to $433,000 for 2008. This increase was primarily due to an increase of $547,000 in other income, offset by a decrease in interest income of $429,000. The Company recognized $547,000 in non-cash other income related to debt relief from settlements with certain vendors in the fourth quarter of 2009. The decrease in interest income is due to lower combined cash, cash equivalents and marketable securities balances and reduced interest rate yields that have occurred as we moved our cash investments solely into money market mutual funds.
Research and Development Expenses
R&D expenses include contracted services relating to our clinical product development activities which include preclinical studies, clinical trials, regulatory affairs and bulk manufacturing scale-up activities and bulk active ingredient purchases for preclinical and clinical trials primarily relating to our two products in clinical development, which are Proellex® and Androxal®. Research and development expenses also include internal operating expenses relating to our general research and development activities. R&D expenses increased 2% or approximately $487,000 to $23.1 million for the year ended 2009 as compared to $22.6 million in 2008. Our primary R&D expenses for 2009 and 2008 are shown in the following table (in thousands):
|
Research and Development
|
December 31,
2009
|
December 31,
2008
|
Variance
|
Change (%)
|
||||||||||||
|
Androxal® clinical development
|
$ | 786 | $ | 2,370 | $ | (1,584 | ) | (67 | )% | |||||||
|
Proellex® clinical development
|
18,376 | 17,788 | 588 | 3 | % | |||||||||||
|
Payroll and benefits
|
1,384 | 1,154 | 230 | 20 | % | |||||||||||
|
Operating and occupancy
|
2,516 | 1,263 | 1,253 | 99 | % | |||||||||||
|
Total
|
$ | 23,062 | $ | 22,575 | $ | 487 | 2 | % | ||||||||
To date through December 31, 2009 we have incurred approximately $14.4 million for the development of Androxal® and approximately $55.2 million for the development of Proellex®. These accumulated costs exclude any internal operating expenses. We are currently developing Androxal® as a treatment for men with low testosterone that want to maintain or improve their fertility and sperm function. In addition, we are exploring the feasibility of developing Androxal® as a treatment for Type 2 diabetes. Prior to 2008, we were developing Androxal® as a treatment for men with low testosterone due to secondary hypogonadism. Before the recent clinical hold on further Proellex® development we were developing Proellex® for three indications which included a pre-surgical treatment of anemia associated with uterine fibroids, a chronic treatment of symptoms associated with uterine fibroids and as a chronic treatment of symptoms associated with endometriosis.
-31-
Androxal®
Androxal® clinical development expenses decreased 67% or approximately $1.6 million to $786,000 for the year ended 2009 as compared to $2.4 million in 2008. The decrease in Androxal® clinical development expenses is shown in the following table (in thousands):
|
Androxal® Clinical
Development
|
December 31,
2009
|
December 31,
2008
|
Variance
|
Change (%)
|
||||||||||||
|
Clinical trials
|
$ | 395 | $ | 1,006 | $ | (611 | ) | (61 | )% | |||||||
|
Preclinical studies
|
282 | 1,178 | (896 | ) | (76 | )% | ||||||||||
|
Formulation and dosage
|
19 | 161 | (142 | ) | (88 | )% | ||||||||||
|
Other
|
90 | 25 | 65 | 260 | % | |||||||||||
|
Total
|
$ | 786 | $ | 2,370 | $ | (1,584 | ) | (67 | )% | |||||||
Prior to 2008 we were developing Androxal® as a treatment for testosterone deficiency due to secondary hypogonadism by restoring normal testosterone production in males with functional testes. As a result of a Type “C” meeting held with the Food and Drug Administration, or FDA, on October 15, 2007 we discontinued clinical efforts for that indication. During 2008 we initiated a clinical development program with Androxal® as a treatment for men being treated for low testosterone that want to maintain their fertility.
Clinical trial expenses during 2009 primarily reflect a Phase 2B proof-of-concept clinical trial. Clinical trial expenses during 2008 primarily reflect long-term Open Label Safety study activities. The decrease in clinical trial expenses in 2009 as compared to 2008 is primarily due to the winding down of the long-term Open Label Safety study in the first quarter of 2009. Preclinical study expenses reflect animal safety activities required by the FDA to file an NDA. The decrease in preclinical expenses in 2009 as compared to 2008 is primarily due to the completion of two carcinogenicity studies in the first quarter of 2009. Formulation and dosage expenses reflect the purchase of active drug to conduct clinical trials and to meet any potential future NDA submission requirements.
Proellex®
Proellex® clinical development expenses increased 3% or approximately $588,000 to approximately $18.4 million for the year ended 2009 as compared to $17.8 million in 2008. The increase in Proellex® clinical development expenses is shown in the following table (in thousands):
|
Proellex® Clinical
Development
|
December 31,
2009
|
December 31,
2008
|
Variance
|
Change (%)
|
||||||||||||
|
Clinical trials
|
$ | 15,202 | $ | 14,547 | $ | 655 | 5 | % | ||||||||
|
Preclinical studies
|
500 | 1,679 | (1,179 | ) | (70 | )% | ||||||||||
|
Formulation and dosage
|
2,130 | 1,378 | 752 | 55 | % | |||||||||||
|
Other
|
544 | 184 | 360 | 196 | % | |||||||||||
|
Total
|
$ | 18,376 | $ | 17,788 | $ | 588 | 3 | % | ||||||||
Prior to 2008 we began developing Proellex® for two indications which include a chronic treatment of symptoms associated with uterine fibroids and endometriosis. During the first quarter of 2008 we filed an IND with Proellex® for a new indication as a short course pre-surgical treatment of anemia associated with uterine fibroids. On August 3, 2009, we suspended all ongoing clinical trials of Proellex® pending resolution of certain safety issues relating to such trials as described more fully above. Proellex® clinical expenses for the year ended December 31, 2009 include Phase 1, Phase 2, Phase 3 and long-term Open Label Safety study activities and costs to close out all clinical trials of Proellex®. Of the $15.2 million in clinical trial expenses for 2009, $14.5 million was incurred through September 30, 2009. The increase in clinical trials expenses for Proellex® as compared to 2008 is primarily due to increased activity in Phase 3 studies prior to the suspension of such trials and approximately $700,000 in costs to close out the clinical trials in the third and fourth quarters of 2009.
Preclinical study expenses reflect animal safety activities required by the FDA to file a NDA. Formulation and dosage expenses reflect activities associated with the bulk scale-up and purchase of active drug to conduct clinical trials and to meet any potential future NDA submission requirements.
Formulation and dosage expenses for the year ended December 31, 2009 include a charge for $1.5 million previously reflected in Prepaid Expense and Other Current Assets in conjunction with our commitment to purchase the bulk active ingredient of Proellex® from Gedeon Richter under a new scaled-up amended manufacturing process. As of September 4, 2009 this agreement was terminated and Repros accepted the material produced through this date and as a result expensed the $1.5 million prepaid asset to R&D Expense during the third quarter of 2009.
-32-
Payroll and Benefits
R&D payroll and benefit expense include salaries, non-cash stock option compensation expense and fringe benefits which increased 20% or approximately $230,000 to $1.4 million for the year ended 2009 as compared to $1.2 million in 2008. This increase is primarily due to an increase in headcount and an increase in non-cash stock option compensation of $96,000. Included in payroll and benefit expense is a charge for non-cash stock option expense of $485,000 for the year ended 2009 as compared to $389,000 in the year 2008.
Operating and Occupancy
R&D operating and occupancy increased 99% or approximately $1.3 million to approximately $2.5 million for the year ended 2009 as compared to $1.3 million in 2008. Due to the clinical hold on Proellex® and the uncertainty of future cash flows related to the Proellex® patent applications, the Company recorded an impairment charge of approximately $957,000 in 2009 related to these patent applications. Additionally, the Company concluded that it will no longer seek to protect the specific matter covered in certain Androxal® patent applications and recorded an impairment charge of approximately $318,000 to abandon these patent applications in 2009.
General and Administrative Expenses
G&A increased 54% to approximately $4.7 million for 2009 as compared to $3.1 million for 2008. Our primary G&A expenses for 2009 and 2008 are shown in the following table (in thousands):
|
General and Administrative
|
December 31,
2009
|
December 31,
2008
|
Variance
|
Change (%)
|
||||||||||||
|
Payroll and benefits
|
$ | 2,039 | $ | 1,478 | $ | 561 | 38 | % | ||||||||
|
Operating and occupancy
|
2,684 | 1,582 | 1,102 | 70 | % | |||||||||||
|
Total
|
$ | 4,723 | $ | 3,060 | $ | 1,663 | 54 | % | ||||||||
G&A payroll and benefit expense for both 2009 and 2008, include salaries, bonuses, non-cash stock option compensation expense and fringe benefits. Included in payroll and benefit expense is a charge for non-cash stock option expense of $799,000 for the year ended 2009 as compared to $482,000 in the year 2008. Additionally, salaries for the year ended 2009 were $949,000 as compared to $891,000 for 2008.
G&A operating and occupancy expenses, which include expenses to operate as a public company, increased 70% or approximately $1.1 million to $2.7 million in 2009 as compared to $1.6 million in 2008. The increase is primarily due to an increase in professional services.
Off-Balance Sheet Arrangements
As of December 31, 2010, we did not have any off-balance sheet arrangements except the operating lease relating to our facility.
Liquidity and Capital Resources
Since our inception, we have financed our operations primarily with proceeds from private placements and public offerings of equity securities and with funds received under collaborative agreements. We have experienced negative cash flows from operations since inception. We will require substantial funds for research and development, including preclinical studies and clinical trials of our product candidates, and to commence sales and marketing efforts if appropriate, if the FDA or other regulatory approvals are obtained. Based on our existing and projected accounts payable and commitments, we believe we will need to raise additional capital by the end of the second quarter of 2012 in order to continue operations on a normal basis. If our expenses are greater than expected or our revenues are less than expected, we may be required to raise additional funds prior to that time. We believe we can secure additional cash resources through the sale of our equity securities; however, there can be no assurance that the Company will be able to raise sufficient capital.
On February 12, 2010, we entered into the Equity Distribution Agreement with Ladenburg , pursuant to which we may issue and sell from time to time through Ladenburg, as sales agent and/or principal, shares of our common stock having an aggregate offering price of up to $10 million (the “ATM Shares”). Ladenburg is not required to sell on our behalf any specific number or dollar amount of the ATM Shares, but Ladenburg, upon acceptance of written instructions from us, agreed to use its commercially reasonable efforts consistent with its customary trading and sales practices, to sell the ATM Shares up to the amount specified, and otherwise in accordance with the terms of a placement notice delivered to Ladenburg. We have no obligation to sell any ATM Shares under the Equity Distribution Agreement, and may at any time suspend sales under the Equity Distribution Agreement, provided that such suspension shall not affect either party’s obligations with respect to the ATM Shares sold prior to the receipt of notice of such suspension. Ladenburg receives a commission of 4% of the gross sales price of all ATM Shares sold through it under the Equity Distribution Agreement. The ATM Shares are issued pursuant to our shelf registration statement on Form S-3, as amended (File No. 333-163648). Cumulative through December 31, 2010, we have sold 2,448,572 ATM Shares at a weighted average share price of $2.77, for proceeds of approximately $6.4 million, net of expenses. Pursuant to General Instruction I.B.6. of Form S-3, we may not sell more than one-third of the aggregate market value of our common stock held by non-affiliates during a period of 12 calendar months immediately prior to, and including, the date of such sale of such common stock. Due to this limitation, we announced on August 3, 2010 that we had suspended this ATM offering of Company securities. Additionally, on December 30, 2010 we announced our decision to reinstate our offering of the ATM shares. As a result, we sold 286,187 ATM Shares at a weighted average share price of $2.90 in January 2011, for proceeds of approximately $831,000, net of expenses.
-33-
On February 8, 2011, we completed an underwritten public offering of 690,000 units (including the exercise of the underwriter’s over-allotment option), consisting of an aggregate of 2,760,000 shares of our common stock, Series A Warrants to purchase 2,070,000 shares of our common stock and Series B warrants to purchase 1,690,500 shares of our common stock, at a price per unit of $17.15. Each unit consisted of four shares of our common stock, Series A Warrants exercisable for three shares of our common stock at an exercise price of $0.01 per share and Series B Warrants exercisable for 2.45 shares of our common stock at an exercise price of $2.49 per share. Gross proceeds to us, after the underwriting discount and before offering expenses, were approximately $11 million.
Our primary use of cash to date has been in operating activities to fund research and development, including preclinical studies and clinical trials, and general and administrative expenses. We had cash and cash equivalents of approximately $3.0 million as of December 31, 2010 as compared to $1.9 million as of December 31, 2009. Additionally, we had accounts payable and accrued expenses of $1.3 million as of December 31, 2010 as compared to $2.4 million as of December 31, 2009.
Net cash of approximately $5.0 million, $22.1 million and $21.7 million was used in operating activities during 2010, 2009 and 2008, respectively. The major use of cash for operating activities during 2010 was to fund our clinical development programs and associated administrative costs and pay down our accounts payable and accrued expenses. Cash used in investing activities was $371,000 and $502,000 during 2010 and 2009, respectively, and cash provided by investing activities was $23.6 million in 2008. The major use of cash for investing activities during 2010 was primarily for capitalized patent and patent application costs for Androxal®. Cash provided by financing activities was $6.4 million, $5.0 million and $15.9 million during 2010, 2009 and 2008, respectively. Cash provided by financing activities during 2010 was due to the 2,448,572 ATM Shares sold at a weighted average share price of $2.77.
Our capital requirements will depend on many factors, including: the costs and timing of seeking regulatory approvals of our products; the problems, delays, expenses and complications frequently encountered by development stage companies; the progress of our preclinical and clinical activities; the costs associated with any future collaborative research, manufacturing, marketing or other funding arrangements; our ability to obtain regulatory approvals; the success of our potential future sales and marketing programs; the cost of filing, prosecuting and defending and enforcing any patent claims and other intellectual property rights; changes in economic, regulatory or competitive conditions of our planned business; and additional costs associated with being a publicly-traded company. To satisfy our capital requirements, we are exploring ways to raise additional funds by the end of the second quarter of 2012. There can be no assurance that any such funding will be available to us on favorable terms or at all. If we are successful in obtaining additional financing, we anticipate that such financing will result in significant dilution of the ownership interests of our current stockholders and may provide certain rights to the new investors senior to the rights of our current stockholders, including but not limited to voting rights and rights to proceeds in the event of a sale or liquidation of the Company. The uncertainties relating to the foregoing and other matters raise substantial doubt about our ability to continue as a going concern.
Contractual Obligations and Commercial Commitments
The Company leases laboratory and office space, and equipment pursuant to leases accounted for as operating leases. The lease for the Company’s laboratory and office space expires in June 2015. Rental expense for the years ended December 31, 2010, 2009 and 2008, was approximately $63,000, $60,000 and $59,000, respectively. Future minimum lease payments under non-cancelable leases with original terms in excess of one year as of December 31, 2010, are as follows (in thousands):
|
2011
|
$ | 49 | ||
|
2012
|
50 | |||
|
2013
|
52 | |||
|
2014
|
53 | |||
|
2015
|
27 | |||
|
Total
|
$ | 231 |
ITEM 7A.QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate Risk. We had cash and cash equivalents of approximately $3.0 million as of December 31, 2010 which is primarily held in a money market mutual fund backed by U.S. government securities. Although this cash account is subject to fluctuations in interest rates and market conditions, no significant gain or loss on this account is expected to be recognized in earnings. We do not invest in derivative securities.
-34-
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The financial statements required by this item are set forth in Item 15 of this Report.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
Not applicable.
ITEM 9A.CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our reports filed with the Securities and Exchange Commission, or SEC, pursuant to the Securities Exchange Act of 1934, or the Exchange Act, is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the Commission and that such information is accumulated and communicated to our management, including our Chief Executive Officer (CEO) and Principal Financial Officer (PFO), as appropriate, to allow timely decisions regarding required disclosures.
Management, with the participation of our CEO and PFO, has evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) of the Exchange Act) as of the end of the period covered by this report. Based on such evaluation, our CEO and PFO have each concluded that as of the end of such period, our disclosure controls and procedures were effective to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and that such information is accumulated and communicated to our management, including the CEO and PFO, as appropriate, to allow timely decisions regarding required disclosures.
Management’s Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) under the Exchange Act. Our internal control over financial reporting was designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Management evaluated the effectiveness of internal control over financial reporting based on the criteria in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. Due to its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Based on management’s evaluation, management has concluded that internal control over financial reporting was effective as of December 31, 2010.
Changes in Internal Control
There have been no changes in our internal control over financial reporting during our quarter ended December 31, 2010 that have materially affected, or is reasonable likely to materially affect, our internal control over financial reporting.
|
ITEM9B.
|
OTHER INFORMATION
|
Not applicable.
-35-
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this item is hereby incorporated by reference from the information in our proxy statement for our 2011 annual meeting of stockholders. Such proxy statement will be filed with the SEC pursuant to the Exchange Act within 120 days of the end of our fiscal year ended December 31, 2010.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this item is hereby incorporated by reference from the information in our proxy statement for our 2011 annual meeting of stockholders. Such proxy statement will be filed with the SEC pursuant to the Exchange Act within 120 days of the end of our fiscal year ended December 31, 2010.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this item is hereby incorporated by reference from the information in our proxy statement for our 2011 annual meeting of stockholders. Such proxy statement will be filed with the SEC pursuant to the Exchange Act within 120 days of the end of our fiscal year ended December 31, 2010.
ITEM 13. CERTAIN RELATIONSHIP AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this item is hereby incorporated by reference from the information in our proxy statement for our 2011 annual meeting of stockholders. Such proxy statement will be filed with the SEC pursuant to the Exchange Act within 120 days of the end of our fiscal year ended December 31, 2010.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this item is hereby incorporated by reference from the information in our proxy statement for our 2011 annual meeting of stockholders. Such proxy statement will be filed with the SEC pursuant to the Exchange Act within 120 days of the end of our fiscal year ended December 31, 2010.
-36-
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a) Documents Filed as a Part of this Report.
|
Financial Statements
|
Page
|
|
|
Report of Independent Registered Public Accounting Firm
|
F-1
|
|
|
Reports of Independent Public Accountants
|
F-2
|
|
|
Consolidated Balance Sheets as of December 31, 2010 and 2009
|
F-8
|
|
|
Consolidated Statements of Operations for the Years Ended
December 31, 2010, 2009 and 2008 and (unaudited)
from Inception (August 20, 1987) through December 31, 2010
|
F-9
|
|
|
Consolidated Statement of Stockholders' Equity (from inception)
|
F-10
|
|
|
Consolidated Statements of Cash Flows for the Years Ended
December 31, 2010, 2009 and 2008 and (unaudited) from Inception
(August 20, 1987) through December 31, 2010
|
F-16
|
|
|
Notes to Consolidated Financial Statements
|
F-17
|
|
All financial statement schedules are omitted because they are not applicable, not required, or because the required information is included in the financial statements or the notes thereto.
(b) Exhibits.
Exhibits to the Form 10-K have been included only with the copies of the Annual Report on Form 10-K filed with the Securities and Exchange Commission. Upon request to the Company and payment of a reasonable fee, copies of the individual exhibits will be furnished.
|
Exhibit Number
|
Identification Of Exhibit
|
|
|
3.1(a)
|
Restated Certificate of Incorporation. Exhibit 3.3 to the Company's Registration Statement on Form SB-2 (No. 33-57728-FW), as amended ("Registration Statement"), is incorporated herein by reference.
|
|
|
3.1(b)
|
Certificate of Amendment to the Company's Restated Certificate of Incorporation, dated as of May 2, 2006. Exhibit 3.1 to the Company's Current Report on Form 8-K as filed with the Commission on May 2, 2006 is incorporated herein by reference.
|
|
|
3.1(c)
|
Certificate of Designation of Series One Junior Participating Preferred Stock dated September 2, 1999. Exhibit A to Exhibit 4.1 to the Company's Registration Statement on Form 8-A as filed with the Commission on September 3, 1999 (the "Rights Plan Registration Statement"), is incorporated herein by reference.
|
|
|
3.1(d)
|
Certificate of Amendment to Restated Certificate of Incorporation, dated as of December 16, 2008. Exhibit 3.1(d) to the Company’s Current Report on Form 8-K as filed with the Commission on December 23, 2008 is incorporated herein by reference.
|
|
|
3.1(e)
|
Certificate of Amendment to Restated Certificate of Incorporation, dated as of November 18, 2009. Exhibit 3.1(e) to the Company’s Current Report on Form 8-K dated November 19, 2009 is incorporated herein by reference.
|
|
|
3.1(f)
|
Certificate of Amendment to Restated Certificate of Incorporation, dated October 14, 2010. Exhibit 3.1(f) to the Company’s Current Report on Form 8-K dated October 14, 2010 is incorporated herein by reference.
|
|
|
3.2
|
Restated Bylaws of the Company. Exhibit 3.4 to the Registration Statement is incorporated herein by reference.
|
|
|
4.1
|
Specimen Certificate of Common Stock, $.001 par value, of the Company. Exhibit 4.1 to the Registration Statement is incorporated herein by reference.
|
|
|
4.2
|
Rights Agreement dated September 1, 1999 between the Company and Computershare Investor Services LLC (as successor in interest to Harris Trust & Savings Bank), as Rights Agent. Exhibit 4.1 to the Rights Plan Registration Statement is incorporated herein by reference.
|
|
|
4.3
|
First Amendment to Rights Agreement, dated as of September 6, 2002, between the Company, Harris Trust & Savings Bank and Computershare Investor Services LLC. Exhibit 4.3 to Amendment No. 1 to the Rights Plan Registration Statement on Form 8-A/A as filed with the Commission on September 11, 2002 is incorporated herein by reference.
|
-37-
|
4.4
|
Second Amendment to Rights Agreement, dated as of October 30, 2002, between the Company and Computershare Investor Services LLC. Exhibit 4.4 to Amendment No. 2 to the Rights Plan Registration Statement on Form 8-A/A as filed with the Commission on October 31, 2002 is incorporated herein by reference.
|
|
|
4.5
|
Third Amendment to Rights Agreement, dated as of June 30, 2005, between the Company and Computershare Trust Company, Inc. (as successor in interest to Computershare Investor Services, LLC). Exhibit 4.4 to the Company's Current Report on Form 8-K as filed with the Commission on June 30, 2005 is incorporated herein by reference.
|
|
|
4.6
|
Fourth Amendment to Rights Agreement, dated as of January 9, 2008, between the Company and Computershare Trust Company, Inc. (as successor in interest to Computershare Investor Services, LLC). Exhibit 4.5 to the Company's Current Report on Form 8-K as filed with the Commission on January 10, 2008 is incorporated herein by reference.
|
|
|
4.7
|
Fifth Amendment to Rights Agreement, dated as of October 10, 2008, between the Company and Computershare Trust Company, Inc. (as successor in interest to Computershare Investor Services, LLC). Exhibit 4.6 to the Company’s Current Report on Form 8-K as filed with the Commission on January 10, 2008 is incorporated herein by reference.
|
|
|
4.8
|
Sixth Amendment to Rights Agreement, dated as of September 9, 2010, between the Company and Computershare Trust Company, Inc. (as successor in interest to Computershare Investor Services, LLC). Exhibit 4.7 to the Company’s Current Report on Form 8-K as filed with the Commission on September 10, 2010 is incorporated herein by reference.
|
|
|
4.9
|
Form of Rights Certificate. Exhibit B to Exhibit 4.1 to the Rights Plan Registration Statement is incorporated herein by reference.
|
|
|
4.10
|
Form of Series A Warrant Certificate. Exhibit 4.10 to the Company’s Registration Statement on Form S-1/A (No. 333-171196) as filed with the Commission on February 2, 2011 is incorporated herein by reference.
|
|
|
4.11
|
Form of Series B Warrant Certificate. Exhibit 4.11 to the Company’s Registration Statement on Form S-1/A (No. 333-171196) as filed with the Commission on February 2, 2011 is incorporated herein by reference
|
|
|
4.12
|
Series A Warrant Agreement dated February 8, 2011 by and among the Company and Computershare Inc. and its wholly-owned subsidiary, Computershare Trust Company, N.A. Exhibit 4.1 to the Company’s Current Report on Form 8-K as filed with the Commission on February 9, 2011 is incorporated herein by reference.
|
|
|
4.13
|
Series B Warrant Agreement dated February 8, 2011 by and among the Company and Computershare Inc. and its wholly-owned subsidiary, Computershare Trust Company, N.A. Exhibit 4.2 to the Company’s Current Report on Form 8-K as filed with the Commission on February 9, 2011 is incorporated herein by reference.
|
|
|
10.1+
|
Amended and Restated 1993 Employee and Consultant Stock Option Plan. Exhibit 10.3 to the Registration Statement is incorporated herein by reference.
|
|
|
10.2+
|
First Amendment to the Repros Therapeutics Inc. Amended and Restated 1993 Stock Option Plan. Exhibit 10.22 to the Company's Annual Report on Form 10-K for the year ended December 31, 1999 is incorporated herein by reference.
|
|
|
10.3+
|
1994 Employee and Consultant Stock Option Plan. Exhibit 4.2 to the Company's Registration Statement on Form S-8 (File No. 033-83406) as filed with the Commission on August 29, 1994 is incorporated herein by reference.
|
|
|
10.4+
|
2000 Non-Employee Directors' Stock Option Plan. Appendix B to the Company's Definitive Proxy Statement filed on April 26, 2000 is incorporated herein by reference.
|
|
|
10.5+
|
First Amendment to the Repros Therapeutics Inc. 2000 Non-Employee Directors' Stock Option Plan. Exhibit 10.21 to the 2000 Form 10-K is incorporated herein by reference.
|
|
|
10.6+
|
Second Amendment to 2000 Non-Employee Directors' Stock Option Plan. Exhibit 10.6 to the Company's Annual Report on Form 10-K for the year ended December 31, 2002 (the "2002 Form 10-K") is incorporated herein by reference.
|
-38-
|
10.7+
|
Repros Therapeutics Inc. 2004 Stock Option Plan. Exhibit 10.17 to the Company's Registration Statement on Form S-1 (No. 333-119861), as amended, is incorporated herein by reference.
|
|
|
10.8+
|
Employment Agreement between the Company and Joseph S. Podolski. Exhibit 10.5 to the Registration Statement is incorporated herein by reference.
|
|
|
10.9+
|
First Amendment to Employment Agreement between the Company and Joseph S. Podolski. Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2001 is incorporated herein by reference.
|
|
|
10.10+
|
Second Amendment to Employment Agreement between the Company and Joseph S. Podolski. Exhibit 10.17 to the 2002 Form 10-K is incorporated herein by reference.
|
|
|
10.11+
|
Third Amendment to Employment Agreement dated effective March 11, 2009, between the Company and Joseph S. Podolski. Exhibit 10.1 to the Company’s Current Report on Form 8-K as filed with the Commission on March 17, 2009 is incorporated herein by reference.
|
|
|
10.12+
|
Fourth Amendment to Employment Agreement effective March 10, 2010 between the Company and Joseph S. Podolski. Exhibit 10.1 to the Company’s Current Report on Form 8-K as filed with the Commission on March 11, 2010 is incorporated herein by reference.
|
|
|
10.13+
|
Consulting Agreement dated October 29, 2009 by and between the Company and Katherine Anderson. Exhibit 10.2 to the Company’s Current Report on Form 8-K as filed with the Commission on November 3, 2009 is incorporated herein by reference.
|
|
|
10.14
|
Lease Agreement dated May 11, 2004 between the Company and Sealy Woodlands, L.P. Exhibit 10.14 to the Company's Annual Report on Form 10-K for the year ended December 31, 2004 is incorporated herein by reference.
|
|
|
10.15
|
Amendment to Lease Agreement between the Company and Sealy Woodlands, L.P., dated May 17, 2006. Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2006 is incorporated herein by reference.
|
|
|
10.16
|
Second Amendment to Lease, effective as of July 1, 2010, between the Company and Columbia Texas 2408 Timberloch Industrial, L.P. Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the fiscal quarter ended September 30, 2010 is incorporated herein by reference.
|
|
|
10.17++
|
Letter Agreement dated July 15, 2002 between the Company, Schering Plough Ltd. and Schering-Plough Corporation. Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the fiscal quarter ended June 30, 2002 is incorporated herein by reference.
|
|
|
10.18++
|
PHS Patent License Agreement dated April 16, 1999 between the Company and certain agencies of the United States Public Health Service within the Department of Health and Human Services, with amendments. Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q for the fiscal quarter ended September 30, 2003 is incorporated herein by reference.
|
|
|
10.19
|
Waiver to PHS Patent License Agreement, as amended, dated March 8, 2007 between the Company and certain agencies of the United States Public Health Service within the Department of Health and Human Services. Exhibit 10.2 to the Company’s Current Report on Form 8-K as filed with the Commission on March 19, 2007 is incorporated herein by reference.
|
|
|
10.20++
|
Sixth Amendment to PHS Patent License Agreement, as amended, dated July 7, 2009 between the Company and certain agencies of the United States Public Health Service within the Department of Health and Human Services. Exhibit 10.1 to the Company’s Current Report on Form 8-K/A as filed with the Commission on December 22, 2009 is incorporated herein by reference.
|
|
|
10.21++
|
Seventh Amendment to PHS Patent License Agreement, as amended, dated October 28, 2009 between the Company and certain agencies of the United States Public Health Service within the Department of Health and Human Services. Exhibit 10.21 to the Company’s Annual Report on Form 10-K as filed with the Commission on March 15, 2010 is incorporated herein by reference.
|
-39-
|
10.22
|
Master Settlement Agreement and Releases dated October 29, 2009 by and among the Company and its creditors signatory thereto. Exhibit 10.1 to the Company’s Current Report on Form 8-K as filed with the Commission on November 3, 2009 is incorporated herein by reference.
|
|
|
10.23
|
Securities Purchase Agreement dated October 7, 2009, among the Company and the purchasers identified on the signature pages thereto. Exhibit 10.1 to the Company’s Current Report on Form 8-K as filed with the Commission on October 14, 2009 is incorporated herein by reference.
|
|
|
10.24
|
Securities Purchase Agreement between the Company and Enable Growth Partners LP dated September 8, 2009. Exhibit 10.1 to the Company’s Current Report on Form 8-K as filed with the Commission on September 10, 2009 is incorporated herein by reference.
|
|
|
10.25
|
Form of Indemnification Agreement entered into between the Company and each of its directors. Exhibit 10.1 to the Company’s Current Report on Form 8-K as filed with the Commission on May 20, 2009 is incorporated herein by reference.
|
|
|
10.26
|
Equity Distribution Agreement dated February 12, 2010 between the Company and Ladenburg Thalmann & Co. Inc. Exhibit 10.1 to the Company’s Current Report on Form 8-K as filed with the Commission on February 19, 2010 is incorporated herein by reference.
|
|
|
23.1*
|
Consent of PricewaterhouseCoopers LLP
|
|
|
31.1*
|
Certification Pursuant to Rule 13(a)-14(a) or 15(d)-14(a) of the Exchange Act, As Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 (Chief Executive Officer)
|
|
|
31.2*
|
Certification Pursuant to Rule 13(a)-14(a) or 15(d)-14(a) of the Exchange Act, As Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 (Principal Financial Officer)
|
|
|
32.1*
|
Certification Furnished Pursuant to 18 U.S.C. Section 1350, As Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (Chief Executive Officer)
|
|
|
32.2*
|
Certification Furnished Pursuant to 18 U.S.C. Section 1350, As Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (Principal Financial Officer)
|
_________________
|
*
|
Filed herewith.
|
|
+
|
Management contract or compensatory plan.
|
|
++
|
Portions of this exhibit have been omitted based on a request for confidential treatment pursuant to Rule 24b-2 of the Exchange Act. Such omitted portions have been filed separately with the Commission.
|
-40-
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
REPROS THERAPEUTICS INC.
|
||
|
By:
|
/s/ Joseph S. Podolski
|
|
|
Joseph S. Podolski
|
||
|
President and Chief Executive Officer
|
||
Dated: March 28, 2011
|
Signature
|
Title
|
Date
|
||
|
/s/ Joseph S. Podolski
|
President, Chief Executive Officer and Director
|
March 28, 2011
|
||
|
Joseph S. Podolski
|
(Principal Executive Officer)
|
|||
|
/s/ Katherine A. Anderson
|
Chief Accounting Officer and Secretary
|
March 28, 2011
|
||
|
Katherine A. Anderson
|
(Principal Financial Officer and
|
|||
|
Principal Accounting Officer)
|
||||
|
/s/ Nola Masterson
|
Chair of the Board
|
March 28, 2011
|
||
|
Nola Masterson
|
||||
|
/s/ Daniel F. Cain
|
Director
|
March 28, 2011
|
||
|
Daniel F. Cain
|
||||
|
/s/ Jean L. Fourcroy, M.D., Ph.D., M.P.H.
|
Director
|
March 28, 2011
|
||
|
Jean L. Fourcroy, M.D., Ph.D., M.P.H.
|
||||
|
/s/ Jaye Thompson, Ph.D.
|
Director
|
March 28, 2011
|
||
|
Jaye Thompson, Ph.D.
|
-41-
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Repros Therapeutics, Inc.:
In our opinion, the accompanying consolidated balance sheets as of December 31, 2010 and 2009, the related consolidated statements of operations and cash flows for each of the three years in the period ended December 31, 2010, and the statements of stockholders' equity for each of the nine years in the period ended December 31, 2010 present fairly, in all material respects, the financial position of Repros Therapeutics, Inc. and subsidiary (collectively, the "Company"), a development stage company, at December 31, 2010 and 2009, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2010, and cumulatively for the period January 1, 2002 through December 31, 2010 (not separately presented) in conformity with accounting principles generally accepted in the United States of America. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits. We did not audit the cumulative totals of the Company for the period from August 20, 1987 (date of inception) to December 31, 2001, which totals reflect a deficit of $75.8 million accumulated during the development stage. The cumulative totals for the period January 1, 1994 to December 31, 2001 were audited by other auditors who have ceased operations. Those auditors expressed unqualified opinions on the consolidated financial statements for the three years in the period ended December 31, 2001, the three years in the period ended December 31, 2000, the three years in the period ended December 31, 1999, the three years in the period ended December 31, 1998, the three years in the period ended December 31, 1997, and the three years in the period ended December 31, 1996 dated February 6, 2002, February 2, 2001, February 2, 2000, January 26, 1999, March 24, 1998, and March 11, 1997, respectively. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company is a development stage company, has an accumulated deficit, projects it will need to raise additional capital and there can be no assurance that the Company will be able to raise sufficient capital. These and other matters raise substantial doubt about the Company's ability to continue as a going concern. Management's plans in regard to this matter are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ PricewaterhouseCoopers LLP
Houston, Texas
March 28, 2011
F-1
THE FOLLOWING REPORT IS A COPY OF A REPORT PREVIOUSLY ISSUED BY AR THUR ANDERSEN LLP AND HAS NOT BEEN REISSUED BY ARTHUR ANDERSEN LLP.
REPORT OF INDEPENDENT PUBLIC ACCOUNTANTS
To Zonagen, Inc.:
We have audited the accompanying consolidated balance sheets of Zonagen, Inc. (a Delaware corporation in the development stage), and subsidiary (collectively, "the Company") as of December 31, 2001 and 2000, and the related consolidated statements of operations, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2001. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with auditing standards generally accepted in the United States. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Zonagen, Inc., and subsidiary as of December 31, 2001 and 2000, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2001, in conformity with accounting principles generally accepted in the United States.
As explained in Note 2 to the consolidated financial statements, effective January 1, 2000, the Company changed its method of accounting for revenue recognition.
/S/ ARTHUR ANDERSEN LLP
Houston, Texas
February 6, 2002
F-2
THE FOLLOWING REPORT IS A COPY OF A REPORT PREVIOUSLY ISSUED BY AR THUR ANDERSEN LLP AND HAS NOT BEEN REISSUED BY ARTHUR ANDERSEN LLP.
REPORT OF INDEPENDENT PUBLIC ACCOUNTANTS
To Zonagen, Inc.:
We have audited the accompanying consolidated balance sheets of Zonagen, Inc. (a Delaware corporation in the development stage), and subsidiary (collectively, "the Company") as of December 31, 2000 and 1999, and the related consolidated statements of operations, stockholders' equity and cash flows for each of the three years in the period ended December 31, 2000. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with auditing standards generally accepted in the United States. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Zonagen, Inc., and subsidiary as of December 31, 2000 and 1999, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2000, in conformity with accounting principles generally accepted in the United States.
As explained in Note 2 to the consolidated financial statements, effective January 1, 2000, the Company changed its method of accounting for revenue recognition.
/S/ ARTHUR ANDERSEN LLP
Houston, Texas
February 2, 2001
F-3
THE FOLLOWING REPORT IS A COPY OF A REPORT PREVIOUSLY ISSUED BY AR THUR ANDERSEN LLP AND HAS NOT BEEN REISSUED BY ARTHUR ANDERSEN LLP.
REPORT OF INDEPENDENT PUBLIC ACCOUNTANTS
To Zonagen, Inc.:
We have audited the accompanying consolidated balance sheets of Zonagen, Inc. (a Delaware corporation in the development stage), and subsidiary (collectively, "the Company") as of December 31, 1999 and 1998, and the related consolidated statements of operations, stockholders' equity and cash flows for each of the three years in the period ended December 31, 1999. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with auditing standards generally accepted in the United States. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Zonagen, Inc., and subsidiary as of December 31, 1999 and 1998, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 1999, in conformity with accounting principles generally accepted in the United States.
/S/ ARTHUR ANDERSEN LLP
Houston, Texas
February 2, 2000
F-4
THE FOLLOWING REPORT IS A COPY OF A REPORT PREVIOUSLY ISSUED BY AR THUR ANDERSEN LLP AND HAS NOT BEEN REISSUED BY ARTHUR ANDERSEN LLP.
REPORT OF INDEPENDENT PUBLIC ACCOUNTANTS
To Zonagen, Inc.:
We have audited the accompanying balance sheets of Zonagen, Inc. (a Delaware corporation in the development stage), and subsidiary (collectively, "the Company") as of December 31, 1998 and 1997, and the related statements of operations, stockholders' equity and cash flows for each of the three years in the period ended December 31, 1998. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with generally accepted auditing standards. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Zonagen, Inc., and subsidiary as of December 31, 1998 and 1997, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 1998, in conformity with generally accepted accounting principles.
/S/ ARTHUR ANDERSEN LLP
Houston, Texas
January 26, 1999
F-5
THE FOLLOWING REPORT IS A COPY OF A REPORT PREVIOUSLY ISSUED BY AR THUR ANDERSEN LLP AND HAS NOT BEEN REISSUED BY ARTHUR ANDERSEN LLP.
REPORT OF INDEPENDENT PUBLIC ACCOUNTANTS
To Zonagen, Inc.:
We have audited the accompanying balance sheets of Zonagen, Inc. (a Delaware corporation in the development stage), and subsidiary (collectively, "the Company") as of December 31, 1997 and 1996, and the related statements of operations, stockholders' equity and cash flows for each of the three years in the period ended December 31, 1997. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with generally accepted auditing standards. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Zonagen, Inc., and subsidiary as of December 31, 1997 and 1996, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 1997, in conformity with generally accepted accounting principles.
/S/ ARTHUR ANDERSEN LLP
Houston, Texas
March 24, 1998
F-6
THE FOLLOWING REPORT IS A COPY OF A REPORT PREVIOUSLY ISSUED BY AR THUR ANDERSEN LLP AND HAS NOT BEEN REISSUED BY ARTHUR ANDERSEN LLP.
REPORT OF INDEPENDENT PUBLIC ACCOUNTANTS
To Zonagen, Inc.:
We have audited the accompanying consolidated balance sheets of Zonagen, Inc. (a Delaware corporation in the development stage), and subsidiary as of December 31, 1996 and 1995, and the related consolidated statements of operations, stockholders' equity and cash flows for each of the three years in the period ended December 31, 1996. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with generally accepted audited standards. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
As discussed in Note 1 to the consolidated financial statements, the Company has operated as a development stage enterprise since its inception by devoting substantially all of its efforts to raising capital and performing research and development. In order to complete the research and development and other activities necessary to commercialize its products, additional financing will be required. Management's current projections indicate that the Company can conserve its cash resources to maintain the Company's operations through 1997. Management's plans in regard to those matters are also described in Note 1.
In our opinion, based on our audits, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Zonagen, Inc., and subsidiary as of December 31, 1996 and 1995, and the results of their operations and cash flows for each of the three years in the period ended December 31, 1996, in conformity with generally accepted accounting principles.
/S/ ARTHUR ANDERSEN LLP
Houston, Texas
March 11, 1997
F-7
REPROS THERAPEUTICS INC. AND SUBSIDIARY
(A development stage company)
CONSOLIDATED BALANCE SHEETS
(in thousands except share and per share amounts)
|
December 31,
|
December 31,
|
|||||||
|
2010
|
2009
|
|||||||
|
ASSETS
|
||||||||
|
Current Assets
|
||||||||
|
Cash and cash equivalents
|
$ | 2,957 | $ | 1,886 | ||||
|
Prepaid expenses and other current assets
|
328 | 177 | ||||||
|
Total current assets
|
3,285 | 2,063 | ||||||
|
Fixed Assets, net
|
7 | 12 | ||||||
|
Other Assets, net
|
1,173 | 885 | ||||||
|
Total assets
|
$ | 4,465 | $ | 2,960 | ||||
|
LIABILITIES AND STOCKHOLDERS' EQUITY
|
||||||||
|
Current Liabilities
|
||||||||
|
Accounts payable
|
$ | 1,151 | $ | 2,043 | ||||
|
Accrued expenses
|
147 | 355 | ||||||
|
Total current liabilities
|
1,298 | 2,398 | ||||||
|
Commitments and Contingencies (note 10)
|
||||||||
|
Stockholders' Equity
|
||||||||
|
Preferred Stock, $.001 par value, 5,000,000 shares authorized, none issued and outstanding
|
- | - | ||||||
|
Common Stock, $.001 par value, 75,000,000 shares authorized, 9,042,372 and 6,496,999 shares issued, respectively; 8,930,022 and 6,384,649 shares outstanding, respectively
|
9 | 6 | ||||||
|
Additional paid-in capital
|
183,782 | 176,412 | ||||||
|
Cost of treasury stock, 112,350 shares
|
(1,380 | ) | (1,380 | ) | ||||
|
Deficit accumulated during the development stage
|
(179,244 | ) | (174,476 | ) | ||||
|
Total stockholders' equity
|
3,167 | 562 | ||||||
|
Total liabilities and stockholders' equity
|
$ | 4,465 | $ | 2,960 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-8
REPROS THERAPEUTICS INC. AND SUBSIDIARY
(A development stage company)
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands except per share amounts)
|
From Inception
|
||||||||||||||||
|
(August 20, 1987)
|
||||||||||||||||
|
through
|
||||||||||||||||
|
For the Year Ended December 31,
|
December 31,
|
|||||||||||||||
|
2010
|
2009
|
2008
|
2010
|
|||||||||||||
|
(unaudited)
|
||||||||||||||||
|
Revenues and other income
|
||||||||||||||||
|
Licensing fees
|
$ | - | $ | - | $ | - | $ | 28,755 | ||||||||
|
Product royalties
|
- | - | - | 627 | ||||||||||||
|
Research and development grants
|
- | - | - | 1,219 | ||||||||||||
|
Interest income
|
- | 4 | 433 | 16,297 | ||||||||||||
|
Gain on disposal of fixed assets
|
- | - | - | 102 | ||||||||||||
|
Other income
|
421 | 547 | - | 1,003 | ||||||||||||
|
Total revenues and other income
|
421 | 551 | 433 | 48,003 | ||||||||||||
|
Expenses
|
||||||||||||||||
|
Research and development
|
2,904 | 23,062 | 22,575 | 173,234 | ||||||||||||
|
General and administrative
|
2,285 | 4,723 | 3,060 | 44,282 | ||||||||||||
|
Other expense
|
- | - | - | 388 | ||||||||||||
|
Total expenses
|
5,189 | 27,785 | 25,635 | 217,904 | ||||||||||||
|
Loss from continuing operations
|
(4,768 | ) | (27,234 | ) | (25,202 | ) | (169,901 | ) | ||||||||
|
Loss from discontinued operations
|
- | - | - | (1,828 | ) | |||||||||||
|
Gain on disposal of discontinued operations
|
- | - | - | 939 | ||||||||||||
|
Net loss before cumulative effect of changes in accounting principles
|
(4,768 | ) | (27,234 | ) | (25,202 | ) | (170,790 | ) | ||||||||
|
Cumulative effect of changes in accounting principles
|
- | - | - | (8,454 | ) | |||||||||||
|
Net loss
|
$ | (4,768 | ) | $ | (27,234 | ) | $ | (25,202 | ) | $ | (179,244 | ) | ||||
|
Loss per share - basic and diluted
|
$ | (0.59 | ) | $ | (6.28 | ) | $ | (7.54 | ) | |||||||
|
Shares used in loss per share calculation:
|
||||||||||||||||
|
Basic
|
8,057 | 4,336 | 3,343 | |||||||||||||
|
Diluted
|
8,057 | 4,336 | 3,343 | |||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-9
(A development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(in thousands except share and per share amounts)
|
Deficit
|
||||||||||||||||||||||||||||||||||||||||
|
Accumulated
|
||||||||||||||||||||||||||||||||||||||||
|
Additional
|
During the
|
Total
|
||||||||||||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Deferred
|
Treasury Stock
|
Development
|
Stockholders'
|
||||||||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Compensation
|
Shares
|
Amount
|
Stage
|
Equity
|
|||||||||||||||||||||||||||||||
|
Exchange of common stock ($.016 per share) for technology rights and services from founding stockholders
|
- | $ | - | 61,342 | $ | - | $ | 1 | $ | - | - | $ | - | $ | - | $ | 1 | |||||||||||||||||||||||
|
Net Loss
|
- | - | - | - | - | - | - | - | (28 | ) | (28 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 1987 (unaudited)
|
- | - | 61,342 | - | 1 | - | - | - | (28 | ) | (27 | ) | ||||||||||||||||||||||||||||
|
Net Loss
|
- | - | - | - | - | - | - | - | (327 | ) | (327 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 1988 (unaudited)
|
- | - | 61,342 | - | 1 | - | - | - | (355 | ) | (354 | ) | ||||||||||||||||||||||||||||
|
Proceeds from issuance of common stock
|
- | - | 16,358 | - | 3 | - | - | - | - | 3 | ||||||||||||||||||||||||||||||
|
Net Loss
|
- | - | - | - | - | - | - | - | (967 | ) | (967 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 1989 (unaudited)
|
- | - | 77,700 | - | 4 | - | - | - | (1,322 | ) | (1,318 | ) | ||||||||||||||||||||||||||||
|
Proceeds from issuance of common stock
|
- | - | 117 | - | - | - | - | - | - | - | ||||||||||||||||||||||||||||||
|
Net Loss
|
- | - | - | - | - | - | - | - | (1,426 | ) | (1,426 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 1990 (unaudited)
|
- | - | 77,816 | - | 4 | - | - | - | (2,748 | ) | (2,744 | ) | ||||||||||||||||||||||||||||
|
Net Loss
|
- | - | - | - | - | - | - | - | (1,820 | ) | (1,820 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 1991 (unaudited)
|
- | - | 77,816 | - | 4 | - | - | - | (4,568 | ) | (4,564 | ) | ||||||||||||||||||||||||||||
|
Conversion of 391,305 shares of Series C preferred stock into common stock
|
- | - | 22,861 | - | 360 | - | - | - | - | 360 | ||||||||||||||||||||||||||||||
|
Purchase of retirement of common stock
|
- | - | (5,889 | ) | - | (1 | ) | - | - | - | - | (1 | ) | |||||||||||||||||||||||||||
|
Proceeds from issuance of common stock
|
- | - | 4,236 | - | 7 | - | - | - | - | 7 | ||||||||||||||||||||||||||||||
|
Net Loss
|
- | - | - | - | - | - | - | - | (1,583 | ) | (1,583 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 1992 (unaudited)
|
- | - | 99,024 | - | 370 | - | - | - | (6,151 | ) | (5,781 | ) | ||||||||||||||||||||||||||||
|
Issuance of common stock for cash, April 1, 1993, and May 12, 1993 ($22.00 per share), net of offering costs of $1,403
|
- | - | 383,749 | - | 7,039 | - | - | - | - | 7,039 | ||||||||||||||||||||||||||||||
|
Issuance of common stock for cash and license agreement, December 9, 1993 ($41.68 per share), net of offering costs of $47
|
- | - | 59,983 | - | 2,453 | - | - | - | - | 2,453 | ||||||||||||||||||||||||||||||
|
Conversion of Series A preferred stock to common stock
|
- | - | 44,984 | - | 600 | - | - | - | - | 600 | ||||||||||||||||||||||||||||||
|
Conversion of Series B preferred stock to common stock
|
- | - | 24,003 | - | 378 | - | - | - | - | 378 | ||||||||||||||||||||||||||||||
|
Conversion of Series C preferred stock to common stock
|
- | - | 219,078 | - | 3,444 | - | - | - | - | 3,444 | ||||||||||||||||||||||||||||||
|
Conversion of Series D preferred stock to common stock
|
- | - | 70,062 | - | 600 | - | - | - | - | 600 | ||||||||||||||||||||||||||||||
|
Conversion of bridge loan to common stock
|
- | - | 16,000 | - | 256 | - | - | - | - | 256 | ||||||||||||||||||||||||||||||
|
Net Loss
|
- | - | - | - | - | - | - | - | (2,532 | ) | (2,532 | ) | ||||||||||||||||||||||||||||
F-10
(A development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(in thousands except share and per share amounts)
|
Deficit
|
||||||||||||||||||||||||||||||||||||||||
|
Accumulated
|
||||||||||||||||||||||||||||||||||||||||
|
Additional
|
During the
|
Total
|
||||||||||||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Deferred
|
Treasury Stock
|
Development
|
Stockholders'
|
||||||||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Compensation
|
Shares
|
Amount
|
Stage
|
Equity
|
|||||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 1993 (unaudited)
|
- | $ | - | 916,884 | $ | - | $ | 15,140 | $ | - | - | $ | - | $ | (8,683 | ) | $ | 6,457 | ||||||||||||||||||||||
|
Deferred compensation resulting from grant of options
|
- | - | - | - | 188 | (188 | ) | - | - | - | - | |||||||||||||||||||||||||||||
|
Amortization of deferred compensation
|
- | - | - | 38 | - | - | - | 38 | ||||||||||||||||||||||||||||||||
|
Exercise of warrants to purchase common stock for cash, June 30, 1994 ($15.76 per share)
|
- | - | 9,906 | - | 156 | - | - | - | - | 156 | ||||||||||||||||||||||||||||||
|
Issuance of common stock for purchase of FTI, October 13, 1994
|
- | - | 27,778 | - | 1,567 | - | - | - | - | 1,567 | ||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (3,970 | ) | (3,970 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 1994
|
- | - | 954,567 | - | 17,051 | (150 | ) | - | - | (12,653 | ) | 4,248 | ||||||||||||||||||||||||||||
|
Amortization of deferred compensation
|
- | - | - | - | - | 37 | - | - | - | 37 | ||||||||||||||||||||||||||||||
|
Exercise of options to purchase common stock for cash, January and April 1995 ($.40 to $24.52 per share)
|
- | - | 1,136 | - | 14 | - | - | - | - | 14 | ||||||||||||||||||||||||||||||
|
Issuance of common stock for cash and a financing charge, March 9, 1995
|
- | - | 4,000 | - | 76 | - | - | - | - | 76 | ||||||||||||||||||||||||||||||
|
Issuance of Series A preferred stock for cash, October 4, 1995, and October 19, 1995 ($10.00 per share), net of offering costs of $651
|
598,850 | 1 | - | 5,336 | - | - | - | - | 5,337 | |||||||||||||||||||||||||||||||
|
Conversion of warrants to purchase common stock as a result of offering under antidilution clause,
|
||||||||||||||||||||||||||||||||||||||||
|
October 19, 1995 ($14.52 per share)
|
- | - | - | - | - | - | - | - | - | |||||||||||||||||||||||||||||||
|
Conversion of Series A preferred stock into common stock, November and December 1995
|
(94,000 | ) | - | 64,827 | - | - | - | - | - | - | - | |||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (4,287 | ) | (4,287 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 1995
|
504,850 | 1 | 1,024,530 | - | 22,477 | (113 | ) | - | - | (16,940 | ) | 5,425 | ||||||||||||||||||||||||||||
|
Deferred compensation resulting from grant of options
|
- | - | - | - | 86 | (86 | ) | - | - | - | - | |||||||||||||||||||||||||||||
|
Amortization of deferred compensation
|
- | - | - | - | - | 54 | - | - | - | 54 | ||||||||||||||||||||||||||||||
|
Exercise of warrants to purchase common stock for cash, January through December 1996 ($14.52 per share)
|
- | - | 56,944 | - | 827 | - | - | - | - | 827 | ||||||||||||||||||||||||||||||
|
Conversion of Series A preferred stock into common stock, January through November 1996
|
(507,563 | ) | (1 | ) | 349,206 | - | 1 | - | - | - | - | - | ||||||||||||||||||||||||||||
|
Issuance of options for services, January 12, 1996
|
- | - | - | 99 | - | - | - | - | 99 | |||||||||||||||||||||||||||||||
|
Exercise of options to purchase common stock for cash, February through November 1996 ($.004 to $22.00 per share)
|
- | - | 5,775 | - | 75 | - | - | - | - | 75 | ||||||||||||||||||||||||||||||
|
Issuance of common stock for agreement not to compete, April 13, 1996
|
- | - | 4,878 | - | 200 | - | - | - | - | 200 | ||||||||||||||||||||||||||||||
|
Exercise of warrants to purchase Series A preferred stock under cashless exercise provision, June 5, 1996
|
2,713 | - | - | - | - | - | - | - | - | |||||||||||||||||||||||||||||||
|
Issuance of Series B preferred stock for cash, September 30, 1996, and October 11, 1996 ($10.00 per share), net of offering costs of $2,557
|
1,692,500 | 2 | - | 14,366 | - | - | - | - | 14,368 | |||||||||||||||||||||||||||||||
|
Conversion of Series B preferred stock into common stock, November through December 1996
|
(177,594 | ) | - | 67,014 | - | - | - | - | - | - | - | |||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (9,470 | ) | (9,470 | ) | ||||||||||||||||||||||||||||
F-11
(A development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(in thousands except share and per share amounts)
|
Deficit
|
||||||||||||||||||||||||||||||||||||||||
|
Accumulated
|
||||||||||||||||||||||||||||||||||||||||
|
Additional
|
During the
|
Total
|
||||||||||||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Deferred
|
Treasury Stock
|
Development
|
Stockholders'
|
||||||||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Compensation
|
Shares
|
Amount
|
Stage
|
Equity
|
|||||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 1996
|
1,514,906 | $ | 2 | 1,508,347 | $ | - | $ | 38,131 | $ | (145 | ) | - | $ | - | $ | (26,410 | ) | $ | 11,578 | |||||||||||||||||||||
|
Deferred compensation resulting from grant of options
|
- | - | - | - | 2,110 | (2,110 | ) | - | - | - | - | |||||||||||||||||||||||||||||
|
Amortization of deferred compensation
|
- | - | - | - | - | 854 | - | - | - | 854 | ||||||||||||||||||||||||||||||
|
Exercise of options to purchase common stock for cash, January through December 1997 ($0.00 to $89.00 per share)
|
- | - | 22,739 | - | 522 | - | - | - | - | 522 | ||||||||||||||||||||||||||||||
|
Exercise of warrants to purchase common stock for cash, January through December 1997 ($14.52 and $12.28 per share)
|
- | - | 5,592 | - | 75 | - | - | - | - | 75 | ||||||||||||||||||||||||||||||
|
Issuance of common stock for a cashless exercise of Series A preferred stock warrants, February through September 1997
|
- | - | 20,324 | - | - | - | - | - | - | - | ||||||||||||||||||||||||||||||
|
Exercise of Series A preferred stock warrants to purchase common stock for cash, April 1997 ($11.00 per share)
|
- | - | 205 | - | 3 | - | - | - | - | 3 | ||||||||||||||||||||||||||||||
|
Issuance of common stock for a cashless exercise of Series B preferred stock warrants, April through November 1997
|
- | - | 22,056 | - | - | - | - | - | - | - | ||||||||||||||||||||||||||||||
|
Exercise of Series B preferred stock warrants to purchase common stock for cash, April through July 1997 ($11.00 per share)
|
- | - | 4,292 | - | 125 | - | - | - | - | 125 | ||||||||||||||||||||||||||||||
|
Issuance of common stock as final purchase price for acquisition of FTI, January 31, 1997 ($39.332 per share)
|
- | - | 76,274 | - | 1 | - | - | - | - | 1 | ||||||||||||||||||||||||||||||
|
Issuance of common stock as final debt payment on FTI acquisition, January 31, 1997 ($39.332 per share)
|
- | - | 4,961 | - | 94 | - | - | - | - | 94 | ||||||||||||||||||||||||||||||
|
Conversion of Series B preferred stock into common stock, January through October 1997
|
(1,514,906 | ) | (2 | ) | 573,816 | 1 | - | - | - | - | - | (1 | ) | |||||||||||||||||||||||||||
|
Issuance of common stock for cash, July 25, 1997 ($120.00 per share), net of offering costs of $5,439
|
- | - | 646,875 | 1 | 72,185 | - | - | - | - | 72,186 | ||||||||||||||||||||||||||||||
|
Purchase of treasury stock, December 1997
|
- | - | - | - | - | - | 15,375 | (1,287 | ) | - | (1,287 | ) | ||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (13,174 | ) | (13,174 | ) | ||||||||||||||||||||||||||||
F-12
(A development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(in thousands except share and per share amounts)
|
Deficit
|
||||||||||||||||||||||||||||||||||||||||
|
Accumulated
|
||||||||||||||||||||||||||||||||||||||||
|
Additional
|
During the
|
Total
|
||||||||||||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Deferred
|
Treasury Stock
|
Development
|
Stockholders'
|
||||||||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Compensation
|
Shares
|
Amount
|
Stage
|
Equity
|
|||||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 1997
|
- | $ | - | 2,885,481 | $ | 2 | $ | 113,246 | $ | (1,401 | ) | 15,375 | $ | (1,287 | ) | $ | (39,584 | ) | $ | 70,976 | ||||||||||||||||||||
|
Deferred compensation resulting from grant of options
|
- | - | - | - | 55 | - | - | - | - | 55 | ||||||||||||||||||||||||||||||
|
Amortization of deferred compensation
|
- | - | - | - | - | 422 | - | - | - | 422 | ||||||||||||||||||||||||||||||
|
Forfeiture of stock options, December 1998
|
- | - | - | - | (21 | ) | 21 | - | - | - | - | |||||||||||||||||||||||||||||
|
Exercise of options to purchase common stock for cash, January through October 1998 ($1.72 to $89.00 per share)
|
- | - | 15,755 | - | 344 | - | - | - | - | 344 | ||||||||||||||||||||||||||||||
|
Issuance of common stock for services, January 15, 1998
|
- | - | 1,250 | - | 103 | - | - | - | - | 103 | ||||||||||||||||||||||||||||||
|
Issuance of common stock for a cashless exercise of Series B preferred stock warrants, May through July 1998
|
- | - | 2,799 | - | - | - | - | - | - | - | ||||||||||||||||||||||||||||||
|
Purchase of treasury stock, January through September 1998 ($52.00 to $82.60 per share)
|
- | - | - | - | - | - | 88,450 | (6,197 | ) | - | (6,197 | ) | ||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (12,316 | ) | (12,316 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 1998
|
- | - | 2,905,285 | 2 | 113,727 | (958 | ) | 103,825 | (7,484 | ) | (51,900 | ) | 53,387 | |||||||||||||||||||||||||||
|
Deferred compensation resulting from grant of options
|
- | - | - | - | (229 | ) | 229 | - | - | - | - | |||||||||||||||||||||||||||||
|
Amortization of deferred compensation
|
- | - | - | - | - | 239 | - | - | - | 239 | ||||||||||||||||||||||||||||||
|
Exercise of options to purchase common stock for cash, February through September 1999 ($0.16 to $33.50 per share)
|
- | - | 7,966 | - | 72 | - | - | - | - | 72 | ||||||||||||||||||||||||||||||
|
Issuance of common stock for a cashless exercise of common stock warrants, February 1999
|
- | - | 1,194 | - | - | - | - | - | - | - | ||||||||||||||||||||||||||||||
|
Issuance of common stock for a cashless exercise of Series A preferred stock warrants, April 1999
|
- | - | 5,533 | - | - | - | - | - | - | - | ||||||||||||||||||||||||||||||
|
Issuance of common stock for a cashless exercise of Series B preferred stock warrants, March through April 1999
|
- | - | 219 | - | - | - | - | - | - | - | ||||||||||||||||||||||||||||||
|
Exercise of Series B preferred stock warrants to purchase common stock for cash, January 1999 ($11.00 per share)
|
- | - | 134 | - | 4 | - | - | - | - | 4 | ||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (11,952 | ) | (11,952 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 1999
|
- | - | 2,920,331 | 2 | 113,574 | (490 | ) | 103,825 | (7,484 | ) | (63,852 | ) | 41,750 | |||||||||||||||||||||||||||
|
Deferred compensation resulting from grant of options
|
- | - | - | - | 77 | (34 | ) | - | - | - | 43 | |||||||||||||||||||||||||||||
|
Amortization of deferred compensation
|
- | - | - | - | - | 283 | - | - | - | 283 | ||||||||||||||||||||||||||||||
|
Exercise of options to purchase common stock for cash, March through September 2000 ($1.72 to $33.50 per share)
|
- | - | 12,354 | - | 112 | - | - | - | - | 112 | ||||||||||||||||||||||||||||||
|
Issuance of common stock through employee stock purchase plan for cash, December 2000
|
- | - | 2,345 | - | 21 | - | - | - | - | 21 | ||||||||||||||||||||||||||||||
|
Issuance of common stock to Board of Director members for services, May through December 2000
|
- | - | 509 | - | 6 | - | - | - | - | 6 | ||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (11,155 | ) | (11,155 | ) | ||||||||||||||||||||||||||||
F-13
REPROS THERAPEUTICS INC. AND SUBSIDIARY
(A development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(in thousands except share and per share amounts)
|
Deficit
|
||||||||||||||||||||||||||||||||||||||||
|
Accumulated
|
||||||||||||||||||||||||||||||||||||||||
|
Additional
|
During the
|
Total
|
||||||||||||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Deferred
|
Treasury Stock
|
Development
|
Stockholders'
|
||||||||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Compensation
|
Shares
|
Amount
|
Stage
|
Equity
|
|||||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 2000
|
- | $ | - | 2,935,539 | $ | 2 | $ | 113,790 | $ | (241 | ) | 103,825 | $ | (7,484 | ) | $ | (75,007 | ) | $ | 31,060 | ||||||||||||||||||||
|
Compensation resulting from grant of options
|
- | - | - | - | 36 | - | - | - | - | 36 | ||||||||||||||||||||||||||||||
|
Compensation resulting from extension of warrants
|
- | - | - | - | 23 | - | - | - | - | 23 | ||||||||||||||||||||||||||||||
|
Amortization of deferred compensation
|
- | - | - | - | - | 230 | - | - | - | 230 | ||||||||||||||||||||||||||||||
|
Exercise of options to purchase common stock for cash, February through December 2001 ($2.56 to $16.00 per share)
|
- | - | 3,060 | - | 25 | - | - | - | - | 25 | ||||||||||||||||||||||||||||||
|
Issuance of common stock through employee stock purchase plan for cash, June and December 2001
|
- | - | 2,108 | - | 25 | - | - | - | - | 25 | ||||||||||||||||||||||||||||||
|
Issuance of common stock to Board of Director members for services, February through December 2001
|
- | - | 673 | - | 9 | - | - | - | - | 9 | ||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (839 | ) | (839 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 2001
|
- | $ | - | 2,941,380 | $ | 2 | $ | 113,908 | $ | (11 | ) | 103,825 | $ | (7,484 | ) | $ | (75,846 | ) | $ | 30,569 | ||||||||||||||||||||
|
Amortization of deferred compensation
|
- | - | - | - | - | 11 | - | - | - | 11 | ||||||||||||||||||||||||||||||
|
Exercise of options to purchase common stock for cash, January and February 2002 ($2.56 to $11.76 per share)
|
- | - | 7,816 | - | 21 | - | - | - | - | 21 | ||||||||||||||||||||||||||||||
|
Issuance of common stock through employee stock purchase plan for cash, June 2002
|
- | $ | - | 1,206 | - | 6 | $ | - | 0 | $ | - | $ | - | 6 | ||||||||||||||||||||||||||
|
Issuance of common stock to Employees
|
- | - | 26,250 | - | 111 | - | - | - | - | 111 | ||||||||||||||||||||||||||||||
|
Issuance of common stock to Board of Director members for services, March through December 2002
|
- | - | 2,893 | - | 15 | - | - | - | - | 15 | ||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (3,882 | ) | (3,882 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 2002
|
- | $ | - | 2,979,545 | $ | 2 | $ | 114,061 | $ | - | 103,825 | $ | (7,484 | ) | $ | (79,728 | ) | $ | 26,851 | |||||||||||||||||||||
|
Issuance of common stock to Board of Director members for services, February through May 2003
|
- | - | 2,718 | - | 14 | - | - | - | - | 14 | ||||||||||||||||||||||||||||||
|
Purchase of treasury stock April ($5.48 to $6.00 per share)
|
- | - | - | - | - | - | 8,525 | (49 | ) | - | (49 | ) | ||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (3,329 | ) | (3,329 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 2003
|
- | $ | - | 2,982,263 | $ | 2 | $ | 114,075 | $ | - | 112,350 | $ | (7,533 | ) | $ | (83,057 | ) | $ | 23,487 | |||||||||||||||||||||
|
Self Tender Offer of 1,636,909 shares at $8.40 including 15,222 exercised options
|
- | - | 15,222 | - | - | - | 1,636,909 | (13,665 | ) | - | (13,665 | ) | ||||||||||||||||||||||||||||
|
Costs associated with self tender offer
|
- | - | - | - | - | - | - | (289 | ) | - | (289 | ) | ||||||||||||||||||||||||||||
|
Noncash stock compensation related to stock option bonus program
|
- | - | - | - | 78 | - | - | - | - | 78 | ||||||||||||||||||||||||||||||
|
Issuance of 88,618 stock options to employees on March 29, 2004 and approved on September 29, 2004 (issue price of $10.88, fmv when approved $14.40)
|
- | - | - | - | 312 | (312 | ) | - | - | - | - | |||||||||||||||||||||||||||||
|
Amortization of deferred compensation
|
- | - | - | - | - | 78 | - | - | - | 78 | ||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (3,697 | ) | (3,697 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 2004
|
- | $ | - | 2,997,485 | $ | 2 | $ | 114,465 | $ | (234 | ) | 1,749,259 | $ | (21,487 | ) | $ | (86,754 | ) | $ | 5,992 | ||||||||||||||||||||
|
Issuance of 1,265,000 shares of treasury stock at $16.00 per share February 1, 2005
|
- | - | - | - | 2,641 | - | (1,265,000 | ) | 15,539 | - | 18,180 | |||||||||||||||||||||||||||||
|
Exercise of options to purchase common stock for cash, January and February 2005 ($11.76 to $13.88 per share)
|
- | - | 6,675 | - | 85 | - | - | - | - | 85 | ||||||||||||||||||||||||||||||
|
Noncash stock compensation related to stock option bonus program
|
- | - | - | - | (15 | ) | - | - | - | - | (15 | ) | ||||||||||||||||||||||||||||
|
Amortization of deferred compensation
|
- | - | - | - | - | 104 | - | - | - | 104 | ||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (7,391 | ) | (7,391 | ) | ||||||||||||||||||||||||||||
|
BALANCE AT DECEMBER 31, 2005
|
- | $ | - | 3,004,160 | $ | 2 | $ | 117,176 | $ | (130 | ) | 484,259 | $ | (5,948 | ) | $ | (94,145 | ) | $ | 16,955 | ||||||||||||||||||||
|
Exercise of options to purchase common stock for cash, January and July 2006 ($6.80 to $30.00 per share)
|
- | - | 17,840 | - | 241 | - | - | - | - | 241 | ||||||||||||||||||||||||||||||
|
Reclassification of previous deferred compensation due to the adoption of FAS 123(R)
|
- | - | - | - | (130 | ) | 130 | - | - | - | - | |||||||||||||||||||||||||||||
|
Stock option compensation
|
- | - | - | - | 789 | - | - | - | - | 789 | ||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (14,195 | ) | (14,195 | ) | ||||||||||||||||||||||||||||
F-14
(A development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(in thousands except share and per share amounts)
|
Deficit
|
||||||||||||||||||||||||||||||||||||||||
|
Accumulated
|
||||||||||||||||||||||||||||||||||||||||
|
Additional
|
During the
|
Total
|
||||||||||||||||||||||||||||||||||||||
|
Preferred Stock
|
Common Stock
|
Paid-in
|
Deferred
|
Treasury Stock
|
Development
|
Stockholders'
|
||||||||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Compensation
|
Shares
|
Amount
|
Stage
|
Equity
|
|||||||||||||||||||||||||||||||
|
Balance at December 31, 2006
|
- | $ | - | 3,022,000 | $ | 2 | $ | 118,076 | $ | - | 484,259 | $ | (5,948 | ) | $ | (108,340 | ) | $ | 3,790 | |||||||||||||||||||||
|
Exercise of options to purchase common stock for cash, January and April @ $9.60 & $32.00 per share
|
- | - | 3,485 | - | 37 | - | - | - | - | 37 | ||||||||||||||||||||||||||||||
|
Issuance of 652,500 shares of common stock at $55.00 per share February 5, 2007, net of offering costs of $2,835
|
- | - | 652,500 | 1 | 33,052 | - | - | - | - | 33,053 | ||||||||||||||||||||||||||||||
|
Stock option compensation
|
- | - | - | - | 880 | - | - | - | - | 880 | ||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (13,700 | ) | (13,700 | ) | ||||||||||||||||||||||||||||
|
Balance at December 31, 2007
|
- | $ | - | 3,677,985 | $ | 3 | $ | 152,045 | $ | - | 484,259 | $ | (5,948 | ) | $ | (122,040 | ) | $ | 24,060 | |||||||||||||||||||||
|
Stock based option compensation
|
- | - | - | - | 871 | - | - | - | - | 871 | ||||||||||||||||||||||||||||||
|
Proceeds from a shareholder transaction
|
- | - | - | - | 327 | - | - | - | - | 327 | ||||||||||||||||||||||||||||||
|
Issuance of 600,000 shares of common stock at $26.00 per share October 2, 2008, net of offering costs of $41,458
|
- | - | 600,000 | 1 | 15,557 | - | - | - | - | 15,558 | ||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (25,202 | ) | (25,202 | ) | ||||||||||||||||||||||||||||
|
Balance at December 31, 2008
|
- | $ | - | 4,277,985 | $ | 4 | $ | 168,800 | $ | - | 484,259 | $ | (5,948 | ) | $ | (147,242 | ) | $ | 15,614 | |||||||||||||||||||||
|
Exercise of stock option to purchase common stock for cash @ $14.84 per share
|
- | - | 625 | - | 9 | - | - | - | - | 9 | ||||||||||||||||||||||||||||||
|
Issuance of 375,000 shares of common stock at $2.60 per share September 11, 2009, net of offering costs of $106
|
- | - | 375,000 | - | 869 | - | - | - | - | 869 | ||||||||||||||||||||||||||||||
|
Issuance of 875,000 shares of common stock at $5.08 per share October 13, 2009, net of offering costs of $323
|
- | - | 875,000 | 1 | 4,120 | - | - | - | - | 4,121 | ||||||||||||||||||||||||||||||
|
Issuance of 1,340,298 shares of common stock at $4.40 per share October 29, 2009, as settlement with trade creditors
|
- | - | 968,389 | 1 | 1,330 | (371,909 | ) | 4,568 | - | 5,899 | ||||||||||||||||||||||||||||||
|
Stock based option compensation
|
- | - | - | - | 1,284 | - | - | - | - | 1,284 | ||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (27,234 | ) | (27,234 | ) | ||||||||||||||||||||||||||||
|
Balance at December 31, 2009
|
- | $ | - | 6,496,999 | $ | 6 | $ | 176,412 | $ | - | 112,350 | $ | (1,380 | ) | $ | (174,476 | ) | $ | 562 | |||||||||||||||||||||
|
Stock based option compensation
|
- | - | - | - | 609 | - | - | - | - | 609 | ||||||||||||||||||||||||||||||
|
Issuance of 96,836 shares of common stock at $2.88 to $4.40 per share, as settlement with trade creditors
|
- | - | 96,836 | - | 370 | - | - | - | - | 370 | ||||||||||||||||||||||||||||||
|
Issuance of 2,448,537 shares of common stock at a weighted average share price of $2.77, net of offering costs of $381
|
- | - | 2,448,537 | 3 | 6,391 | - | - | - | - | 6,394 | ||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (4,768 | ) | (4,768 | ) | ||||||||||||||||||||||||||||
|
Balance at December 31, 2010
|
- | $ | - | 9,042,372 | $ | 9 | $ | 183,782 | $ | - | 112,350 | $ | (1,380 | ) | $ | (179,244 | ) | $ | 3,167 | |||||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-15
REPROS THERAPEUTICS INC. AND SUBSIDIARY
(A development stage company)
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
|
From Inception
|
||||||||||||||||
|
(August 20, 1987)
|
||||||||||||||||
|
through
|
||||||||||||||||
|
For the Year Ended December 31,
|
December 31,
|
|||||||||||||||
|
2010
|
2009
|
2008
|
2010
|
|||||||||||||
|
(unaudited)
|
||||||||||||||||
|
Cash Flows from Operating Activities
|
||||||||||||||||
|
Net loss
|
$ | (4,768 | ) | $ | (27,234 | ) | $ | (25,202 | ) | $ | (179,244 | ) | ||||
|
Gain on disposal of discontinued operations
|
- | - | - | (939 | ) | |||||||||||
|
Gain on disposal of fixed assets
|
- | - | - | (102 | ) | |||||||||||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
||||||||||||||||
|
Noncash financing costs
|
- | - | - | 316 | ||||||||||||
|
Noncash inventory impairment
|
- | - | - | 4,417 | ||||||||||||
|
Noncash patent impairment
|
- | 1,275 | - | 2,614 | ||||||||||||
|
Noncash other income
|
(162 | ) | (547 | ) | - | (709 | ) | |||||||||
|
Noncash decrease in accounts payable
|
- | - | - | (1,308 | ) | |||||||||||
|
Depreciation and amortization
|
87 | 72 | 50 | 4,041 | ||||||||||||
|
Noncash stock-based compensation
|
609 | 1,284 | 871 | 7,250 | ||||||||||||
|
Common stock issued for agreement not to compete
|
- | - | - | 200 | ||||||||||||
|
Series B Preferred Stock issued for consulting services
|
- | - | - | 18 | ||||||||||||
|
Changes in operating assets and liabilities (net effects of purchase of businesses in 1988 and 1994):
|
||||||||||||||||
|
Increase in receivables
|
- | - | - | (199 | ) | |||||||||||
|
Increase in inventory
|
- | - | - | (4,447 | ) | |||||||||||
|
(Increase) decrease in prepaid expenses and other current assets
|
(150 | ) | 1,215 | (913 | ) | (25 | ) | |||||||||
|
Increase (decrease) in accounts payable and accrued expenses
|
(568 | ) | 1,854 | 3,449 | 9,470 | |||||||||||
|
Net cash used in operating activities
|
(4,952 | ) | (22,081 | ) | (21,745 | ) | (158,647 | ) | ||||||||
|
Cash Flows from Investing Activities
|
||||||||||||||||
|
Change in trading marketable securities
|
- | - | 24,124 | (191 | ) | |||||||||||
|
Capital expenditures
|
(7 | ) | - | (4 | ) | (2,378 | ) | |||||||||
|
Purchase of other assets
|
(364 | ) | (502 | ) | (569 | ) | (4,636 | ) | ||||||||
|
Proceeds from sale of fixed assets
|
- | - | - | 225 | ||||||||||||
|
Cash acquired in purchase of FTI
|
- | - | - | 3 | ||||||||||||
|
Proceeds from sale of subsidiary, less $12,345 for operating losses during 1990 phase-out period
|
- | - | - | 138 | ||||||||||||
|
Proceeds from sale of the assets of FTI
|
- | - | - | 2,250 | ||||||||||||
|
Increase in net assets held for disposal
|
- | - | - | (213 | ) | |||||||||||
|
Net cash provided by (used in) investing activities
|
(371 | ) | (502 | ) | 23,551 | (4,802 | ) | |||||||||
|
Cash Flows from Financing Activities
|
||||||||||||||||
|
Proceeds from issuance of common stock, net of offering costs
|
6,394 | 4,990 | 15,558 | 162,399 | ||||||||||||
|
Exercise of stock options
|
- | 9 | - | 372 | ||||||||||||
|
Proceeds from a shareholder transaction
|
- | - | 327 | 327 | ||||||||||||
|
Proceeds from issuance of preferred stock
|
- | - | - | 23,688 | ||||||||||||
|
Purchase of treasury stock
|
- | - | - | (21,487 | ) | |||||||||||
|
Proceeds from issuance of notes payable
|
- | - | - | 2,839 | ||||||||||||
|
Principal payments on notes payable
|
- | - | - | (1,732 | ) | |||||||||||
|
Net cash provided by financing activities
|
6,394 | 4,999 | 15,885 | 166,406 | ||||||||||||
|
Net increase (decrease) in cash and cash equivalents
|
1,071 | (17,584 | ) | 17,691 | 2,957 | |||||||||||
|
Cash and cash equivalents at beginning of period
|
1,886 | 19,470 | 1,779 | - | ||||||||||||
|
Cash and cash equivalents at end of period
|
$ | 2,957 | $ | 1,886 | $ | 19,470 | $ | 2,957 | ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-16
1. ORGANIZATION AND OPERATIONS:
Repros Therapeutics Inc. (the “Company”, “RPRX,” “Repros,” or “we,” “us” or “our”) was organized on August 20, 1987. We are a development stage biopharmaceutical company focused on the development of new drugs to treat hormonal and reproductive system disorders.
Our primary product candidate, Androxal®, is a single isomer of clomiphene citrate and is an orally active proprietary small molecule compound. We are developing Androxal® for men of reproductive age with low testosterone levels. Androxal® treats the underlying mechanism that causes secondary hypogonadism and restores normal testicular function. In addition, we are conducting a Phase 2 clinical trial of Androxal® as a potential treatment for Type 2 diabetes.
On November 8, 2010, we held a Type B meeting with the Food and Drug Administration (“FDA”) to discuss whether the FDA would review our protocols for a Phase 3 trial of Androxal® in men with secondary hypogonadism under a Special Protocol Assessment (“SPA”). In the meeting, the FDA recommended that a Phase 2B study in men with secondary hypogonadism but naïve to testosterone treatment be conducted if the Company desired the protocols to be reviewed under an SPA. The FDA further opined that such Phase 2B study would provide for a more solid data base for design of Phase 3 studies and eventual approval of such studies under an SPA. In our 24-patient Phase 2B proof-of-concept clinical trial which was initiated in the second quarter of 2008, we monitored the effects of Androxal® on male fertility and testicular function in patients being treated for low testosterone as compared to Testim®, a popular marketed topical testosterone medication. This trial showed that Androxal® was able to maintain sperm counts in men being treated for their low testosterone levels, whereas Testim® resulted in suppressed sperm levels. On January 3, 2011, we announced that we have received Investigational Review Board (“IRB”) approval to commence the Phase 2B study of Androxal® in men with secondary hypogonadism, and we have begun enrolling patients. Depending on the rate of subject enrollment, we hope to have the study completed by the end of 2011.
Our findings from a retrospective review of the clinical data from our 200 patient non-pivotal Phase 2 clinical trial showed that Androxal® therapy resulted in a significant reduction in mean fasting plasma glucose levels in men with glucose levels greater than 104 mg/dL at the outset of the trial, an outcome not seen in the placebo or AndroGel® arms of this study. Based on these results, in April 2008, we submitted a White Paper to the Division of Reproductive and Urology Products. The data demonstrated that among subjects with a serum glucose of greater than or equal to 105 mg/dL, there was a higher response rate to treatment in the Androxal® group than the placebo or AndroGel® groups, and the reduction in fasting serum glucose in this group was statistically significant. In November 2008, after the FDA reviewed this paper we received guidance from them suggesting that we open a new Investigational New Drug Application (“IND”) with the Division of Metabolic and Endocrine Products, (“DMEP”), for the investigation of Androxal® as a potential treatment for Type 2 diabetes. In December 2009, we submitted an IND to DMEP for the investigation of Androxal® for such purpose. On February 1, 2010, we received confirmation from DMEP that our IND was accepted and, as a result, we have initiated our Phase 2 trial. We hope that the study will be fully enrolled by the end of the third quarter 2011.
Proellex®, our product candidate for female reproductive health, is a new chemical entity that acts as a selective blocker of the progesterone receptor and is being developed for the treatment of symptoms associated with uterine fibroids and endometriosis. We have conducted numerous studies with Proellex® enrolling over 750 women, roughly 700 of whom were dosed with the drug. Up until the summer of 2009, all side effects exhibited in the studies were considered manageable and the benefit of Proellex® far outweighed the risk. However, in Phase 3 efficacy and larger Phase 3 safety studies in diverse populations, a small number of subjects exhibited serious adverse effects associated with elevated liver enzymes. As a result of these findings, we elected to stop the trials and the FDA subsequently placed Proellex® on full clinical hold. All women that experienced elevated liver enzymes and returned for follow-up visits returned to baseline conditions with no overnight hospitalization necessary. An analysis of all the subjects that experienced such serious adverse effects showed that the effect only occurred in a small percentage of subjects that were exposed to the 50 mg dose of the drug for any period of time. Based on these findings, the Company petitioned the FDA to allow it to conduct a low dose study to demonstrate both safety and signals of efficacy in low oral doses of Proellex®, up to 12 mg administered per day. The FDA upgraded the full clinical hold to a partial hold to allow the low dose study to be conducted, which we have since commenced. In addition, the Company has undertaken two related initiatives presently at the preclinical stage. The first is the exploration of vaginal delivery as an alternative administrative route to bypass first-pass liver effects and reduce systemic exposure. The second is the screening of second generation molecules that do not possess the specific structures the Company believes induced the liver toxicity exhibited at higher doses of Proellex®.
We continue limited out-licensing efforts for our phentolamine-based product candidates, including VASOMAX®, which had previously been approved for marketing in several countries in Latin America for the treatment of male erectile dysfunction under the brand name, Z-Max. VASOMAX® has been on partial clinical hold in the U.S. since 1998, and no further development activities are planned.
F-17
On February 12, 2010, we entered into an Equity Distribution Agreement (the “Equity Distribution Agreement”) with Ladenburg Thalmann & Co. Inc. (“Ladenburg”), pursuant to which we may issue and sell from time to time through Ladenburg, as sales agent and/or principal, shares of our common stock having an aggregate offering price of up to $10 million (the “ATM Shares”). Ladenburg is not required to sell on our behalf any specific number or dollar amount of the ATM Shares, but Ladenburg, upon acceptance of written instructions from us, agreed to use its commercially reasonable efforts consistent with its customary trading and sales practices, to sell the ATM Shares up to the amount specified, and otherwise in accordance with the terms of a placement notice delivered to Ladenburg. We have no obligation to sell any ATM Shares under the Equity Distribution Agreement, and may at any time suspend sales under the Equity Distribution Agreement, provided that such suspension shall not affect either party’s obligations with respect to the ATM Shares sold prior to the receipt of notice of such suspension. Ladenburg receives a commission of 4% of the gross sales price of all ATM Shares sold through it under the Equity Distribution Agreement. The ATM Shares are issued pursuant to our shelf registration statement on Form S-3, as amended (File No. 333-163648). Cumulative through December 31, 2010, we have sold 2,448,572 ATM Shares at a weighted average share price of $2.77, for proceeds of approximately $6.4 million, net of expenses. Pursuant to General Instruction I.B.6. of Form S-3, we may not sell more than one-third of the aggregate market value of our common stock held by non-affiliates during a period of 12 calendar months immediately prior to, and including, the date of such sale of such common stock. Due to this limitation, we announced on August 3, 2010 that we had suspended this ATM offering of Company securities. Additionally, on December 30, 2010 we announced our decision to reinstate our offering of the ATM shares. As a result, we sold 286,187 ATM Shares at a weighted average share price of $2.90 in January 2011, for proceeds of approximately $831,000, net of expenses.
On October 14, 2010, the Company effected a one-for-four reverse split of its common stock. The split-adjusted shares of the Company’s common stock began trading on the Nasdaq Capital Market on October 15, 2010. The one-for-four reverse split converted all shares of the Company’s common stock issued and outstanding, plus all outstanding stock options and the number of shares of common stock available for issuance under the Company’s approved stock plans. The number of authorized shares of common stock was not affected by the reverse split. The reverse split enabled the Company to meet the continued listing rules of the Nasdaq Capital Market as evidenced by the Compliance Letter received from Nasdaq on October 29, 2010. All share and per share amounts have been retroactively adjusted to reflect the reverse stock split for all periods presented.
On February 8, 2011, we completed an underwritten public offering of 690,000 units (including the exercise of the underwriter’s over-allotment option), consisting of an aggregate of 2,760,000 shares of our common stock, Series A Warrants to purchase 2,070,000 shares of our common stock and Series B warrants to purchase 1,690,500 shares of our common stock, at a price per unit of $17.15. Each unit consisted of four shares of our common stock, Series A Warrants exercisable for three shares of our common stock at an exercise price of $0.01 per share and Series B Warrants exercisable for 2.45 shares of our common stock at an exercise price of $2.49 per share. Gross proceeds to us, after the underwriting discount and before offering expenses, were approximately $11 million.
As of December 31, 2010, we had accumulated losses of $179.2 million, approximately $3.0 million in cash and cash equivalents, and our accounts payable and accrued expenses were approximately $1.3 million. We believe we have sufficient funding to complete all of the Phase 2 and 2B clinical trials currently planned or underway; however, significant additional capital will be required for us to complete development of either of our product candidates. We continue to explore potential additional financing alternatives (including corporate partnering opportunities) that would provide sufficient funds to enable us to continue to develop our two product candidates through completion of the outlined clinical trials; however, there can be no assurance that we will be successful in raising any such additional funds on a timely basis or at all. The foregoing and other matters raise substantial doubt about our ability to continue as a going concern.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES:
USE OF ESTIMATES
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
CERTAIN RISKS AND UNCERTAINTIES
Our product candidates under development require approval from the FDA or other international regulatory agencies prior to commercial sales. There can be no assurance our product candidates will receive the necessary clearance. If we are denied clearance or clearance is delayed, it may have a material adverse impact on us.
Our product candidates are concentrated in rapidly changing, highly competitive markets, which are characterized by rapid technological advances, evolving regulatory requirements and industry standards. Any failure by us to anticipate or to respond adequately to technological developments in our industry, changes in regulatory requirements or industry standards, or any significant delays in the development or introduction of products or services, could have a material adverse effect on our business, operating results and future cash flows.
F-18
CASH AND CASH EQUIVALENTS
The Company considers all cash accounts and highly liquid investments having original maturities of three months or less to be cash and cash equivalents.
PREPAID EXPENSES AND OTHER CURRENT ASSETS
Prepaid expenses and other current assets primarily consist of prepaid insurance, prepaid operating expenses and other miscellaneous assets, interest and other receivables.
FIXED ASSETS
Fixed assets include lab equipment, furniture and leasehold improvements and are recorded at cost, less accumulated depreciation and amortization. Depreciation is computed on the straight-line method over an estimated useful life of three to five years or, in the case of leasehold improvements, amortized over the shorter of the useful life or the remaining term of the lease. Maintenance and repairs that do not improve or extend the life of assets are expensed as incurred. When assets are sold or retired, the cost and accumulated depreciation are removed from the accounts and the resulting gain or loss is included in income during the period in which the transaction occurred.
OTHER ASSETS
The Company capitalizes the cost associated with building its patent library for its Androxal®. As of December 31, 2010 and 2009, other assets consist of capitalized patent costs in the amount of $1.2 million and $885,000 respectively. Patent costs, which include legal and application costs related to the patent portfolio, are being amortized over the lesser of 20 years or the estimated economic life of the patent. Amortization of patent cost expense was $76,000, $54,000 and $27,000 in 2010, 2009 and 2008, respectively. All of the $1.2 million in capitalized patent costs as of December 31, 2010 related to Androxal® patent and patent application costs.
We review capitalized patent and patent application costs for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. An impairment exists when estimated undiscounted cash flows expected to result from the patent are less than its carrying amount. The impairment loss recognized represents the excess of the patent cost as compared to its estimated fair value.
Due to the clinical hold on Proellex® and the uncertainty of future cash flows related to the Proellex® patent applications, the Company recorded an impairment charge of approximately $957,000 in 2009 related to these patent applications. Additionally, the Company concluded that it will no longer seek to protect the specific matter covered in certain Androxal® patent applications and recorded an impairment charge of approximately $318,000 in 2009 to abandon these patent applications. These charges were recorded in Research and Development expenses on the consolidated statement of operations. The remaining capitalized patent and patent application costs relating to Androxal® can continue to be used, outlicensed or sold to third parties for at least an amount management believes is sufficient to recover the carrying value of the capitalized patent costs.
Should the Company not continue development of Androxal® or should the Company not continue as a going concern, the remaining capitalized patent and patent application costs may not be recoverable, which would result in charges to operating results in future periods.
RESEARCH AND DEVELOPMENT EXPENSE
Research and development, or R&D, expenses include salaries and related employee expenses, contracted regulatory affairs activities, insurance coverage for clinical trials and prior product sales, contracted research and consulting fees, facility costs, amortization of capitalized patent costs and internal research and development supplies. We expense research and development costs in the period they are incurred. These costs consist of direct and indirect costs associated with specific projects as well as fees paid to various entities that perform research on our behalf.
We estimate accrued expenses as part of our process of preparing financial statements. Examples of areas in which subjective judgments may be required include costs associated with services provided by contract organizations for clinical trials, preclinical development and manufacturing of clinical materials. We accrue for costs incurred as the services are being provided by monitoring the status of the trials or services provided and the invoices received from our external service providers. In the case of clinical trials, a portion of the estimated cost normally relates to the projected cost to treat a patient in our trials, and we recognize this cost over the estimated term of the study based on the number of patients enrolled in the trial on an ongoing basis, beginning with patient enrollment. As actual costs become known to us, we adjust our accruals. To date, our estimates have not differed significantly from the actual costs incurred. However, subsequent changes in estimates may result in a material change in our accruals, which could also materially affect our balance sheet and results of operations.
F-19
LOSS PER SHARE
Basic loss per share is computed by dividing net loss by the weighted average number of shares of common stock outstanding during the year. Diluted loss per share is computed using the average share price for the period and applying the treasury stock method to potentially dilutive outstanding options. In applicable years all potential common stock equivalents were antidilutive and accordingly were not included in the computation.
SHARE-BASED COMPENSATION
We had two stock-based compensation plans at December 31, 2010. the 2000 Non-Employee Directors' Stock Option Plan, or 2000 Director Plan and the 2004 Stock Option Plan, or 2004 Plan. Accounting for stock based compensation generally requires the recognition of the cost of employee services for share-based compensation based on the grant date fair value of the equity or liability instruments issued. We use the Black-Scholes option pricing model to estimate the fair value of our stock options. Expected volatility is determined using historical volatilities based on historical stock prices for a period equal to the expected term. The expected volatility assumption is adjusted if future volatility is expected to vary from historical experience. The expected term of options represents the period of time that options granted are expected to be outstanding and falls between the options' vesting and contractual expiration dates. The risk-free interest rate is based on the yield at the date of grant of a zero-coupon U.S. Treasury bond whose maturity period equals the option's expected term.
INCOME TAXES
Our net operating losses from inception to date have resulted principally from costs incurred in conducting clinical trials and in research and development activities related to efforts to develop our products and from the associated administrative costs required to support those efforts. We have recorded a deferred tax asset for our net operating losses (“NOL”); however, as the Company has incurred net operating losses since inception, and since there is no certainty of future profits, a valuation allowance has been provided in full on our deferred tax assets in the accompanying consolidated financial statements. If the Company has an opportunity to use this NOL to off-set tax liabilities in the future, the use of this asset would be restricted based on Internal Revenue Service, state and local NOL use guidelines. The Company’s public offerings completed on February 5, 2007, October 2, 2008, September 11, 2009, October 13, 2009, February 8, 2011, the sale and issuance of our ATM Shares and the issuances of unregistered shares as part of the October 29, 2009 Settlement Agreement and Subsequent Settlement Agreements may have created a change of ownership for Federal Income tax purposes. The Company has not undertaken a study to determine if this has occurred. A change in ownership for Federal Income tax purposes may result in a limitation on the use of net operating loss and tax credit carryforwards in future periods.
3. FIXED ASSETS:
Fixed assets are as follows (in thousands):
|
December 31,
|
||||||||
|
|
2010
|
2009
|
||||||
|
Laboratory equipment
|
$ | 20 | $ | 20 | ||||
|
Office equipment
|
51 | 44 | ||||||
|
Leasehold improvements
|
38 | 38 | ||||||
|
Total fixed assets
|
109 | 102 | ||||||
|
Less — Accumulated depreciation and amortization
|
102 | 90 | ||||||
|
Net Fixed Assets
|
$ | 7 | $ | 12 | ||||
Depreciation and amortization was $11,000, $16,000 and $22,000 for the years ended December 31, 2010, 2009 and 2008, respectively.
4. OPERATING LEASES:
The Company leases laboratory and office space, and equipment pursuant to leases accounted for as operating leases. The lease for the Company’s laboratory and office space expires in June 2015. Rental expense for the years ended December 31, 2010, 2009 and 2008, was approximately $63,000, $60,000 and $59,000, respectively. Future minimum lease payments under non-cancelable leases with original terms in excess of one year as of December 31, 2010, are as follows (in thousands):
|
2011
|
$ | 49 | ||
|
2012
|
50 | |||
|
2013
|
52 | |||
|
2014
|
53 | |||
|
2015
|
27 | |||
|
Total
|
$ | 231 |
F-20
5. ACCRUED EXPENSES:
Accrued expenses consist of the following (in thousands):
|
December 31,
|
||||||||
|
|
2010
|
2009
|
||||||
|
Personnel related costs
|
$ | 87 | $ | 181 | ||||
|
Other
|
34 | 159 | ||||||
|
Patent costs
|
18 | 15 | ||||||
|
Research and development costs
|
8 | — | ||||||
|
Total
|
$ | 147 | $ | 355 | ||||
6. FEDERAL INCOME TAXES:
The Company has had net operating losses since inception and, therefore, has not been subject to federal income taxes. As of December 31, 2010, the Company has accumulated approximately $2.4 million of research and development tax credits. As of December 31, 2010, the Company had approximately $153 million of net operating loss (“NOL”) carryforwards for federal income tax purposes. Additionally, approximately $9.1 million of NOLs, and approximately $236,000 of research and development tax credits will expire in 2011.
The Tax Reform Act of 1986 provided for a limitation on the use of NOL and tax credit carryforwards following certain ownership changes that could limit the Company’s ability to utilize these NOLs and tax credits. The sale of preferred stock, together with changes in stock ownership, resulted in multiple ownership changes for federal income tax purposes. The Company estimates that the amount of pre-2007 NOL carryforwards and the credits available to offset taxable income is limited to approximately $7.6 million per year on a cumulative basis. Accordingly, if the Company generates taxable income in any year in excess of its then cumulative limitation, the Company may be required to pay federal income taxes even though it has unexpired NOL carryforwards. Additionally, because U.S. tax laws limit the time during which NOLs and tax credit carryforwards may be applied against future taxable income and tax liabilities, the Company may not be able to take full advantage of its NOLs and tax credit carryforwards for federal income tax purposes.
The Company’s public offerings completed on February 5, 2007, October 2, 2008, September 11, 2009, October 13, 2009, February 8, 2011, the sale and issuance of the ATM Shares and the issuance of unregistered shares as part of the October 29, 2009 Settlement Agreement and Subsequent Settlement Agreements may have created a change of ownership for Federal Income tax purposes. The Company has not undertaken a study to determine if this has occurred. A change in ownership for Federal income tax purposes may result in a limitation on the use of net operating loss and tax credit carryforwards in future periods.
Accounting standards require the recognition of a deferred tax asset for NOLs. As the Company has incurred net operating losses since inception, and there is no certainty of future revenues, a valuation allowance has been provided in full in the accompanying consolidated financial statements.
The tax effects of temporary differences that give rise to significant portions of the deferred tax assets are as follows (in thousands):
|
December 31,
|
||||||||
|
|
2010
|
2009
|
||||||
|
Net operating loss carryforwards
|
$ | 51,907 | $ | 51,714 | ||||
|
Research and development tax credits
|
2,434 | 2,483 | ||||||
|
Accrued expenses
|
1,510 | 1,510 | ||||||
|
Total deferred tax assets
|
55,851 | 55,707 | ||||||
|
Capitalized patent costs
|
(399 | ) | (301 | ) | ||||
|
Total deferred tax liabilities
|
(399 | ) | (301 | ) | ||||
|
Less — Valuation allowance
|
(55,452 | ) | (55,406 | ) | ||||
|
Net deferred tax assets
|
$ | — | $ | — | ||||
F-21
7. STOCKHOLDERS’ EQUITY:
PUBLIC OFFERINGS
On February 12, 2010, we entered into an Equity Distribution Agreement (the “Equity Distribution Agreement”) with Ladenburg Thalmann & Co. Inc. (“Ladenburg”), pursuant to which we may issue and sell from time to time through Ladenburg, as sales agent and/or principal, shares of our common stock having an aggregate offering price of up to $10 million (the “ATM Shares”). Ladenburg is not required to sell on our behalf any specific number or dollar amount of the ATM Shares, but Ladenburg, upon acceptance of written instructions from us, agreed to use its commercially reasonable efforts consistent with its customary trading and sales practices, to sell the ATM Shares up to the amount specified, and otherwise in accordance with the terms of a placement notice delivered to Ladenburg. We have no obligation to sell any ATM Shares under the Equity Distribution Agreement, and may at any time suspend sales under the Equity Distribution Agreement, provided that such suspension shall not affect either party’s obligations with respect to the ATM Shares sold prior to the receipt of notice of such suspension. Ladenburg receives a commission of 4% of the gross sales price of all ATM Shares sold through it under the Equity Distribution Agreement. The ATM Shares are issued pursuant to our shelf registration statement on Form S-3, as amended (File No. 333-163648). Cumulative through December 31, 2010, we have sold 2,448,572 ATM Shares at a weighted average share price of $2.77, for proceeds of approximately $6.4 million, net of expenses. Pursuant to General Instruction I.B.6. of Form S-3, we may not sell more than one-third of the aggregate market value of our common stock held by non-affiliates during a period of 12 calendar months immediately prior to, and including, the date of such sale of such common stock. Due to this limitation, we announced on August 3, 2010 that we have suspended this ATM offering of Company securities. Additionally, on December 30, 2010 we announced our decision to reinstate our offering of the ATM shares. As a result, we sold 286,187 ATM Shares at a weighted average share price of $2.90 in January 2011, for proceeds of approximately $831,000, net of expenses.
On October 13, 2009, we completed a direct registered offering of 875,000 shares of our common stock at a purchase price of $5.08 per share for aggregate proceeds after expenses of approximately $4.1 million.
On September 11, 2009, we completed a direct registered offering of 375,000 shares of our common stock at a purchase price of $2.60per share for aggregate proceeds after expenses of approximately $869,000.
SETTLEMENT WITH TRADE CREDITORS
On October 29, 2009, we entered into a Master Settlement Agreement and Releases (the “October Settlement Agreement”) with certain trade creditors, pursuant to which we issued 1,340,298 shares of our common stock, at $4.40 per share, and paid approximately $2.77 million in cash to such creditors as payment in full for our then-outstanding liabilities of approximately $8.7 million and for the release of the claims held by and the dismissal of the litigation commenced by such creditors against the Company. Pursuant to the terms of the October Settlement Agreement, we filed a registration statement on Form S-3 (File No. 333-163510), which was filed with the Securities and Exchange Commission on December 4, 2009, to register the resale of such shares by such creditors. Such registration statement was declared effective by the Securities and Exchange Commission on January 7, 2010.
Between November 30, 2009 and March 31, 2010, we entered into settlement agreements and mutual releases (the “Prior Settlement Agreements”) with certain of our creditors, pursuant to which we issued an aggregate of 88,115 shares of common stock and paid an aggregate of $140,572 in cash as payment in full for our then-outstanding liabilities to such creditors. On April 8, 2010, we entered into an additional settlement agreement and mutual release (together with the Prior Settlement Agreements, the “Settlement Agreements”) with a creditor, pursuant to which we issued 8,721 shares of common stock (together with the shares issued under the Prior Settlement Agreements, the “Settlement Shares”) and paid $8,721 in cash as payment in full for our then-outstanding liability to such creditor. The Settlement Shares were issued by the Company pursuant to Section 4(2) and /or Rule 506 of Regulation D promulgated under the Securities Act of 1933, as amended. Pursuant to the Settlement Agreements, we filed a registration statement to register the Settlement Shares on June 9, 2010, which was declared effective by the SEC on June 25, 2010, and we agreed to use our best efforts to maintain such registration statement until all such Settlement Shares registered thereunder to such creditors have been sold or for a period of one year, whichever comes first.
In addition to the October Settlement Agreement and Subsequent Settlement Agreements, the Company has settled with several of its creditors during 2010 in an amount less than its then-outstanding liabilities to such creditors. These settlements resulted in recognition of $177,000 in other income on the Consolidated Statement of Operations.
LOSS PER SHARE
The following table presents information necessary to calculate loss per share for the three years ended December 31, 2010, 2009 and 2008 (in thousands, except per share amounts):
|
2010
|
2009
|
2008
|
||||||||||
|
Net loss
|
$ | (4,768 | ) | $ | (27,234 | ) | $ | (25,202 | ) | |||
|
Weighted average common shares outstanding
|
8,057 | 4,336 | 3,343 | |||||||||
|
Basic loss per share
|
$ | (0.59 | ) | $ | (6.28 | ) | $ | (7.54 | ) | |||
|
Weighted average common and dilutive potential common shares outstanding:
|
||||||||||||
|
Weighted average common shares outstanding
|
8,057 | 4,336 | 3,343 | |||||||||
|
Assumed exercise of stock options
|
— | — | — | |||||||||
| 8,057 | 4,336 | 3,343 | ||||||||||
|
Diluted earnings per share
|
$ | (0.59 | ) | $ | (6.28 | ) | $ | (7.54 | ) | |||
F-22
Other potential common stock of 613,869, 456,053 and 707,558 for the years ended December 31, 2010, 2009 and 2008, respectively, were excluded from the above calculation of diluted loss per share since they were antidilutive.
8. STOCK OPTION PLANS:
During 2010 the Company had two stock option plans available which were the 2000 Non-Employee Directors’ Stock Option Plan, or 2000 Director Plan; and the 2004 Stock Option Plan, or 2004 Plan.
As of December 31, 2010, there were 100,781 options available under the 2004 Plan and 112,354 options available options under the 2000 Director Plan. The 2000 Director Plan has an evergreen provision pursuant to which the number of shares available under such plan are automatically increased each year on the day after the Company’s Annual Shareholders’ Meeting by the number of shares granted during the prior year under such plan (or by one-half percent of the Company’s then outstanding common stock, if greater). At the Annual Meeting of Stockholders held on May 20, 2009, the stockholders approved amendments of the 2004 Plan and 2000 Director Plan to increase the number of shares of common stock issuable by 250,000 and 125,000, respectively. Additionally, the stockholders approved an amendment to the 2000 Director Plan which extends the term of such plan by nine years to April 17, 2019. There are no significant differences between the provisions of the two remaining plans other than with respect to the period of exercisability following termination of services. Typically, options are granted with an exercise price per share which is equal to the fair market value per share of common stock on the date of grant. Vesting provisions for each grant are determined by the board of directors and typically vest quarterly over a three year period. All options expire no later than the tenth anniversary of the grant date.
A summary of the status of the Company’s option plans at December 31, 2010, 2009, and 2008 and changes during the years then ended is presented in the tables below:
|
Stock
Options
|
Weighted
Average
Exercise
Price
|
Remaining
Weighted
Average
Contractual
Term
(Years)
|
||||||||||
|
Outstanding at December 31, 2007
|
388,891 | $ | 18.80 | |||||||||
|
Granted
|
60,000 | 36.04 | ||||||||||
|
Exercised
|
— | — | ||||||||||
|
Forfeited
|
(1,750 | ) | 79.80 | |||||||||
|
Outstanding at December 31, 2008
|
447,141 | 20.88 | ||||||||||
|
Granted
|
184,162 | 24.80 | ||||||||||
|
Exercised
|
(625 | ) | 14.84 | |||||||||
|
Forfeited
|
(174,625 | ) | 31.92 | |||||||||
|
Outstanding at December 31, 2009
|
456,053 | 18.24 | ||||||||||
|
Granted
|
224,872 | 2.16 | ||||||||||
|
Exercised
|
— | — | ||||||||||
|
Forfeited
|
(67,056 | ) | 16.62 | |||||||||
|
Outstanding at December 31, 2010
|
613,869 | 12.53 | 5.99 | |||||||||
|
Exercisable at December 31, 2010
|
389,174 | 15.75 | 5.14 | |||||||||
The following table summarizes information about stock options outstanding at December 31, 2010:
|
Range Of Exercise Prices
|
Number
Outstanding
|
Weighted
Average
Remaining
Life
|
Weighted
Average
Exercise
Price
|
Number
Exercisable
|
Weighted
Average
Exercise
Price
|
|||||||||||||||||||||
| $ | 1.33 to | $ | 5.00 | 274,033 | 9.4 | $ | 2.30 | 113,294 | $ | 2.92 | ||||||||||||||||
|
5.01 to
|
10.00 | 20,000 | 3.0 | 9.60 | 20,000 | 9.60 | ||||||||||||||||||||
|
10.01 to
|
15.00 | 157,087 | 2.7 | 11.91 | 157,087 | 11.91 | ||||||||||||||||||||
|
15.01 to
|
20.00 | 75,000 | 1.5 | 17.27 | 18,750 | 16.99 | ||||||||||||||||||||
|
25.01 to
|
35.00 | 22,499 | 4.2 | 30.82 | 22,499 | 30.82 | ||||||||||||||||||||
|
35.01 to
|
50.00 | 57,750 | 6.7 | 44.05 | 50,044 | 45.05 | ||||||||||||||||||||
|
50.01 to
|
133.00 | 7,500 | 2.8 | 62.49 | 7,500 | 62.49 | ||||||||||||||||||||
| 613,869 | 389,174 | |||||||||||||||||||||||||
The intrinsic value of options exercised during the years ended December 31, 2009 was approximately $10,000.
Stock-based compensation is outlined in the following table (in thousands):
F-23
|
|
2010
|
2009
|
2008
|
|||||||||
|
R&D expense
|
$ | 241 | $ | 485 | $ | 389 | ||||||
|
G&A expense
|
368 | 799 | 482 | |||||||||
|
Total expense
|
$ | 609 | $ | 1,284 | $ | 871 | ||||||
At December 31, 2010, there was approximately $347,000 of total unrecognized compensation cost related to non-vested stock options. This compensation cost is expected to be recognized over a weighted-average period of approximately six months.
Estimated fair values of stock options granted have been determined using the Black-Scholes option pricing model with the following assumptions:
|
2010
|
2009
|
2008
|
||||||||||
|
Risk-free interest rate
|
2.0 | % | 2.4 | % | 3.2 | % | ||||||
|
Expected term
|
5 years
|
7 years
|
7 years
|
|||||||||
|
Volatility
|
91 | % | 74 | % | 77 | % | ||||||
|
Dividend yield
|
— | — | — | |||||||||
|
Fair value
|
$ | 1.54 | $ | 16.96 | $ | 26.04 | ||||||
Due to our net operating loss position there are no anticipated windfall tax benefits upon exercise of options.
The Black-Scholes option pricing model and other existing models were developed for use in estimating the fair value of traded options that have no vesting restrictions and are fully transferable. In addition, option valuation models require the input of and are highly sensitive to subjective assumptions including the expected stock price volatility. The Company’s employee stock options have characteristics significantly different from those of traded options and changes in the subjective input assumptions can materially affect the fair value estimate.
9. LICENSE, RESEARCH AND DEVELOPMENT AGREEMENTS:
NATIONAL INSTITUTES OF HEALTH (NIH)
In 1999, we licensed rights to Proellex® from the National Institutes of Health, or NIH, under an exclusive, worldwide license in the field of treatment of human endocrinologic pathologies or conditions in steroid-sensitive tissues which expires upon the expiration of the last licensed patent. Under the terms of the agreement, we are obligated to meet certain developmental milestones as outlined in a commercial development plan, which has been amended and revised from time to time as circumstances warrant. We have recently amended the agreement to provide us with rights to certain second generation compounds under certain circumstances.
We provide annual updates to the NIH on the progress of our development of Proellex®. The NIH has the ability to terminate the agreement for lack of payment or if we are not meeting milestones as outlined in the commercial development plan and for other reasons as outlined in the agreement. Although we believe that we have a good working relationship with the NIH, there can be no assurance that all of the objectives and conditions in the commercial development plan will be met on a timely basis or at all, or that, if we fail to meet any of such objectives, the NIH will agree to amend this agreement to our satisfaction. Failure to comply with the material terms contained in the license agreement could result in termination of such agreement, which would prohibit us from further development of Proellex® and severely harm our business prospects. The NIH retains, on behalf of the government, a nonexclusive, nontransferable, worldwide license to practice the inventions licensed under the licensed patents by or on behalf of the government. For the purpose of encouraging basic research, the NIH retains the right to grant nonexclusive research licenses to third parties. Due to the work that was done on Proellex® at the NIH prior to our license agreement, the government also has certain rights to use the product in the event of a national emergency pursuant to the Patent and Trademark Laws Amendments Act of 1980, as amended.
10. COMMITMENTS AND CONTINGENCIES:
See footnote 4 for a discussion of our operating lease commitments.
Therapeutic uses of our Androxal® product candidate are covered in the United States by four issued U.S. patents and four pending patent applications. Foreign coverage of therapeutic uses of our Androxal® product candidate includes 42 issued foreign patents and 68 foreign pending patent applications. The issued patents and pending applications relate to methods for treating certain conditions including the treatment of testosterone deficiency in men, the treatment of metabolic syndrome and conditions associated therewith, and the treatment of infertility in hypogonadal men. Androxal® (the trans-isomer of clomiphene) is purified from clomiphene citrate. A third party individual holds two issued patents related to the use of an anti-estrogen such as clomiphene citrate and others for use in the treatment of androgen deficiency and disorders related thereto. In our prior filings with the SEC, we have described our request to the U.S. Patent and Trademark Office, or PTO, for re-examination of one of these patents based on prior art. The third party amended the claims in the re-examination proceedings, which led the PTO to determine that the amended claims are patentable in view of those publications under consideration and a re-examination certificate was issued. However, we believe that the amended claims are invalid based on additional prior art publications, and we filed a second request for re-examination by the PTO in light of a number of these additional publications and other publications cited by the PTO. The request was granted and all of the claims were finally rejected by the PTO in the re-examination. The patent holder appealed the rejections to the PTO Board of Patent Appeals and Interferences (“the Board”) which affirmed the rejection of all of the claims. The patent holder subsequently filed a request for rehearing, which led the Board to reverse the rejections of several dependent claims in view of those publications under consideration. The patent holder filed a Notice of Appeal to the Federal Circuit on September 28, 2010 contesting the rejections maintained by the Board and submitted his brief on February 8, 2011. We also believe that the second of these two patents is invalid in view of published prior art not considered by the PTO. Nevertheless, there is no assurance that either patent will ultimately be found invalid over the prior art. If such patents are not invalidated by the PTO we may be required to obtain a license from the holder of such patents in order to develop Androxal® further or attempts may be made to undertake further legal action to invalidate such patents. If such licenses were not available on acceptable terms, or at all, we may not be able to successfully commercialize or out-license Androxal®.
F-24
In August and September of 2009, several securities fraud class action lawsuits were filed in federal court for the Southern District of Texas against the Company and various of its current or former officers and directors. The lawsuits alleged that the defendants made certain misleading statements related to the Company's Proellex drug. Among other claims, the lawsuits alleged that the defendants misrepresented the side effects of the drug related to liver function, and the risk that these side effects could cause a suspension of clinical trials on Proellex. The lawsuits were consolidated under the caption In re Repros Therapeutics, Inc. Securities Litigation, Civil Action No. 09 Civ. 2530 (VDG), and the court appointed lead plaintiffs and class counsel. Lead plaintiffs filed a consolidated amended complaint making essentially the same allegations as had been made in the prior complaints. Lead plaintiffs sought to represent a class of all persons who purchased or otherwise acquired Repros common stock between July 1, 2009 and August 2, 2009, and asserted claims under the Securities Exchange Act of 1934. Defendants filed a motion to dismiss the complaint. On January 19, 2011, the court granted the defendants' motion to dismiss and entered a final judgment dismissing the case. The time for plaintiffs to file an appeal of that order expired on February 18, 2011.
On March 1, 2010, we were served with a lawsuit where we were named as a co-defendant along with one of our clinical regulatory service providers (“CRO”) relating to the Proellex® clinical trial study. The lawsuit was filed in the State of Tennessee, 30th Judicial District Chancery Court at Memphis by an investigator and claims that the CRO did not pay it amounts owing to it relating to the Proellex® study. We did not engage the investigator and under our agreement with the CRO, we believe the CRO is responsible for any such costs or damages regarding such lawsuit. Pursuant to a Settlement Agreement and Mutual Release entered into in October 2009, such CRO, on behalf of itself and its agents, released us from all claims which could be asserted by them against us. We believe such release covers the claims set forth in this lawsuit. The CRO failed to respond to the lawsuit, and a default judgment was entered against it in the amount of $172,901.29. We intend to vigorously defend any and all claims asserted by the investigator. An estimate of the possible costs or expenses to defend ourselves in this matter or risk of exposure under the litigation cannot be made at this time.
Rights Plan
We are party to a rights agreement, as amended, pursuant to which a dividend consisting of one preferred stock purchase right was distributed for each share of our common stock held as of the close of business on September 13, 1999, and to each share of common stock issued thereafter until the earlier of (i) the distribution date which is defined in the rights plan, (ii) the redemption date which is defined in the rights plan or (iii) September 13, 2015. The rights plan is designed to deter coercive takeover tactics and to prevent an acquirer from gaining control of us without offering fair value to our stockholders. The rights will expire on September 13, 2015, subject to earlier redemption or exchange as provided in the rights plan. Each right entitles its holder to purchase from us one one-hundredth of a share of a new series of Series One Junior Participating Preferred Stock at a price of $20.00 per one one-hundredth of a share, subject to adjustment. The rights are generally exercisable only if a person acquires beneficial ownership of 20% or more of our outstanding common stock.
|
|
11.
|
RELATED PARTY TRANSACTIONS:
|
A former member of our Board of Directors is also a shareholder of a law firm that provides legal services to the Company. He served on the Board of Directors for a portion of 2009 and all of 2008, and total fees charged by the firm to the Company during the years ended 2009 and 2008 were $345,000 and $275,000, respectively. He did not serve on the Board of Directors during 2010.
|
|
12.
|
QUARTERLY FINANCIAL INFORMATION (UNAUDITED):
|
|
First Quarter
Ended
March 31,
2010
|
Second Quarter
Ended
June 30,
2010
|
Third Quarter
Ended
September 30,
2010
|
Fourth Quarter
Ended
December 31,
2010
|
|||||||||||||
|
(In thousands except per share amounts)
|
||||||||||||||||
|
Revenues and other income:
|
||||||||||||||||
|
Interest income
|
$ | — | $ | — | $ | — | $ | — | ||||||||
|
Other income
|
— | 53 | 85 | 283 | ||||||||||||
|
Total revenues and other income
|
— | 53 | 85 | 283 | ||||||||||||
|
Expenses:
|
||||||||||||||||
|
Research and development
|
458 | 756 | 736 | 954 | ||||||||||||
|
General and administrative
|
669 | 570 | 533 | 513 | ||||||||||||
|
Total expenses
|
1,127 | 1,326 | 1,269 | 1,467 | ||||||||||||
|
Net loss
|
$ | (1,127 | ) | $ | (1,273 | ) | $ | (1,184 | ) | $ | (1,184 | ) | ||||
|
Net loss per share – basic and diluted
|
$ | (0.17 | ) | $ | (0.16 | ) | $ | (0.13 | ) | $ | (0.13 | ) | ||||
|
Shares used in loss per share calculation
|
6,457 | 7,931 | 8,875 | 8,930 | ||||||||||||
F-25
|
First Quarter
Ended
March 31,
2009
|
Second Quarter
Ended
June 30,
2009
|
Third Quarter
Ended
September 30,
2009
|
Fourth Quarter
Ended
December 31,
2009
|
|||||||||||||
|
(In thousands except per share amounts)
|
||||||||||||||||
|
Revenues and other income:
|
||||||||||||||||
|
Interest income
|
$ | 3 | $ | 1 | $ | — | $ | — | ||||||||
|
Other income
|
— | — | — | 547 | ||||||||||||
|
Total revenues and other income
|
3 | 1 | — | 547 | ||||||||||||
|
Expenses:
|
||||||||||||||||
|
Research and development
|
5,698 | 7,784 | 8,282 | 1,298 | ||||||||||||
|
General and administrative
|
1,060 | 1,105 | 1,962 | 596 | ||||||||||||
|
Total expenses
|
6,758 | 8,889 | 10,244 | 1,894 | ||||||||||||
|
Net loss
|
$ | (6,755 | ) | $ | (8,888 | ) | $ | (10,244 | ) | $ | (1,347 | ) | ||||
|
Net loss per share – basic and diluted
|
$ | (1.78 | ) | $ | (2.34 | ) | $ | (2.64 | ) | $ | (0.23 | ) | ||||
|
Shares used in loss per share calculation
|
3,794 | 3,794 | 3,876 | 5,863 | ||||||||||||
13. SUBSEQUENT EVENTS
On December 30, 2010 we announced our decision to reinstate our offering of the ATM shares. As a result, we sold 286,187 ATM Shares at a weighted average share price of $2.90 in January 2011, for proceeds of approximately $831,000, net of expenses.
On February 8, 2011, we completed an underwritten public offering of 690,000 units (including the exercise of the underwriter’s over-allotment option), consisting of an aggregate of 2,760,000 shares of our common stock, Series A Warrants to purchase 2,070,000 shares of our common stock and Series B warrants to purchase 1,690,500 shares of our common stock, at a price per unit of $17.15. Each unit consisted of four shares of our common stock, Series A Warrants exercisable for three shares of our common stock at an exercise price of $0.01 per share and Series B Warrants exercisable for 2.45 shares of our common stock at an exercise price of $2.49 per share. Gross proceeds to us, after the underwriting discount and before offering expenses, were approximately $11 million.
On February 28, 2011, the Compensation Committee of the Board of Directors of Repros Therapeutics Inc. (the "Company") approved the Company's 2011 Equity Incentive Plan (the "Plan") in order to (i) simplify the administration of the 2000 Non-Employee Director's Stock Option Plan and the 2004 Stock Option Plan (collectively, the "Prior Plans") by combining the share reserves of the Prior Plans that remain available for issuance and rolling those shares forward to the Plan. The Plan will be included in the Company's Proxy Statement for its 2011 Annual Meeting of Stockholders (the "Annual Meeting") and will be subject to stockholder approval at such meeting. Additionally, the Compensation Committee of the Board of Directors of the Company approved grants of stock options to certain key employees and directors of the Company. The stock options will automatically expire and terminate if the Plan is not approved by its stockholders at the 2011 Annual Meeting.
On March 3, 2011, we announced the dismissal of our class action lawsuits. In August and September of 2009, several securities fraud class action lawsuits were filed in federal court for the Southern District of Texas against the Company and various of its current or former officers and directors. The lawsuits alleged that the defendants made certain misleading statements related to the Company's Proellex drug. Among other claims, the lawsuits alleged that the defendants misrepresented the side effects of the drug related to liver function, and the risk that these side effects could cause a suspension of clinical trials on Proellex. The lawsuits were consolidated under the caption In re Repros Therapeutics, Inc. Securities Litigation, Civil Action No. 09 Civ. 2530 (VDG), and the court appointed lead plaintiffs and class counsel. Lead plaintiffs filed a consolidated amended complaint making essentially the same allegations as had been made in the prior complaints. Lead plaintiffs sought to represent a class of all persons who purchased or otherwise acquired Repros common stock between July 1, 2009 and August 2, 2009, and asserted claims under the Securities Exchange Act of 1934. Defendants filed a motion to dismiss the complaint. On January 19, 2011, the court granted the defendants' motion to dismiss and entered a final judgment dismissing the case. The time for plaintiffs to file an appeal of that order expired on February 18, 2011.
F-26
On March 15, 2011, we received notice from the Nasdaq Stock Market that the Company’s Series A Warrants (RPRXW) and Series B Warrants (RPRXZ) (collectively, the “Warrants”) have not had a minimum of two active and registered market makers, as required for continued inclusion by Listing Rule 5560(a) (the “Rule”). We have been provided until April 14, 2011 to regain compliance with the Rule. If, at any time before such date, the Warrants have at least two active and registered market makers for 10 consecutive trading days, we will have achieved compliance with the Rule. If compliance is not demonstrated by the Company by such date, the Warrants will be delisted from the Nasdaq Capital Market. We believe that, due to certain technical issues, relevant information related to the active and registered market makers for the Warrants was not being disseminated regarding quotes, which resulted in the issuance of such notice. We believe that such technical issues have been resolved and, therefore, we anticipate that it will demonstrate compliance with the Rule within the requisite time period.
F-27