Attached files
| file | filename |
|---|---|
| EX-23.1 - EXHIBIT 23.1 - ARIAD PHARMACEUTICALS INC | a6212068ex231.htm |
| EX-10.13 - EXHIBIT 10.13 - ARIAD PHARMACEUTICALS INC | a6212068ex1013.htm |
| EX-10.2.6 - EXHIBIT 10.2.6 - ARIAD PHARMACEUTICALS INC | a6212068ex1026.htm |
| EX-32.1 - EXHIBIT 32.1 - ARIAD PHARMACEUTICALS INC | a6212068ex321.htm |
| EX-31.2 - EXHIBIT 31.2 - ARIAD PHARMACEUTICALS INC | a6212068ex312.htm |
| EX-31.1 - EXHIBIT 31.1 - ARIAD PHARMACEUTICALS INC | a6212068ex311.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
(Mark One)
|
[X] ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
|
|
SECURITIES EXCHANGE ACT OF 1934
|
|
For the fiscal year ended December 31, 2009
|
|
OR
|
|
[ ] TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF
|
|
THE SECURITIES EXCHANGE ACT OF 1934
|
For the transition period from ________ to ________
Commission file number 0-21696
ARIAD Pharmaceuticals, Inc.
(Exact name of registrant as specified in its charter)
|
Delaware
(State or other jurisdiction of
incorporation or organization)
|
22-3106987
(I.R.S. Employer Identification No.)
|
26 Landsdowne Street, Cambridge, Massachusetts 02139-4234
|
(Address of principal executive offices) (Zip Code)
|
Registrant’s telephone number, including area code: (617) 494-0400
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class
|
Name of each exchange on which registered
|
|
Common Stock, $.001 par value
Rights to Purchase Series A Preferred Stock
|
The Nasdaq Global Market
|
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes [ ] No [ X ]
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act.
Yes [ ] No [ X ]
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes [ X ] No [ ]
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes [ ] No [ ]
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. [ ]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer [ ] Accelerated filer [ X ]
Non-accelerated filer [ ] (Do not check if a smaller reporting company) Smaller reporting company [ ]
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes [ ] No [ X ]
The aggregate market value of the registrant’s common stock held by nonaffiliates of the registrant (without admitting that any person whose shares are not included in such calculation is an affiliate), computed by reference to the price at which the common stock was last sold, as of the last business day of the registrant’s most recently completed second fiscal quarter was approximately $132 million.
As of March 12, 2010, the registrant had 109,224,265 shares of common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
The following documents (or parts thereof) are incorporated by reference into the following parts of this Form 10-K: Certain information required in Part III of this Annual Report on Form 10-K is incorporated from the Registrant’s Definitive Proxy Statement for the 2010 Annual Meeting of Stockholders.
TABLE OF CONTENTS
The following Business Section contains forward-looking statements, which involve risks and uncertainties. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of certain factors (see Part I, Item 1A: Risk Factors). Unless the content requires otherwise, references to “ARIAD,” “we,” “our,” and “us,” in this Annual Report on Form 10-K refer to ARIAD Pharmaceuticals, Inc. and our subsidiaries.
Overview
Our Business and Strategy
ARIAD’s vision is to transform the lives of cancer patients with breakthrough medicines. Our mission is to discover, develop and commercialize small-molecule drugs to treat cancer in patients with the greatest and most urgent unmet medical need – aggressive cancers where current therapies are inadequate. Our goal is to build a fully integrated oncology company focused on novel, molecularly targeted therapies to treat solid tumors and hematologic cancers, as well as the spread of primary tumors to distant sites. Our business strategy is to:
|
●
|
build a fully integrated oncology company and become a leader in the discovery, development and commercialization of molecularly targeted oncology therapies;
|
|
●
|
broadly develop our lead oncology product candidates and build a pipeline of innovative follow-on product candidates;
|
|
●
|
enter into collaborations with major pharmaceutical or biotechnology companies, after obtaining definitive clinical data, to assist in developing our cancer product candidates and commercializing them in selected markets; and
|
|
●
|
license our cell-signaling regulation technologies to pharmaceutical and biotechnology companies.
|
Our Product Candidates
Our lead cancer product candidate, ridaforolimus (previously known as deforolimus and, prior to that, AP23573), is an internally discovered, potent inhibitor of the protein mTOR. mTOR acts as a central regulator of protein synthesis, cell proliferation, cell cycle progression and cell survival. Blocking mTOR creates a starvation-like effect in cancer cells by interfering with cell growth, division, metabolism and angiogenesis.
We are developing ridaforolimus in partnership with Merck & Co., Inc., or Merck, under a collaboration agreement signed in July 2007. The collaboration agreement provides that we, together with Merck, will conduct a broad-based development program in multiple cancer indications. The collaboration agreement establishes responsibilities for development, annual budgeting and planning, manufacturing, promotion, distribution and sales of the product, governance of the collaboration, dispute resolution, termination provisions and other matters.
The collaboration agreement provides for (i) an up-front payment of $75 million which was paid to us in July 2007, (ii) sharing of the costs of development, (iii) up to $652 million in milestone payments based on successful development of and achievement of specific sales thresholds related to ridaforolimus, of which $53.5 million in milestones have been received to date, and (iv) the availability of up to $200 million of repayable advances to fund our share of ongoing development of ridaforolimus upon obtaining regulatory approval to market ridaforolimus and our having spent at least $150 million for our share of development costs. The collaboration agreement also provides for profit-sharing and royalties upon successful commercialization of ridaforolimus. See “Our Licenses to Third Parties” under this Part I for a detailed description of our collaboration agreement with Merck.
1
Pursuant to a joint global development plan established by us and Merck, we are developing ridaforolimus in multiple cancer indications, both as a single agent and in combination with other targeted agents. We have completed enrollment in a Phase 3 clinical trial of ridaforolimus in patients with metastatic sarcomas and expect results from a second interim analysis and the final analysis of progression free survival, the primary endpoint of this trial in 2010. We and Merck are conducting Phase 2 clinical trials of oral ridaforolimus in patients with endometrial, breast, prostate and non-small cell lung cancers, and Phase 1 studies of ridaforolimus in combination with other agents, all as part of the joint global development plan.
As of the time of filing of this Annual Report on Form 10-K, we are engaged in advanced negotiations with Merck regarding the terms of a revised collaboration agreement for the development and commercialization of ridaforolimus. Although there can be no assurance that we will reach agreement with respect to such revisions on terms acceptable to us, our current expectation is that a revised agreement will be entered into in the second fiscal quarter of 2010. If we do not finalize such a revised agreement as currently anticipated, we will seek to raise the additional capital necessary to fund our operations through equity offerings, debt financings and pursuit of other strategic options, and reduce our operating expenses.
Ridaforolimus is also being developed pursuant to license agreements with medical device companies for use on drug-eluting stents to prevent restenosis, or reblockage, of injured vessels following interventions in which stents are used in conjunction with balloon angioplasty. We have entered into two such license agreements to date, one with Medinol Ltd., or Medinol, and another with ICON Medical Corp., or ICON, and have retained the right to enter into one additional non-exclusive agreement in this area.
Our second product candidate, AP24534, is an investigational, pan BCR-ABL inhibitor that we believe has potential applications in various hematological cancers and solid tumors and is wholly owned by us. We are currently conducting a Phase 1 clinical trial of oral AP24534 in patients with advanced hematological cancers. Preliminary data from the Phase 1 trial show evidence of hematologic, cytogenetic and molecular anti-cancer activity of AP24534 in heavily pre-treated patients with resistant and refractory chronic myeloic leukemia, or CML, including those with the T315I mutation of the target protein, BCR-ABL. Pending further analysis of the results of this trial and discussions with regulatory authorities, and subject to available funding, we believe that we will be able to initiate a pivotal registration trial of AP24534 in patients with CML in the second half of 2010. In preclinical studies, AP24534 has also demonstrated potent inhibition of kinase targets associated with acute myeloid leukemia, or AML, as well as proliferation and angiogenesis in solid tumors.
Our third product candidate, AP26113, is an investigational anaplastic lymphoma kinase, or ALK, inhibitor that we believe has the potential to regulate multiple cancer pathways and to be used in the treatment of certain patients with various cancers, including non-small cell lung cancer, lymphoma and neuroblastoma. We have commenced preclinical testing and investigational new drug, or IND, enabling studies of this product candidate.
We have a focused drug discovery program centered on small-molecule therapies, molecularly targeted to cell-signaling pathways implicated in cancer. Our drug discovery program builds on our expertise in cell signaling, cancer biology, structure-based drug design and computational chemistry in designing and characterizing small-molecule drugs, such as ridaforolimus, AP24534 and AP26113, to treat life-threatening diseases.
See the section entitled “Risk Factors” in Part I, Item 1A of this Annual Report on Form 10-K for a description of the risks related to our business and our clinical and preclinical programs.
2
Our Technologies
We are the exclusive licensee of a family of patents, three in the U.S. and one in Europe, including a pioneering U.S. patent covering methods of treating human disease by regulating NF-κB cell-signaling activity, hereinafter referred to as the ‘516 Patent, awarded to a team of inventors from The Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology and Harvard University. NF-κB is a protein that can be generally thought of as a “biological switch” that can be turned off using these treatment methods to treat disorders such as inflammation, cancer, sepsis and osteoporosis. We permit broad use of our NF-κB intellectual property, at no cost, by investigators at academic and not-for-profit institutions to conduct non-commercial research. Our goal is to license our NF-κB technology to pharmaceutical and biotechnology companies that are conducting research to discover and develop drugs that modulate NF-κB cell signaling and/or that are marketing such drugs. We have entered into two license agreements for use of our NF-κB cell-signaling technology for research and development purposes. However, the ‘516 Patent is the subject of an outstanding lawsuit and a proceeding before the United States Patent and Trademark Office, or PTO. See Part I, Item 3 entitled “Legal Proceedings” and Part I, Item 1A entitled “Risk Factors” of this Annual Report on Form 10-K for a description of the status of these proceedings and related risks.
We have also developed a proprietary portfolio of cell-signaling regulation technologies, our ARGENT technology, to control intracellular processes with small molecules, which may be useful in the development of therapeutic vaccines and gene and cell therapy products and which provide versatile tools for applications in cell biology, functional genomics and drug discovery research. We distribute our ARGENT technologies at no cost to academic investigators in the form of our Regulation Kits to use in various research applications in an academic setting. In addition, we have licensed the ARGENT technology to several pharmaceutical and biotechnology companies for research and development and/or commercial purposes.
Our Lead Development Programs
Potential Oncology Indications of our mTOR Inhibitor, Ridaforolimus
Human cells, both healthy and malignant, share an elaborate system of molecular pathways that carry signals back and forth from the cell surface to the nucleus and within the cell. Such signaling is essential to cell functioning and viability. When disrupted or over-stimulated, such pathways may trigger diseases such as cancer. For example, growth and proliferation of cancer cells are dependent on signals from external growth factors, as well as signals indicating the availability of sufficient nutrients and blood supply. These signals are conveyed along well-defined pathways, several of which are regulated by the protein called the mammalian target of rapamycin, or mTOR.
Our lead cancer product candidate, ridaforolimus, is an internally discovered, potent mTOR inhibitor. mTOR acts as a central regulator of protein synthesis, cell proliferation, cell cycle progression and cell survival. Blocking mTOR creates a starvation-like effect in cancer cells by interfering with cell growth, division, metabolism and angiogenesis.
As part of our global clinical development plan and registration strategy, we have studied ridaforolimus as a single agent in multiple Phase 1 and Phase 2 clinical trials in the U.S. and Europe in patients with solid tumors, including sarcomas, hormone refractory prostate cancer, endometrial cancer, brain cancer and certain leukemias and lymphomas. We have also conducted several Phase 1b trials of ridaforolimus in combination with other anti-cancer therapies. These trials focused primarily on patients with various types of solid tumors. Intravenous and oral tablet formulations of ridaforolimus have been studied in these trials.
In clinical trials to date, ridaforolimus has been well tolerated at the doses administered, and adverse events were generally mild to moderate in severity and manageable. The most common treatment-related adverse events experienced by patients in the trials were mouth sores, rash, fatigue, anemia, nausea and lipid abnormalities.
3
We are developing ridaforolimus in partnership with Merck pursuant to our collaboration agreement signed in July 2007. Our most advanced potential indication and initial registration path for ridaforolimus is in patients with soft-tissue and bone sarcomas. In a multi-center Phase 2 trial of 212 patients with advanced sarcomas, at least 90 percent of whom had progressive disease, ridaforolimus demonstrated efficacy and was well tolerated. The primary endpoint of the trial – evidenced by clinical-benefit response, or CBR, rates – was achieved in the most common types of sarcoma. Progression free survival in patients treated with ridaforolimus was more than twice as long as progression free survival estimated from historical control data published by the European Organization for Research and Treatment of Cancer, or EORTC.
In September 2007, we initiated our first Phase 3 clinical trial of ridaforolimus in patients with metastatic soft-tissue and bone sarcomas. The SUCCEED (Sarcoma Multi-Center Clinical Evaluation of the Efficacy of Ridaforolimus) trial is a randomized, double-blind, placebo-controlled study designed to assess the impact of oral ridaforolimus on progression-free survival, or PFS, the primary endpoint of the trial, and several secondary endpoints, in metastatic soft-tissue and bone sarcoma patients who benefited from prior chemotherapy. Continued treatment with traditional chemotherapeutic drugs has not been shown to provide additional clinical benefit to such patients after such previous benefit. Thus, absent other alternatives, physicians generally either continue potentially toxic chemotherapy until the side effects become unacceptable or, more commonly, monitor patients carefully for disease progression, or tumor growth, prior to initiating another line of chemotherapy. Therefore, the placebo arm represents a current standard of care for patients in this clinical setting.
The SUCCEED trial is designed to evaluate approximately 650 patients who are randomized one-to-one to oral ridaforolimus or placebo at over 125 sites worldwide. The trial is 90 percent powered to detect a 33 percent increase in median PFS comparing the ridaforolimus arm with the placebo arm. We have agreement with the U.S. Food and Drug Administration, or FDA, on a Special Protocol Assessment, or SPA, for the SUCCEED trial. The European Medicines Agency, or EMEA, has provided protocol advice consistent with that of the FDA regarding the trial design as part of its Protocol Assistance program.
In September 2009, we received the results of the first of two interim efficacy analyses provided for in the protocol for the SUCCEED trial. Based on its review of approximately one-third of the number of PFS events anticipated in the trial, the independent Data Monitoring Committee, or DMC, recommended that the trial continue to full patient enrollment and completion. The DMC indicated that the safety data from this first interim analysis were consistent with the known safety profile of ridaforolimus and recommended no changes to the study protocol. We completed enrollment of 650 patients in this clinical trial in December 2009. In the first quarter of 2010, two-thirds of the number of PFS events anticipated in the trial was reached, triggering the second interim efficacy analysis. We expect to receive the results of a second interim efficacy analysis in the second quarter of 2010 and the results of the final efficacy analysis in the second half of 2010.
The FDA and the EMEA have designated ridaforolimus as an orphan drug for treatment of soft-tissue and bone sarcomas. The FDA has also designated ridaforolimus as a fast-track product for the same potential indication.
In addition to the SUCCEED clinical trial, pursuant to the global development plan established with Merck, we and Merck are conducting multiple clinical trials, including Phase 2 trials in endometrial, breast, prostate and non-small cell lung cancers, and Phase 1 clinical trials of ridaforolimus in combination with other agents. The global development plan also includes a focused biomarker research program that exploits the companies' expertise in cell-signaling, mTOR biology and diverse state-of-the-art molecular profiling technologies. We believe this program will help characterize and identify rational combinations with ridaforolimus, identify responder profiles and inform decisions in alignment with the development plan.
4
As noted above under “Business – Overview – Our Product Candidates,” we are currently engaged in advanced negotiations with Merck regarding the terms of a revised collaboration agreement.
Potential Cardiovascular Indications of our mTOR Inhibitor, Ridaforolimus
As an mTOR inhibitor, ridaforolimus has also been shown to block the proliferation and migration of vascular smooth muscle cells, the primary cause of narrowing and blockage of injured arteries, and is an analog of sirolimus, another mTOR inhibitor that has been approved for use in drug-eluting stents. Recent clinical studies have found lower reblockage rates in patients treated with stents that deliver small-molecule drugs, such as sirolimus or paclitaxel, a cytotoxic agent, locally to the site of vascular injury. Such stents have become the standard of care for many patients undergoing interventional procedures to open narrowed coronary arteries.
We entered into license agreements with Medinol, a leading innovator in stent technology, in January 2005, and with ICON, an emerging medical device company, in October 2007, to develop and commercialize stents and other medical devices to deliver ridaforolimus to prevent restenosis, or reblockage, of injured vessels following interventions in which stents are used in conjunction with balloon angioplasty. We have retained the right to enter into one additional non-exclusive license agreement, in addition to the licenses granted to ICON and Medinol, to develop and commercialize medical devices delivering ridaforolimus for use in vascular disease.
Our Pan BCR-ABL Inhibitor, AP24534
Our second oncology product candidate, AP24534, is an investigational pan BCR-ABL inhibitor that we believe has broad potential applications in various hematological cancers and solid tumors. AP24534 was internally discovered and is wholly owned by us. We are currently in Phase 1 clinical development of AP24534. Results to date from preclinical studies and preliminary clinical data from the Phase 1 clinical trial show that AP24534 potently inhibited BCR-ABL, a target protein associated with drug-resistant chronic myeloid leukemia, or CML, as well as various mutants of BCR-ABL.
Preclinical studies showed that AP24534 demonstrated efficacy and oral dosing flexibility in animal models of CML, including forms of CML caused by clinically relevant mutants of BCR-ABL. Significantly, AP24534 potently inhibited a specific mutant, T315I, which is resistant to all currently marketed drugs. Additional preclinical studies demonstrated that AP24534 also inhibits Flt3, a target associated with acute myeloid leukemia, or AML.
In addition, AP24534 has demonstrated in preclinical studies potent inhibition of additional targets that control the process of angiogenesis, or blood vessel growth, including the receptors for vascular endothelial growth factors, or VEGFRs, fibroblast growth factors, or FGFRs, and angiopoietin, or Tie2. Inhibiting angiogenesis is a clinically validated approach to treating multiple solid tumors. Based on AP24534’s differentiated profile, we believe these findings support the broad potential of the drug not only in drug-resistant CML, but also in other hematological cancers, such as AML, and various solid tumors.
In 2008, we initiated a Phase 1 clinical trial of AP24534 in heavily pretreated patients with drug-resistant and refractory CML and other hematologic malignancies. This multi-center, sequential dose-escalation study in approximately 50 patients is designed to determine the safety, tolerability and initial evidence of the anti-leukemic activity of AP24534, as well as its pharmacokinetics (the behavior of AP24534 in patients) and its pharmacodynamics (the effects of AP24534 on patients’ cells).
In December 2009, we announced positive preliminary clinical data from this ongoing Phase 1 trial. The data indicate that AP24534 is well tolerated with no dose limiting toxicities observed at dose levels lower than 60 mg administered orally once per day. Four of the twelve patients at the 60 mg dose level had dose limiting toxicities, consisting of chemical or clinical pancreatitis which are considered manageable and treatable. This study is continuing enrollment of patients at the 45 mg dose level, the midpoint of the 30 mg and 60 mg dose levels.
5
We believe that the preliminary data from this study also show strong evidence of hematologic, cytogenetic and molecular anti-cancer activity, including in patients with mutations of the BCR-ABL protein that are resistant to currently marketed drugs. Pending further analysis of the results of this trial and discussions with regulatory authorities, and subject to available funding, we believe that we will be able to initiate a registration trial of this product candidate in patients with CML in the second half of 2010.
The FDA has designated AP24534 as an orphan drug for the treatment of CML and Philadelphia chromosome-positive acute lymphoblastic leukemia and the EMEA has designated it as an orphan drug for CML and acute lymphoblastic anemia.
Our Anaplastic Lymphoma Kinase Inhibitor, AP26113
Our third oncology product candidate, AP26113, is an internally discovered small-molecule anaplastic lymphoma kinase, or ALK, inhibitor that targets a unique genetic feature of cancer cells similar to AP24534.
ALK was first identified as a chromosomal rearrangement in anaplastic large cell lymphoma, or ALCL. Genetic studies now indicate that abnormal expression of ALK is a key driver of certain types of non-small cell lung cancer and neuroblastoma, as well as ALCL. Since ALK is generally not expressed in normal adult tissues, it represents a highly promising target for molecularly targeted cancer therapy.
In preclinical studies to date, AP26113 has been demonstrated to potently inhibit tumor cells that express ALK while having no effects on cells that do not express ALK. In addition, this compound is active when administered orally in in vivo animal models of lymphoma and lung cancer, with minimal effects on insulin or glucose levels. Subject to available funding, we plan to continue to perform the pre-clinical studies necessary to file an IND with regulatory authorities, which we expect to file in 2011.
Our Discovery Programs
Our research and development programs are focused on discovering and developing small-molecule drugs that regulate cell signaling. Many of the critical functions of cells, such as cell growth, differentiation, gene transcription, metabolism, motility and survival, are dependent on signals carried back and forth from the cell surface to the nucleus and within the cell through a system of molecular pathways. When disrupted or over-stimulated, such pathways may trigger diseases such as cancer. From our inception, our research has focused on exploring cell-signaling pathways, identifying their role in specific diseases, and discovering drug candidates to treat those diseases by interfering with the aberrant signaling pathways of cells. The specific cellular proteins blocked by our product candidates have been well characterized and validated as targets. Product candidates like ridaforolimus, AP24534 and AP26113 have been developed in-house through the integrated use of structure-based drug design and computational chemistry, and their targets have been validated with techniques such as functional genomics, proteomics, and chemical genetics.
Our Proprietary Technologies
NF-kB Cell-signaling Technology
Dr. David Baltimore, former director of the Whitehead Institute for Biomedical Research, Dr. Phillip Sharp of the Massachusetts Institute of Technology, and Dr. Thomas Maniatis of Harvard University, together with a team of scientists in their respective laboratories, discovered a family of genes that encode proteins they called NF-κB and I-κB, its inhibitor; the critical role played by NF-κB cell signaling in regulating cellular processes involved in various difficult-to-treat diseases; methods to identify compounds to regulate NF-κB cell-signaling activity; and methods of treating disease by inhibiting NF-κB activity. NF-κB can be generally thought of as a “biological switch” that can be turned off using these methods to treat disorders, such as inflammation, cancer, sepsis and osteoporosis.
6
We have an exclusive license from these academic institutions to pioneering technology and patents related to methods of treating human disease by regulating NF-κB cell-signaling activity, and the discovery and development of drugs to regulate NF-κB cell-signaling activity. We have a program to license this technology and these treatment methods to pharmaceutical and biotechnology companies that are conducting research to discover and develop drugs that modulate NF-κB cell-signaling and/or that are marketing such drugs. One of the NF-κB patents is the subject of reexamination proceedings in the U.S. Patent and Trademark Office, or PTO, and a patent infringement lawsuit filed in 2002 by us and the academic institutions against Eli Lilly and Company, or Lilly. See Part I, Item 3 entitled “Legal Proceedings” and Part I, Item 1A entitled “Risk Factors” of this Annual Report on Form 10-K for a description of the status of these proceedings and related risks.
ARGENT Cell-signaling Regulation Technology
Our proprietary portfolio of cell-signaling regulation technologies includes the ARGENT signaling and transcription technologies. Our ARGENT technologies allow intracellular processes to be controlled with small molecules, which may be useful in the development of therapeutic vaccines and gene and cell therapy products, and which provide versatile tools for applications in cell biology, functional genomics and drug-discovery research, including three-hybrid screening approaches to discover and characterize targets and lead molecules. To maximize their use by the scientific community, we distribute our technologies at no cost to academic investigators in the form of our Regulation Kits. As of February 28, 2010, we have entered into more than 1,700 material transfer agreements with more than 570 different institutions in 35 countries for the use of this technology in diverse areas of research, and more than 300 scientific papers describing their use have been published. In addition, we have licensed the ARGENT technology to several pharmaceutical and biotechnology companies for research and development and/or commercial purposes.
Our Intellectual Property
Patents and other intellectual property rights are essential to our business. We file patent applications to protect our technology, inventions and improvements to our inventions that are considered important to the development of our business.
As of February 28, 2010, we owned, co-owned or held an exclusive license to 62 U.S. patents and 35 pending U.S. patent applications together with their various foreign counterparts. We also have several nonexclusive technology licenses from certain institutions in support of our research programs, and may seek additional such licenses where applicable technology complements our research and development efforts.
Our mTOR inhibitor, ridaforolimus, and its production are covered by two of our issued U.S. patents. Those patents and foreign counterparts are expected to expire in 2023. Patents that may issue based on other pending applications would provide in some cases up to five years of additional patent protection for covered therapeutic uses of ridaforolimus.
Our multi-targeted kinase inhibitor, AP24534, and our ALK inhibitor, AP26113, are each covered by pending patent applications that are expected to provide patent protection through late 2026 and mid-2029, respectively. In both cases, other pending patent applications relating to therapeutic uses of the respective inhibitors are expected to provide additional patent protection into 2030.
The remainder of our patent portfolio is focused primarily on inventions involving additional classes of chemical compounds; the mTOR gene; the components, configurations and use of our ARGENT regulation technologies; and the NF-κB cell-signaling technology.
7
We also rely on unpatented trade secrets and proprietary know-how, some of which is not believed to be adequately protectable through patents. In order to protect our trade secrets, we enter into confidentiality agreements with our employees, consultants, investigators, clinical trial sites, contractors, collaborators and other third parties to whom we disclose confidential information, although protection of trade secrets is generally recognized as challenging.
Our Licenses to Third Parties
Our Collaboration with Merck & Co., Inc.
On July 11, 2007, we entered into a collaboration agreement with Merck for the joint global development and commercialization of ridaforolimus. We are currently engaged in advanced negotiations with Merck regarding the terms of a revised collaboration agreement. Although there can be no assurance that we will reach agreement with respect to such revisions on terms acceptable to us, our current expectation is that a revised agreement will be entered into in the second fiscal quarter of 2010.
Under the terms of the collaboration agreement, as currently in effect, we and Merck are conducting a broad-based development program in which clinical trials, preclinical studies and biomarker studies are being conducted concurrently in multiple potential cancer indications, pursuant to a global development plan agreed upon by the parties. Each party funds 50 percent of the global development costs, except that Merck funds 100 percent of any cost of development that is specific to development or commercialization of ridaforolimus outside the United States. The agreement provides that, in certain circumstances, either party may opt out of conducting and funding certain late-stage clinical trials, which would result in changes in development and commercialization responsibilities and compensation arrangements. We are responsible for supplying the active pharmaceutical ingredient used in ridaforolimus drug product, and Merck is responsible for the formulation of the finished product, all under a separate supply agreement between the parties.
The collaboration agreement currently provides that, in the United States, we and Merck will co-promote ridaforolimus, we will distribute and sell ridaforolimus for all cancer indications and record all sales, and each party will receive 50 percent of the net profit from such sales. Outside the United States, Merck will distribute, sell and promote ridaforolimus and book all sales, and Merck will pay us tiered double-digit royalties on such sales. Royalties are payable by Merck, on a country by country basis, until the later of (i) the expiration of the last valid claim of any patent rights owned by either us or Merck that cover ridaforolimus, (ii) a specified number of years from first commercial sale, or (iii) the last date upon which we supply active pharmaceutical ingredient to Merck under the supply agreement, subject to partial reduction in certain circumstances.
Under the terms of the collaboration agreement, Merck paid us an initial up-front payment of $75 million in July 2007, and has agreed to pay up to $452 million in milestone payments based on the successful development of ridaforolimus in multiple potential cancer indications, of which $53.5 million has been paid to us through December 31, 2009, and up to $200 million in milestone payments based on achievement of specified product sales thresholds. Merck has also agreed to provide us with up to $200 million in interest-bearing, repayable, development cost advances to cover a portion of our share of global development costs, after we have paid $150 million in global development costs and have obtained regulatory approval to market ridaforolimus from the FDA in the United States or similar regulatory authorities in Europe or Japan. All amounts to be paid to us by Merck, with the exception of any development cost advances, are non-refundable.
The collaboration agreement may be terminated (i) by either party based on insolvency or uncured breach by the other party, (ii) by Merck on or after the third anniversary of the effective date by providing at least 12 months prior written notice, (iii) by Merck upon the failure of ridaforolimus to meet certain developmental and safety requirements, or (iv) after discussions between the parties, in the event Merck concludes it is not advisable to continue the development of ridaforolimus for use in a potential cancer indication. Upon termination of the collaboration agreement, depending upon the circumstances, the parties have varying rights and obligations with respect to the continued development and commercialization of ridaforolimus and continuing royalty obligations.
8
Under the terms of the collaboration agreement, we and Merck have established a series of joint committees which are responsible for the development and commercialization of ridaforolimus. Under the committee structure, if the committees are unable to reach a decision, the matter is referred to senior executives of the parties. Each party has ultimate decision making authority with respect to a specified limited set of issues, and for all other issues, the matter must be resolved by consensus of the parties. Either party may choose not to appoint members to any of the joint committees and such a determination by either party has no impact on the financial or other terms of the collaboration.
Our Stent Collaborations
Medinol Ltd.
In January 2005, we entered into a license agreement with Medinol Ltd., or Medinol, a cardiovascular medical device company, to develop and commercialize ridaforolimus-eluting stents and other medical devices to prevent restenosis, or reblockage, of injured vessels following interventions in which stents are used in conjunction with balloon angioplasty. Under the agreement, we granted to Medinol a non-exclusive, world-wide, royalty-bearing license, under our patents and technology relating to ridaforolimus, to develop, manufacture and sell the stents and certain other medical devices that deliver ridaforolimus. We are responsible for supplying Medinol with, and Medinol agreed to purchase from us, certain quantities of ridaforolimus for use in its development, manufacture and sale of the stents and other medical devices. The agreement allows Medinol to distribute products resulting from the agreement worldwide through distributors authorized by us. We have entered into a similar non-exclusive license agreement with ICON Medical Corp., as described further below, and we have retained the right to enter into one additional non-exclusive license agreement with an additional third party to develop and commercialize stents and certain other medical devices to deliver ridaforolimus for use in vascular disease.
The agreement provides for the payment by Medinol to us of up to $39.3 million, which includes an upfront license fee and payments based upon achievement of development, regulatory and commercial milestones, if two products are developed. Through December 31, 2009, we have received $750,000 under the agreement. In addition, we are eligible to receive tiered single-digit royalties based on various minimum levels of stents or other medical devices sold under the agreement. As of December 31, 2009, no products have been approved by regulatory authorities for sale under this agreement.
The term of the agreement extends to the later to occur of the expiration of our patents relating to the rights licensed to Medinol under the agreement or 15 years after the first commercial sale of a product. The agreement may be terminated by either party for breach following the failure to cure after a 90-day cure period. In addition, Medinol may terminate the agreement upon 30 days notice to us upon certain events, including if it determines, in its reasonable business judgment, that it is no longer in its business interest to continue the development of a medical device to deliver ridaforolimus. We may terminate the agreement upon 30 days notice to Medinol, if we determine that it is no longer in our business interest to continue our development and regulatory approval efforts with respect to ridaforolimus.
The agreement also provides for periodic reporting of progress, sharing of relevant clinical and non-clinical data, assistance in resolution of technical and regulatory issues and, if a product is approved, timely reporting of sales and remittance of royalty payments.
ICON Medical Corp.
In October 2007, we entered into a license agreement with ICON Medical Corp., or ICON, a cardiovascular medical device company, to develop and commercialize ridaforolimus-eluting stents and other medical devices to prevent restenosis, or reblockage, of injured vessels following interventions in which stents are used in conjunction with balloon angioplasty. Under the agreement, we granted to ICON a non-exclusive, world-wide, royalty-bearing license, under our patents and technology relating to ridaforolimus, to develop, manufacture and sell the stents and certain other medical devices that deliver ridaforolimus. We are responsible for supplying ICON with, and ICON agreed to purchase from us, certain quantities of ridaforolimus for use in its development, manufacture and sale of the stents and other medical devices. We have entered into a similar non-exclusive license agreement with Medinol, as described above, and we have retained the right to enter into one additional non-exclusive license agreement with an additional third party to develop and commercialize stents and certain other medical devices to deliver ridaforolimus for use in vascular disease.
9
Concurrent with the execution of the agreement, we received shares of ICON common stock equal to an ownership interest in ICON of less than 10% and certain percentage maintenance, anti-dilution, registration and other rights. The agreement provides for the payment by ICON to us of up to $27.4 million based upon achievement of certain clinical, regulatory and commercial milestones, if two products are developed. Through December 31, 2009, we have received no such payments under the agreement. In addition, we are eligible to receive single-digit royalties based on net sales of stents or other medical devices sold under the agreement. As of December 31, 2009, no products have been approved by regulatory authorities for sale under this agreement.
The term of the agreement extends to the later to occur of the expiration of our patents relating to the rights licensed to ICON under the agreement or 15 years after the first commercial sale of a product. The agreement may be terminated by either party for breach following the failure to cure after a 90-day cure period. In addition, ICON may terminate the agreement upon 30 days notice to us upon certain events, including if it determines, in its reasonable business judgment, that it is no longer in its business interest to continue the development of a medical device to deliver ridaforolimus. We may terminate the agreement upon 30 days notice to ICON, if we determine that it is no longer in our business interest to continue our development and regulatory approval efforts with respect to ridaforolimus.
The agreement also provides for periodic reporting of progress, sharing of relevant clinical and non-clinical data, assistance in resolution of technical and regulatory issues and, if a product is approved, timely reporting of sales and remittance of royalty payments.
Other Licenses to Third Parties
We have a program to license our NF-κB cell-signaling technology and treatment methods to pharmaceutical and biotechnology companies conducting research to discover and develop drugs that modulate NF-κB cell-signaling and/or marketing such drugs. To date, we have entered into several licenses for this technology with pharmaceutical companies and companies manufacturing and commercializing kits, technologies and tools for research applications.
We also have a program to license our ARGENT cell-signaling regulation technologies to pharmaceutical and biotechnology companies to develop and commercialize innovative therapeutic products and to conduct drug discovery research. To date, we have entered into several licenses for use of our ARGENT cell-signaling regulation technologies for a variety of applications, including the development of therapeutic vaccines and gene and cell therapy products and for use in drug discovery. In addition, several biotechnology companies have conducted collaborative studies of these technologies for use in gene and cell therapy applications.
Our Licenses from Third Parties
NF-κB Cell-Signaling Technologies
In August 1991, we entered into an exclusive license agreement with Massachusetts Institute of Technology and the Whitehead Institute (on behalf of themselves and Harvard University, collectively “MIT”), pursuant to which we are the exclusive licensee of pioneering technology and patents relating to NF-κB cell-signaling technologies and methods of treating human disease through modulation of NF-κB cell-signaling activity. This license agreement, as amended in 1995, required us to pay MIT an up-front license issue fee and a one-time milestone fee, as well as annual license maintenance fees. In addition, we are obligated to pay MIT low-single-digit royalties based on net sales of any products and processes developed using the NF-κB cell-signaling technologies and treatment methods. Through December 31, 2009, we have paid MIT approximately $773,000 under this agreement.
10
After obtaining the patent rights related to the NF-κB cell-signaling activity, we began a program to generate revenues by outlicensing this technology and these treatment methods to pharmaceutical and biotechnology companies that are conducting research to discover and develop drugs that modulate NF-κB cell-signaling and/or that are marketing related drugs. To date, we have entered into several outlicenses of this technology with pharmaceutical companies and companies manufacturing and commercializing kits, technologies and tools for research applications.
The license agreement also grants us the right to undertake the enforcement and/or defense of these patent rights at our sole expense, subject to our right to withhold a significant percentage of any royalties otherwise due to the academic institutions to be applied toward reimbursement of our fees and expenses in connection with any such litigation. The license agreement also provides that we will share a percentage of any damages, net of fees and expenses, awarded in such litigations with the academic institutions. We have been engaged in litigation with Eli Lilly and Company and Amgen Inc. and certain affiliated entities with respect to one of the NF-κB patents, as well as a proceeding in the U.S. Patent and Trademark Office, as disclosed in Part I, Item 3 entitled “Legal Proceedings” of this Annual Report on Form 10-K.
The term of the agreement extends to 12 years after the first commercial sale of a product resulting from technology licensed under the agreement. The agreement may be terminated by MIT upon a failure by us to pay MIT any royalties due under the agreement after a 30-day notice of termination or upon a breach by us following our failure to cure after a 90-day cure period. We may terminate the agreement upon six months notice to MIT and payment of all amounts due to MIT through the effective date of termination, or we may terminate our license with respect to any particular patents upon written notice to MIT.
ARGENT Cell-Signaling Regulation Technologies
In December 1997, we entered into an amended and restated exclusive license agreement with Stanford University (on behalf of itself and Harvard University, collectively “Stanford”), pursuant to which we became the exclusive licensee of certain technology and patent rights to our ARGENT cell-signaling regulation technologies, which includes materials and methods for regulating the transcription of specific genes in vivo.
Concurrent with our execution of this agreement, we issued an aggregate of 128,571 shares of common stock of our former subsidiary AGTI, which was merged into us in September 2008, to Stanford. The agreement required us to pay Stanford an up-front license issue fee, as well as payments based upon achievement of certain clinical, regulatory and commercial milestones. Through December 31, 2009, we have paid Stanford $840,000 under this agreement. In addition, we are obligated to pay Stanford single-digit royalties based on net sales of any products and processes developed using the ARGENT cell-signaling regulation technologies, including therapies and research reagents. As of December 31, 2009, no products or processes have been developed using the ARGENT cell-signaling regulation technologies and approved for sale.
The initial term of the agreement extends to 12 years after the first commercial sale of a product resulting from technology licensed under the agreement, augmented by any patent term extension awarded in connection with the patents licensed under the agreement. The agreement further extends for multi-year terms, unless Stanford demonstrates that we are not diligently pursuing the commercialization of the technologies licensed under the agreement. The agreement may be terminated by Stanford upon a material breach by us, including failure to pay royalties owed under the agreement, following our failure to cure after a 60-day cure period. We may terminate the agreement upon 30 days’ written notice to Stanford and payment of all amounts due to Stanford through the effective date of termination.
11
We have also entered into other license agreements with various institutions and universities pursuant to which we are the licensees of certain technologies relating to our research and development programs. In some instances, our license agreements from third parties also impose insurance, development, sublicensing and other obligations on us. Failure by us to comply with these requirements could result in the termination of the applicable agreement, which, depending upon the technologies which are the subject of the applicable agreement, could have a material adverse effect on our business, financial condition, and results of operations.
Research and Development Spending
During each of the three years ended December 31, 2009, 2008 and 2007, we spent approximately $63.4 million, $50.8 million and $39.6 million, respectively, on our research and development activities.
Manufacturing
Our drug candidates and preclinical compounds are small molecules that can be readily synthesized by processes that we have developed. We are able to manufacture in-house the quantities of our product candidates necessary for certain preclinical studies. We do not own or operate manufacturing facilities for the production of clinical or commercial quantities of our product candidates. We contract with third- party manufacturers to assist in the development and optimization of our manufacturing processes and methods and to supply our product candidates in sufficient bulk quantities and in suitable dosage forms for use in our clinical trials. We also expect to depend on third-party manufacturers for the supply of our products upon commercialization.
Our lead product candidate, ridaforolimus, is produced by an established manufacturing process using conventional synthetic and natural-product fermentation techniques. The production of ridaforolimus is based in part on technology that we believe is proprietary to us. Pursuant to our collaboration agreement and a separate supply agreement with Merck, we are responsible for supplying the active pharmaceutical ingredient used in ridaforolimus drug product and Merck is responsible for the supply of ridaforolimus as a finished drug product. We, with Merck, may license this technology to contract manufacturers to enable them to manufacture ridaforolimus for us. In addition, a contract manufacturer may develop process technology related to the manufacture of our drug candidates that the manufacturer owns either independently or jointly with us. This would increase our reliance on that manufacturer or require us to obtain a license from that manufacturer in order to have our product manufactured by other parties. We are negotiating with our existing suppliers and other third-party manufacturers to secure the long-term supply and manufacture of the active pharmaceutical ingredient used in ridaforolimus at commercially reasonable costs with appropriate redundancy for commercialization.
Contract manufacturers are subject to extensive governmental regulation and we depend on them to manufacture our product candidates in accordance with the FDA’s current good manufacturing practice regulations, or cGMPs. We have an established quality assurance program intended to ensure that third-party manufacturers under contract produce our compounds in accordance with cGMPs, and other applicable domestic and foreign regulations. We believe that our current contractors comply with such regulations.
Competition
The pharmaceutical and biotechnology industries are intensely competitive. We compete directly and indirectly with other pharmaceutical companies, biotechnology companies and academic and research organizations, many of whom have greater resources than us. We compete with companies who have products on the market or in development for the same indications as our product candidates. We may also complete with organizations that are developing similar technology platforms.
12
In the area of oncology, pharmaceutical and biotechnology companies such as Amgen Inc., AstraZeneca PLC, Bristol-Myers Squibb Company, Eli Lilly and Company, the Roche Group, GlaxoSmithKline plc, Johnson & Johnson, Merck & Co., Inc., Merck KGaA, Novartis AG, and Pfizer, Inc., are developing and marketing drugs to treat cancer, including mTOR inhibitors. Specifically, Pfizer, Inc. and Novartis AG are developing mTOR inhibitors for use in cancer. Pfizer’s mTOR inhibitor, temsirolimus, and Novartis’ mTOR inhibitor, everolimus are both approved to treat patients with advanced kidney cancer. Biotechnology companies such as Amgen Inc., Biogen-Idec, Inc., Onyx Pharmaceuticals, Inc. and OSI Pharmaceuticals, Inc., are developing and, in some cases, marketing drugs to treat various diseases, including cancer, by inhibiting cell-signaling pathways. Other companies have products on the market or in development against which our drug candidates, if approved, may have to compete. Specifically, PharmaMar, a wholly owned subsidiary of Zeltia Group, has a product, trabectedin, approved for the treatment of soft-tissue sarcomas in Europe, and Takeda Pharmaceutical Co., Ltd. has mifamurtide, an immunotherapy product approved in Europe for treatment of bone sarcomas. In addition, Chemgenex Pharmaceuticals Limited has a product candidate, omacetaxine, for the treatment of patients with CML who have failed prior therapy with imatinib and who have developed the T315I mutation of BCR-ABL. We may also experience competition from companies that have acquired or may acquire technology from companies, universities, and other research institutions. As these companies develop their technologies, they may develop proprietary positions that may materially and adversely affect us.
Government Regulation
Government authorities in the United States, at the federal, state and local level, and in other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, marketing and export and import of products such as those we are developing. The process of obtaining regulatory approvals in the United States and in foreign countries, along with subsequent compliance with applicable statutes and regulations, require the expenditure of substantial time and financial resources.
United States Drug Development Process
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and implementing regulations. Our products must be approved by the FDA through the new drug application, or NDA, process before they may be legally marketed in the United States, which generally involves the following:
|
●
|
completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices or other applicable regulations;
|
|
●
|
submission to the FDA of an investigational new drug application, or IND, which must become effective before human clinical trials may begin;
|
|
●
|
performance of adequate and well-controlled human clinical trials according to Good Clinical Practices to establish the safety and efficacy of the proposed drug for its intended use;
|
|
●
|
submission to the FDA of an NDA;
|
|
●
|
satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with cGMP to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and
|
|
●
|
FDA review and approval of the NDA.
|
13
As part of the IND, an IND sponsor must submit to the FDA the results of preclinical tests, which may include laboratory evaluations and animal studies, together with manufacturing information and analytical data, and the proposed clinical protocol for the first phase of the clinical trial of the drug. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, places the clinical trial on a “clinical hold,” pending resolution between the IND sponsor and the FDA of any outstanding concerns. Clinical holds may be imposed by the FDA at any time before or during clinical studies due to safety concerns or non-compliance by the sponsor.
All clinical trials must be conducted under the supervision of a qualified investigator(s) in accordance with good clinical practice regulations. An institutional review board, or IRB, must also review and approve each new clinical protocol and patient informed consent form prior to commencement of the corresponding clinical trial at each institution where a trial is performed. Protocols detail, among other things, the objectives of the study, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor patient safety.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
|
●
|
Phase 1: The drug is introduced into healthy human subjects or patients (in the case of certain inherently toxic products for severe or life-threatening diseases) and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion.
|
|
●
|
Phase 2: Involves studies in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage.
|
|
●
|
Phase 3: Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical study sites. These studies are intended to establish the overall risk-benefit ratio of the product and provide, if appropriate, an adequate basis for product labeling.
|
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events. The FDA or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the study participants are being exposed to an unacceptable health risk.
If a drug is intended to treat a serious or life threatening condition for which there is an unmet medical need, a company may request that the FDA consider the drug for a fast track development program at the time of submitting its IND or at any time prior to receiving marketing approval. The fast track program is designed to facilitate the development and expedite the review of a new drug for the treatment of the specific conditions. If the FDA agrees that the drug meets the criteria for fast track development for treatment of one or more conditions, it will grant fast track status. The FDA granted fast track status to ridaforolimus for treatment of soft tissue sarcomas and bone sarcomas.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition which is defined as one affecting fewer than 200,000 individuals in the United States or more than 200,000 individuals where there is no reasonable expectation that the product development cost will be recovered from product sales in the United States. Orphan drug designation must be requested before submitting an NDA and does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
If an orphan drug-designated product subsequently receives the first FDA approval for the disease for which it was designed, the product will be entitled to seven years of product exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for seven years. If a competitor obtains approval of the same drug, as defined by the FDA, or if our product candidate is determined to be contained within the competitor’s product for the same indication or disease, the competitor’s exclusivity could block the approval of our product candidate in the designated orphan indication for seven years. The FDA granted two orphan drug designations for ridaforolimus; the first for the treatment of soft tissue sarcoma and the second for the treatment of bone sarcoma. The FDA also granted orphan drug designation for AP24534 for the treatment of CML and Philadelphia chromosome-positive acute lymphoblastic leukemia.
14
Special Protocol Assessment
A sponsor of an IND may request that the FDA evaluate within 45 days certain protocols and issues relating to the protocols to assess whether they are adequate to meet scientific and regulatory requirements identified by the sponsor. Such special protocol assessments, or SPAs, may be requested for clinical protocols for Phase 3 trials whose data will form the primary basis for an efficacy claim if the trials had been the subject of discussion at an end-of-Phase 2/pre-Phase 3 meeting with the FDA. If the sponsor and the FDA reach a written agreement regarding the protocol, the SPAs will be considered binding on the FDA and will not be changed unless the sponsor fails to follow the agreed-upon protocol, data supporting the request are found to be false or incomplete, or the FDA determines that a substantial scientific issue essential to determining the safety or effectiveness of the drug was identified after the testing began. Even if a SPA is agreed to, approval of the NDA is not guaranteed since a final determination that an agreed-upon protocol satisfies a specific objective, such as the demonstration of efficacy, or supports an approval decision, will be based on a complete review of all the data in the NDA. We have an SPA with the FDA for our Phase 3 SUCCEED trial.
United States Review and Approval Processes
The results of product development, preclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the drug, proposed labeling, and other relevant information are submitted to the FDA as part of an NDA requesting approval to market the product.
The FDA reviews all NDAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. Once the submission is accepted for filing, the FDA begins an in-depth and substantive review. The FDA may seek advice and a recommendation from an external advisory committee as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee. The FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require submission of additional clinical or other data and information which, upon agency review and interpretation, may or may not be deemed by the FDA to satisfy the criteria for approval. The FDA may also issue a “complete response” letter, which may require additional clinical or other data or impose other conditions that must be met in order to secure final approval of the NDA.
NDAs receive either standard or priority review. A drug representing a significant improvement in treatment, prevention or diagnosis of disease may receive priority review. In addition, products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval and may be approved on the basis of adequate and well-controlled clinical trials establishing that the drug product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials. Priority review and accelerated approval do not change the standards for approval, but may expedite the approval process.
If approved by the FDA, the product’s use may be limited to specific diseases, dosages or indications. In addition, the FDA may require us to conduct post-NDA approval, or Phase 4, testing which involves further nonclinical studies or clinical trials designed to further assess the drug’s safety and effectiveness and may require additional testing and surveillance programs to monitor the safety of the drug in the marketplace.
15
Marketing Exclusivity
Market exclusivity provisions under the FDCA can delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages, or strengths of an existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA; however, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Pediatric exclusivity is another type of exclusivity in the United States. Pediatric exclusivity, if granted, provides an additional six months to an existing exclusivity or statutory delay in approval resulting from a patent certification. This six-month exclusivity, which runs from the end of other exclusivity protection or patent delay, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study. The current pediatric exclusivity provision under the FDCA will expire on October 1, 2012.
Post-Approval Requirements
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval. Manufacturers and other entities involved in the manufacture and distribution of approved FDA-regulated products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies. We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our products. Future FDA and state inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct.
Any drug products manufactured or distributed by us or our partners pursuant to FDA approvals are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the drug, providing the FDA with updated safety and efficacy information, drug sampling and distribution requirements, and complying with FDA promotion and advertising requirements. The FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market. Products may be promoted only for the approved indications and in accordance with the provisions of the approved label.
16
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. For example, the Food and Drug Administration Amendments Act of 2007, or FDAAA, gave the FDA enhanced post-market authority, including the authority to require post-market studies and clinical trials, labeling changes based on new safety information, and compliance with a risk evaluation and mitigation strategy approved by the FDA. Additionally, the law expands the clinical trial registry so that sponsors of all clinical trials, except for Phase I trials, are required to submit certain clinical trial information for inclusion in the clinical trial registry data bank. Failure to comply with any requirements under FDAAA may result in significant penalties. In addition to new legislation, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether further legislative changes will be enacted, or FDA regulations, guidance or interpretations changed or what the impact of such changes, if any, may be. Failure to comply with applicable regulatory requirements can result in enforcement action by the FDA, which may include any of the following sanctions:
|
●
|
warning letters;
|
|
●
|
fines, injunctions, and civil penalties;
|
|
●
|
recall or seizure of our products;
|
|
●
|
operating restrictions, partial suspension or total shutdown of production;
|
|
●
|
refusal to approvals of new products;
|
|
●
|
withdrawal approvals; and
|
|
●
|
criminal prosecution.
|
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
As in the U.S., the European Union may grant orphan drug status for specific indications if the request is made before an application for marketing authorization is made. The European Union considers an orphan medicinal product to be one that affects less than five of every 10,000 people in the European Union. A company whose application for orphan drug designation in the European Union is approved is eligible to receive, among other benefits, regulatory assistance in preparing the marketing application, protocol assistance and reduced application fees. Orphan drugs in the European Union also enjoy economic and marketing benefits, including up to ten years of market exclusivity for the approved indication, unless another applicant can show that its product is safer, more effective or otherwise clinically superior to the orphan designated product. We have been granted orphan designation in the European Union for ridaforolimus for the treatment of soft tissue sarcoma and bone sarcoma, and for AP24534 for the treatment of CML and ALL.
Reimbursement
Sales of pharmaceutical products depend in significant part on the availability of third-party reimbursement. Third-party payors include government health programs such as Medicare and Medicaid, managed care organizations and commercial health insurers. We anticipate that third-party payors, including Medicare, will provide coverage for our products. However, the amount of coverage may not be sufficient to allow us to sell our products on a competitive and profitable basis.
17
We expect that there will continue to be a number of federal and state proposals, including changes to the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or MMA, to implement governmental pricing controls and limit the growth of healthcare costs, including the cost of prescription drugs. While we cannot predict whether such legislative or regulatory proposals will be adopted, the adoption of such proposals could have a material adverse effect on our business, financial condition and profitability.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products.
Our Employees
As of February 28, 2010, we had 149 employees, 81 of whom hold post-graduate medical or science degrees, including 49 with a Ph.D. or M.D. Most of our employees are engaged directly in research and development. We have entered into confidentiality, assignment of inventions and non-disclosure agreements with all of our employees and non-competition agreements with all of our senior level employees. None of our employees are covered by a collective bargaining agreement, and we consider relations with our employees to be good.
Our Company
ARIAD was organized as a Delaware corporation in April 1991. Our principal executive offices are located at 26 Landsdowne Street, Cambridge, Massachusetts 02139-4234, and our telephone number is (617) 494-0400. We maintain an internet website at http://www.ariad.com, the contents of which are not incorporated herein. Our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q and Current Reports on Form 8-K and all amendments to such reports are made available free of charge through www.sec.gov and the Investor Relations section of our website as soon as reasonably practicable after they have been electronically filed with or furnished to the United States Securities and Exchange Commission, or SEC.
ARIAD and the ARIAD logo are our registered trademarks. ARGENT is our trademark. Other service marks, trademarks and trade names appearing in this report are the property of their respective owners.
Merger of ARIAD Gene Therapeutics, Inc. into ARIAD Pharmaceuticals, Inc.
On September 11, 2008, ARIAD Pharmaceuticals, Inc. or ARIAD, and ARIAD Gene Therapeutics, Inc., or AGTI, entered into an agreement pursuant to which AGTI was merged with and into ARIAD on September 12, 2008, with ARIAD as the surviving company. Prior to the merger, AGTI was an 80 percent owned subsidiary of ARIAD. AGTI owned or licensed from others the intellectual property related to our ARGENT technology and know-how, as well as the product candidates developed from the application of this technology, including ridaforolimus. We effectuated the merger to eliminate conflicts of interest between ARIAD and AGTI, to ensure that ARIAD will receive benefits from the successful commercialization of its products proportionate to its investment and to create additional value for our stockholders.
Under the terms of the merger agreement, each outstanding share of AGTI common stock owned by AGTI’s minority stockholders, a total of 1,126,064 AGTI shares, was converted into the right to receive two shares of ARIAD common stock. Under Delaware law, any of the AGTI minority stockholders had the right to demand appraisal of his or her AGTI shares and to seek judicial determination of the fair value of such shares. Four AGTI stockholders holding a total of 226,426 shares of AGTI common stock notified us of their intent to pursue appraisal of their shares. We reached a settlement with such AGTI stockholders in January 2009 pursuant to which these AGTI stockholders received two shares of ARIAD common stock plus approximately $2.43 in cash for each share of AGTI common stock they owned. In total, in exchange for all of the AGTI common stock owned by the AGTI minority stockholders, we issued 2,252,128 shares of ARIAD common stock, or approximately 3.1 percent of the outstanding common stock of ARIAD at the time of the merger, and $550,000 in cash. The total cost of the acquisition of the 20 percent minority interest of AGTI was approximately $5.9 million.
18
THE RISKS AND UNCERTAINTIES DESCRIBED BELOW ARE THOSE THAT WE CURRENTLY BELIEVE MAY MATERIALLY AFFECT OUR COMPANY. IF ANY OF THE FOLLOWING RISKS ACTUALLY OCCUR, THEY MAY MATERIALLY HARM OUR BUSINESS, OUR FINANCIAL CONDITION AND OUR RESULTS OF OPERATIONS.
Risks Relating to Our Business
Based on our current financial resources and liquidity and projected cash requirements to fund our operations, there is substantial risk related to our ability to continue as a going concern.
At December 31, 2009, we had cash and cash equivalents totaling $40.4 million and working capital of $8.2 million, compared to cash, cash equivalents and marketable securities totaling $38.4 million and working capital of $13.5 million at December 31, 2008. For the year ended December 31, 2009, we incurred a net loss of $80.0 million and reported cash used in operating activities of $51.9 million. We expect to continue to incur significant operating expenses and net losses through at least 2011, and therefore, we will require substantial additional funding to support our research and development programs, including preclinical development and clinical trials, for operating expenses including intellectual property protection and enforcement, for the pursuit of regulatory approvals, and for establishing manufacturing, marketing and sales capabilities.
In order to conserve our financial resources and extend our liquidity, since mid-2009, we have reduced, deferred or eliminated previously planned spending on activities and initiatives that are not considered directly related to our highest priority programs and objectives or are committed obligations. These actions have had a positive effect on our financial resources but will not be sufficient by themselves to eliminate our need for additional funding.
We are pursuing various potential sources of additional funding. We are currently in advanced negotiations with Merck regarding the terms of a revised collaboration agreement for the development and commercialization of ridaforolimus. Although there can be no assurance that we will complete negotiation of a revised collaboration agreement with Merck on terms acceptable to us, our current expectation is that the revised agreement will be finalized and executed by the parties in the second quarter of 2010. We are also pursuing partnering opportunities with our earlier stage product candidates, AP24534 and AP26113, which could generate up-front and milestone payments as well as funding of on-going development costs, and other licensing possibilities with our product candidates and technologies.
If we are unable to execute a revised collaboration agreement with Merck on a timely basis, we will need to raise funds from other sources and further reduce our operating expenses in order to continue operating our business. In such circumstances, we may seek to raise funds by issuing common stock or other securities in one or more private placements or public offerings, or through the issuance of debt, or by out-licensing our product candidates and technologies. There can be no assurance that we will be able to raise additional funding from the above sources or enter into license agreements on terms acceptable to us or at all. Depending upon the success of such financing and licensing efforts, our expense reductions could include the discontinuance of most development activities and a substantial reduction in our staff.
If we are not successful in our efforts to secure additional funding as described above or otherwise reduce spending to conserve our cash resources, our cash and cash equivalents as of December 31, 2009 will not be sufficient to fund our operations beyond the second quarter of 2010, and thus there is substantial risk related to our ability to continue as a going concern through 2010.
19
Insufficient funding may jeopardize our research and development programs and may require us to reduce our operations or prevent commercialization of our products and technologies.
We have funded our operations to date through sales of equity securities, debt, the upfront and milestone payments received from Merck since July 2007, and, to a limited extent, operating revenues. Most of our operating revenue to date has been generated through previous collaborative research and development agreements and existing licenses.
Although our collaboration agreement with Merck for the global development and commercialization of ridaforolimus was structured to provide us with substantial funding for the remaining development of ridaforolimus if the collaboration was successful in meeting specified milestones, in 2009 we reported that the expected initiation of certain Phase 3 clinical trials and the associated significant milestone payments from Merck would be delayed for at least one year, negatively impacting a significant source of our funding. We are currently engaged in advanced negotiations with Merck regarding the terms of a revised collaboration agreement. Although there can be no assurance that we will successfully conclude our negotiations with Merck on terms acceptable to us, our current expectation is that a revised agreement will be entered into in the second fiscal quarter of 2010.
We will require substantial additional funding for our other research and development programs (including pre-clinical development and clinical trials), for the pursuit of regulatory approvals and for establishing or accessing manufacturing, marketing and sales capabilities related to our other product candidates, and for other operating expenses (including intellectual property protection and enforcement) as well as capital expenditures to maintain and improve our facility, equipment and systems. We may from time to time access funding by issuing common stock or other securities in private placements or public offerings, including under our universal shelf registration statement under which we currently have $125 million in securities available for issuance. We may also from time to time seek additional funding from other product-based collaborations such as partnering of our product candidates AP24534 and AP26113, technology licensing, or the issuance of debt. However, such additional funding may not be available on terms acceptable to us, or at all. Accordingly, we may not be able to secure the significant funding which is required to maintain our operations or continue to fund current or future research and development programs at their current levels or at levels that may be required in the future. If we cannot secure adequate financing, we may be required to reduce our operations, to delay, scale back, eliminate or terminate clinical trials for one or more of our other research and development programs, or to enter into licenses, settlements or other arrangements with third parties on terms that may be unfavorable to us to purchase, commercialize or otherwise obtain rights in our product candidates, approved products, technologies or intellectual property.
Raising additional capital by issuing securities or through collaboration and licensing arrangements may cause dilution to existing stockholders, restrict our operations or require us to relinquish proprietary rights.
As noted above, we may seek to raise the additional capital necessary to fund our operations through public or private equity offerings, debt financings, and collaboration and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, our stockholders’ ownership interest will be diluted, and the terms of such securities may include liquidation or other preferences that adversely affect our stockholders’ rights. If we raise additional funds through collaboration and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us.
20
We have incurred significant losses to date and may never be profitable.
We have incurred significant losses in each year since our formation in 1991, including a net loss of $80.0 million in 2009, and have an accumulated deficit of $518.6 million through December 31, 2009. Our losses have resulted principally from costs incurred in research and development of our product candidates, including clinical development of ridaforolimus and AP24534, and from general and administrative costs, including costs incurred to prosecute and protect our intellectual property, associated with our operations. It is likely that we will incur significant operating losses for the foreseeable future, as we continue our research and development activities and begin to build a sales and marketing organization in anticipation of obtaining regulatory approval to market ridaforolimus in the United States, which approval may never occur. We currently have no product revenues, limited license revenues and limited commitments for future licensing revenues, and may not be able to generate such revenues in the future. If our losses continue and we and our existing partners or potential future partners are unable to successfully develop, commercialize, manufacture and market our product candidates and/or we are unable to enter into agreements and licenses of our intellectual property, we may never generate sufficient revenues to achieve profitability. Even if we and our partners are able to commercialize products and we are able to enter into agreements or licenses in the future, we may never generate sufficient revenues to have profitable operations.
Significant additional losses or insufficient funding may cause us to default on certain covenants of our loan documents.
At December 31, 2009, we had $11.6 million outstanding under a term loan agreement with a bank, pursuant to which we are required to maintain certain financial and non-financial covenants, including minimum cash, cash equivalents and investments of $15 million, a default of any of which would allow the bank to demand payment of its loan. In addition, a covenant in the loan agreement requires that we not receive an audit report on our annual audited financial statements that includes a “going concern” explanatory paragraph within the audit report. We have obtained a waiver from the bank related to this requirement for the year ended December 31, 2009. Because we are unable to conclude that it is not probable that future covenant violations would not occur within the next twelve months, the entire term loan balance has been classified as a current liability as of December 31, 2009. We currently have sufficient liquidity to fund payment of this loan if demand for payment were made. However, if we are unable to raise adequate financing to fund continuing operations or otherwise to refinance our loan, we may not be able to maintain compliance with loan covenants, may be required to pay off the loan and may be required to reduce our spending on operations.
We have no product candidates that have been approved by the FDA or any foreign regulatory authority, and we and our partners may never succeed in obtaining regulatory approval for any products, developing marketable products or generating product revenues.
We are a biopharmaceutical company focused on the discovery and development of drugs to provide therapeutic intervention in treating human diseases at the cellular level. As with all scientific endeavors, we face much trial and error, and we may fail at numerous stages along the way, which would inhibit us from successfully developing, manufacturing and marketing our drug candidates.
Our lead product candidate, ridaforolimus, is currently being developed by us in collaboration with Merck for potential cancer indications and by our partners, Medinol and ICON, for use in stents or other medical devices to reduce reblockage of injured vessels following interventions in which stents are used in conjunction with balloon angioplasty. Ridaforolimus is currently being studied in a Phase 3 clinical trial in patients with metastatic sarcomas and in multiple Phase 1 and Phase 2 clinical trials in various other cancer indications and in combination with other agents. Our second product candidate, AP24534, is currently being studied in a Phase 1 clinical trial in patients with hematologic malignancies. Our third product candidate, AP26113, is currently in preclinical testing and investigational new drug, or IND, enabling studies. We do not currently have any products on the market and have no product revenues. Therefore, our success is substantially dependent on (1) our ability to work in collaboration with Merck to obtain marketing approval for ridaforolimus for metastatic sarcoma and other cancer indications, (2) the ability of our partners, Medinol and ICON, to obtain marketing approval for stents or other medical devices delivering ridaforolimus, and (3) our ability to successfully complete clinical development and obtain marketing approval for AP24534, AP26113 and our other product candidates, or enter into collaboration agreements for these product candidates on terms favorable to us.
21
Neither we nor our partners have submitted any new drug applications for ridaforolimus, AP24534 or any other product candidate of ours to the FDA or foreign regulatory authorities for marketing approval. Factors which could affect the ability to obtain regulatory approval and to achieve market acceptance and gain market share for ridaforolimus, AP24534 and any other product candidate of ours include, among other factors, product formulation, dose, dosage regimen, the ability to obtain timely and sufficient patient enrollment in clinical trials, the risk of occurrence of adverse side effects in patients participating in clinical trials, the ability to manufacture, directly or indirectly, sufficient quantities of product candidates at commercially reasonable costs, the ability to fund commercial development and to build or access a sales force in the marketplace, the ability to successfully differentiate product candidates from competitive product(s), the ability to educate physicians and build awareness about our product candidates, and the ability to sell, market and distribute, directly or indirectly, such product candidates.
In addition, positive results from early-stage clinical trials may not be replicated in later-stage Phase 2 or Phase 3 clinical trials. Similarly, positive results from preclinical studies of a product candidate may not be predictive of similar results in humans during clinical trials. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials even after achieving promising results in early-stage development. Accordingly, the results from the completed preclinical studies and clinical trials for ridaforolimus may not be predictive of the results to be obtained in the SUCCEED Phase 3 clinical trial, preliminary results of ongoing Phase 2 or completed Phase 1 trials of ridaforolimus may not be predictive of the results obtained in subsequent clinical trials, if conducted, and the promising activity we have seen in AP24534 in preclinical studies and initial results from our Phase 1 clinical trial may not be predictive of the results obtained in the remainder of the Phase 1 clinical trial or in subsequent clinical trials. Furthermore, potential competitive commercial factors may influence future decisions and directions on which clinical indications to pursue and when.
Although we have entered into a collaboration agreement with Merck for the joint global development and commercialization of ridaforolimus, we are currently in advanced negotiations with Merck regarding the terms of a revised collaboration agreement. In addition, we do not currently have any partners to assist in developing and commercializing our other cancer product candidates. We will depend heavily on Merck for the successful development and commercialization of ridaforolimus, particularly with respect to the commercialization of ridaforolimus outside of the United States. We would expect to be dependent upon other partners, if we enter into arrangements with one or more of them, to successfully develop and commercialize our other cancer product candidates, including AP24534. There can be no assurance that we will successfully conclude our negotiations with Merck on terms acceptable to us, that our collaboration with Merck will be successful or that we will be able to secure any other partners on terms favorable to us, or at all.
We and our medical device partners have limited experience in designing, conducting and managing the clinical trials necessary to obtain regulatory approval of drug-eluting stents or other combination products that use a medical device to deliver small-molecule drugs to reduce blockage of injured vessels following interventions in which stents are used in conjunction with balloon angioplasty. We are dependent upon the success of Medinol and ICON and any future medical device partner to successfully develop, manufacture and market stents or other medical devices to deliver ridaforolimus. If Medinol or ICON is not successful and/or if we are not able to enter into an agreement with an additional medical device company experienced in the development, manufacture, and marketing of medical devices to deliver ridaforolimus, we will not be able to generate revenues from the marketing of stents or other medical devices that deliver ridaforolimus.
22
We do not expect to have any products on the market before 2011, at the earliest, and, ultimately, we and our partners may not succeed in developing or commercializing any products which will generate product revenues for our company. If we and our partners are not successful in developing or marketing ridaforolimus or other product candidates, we will not be profitable.
If our collaboration with Merck relating to the development and commercialization of ridaforolimus is unsuccessful, our ability to commercialize ridaforolimus on a timely basis, or at all, could be affected and our business could be materially harmed.
In July 2007, we entered into a collaboration agreement with Merck for the joint global development and commercialization of ridaforolimus, our lead product candidate, for use in cancer. Other than with respect to our collaborative efforts in developing ridaforolimus to date, we do not have a history of working together with Merck and cannot predict the success of this collaboration. The collaboration involves a complex allocation of responsibilities, costs and benefits and provides for milestone payments to us upon the achievement of specified clinical, regulatory and sales milestones. Moreover, as noted above, we are currently engaged in advanced negotiations to revise the collaboration agreement, and there is no assurance that we will successfully conclude those negotiations on terms acceptable to us.
With respect to responsibilities and control over decisions as provided in the collaboration agreement in its current form, we and Merck have established a series of joint committees which are responsible for the development and commercialization of ridaforolimus. Under the committee structure, if the committees are unable to reach a decision, the matter is referred to senior executives of the parties. Each party has ultimate decision making authority with respect to a specified limited set of issues, and for all other issues, the matter must be resolved by consensus of the parties. Accordingly, Merck’s failure to devote sufficient resources to the development and commercialization of ridaforolimus or the failure of the parties to reach consensus on development or commercialization activities for ridaforolimus may delay its clinical development, which could lead to the delay in payment of, or the failure to earn, clinical and regulatory milestones under the collaboration agreement and may delay commercialization of ridaforolimus.
The collaboration agreement provides that, in certain circumstances, either party may opt out of conducting and funding certain late-stage clinical trials, which would result in changes in development and commercialization responsibilities and compensation arrangements. Furthermore, the collaboration agreement may be terminated by Merck (i) based on an uncured breach by us, (ii) on or after the third anniversary of the effective date of the agreement by providing at least 12 months prior written notice, (iii) upon the failure of ridaforolimus to meet certain developmental and safety requirements, or (iv) after discussions between the parties, in the event Merck concludes that it is not advisable to continue the development of ridaforolimus for use in a potential cancer indication. In addition, unrelated to our ridaforolimus collaboration, Merck’s research and development plans may be affected by its corporate, business or other developments, such as its recently completed merger with Schering-Plough Corporation, which may impact the joint development plans for ridaforolimus. Any loss of Merck as a collaborator in the development or commercialization of ridaforolimus, our failure to successfully renegotiate revisions to the collaboration agreement or any dispute over the terms of, or decisions regarding, the collaboration, or any other adverse developments in our relationship with Merck could result in our inability to fully develop or commercialize ridaforolimus, if at all, or could materially harm our business and could accelerate our need for additional capital.
23
We have limited manufacturing experience and are dependent upon the ability of third parties, including Merck, to manufacture our product candidates.
Under our collaboration with Merck, as currently in effect, we are responsible for providing the active pharmaceutical ingredient used in ridaforolimus drug product and Merck is responsible for the formulation of the finished product. Under our agreements with Medinol and ICON, we are responsible for providing the ridaforolimus to be delivered by the stents or medical devices being developed by Medinol and ICON. We have no experience in manufacturing any of our product candidates on a large scale and have contracted and expect to continue to contract with third-party manufacturers, including Merck, to provide material for clinical trials and potential commercial launch, and to assist in the development and optimization of our manufacturing processes and methods. Our ability to conduct clinical trials and commercialize our product candidates will depend on the ability of such third parties to manufacture our products on a large scale at a competitive cost and in accordance with current good manufacturing practices, or cGMPs, and other regulatory requirements. If we are not able to obtain contract manufacturing on commercially reasonable terms, obtain or develop the necessary materials and technologies for manufacturing, or obtain intellectual property rights necessary for manufacturing, or if our contract manufacturers fail to provide us with the quantities and quality of the products we require in a timely manner, we may not be able to conduct or complete clinical trials or commercialize our product candidates, including ridaforolimus. There can be no assurance that we will be able to obtain the materials, technologies and intellectual property necessary to successfully manufacture our product candidates for clinical trials and commercialization.
We have limited experience in conducting clinical trials and are dependent upon the ability of third parties, including Merck, contract research organizations, collaborative academic groups, clinical trial sites and investigators, to conduct or to assist us in conducting clinical trials for our product candidates.
We have limited experience in designing, initiating, conducting and monitoring the clinical trials necessary to obtain regulatory approval of our product candidates. Our collaboration agreement with Merck provides that the development and commercialization of ridaforolimus, our lead product candidate, will be jointly conducted pursuant to a global development plan. Pursuant to the global development plan, we are conducting multiple clinical trials of ridaforolimus in multiple cancer indications. As noted above, we are currently engaged in advanced negotiations with Merck regarding the terms of a revised collaboration agreement. Together with the efforts of Merck, contract research organizations, advisory boards, review committees, collaborative academic groups, clinical trial sites and investigators, we are heavily dependent on our and their ability to successfully initiate, enroll, conduct and monitor our SUCCEED Phase 3 clinical trial and other clinical trials of ridaforolimus, particularly outside the United States. In particular, we are dependent upon the review, advice and/or services of several independent committees, consultants and contractors with respect to protocol design, patient enrollment, data monitoring, radiology review, pathology and drug distribution to clinical trial sites for our SUCCEED trial and other clinical trials of ridaforolimus. We are also dependent upon our ability and the ability of Merck and our contractors to coordinate with us and to timely and accurately collect and report to regulatory authorities worldwide the patient data generated in our SUCCEED trial and other clinical trials of ridaforolimus. We, Merck, and our respective contractors, collaborative academic groups, clinical trial sites or investigators may lack sufficient personnel, technology, expertise, experience or resources to effectively initiate clinical trial sites, recruit and enroll patients, conduct and monitor clinical trials, and to collect and report patient data relating to our SUCCEED trial or other clinical trials of ridaforolimus, either generally or in specific countries.
We are conducting a Phase 1 clinical trial of AP24534 in patients with hematologic malignancies. We do not currently have a partner for the development and commercialization of AP24534 and are dependent upon our ability and/or the ability of our contractors, collaborative academic groups, clinical trial sites and investigators, to successfully design, initiate, conduct and monitor clinical trials of AP24534, including the ongoing Phase 1 trial. Failure by us or our partners, contractors, collaborative academic groups, clinical trial sites or investigators to timely and effectively initiate, conduct and monitor our clinical trials could significantly delay or materially impair our ability to complete clinical trials and/or obtain regulatory approval of ridaforolimus, AP24534 or our other product candidates and, consequently, could delay or materially impair our ability to generate revenues therefrom.
24
We will continue to expend resources on the enforcement and licensing of our NF-κB patent portfolio and may be unable to generate material revenues from these efforts if we are unable to enforce against, or license our NF-κB patents to, pharmaceutical and biotechnology companies.
We are the exclusive licensee of a family of patents, three in the U.S. and one in Europe, including a pioneering U.S. patent covering methods of treating human disease by regulating NF-κB cell-signaling activity, hereinafter referred to as the ’516 Patent, awarded to a team of inventors from The Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology and Harvard University. Dr. David Baltimore, the former president of the California Institute of Technology and one of our consultants and scientific founders, is a lead inventor of the ’516 Patent and a member of the board of directors of Amgen Inc. We have a licensing program to generate revenues from the discovery, development, manufacture and sale of products covered by our NF-κB patent portfolio. These patents have been, and in the future may be, challenged and may be subsequently narrowed, invalidated, declared unenforceable or circumvented, any of which could materially impact our ability to generate licensing revenues from them.
We have been engaged in litigation concerning the ‘516 Patent. In the case against Amgen, on June 1, 2009 the Court of Appeals for the Federal Circuit, or CAFC, affirmed a lower court’s claim interpretation and summary judgment of non-infringement with respect to the Amgen product, Enbrel. The plaintiffs did not file a petition for rehearing in Amgen case. The matter was formally concluded on December 28, 2009 with the withdrawal of all pending motions. No issues of claim validity were decided in that case. In the case against Lilly, a lower court decision in our favor was overturned on appeal by the CAFC based on an adverse decision on a claim validity issue. The CAFC granted the plaintiffs’ subsequent petition requesting en banc rehearing of this matter before the full CAFC. The CAFC heard oral arguments on December 7, 2009 and its ruling on the matter is pending. The ‘516 Patent was also the subject of reexamination at the United States Patent and Trademark Office, or PTO, which issued a final rejection of a number of the claims, including those involved in the Amgen and Lilly cases. An appeal of that decision is pending at the Patent Office Board of Appeals and Interferences.
We cannot provide any assurance that other third parties, who may be infringing our NF-κB patents, will not seek to initiate similar, further proceedings for declaratory relief or reexamination with regard to the ’516 Patent or other NF-κB patents. As exclusive licensee of the ’516 Patent, we are obligated for the costs expended for its prosecution in the PTO, for its enforcement in the above noted litigations and otherwise. If other third parties initiate proceedings with regard to the ‘516 Patents, we may need to expend additional significant capital and management resources pursuing these matters in court or in the PTO. In addition, if no claims which cover commercially significant subject matter survive, we will not realize any further sublicensing revenues based on the ‘516 Patent, and could be liable under certain limited circumstances for litigation costs and potentially attorneys’ fees. Those financial consequences could adversely impact our ability to further our clinical programs and our research and development programs at the current levels or at levels that may be required in the future.
The loss of key members of our scientific and management staff could delay and may prevent the achievement of our research, development and business objectives.
Our performance as a specialized scientific business is substantially dependent on our key officers and members of our scientific staff responsible for areas such as drug development, clinical trials, regulatory affairs, drug discovery, manufacturing, marketing, business development and intellectual property protection and licensing. We also are dependent upon certain of our scientific advisors to assist in formulating our research and development strategy. While we have entered into employment agreements with all of our executive officers, these officers may terminate their employment with us at any time. The loss of, and failure to promptly replace, any member of our management team could significantly delay and may prevent the achievement of our research, development and business objectives.
25
We are dependent upon the ability of our medical device partners to develop, manufacture, test and market stents or other medical devices to deliver ridaforolimus.
We have no experience in the development of medical devices and do not intend ourselves to develop stents or other medical devices to deliver ridaforolimus. Instead, we have granted two licenses (to Medinol and to ICON) and, under those license agreements, we may grant one additional license, under our rights to ridaforolimus to a medical device company for its use in developing and commercializing such medical devices to reduce blockage of injured vessels following stent-assisted angioplasty.
While we expect to supply ridaforolimus to our medical device partners and any additional partner, we will be otherwise dependent upon them to develop and commercialize stents or other medical devices to deliver ridaforolimus. Such medical device partners have varying degrees of scientific, technical, medical and regulatory experience and resources to, directly or through third parties, develop, manufacture, test or market stents or other medical devices to deliver ridaforolimus. Their ability to conduct clinical trials and commercialize such medical devices will be dependent on both the safety profile of their medical devices and ridaforolimus, as well as their ability to manufacture and supply medical devices for clinical trials and marketing purposes and our ability to manufacture and supply ridaforolimus, either directly or through third parties, at a competitive cost and in accordance with cGMPs and other regulatory requirements. Although, under our collaboration with Merck, Merck is responsible for the supply of ridaforolimus as a finished drug product for potential indications covered by the collaboration, we depend upon third-party manufacturers or collaborative partners for the production of ridaforolimus for clinical trials to be conducted by our medical device partners, and we intend to use third-party manufacturers to produce ridaforolimus on a commercial scale, if any partner receives regulatory approval. Our reliance on third-party manufacturers and their potential inability to meet our supply commitments to one or more of our partners could adversely impact the ability of our partners to commercialize stents or other medical devices to deliver ridaforolimus.
We anticipate that our partners will seek to develop and commercialize stents or other medical devices to deliver ridaforolimus that do not infringe third-party patents. However, there can be no assurance that the devices delivering ridaforolimus marketed by our partners will not be subject to third-party claims. Furthermore, the patents issued to us or our partners covering ridaforolimus and/or medical devices, including stents, may be subject to challenge and may be subsequently narrowed, invalidated or circumvented. Any such event would adversely impact the ability of one or more of our partners to market their stents or other medical devices to deliver ridaforolimus.
Our existing license agreements with Medinol and ICON allow either party to terminate its agreement with us under certain circumstances, including our partner’s reasonable business judgment that development of a medical device to deliver ridaforolimus is not feasible. Medinol or ICON may be unable to develop a medical device to deliver ridaforolimus and we may not be able to enter into any additional licensing agreements with other medical device companies to develop such devices on terms that are acceptable to us, or at all. Our inability to enter into such arrangements, or the inability of one or more of our partners to develop or commercialize stents or other medical devices to deliver ridaforolimus for any reason, will adversely impact our ability to generate revenues from any licenses of ridaforolimus.
26
We may not be able to protect our intellectual property relating to our research programs, technologies and product candidates.
We and our licensors have issued patents and pending patent applications covering research methods useful in drug discovery, new chemical compounds discovered in our drug discovery programs including, among others, ridaforolimus, certain components, configurations and uses of our cell-signaling regulation technologies and products-in-development, methods and materials for manufacturing our products-in-development and other pharmaceutical products and methods and materials for conducting pharmaceutical research. Pending patent applications may not issue as patents and may not issue in all countries in which we develop, manufacture or sell our products or in countries where others develop, manufacture and sell products using our technologies. In addition, patents issued to us or our licensors may be challenged, as is the case with the PTO proceeding and the Lilly and Amgen litigations regarding the NF-κB ’516 Patent, and they may be subsequently narrowed, invalidated or circumvented. In that event, such patents may not afford meaningful protection for our technologies or product candidates, which would materially impact our ability to develop and market our product candidates and to generate licensing revenues from our patent portfolio. Certain technologies utilized in our research and development programs are already in the public domain. Moreover, a number of our competitors have developed technologies, filed patent applications or obtained patents on technologies, compositions and methods of use that are related to our business and may cover or conflict with our patent applications, technologies or product candidates. Such conflicts could limit the scope of the patents that we may be able to obtain or may result in the denial of our patent applications. If a third party were to obtain intellectual property protection for any of the foregoing, we may be required to challenge such protection, terminate or modify our programs impacted by such protection or obtain licenses from such third parties, which might not be available on acceptable terms, or at all. Also, if a third party were to introduce a product into the market which we believe infringes our patents, we may be required to enforce our patent rights or seek to obtain an injunction or other relief, which could be time-consuming and expensive.
We may be unable to develop or commercialize our product candidates if we are unable to obtain or maintain certain licenses on commercial terms or at all.
We have entered, and will continue to enter, into agreements with third parties to test compounds, blood and tissue samples, to perform gene expression analysis and to develop biological tests for use with our product candidates, which testing may yield new inventions and discoveries requiring us to obtain licenses in order to exclusively develop or market new products, alone or in combination with our product candidates, or to develop or market our product candidates for new indications. We have also entered into license agreements for some of our technologies. We use third parties to test blood and tissue samples and other biological materials in our clinical programs and to develop biological tests, with respect to which we may be required to obtain licenses or pay royalties or other fees in order to commercialize such tests for use with our product candidates. We also use gene sequences or proteins encoded by those sequences and other biological materials in each of our research programs which are, or may become, patented by others and to which we would be required to obtain licenses in order to develop or market our product candidates. Manufacturing and/or use of our products may also require licensing biological materials, technologies and intellectual property from third parties. Our inability to obtain any one or more of these licenses, on commercially reasonable terms, or at all, or to circumvent the need for any such license, could cause significant delays and cost increases and materially affect our ability to develop and commercialize or prevent us from developing and commercializing our product candidates. Obtaining licenses for these discoveries, materials and technologies may require us to make cumulative royalty payments or other payments to several third parties, potentially reducing amounts paid to us or making the cost of our products commercially prohibitive.
Some of our licenses obligate us to exercise diligence in pursuing the development of product candidates, to make specified milestone payments and to pay royalties. In some instances, we are responsible for the costs of filing and prosecuting patent applications and actions to enforce our rights against infringers. These licenses generally expire upon the earlier of a fixed term of years after the date of the license or the expiration of the applicable patents, but each license is also terminable by the other party upon default by us of our obligations. Our inability or failure to meet our diligence requirements or make any payments required under these licenses would result in a reversion to the licensor of the rights granted which, with respect to the licenses pursuant to which we have obtained exclusive rights, would materially and adversely affect our ability to develop and market products based on our licensed technologies.
27
Competing technologies may render some or all of our programs or future products noncompetitive or obsolete.
Many well-known pharmaceutical, healthcare and biotechnology companies, academic and research institutions and government agencies, which have substantially greater capital, research and development capabilities and experience than us or our potential partners, are presently engaged in one or more of the following activities:
|
●
|
developing products based on cell signaling, cancer biology, and computational chemistry;
|
|
●
|
conducting research and development programs for the treatment of the various potential disease indications in which we are focused; and
|
|
●
|
manufacturing, promoting, marketing and selling pharmaceutical or medical device products for treatment of diseases in all of the various disease indications in which we or our current or possible future partners are focused.
|
Some of these entities already have competitive products on the market or product candidates in clinical trials or in more advanced preclinical studies than we do. Many of these entities also have substantially greater research, development, manufacturing and marketing resources and experience than us. In particular, we are aware that Pfizer and Novartis have mTOR inhibitors on the market which are competitive with ridaforolimus, our lead product candidate. Decisions taken by either of these parties regarding clinical initiatives, including Phase 3 trials, of their mTOR inhibitors may impact on or block the clinical and commercial opportunities available to us and Merck for ridaforolimus. Additionally, PharmaMar has a marine derived antitumoral agent currently approved for the treatment of soft tissue sarcomas in Europe and Takeda Pharmaceutical Co., Ltd. has mifamurtide, an immunotherapy product approved for treatment of bone sarcomas in Europe. By virtue of having or introducing competitive products on the market before us, these entities may gain a competitive advantage. Competing technologies may render some or all of our programs or future products noncompetitive or obsolete, and we may not be able to make the enhancements to our technology necessary to compete successfully with newly emerging technologies. Competing products on the market or in development may also lead us and our collaborators to revise or cease development of our product candidates in one or more indications for commercial reasons, even where clinical data may be promising. If we are unable to successfully compete in our chosen markets, we will not become profitable.
If our product candidates are not accepted by patients, physicians and insurers, we will not be successful.
Our success is dependent on the acceptance of any approved products. Our product candidates may not achieve market acceptance among patients, physicians or third-party payors, even if we obtain necessary regulatory and reimbursement approvals. Physicians and health care payors may conclude that any of our product candidates are not as safe and/or effective as competing therapies or are not as attractive based on a cost/benefit analysis as alternative treatments. For example, physicians may elect not to prescribe our drugs, and patients may elect not to request or take them, for a variety of reasons including: lower demonstrated clinical safety and efficacy compared to other drugs; prevalence and severity of adverse side effects; lack of cost-effectiveness; lack of reimbursement availability from third-party payors; a decision to wait for the approval of other therapies that have significant perceived advantages over our drug candidates; convenience and ease of administration; other potential advantages of alternative treatment methods; and ineffective marketing and distribution support. Failure to achieve significant market acceptance of our product candidates, or to be paid and adequate amount for our product candidates, will harm our business. We believe that recommendations by physicians and health care payors will be essential for market acceptance of any product candidates. If our product candidates are approved and fail to achieve market acceptance, we will not be able to generate significant revenues.
28
If we are unable to establish sales, marketing and distribution capabilities or to enter into agreements with third parties to do so, we may be unable to successfully market and sell any products, even if we are able to obtain regulatory approval.
Pursuant to our collaboration agreement with Merck as currently in effect, we will distribute, sell and with Merck co-promote ridaforolimus for all cancer indications in the United States, and Merck will distribute, sell and promote ridaforolimus outside the United States. We are currently establishing a commercial oncology organization, but we have no experience in marketing or selling any products or with respect to pricing and obtaining adequate third-party reimbursement for drugs. In order to market ridaforolimus in the United States if it is approved, we are building a marketing organization and will need to build a specialized sales force, which requires substantial efforts and significant management and financial resources. While we intend to stage our commitments to the extent possible in consideration of the development timelines, in order to support an effective launch of ridaforomlimus, we will need to make significant financial commitments to our marketing organization prior to receiving regulatory approval. We will need to devote significant effort, in particular, to recruiting individuals with experience in the sales and marketing of pharmaceutical products. Competition for personnel with these skills is very high and may be particularly difficult for us since ridaforolimus is still an investigational drug candidate and we will be competing with companies that are currently marketing approved, successful drugs. Accordingly, we may be unable to successfully, directly or indirectly, sell ridaforolimus or any other product candidates that we obtain marketing approval to sell. If we are unable to effectively sell our products, our ability to generate revenues will be materially adversely affected. We may not be able to hire, in a timely manner, the qualified sales and marketing personnel we need, if at all. In addition, we may not be able to enter into any marketing or distribution agreements on acceptable terms, if at all. If we cannot establish sales, marketing and distribution capabilities as we intend, either by developing our own capabilities or entering into agreements with third parties, sales of future products, if any, may be harmed.
If we develop a product for commercial use, a subsequent product liability-related claim or recall could have an adverse effect on our business.
Our business exposes us to potential product liability risks inherent in the testing, manufacturing and marketing of pharmaceutical products. Prior to obtaining regulatory approval to market our products, we or our partners are required to test such products in human clinical trials at health care institutions pursuant to agreements which indemnify such institutions in case of harm caused to patients by our products. We may not be able to avoid significant product liability exposure resulting from use of our products. A product liability-related claim or recall could be detrimental to our business. In addition, except for insurance covering product use in our clinical trials, we do not currently have any product liability insurance, and we may not be able to obtain or maintain such insurance on acceptable terms, or we may not be able to obtain any insurance to provide adequate coverage against potential liabilities, including liabilities arising from our clinical trials. Our inability to obtain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims could prevent or limit the commercialization of any products that we or our partners may develop.
Risks Relating to Regulatory Approvals, Pricing and Reimbursement
We have limited experience in conducting clinical trials, which may cause delays in commencing and completing clinical trials of our product candidates.
Clinical trials must meet applicable FDA and foreign regulatory requirements. We have limited experience in designing, conducting and managing the preclinical studies and clinical trials necessary to obtain regulatory approval for our product candidates in any country and no experience in conducting and managing post-approval studies of any products. We or our collaborative partners may encounter problems in clinical trials that may cause us or the FDA or foreign regulatory agencies to delay, suspend or terminate our clinical trials at any phase. These problems could include the possibility that we may not be able to manufacture sufficient quantities of cGMP materials for use in our clinical trials, conduct clinical trials at our preferred sites, enroll a sufficient number of patients for our clinical trials at one or more sites, or begin or successfully complete clinical trials in a timely fashion, if at all. Furthermore, we, our partners, the FDA or foreign regulatory agencies may suspend clinical trials of our product candidates at any time if we or they believe the subjects participating in the trials are being exposed to unacceptable health risks, whether as a result of adverse events occurring in our trials or otherwise, or if we or they find deficiencies in the clinical trial process or conduct of the investigation. If clinical trials of any of our product candidates fail, we or our partners will not be able to market the product candidate which is the subject of the failed clinical trials. The FDA and foreign regulatory agencies could also require additional clinical trials before or after granting of marketing approval for any products, which would result in increased costs and significant delays in the development and commercialization of such products and could result in the withdrawal of such products from the market after obtaining marketing approval. Our failure, or the failure of our partners, to adequately demonstrate the safety and efficacy of a product candidate in clinical development could delay or prevent obtaining marketing approval of the product candidate and, after obtaining marketing approval, data from post-approval studies could result in the product being withdrawn from the market, either of which would likely have a material adverse effect on our business.
29
We may not be able to obtain government regulatory approval to market our product candidates.
To date, neither we nor our partners have submitted a marketing application for any of our product candidates to the FDA or any foreign regulatory agency, and none of our product candidates has been approved for commercialization in any country. Prior to commercialization, each product candidate will be subject to an extensive and lengthy governmental regulatory approval process in the United States and in other countries. We or our partners may not be able to obtain regulatory approval for any product candidates, or even if approval is obtained, the labeling for such products may place restrictions on their use that could materially impact the marketability and profitability of the product subject to such restrictions. Satisfaction of these regulatory requirements, which includes satisfying the FDA and foreign regulatory authorities that the product is both safe and effective for its intended uses, typically takes several years or more depending upon the type, complexity, novelty and safety profile of the product and requires the expenditure of substantial resources. Uncertainty with respect to meeting the regulatory requirements governing our product candidates may result in excessive costs or extensive delays in the regulatory approval process, adding to the already lengthy review process. If regulatory approval of a product is granted, such approval will be limited to those disease states and conditions for which the product is proven safe and effective, as demonstrated by clinical trials, and may not include all of the indications necessary to successfully market the product. Even though we have obtained orphan drug designation from the FDA and EMEA for ridaforolimus in bone and soft-tissue sarcomas and for AP24534 in certain indications, the designation-related marketing exclusivity periods may be challenged by others or may prove to be of no practical benefit. In addition, even though we have reached agreement on a Special Protocol Assessment, or SPA, with the FDA with respect to our SUCCEED Phase 3 trial of ridaforolimus for metastatic sarcoma, the FDA is not obligated to approve ridaforolimus as a result of the SPA, even if the clinical outcome of the SUCCEED trial is positive. Therefore, we cannot provide assurance that positive results in the SUCCEED trial will be sufficient for FDA approval of ridaforolimus.
We will not be able to sell our product candidates if we, Merck or our third-party manufacturers fail to comply with FDA manufacturing and quality requirements.
Under our collaboration with Merck, we are responsible for providing the active pharmaceutical ingredient used in ridaforolimus drug product, and Merck will be responsible for the formulation of the finished drug product. Under our agreements with Medinol and ICON, we are responsible for providing the ridaforolimus to be delivered by the stents or other medical devices being developed by Medinol and ICON. Before approving any of our product candidates, the FDA will inspect the facility or facilities at which the drug product is manufactured and will not approve the drug candidate unless it is satisfied with our or our third-party manufacturer’s compliance with manufacturing and quality requirements. The manufacturing of our product candidates must comply with cGMP requirements of the FDA and similar requirements of regulatory agencies in other countries. These requirements govern, among other things, quality control and documentation procedures. We, Merck or any third-party manufacturer of product candidates, may not be able to comply with these requirements, which would prevent us from obtaining approval for or selling such products. Material changes to the manufacturing processes of products after approvals have been granted are also subject to review and approval by the FDA or other regulatory agencies. Following approval, such facilities are subject to continuing FDA and foreign regulatory inspections and failure to comply with cGMPs or similar regulations can result in regulatory action up to and including cessation of shipment of product.
30
Even if we or our partners bring products to market, we or they may be unable to effectively price the products or obtain adequate reimbursement for sales of the products, which would prevent the products from becoming profitable.
If we or our partners succeed in bringing any product candidates to the market, they may not be considered cost-effective, and coverage and adequate payments may not be available or may not be sufficient to allow us to sell such products on a competitive basis. In both the United States and elsewhere, sales of medical products and the availability or acceptance of treatments are dependent, in part, on the availability of reimbursement from third-party payors, such as health maintenance organizations and other commercial insurance plans and governmental programs such as Medicare. Third-party payors are increasingly challenging the prices charged for pharmaceutical products and medical procedures. Our business may be affected by the efforts of government and third-party payors to contain or reduce the cost of health care through various means. In the United States, there have been and will continue to be a number of federal and state proposals to implement government controls on pricing. Similar government pricing controls exist in varying degrees in other countries. In addition, the emphasis on managed care in the United States has increased and will continue to increase the pressure on the pricing of pharmaceutical products. We cannot predict whether any legislative or regulatory proposals will be adopted or the effect these proposals or managed care efforts may have on our business.
On February 17, 2009, President Obama signed into law the American Recovery and Reinvestment Act of 2009. This law provides funding for the federal government to compare the effectiveness of different treatments for the same illness. A plan for the research will be developed by the Department of Health and Human Services, the Agency for Healthcare Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures will be made to Congress. Although the results of the comparative effectiveness studies are not intended to mandate any policies for public or private payors, it is not clear what if any effect the research will have on the sales of our product candidates if any such product or the condition that it is intended to treat is the subject of a study. Decreases in third-party reimbursement for our product candidates or a decision by a third-party payor to not cover our product candidates could reduce physician usage of the product candidate and have a material adverse effect on our sales, results of operations and financial condition.
In addition, President Obama has been advocating change in the way in which healthcare is provided in the United States, and several proposals intended to achieve this goal are being actively debated by Congress. It is not clear how such changes, if enacted into law, would affect the commercialization of our product candidates. We cannot predict whether any other legislative or regulatory proposals will be adopted or the effect these proposals or healthcare reform efforts may have on our business.
31
Each of our product candidates will remain subject to ongoing regulatory review even if it receives marketing approval. If we or our collaborators or contractors fail to comply with continuing regulations, we or they may be subject to enforcement action that could adversely affect us.
We and our collaborators and contractors will continue to be subject to pervasive regulation by the FDA and other regulatory authorities even after our product candidates become approved products. We and our collaborators and contractors will continue to be subject to FDA requirements governing among other things the manufacture, packaging, sale, promotion adverse event reporting, storage and recordkeeping of our approved products. The Commissioner of the FDA, who was appointed in calendar year 2009, has put FDA-regulated entities on notice that they should expect to see more enforcement actions in all areas regulated by the FDA. Although we have not received any notice that we are the subject of any such enforcement action, it is possible that we may be in the future and that could have a material adverse effect on our business. We or any applicable collaborator of ours may be slow to adapt, or may never adapt, to changes in existing regulatory requirements or adoption of new regulatory requirements.
If we or any applicable collaborator of ours fails to comply with the requirements of the FDA and other applicable U.S. or foreign governmental or regulatory authorities or previously unknown problems with our products, manufacturers or manufacturing processes are discovered, we or the collaborator could be subject to administrative or judicially imposed sanctions, including:
|
●
|
restrictions on the products, manufacturers or manufacturing processes;
|
|
●
|
warning letters;
|
|
●
|
civil or criminal penalties;
|
|
●
|
fines;
|
|
●
|
injunctions;
|
|
●
|
product seizures or detentions;
|
|
●
|
import bans;
|
|
●
|
voluntary or mandatory product recalls and publicity requirements;
|
|
●
|
suspension or withdrawal of regulatory approvals;
|
|
●
|
total or partial suspension of production; and
|
|
●
|
refusal to approve pending applications for marketing approval of new drugs or supplements to approved applications.
|
Risks Relating to Our Common Stock
Results of our operations, general market conditions for biotechnology stocks and other factors could result in a sudden change in the value of our stock.
As a biopharmaceutical company with no products currently on the market, we continue to experience significant volatility in the price of our common stock. In 2009, our stock price ranged from a high of $3.48 to a low of $0.83. Factors that contribute to such volatility may include: announcements regarding results and timing of preclinical studies and clinical trials for our product candidates; announcements regarding our collaborations and partnerships, including developments or changes in our collaboration with Merck; evidence of the safety or efficacy of our product candidates; announcements regarding product developments or regulatory approvals obtained by companies developing competing products; decisions by regulatory agencies that impact or may impact our product candidates; the results and timing of efforts by our partner or future partners to develop stents or other medical devices to deliver ridaforolimus; announcements of new collaborations or failure to enter into collaborations; our funding resources and requirements; announcements of new equity or debt financings; announcements of technological innovations or new therapeutic product candidates; developments relating to intellectual property rights, including licensing, litigation and governmental regulation and, in particular, our ongoing patent litigation and PTO reexamination proceedings with respect to the ‘516 Patent; healthcare or cost-containment legislation; general market trends for the biotechnology industry and related high-technology industries; the impact of exchange rates for the U.S. dollar; the impact of changing interest rates and policies of the Federal Reserve; and public policy pronouncements. These and other factors could have a significant impact on the value and volatility of our common stock in future periods.
32
Anti-takeover provisions of Delaware law, provisions in our charter and bylaws and our stockholders’ rights plan, or poison pill, could make a third-party acquisition of us difficult.
Because we are a Delaware corporation, the anti-takeover provisions of Delaware law could make it more difficult for a third party to acquire control of us, even if the change in control would be beneficial to stockholders. We are subject to the provisions of Section 203 of the General Corporation Law of Delaware, which prohibits us from engaging in certain business combinations, unless the business combination is approved in a prescribed manner. In addition, our certificate of incorporation and our bylaws, each as currently in effect, also contain certain provisions that may make a third-party acquisition of us difficult, including:
|
●
|
a classified board of directors, with three classes of directors each serving a staggered three-year term;
|
|
●
|
the ability of the board of directors to issue preferred stock; and
|
|
●
|
the inability of our stockholders to call a special meeting.
|
We also have implemented a stockholders’ rights plan, also called a poison pill, which could make it uneconomical for a third party to acquire our company on a hostile basis. These provisions, as well as Section 203, may discourage certain types of transactions in which our stockholders might otherwise receive a premium for their shares over the current market price, and may limit the ability of our stockholders to approve transactions that they think may be in their best interests.
33
None.
We have leased approximately 100,000 square feet of laboratory and office space at 26 Landsdowne Street, Cambridge, Massachusetts through July 2012, with two consecutive five-year renewal options. We believe that our currently leased facility will be adequate for our research and development and other business activities at least through the year 2011. We believe that any additional space we may require will be available on commercially reasonable terms.
We are from time to time involved in legal proceedings regarding patent, contract and other matters. Legal proceedings are summarized below and described more fully in Note 16 of the Notes to Consolidated Financial Statements included elsewhere in this Annual Report on Form 10-K, which is incorporated herein by reference.
Certain of our patents are the subject of a patent infringement lawsuit filed in June 2002 in the U.S. District Court in Massachusetts by us and others against Eli Lilly & Company. One of these patents is also the subject of reexamination proceedings in the PTO.
A shareholder derivative complaint was filed in the Delaware Court of Chancery in February 2009 by a stockholder alleging breaches of fiduciary duties and naming each of the members of our board of directors as a defendant and ARIAD as a nominal defendant. On February 22, 2010, the Court dismissed the complaint with prejudice.
34
|
MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
|
Market Information
Our common stock is traded on The NASDAQ Global Market under the symbol “ARIA”. The following table sets forth the high and low sales prices of our common stock as quoted on The NASDAQ Global Market for the periods indicated.
|
2009:
|
High
|
Low
|
||||||
|
First Quarter
|
$ | 2.95 | $ | 0.83 | ||||
|
Second Quarter
|
1.93 | 1.15 | ||||||
|
Third Quarter
|
3.48 | 1.46 | ||||||
|
Fourth Quarter
|
2.69 | 1.70 | ||||||
|
2008:
|
||||||||
|
First Quarter
|
$ | 4.48 | $ | 2.66 | ||||
|
Second Quarter
|
3.72 | 2.37 | ||||||
|
Third Quarter
|
3.55 | 2.10 | ||||||
|
Fourth Quarter
|
2.49 | 0.72 | ||||||
On March 12, 2010, the last reported sale price of our common stock was $3.23.
Stock Performance Graph
The following graph compares the yearly percentage change in the cumulative total stockholder return on our common stock since December 31, 2004, with the total cumulative return of the NASDAQ Biotechnology Index and the Russell 2000® Index, each of which ARIAD is a member. The Russell 2000 Index is a market capitalization-weighted index of stock price performance for the 2,000 smallest companies in the Russell 3000® Index. Since the Russell 2000 Index is specifically designed to measure the stock price trends of smaller companies, we believe it is a meaningful index against which to compare our stock price performance.
The price of a share of common stock is based upon the closing price per share as quoted on The NASDAQ Global Market on the last trading day of the year shown. The graph lines merely connect year-end values and do not reflect fluctuations between those dates. The comparison assumes $100 was invested on December 31, 2004 in our common stock and in each of the foregoing indices. We did not declare or pay any dividends during the comparison period. The stock price performance as shown in the graph below is not necessarily indicative of future stock price performance.
35
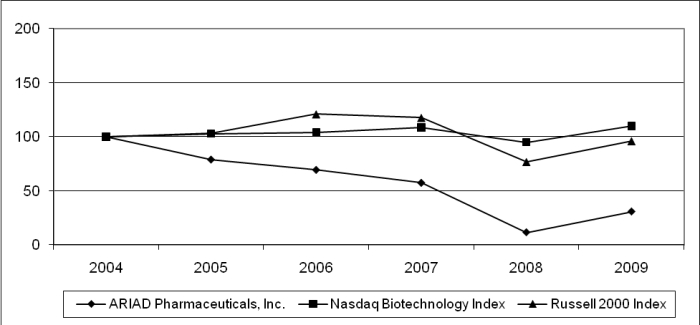
|
2004
|
2005
|
2006
|
2007
|
2008
|
2009
|
|||||||||||||||||||
|
ARIAD Pharmaceuticals, Inc.
|
100.00 | 78.73 | 69.18 | 57.20 | 11.44 | 30.69 | ||||||||||||||||||
|
NASDAQ Biotechnology Index
|
100.00 | 102.84 | 103.89 | 108.65 | 94.93 | 109.77 | ||||||||||||||||||
|
Russell 2000 Index
|
100.00 | 103.32 | 120.89 | 117.57 | 76.65 | 95.98 | ||||||||||||||||||
The performance graph shall not be deemed to be incorporated by reference by means of any general statement incorporating by reference this Form 10-K into any filing under the Securities Act of 1933, as amended or the Securities Exchange Act of 1934, as amended, except to the extent that we specifically incorporate such information by reference, and shall not otherwise be deemed filed under such acts.
Stockholders
As of February 28, 2010, the approximate number of holders of record of our common stock was 470, and the approximate total number of beneficial holders of our common stock was 49,000.
Dividends
We have not declared or paid dividends on our common stock in the past and do not intend to declare or pay such dividends in the foreseeable future. Our long-term debt agreement prohibits the payment of cash dividends.
Unregistered Sales of Securities
Not applicable.
Issuer Purchases of Equity Securities
Not applicable.
36
The selected financial data set forth below as of December 31, 2009, 2008, 2007, 2006 and 2005 and for each of the years then ended have been derived from the audited consolidated financial statements of the Company, of which the financial statements as of December 31, 2009 and 2008 and for the years ended December 31, 2009, 2008 and 2007 are included elsewhere in this Annual Report on Form 10-K, and are qualified by reference to such financial statements. The information set forth below should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the audited consolidated financial statements, and the notes thereto, and other financial information included herein. The report of our independent auditors on our consolidated financial statements as of December 31, 2009 expresses an unqualified opinion on such consolidated financial statements and includes an explanatory paragraph relating to substantial doubt about our ability to continue as a going concern.
|
Years Ended December 31,
|
||||||||||||||||||||
|
In thousands, except share and per share data
|
2009
|
2008
|
2007
|
2006
|
2005
|
|||||||||||||||
| Consolidated Statements of Operations Data: | ||||||||||||||||||||
|
License and collaboration revenue
|
$ | 8,302 | $ | 7,082 | $ | 3,583 | $ | 896 | $ | 1,217 | ||||||||||
| Operating expenses: | ||||||||||||||||||||
|
Research and development
|
63,447 | 50,841 | 39,565 | 43,312 | 45,916 | |||||||||||||||
|
General and administrative
|
16,888 | 28,092 | 24,712 | 21,251 | 12,261 | |||||||||||||||
|
Operating expenses
|
80,335 | 78,933 | 64,277 | 64,563 | 58,177 | |||||||||||||||
|
Loss from operations
|
(72,033 | ) | (71,851 | ) | (60,694 | ) | (63,667 | ) | (56,960 | ) | ||||||||||
|
Other income (expense):
|
||||||||||||||||||||
|
Interest income
|
116 | 1,349 | 2,509 | 2,222 | 1,900 | |||||||||||||||
|
Interest expense
|
(287 | ) | (550 | ) | (337 | ) | (483 | ) | (422 | ) | ||||||||||
|
Revaluation of warrant liability
|
(7,804 | ) | --- | --- | --- | --- | ||||||||||||||
|
Other income (expense), net
|
(7,975 | ) | 799 | 2,172 | 1,739 | 1,478 | ||||||||||||||
|
Net loss
|
$ | (80,008 | ) | $ | (71,052 | ) | $ | (58,522 | ) | $ | (61,928 | ) | $ | (55,482 | ) | |||||
|
Net loss per share
|
$ | (0.86 | ) | $ | (1.02 | ) | $ | (0.86 | ) | $ | (0.99 | ) | $ | (0.99 | ) | |||||
|
Weighted average number of shares of common stock outstanding
|
93,330,308 | 69,790,784 | 68,215,803 | 62,679,807 | 56,283,948 | |||||||||||||||
|
As of December 31,
|
||||||||||||||||||||
|
In thousands
|
2009
|
2008
|
2007
|
2006
|
2005
|
|||||||||||||||
|
Consolidated Balance Sheet Data:
|
||||||||||||||||||||
|
Cash, cash equivalents and marketable securities
|
$ | 40,362 | $ | 38,369 | $ | 84,499 | $ | 39,476 | $ | 81,188 | ||||||||||
|
Working capital
|
8,212 | 13,475 | 63,892 | 25,531 | 65,643 | |||||||||||||||
|
Total assets
|
65,010 | 68,188 | 101,105 | 51,043 | 96,174 | |||||||||||||||
|
Total deferred revenue
|
111,611 | 97,264 | 85,845 | 454 | 875 | |||||||||||||||
|
Long-term debt and capital lease obligations
|
142 | 11,622 | 0 | 3,815 | 5,735 | |||||||||||||||
|
Accumulated deficit
|
(518,608 | ) | (438,600 | ) | (367,549 | ) | (309,026 | ) | (247,098 | ) | ||||||||||
|
Stockholders’ equity (deficit)
|
(89,016 | ) | (69,198 | ) | (7,900 | ) | 30,262 | 71,378 | ||||||||||||
37
|
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
|
The information set forth below should be read in conjunction with the audited consolidated financial statements, and the notes thereto, and other financial information included herein. The report of our independent auditors on our consolidated financial statements as of December 31, 2009 expresses an unqualified opinion on such consolidated financial statements and includes an explanatory paragraph related to substantial doubt about our ability to continue as a going concern.
Overview
Our vision is to transform the lives of cancer patients with breakthrough medicines. Our mission is to discover, develop and commercialize small-molecule drugs to treat cancer in patients with the greatest and most urgent unmet medical need – aggressive cancers where current therapies are inadequate. Our goal is to build a fully integrated oncology company focused on novel, molecularly targeted therapies to treat solid tumors and hematologic cancers, as well as the spread of primary tumors to distant sites.
Product Development and Discovery
Our lead cancer product candidate, ridaforolimus, previously known as deforolimus, is being studied in multiple clinical trials in patients with various types of cancers. In July 2007, we entered into a global collaboration with Merck & Co., Inc., or Merck, to jointly develop and commercialize ridaforolimus for use in cancer. We initiated patient enrollment in our initial Phase 3 clinical trial of ridaforolimus in patients with metastatic sarcoma in the third quarter of 2007. We completed patient enrollment in this Phase 3 clinical trial in the fourth quarter of 2009. We expect to obtain the results of the second interim analysis of progression-free survival or PFS, the primary endpoint of the trial in the second quarter of 2010 and the final analysis of PFS in the second half of 2010. In addition, in 2008 and 2009 we and Merck initiated patient enrollment in Phase 2 clinical trials in patients with metastatic breast cancer, metastatic endometrial cancer, metastatic non-small-cell lung cancer and advanced prostate cancer, and Phase 1 clinical trials of ridaforolimus in combination with other agents, all as part of our joint global development plan with Merck. These various trials are ongoing at this time.
Our collaboration with Merck for the global development and commercialization of ridaforolimus anticipates that we together with Merck will conduct a broad-based development program in multiple potential indications. The collaboration agreement provides that each party will fund 50 percent of global development costs, except for certain specific costs to be funded 100 percent by Merck. The collaboration agreement establishes responsibilities for supply of the product for development and commercial purposes, promotion, distribution and sales of the product, governance of the collaboration, termination provisions and other matters.
In addition to cost-sharing provisions, the collaboration agreement provided for an up-front payment by Merck of $75 million, which was paid to us in July 2007, and currently provides up to $452 million in milestone payments based on the successful development of ridaforolimus in multiple potential cancer indications, of which $53.5 million has been paid to us through December 31, 2009, and up to $200 million in milestone payments based on achievement of specified product sales thresholds. The upfront payment and milestone payments, when earned by us and paid by Merck, are non-refundable. Merck has also agreed to provide us with up to $200 million in interest-bearing, repayable, development cost advances to cover a portion of our share of global development costs, after we have paid $150 million in global development costs and have obtained regulatory approval to market ridaforolimus from the FDA in the United States or similar regulatory authorities in Europe or Japan. The collaboration agreement provides that each party will receive 50 percent of the profit from the sales of ridaforolimus in the United States, and Merck will pay us tiered double-digit royalties on sales of ridaforolimus outside the United States.
Although our collaboration agreement with Merck for the global development and commercialization of ridaforolimus was structured to provide us with substantial funding for the remaining development of ridaforolimus if the collaboration was successful in meeting specified milestones, in 2009 we experienced significant shortfalls in anticipated funding when the expected initiation of certain Phase 3 clinical trials and the associated significant milestone payments from Merck were delayed. We are currently engaged in advanced negotiations with Merck regarding the terms of a revised collaboration agreement. Although there can be no assurance that we will reach agreement with respect to such revisions on terms acceptable to us, our current expectation is that a revised agreement will be entered into in the second fiscal quarter of 2010. If we do not finalize such a revised agreement as currently anticipated, we will seek to raise the additional capital necessary to fund our operations through equity offerings, debt financings and pursuit of other strategic options, and reduce our operating expenses.
38
Our second product candidate, AP24534, is an investigational pan BCR-ABL inhibitor for which we initiated a Phase 1 clinical trial in the second quarter of 2008 in patients with chronic myeloid leukemia, or CML, acute myeloid leukemia, or AML, and other hematologic cancers, which is on-going at this time. Pending further analysis of the results of this trial and discussions with regulatory authorities, and subject to available funding, we believe that we will be able to proceed to a registration trial of this product candidate in the second half of 2010.
In the second quarter of 2009, we designated our third product candidate, AP26113, an investigational anaplastic lymphoma kinase, or ALK, inhibitor, as a development candidate. We have commenced preclinical testing and investigational new drug, or IND, enabling studies of this product candidate.
In addition to our lead development programs, we have a focused drug discovery program centered on small-molecule, molecularly targeted therapies and cell-signaling pathways implicated in cancer. We also have an exclusive license to a family of patents, three in the United States and one in Europe, including a pioneering U.S. patent covering methods of treating human disease by regulating NF-κB cell-signaling activity. Additionally, we have developed a proprietary portfolio of cell-signaling regulation technologies, our ARGENT technology, to control intracellular processes with small molecules, which may be useful in the development of therapeutic vaccines and gene and cell therapy products and which provide versatile tools for applications in cell biology, functional genomics and drug discovery research.
Liquidity and Sources and Uses of Funding
As of December 31, 2009, we had cash and cash equivalents of $40.4 million, working capital of $8.2 million, deferred revenue of $111.6 million related to non-refundable up-front and milestone payments from Merck, and total stockholders’ deficit of $89.0 million. We expect to continue to incur significant operating expenses and net losses through at least 2011, and, therefore, we will require substantial additional funding to support our operations both in the short term and longer term. The Company is pursuing various potential sources of additional funding. We are currently engaged in advanced negotiations with Merck regarding the terms of a revised collaboration agreement for the development and commercialization of ridaforolimus. Although there can be no assurance that the parties will reach agreement with respect to such revisions, we expect that a revised agreement will be entered into in the second quarter of 2010. We are also pursuing partnering opportunities with our earlier stage product candidates, AP24534 and AP26113, which could generate up-front and milestone payments as well as funding of on-going development costs, and other licensing possibilities with our technologies. If we are unable to execute a revised collaboration with Merck on a timely basis, we will seek to raise funding by issuing common stock or other securities in a private placement or a public offering, or through the issuance of debt, and reduce our operating expenses.
If we are not successful in our efforts to secure additional funding as described above or otherwise reduce spending to conserve our cash resources, our cash and cash equivalents as of December 31, 2009 will not be sufficient to fund our operations beyond the second quarter of 2010, and thus there is substantial uncertainty about our ability to continue as a going concern through 2010.
39
Our operating losses are primarily due to the costs associated with our pharmaceutical product development programs, personnel and intellectual property protection and enforcement. As our product development programs progress, we incur significant costs for toxicology and pharmacology studies, product development, manufacturing, clinical trials and regulatory support. We also incur costs related to planning for potential regulatory approval and commercial launch of products, including market research and assessment. These costs can vary significantly from quarter to quarter depending on the number of product candidates in development, the stage of development of each product candidate, the number of patients enrolled in and complexity of clinical trials and other factors. Costs associated with our intellectual property include legal fees and other costs to prosecute, maintain, protect and enforce our intellectual property, which can fluctuate from quarter to quarter depending on the status of patent issues being pursued, including our on-going patent litigation.
Historically, we have relied primarily on the capital markets as our source of funding. We may also obtain funding from collaborations with pharmaceutical, biotechnology and/or medical device companies for development and commercialization of our product candidates, such as our collaboration with Merck for the global development and commercialization of ridaforolimus. These collaborations can take the form of licensing arrangements, co-development or joint venture arrangements or other structures. In addition, we utilize long-term debt and leases to supplement our funding, particularly as a means of funding investment in property and equipment and infrastructure needs. If funding from these various sources is unavailable on reasonable terms, we may be required to reduce our operations in order to conserve cash and capital by delaying, scaling back or eliminating one or more of our product development programs or enter into licenses or other arrangements with third parties on terms that may be unfavorable to us. Please see additional information under the caption “Liquidity and Capital Resources” below.
Each of our potential sources of funding is subject to numerous risks and uncertainties, and there is no assurance that such funding will become available in 2010, or at all, as discussed further in the “Risk Factors” in Part I, Item 1A of this Annual Report on Form 10-K for the year ended December 31, 2009.
Critical Accounting Policies and Estimates
Our financial position and results of operations are affected by subjective and complex judgments, particularly in the areas of revenue recognition, the carrying value of intangible assets, stock-based compensation and the fair value of warrants to purchase our common stock.
Revenue Recognition
For the year ended December 31, 2009, we reported license and collaboration revenue of $8.3 million. License and collaboration revenue is recorded based on up-front payments, periodic license payments and milestone payments received or deemed probable of receipt, spread over the estimated performance period of the license or collaboration agreement. Regarding our collaboration with Merck for the development and commercialization of ridaforolimus, as of December 31, 2009, we have received an up-front payment of $75 million and milestone payments of $53.5 million related to the start of Phase 2 and Phase 3 clinical trials of ridaforolimus. We are recognizing revenues related to such payments on a straight-line basis through 2023, the estimated patent life of the underlying technology. Changes in estimated performance periods, including changes in patent lives of underlying technology, could impact the rate of revenue recognition in any period. Such changes in revenue could have a material impact on our statement of operations.
Intangible Assets
At December 31, 2009, we reported $9.6 million of intangible assets, consisting of capitalized costs related primarily to purchased and issued patents, patent applications and licenses and the recorded value of the technology associated with our acquisition in September 2008 of the 20-percent minority interest of ARIAD Gene Therapeutics, Inc. that we did not previously own, net of accumulated amortization. The carrying value of these intangible assets is evaluated for possible impairment, and losses are recorded when the analysis indicates that the carrying value is not recoverable. This analysis involves estimates of future net cash flows expected to be generated by the asset. Such estimates require judgment regarding future events and probabilities. Changes in these estimates, including decisions to discontinue using the technologies, could result in material changes to our balance sheet and statements of operations. If we were to abandon the ongoing development of the underlying product candidates or technologies or terminate our efforts to pursue collaborations or license agreements, we may be required to write off a portion of the carrying value of our intangible assets. The net book value as of December 31, 2009 of intangible assets related to our NF-κB technology is $306,000. If the patentability of our NF-κB patents, one of which is currently the subject of litigation and reexamination proceedings, is successfully challenged and such patents are subsequently narrowed, invalidated or circumvented, we may be required to write off some or all of the net book value related to such technology.
40
Stock-Based Compensation
In determining expense related to stock-based compensation, we utilize the Black-Scholes option valuation model to estimate the fair value of stock options granted to employees, consultants and directors. Application of the Black-Scholes option valuation model requires the use of factors such as the market value and volatility of our common stock, a risk-free discount rate and an estimate of the life of the option contract. Fluctuations in these factors can result in adjustments to our statements of operations. If, for example, the volatility of our common stock, or the expected life of stock options granted during the year ended December 31, 2009 were 10% higher or lower than used in the valuation of such stock options, our valuation of, and total stock-based compensation expense to be recognized for, such awards would have increased or decreased by up to $94,000, or $54,000 respectively.
Fair Value of Warrants
Warrants to purchase 10,784,024 shares of our common stock, issued on February 25, 2009 in connection with a registered direct offering of 14,378,698 shares of our common stock, are classified as a derivative liability. Accordingly, the fair value of the warrants is recorded on our consolidated balance sheet as a liability, and such fair value is adjusted at each financial reporting date with the adjustment to fair value reflected in our consolidated statement of operations. The fair value of the warrants is determined using the Black-Scholes option valuation model. Fluctuations in the assumptions and factors used in the Black-Scholes model would result in adjustments to the fair value of the warrants reflected on our balance sheet and, therefore, our statement of operations. If, for example, the volatility of our common stock at December 31, 2009 were 10% higher or lower than used in the valuation of such warrants, our valuation of the warrants would have increased or decreased by up to $934,000 with such difference reflected in our statement of operations.
Results of Operations
Years Ended December 31, 2009 and 2008
Revenue
We recognized license and collaboration revenue of $8.3 million for the year ended December 31, 2009, compared to $7.1 million for the year ended December 31, 2008. The increase in license and collaboration revenue was due primarily to an increase in the revenue recognized from the Merck collaboration, based on the non-refundable up-front and milestone payments, totaling $128.5 million, paid by Merck through December 31, 2009, including $22.5 million in milestone payments paid by Merck during the year ended December 31, 2009, in accordance with our revenue recognition policy. We expect that license and collaboration revenue that we recognize will increase in 2010 reflecting the full-year impact of milestone payments received during 2009.
41
Operating Expenses
Research and Development Expenses
Research and development expenses increased by $12.6 million, or 25 percent, to $63.4 million in 2009, compared to $50.8 million in 2008. The research and development process necessary to develop a pharmaceutical product for commercialization is subject to extensive regulation by numerous governmental authorities in the United States and other countries. This process typically takes years to complete and requires the expenditure of substantial resources. Current requirements include:
|
●
|
preclinical toxicology, pharmacology and metabolism studies, as well as in vivo efficacy studies in relevant animal models of disease;
|
|
●
|
manufacturing of drug product for preclinical studies and clinical trials and ultimately for commercial supply;
|
|
●
|
submission of the results of preclinical studies and information regarding manufacturing and control and proposed clinical protocol to the U.S. Food and Drug Administration, or FDA, in an Investigational New Drug application, or IND (or similar filings with regulatory agencies outside the United States);
|
|
●
|
conduct of clinical trials designed to provide data and information regarding the safety and efficacy of the product candidate in humans; and
|
|
●
|
submission of all the results of testing to the FDA in a New Drug Application, or NDA (or similar filings with regulatory agencies outside the United States).
|
Upon approval by the appropriate regulatory authorities, including in some countries approval of product pricing, we may commence commercial marketing and distribution of the product.
We group our research and development, or R&D, expenses into two major categories: direct external expenses and all other R&D expenses. Direct external expenses consist of costs of outside parties to conduct laboratory studies, to develop manufacturing processes and manufacture product candidates, to conduct and manage clinical trials and similar costs related to our clinical and preclinical studies. These costs are accumulated and tracked by product candidate. All other R&D expenses consist of costs to compensate personnel, to purchase lab supplies and services, to lease, operate and maintain our facility, equipment and overhead and similar costs of our research and development efforts. These costs apply to work on our clinical and preclinical candidates as well as our discovery research efforts. These costs have not been tracked by product candidate because the number of product candidates and projects in R&D may vary from time to time and because we utilize internal resources across multiple projects at the same time.
Direct external expenses are further categorized as costs for clinical programs and costs for preclinical programs. Preclinical programs include product candidates undergoing toxicology, pharmacology, metabolism and efficacy studies and manufacturing process development required before testing in humans can begin. Product candidates are designated as clinical programs once we have filed an IND with the FDA, or a similar filing with regulatory agencies outside the United States, for the purpose of commencing clinical trials in humans.
Our R&D expenses for 2009 as compared to 2008 were as follows:
| Year ended December 31, | Increase / | |||||||||||
|
In thousands
|
2009
|
2008
|
(decrease)
|
|||||||||
|
Direct external expenses:
|
||||||||||||
|
Clinical programs
|
$ | 35,406 | $ | 24,168 | $ | 11,238 | ||||||
|
Preclinical programs
|
61 | --- | 61 | |||||||||
|
All other R&D expenses
|
27,980 | 26,673 | 1,307 | |||||||||
| $ | 63,447 | $ | 50,841 | $ | 12,606 | |||||||
In 2009, our clinical programs consisted of our lead product candidate ridaforolimus, for which we initiated clinical development in 2003, and AP24534, for which we initiated clinical development in 2008. The direct external expenses for ridaforolimus reflect our share of such expenses, including our share of Merck’s costs, pursuant to the cost-sharing arrangement of our collaboration with Merck.
Direct external expenses for ridaforolimus were $30.0 million in 2009, an increase of $9.4 million, as compared to 2008, primarily reflecting our share of increases in clinical trial costs ($6.4 million) and manufacturing costs ($2.2 million) and our share of costs for Merck’s services ($1.6 million). Clinical trial costs and contract manufacturing costs increased due primarily to increasing enrollment in our Phase 3 clinical trial of ridaforolimus in patients with metastatic sarcomas and in Phase 2 clinical trials of ridaforolimus in patients with breast cancer, endometrial cancer, prostate cancer and non-small cell lung cancer. Merck’s services provided to the collaboration increased as a result of Merck’s increasing activities in support of clinical trials and other activities for which Merck is responsible. Our direct external expenses for ridaforolimus in 2010 will depend upon the outcome of our current negotiations with Merck regarding the terms of a revised collaboration agreement.
Direct external expenses for our second clinical program, AP24534, were $5.5 million in 2009, an increase of $1.8 million as compared to 2008. The increase is due primarily to an increase in clinical costs of $548,000 and toxicology costs of $1.3 million. Clinical costs increased due to increasing enrollment in our Phase 1 clinical trial in patients with hematologic malignancies. Toxicology costs increased due to the conduct of long-term toxicology studies necessary to support development of this product candidate. Subject to sufficient funding, we expect that our direct external expenses for AP24534 will increase in 2010 as we expect to initiate additional clinical trials for this product candidate, including a registration trial in patients with chronic myeloid leukemia, pending further analysis of the results of the Phase 1 trial and discussions with regulatory authorities.
The direct external expenses incurred in our preclinical program relate to costs for toxicology studies for our third product candidate, AP26113. We incurred no direct external expenses for preclinical programs in 2008. Prior to the designation of our third product candidate, all programs other than clinical programs were designated as discovery research and are included in “all other R&D expenses” in the above table. Subject to sufficient funding, we expect that our direct external expenses for our preclinical program, AP26113, will increase in 2010 as we conduct additional pharmacology, toxicology and other studies designed to support the filing of an IND for this product candidate which we expect in 2011.
All other R&D expenses increased by $1.3 million in 2009 as compared to the corresponding period in 2008. This increase is due to an increase in 2009 in personnel expenses of $3.6 million related to the hiring of additional R&D personnel, primarily in our clinical, regulatory and manufacturing areas to support the expanding development of our product candidates, and an increase in overhead and general expenses of $1.1 million due to increased depreciation and amortization related to capital expenditures, offset in part by a decrease in lab supplies and services of $594,000 and legal and consulting costs of $833,000, due to a focus on cost reduction, and an increase in Merck’s allocated share of our internal expenses under the terms of the collaboration agreement of $1.9 million. All other R&D expenses in 2010 will depend upon the outcome of our current negotiations with Merck regarding the terms of a revised collaboration agreement.
The successful development of our product candidates is uncertain and subject to a number of risks. We cannot be certain that any of our product candidates will prove to be safe and effective or will meet all of the applicable regulatory requirements needed to receive and maintain marketing approval. Data from preclinical studies and clinical trials are susceptible to varying interpretations that could delay, limit or prevent regulatory clearance. We, the FDA or other regulatory authorities may suspend clinical trials at any time if we or they believe that the subjects participating in such trials are being exposed to unacceptable risks or if such regulatory agencies find deficiencies in the conduct of the trials or other problems with our products under development. Delays or rejections may be encountered based on additional governmental regulation, legislation, administrative action or changes in FDA or other regulatory policy during development or the review process. Other risks associated with our product development programs are described in the section entitled “Risk Factors” in Part I, Item 1A of this Annual Report on Form 10-K. Due to these uncertainties, accurate and meaningful estimates of the ultimate cost to bring a product to market, the timing of completion of any of our drug development programs and the period in which material net cash inflows from any of our drug development programs will commence are unavailable.
42
General and Administrative Expenses
General and administrative expenses decreased by $11.2 million, or 40 percent, from $28.1 million in 2008 to $16.9 million in 2009. Professional fees decreased by $11.3 million to $7.6 million in 2009 as compared to $18.9 million in 2008, due primarily to reduced costs related to corporate and commercial development initiatives, including reduced costs related to the development of systems and processes to support growth, and to our patent infringement litigations against Eli Lilly and Company, or Lilly, and Amgen Inc., or Amgen. General and administrative expenses in 2010 will depend upon the outcome of our current negotiations with Merck regarding the terms of a revised collaboration agreement.
Other Income /Expense
Interest Income/Expense
Interest income decreased by 91 percent to $116,000 in 2009 from $1.3 million in 2008, as a result of lower interest yields from our cash equivalents and marketable securities and a lower average balance of funds invested in 2009.
Interest expense decreased by 48 percent to $287,000 in 2009 from $550,000 in 2008, due to lower average loan balances and lower interest rates in 2009.
Revaluation of Warrant Liability
The fair value of our warrant liability at December 31, 2009 was $7.8 million higher than its fair value at its inception in February 2009, resulting in a non-cash charge of $7.8 million for the year ended December 31, 2009. The increase in value of the warrant liability is primarily due to the impact of the increase in the market price of our common stock since inception of the warrant. Potential future increases or decreases in our stock price, or other changes in the factors that impact the valuation of the warrant liability, will result in charges or credits, recognized in our consolidated statement of operations in future periods. Such charges or credits will not have any impact on our cash balances, current liquidity or cash flows from operations.
Operating Results
We reported a loss from operations of $72.0 million in 2009 compared to a loss from operations of $71.9 million in 2008, an increase in loss of $182,000, or less than 1 percent. The increase in loss from operations is largely due to the net impact of increases in R&D expenses and decreases in general and administrative expenses described above. We also reported a net loss of $80.0 million in 2009 compared to a net loss of $71.1 million in 2008, an increase in net loss of $8.9 million or 13 percent, and a net loss per share of $0.86 and $1.02, in 2009 and 2008, respectively. The increase in net loss is largely due to the revaluation of our warrant liability described above. The decrease in net loss per share is largely due to the increase in the weighted average number of shares of common stock outstanding as a result of sales of common stock completed in February 2009 and August 2009. Our results of operations for 2010 will vary from those of 2009, and actual losses will depend on a number of factors, including the outcome of our negotiations with Merck regarding revisions to the collaboration agreement, the progress of our product development programs, the progress of our discovery research programs, the impact of commercial and business development activities, developments in our legal proceedings, and changes in the valuation of our warrant liability, among other factors. The extent of such losses will also depend on the sufficiency of funds on hand or available from time to time, which will influence the amount we will spend on operations and capital expenditures as well as the development timelines for our product candidates.
43
Years Ended December 31, 2008 and 2007
Revenue
We recognized license and collaboration revenue of $7.1 million for the year ended December 31, 2008, compared to $3.6 million for the year ended December 31, 2007. The increase in license and collaboration revenue was due primarily to an increase in the revenue recognized from the Merck collaboration, based on the non-refundable up-front and milestone payments, totaling $106.0 million, paid by Merck through December 31, 2008, in accordance with our revenue recognition policy. We entered into this collaboration with Merck in July 2007, and thus our statement of operations reflects a full year of revenue recognition in 2008 as compared to a partial year in 2007.
Operating Expenses
Research and Development Expenses
Research and development expenses increased by $11.2 million, or 28 percent, to $50.8 million in 2008, compared to $39.6 million in 2007, as follows:
|
Year ended December 31,
|
Increase /
|
|||||||||||
|
In thousands
|
2008
|
2007
|
(decrease)
|
|||||||||
|
Direct external expenses:
|
||||||||||||
|
Clinical programs
|
$ | 24,168 | $ | 10,026 | $ | 14,142 | ||||||
|
Preclinical programs
|
--- | 3,881 | (3,881 | ) | ||||||||
|
All other R&D expenses
|
26,673 | 25,658 | 1,015 | |||||||||
| $ | 50,841 | $ | 39,565 | $ | 11,276 | |||||||
In 2008, our clinical programs consisted of ridaforolimus and AP24534. Prior to 2008, we classified AP24534 as a preclinical program.
Direct external expenses for ridaforolimus were $20.4 million in 2008, an increase of $10.4 million, as compared to the corresponding period in 2007. This increase is due to an increase in clinical trial costs of $9.8 million, costs of non-clinical studies of $3.6 million and manufacturing costs of $1.9 million in 2008 as compared to 2007, offset in part by an increase in Merck’s share of expenses of $10.4 million in 2008. In addition, costs for Merck’s services provided to the collaboration increased by $4.5 million in 2008 as compared to 2007. Clinical trial costs and contract manufacturing costs increased due primarily to increasing enrollment in our Phase 3 clinical trial of ridaforolimus in patients with metastatic sarcomas and initiation of enrollment in 2008 in Phase 2 clinical trials of ridaforolimus in patients with breast cancer and endometrial cancer. Costs of non-clinical studies increased due to the initiation and conduct of toxicology studies of ridaforolimus required to support regulatory filings with the FDA.
Direct external expenses for our second clinical program, AP24534, were $3.7 million in 2008, which consisted primarily of clinical trial costs of $923,000, toxicology costs of $812,000 and contract manufacturing costs of $1.8 million as we initiated enrollment in our first Phase 1 clinical trial of this product candidate.
We incurred no direct external expenses for preclinical programs in 2008 as, during that year, no R&D programs were designated as preclinical programs. All programs other than clinical and preclinical programs are designated as discovery research and are included in “all other R&D expenses” in the above table. Direct external expenses for preclinical programs for the period ended December 31, 2007 relate primarily to costs for toxicology and contract manufacturing studies for AP24534 in support of the filing of the IND for this product candidate in late 2007.
44
All other R&D expenses increased by $1.0 million in 2008 as compared to 2007. This increase is due primarily to an increase in personnel expenses of $3.1 million, related to the hiring of additional R&D personnel and related expenses ($3.9 million) offset in part by reduced stock-based compensation expense ($796,000) resulting from previous year stock-option awards fully vested prior to 2008 and forfeitures in 2008, and an increase in overhead expenses of $1.8 million due to the expiration of a sub-lease agreement for a portion of our office and laboratory facility in July 2007, as well as miscellaneous increases in lab supplies and services and professional services. These variances were offset in part by an increase in Merck’s allocated share of such expenses under the terms of the collaboration agreement of $5.3 million in 2008.
General and Administrative Expenses
General and administrative expenses increased by $3.4 million, or 14 percent, from $24.7 million in 2007 to $28.1 million in 2008. Professional fees increased by $2.1 million to $18.9 million in 2008 as compared to $16.8 million in 2007, due primarily to costs related to corporate and commercial development initiatives, including costs related to the development of systems and processes to support growth, and to our patent infringement litigations against Lilly and Amgen. Personnel and related costs increased by $1.8 million due to an increase in the number of personnel and salary adjustments ($2.1 million), offset in part by reduced stock-based compensation expense ($349,000) resulting from previous year stock-option awards fully vested prior to 2008 and forfeitures in 2008. These increases were partially offset by an increase in Merck’s allocated share of such expenses under the terms of the collaboration agreement.
Interest Income / Expense
Interest income decreased by 46 percent to $1.3 million in 2008 from $2.5 million in 2007, as a result of lower interest yields from our cash equivalents and marketable securities and a lower average balance of funds invested in 2008.
Interest expense increased by 63 percent to $550,000 in 2008 from $337,000 in 2007, due to higher average loan balances in 2008, as a result of our amendment of our loan agreement in March 2008 which, among other things, provided us an additional $10 million in loan proceeds, offset in part by lower interest rates in 2008.
Operating Results
We reported a loss from operations of $71.9 million in 2008 compared to a loss from operations of $60.7 million in 2007, an increase in loss of $11.2 million, or 18 percent. We also reported a net loss of $71.1 million in 2008 compared to a net loss of $58.5 million in 2007, an increase in net loss of $12.5 million or 21 percent, and a net loss per share of $1.02 and $0.86, in 2008 and 2007, respectively. Such increases were due primarily to the net effect of changes in R&D expenses and general and administrative expenses noted above.
45
Selected Quarterly Financial Data
Summarized unaudited quarterly financial data are as follows:
|
2009
|
||||||||||||||||
|
In thousands, except per share amounts
|
First
|
Second
|
Third
|
Fourth
|
||||||||||||
|
Total license and collaboration revenue
|
$ | 1,900 | $ | 2,094 | $ | 2,155 | $ | 2,153 | ||||||||
|
Net loss
|
(20,234 | ) | (20,954 | ) | (20,809 | ) | (18,011 | ) | ||||||||
|
Net loss per share – basic and diluted
|
(0.26 | ) | (0.24 | ) | (0.21 | ) | (0.17 | ) | ||||||||
|
2008
|
||||||||||||||||
|
In thousands, except per share amounts
|
First
|
Second
|
Third
|
Fourth
|
||||||||||||
|
Total license and collaboration revenue
|
$ | 1,495 | $ | 1,450 | $ | 1,536 | $ | 2,601 | ||||||||
|
Net loss
|
(17,011 | ) | (17,267 | ) | (19,993 | ) | (16,781 | ) | ||||||||
|
Net loss per share – basic and diluted
|
(0.25 | ) | (0.25 | ) | (0.29 | ) | (0.24 | ) | ||||||||
Liquidity and Capital Resources
We have financed our operations and investments to date primarily through sales of our common stock to institutional investors, collaborations with pharmaceutical companies and, to a lesser extent, through issuances of our common stock pursuant to our stock option and employee stock purchase plans, supplemented by the issuance of long-term debt. We sell securities and incur debt when the terms of such transactions are deemed favorable to us and as necessary to fund our current and projected cash needs. Our collaboration with Merck for the development and commercialization of ridaforolimus, as currently in effect, provides for funding in the form of up-front and potential milestone payments, as well as the sharing of development costs for ridaforolimus. We seek to balance the level of cash, cash equivalents and marketable securities on hand with our projected needs and to allow us to withstand periods of uncertainty relative to the availability of funding on favorable terms.
Sources of Funds
During the years ended December 31, 2009, 2008 and 2007, our sources of funds were as follows:
|
In thousands
|
2009
|
2008
|
2007
|
|||||||||
|
Sales/issuances of common stock:
|
||||||||||||
|
In common stock offerings
|
$ | 58,370 | $ | --- | $ | 12,300 | ||||||
|
Pursuant to stock option and employee stock purchase plans
|
568 | 385 | 2,071 | |||||||||
|
Proceeds from long-term borrowings
|
--- | 10,505 | --- | |||||||||
|
Up-front payment from Merck, included in cash provided by operating activities
|
--- | --- | 75,000 | |||||||||
| $ | 58,938 | $ | 10,890 | $ | 89,371 | |||||||
The amount of funding we raise through sales of our common stock depends on many factors, including, but not limited to, the status and progress of our product development programs, projected cash needs, availability of funding from other sources, our stock price and the status of the capital markets.
In March 2007, we sold 3,072,393 shares of our common stock to Azimuth Opportunity Ltd., pursuant to an equity financing facility between the parties dated February 14, 2007, at an average purchase price of $4.07 per share. We received aggregate gross proceeds from this sale of $12.5 million, or $12.3 million net of issuance expenses. The equity financing facility expired on September 1, 2008.
46
On February 25, 2009, we sold 14,378,698 shares of our common stock in a registered direct offering to institutional investors, at a purchase price of $1.69 per share, resulting in net proceeds after fees and expenses of $22.8 million. The investors also received warrants to purchase an additional 10,784,024 shares of our common stock exercisable at a price of $2.15 per share in cash or pursuant to the net exercise provisions of the warrants. At the election of the warrant holder, upon certain transactions, including a merger, tender offer or sale of all or substantially all of our assets, the holder may receive cash in exchange for the warrant, in an amount determined by application of the Black-Scholes option valuation model at the time of any such event, if the consideration received by the stockholders from such transaction is less than $2.15 per share. The warrants became exercisable on August 25, 2009 and will expire on February 25, 2012 if not exercised by that date. As of March 15, 2010, no warrants have been exercised.
On August 7, 2009, we sold 21,850,000 shares of our common stock in an underwritten public offering, including 2,850,000 shares of common stock upon exercise by the underwriters of their over-allotment option, at a purchase price of $1.75 per share. Net proceeds of this offering, after underwriting discounts and commissions and direct expenses, were $35.6 million. Following this offering, we had approximately $1.8 million of securities remaining available under its shelf registration statement which expired on February 6, 2010, pursuant to SEC rules.
We have filed shelf registration statements with the SEC, from time to time, to register shares of our common stock or other securities for sale, giving us the opportunity to raise funding when needed or otherwise considered appropriate. On January 11, 2010, we filed a shelf registration statement with the SEC for the issuance of common stock, preferred stock, various series of debt securities and/or warrants or rights to purchase any of such securities, either individually or in units, with a total value of up to $125 million, from time to time at prices and on terms to be determined at the time of any such offering. This filing was declared effective on January 21, 2010.
In March 2008, we amended our existing term loan with a bank. The amendment provided for an increase of $10.5 million in our loan balance to $14.0 million, the extension of the maturity date from March 31, 2008 to March 31, 2013, and changes to the repayment provisions. The amended terms of the loan require us to maintain at least $15.0 million in unrestricted cash, cash equivalents and investments. The agreement also contains certain covenants that restrict additional indebtedness, additional liens, and sales of assets, and dividends, distributions or repurchases of common stock. At December 31, 2009, the balance outstanding on our term loan agreement was $11.6 million which has been classified as a current liability on our balance sheet.
Our up-front payment from Merck of $75 million was received pursuant to our collaboration agreement for the development and commercialization of ridaforolimus. This up-front payment is included in cash provided by operating activities in our consolidated statement of cash flows for the year ended December 31, 2007 but is presented separately in this analysis due to the non-recurring nature of this payment. The agreement also provides for, among other things, the payment by Merck of up to $452 million in development and regulatory milestones during the remaining development of ridaforolimus, including $53.5 million in milestone payments received through December 31, 2009 related to the start of various Phase 2 and the Phase 3 clinical trials, and up to $200 million based on the achievement of specified product sales thresholds. Milestone payments, including the $53.5 million in payments referred to above, are reflected as a reduction of cash used in operating activities in ”Uses of Funds” later in this analysis.
47
Uses of Funds
The primary uses of our cash are to fund our operations and working capital requirements and, to a lesser degree, to repay our long-term debt, to invest in intellectual property and to invest in property and equipment as needed for our business. Our uses of cash during the years ended December 31, 2009, 2008 and 2007 were as follows:
|
In thousands
|
2009
|
2008
|
2007
|
|||||||||
|
Net cash used in (provided by) operating activities
|
$ | 51,904 | $ | 48,251 | $ | (33,988 | ) | |||||
|
Less up-front payment from Merck
|
--- | --- | 75,000 | |||||||||
|
Adjusted net cash used in operating activities
|
51,904 | 48,251 | 41,012 | |||||||||
|
Repayment of long-term borrowings
|
1,400 | 1,370 | 1,920 | |||||||||
|
Investment in intangible assets
|
1,308 | 1,091 | 497 | |||||||||
|
Investment in property and equipment
|
2,198 | 6,651 | 1,346 | |||||||||
| $ | 56,810 | $ | 57,363 | $ | 44,775 | |||||||
Net cash used in (provided by) operating activities is comprised of our net losses, adjusted for non-cash expenses, deferred revenue, including deferrals of the up-front and milestone payments received from Merck, and working capital requirements. Adjusted net cash used in operating activities excludes the favorable impact of the non-recurring, up-front payment from Merck of $75.0 million in the third quarter of 2007 pursuant to our collaboration agreement. As noted above, our net loss increased in 2009 and 2008, due primarily to the increased costs of advancing our product candidates through clinical phases of development and expansion of business and commercial development initiatives and in 2008 patent litigation. Our adjusted net cash used in operating activities as presented above varied from year to year for the same reasons, including the favorable impact of milestone payments received from Merck of $13.5 million in 2007, $17.5 million in 2008 and $22.5 million in 2009. As noted above, subject to available funding, we expect that our net loss will increase in 2010 due to ongoing development of our product candidates; that our investment in intangible assets, consisting of our intellectual property, will increase in 2010 in support of our product development activities; and that our investment in property and equipment will increase in 2010 to support growth of our R&D and general and administrative functions.
Off-Balance Sheet Arrangements
As part of our ongoing business, we do not participate in transactions that generate relationships with unconsolidated entities for financial partnerships, such as entities often referred to as structured finance or special purpose entities which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. As of December 31, 2009, we maintained outstanding letters of credit of $749,000 in accordance with the terms of our long-term lease for our office and laboratory facility and for other purposes.
Contractual Obligations
We have substantial fixed contractual obligations under our long-term debt agreement, operating and capital lease agreements, employment agreements and benefit plans. These non-cancellable contractual obligations were comprised of the following as of December 31, 2009:
|
In thousands
|
Payments Due By Period
|
|||||||||||||||||||
|
Total
|
In 2010
|
2011
through
2013
|
2014
through
2015
|
After
2015
|
||||||||||||||||
|
Long-term debt
|
$ | 11,550 | $ | 1,925 | $ | 9,625 | $ | --- | $ | --- | ||||||||||
|
Leases
|
5,680 | 2,215 | 3,465 | --- | --- | |||||||||||||||
|
Employment agreements
|
10,490 | 6,040 | 4,450 | --- | --- | |||||||||||||||
|
Other long-term obligations
|
3,727 | 655 | 2,409 | 663 | --- | |||||||||||||||
| $ | 31,447 | $ | 10,835 | $ | 19,949 | $ | 663 | $ | --- | |||||||||||
48
Long-term debt consists of scheduled principal payments on such debt. Interest on our long-term debt is based on variable interest rates. Assuming a constant interest rate of 1.70 percent, the interest rate on our debt at December 31, 2009, over the remaining term of the debt, our interest expense would total approximately $401,000 in 2010. If we are not able to maintain compliance with our loan covenants in 2010, we will be required to repay in 2010 the remaining principal balance of the loan, which was $11.6 million at December 31, 2009. In addition, a covenant in the loan agreement requires that we not receive an audit report on our annual audited financial statements that includes a “going concern” explanatory paragraph within the audit report. We have obtained a waiver from the bank related to this requirement for the year ended December 31, 2009. Because we are unable to conclude that it is not probable that future covenant violations would not occur within the next twelve months, the entire term loan balance has been classified as a current liability in our consolidated balance sheet as of December 31, 2009.
Leases consist of payments to be made on our lease for our office and laboratory facility, the term of which extends to July 2012, and on agreements for certain assets acquired under capital leases which expire at various dates into 2013. Employment agreements represent base salary payments under agreements with officers that extend for terms ranging from one to four years. Other long-term obligations are comprised primarily of our obligations under our deferred executive compensation plans.
Liquidity
At December 31, 2009, we had cash and cash equivalents totaling $40.4 million and working capital of $8.2 million, compared to cash, cash equivalents and marketable securities totaling $38.4 million and working capital of $13.5 million at December 31, 2008. For the year ended December 31, 2009, we incurred a net loss of $80.0 million and reported cash used in operating activities of $51.9 million which reflects the favorable impact of $22.5 million in milestone payments received from Merck related to the start of Phase 2 clinical trials of ridaforolimus in patients with advanced prostate cancer and metastatic non-small-cell lung cancer.
On February 25, 2009, we raised net proceeds of $22.8 million from the sale of 14,378,698 shares of our common stock and warrants to purchase 10,784,024 shares of our common stock from our existing shelf registration statement at a price of $1.69 per unit. On August 7, 2009, we raised net proceeds of $35.6 million from an underwritten public offering and sale of 21,850,000 shares of our common stock, including 2,850,000 shares of common stock from the exercise by the underwriters of their over-allotment option, at $1.75 per share. On January 11, 2010, we filed a shelf registration statement with the SEC for the issuance of common stock, preferred stock, various series of debt securities, and/or warrants or rights to purchase any of such securities, either individually or in units, with a total value of up to $125 million, from time to time at prices and on terms to be determined at the time of any such offerings. This filing was declared effective on January 21, 2010.
As of December 31, 2009, we had a warrant liability recorded of $11.4 million which relates to warrants issued in February 2009 that are accounted for as a liability. The warrants are classified as a derivative as the warrants do not qualify to be classified as an equity instrument as a result of the potential for cash settlement of the warrants. Our accounts payable balance is $4.6 million lower at December 31, 2009 than December 31, 2008 as a result of the timing of payments made to our vendors.
Subject to available funding, we expect to continue to incur significant operating expenses and net losses through at least 2011 as we advance our product development programs through clinical trials and non-clinical studies. There are numerous factors that are likely to affect our spending levels, including the terms of a potential revision to the collaboration agreement with Merck for the development and commercialization of ridaforolimus, the extent of clinical trials and other development activities for ridaforolimus in collaboration with Merck, the timing and amount of milestone payments to be received from Merck, the progress of our other product development, discovery research and preclinical programs, including activities related to AP24534 and AP26113, and the impact of potential business development activities, among other factors. In any case, we will require additional funding in 2010 to support our continuing research and development programs and related activities.
49
In recognition of our current financial resources and liquidity, since mid-2009, we have reduced, deferred or eliminated previously planned spending on activities and initiatives that are not considered directly related to our highest priority programs and objectives or are committed obligations. These actions have had a positive effect on our financial resources but will not be sufficient by themselves to eliminate the need to raise additional funding.
We are pursuing various potential sources of additional funding. We are currently in advanced negotiations with Merck regarding the terms of a revised collaboration agreement. Although there can be no assurance that we will ultimately execute a revised collaboration agreement with Merck on terms acceptable to us, our current expectation is that the revised agreement will be entered into in the second quarter of 2010. If we do not finalize such a revised agreement as currently anticipated, we will seek to raise the additional capital necessary to fund our operations through equity offerings, debt financings and pursuit of other strategic options. We are also pursuing partnering opportunities with our earlier stage product candidates, AP24534 and AP26113, which could generate up-front and milestone payments as well as funding of on-going development costs and other licensing possibilities with our technologies.
If we are unable to execute a revised collaboration agreement with Merck on a timely basis, we will need to raise funds from other sources and further reduce our operating expenses in order to continue operating our business. In such circumstances, we may seek to raise funds by issuing common stock or other securities in one or more private placements or public offerings, or through the issuance of debt, or through licensing of our product candidates and other technologies. There can be no assurance that we will be able to raise additional funding from the above sources on terms acceptable to us or at all. Depending upon the success of such financing efforts, our expense reductions could include the discontinuance of most development activities other than those for which we have contractual commitments and a substantial reduction in our staff.
To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interest of our existing stockholders will be diluted, and the terms may include liquidation or other preferences that adversely affect the rights of our stockholders. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring debt, making capital expenditures or declaring dividends. If we raise additional funds through collaborations, strategic alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us.
If we are not successful in our efforts to secure additional funding as described above or otherwise reduce spending to conserve our cash resources, our cash and cash equivalents as of December 31, 2009 will not be sufficient to fund our operations beyond the second quarter of 2010, and thus there is substantial uncertainty about our ability to continue as a going concern.
Recently Adopted or Issued Accounting Pronouncements
In September 2009, the Financial Accounting Standards Board (“FASB”) issued authoritative guidance that modifies the accounting for multiple element revenue arrangements. This guidance requires an entity to allocate revenue to each unit of accounting in multiple deliverable arrangements based on the relative selling price of each deliverable. It also changes the level of evidence of stand-alone selling prices required to separate deliverables by requiring an entity to make its best estimate of the stand-alone selling price of the deliverables when more objective evidence of selling price is not available. Implementation of this guidance is required no later than fiscal years beginning after June 15, 2010 and this guidance may be applied prospectively to new or materially modified arrangements after the effective date or retrospectively. Early application is permitted. This guidance may impact our determination of the separation of deliverables for future arrangements or for material modifications to its current collaboration with Merck.
50
We invest our available funds in accordance with our investment policy to preserve principal, maintain proper liquidity to meet operating needs and maximize yields. Our investment policy specifies credit quality standards for our investments and limits the amount of credit exposure to any single issue, issuer or type of investment.
We invest cash balances in excess of operating requirements in short-term, highly liquid securities, with original maturities of 90 days or less, and money market accounts. Depending on our level of available funds and our expected cash requirements, we may invest a portion of our funds in marketable securities, consisting generally of corporate debt and U.S. government and agency securities. Maturities of our marketable securities are generally limited to periods necessary to fund our liquidity needs and may not in any case exceed three years. These securities are classified as available-for-sale.
Our investments are sensitive to interest rate risk. We believe, however, that the effect, if any, of reasonably possible near-term changes in interest rates on our financial position, results of operations and cash flows generally would not be material due to the current short-term nature of these investments. In particular, at December 31, 2009, because our available funds were invested solely in cash equivalents, our risk of loss due to changes in interest rates is not material.
We record as a liability the fair value of warrants to purchase 10,784,024 shares of our common stock issued to investors in connection with a registered direct offering of our common stock on February 25, 2009. The fair value of this warrant liability is determined using the Black-Scholes option valuation model and is therefore sensitive to changes in the market price and volatility of our common stock among other factors. In the event of a hypothetical 10% increase in the market price ($0.23 based on the market price of our stock at December 31, 2009) of our common stock on which the December 31, 2009 valuation was based, the value would have increased by $1.9 million. Such increase would have been reflected as additional loss on revaluation of the warrant liability in our statement of operations.
At December 31, 2009, we had $11.6 million outstanding under a bank term note which bears interest at prime or, alternatively, LIBOR plus 1.25 to 2.25 percent. This note is sensitive to changes in interest rates. In the event of a hypothetical 10 percent increase in the interest rate on which the loan is based (17.0 basis points at December 31, 2009), we would incur approximately $18,000 of additional interest expense per year based on expected balances over the next twelve months.
Certain Factors That May Affect Future Results of Operations
The SEC encourages companies to disclose forward-looking information so that investors can better understand a company's future prospects and make informed investment decisions. This Annual Report on Form 10-K contains such "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be made directly in this Annual Report, and they may also be made a part of this Annual Report by reference to other documents filed with the SEC, which is known as "incorporation by reference." Such statements in connection with any discussion of future operating or financial performance are identified by use of words such as "may," "anticipate," "estimate," "expect," "project," "intend," "plan," "believe," and other words and terms of similar meaning. Such statements are based on management's expectations and are subject to certain factors, risks and uncertainties that may cause actual results, outcome of events, timing and performance to differ materially from those expressed or implied by such forward-looking statements. These risks include, but are not limited to, the costs associated with our research, development, manufacturing and other activities, the conduct and results of preclinical and clinical studies of our product candidates, difficulties or delays in obtaining regulatory approvals to market products resulting from our development efforts, our reliance on our strategic partners and licensees and other key parties for the successful development, manufacturing and commercialization of products, the adequacy of our capital resources and the availability of additional funding, patent protection and third-party intellectual property claims relating to our and any partner's product candidates, the timing, scope, cost and outcome of legal proceedings, future capital needs, risks related to key employees, markets, economic conditions, prices, reimbursement rates and competition, and other factors. Please also see the discussion under “Risk Factors” in Part I, Item 1A appearing elsewhere in this Annual Report on Form 10-K for more details regarding these and other risks.
In light of these assumptions, risks and uncertainties, the results and events discussed in the forward-looking statements contained in this Annual Report or in any document incorporated by reference might not occur. Stockholders are cautioned not to place undue reliance on the forward-looking statements, which speak only as of the date of this report or the date of the document incorporated by reference in this Annual Report. We are not under any obligation, and we expressly disclaim any obligation, to update or alter any forward-looking statements, whether as a result of new information, future events or otherwise. All subsequent forward-looking statements attributable to us or to any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section.
51
Management’s Report on Internal Control over Financial Reporting
The management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934, as amended. The Company’s internal control system was designed to provide reasonable assurance to the Company’s management and board of directors regarding the preparation and fair presentation of published financial statements. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The Company’s management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2009. In making this assessment, it used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework. Based on our assessment we believe that, as of December 31, 2009, the Company’s internal control over financial reporting is effective based on those criteria.
Deloitte & Touche LLP, the independent registered public accounting firm that audited the Company’s consolidated financial statements, has issued an attestation report on the Company’s internal control over financial reporting as of December 31, 2009, which is included below.
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
ARIAD Pharmaceuticals, Inc.
Cambridge, Massachusetts
We have audited the internal control over financial reporting of ARIAD Pharmaceuticals, Inc. and subsidiaries (the “Company”) as of December 31, 2009, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission. The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed by, or under the supervision of, the company’s principal executive and principal financial officers, or persons performing similar functions, and effected by the company’s board of directors, management, and other personnel to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of the inherent limitations of internal control over financial reporting, including the possibility of collusion or improper management override of controls, material misstatements due to error or fraud may not be prevented or detected on a timely basis. Also, projections of any evaluation of the effectiveness of the internal control over financial reporting to future periods are subject to the risk that the controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2009, based on the criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated financial statements as of and for the year ended December 31, 2009 of the Company and our report dated March 15, 2010 expressed an unqualified opinion on those financial statements and included an explanatory paragraph regarding substantial doubt about the Company’s ability to continue as a going concern.
/s/ DELOITTE & TOUCHE LLP
Boston, Massachusetts
March 15, 2010
52
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
ARIAD Pharmaceuticals, Inc.
Cambridge, Massachusetts
We have audited the accompanying consolidated balance sheets of ARIAD Pharmaceuticals, Inc. and subsidiaries (the “Company”) as of December 31, 2009 and 2008, and the related consolidated statements of operations, stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2009. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such consolidated financial statements present fairly, in all material respects, the financial position of ARIAD Pharmaceuticals, Inc. and subsidiaries as of December 31, 2009 and 2008, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2009, in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements for the year ended December 31, 2009 have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company’s recurring losses from operations and negative operating cash flows raise substantial doubt about its ability to continue as a going concern. Management’s plans concerning these matters are also discussed in Note 1 to the financial statements. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the Company’s internal control over financial reporting as of December 31, 2009, based on the criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 15, 2010 expressed an unqualified opinion on the Company’s internal control over financial reporting.
/s/ DELOITTE & TOUCHE LLP
Boston, Massachusetts
March 15, 2010
53
ARIAD PHARMACEUTICALS, INC. AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
|
December 31,
|
||||||||
|
In thousands, except share and per share data
|
2009
|
2008
|
||||||
|
ASSETS
|
||||||||
|
Current assets:
|
||||||||
|
Cash and cash equivalents
|
$ | 40,362 | $ | 23,544 | ||||
|
Marketable securities (Note 4)
|
--- | 14,825 | ||||||
|
Inventory and other current assets
|
1,951 | 4,055 | ||||||
|
Amounts due under collaboration agreement (Note 2)
|
3,583 | 5,580 | ||||||
|
Total current assets
|
45,896 | 48,004 | ||||||
|
Restricted cash
|
749 | 699 | ||||||
|
Property and equipment, net (Note 5)
|
8,738 | 9,593 | ||||||
|
Intangible and other assets, net (Note 6)
|
9,627 | 9,892 | ||||||
|
Total assets
|
$ | 65,010 | $ | 68,188 | ||||
|
LIABILITIES AND STOCKHOLDERS’ DEFICIT
|
||||||||
|
Current liabilities:
|
||||||||
|
Accounts payable
|
$ | 4,806 | $ | 9,370 | ||||
|
Current portion of long-term debt and capital lease obligations (Note 7)
|
11,669 | 1,470 | ||||||
|
Accrued compensation and benefits
|
1,050 | 817 | ||||||
|
Accrued product development expenses
|
8,072 | 9,936 | ||||||
|
Other accrued expenses
|
2,708 | 3,990 | ||||||
|
Current portion of deferred executive compensation (Note 8)
|
655 | 941 | ||||||
|
Current portion of deferred revenue (Note 2)
|
8,592 | 6,982 | ||||||
|
Other current liabilities
|
132 | --- | ||||||
|
Accrued merger consideration (Note 3)
|
--- | 1,023 | ||||||
|
Total current liabilities
|
37,684 | 34,529 | ||||||
|
Long-term debt and capital lease obligations (Note 7)
|
142 | 11,622 | ||||||
|
Other long-term liabilities
|
454 | --- | ||||||
|
Deferred revenue (Note 2)
|
103,019 | 90,282 | ||||||
|
Deferred executive compensation (Note 8)
|
1,364 | 953 | ||||||
|
Warrant liability
|
11,363 | --- | ||||||
|
Commitments and contingent liabilities (Notes 9 and 16)
|
||||||||
|
Stockholders’ deficit (Notes 10, 12 and 13):
|
||||||||
|
Preferred stock, $.01 par value, authorized 10,000,000 shares, none issued and outstanding
|
||||||||
|
Common stock, $.001 par value, authorized 145,000,000 shares, issued and outstanding 109,042,782 shares in 2009, 71,365,339 shares in 2008
|
109 | 71 | ||||||
|
Additional paid-in capital
|
429,483 | 369,313 | ||||||
|
Accumulated other comprehensive income
|
--- | 18 | ||||||
|
Accumulated deficit
|
(518,608 |
)
|
(438,600 | ) | ||||
|
Total stockholders’ deficit
|
(89,016 |
)
|
(69,198 | ) | ||||
|
Total liabilities and stockholders’ deficit
|
$ | 65,010 | $ | 68,188 | ||||
See notes to consolidated financial statements.
54
ARIAD PHARMACEUTICALS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS
|
Years Ended December 31,
|
||||||||||||
|
In thousands, except share and per share data
|
2009
|
2008
|
2007
|
|||||||||
|
License and collaboration revenue (Note 2)
|
$ | 8,302 | $ | 7,082 | $ | 3,583 | ||||||
|
Operating expenses:
|
||||||||||||
|
Research and development
|
63,447 | 50,841 | 39,565 | |||||||||
|
General and administrative
|
16,888 | 28,092 | 24,712 | |||||||||
|
Operating expenses
|
80,335 | 78,933 | 64,277 | |||||||||
|
Loss from operations
|
(72,033 | ) | (71,851 | ) | (60,694 | ) | ||||||
|
Other income (expense):
|
||||||||||||
|
Interest income
|
116 | 1,349 | 2,509 | |||||||||
|
Interest expense
|
(287 | ) | (550 | ) | (337 | ) | ||||||
|
Revaluation of warrant liability
|
(7,804 | ) | --- | --- | ||||||||
|
Other income (expense), net
|
(7,975 | ) | 799 | 2,172 | ||||||||
|
Net loss
|
$ | (80,008 | ) | $ | (71,052 | ) | $ | (58,522 | ) | |||
|
Net loss per share – basic and diluted
|
$ | (0.86 | ) | $ | (1.02 | ) | $ | (0.86 | ) | |||
|
Weighted average number of shares of common stock outstanding – basic and diluted
|
93,330,308 | 69,790,784 | 68,215,803 | |||||||||
See notes to consolidated financial statements.
55
ARIAD PHARMACEUTICALS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
|
Accumulated
|
||||||||||||||||||||||||
|
Additional
|
Other
|
|||||||||||||||||||||||
|
Common Stock
|
Paid-in
|
Comprehensive
|
Accumulated
|
Stockholders’
|
||||||||||||||||||||
|
In thousands, except share data
|
Shares
|
Amount
|
Capital
|
Income (Loss)
|
Deficit
|
Equity (Deficit)
|
||||||||||||||||||
|
Balance, January 1, 2007
|
65,391,347 | $ | 65 | $ | 339,220 | $ | 3 | $ | (309,026 | ) | $ | 30,262 | ||||||||||||
|
Issuance of common stock, net of issuance costs
|
3,072,393 | 3 | 12,297 | 12,300 | ||||||||||||||||||||
|
Issuance of shares pursuant to ARIAD stock plans
|
777,750 | 1 | 2,070 | 2,071 | ||||||||||||||||||||
|
Stock-based compensation
|
5,989 | 5,989 | ||||||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||
|
Net loss and total comprehensive loss
|
(58,522 | ) | (58,522 | ) | ||||||||||||||||||||
|
Balance, December 31, 2007
|
69,241,490 | 69 | 359,576 | 3 | (367,548 | ) | (7,900 | ) | ||||||||||||||||
|
Issuance of common stock, net of issuance costs
|
||||||||||||||||||||||||
|
Issuance of shares pursuant to ARIAD stock plans
|
324,573 | 385 | 385 | |||||||||||||||||||||
|
Issuance of shares to minority shareholders of AGTI
|
1,799,276 | 2 | 4,601 | 4,603 | ||||||||||||||||||||
|
Stock-based compensation
|
4,751 | 4,751 | ||||||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||
|
Net loss
|
(71,052 | ) | (71,052 | ) | ||||||||||||||||||||
|
Net unrealized gains on marketable securities
|
15 | 15 | ||||||||||||||||||||||
|
Total comprehensive loss
|
(71,037 | ) | ||||||||||||||||||||||
|
Balance, December 31, 2008
|
71,365,339 | 71 | 369,313 | 18 | (438,600 | ) | (69,198 | ) | ||||||||||||||||
|
Issuance of shares pursuant to ARIAD stock plans
|
995,893 | 1 | 567 | 568 | ||||||||||||||||||||
|
Issuance of shares to minority shareholders of AGTI
|
452,852 | 1 | 473 | 474 | ||||||||||||||||||||
|
Issuance of common stock, net of issuance costs
|
36,228,698 | 36 | 58,334 | 58,370 | ||||||||||||||||||||
|
Issuance of warrants
|
(3,559 | ) | (3,559 | ) | ||||||||||||||||||||
|
Stock-based compensation
|
4,355 | 4,355 | ||||||||||||||||||||||
|
Comprehensive loss:
|
||||||||||||||||||||||||
|
Net loss
|
(80,008 | ) | (80,008 | ) | ||||||||||||||||||||
|
Net unrealized gains on marketable securities
|
(18 | ) | (18 | ) | ||||||||||||||||||||
|
Total comprehensive loss
|
(80,026 | ) | ||||||||||||||||||||||
|
Balance, December 31, 2009
|
109,042,782 | $ | 109 | $ | 429,483 | $ | --- | $ | (518,608 | ) | $ | (89,016 | ) | |||||||||||
See notes to consolidated financial statements.
56
ARIAD PHARMACEUTICALS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
Years Ended December 31,
|
||||||||||||
|
In thousands
|
2009
|
2008
|
2007
|
|||||||||
|
Cash flows from operating activities:
|
||||||||||||
|
Net loss
|
$ | (80,008 | ) | $ | (71,052 | ) | $ | (58,522 | ) | |||
|
Adjustments to reconcile net loss to net cash provided by (used in) operating activities:
|
||||||||||||
|
Depreciation and amortization
|
4,219 | 3,016 | 2,307 | |||||||||
|
Accretion of discount on marketable securities
|
(20 | ) | (381 | ) | (798 | ) | ||||||
|
Stock-based compensation
|
4,355 | 4,751 | 5,989 | |||||||||
|
Deferred executive compensation expense
|
1,134 | 402 | 955 | |||||||||
|
Revaluation of warrant liability
|
7,804 | |||||||||||
|
Increase (decrease) from:
|
||||||||||||
|
Inventory and other current assets
|
2,104 | (1,681 | ) | (535 | ) | |||||||
|
Amounts due under collaboration agreement
|
1,997 | (992 | ) | (4,588 | ) | |||||||
|
Other assets
|
(12 | ) | 7 | (9 | ) | |||||||
|
Accounts payable
|
(4,564 | ) | 4,321 | 1,046 | ||||||||
|
Accrued compensation and benefits
|
233 | 294 | 95 | |||||||||
|
Accrued product development expenses
|
(1,864 | ) | 2,649 | 675 | ||||||||
|
Other accrued expenses
|
(1,282 | ) | (341 | ) | 2,490 | |||||||
|
Other liabilities
|
662 | --- | --- | |||||||||
|
Deferred revenue
|
14,347 | 11,419 | 85,391 | |||||||||
|
Deferred executive compensation paid
|
(1,009 | ) | (663 | ) | (508 | ) | ||||||
|
Net cash provided by (used in) operating activities
|
(51,904 | ) | (48,251 | ) | 33,988 | |||||||
|
Cash flows from investing activities:
|
||||||||||||
|
Acquisitions of marketable securities
|
(7,599 | ) | (57,264 | ) | (67,657 | ) | ||||||
|
Proceeds from maturities of marketable securities
|
22,426 | 60,169 | 59,197 | |||||||||
|
Change in restricted cash
|
(50 | ) | --- | (371 | ) | |||||||
|
Investment in property and equipment
|
(2,198 | ) | (6,651 | ) | (1,346 | ) | ||||||
|
Investment in intangible assets
|
(1,308 | ) | (1,091 | ) | (497 | ) | ||||||
|
Net cash provided by (used in) investing activities
|
11,271 | (4,837 | ) | (10,674 | ) | |||||||
|
Cash flows from financing activities:
|
||||||||||||
|
Proceeds from long-term borrowings
|
10,505 | |||||||||||
|
Repayment of long-term borrowings
|
(1,400 | ) | (1,370 | ) | (1,920 | ) | ||||||
|
Proceeds from issuance of common stock, net of issuance costs
|
58,370 | 12,300 | ||||||||||
|
Principal payments under capital lease obligation
|
(87 | ) | (53 | ) | ||||||||
|
Proceeds from issuance of common stock pursuant to
stock option and purchase plans
|
568 | 385 | 2,071 | |||||||||
|
Net cash provided by financing activities
|
57,451 | 9,467 | 12,451 | |||||||||
|
Net increase (decrease) in cash and cash equivalents
|
16,818 | (43,621 | ) | 35,765 | ||||||||
|
Cash and cash equivalents, beginning of year
|
23,544 | 67,165 | 31,400 | |||||||||
|
Cash and cash equivalents, end of year
|
$ | 40,362 | $ | 23,544 | $ | 67,165 | ||||||
|
Interest paid
|
$ | 302 | $ | 511 | $ | 379 | ||||||
|
Supplemental disclosure on non-cash activities:
|
||||||||||||
|
Property and equipment acquired through capital lease
|
$ | 206 | $ | 195 | $ | --- | ||||||
See notes to consolidated financial statements.
57
ARIAD PHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Nature of Business and Summary of Significant Accounting Policies
Nature of Business
ARIAD’s vision is to transform the lives of cancer patients with breakthrough medicines. The Company’s mission is to discover, develop and commercialize small-molecule drugs to treat cancer in patients with the greatest and most urgent unmet medical need – aggressive cancers where current therapies are inadequate. The Company’s goal is to build a fully integrated oncology company focused on novel, molecularly targeted therapies to treat solid tumors and hematologic cancers, as well as the spread of primary tumors to distant sites. The Company’s lead cancer product candidate, ridaforolimus, previously known as deforolimus, is being studied in multiple clinical trials in patients with various types of cancers, including sarcomas, breast cancer, prostate cancer, non-small cell lung cancer and endometrial cancer. The Company entered into a global collaboration in July 2007 with Merck & Co., Inc. (“Merck”) to jointly develop and commercialize ridaforolimus for use in cancer. The Company is currently engaged in active, later-stage negotiations with Merck regarding the terms of a revised collaboration agreement. The Company also has partnerships with two medical device companies to develop and commercialize stents to deliver ridaforolimus to prevent restenosis, or reblockage, of injured vessels following interventions in which stents are used in conjunction with balloon angioplasty. The Company’s second product candidate, AP24534, is in a Phase 1 clinical trial in patients with hematologic cancers. The Company’s third product candidate, AP26113, is in the preclinical stage of development.
The Company has a focused drug discovery program centered on small molecule, molecularly targeted therapies and cell-signaling pathways implicated in cancer. The Company also has an exclusive license to pioneering technology and patents related to certain NF-κB cell-signaling activity, which may be useful in treating certain diseases. Additionally, the Company has developed a proprietary portfolio of cell-signaling regulation technologies, the Company’s ARGENT technology, to control intracellular processes with small molecules, which may be useful in the development of therapeutic vaccines and gene and cell therapy products, and which provide versatile tools for use in cell biology, functional genomics and drug discovery research.
Since its inception, the Company has incurred significant operating losses related to its research and development programs and supporting activities. The Company has funded its losses through the sale of equity securities, debt and cash received pursuant to collaboration agreements, including its collaboration agreement with Merck for the development and commercialization of ridaforolimus. At December 31, 2009, the Company had $40.4 million of cash and cash equivalents.
Subject to available funding, the Company expects to continue to incur significant operating expenses and net losses related to its research and development programs and supporting activities through at least 2011. There are numerous factors that affect the Company’s level of spending on its research and development programs and supporting activities, including the outcome of negotiations with Merck regarding the terms of a revised collaboration agreement, the timing and amount of milestone payments to be received from Merck, the progress of our product development and discovery research programs, including AP24534 and AP26113, and the impact of business development activities, among other factors.
Going Concern Presentation
The Company expects to continue to incur significant operating expenses and net losses through at least 2011 and, therefore, will require substantial additional funding to support its research and development programs, including preclinical development and clinical trials, for operating expenses, including intellectual property protection and enforcement, for the pursuit of regulatory approvals, and for establishing manufacturing, marketing and sales capabilities.
58
The Company is pursuing various potential sources of additional funding. The Company is currently engaged in advanced negotiations with Merck regarding the terms of a revised collaboration agreement for the development and commercialization of ridaforolimus. Although there can be no assurance that the parties will reach agreement with respect to such revisions on terms acceptable to the Company, the Company expects that a revised agreement will be entered into in the second quarter of 2010. The Company is also pursuing partnering opportunities with its earlier stage product candidates, AP24534 and AP26113, which could generate up-front and milestone payments as well as funding of on-going development costs, and other licensing possibilities with its technologies. If the Company is unable to execute a revised collaboration with Merck on a timely basis, it will seek to raise funding through equity offerings, debt financings and pursuit of other strategic options, and reduce operating expenses.
There can be no assurance that the Company will be able to raise additional funding from the above sources on acceptable terms or at all. If the Company is not successful in its efforts to secure additional funding from these or other sources or otherwise reduce spending to conserve cash and capital, its cash and cash equivalents as of December 31, 2009 will not be sufficient to fund its operations beyond the second quarter of 2010. These factors give rise to substantial uncertainty about the Company’s ability to continue as a going concern. The accompanying financial statements have been prepared on the basis of a going concern assumption and do not reflect any adjustments that might result from the outcome of this uncertainty.
Principles of Consolidation
The consolidated financial statements include the accounts of ARIAD Pharmaceuticals, Inc. and its wholly-owned subsidiaries, ARIAD Corporation, ARIAD Pharma S.A. and ARIAD Pharma Ltd. Intercompany accounts and transactions have been eliminated in consolidation. Until September 12, 2008, the consolidated financial statements also included the accounts of ARIAD Gene Therapeutics, Inc. (“AGTI”), an 80 percent owned subsidiary of ARIAD Pharmaceuticals, Inc. which was merged with and into ARIAD Pharmaceuticals, Inc. on that date (see Note 3).
Accounting Estimates
The preparation of the consolidated financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts and disclosure of assets and liabilities at the date of the consolidated financial statements and the reported amounts and disclosure of revenue and expenses during the reporting period. Actual results could differ from those estimates.
Cash Equivalents
Cash equivalents include short-term, highly liquid investments, which consist principally of United States government and agency securities, purchased with remaining maturities of 90 days or less, and money market accounts.
Marketable Securities
The Company has classified its marketable securities as “available-for-sale” and, accordingly, carries such securities at aggregate fair value. The difference between fair value and original cost is reflected as a component of accumulated other comprehensive income. Fair value has been determined based on quoted market prices, in a dealer market, at the closing bid price for each individual security held.
Inventory
Inventory consists of bulk pharmaceutical material to be used for multiple development programs. Inventories are carried at cost using the first-in, first-out method and are charged to research and development expense when consumed. The carrying value of inventory amounted to $130,000 and $1.1 million at December 31, 2009 and 2008, respectively.
59
Restricted Cash
Restricted cash consists of cash balances held as collateral for outstanding letters of credit related to the lease of the Company’s laboratory and office facility and other purposes.
Property and Equipment
Property and equipment are recorded at cost. Depreciation is recorded using the straight-line method over the estimated useful lives of the assets (3 to 10 years). Leasehold improvements and assets under capital leases are amortized over the shorter of their useful lives or lease term using the straight-line method.
Intangible and Other Assets
Intangible and other assets consist primarily of purchased technology and capitalized patent and license costs. The cost of purchased technology, patents and patent applications, costs incurred in filing patents and certain license fees are capitalized. Capitalized costs related to purchased technology are amortized over the estimated useful life of the technology. Capitalized costs related to issued patents are amortized over a period not to exceed seventeen years or the remaining life of the patent, whichever is shorter, using the straight-line method. Capitalized license fees are amortized over the periods to which they relate. In addition, capitalized costs are expensed when it becomes determinable that the related patents, patent applications or technology will not be pursued.
Impairment of Long-Lived Assets
The Company reviews its long-lived assets, including the above-mentioned intangible assets, for impairment when events or changes in circumstances indicate that the carrying amount of a long-lived asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to future net cash flows expected to be generated by the asset. If such assets are considered to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the assets exceeds the fair value of the assets.
Revenue Recognition
The Company generates revenue from license and collaboration agreements with third parties related to use of the Company’s technology and/or development and commercialization of product candidates. Such agreements may provide for payment to the Company of up-front payments, periodic license payments, milestone payments and royalties.
Revenue is recognized when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed and determinable and collection is reasonably assured. Revenue arrangements with multiple elements are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered items. When deliverables are separable, consideration received is allocated to the separate units of accounting based on the fair value of each unit and the appropriate revenue recognition principles are applied to each unit.
Up-front and annual license fees associated with collaboration and license agreements are recorded as deferred revenue upon receipt and recognized as revenue on a systematic basis over the period of time they are earned in accordance with the terms of the agreements. Milestone payments are also recognized as revenue on a systematic basis over the remaining performance period of the agreements, commencing when the milestone has been achieved or is probable of achievement. Royalty payments will be recognized as revenue based on contract terms and reported sales of licensed products, when reported sales are reliably measurable and collectability is reasonably assured.
60
Income Taxes
The Company accounts for income taxes using an asset and liability approach to financial accounting and reporting for income taxes. Deferred income tax assets and liabilities are computed annually for differences between the financial statement basis and the income tax basis of assets and liabilities that will result in taxable or deductible amounts in the future. Such deferred income tax computations are based on enacted tax laws and rates applicable to the years in which the differences are expected to affect taxable income. A valuation allowance is established when it is necessary to reduce deferred income tax assets to the expected realized amounts.
The Company does not recognize a tax benefit unless it is more likely than not that the tax position will be sustained upon examination, including resolution of any related appeals or litigation processes, based on the technical merits of the position. The tax benefit that is recorded for these positions is measured at the largest amount of benefit that is greater than 50 percent likely of being realized upon ultimate settlement.
Segment Reporting
The Company organizes itself into one operating segment reporting to the chief executive officer. No revenues from product sales or services occurred in 2009, 2008 or 2007.
Stock-Based Compensation
The Company awards stock options and other equity-based instruments to its employees, directors and consultants and provides employees the right to purchase common stock (collectively “share-based payments”), pursuant to stockholder approved plans. Compensation cost related to such awards is measured based on the fair value of the instrument on the grant date and is recognized on a straight-line basis over the requisite service period, which generally equals the vesting period. All of the Company’s stock-based compensation is based on grants of equity instruments and no liability awards have been granted.
Executive Compensation Plan
The Company has an unfunded deferred executive compensation plan that defers the payment of annual bonus awards to officers to future periods as specified in each award. The value of the awards is indexed to the value of specified mutual funds. The Company accrues a liability based on the value of the awards ratably over the vesting period (generally four years). The recorded balances of such awards are increased or decreased based on the actual total return and quoted market prices of the specified mutual funds.
Subsequent Events
The Company considers events or transactions that occur after the balance sheet date but before the financial statements are issued to provide additional evidence relative to certain estimates or to identify matters that require additional disclosure.
Reclassifications
Certain amounts in prior period financial statements have been reclassified to conform to the current period presentation. Restricted cash was previously included in cash and cash equivalents on the balance sheet but is now properly reflected as a separate line item on the balance sheet and changes in restricted cash has been reflected within investing activities in the statement of cash flows. The amount was immaterial for all periods presented. In addition, the current and long-term portion of capital lease obligations are now combined with the current and long-term portion of long-term debt. These reclassifications did not change previously reported totals of assets, liabilities or stockholders’ deficit or previously reported net loss.
61
Recently Issued Accounting Pronouncements
In September 2009, the Financial Accounting Standards Board (“FASB”) issued authoritative guidance that modifies the accounting for multiple element revenue arrangements. This guidance requires an entity to allocate revenue to each unit of accounting in multiple deliverable arrangements based on the relative selling price of each deliverable. It also changes the level of evidence of stand-alone selling prices required to separate deliverables by requiring an entity to make its best estimate of the stand-alone selling price of the deliverables when more objective evidence of selling price is not available. Implementation of this guidance is required no later than fiscal years beginning after June 15, 2010 and this guidance may be applied prospectively to new or materially modified arrangements after the effective date or retrospectively. Early application is permitted. This guidance may impact the Company’s determination of the separation of deliverables for future arrangements or material modifications to its current collaboration.
2. Collaboration Agreement with Merck & Co., Inc.
In July 2007, the Company entered into a collaboration agreement with Merck & Co., Inc. (“Merck”) for the joint global development and commercialization of ridaforolimus, the Company’s lead product candidate, for use in cancer (the “Collaboration Agreement”). The Company is currently engaged in advanced negotiations with Merck regarding the terms of a revised collaboration agreement. Although there can be no assurance that the parties will reach agreement with respect to such revisions on terms acceptable to the Company, the Company’s current expectation is that a revised agreement will be entered into in the second fiscal quarter of 2010.
Under the terms of the Collaboration Agreement, as currently in effect, Merck and the Company will conduct a broad-based development program in multiple types of cancer, pursuant to a global development plan agreed upon by the parties. Each party will fund 50 percent of the global development costs, except that Merck will fund 100 percent of any cost of development that is specific to development or commercialization of ridaforolimus outside the United States. The Collaboration Agreement provides that, in certain circumstances, either party may opt out of conducting and funding certain late-stage clinical trials, which would result in changes in development and commercialization responsibilities and compensation arrangements. The Company is responsible for supplying the active pharmaceutical ingredient used in the product and Merck is responsible for the formulation of the finished product, all under a separate supply agreement between the parties entered into in May 2008.
The Collaboration Agreement currently provides that, in the United States, the Company and Merck will co-promote the product, the Company will distribute and sell the product for all cancer indications and record all sales, and each party will receive 50 percent of the profit from such sales. Outside the United States, Merck will distribute, sell and promote the product and book all sales, and Merck will pay the Company tiered double-digit royalties on such sales. Royalties are payable by Merck, on a country by country basis, until the later of (i) the expiration of the last valid claim of any patent rights owned by either the Company or Merck that cover the product, (ii) a specified number of years from first commercial sale, or (iii) the last date upon which the Company supplies the active pharmaceutical ingredient to Merck under the supply agreement, subject to partial reduction in certain circumstances.
Under the terms of the Collaboration Agreement, Merck paid the Company an initial up-front payment of $75 million in July 2007, and has agreed to pay up to $452 million in milestone payments, of which $53.5 million has been paid through December 31, 2009 (including $22.5 million in 2009), based on the successful development of ridaforolimus in multiple potential cancer indications, and up to $200 million in milestone payments based on achievement of specified product sales thresholds. The Company assessed each of the deliverables related to the Collaboration Agreement against the separation criteria for multiple element arrangements and concluded that the deliverables constitute one unit of accounting. The up-front and milestone payments received through December 31, 2009 have been deferred and are being recognized as revenue on a straight-line basis through 2023, the estimated expiration of the patents related to the underlying technology.
62
Under the existing agreement, Merck has also agreed to provide the Company with up to $200 million in interest-bearing, repayable, development cost advances to cover a portion of the Company’s share of global development costs, after the Company has paid $150 million in global development costs and has obtained regulatory approval to market ridaforolimus from the FDA in the United States or similar regulatory authorities in Europe or Japan. All amounts to be paid to the Company by Merck, with the exception of any development cost advances, are non-refundable.
Development costs under the Collaboration Agreement are aggregated and split between the Company and Merck in accordance with the terms of the agreement. The Company’s share of such development costs are reflected in operating expenses in the Company’s statement of operations. Any amounts due to or from Merck in respect of such development costs and milestone payments earned but not received are recorded as such on the Company’s balance sheet. At December 31, 2009, the Company has recorded an amount due from Merck under the collaboration agreement of $3.6 million.
3. Merger of AGTI into ARIAD Pharmaceuticals, Inc.
On September 11, 2008, ARIAD and AGTI entered into a merger agreement, pursuant to which AGTI was merged with and into ARIAD on September 12, 2008, with ARIAD as the surviving company. Prior to the merger, AGTI was an 80 percent owned subsidiary of ARIAD. The minority stockholders of AGTI included the Company’s chairman and chief executive officer, a member of the Board of Directors of the Company, several of the Company’s current and former officers and scientific advisors, Harvard University, and Stanford University. ARIAD effectuated the merger to eliminate conflicts of interest between ARIAD and AGTI, to ensure that ARIAD will receive benefits from the successful commercialization of its products proportionate to its investment and to create additional value for its stockholders.
Under the terms of the merger agreement, each outstanding share of AGTI common stock owned by AGTI’s minority stockholders, a total of 1,126,064 AGTI shares, was converted into the right to receive two shares of ARIAD common stock. Under Delaware law, any of the AGTI minority stockholders had the right to demand appraisal of his or her AGTI shares and to seek judicial determination of the fair value of such shares. Four AGTI stockholders holding a total of 226,426 shares of AGTI common stock notified the Company of their intent to pursue appraisal of their shares. The Company reached a settlement with such AGTI stockholders in January 2009 pursuant to which these AGTI stockholders received two shares of ARIAD common stock plus approximately $2.43 in cash for each share of AGTI common stock they owned. In total, in exchange for all of the AGTI common stock owned by the AGTI minority stockholders, ARIAD issued 2,252,128 shares of ARIAD common stock, or approximately 3.1 percent of the outstanding common stock of ARIAD at the time of the merger, and $550,000 in cash. The total value of the acquisition of the 20 percent minority interest of AGTI was approximately $5.9 million.
The total cost of the acquisition of the 20 percent minority interest of $5.9 million was accounted for using the purchase method of accounting. The cost has been allocated to intangible assets and will be amortized over approximately fifteen years, the remaining life of the patents related to AGTI’s technology. The cost of the settlement reached in January 2009 with the dissenting stockholders of AGTI is reflected in the cost of the intangible asset and was recorded as a liability at December 31, 2008.
4. Marketable Securities
The Company has classified its marketable securities as available-for-sale and, accordingly, carries such securities at aggregate fair value. At December 31, 2008, all of the Company’s marketable securities consisted of United States government agency securities. At December 31, 2009, the Company had no marketable securities. At December 31, 2008, the aggregate fair value and amortized cost of the Company’s marketable securities were $14,825,000 and $14,807,000, respectively. Gross unrealized gains and losses were $18,000 and $0, respectively, at December 31, 2008.
63
Realized gains and losses on investment security transactions are reported on the specific-identification method. There were no realized gains and losses on sales of marketable securities in 2009, 2008 and 2007. Changes in market values resulted in a decrease in net unrealized losses or increase in net unrealized gains of $18,000, $15,000 and $1,000 in 2009, 2008 and 2007, respectively.
5. Property and Equipment, Net
Property and equipment, net, was comprised of the following at December 31:
|
In thousands
|
2009
|
2008
|
||||||
|
Leasehold improvements
|
$ | 22,027 | $ | 22,004 | ||||
|
Equipment and furniture
|
15,542 | 14,991 | ||||||
| 37,569 | 36,995 | |||||||
|
Less accumulated depreciation and amortization
|
(28,831 | ) | (27,402 | ) | ||||
| $ | 8,738 | $ | 9,593 | |||||
Depreciation and amortization expense for the years ended December 31, 2009, 2008 and 2007 was $3.2 million, $2.3 million and $1.4 million, respectively.
The Company leases certain assets under capital leases having terms up to three years. Assets under capital leases included in property and equipment were as follows at December 31:
|
In thousands
|
2009
|
2008
|
||||||
|
Equipment and furniture
|
$ | 401 | $ | 195 | ||||
|
Less accumulated depreciation and amortization
|
(87 | ) | (23 | ) | ||||
| $ | 314 | $ | 172 | |||||
6. Intangible and Other Assets, Net
Intangible and other assets, net, were comprised of the following at December 31:
|
In thousands
|
2009
|
2008
|
||||||
|
Capitalized patent and license costs
|
$ | 11,817 | $ | 11,107 | ||||
|
Purchased technology (see Note 3)
|
5,901 | 5,901 | ||||||
| 17,718 | 17,008 | |||||||
|
Less accumulated amortization
|
(8,127 | ) | (7,142 | ) | ||||
| 9,591 | 9,866 | |||||||
|
Other assets
|
36 | 26 | ||||||
| $ | 9,627 | $ | 9,892 | |||||
Amortization expense for intangible assets amounted to $1,031,000, $749,000 and $851,000 in 2009, 2008 and 2007, respectively. The weighted average amortization period for intangible assets was 15.2 years, 14.8 years and 13.8 years in 2009, 2008 and 2007, respectively. In addition, the Company expensed unamortized patent and license costs of $47,000, $1,000 and $36,000 in 2009, 2008 and 2007, respectively, related to patent applications or technology no longer being pursued. The estimated future amortization expenses for capitalized patent and license costs and purchased technology are $880,000 for 2010, $732,000 for 2011, $732,000 for 2012, $732,000 for 2013 and $732,000 for 2014.
64
7. Long-term Debt and Capital Lease Obligations
Long-term debt and capital lease obligations were comprised of the following at December 31:
|
In thousands
|
2009
|
2008
|
||||||
|
Bank term loan
|
$ | 11,550 | $ | 12,950 | ||||
|
Capital lease obligations
|
261 | 142 | ||||||
| 11,811 | 13,092 | |||||||
|
Less current portion
|
(11,669 | ) | (1,470 | ) | ||||
| $ | 142 | $ | 11,622 | |||||
The term loan provides for quarterly payments of principal and interest with final maturity in 2013. The loan bears interest at LIBOR plus 1.25 to 2.25 percent, depending on the percentage of the Company’s liquid assets on deposit with or invested through the bank, or at the prime rate, as provided in the amendment. The effective interest on the loan was 1.70% at December 31, 2009. The loan is secured by a lien on all assets of the Company excluding intellectual property, which the Company has agreed not to pledge to any other party. The loan, as amended, requires the Company to maintain a minimum of $15.0 million in unrestricted cash, cash equivalents and investments. The loan also contains certain covenants that restrict additional indebtedness, additional liens and sales of assets, and dividends, distributions or repurchases of common stock. In addition, a covenant in the loan agreement requires that the Company not receive an audit report on its annual audited financial statements that includes a “going concern” explanatory paragraph within the audit report. The Company has obtained a waiver from the bank related to this requirement for the year ended December 31, 2009. Because the Company is unable to conclude that it is not probable that future covenant violations would not occur within the next twelve months, the entire term loan balance has been classified as a current liability as of December 31, 2009.
In addition, the Company leases certain equipment under capital leases with original terms of generally three years. These leases have effective interest rates ranging from 5.6% to 7.2% and are secured by the underlying leased asset.
The future principal payments due under these financing obligations, absent any future debt covenant violations that would require early repayment of the loan, were as follows at December 31, 2009:
|
In thousands
|
Bank Term
Loan
|
Capital
Lease
Obligations
|
||||||
|
Year ended December 31:
|
||||||||
|
2010
|
$ | 1,925 | $ | 117 | ||||
|
2011
|
3,675 | 69 | ||||||
|
2012
|
4,725 | 59 | ||||||
|
2013
|
1,225 | 16 | ||||||
| 11,550 | 261 | |||||||
|
Less current portion
|
(11,550 | ) | (119 | ) | ||||
|
Long-term portion
|
$ | --- | $ | 142 | ||||
8. Executive Compensation Plans
Under the Company’s deferred executive compensation plan, the Company accrues a liability for the value of the awards ratably over the vesting period. The value of awards made in 2009, 2008 and 2007 were $1,121,000, $812,000 and $1,535,000, respectively. The net expense for this plan was $1,134,000, $402,000 and $955,000 in 2009, 2008 and 2007, respectively. The estimated future expenses for awards made through December 31, 2009, assuming no change in the value of the underlying mutual funds, are $768,000, $567,000, and $374,000 for 2010, 2011 and 2012, respectively.
65
9. Leases, Licensed Technology and Other Commitments
Facility Lease
The Company conducts its operations in a 100,000 square foot office and laboratory facility under a non-cancelable operating lease. The lease was amended in 2006 and provides that the current lease term extends to July 2012 with two consecutive five-year renewal options. The Company maintains an outstanding letter of credit of $699,000 in accordance with the terms of the amended lease. The Company subleased approximately 31,000 square feet of space to one tenant and such sublease expired in July 2007. Rent expense, net of sublease income of $710,000 in 2007, amounted to $2.1 million, $2.1 million and $1.3 million in 2009, 2008 and 2007, respectively. Future minimum annual rental payments through July 2012 are $2.1 million in 2010, $2.1 million in 2011 and $1.2 million in 2012.
Licensed Technology
The Company has entered into agreements with several universities under the terms of which the Company has received exclusive licenses to technology and intellectual property. The agreements, which are generally cancelable by the Company, provide for the payment of license fees and/or minimum payments, which are generally creditable against future royalties. Fees paid by the Company amounted to $145,000 in each of 2009, 2008 and 2007, and are expected to amount to approximately $145,000 annually in 2010 and thereafter. In addition, the agreements provide for payments upon the achievement of certain milestones in product development. The agreements also require the Company to fund certain costs associated with the filing and prosecution of patent applications.
Other Commitments
The Company has entered into various employment agreements with eighteen officers of the Company. The agreements provide for aggregate annual base salaries of $6.0 million for 2010, $2.5 million for 2011, $1.3 million for 2012, and $608,000 for 2013, and remaining terms of employment of up to four years.
10. Stockholders’ Equity and Warrants
Preferred Stock
The Company has authorized 10,000,000 shares of preferred stock which the Board of Directors is authorized to designate and issue in different series. At December 31, 2009, the Board of Directors had designated 500,000 shares as series A preferred stock for potential issuance under the Company’s stockholder rights plan and 9,500,000 shares remained undesignated.
Common Stock and Warrants
At December 31 2009, the Company had 145,000,000 shares of common stock authorized. On January 20, 2010, following approval at a special meeting of stockholders of the Company, the Company filed a certificate of amendment to its certificate of incorporation to increase the number of authorized shares of common stock of the Company to 240,000,000 shares.
In March 2007, the Company sold 3,072,393 shares of its common stock to Azimuth Opportunity Ltd. pursuant to an equity financing facility between the parties dated February 14, 2007. The Company received aggregate gross proceeds from this sale of $12.5 million, or $12.3 million net of issuance expenses. The equity financing facility expired on September 1, 2008.
66
On February 25, 2009, the Company sold 14,378,698 shares of its common stock in a registered direct offering to institutional investors, at a purchase price of $1.69 per share, resulting in net proceeds after fees and expenses of $22.8 million. The investors also received warrants to purchase an additional 10,784,024 shares of the Company’s common stock exercisable at a price of $2.15 per share in cash or pursuant to the net exercise provisions of the warrants. At the election of the warrant holder, upon certain transactions, including a merger, tender offer or sale of all or substantially all of the assets of the Company, the holder may receive cash in exchange for the warrant, in an amount determined by application of the Black-Scholes option valuation model at the time of any such event, if the consideration received by the stockholders from such transaction is less than $2.15 per share. The warrants became exercisable on August 25, 2009 and will expire on February 25, 2012 if not exercised by that date. No warrants have been exercised through December 31, 2009.
As a result of the potential cash settlement provision, the warrants do not qualify to be classified as an equity instrument but instead are classified as a derivative liability. Accordingly, the fair value of the warrants is reflected on the consolidated balance sheet as a liability and such fair value is adjusted at each financial reporting date with the adjustment reflected in the consolidated statement of operations. The Company has classified the warrant obligation as a long-term liability as there is no indication that a merger, tender offer or similar transaction is probable.
On August 7, 2009, the Company sold 21,850,000 shares of its common stock in an underwritten public offering, including 2,850,000 shares of common stock upon exercise by the underwriters of their over-allotment option, at a purchase price of $1.75 per share. Net proceeds of this offering, after underwriting discounts and commissions and direct expenses, were $35.6 million.
On January 11, 2010, the Company filed a shelf registration statement with the SEC for the issuance of common stock, preferred stock, various series of debt securities and/or warrants or rights to purchase any of such securities, either individually or in units, with a total value of up to $125 million, from time to time at prices and on terms to be determined at the time of any such offering. This filing was declared effective on January 21, 2010.
Stockholder Rights Plan
The Board of Directors of the Company adopted a Rights Agreement, dated as of June 8, 2000 (the “Rights Agreement”), between the Company and State Street Bank and Trust Company, as Rights Agent, and approved the declaration of a dividend distribution of one Preferred Share Purchase Right (a “Right”) on each outstanding share of its Common Stock. In general, the Rights become exercisable if a person or group hereafter acquires 15 percent or more of the Common Stock of the Company or announces a tender offer for 15 percent or more of the Common Stock. The Board of Directors will, in general, be entitled to redeem the Rights at one cent per Right at any time before any such person hereafter acquires 15 percent or more of the outstanding Common Stock. The plan is designed to protect the Company’s stockholders in the event that an attempt is made to acquire the Company without an offer of fair value.
If a person hereafter acquires 15 percent or more of the outstanding Common Stock of the Company (the “Acquiring Person”), each Right will entitle its holder to purchase, for an initial exercise price of $65, a number of shares of Common Stock having a market value at that time of twice the Right’s exercise price. Rights held by the Acquiring Person will become void. If the Company is acquired in a merger or other business combination transaction after a person acquires 15 percent or more of the Company’s Common Stock, each Right will entitle its holder to purchase, at the Right’s then-current exercise price, a number of the acquiring company’s common shares having a market value at that time of twice the Right’s exercise price.
The dividend distribution of Rights was payable on July 19, 2000 to shareholders of record on June 19, 2000. The Rights Agreement will expire on June 8, 2010.
67
11. Fair Value of Financial Instruments
The Company provides disclosure of financial assets and financial liabilities that are carried at fair value based on the price that would be received upon sale of an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. Fair value measurements may be classified based on the amount of subjectivity associated with the inputs to fair valuation of these assets and liabilities using the following three levels:
Level 1—Inputs are unadjusted quoted prices in active markets for identical assets or liabilities that the Company has the ability to access at the measurement date.
Level 2—Inputs include quoted prices for similar assets and liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets that are not active, inputs other than quoted prices that are observable for the asset or liability (i.e., interest rates, yield curves, etc.) and inputs that are derived principally from or corroborated by observable market data by correlation or other means (market corroborated inputs).
Level 3—Unobservable inputs that reflect the Company's estimates of the assumptions that market participants would use in pricing the asset or liability. The Company develops these inputs based on the best information available, including its own data.
The following table presents information about the Company’s assets and liabilities as of December 31, 2009 and 2008 that are measured at fair value on a recurring basis and indicates the fair value hierarchy of the valuation techniques the Company utilized to determine such fair value:
|
December 31, 2009
|
||||||||||||||||
|
In thousands
|
Total
|
Level 1
|
Level 2
|
Level 3
|
||||||||||||
|
Assets:
|
||||||||||||||||
|
Cash equivalents
|
$ | 17,955 | $ | --- | $ | 17,955 | $ | --- | ||||||||
| $ | 17,955 | $ | --- | $ | 17,955 | $ | --- | |||||||||
|
Liabilities:
|
||||||||||||||||
|
Warrant liability
|
$ | 11,363 | $ | --- | $ | --- | $ | 11,363 | ||||||||
| $ | 11,363 | $ | --- | $ | --- | $ | 11,363 | |||||||||
|
December 31, 2008
|
||||||||||||||||
|
In thousands
|
Total
|
Level 1
|
Level 2
|
Level 3
|
||||||||||||
|
Assets:
|
||||||||||||||||
|
Cash equivalents
|
$ | 3,116 | $ | --- | $ | 3,116 | $ | --- | ||||||||
|
Marketable securities
|
14,825 | --- | 14,825 | --- | ||||||||||||
| $ | 17,941 | $ | --- | $ | 17,941 | $ | --- | |||||||||
The Company’s warrant liability is carried at fair value and is classified as Level 3 in the fair value hierarchy due to the use of significant unobservable inputs. The fair value of the warrants on the date of their issuance was determined to be $3.6 million using the Black-Scholes option valuation model applying the following assumptions: (i) a risk-free rate of 1.5%, (ii) an expected term of 3 years, (iii) no dividend yield, and (iv) a volatility of 66%. As of December 31, 2009, the fair value of the warrants was determined to be $11.4 million using the Black-Scholes option valuation model applying the following assumptions: (i) a risk-free rate of 1.23%, (ii) an expected term of 2.2 years, (iii) no dividend yield and (iv) a volatility of 79%. The increase in the fair value of the warrants was recognized in other income (expense) in the consolidated statement of operations. The change in the fair value of the warrant liability since its initial measurement on February 25, 2009 was as follows (in thousands):
68
|
Initial measurement
|
$ | 3,559 | ||
|
Revaluation of warrant liability
|
7,804 | |||
|
Balance at December 31, 2009
|
$ | 11,363 |
The carrying amounts of cash, cash equivalents, accounts payable and accrued liabilities approximate fair value because of their short-term nature. The carrying amount of the Company’s bank term note of $11.6 million at December 31, 2009 approximates fair value due to its variable interest rate. The Company’s obligation under its executive compensation plans is based in part on the current fair market value of underlying securities, which is therefore stated at its estimated fair value.
12. Stock Plan
ARIAD Stock Option and Stock Plans
The Company’s 1991, 1994, 2001 and 2006 stock option and stock plans (the “Plans”) provide for the awarding of nonqualified and incentive stock options, stock grants and restricted stock units to officers, directors, employees and consultants of the Company. Stock options become exercisable as specified in the related option certificate, typically over a four-year period, and expire ten years from the date of grant. Stock grants and restricted stock units provide the recipient with ownership of common stock subject to any rights the Company may have to repurchase the shares granted or other restrictions. The 1991 and 1994 Plans have expired according to their terms and the 2001 Plan has no shares remaining available for grant, although existing stock options granted under these Plans remain outstanding. As of December 31, 2009, there are 7,119,167 shares available for awards under the 2006 Plan.
Employee Stock Purchase Plan
In 1997, the Company adopted the 1997 Employee Stock Purchase Plan and reserved 500,000 shares of common stock for issuance under this plan. In June 2008, the Plan was amended to reserve an additional 500,000 shares of common stock for issuance. Under this plan, substantially all of the Company’s employees may, through payroll withholdings, purchase shares of the Company’s common stock at a price of 85 percent of the lesser of the fair market value at the beginning or end of each three-month withholding period. In 2009, 2008 and 2007, 401,797, 92,698 and 52,113 shares of common stock were issued under the plan, respectively.
13. Stock-Based Compensation
The Company awards stock options and other equity-based instruments to its employees, directors and consultants and provides employees the right to purchase common stock (collectively “share-based payments”), pursuant to stockholder approved plans. The Company’s statement of operations included total compensation cost from share-based payments for the years ended December 31, as follows:
|
In thousands
|
2009
|
2008
|
2007
|
|||||||||
|
Compensation cost from:
|
||||||||||||
|
Stock options
|
$ | 3,068 | $ | 3,798 | $ | 5,254 | ||||||
|
Stock and stock units
|
1,096 | 900 | 662 | |||||||||
|
Purchases of common stock at a discount
|
191 | 53 | 73 | |||||||||
| $ | 4,355 | $ | 4,751 | $ | 5,989 | |||||||
|
Compensation cost included in:
|
||||||||||||
|
Research and development expenses
|
$ | 2,123 | $ | 2,441 | $ | 3,237 | ||||||
|
General and administrative expenses
|
2,232 | 2,310 | 2,752 | |||||||||
| $ | 4,355 | $ | 4,751 | $ | 5,989 | |||||||
69
Stock Options
Stock options are granted with an exercise price equal to the closing market price of the Company’s common stock on the date of grant. Stock options generally vest ratably over four years and have contractual terms of ten years. Stock options are valued using the Black-Scholes option valuation model and compensation cost is recognized based on such fair value over the period of vesting on a straight-line basis.
The following table summarizes information about stock options as of and for the years ended December 31, 2009, 2008 and 2007:
|
In thousands, except per share amounts
|
2009
|
2008
|
2007
|
|||||||||
|
Weighted average fair value of options granted, per share
|
$ | 1.03 | $ | 1.83 | $ | 3.24 | ||||||
|
Total cash received from exercises of stock options
|
206 | 154 | 1,872 | |||||||||
|
Total intrinsic value of stock options exercised
|
271 | 90 | 1,388 | |||||||||
The weighted average fair value of options granted in the years ended December 31, 2009, 2008 and 2007, reflect the following weighted-average assumptions:
|
2009
|
2008
|
2007
|
||||||||||
|
Expected life of options granted (in years)
|
7.04 | 7.04 | 7.54 | |||||||||
|
Expected volatility
|
70.68 | % | 69.37 | % | 68.03 | % | ||||||
|
Risk-free rate
|
2.75 | % | 3.04 | % | 4.41 | % | ||||||
|
Expected dividends
|
0 | % | 0 | % | 0 | % | ||||||
The expected life assumption is based on an analysis of historical behavior of participants related to options awarded over time. The expected volatility assumption for the years ended December 31, 2009, 2008 and 2007 is based on the implied volatility of the Company’s common stock, derived from analysis of historical traded and quoted options on the Company’s common stock over the period commensurate with the expected life of the options granted. The risk-free rate is based on the forward U.S. Treasury yield curve. The expected dividends reflect the Company’s current and expected future policy for dividends on its common stock.
Stock option activity under the Company’s stock plans for the year ended December 31, 2009 was as follows:
|
Number of
Shares
|
Weighted Average
Exercise Price
Per Share
|
|||||||
|
Options outstanding, January 1, 2009
|
7,424,428 | $ | 4.98 | |||||
|
Granted
|
1,372,140 | $ | 1.50 | |||||
|
Forfeited
|
(878,301 | ) | $ | 4.09 | ||||
|
Exercised
|
(234,096 | ) | $ | 0.88 | ||||
|
Options outstanding, December 31, 2009
|
7,684,171 | $ | 4.58 | |||||
The following table summarizes information about stock options outstanding as of December 31, 2009:
|
Options
Outstanding
|
Options
Exercisable
|
Remaining
Options Expected
To Vest
|
||||||||||
|
Number of options
|
7,684,171 | 4,922,839 | 2,579,574 | |||||||||
|
Weighted-average exercise price per share
|
$ | 4.58 | $ | 5.58 | $ | 2.89 | ||||||
|
Aggregate intrinsic value (in 000’s)
|
$ | 1,270 | $ | 132 | $ | 983 | ||||||
|
Weighted average remaining contractual term (in years)
|
5.97 | 4.61 | 8.33 | |||||||||
Options expected to vest consist of options scheduled to vest in the future less expected forfeitures.
70
At December 31, 2009, total unrecognized compensation cost related to non-vested stock options outstanding amounted to $3.6 million. That cost is expected to be recognized over a weighted-average period of 2.1 years.
Stock and Stock Unit Grants
Stock and stock unit grants are provided to non-employee directors as compensation and generally carry no restrictions as to resale or are fully vested upon grant. Stock and stock unit grants to officers carry restrictions as to resale for periods of time or vesting provisions over time as specified in the grant. Stock and stock unit grants are valued at the closing market price of the Company’s common stock on the date of grant and compensation expense is recognized over the requisite service period, vesting period or period during which restrictions remain on the common stock or stock units granted.
Stock and stock unit activity under the Company’s stock plans for the year ended December 31, 2009 was as follows:
|
Number of
Shares
|
Weighted Average
Grant Date
Fair Value
|
|||||||
|
Outstanding, January 1, 2009
|
553,500 | $ | 3.75 | |||||
|
Granted
|
1,143,000 | $ | 1.36 | |||||
|
Vested or restrictions lapsed
|
(207,500 | ) | $ | 2.77 | ||||
|
Outstanding, December 31, 2009
|
1,489,000 | $ | 2.05 | |||||
At December 31, 2009, total unrecognized compensation cost related to stock and stock unit awards amounted to $1.4 million.
Purchase of Common Stock Pursuant to Employee Stock Purchase Plan
Purchases of common stock by employees are provided pursuant to the Company’s employee stock purchase plan. Purchase price is calculated as 85 percent of the lower of the closing price of our common stock on the first trading day or last trading day of each calendar quarter. Compensation cost is equal to the fair value of the discount on the date of grant and is recognized as compensation in the period of purchase.
14. Net Loss Per Share
Net loss per share amounts have been computed based on the weighted-average number of common shares outstanding during each period. Because of the net loss reported in each period, diluted and basic net loss per share amounts are the same. For the years ended December 31, 2009, 2008 and 2007, the following potentially dilutive securities were not included in the computation of net loss per share because the effect would be anti-dilutive:
|
2009
|
2008
|
2007
|
||||||||||
|
Outstanding stock options
|
7,684,171 | 7,424,428 | 7,568,044 | |||||||||
|
Outstanding restricted stock and restricted stock units
|
1,489,000 | 553,500 | 90,000 | |||||||||
|
Warrants to purchase common stock
|
10,784,024 | --- | --- | |||||||||
| 19,957,195 | 7,977,928 | 7,658,044 | ||||||||||
71
15. Income Taxes
The components of deferred income taxes were as follows at December 31:
|
In thousands
|
2009
|
2008
|
||||||
|
Deferred tax liabilities:
|
||||||||
|
Intangible and other assets
|
$ | 3,836 | $ | 3,946 | ||||
|
Deferred tax assets:
|
||||||||
|
Net operating loss carryforwards
|
142,604 | 124,991 | ||||||
|
Federal and State tax credit carryovers
|
25,274 | 22,043 | ||||||
|
Depreciation
|
4,739 | 4,422 | ||||||
|
Deferred revenue
|
36,113 | 32,011 | ||||||
|
Stock-based compensation
|
2,518 | 1,799 | ||||||
|
Other
|
1,058 | 871 | ||||||
|
Total deferred tax assets
|
212,306 | 186,137 | ||||||
|
Deferred tax assets, net
|
208,470 | 182,191 | ||||||
|
Valuation allowance
|
(208,470 | ) | (182,191 | ) | ||||
|
Total deferred taxes
|
$ | --- | $ | --- | ||||
At December 31, 2009, the Company had available estimated net operating loss carryforwards and research and development credit carryforwards for federal and state tax reporting purposes as follows:
|
Amount
|
Expiring if not utilized
|
||||
|
(in 000s)
|
|||||
|
Net operating loss carryforwards:
|
|||||
|
Federal
|
$ | 403,521 |
2010 through 2028
|
||
|
State
|
$ | 107,387 |
2010 through 2014
|
||
|
Research and development credit carryforwards:
|
|||||
|
Federal
|
$ | 17,127 |
2010 through 2029
|
||
|
State
|
$ | 7,717 |
2010 through 2024
|
||
Since the Company has not yet achieved sustained profitable operations, management believes the tax benefits do not satisfy the more-likely-than-not realization criteria and has recorded a valuation allowance for the entire net deferred tax asset. The increase in the valuation allowance was $26.3 million, $21.9 million and $21.8 million in 2009, 2008 and 2007, respectively. The Company does not have any significant uncertain tax positions.
16. Legal Proceedings
NF-κB Patent Infringement Litigation and Reexamination
Lilly Litigation
In 2002, the Company, together with Massachusetts Institute of Technology (“MIT”), The Whitehead Institute for Biomedical Research (“Whitehead”) and Harvard University (“Harvard”) (collectively, the Plaintiffs) filed a lawsuit in the United States District Court for the District of Massachusetts (the “U.S. District Court“) against Eli Lilly and Company (“Lilly”) alleging infringement of four claims (the “NF-κB ‘516 Claims”) of the Plaintiffs’ U.S. Patent No. 6,410,516 (the “‘516 Patent”), covering methods of treating human disease by regulating NF-κB cell-signaling activity through sales of Lilly’s drugs, Evista® and Xigris®. In 2006, a jury rendered a verdict in favor of the Plaintiffs and awarded damages of $65.2 million to the Plaintiffs, plus further damages equal to 2.3 percent of U.S. sales of Evista and Xigris from February 28, 2006 through the year 2019, when the patent expires.
72
Lilly appealed several of the District Court’s rulings to the U.S. Court of Appeals for the Federal Circuit (the “CAFC”) and, on April 3, 2009, the CAFC ruled in Lilly’s favor, finding that the four claims of the ‘516 Patent asserted in this lawsuit are not supported by adequate written description and are therefore invalid. The CAFC did not rule on other validity issues raised by Lilly or on the findings of infringement. In addition, the CAFC affirmed the District Court’s ruling that the patent’s enforceability is not impaired by inequitable conduct during its prosecution. The CAFC granted Plaintiffs subsequent petition requesting en banc rehearing of this matter before the full CAFC. The CAFC heard oral arguments on December 7, 2009 and its ruling on the matter is pending.
Amgen Litigation
In April 2006, Amgen Inc. and certain affiliated entities (“Amgen“) filed a lawsuit against the Company in the U.S. District Court for the District of Delaware (the “Delaware Court“) seeking a declaratory judgment that each of the claims contained in the ‘516 Patent is invalid and that Amgen has not infringed any of the claims of the ‘516 Patent based on activities related to Amgen’s products, Enbrel® and Kineret®. In April 2007, the Company, together with MIT, Whitehead, and Harvard, filed a counterclaim against Amgen, alleging infringement of the ‘516 Patent based on activities related to Enbrel and Kineret, as well as the Company’s answer to Amgen’s complaint, counter-claim and demand for jury trial.
On September 19, 2008, the Delaware Court issued a series of rulings that, among other things: (i) granted Amgen’s motion for summary judgment of non-infringement of the asserted seven (7) claims of the ‘516 Patent based on the Delaware Court’s interpretation of these claims to exclude extracellular methods of reducing NF-κB activity, (ii) granted the Company’s motion seeking to dismiss for lack of jurisdiction under the Declaratory Judgment Act Amgen’s challenges to the validity of claims of the ‘516 Patent that are not being asserted against Enbrel, and (iii) granted in part and denied in part the Company’s motion for partial summary judgment with respect to Amgen’s inequitable conduct defense.
On June 1, 2009, the CAFC affirmed the Delaware Court’s claim interpretation and summary judgment of non-infringement with respect to Enbrel. Plaintiffs did not file a petition for rehearing in this case. The matter was formally concluded on December 28, 2009 with the withdrawal of all pending motions.
PTO Reexamination
A reexamination at the United States Patent and Trademark Office (“PTO”) of aspects of patentability of the ‘516 Patent had been initiated by Lilly in April 2005. On October 16, 2008, the PTO issued a final office action confirming that 53 claims of the ‘516 patent are patentable, while rejecting 45 of the remaining claims, including claims relating to the Lilly litigation and claims relating to the Amgen litigation. The Company has appealed that decision to the Patent Office Board of Appeals and Interferences and filed its appeal brief on May 18, 2009.
Shareholder Derivative Suit
On February 13, 2009, a shareholder derivative complaint alleging breaches of fiduciary duties was filed in the Delaware Court of Chancery (the “Court”), naming each member of the Company's board of directors as a defendant and the Company as a nominal defendant. The complaint, filed by a stockholder of the Company, alleges breaches of fiduciary duties by the defendants related to the merger of AGTI with and into ARIAD, the departure of the Company’s former chief legal officer and changes in the by-laws of the Company and roles and responsibilities of members of the Board of Directors, and seeks unspecified damages plus reimbursement of the legal and other costs of the plaintiffs. On February 22, 2010, the Court dismissed the complaint with prejudice.
73
17. Related Party Transactions
In June 2007, the Company entered into an agreement with its chief executive officer and with a member of its Board of Directors in their individual capacities as shareholders of AGTI. The agreement contains provisions regarding (i) confidentiality of material non-public information provided to them and their advisors in the course of evaluation of any potential transaction to acquire the 20 percent interest in AGTI that the Company did not own, (ii) reimbursement by the Company of certain reasonable expenses incurred by them to retain financial advisors and legal counsel to advise them in connection with any potential transaction, (iii) indemnification of them by the Company for claims arising out of or relating to any potential transaction and (iv) the maintenance by the Company of liability insurance for their benefit. For the years ended December 31, 2008 and 2007, the Company reimbursed $259,000 and $290,000, respectively, in expenses pursuant to this agreement. AGTI was merged with and into ARIAD Pharmaceuticals, Inc. on September 12, 2008 and the 20 percent minority interest in AGTI was acquired by the Company consequent to the merger (see Note 3).
In the offering and sale by the Company of 21,850,000 shares of its common stock on August 7, 2009 (see Note 10), the Company’s chief executive officer purchased 1,714,286 shares, at the offering price of $1.75 per share, for $3 million.
74
Not applicable.
(a) Evaluation of Disclosure Controls and Procedures. Our principal executive officer and principal financial officer, after evaluating the effectiveness of our disclosure controls and procedures (as defined in paragraph (e) of Rules 13a-15 and 15d-15 under the Securities Exchange Act of 1934) as of the end of the period covered by this Annual Report on Form 10-K, have concluded that, based on such evaluation, our disclosure controls and procedures were effective to ensure that information required to be disclosed by us in the reports that we file or submit under the Securities Exchange Act of 1934 is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms and is accumulated and communicated to management, including the Chief Executive Officer and Chief Financial Officer, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure, particularly during the period in which this Annual Report on Form 10-K was being prepared.
Our disclosure controls and procedures are designed to provide reasonable assurance of achieving their desired control objectives. Our principle executive officer and principle financial officer have concluded that our controls and procedures are effective at that reasonable assurance level.
(b) Changes in Internal Controls. There were no changes in our internal control over financial reporting, identified in connection with the evaluation of such internal control that occurred during our last fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
On January 20, 2010, at a special meeting of our stockholders, our stockholders approved a certificate of amendment to our certificate of incorporation, as amended, to increase our authorized common stock from 145,000,000 shares to 240,000,000 shares. A total of 91,437,485 shares were represented at the meeting. Of the 109,042,782 shares of common stock outstanding on the record date, 65,712,520 shares, or 60.3%, voted in favor of approving the certificate of amendment, 25,366,265 voted against approving the certificate of amendment and 358,600 abstained. The certificate of amendment was filed with the Secretary of State of the State of Delaware on January 20, 2010.
75
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Board of Directors,” “Executive Officers,“ “Section 16(a) Beneficial Ownership Reporting Compliance” and “Corporate Code of Conduct and Ethics” in the Company’s Definitive Proxy Statement for the 2010 Annual Meeting of Stockholders.
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Executive Compensation,” “Compensation Discussion and Analysis”, “Compensation Committee Report”, “Board of Directors” and “Compensation Practices and Policies Relating to Risk Management” in the Company’s Definitive Proxy Statement for the 2010 Annual Meeting of Stockholders.
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” in the Company’s Definitive Proxy Statement for the 2010 Annual Meeting of Stockholders.
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Board of Directors” and “Certain Relationships and Related Transactions” in the Company’s Definitive Proxy Statement for the 2010 Annual Meeting of Stockholders.
The response to this item is incorporated by reference from the discussion responsive thereto under the caption “Proposal 2: Ratification of Selection of Independent Registered Public Accounting Firm” in the Company’s Definitive Proxy Statement for the 2010 Annual Meeting of Stockholders.
76
|
|
(a)(1)
|
The following Consolidated Financial Statements, Notes thereto and Report of Independent Registered Public Accounting Firm have been presented in Item 8:
|
Report of Independent Registered Public Accounting Firm
Consolidated Balance Sheets
Consolidated Statements of Operations
Consolidated Statements of Stockholders’ Equity (Deficit)
Consolidated Statements of Cash Flows
Notes to Consolidated Financial Statements
|
|
(a)(2)
|
Financial Statement Schedules:
|
Schedules have been omitted because of the absence of conditions under which they are required or because the required information is included in the financial statements or notes thereto.
|
|
(a)(3)
|
The Exhibits listed in the Exhibit Index are filed herewith in the manner set forth therein.
|
|
|
(b)
|
See (a) (3) above.
|
|
|
(c)
|
See (a) (2) above.
|
77
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Cambridge and Commonwealth of Massachusetts on the 16th day of March, 2010.
| ARIAD PHARMACEUTICALS, INC. | |||
|
|
By:
|
/s/ Harvey J. Berger, M.D. | |
| Name: | Harvey J. Berger, M.D. | ||
| Title: | Chairman, Chief Executive Officer and President | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Signature
|
Title
|
Date
|
||
|
/s/ Harvey J. Berger, M.D.
Harvey J. Berger, M.D.
|
Chairman of the Board of Directors, Chief Executive
Officer and President (Principal Executive Officer)
|
March 16, 2010
|
||
|
/s/ Edward M. Fitzgerald
Edward M. Fitzgerald
|
Senior Vice President, Chief Financial Officer and
Treasurer (Principal Financial Officer and Principal
Accounting Officer)
|
March 16, 2010
|
||
|
/s/ Jay R. LaMarche
Jay R. LaMarche
|
Director
|
March 16, 2010
|
||
|
/s/ Athanase Lavidas, Ph.D.
Athanase Lavidas, Ph.D.
|
Director
|
March 16, 2010
|
||
|
/s/ Massimo Radaelli, Ph.D.
Massimo Radaelli, Ph.D.
|
Director
|
March 16, 2010
|
||
|
/s/ A. Collier Smyth, M.D.
A. Collier Smyth, M.D.
|
Director
|
March 16, 2010
|
||
|
/s/ Wayne Wilson
Wayne Wilson
|
Director
|
March 16, 2010
|
78
ARIAD Pharmaceuticals, Inc.
Form 10-K for the year ended December 31, 2009
Exhibit List
|
Exhibit
Number
|
Exhibit Description
|
Filed
with
this
Report
|
Incorporated by
Reference
herein from
Form or
Schedule
|
Filing Date
|
SEC File/
Reg. Number
|
|
|
3.1
|
.1
|
Certificate of Incorporation of ARIAD Pharmaceuticals, Inc., as amended
|
S-8
(Exhibit 4.2)
|
06/30/04
|
333-116996
|
|
|
.2
|
Certificate of Amendment to Certificate of Incorporation of ARIAD Pharmaceuticals, Inc., as amended
|
8-K
(Exhibit 3.1)
|
01/22/10
|
000-21696
|
||
|
3.2
|
Amended and Restated By-laws of ARIAD Pharmaceuticals, Inc.
|
8-K
(Exhibit 3.1)
|
08/27/09
|
000-21696
|
||
|
4.1
|
Specimen common stock certificate of ARIAD Pharmaceuticals, Inc.
|
S-3
(Exhibit 4.5)
|
10/14/94
|
33-85166
|
||
|
4.2
|
Rights Agreement, dated as of June 8, 2000, between the ARIAD Pharmaceuticals, Inc. and State Street Bank and Trust Company, which includes the Form of Certificate of Designations in respect of the Series A Preferred Stock, as Exhibit A, the Form of Right Certificate as Exhibit B and the Summary of Rights to Purchase Series A Preferred Stock as Exhibit C
|
8-A
(Exhibit 1)
|
06/19/00
|
000-21696
|
||
|
4.3
|
Form of Warrant to Purchase Common Stock dated February 25, 2009
|
8-K
(Exhibit 10.2)
|
02/20/09
|
000-21696
|
79
|
Exhibit
Number
|
Exhibit Description
|
Filed
with
this
Report
|
Incorporated by
Reference
herein from
Form or
Schedule
|
Filing Date
|
SEC File/
Reg. Number
|
|
|
Leases and Credit Agreements
|
||||||
|
10.1
|
.1
|
Lease Agreement, dated January 8, 1992, between ARIAD Pharmaceuticals, Inc. and Forest City Cambridge, Inc.
|
10
(Exhibit 10.1)
|
04/30/93
|
000-21696
|
|
|
.2
|
Eighth Amendment to Lease dated October 30, 2006
|
10-K
(Exhibit 10.57)
|
03/14/07
|
000-21696
|
||
|
10.2
|
.1
|
Credit Agreement, dated as of March 12, 2003, by and among ARIAD Pharmaceuticals, Inc., ARIAD Corporation and ARIAD Gene Therapeutics, Inc. and Citizens Bank of Massachusetts
|
10-Q
(Exhibit 10.1)
|
05/13/03
|
000-21696
|
|
|
.2
|
Amendment No. 1 to Credit Agreement, dated as of December 31, 2003
|
10-K
(Exhibit 10.57)
|
03/02/04
|
000-21696
|
||
|
.3
|
Amendment No. 2 to Credit Agreement dated as of December 31, 2004
|
10-K
(Exhibit 10.52)
|
02/18/05
|
000-21696
|
||
|
.4
|
Amendment No. 3 to Credit Agreement, dated as of March 26, 2008, by and among ARIAD Pharmaceuticals, Inc., ARIAD Corporation and ARIAD Gene Therapeutics, Inc. and RBS Citizens, National Association, successor by merger to Citizens Bank of Massachusetts
|
8-K
(Exhibit 10.2.4)
|
03/27/08
|
000-21696
|
||
|
.5
|
Waiver and Amendment No. 4 to Credit Agreement dated as of June 19, 2009, by and among ARIAD Pharmaceuticals, Inc., ARIAD Corporation and RBS Citizens, National Association
|
10-Q
(Exhibit 10.3)
|
08/10/09
|
000-21696
|
||
|
.6
|
Waiver and Amendment No. 5 to Credit Agreement dated as of December 14, 2009
|
X
|
||||
|
10.3
|
Security Agreement - All Assets, dated as of March 12, 2003, by and between ARIAD Pharmaceuticals, Inc. and Citizens Bank of Massachusetts
|
10-Q
(Exhibit 10.3)
|
05/13/03
|
000-21696
|
||
|
10.4
|
Security Agreement - All Assets, dated as of March 12, 2003, by and between ARIAD Corporation and Citizens Bank of Massachusetts
|
10-Q
(Exhibit 10.4)
|
05/13/03
|
000-21696
|
||
|
10.5
|
Third Amended and Restated Term Note, dated March 26, 2008, issued by ARIAD Pharmaceuticals, Inc., ARIAD Corporation and ARIAD Gene Therapeutics, Inc. to RBS Citizens, National Association, successor by merger to Citizens Bank of Massachusetts
|
8-K
(Exhibit 10.2.4)
|
03/27/08
|
000-21696
|
||
80
|
Exhibit
Number
|
Exhibit Description
|
Filed
with
this
Report
|
Incorporated by
Reference
herein from
Form or
Schedule
|
Filing Date
|
SEC File/
Reg. Number
|
|
|
Agreements with Respect to Collaborations, Licenses, Research and Development
|
||||||
|
10.6
|
License Agreement dated August 19, 1991 by and among The Massachusetts Institute of Technology, The Whitehead Institute and ARIAD Pharmaceuticals, Inc.*
|
10-Q
(Exhibit 10.1)
|
05/10/06
|
000-21696
|
||
|
10.7
|
Amended and Restated Agreement, dated as of December 12, 1997, between The Board of Trustees of The Leland Stanford Junior University and ARIAD Gene Therapeutics, Inc.*
|
10-K
(Exhibit 10.14)
|
03/10/98
|
000-21696
|
||
|
10.8
|
Revised and Restated Research and Development Agreement, dated as of March 15, 2002, by and between ARIAD Pharmaceuticals, Inc. and ARIAD Corporation
|
10-K
(Exhibit 10.53)
|
03/22/02
|
000-21696
|
||
|
10.9
|
License Agreement, effective January 26, 2005, by and between ARIAD Pharmaceuticals, Inc. and Medinol Ltd.*
|
10-Q
(Exhibit 10.1)
|
05/10/05
|
000-21696
|
||
|
10.10
|
Supply Agreement, entered into as of January 26, 2005, by and between ARIAD Pharmaceuticals, Inc. and Medinol Ltd.*
|
10-Q
(Exhibit 10.2)
|
05/10/05
|
000-21696
|
||
|
10.111
|
Collaboration Agreement, dated July 11, 2007, by and among ARIAD Pharmaceuticals, Inc., ARIAD Gene Therapeutics, Inc. and Merck & Co., Inc.*
|
10-Q
(Exhibit 10.1)
|
11/09/07
|
000-21696
|
||
|
10.12
|
Ridaforolimus API and Tablet Supply Agreement dated May 7, 2008 among ARIAD Pharmaceuticals, Inc., ARIAD Gene Therapeutics, Inc. and Merck & Co., Inc.*
|
10-Q
(Exhibit 10.2)
|
08/11/08
|
000-21696
|
||
|
10.13
|
License Agreement, dated October 9, 2007, among ARIAD Pharmaceuticals, Inc., ARIAD Gene Therapeutics, Inc. and ICON Medical Corp.**
|
X
|
||||
81
|
Exhibit
Number
|
Exhibit Description
|
Filed
with
this
Report
|
Incorporated by
Reference
herein from
Form or
Schedule
|
Filing Date
|
SEC File/
Reg. Number
|
|
|
Agreements with Executive Officers and Directors
|
||||||
|
10.14
|
.1
|
Executive Employment Agreement, dated as of January 1, 1992, between ARIAD Pharmaceuticals, Inc. and Harvey J. Berger, M.D.+
|
10
(Exhibit 10.3)
|
04/30/93
|
000-21696
|
|
|
.2
|
Amendment to Executive Employment Agreement, dated April 19, 1994+
|
S-1
(Exhibit 10.25)
|
05/10/94
|
33-76414
|
||
|
.3
|
Amendment to Executive Employment Agreement, dated June 30, 1994+
|
10-K
(Exhibit 10.23)
|
03/31/95
|
000-21696
|
||
|
.4
|
Amendment to Executive Employment Agreement, dated as of January 1, 2006+
|
10-K
(Exhibit 10.56)
|
03/16/06
|
000-21696
|
||
|
.5
|
Amendment to Executive Employment Agreement, dated October 14, 2008 (solely to extend term)+
|
10-K
(Exhibit 10.23)
|
03/16/09
|
000-21696
|
||
|
.6
|
Amendment to Executive Employment Agreement, dated December 31, 2008 (related to §409A)+
|
10-K
(Exhibit 10.24)
|
03/16/09
|
000-21696
|
||
|
10.15
|
.1
|
Executive Employment Agreement dated May 1, 1992, between ARIAD Pharmaceuticals, Inc. and John Iuliucci, Ph.D., as amended March 2, 1994, January 1, 1997, January 1, 1999 and June 8, 2000+
|
10-Q
(Exhibit 10.3)
|
08/10/00
|
000-21696
|
|
|
.2
|
Amendment to Executive Employment Agreement, dated September 2, 2003+
|
10-Q
(Exhibit 10.6)
|
11/04/03
|
000-21696
|
||
|
.3
|
Amendment to Executive Employment Agreement, dated April/May 2007+
|
10-Q
(Exhibit 10.2)
|
08/09/07
|
000-21696
|
||
|
.4
|
Amendment to Executive Employment Agreement, dated December 31, 2008 (related to §409A)+
|
10-K
(Exhibit 10.24)
|
03/16/09
|
000-21696
|
||
|
10.16
|
.1
|
Executive Employment Agreement, dated August 1, 1993, between ARIAD Pharmaceuticals, Inc. and David L. Berstein, J.D., as amended March 2, 1994, January 1, 1997 and June 8, 2000+
|
10-Q
(Exhibit 10.4)
|
08/10/00
|
000-21696
|
|
|
.2
|
Amendment to Executive Employment Agreement, dated as of January 1, 2001+
|
10-Q
(Exhibit 10.2)
|
05/14/01
|
000-21696
|
||
|
.3
|
Amendment to Executive Employment Agreement, dated September 2, 2003+
|
10-Q
(Exhibit 10.3)
|
11/04/03
|
000-21696
|
||
|
.4
|
Amendment to Employment Agreement, dated April 14, 2008+
|
10-K
(Exhibit 10.16.4)
|
03/16/09
|
000-21696
|
||
|
.5
|
Amendment to Executive Employment Agreement, dated October 14, 2008 (solely to extend term)+
|
10-K
(Exhibit 10.23)
|
03/16/09
|
000-21696
|
||
|
.6
|
Amendment to Executive Employment Agreement, dated December 31, 2008 (related to §409A)+
|
10-K
(Exhibit 10.24)
|
03/16/09
|
000-21696
|
||
|
10.17
|
.1
|
Executive Employment Agreement, dated May 6, 2002, between ARIAD Pharmaceuticals, Inc. and Edward M. Fitzgerald+
|
10-Q
(Exhibit 10.1)
|
05/09/02
|
000-21696
|
|
|
.2
|
Amendment to Executive Employment Agreement, dated September 2, 2003+
|
10-Q
(Exhibit 10.5)
|
11/04/03
|
000-21696
|
||
82
|
Exhibit
Number
|
Exhibit Description
|
Filed
with
this
Report
|
Incorporated by
Reference
herein from
Form or
Schedule
|
Filing Date
|
SEC File/
Reg. Number
|
|
.3
|
Amendment to Executive Employment Agreement, dated October 14, 2008 (solely to extend term)+
|
10-K
(Exhibit 10.23)
|
03/16/09
|
000-21696
|
||
|
.4
|
Amendment to Executive Employment Agreement, dated December 31, 2008 (related to §409A)+
|
10-K
(Exhibit 10.24)
|
03/16/09
|
000-21696
|
||
|
10.18
|
.1
|
Executive Employment Agreement, dated June 8, 2000, between ARIAD Pharmaceuticals, Inc. and Timothy Clackson, Ph.D.+
|
10-K
(Exhibit 10.49)
|
03/14/03
|
000-21696
|
|
|
.2
|
Amendment to Executive Employment Agreement, dated July 1, 2001+
|
10-K
(Exhibit 10.50)
|
03/14/03
|
000-21696
|
||
|
.3
|
Amendment to Executive Employment Agreement, dated June 12, 2002+
|
10-K
(Exhibit 10.51)
|
03/14/03
|
000-21696
|
||
|
.4
|
Amendment to Executive Employment Agreement, dated September 2, 2003+
|
10-Q
(Exhibit 10.4)
|
11/04/03
|
000-21696
|
||
|
.5
|
Amendment to Executive Employment Agreement, dated October 14, 2008 (solely to extend term)+
|
10-K
(Exhibit 10.23)
|
03/16/09
|
000-21696
|
||
|
.6
|
Amendment to Executive Employment Agreement, dated December 31, 2008 (related to §409A)+
|
10-K
(Exhibit 10.24)
|
03/16/09
|
000-21696
|
||
|
10.19
|
.1
|
Executive Employment Agreement, dated May 29, 2007, by and between ARIAD Pharmaceuticals, Inc. and Pierre F. Dodion+
|
10-Q
(Exhibit 10.1)
|
08/09/07
|
000-21696
|
|
|
.2
|
Amendment to Executive Employment Agreement, dated October 14, 2008 (solely to extend term)+
|
10-K
(Exhibit 10.23)
|
03/16/09
|
000-21696
|
||
|
.3
|
Amendment to Executive Employment Agreement, dated December 31, 2008 (related to §409A)+
|
10-K
(Exhibit 10.24)
|
03/16/09
|
000-21696
|
||
|
10.20
|
.1
|
Executive Employment Agreement, dated November 4, 2008, between ARIAD Pharmaceuticals, Inc. and Daniel M. Bollag, Ph.D.+
|
10-K
(Exhibit 10.20.1)
|
03/16/09
|
000-21696
|
|
|
.2
|
Amendment to Executive Employment Agreement, dated December 31, 2008 (related to §409A)+
|
10-K
(Exhibit 10.24)
|
03/16/09
|
000-21696
|
||
|
10.21
|
.1
|
Executive Employment Agreement, dated February 1, 2008, between ARIAD Pharmaceuticals, Inc. and Raymond T. Keane, Esq.+
|
10-K
(Exhibit 10.21.1)
|
03/16/09
|
000-21696
|
|
|
.2
|
Amendment to Executive Employment Agreement, dated October 14, 2008 (solely to extend term)+
|
10-K
(Exhibit 10.23)
|
03/16/09
|
000-21696
|
||
|
.3
|
Amendment to Executive Employment Agreement, dated December 31, 2008 (related to §409A)+
|
10-K
(Exhibit 10.24)
|
03/16/09
|
000-21696
|
||
|
10.22
|
.1
|
Executive Employment Agreement, dated October 25, 2007, between ARIAD Pharmaceuticals, Inc. and Matthew E. Ros+
|
10-K
(Exhibit 10.22.1)
|
03/16/09
|
000-21696
|
83
|
Exhibit
Number
|
Exhibit Description
|
Filed
with
this
Report
|
Incorporated by
Reference
herein from
Form or
Schedule
|
Filing Date
|
SEC File/
Reg. Number
|
|
.2
|
Amendment to Executive Employment Agreement, dated October 14, 2008 (solely to extend term)+
|
10-K
(Exhibit 10.23)
|
03/16/09
|
000-21696
|
||
|
.3
|
Amendment to Executive Employment Agreement, dated December 31, 2008 (related to §409A)+
|
10-K
(Exhibit 10.24)
|
03/16/09
|
000-21696
|
||
|
.4
|
Amendment to Employment Agreement dated January 8, 2009+
|
10-K
(Exhibit 10.22.4)
|
03/16/09
|
000-21696
|
||
|
10.23
|
Amendments to Executive Employment Agreements, dated October 14, 2008 (solely to extend term)+
|
10-K
(Exhibit 10.23)
|
03/16/09
|
000-21696
|
||
|
10.24
|
Amendments to Executive Employment Agreements, dated December 31, 2008 (related to §409A)+
|
10-K
(Exhibit 10.24)
|
03/16/09
|
000-21696
|
||
|
10.25
|
.1
|
Executive Employment Agreement, dated as of March 4, 2002, between ARIAD Pharmaceuticals, Inc. and Laurie A. Allen, Esq.+
|
10-K
(Exhibit 10.56)
|
03/22/02
|
000-21696
|
|
|
.2
|
Amendment to Executive Employment Agreement, dated September 2, 2003+
|
10-Q
(Exhibit 10.2)
|
11/04/03
|
000-21696
|
||
|
.3
|
Amendment to Executive Employment Agreement, dated April/May 2007+
|
10-Q
(Exhibit 10.2)
|
08/09/07
|
000-21696
|
||
|
.4
|
Amendment to Executive Employment Agreement, dated September 11, 2008+
|
8-K
(Exhibit 10.3)
|
09/17/08
|
000-21696
|
||
|
10.26
|
Indemnity Agreement, dated September 11, 2008, by and between ARIAD Pharmaceuticals, Inc. and Laurie A. Allen, Esq.+
|
8-K
(Exhibit 10.1)
|
09/17/08
|
000-21696
|
||
|
10.27
|
Indemnity Agreement, dated September 11, 2008, by and between ARIAD Gene Therapeutics, Inc. and Laurie A. Allen, Esq.+
|
8-K
(Exhibit 10.2)
|
09/17/08
|
000-21696
|
||
|
10.28
|
Guarantee, dated September 11, 2008, by and between ARIAD Gene Therapeutics, Inc. and Laurie A. Allen, Esq.+
|
8-K
(Exhibit 10.4)
|
09/17/08
|
000-21696
|
||
|
10.29
|
Consulting Agreement, dated September 11, 2008, by and between ARIAD Gene Therapeutics, Inc. and Laurie A. Allen, Esq.+
|
8-K
(Exhibit 10.5)
|
09/17/08
|
000-21696
|
||
|
10.30
|
.1
|
ARIAD Pharmaceuticals, Inc. 1997 Executive Compensation Plan+
|
10-K
(Exhibit 10.41)
|
03/10/98
|
000-21696
|
|
|
.2
|
Amendment to ARIAD Pharmaceuticals, Inc. 1997 Executive Compensation Plan+
|
10-Q
(Exhibit 10.2)
|
11/09/05
|
000-21696
|
||
|
10.31
|
ARIAD Pharmaceuticals, Inc. 2005 Executive Compensation Plan (as amended and restated effective October 1, 2008)+
|
10-K
(Exhibit 10.31)
|
03/16/09
|
000-21696
|
||
|
10.32
|
Director Compensation Arrangements+
|
10-Q
(Exhibit 10.1)
|
05/11/09
|
000-21696
|
||
|
10.33
|
Form of Indemnity Agreement between ARIAD Pharmaceuticals, Inc. and its directors and officers+
|
10-K
(Exhibit 10.33)
|
03/16/09
|
000-21696
|
||
|
10.34
|
Letter Agreement, dated June 19, 2007, by and among ARIAD Pharmaceuticals, Inc. Harvey J. Berger, M.D. and Jay LaMarche+
|
8-K
(Exhibit 10.1)
|
06/21/07
|
000-21696
|
84
|
Exhibit
Number
|
Exhibit Description
|
Filed
with
this
Report
|
Incorporated by
Reference
herein from
Form or
Schedule
|
Filing Date
|
SEC File/
Reg. Number
|
|
|
Equity Compensation Plans
|
||||||
|
10.35
|
.1
|
ARIAD Pharmaceuticals, Inc. 1991 Stock Option Plan for Employees and Consultants, as amended+
|
10-K
(Exhibit 10.13)
|
03/31/95
|
000-21696
|
|
|
.2
|
Amendment to the 1991 Stock Option Plan for Employees and Consultants+
|
10-Q
(Exhibit 10.36)
|
08/12/97
|
000-21696
|
||
|
10.36
|
ARIAD Pharmaceuticals, Inc. 1991 Stock Option Plan for Directors+
|
10
(Exhibit 10.15)
|
04/30/93
|
000-21696
|
||
|
10.37
|
.1
|
ARIAD Pharmaceuticals, Inc. 1994 Stock Option Plan for Non-Employee Directors+
|
10-K
(Exhibit 10.24)
|
03/31/95
|
000-21696
|
|
|
.2
|
Amendment to the 1994 Stock Option Plan for Non-Employee Directors.+
|
10-Q
(Exhibit 10.37)
|
08/12/97
|
000-21696
|
||
|
10.38
|
Amended and Restated ARIAD Pharmaceuticals, Inc. 1997 Employee Stock Purchase Plan+
|
Def 14A
(Appendix A)
|
04/30/09
|
000-21696
|
||
|
10.39
|
ARIAD Pharmaceuticals, Inc. 2001 Stock Plan, as amended and restated+
|
10-Q
(Exhibit 10.3)
|
11/09/05
|
000-21696
|
||
|
10.40
|
.1
|
ARIAD Pharmaceuticals, Inc. 2006 Long-Term Incentive Plan, as amended+
|
Def 14A
(Appendix A)
|
04/30/09
|
000-21696
|
|
|
.2
|
Form of Stock Option Certificate under the ARIAD Pharmaceuticals, Inc. 2006 Long-Term Incentive Plan+
|
10-Q
(Exhibit 10.2)
|
08/08/06
|
000-21696
|
||
|
.3
|
Form of Stock Grant Certificate under the ARIAD Pharmaceuticals, Inc. 2006 Long-Term Incentive Plan+
|
10-Q
(Exhibit 10.3)
|
08/08/06
|
000-21696
|
||
|
.4
|
Form of Restricted Stock Unit Certificate under the ARIAD Pharmaceuticals, Inc. 2006 Long-Term Incentive Plan+
|
10-Q
(Exhibit 10.4)
|
08/08/06
|
000-21696
|
||
|
21.1
|
Subsidiaries of ARIAD Pharmaceuticals, Inc.
|
10-K
(Exhibit 21.1)
|
03/16/09
|
000-21696
|
||
|
23.1
|
Consent of Deloitte & Touche LLP
|
X
|
||||
|
31.1
|
Certification of the Chief Executive Officer
|
X
|
||||
|
31.2
|
Certification of the Chief Financial Officer
|
X
|
||||
|
32.1
|
Certification pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
|
X
|
||||
|
(+)
|
Management contract or compensatory plan or arrangement.
|
|
(*)
|
Confidential treatment has been granted by the Securities and Exchange Commission as to certain portions.
|
|
(**)
|
Confidential treatment has been requested from the Securities and Exchange Commission as to certain portions.
|
85