Attached files
U.S. SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(MARK ONE)
|
[X]
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES
EXCHANGE ACT OF 1934
|
For the Fiscal Year Ended February 28, 2018
OR
|
[ ]
|
TRANSITION REPORT PURSUANT TO SECTION 13 or 15(d) OF THE SECURITIES
EXCHANGE ACT OF 1934
|
For the Transition Period from _______________ to
_________________
Commission file number: 000-52735
METASTAT, INC.
(Exact name of Registrant as Specified in Its Charter)
|
NEVADA
|
20-8753132
|
|
(State or Other Jurisdiction
of Incorporation or Organization)
|
(I.R.S. Employer Identification No.)
|
|
|
|
|
27 Drydock Ave., 2nd Floor
|
|
|
Boston, Massachusetts
|
02210
|
|
(Address of principal executive offices)
|
(Zip Code)
|
Registrant’s telephone number, including area code:
(617)
531-6500
SECURITIES REGISTERED PURSUANT TO SECTION 12 (B) OF THE
ACT: NONE
SECURITIES REGISTERED PURSUANT TO SECTION 12 (G) OF THE
ACT:
COMMON STOCK, PAR VALUE $0.0001 PER SHARE
Name of each exchange on which registered: The OTCQB
marketplace
Indicate by check mark if the registrant is a well-known seasoned
issuer, as defined in Rule 405 of the Securities
Act. Yes ☐
No ☒
Indicate by check mark if the registrant is not required to file
reports pursuant to Section 13 or Section 15(d) of the
Act. Yes ☐
No ☒
Indicate by check mark whether the registrant (1) has filed
all reports required to be filed by Section 13 or 15(d) of the
Exchange Act of 1934 during the preceding 12 months (or for such
shorter period that the registrant was required to file such
reports), and (2) has been subject to such filing requirements
for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted
electronically and posted on its corporate Web site, if any, every
Interactive Data File required to be submitted and posted pursuant
to Rule 405 of Regulation S-T (§229.405 of this chapter)
during the preceding 12 months (or for such shorter period that the
registrant was required to submit and post such files).
Yes ☒
No ☐
Indicate by check mark if disclosure of delinquent filers pursuant
to Item 405 of Regulation S-K (§ 229.405) is not contained
herein, and will not be contained, to the best of
registrant’s knowledge, in definitive proxy or information
statements incorporated by reference in Part III of this Form 10-K
or any amendment to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large
accelerated filer, an accelerated filer, a non-accelerated filer,
smaller reporting company, or an emerging growth company.
See the definitions of “large
accelerated filer,” “accelerated filer,”
“smaller reporting company,” and “emerging growth
company” in Rule 12b-2 of the Exchange Act. (Check
one):
|
Large accelerated filer
|
☐
|
Non-accelerated filer
|
☐
|
|
Accelerated filer
|
☐
|
Smaller reporting company
|
☒
|
|
|
|
Emerging
growth company
|
☐
|
(Do not check if a smaller reporting company)
If an emerging growth company, indicate by check mark if the
registrant has elected not to use the extended transition period
for complying with any new or revised financial accounting
standards provided pursuant to Section 13(a) of the Exchange
Act. ☐
Indicate by check mark whether the registrant is a shell company
(as defined in Rule 12b-2 of the Exchange
Act). Yes ☐ No ☒
The aggregate market value of the shares of common stock, par value
$0.0001 per share, of the registrant held by non-affiliates on
August 31, 2016 was approximately $4.2 million, which was computed
upon the basis of the closing price on that date.
There were 5,877,383 shares of common stock of the registrant
outstanding as of May 18, 2018.
TABLE OF
CONTENTS
|
|
|
||
|
|
|
|
|
|
|
2
|
||
|
|
35
|
||
|
|
69
|
||
|
|
69
|
||
|
|
69
|
||
|
|
69
|
||
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
70
|
||
|
|
73
|
||
|
|
73
|
||
|
|
81
|
||
|
|
81
|
||
|
|
81
|
||
|
|
82
|
||
|
|
82
|
||
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
83
|
||
|
|
88
|
||
|
|
94
|
||
|
|
95
|
||
|
|
95
|
||
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
96
|
||
INTRODUCTORY NOTE
Except as otherwise indicated by the context, references in this
Annual Report on Form 10-K (this “Form 10-K”) to the
“Company,” “MetaStat,” “we,”
“us” or “our” are references to the
combined business of MetaStat, Inc., a Nevada corporation, and its
consolidated subsidiary.
Special Note Regarding Forward-Looking Statements
The statements contained in this Form 10-K, including under the
section titled "Management's Discussion and Analysis of Financial
Condition and Results of Operations" and other sections of this
Form 10-K, include forward-looking statements within the meaning of
Section 27A of the Securities Act of 1933, as amended, and Section
21E of the Securities Exchange Act of 1934, as amended, including,
without limitation, statements regarding our or our management's
expectations, hopes, beliefs, intentions or strategies regarding
the future. The words "believe," "may," "will," "estimate,"
"continue," "anticipate," "intend," "expect," "plan" and similar
expressions may identify forward-looking statements, but the
absence of these words does not mean that a statement is not
forward-looking. The forward-looking statements contained in this
Form 10-K are based on our current expectations and beliefs
concerning future developments and their potential effects on us.
There can be no assurance that future developments affecting us
will be those that we have anticipated. These forward-looking
statements involve a number of risks, uncertainties (some of which
are beyond our control) or other assumptions that may cause actual
results or performance to be materially different from those
expressed or implied by these forward-looking statements. These
risks and uncertainties include, but are not limited to, those
factors described in the section titled "Risk Factors." Should one
or more of these risks or uncertainties materialize, or should any
of our assumptions prove incorrect, actual results may vary in
material respects from those projected in these forward-looking
statements. We undertake no obligation to update or revise any
forward-looking statements, whether as a result of new information,
future events or otherwise, except as may be required under
applicable securities laws.
PART I
|
Item 1.
|
BUSINESS
|
Overview
MetaStat
is a precision medicine company dedicated to improving survival of
patients with aggressive cancer. Despite significant advances in
the war against cancer, effective treatment and prevention of
cancer metastasis remains an unmet medical need. The metastatic spread of cancer is
responsible for over 90% of solid tumor cancer-related deaths. Our
therapeutic focus targets a critical metastatic pathway in solid
tumors responsible for driving tumor resistance and the spread of
aggressive cancer. We are leveraging our core expertise in
understanding the tumor microenvironment and drivers of the
metastatic cascade to develop anti-metastatic therapeutics pared
with companion diagnostics tests. MetaStat’s goal is to
transform the treatment of aggressive cancer into a manageable
disease through therapies that target
the metastatic spread of cancer and improve
survival.
MetaStat’s
drug discovery program is focused on developing novel
first-in-class drug candidates that target the MENA pathway.
Elevated expression of the MENA protein splice-derived variants or
isoforms in tumors have been shown in clinical studies to be
significantly associated with metastasis and decreased survival. We
are discovering and developing therapeutic product candidates based
on a different approach making the MENA protein a druggable target.
We are not targeting MENA directly, but rather the MAPKAPK2 pathway
responsible for activation of MENA. Our diagnostic tests are
clinically validated as prognostic markers of metastasis and
identify patients at risk for recurrence who are likely to benefit
from our anti-metastatic therapy.
In
2017, we demonstrated inhibition of the MAPKAPK2 kinase reverses
MENA-induced metastatic phenotypes to MENA-null levels in studies
of metastatic Triple Negative Breast Cancer (TNBC) and fibroblast
cell lines. In February 2018, we announced top-line results from
preclinical studies showing dissemination and cancer metastasis was
reduced by 50% or more in
vivo following inhibition of MAPKAPK2 in preclinical models
of aggressive breast cancer. Administration of the MAPKAPK2
inhibitor alone significantly blocked distant metastasis similar to
the published effects of MENA deficiency in the MMTV-PyMT mouse
model. MAPKAPK2 is a downstream terminal serine-threonine kinase in
the p38/MAPK signaling pathway and is associated with inflammatory
disease, metastasis and in the resistance mechanism to antitumor
agents. Several biopharmaceutical companies have advanced p38
inhibitors into clinical studies, however, none have been approved
due to adverse side effects likely associated with the role of p38
in the regulation of more than 60 substrates with different
physiological functions. We believe MAPKAPK2 is a more attractive
drug target because it is a terminal kinase with fewer substrates
and is downstream of p38 in the p38/MAPK pathway. The goal of our
small molecule discovery program over the next 12-18 months is to
identify 1-2 lead candidates to advance into investigational new
drug (IND)-enabling studies for clinical development. We
intend to develop our drugs for use as a monotherapy or in
combination with other medications to treat patients with
MENA-positive solid tumors. Our MENA diagnostic assay will be used to
screen patient’s tumors for expression of MENA INV, the
pro-metastatic MENA splice-variant for inclusion in clinical
studies of our anti-metastatic drug candidates. We plan to
study a range of MENA-positive cancers including pancreatic,
glioblastoma, bladder, colorectal and triple negative breast
cancer.
The
MENA diagnostic assay is
a tissue-based quantitative immunofluorescence (QIF) test that
measures expression of the pro-metastatic MENA splice-variant,
which is significantly associated with poor outcomes and metastasis
in Early Stage Breast Cancer (ESBC) and Squamous Cell Carcinoma of
the Lung. We intend to use this test as a companion precision
medicine diagnostic assay to select patients for treatment with our
anti-metastatic therapeutic. In April 2018, we completed methods
development and optimization utilizing our proprietary monoclonal
antibody (mAbs) specific for the pro-metastatic MENA protein
splice-valiant. In January 2017, results from a preclinical study
were published in Molecular Cancer
Therapeutics (Oudin et
al., 2017) demonstrating a novel mechanism for taxane
resistance in MENA-positive metastatic TNBC. Results from this
study demonstrated MENA expression conferred resistance to
the taxane paclitaxel and treatment failed to attenuate growth of
MENA driven metastatic lesions. The MENA biomarker is common to
most epithelial solid tumors. Analysis of data from The Cancer
Genome Atlas (TCGA) RNAseq database show expression of
pro-metastatic MENA protein splice-variant is common to many solid
tumors and higher levels are found in more aggressive tumors with
poor 5-year survival rates including pancreatic adenocarcinoma and
BRCA mutation-positive breast cancer.
Our CLIA-certified diagnostic, MetaSite
Breast™, is an immunohistochemistry test (IHC)
that measures micro-anatomical intravasation sites (TMEM) within
the tumor microenvironment which are clinically validated as
prognostic markers of metastasis in hormone receptor-positive (HR+)
ESBC. In November 2017, results a second
prospective-retrospective validation study were published in
npj Breast Cancer
(Sparano et al., 2017)
demonstrating the clinical validity of MetaSite Breast™ assay in patients with HR+
HER2-negative breast cancer for early recurrence within 5 years of
diagnosis. In addition, the study demonstrated MetaSite
Breast™ provides complementary prognostic
information to Oncotype Dx Recurrence Score. High MetaSite Scores
were associated with a 9.7-fold higher risk of distant recurrence
and 6.1-fold higher risk of overall recurrence if the Oncotype Dx
Recurrence Score was low. In July
2017, results from a study published in Science Translational
Medicine (Karagiannis
et
al., 2017) showed neoadjuvant
chemotherapy (NAC) can induce breast cancer metastasis through a
TMEM-mediated mechanism. This study suggests that MetaSite
Breast™ might also be useful in predicting
the development of pro-metastatic changes in the tumor
microenvironment in response to NAC. There are currently no tests
that can monitor or predict response to NAC.
Business Strategy
Our goal is to
become a leading precision medicine company focused on improving
the survival of patients with aggressive cancer. Our research team
is discovering and developing and therapeutics and companion
diagnostics that target a critical metastatic pathway responsible
for driving tumor resistance and the spread of aggressive cancer.
Key elements of our integrated therapeutic and companion diagnostic (Rx/Dx)
development strategy are to:
●
Continue advancing our
therapeutic discovery and development program focused on the MENA
protein isoform drug target. Our therapeutic strategy is
based on directly targeting the MENA pathway through inhibition of
MENA expression or activity and indirectly, through inhibition of
the MAPKAPK2 pathway responsible for activation of
MENA.
●
Explore business
development and collaborative research opportunities with
innovative companies developing compelling new modalities.
We intend to seek out and work with groups developing new
technologies that address previously considered undruggable targets
including RNA interference (RNAi), CRISPR, and targeted protein
degradation.
●
Pursue development and
regulatory strategy-based treatment of patients with MENA-positive
cancer. We intend to pursue a pan-cancer development and
regulatory strategy for our anti-metastatic therapeutics based on
seeking accelerated approval for treatment of patients whose cancer
are positive for MENA expression rather than the location of the
body in which the tumor originated.
●
Develop
the MENA diagnostic assay as a
companion diagnostic test to identify and select appropriate
patient populations with MENA-positive solid tumors most likely to
benefit from our targeted anti-metastatic
therapy. Companion
diagnostic tests are used as a companion to a therapeutic drug to
determine the applicability to a specific patient and if that
patient is likely to respond. The MENA diagnostic assay will enable
accelerated drug development and reduced risk by allowing for
selection of responder patient populations to ant-MENA drug
therapy.
●
Expand the market opportunity for the MENA diagnostic assay to
include prognostic diagnostic and companion diagnostic
applications. Complete clinical validation of MENA
diagnostic assay as a prognostic marker for risk of distant
metastasis in patients with early stage solid cancers and for use
as a predictive marker for drug response to Receptor Tyrosine
Kinase (RTK) inhibitors that target EGFR, HGFR, IGF-R1 and other
key receptors, as well as cytotoxic chemotherapeutics, including
anti-microtubule agents, such as docetaxel, paclitaxel and
albumin-bound paclitaxel.
●
Commercialize our therapeutics and diagnostic assays .
Through our exclusive license agreements and company sponsored
patent applications we own the world-wide rights to our products.
We seek commercialization partners that have existing
commercialization infrastructure, established distribution
channels, and strong relationships with our target audience in the
medical community. Our goal is to avoid the cost and risk
associated with building a new sales and marketing infrastructure.
Initially, we plan to build the necessary commercial infrastructure
only when needed to supplement partnerships and not economically
available through third party vendors. As profitability and market
penetration grow, we may supplement our strategic partnership or
contract sales organization (CSO) strategy with a phased-in
internal sales and marketing efforts.
Scientific Background and Technology
Our product
candidates target the MENA pathway, where MENA INV, the
pro-metastatic MENA splice-variant, MENA INV, plays a critical role
in solid tumors by promoting tumor dissemination, metastasis and
cancer cell resistance. The MENA pathway is a novel mechanism and
represents first-in-class target for drug development.
MENA is a naturally
occurring pro-motility protein which plays a key role in cell
migration and embryogenesis. It belongs to the Ena/VASP
(enabled/VASO) family being encoded by the MENA gene located on
chromosome 1. MENA facilitates and organizes formation, extension
and navigation of growing nerve fibers through tissue to link with
other neurons, forming the proper circuits needed for a functional
nervous system. Ena/VASP proteins such as MENA are involved in
these embryonic processes where dynamic actin reorganization is
necessary, such as neural closure, phagocytosis, hemostasis,
chemotaxis, cell migration, cell-cell and cell-matrix adhesions,
fibrillogenesis, or fibroblastic motility. MENA expression
decreases from embryonic to adult life and MENA INV is largely
absent from normal tissue.
The MENA gene can
be alternatively spliced to produce multiple splice-variants or
isoforms of which the MENA 11a and MENA INV isoforms dominate.
Alternative splicing is the process by which exons within a
pre-mRNA transcript of a gene are differentially spliced, resulting
in multiple protein isoforms being encoded by a single gene.
Post-transcriptional processing of the MENA gene provides an
opportunity for gene regulation and increases the functional
informational capacity of the gene. These small differences in MENA
structure produce large differences in MENA protein function. The
MENA gene corresponds to a 570-amino acid protein with the MENA INV
isoform containing a supplementary exon corresponding to a 19-amino
acid addition to the EVH1 domain of the protein. MENA 11a contains
a supplementary exon corresponding to a 21-amino acid addition to
the EVH2 domain of the protein. The invasive MENA isoform, MENA
INV, and the less dangerous MENA isoform, MENA 11a, play distinct
roles in cancer morphology. MENA INV promotes invasion and
metastasis by helping cancer cells subvert normal regulatory
networks regulating cell motility and increases sensitivity to the
chemo-attractant EGF by up to forty (40x) times. MENA INV
attenuates RTK signaling allowing cancer cells to respond to lower
EGF concentrations. MENA INV expressing tumor cells are
significantly less cohesive and have discontinuous cell-cell
contacts compared to MENA 11a expressing tumor cells. In
preclinical studies published in Breast Cancer Research (Roussos
et al., 2010), PyMT mice
that express MENA all succumb to systemic metastasis while the
majority of MENA-null knockout mice that lack expression of the
MENA protein and its splice-variants survive. Illustrated below is
Kaplan-Meir survival curve showing percentage of mice from each
genotype (n=12-25) that are not moribund (mice that did not
developed tumors or mice that had small tumors that did not
immobilized them). MENA knockout mice had significantly improved
survival compared to wild type mice that express MENA
(p=0.01).
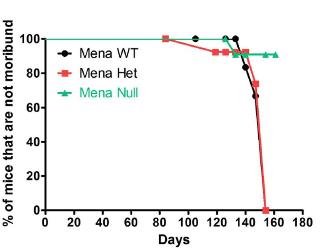
Source: Breast Cancer
Research 2010
12:R101 (Roussos et
al.; licensee
BioMed Central Ltd. 2010;https://doi.org/10.1186/bcr2784)
Aggressive tumors
hijack this embryonic motility mechanism through upregulation of
MENA and overexpression of the alternatively spliced mRNA
pro-metastatic MENA INV isoform. Consequently, MENA is
overexpressed in primary and metastatic cancers, and is
particularly overexpressed in migratory and disseminating
subpopulations of tumor cells. Until now the development of
anti-metastatic therapies has been slow and historically hampered
by the lack of understanding of the biology behind the metastatic
cascade. Our proprietary and patented platform technologies are
derived from novel ways of observing cancer cell behavior in living
functioning tumors in live animals and are based on the discovery
of a common pathway for the development of aggressive or metastatic
cancer in solid epithelial-based tumors. These discoveries are the
result of nearly 20 years of study and collaboration among four
scientific/academic institutions, including Massachusetts Institute
of Technology (MIT), Albert Einstein College of Medicine (AECOM),
Cornell University (Cornell), and the IFO-Regina Elena Cancer
Institute in Rome, Italy (IFO-Regina). This collaboration has
enabled us to understand the underlying biology, including the
direct mechanisms of action and specific micro-environmental
factors that drive systemic metastasis and drug resistance to
certain cancer treatments.
Recently, it was
discovered MENA is activated through the p38/MAPK pathway and
inhibition of MAPKAPK2 kinase results in a reversal of the
MENA-dependent phenotypes in
vitro and in vivo.
In 2017, we demonstrated inhibition of the p38/MAPK pathway
reverses MENA-dependent phenotypes of invasion, fibronectin
deposition, and metastasis in
vitro in cell line studies using MDA-MB-231 metastatic
Triple Negative Breast Cancer (TNBC) and MVD7 fibroblasts
engineered to express MENA at levels equivalent to that found
in vivo. In February 2018,
we announced cancer metastasis was reduced by 50% or more
in vivo following
inhibition of MAPKAPK2 in studies using orthotopic MDA-MD-231
transplant and syngeneic MMTV-PyMT models of MENA-positive
aggressive breast cancer.
Novelty and Innovative Medicine Targeting First-in-Class
Mechanism
Novel first-in-class drugs are innovative medicines that use a
novel mechanisms of action that serve unmet medical need. MENA and
its splice variants are found in the cytoplasm and not on the
surface of cells and have historically been considered to be
undrugable by conventional means such as mAbs and small molecules.
Our ability to target this novel first-in-class mechanism in made
possible through indirect inhibition of MAPKAPK2-dependent MENA
activation. Our unique
therapeutic approach is to target this critical metastatic pathway
and block tumor cell dissemination and the metastatic spread of
cancer which is responsible for more than 90% of solid tumor
cancer-related deaths. Despite the number and variety of clinically
available anticancer therapies there are currently no approved
MAPKAPK2 inhibitors or anti-metastatic drugs.
The
biopharmaceutical industry has increasingly focused on the same
core set of targets as evidenced by the exploding immune-oncology
space, with 2,000 active clinical programs and greater than 150
PD-L1/PD-1 checkpoint inhibitor mAbs in development. Current cancer
therapies, including newer targeted therapies and immunotherapies
are still evaluated for efficacy predominantly by their ability to
reduce tumor size and induce pathologic complete response (pCR).
Unfortunately, reduction of tumor size is not predictive of
anti-metastatic effect which explains why patients with pathologic
complete response (pCR) can undergo disease recurrence. We are
addressing an untapped field in oncology through discovering and
developing first-in-class anti-metastatic therapies targeting the
novel MENA pathway.
Kinases
and their Role in Cancer
Kinases
are enzymes that play an important role in regulating cellular
functions, signaling pathways and communication of cells with their
environments. Dysregulated protein kinases are implicated in a
large number of diseases including cancer and autoimmune diseases.
Many cancers are driven by genetic mutations that cause abnormal
activity of kinases which contribute to uncontrolled cell growth.
Kinase inhibitors act by blocking the abnormal activity of these
enzymes and has proven to be a highly successful therapeutic
strategy. Since the first approval in 2001, kinase inhibitors have
become an important therapeutic class playing a central role in
precision medicine and forming the backbone of targeted therapies
in oncology. A total of 39 kinase inhibitors have been
approved by the Federal Drug Administration or FDA with an
estimated 2016 combined annual sales of $20 billion.
The p38/MAPK signaling pathway has been a therapeutic target
for inflammatory disease and cancer for many years because of its
involvement in pro-inflammatory cytokine production, metastasis and
cell-cycle regulation. Several biopharmaceutical companies have
advanced p38 inhibitors into clinical studies, however, none have
been approved due to adverse side effects likely associated with
the role of p38 in the regulation of more than 60 substrates with
different physiological functions. Recently, p38/MAPK has been
implicated in activation of the MENA pathway. The MAPKAPK2
serine/threonine-specific terminal kinase is a more favorable
downstream target in the p38/MARK signaling pathway. Compared to
upstream kinases in the pathway, MAPKAPK2 has considerably fewer
substrates and fewer off-target effects. Unlike preclinical studies
of p38 knockout mice, the health and fertility of MAPKAPK2 mice are
unaffected. In addition, MAPKAPK2 deficient mice were shown to have
reduced joint damage in a collagen-induced arthritis model and no
indication of asthma in a lung ovalbumin sensitization
model.
The
success of individual kinase inhibitors can be limited by the
development of drug resistance. The mechanisms of acquired drug
resistance include target receptor mutation, activation of
secondary bypass pathways or histologic transformation. Our
therapeutic strategy is likely to bypass the selection pressures
responsible for driving resistance since neither MAPKAPK2 nor MENA
gene knockouts are embryonically lethal. Furthermore, our
inhibitors target tumor cell phenotypes associated with the
metastatic spread of cancer unlike most cytotoxic chemotherapeutic
drugs which are used to kill tumor cells.
For
these reasons we believe developing MAPKAPK2 kinase inhibitors
represent a particularly promising approach as a therapeutic
strategy to target the MENA pathway.
MENA and its Role in Driving Resistance Mechanisms to Anti-tumor
Therapies
Taxane-based anti-microtubule drugs, including paclitaxel and
docetaxel are widely used and highly efficacious as single
chemotherapy agents or in combination with other chemotherapeutic
drugs to treat breast, lung, ovarian, pancreatic and other cancers.
Taxanes interfere with the normal breakdown of microtubules which
are critical cytoskeletal structures that mediate cell
division. In vitro and in vivo studies have demonstrated increased expression of
the pro-metastatic MENA splice-variant desensitize cells to
taxane-based treatments. In January 2017, results from a
preclinical study published in Molecular Cancer Therapeutics (Oudin et
al., 2017) demonstrated a novel mechanism for taxane resistance in
MENA-positive metastatic TNBC. These results showed
MENA INV
expression conferred resistance to the taxane paclitaxel and
treatment failed to attenuate growth of MENA driven metastatic
lesions. MENA INV expression alters
the ratio of dynamic and stable microtubule populations in
paclitaxel-treated cells. MENA INV expression also increases MAPK
signaling in response to paclitaxel treatment. Decreasing MAPK
signaling through administration of a MAPKAPK2 inhibitor could
restore paclitaxel sensitivity by driving microtubule stabilization
in MENA INV-expressing cells. These results reveal a novel
mechanism of taxane resistance in highly metastatic breast cancer
cells and identify a potential combination therapy to overcome such
resistance.
In July 2017, results published in Science Translational
Medicine (Karagiannis
et
al., 2017) from a preclinical
study of mice transplanted with MMTV-PyMT breast cancers
demonstrated paclitaxel treatment promotes dissemination of
circulating tumor cells (CTCs) and metastasis in a MENA-dependent
manner.
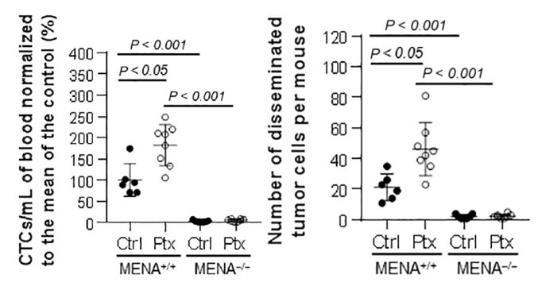
From Neoadjuvant chemotherapy induces breast
cancer metastasis through a TMEM-mediated mechanism,
By Karagiannis et al., Science Translational
Medicine 05 Jul 2017;Vol. 9,
Issue 397, eaan0026 DOI: 10.1126/scitranslmed.aan0026. Reprinted with
permission from American Association for the Advancement of Science
(AAAS).
As shown above MENA knockout recipient mice (MENA-/-) grafted with
MENA-positive MMTV-PyMT tumors do not develop CTCs or lung
metastases after treatment with vehicle control or paclitaxel. In
comparison, recipient mice (MENA+/+) grafted with MENA-positive
MMTV-PyMT tumors develop CTCs and lung metastasis with significant
increases in CTC dissemination and lung metastasis after paclitaxel
treatment. This study also demonstrated patients treated with
neoadjuvant chemotherapy consisting of paclitaxel followed by
doxorubicin plus cyclophosphamide undergo pro-metastatic changes in
their tumor microenvironment that can induce breast cancer
metastasis through a MetaSite (TMEM)-mediated mechanism. We believe
there is a substantial opportunity for anti-MENA therapeutics to be
used in combination with taxane therapy to overcome MENA-dependent
paclitaxel resistance and/or block paclitaxel-induced tumor cell
dissemination. Further, we believe our MetaSite Breast™ assay and MENA diagnostic assay could be
used to monitor or predict response to taxane-based
NAC.
MENA has also been shown to constitutively associate with the
tyrosine phosphatase PTP1B to mediate a novel negative feedback
mechanism that attenuates RTK signaling. On EGF stimulation,
complexes containing MENA and PTP1B are recruited to the EGF
receptor, or EGFR, causing receptor dephosphorization (the removal
of phosphate groups that can prevent ligation) and leading to
decreased motility responses. When the pro-metastatic MENA protein
splice-variant is expressed, PTP1B recruitment to the EGFR is
impaired, providing a mechanism for growth factor sensitization to
EGF, as well as HGF and IGF, and increased resistance to EGFR and
Met inhibitors. MENA INV disrupts this negative feedback mechanism
to drive sensitivity to EGF, HGF, and IGF growth factors and
resistance to RTK inhibitors that target EGFR, HGFR (c-Met), and
IGF-R1. Disruption of this attenuation by MENA INV sensitizes tumor
cells to low growth factor concentrations, thereby increasing the
migration and invasion responses that contribute to metastasis. We
believe there is a substantial opportunity for anti-MENA
therapeutics to be used in combination with EGFR, HGFR and IGF-R1
inhibitors to overcome drug resistance and/or increase the
proportion of patients who respond to these medications. Further,
we believe our MENA diagnostic assay could be used to predict
initial response to RTK inhibitors and monitor for the development
of therapeutic resistance.
Cancer Background
Cancer is a complex
disease characterized most simply by uncontrolled growth and spread
of abnormal cells. Cancer remains one of the world's most serious
health problems and is the second most common cause of death in the
United States after heart disease. The American Cancer Society, or
ACS estimates in 2018, there will be nearly 1.74 million new cases
of cancer and approximately 610,000 deaths from cancer in the
United States alone.
When
dealing with cancer, patients and physicians need to develop
strategies for local, regional, and distant control of the disease.
Ultimately, however, aggressive cancer that spreads or
“metastasizes" to other parts of the body is responsible for
more than 90% of all cancer related deaths in patients with such
common types of solid tumors as breast, lung, prostate and colon.
The most common methods of treating cancer are surgery, radiation
and drug therapy, or a combination of these methods, with varying
degrees of benefit and side effects that may not always justify the
expense and burden of the therapy. Our therapeutic drugs target the
MENA pathway, common to a type of cancer called carcinoma which are
malignancies of epithelial tissue and represent approximately
80-90% of all cancer cases. Our product candidates have the
potential to have broad pan-cancer applicability.
Prior
to the advent of personalized medicine, most cancer patients with a
specific type and stage of cancer received the same treatment,
which historically consisted of cytotoxic chemotherapies, including
taxanes, such as paclitaxel and docetaxel. These chemotherapies
kill rapidly proliferating cancer cells through non-specific
mechanisms, such as deterring cell metabolism or causing damage to
cellular components required for survival and rapid growth. While
these drugs have been effective in the treatment of some cancers,
many unmet medical needs remain. This approach is not optimal as
some treatments worked well for some patients but not for others.
Differences in the genome and how these genes are expressed, called
the expressome, explain many of these differences in response to
treatment. The convergence between understanding the expressome and
our ability to identify and develop biomarkers for certain disease
is accelerating growth and interest in personalized medicine and
the attractiveness of our intergraded Rx/Dx strategy.
Advances
in personalized medicine and cancer treatment are progressing
rapidly and are enabling a shift in clinical treatment from a
one-size-fits-all approach to one that is highly individualized.
Recently, more targeted therapies and immunotherapies have
represented some of the most promising agents in development for
the treatment of cancer. Targeted therapies are drugs that block
the growth and spread of cancer by interfering with specific
molecules that are involved in the growth, progression and spread
of cancer. Targeted therapies, such as RTK inhibitors that
selectively target kinase signaling pathways, are designed to
preferentially kill cancer cells and spare normal cells, improve
efficacy and minimize side effects. RTK inhibitors typically have
less severe side effects than cytotoxic chemotherapies, however, a
main limitation is that a significant number of patients do not
respond to treatment, and the emergence of secondary drug
resistance for those patients that do show an initial benefit. The
use of predictive biomarkers allows oncologists to better
understand and overcome drug resistance through the clinical
assessment of rational therapeutic drug combinations. We believe identification of patients with our
MetaSite Breast™ and MENA diagnostic tests followed by
combination therapy with our MAPKAPK2 inhibitor anti-MENA
therapeutics have the potential to overcome drug resistance and
protect patients from chemotherapy-induced tumor cell
dissemination.
Product Pipeline
MetaStat’s
product development programs are focused on developing novel
first-in-class drug candidates and companion diagnostic tests that
target the MENA pathway.
Therapeutics (Rx)
MENA is activated through the p38/MAPK pathway and inhibition of
MAPKAPK2 kinase results in a reversal of the MENA-dependent
phenotypes in vitro and in vivo in studies of metastatic TNBC and
fibroblast cell lines. In February 2018, we announced top-line
results from preclinical studies showing cancer metastasis was
reduced by 50% or more in
vivo following inhibition of MAPKAPK2 in models of
aggressive breast cancer. MetaStat will present results from these
in vitro and in vivo studies at the American
Association of Cancer Research (AACR) Cancer Dormancy and Residual
Disease conference to be held June 19-22, 2018 in Montreal, QC,
Canada.
MAPKAPK2
is a downstream terminal serine/threonine-specific kinase in the
p38/MAPK signaling pathway and is associated with inflammatory
disease, metastasis and in the resistance mechanism to antitumor
agents. Currently our discovery efforts are focused on
lead-optimization of non-ATP competitive inhibitors screened for
efficacy in inhibiting MENA activity with superior selectivity and
potency to the ATP-competitive research tools used in our
preclinical proof-of-concept studies. The goal of our small
molecule discovery program over the next 12-18 months is to
identify 1-2 lead candidates to advance into investigational new
drug (IND)-enabling studies for clinical
development.
The
goal of treatment with anti-MENA therapeutics is to reduce the
activity or expression of the pro-metastatic MENA splice variant
and thereby reverse the MENA-dependent cancer phenotypes of drug
resistance, tumor cell invasion, dissemination, and metastasis. In
targeting the movement of tumor cells from the primary site to
distant sites, we are directly addressing the major contributor to
the deaths of cancer patients. Since elevated expression of
pro-metastatic MENA splice variant also drives resistance to
certain RTK inhibitors and cytotoxic chemotherapeutics,
co-administration of an effective anti-MENA therapeutic provides an
opportunity to expand the utility and effectiveness of these drugs
by addressing a significant challenge to the clinical management of
advanced cancers. We intend to develop our drugs for use as a
monotherapy or in combination with other medications to treat
patients with MENA-positive solid tumors. Our MENA diagnostic assay
will be used to screen patient’s tumors for expression of the
pro-metastatic MENA splice-variant for inclusion in clinical
studies of our anti-metastatic drug candidates. We plan to study a range of
MENA-positive cancers including pancreatic, glioblastoma, bladder,
colorectal and triple negative breast cancer.
Diagnostics (Dx)
MENA Diagnostic Assay
The
MENA diagnostic assay is
a tissue-based quantitative immunofluorescence (QIF) test that
measures expression of the pro-metastatic MENA protein
splice-variant which is significantly associated with poor outcomes
and metastasis in Early Stage Breast Cancer (ESBC) and Squamous
Cell Carcinoma of the Lung.
We
intend to use the MENA diagnostic assay as a companion diagnostic
test to select patients for treatment with our anti-metastatic
therapeutic. In April 2018, we completed methods development and
optimization utilizing our proprietary monoclonal antibody specific
for the pro-metastatic MENA protein splice-valiant. In January
2017, Oudin et al.,
published results from a preclinical study demonstrating a novel
mechanism for taxane resistance in MENA-positive metastatic
Triple-negative Breast Cancer (TNBC). Results from this study
demonstrated MENA expression conferred resistance to
the taxane paclitaxel and treatment failed to attenuate growth of
MENA driven metastatic lesions. Data from an analysis of TCGA
RNAseq database show expression of the pro-metastatic MENA protein
splice-variant is common to many solid tumors and higher levels are
found in more aggressive tumors with poor 5-year survival rates
including pancreatic adenocarcinoma and BRCA mutation-positive
breast cancer.
Companion diagnostic to predict RTK inhibitor drug
response
MENA
participates in a mechanism that attenuates RTK signaling by
interacting with the tyrosine phosphatase PTP1B and the 5‟
inositol phosphatase SHIP2. Elevated expression of MENA INV
disrupts this regulation, and results in a pro-metastatic phenotype
characterized by increased RTK activation signaling from low ligand
stimulation and resistance to targeted RTK inhibitors. A main
limitation of therapies that selectively target kinase signaling
pathways is a significant number of patients do not respond and for
those patients that do respond the emergence of secondary drug
resistance after an initial benefit. We believe the MENA diagnostic
assay has the potential to be used as a highly actionable clinical
biomarker and/or companion diagnostic to predict response to
targeted RTK inhibitors.
Companion diagnostic to predict anti-microtubule drug
response
In
January 2017, results from a preclinical study published in
Molecular Cancer
Therapeutics (Oudin et al., 2017) demonstrated a novel
mechanism for taxane resistance in MENA-positive metastatic TNBC.
These results demonstrated MENA expression
conferred resistance to the taxane paclitaxel and treatment failed
to attenuate growth of MENA driven metastatic lesions. There is a
significant clinical need to develop biomarkers that predict
response to initial treatment or the development of secondary
resistance to taxane-based chemotherapy, while minimizing the risk
of unnecessary side effects. We believe the MENA diagnostic assay has the potential to be used as a
highly actionable clinical biomarker and/or companion diagnostic to
predict response to taxane-based drugs.
Liquid blood-based biopsy
There is excitement within the oncology community about the promise
of liquid biopsy assays for their potential to improve cancer
diagnosis and optimize patient care. Should the prognostic and
predictive role of the MENA diagnostic assay be clinically validated using FFPE tissue for
patients treated with RTKs and taxane-based chemotherapies, we
believe there will be a compelling need for the development of a
blood-based version of the MENA diagnostic assay. In addition to allowing for repeat
non-invasive testing, a blood-based MENA diagnostic
assay would be especially useful for
patients with advanced cancer undergoing multiple cycles of
treatment to predict initial drug response or the development of
secondary resistance. We intend to evaluate the potential for
developing the blood-based version of the MENA diagnostic
assay through collaborative research
and development partnerships with companies developing compatible
exosome and/or CTC technology platforms.
MetaSite Breast™ Assay
Our
CLIA-certified diagnostic, MetaSite Breast™, is an
immunohistochemistry test (IHC) that measurs micro-anatomical
intravasation sites (TMEM) at blood
vessels within the tumor microenvironment which are
clinically validated as prognostic markers of metastasis in hormone
receptor-positive (HR+) early stage breast cancer (ESBC). The
MetaSite Breast™ test
has been analytically validated under CLIA, tested in 6 clinical
studies in over 1,700 patients, and is available for clinical use
in most states. The MetaSite Breast™ test is a tissue-based
IHC assay performed on formalin-fixed paraffin-embedded, or
FFPE tissue from a biopsy that directly identifies and
quantifies the active sites of the metastatic process. The
MetaSite Breast™ test
is intended for patients with early stage (stage 1-3) invasive
breast cancer who have node-negative or node-positive (1-3),
ER-positive, HER2-negative disease.
Mechanism of action for use as a prognostic diagnostic to predict
risk of cancer metastasis
In order for breast cancer tumor cells to enter a blood vessel
(intravasate), three types of cells must self-assemble in
apposition to each other in individual three-cell structures
located at blood vessels within the tumor
microenvironment. This structure termed a
“MetaSite™”, is composed of a M2 tumor associated
macrophage (protumoral macrophage or type of immune cell), a tumor
cell that expresses the pro-metastatic MENA splice variant and an
endothelial cell (cells that lines blood vessels). We have
demonstrated in clinical studies the number of MetaSites™
correlates with increased risk of cancer metastasis.
This structure termed a “MetaSite™”, is composed
of an endothelial cell (cells that lines blood vessels), a
protumoral tumor associated macrophage (a type of immune cell), and
a tumor cell that expresses the MENA protein. We have demonstrated
in clinical studies that the number of MetaSites™ correlates
with increased risk of cancer metastasis.
MetaSite Breast™ Clinical Studies
In November 2017, the results of a second
prospective-retrospective validation study were published in
npj Breast Cancer
(Sparano et al., 2017)
demonstrating the clinical validity of the MetaSite Breast™ assay in patients with HR+
HER2-negative breast cancer for early recurrence within 5 years of
diagnosis. The ECOG 2197 Cohort Study
is a prospectively designed retrospective study (n=600) in an
independent cohort of ESBC patients treated with surgery, 4 cycles
of adjuvant chemotherapy (doxorubicin 60 mg/m2 and cyclophosphamide
600 mg/m2 (AC) or docetaxel 60 mg/m2 (AT)) and endocrine therapy.
Results from this study revealed a significant positive association
between continuous MetaSite Score and distant recurrence-free
interval (DRFI) p=0.001 and recurrence-free interval (RFI)
p=0.00006 in HR-positive HER2-negative disease in years 0-5 and by
MetaSite Score tertiles for DRFI (p=0.04) and RFI (p=0.01).
Proportional hazards models including clinical covariates (N0 vs.
N1; T1 vs. T2; high vs. int. vs. low grade) also revealed
significant positive associations for continuous MetaSite Score
with RFI (p=0.04), and borderline association with DRFI
(p=0.08). In addition, the study demonstrated MetaSite
Breast™ provides complementary prognostic
information to Oncotype Dx Recurrence Score. High MetaSite Scores
were associated with a 9.7-fold higher risk of distant recurrence
(95% confidence intervals [CI] 1.8, 54.1) and 6.1-fold higher risk
of overall recurrence (95% CI 1.3, 27.8) if the Oncotype Dx
Recurrence Score was low (RS<18). Patients with intermediate MetaSite Score
(MS=6-17) and low Recurrence Score (RS<18) results had
approximately 4.7-fold greater risk (HR=4.7, 95%CI=0.9-24.2) of
distant metastasis compared to patients with low MetaSite Score
(MS<6) results.
In July 2017, results from a study published in Science Translational
Medicine (Karagiannis
et
al., 2017) showed neoadjuvant
chemotherapy (NAC) can induce breast cancer metastasis through a
TMEM-mediated mechanism. The result of this study suggests
that MetaSite Breast™ might also be useful in predicting
the development of pro-metastatic changes in the tumor
microenvironment in response to NAC. There are currently no tests
that can monitor or predict response to NAC.
In
December 2016, we presented results from the Kaiser Permanente
Cohort Study conducted by MetaStat, that demonstrated MetaSite
Score was a statically significant predictor of distant metastasis
and a binary cutpoint was able to discriminate high and low risk
patient groups when adjusted for clinical factors. Independent
verification and clinical validation of MetaStat’s fully
automated and analytically validated tissue-based MetaSite
Breast™ test for risk
of cancer metastasis in HR-positive HER2-negative ESBC. The Kaiser
Permanente Cohort Prognostic Study is a case-control nested cohort
of 3,760 patients diagnosed with ESBC from the Kaiser Permanente
Northwest Health Care System in which 464 tumor samples were tested
using the MetaSite Breast™ assay. MetaSite Score was
a statistically significant predictor of distant metastasis
(p=0.039) in patients with HR-positive HER2-negative disease. Using
predefined cutpoints based on tertiles for the control group in the
overall study population (n=282), MetaSite Score was significantly
associated with distant metastasis for the high (MS>41) versus
low (MS<13) score tertiles (OR=2.94; 95%CI=1.62-5.41, P=0.0005)
and the intermediate (MS=13-41) versus low score tertiles (OR=2.24;
95%CI=1.23-4.13, P=0.009). A binary cut-point for the high-risk
group (MS>14) was significant with a 2-fold higher risk (OR=2.1,
95%CI=1.06-3.96) of distant metastasis versus the low risk group
and adjusted for clinical covariates (P=0.036).
In
December 2015, we presented results from the analytic validation
study of our fully-automated commercial MetaSite Breast™ assay at the Tumor
Metastasis meeting of the American Associations for Cancer Research
(AACR). The reliability of our commercial MetaSite Breast™ test was supported by
confirming the test’s analytical accuracy, reproducibility,
and precision. Reproducibility across operators, instruments and
different sections of a tumor sample ranged from 91% to 97% and
analytical precision was found to be greater than 97% with a mean
percent coefficient of variation (%CV) of 6.6% (n=35). Our
commercial MetaSite Breast™ assay showed a high
degree of analytical accuracy with the reference standard with AUCs
of 0.84 and 0.90 for low and high risk cut-points, respectively.
The gold standard method was originally developed at AECOM, where
results from their study published in August 2014 in the
Journal of the National Cancer
Institute (Rohan et
al., 2014) demonstrated the number of MetaSites™ in
tumors was predictive of metastatic disease in ER-positive breast
cancer.
-10-
In
December 2015, we presented results from the analytic validation
study of our fully-automated commercial MetaSite Breast™ assay at the Tumor
Metastasis meeting of the American Associations for Cancer Research
(AACR). The reliability of our commercial MetaSite Breast™ test was supported by
confirming the test’s analytical accuracy, reproducibility,
and precision. Reproducibility across operators, instruments and
different sections of a tumor sample ranged from 91% to 97% and
analytical precision was found to be greater than 97% with a mean
percent coefficient of variation (%CV) of 6.6% (n=35). Our
commercial MetaSite Breast™ assay showed a high
degree of analytical accuracy with the reference standard with AUCs
of 0.84 and 0.90 for low and high risk cut-points, respectively.
The gold standard method was originally developed at AECOM, where
results from their study published in August 2014 in the
Journal of the National Cancer
Institute (Rohan et
al., 2014) demonstrated the number of MetaSites™ in
tumors was predictive of metastatic disease in ER-positive breast
cancer.
In September 2015,
we announced topline data from a prospectively defined
case-controlled nested cohort of 3,760 patients with invasive
ductal carcinoma of the breast diagnosed between 1980 and 2000
followed through 2010 from the Kaiser Permanente Northwest health
care system. Of the 3,760 patients treated in this cohort, we
received 573 breast cancer tissue blocks of which 481, representing
259 case-controlled pairs, were usable and included in the study.
In this study, the MetaSite Breast™ Score was found to be
significantly and directly associated with increased risk of
distant metastasis in ER-positive, HER2-negative invasive breast
cancer for both high (>35 MetaSites™) versus low (<12
MetaSites™) MetaSite™ scores (OR = 3.4; 95% CI =
2.8-4.1; P=0.0002) as well as between intermediate (12-35
MetaSites™) and low MetaSite™ scores (OR=3.24; 95% CI =
2.6-3.9; P=0.0006). This study demonstrated the MetaSite
Breast™ Score
predicted risk of distant metastasis in ER-positive, HER2-negative
early stage invasive breast cancer independent of traditional
clinical factors. Data from this study was presented at the San
Antonio Breast Cancer Symposium (SABCS) in December
2016.
In August 2014, the
positive results of a 481-patient clinical study demonstrating the
prognostic utility of the MetaSite Breast™ assay was published in
the Journal of the National
Cancer Institute (Rohan et
al., 2014) In a case-controlled nested
prospective-retrospective study, a cohort of 3,760 patients was
examined with invasive ductal breast carcinoma diagnosed between
1980 and 2000 and followed through 2010. The association between
the MetaSite™ score from the MetaSite Breast™ assay and risk of
distant metastasis was prospectively examined. A total
of 481 blocks representing 259 case-controlled pairs were usable
and selected for inclusion in this study. Control and case subjects
had very similar distributions with respect baseline
characteristics such as age and tumor size. Results from
this study demonstrated a statistically significant association
between increasing MetaSite™ score and risk of metastasis in
the ER-positive, HER2-negative subpopulation (N=295) (OR high vs.
low tertile = 2.70, 95% CI=1.39 to 5.26, Ptrend 0.004; OR per
10-unit increase in MetaSite™ score = 1.16, 95% CI = 1.03 to
1.30). The absolute risk of distant metastasis for the low, medium
and high-risk groups was estimated to be 5.9% (95% CI=5.1-6.9%),
14.1% (95% CI=13.0-15.0%), and 30.3% (95% CI=26.1-35.4%),
respectively. Statistical significance was not achieved in the
triple negative (TNC) (N=98) or HER2-positive subpopulations
(N=75). The conclusion from this study was the MetaSite™
score predicted the risk of distant metastasis in ER-positive,
HER2-negative breast cancer patients independently of traditional
clinicopathologic features such as age and tumor size.
In April 2009, the positive results of a clinical
study using the MetaSite Breast™ assay on patient tumor samples with
invasive breast cancer was published in Clinical Cancer
Research (Robinson, et al., 2009). In this case-controlled 5-year
retrospective study, a cohort of 60 patients with invasive ductal
breast carcinoma, including 30 patients who developed metastatic
disease was studied using the MetaSite Breast™ assay. The results from this
study demonstrated MetaSite™ score density was statistically
significantly greater in patients who subsequently developed
systemic metastasis compared with the patients who had only
localized breast cancer (median, 105 vs. 50, respectively;
P=0.00006). For every 10-unit increase in MetaSites™ the odds
ratio of systemic metastasis increased by 1.9 (95% confidence
interval, 1.1-3.4). The number of MetaSites™ observed per
patient ranged from 12 to 240 and the odds of metastasis nearly
doubled for every increase of 10
MetaSites™. Importantly, the MetaSite™ score
density was not correlated with tumor size, lymph node metastasis,
lymphovascular invasion, or hormone receptor
status.
-11-
Competition
The life
sciences, biotechnology and molecular diagnostic industries are
characterized by rapidly advancing technologies, intense
competition and a strong emphasis on proprietary technologies and
products. Any therapeutic, companion diagnostic and prognostic
diagnostic product candidates that we are able to successfully
develop and commercialize will compete with both existing therapies
and diagnostics and new therapies and diagnostics that may become
available in the future. While we believe that our technology and
scientific knowledge provide us with competitive advantages, we
face potential competition from many different sources, including
pharmaceutical, specialty pharmaceutical and biotechnology
companies, both large and small molecular diagnostic companies,
academic institutions and governmental agencies and public and
private research institutions, among others.
We plan to
compete in segments of the pharmaceutical, biotechnology and other
related markets that pursue personalized medicine approaches to
treating cancer. There are many companies presently developing
therapies for cancer in the field of precision medicines, including
divisions of large pharmaceutical companies, specialty
pharmaceutical and biotechnology companies of various sizes,
including Pfizer Inc., Merck & Co., Inc., Novartis
Pharmaceuticals Corp., F. Hoffmann-La Roche Ltd, Bristol-Myers
Squibb Company, Eli Lilly and Company, AstraZeneca, PLC, Amgen,
Inc., Biogen, Inc., Genentech, Inc., Celgene Corp., Bayer AG,
Takeda Pharmaceutical Company Limited, through its wholly owned
subsidiary ARIAD Pharmaceuticals, Inc., Clovis Oncology, Inc.,
Ignyta, Inc., and Deciphera Pharmaceuticals LLC, among many
others.
We believe our
main diagnostic competition will be from a number of private and
public companies that offer molecular diagnostic tests, including
gene profiling and expression in multiple cancers indications,
including companies such as Genomic Health, Inc., Agendia Inc.,
BioTheranostics, Inc., Exact Sciences, Inc. GenomeDx Biosciences
Inc., Hologic Inc., Myriad Genetics, Inc., NanoString Technologies
Inc., NeoGenomics, Inc., Novartis AG, Qiagen N.V., Roche
Diagnostics, a division of Roche Holding, Ltd, Siemens AG, Veridex
LLC, a Johnson & Johnson company, Celera Corporation, and GE
Healthcare, a business unit of General Electric Company, as well as
others. Commercial laboratories, such as Laboratory Corporation of
America Holdings and Quest Diagnostics Incorporated, with strong
distribution networks for diagnostic tests, may also compete with
us. We may also face competition from Illumina, Inc. and Thermo
Fisher Scientific Inc., both of which have announced their
intention to enter the clinical diagnostics market as well as other
companies and academic and research institutions. We may also face
completion from companies focused on liquid biopsies and pan-cancer
clinical diagnostics, such as Danaher Corporation and its Cepheid,
Inc. subsidiary, Foundation Medicine, Inc., Guardant Health, MDx
Health, Inc., Metamark Inc., Natera Inc. and Response Genetics,
Inc., among many others.
Our competitors
may develop and market therapeutic, companion diagnostic and
prognostic diagnostic products or other novel technologies that are
more effective, safer, more convenient or less costly than any that
may be commercialized by us, or may obtain regulatory approval for
their products more rapidly than we may obtain approval for ours.
Many of our present and potential competitors have widespread brand
recognition, distribution and substantially greater financial and
technical resources and development, production and marketing
capabilities than we do. If we are unable to compete successfully,
we may be unable to gain market acceptance and therefore revenue
from our therapeutics and diagnostics may be limited.
-12-
Patents and Intellectual Property
We believe that
clear and extensive patent coverage and protection of the
proprietary nature of our technologies is central to our success.
Our intellectual property strategy is intended to develop and
maintain a competitive position and long-term value through a
combination of patents, patent applications, copyrights,
trademarks, and trade secrets. We have invested and will continue
to invest in our intellectual property portfolio, which has been
partially accomplished in conjunction with the resources of our
Licensors. This applies to both domestic and international patent
coverage.
Four (4) patents
in the United States, and three (3) international patents have been
issued covering key aspects of our core MENA biomarker technologies
for epithelial-based solid tumors including breast, lung, prostate
and colorectal. These patents expire between 2028 and
2031.
We have and
intend to continue to file additional patent applications to
strengthen our therapeutic and diagnostic intellectual property
rights, as well as seek to add to our intellectual property
portfolio through licensing, partnerships, joint development and
joint venture agreements.
Our employees
and key technical consultants working for us are required to
execute confidentiality and assignment agreements in connection
with their employment and consulting relationships. Confidentiality
agreements provide that all confidential information developed or
made known to others during the course of the employment,
consulting or business relationship shall be kept confidential
except in specified circumstances. Additionally, our employment
agreements provide that all inventions conceived by such employee
while employed by us are our exclusive property. We cannot provide
any assurance that employees and consultants will abide by the
confidentiality and assignment terms of these agreements. Despite
measures taken to protect our intellectual property, unauthorized
parties might copy aspects of our technology or obtain and use
information that we regard as proprietary.
As part of our
intellectual property strategy, we are reviewing our license
agreements and related patents and patent applications to determine
applicability with our integrated Rx/Dx product development
strategy.
License Agreements
In August 2010, we
entered into a License Agreement (the “License
Agreement”) with AECOM, MIT, Cornell and IFO-Regina. The
License Agreement covers patents and patent applications, patent
disclosures, cell lines and technology surrounding discoveries in
the understanding of the underlying mechanisms of systemic
metastasis in solid epithelial cancers, including our core
diagnostic technologies, including the MetaSite Breast™ and MENA diagnostic
assays. The License Agreement calls for certain
customary payments such as a license signing fee, reimbursement of
patent expenses, annual license maintenance fees, milestone
payments, and the payment of royalties on sales of products or
services covered under the agreement. See “Contractual
Obligations” in the “Management’s Discussion and
Analysis of Financial Condition and Results of Operations”
section for more information regarding our financial obligations
related to the License Agreement.
Effective March
2012, we entered into a second license agreement (the “Second
License Agreement”) with AECOM. The Second License Agreement
covers patent and patent applications, patent disclosures, and
other technology surrounding discoveries in the understanding of
the underlying mechanisms of systemic metastasis in solid
epithelial cancers, including the isolation (capture of), gene
expression profile (the “Human Invasion Signature”) and
chemotherapeutic resistance of metastatic cells. The Second License
Agreement requires certain customary payments such as a license
signing fee, reimbursement of patent expenses, annual license
maintenance fees, milestone payments, and the payment of royalties
on sales of products or services covered under such agreements. See
“Contractual Obligations” in the
“Management’s Discussion and Analysis of Financial
Condition and Results of Operations” section for more
information regarding our financial obligations related to the
Second License Agreement.
-13-
Pursuant to both
the License Agreement and the Second License Agreement, we have the
right to initiate legal proceedings on our behalf or in the
Licensors’ names, if necessary, against any infringer, or
potential infringer, of a licensed intellectual property who
imports, makes, uses, sells or offers to sell products. Any
settlement or recovery received from any such proceeding shall be
divided eighty percent (80%) to us and twenty percent (20%) to the
Licensors after we deduct from any such settlement or recovery our
actual counsel fees and out-of-pocket expenses relative to any such
legal proceeding. If we decide not to initiate legal proceedings
against any such infringer, then the Licensors shall have the right
to initiate such legal proceedings. Any settlement or recovery
received from any such proceeding initiated by the Licensors shall
be divided twenty percent (20%) to us and eighty percent (80%) to
the Licensors after the Licensors deduct from any such settlement
or recovery their actual counsel fees and out-of-pocket expenses
relative to any such legal proceeding.
Effective
December 2013, we entered into two separate worldwide exclusive
license agreements with MIT and its David H. Koch Institute for
Integrative Cancer Research at MIT and its Department of Biology,
AECOM, and Montefiore Medical Center (“Montefiore” and,
together with MIT and AECOM, the “Alternative Splicing
Licensors”). The diagnostic license agreement (the
“Alternative Splicing Diagnostic License Agreement”)
and the therapeutic license agreement (the “Alternative
Splicing Therapeutic License Agreement” and, together with
the Diagnostic License Agreement, the “2014 Alternative
Splicing License Agreements”) covers pending patent
applications, patent disclosures, and technology surrounding
discoveries of alternatively spliced mRNA and protein isoform
markers for the treatment and/or prevention of cancer through the
epithelial-mesenchymal transition (EMT) in epithelial solid tumor
cancers. The 2014 Alternative Splicing License Agreements call for
certain customary payments such as a license signing fee,
reimbursement of patent expenses, annual license maintenance fees,
milestone payments, and the payment of royalties on sales of
products or services covered under the agreement. See
“Contractual Obligations” in the
“Management’s Discussion and Analysis of Financial
Condition and Results of Operations” section for more
information regarding our financial obligations related to the
Alternative Splicing License Agreements.
Further,
pursuant to the 2014 Alternative Splicing License Agreements, we
have the right to initiate legal proceedings on our behalf or in
the Licensors’ names, if necessary, against any infringer, or
potential infringer, of any licensed intellectual property who
imports, makes, uses, sells or offers to sell products. Any
settlement or recovery received from any such proceeding shall be
divided 80% to us and 20% to the Licensors after we deduct from any
such settlement or recovery our actual counsel fees and
out-of-pocket expenses relative to any such legal proceeding. If we
decide not to initiate legal proceedings against any such
infringer, then the Licensors shall have the right to initiate such
legal proceedings. Any settlement or recovery received from any
such proceeding initiated by the Licensors shall be divided 20% to
us and 80% to the Licensors after the Licensors deduct from any
such settlement or recovery their actual counsel fees and
out-of-pocket expenses relative to any such legal
proceeding.
Effective June
2014, we entered into a License Agreement (the “Antibody
License Agreement”) with MIT. The Antibody License Agreement
covers proprietary technology and know-how surrounding monoclonal
and polyclonal antibodies specific to the Mena protein and its
isoforms. The Antibody License Agreement calls for certain
customary payments such as a license signing fee, reimbursement of
patent expenses, annual license maintenance fees, milestone
payments, and the payment of royalties on sales of products or
services covered under the agreement. See
“Contractual Obligations” in the
“Management’s Discussion and Analysis of Financial
Condition and Results of Operations” section for more
information regarding our financial obligations related to the
Antibody License Agreement. As part of our intellectual property
strategy, we have terminated certain license agreements and patent
applications related to non-core technologies.
Partnerships and Collaborations
In connection
with our business strategy, we may enter into exclusive and/or
non-exclusive research and development and other collaboration or
partnership agreements.
Celgene Corporation
On August 22, 2016,
we executed a pilot materials transfer agreement (the
“MTA”) with Celgene Corporation (“Celgene”)
to conduct a mutually agreed upon pilot research project (the
“Pilot Project”). On September 29, 2016, we entered
into an amendment (the “Amendment”) to the MTA (the
“Amendment,” and together with the MTA, the
“Research Agreement”), which provided for milestone
payments to MetaStat of up to approximately $973,000. Under the
terms of the Research Agreement, Celgene provided certain proprietary materials to the
Company and the Company evaluated Celgene’s proprietary
materials in the Company’s metastatic cell line
(in
vitro) and animal
(in
vivo) nonclinical
models. The Pilot Project was
successfully completed in January 2018. See Note 11 for
accounting treatment related to the Research
Agreement.
-14-
Albert Einstein College of Medicine and Montefiore Medical
Center
Effective January
9, 2015, we executed a collaboration agreement (the
“Collaboration Agreement”) with AECOM and Montefiore
Medical Center (“Montefiore,” and together with AECOM,
the “Institutions”) to collaborate on research projects
(the “Research Projects”) including conducting studies
that establish the clinical validity and clinical utility of
MetaStat’s prognostic diagnostic tests, including the
MetaSite Breast™
test, the MENA diagnostic assay, and a combined test. The term of
the Collaboration Agreement is five years, which may be terminated
by either party with thirty days written notice.
National Institutes of Health, National Cancer
Institute
Effective September
21, 2016, we executed an agreement with the National Institutes of
Health, National Cancer Institute (the “NCI"), whereby the
NCI will contract with MetaStat to perform the MetaSite
Breast™ and MENA
diagnostic analysis of breast cancer tumor tissue as part of a
clinical study. In addition, MetaStat will collaborate with the
Department of Cancer Epidemiology and Genetics (DCEG) at the NCI on
interpretation of the study analysis and dissemination of
results.
Government Regulation
Regulation by
governmental authorities in the United States, at the federal,
state and local level, and in and other countries is a significant
factor in the research and clinical development, testing,
manufacture, commercialization, marketing and advertising,
distribution and post-approval monitoring and reporting, among
others of both pharmaceuticals and diagnostic tests.
Therapeutic Regulation
United States Drug Approval Process
In the United
States, the FDA regulates drugs under the FDCA, and implementing
regulations. The process of obtaining regulatory approvals and the
subsequent compliance with appropriate federal, state, local and
foreign statutes and regulations requires the expenditure of
substantial time and financial resources. Failure to comply with
the applicable U.S. requirements at any time during the product
development process, approval process or after approval, may
subject an applying company to a variety of administrative or
judicial sanctions.
Before a drug may
be marketed in the U.S., the FDA generally requires the
following:
●
completion of
preclinical laboratory tests, animal studies and formulation
studies in compliance with good laboratory practice, or GLP,
regulations;
●
submission of an
Investigational New Drug or IND application to the FDA, which must
become effective before human clinical trials may
begin;
●
approval of each
phase of the proposed clinical trials and related informed consents
by an IRB, at each clinical site where such trial will be
performed;
●
performance of
adequate and well-controlled human clinical trials in accordance
with good clinical practice, or GCP, standards and regulations to
establish the safety and efficacy of the proposed drug for each
indication;
●
submission of a
New Drug Application, or NDA to the FDA;
●
satisfactory
completion of an FDA inspection of the manufacturing facility or
facilities at which the product is produced to assess compliance
with current good manufacturing practice, or cGMP, requirements and
to assure that the facilities, methods and controls are adequate to
preserve the drug’s identity, strength, quality and purity;
and
●
FDA
review and approval of the NDA.
-15-
Preclinical Studies and IND
Preclinical studies
include laboratory evaluation of product chemistry and formulation,
as well as in vitro and
animal studies to assess the potential for AEs and, in some cases,
to establish a rationale for therapeutic use. The conduct of
preclinical studies is subject to federal regulations and
requirements, including GLP regulations for safety/toxicology
studies. An IND sponsor must submit the results of the preclinical
tests, together with manufacturing information, analytical data,
any available clinical data or literature and plans for clinical
trials, among other things, to the FDA as part of an IND. Some
long-term preclinical testing, such as animal tests of reproductive
AEs and carcinogenicity, may continue after the IND is submitted.
An IND automatically becomes effective 30 days after receipt by the
FDA, unless before that time the FDA raises concerns or questions
related to one or more proposed clinical trials and places the
trial on clinical hold. In such a case, the IND sponsor and the FDA
must resolve any outstanding concerns before the clinical trial can
begin. As a result, submission of an IND may not result in the FDA
allowing clinical trials to commence.
Clinical Trials
Clinical trials
involve the administration of the investigational new drug to human
subjects under the supervision of qualified investigators in
accordance with GCP requirements, which include, among other
things, the requirement that all research subjects provide their
informed consent in writing before their participation in any
clinical trial. Clinical trials are conducted under written
protocols detailing, among other things, the objectives of the
trial, the parameters to be used in monitoring safety, and the
safety and effectiveness criteria to be evaluated. A protocol for
each clinical trial and any subsequent protocol amendments must be
submitted to the FDA as part of the IND. In addition, an IRB at
each institution participating in the clinical trial must review
and approve the plan for any clinical trial before it commences at
that institution, and the IRB must conduct continuing review. The
IRB must review and approve, among other things, the study protocol
and informed consent information to be provided to study subjects.
An IRB must operate in compliance with FDA regulations. In
addition, a sponsor must provide information regarding most
clinical trials to be disclosed on http://clinicaltrials.gov, a
website maintained by the National Institutes of
Health.
Human clinical
trials are typically conducted in three sequential phases that may
overlap or be combined:
●
Phase 1: The
drug is initially introduced into healthy human subjects or
patients with the target disease or condition and tested for
safety, dosage tolerance, absorption, metabolism, distribution,
excretion and, if possible, to gain an early indication of its
effectiveness;
●
Phase 2: The
drug is administered to a limited patient population to identify
possible adverse effects and safety risks, to preliminarily
evaluate the efficacy of the product for specific targeted diseases
and to determine dosage tolerance and optimal dosage;
and
●
Phase 3: The
drug is administered to an expanded patient population in adequate
and well-controlled clinical trials to generate sufficient data to
statistically confirm the efficacy and safety of the product for
approval for specified indications, to establish the overall
risk-benefit profile of the product and to provide adequate
information for the labeling of the product.
Progress reports
detailing the results of the clinical trials must be submitted at
least annually to the FDA, and more frequently if serious adverse
events, or AEs occur. The FDA or the sponsor may suspend or
terminate a clinical trial at any time on various grounds,
including a finding that the research subjects are being exposed to
an unacceptable health risk. Similarly, an IRB can suspend or
terminate approval of a clinical trial at its institution if the
clinical trial is not being conducted in accordance with the
IRB’s requirements or if the drug has been associated with
unexpected serious harm to patients.
-16-
Pursuant to the
21st Century Cures Act, which was enacted on December 13, 2016, the
manufacturer of an investigational drug for a serious or
life-threatening disease is required to make available, such as by
posting on its website, its policy on evaluating and responding to
requests for expanded access. This requirement applies on the later
of 60 days after the date of enactment or the first initiation of a
Phase 2 or Phase 3 trial of the investigational drug.
Marketing Approval
Assuming
successful completion of the required clinical testing, the results
of the preclinical studies and clinical trials, together with
detailed information relating to the product’s chemistry,
manufacture, controls and proposed labeling, among other things,
are submitted to the FDA as part of an NDA requesting approval to
market the product for one or more indications. Under federal law,
the submission of most NDAs is subject to a substantial application
user fee.
The FDA
generally conducts a preliminary review of all NDAs to determine if
they are sufficiently complete to permit substantive review within
the first 60 days after submission before accepting them for
filing. The FDA may request additional information in connection
with this preliminary review rather than accept an NDA for filing.
In this event, the application must be resubmitted with the
additional information. The resubmitted application is subject to
further review before the FDA accepts it for filing. Once the
submission is accepted for filing, the FDA begins an in-depth
substantive review. The FDA has agreed to specified performance
goals in the review of NDAs. Under these goals, the FDA has
committed to review most such applications for non-priority
products within 10 months, and most applications for priority
review products, that is, drugs that the FDA determines represent a
significant improvement over existing therapy, within six months.
The FDA may also refer applications for novel drugs or products
that present difficult questions of safety or efficacy to an
advisory committee, typically a panel that includes clinicians and
other experts, for review, evaluation and a recommendation as to
whether the application should be approved. The FDA is not bound by
the recommendations of an advisory committee, but it considers such
recommendations carefully when making decisions. The FDA is not
required to adhere its review time goals, and its review could
experience delays that cause those goals to not be
met.
Before approving
an NDA, the FDA typically will inspect the facility or facilities
where the product is manufactured. The FDA will not approve an
application unless it determines that the manufacturing processes
and facilities are in compliance with cGMP requirements and are
adequate to assure consistent production of the product within
required specifications. In addition, before approving an NDA, the
FDA will typically inspect one or more clinical sites to assure
compliance with GCP and integrity of the clinical data
submitted.
The testing and
approval process for each product candidate requires substantial
time, effort and financial resources, and each may take many years
to complete. Data obtained from preclinical and clinical activities
are not always conclusive and may be susceptible to varying
interpretations, which could delay, limit or prevent regulatory
approval. The FDA may not grant approval of an application for a
product candidate on a timely basis, or at all. Further, applicants
often encounter difficulties or unanticipated costs in their
efforts to develop product candidates and secure necessary
governmental approvals, which could delay or preclude the marketing
of those products.
After the
FDA’s evaluation of the NDA and inspection of the
manufacturing facilities, the FDA may issue an approval letter or a
complete response letter. An approval letter authorizes commercial
marketing of the drug with specific prescribing information for
specific indications. A complete response letter generally outlines
the deficiencies in the submission and may require substantial
additional testing or information in order for the FDA to
reconsider the application. If and when those deficiencies have
been addressed to the FDA’s satisfaction in a resubmission of
the NDA, the FDA may then issue an approval letter. The FDA has
committed to reviewing such resubmissions in two or six months
depending on the type of information included. Even with submission
of this additional information, the FDA ultimately may decide that
the application does not satisfy the regulatory criteria for
approval and refuse to approve the NDA.
-17-
Programs for Expedited Review and Approval
The FDA has
developed certain programs and designations that enable NDAs for
product candidates meeting specified criteria to be eligible for
certain expedited review and approval processes such as fast track
designation, priority review, accelerated approval, and
breakthrough therapy designation. Even if a product qualifies for
one or more of these programs, the FDA may later decide that the
product no longer meets the conditions for qualification or decide
that the time period for FDA review or approval will not be
shortened. These include Fast Track Designation, Priority Review,
Accelerated Approval, and Breakthrough Therapy
Designation.
In addition to
the expedited review and approval programs and designations, the
FDA also recognizes certain other designations and alternative
approval pathways that afford certain benefits, such as the orphan
drug designation and alternative types of NDAs under the
Hatch-Waxman Act.
Under the Orphan
Drug Act, the FDA may grant orphan drug designation to drugs
intended to treat a rare disease or condition, which is generally
defined as a disease or condition that affects fewer than 200,000
individuals in the United States. Orphan drug designation must be
requested before submitting an NDA. After the FDA grants orphan
drug designation, the generic identity of the drug and its
potential orphan use are disclosed publicly by the FDA. Orphan drug
designation does not convey any advantage in, or shorten the
duration of, the regulatory review and approval process. The first
NDA applicant to receive FDA approval for a particular active
moiety to treat a particular disease with FDA orphan drug
designation is entitled to a seven-year exclusive marketing period
in the United States for that product and for that indication.
During the seven-year exclusivity period, the FDA may not approve
any other applications to market the same drug for the same orphan
indication, except in limited circumstances, such as a showing of
clinical superiority to the product with orphan drug exclusivity,
such that it is shown to be safer, more effective or makes a major
contribution to patient care. Orphan drug exclusivity does not
prevent the FDA from approving a different drug for the same
disease or condition, or the same drug for a different disease or
condition. Among the other benefits of orphan drug designation are
tax credits for certain research and a waiver of the NDA
application user fee.
Combination Products
The FDA
regulates combinations of products that cross FDA centers, such as
drug, biologic or medical device components that are physically,
chemically or otherwise combined into a single entity, as a
combination product. The FDA center with primary jurisdiction for
the combination product will take the lead in the premarket review
of the product, with the other center consulting or collaborating
with the lead center. The 21st Century Cures Act, or Cures Act,
amended the provisions of the FDCA relating to the regulation of
combination products to, among other things, require the FDA to
conduct the premarket review of any combination product under a
single application whenever appropriate.
In practice, the
FDA’s Office of Combination Products, or OCP, determines
which center will have primary jurisdiction for the combination
product based on the combination product’s “primary
mode of action.” A mode of action is the means by which a
product achieves an intended therapeutic effect or action. The
primary mode of action is the mode of action that provides the most
important therapeutic action of the combination product, or the
mode of action expected to make the greatest contribution to the
overall intended therapeutic effects of the combination
product.
It is often
difficult for the OCP to determine with reasonable certainty the
most important therapeutic action of the combination product. In
those difficult cases, the OCP will consider consistency with other
combination products raising similar types of safety and
effectiveness questions, or which center has the most expertise to
evaluate the most significant safety and effectiveness questions
raised by the combination product.
If a combination
product sponsor disagrees with OCP’s primary mode of action
determination, the Cures Act permits the sponsor to request that
the FDA provide a substantive rationale for its determination. The
sponsor can then propose one or more studies to establish the
relevance of the chemical action in achieving the product’s
primary mode of action and the FDA and the sponsor will collaborate
to reach agreement on the design of such studies within 90 calendar
days. If the sponsor conducts the agreed-upon studies, the FDA must
consider the resulting data when reevaluating the product’s
primary mode of action.
-18-
Post-Market Drug Regulation
If the FDA
approves a drug product for commercial marketing, it may limit the
approved indications for use of the product, require that
contraindications, warnings or precautions be included in the
product labeling, require that post-approval studies, including
Phase 4 clinical trials, be conducted to further assess a
drug’s safety and/or other factors after approval, require
testing and surveillance programs to monitor the product after
commercialization and/or patients using the product for observation
of the product’s long-term effects, or impose other
conditions, including distribution restrictions or other risk
management mechanisms, including Risk Evaluation and Mitigation
Strategies, or REMS, which can materially affect the potential
market and profitability of the product. Any approved product is
also subject to requirements relating to recordkeeping, periodic
reporting, product sampling and distribution, advertising and
promotion, labeling, and reporting of adverse experiences with the
product. The FDA may prevent or limit further marketing of a
product based on the results of post-market studies or surveillance
programs. After approval, some types of changes to the approved
product, such as adding new indications, manufacturing changes and
additional labeling claims, are subject to further testing
requirements and FDA review and re-approval.
In addition,
drug manufacturers and other entities involved in the manufacture
and distribution of approved drugs are required to register their
establishments with the FDA and state agencies and are subject to
periodic unannounced inspections by the FDA and these state
agencies for compliance with cGMP requirements. Changes to the
manufacturing process are strictly regulated and often require
prior FDA approval before being implemented. FDA regulations also
require investigation and correction of any deviations from cGMP
and impose reporting and documentation requirements upon drug
developers and their manufacturers. Accordingly, manufacturers must
continue to expend time, money and effort in the areas of
production and quality control to maintain cGMP
compliance.
Once an approval
is granted, the FDA may withdraw the approval if compliance with
regulatory requirements and standards is not maintained or if
problems occur after the product reaches the market. Later
discovery of previously unknown problems with a product, including
AEs of unanticipated severity or frequency, or with manufacturing
processes, or failure to comply with regulatory requirements, may
result in revisions to the approved labeling to add new safety
information, imposition of post-market studies or clinical trials
to assess new safety risks or imposition of distribution or other
restrictions under a REMS program. Other potential consequences of
a failure to comply with regulatory requirements during or after
the FDA approval process include, among other things:
●
restrictions on
the marketing or manufacturing of the product, product recalls or
complete withdrawal of the product from the market;
●
fines, warning
or untitled letters or holds on post-approval clinical
trials;
●
refusal of the
FDA to approve pending applications or supplements to approved
applications, or suspension or revocation of product license
approvals;
●
product seizure
or detention, or refusal to permit the import or export of
products; or
●
consent decrees,
injunctions or the imposition of civil or criminal
penalties.
The FDA strictly
regulates marketing, labeling, advertising and promotion of
products that are placed on the market. Drugs may be promoted only
for the approved indications and in accordance with the provisions
of the approved label. The FDA and other agencies actively enforce
the laws and regulations prohibiting the promotion of off label
uses, and a company that is found to have improperly promoted off
label uses may be subject to significant liability.
-19-
Diagnostic Regulation
The United States
Food and Drug Administration, or the FDA, regulates the sale or
distribution, in interstate commerce, of medical devices, including
in vitro diagnostic test
kits or IVDs, such as our companion diagnostics. Devices subject to
FDA regulation must undergo pre-market review prior to
commercialization unless the device is of a type exempted from such
review. Additionally, medical device manufacturers must comply with
various regulatory requirements under the Federal Food, Drug and
Cosmetic Act, or FDCA, and regulations promulgated under that Act,
including quality system review regulations, unless exempted from
those requirements for particular types of devices. Entities that
fail to comply with FDA requirements can be liable for criminal or
civil penalties, such as recalls, detentions, orders to cease
manufacturing and restrictions on labeling and
promotion.
Clinical
laboratory services, such as our prognostic diagnostic tests are
currently not subject to FDA regulation, but IVDs and
analyte-specific reagents and equipment used by these laboratories
may be subject to FDA regulation. Clinical laboratory tests that
are developed and validated by a laboratory for use in examinations
the laboratory performs itself are called “home brew”
tests or more recently, Laboratory Developed Tests, or LDTs. LDTs
are subject to the Clinical Laboratory Improvement Amendments of
1988, or CLIA.
Beginning in
January 2006, the FDA began indicating its belief that LDTs were
subject to FDA regulation as devices and issued a series of
guidance documents intending to establish a framework by which to
regulate certain laboratory tests. In September 2006, the FDA
issued draft guidance on a new class of tests called "In Vitro
Diagnostic Multivariate Index Assays", or IVDMIAs. Under this draft
guidance, specific tests could be classified as either a Class II
or a Class III medical device, which may require varying levels of
FDA pre-market review depending on intended use and the level of
control necessary to assure the safety and effectiveness of the
test. In July 2007, the FDA posted revised draft guidance that
addressed some of the comments submitted in response to the
September 2006 draft guidance. In May 2007, the FDA issued a
guidance document "Class II Special Controls Guidance Document:
Gene Expression Profiling Test System for Breast Cancer Prognosis."
This guidance document was developed to support the classification
of gene expression profiling test systems for breast cancer
prognosis into Class II. In addition, the Secretary of the
Department of Health and Human Services, or HHS, requested that its
Advisory Committee on Genetics, Health and Society make
recommendations about the oversight of genetics testing. A final
report was published in April 2008. In June 2010, the FDA announced
a public meeting to discuss the agency's oversight of LDTs prompted
by the increased complexity of LDTs and their increasingly
important role in clinical decision making and disease management.
The FDA indicated that it is considering a risk-based application
of oversight to LDTs. The public meeting was held in July 2010 and
further public comments were submitted to the FDA in September
2010. In June 2011, the FDA issued draft guidance regarding
"Commercially Distributed In Vitro Diagnostic Products Labeled for
Research Use Only or Investigational Use Only," which was finalized
in November 2013.
In October 2014,
the FDA published two draft guidance documents that, if finalized,
would implement a regulatory approach for most LDTs. In the draft
guidance documents, the FDA stated that it had serious concerns
regarding the lack of independent review of the evidence of
clinical validity of LDTs and asserted that the requirements under
CLIA do not address the clinical validity of any LDT. The draft
guidance documents proposed to impose a risk-based, phased-in
approach for LDTs similar to the existing framework for
in vitro diagnostic
devices. In January 2017, the FDA released a discussion paper
synthesizing public comments on the 2014 draft guidance documents
and outlining a possible approach to regulation of LDTs. The
discussion paper has no legal status and does not represent a final
version of the LDT draft guidance documents. In the discussion
paper, the FDA states that there is “a growing consensus that
additional oversight of LDTs is necessary.” Similar to the
FDA’s 2014 draft guidance, the FDA’s discussion paper
proposes a risk-based framework that would require most LDTs to
comply with most of the FDA’s regulatory requirements for
medical devices. Unlike the draft guidance, however, the discussion
paper proposes to exempt currently marketed LDTs from premarket
review, requiring only new or modified tests to be approved or
cleared by the agency. In addition, the FDA proposed requiring LDTs
to comply with only a subset of the medical device Quality System
Regulation, or QSRs and proposed other changes from the 2014 draft
guidance. We cannot predict whether the FDA will take action to
regulate LDTs under the new administration or what approach the FDA
will seek to take.
-20-
Legislative
proposals have been introduced in Congress or publicly circulated,
each of which would implement differing approaches to the
regulation of LDTs. We cannot predict the ultimate form of any such
guidance or regulation and the potential impact on our prognostic
diagnostic tests or materials used to perform our prognostic
diagnostic tests. If pre-market review is required, our business
could be negatively impacted until such review is completed and
clearance to market or approval is obtained. FDA could
require we seek pre-market clearance or approval for tests
currently under development delaying product commercialization or
following product launch to require that we stop selling our tests.
If our tests are allowed to remain on the market but there is
uncertainty about our tests, if they are labeled investigational by
the FDA, or if labeling claims the FDA allows us to make are
limited, orders or reimbursement may decline. The regulatory
approval process may involve, among other things, successfully
completing additional clinical trials and submitting a pre-market
clearance notice or filing a pre-market application, or PMA with
the FDA. If pre-market review is required by the FDA, there can be
no assurance that our tests will be cleared or approved on a timely
basis, if at all, nor can there be assurance that labeling claims
will be consistent with our current claims or adequate to support
continued adoption of and reimbursement for our tests. Ongoing
compliance with FDA regulations would increase the cost of
conducting our business, and subject us to inspection by the FDA
and to the requirements of the FDA and penalties for failure to
comply with these requirements. We may also decide voluntarily to
pursue FDA pre-market review of our tests if we determine that
doing so would be appropriate.
While we expect
all materials used in our tests to qualify according to CLIA
regulations, we cannot be certain that the FDA might not enact
rules or guidance documents which could impact our ability to
purchase materials necessary for the performance of our tests.
Should any of the reagents obtained by us from vendors and used in
conducting our tests be affected by future regulatory actions, our
business could be adversely affected by those actions, including
increasing the cost of testing or delaying, limiting or prohibiting
the purchase of reagents necessary to perform testing.
Regulation of Medical Devices and In Vitro Diagnostic Devices
(IVDs) – Companion Diagnostics
We may seek to
develop or seek to partner with third parties to develop
in vitro companion
diagnostics for use in selecting the patients that we believe will
respond to certain drugs. We expect our MENA companion diagnostic
tests will be regulated by the FDA as an IVD, companion diagnostic.
As defined by the FDA, an IVD companion diagnostic is a medical
device that provides information that is essential for the safe and
effective use of a corresponding drug or biological product. An IVD
companion diagnostic helps a health care professional determine
whether a therapeutic product’s benefits to patients will
outweigh any potential side effects or risks.
In August 2014, the
FDA issued guidance that addresses issues critical to developing
in vitro companion
diagnostics. The guidance states that if safe and effective use of
a therapeutic product depends on an in vitro diagnostic, then the FDA
generally will require approval or clearance of the diagnostic at
the same time that the FDA approves the therapeutic product. In
July 2016, the FDA issued a draft guidance intended to assist
sponsors of the drug therapeutic and in vitro companion diagnostic device
on issues related to co-development of the products. The FDA
generally requires in
vitro companion diagnostics intended to select the patients
intended to receive a cancer treatment to obtain approval of a PMA
approval, for that diagnostic simultaneously with approval of the
drug.
To be
commercially distributed in the United States, a medical device,
including IVDs, must receive either 510(k) clearance, de novo
authorization, or PMA approval from the FDA prior to marketing.
There are three classes of medical devices recognized by the FDA,
Class I (low risk), Class II (moderate risk), and Class III (high
risk).
Class I devices
are those for which reasonable assurance of safety and
effectiveness can be provided by adherence to the FDA’s
general controls for medical devices, which include applicable
portions of the FDA’s Quality System Regulation, or QSR,
requirements, facility registration and product listing; reporting
of adverse medical events or AEs; and appropriate, truthful, and
non-misleading labeling, advertising and promotional materials.
Many Class I devices are exempt from premarket regulation; however,
some Class I devices require premarket clearance by the FDA through
the 510(k) premarket notification process discussed
below.
Class II devices
are subject to the FDA’s general controls, and any other
special controls, such as performance standards, post-market
surveillance, and FDA guidelines, deemed necessary by the FDA to
provide reasonable assurance of the devices’ safety and
effectiveness. Premarket review and clearance by the FDA for Class
II devices are accomplished through the 510(k) premarket
notification procedure, although some Class II devices are exempt
from the 510(k) requirements. Premarket notifications are subject
to user fees unless a specific exemption applies. To obtain 510(k)
clearance, a manufacturer must submit a premarket notification
demonstrating that the proposed device is “substantially
equivalent” to a predicate device, which is a previously
cleared 510(k) device or a preamendment device that was in
commercial distribution before May 28, 1976, for which the FDA has
not yet called for the submission of a PMA. In determining
substantial equivalence, the FDA assesses whether the proposed
device has the same intended use and technical characteristic as
the predicate device, or whether the proposed device has different
technological characteristics, but the information submitted in the
premarket notification demonstrates the device is as safe and
effective as a legally marketed device and does not raise different
questions of safety and effectiveness than the predicate device.
The FDA may request additional information, including clinical
data. Under the FDCA, a manufacturer must submit a premarket
notification at least 90 days before introducing a device into
interstate commerce, but the FDA’s review of the premarket
notification can take significantly longer. If the FDA determines
that the device is substantially equivalent to the predicate
device(s), the subject device may be marketed. However, if the FDA
determines that a device is not substantially equivalent to the
predicate device(s), then the device would be regulated as a Class
III device, discussed below. If a manufacturer obtains a 510(k)
clearance for its device and then makes a modification that could
significantly affect the device’s safety or effectiveness, a
new premarket notification must be submitted to the
FDA.
-21-
Class III
devices are those deemed by the FDA to pose the greatest risk, such
as those for which reasonable assurance of the device’s
safety and effectiveness cannot be assured solely by the general
controls and special controls described above and that are
life-sustaining or life-supporting. Some preamendment Class III
devices for which the FDA has not yet required a PMA require the
FDA’s clearance of a premarket notification in order to be
marketed. However, most Class III devices are required to undergo
the PMA process in which the manufacturer must demonstrate
reasonable assurance of the safety and effectiveness of the device
to the FDA’s satisfaction. A PMA application must provide
valid scientific evidence, typically extensive preclinical and
clinical trial data, and information about the device and its
components regarding, among other things, device design,
manufacturing, and labeling. PMA applications (and supplemental PMA
applications) are subject to significantly higher user fees than
510(k) premarket notifications. Some PMA applications are exempt
from a user fee, for example, a small business’s first
PMA.
A PMA for an IVD
typically includes data from preclinical studies and
well-controlled clinical trials. Preclinical data for an IVD
includes many different tests, including how reproducible the
results are when the same sample is tested multiple times by
multiple users at multiple laboratories. The clinical data need to
establish that the test is safe and effective for the proposed
intended use in the indicated population. In addition, the PMA must
include information regarding the test’s clinical utility,
meaning that an IVD provides information that is clinically
meaningful. Such information must be provided even if the clinical
significance of the biomarker is obvious. The applicant may also
rely upon published literature or submit data to the FDA to show
clinical utility.
A PMA also must
provide information about the device and its components regarding,
among other things, device design, manufacturing and labeling. The
sponsor must pay an application fee to the FDA upon submission of a
PMA, which is approximately $250,000 for 2017. As part of the PMA
review, the FDA will typically inspect the manufacturer’s
facilities for compliance with QSR requirements, which impose
elaborate testing, control, documentation and other quality
assurance procedures.
Upon submission,
the FDA determines if the PMA is sufficiently complete to permit a
substantive review, and, if so, the FDA accepts the application for
filing. The FDA then commences an in-depth review of the PMA. The
entire process can typically take multiple years from submission of
the PMA to approval but may take longer. The review time is often
significantly extended as a result of the FDA asking for more
information or clarification of information already provided. The
FDA also may respond with a not approvable determination based on
deficiencies in the PMA application and require additional clinical
trial or other data that are often expensive and time-consuming to
generate and can substantially delay approval.
During the
review period, an FDA advisory committee, typically a panel of
clinicians, may be convened to review the PMA application and
recommend to the FDA whether, or upon what conditions, the device
should be approved. Although the FDA is not bound by the advisory
panel decision, the panel’s recommendation is important to
the FDA’s overall decision-making process.
If the
FDA’s evaluation of the PMA is favorable, the FDA typically
issues an approvable letter requiring the applicant’s
agreement to specific conditions, such as changes in labeling, or
specific additional information, such as submission of final
labeling, in order to secure final approval of the PMA. If the FDA
concludes that the applicable criteria have been met, the FDA will
issue a PMA approval for the approved indications, which can be
more limited than those originally sought by the applicant. The PMA
approval can include post-approval conditions that the FDA believes
necessary to ensure the safety and effectiveness of the device,
including, among other things, restrictions on labeling, promotion,
sale and distribution. Failure to comply with the conditions of
approval can result in an enforcement action, including the loss or
withdrawal of the approval.
Even after
approval of a PMA, a new PMA or PMA supplement may be required in
the event of a modification to the device, its labeling or its
manufacturing process. Supplements to a PMA often require the
submission of the same type of information required for an original
PMA, except that the supplement is generally limited to the
information needed to support the proposed change from the product
covered by the original PMA.
-22-
After a PMA
application is submitted and found to be sufficiently complete, the
FDA begins an in-depth review of the submitted information. During
this review period, the FDA may request additional information or
clarification of information already provided. The FDA also may
convene an advisory panel of outside experts to review and evaluate
the application and provide recommendations to the FDA as to the
approvability of the device. In addition, the FDA generally will
conduct a pre-approval inspection of the manufacturing facility to
ensure compliance with the QSR. The FDA can delay, limit, or deny
approval of a PMA application for many reasons.
Failure to
comply with applicable regulatory requirements can result in
enforcement action by the FDA, which may include any of the
following sanctions: public warning letters, fines, injunctions,
civil or criminal penalties, recall or seizure of products,
operating restrictions, partial suspension or total shutdown of
production, delays in or denial of 510(k) clearance or PMA
applications for new products, challenges to existing 510(k)
clearances or PMA applications, and a recommendation by the FDA to
disallow a device manufacturer from entering into government
contracts. The FDA also has the authority to request repair,
replacement, or refund of the cost of any device manufactured or
distributed. In the event that a supplier fails to maintain
compliance with a device manufacturer’s quality requirements,
the manufacturer may have to qualify a new supplier and could
experience manufacturing delays as a result. We believe that
products we may develop in the future for use as companion
diagnostic tests are likely to be regulated as Class III devices
requiring PMA approval.
Clinical Trials and IDEs
A clinical trial
is almost always required to support a PMA. For significant risk
devices, the FDA regulations require that human clinical
investigations conducted in the U.S. be approved via an
Investigational Device Exemption, or IDE, which must be approved
before clinical testing may commence. In some cases, one or more
smaller IDE studies may precede a pivotal clinical trial intended
to demonstrate the safety and efficacy of the investigational
device. A 30-day waiting period after the submission of each IDE is
required prior to the commencement of clinical testing in humans.
The FDA may disapprove, or approve with conditions, the IDE within
the 30-day period. If disapproved, the clinical trial may not begin
until the deficiencies noted by the FDA are addressed, and another
IDE is submitted to the FDA for approval. If approved with
conditions, the sponsor must address the conditions prior to
commencement of the trial. If the FDA does not respond to the
sponsor within the 30-day period, the IDE is deemed approved and
the clinical study may commence.
IVD trials
usually do not require an IDE approval, so long as, among other
things, the results of the IVD test are not used diagnostically
without confirmation of the test results by another, medically
established diagnostic product or procedure. For a trial where the
IVD result directs the therapeutic care of patients with cancer, we
believe that the FDA would likely consider the investigation to
require an IDE application.
An IDE
application must be supported by appropriate data, such as
laboratory test results, showing that it is safe to test the device
in humans and that the testing protocol is scientifically sound.
The IDE application must also include a description of product
manufacturing and controls, and a proposed clinical trial protocol.
The FDA typically grants IDE approval for a specified number of
patients. All clinical studies of investigational devices,
regardless of whether IDE approval is required, require approval
from an institutional review board, or IRB.
During the
clinical trial, the sponsor must comply with the FDA’s IDE
requirements for investigator selection, trial monitoring,
reporting and record keeping. The investigators must obtain patient
informed consent, rigorously follow the investigational plan and
study protocol, control the disposition of investigational devices
and comply with all reporting and record keeping requirements.
These IDE requirements apply to all investigational devices,
whether considered significant or nonsignificant risk. Prior to
granting PMA approval, the FDA typically inspects the records
relating to the conduct of the study and the clinical data
supporting the PMA for compliance with applicable
requirements.
Clinical trials
must be conducted: (i) in compliance with federal regulations; (ii)
in compliance with good clinical practice, or GCP, an international
standard intended to protect the rights and health of patients and
to define the roles of clinical trial sponsors, investigators, and
monitors; and (iii) under protocols detailing the objectives of the
trial, the parameters to be used in monitoring safety, and the
effectiveness criteria to be evaluated.
-23-
The FDA may
order the temporary, or permanent, discontinuation of a clinical
trial at any time, or impose other sanctions, if it believes that
the clinical trial either is not being conducted in accordance with
FDA requirements or presents an unacceptable risk to the clinical
trial patients. An IRB may also require the clinical trial at a
study site to be halted, either temporarily or permanently, for
failure to comply with the IRB’s requirements, or may impose
other conditions.
Although the QSR
does not fully apply to investigational devices, the requirement
for controls on design and development does apply. The sponsor also
must manufacture the investigational device in conformity with the
quality controls described in the IDE application and any
conditions of IDE approval that the FDA may impose with respect to
manufacturing.
Investigational
IVDs may only be distributed for use in an investigation, and the
labeling must prominently contain the statement “For
Investigational Use Only. The performance characteristics of this
product have not been established.”
Expedited Access Pathway Program
In April 2015,
the FDA issued a final guidance document establishing the Expedited
Access Pathway, or EAP program. The EAP program is intended to
speed patient access to devices (including companion diagnostics)
that demonstrate the potential to address unmet medical needs for
life threatening or irreversibly debilitating diseases or
conditions and are subject to PMA approval or de novo
authorization. In order to be accepted into the EAP program, a
sponsor must demonstrate to the FDA’s satisfaction that the
device is intended to treat or diagnose a life-threatening or
irreversibly debilitating disease or condition and that it
addresses an unmet need. The sponsor must also submit an acceptable
draft Data Development Plan. Once accepted into the program, the
FDA intends to engage with sponsors of EAP devices earlier and more
interactively during the device’s development, assessment,
and review. The FDA will also work with the device sponsor to try
to reduce the time and cost from development to an approval
decision. Elements of the EAP program may include priority review,
interactive review, senior management involvement, and assignment
of a case manager.
Post-Market Device Regulation
After a device
obtains FDA approval and is on the market, numerous regulatory
requirements apply. These requirements include the QSR, labeling
regulations, the FDA’s general prohibition against promoting
products for unapproved or “off-label” uses, the
Medical Device Reporting regulation, which requires that
manufacturers report to the FDA if their device may have caused or
contributed to a death or serious injury or malfunctioned in a way
that would likely cause or contribute to a death or serious injury
if it were to recur, and the Reports of Corrections and Removals
regulation, which requires manufacturers to report recalls and
field actions to the FDA if initiated to reduce a risk to health
posed by the device or to remedy a violation of the
FDCA.
The FDA enforces
these requirements by inspection and market surveillance. If the
FDA finds a violation, it can institute a wide variety of
enforcement actions, ranging from a public warning letter to more
severe sanctions such as fines, injunctions and civil penalties;
recall or seizure of products; operating restrictions, partial
suspension or total shutdown of production; refusing requests for
PMA approval of new products; withdrawing PMA approvals already
granted; and criminal prosecution.
Clinical Laboratory Improvement Amendments of 1988, or CLIA –
Prognostic Diagnostics
LDTs, such as
our prognostic diagnostic tests, are subject to the Clinical
Laboratory Improvement Amendments of 1988, or CLIA, and are not
currently regulated as medical devices under the FDCA. Under CLIA,
a laboratory is any facility which performs laboratory testing on
specimens derived from humans for the purpose of providing
information for the diagnosis, prevention or treatment of disease,
or the impairment of, or assessment of health. We have a current
certificate of accreditation under CLIA to perform high complexity
testing of our prognostic diagnostic tests for breast
cancer.
-24-
As a clinical
reference laboratory as defined under CLIA, we are required to hold
a certificate applicable to the type of work we perform and comply
with certain standards. CLIA further regulates virtually all
clinical laboratories by requiring they be certified by the federal
government and comply with various operational, personnel,
facilities administration, quality, and proficiency requirements
intended to ensure that their clinical laboratory testing services
are accurate, reliable, and timely. Laboratories must register and
list their tests with The Centers for Medicare & Medicaid
Services, or CMS, the agency that oversees CLIA. CLIA compliance
and certification is also a prerequisite to be eligible to bill for
services provided to governmental payor program beneficiaries and
for many private payors. CLIA is user-fee funded. Therefore, all
costs of administering the program must be covered by the regulated
facilities, including certification and survey cost.
To renew our
CLIA certificate, we will be subject to survey and inspection every
two years to assess compliance with program standards and may be
subject to additional inspections without prior notice. The
standards applicable to the testing which we perform may change
over time. We cannot assure that we will be able to operate
profitably should regulatory compliance requirements become
substantially costlier in the future. If our clinical reference
laboratory falls out of compliance with CLIA requirements, we may
be subject to sanctions such as suspension, limitation or
revocation of our CLIA certificate, as well as directed plan of
correction, state on-site monitoring, civil money penalties, civil
injunctive suit or criminal penalties. Additionally, we must
maintain CLIA compliance and certification to be eligible to bill
for tests provided to Medicare beneficiaries. If we were to be
found out of compliance with CLIA program requirements and
subjected to sanction, our business would be harmed.
CLIA provides
that a state may adopt laboratory regulations that are more
stringent than those under federal law, and a number of states have
implemented their own more stringent laboratory regulatory
requirements. State laws may require that laboratories meet certain
personnel qualifications, specify certain quality control
procedures, meet facility requirements, or prescribe record
maintenance requirements.
If regulated by
the FDA, we believe that our LDTs would likely be regulated as
either Class II or Class III devices. Accordingly, premarket
review—either a 510(k), de novo application, or a
PMA—would likely be required for our tests if the FDA no
longer applies its enforcement discretion to LDTs and our tests do
not qualify as grandfathered tests that are exempted from premarket
review. While the data requirements are typically greater for Class
III devices, the data required for Class II devices has increased,
and it is likely that some amount of clinical data (retrospective
or prospective or both) would be required for any type of
submission to the FDA. Ongoing compliance with FDA regulations
would increase the cost of conducting our business, subject us to
inspection by the FDA and to the requirements of the FDA and
penalties for failure to comply with the requirements of the FDA.
We cannot assure you that our current prognostic diagnostic
products and other future products will not require 510(k)
clearance or PMA approval in the future, or, in such an event, that
such approval or clearance would be forthcoming. Should any of the
clinical laboratory device reagents obtained by us from vendors and
used in conducting our home brew test be affected by future
regulatory actions, we could be adversely affected by those
actions, including increased cost of testing or delay, limitation
or prohibition on the purchase of reagents necessary to perform
testing.
Massachusetts and Other States’ Laboratory
Testing
Our clinical
reference laboratory is located in Boston, Massachusetts.
Accordingly, we are required to be licensed by Massachusetts, under
Massachusetts laws and regulations, as well as CLIA under CMS
regulations, which both establish standards for:
●
Day-to-day
operation of a clinical laboratory, personnel standards including
training and competency of all laboratory staff;
●
Physical
requirements of a facility, including, policies and procedures; and
safety;
●
Equipment;
and
●
Quality control,
including quality assurance; and proficiency testing.
-25-
In 2015, we
received the necessary certifications and licenses from both CLIA
and Massachusetts for our clinical reference laboratory to perform
testing services of our prognostic diagnostic breast cancer
tests.
If a laboratory
is not in compliance with Massachusetts statutory or regulatory
standards, or CLIA regulations as mandated by CMS, the
Massachusetts State Department of Health and/or CMS may suspend,
limit, revoke or annul the laboratory’s Massachusetts
license, and CLIA certification, censure the holder of the license
or assess civil money penalties. Additionally, statutory or
regulatory noncompliance may result in a laboratory’s
operator being found guilty of a misdemeanor. In the event that we
should be found not to be in compliance with Massachusetts or CLIA
laboratory requirements, we could be subject to such sanctions,
which could harm our business.
California, New
York, Florida, Maryland, Pennsylvania and Rhode Island require
out-of-state laboratories, which accept specimens from those states
to be licensed in each state. We have received licensing from
Massachusetts, California, Florida, Pennsylvania and Rhode Island
and are currently seeking licensing from New York and Maryland.
From time to time, we may become aware of other states that require
out-of-state laboratories to obtain licensure in order to accept
specimens from the state, and it is possible that other states do
have such requirements or will have such requirements in the
future. If we identify any other state with such requirements or if
we are contacted by any other state advising us of such
requirements, we intend to follow instructions from the state
regulators as to how we should comply with such
requirements.
Additional Regulations and Environmental Matters
Health Insurance Portability and Accountability Act (HIPAA) and
HITECH
Under the
administrative simplification provisions the federal Health
Insurance Portability and Accountability Act of 1996, or HIPAA, as
amended by the Health Information Technology for Economic and
Clinical Health Act, or the HITECH Act, the United States
Department of Health and Human Services (HHS) issued regulations
that establish uniform standards governing the conduct of certain
electronic health care transactions and protecting the privacy and
security of protected health information used or disclosed by
health care providers and other covered entities, such as MetaStat.
Three principal regulations with which we are required to comply
have been issued in final form under HIPAA: privacy regulations,
security regulations, and standards for electronic transactions,
which establish standards for common health care transactions. The
privacy and security regulations were extensively amended in 2013
to incorporate requirements from the HITECH Act.
The privacy
regulations cover the use and disclosure of protected health
information by health care providers and other covered entities.
They also set forth certain rights that an individual has with
respect to his or her protected health information maintained by a
health care provider, including the right to access or amend
certain records containing protected health information, or to
request restrictions on the use or disclosure of protected health
information. The security regulations establish requirements for
safeguarding the confidentiality, integrity, and availability of
protected health information that is electronically transmitted or
electronically stored.
The HITECH Act,
among other things, established certain protected health
information security breach notification requirements. A covered
entity must notify affected individual(s) and the HHS when there is
a breach of unsecured protected health information. The HIPAA
privacy and security regulations establish a uniform federal
“floor” that health care providers must meet and do not
supersede state laws that are more stringent or provide individuals
with greater rights with respect to the privacy or security of, and
access to, their records containing protected health information.
Massachusetts, for example, has a state law that protects the
privacy and security of personal information of Massachusetts
residents that is more prescriptive than HIPAA.
These laws
contain significant fines and other include civil and criminal
penalties for wrongful use or disclosure of protected health
information. Additionally, to the extent that we submit electronic
health care claims and payment transactions that do not comply with
the electronic data transmission standards established under HIPAA
and the HITECH Act, payments to us may be delayed or
denied.
We have policies
and procedures to comply with these regulations. The requirements
under these regulations may change periodically and could have an
adverse effect on our business operations if compliance becomes
substantially costlier than under current
requirements.
-26-
In addition to
federal privacy regulations, there are a number of state and
international laws governing confidentiality of health information
that may be applicable to our operations. The United States
Department of Commerce, the European Commission and the Swiss
Federal Data Protection and Information Commissioner have agreed on
a set of data protection principles and frequently asked questions
(the "Safe Harbor Principles") to enable U.S. companies to satisfy
the requirement under European Union and Swiss law that adequate
protection is given to personal information transferred from the
European Union or Switzerland to the United States. The European
Commission and Switzerland have also recognized the Safe Harbor
Principles as providing adequate data protection.
New laws
governing privacy may be adopted in the future as well. We have
taken steps to comply with health information privacy requirements
to which we are aware that we will be subject. However, we cannot
provide assurance that we will be in compliance with diverse
privacy requirements in all of the jurisdictions in which we do
business. Failure to comply with privacy requirements could result
in civil or criminal penalties, which could have a materially
adverse impact on our business.
Federal and State Physician Self-Referral Prohibitions
We will be
subject to the federal physician self-referral prohibitions,
commonly known as the Stark Law, and to similar state restrictions
such as the California's Physician Ownership and Referral Act, or
PORA. Together these restrictions generally prohibit us from
billing a patient or any governmental or private payer for any test
when the physician ordering the test, or any member of such
physician's immediate family, has an investment interest in or
compensation arrangement with us, unless the arrangement meets an
exception to the prohibition. Both the Stark Law and PORA contain
an exception for compensation paid to a physician for personal
services rendered by the physician. We would be required to refund
any payments we receive pursuant to a referral prohibited by these
laws to the patient, the payer or the Medicare program, as
applicable.
Both the Stark
Law and certain state restrictions such as PORA contain an
exception for referrals made by physicians who hold investment
interests in a publicly traded company that has stockholders’
equity exceeding $75 million at the end of its most recent fiscal
year or on average during the previous three fiscal years, and
which satisfies certain other requirements. In addition, both the
Stark Law and certain state restrictions such as PORA contain an
exception for compensation paid to a physician for personal
services rendered by the physician.
However, in the
event that we enter into any compensation arrangements with
physicians, we cannot be certain that regulators would find these
arrangements to be in compliance with Stark, PORA or similar state
laws. In such event, we would be required to refund any payments we
receive pursuant to a referral prohibited by these laws to the
patient, the payer or the Medicare program, as
applicable.
Sanctions for a
violation of the Stark Law include the following:
●
denial of
payment for the services provided in violation of the
prohibition;
●
refunds of
amounts collected by an entity in violation of the Stark
Law;
●
a
civil penalty of up to $15,000 for each service arising out of the
prohibited referral;
●
possible
exclusion from federal healthcare programs, including Medicare and
Medicaid; and
●
a
civil penalty of up to $100,000 against parties that enter into a
scheme to circumvent the Stark Law’s
prohibition.
-27-
These
prohibitions apply regardless of the reasons for the financial
relationship and the referral. No finding of intent to violate the
Stark Law is required for a violation. In addition, under an
emerging legal theory, knowing violations of the Stark Law may also
serve as the basis for liability under the Federal False Claims
Act.
Further, a
violation of PORA is a misdemeanor and could result in civil
penalties and criminal fines. Finally, other states have
self-referral restrictions with which we have to comply that differ
from those imposed by federal and California law. It is possible
that any financial arrangements that we may enter into with
physicians could be subject to regulatory scrutiny at some point in
the future, and we cannot provide assurance that we will be found
to be in compliance with these laws following any such regulatory
review.
Federal, State and International Anti-kickback Laws
The Federal
Anti-Kickback Law makes it a felony for a provider or supplier,
including a laboratory, to knowingly and willfully offer, pay,
solicit or receive remuneration, directly or indirectly, in order
to induce business that is reimbursable under any federal health
care program. A violation of the Anti-kickback Law may result in
imprisonment for up to five years and fines of up to $250,000 in
the case of individuals and $500,000 in the case of organizations.
Convictions under the Anti-kickback Law result in mandatory
exclusion from federal health care programs for a minimum of five
years. In addition, HHS has the authority to impose civil
assessments and fines and to exclude health care providers and
others engaged in prohibited activities from Medicare, Medicaid and
other federal health care programs.
Actions which
violate the Anti-kickback Law or similar laws may also involve
liability under the Federal False Claims Act, which prohibits the
knowing presentation of a false, fictitious or fraudulent claim for
payment to the United States Government. Actions under the Federal
False Claims Act may be brought by the Department of Justice or by
a private individual in the name of the government.
Although the
Anti-kickback Law applies only to federal health care programs, a
number of states have passed statutes substantially similar to the
Anti-kickback Law pursuant to which similar types of prohibitions
are made applicable to all other health plans and third-party
payers.
Federal and
state law enforcement authorities scrutinize arrangements between
health care providers and potential referral sources to ensure that
the arrangements are not designed as a mechanism to induce patient
care referrals and opportunities. The law enforcement authorities,
the courts and the United States Congress have also demonstrated a
willingness to look behind the formalities of a transaction to
determine the underlying purpose of payments between health care
providers and actual or potential referral sources. Generally,
courts have taken a broad interpretation of the scope of the
Anti-kickback Law, holding that the statute may be violated if
merely one purpose of a payment arrangement is to induce future
referrals.
In addition to
statutory exceptions to the Anti-kickback Law, regulations provide
for a number of safe harbors. If an arrangement meets the
provisions of a safe harbor, it is deemed not to violate the
Anti-kickback Law. An arrangement must fully comply with each
element of an applicable safe harbor in order to qualify for
protection.
Among the safe
harbors that may be relevant to us is the discount safe harbor. The
discount safe harbor potentially applies to discounts provided by
providers and suppliers, including laboratories, to physicians or
institutions where the physician or institution bills the payer for
the test, not when the laboratory bills the payer directly. If the
terms of the discount safe harbor are met, the discounts will not
be considered prohibited remuneration under the Anti-kickback Law.
We anticipate that this safe harbor may be potentially applicable
to any agreements that we enter into to sell tests to hospitals
where the hospital submits a claim to the payer.
The personal
services safe harbor to the Anti-kickback Law provides that
remuneration paid to a referral source for personal services will
not violate the Anti-kickback Law provided all of the elements of
that safe harbor are met. One element is that, if the agreement is
intended to provide for the services of the physician on a
periodic, sporadic or part-time basis, rather than on a full-time
basis for the term of the agreement, the agreement specifies
exactly the schedule of such intervals, their precise length, and
the exact charge for such intervals. Failure to meet the terms of
the safe harbor does not render an arrangement illegal. Rather,
such arrangements must be evaluated under the language of the
statute, taking into account all facts and
circumstances.
-28-
In the event
that we enter into relationships with physicians, hospitals and
other customers, there can be no assurance that our relationships
with those physicians, hospitals and other customers will not be
subject to investigation or a successful challenge under such laws.
If imposed for any reason, sanctions under the Anti-kickback Law or
similar laws could have a negative effect on our
business.
Other Federal and State Fraud and Abuse Laws
In addition to
the requirements that are discussed above, there are several other
health care fraud and abuse laws that could have an impact on our
business. For example, provisions of the Social Security Act permit
Medicare and Medicaid to exclude an entity that charges the federal
health care programs substantially in excess of its usual charges
for its services. The terms “usual charge” and
“substantially in excess” are ambiguous and subject to
varying interpretations.
Further, the
Federal False Claims Act prohibits a person from knowingly
submitting a claim, making a false record or statement in order to
secure payment or retaining an overpayment by the federal
government. In addition to actions initiated by the government
itself, the statute authorizes actions to be brought on behalf of
the federal government by a private party having knowledge of the
alleged fraud. Because the complaint is initially filed under seal,
the action may be pending for some time before the defendant is
even aware of the action. If the government is ultimately
successful in obtaining redress in the matter or if the plaintiff
succeeds in obtaining redress without the government’s
involvement, then the plaintiff will receive a percentage of the
recovery. Finally, the Social Security Act includes its own
provisions that prohibit the filing of false claims or submitting
false statements in order to obtain payment. Violation of these
provisions may result in fines, imprisonment or both, and possible
exclusion from Medicare or Medicaid programs.
Corporate Practice of Medicine
Numerous states
have enacted laws prohibiting business corporations, such as
MetaStat, from practicing medicine and employing or engaging
physicians to practice medicine, generally referred to as the
prohibition against the corporate practice of medicine. These laws
are designed to prevent interference in the medical decision-making
process by anyone who is not a licensed physician. For example,
California’s Medical Board has indicated that determining
what diagnostic tests are appropriate for a particular condition
and taking responsibility for the ultimate overall care of the
patient, including providing treatment options available to the
patient, would constitute the unlicensed practice of medicine if
performed by an unlicensed person. Violation of these corporate
practice of medicine laws may result in civil or criminal fines, as
well as sanctions imposed against us and/or the professional
through licensure proceedings. Typically, such laws are only
applicable to entities that have a physical presence in the
state.
Compliance with Environmental Laws
We expect to be
subject to regulation under federal, state and local laws and
regulations governing environmental protection and the use,
storage, handling and disposal of hazardous substances. The cost of
complying with these laws and regulations may be significant. Our
planned activities may require the controlled use of potentially
harmful biological materials, hazardous materials and chemicals. We
cannot eliminate the risk of accidental contamination or injury to
employees or third parties from the use, storage, handling or
disposal of these materials. In the event of contamination or
injury, we could be held liable for any resulting damages, and any
liability could exceed our resources or any applicable insurance
coverage we may have.
Other Regulations
The U.S.
Occupational Safety and Health Administration has established
extensive requirements relating to workplace safety for health care
employers, including requirements to develop and implement programs
to protect workers from exposure to blood-borne pathogens by
preventing or minimizing any exposure through needle stick or
similar penetrating injuries.
-29-
Foreign Regulation
To obtain
marketing approval of a drug under European Union regulatory
systems, we may submit marketing authorization applications, or
MAAs, either under a centralized or decentralized procedure. The
centralized procedure provides for the grant of a single marketing
authorization that is valid for all European Union member states.
The centralized procedure is compulsory for medicines produced by
specified biotechnological processes, products designated as orphan
medicinal products, and products with a new active substance
indicated for the treatment of specified diseases, and optional for
those products that are highly innovative or for which a
centralized process is in the interest of patients. Under the
centralized procedure in the European Union, the maximum timeframe
for the evaluation of an MAA is 210 days, excluding clock stops,
when additional written or oral information is to be provided by
the applicant in response to questions asked by the Scientific
Advice Working Party of the Committee of Medicinal Products for
Human Use, or the CHMP. Accelerated evaluation might be granted by
the CHMP in exceptional cases, when a medicinal product is expected
to be of a major public health interest, defined by three
cumulative criteria comprising the seriousness of the disease, such
as heavy disabling or life-threatening diseases, to be treated; the
absence or insufficiency of an appropriate alternative therapeutic
approach; and anticipation of high therapeutic benefit. In this
circumstance, the European Medicines Agency, or EMA, ensures that
the opinion of the CHMP is given within 150 days.
The EMA grants
orphan drug designation to promote the development of products that
may offer therapeutic benefits for life-threatening or chronically
debilitating conditions affecting not more than five in 10,000
people in the European Union. In addition, orphan drug designation
can be granted if the drug is intended for a life threatening,
seriously debilitating or serious and chronic condition in the
European Union and without incentives it is unlikely that sales of
the drug in the European Union would be sufficient to justify
developing the drug. Orphan drug designation is only available if
there is no other satisfactory method approved in the European
Union of diagnosing, preventing or treating the condition, or if
such a method exists, the proposed orphan drug will be of
significant benefit to patients. Orphan drug designation provides
opportunities for free protocol assistance, fee reductions for
access to the centralized regulatory procedures before and during
the first year after marketing authorization and between 6 and 10
years of market exclusivity following drug approval.
The
decentralized procedure for submitting an MAA provides an
assessment of an application performed by one-member state, known
as the reference member state, and the approval of that assessment
by one or more other member states, known as concerned member
states. Under this procedure, an applicant submits an application,
or dossier, and related materials, including a draft summary of
product characteristics, and draft labeling and package leaflet, to
the reference member state and concerned member states. The
reference member state prepares a draft assessment and drafts of
the related materials within 120 days after receipt of a valid
application. Within 90 days of receiving the reference member
state’s assessment report, each concerned member state must
decide whether to approve the assessment report and related
materials. If a member state cannot approve the assessment report
and related materials on the grounds of potential serious risk to
public health, the disputed points may eventually be referred to
the European Commission, whose decision is binding on all member
states. Prior to submitting an MAA for use of drugs in pediatric
populations, the EMA requires submission of, or a request for
waiver or deferral of, a Pediatric Investigation Plan.
In the European
Union, new chemical entities qualify for eight years of data
exclusivity upon marketing authorization and an additional two
years of market exclusivity. This data exclusivity, if granted,
prevents regulatory authorities in the European Union from
assessing a generic (abbreviated) application for eight years,
after which generic marketing authorization can be submitted but
not approved for two years. Even if a compound is considered to be
a new chemical entity and the sponsor is able to gain the
prescribed period of data exclusivity, another company nevertheless
could also market another version of the drug if such company can
complete a full MAA with a complete human clinical trial database
and obtain marketing approval of its product.
Healthcare Reform
Containing
healthcare expenditures is a major trend in the U.S. and the rest
of the world. Both government authorities and third-party payors
have attempted to control costs by limiting coverage and the amount
of reimbursement for particular medical products, including
therapeutics and diagnostics, implementing reductions in Medicare
and other healthcare funding, and applying new payment
methodologies. For example, in March 2010, the Affordable Care Act
or ACA was enacted, which, among other things, subjected drug
manufacturers to new annual fees based on pharmaceutical
companies’ share of sales to federal healthcare programs;
created a new Patient Centered Outcomes Research Institute to
oversee, identify priorities in, and conduct comparative clinical
effectiveness research, along with funding for such research;
creation of the Independent Payment Advisory Board, which has
authority to recommend certain changes to the Medicare program that
could result in reduced payments for prescription drugs; and
establishment of a Center for Medicare Innovation at the CMS to
test innovative payment and service delivery models to lower
Medicare and Medicaid spending.
-30-
The
recent presidential and congressional elections in the U.S. could
result in significant changes in, and uncertainty with respect to,
legislation, regulation and government policy that could
significantly impact our business and the healthcare industry.
While it is not possible to predict whether and when any such
changes will occur, a variety of initiatives to repeal or
significantly reform key provisions of the ACA have been introduced
in Congress or otherwise proposed. Most notably, Congress enacted
legislation in 2017 that eliminates the ACA’s
“individual mandate” beginning in 2019, which may
significantly impact the number of covered lives participating in
exchange plans. We expect that the new U.S. President and
Congress will continue to seek to modify, repeal, or otherwise
invalidate all, or certain provisions of the ACA. Any changes will
likely take time to unfold, and could have an impact on coverage
and reimbursement for healthcare items and services covered by
plans that were authorized by the ACA. However, we cannot predict
the ultimate content, timing or effect of any healthcare reform
legislation or the impact of potential legislation on
us.
In
addition, other legislative changes have also been proposed and
adopted in the U.S. to reduce healthcare expenditures. These
changes include aggregate reductions of Medicare payments to
providers of 2% per fiscal year that, due to subsequent legislative
amendments, will remain in effect through 2025 unless additional
action is taken by Congress. The American Taxpayer Relief Act of
2012 was signed into law in January 2013, which, among other
things, further reduced Medicare payments to several types of
providers, including hospitals, imaging centers and cancer
treatment centers, and increased the statute of limitations period
for the government to recover overpayments to providers to five
years from three. Recently there has been heightened scrutiny over
the manner in which manufacturers set prices for their marketed
products.
We expect that additional federal and state healthcare reform
measures will be adopted in the future, any of which could limit
the amounts that federal and state governments will pay for
healthcare products and services, which could result in reduced
demand for our therapeutic and diagnostic products or additional
pricing pressures. Other potentially significant changes in policy
include the possibility of modifications and elimination of
programs and reductions in staffing at the FDA and other government
agencies, and initiatives to contain or reduce governmental
spending in the healthcare area, including Medicare and Medicaid
reimbursement. We cannot predict what future healthcare
initiatives will be introduced or implemented at the federal or
state level, or how any future legislation or regulation may affect
us.
Reimbursement
Sales of any of our
therapeutic and companion diagnostic product candidates that may be
approved will depend, in part, on the extent to which the cost of
the products will be covered by government and third-party payers.
Third party payers may limit coverage to an approved list of
products, or formulary, which might not include all drug products
approved by the FDA for an indication. Any product candidates for
which we obtain marketing approval may not be considered medically
necessary or cost-effective by third party payers, and we may need
to conduct expensive pharmacoeconomic studies in the future to
demonstrate the medical necessity and/or cost effectiveness of any
such product.
The reimbursement
environment is evolving as regulators and payors try to establish
new rules and frameworks for the reimbursement of molecular
diagnostic tests. There has been an increased interest in
implementing cost containment programs to limit government-paid
health care costs, including price controls and restrictions on
reimbursement. Continued interest in and adoption of such controls
and measures and tightening of restrictive policies in
jurisdictions with existing controls and measures, could limit
payments for product candidates we are developing.
Our prognostic
diagnostic tests are expected to be offered as a clinical
laboratory service. Revenue for clinical laboratory diagnostics may
come from several sources, including commercial third-party payers,
such as insurance companies and health maintenance organizations
(HMOs), government payers, such as Medicare and Medicaid in the
United States, patient self-pay and, in some cases, from hospitals
or referring laboratories who, in turn, may bill third-party
payers.
The proportion
of private payers compared to government payers such as
Medicaid/Medicare will impact the average selling price
(discounting), length of payables, and losses due to uncollectible
accounts receivable. Working with relevant medical societies and
other appropriate constituents to obtain appropriate reimbursement
amounts by all payers will be key. The objective of this effort
will be to ensure the amount paid by Medicare and other payers for
our assays accurately reflects the technology costs, the benefit
that the analysis brings to patients, and its positive impact on
healthcare economics. In order to gain broad reimbursement
coverage, we expect substantial resources will need to be devoted
to educating payers such as Kaiser Permanente, Aetna, United
Healthcare, and others on the following attributes of our
prognostic diagnostic assays, including, but not limited
to:
●
test
performance (specificity, selectivity, size of the risk
groups);
●
clinical utility
and effectiveness;
●
peer-reviewed
publication and consistent study outcomes;
●
patient and
physician demand; and
●
improved health
economics.
-31-
Billing codes
are the means by which Medicare and private insurers identify
certain medical services that are provided to patients in the
United States. CPT codes are established by the American Medical
Association (AMA). The amounts reimbursed by Medicare for the CPT
codes are established by the Centers for Medicare & Medicaid
Services (CMS) using a relative value system, with recommendations
from the AMA's Relative Value Update Committee and professional
societies representing the various medical
specialties.
Reimbursement
for our prognostic diagnostic tests will be based on:
●
eligibility for
reimbursement under well-established medical billing CPT code
88361;
●
reimbursement
under the CPT miscellaneous procedure code; or
●
qualification
under any applicable new molecular diagnostic codes currently under
consideration.
As part of our
longer-term reimbursement strategy, we or any potential partners
may choose to apply for a unique CPT code once our prognostic
diagnostic assays are commercially available and health economic
data have been established.
Well-established medical billing CPT code 88361
CPT code 88361
is specific to computer-assisted image analysis and went into
effect in 2004. Our prognostic diagnostic tests involve both a
technical and professional component. The technical component
involves preparation of the patient sample and scanning the image,
while the professional component involves the physician's reading
and evaluation of the test results. Since our prognostic diagnostic
tests will be billed as a service, we anticipate payments for both
the professional and technical components. The actual payment
varies based upon a geographic factor index for each state and may
be higher or lower than the Medicare national amounts in particular
cases based on geographic location.
CMS coding
policy defines the unit of service for each IHC stain charge is one
unit per different antigen tested and individually reported, per
specimen. Medicare contractors cannot bill for multiple service
units of CPT code 88361 (Immunohistochemistry, each antibody) for
“cocktail” stains containing multiple antibodies in a
single “vial” applied in a single procedure, even if
each antibody provides distinct diagnostic information. We believe
this CMS policy is not applicable to our procedure because our
multiple stain reaction involves multiple separate steps of
multiple primary antibodies binding followed by
counterstaining.
CPT Miscellaneous Procedure Code
Tests that are
billed under a non-specific, unlisted procedure code are subject to
manual review of each claim. Claims are paid at a rate established
by the local Medicare carrier in Massachusetts and based upon the
development and validation costs of developing the assays, the
costs of conducting the tests, the reimbursement rates paid by
other payers and the cost savings impact of the tests. Because
there is no specific code or national fee schedule rate for the
test, payment rates established by the local Medicare contractor
may be subject to review and adjustment at any time.
Sales
and Marketing
We have not yet
established a sales and marketing infrastructure. For any of our
therapeutic or companion diagnostic product candidates for which we
may in the future receive marketing approvals, we may seek to
commercialize the product ourselves or through one or more
strategic commercialization collaborations.
-32-
Our prognostic
diagnostic tests are expected to be offered as a clinical
laboratory service through our CLIA-certified laboratory located in
Boston, Massachusetts. We plan to implement a de-risked
commercialization strategy based on non-exclusive agreements with
strategic distribution partners and/or CSOs in the U.S. and
distributors in Europe and throughout the
rest-of-world.
We aim to enter
into agreements with commercialization or strategic partners that
have existing commercialization infrastructure, established
distribution channels, and strong relationships with our target
audience in the medical community. We aim to avoid the cost and
risk associated with building a new sales and marketing
infrastructure.
Manufacturing
We do not own or
operate, and currently have no plans to establish, any
manufacturing facilities. We expect to rely on third parties for
the manufacture of any therapeutic product candidates for
preclinical and clinical testing, as well as for commercial
manufacture of any products that we may commercialize.
Our
state-of-the-art CLIA-certified reference laboratory is located at
27 Drydock Avenue in Boston, MA. Our CLIA-certified laboratory is
our primary location for our diagnostic testing and data analysis
of patient tumor samples. Although the science behind our
diagnostic technology is cutting edge and sophisticated, a key
competitive advantage of our approach is that we have simplified
our testing methods and procedures based on established
immunohistochemical, or IHC, and quantitative immunofluorescence,
or QIF techniques and utilize common inexpensive
materials.
The MENA diagnostic
assays use widely available QIF techniques to identify individual
cell types, allowing the test to interrogate tumor cells separately
within tumor microenvironment rather than measuring homogenous
biopsies containing tumor and non-tumor cell types. This staining
technique uses antibodies that recognize or detect MENA. The
antibodies used are detected by labeling the different antibody
types different fluorescent dyes that allow the operator to measure
and quantify the levels selectively within the tumor cells on the
slide. We believe this approach to diagnosis and prognosis of
cancer is more cost effective than many genomic-based approaches
currently on the market that utilize heterogeneous mixtures of
tumor and stromal cells in patient samples. We believe the most
economical way to enter the market with the MENA diagnostic assay
will be through contract manufacturing the manufacturing of MENA
mAbs and other QIFs.
The MetaSite
Breast™ assay uses
widely available IHC dyeing techniques to identify individual cell
types. This staining technique uses antibodies that recognize
individual cell types. By attaching different dye colors to
different antibody types, the operator can view different cell
types on a single slide. We believe this approach to diagnosis and
prognosis of cancer is more cost effective than many genomic-based
approaches currently on the market. We believe the most economical
way to enter the market with the MetaSite Breast™ test will be through
contract manufacturing for these IHCs.
Employees
We currently
have six full-time employees. In addition, we utilize outside
consultants to support certain elements of our research and
development, information technology, and general and administrative
operations. From time to time we have also engaged several
consulting firms involved with public relations, investor relations
and other functions.
Insurance
We have general
and umbrella liability insurance, employment practices liability
insurance as well as directors and officers (D&O) insurance in
amounts that we believe comply with industry
standards.
Legal Proceedings
We are not
engaged in any material litigation, arbitration or claim, and no
material litigation, arbitration or claim is known by our
management to be pending or threatened by or against us that would
have a material adverse effect on our results from operations or
financial condition.
-33-
Corporate Structure
We were
incorporated on March 28, 2007 under the laws of the State of
Nevada. From inception until November of 2008, our business plan
was to produce and market inexpensive solar cells and in November
2008, our board of directors determined that the implementation of
our business plan was no longer financially feasible. At such time,
we discontinued the implementation of our prior business plan and
pursued an acquisition strategy, whereby we sought to acquire a
business. Based on these business activities, until February 27,
2012, we were considered a "blank check" company, with no or
nominal assets (other than cash) nor operations.
MetaStat
BioMedical, Inc. (“MBM”) (formerly known as MetaStat,
Inc.), our wholly owned Delaware subsidiary, was incorporated in
the State of Texas on July 22, 2009 and re-incorporated in the
State of Delaware on August 26, 2010. MBM was formed to allow
cancer patients to benefit from the latest discoveries in how
cancer spreads to other organs in the body. The Company’s
mission is to become an industry leader in the emerging field of
personalized cancer therapy.
On February 27,
2012 (the “Closing Date”), we consummated a share
exchange as more fully described below, whereby we acquired all the
outstanding shares of MBM and, MBM became our wholly owned
subsidiary. From and after the share exchange, our business is
conducted through our wholly owned subsidiary, MBM, and the
discussion of our business is that of our current business which is
conducted through MBM.
Prior to April
9, 2012, our company name was Photovoltaic Solar Cells, Inc. For
the sole purpose of changing our name, on April 9, 2012, we merged
with a newly-formed, wholly owned subsidiary incorporated under the
laws of Nevada called MetaStat, Inc. As a result of the merger, our
corporate name was changed to MetaStat, Inc. In May 2012, we
changed the name of our Delaware subsidiary to MetaStat BioMedical,
Inc. from MetaStat, Inc.
Share Exchange
On the Closing
Date, we entered into a Share Exchange Agreement (the
“Exchange Agreement”) by and among us, MBM, the holders
of all outstanding shares of MBM (the “MBM
Shareholders”) and Waterford Capital Acquisition Co IX, LLC,
our principal shareholder (the “Company Principal
Shareholder”), whereby we acquired all of the outstanding
shares of MBM (the “MBM Shares”) from the MBM
Shareholders. In exchange, we issued to the MBM Shareholders an
aggregate of 1,224,629 shares of our common stock (the
“Exchange Shares”), equal to 95.6% of our outstanding
shares of common stock after such issuance. As a result of the
transactions contemplated by the Exchange Agreement (collectively,
the “Share Exchange”), MBM became our wholly owned
subsidiary. Pursuant to the Exchange Agreement, we assumed warrants
to purchase up to 52,035 shares of MBM’s common stock, with
exercise prices ranging between $22.50 and $30.00 per share on a
2.2-for-1 basis, equivalent to 114,475 shares of our common stock
with exercise prices ranging from $10.20 to $13.65 per share.
Immediately prior to the Share Exchange, we converted approximately
$336,075 of debt owed to the Company Principal Shareholder into
20,640 shares of our common stock (the “Debt
Conversion”) and issued an aggregate of 2,400 shares of our
common stock to certain of our officers, directors and consultants
in consideration for services rendered to us, leaving 56,000 shares
of our common stock outstanding immediately prior to the issuance
of the Exchange Shares. Additionally, immediately prior to the
Share Exchange, we issued five-year warrants to purchase up to an
aggregate of 23,334 shares of our common stock at an exercise price
of $21.00 per share, of which warrants to purchase 22,500 shares
were issued for a purchase price of $21,000 and warrants to
purchase 834 shares were issued for services rendered to us prior
to the Share Exchange (the “Warrant Financing”). We
used the proceeds of the Warrant Financing to pay off all of our
liabilities prior to the Share Exchange. On the Closing Date, we
assumed MBM’s 2012 Omnibus Securities and Incentive Plan (the
“2012 Incentive Plan”) and reserved 74,453 shares of
our common stock for the benefit of our employees, nonemployee
directors and consultants. All 33,834 options outstanding under the
2012 Incentive Plan were converted, on a 2.2-for-1 basis, into the
right to receive options to purchase up to 74,434 shares of our
common stock with an exercise price of $10.20 per
share.
Principal Executive Offices
Our principal
executive office and clinical reference laboratory are located at
27 Drydock Ave., 2nd Floor, Boston, Massachusetts 02210, We have
additional executive offices at 401 Park Ave. South, 10th Floor,
New York, New York 10016. Our corporate telephone number is (617)
531-6500 and our website is http://www.metastat.com. Information
contained on our website does not constitute part of, and is not
deemed incorporated by reference into, this Form 10-K.
|
Item 1A.
|
RISK FACTORS
|
We operate in a dynamic and rapidly changing environment that
involves numerous risks and uncertainties. Certain factors may have
a material adverse effect on our business, prospects, results of
operations, financial condition and cash flows, and you should
carefully consider them. Accordingly, in evaluating our business,
we encourage you to consider the following discussion of risk
factors, in its entirety, in addition to other information
contained in this Annual Report on Form 10-K and our other public
filings with the SEC. Other events that we do not currently
anticipate or that we currently deem immaterial may also affect our
business, prospects, financial condition and results of operations.
If any of the following risks actually occur, our business,
prospects, operating results and financial condition could suffer
materially. In such event, the trading price of our common stock
could decline and you might lose all or part of your
investment.
Risks Relating to Our Financial Condition and Capital
Resources
If we are unable to continue as a going concern, our securities
will have little or no value.
The report of our independent registered public accounting firm
that accompanies our audited consolidated financial statements
for the years ended February 28, 2018 and February 28, 2017 contain
a going concern qualification in which such firm expressed
substantial doubt about our ability to continue as a going concern.
As of February 28, 2018, and February 28, 2017, we had an
accumulated deficit of approximately $29.5 million and $26.3
million, respectively. At February 28, 2018, we have a negative
working capital. We currently anticipate that our cash and cash
equivalents will not be sufficient to fund our operations for
the next twelve months, without raising additional capital. Our
continuation as a going concern is dependent upon continued
financial support from our shareholders, the ability of us to
obtain necessary equity and/or debt financing to continue
operations, and the attainment of profitable operations. These
factors raise substantial doubt regarding our ability to continue
as a going concern. Although we are actively working to obtain
additional funding, we cannot make any assurances that additional
financings will be available to us and, if available, completed on
a timely basis, on acceptable terms or at all. If we are unable to
complete an equity or debt offering, or otherwise obtain sufficient
financing when and if needed, it would negatively impact our
business and operations, which would likely cause the price of our
common stock to decline. It could also lead to the reduction or
suspension of our operations and ultimately force us to cease our
operations.
We are at an early stage of development as a company and do not
have, and may never have, any products that generate
revenue.
We
are a pre-commercial precision medicine company. At this time,
we do not have any commercial products or laboratory services that
generate revenue. Our business has evolved into a fully
integrated precision medicine company focused on discovering and
developing anti-metastatic drugs and paired companion diagnostics
based on the MENA pathway from a pure play prognostic diagnostic
company. We are not profitable and have incurred losses in each
year since our inception and expect to continue to incur losses for
the foreseeable future.
Our therapeutic and companion diagnostic product candidates are at
early stages of development, have not obtained regulatory marketing
approval, have never generated any sales and require extensive
testing before commercialization. Our limited operating history may
adversely affect our ability to implement our business strategy and
achieve our business goals, which include, among others, the
following activities:
●
develop our therapeutic or diagnostic product
candidates;
●
obtain the human and financial resources necessary to develop,
test, commercialize and market our product candidates;
●
continue to build and maintain an intellectual property portfolio
covering our technology and our product candidates;
●
satisfy the requirements of clinical trial protocols, including
patient enrollment, establish and demonstrate the clinical
efficacy, safety and utility of our product candidates and obtain
necessary regulatory approvals;
●
commercialize and market our product candidates that receive
regulatory approvals to achieve acceptance and use by the medical
community in general;
●
develop and maintain successful collaboration, strategic and other
relationships for the development and commercialization of our
product candidates; and
●
manage our cash flows and any growth we may experience in an
environment where costs and expenses relating to clinical trials,
regulatory approvals and commercialization continue to
increase.
Additionally, our prognostic diagnostic product candidates will
require additional development, analytical validation, clinical
evaluation, additional state and CLIA licensing, potential
regulatory review, significant sales and marketing efforts and
substantial investment or collaboration before they could provide
any product revenue.
If we are unsuccessful in accomplishing these objectives, we may
not be able to successfully develop product candidates, raise
capital, generate significant revenue, or any revenue at
all, grow our business or continue our
operations. Even if we achieve profitability in the future,
we may not be able to sustain profitability in subsequent periods.
Our prior losses, combined with expected future losses, have had
and will continue to have an adverse effect on our
stockholders’ equity and working capital.
We will require additional debt or equity financing to fund our
operations, but may not be able to obtain such financing on
satisfactory terms or at all.
We have
financed our operations primarily through equity and debt
financings. We will require additional debt or equity financing in
the future to maintain our business and fund our operations. Our
ability to secure additional financing will depend on a variety of
factors, many of which are beyond our control, including our
operational performance, economic conditions of the U.S., other
companies that may also seek funding and investors’ and
lenders’ perceptions of, and demand for, debt and equity
securities of our company. As a result, we cannot assure you that
we will be able to access capital from external sources on
satisfactory terms and conditions, or at all. If we are unable to
promptly obtain additional financing, we will not be able to
maintain our business as anticipated or to fund future operations,
and our business, financial condition and results of operations
would be materially and adversely affected.
We have a history of net losses, and we expect to incur net losses
for the foreseeable future and we expect to continue to incur
significant expenses to develop and commercialize our
products.
We have
incurred substantial net losses since our inception. For the fiscal
year ended February 28, 2018 and February 28, 2017, we incurred net
losses of approximately $3.2 million and approximately $2.9
million, respectively. From our inception in July 2009 through
February 28, 2018, we had an accumulated deficit of approximately
$29.5 million. To date, we have not achieved, and we may never
achieve, revenue sufficient to offset expenses. We expect to devote
substantially all of our resources to research and development and
commercialization of our therapeutic and companion diagnostic
product offerings. We expect to incur additional losses in the
future, and we may never achieve profitability.
We expect our losses to continue as a result of costs relating to
ongoing research and development primarily for our therapeutic drug
discovery and companion diagnostic programs, clinical studies,
operational expenses, and other commercialization costs. These
losses have had, and will continue to have, an adverse effect on
our working capital, total assets and stockholders’ equity.
Because of the numerous risks and uncertainties associated with our
commercialization efforts, we are unable to predict when we will
become profitable, if ever. Even if we do achieve profitability, we
may not be able to sustain or increase profitability on a quarterly
or annual basis.
We have incurred significant indebtedness under our non-convertible
promissory bridge notes with shareholders.
On
March 30, 2018, we issued (i) senior non-convertible promissory
bridge notes in the aggregate principal amount of $2,084,028, of
which $834,027 was paid through the exchange of an outstanding
promissory note (the “Senior Notes”) and (ii) junior
non-convertible promissory bridge notes in the aggregate principal
amount of $1,294,900 (the “Junior Notes” and, together
with the Senior Notes, the “Notes”). The Notes mature
on September 30, 2018, accrue interest at a rate of ten percent
(10%) per annum and may not be prepaid by the Company prior to the
maturity without the consent of the holder. The
principal amount plus all accrued and unpaid interest thereon shall
automatically exchange (the “Automatic Exchange”),
without any action of the holder, into such number of fully paid
and non-assessable securities (e.g. shares and warrants) to be
issued in a Qualified Offering. “Qualified Offering”
means one or a series of offerings of equity or equity-linked
securities resulting in aggregate gross proceeds of at least
$6,628,927 to the Company, including the Automatic Exchange of the
Notes into the Qualified Offering.
Our ability to complete the Qualified Financing or repay or to
refinance our indebtedness depends on our future performance and
ability to raise additional sources of cash, which is subject to
economic, financial, competitive and other factors beyond our
control. If we are unable to generate sufficient cash to service
our indebtedness we may be required to adopt one or more
alternatives, such as restructuring our debt or obtaining
additional equity capital on terms that may be onerous or highly
dilutive or selling assets. If we desire to refinance our
indebtedness, our ability to do so will depend on the capital
markets and our financial condition at such time. We may not be
able to engage in any of these activities or engage in these
activities on desirable terms, which could result in a default on
our debt obligations.
Risks Relating to Our Business and
Strategy
We expect to continue to incur significant research and development
expenses, which may make it difficult for us to achieve and
maintain profitability.
Our operations have consumed substantial amounts of cash since
inception. We expect to need substantial additional funding to
pursue our development programs and launch and commercialize any
product candidates for which we receive regulatory approval, which
may include building internal sales and marketing forces to address
certain markets.
In past years, we have incurred significant costs in connection
with the development of our prognostic diagnostic tests and in
recent years, we have incurred significant costs in connection with
the development of our driver-based biomarkers for
anti-metastatic drugs and companion diagnostics. Our research and development expenses were
approximately $1.3 million and
$1.0 million for the fiscal years ended February 28, 2018 and
February 28, 2017, respectively.
We expect our research and development expenses to increase and
remain high for the foreseeable future as we continue to
develop our therapeutics and companion diagnostics and move them
into the clinic. Additionally, we may not be able to
successfully monetize or commercialization our prognostic diagnostic tests. As a result,
we will need to generate significant revenue in order to achieve
profitability. Our failure to achieve revenue or profitability
in the future could cause the market price of our common stock to
decline. Our prior losses, combined with expected future losses,
have had and will continue to have an adverse effect on our
stockholders’ equity and working capital.
Our research and development efforts are based on a rapidly
evolving area of science, and our approach to development is novel
and may never lead to marketable products.
Biopharmaceutical
product development is generally a highly speculative undertaking
and involves substantial risk. The field of personalized medicine,
in which we engage, is an emerging field, and the scientific
discoveries that form the basis of our therapeutic and companion
diagnostic product development efforts are relatively new. Further,
the scientific evidence to support the feasibility of developing
product candidates based on those discoveries is both preliminary
and limited. The failure of the scientific underpinnings of our
business model to produce viable product candidates would
substantially harm our operations and prospects.
If we are unable to commercialize and generate sales from our
products or successfully develop and commercialize other product
candidates, our revenue will be insufficient for us to achieve
profitability.
We are a pre-commercial biotechnology company. We do not have
any commercial products or laboratory services that generate
revenue. Our therapeutic and companion diagnostics programs
are in an early stage of development. Our future success is
substantially dependent on our ability to successfully develop,
obtain regulatory approval for, and then successfully commercialize
our therapeutic and companion diagnostic product products resulting
from these programs and any others we may develop or acquire in the
future, which may never occur.
Before
we could generate any revenues from our therapeutic and companion
diagnostic product candidates, we must complete the following
activities for each of them, any one of which we may not be able to
successfully complete:
●
conduct substantial preclinical development;
●
manage preclinical and clinical activities;
●
achieve regulatory approval;
●
manage manufacturing activates and establish manufacturing
relationships;
●
develop our companion diagnostics and conduct clinical testing and
achieve regulatory approvals for use of our companion diagnostics
with corresponding therapeutics;
●
build a commercial sales and marketing infrastructure, if we choose
to market any such products ourselves, or enter into a
collaboration to access sales and marketing functions;
●
develop and implement marketing and reimbursement strategies;
and
●
invest significant additional cash in each of the above
activities.
If we
are unable to commercialize and generate sales of our product
candidates, or successfully develop, monetize or commercialization
our prognostic diagnostic
tests, we will not produce sufficient revenue to become
profitable.
We may not be successful in our efforts to build a pipeline of
product candidates.
A key element of our product development strategy is to exploit the
MENA platform to build a pipeline of drug candidates and paired
companion diagnostic tests and progress those product candidates
through preclinical and clinical development for the treatment
patients with aggressive cancer in solid tumors indications. We may
not be able to develop product candidates that are safe and
effective. Even if we are successful in building a product
pipeline, the potential product candidates that we identify may not
be suitable for clinical development for a number of reasons,
including causing harmful side effects or demonstrating other
characteristics that indicate a low likelihood of receiving
marketing approval or achieving market acceptance. If our methods
of identifying potential product candidates fail to produce a
pipeline of potentially viable product candidates, then our success
as a business will be dependent on the success of fewer potential
product candidates, which introduces risks to our business model
and potential limitations to any success we may
achieve.
We may expend our limited resources to pursue a particular product
candidate or indication that does not produce any commercially
viable products and may fail to capitalize on product candidates or
indications that may be more profitable or for which there is a
greater likelihood of success.
Because
we have limited financial and operational resources, we must focus
our efforts on particular product candidates for specific
indications. As a result, we may forego or delay pursuit of
opportunities with other product candidates or for other
indications that later prove to have greater commercial potential.
Further, our resource allocation decisions may result in our use of
funds for research and development programs and product candidates
for specific indications that may not yield any commercially viable
products. If we do not accurately evaluate the commercial potential
or target market for a particular product candidate, we may
relinquish valuable rights to that product candidate through
collaboration, licensing or other royalty arrangements in cases in
which it would have been more advantageous for us to retain sole
development and commercialization rights to such product candidate.
Any such failure to properly assess potential product candidates
could result in missed opportunities and/or our focus on product
candidates with low market potential, which would harm our business
and financial condition.
Drug development involves a lengthy and expensive process with
uncertain outcomes, and any of our preclinical development and
clinical trials or studies could produce unsuccessful results or
fail at any stage in the testing process.
Preclinical
development and clinical testing is expensive and can take many
years to complete. The outcomes are inherently uncertain, and
failure can occur at any time during the preclinical development
and clinical trial process. Additionally, any positive results of
preclinical studies and early clinical trials of a product
candidate may not be predictive of the results of later-stage
clinical trials, such that product candidates may reach later
stages of clinical trials and fail to show the desired safety and
efficacy traits despite having shown indications of those traits in
earlier studies. A number of companies in the pharmaceutical and
biopharmaceutical industry have suffered significant setbacks in
advanced clinical trials due to lack of efficacy or adverse safety
profiles, notwithstanding promising results in earlier trials. The
results of any preclinical development and clinical trials we
conduct may be delayed or unsuccessful for a variety of
reasons.
If we experience delays or difficulties in the enrollment of
patients in clinical trials, those clinical trials could take
longer than expected to complete and our receipt of necessary
regulatory approvals could be delayed or prevented.
If any of our product candidates enter the clinical development
stage, we may not be able to initiate or continue clinical trials
for our product candidates if we are unable to locate and enroll a
sufficient number of eligible patients to participate in these
trials as required by the FDA or similar regulatory authorities
outside the United States. In particular, because we are primarily
focused on patients with molecularly defined cancers, which may
have relatively low incidence rates, our pool of suitable patients
may be smaller and more selective and our ability to enroll a
sufficient number of suitable patients may be limited or take
longer than anticipated. Additionally, some of our competitors have
ongoing clinical trials for product candidates that treat the same
indications that our product candidates target, and patients who
would otherwise be eligible for our clinical trials may instead
enroll in clinical trials of our competitors’ product
candidates.
The
inability to enroll a sufficient number of patients for our
clinical trials would result in significant delays and could
require us to abandon our clinical trials altogether. Enrollment
delays in our clinical trials would likely result in increased
development costs, and we may not have or be able to obtain
sufficient cash to fund such increased costs when needed, which
could result in the further delay or termination of the
trials.
The approval processes of regulatory authorities are lengthy, time
consuming, expensive and inherently unpredictable. If we are unable
to obtain approval for our product candidates from applicable
regulatory authorities, we will not be able to market and sell
those product candidates in those countries or regions and our
business will be substantially harmed.
The
time required to obtain approval by the FDA and comparable foreign
authorities is unpredictable, but typically takes many years
following the commencement of clinical trials and depends upon
numerous factors, including the substantial discretion of the
regulatory authorities. We have not submitted an NDA or similar
filing or obtained regulatory approval for any product candidate in
any jurisdiction and it is possible that none of our existing
product candidates or any product candidates we may seek to develop
in the future will ever obtain regulatory approval.
Our
product candidates could fail to receive regulatory approval for
many reasons, including any one or more of the
following:
●
the FDA or comparable foreign regulatory authorities may disagree
with the design or implementation of our clinical
trials;
●
we may be unable
to demonstrate to the satisfaction of the FDA or comparable foreign
regulatory authorities that a product candidate is safe and
effective for its proposed indication;
●
the results of
clinical trials may not meet the level of statistical significance
required by the FDA or comparable foreign regulatory authorities
for approval;
●
we may be unable
to demonstrate that a product candidate’s clinical and other
benefits outweigh its safety risks;
●
the FDA or
comparable foreign regulatory authorities may disagree with our
interpretation of data from preclinical studies or clinical
trials;
●
the data
collected from clinical trials of our product candidates may not be
sufficient to support the submission of an NDA or other submission
or to obtain regulatory approval in the United States or
elsewhere;
●
the FDA or
comparable foreign regulatory authorities may fail to hold to
previous agreements or commitments;
●
the FDA or
comparable foreign regulatory authorities may fail to approve the
manufacturing processes or facilities of third-party manufacturers
with which we contract for clinical and commercial
supplies;
●
the FDA or
comparable foreign regulatory authorities may fail to approve our
companion diagnostics;
●
invest significant additional cash in each of the above activities;
and
●
the approval
policies or regulations of the FDA or comparable foreign regulatory
authorities may significantly change in a manner rendering our
clinical data insufficient for approval.
The
time and expense of the approval process, as well as the
unpredictability of clinical trial results and other contributing
factors, may result in our failure to obtain regulatory approval to
market, in one or more jurisdictions, any of our or future product
candidates, which would significantly harm our business, results of
operations and prospects.
In
order to market and sell our products in any jurisdiction, we or
our third-party collaborators must obtain separate marketing
approvals in that jurisdiction and comply with its regulatory
requirements. The approval procedure can vary drastically among
countries, and each jurisdiction may impose different testing and
other requirements to obtain and maintain marketing approval.
Further, the time required to obtain those approvals may differ
substantially among jurisdictions. Approval by the FDA or an
equivalent foreign authority does not ensure approval by regulatory
authorities in any other countries or jurisdictions. As a result,
the ability to market and sell a product candidate in more than one
jurisdiction can involve significant additional time, expense and
effort to undertake separate approval processes, and could subject
us and our collaborators to the numerous and varying post-approval
requirements of each jurisdiction governing commercial sales,
manufacturing, pricing and distribution of our product candidates.
We or any third parties with whom we may collaborate may not have
the resources to pursue those approvals, and we or they may not be
able to obtain any approvals that are pursued. The failure to
obtain marketing approval for our product candidates in foreign
jurisdictions could severely limit their potential markets and our
ability to generate revenue.
In
addition, even if we were to obtain regulatory approval in one or
more jurisdictions, regulatory authorities may approve any of our
product candidates for fewer or more limited indications than we
request, may not approve the prices we may propose to charge for
our products, may grant approval contingent on the performance of
costly post-marketing clinical trials, or may approve a product
candidate with labeling that does not include the claims necessary
or desirable for the successful commercialization of that product
candidate. Any of the foregoing circumstances could materially harm
the commercial prospects for our product candidates.
Our product candidates may cause undesirable side effects or have
other properties that could delay or prevent their regulatory
approval, limit the commercial profile of the approved labeling, or
result in significant negative consequences following marketing
approval, if any.
Results of future clinical trials of our product candidates could
reveal a high and/or unacceptable severity and frequency of these
or other side effects. In such an event, our trials could be
suspended or terminated, and the FDA or comparable foreign
regulatory authorities could order us to cease further development
of, or deny approval of, our product candidates for any or all
targeted indications. Further, any observed drug-related side
effects could affect patient recruitment or the ability of enrolled
patients to complete the trial or result in potential product
liability claims. Any of these occurrences could materially harm
our business, financial condition and
prospects.
Additionally,
if any of our product candidates receives marketing approval, and
we or others later identify undesirable side effects caused by our
products, a number of potentially significant negative consequences
could result, including:
●
regulatory
authorities may withdraw approvals of such product;
●
regulatory
authorities may require additional warnings in the product’s
labeling;
●
we may be
required to create a medication guide for distribution to patients
that outlines the risks of such side effects;
●
we could be sued
and held liable for harm caused to patients; and
●
our reputation
may suffer.
Any of
these events could prevent us from achieving or maintaining market
acceptance of the particular product, if approved, and could
significantly harm our business, results of operations and
prospects.
Failure to successfully validate, develop and obtain regulatory
approval for our companion diagnostics could harm our business
strategy and operational results.
As one
of the central elements of our business strategy, we seek to
identify appropriate patient
populations most likely to benefit from our anti-metastatic drugs
as well as next generation RTK inhibitors and anti-microtubule
drugs being developed by potential pharmaceutical and biotechnology
partners. In order to assist in identifying those subsets of
patients, a companion diagnostic, which is a test or measurement
that evaluates the presence of biomarkers in a patient, could be
used. We anticipate that the development of companion diagnostics
concurrently with our therapeutic
agents or with our drugs from potential strategic partners
will help us more accurately identify the patients who
belong to the target subset, both during the clinical trials and in
connection with the commercialization of product
candidates.
Companion
diagnostics are subject to regulation by the FDA and comparable
foreign regulatory authorities as medical devices and require
separate regulatory clearance or approval prior to their
commercialization. We may be dependent on the sustained cooperation
and effort of any third-party collaborators with whom we may
partner in the future to develop and obtain clearance or approval
for these companion diagnostics, and we may not be able to
establish arrangements with any such third-party collaborators for
the development and production of companion diagnostics when needed
or on terms that are beneficial to us, or at all. We and our
potential future collaborators may encounter difficulties in
developing and obtaining approval for these companion diagnostics,
including issues relating to the selectivity and/or specificity of
the diagnostic, analytical validation, reproducibility, or clinical
validation.
Since
the FDA generally requires concurrent approval of a companion
diagnostic and therapeutic product, any delay or failure by us or
our potential future collaborators to develop or obtain regulatory
clearance or approval of any companion diagnostics could delay or
prevent approval of our related product candidates. The occurrence
of any delay or failure could adversely affect and/or delay the
development or commercialization of our product
candidates.
If we are unable to execute our sales and marketing strategy for
our products and are unable to gain market acceptance, we may be
unable to generate sufficient revenue to sustain our
business.
We are
a pre-commercial precision medicine company and have yet to begin
to generate revenue from our product candidates. Our therapeutic
and companion diagnostic product candidates are in an early stage
of development, and, if we obtain marketing approval for any of
products in the future, which we anticipate would not occur for
several years, if at all. Additionally, we are seeking to monetize
or commercialize through
strategic partnerships our prognostic diagnostic tests
through our CLIA-certified laboratory, located in Boston,
Massachusetts.
Although we believe that our prognostic diagnostic tests
represent a promising commercial
opportunity, we may never gain significant market acceptance and
therefore may never generate substantial revenue or profits for us.
We will need to establish a market for our prognostic diagnostic
tests and build that market through physician education, awareness
programs and the publication of clinical data. Gaining acceptance
in medical communities requires, among other things, publication in
leading peer-reviewed journals of results from studies using our
current tests and/or our planned cancer tests. The process of
publication in leading medical journals is subject to a peer review
process and peer reviewers may not consider the results of our
studies sufficiently novel or worthy of publication. Failure to
have our studies published in peer-reviewed journals would limit
the adoption of our current tests and our planned tests. Our
ability to successfully market our prognostic diagnostic tests that
we may develop will depend on numerous factors,
including:
●
conducting validation studies of such tests in collaboration with
key thought leaders to demonstrate their use and value in important
medical decisions such as treatment selection;
●
conducting clinical utility studies of such tests to demonstrate
economic usefulness to providers and payers;
●
whether our current or future partners, support our
offerings;
●
the success of the sales force and marketing effort;
●
whether healthcare providers believe such diagnostic tests provide
clinical utility;
●
whether the medical community accepts that such diagnostic tests
are sufficiently sensitive and specific to be meaningful in patient
care and treatment decisions; and
●
whether private health insurers, government health programs and
other third-party payers will cover such diagnostic tests and, if
so, whether they will adequately reimburse us.
Failure to achieve significant market acceptance of our products
would materially harm our business, financial condition and results
of operations.
If third-party payers, including managed care organizations and
Medicare, do not provide reimbursement for our products, our
commercial success could be compromised.
Physicians and patients may decide not to order our prognostic
diagnostic tests unless third-party payers, such as managed care
organizations as well as government payers such as Medicare and
Medicaid, pay a substantial portion or all of the test’s
price. There is significant uncertainty concerning third-party
reimbursement of any test incorporating new technology, including
our prognostic diagnostic tests and any of our future diagnostic
tests. Reimbursement by a third-party payer may depend on a
number of factors, including a payer’s determination that
tests using our technologies are:
●
not experimental or investigational;
●
medically necessary;
●
appropriate for the specific patient;
●
cost-effective;
●
supported by peer-reviewed publications; and
●
provide a clinical utility.
Uncertainty surrounds third-party payor coverage and adequate
reimbursement of any diagnostic test incorporating new technology,
including tests developed using our technologies. Technology
assessments of new medical tests conducted by research centers and
other entities may be disseminated to interested parties for
informational purposes. Third-party payors and health care
providers may use such technology assessments as grounds to deny
coverage for a test or procedure. Technology assessments can
include evaluation of clinical utility studies, which define how a
test is used in a particular clinical setting or situation. Because
each payor generally determines for its own enrollees or insured
patients whether to cover or otherwise establish a policy to
reimburse our cancer diagnostic tests, seeking payor approvals is a
time-consuming and costly process. We cannot be certain that
coverage for our current tests and our planned future tests will be
provided in the future by additional third-party payors or that
existing agreements, policy decisions or reimbursement levels will
remain in place or be fulfilled under existing terms and
provisions. If we cannot obtain coverage and adequate reimbursement
from private and governmental payors such as Medicare and Medicaid
for our current tests, or new tests or test enhancements that we
may develop in the future, our ability to generate revenue could be
limited, which may have a material adverse effect on our financial
condition, results of operations and cash flows. Further, we may
experience delays and interruptions in the receipt of payments from
third-party payors due to missing documentation and/or other
issues, which could cause delay in collecting our
revenue.
In addition, to the extent that our testing is ordered for Medicare
inpatients and outpatients, only the hospital may receive payment
from the Medicare program for the technical component of pathology
services and any clinical laboratory services that we perform,
unless the testing is ordered at least 14 days after discharge and
certain other requirements are met. We therefore must look to the
hospital for payment for these services under these circumstances.
If hospitals refuse to pay for the services or fail to pay in a
timely manner, our ability to generate revenue could be limited,
which may have a material adverse effect on our financial
condition, results of operations and cash flows.
Additionally,
there is significant uncertainty related to the third-party
coverage and reimbursement of newly approved drugs and companion
diagnostics. Market acceptance and sales of any of our product
candidates that obtain regulatory approval in domestic or
international markets will depend significantly on the availability
of adequate coverage and reimbursement from third-party payers and
may be affected by existing and future healthcare reform
measures.
In some
foreign countries, particularly in the European Union, the pricing
of prescription pharmaceuticals is subject to governmental control.
In these countries, pricing negotiations with governmental
authorities can be a long and expensive process after the receipt
of marketing approval for a product candidate. To obtain
reimbursement or pricing approval in some countries, we may be
required to conduct additional clinical trials that compare the
cost-effectiveness of our product candidates to other available
therapies. If reimbursement of our product candidates is
unavailable or limited in scope or amount in a particular country,
or if pricing is set at unsatisfactory levels, we may be unable to
achieve or sustain profitability for sales of any of our product
candidates that are approved for marketing in that
country.
Long payment cycles of Medicare, Medicaid and/or other third-party
payers, or other payment delays, could hurt our cash flows and
increase our need for working capital.
Medicare and Medicaid have complex billing and documentation
requirements that we will need to satisfy in order to receive
payment, and the programs can be expected to carefully audit and
monitor our compliance with these requirements. We will also need
to comply with numerous other laws applicable to billing and
payment for healthcare services, including, for example, privacy
laws. Failure to comply with these requirements may result in,
among other things, non-payment, refunds, exclusion from government
healthcare programs, and civil or criminal liabilities, any of
which may have a material adverse effect on our revenue and
earnings. In addition, failure by third-party payors to properly
process our payment claims in a timely manner could delay our
receipt of payment for our products and services, which may have a
material adverse effect on our cash flows and
business.
We expect to rely on third parties to conduct preclinical and
clinical trials of our therapeutic and companion diagnostic product
candidates. If these third parties do not successfully carry out
their contractual duties or meet expected deadlines, we may not be
able to obtain regulatory approval for or commercialize our product
candidates and our business could be substantially
harmed.
We
rely, and expect to continue to rely, upon third-party
collaborators, including CROs to execute our preclinical
development and clinical trials and to monitor and manage data
produced by and relating to those trials. However, we may not be
able to establish arrangements with collaborators and CROs when
needed or on terms that are acceptable to us, or at all, which
could negatively affect our development efforts with respect to our
drug product candidates and materially harm our business,
operations and prospects.
Our research and development and commercialization efforts for our
prognostic diagnostic tests will be hindered if we are not able to
contract with third parties for access to clinical
samples.
Under standard clinical practice, tumor biopsies removed from
patients are typically chemically preserved and embedded in
paraffin wax and stored. Our clinical development relies on our
ability to secure access to these archived tumor biopsy samples, as
well as information pertaining to their associated clinical
outcomes. Generally, the agreements under which we gain access to
archival samples are nonexclusive. Other companies study archival
samples and often compete with us for access. Additionally, the
process of negotiating access to archived samples is lengthy since
it typically involves numerous parties and approval levels to
resolve complex issues such as usage rights, institutional review
board approval, privacy rights, publication rights, intellectual
property ownership and research parameters. If we are not able to
negotiate access to clinical samples with hospitals, clinical
partners, pharmaceutical companies, or companies developing
therapeutics on a timely basis, or at all, or if other laboratories
or our competitors secure access to these samples before us, our
ability to research, develop and commercialize future products will
be limited or delayed. In addition, access to these clinical
samples may be costly, and involve large upfront acquisition costs,
which may have a material adverse effect on our cash flows and
business.
We may experience delays in our clinical studies that could
adversely affect our financial position and our commercial
prospects.
Any delays in completing our preclinical development and clinical
studies for any of our product candidates may delay our ability to
raise additional capital or to generate revenue, and we may have
insufficient capital resources to support our
operations. Even if we have sufficient capital
resources, the ability to become profitable will be delayed if
there are problems with the timing or completion of our clinical
studies.
We are conducting, and expect to conduct, certain preclinical development, validation
studies and clinical studies alone and/or in collaboration with
select academic institutions and other third-party institutions
through services and collaboration agreements. We may experience
delays that are outside of our control in connection with such
services and collaboration agreements, including, but not limited
to, receiving tissue samples, accompanying medical and clinical
data, preparation, review and sign-off of results and/or
manuscripts in a timely fashion. Any delays in completing our
clinical studies and publishing of results in peer-reviewed
journals will delay our commercialization efforts and may
materially harm our business, financial condition and results of
operations.
If we cannot maintain our current clinical collaborations and enter
into new collaborations, our product development could be
delayed.
We rely on, and expect to continue to rely on, clinical
collaborators, including CROs to perform portions of our clinical
trial functions. If any of our collaborators were to breach or
terminate its agreement with us or otherwise fail to conduct the
contracted activities successfully and in a timely manner, the
research, development or commercialization of the products
contemplated by the collaboration could be delayed or terminated.
If any of our collaboration agreements are terminated, or if we are
unable to renew those agreements on acceptable terms, we would be
required to seek alternatives. We may not be able to negotiate
additional collaborations on acceptable terms, if at all, and these
collaborations may not be successful.
Our success in the future depends in part on our ability to enter
into agreements with other leading cancer organizations. This can
be difficult due to internal and external constraints placed on
these organizations. Some organizations may limit the number of
collaborations they have with any one company so as to not be
perceived as biased or conflicted. Organizations may also have
insufficient administrative and related infrastructure to enable
collaborations with many companies at once, which can prolong the
time it takes to develop, negotiate and implement collaboration.
Additionally, organizations often insist on retaining the rights to
publish the clinical data resulting from the collaboration. The
publication of clinical data in peer-reviewed journals is a crucial
step in commercializing and obtaining reimbursement for products
such as ours, and our inability to control when, if ever, results
are published may delay or limit our ability to derive sufficient
revenue from any product that may result from a
collaboration.
From time to time we expect to engage in discussions with potential
clinical collaborators, which may or may not lead to
collaborations. However, we cannot guarantee that any discussions
will result in clinical collaborations or that any clinical
studies, which may result will be completed in a reasonable time
frame or with successful outcomes. If news of discussions regarding
possible collaborations become known in the medical community,
regardless of whether the news is accurate, failure to announce a
collaboration agreement or the entity’s announcement of a
collaboration with an entity other than us could result in adverse
speculation about us, our products or our technology, resulting in
harm to our reputation and our business.
Clinical utility studies are important in demonstrating to both
customers and payers a test’s clinical relevance and value.
If we are unable to identify collaborators willing to work with us
to conduct clinical utility studies, or the results of those
studies do not demonstrate that a product provides clinically
meaningful information and value, commercial adoption of such test
may be slow, which would negatively impact our
business.
Clinical
utility studies show when and how to use a clinical test and
describe the particular clinical situations or settings in which it
can be applied and the expected results. Clinical utility studies
also show the impact of the product results on patient care and
management. Clinical utility studies are typically performed with
collaborating oncologists or other physicians at medical centers
and hospitals, analogous to a clinical trial, and generally result
in peer-reviewed publications. Sales and marketing representatives
use these publications to demonstrate to customers how to use a
clinical test, as well as why they should use it. These
publications are also used with payers to obtain coverage for a
test, helping to assure there is appropriate reimbursement. We
anticipate commencing clinical utility studies for our prognostic
diagnostic tests following product launch. We will need to conduct
additional studies for our prognostic diagnostic tests, and other
tests we plan to introduce, to increase the market adoption and
obtain coverage and adequate reimbursement. Should we not be able
to perform these studies, or should their results not provide
clinically meaningful data and value for oncologists and other
physicians, adoption of our product could be impaired, and we may
not be able to obtain coverage and adequate reimbursement for
them.
If our sole laboratory facility becomes inoperable, we will be
unable to perform our research and development and commercial
activities, and our business will be harmed.
Our state-of-the-art research and development and commercial
laboratory facility located in Boston, Massachusetts received CLIA
certification and licensing from Massachusetts, California,
Florida, Pennsylvania and Rhode Island. We are seeking licensing
from other states including New York and Maryland, in order to
process samples from such states, however we cannot guarantee that
we will receive the necessary certifications and approvals in a
timely fashion. Delays in receiving the necessary state
certifications may delay commercialization efforts in these states
and may materially harm our business, financial condition and
results of operations.
The laboratory facility may be harmed or rendered inoperable by
natural or man-made disasters, including earthquakes, flooding,
fire and power outages, or loss of our commercial lease, which may
render it difficult or impossible for us to perform our testing
services for some period of time. The inability to perform our
research and development and/or commercial activities even for a
short period of time, may result in the loss of customers or harm
our reputation or relationships with scientific or clinical
collaborators, and we may be unable to regain those customers or
repair our reputation in the future. Although we possess
insurance for damage to our property and the disruption of our
business, this insurance may not be sufficient to cover all of our
potential losses and may not continue to be available to us on
acceptable terms, or at all.
In order to rely on a third party to perform our tests, we could
only use another facility with established CLIA certification and
state licensure under the scope of which our diagnostic tests could
be performed following validation and other required
procedures. We cannot assure you that we would be able to find
another CLIA-certified and state-licensed laboratory facility
willing to license, transfer or adopt our diagnostic tests and
comply with the required procedures, or that such partner or
laboratory would be willing to perform the tests for us on
commercially reasonable terms.
In order to establish a redundant laboratory facility, we would
have to spend considerable time and money securing adequate space,
constructing the facility, recruiting and training employees, and
establishing the additional operational and administrative
infrastructure necessary to support a second
facility. Additionally, any new clinical laboratory facility
opened by us would be subject to certification under
CLIA, licensing by several states, including New York,
California, Florida, Maryland, Pennsylvania and Rhode Island, which
can take a significant amount of time and result in delays in our
ability to begin operations.
We may experience limits on our revenue if oncologists and other
physicians decide not to order our prognostic diagnostic tests or
our future tests, we may be unable to generate sufficient revenue
to sustain our business.
If medical practitioners do not order our prognostic diagnostic
assays or any future tests developed by us, we will likely not be
able to create demand for our products in sufficient volume for us
to become profitable. To generate demand, we will need to
continue to make oncologists, surgeons, pathologists and other
health care professionals aware of the benefits, value and clinical
utility of our diagnostic tests and any products we may develop in
the future through published papers, presentations at scientific
conferences and one-on-one education by our sales force. We
need to hire or outsource commercial, scientific, technical and
other personnel to support this process. Some physicians may decide
not to order our test due to its price, part or all of which may be
payable directly by the patient if the applicable payer denies
reimbursement in full or in part. Even if patients recommend their
physicians use our diagnostic tests, physicians may still decide
not to order them, either because they have not been made aware of
their utility or they wish to pursue a particular course of
treatment and/or therapy regardless. If only a small portion
of the physician population decides to use our tests, we will
experience limits on our revenue and our ability to achieve
profitability. In addition, we will need to demonstrate our
ability to obtain adequate reimbursement coverage from third-party
payers.
We may experience limits on our revenue if patients decide not to
use our prognostic diagnostic tests.
Some patients may decide not to order our prognostic diagnostic
tests due to its price, part or all of which may be payable
directly by the patient if the applicable payer denies
reimbursement in full or in part. Even if medical
practitioners recommend that their patients use our test, patients
may still decide not to use our prognostic diagnostic tests, either
because they do not want to be made aware of the likelihood of
metastasis or they wish to pursue a particular course of therapy
regardless of test results. If only a small portion of the patient
population decides to use our test, we will experience limits on
our revenue and our ability to achieve profitability.
If we are unable to develop our product candidates to keep pace
with rapid technological, medical and scientific change, our
operating results and competitive position would be
harmed.
In recent years, there have been numerous advances in technologies
relating to the diagnosis, prognosis and treatment of
cancer. These advances require us to continuously develop new
products and enhance existing products to keep pace with evolving
standards of care. Several new cancer drugs have been
approved, and a number of new drugs in clinical development may
increase patient survival time. There have also been advances in
methods used to identify patients likely to benefit from these
drugs based on analysis of biomarkers. Our tests could become
obsolete unless we continually innovate and expand our products to
demonstrate benefit in the diagnosis, monitoring or prognosis of
patients with cancer. New treatment therapies typically have
only a few years of clinical data associated with them, which
limits our ability to develop cancer diagnostic tests based on for
example, biomarker analysis related to the appearance or
development of resistance to those therapies. If we cannot
adequately demonstrate the applicability of our current tests and
our planned tests to new treatments, by incorporating important
biomarker analysis, sales of our tests could decline, which would
have a material adverse effect on our business, financial condition
and results of operations.
If we become subject to product liability claims, the damages may
exceed insurance coverage levels.
We could be subject to product liability lawsuits based on the use
of our product candidates in clinical testing or, if obtained,
following marketing approval and commercialization. If product
liability lawsuits are brought against us, we may incur substantial
liabilities and may be required to cease clinical testing or limit
commercialization of our product candidates. We plan to obtain
liability insurance for our product candidates as each is entered
into clinical studies, large population validation studies and/or
any other studies where such liability insurance is
needed.
We cannot predict all of the possible harms or side effects that
may result from the use of our products and, therefore, the amount
of insurance coverage we currently hold, or that we or our
collaborators may obtain, may not be adequate to protect us from
any claims arising from the use of our products that are beyond the
limit of our insurance coverage. If we cannot protect against
potential liability claims, we or our collaborators may find it
difficult or impossible to commercialize our products, and we may
not be able to renew or increase our insurance coverage on
reasonable terms, if at all. The marketing, sale and use of our
products and our planned future products could lead to the filing
of product liability claims against us if someone alleges that our
products failed to perform as designed. We may also be subject to
liability for errors in the test results we provide to physicians
or for a misunderstanding of, or inappropriate reliance upon, the
information we provide. A product liability or professional
liability claim could result in substantial damages and be costly
and time-consuming for us to defend.
Any product liability or professional liability claim brought
against us, with or without merit, could increase our insurance
rates or prevent us from securing insurance coverage. Additionally,
any product liability lawsuit could damage our reputation, result
in the recall of tests, or cause current partners to terminate
existing agreements and potential partners to seek other partners,
any of which could impact our results of operations.
Our dependence on commercialization partners for sales of our
prognostic diagnostic tests could limit our success in realizing
revenue growth.
We are seeking to monetize or commercialize through
the use of distribution and
commercialization partners for the sales, marketing and
distribution, billing, collection and reimbursement efforts, and to
do so we must enter into agreements with these partners to sell,
market or commercialize our tests. We may experience launch delays
as a result of the timing of clinical data, establishment of a
final product profile, and the lead time required to
execute commercialization agreements. These agreements may
contain exclusivity provisions and generally cannot be terminated
without cause during the term of the agreement. We may need to
attract additional partners to expand the markets in which we sell
tests. These partners may not commit the necessary resources to
market and sell our cancer diagnostics tests to the level of our
expectations, and we may be unable to locate suitable alternatives
should we terminate our agreement with such partners or if such
partners terminate their agreement with us. Any relationships we
form with commercialization partners are subject to change over
time. If current or future commercialization partners do not
perform adequately, or we are unable to locate commercialization
partners, we may not realize revenue growth.
If we are unable to develop adequate sales, marketing or
distribution capabilities or enter into agreements with third
parties to perform some of these functions, we will not be able to
commercialize our products effectively.
We likely will have a limited infrastructure in sales, marketing
and distribution. Initially, we are not planning to directly market
and distribute our products. We may not be able to enter into
sales, marketing and distribution capabilities of our own or enter
into such arrangements with third parties in a timely manner or on
acceptable terms.
Our sales force collaborator with marketing and distribution rights
to one or more of our products may not commit enough resources to
the marketing and distribution of our products, limiting our
potential revenue from the commercialization of these products.
Disputes may arise delaying or terminating the commercialization or
sales of our diagnostic tests that may result in significant legal
proceedings that may harm our business, limit our revenue and our
ability to achieve profitability.
We depend on third parties for the supply of tissue samples and
other biological materials that we use in our research and
development efforts. If the costs of such tissue samples and
materials increase or our third-party suppliers terminate their
relationship with us, our business may be materially
harmed.
We
have relationships and plan to enter into new relationships with
suppliers and institutions that provide us with tissue samples,
tissue microarrays (TMA’s), and other biological materials
including antibodies that we use in developing and validating our
product candidates. If one or more suppliers terminate their
relationship with us or are unable to meet our requirements for
samples, we will need to identify other third parties to provide us
with samples and biological materials, which could result in a
delay in our research and development activities, clinical studies
and negatively affect our business. In addition, as we grow, our
research and academic institution collaborators may seek additional
financial contributions from us, which may negatively affect our
results of operations.
We rely on a limited number of suppliers or, in some cases, a sole
supplier, for some of our laboratory instruments and materials and
may not be able to find replacements in the event our supplier no
longer supplies that equipment.
We expect to rely on several vendors, including, but not limited to
Perkin Elmer, ThermoFisher Scientific and VisioPharm AS to supply
some of the laboratory equipment and software on which we perform
our diagnostic tests. We will periodically forecast our needs
for laboratory equipment and software and enter into standard
purchase orders or leasing arrangements based on these
forecasts. We believe that there are relatively few equipment
manufacturers that are currently capable of supplying the equipment
necessary for our prognostic diagnostics tests. Even if we
were to identify other suppliers, there can be no assurance that we
will be able to enter into agreements with such suppliers on a
timely basis on acceptable terms, if at all. If we should
encounter delays or difficulties in securing from key vendors the
quality and quantity of equipment and software we require for our
diagnostic tests, we may need to reconfigure our test process,
which would result in delays in commercialization or an
interruption in sales. If any of these events occur, our
business and operating results could be harmed. Additionally,
if key vendors including Perkin Elmer and other vendors deem us to
have become uncreditworthy, they have the right to require
alternative payment terms from us, including payment in
advance. We may also be required to indemnify key vendors
including Perkin Elmer and other vendors against any damages caused
by any legal action or proceeding brought by a third party against
such vendors for damages caused by our failure to obtain required
approval with any regulatory agency.
We may also rely on several sole suppliers for certain laboratory
materials such as reagents, which we use to perform our diagnostic
tests. Although we believe that we will be able to develop
alternate sourcing strategies for these materials, we cannot be
certain that these strategies will be effective. If we should
encounter delays or difficulties in securing these laboratory
materials, delays in commercialization or an interruption in sales
could occur.
We currently rely, and expect to continue to rely, on third-party
suppliers for critical materials needed to perform research and
development activities of our product candidates and our planned
future products and any problems experienced by them could result
in a delay or interruption of their supply to us.
We currently purchase raw materials, including Mabs and testing
reagents for our therapeutic and diagnostic product discovery and
development efforts under purchase orders and do not have long-term
commercial contracts with the suppliers of these materials. If
suppliers were to delay or stop producing our materials or
reagents, or if the prices they charge us were to increase
significantly, or if they elected not to sell to us, we would need
to identify other suppliers. We could experience delays in our
research and development efforts and delays in performing our
prognostic diagnostic tests while finding another acceptable
supplier, which could impact our results of operations. The changes
could also result in increased costs associated with qualifying the
new materials or reagents and in increased operating costs.
Further, any prolonged disruption in a supplier’s operations
could have a significant negative impact on our ability to perform
our prognostic diagnostic tests in a timely manner. Some of the
components used in our current or planned products are currently
sole-source, and substitutes for these components might not be able
to be obtained easily or may require substantial design or
manufacturing modifications. Any significant problem experienced by
one of our sole source suppliers may result in a delay or
interruption in the supply of components to us until that supplier
cures the problem or an alternative source of the component is
located and qualified. Any delay or interruption would likely lead
to a delay or interruption in our operations. The inclusion of
substitute components must meet our product specifications and
could require us to qualify the new supplier with the appropriate
government regulatory authorities.
Our success depends on retention of key personnel and the hiring of
additional key personnel. The loss of key members of our executive
management team could adversely affect our business.
We are dependent on our management team members, including Douglas
A. Hamilton, our president and chief executive officer. Our future
success also will depend in large part on our continued ability to
attract and retain other highly qualified personnel. We intend to
recruit and hire other senior executives, scientific, technical and
management personnel, as well as personnel with expertise in sales
and marketing including reimbursement, clinical testing, and
governmental regulation. Such a management transition subjects us
to a number of risks, including risks pertaining to coordination of
responsibilities and tasks, creation of new management systems and
processes, differences in management style, effects on corporate
culture, and the need for transfer of historical
knowledge.
In addition, Douglas A. Hamilton has not previously been the chief
executive officer of a public or private company. While he has had
experience as a chief financial officer, chief operating officer
and other executive level positions in public companies, a lack of
significant experience in being the chief executive officer of a
public company could have an adverse effect on our ability to
quickly respond to problems or effectively manage issues
surrounding the operation of a public company. Our success in
implementing our business strategy depends largely on the skills,
experience and performance of key members of our executive
management team and others in key management positions. The
collective efforts of our executive management and others working
with them as a team are critical to us as we continue to develop
our technologies, diagnostic tests, research and development
efforts and sales and marketing programs. As a result of the
difficulty in locating qualified new management, the loss or
incapacity of existing members of our executive management team
could adversely affect our operations. If we were to lose one or
more of our key employees, we could experience difficulties in
finding qualified successors, competing effectively, developing our
technologies and implementing our business strategy. We do not
maintain “key person” life insurance on any of our
employees.
In addition, we rely on collaborators, consultants and advisors,
including scientific and clinical advisors, to assist us in our
research and development and commercialization strategy. Our
collaborators, consultants and advisors are generally employed by
employers other than us and may have commitments under agreements
with other entities that may limit their availability to
us.
The loss of a key employee, the failure of a key employee to
perform in his or her current position or our inability to attract
and retain skilled employees could result in our inability to
continue to grow our business or to implement our business
strategy.
There is a scarcity of experienced professionals in our industry.
If we are not able to retain and recruit personnel with the
requisite technical skills, we may be unable to successfully
execute our business strategy.
The specialized nature of our industry results in an inherent
scarcity of experienced personnel in the field. Our future success
depends upon our ability to attract and retain highly skilled
personnel, including scientific, technical, commercial, business,
regulatory and administrative personnel, necessary to support our
anticipated growth, develop our business and perform certain
contractual obligations. Given the scarcity of professionals with
the scientific knowledge that we require and the competition for
qualified personnel among life science businesses, we may not
succeed in attracting or retaining the personnel we require to
continue and grow our operations.
Our operations may involve hazardous materials, and compliance with
environmental laws and regulations is expensive.
Our future research and development and commercial activities may
involve the controlled use of hazardous materials, including
chemicals that cause cancer, volatile solvents, radioactive
materials and biological materials including human tissue samples
that have the potential to transmit diseases. Our operations
may also produce hazardous waste products. We are subject to a
variety of federal, state and local regulations relating to the
use, handling and disposal of these materials. We generally
may contract with third parties for the disposal of such substances
and may store certain low level radioactive waste at our facility
until the materials are no longer considered
radioactive. While we believe that we will comply with then
current regulatory requirements, we cannot eliminate the risk of
accidental contamination or injury from these materials. We
may be required to incur substantial costs to comply with current
or future environmental and safety regulations. If an accident
or contamination occurred, we would likely incur significant costs
associated with civil penalties or criminal fines and in complying
with environmental laws and regulations.
If we use biological and hazardous materials in a manner that
causes injury, we could be liable for damages.
Our activities may require the controlled use of potentially
harmful biological materials, hazardous materials and chemicals and
may in the future require the use of radioactive compounds. We
cannot eliminate the risk of accidental contamination or injury to
employees or third parties from the use, storage, handling or
disposal of these materials. In the event of contamination or
injury, we could be held liable for any resulting damages, and any
liability could exceed our resources or any applicable insurance
coverage we may have. Additionally, we are subject on an
ongoing basis to federal, state and local laws and regulations
governing the use, storage, handling and disposal of these
materials and specified waste products. The cost of compliance
with these laws and regulations might be significant and could
negatively affect our operating results. In the event of an
accident or if we otherwise fail to comply with applicable
regulations, we could lose our permits or approvals or be held
liable for damages or penalized with fines.
Security breaches, loss of data and other disruptions could
compromise sensitive information related to our business or prevent
us from accessing critical information and expose us to liability,
which could adversely affect our business and our
reputation.
We expect to along with certain third-party vendors that we
contract with to collect and store sensitive data, including
legally protected health information, credit card information,
personally identifiable information about our employees, customers
and patients, intellectual property, and our proprietary business
information and that of our customers, payers and collaboration
partners. We expect to manage and maintain our
applications and data utilizing a combination of on-site systems,
managed data center systems and cloud-based data center systems.
These applications and data encompass a wide variety of
business-critical information including research and development
information, commercial information and business and financial
information. We face four primary risks relative to protecting this
critical information, including loss of access risk, inappropriate
disclosure risk and inappropriate modification risk combined with
the risk of our being able to identify and audit our controls over
the first three risks.
The secure processing, storage, maintenance and transmission of
this critical information will be vital to our operations and
business strategy. As such we plan to devote significant resources
to protecting such information. Although we plan to take measures
to protect sensitive information from unauthorized access or
disclosure, our information technology and infrastructure, and that
of our third-party vendors, may be vulnerable to attacks by hackers
or viruses or breached due to employee error, malfeasance or other
disruptions. Any such breach or interruption could compromise our
networks and the information stored there could be accessed by
unauthorized parties, publicly disclosed, lost or stolen. Any such
access, disclosure or other loss of information could result in
legal claims or proceedings, liability under laws that protect the
privacy of personal information, such as the Health Insurance
Portability and Accountability Act of 1996, and regulatory
penalties. Unauthorized access, loss or dissemination could also
disrupt our operations, including our ability to process tests,
provide test results, bill payers or patients, process claims and
appeals, provide customer assistance services, conduct research and
development activities, collect, process and prepare company
financial information, provide information about our tests and
other patient and physician education and outreach efforts through
our website, manage the administrative aspects of our business and
damage our reputation, any of which could adversely affect our
business.
In
addition, the interpretation and application of consumer,
health-related and data protection laws in the U.S., Europe and
elsewhere are often uncertain, contradictory and in flux. It is
possible that these laws may be interpreted and applied in a manner
that is inconsistent with our practices. If so, this could result
in government-imposed fines or orders requiring that we change our
practices, which could adversely affect our business. Complying
with these various laws could cause us to incur substantial costs
or require us to change our business practices and compliance
procedures in a manner adverse to our
business.
We depend on our information technology and telecommunications
systems, and any failure of these systems could harm our
business.
We depend on information technology or IT, and telecommunications
systems for significant aspects of our operations. In addition, we
expect to outsource aspects of our billing and collections to a
third-party provider, whom maybe dependent upon telecommunications
and data systems provided by outside vendors and information we
provide on a regular basis. These information technology and
telecommunications systems will support a variety of functions,
including test processing, sample tracking, quality control,
customer service and support, billing and reimbursement, research
and development activities and our general and administrative
activities. Information technology and telecommunications systems
are vulnerable to damage from a variety of sources, including
telecommunications or network failures, malicious human acts and
natural disasters. Moreover, despite network security and back-up
measures we plan to implement, some or all of our servers are
potentially vulnerable to physical or electronic break-ins,
computer viruses and similar disruptive problems. Despite the
precautionary measures we plan on taking to prevent unanticipated
problems that could affect our information technology and
telecommunications systems, failures or significant downtime of our
information technology or telecommunications systems or those used
by our third-party service providers could prevent us from
processing tests, providing test results to oncologists,
pathologists, billing payers, processing reimbursement appeals,
handling patient or physician inquiries, conducting research and
development activities and managing the administrative aspects of
our business. Any disruption or loss of information technology or
telecommunications systems on which critical aspects of our
operations depend could have an adverse effect on our
business.
We may acquire other businesses or form joint ventures or make
investments in other companies or technologies that could harm our
operating results, dilute our stockholders’ ownership,
increase our debt or cause us to incur significant
expense.
As part of our business strategy, we may pursue acquisitions of
businesses and assets. We also may pursue strategic alliances and
joint ventures that leverage our core technology and expertise to
expand our offerings or distribution. We have minimal experience
with acquiring and integrating other companies or assets and
limited experience with forming strategic alliances and joint
ventures. We may not be able to find suitable partners or
acquisition candidates, and we may not be able to complete such
transactions on favorable terms, if at all. If we make any
acquisitions, we may not be able to integrate these acquisitions
successfully into our existing business, and we could assume
unknown or contingent liabilities. Any future acquisitions also
could result in significant write-offs or the incurrence of debt
and contingent liabilities, any of which could have a material
adverse effect on our financial condition, results of operations
and cash flows. Integration of an acquired company also may disrupt
ongoing operations and require management resources that would
otherwise focus on developing our existing business. We may
experience losses related to investments in other companies, which
could have a material negative effect on our results of operations.
We may not identify or complete these transactions in a timely
manner, on a cost-effective basis, or at all, and we may not
realize the anticipated benefits of any acquisition, technology
license, strategic alliance or joint venture.
To finance any acquisitions or joint ventures, we may choose to
issue shares of our common stock or securities convertible into
shares of our common stock as consideration, which would dilute the
ownership of our stockholders. If the price of our common stock is
low or volatile, we may not be able to acquire other companies or
fund a joint venture project using our stock as consideration.
Alternatively, it may be necessary for us to raise additional funds
for acquisitions through public or private financings. Additional
funds may not be available on terms that are favorable to us, or at
all.
Declining general economic or business conditions may have a
negative impact on our business.
Continuing concerns over United States health care reform
legislation and energy costs, geopolitical issues, the availability
and cost of credit and government stimulus programs in the United
States and other countries have contributed to increased volatility
and diminished expectations for the global economy. These factors,
combined with low business and consumer confidence and high
unemployment, precipitated an economic slowdown and recession. If
the economic climate does not improve, or it deteriorates, our
business, including our access to patient samples and the
addressable market for diagnostic tests that we may successfully
develop, as well as the financial condition of our suppliers and
our third-party payers, could be adversely affected, resulting in a
negative impact on our business, financial condition and results of
operations.
International expansion of our business exposes us to business,
regulatory, political, operational, financial and economic risks
associated with doing business outside of the United
States.
Our business strategy contemplates potential international
expansion, including partnering with academic and commercial
testing partners for research and development and clinical studies,
and commercializing our diagnostic tests outside the United States
and expanding relationships with international payers and
distributors. Doing business internationally involves a number of
risks, including:
●
multiple, conflicting and changing laws and regulations such as tax
laws, export and import restrictions, employment laws, regulatory
requirements and other governmental approvals, permits and
licenses;
●
competition from local and regional product offerings;
●
failure by us or our distributors to obtain regulatory approvals
for the use of our tests in various countries;
●
difficulties in staffing and managing foreign
operations;
●
complexities associated with managing multiple payer reimbursement
regimes, government payers or patient self-pay
systems;
●
logistics and regulations associated with shipping tissue samples,
including infrastructure conditions and transportation
delays;
●
limits in our ability to penetrate international markets if we are
not able to process tests locally;
●
lack of intellectual property protection in certain
markets;
●
financial risks, such as longer payment cycles, difficulty
collecting accounts receivable, the impact of local and regional
financial crises on demand and payment for our tests and exposure
to foreign currency exchange rate fluctuations;
●
natural disasters, political and economic instability, including
wars, terrorism, and political unrest, outbreak of disease,
boycotts, curtailment of trade and other business restrictions;
and
●
regulatory and compliance risks that relate to maintaining accurate
information and control over the activities of our sales force and
distributors that may fall within the purview of the FCPA, its
books and records provisions or its anti-bribery
provisions.
Any of these factors could significantly harm our future
international expansion and operations and, consequently, our
revenue and results of operations.
If we cannot compete successfully with our competitors, we may be
unable to generate, increase or sustain revenue or achieve and
sustain profitability.
We believe our principal competition for our prognostic
diagnostic assays will come from existing diagnostic methods
used by pathologists and oncologists. These methods have been used
for many years and are therefore difficult to change or supplement.
In addition, companies offering capital equipment and kits or
reagents to local pathology laboratories represent another source
of potential competition. These kits are used directly by the
pathologist, which potentially facilitates adoption more readily
than tests like ours that are performed outside the pathology
laboratory.
We
also face competition from companies that offer products or have
conducted research to profile genes, gene expression or protein
expression in breast, lung, prostate and colorectal cancer,
including public companies such as Genomic Health Inc., Agendia
Inc., GE Healthcare, a business unit of General Electric Company,
Hologic Inc., Myriad Genetics Inc., NanoString Technologies Inc.,
Novartis AG, Qiagen N.V., and Response Genetics Inc., and many
other public and private companies. We also face competition from
commercial laboratories with strong distribution networks for
diagnostic tests, such as Laboratory Corporation of America
Holdings and Quest Diagnostics Incorporated. We may also face
competition from Illumina Inc. and ThermoFisher Scientific Inc.,
both of which have announced their intention to enter the clinical
diagnostics market. Other potential competitors include companies
that develop diagnostic tests such as Roche Diagnostics, a division
of Roche Holding Ltd., Siemens AG and Veridex LLC, a Johnson &
Johnson company, as well as other companies and academic and
research institutions.
Others may invent and commercialize technology platforms such as
next generation sequencing approaches that will compete with our
test. Projects related to cancer genomics have received government
funding, both in the United States and internationally. As more
information regarding cancer genomics becomes available to the
public, we anticipate that more products aimed at identifying
targeted treatment options will be developed and that these
products may compete with ours. In addition, competitors may
develop their own versions of our tests in countries where we did
not apply for patents, where our patents have not been issued or
where our intellectual property rights are not recognized and
compete with us in those countries, including encouraging the use
of their test by physicians or patients in other
countries.
The list price of our test may change
as well as the list price of our competitor’s products. Any
increase or decrease in pricing could impact reimbursement of and
demand for our tests. Many of our present and potential competitors
have widespread brand recognition and substantially greater
financial and technical resources and development, production and
marketing capabilities than we do. Others may develop
lower-priced tests that could be viewed by physicians
and payers as functionally equivalent to our tests or offer tests
at prices designed to promote market penetration, which could force
us to lower the list prices of our tests and impact our operating
margins and our ability to achieve sustained profitability. Some
competitors have developed tests cleared for marketing by the FDA.
There may be a marketing differentiation or perception that an
FDA-cleared test is more desirable than our
diagnostic test, and that may discourage adoption of and
reimbursement for our diagnostic test. If we are unable to
compete successfully against current or future competitors, we may
be unable to increase market acceptance for and sales of our tests,
which could prevent us from increasing or sustaining our revenue or
achieving sustained profitability and could cause the market price
of our common stock to decline.
Regulatory Risks Relating to Our Business
Healthcare policy changes, including recently enacted legislation
reforming the U.S. healthcare system, may have a material adverse
effect on our financial condition and results of
operations.
There have been, and may continue to be, legislative and regulatory
proposals at the federal and state levels and in foreign
jurisdictions directed at broadening the availability and
containing or lowering the cost of healthcare. The continuing
efforts of the government, insurance companies, managed care
organizations and other payers to contain or reduce costs of
healthcare may adversely affect our ability to set prices for our
products that would allow us to achieve or sustain profitability.
In addition, governments may impose price controls on any of our
product candidates that obtain marketing approval, which may
adversely affect our future profitability.
In the United States, there have been and continue to be a number
of legislative initiatives to contain healthcare costs. For
example, in March 2010, the Patient Protection and Affordable Care
Act, as amended by the Health Care and Education Reconciliation
Act, or the ACA, was passed, which substantially changed the way
health care is financed by both governmental and private insurers,
and significantly impacted the U.S. pharmaceutical and medical
device industries and clinical laboratories. Among other things,
beginning in 2013 through December 31, 2015, each medical device
manufacturer was required to pay sales tax in an amount equal to
2.3% of the price for which such manufacturer sells its medical
devices that are listed with the FDA. The medical device tax has
been suspended by Congress since 2016 but is scheduled to be
re-imposed in 2020. Various proposals have been put forth,
including by the FDA, to regulate LDTs, as medical devices.
Although none of our diagnostic assays are currently listed with
the FDA, we cannot assure you that the tax will not apply to
services such as our laboratory operations in the
future.
Other significant measures contained in the ACA include, for
example, coordination and promotion of research on comparative
clinical effectiveness of different technologies and procedures,
initiatives to revise Medicare payment methodologies, such as
bundling of payments across the continuum of care by providers and
physicians, and initiatives to promote quality indicators in
payment methodologies. The ACA also includes significant new fraud
and abuse measures, including required disclosures of financial
arrangements with physician customers, lower thresholds for
violations and increasing potential penalties for such violations.
In addition to the ACA, various healthcare reform proposals have
also emerged from federal and state governments.
The current U.S. President and other U.S. lawmakers have made
statements about potentially repealing and/or replacing the
ACA and efforts are currently underway in the U.S. Congress to
consider legislative actions to that end. Notably, Congress enacted
legislation in 2017 that eliminates the ACA’s individual
insurance mandate beginning in 2019, which may significantly impact
the number of covered lives participating in exchange plans. We are
monitoring the impact of the ACA and proposals to repeal, replace
or refine the ACA to enable us to determine the trends and changes
that may potentially impact our business over time.
In
addition, other legislative changes have been proposed and adopted
in the United States since the ACA was enacted. These changes
include aggregate reductions of Medicare payments to providers of
2% per fiscal year, which went into effect on April 1, 2013 and,
due to subsequent legislative amendments to the statute, will
remain in effect through 2025 unless additional Congressional
action is taken. On January 2, 2013, the American Taxpayer Relief
Act of 2012 was signed into law, which, among other things, further
reduced Medicare payments to several types of providers, including
hospitals, imaging centers and cancer treatment centers, and
increased the statute of limitations period for the government to
recover overpayments to providers from three to five years.
Recently there has also been heightened governmental scrutiny over
the manner in which manufacturers set prices for their marketed
products, which has resulted in several Congressional inquiries and
proposed bills designed to, among other things, bring more
transparency to product pricing, review the relationship between
pricing and manufacturer patient programs, and reform government
program reimbursement methodologies for drug
products.
Moreover, payment methodologies, including payment for companion
diagnostics, may be subject to changes in healthcare legislation
and regulatory initiatives. For example, CMS began bundling the
Medicare payments for certain laboratory tests ordered while a
patient received services in a hospital outpatient setting.
Additionally, on April 1, 2014, the Protecting Access to Medicare
Act of 2014, or PAMA, was signed into law, which, among other
things, significantly alters the current payment methodology under
the CLFS. Under the new law, starting January 1, 2017 and every
three years thereafter (or annually in the case of advanced
diagnostic lab tests), clinical laboratories must report laboratory
test payment data for each Medicare-covered clinical diagnostic lab
test that it furnishes during a time period to be defined by future
regulations. The reported data must include the payment rate
(reflecting all discounts, rebates, coupons and other price
concessions) and the volume of each test that was paid by each
private payer (including health insurance issuers, group health
plans, Medicare Advantage plans and Medicaid managed care
organizations). Beginning in 2018, the Medicare payment rate for
each clinical diagnostic lab test, with some exceptions, will be
equal to the weighted median private payer payment for the test, as
calculated using data collected by applicable laboratories during
the data collection period and reported to CMS during a specified
data reporting period. Also, under PAMA, CMS is required to adopt
temporary billing codes to identify new clinical diagnostic
laboratory tests and advanced diagnostic laboratory tests that do
not already have unique diagnostic codes, and that have been
cleared or approved by the FDA.
We
expect that additional federal and state healthcare reform measures
will be adopted in the future, any of which could limit the amounts
that federal and state governments will pay for healthcare products
and services, which could result in reduced demand for our
therapeutic and diagnostic products or additional pricing
pressures. Other potentially significant changes in policy include
the possibility of modifications and elimination of programs and
reductions in staffing at the FDA and CMS, and initiatives to
contain or reduce governmental spending in the healthcare area,
including Medicare and Medicaid reimbursement. We cannot
predict what future healthcare initiatives will be introduced or
implemented at the federal or state level, or how any future
legislation or regulation may affect us. Any taxes imposed by
federal legislation and the expansion of the government’s
role in the U.S. healthcare industry generally, as well as changes
to the reimbursement amounts paid by payors for our existing and
future services, may reduce our profits and have a material adverse
effect on our business, financial condition, results of operations,
and cash flows.
If the FDA were to begin regulating our prognostic diagnostic tests
we could experience significant delays in commercializing our
prognostic diagnostics, be forced to stop our sales, experience
significant delays in commercializing any future products, incur
substantial costs and time delays associated with meeting
requirements for pre-market clearance or approval as well as
experience decreased demand for our prognostic diagnostic tests and
demand for reimbursement of our prognostic diagnostic
tests.
Clinical laboratory tests, like our prognostic diagnostic tests
including the MetaSite Breast™ and MENA diagnostic assays, are regulated under the Clinical
Laboratory Improvement Amendments of 1988, or CLIA, as administered
through the CMS, as well as by applicable state
laws. Diagnostic kits that are sold and distributed through
interstate commerce are regulated as medical devices by
FDA. Clinical laboratory tests that are developed and
validated by a laboratory for its own use are called Laboratory
Development Tests, or “LDTs". Most LDTs currently are
not subject to FDA regulation, although reagents or software
provided by third parties and used to perform LDTs may be subject
to regulation. We believe that our prognostic diagnostic tests
are not a diagnostic kit and we also believe that they are
LDTs. As a result, we believe our prognostic diagnostic tests
should not be subject to regulation under established FDA
policies.
At various times since 2006, the FDA has issued guidance documents
or announced draft guidance regarding initiatives that may require
varying levels of FDA oversight of our tests. In October 2014,
the FDA issued draft guidance that sets forth a proposed risk-based
regulatory framework that would apply varying levels of FDA
oversight to LDTs. In January 2017, the FDA released a discussion
paper synthesizing public comments on the 2014 draft guidance
documents and outlining a possible approach to regulation of LDTs.
The discussion paper has no legal status and does not represent a
final version of the LDT draft guidance documents. It is unclear at
this time if or when the draft guidance will be finalized, and even
then, the new regulatory requirements are proposed to be phased-in
consistent with the schedule set forth in the guidance. If this
draft guidance is finalized as presently written, it includes an
oversight framework that would require pre-market review for high
and moderate risk LDTs
Legislative proposals addressing oversight of genetic testing and
LDTs have been introduced in previous Congresses and we expect that
new legislative proposals will be introduced from time to time in
the future. We cannot provide any assurance that FDA regulation,
including pre-market review, will not be required in the future for
our tests, whether through finalization of guidance issued by the
FDA, new enforcement policies adopted by the FDA or new legislation
enacted by Congress. It is possible that legislation will be
enacted into law or guidance could be issued by the FDA which may
result in increased regulatory burdens for us to continue to offer
our tests or to develop and introduce new tests. If pre-market
review is required, our business could be negatively impacted until
such review is completed and clearance or approval is obtained, and
the FDA could require that we stop selling our tests pending
pre-market clearance or approval. If our tests are allowed to
remain on the market but there is uncertainty about the regulatory
status of our tests, if they are labeled investigational by the
FDA, or if labeling claims the FDA allows us to make are more
limited than the claims we currently make, orders or reimbursement
may decline. The regulatory approval process may involve, among
other things, successfully completing additional clinical studies
and submitting a pre-market clearance notice or filing a pre-market
approval application with the FDA. If pre-market review is required
by the FDA, there can be no assurance that our tests will be
cleared or approved on a timely basis, if at all. Ongoing
compliance with FDA regulations would increase the cost of
conducting our business, and subject us to inspection by and the
regulatory requirements of the FDA, for example registration and
listing and medical device reporting, and penalties for failure to
comply with these requirements. We may also decide voluntarily to
pursue FDA pre-market review of our tests if we determine that
doing so would be appropriate.
We cannot predict the ultimate timing or form of final FDA guidance
or regulations addressing LDTs and the potential impact on our
diagnostic tests, our diagnostic tests in development or the
materials used to perform our tests. While we expect to qualify all
materials used in our tests according to CLIA regulations, we
cannot be certain that the FDA will not enact rules or
guidance documents which could impact our ability to purchase
certain materials necessary for the performance of our tests, such
as products labeled for research use only. Should any of the
reagents obtained by us from suppliers and used in conducting our
tests be affected by future regulatory actions, our business could
be adversely affected by those actions, including increasing the
cost of testing or delaying, limiting or prohibiting the purchase
of reagents necessary to perform testing.
Testing of potential products may be required and there is no
assurance of FDA or any other regulatory approval.
The FDA and comparable agencies in foreign countries impose
substantial requirements upon the introduction of both therapeutic
and diagnostic biomedical products, through lengthy and detailed
laboratory and clinical testing procedures, sampling activities and
other costly and time-consuming procedures. Satisfaction of
these requirements typically takes several years or more and varies
substantially based upon the type, complexity, and novelty of the
product. The effect of government regulation and the need for FDA
approval may be to delay marketing of new products for a
considerable period of time, to impose costly procedures upon our
activities, and to provide an advantage to larger companies that
compete with us. There can be no assurance that FDA or other
regulatory approval for any products developed by us will be
granted on a timely basis or at all. Any such delay in obtaining,
or failure to obtain, such approvals would materially and adversely
affect the marketing of any contemplated products and the ability
to earn product revenue. Further, regulation of manufacturing
facilities by state, local, and other authorities is subject to
change. Any additional regulation could result in limitations or
restrictions on our ability to utilize any of our technologies, thereby adversely
affecting our operations. Human diagnostic and pharmaceutical
products are subject to rigorous preclinical testing and clinical
studies and other approval procedures mandated by the FDA and
foreign regulatory authorities. Various federal and foreign
statutes and regulations also govern or influence the
manufacturing, safety, labeling, storage, record keeping and
marketing of pharmaceutical products. The process of obtaining
these approvals and the subsequent compliance with appropriate
United States and foreign statutes and regulations are
time-consuming and require the expenditure of substantial
resources. In addition, these requirements and processes vary
widely from country to country. Among the uncertainties and risks
of the FDA approval process are the following: (i) the possibility
that studies and clinical studies will fail to prove the safety and
efficacy of the product, or that any demonstrated efficacy will be
so limited as to significantly reduce or altogether eliminate the
acceptability of the product in the marketplace, (ii) the
possibility that the costs of development, which can far exceed the
best of estimates, may render commercialization of the drug
marginally profitable or altogether unprofitable, and (iii) the
possibility that the amount of time required for FDA approval of a
product may extend for years beyond that which is originally
estimated. In addition, the FDA or similar foreign regulatory
authorities may require additional clinical studies, which could
result in increased costs and significant development delays.
Delays or rejections may also be encountered based upon changes in
FDA policy and the establishment of additional regulations during
the period of product development and FDA review. Similar delays or
rejections may be encountered in other
countries.
If we were required to conduct additional clinical studies prior to
marketing our prognostic diagnostic tests, those studies could lead
to delays or failure to obtain necessary regulatory approvals and
harm our ability to become profitable.
If the FDA decides to regulate our prognostic diagnostic tests, it
may require additional pre-market clinical testing before clearing
or approving our prognostic diagnostic tests for commercial sales.
Such pre-market clinical testing could delay the commencement or
completion of clinical testing, significantly increase our test
development costs, delay commercialization of any future tests, and
potentially interrupt sales of our tests. Although, we plan on
performing our future clinical studies at such FDA standards, there
is no assurance that such clinical studies will meet certain FDA
standards. Many of the factors that may cause or lead to a delay in
the commencement or completion of clinical studies may also
ultimately lead to delay or denial of regulatory clearance or
approval. The commencement of clinical studies may be delayed due
to access to adequate tissue samples and corresponding clinical
data, insufficient patient enrollment, which is a function of many
factors, including the size of the patient population, the nature
of the protocol, the proximity of patients to clinical sites and
the eligibility criteria for the clinical trial. Moreover, the
clinical trial process may fail to demonstrate that our breast
cancer tests and our planned future tests are effective for the
proposed indicated uses, which could cause us to abandon a test
candidate and may delay development of other tests.
We may find it necessary to engage CROs to perform data collection
and analysis and other aspects of our clinical studies, which might
increase the cost and complexity of our studies. We may also depend
on clinical investigators, medical institutions, academic
institutions and contract research organizations to perform the
studies. If these parties do not successfully carry out their
contractual duties or obligations or meet expected deadlines, or if
the quality, completeness or accuracy of the clinical data they
obtain is compromised due to the failure to adhere to our clinical
protocols or for other reasons, our clinical studies may have to be
extended, delayed, repeated or terminated. Many of these factors
would be beyond our control. We may not be able to enter into
replacement arrangements without undue delays or considerable
expenditures. If there are delays in testing or approvals as a
result of the failure to perform by third parties, our research and
development costs would increase, and we may not be able to obtain
regulatory clearance or approval for our tests. In addition, we may
not be able to establish or maintain relationships with these
parties on favorable terms, if at all. Each of these outcomes would
harm our ability to market our tests, or to achieve sustained
profitability.
Complying with numerous regulations pertaining to our business is
an expensive and time-consuming process, and any failure to comply
could result in substantial penalties.
We believe our prognostic diagnostic tests are subject to CLIA, a
federal law that regulates clinical laboratories that perform
testing on specimens derived from humans for the purpose of
providing information for the diagnosis, prevention or treatment of
disease. CLIA is intended to ensure the quality and
reliability of clinical laboratories in the United States by
mandating specific standards in the areas of personnel
qualifications, administration, and participation in proficiency
testing, patient test management, quality control, quality
assurance and inspections. Effective October 2015, we received
a certificate of accreditation under CLIA to perform testing of our
prognostic diagnostic tests for breast cancer. In order to
renew the certificate of accreditation, we will be subject to
survey and inspection every two years. Moreover, CLIA
inspectors may make random inspections of our laboratory outside of
the renewal process. The failure to comply with CLIA requirements
can result in enforcement actions, including the revocation,
suspension, or limitation of our CLIA certificate of accreditation,
as well as a directed plan of correction, state on-site monitoring,
civil money penalties, civil injunctive suit and/or criminal
penalties. We must maintain CLIA compliance and certification to be
eligible to bill for tests provided to Medicare beneficiaries. If
we were to be found out of compliance with CLIA program
requirements and subjected to sanctions, our business and
reputation could be harmed. Even if it were possible for us to
bring our laboratory back into compliance, we could incur
significant expenses and potentially lose revenue in doing so.
Additionally, we will seek to have our laboratory accredited by the
College of American Pathologists, or CAP, one of six CLIA-approved
accreditation organizations.
In addition, our laboratory is located in Boston, Massachusetts and
is required by state law to have a Massachusetts state license; as
we expand our geographic focus, we may need to obtain laboratory
licenses from additional states.
In addition, we need to have licenses from other states including
the states of California, New York, Pennsylvania, Florida, Maryland
and Rhode Island among others to test specimens from patients in
those states or received from ordering physicians in those states.
Other states may have similar requirements or may adopt similar
requirements in the future. Finally, we may be subject to
regulation in foreign jurisdictions if we seek to expand
international distribution of our tests outside the United
States.
If we were to lose our CLIA certification or appropriate state
license(s), whether as a result of a revocation, suspension or
limitation, we would no longer be able to sell our prognostic
diagnostic tests, or other diagnostic tests, which would
significantly harm our business. If we were to lose our license in
other states where we are required to hold licenses, we would not
be able to test specimens from those states.
We are subject, directly or indirectly, to federal and state
healthcare fraud and abuse laws, false claims laws, physician
payments transparency and health information privacy and security
laws. If we are unable to comply with any such laws, we could face
substantial penalties.
We are subject to various federal and state fraud and abuse laws,
including, without limitation, anti-kickback and false claims
statutes. Including, but not limited to:
●
Medicare billing and payment regulations applicable to clinical
laboratories;
●
the federal Medicare and Medicaid Anti-kickback Law and state
anti-kickback prohibitions;
●
the federal physician self-referral prohibition, commonly known as
the Stark Law, and the state equivalents;
●
the federal Health Insurance Portability and Accountability Act of
1996;
●
the Medicare civil money penalty and exclusion requirements;
and
●
the federal civil and criminal False Claims Act.
We have and will continue to adopt policies and procedures designed
to comply with these laws, including policies and procedures
relating to financial arrangements between us and physicians who
refer patients to us. In the ordinary course of our business,
we conduct internal reviews of our compliance with these
laws. Our compliance is also subject to governmental
review. The growth of our business and sales organization may
increase the potential of violating these laws or our internal
policies and procedures. The risk of our being found in
violation of these laws and regulations is further increased by the
fact that many of them have not been fully interpreted by the
regulatory authorities or the courts, and their provisions are open
to a variety of interpretations. Any action brought against us
for violation of these laws or regulations, even if we successfully
defend against it, could cause us to incur significant legal
expenses and divert our management’s attention from the
operation of our business. If our operations are found to be
in violation of any of these laws and regulations, we may be
subject to any applicable penalty associated with the violation,
including civil and criminal penalties, damages and fines, we could
be required to refund payments received by us, and we could be
required to curtail or cease our operations. Any of the
foregoing consequences could seriously harm our business and our
financial results.
Our corporate compliance program cannot guarantee that we are in
compliance with all potentially applicable
regulations.
The
development, manufacturing, pricing, sales, and reimbursement of
our products, together with our general operations, are subject to
extensive regulation by federal, state and other authorities within
the United States and numerous entities outside of the United
States. While we have developed and instituted a corporate
compliance program based on what we believe are the current best
practices, we cannot assure you that we are or will be in
compliance with all potentially applicable regulations. If we
fail to comply with any of these regulations, we could be subject
to a range of regulatory actions, including suspension or
termination of clinical studies, the failure to approve a product
candidate, restrictions on our products or manufacturing processes,
withdrawal of products from the market, significant fines, or other
sanctions or litigation.
Risks Relating to our Intellectual Property
If we are unable to protect our intellectual property, we may not
be able to compete effectively.
We rely upon a combination of patents, patent applications, trade
secret protection, and confidentiality agreements to protect the
intellectual property related to our technologies, products and
services. Our success will depend in part on our ability to obtain
or license patents and enforce patent protection of our products
and licensed technologies, as well as the ability of the Licensors
to enforce patent protection covering the patents which we license
pursuant to the License Agreement, Second License Agreement, the
Alternative Splicing License Agreements, and the Antibody License
Agreement or other such license agreements we may enter into both
in the United States and other countries to prevent our competitors
from developing, manufacturing and marketing products based on our
technology.
The
patent positions of biotechnology and molecular diagnostic
companies, such as us, are generally uncertain and involve complex
legal and factual questions. We will be able to protect our
intellectual property rights from unauthorized use by third parties
only to the extent that our licensed technologies are covered by
any valid and enforceable patents or are effectively maintained as
trade secrets. We could incur substantial costs in seeking
enforcement of any eventual patent rights against infringement, and
we cannot guarantee that patents that we obtain, or in-license will
successfully preclude others from using technology that we rely
upon. We have applied and intend to apply for patents in the
United States and other countries covering our technologies and
products as and when we deem appropriate. However, these
applications may be challenged or may fail to result in issued
patents. We cannot predict the breadth of claims that maybe
allowed and issued in patents related to biotechnology
applications. The laws of some foreign countries do not
protect intellectual property rights to the same extent as the laws
of the United States, and many companies have encountered
significant problems in protecting and defending such rights in
foreign jurisdictions. For example, methods of treating humans
are not patentable in many countries outside of the United
States.
The coverage claimed in a patent application can be significantly
narrowed before a patent is issued, both in the United States and
other countries. We do not know whether any of the pending or
future patent applications will result in the issuance of
patents. Any patents we or the Licensors obtain may not be
sufficiently broad to prevent others from using our technologies or
from developing competing therapeutic products based on our
technology or proprietary therapies. Once any such patents
have issued, we cannot predict how the claims will be construed or
enforced. Furthermore, others may independently develop
similar or alternative technologies or design around our
patents.
To the extent patents have been issued or may be issued, we do not
know whether these patents will be subject to further proceedings
that may limit their scope, provide significant proprietary
protection or competitive advantage, or cause them to be
circumvented or invalidated. Furthermore, patents that have or
may issue on our or the Licensors patent applications may become
subject to dispute, including interference, reissue or
reexamination proceedings in the United States, or opposition
proceedings in foreign countries. Any of these proceedings
could result in the limitation or loss of rights.
We may rely on trade secret protection for our confidential and
proprietary information. We have taken measures to protect our
proprietary information and trade secrets, but these measures may
not provide adequate protection. While we seek to protect our
proprietary information by entering into confidentiality agreements
with employees, collaborators and consultants, we cannot assure
that our proprietary information will not be disclosed, or that we
can meaningfully protect our trade secrets. In addition,
competitors may independently develop or may have already developed
substantially equivalent proprietary information or may otherwise
gain access to our trade secrets.
The pending patent applications that we have in-licensed or that we
may in-license in the future may not result in issued patents, and
we cannot assure you that our issued patents or any patents that
might ultimately be issued by the United States Patent and
Trademark Office, or USPTO will protect our technology. Any patents
that may be issued to us might be challenged by third parties as
being invalid or unenforceable, or third parties may independently
develop similar or competing technology that avoids our patents. We
cannot be certain that the steps we have taken will prevent the
misappropriation and use of our intellectual property, particularly
in foreign countries where the laws may not protect our proprietary
rights as fully as in the United States.
Inventions,
and the intellectual property rights covering them, that are
discovered under research, material transfer or other such
collaboration agreements may become solely owned by us in some
cases, jointly owned by us and the other party to such agreements
in some cases and may become the exclusive property of other party
to such agreements in other cases. Under some circumstances, it may
be difficult to determine which party owns a particular invention,
or whether it is jointly owned, and disputes could arise regarding
ownership of those inventions. These disputes could be costly and
time consuming and an unfavorable outcome could have a significant
adverse effect on our business if we were not able to protect or
license rights to these inventions.
Unauthorized uses of our proprietary intellectual property by any
such research collaborators, and publications by our research
collaborators and scientific advisors containing such information,
either with our permission or in contravention of the terms of
their agreements with us, may limit or harm our ability to obtain
patent protection for our product candidates or protect our
proprietary information, which could materially harm our business,
prospects, financial condition and results of
operations.
Patent policy and rule changes could increase the uncertainties and
costs surrounding the prosecution of our patent applications and
the enforcement or defense of our issued patents.
Changes in either the patent laws or interpretation of the patent
laws in the United States and other countries may diminish the
value of our patents or narrow the scope of our patent protection.
The laws of foreign countries may not protect our rights to the
same extent as the laws of the United States. Publications of
discoveries in the scientific literature often lag behind the
actual discoveries, and patent applications in the United States
and other jurisdictions are typically not published until 18 months
after filing, or in some cases not at all. We therefore cannot be
certain that we or our licensors were the first to make the
invention claimed in our owned and licensed patents or pending
applications, or that we or our licensor were the first to file for
patent protection of such inventions. Assuming the other
requirements for patentability are met, in the United States prior
to March 15, 2013, the first to make the claimed invention is
entitled to the patent, while outside the United States, the first
to file a patent application is entitled to the patent. After March
15, 2013, under the Leahy-Smith America Invents Act, or the
Leahy-Smith Act, enacted on September 16, 2011, the United States
has moved to a first to file system. The Leahy-Smith Act also
includes a number of significant changes that affect the way patent
applications will be prosecuted and may also affect patent
litigation. The effects of these changes are currently unclear as
the USPTO must still implement various regulations, the courts have
yet to address any of these provisions and the applicability of the
act and new regulations on specific patents discussed herein have
not been determined and would need to be reviewed. In general, the
Leahy-Smith Act and its implementation could increase the
uncertainties and costs surrounding the prosecution of our patent
applications and the enforcement or defense of our issued patents,
all of which could have a material adverse effect on our business
and financial condition.
Litigation or third-party claims of intellectual property
infringement could impair our ability to develop and commercialize
our products successfully.
Our research, development and commercialization activities, as well
as any product candidates or products resulting from those
activities, may infringe or be alleged to infringe a patent or
other form of intellectual property under which we do not hold a
license or other rights. Third parties may assert that we are
employing their proprietary technology without authorization. Our
success will depend in part on our ability to avoid infringing
patents and proprietary rights of third parties, and not breaching
any licenses that we have entered into with regard to our
technologies. A number of pharmaceutical companies,
biotechnology companies, independent researchers, universities and
research institutions may have filed patent applications or may
have been granted patents that cover technologies similar to the
technologies owned by or licensed to us. For instance, a
number of patents may have issued and may issue in the future on
tests and technologies that we have developed or intend to
develop. If patents covering technologies required by our
operations are issued to others, we may have to rely on licenses
from third parties, which may not be available on commercially
reasonable terms, or at all.
We have no knowledge of any infringement or patent litigation,
threatened or filed at this time. It is possible that we may
infringe on intellectual property rights of others without being
aware of the infringement. If a patent holder believes that
one of our product candidates infringes on our patent, it may sue
we even if we have received patent protection for our
technology. Third parties may claim that we are employing our
proprietary technology without authorization. In addition,
third parties may obtain patents that relate to our technologies
and claim that use of such technologies infringes these
patents. Regardless of their merit, such claims could require
us to incur substantial costs, including the diversion of
management and technical personnel, in defending ourselves against
any such claims or enforcing our patents. In the event that a
successful claim of infringement is brought against us, we may be
required to pay damages and obtain one or more licenses from third
parties. We may not be able to obtain these licenses at a
reasonable cost, or at all. Defense of any lawsuit or failure
to obtain any of these licenses could adversely affect our ability
to develop and commercialize our products. Although we carry
general liability insurance, our insurance may not cover potential
claims of this type. The occurrence of any of the above events
could prevent us from continuing to develop and commercialize one
or more of our product candidates or practice our related methods,
and our business could materially suffer.
Our rights to use technologies licensed from third parties are not
within our control, and we may not be able to develop,
commercialize, and sell our products if we lose our existing rights
or cannot obtain new rights on reasonable terms.
We license technology necessary to develop certain products from
third parties. For example, we license technology from MIT,
AECOM, Cornell and IFO-Regina located in Rome, Italy, that we may
use in certain product candidates and that we may use to develop
certain additional product candidates. As a result, our
current business plans are dependent upon our satisfaction of
certain conditions to the maintenance of those license agreements
and the rights we license under them. Each of the license
agreements provides that we are subject to diligence obligations
relating to the development and commercialization of product
candidates, milestone payments, royalty payments and other
obligations. In addition to these license agreements, we may seek
to enter into additional agreements with other third parties in the
future granting similar license rights with respect to other
potential product candidates. If we fail to comply with any of the
conditions or obligations or otherwise breach the terms of any of
these license agreements, or any future license agreement we may
enter on which our business or product candidates are dependent,
the licensor may have the right to assert a claim for damages
against us or terminate the applicable agreement in whole or in
part and thereby extinguish our rights to the licensed technology
and intellectual property and/or any rights we have acquired to
develop and commercialize certain product candidates. If we become
liable for material damages under any of these license agreements,
this could materially harm our business, prospects, financial
condition and results of operations. Similarly, the loss of the
rights licensed to us under these license agreements, or any future
license agreement that we may enter granting us rights on which our
business or product candidates are dependent, would eliminate our
ability to further develop the applicable product candidates and
would materially harm our business, prospects, financial condition
and results of operations.
Our liquidity issues in the past have sometimes caused a delay in
payment under our existing license agreements. Our business may
suffer if we are unable to meet our obligations, financial or
otherwise, under our existing license agreements and if these
licenses terminate if the licensors fail to abide by the terms of
the licenses or fail to prevent infringement by third parties if
the licensed patents or other rights are found to be invalid or if
we are unable to enter into necessary additional licenses on
acceptable terms.
We may be subject to claims that our employees, consultants, or
independent contractors have wrongfully used or disclosed
confidential information of third parties or that our employees
have wrongfully used or disclosed alleged trade secrets of their
former employers.
We employ certain individuals who were previously employed at
universities, medical institutions, other diagnostic and
biotechnology companies, including potential competitors. Although
we try to ensure that our employees, consultants, and independent
contractors do not use the proprietary information or know-how of
others in their work for us, and we are not currently subject to
any claims that our employees, consultants, or independent
contractors have wrongfully used or disclosed confidential
information of third parties, we may in the future be subject to
such claims. Litigation may be necessary to defend against these
claims. If we fail in defending any such claims, in addition to
paying monetary damages, we may lose valuable intellectual property
rights or personnel, which could adversely impact our business.
Even if we are successful in defending against such claims,
litigation could result in substantial costs and be a distraction
to management and other employees.
We may be subject to claims challenging the inventorship of our
patents and other intellectual property.
Although we are not currently experiencing any claims challenging
the inventorship of our licensed patents, or ownership of our
intellectual property, we may in the future be subject to claims
that former employees, collaborators or other third parties have an
interest in our licensed patents, future patent applications or
other intellectual property as an inventor or co-inventor. For
example, we may have inventorship disputes arise from conflicting
obligations of consultants or others who are involved in developing
our products or services including clinical studies. Litigation may
be necessary to defend against these and other claims challenging
inventorship. If we fail in defending any such claims, in addition
to paying monetary damages, we may lose valuable intellectual
property rights, such as exclusive ownership of, or right to use,
valuable intellectual property. Such an outcome could have a
material adverse effect on our business. Even if we are successful
in defending against such claims, litigation could result in
substantial costs and be a distraction to management and other
employees.
We may desire, or be forced, to seek additional licenses to use
intellectual property owned by third parties, and such licenses may
not be available on commercially reasonable terms or at
all.
A third party may hold intellectual property, including patent
rights, that are important or necessary to the development or
commercialization of our product candidates or to practice our
related methods, in which case we would need to obtain a license
from that third party or develop a different method relating to the
product candidate that does not infringe the applicable
intellectual property, which may not be possible. Additionally, we
may identify product candidates that we believe are promising and
whose composition of matter, use or manufacture are covered by the
intellectual property rights of third parties. In such a case, we
may desire to seek a license to pursue the development and
commercialization of those product candidates. Any license that we
may desire to obtain, or that we may be forced to pursue, may not
be available when needed on commercially reasonable terms, or at
all. Any inability to secure a license that we need, or desire
could have a material adverse effect on our business, financial
condition and prospects.
Changes in U.S. patent law could diminish the value of patents in
general, thereby impairing our ability to protect our
products.
As is the case with other diagnostic and biopharmaceutical
companies, our success is heavily dependent on intellectual
property, particularly patents. Obtaining and enforcing patents in
the diagnostic and biopharmaceutical industry involves both
technological and legal complexity. Therefore, obtaining and
enforcing diagnostic and biotechnology patents is costly, time
consuming, and inherently uncertain. In addition, the United States
has recently enacted and is currently implementing wide-ranging
patent reform legislation. Recent U.S. Supreme Court rulings have
narrowed the scope of patent protection available in certain
circumstances and weakened the rights of patent owners in certain
situations. In addition to increasing uncertainty with regard to
our ability to obtain patents in the future, this combination of
events has created uncertainty with respect to the value of
patents, once obtained. Depending on future actions by the U.S.
Congress, the federal courts, and the USPTO, the laws and
regulations governing patents could change in unpredictable ways
that would weaken our ability to obtain new patents or to enforce
our existing patents and patents that we might obtain in the
future.
Our collaborators may assert ownership or commercial rights to
inventions we develop from our use of the biological materials,
which they provide to us, or otherwise arising from the
collaboration.
We collaborate with several institutions, universities, medical
centers, physicians and researchers in scientific matters and
expect to continue to enter into additional collaboration
agreements. In certain cases, we do not have written agreements
with certain of such collaborators, or the written agreements we
have do not cover intellectual property rights. Also, we rely on
numerous third parties to provide us with tissue samples and
biological materials that we use to develop products. If we cannot
successfully negotiate sufficient ownership and commercial rights
to any inventions that result from our use of a third-party
collaborator’s materials, or if disputes arise with respect
to the intellectual property developed with the use of a
collaborator’s samples, or data developed in a
collaborator’s study, we may be limited in our ability to
capitalize on the market potential of these inventions or
developments.
We may be involved in lawsuits or administrative proceedings to
protect or enforce our patents or the patents of our licensors,
which could be expensive, time-consuming and
unsuccessful.
Competitors may infringe our patents or the patents of our current
or potential licensors. We cannot predict if, when or where a third
party may infringe one or more of our issued patents. To attempt to
stop infringement or unauthorized use, we may need to enforce one
or more of our patents, which can be expensive and time-consuming,
a significant diversion of employee resources, and distract our
management. There is no assurance such action will ultimately be
successful in halting third party infringing activities, for
example, through a permanent injunction, or that we would be fully
or even partially financially compensated for any harm to our
business caused by such third-party infringement. Even if such
action were initially successful, it could be overturned upon
appeal. Even if we are successful in proving in a court of law that
a third party is infringing one or more of our issued patents we
may be forced to enter into a license or other agreement with the
infringing third party on terms less commercially acceptable to us
than if the license or agreement were negotiated under conditions
between those of a willing licensee and a willing licensor. We may
not become aware of a third-party infringer within legal timeframes
that would enable us to seek adequate compensation, or at all,
thereby possibly losing the ability to be compensated for any harm
to our business. Such a third-party may be operating in a foreign
country where the infringer is difficult to locate, where we do not
have issued patents and/or the patent laws may be more difficult to
enforce. If we pursue any litigation, a court may decide that a
patent of ours or our licensor’s is not of sufficient breath,
is invalid, or is unenforceable, or may refuse to stop the other
party from using the relevant technology on the grounds that our
patents do not cover the technology in question. Further, the legal
systems of certain countries, particularly certain developing
countries, do not favor the enforcement of patents, which could
reduce the likelihood of success of any infringement proceeding we
pursue in any such jurisdiction. An adverse result in any patent
litigation could put one or more of our patents at risk of being
invalidated, held unenforceable, or interpreted narrowly and could
put our pending patent applications at risk of not issuing, which
could limit our ability to exclude competitors from directly
competing with us in the applicable jurisdictions.
Certain
administrative proceedings may be provoked by third parties before
the USPTO and certain foreign patent offices, such as interference
proceedings, opposition proceedings, re-examination proceedings,
inter party review, post-grant review, derivation proceedings and
pre-grant submissions, in which third parties may challenge the
validity or breadth of claims contained in our patents or those of
our licensors. An adverse result in any such administrative
proceeding could put one or more of our patents at risk of being
canceled or invalidated or interpreted narrowly and could put our
pending patent applications at risk of not issuing, which could
limit our ability to exclude competitors from directly competing
with us in the applicable jurisdictions.
Interference proceedings provoked by third parties or brought by us
or the USPTO may be necessary to determine the priority of
inventions with respect to our patents or patent applications or
those of our licensors. Derivation proceedings may be brought by us
or a third party to determine whether a patent or application was
filed by the true inventor. An unfavorable outcome in an
interference or derivation proceeding could require us to cease
using the related technology or to attempt to license rights to use
it from the prevailing party. Our business could be harmed if the
prevailing party does not offer us a license on commercially
reasonable terms, or at all. Litigation, interference, or
derivation proceedings may have undesirable outcomes and, even if
successful, may result in substantial costs, be a significant
diversion of employee resources, and distract our
management.
We may not be able to protect our intellectual property rights
throughout the world.
Filing, prosecuting, and defending patents on products and services
in all countries throughout the world would be prohibitively
expensive, and our intellectual property rights in some countries
outside the United States can be less extensive than those in the
United States. In addition, the laws of some foreign countries do
not protect intellectual property rights to the same extent as
federal and state laws in the United States. Consequently, we may
not be able to prevent third parties from practicing our inventions
in all countries outside the United States, or from selling or
importing products made using our inventions in and into the United
States or other jurisdictions. Competitors may use our technologies
in jurisdictions where we have not obtained patent protection to
develop their own products and may also export infringing products
to territories where we have patent protection, but enforcement is
not as strong as that in the United States. These products may
compete with our products and our patents or other intellectual
property rights may not be effective or sufficient to prevent them
from competing.
Many
companies have encountered significant problems in protecting and
defending intellectual property rights in foreign jurisdictions.
The legal systems of certain countries, particularly certain
developing countries, do not favor the enforcement of patents,
trade secrets, and other intellectual property protection,
particularly those relating to biotechnology products, which could
make it difficult for us to stop the infringement of our patents or
marketing of competing products in violation of our proprietary
rights generally. Proceedings to enforce our patent rights in
foreign jurisdictions, whether or not successful, could result in
substantial costs and divert our efforts and attention from other
aspects of our business, could put our patents at risk of being
invalidated or interpreted narrowly and our patent applications at
risk of not issuing and could provoke third parties to assert
claims against us. We may not prevail in any lawsuits that we
initiate, and the damages or other remedies awarded, if any, may
not be commercially meaningful. Accordingly, our efforts to enforce
our intellectual property rights around the world may be inadequate
to obtain a significant commercial advantage from the intellectual
property that we develop or license.
Risks Relating to our Securities
The market price of our common stock may be volatile.
The market price of our common stock has been and will likely
continue to be highly volatile, as is the stock market in general
and the market for OTC or “bulletin board” quoted
stocks in particular. Market prices for securities of
early-stage life sciences companies have historically been
particularly volatile. Some of the factors that may materially
affect the market price of our common stock are beyond our control,
may include, but are not limited to:
●
progress, or lack of progress, in developing and commercializing
our current tests and our planned future cancer diagnostic
tests;
●
favorable or unfavorable decisions about our tests from government
regulators, insurance companies or other third-party
payers;
●
changes in key personnel and our ability to recruit and retain
qualified research and development personnel;
●
changes in investors’ and securities analysts’
perception of the business risks and conditions of our
business;
●
changes in our relationship with key collaborators;
●
changes in the market valuation or earnings of our competitors or
companies viewed as similar to us;
●
depth of the trading market in our common stock;
●
termination of the lock-up agreements or other restrictions on the
ability of our existing stockholders to sell shares;
●
changes in our capital structure, such as future issuances of
securities or the incurrence of additional debt;
●
the granting or exercise of employee stock options or other equity
awards;
●
realization of any of the risks described under this section
entitled “Risk Factors”; and
●
general market and economic conditions.
In addition, the equity markets have experienced significant price
and volume fluctuations that have affected the market prices for
the securities for a number of reasons, including reasons that may
be unrelated to our business or operating performance. These broad
market fluctuations may result in a material decline in the market
price of our common stock and you may not be able to sell your
shares at prices you deem acceptable. In the past, following
periods of volatility in the equity markets, securities class
action lawsuits have been instituted against public companies. Such
litigation, if instituted against us, could result in substantial
cost and the diversion of management attention.
We cannot assure you that our common stock will become liquid or
that it will be listed on a national securities exchange. In
addition, there may
not be sufficient liquidity in the market for our securities in
order for investors to sell their securities.
Currently, our common stock trades on the OTCQB venture stage
marketplace for early stage and developing U.S. and international
companies. Investors may find it difficult to obtain accurate
quotations as to the market value of our common stock. In
addition, if we fail to meet the criteria set forth in SEC
regulations, by law, various requirements would be imposed on
broker-dealers who sell its securities to persons other than
established customers and accredited investors. Consequently,
such regulations may deter broker-dealers from recommending or
selling our common stock, which may further affect its
liquidity. In addition, there is currently only a limited
public market for our common stock and there can be no assurance
that a trading market will develop further or be maintained in the
future.
In order to raise sufficient funds to expand our operations, we may
have to issue additional securities at prices which may result in
substantial dilution to our shareholders.
If we raise additional funds through the sale of equity or
convertible debt, our current stockholders’ percentage
ownership will be reduced. In addition, these transactions may
dilute the value of our outstanding securities. We may have to
issue securities that may have rights, preferences and privileges
senior to our common stock. We cannot provide assurance that
we will be able to raise additional funds on terms acceptable to
us, if at all. If future financing is not available or is not
available on acceptable terms, we may not be able to fund our
future needs, which would have a material adverse effect on our
business plans, prospects, results of operations and financial
condition.
Future sales of our common stock, or the perception that future
sales may occur, may cause the market price of our common stock to
decline, even if our business is doing well.
Sales of substantial amounts of our common stock, or the perception
that these sales may occur, could materially and adversely affect
the price of our common stock and could impair our ability to raise
capital through the sale of additional equity
securities. As of February 28, 2018, we had outstanding
5,877,383 shares of common stock, 3,935,863 which are
restricted securities that may be sold only in accordance with the
resale restrictions under Rule 144 of the Securities Act of 1933,
as amended. In addition, as of February 28, 2018, we had
outstanding 300,000 restricted stock units, outstanding convertible
preferred stock, convertible into 4,298,579 shares of common stock,
outstanding options to purchase 1,286,770 shares of our common
stock, outstanding warrants to purchase 3,060,118 shares of our
common stock, and outstanding convertible debt convertible into
562,082 shares of our common stock. Shares issued upon the exercise
of stock options and warrants will be eligible for sale in the
public market, except that affiliates will continue to be subject
to volume limitations and other requirements of Rule 144 under the
Securities Act. The issuance or sale of such shares could depress
the market price of our common stock.
In the future, we also may issue our securities if we need to raise
additional capital. The number of new shares of our common stock
issued in connection with raising additional capital could
constitute a material portion of the then-outstanding shares of our
common stock.
Rule 144 Related Risk
The SEC adopted amendments to Rule 144 which became effective on
February 15, 2008 that apply to securities acquired both before and
after that date. Under these amendments, a person who has
beneficially owned restricted shares of our common stock for at
least six months would be entitled to sell their securities
provided that: (i) such person is not deemed to have been one of
our affiliates at the time of, or at any time during the three
months preceding a sale, (ii) we are subject to the Exchange Act
periodic reporting requirements for at least 90 days before the
sale and (iii) if the sale occurs prior to satisfaction of a
one-year holding period, we provide current information at the time
of sale. Persons who have beneficially owned restricted shares of
our common stock for at least six months but who are our affiliates
at the time of, or at any time during the three months preceding a
sale, would be subject to additional restrictions, by which such
person would be entitled to sell within any three-month period only
a number of securities that does not exceed the greater of either
of the following:
●
1% of the total number of securities of the same class then
outstanding; or closing or bid price requirements;
●
the average weekly trading volume of such securities during the
four calendar weeks preceding the filing of a notice on Form 144
with respect to the sale;
provided, in each case, that we are subject to the Exchange Act
periodic reporting requirements for at least three months before
the sale. Such sales by affiliates must also comply with the manner
of sale, current public information and notice provisions of Rule
144.
Restrictions on the reliance of Rule 144 by shell companies or
former shell companies.
We are a former shell company. Historically, the SEC staff has
taken the position that Rule 144 is not available for the resale of
securities initially issued by companies that are, or previously
were, blank check companies. The SEC has codified and expanded this
position in amendments to Rule 144 which became effective in
February 2008 by prohibiting the use of Rule 144 for resale of
securities issued by any shell companies (other than
business-combination related shell companies) or any issuer that
has been at any time previously a shell company. The SEC has
provided an important exception to this prohibition, however, if
the following conditions are met:
●
The issuer of the securities that was formerly a shell company has
ceased to be a shell company;
●
The issuer of the securities is subject to the reporting
requirements of Section 13 or 15(d) of the Exchange
Act;
●
The issuer of the securities has filed all Exchange Act reports and
material required to be filed, as applicable, during the preceding
12 months (or such shorter period that the issuer was required to
file such reports and materials), other than Current Reports on
Form 8-K; and
●
At least one year has elapsed from the time that the issuer has
filed current comprehensive disclosure with the SEC reflecting its
status as an entity that is not a shell company.
As a result, it is possible that pursuant to Rule 144, stockholders
may not be able to sell our shares without registration if one of
the aforementioned conditions are not satisfied.
We have not attracted, and do not expect to attract, the attention
of major brokerage firms.
Additional risks may exist since we became public through a
“reverse takeover.” Securities analysts of major
brokerage firms have not provided, and may not in the future
provide coverage of our securities since there is little incentive
to brokerage firms to recommend the purchase of our common
stock. No assurance can be given that brokerage firms will
want to conduct any secondary offerings on our behalf in the
future.
We have incurred and will continue to incur significant increased
costs as a result of operating as a public company, and our
management will be required to devote substantial time to new
compliance initiatives.
As a public company, we are subject to the reporting requirements
of the Securities Exchange Act of 1934, as amended, the Dodd-Frank
Wall Street Reform and Consumer Protection Act, or the Dodd-Frank
Act, the listing requirements of the OTCQB venture stage
marketplace and other applicable securities rules and regulations.
Compliance with these rules and regulations has increased and will
continue to increase our legal and financial compliance costs, make
some activities more difficult, time-consuming or costly, and
increase demand on our systems and resources. The Sarbanes-Oxley
Act requires, among other things, that we maintain effective
disclosure controls and procedures and internal control over
financial reporting. In order to maintain and, if required, improve
our disclosure controls and procedures and internal control over
financial reporting to meet this standard, significant resources
and management oversight may be required. As a result,
management’s attention may be diverted from other business
concerns, which could harm our business and operating results.
Further, there are significant corporate governance and executive
compensation related provisions in the Dodd-Frank Wall Street
Reform and Consumer Protection Act, enacted in 2010, that require
the SEC to adopt additional rules and regulations in these areas
such as “say on pay” and proxy access. Recent
legislation permits smaller “emerging growth companies”
to implement many of these requirements over a longer period. We
intend to take advantage of this new legislation but cannot
guarantee that we will not be required to implement these
requirements sooner than budgeted or planned and thereby incur
unexpected expenses. Stockholder activism, the current political
environment and the current high level of government intervention
and regulatory reform may lead to substantial new regulations and
disclosure obligations, which may lead to additional compliance
costs and impact the manner in which we operate our business in
ways we cannot currently anticipate.
In addition, changing laws, regulations and standards relating to
corporate governance and public disclosure are creating uncertainty
for public companies, increasing legal and financial compliance
costs and making some activities more time consuming. These laws,
regulations and standards are subject to varying interpretations,
in many cases due to their lack of specificity, and, as a result,
their application in practice may evolve over time as new guidance
is provided by regulatory and governing bodies. This could result
in continuing uncertainty regarding compliance matters and higher
costs necessitated by ongoing revisions to disclosure and
governance practices. We intend to invest resources to comply with
evolving laws, regulations and standards, and this investment may
result in increased general and administrative expenses and a
diversion of management’s time and attention from
revenue-generating activities to compliance activities. If our
efforts to comply with new laws, regulations and standards differ
from the activities intended by regulatory or governing bodies due
to ambiguities related to practice, regulatory authorities may
initiate legal proceedings against us and our business may be
harmed.
Additionally, we may be subject to increased corporate governance
requirements in connection with the listing of our common stock on
a national securities exchange, such as the NASDAQ Capital Market,
which may lead to additional compliance costs and impact the manner
in which we operate our business.
If we fail to maintain an effective system of internal control over
financial reporting, we may not be able to accurately report our
financial results. As a result, current and potential investors
could lose confidence in our financial reporting, which could harm
our business and have an adverse effect on our stock
price.
Effective internal controls over financial reporting are necessary
for us to provide reliable financial reports and, together with
adequate disclosure controls and procedures, are designed to
prevent fraud. Any failure to implement required new or improved
controls, or difficulties encountered in their implementation could
cause us to fail to meet our reporting obligations. In addition,
pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, we
are required to annually furnish a report by our management on our
internal control over financial reporting. Such report must
contain, among other matters, an assessment by our principal
executive officer and our principal financial officer on the
effectiveness of our internal control over financial reporting,
including a statement as to whether or not our internal control
over financial reporting is effective as of the end of our fiscal
year. This assessment must include disclosure of any material
weakness in our internal control over financial reporting
identified by management. In addition, under current SEC
rules, we may be required to obtain an attestation from our
independent registered public accounting firm as to our internal
control over financial reporting for our annual report on Form 10-K
covering our next fiscal year. Performing the system and process
documentation and evaluation needed to comply with Section 404 is
both costly and challenging. During the course of our testing
we may identify deficiencies which we may not be able to remediate
in time to meet the deadline imposed by the Sarbanes-Oxley Act of
2002 for compliance with the requirements of Section 404. In
addition, if we fail to maintain the adequacy of our internal
controls, as such standards are modified, supplemented or amended
from time to time, we may not be able to ensure that we can
conclude on an ongoing basis that we have effective internal
controls over financial reporting in accordance with Section 404 of
the Sarbanes-Oxley Act of 2002. Failure to achieve and
maintain an effective internal control environment could also cause
investors to lose confidence in our reported financial information,
which could have a material adverse effect on the price of our
common stock.
Our common stock is considered “penny
stock”.
The SEC has adopted regulations, which generally define
“penny stock” to be an equity security that has a
market price of less than $5.00 per share, subject to specific
exemptions. The market price of our common stock is currently
less than $5.00 per share and therefore may be a “penny
stock.” Brokers and dealers effecting transactions in
“penny stock” must disclose certain information
concerning the transaction, obtain a written agreement from the
purchaser and determine that the purchaser is reasonably suitable
to purchase the securities. These rules may restrict the
ability of brokers or dealers to sell the common stock and may
affect your ability to sell shares.
The market for penny stocks has experienced numerous frauds and
abuses, which could adversely impact investors in our
stock.
Our common stock trades on the OTCQB venture stage marketplace for
early stage and developing U.S. and international companies. OTCQB
securities and other “bulletin board” securities are
frequent targets of fraud or market manipulation, both because of
their generally low prices and because OTCQB and other bulletin
board” reporting requirements are less stringent than those
of national securities exchanges, including the NASDAQ Capital
Market.
Patterns of fraud and abuse include:
●
Control of the market for the security by one or a few
broker-dealers that are often related to the promoter or
issuer;
●
Manipulation of prices through prearranged matching of purchases
and sales and false and misleading press releases;
●
“Boiler room” practices involving high pressure sales
tactics and unrealistic price projections by inexperienced sales
persons;
●
Excessive and undisclosed bid-ask differentials and markups by
selling broker-dealers; and
●
Wholesale dumping of the same securities by promoters and
broker-dealers after prices have been manipulated to a desired
level, along with the inevitable collapse of those prices with
consequent investor losses.
Our quarterly operating results may fluctuate
significantly.
We expect our operating results to be subject to quarterly
fluctuations. Our net loss and other operating results will be
affected by numerous factors, including:
●
the rate of adoption and/or continued use of our current tests and
our planned future tests by healthcare practitioners;
●
variations in the level of expenses related to our development and
commercialization programs;
●
addition or reduction of resources for product
commercialization;
●
addition or termination of clinical validation studies and clinical
utility studies;
●
any intellectual property infringement lawsuit in which we may
become involved;
●
third party payer determinations affecting our tests;
and
●
regulatory developments affecting our tests.
We expect operating results to be subject to quarterly
fluctuations. Our net loss and other operating results will be
affected by numerous factors, including:
If our quarterly operating results fall below the expectations of
investors or securities analysts, the price of our common stock
could decline substantially. Furthermore, any quarterly
fluctuations in our operating results may, in turn, cause the price
of our stock to fluctuate substantially.
Because we do not expect to pay cash dividends to our common
stockholders for the foreseeable future, you must rely on
appreciation of our common stock price for any return on your
investment. Even if we change that policy, we may be restricted
from paying dividends on our common stock.
We do not intend to pay cash dividends on shares of our common
stock for the foreseeable future. Any determination to pay
dividends in the future will be at the discretion of our board of
directors and will depend upon results of operations, financial
performance, contractual restrictions, restrictions imposed by
applicable law and other factors our board of directors deems
relevant. Accordingly, you will have to rely on capital
appreciation, if any, to earn a return on your investment in our
common stock. Investors seeking cash dividends in the foreseeable
future should not purchase our common stock.
Cumulative dividends on the Series B Preferred Stock accrue at the
rate of 8% of the Stated Value per annum, payable quarterly on
March 31, June 30, September 30, and December 31 of each year, from
and after the date of the initial issuance. Dividends
are payable in kind in additional shares of Series B Preferred
Stock valued at the Stated Value or in cash at the sole option of
the Company. At February 28, 2018 and February 28, 2017, the
dividend payable to the holders of the Series B Preferred Stock
amounted to approximately $42,000 and approximately $16,000,
respectively. During the year ended February 28, 2018 and February
28, 2017, the Company issued 13.0520 and 34.5085 shares of Series B
Preferred Stock, respectively, for payment of dividends amounting
to approximately $72,000 and approximately $190,000,
respectively.
Our ability to use our net operating loss carryforwards and certain
other tax attributes may be limited.
Our ability to utilize our federal net operating loss,
carryforwards and federal tax credits may be limited under Sections
382 and 383 of the Internal Revenue Code of 1986, as amended, or
the Code. The limitations apply if an “ownership
change,” as defined by Section 382 of the Code, occurs.
If we have experienced an “ownership change” at any
time since our formation, we may already be subject to limitations
on our ability to utilize our existing net operating losses and
other tax attributes to offset taxable income. In addition, future
changes in our stock ownership (including in connection with this
or future offerings, as well as other changes that may be outside
of our control), may trigger an “ownership change” and,
consequently, limitations under Sections 382 and 383 of the
Code. As a result, if we earn net taxable income, our ability to
use our pre-change net operating loss carryforwards and other tax
attributes to offset United States federal taxable income may be
subject to limitations, which could potentially result in increased
future tax liability to us. As of February 28, 2018, we had federal
net operating loss tax credit carryforwards of approximately
$21.7 million, which could be limited if we have
experienced or do experience any “ownership changes.”
We have not completed a study to assess whether an “ownership
change” has occurred or whether there have been multiple
“ownership changes” since our formation, due to the
complexity and cost associated with such a study, and the fact that
there may be additional ownership changes in the
future.
We could be subject to securities class action
litigation.
In the past, securities class action litigation has often been
brought against a company following a decline in the market price
of its securities. This risk is especially relevant for us because
early-stage life sciences and diagnostic companies have experienced
significant stock price volatility in recent years. If we face such
litigation, it could result in substantial costs and a diversion of
management’s attention and resources, which could harm our
business.
|
Item 1B.
|
UNRESOLVED STAFF
COMMENTS
|
Not applicable.
|
Item 2.
|
PROPERTIES
|
On August 28, 2014, we entered into a lease agreement, subsequently
amended (the “Boston Lease”) for our diagnostic
laboratory and office space located at 27, Drydock Ave, 2nd Floor,
Boston, MA 02210 (the “Boston Property”). We paid
a $40,000 security deposit in connection with entering into the
Boston Lease.
On July 26, 2017, we entered into a second amendment to the Boston
Lease for the Boston Property (the “Second Boston Lease
Amendment”). The Second Boston Lease Amendment extended the
term (the “Second Extension Period”) for five years
from September 1, 2017 through August 31, 2022. Monthly basic rent
payments are approximately $23,000 for the first year of the Second
Extension Period, approximately $24,000 for the second year of the
Second Extension Period, approximately $25,000 for the third year
of the Second Extension Period, approximately $25,000 for the
fourth year of the Second Extension Period, and approximately
$26,000 for the fifth year of the Second Extension
Period.
Effective March 1, 2015, we entered into a lease agreement for
short-term office space in New York, NY. The term of the
lease is month-to-month and may be terminated upon twenty-one (21)
days’ notice. We paid a $2,100 security deposit in connection
with entering into the lease. Effective December 1, 2015, we
amended our lease agreement for the short-term office space in New
York, NY. The basic rent payment increased to $2,400 per month
and we paid an additional $1,500 security deposit in connection
with the amended lease.
|
Item 3.
|
LEGAL PROCEEDINGS
|
We are not engaged in any material litigation, arbitration or
claim, and no material litigation, arbitration or claim is known by
our management to be pending or threatened by or against us that
would have a material adverse effect on our results from operations
or financial condition.
|
Item 4.
|
MINE SAFETY DISCLOSURES
|
Not applicable.
PART II
|
Item 5.
|
MARKET FOR REGISTRANT'S COMMON EQUITY,
RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY
SECURITIES
|
Market Price Information for our Common Stock
Our common stock is quoted on the OTCQB venture stage marketplace
under the symbol “MTST.” The following table sets
forth the high and low bid information for our common stock for the
two most recent fiscal years. The OTCQB quotations reflect
inter-dealer prices, are without retail markup, markdowns or
commissions, and may not represent actual
transactions.
|
|
Common Stock
|
|
|
|
High
|
Low
|
|
March
1, 2016 through May 31, 2016
|
$3.50
|
$1.55
|
|
June
1, 2016 through August 31, 2016
|
$2.19
|
$1.00
|
|
September
1, 2016 through November 30, 2016
|
$3.45
|
$1.40
|
|
December
1, 2016 through February 28, 2017
|
$1.97
|
$1.26
|
|
March
1, 2017 through May 31, 2017
|
$1.61
|
$1.11
|
|
June
1, 2017 through August 31, 2017
|
$1.35
|
$0.60
|
|
September
1, 2017 through November 30, 2017
|
$1.35
|
$0.66
|
|
December
1, 2017 through February 28, 2018
|
$1.35
|
$0.55
|
On May 18, 2018, the last reported price for our common
stock on the OTCQB was $0.80.
Number of Record Holders of Our Common Stock
As of May 18, 2018, we had 5,877,383 shares of our common
stock outstanding and 163 holders of
record of our common stock. The number of record holders was
determined from our records and the records of our transfer
agent.
Dividend Policy
We currently intend to retain all available funds and any future
earnings for use in the operation and expansion of our business and
do not anticipate paying any cash dividends on our Common Stock for
the foreseeable future.
Future cash dividends, if any, will be at the discretion of our
board of directors and will depend upon our future operations and
earnings, capital requirements and surplus, general financial
condition, contractual restrictions and other factors as our board
of directors may deem relevant. We can pay dividends only out of
our profits or other distributable reserves and dividends or
distribution will only be paid or made if we are able to pay our
debts as they fall due in the ordinary course of
business.
Cumulative dividends on the shares of Series B Preferred Stock
accrue at the rate of 8% of the Stated Value per annum, payable
quarterly, on March 31, June 30, September 30, and December 31 of
each year, commencing on March 31, 2015. Dividends are payable in
additional shares of Series B Preferred Stock valued at the Stated
Value or in cash at our sole option.
Securities Authorized for Issuance Under Equity Compensation
Plans
On February 27, 2012, in connection with the Share Exchange, we
assumed the 2012 Incentive Plan from MBM. On June 22, 2015, our
shareholders approved amending our 2012 Incentive Plan to increase
the number of authorized shares of common stock reserved for
issuance under the 2012 Incentive Plan to a number not to exceed
fifteen percent (15%) of the issued and outstanding shares of
common stock on an as converted primary basis (the “As
Converted Primary Shares”) on a rolling basis. For
calculation purposes, the As Converted Primary Shares shall include
all shares of common stock and all shares of common stock issuable
upon the conversion of outstanding preferred stock and other
convertible securities but shall not include any shares of common
stock issuable upon the exercise of options, warrants and other
convertible securities issued pursuant to the 2012 Incentive Plan.
The number of authorized shares of common stock reserved for
issuance under the 2012 Incentive Plan shall automatically be
increased concurrently with the Company’s issuance of fully
paid and non- assessable shares of As Converted Primary Shares.
Shares shall be deemed to have been issued under the 2012 Incentive
Plan solely to the extent actually issued and delivered pursuant to
an award. As of February 28, 2018, there were an aggregate of
1,644,457 shares authorized for issuance under the 2012 Incentive
Plan and 483,647 shares available for issuance under the 2012
Incentive Plan.
The objective of the 2012 Incentive Plan is to maximize the
effectiveness and efficiency of the Company’s operations by
attracting key talent, aligning and incentivizing employees to
corporate goals and reducing the risk of voluntary employee
turn-over. We may issue securities pursuant to the 2012 Incentive
Plan or outside the 2012 Incentive Plan.
|
|
Equity
Compensation Plan Information as of February 28,
2018
|
||
|
Plan category
|
Number of securities to be issued upon exercise of outstanding
options, warrants
and rights
(a) |
Weighted-average exercise
price
of outstanding options,
warrants and rights
(b)
|
Number of securities
remaining
available for
future issuance under equity
compensation plans
(excluding securities
reflected in column (a))
(c)
|
|
|
|
|
|
|
Equity compensation plans approved by security holders
*
|
730,103(1)
|
$5.41
|
483,647
|
|
Equity compensation plans not approved by security
holders
|
-
|
$-
|
-
|
|
|
|
|
|
|
Total
|
730,103
|
$5.41
|
483,647
|
(1)
Does
not include (i) 130,707 restricted shares of common stock issued
under the 2012 Incentive Plan, of which 120,505 have vested and
10,202 are subject to milestone vesting, and (ii) 300,000
restricted stock units issued under the 2012 Incentive Plan, of
which 225,000 have vested and 75,000 are subject to time-based
vesting.
* Additionally, as of February 28, 2018, outside of the 2012
Incentive Plan, we have issued an aggregate of 556,667 stock
options with a weighted-average strike price of $2.65 per share and
an aggregate of 34,989 restricted shares of common stock, of which
33,655 shares have vested and 1,334 are subject to milestone
vesting.
Transfer Agent
The transfer agent for our Common Stock is Transhare Corporation,
15500 Roosevelt Blvd, Suite 301, Clearwater, Florida 33760, Tel.
(303) 662-1112.
Recent Sales of Unregistered Securities
None.
|
Item 6.
|
SELECTED FINANCIAL DATA
|
Not applicable.
|
Item 7.
|
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITIONS AND
RESULTS OF OPERATIONS
|
You should read the following discussion and analysis of our
financial condition and results of operations in conjunction with
our audited consolidated financial statements and the related notes
to the consolidated financial statements included elsewhere in this
Form 10-K. Our audited consolidated financial statements have been
prepared in accordance with U.S. GAAP. In addition, our audited
consolidated financial statements and the financial data included
in this Form 10-K reflect our reorganization and have been prepared
as if our current corporate structure had been in place throughout
the relevant periods. The following discussion and analysis
contains forward-looking statements within the meaning of
Section 27A of the Securities Act of 1933 and Section 21E
of the Securities Exchange Act of 1934, including, without
limitation, statements regarding our expectations, beliefs,
intentions or future strategies that are signified by the words
“expect,” “anticipate,”
“intend,” “believe,” or similar language.
All forward-looking statements included in this document are based
on information available to us on the date hereof, and we assume no
obligation to update any such forward-looking statements. Our
business and financial performance are subject to substantial risks
and uncertainties. Actual results could differ materially from
those projected in the forward-looking statements. In evaluating
our business, you should carefully consider the information set
forth under the heading “Risk Factors” and elsewhere in
this Form 10-K. Readers are cautioned not to place undue reliance
on these forward-looking statements.
Overview
MetaStat is a precision medicine company dedicated to improving
survival of patients with aggressive cancer. Our therapeutic focus
targets a critical metastatic pathway in solid tumors responsible
for driving tumor resistance and the spread of aggressive cancer.
MetaStat’s goal is to transform the treatment of aggressive
cancer into a manageable disease through targeted therapies that
arrest metastatic progression and improve survival. We are
leveraging our core expertise in understanding the tumor
micro-environment and drivers of the metastatic cascade to develop
anti-metastatic therapeutics pared with companion diagnostics. Our
unique approach is to target tumor cell dissemination and cancer
metastasis which is responsible for over 90% of cancer-related
deaths.
Going Concern
Since our inception, we have generated significant net losses. As
of February 28, 2018, we had an accumulated deficit of
approximately $29.5 million. We incurred net losses of
approximately $3.2 million and approximately $2.9 million for the
year ended February 28, 2018 and February 28, 2017,
respectively. We expect our net losses to continue for at
least the next several years. We anticipate that a substantial
portion of our capital resources and efforts will be focused on
research and development, both to develop additional tests for
breast cancer and to develop products for other cancers, to scale
up our commercial organization, and other general corporate
purposes. Our financial results will be limited by a number of
factors, including establishment of coverage policies by
third-party insurers and government payers, and our ability in the
short term to collect from payers often requiring a case-by-case
manual appeals process. Until we receive routine reimbursement
and are able to record revenues as tests are processed and reports
delivered, we are likely to continue reporting net
losses.
We currently anticipate that our cash and cash equivalents will not
be sufficient to fund our operations for the next twelve months,
without raising additional capital. Our continuation as a going
concern is dependent upon continued financial support from our
shareholders, our ability to obtain necessary equity and/or
debt financing to continue operations, and the attainment of
profitable operations. These factors raise substantial doubt
regarding our ability to continue as a going concern. Although we
are actively working to obtain additional funding, we cannot make
any assurances that additional financings will be available to us
and, if available, completed on a timely basis, on acceptable terms
or at all. If we are unable to complete a debt or equity offering,
or otherwise obtain sufficient financing when and if needed, it
would negatively impact our business and operations and could also
lead to the reduction or suspension of our operations and
ultimately force us to cease our operations.
Critical Accounting Policies and Significant Judgments and
Estimates
This discussion and analysis of our financial condition and results
of operations is based on our consolidated financial statements,
which have been prepared in accordance with United
States generally accepted accounting principles. The
preparation of these financial statements requires management to
make estimates and judgments that affect the reported amounts of
assets, liabilities and expenses and the disclosure of contingent
assets and liabilities at the date of the financial statements, as
well as revenues and expenses during the reporting periods. We
evaluate our estimates and judgments on an ongoing basis. We
base our estimates on historical experience and on various other
factors we believe are reasonable under the circumstances, the
results of which form the basis for making judgments about the
carrying value of assets and liabilities that are not readily
apparent from other sources. Actual results could therefore
differ materially from those estimates under different assumptions
or conditions.
Our significant accounting policies are described in Note 2 to
our consolidated financial statements included in this Form
10-K for the year ended February 28, 2018.
We believe the following critical accounting policies reflect our
more significant estimates and assumptions used in the preparation
of our financial statements.
Stock-based
Compensation
We
account for share-based payment awards issued to employees and
members of our Board by measuring the fair value of the award on
the date of grant and recognizing this fair value as stock-based
compensation using a straight-line basis over the requisite service
period, generally the vesting period. For awards issued
to non-employees, the measurement date is the date when the
performance is complete or when the award vests, whichever is the
earliest. Accordingly, non-employee awards are remeasured at each
reporting period until the final measurement date. The fair value
of the award is recognized as stock-based compensation over the
requisite service period, generally the vesting
period.
Debt and Equity
Instruments
We
analyze debt and equity instruments for various features that would
generally require either bifurcation and derivative accounting, or
recognition of a debt discount or premium under authoritative
guidance.
Detachable
warrants issued in conjunction with debt are measured at their
relative fair value, if they are determined to be equity
instrument, or their fair value, if they are determined to be
liability instruments, and recorded as a debt
discount.
Conversion
features that are in the money at the commitment date constitute a
beneficial conversion feature that is measured at its intrinsic
value and recognized as debt discount or deemed dividend. Debt
discount is amortized as interest expense over the maturity period
of the debt using the effective interest method.
Any
contingent beneficial conversion feature would be recognized when
and if the contingent event occurs based on its intrinsic value at
the commitment date.
Derivative Financial Instruments and Fair Value
We
account for certain warrants and exchange features embedded in
notes payable that are not deemed to be indexed to the
Company’s own stock in accordance with the guidance contained
in the FASB ASC Topic 815, Derivatives and Hedging (“ASC
815”) and ASC Topic 480, Distinguishing Liabilities From
Equity (“ASC 480”). Such instruments are classified as
liabilities and measured at their fair values at the time of
issuance and at each reporting period, which change in fair value
being recognized in the statement of operations. The fair values of
these instruments have been estimated using Monte Carlo simulations
and other valuation techniques.
Research and Development Reimbursements
From time to time, we may enter into research and development
agreements in which we share expenses or are reimbursed for
research and development expenses with a collaborative partner. We
record payments received from our collaborative partners as an
offset to research and development expenses, which are discussed in
Note 11, Collaborative and Other Relationships to these
consolidated financial statements.
Financial Operations Overview
Revenue
To
date, we have not generated any revenue from product sales or
otherwise. In the future, we expect that we will seek to generate
revenue primarily from product sales, but we may also generate
non-product revenue from sources including, but not limited to
research funding, development and milestone payments, and royalties
on future product sales in connection with any out-license or other
strategic relationships we may establish. We record these
payments as non-product revenue in the statement of operations and
recognize these non-product revenue upon delivery and acceptance of
the test results or other deliverables.
General and Administrative Expenses
Our
general and administrative expenses primarily consist of personnel
and related costs, including stock-based compensation, legal fees
relating to both intellectual property and corporate matters,
accounting and audit related costs, insurance, corporate
communications and investor relations expenses, information
technology and internet related costs, office and facility rents
and related expenses, and fees for consulting and other
professional services.
We
anticipate that our general and administrative expenses will
increase in the future to support continued research, development
and commercialization activities, including potential partnership
and/or collaboration agreements, intellectual property and
corporate legal expenses, and public company operating costs,
including offering and related expenses in connection with a
potential uplisting to a national stock exchange, SEC and exchange
compliance, insurance and investor relations costs. These increases
will likely include increased costs related to facilities and
information technology expansion, the hiring of additional
personnel and increased fees to outside consultants, lawyers and
accountants, among other expenses.
Research and Development
Expenses
Historically, the majority of research
and development expenses were focused on our prognostic diagnostic
tests for breast cancer. During the year ended February 28, 2018,
our research and development activities primarily focused on our
anti-metastatic drugs and related companion diagnostics which are
based on the MENA pathway.
Research and development activities are central to our business
model and we expect future research and development expenses to be
focused on the development of therapeutics and companion
diagnostics.
We
charge all research and development expenses to operations as they
are incurred. Any nonrefundable advance payments for goods or
services to be received in the future for use in research and
development activities will be deferred and capitalized. Such
capitalized amounts will be expensed as the related goods are
delivered or the services are performed.
We
do not record or maintain information regarding costs incurred in
research and development on a program or project specific
basis. Our research and development staff, outside consultants
and contract research organizations are deployed across several
programs and/or indications. Additionally, many of our costs
are not attributable to individual programs and/or
indications. Therefore, we believe that allocating costs on
the basis of time incurred by our employees does not accurately
reflect the actual costs of a project.
Our
therapeutic and companion diagnostic product development programs
are in early development stages. Since product candidates in later
stages of development generally have higher development costs than
those in earlier stages of development, we expect research and
development costs relating to therapeutic and companion diagnostic
programs to increase significantly for the foreseeable future as
those programs progress. We are unable to determine the duration
and completion costs of our research and development programs or
when, if ever, and to what extent we will receive cash inflows from
the commercialization and sale of any product
candidate.
Results of Operations
Comparison of the Years Ended February 28, 2018 and February 28,
2017
Revenues. Research collaboration revenue in
connection with certain research and development activities was
approximately $23,000 for the year ended February 28, 2018. There
were no revenues for the year ended February 28, 2017.
General and Administrative Expenses. General and
administrative expenses totaled approximately $2.32 million for the
year ended February 28, 2018 as compared to approximately $2.34
million for the year ended February 28, 2017. This represents
a slight decrease of approximately $21,000 or approximately 1% for
the year ended February 28, 2018, as compared to the year ended
February 28, 2017. Stock-based compensation and depreciation
was approximately $455,000 and approximately $5,000, respectively
for the year ended February 28, 2018, as compared to approximately
$495,000 and approximately $15,000, respectively for the year ended
February 28, 2017. Excluding non-cash stock-based compensation
related to stock options and depreciation expenses, general and
administrative expenses slightly increased by approximately $27,000
or approximately 1% to approximately $1.86 million for the year
ended February 28, 2018 from approximately $1.83 million for the
year ended February 28, 2017.
The
approximately $27,000 of increased general and administrative
spending was primarily due to increases in payroll, bonus and
personnel related expense of approximately $156,000, increased rent
of approximately $38,000, increased insurance expenses of
approximately $23,000 and increased state and franchise tax
payments of approximately $10,000. These increased general and
administrative costs were partially offset by decreases related to
public company expenses, including investor relations and corporate
communications of approximately $124,000, as well as decreases in
corporate and intellectual property and patent legal expenses of
approximately $55,000 and accounting and audit expenses of
approximately $16,000. We expect general and administrative
expenses to increase for the next fiscal year ending February 28,
2019 with projected increases in payroll and personnel related
expense, public company expenses, information technology and
related expenses, rent and office related costs, consulting
expenses, and professional fees related to intellectual property
and patents, among others, as compared to the fiscal year ended
February 28, 2018.
Research and Development Expenses. Research and development
totaled approximately $1.3 million for the year ended February 28,
2018, as compared to approximately $1.0 million for the year ended
February 28, 2017. This represents an increase of approximately
$247,000 or approximately 25% for the year ended February 28, 2018,
as compared to the year ended February 28, 2017. Stock-based
compensation and depreciation was approximately $195,000 and
approximately $85,000, respectively for the year ended February 28,
2018, as compared to approximately $85,000 and approximately
$81,000, respectively for the year ended February 28, 2017.
Excluding non-cash stock-based compensation related to stock
options and depreciation expenses, research and development
expenses increased by approximately $134,000, or approximately 16%,
to approximately $977,000 for the year ended February 28, 2018,
from approximately $843,000 for the year ended February 28,
2017.
Increased research and development spending was primarily due to
increases in payroll, bonus and personnel related expense of
approximately $129,000, licensing fees of approximately $61,000,
supplies and materials of approximately $29,000, and rent expenses
of approximately $24,000, partially offset by decreases in
consulting expense of approximately $113,000. We expect research
and development expenses to increase for the next fiscal year
ending February 28, 2019 as we conduct research and development
activities related to the development of our lead anti-metastatic
drug compounds and our MENA diagnostic assay in which we incur
payroll and related expense for new employees and increased
consulting and supplies and materials expenses. Research and
development expenses for the years ended February 28, 2018 and
February 28, 2017 include approximately $664,000 and $309,000,
respectively, of research and development reimbursement earned by
us in connection with our collaborative arrangement as described in
Note 11.
Other Expenses (Income). Other income was approximately
$369,000 for the year ended February 28, 2018, as compared to other
income of approximately $406,000 for the year ended February 28,
2017. This represents a change of approximately $36,000.
Other income for the year ended February 28, 2018 mostly comprised
of approximately $488,000 gain from the change in fair value of the
warrant liability offset by approximately $119,000 of interest
expense on the notes payable. Other income for the year ended
February 28, 2017 mostly comprised of approximately $2.4 million
gain from the change in fair value of the warrant liability, and
approximately $614,000 gain on the change in fair value of the
embedded put feature related to the notes payable, offset by
approximately $1.4 million loss on extinguishment related to the
exchanges of notes payable, approximately $1.1 millions of interest
expense on the notes payable, and approximately $112,000 loss
on sale of ASET note receivable.
Net Loss. As a result of the factors described above, our
net loss increased by approximately $239,000 to approximately $3.2
million for the year ended February 28, 2018 as compared to
approximately $2.9 million for the year ended February 28,
2017.
Liquidity and Capital Resources
Since
our inception, we have incurred significant losses and, as of
February 28, 2018, we had an accumulated deficit of approximately
$29.5 million. We have not yet generated any product revenue
or achieved profitability and anticipate that we will continue to
incur net losses for the foreseeable future. We expect that
our research and development, general and administrative and
commercialization expenses will continue to grow and, as a result,
we will need to generate significant product revenues to achieve
profitability. We may never achieve revenue and/or
profitability.
Sources of Liquidity
Since our inception, substantially all of our operations have been
financed through the sale of our common stock, preferred stock, and
promissory notes. Through February 28, 2018, we had received net
proceeds of approximately $11.78 million through the sale of common
stock and/or Series A-2 Preferred Stock to investors, approximately
$0.26 million through the sale of Series A Preferred Stock to
investors, approximately $3.39 million through the sale of Series B
Preferred Stock to investors, approximately $3.46 million from the
sale of convertible promissory notes and approximately $1.82
million from the sale of non-convertible promissory
notes. As of February 28, 2018, we had cash and cash
equivalents of approximately $0.3 million and debt of approximately
$1.0 million. Through February 28, 2018, we have issued and
outstanding warrants to purchase 3,060,118 shares of our common
stock at a weighted average exercise price of $4.34 per share,
which could result in proceeds to us of approximately $13.3 million
if all outstanding warrants were exercised for
cash. Subsequent to February 28, 2018, we completed an
offering non-convertible promissory bridge notes on March 30, 2018,
which resulted in net proceeds of approximately $1.13 million prior
to the repayment of $0.3 million of then outstanding debt (See Note
13 – Subsequent Events).
Cash Flows
At
February 28, 2018, we had approximately $0.3 million in cash and
cash equivalents, compared to approximately $0.8 million in on
February 28, 2017.
Net cash used in operating activities was approximately $2.7
million for the year ended February 28, 2018, as compared to
approximately $2.3 for the year ended February 28, 2017. The
increase in cash used of approximately $0.4 million was primarily
due to increased operating expenses and the recognition of upfront
payments for research and development reimbursement. We expect
amounts used in operating activities to increase for the next
fiscal year ending February 28, 2019 and beyond as we grow our
corporate operations.
Net cash used in investing activities was approximately $30,000 for
the year ended February 28, 2018, compared to approximately $1,000
of cash used for the year ended February 28, 2017. This increase of
approximately $29,000 was attributed to laboratory equipment
purchases. We expect amounts used in investing activities to
increase for the next fiscal year ending February 28, 2019 and
beyond as we grow our corporate operations, expand research and
development activities and add capacity in our laboratory including
related to information technology, which is expected to result in
an increase of our capital expenditures.
Net
cash provided by financing activities during the year ended
February 28, 2018 was approximately $2.3 million, compared to
approximately $2.7 million for the year ended February 28,
2017. Financing activities consisted primarily of proceeds
from the sale of common stock, Series A-2 preferred stock and
warrants for the year ended February 28, 2018, and primarily from
the issuance of common stock, Series A-2 preferred stock and
warrants, and notes and warrants for the year ended February 28,
2017.
Contractual Obligations
As
of February 28, 2018, we had the following contractual
commitments:
|
|
Payments Due by Period
|
|||
|
Contractual Obligations
|
Total
|
Less than 1 Year
|
1-3 Years
|
4-5 Years
|
|
|
(In thousands)
|
|||
|
License Agreement (1)
|
$700
|
$100
|
$300
|
$200
|
|
|
|
|
|
|
|
Second License Agreement (2)
|
$620
|
$60
|
$260
|
$200
|
|
|
|
|
|
|
|
Alternative Splicing License
Agreements (6)
|
$350
|
$50
|
$150
|
$100
|
|
|
|
|
|
|
|
Alternative Splicing License
Agreements (7)
|
$-
|
$-
|
$-
|
$-
|
|
|
|
|
|
|
|
Antibody License Agreement
(8)
|
$140
|
$20
|
$60
|
$40
|
|
|
|
|
|
|
|
Lease Agreement (5)
|
$1,348
|
$284
|
$906
|
$158
|
|
(1)
|
Amount of additional payments depends on several factors, including
the duration of the License Agreement, which depends on expiration
of the last patent to be issued pursuant to the License Agreement.
That duration is uncertain because the last patent has not yet been
issued.
|
|
|
|
|
(2)
|
Amount of additional payments depends on several factors, including
the duration of the Second License Agreement, which depends on
expiration of the last patent to be issued pursuant to the Second
License Agreement. That duration is uncertain because the last
patent has not yet been issued.
|
|
(3)
|
Amount of additional payments depends on several factors, including
the duration of the Alternative Splicing Diagnostic License
Agreement, which depends on expiration of the last patent to be
issued pursuant to the Alternative Splicing Diagnostic License
Agreement. That duration is uncertain because the last patent has
not yet been issued. No annual license maintenance fee payments are
due on the Alternative Splicing Therapeutic License Agreement as
long as the Alternative Splicing Diagnostic License Agreement is in
effect.
|
|
|
|
|
(4)
|
Amount of additional payments depends on several factors, including
the duration of the Antibody License Agreement, which depends on
expiration of the last patent to be issued pursuant to the Antibody
License Agreement. That duration is uncertain because the last
patent has not yet been issued.
|
|
|
|
|
(5)
|
Only includes basic rent payments through August 31,
2022. Additional monthly payments under the lease agreement
shall include tax payments and operational
costs.
|
License Agreements
Pursuant to the License Agreement, we are required to make annual
license maintenance fee payments beginning August 26,
2011. We have satisfied all license maintenance payments
due through February 28, 2018. We are required to make payments of
$100,000 in 2018 and every year the license is in effect
thereafter. These annual license maintenance fee payments will be
credited to running royalties due on net sales earned in the same
calendar year, if any. We are in compliance with the License
Agreement.
Pursuant to the Second License Agreement, we are required to make
annual license maintenance fee payments beginning on January 3,
2013. In February 2017, we amended the Second License
Agreement to reduce the maintenance payment for 2016 from $30,000
to $5,000, 2017 from $50,000 to $5,000, 2018 from $75,000 to
$5,000, 2019 from $100,000 to $60,000, and 2020 from $100,000 to
$60,000. We are required to make payments of $100,000 in 2021 and
every year the license is in effect thereafter. These annual
license maintenance fee payments will be credited to running
royalties due on net sales earned in the same calendar year, if
any. We are in compliance with the Second License Agreement,
however the maintenance payments for 2017 and 2018 totaling $10,000
are currently outstanding.
Pursuant to the Alternative Splicing Diagnostic License Agreement
and the Alternative Splicing Therapeutic License Agreement, we are
required to make annual license maintenance fee payments for each
license beginning on January 1, 2015. The license maintenance
payments of $37,500 for 2018 is currently outstanding. We are
required to make additional payments of $50,000 in 2019
and every year each license is in effect thereafter. The
parties are currently in discussions regarding amending the
Alternative Splicing Diagnostic License Agreement and the
Alternative Splicing Therapeutic License Agreement and as such, we
are in compliance the Alternative Splicing Diagnostic License
Agreement and the Alternative Splicing Therapeutic License
Agreement.
Pursuant to the Antibody License Agreement, we are required to make
license maintenance fee payments beginning on January 1,
2015. We have satisfied all license maintenance payments due
through February 28, 2018. We are required to make additional
payments of $20,000 in 2019 and every year the license is in effect
thereafter. These annual license maintenance fee payments will be
credited to running royalties due on net sales earned in the same
calendar year, if any. We are in compliance with the Antibody
License Agreement.
Lease Agreements
On August 28, 2014, we entered into a lease agreement, subsequently
amended (the “Boston Lease”) for our diagnostic
laboratory and office space located at 27, Drydock Ave, 2nd Floor,
Boston, MA 02210 (the “Boston Property”). We paid
a $40,000 security deposit in connection with entering into the
Boston Lease.
On July 26, 2017, we entered into a second amendment to the Boston
Lease for the Boston Property (the “Second Boston Lease
Amendment”). The Second Boston Lease Amendment extended the
term (the “Second Extension Period”) for five years
from September 1, 2017 through August 31, 2022. Monthly basic rent
payments are approximately $23,000 for the first year of the Second
Extension Period, approximately $24,000 for the second year of the
Second Extension Period, approximately $25,000 for the third year
of the Second Extension Period, approximately $25,000 for the
fourth year of the Second Extension Period, and approximately
$26,000 for the fifth year of the Second Extension
Period.
Effective March 1, 2015, we entered into a lease agreement for
short-term office space in New York, NY. The term of the
lease is month-to-month and may be terminated upon twenty-one (21)
days’ notice. We paid a $2,100 security deposit in connection
with entering into the lease. Effective December 1, 2015, we
amended our lease agreement for the short-term office space in New
York, NY. The basic rent payment increased to $2,400 per month
and we paid an additional $1,500 security deposit in connection
with the amended lease.
Equipment
We intend to enter into arrangements for the acquisition of
additional laboratory equipment, computer hardware and software,
including data storage, leasehold improvements and office equipment
in fiscal year ending February 28, 2019 as we continue to expand
our research and development activities. We cannot at this
time provide assurances that we will be able to enter into
agreements with vendors on terms commercially favorable to us or
that we will be able to enter into such arrangements without
securing additional financing.
Operating Capital and Capital Expenditure Requirements
We currently anticipate that our cash and cash equivalents will not
be sufficient to fund our operations through for the next twelve
months, without raising additional capital. We expect to continue
to incur substantial operating losses in the future and to make
capital expenditures to keep pace with the expansion of our
research and development programs as we further our preclinical
activities and subsequently advance our drug candidates in
development into clinical trials in conjunction with the
development of our companion diagnostic assays. It may take
several years to move any one of a number of product candidates in
clinical research through the development and validation phases to
commercialization. We expect that our existing cash and cash
equivalents will be used to fund working capital and for capital
expenditures and other general corporate purposes including a
reduction of contractual obligations. We have no current
plans, agreements or commitments with respect to any acquisition or
investment, and we are not currently engaged in any negotiations
with respect to any such transaction.
The amount and timing of actual expenditures may vary significantly
depending upon a number of factors, such as the progress of our
product development, regulatory requirements, commercialization
efforts, the amount of cash used by operations and progress in
reimbursement. We cannot be certain that any of our future
efforts to develop future products will be successful or that we
will be able to raise sufficient additional funds to see these
programs through to a successful result.
Our
future funding requirements will
depend on many factors, including the
following:
●
the timing, cost and progress of our preclinical and clinical
development activities for our drug candidates;
●
the timing, cost and progress of our development activities for our
companion diagnostic tests in support our drug
candidates;
●
successful enrollment in and completion of clinical
trials;
●
the timing and outcome of regulatory review of our drug candidates
and companion diagnostics;
●
our ability to establish agreements with third-party manufacturers
for clinical supply for our clinical trials and, if any of our drug
candidates are approved, commercial manufacturing;
●
the rate of progress and cost of research and development
activities associated with expansion of our current diagnostic
tests and the development of new tests;
●
the cost associated with expanding our laboratory operations for
our prognostic diagnostic tests, including selling and marketing
efforts;
●
the cost and delays in product development and commercialization as
a result of any changes in regulatory oversight applicable to our
products or operations;
●
the costs and timing of future commercialization activities,
including product manufacturing, marketing, sales and distribution,
for any of our drug candidates for which we obtain marketing
approval;
●
the cost related to filing, prosecuting, defending and enforcing
any patent claims and other intellectual property
rights;
●
the addition and retention of key research and development
personnel;
●
our efforts to enhance operational, financial, and information
technology and management systems, and hire additional personnel,
including personnel to support our product development and
commercialization efforts;
●
the economic and other terms in connection
with strategic collaborations, license agreements or
other relationships in which we may enter into or investments or
acquisitions we may seek to enter into; and
●
the impact of changes in Federal, state and international
taxation.
Until we can generate a sufficient amount of revenues to finance
our cash requirements, which we may never do, we expect to finance
future cash needs primarily through a combination of equity and
debt financings, collaborations, strategic alliances and marketing,
distribution or licensing arrangements. We do not currently have
any committed external source of funds. The issuance of equity or
convertible debt securities will result in dilution to
stockholders. Additionally, the terms of these securities may
include liquidation or other preferences that adversely affect the
rights of our stockholders’. Debt and preferred equity
financings, if available, may involve agreements that include
covenants limiting or restricting our ability to take specific
actions, such as incurring additional debt, making acquisitions or
capital expenditures or declaring dividends. If we raise additional
funds through collaborations, strategic alliances or marketing,
distribution or licensing arrangements with third parties, we may
have to relinquish valuable rights to our technologies, future
revenue streams, research programs or drug candidates or grant
licenses on terms that may not be favorable to us. We cannot make
any assurances that additional financings will be completed on a
timely basis, on acceptable terms or at all. If we are unable to
complete a debt or equity offering, or otherwise obtain sufficient
financing when and if needed, it would negatively impact our
business and operations, which could cause the price of our common
stock to decline. It could also lead to the reduction or suspension
of our operations and ultimately force the Company to cease
operations.
Income Taxes
Since inception, we have incurred operating losses and,
accordingly, have not recorded a provision for income taxes for any
of the periods presented. As of February 28, 2018, we had
cumulative net operating loss carryforwards for federal income tax
purposes of approximately $21.7 million. If not utilized, the
federal net operating loss and tax credit carryforwards will expire
between 2029 and 2037. Utilization of net operating loss and
credit carryforwards may be subject to a substantial annual
limitation due to restrictions contained in the Internal Revenue
Code that are applicable if we experience an “ownership
change.” The annual limitation may result in the expiration
of our net operating loss and tax credit carryforwards before they
can be used.
The Tax Cuts and Jobs Act (the “Act”) was enacted into
law in December 2017. Among other things, the Act reduces the U.S.
federal corporate tax rate from 34 percent to 21 percent effective
January 1, 2018 and eliminates the alternative minimum tax for
corporations. The reduction of the corporate tax rate resulted in a
decrease of the Company’s net deferred tax assets of
approximately $3.4 million, and a corresponding decrease of the
valuation allowance.
Recent Accounting Pronouncements
We have implemented all new relevant accounting pronouncements that
are in effect through the date of these financial
statements. These pronouncements did not have any material
impact on the financial statements unless otherwise disclosed. We
are currently assessing the impact of the new accounting
pronouncements disclosed in footnote 2 of our consolidated
financial statements and do not know whether they might have a
material impact on our financial position or results of
operations.
|
Item 7A.
|
QUANTITATIVE AND
QUALITATIVE DISCLOSURES ABOUT MARKET RISK
|
Not applicable.
|
Item 8.
|
FINANCIAL STATEMENTS
AND SUPPLEMENTARY FINANCIAL DATA
|
Consolidated Financial Statements
The financial statements required by this item begin on page F-1
hereof.
|
Item 9.
|
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND
FINANCIAL DISCLOSURE
|
None.
|
Item 9A.
|
CONTROLS AND
PROCEDURES
|
Disclosure Controls and Procedures
Disclosure controls and procedures are designed to ensure that
information required to be disclosed by us in reports filed or
submitted under the Securities Exchange Act of 1934
(“Exchange Act”) is recorded, processed, summarized and
reported within the time periods specified in the SEC’s rules
and forms. Disclosure controls include, without limitation,
controls and procedures designed to ensure that information
required to be disclosed under the Exchange Act is accumulated and
communicated to management, including principal executive and
financial officers, as appropriate, to allow timely decisions
regarding required disclosure. There are inherent limitations to
the effectiveness of any system of disclosure controls and
procedures, including the possibility of human error and the
circumvention or overriding of the controls and procedures.
Accordingly, even effective disclosure controls and procedures can
only provide reasonable assurance of achieving their control
objectives.
Management carried out an evaluation, under the supervision of the
Chief Executive Officer and Vice President, Finance, of the
effectiveness of disclosure controls and procedures as of February
28, 2018. Based upon that evaluation, management, including the
Chief Executive Officer and Vice President, Finance, concluded that
the design and operation of disclosure controls and procedures were
effective.
Management's Report on Internal Control over Financial
Reporting
Management is responsible for establishing and maintaining adequate
internal control over financial reporting, as defined in the
Securities Exchange Act of 1934, as amended. Because of its
inherent limitations, internal control over financial reporting may
not prevent or detect misstatements. Also, projections of any
evaluation of effectiveness to future periods are subject to the
risk that controls may become inadequate because of changes in
conditions, or that the degree of compliance with the policies or
procedures may deteriorate. Management assessed the effectiveness
of internal control over financial reporting as of February 28,
2018. In making this assessment, management used the
criteria set forth by Internal
Control—Integrated Framework (2013 Framework) issued by the Committee of
Sponsoring Organizations of the Treadway Commission. Based on its
assessment using those criteria, management concluded that internal
control over financial reporting was effective as of February 28,
2018.
As a smaller reporting company, we are not required to obtain an
attestation report from our registered public accounting firm
regarding internal controls over financial reporting.
Changes in Internal Controls over Financial Reporting
We have had no changes in internal control over financial reporting
during the quarter ended February 28, 2018 that have materially
affected, or are reasonably likely to materially affect, internal
control over financial reporting have been described
above.
|
Item 9B.
|
OTHER INFORMATION
|
None.
PART
III
|
Item 10.
|
DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
|
Directors and Executive Officers
|
Name
|
Age
|
Position
|
|
Douglas A. Hamilton
|
52
|
President, Chief Executive Officer and Director
|
|
Daniel H. Schneiderman
|
40
|
Vice President of Finance
|
|
Jerome B. Zeldis, M.D., Ph.D.
|
68
|
Chairman of the Board of Directors
|
|
Paul Billings, M.D., Ph.D.
|
65
|
Director
|
Douglas A. Hamilton. Mr.
Hamilton was appointed our president and chief executive officer
effective as of June 17, 2015 and appointed to our board of
directors effective as of May 4, 2017. Mr. Hamilton has
consulted for us as acting chief financial officer since August
2014. Prior to joining the Company, Mr. Hamilton served as partner
at New Biology Ventures, LLC, a life-sciences focused venture
capital incubator founded by Mr. Hamilton since
2007. From January 2012 through January 2014, Mr.
Hamilton was chief financial officer of S.E.A. Medical Systems,
Inc. From 1999 to 2006, Mr. Hamilton served as chief financial
officer and chief operating officer for Javelin Pharmaceuticals,
Inc. (acquired by Hospira, Inc.), in which he led the company to
commercialization and through the private to public transition,
including a successful national markets up-listing. Prior to
Javelin, Mr. Hamilton was the chief financial officer and director
of business development for PolaRx Biopharmaceuticals, Inc.
(acquired by Cell Therapeutics, Inc., now owned by Teva
Pharmaceuticals). Mr. Hamilton also served for several years in
portfolio and project management at Pfizer, Inc. and Amgen, Inc.,
sales and marketing at Pharmacia Biotech (now GE Healthcare Life
Sciences), and research at Connaught Laboratories (now
Sanofi-Pasteur). Mr. Hamilton earned his Bachelor of Science degree
from the Department of Medical Genetics at the University of
Toronto and his MBA from the Ivey Business School at Western
University.
Daniel H. Schneiderman. Mr.
Schneiderman was appointed vice president of finance effective
December 21, 2012 and has served as the Company's vice president,
controller and corporate secretary since February 27, 2012. Mr.
Schneiderman has over fifteen years of investment banking and
corporate finance experience, focusing on private and public small
capitalization companies mainly in the healthcare, life sciences
and technology sectors. Prior to joining the Company, he was vice
president of investment banking for Burnham Hill Partners LLC,
where he worked since 2008. From 2004 through 2008, Mr.
Schneiderman was vice president of investment banking at Burnham
Hill Partners, a division of Pali Capital, Inc. While at Burnham
Hill Partners, Mr. Schneiderman helped raise in excess of
$500 million in capital through private placements, PIPEs and
registered offerings as well as more complex transactions including
restructurings and recapitalizations. Previously, Mr. Schneiderman worked at H.C.
Wainwright & Co., Inc. in 2004 as an investment banking
analyst. Mr. Schneiderman holds a Bachelor's Degree in Economics
from Tulane University. Mr. Schneiderman previously served as a
board member for Unleashed, a not-for-profit organization in New
York dedicated to dog rescue and animal rights.
Jerome B. Zeldis, M.D., Ph.D. Dr. Zeldis was appointed to our board of directors
and vice chairman of the board effective as of April 25, 2016, and
as Chairman of the Board effective as of May 5, 2017. Dr.
Zeldis is currently the Chief Medical Officer and President of
Clinical Research at Sorrento Therapeutics, Inc., positions he has
held since August 2016. Previously, Dr. Zeldis was Chief Medical
Officer of Celgene Corporation and CEO of Celgene Global Health,
until June 2016. Prior to that he was Celgene’s Senior Vice
President of Clinical Research and Medical Affairs and had been at
Celgene since February 1997. He attended Brown University for an
A.B., M.S., followed by Yale University for an M.Phil., M.D., Ph.D.
in Molecular Biophysics and Biochemistry (immunochemistry). Dr.
Zeldis trained in Internal Medicine at the UCLA Center for the
Health Sciences and Gastroenterology at the Massachusetts General
Hospital and Harvard Medical School. He was Assistant Professor of
Medicine at the Harvard Medical School, Associate Professor
of Medicine at University of
California, Davis, Clinical Associate Professor of Medicine at
Cornell Medical School and Professor of Clinical Medicine at the
Robert Wood Johnson Medical School in New Brunswick, New Jersey.
Prior to working at Celgene, Dr. Zeldis worked at Sandoz Research
Institute and Janssen Research Institute in both clinical research
and medical development. He has been a board member of a few
start-up biotechnology companies and is currently Chairman of the
board of Alliqua Biomedical and Trek Therapeutics in addition to
board positions at PTC Therapeutics and Soligenix. He has published
122 peer reviewed articles and is the named inventor on 43 U.S.
patents. Dr. Zeldis’ extensive knowledge of the biotechnology
industry, his extensive role in drug development and clinical
studies as well as his directorships in other life science
companies qualify him to serve as our director and Chairman of the
Board.
Paul Billings,
M.D., Ph.D. Dr. Billings was
appointed to our board of directors effective as of May 24,
2017. Dr. Billings is a
board-certified internist and clinical
geneticist. Dr. Billings is
currently the Chief Medical Officer and Senior Vice President of
Medical Affairs at Natera, Inc., a leading global provider of cell
free DNA tests, positions he has held since April 2018. He is also
a partner at the Bethesda Group Fund L.P., a position he has held
since January 2016. From January 2015 to January 2016, Dr.
Billings served as
Executive-in-Residence at the California Innovation Center of
Johnson and Johnson, Inc. He has also served as the Medical
Director of the IMPACT program at ThermoFisher
Scientific, Inc. (TFS) from 2013 to 2015. From 2010 to 2014,
he served as the first and only Chief Medical Officer at Life
Technologies Corporation (which was acquired by TFS), and the
Genetic Sciences Division of TFS. Dr. Billings has
previously served as a director of Ancestry.com Inc. from February
2012 to May 2013. He serves as an advisor or director for many
private companies, including Fabric Genomics, Inc. (formerly Omicia,
Inc.), ProterixBio, Inc.,
Mission Bio, Inc., Aueon, Inc. and PAX Neuroscience Inc. He
held senior management positions at Cord Blood Registry, Inc,
GeneSage, Inc., Laboratory Corporation of America Holdings
(LabCorp), and CELLective DX Corporation. Dr. Billings’
clinical experience includes senior administrative positions at El
Camino Hospital and the Veteran’s Administration and he
served as a physician at many medical centers. He has held academic
appointments at prestigious universities including Harvard
University, Stanford University, U.C. Berkeley, and U.C. San
Francisco. He is a prolific author with nearly 200 publications and
books on genomic medicine. Dr. Billings holds an M.D. from
Harvard Medical School and a Ph.D. in immunology, also from
Harvard University. Dr. Billings is a nationally
recognized expert on genomic and precision medicine, and his
extensive medical and managerial experience in the field of
personalized medicine qualifies him to serve as our
director.
Other Key Consultants and Employees
Michael J. Donovan, Ph.D., M.D. Dr. Donovan joined the company as a
consultant (acting chief medical officer) as of August 1,
2015. Dr. Donovan is board-certified in anatomic and clinical
pathology and pediatric pathology with extensive experience in
designing and implementing clinical studies. He has spearheaded the
utilization of multiplex tissue and fluid-based assays and coupled
mathematic applications to produce clinically relevant
diagnostic/predictive/prognostic outcome models for a variety of
tumor types and disease states. Dr. Donovan also serves as a
Professor of Experimental Pathology and Director of the
Biorepository and Pathology core at the Icahn School of Medicine at
Mt. Sinai, New York City, New York. In addition to an academic
career at Harvard Medical School and Boston Children’s
Hospital, Dr. Donovan has over 20 years’ experience in the
biotechnology industry, serving in various senior management roles
at Millennium Pharmaceuticals and Incyte Pharmaceuticals. He most
recently served as chief medical officer of Exosome Diagnostics,
Inc. and chief scientific officer for Aureon Biosciences
Corporation. Dr. Donovan graduated from Rutgers University with a
BS in zoology, a MS in endocrinology and a PhD in cell and
developmental biology. He received his MD from the University of
Medicine and Dentistry of New Jersey.
Scientific and Clinical Advisory Board
Effective as of October 24, 2012, the board of directors formally
established a Scientific Advisory Board whose primary
responsibilities include advising our management and the board on
the long-term direction of our scientific and research goals and a
Clinical Advisory Board whose primary responsibilities include
advising our management and the Board on the most efficient
translation of our scientific and research discoveries to clinical
practice. We currently have consulting contracts with Bruce Zetter,
Ph.D. Renato Skerlj and Frank Gertler, Ph.D. for scientific
advisory and other consulting services.
Bruce R. Zetter, Ph.D. Dr.
Bruce Zetter serves as chief scientific officer and vice president
of research at Boston Children's Hospital and the Charles
Nowiszewski Professor of Cancer Biology at Harvard Medical School.
Dr. Zetter serves as a consultant and scientific advisor to major
biotechnology and pharmaceutical companies. He is highly regarded
nationally and internationally as a leader in the research of tumor
angiogenesis, progression, cancer diagnosis, and cancer metastasis.
He served as head of scientific advisors at ProNAi Therapeutics,
Inc. since November 2012. He served as a medical & scientific
advisor of Mersana Therapeutics Inc. He co-founded Predictive
Biosciences Inc. in 2006. Dr. Zetter served as an expert witness
for the United States Senate Cancer Coalition hearings in
Washington, DC. He serves as chairman of Scientific Advisory Board
of the Scientific Advisory Board of SynDevRx, Inc., and Cerulean
Pharma Inc. He served as chairman of Scientific Advisory Board of
Tempo Pharmaceuticals Inc. and Predictive Biosciences, Inc. He
serves as member of Scientific Advisory Board at Blend
Therapeutics, Inc. He serves as member of Scientific & Medical
Advisory Board at ProNAi Therapeutics, Inc. Dr. Zetter serves as a
member of the board of directors and member of Advisory Board of
Attenuon, LLC. Dr. Zetter serves on the Advisory Boards of Angstrom
Pharmaceuticals and GMP Companies. He also serves on several grant
review boards for public agencies such as the American Heart
Association and American Cancer Society and serves on the editorial
board of 11 peer-reviewed journals. Dr. Zetter served as member of
Scientific Advisory Board of Tempo Pharmaceuticals Inc., Synta
Pharmaceuticals Corp., and BioTrove, Inc. His research interests
focus on tumor metastasis and on identifying diagnostic and
prognostic markers that can guide treatment decisions. He has
chaired the grant review board on breast and prostate cancer for
the National Institutes of Health. Dr. Zetter is a pioneer in
understanding how cell movement affects tumor metastasis and is
recognized for his key discovery of the inhibitory effects of alpha
interferon to endothelial cell locomotion. His work led to the use
of interferon alpha to treat hemangiomas. Dr. Zetter serves as a
professor in the Department of Surgery at Harvard Medical School
since 1978. Dr. Zetter has won numerous national and international
awards for his work in the field of cancer research including a
Faculty Research Award from the American Cancer Society and the
prestigious MERIT award from the US National Cancer Institute. He
has also received three teaching awards from the students at
Harvard Medical School for excellence as a teacher and as a course
director. He has authored more than 100 articles and has more than
20 patents to his credit. Dr. Zetter received a B.A. degree in
Anthropology from Brandeis University. Dr. Zetter earned his Ph.D.
from University of Rhode Island and he completed postdoctoral
fellowships at Massachusetts Institute of Technology (MIT) and the
Salk Institute in San Diego.
Renato Skerlj, Ph.D. Dr. Skerlj
serves as Vice President of Drug Discovery and Preclinical
Development at Lysosomal Therapeutics Inc. Dr. Skerlj is a
co-founder and a Member of Scientific Advisory Board of X4
Pharmaceuticals Inc. and co-founder of Noliva Therapeutics. He has
over 25 years of pharmaceutical experience in drug development
resulting in two marketed drugs: Invanz® and
Mozobil® and multiple drugs in clinical development.
Previously, Dr. Skerlj served as the Head of Small Molecule
Discovery at Genzyme, prior to its acquisition by Sanofi. He served
as Vice President of Chemistry for AnorMED Inc., a publicly-traded
company acquired by Genzyme for $580 million in 2006. Prior to
AnorMED Inc., Dr. Skerlj served as a Senior Research Chemist at
Merck & Co., Inc. and Senior Scientist at Johnson Matthey's
biomedical research group. Dr. Skerlj earned his doctorate from the
University of British Columbia and completed a fellowship at the
University of Oxford in Dr. Stephen Davies’
laboratory.
Frank B. Gertler, Ph.D. Dr.
Frank Gertler received his B.S. degree from the University of
Wisconsin-Madison in 1985. During his post-graduate thesis work at
the University of Wisconsin-Madison, Dr. Gertler discovered the
Enabled (Ena) gene in a search for functional downstream targets of
signaling by the Drosophila homolog of the c-Abl proto-oncogene. He
proceeded to demonstrate that Abl and Ena function were key
components of the machinery required to establish normal
connections during development of the nervous system. After
receiving his Ph.D. in Oncology and Genetics in 1992, Dr. Gertler
trained as a Postdoctoral Fellow in the laboratory of Philippe
Soriano at the Fred Hutchinson Center for Cancer Research from 1993
through 1997. During this time, he cloned Mena, the mammalian
homolog of Drosophila Ena, and discovered a family of related
molecules, the “Ena/VASP” proteins. In 1997, Dr.
Gertler joined the Biology Department at the Massachusetts
Institute of Technology (MIT). His laboratory continued to work on
Mena and the related Ena/VASP proteins and described pivotal roles
for these proteins in controlling cell movement, shape and adhesion
during fetal development. In 2005, Dr. Gertler moved to the MIT
Center for Cancer Research and began to work on the role of Mena in
metastatic progression and launched other efforts geared at
understanding how the control of cell motility is dysregulated
during metastatic diseases. Currently, Dr. Gertler is a Full
Professor in the Koch Institute for Integrative Cancer Research at
MIT and a member of the MIT Biology Department.
Family Relationships
There are no family relationships between any of our directors or
executive officers.
Code of Ethics
We adopted a Code of Ethics that applies to all directors, officers
and employees. Our Code of Ethics is available on our website
at www.metastat.com.
A copy of our code of ethics will also be provided to any person
without charge, upon written request sent to us at our offices
located at 27 Drydock Ave., 2nd Floor, Boston, Massachusetts
02210.
Corporate Governance
Board Leadership Structure
Our board of directors (the “Board”) has a chairman,
currently Dr. Zeldis, who has authority, among other things, to
call and preside over board meetings, to set meeting agendas and to
determine materials to be distributed to the board of directors.
Accordingly, the chairman has substantial ability to shape the work
of the board of directors.
The positions of chief executive officer and chairman of our Board
are held by different persons. The chairman of our Board, Dr.
Zeldis, chairs director and any stockholder meetings and
participates in preparing their agendas. Mr. Hamilton serves as a
focal point for communication between management and the Board
between board meetings, although there is no restriction on
communication between directors and management. Mr. Hamilton serves
as our chief executive officer as well as a member of our Board. We
believe that these arrangements afford the other members of our
Board sufficient resources to supervise management effectively,
without being overly engaged in day-to-day operations.
Dr. Zeldis also serves as lead independent director for our Board
and Dr. Billings serves as an independent director.
As a result of the current and ongoing restructuring of our Board,
we intend to appoint independent directors to the Board and to
chair each committee of our Board as soon as reasonably possible.
The Board considers all of its members equally responsible and
accountable for oversight and guidance of its
activities.
Board Committees
Effective as of October 24, 2012, the Board established an Audit
Committee, a Nominating and Corporate Governance Committee and a
Compensation Committee.
Currently, Dr. Zeldis, Dr. Billings, and Mr. Hamilton will serve on
each of the Audit Committee, Nominating and Corporate Governance
Committee and Compensation Committee, until such time that the
Company shall appoint independent directors to fulfil the
requirements of such committees.
Board Practices
Our business and affairs are managed under the direction of our
Board. The primary responsibilities of our Board are to provide
oversight, strategic guidance, counseling and direction to our
management.
Policy Regarding Board Attendance
Our directors are expected to attend meetings of the Board as
frequently as necessary to properly discharge their
responsibilities and to spend the time needed to prepare for each
such meeting. Our directors are expected to attend annual meetings
of stockholders, but we do not have a formal policy requiring them
to do so.
Shareholder Communications
We have a process for shareholders who wish to communicate with our
board of directors. Shareholders who wish to communicate with the
board may write to it at our address given above. These
communications will be reviewed by one or more of our employees
designated by the board, who will determine whether they should be
presented to the board. The purpose of this screening is to allow
the board to avoid having to consider irrelevant or inappropriate
communications.
Section 16(a) Beneficial Ownership Reporting
Compliance
Section 16(a) of the Securities Exchange Act of 1934, as amended,
or the Exchange Act, requires our executive officers, directors and
persons who beneficially own more than 10% of a registered class of
our equity securities to file with the Securities and Exchange
Commission initial reports of ownership and reports of changes in
ownership of our common stock and other equity securities. These
executive officers, directors, and greater than 10% beneficial
owners are required by SEC regulation to furnish us with copies of
all Section 16(a) forms filed by such reporting
persons.
Based solely on our review of such forms furnished to us and
written representations from certain reporting persons, we believe
that during the fiscal year ended February 28, 2018, all filing
requirements applicable to our executive officers, directors and
greater than 10% beneficial owners were filed in a timely manner,
except that except that Dr. Zeldis, Dr. Billings, and Mr. Hamilton
failed to timely file Form 4s in connection with the issuance of
restricted stock units in October 2017.
Nominees to the Board of Directors
The Board will consider director candidates recommended by security
holders. Potential nominees to the Board are required to have such
experience in business or financial matters as would make such
nominee an asset to the Board and may, under certain circumstances,
be required to be “independent”, as such term is
defined under Rule 5605 of the listing standards of NASDAQ and
applicable SEC regulations. Security holders wishing to submit the
name of a person as a potential nominee to the Board must send the
name, address, and a brief (no more than 500 words) biographical
description of such potential nominee to the Board at the following
address: Richard Berman, Chairman of the Board of Directors,
MetaStat, Inc., 27 Drydock Ave., 2nd Floor, Boston, MA 02210.
Potential director nominees will be evaluated by personal
interview, such interview to be conducted by one or more members of
the Board, and/or any other method the Board deems appropriate,
which may, but need not, include a questionnaire. The Board may
solicit or receive information concerning potential nominees from
any source it deems appropriate. The Board need not engage in an
evaluation process unless (i) there is a vacancy on the Board, (ii)
a director is not standing for re-election, or (iii) the Board does
not intend to recommend the nomination of a sitting director for
re-election. A potential director nominee recommended by a security
holder will not be evaluated differently from any other potential
nominee. Although it has not done so in the past, the Board may
retain search firms to assist in identifying suitable director
candidates.
The Board does not have a formal policy on Board candidate
qualifications. The Board may consider those factors it deems
appropriate in evaluating director nominees made either by the
Board or stockholders, including judgment, skill, strength of
character, experience with businesses and organizations comparable
in size or scope to the Company, experience and skill relative to
other Board members, and specialized knowledge or experience.
Depending upon the current needs of the Board, certain factors may
be weighed more or less heavily. In considering candidates for the
Board, the directors evaluate the entirety of each
candidate’s credentials and do not have any specific minimum
qualifications that must be met. “Diversity,” as such,
is not a criterion that the Board considers. The directors will
consider candidates from any reasonable source, including current
Board members, stockholders, professional search firms or other
persons. The directors will not evaluate candidates differently
based on who has made the recommendation.
Limitation of Liability and Indemnification of Officers and
Directors
We are a Nevada corporation and generally governed by the Nevada
Private Corporations Code, Title 78 of the Nevada Revised Statutes,
or NRS. Our officers and directors are indemnified as provided
by NRS and our bylaws.
Section 78.138 of the NRS provides that, unless the
corporation’s articles of incorporation provide otherwise, a
director or officer will not be individually liable unless it is
proven that (i) the director’s or officer’s acts or
omissions constituted a breach of his or her fiduciary duties, and
(ii) such breach involved intentional misconduct, fraud, or a
knowing violation of the law. Our articles of incorporation provide
the personal liability of our directors is eliminated to the
fullest extent permitted under the NRS.
Section 78.7502 of the NRS permits a company to indemnify its
directors and officers against expenses, judgments, fines, and
amounts paid in settlement actually and reasonably incurred in
connection with a threatened, pending, or completed action, suit,
or proceeding, if the officer or director (i) is not liable
pursuant to NRS 78.138, or (ii) acted in good faith and in a manner
the officer or director reasonably believed to be in or not opposed
to the best interests of the corporation and, if a criminal action
or proceeding, had no reasonable cause to believe the conduct of
the officer or director was unlawful. Section 78.7502 of the NRS
requires a corporation to indemnify a director or officer that has
been successful on the merits or otherwise in defense of any action
or suit. Section 78.7502 of the NRS precludes
indemnification by the corporation if the officer or director has
been adjudged by a court of competent jurisdiction, after
exhaustion of all appeals, to be liable to the corporation or for
amounts paid in settlement to the corporation, unless and only to
the extent that the court determines that in view of all the
circumstances, the person is fairly and reasonably entitled to
indemnity for such expenses and requires a corporation to indemnify
its officers and directors if they have been successful on the
merits or otherwise in defense of any claim, issue, or matter
resulting from their service as a director or officer.
Section 78.751 of the NRS permits a Nevada company to indemnify its
officers and directors against expenses incurred by them in
defending a civil or criminal action, suit, or proceeding as they
are incurred and in advance of final disposition thereof, upon
determination by the stockholders, the disinterested board members,
or by independent legal counsel. If so provided in the
corporation’s articles of incorporation, bylaws, or other
agreement, Section 78.751 of the NRS requires a corporation to
advance expenses as incurred upon receipt of an undertaking by or
on behalf of the officer or director to repay the amount if it is
ultimately determined by a court of competent jurisdiction that
such officer or director is not entitled to be indemnified by the
company. Section 78.751 of the NRS further permits the company to
grant its directors and officers additional rights of
indemnification under its articles of incorporation, bylaws, or
other agreement.
Section 78.752 of the NRS provides that a Nevada company may
purchase and maintain insurance or make other financial
arrangements on behalf of any person who is or was a director,
officer, employee, or agent of the company, or is or was serving at
the request of the company as a director, officer, employee, or
agent of another company, partnership, joint venture, trust, or
other enterprise, for any liability asserted against him and
liability and expenses incurred by him in his capacity as a
director, officer, employee, or agent, or arising out of his status
as such, whether or not the company has the authority to indemnify
him against such liability and expenses.
Our bylaws implement the indemnification provisions permitted by
Chapter 78 of the NRS by providing that we shall indemnify our
directors and officers to the fullest extent permitted by the NRS
against expense, liability, and loss reasonably incurred or
suffered by them in connection with their service as an officer or
director. Our bylaws provide shall advance costs and
expenses incurred with respect to any proceeding to which a person
is made a party as a result of being a director or officer in
advance of final disposition of such proceeding upon receipt of an
undertaking by or on behalf of the director or officer to repay
such amount if it is ultimately determined that such person is not
entitled to indemnification. We may purchase and maintain liability
insurance, or make other arrangements for such obligations or
otherwise, to the extent permitted by the NRS.
At the present time, there is no pending litigation or proceeding
involving a director, officer, employee, or other agent of ours in
which indemnification would be required or permitted. We are not
aware of any threatened litigation or proceeding that may result in
a claim for such indemnification.
Summary Compensation Table
The following table sets forth the compensation paid or accrued by
us to our chief executive officer and vice president of finance and
other executive officers. For each of our last two completed fiscal
years, no other officer’s compensation exceeded $100,000 in
each year.
|
|
Fiscal Year
|
|
|
Stock
|
Option
|
All
Other
|
|
|
|
Ended
|
Salary
|
Bonus
|
Awards
|
Awards
|
Compensation
|
Total
|
|
Name and Principal Position
|
February 28
|
($)
|
($)
|
($) (1)
|
($) (1)
|
($)
|
($)
|
|
|
|
|
|
|
|
|
|
|
Douglas A.
Hamilton, President, CEO and Director (2)
|
2018
|
$260,000
|
$153,500
|
$178,000
|
$32,694
|
$-
|
$624,194
|
|
|
2017
|
$260,000
|
$-
|
$-
|
$452,534
|
$-
|
$712,534
|
|
|
|
|
|
|
|
|
|
|
Daniel H.
Schneiderman, VP, Finance (3)
|
2018
|
$165,000
|
$30,938
|
$-
|
$91,081
|
$-
|
$287,019
|
|
|
2017
|
$165,000
|
$-
|
$-
|
$169,701
|
$-
|
$334,701
|
(1)
Represents the aggregate fair value of stock and option awards
computed as of the grant date of each option, common stock or
restricted stock units issuance in accordance with the FASB ASC
Topic 718, rather than amounts actually paid to or realized by the
named individual. There can be no assurance that options will be
exercised (in which case no value will be realized by the
individual), that the value on exercise of options will approximate
the compensation expense we recognized, or that the price of our
common stock when restricted stock units are fully vested and
settled will equal or exceed the price of our common stock on the
date of the applicable restricted stock unit awards.
(2)
Mr.
Hamilton’s
compensation for the year ended February 28,2018 includes a bonus
of $153,500, which payment was deferred incompliance with Section
409A of the Internal Revenue Code, (ii) the issuance of 200,000
fully vested restricted stock units on October11, 2017, and (iii)
the issuance of 30,000 stock options exercisable at $3.00 per share
on April 4, 2017, subject to certaintime-based vesting
milestones.
(3)
Mr.
Schneiderman’s
compensation for the year ended February28, 2018 includes (i) a
bonus of $30,938, which payment was deferred in compliance with
Section 409A of the Internal Revenue Code, (ii) the issuance of
25,000 stock options exercisable at$3.00 per share on April 4,
2017, subject to certain time-based vesting milestones, and (iii)
the issuance of 76,962 stock options exercisable at $0.93 per share
on January 12, 2018, subject to certain time-based vesting
milestones.
Employment Agreements with Executive Officers
Employment Agreement with Douglas A. Hamilton
Effective as of October 31, 2017, we entered into a new employment
agreement with Douglas A. Hamilton to serve as our President and
Chief Executive Officer for a term of two years. Mr. Hamilton had
been serving as the Company’s President and Chief Executive
Officer without an employment agreement since the expiration of his
previous employment agreement on June 17, 2017. The employment
agreement provides for a base salary of $260,000 and an annual
milestone bonus equal to 150% of Mr. Hamilton’s compensation
thereunder, based on his attainment of certain financial, clinical
development, and/or business milestones to be established annually
by the Company’s board of directors or compensation
committee. The employment agreement is terminable by
either party at any time. In the event of termination by the
Company without cause or by Mr. Hamilton for good reason not in
connection with a change of control, as those terms are defined in
the employment agreement, Mr. Hamilton is entitled to six
months’ severance. In the event of termination by the Company
without cause or by Mr. Hamilton for good reason in connection with
a change of control, as those terms are defined in the employment
agreement, he is entitled to twelve months’ severance. The
employment agreement contains standard confidential and proprietary
information, and one-year non-competition and non-solicitation
provisions.
Employment Agreement with Daniel H. Schneiderman
Effective as of May 27, 2013, we entered into an employment
agreement with Daniel H. Schneiderman, to serve as our vice
president of finance and secretary. The employment agreement with
Mr. Schneiderman provides for a base salary of $125,000, and an
annual milestone bonus upon the attainment of certain financial,
clinical development and/or business milestones to be established
annually by our board of directors or compensation
committee. Effective October 1, 2014, the compensation
committee and board of directors authorized an increase to Mr.
Schneiderman’s annual base salary to $165,000. The employment
agreement is terminable by either party at any time. In the event
of termination by us without cause or by Mr. Schneiderman for good
reason not in connection with a change of control, as those terms
are defined in the agreement, Mr. Schneiderman is entitled to six
months’ severance. In the event of termination by us without
cause or by Mr. Schneiderman for good reason in connection with a
change of control, as those terms are defined in the agreement, he
is entitled to twelve months’ severance. The employment
agreement contains standard confidential and proprietary
information, and one-year non-competition and non-solicitation
provisions.
Consulting Agreements with Key Consultants
Consulting Agreement with Michael J. Donovan, Ph.D.,
M.D.
Effective as of August 1, 2015, the Company and Dr. Donovan entered
into a consulting agreement whereby Dr. Donovan serves as the
Company’s acting Chief Medical Officer. The consulting
agreement will terminate on December 31, 2018 and calls for
compensation at a rate of $400 per hour up to a maximum of $2,500
per day. The consulting agreement may be terminated by either party
with thirty days’ notice.
Director Compensation
The following table sets forth certain information concerning
compensation paid or accrued to our non-executive directors during
the year ended February 28, 2018.
|
Name
|
Fees
Earned or Paid in Cash
($)
|
Stock
Awards
($)
(1)
|
Option
Awards
($)
(1)
|
Non-Equity
Incentive Plan Compensation
($)
|
Change
in Pension Value and Nonqualified Deferred Compensation
Earnings
|
All
Other Compensation
($)
|
Total
($)
|
|
Jerome
B. Zeldis (2)
|
$-
|
70,500
|
-
|
-
|
-
|
-
|
$70,500
|
|
Paul
Billings (3)
|
$-
|
44,500
|
-
|
-
|
-
|
-
|
$44,500
|
|
Richard Berman
(4)
|
$-
|
26,000
|
-
|
-
|
-
|
-
|
$26,000
|
|
Martin Driscoll
(5)
|
$-
|
26,000
|
-
|
-
|
-
|
-
|
$26,000
|
|
Oscar Bronsther
(6)
|
$-
|
26,000
|
-
|
-
|
-
|
-
|
$26,000
|
|
H. Philip Goodeve
(7)
|
$-
|
26,000
|
-
|
-
|
-
|
-
|
$26,000
|
(1)
Reflects the
aggregate grant date fair value computed in accordance with FASB
ASC Topic 718.
(2)
Dr.
Zeldis was
issued 20,000 restricted shares of fully vested common stock
approved by the board of directors on April 4, 2017 .
Additionally, Dr. Zeldis was issued 50,000 restricted stock units on October 11,
2017, which vest for active service as follows: (a) 12,500 shares
on January 11, 2018, (b) 12,500 shares on April 11, 2018, (c)
12,500 shares on July 11, 2018, and (d) 12,500 shares on October
11, 2018. Vested restricted
stock units settle on the October 11, 2020, unless
accelerated due to departure from the board of directors or a
change of control.
(3)
Dr. Billings was
issued 50,000 restricted stock
units on October 11, 2017, which vest for active service as
follows: (a) 12,500 shares on January 11, 2018, (b) 12,500 shares
on April 11, 2018, (c) 12,500 shares on July 11, 2018, and (d)
12,500 shares on October 11, 2018. Vested restricted stock units settle on the
October 11, 2020, unless accelerated due to departure from the
board of directors or a change of control.
(4)
Richard
Berman was
issued 20,000 restricted shares of fully vested common stock
approved by the board of directors on April 4, 2017. Mr. Berman
resigned from the board of directors effective May 3,
2017.
(5)
Martin J.
Driscoll was issued 20,000 restricted
shares of fully vested common stock approved by the board of
directors on April 4, 2017. Mr. Driscoll resigned from the board of
directors effective April 25, 2017.
(6)
Dr. Bronsther was
issued 20,000
restricted shares of fully vested common stock approved by the
board of directors on April 4, 2017. Dr. Bronsther resigned from
the board of directors effective May 3, 2017.
(7)
H. Phillip Goodeve was
issued 20,000
restricted shares of fully vested common stock approved by the
board of directors on April 4, 2017. Mr. Goodeve resigned from the
board of directors effective May 3, 2017.
Employee Benefits Plans
Pension Benefits
We do not sponsor any qualified or non-qualified pension benefit
plans.
Nonqualified Deferred Compensation
We do not maintain any non-qualified defined contribution or
deferred compensation plans.
Severance Arrangements
The employment agreements with each of Douglas A. Hamilton and
Daniel H. Schneiderman provide that in the event of termination by
us without cause or by the executives for good reason not in
connection with a change of control, as those terms are defined in
the agreement, such executives are entitled to six months’
severance. In the event of termination by us without cause or by
the executives for good reason in connection with a change of
control, as those terms are defined in the agreement, such
executives are entitled to twelve months’
severance.
Outstanding Equity Awards at Fiscal Year-End
The following table summarizes the number of securities underlying
outstanding 2012 Incentive Plan awards for each named executive
officer as of February 28, 2018.
|
|
Option Awards
|
|
Stock Awards
|
|||||||
|
|
Equity Incentive Plan Awards:
|
|
Equity Incentive Plan Awards:
|
|||||||
|
|
|
|
|
|
|
|
|
|
Number
|
Market or
|
|
|
Number of
|
Number of
|
Number of
|
|
|
|
|
Market
|
of
|
payout value
|
|
|
securities
|
securities
|
securities
|
|
|
|
Number of
|
value of
|
unearned shares
|
of unearned
|
|
|
underlying
|
underlying
|
underlying
|
|
|
|
shares of
|
shares of
|
that
|
shares that
|
|
|
unexercised
|
unexercised
|
unexercised
|
Option
|
Option
|
|
stock that
|
stock that
|
have not
|
have not
|
|
|
options (#)
|
options (#)
|
unearned
|
exercise
|
expiration
|
|
have not vested
|
have not vested
|
vested
|
vested
|
|
Name
|
exercisable
|
unexercisable
|
options (#)
|
price ($)
|
date
|
|
(#)
|
($) (1)
|
(#)
|
($) (1)
|
|
Douglas A. Hamilton
|
30,000
|
-
|
-
|
$3.00
|
4/4/2027
|
|
-
|
$-
|
-
|
$-
|
|
|
20,000
|
-
|
40,000
|
$8.25
|
6/17/2025
|
|
-
|
$-
|
-
|
$-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Daniel H. Schneiderman
|
76,962
|
-
|
-
|
$0.93
|
1/12/2018
|
|
-
|
$-
|
-
|
$-
|
|
|
25,000
|
-
|
-
|
$3.00
|
4/4/2017
|
|
-
|
$-
|
-
|
$-
|
|
|
20,000
|
-
|
-
|
$3.55
|
2/3/2026
|
|
-
|
$-
|
-
|
$-
|
|
|
3,667
|
-
|
-
|
$10.20
|
1/6/2022
|
|
-
|
$-
|
-
|
$-
|
|
|
3,334
|
-
|
-
|
$48.75
|
4/5/2023
|
|
-
|
$-
|
-
|
$-
|
|
|
|
|
|
|
|
|
1,334
|
$1,641
|
-
|
$-
|
(1)
Market value based on closing price of common stock at February 28,
2018.
The following table summarizes the number of securities underlying
awards that fall outside of the 2012 Incentive Plan for each named
executive officer as of February 28, 2018
|
|
Option Awards
|
|
Stock Awards
|
|||||||
|
|
Equity Incentive Awards:
|
|
Equity
Incentive Awards:
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
Market or
|
|
|
Number of
|
Number of
|
Number of
|
|
|
|
|
|
|
payout value
|
|
|
securities
|
securities
|
securities
|
|
|
|
Number of
|
Market value of
|
Number of
|
of unearned
|
|
|
underlying
|
underlying
|
underlying
|
|
|
|
shares of
|
shares of
|
unearned shares
|
shares that
|
|
|
unexercised
|
unexercised
|
unexercised
|
Option
|
Option
|
|
stock that
|
stock that
|
that have
|
have not
|
|
|
options (#)
|
options (#)
|
unearned
|
expiration
|
expiration
|
|
have not vested
|
have not vested
|
not vested
|
vested
|
|
Name
|
exercisable
|
unexercisable
|
options (#)
|
price ($)
|
date
|
|
(#)
|
($) (1)
|
(#)
|
($) (1)
|
|
Douglas
A. Hamilton
|
142,214
|
177,786
|
-
|
$2.00
|
7/7/2026
|
|
-
|
-
|
-
|
-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Daniel
H. Schneiderman
|
53,340
|
66,660
|
-
|
$2.00
|
7/7/2026
|
|
-
|
-
|
-
|
-
|
|
|
11,112
|
8,888
|
-
|
$16.50
|
10/14/2024
|
|
-
|
-
|
-
|
-
|
(1)
Market value based on closing price of common stock at February 28,
2018.
Security Ownership of Certain Beneficial Owners and
Management
The following table sets forth certain information regarding
beneficial ownership of our common stock as of May
18, 2018 by (i) each person (or group of affiliated
persons) who is known by us to own more than five percent of the
outstanding shares of our common stock, (ii) each director and
executive officer, and (iii) all of our directors and executive
officers as a group.
Beneficial ownership is determined in accordance with SEC rules and
generally includes voting or investment power with respect to
securities. Unless otherwise noted, the address of each stockholder
listed below is 27 Drydock Ave. 2nd Floor, Boston, MA
02210.
We had 5,877,383 shares of our common stock outstanding as of May
18, 2018.
|
Name
and Address of Beneficial Owners
|
Amount
and Nature of Beneficial Ownership (1)
|
Percent
of Class (2)
|
|
Douglas
A. Hamilton, President, Chief Executive Officer, and Director
(3)
|
657,013
|
10.4%
|
|
Daniel
H. Schneiderman, Vice President of Finance and Secretary
(4)
|
217,169
|
3.6%
|
|
Jerome
B. Zeldis, M.D., Ph.D., Chairman of the Board of Directors
(5)
|
120,000
|
2.0%
|
|
Paul
Billings, M.D., Ph.D., Director (6)
|
50,000
|
0.9%
|
__________________
* Less than 1%
|
(1)
|
Beneficial ownership is determined in accordance with the rules of
the SEC and generally includes voting or investment power with
respect to securities. Shares of our common stock subject to
securities anticipated to be exercisable or convertible at or
within 60 days of the date hereof, are deemed outstanding for
computing the percentage of the person holding such option or
warrant but are not deemed outstanding for computing the percentage
of any other person. The indication herein that shares are
anticipated to be beneficially owned is not an admission on the
part of the listed stockholder that he, she or it is or will be a
direct or indirect beneficial owner of those shares.
|
|
(2)
|
Based on 5,877,383 shares of our common stock outstanding as of May
18, 2018.
|
|
(3)
|
Consists of (i) 47,013 shares of common stock, (ii) 200,000 fully
vested restricted stock units that settle on the October 11,
2020, unless accelerated due to
departure from the Company or a change of control, (iii) 186,102
shares of common stock underlying vested options, and (iv) 223,898
shares of common stock underlying unvested options subject to
certain time-based and milestone vesting. Excludes 6,241 shares of
Common Stock underlying warrants with 4.9% and 9.9% ownership
blockers.
|
|
(4)
|
Consists of (i) 23,834 shares of common stock, (ii) 1,334
restricted shares of common stock issued pursuant to the 2012
Incentive Plan that vest and become transferable upon the listing
of the common stock on a national securities exchange, (iii) 96,454
shares of common stock underlying vested options, and (iv) 95,547
shares of common stock underlying unvested options subject to
certain time-based and milestone vesting.
|
|
(5)
|
Consists of (i) 20,000 shares of common stock, (ii) 33,334 shares
of common stock underlying vested options, (iii) 16,666 shares of
common stock underlying unvested options subject to annual
time-based vesting. Also includes 25,000 vested restricted stock
units and 25,000 unvested restricted stock units subject to certain
time-based vesting for active service, which 50,000 restricted
stock units settle on the October 11, 2020, unless accelerated due to departure from the
Company or a change of control.
|
|
(6)
|
Consists of 25,000 vested restricted stock units and 25,000
unvested restricted stock units subject to certain time-based
vesting for active service, which 50,000 restricted stock
units settle on the October 11, 2020, unless accelerated due to departure from the
Company or a change of control.
|
|
Item 13.
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR
INDEPENDENCE
|
Related Party Transactions
None.
Director Independence
Jerome B. Zeldis, M.D., Ph.D. and Paul Billings, M.D., Ph.D. have
been determined to be independent as defined by NASDAQ Listing Rule
5605(a)(2) of The NASDAQ Stock Market, LLC and Section 10A(m)(3) of
the Exchange Act. No transactions, relationships or arrangements
were considered by the board of directors in determining that these
directors were independent. Under NASDAQ Listing Rule 5605(a)(2),
an "independent director" is a "person other than an officer or
employee of the company or any other individual having a
relationship which, in the opinion of the company's board of
directors, would interfere with the exercise of independent
judgment in carrying out the responsibilities of a
director."
|
Item 14.
|
PRINCIPAL ACCOUNTING FEES AND SERVICES
|
Public Accounting Fees
The following charts sets forth public accounting fees in
connection with services rendered by EisnerAmper LLP during the two
years ended February 28, 2018.
|
|
Fiscal
Year Ended February 28, 2018
|
Fiscal
Year Ended February 28, 2017
|
|
Audit
Fees
|
$117,383
|
$128,227
|
|
Audit-Related
Fees
|
$-
|
$-
|
|
Tax
Fees
|
$9,200
|
$8,850
|
|
All
Other Fees
|
$-
|
$-
|
|
Total
|
$126,583
|
$137,077
|
Audit fees were for professional services rendered by EisnerAmper
LLP for the audit of our annual financial statements and the review
of the financial statements included in our quarterly reports on
Forms 10-Q for the two years ended February 28, 2018. Audit fees
also includes professional services rendered with the filing of our
registration statements.
Pre-Approval of Services
Our audit committee pre-approved all of the foregoing
services.
PART IV
|
Item 15.
|
EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
|
|
Exhibit
No.
|
Description
|
|
Share
Exchange Agreement dated February 27, 2012 (Incorporated by
reference to our Current Report on Form 8-K filed with the
Commission on May 25, 2012).
|
|
|
|
|
|
Articles
of Incorporation of MetaStat, Inc., as amended (Incorporated by
reference to our Current Report on Form 8-K filed with the
Commission on March 21, 2012).
|
|
|
|
|
|
Certificate
of Designation of Rights and Preferences of the Series A-2
Preferred Stock dated October 5, 2016 (Incorporated by reference to
our Current Report on Form 8-K filed on October 17,
2016).
|
|
|
|
|
|
Amended
and Restated Certificate of Designation of the Preferences, Rights
and Limitations of the Series B Preferred Stock filed on December
31, 2014 (Incorporated by reference to our Current Report on Form
8-K filed with the Commission on April 2, 2015).
|
|
|
|
|
|
Amended
and Restated By-laws (Incorporated by reference to our Current
Report on Form 8-K filed with the Commission on November 23,
2015).
|
|
|
|
|
|
Form
of Warrant issued to Holders of May 2014 Convertible Promissory
Notes (Incorporated by reference to our Annual Report on Form 10-K
filed with the Commission on June 13, 2014).
|
|
|
|
|
|
Form
of Investor Warrant dated June 30, 2014 (Incorporated by reference
to our Current Report on Form 8-K filed with the Commission on July
2, 2014).
|
|
|
|
|
|
Form
of Series A Warrant dated December 31, 2014 (Incorporated by
reference to our Current Report on Form 8-K filed with the
Commission on January 7, 2015).
|
|
|
|
|
|
Form
of Series B Warrant dated December 31, 2014 (Incorporated by
reference to our Current Report on Form 8-K filed with the
Commission on January 7, 2015).
|
|
|
|
|
|
Form
of Amended and Restated Series A Warrant dated March 27, 2015
(Incorporated by reference to our Current Report on Form 8-K filed
with the Commission on April 2, 2015.
|
|
|
|
|
|
Form
of August 2015 Warrant (Incorporated by reference to our Current
Report on Form 8-K filed with the Commission on August 5,
2015).
|
|
|
|
|
|
Form
of OID Warrant (Incorporated by reference to our Current Report on
Form 8-K filed with the Commission on February 19,
2016).
|
|
|
|
|
|
Form
of Convertible Note dated January 17, 2017 (Incorporated by
reference to our Current Report on Form 8-K filed with the
Commission on January 23, 2017).
|
|
|
|
|
|
Form
of Warrant dated January 17, 2017 (Incorporated by reference to our
Current Report on Form 8-K filed with the Commission on January 23,
2017).
|
|
|
|
|
|
Form
of Senior Note (Incorporated by reference to our Current Report on
Form 8-K filed with the Commission on April 5, 2018).
|
|
|
|
|
|
Form
of Junior Note (Incorporated by reference to our Current Report on
Form 8-K filed with the Commission on April 5, 2018).
|
|
|
|
|
|
Form
of Note Warrant (Incorporated by reference to our Current Report on
Form 8-K filed with the Commission on April 5, 2018).
|
|
License
Agreement with AECOM, MIT, Cornell and IFO-Regina dated August 26,
2010 (Incorporated by reference to our Current Report on Form 8-K
filed with the Commission on August 13, 2012).
|
|
|
|
|
|
Second
Amended and Restated 2012 Omnibus Securities and Incentive Plan
(Incorporated by reference to our Definitive Proxy Statement
on Schedule 14A filed with the Commission on May 29,
2015).
|
|
|
|
|
|
Form
of Consultant Non-Qualified Stock Option Agreement (Incorporated by
reference to our Current Report on Form 8-K filed with the
Commission on March 21, 2012).
|
|
|
|
|
|
Form
of Employee Non-Qualified Stock Option Agreement (Incorporated by
reference to our Current Report on Form 8-K filed with the
Commission on March 21, 2012).
|
|
|
|
|
|
“Second”
License Agreement with AECOM effective March 2012 (Incorporated by
reference to our Current Report on Form 8-K filed with the
Commission on August 13, 2012).
|
|
|
|
|
|
“Third”
License Agreement with AECOM effective March 2012 (Incorporated by
reference to our Current Report on Form 8-K filed with the
Commission on August 13, 2012).
|
|
|
|
|
|
Employment
Agreement of Daniel Schneiderman dated May 24, 2013 (Incorporated
by reference to our Annual Report on Form 10-K filed with the
Commission on June 13, 2014).
|
|
|
|
|
|
Diagnostic
License Agreement with the Massachusetts Institute of Technology
and its David H. Koch Institute for Integrative Cancer Research at
MIT and its Department of Biology, AECOM, and Montefiore Medical
Center as of December 7, 2013 (Incorporated by reference to our
Current Report on Form 8-K, as amended, initially filed with the
Commission on December 12, 2013).
|
|
|
|
|
|
Therapeutic
License Agreement with the Massachusetts Institute of Technology
and its David H. Koch Institute for Integrative Cancer Research at
MIT and its Department of Biology, AECOM, and Montefiore Medical
Center as of December 7, 2013 (Incorporated by reference to our
Current Report on Form 8-K, as amended, initially filed with the
Commission on December 12, 2013).
|
|
|
|
|
|
Antibody
License Agreement with MIT dated June 2, 2014 (Incorporated by
reference to our Quarterly Report on Form 10-Q filed with the
Commission on July 15, 2014).
|
|
|
|
|
|
Form
of Note Purchase Agreement dated June 30, 2014 (Incorporated by
reference to our Current Report on Form 8-K filed with the
Commission on August 5, 2015).
|
|
|
|
|
|
Form
of OID Note Purchase Agreement (Incorporated by reference to our
Current Report on Form 8-K filed with the Commission on February
19, 2016).
|
|
|
|
|
|
Form
of Subscription Agreement dated May 26, 2016 (Incorporated by
reference to our Annual Report on Form 10-K filed with the
Commission on May 31, 2016)
|
|
|
|
|
|
Form
of Registration Rights Agreement dated May 26, 2016 (Incorporated
by reference to our Annual Report on Form 10-K filed with the
Commission on May 31, 2016).
|
|
Form
of Subscription Agreement in connection with the 2016 Unit Private
Placement (Incorporated by reference to our Current Report on Form
8-K filed with the Commission on October 17, 2016).
|
|
|
|
|
|
Form
of Registration Rights Agreement in connection with the 2016 Unit
Private Placement (Incorporated by reference to our Current Report
on Form 8-K filed with the Commission on October 17,
2016).
|
|
|
|
|
|
Pilot
Materials Transfer Agreement between MetaStat, Inc. and Celgene
Corporation dated August 22, 2016 (Incorporated by reference to our
Current Report on Form 8-K filed with the Commission on October 6,
2016).
|
|
|
|
|
|
First
Amendment to Pilot Materials Transfer Agreement between MetaStat,
Inc. and Celgene Corporation dated September 29, 2016 (Incorporated
by reference to our Current Report on Form 8-K filed with the
Commission on October 6, 2016).
|
|
|
|
|
|
Form
of Exchange Agreement dated January 17, 2017 (Incorporated by
reference to our Current Report on Form 8-K filed with the
Commission on January 23, 2017).
|
|
|
|
|
|
Form
of Purchase Agreement dated June 23, 2017 (Incorporated by
reference to our Current Report on Form 8-K filed with the
Commission on June 27, 2017).
|
|
|
|
|
|
Employment
Agreement dated October 31, 2017 by and between the Company and
Douglas A. Hamilton (Incorporated by reference to our Current
Report on Form 8-K filed with the Commission on October 31,
2017).
|
|
|
|
|
|
Form
of Note Purchase Agreement (Incorporated by reference to our
Current Report on Form 8-K filed with the Commission on April 5,
2018).
|
|
|
|
|
|
Form
of Exchange Agreement (Incorporated by reference to our Current
Report on Form 8-K filed with the Commission on April 5,
2018).
|
|
|
|
|
|
Subsidiaries
of the Registrant (Incorporated by reference to our Annual Report
on Form 10-K filed with the Commission on May 28,
2013).
|
|
|
|
|
|
Certification
of Chief Executive Officer, pursuant to Section 302 of the
Sarbanes-Oxley Act of 2002.
|
|
|
|
|
|
Certification
of Chief Executive Officer, pursuant to 18 U.S.C. Section 1350, as
adopted pursuant to Section 906 of the Sarbanes-Oxley Act of
2002.
|
|
|
|
|
|
101.INS**
|
XBRL
Instance Document.
|
|
|
|
|
101.SCH**
|
XBRL
Taxonomy Extension Schema.
|
|
|
|
|
101.CAL**
|
XBRL
Taxonomy Extension Calculation Linkbase.
|
|
|
|
|
101.DEF**
|
XBRL
Taxonomy Extension Definition Linkbase.
|
|
|
|
|
101.LAB**
|
XBRL
Taxonomy Extension Label Linkbase.
|
|
|
|
|
101.PRE**
|
XBRL
Taxonomy Extension Presentation Linkbase.
|
†
Confidential
treatment has been granted with respect to portions of this
exhibit.
**
Pursuant
to Rule 406T of Regulation S-T, the XBRL (Extensible Business
Reporting Language) information included in Exhibit 101 hereto is
deemed furnished and not filed or part of a registration statement
or prospectus for purposes of Sections 11 or 12 of the Securities
Act of 1933, as amended, is deemed not filed for purposes of
Section 18 of the Securities Exchange Act of 1934, as amended, and
otherwise is not subject to liability under these
sections.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the
Securities Exchange Act of 1934, the registrant has duly caused
this Report to be signed on its behalf by the undersigned,
thereunto duly authorized.
|
|
METASTAT, INC.
|
|
|
|
|
|
|
May 29,
2018
|
By:
|
/s/ Douglas A. Hamilton
|
|
|
|
Douglas A. Hamilton, President, Chief Executive
Officer,
and Director (Principal
Executive Officer and
Principal Financial and Accounting Officer)
|
Pursuant
to the requirements of the Securities Exchange Act of 1934, this
Report has been signed below by the following persons on behalf of
the registrant and in the capacities and on the date
indicated.
|
Signature
|
|
Capacity
|
Date
|
|
|
|
|
|
|
|
|
/s/ Jerome B. Zeldis
|
|
Chairman of the Board of Directors
|
May 29,
2018
|
|
|
Jerome B. Zeldis, M.D., Ph.D.
|
|
|
|
|
|
|
|
|
|
|
|
/s/ Paul Billings
|
|
Director
|
May 29,
2018
|
|
|
Paul Billings, M.D., Ph.D.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
METASTAT, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED FEBRUARY 28, 2018 AND FEBRUARY 28,
2017
|
|
Page
|
|
|
|
|
F-2
|
|
|
|
|
|
F-3
|
|
|
|
|
|
F-4
|
|
|
|
|
|
F-5
|
|
|
|
|
|
F-6
|
|
|
|
|
|
F-7
|
To the Board of Directors and Stockholders of
MetaStat, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of
MetaStat, Inc. and its subsidiary (the “Company") as of
February 28, 2018 and 2017, and the related consolidated statements
of operations, changes in stockholders’ deficit, and cash
flows for each of the years in the two-year period ended February
28, 2018, and the related notes (collectively referred to as the
“financial statements”). In our opinion, the financial
statements present fairly, in all material respects, the
consolidated financial position of the Company as of February 28,
2018 and 2017, and the consolidated results of their operations and
their cash flows for each of the years in the two-year period ended
February 28, 2018, in conformity with accounting principles
generally accepted in the United States of America.
Going Concern
The accompanying financial statements have been prepared assuming
that the Company will continue as a going concern. As discussed in
Note
[1] to the financial statements, as of February 28, 2018,
the Company has a stockholders’ deficit and a negative
working capital. The Company has sustained cumulative losses and
has not generated positive cash flow from operations. The
aforementioned conditions raise substantial doubt about the
Company’s ability to continue as a going concern.
Management’s plans in regard to these matters are also
described in Note [1]. The financial statements do not include any
adjustments that might result from the outcome of this
uncertainty.
Basis for Opinion
These financial statements are the responsibility of the
Company’s management. Our responsibility is to express an
opinion on the Company’s financial statements based on our
audits. We are a public accounting firm registered with the Public
Company Accounting Oversight Board (United States) ("PCAOB") and
are required to be independent with respect to the Company in
accordance with the U.S. federal securities laws and the applicable
rules and regulations of the Securities and Exchange Commission and
the PCAOB.
We conducted our audits in accordance with the standards of the
PCAOB. Those standards require that we plan and perform the audit
to obtain reasonable assurance about whether the financial
statements are free of material misstatement, whether due to error
or fraud. The Company is not required to have, nor were we engaged
to perform, an audit of its internal control over financial
reporting. As part of our audits we are required to obtain an
understanding of internal control over financial reporting but not
for the purpose of expressing an opinion on the effectiveness of
the Company's internal control over financial reporting.
Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of
material misstatement of the financial statements, whether due to
error or fraud, and performing procedures that respond to those
risks. Such procedures included examining, on a test basis,
evidence regarding the amounts and disclosures in the financial
statements. Our audits also included evaluating the accounting
principles used and significant estimates made by management, as
well as evaluating the overall presentation of the financial
statements. We believe that our audits provide a reasonable basis
for our opinion.
/s/ EisnerAmper LLP
We have served as the Company’s auditor since
2014.
EISNERAMPER LLP
New York, New York
May 29, 2018
METASTAT, INC.
Consolidated Balance
Sheets
As of February 28, 2018, and February 28,
2017
|
|
February 28,
2018
|
February 28,
2017
|
|
ASSETS
|
|
|
|
Current
Assets:
|
|
|
|
Cash
and cash equivalents
|
$316,933
|
$782,707
|
|
Deferred
offering costs
|
10,872
|
-
|
|
Prepaid
expenses
|
4,131
|
20,856
|
|
Total Current Assets
|
331,936
|
803,563
|
|
|
|
|
|
Equipment
|
|
|
|
(net
of accumulated depreciation of $353,467 and $265,234,
respectively)
|
354,098
|
414,635
|
|
Refundable
deposits
|
43,600
|
43,600
|
|
|
|
|
|
TOTAL ASSETS
|
$729,634
|
$1,261,798
|
|
|
|
|
|
LIABILITIES
AND STOCKHOLDERS' DEFICIT
|
|
|
|
|
|
|
|
LIABILITIES
|
|
|
|
Current
Liabilities:
|
|
|
|
Accounts
payable
|
$571,600
|
$572,195
|
|
Accrued
expense
|
342,715
|
179,680
|
|
Deferred
research & development reimbursement
|
-
|
177,517
|
|
Convertible
note payable (net of debt discount of $0 and $10,914,
respectively)
|
1,000,000
|
989,086
|
|
Accrued
interest payable
|
124,163
|
15,890
|
|
Accrued
dividends on Series B Preferred Stock
|
41,819
|
15,638
|
|
Total Current Liabilities
|
2,080,297
|
1,950,006
|
|
|
|
|
|
Deferred
rent liability
|
50,702
|
-
|
|
Warrant
liability
|
147,577
|
2,106,972
|
|
Total Liabilities
|
2,278,576
|
4,056,978
|
|
|
|
|
|
STOCKHOLDERS'
DEFICIT
|
|
|
|
|
|
|
|
Series
A convertible preferred stock ($0.0001 par value; 1,000,000 shares
authorized; 0 and 874,257 issued and outstanding,
respectively)
|
-
|
87
|
|
Series
A-2 convertible preferred stock ($0.0001 par value; 1,000,000
shares authorized; 279,904 and 70,541 shares issued and
outstanding, respectively)
|
28
|
7
|
|
Series
B convertible preferred stock ($0.0001 par value; 1,000 shares
authorized; 226 and 213 shares issued and outstanding,
respectively)
|
-
|
-
|
|
Common
stock ($0.0001 par value; 150,000,000 shares authorized; 5,877,383
and 4,707,942 shares issued and outstanding,
respectively)
|
588
|
471
|
|
Additional
paid-in-capital
|
27,950,351
|
23,523,140
|
|
Accumulated
deficit
|
(29,499,909)
|
(26,318,885)
|
|
Total stockholders' deficit
|
(1,548,942)
|
(2,795,180)
|
|
|
|
|
|
TOTAL LIABILITIES AND STOCKHOLDERS' DEFICIT
|
$729,634
|
$1,261,798
|
The accompanying notes are an integral part of the consolidated
financial statements
METASTAT, INC.
Consolidated Statements of
Operations
For the Years ended February 28, 2018 and February 28,
2017
|
|
Year ended
|
|
|
|
February 28,
2018
|
February 28,
2017
|
|
|
|
|
|
Revenue
- research collaboration
|
$23,300
|
$-
|
|
Total
Revenue
|
23,300
|
-
|
|
|
|
|
|
Operating
Expenses
|
|
|
|
General
& administrative
|
2,317,298
|
2,338,818
|
|
Research
& development
|
1,256,514
|
1,009,134
|
|
Total
Operating Expenses
|
3,573,812
|
3,347,952
|
|
|
|
|
|
Other
Expenses (income)
|
|
|
|
Interest
expense
|
119,282
|
1,062,389
|
|
Other
income, net
|
(637)
|
(965)
|
|
Change
in fair value of warrant liability
|
(488,133)
|
(2,405,985)
|
|
Change
in fair value of put option embedded in notes payable
|
-
|
(614,484)
|
|
Loss
on sale of notes receivable
|
-
|
112,500
|
|
Loss
on extinguishment of debt
|
-
|
1,375,829
|
|
Loss
on settlement of accounts payable
|
-
|
64,323
|
|
Total
Other Expenses (Income)
|
(369,488)
|
(406,395)
|
|
|
|
|
|
Net Loss
|
$(3,181,024)
|
$(2,941,557)
|
|
|
|
|
|
Loss attributable to common shareholders and loss per common
share:
|
|
|
|
|
|
|
|
Net
loss
|
(3,181,024)
|
(2,941,557)
|
|
Deemed
dividend on Series B Preferred Stock issuance
|
-
|
(708,303)
|
|
Accrued
dividend on Series B Preferred Stock
|
(97,968)
|
(227,163)
|
|
Deemed
dividend on Series B Preferred Stock holders exchange of
warrants
|
-
|
(2,340,552)
|
|
Deemed
dividend related to warrants exercise price
modification
|
(31,139)
|
-
|
|
Loss attributable to common shareholders
|
$(3,310,131)
|
$(6,217,575)
|
|
|
|
|
|
Net
loss per share, basic and diluted
|
$(0.61)
|
$(2.10)
|
|
|
|
|
|
Weighted
average of shares outstanding, basic and diluted
|
5,425,284
|
2,965,910
|
The accompanying notes are an integral part of the consolidated
financial statements
METASTAT, INC.
For the years ended February 28, 2018 and February 28,
2017
|
|
Series A Preferred Stock
|
Series A -2 Preferred Stock
|
Series B Preferred Stock
|
Common Stock
|
Paid-in
|
Accumulated
|
Total
|
||||
|
|
Shares
|
Amount
|
Shares
|
Amount
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Deficit
|
Deficit
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at February 29, 2016
|
874,257
|
$87
|
-
|
$-
|
659
|
$-
|
1,851,201
|
$185
|
$21,607,259
|
$(23,377,328)
|
$(1,769,797)
|
|
Issuance
of common stock, preferred stock and warrants for cash, net of
offering costs
|
-
|
-
|
48,300
|
5
|
-
|
-
|
1,065,750
|
107
|
962,550
|
-
|
962,662
|
|
Issuance
of common stock and warrants to convert accounts
payable
|
-
|
-
|
-
|
-
|
-
|
-
|
32,500
|
3
|
212,275
|
-
|
212,278
|
|
Issuance
of common stock and warrants to convert notes payable
|
-
|
-
|
16,000
|
2
|
-
|
-
|
440,500
|
44
|
1,566,755
|
-
|
1,566,801
|
|
Beneficial
conversion feature of Series B Preferred Stock
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
708,303
|
-
|
708,303
|
|
Deemed
dividend to Series B Preferred Stock holders
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(708,303)
|
-
|
(708,303)
|
|
Accrued
dividends on Series B Preferred Stock
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(227,163)
|
-
|
(227,163)
|
|
Series
B PIK Dividend
|
-
|
-
|
-
|
-
|
35
|
-
|
-
|
-
|
191,941
|
-
|
191,941
|
|
Issuance
of common stock, preferred stock and warrants fin exchange for
cancellation of Series B preferred stock and Series A
Warrants
|
-
|
-
|
6,241
|
-
|
(481)
|
-
|
1,292,991
|
129
|
747,486
|
-
|
747,615
|
|
Deemed
dividend to Series B Preferred Stock holders for exchange of
warrants
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(2,340,552)
|
-
|
(2,340,552)
|
|
Issuance
of warrants in connection with OID Notes amendment
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
44,095
|
-
|
44,095
|
|
Issuance
of warrants in connection with convertible note
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
117,632
|
-
|
117,632
|
|
Share-based
compensation
|
-
|
-
|
-
|
-
|
-
|
-
|
25,000
|
3
|
640,862
|
-
|
640,865
|
|
Net
loss
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(2,941,557)
|
(2,941,557)
|
|
Balance at February 28, 2017
|
874,257
|
$87
|
70,541
|
$7
|
213
|
$-
|
4,707,942
|
$471
|
$23,523,140
|
$(26,318,885)
|
$(2,795,180)
|
|
Issuance
of common stock, preferred stock and warrants for cash, net of
offering cost
|
-
|
-
|
229,363
|
23
|
-
|
-
|
783,898
|
78
|
2,308,081
|
-
|
2,308,182
|
|
Issuance
of common stock to convert Series A preferred stock
|
(874,257)
|
(87)
|
-
|
-
|
-
|
-
|
58,283
|
6
|
81
|
-
|
-
|
|
Issuance
of common stock to convert Series A-2 preferred stock
|
-
|
-
|
(20,000)
|
(2)
|
-
|
-
|
200,000
|
20
|
(18)
|
-
|
-
|
|
Issuance
common stock to convert accrued expenses
|
-
|
-
|
-
|
-
|
-
|
-
|
27,260
|
3
|
23,654
|
-
|
23,657
|
|
Accrued
dividends on Series B preferred stock
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(97,968)
|
-
|
(97,968)
|
|
Series
B PIK dividend
|
-
|
-
|
-
|
-
|
13
|
-
|
-
|
-
|
71,787
|
-
|
71,787
|
|
Modification
on warrant exercise price
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
31,139
|
-
|
31,139
|
|
Deemed
dividend related to warrants exercise price
modification
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(31,139)
|
-
|
(31,139)
|
|
Reclassification
between warrant liability and additional
paid-in-capital
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
1,471,262
|
-
|
1,471,262
|
|
Share-based
compensation
|
-
|
-
|
-
|
-
|
-
|
-
|
100,000
|
10
|
650,332
|
-
|
650,342
|
|
Net
loss
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
(3,181,024)
|
(3,181,024)
|
|
Balance as of February 28, 2018
|
-
|
$-
|
279,904
|
$28
|
226
|
$-
|
5,877,383
|
$588
|
$27,950,351
|
$(29,499,909)
|
$(1,548,942)
|
The accompanying notes are an integral part of the consolidated
financial statements
METASTAT, INC.
Consolidated Statements of Cash Flows
For the years ended February 28, 2018 and February 28,
2017
|
|
Year ended
|
|
|
|
February 28,
2018
|
February 28,
2017
|
|
|
|
|
|
Cash
Flows from Operating Activities:
|
|
|
|
Net
loss
|
$(3,181,024)
|
$(2,941,557)
|
|
Adjustments
to reconcile net loss to net cash
used in operating activities:
|
|
|
|
Depreciation
|
89,238
|
95,838
|
|
Share-based
compensation
|
650,342
|
640,865
|
|
Accretion
of debt discount included in interest expense
|
10,914
|
958,053
|
|
Loss
on sale of note receivable and sale of assets
|
-
|
112,500
|
|
Loss
on settlement of accounts payable
|
-
|
64,323
|
|
Loss
on disposal of fixed assets
|
1,399
|
-
|
|
Loss
on extinguishment of debt
|
-
|
1,375,829
|
|
Change
in fair value of warrant liability
|
(488,133)
|
(2,405,985)
|
|
Change
in fair value of put option embedded in notes payable
|
-
|
(614,484)
|
|
Net
changes in assets and liabilities:
|
|
|
|
Prepaid
expenses
|
16,725
|
170,664
|
|
Deferred
rent liability
|
50,702
|
-
|
|
Accounts
payable and accrued expenses
|
175,225
|
(50,095)
|
|
Deferred
research and development reimbursement
|
(177,517)
|
177,517
|
|
Interest
payable
|
108,273
|
101,889
|
|
Net Cash used in Operating Activities
|
(2,743,856)
|
(2,314,643)
|
|
|
|
|
|
Cash
Flows from Investing Activities:
|
|
|
|
Purchase
of equipment
|
(30,100)
|
(13,421)
|
|
Proceeds
from sale of note receivable
|
-
|
12,500
|
|
Net Cash used in Investing Activities
|
(30,100)
|
(921)
|
|
|
|
|
|
Cash
Flows from Financing Activities:
|
|
|
|
Proceeds
from issuance of debt, net
|
-
|
122,790
|
|
Proceeds
from issuance of common stock, preferred stock and warrants,
net
|
2,308,182
|
2,746,688
|
|
Payment
of notes
|
-
|
(8,000)
|
|
Payment
of short-term debt
|
-
|
(126,990)
|
|
Net Cash provided by Financing Activities
|
2,308,182
|
2,734,488
|
|
|
|
|
|
Net
(decrease) increase in cash and cash equivalents
|
(465,774)
|
418,924
|
|
|
|
|
|
Cash
and cash equivalents:
|
|
|
|
Cash
at the beginning of the year
|
782,707
|
363,783
|
|
Cash at the end of the year
|
$316,933
|
$782,707
|
|
|
|
|
|
Supplemental
Disclosure of Non-cash Financing Activities:
|
|
|
|
Warrant
liability associated with note payable
|
$-
|
$15,225
|
|
Issuance
of common stock and warrants as payment of accounts
payable
|
$-
|
$212,278
|
|
Issuance
of common stock and warrants to convert debt and accrued
interest
|
$-
|
$2,326,321
|
|
Financing
of insurance premium through notes payable
|
$-
|
$158,400
|
|
Warrants
issued to placement agents
|
$-
|
$278,223
|
|
Series
B Preferred PIK dividend
|
$71,787
|
$191,941
|
|
Series
B Preferred Stock accrued dividends
|
$97,968
|
$227,163
|
|
Deemed
dividend related to Series B Preferred Stock BCF adjustment for
conversion price adjustment
|
$-
|
$708,303
|
|
Issuance
of common stock , preferred stock and warrants in exchange for
cancellation of Series B preferred stock and Series A
Warrants
|
$-
|
$67,900
|
|
Deemed
dividend to Series B preferred stock holders upon exercising Most
Favorable Nation option
|
$-
|
$2,340,552
|
|
Exchange
OID notes and note payable to convertible debt
|
$-
|
$986,269
|
|
Issuance
of warrants in connection with OID Notes amendment
|
$-
|
$44,095
|
|
Deemed
dividend related to warrants exercise price
modification
|
$31,139
|
$-
|
|
Issuance
of common stock to convert Series A preferred stock
|
$87
|
$-
|
|
Issuance
of common stock to convert Series A-2 preferred stock
|
$2
|
$-
|
|
Issuance
common stock to convert accrued expenses
|
$23,657
|
$-
|
|
Reclassification
between warrant liability and additional
paid-in-capital
|
$1,471,262
|
$-
|
The accompanying notes are an integral part of the consolidated
financial statements
METASTAT, INC.
Notes to Consolidated Financial Statements
February 28, 2018 and February
28, 2017
NOTE 1 – ORGANIZATION, BASIS OF PRESENTATION AND GOING
CONCERN
MetaStat,
Inc. (“we,” “us,” “our,” the
“Company,” or “MetaStat”) is a precision
medicine company dedicated to improving survival of patients with
aggressive cancer. Our therapeutic focus targets a critical
metastatic pathway in solid tumors responsible for driving tumor
resistance and the spread of aggressive cancer. MetaStat’s
goal is to transform the treatment of aggressive cancer into a
manageable disease through targeted therapies that arrest
metastatic progression and improve survival. We are leveraging our
core expertise in understanding the tumor micro-environment and
drivers of the metastatic cascade to develop anti-metastatic
therapeutics pared with companion diagnostics. Our unique approach
is to target tumor cell dissemination and cancer metastasis which
is responsible for over 90% of cancer-related deaths. The Company
was incorporated on March 28, 2007 under the laws of the State of
Nevada.
Basis of Presentation
The accompanying consolidated financial statements include the
accounts of the Company and its wholly-owned subsidiary, MetaStat
Biomedical, Inc., a Delaware corporation and all significant
intercompany balances have been eliminated by
consolidation.
Going Concern
The accompanying financial statements have been prepared on a going
concern basis, which contemplates the realization of assets and the
satisfaction of liabilities in the normal course of
business. The Company has experienced net losses and negative
cash flows from operations since its inception and currently has a
stockholders’ deficit of approximately $1.5 million as of
February 28, 2018. The Company has sustained cumulative
losses of approximately $29.5 million as of February 28, 2018 and
has a negative working capital. The continuation of the
Company as a going concern is dependent upon continued financial
support from its shareholders, the ability of the Company to obtain
necessary equity and/or debt financing to continue operations, and
the attainment of profitable operations. These factors raise
substantial doubt regarding the Company’s ability to continue
as a going concern. Although it is actively working on obtaining
additional funding, the Company cannot make any assurances that
additional financings will be available to it and, if available,
completed on a timely basis, on acceptable terms or at all. If
the Company is unable to complete a debt or equity offering, or
otherwise obtain sufficient financing when and if needed, it would
negatively impact its business and operations and could also lead
to the reduction or suspension of the Company’s operations
and ultimately force the Company to cease operations. These
financial statements do not include any adjustments to the
recoverability and classification of recorded asset amounts and
classification of liabilities that might be necessary should the
Company be unable to continue as a going concern.
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING
POLICIES
The accompanying consolidated financial statements have been
prepared in accordance with the FASB “FASB Accounting
Standard Codification™” or “ASC,” which is
the source of authoritative accounting principles recognized by the
FASB to be applied by nongovernmental entities in the preparation
of financial statements in conformity with generally accepted
accounting principles (“GAAP”) in the United
States.
Use of Estimates
The preparation of financial statements in conformity with GAAP
requires management to make estimates and assumptions that affect
the amounts of assets and liabilities reported and disclosure of
contingent assets and liabilities at the date of the financial
statements and the reported amounts of revenues and expenses during
the reporting period, including contingencies. Accordingly, actual
results could differ from those estimates.
Cash and Cash Equivalents
The Company considers all highly liquid investments with maturities
of three months or less from date of purchase to be cash
equivalents. All cash balances were highly liquid at February 28,
2018 and February 28, 2017.
Concentration of Credit Risk
Financial instruments that potentially expose the Company to
concentrations of credit risk consist primarily of cash and cash
equivalents. The Company primarily maintains its cash balances with
financial institutions in federally insured accounts. The Company
may from time to time have cash in banks in excess of FDIC
insurance limits. The Company has not experienced any losses to
date resulting from this practice. The Company mitigates its risk
by maintaining the majority of its cash and equivalents with high
quality financial institutions.
Debt Instruments
We analyze debt instruments for various features that would
generally require either bifurcation and derivative accounting, or
recognition of a debt discount or premium under authoritative
guidance.
Detachable warrants issued in conjunction with debt are measured at
their relative fair value, if they are determined to be equity
instrument, or their fair value, if they are determined to be
liability instruments, and recorded as a debt
discount. Conversion features that are in the money at
the commitment date constitute a beneficial conversion feature that
is measured at its intrinsic value and recognized as debt discount.
Debt discount is amortized as interest expense over the maturity
period of the debt using the effective interest method. Contingent
beneficial conversion features are recognized when the contingency
has been resolved.
Debt Issuance Costs
Debt issuance costs are recorded as a direct reduction of the
carrying amount of the related debt. Debt issuance costs are
amortized over the maturity period of the related debt instrument
using the effective interest method.
Equipment
Equipment is stated at cost. The cost of equipment is depreciated
over the estimated useful lives of the related assets. Depreciation
is computed using the straight-line method for financial reporting
purposes and accelerated methods for income tax purposes.
Expenditures for major renewals or betterments that extend the
useful lives of equipment are capitalized. Expenditures for
maintenance and repairs are charged to expense as
incurred.
Fair Value Measurements
The Company groups its assets and liabilities measured at fair
value in three levels based on the markets in which the assets and
liabilities are traded and the reliability of the assumptions used
to determine fair value. Fair value is defined as the price that
would be received to sell an asset or paid to transfer a liability
in an orderly transaction between market participants at the
measurement date (an exit price).
Financial instruments with readily available active quoted prices
or for which fair value can be measured from actively quoted prices
generally will have a higher degree of market price observability
and a lesser degree of judgment used in measuring fair
value.
The three levels of the fair value hierarchy are as
follows:
Level 1 – Valuation is based on quoted prices in active
markets for identical assets or liabilities. Valuations are
obtained from readily available pricing sources for market
transactions involving identical assets or
liabilities.
Level 2 – Valuation is based on observable inputs other than
Level 1 prices, such as quoted prices for similar assets or
liabilities; quoted prices in markets that are not active; or other
inputs that are observable or can be corroborated by observable
market data for substantially the full term of the assets or
liabilities.
Level 3 – Valuation is based on unobservable inputs that are
supported by little or no market activity and that are significant
to the fair value of the assets or liabilities. Level 3 assets and
liabilities include financial instruments whose value is determined
using pricing models, some discounted cash flow methodologies, or
similar techniques, as well as instruments for which the
determination of fair value requires significant management
judgment or estimation.
In certain cases, the inputs used to measure fair value may fall
into different levels of the fair value hierarchy. In such cases,
an instrument’s level within the fair value hierarchy is
based on the lowest level of input that is significant to the
overall fair value measurement. The Company’s assessment of
the significance of a particular input to the fair value
measurement in its entirety requires judgment and considers factors
specific to the financial instrument.
The Company recognizes transfers between levels as if the transfers
occurred on the last day of the reporting period.
Income Taxes
Deferred tax assets and liabilities are recognized for the
estimated future tax consequences attributable to net operating
loss carry forwards and temporary differences between the financial
statement carrying amounts of existing assets and liabilities and
their respective tax bases. These assets and liabilities are
measured using enacted tax rates expected to apply to taxable
income in the years in which the temporary differences are expected
to reverse. A valuation allowance is recorded if it is not more
likely than not that some portion or all of the deferred tax assets
will be realized in future periods.
Lease Accounting
Operating leases are accounted for by recording rent expense on a
straight-line basis over the expected life of the lease, commencing
on the date we gain possession of leased property. Tenant
improvement allowances and rent holidays received from landlords
and the effect of any rent escalation clauses are included to
determine the straight-line rent expense over the expected life of
the lease.
Long-lived Assets
Long-lived assets are evaluated for impairment whenever events or
conditions indicate that the carrying value of an asset may not be
recoverable. If the sum of the expected undiscounted cash flows is
less than the carrying value of the related asset or group of
assets, a loss is recognized for the difference between the fair
value and carrying value of the asset or group of assets. There was
no impairment of long-lived assets as of February 28, 2018 and
February 28, 2017.
Net Loss Per Share
Basic net loss per common share is computed based on the weighted
average number of common shares outstanding during the
period. Restricted shares issued with vesting conditions
that have not been met at the end of the period are excluded from
the computation of the weighted average shares. As of February 28,
2018, and February 28, 2017, 11,534 and 11,534, respectively,
unvested restricted shares of common stock were excluded from the
computation of the weighted average shares. For the years ended
February 28, 2018, 225,000 unsettled shares of common stock related
to vested restricted stock units were included in the weighted
average share used for the computation of the basic net loss per
common share.
Diluted net loss per common share is calculated giving effect to
all dilutive potential common shares that were outstanding during
the period. Diluted potential common shares generally consist of
incremental shares issuable upon exercise of stock options and
warrants and conversion of outstanding options and warrants and
shares issuable from convertible securities, as well as nonvested
restricted shares.
In computing diluted loss per share for the years ended February
28, 2018 and February 28, 2017, no effect has been given to the
common shares issuable at the end of the period upon the conversion
or exercise of the following securities as their inclusion would
have been anti-dilutive:
|
|
February 28,
2018
|
February 28,
2017
|
|
Restricted
stock units
|
75,000
|
-
|
|
Preferred
stock
|
4,298,579
|
1,350,109
|
|
Convertible
debt
|
562,082
|
507,946
|
|
Stock
options
|
1,286,770
|
966,474
|
|
Warrants
|
3,060,118
|
2,698,694
|
|
Total
|
9,282,549
|
5,523,223
|
Patent Costs
The Company expenses all costs as incurred in connection with
patent applications (including direct application fees, and the
legal and consulting expenses related to making such applications)
and such costs are included in general and administrative
expenses.
Research and Development Costs
Research and development costs are charged to operations when
incurred and are included in operating expenses. Research and
development costs consist principally of (i) compensation and
related expenses for our employees and consultants that perform our
research activities, (ii) the fees paid to maintain our licenses,
(iii) the payments to third parties for clinical testing and
additional product development including contract research
organizations, (iv) facilities and other expenses, which include
direct and allocated expenses for rent and maintenance of
facilities, and (v) laboratory and other supplies, consumables and
other materials used in research and
development. Research and development costs were
approximately $1.3 million and approximately $1.0 million for the
years ended February 28, 2018 and February 28, 2017, respectively.
During the years ended February 28, 2018 and February 28, 2017, the
Company recorded approximately $664,000 and $309,000, respectively,
of research and development expense reimbursement related to a
research agreement (See Note 11).
In the future, the Company may be required to make advance payments
to vendors for goods or services that will be received in the
future for use in research and development activities. In such
circumstances, the advance payments will be deferred and expensed
when the activity has been performed or when the goods have been
received.
Stock-Based Compensation
We account for share-based payments award issued to employees and
members of our Board of Directors (the “Board”) by
measuring the fair value of the award on the date of grant and
recognizing this fair value as stock-based compensation using a
straight-line method over the requisite service period, generally
the vesting period. For awards issued to non-employees, the
measurement date is the date when the performance is complete or
when the award vests, whichever is the earliest. Accordingly,
non-employee awards are remeasured at each reporting period until
the final measurement date. The fair value of the award is
recognized as stock-based compensation over the requisite service
period, generally the vesting period.
For awards with performance conditions that affect their vesting,
such as the occurrence of certain transactions or the achievement
of certain operating or financial milestones, recognition of fair
value of the award occurs when vesting becomes probable. For awards
with market conditions that affect their vesting, the fair value of
the award is recognized as stock-based compensation over the
requisite service period, generally the vesting
period.
Recently Issued Accounting Pronouncements
In
2016, FASB issued ASU No. 2016-02, Leases (Topic 842) which
supersedes FASB ASC Topic 840, Leases (Topic 840) and provides
principles for the recognition, measurement, presentation and
disclosure of leases for both lessees and lessors. The new standard
requires lessees to apply a dual approach, classifying leases as
either finance or operating leases based on the principle of
whether or not the lease is effectively a financed purchase by the
lessee. This classification will determine whether lease expense is
recognized based on an effective interest method or on a
straight-line basis over the term of the lease, respectively. A
lessee is also required to record a right-of-use asset and a lease
liability for all leases with a term of greater than twelve months
regardless of classification. Leases with a term of twelve months
or less will be accounted for similar to existing guidance for
operating leases. The standard is effective for annual and interim
periods beginning after December 15, 2018, with early adoption
permitted upon issuance. We are currently evaluating the impact of
this guidance on our consolidated financial
statements.
In
2016, the FASB issued ASU No. 2016-09, Improvements to Employee
Share-Based Payment Accounting ("ASU 2016-09"). ASU 2016-09
simplifies certain aspects of accounting for share-based payment
transactions, including the income tax consequences, classification
of awards as either equity or liabilities, an option to recognize
gross stock compensation expense with actual forfeitures recognized
as they occur, as well as certain classifications on the statement
of cash flows. ASU 2016-09 is effective for reporting periods
beginning after December 15, 2016. We adopted this guidance on
March 1, 2017.
In July
2017, the FASB issued ASU No. 2017-11, Earnings Per Share (Topic 260);
Distinguishing Liabilities from
Equity (Topic 480); Derivatives and Hedging (Topic 815):
(Part I) Accounting for Certain Financial Instruments with Down
Round Features. The
amendments in Part I of this ASU change the classification analysis
of certain equity-linked financial instruments (or embedded
features) with down round features. When determining whether
certain financial instruments should be classified as liabilities
or equity instruments, a down round feature no longer precludes
equity classification when assessing whether the instrument is
indexed to an entity’s own stock. As a result, a freestanding
equity-linked financial instrument (or embedded conversion option)
no longer would be accounted for as a derivative liability at fair
value as a result of the existence of a down round feature. For
freestanding equity classified financial instruments, the
amendments require entities that present earnings per share to
recognize the effect of the down round feature when it is
triggered. That effect is treated as a dividend and as a reduction
of income available to common shareholders in basic earnings per
share. For public business entities, the amendments are
effective for fiscal years beginning after December 15, 2018,
including interim periods within those fiscal years. Early
adoption is permitted in any interim or annual period, with any
adjustments reflected as of the beginning of the fiscal year of
adoption. The amendments should be applied retrospectively to
outstanding financial instruments with down round features by means
of either a cumulative-effect adjustment to the consolidated
statement of financial position as of the beginning of the first
fiscal year and interim period of adoption or retrospectively to
each prior reporting period presented in accordance with the
guidance on accounting changes. We are currently in the
process of evaluating the effect this standard will have on our
derivative liabilities and the impact on our consolidated financial
statements.
NOTE 3 – CAPITAL STOCK AND SECURITIES INCENTIVE
PLAN
The Company has authorized 160,000,000 shares of capital stock, par
value $0.0001 per share, of which 150,000,000 are shares of common
stock and 10,000,000 are shares of “blank-check”
preferred stock.
Our Board is empowered, without stockholder approval, to issue
preferred stock with dividend, liquidation, conversion, voting or
other rights, which could adversely affect the voting power or
other rights of the holders of common stock. The preferred stock
could be utilized as a method of discouraging, delaying or
preventing a change in control of us.
Common Stock
The holders of our common stock are entitled to one vote per share.
In addition, the holders of our common stock will be entitled to
receive ratably such dividends, if any, as may be declared by our
Board out of legally available funds; however, the current policy
of our Board is to retain earnings, if any, for operations and
growth. Upon liquidation, dissolution or winding-up, the holders of
our common stock will be entitled to share ratably in all assets
that are legally available for distribution.
Series A Convertible Preferred Stock
Pursuant to the Certificate of Designation of Rights and
Preferences of the Series A Preferred Stock (the “Series A
Certificate of Designation”), the terms of the Series A
Preferred Stock are as follows:
Ranking
The Series A Preferred Stock will rank (i) senior to our common
stock, ii) pari passu with our Series A-2 Preferred Stock (as defined
below) and (iii) junior to our Series B Preferred Stock (as defined
below) with respect to distributions of assets upon the
liquidation, dissolution or winding up of the
Company.
Dividends
The Series A Preferred Stock is not entitled to any
dividends.
Liquidation Rights
In the event of any liquidation, dissolution or winding-up of the
Company, whether voluntary or involuntary, the holders of the
Series A Preferred Stock shall be entitled to receive out of the
assets of the Company, whether such assets are capital or surplus,
for each share of Series A Preferred Stock an amount equal to the
fair market value as determined in good faith by the
Board.
Voluntary Conversion; Anti-Dilution Adjustments
Each fifteen (15) shares of Series A Preferred Stock shall be
convertible into one share of common stock (the “Series A
Conversion Ratio”). The Series A Conversion Ratio is subject
to customary adjustments for issuances of shares of common stock as
a dividend or distribution on shares of the common stock, or
mergers or reorganizations.
Voting Rights
The Series A Preferred Stock has no voting rights. The common stock
into which the Series A Preferred Stock is convertible shall, upon
issuance, have all of the same voting rights as other issued and
outstanding common stock, and none of the rights of the Series A
Preferred Stock.
Series A-2 Convertible Preferred Stock
Pursuant to the Certificate of Designation of Rights and
Preferences of the Series A-2 Convertible Preferred Stock (the
“Series A-2 Preferred Stock” or “Series A-2
Preferred”), the terms of the Series A-2 Preferred Stock are
as follows:
Ranking
The Series A-2 Preferred will rank (i) senior to our common stock,
(ii) pari
passu with our Series A
Preferred Stock, and (iii) junior to our Series B Preferred Stock
(as defined below) with respect to distributions of assets upon the
liquidation, dissolution or winding up of the
Company.
Dividends
The Series A-2 Preferred is not entitled to any
dividends.
Liquidation Rights
In the event of any liquidation, dissolution or winding-up of the
Company, whether voluntary or involuntary, the holders of the
Series A-2 Preferred shall be entitled to receive out of the assets
of the Company, whether such assets are capital or surplus, for
each share of Series A-2 Preferred an amount of cash, securities or
other property to which such holder would be entitled to receive
with respect to each such share of Preferred Stock if such shares
had been converted to common stock immediately prior to such
liquidation, dissolution or winding-up of the Company.
Voluntary Conversion; Anti-Dilution Adjustments
Each share of Series A-2 Preferred shall, at any time, and from
time to time, at the option of the holder, be convertible into ten
(10) shares of common stock (the “Series A-2 Conversion
Ratio”). The Series A-2 Conversion Ratio is subject to
customary adjustments for issuances of shares of common stock as a
dividend or distribution on shares of common stock, or mergers or
reorganizations.
Conversion Restrictions
The holders of the Series A-2 Preferred may not convert their
shares of Series A-2 Preferred into shares of common stock if the
resulting conversion would cause such holder and its affiliates to
beneficially own (as determined in accordance with Section 13(d) of
the Exchange Act, and the rules thereunder) in excess of 4.99% or
9.99% of the common stock outstanding, when aggregated with all
other shares of common stock owned by such holder and its
affiliates at such time; provided, however, that such holder may
elect to waive these conversion restrictions.
Voting Rights
The Series A-2 Preferred has no voting rights. The common stock
into which the Series A-2 Preferred is convertible shall, upon
issuance, have all of the same voting rights as other issued and
outstanding common stock, and none of the rights of the Series A-2
Preferred.
Series B Convertible Preferred Stock
Pursuant to the Certificate of Designation of Rights and
Preferences of the Series B Preferred Stock (the “Series B
Preferred Stock” or “Series B Preferred”), the
terms of the Series B Preferred Stock are as follows:
Ranking
The Series B Preferred Stock ranks senior to our Series A Preferred
Stock, Series A-2 Preferred Stock and common stock with respect to
distributions of assets upon the liquidation, dissolution or
winding up of the Company.
Stated Value
Each shares of Series B Preferred Stock will have a stated value of
$5,500, subject to adjustment for stock splits, combinations and
similar events (the “Stated Value”).
Dividends
Cumulative dividends on the Series B Preferred Stock accrue at the
rate of 8% of the Stated Value per annum, payable quarterly on
March 31, June 30, September 30, and December 31 of each year, from
and after the date of the initial issuance. Dividends
are payable in kind in additional shares of Series B Preferred
Stock valued at the Stated Value or in cash at the sole option of
the Company. At February 28, 2018 and February 28, 2017, the
dividend payable to the holders of the Series B Preferred Stock
amounted to approximately $42,000 and approximately $16,000,
respectively. During the year ended February 28, 2018 and February
28, 2017, the Company issued 13.0520 and 34.5085 shares of Series B
Preferred Stock, respectively, for payment of dividends amounting
to approximately $72,000 and approximately $192,000,
respectively.
Liquidation Rights
If the Company voluntarily or involuntarily liquidates, dissolves
or winds up its affairs, each holder of the Series B Preferred
Stock will be entitled to receive out of the Company’s assets
available for distribution to stockholders, after satisfaction of
liabilities to creditors, if any, but before any distribution of
assets is made on the Series A Preferred Stock or common stock or
any of the Company’s shares of stock ranking junior as to
such a distribution to the Series B Preferred Stock, a liquidating
distribution in the amount of the Stated Value of all such
holder’s Series B Preferred Stock plus all accrued and unpaid
dividends thereon. At February 28, 2018 and February 28, 2017, the
value of the liquidation preference of the Series B Preferred
stocks aggregated to approximately $1.29 million and approximately
$1.19 million, respectively.
Conversion; Anti-Dilution Adjustments
Each share of Series B Preferred Stock will be convertible at the
holder’s option into common stock in an amount equal to the
Stated Value plus accrued and unpaid dividends thereon through the
conversion date divided by the then applicable conversion price.
The initial conversion price was $8.25 per share (the “Series
B Conversion Price”) and is subject to customary adjustments
for issuances of shares of common stock as a dividend or
distribution on shares of common stock, or mergers or
reorganizations, as well as “full ratchet”
anti-dilution adjustments for future issuances of other Company
securities (subject to certain standard carve-outs) at prices less
than the applicable Series B Conversion Price.
The issuance of shares of common stock pursuant to the 2016 Unit
Private Placement (as defined in Note 4) triggered the full ratchet
anti-dilution price protection provision of the Series B Preferred
Stock. Accordingly, the Series B Conversion Price was adjusted from
$8.25 to $2.00 per share. The issuance of shares of common
stock pursuant to the 2017 Common Stock Private Placement (as
defined in Note 4) triggered
the full ratchet anti-dilution price protection provision of the
Series B Preferred Stock. Accordingly, the Series B Conversion
Price was adjusted from $2.00 to $0.83 per share. See Note
4 for the accounting treatment
of the conversion price adjustment.
The Series B Preferred Stock is subject to automatic conversion
(the “Mandatory Conversion”) at such time when the
Company’s common stock has been listed on a national stock
exchange such as the NASDAQ, New York Stock Exchange or NYSE MKT;
provided, that, on the Mandatory Conversion date, a registration
statement providing for the resale of the shares of common stock
underlying the Series B Preferred Stock is effective. In the event
of a Mandatory Conversion, each share of Series B Preferred Stock
will convert into the number of shares of common stock equal to the
Stated Value plus accrued and unpaid dividends divided by the
applicable Series B Conversion Price.
Voting Rights
The holders of the Series B Preferred Stock shall be entitled to
the number of votes equal to the number of shares of common stock
into which such Series B Preferred Stock could be converted for
purposes of determining the shares entitled to vote at any regular,
annual or special meeting of stockholders of the Company, and shall
have voting rights and powers equal to the voting rights and powers
of the common stock (voting together with the common stock as a
single class).
Most Favored Nation
For a period of up to 30 months after March 31, 2015, if the
Company issues any New Securities (as defined below) in a private
placement or public offering (a “Subsequent
Financing”), the holders of Series B Preferred Stock may
exchange all of the Series B Preferred Stock at their Stated Value
plus all Series A Warrants (as defined below) issued to the
Series B Preferred Stock investors in the Series B Private
Placement for the securities issued in the Subsequent Financing on
the same terms of such Subsequent Financing. This right
expires upon the earlier of (i) September 30, 2017 and (ii) the
consummation of a bona fide underwritten public offering in which
the Company receives aggregate gross proceeds of at least $5.0
million. ”New Securities” means shares of the
common stock, any other securities, options, warrants or other
rights where upon exercise or conversion the purchaser or recipient
receives shares of the common stock, or other securities with
similar rights to the common stock, subject to certain standard
carve-outs. This right expired on September 30, 2017 pursuant to
the Certificate of Designation of Rights and Preferences of the
Series B Preferred Stock.
2012 Incentive Plan
Our 2012 Incentive Plan, which is administrated by the compensation
committee of the Board, reserves shares of common stock available
for issuance that the Company
may grant to employees, non-employee directors and consultants,
equity incentives in the form of, among other, stock options,
restricted stock, restricted stock units and stock appreciation
rights. On June 22, 2015, our stockholders approved amending our
2012 Incentive Plan to increase the number of authorized shares of
common stock reserved for issuance under the 2012 Incentive Plan to
a number not to exceed fifteen percent (15%) of the issued and
outstanding shares of common stock on an as converted primary basis
(the “As Converted Primary Shares”) on a rolling basis.
For calculation purposes, the As Converted Primary Shares shall
include all shares of common stock and all shares of common stock
issuable upon the conversion of outstanding preferred stock and
other convertible securities but shall not include any shares of
common stock issuable upon the exercise of options, warrants and
other convertible securities issued pursuant to the 2012 Incentive
Plan. The number of authorized shares of common stock reserved for
issuance under the 2012 Incentive Plan shall automatically be
increased concurrently with the Company’s issuance of fully
paid and non- assessable shares of As Converted Primary Shares.
Shares shall be deemed to have been issued under the 2012 Incentive
Plan solely to the extent actually issued and delivered pursuant to
an award under the 2012 Incentive Plan. As of February 28,
2018, there are an aggregate of 1,610,707 total shares available
under the 2012 Incentive Plan, of which 785,514 are issued and
outstanding and 825,193 shares are available for potential
issuances. The Company may issue shares outside of the 2012
Incentive Plan.
NOTE 4 – EQUITY ISSUANCES
Common stock financing – the 2016 Unit Private
Placement
During the year ended February 28, 2017, the Company entered into a
subscription agreement pursuant to a private placement (the
“2016 Unit Private Placement”) with a number of
accredited investors pursuant to which the Company issued units for
an offering price of $10,000 per unit, with each unit consisting of
(i) 5,000 shares of its common stock, and (ii) five-year warrants
(the “Unit Warrants”) to purchase 2,500 shares of
common stock at an exercise price of $3.00 per
share.
During the year ended February 28, 2017, the Company issued
an aggregate of 49.5 units consisting
of an aggregate of 247,500 shares of common stock and 123,750 Unit
Warrants for an aggregate purchase price of $495,000. After
deducting placement agent fees and other offering expenses,
including legal expenses, net proceeds amounted to approximately
$390,000. Additionally, the Company issued an aggregate of 24,750
placement agent warrants in substantially the same form as the Unit
Warrants.
Registration Rights Agreement
Pursuant to a registration rights agreement entered into by the
parties, the Company agreed to file a registration statement with
the SEC providing for the resale of the shares of common stock and
the shares of common stock underlying the Unit Warrants issued
pursuant to the 2016 Unit Private Placement on or before the date
which is forty-five (45) days after the date of the final closing
of the 2016 Unit Private Placement. The Company will use its
commercially reasonable efforts to cause the registration statement
to become effective within one hundred fifty (150) days from the
filing date. The Company has received a waiver from a majority of
the 2016 Unit Private Placement investors extending the filing date
of the registration statement to no later than December 15,
2016. The Company filed the
Registration Statement on Form S-1 with the SEC on December 14,
2016.
Most Favored Nation Exchange – the MFN Exchange
On July 12, 2016, the Company and one Series B Preferred Stock
shareholder (the “Exchange Purchaser”) entered into an
exchange agreement effective July 1, 2016 (the “Exchange
Agreement”) whereby the Exchange Purchaser elected to
exercise their Most Favored Nation exchange right into the
securities offered pursuant to the 2016 Unit Private Placement (the
“MFN Exchange”). Accordingly, the Exchange
Purchaser tendered all of their 19.4837 shares of Series B
Preferred Stock and approximately $2,000 of accrued and unpaid
dividends for an aggregate exchange amount of approximately
$109,000, plus 9,000 Series A Warrants with an exercise price of
$10.50 per share originally issued in connection with the Series B
Private Placement for an aggregate of 54,652 shares of common stock
and Unit Warrants to purchase 27,326 shares of common stock at an
exercise price of $3.00 per share. Additionally, the parties
entered into a joinder agreement, and the Exchange Purchaser was
granted all rights and benefits under the 2016 Unit Private
Placement financing agreements.
The Company analyzed and determined that the MFN Exchange is a
contingent beneficial conversion feature that should be recognized
upon the occurrence of the contingent event based on its intrinsic
value at the commitment date. Since the Company had fully
recognized all allocated proceeds of the Series B Preferred Stock
in previously recognized beneficial conversion features, no
beneficial conversion was recognized upon the exchange of the
Series B Preferred Stock in the MFN Exchange.
For the year ended February 28, 2017, the Company recorded a
non-cash deemed dividend to Additional Paid-in Capital of
approximately $29,000, in connection with the MFN Exchange equal to
the excess fair value of the shares of common stock and Unit
Warrants received over the carrying value of the exchanged shares
of Series B Preferred and Series A Warrants.
Deemed Dividend due to Conversion Price Adjustment
During the year ended February 28, 2017, as a result of the
adjustment of the Series B Conversion Price from $8.25 to $2.00 per
share due to the 2016 Unit Private Placement, the Company recorded
a non-cash deemed dividend, amounting to approximately
$708,000. The expense was measured at the intrinsic value of
the beneficial conversion feature for each issuance of Series B
Preferred Stock in the Series B Preferred private placement and was
limited to the amount of Series B Preferred Stock allocated
proceeds less previously recognized beneficial conversion
features.
Common stock financing – Additional 2016 Unit Private
Placement
During the year ended February 28, 2017, the Company entered into a
subscription agreement (the “Additional 2016 Unit
Subscription Agreement”) pursuant to a private placement (the
“Additional 2016 Unit Private Placement”) whereby the
Company issued units for an offering price of $10,000 per unit,
with each unit consisting of (i) 5,000 shares of its common stock
at an effective price of $2.00 per share (the “Effective
Price”), and (ii) five-year warrants (the “Additional
Unit Warrants”) to purchase 2,500 shares of common stock at
an exercise price of $3.00 per share. Pursuant to the Additional
2016 Unit Subscription Agreement, for the benefit of certain
investors that would be deemed to have beneficial ownership in
excess of 4.99% or 9.99%, the Company issued shares of Series A-2
Preferred Stock in lieu of issuing shares of common stock to such
investors.
Pursuant to the Additional 2016 Unit Subscription Agreement, for a
period of one hundred eighty (180) days following the final closing
of the Additional 2016 Unit Private Placement , the investors had
“full-ratchet” anti-dilution price protection (the
“Price Protection”) based on certain issuances by the
Company of common stock or securities convertible into shares of
common stock at an effective price per share less than the
Effective Price (a "Down-round Issuance"), whereby the Company
would be required to issue the investors additional shares of
common stock and Additional Unit Warrants. The Price Protection
provision expired in April 2017 without the Company issuing any
additional shares of common stock and Additional Unit
Warrants.
During the year ended February 28, 2017, the Company issued an
aggregate of 260.25 units consisting of an aggregate of 818,250
shares of common stock, 48,300 shares of Series A-2 Preferred Stock
convertible into 483,000 shares of common stock, and Additional
Unit Warrants to purchase 650,625 shares of common stock, for an
aggregate purchase price of approximately $2.6 million. After
deducting placement agent fees and other offering expenses,
including legal expenses, net proceeds amounted to approximately
$2.4 million.
Additionally, in connection with the Additional 2016 Unit Private
Placement, the Company issued placement agent warrants to purchase
an aggregate of 108,958 shares of common stock in substantially the
same form as the Additional Unit Warrants but without the Price
Protection provision.
Exchange of Payables – the Company Payable
Exchange
During the year ended February 28, 2017, the Company entered
into the Additional 2016 Unit
Subscription Agreement with certain accredited vendors of the
Company in connection with the
exchange (the “Company Payable Exchange”) of an
aggregate of $65,000 of accounts payable into the Additional 2016 Unit Private Placement.
Pursuant to the Company Payable Exchange, the Company issued an aggregate of 6.5 units
consisting of an aggregate of 32,500 shares of common stock, and
Additional Unit Warrants to purchase 16,250 shares of common stock,
for the cancellation of $65,000 of accounts payable in the
aggregate. As a result of the Company Payable Exchange, the Company
recognized a loss of approximately $62,000.
Exchange of Promissory Note – the Promissory Note
Exchange
During the year ended February 28, 2017, the Company entered
into the Additional 2016 Unit
Subscription Agreement with the holder of the Promissory Note (the
“Noteholder”) in connection with the exchange (the
“Promissory Note Exchange”) of $600,000 principal
amount of Promissory Notes plus $48,000 of accrued and unpaid
interest into the Additional 2016 Unit
Private Placement. In connection with the Promissory Note
Exchange, the Company issued
64.8 units consisting of 230,000 shares of common stock, 9,400
shares of Series A-2 Preferred, convertible into 94,000 shares of common
stock, and Additional Unit Warrants to
purchase 162,000 shares of common stock in exchange for the
cancellation of $600,000 principal amount plus $48,000 of accrued
and unpaid interest of the Promissory Note (See Note 7).
Exchange of OID Notes – the OID Note Exchange
During the year ended February 28, 2017, the Company entered
into the Additional 2016 Unit
Subscription Agreement with certain holders of OID Notes (the
“OID Noteholders”) in connection with the exchange (the
“OID Note Exchange”) of an aggregate of $553,000
principal amount of OID Notes (the “OID Exchange
Amount”) into the Additional
2016 Unit Private Placement. In connection with the OID Note
Exchange, the Company issued an
aggregate of 55.3 units consisting of 210,500 shares of
common stock, 6,600 shares of
Series A-2 Preferred, convertible into
66,000 shares of common
stock and Additional Unit
Warrants to purchase 138,250 shares of common stock in
exchange for the cancellation of $553,000 of OID Notes (See Note 7).
Most Favored Nation Exchange – the Additional MFN
Exchange
During the year ended February 28, 2017, the Company and certain
Series B Preferred Stockholders (the “Additional Exchange
Purchasers”) entered into exchange agreements (the
“Exchange Agreements”) whereby the Additional Exchange
Purchasers elected to exercise their Most Favored Nation exchange
rights into the securities offered pursuant to the Additional 2016
Unit Private Placement (the “Additional MFN
Exchange”). Accordingly, the Additional Exchange
Purchasers tendered all of their 460.6480 shares of Series B
Preferred Stock and approximately $68,000 of accrued and unpaid
dividends for an aggregate exchange amount of approximately $2.6
million, plus 208,027 Series A Warrants with an exercise price of
$10.50 per share originally issued in connection with the Series B
Private Placement for an aggregate of 1,238,339 shares of common
stock, 6,240.8 shares of Series A-2 Preferred Stock convertible
into 62,408 shares of common stock, and Additional Unit Warrants to
purchase 650,381 shares of common stock. Additionally, the parties
entered into a joinder agreement, and the Exchange Purchasers were
granted all rights and benefits under the Additional 2016 Unit
Private Placement financing agreements.
The Company analyzed and determined that the Additional MFN
Exchange is a contingent beneficial conversion feature that should
be recognized upon the occurrence of the contingent event based on
its intrinsic value at the commitment date. Since the Company had
fully recognized all allocated proceeds of the Series B Preferred
Stock in previously recognized beneficial conversion features, no
beneficial conversion was recognized upon the exchange of the
Series B Preferred Stock in the Additional MFN
Exchange.
For the year ended February 28, 2017, the Company recorded a
non-cash deemed dividend to Additional Paid-in Capital of
approximately $2.3 million in connection with the Additional MFN
Exchange equal to the excess fair value of the shares of common
stocks, shares of Series A-2 Preferred Stock and Additional Unit
Warrants received over the carrying value of the shares of Series B
Preferred Stock and exchanged Series A Warrants.
Accounting for the Price Protection Provision
The Company analyzed the Price Protection provision for embedded
derivatives that require bifurcation. The Company evaluated the
Price Protection provision for both the issuance of additional
shares of common stock and additional warrants in connection with a
down-round issuance in accordance with ASC 480 and ASC 815. In
connection with the potential issuance of additional shares of
common stock, the Company concluded that since the embedded
down-round feature is within the equity host contract, the embedded
Price Protection provision would be considered clearly and closely
related to the equity host under ASC 815-15-25-1(a) and that the
Price Protection provision should not be bifurcated. In connection
with the potential issuance of additional warrants, the Company
concluded that the freestanding Additional Unit Warrants were not
indexed to the Company’s common stock within the scope of ASC
815-40 and therefore were initially bifurcated and measured at fair
value and recorded as a derivative liability in the Consolidated
Balance Sheet. The derivative liability was measured at fair
value on an ongoing basis, with changes in fair value recognized in
the statement of operations until the Price Protection provision
lapses.
Registration Rights Agreement
Pursuant to a registration rights agreement entered into by the
parties, the Company agreed to file a registration statement with
the SEC providing for the resale of the shares of common stock and
the shares of common stock underlying the Additional Unit Warrants
issued pursuant to the Additional 2017 Unit Private Placement on or
before the date which is forty-five (45) days after the date of the
final closing of the Additional 2017 Unit Private Placement, which
occurred on October 30, 2016. The Company will use its
commercially reasonable efforts to cause the registration statement
to become effective within one hundred fifty (150) days from the
filing date. The Company filed the Registration Statement on Form
S-1 with the SEC on December 14, 2016.
Common stock financing – the 2017 Common Stock Private
Placement
During the year ended February 28, 2018, the Company completed
closings of a private placement (the “2017 Common Stock
Private Placement”) with existing and new institutional and
accredited investors pursuant to which the Company issued (i) an
aggregate of 811,158 shares of common stock, (ii) 229,363.2 shares
of Series A-2 Preferred Stock convertible into 2,293,632 shares of common stock
and (iii) reduced the exercise price of outstanding warrants
to purchase 536,434 shares of common
stock from $3.00 to $2.00 per share (see Note 6), for
an aggregate purchase price of
approximately $2.57 million, including the conversion of
approximately $22,000 of compensation payable to our Chief
Executive Officer. After deducting placement agent fees and other
offering expenses, the Company received net proceeds of
approximately $2.31 million. Additionally, the Company issued the
placement agent five-year warrants to purchase an aggregate of
162,486 shares of common stock with an exercise price equal to
$1.27 per share, and a cashless exercise provision. The effective
purchase price of the 2017 Common Stock Private Placement was $0.83
per share.
Conversion Price Adjustment
During the year ended February 28, 2018, as a result of the 2017
Common Stock Private Placement, the Series B Conversion Price was
adjusted from $2.00 to $0.83 per share, the effective price per
share of the 2017 Common Stock Private Placement. No non-cash
deemed dividend was recorded to recognize any contingent beneficial
conversion feature related to this conversion price change as all
proceeds of the Series B Preferred have already been offset by
previously recognized beneficial conversion features.
Issuances of common stock for services
During the year ended February 28, 2017, the Company issued an
aggregate of 25,000 shares of common stock to a consultant for
services that vested over a two-month term and to settle $32,000 of
accounts payable. The fair value of the shares amounted to
approximately $46,000 on the grant date.
During the year ended February 28, 2018, , the Company issued an
aggregate of 100,000 shares of common stock to members of its Board
that vested immediately. The fair value of the shares amounted to
approximately $130,000 on the grant date, which was recognized into
general and administrative expense during the year ended February
28, 2018.
During the year ended February 28, 2018 and February 28, 2017, the
Company recognized approximately $130,000 and approximately
$14,000, respectively, of share-based compensation related to
common stock issued for services, all of which was recognized into
general and administrative expense.
Issuances of restricted stock units
During the year ended February 28, 2018, the Company issued 200,000
restricted stock units to its President and Chief Executive
Officer. Each restricted stock unit represents a right to receive,
at settlement, one share of common stock. These restricted stock
units were granted on October 11, 2017 and were vested immediately.
These restricted stock units settle on the October 11, 2020, unless
accelerated due to departure from the Company or a change of
control. The fair value of these restricted stock units amounted to
approximately $178,000 on the grant date. The Company recognized
approximately $89,000 into general and administrative expense
during the year ended February 28, 2018, and approximately $89,000
into research and development expense during the year ended
February 28, 2018.
During the year ended February 28, 2018, the Company issued an
aggregate of 100,000 restricted stock units to members of the
Board. Each restricted stock unit represents a contingent right to
receive, at settlement, one share of common stock. These restricted
stock units were granted on October 11, 2017 and will vest in equal
quarterly installments for active service over a twelve-month
period. These restricted stock units settle on the October 11,
2020, unless accelerated due to departure from the Company or a
change of control. The fair value of these restricted stock units
amounted to approximately $89,000 on the grant date. The Company
recognized approximately $59,000 into general and administrative
expense during the year ended February 28, 2018.
NOTE 5 – STOCK OPTIONS
During the year ended February 28, 2017, the Company issued options
to purchase 50,000 shares of common stock at $2.19 per share to a
non-executive member of its Board. These 50,000 options vest in
three equal installments on each of May 26, 2017, May 26, 2018, and
May 26, 2019 and expire on May 26, 2026. These options had a total
fair value of approximately $87,000 as calculated using the
Black-Scholes model.
During the year ended February 28, 2017, the Company issued options
to purchase 50,000 shares of common stock at $2.19 per share to a
non-executive member of its Board for performing other services.
These 50,000 options vest upon achieving a certain milestone and
expire on May 26, 2026. These options will be measured and
recognized when vesting becomes probable. The fair value will be
measured using a Black-Scholes model.
During the year ended February 28, 2017, the Company issued options
to purchase an aggregate of 440,000 shares of common stock at an
exercise price of $2.00 per share to members of its management
team. These options expire on July 7, 2026. These options had a
grant date fair value of approximately $622,000 as calculated using
the Black-Scholes model. 73,333 of these options vested immediately
and 146,667 of these options vest in equal monthly installments
over a twenty-four-month period. 220,000 of these options are
subject to certain performance milestone-based vesting. The Company
has not recognized any stock-based compensation for the options
with performance-vesting conditions and expects to recognize the
compensation expense when vesting become probable, which has not
yet occurred.
During the year ended February 28, 2017, the Company issued options
to purchase an aggregate of 100,000 shares of common stock at an
exercise price of $2.00 per share to a non-executive member of its
Board. These options expire on July 7, 2026. These options had a
total fair value of approximately $143,000 as calculated using the
Black-Scholes model. 33,333 of these options vested immediately and
66,667 of these options vest in equal monthly installments over a
twenty-four-month period.
During the year ended February 28, 2017, the Company issued options
to purchase an aggregate of 240,000 shares of common stock at an
exercise price of $2.00 per share to consultants. These options
expire on July 7, 2026. 33,333 of these options, with an aggregate
fair value of approximately $57,000, vest on the first anniversary
date and then 66,667 of these options vest in equal monthly
installments over a twenty-four-month period. 140,000 of these
options are subject to certain milestone-based vesting and the
Company will measure the fair value of these options with vesting
contingent on achieving certain performance-based milestones and
recognize the compensation expense when vesting becomes probable.
The fair value will be measured using a Black-Scholes
model.
During the year ended February 28, 2017, the Company and a member
of its Board voluntarily cancelled options to purchase an aggregate
of 100,000 shares of common stock at an exercise price of $2.00 per
share without replacement. The Company recognized approximately
$69,000 of compensation expense related to the cancellation of
these options.
During the year ended February 28, 2017, the Company issued options
to purchase an aggregate of 21,000 shares of common stock at an
exercise price of $3.00 per share to employees. These options
expire between on November 21, 2026 and December 1, 2026. These
options had a grant date fair value of approximately $29,000 as
calculated using the Black-Scholes model. 7,000 of these options
vest one year following issuance and then 14,000 of these options
vest in equal monthly installments over the following
twenty-four-month period.
During the year ended February 28, 2017, the Company issued options
to purchase 100,000 shares of common stock at $3.00 per share to a
consultant. These options expire on January 13, 2027 and vest upon
achieving certain performance-based milestones. The Company will
measure the fair value of these options with vesting contingent on
achieving certain performance-based milestones and recognize the
compensation expense when vesting becomes probable. The fair
value will be measured using a Black-Scholes model.
For the year ended February 28, 2017, the Company recognized
approximately $580,000 of compensation expense related to stock
options, of which approximately $495,000 was recognized in general
and administrative expenses and approximately $85,000 in research
and development expenses.
During the year ended February 28, 2018, the Company issued options
to purchase an aggregate 55,000 shares of common stock at $3.00 per
share to its President and Chief Executive Officer and a member of
its management team. These options expire on April 4, 2027. 18,334
of these options vest on the first anniversary date of April 4,
2018, and then 36,666 of these options vest in equal monthly
installments over a twenty-four-month period. These options had a
total fair value of approximately $60,000 as calculated using the
Black-Scholes model.
During the year ended February 28, 2018, an aggregate of 39,999
unvested options to purchase shares of common stock at $8.25 per
share to certain members of the Company’s Board were
terminated upon resignation from the board. The Company recognized
a credit of approximately $146,000 for the true-up of forfeitures
related to these unvested options during the year ended February
28, 2018.
During the year ended February 28, 2018, the Company issued options
to purchase an aggregate of 21,000 shares of common stock at $0.89
per share to certain non-executive employees. These options expire
on October 11, 2027. An aggregate of 7,000 of these options
initially vest on dates between February 1, 2018 and November 9,
2018, and then an aggregate of 14,000 of these options vest in
equal monthly installments over a twenty-four-month period
following the vesting of the first tranche. These options had a
total fair value of approximately $17,000 as calculated using the
Black-Scholes model.
During the year ended February 28, 2018, the Company issued options
to purchase an aggregate of 60,000 shares of common stock at $0.89
per share to consultants. These options expire on October 10, 2027.
20,000 of these options vested immediately and 40,000 of these
options vest in quarterly installments commencing on December 1,
2017 for active service. These options had a total grant-date fair
value of approximately $52,000 as calculated using the
Black-Scholes model. The Company also issued 50,000 options to
purchase common stock at $0.89 per share to a consultant that are
subject to certain milestone-based vesting and will measure the
fair value of these options with vesting contingent on achieving
certain performance-based milestones and recognize the compensation
expense when vesting becomes probable.
During the year ended February 28, 2018, the Company issued options
to purchase 100,000 shares of common stock at $0.93 per share to a
consultant. These options expire on January 13, 2028. 50,000 of
these options, with an aggregate grant-date fair value of
approximately $59,000, vest in equal monthly installments over a
twelve-month period following issuance. 50,000 of these options are
subject to certain milestone-based vesting and the Company will
measure the fair value of these options with vesting contingent on
achieving certain performance-based milestones and recognize the
compensation expense when vesting becomes probable. The fair value
will be measured using a Black-Scholes model.
During the year ended February 28, 2018, the Company issued options
to purchase an aggregate of 144,296 shares of common stock at $0.93
per share to certain executive and non-executive employees. These
options expire on January 11, 2028. An aggregate of 48,099 of these
options initially vest on January 12, 2019, and then an aggregate
of 96,197 of these options vest in equal monthly installments over
a twenty-four-month period following the vesting of the first
tranche. These options had a total fair value of approximately
$120,000 as calculated using the Black-Scholes model.
For the year ended February 28, 2018, the Company recognized
approximately $38,000 of compensation expense related to the
vesting of 33,333 stock options granted previously to a consultant
upon achieving a milestone.
For the year ended February 28, 2018, the Company recognized
approximately $121,000 of compensation expense related to stock
options, of which a credit of approximately $41,000 was recognized
in general and administrative expenses and approximately $162,000
in research and development expenses.
The inputs to the Black-Scholes model used to value the stock
options granted during the year ended February 28, 2018 and
February 28, 2017 are as follows:
|
|
February 28,
|
February
28,
|
|
|
2018
|
2017
|
|
Expected
volatility
|
129% - 139%
|
99% - 133%
|
|
Expected
dividend yield
|
0.0%
|
0.0%
|
|
Risk-free
interest rate
|
1.9% - 2.6%
|
1.0% - 1.9%
|
|
Expected
Term
|
7.23
years
|
6.31
years
|
The following table summarizes common stock options issued and
outstanding:
|
|
|
Weighted
|
|
Weighted
|
|
|
|
average exercise
|
Aggregate
|
average remaining
|
|
|
Options
|
price
|
intrinsic value
|
contractual life (years)
|
|
Outstanding
at February 29, 2016
|
426,976
|
$14.45
|
$-
|
7.98
|
|
Granted:
|
1,001,000
|
$2.14
|
$-
|
-
|
|
Expired/
exercised and forfeited:
|
(461,502)
|
$6.05
|
$-
|
-
|
|
Outstanding at February 28, 2017
|
966,474
|
$5.71
|
$-
|
8.87
|
|
Granted:
|
430,296
|
$1.18
|
$-
|
-
|
|
Expired/
exercised and forfeited:
|
(110,000)
|
$5.50
|
$-
|
-
|
|
Outstanding and expected to vest at February 28, 2018
|
1,286,770
|
$4.22
|
$117,829
|
8.50
|
|
Exercisable
at February 28, 2018
|
463,637
|
$7.75
|
$12,130
|
7.68
|
The following table breaks down exercisable and unexercisable
common stock options by exercise price as of February 28,
2018:
|
Exercisable
|
Unexercisable
|
||||
|
Number of Options
|
Exercise Price
|
Weighted Average Remaining Life (years)
|
Number of Options
|
Exercise Price
|
Weighted Average Remaining Life (years)
|
|
32,000
|
$0.89
|
9.62
|
99,000
|
$0.89
|
9.62
|
|
4,167
|
$0.93
|
9.87
|
240,129
|
$0.93
|
9.87
|
|
215,554
|
$2.00
|
8.36
|
264,446
|
$2.00
|
8.36
|
|
16,668
|
$2.19
|
8.24
|
33,332
|
$2.19
|
8.24
|
|
41,995
|
$3.00
|
8.85
|
134,005
|
$3.00
|
8.96
|
|
30,000
|
$3.55
|
7.94
|
-
|
$3.55
|
-
|
|
1,068
|
$8.10
|
6.92
|
-
|
$8.10
|
-
|
|
20,000
|
$8.25
|
7.30
|
40,000
|
$8.25
|
7.30
|
|
41,434
|
$10.20
|
3.86
|
-
|
$10.20
|
-
|
|
3,334
|
$11.25
|
7.22
|
3,333
|
$11.25
|
7.22
|
|
11,112
|
$16.50
|
6.63
|
8,888
|
$16.50
|
6.63
|
|
8,068
|
$22.50
|
6.92
|
-
|
$22.50
|
-
|
|
38,237
|
$48.75
|
5.10
|
-
|
$48.75
|
0
|
|
463,637
|
$7.75
|
7.68
|
823,133
|
$2.22
|
8.97
|
As of February 28, 2018, we had approximately $253,000 of
unrecognized compensation related to employee and consultant stock
options that are expected to vest over a weighted average period of
0.63 years and, approximately $500,000 of unrecognized compensation
related to employee stock options whose recognition is dependent on
certain milestones to be achieved. Additionally, there
were 205,000 stock options with a performance vesting
condition that were granted to consultants which will be measured
and recognized when vesting becomes probable.
NOTE
6 – WARRANTS
For the year ended February 28, 2017, the Company issued warrants
to purchase an aggregate of 9,092 shares of common stock in
connection with the issuance of the OID Notes pursuant to the March
2016 OID Note Purchase Agreements dated between March 3 and 15,
2016, referenced in Note 7. These warrants vested immediately, were
initially exercisable at $8.25 per share and expire between March 3
and 15, 2021. These warrants contained an anti-dilution price
protection provision, which required the warrants to be recorded as
derivative warrant liability. In connection with the issuances of
common stock pursuant to the 2016 Unit Private Placement, the
exercise price of these warrants was adjusted to $2.00 per share.
Such clause will lapse upon completion of a Qualified Offering, as
defined in the warrant agreement. These warrants were recorded as a
debt discount based on their fair value.
For the year ended February 28, 2017, the Company issued Unit
Warrants to purchase an aggregate of 175,826 shares of common
stock to investors in connection with the 2016 Unit Private
Placement and MFN Exchange referenced in Note 4. These Unit
Warrants vested immediately, are exercisable at $3.00 per share and
expire between May 26, 2021 and June 7, 2021. These Unit Warrants
do not contain any provision that would require liability
treatment, therefore they were classified as equity in the
Consolidated Balance Sheet. Additionally, in connection with the
MFN Exchange, the Company cancelled Series A Warrants to purchase
an aggregate of 9,000 shares of common stock that were
exercisable at $10.50 per share and originally issued in connection
with the Series B Private Placement.
For the year ended February 28, 2017, the Company issued warrants
to purchase an aggregate of 45,459 shares of common stock in
connection with the OID Note Amendments referenced in Note 7. These
warrants vested immediately, are exercisable at $2.00 per share and
expire between August 11, 2021 and August 18, 2021. The fair value
of these warrants was determined to be approximately $44,000, as
calculated using the Black-Scholes model and were recorded as a
debt discount based on their fair value.
For the year ended February 28, 2017, the Company issued Additional
Unit Warrants to purchase an aggregate 1,617,506 shares of common
stock in connection with the Additional 2016 Unit Private Placement
including Company Payable Exchange, Promissory Note Exchange, OID
Note Exchange, and Additional MFN Exchange referenced in Note 4.
These Additional Unit Warrants vested immediately, are exercisable
at $3.00 per share and expire between August 30, 2021 and October
29, 2021. As discussed in Note 4, due to the Price Protection
Provision, these Additional Unit Warrants were initially classified
as a derivative liability and measured at fair value.
Additionally, in connection with the
Additional MFN Exchange, the Company cancelled Series A Warrants to
purchase an aggregate of 208,027 shares of common stock that
were exercisable at $10.50 per share and originally issued in
connection with the issuance of Series B Preferred
Stock.
For the year ended February 28, 2017, in connection with the
Additional 2016 Unit Private Placement, the Company issued
placement agent warrants to purchase an aggregate of 108,958 shares
of common stock. These placement agent warrants were issued
between August 30, 2016 and October 28, 2016, vested immediately,
are exercisable at $3.00 per share and expire between August 29,
2021 and October 27, 2021. The fair value of these warrants was
determined to be approximately $259,000, as calculated using the
Black-Scholes model. Weighted-average assumptions used in the
Black-Scholes model included: (1) a discount rate of 1.25%; (2) an
expected term of 5.0 years; (3) an expected volatility of 133% and
(4) zero expected dividends.
For the year ended February 28, 2018, the Company issued warrants
to purchase an aggregate of 75,000 shares of common stock to a
consultant for advisory services. These warrants are exercisable at
$3.00 per share and expire between March 2022 and August 2022.
These warrants vested immediately. The fair value of these warrants
was determined to be approximately $62,000, as calculated using the
Black-Scholes model. Average assumptions used in the Black-Scholes
model included: (1) a discount rate of 1.82%; (2) an expected term
of 5.0 years; (3) an expected volatility of 131%; and (4) zero
expected dividends. For the year ended February 28, 2018, the
Company recognized approximately $62,000 of stock-based
compensation for these warrants.
For the year ended February 28, 2018, the Company reclassified
approximately $1.5 million of derivative warrant liability to
equity in connection with the lapse of a Price Protection provision
referenced in Note 4, that had resulted in these instruments being
classified as a derivative warrant liability at issuance. The fair
value of these warrants at the reclassification date was determined
to be approximately $1.5 million, as calculated using the
Black-Scholes model. Average assumptions used in the Black-Scholes
model included: (1) a discount rate of 1.81%; (2) an expected term
of 4.46 years; (3) an expected volatility of 124%; and (4) zero
expected dividends.
For the year ended February 28, 2018, in connection with the 2017
Common Stock Private Placement, the Company reduced the exercise
price of outstanding warrants to purchase 536,434
shares of common stock from
$3.00 to $2.00 per share. The Company recorded a non-cash deemed
dividend of approximately $31,000 equal to difference in the
aggregated fair value of these
warrants on the measurement dates as calculated using the
Black-Scholes model.
For the year ended February 28, 2018, in connection with the 2017
Common Stock Private Placement, the Company issued placement agent
warrants to purchase an aggregate of 162,486 shares of common
stock. These placement agent warrants were issued between June
2017 and August 2017, are exercisable at $1.27 per share and expire
between June 2022 and August 2022. These placement agent warrants
vest immediately. The fair value of these warrants was determined
to be approximately $185,000, as calculated using the Black-Scholes
model. Weighted-average assumptions used in the Black-Scholes model
included: (1) a discount rate of 1.78 %; (2) an expected term of
5.0 years; (3) an expected volatility of 130%; and (4) zero
expected dividends.
For the year ended February 28, 2018, the Company issued warrants
to purchase an aggregate of 52,500 shares of common stock to a
consultant for advisory services. These warrants are exercisable at
$2.00 per share and expire in September 2022 and December 2022.
These warrants vested immediately. The fair value of these warrants
was determined to be approximately $32,000, as calculated using the
Black-Scholes model. Average assumptions used in the Black-Scholes
model included: (1) a discount rate of 2.06%; (2) an expected term
of 5.0 years; (3) an expected volatility of 136%; and (4) zero
expected dividends. For year ended February 28, 2018, the Company
recognized approximately $32,000 of stock-based compensation for
these warrants.
For the year ended February 28, 2018, the Company issued warrants
to purchase an aggregate of 83,332 shares of common stock to a
consultant for advisory services. These warrants are exercisable at
$0.89 per share and expire between October 2022 and February 2023.
These warrants vested immediately. The fair value of these warrants
was determined to be approximately $67,000, as calculated using the
Black-Scholes model. Average assumptions used in the Black-Scholes
model included: (1) a discount rate of 2.00%; (2) an expected term
of 5.0 years; (3) an expected volatility of 131%; and (4) zero
expected dividends. For year ended February 28, 2018, the Company
recognized approximately $67,000 of stock-based compensation for
these warrants.
The following table summarizes common stock purchase warrants
issued and outstanding:
|
|
|
Weighted
|
Aggregate
|
Weighted
|
|
|
|
average exercise
|
intrinsic
|
average remaining
|
|
|
Warrants
|
price
|
value
|
contractual life (years)
|
|
Outstanding at February 29, 2016
|
913,514
|
$14.56
|
-
|
3.14
|
|
Granted:
|
2,169,959
|
2.97
|
-
|
-
|
|
Cancelled/Expired/Exercised:
|
(384,779)
|
12.94
|
-
|
-
|
|
Outstanding at February 28, 2017
|
2,698,694
|
$5.11
|
$-
|
4.21
|
|
Granted:
|
373,318
|
1.64
|
-
|
-
|
|
Cancelled/Expired/Exercised:
|
(11,894)
|
32.34
|
-
|
-
|
|
Outstanding at February 28, 2018
|
3,060,118
|
$4.34
|
$90,068
|
3.38
|
Warrants exercisable at February 28, 2018 are:
|
Exercise
|
Number
|
Weighted average
|
Exercisable
|
|
Prices
|
of shares
|
remaining life (years)
|
number of shares
|
|
$0.83
|
119,429
|
2.55
|
119,429
|
|
$0.89
|
83,332
|
4.80
|
83,332
|
|
$0.91
|
43,636
|
2.96
|
43,636
|
|
$1.27
|
162,486
|
4.33
|
162,486
|
|
$2.00
|
634,393
|
3.66
|
634,393
|
|
$3.00
|
1,653,974
|
3.69
|
1,653,974
|
|
$8.25
|
9,134
|
2.49
|
9,134
|
|
$10.50
|
126,978
|
2.10
|
126,978
|
|
$15.00
|
556
|
2.25
|
556
|
|
$18.75
|
695
|
2.25
|
695
|
|
$22.50
|
209,754
|
0.38
|
209,754
|
|
$31.50
|
15,684
|
0.88
|
15,684
|
|
$37.50
|
67
|
0.15
|
67
|
|
$4.34
|
3,060,118
|
3.38
|
3,060,118
|
NOTE 7 – NOTES PAYABLE
Promissory Note
In July 2015, the Company entered into a note purchase agreement,
which was subsequently amended, whereby it issued and sold a
non-convertible promissory note in the principal amount of $1.2
million (the “Promissory Note”) and a warrant to
purchase 43,636 shares of the Company’s common stock. In
October 2016, $600,000 principal amount of the Promissory Note plus
$48,000 of accrued and unpaid interest was exchanged into the
Additional 2016 Unit Private Placement. Accordingly, the Company
recorded a loss on extinguishment of approximately $694,000 during
the year ended February 28, 2017. In January 2017, the remaining
unpaid principal balance and accrued interest were exchanged into a
convertible note (see Convertible Note below).
During the year ended February 28, 2017, the Company recognized
approximately $461,000 of interest expense related to the
Promissory Note, as amended, including amortization of debt
discount of approximately $367,000 and accrued interest expense of
approximately $94,000. Additionally, the Company recognized a gain
of approximately $340,000 in the year ended February 28, 2017, due
to the change in estimated fair value of the embedded exchange
provision.
OID Notes
In February 2016, the Company entered into an OID note purchase
agreement dated February 12, 2016 (the “February 2016 OID
Note Purchase Agreement”). Pursuant to the February 2016 OID
Note Purchase Agreement, the Company received an aggregate purchase
price of $500,000 and issued OID promissory Notes (the “OID
Notes”) in the aggregate principal amount of $600,000 and
warrants (the “OID Warrants”) to purchase an aggregate
of 36,367 shares of the Company’s common stock.
The Company entered into OID note purchase agreements between March
4 and 15, 2016 (the “March 2016 OID Note Purchase
Agreements”) with various accredited investors. Pursuant to
the March 2016 OID Note Purchase Agreements, the Company issued OID
Notes with an aggregate purchase price of $125,000 and OID Warrants
to purchase 9,902 shares of the Company’s common stock. The
OID Notes issued in March 2016 have a principal amount equal to
$150,000 or 120% of the purchase price.
Pursuant to the March 2016 closings of the private placement of OID
Notes, the principal amount was first allocated to the fair value
of the OID Warrants in the amount of approximately $15,000, next to
the value of the original issuance discount in the amount of
$25,000, then to the fair value of a bifurcated derivative
liability related to the exchange provision in the OID Notes in the
amount of approximately $33,000, and lastly to the debt discount
related to offering costs of approximately $2,000 with the
difference of approximately $75,000 representing the initial
carrying value of the OID Notes issued in March 2016.
The OID Notes were subsequently amended in August 2016, extending
the maturity date of the OID Notes in exchange for among other, (i)
an increased principal amount of the OID Notes by 10% to $825,000
in the aggregate from $750,000 in the aggregate, and (ii) the
issuance of an aggregate of 45,459 common stock purchase warrants
with an exercise price of $2.00 per share and a term of five
years.
In October 2016, $553,000 principal amount of OID Notes
were exchanged into
the securities issued in the
Additional 2016 Unit Private Placement. Accordingly, the Company recorded a loss on
extinguishment of approximately $555,000 for the year ended
February 28, 2017. Additionally, the Company repaid $8,000 of OID
Notes.
In November 2016, the Company exercised its sole option to further
extend the maturity date to its outstanding OID Note in the
aggregate of $264,000 principal amount of OID Note. In
consideration for the extension, the Company increased the
principal amount of the OID Note by 10% or to $26,400 to $290,400
in the aggregate. In January 2017, the remaining outstanding OID
Note was exchanged into a convertible note (see Convertible Note
below).
During the year ended February 28, 2017, the Company recognized
approximately $583,000 of
interest expense related to the OID Notes, as amended, including
amortization of debt discount. Additionally, the Company recognized
a gain of approximately $275,000 in the year ended February 28,
2017, due to the change in estimated fair value of the embedded
exchange provision.
Convertible Note
In January 2017, the Company entered into an exchange agreement,
pursuant to which the Company issued a new convertible promissory
note in the principal amount of $1,000,000 (the “Convertible
Note”) in exchange (the “Debt Exchange”) for the
cancellation of (i) $600,000 principal amount of the Promissory
Note plus $96,000 of accrued and unpaid interest thereon, and (ii)
$290,400 principal amount of the OID Note. The Convertible Note is
convertible into shares of common stock at $2.00 per share and
accrues interest at a rate of 10% per annum. The Convertible Note
may be prepaid upon 10 days’ advanced written notice by the
Company at any time prior to the maturity date without penalty or
premium (the “Prepayment Option”). The
holder has the right to convert the outstanding principal balance
of the Convertible Note plus all accrued and unpaid interest
thereon into shares of the Company’s common stock at a
conversion price per share of $2.00 (the “Conversion
Option”).
The Convertible Note matured on September 30, 2017 and provided for
a ten (10) business day cure period that expired on October 16,
2017. As of February 28, 2018, the Convertible Note was in default
pursuant to its terms and commencing October 1, 2017 accrues
default interest at a rate of twelve percent (12%) per annum. On
March 30, 2018, the Convertible Note was partially repaid, and the
remaining unpaid balance was exchanged for a new note (See Note
14).
The Company evaluated the Debt Exchange transaction in accordance
with ASC 470-50-40-12 and determined the Debt Exchange constituted
a substantive modification and that the transaction should be
accounted for as an extinguishment.
The Company determined the Prepayment Option feature represents a
contingent call option. The Company evaluated the Prepayment Option
in accordance with ASC 815-15-25. The Company determined that the
Prepayment Option feature is clearly and closely related to the
debt host instrument and is not an embedded derivative requiring
bifurcation. Additionally, the Company determined the Conversion
Option represents an embedded call option. The Company evaluated
the Conversion Option in accordance with ASC 815-15-25. The Company
determined that the Conversion Option feature meets the scope
exception from ASC 815 and is not an embedded derivative requiring
bifurcation.
The Company evaluated the Convertible Note for a beneficial
conversion feature in accordance with ASC 470-20. The Company
determined that the effective conversion price was above the
closing stock price on the commitment date, and the Convertible
Note did not contain a beneficial conversion feature.
The Company recorded the Convertible Note at fair value of
approximately $986,000 with an initial debt discount of $14,000.
Accordingly, in accordance with ASC 470-50-40-2, the Company
recognized a loss on extinguishment of approximately $127,000,
which equals the difference between the reacquisition price of debt
and the net carrying amount of the extinguished debt.
During the year ended February 28, 2017, the Company recognized
approximately $19,000 of interest expense related to the
Convertible Note, including amortization of debt discount of
approximately $3,000 and accrued interest expense of approximately
$16,000.
During the year ended February 28, 2018, the Company recognized
approximately $119,000 of interest expense related to the
Convertible Note, including amortization of debt discount of
approximately $11,000 and accrued interest expense of approximately
$108,000.
The following table summarizes the notes payable:
|
|
Note
|
Convertible Note
|
Debt
|
Put Exchange
|
Debt,
|
|
|
Payable
|
Payable
|
Discount
|
Feature
|
Net
|
|
February 29, 2016 balance
|
$1,800,000
|
$-
|
(743,282)
|
476,402
|
1,533,120
|
|
Issuance of notes
|
150,000
|
-
|
(74,931)
|
32,496
|
107,565
|
|
Repayment of notes
|
(8,000)
|
-
|
-
|
-
|
(8,000)
|
|
Additional debt discount upon notes amendment
|
101,400
|
-
|
(264,812)
|
105,587
|
(57,825)
|
|
Notes conversion
|
(2,043,400)
|
1,000,000
|
114,058
|
-
|
(929,342)
|
|
Amortization of debt discount
|
-
|
-
|
958,053
|
-
|
958,053
|
|
Change in value of voluntary exchange feature
|
-
|
-
|
-
|
(614,485)
|
(614,485)
|
|
February 28, 2017 balance
|
-
|
1,000,000
|
(10,914)
|
-
|
989,086
|
|
Amortization of debt discount
|
-
|
-
|
10,914
|
-
|
10,914
|
|
February 28, 2018 balance
|
$-
|
$1,000,000
|
$-
|
$-
|
$1,000,000
|
NOTE 8 – FAIR VALUE MEASUREMENTS
At February 28, 2018 and February 28, 2017, the warrant liability
balances of approximately $148,000 and $2.1 million, respectively,
were classified as Level 3 instruments.
The following table sets forth the changes in the estimated fair
value for our Level 3 classified derivative warrant
liability:
|
|
Promissory Note Warrants
|
Series B Warrant
|
PPM Warrants
|
Total
|
|
Fair value at February 29, 2016
|
$188,351
|
$46,110
|
$-
|
$234,461
|
|
Addition
|
15,225
|
-
|
4,263,271
|
4,278,496
|
|
Change
in fair value
|
(46,372)
|
(10,420)
|
(2,349,193)
|
(2,405,985)
|
|
Fair value at February 28, 2017
|
$157,204
|
$35,690
|
$1,914,078
|
$2,106,972
|
|
Change
in fair value
|
(35,501)
|
(9,816)
|
(442,816)
|
(488,133)
|
|
Reclassification
of warrant liability to equity
|
-
|
-
|
(1,471,262)
|
(1,471,262)
|
|
Fair value at February 28, 2018
|
$121,703
|
$25,874
|
$-
|
$147,577
|
In connection with the initial issuance of the Series B Preferred
Stock on December 31, 2014, the Company issued a warrant to
purchase an aggregate of 30,334 shares of common stock (the
“Series B Warrant”), originally exercisable at $8.25
per share and expiring on March 31, 2020. The Series B Warrant
contains a full-ratchet anti-dilution price protection provision
that requires liability treatment and the exercise price of the
Series B Warrant was adjusted to $2.00 per share during the year
ended February 28, 2017 and subsequently adjusted to $0.83 per
share during the year ended February 28, 2018. The fair value of
the Series B Warrant at February 28, 2018 and February 28, 2017 was
determined to be approximately $26,000 and $36,000, respectively,
as calculated using the Monte Carlo simulation. The Monte Carlo
simulation as of February 28, 2018 and February 28, 2017 used the
following assumptions: (1) a stock price of $1.15 (the 20-day
volume weighted average price (“VWAP”) of the February
28, 2018 closing stock price) and $1.50, respectively; (2) a
risk-free rate of 2.27% and 1.50% respectively; (3) an expected
volatility of 137% and 131% respectively; and (4) a
fundraising event to occur on July 31, 2018 and May 31, 2017,
respectively, that would result in the issuance of additional
common stock.
In connection with the issuance of the Promissory Note on July 31,
2015, the Company issued a warrant to purchase an aggregate of
43,636 shares of common stock, originally exercisable at $8.25 per
share and expiring on July 31, 2020. This warrant contains a
full-ratchet anti-dilution price protection provision that requires
liability treatment and the exercise price of this warrant was
adjusted to $2.00 per share during the year ended February 28, 2017
and subsequently adjusted to $0.83 per share during the year ended
February 28, 2018. The fair value of the warrant at February 28,
2018 and February 28, 2017 was determined to be approximately
$39,000 and $51,000, respectively, as calculated using the Monte
Carlo simulation. The Monte Carlo simulation as of February 28,
2018 and February 28, 2017 used the following assumptions: (1)
stock price of $1.15 (the 20-day VWAP of the February 28, 2018
closing stock price) and $1.50, respectively; (2) a risk-free rate
of 2.32% and 1.57% respectively; (3) an expected volatility of 137%
and 131%, respectively; and (4) a fundraising event to occur on
July 31, 2018 and May 31, 2017, respectively, that would result in
the issuance of additional common stock.
In connection with the execution of the Note Amendment on
February 12, 2016, the Company issued a warrant to purchase an
aggregate of 43,636 shares of common stock, initially exercisable
at $8.25 per share and expiring on February 11, 2021. This warrant
contains a ratchet anti-dilution price protection provision that
requires liability treatment and the exercise price of this warrant
was adjusted to $2.20 per share during the year ended February 28,
2017 and subsequently adjusted to $0.91 per share during the year
ended February 28, 2018. The fair value of the warrant at February
28, 2018 and February 28, 2017 was determined to be approximately
$41,000 and $51,000, respectively, as calculated using the Monte
Carlo simulation. The Monte Carlo simulation as of February 28,
2018 and February 28, 2017 used the following assumptions: (1)
stock price $1.15 (the 20-day VWAP of the February 28, 2018 closing
stock price) and $1.50, respectively; (2) a risk-free rate of 2.41%
and 1.68%, respectively; (3) an expected volatility of 137% and
131%, respectively; and (4) ) a fundraising event to occur on July
31, 2018 and May 31, 2017, respectively, that would result in the
issuance of additional common stock.
In connection with the issuance of OID Notes in February 2016, the
Company issued warrants to purchase an aggregate of 36,367 shares
of common stock. These warrants were issued between
February 12 and 22, 2016, were initially exercisable at $8.25 per
share and expire between February 11 and 21, 2021. These warrants
contain a full-ratchet anti-dilution price protection provision
that requires liability treatment the exercise price of these
warrants was adjusted to $2.00 per share during the year ended
February 28, 2017 and subsequently adjusted to $0.83 per share
during the year ended February 28, 2018. The fair value of these
warrants at February 28, 2018 and February 28, 2017 was determined
to be approximately $34,000 and $44,000, respectively, as
calculated using the Monte Carlo simulation. The Monte Carlo
simulation as of February 28, 2018 and February 28, 2017 used the
following weighted-average assumptions: (1) stock price of $1.15
(the 20-day VWAP from the February 28, 2018 closing stock price)
and $1.50, respectively; (2) a risk-free rate of 2.41% and 1.68%,
respectively; (3) an expected volatility of 137% and 131%,
respectively; and (4) ) a fundraising event to occur on July 31,
2018 and May 31, 2017, respectively, that would result in the
issuance of additional common stock.
In connection with the issuance of OID Notes in March 2016, the
Company issued warrants to purchase an aggregate of 9,092 shares of
common stock. These warrants were issued between March 4 and
15, 2016, were initially exercisable at $8.25 per share and expire
between March 4 and 15, 2021. These warrants contain a full-ratchet
anti-dilution price protection provision that requires liability
treatment the exercise price of these warrants were adjusted to
$2.00 per share during the year ended February 28, 2017 and
subsequently adjusted to $0.83 per share during the year ended
February 28, 2018. The fair value of these warrants at February 28,
2018 and February 28, 2017 was determined to be approximately
$9,000 and approximately $11,000, respectively, as calculated using
the Monte Carlo simulation. The Monte Carlo simulation as of
February 28, 2018 and February 28, 2017, used the following
weighted-average assumptions: (1) stock price of $1.15 (a 20-day
VWAP from the February 28, 2018 closing stock price) and $1.50,
respectively; (2) a risk-free rate of 2.42% and 1.69%,
respectively; (3) an expected volatility of 137% and 131%,
respectively; and (4) ) a fundraising event to occur on July 31,
2018 and May 31, 2017, respectively, that would result in the
issuance of additional common stock.
In connection with the Additional 2016 Unit Private Placement
including the Company Payable Exchange, the OID Note Exchange, the
Promissory Note Exchange and the Additional 2016 MFN Exchange, the
Company issued warrants to purchase an aggregate of 1,617,506
shares of common stock. These warrants were issued
between August 31, 2016 and October 30, 2016, are exercisable at
$3.00 per share and expire between August 30, 2021 and October 29,
2021. As referenced in Note 6, the Price Protection provision
associated with these warrants required liability treatment. The
fair value of these warrants at February 28, 2017 was determined to
be approximately $1.9 million, as calculated using the Monte Carlo
simulation. The Monte Carlo simulation as of February 28, 2017 used
the following weighted-average assumptions: (1) stock price of
$1.50; (2) a risk-free rate of 1.66%; (3) an expected volatility of
131%; and (4) a fundraising event to occur on May 31, 2017, that
would result in the issuance of additional common stock. The Price
Protection provision expired in April 2017, and the Company
reclassified approximately $1.5 million of derivative warrant
liability to equity, as referenced in Note 4.
NOTE 9 – EQUIPMENT
Equipment consists of the following:
|
|
Estimated
|
February 28,
|
February 28,
|
|
|
Useful Lives
|
2018
|
2017
|
|
Research
equipment
|
7 years
|
$629,416
|
$601,720
|
|
Computer
equipment
|
5 years
|
78,149
|
78,149
|
|
|
707,565
|
679,869
|
|
|
Accumulated
depreciation and amortization
|
|
(353,467)
|
(265,234)
|
|
Equipment, net
|
|
$354,098
|
$414,635
|
Total depreciation and amortization expense was approximately
$89,000 and $96,000 for the years ended February 28, 2018 and
February 28, 2017, respectively.
Depreciation of equipment utilized in research and development
activities is included in research and development expenses and
amounted to approximately $85,000 and $81,000 for the years ended
February 28, 2018 and February 28, 2017, respectively. All other
depreciation is included in general and administrative expense and
amounted to approximately $4,000 and $15,000 for the years ended
February 28, 2018 and February 28, 2017, respectively.
NOTE 10 – LICENSE AGREEMENTS AND COMMITMENTS
License Agreements
Pursuant to the License Agreement, we are required to make annual
license maintenance fee payments beginning August 26,
2011. We have satisfied all license maintenance payments
due through February 28, 2018. We are required to make payments of
$100,000 in 2018 and every year the license is in effect
thereafter. These annual license maintenance fee payments will be
credited to running royalties due on net sales earned in the same
calendar year, if any. We are in compliance with the License
Agreement.
Pursuant to the Second License Agreement, we are required to make
annual license maintenance fee payments beginning on January 3,
2013. In February 2017, we amended the Second License
Agreement to reduce the maintenance payment for 2016 from $30,000
to $5,000, 2017 from $50,000 to $5,000, 2018 from $75,000 to
$5,000, 2019 from $100,000 to $60,000, and 2020 from $100,000 to
$60,000. We are required to make payments of $100,000 in 2021 and
every year the license is in effect thereafter. These annual
license maintenance fee payments will be credited to running
royalties due on net sales earned in the same calendar year, if
any. We are in compliance with the Second License Agreement,
however the maintenance payments for 2017 and 2018 totaling $10,000
are currently outstanding.
Pursuant to the Alternative Splicing Diagnostic License Agreement
and the Alternative Splicing Therapeutic License Agreement, we are
required to make annual license maintenance fee payments for each
license beginning on January 1, 2015. The license maintenance
payments of $37,500 for 2018 is currently outstanding. We are
required to make additional payments of $50,000 in 2019
and every year each license is in effect thereafter. The
parties are currently in discussions regarding amending the
Alternative Splicing Diagnostic License Agreement and the
Alternative Splicing Therapeutic License Agreement and as such, we
are in compliance the Alternative Splicing Diagnostic License
Agreement and the Alternative Splicing Therapeutic License
Agreement.
Pursuant to the Antibody License Agreement, we are required to make
license maintenance fee payments beginning on January 1,
2015. We have satisfied all license maintenance payments due
through February 28, 2018. We are required to make additional
payments of $20,000 in 2019 and every year the license is in effect
thereafter. These annual license maintenance fee payments will be
credited to running royalties due on net sales earned in the same
calendar year, if any. We are in compliance with the Antibody
License Agreement.
Lease Agreements
On August 28, 2014, we entered into a lease agreement, subsequently
amended (the “Boston Lease”) for our diagnostic
laboratory and office space located at 27, Drydock Ave, 2nd Floor,
Boston, MA 02210 (the “Boston Property”). We paid
a $40,000 security deposit in connection with entering into the
Boston Lease.
On July 26, 2017, we entered into a second amendment to the Boston
Lease for the Boston Property (the “Second Boston Lease
Amendment”). The Second Boston Lease Amendment extended the
term (the “Second Extension Period”) for five years
from September 1, 2017 through August 31, 2022. Monthly basic rent
payments are approximately $23,000 for the first year of the Second
Extension Period, approximately $24,000 for the second year of the
Second Extension Period, approximately $25,000 for the third year
of the Second Extension Period, approximately $25,000 for the
fourth year of the Second Extension Period, and approximately
$26,000 for the fifth year of the Second Extension
Period.
Effective March 1, 2015, we entered into a lease agreement for
short-term office space in New York, NY. The term of the
lease is month-to-month and may be terminated upon twenty-one (21)
days’ notice. We paid a $2,100 security deposit in connection
with entering into the lease. Effective December 1, 2015, we
amended our lease agreement for the short-term office space in New
York, NY. The basic rent payment increased to $2,400 per month
and we paid an additional $1,500 security deposit in connection
with the amended lease.
NOTE 11 – COLLABORATIVE AND OTHER RELATIONSHIPS
Research and Development Reimbursements
In connection with our business strategy, we may enter into
research and development and other collaboration agreements.
Depending on the arrangement, we may record payments as advances,
funding receivables, payable balances or non-product income with
our partners, based on the nature of the cost-sharing mechanism and
activity within the collaboration.
On September 29, 2016, the Company entered into an amendment (the
“MTA Amendment”) to a previously executed pilot
materials transfer agreement (the “MTA” and together
with the Amendment, the “Research Agreement”) with
Celgene Corporation (“Celgene”), to conduct a mutually
agreed upon pilot research project (the “Pilot
Project”). The MTA Amendment provides for milestone payments
to the Company of up to approximately $973,000. Under the terms of
the Research Agreement, Celgene provided certain proprietary
materials to the Company and the Company evaluated Celgene’s
proprietary materials in the Company’s metastatic cell line
(in
vitro) and animal
(in
vivo) nonclinical models. The
milestone schedule called for Celgene to pay the Company
approximately $487,000 upon execution of the MTA Amendment, and the
balance in accordance with the completion of three (3) milestones
to Celgene’s reasonable satisfaction, all of which the
Company has received. The term of the Research Agreement was
extended by the parties through January 2018, and has been
successfully completed.
The Company recognized the upfront and each subsequent milestone
payment as a deferred research and development reimbursement in the
Consolidated Balance Sheet and amortized the deferred research and
development reimbursement as incurred over the term of the Research
Agreement.
The Company recorded approximately $664,000 and $309,000 in deferred research and development
reimbursement for the years ended February 28, 2018 and February
28, 2017, respectively. At February 28, 2018 and February 28, 2017,
the Company had a deferred research and development reimbursement
amount of approximately $0, and $178,000,
respectively.
As of February 28, 2018, all four milestones have been successfully
achieved, and the Company has received aggregate milestone payments
of approximately $973,000 or 100% of the total, of which
approximately $487,000 and
$486,000 was received during
the year ended February 28, 2018 and February 28, 2017,
respectively.
Research Collaboration Revenue
We currently do not sell any products and do not have any
product-related revenue. From time to time, we may enter
into research and development
collaboration arrangements, in which we are reimbursed for either
all or a portion of the research and development costs incurred. We
record these payments as revenue in the statement of operations. We
recognize revenue upon delivery and acceptance of the test results
or other deliverables. Approximately $23,000 of research
collaboration revenue was earned during the year ended February 28,
2018.
NOTE 12 – INCOME TAXES
For the years ended February 28, 2018 and 2017, the Company
recorded zero income tax expense. No tax benefit has been recorded
in relation to the pre-tax loss due to a full valuation allowance
to offset any deferred tax asset related to net operating loss
carry forwards attributable to the losses.
The difference between income taxes at the statutory federal income
tax rate and income taxes reported in the statements of operations
are attributable to the following:
|
|
Year
Ended
|
|
|
|
February 28,
2018
|
February 28,
2017
|
|
Federal
income tax rate
|
32%
|
34%
|
|
State
income tax (net of federal benefit)
|
10%
|
15%
|
|
Change
in fair values of warrant and option liabilities
|
5%
|
35%
|
|
Loss
on extinguishment of debt
|
0%
|
(16)%
|
|
Stock
options
|
0%
|
(5)%
|
|
Other
permanent differences and adjustments
|
1%
|
(1)%
|
|
Impact
of tax law change
|
(104)%
|
0%
|
|
Change
in valuation allowance
|
56%
|
(62)%
|
|
Provision
for income tax
|
0%
|
0%
|
The deferred tax assets and liabilities are summarized as
follows:
|
|
February 28,
2018
|
February 28,
2017
|
|
Accrued
compensation
|
$89,731
|
$70,354
|
|
Accrued
interest
|
-
|
6,674
|
|
Net
operating loss carryovers
|
5,789,653
|
7,257,930
|
|
Research
and development credit carryovers
|
298,012
|
253,125
|
|
Capital
loss carryovers
|
11,607
|
16,663
|
|
Deferred
rent
|
13,872
|
-
|
|
Stock
compensation
|
1,143,750
|
1,537,117
|
|
Total gross
deferred tax assets
|
7,346,625
|
9,141,863
|
|
Depreciation
|
(59,313)
|
(90,655)
|
|
|
|
|
|
Less:
Valuation allowance
|
(7,287,312)
|
(9,051,208)
|
|
Net
deferred tax asset
|
$-
|
$-
|
The Tax Cuts and
Jobs Act (the "Act") was enacted in December 2017. Among other
things, the Act reduces the U.S. federal corporate tax rate from 34
percent to 21 percent and eliminates the alternative minimum tax
for corporations. The reduction of the corporate tax rate resulted
in a decrease of the Company’s net deferred tax assets of
approximately $3.3 million, and a corresponding decrease of the
valuation allowance. The accounting for the effects of the Act is
complete as of February 28, 2018.
In assessing the realization of deferred tax assets, management
determines whether it is more likely than not some, or all, of the
deferred tax assets will be realized. The ultimate realization of
deferred tax assets is dependent upon the generation of future
taxable income during the carryforward period as well as the period
in which temporary differences become deductible. Management
considers the reversal of taxable temporary differences, projected
taxable income and tax planning strategies in making this
assessment. Based upon historical losses and the possibility of
continued losses over the periods that temporary differences are
deductible, management believes it is not more likely than not that
the Company will realize the benefits of the deferred tax assets.
Thus, a valuation allowance was recorded against the entire net
deferred tax asset balance. The valuation allowance (decreased) /
increased by approximately $(1.8) million and $1.8 million during the years ended
February 28, 2018 and 2017, respectively. The decrease in the
valuation allowance during the year ended February 28, 2018 was
mainly due to a change in the corporate income tax rate per the
Act. The increase in the valuation allowance during the year ended
February 28, 2017 was mainly due to an increase of the net
operating loss carryforward and other deferred tax
assets.
At February 28, 2018, the cumulative federal and state net
operating loss carryforwards are approximately $21.7
million and $19.3 million,
respectively, and will expire between 2029 and 2037. At February
28, 2018, the Company has research and development credit
carryforwards of approximately $0.3 million that will begin
expiring in 2033.
The amount of net operating loss carryforwards that a company may
use in a given year may be limited in the event of certain
cumulative changes in ownership over a three-year period as
described in Section 382 of the Internal Revenue Code. The Company
has not performed a detailed analysis to determine whether an
ownership change has occurred. Such a change of ownership could
limit the Company’s utilization of the net operating losses
and could be triggered by subsequent sales of securities by the
Company or its stockholders.
The Company records interest and penalties related to unrecognized
tax benefits within income tax expense. The Company had
not accrued any interest or penalties related to unrecognized
benefits. No amounts were provided for unrecognized tax
benefits attributable to uncertain tax positions as of February 28,
2018 and 2017. The Company is no longer subject to Federal income
tax assessments for years prior to 2014. However, since
the Company has incurred net operating losses every year since
inception, all of its income tax returns are subject to examination
and adjustments by the tax authorities for at least three years
following the year in which the tax attributes are
utilized.
NOTE 13 – LICENSE AGREEMENT WITH ASET THERAPEUTICS,
LLC
On August 31, 2016, the Company and ASET Therapeutics, LLC
(“ASET”) entered into a mutual release of claims with
respect to the termination of the Memorandum of Understanding dated
July 14, 2014, as amended, the License and Development and
Commercialization Agreement dated November 25, 2014 and all other
related documents and agreements.
The Company assessed the collectability of its notes receivable in
connection with two past due promissory notes of ASET in the
aggregate principal amount of $125,000 held by the Company (the
“ASET Notes”). The Company determined that the
probability of repayment of the ASET Notes had decreased
significantly and were to be written off. In August 2016, the
Company entered into a sale and assignment agreement with a
non-affiliated shareholder, whereby the Company sold the ASET Notes
for gross proceeds of $12,500. The Company recorded a loss on sale
of notes receivable of $112,500 during the year ended February 28,
2017.
NOTE 14 – SUBSEQUENT EVENTS
Non-Convertible Promissory Bridge Note Private Placement and
Exchange
On March 30, 2018, the Company entered into a note purchase
agreement (the “Note Purchase Agreement”) with a number
of institutional and accredited investors (collectively, the
“Purchasers”), pursuant to which the Company sold in a
private placement (the “Private Placement”), (i) senior
non-convertible promissory bridge notes in the aggregate principal
amount of $2,084,028 (the “Senior Notes”), (ii) junior
non-convertible promissory bridge notes in the aggregate principal
amount of $1,294,900 (the “Junior Notes” and, together
with the Senior Notes, the “Notes”), and (iii) warrants
(the “Note Warrants”) exercisable to purchase ten
thousand (10,000) shares of the Common Stock per share, for each
$100,000 principal amount of Notes issued on a pro rata basis, at
an exercise price equal to $2.00 per share, for a term of five (5)
years.
Pursuant to an exchange agreement dated March 30, 2018 (the
“Exchange Agreement”), the existing holder of the
outstanding Promissory Note as referenced in Note 7, exchanged the
Promissory Note (the “Promissory Note Exchange”) for a
Senior Note with a principal amount of $834,027 and 83,403 Note
Warrants pursuant to the Note Purchase Agreement. Further, the
Company repaid $300,000 of the outstanding balance of the
Promissory Note concurrent with the closing. Additionally, pursuant
to the Exchange Agreement, the holder of (i) all issued and
outstanding shares of Series B Preferred and (ii) certain warrants
to purchase 91,000 shares of Common Stock at an exercise price of
$10.50 per share (the “Series A Warrants”), originally
issued by the Company in the Series B private placement on December
31, 2014, exchanged the Series B Preferred and the Series A
Warrants (the “Series B Exchange” and, together with
the Promissory Note Exchange, the “Exchange”) for a
Junior Note with a principal amount of $1,294,900 and 129,490 Note
Warrants pursuant to the Note Purchase Agreement.
The Notes mature on September 30, 2018, accrue interest at a rate
of ten percent (10%) per annum and may not be prepaid by the
Company prior to the maturity without the consent of the
holder. The principal amount plus all accrued and unpaid
interest thereon shall automatically exchange (the “Automatic
Exchange”), without any action of the holder, into such
number of fully paid and non-assessable securities (e.g. shares and
warrants) to be issued in a Qualified Offering. “Qualified
Offering” means one or a series of offerings of equity or
equity-linked securities resulting in aggregate gross proceeds of
at least $6,628,927 to the Company, including the Automatic
Exchange of the Notes into the Qualified Offering.
The Senior Note and the Junior Note are in substantially similar
form, provided, however, that the Senior Note shall rank senior to
the Junior Note with respect to payment. The Notes shall rank
senior to all future indebtedness of the Company and to the
Company’s issued and outstanding equity securities, except as
otherwise required by applicable law.
Pursuant to the closing of the Private Placement and Exchange, the
Company issued (i) Senior Notes in the aggregate principal amount
of approximately $2,084,028, including $834,027 from the Promissory
Note Exchange, (ii) Junior Notes in the aggregate principal amount
of approximately $1,294,900 solely pursuant to the Series B
Exchange, and (iii) an aggregate of 337,893 Note Warrants for an
aggregate purchase price of approximately $3,378,928, including the
Exchange.
After deducting placement agent fees and other offering expenses,
the Company received net proceeds of approximately $1.13 million
prior to the repayment of $300,000 of the Promissory Note.
Additionally, the Company issued an aggregate of 86,957 placement
agent warrants with a term of five years, an exercise price equal
to $1.27 per share, and a cashless exercise provision.
Pursuant to the Exchange, the Promissory Note and the Series B
Preferred stock have been cancelled and are no longer
outstanding.
F-31