Attached files
UNITED
STATES
SECURITIES
AND EXCHANGE COMMISSION
Washington,
D.C. 20549
______________
FORM
10-K
______________
(MARK
ONE)
x ANNUAL
REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF
1934
For
the fiscal year ended September 30, 2009
OR
¨ TRANSITION
REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF
1934
For
the transition period from ____________ to__________
Commission
File Number 0-50481
______________
AEOLUS
PHARMACEUTICALS, INC.
(Exact
name of registrant as specified in its charter)
______________
|
Delaware
|
56-1953785
|
|
(State
or Other Jurisdiction of
Incorporation
or Organization)
|
(I.R.S.
Employer
Identification
No.)
|
|
26361
Crown Valley Parkway, Suite 150
|
|
|
Mission
Viejo, California
|
92691
|
|
(Address
of principal executive offices)
|
(Zip
Code)
|
Registrant’s
telephone number, including area code: 949-481-9825
Securities
registered pursuant to Section 12(b) of the Act: None
Securities
registered pursuant to Section 12(g) of the Act:
Common
Stock, $.01 par value per share
(Title
of class)
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in
Rule 405 of the Securities Act.
Yes ¨ No ý
Indicate
by check mark if the registrant is not required to file reports pursuant to
Section 13 or Section 15(d) of the Act. Yes ¨ No ý
Indicate
by check mark whether the registrant (1) has filed all reports required to be
filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the
preceding 12 months (or for such shorter period that the registrant was required
to file such reports), and (2) has been subject to such filing requirements for
the past 90 days. Yes ý No ¨
Indicate
by check mark whether the registrant has submitted electronically and posted on
its corporate Web site, if any, every Interactive Data File required to be
submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding
12 months (or for such shorter period that the registrant was required to submit
and post such files). Yes ¨ No ¨
Indicate
by check mark if disclosure of delinquent filers pursuant to Item 405 of
Regulation S-K is not contained herein, and will not be contained, to the best
of registrant’s knowledge, in definitive proxy or information statements
incorporated by reference in Part III of this Form 10-K or any amendment to this
Form 10-K. ¨
Indicate
by check mark whether the registrant is a large accelerated filer, an
accelerated filer, or a non-accelerated filer.
Indicate
by check mark whether the registrant is a shell company (as defined in Rule
12b-2 of the Exchange Act). Yes ¨ No ý
The
aggregate market value of the voting common stock held by non-affiliates of the
registrant based upon the average of the bid and asked price on the OTC Bulletin
Board as of March 31, 2009, the last business day of the registrant’s most
recently completed second fiscal quarter, was approximately
$3,284,000. Shares of common stock held by each executive officer and
director and by each other stockholder who owned 10% or more of the outstanding
common stock as of such date have been excluded in that such stockholder might
be deemed to be an affiliate of ours. This determination of affiliate
status might not be conclusive for other purposes.
As of
December 24, 2009, the registrant had outstanding 48,224,320 shares of common
stock.
INFORMATION
INCORPORATED BY REFERENCE
Portions
of the Company’s definitive Information Statement to be filed pursuant to
Regulation 14C in connection with the registrant’s Written Consent in Lieu of
the 2010 Annual Meeting of Stockholders are incorporated herein by reference
into Part III hereof.
1
AEOLUS
PHARMACEUTICALS, INC.
ANNUAL
REPORT ON FORM 10-K
Table
of Contents
|
Page
|
|||
|
PART
I
|
|||
|
Item
1.
|
|
Business
|
3
|
|
Executive
Officers
|
28
|
||
|
Item
1A.
|
Risk
Factors
|
29
|
|
|
Item
1B.
|
Unresolved
Staff Comments
|
43
|
|
|
Item
2.
|
Properties
|
43
|
|
|
Item
3.
|
Legal
Proceedings
|
43
|
|
|
Item
4.
|
Submission
of Matters to a Vote of Security Holders
|
44
|
|
|
PART
II
|
|||
|
Item
5.
|
Market
for Registrant’s Common Equity, Related Stockholder Matters and Issuer
Purchases of Equity Securities
|
44
|
|
|
Item
6.
|
Selected
Financial Data
|
46
|
|
|
Item
7.
|
Management’s
Discussion and Analysis of Financial Condition and Results of
Operation
|
46
|
|
|
Item
7A.
|
Quantitative
and Qualitative Disclosures About Market Risk
|
53
|
|
|
Item
8.
|
Financial
Statements and Supplementary Data
|
53
|
|
|
Item
9.
|
Changes
in and Disagreements with Accountants on Accounting and Financial
Disclosure
|
72
|
|
|
Item
9A.
|
Controls
and Procedures
|
72
|
|
|
Item
9B.
|
Other
Information
|
73
|
|
|
PART
III
|
|||
|
Item
10.
|
Directors
and Executive Officers of the Registrant
|
73
|
|
|
Item
11.
|
Executive
Compensation
|
73
|
|
|
Item
12.
|
Security
Ownership of Certain Beneficial Owners and Management and Related
Stockholder Matters
|
73
|
|
|
Item
13.
|
Certain
Relationships and Related Transactions
|
73
|
|
|
Item
14.
|
Principal
Accountant Fees and Services
|
73
|
|
|
PART
IV
|
|||
|
Item
15.
|
Exhibits
and Financial Statement Schedules
|
73
|
|
2
PART
I
NOTE
REGARDING FORWARD-LOOKING STATEMENTS
This
Annual Report on Form 10-K contains forward-looking statements within the
meaning of Section 27A of the Securities Act of 1933, as amended, and Section
21E of the Securities Exchange Act of 1934, as amended, that relate to future
events or our future financial performance. You can identify
forward-looking statements by terminology such as “may,” “might,” “will,”
“could,” “should,” “would,” “expect,” “plan,” “anticipate,” “believe,”
“estimate,” “predict,” “intend,” “potential” or “continue” or the negative of
these terms or other comparable terminology. Our actual results might
differ materially from any forward-looking statement due to various risks,
uncertainties and contingencies, including but not limited to those identified
in Item 1A entitled “Risk Factors” beginning on page 29 of this report, as well
as those discussed in our other filings with the Securities and Exchange
Commission and the following:
|
|
·
|
our need for, and our ability
to obtain, additional funds;
|
|
|
·
|
our ability to obtain grants
to develop our drug
candidates;
|
|
|
·
|
uncertainties relating to
non-clinical studies, clinical trials and regulatory reviews and
approvals;
|
|
|
·
|
uncertainties relating to our
pre-clinical trials and regulatory reviews and
approvals;
|
|
|
·
|
our dependence on a limited
number of therapeutic
compounds;
|
|
|
·
|
the early stage of the drug
candidates we are
developing;
|
|
|
·
|
the acceptance of any future
products by physicians and
patients;
|
|
|
·
|
competition with and
dependence on collaborative
partners;
|
|
|
·
|
loss of key consultants,
management or scientific
personnel;
|
|
|
·
|
our ability to obtain adequate
intellectual property protection and to enforce these rights;
and
|
|
|
·
|
our ability to avoid
infringement of the intellectual property rights of
others.
|
Although
we believe that the expectations reflected in the forward-looking statements are
reasonable, we cannot guarantee future results, levels of activity, performance
or achievements. We disclaim any intention or obligation to update or
revise any forward-looking statements, whether as a result of new information,
future events or otherwise.
Item
1. Business.
General
Aeolus
Pharmaceuticals, Inc. (“we” or the “Company”), a Southern California-based
biopharmaceutical company, is developing a new class of catalytic antioxidant
compounds as a medical countermeasure against biological, chemical and
radiological weapons as well as for diseases and disorders of the
central nervous system, respiratory system, autoimmune system and
oncology. Our initial target indications are as a protective agent
against the effects of acute radiation syndrome, sulfur mustard gas exposure and
chlorine gas exposure. We have reported positive safety results from
two Phase I clinical trials of AEOL 10150, our lead drug candidate, with no
serious adverse events noted.
We were
incorporated in the State of Delaware in 1994. Our common stock
trades on the OTC Bulletin Board under the symbol “AOLS.” Our
principal executive offices are located at 26361 Crown Valley Parkway, Suite 150
Mission Viejo, California 92691, and our phone number at that address is (949)
481-9825. Our website address is
www.aeoluspharma.com. However, the information on, or that can be
accessed through, our website is not part of this report. We also
make available free of charge through our website our most recent annual report
on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and
any amendments to those reports, as soon as reasonably practicable after such
material is electronically filed with or furnished to the SEC.
Business
Overview
We are a
pharmaceutical company engaged in development of medical countermeasures against
biological, chemical and radiological weapons. We currently are testing
our lead compound, AEOL 10150, in several indications each in various stages of
development. Our lead indications are:
|
|
·
|
as
a medical countermeasure against the effects of acute radiation syndrome
(“ARS”) in the lungs,
|
|
|
·
|
as
a medical countermeasure against the effects of acute radiation syndrome
in the gastro-intestinal tract,
|
|
|
·
|
as
a medical countermeasure against the effects of sulfur mustard gas,
and
|
|
|
·
|
as
a medical countermeasure against the effects of chlorine
gas.
|
We also
believe that our drug candidates may be an effective agent for preventing or
treating diseases or conditions outside of bio-defense and plan to pursue this
development if we are able to obtain funding specifically for this purpose or
from revenue received from the commercialization of the drug candidates as a
medical countermeasure. We believe the first such
non-bio-defense indication will be as an adjuvant to radiation
therapy. We believe that AEOL 10150 offers the advantages of dual
use
3
(i.e.,
ARS and cancer radiation therapy), the potential ability to address multiple
threats (i.e., ARS, sulfur mustard gas, and chlorine gas) and multiple syndromes
within ARS (pulmonary and gastrointestinal (“GI”)) establish its potential to
offer a revolutionary increase in capability compared with currently available
medical countermeasures (“MCM”).
We also
have two programs underway for the development of our second drug candidate,
AEOL 11207, for the potential treatment of epilepsy and Parkinson’s
disease. These programs are being funded in part by private
foundation and government grants.
Aeolus’
Catalytic Antioxidant Program
The
findings of research on natural antioxidant enzymes and antioxidant scavengers
support the concept of antioxidants as a broad new class of pharmaceuticals if
certain limitations noted below could be overcome. We established our
research and development program to explore and exploit the therapeutic
potential of small molecule catalytic antioxidants. We have achieved
our initial research objectives and have begun to extend our preclinical
accomplishments into non-clinical studies, clinical trials and drug development
programs.
Our
catalytic antioxidant program is designed to:
|
|
·
|
retain
the catalytic mechanism and high antioxidant efficiency of the natural
enzymes, and
|
|
|
·
|
create
and develop stable and small molecule antioxidants without the limitations
of superoxide dismutase (“SOD”) so that
they:
|
|
|
o
|
have
broader antioxidant activity,
|
|
|
o
|
have
better tissue penetration,
|
|
|
o
|
have
a longer life in the body, and
|
|
|
o
|
are
not proteins, which are more difficult and expensive to
manufacture.
|
We have
created a class of small molecules that consume reactive oxygen and nitrogen
species catalytically; that is, these molecules are not themselves consumed in
the reaction. Our class of compounds is a group of manganoporphyrins
(an anti-oxidant containing manganese) that retain the benefits of antioxidant
enzymes, are active in animal models of disease and, unlike the body’s own
enzymes, have properties that make them suitable drug development
candidates. Our most advanced compound, AEOL 10150, is a small
molecule catalytic antioxidant that has shown the ability to scavenge a broad
range of reactive oxygen species, or free radicals. As a catalytic
antioxidant, AEOL 10150 mimics and thereby amplifies the body’s natural
enzymatic systems for eliminating these damaging compounds. Because
oxygen and nitrogen-derived reactive species are believed to have an important
role in the pathogenesis of many diseases, we believe that Aeolus’ catalytic
antioxidants and AEOL 10150 may have a broad range of potential therapeutic
uses. AEOL 10150 has shown efficacy in a variety of animal models,
including sulfur mustard gas exposure, as a protectant against radiation
exposure, ALS, stroke, radiation injury, pulmonary diseases, and
diabetes. We filed an Investigational New Drug Application (“IND”)
for AEOL 10150 in April 2004 under which clinical trials were conducted as more
fully described below under the heading “AEOL 10150 Clinical Development
Program.” For a more detailed description of antioxidants see the
section below titled “Background on Antioxidants.”
AEOL
10150 as a potential medical countermeasure against the effects of acute
radiation syndrome in the lungs
Overview
During
recent years, the threat of nuclear attack on U.S. soil has
increased. The lack of efficient post-exposure treatments for victims
experiencing acute radiation toxicity presents a serious problem should an
attack with a radiological device occur. The most important
components of acute radiation syndrome are the hematopoietic (formation of blood
cellular compounds) and gastrointestinal syndrome. However, both
lethal hematopoietic and gastrointestinal syndromes are potentially avoidable
with proper treatment and complications to later responding tissues may
subsequently become a major problem. A nuclear incident is also
likely to result in a wide inhomogeneous distribution of radiation doses to the
body that allows hematological recovery but a higher exposure to the thorax
leaves open the risk of serious pulmonary complications. Research has
shown that one of the primary concerns associated with the exposure to upper
half body irradiation or total body irradiation is an acute but delayed onset of
radiation pneumonitis (inflammation of lung tissue) with an incidence that rises
very steeply at relatively low radiation doses. Experience points out
that one of the primary complications associated with the exposure to upper half
body irradiation or total body irradiation is pulmonary tissue
toxicity. In situations of accidental exposure, it was initially
assumed that a whole-body dose exceeding 10 Gy was inevitably
fatal. However, experience with nuclear accident victims suggests
that when patients survive gastrointestinal and bone marrow syndromes,
respiratory failure due to radiation pneumonitis and lung fibrosis becomes the
major cause of death. The goal of our research is to complete the
development of a safe and effective medical countermeasure to mitigate
radiation-induced pulmonary injury. AEOL 10150 has been shown in
animal studies to mitigate radiation-induced lung injury when given 2 hours, and
possibly up to 8 weeks after irradiation. AEOL 10150 has demonstrated
statistically significant survival efficacy in an acute radiation-induced lung
injury model. AEOL 10150 has also demonstrated efficacy in validated
animal models for the GI sub-syndrome of ARS. Based on this research,
Aeolus is developing AEOL 10150 as a medical countermeasure (“MCM”) to ARS
related pulmonary injury. AEOL 10150’s pharmacologic mechanism of
action is well supported by findings that correlate efficacy with suppression of
oxidative stress pathways and markers in the lung. Our
current
4
research
is aimed to determine the optimum dose level and duration of treatment to
optimize mitigation of lung injury and to determine the length of the window of
opportunity for initiation of treatment to achieve mitigation of lung
injury.
Pre-
clinical studies
Clinical
experience and experience with nuclear accident victims points out that one of
the primary concerns associated with radiation exposure is an acute but delayed
onset of radiation pneumonitis with an incidence that rises very steeply at
relatively low radiation doses (to 90-percent occurrence at 11 Gray (“Gy”)). To
evaluate AEOL 10150’s ability to mitigate acute radiation-induced lung injury,
mice were exposed to 15 Gy of upper half body irradiation (“UHBI”) and
subsequently treated with AEOL 10150.
In a
study led by Zeljko Vujaskovic, M.D. Ph.D. at Duke University Medical Center,
C57BL/6 female mice were randomized into six groups. Each of the groups was
paired to include irradiated and non-irradiated groups of animals that were
untreated, treated with a low dose (10 mg/kg) of AEOL 10150, or treated with a
high dose of AEOL 10150 (20 mg/kg). Animals received treatments subcutaneously
beginning 2 hours after irradiation (20 and 40 mg/kg initial loading dose,
respectively) followed by a maintenance dose of half the initial dose three
times per week for 4 weeks. Survival, wet lung weights and body weights,
histopathology, and immunohisto-hemistry were used to assess lung damage.
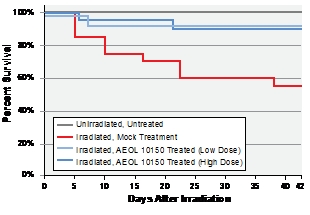
Results demonstrate that treatment with AEOL 10150 increased survival (Fig. 1),
maintained body weight (Fig. 2), protected lung tissue (Fig. 3 and 4), and
reduced oxidative stress (via DNA and protein oxidation analysis) compared with
untreated irradiated animals.
 |
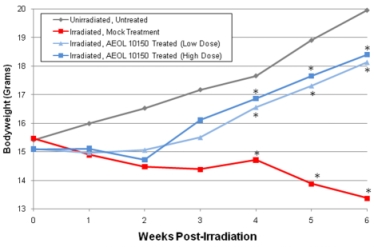 |
| Figure 1. Kaplan Meier survival curves for C57BL/6J mice after upper half body irradiation. The survival data displayed that there were no deaths in the sham-irradiated animals and animals receiving drug alone. In contrast, 9/20 (45%) of the animals that received 15 Gy UHBI died during the 6-week follow-up period. Treatment with low/high doses of AEOL 10150 markedly reduced radiation-induced mortality to only 10% (2/20). | Figure 2: Average body weight changes among groups. UHBI alone mice demonstrated significant weight loss beginning 3 weeks post-exposure compared with UHBI + low/high doses of AEOL 10150 groups. |
 |
 |
|
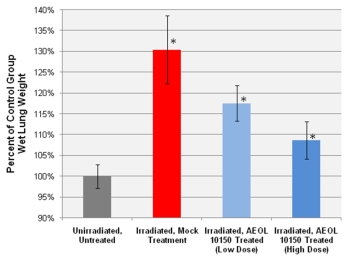
Figure 3. Wet lung weights.
Wet lung weights
were measured as an index of pulmonary edema and consolidation. UHBI alone
mice had significantly higher wet lung weights than did the UHBI +
low/high doses AEOL10150 groups. *=p <
0.05
|
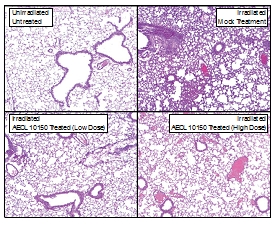
Figure 4. Hematoxylin and Eosin
Staining of Lung Tissue. Lung histology at 6 weeks
revealed a significant decrease in lung structural damage in UHBI +
low/high doses of AEOL 10150 groups, in comparison with UHBI alone. 20x
magnification.
|
Data from
a study in which AEOL 10150 was administered to 40 mice who had been exposed to
radiation also show a statistically significant increase in survival rates among
mice who were treated with AEOL 10150 compared to controls.
5
Additionally,
mice receiving AEOL 10150 experienced a reversal in weight loss seen in the
untreated mice. The six month study, led by Zeljko Vujaskovic, M.D.
Ph.D. at Duke University Medical Center, was designed to test the efficacy of
AEOL 10150 as a treatment for damage to the lungs due to exposure to
radiation. At 45 days, all of the animals in the untreated group had
either died or had to be sacrificed based on animal care rules. The
remaining animals that received AEOL 10150 did not need to be sacrificed based
on animal care rules, but a majority were sacrificed in order to increase the
numbers that could be compared to the untreated animals sacrificed at 45 days,
since there would be no untreated animals for comparison at the end of six
months. A number of treated animals continued to be monitored until the
end of six months and analysis of those animals is currently
underway. In addition to the statistically significant (P< 0.05)
survival advantage, statistically significant differences in body weights and
wet lung weights were seen over the first six weeks of the
study. Untreated mice experienced a steady decline in body weight
over the six weeks, while treated animals experienced weight gain that was just
slightly less than that seen in the controls (animals not receiving
radiation). AEOL 10150 also demonstrated statistically significant
reductions in markers for oxidative stress and inflammation – secondary
endpoints for the study.
A number
of other preclinical studies by Zjelko Vujaskovic, MD, PhD; Mitchell Anscher,
MD, et al at Duke University have demonstrated the efficacy of AEOL 10150 in
radioprotection of normal tissue. Chronic administration of AEOL
10150 by continuous, subcutaneous infusion for 10 weeks has demonstrated a
significant protective effect from radiation-induced lung injury in
rats. Female Fisher 344 rats were randomly divided into four
different dose groups (0, 1, 10 and 30 mg/kg/day of AEOL 10150), receiving
either short (one week) or long-term (ten weeks) drug administration via osmotic
pumps. Animals received single dose radiation therapy of 28 Gy to the
right hemithorax. Breathing rates, body weights, histopathology and
immunohistochemistry were used to assess lung damage. For the long
term administration, functional determinants of lung damage 20 weeks
post-radiation were significantly decreased by AEOL 10150. Lung
histology at 20 weeks revealed a significant decrease in structural damage and
fibrosis. Immunohistochemistry demonstrated a significant reduction
in macrophage accumulation, collagen deposition and fibrosis, oxidative stress
and hypoxia in animals receiving radiation therapy along with AEOL
10150. There were no significant differences between the irradiated
controls, and the 3 groups receiving short-term administration of AEOL 10150 and
single dose radiation therapy. Figure 5 below shows a
semi-quantitative analyses of lung histology at 20 weeks which revealed a
significant decrease in structural damage and its severity in animals receiving
10 and 30 mg/kg/day after radiation in comparison to radiation therapy along
with placebo group or radiation therapy along with 1 mg/kg of AEOL 10150 (p =
0.01).
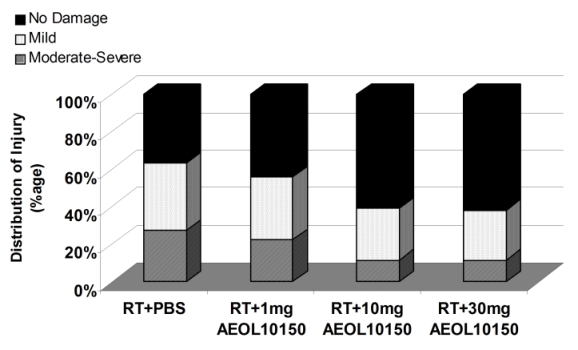
Figure 5 above shows that AEOL
10150 treatment decreases the severity of damage and increases the percentage of
lung tissue with no damage from radiation therapy (Rabbani et al Int J Rad Oncol
Biol Phys 67:573-80, 2007).
Two
additional studies examining the effect of subcutaneous injections of AEOL 10150
on radiation-induced lung injury in rats have been completed. The
compound was administered subcutaneously by a b.i.d. dosing regime (i.e. 2.5
mg/kg or 5.0 mg/kg) on the first day of radiation and daily for five consecutive
weeks. Radiation was fractionated rather than single dose, with 40 Gy
divided in five 8 Gy doses. Preliminary immunohistologic analyses of
the lung tissue from these two studies showed a dose dependent decrease in the
inflammatory response quantified by the number of activated macrophages or areas
of cell damage.
These in
vivo studies employing subcutaneous administration of AEOL 10150, either by
continuous infusion via osmotic pump or BID injection, demonstrate that AEOL
10150 protects healthy lung tissue from radiation injury delivered either in a
single dose or by fractionated radiation therapy doses. AEOL 10150
mediates its protective effect(s) by inhibiting a number of events in the
inflammatory cascade induced by radiation damage. Additional in vivo
studies have been performed that provide support for manganoporphyrin
antioxidant protection of lung tissue from radiation. Treatment with
a related manganoporphyrin compound, AEOL 10113 significantly improved pulmonary
function, decreased histopathologic markers of lung fibrosis, decreased collagen
(hydroxyproline) content, plasma levels of the profibrogenic cytokine,
transforming growth factor beta (TGF-β) and, as demonstrated by immunohistochemistry
of lung tissue, collagen deposition and TGF-β.
6
In
summary, AEOL 10150 has consistently shown a protective effect against the
harmful effects in radiation, including when the drug is administered up to 72
hours after exposure.
We have
recently initiated another study in mice to determine the optimal length of
treatment with AEOL 10150 when used as a MCM to ARS in the
lungs. This study, led by Zeljko Vujaskovic, M.D. Ph.D. at Duke
University, is designed to build on the recently completed study that
demonstrated the efficacy of AEOL 10150 as a treatment for damage to the lungs
due to exposure to radiation (described in detail above), and determine the most
effective duration of delivery for treatment after exposure. We
expect that this study will provide important insight into the most appropriate
length of treatment with AEOL 10150 to protect the lungs following exposure to
radiation and will be the basis for the design of a pivotal study in mice, which
we expect will be one of the two species necessary for approval of AEOL 10150 as
a MCM to ARS under the US FDA’s Animal Rule.
Non-
clinical studies
We have
initiated a study to confirm the efficacy of AEOL 10150 as a countermeasure to
nuclear and radiological exposure in non-human primates. The new
study is designed to test the efficacy of AEOL 10150 as a treatment for damage
to the lungs due to exposure to radiation and to begin establishing an animal
model that can be validated and could be utilized by the U.S. Food and Drug
Administration (“FDA”) for approval of a countermeasure for Pulmonary Acute
Radiation Syndrome under the “Animal Rule”. The FDA “Animal Rule”
enumerates criteria whereby the FDA can rely on animal efficacy data when
“evidence is needed to demonstrate efficacy of new drugs against lethal or
permanently disabling toxic substances when efficacy studies in humans,
ethically cannot be conducted.” The criteria are discussed below.
The new
study builds on results from previous studies in rats and mice where it was
shown that doses in the range of 5 to 30 mg/kg AEOL 10150 given daily up to 24
hours after irradiation and administered for as long as 10 weeks mitigate
functional lung injury in rats and led to a statistically significant survival
advantage in mice. Investigators will examine both the acute and latent effects
of radiation on the lungs and the impact that AEOL 10150 has on mitigating those
effects. In rodents, non-human primates and humans, radiation of the
lungs can cause reduced breathing capacity, pneumonitis, fibrosis, weight loss
and death and is characterized by oxidative stress, inflammation and elevated
macrophage counts. AEOL 10150 has proven to be an effective
countermeasure to radiation exposure of the lungs in mice and rats in published
studies such as Rabbani et al Int J Rad Oncol Biol Phys 67:573-80, 2007, Rabbani
et al Free Rad Res 41:1273-82, 2007 and Gridley et al Anticancer Res 27:3101-9,
2007.
Clinical
studies
We
believe our two previous phase 1 clinical studies can be utilized in any
potential investigational new drug application (an “IND”) and New Drug
Application (“NDA”) filing with the FDA for AEOL 10150 as MCM for
ARS. At this time, we do not have any clinical trials underway but
are in the process of planning additional safety studies.
Future
Development Plans
Aeolus’
objective is to develop AEOL 10150 as a MCM against ARS pulmonary effects, via
the FDA’s “Animal Rule”. This development pathway requires
demonstration of the key study efficacy parameter of AEOL 10150 treatment in two
animal models relevant to the human radiation response and its treatment,
demonstration of safety in humans, demonstration of relevant dosing and
administration in humans, and clear identification of the mechanism of
radiation-induced damage to the lung and its amelioration by the drug
candidate.
AEOL
10150 has several distinct advantages as a MCM, including the
following:
|
|
·
|
Demonstrated
survival increase in animal studies when administered 2 hours after
exposure (P<0.05),
|
|
|
·
|
Demonstrated
reduction in lung fibrosis in animal studies up to 24 hours post exposure
(P<0.05),
|
|
|
·
|
Demonstrated
histological improvement in lung tissue post-radiation
exposure,
|
|
|
·
|
Addresses
an unmet medical need as a MCM to lung
ARS,
|
|
|
·
|
Established
safety profile in both clinical and pre-clinical
studies,
|
|
|
·
|
Subcutaneous
self-administration possible by exposed individuals during
emergency,
|
|
|
·
|
Rapid
administration, allowing large numbers of patients to be treated
quickly,
|
|
|
·
|
Stable
for up to 4½ years at 0–8°C and 1 year at room
temperature,
|
|
|
·
|
Requires
no non-standard storage conditions (i.e., not
photosensitive),
|
|
|
·
|
Currently
in development as an adjunct to radiation therapy; if approved will
provide a pre-existing distribution and stockpile resource at oncology
centers in the event of a radiological
emergency,
|
|
|
·
|
Demonstrated
potential as both a therapeutic and
prophylactic,
|
|
|
·
|
Demonstrated
potential to address multiple sub-syndromes of
ARS,
|
|
|
·
|
Demonstrated
potential to address sulfur mustard gas and chlorine gas exposure,
and
|
|
|
·
|
Potential
dual use as an adjunct treatment for cancer patients receiving radiation
therapy.
|
7
We believe in order to
file an NDA for ARS with the FDA, we will need to demonstrate efficacy in Animal
Models and demonstrate product safety which is based upon the FDA’s “Animal
Rule”. We also plan on pursuing Fast Track submission
status for this indication, enabling priority review and enabling rolling NDA
submission if accepted by the FDA.
The FDA
“Animal Rule” enumerates criteria whereby the FDA can rely on animal
efficacy data when “evidence is needed to demonstrate efficacy of new drugs
against lethal or permanently disabling toxic substances when efficacy studies
in humans, ethically cannot be conducted. The criteria are as
follows:
|
|
·
|
Knowledge
of the mechanism of radiation-induced damage to the lung and its
amelioration by the candidate drug.
|
|
|
·
|
Pharmacokinetic
and pharmacodynamic analysis to provide information on relevant dose and
administration schedule.
|
|
|
·
|
Direct
correlation of key study parameters (e.g., survival or major morbidity)
with the desired clinical benefit in
humans.
|
|
|
·
|
Collection
of efficacy data in two species relevant to the human radiation response
and its treatment unless otherwise justified under GLP-compliant
conditions.
|
|
|
·
|
A
Phase 1 safety trial using the same product and formulation as used in the
pivotal trial(s) required.
|
Demonstrate
Efficacy in Animal Models
Our
efficacy plan is designed to accomplish two key goals: the validation of two
animal models for acute radiation-induced lung injury and the generation of
pivotal efficacy data in these species. The efficacy data produced in pivotal
studies using these validated models will provide the data required to
demonstrate efficacy of AEOL 10150 at the dose and schedule proposed for
licensure. A second criterion of the “Animal Rule” is that the models must be
reflective of “real world” conditions to which a human is likely to be exposed.
The proposed models have been designed to reflect these real world conditions.
Initial studies have been conducted with whole thorax exposure models to
irradiate the total lung parenchym, and will be followed by studies with Total
Body Irradiation (“TBI”) with shielding of roughly 5 percent of bone marrow.
This study design mimics real world conditions in which it is anticipated that
many of those exposed to radiation will benefit from some shielding (e.g., from
cars, buildings, etc.), which will protect some bone marrow and allow for
survival without a bone marrow transplant. This shielding approach has been used
to develop both murine and NHP models for GI ARS and in the NHP for
radiation-induced lung injury.
Demonstrate
Product Safety
For
product approval under the Animal Rule, we will also demonstrate product safety
using the same product and formulation used in the animal efficacy trials and
proposed for use in humans. Demonstration of safety includes preclinical
demonstration of safety via the standard pre-clinical studies and analyses
methods and Phase I safety trials sufficient to demonstrate product safety in
the target patient population. We believe our safety studies
completed as a therapy for ALS may be utilized to demonstrate safety for this
indication. As a result, we believe it may only be necessary to
conduct mutagenicity and teratogenicity studies for completion of the
pre-clinical package required for NDA filing and product
licensure. We also plan to conduct two additional Phase I clinical
safety studies in patients receiving radiation therapy. This will
provide safety data in a population similar to that for which the final product
indication will be targeted (i.e., patients receiving high radiation doses
sufficient to cause radiation-related lung damage) for a longer period of
treatment with our compound. A secondary benefit of using this population is
that prophylactic effects of the product can also be demonstrated; efficacy
measures toward achieving this end will be incorporated into the study
design.
Competition
Currently
there are no FDA-approved drugs for the treatment of ARS related pulmonary
injury. We are also not aware of any other drug candidates that
have demonstrated the ability to protect the lungs from radiation given post
exposure which we believe is a critical aspect of the development of a medical
countermeasure against the effects of acute radiation syndrome.
However,
in general, we face significant competition for U.S. government funding for both
development and procurement of medical countermeasures for biological, chemical
and nuclear threats, diagnostic testing systems and other emergency preparedness
countermeasures. The U.S. federal government has currently allocated
a significant amount of research funding to the development of countermeasures
against the effects of radiation exposure. As a result, there are
many drug candidates under development as a possible countermeasure against the
effects of radiation exposure.
Funding
Options
We are
currently internally funding the development of AEOL 10150 as a MCM for ARS
related pulmonary injury, however, AEOL 10150 was recently chosen by the
NIH/NIAID for evaluation in their Medical Countermeasures Against Radiological
Threats (“MCART”) program for the treatment of ARS related GI and pulmonary
injury.
We have
also submitted a white paper to the Biomedical Advanced Research and Development
Authority (“BARDA”) in response to Special Instructions Amendment 4 to a Broad
Agency Announcement (BAA-BARDA-09-34) for advanced research
8
and
development of medical countermeasures for chemical, biological, radiological
and nuclear threats. The purpose of the special instructions amendment is to
specifically solicit solutions for treating cutaneous and/or pulmonary
(life-threatening pneumonitis and fibrosis) injuries resulting from exposure to
ionizing radiation. BARDA is interested in advanced development and eventual
licensure/approval of medical countermeasures for cutaneous and/or pulmonary
injuries resulting from an acute exposure to radiation from a
radiological/nuclear accident or attack, particularly injuries associated with
ARS or Delayed Effects of Acute Radiation Exposure (“DEARE”). BARDA
is seeking safe and effective medical countermeasures that mitigate, treat,
affect, delay, or interrupt the progression of cutaneous and/or pulmonary injury
caused by ionizing radiation. Aeolus is requesting funding to further develop
AEOL 10150 for the treatment and prevention of such pulmonary injuries,
including pneumonitis and fibrosis.
We are
also exploring other possible funding sources such as the Department of Defense
(the “DoD”) and the Chemical Biological Medical Systems Joint Project Management
Office (“CBMS-JPMO”).
AEOL
10150 as a potential medical countermeasure against the effects of radiation on
the gastro-intestinal tract
Overview
Acute
radiation syndrome of the gastro-intestinal tract (“GI-ARS”) is a massive,
currently untreatable, problem following high-dose, potentially lethal radiation
exposure. Agents that mitigate these effects would reduce sickness and hopefully
prevent fatalities. The intestinal epithelium, a single layer of
cells lining the surface of the GI lumen, is responsible for vital functions of
nutrient absorption, maintaining fluid and electrolyte balance and protection of
the body from bacteria, bacterial toxins and non absorbed materials. The
functional integrity of the GI system is maintained via incessant production of
epithelial cells from specialized stem cells located in crypts at the base of
the epithelium. High-dose, total-body irradiation can result in a
lethal GI syndrome that results in significant morbidity and mortality within
days consequent to killing of the crypt stem cells and loss of the protective
and absorptive epithelial barrier. There are no FDA-approved drugs or biologics
to treat the acute GI-ARS syndrome.
Pre-
clinical studies
The
National Institutes of Health’s (“NIH”), National Institute of Allergy and
Infectious Diseases (“NIAID”) Radiation/Nuclear Medical Countermeasures
development program is currently testing AEOL 10150 as a countermeasure for
radiation exposure to the GI tract. The study is being funded by the NIAID and
is designed to test the efficacy of AEOL 10150 as a treatment for damage to the
GI tract due to exposure to radiation. The study protocol calls for
the examination of both histological and survival endpoints in C57BL/6 mice in a
multi-armed vehicle-controlled trial. For the histological portion,
crypt histology will be assessed with crypt number and crypt width being the
primary endpoint. Animals receiving AEOL 10150 began dosing 24 hours
after radiation exposure and receive one dose per day for the remainder of the
study. Preliminary results have demonstrated that AEOL 10150 can
effectively increase regeneration of GI stem cells, reduce the severity and
duration of diarrhea and improve survival when administered at 24 hours after
doses of total-body irradiation that produce the lethal GI
syndrome. The study is being conducted by Epistem in compliance
with criteria of the FDA that are a pre-requisite for movement of the Aeolus
drug along the pathway for FDA licensure to treat lethally irradiated persons in
the event of a terrorist nuclear act. Epistem operates a major
contract research organization and provides services to identify novel drugs
that can protect or improve the repair of the GI tract following exposure to
irradiation and performed these studies as part of NIH’s program for the
screening of a novel agents for bio-defense applications.
We are
unaware of any published studies of agents that accomplish this enhanced stem
cell regenerative effect while maintaining GI function and improving survival
when administered post irradiation.
Future
Development Plans
In
collaboration with the NIH NIAID, we are planning additional studies to confirm
the efficacy results demonstrated in the first study described
above. We also expect to perform additional studies which could be
funded by NIH, NIAID to optimize dose and duration of delivery, and to evaluate
the window of opportunity for treatment after exposure.
Upon
completion of these studies we would need to demonstrate efficacy in Animal
Models and demonstrate product safety based upon the FDA’s “Animal
Rule”. We also plan on pursuing Fast Track submission
status for this indication, enabling priority review and enabling rolling NDA
submission if accepted by the FDA. Under the ‘Animal Rule’, we would
need to complete pivotal studies in two species relevant to the human radiation
response and its treatment. We believe that these studies can be
completed using existing validated models for both murine and
NHP. This study design would also mimic real world conditions in
which it is anticipated that many of those exposed to radiation will benefit
from some shielding (e.g., from cars, buildings, etc.), which will protect some
bone marrow and allow for survival without a bone marrow
transplant.
We will
also demonstrate product safety using the same product and formulation used in
the animal efficacy trials and proposed for use in humans. Demonstration of
safety includes preclinical demonstration of safety via the standard
pre-clinical studies and analyses methods and Phase I safety trials sufficient
to demonstrate product safety in the target patient population. We
believe our safety studies completed as a therapy for ALS may be utilized to
demonstrate safety for this indication. As a result,
9
we
believe it may only be necessary to conduct mutagenicity and teratogenicity
studies for completion of the pre-clinical package required for NDA filing and
product licensure. We also plan to conduct two additional Phase I
clinical safety studies in patients receiving radiation therapy. This
will provide safety data in a population similar to that for which the final
product indication will be targeted (i.e., patients receiving high radiation
doses sufficient to cause radiation-related lung damage) for a longer period of
treatment with our compound. A secondary benefit of using this population is
that prophylactic effects of the product can also be demonstrated; efficacy
measures toward achieving this end will be incorporated into the study
design.
Competition
We are
unaware of any compounds that protect crypt cells and that increase survival
when given to animals exposed to radiation at levels greater than 10 Gys and
given after exposure. There are several companies developing drug
candidates that have shown efficacy when given prior to exposure or at lower
levels of radiation.
However,
in general, we face significant competition for U.S. government funding for both
development and procurement of medical countermeasures for biological, chemical
and nuclear threats, diagnostic testing systems and other emergency preparedness
countermeasures. The U.S. federal government has currently allocated
a significant amount of research funding to the development of countermeasures
against the effects of radiation exposure. As a result, there are
many drug candidates under development as a possible countermeasure against the
effects of radiation exposure.
Funding
Options
AEOL
10150 as a MCM for GI-ARS is being tested by the NIH NIAID Medical
Countermeasures Against Radiological Threats (“MCART”). The NIH NIAID
MCART development program leads the U.S. effort to develop treatments for
radiation sickness following a nuclear terrorist attack. GI-ARS is a
potentially fatal, currently untreatable, problem following high-dose radiation
exposure. Agents that mitigate these effects could reduce sickness and hopefully
prevent fatalities. There are currently no medications approved by the FDA to
treat this syndrome.
We are
also exploring other possible funding sources such as BARDA and the
DoD.
AEOL
10150 as a potential medical countermeasure against the effects of sulfur
mustard gas
Overview
Sulfur
mustards, of which sulfur mustard gas is a member, are a class of related
cytotoxic, vesicant chemical warfare agents with the ability to form large
blisters on exposed skin and cause pneumonitis and fibrosis in the
lungs. In their pure form most sulfur mustards are colorless,
odorless, viscous liquids at room temperature. When used as warfare
agents they are usually yellow-brown in color and have an odor resembling
mustard plants, garlic or horseradish. Mustard agents, including
sulfur mustard, are regulated under the 1993 Chemical Weapons
Convention. Three classes of chemicals are monitored under this
Convention, with sulfur and nitrogen mustard grouped in the highest risk class,
"schedule 1". However, concerns about its use in a terrorist attack
have lead to resurgence in research to develop a protectant against
exposure.
The
increased risk of a terrorist attack in the United States involving chemical
agents has created new challenges for many departments and agencies across the
federal government. Within the Department of Health and Human
Services, the NIH is taking a leadership role in pursuing the development of new
and improved medical countermeasures designed to prevent, diagnose, and treat
the conditions caused by potential and existing chemical agents of
terrorism. In addition, many of the same chemicals posing a threat as
terrorist agents may also be released from transportation and storage facilities
by industrial accidents or during a natural disaster. The NIH has
developed a comprehensive Countermeasures Against Chemical Threats
(“CounterACT”) Research Network that includes Research Centers of Excellence,
individual research projects, SBIRs, contracts and other
programs. The CounterACT network is conducting basic, translational,
and clinical research aimed at the discovery and/or identification of better
therapeutic and diagnostic medical countermeasures against chemical threat
agents, and their movement through the regulatory process. The
overarching goal of this research program is to enhance our diagnostic and
treatment response capabilities during an emergency.
Another
critical goal of the CounterACT program is to assist in the development of safe
and effective medical countermeasures designed to prevent, diagnose, and treat
the conditions caused by potential and existing chemical agents of terrorism
which can be added to the Nation’s Strategic National Stockpile
(“SNS”). The SNS is maintained by the Centers for Disease Control and
Prevention (“CDC”). The SNS now contains CHEMPACKS which are located
in secure, environmentally controlled areas throughout the United States
available for rapid distribution in case of emergency. The CDC has
established a diagnostic response network for the detection of nerve agents,
mustard, cyanide and toxic metals. The NIH will continue to research,
develop and improve medical products that include chemical antidotes, drugs to
reduce morbidity and mitigate injury, drugs to reduce secondary chemical
exposure and diagnostic tests and assessment tools to be used in mass casualty
situations.
10
Sulfur
mustard gas is a strong vesicant (blister-causing agent). Due to its
alkylating properties, it is also strongly mutagenic (causing damage to the DNA
of exposed cells) and carcinogenic (cancer causing). Those exposed
usually suffer no immediate symptoms. Within 4 to 24 hours the
exposure develops into deep, itching or burning blisters wherever the mustard
contacted the skin; the eyes (if exposed) become sore and the eyelids swollen,
possibly leading to conjunctivitis and blindness. At very high
concentrations, if inhaled, it causes bleeding and blistering within the
respiratory system, damaging the mucous membrane and causing pulmonary
edema. Blister agent exposure over more than 50% body surface area is
usually fatal.
The NIH
awarded a five-year, $7.8 million grant to National Jewish Health and the
University of Colorado Health Sciences Center, both in Denver,
Colorado. This Center of Excellence was developed to focus on sulfur
mustard toxicity in the lung and skin and the long-term goal is to develop an
effective treatment for sulfur mustard gas induced injury in lung and
skin. Members of the Center have been testing numerous compounds with
the goal to identify optimal compounds, route and mode of delivery. Research
projects are ongoing to determine countermeasures that will help establish
specific interventions needed to treat sulfur mustard gas-induced
injury. After three years of research, AEOL 10150 has been
identified by the National Jewish Health Center of Excellence as the lead
compound for its center, and research work there has been focused on further
testing and studies of AEOL 10150.
Research
in the area of sulfur mustard gas-mediated lung injury has provided experimental
evidence that the mechanisms of these injuries are directly linked to the
formation of reactive oxygen and nitrogen species and that superoxide dismutase
and catalase can improveinjury responses. This theory has led to the
hypothesis that the administration of catalytic antioxidant therapy can protect
against sulfur mustard gas-induced acute lung and dermal injury. AEOL
10150 has already been shown to be well tolerated in humans and could be rapidly
developed as a drug candidate in this area pending animal efficacy
data.
Researchers
have found that the chemical warfare agent analog, 2-chloroethyl ethyl sulfide
(“CEES”)-induced lung injury could be improved by both exogenous superoxide
dismutase and catalase. Both of these natural enzymes are important
catalytic antioxidants and both of these reactions are exhibited by
metalloporphyrins. CEES-induced lung injury is dependent in part upon
blood neutrophils. Activated neutrophils are an important source of
reactive oxygen species that are known to contribute to lung injury
responses. Antioxidants have also been shown to protect against
CEES-induced dermal injury. Mustard exposure is often associated with
producing acute respiratory distress syndrome (“ARDS”) that requires
supplemental oxygen therapy to maintain adequate tissue
oxygenation.
Pre-clinical
studies
A study
performed by researchers from National Jewish Health demonstrated that AEOL
10150 showed statistically significant protection of lung tissue in animals
exposed to CEES or half-mustard. In a study sponsored by the
NIH CounterACT program, AEOL 10150 was tested along with 19 other compounds to
determine effectiveness in protecting lung tissue against edema and hemorrhage
resulting from exposure to sulfur mustard gas.
AEOL
10150 was given to rats one hour after CEES exposure and again 6 hours later.
Eighteen hours after exposure, lung edema and hemorrhage was assessed by changes
in the bronchoalveolar lavage protein and red blood cell levels. AEOL
10150 significantly reduced (p<0.05) sulfur mustard gas-induced lung edema
and hemorrhage. These results suggest that AEOL 10150 rescues the lung from
sulfur mustard gas exposure and may provide a countermeasure against sulfur
mustard gas-induced lung injury. Further studies at National Jewish
Health and the University of Colorado showed that doses in the range of 5 to 30
mg/kg of AEOL 10150 given at one and eight hours after exposure mitigate both
lung and skin injury in animal models. Doses in the range of 5 to 10
mg/kg/d showed the most potent effect including significant mitigation as
assessed by histopathology and immunohistochemistry.
Non-clinical
studies
In 2009,
several studies were launched to test the efficacy of AEOL 10150 as a treatment
for damage to the skin and lungs due to exposure to sulfur mustard
gas and to examine potential effective doses, duration of delivery and the
window of opportunity for treatment after exposure. The studies are
being conducted using “whole” sulfur mustard gas at Lovelace Respiratory
Research Institute, another CounterACT Center of Excellence, and using CEES at
National Jewish Health and build on results from previous studies using CEES
conducted at National Jewish Health and the University of Colorado.
The first
whole sulfur mustard study was completed in October 2009. The study
demonstrated that AEOL 10150 protects lungs from whole sulfur mustard gas
exposure in rats. The data affirmed our earlier studies
where AEOL 10150 protected the lung against the half-mustard,
CEES. The primary objective of the studies was to determine whether
administration of AEOL 10150, after exposure, reduces the severity of acute lung
injury induced by sulfur mustard gas. AEOL 10150 was given to
rats one hour after sulfur mustard exposure and repeated every 6
hours. Twenty-four hours after exposure, lung edema was assessed by
changes in the bronchoalveolar lavage (“BAL”) protein levels. AEOL 10150
significantly reduced (p<0.05) sulfur mustard gas-induced lung edema as
measured by BAL protein levels. In addition, AEOL 10150 decreased
SM-induced increase in the numbers of BAL neutrophils. These results indicate
that AEOL 10150 can attenuate lung injury from sulfur mustard gas exposure and
may provide an effective countermeasure against sulfur mustard gas-induced lung
injury.
11
Future
Development Plans
Upon
completion of the ongoing studies, we expect to perform additional studies to
confirm the results obtained in CEES and whole sulfur mustard
studies.
Following
these confirmatory studies, we seek to launch the two pivotal efficacy studies
required for approval by the FDA under the ‘Animal Rule’ as well as complete the
necessary safety studies as further described under the heading AEOL 10150 as a potential medical countermeasure
against the effects of acute radiation syndrome in the lungs – Future
Development Plans – Demonstrate Product Safety.
Competition
There are
currently no effective treatments for sulfur mustard gas exposure and AEOL 10150
is a major focus of a sponsored research grant awarded by the NIH CounterAct
program to National Jewish Health to identify an effective
treatment.
However,
in general, we face significant competition for U.S. government funding for both
development and procurement of medical countermeasures for biological, chemical
and nuclear threats, diagnostic testing systems and other emergency preparedness
countermeasures. The U.S. federal government has currently allocated
a significant amount of research funding to the development of countermeasures
against bioterrorism. As a result, there are many drugs candidates
under development as a possible countermeasure against chemical threat
agents.
Funding
Options
This
development program to date has been funded under the NIH CounterAct Program and
we expect that future efficacy studies necessary for approval by the FDA will
also be funded by the NIH CounterAct program. We also believe funding
could be available from BARDA.
AEOL
10150 as a potential medical countermeasure against the effects of chlorine
gas
Overview
Like
sulfur mustard, chlorine gas is a toxic gas that confers airway injury through
primary oxidative stress and secondary inflammation. Chlorine
inhalation was recently used in terrorist/insurgent attacks on military and
civilian populations, and has caused numerous industrial, transportation,
swimming pool, and household accidents, as well as deaths to members of the US
military in the past. Chlorine gas, also known as bertholite, was
first used as a weapon in World War I. Chlorine gas was also used against the
local population and coalition forces in the Iraq War in the form of Chlorine
bombs.
Worldwide,
independent of warfare and chemical terrorism, chlorine is the greatest single
cause of major toxic release incidents. In the US, there are about
5-6,000 exposures per year resulting in, on average, about one death, 10 major,
400-500 moderate, and 3-4,000 minor adverse outcomes. Like mustard,
chlorine causes damage to upper and lower respiratory tracts. While
chlorine is an irritant, its intermediate water solubility may delay emergence
of upper airway symptoms for several minutes. Aqueous decomposition
of chlorine gas forms hydrochloric acid and hypochlorous acid, itself also a
product of inflammation. Cell injury is thought to result from
oxidation of functional groups in cell components, from tissue formation of
hydrochloric acid and hypochlorous acid, and possibly from formation of other
reactive oxygen species (“ROS”). For treatment of acute exposures in
humans, decontamination, supplemental oxygen, treatment of bronchospasm and/or
laryngospasm, and supportive care are the only accepted therapies, while use of
nebulized sodium bicarbonate and parenteral and/or inhaled steroids remain quite
controversial. No specific beneficial therapies are
available. We expect that AEOL 10150 will decrease airway injury,
inflammation, oxidative damage, hyperreactivity, and cell proliferation after
acute chlorine gas inhalation in mice and therefore could be a possible
beneficial therapy for chlorine gas inhalation injury to the
airways.
Pre-clinical
studies
Researchers
from National Jewish Health and McGill University have completed a series of
preliminary studies demonstrating that AEOL 10150 protects lungs from chlorine
gas exposure in mice. The primary objective of these studies
was to determine whether administration of AEOL 10150, after exposure, reduces
the severity of acute lung injury and asthma-like symptoms induced by chlorine
gas. AEOL 10150 was given to mice at a 5 mg/kg subcutaneous
dose one hour after chlorine gas exposure (100 ppm for 5 minutes) and repeated
every 6 hours. Twenty-four hours after exposure, lung inflammation
was assessed by changes in BAL cellularity and neutrophil influx. AEOL 10150
significantly reduced (p<0.05, n=6/group) chlorine gas-induced lung
inflammation as measured by BAL fluid cellularity levels by 40% that appeared to
be due to limiting neutrophil influx. AEOL 10150 also significantly
attenuated (p<0.05, n=6) the degree of asthma-like airway reactivity induced
by chlorine gas exposure by 40%. These results indicate that AEOL 10150 can
attenuate lung injury and asthma-like symptoms from chlorine gas exposure and
may provide an effective countermeasure against chlorine gas-induced lung
injury.
12
Future
Development Plans
We expect
that the development plan will mirror the development of AEOL 10150 as a
potential medical countermeasure against the effects of sulfur mustard gas
above.
Competition
There are
currently no effective treatments for chlorine gas exposure and AEOL 10150 is a
major focus of the NIH CounterACT program to identify an effective
treatment.
However,
in general, we face significant competition for U.S. government funding for both
development and procurement of medical countermeasures for biological, chemical
and nuclear threats, diagnostic testing systems and other emergency preparedness
countermeasures. The U.S. federal government has currently allocated
a significant amount of research funding to the development of countermeasures
against bioterrorism. As a result, there are many drugs candidates
under development as a possible countermeasure against chemical threat
agents.
Funding
Options
We expect
that the development of AEOL 10150 as a potential countermeasure against the
effects of Chlorine gas will be funded by the NIH CounterACT
program.
AEOL
10150 in Radiation Therapy
Overview
According
to the American Cancer Society, cancer is the second leading cause of death by
disease representing one out of every four deaths in the United States with an
expected 562,000 Americans expected to die of cancer in 2009. In
2009, nearly 1.5 million new cancer cases are expected to be diagnosed in the
United States. The NIH estimates overall costs of cancer in 2008 in
the United States at $228.1 billion: $93.2 billion for direct medical costs,
$18.8 billion for indirect morbidity costs (costs of lost productivity due to
illness) and $116.1 billion for indirect mortality costs (cost of lost
productivity due to premature death).
Combinations
of surgery, chemotherapy and radiation treatments are the mainstay of modern
cancer therapy. Success is often determined by the ability of
patients to tolerate the most aggressive, and most effective, treatment
regimens. Radiation therapy-induced toxicity remains a major factor
which limits the ability to escalate radiation doses in the treatment of
tumors. The ability to deliver optimal radiation therapy for
treatment of tumors without injury to surrounding normal tissue has important
implications in oncology because higher doses of radiation therapy may improve
both local tumor control and patient survival. Advances in the tools
of molecular and cellular biology have enabled researchers to develop a better
understanding of the underlying mechanisms responsible for radiation
therapy-induced normal tissue injury. For decades ionizing radiation
has been known to increase production of free radicals, which is reflected by
the accumulation of oxidatively damaged cellular macromolecules. As
one example of radiation-induced damage to adjacent normal tissue, radiation
therapy may injure pulmonary tissue either directly via generation of ROS or
indirectly via the action on parenchymal and inflammatory cells through
biological mediators such as transforming growth factor beta (TGF B) and
pro-inflammatory cytokines. Since the discovery of SOD, it has become
clear that these enzymes provide an essential line of defense against
ROS. SODs and SOD mimics, such as AEOL 10150, act by catalyzing the
degradation of superoxide radicals into oxygen and hydrogen
peroxide. SODs are localized intra/extracellularly, are widely
expressed throughout the body, and are important in maintenance of redox status
(the balance between oxidation and reduction). Previous studies have
demonstrated that treating irradiated animal models with SOD delivered by
injection of the enzyme through liposome/viral-mediated gene therapy or
insertion of human SOD gene can ameliorate radiation therapy-induced
damage. For an illustrative example of the radiation therapy reaction
see Figure 6 below.
13
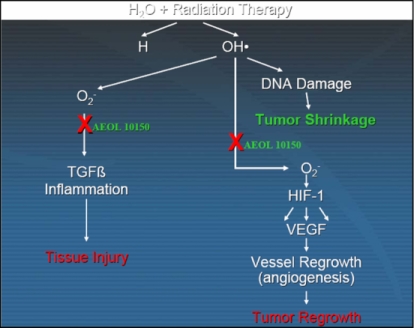
Figure 6 above shows the dual
mechanism of action of radiation therapy and the application of AEOL 10150 to
the process.
In vitro
studies have demonstrated that AEOL 10150 reduces the formation of lipid
peroxides and that it inactivates biologically important ROS molecules such as
superoxide, hydrogen peroxide, and peroxynitrite. AEOL 10150
inactivates these ROS by one or two electron oxidation or reduction reactions in
which the oxidation state of the manganese moiety in AEOL 10150
changes. AEOL 10150 is not consumed in the reaction and it continues
to inactivate such ROS molecules as long as it is present at the target
site.
Pre-
clinical studies
Due to
the similar mechanisms of actions between radiation therapy and radiation
exposure, we believe that the pre-clinical studies performed for the development
of AEOL 10150 as a potential medical countermeasure against the effects of acute
radiation syndrome in the lungs as described above also provides support for the
development of AEOL 10150 as an adjuvant in radiation therapy.
In
addition, we have performed several additional studies specifically for this
indication to ensure the use of an antioxidant in radioprotection of normal
adjacent tissue does not interference with the efficacy of tumor
radiotherapy. A number of preclinical in vivo studies have addressed
this issue and have demonstrated that AEOL 10150 does not negatively affect
tumor radiotherapy.
In one
study (Vujaskovic, et al. of Duke University), human prostate tumors (PC3) grown
in nude mice to substantial size were fraction irradiated with 5 Gy per day for
3 days for a total of 15 Gy. AEOL 10150 at 7.5 mg/kg/bid was
administered subcutaneously on the first day of radiation and continued for
either of two time courses: when tumor volume reached 5 times the initial volume
or for twenty days. The receding tumor volume curves for irradiation
only and for irradiation plus AEOL 10150 were
super-imposable. Therefore AEOL 10150 did not interfere with the
radiation effect on xenogenic prostate tumor.
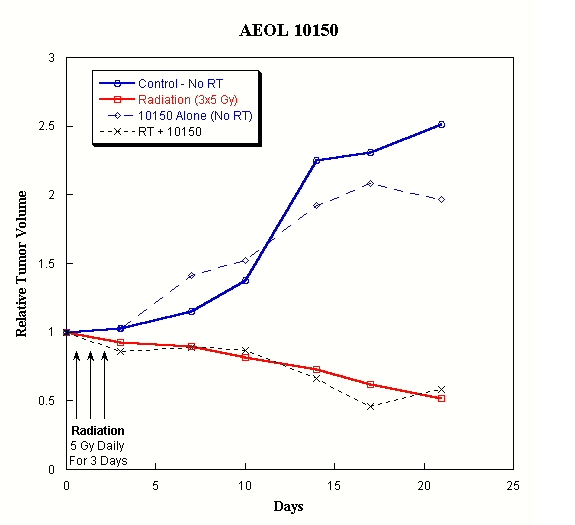
14
Figure 7. Relative
tumor volumes of human prostate tumor implants in nude mice: Implants of
well-vascularized PC3 tumors were grown to substantial size prior to receiving
fractionated radiation (5 Gy daily for three days). AEOL 10150 (7.5
mg/kg/bid) was administered subcutaneously commencing on the first day of
irradiation and continued for 20 days. Other groups of mice received
either no irradiation, irradiation only or AEOL 10150 without
irradiation.
In
another study of prostate cancer tumors (Gridley, et al of Loma Linda
University), mouse prostate cancer cell line RM-9 was injected subcutaneously
into C57/Bl6 mice, followed by up to 16 days of AEOL 10150 delivered
intraperitonealy at 6 mg/kg/day. On day seven, a single
non-fractionated dose of radiation (10 Gy) was delivered. Therefore,
the mice received compound for seven days prior to radiation. The
results of this study demonstrated that AEOL 10150 does not protect the prostate
tumor against radiation and in fact AEOL 10150 showed a trend towards increasing
the effectiveness of the radiation treatment. The primary effect
appears to be in down-regulation of radiation induced HIF-1 expression and VEGF
and up-regulation of IL-4. Thus, AEOL 10150, through its
down-regulation of VEGF, may inhibit formation of blood vessels (i.e.
angiogenisis) required for tumor re-growth and protects normal tissues from
damage induced by radiation and chemotherapy.
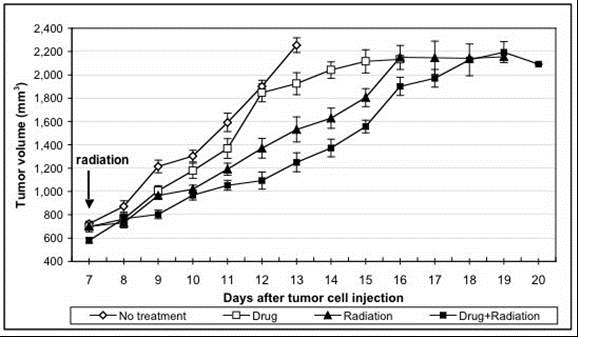
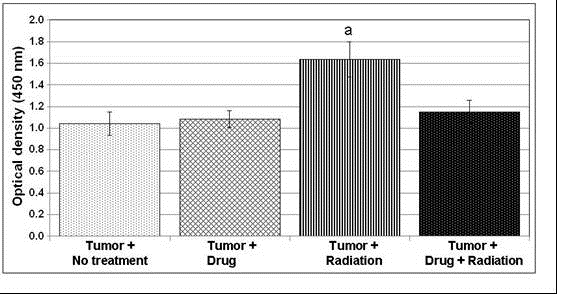
Figure 8 above measures tumor
volume against time after implantation of RM-9 tumor cells and shows that AEOL
10150 treatment resulted in inhibition of tumor re-growth in a study performed
by Dr. Gridley of Loma Linda University. Daily intraperitoneal
injections of AEOL 10150 were initiated on day 1. At 12 days,
approximately one half of each tumor-bearing group and control mice with no
tumor were euthanized for in vitro analyses; remaining mice/group were followed
for tumor growth and euthanized individually when maximum allowed tumor volume
was attained. Each point represents the mean +/- standard error of
the mean. Two-way analysis of the variance for days 8 to 14 revealed
that group and time had highly significant main effects (Ps<0.001) and a
group x time interaction was noted (P<0.001).
15
Figure 9 above shows the HIF-1
Expression in prostate tumors and the impact of the treatment of AEOL 10150 in a
study by Dr. Gridley of Loma Linda University.
In
summary, the data obtained in these preclinical studies suggest that the post
irradiation long term delivery of AEOL 10150 may be protective against
radiation-induced lung injury, as assessed by histopathology and
immunohistochemistry. Oxidative stress, inflammation and hypoxia,
which play important roles in the pathogenesis of radiation mediated fibrosis,
were also shown to be reduced in animals treated with higher doses of AEOL
10150. Studies have also shown that AEOL 10150 does not adversely
affect tumor response to radiation therapy. Thus, treatment with AEOL
10150 does not significantly protect tumors from the cell killing effects of
radiation therapy. This combined with other studies that have shown
that AEOL 10150 significantly prevents radiation induced normal tissue injury
suggests that AEOL 10150 has the potential to achieve normal tissue protection
without protection of tumor tissue.
Future
Development Plans
We plan
to leverage off the development of AEOL 10150 as a potential medical
countermeasure against the effects of acute radiation syndrome in the lungs and
GI Tract to develop AEOL 10150 as an adjuvant for radiation
therapy. We believe we would be able to use many of the non-clinical
studies as support for efficacy and will be running the required safety studies
in radiation therapy patients and therefore can use the studies for support of
an NDA filing for both indications.
Competition
There are
currently three drugs approved for the treatment of the side effects of
radiation therapy. Amifostine (Ethyol®) is approved by the FDA as a
radioprotector. Amifostine (Ethyolâ) is marketed by
MedImmune, Inc. for use in reduction of chemotherapy-induced kidney toxicity
associated with repeated administration of cisplatin in patients with advanced
ovarian cancer and radiation-induced xerostomia (damage to the salivary gland)
in patients undergoing post-operative radiation treatment for head and neck
cancer. MedImmune is studying amifostine in other indications of
radiation therapy. KepivanceTM
(palifermin) is marketed by Amgen, Inc. for use in the treatment of
severe oral mucositis (mouth sores) in patients with hematologic (blood) cancers
who are undergoing high-dose chemotherapy followed by bone
transplant. Amgen is also studying Kepivance as an antimucositis
agent in patients with head and neck cancer, non small cell lung cancer and
colon cancer. Salagen Tablets (pilocarpine hydrochloride) (“Salagen”)
is marketed by MGI Pharma in the United States as a treatment for the symptoms
of xerostomia induced by radiation therapy in head and neck cancer
patients. In addition, there are many drugs under development to
treat the side effects of radiation therapy.
Funding
Options
We plan
on funding the development of AEOL 10150 as an adjuvant in radiation therapy
using internally generated funds and will most likely follow the development of
AEOL 10150 as a medical countermeasure. We have also been contacted
by the National Cancer Institute (NCI) about the possibility of NCI funded phase
1 or phase 2 studies of the compound as a potential treatment for cancer
radiation therapy.
AEOL
10150 in Amyotrophic Lateral Sclerosis
Overview
Amyotrophic
Lateral Sclerosis (“ALS”), commonly referred to as “Lou Gehrig’s disease,” the
most common motor neuron disease, results from progressive degeneration of both
upper and lower motor neurons. According to the ALS Association
(“ALSA”), the incidence of ALS is two per 100,000 people. ALS occurs
more often in men than women, with typical onset between 40 and 70 years of
age. ALS is a progressive disease and approximately 80% of ALS
patients die within five years of diagnosis, with only 10% living more than 10
years. The average life expectancy is two to five years after
diagnosis, with death from respiratory and/or bulbar muscle
failure. The International Alliance of ALS/MND Associations reports
there are over 350,000 patients with ALS/MND worldwide and 100,000 people die
from the disease each year worldwide. In the United States, ALSA
reports that there are approximately 30,000 patients with ALS with 5,600 new
patients diagnosed each year.
Sporadic
(i.e., of unknown origin) ALS is the most common form, accounting for
approximately 90% of cases. The cause of sporadic ALS is
unclear. Familial ALS comprises the remainder of cases and 5-10% of
these patients have a mutated superoxide dismutase 1 (“SOD1”)
gene. More than 90 point mutations have been identified, all of which
appear to associate with ALS, and result in motor neuron disease in
corresponding transgenic mice. SOD mutations have been observed in
both familial and sporadic ALS patients, although the nature of the dysfunction
produced by the SOD1 mutations remains unclear. The clinical and
pathological manifestations of familial ALS and sporadic ALS are
indistinguishable suggesting common pathways in both types of
disease.
In
November 2003, the U.S. Food and Drug Administration (the “FDA”) granted orphan
drug designation for our ALS drug candidate. Orphan drug designation
qualifies a product for possible funding to support clinical trials, study
design assistance from
16
the FDA
during development and for financial incentives, including seven years of
marketing exclusivity upon FDA approval.
Pre-clinical
studies
John P.
Crow, Ph.D., and his colleagues at the University of Alabama at Birmingham
tested AEOL 10150 in an animal model of ALS (SOD1 mutant G93A transgenic
mice). The experiments conducted by Dr. Crow (now at the
University of Arkansas College of Medicine) were designed to be clinically
relevant by beginning treatment only after the onset of symptoms in the animals
is observed. Twenty-four confirmed transgenic mice were alternately
assigned to either a control group or AEOL 10150-treatment on the day of symptom
onset, which was defined as a noticeable hind-limb
weakness. Treatment began on the day of symptom onset. The
initial dose of AEOL 10150 was 5 mg/kg, with continued treatment at a dose of
2.5 mg/kg once a day until death or near death.
|
Treatment
|
Age
at Symptom onset mean days + SD(range)
|
Survival
Interval mean days + SD(range)
|
P-value
Log-rank (v. control)
|
P-value
Wilcoxon (v. control)
|
||||
|
Control
|
104.8
+ 1.43
|
12.8
+ 0.79
|
||||||
|
(100-112
|
)
|
(9-16
|
)
|
|||||
|
AEOL
10150
|
106.1
+ 1.5
|
32.2
+ 2.73
|
||||||
|
(100-115
|
)
|
(15-46
|
)
|
<
0.0001
|
0.0002
|
Table 1. Effect of AEOL 10150 on
survival of G93A transgenic mice
Figure 10.
Table 1
and Figure 10 above show that AEOL 10150 treatment resulted in a greater than
2.5 times mean survival interval, compared to control. AEOL
10150-treated mice were observed to remain mildly disabled until a day or two
before death. In contrast, control mice experienced increased
disability daily.
Dr. Crow
has repeated the ALS preclinical experiment a total of four times, in each case
with similar results. The efficacy of AEOL 10150 in the G93A mouse
model of ALS has also been evaluated by two additional
laboratories. One of these laboratories verified an effect of AEOL
10150 in prolonging survival of the G93A mouse, while no beneficial effect of
the drug was identified in the other laboratory.
Future
Development Plans
We do not
currently have any plans to pursue the development of AEOL 10150 for the
treatment of ALS unless we are able to obtain funding specifically for this
purpose.
Competition
Rilutek®
(riluzole), marketed by Sanofi-Aventis SA, is the only commercially approved
treatment for ALS in the United States and the European
Union. Administration of Rilutek prolongs survival of ALS patients by
an average of 60-90 days, but has little or no effect on the progression of
muscle weakness, or quality of life. Rilutek was approved in the
United States in 1995, and in 2001 in the European Union. However,
there are at least twenty drug candidates reported to be in clinical development
for the treatment of ALS.
17
In addition, ALS belongs
to a family of diseases called neurodegenerative diseases, which includes
Alzheimer’s, Parkinson’s and Huntington’s disease. Due to
similarities between these diseases, a new treatment for one ailment potentially
could be useful for treating others. There are many companies that
are producing and developing drugs used to treat neurodegenerative diseases
other than ALS.
AEOL
10150 Clinical Development Program
AEOL
10150 has been tested for safety, tolerability and pharmacokinetics with no
serious or clinically significant adverse effects observed. To date,
38 patients have received AEOL 10150 in three clinical trials designed to test
the safety and tolerability of the drug candidate.
In
September 2005, we completed a multi-center, double-blind, randomized,
placebo-controlled, Phase I clinical trial. This escalating single
dose study was conducted to evaluate the safety, tolerability and
pharmacokinetics of AEOL 10150 administered by twice daily subcutaneous
injections in patients with ALS.
In the
Phase Ia study, 4-5 patients diagnosed with ALS were placed in a dosage cohort
(3 or 4 receiving AEOL 10150 and 1 receiving placebo). Each dose
cohort was evaluated at a separate clinical center. In total, seven
separate cohorts were evaluated in the study, and 25 ALS patients received AEOL
10150. Based upon an analysis of the data, it was concluded that
single doses of AEOL 10150 ranging from 3 mg to 75 mg were safe and well
tolerated. In addition, no serious or clinically significant adverse
clinical events were reported, nor were there any significant laboratory
abnormalities. Based upon extensive cardiovascular monitoring (i.e.,
frequent electrocardiograms and continuous Holter recordings for up to 48 hours
following dosing), there were no compound-related cardiovascular
abnormalities.
The most
frequently reported adverse events in this Phase I clinical trial were injection
site reactions, followed by dizziness and headache. Adverse events
were primarily mild in severity, and approximately one-half of the events were
considered to have a possible relationship to the study
medication. In addition, no clinically meaningful findings were noted
in the safety, laboratory, vital sign, the Unified Parkinson’s Disease Rating
Scale (“UPDRS”), functional ALS, or electro cardiogram (“ECG”)
data. All cohorts exhibited dose-related peak plasma drug
concentrations and consistent disappearance half-lives.
In
October 2006, we completed a multi-center, double-blind, randomized,
placebo-controlled, Phase Ib clinical trial. This multiple dose study
was conducted to evaluate the safety, tolerability and pharmacokinetics of AEOL
10150 administered by subcutaneous injection and infusion pump in patients with
ALS. Under the multiple dose protocol, three groups of six ALS
patients (four receiving AEOL 10150 and two receiving placebo) were enrolled,
based upon patients who meet the El Escorial criteria for Clinically Definite
ALS, Clinically Probable ALS, Clinically Probable-Laboratory Supported ALS, or
Definite Familial-Laboratory Supported ALS (i.e., Clinically Possible ALS with
an identified SOD gene mutation).
The first
two cohorts of the Phase Ib multiple dose study received a fixed daily dose of
AEOL 10150 twice a day by subcutaneous injection. In the first
cohort, each patient received twice daily subcutaneous injections of 40 mg of
AEOL 10150 or placebo, for six consecutive days, followed by a single
subcutaneous injection on the seventh day, for a total of 13
injections. In the second cohort, each patient received twice daily
subcutaneous injections of 60 mg of AEOL 10150 or placebo, for six consecutive
days, followed by a single subcutaneous injection on the seventh day, for a
total of 13 injections.
In
contrast, the third cohort received a weight adjusted dose (i.e. mg per kilogram
of body weight per day) delivered subcutaneously over twenty four hours by
continuous infusion pump. In the third cohort, each patient received
AEOL 10150 via continuous infusion pump for six and one half consecutive days
for a total of 2.0 mg per patient kilogram per day. Each patient in
all three cohorts completed the study and follow-up evaluation at 14
days.
The Phase
Ib study was conducted at five academic clinical ALS centers. Male
and female ALS patients, 18 to 70 years of age, who were ambulatory (with the
use of a walker or cane, if needed) and capable of orthostatic blood pressure
assessments were enrolled in the study. Clinical signs/symptoms,
laboratory values, cardiac assessments, and pharmacokinetics (PK) were
performed.
Based
upon an analysis of the data, it was concluded that multiple doses of AEOL 10150
for a period of six and one half consecutive days in the amount of 40 mg per
day, 60 mg per day and 2 mg per kilogram per day were safe and well
tolerated. No serious or clinically significant adverse events were
reported or observed. The most frequent adverse events related to
study compound were injection site observations related to compound
delivery. There were no significant laboratory
abnormalities. Based upon extensive cardiovascular monitoring (i.e.,
frequent electrocardiograms and continuous Holter recordings throughout the six
and one half days of dosing), there were no compound-related cardiovascular
abnormalities.
Pharmacokinetic
findings from the Phase Ib study to data are as follows:
|
|
·
|
Increases
in Cmax and AUC(0-8) appear to correlate with increases in dose, but the
correlation is not strong.
|
|
|
·
|
The
mean Cmax for the 40 mg cohort was 1,735 ng/mL; 2,315 ng/mL for the 60 mg
cohort and 1,653 ng/ml for the
|
18
|
|
2
mg/kg cohort.
|
|
|
·
|
There
were probable linear correlations between both Cmax and AUC(0-8) and dose
based on body weight.
|
|
|
·
|
The
terminal half life (a measurement of the time period for which a compound
stays in the body) as determined from Day 7 data was approximately 8 to 9
hours.
|
|
|
·
|
Steady-state
occurs within three days of multiple dosing. There was no
evidence for a third longer half life that would be associated with long
term accumulation. Thus, compound accumulation is not expected
beyond the third day with multiple
dosing.
|
|
|
·
|
From
48 hours to the end of the infusion, the plasma concentrations of AEOL
10150 during the infusion showed little variability, indicating a smoother
delivery of the drug than with twice-daily
injections.
|
During
2008, we completed a follow-on Phase I open label compassionate use multiple
dose study of AEOL 10150 in a patient diagnosed with progressive and
debilitating amyotrophic ALS. The study was conducted at the
University of California, Los Angeles by Martina Wiedau-Pazos, M.D., and was
designed to evaluate the safety and efficacy of AEOL 10150 in an ALS patient
over an extended period of time. The patient received a subcutaneous
injection of 75mg of AEOL 10150 two times each day for 34
days. Efficacy and safety data was monitored for the duration of the
study. The primary objective of this study was to assess the clinical
efficacy of AEOL 10150 with respect to the patient’s baseline assessment of
functional status. Secondary objectives included the assessments of
muscle strength, respiratory function, quality of life and
safety. The patent’s baseline efficacy results were an ALS Functional
Rating Scale (“ALSFRS-R”) rating of 19, Muscle strength Manual Muscle Testing
Scale (“MMTS”) of 68 and a forced vital capacity (“FVC”)
of 30%. The patient’s results after 2 months were an ALSFRS-R rating
of 22, a MMTS rating of 86 and an FVC of 28%. It should be
noted that the subject began using breathing assistance (BiPAP) approximately
two weeks after the study started. The patient discontinued treatment
due to nausea and moderately increased liver transaminases. Other
drug-associated adverse events included mild skin irritation at the injection
site and mild urine discoloration.
We do not
currently have any plans to launch any additional clinical trials of AEOL 10150
for the treatment of ALS unless we are able to obtain funding specifically for
this purpose. However, we believe our safety studies completed as a
therapy for ALS may be utilized to demonstrate safety for our primary
indications.
AEOL
11207
Overview
We have
selected AEOL 11207 as our second development candidate based upon results from
data obtained from our pre-clinical testing of our pipeline drug
candidates. Because of the wide-ranging therapeutic opportunities
that the compound evidenced in diverse pre-clinical models of human diseases, we
have not yet ascertained what the most robust therapeutic use of AEOL 11207
might be. However, data collected to date suggest that AEOL 11207 may
be useful as a potential once-every-other-day oral therapeutic treatment option
for central nervous system (“CNS”) disorders, most likely Parkinson’s
disease.
Parkinson’s
disease is a common neurodegenerative disorder, second in occurrence among these
disorders only to Alzheimer’s disease. According to the National
Parkinson Foundation, Parkinson’s affects as many as one million people in the
United States, with approximately 60,000 new cases diagnosed in the United
States each year.
Parkinson’s
specifically involves the progressive destruction of the nerves that secrete
dopamine and control the basal ganglia, an area of the brain involved in the
regulation of movement. Dopamine turnover has been shown to elevate
the levels of ROS in the brain. In addition, a street-drug
contaminant has appeared that can cause parkinsonism in drug
abusers. The compound N-methyl-4-phenyl-1, 2, 3, 6tetrahydropyridine
(“MPTP”) has been identified in underground laboratory preparations of a potent
analog of meperidine (Demerol). MPTP-containing powder, sometimes
sold as a new “synthetic heroin,” can be dissolved in water and administered
intravenously or taken by the intranasal route. MPTP has been
documented to produce irreversible chronic Parkinson symptoms in drug
abusers. Agents such as MPTP overproduce ROS in the basal
ganglia. Therefore, ROS mediated neuronal dysfunction may play a key
role in the development of Parkinson’s disease. Symptoms of this
disease include tremors, rigidity and bradykinesia (i.e., slowness of
movement). In the more advanced stages, it can cause fluctuations in
motor function, sleep problems and various neuro-psychiatric
disorders. A biological hallmark of Parkinson’s disease is a
reduction in brain dopamine levels. Preventing or slowing the
destruction of brain cells that lead to the depletion of dopamine levels in the
brain is an important therapeutic approach for the treatment of this
disease.
Pre-clinical
studies
Data
developed by our scientists and Dr. Manisha Patel at University of Colorado
Health Sciences Center and Department of Medicine, indicate that when
administered orally, AEOL 11207 is greater than 80% bioavailable, meaning that
it is readily absorbed and reaches both the circulatory system and the brain in
sufficient amounts to demonstrate biological
activity. Data developed with AEOL 11207 in a widely used animal
model of Parkinson’s disease (the “MPTP model”) showed that when administered
orally, AEOL 11207 crosses the blood brain barrier and protected dopamine
neurons in a dose-dependent manner. Further data suggest that the
compound has a half life (a measurement of the time period for which a compound
stays in the body) of about 3 days in both the circulatory system and the brain,
and that prior to stopping administration of the compound, the
levels
19
of AEOL
11207 in both the circulatory system and brain reach a steady state (a valuable
measurement of when the levels of the drug in the body remain substantially
constant, neither increasing nor decreasing) after 2 days of
dosing. Data have also been developed that indicate that when dosing
of AEOL 11207 is stopped, the compound is excreted from the body.
Future
Development Plans
For this
and other reasons, we believe that the therapeutic rationale for developing AEOL
11207 as a neuroprotectant, may substantially change the course of therapeutic
treatment options for Parkinson’s disease if AEOL 11207 were to achieve
regulatory approval for commercialization. However, we are unable to
determine at this time that such regulatory approval for AEOL 11207 can be or
will be secured and we will not be able to further develop AEOL 11207 until
funding for this purpose is obtained.
AEOL
11207 is patent-protected and has the same chemical core structure as AEOL
10150. Because of this common structural feature, it is anticipated
that AEOL 11207 will evidence substantially the same safety profile in clinical
evaluations as observed with AEOL 10150, making clinical trial design and
testing of AEOL 11207 more robust and facile. Furthermore, all of the
Aeolus compounds evidence the ability to scavenge and decrease ROS and reactive
nitrogen species (RNS), all of which are implicated in a variety of CNS
diseases.
Funding
Options
The
University of Colorado, our research provider for the development of AEOL 11207
for the treatment of Parkinson’s Disease, recently received funding from the
Michael J. Fox Foundation to further test AEOL 11207.
Background
on Antioxidants
Oxygen
Stress and Disease
Oxygen
plays a pivotal role in supporting life by enabling energy stored in food to be
converted to energy that living organisms can use. The ability of
oxygen to participate in key metabolic processes derives from its highly
reactive nature. This reactivity is necessary for life, but also
generates different forms of oxygen that can react harmfully with living
organisms. In the body, a small proportion of the oxygen we consume
is converted to superoxide, a free radical species that gives rise to hydrogen
peroxide, hydroxyl radical, peroxynitrite and various other
oxidants.
Oxygen-derived
free radicals can damage DNA, proteins and lipids resulting in inflammation and
both acute and delayed cell death. The body protects itself from the
harmful effects of free radicals and other oxidants through multiple antioxidant
enzyme systems such as superoxide dismutase (“SOD”). These natural
antioxidants convert the reactive molecules into compounds suitable for normal
metabolism. When too many free radicals are produced for the body’s
normal defenses to convert, “oxidative stress” occurs with a cumulative result
of reduced cellular function and, ultimately, disease.
Data also
suggests that oxygen-derived free radicals are an important factor in the
pathogenesis of a large variety of diseases, including neurological disorders
such as ALS, Parkinson’s disease, Alzheimer’s disease and stroke, and in
non-neurological disorders such as cancer radiation therapy damage, emphysema,
asthma and diabetes.
Antioxidants
as Therapeutics
Because
of the role that oxygen-derived free radicals play in disease, scientists are
actively exploring the possible role of antioxidants as a treatment for related
diseases. Preclinical and clinical studies involving treatment with
SOD, the body’s natural antioxidant enzyme, or more recently, studies involving
over-expression of SOD in transgenic animals, have shown promise of therapeutic
benefit in a broad range of disease therapies. Increased SOD function
improves outcome in animal models of conditions including stroke,
ischemia-reperfusion injury (a temporary cutoff of blood supply to tissue) to
various organs, harmful effects of radiation and chemotherapy for the treatment
of cancer, and in neurological and pulmonary diseases. Clinical
studies with bovine SOD, under the brand Orgotein, or recombinant human SOD in
several conditions including arthritis and protection from limiting side effects
of cancer radiation or chemotherapy treatment, have also shown promise of
benefit. The major limitations of enzymatic SOD as a therapeutic are
those found with many proteins, most importantly limited cell penetration and
allergic reactions. Allergic reactions led to the withdraw of
Orgotein from almost every worldwide market.
Catalytic
Antioxidants vs. Antioxidant Scavengers
From a
functional perspective, antioxidant therapeutics can be divided into two broad
categories, scavengers and catalysts. Antioxidant scavengers are
compounds where one antioxidant molecule combines with one reactive oxygen
molecule and both are consumed in the reaction. There is a one-to-one
ratio of the antioxidant and the reactive molecule. With catalytic
antioxidants, in contrast, the antioxidant molecule can repeatedly inactivate
reactive oxygen molecules, which could result in multiple reactive oxygen
molecules combining with each antioxidant molecule.
20
Vitamin derivatives that
are antioxidants are scavengers. The SOD enzymes produced by the body
are catalytic antioxidants. Catalytic antioxidants are typically much
more potent than antioxidant scavengers, in some instances by a multiple of up
to 10,000.
Use of
antioxidant scavengers, such as thiols or vitamin derivatives, has shown promise
of benefit in preclinical and clinical studies. Ethyol, a
thiol-containing antioxidant, is approved for reducing radiation and
chemotherapy toxicity during cancer treatment, and clinical studies have
suggested benefit of other antioxidants in kidney and neurodegenerative
diseases. However, large sustained doses of the compounds are
required as each antioxidant scavenger molecule is consumed by its reaction with
the free radical. Toxicities and the inefficiency of scavengers have
limited the utility of antioxidant scavengers to very specific
circumstances.
Contracts
and Grants
We seek
to advance development of our drug candidates through external funding
arrangements. We may slow down development programs or place them on hold during
periods that are not covered by external funding. We have received external
funding awards for the development of AEOL 10150 as a MCM for acute radiation
syndrome in the GI, sulfur mustard gas and chlorine gas exposure.
On August
31, 2009, we submitted a white paper to BARDA in response to Special
Instructions Amendment 4 to a Broad Agency Announcement (BAA-BARDA-09-34) for
advanced research and development of medical countermeasures for chemical,
biological, radiological and nuclear threats. The purpose of the special
instructions amendment was to specifically solicit solutions for treating
cutaneous and/or pulmonary (life-threatening pneumonitis and fibrosis) injuries
resulting from exposure to ionizing radiation. BARDA is interested in advanced
development and eventual licensure/approval of MCMs for cutaneous and/or
pulmonary injuries resulting from an acute exposure to radiation from a
radiological/nuclear accident or attack, particularly injuries associated with
ARS or DEARE.
We
continue to actively pursue additional government or foundation sponsored
development contracts and grants and to encourage both governmental,
non-governmental agencies and philanthropic organizations to provide development
funding or to conduct clinical studies of our drug candidates.
Collaborative
and Licensing Arrangements
Duke
Licenses
Through
our wholly owned subsidiary, Aeolus Sciences, Inc., we have obtained exclusive
worldwide rights from Duke University (“Duke”) to products using antioxidant
technology and compounds developed by Dr. Irwin Fridovich and other
scientists at Duke. We must pay royalties to Duke on net product
sales during the term of the Duke licenses, and must make payments upon the
occurrence of development milestones. In addition, we are obligated
under the Duke licenses to pay patent filing, prosecution, maintenance and
defense costs. The Duke licenses are terminable by Duke in the event
of breach by us and otherwise expire when the last licensed patent
expires.
National
Jewish Health
We have
obtained an exclusive worldwide license from the National Jewish Health (the
“NJH”) to develop, make, use and sell products using proprietary information and
technology developed under a previous Sponsored Research Agreement within the
field of antioxidant compounds and related discoveries. We must make
milestone payments to the NJH upon the occurrence of development milestones and
pay royalties on net sales. We are also obligated to pay patent
filing, prosecution, maintenance and defense costs. The NJH agreement
is terminable by the NJH in the event of breach and otherwise expires when the
last licensed patent expires.
In 2009,
we obtained an additional exclusive worldwide license from the NJH to develop,
make, use and sell products using proprietary information and technology
developed at the NJH related to certain compounds as a MCM against sulfur
mustard gas exposure. We must make milestone payments to the NJH upon
the occurrence of development milestones and pay royalties on net
sales. We are also obligated to pay patent filing, prosecution,
maintenance and defense costs. The NJH agreement is terminable by the
NJH in the event of breach and otherwise expires when the last licensed patent
expires.
Research
and Development Expenditures
Expenditures
for research and development activities were $711,000, $977,000, and $1,381,000
during the years ended September 30, 2009, 2008 and 2007,
respectively. Research and development expenses for fiscal 2009
related primarily to the advancement of our lead compound, AEOL
10150.
21
Manufacturing
We
currently do not have the capability to manufacture any of our drug candidates
on a commercial scale. Assuming the successful development of one or
more of our catalytic antioxidant compounds, we plan to contract with third
parties to manufacture them.
Commercialization
Assuming
successful development and FDA approval of one or more of our compounds, to
successfully commercialize our catalytic antioxidant programs, we must seek
corporate partners with expertise in commercialization or develop this expertise
internally. However, we may not be able to successfully commercialize
our catalytic antioxidant technology, either internally or through collaboration
with others.
Marketing
Our
potential catalytic antioxidant products are being developed for large
therapeutic markets. We believe these markets are best approached by
partnering with established biotechnology or pharmaceutical companies that have
broad sales and marketing capabilities. We are pursuing
collaborations of this type as part of our search for development
partners. However, we may not be able to enter into any marketing
arrangements for any of our products on satisfactory terms or at
all.
Biodefense
Industry
Market
Overview
The
market for biodefense countermeasures has grown dramatically as a result of the
increased awareness of the threat of global terror activity in the wake of the
September 11, 2001 terrorist attacks. The U.S. government is the
principal source of worldwide biodefense spending. Most U.S. government spending
on biodefense programs is in the form of development funding from NIAID, BARDA
and the DoD, and procurement of countermeasures by BARDA, the CDC and the DoD.
The U.S. government is now the largest source of development and procurement
funding for academic institutions and biotechnology companies conducting
biodefense research or developing vaccines and immunotherapies directed at
potential agents of bioterror or biowarfare.
We
analyze the biodefense market in three segments; the United States military
market, United States commercial market and non U.S. markets. With
the U.S. government funding representing the vast majority of the worldwide
market. According to the Center for Biosecurity at the University of
Pittsburgh Medical Center the U.S. government biodefense military and civilian
spending approximated $8 billion in fiscal 2009 and has averaged around $5.5
billion from fiscal years 2001 to 2009. Funding in fiscal year 2010 is
expected to be $6 billion.
|
|
·
U.S.
Civilian: The U.S. civilian market includes funds to protect the
U.S. population from biological agents and is largely funded by the
Project BioShield Act of 2004. Project BioShield, the U.S.
government’s largest biodefense initiative, governs and funds with $5.6
billion the procurement of biodefense countermeasures for the SNS for the
period from July 2004 through 2013. A total of $3.7 billion
remains in the Project BioShield Special Reserve Funds for future grants
as of March 2009.
|
|
|
·
U.S.
Military: The DoD is responsible for the development and
procurement of countermeasures for the military segment which focuses on
providing protection for military personnel and civilians who are on
active duty. Funding in fiscal year 2009 is expected to be approximately
$737 million compared to estimated spending of $622 million for
2008. We anticipate that annual funding for the programs
through 2013 will continue in a comparable
range.
|
|
|
·
Non-U.S.
Markets: Non-U.S. markets address protection against
biowarfare agents for both civilians and military personnel in foreign
countries. We anticipate that foreign countries will want to procure
biodefense products as they are developed and validated by procurement by
the U.S. government.
|
|
|
|
Project
BioShield and the Pandemic and All-Hazards Preparedness Act
The
Project BioShield Act, which became law in 2004, authorizes the procurement of
countermeasures for chemical, biological, radiological and nuclear attacks for
the Strategic National Stockpile (“SNS”) which is a national repository of
medical assets and countermeasures designed to provide federal, state and local
public health agencies with medical supplies needed to treat those affected by
terrorist attacks, natural disasters, industrial accidents and other public
health emergencies. Project BioShield provided appropriations of $5.6 billion to
be expended over ten years into a special reserve fund. As
March 2009, a total of $3.7 billion remains in the Project BioShield Special
Reserve Funds for future grants.
22
The
Pandemic and All-Hazards Preparedness Act, passed in 2006, established BARDA as
the agency responsible for awarding procurement contracts for biomedical
countermeasures and providing development funding for advanced research and
development in the biodefense arena, and supplements the funding available under
Project BioShield for chemical, biological, radiological and nuclear
countermeasures, and provides funding for infectious disease pandemics. Funding
for BARDA is provided by annual appropriations by Congress. Congress also
appropriates annual funding for the CDC for the procurement of medical assets
and countermeasures for the SNS and for NIAID to conduct biodefense research.
This appropriation funding supplements amounts available under Project
BioShield.
Currently,
the U.S. government may, at its discretion, purchase critical biodefense
products for the SNS prior to FDA approval based on Emergency Use Authorization
(“EUA”) enabled under the Project Bioshield legislation. On an ongoing
basis we monitor notices for requests for proposal, grants and other potential
sources of government funding that could potentially support the development of
our drug candidates. Nevertheless, changes in government budgets,
priorities and agendas as well as political pressures could result in a
reduction in overall government financial support for the biodefense sector in
general and/or specifically the drug candidates we are developing. Due to
the current economic downturn, the accompanying fall in tax revenues and the
U.S. government’s efforts to stabilize the economy, the U.S. government may be
forced or choose to reduce or delay spending in the biodefense field, which
could decrease the likelihood of future government contract awards, the
likelihood that the government will exercise its right to extend any of its
existing contracts and/or the likelihood that the government would procure
products from us. (For further information, see “Risk Factors — Risks Related to Our
Dependence on U.S. Government Contracts — U.S. government agencies have special
contracting requirements which give them the ability to unilaterally control
contracts.”) As a result, further development of our drug candidates and
ultimate product sales to the government, if any, could be delayed or stopped
altogether.
Competition
General
Competition
in the pharmaceutical industry is intense and we expect it to
increase. Technological developments in our field of research and
development occur at a rapid rate and we expect competition to intensify as
advances in this field are made. We will be required to continue to
devote substantial resources and efforts to research and development
activities. Our most significant competitors, among others, are fully
integrated pharmaceutical companies and more established biotechnology
companies, which have substantially greater financial, technical, sales,
marketing, and human resources than we do. These companies may
succeed in developing and obtaining regulatory approval for competitive products
more rapidly than we can for our drug candidates. In addition,
competitors may develop technologies and products that are, or are perceived as
being, cheaper, safer or more effective than those being developed by us or that
would render our technology obsolete.
We expect
that important competitive factors in our potential product markets will be the
relative speed with which we and other companies can develop products, complete
the clinical testing and approval processes, and supply commercial quantities of
a competitive product to the market. With respect to clinical
testing, competition might result in a scarcity of clinical investigators and
patients available to test our potential products, which could delay
development.
We are
aware of products in research or development by our competitors that address the
diseases and therapies being targeted by us. In addition, there may
be other competitors of whom we are unaware with products which might be more
effective or have fewer side effects than our products and those of our known
competitors. The following discussion is a summary of information
known to us and is not meant to be an exhaustive list of our competitors or
their products or drug candidates.
Antioxidants
Several
companies have explored the therapeutic potential of antioxidant compounds in
numerous indications. Historically, most of these companies have
focused on engineered versions of naturally occurring antioxidant enzymes, but
with limited success, perhaps because the large size of these molecules makes
delivery into the cells difficult. Antioxidant drug research
continues at a rapid pace despite previous clinical setbacks.
Patents
and Proprietary Rights
We
currently license rights to our potential products from third
parties. We generally seek patent protection in the United States and
other jurisdictions for the potential products and proprietary technology
licensed from these third parties. The process for preparing and
prosecuting patents is lengthy, uncertain and costly. Patents may not
issue on any of the pending patent applications owned by us or licensed by us
from third parties. Even if patents issue, the claims allowed might
not be sufficiently broad to protect our technology or provide us protection
against competitive products or otherwise be commercially
valuable. Patents issued to or licensed by us could be challenged,
invalidated, infringed, circumvented or held unenforceable. Even if
we successfully defend our patents for our products, the costs of defense can be
significant.
23
As of
December 24, 2009, our catalytic antioxidant small molecule technology base is
described in thirteen issued United States patents and five United States
pending patent applications. These patents and patent applications
belong in whole or in part to Duke or the NJH and are licensed to
us. These patents and patent applications cover soluble manganic
porphyrins as antioxidant molecules as well as targeted compounds obtained by
coupling such antioxidant compounds to molecules that bind to specific
extracellular elements. The pending U.S. patent applications and
issued U.S. patents include composition of matter claims and method claims for
several series of compounds. Corresponding international patent
applications have been filed, 75 of which have issued as of December 24,
2009.
In
addition to patent protection, we rely upon trade secrets, proprietary know-how
and technological advances that we seek to protect in part through
confidentiality agreements with our collaborative partners, employees and
consultants. Our employees and consultants are required to enter into
agreements providing for confidentiality and the assignment of rights to
inventions made by them while in our service. We also enter into
non-disclosure agreements to protect our confidential information furnished to
third parties for research and other purposes.
Government
Regulation
Our
research and development activities and the manufacturing and marketing of our
future products are subject to regulation by numerous governmental agencies in
the United States and in other countries. The FDA and comparable
agencies in other countries impose mandatory procedures and standards for the
conduct of clinical trials and the production and marketing of products for
diagnostic and human therapeutic use. Before obtaining regulatory
approvals for the commercial sale of any of our products under development, we
must demonstrate through preclinical studies and clinical trials that the
product is safe and efficacious for use in each target
indication. The results from preclinical studies and early clinical
trials might not be predictive of results that will be obtained in large-scale
testing. Our clinical trials might not successfully demonstrate the
safety and efficacy of any products or result in marketable
products.
The
United States system of drug approvals is considered to be the most rigorous in
the world. It takes an average of 8.5 years for a drug candidate to
move through the clinical and approval phases in the United States according to
a November 2005 study by the Tufts Center for the Study of Drug
Development. Only five in 5,000 drug candidates that enter
preclinical testing make it to human testing and only one of those five is
approved for commercialization. On average it costs a company $897
million to get one new drug candidate from the laboratory to United States
patients according to a May 2003 report by Tufts Center for the Study of Drug
Development. A November 2006 study by Tufts Center for the Study of
Drug Development reported that the average cost of developing a new
biotechnology product was $1.2 billion and took on average slightly more than
eight years to be approved by the FDA.
The steps
required by the FDA before new drug products may be marketed in the United
States include:
|
|
·
|
completion
of preclinical studies;
|
|
|
·
|
the
submission to the FDA of a request for authorization to conduct clinical
trials on an IND, which must become effective before clinical trials may
commence;
|
|
|
·
|
adequate
and well-controlled Phase I clinical trials which typically involves
normal, healthy volunteers. The test study a drug candidate’s
safety profile, including the safe dosage range. The studies
also determine how a drug is absorbed, distributed, metabolized and
excreted as well as the duration of its
action;
|
|
|
·
|
adequate
and well-controlled Phase II clinical trials which typically
involve treating patients with the targeted disease with the drug
candidate to assess a drug’s
effectiveness;
|
|
|
·
|
adequate
and well-controlled Phase III clinical trials involving a larger
population of patients with the targeted disease are treated with the drug
candidate to confirm efficacy of the drug candidate in the treatment of
the targeted indication and to identify adverse
events;
|
|
|
·
|
submission
to the FDA of an NDA; and
|
|
|
·
|
review
and approval of the NDA by the FDA before the product may be shipped or
sold commercially.
|
In
addition to obtaining FDA approval for each product, each product manufacturing
establishment must be registered with the FDA and undergo an inspection prior to
the approval of an NDA. Each manufacturing facility and its quality
control and manufacturing procedures must also conform and adhere at all times
to the FDA’s current good manufacturing practices (“cGMP”)
regulations. In addition to preapproval inspections, the FDA and
other government agencies regularly inspect manufacturing facilities for
compliance with these requirements. Manufacturers must expend
substantial time, money and effort in the area of production and quality control
to ensure full technical compliance with these standards.
Preclinical
testing includes laboratory evaluation and characterization of the safety and
efficacy of a drug and its formulation. Preclinical testing results
are submitted to the FDA as a part of an IND which must become effective prior
to commencement of clinical trials. Clinical trials are typically
conducted in three sequential phases following submission of an IND. Phase I
represents the initial administration of the drug to a small group of humans,
either patients or healthy volunteers, typically to test for safety (adverse
effects), dosage tolerance, absorption, distribution, metabolism, excretion and
clinical pharmacology, and, if possible, to gain early evidence of
effectiveness. Phase II involves studies in a small sample of the
actual intended patient population to assess the efficacy of the drug for a
specific indication, to determine dose tolerance and the optimal dose range
and
24
to gather
additional information relating to safety and potential adverse
effects. Once an investigational drug is found to have some efficacy
and an acceptable safety profile in the targeted patient population, Phase III
studies are initiated to further establish clinical safety and efficacy of the
therapy in a broader sample of the general patient population, in order to
determine the overall risk-benefit ratio of the drug and to provide an adequate
basis for any physician labeling. During all clinical studies, we
must adhere to good clinical practice (“GCP”) standards. The results
of the research and product development, manufacturing, preclinical studies,
clinical studies and related information are submitted in an NDA to the
FDA.
The
process of completing clinical testing and obtaining FDA approval for a new drug
is likely to take a number of years and require the expenditure of substantial
resources. If an application is submitted, there can be no assurance
that the FDA will review and approve the NDA. Even after initial FDA
approval has been obtained, further studies, including post-market studies,
might be required to provide additional data on safety and will be required to
gain approval for the use of a product as a treatment for clinical indications
other than those for which the product was initially tested and
approved. Also, the FDA will require post-market reporting and might
require surveillance programs to monitor the side effects of the
drug. Results of post-marketing programs might limit or expand the
further marketing of the products. Further, if there are any
modifications to the drug, including changes in indication, manufacturing
process, labeling or a change in manufacturing facility, an NDA supplement might
be required to be submitted to the FDA.
The rate
of completion of any clinical trials will be dependent upon, among other
factors, the rate of patient enrollment. Patient enrollment is a
function of many factors, including the size of the patient population, the
nature of the trial, the availability of alternative therapies and drugs, the
proximity of patients to clinical sites and the eligibility criteria for the
study. Delays in planned patient enrollment might result in increased
costs and delays, which could have a material adverse effect on us.
Failure
to comply with applicable FDA requirements may result in a number of
consequences that could materially and adversely affect us. Failure
to adhere to approved trial standards and GCPs in conducting clinical trials
could cause the FDA to place a clinical hold on one or more studies which would
delay research and data collection necessary for product
approval. Noncompliance with GCPs could also have a negative impact
on the FDA’s evaluation of an NDA. Failure to adhere to GMPs and
other applicable requirements could result in FDA enforcement action and in
civil and criminal sanctions, including but not limited to fines, seizure of
product, refusal of the FDA to approve product approval applications, withdrawal
of approved applications, and prosecution.
Whether
or not FDA approval has been obtained, approval of a product by regulatory
authorities in foreign countries must be obtained prior to the commencement of
marketing of the product in those countries. The requirements
governing the conduct of clinical trials and product approvals vary widely from
country to country, and the time required for approval might be longer or
shorter than that required for FDA approval. Although there are some
procedures for unified filings for some European countries, in general, each
country at this time has its own procedures and requirements. There
can be no assurance that any foreign approvals would be obtained.
In
addition to the regulatory framework for product approvals, we and our
collaborative partners must comply with laws and regulations regarding
occupational safety, laboratory practices, the use, handling and disposition of
radioactive materials, environmental protection and hazardous substance control,
and other local, state, federal and foreign regulation. The impact of
such regulation upon us cannot be predicted and could be material and
adverse.
Legislation
and Regulation Related to Bioterrorism Counteragents
Because
some of our drug candidates are intended for the treatment of diseases that may
result from acts of bioterrorism, they may be subject to the specific
legislation and regulation described below.
Project
BioShield
The
Project BioShield Act of 2004 provides expedited procedures for bioterrorism
related procurement and awarding of research grants, making it easier for HHS to
quickly commit funds to countermeasure projects. Project BioShield relaxes
procedures under the Federal Acquisition Regulation for procuring property or
services used in performing, administering or supporting biomedical
countermeasure research and development. In addition, if the Secretary of HHS
deems that there is a pressing need, Project BioShield authorizes the Secretary
to use an expedited award process, rather than the normal peer review process,
for grants, contracts and cooperative agreements related to biomedical
countermeasure research and development activity.
Under
Project BioShield, the Secretary of HHS, with the concurrence of the Secretary
of the Department of Homeland Security (“DHS”), and upon the approval of the
President, can contract to purchase unapproved countermeasures for the SNS in
specified circumstances. Congress is notified of a recommendation for a
stockpile purchase after Presidential approval. Project BioShield specifies that
a company supplying the countermeasure to the SNS is paid on delivery of a
substantial portion of the countermeasure. To be eligible for purchase under
these provisions, the Secretary of HHS must determine that there is sufficient
and satisfactory clinical results or research data, including data, if
available, from preclinical and clinical trials, to support a
25
reasonable
conclusion that the countermeasure will qualify for approval or licensing within
eight years. Project BioShield also allows the Secretary of HHS to authorize the
emergency use of medical products that have not yet been approved by the FDA. To
exercise this authority, the Secretary of HHS must conclude that:
|
•
|
the
agent for which the countermeasure is designed can cause serious or
life-threatening disease;
|
|
•
|
the
product may reasonably be believed to be effective in detecting,
diagnosing, treating or preventing the
disease;
|
|
•
|
the
known and potential benefits of the product outweigh its known and
potential risks; and
|
|
•
|
there
is no adequate alternative to the product that is approved and
available.
|
Although
this provision permits the Secretary of HHS to circumvent the FDA approval
process, its use would be limited to rare circumstances.
Safety
Act
The
Support Anti-Terrorism by Fostering Effective Technologies Act, or Safety Act,
enacted by the U.S. Congress in 2002 creates product liability limitations for
qualifying anti-terrorism technologies for claims arising from or related to an
act of terrorism. In addition, the Safety Act provides a process by which an
anti-terrorism technology may be certified as an “approved product” by the
Department of Homeland Security and therefore entitled to a rebuttable
presumption that the government contractor defense applies to sales of the
product. The government contractor defense, under specified circumstances,
extends the sovereign immunity of the United States to government contractors
who manufacture a product for the government. Specifically, for the government
contractor defense to apply, the government must approve reasonably precise
specifications, the product must conform to those specifications and the
supplier must warn the government about known dangers arising from the use of
the product.
Public
Readiness and Emergency Preparedness Act
The
Public Readiness and Emergency Preparedness Act (the “PREP Act”), enacted by
Congress in 2005 provides immunity for manufacturers from all claims under state
or federal law for “loss” arising out of the administration or use of a “covered
countermeasure”. However, injured persons may still bring a suit for “willful
misconduct” against the manufacturer under some circumstances. “Covered
countermeasures” include security countermeasures and “qualified pandemic or
epidemic products”. For these immunities to apply, the Secretary of
HHS must issue a declaration in cases of public health emergency or “credible
risk” of a future public health emergency. We cannot predict whether Congress
will fund the relevant PREP Act compensation programs; or whether the necessary
prerequisites for immunity would be triggered with respect to our drug
candidates.
Foreign
Regulation
In
addition to regulations in the United States, we will be subject to a variety of
foreign regulations governing clinical trials and commercial sales and
distribution of our products. Whether or not we obtain FDA approval for a
product, we must obtain approval of a product by the comparable regulatory
authorities of foreign countries before we can commence clinical trials or
marketing of the product in those countries. The actual time required to obtain
clearance to market a product in a particular foreign jurisdiction may vary
substantially, based upon the type, complexity and novelty of the pharmaceutical
product candidate and the specific requirements of that jurisdiction. The
requirements governing the conduct of clinical trials, marketing authorization,
pricing and reimbursement vary from country to country.
Reimbursement
and Pricing Controls
In many
of the markets where we could commercialize a drug candidate following
regulatory approval, the prices of pharmaceutical products are subject to direct
price controls by law and to reimbursement programs with varying price control
mechanisms.
In the
United States, there is an increasing focus on drug pricing in recent years.
There are currently no direct government price controls over private sector
purchases in the United States. However, the Veterans Health Care Act
establishes mandatory price discounts for certain federal purchasers, including
the Veterans Administration, Department of Defense, and the Public Health
Service; the discounts are based on prices charged to other
customers.
Under the
Medicaid program (a joint federal/state program that provides medical coverage
to certain low income families and individuals), pharmaceutical manufacturers
must pay prescribed rebates on specified drugs to enable them to be eligible for
reimbursement. Medicare (the federal program that provides medical
coverage for the elderly and disabled) generally reimburses for
physician-administered drugs and biologics on the basis of the product’s average
sales price. Outpatient drugs may be reimbursed under Medicare Part
D. Part D is administered through private entities that attempt to negotiate
price concessions from pharmaceutical manufacturers. Various states have adopted
further mechanisms that seek to control drug prices, including
by
26
disfavoring
higher priced products and by seeking supplemental rebates from manufacturers.
Managed care has also become a potent force in the market place that increases
downward pressure on the prices of pharmaceutical products.
Public
and private health care payors control costs and influence drug pricing through
a variety of mechanisms, including through negotiating discounts with the
manufacturers and through the use of tiered formularies and other mechanisms
that provide preferential access to particular products over others within a
therapeutic class. Payors also set other criteria to govern the uses of a drug
that will be deemed medically appropriate and therefore reimbursed or otherwise
covered. In particular, many public and private health care payors limit
reimbursement and coverage to the uses that are either approved by the FDA or
that are supported by other appropriate evidence, such as published medical
literature, and appear in a recognized compendium. Drug compendia are
publications that summarize the available medical evidence for particular drug
products and identify which uses are supported or not supported by the available
evidence, whether or not such uses have been approved by the FDA.
Different
pricing and reimbursement schemes exist in other countries. In the European
Community, governments influence the price of pharmaceutical products through
their pricing and reimbursement rules and control of national health care
systems that fund a large part of the cost of those products to consumers. Some
jurisdictions operate positive and negative list systems under which products
may only be marketed once a reimbursement price has been agreed. Other member
states allow companies to fix their own prices for medicines, but monitor and
control company profits. The downward pressure on health care costs in general,
particularly prescription drugs, has become very intense. As a result,
increasingly high barriers are being erected to the entry of new products. In
addition, in some countries cross-border imports from low-priced markets exert a
commercial pressure on pricing within a country.
Regulations
Regarding Government Contracting
We may
become a government contractor in the United States and elsewhere which would
mean that we would be subject to various statutes and regulations, including the
Federal Acquisition Regulation, which govern the procurement of goods and
services by agencies of the United States and other countries. These governing
statutes and regulations can impose stricter penalties than those normally
applicable to commercial contracts, such as criminal and civil damages liability
and suspension and debarment from future government contracting. In addition,
pursuant to various statutes and regulations, our government contracts can be
subject to unilateral termination or modification by the government for
convenience in the United States and elsewhere, detailed auditing requirements
and accounting systems, statutorily controlled pricing, sourcing and
subcontracting restrictions and statutorily mandated processes for adjudicating
contract disputes.
Hazardous
Materials and Select Agents
Our
development and manufacturing processes involve the use of hazardous materials,
including chemicals and radioactive materials, and produce waste products.
Accordingly, we are subject to federal, state and local laws and regulations
governing the use, manufacture, storage, handling and disposal of these
materials. In addition to complying with environmental and occupational health
and safety laws, we must comply with special regulations relating to biosafety
administered by the CDC, HHS and the DoD.
Other
Regulations
In the
United States and elsewhere, the research, manufacturing, distribution, sale and
promotion of drug and biological products are subject to regulation by various
federal, state and local authorities in addition to the FDA, including the
Centers for Medicare and Medicaid Services; other divisions of HHS, such as the
Office of Inspector General: the U.S. Department of Justice and individual U.S.
Attorney offices within the Department of Justice and state and local
governments. For example, sales, marketing and scientific and educational grant
programs must comply with the anti-kickback and fraud and abuse provisions of
the Social Security Act, the False Claims Act, the privacy provisions of the
Health Insurance Portability and Accountability Act and similar state laws.
Pricing and rebate programs must comply with the Medicaid rebate requirements of
the Omnibus Budget Reconciliation Act of 1990 and the Veterans Health Care Act
of 1992. All of these activities are also potentially subject to federal and
state consumer protection and unfair competition laws.
CPEC,
LLC
We were
previously developing bucindolol for the treatment of heart failure, but
development was discontinued in 1999. Commercial rights to bucindolol
are owned by CPEC, LLC, a limited liability company, of which we own 35% and
Indevus Pharmaceuticals, Inc. owns 65%.
In July
1999, the Department of Veterans Affairs and the National Heart, Lung, and Blood
Institute, a division of the NIH, terminated the Phase III heart failure study
of bucindolol earlier than scheduled, based on an interim analysis that revealed
a reduction in mortality in subpopulations that had been reported in other
trials and who constituted the majority of patients in the trial, but no
efficacy in some other subpopulations that had not been previously investigated
in beta-blocker heart failure trials. As a result, we discontinued development
of bucindolol for heart failure in 1999.
27
ARCA
Biopharma, Inc. (“ARCA”) of Broomfield, Colorado, and its academic
collaborators, have reexamined this clinical trial data and have identified a
genetic marker that highly correlates with patients who did not respond to
bucindolol. ARCA believes that bucindolol’s unique pharmacology is
suitable for therapy of most heart failure patients who do not exhibit this
genetic marker, in other pharmacogenetically-identified subpopulations that are
ideally suited for bucindolol’s novel therapeutic action, and for the treatment
of ischemia in the setting of left ventricular dysfunction. In
October 2003, CPEC outlicensed bucindolol to ARCA. Terms of the
license call for future royalty and milestone payments to CPEC upon the
development and commercialization of bucindolol.
During
fiscal 2006, CPEC agreed to modify the license agreement between CPEC and ARCA
and received 400,000 shares of ARCA common stock as consideration for the
amendment. In addition, during fiscal 2006, CPEC received a milestone
payment of $1,000,000 as a result of ARCA completing a financing.
During
fiscal 2008, CPEC received a milestone payment from ARCA of
$500,000. The milestone payment was triggered by the acceptance by
the FDA of a NDA for bucindolol. Future milestone payments and
royalty payments to CPEC and Aeolus, if any, while provided for under the
agreement between CPEC and ARCA, cannot be assured or
guaranteed. Also as a result of the filing of the NDA with the FDA,
we are obligated to pay $412,500 in the form of cash or stock at our election to
the majority owner of CPEC who will in turn pay the original licensors of
bucindolol per the terms of the 1994 Purchase Agreement of CPEC. The
obligation is included in our financial statements under the heading “Accounts
Payable.”
During
fiscal 2009, we sold our holdings of ARCA generating a gain of
$133,000. In addition during fiscal 2009, ARCA received a Complete
Response letter from the FDA for its NDA for bucindolol for the treatment of
patients with chronic heart failure. In the Complete Response
letter, the FDA states that it cannot approve the NDA in its current form and
specifies additional actions and information required for approval of the
bucindolol NDA.
Employees
At
December 24, 2009, we had two employees; David C. Cavalier, our Chairman of the
Board and John L. McManus, our President and Chief Executive
Officer. Mr. Cavalier and Mr. McManus are not represented by a labor
union. Each of our other executive officers and service providers are
consultants.
Executive
Officers
Our
executive officers and their ages as of December 24, 2009 were as
follows:
|
Name
|
Age
|
Position(s)
|
||
|
John
L. McManus
|
45
|
President
and Chief Executive Officer
|
||
|
Brian
J. Day, Ph.D.
|
49
|
Chief
Scientific Officer
|
||
|
Michael
P. McManus
|
40
|
Chief
Financial Officer, Treasurer and
Secretary
|
John L.
McManus. Mr. McManus began as a consultant to the Company in
June 2005 as President. He became employed as our President and Chief
Operating Officer in July 2006 and was appointed President and Chief Executive
Officer in March 2007. Mr. McManus, who received his degree in
business administration from the University of Southern California in 1986, is
the founder and president of McManus Financial Consultants, Inc. (“MFC”), which
provides strategic, financial and investor relations advice to senior
managements and boards of directors of public companies, including advice on
mergers and acquisitions. These companies have a combined value of
over $25 billion. He has served as president of MFC since
1997. In addition, Mr. McManus previously served as Vice President,
Finance and Strategic Planning to Spectrum Pharmaceuticals, Inc. where he had
primary responsibility for restructuring Spectrum’s operations and finances,
including the design of strategic and financial plans to enhance Spectrum’s
corporate focus, and leading the successful implementation of these
plans. The implementation of these plans led to an increase in
Spectrum’s market value from $1 million to more than $125 million at the time of
Mr. McManus’ departure.
Brian J. Day, Ph.D. Dr. Day
is a part-time consultant and was appointed Chief Scientific Officer of Aeolus
in September 2004. Dr. Day has extensive training in both
pharmacology and toxicology with over 14 years experience. Since 1994
he has helped guide the design and synthesis of metalloporphyrins and has
discovered a number of their novel activities in biological
systems. Dr. Day has authored over 70 original scientific
publications and served as a consultant to biotechnology companies for over 10
years. He is an active member of a number of scientific societies
including the American Chemical Society, Society for Free Radicals in Biology
and Medicine, and Society of Toxicology, where he served on the Board of
Publications. Dr. Day has been at the NJH since 1997 and currently is
a Professor in the Environmental and Occupational Health Sciences
Division. He is one of the scientific co-founders of Aeolus and an
inventor on a majority of the catalytic antioxidant program’s
patents.
28
Michael P.
McManus. Mr. McManus began as a consultant to the Company in
June 2005, serving as Chief Accounting Officer, Treasurer and
Secretary. In July 2006, Mr. McManus was appointed Chief Financial
Officer, Treasurer and Secretary. Mr. McManus served as the Executive
Vice President of MFC from 1995 to 2008. MFC is a leading provider of
financial, management and investor relations consulting and support services to
publicly traded companies. From 2001 to 2003, Mr. McManus also served
as Controller and Principal Accounting Officer of Spectrum Pharmaceuticals,
Inc., where he was responsible for restructuring Spectrum’s accounting and
administration functions. Prior to joining MFC, from 1991 to 1995, he
worked at Price Waterhouse LLP (now PricewaterhouseCoopers LLP) as an audit
manager for healthcare and financial services companies. Mr. McManus
is a retired Certified Public Accountant and holds a B.S. in Accounting from the
University of Southern California.
Item
1A. Risk Factors.
You
should carefully consider the following information about risks described below,
together with the other information contained in this annual report on
Form 10-K and in our other filings with the SEC, before you decide to buy or
maintain an investment in our common stock. We believe the risks
described below are the risks that are material to us as of the date of this
annual report. If any of the following risks actually occur, our
business, financial condition, results of operations and future growth prospects
would likely be materially and adversely affected. In these
circumstances, the market price of our common stock could decline, and you may
lose all or part of the money you paid to buy our capital stock.
Risks
Related to Our Business
We
have operated at a loss and will likely continue to operate at a loss for the
foreseeable future.
We have
incurred significant losses over the past five years, including net losses of
$2.3 million, $3.0 million and $3.0 million for the years ended September 30,
2009, 2008 and 2007, respectively, and we had an accumulated deficit of
approximately $161.2 million as of September 30, 2009. Our operating
losses have been due primarily to our expenditures for research and development
on our drug candidates and for general and administrative expenses and our lack
of significant revenues. We are likely to continue to incur operating
losses until such time, if ever, that we generate significant recurring
revenues. We anticipate it will take a minimum of four years (and
possibly longer) for us to generate recurring revenues, since we expect that it
will take at least that long before the development of any of our licensed or
other current potential products is completed, marketing approvals are obtained
from the FDA and commercial sales of any of these products can
begin.
We
need substantial additional funding to continue our operations and may be unable
to raise capital when needed, or at all, which would force us to delay, curtail
or eliminate our clinical programs and our product development
programs.
We need
to raise substantial additional capital to fund our operations and clinical
trials and continue our research and development. In addition, we may
need to raise substantial additional capital to enforce our proprietary rights,
defend, in litigation or otherwise, any claims that we infringe third party
patents or other intellectual property rights; and commercialize any of our
products that may be approved by the FDA or any international regulatory
authority.
As of
September 30, 2009, we had cash of approximately $646,000. In
addition, we raised $1,650,000 in October 2009 through the sale of our common
stock and warrants. We expect to use these funds, including any
additional funds received pursuant to the issuance of additional Common Stock
and the exercise of outstanding warrants to purchase our capital stock, to
continue the development of our drug candidates, to expand the development of
our drug pipeline and for working capital.
We
believe we have adequate financial resources to fund our current operations into
the fourth quarter of fiscal year 2010. However, in order to fund
on-going cash requirements beyond that point, or to further accelerate or expand
our programs, we will need to raise additional funds. We are
considering strategic and financial options available to us, including public or
private equity offerings, debt financings and collaboration
arrangements. If we raise additional funds by issuing securities, our
stockholders will experience dilution of their ownership
interest. Debt financings, if available, may involve restrictive
covenants and require significant interest payments. If we do not
receive additional financing to fund our operations through the fourth quarter
of fiscal 2010, we would have to discontinue some or all of our activities,
merge with or sell some or all of our assets to another company, or cease
operations entirely, and our stockholders might lose all or part of their
investments.
In
addition, if our catalytic antioxidant program shows scientific progress, we
will need significant additional funds to move therapies through the preclinical
stages of development and clinical trials. If we are unable to raise
the amount of capital necessary to complete development and reach
commercialization of any of our catalytic antioxidant products, we will need to
delay or cease development of one or more of these products or partner with
another company for the development and commercialization of these
products.
29
Our
independent registered public accounting firm has expressed substantial doubt
about our ability to continue as a going concern.
In its
audit opinion issued in connection with our consolidated balance sheets as of
September 30, 2009 and 2008 and our consolidated statements of operations,
stockholder’s equity and cash flows for the years ended September 30, 2009, 2008
and 2007, our independent registered public accounting firm has expressed
substantial doubt about our ability to continue as a going concern given our
recurring net losses, negative cash flows from operations and working capital
deficiency. The accompanying financial statements have been prepared
on a going concern basis, which contemplates the realization of assets and the
satisfaction of liabilities and commitments in the normal course of
business. The financial statements do not include any adjustments
relating to the recoverability and classification of recorded asset amounts or
amounts of liabilities that might be necessary should we be unable to continue
in existence.
We
have a history of operating losses, expect to continue to incur substantial
losses and may never become profitable.
We have
no products approved for commercialization in the United States or
abroad. Our drug candidates are still being developed, and all but
our AEOL 10150 candidate are still in early stages of
development. Our drug candidates will require significant additional
development, clinical trials, regulatory clearances or approvals by the FDA and
additional investment before they can be commercialized in the United
States.
Our
likelihood for achieving profitability will depend on numerous factors,
including success in:
|
|
·
|
developing
our existing drug candidates and developing and testing new drug
candidates;
|
|
|
·
|
carrying
out our intellectual property
strategy;
|
|
|
·
|
establishing
our competitive position;
|
|
|
·
|
achieving
third-party collaborations;
|
|
|
·
|
receiving
regulatory approvals;
|
|
|
·
|
manufacturing
and marketing products; and
|
|
|
·
|
receiving
government funding and identifying new government funding
opportunities.
|
Many of
these factors will depend on circumstances beyond our control. We
cannot guarantee that we will achieve sufficient revenues for
profitability. Even if we do achieve profitability, we cannot
guarantee that we can sustain or increase profitability on a quarterly or annual
basis in the future. If revenues grow more slowly than we anticipate,
or if operating expenses exceed our expectations or cannot be adjusted
accordingly, then our business, results of operations, financial condition and
cash flows will be materially and adversely affected.
The
current turmoil affecting the financial markets and the possibility that
financial institutions may consolidate or cease operations has resulted in a
tightening in the credit markets, a low level of liquidity in many financial
markets and extreme volatility in fixed income, credit, currency and equity
markets. As a result, there can be no assurances that we will be
successful in obtaining sufficient financing on commercially reasonable terms or
at all. Our requirements for additional capital may be substantial
and will be dependent on many factors, including the success of our research and
development efforts, our ability to commercialize and market products, our
ability to successfully pursue our licensing and collaboration strategy, the
receipt of government funding, competing technological developments, costs
associated with the protection of our intellectual property and any future
change in our business strategy.
As of
September 30, 2009, we had an accumulated deficit of $161.2 million from our
research, development and other activities. We have not generated
material revenues from product sales and do not expect to generate product
revenues sufficient to support us for at least several more years.
Our
research and development (“R&D”) activities are at an early stage and
therefore might never result in viable products.
Our
catalytic antioxidant program is in the early stages of development, involves
unproven technology, requires significant further R&D and regulatory
approvals and is subject to the risks of failure inherent in the development of
products or therapeutic procedures based on innovative
technologies. These risks include the possibilities
that:
|
|
·
|
any
or all of these proposed products or procedures are found to be unsafe or
ineffective or otherwise fail to receive necessary regulatory
approvals;
|
· the
proposed products or procedures are not economical to market or do not achieve
broad market acceptance;
· third
parties hold proprietary rights that preclude us from marketing the proposed
products or procedures; and
· third
parties market a superior or equivalent product.
Further,
the timeframe for commercialization of any product is long and uncertain because
of the extended testing and regulatory review process required before marketing
approval can be obtained. There can be no assurance that we will be
able to successfully develop or market any of our proposed products or
procedures.
30
If
our products are not successfully developed and eventually approved by the FDA,
we may be forced to reduce or terminate our operations.
All of
our drug candidates are at various stages of development and must be approved by
the FDA or similar foreign governmental agencies before they can be
marketed. The process for obtaining FDA and foreign regulatory
approval is both time-consuming and costly, with no certainty of a successful
outcome. This process typically requires extensive preclinical and
clinical testing, which may take longer or cost more than we anticipate, and may
prove unsuccessful due to numerous factors. Drug candidates that may
appear to be promising at early stages of development may not successfully reach
the market for a number of reasons. The results of preclinical and
initial clinical testing of these drug candidates may not necessarily indicate
the results that will be obtained from later or more extensive
testing. A number of companies in the pharmaceutical and
biotechnology industries have suffered significant setbacks in advanced clinical
trials, even after obtaining promising results in earlier trials.
Numerous
factors could affect the timing, cost or outcome of our drug development
efforts, including the following:
· Difficulty
in securing research laboratories to conduct research activities;
· Difficulty
in securing centers to conduct trials;
· Difficulty
in enrolling patients in conformity with required protocols or projected
timelines;
· Unexpected
adverse reactions by patients in trials;
· Difficulty
in obtaining clinical supplies of the product;
· Changes
in the FDA’s or other regulatory body’s requirements for our testing during the
course of that testing;
· Inability
to generate statistically significant data confirming the efficacy of the
product being tested;
· Modification
of the drug during testing; and
· Reallocation
of our limited financial and other resources to other clinical
programs.
It is
possible that none of the products we develop will obtain the regulatory
approvals necessary for us to begin commercializing them. The time
required to obtain FDA and other approvals is unpredictable but often can take
years following the commencement of clinical trials, depending upon the nature
of the drug candidate. Any analysis we perform of data from clinical
activities is subject to confirmation and interpretation by regulatory
authorities, which could delay, limit or prevent regulatory
approval. Any delay or failure in obtaining required approvals could
have a material adverse effect on our ability to generate revenues from the
particular drug candidate and we may not have the financial resources to
continue to develop our drug candidates and, as a result, may have to terminate
our operations.
If
we do not reach the market with our products before our competitors offer
products for the same or similar uses, or if we are not effective in marketing
our products, our revenues from product sales, if any, will be
reduced.
We face
intense competition in our development activities. Many of our
competitors are fully integrated pharmaceutical companies and more established
biotechnology companies, which have substantially greater financial, technical,
sales and marketing and human resources than we do. These companies
might succeed in obtaining regulatory approval for competitive products more
rapidly than we can for our products. In addition, competitors might
develop technologies and products that are less expensive and perceived to be
safer or more effective than those being developed by us, which could impair our
product development and render our technology obsolete.
We
are and expect to remain dependent upon collaborations with third parties for
the development of new products, and adverse events involving these
collaborations could prevent us from developing and commercializing our drug
candidates and achieving profitability.
We
currently license from third parties, and do not own, rights under patents and
certain related intellectual property for the development of our drug
candidates. In addition, we expect to enter into agreements with
third parties to license rights to our drug candidates. We might not
be able to enter into or maintain these agreements on terms favorable to us, if
at all. Further if any of our current licenses were to expire or
terminate, our business, prospects, financial condition and results of
operations could be materially and adversely affected.
Our
research and development activities rely on technology licensed from third
parties, and termination of any of those licenses would result in loss of
significant rights to develop and market our products, which would impair our
business, prospects, financial condition and results of operations.
We have
exclusive worldwide rights to our antioxidant small molecule technology through
license agreements with Duke University and the National Jewish
Health. Each license generally may be terminated by the licensor if
we fail to perform our obligations under the agreement, including obligations to
develop the compounds and technologies under license. If terminated,
we would lose the right to develop the products, which could adversely affect
our business, prospects, financial condition and results of
operations. The license agreements also generally require us to meet
specified milestones or show reasonable diligence in development of the
technology. If disputes arise over the definition of these
requirements or whether we have satisfied the requirements in a timely manner,
or if any other obligations in the license agreements are disputed by the other
party, the other
31
party
could terminate the agreement, and we could lose our rights to develop the
licensed technology.
If new
technology is developed from these licenses, we may be required to negotiate
certain key financial and other terms, such as royalty payments, for the
licensing of this future technology with these research institutions, and it
might not be possible to obtain any such license on terms that are satisfactory
to us, or at all.
We
now rely, and will continue to rely, heavily on third parties for product and
clinical development, manufacturing, marketing and distribution of our
products.
We
currently depend heavily and will depend heavily in the future on third parties
for support in product development, clinical development, manufacturing,
marketing and distribution of our products. The termination of some
or all of our existing collaborative arrangements, or our inability to establish
and maintain collaborative arrangements, could have a material adverse effect on
our ability to continue or complete clinical development of our
products.
We rely
on contract clinical research organizations (“CROs”) for various aspects of our
clinical development activities including clinical trial monitoring, data
collection and data management. As a result, we have had and continue
to have less control over the conduct of clinical trials, the timing and
completion of the trials, the required reporting of adverse events and the
management of data developed through the trial than would be the case if we were
relying entirely upon our own staff. Although we rely on CROs to
conduct our clinical trials, we are responsible for confirming that each of our
clinical trials is conducted in accordance with the investigational plan and
protocol. Moreover, the FDA and foreign regulatory agencies require
us to comply with good clinical practices (“GCPs”) for conducting, recording and
reporting the results of clinical trials to assure that the data and results are
credible and accurate and that the trial participants are adequately
protected. Our reliance on third parties does not relieve us of these
responsibilities and requirements.
The third
parties on which we rely may have staffing difficulties, may undergo changes in
priorities or may become financially distressed, adversely affecting their
willingness or ability to conduct our trials. We may experience
unexpected cost increases that are beyond our control. Any failure of
such CROs to successfully accomplish clinical trial monitoring, data collection
and data management and the other services they provide for us in a timely
manner and in compliance with regulatory requirements could have a material
adverse effect on our ability to complete clinical development of our products
and obtain regulatory approval. Problems with the timeliness or
quality of the work of a CRO may lead us to seek to terminate the relationship
and use an alternate service provider. However, making such changes
may be costly and would likely delay our trials, and contractual restrictions
may make such a change difficult or impossible. Additionally, it may
be difficult to find a replacement organization that can conduct our trials in
an acceptable manner and at an acceptable cost.
If
we do not receive the contract from the Biomedical Advanced Research and
Development Authority (“BARDA”) for advanced research and development of medical
countermeasures for chemical, biological, radiological and nuclear threats, we
would be dependent upon grants from NIH for continued development of AEOL 10150
for Pulmonary ARS, or we would need to curtail our development
program in this area significantly and we may be placed at a
competitive disadvantage in addressing this market opportunity.
On March
4, 2009, BARDA issued a Special Instructions Amendment 4 to a Broad Agency
Announcement (BAA-BARDA-09-34) for advanced research and development of medical
countermeasures for chemical, biological, radiological and nuclear
threats. We submitted a white paper to this request on August 31,
2009. We are currently aware of at least one other company who
has submitted a white paper. Timing for an award under this
announcement remains uncertain. There can be no assurance that we
will be awarded a contract under that announcement or if we receive a contract,
that it will not include substantial conditions, that we can satisfy any of the
conditions or that we can begin to receive any proceeds from any such contract
within any specific period of time.
Necessary
Reliance on the Animal Rule in Conducting Trials is Time-Consuming and
Expensive.
To obtain
FDA approval for our drug candidate for a bioterrorism indication under current
FDA regulations, we are required to utilize animal model studies for efficacy
and provide animal and human safety data under the “Animal Rule.” For
many of the biological and chemical threats, animal models are not yet
available, and as such we are developing, or will have to develop, appropriate
animal models, which is a time-consuming and expensive research
effort. Further, we may not be able to sufficiently demonstrate the
animal correlation to the satisfaction of the FDA, as these corollaries are
difficult to establish and are often unclear. The FDA may decide that
our data are insufficient for approval and require additional preclinical,
clinical or other studies, refuse to approve our products, or place restrictions
on our ability to commercialize those products. Further, other
countries do not, at this time, have established criteria for review and
approval of these types of products outside their normal review process; i.e.,
there is no “Animal Rule” equivalent, and consequently there can be no assurance
that we will be able to make a submission for marketing approval in foreign
countries based on such animal data.
Additionally,
few facilities in the U.S. and internationally have the capability to test
animals with radiation, nerve agents, or other lethal biotoxins or chemical
agents or otherwise assist us in qualifying the requisite animal
models. We have to compete
32
with
other biodefense companies for access to this limited pool of highly specialized
resources. We therefore may not be able to secure contracts to
conduct the testing in a predictable timeframe, cost-effectively or at
all.
Even
if we succeed in commercializing our drug candidates, they may not become
profitable and manufacturing problems or side effects discovered at later stages
can further increase costs of commercialization.
We cannot
assure you that any drugs resulting from our research and development efforts
will become commercially available. Even if we succeed in developing
and commercializing our drug candidates, we may never generate sufficient or
sustainable revenues to enable us to be profitable. Even if
effective, a product that reaches market may be subject to additional clinical
trials, changes to or re-approvals of our manufacturing facilities or a change
in labeling if we or others identify side effects or manufacturing problems
after a product is on the market. This could harm sales of the
affected products and could increase the cost and expenses of commercializing
and marketing them. It could also lead to the suspension or
revocation of regulatory approval for the products.
We and
our CMOs will also be required to comply with the applicable FDA current cGMP
regulations. These regulations include requirements relating to
quality control and quality assurance as well as the corresponding maintenance
of records and documentation. Manufacturing facilities are subject to
inspection by the FDA. These facilities must be approved to supply
licensed products to the commercial marketplace. We and our contract
manufacturers may not be able to comply with the applicable cGMP requirements
and other FDA regulatory requirements. Should we or our contract
manufacturers fail to comply, we could be subject to fines or other sanctions or
could be prohibitedfrom marketing any products we develop.
Risks
Related to Our Dependence on U.S. Government Grants and Contracts
Most
of our immediately foreseeable future revenues are contingent upon grants and
contracts from the U.S. government and we may not achieve sufficient, if any,
revenues from these agreements to attain profitability.
For the
foreseeable future, we believe our main customer, if any, will be national
governments, primarily the U.S. government. There can be no
assurances that we will receive any grants. The process of obtaining
government contracts is lengthy and uncertain and we will have to compete with
other companies for each contract. There can be no assurances that we
will be awarded any contracts to supply the U.S. or other governments with our
drug candidates as such awards may be made, in whole or in part, to our
competitors. If the U.S. government makes significant future contract
awards for the supply to the U.S. emergency stockpile of a competing product,
our business will be harmed and it is unlikely that we will ultimately be able
to supply that particular treatment or product to foreign governments or other
third parties. Further, changes in government budgets and agendas, or
advances by our competitors, may result in a decreased and de-prioritized
emphasis on procuring the biodefense products we are developing.
Due to
the current economic downturn, the accompanying fall in tax revenues and the
U.S. government’s efforts to stabilize the economy, the U.S. government may be
forced or choose to reduce or delay spending in the biodefense field, which
could decrease the likelihood of future government contract awards or that the
government would procure products from us.
The
U.S. government’s determination to award any contracts may be challenged by an
interested party, such as another bidder, at the GAO or in federal
court. If such a challenge is successful, a contract may be
terminated.
The laws and regulations governing the procurement of goods and services by the U.S. government provide procedures by which other bidders and other interested parties may challenge the award of a government contract. If we are awarded a government contract, such challenges or protests could be filed even if there are not any valid legal grounds on which to base the protest. If any such protests are filed, the government agency may decide to suspend our performance under the contract while such protests are being considered by the GAO or the applicable federal court, thus potentially delaying delivery of goods and services and payment. In addition, we could be forced to expend considerable funds to defend any potential award. If a protest is successful, the government may be ordered to terminate the contract and reselect bids. The government could even be directed to award a potential contract to one of the other bidders.
Our
business may become subject to audit by the U.S. government and a negative audit
could adversely affect our business.
U.S.
government agencies such as the Defense Contract Audit Agency (the “DCAA”),
routinely audit and investigate government contractors. These
agencies review a contractor’s performance under its contracts, cost structure
and compliance with applicable laws, regulations and standards.
The DCAA
also reviews the adequacy of, and a contractor’s compliance with, its internal
control systems and policies, including the contractor’s purchasing, property,
estimating, compensation and management information systems. Any
costs found to be improperly allocated to a specific contract will not be
reimbursed, while such costs already reimbursed must be refunded. If
an audit uncovers improper or illegal activities, we may be subject to civil and
criminal penalties and administrative sanctions, including:
33
|
|
·
|
termination
of contracts;
|
|
|
·
|
forfeiture
of profits;
|
|
|
·
|
suspension
of payments;
|
|
|
·
|
fines;
and
|
|
|
·
|
suspension
or prohibition from conducting business with the U.S.
government.
|
In
addition, we could suffer serious reputational harm if allegations of
impropriety were made against us.
Laws
and regulations affecting government contracts make it more costly and difficult
for us to successfully conduct our business.
We must
comply with numerous laws and regulations relating to the formation,
administration and performance of government contracts, which can make it more
difficult for us to retain our rights under these contracts. These
laws and regulations affect how we conduct business with government
agencies. Among the most significant government contracting
regulations that affect our business are:
|
|
·
|
the
Federal Acquisition Regulations (“FAR”), and agency-specific regulations
supplemental to the Federal Acquisition Regulations, which comprehensively
regulate the procurement, formation, administration and performance of
government contracts;
|
|
|
·
|
the
business ethics and public integrity obligations, which govern conflicts
of interest and the hiring of former government employees, restrict the
granting of gratuities and funding of lobbying activities and incorporate
other requirements such as the Anti-Kickback Act and Foreign Corrupt
Practices Act;
|
|
|
·
|
export
and import control laws and regulations;
and
|
|
|
·
|
laws,
regulations and executive orders restricting the use and dissemination of
information classified for national security purposes and the exportation
of certain products and technical
data.
|
Foreign governments typically also have laws and regulations governing contracts with their respective agencies. These foreign laws and regulations could affect how we conduct business and, in some instances, impose added costs on our business. Any changes in applicable laws and regulations could restrict our ability to obtain contracts, which could limit our ability to conduct our business and materially adversely affect our revenues and results of operations.
Because
we depend on clinical research centers and other contractors for clinical and
non-clinical testing, including testing under the Animal Rule, and for certain
research and development activities, the results of our clinical trial,
non-clinical animal efficacy studies, and research and development activities
are largely beyond our control.
The
nature of studies, clinical trials and our business strategy of outsourcing
substantially all of our research and development and manufacturing work require
that we rely on clinical research centers and other contractors to assist us
with research and development, clinical and non-clinical testing (including
animal efficacy studies under the Animal Rule), patient enrollment and other
activities. As a result, our success depends largely on the success
of these third parties in performing their responsibilities. Although
we prequalify our contractors and believe that they are fully capable of
performing their contractual obligations, we cannot directly control the
adequacy and timeliness of the resources and expertise that they apply to these
activities. Furthermore, we have to compete with other biodefense
companies for access to this limited pool of highly specialized
resources. If our contractors do not perform their obligations in an
adequate and timely manner or we are unable to enter into contracts with them
because of prior commitments to our competitors, the pace of clinical or
non-clinical development, regulatory approval and commercialization of our drug
candidates could be significantly delayed and our prospects could be adversely
affected.
Data
obtained from clinical trials is susceptible to varying interpretations, which
could delay, limit or prevent regulatory clearances.
Data
already obtained, or obtained in the future, from pre-clinical studies,
non-clinical studies and clinical trials does not necessarily predict the
results that will be obtained from later pre-clinical studies and clinical
trials. Moreover, pre-clinical and clinical data is susceptible to varying
interpretations, which could delay, limit or prevent regulatory approval.
A number of companies in the pharmaceutical industry have suffered significant
setbacks in advanced clinical trials, even after promising results in earlier
trials. The failure to adequately demonstrate the safety and effectiveness
of an intended product under development could delay or prevent regulatory
clearance of the drug candidate, which would result in delays to
commercialization and could materially harm our business. Our studies and
clinical trials may not demonstrate sufficient levels of safety and efficacy
necessary to obtain the requisite regulatory approvals for our drugs, and our
proposed drugs may not be approved for marketing.
We may
encounter delays or rejections based on additional government regulation from
future legislation or administrative action or changes in FDA policy during the
period of development, clinical trials and FDA regulatory review. We may
encounter similar delays in foreign countries. If any of our products are
approved for commercialization, sales of the products outside
the
34
U.S.
would be subject to foreign regulatory approvals that vary from country to
country. The time required to obtain approvals from foreign countries may
be shorter or longer than that required for FDA approval, and requirements for
foreign licensing may differ from FDA requirements. We may be unable to
obtain requisite approvals from the FDA or foreign regulatory authorities, and
even if obtained, such approvals may not be on a timely basis, or they may not
cover the uses that we request.
Even if
we do ultimately receive FDA approval for any of our drug candidates, these drug
candidates will be subject to extensive ongoing regulation, including
regulations governing manufacturing, labeling, packaging, testing, dispensing,
prescription and procurement quotas, record keeping, reporting, handling,
shipment and disposal of any such drug. Failure to obtain and maintain
required registrations or to comply with any applicable regulations could
further delay or preclude development and commercialization of our drugs and
subject us to enforcement action.
Unfavorable provisions in government
contracts, some of which may be customary, may harm our business, financial
condition and operating results.
Government
contracts customarily contain provisions that give the government substantial
rights and remedies, many of which are not typically found in commercial
contracts, including provisions that allow the government to:
|
•
|
terminate
existing contracts, in whole or in part, for any reason or no
reason;
|
|
•
|
unilaterally
reduce or modify contracts or subcontracts, including equitable price
adjustments;
|
|
•
|
cancel
multi-year contracts and related orders if funds for contract performance
for any subsequent year become
unavailable;
|
|
•
|
decline
to exercise an option to renew a
contract;
|
|
•
|
exercise
an option to purchase only the minimum amount specified in a
contract;
|
|
•
|
decline
to exercise an option to purchase the maximum amount specified in a
contract;
|
|
|
•
|
claim
rights to products, including intellectual property, developed under the
contract;
|
|
•
|
take
actions that result in a longer development timeline than
expected;
|
|
•
|
direct
the course of a development program in a manner not chosen by the
government contractor;
|
|
•
|
suspend
or debar the contractor from doing business with the government or a
specific government agency;
|
|
•
|
pursue
criminal or civil remedies under the False Claims Act and False Statements
Act; and
|
|
•
|
control
or prohibit the export of products.
|
Generally,
government contracts contain provisions permitting unilateral termination or
modification, in whole or in part, at the government’s convenience. Under
general principles of government contracting law, if the government terminates a
contract for convenience, the terminated company may recover only its incurred
or committed costs, settlement expenses and profit on work completed prior to
the termination.
If the
government terminates a contract for default, the defaulting company is entitled
to recover costs incurred and associated profits on accepted items only and may
be liable for excess costs incurred by the government in procuring undelivered
items from another source. Some government contracts grant the
government the right to use, for or on behalf of the U.S. government, any
technologies developed by the contractor under the government contract. If we
were to develop technology under a contract with such a provision, we might not
be able to prohibit third parties, including our competitors, from using that
technology in providing products and services to the government.
Political
or social factors may delay or impair our ability to market our products and our
business may be materially adversely affected.
Products
developed to treat diseases caused by, or to combat the threat of, bioterrorism
will be subject to changing political and social environments. The
political and social responses to bioterrorism have been
unpredictable. Political or social pressures may delay or cause
resistance to bringing our products to market or limit pricing of our products,
which would harm our business.
Legislation
limiting or restricting liability for medical products used to fight
bioterrorism is new, and we cannot be certain that any such protection will
apply to our products or if applied what the scope of any such coverage will
be.
The U.S.
Public Readiness Act was signed into law in December 2005 and creates general
immunity for manufacturers of countermeasures, including security
countermeasures (as defined in Section 319F-2(c)(1)(B) of that act), when the
U.S. Secretary of Health and Human Services issues a declaration for their
manufacture, administration or use. The declaration is meant to
provide general immunity from all claims under state or federal law for loss
arising out of the administration or use of a covered
countermeasure. Manufacturers are excluded from this protection in
cases of willful misconduct. There can be no assurance that the
Secretary of Health and Human Services will make declarations that would cover
any of our other drug candidates or that the U.S. Congress will not act in the
future to reduce coverage under the Public Readiness Act or to repeal it
altogether.
35
Upon a
declaration by the Secretary of Health and Human Services, a compensation fund
would be created to provide “timely, uniform, and adequate compensation to
eligible individuals for covered injuries directly caused by the administration
or use of a covered countermeasure.” The “covered injuries” to which
the program applies are defined as serious physical injuries or
death. Individuals are permitted to bring a willful misconduct action
against a manufacturer only after they have exhausted their remedies under the
compensation program. A willful misconduct action could be brought
against us if an individual(s) has exhausted their remedies under the
compensation program which thereby could expose us to
liability. Furthermore, there is no assurance that the Secretary of
Health and Human Services will issue under this act a declaration to establish a
compensation fund. We may also become subject to standard product
liability suits and other third party claims if products we develop which fall
outside of the Public Readiness Act cause injury or if treated individuals
subsequently become infected or otherwise suffer adverse effects from such
products.
Healthcare
reform measures and other statutory or regulatory changes could adversely affect
our business.
The
pharmaceutical and biotechnology industries are subject to extensive regulation,
and from time to time legislative bodies and governmental agencies consider
changes to such regulations that could have significant impact on industry
participants. For example, in light of certain highly-publicized safety issues
regarding certain drugs that had received marketing approval, the U.S. Congress
is considering various proposals regarding drug safety, including some which
would require additional safety studies and monitoring and could make drug
development more costly. We are unable to predict what additional legislation or
regulation, if any, relating to safety or other aspects of drug development may
be enacted in the future or what effect such legislation or regulation would
have on our business.
The
business and financial condition of pharmaceutical and biotechnology companies
are also affected by the efforts of governments, third-party payors and others
to contain or reduce the costs of healthcare to consumers. In the United States
and various foreign jurisdictions there have been, and we expect that there will
continue to be, a number of legislative and regulatory proposals aimed at
changing the healthcare system, such as proposals relating to the reimportation
of drugs into the U.S. from other countries (where they are then sold at a lower
price) and government control of prescription drug pricing. The pendency or
approval of such proposals could result in a decrease in our share price or
limit our ability to raise capital or to obtain strategic collaborations or
licenses.
The
current disruptions in the financial markets could affect our ability to obtain
additional debt financing on favorable terms (or at all) and have other adverse
effects on us.
The
United States credit markets have recently experienced historic dislocations and
liquidity disruptions which have caused financing to be unavailable in many
cases and even if available caused spreads on prospective debt financings to
widen considerably. These circumstances have materially impacted
liquidity in the debt markets, making financing terms for borrowers able to find
financing less attractive, and in many cases have resulted in the unavailability
of certain types of debt financing. Continued uncertainty in the
credit markets may negatively impact our ability to access additional debt
financing on favorable terms or at all. In addition, Federal
legislation to deal with the current disruptions in the financial markets could
have an adverse affect on our financial condition and results of
operations.
We
will need to enter into collaborative arrangements for the manufacturing and
marketing of our drug candidates, or we will have to develop the expertise,
obtain the additional capital and invest the resources to perform those
functions internally.
We do not
have the staff or facilities to manufacture or market any of the drug candidates
being developed in our catalytic antioxidant program. As a result, we
will need to enter into collaborative arrangements to develop, commercialize,
manufacture and market products that we expect to emerge from our catalytic
antioxidant program, or develop the expertise within the company. We
might not be successful in entering into such third party arrangements on terms
acceptable to us, if at all. If we are unable to obtain or retain
third-party manufacturing or marketing on acceptable terms, we may be delayed in
our ability to commercialize products, which could have a material adverse
effect on our business, prospects, financial condition and results of
operations. Substantial additional funds and personnel would be
required if we needed to establish our own manufacturing or marketing
operations. We may not be able to obtain adequate funding or
establish these capabilities in a cost-effective or timely manner, which could
have a material adverse effect on our business, prospects, financial condition
and results of operations.
A
failure to obtain or maintain patent and other intellectual property rights
would allow others to develop and sell products similar to ours, which could
impair our business, prospects, financial condition and results of
operations.
The
success of our business depends, in part, on our ability to establish and
maintain adequate protection for our intellectual property, whether owned by us
or licensed from third parties. We rely primarily on patents in the
United States and in other key markets to protect our intellectual
property. If we do not have adequate patent protection, other
companies could develop and sell products that compete directly with ours,
without incurring any liability to us. Patent prosecution,
maintenance and enforcement on a global basis are time-consuming and expensive,
and many of these costs must be incurred before we know whether a product
covered by the claims can be successfully developed or marketed.
36
Even if
we expend considerable time and money on patent prosecution, a patent
application may never issue as a patent. We can never be certain that
we were the first to invent the particular technology or that we were the first
to file a patent application for the technology because patent applications in
the United States and elsewhere are not typically published for public
inspection for at least 18 months from the date when they are
filed. It is always possible that a competitor is pursuing a patent
for the same invention in the United States as we are and has an earlier
invention date. Outside the United States in some jurisdictions,
priority of invention is determined by the earliest effective filing date, not
the date of invention. Consequently, if a third party pursues the
same invention and has an earlier filing date, patent protection outside the
United States would be unavailable to us. Also, outside the United
States, an earlier date of invention cannot overcome a date of publication that
precedes the earliest effective filing date. Accordingly, the
patenting of our proposed products would be precluded outside the United States
if a prior publication anticipates the claims of a pending application, even if
the date of publication is within a year of the filing of the pending
application.
Even if
patents issue, the patent claims allowed might not be sufficiently broad to
offer adequate protection for our technology against competitive
products. Patent protection differs from country to country, giving
rise to increased competition from other products in countries where patent
coverage is either unavailable, weak or not adequately enforced, if enforced at
all. Once a patent issues, we still face the risk that others will
try to design around our patent or will try to challenge the validity of the
patent. The cost of defending against a challenge to one or more of
our patents could be substantial and even if we prevailed, there could be no
assurance that we would recover damages.
If
a third party were to bring an infringement claim against us, we would incur
significant costs in our defense; if the claim were successful, we would need to
develop non-infringing technology or obtain a license from the successful patent
holder, if available.
Our
business also depends on our ability to develop and market products without
infringing on the proprietary rights of others or being in breach of our license
agreements. The pharmaceutical industry is characterized by a large
number of patents, patent filings and frequent and protracted litigation
regarding patent and other intellectual property rights. Many
companies have numerous patents that protect their intellectual property
rights. Third parties might assert infringement claims against us
with respect to our drug candidates and future products. If
litigation were required to determine the validity of a third party’s claims, we
could be required to spend significant time and financial resources, which could
distract our management and prevent us from furthering our core business
activities, regardless of the outcome. If we did not prevail in the
litigation, we could be required to pay damages, license a third party’s
technology, which may not be possible on terms acceptable to us, or at all, or
discontinue our own activities and develop non-infringing technology, any of
which could prevent or significantly delay pursuit of our development
activities.
Protection
of trade secret and confidential information is difficult, and loss of
confidentiality could eliminate our competitive advantage.
In
addition to patent protection, we rely on trade secrets, proprietary know-how
and confidential information to protect our technology. We use
confidentiality agreements with our employees, consultants and collaborators to
maintain the proprietary nature of this technology. However,
confidentiality agreements can be breached by the other party, which would make
our trade secrets and proprietary know-how legally available for use by
others. There is generally no adequate remedy for breach of
confidentiality obligations. In addition, the competitive advantage
afforded by trade secrets is limited because a third party can independently
discover or develop something identical to our own trade secrets or know-how,
without incurring any liability to us.
If our
current or former employees, consultants or collaborators were to use
information improperly obtained from others (even if unintentional), we may be
subject to claims as to ownership and rights in any resulting know-how or
inventions.
If
we cannot retain or hire qualified personnel or maintain our collaborations, our
programs could be delayed and may be discontinued.
As of
December 24, 2009, we had one full-time employee, our President and Chief
Executive Officer. We utilize consultants to assist with our
operations and are highly dependent on the services of our executive
officers. We also are dependent on our collaborators for our research
and development activities. The loss of key executive officers or
collaborators could delay progress in our research and development activities or
result in their termination entirely.
We
believe that our future success will depend in large part upon our ability to
attract and retain highly skilled scientific and managerial
personnel. We face intense competition for these kinds of personnel
from other companies, research and academic institutions, government entities
and other organizations. If we fail to identify, attract and retain
personnel, we may be unable to continue the development of our drug candidates,
which would have a material adverse effect on our business, prospects, financial
condition and results of operations.
37
We
face the risk of product liability claims which could exceed our insurance
coverage and deplete our cash resources.
The
pharmaceutical and biotechnology industries expose us to the risk of product
liability claims alleging that use of our drug candidates caused an injury or
harm. These claims can arise at any point in the development,
testing, manufacture, marketing or sale of pharmaceutical products and may be
made directly by patients involved in clinical trials of our products, by
consumers or healthcare providers or by organizations selling our
products. Product liability claims can be expensive to defend, even
if the product did not actually cause the alleged injury or harm.
Insurance
covering product liability claims becomes increasingly expensive as a product
candidate moves through the development pipeline to
commercialization. We have limited product liability insurance
coverage for our clinical trials and this coverage may not be sufficient to
cover us against some or all potential losses due to liability, if any, or to
the expenses associated with defending against liability claims. A
product liability claim successfully asserted against us could exceed our
insurance coverage, require us to use our own cash resources and have a material
adverse effect on our business, financial condition and results of
operations.
In
addition, some of our licensing and other agreements with third parties require
or might require us to maintain product liability insurance. If we
cannot maintain acceptable amounts of coverage on commercially reasonable terms
in accordance with the terms set forth in these agreements, the corresponding
agreements would be subject to termination.
The
costs of compliance with environmental, safety and similar laws could increase
our cost of doing business or subject us to liability in the event of
noncompliance.
Our
business is subject to regulation under state and federal laws regarding
occupational safety, laboratory practices, environmental protection and the use,
generation, manufacture, storage and disposal of hazardous
substances. We may be required to incur significant costs in the
future to comply with existing or future environmental and health and safety
regulations. Our research activities involve the use of hazardous
materials, chemicals and radioactive compounds. Although we believe
that our procedures for handling such materials comply with applicable state and
federal regulations, we cannot eliminate the risk of contamination or injury
from these materials. In the event of contamination, we could be
liable for any resulting damages, which could have a material adverse effect on
our business, financial condition and results of operations.
We
are subject to intense competition that could materially impact our operating
results.
We may be
unable to compete successfully against our current or future
competitors. The pharmaceutical, biopharmaceutical and biotechnology
industry is characterized by intense competition and rapid and significant
technological advancements. Many companies, research institutions and
universities are working in a number of areas similar to our primary fields of
interest to develop new products. There also is intense competition
among companies seeking to acquire products that already are being
marketed. Many of the companies with which we compete have or are
likely to have substantially greater research and product development
capabilities and financial, technical, scientific, manufacturing, marketing,
distribution and other resources than at least some of our present or future
strategic partners or licensees.
As a
result, these competitors may:
· Succeed
in developing competitive products sooner than us or our strategic partners or
licensees;
· Obtain
FDA and other regulatory approvals for their products before approval of any of
our products;
· Obtain
patents that block or otherwise inhibit the development and commercialization of
our drug candidates;
· Develop
products that are safer or more effective than our products;
· Devote
greater resources to marketing or selling their products;
· Introduce
or adapt more quickly to new technologies or scientific advances;
· Introduce
products that render our products obsolete;
· Withstand
price competition more successfully than us or our strategic partners or
licensees;
· Negotiate
third-party strategic alliances or licensing arrangements more effectively;
or
· Take
advantage of other opportunities more readily.
Currently,
there are three drugs approved as radiation protection
agents. Amifostine (Ethyol®) is marketed by MedImmune, Inc. for use
in reduction of chemotherapy-induced kidney toxicity and radiation-induced
xerostomia (damage to the salivary gland). KepivanceTM (palifermin)
is marketed by Amgen, Inc. for use in the treatment of severe oral mucositis
(mouth sores) in patients with hematologic (blood) cancers. Salagen
Tablets (pilocarpine hydrochloride) is marketed by MGI Pharma in the United
States as a treatment for the symptoms of xerostomia induced by radiation
therapy in head and neck cancer patients. However, there are also
many companies working to develop pharmaceuticals that act as a radiation
protection agent.
38
Acceptance
of our products in the marketplace is uncertain, and failure to achieve market
acceptance will harm our business.
Even if
approved for marketing, our products may not achieve market
acceptance. The degree of market acceptance will depend upon a number
of factors, including:
|
|
·
|
the
receipt of regulatory approvals for the indications that we are
studying;
|
|
|
·
|
the
establishment and demonstration in the medical community of the safety,
clinical efficacy and cost-effectiveness of our products and their
potential advantages over existing therapeutic
products;
|
|
|
·
|
marketing
and distribution support;
|
|
|
·
|
the
introduction, market penetration and pricing strategies of competing and
future products; and
|
|
|
·
|
coverage
and reimbursement policies of governmental and other third-party payors
such as insurance companies, health maintenance organizations and other
plan administrators.
|
Physicians,
patients, payors or the medical community in general may be unwilling to accept,
purchase, utilize or recommend any of our products.
We
may be required to pay milestone and other payments relating to the
commercialization of our products.
Our
agreements by which we acquired rights to our drug candidates provide for
milestone payments by us upon the occurrence of certain regulatory filings and
approvals related to the acquired products. In the event that we
successfully develop our drug candidates, these milestone payments could be
significant. In addition, our agreements require us to pay a royalty
interest on worldwide sales. Also, any future license, collaborative
or other agreements we may enter into in connection with our development and
commercialization activities may require us to pay significant milestone,
license and other payments in the future.
We are continually evaluating our
business strategy, and may modify this strategy in light of developments in our
business and other factors.
We
continue to evaluate our business strategy and, as a result, may modify this
strategy in the future. In this regard, we may, from time to time, focus our
development efforts on different drug candidates or may delay or halt the
development of our drug candidates. In addition, as a result of
changes in our strategy, we may also change or refocus our existing drug
discovery, development, commercialization and manufacturing
activities.
Our short-term investments,
marketable securities and restricted investments are subject to certain risks
which could materially adversely affect our overall financial
position.
We
invest our cash in accordance with an established internal policy and
customarily in instruments which historically have been highly liquid and
carried relatively low risk. However, the capital and credit markets have been
experiencing extreme volatility and disruption. Over the past two years, the
volatility and disruption have reached unprecedented levels. We maintain a
portfolio of investments in short-term investments, marketable debt securities
and restricted investments, which are recorded at fair value. Certain of these
transactions expose us to credit risk in the event of default of the issuer. To
minimize our exposure to credit risk, we invest in securities with strong credit
ratings. Should any of our short-term investments, marketable securities or
restricted investments lose value or have their liquidity impaired, it could
materially and adversely affect our overall financial position by imperiling our
ability to fund our operations and forcing us to seek additional financing
sooner than we would otherwise. Such financing may not be available on
commercially attractive terms or at all.
Our insurance policies are expensive
and protect us only from some business risks, which could leave us exposed to
significant, uninsured liabilities.
We
do not carry insurance for all categories of risk that our business may
encounter. We currently maintain general liability, property, auto, workers’
compensation, products liability, fiduciary and directors’ and officers’
insurance policies. We do not know, however, if we will be able to maintain
existing insurance with adequate levels of coverage. For example, the premiums
for our directors’ and officers’ insurance policy have increased in the past and
may increase in the future, and this type of insurance may not be available on
acceptable terms or at all in the future. Any significant uninsured liability
may require us to pay substantial amounts, which would adversely affect our cash
position and results of operations.
We
may have a limitation on the use of Net Operating Loss Carryforwards and Tax
Credits.
Our
ability to utilize our net operating loss carryforwards, or NOLs, and tax
credits may be limited if we undergo or have undergone an ownership change, as
defined in section 382 of the Internal Revenue Code, as a result of changes in
the ownership of outstanding stock. An ownership change generally occurs if the
percentage of stock owned by one or more stockholders who own, directly or
indirectly, 5% or more of the value of our outstanding stock (or are otherwise
treated as 5% stockholders under section 382 and the regulations promulgated
thereunder) has increased by more than 50 percentage points over the lowest
percentage of our outstanding stock owned by these stockholders at any time
during the testing period, which is generally the
39
three-year
period preceding the potential ownership change. In the event of an ownership
change, section 382 imposes an annual limitation on the amount of post-ownership
change taxable income a corporation may offset with pre-ownership change
NOLs.
We
are exposed to risks if we are unable to comply with changes to laws affecting
public companies, including the Sarbanes-Oxley Act of 2002, and also to
increased costs associated with complying with such laws.
Laws and
regulations affecting public companies, including the provisions of the
Sarbanes-Oxley Act of 2002 in the U.S., will cause us to incur increased costs
as we evaluate the implications of new rules and respond to new
requirements. Delays or a failure to comply with the new laws, rules
and regulations could result in enforcement actions, the assessment of other
penalties and civil suits. These laws and regulations make it more
expensive for us under indemnities provided by the Company to our officers and
directors and may make it more difficult for us to obtain certain types of
insurance, including liability insurance for directors and officers; as such, we
may be forced to accept reduced policy limits and coverage or incur
substantially higher costs to obtain the same or similar
coverage. The impact of these events could also make it more
difficult for us to attract and retain qualified persons to serve on our Board
of Directors, or as executive officers. We may be required to hire
additional personnel and utilize additional outside legal, accounting and
advisory services — all of which could cause our general and administrative
costs to increase beyond what we currently have planned.
We
have reported a material weakness in the effectiveness of our internal control
over financial reporting, and if we cannot maintain effective internal controls
or provide reliable financial and other information, investors may lose
confidence in our SEC reports.
In this
Annual Report, we are reporting a material weakness in the effectiveness of our
internal control over financial reporting related to adequate segregation of
duties, which are described in more detail below under the heading “Controls and
Procedures.” Based on the material weakness, we are also reporting in this
Annual Report that our disclosure controls and procedures were not effective as
of September 30, 2009.
Effective
internal control over financial reporting and disclosure controls and procedures
are necessary for us to provide reliable financial and other reports and
effectively prevent fraud. If we cannot maintain effective internal
control or disclosure controls and procedures, or provide reliable financial or
SEC reports or prevent fraud, investors may lose confidence in our SEC reports,
our operating results and the trading price of our common stock could suffer and
we may become subject to litigation.
Our
corporate compliance program cannot guarantee that we are in compliance with all
potentially applicable regulations.
The
development, manufacturing, pricing, sales, coverage and reimbursement of our
products, together with our general operations, are subject to extensive
regulation by federal, state and other authorities within the United States and
numerous entities outside of the United States. While we have
developed and instituted a corporate compliance program based on what we believe
are the current best practices, we cannot provide any assurance that
governmental authorities will find that our business practices comply with
current or future administrative or judicial interpretations of potentially
applicable laws and regulations. If we fail to comply with any of
these laws and regulations, we could be subject to a range of regulatory
actions, including suspension or termination of clinical trials, the failure to
approve a product candidate, restrictions on our products or manufacturing
processes, withdrawal of products from the market, significant fines, or other
sanctions or litigation.
We
may be unable to repay our substantial indebtedness and other
obligations.
Under the
terms of the indentures governing our debt instruments, we are obligated to
repay in cash the aggregate principal balance of any such notes upon maturity
and in some cases upon demand. As of the filing date of this report,
we do not have available cash, cash equivalents and investments sufficient to
repay the note, if presented. In addition, there are no restrictions
on our use of this cash and the cash available to repay indebtedness may decline
over time. If we do not have sufficient funds available to repay the
principal balance of note, we will be required to raise additional
funds. Because the financing markets may be unwilling to provide
funding to us or may only be willing to provide funding on terms that we would
consider unacceptable, we may not have cash available or be able to obtain
funding to permit us to meet our repayment obligations, thus adversely affecting
the market price for our securities.
We
may become involved in the future in legal proceedings that, if adversely
adjudicated or settled, could materially impact our financial
condition.
As a
pharmaceutical company, we may become a party to litigation in the ordinary
course of our business, including, among others, matters alleging product
liability, patent or other intellectual property rights infringement, patent
invalidity or breach of commercial contract. In general, litigation
claims can be expensive and time consuming to bring or defend against and could
result in settlements or damages that could significantly impact results of
operations and financial condition.
40
We
are prohibited from taking certain actions and entering into certain
transactions without the consent of certain holders of our Common Stock and
warrants
Until the
earliest to occur of the date Goodnow Capital L.L.C. owns less than 20% of our
outstanding common stock or we complete, to the absolute satisfaction of
Goodnow, initial clinical safety studies of AEOL 10150, and analysis of the data
developed based upon such studies with results satisfactory to Goodnow, in its
absolute discretion, to initiate efficacy studies of AEOL 10150 or we initiate
the dosing of the first human patient in an efficacy-based study of AEOL 10150,
we are prohibited from taking certain actions or entering into certain
transactions without the prior consent of Goodnow. We are prohibited
from paying dividends to common stockholders, amending our certificate of
incorporation, issuing any equity security or any security convertible into or
exercisable for any equity security, changing our business or operations,
merging with or selling or leasing all or a substantial portion of our assets to
any entity, incurring debt from any third party, placing a lien on any of our
properties, increasing the compensation we pay our employees, cancelling any
debt except for full value, or making any expenditure or series of related
expenditures in excess of $25,000, except as specified in a budget approved in
writing in advance by Goodnow and our board of directors or directly relating to
the development of AEOL 10150 for the treatment of ALS.
For as
long as certain warrants that were issued on October 6, 2009 are outstanding, we
are prohibited from taking certain actions or entering into certain transactions
without the prior consent of the warrant holders of the warrants issued on
October 6, 2009 (the “October 2009 Warrant Holders”). Under these
warrants, we are also prohibited from selling the Company to an entity other
than one that is publicly traded.
Even
though our board of directors may determine that any of these actions are in the
best interest of the Company or our shareholders, we may be unable to complete
them if we do not get the approval of Goodnow or the October 2009 Warrant
Holders, either of which may withheld consent of any transaction in its sole
discretion. The interests of Goodnow or the October 2009 Warrant
Holders may differ from those of our stockholders generally. If we
are unable to obtain consent Goodnow or the October 2009 Warrant Holders, we may
be unable to complete actions or transactions that our board of directors
determines are in the best interest of the Company and its
shareholders.
Risks
Related to Owning Our Stock
Our
principal stockholders own a significant percentage of our outstanding common
stock and are, and will continue to be, able to exercise significant influence
over our affairs.
As of
December 24, 2009, Xmark Opportunity Partners, LLC (“Xmark”) possessed voting
power over 30,482,970 shares, or 63.2%, of our common stock outstanding as of
such date, through its management of Goodnow Capital, L.L.C. (“Goodnow”), Xmark
Opportunity Fund, L.P., Xmark Opportunity Fund, Ltd. and Xmark JV Investment
Partners, LLC (collectively, the “Xmark Funds”), and through a voting trust
agreement by and among Biomedical Value Fund, L.P., Biomedical Value Fund, Ltd.,
Xmark Opportunity Partners, LLC and the Company (the “Xmark Voting
Trust”) with respect to 1,000,000 shares. As a result, Xmark is able
to determine a significant part of the composition of our board of directors,
holds significant voting power with respect to matters requiring stockholder
approval and is able to exercise significant influence over our
operations. The interests of Xmark may be different than the
interests of other stockholders on these and other matters. This
concentration of ownership also could have the effect of delaying or preventing
a change in our control or otherwise discouraging a potential acquirer from
attempting to obtain control of us, which could reduce the price of our common
stock.
David
Cavalier, our Chairman of the Board of Directors, and Xmark with which he is
affiliated, possess voting power of 63.2% of our
outstanding common stock as of December 24, 2009. Accordingly,
Mr. Cavalier currently has, and will continue to have, a significant
influence over the outcome of all corporate actions requiring stockholder
approval.
Also as
of December 24, 2009, Efficacy Capital Ltd. (“Efficacy Capital”) owned 9,800,000
shares, or 20.3%, of our outstanding common stock as of such date, through its
management of Efficacy Biotech Master Fund Ltd. As a result, Efficacy
Capital is able to determine a part of the composition of our board of
directors, holds significant voting power with respect to matters requiring
stockholder approval and is able to exercise significant influence over our
operations. The interests of Efficacy Capital may be different than
the interests of other stockholders on these and other matters. This
concentration of ownership could also have the effect of delaying or preventing
a change in our control or otherwise discouraging a potential acquirer from
attempting to obtain control of us, which could reduce the price of our common
stock.
Our
executive officers and directors and holders of greater than five percent of our
outstanding common stock, together with entities that may be deemed affiliates
of, or related to, such persons or entities, owned greater than 83 percent of
our common stock as of December 24, 2009. As a result, these stockholders,
acting together, may be able to control our management and affairs and matters
requiring stockholder approval, including the election of directors and approval
of significant corporate transactions, such as mergers, consolidations or the
sale of substantially all of our assets. The interests of our current major
stockholders may not always coincide with the interests of other stockholders
and they may take actions to advance their respective interests to the detriment
of other stockholders.
41
We
may need to sell additional shares of our common stock, preferred stock or other
securities to meet our capital requirements and these future sales could cause
dilution and adversely affect our stock price.
Sales of
substantial amounts of capital stock, or the perception that such sales could
occur, could adversely affect the prevailing market price of the common stock
and our ability to raise capital. We may issue additional common
stock in future financing transactions or as incentive compensation for our
executive management and other key personnel, consultants and
advisors. Issuing any equity securities would be dilutive to the
equity interests represented by our then-outstanding shares of common
stock. The market price for our common stock could decrease as the
market takes into account the dilutive effect of any of these
issuances.
In
the event of the conversion of our preferred stock and exercises of currently
outstanding options and warrants, the ownership interests of our current
stockholders could be substantially diluted, which would reduce the market price
of our common stock and could make it more difficult for us to raise funds in
the future.
As of
December 24, 2009, we had 48,224,320 shares of common stock
outstanding. We may grant to our employees, directors and consultants
options to purchase shares of our common stock under our 2004 Stock Option
Plan. In addition, as of December 24, 2009, options to purchase
6,290,015 shares were outstanding at exercise prices ranging from $0.29 to
$51.25 per share, with a weighted average exercise price of $1.56 per share, and
637,159 shares were reserved for issuance under the 2004 Stock Option
Plan. In addition, as of December 24, 2009, warrants to purchase
53,390,953 shares of common stock were outstanding at exercise prices ranging
from $0.28 to $2.50 per share, with a weighted exercise price of $0.35 per
share. We have also reserved 475,087 shares of common stock for the
conversion of our outstanding Series B Preferred stock.
In
connection with prior collaborations and financing transactions, we also have
issued Series B preferred stock and a promissory note convertible into Series B
preferred stock to affiliates of Elan Corporation, plc
(“Elan”). These securities generally are exercisable and convertible
at the option of the Elan affiliates. The exercise or conversion of
all or a portion of these securities would dilute the ownership interests of our
stockholders.
Our
common stock is not listed on a national exchange, is illiquid and is
characterized by low and/or erratic trading volume, and the per share price of
our common stock has fluctuated from $0.15 to $0.55 during the last two
years.
Our
common stock is quoted on the OTC Bulletin Board under the symbol
“AOLS.” An active public market for our common stock is unlikely to
develop as long as we are not listed on a national securities
exchange. Even if listed, the market for our stock may be impaired
because of the limited number of investors, the significant ownership stake of
Efficacy Capital and Xmark (through its management of Goodnow and the Xmark
Funds), and our small market capitalization, which is less than that authorized
for investment by many institutional investors.
Historically,
the public market for our common stock has been characterized by low and/or
erratic trading volume, often resulting in price volatility. The
market price of our common stock is subject to wide fluctuations due to factors
that we cannot control, including the results of preclinical and clinical
testing of our products under development, decisions by collaborators regarding
product development, regulatory developments, market conditions in the
pharmaceutical and biotechnology industries, future announcements concerning our
competitors, adverse developments concerning proprietary rights, public concern
as to the safety or commercial value of any products and general economic
conditions.
Furthermore,
the stock market has experienced significant price and volume fluctuation
unrelated to the operating performance of particular companies. These
market fluctuations can adversely affect the market price and volatility of our
common stock.
If
registration rights that we have previously granted are exercised, or if we
grant additional registration rights in the future, the price of our common
stock may be adversely affected.
Upon
receiving notice from Xmark, we are obligated to register with the SEC shares of
common stock and the common stock underlying the warrants to purchase common
stock held by the Xmark. If these securities are registered with the
SEC, they may be sold in the open market. In addition, upon receiving
notice from Elan, we are obligated to register with the SEC shares of common
stock underlying the Series B preferred stock, warrants to purchase Series B
preferred stock and a promissory note held by the Elan affiliates. If
these securities are registered with the SEC, they may be sold in the open
market. We expect that we also will be required to register any
securities sold in future private financings. The sale of a
significant amount of shares in the open market, or the perception that these
sales may occur, could cause the trading price of our common stock to decline or
become highly volatile.
42
Anti-takeover
provisions in our charter documents and under Delaware law could make an
acquisition of us, which may be beneficial to our stockholders, more difficult
and may prevent attempts by our stockholders to replace or remove our current
management.
Provisions
in our amended and restated certificate of incorporation and bylaws may delay or
prevent an acquisition of us or a change in our management. These
provisions include a prohibition on actions by written consent of our
stockholders and the ability of our board of directors to issue preferred stock
without stockholder approval. In addition, because we are
incorporated in Delaware, we are governed by the provisions of Section 203 of
the Delaware General Corporation Law, which prohibits stockholders owning in
excess of 15% of our outstanding voting stock from merging or combining with
us. These provisions may frustrate or prevent any attempts by our
stockholders to replace or remove our current management by making it more
difficult for stockholders to replace members of our board of directors, which
is responsible for appointing the members of our management.
We
do not expect to pay cash dividends on our common stock for the foreseeable
future.
We have
never paid cash dividends on our common stock and do not anticipate that any
cash dividends will be paid on the common stock for the foreseeable
future. The payment of any cash dividend by us will be at the
discretion of our board of directors and will depend on, among other things, our
earnings, capital, regulatory requirements and financial
condition. Furthermore, the terms of some of our financing
arrangements directly limit our ability to pay cash dividends on our common
stock.
We
may experience significant and unpredictable changes to the liability for
warrants and record significant gains or losses to our statement of operations
in each period the warrants are outstanding
In June
2008, the Financial Accounting Standards Board ("FASB") ratified Emerging Issues
Task Force (“EITF”) Issue No. 07-5, which contains guidance for determining
whether an Instrument (or embedded feature) is indexed to an entity’s own
stock. Equity-linked instruments (or embedded features) that
otherwise meet the definition of a derivative are not accounted for as
derivatives if certain criteria are met, one of which is that the instrument (or
embedded feature) must be indexed to the entity’s own stock. The
Company adopted this guidance on October 1, 2009 and began applying its
provisions to outstanding instruments as f that date. The fair value
of the warrants impacted by the revised guidance at their dates of issuance
totaled $8,282,000 and was initially recorded as a component of additional
paid-in capital. Upon adoption of the new guidance in the first quarter of 2010,
the Company recorded a decrease to the opening balance of additional-paid-in
capital of $8,142,000 and recorded a decrease to accumulated deficit totaling
$4,353,000, representing the decrease in the fair value of the warrants from
their date of issuance to October 1, 2009. The fair value of the
warrants at October 1, 2009 of $3,789,000 will be included as a current
liability in the balance sheet as of that date. Any future increase
or decrease in fair value of the warrants will be included as a component of
other income in the accompanying statement of operations for the respective
period.
In
connection with a financing completed on October 6, 2009 and previously
disclosed on Form 8-K dated October 6, 2009, the Company issued warrants to
purchase up to an aggregate of 43,614,285 shares of its common stock and
cancelled warrants to purchase up to an aggregate of 17,542,857 shares of its
common stock. Based on EITF 07-5, this resulted in a charge to our
statement of operations of $6,213,000 and increased the liability for warrants
to $11,022,000.
As of
October 31, 2009, the liability for warrants increased to $17,272,000 resulting
in an additional charge to the statement of operations of
$6,250,000. As of November 30, 2009, the liability for warrants
decreased to $15,389,000 resulting in a gain in the statement of operations of
$1,882,000.
The Company’s outstanding
warrants will continue to be revalued at each balance sheet date, which could
result in significant and unpredictable changes to our reported liabilities and
significant additional gains or losses charged to the statement of operations
for each period regardless of any changes to the Company’s working capital,
liquidity, or business operations.
Item
1B. Unresolved Staff Comments.
None.
Item
2. Properties.
None.
Item
3. Legal Proceedings.
None.
43
Item
4. Submission of Matters to a Vote of Security Holders.
No
matters were submitted to us by a vote of the security holders during the
quarter ended September 30, 2009.
PART
II
Item
5. Market for Registrant’s Common Equity, Related Stockholder Matters
and Issuer Purchases of Equity Securities.
(a)
Price Range of Common Stock
Our
common stock is traded on the OTC Bulletin Board under the symbol
“AOLS.” The following sets forth the quarterly high and low trading
prices as reported by the OTC Bulletin Board for the periods
indicated. These prices are based on quotations between dealers,
which do not reflect retail mark-up, markdown or commissions, and do not
necessarily represent actual transactions.
|
High
|
Low
|
|||||
|
Fiscal
Year Ended September 30, 2008
|
||||||
|
October
1, 2007 through December 31, 2007
|
$
|
0.55
|
$
|
0.33
|
||
|
January
1, 2008 through March 31, 2008
|
$
|
0.40
|
$
|
0.31
|
||
|
April
1, 2008 through June 30, 2008
|
$
|
0.48
|
$
|
0.32
|
||
|
July
1, 2008 through September 30, 2008
|
$
|
0.45
|
$
|
0.25
|
||
|
Fiscal
Year Ended September 30, 2009
|
||||||
|
October
1, 2008 through December 31, 2008
|
$
|
0.46
|
$
|
0.20
|
||
|
January
1, 2009 through March 31, 2009
|
$
|
0.46
|
$
|
0.15
|
||
|
April
1, 2009 through June 30, 2009
|
$
|
0.40
|
$
|
0.20
|
||
|
July
1, 2009 through September 30, 2009
|
$
|
0.39
|
$
|
0.22
|
||
(b)
Approximate Number of Equity Security Holders
As of
December 24, 2009, the number of record holders of our common stock was 127 and
we estimate that the number of beneficial owners was approximately
2,300.
(c)
Dividends
We have
never paid a cash dividend on our common stock and we do not anticipate paying
cash dividends on our common stock in the foreseeable future. If we
pay a cash dividend on our common stock, we also must pay the same dividend on
an as converted basis on our Series B preferred stock. Moreover, any
additional preferred stock to be issued and any future credit facilities might
contain restrictions on our ability to declare and pay dividends on our common
stock. We plan to retain all earnings, if any, for the foreseeable
future for use in the operation of our business and to fund future
growth.
In
addition, we cannot pay a dividend on our common stock without the prior
approval of Goodnow Capital pursuant to the terms of the Debenture and Warrant
Purchase Agreement dated September 16, 2003 between us and
Goodnow. This restriction will expire on the earliest
of:
|
|
·
|
the
date that Goodnow owns less than 20% of our outstanding common stock on an
as converted basis;
|
|
|
·
|
the
completion, to the absolute satisfaction of Goodnow, of initial clinical
safety studies of AEOL 10150 and analysis of the data developed based upon
such studies with the results satisfactory to Goodnow, in its absolute
discretion, to initiate efficacy studies of AEOL 10150 in humans;
or
|
|
|
·
|
the
initiation of dosing of the first human patient in an efficacy-based study
of AEOL 10150.
|
In
addition, under the terms of the warrants to purchase up to 43,614,285 shares of
our common stock issued to Xmark on October 6, 2009, if we were to pay a
dividend on our common stock the exercise price of these warrants would be reset
from $0.28 per share to $0.01 per share and the warrantholders would also
receive any such dividend paid.
44
(d)
Equity Compensation Plan and Additional Equity Information as of September 30,
2009
|
Plan
category
|
(a)Number
of securities to be issued upon exercise of outstanding options, warrants
and rights
|
(b)Weighted-average
exercise price of outstanding options, warrants and rights
|
(c)Number
of securities remaining available for future issuance under equity
compensation plans (excluding securities reflected in column
(a))
|
|||
|
Equity
compensation plans approved by our stockholders:
|
|
|
|
|||
|
2004
Stock Option Plan
|
4,249,091
|
$0.53
|
750,909
|
|||
|
1994
Stock Option Plan
|
1,927,124
|
$3.92
|
0
|
|||
|
Equity
compensation plans and securities not approved by our
stockholders:
|
||||||
|
Warrant
to Purchase Common Stock Issued to Dan Delmonico
|
50,000
|
$0.49
|
Not
applicable
|
|||
|
Warrant
to Purchase Common Stock Issued to Brookstreet Securities
Corporation
|
250,000
|
$1.50
|
Not
applicable
|
|||
|
Total
– Common Stock
|
6,476,215
|
750,909
|
||||
|
Convertible
Promissory Note convertible into shares of Series B Preferred Stock Issued
to Elan Pharma International Limited (as of September 30,
2009)(1)(2)
|
65,985
|
$9.00
|
10,614
|
|||
|
Total
– Series B Preferred Stock
|
65,985
|
10,614
|
——————
(1) As
of September 30, 2009, each share of Series B preferred stock was convertible
into one share of common stock.
(2) The
conversion value of the note will increase by its 11% interest rate until its
maturity on February 8, 2011.
Description
of Equity Compensation Plans and Equity Securities Not Approved by Our
Stockholders
The
warrants to purchase shares of our common stock issued to Brookstreet Securities
Corporation (“Brookstreet”) have not been approved by our
stockholders. In May 2006, we entered into an agreement with
Brookstreet to provide us with financial advisory services for a one-year
period. For these services, we issued five warrants each to purchase
up to 50,000 shares of our common stock with an exercise price of $0.50, $1.00,
$1.50, $2.00 and $2.50 and vesting dates of on May 24, 2006, August 22, 2006,
November 20, 2006, February 18, 2007 and May 19, 2007,
respectively. The warrants are exercisable for five
years.
The
warrants to purchase shares of our common stock issued to Dan Delmonico in
September 2009 have not been approved by our stockholders. We issued
three warrants each to purchase up to 20,000, 15,000 and 15,000 shares of our
common stock with an exercise price of $0.39, $.50 and $0.60,
respectively. The warrants are exercisable for five
years.
(e)
Recent Sales of Unregistered Securities
None
(f)
Purchase of Equity Securities by the Issuer and Affiliated
Purchases
None.
45
Item
6. Selected Financial Data.
You
should read the following selected financial data in conjunction with our
consolidated financial statements and the notes to those statements and
“Management’s Discussion and Analysis of Financial Condition and Results of
Operations” included elsewhere in this Form 10-K. We derived the
consolidated statements of operations data for the five fiscal years ended
September 30, 2009 and the related consolidated balance sheet data at those
dates from our audited consolidated financial statements. Except for
the consolidated statements of operations for the fiscal years ended September
30, 2006 and 2005 and the consolidated balance sheet data at September 30, 2007,
2006 and 2005, each of these consolidated financial statements are included
elsewhere in this Form 10-K.
Statement
of Operations Data:
|
Year
Ended September 30,
|
|||||||||||||||||||
|
2009
|
2008
|
2007
|
2006
|
2005
|
|||||||||||||||
|
(in
thousands, except per share data)
|
|||||||||||||||||||
|
Revenue:
|
|
|
|
|
|
|
|
|
|
||||||||||
|
Grant
income and contract revenue
|
$
|
—
|
$
|
—
|
$
|
—
|
$
|
92
|
$
|
252
|
|||||||||
|
Costs
and expenses:
|
|||||||||||||||||||
|
Research
and development
|
711
|
977
|
1,381
|
3,480
|
4,515
|
||||||||||||||
|
General
and administrative
|
1,292
|
1,540
|
1,919
|
2,216
|
2,674
|
||||||||||||||
|
Total
costs and expenses
|
2,003
|
2,517
|
3,300
|
5,696
|
7,189
|
||||||||||||||
|
Loss
from operations
|
(2,003
|
)
|
(2,517
|
)
|
(3,300
|
)
|
(5,604
|
)
|
(6,937
|
)
|
|||||||||
|
Other
income (expenses), net
|
144
|
(405)
|
225
|
(118
|
)
|
63
|
|||||||||||||
|
Interest
income (expense), net
|
(437
|
)
|
(51
|
)
|
51
|
(6
|
)
|
(31
|
)
|
||||||||||
|
Net
loss
|
(2,296
|
)
|
(2,973
|
)
|
(3,024
|
)
|
(5,728
|
)
|
(6,905
|
)
|
|||||||||
|
Preferred
stock dividend and accretion
|
—
|
—
|
—
|
(81
|
)
|
—
|
)
|
||||||||||||
|
Net
loss attributable to common stockholders
|
$
|
(2,296
|
)
|
$
|
(2,973
|
)
|
$
|
(3,024
|
)
|
$
|
(5,809
|
)
|
$
|
(6,905
|
)
|
||||
|
Basic
net loss per share attributable to common stockholders
|
$
|
(0.07
|
)
|
$
|
(0.09
|
)
|
$
|
(0.10
|
)
|
$
|
(0.31
|
)
|
$
|
(0.49
|
)
|
||||
|
Diluted
net loss per share attributable to common stockholders
|
$
|
(0.07
|
)
|
$
|
(0.11
|
)
|
$
|
(0.10
|
)
|
$
|
(0.31
|
)
|
$
|
(0.49
|
)
|
||||
|
Weighted
average common shares outstanding:
|
|||||||||||||||||||
|
Basic
|
34,789
|
31,953
|
30,239
|
18,926
|
13,976
|
||||||||||||||
|
Diluted
|
34,789
|
32,217
|
30,239
|
18,926
|
13,976
|
||||||||||||||
Balance
Sheet Data:
|
September
30,
|
|||||||||||||||||||
|
2009
|
2008
|
2007
|
2006
|
2005
|
|||||||||||||||
|
(in
thousands)
|
|||||||||||||||||||
|
Cash
and cash equivalents and marketable securities
|
$
|
646
|
$
|
399
|
$
|
1,727
|
$
|
3,324
|
$
|
626
|
|||||||||
|
Working
capital (deficiency)
|
$
|
5
|
$
|
(1,336
|
)
|
$
|
1,538
|
$
|
1,581
|
$
|
(73
|
)
|
|||||||
|
Total
assets
|
$
|
811
|
$
|
1,120
|
$
|
1,931
|
$
|
3,554
|
$
|
937
|
|||||||||
|
Long-term
portion of capital lease obligations and notes payable
|
$
|
1,194
|
$
|
266
|
$
|
483
|
$
|
—
|
$
|
867
|
|||||||||
|
Total
liabilities
|
$
|
1,968
|
$
|
2,157
|
$
|
751
|
$
|
1,847
|
$
|
1,869
|
|||||||||
|
Total
stockholders’ equity (deficit)
|
$
|
(1,157
|
)
|
$
|
(1,037
|
)
|
$
|
1,180
|
$
|
1,707
|
$
|
(932
|
)
|
||||||
Item
7. Management’s Discussion and Analysis of Financial Condition and
Results of Operation.
Introduction
You
should read the following discussion in conjunction with our consolidated
financial statements and the notes appearing elsewhere in this Form
10-K. The following discussion contains forward-looking statements
that involve risks and uncertainties. Our actual results could differ
materially from those anticipated in the forward-looking statements as a result
of various factors, including those discussed in “Item 1A. Risk
Factors” and elsewhere in this Form 10-K.
46
Overview
We are
developing a new class of catalytic antioxidant compounds as a medical
countermeasure against biological, chemical and radiological
weapons as well as for diseases and disorders of the central nervous
system, respiratory system, autoimmune system and oncology. Our
initial target indications are as a protective agent against the effects of
acute radiation syndrome, sulfur mustard gas exposure and chlorine gas
exposure. We have reported positive safety results from two Phase I
clinical trials of AEOL 10150, our lead drug candidate, with no serious adverse
events noted.
We do not
have any recurring revenue and therefore we must rely on public or private
equity offerings, debt financings, collaboration arrangements or grants to
finance our operations.
We had
net losses of $2,296,000 and $2,973,000 for the fiscal years ended September 30,
2009 and 2008, respectively. We had an accumulated deficit of
$161,195,000 at September 30, 2009. We have not yet generated any
revenue from product sales and do not expect to receive any product revenue in
the foreseeable future, if at all.
Corporate
Matters
On May
22, 2007, we completed a private placement of 2,666,667 shares of our Common
Stock and warrants to purchase up to an aggregate of 2,000,001 shares of common
stock with an exercise price of $0.75 per share and a five year term for
aggregate net proceeds of approximately $1,761,000 (the “2007
Financing”).
On August
1, 2008, we completed a private placement pursuant to which the Company agreed
to sell to the investors (the “Investors”) units comprised of senior unsecured
convertible notes of the Company (the "Notes"), in an aggregate principal amount
of up to $5,000,000, which bear interest at a rate of 7% per year and matured on
the 30-month anniversary of their date of issuance, and warrants to purchase up
to an aggregate of 10,000,000 additional shares of Common Stock, each with an
initial exercise price of $0.50 per share, subject to adjustment pursuant to the
warrants. Each unit (collectively, the "Units") is comprised of
$1,000 in Note principal and Warrants to purchase up to 2,000 shares of the
Company's common stock, par value $0.01 per share (the "Common Stock"), and has
a purchase price of $1,000. On August 1, 2008, the Company sold and
issued to the Investors 500 Units comprised of Notes in the aggregate principal
amount of $500,000 and Warrants to purchase up to 1,000,000 shares of Common
Stock for an aggregate purchase price of $500,000 (the
"Financing"). On each of September 4, 2008, October 1, 2008, November
3, 2008 and December 1, 2008, we sold and issued to the Investors 125 Units
comprised of Notes in the aggregate principal amount of $125,000 and Warrants to
purchase up to 250,000 shares of Common Stock for an aggregate purchase price of
$125,000 (the "Subsequent Financing").
On March
30, 2009, we completed a private placement of 5,357,143 shares of common stock
of the Company and warrants to purchase up to an aggregate of 13,392,857 shares
of common stock with an initial exercise price of $0.35 per share and a five
year term for aggregate net proceeds of $1,409,000 (the “March 2009
Financing”).
On
October 6, 2009, we completed a private placement of 5,892,857 shares of common
stock of the Company and warrants to purchase up to an aggregate of 11,785,714
additional shares of Common Stock, with an initial exercise price of $0.28 per
share and a seven year term for aggregate proceeds of $1,650,000 (the “October
2009 Financing”). The Company also granted to the Investors the
option to acquire, collectively, up to an additional 5,892,857 additional Units
(the “Additional Units”), comprised of an aggregate of 5,892,857 shares of
common stock of the Company and warrants to purchase up to an aggregate of
11,785,714 additional shares of common stock at the per Additional Unit purchase
price of $0.28 (the “Call Option”). In addition, the Investors granted to the
Company the option to require these Investors, severally and not jointly, to
acquire up to 5,892,857 Additional Units, less any Additional Units acquired
under the Call Option, at the per Additional Unit purchase price of $0.28 (the
“Put Option”). The Call Option is exercisable at any time, and from
time to time, on or prior to June 30, 2010. The Put Option is
exercisable at any time from June 30, 2010 to July 30, 2010. However,
the Investors shall have the right to terminate the Put Option if they
reasonably determine that a material adverse event, condition or circumstance
has occurred with respect to the prospects of the Company’s AEOL 10150 drug
candidate for acute radiation syndrome; provided that the Company’s failure to
receive a grant or financing shall not, by itself, constitute a material adverse
event, condition or circumstance with respect thereto.
In
addition in connection with the October 2009 Financing, the Investors agreed to
convert all $1,000,000 of the Company’s Notes into common stock at a conversion
rate of $0.35 per share and to exchange their remaining option to purchase an
additional $4,000,000 in Senior Convertible Notes for warrants to purchase up to
14,285,714 shares of common stock in substantially the same of form and terms of
the Warrants issued in the October 2009 Financing, including an initial exercise
price of $0.28 per share. As consideration for the investors to
convert the Notes, the Company agreed to exchange warrants to purchase up to
2,000,000 shares of common stock issued to the Investors in connection with the
sale of the Notes, warrants to purchase up to 2,150,000 shares of common stock
issued to the Investors and one of their affiliates in connection with a
financing completed in November 2005 and warrants to purchase up to 13,392,857
shares of common stock issued to the Investors in connection with the March 2009
Financing for warrants to purchase up to 17,542,857 shares of Common Stock in
substantially the same form and terms of the warrants issued in the October 2009
Financing, including an initial exercise price of $0.28 per share, subject to
adjustment pursuant to the warrants.
47
Results
of Operations
Fiscal
Year Ended September 30, 2009 Compared to Fiscal Year Ended September 30,
2008
We had a
net loss of $2,296,000 for the fiscal year ended September 30, 2009, versus a
net loss of $2,973,000 for fiscal year ended September 30, 2008.
We did
not generate any revenues through the sale of our drug candidates or grants
during fiscal 2009 or fiscal 2008.
Research
and Development
Research
and development (“R&D”) expenses decreased $266,000, or 27%, to $711,000 for
fiscal 2009 from $977,000 for fiscal 2008. The lower level of R&D
expenses during the current period reflects a lower amount of manufacturing and
research expenses. During fiscal 2008, we were in the process of
manufacturing small quantities of our drug candidates to support our development
program whereas during fiscal 2009 we had no manufacturing underway and were
only running stability studies on existing drug supplies resulting in a decrease
of $105,000 in manufacturing expenses. Research expenses also
declined during fiscal 2009 as more of the Company’s research programs were
funded by external grants. During fiscal 2009, outside research
expenses were $240,000 compared to $458,000 during fiscal 2008. The
Company currently has seven studies in progress: the study of its drug
candidates as a potential countermeasure against the effects of sulfur mustard
gas on the lung and skin, as a protectant against the effects of radiation on
the lungs and on the gastro-intestinal tract, as a countermeasure against the
effects of chlorine gas, as a potential treatment for epilepsy and for the
potential treatment for Parkinson’s disease.
R&D
expenses for our antioxidant program have totaled $35,222,000 from inception
through September 30, 2009. Because of the uncertainty of our
research and development and clinical studies, we are unable to predict the
total level of spending on the program or the program completion
date. However, we expect R&D expenses during fiscal year 2010
will be higher than fiscal 2009 as we may initiate additional studies of AEOL
10150. Our ongoing cash requirements will also depend on numerous
factors, particularly the progress of our R&D programs, our ability to
obtain grants from governmental or other organizations and our ability to
negotiate and complete collaborative agreements.
General
and Administrative
General
and administrative (“G&A”) expenses include corporate costs required to
support our company, our employees and consultants and our
stockholders. These costs include personnel and outside costs in the
areas of legal, human resources, investor relations and
finance. Additionally, we include in general and administrative
expenses such costs as rent, repair and maintenance of equipment, depreciation,
utilities, information technology and procurement costs that we need to support
the corporate functions listed above.
G&A
expenses decreased $248,000, or 16%, to $1,292,000 for fiscal year 2009 from
$1,540,000 for fiscal year 2008. G&A expenses were lower during
fiscal year 2009 versus fiscal year 2008 due to a decline in stock based
compensation expense, Board of Directors expense and legal
fees. Stock based compensation expense decreased by $50,000 as
a result of lower valuations assigned to our stock option grants in the current
year when compared to the prior year. Board of Directors expense
decreased by $101,000 as the Board of Directors adopted a stock based
compensation plan in July 2008 whereas during the prior year, the Board of
Directors received cash and stock based compensation. Legal fees
declined by $84,000 due to management’s continued efforts to reduce costs by
performing regulatory and compliance activities in-house.
During
fiscal year 2009, we recorded a gain on the sale of our holdings of Arca
Biopharma, Inc. (“ARCA”) common stock of $133,000.
During
fiscal 2008, CPEC LLC (“CPEC”) received a milestone payment from
ARCA. In 2003, CPEC, of which we own 35%, out-licensed all rights to
a potential therapeutic compound referred to as “bucindolol” to
ARCA. During fiscal 2008, CPEC received a milestone payment of
$500,000 as a result of ARCA filing a New Drug Application for
bucidnolol. We recorded $175,000 of income during fiscal 2008 as a
result of our equity ownership of CPEC. Also as a result of the
filing of the New Drug Application with the US Food and Drug Administration, we
are obligated to pay $413,000 in the form of cash or stock at our election to
the majority owner of CPEC who will in turn pay the original licensors of
bucindolol per the terms of the 1994 Purchase Agreement of CPEC. The
obligation is included in our financial statements under the heading “Accounts
Payable.”
We
incurred net interest expense of $437,000 during fiscal 2009 compared to net
interest expense of $51,000 for fiscal 2008. The change reflects
higher interest expense as a result of the issuance of the Senior Convertible
Notes which bear interest at a rate of 7% as well as the related amortization of
debt issuance costs and a note issuance discount. Interest expense
also increased as a result of a higher average balance of our note payable with
Elan. Interest income on our investments also decreased due to a
lower level of investable assets as well as a lower level of interest rates
during fiscal 2009 versus fiscal 2008.
48
During
fiscal 2009 as a result of our March 2009 Financing, we were required to lower
the exercise price of 4,687,000 warrants issued in our November 2005 Financing
and in our 2007 Financing to $0.28 per share, the purchase price of the common
stock and warrants issued on March 30, 2009. As a result of the
change in the exercise price, these warrants were revalued resulting in an
increase in the value of $38,000 which was charged to the statement of
operations. During fiscal 2008 as a result of the issuance of Senior
Convertible Notes, we were required to lower the exercise price of 4,687,000
warrants previously issued in our November 2005 Financing and in our 2007
Financing to $0.35 per share, the conversion price of the Senior Convertible
Notes issued on August 1, 2008. As a result of the change in the
exercise price, these warrants were revalued resulting in an increase in the
value of $118,000 which was charged to the statement of operations.
During
fiscal 2009, we recorded a gain of $49,000 related to the net increase in the
market value of our trading securities. While during fiscal 2008, we
recorded an “other-than-temporary” impairment charge of $49,000 based upon
reduced market values of these securities.
Fiscal
Year Ended September 30, 2008 Compared to Fiscal Year Ended September 30,
2007
We had a
net loss of $2,973,000 for the fiscal year ended September 30, 2008, versus a
net loss of $3,024,000 for fiscal 2007.
We did
not generate any revenues through the sale of our drug candidates or grants
during fiscal 2008 or fiscal 2007.
Research
and Development
Research
and development (“R&D”) expenses decreased $404,000, or 29%, to $977,000 for
fiscal 2008 from $1,381,000 for fiscal 2007. The lower level of
R&D expenses during the current period reflects a lower amount of
employment, consulting and manufacturing expenses offset by a higher level of
pre-clinical and patent expenses. Employment and consulting expenses
were $152,000 during fiscal 2008 versus $434,000 during fiscal
2007. The decline in employment and consulting expenses reflects that
we were completing our multiple dose clinical trail and were in the process of
manufacturing bulk quantities of our lead drug candidate, AEOL 10150, during
fiscal 2007, whereas during the current year we had restructured our research
program to utilize outside research institutions and grants to perform our
research activities and therefore had a lower level of employment and consulting
expenses. During fiscal 2008, manufacturing costs were $136,000
compared to $526,000 during fiscal 2007. Offsetting these declines
was an increase of $335,000 in outside research services as a result of our
transition to outsourcing of research activities during the current period and
$130,000 in patent fees as a result of an increase in patent filing
activity.
General
and Administrative
G&A
expenses decreased $379,000, or 20%, to $1,540,000 for fiscal year 2008 from
$1,919,000 for fiscal year 2007. G&A expenses were lower during
fiscal year 2008 versus fiscal year 2007 due to a decline in employment costs,
stock compensation expense and investor relations expense. Employment
costs declined by $156,000 during fiscal 2008 compared to fiscal 2007, as the
current year reflects employment costs of our sole employee, our Chief Executive
Officer, whereas the prior year includes employment costs of two additional
executive officers as well as severance and bonus costs to three executive
officers. Stock compensation expense decreased by $216,000 as a
result of the lower headcount during the current year. Investor
relations expenses declined by $54,000, as a result of a decrease in the level
of activity for our investor relations program. Offsetting these
declines were increased consulting expenses in the amount of $65,000 for market
analysis and out-licensing analysis for our drug candidates.
During
fiscal 2008, CPEC received a milestone payment from ARCA. We recorded
$175,000 of income during fiscal 2008 as a result of our equity ownership of
CPEC. Also as a result of the filing of the NDA with the FDA, we are
obligated to pay $413,000 in the form of cash or stock at our election to the
majority owner of CPEC who will in turn pay the original licensors of bucindolol
per the terms of the 1994 Purchase Agreement of CPEC. The obligation
is included in our financial statements under the heading “Accounts
Payable.”
We
incurred net interest expense of $51,000 during fiscal year 2008 compared to net
interest income of $51,000 for fiscal year 2007. The change reflects
higher interest expense as a result of the issuance of the Senior Convertible
Note which bear interest at a rate of 7% as well as the related amortization of
debt issuance costs and a note issuance discount. Interest expense
also increased as a result of a higher average balance of our note payable with
Elan and the draws on our margin loan with UBS Financial Services,
Inc. Interest income on our investments also decreased due to a lower
level of investable assets as well as a lower level of interest rates during
fiscal 2008 versus fiscal 2007.
During
fiscal 2008 as a result of the issuance of Senior Convertible Notes, we were
required to lower the exercise price of 4,687,000 warrants previously issued in
our November 2005 Financing and in our 2007 Financing to $0.35 per share, the
conversion price of the Senior Convertible Notes issued on August 1,
2008. As a result of the change in the exercise price, these warrants
were revalued resulting in an increase in the value of $118,000 which was
charged to the statement of operations.
49
During
fiscal 2008, we recorded an “other-than-temporary” impairment charge of $49,000
based upon reduced market values of our auction-rate securities as determined
based upon investment statements received from UBS Financial Services,
Inc. During fiscal 2008, the auction rate securities which the
Company has invested in have experienced auction failures as a result of the
current disruptions in the credit markets. This is the first time the
Company has experienced this type of event for its holdings of auction-rate
securities and the Company believes that prior to February 13, 2008, the
Company’s investment advisor, UBS, had not had a failed auction. The
Company understands that the failure of auctions is broad based and not limited
to those securities held by the Company. As a result of the failed
auctions, our auction-rate securities are currently not
liquid. Furthermore, the Company cannot predict how long they will
remain illiquid.
During
fiscal 2007, we recognized $225,000 in income as a result of the forgiveness of
a portion of the principal balance of a note payable from Elan Corporation,
plc. (“Elan”). In connection with the termination of a
note payable and issuance of a new note payable, we paid $300,000 in cash to
Elan, Elan and the Company entered into a new note payable in the amount of
$453,000 for a period of two years under substantially the same terms as the
original note and Elan forgave $225,000 of the original note
payable.
Liquidity
and Capital Resources
At
September 30, 2009, we had $646,000 of cash, an increase of $247,000 from
September 30, 2008. The increase in cash from September 30, 2008 to
2009 was primarily due to the proceeds from the issuance of Senior Convertible
Notes and our March 2009 Financing, offset by fiscal 2009 operating
expenses. During fiscal 2009, we sold $375,000 senior convertible
notes and raised $1,409,000 through the sale of our common stock and warrants to
purchase shares of common stock. In addition, we raised $1,650,000 in
October 2009 through the sale of our common stock and warrants and may receive
an additional $1,650,000 through a call and put option from these investors if
certain conditions and circumstances are met.
We
believe we have adequate financial resources (not including any possible
exercise of the Call or Put Option) to conduct operations into the fourth
quarter of fiscal year 2010. This raises substantial doubt about our
ability to continue as a going concern, which will be dependent on our ability
to generate sufficient cash flows to meet our obligations on a timely basis, to
obtain additional financing and, ultimately, to achieve operating
profit.
We
incurred operational losses of $2,003,000 during fiscal 2009. Our
ongoing cash requirements will depend on numerous factors, particularly the
progress of our catalytic antioxidant program and clinical trials and our
ability to negotiate and complete collaborative agreements or out-licensing
arrangements or receive grants to fund our development programs. In
order to help fund our on-going operating cash requirements, we intend to seek
new collaborations for our antioxidant research program that include initial
cash payments and on-going research support. In addition, we will
need to raise additional funds and explore other strategic and financial
alternatives, including a merger with another company and the establishment of
new collaborations for current research programs that include initial cash
payments and ongoing research support, or the out-licensing of our compounds for
development by a third party.
There are
significant uncertainties as to our ability to access potential sources of
capital. We may not be able to enter into any collaboration on terms
acceptable to us, or at all, due to conditions in the pharmaceutical industry or
in the economy in general or based on the prospects of our catalytic antioxidant
program. Even if we are successful in obtaining a collaboration for
our antioxidant program, we may have to relinquish rights to technologies, drug
candidates or markets that we might otherwise develop
ourselves. These same risks apply to any attempt to out-license our
compounds.
Similarly,
due to market conditions, the illiquid nature of our stock and other possible
limitations on equity offerings, we may not be able to sell additional
securities or raise other funds on terms acceptable to us, if at
all. It generally is difficult for small biotechnology companies like
us to raise funds in the equity markets. Any additional equity
financing, if available, would likely result in substantial dilution to existing
stockholders.
Our
forecast of the period of time through which our financial resources will be
adequate to support our operations is forward-looking information, and actual
results could vary.
50
Contractual
Obligations
Our
contractual obligations (in thousands) as of September 30, 2009 were as
follows:
|
Payments
due by period
|
|||||||||||||||
|
Contractual
Obligations
|
Total
|
Less
than 1 Year
|
1-3
Years
|
3-5
Years
|
More
than 5 Years
|
||||||||||
|
Long-term
debt
|
|
$
|
1,689
|
|
$
|
—
|
|
$
|
1,689
|
|
$
|
—
|
|
$
|
—
|
|
Capital
lease obligations
|
—
|
—
|
—
|
—
|
—
|
||||||||||
|
Operating
leases
|
—
|
—
|
—
|
—
|
—
|
||||||||||
|
Purchase
obligations
|
1,837
|
1,837
|
—
|
—
|
—
|
||||||||||
|
Total
|
$
|
3,526
|
$
|
1,837
|
$
|
1,689
|
$
|
—
|
$
|
—
|
|||||
Off
Balance Sheet Arrangements
We do not
have any off-balance sheet arrangements that have or are reasonably likely to
have a current or future effect on our financial condition, changes in financial
condition, revenues or expenses, results of operations, liquidity, capital
expenditures or capital resources as defined under the rules of SEC Release No.
FR-67. We do not have operating or capital leases.
Relationship
with Goodnow Capital and Xmark Opportunity Partners, LLC
In July
2003, we initiated a series of transactions that led to our corporate
reorganization and recapitalization. We obtained an aggregate of $8.0
million in secured bridge financing in the form of convertible promissory notes
we issued to Goodnow Capital, L.L.C. A portion of this financing
allowed us to pay our past due payables and become current. We used
the remainder for our operations, including a toxicology study for our catalytic
antioxidant compounds under development as a treatment for ALS.
We
completed our corporate reorganization on November 20, 2003. The
reorganization involved the merger of our former parent company into one of its
wholly owned subsidiaries. Upon consummation of the merger, a $3.0
million note held by Goodnow, including accrued interest, converted into
3,060,144 shares of our common stock. On April 19, 2004, we sold
$10.26 million of our common stock in a private placement. In
conjunction with the private placement, Goodnow voluntarily converted a $5.0
million debenture, including accrued interest thereon, into 5,046,875 shares of
our common stock, which, along with the 3,060,144 shares issued in the merger
and the 20 shares that Goodnow owned before the consummation of the merger,
represented 58.1% of the shares of our common stock outstanding on November 30,
2004. As of December 24, 2009, Xmark Opportunity Partners, LLC,
through its management of Goodnow and the Xmark Funds, and through the Xmark
Voting Trust, had beneficial ownership of approximately 80.7% of our outstanding
common stock. As a result of this significant ownership, Goodnow is
able to significantly influence, if not control, future actions voted on by
stockholders of our company.
As part
of the $8.0 million financing from Goodnow, we agreed:
|
|
·
|
to
secure the $8.0 million debt with liens on all of our assets, which liens
expired on April 19, 2004 when the remaining debt converted to shares of
common stock;
|
|
|
·
|
to
spend the financing proceeds only in accordance with a budget and
development plan agreed to by
Goodnow;
|
|
|
·
|
to
not enter into any arrangement with a party other than Goodnow in which we
would raise capital through the issuance of our securities other than the
raising of up to an aggregate of $20,000,000 through the issuance of
shares of our common stock at a price of greater than $3.00 per share and
which would represent 25% or less of our then outstanding common stock on
an as-converted to common and fully diluted basis. If we agree
to or consummate a financing transaction with someone other than Goodnow
that exceeds these limitations, we will pay Goodnow a break-up fee of
$500,000. Goodnow approved the April 2004 private placement,
which exceeded these limitations, and waived the fee. However,
the $20,000,000 limitation was lowered to $9,740,000 and the 25%
limitation was reduced to zero. Goodnow also approved the 2005
Financing, the 2006 Financing, the 2007 Financing, the SCN Financing, the
March 2009 Financing and the October 2009 Financing, each of which
exceeded these limitations and waived the fee;
and
|
|
|
·
|
to
allow Goodnow to appoint one director to our board of directors, provided
Goodnow owns at least 10%, but less than 20%, of our outstanding common
stock, on an as-converted to common and fully diluted basis, and two
directors if Goodnow owns more than 20% of our outstanding common
stock.
|
51
In
addition, without Goodnow’s prior approval, we have agreed to not:
|
|
·
|
make
any expenditure or series of related expenditures in excess of $25,000,
except (i) expenditures pursuant to the SBIR grant from the U.S. Small
Business Administration, (ii) specified in a budget approved in writing in
advance by Goodnow and our Board, and (iii) directly relating to the
development of AEOL 10150 for the treatment of
ALS;
|
|
|
·
|
change
our business or operations;
|
|
|
·
|
merge
with or sell or lease a substantial portion of our assets to any
entity;
|
|
|
·
|
incur
debt from any third party or place a lien on any of our
properties;
|
|
|
·
|
amend
our certificate of incorporation or
bylaws;
|
|
|
·
|
increase
the compensation we pay our
employees;
|
|
|
·
|
pay
dividends on any class of our capital
stock;
|
|
|
·
|
cancel
any debt except for full value; or
|
|
|
·
|
issue
any capital stock except pursuant to agreements with or as agreed to by
Goodnow.
|
The
affirmative covenants expire on the earliest of:
|
|
·
|
the
date that Goodnow owns less than 20% of our outstanding common stock on an
as converted basis;
|
|
|
·
|
the
completion, to the absolute satisfaction of Goodnow, of initial clinical
safety studies of AEOL 10150, and analysis of the data developed based
upon such studies with results satisfactory to Goodnow, in its absolute
discretion, to initiate efficacy studies of AEOL 10150;
or
|
|
|
·
|
the
initiation of dosing of the first human patient in an efficacy-based study
of AEOL 10150.
|
In
addition, under the terms of the warrants to purchase up to 43,614,285 shares of
our common stock issued to Xmark on October 6, 2009 (the “Xmark Warrants”) if we
were to pay a dividend on our common stock the exercise price of these warrants
would be reset from $0.28 per share to $0.01 per share and the warrantholders
would also receive any such dividend paid. The Xmark Warrants also
contain a provision that provides for the reduction of the exercise price to
$0.01 upon a change of control and contain anti-dilution provisions in the event
of a stock dividend or split, dividend payment or other issuance,
reorganization, recapitalization or similar event. In addition, the
Xmark Warrants prohibit the sale of the Company to an entity other than one that
is publicly traded.
Critical
Accounting Policies and Estimates
Our
consolidated financial statements have been prepared in accordance with
accounting principles generally accepted in the United States of America, which
require us to make estimates and judgments that affect the reported amounts of
assets, liabilities, revenues, expenses and related disclosure of contingent
assets and liabilities. We evaluate our estimates, judgments and the
policies underlying these estimates on a periodic basis as the situation
changes, and regularly discuss financial events, policies, and issues with our
independent registered public accounting firm and members of our audit
committee. We routinely evaluate our estimates and policies regarding
revenue recognition; clinical trial, preclinical, manufacturing and patent
related liabilities; license obligations; inventory; intangible assets;
share-based payments; and deferred tax assets.
We
generally enter into contractual agreements with third-party vendors to provide
clinical, preclinical and manufacturing services in the ordinary course of
business. Many of these contracts are subject to milestone-based
invoicing and the contract could extend over several years. We record
liabilities under these contractual commitments when we determine an obligation
has been incurred, regardless of the timing of the
invoice. Patent-related liabilities are recorded based upon various
assumptions or events that we believe are the most reasonable to each individual
circumstance, as well as based upon historical experience. License
milestone liabilities and the related expense are recorded when the milestone
criterion achievement is probable. We have not recognized any assets
for inventory, intangible items or deferred taxes as we have yet to receive
regulatory approval for any of our compounds. Any potential asset
that could be recorded in regards to any of these items is fully
reserved. In all cases, actual results may differ from our estimates
under different assumptions or conditions.
New
Accounting Pronouncements
In June
2008, the Financial Accounting Standards Board ("FASB") issued revised guidance
for the determining whether an Instrument (or Embedded Feature) is indexed to an
entity’s own stock. Equity-linked instruments (or embedded features)
that otherwise meet the definition of a derivative are not accounted for as
derivatives if certain criteria are met, one of which is that the instrument (or
embedded feature) must be indexed to the entity’s own stock. The
Company adopted this guidance on October 1, 2009 and began applying its
provisions to outstanding instruments as of that date. The fair value
of the warrants affected at their dates of issuance totaled $8,282,000 and was
initially recorded as a component of additional paid-in capital. Upon adoption
of the new guidance in the first quarter of 2010, the Company recorded a
decrease to the opening balance of additional-paid-in capital of $8,142,000 and
recorded a decrease to accumulated deficit totaling $4,353,000, representing the
decrease in the fair value of the warrants from the date of issuance to October
1, 2009. The fair value of the warrants at October 1, 2009 of
$3,789,000 will be included as a current liability in the balance sheet as of
that date. Any future increase or decrease in fair value of the
warrants will be included as a component of other income in the accompanying
statement of operations for the respective period.
52
In June
2008, the FASB issued revised guidance for determining whether instruments
granted in share-based payment transactions are participating securities. The
new guidance addresses whether instruments granted in share-based payment
transactions are participating securities prior to vesting, and therefore need
to be included in the computation of earnings per share under the two-class
method for calculating earnings per share. The new guidance is
effective for financial statements issued for fiscal years beginning on or after
December 15, 2008 and earlier adoption is prohibited. The adoption of this new
guidance is not expected to have a material effect on our financial
statements.
In May
2008, the FASB issued new guidance for the accounting for convertible debt
instruments that may be settled in cash upon conversion. This new
guidance applies to convertible debt instruments that may be settled in cash, or
other assets, upon conversion and are not addressed by other accounting
guidance. This new guidance is effective for financial statements
issued for fiscal years beginning after December 15, 2008, and interim periods
within those fiscal years. Early adoption is prohibited. This new guidance is
not expected to have a material impact on our financial position, results of
operations or liquidity.
Item
7A. Quantitative and Qualitative Disclosures About Market
Risk.
We are
exposed to market risk related to changes in interest rates. Our
current investment policy is to maintain an investment portfolio consisting of
U.S. government treasury and agency notes, corporate debt obligations, municipal
debt obligations, auction rate securities and money market funds, directly or
through managed funds. Our cash is deposited in and invested through
highly rated financial institutions in North America. Our investments
are subject to interest rate risk and will fall in value if market interest
rates increase. We could be exposed to losses related to these
securities should one of our counterparties default. We attempt to
mitigate this risk through credit monitoring procedures.
At
September 30, 2009, we had two fixed-rate notes payable.
Item
8. Financial Statements and Supplementary Data.
INDEX
TO FINANCIAL STATEMENTS
|
Page
|
|
|
Report
of Independent Registered Public Accounting Firm
|
54
|
|
Consolidated
Balance Sheets – As of September 30, 2009 and 2008
|
55
|
|
Consolidated
Statements of Operations – For the fiscal years ended September 30, 2009,
2008 and 2007
|
56
|
|
Consolidated
Statements of Stockholders’ Equity (Deficit) – For the fiscal years ended
September 30, 2009, 2008 and 2007
|
57
|
|
Consolidated
Statements of Cash Flows – For the fiscal years ended September 30, 2009,
2008 and 2007
|
58
|
|
Notes
to Consolidated Financial Statements
|
59
|
53
REPORT
OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the
Board of Directors
Aeolus
Pharmaceuticals, Inc.
We have
audited the accompanying consolidated balance sheets of Aeolus Pharmaceuticals,
Inc. (the “Company”) as of September 30, 2009 and 2008, and the related
consolidated statements of operations, stockholders’ equity (deficit), and cash
flows for each of the years ended September 30, 2009, 2008 and
2007. These financial statements are the responsibility of the
Company’s management. Our responsibility is to express an opinion on
these financial statements based on our audits.
We
conducted our audits in accordance with the standards of the Public Company
Accounting Oversight Board (United States). Those standards require
that we plan and perform the audits to obtain reasonable assurance about whether
the financial statements are free of material misstatement. The
Company is not required to have, nor were we engaged to perform, an audit of its
internal control over financial reporting. Our audits included
consideration of internal control over financial reporting as a basis for
designing audit procedures that are appropriate in the circumstances, but not
for the purpose of expressing an opinion on the effectiveness of the Company’s
internal control over financial reporting. Accordingly, we express no
such opinion. An audit also includes examining, on a test basis,
evidence supporting the amounts and disclosures in the financial statements,
assessing the accounting principles used and significant estimates made by
management, as well as evaluating the overall financial statement presentation.
We believe that our audits provide a reasonable basis for our
opinion.
In our
opinion, the consolidated financial statements referred to above present fairly,
in all material respects, the consolidated financial position of Aeolus
Pharmaceuticals, Inc. as of September 30, 2009 and 2008, and the consolidated
results of its operations and its cash flows for each of the years ended
September 30, 2009, 2008 and 2007, in conformity with accounting principles
generally accepted in the United States of America.
The
accompanying consolidated financial statements have been prepared assuming that
the Company will continue as a going concern. As discussed in Note B
of the consolidated financial statements, the Company has suffered recurring
losses, negative cash flows from operations and does not currently possess
sufficient working capital to fund its operations throughout the next fiscal
year. These matters raise substantial doubt about the Company’s
ability to continue as a going concern. Management’s plans in regard
to these matters are also described in Note B. The consolidated financial
statements do not include any adjustments that might result from the outcome of
this uncertainty.
/s/ Haskell & White
LLP
HASKELL & WHITE LLP
Irvine,
California
December
28, 2009
54
AEOLUS
PHARMACEUTICALS, INC.
CONSOLIDATED
BALANCE SHEETS
(Dollars
in thousands, except per share data)
|
September
30,
|
||||||||
|
2009
|
2008
|
|||||||
|
ASSETS
|
||||||||
|
Current
assets:
|
||||||||
|
Cash
and cash equivalents
|
$ | 646 | $ | 399 | ||||
|
Prepaids
and other current assets
|
133 | 156 | ||||||
|
Total
current assets
|
779 | 555 | ||||||
|
Investments,
available for sale
|
-- | 440 | ||||||
|
Investment
in CPEC LLC
|
32 | 125 | ||||||
|
Total
assets
|
$ | 811 | $ | 1,120 | ||||
|
LIABILITIES
AND STOCKHOLDERS' EQUITY (DEFICIT)
|
||||||||
|
Current
liabilities:
|
||||||||
|
Accounts
payable
|
$ | 774 | $ | 991 | ||||
|
Margin
loan (Note F)
|
-- | 366 | ||||||
|
Current
maturity of long-term note payable
|
-- | 534 | ||||||
|
Total
current liabilities
|
774 | 1,891 | ||||||
|
Senior
convertible notes to related parties, net (redemption value of $1,000,000
and $625,000 as of September 30, 2009 and 2008, respectively) (Note
F)
|
600 | 266 | ||||||
|
Long-term
note payable
|
594 | — | ||||||
|
Total
liabilities
|
1,968 | 2,157 | ||||||
|
Commitments
and Contingencies (Note E and J)
|
||||||||
|
Stockholders'
equity (deficit):
|
||||||||
|
Preferred
stock, $.01 par value per share, 10,000,000 shares
authorized:
|
||||||||
|
Series
B nonredeemable convertible preferred stock, 600,000 shares
authorized; 475,087 shares issued and outstanding as of
September 30, 2009 and 2008
|
5 | 5 | ||||||
|
Common
stock, $.01 par value per share, 200,000,000 shares authorized; 37,563,392
and 31,952,749 shares issued and outstanding at September 30, 2009 and
2008, respectively
|
376 | 320 | ||||||
|
Additional
paid-in capital
|
159,657 | 157,573 | ||||||
|
Unrealized
losses on investments, available for sale
|
-- | (36 | ) | |||||
|
Accumulated
deficit
|
(161,195 | ) | (158,899 | ) | ||||
|
Total
stockholders' equity (deficit)
|
(1,157 | ) | (1,037 | ) | ||||
|
Total
liabilities and stockholders' equity (deficit)
|
$ | 811 | $ | 1,120 | ||||
The
accompanying notes are an integral part of these consolidated financial
statements.
55
AEOLUS
PHARMACEUTICALS, INC.
CONSOLIDATED
STATEMENTS OF OPERATIONS
(In
thousands, except per share data)
|
Fiscal
Year Ended September 30,
|
|||||||||||
|
2009
|
2008
|
2007
|
|||||||||
|
Revenue
|
|
|
|
|
|
||||||
|
Grant
income
|
$
|
—
|
$
|
—
|
$
|
—
|
|||||
|
Costs
and expenses:
|
|||||||||||
|
Research
and development
|
711
|
977
|
1,381
|
||||||||
|
General
and administrative
|
1,292
|
1,540
|
1,919
|
||||||||
|
Total
costs and expenses
|
2,003
|
2,517
|
3,300
|
||||||||
|
Loss
from operations
|
(2,003
|
)
|
(2,517
|
)
|
(3,300
|
)
|
|||||
|
Equity
in income of CPEC LLC ($175 dividend received in 2008)
|
—
|
175
|
—
|
||||||||
|
Interest
expense
|
(441
|
)
|
(93
|
)
|
(51
|
) | |||||
|
Interest
income
|
4
|
42
|
102
|
||||||||
|
Warrant
repricing charges
|
(38
|
)
|
(118
|
)
|
—
|
||||||
|
Collaboration
expense
|
—
|
(413
|
)
|
—
|
|||||||
|
Gain
(loss) on marketable investments
|
49
|
(49
|
)
|
—
|
|||||||
|
Gain
on sale of investments, available for sale
|
133
|
—
|
—
|
||||||||
|
Gain
on forgiveness of note payable
|
—
|
—
|
225
|
||||||||
|
Net
loss
|
$
|
(2,296
|
)
|
$
|
(2,973
|
)
|
$
|
(3,024
|
)
|
||
|
Basic
net loss per common share
|
$
|
(0.07
|
)
|
$
|
(0.09
|
)
|
$
|
(0.10
|
)
|
||
|
Diluted
net loss per common share
|
$
|
(0.07
|
)
|
$
|
(0.11
|
)
|
$
|
(0.10
|
)
|
||
|
Weighted
average common shares outstanding:
|
|||||||||||
|
Basic
|
34,789
|
31,953
|
30,239
|
||||||||
|
Diluted
|
34,789
|
32,217
|
30,239
|
||||||||
The
accompanying notes are an integral part of these consolidated financial
statements.
56
AEOLUS
PHARMACEUTICALS, INC.
CONSOLIDATED
STATEMENTS OF STOCKHOLDERS' EQUITY (DEFICIT)
(Dollars
in thousands)
|
Series
B Preferred Stock
|
Common
Stock
|
Additional
Paid-in Capital
|
Unrealized
Losses
|
Accumulated
Deficit
|
Total
Stockholders’ Equity (Deficit)
|
|||||||||||||||||
|
Number
of Shares
|
Par
Value
|
Number
of Shares
|
Par
Value
|
|||||||||||||||||||
|
Balance
at September 30, 2006
|
475,087
|
$
|
5
|
29,265,249
|
$
|
293
|
$
|
154,311
|
$
|
—
|
$
|
(152,902
|
)
|
$
|
1,707
|
|||||||
|
Sale
of common stock pursuant to stock offering, net of issuance costs of
$239,000
|
—
|
—
|
2,666,667
|
27
|
1,734
|
—
|
—
|
1,761
|
||||||||||||||
|
Exercise
of common stock options
|
—
|
—
|
20,833
|
—
|
20
|
—
|
—
|
20
|
||||||||||||||
|
Stock-based
compensation and amortization of warrants
|
—
|
—
|
—
|
—
|
716
|
—
|
—
|
716
|
||||||||||||||
|
Net
loss for the fiscal year ended September 30, 2007
|
—
|
—
|
—
|
—
|
—
|
—
|
(3,024
|
)
|
(3,024
|
)
|
||||||||||||
|
Balance
at September 30, 2007
|
475,087
|
5
|
31,952,749
|
320
|
156,781
|
—
|
(155,926
|
)
|
1,180
|
|||||||||||||
|
Sale
of Senior Convertible Notes and warrants, net of issuance costs of
$156,000
|
—
|
—
|
—
|
—
|
451
|
—
|
—
|
451
|
||||||||||||||
|
Stock-based
compensation
|
—
|
—
|
—
|
—
|
341
|
—
|
—
|
341
|
||||||||||||||
|
Unrealized
loss on marketable securities held for sale
|
—
|
—
|
—
|
—
|
—
|
(36
|
)
|
—
|
(36
|
)
|
||||||||||||
|
Net
loss for the fiscal year ended September 30, 2008
|
—
|
—
|
—
|
—
|
—
|
—
|
(2,973
|
)
|
(2,973
|
)
|
||||||||||||
|
Balance
at September 30, 2008
|
475,087
|
5
|
31,952,749
|
320
|
157,573
|
(36
|
)
|
(158,899
|
)
|
(1,037
|
)
|
|||||||||||
|
Sale
of Senior Convertible Notes and warrants
|
—
|
—
|
—
|
—
|
323
|
—
|
—
|
323
|
||||||||||||||
|
Stock-based
compensation
|
—
|
—
|
—
|
—
|
283
|
—
|
—
|
283
|
||||||||||||||
|
Issuance
of common stock pursuant to a consulting agreement
|
—
|
—
|
78,125
|
1
|
24
|
—
|
—
|
25
|
||||||||||||||
|
Payment
of interest on Senior Convertible Notes in the form of common
stock
|
—
|
—
|
175,375
|
2
|
61
|
—
|
—
|
63
|
||||||||||||||
|
Sale
of common stock pursuant to stock offering, net of issuance costs of
$91,000
|
—
|
—
|
5,357,143
|
53
|
1,393
|
—
|
—
|
1,446
|
||||||||||||||
|
Unrealized
gain on marketable securities held for sale
|
—
|
—
|
—
|
—
|
—
|
36
|
—
|
36
|
||||||||||||||
|
Net
loss for the fiscal year ended September 30, 2009
|
—
|
—
|
—
|
—
|
—
|
—
|
(2,296
|
)
|
(2,296
|
)
|
||||||||||||
|
Balance
at September 30, 2009
|
475,087
|
$
|
5
|
37,563,392
|
$
|
376
|
$
|
159,657
|
$
|
—
|
$
|
(161,195
|
)
|
$
|
(1,157
|
)
|
||||||
The
accompanying notes are an integral part of these consolidated financial
statements.
57
AEOLUS
PHARMACEUTICALS, INC.
CONSOLIDATED
STATEMENTS OF CASH FLOWS
(In
thousands)
|
|
|||||||||||
|
Fiscal
Year Ended September 30,
|
|||||||||||
|
2009
|
2008
|
2007
|
|||||||||
|
Cash
flows from operating activities:
|
|
|
|||||||||
|
Net
loss
|
$
|
(2,296
|
)
|
$
|
(2,973
|
)
|
$
|
(3,024
|
)
|
||
|
Adjustments
to reconcile net loss to net cash used in operating
activities:
|
|||||||||||
|
Noncash
compensation
|
283
|
341
|
716
|
||||||||
|
Noncash
interest and financing costs
|
433
|
89
|
52
|
||||||||
|
Warrant
repricing charges
|
38
|
118
|
—
|
||||||||
|
Equity
income in CPEC LLC
|
(175
|
)
|
—
|
||||||||
|
Noncash
consulting and license fee
|
25
|
—
|
—
|
||||||||
|
(Gain)
loss on marketable investments
|
(49
|
)
|
49
|
—
|
|||||||
|
(Gain)
on forgiveness of note payable
|
—
|
—
|
(225
|
)
|
|||||||
|
(Gain)
on sale of investments, available for sale
|
(133
|
)
|
—
|
—
|
|||||||
|
Change
in assets and liabilities:
|
|||||||||||
|
Accounts
receivable
|
—
|
—
|
1
|
||||||||
|
Prepaid
expenses and other assets
|
(7
|
)
|
15
|
24
|
|||||||
|
Accounts
payable and accrued expenses
|
(217
|
)
|
723
|
(623
|
)
|
||||||
|
Net
cash used in operating activities
|
(1,923
|
)
|
(1,813
|
)
|
(3,079
|
)
|
|||||
|
Cash
flows from investing activities:
|
|||||||||||
|
Proceeds
from dividend from CPEC LLC
|
—
|
175
|
—
|
||||||||
|
Sale
(purchase) of investments
|
751
|
(525
|
)
|
—
|
|||||||
|
Net
cash provided by (used in) investing activities
|
751
|
(350
|
)
|
—
|
|||||||
|
Cash
flows from financing activities:
|
|||||||||||
|
Repayment
of Note Payable
|
—
|
—
|
(300
|
)
|
|||||||
|
Proceeds
from the issuance of Senior Convertible Notes and Warrants
|
375
|
625
|
—
|
||||||||
|
Costs
related to the issuance of Senior Convertible Notes and
Warrants
|
—
|
(156
|
)
|
—
|
|||||||
|
Proceeds
from short term note payable
|
3
|
372
|
—
|
||||||||
|
Repayments
of short term note payable
|
(368
|
)
|
(6
|
)
|
—
|
||||||
|
Proceeds
from issuance of common stock and warrants
|
1,500
|
—
|
2,000
|
||||||||
|
Costs
related to the issuance of common stock and warrants
|
(91
|
)
|
—
|
(239
|
)
|
||||||
|
Proceeds
from exercise of stock options
|
—
|
—
|
21
|
||||||||
|
Net
cash provided by financing activities
|
1,419
|
835
|
1,482
|
||||||||
|
Net
(decrease) increase in cash and cash equivalents
|
247
|
(1,328
|
)
|
(1,597
|
)
|
||||||
|
Cash
and cash equivalents at beginning of year
|
399
|
1,727
|
3,324
|
||||||||
|
Cash
and cash equivalents at end of year
|
$
|
646
|
$
|
399
|
$
|
1,727
|
|||||
|
Supplemental
disclosure of cash flow information:
|
|||||||||||
|
Cash
payments of interest
|
$
|
—
|
$
|
4
|
$
|
—
|
|||||
|
Supplemental
disclosure of non-cash investing and financing activities:
|
|||||||||||
|
Common
stock issued for payment of interest on Senior Convertible
Notes
|
$
|
63
|
$
|
—
|
$
|
—
|
|||||
The
accompanying notes are an integral part of these consolidated financial
statements.
58
AEOLUS
PHARMACEUTICALS, INC.
NOTES
TO CONSOLIDATED FINANCIAL STATEMENTS
September
30, 2009
A.
Nature of the Business
Aeolus
Pharmaceuticals, Inc. is a biopharmaceutical company that is developing a new
class of catalytic antioxidant compounds as a medical countermeasure against
biological, chemical and radiological weapons as well as for diseases
and disorders of the central nervous system, respiratory system, autoimmune
system and oncology. The Company’s initial target indications are as
a protective agent against the effects of acute radiation syndrome, sulfur
mustard gas exposure and chlorine gas exposure. The Company has
reported positive safety results from two Phase I clinical trials of AEOL 10150,
our lead drug candidate, with no serious adverse events noted.
The
“Company” or “Aeolus” refers collectively to Aeolus Pharmaceuticals, Inc., a
Delaware corporation (“Aeolus”) and its wholly owned subsidiary, Aeolus
Sciences, Inc., a Delaware corporation (“Aeolus Sciences”). As of
September 30, 2009, Aeolus also owned a 35.0% interest in CPEC LLC, a Delaware
limited liability company (“CPEC”). The Company’s primary operations
are located in Mission Viejo, California.
B.
Liquidity
The
Company has incurred significant operating losses and cash outflows from
operations of $2,003,000 and $1,923,000 for the fiscal year ended September 30,
2009, respectively. The Company expects to incur additional losses
and negative cash flow from operations in fiscal 2010 and for several more
years. Management believes the Company has adequate financial
resources to conduct operations into the fourth quarter of fiscal year
2010. This raises substantial doubt about our ability to continue as
a going concern, which will be dependent on our ability to generate sufficient
cash flows to meet our obligations on a timely basis, to obtain additional
financing and, ultimately, to achieve operating profit.
The
Company intends to explore strategic and financial alternatives, including a
merger with another company, the sale of shares of stock, the establishment of
new collaborations for current research programs that include initial cash
payments and on-going research support and the out-licensing of our compounds
for development by a third party. The Company believes that without
additional investment capital it will not have sufficient cash to fund its
activities in the near future, and will not be able to continue
operating. As such, the Company’s continuation as a going concern is
dependent upon its ability to raise additional financing. The Company
is actively pursuing additional equity financing to provide the necessary funds
for working capital and other planned activities.
If the
Company is unable to obtain additional financing to fund operations beyond the
fourth quarter of fiscal year 2010, it will need to eliminate some or all of its
activities, merge with another company, sell some or all of its assets to
another company, or cease operations entirely. There can be no
assurance that the Company will be able to obtain additional financing on
favorable terms or at all, or that the Company will be able to merge with
another Company or sell any or all of its assets.
C.
Summary of Significant Accounting Policies
Basis
of Presentation
The
consolidated financial statements include the accounts of Aeolus and its wholly
owned subsidiary. All significant intercompany accounts and
transactions have been eliminated. The Company uses the equity method
to account for its 35.0% ownership interest in CPEC.
Use
of Estimates
The
preparation of financial statements in conformity with accounting principles
generally accepted in the United States of America requires management to make
estimates and assumptions that affect the reported amounts of assets and
liabilities and disclosures of contingent assets and liabilities at the date of
the financial statements and the reported amounts of revenues and expenses
during the reporting period. Actual results could differ from those
estimates.
Cash
and Cash Equivalents
The
Company invests available cash in short-term bank deposits, money market funds,
commercial paper and U.S. Government securities. Cash and cash
equivalents include investments with maturities of three months or less at the
date of purchase. The carrying value of cash and cash equivalents
approximate their fair market value at September 30, 2009 and 2008 due to their
short-term nature.
59
Investments
Investments
consist of equity securities and auction rate securities, each of
investment-grade quality, which have an original maturity dates greater than 90
days. These investments are recorded at fair value and accounted for
as available-for-sale securities. The unrealized gain (loss) during
the period is recorded within accumulated other comprehensive loss unless it is
determined to be other-than-temporary. During the year ended
September 30, 2009, the Company recorded a net unrealized gain on investments of
$36,000 and a realized gain on the auction-rate securities of
$49,000. During the year ended September 30, 2008, the Company
recorded a net unrealized loss on investments of $36,000 and also recorded an
other than temporary impairment on the auction-rate securities of
$49,000.
Revenue
Recognition
Grant
income is recognized as revenue as work under the grant is performed and the
related expenses are incurred.
Research
and Development
Research
and development costs are expensed in the period incurred. Payments
related to the acquisition of in-process research and development are expensed
due to the stage of development of the acquired compound or technology at the
date of acquisition.
Income
Taxes
Deferred
tax assets and liabilities are determined based on the difference between the
financial statement and tax basis of assets and liabilities using enacted tax
rates in effect for the year in which the differences are expected to affect
taxable income. Valuation allowances are established when necessary
to reduce net deferred tax assets to the amounts expected to be
realized.
Net
Loss Per Common Share
The
Company computes basic net loss per weighted share attributable to common
stockholders using the weighted average number of shares of common stock
outstanding during the period. The Company computes diluted net loss
per weighted share attributable to common stockholders using the weighted
average number of shares of common and dilutive potential common shares
outstanding during the period. Potential common shares consist of
stock options, convertible debt, warrants and convertible preferred stock using
the treasury stock method and are excluded if their effect is
anti-dilutive. Diluted weighted average common shares included
incremental shares issuable upon conversion of the Senior Convertible Notes and
shares expected to be issued to satisfy a payable in fiscal year
2008. Diluted weighted average shares excluded incremental shares of
approximately 36,954,000, 20,010,000 and 18,428,000 as of September 30, 2009,
2008 and 2007, respectively, related to stock options, convertible debt,
convertible preferred stock and warrants to purchase common
stock. These shares are excluded due to their anti-dilutive effect as
a result of the Company’s net loss.
Accounting
for Stock-Based Compensation
The
Company recognizes stock based compensation expense in the statement of
operations based upon the fair value of the equity award amortized over the
vesting period.
Segment
Reporting
The
Company currently operates in only one segment.
New
Accounting Pronouncements
In June
2008, the Financial Accounting Standards Board ("FASB") issued revised guidance
for determining whether an Instrument (or Embedded Feature) is indexed to an
entity’s own stock. Equity-linked instruments (or embedded features)
that otherwise meet the definition of a derivative are not accounted for as
derivatives if certain criteria are met, one of which is that the instrument (or
embedded feature) must be indexed to the entity’s own stock. The
Company will adopt this guidance on October 1, 2009 and apply its provisions to
outstanding instruments as of that date. The fair value of the
warrants affected at their dates of issuance totaled $8,282,000 and was
initially recorded as a component of additional paid-in capital. Upon adoption
of the new guidance in the first quarter of 2010, the Company recorded a
decrease to the opening balance of additional-paid-in capital of $8,142,000 and
recorded a decrease to accumulated deficit totaling $4,353,000, representing the
decrease in the fair value of the warrants from the date of issuance to October
1, 2009. The fair value of the warrants at October 1, 2009 of
$3,789,000 will be included as a current liability in the balance sheet as of
that date. Any future increase or decrease in fair value of the
warrants will be included as a component of other income in the accompanying
statement of operations for the respective period.
In June
2008, the FASB issued revised guidance for determining whether instruments
granted in share-based payment transactions are participating securities. The
new guidance addresses whether instruments granted in share-based payment
transactions are participating securities prior to vesting, and therefore need
to be included in the computation of earnings per share under the two-class
method for calculating earnings per share. The new guidance is
effective for financial statements issued for
60
fiscal
years beginning on or after December 15, 2008 and earlier adoption is
prohibited. The adoption of this new guidance is not expected to have a material
effect on the Company's financial statements.
In May
2008, the FASB issued new guidance for the accounting for convertible debt
instruments that may be settled in cash upon conversion. This new
guidance applies to convertible debt instruments that may be settled in cash, or
other assets, upon conversion and are not addressed by other accounting
guidance. This new guidance is effective for financial statements
issued for fiscal years beginning after December 15, 2008, and interim periods
within those fiscal years. Early adoption is prohibited. This new guidance is
not expected to have a material impact on the Company’s financial position,
results of operations or liquidity.
D. Investments
Investments
in Auction-Rate Securities
In fiscal
year 2008, the Company invested in auction-rate securities with a par value of
$525,000. The auction-rate securities are debt obligations secured by
student loans, which loans are generally guaranteed by the U.S. Government under
the Federal Family Education Loan Program (FFELP). In addition to the
U.S. Government guarantee on such student loans, many of the securities also
have separate insurance policies guaranteeing both the principal and accrued
interest. The final maturity dates of the auction-rate securities
which the Company owned was between 2029 and 2038. Liquidity
for these securities has historically been provided by an auction process that
resets the applicable interest rate at pre-determined intervals for up to 35
days. In the past, the auction process has generally allowed
investors to obtain immediate liquidity if so desired by selling the securities
at their face amounts. However, disruptions in the credit markets
adversely affected the auction market for these types of
securities. From February 26, 2008 to January 2, 2009, all auctions
scheduled with respect to the Company’s auction-rate securities failed to
close.
The
Company recorded an “other-than-temporary” impairment charge during the fiscal
year ended September 30, 2008 of $49,000 based upon reduced market values as
determined based upon investment statements received from UBS. The
Company also recorded these investments as available for sale and accordingly
recorded their value on the balance sheet at market value as determined by
investment statements provided by UBS.
In
October 2008, the Company entered into an agreement (the “UBS Agreement”) with
UBS Financial Services, Inc. (“UBS”) for which the Company received the right
(“UBS Put Option”) to sell all four of the auction rate securities back to UBS
at par, at its sole discretion, anytime during the period from January 2, 2009
through January 4, 2011, and gave UBS the right to purchase these auction rate
securities or sell them on the Company’s behalf at par any time after the
execution of the Agreement through January 4, 2011. The UBS Put
Option was not transferable, tradable or marginable, and was not listed or
quoted on any securities exchange or any electronic communications
network. Since the UBS Agreement is a legally enforceable firm
commitment, the UBS Put Option was recognized as a financial asset at fair value
in the financial statements, and accounted for separately from the associated
securities as a noncurrent asset. Since the Company intended to and subsequently
exercised the UBS Put Option on January 2, 2009, the Company did not have the
intent to hold the associated auction rate securities until recovery or
maturity. Therefore, the Company classified these securities as trading which
required changes in the fair value of these securities to be recorded in current
period earnings. As a result of this transfer, the Company recognized
in the Statement of Operations a change in market value of $65,000, reflecting a
reversal of the related temporary valuation allowance that was previously
recorded in other comprehensive loss.
Prior to
entering into the UBS Agreement, the Company recorded the auction rate
securities as investments available-for-sale. The Company recorded
unrealized gains and losses on the available-for-sale debt securities in
accumulated other comprehensive income in the shareholders’ equity section of
the balance sheets. Such an unrealized loss did not reduce net income
for the applicable accounting period.
The
Company measured the UBS Put Option at fair value and thus recorded income of
approximately $114,000 and recorded a corresponding long term investment during
the first quarter of fiscal year 2009.
Following
the reclassification of the auction rate securities to trading, the Company
recorded an additional charge to the Statement of Operations of $16,000 to
reduce the value of the auction rate securities, offset by the gain on the Put
Option of $16,000.
On
January 2, 2009, the Company exercised its rights under the UBS Put Option and
sold its four auction rate securities for their par value of $525,000 to
UBS.
61
Investments
in ARCA Biopharma, Inc.
The
Company also held an investment in equity securities of ARCA BioPharma, Inc.
(“ARCA”). The aggregate carrying amount of the holdings in ARCA was
zero and $93,000 as of September 30, 2009 and 2008, respectively. The
Company accounted for the investment on cost basis and reviewed the investment
for impairment whenever there are identified events or changes in circumstances
that may have a significant adverse effect on the fair value of the
investment. During fiscal years ended September 30, 2009, 2008 and
2007, the Company did not record an impairment on the investment in
ARCA. During April 2009, the Company sold all of its 23,377 shares of
ARCA generating net proceeds of $226,000 and a net gain of
$133,000.
Investments
in CPEC LLC
The
Company uses the equity method to account for its 35.0% ownership interest in
CPEC. During fiscal 2003, CPEC licensed bucindolol, a drug previously
under development by the Company for the treatment of heart failure, to ARCA in
return for possible future royalty and milestone payments. During
fiscal 2008, CPEC declared and paid a dividend of which the Company received
$175,000. The dividend was paid upon receipt of a milestone payment
by CPEC from ARCA which was triggered upon the filing of a New Drug Application
for bucidndolol. Also as a result of the filing of the New Drug
Application with the US Food and Drug Administration, the Company is obligated
to pay $413,000 in the form of cash or stock at the Company’s election to the
majority owner of CPEC who will in turn pay the original licensors of bucindolol
per the terms of the 1994 Purchase Agreement of CPEC. The obligation
is included in our financial statements under the heading “Accounts
Payable.”
CPEC had
$91,000 of net assets at September 30, 2009 and 2008. Aeolus’ share
of CPEC’s net assets is included in other assets and the Company has no
operations or activities unrelated to the out licensing of
buicndolol.
E.
Commitments
The
Company acquires assets still in development and enters into research and
development arrangements with third parties that often require milestone and
royalty payments to the third party contingent upon the occurrence of certain
future events linked to the success of the asset in
development. Milestone payments may be required, contingent upon the
successful achievement of an important point in the development life-cycle of
the pharmaceutical product (e.g., approval of the product for marketing by a
regulatory agency). If required by the arrangement, the Company may
have to make royalty payments based upon a percentage of the sales of the
pharmaceutical product in the event that regulatory approval for marketing is
obtained. Because of the contingent nature of these payments, they
are not included in the table of contractual obligations.
These
arrangements may be material individually, and in the unlikely event that
milestones for multiple products covered by these arrangements were reached in
the same period, the aggregate charge to expense could be material to the
results of operations in any one period. In addition, these
arrangements often give Aeolus the discretion to unilaterally terminate
development of the product, which would allow Aeolus to avoid making the
contingent payments; however, Aeolus is unlikely to cease development if the
compound successfully achieves clinical testing objectives.
F.
Notes Payable
The
Company has two debt obligations outstanding as of September 30, 2009 both of
which are due for a combined redemption amount of
$1,689,000. However, in October 2009, the holders of the Senior
Convertible Notes converted all $1,000,000 of the outstanding Senior Convertible
Notes to 2,857,143 shares of common stock (Note L).
Senior
Convertible Notes to Related Parties
On August
1, 2008, the Company entered into a Securities Purchase Agreement (the "SCN
Purchase Agreement") with two accredited institutional investors (the
"Investors") pursuant to which the Company agreed to sell to the Investors units
comprised of senior unsecured convertible notes of the Company (the "Notes"), in
an aggregate principal amount of up to $5,000,000, which shall bear interest at
a rate of 7% per year and mature on the 30-month anniversary of their date of
issuance, and warrants to purchase up to an aggregate of 10,000,000 additional
shares of Common Stock (the "Warrant Shares"), each with an initial exercise
price of $0.50 per share, subject to adjustment pursuant to the warrants (the
"Warrants") (collectively the “SCN Financing”). Each unit (collectively, the
"Units") is comprised of $1,000 in Note principal and Warrants to purchase up to
2,000 shares of the Company's common stock, par value $0.01 per share (the
"Common Stock"), and has a purchase price of $1,000.
On August
1, 2008, the Company sold and issued to the Investors 500 Units comprised of
Notes in the aggregate principal amount of $500,000 and Warrants to purchase up
to 1,000,000 shares of Common Stock for an aggregate purchase price of $500,000
(the "Financing").
62
On each
of September 4, 2008, October 1, 2008, November 3, 2008 and December 1, 2008,
the Company sold and issued to the Investors 125 Units comprised of Notes in the
aggregate principal amount of $125,000 and Warrants to purchase up to 250,000
shares of Common Stock for an aggregate purchase price of $125,000 (the
"Subsequent Financings").
The Notes
issued in the Financing and the Subsequent Financings have an initial conversion
price of $0.35 per share, subject to adjustment pursuant to the
Notes. In addition, the Investors had the option to purchase up to an
additional 4,000 Units, in one or more closings (each, an "Election Closing"),
and at their sole option at any time on or before December 31,
2013. However, on October 6, 2009, this option was cancelled (for
more information, see Note L – Subsequent Events).
The Notes
will be convertible, at the Investors' sole election, into shares of Common
Stock at any time and from time to time. As of September 30, 2009,
the Notes would be convertible into 2,857,143 shares of Common Stock with a fair
value of $1,114,000. The conversion price of the Notes and the
exercise price of the Warrants are subject to adjustment in the event of a stock
dividend or split, reorganization, recapitalization or similar
event. Additionally, the exercise price of the Warrants may be
reduced in the event the Company issues securities at a price per share lower
than the then current exercise price of the Warrants. The Notes are
due and payable in cash at the aggregate principal value plus accrued interest
30 months from the date of issuance if not converted earlier by the
Investors.
Interest
on the Notes accrued at the rate of 7.0% per annum from the date of issuance,
and is payable semi-annually, on January 31 and July 31 of each
year. Interest shall be payable, at the Company's sole election, in
cash or shares of Common Stock, to holders of Notes on the record date for such
interest payments, with the record dates being each January 15 and July 15
immediately preceding an interest payment date.
The
Warrants were exercisable for a five year period from the date of issuance and
contain a "cashless exercise" feature that allows the Investors to exercise the
Warrants without a cash payment to the Company under certain
circumstances.
The net
proceeds to the Company from the sale of 1,000 Units in the Financing and
Subsequent Financing, after deducting for expenses, were approximately
$844,000. The Company used the net proceeds to fund the development
of AEOL 10150 and to fund ongoing operations of the Company. Offering
costs of the private placement were $156,000 and were allocated to the Notes and
Warrants based upon their respective fair values. The offering costs
attributed to the Notes in the amount of $100,000 were capitalized as Debt
Issuance Costs and are included in Prepaid and other current assets in the
Consolidated Balance Sheet. The Debt Issuance Costs are being
amortized over the 30-month life of the Notes in the Financing.
As of
September 30, 2009, the carrying value of the Notes was $588,000 and the
redemption value was $1,000,000. In connection with the Financing and
the Subsequent Financings, the Company recorded a note discount in the amount of
$365,000 based on the relative fair value of the warrants issued in connection
with the Notes. In addition, the Company determined that the Notes
contained an embedded beneficial conversion feature with a computed value of
$347,000 which was also recorded as a discount to the Notes. The note
discounts are being amortized to interest expense over the thirty-month term of
the Notes. The effective interest rate of the Note including the
effect of the amortization of the embedded conversion feature and the note
discount is 39.5 percent.
The
maturity of the Notes may be accelerated upon the occurrence of an event of
default, which includes, subject to certain grace periods, exceptions and
qualifications as set forth in the Notes, the failure by the Company to maintain
the listing of the Common Stock, the failure of the Company to deliver shares
Common Stock in a timely manner following a conversion, the failure of the
Company to have reserved a sufficient number shares of Common Stock to issue
upon conversion of the Notes, the failure by the Company to make payments on the
Notes in a timely manner, payment defaults by the Company or any of its
significant subsidiaries on debt or other obligations in excess of $100,000, the
occurrence of certain bankruptcy events with respect to the Company or its
significant subsidiaries, judgments rendered against the Company or its
significant subsidiaries in excess of $100,000 and breaches of material
representations, warranties or covenants by the Company under the Amended
Purchase Agreement or the Notes. Upon the occurrence of an event of default
related to a bankruptcy of the Company, the Notes shall immediately become due
and payable. Upon the occurrence of any event default other than a
bankruptcy event of default, any holder of the Notes, in its sole discretion,
may declare this Note to be immediately due and payable and the Company shall
pay to the holder in cash the sum of all outstanding principal multiplied by
115%, plus accrued and unpaid interest and late charges, if any,
thereon. The Notes are unsubordinated obligations of the Company and
all payments due under the Notes rank pari passu with the Elan Note and shall
not be subordinated to any other Indebtedness of the Company.
The Notes
also provide that the Company shall not issue any equity securities other than
certain exempt securities as defined in the Notes, incur any indebtedness other
than certain exempt indebtedness as defined in the Notes, allow the incurrence
of any liens on its assets, repay any indebtedness other than the Notes, pay a
dividend on its common stock or make an investment other than ordinary investing
activities without the consent of the holders of Notes representing a majority
of the then-outstanding principal subject to the Notes.
Affiliates
of Xmark Opportunity Partners, LLC are the sole investors in the SCN Financing.
Together with its affiliates, Xmark Opportunity Partners, LLC beneficially owned
approximately 52% of the Company's outstanding common stock prior
to
63
the SCN
Financing. Xmark Opportunity Partners, LLC is the sole manager of Goodnow
Capital, L.L.C. and possesses sole power to vote and direct the disposition of
all securities of the Company held by Goodnow. Goodnow has the right to
designate up to two directors for election to the Company's Board of Directors
pursuant to the terms of a purchase agreement between Goodnow and the Company.
David C. Cavalier, a current director of the Company, is President of
Goodnow. The transaction was evaluated by Management and the Board of
Directors for fairness to ensure the terms were reasonable given the related
party nature of the SCN Financing by providing an option for non-related party
investors to participate in the transaction.
Elan
Note Payable
In August
2002, Aeolus borrowed from Elan Corporation, plc. (“Elan”)
$638,000. The note payable accrued interest at 10% compounded
semi-annually. The note was convertible at the option of Elan into shares of the
Company’s Series B non-voting convertible preferred stock (“Series B Stock”) at
a rate of $43.27 per share. The original note matured on December 21,
2006. However, in February 2007, the Company and Elan terminated the
note, the Company paid $300,000 in cash to Elan, Elan forgave
$225,000 of the note payable and Elan and the Company entered into a new
two-year note payable in the amount of $453,000 under substantially the same
terms as the original note. In February 2009, the Company and Elan
agreed to amend the note payable to extend the maturity date of the convertible
promissory note from February 7, 2009 to February 7, 2011 and increased the
interest rate of the convertible promissory note from 10% to 11% effective
February 7, 2009. As of the date of the Amendment, an aggregate of $553,000 in
principal and interest was outstanding under the convertible promissory note. In
the event of an event of default under the convertible promissory note, Elan may
demand immediate payment of all amounts outstanding under the note. For purposes
of the note, an event of default includes, among other items, a default in the
payment of the note principal or interest when due and payable, an uncured
breach by the Company of its obligations to Elan pursuant the agreements under
which the convertible promissory note was issued, an inability of the Company to
pay its debts in the normal course of business, the cessation of business
activities by the Company (other than as a result of a merger or consolidation
with a third party) without Elan’s prior written consent and the appointment of
a liquidator, receiver, administrator, examiner, trustee or similar officer of
the Company or over all or substantially all of its assets under the
law.
During
the term of the note payable, Elan has the option to convert the note into
shares of Series B Preferred Stock at a rate of $9.00 per share. Upon
the maturity of the note payable, Aeolus has the option to repay the note either
in cash or in shares of Series B Stock and warrants having a then fair market
value of the amount due; provided that the fair market value used for
calculating the number of shares to be issued will not be less than $13.00 per
share. As of September 30, 2009, the outstanding balance, including interest, on
the note payable to Elan was $594,000.
Margin
Loan with UBS Financial Services, Inc.
Aeolus
entered into a secured credit agreement (the “Margin Agreement”) with UBS
Financial Services, Inc and subsequently drew $375,000 under the Margin
Agreement. The Margin Agreement bore interest at the per annum rate
of LIBOR plus 0.25 percent. The Company repaid the loan in full on
January 2, 2009. The weighted average interest rate for the loan was
2.4% during the period the loan was outstanding in the fiscal year ended
September 30, 2009 and 2008. The average balance of the loan and the
total interest paid on the loan during the period the loan was outstanding in
the fiscal year ended September 30, 2008 was $351,000 and $4,000,
respectively. The average balance of the loan and the total interest
paid on the loan during the period the loan was outstanding in the fiscal year
ended September 30, 2009 was $366,000 and zero, respectively.
G.
Stockholders’ Equity (Deficit)
Preferred
Stock
The
Certificate of Incorporation of Aeolus authorizes the issuance of up to
10,000,000 shares of Preferred Stock, at a par value of $.01 per share. The
Board of Directors has the authority to issue Preferred Stock in one or more
series, to fix the designation and number of shares of each such series, and to
determine or change the designation, relative rights, preferences, and
limitations of any series of Preferred Stock, without any further vote or action
by the stockholders of the Company.
In
January 2001, Aeolus issued to Elan 28,457 shares of Series B Stock. In February
2002, the Company issued 58,883 shares of Series B Stock and 480,000 shares of
common stock to Elan in exchange for the retirement of a $1,400,000 note payable
to Elan. In May 2002, the Company sold 416,204 shares of Series B Stock to Elan
for $3,000,000. On January 14, 2005, Elan converted 28,457 shares of the Series
B Stock into 28,457 shares of common stock. As of September 30, 2009, 475,087
shares of Series B Stock were outstanding. Each share of Series B Stock is
convertible into one share of common stock.
Common
Stock
On May
22, 2007, Aeolus Pharmaceuticals, Inc. entered into a Securities Purchase
Agreement with certain accredited investors (the “Investors”) pursuant to which
the Company sold to the Investors an aggregate of 2,666,667 shares of the
Company’s common stock at a purchase price of $0.75 per share for aggregate
gross proceeds of $2,000,000 and issued to the
64
Investors
warrants (the “2007 Investor Warrants”) to purchase up to an aggregate of
2,000,001 shares of common stock of the Company with an exercise price of $0.75
per share (collectively, the “May 2007 Private Placement”). The 2007 Investor
Warrants are exercisable until May 22, 2012. In addition, the Company
issued to a placement agent a warrant to purchase up to an aggregate of 186,667
shares of common stock with an exercise price of $0.75 per share.
The
aggregate net proceeds to the Company from the May 2007 Private Placement, after
deducting for expenses related to finders fees, legal and accounting fees, were
approximately $1,761,000.
The fair
value of the 2007 Investor Warrants on May 22, 2007 was estimated to be
$1,428,000 using the Black-Scholes option pricing model with the following
assumptions: dividend yield of 0%; risk free interest rate of 4.8%; expected
volatility of 132%; and an expected life of five years.
Pursuant
to the terms of the Securities Purchase Agreement, the Company filed a
registration statement which was declared effective on July 19,
2007. The Securities Purchase Agreement further provides that if a
registration statement is not filed, declared effective within specified time
periods or its effectiveness maintained, the Company is required to pay each
holder an amount in cash, as liquidated damages, equal to 1.5% per month of the
aggregate purchase price paid by such holder in the private placement for the
common stock and warrants then held.
On March
30, 2009, Aeolus entered into a Securities Purchase Agreement (the "Purchase
Agreement") with two accredited institutional investors (the "March
2009 Investors") pursuant to which the Company sold and issued to the March 2009
Investors in a private placement an aggregate of 5,357,143 units (the “March
2009 Units”), comprised of an aggregate of 5,357,143 shares of Common Stock of
the Company (the “Shares”) and warrants to purchase up to an aggregate of
13,392,857 additional shares of Common Stock (the “March 2009 Warrants”), with
an initial exercise price of $0.35 per share, subject to adjustment pursuant to
the March 2009 Warrants, with each March 2009 Unit representing one share of
Common Stock and a March 2009 Warrant to purchase two-and-one-half shares of
Common Stock, at a purchase price of $0.28 per March 2009 Unit for aggregate
gross proceeds of $1,500,000 (collectively, the “March 2009
Financing”). The March 2009 Warrants are exercisable for a five year
period from their date of issuance; contain a “cashless exercise” feature that
allows the holder to exercise the March 2009 Warrants without a cash payment to
the Company under certain circumstances; contain a dividend participation right
which allows the holder to receive any cash dividends paid on the Common Stock
without exercising the March 2009 Warrant and contain a provision that provides
for the reduction of the exercise price to $0.01 in the event of any such
payment of cash dividends by the Company; and contain standard anti-dilution
provisions that provide for the adjustment of the exercise price and the number
of shares of common stock that can be purchased in the event of a financing at a
price per share below the exercise price, a stock dividend or split, dividend
payment or other issuance, reorganization, recapitalization or similar
event.
The fair
value of the March 2009 Warrants was estimated to be $4,129,000 using the
Black-Scholes option pricing model with the following assumptions: dividend
yield of 0%; risk free interest rate of 1.7%; expected volatility of 164%; and
an expected life of five years.
In
connection with the March 2009 Financing, the Company also entered into a
Registration Rights Agreement (the “Rights Agreement”) with the March 2009
Investors. Pursuant to the Rights Agreement, the Company agreed to
file one or more registration statements (collectively, the “Registration
Statements”) with the Securities and Exchange Commission (the “SEC”) covering
the resale of the Shares and all shares of common stock issuable upon exercise
of the March 2009 Warrants (together with the Shares, the “Registrable
Securities”) upon demand of the holders of a majority of the Registrable
Securities (a “Demand Registration”). Such holders have
the right to two (2) Demand Registrations; provided, however, that the Company
is not obligated to effect more than one (1) Demand Registration within any
period of 12 consecutive months, subject to certain exceptions. In
the event the holders exercise their right to a Demand Registration, the Company
has agreed to file a Registration Statement to register the resale of the
Registrable Securities within a certain numbers of days after the request and to
use commercially reasonable efforts to cause the Registration Statement to be
declared effective by the SEC as soon as practicable after the filing
thereof. The Company also agreed to use its commercially reasonable
efforts to keep the Registration Statements effective for a specified
period.
Offering
costs of the private placement were $91,000 resulting in net proceeds to the
Company from the March 2009 Financing, after deducting for expenses, of
approximately $1.4 million. The Company intends to use the net proceeds from the
March 2009 Financing to finance the development of AEOL 10150 and to fund
ongoing operations of the Company.
Affiliates
of Xmark Opportunity Partners, LLC were the sole investors in the Financing.
Together with its affiliates, Xmark Opportunity Partners, LLC beneficially owned
approximately 57% of the Company's outstanding common stock prior to the March
2009 Financing.
Dividends
The
Company has never paid a cash dividend on its common stock and does not
anticipate paying cash dividends on its common stock in the foreseeable future.
If the Company pays a cash dividend on its common stock, it also must pay the
same
65
dividend
on an as converted basis on our Series B preferred stock. In
addition, Aeolus cannot pay a dividend on its common stock without the prior
approval of Goodnow Capital pursuant to the terms of the Debenture and Warrant
Purchase Agreement dated September 16, 2003 between the Company and Goodnow and
Xmark Opportunity Partners, LLC pursuant to the SCN Purchase
Agreement.
Warrants
As a
result of the March 2009 Financing, the Company was required to lower the
exercise price of 4,687,000 warrants previously issued in November 2005 and May
2007 to $0.28 per share, the purchase price of the March 2009 Units issued in
the March 2009 Financing. As a result of the change in the
exercise price, these warrants were revalued resulting in an increase in the
value of $38,000 which was charged to the statement of operations.
In
connection with the SCN Financing (Note F), Aeolus issued warrants to purchase
2,000,000 shares at an exercise price of $0.50 per share with a five year
term. The fair value of the warrants issued on August 1, 2008 was
estimated to be $282,000 using the Black-Scholes option pricing model with the
following assumptions: dividend yield of 0%; expected volatility of 128% risk
free interest rate of 3.2%; and an expected life of five years. The
fair value of the warrants issued on September 4, 2008 was estimated to be
$53,000 using the Black-Scholes option pricing model with the following
assumptions: dividend yield of 0%; expected volatility of 130% risk free
interest rate of 3.0%; and an expected life of five
years. The fair value of the warrants issued on October
1, 2008 was estimated to be $93,000 using the Black-Scholes option pricing model
with the following assumptions: dividend yield of 0%; expected volatility of
133% risk free interest rate of 2.9%; and an expected life of five
years. The fair value of the warrants issued on November
3, 2008 was estimated to be $76,000 using the Black-Scholes option pricing model
with the following assumptions: dividend yield of 0%; expected volatility of
139% risk free interest rate of 2.7%; and an expected life of five
years. The fair value of the warrants issued on December
1, 2008 was estimated to be $75,000 using the Black-Scholes option pricing model
with the following assumptions: dividend yield of 0%; expected volatility of
138% risk free interest rate of 1.7%; and an expected life of five
years.
As of
September 30, 2009, warrants for common stock outstanding were as
follows:
|
Number
of Shares
|
Exercise
Price
|
Expiration
Date
|
|||||
| 2,500,000 |
$
|
0.28
|
November
2010
|
||||
| 2,186,668 |
$
|
0.28
|
May
2012
|
||||
| 13,392,857 |
$
|
0.35
|
March
2014
|
||||
| 20,000 |
$
|
0.39
|
September
2014
|
||||
| 50,000 |
$
|
0.50
|
May
2011
|
||||
| 1,000,000 |
$
|
0.50
|
August
2013
|
||||
| 250,000 |
$
|
0.50
|
September
2013
|
||||
| 250,000 |
$
|
0.50
|
October
2013
|
||||
| 250,000 |
$
|
0.50
|
November
2013
|
||||
| 250,000 |
$
|
0.50
|
December
2013
|
||||
| 15,000 |
$
|
0.50
|
September
2014
|
||||
| 15,000 |
$
|
0.60
|
September
2014
|
||||
| 7,000,000 |
$
|
0.75
|
June
2011
|
||||
| 50,000 |
$
|
1.00
|
May
2011
|
||||
| 50,000 |
$
|
1.50
|
May
2011
|
||||
| 50,000 |
$
|
2.00
|
May
2011
|
||||
| 50,000 |
$
|
2.50
|
May
2011
|
||||
| 27,379,525 |
$
|
0.46
|
|||||
H.
Stock Compensation Plans
Stock
Option Plans
As an
integral component of a management and employee retention program designed to
motivate, retain and provide incentive to the Company’s management, employees
and key consultants, the Board of Directors approved the 2004 Stock Option Plan
(the “2004 Plan”) and reserved 5,000,000 shares of common stock for issuance
under the 2004 Plan. As of September 30, 2009, 750,909 shares were available to
be granted under the 2004 Plan. The exercise price of the incentive stock
options (“ISOs”) granted under the 2004 Plan must not be less than the fair
market value of the common stock as determined on the date of the grant. The
options may have a term up to 10 years. Options typically vest immediately or up
to one year following the date of the grant.
Under the
Company’s 1994 Stock Option Plan (the “1994 Plan”), incentive stock options or
non-qualified stock options to purchase 2,500,000 shares of Aeolus’ common stock
may be granted to employees, directors and consultants of the Company.
As
66
of
September 30, 2008, there were no shares available to be granted under the 1994
Plan. The exercise price of the ISOs granted under the 1994 Plan must
not be less than the fair market value of the common stock as determined on the
date of the grant. The options may have a term up to 10 years. Options typically
vest over three years following the date of the grant.
Stock
option activity under the 2004 Plan and 1994 Plan were as follows:
|
Shares
|
Weighted
Average Exercise Price
|
Weighted
Average Contractual Life
|
Intrinsic
Value
|
|||||||
|
Outstanding
at September 30, 2006
|
3,071,806
|
$
|
3.25
|
7.7
years
|
$
|
22
|
||||
|
Granted
|
1,315,000
|
$
|
0.68
|
10.0
years
|
---
|
|||||
|
Exercised
|
(20,833
|
)
|
$
|
1.00
|
8.6
years
|
(8)
|
||||
|
Cancelled
|
(492,356
|
)
|
$
|
0.65
|
||||||
|
Outstanding
at September 30, 2007
|
3,873,617
|
$
|
2.72
|
7.3
years
|
$
|
2
|
||||
|
Granted
|
475,000
|
$
|
0.35
|
10.0
years
|
---
|
|||||
|
Exercised
|
----
|
|||||||||
|
Cancelled
|
(113,336
|
)
|
$
|
0.87
|
||||||
|
Outstanding
at September 30, 2008
|
4,235,281
|
$
|
2.50
|
6.7
years
|
$
|
46
|
||||
|
Granted
|
1,992,750
|
$
|
0.33
|
10.0
years
|
117
|
|||||
|
Exercised
|
----
|
|||||||||
|
Cancelled
|
(51,766
|
)
|
$
|
32.36
|
---
|
|||||
|
Outstanding
at September 30, 2009
|
6,176,265
|
$
|
1.58
|
7.0
years
|
$
|
138
|
||||
|
Exercisable
at September 30, 2009
|
4,876,524
|
$
|
1.92
|
6.3
years
|
$
|
61
|
||||
Stock
options granted to consultants during fiscal year 2009, 2008 and 2007 were fully
vested when issued or vested over a twelve month period. Stock option
expense for stock options granted to consultants was $69,000, $77,000 and
$248,000 for fiscal year 2009, 2008 and 2007, respectively. For the
fiscal years ended September 30, 2009, 2008 and 2007, all stock options were
issued at or above the fair market value of a share of common
stock. The weighted-average grant-date fair value of options granted
during the fiscal years 2009, 2008 and 2007 was $0.30, $0.35 and $0.68,
respectively.
A summary
of the status of nonvested shares during the three years ended September 30,
2009 was:
|
Shares
|
|||
|
Nonvested
at September 30, 2006
|
539,586
|
||
|
Granted
|
1,315,000
|
||
|
Forfeited
|
(450,000
|
)
|
|
|
Vested
|
(867,086
|
)
|
|
|
Nonvested
at September 30, 2007
|
537,500
|
||
|
Granted
|
475,000
|
||
|
Vested
|
(701,667
|
)
|
|
|
Nonvested
at September 30, 2008
|
310,833
|
||
|
Granted
|
1,992,750
|
||
|
Vested
|
(1,003,842
|
)
|
|
|
Nonvested
at September 30, 2009
|
1,229,741
|
The total
deferred compensation expense for outstanding stock options was $335,000 as of
September 30, 2009, which will be recognized over a weighted average period of
eight months. The total fair value of shares vested during fiscal
years 2009, 2008 and 2007 was $321,000, $341,000 and $689,000,
respectively.
67
The
details of stock options outstanding at September 30, 2009 were as
follows:
|
Options
Outstanding
|
Options
Exercisable
|
|||||||||||
|
Range
of Exercise Prices
|
Number
Outstanding at September 30, 2009
|
Weighted
Average Exercise Price
|
Weighted
Average Remaining Contractual Life
|
Number
Exercisable at September 30, 2009
|
Weighted
Average Exercise Price
|
|||||||
|
$0.29
- $0.32
|
1,476,250
|
|
$
|
0.30
|
|
9.5
years
|
|
671,564
|
|
$
|
0.31
|
|
|
$0.33
- $0.45
|
993,500
|
|
$
|
0.39
|
|
9.4
years
|
|
498,445
|
|
$
|
0.39
|
|
|
$0.55
- $0.75
|
771,861
|
$
|
0.65
|
7.2
years
|
771,861
|
$
|
0.65
|
|||||
|
$0.78
- $0.90
|
889,335
|
$
|
0.87
|
6.7
years
|
889,335
|
$
|
0.87
|
|||||
|
$0.91
- $1.45
|
223,500
|
$
|
1.04
|
6.6
years
|
223,500
|
$
|
1.04
|
|||||
|
$1.50
|
1,256,019
|
$
|
1.50
|
3.8
years
|
1,256,019
|
$
|
1.50
|
|||||
|
$1.52
- $1.85
|
212,500
|
$
|
1.84
|
5.0
years
|
212,500
|
$
|
1.84
|
|||||
|
$2.10
- $8.13
|
173,018
|
$
|
4.20
|
4.2
years
|
173,018
|
$
|
4.20
|
|||||
|
$11.50
- $19.38
|
90,860
|
$
|
13.95
|
2.1
years
|
90,860
|
$
|
13.95
|
|||||
|
$20.00
- $51.25
|
89,422
|
$
|
35.54
|
0.9
years
|
89,422
|
$
|
35.54
|
|||||
|
$0.29
- $51.25
|
6,176,265
|
$
|
1.58
|
7.0
years
|
4,876,524
|
$
|
1.92
|
|||||
For
fiscal 2009, 2008 and 2007, stock-based compensation expense recognized in the
statement of operations is as follows (in thousands):
|
For
the fiscal year ended September 30,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
Research
and development expenses
|
$ | 38 | $ | 45 | $ | 177 | ||||||
|
General
and administrative expenses
|
245 | 296 | 539 | |||||||||
|
Total
stock-based compensation expense
|
$ | 283 | $ | 341 | $ | 716 | ||||||
The fair
value of the options associated with the above compensation expense for fiscal
2009, 2008 and 2007, was determined at the date of the grant using the
Black-Scholes option pricing model with the following weighted average
assumptions:
|
For
the fiscal year ended September 30,
|
||||||
|
2009
|
2008
|
2007
|
||||
|
Dividend
yield
|
0%
|
0%
|
0%
|
|||
|
Expected
volatility
|
111%
|
197%
- 198%
|
191%
- 195%
|
|||
|
Risk-free
interest rate
|
3.0%
|
3.8%
- 4.6%
|
4.5%
- 5.1%
|
|||
|
Expected
option life after shares are vested
|
9.3
years
|
10
years
|
10
years
|
|||
Effective
April 1, 2009, the Company began to use a peer group to determine an expected
volatility rate for stock option valuation. Prior to April 1, 2009,
the Company used its historical stock price to calculate
volatility. In addition, the Company changed its method
of amortization of stock based compensation from the multiple attribute method
to straight line for grants made subsequent to April 1, 2009. There
was no material impact on the financial statements as a result of these changes
as of April 1, 2009. The Company believes the use of the peer group
and straight line amortization results in a better estimate of stock based
compensation expense for the Company.
I.
Income Taxes
As of
September 30, 2009 and 2008, the Company had federal net operating loss (“NOL”)
carryforwards of $105,303,000 and $104,015,000, respectively and state operating
loss carryforwards of $28,715,000 and $27,429,000, respectively. The
use of these federal NOL carryforwards might be subject to limitation under the
rules regarding a change in stock ownership as determined by the Internal
Revenue Code (the “Code”). The Company may have had a change of control under
Section 382 of the Code during fiscal 2004 and 2006; however, a complete
analysis of the limitation of the NOL carryforwards will not be completed until
the time the Company projects it will be able to utilize such
NOLs. The federal net operating losses will begin to expire in 2010.
The state net operating losses begin to expire in fiscal year 2010.
Additionally, the Company had federal research and development carryforwards as
of September 30, 2009 and 2008 of $3,078,000 and $3,060,000,
respectively. The Company had
68
state
research and development carryforwards as of September 30, 2009 and 2008 of
$369,000 and $350,000, respectively.
Significant
components of the Company’s deferred tax assets at September 30, 2009 and 2008
consisted of the following (in thousands):
|
2009
|
2008
|
||||||
|
Net
operating loss carryforwards
|
$
|
38,341
|
|
$
|
37,790
|
||
|
Research
and development credit
carryforwards
|
3,446
|
3,410
|
|||||
|
Accrued
payroll related liabilities
|
2,542
|
2,420
|
|||||
|
Note
discount
|
322
|
10
|
|||||
|
Impairment
on marketable securities
|
—
|
21
|
|||||
|
Total
deferred tax assets
|
44,651
|
43,651
|
|||||
|
Deferred
tax liabilities
|
—
|
—
|
|||||
|
Valuation
allowance for deferred assets
|
(44,651
|
)
|
(43,651
|
)
|
|||
|
Net
deferred tax asset
|
$
|
—
|
$
|
—
|
|||
Due to
the uncertainty surrounding the realization of the favorable tax attributes in
future tax returns, all of the deferred tax assets have been fully offset by a
valuation allowance. The change in the valuation allowance is
primarily a result of the net operating loss carryforwards.
Taxes
computed at the statutory federal income tax rate of 34% are reconciled to the
provision for income taxes as follows (dollars in thousands):
|
2009
|
2008
|
2007
|
|||||||
|
Effective
income tax rate
|
0
|
%
|
0
|
%
|
0
|
%
|
|||
|
United
States Federal income tax at statutory rate
|
$
|
(781
|
)
|
$
|
(1,011
|
)
|
$
|
(1,028
|
)
|
|
State
income taxes (net of federal benefit)
|
(75
|
)
|
(142
|
)
|
(170
|
)
|
|||
|
Warrant
expense
|
13
|
40
|
—
|
||||||
|
Change
in valuation reserves
|
857
|
1,145
|
1,244
|
||||||
|
Other
|
(14)
|
(32)
|
(46)
|
||||||
|
Provision
for income taxes
|
$
|
—
|
$
|
—
|
$
|
—
|
|||
On
October 1, 2007, the Company adopted new accounting guidance which altered the
framework for recognizing income tax contingencies. Previously, the focus was on
the subsequent liability recognition for estimated losses from tax contingencies
where such losses were probable and the related amounts could be reasonably
estimated. Under this new guidance, a contingent tax asset (i.e., an uncertain
tax position) may only be recognized if it is more likely than not that it will
ultimately be sustained upon audit. The Company has evaluated its tax positions
for all jurisdictions and all years for which the statute of limitations remains
open and determined that no additional liability for unrecognized tax benefits
and interest was necessary.
J.
Agreements
Duke
Licenses
Aeolus
has obtained exclusive worldwide licenses (the “Duke Licenses”) from Duke
University (“Duke”) to develop, make, have made, use and sell products using
certain technology in the field of free radical and antioxidant research,
developed by certain scientists at Duke. Future discoveries in the
field of antioxidant research from these scientists’ laboratories at Duke are
also covered by the Duke Licenses. The Duke Licenses require Aeolus to use its
best efforts to pursue development of products using the licensed technology and
compounds. These efforts are to include the manufacture or production
of products for testing, development and sale. Aeolus is also obligated to use
its best efforts to have the licensed technology cleared for marketing in the
United States by the U.S. Food and Drug Administration and in other countries in
which Aeolus intends to sell products using the licensed
technology. Aeolus will pay royalties to Duke on net product sales
during the terms of the Duke Licenses, and milestone payments upon certain
regulatory approvals and annual sales levels. In addition, Aeolus is obligated
under the Duke Licenses to pay all or a portion of patent prosecution,
maintenance and defense costs. Unless earlier terminated, the Duke
Licenses continue until the expiration of the last to expire issued patent on
the licensed technology.
69
National
Jewish Medical and Research Center Agreements
Aeolus
has an exclusive worldwide license (“NJH License”) from National Jewish Health
(“NJH”) to develop, make, have made, use and sell products using certain
technology developed by certain scientists at NJH. The NJH License
requires Aeolus to use commercially reasonable efforts to diligently pursue the
development and government approval of products using the licensed
technology. Aeolus will pay royalties to NJH on net product sales
during the term of the NJH License and a milestone payment upon regulatory
approval. In addition, Aeolus is obligated under the NJH License to
pay all or a portion of patent prosecution, maintenance and defense
costs. Unless earlier terminated, the NJH License continues until the
expiration of the last to expire issued patent on the licensed
technology.
Elan
Corporation, plc
In May
2002, the Company entered into a collaboration transaction with affiliates of
Elan Corporation, plc for the development of our catalytic antioxidant compounds
as a treatment for tissue damage from cancer radiation and
chemotherapy. Although Elan and the Company terminated this
collaboration in January 2003, the Company will pay Elan a royalty on net sales
of our catalytic antioxidant products sold, if any, for the prevention and
treatment of radiation-induced and chemotherapy-induced tissue
damage.
K.
Quarterly Financial Data (unaudited)
|
First
Quarter
|
Second
Quarter
|
Third
Quarter
|
Fourth
Quarter
|
Total
Year
|
||||||||||||||||
|
(in
thousands, except per share amounts)
|
||||||||||||||||||||
|
Fiscal
2009
|
||||||||||||||||||||
|
Total
revenue
|
$ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||
|
Net
loss attributable to common stockholders
|
$ | (459 | ) | $ | (665 | ) | $ | (426 | ) | $ | (746 | ) | $ | (2,296 | ) | |||||
|
Basic
net loss per common share attributable to common
stockholders
|
$ | (0.01 | ) | $ | (0.02 | ) | $ | (0.01 | ) | $ | (0.02 | ) | $ | (0.07 | ) | |||||
|
Diluted
net loss per common share attributable to common
stockholders
|
$ | (0.02 | ) | $ | (0.02 | ) | $ | (0.01 | ) | $ | (0.02 | ) | $ | (0.07 | ) | |||||
|
Fiscal
2008
|
||||||||||||||||||||
|
Total
revenue
|
$ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||
|
Net
loss attributable to common stockholders
|
$ | (641 | ) | $ | (697 | ) | $ | (580 | ) | $ | (1,055 | ) | $ | (2,973 | ) | |||||
|
Basic
net loss per common share attributable to common
stockholders
|
$ | (0.02 | ) | $ | (0.02 | ) | $ | (0.02 | ) | $ | (0.03 | ) | $ | (0.09 | ) | |||||
|
Diluted
net loss per common share attributable to common
stockholders
|
$ | (0.02 | ) | $ | (0.02 | ) | $ | (0.02 | ) | $ | (0.05 | ) | $ | (0.11 | ) | |||||
L.
Subsequent Events
On
October 6, 2009, the Company entered into a Securities Purchase and Exchange
Agreement (the "October 2009 Purchase Agreement") with several accredited
institutional investors (the "October 2009 Investors") pursuant to
which the Company sold and issued to the October 2009 Investors in a private
placement an aggregate of 5,892,857 units (the “Units”), comprised of an
aggregate of 5,892,857 shares of Common Stock (the “Shares”) and warrants to
purchase up to an aggregate of 11,785,714 additional shares of Common Stock (the
“October 2009 Warrants”), with an initial exercise price of $0.28 per share,
subject to adjustment pursuant to the October 2009 Warrants, with each Unit
representing one share of Common Stock and a October 2009 Warrant to purchase
two shares of Common Stock, at a purchase price of $0.28 per Unit for aggregate
gross proceeds of $1,650,000 (collectively, the “October 2009
Financing”). The October 2009 Warrants are exercisable for a seven
year period from their date of issuance; contain a “cashless exercise” feature
that allows the holder to exercise the October 2009 Warrants without a cash
payment to the Company under certain circumstances; contain a dividend
participation right which allows the holder to receive any cash dividends paid
on the Common Stock without exercising the October 2009 Warrant and contain a
provision that provides for the reduction of the exercise price to $0.01 in the
event of any such payment of cash dividends by the Company or upon a change of
control and contain anti-dilution provisions in the event of a stock dividend or
split, dividend payment or other issuance, reorganization, recapitalization or
similar event.
The
Company also granted to the October 2009 Investors the option to acquire,
collectively, up to an additional 5,892,857 additional Units (the “Additional
Units”), comprised of an aggregate of 5,892,857 shares of Common Stock and
warrants to purchase up to an aggregate of 11,785,714 additional shares of
common stock at the per Additional Unit purchase price of $0.28 (the “Call
Option”). In addition, the October 2009 Investors granted to the Company the
option to require these October 2009 Investors, severally and not jointly, to
acquire up to 5,892,857 Additional Units, less any Additional Units
acquired
70
under the
Call Option, at the per Additional Unit purchase price of $0.28 (the “Put
Option”). The Call Option is exercisable at any time, and from time
to time, on or prior to June 30, 2010. The Put Option is exercisable
at any time from June 30, 2010 to July 30, 2010. However, the
Investors shall have the right to terminate the Put Option if they reasonably
determine that a material adverse event, condition or circumstance has occurred
with respect to the prospects of the Company’s AEOL 10150 drug candidate for
acute radiation syndrome; provided that the Company’s failure to receive a grant
or financing shall not, by itself, constitute a material adverse event,
condition or circumstance with respect thereto.
In
addition, the October 2009 Investors agreed to convert all $1,000,000 of the
Company’s Senior Convertible Notes issued in 2008 (the “Notes”) into Common
Stock at a conversion rate of $0.35 per share (the “Conversion Shares”) and to
exchange their remaining option to purchase an additional $4,000,000 in Senior
Convertible Notes for warrants to purchase up to 14,285,714 shares of Common
Stock in substantially the same of form and terms of the October 2009 Warrants
issued in the October 2009 Financing, including an initial exercise price of
$0.28 per share, subject to adjustment pursuant to the warrants (the “Note
Warrants”). As consideration for the October 2009 Investors to
convert the Notes, the Company agreed to exchange warrants to purchase up to
2,000,000 shares of Common Stock issued to the October 2009 Investors in
connection with the sale of the Notes, warrants to purchase up to 2,150,000
shares of Common Stock issued to the October 2009 Investors and one of their
affiliates in connection with a financing completed in November 2005 and
warrants to purchase up to 13,392,857 shares of Common Stock issued to the
October 2009 Investors in connection with a financing completed in March 2009
(collectively, the “Prior Warrants”) for warrants to purchase up to 17,542,857
shares of Common Stock in substantially the same form and terms of the October
2009 Warrants issued in the October 2009 Financing, including an initial
exercise price of $0.28 per share, subject to adjustment pursuant to the
warrants (the “Exchange Warrants”) (collectively, the
“Conversion”).
In
connection with the October 2009 Financing and the Conversion, the Company also
entered into a Registration Rights Agreement (the “October 2009 Rights
Agreement”) with the Investors. In addition, the Investors agreed to
terminate the Company’s Registration Rights Agreements dated November 21, 2005
and March 30, 2009. Pursuant to the October 2009 Rights Agreement,
the Company agreed to file one or more registration statements (collectively,
the “Registration Statements”) with the Securities and Exchange Commission (the
“SEC”) covering the resale of the Shares, the Conversion Shares and all shares
of common stock issuable upon exercise of the Warrants, the Note Warrants and
the Exchange Warrants (collectively, the “Registrable Securities”) upon demand
of the holders of a majority of the Registrable Securities (a “Demand
Registration”). Such holders have the right to two Demand
Registrations, subject to certain exceptions. In the event the
holders exercise their right to a Demand Registration, the Company has agreed to
file a Registration Statement to register the resale of the Registrable
Securities within a certain number of days after the request and to use
commercially reasonable efforts to cause the Registration Statement to be
declared effective by the SEC as soon as practicable after the filing
thereof. The Company also agreed to use its commercially reasonable
efforts to keep the Registration Statements effective for a specified
period.
The net
proceeds to the Company from the October 2009 Financing, after deducting for
expenses, were approximately $1.6 million. The Company intends to use the net
proceeds from the Financing to finance animal efficacy studies in Acute
Radiation Syndrome, the development of AEOL 10150 and ongoing operations of the
Company.
Affiliates
of Xmark Opportunity Partners, LLC are the sole investors in the Financing and,
together with the Company, are the sole participants in the Conversion. Together
with its affiliates, Xmark Opportunity Partners, LLC beneficially owned
approximately 71% of the Company's outstanding common stock prior to the
Financing and the Conversion. Xmark Opportunity Partners, LLC is the
sole manager of Goodnow Capital, L.L.C. and possesses sole power to vote and
direct the disposition of all securities of the Company held by Goodnow. Goodnow
has the right to designate up to two directors for election to the Company's
Board of Directors pursuant to the terms of a purchase agreement between Goodnow
and the Company. David C. Cavalier, a current director of the Company, is
President of Goodnow.
On
October 1, 2009, the Company adopted EITF 07-5 and applied its provisions to
outstanding instruments as of that date. The fair value of the
warrants affected by EITF 07-05 at the dates of issuance totaling $8,282,000 was
initially recorded as a component of additional paid-in capital. Upon adoption
of EITF 07-5, in the first quarter of 2010, the Company recorded a decrease to
the opening balance of additional-paid-in capital of $8,142,000 and recorded a
decrease to accumulated deficit totaling $4,353,000, representing the decrease
in the fair value of the warrants from the date of issuance to October 1,
2009. The fair value of the warrants at October 1, 2009 of $3,789,000
will be included as a current liability in the balance sheet as of that
date. Any future increase or decrease in fair value of the warrants
will be included as a component of other income in the accompanying statement of
operations for the respective period. As of October 31, 2009, the
liability for warrants increased to $17,272,000 resulting in an additional
charge to the statement of operations of $6,250,000. As of November
30, 2009, the liability for warrants decreased to $15,389,000 resulting in a
gain in the statement of operations of $1,882,000.
On
December 24, 2009, the Company entered into an amendment (the “Amendment”) to
the Securities Purchase and Exchange Agreement dated October 6, 2009 (the
“Agreement”) pursuant to which the Company agreed to lower the
conversion price of the Company’s Senior Convertible Notes issued in 2008 (the
“Notes”) from $0.35 per share to $0.28 per share and as a result, issued to the
investors in the Company’s October 2009 financing (the “Financing”) an
additional 714,286 shares of the Company’s Common Stock upon conversion of the
Notes (the “Issuance”). The Agreement was executed to resolve a
misunderstanding regarding one of the Financing terms between the Company and
the investors in the Financing. The Company
71
will not
receive any proceeds from the Issuance.
We have
evaluated subsequent events through the issuance of the financial statements,
which occurred on December 28, 2009.
Item
9. Changes in and Disagreements with Accountants on Accounting and Financial
Disclosure.
None
Item
9A. Controls and Procedures.
(a) As
of the end of the period covered by this report, the Company carried out an
evaluation, under the supervision and with the participation of the Company’s
management, including the Company’s President and Chief Executive Officer (the
Company’s Principal Executive Officer) and Chief Financial Officer (the
Company’s Principal Financial and Accounting Officer), of the effectiveness of
the Company’s disclosure controls and procedures required by Exchange Act Rule
13a-15. Based upon that evaluation, the Company’s Principal Executive
Officer and Principal Financial and Accounting Officer have concluded that the
Company’s disclosure controls and procedures were not effective as of September
30, 2009 to provide reasonable assurance that information required to be
disclosed by us in reports that we file or submit under the Exchange Act is
recorded, processed, summarized and reported within the time periods specified
in SEC rules and forms because of the material weakness discussed
below.
(b) During
the most recent fiscal quarter, there were no significant changes in the
Company’s internal control over financial reporting or in other factors that
materially affected or are reasonably likely to materially affect the Company’s
internal control over financial reporting.
(c) Management’s Report on Internal
Control over Financial Reporting
Management
of Aeolus Pharmaceuticals, Inc. (“Aeolus” or the “Company”) is
responsible for establishing and maintaining adequate internal control over
financial reporting as defined in Rule 13A-15 (f) and 15d-15 (f) of the
Securities and Exchange Act of 1934, as amended. The Company’s
internal control over financial reporting is a process designed to provide
reasonable assurance to the Company’s Management and Board of Directors
regarding the reliability of financial reporting and the preparation of
financial statements for external purposes in accordance with generally accepted
accounting principles and includes policies and procedures that:
|
|
·
|
pertain
to the maintenance of records that, in reasonable detail, accurately and
fairly reflect the transactions and dispositions of the assets of the
Company;
|
|
|
·
|
provide
reasonable assurance that transactions are recorded as necessary to permit
preparation of financial statements in accordance with generally accepted
accounting principles, and that receipts and expenditures of the Company
are being made only in accordance with authorizations of management and
directors of the Company; and
|
|
|
·
|
provide
reasonable assurance regarding prevention or timely detection of
unauthorized acquisition, use, or disposition of the company’s assets that
could have a material effect on the financial
statements.
|
Because
of its inherent limitations, internal control over financial reporting may not
prevent or detect misstatements, errors or fraud. Also, projections
of any evaluations of effectiveness to future periods are subject to the risk
that controls may become inadequate because of changes in conditions, or that
the degree of compliance with the policies or procedures may
deteriorate.
Management
assessed the effectiveness of the Company’s internal control over financial
reporting as of September 30, 2009. In making this assessment,
management used criteria set forth by the Committee of Sponsoring Organizations
of the Treadway Commission (COSO) in an Internal Control Integrated
Framework. As a result of the lack of segregation of duties,
management has determined that a material weakness in internal control
over financial reporting related to the segregation of duties existed as of
September 30, 2009, and based on the criteria set forth by COSO, concluded that
the Company’s internal control over financial reporting was not effective as of
September 30, 2009.
A
“material weakness,” as defined by the Public Company Accounting Oversight Board
(PCAOB) is a deficiency, or a combination of deficiencies, in internal control
over financial reporting, such that there is a reasonable possibility that a
material misstatement of the Company’s annual or interim financial statements
will not be prevented or detected on a timely basis.
To
address the material weakness, management has established mitigating controls to
minimize the potential for material misstatements in the financial
statements. Management has determined that given the Company’s size,
level of operations and financial resources, it is not practicable for the
Company to eliminate the segregation of controls weakness and therefore
management has established mitigating controls to minimize the impact of the
lack of segregation of duties.
This
annual report does not include an attestation report of the company's registered
public accounting firm regarding internal control over financial reporting.
Management's report was not subject to attestation by the company's registered
public accounting firm pursuant to temporary rules of the Securities and
Exchange Commission that permit the company to provide only management's report
in this annual report.
72
Item
9B. Other Information.
None.
PART
III
Certain
information required by Part III is omitted from this report because the
Registrant expects to file a definitive information statement in connection with
the registrant’s Written Consent in Lieu of 2010 Annual Meeting of Stockholders
(the “Information Statement”) within 120 days after the end of its fiscal year
pursuant to Regulation 14C promulgated under the Securities Exchange Act of
1934, as amended, and the information included therein is incorporated herein by
reference to the extent provided below.
Item
10. Directors and Executive Officers of the Registrant.
The
information set forth under the headings "Proposal
No. 1: Election of Directors," "Corporate Governance" and
"Section 16(a) Beneficial Ownership Reporting Compliance" in our definitive
Information Statement is incorporated herein by reference. The
information required by this Item 10 concerning the Registrant’s executive
officers is set forth under the heading “Executive Officers” located at the end
of Part I Item 1 of this Form 10-K.
Code
of Ethics.
We have
adopted a code of ethics that applies to our principal executive officer,
principal financial officer, principal accounting officer and persons performing
similar functions. We have posted the text of Code of Ethics on our
Internet website at www.aeoluspharma.com. A
copy of the Code of Ethics can also be obtained free of charge by writing to
Michael P. McManus, Corporate Secretary, Aeolus Pharmaceuticals, Inc., 26361
Crown Valley Parkway, Suite 150 Mission Viejo, CA 92691.
Item
11. Executive Compensation.
The
information set forth under the headings "Compensation Discussion and Analysis,"
"Compensation Committee Report," "Compensation Committee Interlocks and Insider
Participation," "Executive Compensation" and "Director Compensation" in our
definitive Information Statement is incorporated herein by
reference.
Item
12. Security Ownership of Certain Beneficial Owners and Management and Related
Stockholder Matters.
The
information set forth under the headings “Other Information — Principal
Stockholders” and "Equity Compensation Plan Information" in our definitive
Information Statement is incorporated herein by reference.
Item
13. Certain Relationships and Related Transactions.
The
information set forth under the headings "Information Concerning the Board of
Directors and its Committees" and "Certain Related Transactions” in our
definitive Information Statement is incorporated herein by
reference.
Item
14. Principal Accountant Fees and Services.
The information set forth under the
heading “Independent Registered Public Accounting Firm — Fees” in our definitive
Information Statement is incorporated herein by reference.
PART
IV
Item
15. Exhibits and Financial Statement Schedules.
(a) The
following financial statement schedules and exhibits are filed as part of this
report or incorporated herein by reference:
(1) Financial
Statement Schedules.
All
financial statement schedules for which provision is made in Regulation S-X are
omitted because they are not required under the related instructions, are
inapplicable, or the required information is given in the financial statements,
including the notes thereto.
73
(2) Exhibits.
|
Incorporated
by Reference To
|
|||||||||||||||
|
Exhibit
Number
|
Description
of Document
|
Registrant’s
Form
|
Dated
|
Exhibit
Number
|
Filed
Herewith
|
||||||||||
|
2.1
|
|
Agreement
and Plan of Merger and Reorganization dated September 16, 2003 between
Incara, Inc. and Incara Pharmaceuticals Corporation
|
S-4
|
09/19/03
|
2.1
|
||||||||||
|
3.1
|
Certificate
of Incorporation, as amended
|
10-Q
|
06/30/04
|
3.1
|
|||||||||||
|
3.2
|
Certificate
of Amendment of Amended and Restated Certificate of
Incorporation
|
8-K
|
3/27/06
|
3.1
|
|||||||||||
|
3.3
|
Certificate
of Amendment of Amended and Restated Certificate of
Incorporation
|
8-K
|
10/27/06
|
3.1
|
|||||||||||
|
3.4
|
Bylaws,
as amended
|
8-K
|
10/25/05
|
3.1
|
|||||||||||
|
3.5
|
Certificate
of Designations, Preferences and Rights of Series A Convertible Preferred
Stock of the Company dated November 18, 2005
|
8-K
|
11/23/05
|
3.1
|
|||||||||||
|
4.1
|
Form
of Common Stock Certificate
|
10-Q
|
06/30/04
|
4.1
|
|||||||||||
|
4.2
|
Form
of Series B Preferred Stock Certificate
|
S-4
|
09/19/03
|
4.8
|
|||||||||||
|
4.3
|
Form
of Warrant to Purchase Common Stock dated November 21,
2005
|
8-K
|
11/23/05
|
10.2
|
|||||||||||
|
4.4
|
Form
of Warrant to Purchase Common Stock dated June 5, 2006.
|
8-K
|
6/5/06
|
10.3
|
|||||||||||
|
4.5
|
Warrant
to Purchase Common Stock dated June 5, 2006 issued to Efficacy Biotech
Master Fund Ltd.
|
8-K
|
6/5/06
|
10.4
|
|||||||||||
|
4.6
|
Registration
Rights Agreement dated May 22, 2007 by and among the Company and each of
the Purchasers whose names appear on the Schedule attached
thereto.
|
8-K
|
5/22/07
|
4.1
|
|||||||||||
|
4.7
|
Registration
Rights Agreement dated October 6, 2009 by and among the Company and the
investors whose names appear on the signature pages
thereof.
|
8-K
|
10/6/09
|
4.1
|
|||||||||||
|
4.8
|
Form
of Warrant to Purchase Common Stock dated May 22, 2007.
|
8-K
|
5/22/07
|
10.2
|
|||||||||||
|
4.9
|
Form
of Warrant to Purchase Common Stock
|
8-K
|
10/6/09
|
10.2
|
|||||||||||
|
10.1*
|
License
Agreement between Duke University and Aeolus Pharmaceuticals, Inc., dated
July 21, 1995
|
S-1
|
12/08/95
|
10.4
|
|||||||||||
|
10.2
|
Amended
and Restated Limited Liability Company Agreement of CPEC LLC dated July
15, 1999, among CPEC LLC, Intercardia, Inc. and Interneuron
Pharmaceuticals, Inc.
|
8-K
|
07/23/99
|
10.42
|
|||||||||||
|
10.3
|
Assignment,
Assumption and License Agreement dated July 15, 1999, between CPEC LLC and
Intercardia, Inc.
|
8-K
|
07/23/99
|
10.43
|
|||||||||||
|
10.4*
|
License
Agreement dated January 19, 2001 between Incara Pharmaceuticals
Corporation and Incara Development, Ltd.
|
10-Q
|
12/31/00
|
10.59
|
|||||||||||
|
10.5*
|
License
Agreement dated January 19, 2001 between Elan Corporation, plc, Elan
Pharma International Ltd. and Incara Development, Ltd.
|
10-Q
|
12/31/00
|
10.60
|
|||||||||||
|
10.6
|
Registration
Rights Agreement dated December 21, 2000 among Incara Pharmaceuticals
Corporation, Elan International Services, Ltd. and Elan Pharma
International Ltd.
|
10-Q
|
12/31/00
|
10.62
|
|||||||||||
|
10.7
|
Agreement
and Amendment, effective as of January 22, 2001, by and among Incara
Pharmaceuticals Corporation, Elan International Services, Ltd. and Elan
Pharma International Limited
|
10-Q
|
03/31/01
|
10.64
|
|||||||||||
|
10.8
|
Second
Agreement and Amendment, effective as of January 22, 2001, by and among
Incara Pharmaceuticals Corporation, Elan International Services, Ltd. and
Elan Pharma International Limited
|
10-Q
|
03/31/01
|
10.65
|
|||||||||||
|
10.9
|
Third
Agreement and Amendment, effective as of January 22, 2001, by and among
Incara Pharmaceuticals Corporation, Elan International Services, Ltd. and
Elan Pharma International Limited
|
8-K
|
06/01/01
|
10.66
|
|||||||||||
|
10.10
|
Agreement
and Fourth Amendment, effective February 13, 2002, by and among Incara
Pharmaceuticals Corporation, Elan International Services, Ltd., Elan
Pharma International Limited and Elan Pharmaceutical Investments III,
Ltd.
|
10-Q
|
12/31/01
|
10.75
|
|||||||||||
|
10.11*
|
License
Agreement dated June 25, 1998 between Duke University and Aeolus
Pharmaceuticals, Inc.
|
10-Q
|
03/31/02
|
10.82
|
|||||||||||
|
10.12*
|
License
Agreement dated May 7, 2002 between Duke University and Aeolus
Pharmaceuticals, Inc.
|
10-Q
|
03/31/02
|
10.83
|
|||||||||||
|
10.13*
|
License
Agreement dated November 17, 2000 between National Jewish Medical and
Research Center and Aeolus Pharmaceuticals, Inc.
|
10-Q
|
12/31/00
|
10.56
|
|||||||||||
|
10.14*
|
Securities
Purchase Agreement dated as of May 15, 2002, among Incara Pharmaceuticals
Corporation, Aeolus Pharmaceuticals, Inc., Elan Pharma International
Limited and Elan International Services, Ltd.
|
8-K
|
07/03/02
|
10.84
|
|||||||||||
|
10.15*
|
Development
and Option Agreement dated May 15, 2002, among Elan Pharma International
Limited, Incara Pharmaceuticals Corporation and Aeolus Pharmaceuticals,
Inc.
|
8-K
|
07/03/02
|
10.85
|
|||||||||||
|
10.16
|
Amended
and Restated Registration Rights Agreement dated as of May 15, 2002, among
Incara Pharmaceuticals Corporation, Elan International Services, Ltd. and
Elan Pharma International Limited
|
8-K
|
07/03/02
|
10.86
|
|||||||||||
|
10.17
|
Amendment
No. 1 to License Agreement dated May 14, 2002, between Aeolus
Pharmaceuticals, Inc. and Duke University (amending License Agreement
dated July 21, 1995)
|
8-K
|
07/03/02
|
10.87
|
|||||||||||
|
10.18
|
Amendment
No. 1 to License Agreement dated May 14, 2002, between Aeolus
Pharmaceuticals, Inc. and Duke University (amending License Agreement
dated June 25, 1998)
|
8-K
|
07/03/02
|
10.88
|
|||||||||||
|
10.19
|
Amendment
No. 1 to License Agreement dated May 14, 2002, between Aeolus
Pharmaceuticals, Inc. and National Jewish Medical and Research Center
(amending License Agreement dated November 17, 2000)
|
8-K
|
07/03/02
|
10.89
|
|||||||||||
|
10.20
|
Convertible
Secured Promissory Note dated July 28, 2003 issued by Incara, Inc. to
Goodnow Capital, Inc.
|
10-Q
|
06/30/03
|
10.97
|
|||||||||||
|
10.21
|
Guaranty
dated July 28, 2003 issued by Incara Pharmaceuticals Incorporation to
Goodnow Capital, Inc.
|
10-Q
|
06/30/03
|
10.98
|
|||||||||||
|
10.22
|
Security
Agreement dated July 28, 2003 issued by Incara Pharmaceuticals
Incorporation to Goodnow Capital, Inc.
|
10-Q
|
06/30/03
|
10.90
|
|||||||||||
|
10.23
|
Debenture
and Warrant Purchase Agreement dated September 16, 2003 among Incara
Pharmaceuticals Corporation, Incara, Inc. and Goodnow Capital,
L.L.C.
|
S-4
|
09/19/03
|
10.100
|
|||||||||||
|
10.24
|
Registration
Rights Agreement dated September 16, 2003 among Incara Pharmaceuticals
Corporation, Incara, Inc. and Goodnow Capital, L.L.C.
|
S-4
|
09/19/03
|
10.101
|
|||||||||||
|
10.25
|
Amendment
No. 1 to Debenture and Warrant Purchase Agreement dated September 16, 2003
among Incara Pharmaceuticals Corporation, Incara, Inc. and Goodnow
Capital, L.L.C.
|
8-K
|
04/21/04
|
10.104
|
|||||||||||
|
10.26
|
Letter
dated May 17, 2004 from Elan International Services, Limited and Elan
Pharma International Limited to Incara Pharmaceuticals
Corporation
|
10-Q
|
06/30/04
|
10.106
|
|||||||||||
|
10.27+
|
Aeolus
Pharmaceuticals, Inc. 1994 Stock Option Plan, as amended
|
10-Q
|
06/30/04
|
10.109
|
|||||||||||
|
10.28+
|
Aeolus
Pharmaceuticals, Inc. Amended and Restated 2004 Stock Incentive
Plan
|
S-8
POS
|
3/31/08
|
99.1
|
|||||||||||
|
10.29+
|
Employment
Agreement dated July 14, 2006 between Aeolus Pharmaceuticals, Inc. and
John L. McManus
|
8-K
|
7/14/06
|
10.1
|
|||||||||||
|
10.30+
|
Letter
Agreement dated July 10, 2006 between Aeolus Pharmaceuticals, Inc. and
McManus & Company, Inc.
|
8-K
|
7/10/06
|
10.2
|
|||||||||||
|
10.31+
|
Form
of Indemnity Agreement
|
8-K
|
2/18/05
|
10.118
|
|||||||||||
|
10.32
|
Terms
of Outside Director Compensation
|
10-K
|
12/17/04
|
10.114
|
|||||||||||
|
10.33+
|
Form
of Incentive Stock Option Agreement
|
10-Q
|
2/8/05
|
10.115
|
|||||||||||
|
10.34+
|
Form
of Nonqualified Stock Option Agreement
|
10-Q
|
2/8/05
|
10.116
|
|||||||||||
|
10.35
|
Purchase
Agreement dated November 21, 2005 by and among the Company and the
investors whose names appear on the signature pages
thereof
|
8-K
|
11/23/05
|
10.1
|
|||||||||||
|
10.36
|
Subscription
Agreement dated June 5, 2006 by and between the Company and the investors
whose names appear on the signature pages thereof.
|
8-K
|
6/5/06
|
10.1
|
|||||||||||
|
10.37
|
Board
Observer Letter dated June 5, 2006 by and among the Company and Efficacy
Biotech Master Fund Ltd.
|
8-K
|
6/5/06
|
10.6
|
|||||||||||
|
10.38
|
Securities
Purchase Agreement dated May 22, 2007 by and among the Company and the
investors whose names appear on the signature pages
thereof.
|
8-K
|
5/22/07
|
10.1
|
|||||||||||
|
10.39
|
Convertible
Promissory Note dated February 7, 2007 issued by Aeolus Pharmaceuticals,
Inc. to Elan Pharma International Ltd.
|
S-1
|
6/4/07
|
10.43
|
|||||||||||
|
10.40
|
Amendment
No. 1 To Convertible Promissory Note dated February 7, 2009 by and between
Aeolus Pharmaceuticals, Inc. and Elan Pharma International
Limited
|
8-K
|
3/16/09
|
10.1
|
|||||||||||
|
10.41+
|
Form
of Restricted Share Award Agreement
|
S-8
POS
|
3/31/08
|
99.2
|
|||||||||||
|
10.42
|
Securities
Purchase and Exchange Agreement dated October 6, 2009 by and among the
Company and the investors whose names appear on the signature pages
thereof
|
8-K
|
10/6/09
|
10.1
|
|||||||||||
| 10.43 | Amendment Agreement to the Securities Purchase and Exchange Agreement dated December 24, 2009 by and among the Company and the investors whose names appear on the signature pages thereof | 8-K | 12/28/09 | 10.1 | |||||||||||
|
14.1
|
Aeolus
Pharmaceuticals, Inc. Code of Ethics for Chief Executive Officer and
Senior Financial Officers, as amended on December 13, 2004
|
8-K
|
12/14/04
|
10.113
|
|||||||||||
|
21.1
|
List
of Subsidiaries
|
X
|
|||||||||||||
|
23.1
|
Consent
of Haskell & White LLP, Independent Registered Public Accounting
Firm
|
X
|
|||||||||||||
|
31.1
|
Certification
of the Principal Executive Officer pursuant to Rule 13a-14(a)
and 15d-14(a)
|
X
|
|||||||||||||
|
31.2
|
Certification
of the Principal Financial and Accounting Officer pursuant to Rule
13a-14(a) and 15d-14(a)
|
X
|
|||||||||||||
|
32.1
|
Certification
by the Principal Executive Officer and Principal Financial and Accounting
Officer pursuant to 18 U.S.C. 1350 as adopted pursuant to Section 906 of
the Sarbanes-Oxley Act of 2002
|
X
|
|||||||||||||
——————
* The
Company has received confidential treatment of certain portions of this
agreement which have been omitted and filed separately with the U.S. Securities
and Exchange Commission.
+ Indicates
management contract or compensatory plan or arrangement.
SIGNATURES
Pursuant
to the requirements of Section 13 or 15(d) of the Securities Exchange Act of
1934, the registrant has duly caused this report to be signed on its behalf by
the undersigned, thereunto duly authorized.
|
AEOLUS
PHARMACEUTICALS, INC.
|
||
|
By:
|
/s/
John L. McManus
|
|
|
John
L. McManus
President
and Chief Executive Officer
|
Date:
December 28, 2009
Pursuant
to the requirements of the Securities Exchange Act of 1934, this report has been
signed below by the following persons on behalf of the registrant and in the
capacities and on the dates indicated.
|
Signatures
|
|
Title
|
|
Date
|
|
/s/
John L. McManus
|
President
and Chief Executive Officer
|
December
28, 2009
|
||
|
John
L. McManus
|
(Principal
Executive Officer)
|
|||
|
/s/
Michael P. McManus
|
Chief
Financial Officer, Treasurer and Secretary
|
December
28, 2009
|
||
|
Michael
P. McManus
|
(Principal
Financial and Accounting Officer)
|
|||
|
/s/
David C. Cavalier
|
Chairman
of the Board of Directors
|
December
28, 2009
|
||
|
David
C. Cavalier
|
||||
|
/s/
John M. Farah, Jr.
|
Director
|
December
28, 2009
|
||
|
John
M. Farah, Jr., Ph.D.
|
||||
|
/s/
Joseph J. Krivulka
|
Director
|
December
28, 2009
|
||
|
Joseph
J. Krivulka
|
||||
|
/s/
Amit Kumar
|
Director
|
December
28, 2009
|
||
|
Amit
Kumar, Ph.D.
|
||||
|
/s/
Michael E. Lewis
|
Director
|
December
28, 2009
|
||
|
Michael
E. Lewis, Ph.D.
|
||||
|
/s/
Chris A. Rallis
|
Director
|
December
28, 2009
|
||
|
Chris
A. Rallis
|
||||
|
/s/
Peter D. Suzdak
|
Director
|
December
28, 2009
|
||
|
Peter
D. Suzdak, Ph.D.
|
78