UNITED STATES SECURITIES AND
EXCHANGE COMMISSION
Washington, D.C.
20549
Form 10-K
| |
|
|
|
þ
|
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the fiscal year ended
December 31,
2010
|
|
or
|
|
o
|
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
|
|
|
|
For the transition period
from to .
|
Commission File
No. 1-11181
IRIS INTERNATIONAL,
INC.
(Exact name of Registrant as
Specified In Its Charter)
| |
|
|
|
Delaware
|
|
94-2579751
|
(State or other jurisdiction
of
Incorporation or Organization)
|
|
(I.R.S. Employer
Identification No.)
|
9158 Eton Avenue, Chatsworth, California 91311
(Address of principal executive
offices) (Zip Code)
(818) 709-1244
(Registrant’s telephone
number, including area code)
Securities registered pursuant to Section 12(b) of the
Act:
| |
|
|
|
Title of Each Class
|
|
Name of Each Exchange on Which Registered
|
|
Common Stock, par value $0.01 per share
|
|
NASDAQ Global Market
|
Securities registered pursuant to Section 12(g) of the
Act:
None
Indicate by check mark if the registrant is a well-known
seasoned issuer, as defined in Rule 405 of the Securities
Act. Yes o No þ
Indicate by check mark if the registrant is not required to file
reports pursuant to Section 13 or Section 15(d) of the
Exchange
Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed
all reports required to be filed by Section 13 or 15(d) of
the Securities Exchange Act of 1934 during the preceding
12 months (or for such shorter period that the registrant
was required to file such reports), and (2) has been
subject to such filing requirements for the past
90 days. Yes þ No o
Indicate by check mark whether the registrant has submitted
electronically and posted on its corporate Web site, if any,
every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of
Regulation S-T
during the preceding 12 months (or for such shorter period
that the registrant was required to submit and post such
files). Yes o No o
Indicate by check mark if disclosure of delinquent filers
pursuant to Item 405 of
Regulation S-K
is not contained herein, and will not be contained, to the best
of registrant’s knowledge, in definitive proxy or
information statements incorporated by reference in
Part III of this
Form 10-K
or any amendment to this
Form 10-K. o
Indicate by check mark whether the registrant is a large
accelerated filer, an accelerated filer, a non-accelerated
filer, or a smaller reporting company. See the definitions of
“large accelerated filer,” “accelerated
filer” and “smaller reporting company” in Rule
12b-2 of the
Exchange Act. (Check one):
|
|
|
|
| Large
accelerated
filer o
|
Accelerated
filer þ
|
Non-accelerated
filer o
|
Smaller
reporting
company o
|
(Do not check if a smaller reporting company)
Indicate by check mark whether the registrant is a shell company
(as defined in
Rule 12b-2
of the Exchange
Act. Yes o No þ
The aggregate market value of the shares of common stock held by
non-affiliates of the Registrant on March 7, 2011 was
approximately $166 million based upon the closing price of
$9.30 per share of its common stock as reported on the NASDAQ
Global Market on such date.
The registrant had 17,802,163 shares of common stock
outstanding on March 7, 2011.
DOCUMENTS
INCORPORATED BY REFERENCE
Portions of the registrant’s definitive Proxy Statement for
the 2011 Annual Meeting of Stockholders to be filed with the
Securities and Exchange Commission, or SEC, pursuant to
Regulation 14A not later than 120 days after the end
of the fiscal year covered on this
Form 10-K
are incorporated by reference into Part III,
Items 10-14
of this
Form 10-K.
IRIS
INTERNATIONAL, INC.
ANNUAL
REPORT ON
FORM 10-K
Fiscal
Year Ended December 31, 2010
i
PART I
Forward-Looking
Statements
This report includes “forward-looking statements”
within the meaning of Section 27A of the Securities Act of
1933, as amended, and Section 21E of the Securities and
Exchange Act of 1934, as amended. All statements other than
statements of historical facts are “forward-looking
statements” for purposes of these provisions, including any
projections of earnings, revenues or other financial items, any
statement of the plans and objectives of management for future
operations, any statements concerning proposed new products or
strategic arrangements, any statements regarding future economic
conditions or performance, and any statement of assumptions
underlying any of the foregoing. In some cases, forward-looking
statements can be identified by the use of terminology such as
“may,” “will,” “expects,”
“plans,” “anticipates,”
“estimates,” “potential,”
“intends”, or “continue” or the negative
thereof or other comparable terminology. Although we believe
that the expectations reflected in the forward-looking
statements contained herein are reasonable, there can be no
assurance that such expectations or any of the forward-looking
statements will prove to be correct, and actual results could
differ materially from those projected or assumed in the
forward-looking statements. Our future financial condition and
results of operations, as well as any forward-looking
statements, are subject to inherent risks and uncertainties,
including but not limited to the Risk Factors set forth under
Item 1A, and for the reasons described elsewhere in this
report. All forward-looking statements and reasons why results
may differ included in this report are made as of the date
hereof, and we assume no obligation to update these
forward-looking statements or reasons why actual results might
differ.
Company
Overview
We are a leading manufacturer of automated in-vitro
diagnostics, or IVD, systems and consumables for use in
hospitals and commercial laboratories worldwide, and a provider
of high value personalized diagnostics testing services through
a high complexity molecular diagnostics laboratory. We offer
products that analyze particles and living cell forms and
structures, or morphology of a variety of body fluids. Our IVD
products leverage our strengths in flow imaging technology,
particle recognition and automation to bring efficiency to the
hospital and commercial laboratories, where a shortage of
qualified laboratory technicians and labor-intensive processes
result in significant cost and workflow challenges for our
customers. The initial applications for our technology have been
in the urinalysis market and we are the leading worldwide
provider of automated urine microscopy systems, with
approximately 3,000 systems sold in over 50 countries. In this
market, we also provide integrated solutions comprising urine
microscopy and urine chemistry products as well as consumable
supplies, system support services and sample preparation
products. We intend to expand into related market segments that
can clearly benefit from automated morphology solutions,
including hematology. Also, we have an active research and
development platform in molecular diagnostics based on our
Nucleic Acid Detection Immunoassay, or
NADiA®,
platform, which we are developing for various applications in
personalized diagnostics for oncology. In July 2010, we acquired
a high complexity CLIA-certified molecular pathology laboratory
offering differentiated, high value molecular diagnostic
services in the rapidly growing field of personalized medicine
with emphasis in cancer diagnostics. In addition to its current
testing services, the laboratory, Arista Molecular, Inc. (Arista
or Arista Molecular), will provide a direct commercial channel
for our NADiA platform.
Historically, we have predominantly focused on developing,
manufacturing and commercializing in vitro
diagnostics instruments and consumables for urinalysis,
including our flagship
iQ®
analyzers, a family of fully-automated, image-based bench-top
analyzers for urine microscopy. Urine microscopy is the
visualization and identification of cells and other sediments in
urine. The iQ analyzer uses proprietary flow microscope and
image-analysis software that captures the morphology of cells
and sediments in urine and serous fluids, and assists in their
identification and classification. Our systems are designed to
provide users with faster, more complete and more consistent
results, while substantially reducing hands-on time spent by
laboratory technicians and turnaround time, as compared to
traditional manual methods.
The iQ analyzer can be seamlessly integrated with an automated
urine chemistry analyzer to simultaneously perform urine
microscopy and chemistry testing in a fully automated manner. In
September 2008, we released our
1
proprietary
iChem®VELOCITYtm
automated urine chemistry analyzer and a fully integrated urine
microscopy and urine chemistry work-cell, called the iRICELL in
some international markets. We plan to begin selling these
instruments in the United States contingent upon attaining
clearance from the U.S. Food and Drug Administration (FDA)
on our 510(k) application. Historically in the US, we sold our
family of iQ analyzers integrated with an automated chemistry
analyzer, the AUTION MAX
AX-4280,
which we sourced from a Japanese manufacturer.
We intend to solidify our leadership position in the urinalysis
market, as well as enter into several adjacent markets with our
product pipeline under development. To maintain our market
position in urinalysis, we continue to implement improvements to
our existing product lines, including enhancing our data
analysis and productivity tools for the iQ analyzer, such as
Edit Free Release Technology to improve turnaround time and
iWare to enhance the laboratory information system’s, or
LIS, communication protocol for more effective management of
resources and costs in the screening for negative urine cultures.
We are also developing our 3GEMS (Third Generation Morphology)
platform, which will serve as the basis for our next generation
products in urinalysis and an emerging pipeline of hematology
products. These 3GEMS hematology products, currently in
feasibility testing, use image-based technology to automate the
identification and characterization of blood cells, in
particular, abnormal white blood cells. We believe an automated
hematology analyzer using our proprietary imaging technology and
software recognition capabilities will provide improvements in
the identification of abnormal blood cells, including an
automated, image-based expanded white blood cell differential
analysis. Like our urine microscopy products, these new
hematology products by virtue of IRIS’s inherent
capabilities to capture and analyze images are expected to
significantly reduce the need for manual slide preparation and
reviews, increasing the efficiency of what is currently a highly
labor-intensive process.
We believe a significant driver in the future growth of
diagnostics will be in personalized medicine, meaning the
ability to analyze the molecular
make-up of
an individual patient’s cancer and assess disease
progression in order to more precisely prescribe appropriate
treatment. In order to be in a position to realize the potential
of this segment, in July 2010 we acquired a high complexity CLIA
certified molecular pathology laboratory, renamed Arista
Molecular, to complement our active research and development
program for our proprietary molecular diagnostics platform
called NADiA.
NADiA has the ability to measure proteins below the detection
thresholds of current immunoassay and molecular diagnostic
methods. We believe our proprietary diagnostic products will
address the need for increased sensitivity in the monitoring of
disease enabling personalized treatment of cancers. Our first
product pending regulatory clearance, NADiA
ProsVuetm,
is an ultra-sensitive, blood-based test designed to be a
prognostic indicator by using a threshold based on the slope of
three successive test measurements of residual amounts of
prostate specific antigen, or PSA, in prostate cancer patients
following radical prostatectomy. In April 2010, we submitted our
NADiA ProsVue 510(k) pre-market notification application to the
FDA requesting clearance of the product with a prognostic claim.
In February 2011, we submitted additional data requested by the
FDA for this 510(k) application.
Our Arista Molecular laboratory is oncology focused and offers
differentiated, high value molecular diagnostic services in the
rapidly growing field of personalized medicine. With a
comprehensive test menu, we believe Arista is able to meet the
evolving needs of pathologists and cancer-focused physicians. In
addition, the laboratory will provide us more control over the
commercialization of our NADiA pipeline products, including
sales, marketing and communications programs and value-based
pricing strategies. Further, the laboratory should accelerate
the development efforts of NADiA-based products and enable more
direct access to the clinical end-users. Recently, Arista
Molecular expanded its solid tumor test menu for breast, lung
and colorectal cancer adding flow cytometry for detection and
monitoring of leukemia and lymphoma and fluorescence in situ
hybridization, or FISH, testing for certain cancers. We
believe a broad menu of high value tests along with the
additions of proprietary NADiA assays will provide Arista
Molecular with a significant competitive advantage in the
marketplace.
Our Sample Processing group markets and develops centrifuges,
semi-automated DNA processing workstations and sample processing
consumables. Our StatSpin brand bench-top centrifuges are used
for specimen preparation in coagulation, cytology, chemistry and
urinalysis. Our worldwide markets include medical institutions,
commercial laboratories, clinics, doctors’ offices,
veterinary laboratories and research facilities.
2
Market
Overview and Opportunity
The global market for IVD in 2010 is estimated at approximately
$44 billion and is expected to grow in the mid-single
digits annually. IVD manufacturers provide products and services
to the clinical laboratory industry, which is confronted with
significant challenges in the current market. Healthcare
professionals are demanding improved turnaround time for
diagnostic tests, greater sensitivity and lower costs. To
improve accuracy, productivity and efficiency, many laboratories
are turning to automated methods to perform these tests.
Moreover, automated testing solutions better position
laboratories to cope with the declining number of certified
medical technologists available to perform tests.
Currently, we participate primarily in the urinalysis segment of
the IVD market which we estimate is approximately
$650 million. With our expansion into molecular
diagnostics, we have the opportunity to participate in the
personalized medicine market. PricewaterhouseCoopers estimates
the market for esoteric services and test sales was
$11 billion in 2009, including $3 billion related to
the molecular diagnostics market with a 15% growth rate. We
believe we are well positioned to experience growth in this
segment with our focus on a personalized medicine test menu at
Arista Molecular, which will include our proprietary molecular
tests utilizing our NADiA technology platform.
Urinalysis
Urinalysis is performed as part of most routine medical
examinations and is necessary for the diagnosis and monitoring
of conditions such as urinary tract infection, and kidney and
bladder disease. Traditionally, urinalysis comprises urine
chemistry and urine microscopy tests, while urine cultures are
considered part of microbiology. We believe that the advancement
of automated technologies will blur this distinction, with urine
cultures being performed increasingly in the same laboratories
as urine chemistry and urine microscopy tests and eventually
becoming part of the urinalysis market.
Urine
Chemistry and Microscopy Market Overview
Urine chemistry consists of a panel of tests that identifies
various chemical analytes in urine, while urine microscopy
analyzes the microscopic solid particles and cells suspended in
urine. Urine chemistry comprises the majority of the urinalysis
market and is broadly used, with limited differentiation between
products. Traditional urine microscopy is used less routinely
because as a manual process it is time consuming and requires a
trained medical technician to characterize particles and cells
based on their morphology. In order to reduce costs, many
laboratories perform manual urine microscopy only in response to
results from an initial urine chemistry test despite evidence
that urine microscopy can provide a more reliable clinical
diagnosis. The commercial success of our iQ analyzer is
attributable to its capability to image and accurately identify
particles and cells suspended in urine in a time-efficient
manner eliminating manual microscopic examination.
Of the $650 million urinalysis segment, urine chemistry
represented approximately $490 million and automated urine
microscopy represented approximately $160 million, but
growing at a much faster rate than the other urinalysis
sub-segments.
We estimate there are 13,200 sites performing greater than 40
microscopy tests per day on a global basis including
approximately 6,000 in China. These higher volume sites
represent a significant opportunity for us, because they would
benefit from the automation and consolidation of their urine
chemistry and microscopy procedures. We believe the full
automation and integration of results brought by the iQ product
platform has accelerated the adoption of automated urine
microscopy as a routine test. We believe approximately 40% of
these targeted domestic sites continue to perform manual
microscopy procedures. The penetration of automated urine
microscopy analyzers varies significantly from country to
country.
Limitations
in Urine Chemistry and Microscopy
Current manual testing of urine and body fluids requires the
clinical laboratory to split samples, perform automated and
manual procedures and consolidate the separate results into one
report. Moreover, the manual procedure for microscopy requires a
qualified medical technician to accurately categorize particles
and cells observed under the microscope. Therefore, these tests
represent both time and cost intensive procedures for the
clinical laboratory. Further, the inherent variability in sample
preparation limits the quantitative and qualitative
3
accuracy of the diagnostic result. The manual nature of
performing urine microscopy and the lack of qualified personnel
represents a significant opportunity. However, the challenge
remains to compete for capital for urinalysis automation versus
other disciplines of the laboratory.
Laboratories typically perform microscopy and chemistry tests
separately and generally perform microscopy only in the case of
an abnormal chemistry result because urine microscopy is a very
tedious process. If both tests are performed, the separate
results must then be manually consolidated into one report or
file. Without the automatic integration of both the microscopy
and chemistry results, valuable clinical information may be
overlooked. By reducing the amount of manual labor spent
conducting these tests and by automatically integrating the
chemistry and microscopy results, we believe we can improve the
consistency, reliability and value of the combined results and
improve specimen turnaround time.
Hematology
Hematology
Market Overview
The enumeration of the various cellular components of blood is
an essential part of routine medical examinations. A complete
blood count, or CBC, is the most common type of blood test
performed and measures the number of specific types of blood
cells, including red blood cells (RBC), white blood cells (WBC),
platelets, and other blood components, such as hemoglobin. In
most instances, a white cell differential count, which measures
the percentage of five types of white blood cells, is added to
the CBC test. Variations from concentration, size, or maturity
of the blood cells can be used to indicate an infection or
illness. CBC tests and differential WBC counts are performed
primarily in hospitals and clinical reference laboratories.
According to industry sources, the hematology market is
approximately $2.1 billion and growing approximately 4% per
year driven primarily by system replacement sales and a small
increase in test volume. Over the past 15 years, innovation
within this market has been limited to automation of slide
making and staining and algorithmic improvements to aid in the
interpretation of results.
Limitations
in Hematology
Traditional CBC test instruments use indirect means to measure
the type and number of blood cells rather than direct
observation of the blood cells. Despite the high number of these
automated CBC analyzers in use, a significant percentage of the
samples require a manual cell differential count of the blood
specimen under a microscope. Frequently, a manual count is
required due to the inability of automated CBC and differential
analyzers to discriminate the complex morphology, especially the
shape, of abnormal cells, such as immature white blood cells, or
the presence of diseased cells, as in the case of sickle-cell
anemia. The presence of immature white blood cells is often
associated with conditions such as leukemia, infection,
inflammation or tissue injury. However, a manual differential
count of a blood specimen must be performed by a medical
technologist trained in cytology or a pathologist under a
microscope — a time consuming and subjective process,
resulting in longer specimen turnaround times and higher cost.
According to a 2006 study conducted by the College of American
Pathologists, of the 263 US laboratories surveyed, an average of
29% of automated CBCs required a manual review, scan or
differential and this percentage dramatically increased
depending on the pathology of the patient population. IRIS has
recently confirmed this manual review rate, independently. Since
the hematology market is dominated by a few large companies that
typically compete on their ability to marginally reduce manual
review rates, we believe there is significant opportunity to
offer an automated image-based instrument that has the ability
to identify immature white blood cells and other anomalies in a
systematic fashion and to reduce significantly the number of
manual reviews performed.
Personalized
Medicine
Personalized medicine may be defined as giving the right
treatment to the right patient at the right time. The
implementation of personalized medicine has been made possible
by state of the art molecular diagnostic testing. By examining
the molecular make up of an individual patient’s cancer,
therapy can be tailored for that specific patient, rather than
blindly treating all patients with “one size fits all”
drug cocktails. This leads to better
4
management of the individual patient’s cancer, with fewer
potentially ineffective drugs, reduced incidence of side
effects, and overall cost savings for the healthcare industry.
With the advancement in genomics and proteomics, there is an
accelerating offering of better targeted diagnostics and
therapeutics to aid clinicians in patient management, which is
improving health outcomes. PricewaterhouseCoopers estimates the
market for esoteric services and test sales addressing this
segment was $11 billion in 2009 and is expected to grow to
$21 billion in 2015 with an 11% annual growth rate.
The acquisition of our cancer focused CLIA-certified laboratory,
Arista Molecular, provides us with differentiated, high value
molecular diagnostic services in the rapidly growing field of
personalized medicine. Our menu includes tests for solid tumor
cancers such as lung, colon, breast and prostate, as well as
hematological cancers such as leukemia and lymphoma. According
to the American Cancer Society, there were an estimated 752,210
cases of these solid tumor cancers and approximately 137,260
cases of hematological cancers in 2010 in the US. The number of
these cases is expected to increase due to the growth and aging
of the population and the increase in unhealthy lifestyle
behaviors.
Many smaller community-based pathology groups, hospitals and
laboratories are unable to offer molecular diagnostics as they
lack the required technical expertise, financial resources to
fund the high costs of equipment, and sufficient test volume
necessary to justify their investment in this high complexity
test modality. As a result, the majority of these laboratories
in the US do not offer molecular diagnostic services.
Consequently, this provides a significant opportunity for our
laboratory to offer these complementary high value tests to
broaden these groups’ menu.
Molecular
Diagnostics Market Overview
Molecular diagnostic tests examine nucleic acids, including DNA
and RNA, and protein biomarkers, to identify a disease,
determine prognosis, monitor its progression and response to
treatment, or predict individual predisposition to a disease or
genetic disorder. These biomarkers can also provide information
in drug discovery, preclinical drug development and patient
monitoring during clinical trials. Currently, the clinical
market for molecular diagnostics is primarily nucleic acid
testing performed by real-time polymerase chain reaction, or
PCR, instruments that amplify and detect nucleic acid targets
for diseases or infections. These tests require a
high-complexity CLIA (Clinical Laboratory Improvement Act)
license and are generally performed in commercial reference
laboratories and large academic and research hospital
laboratories due to the complexity and cost of the tests. It is
expected that as more highly automated solutions become
available, these tests may migrate to smaller laboratories and
potentially even to
point-of-care
sites.
The analysis of DNA expression, presence of cell surface
receptors, or the production of specific proteins in cells,
provides the ability to characterize diseases, such as cancer
and infectious diseases. As a result, the detection and
identification of DNA and proteins can provide physicians with a
means to tailor therapy, monitor disease progression and detect
relapse.
Currently, the ability to detect specific proteins in cells is
limited by sensitivity. We believe that development of an
ultra-sensitive detection method for proteins with the
capability to measure concentrations hundreds of times below the
detection limits of currently available immunoassays, may
provide a reliable method to detect disease at an earlier stage,
which may result in improved patient care.
According to PricewaterhouseCoopers, the worldwide molecular
diagnostics market in 2009 was approximately $3 billion and
growing at 15% annually. We believe growth in the market is
being driven primarily by an increase in the number of
personalized diagnostic tests available to treat cancers and
infectious diseases.
Limitations
with Current Methods
Proteins are critical to understanding diseases and current
testing methods lack the degree of sensitivity necessary to
detect minute amounts of protein and the precision to monitor
serial determinations for disease progression. Traditional
methods to detect proteins, including enzyme-linked
immunosorbent assay (ELISA) and chemiluminescence immunoassay
(CIA), are unable to quantify protein biomarkers in extremely
low concentrations. These methods become effective only after a
disease has progressed to a more advanced stage and the
5
concentration of the protein biomarker has increased to reach
the lower limit of detection of those conventional methods. We
believe there is a significant market opportunity for an
ultra-sensitive detection technology that measures concentration
as low as one femtogram per milliliter
(10-15
gram/milliliter) compared to today’s technologies that are
limited to measuring concentrations of greater than 50,000
femtograms per milliliter.
Sample
Processing
Sample
Processing Market Overview
Nearly every sample presented to a clinical laboratory for
testing requires some sort of sample processing before analysis.
These samples include blood, urine and other body fluids,
tissue, stool and other materials which may need to be separated
into its different constituents. In the United States, there are
over 180,000 testing sites where sample processing occurs,
including hospital laboratories, independent laboratories,
doctor’s offices, health maintenance organizations and
community clinics.
Although testing is performed on many different types of
samples, most tests are performed on blood specimens that
require separation in a centrifuge. The centrifuge market
comprises five segments including non-refrigerated bench-top,
refrigerated bench-top, floor, high-speed and ultra-centrifuges.
According to Strategic Directions International, the worldwide
market for sample processing centrifuges in 2010 is estimated at
$660 million, of which the market for non-refrigerated
bench-top centrifuges, the market segment we serve, is estimated
at approximately $100 million. To improve laboratory
productivity and sample turnaround time, the current trend is
toward smaller and faster bench-top models and away from large
capacity floor models which have longer processing time per
batch. This represents a significant opportunity for our Express
line of bench-top centrifuges.
Limitations
with Current Sample Processing Methods
The time it takes to process a sample is critical for clinical
laboratories as the volume of samples to be tested increases.
Each day, laboratory technicians are expected to handle
thousands of samples with minimal error in a defined amount of
time with limited laboratory space. In most laboratories, sample
processing occurs in a central location where blood specimens
are sorted and centrifuged in batches. The entire process can
take up to an hour and requires dedicated resources to manage
the sample flow. Once processed, the samples are often split and
then sent to the various stations within the laboratory for
analysis. The centralized processing of samples is thus quite
inefficient as samples wait to enter the floor model centrifuge
in large batches followed by long centrifugation times. In fact,
many sample processing procedures create significant delays in
specimen turnaround time.
Our
Products
Our commercialized products and product pipeline comprise three
main categories: morphology, personalized medicine and sample
processing. Our morphology category includes all urinalysis and
hematology products consisting of our commercialized urine
chemistry and microscopy products, as well as our
development-stage products such as our 3GEMS urinalysis and
hematology analyzers. Our personalized medicine category
consists of our development-stage products that utilize our
NADiA technology for ultra-sensitive detection of proteins for
monitoring cancer and infectious disease applications and our
recently acquired CLIA-certified oncology laboratory. Our sample
processing category develops and markets small centrifuges and
other processing equipment and accessories for rapid specimen
processing. The table below is a summary of our major
commercialized and in-development products:
| |
|
|
|
|
|
Major Products
|
|
Status
|
|
Description
|
|
|
|
Morphology and Related Products
|
|
|
|
|
|
iQ analyzers (200, Sprint, Elite, Select, Pro and Plus)
|
|
Marketed
|
|
Fully-automated urine microscopy and
body fluids analyzer
|
|
iQ Body Fluids Module
|
|
Marketed
|
|
|
|
Optional iWare Software
|
|
Marketed
|
|
|
6
| |
|
|
|
|
|
Major Products
|
|
Status
|
|
Description
|
|
|
|
iChemVELOCITY
|
|
Launched internationally: 3Q2008
U.S.: pending 510(k) clearance
|
|
Fully-automated urine chemistry analyzer
|
|
iRICELL
|
|
Launched internationally: 3Q2008
U.S.: pending 510(k) clearance
|
|
Integrated iQ and iChemVELOCITY
workcell
|
|
3GEMS Urinalysis and Body Fluids
|
|
In feasibility
|
|
Next generation urine microscopy and
body fluids analyzer
|
|
3GEMS Hematology
|
|
In feasibility
|
|
Complete blood count, white blood cell
count with expanded differentials and red
blood cell and platelet morphology
|
|
Personalized Medicine
|
|
|
|
|
|
Arista Molecular Tests — Molecular pathology
|
|
Marketed
|
|
Application of molecular biology and
molecular genetics at the level of basic
molecules such as DNA, RNA, and
proteins, to aid in the diagnosis and
prognostication of pathologic entities
|
|
— Flow cytometry
|
|
Marketed
|
|
A cell analysis platform using
fluorescence tagged antibodies for enumeration &
characterization of cells
for diagnosis of hematological disorders
|
|
— FISH
|
|
Marketed
|
|
An enhanced molecular cytogenetic
method using fluorescent probes for
detection of DNA on chromosomes in the
intact cell (‘in-situ’)
|
|
NADiA ProsVue
|
|
510(k) submitted 2Q2010
|
|
Prognosticate stable patients after
radical prostatectomy
|
|
NADiA HIV
|
|
Feasibility complete Pursuing licensing partners
|
|
Monitoring HIV viral load during
anti-retroviral therapy
|
|
NADiA CECs
|
|
In feasibility
|
|
Detect circulating epithelial cells to
monitor cancer progression
|
|
Sample Processing
|
|
|
|
|
|
Express centrifuge line
|
|
Marketed
|
|
Centrifuges for clinical diagnostic market
|
|
ThermoBrite
|
|
Marketed
|
|
DNA workstation for FISH procedures
|
|
Cytofuge 2
|
|
Marketed
|
|
Centrifuge used for thin layer cell preparation
|
|
Cytofuge 12
|
|
Marketed
|
|
12 placement centrifuge used for thin layer cell preparation
|
|
IDEXX Drive
|
|
Marketed
|
|
For internal use in IDEXX chemistry analyzers
|
|
IDEXX whole blood separator
|
|
Manufacturing rights licensed to IDEXX
|
|
Consumable used in IDEXX chemistry analyzers
|
|
OvaTube
|
|
Marketed
|
|
Ova and parasite testing for veterinarian market
|
|
Slide based sample preparation system
|
|
In development
|
|
Benchtop platform for automating slide based procedures
including FISH testing
|
Morphology
and Related Products
Cell morphology is the science of cell form and structure. Our
morphology segment utilizes our proprietary imaging technology
to identify cells and particles in a fully automated manner. In
the urinalysis market, we offer urine microscopy analyzers and
related urine chemistry instruments. As part of our 3GEMS Third
Generation Morphology program, we are developing a
next-generation urine microscopy analyzer and an image-based
hematology analyzer.
7
Automated
Urine Microscopy Analyzers
Our flagship product is the family of iQ urine microscopy
analyzers, which was first launched in 2003. Our iQ technology
platform utilizes proprietary image flow cytometry and software
to achieve significant reductions in cost and processing time as
compared to manual urine microscopy. Our technology enables high
speed digital processing to classify and display images of
microscopic particles in an
easy-to-view
graphical user interface. We believe our iQ product line has
numerous benefits over competing products, including increased
accuracy, digital imaging of particles and fully automated
analysis of urine and body fluids and lower manual review rates
of abnormal samples.
The iQ microscopy product line comprises the iQ SELECT, a
fully-automated instrument capable of analyzing 40 samples an
hour and enabling partial automation at laboratory sites with
lower test volumes; the iQ ELITE, a fully-automated instrument
capable of analyzing 70 samples an hour that is appropriate for
mid-sized hospital laboratories; and the iQ SPRINT, a
fully-automated instrument capable of analyzing 101 samples an
hour that is appropriate for large volume hospital and
commercial laboratories. By utilizing our urinalysis system, we
believe the average laboratory, which we define as those
laboratories that typically perform 60 microscopy tests per day,
can re-assign one medical technician currently performing these
tests to another function, with a payback period of
approximately two years. We also offer the iQ Body Fluids Module
as an addition to the iQ urinalysis test menu, which enables the
rapid diagnosis for the presence of nucleated cells, red blood
cells, bacteria and crystals in body fluid samples. In 2010, we
attained 510(k) clearance for the synovial fluid application and
added it to this optional software module.
In 2010, we also introduced the
iRICELL®Plus
and the
iRICELL®
Pro which we believe delivers significant workflow enhancements
and productivity to the laboratory. In addition, we launched
iWAREtm,
an optional, expert software product that further enhances
laboratory productivity by enabling real-time patient validation
based on lab-defined verification rules for urine chemistry and
microscopy results. The iWARE product includes an enhanced LIS
communication protocol allowing for direct electronic
communication between urinalysis and microbiology laboratories
for more effective management of resources and costs in the
screening for negative urine cultures.
Urine
Chemistry Analyzers
We market our proprietary fully-automated urine chemistry
analyzer, the iChemVELOCITY, which can be seamlessly connected
to the higher throughput iQ automated urine microscopy analyzers
and provide laboratories with walk-away solutions for chemistry
and microscopy urinalysis with results combined and displayed in
a single report. The iChemVELOCITY is designed for medium to
high volume laboratories that typically process more than 100
urine chemistry samples per day. We offer the iChemVELOCITY as a
stand-alone analyzer or as part of our integrated urinalysis
workcell solution, the iRICELL.
Internationally, we began selling the iChemVELOCITY in September
2008 following CE Mark certification. In the United States,
sales of our iChemVELOCITY chemistry analyzer and iRICELL
workstations will be contingent upon clearance of our 510(k)
application with the FDA. Historically in the U.S., we sold a
fully-automated urine chemistry analyzers manufactured by
ARKRAY, a Japanese IVD company. Our agreement with ARKRAY allows
us to sell consumable strips for our existing installed base of
AX-4280
analyzers through 2013. We plan to continue to support and
service these instruments through the life of the
customers’ contracts.
Consumables
and Service
We generate significant revenue from the sale of consumables and
service contracts for our urine microscopy and urine chemistry
analyzers. For the year ended December 31, 2010, revenue
derived from consumables and service contracts accounted for 57%
of our total consolidated revenues and 66% of our IDD urinalysis
segment’s revenue. Consumables include urine and body
fluids reagents, calibrators and controls for our microscopy
systems and test strips, calibrators, controls, and other
solutions for the urine chemistry analyzers we manufacture and
distribute. After the initial year of sale, which is covered by
product warranty, we offer annual service contracts for our
domestic and other direct customers in select international
markets. To our distributors, we offer spare parts who in turn
service the end-use customer.
8
3GEMS
Platform
Our 3GEMS platform combines our core imaging technology with
improved software and sample processing to enhance the
identification of various cell types and particles found in
urine, blood and other body fluids. We believe the increased
sensitivity and specificity, reliability, ease of use and
improved imaging of our 3GEMS platform will increase the
clinical utility of our diagnostic tests and allow physicians
and other caregivers to make more informed treatment decisions.
The 3GEMS Platform will be the basis for our next generation
urine microscopy analyzer and improved body fluids module and a
new hematology analyzer product line. The following describes
our 3GEMS products under development:
|
|
|
| |
•
|
Next Generation Urine Microscopy
Analyzers. Our next generation urine microscopy
analyzer will include advances in electronics and optics,
including very high speed color cameras with higher resolution.
We anticipate these advances will improve the clinical utility
of the instrument as a greater number of cell types will be able
to be identified with greater precision.
|
| |
| |
•
|
Next Generation Body Fluids Module. We
currently offer products that utilize our proprietary technology
for morphology analysis of other body fluids, including
cerebrospinal, synovial and serous fluids. We are developing a
next generation body fluids module that we believe will possess
improved diagnostic capabilities relative to our current product
offering.
|
| |
| |
•
|
Hematology Analyzers. We are developing a
portfolio of hematology analyzers to automate the identification
and characterization of cells in blood. Our initial hematology
analyzer will conduct a complete blood count, or CBC, the most
common type of blood test, as well as automatically complete an
expanded white cell differential analysis. This differential
analysis will identify the presence and quantity of immature
white blood cells, whose presence is often associated with
conditions such as leukemia, infection, inflammation or tissue
injury. Importantly, the automation of the white cell
differential will eliminate the need for a medical technologist
trained in cytology or pathologist to manually identify and
count cells under a microscope — a time consuming and
subjective process. Finally, since our analyzer captures digital
images of individual cells, the creation of slides to enable the
review of a blood smear under a microscope and to retain
physical evidence of a particular sample would be significantly
reduced. Our virtual slides will be stored digitally and
transmitted electronically between laboratories or healthcare
providers.
|
Personalized
Medicine
NADiA
(Nucleic Acid Detection Immunoassay) Platform
Our Personalized Medicine category is leveraging our proprietary
NADiA technology platform to develop ultra-sensitive and precise
diagnostic tests. NADiA technology has the ability to measure
proteins in extremely low concentrations below the detection
thresholds of current immunoassay and molecular diagnostic
methods. NADiA combines immunoassay and PCR methodologies, or
Immuno-PCR, with the potential to detect proteins with
femtogram/milliliter sensitivity
(10-15
gram/milliliter). The Immuno-PCR approach is similar to that of
an enzyme immunoassay, which makes use of antibody binding
reactions and washing steps, but in the NADiA method, the enzyme
label is replaced with a double-strand of DNA which is used to
detect and quantify the target protein using PCR amplification.
We believe diagnostic tests that utilize our NADiA technology
will aid in the early detection of disease relapse and
potentially provide better therapeutic outcomes for patients.
Our molecular diagnostics pipeline includes the following
products under development:
|
|
|
| |
•
|
Prostate Cancer: NADiA ProsVue. PSA, or
prostate-specific antigen, is the most widely used cancer marker
for the diagnosis and clinical management of prostate cancer in
men. We have developed an ultra-sensitive blood-based diagnostic
test, called NADiA ProsVue, designed to be a prognostic
indicator by taking the slope of three successive test
measurements of residual levels of PSA that currently are
undetectable with current testing methods, in men with prostate
cancer who have undergone radical prostatectomy, or removal of
the prostate gland. In April 2010, we submitted our NADiA
ProsVue 510(k)
|
9
|
|
|
| |
|
pre-market notification application to the FDA requesting
clearance of a prognostic claim. At the American Society for
Clinical Oncology (ASCO) 2011 Genitourinary Cancers Symposium,
the results of a multi-center clinical study were presented. The
study’s results demonstrate a negative predictive value
(NPV), or the proportion of patients correctly identified as
stable, of 95.2% and a positive predictive value (PPV) of 81.4%.
We believe the data supports our hypothesis that NADiA ProsVue
can contribute useful information as a prognostic indicator in
identifying post-prostatectomy patients with low risk of cancer
recurrence.
|
|
|
|
| |
•
|
HIV: Viral Load Test. Current HIV viral load
tests measure the quantity of HIV RNA in the blood of an
individual infected with HIV. The presence of RNA indicates that
the virus is actively replicating and increasing levels of RNA
can indicate more serious or advanced disease. Viral load
testing is one of the most valuable measures for predicting HIV
disease progression and gauging how well anti-retroviral
treatment is working. Effective anti-retroviral treatment can
often reduce RNA viral load to levels that are undetectable by
current diagnostic tests. Unlike current tests measuring HIV
RNA, our NADiA technology measures a specific HIV viral protein,
p24, which is present in greater numbers relative to HIV RNA.
Specifically, per unit of sample, there are 3,000 p24 molecules
while only two copies of HIV RNA. We have developed an
experimental blood-based HIV viral load p24 assay utilizing
NADiA technology that achieved a sensitivity of one
femtogram/mL, which is below the limit of detection of the most
sensitive FDA cleared HIV RNA-based assay. As a result of our
decision to focus on cancer diagnostics, we are currently
pursuing a development and commercial partner for our NADiA HIV
assay.
|
| |
| |
•
|
Cancer Progression: Circulating Epithelial
Cells. In addition to our NADiA technology, we
have a novel cell isolation technology that makes it possible to
isolate rare cells in the presence of billions of non-target
cells using antibody-coated albumin micro-bubbles to bind to
target cells. We believe our albumin bubbles have significant
competitive advantages in terms of affinity. In addition, the
albumin bubbles can easily collapse and disappear without the
cumbersome step of separating glass bubbles or magnetic beads
from targeted cells, as is necessary with current commercial
isolation technologies. Utilizing our bubble isolation
technology and NADIA, we are developing a test method to
identify, quantify and characterize circulating epithelial cells
present in blood to aid in monitoring cancer progression.
|
Arista
Molecular
In July 2010, we acquired a high complexity CLIA-certified
molecular pathology laboratory, renamed Arista Molecular, in
order to provide a direct commercial channel for NADiA. The
laboratory is oncology focused and offers differentiated, high
value molecular diagnostic services in the rapidly growing field
of personalized medicine. With a comprehensive test menu, we
believe Arista is able to meet the evolving needs of
pathologists and cancer-focused physicians. In addition, we
believe the laboratory will provide us more control over the
commercialization of our NADiA pipeline products, including
sales, marketing and communications programs and value-based
pricing strategies. Further, the laboratory should accelerate
the development efforts of NADiA-based products and enable more
direct access to the clinical end-users. We anticipate that
ProsVue will be the first NADiA test offered by Arista Molecular
following FDA 510(k) clearance.
We are expanding Arista’s test menu to provide a broader
menu of diagnostic panels useful for the diagnosis, disease
characterization, treatment and monitoring of cancer. Currently,
Arista Molecular offers companion molecular tests used to
characterize and tailor cancer therapy for solid tumors,
including lung, colorectal, breast and prostate cancer, flow
cytometry for detection and monitoring of leukemia and lymphoma
and FISH testing for certain cancers. We believe having a broad
menu of high-value tests along with the additions of our
proprietary NADiA assays will provide Arista Molecular with a
significant competitive advantage in the marketplace.
In addition to its menu of high-value tests, Arista Molecular is
striving to differentiate itself in the marketplace by providing
industry-leading turnaround times of less than 72 hours for
these test results, exceptional service to clients and
competitive pricing. By providing valuable diagnostic
information in a rapid manner, the laboratory is able to aid
clinicians in improving patient care. In addition, upon request
the laboratory has the ability to provide pathology consultation
and pathology-guided diagnostic services.
Arista’s primary customers include anatomic pathology
groups, hospitals and regional and community laboratories, many
of which are unable to integrate molecular diagnostics into
their test menu as they lack the
10
required technical expertise, financial resources to fund the
high costs of equipment, and sufficient test volume necessary to
justify the investment in this high complexity test modality. As
a result, the sales strategy of the laboratory is to become
partners with these pathology groups and community-based
hospitals and laboratories by providing complementary high value
services to their current offering. These groups benefit by
being able to broaden their test menu with specialized tests,
without the investment in equipment and technically skilled
personnel necessary to perform the tests.
Arista Molecular continues to build a national sales force to
offer high value testing services directly to our customers. Our
sales people typically have experience in the laboratory
services market focusing on molecular diagnostics with strong
technical backgrounds.
Sample
Processing
Our sample processing group markets and develops centrifuges,
semi-automated DNA processing workstations and sample processing
consumables. Our StatSpin brand bench-top centrifuges are used
for specimen preparation in coagulation, cytology, chemistry and
urinalysis. Our worldwide markets include medical institutions,
commercial laboratories, clinics, doctors’ offices,
veterinary laboratories and research facilities.
With our sample processing products, we believe we offer
laboratories the ability to increase their efficiency by
processing samples as they arrive rather than in a batch mode.
Our bench-top centrifuges offer a significant advantage with two
to three minute cycles compared to conventional centrifuges
taking up to
15-30
minutes, depending on batch size. Further, our bench-top models
are small enough to sit along side an analyzer eliminating the
need for a separate central sample processing area.
Our Express 4 centrifuge employs a unique high speed horizontal
rotor for separating samples and replaces larger and slower
batch centrifuges by reducing sample preparation time and
streamlining laboratory workflow. This product platform enables
us to serve the high-volume chemistry market.
In November of 2010, we acquired the assets of a multi-purpose,
bench-top instrument platform for automating highly repetitive,
manual laboratory protocols for FISH (fluorescence
in-situ hybridization) testing and other slide-based
cytogenetic applications. The product acquisition is a natural
extension to our successful
ThermoBrite®
DNA Hybridization System and consistent with our entry into
personalized medicine with emphasis on cancer diagnostics. The
new product platform will be integrated into our Iris Sample
Processing Division and it is expected to position us as a major
competitor in the high growth cytogenetic instrumentation market.
In November of 2009, we initiated shipments of our StatSpin
Ovatube System for use in veterinary hospitals, clinics and
reference laboratories to process fecal samples for the
detection of ova and parasites. Based on our research, there are
some 20 million ova parasite tests performed on animals
each year in the U.S. alone, with some 15 million of
these tests being performed in 25,000 veterinary clinics.
We have been collaborating with Becton Dickinson, or BD, in
their Lean-Six Sigma initiatives with our Express 3 centrifuge
utilizing BD’s blood collection tubes. Our collaboration
study showed numerous benefits from utilizing our combined
products in the laboratory including decreased turn-around time,
increased productivity and decreased costs. As a result in 2008,
our sample processing group and BD entered into a three-year
distribution agreement. The agreement provides for BD
distributing Express 3 centrifuges on a co-branded basis for use
with its tubes in certain international markets.
Our
Strategy
Our goal is to maintain our leadership position in automated
in vitro diagnostics for urinalysis and sample
processing while becoming a global leader in hematology and
molecular diagnostics by offering products and solutions that
increase laboratory productivity and efficiency and diagnostic
tests that improve patient care. To achieve this goal, we intend
to:
Increase the market adoption of the automated urinalysis
platform in the in vitro diagnostics
market. We strive to develop automated diagnostic
instrumentation and solutions that enable increased laboratory
productivity and efficiency. As we continue to market the
clinical value of urinalysis, specifically in microscopy, we
expect to
11
increase market awareness and demand for our automated
urinalysis products. In addition, we continue to pursue large
multi-unit,
multi-site contracts with large volume laboratories and to
invest in emerging markets to increase our installed base of
instruments.
Broaden our product offerings in the urinalysis IVD
market. We intend to broaden our offerings in the
urinalysis IVD market by developing and commercializing new
products and solutions that improve laboratory efficiency and
provide healthcare providers with more timely and more valuable
information. The iChemVELOCITY, an automated urine chemistry
analyzer, and iRICELL workstation represent key new product
offerings that we launched in the international market in 2008
and for which we will launch domestically contingent upon
attaining FDA clearance. The iChemVELOCITY, when linked with our
iQ microscopy analyzer, allows us to offer an integrated system,
called the iRICELL. In addition, we are leveraging our 3GEMS
imaging technology to develop next-generation urinalysis
instruments to provide our customers with innovative products.
Use our imaging technology expertise to enter the automated
hematology in vitro diagnostics market. We
intend to leverage our imaging technology, which forms the basis
of our market-leading urinalysis products, into other IVD
markets where customers will value automation and increased
efficiency. We are developing a hematology product portfolio
that utilizes our next-generation 3GEMS imaging technology to
reduce the need to prepare manual slides and perform subjective
assessments of those slides under a microscope. We believe our
product, by using a digitally-imaged virtual slide, will
significantly decrease labor spent in laboratories by reducing
the manual examination of abnormal blood samples while improving
standardization.
Enter higher value segments of the market focused on
personalized medicine. We believe that NADiA, our
proprietary molecular diagnostic technology, has the potential
to improve patient care management by allowing for earlier
monitoring of disease and detection of relapse and to improve
patient outcomes due to its ultra-sensitive detection of
proteins. Our portfolio of emerging molecular diagnostic tests
for cancer is designed to provide physicians and patients with
more valuable information that may impact the treatment decision
in managing the course of disease. As a result, we believe our
NADiA platform is well-positioned for growth with the advent of
personalized medicine utilizing genomic and molecular data to
better determine targeted therapies for patients. With the
recent acquisition of our Arista Molecular diagnostics
laboratory focused on oncology, we have a direct commercial
channel for our NADiA products starting with our ProsVue test as
a prognostic indicator for prostate cancer. Currently, the
laboratory provides customers a broad menu of high value tests
focused on personalized medicine, which will be supplemented
with products from our NADiA pipeline.
Pursue selective acquisitions and technology
in-licensing. We will continue to pursue
selective acquisitions to augment our organic growth. Our
acquisition strategy is to target companies and product lines
that complement our business and provide additional
infrastructure necessary for the commercialization of our
pipeline. In 2010, we closed three acquisitions. We acquired a
CLIA-certified laboratory to provide a direct commercial channel
for our NADiA product pipeline. Our Sample Processing division
acquired an automated bench-top instrument for FISH testing.
Lastly, our Iris Diagnostics division acquired the assets from
our European distributor relating to its distribution of IRIS
products in Germany and the UK to provide for an expanded
international direct sales channel for current and future
products.
Competition
Competition in the IVD industry is intense. Many of our
competitors are substantially larger than we are and have
greater financial, research, manufacturing, marketing, sales and
other resources than we do. As a result, our competitors may
develop technologies or products that could render our products
or products under development obsolete or noncompetitive.
Urinalysis
The principal competitive factors in the urinalysis market are
cost-per-test,
ease of use and quality of results. In the automated urine
microscopy segment, Sysmex Corporation markets its automated
non-imaging urine sediment analyzers globally and remains our
principal competitor in the urine microscopy segment.
Elektronika 77, a Hungarian company, offers a slide-based
automated microscopy analyzer that can be run as stand-alone or
in combination with a urine chemistry system. In the urine
chemistry segment, Siemens Healthcare Diagnostics,
12
ARKRAY and Roche Diagnostics are our principal competitors
selling urine analyzers and test strips used in determining the
concentration of various chemical substances found in urine. We
believe our systems provide the highest level of integration of
urine chemistry and microscopy and the broadest menu available
to provide digital images of urine and other body fluids
particles, which provides significant competitive advantages.
We are experiencing increased domestic and international pricing
pressures in the urinalysis market due to the ongoing
consolidation of both hospitals and medical device suppliers,
increasing competition and decreasing reimbursement. Competitors
are attempting to offer one-stop shopping for a variety of
laboratory instruments, supplies and service with price
discounts based on the hospital’s aggregate volume of
business. We have been successful in countering this type of
strategy by our large competitors by negotiating contracts with
group purchasing organizations, or GPOs, in the United States
allowing GPO members to purchase our products at competitive
pricing.
Hematology
Hematology is a mature segment of the in vitro
market in which there are a number of large competitors who
already have an established market presence and significantly
greater resources than we have. The major competitors in the
hematology market include Abbott Laboratories, Beckman Coulter,
Inc., Siemens Healthcare Diagnostics, Sysmex Corporation and
Horiba ABX. Our hematology analyzer currently under development
represents a significant advancement in this market by combining
an automated instrument with image-based expanded white cell
differentials with complete blood counts. We believe this
differentiates us from currently existing products, and will
allow us to make a strong entry into the hematology testing
market.
Personalized
Medicine
In the ultra-sensitive protein detection market, we may
experience competition from companies that utilize enzyme-linked
immunosorbent assay, or ELISA, chemiluminescence and
fluorescence technologies, including Abbott Diagnostics, Beckman
Coulter, Inc., Ortho-Clinical Diagnostics, Inc., Roche
Diagnostics and Siemens Healthcare Diagnostics. Our technology
detects proteins at lower concentrations, which we believe will
enable earlier detection of relapse of disease. Many of these
companies market instruments and reagents for measuring serum
markers in concentrations greater than 50,000 femtograms per
milliliter, while we believe our technology detects
concentrations as low as one femtogram per milliliter.
Arista Molecular participates in the significantly competitive
medical testing laboratory segment. There are several factors
driving competition in the industry including breadth of test
menu, accuracy of results, turnaround time, reputation of
laboratory, quality of service including analysis and pricing.
The segment is dominated by two large national laboratories,
Laboratory Corporation of America and Quest Diagnostics that
have significant market share and resources. There are many
other laboratories competing in the molecular and genetic
testing segment of the market including academic institutions
and specialized laboratories, with new competitors expected to
enter the market.
Sample
Processing
The primary competitive factors in the centrifuge market include
speed, ease of use, size and cost. The major competitors in the
bench-top centrifuge market include the Drucker Company, LW
Scientific and Hettich. With the industry trend moving away from
bulky floor models to smaller, faster, more efficient bench-top
models, we are facing competition from a number of U.S. and
foreign competitors. We believe our products are differentiated
due to innovative design, single push button operation, small
footprint, quiet cycle and rapid separation time.
Intellectual
Property
We have a long history of innovation. Our diversified core
technology spans through a number of scientific endeavors, which
include IVD, immuno-assay, rare cell separation technology,
specimen processing and handling, pattern recognition and image
analysis. Our commercial success depends on our ability to
protect and maintain our proprietary rights. We protect our
proprietary technology by filing various patent applications
domestically and in
13
many foreign countries. We own various active patents and have
pending patent applications for our technologies domestically
and internationally.
These patents cover developments in imaging analysis and
processing software, blood processing, digital refractometers,
fluidics, centrifuges, immuno-PCR processes, rare cell
separation, automated slide handling and disposable urinalysis
products sold by us. In addition, we have various patents
related to products of our sample processing business segment.
These patents have various useful lives ranging from five to
15 years with expirations ranging from one to
15 years. Our core IVD patents in the United States will
start to expire in 2017.
For our molecular platform technologies, we have a license for
three patents from the University of California that cover the
use of nucleic acid labeled antibodies in immunoassays. In
addition, we filed a patent on the improved use of DNA labeled
antibodies and obtained patents covering NADiA and our bubble
isolation technology methods. In October of 2010, Iris Molecular
Diagnostic was granted a patent by the European Patent Office,
which covers important aspects of the NADiA technology. This
patent will be effective until November of 2024 and will be in
force in numerous countries throughout Europe. In addition to
this patent, Iris Molecular Diagnostics has also applied for
numerous other patents throughout the world to obtain additional
patent protection for our NADiA platform.
We have trade secrets, unpatented technology and proprietary
knowledge about the sale, promotion, operation, development and
manufacturing of our products. We have confidentiality
agreements with our employees and consultants to protect these
rights.
We claim copyright in our source code and have a patent in the
ways in which our software displays images, and have filed
copyright registrations with the United States Copyright Office.
We also own various federally registered trademarks, including
“IRIS”, “iChem”, “iQ”,
“NADiA”, “ThermoBrite”, and
“StatSpin.” We own other registered and unregistered
trademarks, and have certain trademark rights in foreign
jurisdictions. We intend to aggressively protect our patents,
copyrights and trademarks.
Third-Party
Payor Reimbursements
Successful sales of our products in the United States and other
countries will depend on the availability of reimbursement from
third-party payors such as private insurance plans, managed care
organizations, and Medicare and Medicaid. In the United States,
the American Medical Association assigns to diagnostics tests
specific Current Procedural Terminology, or CPT, codes, which
are necessary for reimbursement of those tests. Once the CPT
code is established, the Center for Medicare and Medicaid
Services, or CMS, establish reimbursement payment levels and
coverage rules under Medicaid and Medicare, and private payors
establish rates and coverage rules independently. Our urinalysis
tests are covered by established CPT codes and are therefore
approved for reimbursement by Medicare and Medicaid as well as
most third-party payors.
However, we may develop tests in the future that do not relate
to previously established CPT codes and we may need to obtain
new CPT codes in order to obtain reimbursement. Reimbursement by
a third-party payor depends on a number of factors, including
the level of demand by health care providers and the
payor’s determination that the use of the product
represents a clinical advance or a reduction in the overall cost
of treatment. In addition, in the United States, third-party
payors and state governments routinely review reimbursement
coverage for diagnostic tests considering budgetary constraints
vis-à-vis demonstrated clinical efficacy. Also, outside of
the United States, health care reimbursement systems vary from
country to country, and to the extent we sell our products
outside the United States, we may not be able to obtain adequate
reimbursement coverage, if any, for our products.
Arista accepts assignment for Medicare patients as payment in
full on Medicare covered tests. Reimbursement under the Medicare
program for most of our laboratory testing is subject to the
national Medicare clinical laboratory fee schedule. Payment
under the clinical laboratory fee schedule has been limited from
year-to-year
by Congressional action such as imposition of national
limitation amounts and freezes on the otherwise applicable
annual consumer price index, or CPI, updates. Reimbursement from
third-party insurance companies varies widely, even from a
single payor in a given geographic area and population.
Insurance companies often follow the lead of Medicare in
determining whether a clinical laboratory test is covered and
reimbursable. The amount that we are able to be reimbursed from
private health insurers is based on several factors, including
the type of health insurance
14
coverage (for example, health maintenance organization or
preferred provider organization), whether the services are
considered to be in network or out of network by the health
insurance provider, and the amount of any co-pays or deductibles
for which the patient is responsible.
Depending on our billing arrangement with each third party payor
and applicable law, we are often obligated to bill in the
specific manner prescribed by the various payors, such as
private insurance companies, managed care companies,
governmental payors such as Medicare and Medicaid, physicians,
hospitals and employer groups, each of which may have different
billing requirements. Additionally, the audit requirements we
must meet to ensure compliance with applicable laws and
regulations as well as our internal compliance policies and
procedures add further complexity to the billing process.
Government
Regulations
Our products are subject to stringent government regulation in
the United States and other countries. These laws and
regulations govern product testing, manufacture, labeling,
storage, record keeping, distribution, sale, marketing,
advertising and promotion. The regulatory process can be
lengthy, expensive and uncertain, and securing clearances or
approvals often requires the submission of extensive testing and
other supporting information. If we do not comply with
regulatory requirements, we may be subject to fines, recall or
seizure of products, total or partial suspension of production,
withdrawal of existing product approvals or clearances, refusal
to approve or clear new applications or notices and criminal
prosecution. Further, any change in existing federal, state or
foreign laws or regulations, or in their interpretation or
enforcement, or the enactment of any additional laws or
regulations, could affect us both materially and adversely.
In the United States, the FDA regulates medical devices under
the Food, Drug, and Cosmetics Act. Before a new medical device
can be commercially introduced in the United States, the
manufacturer usually must obtain FDA clearance by filing a
pre-market notification, or PMA, under Section 510(k) of
the Food, Drug, and Cosmetics Act, or obtain FDA approval by
filing a PMA application. The PMA application process is
significantly more complex, expensive, time-consuming and
uncertain than the 510(k) notification process. To date, we have
cleared all of our regulated products with the FDA through the
510(k) notification process. We cannot guarantee that we will be
able to use the 510(k) notification process for future products.
Furthermore, FDA clearance of a 510(k) notification or approval
of a PMA application is subject to continual review, and the
subsequent discovery of previously unknown facts may result in
restrictions on a product’s marketing or withdrawal of the
product from the market.
We are also required to register as a medical device
manufacturer with the FDA and comply with FDA regulations
concerning good manufacturing practices for medical devices, or
GMP Standards. In 1997, the FDA expanded the scope of the GMP
Standards with new regulations requiring medical device
manufacturers to maintain control procedures for the design
process, component purchases and instrument servicing (Quality
System Regulation or QSR). The FDA biannually inspects our
manufacturing facilities for compliance with GMP Standards. We
believe that we are in substantial compliance with the QSR.
Labeling, advertising and promotional activities for medical
devices are subject to scrutiny by the FDA and, in certain
instances, by the U.S. Federal Trade Commission. The FDA
also enforces statutory and policy prohibitions against
promoting or marketing medical devices for unapproved uses.
Clinical laboratory diagnostic tests that are developed and
validated by a laboratory for use in testing that the laboratory
performs itself are called “home brew” or laboratory
developed tests (“LDT”). Like many other laboratories,
Arista has developed and validated certain “home brew”
tests. The FDA maintains that it has authority to regulate the
development and use of “home brews” as diagnostic
medical devices, but historically has elected not to exercise
its enforcement authority with regard to most “home
brew” tests. The FDA indicated in June 2010 that it is
considering exercising greater oversight of laboratory developed
tests using a risk-based approach, but has taken no further
action at this time.
Many states have also enacted statutory provisions regulating
medical devices. The State of California’s requirements in
this area, in particular, are extensive, and require
registration with the state and compliance with regulations
identical to the GMP Standards established by the FDA. While the
impact of such laws and regulations
15
has not been significant to date, it is possible that future
developments in this area could affect us both materially and
adversely.
In addition to domestic regulation of medical devices, many of
our products are subject to regulations in foreign
jurisdictions. The requirements for the sale of medical devices
in foreign markets vary widely from country to country, ranging
from simple product registrations to detailed submissions
similar to those required by the FDA. Our business strategy
includes expanding the geographic distribution of these and
other products, and we cannot guarantee that we will be able to
secure the necessary clearances and approvals in the relevant
foreign jurisdictions. Furthermore, the regulations in certain
foreign jurisdictions continue to develop and we cannot be sure
that new laws or regulations will not have a material adverse
effect on our existing business or future plans. Among other
things, CE Mark certifications are required for the sale of many
products in certain international markets such as the European
Community. We have secured CE Mark certification for our
existing product lines.
We have obtained European Norm ISO 13485:2003 certification for
our manufacturing facilities and are subject to surveillance by
European notified bodies. In addition, our Chatsworth
manufacturing facility is certified to ISO 13485:2003 with the
Canadian Medical Devices Conformity Assessment System, or
CMDCAS, which is the regulatory protocol used by Health Canada
to certify manufacturers.
Our products are also subject to regulation by the
U.S. Department of Commerce export controls, primarily as
they relate to the associated computers and peripherals. We have
not experienced any material difficulties in obtaining necessary
export licenses to date.
Federal
and State Clinical Laboratory Certification and
Licensing
Our Arista laboratory, similar to almost all clinical
laboratories operating in the United States, is required to
maintain federal certification pursuant to the Clinical
Laboratory Improvement Act, as amended, commonly known, together
with its implementing regulations, as CLIA. The CLIA regulations
have established three levels of regulatory control based on
test complexity: “waived,” “moderate
complexity” and “high complexity.” Arista has
staffed and organized its San Diego, California clinical
laboratory facility to meet the standards for a “high
complexity” test laboratory, the most rigorous level of
quality. CLIA imposes requirements relating to test processes,
personnel qualifications, facilities and equipment, record
keeping, quality assurance and participation in proficiency
testing, which involve comparing the results of testing of
specimens that have been specifically prepared for our
laboratory to the known values of those specimens. Compliance
with CLIA standards is verified by periodic
on-site
inspections. CLIA accreditation is maintained through regular
inspections by the College of American Pathologists, and
therefore, subject to its requirements and evaluation. The CLIA
requirements also apply as a condition for participation for
clinical laboratories in the Medicare and Medicaid programs.
CLIA does not preempt state laws that are more stringent than
federal law. Therefore, similar requirements also apply to our
laboratory under California’s clinical laboratory licensure
laws. The State of California Department of Health and Human
Services — Laboratory Field Services enforces the
state’s requirements to apply for and maintain licensure,
CLIA certification, and proficiency testing. Our facilities have
been inspected by these authorities and have been issued
licenses to manufacture medical devices and provide laboratory
diagnostic services in California. These licenses must be
renewed every year. The State of California could prohibit our
provision of laboratory services if we failed to maintain these
licenses. Sanctions for failure to meet these certification,
accreditation and licensure requirements may include suspension,
limitation or revocation of certification, accreditation or
licensure, civil penalties, criminal penalties, injunctive
actions, and the imposition of plans of correction to remedy
deficiencies. If a laboratory’s CLIA certificate or
California license is revoked or suspended, the laboratory must
then cease performing testing, until licensure is reinstated.
Antifraud
Laws/Overpayments.
Federal Medicare/Medicaid laws apply a wide array of fraud and
abuse provisions to laboratories that participate in these
programs. These laws include the federal False Claims Act, and
prohibit, among other things: the submission of false claims or
false information to government programs; deceptive or
fraudulent conduct; and the provision and billing for excessive
or unnecessary services. Federal law also prohibits fraud on
private sector health insurers. Penalties for violating these
laws may include exclusion from participation in the
Medicare/Medicaid
16
programs, asset forfeitures, civil penalties and criminal
penalties. If an entity is determined to have violated the
federal False Claims Act, it may be required to pay up to three
times the actual damages sustained by the government, plus civil
penalties ranging from $6,000 to $11,000 for each separate false
claim. While there are many potential bases for liability under
the federal False Claims Act, such liability primarily arises
when an entity knowingly submits, or causes another to submit, a
false claim for reimbursement to the federal government.
Submitting a claim or failing to repay an overpayment with
reckless disregard or deliberate ignorance of its validity could
result in substantial civil liability. Exclusion from the
Medicare and Medicaid programs is mandatory for certain
offenses, and regulators also have the authority to impose
permissive exclusions from Medicare and Medicaid programs in
response to a wide range of less serious misconduct. In
addition, the CMS may suspend Medicare payments to any provider
it believes has engaged in fraudulent billing practices. Because
the financial consequences to a laboratory of exclusion from
participation in federal health care payment programs would
typically be devastating, fear of such exclusion has been a
motivating factor in the settlement of many fraud investigations.
California law extends similar penalties beyond Medicare to
punish laboratories engaged in conduct which defrauds the
Medi-Cal program, private insurers or patients. California law
also denies Medi-Cal enrollment to any provider that has entered
into a settlement in lieu of conviction for fraud or abuse in
any government program and further provides that a provider
under investigation by certain governmental agencies for fraud
or abuse will be subject to a temporary suspension from Medi-Cal
pending investigation.
Independent of fraud allegations, Medicare and Medicaid programs
and private payors may also retroactively determine that certain
payments for services must be repaid due to failure of a
laboratory to satisfy applicable payor requirements.
Federal
and California “Self-Referral”
Restrictions.
A federal “self-referral” law, commonly referred to as
the “Stark” law, prohibits Medicare payments for
laboratory tests referred by physicians who, personally or
through a family member, have ownership interests in or
compensation arrangements with the testing laboratory. Sanctions
for violations of the self-referral prohibitions include denial
of payment, refunds, civil money penalties of up to $15,000 for
each service billed in violation of the prohibitions and
exclusion from the Medicare program. In addition, claims
submitted in violation of the Stark Law may also give rise to
liability under the federal False Claims Act and its
“whistleblower” provisions. Several states, including
California, have enacted legislation that prohibits physician
self-referral arrangements
and/or
requires physicians to disclose to their patients any financial
interest they may have with a healthcare provider when referring
patients to that provider. Some of these statutes, including
California’s, cover all patients and are not limited to
beneficiaries of Medicare, Medicaid or other federal healthcare
programs. Possible sanctions for violating state physician
self-referral laws vary, but may include loss of license and
civil and criminal sanctions.
Anti-Kickback
Laws
Existing federal laws governing Medicare and Medicaid, and other
similar state laws, impose a variety of broadly described
restrictions on financial relationships among healthcare
providers, including clinical laboratories. The
Medicare/Medicaid antikickback statute prohibits laboratories
from paying for patient or specimen referrals for testing paid
for by Medicare or Medicaid. Violation of the Medicare/Medicaid
antikickback statute can result in criminal penalties pursuant
to the U.S. sentencing guidelines, civil monetary penalties
of $50,000 per violation plus treble damages, and exclusion from
Medicare and Medicaid participation. The OIG has criticized a
number of additional business practices in the clinical
laboratory industry as potentially implicating the antikickback
statute, including providing phlebotomy staff to clients who
perform clerical or other functions for the client which are not
solely related to the collection or processing of laboratory
specimens, providing computers or fax machines to clients which
are not used exclusively in connection with performance of the
laboratory’s work, the lease of space in a physician’s
office for rent which exceeds the fair rental value of such
space, certain acquisition agreements where the sellers may make
referrals to the buyer after the sale and various other
compensation relationships between laboratories and entities
from which they receive referrals, or to which they make
referrals, if such relationships are intended to induce
referrals. In addition, the OIG has indicated that discounts
given by
17
laboratories to clients concerning their private pay patients
and/or HMO
patients must not be intended to induce a client to refer
Medicare or Medicaid patients to the laboratory.
Health
Insurance Portability and Accountability Act and HITECH
Act
The Health Insurance Portability and Accountability Act of 1996,
or HIPAA, protects the security and privacy of individually
identifiable health information. HIPAA standards govern the
conduct of certain electronic transmission of health care
information and to protect the security and privacy of
individually identifiable health information maintained or
transmitted by health care providers. These standards include:
1) The Standards for Electronic Transactions establishes
standards for common health care transactions such as claims
information, plan eligibility, and payment information. It also
establishes standards for the use of electronic signatures,
unique identifiers for providers, employers, health plans, and
individuals.
2) The Standards for Privacy of Individually Identifiable
Information restricts the use and disclosure of certain
individually identifiable health information. The HIPAA privacy
and security regulations establish a uniform federal
“floor” and do not supersede state laws that are more
stringent or provide individuals with greater rights with
respect to the privacy or security of, and access to, their
records containing patient/private health information (PHI). As
a result, we are required to comply with both HIPAA privacy
regulations and varying state privacy and security laws, which
include physical and electronic safeguard requirements. These
laws contain significant fines and other penalties for wrongful
use or disclosure of PHI.
The federal Health Information Technology for Economic and
Clinical Health Act, or HITECH, enacted in February 2009,
strengthens and expands the HIPAA privacy and security rules and
its restrictions on use and disclosure of PHI. HITECH includes,
but is not limited to, prohibitions on exchanging patient
identifiable health information for remuneration, restrictions
on marketing to individuals and obligations to agree to provide
individuals an accounting of virtually all disclosures of their
health information. Violation of HITECH may result in penalties
ranging from $100 — $50,000 per violation depending
upon the violation category, subject to a $1.5 million cap
for multiple violations of an identical requirement or
prohibition in a calendar year.
Other
Regulatory Requirements
Our laboratory is subject to federal, state and local
regulations relating to the handling and disposal of regulated
medical waste, hazardous waste and biohazardous waste, including
chemical, biological agents and compounds, blood and bone marrow
samples and other human tissue. Typically, we use outside
vendors who are contractually obligated to comply with
applicable laws and regulations to dispose of such waste. These
vendors are licensed or otherwise qualified to handle and
dispose of such waste. Historically, our costs associated with
handling and disposal of such wastes have not been material.
The Occupational Safety and Health Administration, or OSHA, has
established extensive requirements relating to workplace safety
for healthcare employers, including requirements to develop and
implement programs to protect workers from exposure to
blood-borne pathogens by preventing or minimizing any exposure
through needle stick or similar penetrating injuries.
Summary
of Revenues by Product Line
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
|
|
|
2010
|
|
|
|
|
|
2009
|
|
|
|
|
|
2008
|
|
|
|
|
|
|
|
($ in thousands)
|
|
|
|
|
Revenues
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
IDD instruments
|
|
$
|
32,083
|
|
|
|
30
|
%
|
|
$
|
26,018
|
|
|
|
28
|
%
|
|
$
|
35,128
|
|
|
|
37
|
%
|
|
IDD consumables and service
|
|
|
61,112
|
|
|
|
57
|
%
|
|
|
52,213
|
|
|
|
56
|
%
|
|
|
46,643
|
|
|
|
49
|
%
|
|
Sample Processing instruments and supplies
|
|
|
14,408
|
|
|
|
13
|
%
|
|
|
14,335
|
|
|
|
16
|
%
|
|
|
13,731
|
|
|
|
14
|
%
|
|
Personalized Medicine services
|
|
|
69
|
|
|
|
0
|
%
|
|
|
—
|
|
|
|
0
|
%
|
|
|
—
|
|
|
|
0
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenues
|
|
$
|
107,672
|
|
|
|
100
|
%
|
|
$
|
92,566
|
|
|
|
100
|
%
|
|
$
|
95,502
|
|
|
|
100
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
18
See Note 19 to the Consolidated Financial Statements,
“Segment and Geographic Information,” for financial
information regarding our operating segments and geographic
areas.
Backlog
We did not have a material amount of backlog as of
December 31, 2010. Our products usually ship within 30 to
60 days of receipt of sales orders. We do not believe that
backlog is necessarily indicative of sales for any succeeding
period.
Employees
At March 1, 2011, we had 377 full-time employees,
including 329 in the United States and 48 internationally. We
also use outside consultants and part-time and temporary
employees in production, administration, marketing, research and
development and engineering. None of our employees are covered
by collective bargaining agreements. We consider our relations
with our employees to be satisfactory.
Corporate
Information
We incorporated in California in 1979 and reincorporated in
Delaware in 1987. We currently have ten facilities, three in
California, one in Massachusetts, one in Marburg, Germany, and
sales offices in France, Germany, the United Kingdom, Japan and
Hong Kong.
Available
Information
We make available our annual reports on
Form 10-K,
quarterly reports on
Form 10-Q,
current reports on
Form 8-K,
along with any related amendments and supplements on our website
as soon as reasonably practicable after we electronically file
or furnish such materials with or to the Securities and Exchange
Commission, or the SEC. These reports are available, free of
charge, at www.proiris.com. Our website and the
information contained in it and connected to it do not
constitute part of this annual report or any other report we
file with, or furnish, to the SEC.
We have identified the following additional risks and
uncertainties that may have a material adverse effect on our
business, financial condition or results of operations.
Investors should carefully consider the risks described below
before making an investment decision. The risks described below
are not the only ones we face. Additional risks not presently
known to us or that we currently believe are immaterial may also
significantly impair our business operations. Our business could
be harmed by any of these risks. The trading price of our common
stock could decline due to any of these risks, and investors may
lose all or part of their investment.
Risks
Related to Our Business
Adverse
conditions in the global economy and disruption of financial
markets could negatively impact our customers and therefore our
results of operations.
An economic downturn in the businesses or geographic areas in
which we sell our products could reduce demand for these
products and result in a decrease in sales volume that could
have a negative impact on our results of operations. Volatility
and disruption of financial markets could limit our
customers’ ability to obtain adequate financing or credit
to purchase and pay for our products in a timely manner, or to
maintain operations, and result in a decrease in sales volume
that could have a negative impact on our results of operations.
In the past few years, we have faced adverse macro economic
forces, which have impacted our selling markets and the credit
markets of our customers. At this point the impact from these
forces are relatively mild, but in the future we may face the
following challenges: deferrals of purchases due to decreases in
capital budgets of our customers, delays in the purchasing cycle
due to greater scrutiny of deals and increased internal
competition for limited capital dollars, and a significant
increase in requests for quotes for operating leases. The
aforementioned
19
factors may lead to a decrease in revenue, an increase of
deferred revenue, or could lead to installment cash collection.
Our
success depends largely on the continued acceptance of our iQ
and iChem product lines.
Our current strategy assumes that our instrument platforms will
be adopted by a large number of end-users. We have invested and
continue to invest a substantial amount of our resources in
promotion and marketing of the iQ and iChem product lines in
order to increase their market penetration, expand sales into
new geographic areas and enhance and expand the system features.
Failure of our instrument operating platforms to achieve and
maintain a significant market presence, or the failure to
successfully implement our promotion and marketing strategy,
will have a material adverse effect on our financial condition
and results of operations.
If we
fail to obtain, or experience significant delays in obtaining,
regulatory clearances or approvals for our products or product
enhancements, our ability to commercially distribute and market
our products could suffer.
Our products are subject to rigorous regulation by the
U.S. Food and Drug Administration, or FDA, and numerous
other Federal, state and foreign governmental authorities. Our
failure to comply with such regulations could lead to the
imposition of injunctions, suspensions or loss of regulatory
clearances or approvals, product recalls, termination of
distribution, product seizures or civil penalties. In the most
egregious cases, criminal sanctions or closure of our
manufacturing facilities are possible. The process of obtaining
regulatory authorizations to market a medical device,
particularly from the FDA, can be costly and time consuming, and
there can be no assurance that such authorizations will be
granted on a timely basis, if at all. In particular, the FDA
permits commercial distribution of a new class II
or III medical device only after the device has received
510(k) clearance or is the subject of an approved pre-market
approval, or PMA, application. The FDA will clear marketing of a
medical device through the 510(k) process if it is demonstrated
that the new product is substantially equivalent to other
510(k)-cleared products. The PMA approval process is more
costly, lengthy and uncertain than the 510(k) clearance process.
Introduction to the market of products we develop that require
regulatory clearance or approval may be delayed. In addition,
because we cannot assure you that any new products or any
product enhancements we develop will be subject to the shorter
510(k) clearance process, the regulatory approval process for
our products or product enhancements may take significantly
longer than anticipated. There is no assurance that the FDA will
not require that a new product or product enhancement go through
the lengthy and expensive PMA approval process. To date, all of
our class II or III products have been cleared through
the 510(k) process. We anticipate several of our molecular
diagnostics products under development will be reviewed by the
FDA under a PMA filing.
Foreign governmental authorities that regulate the manufacture
and sale of medical devices have become increasingly stringent,
and to the extent we continue to market and sell our products in
foreign countries, we will be subject to rigorous regulation in
the future. In such circumstances, we would rely significantly
on our distributors to comply with the varying regulations, and
any failures on their part could result in restrictions on the
sale of our products in foreign countries.
There is no assurance that U.S. or foreign regulatory
bodies will ultimately allow market clearance or approval for
these products. Regulatory delays or failures to obtain
clearances and approvals could disrupt our business, harm our
reputation and adversely affect our sales.
Modifications
to our products may require new regulatory clearances or
approvals or may require us to recall or cease marketing our
products until clearances are obtained.
Modifications to our products may require new 510(k) clearances
or PMA approvals or require us to recall or cease marketing the
modified devices until these clearances or approvals are
obtained. The FDA requires device manufacturers to initially
make and document a determination of whether or not a
modification requires a new approval, supplement or clearance. A
manufacturer may determine that a modification could not
significantly affect safety or efficacy and does not represent a
major change in its intended use, so that no new 510(k) is
necessary. However, the FDA can review a manufacturer’s
decision and may disagree. The FDA may also on its own
initiative determine that a new clearance or approval is
required. We have made modifications to our products in the past
and
20
may make additional modifications in the future that we believe
do not or will not require additional clearances or approvals.
If the FDA disagrees and requires new clearances or approvals
for the modifications, we may be required to recall and to stop
marketing our products as modified, which could require us to
redesign our products and harm our operating results. In these
circumstances, we may be subject to significant enforcement
actions.
If a manufacturer determines that a modification to an
FDA-cleared device could significantly affect its safety or
efficacy, or would constitute a major change in its intended
use, then the manufacturer must file for a new 510(k) clearance
or possibly a PMA approval. Where we determine that
modifications to our products require a new 510(k) clearance or
PMA approval, we may not be able to obtain those additional
clearances or approvals for the modifications or additional
indications in a timely manner, or at all. For those products
sold in the European Union, we must notify our E.U. competent
authority by means of revision of our technical file, if
significant changes are made to the products or if there are
substantial changes to our quality assurance systems affecting
those products. Delays in obtaining required future clearances
or approvals would adversely affect our ability to introduce new
or enhanced products in a timely manner, which in turn would
harm our future growth.
Any
failure to introduce our future products and systems
successfully into the market could adversely affect our
business.
Our commercial success depends on the timely development of new
products that are needed for future growth. These new products
depend on our success in demonstrating technical feasibility and
achieving cost targets and functionality demanded by the market.
Even if our product development efforts are successful and even
if the requisite regulatory approvals are obtained, our products
may not gain market acceptance among physicians, healthcare
payers and the medical community. A number of additional factors
may limit the market acceptance of products including the
following:
|
|
|
| |
•
|
rate of adoption by healthcare providers;
|
| |
| |
•
|
rate of our products’ acceptance by the target market;
|
| |
| |
•
|
timing of market entry relative to competitive products;
|
| |
| |
•
|
availability of alternative products;
|
| |
| |
•
|
price of our product relative to alternative products;
|
| |
| |
•
|
availability of third-party reimbursement; and
|
| |
| |
•
|
extent of marketing efforts by us and third-party distributors
or agents retained by us.
|
Failure to achieve clinical acceptance will adversely impact our
financial condition and results of operations.
We
face intense competition and our failure to compete effectively,
particularly against larger, more established companies will
cause our business to suffer.
The healthcare industry is highly competitive. We compete in
this industry based primarily on product performance, service
and price. Many of our competitors have substantially greater
financial, technical and human resources than we do, and may
also have substantially greater experience in developing
products, obtaining regulatory approvals and manufacturing and
marketing and distribution. As a result, they may be better able
to compete for market share, even in areas in which our products
may be superior. Further, our competitive position could be
harmed by the establishment of patent protection by our
competitors or other companies. The existing competitors or
other companies may succeed in developing technologies and
products that are more effective or affordable than those being
developed by us or that would render our technology and products
less competitive or obsolete. If we are unable to effectively
compete in our market, our financial condition and results of
operation will materially suffer.
21
If we
choose to acquire new businesses, products or technologies, we
may experience difficulty in the identification or integration
of any such acquisition, and our business may
suffer.
Our commercial success depends on our ability to continually
enhance and broaden our product offerings in response to
changing customer demands, competitive pressures and
technologies. Accordingly, we may in the future pursue the
acquisition of complementary businesses, products or
technologies instead of developing them ourselves. We do not
know if we will be able to identify or complete any
acquisitions, or whether we will be able to successfully
integrate any acquired business, product or technology or retain
key employees. Integrating any business, product or technology
we acquire could be expensive and time consuming, disrupt our
ongoing business and distract our management. Moreover, we may
fail to realize the anticipated benefits of any acquisition. If
we are unable to integrate any acquired businesses, products or
technologies effectively, our business will suffer. In addition,
any amortization or charges resulting from acquisitions could
adversely affect our operating results.
If we
do not establish strategic partnerships to commercialize our
products under development, we will have to undertake
commercialization efforts on our own, which could be costly and
may ultimately be unsuccessful.
We may selectively partner with other companies to obtain
assistance for the commercialization of certain of our products.
We may enter into strategic partnerships with third parties to
develop and commercialize some of our products that are intended
for larger markets or that otherwise require a large,
specialized sales and marketing organization, and we may enter
into strategic partnerships for products that are targeted
beyond our selected target markets. We face competition in
seeking appropriate strategic partners, and these strategic
partnerships can be intricate and time consuming to negotiate
and document. We may not be able to negotiate strategic
partnerships on acceptable terms, or at all. We are unable to
predict when, if ever, we will enter into any strategic
partnerships because of the numerous risks and uncertainties
associated with establishing strategic partnerships. If we are
unable to negotiate strategic partnerships for our products
under development, we may be forced to reduce the scope of our
sales or marketing activities or undertake commercialization
activities at our own expense. In addition, we will bear the
entire risk related to the commercialization of these products.
If we elect to increase our expenditures to fund
commercialization activities on our own, we may need to obtain
additional capital, which may not be available to us on
acceptable terms, or at all.
Changes
in reimbursement fees or lower than anticipated reimbursement
for diagnostics tests could reduce demand and the price at which
we can sell our products.
Successful sales of our products will depend on the availability
of adequate coverage and reimbursement from third-party payors
both domestically and internationally. Healthcare providers that
purchase medical devices generally rely on third-party payors to
reimburse all or part of the costs and fees associated with the
procedures performed with these devices. Both public and private
insurance coverage and reimbursement plans are central to new
product acceptance. Customers are unlikely to use our products
if they do not receive reimbursement adequate to cover the cost
of our products.
To date, reimbursement has generally been available for the
diagnostic tests that our products perform. However, all
third-party coverage and reimbursement programs, whether
government funded or insured commercially, whether inside the
United States or outside, are developing increasingly
sophisticated methods of controlling healthcare costs through
limitations on covered items and services, prospective
reimbursement and capitation programs, group purchasing,
redesign of benefits, second opinions required prior to major
surgery, careful review of bills, encouragement of healthier
lifestyles and exploration of more cost-effective methods of
delivering healthcare. These types of programs and legislative
changes to coverage and reimbursement policies could potentially
limit the amount which healthcare providers may be willing to
pay for medical devices.
We believe that future coverage and reimbursement may be subject
to increased restrictions both in the United States and in
international markets. Third-party reimbursement and coverage
for our products may not be available or adequate in either the
United States or international markets. Future legislation,
regulation, coverage or reimbursement policies of third-party
payors may adversely affect the demand for our existing products
or our products currently under development, and limit our
ability to sell our products on a profitable basis.
22
If we
fail to meet changing demands of technology, we may not continue
to be able to compete successfully with our
competitors.
The market for our products and systems is characterized by
rapid technological advances, changes in customer requirements,
and frequent new product introductions and enhancements. Our
future success depends upon our ability to introduce new
products that keep pace with technological developments, enhance
current product lines and respond to evolving customer
requirements. Our failure to meet these demands could result in
a loss of our market share and competitiveness and could harm
our revenues and results of operations.
Our
ability to protect our intellectual property and proprietary
technology through patents and other means is
uncertain.
Our success depends significantly on our ability to protect our
intellectual property and proprietary technologies. We rely on
patent protection, as well as a combination of copyright, trade
secret and trademark laws, and nondisclosure, confidentiality
and other contractual restrictions to protect our proprietary
technology. However, these legal means afford only limited
protection and may not adequately protect our rights or permit
us to gain or keep any competitive advantage.
Our pending U.S. and foreign patent applications may not
issue as patents or may not issue in a form that will be
advantageous to us. Any patents we have obtained or do obtain
may be challenged by re-examination, opposition or other
administrative proceeding, or may be challenged in litigation,
and such challenges could result in a determination that the
patent is invalid. In addition, competitors may be able to
design alternative methods or devices that avoid infringement of
our patents. To the extent our intellectual property protection
offers inadequate protection, or is found to be invalid, we are
exposed to a greater risk of direct competition. If our
intellectual property does not provide adequate protection
against our competitors’ products, our competitive position
could be adversely affected, as could our business. Both the
patent application process and the process of managing patent
disputes can be time consuming and expensive. Furthermore, the
laws of some foreign countries may not protect our intellectual
property rights to the same extent as do the laws of the United
States.
In addition to pursuing patents on our technology, we have taken
steps to protect our intellectual property and proprietary
technology by entering into confidentiality agreements and
intellectual property assignment agreements with our employees,
consultants, corporate partners and, when needed, our advisors.
Such agreements may not be enforceable or may not provide
meaningful protection for our trade secrets or other proprietary
information in the event of unauthorized use or disclosure or
other breaches of the agreements, and we may not be able to
prevent such unauthorized disclosure. Monitoring unauthorized
disclosure is difficult, and we do not know whether the steps we
have taken to prevent such disclosure are, or will be, adequate.
In the event a competitor infringes upon our patents or other
intellectual property rights, litigation to enforce our
intellectual property rights or to defend our patents against
challenge, even if successful, could be expensive and time
consuming and could require significant time and attention from
our management. We may not have sufficient resources to enforce
our intellectual property rights or to defend our patents
against challenges from others. Additionally, our earliest
patents begin to expire in 2010, which may allow our
competitors, many of which may have substantial resources and
have made substantial investments in competing technologies, to
develop technologies previously protected by these expiring
patents
The
medical device industry is characterized by patent litigation,
and we could become subject to litigation that could be costly,
result in the diversion of our management’s time and
efforts, require us to pay damages or prevent us from selling
our products.
The medical device industry is characterized by extensive
litigation and administrative proceedings over patent and other
intellectual property rights. Whether or not a product infringes
a patent involves complex legal and factual issues, the
determination of which is often uncertain. Our competitors may
assert that they own U.S. or foreign patents containing
claims that cover our products, their components or the methods
we employ in the manufacture or use of our products. In
addition, we may become a party to an interference proceeding
declared by the U.S. Patent and Trademark Office to
determine the priority of invention. Because patent applications
can take many years to issue and in many instances at least
18 months to publish, there may be applications now pending
of
23
which we are unaware, which may later result in issued patents
that contain claims that cover our products. There could also be
existing patents, of which we are unaware, that contain claims
that cover one or more components of our products. As the number
of participants in our industry increases, the possibility of
patent infringement claims against us also increases.
Any interference proceeding, litigation or other assertion of
claims against us may cause us to incur substantial costs, could
place a significant strain on our financial resources, divert
the attention of our management from our core business and harm
our reputation. If the relevant patents were upheld as valid and
enforceable and we were found to be infringing, we could be
required to pay substantial damages
and/or
royalties and could be prevented from selling our products
unless we could obtain a license or were able to redesign our
products to avoid infringement. Any such license may not be
available on reasonable terms, if at all. If we fail to obtain
any required licenses or make any necessary changes to our
products or technologies, we may be unable to make, use, sell or
otherwise commercialize one or more of our products. In
addition, if we are found to willfully infringe, we could be
required to pay treble damages, among other penalties.
We
operate in a consolidating industry that creates barriers to our
market penetration.
The healthcare industry in recent years has been characterized
by consolidation. Large hospital chains and groups of affiliated
hospitals prefer to negotiate comprehensive supply contracts for
all of their supply needs at once. Large suppliers can often
equip an entire laboratory and offer hospital chains and groups
one-stop shopping for laboratory instruments, supplies and
service. Larger suppliers also typically offer pricing discounts
to their customers based on the customer’s total volume of
business with the supplier. The convenience and discounts
offered by these large suppliers are administrative and
financial incentives that we do not offer our customers. Our
plans for further market penetration in the urinalysis market
and penetration into the hematology and molecular diagnostics
markets will depend in part on our ability to overcome these and
any new barriers resulting from continued consolidation in the
healthcare industry. The failure to overcome such barriers could
have a material adverse effect on our financial condition or
results of operation.
Changes
in government regulation of the healthcare industry could
adversely affect our business.
Federal and state legislative proposals are periodically
introduced or proposed that would affect major changes in the
healthcare system, nationally, at the state level or both.
Future legislation, regulation or payment policies of Medicare,
Medicaid, private health insurance plans, health maintenance
organizations and other third-party payors could adversely
affect the demand for our current or future products and our
ability to sell our products on a profitable basis. Moreover,
healthcare legislation is an area of extensive and dynamic
change, and we cannot predict future legislative changes in the
healthcare field or their impact on our industry or our
business. Any impairment in our ability to market our products
could have a material adverse effect on our financial condition
and results of operation.
We may
not be able to fully realize deferred tax assets.
As of December 31, 2010, we have deferred tax assets of
approximately $5.8 million resulting from the differences
between the financial statement and the tax bases of assets and
liabilities using enacted tax rates in effect for the year in
which the differences are expected to reverse. Management
believes it is more likely than not that the deferred tax assets
will be realized through future taxable income or alternative
tax strategies. However, the net deferred tax assets could be
reduced in the near term if management’s estimates of
taxable income during the carryforward period are not realized
or are significantly reduced or alternative tax strategies are
not available. Although we believe that the deferred tax asset
is recoverable, there is no assurance that we will be able to
generate taxable income in the years that the differences
reverse.
24
We
rely on independent and some single-source suppliers for key
components of our instruments. Any delay or disruption in the
supply of components may prevent us from selling our products
and negatively impact our operations.
Certain of our components are obtained from outside vendors, and
the loss or breakdown of our relationships with these outside
vendors could subject us to substantial delays in the delivery
of our products to our customers. Furthermore, certain key
components of our instruments and consumables are manufactured
by only one supplier. Because this supplier is the only vendor
with which we have a relationship for a particular component, we
may be unable to sell products this supplier becomes unwilling
or unable to deliver components meeting our specifications. Our
inability to sell products to meet delivery schedules could have
a material adverse effect on our reputation in the industry, as
well as our financial condition and results of operation.
We may
not be able to maintain contracts with Group Purchasing
Organizations, or GPOs.
A significant portion of our domestic sales are affected through
agreements with participant members of GPOs. A failure to renew
one or more GPO contracts may have a material effect on our
domestic sales.
Our
success depends on our ability to attract, retain and motivate
management and other skilled employees.
Our success depends in significant part upon the continued
services of key management and skilled personnel. Competition
for qualified personnel is intense and there are a limited
number of people with knowledge of, and experience in, our
industry. We do not have employment agreements with most of our
key employees nor maintain life insurance policies on them. The
loss of key personnel, especially without advance notice, or our
inability to hire or retain qualified personnel, could have a
material adverse effect on our ability to maintain our
technological edge and ultimately our financial condition and
results of operations. We cannot guarantee that we will continue
to retain our key management and skilled personnel, or that we
will be able to attract, assimilate and retain other highly
qualified personnel in the future.
The
expense and potential unavailability of insurance coverage for
us, our customers or our products may have an adverse effect on
our financial position and results of operations.
Our products are used to gather information for medical
decisions and diagnosis. Accordingly, a defect in the design or
manufacture of our products, or a failure of our products to
perform for the use that we specify, could have a material
adverse effect on our reputation in the industry and subject us
to claims of liability arising from inaccurate or allegedly
inaccurate test results. Misuse of our products by a technician
that results in inaccurate or allegedly inaccurate test results
could similarly subject us to claims of liability. While we
currently have insurance for our business, property, directors
and officers, and products, insurance is increasingly costly and
the scope of coverage is more narrow, and we may be required to
assume more risk in the future. If we are subject to claims or
suffer a loss or damage in excess of our insurance coverage, we
will be required to cover the amounts in excess of our insurance
limits. If we are subject to claims or suffer a loss or damage
that is outside of our insurance coverage, we may incur
significant costs associated with loss or damage that would have
a material adverse effect on our financial position and results
of operations. Furthermore, any claims made on our insurance
policies may impact our ability to obtain or maintain insurance
coverage at reasonable costs, or at all. We do not have the
financial resources to self-insure, and it is unlikely that we
will have these financial resources in the foreseeable future.
We
have product liability insurance that covers our products and
business operation, but we may need to increase and expand this
coverage commensurate with our expanding business.
Any product liability claims brought against us, with or without
merit, could result in:
|
|
|
| |
•
|
substantial costs of related litigation or regulatory action;
|
| |
| |
•
|
substantial monetary penalties or awards;
|
| |
| |
•
|
decreased demand for our products;
|
25
|
|
|
| |
•
|
reduced revenue or market penetration;
|
| |
| |
•
|
injury to our reputation;
|
| |
| |
•
|
an inability to establish new strategic relationships;
|
| |
| |
•
|
increased product liability insurance rates; and
|
| |
| |
•
|
an inability to secure continuing coverage.
|
Furthermore, any impairment of our reputation could have a
material adverse effect on our sales and prospects for future
business. We have not received any indication that our insurance
carrier will not renew our product liability insurance at or
near current premiums; however, we cannot guarantee that this
will continue to be the case. In addition, any failure to comply
with FDA regulations governing manufacturing practices could
hamper our ability to defend against product liability lawsuits.
Our
facilities are located near known earthquake fault zones, and
the occurrence of an earthquake or other catastrophic disaster
could damage our facilities and equipment, which could cause us
to curtail or cease operations.
Instruments for our diagnostics division are manufactured in a
single facility in the San Fernando Valley, Arista
Molecular is located in San Diego, CA and Iris Molecular
Diagnostics is located in Carlsbad, California, all of which are
near known earthquake fault zones and, therefore, are vulnerable
to damage from earthquakes. We are also vulnerable to damage
from other types of disasters, such as power loss, fire, and
similar events. If any disaster were to occur, our ability to
operate our business could be seriously impaired. We currently
may not have adequate insurance to cover our losses resulting
from disasters or other similar significant business
interruptions, and we do not plan to purchase additional
insurance to cover such losses due to the cost of obtaining such
coverage. Any significant losses that are not recoverable under
our insurance policies could seriously impair our business,
financial condition and prospects.
If we
are unable to manage our growth, our results could
suffer.
We have been experiencing significant growth in the scope of our
operations. This growth has placed significant demands on our
management, as well as operational resources. In order to
achieve our business objectives, we anticipate that we will need
to continue to grow. If this growth occurs, it will continue to
place additional significant demands on our management and our
operational resources, and will require that we continue to
develop and improve our operational, financial and other
internal controls both in the United States and internationally.
In particular, if our growth continues, it will increase the
challenges in implementing appropriate control systems,
expanding our sales and marketing infrastructure capabilities,
providing adequate training and supervision to maintain high
quality standards, and preserving our cultural values. The main
challenge associated with our growth has been the management of
our expenses. Our inability to scale our business appropriately
or otherwise adapt to growth, could cause our business,
financial condition and results of operations to suffer.
To
market and sell our products, we depend on third-party
distributors, and they may not be successful.
We currently depend on third-party distributors to sell our IVD
products in most markets outside of the United States as
well as all Iris Sample Processing products. The quarterly and
yearly demand from distributors depends on factors such as
buying patterns, cash availability, currency exchange rates,
etc. If these distributors are not successful in selling our
products, we may be unable to increase or maintain our level of
revenue. Our distributors may not commit the necessary resources
to market and sell our products. If current or future
distributors do not perform adequately or if we are unable to
locate distributors in particular geographic areas, we may not
realize revenue growth internationally. Over the long term, we
intend to grow our business internationally, and to do so we
will need to attract additional distributors to expand into new
territories or to sell new products.
26
Our
sales in international markets are subject to a variety of laws
and political and economic risks that may adversely impact our
sales and results of operations in certain
regions.
Our ability to capitalize on growth in international markets is
subject to risks including:
|
|
|
| |
•
|
changes in currency exchange rates that impact the price to
international consumers;
|
| |
| |
•
|
the burdens of complying with a variety of foreign laws and
regulations;
|
| |
| |
•
|
unexpected changes in local regulatory requirements; and
|
| |
| |
•
|
the difficulties associated with promoting products in
unfamiliar cultures;
|
| |
| |
•
|
subject to general, political and economic risks in connection
with our international sales operations, including:
|
| |
| |
•
|
political instability;
|
| |
| |
•
|
changes in diplomatic and trade relationships; and
|
| |
| |
•
|
general economic fluctuations in specific countries or markets.
|
Any of the above mentioned factors could adversely affect our
sales and results of operations in international markets.
We are
subject to currency fluctuations.
We are exposed to certain foreign currency risks in
international markets where we source, produce, market or sell
product. Some consumable chemistry strips and spare parts for
our installed base of chemistry analyzers are sourced from
ARKRAY, a supplier located in Kyoto, Japan. Our purchases from
this supplier are denominated in Japanese Yen and any
fluctuations in the U.S. Dollar/Japanese Yen exchange rate
could result in increased costs for these key components. We
anticipate the exposure to the Japanese Yen to decrease as we no
longer source instruments from Japan. For consumables, the
exposure will gradually decrease as we begin replacing
AX-4280
machines with our iChemVELOCITY domestically, contingent upon us
attaining FDA clearance.
We are also exposed to currency fluctuations with respect to the
exportation of our products. With the exception of France,
Germany and the United Kingdom where our operations are
denominated in Euros and British Pounds, most of our sales are
denominated in U.S. Dollars. Accordingly, any fluctuation
in the exchange rate between the U.S. Dollar and the
currency of the country with which we are exporting products
could also affect our ability to sell internationally. To the
extent we conduct operations abroad in non-local currency,
fluctuations in the exchange rate between functional currency of
those operations and the local currency may lead to significant
increased costs and could negatively impact our profitability.
Risks
Related to Our Laboratory Business
We
have a limited operating history in our laboratory business and
have experienced operating losses making it difficult to
evaluate whether we will operate profitably.
Arista, formerly AlliedPath, was established in March 2008 and
commenced operation of its clinical laboratory business in
November of 2008. Arista began testing services in August 2009
upon receiving its CLIA license. Because Arista only recently
commenced its principal operations, we do not have a meaningful
historical record of sales and revenues nor an established
business track record. While we believe that we have the
opportunity to be successful in the specialized, cancer-focused
molecular diagnostics segment of the clinical laboratory
industry, there can be no assurance that we will be successful
in accomplishing our business initiatives, or that we will be
able to achieve any significant levels of revenues or net
income, from the sale of Arista’s products and services.
Unanticipated problems, expenses and delays are frequently
encountered in increasing marketing and sales efforts and
developing new products, especially in the current stages of our
business. Our ability to continue to
27
successfully develop, produce and sell our products and to
generate significant operating revenues will depend on our
ability to, among other matters, successfully market and sell
our services to medical providers.
Changes
in healthcare laws, regulations or policies, or third-party
payor policies could affect coverage or reimbursement for our
specialized laboratory tests and adversely affect our financial
results.
Government payors, including Medicare and Medicaid and
third-party payors, such as insurance companies, have
historically implemented measures to control the cost,
utilization and delivery of healthcare services. In the past,
Congress has implemented changes to healthcare laws, which have
resulted in amendments to the Medicare fee schedule, new
administrative requirements and added costs for healthcare
service providers. In the future, additional Congressional
intervention may results in further reductions of reimbursement
rates and additional costs to operate clinical laboratories like
Arista. These changes have, and in the future may have,
adversely affected coverage for the specialized tests that we
offer.
Any
changes to the current healthcare laws and regulations may
necessitate a change to our business model.
The clinical laboratory industry is a highly regulated industry.
Our clinical laboratory is subject to numerous federal and state
laws and regulations, including with respect to anti-kickback,
billing and claims, privacy, security and electronic
transactions (including HIPAA), and the regulation of
laboratories, including CLIA. While we believe that we are
currently in compliance with all of these laws and regulations,
they may be, or the way the courts interpret these laws and
regulations, may be, changed or amended at anytime. Any such
changes may necessitate a change to our business model or result
in increased costs of compliance, which may adversely affect our
business, prospects and the results of our operations.
U.S.
healthcare reform legislation may result in significant changes,
and our business could be adversely impacted if we fail to
adapt.
Government oversight of and attention to the healthcare industry
in the United States is significant and increasing. In March
2010, U.S. federal legislation was enacted to reform
healthcare. The legislation provides for reductions in the
Medicare clinical laboratory fee schedule of 1.75% for five
years beginning in 2011 and also includes a productivity
adjustment that reduces the CPI market basket update beginning
in 2011. The legislation imposes an excise tax on the seller for
the sale of certain medical devices in the United States,
including those purchased and used by laboratories, beginning in
2013. The legislation establishes the Independent Payment
Advisory Board, which will be responsible, beginning in 2014,
annually to submit proposals aimed at reducing Medicare cost
growth while preserving quality. These proposals automatically
will be implemented unless Congress enacts alternative proposals
that achieve the same savings targets. Further, the legislation
calls for a Center for Medicare and Medicaid Innovation that
will examine alternative payment methodologies and conduct
demonstration programs. The legislation provides for extensive
health insurance reforms, including the elimination of
pre-existing condition exclusions and other limitations on
coverage, fixed percentages on medical loss ratios, expansion in
Medicaid and other programs, employer mandates, individual
mandates, creation of state and regional health insurance
exchanges, and tax subsidies for individuals to help cover the
cost of individual insurance coverage. The legislation also
permits the establishment of accountable care organizations, a
new healthcare delivery model. While the ultimate impact of the
legislation on the healthcare industry is unknown, it is likely
to be extensive and may result in significant change. Our
failure to adapt to these changes could have a material adverse
effect on our business.
We may
incur substantial penalties including fines or possible
debarment from Medicare or Medicaid if we violate certain
government payor laws and regulations.
We are required to comply with a number of different government
regulations relating to billing practices and relationships with
physicians and hospitals. Violations of these regulations can
lead to civil penalties, criminal liability and possible
disbarment from participation in Medicare and Medicaid. If we
were ever determined to be in violation of these regulations our
relationship with many of our clients would be severely affected
and we could face a material adverse effect to our financial
condition or results of operations.
28
Our
laboratory business may be harmed if we are unable to comply
with certain healthcare related laws and regulations, including
the Clinical Laboratory Improvement Act of 1967 and the Clinical
Laboratory Improvement Amendments of 1988 (collectively,
CLIA).
Arista is regulated and licensed by a number of federal and
state statutes and regulations, including CLIA. If a clinical
laboratory fails to comply with CLIA requirement or state
healthcare and licensing laws and regulations, it can have its
license suspended or revoked. At this time we believe that we
comply with these laws and regulations. If these laws or
regulations are amended, we may have to change the manner in
which we conduct our laboratory business, which could lead to
additional expenses in running our laboratory business. If we
are unable to properly comply with these changes, we could have
our licenses suspended or revoked, which would adversely affect
our laboratory operations.
We
face possible litigation, penalties and damage to our reputation
with payors, physicians and patients if we are unable to
properly protect customer-related information.
Arista receives a variety of personal information about our
customers. We believe that we have a strong security system in
place and that we comply with all federal and state laws and
regulations relating to the protection of personal information
(including HIPAA). However, if there was a breach of our
security system or we inadvertently released protected patient
information, we face possible monetary fines, litigation and
damage to our reputation.
We
face possible litigation, penalties and medical malpractice for
errors in laboratory testing results.
Arista utilizes the best practices in quality assurance to avoid
and prevent errors in test results. However, if the laboratory
produces an inadvertent erroneous result, this can adversely
affect a patient’s diagnosis and treatment and may result
in medical malpractice litigation for which fines and damages
can be substantial and could exceed the limits of our medical
malpractice insurance coverage, causing substantial damage to
the our financial condition.
The
nature of the testing performed in our laboratory is complex and
often depends on our ability to obtain specialized reagents from
a sole source provider. Any disruption of supply, or
discontinuance of products by a sole source may severely impact
Arista’s ability to provide testing services.
Many of the tests performed by our laboratory are not
commercially available and are laboratory-developed tests (LDT).
Often LDTs rely on specialized reagents or products that are
supplied by a single source. Any disruption in our ability to
obtain these specialized reagents may result in the
laboratory’s inability to continue to perform such testing,
or may require that the laboratory change its testing protocol
to perform the testing using a different reagent. In either
case, this can severely affect our financial performance or harm
our relationship with our customers.
Federal,
state and CLIA regulations define certain personnel standards
that our laboratory must adhere to, including education,
background and experience requirements. Our ability to attract,
hire and maintain critical personnel may limit the testing our
laboratory can perform.
The laboratory relies on its ability to attract, hire and keep
certain critical personnel such as Medical Directors and
Pathologists, and licensed laboratory personnel who have
appropriate advanced degrees and years of experience necessary
to satisfy CLIA requirements and certain state requirements. If
the laboratory cannot attract, hire and maintain these critical
employees, then the laboratory’s ability to perform certain
testing may be severely limited. This can damage the
laboratory’s reputation with its customers and severely
impact our financial performance.
The
laboratory industry is highly competitive and many other
laboratories offer the same testing that our laboratory offers.
Our ability to discriminate our laboratory in a cluttered
marketplace may limit our ability to attract and maintain
customers and will have a material adverse effect on our
financial performance.
There are many competitors currently offering the same menu of
testing and in some cases a larger menu of tests than Arista. We
primarily compete with large national laboratories that have
significant market share and
29
resources. We also compete with many other laboratories,
including academic institutions and specialized laboratories,
with new competitors expected to enter the market. If we fail to
compete effectively, our business could be adversely affected
and our revenues and profitability could be damaged. Our ability
to distinguish our laboratory from our competitors is critical
to our financial performance.
Discontinuation
or recalls of existing testing products or failure to develop,
or acquire, licenses for new or improved testing technologies
could adversely affect the Company’s
business.
From time to time, manufacturers discontinue or recall reagents,
test kits or instruments used by the Company to perform
laboratory testing. Such discontinuations or recalls could
adversely affect the Company’s costs, testing volume and
revenue.
The clinical laboratory industry is subject to changing
technology and new product introductions. The Company’s
success in maintaining a leadership position in genomic and
other advanced testing technologies will depend, in part, on its
ability to develop, acquire or license new and improved
technologies on favorable terms and to obtain appropriate
coverage and reimbursement for these technologies. The Company
may not be able to negotiate acceptable licensing arrangements
and it cannot be certain that such arrangements will yield
commercially successful diagnostic tests. If the Company is
unable to license these testing methods at competitive rates,
its research and development costs may increase as a result. In
addition, if the Company is unable to license new or improved
technologies to expand its esoteric testing operations, its
testing methods may become outdated when compared with the
Company’s competition and testing volume and revenue may be
materially and adversely affected.
Risks
Related to Ownership of Our Common Stock
We
have adopted a number of anti-takeover measures that may depress
the price of our common stock.
Our stockholders rights plan, our ability to issue additional
shares of preferred stock and some provisions of our certificate
of incorporation and bylaws could make it more difficult for a
third party to make an unsolicited takeover attempt of us.
Additionally, we are subject to the anti-takeover provisions of
Section 203 of the Delaware General Corporation Law, which
regulates corporate acquisitions. These anti-takeover measures
may depress the price of our common stock by making it more
difficult for third parties to acquire us by offering to
purchase shares of our stock at a premium to its market price
without approval of our board of directors. They could also have
the effect of preventing changes in our management.
Our
quarterly sales and operating results may fluctuate in future
periods, and if we fail to meet expectations the price of our
common stock may decline.
Our quarterly sales and operating results have fluctuated
significantly in the past and are likely to do so in the future
due to a number of factors, many of which are not within our
control. If our quarterly sales or operating results fall below
the expectations of investors or securities analysts, the price
of our common stock could decline substantially. Factors that
might cause quarterly fluctuations in our sales and operating
results include the following:
|
|
|
| |
•
|
variation in demand for our products, including seasonality;
|
| |
| |
•
|
our ability to develop, introduce, market and gain market
acceptance of new products and product enhancements in a timely
manner;
|
| |
| |
•
|
our ability to manage inventories, accounts receivable and cash
flows;
|
| |
| |
•
|
our ability to control costs;
|
| |
| |
•
|
the size, timing, rescheduling or cancellation of orders from
consumers and distributors; and
|
| |
| |
•
|
our ability to forecast future sales and operating results and
subsequently attain them.
|
The amount of expenses we incur depends, in part, on our
expectations regarding future sales. In particular, we expect to
continue incurring substantial expenses relating to the
marketing and promotion of our products. Since
30
many of our costs are fixed in the short term, if we have a
shortfall in sales, we may be unable to reduce expenses quickly
enough to avoid losses. If this occurs, we will not be
profitable. Accordingly, you should not rely on
quarter-to-quarter
comparisons of our operating results as an indication of our
future performance.
|
|
|
Item 1B.
|
Unresolved
Staff Comments.
|
None.
We lease all of our facilities. Our headquarters are located at
9158 Eton Avenue, Chatsworth, California 91311. The table below
sets forth certain information regarding our leased properties
as of March 1, 2011.
| |
|
|
|
|
|
|
|
|
|
|
|
Approximate Floor
|
|
Monthly
|
|
|
|
Location
|
|
Space (Sq. Ft.)
|
|
Rent
|
|
Use
|
|
|
|
Chatsworth, CA
|
|
98,446
|
|
$
|
74,725
|
|
|
Corporate headquarters and manufacturing (Corporate and Iris
Diagnostics Division)
|
|
Westwood, MA
|
|
29,220
|
|
$
|
34,967
|
|
|
Sample Processing division
|
|
Carlsbad, CA
|
|
14,941
|
|
$
|
17,675
|
|
|
IRIS Molecular Diagnostic research division (Personalized
Medicine)
|
|
San Diego, CA
|
|
12,716
|
|
$
|
20,354
|
|
|
Arista Molecular laboratory and testing facility. (Personalized
Medicine)
|
|
Marburg, Germany
|
|
8,539
|
|
$
|
16,775
|
|
|
Urine chemistry strip manufacturing facility (Iris Diagnostics
Division)
|
|
Marburg, Germany
|
|
4,844
|
|
$
|
2,115
|
|
|
Urine chemistry strip manufacturing warehouse (Iris Diagnostics
Division)
|
|
Marburg, Germany
|
|
953
|
|
$
|
1,734
|
|
|
Urine chemistry strip R&D facility (Iris Diagnostics
Division)
|
|
Paris, France
|
|
2,684
|
|
$
|
3,117
|
|
|
Sales office (Iris Diagnostics Division)
|
|
Causeway Bay, HK
|
|
120
|
|
$
|
1,744
|
|
|
Sales office (Iris Diagnostics Division)
|
|
Cologne, Germany
|
|
130
|
|
$
|
2,520
|
|
|
Sales office (Iris Diagnostics Division)
|
|
Cambridge, UK
|
|
323
|
|
$
|
1,452
|
|
|
Sales office (Iris Diagnostics Division)
|
We believe our facilities are adequate to meet our current and
near-term needs.
|
|
|
Item 3.
|
Legal
Proceedings.
|
From time to time, we are party to certain litigation arising in
the normal course of business. Management believes that the
resolution of such matters will not have a material adverse
effect on our financial position, results of operations or cash
flows. We are not currently involved in any litigation that
requires disclosure in this report.
31
PART II
|
|
|
Item 5.
|
Market
for Registrant’s Common Equity, Related Stockholder Matters
and Issuer Purchases of Equity Securities.
|
Our common stock is listed on the NASDAQ Global Market under the
symbol “IRIS.” The closing price of our common stock
on March 7, 2011 was $9.30 per share. The table below sets
forth the high and low closing prices of our common stock on the
NASDAQ Global Market for the periods ended:
| |
|
|
|
|
|
|
|
|
|
|
|
Price per Share
|
|
|
|
Low
|
|
High
|
|
|
|
Fiscal 2010:
|
|
|
|
|
|
|
|
|
|
First Fiscal Quarter
|
|
$
|
10.21
|
|
|
|
12.08
|
|
|
Second Fiscal Quarter
|
|
|
9.79
|
|
|
|
11.86
|
|
|
Third Fiscal Quarter
|
|
|
7.34
|
|
|
|
10.12
|
|
|
Fourth Fiscal Quarter
|
|
|
8.80
|
|
|
|
10.44
|
|
|
Fiscal 2009:
|
|
|
|
|
|
|
|
|
|
First Fiscal Quarter
|
|
$
|
8.60
|
|
|
|
13.33
|
|
|
Second Fiscal Quarter
|
|
|
10.03
|
|
|
|
12.94
|
|
|
Third Fiscal Quarter
|
|
|
9.50
|
|
|
|
11.81
|
|
|
Fourth Fiscal Quarter
|
|
|
9.99
|
|
|
|
12.62
|
|
As of March 7, 2011 we had approximately 1,914 holders of
record of our common stock.
Dividend
Policy
We have never paid any cash dividends on our common stock. We
currently expect to retain any future earnings for use in the
operation and expansion of our business and do not anticipate
paying any cash dividends on our common stock in the foreseeable
future. Furthermore, we may not pay any cash dividends on the
common stock without the written consent of our lender.
32
FIVE-YEAR
STOCK PRICE PERFORMANCE COMPARISON (1)
The following graph and table compare the cumulative total
return on our common stock with the cumulative total return
(including reinvested dividends) of the Standard &
Poor’s 500 Index (S&P 500), the NASDAQ Medical
Devices, Instruments and Supplies, Manufacturers and
Distributors Stocks Index and the Standard &
Poor’s HealthCare Equipment Index for the five years ending
December 31, 2010, assuming that the relative value of the
common stock and each index was $100 on December 31, 2005.
Amounts below have been rounded to the nearest dollar. The stock
price performance on the following graph is not necessarily
indicative of future stock price performance.
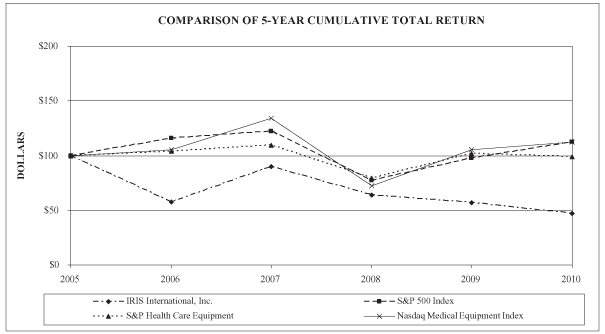
|
|
|
|
(1) |
|
This section is not “soliciting material,” is not
deemed “filed” with the SEC and is not to be
incorporated by reference into any filing of IRIS International,
Inc. under the Securities Act of 1933, as amended, or the
Securities Exchange Act of 1934, as amended, whether made before
or after the date hereof and irrespective of any general
incorporation language in any such filing. |
33
|
|
|
Item 6.
|
Selected
Financial Data.
|
The following selected financial data should be read in
conjunction with our consolidated financial statements. The
information set forth below is not necessarily indicative of
results of future operations, and should be read in conjunction
with Item 7, “Management’s Discussion and
Analysis of Financial Condition and Results of Operations”
and the consolidated financial statements and notes thereto
included in Item 8, “Financial Statements and
Supplementary Data” of this
Form 10-K
in order to understand fully factors that may affect the
comparability of the financial data presented below.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
2010
|
|
2009
|
|
2008
|
|
2007
|
|
2006
|
|
|
|
(In thousands, except per share data)
|
|
|
|
Statement of Operations Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues
|
|
$
|
107,672
|
|
|
$
|
92,566
|
|
|
$
|
95,502
|
|
|
$
|
84,306
|
|
|
$
|
72,067
|
|
|
Operating income
|
|
|
3,108
|
|
|
|
7,811
|
|
|
|
10,967
|
|
|
|
8,953
|
|
|
|
1,049
|
|
|
Other income
|
|
|
1,022
|
|
|
|
894
|
|
|
|
2,409
|
|
|
|
1,440
|
|
|
|
1,089
|
|
|
Net income (loss)
|
|
|
3,042
|
|
|
|
6,251
|
|
|
|
9,013
|
|
|
|
7,549
|
|
|
|
(175
|
)
|
|
Basic net income (loss) per share
|
|
|
0.17
|
|
|
|
0.35
|
|
|
|
0.49
|
|
|
|
0.42
|
|
|
|
(0.01
|
)
|
|
Diluted net income (loss) per share
|
|
|
0.17
|
|
|
|
0.35
|
|
|
|
0.48
|
|
|
|
0.40
|
|
|
|
(0.01
|
)
|
|
Balance Sheet Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Working capital
|
|
|
48,435
|
|
|
|
58,988
|
|
|
|
51,553
|
|
|
|
49,685
|
|
|
|
37,283
|
|
|
Total assets
|
|
|
106,609
|
|
|
|
97,790
|
|
|
|
90,638
|
|
|
|
86,390
|
|
|
|
74,317
|
|
|
Total debt
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Total liabilities
|
|
|
17,958
|
|
|
|
12,568
|
|
|
|
14,728
|
|
|
|
11,456
|
|
|
|
11,751
|
|
|
Stockholders’ equity
|
|
|
88,651
|
|
|
|
85,222
|
|
|
|
75,910
|
|
|
|
74,934
|
|
|
|
62,566
|
|
Our financial results for the years set forth above were
impacted by the following events:
1. 2010 results include the operating losses, including
acquisition related costs, of approximately $3.7 million
related to our acquisition in July 2010 of Arista Molecular,
Inc. In addition, we incurred $923,000 in CFO and related
transition costs, $1.1 million unfavorable foreign currency
fluctuation and losses and approximately $800,000 related to
higher instrument cost of goods due to a price premium on our
final purchase of automated chemistry analyzers from the
Japanese manufacturer.
2. 2009 results include the following expenses totaling
$1.1 million; an accrual of $475,000 for payroll taxes
attributable to the exercise of stock options, $350,000 in
start-up
expenses for the initiation of direct sales operations in the UK
and Germany and $250,000 for product retrofit costs related to
the voluntary recall of approximately 1,565 StatSpin Express 4
Centrifuges.
3. 2008 results include the non recurring other income
related to the $1.2 million net payment to us as part of
the manufacturing transition rights agreement signed with IDEXX
Laboratories in December 2008.
4. 2007 results include the effects of closing our Advanced
Digital Imaging Research subsidiary’s operations which
resulted in a $163,000 write-off.
5. 2006 results include the effect of the acquisition in
April 2006 of Leucadia Technologies, Inc. We recorded a
$5.2 million charge for purchased in-process research and
development. In addition we incurred additional research and
development expenses of $1.8 million as a result of the
acquisition.
|
|
|
Item 7.
|
Management’s
Discussion and Analysis of Financial Condition and Results of
Operations.
|
Overview
IRIS International, Inc. consists of three operating units in
three business segments as determined in accordance with FASB
ASC Topic 280, Segment Reporting. Our in-vitro
diagnostics segment also called Iris Diagnostics Division
(“IDD”), designs, manufactures and markets systems,
consumables and supplies for urinalysis
34
and body fluids. Our Personalized Medicine segment combines the
research and development operations of our Iris Molecular
Diagnostics and Arista Molecular, Inc. subsidiaries. Under this
new segment we consolidate all operations for the development
and commercialization of cancer diagnostic testing services and
related products. Our Sample Processing segment markets small
centrifuges and other processing equipment and accessories for
rapid specimen processing, as well as, equipment for fluorescent
in-situ hybridization (FISH).
Iris
Diagnostics Division
Our core business is in the urinalysis market and we are the
leading worldwide provider of automated urine microscopy
systems, with more than 3,000 iQ microscopy analyzers shipped to
date in over 50 countries. We generate revenues primarily from
sales of instruments, consumables and service. Revenues from
instruments include global sales of urine microscopy analyzers
and sales of chemistry analyzers. In September 2008, we released
our proprietary iChemVELOCITY automated urine chemistry analyzer
and a fully integrated urine microscopy and urine chemistry
work-cell, called the iRICELL in some international markets. We
plan to commence selling the iChemVELOCITY and iRICELL in the
United States contingent upon attaining clearance from the
U.S. Food and Drug Administration (FDA) on our 510(k)
application. Historically we sold our family of iQ analyzers
integrated with an automated chemistry analyzer, the AUTION MAX
AX-4280, which we have sourced from a Japanese manufacturer.
Our consumables offering includes products such as chemical
reagents, urine test strips, calibrators and controls. Service
revenues are derived primarily from annual service contracts
purchased by our domestic customers after the initial year of
sale, which is covered by product warranty, and spare parts
purchased by international customers. Once the analyzers are
installed, we generate recurring revenue from sales of
consumables. Consumable and service revenue should continue to
expand as the installed base of related instruments increases.
In the United States, France, Germany, the United Kingdom and
Puerto Rico sales of our urinalysis systems are direct to the
end-user through our sales force. All other international sales
are through independent distributors. In January 2010, we
completed the purchase from certain European distributors of
assets relating to the distribution of products in the United
Kingdom, Ireland and Germany. The purchased assets consisted
primarily of installed instruments leased to customers and the
related lease agreements and service contracts. Our incremental
revenue relating to the acquired distributors’ businesses
was $2.4 million for the year ended December 31, 2010.
International sales represented 34% of consolidated revenues for
the years ended December 31, 2010 and 2009. Since the
majority of international sales are made through independent
distributors, gross profit margin is lower than domestic sales
of the same products, but we incur minimal sales and marketing
costs for such sales.
Personalized
Medicine
On July 28, 2010, we acquired Arista Molecular, a high
complexity CLIA-certified molecular pathology laboratory
offering differentiated, high value molecular diagnostic
services in the rapidly growing field of personalized medicine.
Pursuant to the terms of the merger agreement dated
July 26, 2010, we acquired all the issued and outstanding
stock for an amount in cash equal to $4.7 million less
certain indebtedness existing at the closing, with an additional
earn-out of up to $1.3 million subject to the achievement
of specific sales and earnings targets through December 2013. We
did not assume any outstanding options or warrants in connection
with the acquisition. Arista will operate under the Personalized
Medicine reporting segment of the consolidated financial
statements.
At the time of acquisition, Arista Molecular was an early-stage
laboratory with limited commercial operations. In 2010, we
focused the laboratory’s efforts to broaden its menu of
diagnostic panels useful for the diagnosis, disease
characterization, treatment and monitoring of cancer. We added
testing for breast and prostate solid tumors beyond the existing
lung and colorectal offering. In addition, we now offer flow
cytometry for detection and monitoring of leukemia and lymphoma
and FISH testing. Further, we are building the commercial
infrastructure of the laboratory by adding sales and marketing
personnel. As a result, 2010 results included the dilutive
impact from Arista of $3.7 million, or approximately $0.15
per share with negligible revenue. With the expansion of the
test menu and sales force, we expect the revenue contribution
from this segment to become more meaningful in 2011, but
continue to have a dilutive impact for the year.
35
Sample
Processing
Our IRIS Sample Processing group markets and develops
centrifuges, semi-automated DNA processing workstations and
sample processing consumables. Our
StatSpin®
brand bench-top centrifuges are used for specimen preparation in
coagulation, cytology, chemistry and urinalysis. Our worldwide
markets include medical institutions, commercial laboratories,
clinics, doctors’ offices, veterinary laboratories and
research facilities. Our Sample Processing products are sold
worldwide primarily through distributors and incorporated into
our OEM partners products.
On November 22, 2010, we acquired the assets of a
multi-purpose, bench-top instrument platform for automating
highly repetitive, manual laboratory protocols for FISH
(fluorescence in-situ hybridization) testing and other
slide-based cytogenetic applications. The product acquisition is
a natural extension to the successful
ThermoBrite®
DNA Hybridization System and in line with our entry into
personalized medicine with emphasis on cancer diagnostics. The
product prototypes and proprietary technology assets were
purchased for $3.2 million in cash from BioMicro Systems,
Inc. Although BioMicro Systems built and tested a working
prototype, which proved the technical feasibility of the base
technology, several elements of this technology platform
requires continued development in order to reach salability for
its stated purpose. Once development is completed, a useful life
will be determined and the assets will be amortized over the
useful life determined at that time.
Research
and Development
We have invested significant capital to acquire technologies and
to increase our investment in research and development both to
sustain our technological leadership in our existing core
product lines in urinalysis and sample processing and as part of
our growth strategy to broaden our product pipeline into new
high value platforms in hematology and molecular diagnostics.
The decision to diversify into IVD market segments in which we
do not presently participate has adversely affected our earnings
growth over the last four years, as we have invested
significantly in research and development without any
corresponding revenues from new products still in development.
Our investment in new product platforms, however, is of
strategic importance and is necessary to position the company
for significant revenue and earnings acceleration in future
years. Research and development expense increased to
$14.6 million, or 14% of revenue, in 2010 compared to
$11.4 million, or 12% of revenue, in 2009. Of these
expenditures, approximately $7.0 million and
$6.0 million was invested in 2010 and 2009, respectively,
in our NADiA and hematology platforms. For 2011, we anticipate
that research and development expense will be approximately 14%
of revenue.
The following table summarizes product technology expenditures
for the periods indicated:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Total product technology expenditures during year
|
|
$
|
15,357
|
|
|
$
|
12,331
|
|
|
$
|
11,699
|
|
|
Less: amounts capitalized during year to software development
costs as reported in the consolidated statements of cash flow
|
|
|
(754
|
)
|
|
|
(835
|
)
|
|
|
(1,178
|
)
|
|
Less: amounts reimbursed through grants for government sponsored
research and development
|
|
|
(41
|
)
|
|
|
(85
|
)
|
|
|
(164
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development expense as reported in the consolidated
statements of income
|
|
$
|
14,562
|
|
|
$
|
11,411
|
|
|
$
|
10,357
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Critical
Accounting Policies
The preparation of financial statements and related disclosures
in conformity with U.S. generally accepted accounting
principles and our discussion and analysis of our financial
condition and results of operations require us to make
judgments, assumptions, and estimates that affect the amounts
reported in our consolidated financial statements and
accompanying notes. Note 2 of the Notes to Consolidated
Financial Statements of this
Form 10-K
describes the significant accounting policies and methods used
in the preparation of our consolidated financial statements. We
base our estimates on historical experience and on various other
assumptions we believe to be reasonable under the circumstances,
the results of which form the basis for making judgments about
the carrying
36
values of assets and liabilities. We regularly discuss with our
audit committee the basis of our estimates. Actual results may
differ from these estimates and such differences may be material.
We believe the following critical accounting policies, among
others, affect our more significant judgments and estimates used
in the preparation of our consolidated financial statements:
Revenue recognition: IDD and Sample
Processing— Revenues are primarily derived from
the sale of IVD instruments, sales of consumable supplies and
services for IVD systems as well as sales of sample processing
instruments and related supplies. Revenue is recognized once all
of the following conditions have been met: (i) an
authorized purchase order has been received in writing with a
fixed and determinable sales price; (ii) customer credit
worthiness has been established; and (iii) delivery of the
product based on shipping terms.
Revenue is recorded in accordance with the provisions of FASB
Accounting Standards Codification (“ASC”) Topic 605
“Revenue Recognition”, which generally require revenue
earned on product sales involving multiple-elements to be
allocated to each element based on the relative fair values of
those elements if sold separately. Multiple elements of certain
domestic product sales include IVD instruments, training,
consumables and service.
Accordingly, we allocate revenue to each element in a multiple
element arrangement based on the element’s respective fair
value, with the fair value determined by the price charged when
that element is sold separately and specifically defined in a
quotation or contract.
A portion of our revenues are derived from sales-type leases as
we provide lease financing to certain customers that purchase
our diagnostic instruments. Leases under these arrangements are
classified as investment in sales-type leases. These leases
typically have terms of five years. Revenue from sales-type
leases is recognized when collectability of the minimum lease
payments is reasonably predictable and no important
uncertainties surround the amount of unreimbursable costs yet to
be incurred by us as lessor under the lease. The minimum lease
payments that accrue to our benefit as lessor are recorded as
the gross investment in the lease. The difference between the
gross investment in the lease and the sum of the present value
of the minimum lease payments and unguaranteed residual value,
accruing to our benefit as lessor, are recorded as unearned
income.
We have certain government contracts with cancellation clauses
or renewal provisions that are generally required by law, such
as (i) those dependant on fiscal funding outside of a
governmental unit’s control; (ii) those that can be
cancelled if deemed in the tax payers’ best interest; and
(iii) those that must be renewed each fiscal year, given
limitations that may exist on multi-year contracts that are
imposed by statute. Under these circumstances and in accordance
with the relevant accounting literature, as well as considering
our historical experience, a thorough evaluation of these
contracts is performed to assess whether cancellation is remote
or whether exercise of the renewal option is reasonably assured.
We recognize revenues from service contracts ratably over the
term of the service period, which typically ranges from twelve
to sixty months. Payments for service contracts are generally
received in advance. Deferred revenue represents the revenues to
be recognized over the remaining term of the service contracts.
Revenue recognition: Personalized
Medicine — Revenues related to our Personalized
Medicine segment are recorded in accordance with FASB
ASC 605-10-S99-1,
“Revenue Recognition”, when (i) the price is
fixed or determinable, (ii) persuasive evidence of an
arrangement exists, (iii) the service is performed and
(iv) collectability of the resulting receivable is
reasonably assured.
Our specialized diagnostic services are performed based on a
written test requisition form and revenues are recognized once
the diagnostic services have been performed, the results have
been delivered to the ordering physician, the payor has been
identified and eligibility and insurance have been verified.
These diagnostic services are billed to various payors,
including Medicare, commercial insurance companies, other
directly billed healthcare institutions such as hospitals and
clinics, and individuals. We report revenues from contracted
payors, including Medicare, certain insurance companies and
certain healthcare institutions, based on the contractual rate,
or in the case of Medicare, published fee schedules. For
non-contracted payors, including certain insurance companies and
individuals, we report revenues based on the amount which we
expect to be collected. The difference between the amount billed
and the amount expected to be collected from non-contracted
payors is recorded as a contractual allowance to arrive at the
reported revenues. The expected revenues from non-contracted
payors are based on the
37
historical collection experience of each payor or payor group,
as appropriate. In each reporting period, we review our
historical collection experience for non-contracted payors and
adjust our expected revenues for current and subsequent periods
accordingly.
Inventory valuation — We value inventories at
the lower of cost or market value on a
first-in,
first-out basis. Provision for potentially obsolete or
slow-moving inventory is made based on management’s
analysis of inventory levels and future sales forecasts.
Goodwill and Intangible Assets — Our intangible
assets consist of goodwill and intangible assets with indefinite
lives such as a CLIA license, which are not being amortized and
intangible assets with a finite life, which are being amortized
over useful lives ranging from 3 to 20 years. All
intangible assets are subject to impairment tests on an annual
or periodic basis. Goodwill and intangible assets with
indefinite lives are evaluated in accordance with FASB ASC Topic
350, “The Intangibles-Goodwill and Other”, based on
various analyses, including a comparison of the carrying value
of the reporting unit to its estimated fair value and discounted
cash flow. The analysis necessarily involves significant
management judgment to evaluate the capacity of an acquired
business to perform within projections. Intangible assets with a
finite life are evaluated for impairment using the methodology
set forth in FASB ASC Topic 360, “Property, Plant and
Equipment.” Recoverability of these assets is assessed only
when events have occurred that may give rise to a potential
impairment. When a potential impairment has been identified,
forecasted undiscounted net cash flows of the operations to
which the asset relates are compared to the current carrying
value of the long-lived assets present in that operation. If
such cash flows are less than such carrying amounts, long-lived
assets, including such intangibles, are written down to their
respective fair values.
At December 31, 2010, we evaluated goodwill and intangible
assets and determined that fair value had not decreased below
carrying value and no adjustment to impair goodwill was
necessary in accordance with ASC Topic 350. Substantially all of
the goodwill resides in the Personalized Medicine reporting unit.
Capitalized software — We capitalize certain
software development costs in connection with our development of
our urine analyzers in accordance with FASB ASC Topic 985,
“Software.” We capitalize software development costs
once technological feasibility is established and such costs are
determined to be recoverable against future revenues.
Capitalized software development costs are expensed to cost of
sales over periods of up to five years. When, in
management’s estimate, future revenues will not be
sufficient to recover previously capitalized software
development costs, we will expense such items as additional
software development amortization in the period the impairment
is identified. Such adjustments are normally attributable to
changes in market conditions or product quality considerations.
Income taxes — We account for income taxes in
accordance with FASB ASC Topic 740, “Income Taxes,”
which requires recognition of deferred tax liabilities and
assets for the expected future tax consequences of events that
have been included in the financial statements or tax returns.
Under this method, deferred tax liabilities and assets are
determined based on the differences between the financial
statement and the tax bases of assets and liabilities using
enacted tax rates in effect for the year in which the
differences are expected to reverse. Valuation allowances are
established when necessary to reduce deferred tax assets to the
amount expected to be realized. Income tax expense represents
the tax payable for the period and the change during the period
in deferred tax assets and liabilities.
We account for uncertain tax positions in accordance with FASB
ASC Topic
740-10 which
prescribes a recognition threshold and measurement attribute for
financial statement recognition and measurement of a tax
position taken or expected to be taken in a tax return and also
provides guidance on various related matters such as
derecognition, interest, penalties and disclosures required. We
recognize interest and penalties, if any, related to
unrecognized tax benefits in income tax expense.
Stock based compensation — The Company accounts
for stock based compensation under FASB ASC Topic 718,
Compensation- Stock Compensation (“ASC 718”)
which requires compensation costs related to share-based
transactions, including employee stock options, to be recognized
in the financial statements based on fair value.
38
Results
of Operations
The following table summarizes results of operations data for
the periods indicated. The percentages in the table are based on
total revenues with the exceptions of percentages for gross
profit margins, which are computed on related revenue, and
percentages for income taxes, which are computed based on the
relationship of income taxes to pre-tax income.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended December 31,
|
|
|
|
|
|
2010
|
|
|
|
2009
|
|
|
|
2008
|
|
|
|
|
|
(In thousands)
|
|
|
|
|
|
Revenues
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
IDD instruments
|
|
$
|
32,083
|
|
|
|
30
|
%
|
|
$
|
26,018
|
|
|
|
28
|
%
|
|
$
|
35,128
|
|
|
|
37
|
%
|
|
IDD consumables and service
|
|
|
61,112
|
|
|
|
57
|
%
|
|
|
52,213
|
|
|
|
56
|
%
|
|
|
46,643
|
|
|
|
49
|
%
|
|
Sample Processing instruments and supplies
|
|
|
14,408
|
|
|
|
13
|
%
|
|
|
14,335
|
|
|
|
16
|
%
|
|
|
13,731
|
|
|
|
14
|
%
|
|
Personalized Medicine services
|
|
|
69
|
|
|
|
0
|
%
|
|
|
—
|
|
|
|
0
|
%
|
|
|
—
|
|
|
|
0
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenues
|
|
|
107,672
|
|
|
|
100
|
%
|
|
|
92,566
|
|
|
|
100
|
%
|
|
|
95,502
|
|
|
|
100
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gross Profit(1)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
IDD instruments
|
|
|
10,846
|
|
|
|
34
|
%
|
|
|
9,240
|
|
|
|
36
|
%
|
|
|
15,523
|
|
|
|
44
|
%
|
|
IDD consumable and service
|
|
|
36,623
|
|
|
|
60
|
%
|
|
|
32,055
|
|
|
|
61
|
%
|
|
|
26,648
|
|
|
|
57
|
%
|
|
Sample Processing instruments and supplies
|
|
|
7,789
|
|
|
|
54
|
%
|
|
|
7,370
|
|
|
|
51
|
%
|
|
|
6,868
|
|
|
|
50
|
%
|
|
Personalized Medicine services
|
|
|
(372
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
0
|
%
|
|
|
—
|
|
|
|
0
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gross profit
|
|
|
54,886
|
|
|
|
51
|
%
|
|
|
48,665
|
|
|
|
53
|
%
|
|
|
49,039
|
|
|
|
51
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating expenses
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Marketing and selling
|
|
|
19,829
|
|
|
|
18
|
%
|
|
|
16,122
|
|
|
|
17
|
%
|
|
|
15,706
|
|
|
|
16
|
%
|
|
General and administrative
|
|
|
17,387
|
|
|
|
16
|
%
|
|
|
13,321
|
|
|
|
14
|
%
|
|
|
12,009
|
|
|
|
13
|
%
|
|
Research and development, net
|
|
|
14,562
|
|
|
|
14
|
%
|
|
|
11,411
|
|
|
|
12
|
%
|
|
|
10,357
|
|
|
|
11
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
51,778
|
|
|
|
48
|
%
|
|
|
40,854
|
|
|
|
44
|
%
|
|
|
38,072
|
|
|
|
40
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating income
|
|
|
3,108
|
|
|
|
3
|
%
|
|
|
7,811
|
|
|
|
9
|
%
|
|
|
10,967
|
|
|
|
11
|
%
|
|
Other income
|
|
|
1,022
|
|
|
|
|
|
|
|
894
|
|
|
|
|
|
|
|
2,409
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Income before provision for income taxes
|
|
|
4,130
|
|
|
|
4
|
%
|
|
|
8,705
|
|
|
|
9
|
%
|
|
|
13,376
|
|
|
|
14
|
%
|
|
Provision for income taxes(2)
|
|
|
1,088
|
|
|
|
26
|
%
|
|
|
2,454
|
|
|
|
28
|
%
|
|
|
4,363
|
|
|
|
33
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income
|
|
$
|
3,042
|
|
|
|
3
|
%
|
|
$
|
6,251
|
|
|
|
7
|
%
|
|
$
|
9,013
|
|
|
|
9
|
%
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Gross profit margin percentages are based on the related sales
of each category. |
| |
|
(2) |
|
Income tax percentage is computed based on the relationship of
income taxes to pre-tax income. |
Comparison
of Year Ended December 31, 2010 to 2009
Consolidated revenues for the year ended December 31, 2010
increased 16% over the prior year to $107.7 million from
$92.6 million in 2009. International revenues accounted for
approximately 34% of consolidated revenue in 2010 compared to
33% in 2009. Revenues in the IDD urinalysis segment increased
19% to $93.2 million in 2010, from $78.2 million in
the prior year. The current year IDD revenues include
approximately $2.4 million incremental revenue related to
the acquisition of European distributor operations earlier in
the year. Sales of IDD instruments increased 23% to
$32.1 million from $26.0 million in the prior year.
The continued increase in IDD instrument sales is primarily
driven by strong domestic sales versus a year ago period due to
new marketing and sales initiatives and improvement in the
U.S. medical capital equipment market environment.
IDD consumables and service increased during the year to
$61.1 million from $52.2 million, an increase of 17%
over 2009, primarily due to the larger installed base of
instruments both domestically and internationally and
39
increased sales of iChemVELOCITY test strips due to increased
placements of our iChemVELOCITY analyzer in the international
market.
Revenues derived from Sample Processing instruments and supplies
modestly increased to $14.4 million, a 1% increase over
2009 revenues of $14.3 million. Revenues were favorably
impacted by the following factors: a) continued strong
sales of our ThermoBrite line of instruments,
b) introduction of our new Cytofuge 12 instrument and
c) OvaTube consumables. Revenues were unfavorably impacted
by the completion of the transition of the separator consumable
business to IDEXX. We will receive royalties on these units
beginning in 2014.
Personalized Medicine revenues in 2010 totaled $69,000.
Consolidated gross profit margin decreased to 51% in 2010
compared to 53% in 2009, primarily the result of higher cost of
goods due to a price premium on our final purchase of automated
chemistry analyzers from Arkray, the negative effect of foreign
currency fluctuations, and increased service costs relating to
our start up of direct commercial operations in Europe.
The gross profit margin of our IDD instruments was 34% in 2010
compared to 36% in 2009. Instrument gross margins were impacted
by increased costs from foreign-sourced products due to the
weakening of the U.S. dollar, in particular on our
purchases of AUTION MAX AX-4280 chemistry analyzers which are
denominated in Japanese Yen, partially offset by higher
instrument volume and a greater mix of direct domestic sales
versus international sales through distributors.
The gross profit margin of our IDD consumables and service
decreased to 60% in 2010 compared to 61% in 2009, primarily the
result of increased costs of foreign-sourced products due to the
weaker U.S. dollar, increased service costs related to
iChemVELOCITY and an increase in service costs related to our
direct commercial operations in the United Kingdom and Germany,
partially offset by the higher margins due to manufacturing
efficiencies resulting from higher volume in consumables sold.
Gross profit margin for our Sample Processing instruments and
supplies segment increased to 54% in 2010 from 51% in 2009 as a
result of product mix, favorable purchase price discounts, and
cost reduction initiatives such as lean manufacturing.
Marketing and selling expenses totaled $19.8 million, or
18% of revenues, in 2010 as compared to $16.1 million, or
17% of revenues in 2009. The increase primarily results from
$1.3 million of expenses related to Arista’s
operations, additional personnel and related costs of
$2.0 million related primarily to our direct operations in
the United Kingdom and Germany, travel and entertainment,
excluding UK and Germany of $192,000 as well as higher
commissions and GPO fees of $210,000.
General and administrative expenses increased to
$17.4 million, or 16% of revenues, in 2010, from
$13.3 million or 14% of revenues in 2009. This increase
primarily relates to $1.9 million in acquisition and
operating expenses relating to Arista, $923,000 of CFO severance
and related transition expenses, $796,000 of additional
personnel and related costs in information technology, quality
assurance and regulatory affairs and $455,000 of recruiting and
relocation expenses.
Research and development expense increased to
$14.6 million, or 14% of revenues, in 2010 from
$11.4 million, or 12% of revenues in 2009. The increase was
primarily attributable to $1.2 million in research
materials, consulting, clinical development expenses,
$1.6 million in personnel and related costs and other
expenses of approximately $400,000 as we continue to invest
heavily in research and development for the continued
development of our diagnostics product pipeline, including our
NADiA platform and 3GEMS urinalysis and hematology programs.
Interest income during 2010 amounted to $1.1 million, a
$266,000 increase over 2009 mainly due to a higher cash balance
through part of 2010 as well as an increase in in-house
sales-type leases.
Income tax expense during the year amounted to 26.4% of pre-tax
income as compared to 28% during the prior year. The decrease in
the income tax provision resulted from an increase in research
and development tax credits due to an increase in qualifying
research and development expenses. We have federal and state
research and development and other tax credit carryovers
totaling $3.6 million.
40
Comparison
of Year Ended December 31, 2009 to 2008
Revenues for the year ended December 31, 2009 decreased by
3% over the prior year to $92.6 million from
$95.5 million in 2008. International revenues accounted for
approximately 33% of consolidated revenue for both 2009 and
2008. Revenues in the IDD urinalysis segment decreased 4% to
$78.2 million in 2009, from $81.8 million in the prior
year. Sales of IDD instruments decreased 26% to
$26.0 million from $35.1 million in the prior year as
a result of a significant shortage of capital in the global
markets.
Sales of IDD consumables and service increased during the year
to $52.2 million from $46.6 million, an increase of
12% over 2008, primarily due to the larger installed base of
instruments and the success of converting expiring warranty
agreements to service agreements.
Revenues derived from Sample Processing instruments and supplies
increased to $14.3 million, a 4% increase over 2008
revenues of $13.7 million. This growth was driven by
continued strong sales of our new Express 4 centrifuge along
with an increase in orders from our international distributors,
most notably our distributor in the United Kingdom.
Overall gross profit margins increased to 53% in 2009 compared
to 51% in 2008. The gross profit margin of our IDD instruments
was 36% in 2009 versus 44% in 2008.
As compared to the prior year period, IDD instrument gross
margin in 2009 was impacted by $368,000 in sales promotions,
$1,193,000 in costs related to the iChemVELOCITY, which included
Velocity retrofit costs of $373,000, volume and other costs of
$320,000, severance costs of approximately $40,000 with an
offset of a reduction in employee bonuses of $76,000. In
addition, our gross margins on IDD instruments were negatively
impacted by foreign currency fluctuations on our purchases of
AUTION MAX AX-4280 chemistry analyzers which are denominated in
Japanese Yen.
The gross margin of our IDD consumables and service increased to
61% during 2009 compared to 57% in 2008, respectively, and
included $501,000 in international sales discounts. The
consumable gross margin improvement primarily resulted from
economies of scale generated by our increasing volume of urine
microscopy consumables and spare parts, improved efficiencies in
our domestic service business, a reduction in unfavorable
foreign currency fluctuations on purchases of dry chemistry
strips denominated in Japanese Yen and a reduction of employee
bonuses of $160,000.
Gross profit margin for our Sample Processing instruments and
supplies segment increased to 51% in 2009 from 50% in 2008 as a
result of product mix, price increases and reduced headcount
related to the reduction in force in the second quarter of 2009.
In addition, the gross margin was affected by our Sample
Processing division’s voluntarily recall of approximately
1,565 centrifuges, requiring a $250,000 accrual to cover the
product retrofit costs during the fourth quarter of 2009.
Marketing and selling expenses totaled $16.1 million, or
17% of revenues, in 2009 as compared to $15.7 million, or
16% of revenues in 2008. The increase includes personnel and
related costs of $967,000, partially offset by lower commission
and GPO fees of $319,000, travel and entertainment of $198,000,
a reduction of employee bonuses of $134,000 and professional
fees of $151,000. Our $967,000 increase in personnel and related
costs includes $350,000 in
start-up
expenses relating to the initiation of direct sales in the
United Kingdom and Germany.
General and administrative expenses increased to
$13.3 million, or 14% of revenues, in 2009, from
$12.0 million or 13% of revenues in 2008. This increase
includes additional personnel and related costs of $619,000,
stock-based compensation expense of $804,000, professional fees
of $108,000, board compensation expense of $64,000, an accrual
of $475,000 for payroll taxes attributable to the exercise of
stock options, other corporate related expenses of approximately
$300,000 and an offset of $541,000 for a reduction in employee
bonuses.
Research and development expense increased to
$11.4 million, or 12% of revenues, in 2009 from
$10.4 million, or 11% of revenues in 2008. The increase
includes research materials, consulting, clinical development
expenses and software capitalization of $1.1 million as we
continue to invest heavily in research and development for the
continued development of our diagnostics product pipeline,
including our NADiA platform
41
and 3GEMS urinalysis and hematology programs. These costs were
partially offset by a reduction in employee bonuses of $431,000.
Interest income during 2009 amounted to $857,000, a $323,000
decrease over 2008 mainly due to a lower interest rate
environment on our cash balances, and in spite of a higher cash
balance.
Income tax expense during the year amounted to 28% of pre-tax
income as compared to 33% during the prior year. The decrease in
the income tax provision resulted from an increase in research
and development tax credits. At December 31, 2009 we had
federal and state research and development and other tax credit
carryovers totaling $5.3 million.
Contractual
Obligations and Commercial Commitments
The following table aggregates our expected minimum contractual
obligations and commitments subsequent to December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Less Than
|
|
|
1-3
|
|
|
3-5
|
|
|
More Than
|
|
|
Contractual Obligations
|
|
Total
|
|
|
1 Year
|
|
|
Years
|
|
|
Years
|
|
|
5 Years
|
|
|
|
|
(In thousands)*
|
|
|
|
|
Operating lease commitments
|
|
$
|
9,390
|
|
|
$
|
2,358
|
|
|
$
|
4,200
|
|
|
$
|
2,718
|
|
|
$
|
114
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* |
|
Not included in the table above are normal recurring accounts
payable or accrued expenses which are presented on the
accompanying consolidated financial statements. |
Off-Balance
Sheet Arrangements
At December 31, 2010 and 2009, we did not have any
relationships with unconsolidated entities or financial
partnerships, such as entities often referred to as structured
finance or special purpose entities, which would have been
established for the purpose of facilitating off-balance sheet
arrangements or other contractually narrow or limited purposes.
As such, we are not exposed to any financing, liquidity, market
or credit risk that could arise if we had engaged in such
relationships.
Liquidity
and Capital Resources
Our primary source of liquidity is cash from operations, which
depends heavily on sales of our IDD instruments, consumables and
service, as well as sales of sample processing instruments and
supplies. At December 31, 2010, our cash and cash
equivalents amounted to $25.5 million compared to
$34.3 million at December 31, 2009.
In the past few years, we have faced adverse macro economic
forces, which have impacted our selling markets and the credit
markets of our customers. At this point the impact from these
forces are relatively mild, but in the future we may face the
following challenges: deferrals of purchases due to decreases in
capital budgets of our customers, delays in the purchasing cycle
due to greater scrutiny of deals and increased internal
competition for limited capital dollars, and a significant
increase in requests for quotes for operating leases. The
aforementioned factors may lead to a decrease in revenue, an
increase of deferred revenue, or could lead to installment cash
collection.
Operating Cash Flows. Cash provided by
operations for the year ended December 31, 2010 was
$8.8 million, which included net income of
$3.0 million, a decrease in inventory of $0.5 million,
an increase in operating liabilities of $3.6 million,
depreciation and amortization of $4.2 million and stock
based compensation of $4.2 million. Cash provided by
operations was offset by an increase in accounts receivable of
$3.1 million, an increase in investment in sales-type
leases of $2.8 million and an increase in prepaid expenses
and other assets of $0.8 million.
The number of days sales in accounts receivable remained at
70 days at the end of 2010 compared to the prior year end.
The number of days sales in accounts receivable varies and
extends due to extended payment terms for our international
customers and the granting of an increased volume of extended
payment terms for certain customers of our instrument sales and
instrument, consumables and service mix.
42
Our cash flow has been favorably affected by tax credit
carryforwards. As of December 31, 2010, we had federal and
state tax credit carryforwards of approximately $666,000 and
$2.9 million, respectively. We continue to realize tax
deductions from the exercise of certain stock options. During
the year ended December 31, 2010, we realized tax
deductions of approximately $201,000 relating to this item.
During the year ended December 31, 2010, we paid federal
and state taxes of $1.7 million and $820,000, respectively.
Investing Activities. Cash used in investing
activities totaled $13.9 million in 2010 primarily as the
result of the acquisition of Arista of $4.6 million, an
increase in the purchase of assets from a European distributor
of $0.7 million, an acquisition of product technology
totaling $3.3 million, an increase in the purchase of
property and equipment of $4.6 million and an increase in
software development costs capitalized of $0.8 million.
Financing Activities. Cash used in financing
activities totaled $3.2 million in 2010, which was related
primarily to repurchase of common stock.
We currently have a credit facility with a commercial bank
consisting of a $6.5 million revolving line of credit for
working capital and a $10.0 million line of credit for
acquisitions and product opportunities. The credit facility has
variable interest rates, which will change from time to time
based on changes to either the LIBOR rate or the lender’s
prime rate. As of December 31, 2010, there were no
borrowings under the new credit facility. We are subject to
certain financial and non-financial covenants under the credit
facility with the bank and as of December 31, 2010, we were
in compliance with these covenants.
In August 2010, the Company’s board of directors authorized
a share repurchase and retirement plan of up to $10 million
of the Company’s common stock over a
12-month
period. During the year ended December 31, 2010, the
Company repurchased 330,454 shares of common stock at an
average price per share of approximately $9.10, for an aggregate
amount of approximately $3.0 million.
In 2008, our board of directors authorized two stock repurchase
plans, which resulted in our purchase of an aggregate of
983,579 shares of our common stock for approximately
$11.9 million during 2008, and an additional
250,800 shares of our common stock for approximately
$2.5 million during 2009.
We believe that our current cash on hand, together with cash
generated from operations and cash available under the credit
facility with the bank will be sufficient to fund normal
operations for the foreseeable future. However, additional
funding may be required to fund expansion of our business. There
is no assurance that such funding will be available on terms
acceptable to us.
Recent
Accounting Pronouncements
In December 2010, the FASB issued Accounting Standards Update
(“ASU”) ASU
2010-29,
Business Combinations- Disclosure of Supplementary Pro Forma
Information, which specifies that if a public entity
presents comparative financial statements, the entity should
disclose revenue and earnings of the combined entity as though
the business combination(s) that occurred during the current
year had occurred as of the beginning of the comparable prior
annual reporting period only.
ASU 2010-29
is effective on a prospective basis for business combinations
for which the acquisition date is on or after the beginning of
the first annual reporting period beginning on or after
December 15, 2010 with early adoption permitted. We are
currently evaluating both the timing and the impact of the
adoption of the ASU on our consolidated financial statements.
In July 2010, the FASB issued ASU
2010-20,
Disclosures about the Credit Quality of Financing Receivables
and the Allowance for Credit Losses which amends ASC Topic
310, Receivables. ASU
2010-20
requires disclosures about the nature of the credit risk in an
entity’s financing receivables, how that risk is
incorporated into the allowance for credit losses, and the
reasons for any changes in the allowance. Disclosure is required
to be disaggregated, primarily at the level at which an entity
calculates its allowance for credit losses.
ASU 2010-20
is applicable to both private and public companies, and will
affect any entity that has financing receivables on its balance
sheet, not including short-term trade accounts receivable.
Public entities must apply the disclosure requirements
applicable to period-end balances beginning with the first
interim or annual reporting period ending on or after
December 15, 2010 (December 31, 2010 for a calendar
year-end entity). The adoption of this guidance resulted in
additional disclosures (see Note 7 in the Notes to
Consolidated Financial Statements) but did not have an impact on
our consolidated financial statements.
43
In October 2009, the FASB issued Accounting Standards Update ASU
2009-14,
Certain Revenue Arrangements That Include Software
Elements, now codified under FASB ASC Topic 985,
Software. ASU
2009-14,
removes tangible products from the scope of software revenue
guidance and provides guidance on determining whether software
deliverables in an arrangement that includes a tangible product
are covered by the scope of the software revenue guidance. ASU
2009-14 is
to be applied on a prospective basis for revenue arrangements
entered into or materially modified in fiscal years beginning on
or after June 15, 2010, with early adoption permitted. We
are currently evaluating both the timing and the impact of the
adoption of the ASU on our consolidated financial statements.
In October 2009, the FASB issued Accounting Standards Update
(“ASU”)
2009-13,
Multiple-Deliverable Revenue Arrangements, which amends
ASC Topic 605, Revenue Recognition, to require companies
to allocate revenue in multiple-element arrangements based on an
element’s estimated selling price if vendor-specific or
other third-party evidence of value is not available. ASU
2009-13 is
effective for fiscal years beginning on or after June 15,
2010. Earlier application is permitted. The adoption of ASU
2009-13 did
not have a material impact on our consolidated financial
statements.
In December 2010, the FASB issued Accounting Standards Update
(“ASU”)
No. 2010-28 —
When to Perform Step 2 of the Goodwill Impairment Test for
Reporting Units with Zero or Negative Carrying Amounts. This
update provides amendments to ASC Topic 350 —
Intangibles, Goodwill and Other that requires an entity to
perform Step 2 impairment test even if a reporting unit has zero
or negative carrying amount. Step 1 tests whether the carrying
amount of a reporting unit exceeds its fair value. Previously
reporting units with zero or negative carrying value passed Step
1 because the fair value was generally greater than zero. Step 2
requires impairment testing and impairment valuation be
calculated in between annual tests if an event or circumstances
indicate that it is more likely than not that goodwill has been
impaired. ASU
2010-28 is
effective beginning January 1, 2011. As a result of this
standard, goodwill impairments may be reported sooner than under
current practice. We do not expect ASU
2010-28 to
have a material impact on our financial statements.
|
|
|
Item 7A.
|
Quantitative
and Qualitative Disclosures About Market Risk.
|
Market
Risk
Our business is exposed to various market risks, including
changes in interest rates and foreign currency exchange rates.
Market risk is the potential loss arising from adverse changes
in market rates and prices, such as interest rates and foreign
currency exchange rates. We do not invest in derivatives,
foreign currency forward contracts or other financial
instruments for trading or speculative purposes. We had no debt
at December 31, 2010, thus were not subject to market risk
for changes in interest rates on debt obligations. We are
subject to market risk for changes in interest rates on our
short-term investment portfolio. We invest our excess cash in
certificates of deposit and the market value of these
investments fluctuate based on changes in interest rates.
Foreign
Currencies
We conduct business in certain foreign markets, primarily in the
European Union and Asia. Our primary exposure to foreign
currency risk relates to investments in foreign subsidiaries
that transact business in a functional currency other than the
U.S. Dollar, primarily the Euro. We are also subject to
certain foreign currency risks in the importation of goods from
Japan and as a result of commercial operations in Europe and
Asia. Our purchases from a major Japanese IVD supplier are
denominated in Japanese Yen. These components represent a
significant portion of our material costs, but our Yen exposure
should decrease with the discontinuation of Japanese-sourced
instruments beginning in January 2011. All of our sales are
denominated in U.S. Dollars with the exception of France
and Germany, where sales are denominated in Euros and the United
Kingdom, where sales are denominated in British Pounds.
Fluctuations in the U.S. Dollar exchange rate for Japanese
Yen and Euros could result in increased costs for our key
components and increased costs for commercial operations in
Europe.
To mitigate the potential impact of adverse fluctuations in the
U.S. Dollar exchange rate for these currencies, we have
periodically purchased foreign currency forward contracts in the
past for Euros and Japanese Yen. During the years ended
December 31, 2009 and 2010, we did not enter into any such
contracts, and no such contracts existed at December 31,
2010.
44
|
|
|
Item 8.
|
Financial
Statements and Supplementary Data.
|
Index
to Financial Statements
45
Report of
Independent Registered Public Accounting Firm
Board of Directors and Shareholders of IRIS International, Inc.
Chatsworth, California
We have audited the accompanying consolidated balance sheets of
IRIS International, Inc.’s as of December 31, 2010 and
2009 and the related consolidated statements of operations,
shareholders’ equity, cash flows and other comprehensive
income for each of the three years in the period ended
December 31, 2010. These financial statements are the
responsibility of the Company’s management. Our
responsibility is to express an opinion on these financial
statements based on our audits.
We conducted our audits in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are
free of material misstatement. An audit includes examining, on a
test basis, evidence supporting the amounts and disclosures in
the financial statements, assessing the accounting principles
used and significant estimates made by management, as well as
evaluating the overall financial statement presentation. We
believe that our audits provide a reasonable basis for our
opinion.
In our opinion, the consolidated financial statements referred
to above present fairly, in all material respects, the financial
position of IRIS International, Inc. at December 31, 2010
and 2009, and the results of its operations, cash flows and its
other comprehensive income for each of the three years in the
period ended December 31, 2010, in conformity with
accounting principles generally accepted in the United States of
America.
We also have audited, in accordance with the standards of the
Public Company Accounting Oversight Board (United States), IRIS
International Inc.’s internal control over financial
reporting as of December 31, 2010, based on criteria
established in Internal Control — Integrated Framework
issued by the Committee of Sponsoring Organizations of the
Treadway Commission (COSO) and our report dated March 16,
2011, expressed an unqualified opinion thereon.
Los Angeles, California
March 16, 2011
46
Report of
Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders of IRIS
International, Inc.
Chatsworth, California
We have audited IRIS International, Inc.’s internal control
over financial reporting as of December 31, 2010, based on
criteria established in Internal Control —
Integrated Framework issued by the Committee of Sponsoring
Organizations of the Treadway Commission (the COSO criteria).
IRIS International, Inc.’s management is responsible for
maintaining effective internal control over financial reporting
and for its assessment of the effectiveness of internal control
over financial reporting, included in the accompanying
“Item 9A, Management’s Report on Internal Control
over Financial Reporting”. Our responsibility is to express
an opinion on the company’s internal control over financial
reporting based on our audit.
We conducted our audit in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether effective internal control
over financial reporting was maintained in all material
respects. Our audit included obtaining an understanding of
internal control over financial reporting, assessing the risk
that a material weakness exists, and testing and evaluating the
design and operating effectiveness of internal control based on
the assessed risk. Our audit also included performing such other
procedures as we considered necessary in the circumstances. We
believe that our audit provides a reasonable basis for our
opinion.
A company’s internal control over financial reporting is a
process designed to provide reasonable assurance regarding the
reliability of financial reporting and the preparation of
financial statements for external purposes in accordance with
generally accepted accounting principles. A company’s
internal control over financial reporting includes those
policies and procedures that (1) pertain to the maintenance
of records that, in reasonable detail, accurately and fairly
reflect the transactions and dispositions of the assets of the
company; (2) provide reasonable assurance that transactions
are recorded as necessary to permit preparation of financial
statements in accordance with generally accepted accounting
principles, and that receipts and expenditures of the company
are being made only in accordance with authorizations of
management and directors of the company; and (3) provide
reasonable assurance regarding prevention or timely detection of
unauthorized acquisition, use, or disposition of the
company’s assets that could have a material effect on the
financial statements.
Because of its inherent limitations, internal control over
financial reporting may not prevent or detect misstatements.
Also, projections of any evaluation of effectiveness to future
periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree
of compliance with the policies or procedures may deteriorate.
In our opinion, IRIS International, Inc. maintained, in all
material respects, effective internal control over financial
reporting as of December 31, 2010, based on the COSO
criteria.
We also have audited, in accordance with the standards of the
Public Company Accounting Oversight Board (United States), the
consolidated balance sheets of IRIS International Inc. as of
December 31, 2010 and 2009, and the related consolidated
statements of operations, shareholders’ equity, cash flows
and other comprehensive income for each of the three years in
the period ended December 31, 2010 and our report dated
March 16, 2011 expressed an unqualified opinion thereon.
Los Angeles, California
March 16, 2011
47
IRIS
INTERNATIONAL, INC.
| |
|
|
|
|
|
|
|
|
|
|
|
At December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
(In thousands, except per share data)
|
|
|
|
|
ASSETS
|
|
Current assets:
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
25,531
|
|
|
$
|
34,253
|
|
|
Accounts receivable, net of allowance for doubtful accounts and
sales returns of $453 and $417 at December 31, 2010 and
2009, respectively
|
|
|
20,733
|
|
|
|
17,715
|
|
|
Inventories
|
|
|
10,310
|
|
|
|
10,866
|
|
|
Prepaid expenses and other current assets
|
|
|
1,661
|
|
|
|
1,045
|
|
|
Investment in sales-type leases, current portion
|
|
|
3,578
|
|
|
|
3,397
|
|
|
Deferred tax asset
|
|
|
3,135
|
|
|
|
4,238
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current assets
|
|
|
64,948
|
|
|
|
71,514
|
|
|
Property and equipment, net of accumulated depreciation of
$14,491 and $11,713 at December 31, 2010 and 2009,
respectively
|
|
|
12,035
|
|
|
|
9,667
|
|
|
Goodwill
|
|
|
3,957
|
|
|
|
2,450
|
|
|
Intangible assets, net of accumulated amortization of $529 and
$336 at December 31, 2010 and 2009, respectively
|
|
|
9,345
|
|
|
|
1,454
|
|
|
Software development costs, net of accumulated amortization of
$4,226 and $3,365 at December 31, 2010 and 2009,
respectively
|
|
|
2,637
|
|
|
|
2,534
|
|
|
Deferred tax asset
|
|
|
2,615
|
|
|
|
1,898
|
|
|
Investment in sales-type leases, non-current portion
|
|
|
10,002
|
|
|
|
7,441
|
|
|
Other assets
|
|
|
1,070
|
|
|
|
832
|
|
|
|
|
|
|
|
|
|
|
|
|
Total assets
|
|
$
|
106,609
|
|
|
$
|
97,790
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
|
Current liabilities:
|
|
|
|
|
|
|
|
|
|
Accounts payable
|
|
$
|
5,795
|
|
|
$
|
4,479
|
|
|
Accrued expenses
|
|
|
7,513
|
|
|
|
5,761
|
|
|
Deferred service contract revenue, current portion
|
|
|
3,205
|
|
|
|
2,286
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current liabilities
|
|
|
16,513
|
|
|
|
12,526
|
|
|
Deferred service contract revenue, non-current portion
|
|
|
71
|
|
|
|
42
|
|
|
Other long term liabilities
|
|
|
1,374
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities
|
|
|
17,958
|
|
|
|
12,568
|
|
|
Commitments and contingencies
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity:
|
|
|
|
|
|
|
|
|
|
Common stock, $0.01 par value
|
|
|
|
|
|
|
|
|
|
Authorized: 50 million shares; Issued and outstanding:
17,791 shares and 18,111 shares at December 31,
2010 and 2009, respectively
|
|
|
178
|
|
|
|
181
|
|
|
Preferred Stock, $0.01 par value; Authorized
1.0 million shares: Callable Series C shares issued
and outstanding: none
|
|
|
—
|
|
|
|
—
|
|
|
Additional paid-in capital
|
|
|
89,703
|
|
|
|
87,692
|
|
|
Other comprehensive income
|
|
|
140
|
|
|
|
560
|
|
|
Accumulated deficit
|
|
|
(1,370
|
)
|
|
|
(3,211
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total stockholders’ equity
|
|
|
88,651
|
|
|
|
85,222
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities and stockholders’ equity
|
|
$
|
106,609
|
|
|
$
|
97,790
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these
consolidated financial statements.
48
IRIS
INTERNATIONAL, INC.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(in thousands, except per share data)
|
|
|
|
|
Revenues
|
|
|
|
|
|
|
|
|
|
|
|
|
|
IDD instruments
|
|
$
|
32,083
|
|
|
$
|
26,018
|
|
|
$
|
35,128
|
|
|
IDD consumables and service
|
|
|
61,112
|
|
|
|
52,213
|
|
|
|
46,643
|
|
|
Sample processing instruments and supplies
|
|
|
14,408
|
|
|
|
14,335
|
|
|
|
13,731
|
|
|
Personalized medicine services
|
|
|
69
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenues
|
|
|
107,672
|
|
|
|
92,566
|
|
|
|
95,502
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of Revenue
|
|
|
|
|
|
|
|
|
|
|
|
|
|
IDD instruments
|
|
|
21,237
|
|
|
|
16,778
|
|
|
|
19,605
|
|
|
IDD consumables and service
|
|
|
24,489
|
|
|
|
20,158
|
|
|
|
19,995
|
|
|
Sample processing instruments and supplies
|
|
|
6,619
|
|
|
|
6,965
|
|
|
|
6,863
|
|
|
Personalized medicine services
|
|
|
441
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total cost of revenue
|
|
|
52,786
|
|
|
|
43,901
|
|
|
|
46,463
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gross profit
|
|
|
54,886
|
|
|
|
48,665
|
|
|
|
49,039
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Marketing and selling
|
|
|
19,829
|
|
|
|
16,122
|
|
|
|
15,706
|
|
|
General and administrative
|
|
|
17,387
|
|
|
|
13,321
|
|
|
|
12,009
|
|
|
Research and development, net
|
|
|
14,562
|
|
|
|
11,411
|
|
|
|
10,357
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
51,778
|
|
|
|
40,854
|
|
|
|
38,072
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating income
|
|
|
3,108
|
|
|
|
7,811
|
|
|
|
10,967
|
|
|
Other income (expense):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income
|
|
|
1,123
|
|
|
|
857
|
|
|
|
1,180
|
|
|
Interest expense
|
|
|
(10
|
)
|
|
|
(21
|
)
|
|
|
(11
|
)
|
|
Manufacturing transition rights
|
|
|
—
|
|
|
|
—
|
|
|
|
1,232
|
|
|
Foreign currency transaction (loss) gain and other
|
|
|
(91
|
)
|
|
|
58
|
|
|
|
8
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Income before provision for income taxes
|
|
|
4,130
|
|
|
|
8,705
|
|
|
|
13,376
|
|
|
Provision for income taxes
|
|
|
1,088
|
|
|
|
2,454
|
|
|
|
4,363
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income
|
|
$
|
3,042
|
|
|
$
|
6,251
|
|
|
$
|
9,013
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income per share — basic
|
|
$
|
0.17
|
|
|
$
|
0.35
|
|
|
$
|
0.49
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income per share — diluted
|
|
$
|
0.17
|
|
|
$
|
0.35
|
|
|
$
|
0.48
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average common shares outstanding — basic
|
|
|
17,903
|
|
|
|
17,727
|
|
|
|
18,246
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average common shares outstanding — diluted
|
|
|
18,019
|
|
|
|
17,874
|
|
|
|
18,728
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these
consolidated financial statements.
49
IRIS
INTERNATIONAL, INC.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Accumulated Other
|
|
|
|
|
|
|
|
|
|
|
Common Stock
|
|
|
Paid-In
|
|
|
Comprehensive
|
|
|
Accumulated
|
|
|
|
|
|
|
|
Shares*
|
|
|
Amount
|
|
|
Capital
|
|
|
Income
|
|
|
Deficit
|
|
|
Total
|
|
|
|
|
(In thousands)
|
|
|
|
|
Balance, January 1, 2008
|
|
|
18,601
|
|
|
$
|
186
|
|
|
$
|
84,289
|
|
|
$
|
345
|
|
|
$
|
(9,886
|
)
|
|
$
|
74,934
|
|
|
Common stock issued on exercise of options
|
|
|
369
|
|
|
|
4
|
|
|
|
1,808
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,812
|
|
|
Restricted stock grants to employees
|
|
|
119
|
|
|
|
1
|
|
|
|
(1
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Tax benefit from stock option exercises
|
|
|
—
|
|
|
|
—
|
|
|
|
760
|
|
|
|
—
|
|
|
|
—
|
|
|
|
760
|
|
|
Translation adjustment, net of tax
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
70
|
|
|
|
—
|
|
|
|
70
|
|
|
Stock based compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
2,467
|
|
|
|
—
|
|
|
|
—
|
|
|
|
2,467
|
|
|
Purchase of common stock for retirement
|
|
|
(1,051
|
)
|
|
|
(11
|
)
|
|
|
(5,655
|
)
|
|
|
—
|
|
|
|
(7,458
|
)
|
|
|
(13,124
|
)
|
|
Settlement on restricted stock tax withholding
|
|
|
(2
|
)
|
|
|
|
|
|
|
(22
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(22
|
)
|
|
Net income for year
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
9,013
|
|
|
|
9,013
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance, December 31, 2008
|
|
|
18,036
|
|
|
|
180
|
|
|
|
83,646
|
|
|
|
415
|
|
|
|
(8,331
|
)
|
|
|
75,910
|
|
|
Common stock issued on exercise of options
|
|
|
221
|
|
|
|
2
|
|
|
|
1,448
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,450
|
|
|
Common stock issued on exercise of restricted stock units
|
|
|
17
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Restricted stock grants to employees
|
|
|
113
|
|
|
|
2
|
|
|
|
(2
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Tax benefit from stock option exercises
|
|
|
—
|
|
|
|
—
|
|
|
|
416
|
|
|
|
—
|
|
|
|
—
|
|
|
|
416
|
|
|
Translation adjustment, net of tax
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
145
|
|
|
|
—
|
|
|
|
145
|
|
|
Stock based compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
3,729
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,729
|
|
|
Cancellation of restricted stock
|
|
|
(9
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Purchase of common stock for retirement
|
|
|
(251
|
)
|
|
|
(3
|
)
|
|
|
(1,366
|
)
|
|
|
—
|
|
|
|
(1,131
|
)
|
|
|
(2,500
|
)
|
|
Settlement on restricted stock tax withholding
|
|
|
(16
|
)
|
|
|
—
|
|
|
|
(179
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(179
|
)
|
|
Net income for year
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
6,251
|
|
|
|
6,251
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance, December 31, 2009
|
|
|
18,111
|
|
|
|
181
|
|
|
|
87,692
|
|
|
|
560
|
|
|
|
(3,211
|
)
|
|
|
85,222
|
|
|
Common stock issued on exercise of options
|
|
|
22
|
|
|
|
—
|
|
|
|
31
|
|
|
|
—
|
|
|
|
—
|
|
|
|
31
|
|
|
Common stock issued on exercise of restricted stock units
|
|
|
10
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Restricted stock grants to employees
|
|
|
23
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Tax shortfall from stock option exercises
|
|
|
—
|
|
|
|
—
|
|
|
|
(61
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(61
|
)
|
|
Translation adjustment, net of tax
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(420
|
)
|
|
|
—
|
|
|
|
(420
|
)
|
|
Stock based compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
4,157
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,157
|
|
|
Cancellation of restricted stock
|
|
|
(15
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Purchase of common stock for retirement
|
|
|
(330
|
)
|
|
|
(3
|
)
|
|
|
(1,801
|
)
|
|
|
—
|
|
|
|
(1,201
|
)
|
|
|
(3,005
|
)
|
|
Settlement on restricted stock tax withholding
|
|
|
(30
|
)
|
|
|
|
|
|
|
(315
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
(315
|
)
|
|
Net income for year
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
3,042
|
|
|
|
3,042
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance, December 31, 2010
|
|
|
17,791
|
|
|
$
|
178
|
|
|
$
|
89,703
|
|
|
$
|
140
|
|
|
$
|
(1,370
|
)
|
|
$
|
88,651
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* |
|
Shares outstanding (does not include unvested restricted stock
units or vested restricted stock units which have not been
settled) |
The accompanying notes are an integral part of these
consolidated financial statements
50
IRIS
INTERNATIONAL, INC.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Cash flows from operating activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income
|
|
$
|
3,042
|
|
|
$
|
6,251
|
|
|
$
|
9,013
|
|
|
Adjustments to reconcile net income to net cash provided by
operating activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss on disposal of fixed assets
|
|
|
41
|
|
|
|
74
|
|
|
|
185
|
|
|
Loss on foreign currency remeasurement
|
|
|
155
|
|
|
|
—
|
|
|
|
—
|
|
|
Deferred taxes
|
|
|
(217
|
)
|
|
|
(182
|
)
|
|
|
2,158
|
|
|
Tax benefit from stock option exercises
|
|
|
(99
|
)
|
|
|
(416
|
)
|
|
|
(760
|
)
|
|
Depreciation and amortization
|
|
|
4,164
|
|
|
|
3,523
|
|
|
|
3,127
|
|
|
Stock based compensation
|
|
|
4,157
|
|
|
|
3,729
|
|
|
|
2,467
|
|
|
Changes in operating assets and liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accounts receivable
|
|
|
(3,062
|
)
|
|
|
2,546
|
|
|
|
(4,186
|
)
|
|
Inventories
|
|
|
512
|
|
|
|
(909
|
)
|
|
|
(71
|
)
|
|
Prepaid expenses and other assets
|
|
|
(779
|
)
|
|
|
1,280
|
|
|
|
(1,890
|
)
|
|
Investment in sales-type leases
|
|
|
(2,790
|
)
|
|
|
(1,677
|
)
|
|
|
112
|
|
|
Accounts payable
|
|
|
1,100
|
|
|
|
(1,820
|
)
|
|
|
2,010
|
|
|
Accrued expenses
|
|
|
1,593
|
|
|
|
(713
|
)
|
|
|
762
|
|
|
Deferred service contract revenue
|
|
|
938
|
|
|
|
373
|
|
|
|
500
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by operating activities
|
|
|
8,755
|
|
|
|
12,059
|
|
|
|
13,427
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from investing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Acquisition of business
|
|
|
(4,630
|
)
|
|
|
—
|
|
|
|
—
|
|
|
Purchase of assets from European distributor
|
|
|
(660
|
)
|
|
|
—
|
|
|
|
—
|
|
|
Acquisition of property and equipment
|
|
|
(4,564
|
)
|
|
|
(2,905
|
)
|
|
|
(3,588
|
)
|
|
Purchase of core technology
|
|
|
(3,284
|
)
|
|
|
—
|
|
|
|
—
|
|
|
Software development costs capitalized
|
|
|
(754
|
)
|
|
|
(835
|
)
|
|
|
(1,178
|
)
|
|
Purchase of short-term investments in marketable securities
|
|
|
—
|
|
|
|
—
|
|
|
|
(1,857
|
)
|
|
Sale of short-term investments in marketable securities
|
|
|
—
|
|
|
|
2,157
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in investing activities
|
|
|
(13,892
|
)
|
|
|
(1,583
|
)
|
|
|
(6,623
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from financing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Issuance of common stock and warrants for cash
|
|
|
31
|
|
|
|
1,450
|
|
|
|
1,812
|
|
|
Settlement on restricted stock tax withholding
|
|
|
(315
|
)
|
|
|
(179
|
)
|
|
|
(22
|
)
|
|
Repurchase of common stock
|
|
|
(3,005
|
)
|
|
|
(2,500
|
)
|
|
|
(13,124
|
)
|
|
Tax benefit from stock option exercises
|
|
|
99
|
|
|
|
416
|
|
|
|
760
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in financing activities
|
|
|
(3,190
|
)
|
|
|
(813
|
)
|
|
|
(10,574
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net foreign currency translation adjustments
|
|
|
(395
|
)
|
|
|
145
|
|
|
|
70
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net increase (decrease) in cash and cash equivalents
|
|
|
(8,722
|
)
|
|
|
9,808
|
|
|
|
(3,700
|
)
|
|
Cash and cash equivalents at beginning of year
|
|
|
34,253
|
|
|
|
24,445
|
|
|
|
28,145
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents at end of year
|
|
$
|
25,531
|
|
|
$
|
34,253
|
|
|
$
|
24,445
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
51
IRIS
INTERNATIONAL, INC.
CONSOLIDATED
STATEMENTS OF CASH FLOWS — (Continued)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Supplemental schedule of non-cash investing and financing
activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
During the year ended December 31, 2010, the Company
disposed of property and equipment with a cost and accumulated
depreciation of $666 and $625, respectively.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fair value of contingent consideration in connection with
business acquisition completed in the year ended
December 31, 2010 was $1.2 million
|
|
|
|
|
|
|
|
|
|
|
|
|
|
During the year ended December 31, 2009, the Company
disposed of property and equipment with a cost and accumulated
depreciation of $122 and $48, respectively.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
During the year ended December 31, 2008, the Company
disposed of property and equipment with a cost and accumulated
depreciation of $957 and $773, respectively.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental disclosure of cash flow information:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for income taxes
|
|
$
|
2,523
|
|
|
$
|
2,729
|
|
|
$
|
2,200
|
|
|
Cash paid for interest
|
|
$
|
9
|
|
|
$
|
21
|
|
|
$
|
11
|
|
The accompanying notes are an integral part of these
consolidated financial statements.
52
IRIS
INTERNATIONAL, INC.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Net income
|
|
$
|
3,042
|
|
|
$
|
6,251
|
|
|
$
|
9,013
|
|
|
Foreign currency translation, net of tax
|
|
|
(420
|
)
|
|
|
145
|
|
|
|
70
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive income
|
|
$
|
2,622
|
|
|
$
|
6,396
|
|
|
$
|
9,083
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these
consolidated financial statements.
53
IRIS
INTERNATIONAL, INC.
|
|
|
1.
|
Description
of Business
|
IRIS International, Inc. (the “Company”) was
incorporated in California in 1979 and reincorporated in 1987 in
Delaware. IRIS International, Inc. consists of three operating
units. Our in-vitro diagnostics segment also called Iris
Diagnostics Division (“IDD”), designs, manufactures
and markets systems, consumables and supplies for urinalysis and
body fluids. Our Sample Processing segment markets small
centrifuges and other processing equipment and accessories for
rapid specimen processing, as well as, equipment for fluorescent
in-situ hybridization (FISH). Our Personalized Medicine segment
combines the research and development operations of our Iris
Molecular Diagnostics and Arista Molecular, Inc. subsidiaries.
Under this new segment we consolidate all operations for the
development and commercialization of cancer diagnostic testing
services and related products.
|
|
|
2.
|
Summary
of Significant Accounting Policies
|
Use of
Estimates
The preparation of financial statements in conformity with
accounting principles generally accepted in the United States of
America requires management to make estimates and assumptions
that affect the reported amounts of assets and liabilities and
disclosure of contingent assets and liabilities at the date of
the financial statements and the reported amounts of revenues
and expenses during the reporting periods. The significant
estimates in the preparation of the consolidated financial
statements relate to the assessment of the carrying allowance
for doubtful accounts, inventory reserves, the useful lives,
fair value and recoverability of carrying value of long-lived
and intangible assets, including goodwill, unearned income on
sales-type leases, estimated provisions for warranty costs,
laboratory information system implementations, currency hedges
for foreign purchases and deferred tax assets. Actual results
and outcomes may differ from management’s estimates and
assumptions.
Principles
of Consolidation
The Company’s consolidated financial statements include the
accounts of IRIS International, Inc. and its wholly-owned
subsidiaries. Inter-company accounts and transactions have been
eliminated in the consolidated financial statements.
Cash
Equivalents
The Company considers highly liquid investments with maturities
of three months or less from the date of purchase to be cash
equivalents. These investments are carried at cost, which
approximates fair value.
Investments
in Marketable Securities
In 2008, the Company adopted FASB ASC Topic 820, “Fair
Value Measurement and Disclosures” (“ASC 820”),
for assets and liabilities measured at fair value on a recurring
basis. ASC 820 defines fair values as the price that would be
received to sell an asset or paid to transfer a liability in an
orderly transaction between market participants at the
measurement date. ASC 820 establishes a fair value hierarchy,
which prioritizes the inputs used in measuring fair value into
three broad levels as follows:
|
|
|
| |
•
|
Level 1 inputs are quoted prices (unadjusted) in
active markets for identical assets or liabilities that the
reporting entity has the ability to access at the measurement
date.
|
| |
| |
•
|
Level 2 inputs include quoted prices for similar
assets or liabilities in active markets, quoted prices for
identical or similar assets or liabilities in markets that are
not active, inputs other than quoted prices that are observable
for the asset or liability and inputs that are derived
principally from or corroborated by observable market data by
correlation or other means.
|
| |
| |
•
|
Level 3 inputs are unobservable inputs for the asset
or liability
|
54
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Accounts
Receivable
The Company sells predominantly to entities in the health care
industry. The Company grants uncollateralized credit to
customers, primarily hospitals, clinical and research
laboratories, and distributors. The Company performs ongoing
credit evaluations of customers’ financial condition before
granting uncollateralized credit. Although the Company generally
does not require collateral, letters of credit may be required
from its customers in certain circumstances. No single customer
accounts for 10% or more of the Company’s consolidated
revenues or 10% or more of the Company’s accounts
receivable at the balance sheet date.
Accounts receivable are carried at original invoice amount less
allowances made for sales markdowns, returns and doubtful
accounts. Accounts receivables are customer obligations due
under normal trade terms. Despite the Company’s stated
trade terms, several customers are subject to reimbursement
delays attributed to government and third party payer compliance
and regulation issues. Management determines the allowance for
doubtful accounts by identifying troubled accounts and by using
historical experience applied to an aging of accounts.
Receivables are written off when deemed uncollectible.
Inventories
Inventories are carried at the lower of cost or market on a
first in, first out basis. Provision for potentially obsolete or
slow-moving inventory is made based on management’s
analysis of inventory levels and future sales forecasts. Other
inventory that is considered excess inventory is fully reserved.
Property
and Equipment
Property and equipment are recorded at cost, less accumulated
depreciation and amortization. Depreciation is generally
computed using the straight-line method over three to seven
years, the estimated useful lives of the assets. Leasehold
improvements are amortized over the lesser of their useful life
or the remaining term of the lease.
Goodwill
and Intangible Assets
Goodwill represents the excess of the aggregate purchase price
over the fair value of the tangible and identifiable intangible
assets acquired by the Company. Goodwill and intangible assets
with indefinite lives, which consists of a CLIA license, are not
amortized. Goodwill and intangible assets with indefinite lives
are subject to impairment tests on an annual basis or more
frequently if facts and circumstances warrant such a review.
Goodwill and intangible assets with indefinite lives are
evaluated in accordance with FASB ASC Topic 350, Intangibles-
Goodwill and Other (“ASC 350”), based on various
analyses, including a comparison of the carrying value of the
reporting unit to its estimated fair value and discounted cash
flows. The analysis necessarily involves significant management
judgment to evaluate the capacity of an acquired business to
perform within projections. If the carrying amount of a
reporting unit exceeds its fair value, the goodwill impairment
test is performed to measure the amount of the impairment loss,
if any. During the years ended December 31, 2010, 2009 and
2008, the Company did not record any impairment charges related
to goodwill or intangible assets with indefinite lives.
Intangible assets are initially measured at their fair value,
determined either by the fair value of the consideration
exchanged for the intangible asset, or the estimated discounted
cash flows expected to be generated from the intangible asset.
Intangible assets with a finite life, such as core technology,
customer relationships and non-compete agreements are amortized
on a straight-line basis over their estimated useful life,
ranging from 3 to 20 years. Intangible assets with a finite
life are evaluated for impairment using the methodology set
forth in FASB ASC Topic 360, Property, Plant and
Equipment. Recoverability of these assets is assessed only
when events have occurred that may give rise to a potential
impairment. When a potential impairment has been identified,
forecasted undiscounted net cash flows of the operations to
which the asset relates are compared to the current carrying
value of the long-lived assets present in that operation. If
such cash flows are less than such carrying amounts, long-lived
55
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
assets, including such intangibles, are written down to their
respective fair values. During the years ended December 31,
2010, 2009 and 2008, no intangible asset impairment was recorded.
In determining the useful lives of intangible assets, the
Company considers the expected use of the assets and the effects
of obsolescence, demand, competition, anticipated technological
advances, market influences and other economic factors. For
technology based intangible assets, the Company considers the
expected life cycles of products which incorporate the
corresponding technology.
Goodwill and intangible assets as of December 31, 2010
consisted of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Average
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Amortization
|
|
|
Gross
|
|
|
|
|
|
|
|
|
|
|
Period (in
|
|
|
Carrying
|
|
|
Accumulated
|
|
|
Intangible
|
|
|
|
|
Years)
|
|
|
Amount
|
|
|
Amortization
|
|
|
Assets, Net
|
|
|
|
|
(Dollars in thousands)
|
|
|
|
|
Intangible assets subject to amortization:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Core technology
|
|
|
17
|
|
|
$
|
8,164
|
|
|
$
|
(513
|
)
|
|
$
|
7,651
|
|
|
Customer relationships
|
|
|
5
|
|
|
|
6
|
|
|
|
(1
|
)
|
|
|
5
|
|
|
Non-compete agreements
|
|
|
3
|
|
|
|
100
|
|
|
|
(15
|
)
|
|
|
85
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Subtotal
|
|
|
|
|
|
$
|
8,270
|
|
|
$
|
(529
|
)
|
|
|
7,741
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Intangible assets not subject to amortization:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CLIA License
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1,604
|
|
|
Goodwill
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3,957
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total intangible assets, net
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
13,302
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Core technology as of December 31, 2010, consists of
technology acquired in the acquisition of Leucadia Technologies,
Inc. in 2006, technology acquired in the acquisition of
AlliedPath, Inc. in July 2010 (see Note 3) and
technology acquired from BioMicro Systems, Inc. in November 2010
(see (Note 3).
Goodwill and intangible assets as of December 31, 2009
consisted of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Average
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Amortization
|
|
|
Gross
|
|
|
|
|
|
|
|
|
|
|
Period (in
|
|
|
Carrying
|
|
|
Accumulated
|
|
|
Intangible
|
|
|
|
|
Years)
|
|
|
Amount
|
|
|
Amortization
|
|
|
Assets, Net
|
|
|
|
|
(In thousands)
|
|
|
|
|
Intangible assets subject to amortization:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Core technology
|
|
|
20
|
|
|
$
|
1,790
|
|
|
$
|
(336
|
)
|
|
$
|
1,454
|
|
|
Intangible assets not subject to amortization:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Goodwill
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2,450
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
3,904
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total expense related to the amortization of intangible assets
was $193,000, $89,000 and $90,000 for the years ended
December 31, 2010, 2009 and 2008, respectively. In-process
research and development will be amortized beginning on the
completion date of the acquired product and will be amortized
over the estimated useful life determinable at that time.
56
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Total future amortization expense related to intangible assets
subject to amortization at December 31, 2010 is set forth
in the table below (in thousands):
| |
|
|
|
|
|
2011
|
|
$
|
330
|
|
|
2012
|
|
|
330
|
|
|
2013
|
|
|
316
|
|
|
2014
|
|
|
297
|
|
|
2015
|
|
|
296
|
|
|
Thereafter
|
|
|
2,888
|
|
|
|
|
|
|
|
|
|
|
$
|
4,457
|
|
|
|
|
|
|
|
The future amortization expense shown above does not include
amortization of core technology acquired from BioMicro Systems,
Inc. since a useful life has not yet been determined (see
Note 3).
The change in goodwill during the year ended December 31,
2010 is comprised of the following (in thousands):
| |
|
|
|
|
|
Balance at December 31, 2009
|
|
$
|
2,450
|
|
|
Addition recorded in connection with acquisition
|
|
|
1,507
|
|
|
|
|
|
|
|
|
Balance at December 31, 2010
|
|
$
|
3,957
|
|
|
|
|
|
|
|
Of the balance of goodwill and intangibles assets as of
December 31, 2010, $10 million relates to the
Personalized Medicine segment and $3.3 million relates to
the Sample Processing segment. The entire balance of goodwill
and intangible assets as of December 31, 2009 relates to
the Personalized Medicine segment.
Software
Development Costs
The Company capitalizes certain software development costs for
new products and product enhancements once all planning,
designing, coding and testing activities necessary to establish
that the product can be produced to meet our design
specifications are completed, and concludes capitalization when
the product is ready for general release. Research and
development costs relating to software development are expensed
as incurred. Amortization of capitalized software development
costs is provided on a
product-by-product
basis at the greater of the amount computed using (i) the
ratio of current revenues for a product to the total of current
and anticipated future revenues or (ii) the straight-line
method over the remaining estimated economic life of the product
up to five years.
Total software development costs capitalized totaled $754,000,
$835,000 and $1,178,000 for the years ended December 31,
2010, 2009 and 2008, respectively. Amortization expense of
software development costs totaled $659,000, $592,000 and
$651,000 for the years ended December 31, 2010, 2009 and
2008, respectively.
Impairment
of Long-Lived Assets
The Company will identify and record impairment losses for
long-lived assets whenever events or changes in circumstances
indicate that the carrying amount of an asset may not be
recoverable. Recoverability of assets to be held and used is
measured by a comparison of the carrying amount of an asset to
future undiscounted net cash flows expected to be generated by
the asset. If such assets are considered to be impaired, the
impairment to be recognized is measured by the amount by which
the carrying amount of the assets exceeds the fair value of the
assets. Assets to be disposed of are reported at the lower of
the carrying amount or fair value less costs to sell. There were
no impairments of long-lived assets at December 31, 2010,
2009 and 2008.
57
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Revenue
recognition
For products, revenue is recognized when risk of loss transfers,
when persuasive evidence of an arrangement exists, the price to
the buyer is fixed and determinable and collectability is
reasonably assured. When a customer enters into an
operating-type lease agreement, hardware revenue is recognized
on a straight-line basis over the life of the lease, while the
cost of the leased equipment is carried in customer leased
equipment within property, plant and equipment and amortized
over its estimated useful life. Under a sales-type lease
agreement, hardware revenue is generally recognized at the time
of shipment based on the present value of the minimum lease
payments with interest income recognized over the life of the
lease using the interest method. Instrument costs under a
sales-type lease agreement are recognized at the time of
shipment. Service revenues on maintenance contracts are
recognized ratably over the life of the service agreement or as
service is performed, if not under a contract. For those
instrument sales that include multiple deliverables, such as
instruments, training, consumables and service, the Company
allocated revenue based on the relative fair values of the
individual component sold separately, as determined in
accordance with FASB ASC Topic 605, Revenue Recognition.
The Company’s accounting for leases involves specific
determinations under FASB ASC Topic 840 Leases (“ASC
840”), which often involve complex provisions and
significant judgments. The four criteria of ASC 840 that
the Company uses in the determination of a sales-type lease or
operating-type lease are: (i) a review of the lease term to
determine if it is equal to or greater than 75 percent of
the economic life of the equipment; (ii) a review of the
minimum lease payments to determine if they are equal to or
greater than 90 percent of the fair market value of the
equipment; (iii) a determination of whether or not the
lease transfers ownership to the lessee at the end of the lease
term; and (iv) determination of whether or not the lease
contains a bargain purchase option.
Additionally, before classifying a lease as a sales-type lease,
the Company assesses whether collectability of the lease
payments is reasonably assured and whether there are any
significant uncertainties related to costs that the Company has
yet to incur with respect to the lease. Generally, the
Company’s leases that qualify as sales-type lease are
non-cancelable leases with a term of 75 percent or more of
the economic life of the equipment. Certain of the
Company’s lease contracts are customized for larger
customers and often result in complex terms and conditions that
typically require judgment in applying the lease accounting
criteria.
The economic life of the Company’s leased instruments and
their fair value require significant accounting estimates and
judgment. These estimates are based on the Company’s
historical experience. The most objective measure of the
economic life of the Company’s leased instrument is the
original term of a lease, which is typically five years, since a
majority of the instruments are returned by the lessee at or
near the end of the lease term and there is not a significant
after-market for our used instruments without substantial
remanufacturing. The Company believes that this is
representative of the period during which the instrument is
expected to be economically usable, with normal service, for the
purpose for which it is intended. The Company regularly
evaluates the economic life of existing and new products for
purposes of this determination.
The fair value of the Company’s leased instruments is
determined by a range of cash selling prices which the Company
deemed to be verifiable objective evidence. The Company
regularly evaluates available objective evidence of instrument
fair values using historical data.
The Company has certain government contracts with cancellation
clauses or renewal provisions that are generally required by
law, such as: (i) those dependant on fiscal funding outside
of a governmental unit’s control; (ii) those that can
be cancelled if deemed in the tax payers’ best interest; or
(iii) those that must be renewed each fiscal year, given
limitations that may exist on multi-year contracts that are
imposed by statute. Under these circumstances and in accordance
with the relevant accounting literature, as well as considering
the Company’s historical experience, a thorough evaluation
of these contracts is performed to assess whether cancellation
is remote or whether exercise of the renewal option is
reasonably assured.
58
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
The Company recognizes revenues from service contracts ratably
over the term of the service period, which typically ranges from
twelve to sixty months. Payments for service contracts are
generally received in advance. Deferred revenue represents the
revenues to be recognized over the remaining term of the service
contracts.
Shipping
and Handling Costs
The Company records shipping and handling costs billed to
customers as a component of revenue. Costs to distribute
products to customers, including inbound and outbound freight,
and other shipping and handling activities are included in cost
of goods sold in accordance with FASB ASC Topic
605-45-45-20.
Total shipping and handling costs included as a component of
revenue for the years ended December 31, 2010, 2009 and
2008 amounted to approximately $697,000, $707,000 and $964,000,
respectively. Total shipping and handling costs included as a
component of cost of sales amounted to $2,490,000, $1,871,000
and $2,343,000 for years ended December 31, 2010, 2009 and
2008, respectively.
Warranties
The Company recognizes warranty expense, based on
management’s estimate of expected cost, as an accrued
liability at the time of sale. The Company regularly reevaluates
its estimates to assess the adequacy of the recorded warranty
liabilities and adjust the amounts as necessary. Warranty
expense was approximately $797,000, $1,137,000 and $921,000
during the years ended December 31, 2010, 2009 and 2008,
respectively.
Advertising &
Literature Expenditures
Advertising and literature costs are charged to expense as
incurred. Advertising and literature expense for the years ended
December 31, 2010, 2009 and 2008 amounted to $360,000,
$244,000 and $234,000, respectively.
Research
and Development Expenditures
Except for certain software development costs capitalized as
described above, research and development expenditures are
charged to operations as incurred. Net research and development
expense includes total research and development costs incurred,
including costs incurred under research and development grants
and contracts, less costs reimbursed under research and
development contracts.
From time to time the Company receives grants from agencies of
the U.S. Government. The Company does not recognize any
revenue from such grants since they are cost reimbursement
grants whereby the Company submits requests for reimbursement
for certain costs incurred. There are no ongoing obligations or
requirements with respect to the grants received, the Company
retains ownership of any intellectual property that results from
the research and development, and the U.S. Government
agency receives a right to use the results of the research for
government projects. The Company received cost reimbursements of
$41,000, $85,000 and $164,000 during the years ended
December 31, 2010, 2009 and 2008, respectively.
Income
Taxes
The Company accounts for income taxes in accordance with FASB
ASC Topic
740-10,
Income Taxes, (“ASC 740”) which requires
recognition of deferred tax liabilities and assets for the
expected future tax consequences of events that have been
included in the financial statements or tax returns. Under this
method, deferred tax liabilities and assets are determined based
on the differences between the financial statement and the tax
bases of assets and liabilities using enacted tax rates in
effect for the year in which the differences are expected to
reverse. Valuation allowances are established when necessary to
reduce deferred tax assets to the amount expected to be
realized. Income tax expense represents the tax payable for the
period and the change during the period in deferred tax assets
and liabilities.
59
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
The Company accounts for uncertain tax positions in accordance
with ASC 740, which prescribes a recognition threshold and
measurement attribute for financial statement recognition and
measurement of a tax position taken or expected to be taken in a
tax return and also provides guidance on various related matters
such as derecognition, interest, penalties and disclosures
required. The Company recognizes interest and penalties, if any,
related to unrecognized tax benefits in income tax expense.
Foreign
Currency Exchange Translation
The Company’s functional currency is the U.S. dollar.
The functional currencies of the Company’s foreign
subsidiaries are primarily accounted for in their respective
local currencies. The statements of operations of foreign
operations are translated into U.S. dollars at rates of
exchange in effect each month. The balance sheets of these
subsidiaries are translated at period-end exchange rates, and
the differences from historical exchanges rates are reflected in
stockholders’ equity as other comprehensive income (loss).
Foreign currency transaction gains and losses from certain
intercompany transactions are recorded in foreign currency
transaction gain (loss) and other. Transactions denominated in
currencies other than the functional currency are recorded based
on rates in effect at the time such transactions arise.
Subsequent changes in exchange rates result in transaction gains
and losses and are reflected in the accompanying consolidated
statements of operations as unrealized (based on the applicable
period-end exchange rate) or realized upon settlement of the
transactions. All other foreign currency gains and losses are
also recorded on foreign currency transaction gain (loss) and
other.
Fair
Value of Financial Instruments
The carrying amounts of financial instruments including cash and
cash equivalents, short term investments in marketable
securities, accounts receivable, investment in sales-type
leases, accounts payable, accrued expenses and deferred service
contract revenues approximate fair value due to their short
maturity. The carrying amount of our long-term liabilities also
approximates fair value based on interest rates currently
available to us for debt of similar terms and remaining
maturities.
Earnings
Per Share
The Company computes and presents earnings per share in
accordance with FASB ASC Topic 260, Earnings Per Share.
Basic earnings per share are computed by dividing net income or
loss by the weighted average number of common shares
outstanding, including vested restricted shares and restricted
stock units, during each period. Diluted earnings per share is
computed by dividing net income by the weighted average number
of common shares and common stock equivalents outstanding,
calculated on the treasury stock method for options and warrants
using the average market prices during the period. The weighted
average number of outstanding antidilutive common stock options
and warrants excluded from the computation of diluted net income
per common share for the years ended December 31, 2010,
2009 and 2008 were 1,777,000, 1,479,000 and 715,000,
respectively.
60
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
A reconciliation of the shares used in the calculation of basic
and diluted earnings per common share is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Weighted average common shares outstanding — basic
|
|
|
17,903
|
|
|
|
17,727
|
|
|
|
18,246
|
|
|
Effect of dilutive securities
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock options
|
|
|
94
|
|
|
|
111
|
|
|
|
366
|
|
|
Restricted common shares and restricted stock units
|
|
|
22
|
|
|
|
36
|
|
|
|
80
|
|
|
Warrants
|
|
|
—
|
|
|
|
—
|
|
|
|
36
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average common shares outstanding — diluted
|
|
|
18,019
|
|
|
|
17,874
|
|
|
|
18,728
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock
Based Compensation
The Company accounts for stock based compensation under FASB ASC
Topic 718, Compensation — Stock Compensation
(“ASC 718”) which requires compensation costs
related to share-based transactions, including employee stock
options, to be recognized in the financial statements based on
fair value.
The Company’s results for the years ended December 31,
2010, 2009 and 2008 include share-based compensation expense
totaling $4,157,000, $3,729,000 and $2,464,000, respectively. In
addition, the Company’s results include stock based
compensation expense of $0, $0 and $3,000 relating to stock
issuances under our Employee Stock Purchase Plan for the years
ended December 31, 2010, 2009 and 2008, respectively.
Accordingly, the Company’s stock based compensation expense
totaled $4,157,000, $3,729,000 and $2,467,000 for the years
ended December 31, 2010, 2009 and 2008, respectively. The
total income tax benefit recognized in the income statement for
stock based compensation arrangements amounted to $1,369,000,
$1,188,000 and $996,000 for the years ended December 31,
2010, 2009, and 2008, respectively.
Foreign
Currency Hedge
The Company conducts business in certain foreign markets,
primarily in the European Union and Asia. To mitigate the
potential impact of adverse fluctuations in the U.S. Dollar
exchange rate for these currencies, the Company may periodically
purchases foreign currency forward contracts. The Company does
not speculate in these hedging instruments in order to profit
from foreign currency exchanges; nor does it enter into trades
for which there are no underlying exposures.
Under FASB ASC Topic 815, Accounting for Derivatives
Instruments and Hedging Activities, the Company documents
all relationships between hedging instruments and hedged items,
as well as its risk management objective for undertaking these
hedging transactions. This process includes relating the forward
contracts that are designated as fair value or cash flow hedges
to specific assets and liabilities on the balance sheet or to
specific firm commitments or forecasted transactions. The
Company also formally assesses, both at the inception of the
hedge and on an ongoing basis, whether each derivative is highly
effective in offsetting changes in fair values or cash flows of
the hedged of the hedged items.
At December 31, 2010, the Company did not have any foreign
currency forward contracts outstanding. During the year ended
December 31, 2008, the Company entered into such contracts
for Euros and Japanese Yen totaling $404,000 and
$7.6 million, respectively, which expired by
December 31, 2008. The contracts were to hedge purchases of
product in those countries and the results of these forwards are
included in cost of sales.
61
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Segment
Reporting
The Company determines and discloses industry segments in
accordance with FASB ASC Topic 280: Segment Reporting
(“ASC 280”) which uses a “management”
approach for determining business segments. The management
approach designates the internal organization that is used by
management for making operating decisions and assessing
performance as the source of the Company’s reportable
segments. ASC 280 also requires disclosures about products and
services, geographic areas, and major customers. See
Note 19 — “Segment and Geographic
Information”.
Reclassifications
In 2009, the Company reclassified employee bonus expenses
between cost of goods and operating expense categories in the
consolidated statement of income to conform to the presentation
used in the current year. The accompanying 2008 consolidated
statements of income contain these reclassification adjustments.
This resulted in an increase to cost of goods sold and a
decrease to operating expenses of $211,000 for the year ended
December 31, 2008.
These reclassifications had no impact on the Company’s
previously reported consolidated operating income, net income or
basic or diluted earnings per share.
Certain
Risks and Uncertainties
Financial instruments, which potentially expose the Company to
concentration of credit risk, consist primarily of cash and cash
equivalents, accounts receivable and investment in sales-type
leases. Concentration of credit risk with respect to accounts
receivable and investment in sales-type leases is mitigated by
the Company’s performance of on-going credit evaluations of
its customers and the Company maintains an allowance for
doubtful accounts. Investments in sales-type leases are secured
by the underlying instruments.
At December 31, 2010, the amount of the Company’s cash
deposited in demand deposit accounts which are fully guaranteed
by the Federal Deposit Insurance Corporation was $3.7M. The rest
of the cash balances on deposit with banks are guaranteed by the
Federal Deposit Insurance Corporation up to $250,000. The
Company may be exposed to risk for the amount of funds held in
one bank in excess of the insurance limit. In assessing the
risk, the Company’s policy is to maintain cash balances
with high quality financial institutions.
The Company derives most of its revenues from the sale of the
urinalysis analyzers, and related supplies and services.
Declines in unit sales or gross margins could have a material
adverse effect on the Company’s revenues and profits,
respectively.
Certain of the Company’s components are obtained from
outside vendors, and the loss or breakdown of the Company’s
relationships with these outside vendors could subject us to
substantial delays in the delivery of its products to its
customers. Furthermore, certain key components of the
Company’s instruments and certain consumables are
manufactured by only one supplier. The Company’s inability
to sell products to meet delivery schedules could have a
material adverse effect on its reputation in the industry, as
well as its financial condition and results of operation.
Recent
Accounting Pronouncements
In December 2010, the FASB issued Accounting Standards Update
(“ASU”) ASU
2010-29,
Business Combinations- Disclosure of Supplementary Pro Forma
Information, which specifies that if a public entity
presents comparative financial statements, the entity should
disclose revenue and earnings of the combined entity as though
the business combination(s) that occurred during the current
year had occurred as of the beginning of the comparable prior
annual reporting period only.
62
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
ASU 2010-29
is effective on a prospective basis for business combinations
for which the acquisition date is on or after the beginning of
the first annual reporting period beginning on or after
December 15, 2010 with early adoption permitted. We are
currently evaluating both the timing and the impact of the
adoption of the ASU on our consolidated financial statements.
In July 2010, the FASB issued ASU
2010-20,
Disclosures about the Credit Quality of Financing Receivables
and the Allowance for Credit Losses which amends ASC Topic
310, Receivables. ASU
2010-20
requires disclosures about the nature of the credit risk in an
entity’s financing receivables, how that risk is
incorporated into the allowance for credit losses, and the
reasons for any changes in the allowance. Disclosure is required
to be disaggregated, primarily at the level at which an entity
calculates its allowance for credit losses.
ASU 2010-20
is applicable to both private and public companies, and will
affect any entity that has financing receivables on its balance
sheet, not including short-term trade accounts receivable.
Public entities must apply the disclosure requirements
applicable to period-end balances beginning with the first
interim or annual reporting period ending on or after
December 15, 2010 (December 31, 2010 for a calendar
year-end entity). The adoption of this guidance resulted in
additional disclosures (see Note 7) but did not have
an impact on our consolidated financial statements.
In October 2009, the FASB issued Accounting Standards Update
(“ASU”) ASU
2009-14,
Certain Revenue Arrangements That Include Software
Elements, now codified under FASB ASC Topic 985,
Software. ASU
2009-14,
removes tangible products from the scope of software revenue
guidance and provides guidance on determining whether software
deliverables in an arrangement that includes a tangible product
are covered by the scope of the software revenue guidance. ASU
2009-14 is
to be applied on a prospective basis for revenue arrangements
entered into or materially modified in fiscal years beginning on
or after June 15, 2010, with early adoption permitted. We
are currently evaluating both the timing and the impact of the
adoption of the ASU on our consolidated financial statements.
In October 2009, the FASB issued Accounting Standards Update
(“ASU”)
2009-13,
Multiple-Deliverable Revenue Arrangements, which amends
ASC Topic 605, Revenue Recognition, to require companies
to allocate revenue in multiple-element arrangements based on an
element’s estimated selling price if vendor-specific or
other third-party evidence of value is not available. ASU
2009-13 is
effective beginning June 15, 2010. Earlier application is
permitted. The adoption of ASU
2009-13 did
not have a material impact on our consolidated financial
statements.
In December 2010, the FASB issued Accounting Standards Update
(“ASU”)
No. 2010-28 —
When to Perform Step 2 of the Goodwill Impairment Test for
Reporting Units with Zero or Negative Carrying Amounts. This
update provides amendments to ASC Topic 350 —
Intangibles, Goodwill and Other that requires an entity to
perform Step 2 impairment test even if a reporting unit has zero
or negative carrying amount. Step 1 tests whether the carrying
amount of a reporting unit exceeds its fair value. Previously
reporting units with zero or negative carrying value passed Step
1 because the fair value was generally greater than zero. Step 2
requires impairment testing and impairment valuation be
calculated in between annual tests if an event or circumstances
indicate that it is more likely than not that goodwill has been
impaired. ASU
2010-28 is
effective beginning January 1, 2011. As a result of this
standard, goodwill impairments may be reported sooner than under
current practice. We do not expect ASU
2010-28 to
have a material impact on our financial statements.
On January 1, 2010, the Company purchased certain assets
relating to its current distribution of IRIS products in the
United Kingdom, Ireland and Germany from one of the
Company’s European distributors for a cash payment of
$660,000. The assets purchased consist primarily of customer
leases for installed IRIS instruments and service contracts
valued at inventory book value. This purchase increased the
Company’s direct sales presence within its international
sales channels, and will serve as a template for further
potential transactions in territories that represent new
business opportunities for the Company.
63
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
On July 28, 2010, the Company acquired AlliedPath, Inc.
AlliedPath is a high complexity CLIA-certified molecular
pathology laboratory offering differentiated, high value
molecular diagnostic services in the rapidly growing field of
personalized medicine. Pursuant to the terms of the merger
agreement dated July 26, 2010, the Company acquired all the
issued and outstanding stock of AlliedPath for an amount in cash
equal to $4.6 million less certain indebtedness existing at
the closing, with an additional earn-out of up to
$1.3 million subject to the achievement of specific sales
and earnings targets through December 2013. We did not assume
any outstanding options or warrants of AlliedPath in connection
with the acquisition. AlliedPath is now called Arista Molecular,
Inc. (“Arista”) as of January 2011 and will operate
under the Personalized Medicine reporting segment of the
consolidated financial statements
Through the acquisition of Arista, the Company seeks to achieve
the following goals:
|
|
|
| |
•
|
to expand beyond its initial molecular pathology test menu by
adding other molecular panels, flow cytometry for the detection
and monitoring of leukemia and lymphoma, FISH testing, and
proprietary new tests based on the Company’s NADiA
technology platform;
|
| |
| |
•
|
to have better control of all aspects of the commercial
operations of the NADiA platform, starting with NADiA ProsVue;
|
| |
| |
•
|
to enable the acceleration of the development efforts of the
NADiA technology product pipeline; and
|
| |
| |
•
|
to enter the attractive personalized medicine market due to its
significant growth potential.
|
The aggregate consideration paid for the acquisition of Arista
is as follows:
| |
|
|
|
|
|
|
|
(In thousands)
|
|
|
|
|
Cash
|
|
$
|
4,630
|
|
|
Fair value of contingent consideration
|
|
|
1,210
|
|
|
|
|
|
|
|
|
Total purchase price
|
|
$
|
5,840
|
|
|
|
|
|
|
|
Acquisition related expenses included in marketing and sales and
general and administrative total $525,000 for the year ended
December 31, 2010.
The following table summarizes the estimated fair values of
assets acquired and liabilities assumed as of July 28, 2010.
| |
|
|
|
|
|
|
|
(In thousands)
|
|
|
|
|
Current assets
|
|
$
|
74
|
|
|
Property and equipment
|
|
|
523
|
|
|
Core technology
|
|
|
3,090
|
|
|
CLIA License
|
|
|
1,604
|
|
|
Customer relationships
|
|
|
6
|
|
|
Non-compete agreements
|
|
|
100
|
|
|
Goodwill
|
|
|
1,507
|
|
|
Other assets
|
|
|
31
|
|
|
Current liabilities
|
|
|
(316
|
)
|
|
Lease obligations
|
|
|
(178
|
)
|
|
Other liabilities
|
|
|
(61
|
)
|
|
Deferred tax liability, net
|
|
|
(540
|
)
|
|
|
|
|
|
|
|
Total purchase price
|
|
$
|
5,840
|
|
|
|
|
|
|
|
64
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
The purchase price allocations and pro forma adjustments are
based on preliminary estimates, available information and
certain assumptions, and may be revised as additional
information becomes available.
In determining the purchase price allocation, the Company
considered, among other factors, historical demand for products,
estimates of future demand for those services, customer
relationships, the revenue generating potential of core
technology, the assets’ useful lives, and agreements not to
compete. The market, income and cost approaches were used to
determine fair values of these intangibles. The rate used to
discount the net cash flows to their present value was a 16.5%
weighted average cost of capital for the business as a whole,
and from 16.5% to 17.5% for the individual intangible assets
depending on the risk associated with the asset’s potential
to generate revenues and its projected remaining useful economic
life. The weighted average cost of capital was determined after
consideration of market rates of return on debt and equity
capital of comparable companies, the weighted average return on
invested capital and the risk associated with achieving
forecasted sales related to technology and assets acquired. The
fair value of the contingent consideration was determined
considering the probability of payout and using a 3% discount
rate. Property and equipment net book value was evaluated at
approximately fair value on the acquisition date due to the
nature and relative age of the assets acquired.
Acquired property and equipment are being depreciated on a
straight-line basis with estimated remaining useful lives
ranging from 1 year to 5 years. Intangible assets
except the CLIA license are being amortized on a straight-line
basis with estimated remaining useful lives ranging from
3 years to 15 years reflecting the expected future
value. The CLIA license is considered to have an indefinite
useful life. The purchase was structured as a stock purchase
therefore the value assigned to the core technology, CLIA
license, customers relationships, non-compete agreements and
goodwill is not deductible for tax purposes.
Revenue and net loss of Arista included in the Company’s
consolidated financial statements since the laboratory’s
acquisition on July 28, 2010 totaled $69,000 and
$3,672,000, respectively, for the year ended December 31,
2010
The following table summarizes unaudited pro forma financial
information assuming the acquisition of Arista had occurred on
January 1, 2009, in the corresponding period of the fiscal
year immediately preceding the acquisition. This unaudited pro
forma financial information does not necessarily represent what
would have occurred if the transaction had taken place on
January 1, 2009 and should not be taken as representative
of the Company’s future consolidated results of operations
or financial position.
| |
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31,
|
|
|
|
2010
|
|
2009
|
|
|
|
(In thousands)
|
|
|
|
Revenue
|
|
$
|
107,697
|
|
|
$
|
92,570
|
|
|
Net income
|
|
|
1,720
|
|
|
|
3,803
|
|
|
Net income per basic and diluted share
|
|
$
|
0.10
|
|
|
$
|
0.21
|
|
On November 22, 2010, the Company acquired the assets of a
multi-purpose, bench-top instrument platform for automating
highly repetitive, manual laboratory protocols for FISH
(fluorescence in-situ hybridization) testing and other
slide-based cytogenetic applications. The product acquisition is
a natural extension to the successful
ThermoBrite®
DNA Hybridization System and in line with the Company’s
entry into personalized medicine with emphasis on cancer
diagnostics. The product prototypes and proprietary technology
assets were purchased for $3.2 million in cash from
BioMicro Systems, Inc. The new product platform will be
integrated into the Iris Sample Processing Division and it is
expected to position IRIS as a major competitor in the high
growth cytogenetic instrumentation market. This acquisition was
recorded as an acquisition of assets determined not to be a
business, since no workforce nor strategic management,
operational or resource management processes were included in
the purchase.
65
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
The purchase price of $3.2 million plus related asset
acquisition costs of $84,000 was allocated to core technology
and is recorded in intangible assets on the Company’s
consolidated balance sheet as of December 31, 2010. The
purchase price allocation was based on preliminary estimates and
available information, and may be revised as additional
information becomes available. Although BioMicro Systems built
and tested a working prototype, which proved the technical
feasibility of the base technology, several elements of this
technology platform requires continued development in order to
reach salability for its stated purpose. Once development is
completed, a useful life will be determined and the assets will
be amortized over the useful life determined at that time.
|
|
|
4.
|
Investments
in Marketable Securities
|
In 2008, the Company adopted FASB ASC Topic 820, Fair Value
Measurement (“ASC 820”), for assets and
liabilities measured at fair value on a recurring basis.
ASC 820, defines fair values as the price that would be
received to sell an asset or paid to transfer a liability in an
orderly transaction between market participants at the
measurement date. ASC 820 establishes a fair value
hierarchy, which prioritizes the inputs used in measuring fair
value into three broad levels as follows:
Level 1 inputs are quoted prices (unadjusted) in
active markets for identical assets or liabilities that the
reporting entity has the ability to access at the measurement
date.
• Level 2 inputs
include quoted prices for similar assets or liabilities in
active markets, quoted prices for identical or similar assets or
liabilities in markets that are not active, inputs other than
quoted prices that are observable for the asset or liability and
inputs that are derived principally from or corroborated by
observable market data by correlation or other means.
Level 3 inputs are unobservable inputs for the asset
or liability
At December 31, 2010 and 2009, the Company did not hold any
investments in marketable securities.
Inventories consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
At December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
(In thousands)
|
|
|
|
|
Finished goods
|
|
$
|
3,423
|
|
|
$
|
2,595
|
|
|
Work-in-process
|
|
|
181
|
|
|
|
214
|
|
|
Raw materials, parts and
sub-assemblies
|
|
|
6,706
|
|
|
|
8,057
|
|
|
|
|
|
|
|
|
|
|
|
|
Inventories
|
|
$
|
10,310
|
|
|
$
|
10,866
|
|
|
|
|
|
|
|
|
|
|
|
66
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
|
|
|
6.
|
Property
and Equipment
|
Property and equipment consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
At December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
(In thousands)
|
|
|
|
|
Machinery and equipment
|
|
$
|
13,894
|
|
|
$
|
11,145
|
|
|
Leasehold improvements
|
|
|
8,021
|
|
|
|
6,385
|
|
|
Tooling, dies and molds
|
|
|
1,224
|
|
|
|
2,158
|
|
|
Furniture and fixtures
|
|
|
2,088
|
|
|
|
1,152
|
|
|
Rental units
|
|
|
1,299
|
|
|
|
540
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
26,526
|
|
|
|
21,380
|
|
|
Less: accumulated depreciation
|
|
|
(14,491
|
)
|
|
|
(11,713
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
12,035
|
|
|
$
|
9,667
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation expense for Property and Equipment, for the years
ended December 31, 2010, 2009 and 2008 was
$3.3 million, $2.8 million, and $2.4 million,
respectively.
The components of net investment in sales-type leases consist of
the following:
| |
|
|
|
|
|
|
|
|
|
|
|
At December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
(In thousands)
|
|
|
|
|
Total minimum lease payments
|
|
$
|
16,044
|
|
|
$
|
12,735
|
|
|
Less: unearned income
|
|
|
(2,464
|
)
|
|
|
(1,897
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Net investment in sales-type leases
|
|
|
13,580
|
|
|
|
10,838
|
|
|
Less: current portion
|
|
|
(3,578
|
)
|
|
|
(3,397
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Net investment in sales-type leases, non-current portion
|
|
$
|
10,002
|
|
|
$
|
7,441
|
|
|
|
|
|
|
|
|
|
|
|
Future minimum lease payments due from customers under
sales-type leases for each of the five succeeding years:
| |
|
|
|
|
|
|
|
(In thousands)
|
|
|
|
|
Year Ending December 31,
|
|
|
|
|
|
2011
|
|
$
|
3,578
|
|
|
2012
|
|
|
3,151
|
|
|
2013
|
|
|
2,736
|
|
|
2014
|
|
|
2,366
|
|
|
2015
|
|
|
1,509
|
|
|
Thereafter
|
|
|
240
|
|
|
|
|
|
|
|
|
|
|
$
|
13,580
|
|
|
|
|
|
|
|
Our leases are primarily to customers in the health care
industry or to governments. We assess credit risk for all of our
customers including those who lease equipment. Credit risk is
assessed using an internally developed model which incorporates
credit scores from third party providers and our own custom risk
ratings and is updated on a quarterly basis. The external credit
scores are developed based on the customer’s historical
payment patterns and an
67
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
overall assessment of the likelihood of delinquent payments. Our
internal ratings are weighted based on company size, years in
business, and other credit related factors (i.e. profitability,
cash flow, liquidity, tangible net worth, etc.). Any one of the
following factors may result in a customer being classified as
high risk: i) the customer has a history of late payments;
ii) the customer has open lawsuits, liens or judgments; and
iii) the customer has been in business less than three
years. Our lease receivables are collateralized by the
equipment’s fair value, which mitigates our credit risk.
The following table presents the risk profile by
creditworthiness category of our sales-type lease receivables at
December 31, 2010:
| |
|
|
|
|
|
|
|
(In thousands)
|
|
|
|
|
Low risk
|
|
$
|
12,128
|
|
|
Moderate risk
|
|
|
785
|
|
|
High Risk
|
|
|
667
|
|
|
|
|
|
|
|
|
|
|
$
|
13,580
|
|
|
|
|
|
|
|
The balance of the allowance for uncollectible accounts for our
sales-type leases was zero as of December 31, 2010. We
determine the adequacy of our allowance for uncollectible
accounts for sales-type leases based on an analysis of
historical write-offs. There have been no write-offs of
sales-type lease receivables for the years ended
December 31, 2010, 2009 or 2008. As of December 31,
2010, the amount of sales-type leases which were past due was
not significant and there were no impaired sales-type leases.
Accordingly, there was no material risk of default with respect
to sales-type leases as of December 31, 2010.
The Company has a credit facility with a commercial bank. The
credit facility consists of a $6.5 million revolving line
of credit for working capital and a $10.0 million line of
credit for acquisitions and product opportunities. The credit
facility has variable interest rates, which will change from
time to time based on changes to either the LIBOR rate or the
lender’s prime rate. Borrowings under the credit facility
are secured by all of the Company’s assets and mature in
June 2012 and June 2015, respectively.
As of December 31, 2010 and 2009, there were no borrowings
under the credit facility. However, the Company is subject to
certain financial and non financial covenants under the credit
facility with the bank and as of December 31, 2010, the
Company was in compliance with these covenants.
Accrued expenses consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
At December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
(In thousands)
|
|
|
|
|
Accrued bonuses
|
|
$
|
1,762
|
|
|
$
|
197
|
|
|
Accrued commissions
|
|
|
824
|
|
|
|
607
|
|
|
Accrued payroll
|
|
|
1,059
|
|
|
|
1,334
|
|
|
Accrued vacation
|
|
|
1,624
|
|
|
|
1,234
|
|
|
Accrued professional fees
|
|
|
499
|
|
|
|
416
|
|
|
Accrued warranty
|
|
|
705
|
|
|
|
946
|
|
|
Accrued laboratory information system implementations
|
|
|
487
|
|
|
|
335
|
|
|
Accrued — other
|
|
|
553
|
|
|
|
692
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
7,513
|
|
|
$
|
5,761
|
|
|
|
|
|
|
|
|
|
|
|
68
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Changes in accrued warranty were as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
At December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
(In thousands)
|
|
|
|
|
Balance — beginning of year
|
|
$
|
946
|
|
|
$
|
808
|
|
|
Additions for provisions during year
|
|
|
797
|
|
|
|
1,137
|
|
|
Reductions during year
|
|
|
(1,038
|
)
|
|
|
(999
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Balance — end of year
|
|
$
|
705
|
|
|
$
|
946
|
|
|
|
|
|
|
|
|
|
|
|
The provision for income taxes from operations consists of the
following:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Current:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal
|
|
$
|
514
|
|
|
$
|
1,879
|
|
|
$
|
2,323
|
|
|
Foreign
|
|
|
63
|
|
|
|
—
|
|
|
|
—
|
|
|
State
|
|
|
665
|
|
|
|
757
|
|
|
|
644
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1,242
|
|
|
|
2,636
|
|
|
|
2,967
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deferred:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal
|
|
|
243
|
|
|
|
364
|
|
|
|
1,182
|
|
|
Foreign
|
|
|
170
|
|
|
|
(32
|
)
|
|
|
(55
|
)
|
|
State
|
|
|
(567
|
)
|
|
|
(514
|
)
|
|
|
269
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(154
|
)
|
|
|
(182
|
)
|
|
|
1,396
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
1,088
|
|
|
$
|
2,454
|
|
|
$
|
4,363
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Income taxes have been based on the following components of
pre-tax income (loss):
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Domestic
|
|
$
|
3,541
|
|
|
$
|
8,821
|
|
|
$
|
13,557
|
|
|
Foreign
|
|
|
589
|
|
|
|
(116
|
)
|
|
|
(181
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
4,130
|
|
|
$
|
8,705
|
|
|
$
|
13,376
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
69
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
The provision for income taxes differs from the amount obtained
by applying the federal statutory income tax rate to income
before provision for income taxes as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Tax provision computed at Federal statutory rate
|
|
$
|
1,404
|
|
|
$
|
2,960
|
|
|
$
|
4,549
|
|
|
State taxes, net of federal benefit
|
|
|
252
|
|
|
|
544
|
|
|
|
499
|
|
|
R&D tax credits
|
|
|
(790
|
)
|
|
|
(258
|
)
|
|
|
(507
|
)
|
|
Incentive stock options
|
|
|
293
|
|
|
|
303
|
|
|
|
(10
|
)
|
|
Nondeductible expenses
|
|
|
72
|
|
|
|
(83
|
)
|
|
|
(120
|
)
|
|
Change in valuation allowance
|
|
|
(45
|
)
|
|
|
(269
|
)
|
|
|
(260
|
)
|
|
Rate differential on Foreign Income
|
|
|
(9
|
)
|
|
|
(32
|
)
|
|
|
(55
|
)
|
|
Prior year return to provision adjustment
|
|
|
(61
|
)
|
|
|
(772
|
)
|
|
|
—
|
|
|
Other
|
|
|
(28
|
)
|
|
|
61
|
|
|
|
267
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
1,088
|
|
|
$
|
2,454
|
|
|
$
|
4,363
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The primary components of temporary differences, which give rise
to the Company’s net deferred tax asset at
December 31, 2010 and 2009, are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
At December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
(In thousands)
|
|
|
|
|
Depreciation and amortization
|
|
$
|
(2,738
|
)
|
|
$
|
(998
|
)
|
|
Allowance for doubtful accounts
|
|
|
187
|
|
|
|
166
|
|
|
Accrued liabilities
|
|
|
1,382
|
|
|
|
1,562
|
|
|
Inventory
|
|
|
145
|
|
|
|
196
|
|
|
Stock compensation
|
|
|
2,132
|
|
|
|
1,449
|
|
|
Net operating loss carryforwards
|
|
|
2,740
|
|
|
|
1,264
|
|
|
Tax credits
|
|
|
3,603
|
|
|
|
3,585
|
|
|
Valuation allowance
|
|
|
(598
|
)
|
|
|
(335
|
)
|
|
State deferred taxes
|
|
|
(1,103
|
)
|
|
|
(753
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
5,750
|
|
|
$
|
6,136
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31, 2010, withholding and U.S. taxes
have not been provided on approximately $200,000 of cumulative
undistributed earnings of the Company’s
non-U.S. subsidiaries
because the Company intends to indefinitely reinvest these
earnings in its
non-U.S. subsidiaries.
At December 31, 2010, the Company has Federal, state and
foreign net operating loss carryforwards of approximately
$5.2 million, $3.9 million, and $1.9 million,
respectively expiring beginning in 2011 through 2030. The
federal and state net operating loss carryforwards are subject
to limitations on their utilization. The Company also has
federal and state tax credit carryforwards of $666,000 and
$2.9 million, respectively, net of valuation allowances.
Realization of deferred tax assets associated with foreign net
operating losses (“NOL”) and tax credit carryforwards
is dependent upon the Company’s ability to generate
sufficient taxable income prior to their expiration. Management
believes it is more likely than not that the deferred tax assets
will be realized through future taxable income or alternative
tax strategies. However, the net deferred tax assets could be
reduced in the near term if
70
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
management’s estimates of taxable income during the
carryforward period are not realized or are significantly
reduced or alternative tax strategies are not available. The
company has recorded a valuation allowance of $308,000 related
to net operating loss carryforwards of Arista which will likely
expire due to a section 382 limitation on their
utilization. The Company has reversed a $45,000 valuation
allowance related to foreign net operating losses based on its
estimate of future utilization. The Company will continue to
review estimates of taxable income and will adjust the valuation
allowance, when necessary.
In connection with the acquisition of Arista, the company
recorded deferred tax liabilities of $540,000 net of the
aforementioned valuation allowance.
The Company adopted the provisions of FASB ASC Topic
740-10,
Income Taxes, which requires that the Company recognize
the impact of a tax position in its financial statements if that
position is more likely than not of being sustained upon audit,
based on the technical merits of the position.
The Company files income tax returns in the U.S. federal
jurisdiction and various state and foreign jurisdictions. In
general, the U.S. federal statute for the assessment of
taxes for the Company is no longer open for tax years prior to
2007 and for state income tax purposes for tax years prior to
2006. The Company is not under examination by any taxing
authorities other than in Germany, which is examining the
Company’s 2006 through 2009 income tax returns. The Company
does not believe any additional taxes will be due in connection
with the examination.
As of December 31, 2010, the total amount of unrecognized
tax benefits was $1,908,000. The unrecognized tax benefits
primarily relate to uncertainties with respect to tax credit
carryovers and all future reductions to the unrecognized tax
benefits would result in a reduction to the future effective tax
rate.
The following changes occurred in the amount of unrecognized tax
benefits (including related interest and penalties) during the
years ended December 31, 2010, 2009 and 2008 as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Balance at January 31
|
|
$
|
1,282
|
|
|
$
|
998
|
|
|
$
|
616
|
|
|
Additions for current year tax positions
|
|
|
626
|
|
|
|
284
|
|
|
|
382
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31
|
|
$
|
1,908
|
|
|
$
|
1,282
|
|
|
$
|
998
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The Company does not expect any reduction to the unrecognized
tax benefits in the next twelve months. Currently, there are no
amounts of interest or penalties recorded in the financial
statements as the unrecognized tax benefits relate to deferred
tax assets. The Company will recognize potential interest and
penalties related to income tax positions as a component of the
provision for income taxes on the consolidated statements of
income in any future periods in which the Company must record a
liability.
71
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
|
|
|
11.
|
Stock-Based
Compensation
|
The Company accounts for stock-based compensation pursuant to
FASB ASC Topic 718, Stock Compensation, which requires
compensation costs related to share-based transactions,
including employee stock options, to be recognized in the
financial statements based on fair value. Share-based
compensation expense for the years ended December 31, 2010,
2009 and 2008 is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended
|
|
|
|
|
December 31,
|
|
|
|
|
2009
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(In thousands)
|
|
|
|
|
Cost of sales
|
|
$
|
357
|
|
|
$
|
417
|
|
|
$
|
341
|
|
|
Marketing and selling
|
|
|
688
|
|
|
|
631
|
|
|
|
398
|
|
|
General and administrative
|
|
|
2,233
|
|
|
|
2,026
|
|
|
|
1,210
|
|
|
Research and development
|
|
|
879
|
|
|
|
655
|
|
|
|
518
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock-based compensation
|
|
$
|
4,157
|
|
|
$
|
3,729
|
|
|
$
|
2,467
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock
Options
As of December 31, 2010, the Company had a stock option
plan under which the Company may grant future non-qualified
stock options, incentive stock options and stock appreciation
rights. No stock appreciation rights have been granted under any
of the Company’s stock option plans. On July 13, 2007,
the Company’s stockholders approved the adoption of the
IRIS International, Inc. 2007 Stock Incentive Plan, which
authorizes the issuance of up to 1,750,000 shares of common
stock pursuant to equity awards granted under the plan. On
May 22, 2009, the Company’s stockholders approved an
increase of 1,550,000 shares to the 2007 Stock Incentive
Plan for a total of 3,300,000 authorized shares.
The following schedule sets forth options authorized, exercised,
outstanding and available for grant under the Company’s
existing stock option plans as of December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Number of Option Shares
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Available
|
|
|
Plan
|
|
Authorized
|
|
|
Exercised
|
|
|
Outstanding
|
|
|
for Grant
|
|
|
|
|
(In thousands)
|
|
|
|
|
1994 Plan
|
|
|
700
|
|
|
|
680
|
|
|
|
20
|
|
|
|
—
|
|
|
1998 Plan
|
|
|
4,100
|
|
|
|
2,717
|
|
|
|
899
|
|
|
|
—
|
|
|
2007 Plan
|
|
|
3,300
|
|
|
|
—
|
|
|
|
1,849
|
|
|
|
794
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8,100
|
|
|
|
3,397
|
|
|
|
2,768
|
|
|
|
794
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
72
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
The following table sets forth certain information relative to
stock options during the three years ended December 31,
2010.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted Average
|
|
|
Average
|
|
|
|
|
Shares
|
|
|
Exercise Price
|
|
|
Intrinsic Value
|
|
|
|
|
(In thousands, except for per share)
|
|
|
|
|
Outstanding at January 1, 2008
|
|
|
1,861
|
|
|
$
|
11.62
|
|
|
|
|
|
|
Granted
|
|
|
502
|
|
|
$
|
13.03
|
|
|
|
|
|
|
Exercised
|
|
|
(369
|
)
|
|
$
|
4.90
|
|
|
|
|
|
|
Canceled or expired
|
|
|
(20
|
)
|
|
$
|
11.47
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at December 31, 2008
|
|
|
1,974
|
|
|
$
|
13.24
|
|
|
|
|
|
|
Granted
|
|
|
571
|
|
|
$
|
9.02
|
|
|
|
|
|
|
Exercised
|
|
|
(197
|
)
|
|
$
|
7.34
|
|
|
|
|
|
|
Canceled or expired
|
|
|
(143
|
)
|
|
$
|
13.10
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at December 31, 2009
|
|
|
2,205
|
|
|
$
|
12.97
|
|
|
|
|
|
|
Granted
|
|
|
827
|
|
|
$
|
10.74
|
|
|
|
|
|
|
Exercised
|
|
|
(22
|
)
|
|
$
|
1.44
|
|
|
|
|
|
|
Canceled or expired
|
|
|
(241
|
)
|
|
$
|
14.52
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at December 31, 2010 average remaining
life — 4.8 years
|
|
|
2,769
|
|
|
$
|
12.26
|
|
|
$
|
1,564
|
|
|
Exercisable at December 31, 2010, average remaining
life — 3.2 years
|
|
|
1,642
|
|
|
$
|
13.30
|
|
|
$
|
1,217
|
|
The weighted average grant-date fair value of options granted
during the years ended December 31, 2010, 2009 and 2008 was
$5.14, $4.91, and $4.48, respectively. The intrinsic value of
options exercised during the years ended December 31, 2010,
2009 and 2008 was $201,000, $729,000, and $4,073,000,
respectively. Of the total outstanding stock options shown
above, 1,068,000, 790,000 and 663,000 were incentive stock
options as of December 31, 2010, 2009 and 2008,
respectively.
The aggregate intrinsic value in the table above represents the
total pretax intrinsic value (the difference between the
Company’s closing stock price on December 31, 2010 and
the exercise price, multiplied by the number of
in-the-money
options) that would have been received by the option holders,
had all option holders exercised their options on
December 31, 2010. As of December 31, 2010, total
unrecognized stock-based compensation expense related to
non-vested
stock options was $4,547,000, which is expected to be recognized
over the remaining weighted average period of approximately
2.8 years.
The Compensation Committee of the board of directors determines
the total value of the stock based compensation grants. The
exercise price of options is the closing price on the date the
options are granted. Payment of the exercise price may be made
either in cash or with shares of common stock that have been
held at least six months. The options generally vest over four
years and expire between five or ten years from the date of
grant. The
73
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
fair value of each option grant is estimated on the date of the
grant using the Black-Scholes option-pricing model with the
following weighted average assumptions:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For the Years Ended December 31,
|
|
|
|
2010
|
|
2009
|
|
2008
|
|
|
|
(In thousands)
|
|
|
|
Risk free interest rate
|
|
|
1.75
|
%
|
|
|
2.0
|
%
|
|
|
2.4
|
%
|
|
Expected lives (years)
|
|
|
4.35
|
|
|
|
4.0
|
|
|
|
3.0
|
|
|
Expected volatility
|
|
|
56.57
|
%
|
|
|
60.5
|
%
|
|
|
48
|
%
|
|
Expected dividend yield
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
The expected volatilities are based on the historical volatility
of the Company’s stock. The observation is made on a weekly
basis. The expected terms of the stock options are based on the
average vesting period on a basis consistent with the historical
experience for similar option grants. The risk-free interest
rate is consistent with the expected terms of the stock options
and based on the U.S. Treasury yield curve in effect at the
time of grant. The Company estimates forfeiture rates based on
historical data.
A summary of the Company’s non-vested stock options during
the year ended December 31, 2010 is presented below:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted
|
|
|
|
|
|
|
|
Average
|
|
|
|
|
|
|
|
Grant Date
|
|
|
|
|
Shares
|
|
|
Fair Value
|
|
|
|
|
(In thousands)
|
|
|
|
|
Non-vested options at January 1, 2010
|
|
|
933
|
|
|
$
|
11.26
|
|
|
Granted
|
|
|
827
|
|
|
$
|
11.91
|
|
|
Vested
|
|
|
(524
|
)
|
|
$
|
11.40
|
|
|
Forfeited or expired
|
|
|
(109
|
)
|
|
$
|
5.88
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-vested options at December 31, 2010
|
|
|
1,127
|
|
|
$
|
10.74
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31, 2010, there was approximately $4,547,000
of accumulated unrecognized stock compensation based on fair
value on the grant date related to non-vested options granted
under the stock option plans. That cost is expected to be
recognized during the weighted average service period of
2.8 years. The share-based compensation will be amortized
based on the straight line method over the vesting period and
the expense includes an estimate of the awards that will be
forfeited. The total fair value of shares vested during the year
ended December 31, 2010 was $266,000.
Restricted
Shares
The Company began awarding restricted shares of its common stock
in 2006. In March 2009, the Company began to grant restricted
stock units to its non-employee directors and to certain
employees. Such awards generally require that certain
performance conditions and service conditions be met before the
awards vest. Restricted shares currently vest 25% after one year
and
61/4%
quarterly thereafter. Restricted stock units currently vest 25%
after thirteen months and
61/4%
quarterly thereafter. However, awards to non-employee directors
are immediately vested on the grant date. Unvested restricted
shares are forfeited if the recipient’s employment
terminates for any reason other than death, disability, or
special circumstances as determined by the Compensation
Committee of the
74
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
Company’s board of directors. Restricted share and
restricted stock unit activity during the year ended
December 31, 2010 is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted
|
|
|
|
|
Shares
|
|
|
Average Grant
|
|
|
|
|
and
|
|
|
Date Fair Value
|
|
|
|
|
Units
|
|
|
per Share
|
|
|
|
|
(In thousands, except for per share)
|
|
|
|
|
Unvested at January 1, 2010
|
|
|
248
|
|
|
$
|
11.40
|
|
|
Granted
|
|
|
193
|
|
|
$
|
10.80
|
|
|
Vested during period
|
|
|
(134
|
)
|
|
$
|
11.56
|
|
|
Cancelled during period
|
|
|
(21
|
)
|
|
$
|
11.73
|
|
|
|
|
|
|
|
|
|
|
|
|
Unvested at December 31, 2010
|
|
|
286
|
|
|
$
|
10.89
|
|
|
|
|
|
|
|
|
|
|
|
Fair value of the Company’s restricted shares and
restricted stock units is based on the Company’s closing
stock price on the date of grant. As of December 31, 2010,
total unrecognized stock-based compensation expense related to
nonvested restricted share grants was $2,863,000 which is
expected to be recognized over the remaining weighted average
period of approximately 2.8 years.
From time to time, the company retains shares of common stock
from employees upon vesting of restricted shares and restricted
stock units to cover income tax withholding. The impact of such
withholding totaled $315,000, $179,000 and $22,000 for the years
ended December 31, 2010, 2009 and 2008, respectively, and
was recorded as settlement on restricted stock tax withholding
in the accompanying consolidated statements of
stockholders’ equity.
At December 31, 2010, there were no warrants outstanding.
During the year ended December 31, 2009, 24,016 warrants
were exercised prior to their expiration. At December 31,
2008, there were warrants outstanding to purchase
74,300 shares of common stock at $7.80 per share, which
expired on April 23, 2009. During the year ended
December 31, 2008, no warrants were issued, exercised,
cancelled or expired.
The Company has 1,000,000 shares of Callable Series C
preferred stock authorized, none of which has been issued.
|
|
|
13.
|
Common
Stock Repurchase Plan
|
In August 2010, the Company’s board of directors authorized
a share repurchase and retirement plan of up to $10 million
of the Company’s common stock over a
12-month
period. During the year ended December 31, 2010, the
Company repurchased 330,454 shares of common stock at an
average price per share of approximately $9.10, for an aggregate
amount of approximately $3.0 million.
In 2008, the Company’s board of directors authorized two
stock repurchase plans. On March 3, 2008, the
Company’s board of directors authorized the first share
repurchase and retirement plan of up to $15 million of the
Company’s common stock over a
12-month
period. Under this first plan the Company repurchased
492,068 shares of common stock for approximately
$5.7 million. On July 25, 2008, the Company’s
board of directors terminated the first share repurchase and
retirement plan.
On November 21, 2008, the Company’s board of directors
authorized a second share repurchase and retirement plan of up
to $10 million of the Company’s common stock over a
12-month
period. During the year ended December 31, 2008, the
Company repurchased 491,511 shares of common stock for
approximately $6.2 million against this second
authorization. During the year ended December 31, 2009, the
Company repurchased an additional 250,800 shares of common
stock for approximately $2.5 million. As of
December 31, 2009, under this
75
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
second repurchase plan, the Company had repurchased a cumulative
total of 742,311 shares of common stock for approximately
$8.7 million.
Cumulatively between the three plans mentioned above, the
Company purchased a total of 1,564,833 shares of common
stock for approximately $17.4 million in 2010, 2009 and
2008.
On September 11, 2008, the Company’s Chief Executive
Officer exercised a stock option to purchase an aggregate of
130,000 shares of common stock with an exercise price of
$4.34 per share, on a net issue basis, in a transaction approved
by the compensation committee of the board of directors. The
Company issued 62,081 shares of common stock to the Chief
Executive Officer, and retained 67,919 shares of common
stock with an aggregate market value of $1,219,000 based on the
last closing price of the Company’s common stock
immediately prior to exercise of $17.95 per share. Of this
amount, $564,000 was applied in payment of the aggregate
exercise price of the stock options and $655,000 was applied in
payment of payroll taxes arising from the option exercise.
All employees are also eligible to participate in a 401(k) Plan
at the beginning of the first quarter following their employment
start date. The Company’s contributions are discretionary.
Effective January 1, 2006 the Company commenced matching
$0.50 per $1.00 contributed by the employees up to 4% of the
employee’s annual salary; prior to 2006, the Company’s
practice was to match $0.25 per $1.00 contributed by the
employees up to 4% of the employee’s annual salary.
Employees vest in amounts contributed by the Company
immediately. The Company contributed $408,000, $362,000 and
$312,000 to the 401(k) Plan for the years ended
December 31, 2010, 2009, and 2008, respectively.
|
|
|
15.
|
Manufacturing
Transition Rights
|
On December 30, 2008, the Company entered into a
manufacturing, supply and transition agreement
(“manufacturing transition rights”) with IDEXX
Operations, Inc. Under this agreement, the Company sold its
exclusive rights to manufacture and distribute rotors compatible
with the drives of IDEXX machines to IDEXX. The Company received
a nonrecurring $1.5 million payment, which was offset by
$268,000 related to costs associated with the manufacturing
transition rights. The $1.5 million payment was classified
as other income and did not impact revenue from operations, as
this payment was not contingent upon any significant action by
the Company.
During the transition period, the Company will provide IDEXX
with its know-how related to the manufacturing of the rotors and
will manufacture any rotors needed by IDEXX. Prices for these
transition services and rotors are exclusive of the
aforementioned nonrecurring payment. As of December 31,
2010, the Company has continued to manufacture and sell the
rotors to IDEXX at a set price.
In addition, IDEXX owns the rights to manufacture the rotors
without any further involvement or commitment from the Company.
Commencing January 2014 and continuing through December 2020, if
IDEXX uses this manufacturing technology, a royalty fee for each
rotor unit sold will be assessed and paid to the Company.
In November 2010, the Company filed a
Form S-3
shelf registration statement (“$125 million
shelf”) to provide for financial flexibility. The
$125 million shelf allows the Company to issue common
stock, preferred stock and debt securities of the Company. Under
the $125 million shelf, all of the securities available for
issuance may be offered from time to time with terms to be
determined at the time of issuance. In December 2010 the
Form S-3
registration statement was declared effective. As of
December 31, 2010, no securities had been issued under the
$125 million shelf which will expire in December 2013.
76
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
|
|
|
17.
|
Commitments
and Contingencies
|
Leases
The Company leases real property, automobiles and equipment
under operating lease agreements, which expire at various times
through 2018. Certain leases contain renewal options and
generally require us to pay utilities, insurance, taxes and
other operating expenses.
Future minimum rental payments required under operating lease
agreements that have an initial term in excess of one year as of
December 31, 2010, are as follows:
| |
|
|
|
|
|
|
|
Operating
|
|
|
|
|
Leases
|
|
|
|
|
(In thousands)
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
|
2011
|
|
$
|
2,358
|
|
|
2012
|
|
|
2,148
|
|
|
2013
|
|
|
2,052
|
|
|
2014
|
|
|
1,528
|
|
|
2015
|
|
|
1,190
|
|
|
Thereafter
|
|
|
114
|
|
|
|
|
|
|
|
|
|
|
$
|
9,390
|
|
|
|
|
|
|
|
Consolidated rental expense under all operating leases during
the years ended December 31, 2010, 2009 and 2008 was
$2,424,000 $1,997,000 and $1,787,000, respectively.
Litigation
From time to time, the Company is party to certain litigation
arising in the normal course of business. Management believes
that the resolution of such matters will not have a material
adverse effect on the Company’s financial position, results
of operations or cash flows.
Guarantees
The Company enters into indemnification provisions under
(i) agreements with other companies in its ordinary course
of business, typically with business partners, contractors, and
customers, landlords and (ii) agreements with investors.
Under these provisions, the Company generally indemnifies and
holds harmless the indemnified party for losses suffered or
incurred by the indemnified party as a result of its activities
or, in some cases, as a result of the indemnified party’s
activities under the agreement. These indemnification provisions
often include indemnifications relating to representations made
by the Company with regard to intellectual property rights.
These indemnification provisions generally survive termination
of the underlying agreement. In addition, in some cases, the
Company has agreed to reimburse employees for certain expenses
and to provide salary continuation during short-term disability.
The maximum potential amount of future payments the Company
could be required to make under these indemnification provisions
is unlimited. The Company reviews its exposure under these
agreements no less than annually, or more frequently when events
indicate. The Company has not incurred material costs to defend
lawsuits or settle claims related to these indemnification
agreements. As a result, the Company believes the estimated fair
value of its obligations under these agreements is minimal.
Accordingly, the Company has not recorded any liabilities for
these agreements as of December 31, 2010 and 2009.
77
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
|
|
|
18.
|
Supplier
Concentration
|
One supplier comprised greater than 10% of the Company’s
consolidated purchases. Consolidated purchases from this
supplier amounted to 17%, 12% and 15% for the years end
December 31, 2010, 2009, and 2008, respectively.
|
|
|
19.
|
Segments
and Geographic Information
|
The Company’s operations are organized on the basis of
products and related services, and under FASB ASC Topic 280,
Segment Reporting, the Company operates in three
segments: (1) Iris Diagnostics Division (“IDD”),
(2) Sample Processing and (3) Personalized Medicine.
The IDD segment designs, develops, manufactures, markets and
distributes in-vitro diagnostic systems based on patented and
proprietary technology for automating microscopic and clinical
chemistry procedures for urinalysis. The segment also provides
ongoing sales of consumables and services necessary for the
operation of installed urinalysis workstations. In the United
States, these products are mostly sold through a direct sales
and service force. Internationally, these products are sold and
serviced through distributors, with the exception of France,
Germany, United Kingdom and Puerto Rico.
The Sample Processing segment designs, develops, manufactures
and markets a variety of benchtop centrifuges, small instruments
and supplies. These products are used primarily for manual
specimen preparation and dedicated applications in coagulation,
cytology, hematology, urinalysis and DNA processing. These
products are sold worldwide through distributors.
The Personalized Medicine segment operates a CLIA-certified
laboratory focused on oncology and molecular diagnostics
services in personalized medicine. This segment also includes
the research and development operations of Iris Molecular
Diagnostics, or IMD.
The accounting policies of the segments are the same as those
described in the “Summary of Significant Accounting
Policies”. The Company evaluates the performance of its
segments and allocates resources to them based on earnings
before income taxes, excluding corporate charges.
78
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
The tables below present information about reported segments for
the three years ended December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Unallocated
|
|
|
|
|
|
|
|
|
|
|
Sample
|
|
|
Personalized
|
|
|
Corporate
|
|
|
|
|
|
|
|
IDD
|
|
|
Processing
|
|
|
Medicine
|
|
|
Expenses
|
|
|
Total
|
|
|
|
|
(In thousands)
|
|
|
|
|
For the Year Ended December 31, 2010
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues
|
|
$
|
93,195
|
|
|
$
|
14,408
|
|
|
$
|
69
|
|
|
$
|
—
|
|
|
$
|
107,672
|
|
|
Gross profit (loss)
|
|
|
47,469
|
|
|
|
7,789
|
|
|
|
(372
|
)
|
|
|
—
|
|
|
|
54,886
|
|
|
Marketing and selling
|
|
|
17,375
|
|
|
|
1,189
|
|
|
|
1,265
|
|
|
|
—
|
|
|
|
19,829
|
|
|
General and administrative
|
|
|
6,386
|
|
|
|
1,360
|
|
|
|
1,872
|
|
|
|
7,769
|
|
|
|
17,387
|
|
|
Research and development, net
|
|
|
8,840
|
|
|
|
619
|
|
|
|
5,103
|
|
|
|
—
|
|
|
|
14,562
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
32,601
|
|
|
|
3,168
|
|
|
|
8,240
|
|
|
|
7,769
|
|
|
|
51,778
|
|
|
Operating income (loss)
|
|
|
14,868
|
|
|
|
4,621
|
|
|
|
(8,612
|
)
|
|
|
(7,769
|
)
|
|
|
3,108
|
|
|
Interest income
|
|
|
141
|
|
|
|
26
|
|
|
|
—
|
|
|
|
956
|
|
|
|
1,123
|
|
|
Interest expense
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(10
|
)
|
|
|
(10
|
)
|
|
Depreciation and amortization
|
|
|
3,612
|
|
|
|
181
|
|
|
|
357
|
|
|
|
14
|
|
|
|
4,164
|
|
|
Segment pre-tax income (loss)
|
|
|
15,744
|
|
|
|
4,545
|
|
|
|
(8,620
|
)
|
|
|
(7,539
|
)
|
|
|
4,130
|
|
|
Segment assets
|
|
|
76,981
|
|
|
|
12,683
|
|
|
|
11,195
|
|
|
|
5,750
|
|
|
|
106,609
|
|
|
Investment in long-lived assets
|
|
|
24,281
|
|
|
|
3,720
|
|
|
|
11,045
|
|
|
|
—
|
|
|
|
39,046
|
|
|
For the Year Ended December 31, 2009
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues
|
|
$
|
78,231
|
|
|
$
|
14,335
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
92,566
|
|
|
Gross profit
|
|
|
41,295
|
|
|
|
7,370
|
|
|
|
—
|
|
|
|
—
|
|
|
|
48,665
|
|
|
Marketing and selling
|
|
|
14,999
|
|
|
|
1,123
|
|
|
|
—
|
|
|
|
—
|
|
|
|
16,122
|
|
|
General and administrative
|
|
|
5,077
|
|
|
|
1,345
|
|
|
|
—
|
|
|
|
6,899
|
|
|
|
13,321
|
|
|
Research and development, net
|
|
|
6,275
|
|
|
|
824
|
|
|
|
4,312
|
|
|
|
—
|
|
|
|
11,411
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
26,351
|
|
|
|
3,292
|
|
|
|
4,312
|
|
|
|
6,899
|
|
|
|
40,854
|
|
|
Operating income (loss)
|
|
|
14,944
|
|
|
|
4,078
|
|
|
|
(4,312
|
)
|
|
|
(6,899
|
)
|
|
|
7,811
|
|
|
Interest income
|
|
|
663
|
|
|
|
25
|
|
|
|
—
|
|
|
|
169
|
|
|
|
857
|
|
|
Interest expense
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(21
|
)
|
|
|
(21
|
)
|
|
Depreciation and amortization
|
|
|
3,051
|
|
|
|
230
|
|
|
|
226
|
|
|
|
16
|
|
|
|
3,523
|
|
|
Segment pre-tax income (loss)
|
|
|
16,787
|
|
|
|
4,014
|
|
|
|
(4,262
|
)
|
|
|
(7,834
|
)
|
|
|
8,705
|
|
|
Segment assets
|
|
|
78,453
|
|
|
|
8,784
|
|
|
|
4,417
|
|
|
|
6,136
|
|
|
|
97,790
|
|
|
Investment in long-lived assets
|
|
|
19,681
|
|
|
|
399
|
|
|
|
4,298
|
|
|
|
—
|
|
|
|
24,378
|
|
79
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Unallocated
|
|
|
|
|
|
|
|
|
|
|
Sample
|
|
|
Personalized
|
|
|
Corporate
|
|
|
|
|
|
|
|
IDD
|
|
|
Processing
|
|
|
Medicine
|
|
|
Expenses
|
|
|
Total
|
|
|
|
|
(In thousands)
|
|
|
|
|
For the Year Ended December 31, 2008
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues
|
|
$
|
81,771
|
|
|
$
|
13,731
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
95,502
|
|
|
Gross profit
|
|
|
42,171
|
|
|
|
6,868
|
|
|
|
—
|
|
|
|
—
|
|
|
|
49,039
|
|
|
Marketing and selling
|
|
|
14,687
|
|
|
|
1,019
|
|
|
|
—
|
|
|
|
—
|
|
|
|
15,706
|
|
|
General and administrative
|
|
|
5,459
|
|
|
|
1,183
|
|
|
|
—
|
|
|
|
5,367
|
|
|
|
12,009
|
|
|
Research and development, net
|
|
|
5,443
|
|
|
|
658
|
|
|
|
4,256
|
|
|
|
—
|
|
|
|
10,357
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses
|
|
|
25,589
|
|
|
|
2,860
|
|
|
|
4,256
|
|
|
|
5,367
|
|
|
|
38,072
|
|
|
Operating income (loss)
|
|
|
16,582
|
|
|
|
4,008
|
|
|
|
(4,256
|
)
|
|
|
(5,367
|
)
|
|
|
10,967
|
|
|
Interest income
|
|
|
28
|
|
|
|
—
|
|
|
|
—
|
|
|
|
1,152
|
|
|
|
1,180
|
|
|
Interest expense
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(11
|
)
|
|
|
(11
|
)
|
|
Depreciation and amortization
|
|
|
2,784
|
|
|
|
218
|
|
|
|
109
|
|
|
|
16
|
|
|
|
3,127
|
|
|
Segment pre-tax income (loss)
|
|
|
16,479
|
|
|
|
5,113
|
|
|
|
(4,256
|
)
|
|
|
(3,960
|
)
|
|
|
13,376
|
|
|
Segment assets
|
|
|
75,100
|
|
|
|
6,050
|
|
|
|
3,950
|
|
|
|
5,538
|
|
|
|
90,638
|
|
|
Investment in long-lived assets
|
|
|
18,229
|
|
|
|
419
|
|
|
|
3,916
|
|
|
|
—
|
|
|
|
22,564
|
|
The Company ships products from two locations in the United
States and one location in Germany. Substantially all long-lived
assets are located in the United States. Sales to international
customers amounted to approximately $36.0 million in 2010,
$30.2 million in 2009 and $31.6 million in 2008. For
the year ended December 31, 2010, one customer represented
13% of international sales. For the year ended December 31,
2009, three customers represented 19%, 12% and 12% of
international sales, respectively. For the year ended
December 31, 2008, two customers represented 21% and 10% of
international sales, respectively.
Segment assets attributed to corporate unallocated expenses are
deferred taxes. Long-lived assets include property and
equipment, intangible assets, long-term portion of inventory and
other long-term assets. Deferred income taxes are excluded from
long-lived assets.
80
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
|
|
|
20.
|
Valuation
and Qualifying Accounts
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additions
|
|
|
|
|
|
|
|
|
|
Charged to
|
|
Charged to
|
|
|
|
|
|
|
|
Beginning
|
|
Costs and
|
|
Other
|
|
|
|
Ending
|
|
|
|
Balance
|
|
Expenses
|
|
Accounts
|
|
Deductions
|
|
Balance
|
|
|
|
(In thousands)
|
|
|
|
Year Ended December 31, 2010
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Allowance for doubtful accounts
|
|
$
|
369
|
|
|
$
|
—
|
|
|
$
|
43
|
|
|
$
|
(7
|
)(1)
|
|
$
|
405
|
|
|
Allowance for sales returns
|
|
|
48
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
(1)
|
|
|
48
|
|
|
Reserve for inventory obsolescence
|
|
|
983
|
|
|
|
131
|
|
|
|
—
|
|
|
|
(969
|
)(1)
|
|
|
145
|
|
|
Valuation of deferred tax assets
|
|
|
335
|
|
|
|
—
|
|
|
|
308
|
|
|
|
(45
|
)(2)
|
|
|
598
|
|
|
Year Ended December 31, 2009
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Allowance for doubtful accounts
|
|
$
|
410
|
|
|
$
|
—
|
|
|
$
|
(20
|
)
|
|
$
|
(21
|
)(1)
|
|
$
|
369
|
|
|
Allowance for sales returns
|
|
|
35
|
|
|
|
—
|
|
|
|
13
|
|
|
|
—
|
|
|
|
48
|
|
|
Reserve for inventory obsolescence
|
|
|
846
|
|
|
|
255
|
|
|
|
—
|
|
|
|
(118
|
)(1)
|
|
|
983
|
|
|
Valuation of deferred tax assets
|
|
|
596
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(261
|
)(2)
|
|
|
335
|
|
|
Year Ended December 31, 2008
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Allowance for doubtful accounts
|
|
$
|
401
|
|
|
$
|
26
|
|
|
$
|
—
|
|
|
$
|
(17
|
)(1)
|
|
$
|
410
|
|
|
Allowance for sales returns
|
|
|
35
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
(1)
|
|
|
35
|
|
|
Reserve for inventory obsolescence
|
|
|
623
|
|
|
|
462
|
|
|
|
—
|
|
|
|
(239
|
)(1)
|
|
|
846
|
|
|
Valuation of deferred tax assets
|
|
|
856
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(260
|
)(2)
|
|
|
596
|
|
|
|
|
|
(1) |
|
Relates to the write-off of accounts receivable, return of
merchandise, disposal of obsolete inventory or specific portion
of the accounts receivable reserve or reserve for sales returns
no longer needed. |
| |
|
(2) |
|
Valuation adjustment relating to realization of deferred tax
assets. |
|
|
|
21.
|
Selected
Quarterly Data (Unaudited)
|
The following table summarizes certain financial information by
quarter:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2010 Quarter Ended
|
|
|
|
March 31
|
|
June 30
|
|
September 30
|
|
December 31
|
|
|
|
(In thousands, except per share)
|
|
|
|
Net revenues
|
|
$
|
25,980
|
|
|
$
|
26,688
|
|
|
$
|
25,726
|
|
|
$
|
29,278
|
|
|
Gross profit
|
|
|
13,254
|
|
|
|
14,495
|
|
|
|
13,159
|
|
|
|
13,978
|
|
|
Net income
|
|
|
1,042
|
|
|
|
624
|
|
|
|
924
|
|
|
|
452
|
|
|
Net income per share — basic
|
|
|
0.06
|
|
|
|
0.03
|
|
|
|
0.05
|
|
|
|
0.03
|
|
|
Net income per share — diluted
|
|
|
0.06
|
|
|
|
0.03
|
|
|
|
0.05
|
|
|
|
0.03
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2009 Quarter Ended
|
|
|
|
March 31
|
|
June 30
|
|
September 30
|
|
December 31
|
|
|
|
(In thousands, except per share)
|
|
|
|
Net revenues
|
|
$
|
21,575
|
|
|
$
|
22,342
|
|
|
$
|
22,186
|
|
|
$
|
26,463
|
|
|
Gross profit
|
|
|
11,695
|
|
|
|
11,332
|
|
|
|
12,182
|
|
|
|
13,456
|
|
|
Net income
|
|
|
1,392
|
|
|
|
1,012
|
|
|
|
1,916
|
|
|
|
1,931
|
|
|
Net income per share — basic
|
|
|
0.08
|
|
|
|
0.06
|
|
|
|
0.11
|
|
|
|
0.11
|
|
|
Net income per share — diluted
|
|
|
0.08
|
|
|
|
0.06
|
|
|
|
0.11
|
|
|
|
0.11
|
|
81
IRIS
INTERNATIONAL, INC.
NOTES TO
CONSOLIDATED FINANCIAL
STATEMENTS — (Continued)
|
|
|
|
(1) |
|
2010 results include the operating losses, including acquisition
related costs, of approximately $3.7 million related to our
acquisition in July 2010 of Arista Molecular, Inc. In addition,
we incurred $923,000 in CFO and related transition costs,
$1.1 million for foreign currency translation loss and
$800,000 related to higher instrument cost of goods due to a
price premium on the last purchase of automated chemistry
analyzers from Arkray. |
| |
|
(2) |
|
2009 results include the following expenses totaling
$1.1 million; an accrual of $475,000 for payroll taxes
attributable to the exercise of stock options, $350,000 in
start-up
expenses for the initiation of direct sales operations in the UK
and Germany and $250,000 for product retrofit costs related to
the voluntary recall of approximately 1,565 StatSpin Express 4
Centrifuges. |
|
|
|
21.
|
Shareholders’
Rights Plan
|
On September 24, 2010, the Company’s board of
directors adopted a shareholders’ rights plan under which
each holder of a share of common stock also has one right to
purchase one one-thousandth of a newly created Series A
preferred share at $100 per one one-thousandth of a share.
The rights are not presently exercisable. Upon the occurrence of
certain “flip-in” events, each right becomes
exercisable which then entitles its holder to receive the number
of Series A preferred shares having an aggregate per share
market value price equal to two times the purchase price. Upon
certain “flip-over” events, each right when exercised
entitles its holder to receive equity securities of the
acquiring company having a value equal to two times the purchase
price. A flip-in event includes the acquisition by a person or
group (an “acquiring person”) of 20 percent or
more of the Company’s common stock. Rights held by an
acquiring person are void. The Company may redeem the rights for
$0.001 per right and may amend the rights under the plan at any
time prior to a triggering event. The rights expire on
September 24, 2020.
82
|
|
|
Item 9.
|
Changes
in and Disagreements with Accountants on Accounting and
Financial Disclosure.
|
None.
|
|
|
Item 9A.
|
Controls
and Procedures.
|
Disclosure
Controls and Procedures.
Based on their evaluation as of December 31, 2010, our
Chief Executive Officer and Chief Financial Officer, with the
participation of management, have concluded that our disclosure
controls and procedures (as defined in
Rules 13a — 15(e) and 15d — 15(e) of
the Securities Exchange Act of 1934) were effective.
Management’s
Annual Report on Internal Control over Financial
Reporting.
Our management is responsible for establishing and maintaining
adequate internal control over financial reporting, as defined
in
Rules 13a-15(f)
and
15d-15(f)
under the Securities and Exchange Act of 1934. Internal control
over financial reporting is a process designed to provide
reasonable assurance regarding the reliability of financial
reporting and the preparation of financial statements for
external purposes in accordance with generally accepted
accounting principles in the United States.
Under the supervision and with the participation of our
management, including our Chief Executive Officer and Chief
Financial Officer, we conducted an evaluation of the
effectiveness of our internal control over financial reporting
as of December 31, 2010 using the criteria set forth by the
Committee of Sponsoring Organizations of the Treadway Commission
(COSO) in Internal Control — Integrated
Framework. Based on this evaluation, our management
concluded that as of December 31, 2010, our internal
control over financial reporting was effective.
Attestation
Report of the Registered Public Accounting Firm.
BDO USA LLP, our independent registered public accounting firm
that has audited our financial statements included herein, has
issued an attestation report on our internal control over
financial reporting, which report is included under Item 8
of this Annual Report on
Form 10-K.
Changes
in Internal Control over Financial Reporting.
There were no changes in our internal control over financial
reporting during the quarter ended December 31, 2010 that
have materially affected, or are reasonably likely to materially
affect, our internal control over financial reporting.
Limitations
on the Effectiveness of Controls.
Our disclosure controls and procedures provide our Chief
Executive Officer and Chief Financial Officer reasonable
assurances that our disclosure controls and procedures will
achieve their objectives. However, company management, including
our Chief Executive Officer and Chief Financial Officer, does
not expect that our disclosure controls and procedures or our
internal control over financial reporting can or will prevent
all human error. A control system, no matter how well designed
and implemented, can provide only reasonable, not absolute,
assurance that the objectives of the control system are met.
Furthermore, the design of a control system must reflect the
fact that there are internal resource constraints, and the
benefit of controls must be weighed relative to their
corresponding costs. Because of the limitations in all control
systems, no evaluation of controls can provide complete
assurance that all control issues and instances of error, if
any, within our company are detected. These inherent limitations
include the realities that judgments in decision-making can be
faulty, and that breakdowns can occur due to human error or
mistake. Additionally, controls, no matter how well designed,
could be circumvented by the individual acts of specific persons
within the organization. The design of any system of controls is
also based in part upon certain assumptions about the likelihood
of future events, and there can be no assurance that any design
will succeed in achieving its stated objectives under all
potential future conditions.
|
|
|
Item 9B.
|
Other
Information.
|
None.
83
PART III
Certain information required by Part III is omitted from
this Annual Report on
Form 10-K
because the registrant will file with the U.S. Securities
and Exchange Commission a definitive proxy statement pursuant to
Regulation 14A in connection with the solicitation of
proxies for our Annual Meeting of Stockholders expected to be
held in May 2011 (the “Proxy Statement”) not later
than 120 days after the end of the fiscal year covered by
this Annual Report on
Form 10-K,
and certain information included therein is incorporated herein
by reference.
|
|
|
Item 10.
|
Directors,
Executive Officers and Corporate Governance.
|
The information required by this item with respect to directors
and executive officers may be found in the section
“Election of Directors” appearing in the Proxy
Statement. Such information is incorporated herein by reference.
The information required by this Item with respect to our audit
committee and audit committee financial expert may be found in
the section entitled “Election of Directors —
Audit Committee” appearing in the Proxy Statement. Such
information is incorporated herein by reference.
The information required by this Item with respect to compliance
with Section 16(a) of the Securities Exchange Act of 1934
and our code of ethics may be found in the sections entitled
“Section 16(a) Beneficial Ownership Reporting
Compliance” and “Election of Directors —
Code of Business Conduct and Ethics,” respectively,
appearing in the Proxy Statement. Such information is
incorporated herein by reference.
|
|
|
Item 11.
|
Executive
Compensation.
|
The information required by this Item with respect to director
and executive officer compensation is incorporated herein by
reference to the information from the Proxy Statement under the
section entitled “Executive Compensation.”
The information required by this Item with respect to
Compensation Committee interlocks and insider participation is
incorporated herein by reference to the information from the
Proxy Statement under the section entitled “Election of
Directors — Compensation Committee Interlocks and
Insider Participation.”
The information required by this Item with respect to our
Compensation Committee’s review and discussion of the
Compensation Discussion and Analysis included in the Proxy
Statement is incorporate herein by reference to the information
from the Proxy Statement under the section entitled
“Election of Directors — Compensation Committee
Report.”
|
|
|
Item 12.
|
Security
Ownership of Certain Beneficial Owners and Management and
Related Stockholder Matters.
|
The information required by this Item with respect to security
ownership of certain beneficial owners and management is
incorporated herein by reference to the information from the
Proxy Statement under the section entitled “Security
Ownership of Certain Beneficial Owners and Management.”
The information required by this Item with respect to securities
authorized for issuance under our equity compensation plans is
incorporated herein by reference to the information from the
Proxy Statement under the section entitled “Equity
Compensation Plan Information.”
|
|
|
Item 13.
|
Certain
Relationships and Related Transactions, and Director
Independence.
|
The information required by this Item with respect to related
party transactions is incorporated herein by reference to the
information from the Proxy Statement under the section entitled
“Certain Relationships and Related Transactions.”
The information required by this Item with respect to director
independence is incorporated herein by reference to the
information from the Proxy Statement under the section entitled
“Election of Directors — Independence of the
Board of Directors.”
|
|
|
Item 14.
|
Principal
Accounting Fees and Services.
|
The information required by this Item is incorporated herein by
reference to the information from the Proxy Statement under the
section entitled “Ratification of Selection of Independent
Registered Public Accounting Firm.”
84
PART IV
|
|
|
Item 15.
|
Exhibits,
Financial Statement Schedules.
|
(a) Documents
filed as part of this report
1. Financial Statements
See Index to Financial Statements in Item 8 of this Annual
Report on
Form 10-K,
which is incorporated herein by reference.
2. Financial Statement Schedules
All financial statement schedules are omitted because the
information is inapplicable or presented in the Notes to
Financial Statements.
The following exhibits are included herein or incorporated
herein by reference:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exhibit
|
|
|
|
Incorporated by Reference
|
|
Filed
|
|
Number
|
|
Description
|
|
Form
|
|
File Number
|
|
Exhibit
|
|
Filing Date
|
|
Herewith
|
|
|
|
|
2
|
.1
|
|
Merger Agreement, dated July 26, 2010, by and among Iris
International, Inc., AlliedPath Inc., and API Acquisition Corp.
|
|
8-K
|
|
001-11181
|
|
2.1
|
|
July 30, 2010
|
|
—
|
|
|
3
|
.1(a)
|
|
Certificate of Incorporation, filed June 9, 1987
|
|
8-K
|
|
001-11181
|
|
3.1(a)
|
|
September 29, 2010
|
|
—
|
|
|
3
|
.1(b)
|
|
Certificate of Amendment of Certificate of Incorporation, filed
July 9, 1993
|
|
8-K
|
|
001-11181
|
|
3.1(b)
|
|
September 29, 2010
|
|
—
|
|
|
3
|
.1(c)
|
|
Certificate of Amendment of Certificate of Incorporation, filed
June 6, 2001
|
|
8-K
|
|
001-11181
|
|
3.1(c)
|
|
September 29, 2010
|
|
—
|
|
|
3
|
.1(d)
|
|
Certificate of Ownership and Merger, filed November 26, 2003
|
|
8-K
|
|
001-11181
|
|
3.1(d)
|
|
September 29, 2010
|
|
—
|
|
|
3
|
.1(e)
|
|
Certificate of Correction of Certificate of Ownership and
Merger, filed December 11, 2003
|
|
8-K
|
|
001-11181
|
|
3.1(e)
|
|
September 29, 2010
|
|
—
|
|
|
3
|
.1(f)
|
|
Certificate of Designation of Rights, Preferences and Privileges
of Series A Preferred Stock, filed September 27, 2010
|
|
8-K
|
|
001-11181
|
|
3.1(f)
|
|
September 29, 2010
|
|
—
|
|
|
3
|
.2(a)
|
|
Restated Bylaws
|
|
10-KSB
|
|
001-11181
|
|
3.2
|
|
March 26, 2004
|
|
—
|
|
|
3
|
.2(b)
|
|
Amendment to Amended and Restated Bylaws
|
|
8-K
|
|
001-11181
|
|
3.2
|
|
July 18, 2007
|
|
—
|
|
|
3
|
.2(c)
|
|
Amendment to Amended and Restated Bylaws
|
|
8-K
|
|
001-11181
|
|
3.3
|
|
January 20, 2010
|
|
—
|
|
|
4
|
.1
|
|
Rights Agreement, dated as of September 24, 2010, between the
Registrant and Continental Stock Transfer & Trust Company,
as Rights Agent, including the Certificate of Designation of
Rights, Preferences and Privileges of Series A Preferred Stock,
the Form of Rights Certificate, and the Summary of Rights to
Purchase Preferred Stock, attached thereto as Exhibits A, B and
C, respectively.
|
|
8-K
|
|
001-11181
|
|
4.1
|
|
September 29, 2010
|
|
—
|
85
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exhibit
|
|
|
|
Incorporated by Reference
|
|
Filed
|
|
Number
|
|
Description
|
|
Form
|
|
File Number
|
|
Exhibit
|
|
Filing Date
|
|
Herewith
|
|
|
|
|
10
|
.1(a)†
|
|
Key Employee Agreement, dated February 13, 2004, between the
Registrant and Cesar M Garcia.
|
|
10-K
|
|
001-11181
|
|
10.8(i)
|
|
March 26, 2004
|
|
—
|
|
|
10
|
.1(b)†
|
|
First Amendment to Key Employee Agreement, dated December 21,
2006, between the Registrant and Cesar M Garcia.
|
|
10-K
|
|
001-11181
|
|
10.9(b)
|
|
March 23, 2007
|
|
—
|
|
|
10
|
.1(c)†
|
|
Second Amendment to Key Employee Agreement, dated November 14,
2007, between the Registrant and Cesar M. Garcia.
|
|
8-K
|
|
001-11181
|
|
10.1
|
|
November 14, 2007
|
|
|
|
|
10
|
.1(d)†
|
|
Third Amendment to Key Employee Agreement for Cesar M. Garcia
effective May 14, 2010 between IRIS International, Inc. and
Cesar M. Garcia.
|
|
8-K
|
|
001-11181
|
|
10.1
|
|
September 7, 2010
|
|
—
|
|
|
10
|
.2(a)†
|
|
Key Employee Agreement, dated March 1, 2007, between the
Registrant and Thomas E. Warekois.
|
|
10-K
|
|
001-1181
|
|
10.12
|
|
March 14, 2008
|
|
—
|
|
|
10
|
.2(b)†
|
|
Amendment to Key Employee Agreement for Thomas Warekois,
effective May 14, 2010 between IRIS International, Inc. and
Thomas Warekois.
|
|
8-K
|
|
001-11181
|
|
10.3
|
|
September 7, 2010
|
|
—
|
|
|
10
|
.3(a)†
|
|
Key Employee Agreement, dated November 7, 2007, between the
Registrant and Robert Mello.
|
|
8-K
|
|
001-11181
|
|
10.2
|
|
November 14, 2007
|
|
|
|
|
10
|
.3(b)†
|
|
Amendment to Key Employee Agreement for Robert Mello, effective
May 14, 2010 between IRIS International, Inc. and Robert Mello.
|
|
8-K
|
|
001-11181
|
|
10.2
|
|
September 7, 2010
|
|
—
|
|
|
10
|
.4†
|
|
Key Employee Agreement for Thomas H. Adams, PhD, dated September
2, 2010 between IRIS International, Inc. and Thomas H.
Adams, Ph.D.
|
|
8-K
|
|
001-11181
|
|
10.5
|
|
September 7, 2010
|
|
—
|
|
|
10
|
.5†
|
|
Key Employee Agreement for Amin Khalifa
|
|
8-K
|
|
001-11181
|
|
10.1
|
|
October 13, 2010
|
|
—
|
|
|
|
|
|
dated October 11, 2010 between IRIS International, Inc. and Amin
Khalifa.
|
|
|
|
|
|
|
|
|
|
|
|
|
10
|
.6†
|
|
Key Employee Agreement for Richard A. O’Leary, dated
February 14, 2011, between IRIS International, Inc. and Richard
A. O’Leary
|
|
8-K
|
|
001-11181
|
|
10.1
|
|
February 17, 2011
|
|
—
|
|
|
10
|
.7(a)†
|
|
Key Employee Agreement, dated November 7, 2007, between the
Registrant and John Yi.
|
|
8-K
|
|
001-11181
|
|
10.3
|
|
November 14, 2007
|
|
|
86
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exhibit
|
|
|
|
Incorporated by Reference
|
|
Filed
|
|
Number
|
|
Description
|
|
Form
|
|
File Number
|
|
Exhibit
|
|
Filing Date
|
|
Herewith
|
|
|
|
|
10
|
.7(b)†
|
|
Amendment to Key Employee Agreement for John Yi, effective May
14, 2010 between IRIS International, Inc. and John Yi.
|
|
8-K
|
|
001-11181
|
|
10.4
|
|
September 7, 2010
|
|
—
|
|
|
10
|
.8†
|
|
Key Employee Agreement for Philip Ginsburg, dated July 28, 2010,
by and between Iris International, Inc. and Philip Ginsburg.
|
|
8-K
|
|
001-11181
|
|
10.1
|
|
July 30, 2010
|
|
—
|
|
|
10
|
.9†
|
|
Key Employee Agreement for Vance Randal White, dated July 28,
2010, by and between Iris International, Inc. and Vance Randal
White.
|
|
8-K
|
|
001-11181
|
|
10.2
|
|
July 30, 2010
|
|
—
|
|
|
10
|
.10†
|
|
1994 Stock Option Plan and forms of Stock Option Agreements.
|
|
S-8
|
|
33-82560
|
|
n/a
|
|
|
|
—
|
|
|
10
|
.11†
|
|
1997 Stock Option Plan and form of Stock Option Agreement.
|
|
S-8
|
|
333-31393
|
|
4.2(a),
4.2(b)
|
|
July 16, 1997
|
|
—
|
|
|
10
|
.12†
|
|
Amended and Restated 1998 Stock Option Plan.
|
|
10-K
|
|
001-11181
|
|
10.6
|
|
March 23, 2007
|
|
—
|
|
|
10
|
.13(a)†
|
|
2007 Stock Incentive Plan.
|
|
S-8
|
|
333-145635
|
|
4.3
|
|
August 22, 2007
|
|
—
|
|
|
10
|
.13(b)†
|
|
Amendment No. 1 to 2007 Stock Incentive Plan
|
|
8-K
|
|
001-11181
|
|
10.1
|
|
May 29, 2009
|
|
—
|
|
|
10
|
.14†
|
|
Form of Restricted Stock Unit Agreement.
|
|
10-K
|
|
001-11181
|
|
10.18
|
|
March 16, 2010
|
|
—
|
|
|
10
|
.15†
|
|
Form of Non-Qualified Stock Option Agreement.
|
|
10-K
|
|
001-11181
|
|
10.19
|
|
March 16, 2010
|
|
—
|
|
|
10
|
.16†
|
|
Form of Incentive Stock Option Agreement.
|
|
10-K
|
|
001-11181
|
|
10.20
|
|
March 16, 2010
|
|
—
|
|
|
10
|
.17†
|
|
Form of Restricted Stock Unit Deferral Election.
|
|
10-K
|
|
001-11181
|
|
10.21
|
|
March 16, 2010
|
|
—
|
|
|
10
|
.18†
|
|
Letter Agreement, May 11, 2010, between IRIS International, Inc.
and Avant Advisory Group.
|
|
10-Q
|
|
001-11181
|
|
10.1
|
|
August 6, 2010
|
|
—
|
|
|
10
|
.19(a)
|
|
Lease for Property Located at 9172 Eton Avenue, Chatsworth,
California, dated November 29, 2001.
|
|
10-K
|
|
001-11181
|
|
10.1(a)
|
|
April 1, 2002
|
|
—
|
|
|
10
|
.19(b)
|
|
Amendment No. 1, dated October 17, 2005, to the Lease for
Property Located at 9172 Eton Avenue, Chatsworth, California,
dated November 28, 2001.
|
|
8-K
|
|
001-11181
|
|
10.2
|
|
November 18, 2005
|
|
—
|
|
|
10
|
.20
|
|
Lease for Property Located at 9158-9162 Eton Avenue, Chatsworth,
California, dated October 17, 2005.
|
|
8-K
|
|
001-11181
|
|
10.1
|
|
November 18, 2005
|
|
—
|
|
|
10
|
.21
|
|
Lease for Property Located at 9232 Eton Avenue, Chatsworth,
California, dated February 8, 2010.
|
|
10-K
|
|
001-11181
|
|
10.17
|
|
March 16, 2010
|
|
—
|
|
|
10
|
.22(a)
|
|
Business Loan Agreement dated May 25, 2004 by and between the
Registrant and California Bank & Trust.
|
|
10-K
|
|
001-11181
|
|
10.3(b)
|
|
March 16, 2005
|
|
—
|
87
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exhibit
|
|
|
|
Incorporated by Reference
|
|
Filed
|
|
Number
|
|
Description
|
|
Form
|
|
File Number
|
|
Exhibit
|
|
Filing Date
|
|
Herewith
|
|
|
|
|
10
|
.22(b)
|
|
Change in Terms Agreement dated March 24, 2006 by and between
the Registrant and California Bank & Trust.
|
|
10-K
|
|
001-11181
|
|
10.6(b)
|
|
March 16, 2010
|
|
—
|
|
|
10
|
.22(c)
|
|
Change in Terms Agreement dated May 1, 2008 by and between the
Registrant and California Bank & Trust.
|
|
8-K/A
|
|
001-11181
|
|
10.2
|
|
July 14, 2008
|
|
—
|
|
|
10
|
.22(d)
|
|
Letter Agreement, dated June 9, 2010, between IRIS
International, Inc. and California Bank & Trust.
|
|
10-Q
|
|
001-11181
|
|
10.2
|
|
August 6, 2010
|
|
—
|
|
|
10
|
.23
|
|
Commercial Guaranty Agreement dated May 25, 2004 by and between
the Registrant, StatSpin, Inc (the Registrant’s affiliate)
and California Bank & Trust.
|
|
10-K
|
|
001-11181
|
|
10.3(f)
|
|
March 16, 2005
|
|
—
|
|
|
10
|
.24
|
|
Commercial Security Agreements dated May 25, 2004 by and between
the Registrant and California Bank & Trust.
|
|
10-K
|
|
001-11181
|
|
10.3(d)
|
|
March 16, 2005
|
|
—
|
|
|
10
|
.25(a)
|
|
Business Loan Agreement dated March 24, 2006 by and between the
Registrant and California Bank & Trust.
|
|
10-K
|
|
001-11181
|
|
10.8(b)
|
|
March 16, 2010
|
|
—
|
|
|
10
|
.25(b)
|
|
Change in Terms Agreement dated March 24, 2006 by and between
the Registrant and California Bank & Trust.
|
|
10-K
|
|
001-11181
|
|
10.8(c)
|
|
March 16, 2010
|
|
—
|
|
|
10
|
.25(c)
|
|
Change in Terms Agreement dated May 1, 2008 by and between the
Registrant and California Bank & Trust.
|
|
8-K/A
|
|
001-11181
|
|
10.1
|
|
July 14, 2008
|
|
—
|
|
|
10
|
.25(d)
|
|
Change in Terms Agreement, dated August 31, 2010, by and between
IRIS International, Inc. and California Bank & Trust.
|
|
8-K
|
|
001-11181
|
|
10.1
|
|
September 2, 2010
|
|
—
|
|
|
10
|
.26
|
|
Commercial Guaranty, dated August 31, 2010, by IRIS Molecular
Diagnostics, Inc.
|
|
8-K
|
|
001-11181
|
|
10.2
|
|
September 2, 2010
|
|
—
|
|
|
10
|
.27
|
|
Commercial Security Agreement, dated August 31, 2010, by and
among IRIS Molecular Diagnostics, Inc., IRIS International, Inc.
and California Bank & Trust.
|
|
8-K
|
|
001-11181
|
|
10.3
|
|
September 2, 2010
|
|
—
|
|
|
21
|
|
|
List of Subsidiaries.
|
|
|
|
|
|
|
|
|
|
*
|
|
|
23
|
.1
|
|
Consent of BDO USA, LLP.
|
|
|
|
|
|
|
|
|
|
*
|
|
|
24
|
.1
|
|
Power of Attorney (included on signature page)
|
|
|
|
|
|
|
|
|
|
*
|
|
|
31
|
.1
|
|
Certification of Principal Executive officer pursuant to
Securities Exchange Act Rules 13a-14 and 15d-14 as adopted
pursuant to Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
|
|
|
|
*
|
88
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exhibit
|
|
|
|
Incorporated by Reference
|
|
Filed
|
|
Number
|
|
Description
|
|
Form
|
|
File Number
|
|
Exhibit
|
|
Filing Date
|
|
Herewith
|
|
|
|
|
31
|
.2
|
|
Certification of Principal Financial officer pursuant to
Securities Exchange Act Rules 13a-14 and 15d-14 as adopted
pursuant to Section 302 of the Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
|
|
|
|
*
|
|
|
32
|
.1
|
|
Certification of Principal Executive officer pursuant to
Securities Exchange Act Rules 13a-14(b) of the Exchange Act and
18 U.S.C. Section 1350 as adopted pursuant to Section 906
of the Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
|
|
|
|
*
|
|
|
32
|
.2
|
|
Certification of Principal Financial officer pursuant to
Securities Exchange Act Rules 13a-14(b) of the Exchange Act and
18 U.S.C. Section 1350 as adopted pursuant to Section 906
of the Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
|
|
|
|
*
|
|
|
|
|
† |
|
Each a management contract or compensatory plan or arrangement
required to be filed as an exhibit to this annual report on
Form 10-K. |
89
SIGNATURES
Pursuant to Section 13 or 15(d) of the Securities Exchange
Act of 1934, the Registrant has duly caused this report on
Form 10-K
to be signed on its behalf by the undersigned, thereunto duly
authorized, in Chatsworth, California, on March 16, 2011.
IRIS INTERNATIONAL, INC.
Cesar M. García,
President and Chief Executive Officer
POWER OF
ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose
signature appears below constitutes and appoints Cesar M. Garcia
and Amin I. Khalifa, and each of them, as his true and lawful
attorneys-in-fact and agents, with full power of substitution
for him, and in his name in any and all capacities, to sign any
and all amendments to this Annual Report on
Form 10-K,
and to file the same, with exhibits thereto and other documents
in connection therewith, with the Securities and Exchange
Commission, granting unto said attorneys-in-fact and agents, and
each of them, full power and authority to do and perform each
and every act and thing requisite and necessary to be done
therewith, as fully to all intents and purposes as he might or
could do in person, hereby ratifying and confirming all that
said attorneys-in-fact and agents, and any of them or his or her
substitute or substitutes, may lawfully do or cause to be done
by virtue hereof.
Pursuant to the requirements of the Securities Act of 1934, this
report has been signed by the following persons on behalf of the
Registrant in the capacities indicated and on the date indicated:
| |
|
|
|
|
|
|
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
|
|
/s/ CESAR
M. GARCIA
Cesar
M. Garcia
|
|
President, Chief Executive Officer and Chairman of the Board
(Principal Executive Officer)
|
|
March 16, 2011
|
|
|
|
|
|
|
|
/s/ AMIN
I. KHALIFA
Amin
I. Khalifa
|
|
Corporate Vice President of Finance and Chief Financial Officer
(Principal Financial and Accounting Officer)
|
|
March 16, 2011
|
|
|
|
|
|
|
|
/s/ THOMAS
H. ADAMS
Thomas
H. Adams
|
|
Director
|
|
March 16, 2011
|
|
|
|
|
|
|
|
/s/ STEVEN
M. BESBECK
Steven
M. Besbeck
|
|
Director
|
|
March 16, 2011
|
|
|
|
|
|
|
|
/s/ DAVID
T. DELLA PENTA
David
T. Della Penta
|
|
Director
|
|
March 16, 2011
|
|
|
|
|
|
|
|
/s/ BETH
Y. KARLAN
Beth
Y. Karlan
|
|
Director
|
|
March 16, 2011
|
90
| |
|
|
|
|
|
|
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
|
|
/s/ MICHAEL
D. MATTE
Michael
D. Matte
|
|
Director
|
|
March 16, 2011
|
|
|
|
|
|
|
|
/s/ RICHARD
G. NADEAU
Richard
G. Nadeau
|
|
Director
|
|
March 16, 2011
|
|
|
|
|
|
|
|
/s/ RICK
TIMMINS
Rick
Timmins
|
|
Director
|
|
March 16, 2011
|
|
|
|
|
|
|
|
/s/ EDWARD
F. VOBORIL
Edward
F. Voboril
|
|
Director
|
|
March 16, 2011
|
|
|
|
|
|
|
|
/s/ STEPHEN
E. WASSERMAN
Stephen
E. Wasserman
|
|
Director
|
|
March 16, 2011
|
91