Attached files
| file | filename |
|---|---|
| EX-32.2 - Adhera Therapeutics, Inc. | ex32-2.htm |
| EX-32.1 - Adhera Therapeutics, Inc. | ex32-1.htm |
| EX-31.2 - Adhera Therapeutics, Inc. | ex31-2.htm |
| EX-31.1 - Adhera Therapeutics, Inc. | ex31-1.htm |
| EX-21.1 - Adhera Therapeutics, Inc. | ex21-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
Form 10-K
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES
EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2017
Commission File Number 000-13789
MARINA BIOTECH, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 11-2658569 | |
| (State or other jurisdiction of | (I.R.S. Employer | |
| incorporation or organization) | Identification No.) |
| 17870 Castleton Street, Suite 250 | ||
| City of Industry, California | 91748 | |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code:
(626) 964-5788
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, $0.006 par value
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes [ ] No [X]
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes [ ] No [X]
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [X] No [ ]
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes [X] No [ ]
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. [ ]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | [ ] | Accelerated filer | [ ] |
| Non-accelerated filer | [ ] | Smaller reporting company | [X] |
| (Do not check if a smaller reporting company) | Emerging Growth Company | [ ] | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. [ ]
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes [ ] No [X].
The aggregate market value of the voting stock held by non-affiliates of the registrant was approximately $19.5 million as of June 30, 2017 based upon the closing price of $3.80 per share on the OTCQB tier of the OTC Markets on June 30, 2017.
As of April 16, 2018, there were 10,521,278 shares of the registrant’s $0.006 par value common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement for the registrant’s fiscal year ended December 31, 2017, to be filed by the registrant with the Securities and Exchange Commission not later than 120 days from the end of the registrant’s fiscal year ended December 31, 2017, in conjunction with the registrant’s annual meeting of stockholders, are incorporated by reference in Part III of this Annual Report on Form 10-K.
MARINA BIOTECH, INC.
Table of Contents
| 2 |
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995 and regulations promulgated thereunder. These forward-looking statements reflect our current views with respect to future events or our financial performance, and involve certain known and unknown risks, uncertainties and other factors, including those identified below, those discussed in Item 1A of this report under the heading “Risk Factors,” and those discussed in our other filings with the Securities and Exchange Commission, which may cause our or our industry’s actual or future results, levels of activity, performance or achievements to differ materially from those expressed or implied by any forward-looking statements or from historical results. We intend such forward-looking statements to be covered by the safe harbor provisions for forward-looking statements contained in the Private Securities Litigation Reform Act. Forward-looking statements include information concerning our possible or assumed future results of operations and statements preceded by, followed by, or that include the words “may,” “will,” “could,” “would,” “should,” “believe,” “expect,” “plan,” “anticipate,” “intend,” “estimate,” “predict,” “potential” or similar expressions.
Forward-looking statements are inherently subject to risks and uncertainties, many of which we cannot predict with accuracy and some of which we might not even anticipate. Although we believe that the expectations reflected in such forward-looking statements are based upon reasonable assumptions at the time made, we can give no assurance that such expectations will be achieved. Future events and actual results, financial and otherwise, may differ materially from the results discussed in the forward-looking statements. Readers are cautioned not to place undue reliance on these forward-looking statements. We undertake no obligation to publicly update or revise any forward-looking statements after the date of this Annual Report on Form 10-K or to conform them to actual results, new information, future events or otherwise, except as otherwise required by securities and other applicable laws.
The following factors, among others, could cause our or our industry’s future results to differ materially from historical results or those anticipated:
| ● | our ability to obtain additional and substantial funding for our company on an immediate basis, whether pursuant to a capital raising transaction arising from the sale of our securities, a strategic transaction or otherwise; | |
| ● | our ability to attract and/or maintain research, development, commercialization and manufacturing partners; | |
| ● | the ability of our company and/or a partner to successfully complete product research and development, including pre-clinical and clinical studies and commercialization; | |
| ● | the ability of our company and/or a partner to obtain required governmental approvals, including product and patent approvals; | |
| ● | the ability of our company and/or a partner to develop and commercialize products that can compete favorably with those of our competitors; | |
| ● | the timing of costs and expenses related to the research and development programs of our company and/or our partners; | |
| ● | the timing and recognition of revenue from milestone payments and other sources not related to product sales; | |
| ● | our ability to obtain suitable facilities in which to conduct our planned business operations on acceptable terms and on a timely basis; | |
| ● | our ability to satisfy our disclosure obligations under the Securities Exchange Act of 1934, as amended, and to maintain the registration of our common stock thereunder; | |
| ● | our ability to attract and retain qualified officers, employees and consultants as necessary; and | |
| ● | costs associated with any product liability claims, patent prosecution, patent infringement lawsuits and other lawsuits. |
| 3 |
These factors are the important factors of which we are currently aware that could cause actual results, performance or achievements to differ materially from those expressed in any of our forward-looking statements. We operate in a continually changing business environment, and new risk factors emerge from time to time. Other unknown or unpredictable factors also could have material adverse effects on our future results, performance or achievements. We cannot assure you that projected results or events will be achieved or will occur.
Overview
We are a fully integrated, commercial stage biopharmaceutical company delivering proprietary drug therapeutics for significant unmet medical needs in the U.S., Europe and certain additional international markets. Our portfolio of products currently focuses on fixed dose combinations (“FDC”) in hypertension, arthritis, pain and oncology allowing for innovative solutions to such unmet medical needs. Our mission is to provide effective and patient centric treatment for hypertension – including resistant hypertension – through our patented total care platform. In this connection, we acquired from Symplmed Pharmaceuticals LLC and its wholly-owned subsidiary, Symplmed Technologies, LLC, certain of the intellectual property assets related to the patented technology platform known as DyrctAxess, also called Total Care, that offers enhanced efficiency, control and information to empower patients, physicians and manufacturers to help achieve optimal care.
In doing so, we have created a universal platform for the effective treatment of hypertension as well as for the distribution of FDC hypertensive drugs such as our FDA-approved product Prestalia, and the other products in our pipeline, devices for Therapeutic Drug Monitoring (TDM), Blood Pressure (BP), and other cardiac monitors, as well as services such as counseling and prescription reminders.
We currently have one commercial and three clinical development programs underway: (i) Prestalia®, a single-pill FDC of perindopril argentine (“perindopril”), an angiotensin-converting-enzyme (“ACE”) inhibitor and amlodipine besylate (“amlodipine”), a calcium channel blocker (“CCB”), which has been approved by the U.S. Food and Drug Administration (“FDA”) and is actively marketed in the U.S.; (ii) our next generation celecoxib program drug candidates for the treatment of acute and chronic pain, IT-102 and IT-103, each of which is an FDC of celecoxib, a COX-2 selective nonsteroidal anti-inflammatory drug (“NSAID”) and either lisinopril (IT-102) or olmesartan (IT-103) – both lisinopril and olmesartan are antihypertension drugs; (iii) CEQ508, an oral delivery of small interfering RNA (“siRNA”) against beta-catenin, combined with IT-102 to suppress polyps in the precancerous syndrome and orphan indication Familial Adenomatous Polyposis (“FAP”); and (iv) CEQ508 combined with IT-103 to treat Colorectal Cancer.
On April 16, 2018, we raised in excess of $10 million, net of fees and expenses from a private placement of our newly created Series E Convertible Preferred Stock (See Recent Developments: Series E Convertible Preferred Share Private Placement Offering below). The use of funds from the raise will primarily be on the commercialization of Prestalia, funding working capital and capex needs and other general corporate requirements. For the development of IT-102 and IT-103, we will seek partners or raise additional funds to advance the development programs. We believe that by combining a COX-2 inhibitor with an antihypertensive in a single FDC oral tablet, IT-102 and IT-103 will each offer improved safety profiles as compared to currently available and previously marketed COX-2 inhibitors as well as address patients with chronic pain who are commonly taking antihypertension drugs concurrently. We further believe that the current opioid addiction epidemic in the U.S. has been driven in part by the withdrawal from the market of certain COX-2 inhibitors due to their associated risk of cardiovascular-related adverse events. We plan to license or divest our other assets since they no longer align with our focus on the treatment of hypertension.
We intend to create value through the continued commercialization of our FDA-approved product, Prestalia, while moving our FDC development programs forward to further strengthen our commercial presence. We intend to retain ownership and control of all of our product candidates, but in the interest of accelerated growth and market penetration, we will also consider partnerships with pharmaceutical or biotechnology companies in order to reduce time to market and to balance development risks, both clinically and financially.
| 4 |
As our strategy is to be a fully integrated biopharmaceutical company, we will drive a primary corporate focus on revenue generation through our commercial assets, with a secondary focus on advancing our FDC pipeline to further enhance our commercial presence.
Background
As further described below under “Merger with IThenaPharma”, on November 15, 2016, Marina entered into an Agreement and Plan of Merger with IThenaPharma, Inc., a Delaware corporation (“IThena” or “IThenaPharma”), IThena Acquisition Corporation, a Delaware corporation and a wholly owned subsidiary of IThena (“Merger Sub”), and Vuong Trieu, Ph.D. as the IThena Representative (the “Merger Agreement”), pursuant to which, among other things, Merger Sub merged with and into IThena, with IThena surviving as a wholly owned subsidiary of Marina (such transaction, the “Merger”). As a result of the Merger, the former holders of IThena common stock immediately prior to the completion of the Merger owned approximately 65% of the issued and outstanding shares of Marina common stock immediately following the completion of the Merger.
Marina was incorporated under the laws of the State of Delaware under the name Nastech Pharmaceutical Company on September 23, 1983, and IThena was incorporated under the laws of the State of Delaware on September 3, 2014. IThena is deemed to be the accounting acquirer in the Merger, and thus the historical financial statements of IThena will be treated as the historical financial statements of our company and will be reflected in our quarterly and annual reports for periods ending after the effective time of the Merger. Accordingly, beginning with our Annual Report on Form 10-K for the fiscal year ended December 31, 2016, we started to report the results of IThena and Marina and their respective subsidiaries on a consolidated basis. The December 31, 2017 statement of operations include the full year for Marina, Ithena and their subsidiaries whereas the December 31, 2016 statement of operations include the results of Ithena alone from January 1, 2016 to November 15, 2016 and combined with Marina and its subsidiaries from November 16, 2016 through December 31, 2016. As such, the statements of operations may not be comparable for the two years ended December 31, 2017.
Prior to the Merger, Marina’s pipeline consisted of oligonucleotide-based therapeutics. That pipeline included CEQ508, a product in clinical development for the treatment of FAP, for which Marina received both Orphan Drug Designation (“ODD”) and Fast Track Designation (“FTD”) from the FDA, as well as preclinical programs for the treatment of type 1 myotonic dystrophy (“DM1”) and DMD. The IThena pipeline of celecoxib FDCs is now incorporated into the combined company. We plan to develop IT-102/IT-103 – next generation celecoxib – together with CEQ508, as a therapeutic enhancer for therapies against FAP and CRC. We are also developing IT-102/IT-103 for the treatment of combined arthritis / hypertension and the treatment of pain requiring a high dose of celecoxib.
Following the Merger, we have reorganized the acquired Marina platform into a strong pipeline of preclinical and clinical drug candidates, which we believe will unlock their value. We plan to divest this asset as soon as reasonably practicable. At the same time, we have divested the SMARTICLES asset as it is no longer a strategic fit, as described under “Recent Developments – Sale of Smarticles Assets” below.
On March 16, 2018, Marina entered into an Licensing Agreement with Agenovir Corporation, whereby Marina granted Agenovir exclusive rights to the company’s DiLA2 delivery system in exchange for an upfront payment of $200,000 and further potential future consideration dependent upon event and sales-based milestones. Under the terms of the agreement, Marina has agreed to assign ownership of the intellectual property associated with the DiLA2 delivery system to Agenovir Corporation.
Subsequent to the Merger, we executed on our strategy to become a commercial stage company by acquiring Prestalia from Symplmed. Specifically, and as described under “Acquisition of Assets from Symplmed” below, on June 6, 2017 we entered into an Asset Purchase Agreement with Symplmed for the purchase of Prestalia. Prestalia is an FDA-approved and marketed anti-hypertensive drug. Prestalia is an FDC of perindopril arginine, which is an ACE inhibitor, and amlodipine besylate, which is a CCB, and is indicated as a first line therapy for hypertension control.
The acquisition of Prestalia transitions our company from a clinical stage company to a commercial organization. Prestalia was approved by the FDA in January 2015 and has been marketed in select U.S. states since then by Symplmed. Prestalia sales saw solid growth through September of 2016, via new patient acquisition and strong patient retention. Due to funding circumstances, further sales promotion of Prestalia was ceased by the end of calendar year 2016. In the near term our focus will be dedicated to re-acquiring prior Prestalia patients, with subsequent efforts dedicated to building a strong sales team to fully promote the product.
| 5 |
We believe that the Prestalia acquisition will make us a revenue-stage company. We also believe that the marketing, distribution and sales network that we will build will pave a strong foundation for the promotion and commercialization of our two other hypertension pipeline products – namely IT-102 and IT-103.
Recent Developments
Series E Convertible Preferred Share Private Placement Offering
On April 16, 2018, we entered into Subscription Agreements (the “Purchase Agreements”) with certain accredited investors and conducted a closing pursuant to which we sold 2,334 shares of our Series E convertible preferred stock, par value of $0.01 per share (the “Preferred Stock”), at a purchase price of $5,000 per share of Preferred Stock. Each share of Preferred Stock is initially convertible into shares of our common stock, par value $0.006 per share (the “Common Stock”), at a conversion price of $0.50 per share of Common Stock. In addition, each investor received a 5-year warrant (the “Warrants”, and collectively with the Preferred Stock, the “Securities”) to purchase 0.75 shares of Common Stock for each share of Common Stock issuable upon the conversion of the Preferred Stock purchased by such investor at an exercise price equal to $0.55 per share of Common Stock, subject to adjustment thereunder. The closing is the initial closing (the “Initial Closing”) of our private placement (the “Private Placement”) of up to $10,000,000 of Securities, which amount may be increased to $15,000,000 at the discretion of our company and its placement agent in the Private Placement (the “Maximum Offering Amount”). The Preferred Stock has voting rights, dividend rights, liquidation preferences, conversion rights and anti-dilution rights as described in the Certificate of Designation of Preferences, Rights and Limitations of the Preferred Stock. The Warrants have full-ratchet anti-dilution protection, are exercisable for a period of five years following the final closing of the Private Placement and contain customary exercise limitations.
We received total gross proceeds of approximately $11.7 million from the Initial Closing, prior to deducting placement agent fees and estimated expenses payable by us associated with the Initial Closing of $1.25 million. We intend to use the proceeds of the Private Placement for funding our commercial operations to the sale and promotion of our Prestalia product, working capital needs, capital expenditures, the repayment of certain liabilities and other general corporate purposes.
The outstanding balance related to the notes that we issued in June 2016 in the amount of $357,300 (plus certain additional “consideration securities” in the amount of $375,000 that we committed to issue to the holders of such notes pursuant to amendments that we entered into with such noteholders), the outstanding balance related to the notes that we issued in June 2017 in the amount of $417,173, the outstanding balance related to the lines of credit from our Executive Chairman and from Autotelic Inc. in the amounts of $573,174 and $94,981, respectively, the outstanding balance related to the note that we issued to a trust affiliated with Isaac Blech in November 2017 in the amount of $510,740, in each case inclusive of accrued but unpaid interest, as well as payables due to Autotelic Inc. in the amount of $812,967 pursuant to our Master Services Agreement with Autotelic Inc., have all been converted into our Series E Preferred Stock and Warrants, at the same terms mentioned above with respect to investors in the Private Placement, in full and complete satisfaction of the outstanding debt and as such the notes, lines of credit or payables are no longer outstanding. In addition, $291,250 of accrued and unpaid Board of Director fees due to four current and one former Board member for services rendered during the period prior to January 1, 2018 have also been converted into our Series E Preferred Stock and Warrants, at the same terms mentioned above with respect to investors in the Private Placement, in full and complete satisfaction of the outstanding fees owed to such directors, and as such the accrual is no longer outstanding.
Binding Term Sheet Agreement with Autotelic BIO
On January 11, 2018, we entered into a binding term sheet agreement with Autotelic BIO (“ATB”), as described below, pursuant to which, among other things, and subject to the satisfaction of certain conditions on or prior to January 15, 2019 (the “Termination Date”), we shall grant to ATB a perpetual exclusive right of development and marketing of our IT-103 product candidate, which is a fixed dose combination of celecoxib and olmesartan medoxomil (the “Product”), at the currently approved dose/approved indications only for celecoxib (100 mg, 200mg and 400mg) for combined hypertension and arthritis only, with such right extending throughout the entire world (excluding the United States and Canada and their territories) (the “Territory”). The grant of the license would be memorialized in a definitive license agreement to be entered into between the parties following the satisfaction of the applicable conditions.
The conditions to the grant of the license include, without limitation, that prior to the Termination Date, ATB shall satisfy one of the following conditions: (i) ATB shall obtain funding in a certain specified amount to perform thirteen (13) weeks of repeated toxicity testing with rats and to obtain Investigational New Drug Application approval in the Republic of Korea (the “Fundraising”); (ii) ATB shall obtain a co-development and licensing deal with other third-party pharmaceutical companies with respect to the Product; and (iii) ATB shall obtain a government-sponsored research and development project in the Republic of Korea with respect to the Product. There can be no assurance that all or any of the foregoing conditions to the grant of the license will be satisfied on or prior to the Termination Date, or at all. Such failure will cause the term sheet agreement to expire, and will result in a definitive agreement with respect to the grant of the license not being entered into, in which event we will retain full rights to the Product and full responsibility for its development and commercialization.
The term sheet agreement provides that, following the date on which the license is granted: (A) if ATB should sub-license the Product, we and ATB would share all proceeds of such sub-license equally; and (B) if ATB markets the Product on its own, ATB would provide us with a royalty equal to a percentage of net profits in the mid-single digits. The term sheet agreement also provides that ATB will make a payment to us in the amount of $100,000 upon the successful completion of the Fundraising, and a payment to us in the amount of $300,000 following the date on which we have transferred certain specified technology and provided certain assistance regarding the manufacturing and production of the Product. ATB will have the right to provide us with the Product at the price of Cost of Goods +30% in the event ATB can meet current good manufacturing practices, including obligations to obtain marketing approval in the United States, so long as ATB is able to meet our timeline as to production. Once we have initiated tech transfer for commercial production, if ATB is not providing us with Product, the term sheet provision under which if ATB markets the Product on its own, ATB would provide us with a royalty equal to a percentage of net profits in the mid-single digits, is null and void. We will be entitled to the clinical trial data and any enhancements and inventions developed by ATB during this process. ATB will have intellectual property rights and bear the cost of the Product worldwide, excluding the United States and Canada and their territories. We will have intellectual property rights and bear the cost of the Product in the United States and Canada and their territories. ATB is to conduct clinical trials in full gene therapy medicinal product / good clinical practice compliance with full consultation and approval of our company to be submissible to the U.S. FDA.
Autotelic LLC, an entity that owns approximately 22 % of our issued and outstanding shares of common stock and of which Dr. Trieu, our Executive Chairman, serves as Chief Executive Officer, owns approximately 19% of the issued and outstanding shares of the common stock of ATB.
| 6 |
Isaac Blech Investment / Board Appointment / Option Grant
On November 22, 2017, we entered into a Note Purchase Agreement (the “Purchase Agreement”) with a trust affiliated with Mr. Isaac Blech (the “Purchaser”) pursuant to which we issued to the Purchaser a secured convertible promissory note in the aggregate principal amount of $500,000 ( “Note”). The Note became due and payable on March 31, 2018. The unpaid principal balance of the Note, together with accrued and unpaid interest thereon, automatically converted into 103.7 shares of Preferred Stock and Warrants to purchase up to 777,750 shares of Common Stock upon the Initial Closing of the Private Placement described above. As a result of the conversion of the Note, all obligations of the Company to the Purchaser under the Note have been satisfied and the Note is no longer outstanding.
Reverse Stock Split
On August 1, 2017, we filed a Certificate of Amendment of our Amended and Restated Certificate of Incorporation to effect a one-for-ten reverse split of our issued and outstanding shares of common stock. Our common stock commenced trading on the OTCQB tier of the OTC Markets on a split-adjusted basis on Thursday, August 3, 2017. There were no changes to the authorized shares of our common stock as a result of the reverse split. No fractional shares were issued in connection with the reverse split; any fraction of a share of common stock that would otherwise have resulted from the reverse split was rounded up to the nearest whole share of common stock. Unless indicated otherwise, all share and per share information included in this report give effect to the reverse split.
Acquisition of Prestalia and DyrctAxess from Symplmed
On June 5, 2017, we entered into an Asset Purchase Agreement (the “Purchase Agreement”) with Symplmed pursuant to which we purchased from Symplmed, for aggregate consideration of approximately $620,000 (consisting of $300,000 in cash plus the assumption of certain liabilities of Symplmed in the amount of approximately $320,000), Symplmed’s assets relating to a single-pill FDC of perindopril arginine and amlodipine besylate known as Prestalia, that has been approved by the FDA for the treatment of hypertension. In addition, as part of the transactions contemplated by the Purchase Agreement: (i) Symplmed agreed to transfer to us, not later than 150 days following the closing date, the New Drug Applications for the approval of Prestalia as a new drug by the FDA; and (ii) Symplmed assigned to us all of its rights and obligations under that certain Amended and Restated License and Commercialization Agreement by and between Symplmed and Les Laboratoires Servier (“Servier”) dated January 11, 2012, pursuant to which Symplmed has an exclusive license from Servier to manufacture, have manufactured, develop, promote, market, distribute and sell Prestalia in the U.S. (and its territories and possessions) in consideration of regulatory and sales-based milestone payments and royalty payments based on net sales.
Further, we entered into an offer letter with Erik Emerson, the President and Chief Executive Officer of Symplmed, pursuant to which we hired Mr. Emerson to serve as our Chief Commercial Officer, which appointment became effective on June 22, 2017. We also agreed in such offer letter to issue to Mr. Emerson 60,000 restricted shares of our common stock under our 2014 Long-Term Incentive Plan, which shares vested in December 2017.
| 7 |
In furtherance of the acquisition and commercialization of Prestalia, on July 21, 2017, we acquired from Symplmed and its wholly-owned subsidiary, Symplmed Technologies, LLC, certain of the intellectual property assets related to the patented technology platform known as DyrctAxess that offers enhanced efficiency, control and information to empower patients, physicians and manufacturers to help achieve optimal care. This acquisition is the basis for our opportunity to build out the total care platform through patient support, management and counseling.
Amendment of Notes and Warrants
On July 3, 2017, we entered into an amendment agreement (the “Amendment Agreement”) with respect to those certain promissory notes in the aggregate principal amount of $300,000 (each a “Note” and collectively the “Notes”) that we issued to two accredited investors (the “Purchasers”) pursuant to that certain Note Purchase Agreement dated June 20, 2016 by and among us and the Purchasers (the “Purchase Agreement”), and those certain warrants to purchase up to an aggregate of 951,263 shares of our common stock that were originally issued pursuant to that certain Note and Warrant Purchase Agreement dated as of February 10, 2012 by and among Marina, certain of its wholly-owned subsidiaries and the purchasers identified on the signature pages thereto (as amended from time to time), that are currently held by the Purchasers, and that were amended concurrently with the Purchase Agreement to, among other things, extend the price protection with respect to dilutive offerings afforded thereunder to June 19, 2017 (such warrants, as so amended, the “Amended Prior Warrants”).
Pursuant to the Amendment Agreement, among other things:
| (i) | the maturity date of the Notes was extended from June 20, 2017 to December 31, 2017; | |
| (ii) | the Purchasers agreed, upon the closing of any financing transaction yielding aggregate gross proceeds to us of not less than $3 million that occurs while the Notes are outstanding (any such financing transaction, the “Qualifying Financing Transaction”), to convert the outstanding principal balance and any accrued interest thereon into the securities of our company to be issued and sold at the closing of the Qualifying Financing Transaction at the most favorable price and terms at which our securities are sold to investors in the Qualifying Financing Transaction; | |
| (iii) | the parties agreed to extend the price protection with respect to the Amended Prior Warrants resulting from dilutive issuances until the expiration of the term of the Amended Prior Warrants (currently February 10, 2020); provided, that such protection shall not apply to the Qualifying Financing Transaction; | |
| (iv) | we agreed to issue to the Purchasers, on a pro rata basis, such number of our securities as are being issued to investors in the Qualifying Financing Transaction as have an aggregate purchase price equal to $375,000 (such securities, the “Consideration Securities”); | |
| (v) | the Purchasers agreed to waive any claim that the exercise price of the Amended Prior Warrants should be reduced to an amount less than $2.80 as a result of any issuance of securities that occurred while the Amended Prior Warrants were outstanding and prior to the date of the Amendment Agreement; | |
| (vi) | the Purchasers agreed that they shall not, for a period of 90 days after the closing of the Qualifying Financing Transaction, sell any Consideration Securities (or any securities issuable upon exercise or conversion of the Consideration Securities) without the prior written consent of the placement agent with respect to such financing transaction; | |
| (vii) | the Purchasers agreed that they shall not, beginning ninety (90) days following the closing of the Qualifying Financing Transaction, sell, in the aggregate, on any given trading day: (x) for so long as the closing price of our common stock is less than or equal to 200% of the per share purchase price of the Consideration Securities in the Qualifying Financing Transaction on the immediately preceding trading day, such number of Consideration Securities (or shares of common stock issuable upon exercise or conversion of the Consideration Securities) as is equal to more than 5% of the total number of shares of common stock traded on such trading day; and (y) for so long as the closing price of our common stock is greater than 200% of the per share purchase price of the Consideration Securities in the Qualifying Financing Transaction on the immediately preceding trading day, such number of Consideration Securities (or shares of common stock issuable upon exercise or conversion of the Consideration Securities) as is equal to more than 10% of the total number of shares of common stock traded on such trading day; and | |
| (viii) | each Purchaser agreed that, prior to one year before the termination date of the Prior Amended Warrants, such Purchaser shall not exercise any of the Prior Amended Warrants at such time as such Purchaser holds any Consideration Securities (or any securities issued upon the exercise or conversion of any Consideration Securities). |
The unpaid principal balance of the Notes, together with accrued and unpaid interest thereon, automatically converted into 71.46 shares of Preferred Stock and Warrants to purchase up to 535,950 shares of common stock upon the Initial Closing of the Private Placement described above. As a result of the conversion of the Notes, all obligations of the Company to the Purchasers under the Notes have been satisfied and the Notes are no longer outstanding. Also, upon the issuance by us to the Purchasers of 75 shares of Preferred Stock and Warrants to purchase up to 562,500 shares of our common stock upon the Initial Closing of the Private Placement described above, our obligations to issue the Consideration Securities to the Purchasers has been satisfied in full.
| 8 |
Merger with IThenaPharma
On November 15, 2016, Marina entered into the Merger Agreement with IThenaPharma, Merger Sub and Vuong Trieu, as the IThena representative, pursuant to which, among other things, Merger Sub merged with and into IThenaPharma, with IThenaPharma surviving as a wholly owned subsidiary of Marina.
Pursuant to the Merger Agreement, at the effective time of the Merger, without any action on the part of any shareholder, each issued and outstanding share of IThenaPharma’s common stock, other than shares to be cancelled pursuant to the Merger Agreement, was converted into the right to receive shares of Marina common stock at the exchange ratio set forth therein (the “Exchange Ratio”). In addition, each outstanding IThenaPharma warrant was assumed by Marina and converted into a warrant representing the right to purchase shares of Marina common stock, with the number of shares underlying such warrant and the exercise price thereof being adjusted by the Exchange Ratio, with any fractional shares rounded down to the next lowest number of whole shares.
As a result of the Merger, the former holders of IThenaPharma common stock immediately prior to the completion of the Merger owned approximately 65% of the issued and outstanding shares of Marina common stock immediately following the completion of the Merger.
Sale of DiLA2 Assets
On March 16, 2018, Marina entered into an Licensing Agreement with Agenovir Corporation, an unrelated entity, whereby Marina granted Agenovir exclusive rights to the company’s DiLA2 delivery system in exchange for an upfront payment of $200,000 and further potential future consideration dependent upon event and sales-based milestones. Under the terms of the agreement, Marina has agreed to assign ownership of the intellectual property associated with the DiLA2 delivery system to Agenovir Corporation.
Liquidity
We have sustained recurring losses and negative cash flows from operations. At December 31, 2017, we had an accumulated deficit of approximately $8.0 million, negative working capital of approximately $5.6 million, and $106,378 in cash. We have been funded primarily through a combination of licensing payments and debt and equity offerings.
On April 16, 2018, we raised in excess of $10 million from a private placement of our recently created Series E, Convertible Preferred Shares. The funds from this raise will be used primarily to commercialize our FDA approved product Prestalia, fund operations, working capital and capex needs and fund general corporate needs. For the development of our pipeline products, we will seek partners or raise additional funds to advance the development programs.
The volatility in our stock price, as well as market conditions in general, could make it difficult for us to raise capital on favorable terms, or at all. If we fail to obtain additional capital when required, we may have to modify, delay or abandon some or all of our planned activities, or terminate our operations. There can be no assurance that we will be successful in any such endeavors. The accompanying consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
| 9 |
Our Strategy
Our mission is to provide effective and patient centric treatment for hypertension – including resistant hypertension. In doing so we plan to create a universal platform for the effective treatment of hypertension as well as for the distribution of FDC hypertensive drugs, such as our approved product Prestalia and the other products in our pipeline, devices for TDM, BP, and other cardiac monitors, as well as services such as counseling and prescription reminders.
Total Care Platform for Hypertension
Despite the availability of highly effective, safe and cheap generics, hypertension and especially resistant hypertension is still an unmet medical need. These are some of the fact about hypertension in the US (https://www.cdc.gov/bloodpressure/facts.htm). About 75 million American adults (29%) have high blood pressure. Only about half (54%) of people with high blood pressure have their condition under control. High blood pressure costs the nation $46 billion each year. This total includes the cost of health care services, medications to treat high blood pressure, and missed days of work. Worldwide, more than 1 billion hypertensive patients remain with uncontrolled BP. Even among hypertensive patients who receive treatment, in most countries at least half of them fail to reach currently recommended BP targets [Wolf-Maier K et al., Hypertension Treatment and control in five European countries, Canada, and the United States. Hypertension. 2004;43:10 –17].
As defined by the American Heart Association Scientific Statement on Resistant Hypertension in 2008, resistant hypertension refers to patients having uncontrolled BP (>140/90mmHg) despite use of three or more antihypertensive medications, including a diuretic. The prevalence of resistant hypertension from various cohorts is estimated to be around 10–20% of patients being treated for hypertension [Mohammed Siddiqui and David A. Calhoun. Refractory versus resistant hypertension. Curr Opin Nephrol Hypertens 2017, 26:14–19]. As would be expected with a history of poorly controlled, often severe hypertension, patients with resistant hypertension have a worse cardiovascular disease prognosis, including coronary heart disease, stroke, congestive heart failure, and peripheral artery disease, compared with patients with more easily controlled hypertension. Similarly, patients with resistant hypertension are more likely to develop chronic kidney disease. Not surprisingly, given this increased cardiovascular risk, having resistant hypertension is associated with an overall higher mortality compared with nonresistant hypertension [Mohammed Siddiqui and David A. Calhoun. Refractory versus resistant hypertension. Curr Opin Nephrol Hypertens 2017, 26:14–19].
Hypertension (HTN) affects approximately 1 billion people worldwide and the number of patients is projected to increase to 1.56 billion people by 2025 [http://www.world-heart-federation.org/cardiovascular-health/cardiovascular-disease-risk-factors/hypertension/]. While HTN can be controlled with drugs and lifestyle changes in the majority of patients, uncontrolled or resistant HTN is a significant unmet clinical need in 22% of the HTN population [Persell, S. D. (2011). Prevalence of Resistant Hypertension in the United States, 2003-2008. Hypertension, 57: 1076-108]. Resistant HTN is defined as the failure to reach controlled BP with at least a three drug regimen at optimal dosage, including at least one diuretic [The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: The JNC 7 report. JAMA. 2003;289:2560–72.]. Approximately 22% of the 1 billion HTN patients worldwide are affected by resistant HTN. Assuming a 4% penetration rate and an estimated price of $100 per 30 pills, there is over a $1 billion resistant HTN market. The recent clinical failure of renal denervation means limited competition “Despite meeting primary safety endpoints, SYMPLICITY HTN-3 – the pivotal U.S. trial examining renal denervation for treatment-resistant hypertension – has fallen short of its secondary efficacy goals, and failed to reach its primary efficacy endpoint as announced earlier this year by the study’s sponsor.” [SYMPLICITY HTN-3: Renal Artery Denervation Fails for Resistant HTN. March 29, 2014]. Without renal denervation, there are limited treatment options for resistant HTN except for potentially adding chlorthalidone and spironolactone if there is an underlying fluid retention problem.
Adherence to medications is a major challenge clinicians often face in treating hypertension. An increasing number of studies show TDM is reliable for detecting medication nonadherence in patients who seem to have resistant hypertension (RH) [Jung O, Gechter JL, Wunder C, et al. Resistant hypertension? Assessment of adherence by toxicological urine analysis. J Hypertens. 2013; 31:766–774; Ceral J, Habrdova V, Vorisek V, Bima M, Pelouch R, Solar M. Difficult-to-control arterial hypertension or uncooperative patients? The assessment of serum antihypertensive drug levels to differentiate non-responsiveness from non-adherence to recommended therapy. Hypertens Res. 2011; 34:87–90.]. Strauch et al. [Strauch B, Petrak O, Zelinka T, et al. Precise assessment of noncompliance with the antihypertensive therapy in patients with resistant hypertension using toxicological serum analysis. J Hypertens 2013; 31:2455–2461] found medication nonadherence among a cohort of patients with resistant hypertension to be 47%, also having directly measured drug or appropriate metabolite levels by liquid chromatography–mass spectrometry. In fact Brinker et al [Stephanie Brinker et al., Therapeutic Drug Monitoring Facilitates Blood Pressure Control in Resistant Hypertension. J Am Coll Cardiol. 2014 March 4; 63(8): 834–835] found that over one-half (54%) of patients who underwent TDM were found to be nonadherent to treatment and when patients were informed of their undetectable serum drug levels and provided additional counseling, BP control was markedly improved without increasing treatment intensity.
| 10 |
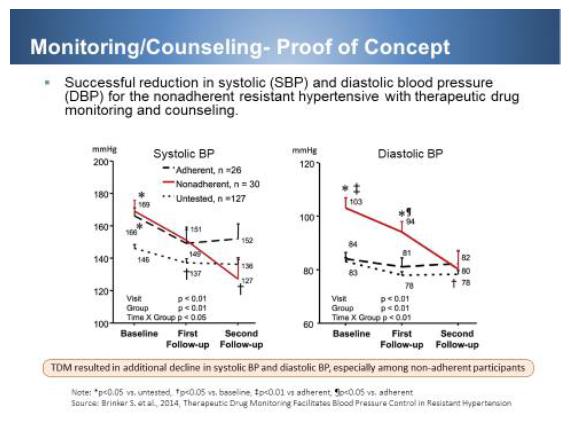
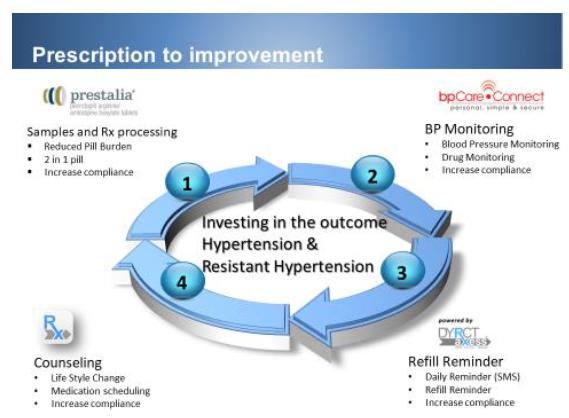
To this effect, we have developed a total care program consisting of four pillars to achieve improved compliance and therefore targeted BP: 1) FDCs. Hypertension is most effectively treated with combination therapy. Furthermore, hypertensive patients frequently suffer from other diseases such as hypercholesterolemia, arthritis, diabetes, dementia, Alzheimer, etc. Their pill burden can be upward of 10-15 pills per day. Any reduction in pill burden would increase compliance. 2) Monitoring- TDM as well as BP monitoring has been shown to improve compliance. With our current bpCareConnect, patients are provided with a BP monitoring system which allows both patients and health care provider to track treatment progress. We plan to upgrade the system to include a point of care TDM device developed by Autotelic Inc. and other cardiac monitoring devices as we further refine and enhance our platform. 3) Our current DyrctAxess platform reminds patients to take their medication through various mechanisms, including texting. More importantly, our call office center will call the patients and remind them to refill their prescriptions. This results in a refill rate of 80%, which is higher than the industry standard of 50%. This low turnover of patients allows us to build prescription rapidly during our launch of Prestalia. 4) As shown by Brinker et al, counseling coupled with monitoring effectively treats resistant hypertension. This arm of the total care program will be implemented using our current call center staff in conjunction with our medical and scientific team. Together this total care platform will transform care not only for hypertension but possibly for other chronic diseases such as diabetes and hypercholesterolemia. Below is the summary of our total care program, which focus on patient compliance to achieve target BP control.
| 11 |
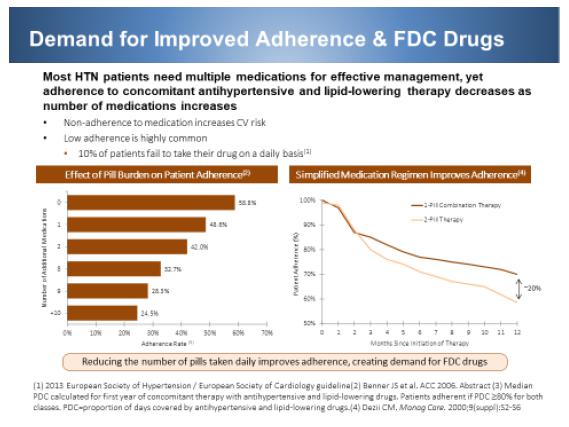
The demand for improved adherence will continue to drive the development of FDCs, which is one of the pillars for our total care program. The need for FDCs in total care program for hypertensive patients is shown below. Combination therapy administered as an FDC has superior efficacy and better tolerance, which is supplemented by higher adherence. Therefore, healthcare providers should not be reluctant and prioritize the use of FDCs over uptitration and switching strategies for addressing efficacy-related issues, particularly if a patient has a history of poor adherence. Compared with free-drug combinations, the use of FDCs of hypertensive agents is associated with a significant improvement in compliance and persistence with therapy and with possible beneficial trends on BP levels and reported adverse effects [Gupta AK et al., Compliance, Safety, and Effectiveness of Fixed-Dose Combinations of antihypertensive agents. A meta-analysis. Hypertension 2010; 55:399-407]. A recent meta-analysis showed that BP reduction by using two drugs in combination is approximately five times greater than doubling the dose of one drug [Krousel-Wood M et al., Medication adherence: a key factor in achieving blood pressure control and good clinical outcomes in hypertensive patients. Curr Opin Cardiol. 2004;19:357–362]. In this context, Prestalia is ideally suited to the total care program: 1) it is approved for 1st line therapy; 2) clinical data has shown that it can achieve rapid and sustained BP reduction; and 3) it is approved only in three strengths – therefore simplifying dose titration versus complicated dose titration scheme of single agent and then double agent.
| 12 |
To further expand our pipeline of FDCs, we will continue to acquire, inlicense, or internally develop FDCs that lend themselves to the total care program. A list of therapeutic agents in our pipeline is shown below.
| a. | Perindopril- ACEON (Perindopril) NDA- Ready to relaunch | |
| i. | Hypertension- monotherapy | |
| ii. | Reduction of cognitive decline in dementia | |
| iii. | Reduction in motor decline in DMD | |
| b. | Prestalia- Ready to relaunch. | |
| i. | Hypertension- combination therapy | |
| ii. | Perindopril/Amlodipine | |
| iii. | 1st line hypertension | |
| c. | Atorvastatin/Perindopril | |
| i. | Hypertension & hypercholesterolemia | |
| ii. | Actively developed by our partner Servier | |
| d. | Pipeline drug | |
| i. | Hypertension & Pain | |
| ii. | FDC developed by our strategic partner – NDA approved/ approval 2018 | |
| iii. | Celecoxib/Lisinopril- planned approval 2019 | |
| iv. | Celecoxib/Olmesartan- planned approval 2020 | |
We intend to initially advance sales of Prestalia in the U.S. through the total care platform. Of secondary priority, we plan push forward with the approval and launch of IT-102 and IT-103.
In our effort to focus and revitalize our company as well as increase shareholder value we intend to explore appropriate opportunities to divest our oligotherapeutics assets through either a spin off to our shareholders or the sale of, or the grant of licenses to, our assets related to these technologies. This will allow us to focus exclusively on Prestalia, as well as IT-102 and IT-103. We also intend to be opportunistic in acquiring assets/asset classes that further complement our product offering.
| 13 |
Prestalia
Acquired in June 2017, Prestalia is a commercially available product. Following the Initial Closing, we plan to integrate the distribution, marketing and sales platform of the acquired assets into our company. This will be concurrent with efforts to mobilize a sales force and build on the existing patient/prescription base of Prestalia and build a strong revenue base. The non-U.S. market for FDCs of ACE inhibitor and CCB is over $300 million and we believe that this market is underplayed in the U.S.
Prestalia was developed in conjunction with Les Laboratories, Servier. It was launched in October of 2015, driven to 1615 prescriptions per month with only 10 sales representatives. It is available in three doses: 3.5/2.5, 7/5 and 14/10 and is promoted worldwide ex-US as Coveram and/or Viacoram by Servier with >$1billion turnover in 2016 from perindopril franchise, WW, for Servier.
Prestalia is a unique FDC drug that simplified dose titration to only three dose strengths. Prestalia is approved for fist line hypertension and titration can be done with just Prestalia with only three dose levels to adjust unlike performing titration of each drug alone where there are at least three strengths for each drug requiring titrating through at least six different strengths and strength combinations. Lisinopril alone has six dose strengths. Per package insert information: Initiate treatment at 3.5/2.5 mg, once daily. Adjust dose according to blood pressure goals waiting 1 to 2 weeks between titration steps. DOSAGE FORMS AND STRENGTHS: Tablets (perindopril arginine/amlodipine): 3.5/2.5 mg, 7/5 mg and14/10 mg.
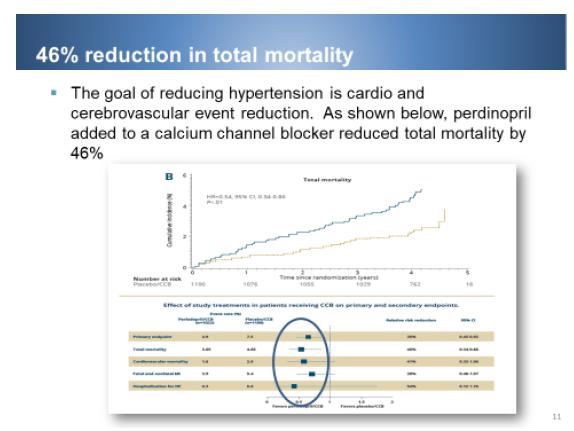
Prestalia is the only product with an active component to demonstrate event reduction across the CV continuum. In aggregate there was reduction in cardiovascular morbidity-mortality in randomised clinical trials with more than 54,000 patients. Various clinical trials have shown that despite lowering blood pressure not all drugs effectively reduce heart attack, stroke and death. In the CAMELOT trial, which compared amlodipine vs. enalapril – another ACEI – for 24 months, there was 31% vs 15% fewer events with amlodipine. In the EUROPA trial, using perindopril, compared to placebo in12,000 ACS (64% had previous MI) reduced CV events by 20%. Post-hoc analysis of EUROPA showed subjects on any CCB for 4.2 yrs. and either on perindopril or placebo, the event rate was 46% less for patients on perindopril and CCB than for patients on a CCB and placebo.
Key Prestalia messages: 1) Rapid and sustained blood pressure control - patients achieve better blood pressure reduction at day 21 on combination than at day 42 on monotherapy. 2) One pill, one time per day. 3) No out of pocket expense for patients with commercial insurance. Six out of 10 patients have unrestricted access to Prestalia with 100% coverage by Express Scripts PBM, 55% coverage by CVS Caremark RX, and 90% coverage by Anthem Inc. And finally as shown below, Prestalia combination reduced total mortality by 46%.
| 14 |
Hypertension Market
Approximately one in three adults, or roughly 75 million people, in the U.S. have high blood pressure, and only about half (54%) have their blood pressure under control. The population of hypertensive patients will continue to increase through 2050 as the current population ages and advances in treatment allow patients already diagnosed to live longer. Although many products within the market will become generic, the focus on combination therapies will prevent a significant decline in the market. Moreover, the launch of new disease-modifying therapies is expected to bolster the growth prospects of the market during the forecast period. The rise in the prevalence of hypertension, from a population of 181 million to 190 million, combined with the anticipated launches of FDCs will continue to create a valuable market opportunity.
In the U.S., 37 of the 50 states have a hypertension rate of greater than 30% of their residents. According to JNC VII (Joint national committee on hypertension), only 1/3 of patients will be controlled by a single product, meaning that 2/3 of all patients will require more than one class of medication to control their high blood pressure (reference https://www.ncbi.nlm.nih.gov/books/NBK9626/). First line therapy, initial treatment, for all hypertension patients should include either a CCB, angiotensin converting enzyme inhibitor (“ACEi”), Angiotensin receptor blocker or diuretic. According to a market share study on hypertension therapies from 2009 to 2019, it was estimated that market share of combination therapies of Renin Angiotensin Aldosterone System inhibitors (RAASi) (of which ACEi is a type) and CCBs would increase from 6% in 2009 to 27% in 2019 (Decision Resources).
In a national prescription audit conducted by IMS health in 2011, it was reported that ACEi were the most prescribed anti-hypertensive category at 163 million prescriptions and CCBs were the third most prescribed antihypertensive category with 98.1 million prescriptions. Ultimately, the market of hypertension patients will continue to expand, but with the proliferation of generics, the pharmaceutical participation in sales and promotion has declined. For our company, the opportunity exists to promote a branded combination, comprised of the two highest prescribed categories in hypertension. Only one product of a similar type, an ACEi and Amlodipine (the CCB that controls over 90% of the CCB market) has ever been promoted and sold in the U.S. That product, Lotrel®, had peak sales of $1.3 billion in the U.S. alone. This level of sales was experienced by Novartis in 2008, and achieved when there were over 15 large pharmaceutical companies, including Wyeth, Novartis, AstraZeneca, King, Forrest, Takeda, Merck, Sanofi and multiple others fighting for share of voice and positioning with patients and physicians. Currently, only two companies other than us compete in the hypertension market - Actavis, selling a beta blocker, Bystolic, with over $500 million in sales in 2015, and Arbor, selling an Angiotensin Receptor Blocker, Edarbi and EdarbiChlor – an FDC of Edarbi and chlortahlidone. The edarbi franchise is selling in excess of $100 million annually.
| 15 |
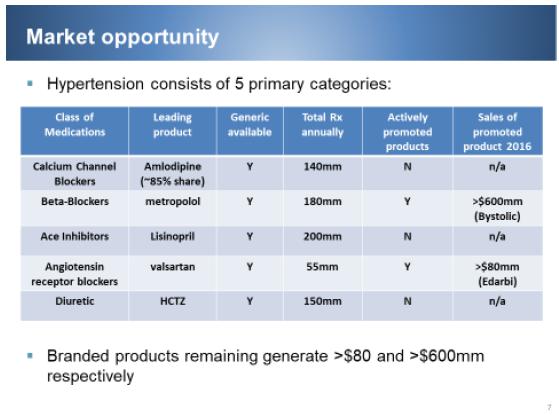
The benchmark for our hypertension product, Prestalia, is Lotrel. Lotrel is the only FDC of an ACEi and amlodipine, and is currently responsible as a brand and generic combined for a total prescription volume in excess of 11 million annually. The product is not promoted, and through analysis of the Prestalia clinical data in market research, Prestalia has been termed by physicians as ‘better than Lotrel’. The success of the Lotrel brand was driven by the combination of two classes of medication that had not only shown the ability to reduce blood pressure but, via the Camelot study for amlodipine and the HOPE and Europa study for ramipril and perindopril, the ability to lower cardiovascular events beyond the effect of lowering blood pressure. The potential success of Prestalia in the U.S. is further supported by the data outside the U.S., which has shown that our worldwide partner, Servier, has produced in excess of €400 million in annual sales of perindopriil and amlodipine as an FDC.
In summary, as one looks at the opportunity for our Prestalia product, there are four key concepts:
| 1) | The market of patient opportunity continues to grow and the top two dispensed categories are ACEi and Amlodipine; |
| 2) | The competitive landscape regarding promotion to physicians leaves us as the only active promoter; |
| 3) | As a branded, patent-protected product, there is no generic alternative to Prestalia; and |
| 4) | The category of ACEi/CCB combination has been proven through the success of Lotrel with $1.3 billion in peak sales, and further validated for Prestalia with sales by Servier in excess of €400 million outside the U.S. |
Commercialization Plan
The two main challenges to the uptake of a drug are getting physicians to prescribe it and getting insurance to reimburse for its use. Prestalia has an existing patient base of 1,500 patients and 400 prescribers that was achieved over a year. It has established unrestricted access to 60% of all commercial plans in the U.S. It has Medicaid coverage nationwide (with the exception of Oregon) and is preferred without step edits and prior authorizations in a few states. With the demonstrated history of physician acceptance and growing acceptance by insurers, combined with the fact that combination of ACEi and CCB are well characterized and understood, we believe that our ability to penetrate the market is dictated by the number of people we are able to engage to assist in our commercialization efforts.
| 16 |
In terms of execution, we plan to take a targeted approach looking to capitalize on geographies with a strong reimbursement position for Prestalia, along with a high degree of hypertension within the territory. Our targeting efforts from the physician perspective will be focused on identifying high prescribers of FDC therapy that includes ACEi and CCB use, along with those physicians writing ACE/Diuretic combinations. An additional key to our targeting will be high prescribers of each of these as concomitant monotherapy, or individual use. This approach will allow us to focus our message on physicians that are already committed to the components, and do not require a clinical communication to convert them from use of other classes of medications.
These targeting approaches will be supplemented by a distribution program, “Prestalia Direct”, which will be operated through the patented DyrctAxess platform that we acquired from Symplmed. The concepts here revolve around fulfillment via mail through our partner pharmacies, and support through a patient engagement call center that drives an emphasis on compliance, persistence and high conversion of initial prescriptions. This marketing platform is what was previously used in the execution of Prestalia fulfillment and led to patient retention greater than 80% after one year. As we execute our commercialization plan in this manner, and drive new prescription volume in a supported and focused fashion, we believe that our ability to maintain our existing patients will create a strong growth of patient accounts as each new patient will be incremental to our growing total. Furthermore as we continue to demonstrate value through the total care program for the effective treatment of hypertension we expect to gain traction and market share within the hypertension space.
Finally, to ensure that we convert the highest percentage of prescriptions generated to new patients, along with keeping our existing patients on medication, we plan to run an expansive patient co-pay support program through our pharmacy partners. This program will be specifically dedicated to ensuring all patients receive medication, covered or not, so that we can ensure patients and physicians are satisfied, as well as put us in a position to continue expanding insurance coverage through continued demand growth.
The final piece to our promotional strategy will be the implementation of our sales team. We plan to have approximately 20 sales people promoting Prestalia in key geographies by the end of the second quarter of 2018 and increasing to 35 by end of the second quarter of 2019. It is intended that these representatives will be trained and implemented by management, and that many of them will be young business-to-business professionals that are looking to get a start in the pharmaceutical industry.
Summary of Follow on Products
To build on commercialization capabilities of Prestalia, we plan to further increase the menu of product offerings through acquisition or internal development. We will be opportunistic in acquiring products/ product lines that complement Prestalia and align with our goal to become a strong player in the hypertension space. We believe we have assembled a strong team with in-depth domain knowledge in drug development and commercialization to have a substantial internal product development program. However, our internal program would be secondary to the primary goal of total care platform and marketing of Prestalia through that platform. With respect to the programs in our internal pipeline described below, items 2 through 5 are part of the therapeutic microbiome program which we are actively discussing to either outlicensing or spin off:
| 1) | IT-102 and IT013 as our next generation celecoxib for management of arthritis pain. IT-102 targets a population requiring angiotensin converting enzyme (“ACE”) inhibitors such as lisinopril and IT-103 targets a population requiring olmesartan. The initial approval based on pivotal bioequivalence (“BE”) trial and a small phase III trial will be for combined arthritis pain and hypertension for patients already taking both drugs. Exploiting the suppression of celecoxib induced edema, we anticipate that these FDCs can eventually replace all of celecoxib prescriptions with or without hypertension once our phase III trial is completed with positive demonstration of edema suppression. This trial will be conducted post approval for label change and will target the highest edema risk patients- the elderly patients whose pill burden is greater than five per day. The inherent lower risk of gastrointestinal (“GI”) bleeding with celecoxib makes it likely that IT-102 and IT-103 can also capture market shares of other pain medications such as ibuprofen and indomethacin. |
| 2) | M101 as beta-catenin short-hairpin RNA (“shRNA”) combination against FAP. This is a combination of IT-102 and CEQ508 (tkRNAi beta-catenin shRNA). Celecoxib was originally approved for FAP, however, it was removed from the market due to fear of cardiovascular risks during the VIOXX withdrawal. But with the PRECISION trial showing that celecoxib is as safe as ibuprofen and naproxen, we anticipate more acceptance of celecoxib. Furthermore, having lisinopril on board to control edema and hypertension, we anticipate that high doses of celecoxib would be safe and effective against FAP. Additionally, the systemic suppression of COX-2 directly and beta-catenin indirectly with celecoxib will be augmented by targeted and local suppression of beta-catenin by beta-catenin shRNA (CEQ508). Together we anticipate a synergistic, safe and effective suppression of polyps in FAP. Since we have completed the phase I proof of concept study for CEQ508, we will move forward to registration phase III trial once we have FDA acceptance of Special Protocol Assessment (“SPA”). |
| 17 |
| 3) | M102 as beta-catenin shRNA combination against CRC. This is a combination of IT-103 and CEQ508 (tkRNAi beta-catenin shRNA). Olmesartan has been shown to improve overall survival (“OS”) among various cancer types, the combination of systemic suppression of ARB/COX-2/Catenin by IT-103 augmented by targeted and local suppression of beta-catenin by CEQ508 is expected to significantly improve the outcome for CRC patients. Additionally, the potential of using CEQ508 to manipulate the microbiome such that it is therapeutic will be investigated. We have termed this “therapeutic microbiome”. |
| 4) | M300 series as IL-6Ra/ Claudin-2/ MIP3a as specific tkRNA/shRNAs against IBD. We evaluated live attenuated bacterial delivery of shRNAs against selected IBD gene targets to achieve specificity, efficacy, and safety. The in vitro efficacy was assessed by an invasion assay using the CMT-93 mouse colon epithelial cells (or RAW264.7 macrophages for TNF-a) and qRT-PCR measurement of mRNA reduction vs. b-actin control. Three gene targets (IL-6Ra, Claudin-2, and MIP3a) and two tkRNAi delivery strains were tested in vivo using an oxazolone or dextran sulfate sodium (DSS) acute murine colitis model. Oral delivery of IL-6Ra tkRNAi strains (CEQ608 and CEQ609) led to a significant reduction in colon length and abolished IL-6Rα message in proximal ileum in DSS exposed groups. Claudin-2 strains (CEQ621 and CEQ626) caused a significant reduction in Claudin-2 mRNA expression and protein levels in the colon as well as attenuation of the disease phenotype and enhanced survival. Treatment with MIP3a therapeutic strains CEQ631 and CEQ632 also resulted in a significant reduction in sum pathology scores and reduction in MIP3a mRNA expression. These findings suggest that tkRNAi-mediated gene silencing of pro-inflammatory targets represents a potential therapeutic development avenue for IBD therapy. |
| 5) | M400 series as surviving/PLK1 as specific DiLA2 (Di-Alkylated Amino Acid)/siRNA against bladder cancer . This program was originally licensed to Debiopharm. A range of RNA interference (“RNAi”) triggers against the cancer-related genes polo-like kinase 1 (PLK1) and survivin were able to knockdown efficacies with IC50 values in the 10 to 30 pM range in cell based assays. This triggered widespread apoptosis and, in the case of PLK1, a strong reduction in cell viability. The selected siRNAs were formulated into positively charged multilamellar liposomes of around 100nm. Due to the negatively charged proteoglycan-rich urothelium, a formulation with a lipid containing a guanidinium group was deemed particularly promising in being able to penetrate the 6-7 cell-layered urothelium. Accordingly, these formulations, when instilled into the bladder, were able to very efficiently suppress the growth of nonmuscle invasive bladder cancers in mouse models of the disease. Highly efficient in vivo knockdowns were found, 90-95% with 1mg/kg dose level. |
Product Candidates
We currently have two late stage arthritis pain/hypertension drug candidates, IT-102 and IT-103. These programs will be conducted secondary to the launch of Prestalia and are dependent on partnering with other entities or fund raising for their development.
Clinical Program for M101: Combination of celecoxib and CEQ508
We believe that the effectiveness of CEQ508 and celecoxib justify the combination as M101. We plan to meet with the FDA to discuss an SPA with a clearly defined clinical design and endpoints for regulatory approval. Depending on being able to partner with other entities or raise additional funding, we plan to meet the FDA sometime in 2018 and the trial may start in 2019. If successful, we could anticipate a potential launch of the product in 2024.
The oncology programs targeting beta-catenin against FAP and CRC will progress along their developmental timeline following a meeting with the FDA to obtain concurrence on trial design and endpoints. Additionally we have programs for IBD and bladder cancer with completed animal proof of concept.
These programs will be developed as resources allow. In subsequent sections we will discuss in detail our three leading clinical programs (IT-102, IT-103, and M101). The M101 program no longer aligns with our focus and we are actively engaged in efforts to outlicense or spin off the therapeutic microbiome asset.
| 18 |

The potential market size of IT-102 and IT-103 was projected to be $170 million and $250 million, respectively. With the FAP potential market size of $400 million, we are projecting the total addressable market for these product candidates to be approximately $820 million annually.
IT-102/IT-103
IT-102 is a FDC formulation of celecoxib, a cyclooxygenase (COX) - 2 selective inhibitor, and lisinopril, an ACE inhibitor, indicated in patients for whom treatment with both celecoxib and lisinopril is appropriate. IT-103 is the same as IT-102, except lisinopril was replaced by olmesartan- an Angiotensin II receptor blockers (“ARB”). IT-103 is for patients for whom treatment with both celecoxib and olmesartan is appropriate. These FDCs will allow rapid access to market through a short clinical program. The initial approval based on pivotal BE trial and a small phase III trial will be for combined arthritis pain and hypertension for patients already taking both drugs. Exploiting the suppression of celecoxib induced edema, we anticipate that they can eventually replace all of celecoxib prescriptions with or without hypertension once our phase III trial is completed with positive demonstration of edema suppression. This trial will be conducted post approval for label change and will target the highest edema risk patients- the elderly patients whose pill burden is greater than five per day. The inherent lower risk of GI bleeding with celecoxib can push IT-102 and IT-103 to also capture market shares of other pain medications such as ibuprofen and indomethacin.
The rationale for IT-102/IT-103 drug development is based on the coexistence of arthritis pain and hypertension in populations, as well as association of hypertension and edema with celecoxib treatment. Additionally, the preference for and improved compliance with a single tablet makes the proposed FDC formulation a very useful drug for treatment of two common conditions of increasing frequency in the aging population.
Arthritis/Hypertension
Arthritis and hypertension often coexist due to common risk factors. Firstly, both conditions are age related. The risk of developing osteoarthritis (“OA”) increases from the age of 40 onwards, with 25% of the population over the age of 45 presenting with clinical symptoms (Hunter et al, 2006). It has been reported that approximately 50% of patients with OA suffer from hypertension. Data from the 2009 Behavioral Risk Factor Surveillance System indicated that the top 2 most prevalent conditions in those over 70 years of age were hypertension (60.7%) and arthritis (55%) (Hunter et al, 2011). The prevalence of hypertension in rheumatoid arthritis (“RA”) in most large studies lies between 52% and 73%, with the age ranging from 51 to 66 years (Fernandes et al, 2015).
| 19 |
Hypertension is one of the most important modifiable risk factors for the development of cardiovascular disease in the general population (Yusuf et al, 2004). It affects about 1 billion individuals worldwide (Kearney et al, 2005) and about 30% of the adult population in the United States (Nwankwo et al, 2013). Despite its high prevalence and the impact of its complications, control of hypertension is far from adequate both in the general population (Chobanian et al, 2003; Oliveria et al, 2002; Primatesta et al, 2006; Luepker et al, 2006) and in arthritis patients (Panoulas et al, 2007). The poor control rates in the general population, where only a third of the people with hypertension have their blood pressure under control (Wang et al, 2005), is attributed to poor access to health care and medications, as well as a lack of adherence to long-term therapy for a usually asymptomatic condition. In the general population, anti-hypertensive therapy has been associated with a reduction of 40% in strokes, 20% in myocardial infarction and >50% in heart failure (Neal et al, 2000), which emphasizes the importance of optimal blood pressure control in any population, including arthritis patients.
Effective simultaneous control of arthritis and hypertension is greatly facilitated by FDC, as most hypertension patients require multiple medications for effective management. However, adherence to concomitant hypertension therapy decreases as the number of medications increases. As the pill burden increases from 1 to ≥10, patient adherence rapidly decreases from 58.8% to 24.5%, respectively (Resnic et al., 2006). A single FDC tablet results in 20% higher patient adherence than observed with a 2-tablet combination therapy (Dezii et al, 2009). In addition, coupling the treatment for asymptomatic hypertension with painful arthritis will not only improve compliance to the long-term therapy of hypertension, but also reduce the renal adverse events associated with NSAIDs/celecoxib treatment. So far, there is no such FDC available in the U.S. Therefore, there is an urgent need for a celecoxib/anti-hypertensive FDC such as IT-102.
Celecoxib side effects
Hypertension and other cardiovascular risks are associated with celecoxib treatment. Clinical trials and observational studies have shown that nonselective and COX-2 selective NSAIDs are associated with increased cardiovascular risks and events (Cheng et al, 2002; Boers et al, 2001; Mukherjee et al, 2001; Solomon et al, 2005). That is why cardiovascular thrombotic events, hypertension, congestive heart failure and edema are listed in the warnings and precautions of the CELEBREXÒ package insert (CELEBREX® Package Insert, 2016). Two randomized, placebo-controlled trials, Adenoma Prevention with Celecoxib (APC) trial and Prevention of Spontaneous Adenomatous Polyps (PreSAP) trial, showed a nearly 2-fold-increased cardiovascular risk in celecoxib treatment groups compared with the control group. Both dose groups in APC trial, celecoxib at 200 or 400mg twice daily, showed significant systolic blood pressure (SBP) elevations at 1 and 3 years from 2 to 5.2 mmHg; however, no significant elevation of SBP was observed in the 400 mg once daily group in the PreSAP trial (Solomon et al, 2006). This trend for a dose-related increase in cardiovascular events and blood pressure raises the possibility that lower doses or other dose intervals may be associated with less cardiovascular risk.
Celecoxib has been intensively evaluated on its blood pressure effects. A post hoc analysis on the renal safety of celecoxib with data from more than 50 clinical studies involving more than 13,000 subjects showed that celecoxib had no clinically detectable effect on blood pressure (Whelton 2000). In the Celecoxib Long-term Arthritis Safety Study (CLASS) with more than 8000 OA and RA patients, there were 2.7% of patients in the celecoxib group (400 mg, b.i.d, N=3987) that showed either new-onset or aggravated hypertension (Whelton 2006). A meta-analysis on the adverse events of celecoxib in OA and RA patients, which included data from 39,605 randomized patients in 31 trials, showed that the proportion of any patient having hypertension or aggregated hypertension was only 1-2% with celecoxib and there was no significant difference between celecoxib and placebo group (Moore 2005).
The large meta-analysis of 31 randomized controlled trials in patients with OA or RA found that celecoxib was associated with a significantly higher incidence of edema (at any site) than placebo (2.6% vs 1.4%: RR 1.9, 95% CI 1.4, 2.7) (Moore et al, 2005). Similarly, a pooled analysis of renal adverse event data from seven 12-week North American trials involving 9,666 patients with OA or RA found that the overall incidence of renal adverse events with celecoxib (4.3%) was greater than that with placebo (2.5%; p<0.05) and was not significantly different from that with NSAIDs (4.1%) (Whelton et al, 2000). The most common renal adverse events with celecoxib were peripheral edema (2.1%), hypertension (0.8%) and aggravated hypertension (0.6%) (Whelton et al, 2000).
| 20 |
Proprietary Patient Level Data Analyses
We have also compared the edema in patient populations receiving celecoxib alone and celecoxib in combination with a variety of antihypertensives. To support this study a proprietary database was created which contains: 1) Claims data from Symphony pertaining to anti-hypertensives, Statins, COX-2 inhibitors, and NSAIDs. The data span the most recent 36 months and 2) registry data from the ACC reporting blood pressure (systolic/diastolic), peripheral edema flags (yes, no, missing), heart rate, LDL, glucose level, ejection fraction, glomerular filtration rate, height, weight, body mass index, and the like.
Symphony dataset is True Patient Level data - All Data Sources be it RX or MX claims is tied back to individual patients which is tracked and then encrypted based on first name, last name, gender, date of birth and zip code to give an accurate picture of patient level informatics year over year regardless of insurance changes. The source of Managed Markets prescription claims data comes from various providers, including Intelligent network services (Switch Data) as well as direct data feeds from pharmacies that do not use Switches so it does not create payer biases.
The definition of the Symphony database is as follow: 1) Takes Celebrex, Anti-hypertensive (“AH”), Statin or NSAID or have OA, RA or some other form of arthritis for 36 months, 2) Time Frame of Jan 1, 2012 – Dec 31, 2014 (3 years), 3) Number of files: 201, 4) File Size: 561 GB zipped (~ 2.5 TB), 5) Unique Patients: 162 million, 6) Patients on Celebrex: 4.3 million, 7) Patients that have OA 16.3 million (15.4 million only OA), 8) Patients that have RA :2.3 million (1.4 million only RA).
The definition for the ACC registry is as follow: 1) Have 3+ BP readings, 2) Time Frame: Jan 1, 2012 – Dec 31, 2014 (3 years), 3) Number of files: 2, 4) Size: 590 MB, 51 MB, 5) Unique Patients: 1.58 million, 6) Patients with BP readings: 1.58 million, 7) Patients with Edema Flag True:870K.
The analysis also showed that there was no impact of celecoxib consumed on the change in blood pressure readings, even at a dose of 400 mg/day (Qazi 2017). Therefore, we confirmed that celecoxib has minimum impact on blood pressure at doses in treatment of arthritis pain. However, the effect of celecoxib on edema is higher than reported in controlled clinical trials. Incidence of edema increased from 20-25% for celecoxib alone to 25-35% when celecoxib was combined with any drug suggestive of drug induced edema. Coadministration with either ACE (i.e. lisinopril) or ARB (i.e. olmesartan) reduced the edema to 10-15%. The edema rate was then measured in the aforementioned database. The incidence of edema was higher for OA patients than RA, other arthritis, or arthritis free patients. The incidence of edema increased when patients were taking Celebrex for all groups except for RA and no arthritis free patients. Overall OA seems to be susceptible to Celebrex induced edema- the frequency of which is higher among patients on the ACC registry which would have prior cardiovascular history.
| 21 |
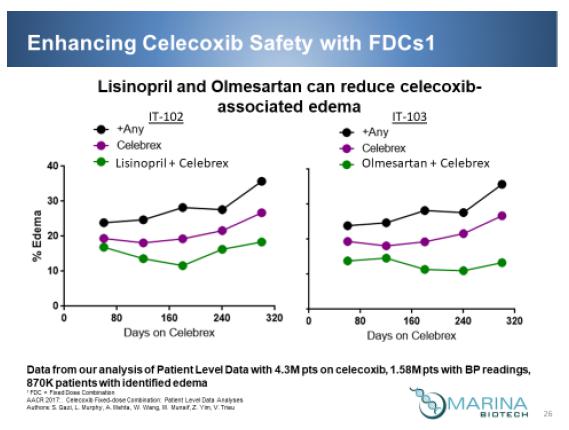
Manufacturing
Formulation work up for IT-102 is completed. The formulation was designed by considering the following characteristics: 1) Single free dose of individual drugs is already marketed in form of hard capsule and tablet, 2) Reference Listed Drug of Celecoxib (Celecoxib®) is hard capsule in High Density Polyethylene (“HDPE”) bottles, 3) Reference Listed Drug of Lisinopril (Zestril®) is uncoated tablet in HDPE bottles. Based on these characteristics, an oral dosage form suitable for administration to the adult was favored. Tablets were preferred to capsules in order to increase the quantity of drug substance available per unit. Celecoxib/Lisinopril FDC tablets are white circular biconvex bi-layered tablets of different size. Consequently, the size and weight increase with dosage strength. Celecoxib/Lisinopril FDC tablet drug products are supplied as 100/2.5 mg, 100/5.0 mg, 100/10.0 mg, 100/20.0 mg, 200/2.5 mg, 200/5.0 mg, 200/10.0 mg, and 200/20.0 mg tablets in HDPE bottles. The constituents were chosen to achieve the following objectives: 1) using well-known and compatible excipients, which allow a world-wide registration of the product, 2) satisfactory chemical stability of the active substance, 3) satisfactory dissolution rate. The objective was to obtain a mean upper than 75% at 30 minutes with slight variation inter and intra batches, and 4) a quantity of excipients as low as possible to obtain a tablet as small as possible.
Pilot scale manufacturing at 10,000 tablets per batch was performed. The dissolution profiles of RLDs and pilot product (200/20 mg FDC bi-layer tablet) were similar with the results of celecoxib and lisinopril. Additionally, the dissolution profiles were considered similar using the similarity factor (f2) following the guidance for industry “Dissolution testing of immediate release solid oral dosage forms”, FDA, CDER, August 1997. With similarity in dissolution we are expecting to have similarity in BE trial.
IT-102 manufacturing has been transferred to an FDA inspected current good manufacturing practices (“cGMP”) contract manufacturing organization and the exhibit batches and the clinical trial materials batches are being manufactured for clinical trial. This would complete the manufacturing portion of the NDA dossier to be submitted to the FDA to support the marketing approval of IT-102. IT-103 is at the beginning of this process.
| 22 |
Clinical Study Plan for IT-102 (Celebrex/lisinopril)
One BE study is planned to compare and assess the safety and pharmacokinetic characteristics between co-administered 200 mg celecoxib and 20 mg or 2.5 mg lisinopril and IT-102 (200/20 mg or 200/2.5 mg celecoxib/lisinopril) monotherapy in healthy volunteers aged between 20 and 50 years. The study could be completed in six months.
This is going to be a single-dose, cross-over study of 60 subjects divided into two cohorts and treated over four periods. One tablet of IT-102 at the highest dose (FDC tablet of 200 mg celecoxib/20 mg lisinopril) or the lowest dose (FDC tablet of 200 mg celecoxib/2.5 mg lisinopril) will be administered once orally as the test drug. Co-administration once daily of one tablet each of 200 mg celecoxib and 20 mg lisinopril or one tablet each of 200 mg celecoxib and 2.5 mg lisinopril will be used as the comparator for the highest and lowest dose of the FDC, respectively.
For comparison of IT-102 with the comparator (co-administration of dose matched celecoxib and lisinopril), the 90% confidence intervals of the geometric mean ratios for the primary pharmacokinetic parameters (AUC and Cmax) will be used for determination of BE according to the FDA’s criteria. Adverse events (AEs) will also be assessed and compared between IT-102 and the comparator.
Phase III Safety and Efficacy Study Plan. The purpose of this study is to evaluate the effect of celecoxib on the efficacy and safety of lisinopril in subjects with OA and hypertension requiring antihypertensive therapy. One multi-site, randomized, double-blind, placebo-controlled, 4-arm, 2-week phase 3 clinical study is planned to demonstrate the efficacy and safety of IT-102. It is planned to recruit 150 to 250 patients with OA and hypertension randomized into four arms, which are IT-102 (200 mg celecoxib/20 mg lisinopril), celecoxib (200 mg), lisinopril (20 mg) and placebo. The primary endpoint is demonstration that the reduction in blood pressure by IT-102 (200 mg celecoxib/20 mg lisinopril) is at least 50% of the reduction by lisinopril (20 mg) alone in the patients with OA and hypertension. The study is not planned to demonstrate pain reduction. AEs will also be assessed and compared among IT-102 and the comparator groups. The study could be completed in 12 months.
Both BE and phase III studies are to be conducted concurrently and followed by NDA submission of IT-102 for FDA approval by the 505(b)2 pathway.
IT-103 will undergo the same clinical developmental plan as IT-102, as summarized below:

| 23 |
M101- Outlicensing or spin off asset
M101 is being developed as beta-catenin siRNA/celecoxib combination against FAP. This is a combination of IT-102 and CEQ508 (tkRNAi beta-catenin shRNA). Celecoxib was originally approved for FAP, however, it was removed from the market due to fear of cardiovascular risks during the VIOXX withdrawal. But with the PRECISION trial showing that celecoxib is as safe as ibuprofen and naproxen, we anticipate higher acceptance of celecoxib. Furthermore, with the addition of lisinopril to control edema and hypertension, we anticipate that high doses of celecoxib would be safe and effective against FAP. Additionally, the systemic suppression of COX-2 directly and beta-catenin indirectly with celecoxib will be augmented by targeted and local suppression of beta-catenin by beta-catenin shRNA (CEQ508). Together we anticipate a synergistic and safe and effective suppression of polyps in FAP.
FAP is an autosomal dominant disorder with an estimated incidence of approximately 1:10,000 persons and is a well described form of hereditary colorectal cancer (Bisgaard 1994, Neklason 2008, Steinbach 2000). FAP is caused by a heterozygous mutation in the Adenomatous Polyposis Coli (APC) gene located on chromosome 5, which results in low levels of functional APC protein required to regulate intracellular levels of beta-catenin. This dysregulation and accumulation of beta-catenin initiates an activation of downstream target genes, resulting in uncontrolled cellular proliferation, hyperplasia, adenoma formation, and an increased risk of colon cancer development (Kinzler 1996). In addition, the APC gene also plays a role in chromosome segregation through microtubule binding and cell polarity. Almost all of the cancer- causing mutations in the APC gene create a truncated gene devoid of its C-terminal region. Loss of the C-terminal region leads to chromosome instability, a hallmark of cancer (Kinzler 1996, Hanahan 2000). Typically, FAP results in the formation of hundreds to thousands of polyps in the large and small intestine. While these polyps start out benign, malignant transformation into colon cancer occurs 100% of the time when untreated. When the frequency of polyp formation exceeds the criteria for polypectomy as assessed by the physician, surgical intervention including a partial or complete colectomy is performed. Colectomies are typically performed in the late teenage years or early twenties. By age 35, 95% of individuals with FAP have developed polyps. Without surgical intervention, the mean age of colon cancer onset is 39 years of age (range of 34-43 years) (Trimbath 2002). In Attenuated FAP, the APC mutation resides in the 3’UTR (untranslated region) of the APC gene, resulting in a less severe phenotype of FAP. Patients usually develop fewer (<100) polyps, and the age at which polyp formation occurs is later than FAP. Colon cancer develops in these individuals as well but at a slower rate, typically after 40 years of age.
Duodenal/periampullary adenocarcinoma is the next leading cause of death in FAP patients following colorectal cancer (Vasen 2008). FAP patients are also at increased risk of developing other malignancies, including hepatoblastoma, pancreatic, thyroid, biliary tree, and brain tumors. Additionally, the risk of cancer forming in the remaining stump of the rectum and small intestine, after colectomy, remains high (Trimbath 2002, Vasen 2008).
Celecoxib for treatment of FAP
Cyclooxygenase (COX) inhibiting NSAIDs has been thoroughly investigated as a potential chemopreventive drug. Overexpression of COX-2 has been identified in colorectal adenomas and carcinomas. This overexpression was linked with reduced apoptosis, enhanced cell growth, tumor angiogenesis, tissue invasion and metastasis. This is likely attributed to the mechanism of COX-2, whereby expression of COX-2 prevents degradation of β-catenin protein increasing proliferation and survival. As such, the COX-2 specific inhibitor, celecoxib, has been utilized for the treatment of FAP patients.
In a randomized, double-blind, placebo-controlled study, treatment of celecoxib at 100 mg or 400 mg twice daily was compared against placebo for six months. Significant reduction in mean number of colorectal polyps (28% vs 4.5%, p = 0.003) and polyp burden assessed as the sum of polyp diameters (30.7% vs 4.9%, p = 0.001) was observed in patients treated with 400 mg twice daily compared with placebo (Steinbach et al. 2000). No significant reduction was observed in patients treated with 100 mg twice daily in terms of mean number of colorectal polyps (11.9%, p = 0.33) and polyp burden (14.6%, p = 0.09). In a similar study, 400 mg twice daily treatment of celecoxib showed significant reduction in area of duodenal polyposis (30.8% vs 8.3%, p = 0.049) of patients with >5% coverage at baseline compared to placebo (Phillips et al. 2002). No significant reduction was observed from treatment at 100 mg (26.6%, p = 0.252).
| 24 |
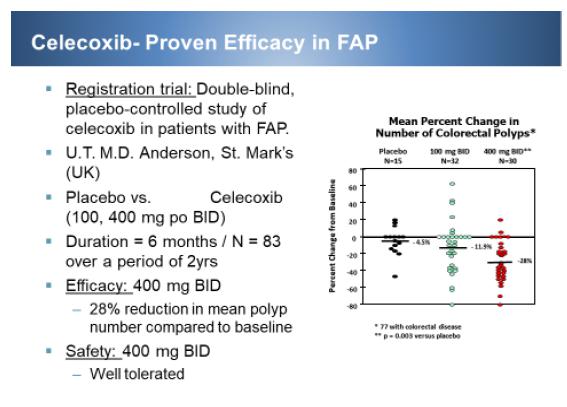
Evaluation of celecoxib for treatment of FAP was also evaluated in children with APC gene mutations and/or adenomas with a family history of FAP. Children were evaluated in a phase I, dose-escalation trial in three successive cohorts of six children (Lynch et al. 2010). Random assignment of subjects in a 2:1 placebo ratio was conducted for cohort 1 (4 mg/kg/day) to cohort 2 (8 mg/kg/day) to cohort 3 (16 mg/kg/day). Colonoscopies were performed at baseline and month 3. At month 3, a 39.1% increase in number of polyps was observed in placebo patients whereas a 44.2% reduction was seen in the highest dose celecoxib group (p = 0.01). This corresponds with the adult dose of 400 mg BID and was shown to be safe and well tolerated. Reduction in number of polyps was also observed in the 8 mg/kg/day group (adult dose of 200 mg BID), with a 44.2% decrease. However, an increase of 69.7% was observed in patients treated at 4 mg/kg/day (adult dose 100 mg BID). In line with results observed in adult patients, high dose of celecoxib is most effective in treating patients with FAP.
Together, these studies have shown that celecoxib is effective in treating FAP at high doses (400 mg twice daily). However, although therapeutically effective, the high dose of celecoxib results in higher risk of cardiovascular adverse events. The increased risk has lowered the attractiveness of celecoxib as an effective treatment for FAP. However, should the risk be diminished, a novel and previously FDA approved drug would be available for treatment of a disease that currently has no effective treatment. This gap can be filled by IT-102.
IT-102 is a FDC of celecoxib, a COX-2 selective inhibitor, and lisinopril, an ACE inhibitor. The combination of an antihypertensive agent, e.g. lisinopril, with celecoxib is intended to suppress the cardiovascular side effects associated with high dose of celecoxib to offer a safe and tolerable therapeutic option for the medical management of FAP patients. Coupled with local and target suppression of beta-catenin by CEQ508 – we believe that M101 should be a safe and effective agent against FAP.
CEQ508 for treatment of FAP
TransKingdom RNA™ interference (tkRNAi) platform. tkRNAi is a broad-reaching platform that can be used to develop highly specific drug products for a diverse set of diseases. The tkRNAi platform involves the modification of bacteria to deliver shRNA to cells of the gastrointestinal tract. A significant advantage of the tkRNAi platform is oral (by mouth) delivery making this platform extremely patient friendly while harnessing the full potential of the RNAi process. The tkRNAi platform has demonstrated in vivo mRNA down-regulation of both inflammatory and cancer targets, thus providing a unique opportunity to develop RNAi-based therapeutics against inflammation and oncology diseases such as Crohn’s Disease, ulcerative colitis and colon cancer. For our own clinical pipeline, we have used the tkRNAi platform to discover and develop CEQ508 for the treatment of FAP as a beta-catenin siRNA knockdown.
| 25 |
Phase I proof of concept (“POC”) was conducted and the trial closed at meeting both its primary and secondary endpoints. START-FAP is a phase I dose-escalating study to evaluate safety and tolerability of single daily doses of CEQ508 in adult patients with FAP. Six patients with FAP were orally administered (3 each in Cohort 1 and 2) with CEQ-508 (108 and 109 colony forming units [CFU]/day for 28 days). The primary objective was to establish general safety for orally administered CEQ508 and to determine the maximum tolerated dose. The secondary objective was the effectiveness of CEQ508 on the gene expression of the target gene beta-catenin. Gene expression was evaluated in GI tissues (duodenum, ileum, right and left colon, antrum) taken during endoscopy examinations at baseline and at end-of-treatment (EOT). Expression levels were measured using qPCR and analyzed with ViiA™ 7 Real-Time system (Life Technologies, Carlsbad, CA). Ct values of β-catenin were normalized to two of three housekeeping genes (EIF2B1, HPRT1, GUSβ). A mixed Nested-ANOVA model was used to evaluate beta-catenin knockdown in normal mucosa and polyps. This phase I trial of bacterial delivery of RNAi investigational agent CEQ508 in FAP patients demonstrated an acceptable safety profile and was well-tolerated at the two bacterial dose levels tested, with no MTD having been identified. Without hitting MTD, START-FAP achieved both primary endpoint of safety and secondary endpoint of beta-catenin knockdown.
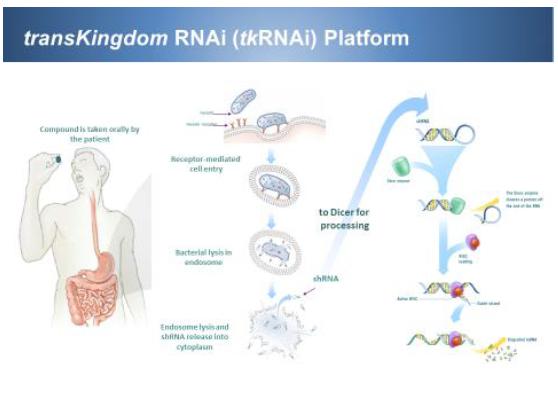
Analysis of Ct values stratifying by tissue type showed a decrease in β-catenin expression moving down the gastrointestinal tract (Duodenum > Ileum > Colon > Antrum). A Mixed nested-ANOVA model was developed to compare the levels of β-catenin in the normal mucosa and polyps taken at baseline and EOT.
A statistical model was generated to test whether CEQ508 was successful in suppressing beta-catenin expression. The model developed by pooling data from both cohorts for normal mucosa samples explained a significant proportion of variation (R-squared = 0.64, P<0.0001; Patient to patient variation accounted for 48% of the total variation). No significant reduction in overall β-catenin expression was observed in EOT samples. Modeling of pooled data for polyp samples explained a significant proportion of variation (R-squared = 0.54, P<0.0001; Patient to patient variation accounted for 6% of the total variation). Significant reduction was observed in overall β-catenin expression in EOT samples (F1,113.1 = 6.87, P=0.01). Furthermore, significant reduction of β-catenin expression in the Duodenum was observed in EOT samples (Linear Contrast, Effect size = 0.363, 22.2% decrease, T = 2.75, P=0.007).
| 26 |
Evaluation of individual cohorts was also examined. The model for cohort 1 in normal mucosa explained a significant proportion of variation (R-squared = 0.84, P<0.0001; Patient to patient variation accounted for 80% of the total variation). Nonstatistical significant reduction was observed in overall β-catenin expression in EOT samples (F1,46 = 6.03, P=0.018). Therefore, component analyses were deemed irrelevant and cohort 1 was declared not effective. The model for cohort 2 in polyps explained a significant proportion of variation (R-squared = 0.69, P<0.0001; Patient to patient variation accounted for 8.2% of the total variation). Significant reduction was observed in overall β-catenin expression at EOT (; F1,86.06 = 13.13, P=0.0005). There was a significant reduction of β-catenin expression in the Duodenum (Linear Contrast, Effect size = 0.72, 39.3% decrease, T = 5.3, P<0.0001) and Ileum (Effect size = 0.49, 28.8% decrease, T = 2.57, P=0.012).

Together, the data indicate a higher expression of β-catenin in the small intestine (duodenum, ileum) compared to the large intestine (colon, antrum). Additionally, the mixed nest-ANOVA model shows effective decrease in β-catenin mRNA from treatment of CEQ508. The models indicate that treatment at 108 CFU/day may not be effective enough at lowering β-catenin expression. However, significant effects were observed within the Duodenum and Ileum after treatment of 109 CFU/day of CEQ508. Furthermore, decrease in β-catenin was observed only in the polyps while insignificant effects were observed within the normal mucosa. This shows that CEQ508 is a therapeutically effective and specifically targeted novel treatment for patients with FAP. In addition, CEQ508 was granted orphan drug designation and fast track designation by the FDA.
Clinical Program for M101: Combination of celecoxib and CEQ508
We believe that the effectiveness of CEQ508 and celecoxib justify the combination as M101. We plan to meet with the FDA to discuss an SPA with a clearly defined clinical design and endpoints for regulatory approval. The meeting is planned for 2018 and the trial may start in 2019. If successful, we anticipate a potential launch of the product in 2024.
| 27 |
Partnering and Licensing Agreements
Oncotelic Inc.
On July 17, 2017, we entered into a License Agreement with Oncotelic, Inc. pursuant to which, among other things, we provided to Oncotelic a license to our SMARTICLES platform for the delivery of antisense DNA therapeutics, as well as a license to our conformationally restricted nucleotide (“CRN”) technology with respect to TGF-Beta. Under the terms of the License Agreement, Oncotelic also agreed to purchase 49,019 shares of our common stock for an aggregate purchase price of $250,000 ($5.10 per share), with such purchase and sale to be made pursuant to a Stock Purchase Agreement to be entered into between us and Oncotelic within thirty (30) days following the date of the License Agreement. Under the terms of the License Agreement, we could receive up to $90 million in success-based milestones based on commercial sales of licensed products. In addition, if Oncotelic determines to pursue further development and commercialization of products under the License Agreement, Oncotelic agreed, in connection therewith, to purchase shares of our common stock for an aggregate purchase price of $500,000, with the purchase price for each share of common stock being the greater of $5.10 or the volume weighted average price of our common stock for the thirty (30) trading days immediately preceding the date on which Oncotelic notifies us that it intends to pursue further development or commercialization of a licensed product. As of the date of this filing, Oncotelic has not yet completed its obligations to purchase the stock and Oncotelic, and we are still in discussions to complete such purchase. Dr. Trieu, our Executive Chairman, is the principal stockholder and Chief Executive Officer of Oncotelic.
LipoMedics Inc.
On February 6, 2017, we entered into a License Agreement with LipoMedics, Inc. pursuant to which, among other things, we provided to LipoMedics a license to our SMARTICLES platform for the delivery of nanoparticles including small molecules, peptides, proteins and biologics. On the same date, we also entered into a Stock Purchase Agreement with LipoMedics pursuant to which we issued to LipoMedics an aggregate of 86,206 shares of our common stock for a total purchase price of $250,000. Under the terms of the License Agreement, we could receive up to $90 million in success-based milestones based on commercial sales of licensed products. In addition, if LipoMedics determines to pursue further development and commercialization of products under the License Agreement, LipoMedics agreed, in connection therewith, to purchase shares of our common stock for an aggregate purchase price of $500,000, with the purchase price for each share of common stock being the greater of $2.90 or the volume weighted average price of our common stock for the thirty (30) trading days immediately preceding the date on which LipoMedics notifies us that it intends to pursue further development or commercialization of a licensed product. Dr. Trieu, our Executive Chairman, is the Chairman of the Board and Chief Operating Officer of LipoMedics.
Autotelic LLC
In connection with the Merger Agreement and the closing of the Merger, on November 15, 2016, Marina entered into a License Agreement with Autotelic LLC, a stockholder of IThenaPharma that became the holder of 2,312,355 shares of Marina common stock as a result of the Merger, and an entity of which Dr. Trieu, our Executive Chairman, serves as Chief Executive Officer, pursuant to which (A) Marina licensed to Autotelic LLC certain patent rights, data and know-how relating to FAP and nasal insulin, for human therapeutics other than for oncology-related therapies and indications, and (B) Autotelic LLC licensed to Marina certain patent rights, data and know-how relating to IT-102 and IT-103, in connection with individualized therapy of pain using a non-steroidal anti-inflammatory drug and an anti-hypertensive without inducing intolerable edema, and treatment of certain aspects of proliferative disease, but not including rights to IT-102/IT-103 for Therapeutic Drug Monitoring (TDM) guided dosing for all indications using an Autotelic Inc. TDM Device. Marina also granted a right of first refusal to Autotelic LLC with respect to any license by Marina of the rights licensed by or to Marina under the License Agreement in any cancer indication outside of gastrointestinal cancers.
The License Agreement shall immediately terminate, all rights granted by a licensor under the License Agreement shall immediately revert forthwith to the applicable licensor, all benefits which have accrued under the License Agreement shall automatically be transferred to the applicable licensor, and all rights, title and interest in the licensed intellectual property shall immediately revert back to the applicable licensor if: (i) the applicable licensee makes a general assignment for the benefit of its creditors prior to the two (2) year anniversary of the date of the License Agreement; (ii) the applicable licensee applies for or consents to the appointment of a receiver, a custodian, a trustee or liquidator of all or a substantial part of its intellectual property prior to the two (2) year anniversary of the date of the License Agreement; (iii) prior to the two (2) year anniversary of the date of the License Agreement, and without the consent of the applicable licensor, the applicable licensee effects a Change of Control Transaction (as defined in the License Agreement); (iv) the applicable licensee ceases operations; or (v) the applicable licensee fails to take any material steps, as reasonably determined by the applicable licensor, to develop the licensed intellectual property prior to the one (1) year anniversary of the date of the License Agreement (each of the foregoing items (i) through (v), a “Termination Event”). Upon the occurrence of any Termination Event, the applicable licensee shall immediately discontinue all use of the licensed intellectual property.
| 28 |
Hongene Biotechnology
In September 2015, Marina entered into a license agreement with Hongene, a leader in process development and analytical method development of oligonucleotide therapeutics, regarding the development and supply of certain oligonucleotide constructs using our CRN technology. We could receive double digit percentage royalties on the sales of research reagents using our CRN technology.
MiNA
On December 17, 2014, Marina entered into a license agreement with MiNA Therapeutics, Inc. (“MiNA”) regarding the development and commercialization of small activating RNA-based therapeutics utilizing MiNA’s proprietary oligonucleotides and our SMARTICLES nucleic acid delivery technology. MiNA will have full responsibility for the development and commercialization of any products arising under the agreement. MiNA paid an upfront fee of $0.5 million in January 2015 and an accelerated milestone payment of $200,000 in November 2015. We could receive up to an additional $49 million in clinical and commercialization milestone payments, as well as royalties on sales, based on the successful development of MiNA’s potential product candidates.
Rosetta
On April 1, 2014, Marina entered into a strategic alliance with Rosetta to identify and develop microRNA- (“miRNA”) based products designed to diagnose and treat various neuromuscular diseases and dystrophies. Under the terms of the alliance, Rosetta will apply its industry leading miRNA discovery expertise for the identification of miRNAs involved in the various dystrophy diseases. If the miRNA is determined to be correlative to the disease, Rosetta may further develop the miRNA into a diagnostic for patient identification and stratification. If the miRNA is determined to be involved in the disease pathology and represents a potential therapeutic target, Marina may develop the resulting miRNA-based therapeutic for clinical development. The alliance is exclusive as it relates to neuromuscular diseases and dystrophies, with both companies free to develop and collaborate outside this field both during and after the terms of the alliance.
Novartis
On August 2, 2012, Marina and Novartis entered into a worldwide, non-exclusive License Agreement for the CRN technology for the development of both single and double-stranded oligonucleotide therapeutics. Novartis made a $1.0 million one-time payment for the non-exclusive license. In addition, in March 2009, Marina granted to Novartis a worldwide, non-exclusive, irrevocable, perpetual, royalty-free, fully paid-up license, with the right to grant sublicenses, to the DiLA2-based siRNA delivery platform in consideration of a one-time, non-refundable fee of $7.25 million. Novartis may terminate this agreement immediately upon written notice.
Monsanto
On May 3, 2012, Marina and Monsanto entered into a worldwide exclusive Intellectual Property License Agreement for Marina’s delivery and chemistry technologies. Marina and Monsanto also entered into a Security Agreement pursuant to which Marina granted to Monsanto a security interest in that portion of its IP that is the subject of the License Agreement in order to secure the performance of Marina’s obligations under the License Agreement. Monsanto paid $1.5 million in initiation fees, and may be required to pay royalties on product sales in the low single digit percentages. Monsanto may terminate the License Agreement at any time in whole or as to any rights granted thereunder upon three months’ prior written notice.
ProNAi
On March 13, 2012, Marina entered into an Exclusive License Agreement with ProNAi therapeutics, Inc. (“ProNAi”) regarding the development and commercialization of ProNAi’s proprietary DNAi-based therapeutics utilizing SMARTICLES. The License Agreement provided that ProNAi would have full responsibility for the development and commercialization of any products arising under the License Agreement. ProNAi’s clinical compound utilizing SMARTICLES, PNT2258, is a first-in-class, 24-base, single-stranded, chemically-unmodified DNA oligonucleotide drug targeting BCL2, which proceeded to a phase 2 clinical study. In June 2016, ProNAi suspended the development of PNT2258 based on its review of the interim results from a phase 2 trial of PNT2258. Thereafter, the License Agreement has been terminated.
| 29 |
Novosom
On July 27, 2010, Marina acquired the intellectual property of Novosom for SMARTICLES. As per the terms of the acquisition agreement (the “Original Purchase Agreement”), Marina was required to pay to Novosom an amount equal to 30% of the value of each upfront (or combined) payment actually received in respect of the license of liposomal-based delivery technology or related product or disposition of the liposomal-based delivery technology by Marina, up to $3.3 million, which amount was to be paid in shares of common stock, or a combination of cash and shares of common stock, at Marina’s discretion. On September 8, 2017, we entered into an Intellectual Property Purchase Agreement (the “IP Purchase Agreement”) with Novosom pursuant to which we sold to Novosom substantially all of our intellectual property estate relating to SMARTICLES (the “Smarticles IP”), including that acquired pursuant to the Original Purchase Agreement, for an aggregate purchase price of $1.00. As per the IP Purchase Agreement, we will retain rights to any future payments that may be due to us pursuant to those agreements that we entered into with third parties pursuant to which we provided to such third parties certain licenses and rights with respect to the Smarticles IP, including milestone and royalty payments, if any, and Novosom relinquished any rights that it may have under the Original Purchase Agreement to any portion of such payments.
Valeant Pharmaceuticals
On March 23, 2010, Marina acquired intellectual property related to the CRN chemistry from Valeant Pharmaceuticals North America (“Valeant”). Subject to meeting certain milestones triggering the obligation to make any such payments, we may be obligated to make a product development milestone payment of $5.0 million and $2.0 million within 180 days of FDA approval of a NDA for our first and second CRN related product, respectively. To date, we had not made any such milestone payments but have milestone obligations of $0.1 million based on CRN licenses to date. Valeant is entitled to receive earn-outs based upon a percentage in the low single digits of future commercial sales and earn-outs based upon a percentage in the low double digits of future revenue from sublicensing. We are required to pay Valeant an annual amount equal to $50,000 per assigned patent which shall be creditable against other payment obligations. The term of our financial obligations under the agreement shall end, on a country-by-country basis, when there no longer exists any valid claim in such country. We may terminate the agreement upon 30-day notice, or upon 10-day notice in the event of adverse results from clinical studies.
Proprietary Rights and Intellectual Property
We rely primarily on patents and contractual obligations with employees and third parties to protect our proprietary rights. We have sought, and intend to continue to seek, appropriate patent protection for important and strategic components of our proprietary technologies by filing patent applications in the U.S. and certain foreign countries. There can be no assurance that any of our patents will guarantee protection or market exclusivity for our products and product candidates. We also use license agreements both to access external technologies and to convey certain intellectual property rights to others. Our financial success will be dependent in part on our ability to obtain commercially valuable patent claims and to protect our intellectual property rights and to operate without infringing upon the proprietary rights of others. As of December 31, 2017, we owned or controlled the issued or allowed patents, and the U.S. and foreign patent applications, to protect our proprietary technologies, as set forth in the table below (which table does not include the intellectual property relating to our DiLA2 delivery system, which we are seeking to divest).
| Estimated Expiration | No.
of Issued/Allowed Patents | Jurisdiction | No.
of Pending Patents | Jurisdiction | ||||
| 2019 | 3 | U.S. | ||||||
| 2020 | 1 | Germany | ||||||
| 2021 | 1 | U.S. | ||||||
| 2023 | 1 | U.S. | ||||||
| 2024 | 1 | China | ||||||
| 3 | U.S. | |||||||
| 2025 | 2 | U.S. | 1 | U.S. | ||||
| 2026 | 1 each | China, Japan | 1 | Europe | ||||
| 2027 | 2 | U.S. | 1 | U.S. | ||||
| 2028 | 1 each | New Zealand, China, Japan, Australia | 1 each | China, U.S. Europe | ||||
| 2 | Europe | |||||||
| 4 | U.S. | |||||||
| 2029 | 1 | U.S. | ||||||
| 2031 | 2 | U.S. | 1 | Europe | ||||
| 2032 | 1 each | Singapore, Australia, Europe, U.S. | 1 each | Hong Kong, India | ||||
| 2035 | 1 each | Taiwan, Korea, India, Europe, Australia, Canada, China, Japan, U.S. |
| 30 |
The patents listed in the table above will expire generally between 2019 and 2035, subject to any potential patent term extensions and/or supplemental protection certificates that would extend the terms of the patents in countries where such extensions may become available.
Competition
The biopharmaceutical industry is characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. The key competitive factors affecting the success of all of our products and product candidates are their efficacy, safety, convenience, price, the level of generic competition and the availability of reimbursement from government and other third-party payors. While we believe that our technology, knowledge, experience and scientific resources provide us with certain competitive advantages, we face potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions, governmental agencies, and public and private research institutions. Our products, and any product candidates that we successfully develop and commercialize, will compete with existing therapies and new therapies that may become available in the future.
Many of the companies against which we are competing or against which we may compete in the future have significantly greater financial and other resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the biopharmaceutical industry may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete, or may compete, with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or that may be necessary for, our programs.
The commercial opportunity for our product candidates could be reduced or eliminated if our competitors develop and commercialize drugs that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any drugs that we may develop. Our competitors also may obtain FDA or other regulatory approval for their product candidates more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. In addition, our ability to compete may be affected in many cases by insurers or other third-party payors seeking to encourage the use of generic drugs.
Government Regulation
Government authorities in the U.S. and other countries extensively regulate the research, development, testing, manufacture, labeling, promotion, advertising, distribution and marketing, among other things, of drugs and biologic products. Our Prestalia product is, and all of our foreseeable product candidates are expected to be, regulated as drug products.
| 31 |
In the U.S., the FDA regulates drug products under the Federal Food, Drug and Cosmetic Act (the “FDCA”), and other laws within the Public Health Service Act. Failure to comply with applicable U.S. requirements, both before and after approval, may subject us to administrative and judicial sanctions, such as a delay in approving or refusal by the FDA to approve pending applications, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, and/or criminal prosecutions. Before our drug products are marketed they must be approved by the FDA. The steps required before a novel drug product is approved by the FDA include: (1) pre-clinical laboratory, animal, and formulation tests; (2) submission to the FDA of an Investigational New Drug Application (“IND”) for human clinical testing, which must become effective before human clinical trials may begin; (3) adequate and well-controlled clinical trials to establish the safety and effectiveness of the product for each indication for which approval is sought; (4) submission to the FDA of a New Drug Application (“NDA”); (5) satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug product is produced to assess compliance with cGMP and FDA review; and finally (6) approval of an NDA.
Pre-clinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies. The results of the pre-clinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND, which must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions, such as the conduct of the trials as outlined in the IND. In such a case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. There can be no assurance that submission of an IND will result in FDA authorization to commence clinical trials. Once an IND is in effect, each clinical trial to be conducted under the IND must be submitted to the FDA, which may or may not allow the trial to proceed.
Clinical trials involve the administration of the investigational drug to human subjects under the supervision of qualified physician-investigators and healthcare personnel. Clinical trials are typically conducted in three defined phases, but the phases may overlap or be combined. Phase 1 usually involves the initial administration of the investigational drug or biologic product to healthy individuals to evaluate its safety, dosage tolerance and pharmacodynamics. Phase 2 usually involves trials in a limited patient population, with the disease or condition for which the test material is being developed, to evaluate dosage tolerance and appropriate dosage; identify possible adverse side effects and safety risks; and preliminarily evaluate the effectiveness of the drug or biologic for specific indications. Phase 3 trials usually further evaluate effectiveness and test further for safety by administering the drug or biologic candidate in its final form in an expanded patient population. Our product development partners, the FDA, or we may suspend clinical trials, if any, at any time on various grounds, including any situation where we or our partners believe that patients are being exposed to an unacceptable health risk or are obtaining no medical benefit from the test material.
Assuming successful completion of the required clinical testing, the results of the pre-clinical trials and the clinical trials, together with other detailed information, including information on the manufacture and composition of the product, are submitted to the FDA in the form of an NDA requesting approval to market the product for one or more indications. Before approving an application, the FDA will usually inspect the facilities where the product is manufactured, and will not approve the product unless cGMP compliance is satisfactory. If the FDA determines the NDA is not acceptable, the FDA may outline the deficiencies in the NDA and often will request additional information. If the FDA approves the NDA, certain changes to the approved product, such as adding new indications, manufacturing changes or additional labeling claims are subject to further FDA review and approval. The testing and approval process requires substantial time, effort and financial resources, and we cannot be sure that any approval will be granted on a timely basis, if at all.
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the U.S. and for which there is no reasonable expectation that the cost of developing and making available in the U.S. a drug for this type of disease or condition will be recovered from sales in the U.S. for that drug. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. If a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications, including a full BLA, to market the same drug for the same indication, except in very limited circumstances, for seven years. The FDA granted orphan drug designation to CEQ508 for the treatment of FAP in December 2010.
| 32 |
In addition, regardless of the type of approval, we and our partners are required to comply with a number of FDA requirements both before and after approval. For example, we and our partners are required to report certain adverse reactions and production problems, if any, to the FDA, and to comply with certain requirements concerning advertising and promotion for products. In addition, quality control and manufacturing procedures must continue to conform to cGMP after approval, and the FDA periodically inspects manufacturing facilities to assess compliance with cGMP. Accordingly, manufacturers must continue to expend time, money and effort in all areas of regulatory compliance, including production and quality control to comply with cGMP. In addition, discovery of problems, such as safety problems, may result in changes in labeling or restrictions on a product manufacturer or NDA holder, including removal of the product from the market.
Product Liability
We currently do not carry product liability insurance as no patients are currently being treated with our products. However, we intend to obtain product liability insurance in connection with our commercialization efforts for Prestalia and our clinical efforts for our other product candidates.
Environmental Compliance
Our research and development activities involve the controlled use of potentially harmful biological materials as well as hazardous materials, chemicals and various radioactive compounds. We are subject to federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and specific waste products. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of bio-hazardous materials. The cost of compliance with these laws and regulations could be significant and may adversely affect capital expenditures to the extent we are required to procure expensive capital equipment to meet regulatory requirements. At this time, we are not conducting any R&D activities that require compliance with federal, state or local laws.
Employees and Business Services
As of the date of this report, we have seven employees, all of whom are officers of our company, and all of whom – other than Mr. Ramelli, our Chief Executive Officer – spend a portion of their time working for other entities. To the extent that compensation is currently being paid to certain of our employees, such compensation is being paid with respect to work performed for our company by Autotelic Inc. as per the terms of the Master Services Agreement (as described below). None of our employees are covered by collective bargaining agreements.
In connection with the closing of the Merger, on November 15, 2016, Marina entered into a Master Services Agreement (the “MSA”) with Autotelic Inc., a stockholder of IThenaPharma that became the holder of 525,535 shares of Marina common stock as a result of the Merger, and an entity of which Dr. Trieu serves as Chairman of the Board, pursuant to which Autotelic Inc. agreed to provide certain business functions and services from time to time at Marina’s request. The MSA has a term of ten years, though it can be terminated by either party upon ninety (90) days’ prior written notice to the other party. The resources available to us through Autotelic Inc. include, without limitation, regulatory, clinical, preclinical, manufacturing, formulation, legal, accounting and IT.
As partial consideration for the services to be performed by Autotelic Inc. under the MSA, during the period prior to the date on which we have completed an equity offering of either common or preferred stock in which the gross proceeds therefrom is no less than $10 million, we shall issue to Autotelic Inc. warrants to purchase shares of our common stock (the “MSA Warrants”), with the exercise price for such MSA Warrants being based on the closing price of our common stock at the time the MSA Warrants are issued; provided, that in no event shall such price be lower than the lower of (x) $2.80 per share or (y) the lowest exercise price of any warrants that have been issued by us in a capital raising transaction (and that would otherwise reduce the exercise price of any other outstanding warrants issued by us) during the period beginning on November 15, 2016 and ending on the date of the issuance of the applicable MSA Warrants. The number of shares of common stock for which the MSA Warrants are exercisable shall be equal to the quotient obtained by dividing the actual costs to Autotelic Inc. of providing the services under the MSA by the exercise price for the MSA Warrants.
| 33 |
Company Information
We are a reporting company and are required to file annual, quarterly and current reports, proxy statements and other information with the SEC. You may read and copy these reports, proxy statements and other information at the SEC’s Public Reference Room at 100 F Street N.E., Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 or e-mail the SEC at publicinfo@sec.gov for more information on the operation of the public reference room. Our SEC filings are also available at the SEC’s website at http://www.sec.gov. Our Internet address is http://www.marinabio.com. There we make available, free of charge, our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and any amendments to those reports, as soon as reasonably practicable after we electronically file such material with, or furnish such material to, the SEC.
Risks Relating to Our Financial Condition and Business Operations
Our current cash and other sources of liquidity are not sufficient to fund our intended operations. We will require substantial additional funding to continue our operations beyond that date. If additional capital is not available, we may have to curtail or cease operations, or take other actions that could adversely impact our shareholders.
Our business does not generate the cash necessary to finance our operations. We incurred net operating losses of approximately $6.2 million and $0.84 million in the years ended December 31, 2017 and 2016, respectively. We will require significant additional capital to:
| ● | ramp up commercialization efforts with respect to our FDA-approved Prestalia product; | |
| ● | fund research and development activities relating to the development of our product candidates, including clinical and pre-clinical trials; | |
| ● | obtain regulatory approval for our product candidates; | |
| ● | pursue licensing opportunities for our technologies, products and product candidates; | |
| ● | protect our intellectual property; | |
| ● | attract and retain highly-qualified personnel; | |
| ● | respond effectively to competitive pressures; and | |
| ● | acquire complementary businesses or technologies. |
Our future capital needs depend on many factors, including:
| ● | the scope, duration and expenditures associated with our research, development and commercialization efforts (including the costs of clinical and pre-clinical trials); | |
| ● | continued scientific progress in our programs; | |
| ● | the outcome of potential partnering or licensing transactions, if any; | |
| ● | competing technological developments; | |
| ● | our proprietary patent position, if any, in our products; and | |
| ● | the regulatory approval process for our products. |
| 34 |
We believe that our currently available cash resources (including the remaining amount available to us under our Line Letter with Autotelic Inc.) will not be sufficient to fund our intended operations. We will need to raise additional funds through public or private equity offerings, debt financings or additional strategic alliances and licensing arrangements to continue our operations past the foregoing dates. We may not be able to obtain additional financing on terms favorable to us, if at all. General market conditions, as well as market conditions for companies at our stage of development, may make it difficult for us to seek financing from the capital markets, and the terms of any financing may adversely affect the holdings or the rights of our stockholders. For example, if we raise additional funds by issuing equity securities, further dilution to our stockholders will result, which may substantially dilute the value of their investment. In addition, as a condition to providing additional funds to us, future investors may demand, and may be granted, rights superior to those of existing stockholders. Debt financing, if available, may involve restrictive covenants that could limit our flexibility to conduct future business activities and, in the event of insolvency, could be paid before holders of equity securities received any distribution of corporate assets. We may be required to relinquish rights to our technologies or drug candidates, or grant licenses through alliance, joint venture or agreements on terms that are not favorable to us, in order to raise additional funds. If adequate funds are not available, we may have to further delay, reduce or eliminate one or more of our planned activities with respect to our business, or terminate our operations. These actions would likely reduce the market price of our common stock.
We have no history of profitability and there is a potential for fluctuation in operating results.
We have experienced significant operating losses since inception. We currently have no revenues from product sales, and although we anticipate generating revenues from the sale of our Prestalia product and such other products that may obtain regulatory approval, the amount of such revenues is uncertain. We expect that the continued operation of our business will cause us to continue to experience losses in the near term as we pursue our plans to commercialize Prestalia and continue our development plans with respect to our product candidates. See the sections entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Cautionary Statement Regarding Forward-Looking Statements” contained elsewhere in this report.
We and our partners are engaged in the business of developing and commercializing novel therapeutic products. The process of developing such products requires significant research and development efforts, including basic research, pre-clinical and clinical development, and regulatory approval. These activities, together with our sales, marketing, general and administrative expenses, have resulted in operating losses in the past, and there can be no assurance that we can achieve profitability in the future. Our ability to achieve profitability depends on our ability, alone or with our partners, to commercialize and develop drug candidates, conduct pre-clinical development and clinical trials, obtain necessary regulatory approvals, and manufacture, distribute, market and sell drug products. We cannot assure you of the success of any of these activities or predict if or when we will ever become profitable.
There is substantial doubt about our ability to continue as a going concern, which may affect our ability to obtain future financing or engage in strategic transactions, and may require us to curtail our operations.
Our financial statements as of December 31, 2017 were prepared under the assumption that we will continue as a going concern. The independent registered public accounting firm that audited our 2017 consolidated financial statements, in their report, included an explanatory paragraph referring to our recurring losses and expressing substantial doubt in our ability to continue as a going concern. Our financial statements do not include any adjustments that might result from the outcome of this uncertainty. Our ability to continue as a going concern depends on our ability to raise substantial additional funds through public or private equity offerings, debt financings or additional strategic alliances and licensing arrangements. There can be no assurance that we will be successful in any such endeavors.
On April 16, 2018, we held the initial closing of our private placement of shares of our Series E Convertible Preferred Stock, and warrants to purchase shares of our common stock, as a result of which we raised over $10 million in net proceeds to our company.
| 35 |
If we are unable to raise sufficient additional capital, we may seek to merge with or be acquired by another entity, or to sell our assets to another entity, and that transaction may adversely affect our business and the value of our securities.
If we are unable to raise sufficient additional capital to continue our business or to execute on our business plan, we may seek to merge or combine with, or otherwise be acquired by, another entity with a stronger cash position, complementary work force, or product candidate portfolio or for other reasons. There are numerous risks associated with merging, combining or otherwise being acquired, whether in whole or in part. These risks include, among others, incorrectly assessing the quality of a prospective acquirer or merger-partner, encountering greater than anticipated costs in integrating businesses, facing resistance from employees and being unable to profitably deploy the assets of the new entity. The operations, financial condition, and prospects of the post-transaction entity depend in part on our and our acquirer/merger-partner’s ability to successfully integrate the operations related to our products, product candidates, business and technologies. We may be unable to integrate operations successfully or to achieve expected cost savings, and any cost savings that are realized may be offset by losses in revenues or other charges to operations. As a result, our stockholders may not realize the full value of their investment.
We are dependent on our key personnel, and if we are unable to retain such personnel, or to attract and retain other highly qualified personnel, then we may be unable to successfully develop our business.
Our ability to compete in the highly competitive biotechnology and pharmaceutical industries depends upon our ability to attract and retain highly qualified personnel. We are dependent on our management and scientific personnel, including Vuong Trieu, Ph.D., our Executive Chairman, Joseph W. Ramelli, our Chief Executive Officer, Larn Hwang, Ph.D., our Chief Scientific Officer, Mihir Munsif, our Chief Operating Officer, Erik Emerson, our Chief Commercial Officer, Amit Shah, our Chief Financial Officer, and Peter Weinstein, our Chief Legal Officer. There can be no assurance that we will be able to retain the services of any of the foregoing persons, or of any of our other current and future personnel, regardless of whether or not we have entered into employment agreements with such persons.
If we are unable to attract or retain qualified personnel, or if we are unable to adequately replace such personnel if we lose their services for any reason, our business could be seriously harmed. In addition, if we have to replace any of these individuals, we may not be able to replace the knowledge that they have about our operations.
If we make strategic acquisitions, we will incur a variety of costs and might never realize the anticipated benefits.
We have limited experience in independently identifying acquisition candidates and integrating the operations of acquisition candidates with our company. If appropriate opportunities become available, and we have sufficient resources to do so, we might attempt to acquire approved products, additional drug candidates, technologies or businesses that we believe are a strategic fit with our business, such as our acquisition of Prestalia from Symplmed. If we pursue any transaction of that sort, the process of negotiating the acquisition and integrating an acquired product, drug candidate, technology or business might result in operating difficulties and expenditures and might require significant management attention that would otherwise be available for ongoing development of our business, whether or not any such transaction is ever consummated. Moreover, we might never realize the anticipated benefits of any acquisition. Future acquisitions could result in, among other things, dilutive issuances of equity securities, the incurrence of debt, contingent liabilities, or impairment expenses related to goodwill, and impairment or amortization expenses related to other intangible assets, which could harm our financial condition.
Failure of our internal control over financial reporting could harm our business and financial results.
Our management is responsible for establishing and maintaining effective internal control over financial reporting. Internal control over financial reporting is a process to provide reasonable assurance regarding the reliability of financial reporting for external purposes in accordance with accounting principles generally accepted in the United States. Internal control over financial reporting includes maintaining records that in reasonable detail accurately and fairly reflect our transactions; providing reasonable assurance that transactions are recorded as necessary for preparation of the financial statements; providing reasonable assurance that receipts and expenditures of our assets are made in accordance with management authorization; and providing reasonable assurance that unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements would be prevented or detected on a timely basis. Any failure to maintain an effective system of internal control over financial reporting could limit our ability to report our financial results accurately and timely or to detect and prevent fraud.
| 36 |
In connection with the evaluation of our internal control over financial reporting as of December 31, 2016 that was undertaken by management in connection with the preparation of our Annual Report on Form 10-K for the fiscal year ended December 31, 2016, management determined that our lack of a sufficient complement of personnel with an appropriate level of knowledge and experience in the performance of an audit of a public company that is commensurate with our financial reporting requirements constituted a material weakness as of December 31, 2016. To remediate the foregoing material weakness, we hired additional experienced accounting and other personnel to assist with filings and financial record keeping, and took additional steps to improve our financial reporting systems and enhance our existing policies, procedures and controls, which efforts included the hiring of Amit Shah in October 2017 to serve as our Chief Financial Officer. Despite these remedial measures undertaken during 2017, we were not be able to adequately address the weaknesses that we identified , and thus management determined that the same weaknesses existed as of December 31, 2017.
We depend on our information technology and infrastructure.
We rely on the efficient and uninterrupted operation of information technology systems to manage our operations, to process, transmit and store electronic and financial information, and to comply with regulatory, legal and tax requirements. We also depend on our information technology infrastructure for electronic communications among our personnel, contractors, consultants and vendors. System failures or outages could compromise our ability to perform these functions in a timely manner, or could result in the loss of information, which could harm our ability to conduct business or delay our financial reporting. Such failures could materially adversely affect our operating results and financial condition.
In addition, we depend on third parties and applications on virtualized (cloud) infrastructure to operate and support our information systems. These third parties vary from multi-disciplined to boutique providers. Failure by these providers to adequately deliver the contracted services could have an adverse effect on our business, which in turn may materially adversely affect our operating results and financial condition. All information systems, despite implementation of security measures, are vulnerable to disability, failures or unauthorized access. If our information systems were to fail or be breached, such failure or breach could materially adversely affect our ability to perform critical business functions and sensitive and confidential data could be compromised.
Our business and operations could suffer in the event of system failures.
Our internal computer systems and those of our contractors, consultants and partners are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. Such events could cause interruption of our operations, and could result in a material disruption of our development programs and commercialization efforts. For example, the loss of pre-clinical trial data or data from completed or ongoing clinical trials for our product candidates, if any, could result in delays in our regulatory filings and development efforts and significantly increase our costs. To the extent that any disruption or security breach were to result in a loss of or damage to our data, or inappropriate disclosure of confidential or proprietary information, we could incur liability and our business operations could be delayed.
We may be unable to adequately protect our information technology systems from cyber-attacks, which could result in the disclosure of confidential information, damage our reputation, and subject us to significant financial and legal exposure.
Cyber-attacks are increasing in their frequency, sophistication and intensity, and have become increasingly difficult to detect. Cyber-attacks could include wrongful conduct by hostile foreign governments, industrial espionage, the deployment of harmful malware, denial-of-service, and other means to threaten data confidentiality, integrity and availability. A successful cyber-attack could cause serious negative consequences for our company, including the disruption of operations, the misappropriation of confidential information (including patient information) and trade secrets, and the disclosure of corporate strategic plans and results. To date, we have not experienced threats to our data and information technology systems. However, although we devote resources to protect our information technology systems, we realize that cyber-attacks are a threat, and there can be no assurance that our efforts will prevent information security breaches that would result in business, legal or reputational harm to us, or would have a material adverse effect on our operating results and financial condition.
| 37 |
Risks Related to the Development and Regulatory Approval of Our Drug Candidates
The development of pharmaceutical products is uncertain and may never lead to marketable products.
The future success of our operations will depend on the successful development, by us or our partners, of products based on our proprietary technologies. Our product candidates may not demonstrate in patients the chemical and pharmacological properties ascribed to them in laboratory studies, and they may interact with human biological systems in unforeseen, ineffective or harmful ways. We may make significant expenditures developing product candidates without success, and thus may never develop a marketable product utilizing our technologies. If neither we nor any of our partners develops and commercializes drugs based upon our technologies, we may be required to change the scope and direction of, or cease pursuing, our product candidates based on such technologies.
If we or our partners are unable to develop and commercialize product candidates utilizing our technologies, our business will be adversely affected.
A key element of our business strategy is to discover, develop and commercialize a portfolio of new products through internal efforts and through those of our partners. Whether or not any product candidates are ultimately identified, research programs to identify new disease targets and product candidates require substantial technical, financial and human resources. These research programs may initially show promise in identifying potential product candidates, yet fail to yield a successful commercial product for many reasons, including the following:
| ● | Partner with other entities or raise additional funds to advance the development of new products ; | |
| ● | competitors may develop alternatives that render our product candidates (or those of our partners) obsolete; | |
| ● | a product candidate may not have a sustainable intellectual property position in major markets; | |
| ● | a product candidate may, after additional studies, be shown to have harmful side effects or other characteristics that indicate it is unlikely to be effective; | |
| ● | a product candidate may not receive regulatory approval; | |
| ● | a product candidate may not be capable of production in commercial quantities at an acceptable cost, or at all; or | |
| ● | a product candidate may not be accepted by patients, the medical community or third-party payors. |
Clinical trials of product candidates utilizing our technologies would be expensive and time-consuming, and the results of any of these trials would be uncertain.
Before obtaining regulatory approval for the sale of any product candidates, we and our partners must conduct expensive and extensive pre-clinical tests and clinical trials to demonstrate the safety and efficacy of such product candidates. Pre-clinical and clinical testing is a long, expensive and uncertain process, and the historical failure rate for product candidates is high. The length of time generally varies substantially according to the type of drug, complexity of clinical trial design, regulatory compliance requirements, intended use of the drug candidate and rate of patient enrollment for the clinical trials.
A failure of one or more pre-clinical studies or clinical trials can occur at any stage of testing. We and our partners may experience numerous unforeseen events during, or as a result of, the pre-clinical testing and the clinical trial process that could delay or prevent the receipt of regulatory approval or the commercialization of our product candidates, or that could render such process substantially more expensive, including:
| ● | regulators may not authorize us to commence a clinical trial or conduct a clinical trial at a prospective trial site; | |
| ● | pre-clinical tests or clinical trials may produce negative or inconclusive results, and we or a partner may decide, or a regulator may require us, to conduct additional pre-clinical testing or clinical trials, or we or a partner may abandon projects that were previously expected to be promising; |
| 38 |
| ● | enrollment in clinical trials may be slower than anticipated or participants may drop out of clinical trials at a higher rate than anticipated, in each case for a variety of reasons, resulting in significant delays; | |
| ● | third party contractors may fail to comply with regulatory requirements or meet their contractual obligations in a timely manner; | |
| ● | product candidates may have very different chemical and pharmacological properties in humans than in laboratory testing and may interact with human biological systems in unforeseen, ineffective or harmful ways; | |
| ● | the suspension or termination of clinical trials for a variety of reasons, including if the participants are being exposed to unacceptable health risks or if such trials are not being conducted in accordance with applicable regulatory requirements; |
| ● | regulators, including the FDA, may require that clinical research be held, suspended or terminated for various reasons, including noncompliance with regulatory requirements; | |
| ● | the costs of clinical trials (or the components thereof) may be greater than anticipated; | |
| ● | the supply or quality of drug candidates or other materials necessary to conduct clinical trials may be insufficient or inadequate; and | |
| ● | product candidates may not have the desired effects or may include undesirable side effects or the product candidates may have other unexpected characteristics. |
Further, even if the results of pre-clinical studies or clinical trials are initially positive, it is possible that different results will be obtained in the later stages of drug development or that results seen in clinical trials will not continue with longer term treatment. Drugs in late stages of clinical development may fail to show the desired safety and efficacy traits despite having progressed through initial clinical testing. For example, positive results in early phase 1 or phase 2 clinical trials may not be repeated in larger phase 2 or phase 3 clinical trials. It is expected that all of the drug candidates that may be developed by us or our partners based on our technologies will be prone to the risks of failure inherent in drug development. The clinical trials of any or all of the drug candidates of us or our partners could be unsuccessful, which would prevent the commercialization of these drugs. The FDA conducts its own independent analysis of some or all of the pre-clinical and clinical trial data submitted in a regulatory filing and often comes to different and potentially more negative conclusions than the analysis performed by the drug sponsor. The failure to develop safe, commercially viable drugs approved by the FDA or another applicable regulatory body would substantially impair our ability to generate product sales and sustain our operations and would materially harm our business and adversely affect our stock price. In addition, significant delays in pre-clinical studies and clinical trials will impede the regulatory approval process, the commercialization of drug candidates and the generation of revenue, as well as substantially increase development costs.
Our product candidates may cause undesirable side effects or have other properties that could halt their development, prevent their regulatory approval, limit their commercial potential or result in significant negative consequences.
It is possible that the FDA or foreign regulatory authorities may not agree with any future assessment of the safety profile of our product candidates. Undesirable side effects caused by any of our product candidates could cause us or our partners to interrupt, delay or discontinue development of our product candidates, could result in a clinical hold on any clinical trial, or could result in the denial of regulatory approval of our product candidates by the FDA or foreign regulatory authorities. This, in turn, could prevent us from commercializing our product candidates and generating revenues from their sale. In addition, if any of our products cause serious or unexpected side effects or are associated with other safety risks after receiving marketing approval, a number of potential significant negative consequences could result, including: (i) regulatory authorities may withdraw their approval of the product; (ii) we may be required to recall the product, change the way it is administered, conduct additional clinical trials or change the labeling of the product; (iii) the product may be rendered less competitive and sales may decrease; (iv) our reputation may suffer generally both among clinicians and patients; (v) regulatory authorities may require certain labeling statements, such as warnings or contraindications or limitations on the indications for use, or impose restrictions on distribution in connection with approval, if any; or (vi) we may be required to change the way the product is administered or conduct additional preclinical studies or clinical trials.
| 39 |
If preliminary data demonstrate that any of our product candidates has an unfavorable safety profile and is unlikely to receive regulatory approval or be successfully commercialized, we may voluntarily suspend or terminate future development of such product candidate.
Any one or a combination of these events could prevent us from obtaining approval and achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing the product candidate, which in turn could delay or prevent us from generating significant revenues from the sale of the product.
Even if regulatory approvals are obtained for our products, including our approved Prestalia product, such products will be subject to ongoing regulatory obligations and continued regulatory review. If we or a partner fail to comply with continuing U.S. and foreign regulations, the approvals to market drugs could be lost and our business would be materially adversely affected.
Following any initial FDA or foreign regulatory approval of any drugs we or a partner may develop, such drugs will continue to be subject to extensive and ongoing regulatory review, including the review of adverse drug experiences and clinical results that are reported after such drugs are made available to patients. This would include results from any post marketing studies or vigilance required as a condition of approval. The manufacturer and manufacturing facilities used to make any drug candidates will also be subject to periodic review and inspection by regulatory authorities, including the FDA. The discovery of any new or previously unknown problems with the product, manufacturer or facility may result in restrictions on the drug or manufacturer or facility, including withdrawal of the drug from the market. Marketing, advertising and labeling also will be subject to regulatory requirements and continuing regulatory review. The failure to comply with applicable continuing regulatory requirements may result in fines, suspension or withdrawal of regulatory approval, product recalls and seizures, operating restrictions and other adverse consequences.
We and our partners are subject to extensive U.S. and foreign government regulation, including the requirement of approval before products may be marketed.
We, our present and future collaborators, and the drug product candidates developed by us or in collaboration with partners are subject to extensive regulation by governmental authorities in the U.S. and other countries. Failure to comply with applicable requirements could result in, among other things, any of the following actions: warning letters, fines and other civil penalties, unanticipated expenditures, delays in approving or refusal to approve a product candidate, product recall or seizure, interruption of manufacturing or clinical trials, operating restrictions, injunctions and criminal prosecution.
Our product candidates and those of our partners cannot be marketed in the U.S. without FDA approval or clearance, and they cannot be marketed in foreign countries without applicable regulatory approval. Neither the FDA nor any foreign regulatory authority has approved any of the product candidates being developed by us or of our partners based on our technologies, other than our Prestalia product. These product candidates (other than Prestalia) are in pre-clinical and clinical development and will have to be approved by the FDA or applicable foreign regulatory authorities before they can be marketed in the U.S. or abroad. Obtaining regulatory approval requires substantial time, effort and financial resources, and may be subject to both expected and unforeseen delays, including, without limitation, citizen’s petitions or other filings with the FDA or applicable foreign regulatory authority, and there can be no assurance that any approval will be granted on a timely basis, if at all, or that delays will be resolved favorably or in a timely manner. If our product candidates are not approved in a timely fashion, or are not approved at all, our business and financial condition may be adversely affected.
In addition, both before and after regulatory approval, we, our collaborators and our product candidates are subject to numerous requirements by the FDA and foreign regulatory authorities covering, among other things, testing, manufacturing, quality control, labeling, advertising, promotion, distribution and export. These requirements may change and additional government regulations may be promulgated that could affect us, our collaborators or our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the U.S. or abroad. There can be no assurance that neither we nor any of our partners will be required to incur significant costs to comply with such laws and regulations in the future or that such laws or regulations will not have a material adverse effect upon our business and prospects.
| 40 |
We may use hazardous chemicals and biological materials in our business. Any disputes relating to improper use, handling, storage or disposal of these materials could be time-consuming and costly.
Our research and development operations may involve the use of hazardous and biological, potentially infectious, materials. Such use subjects us to the risk of accidental contamination or discharge or any resultant injury from these materials. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of these materials and specific waste products. We could be subject to damages, fines or penalties in the event of an improper or unauthorized release of, or exposure of individuals to, these hazardous materials, and our liability could be substantial. The costs of complying with these current and future environmental laws and regulations may be significant, thereby impairing our business.
We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of biohazardous materials. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials. Additional federal, state and local laws and regulations affecting our operations may be adopted in the future. We may incur substantial costs to comply with, and substantial fines or penalties if we violate, any of these laws or regulations.
Failure to comply with foreign regulatory requirements governing human clinical trials and marketing approval for drugs could prevent the sale of drug candidates based on our technologies in foreign markets, which may adversely affect our operating results and financial condition.
The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement for marketing drug candidates based on our technologies outside the U.S. vary greatly from country to country. We have, and our partners may have, limited experience in obtaining foreign regulatory approvals. The time required to obtain approvals outside the U.S. may differ from that required to obtain FDA approval. We may not be able to obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other countries or by the FDA. Failure to comply with these regulatory requirements or obtain required approvals could restrict the development of foreign markets for our drug candidates and may have a material adverse effect on our financial condition or results of operations.
Risks Related to our Dependence on Third Parties
We may become dependent on our collaborative arrangements with third parties for a substantial portion of our revenue, and our development and commercialization activities may be delayed or reduced if we fail to initiate, negotiate or maintain successful collaborative arrangements.
We are, in part, dependent on partners to develop and commercialize products based on our technologies and to provide the regulatory compliance, sales, marketing and distribution capabilities required for the success of our business. If we fail to secure or maintain successful collaborative arrangements, our development and commercialization activities will be delayed, reduced or terminated, and our revenues could be materially and adversely impacted.
The potential future milestone and royalty payments and cost reimbursements from collaboration agreements could provide an important source of financing for our research and development programs, thereby facilitating the application of our technology to the development and commercialization of our products. These collaborative agreements might be terminated either by us or by our partners upon the satisfaction of certain notice requirements. Our partners may not be precluded from independently pursuing competing products and drug delivery approaches or technologies. Even if our partners continue their contributions to our collaborative arrangements, of which there can be no assurance, they may nevertheless determine not to actively pursue the development or commercialization of any resulting products. Our partners may fail to perform their obligations under the collaborative arrangements or may be slow in performing their obligations. In addition, our partners may experience financial difficulties at any time that could prevent them from having available funds to contribute to these collaborations. If our collaborators fail to conduct their commercialization, regulatory compliance, sales and marketing or distribution activities successfully and in a timely manner, or if they terminate or materially modify their agreements with us, the development and commercialization of one or more product candidates could be delayed, curtailed or terminated because we may not have sufficient financial resources or capabilities to continue such development and commercialization on our own.
| 41 |
An interruption in the supply of raw and bulk materials needed for the development of our product candidates, or the manufacture of our approved products, could cause product development and/or sales to be slowed or stopped.
We and our partners may obtain supplies of critical raw and bulk materials used in research and development efforts from several suppliers, and long-term contracts may not be in place with any or all of these suppliers. There can be no assurance that sufficient quantities of product candidates or approved products could be manufactured if our suppliers are unable or unwilling to supply such materials. Any delay or disruption in the availability of raw or bulk materials could slow or stop research and development, or sales, of the relevant product.
We rely on third parties to conduct clinical trials, and those third parties may not perform satisfactorily, including failing to meet established timelines for the completion of such clinical trials.
We are, and anticipate that we and certain of our partners will continue to be, dependent on contract research organizations, third-party vendors and investigators for performing or managing pre-clinical testing and clinical trials related to drug discovery and development efforts. These parties are not employed by us or our partners, and neither we nor our partners can control the amount or timing of resources that they devote to our programs. If they fail to devote sufficient time and resources to our drug development programs or if their performance is substandard, it will delay, and potentially materially adversely affect, the development and commercialization of our product candidates. Moreover, these parties also may have relationships with other commercial entities, some of which may compete with us and our partners. If they assist our competitors, it could harm our competitive position.
If we or our partners lose our relationship with any one or more of these parties, there could be a significant delay in both identifying another comparable provider and then contracting for its services. An alternative provider may not be available on reasonable terms, if at all. Even if we locate an alternative provider, is it likely that this provider may need additional time to respond to our needs and may not provide the same type or level of service as the original provider. In addition, any alternative provider will be subject to current Good Laboratory Practices (“cGLP”) and similar foreign standards and neither we nor our partners have control over compliance with these regulations by these providers. Consequently, if these providers do not adhere to these practices and standards, the development and commercialization of our product candidates could be delayed.
We have limited manufacturing experience or resources, and we must incur significant costs to develop this expertise or rely on third parties to manufacture our products.
We have limited manufacturing experience. Some of our products and product candidates utilize specialized formulations whose scale-up and manufacturing could be very difficult. We also have limited experience in such scale-up and manufacturing, requiring us to depend on a limited number of third parties, who might not be able to deliver in a timely manner, on acceptable terms, or at all. In order to develop products, apply for regulatory approvals and commercialize our products, we will need to develop, contract for, or otherwise arrange for the necessary manufacturing capabilities.
There are a limited number of manufacturers that supply the materials needed for the development of our products and product candidates. There are risks inherent in pharmaceutical manufacturing that could affect the ability of our contract manufacturers to meet our delivery requirements or provide adequate amounts of material to meet our needs. Included in these risks are synthesis and purification failures and contamination during the manufacturing process, which could result in unusable product and cause delays in our development process, as well as additional expense to us. To fulfill our supply requirements, we may also need to secure alternative suppliers. In addition to the manufacture of the materials necessary to develop our products, we may have additional manufacturing requirements related to the technology required to deliver certain of our product candidates to the relevant cell or tissue type. In some cases, the delivery technology we utilize is highly specialized or proprietary, and for technical and legal reasons, we may have access to only one or a limited number of potential manufacturers for such delivery technology. Failure by these manufacturers to properly formulate our product candidates for delivery could also result in unusable product and cause delays in our discovery and development process, as well as additional expense to us.
| 42 |
The manufacturing process for any products based on our technologies that we or our partners may develop is subject to the FDA and foreign regulatory authority approval process, and we or our partners will need to contract with manufacturers who can meet all applicable FDA and foreign regulatory authority requirements on an ongoing basis. If we are unable to obtain or maintain contract manufacturing for these product candidates or approved products, or to do so on commercially reasonable terms, we may not be able to successfully develop and commercialize our products.
To the extent that we enter into manufacturing arrangements with third parties, we will depend on these third parties to perform their obligations in a timely manner and consistent with regulatory requirements, including those related to quality control and quality assurance. The failure of a third-party manufacturer to perform its obligations as expected could adversely affect our business in a number of ways.
If a third-party manufacturer with whom we contract fails to perform its obligations, we may be forced to manufacture the materials ourselves, for which we may not have the capabilities or resources, or enter into an agreement with a different third-party manufacturer, which we may not be able to do on acceptable terms, if at all. In addition, if we are required to change manufacturers for any reason, we will be required to verify that the new manufacturer maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. The delays associated with the verification of a new manufacturer could negatively affect our ability to develop product candidates in a timely manner or within budget, or to sell approved products in sufficient quantities. Furthermore, a manufacturer may possess technology related to the manufacture of our product candidates or approved products that such manufacturer owns independently. This would increase our reliance on such manufacturer or require us to obtain a license from such manufacturer in order to have another third party manufacture our products.
Risks Related to our Intellectual Property and Other Legal Matters
If we are unable to adequately protect our proprietary technology from legal challenges, infringement or alternative technologies, our competitive position may be hurt and our operating results may be negatively impacted.
Our business is based upon the development and delivery of novel therapeutics, and we rely on the issuance of patents, both in the U.S. and internationally, for protection against competitive technologies. Although we believe we exercise the necessary due diligence in our patent filings, our proprietary position is not established until the appropriate regulatory authorities actually issue a patent, which may take several years from initial filing or may never occur.
Moreover, even the established patent positions of pharmaceutical companies are generally uncertain and involve complex legal and factual issues. Although we believe our issued patents are valid, third parties may infringe our patents or may initiate proceedings challenging the validity or enforceability of our patents. The issuance of a patent is not conclusive as to its claim scope, validity or enforceability. Challenges raised in patent infringement litigation we initiate or in proceedings initiated by third parties may result in determinations that our patents have not been infringed or that they are invalid, unenforceable or otherwise subject to limitations. In the event of any such determinations, third parties may be able to use the discoveries or technologies claimed in our patents without paying us licensing fees or royalties, which could significantly diminish the value of these discoveries or technologies. As a result of such determinations, we may be enjoined from pursuing commercialization of potential products or may be required to obtain licenses, if available, to the third party patents or to develop or obtain alternative technology. Responding to challenges initiated by third parties may require significant expenditures and divert the attention of our management and key personnel from other business concerns.
Furthermore, it is possible that others will infringe or otherwise circumvent our issued patents and that we will be unable to fund the cost of litigation against them or that we would elect not to pursue litigation. In addition, enforcing our patents against third parties may require significant expenditures regardless of the outcome of such efforts. We also cannot assure you that others have not filed patent applications for technology covered by our pending applications or that we were the first to invent the technology. There may also exist third party patents or patent applications relevant to our potential products that may block or compete with the technologies covered by our patent applications and third parties may independently develop IP similar to our patented IP, which could result in, among other things, interference proceedings in the U.S. Patent and Trademark Office to determine priority of invention.
| 43 |
In addition, we may not be able to protect our established and pending patent positions from competitive technologies, which may provide more effective therapeutic benefit to patients and which may therefore make our products, technology and proprietary position obsolete.
We also rely on copyright and trademark protection, trade secrets, know-how, continuing technological innovation and licensing opportunities. In an effort to maintain the confidentiality and ownership of our trade secrets and proprietary information, we have typically required our employees, consultants, advisors and others to whom we disclose confidential information to execute confidentiality and proprietary information agreements. However, it is possible that these agreements may be breached, invalidated or rendered unenforceable, and if so, there may not be an adequate corrective remedy available. Furthermore, like many companies in our industry, we may from time to time hire scientific personnel formerly employed by other companies involved in one or more areas similar to the activities we conduct. In some situations, our confidentiality and proprietary information agreements may conflict with, or be subject to, the rights of third parties with whom our employees, consultants or advisors have prior employment or consulting relationships. Although we have typically required our employees and consultants to maintain the confidentiality of all confidential information of previous employers, we or these individuals may be subject to allegations of trade secret misappropriation or other similar claims as a result of their prior affiliations. Finally, others may independently develop substantially equivalent proprietary information and techniques, or otherwise gain access to our trade secrets. Our failure to protect our proprietary information and techniques may inhibit or limit our ability to exclude certain competitors from the market and execute our business strategies.
If we are unable to adequately protect our proprietary intellectual property from legal challenges, infringement or alternative technologies, we will not be able to compete effectively in the drug discovery, development and commercialization business.
Because intellectual property rights are of limited duration, expiration of intellectual property rights and licenses will negatively impact our operating results.
Intellectual property rights, such as patents and license agreements based on those patents, generally are of limited duration. Therefore, the expiration or other loss of rights associated with IP and IP licenses can negatively impact our business, and the future sales of our approved products, if any.
Our patent applications may be inadequate in terms of priority, scope or commercial value.
We apply for patents covering our discoveries and technologies as we deem appropriate and as our resources permit. However, we or our partners may fail to apply for patents on important discoveries or technologies in a timely fashion or at all. Also, our pending patent applications may not result in the issuance of any patents. These applications may not be sufficient to meet the statutory requirements for patentability, and therefore we may be unable to obtain enforceable patents covering the related discoveries or technologies we may want to commercialize. In addition, because patent applications are maintained in secrecy for approximately 18 months after filing, other parties may have filed patent applications relating to inventions before our applications covering the same or similar inventions. In addition, foreign patent applications are often published initially in local languages, and until an English language translation is available it can be impossible to determine the significance of a third party invention. Any patent applications filed by third parties may prevail over our patent applications or may result in patents that issue alongside patents issued to us, leading to uncertainty over the scope of the patents or the freedom to practice the claimed inventions.
Although we have acquired and in-licensed a number of issued patents, the discoveries or technologies covered by these patents may not have any therapeutic or commercial value. Also, issued patents may not provide commercially meaningful protection against competitors. Other parties may be able to design around our issued patents or independently develop products having effects similar or identical to our patented product candidates or approved products. In addition, the scope of our patents is subject to considerable uncertainty and competitors or other parties may obtain similar patents of uncertain scope.
| 44 |
We have depended on technologies we license, and if we lose the right to license such technologies or we fail to license new technologies in the future, our ability to develop or commercialize new or existing products would be harmed.
We have depended on licenses from third parties for certain of our key technologies, products and product candidates. Our licenses impose various development, funding, royalty, diligence, sublicensing, insurance and other obligations on us. If our license with respect to any of these technologies or products is terminated for any reason, the development and/or commercialization of the products contemplated by the licenses would be delayed, or suspended altogether, while we seek to license similar products or technology (which licenses may not be available on commercially acceptable terms or at all) or develop new non-infringing products or technology. If our existing license is terminated, the development and/or commercialization of the products contemplated by the licenses could be delayed or terminated and we may not be able to negotiate additional licenses on acceptable terms, if at all, which would have a material adverse effect on our business.
We may be required to defend lawsuits or pay damages for product liability claims.
Our business inherently exposes us to potential product liability claims. We may face substantial product liability exposure in human clinical trials that we may initiate and for products that we sell, or manufacture for others to sell, after regulatory approval. The risk exists even with respect to those drugs that are approved by regulatory agencies for commercial distribution and sale and are manufactured in facilities licensed and regulated by regulatory agencies. Any product liability claims, regardless of their merits, could be costly, divert management’s attention, delay or prevent completion of our clinical development programs, and adversely affect our reputation, the demand for our products and our stock price. We currently do not have product liability insurance. We intend to obtain such insurance as we deem appropriate as we move forward with the commercialization of our Prestalia product and with further clinical development of our product candidates. Any product liability insurance we have or may obtain may not provide sufficient coverage against potential liabilities. Furthermore, clinical trial and product liability insurance is becoming increasingly expensive. As a result, we may be unable to obtain sufficient insurance at a reasonable cost to protect us against losses caused by product liability claims that could have a material adverse effect on our business.
An unfavorable outcome in the pending legal action filed by Vaya Pharma, Inc. in which we are a named defendant could have a material adverse effect on our company.
We have been named in a complaint filed in the Supreme Court of the State of New York as a defendant in the matter entitled Vaya Pharma, Inc. v. Symplmed Technologies, Inc., Symplmed Pharmaceuticals, Inc., Erik Emerson and Marina Biotech, Inc. Although the complaint has been filed, we have not been legally served. The complaint alleges, in relevant part, that: (i) the sale by Symplmed Pharmaceuticals, Inc. of its assets related to its Prestalia product, and the sale by Symplmed Technologies, Inc. of its assets related to its DyrctAxess platform, should be set aside pursuant to New York law as they were consummated without fair consideration to the sellers (the “Symplmed Defendants”), and thereby had the effect of fraudulently depriving the creditors of the Symplmed Defendants, including Vaya Pharma, Inc., of funds that could have been used to pay their debts; and (ii) we are liable, as successor, for any and all claims by Vaya Pharma, Inc. against the Symplmed Defendants, though pursuant to the agreement we are only contractually responsible for liabilities that accrue after the parties entered into the agreement for Prestalia and any liabilities that existed prior to the agreement are contractually held by Symplmed. If and when we are legally served, we intend to dispute jurisdiction, the sufficiency of the pleading and the claims set forth in this complaint, and to defend this matter, vigorously. However, due to the inherent uncertainties of litigation, the ultimate outcome of this matter is uncertain. The Company reached a tentative settlement agreement and, although such settlement has not been finalized, the Company accrued a total provision of $250,000 for this complaint, which is included in accrued expenses on the accompanying consolidated balance sheet.
Our Prestalia product is currently involved in a paragraph IV challenge. Our failure to resolve the challenge in the manner currently proposed, or to expand our product offerings prior to the date before the challenger launches a generic version of Prestalia to derive meaningful revenues from such additional product offerings, could materially adversely affect us and our business operations.
| 45 |
Our Prestalia product is currently involved in a paragraph IV challenge regarding patents issued to perindopril arginine. This challenge, which is currently pending in the United States District Court for the District of Delaware (No. 1:17-cv-00276), is captioned Apotex Inc. and Apotex Corp. v. Symplmed Pharmaceuticals, LLC and Les Laboratoires Servier. The challengers (Apotex Inc. and Apotex Corp. (“Apotex”)) have filed an Abbreviated New Drug Application seeking FDA approval to market a generic version of Prestalia and included a Paragraph (IV) certification. In the litigation, Apotex seeks a declaratory judgment that no valid claims of the two patents Symplmed listed in the FDA Orange Book as having claims covering Prestalia, U.S. Patent No. 6,696,481 and 7,846,961, will be infringed by the Apotex proposed generic version of Prestalia and that the claims of those patents are invalid. The challenge is designed to provide Apotex with an opportunity to enter the market with a generic version of Prestalia, ahead of the expiration of the patents with claims covering that product. Apotex entered into negotiations with Symplmed Pharmaceuticals, LLC (which entity sold its assets relating to Prestalia to us in June 2017, including its License and Commercialization Agreement with Les Laboratories Servier) and Les Laboratories Servier (which entity owns or controls intellectual property rights relating to pharmaceutical products containing as an active pharmaceutical ingredient perindopril in combination with other active pharmaceutical ingredients, which rights have been licensed to Symplmed Pharmaceuticals) to resolve the challenge in the second quarter of 2017, and such parties, along with us, have come to a general agreement on terms that will result in a delay to the challengers’ ability to enter the market with a generic version of Prestalia, while still providing the challenger with the right to enter the market prior to the expiration of the patent covering such product. The term sheet memorializing such terms is pending execution in a final settlement agreement. In the meantime, the District Court has entered an order extending the time for the defendants to respond to Apotex’s Complaint until January 9, 2018 .
We do not anticipate that the proposed settlement terms – which would provide us with the ability to exclusively market Prestalia for a specified period of time without incurring the risks or costs of litigation – will have a material adverse impact on our long term business as a whole. We hold this expectation because it is our intention that Prestalia will be a building block toward the development of our company into a larger organization, which we hope will include the commercial launch by us of Prestalia and the expansion of our commercial capabilities in connection with such activities, followed by the launch of products that we license for commercialization purposes from our partners and the commercial launch of products that we develop internally. We anticipate that the aforementioned steps to expand and strengthen our commercial capabilities and diversify our product offerings so as to generate meaningful revenues in excess of those revenues generated by sales of Prestalia will occur during our exclusivity period with respect to Prestalia (i.e., prior to the date on which Apotex enters the market with a generic version of Prestalia). If we are able to achieve this growth, the overall economic impact that would result to us even if Apotex markets a generic version of Prestalia and captures a significant percentage of the market for that drug following our exclusivity period would be reduced.
However, there can be no assurance that the paragraph IV challenge will be resolved without litigation or resolved on the terms currently proposed. There also cannot be any assurance that our plans to commercialize Prestalia (or any of the products that it may license from its partners or that it may develop internally) and expand our product offerings and the revenue generated therefrom so as to lessen our reliance on sales of Prestalia will be achieved. Any failure of our expectations regarding the resolution of the paragraph IV challenge and the expansion of our commercial activities (and the revenues to be derived therefrom) could have a material adverse effect on us, our prospects and our results of operations.
Risks Related to the Commercialization of our Product Candidates
Our product development efforts may not result in commercial products.
The results of our operations depend, to a significant degree, upon the ability of us and our collaborators to successfully develop and commercialize pharmaceutical products. The development and commercialization process for pharmaceutical products is both time consuming and costly and involves a high degree of business risk. Successful product development in the pharmaceutical industry is highly uncertain, and very few research and development projects result in a commercial product. Product candidates that appear promising in the early phases of development, such as in preclinical testing or in early human clinical trials, may fail to reach the market for a number of reasons, such as:
| 46 |
| ● | a product candidate may not perform as expected in later or broader trials in humans and limit marketability of such product candidate; | |
| ● | necessary regulatory approvals may not be obtained in a timely or cost-effective manner, if at all; | |
| ● | a product candidate may not be able to be successfully and profitably produced and marketed; | |
| ● | third parties may have proprietary rights to a product candidate, and do not allow sale on reasonable terms; or | |
| ● | a product candidate may not be financially successful because of existing therapeutics or treatments that offer, or that are perceived to offer, equivalent or better treatments. |
There can be no assurance that any of our products or product candidates will ever be successfully commercialized by us or by one of our partners, and delays or additional expenses in any part of the process or the inability to obtain regulatory approval in a timely or cost-effective manner could adversely affect our operating results by restricting introduction of new products by us and/or our partners.
It is possible that the commercial opportunity for our products and product candidates will be limited.
Our products and product candidates are based on novel technologies and therapeutic approaches. Key participants in pharmaceutical marketplaces, such as physicians, third-party payors and consumers, may not accept such products. Accordingly, while we believe there will be a commercial market for our products and product candidates, there can be no assurance that this will be the case.
Risks Related to our Industry
If we or any of our independent contractors, consultants, collaborators, manufacturers, vendors or service providers fail to comply with healthcare laws and regulations, we or they could be subject to enforcement actions, which could result in penalties and affect our ability to develop, market and sell our products and product candidates and may harm our reputation.
We are or may in the future be subject to federal, state and foreign healthcare laws and regulations pertaining to, among other things, fraud and abuse and patients’ rights. These laws and regulations include:
| ● | the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual for a healthcare item or service, or the purchasing or ordering of an item or service, for which payment may be made under a federal healthcare program such as Medicare or Medicaid; | |
| ● | the U.S. federal false claims and civil monetary penalties laws, including the federal civil False Claims Act, which prohibit, among other things, individuals or entities from knowingly presenting or causing to be presented, claims for payment by government funded programs such as Medicare or Medicaid that are false or fraudulent, and which may apply to us by virtue of statements and representations made to customers or third parties; | |
| ● | the U.S. federal Health Insurance Portability and Accountability Act (HIPAA), which created additional federal criminal statutes that prohibit, among other things, knowingly and willfully executing or attempting to execute a scheme to defraud healthcare programs; | |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act (HITECH), which imposes requirements on certain types of people and entities relating to the privacy, security, and transmission of individually identifiable health information, and requires notification to affected individuals and regulatory authorities of certain breaches of security of individually identifiable health information; |
| 47 |
| ● | the federal Physician Payment Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, to report annually to the Centers for Medicare & Medicaid Services information related to payments and other transfers of value to physicians, other healthcare providers and teaching hospitals, and ownership and investment interests held by physicians and other healthcare providers and their immediate family members, which is published in a searchable form on an annual basis; and |
| ● | state laws comparable to each of the above federal laws, such as, for example, anti-kickback and false claims laws that may be broader in scope and also apply to commercial insurers and other non-federal payors, requirements for mandatory corporate regulatory compliance programs, and laws relating to patient data privacy and security. Other state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government; require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state and foreign laws govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
If our operations are found to be in violation of any such health care laws and regulations, we may be subject to penalties, including administrative, civil and criminal penalties, monetary damages, disgorgement, imprisonment, the curtailment or restructuring of our operations, loss of eligibility to obtain approvals from the FDA or foreign regulatory authorities, or exclusion from participation in government contracting, healthcare reimbursement or other government programs, including Medicare and Medicaid, any of which could adversely affect our financial results. Although effective compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, these risks cannot be entirely eliminated. Any action against us for an alleged or suspected violation could cause us to incur significant legal expenses and could divert our management’s attention from the operation of our business, even if our defense is successful. In addition, achieving and sustaining compliance with applicable laws and regulations may be costly to us in terms of money, time and resources.
Any drugs based on our technologies that we or any of our partners develop may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, which could have a material adverse effect on our business and financial results.
Our ability to commercialize any products successfully also will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other third-party payors. In many jurisdictions, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. Obtaining coverage and reimbursement approval of a product from a government or other third-party payor is a time-consuming and costly process that could require us to provide to the payor supporting scientific, clinical and cost-effectiveness data for the use of our products. If we are not currently capturing the scientific and clinical data that will be required for reimbursement approval, we may be required to conduct additional trials, which may delay or suspend reimbursement approval. Additionally, in the United States, no uniform policy of coverage and reimbursement for products exists among third-party payors. Therefore, coverage and reimbursement for products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our product candidates and approved products to each payor separately, with no assurance that coverage and adequate reimbursement will be obtained.
It is possible that our approved products and any other product candidates that we bring to the market may not be considered cost-effective, and the amount reimbursed for any products may be insufficient to allow us to sell our products on a competitive basis. Because many of our programs are in the early stages of development, we are unable at this time to determine their cost effectiveness or the likely level or method of reimbursement. Increasingly, the third-party payors, such as government and private insurance plans, who reimburse patients or healthcare providers, are requiring that drug companies provide them with predetermined discounts from list prices, and are seeking to reduce the prices charged or the amounts reimbursed for pharmaceutical products. If the coverage provided for any products we develop or commercialize is inadequate in light of our development and other costs, our return on investment could be adversely affected.
| 48 |
It is possible that certain of the drugs based on our technologies that we or a partner develop may need to be administered under the supervision of a physician on an outpatient basis. Under currently applicable law, drugs that are not usually self-administered may be eligible for coverage by Medicare if they:
| ● | are “incidental” to a physician’s services; | |
| ● | are “reasonable and necessary” for the diagnosis or treatment of the illness or injury for which they are administered according to accepted standards of medical practice; | |
| ● | are not excluded as immunizations; and | |
| ● | have been approved by the FDA. |
There may be significant delays in obtaining coverage for newly-approved products, and coverage may be more limited than the purposes for which the drug is approved by the FDA or foreign regulatory authorities. Moreover, eligibility for coverage does not imply that any drug will be reimbursed in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim payments for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent.
Reimbursement may be based on payments allowed for lower-cost products that are already reimbursed, may be incorporated into existing payments for other services and may reflect budgetary constraints or imperfections in Medicare data. Net prices for products may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of products from countries where they may be sold at lower prices than in the United States. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement rates. However, no uniform policy requirement for coverage and reimbursement for products exists among third-party payors in the United States. Therefore, coverage and reimbursement for products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. Our inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for new drugs that we develop, for which we obtain regulatory approval and/or that we seek to commercialize could have a material adverse effect on our operating results, our ability to raise capital and our financial condition.
We believe that the efforts of governments and third-party payors to contain or reduce the cost of healthcare and legislative and regulatory proposals to broaden the availability of healthcare will continue to affect the business and financial condition of pharmaceutical and biopharmaceutical companies. A number of legislative and regulatory changes in the healthcare system in the United States and other major healthcare markets have been proposed in recent years, and such efforts have expanded substantially in recent years. These developments have included prescription drug benefit legislation that was enacted and took effect in January 2006, healthcare reform legislation recently enacted by certain states, and major healthcare reform legislation that was passed by Congress and enacted into law in the United States in 2010. The U.S. Congress and the Trump administration have similarly expressed concerns over the pricing of pharmaceutical products and there can be no assurance as to how this scrutiny will impact future pricing of pharmaceutical products generally. Future developments could, directly or indirectly, affect our ability to sell our products, if approved, at a favorable price.
For example, the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act (PPACA), contains provisions that affect companies in the pharmaceutical industry and other healthcare related industries by imposing additional costs and changes to business practices. Provisions affecting pharmaceutical companies include the following:
| ● | mandatory rebates for drugs sold into the Medicaid program were increased, and the rebate requirement was extended to drugs used in risk-based Medicaid managed care plans; |
| 49 |
| ● | the 340B Drug Pricing Program under the Public Health Services Act was extended to require mandatory discounts for drug products sold to certain critical access hospitals, cancer hospitals and other covered entities; | |
| ● | expansion of eligibility criteria for Medicaid programs; | |
| ● | expansion of entities eligible for discounts under the Public Health Service pharmaceutical pricing program; | |
| ● | a new Patient Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; | |
| ● | pharmaceutical companies are required to offer discounts on brand-name drugs to patients who fall within the Medicare Part D coverage gap, commonly referred to as the “donut hole”; and | |
| ● | pharmaceutical companies are required to pay an annual non-tax deductible fee to the federal government based on each company’s market share of prior year total sales of branded products to certain federal healthcare programs, such as Medicare, Medicaid, Department of Veterans Affairs and Department of Defense. Since we expect our branded pharmaceutical sales, if any of our products are approved, to constitute a small portion of the total federal health program pharmaceutical market, we do not expect this annual assessment to have a material impact on our financial condition. |
There have been judicial and Congressional challenges, and amendments to certain aspects of the PPACA. More recently, Congress has sought to repeal all or portions of the PPACA and to replace the PPACA with new legislation. We expect there will be additional challenges and amendments to the PPACA in the future, including potential additional attempts to repeal the PPACA in full or in part. The full effect of the U.S. healthcare reform legislation on our business activities, both in the near and long term, is unknown. The financial impact of the U.S. healthcare reform legislation will depend on a number of factors, including, but not limited to, the policies reflected in implementing regulations and guidance and changes in sales volumes for products affected by the new system of rebates, discounts and fees. The legislation may also have a positive impact on our future net sales, if any, by increasing the aggregate number of persons with healthcare coverage in the United States.
Moreover, we cannot predict what healthcare reform initiatives may be adopted in the future, and whether (or to what extent) existing legislation may be modified or repealed. Further federal and state legislative and regulatory developments are likely, and we expect ongoing initiatives in the United States to increase pressure on drug pricing. Such reforms could have an adverse effect on anticipated revenues from our approved products and any other product candidates based on our technologies that are successfully developed and for which regulatory approval is obtained, and may affect our overall financial condition and ability to develop drug candidates.
The pharmaceutical market is intensely competitive. If we are unable to compete effectively with existing drugs, and existing and new treatment methods and technologies, we may be unable to commercialize successfully any drugs that we develop.
The pharmaceutical market is intensely competitive and rapidly changing. Many large pharmaceutical and biotechnology companies, academic institutions, governmental agencies and other public and private research organizations are pursuing the development of novel drugs or treatments for the same diseases and conditions that we are targeting or expect to target. Many of our competitors have:
| ● | much greater financial, technical and human resources than we have at every stage of the discovery, development, manufacture and commercialization of products; | |
| ● | more extensive experience in pre-clinical testing, conducting clinical trials, obtaining regulatory approvals, and in manufacturing, marketing and selling pharmaceutical products; | |
| ● | product candidates that are based on previously tested or accepted technologies; | |
| ● | products that have been approved or are in late stages of development; and | |
| ● | collaborative arrangements in our target markets with leading companies and research institutions. |
| 50 |
Our products may face intense competition from drugs or treatments that have already been approved and accepted by the medical community for the treatment of the conditions for which we may develop drugs or treatments, or from new drugs that enter the market. We believe a significant number of drugs are currently under development, and may become commercially available in the future, for the treatment of conditions for which we and our partners may try to develop drugs. These drugs may be more effective, safer, less expensive, or marketed and sold more effectively, than any products we and our partners develop.
We anticipate that we and our partners will face competition based on many different factors regarding products that we successfully develop, including:
| ● | safety and effectiveness of such products; | |
| ● | ease with which such products can be administered and the extent to which patients accept relatively new routes of administration; | |
| ● | timing and scope of regulatory approvals for these products; | |
| ● | availability and cost of manufacturing, marketing and sales capabilities; | |
| ● | price; | |
| ● | reimbursement coverage; and | |
| ● | patent position. |
Our competitors may develop or commercialize products or treatments with significant advantages over any products we develop based on any of the factors listed above or on other factors. Our competitors may therefore be more successful in commercializing their products than we are, which could adversely affect our competitive position and business. Competitive products or treatments may make any products we develop obsolete or noncompetitive before we can recover the expenses of developing and commercializing our product candidates. Such competitors could also recruit our future employees, which could negatively impact our level of expertise and the ability to execute on our business plan. Furthermore, we also face competition from existing and new treatment methods that reduce or eliminate the need for drugs, such as the use of advanced medical devices. The development of new medical devices or other treatment methods for the conditions we are targeting could make our product candidates noncompetitive, obsolete or uneconomical.
We may be unable to compete successfully against other companies that are working to develop novel drugs and technology platforms using technology similar to ours.
In addition to the competition we face from competing drugs in general with respect to our operations, we have also faced competition from other biotechnology and pharmaceutical companies and medical institutions that are working to develop novel drugs using technology that competes more directly with our technologies. For example, there are a number of companies with programs in the nucleic acid therapeutics field, including: Alnylam Pharmaceuticals, Arbutus, Arcturus Therapeutics, Benitec Biopharma, Dicerna Pharmaceuticals, Isis Pharmaceuticals, miRagen Therapeutics, Mirna, PhaseRx Pharmaceuticals, Quark Pharmaceuticals, Regulus Therapeutics, RXi Pharmaceuticals, Sarepta Therapeutics and Silence Therapeutics. With respect to IT-102/IT-103, our primary competitor is Kitov, which is developing a celecoxib/amlodipine FDC using the same regulatory pathway that we are using for IT-102/IT-103. Any of the aforementioned companies may develop its technology more rapidly and more effectively than us.
| 51 |
In addition to competition with respect to our technology and with respect to specific products, we and our partners face substantial competition from third parties, both in academic laboratories and in the corporate sector, to discover and develop safe and effective means to deliver the drugs based on our technologies that are developed to the relevant cell and tissue types. If safe and effective means of delivery were developed by our competitors, our ability to successfully commercialize a competitive product would be adversely affected.
Many of our competitors, either alone or together with their partners, have substantially greater R&D capabilities and financial, scientific, technical, manufacturing, sales, marketing, distribution, regulatory and other resources and experience than us. They may also have more established relationships with pharmaceutical companies. In order to compete successfully with respect to our approved products, we may need to be first to obtain IP protection for, or to commercialize, such products, or we may need to demonstrate that such products are superior to, or more cost effective than, products developed by our competitors (including therapies that are based on different technologies). If we are not first to protect or market our products, or if we are unable to differentiate our products from those offered by our competitors, any products for which we are able to obtain approval may not be successful.
Universities and public and private research institutions are also potential competitors. While these organizations primarily have educational objectives, they may develop proprietary technologies related to the drug delivery field or secure protection that we may need for development of our technologies and products. We may attempt to license one or more of these proprietary technologies, but these licenses may not be available to us on acceptable terms, if at all.
Risks Related to our Common Stock
The trading price of our common stock has been volatile, and investors in our common stock may experience substantial losses.
The trading price of our common stock has been volatile and may become volatile again in the future. The trading price of our common stock could decline or fluctuate in response to a variety of factors, including:
| ● | our general financial condition and ability to maintain sufficient capital to continue operations; | |
| ● | our ability to enter into and maintain collaborative arrangements with third parties; | |
| ● | our ability to meet the performance estimates of securities analysts; | |
| ● | changes in buy/sell recommendations by securities analysts; | |
| ● | negative results from clinical and pre-clinical trials; |
| ● | fluctuation in our quarterly operating results; | |
| ● | reverse splits or increases in authorized shares; | |
| ● | substantial sales of our common stock; | |
| ● | general stock market conditions; or | |
| ● | other economic or external factors. |
The stock markets in general, and the markets for the securities of companies of our size and in our industry in particular, have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. These broad market fluctuations may adversely affect the trading price of our common stock.
Our executive officers and directors control a large percentage of the outstanding shares of our common stock, and thus can influence our corporate actions.
As of the date of this report, and after giving effect to the closing of our private placement of Series E Convertible Preferred Stock, Dr. Vuong Trieu, our Executive Chairman, directly or indirectly beneficially owns approximately 40% of the issued and outstanding shares of our common stock. Further, as of the date of this report, and after giving effect to the closing of our private placement of Series E Convertible Preferred Stock, our executive officers and directors as a group (including Dr. Trieu) beneficially own approximately half of the issued and outstanding shares of our common stock. Accordingly, and subject to the voting and director appointment rights of the holders of the Series E Convertible Preferred Stock, Dr. Trieu individually, and our executive officers and directors as a group, can significantly influence many, if not most, of our corporate actions. The concentration of the ownership of the issued and outstanding shares of our common stock by our executive officers and directors may also serve to limit the trading volume of our common stock.
| 52 |
We may not be able to achieve secondary trading of our stock in certain states because our common stock is not nationally traded.
Because our common stock is not listed for trading on a national securities exchange, our common stock is subject to the securities laws of the various states and jurisdictions of the U.S. in addition to federal securities law. This regulation covers any primary offering we might attempt and all secondary trading by our stockholders. If we fail to take appropriate steps to register our common stock or qualify for exemptions for our common stock in certain states or jurisdictions of the U.S., the investors in those jurisdictions where we have not taken such steps may not be allowed to purchase our stock or those who presently hold our stock may not be able to resell their shares without substantial effort and expense. These restrictions and potential costs could be significant burdens on our stockholders.
Our common stock is quoted on the OTCQB, which may limit the ability of our stockholders to sell their securities, and may cause volatility in the price of our common stock.
Our common stock is currently quoted on the OTCQB. Securities quoted on the OTCQB often experience a lack of liquidity as compared to securities trading on a national securities exchange. Such securities also have experienced extreme price and volume fluctuations in recent years, which have particularly affected the market prices of many smaller companies like ours. We anticipate that our common stock will be subject to the lack of liquidity and this volume and price volatility that is characteristic of the OTCQB.
Our common stock is considered a “penny stock,” and thereby is subject to additional sale and trading regulations that may make it more difficult to sell.
Our common stock is considered to be a “penny stock” since it does not qualify for one of the exemptions from the definition of “penny stock” under Section 3a51-1 of the Exchange Act. The principal result or effect of being designated a “penny stock” is that securities broker-dealers participating in sales of our common stock are subject to the “penny stock” regulations set forth in Rules 15-2 through 15g-9 promulgated under the Exchange Act. For example, Rule 15g-2 requires broker-dealers dealing in penny stocks to provide potential investors with a document disclosing the risks of penny stocks and to obtain a manually signed and dated written receipt of the document at least two business days before effecting any transaction in a penny stock for the investor’s account.
Moreover, Rule 15g-9 requires broker-dealers in penny stocks to approve the account of any investor for transactions in such stocks before selling any penny stock to that investor. This procedure requires the broker-dealer to (i) obtain from the investor information concerning his or her financial situation, investment experience and investment objectives; (ii) reasonably determine, based on that information, that transactions in penny stocks are suitable for the investor and that the investor has sufficient knowledge and experience as to be reasonably capable of evaluating the risks of penny stock transactions; (iii) provide the investor with a written statement setting forth the basis on which the broker-dealer made the determination in (ii) above; and (iv) receive a signed and dated copy of such statement from the investor, confirming that it accurately reflects the investor’s financial situation, investment experience and investment objectives. Compliance with these requirements may make it more difficult and time consuming for holders of our common stock to resell their shares to third parties or to otherwise dispose of them in the market or otherwise.
Various restrictions in our charter documents and Delaware law could prevent or delay a change in control of us that is not supported by our board of directors.
We are subject to a number of provisions in our charter documents and Delaware law that may discourage, delay or prevent a merger, acquisition or change of control that a stockholder may consider favorable. These anti-takeover provisions include:
| 53 |
| ● | advance notice procedures for nominations of candidates for election as directors and for stockholder proposals to be considered at stockholders’ meetings; and | |
| ● | the Delaware anti-takeover statute contained in Section 203 of the Delaware General Corporation Law. |
Section 203 of the Delaware General Corporation Law prohibits a merger, consolidation, asset sale or other similar business combination between us and any stockholder of 15% or more of our voting stock for a period of three years after the stockholder acquires 15% or more of our voting stock, unless (1) the transaction is approved by our board of directors before the stockholder acquires 15% or more of our voting stock, (2) upon completing the transaction the stockholder owns at least 85% of our voting stock outstanding at the commencement of the transaction, or (3) the transaction is approved by our board of directors and the holders of 66 2/3% of our voting stock, excluding shares of our voting stock owned by the stockholder.
We have never paid dividends on our common stock and do not anticipate paying cash dividends in the foreseeable future.
We have not paid any dividends on our common stock and do not expect to do so in the foreseeable future. In addition, the terms of any financing arrangements that we may enter into may restrict our ability to pay any dividends.
A significant number of shares of our common stock are subject to options, warrants and conversion rights, and we expect to sell additional shares of our common stock in the future. The issuance of these shares, which in some cases may occur on a cashless basis, will dilute the interests of other security holders and may depress the price of our common stock.
At December 31, 2017, there were outstanding warrants to purchase up to approximately 2.56 million shares common stock. If any of these warrants are exercised on a cashless basis, we will not receive any cash as a result of such exercises. At December 31, 2017, there were also outstanding 750 shares of Series C Convertible Preferred Stock, which shares are convertible into 0.5 million shares of common stock at an assumed conversion price of $7.50 per share of common stock, and 60 shares of Series D Stock, which shares are convertible into 75,000 shares of common stock at an assumed conversion price of $4.00 per share of common stock. On April 16, 2018, we issued 3,022 shares of Series E Convertible Preferred Stock, which shares are convertible into 30,220,000 shares of common stock at an assumed conversion price of $0.50 and 22,665,525 million warrants to purchase our common stock at a price of $0.55 per common share. In addition, we may issue a significant number of additional shares of common stock (and securities convertible into or exercisable for common stock) from time to time to finance our operations, to fund potential acquisitions, or in connection with additional stock options or restricted stock granted to our employees, officers, directors and consultants. The issuance of common stock (or securities convertible into or exercisable for common stock), and the exercise or conversion of securities exercisable for or convertible into common stock, will have a dilutive impact on other stockholders and could have a material negative effect on the market price of our common stock.
There are outstanding a significant number of shares available for future sales under Rule 144.
Many shares of our common stock may be deemed “restricted shares” and, in the future, may be sold in compliance with Rule 144 promulgated under the Securities Act of 1933, as amended (the “Securities Act”). Any sales of such shares of our common stock under Rule 144 could have a depressive effect on the market price of our common stock. In general, under Rule 144, a person who is not deemed to have been an affiliate of ours at any time during the three months preceding a sale, and who has beneficially owned restricted securities within the meaning of Rule 144 for at least six months (including any period of consecutive ownership of preceding non-affiliated holders) would be entitled to sell those shares, subject only to the availability of current public information about us. A non-affiliated person who has beneficially owned restricted securities within the meaning of Rule 144 for at least one year would be entitled to sell those shares without regard to the provisions of Rule 144. A person who is deemed to be an affiliate of ours and who has beneficially owned restricted securities within the meaning of Rule 144 for at least six months would be entitled to sell within any three-month period a number of shares that does not exceed the greater of one percent of the then outstanding shares of our common stock or the average weekly trading volume of our common stock during the four calendar weeks preceding such sale. Such sales are also subject to certain manner of sale provisions, notice requirements and the availability of current public information about us.
| 54 |
Our Board of Directors has the ability to issue “blank check” Preferred Stock.
Our Certificate of Incorporation authorizes the issuance of up to 100,000 shares of “blank check” preferred stock, with such designation rights and preferences as may be determined from time to time by our Board of Directors. At December 31, 2017, 90,000 shares had been designated as Series A Junior participating preferred stock and 1,000 shares had been designated as Series B Preferred Stock, none of which are issued and outstanding. Also at December 31, 2017, 1,200 shares had been designated as Series C Convertible Preferred Stock (of which 750 were outstanding as of such date) and 220 shares had been designated as Series D Convertible Preferred Stock (of which 60 were outstanding as of such date). As of the date of this report, 3,500 shares have been designated as Series E Convertible Preferred Stock, of which 3,022 are issued and outstanding. Our Board is empowered, without shareholder approval, to issue shares of preferred stock with dividend, liquidation, conversion, voting or other rights which could adversely affect the voting power or other rights of the holders of our common stock. In the event of such issuances, the preferred stock could be utilized, under certain circumstances, as a method of discouraging, delaying or preventing a change in control of our company. Although we have no present intention to issue any additional shares of our preferred stock, there can be no assurance that we will not do so in the future.
ITEM 1B. Unresolved Staff Comments.
Not applicable.
We do not own or lease any real property or facilities that are material to our current business operations. If facilities are necessary to conduct our current business operations, we utilize the facilities of Autotelic Inc. through the Master Services Agreement to which we are a party with Autotelic Inc. As we seek to expand our business operations, we may seek to lease facilities of our own in order to support our development, operational and administrative needs under our current operating plan. There can be no assurance that such facilities will be available, or that they will be available on suitable terms. Our inability to obtain such facilities could have a material adverse effect on our future plans and operations.
Legal Proceedings Regarding Vaya Pharma
We have been named in a complaint filed in the Supreme Court of the State of New York as a defendant in the matter entitled Vaya Pharma, Inc. v. Symplmed Technologies, Inc., Symplmed Pharmaceuticals, Inc., Erik Emerson and Marina Biotech, Inc. Although the complaint has been filed, we have not been legally served. The complaint alleges, in relevant part, that: (i) the sale by Symplmed Pharmaceuticals, Inc. of its assets related to its Prestalia product, and the sale by Symplmed Technologies, Inc. of its assets related to its DyrctAxess platform, should be set aside pursuant to New York law as they were consummated without fair consideration to the sellers (the “Symplmed Defendants”), and thereby had the effect of fraudulently depriving the creditors of the Symplmed Defendants, including Vaya Pharma, Inc., of funds that could have been used to pay their debts; and (ii) we are liable, as successor, for any and all claims by Vaya Pharma, Inc. against the Symplmed Defendants, though pursuant to the agreement we are only contractually responsible for liabilities that accrue after the parties entered into the agreement for Prestalia and any liabilities that existed prior to the agreement are contractually held by Symplmed. If and when we are legally served , we intend to dispute jurisdiction, the sufficiency of the pleading and the claims set forth in this complaint, and to defend this matter, vigorously. However, due to the inherent uncertainties of litigation, the ultimate outcome of this matter is uncertain. The Company reached a tentative settlement agreement and, although such settlement has not been finalized, the Company accrued a total provision of $250,000 for this complaint, which is included in accrued expenses on the accompanying consolidated balance sheet.
Paragraph IV Challenge
Our Prestalia product is currently involved in a paragraph IV challenge regarding patents issued to perindopril arginine. This challenge, which is currently pending in the United States District Court for the District of Delaware (No. 1:17-cv-00276), is captioned Apotex Inc. and Apotex Corp. v. Symplmed Pharmaceuticals, LLC and Les Laboratoires Servier. The challengers (Apotex Inc. and Apotex Corp. (“Apotex”)) have filed an Abbreviated New Drug Application seeking FDA approval to market a generic version of Prestalia and included a Paragraph (IV) certification. In the litigation, Apotex seeks a declaratory judgment that no valid claims of the two patents Symplmed listed in the FDA Orange Book as having claims covering Prestalia, U.S. Patent No. 6,696,481 and 7,846,961, will be infringed by the Apotex proposed generic version of Prestalia and that the claims of those patents are invalid. The challenge is designed to provide Apotex with an opportunity to enter the market with a generic version of Prestalia, ahead of the expiration of the patents with claims covering that product. Apotex entered into negotiations with Symplmed Pharmaceuticals, LLC (which entity sold its assets relating to Prestalia to us in June 2017, including its License and Commercialization Agreement with Les Laboratories Servier) and Les Laboratories Servier (which entity owns or controls intellectual property rights relating to pharmaceutical products containing as an active pharmaceutical ingredient perindopril in combination with other active pharmaceutical ingredients, which rights have been licensed to Symplmed Pharmaceuticals) to resolve the challenge in the second quarter of 2017, and such parties, along with us, have come to a general agreement on terms that will result in a delay to the challengers’ ability to enter the market with a generic version of Prestalia, while still providing the challenger with the right to enter the market prior to the expiration of the patent covering such product. The term sheet memorializing such terms is pending execution in a final settlement agreement. In the meantime, the District Court has entered an order extending the time for the defendants to respond to Apotex’s Complaint until January 9, 2018.
| 55 |
We do not anticipate that the proposed settlement terms – which would provide us with the ability to exclusively market Prestalia for a specified period of time without incurring the risks or costs of litigation – will have a material adverse impact on our long term business as a whole. We hold this expectation because it is our intention that Prestalia will be a building block toward the development of our company into a larger organization, which we hope will include the commercial launch by us of Prestalia and the expansion of our commercial capabilities in connection with such activities, followed by the launch of products that we license for commercialization purposes from our partners and the commercial launch of products that we develop internally. We anticipate that the aforementioned steps to expand and strengthen our commercial capabilities and diversify our product offerings so as to generate meaningful revenues in excess of those revenues generated by sales of Prestalia will occur during our exclusivity period with respect to Prestalia (i.e., prior to the date on which Apotex enters the market with a generic version of Prestalia). If we are able to achieve this growth, the overall economic impact that would result to us even if Apotex markets a generic version of Prestalia and captures a significant percentage of the market for that drug following our exclusivity period would be reduced.
However, there can be no assurance that the paragraph IV challenge will be resolved without litigation or resolved on the terms currently proposed. There also cannot be any assurance that our plans to commercialize Prestalia (or any of the products that it may license from its partners or that it may develop internally) and expand our product offerings and the revenue generated therefrom so as to lessen our reliance on sales of Prestalia will be achieved. Any failure of our expectations regarding the resolution of the paragraph IV challenge and the expansion of our commercial activities (and the revenues to be derived therefrom) could have a material adverse effect on us, our prospects and our results of operations.
General
Currently, there is no material litigation pending against our company other than as disclosed above. From time to time, we may become a party to litigation and subject to claims incident to the ordinary course of our business. Although the results of such litigation and claims in the ordinary course of business cannot be predicted with certainty, we believe that the final outcome of such matters will not have a material adverse effect on our business, results of operations or financial condition. Regardless of outcome, litigation can have an adverse impact on us because of defense costs, diversion of management resources and other factors.
ITEM 4. Mine Safety Disclosures.
Not applicable.
| 56 |
ITEM 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information
The common stock of Marina is quoted on the OTCQB under the symbol “MRNA”. The table below sets forth, for each of the quarterly periods indicated, the range of high and low bid prices of Marina’s common stock, as reported by the OTC Markets. The prices reflect inter-dealer prices, without retail mark-up, mark-down or commission, and may not represent actual transactions. The prices also reflect the reverse split of Marina’s common stock that was effective August 3, 2017. Prior to the Merger, which occurred on November 15, 2016, the common stock of IThena did not trade on any market or exchange.
| High | Low | |||||||
| Fiscal 2016: | ||||||||
| First Quarter | $ | 2.60 | $ | 1.20 | ||||
| Second Quarter | 5.20 | 1.20 | ||||||
| Third Quarter | 1.80 | 1.10 | ||||||
| Fourth Quarter (through November 15, 2016) | 1.40 | 0.80 | ||||||
| Fourth Quarter (after November 15, 2016) | 2.40 | 0.80 | ||||||
| Fiscal 2017: | ||||||||
| First Quarter | $ | 2.80 | $ | 1.20 | ||||
| Second Quarter | 4.80 | 2.60 | ||||||
| Third Quarter | 5.00 | 2.00 | ||||||
| Fourth Quarter | 2.70 | 1.33 | ||||||
| Fiscal 2018: | ||||||||
| First Quarter (through March 31, 2018) | $ | 1.97 | $ | 1.33 | ||||
On April 16, 2018, the closing price of our common stock reported by the OTC Markets was $1.22 per share.
Holders
As of April 16, 2018, there were 43 holders of record of our common stock.
Dividends
Payment of dividends and the amount of dividends depend on matters deemed relevant by our Board, such as our results of operations, financial condition, cash requirements, future prospects and any limitations imposed by law, credit agreements and debt securities. To date, we have not paid any cash dividends or stock dividends on our common stock. In addition, we currently anticipate that we will not pay any cash dividends in the foreseeable future. Furthermore, the terms of any financing arrangements that we may enter into may restrict our ability to pay any dividends.
ITEM 6. Selected Financial Data.
Not applicable.
| 57 |
ITEM 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
Overview
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations is intended to provide information necessary to understand our audited consolidated financial statements for the two year period ended December 31, 2017 and highlight certain other information which, in the opinion of management, will enhance a reader’s understanding of our financial condition, changes in financial condition and results of operations. In particular, the discussion is intended to provide an analysis of significant trends and material changes in our financial position and the operating results of our business during the year ended December 31, 2017, as compared to the year ended December 31, 2016. The December 31, 2017 statement of operations include the full year for Marina, Ithena and their subsidiaries whereas the December 31, 2016 statement of operations include the results of Ithena alone from January 1, 2016 to November 15, 2016 and combined with Marina and its subsidiaries from November 16, 2016 through December 31, 2016. As such, the statements of operations may not be comparable for the two years ended December 31, 2017. This discussion should be read in conjunction with our consolidated financial statements for the two-year period ended December 31, 2017 and related notes included elsewhere in this annual report on Form 10-K. These historical financial statements may not be indicative of our future performance. This Management’s Discussion and Analysis of Financial Condition and Results of Operations contains a number of forward-looking statements, all of which are based on our current expectations and could be affected by the uncertainties and risks described throughout this filing, particularly in “Item 1A. Risk Factors.”
Background and Merger
Overview
We are a fully integrated, commercial stage biopharmaceutical company delivering proprietary drug therapeutics for significant unmet medical needs in the U.S., Europe and certain additional international markets. Our portfolio of products currently focuses on fixed dose combinations (“FDC”) in hypertension, arthritis, pain and oncology allowing for innovative solutions to such unmet medical needs. Our mission is to provide effective and patient centric treatment for hypertension – including resistant hypertension – through our patented total care platform. In this connection, we acquired from Symplmed Pharmaceuticals LLC and its wholly-owned subsidiary, Symplmed Technologies, LLC, certain of the intellectual property assets related to the patented technology platform known as DyrctAxess, also called Total Care, that offers enhanced efficiency, control and information to empower patients, physicians and manufacturers to help achieve optimal care.
In doing so, we have created a universal platform for the effective treatment of hypertension as well as for the distribution of FDC hypertensive drugs such as our FDA-approved product Prestalia, and the other products in our pipeline, devices for Therapeutic Drug Monitoring (TDM), Blood Pressure (BP), and other cardiac monitors, as well as services such as counseling and prescription reminders.
We currently have one commercial and three clinical development programs underway: (i) Prestalia®, a single-pill FDC of perindopril, an angiotensin-converting-enzyme (“ACE”) inhibitor and amlodipine, a calcium channel blocker (“CCB”), which has been approved by the U.S. Food and Drug Administration (“FDA”) and is actively marketed in the U.S.; (ii) our next generation celecoxib program drug candidates for the treatment of acute and chronic pain, IT-102 and IT-103, each of which is an FDC of celecoxib, a COX-2 selective nonsteroidal anti-inflammatory drug (“NSAID”) and either lisinopril (IT-102) or olmesartan (IT-103) – both Lisinopril and olmesartan are antihypertension drugs; (iii) CEQ508, an oral delivery of small interfering RNA (“siRNA”) against beta-catenin, combined with IT-102 to suppress polyps in the precancerous syndrome and orphan indication Familial Adenomatous Polyposis (“FAP”); and (iv) CEQ508 combined with IT-103 to treat Colorectal Cancer.
Our current focus is primarily on the commercialization of Prestalia and secondarily the development of IT-102 and IT-103. We believe that by combining a COX-2 inhibitor with an antihypertensive in a single FDC oral tablet, IT-102 and IT-103 will each offer improved safety profiles as compared to currently available and previously marketed COX-2 inhibitors as well as address patients with chronic pain who are commonly taking antihypertension drugs concurrently. We further believe that the current opioid addiction epidemic in the U.S. has been driven in part by the withdrawal from the market of certain COX-2 inhibitors due to their associated risk of cardiovascular-related adverse events. We plan to license or divest our other assets since they no longer align with our focus on the treatment of hypertension.
| 58 |
We intend to create value through the continued commercialization of our FDA-approved product, Prestalia, while moving our FDC development programs forward to further strengthen our commercial presence. We intend to retain ownership and control of all of our product candidates, but in the interest of accelerated growth and market penetration, we will also consider partnerships with pharmaceutical or biotechnology companies in order to reduce time to market and to balance development risks, both clinically and financially.
As our strategy is to be a fully integrated biopharmaceutical company, we will drive a primary corporate focus on revenue generation through our commercial assets, with a secondary focus on advancing our FDC pipeline to further enhance our commercial presence.
Merger with IThenaPharma
On November 15, 2016, Marina entered into the Merger Agreement with IThenaPharma, Merger Sub and Vuong Trieu, as the IThena representative, pursuant to which, among other things, Merger Sub merged with and into IThenaPharma, with IThenaPharma surviving as a wholly owned subsidiary of Marina.
Pursuant to the Merger Agreement, at the effective time of the Merger, without any action on the part of any shareholder, each issued and outstanding share of IThenaPharma’s common stock, other than shares to be cancelled pursuant to the Merger Agreement, was converted into the right to receive shares of Marina common stock at the exchange ratio set forth therein (the “Exchange Ratio”). In addition, each outstanding IThenaPharma warrant was assumed by Marina and converted into a warrant representing the right to purchase shares of Marina common stock, with the number of shares underlying such warrant and the exercise price thereof being adjusted by the Exchange Ratio, with any fractional shares rounded down to the next lowest number of whole shares.
As a result of the Merger, the former holders of IThenaPharma common stock immediately prior to the completion of the Merger owned approximately 65% of the issued and outstanding shares of Marina common stock immediately following the completion of the Merger.
IThena was deemed to be the accounting acquirer in the Merger, and thus the historical financial statements of IThena are treated as the historical financial statements of our company and are reflected in our quarterly and annual reports for periods ending after the effective time of the Merger. Accordingly, beginning with our Annual Report on Form 10-K for the fiscal year ended December 31, 2016, we reported the results of IThena and Marina and their respective subsidiaries on a consolidated basis. The December 31, 2017 statement of operations include the full year for Marina, Ithena and their subsidiaries whereas the December 31, 2016 statement of operations include the results of Ithena alone from January 1, 2016 to November 15, 2016 and combined with Marina and its subsidiaries from November 16, 2016 through December 31, 2016. As such, the statements of operations may not be comparable for the two years ended December 31, 2017.
Results of Operations
Comparison of the Year Ended December 31, 2017 to December 31, 2016
Our loss before income taxes for the year ended December 31, 2017 is summarized as follows in comparison to the year ended December 31, 2016. The December 31, 2017 statement of operations include the full year for Marina, Ithena and their subsidiaries whereas the December 31, 2016 statement of operations include the results of Ithena alone from January 1, 2016 to November 15, 2016 and combined with Marina and its subsidiaries from November 16, 2016 through December 31, 2016. As such, the statements of operations may not be comparable for the two years ended December 31, 2017. As such, the results of operations for the years ended December 31, 2017 and 2016, respectively, are not comparable.
| 59 |
| Years Ended | ||||||||
| December 31, 2017 | December 31, 2016 | |||||||
| Revenues | $ | - | $ | - | ||||
| Research and development | 907,493 | 132,513 | ||||||
| General and administrative expenses | 4,732,221 | 576,028 | ||||||
| Amortization | 450,903 | 49,189 | ||||||
| Other income (expense), net | (127,931 | ) | (78,613 | ) | ||||
| Loss before provision for income taxes | $ | (6,218,548 | ) | $ | (836,343 | ) | ||
Revenues
We had no revenues in the year ended December 31, 2017 or 2016. The majority of our licensing deals provide for clinical and regulatory milestones, so significant revenues could result from the existing licenses, but are uncertain as to timing or probability. We will continue to seek research and development collaborations as well as licensing transactions to fund business operations.
Expenses
Our expenses for the year ended December 31, 2017 are summarized as follows in comparison to our expenses for the year ended December 31, 2016. As stated above, as a result of the November 2016 Merger with IThena, the December 31, 2017 statement of operations include the full year for Marina, Ithena and their subsidiaries whereas the December 31, 2016 statement of operations include the results of Ithena alone from January 1, 2016 to November 15, 2016 and combined with Marina and its subsidiaries from November 16, 2016 through December 31, 2016. As such, the statements of operations may not be comparable for the two years ended December 31, 2017.
Research and Development
Research and development (“R&D”) expense increased by $0.8 million, as compared to the year ended December 31, 2016, primarily due to costs related to the MSA with Autotelic Inc. We incurred $0.7 million of costs related to the MSA, of which $0.35 million was on a non-cash basis through the issuance of warrants. Other R&D expenses consist of costs of sublicensing fees, clinical development, pre-clinical studies, consulting, other outside services, and other costs.
General and Administrative
General and administrative (“G&A”) expense increased by $4.2 million for the year ended December 31, 2017, as compared to the year ended December 31, 2016, primarily due to G&A expenses of advisory services of $1.35 million, legal costs of approximately $0.9 million, $0.25 million to settle litigation (described in more detail above under Part 3: Legal Proceedings), accounting and auditing and related services fees of approximately $0.2 million, approximately $0.2 million for board member fees and approximately $0.2 million for insurance costs. In addition, we incurred about $0.4 million of costs related to the MSA, of which $0.2 million was on a non-cash basis through the issuance of warrants. No similar expenses were recorded during the year ended December 31, 2016.
Amortization Expense
Amortization expenses relates to amortization of intangible assets acquired in the November 2016 Merger and the asset purchases on June 5, 2017 and July 21, 2017, with a combined estimated fair value of $3,056,066.
| 60 |
Other Income (Expense)
| Years ended | ||||||||
| December 31, 2017 | December 31, 2016 | |||||||
| Interest expense | $ | (78,890 | ) | $ | (3,513 | ) | ||
| Change in fair value liability of warrants | - | (75,100 | ) | |||||
| Amortization of debt discount | (49,041 | ) | - | |||||
| Total other expense, net | $ | (127,931 | ) | $ | (78,613 | ) | ||
Total net other expense for the year ended December 31, 2017 increased $49,318 compared to the year ended December 31, 2016. The increase in interest expense is primarily attributable to an increase in notes payable during 2017, partially offset by a decrease in the change in fair value of the Company’s price adjustable warrants. The amortization of debt discount for 2017 is a result of the debt discount arising from the issuance of convertible debt in November of 2017. The fair value liability is revalued each balance sheet date utilizing probability-weighted Black-Scholes computations, with the decrease or increase in fair value being reported in the statement of operations as other income or expense, respectively.
Liquidity & Capital Resources
Working Capital Deficiency
| December 31, 2017 | December 31, 2016 | |||||||
| Current assets | $ | 124,943 | $ | 316,480 | ||||
| Current liabilities | (5,735,503 | ) | (2,967,669 | ) | ||||
| Working capital deficiency | $ | (5,610,560 | ) | $ | (2,651,189 | ) | ||
Current assets decreased by $191,537, which was attributable to a decrease in prepaid expenses of $192,568.
Current liabilities increased by $2,767,834, which was primarily attributable to an increase of accounts payable of $370,092, an increase in amounts due to related party of $1,205,561, an increase of $320,000 in accrued fee payable to the FDA, an increase of $1,212,040 in convertible notes to related and unrelated parties. This was partially offset by a decrease in the fair value of price adjustable warrant liabilities of $141,723 issuable in connection with the MSA with Autotelic Inc.
Cash Flows
| Year ended | ||||||||
| December 31, 2017 | December 31, 2016 | |||||||
| Net cash (used in) provided by operating activities | $ | (1,260,685 | ) | $ | (522,314 | ) | ||
| Net cash (used in) provided by investing activities | (375,000 | ) | 5,867 | |||||
| Net cash provided by financing activities | 1,636,716 | 359,946 | ||||||
| Increase (decrease) in cash and cash equivalents | $ | 1,031 | $ | (156,501 | ) | |||
The increase in net cash used in operating activities during the year ended December 31, 2017, compared to 2016, was mainly due to increased operating expenses subsequent to the Merger, offset by non-cash share-based compensation of $1,828,941, increase in amortization of intangibles of $401,714, an increase in accrued expenses of $837,714, an increase in accounts payable of $425,339, and an increase in amounts due to related party of $1,205,561.
The Company used cash of $300,000 in investing activities for payments towards the July 21, 2017 Prestalia acquisition and $75,000 towards the DyrctAxess acquisition during the year ended December 31, 2017. This investment was made to transform the Company to be a commercial stage company from a development stage company.
| 61 |
The $1,276,770 increase in net cash provided by financing activities during the year ended December 31, 2017, compared to 2016, is primarily attributable to proceeds of $250,000 from the sale of stock, $1,240,073 from additional borrowings on related party notes and $170,643 received from the conversion of warrants to common stock.
We will need to raise additional operating capital in calendar year 2018 in order to maintain our operations and to realize our business plan. Without additional sources of cash and/or the deferral, reduction, or elimination of significant planned expenditures, we will not have the cash resources to continue as a going concern.
Going Concern
The consolidated financial statements contained in this report have been prepared assuming that the Company will continue as a going concern. We have an accumulated deficit for the period from inception through December 31, 2017 in excess of $8 million, as well as negative cash flows from operating activities. We had obtained a line of credit from Autotelic Inc. of $500,000, of which we have utilized $90,816, and such line of credit has since expired. Our ability to continue as a going concern depends on our ability to raise substantial additional funds through public or private equity offerings, debt financings or additional strategic alliances and licensing arrangements. On April 16, 2018, we held the initial closing of our private placement of shares of our Series E Convertible Preferred Stock, and warrants to purchase shares of our common stock, as a result of which we raised over $10 million in net proceeds to our company. For our assessment as of December 31, 2017, we have considered the amount raised and we will continue to reassess our ability to address the going concern. We may raise further capital or partner with other entities to further the development of our pipeline products.
Our continuation as a going concern is dependent on our ability to obtain additional financing as may be required and ultimately to attain profitability. If we raise additional funds through the issuance of equity or equity-linked securities, the percentage ownership of current shareholders could be reduced, and such securities might have rights, preferences or privileges senior to our common stock. Additional financing may not be available upon acceptable terms, or at all. If adequate funds are not available or are not available on acceptable terms, we may not be able to take advantage of prospective business endeavors or opportunities, which could significantly and materially restrict our future plans for developing our business and achieving commercial revenues. If we are unable to obtain the necessary capital when needed, we may have to cease operations.
During 2016 and 2017, we have funded our losses primarily through the sale of common stock and warrants, loans provided by Dr. Trieu and Autotelic Inc. pursuant to the Line Letters, and the issuance of convertible notes and/or secured loans. During the year ended December 31, 2017, we raised $250,000 from the private placement of our equity securities, received $170,643 from the conversion of warrants to common stock, and raised $1,240,073 from the issuance of notes and convertible notes, including $380,888 borrowed under the Line Letter from Dr. Trieu and Autotelic Inc. In addition, in April 2017, we entered into an additional credit agreement with Autotelic Inc., pursuant to which Autotelic Inc. offered to the Company an unsecured line of credit in an amount not to exceed $500,000, to be used for current operating expenses of the Company. We have utilized $90,819, which amount is outstanding at December 31, 2017. No additional advances are available under this line as it had expired by December 31, 2017.
Future Financing
The current financing through the issuance of our newly created Series E Convertible Preferred shares will allow us to commercialize Prestalia. We will require additional funds to implement the growth strategy for our business. As mentioned above, we have, in the past, raised additional capital to both supplement our commercialization, clinical development and operational needs. We may need to raise additional funds required to further our commercialization efforts through equity financing, debt financing, strategic alliances or other sources, which may result in further dilution in the equity ownership of our shares. We continue to pursue options available to us to raise additional funds. There can be no assurance that additional financing will be available when needed or, if available, that it can be obtained on commercially reasonable terms. If we will not be able to obtain the additional financing on a timely basis as required, or generate material revenues from operations, we will not be able to meet our other obligations as they become due and will be forced to scale down or perhaps even cease our operations.
Off-Balance Sheet Arrangements
As of December 31, 2017, we did not have any off-balance sheet arrangements, as defined in Item 303(a)(4)(ii) of SEC Regulation S-K.
| 62 |
Critical Accounting Policies and Estimates
Our significant accounting policies are more fully described in the notes to our financial statements included herein for the year ended December 31, 2017.
New and Recently Adopted Accounting Pronouncements
Any new and recently adopted accounting pronouncements are more fully described in Note 1 to our financial statements included herein for the year ended December 31, 2017.
ITEM 7A. Quantitative and Qualitative Disclosures About Market Risk.
Not applicable.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The information called for by Item 8 is included following the “Index to Financial Statements” on page F-1 contained in this annual report on Form 10-K.
ITEM 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
ITEM 9A. Controls and Procedures.
(a) Disclosure Controls and Procedures. As of the end of the period covered by this Annual Report on Form 10-K, we carried out an evaluation, under the supervision and with the participation of senior management, including our principal executive officer (“PEO”) and principal financial officer (“PFO”), of the effectiveness of the design and operation of our disclosure controls and procedures (as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act). Management identified material weaknesses in internal control over financial reporting as described below in “Management Report on Internal Control” and therefore, our PEO and PFO concluded that, as of December 31, 2017, our disclosure controls and procedures were not effective.
(b) Internal Control over Financial Reporting. Management has reported to our Board of Directors and the Audit Committee thereof the material weaknesses described below. Other than the material weaknesses discussed in management’s assessment, which initially arose during the fourth fiscal quarter of the fiscal year ended December 31, 2016 and thus were described in our Annual Report on form 10-K for the fiscal year ended December 31, 2016, and certain remediation efforts that have been undertaken in response thereto, there have been no changes in our internal control over financial reporting or in other factors during the fiscal quarter ended December 31, 2017 that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
(c) Management Report on Internal Control. Management is responsible for establishing and maintaining effective internal control over financial reporting. Internal control over financial reporting, as such term is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act, is a process designed by, or under the supervision of, our PEO and PFO, or persons performing similar functions, and effected by our Board, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles (GAAP). Our management, with the participation of our PEO and PFO, has established and maintained policies and procedures designed to maintain the adequacy of our internal control over financial reporting, and include those policies and procedures that:
1) Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets;
2) Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and
3) Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
| 63 |
Management has evaluated the effectiveness of our internal control over financial reporting as of December 31, 2017, based on the control criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in a report entitled Internal Control — Integrated Framework (2013), and identified material weaknesses which are described below. Because of these material weaknesses, which initially arose during the fourth fiscal quarter of the fiscal year ended December 31, 2016 and thus were described in our Annual Report on form 10-K for the fiscal year ended December 31, 2016, management has concluded that we did not maintain effective internal control over financial reporting for a majority of 2017 and as of December 31, 2017 with respect to the preparation of these financial statements.
A material weakness is a deficiency or a combination of deficiencies in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis. In connection with the evaluation described above, management determined that our lack of a sufficient complement of personnel with an appropriate level of knowledge and experience in the performance of an audit of a public company that is commensurate with our financial reporting requirements constituted a material weakness as of December 31, 2017. To remediate the foregoing material weakness, we have recently hired additional external experienced accounting and other personnel to assist with filings and financial record keeping, including the hiring of a Chief Financial Officer, Amit Shah, in October 2017, and have taken additional steps to improve our financial reporting systems, enhance our existing policies, procedures and controls. We believe that, as a result of the foregoing efforts, the material weaknesses that were identified will be remediated in 2018.
This annual report does not include an attestation report of our registered public accounting firm regarding internal control over financial reporting. Such a report is not required for smaller reporting companies such as us pursuant to The Dodd-Frank Wall Street Reform and Consumer Protection Act that Congress enacted in July 2010, which permanently exempts companies with less than $75 million in market capitalization from Section 404(b) of the Sarbanes-Oxley Act of 2002 requiring an outside auditor to attest annually to a company’s internal-control evaluations.
(d) Because of its inherent limitations, internal control over financial reporting may not prevent or detect all errors or misstatements and all fraud. Therefore, even those systems determined to be effective can provide only reasonable, not absolute, assurance that the objectives of the policies and procedures are met. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
None.
ITEM 10. Directors, Executive Officers and Corporate Governance.
The information required by this Item is incorporated by reference to our Definitive Proxy Statement prepared in connection with our 2018 Annual Meeting of Stockholders to be filed not later than 120 days after the end of our 2017 fiscal year.
ITEM 11. Executive Compensation.
The information required by this Item is incorporated by reference to our Definitive Proxy Statement prepared in connection with our 2018 Annual Meeting of Stockholders to be filed not later than 120 days after the end of our 2017 fiscal year.
ITEM 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The information required by this Item is incorporated by reference to our Definitive Proxy Statement prepared in connection with our 2018 Annual Meeting of Stockholders to be filed not later than 120 days after the end of our 2017 fiscal year.
ITEM 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by this Item is incorporated by reference to our Definitive Proxy Statement prepared in connection with our 2018 Annual Meeting of Stockholders to be filed not later than 120 days after the end of our 2017 fiscal year.
ITEM 14. Principal Accounting Fees and Services.
The information required by this Item is incorporated by reference to our Definitive Proxy Statement prepared in connection with our 2018 Annual Meeting of Stockholders to be filed not later than 120 days after the end of our 2017 fiscal year.
| 64 |
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
| 65 |
| 66 |
| 67 |
| 68 |
| 69 |
| (1) | Portions of this exhibit have been omitted pursuant to a request for confidential treatment under Rule 24b-2 of the Securities Exchange Act of 1934, amended, and the omitted material has been separately filed with the SEC. |
| (2) | Filed herewith. |
| (3) | Furnished herewith. |
| (4) | To be filed by amendment. |
| # | Previously filed or furnished. |
| ** | Indicates management contract or compensatory plan or arrangement. |
(b) Financial Statement Schedules. All financial statement schedules are omitted because they are not applicable or not required or because the required information is included in the financial statements or notes thereto.
| 70 |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on April 16 2018.
| MARINA BIOTECH, INC. | ||
| By: | /s/ Vuong Trieu, Ph.D. | |
| Vuong Trieu, Ph.D. | ||
| Executive Chairman | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this report has been signed by the following persons on behalf of the Registrant and in the capacities indicated on March __, 2018.
| Signature | Title | |
| /s/ Vuong Trieu, Ph.D. | Executive Chairman | |
| Vuong Trieu, Ph.D. | (Principal Executive Officer) | |
| /s/ Joseph Ramelli | Chief Executive Officer | |
| Joseph Ramelli | (Principal Executive Officer) | |
| /s/ Amit Shah | Chief Financial Officer | |
| Amit Shah | (Principal Financial Officer) | |
| /s/ Isaac Blech | Director | |
| Isaac Blech | ||
| /s/ Philippe P. Calais, Ph.D. | Director | |
| Philippe P. Calais, Ph.D. | ||
| /s/ Philip C. Ranker | Director | |
| Philip C. Ranker | ||
| /s/ Donald A. Williams | Director | |
| Donald A. Williams | ||
/s/ Joseph Ramelli |
Chief Executive Officer | |
| Joseph Ramelli | (Principal Executive Officer) |
| 71 |
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
MARINA BIOTECH, INC.
CONSOLIDATED FINANCIAL STATEMENTS AS OF DECEMBER 31, 2017 AND 2016
TABLE OF CONTENTS
| Page | |
| REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM | F-2 |
| CONSOLIDATED FINANCIAL STATEMENTS: | |
| Consolidated Balance Sheets | F-3 |
| Consolidated Statements of Operations | F-4 |
| Consolidated Statements of Stockholders’ Equity | F-5 |
| Consolidated Statements of Cash Flows | F-6 |
| Notes to Consolidated Financial Statements | F-7 |
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of Marina Biotech, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Marina Biotech, Inc. and its subsidiaries (the Company) as of December 31, 2017 and 2016, the related consolidated statements of operations, stockholders’ equity and cash flows for the years then ended, and the related notes to the consolidated financial statements (collectively, the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern Uncertainty
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has suffered recurring losses from operations and negative cash flows from operations and has had recurring negative working capital. This raises substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters also are described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Squar Milner LLP
We have served as the Company’s auditor since 2015.
Los Angeles, California
April 17, 2017
| F-2 |
MARINA BIOTECH, INC.
| December 31, 2017 | December 31, 2016 | |||||||
| ASSETS | ||||||||
| Current assets | ||||||||
| Cash | $ | 106,378 | $ | 105,347 | ||||
| Prepaid expenses and other assets | 18,565 | 211,133 | ||||||
| Total current assets | 124,943 | 316,480 | ||||||
| Intangible assets, net of amortization | 2,555,974 | 2,311,877 | ||||||
| Goodwill | 3,502,829 | 3,558,076 | ||||||
| 6,058,803 | 5,869,953 | |||||||
| Total assets | $ | 6,183,746 | $ | 6,186,433 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current liabilities | ||||||||
| Accounts payable | $ | 1,033,353 | $ | 663,261 | ||||
| Accrued expenses | 1,139,369 | 1,345,730 | ||||||
| Due to related party | 1,336,518 | 130,957 | ||||||
| Accrued fee payable | 320,000 | - | ||||||
| Notes payable | 444,223 | 435,998 | ||||||
| Notes payable - related parties | 1,462,040 | 250,000 | ||||||
| Fair value of liabilities for price adjustable warrants (Note 1) | - | 141,723 | ||||||
| Total current liabilities | 5,735,503 | 2,967,669 | ||||||
| Commitments and contingencies (Note 9) | ||||||||
| Stockholders’ equity | ||||||||
| Preferred stock, $0.01 par value; 100,000 shares authorized | ||||||||
| Series C convertible preferred stock, $0.01 par value; $5,100 liquidation preference; 1,200 shares authorized; 750 and 1,020 shares issued and outstanding as of September 30, 2017 and December 31, 2016, respectively | - | - | ||||||
| Series D convertible preferred stock, $0.01 par value; $300 liquidation preference; 220 shares authorized; 60 shares issued and outstanding as of December 31, 2017 and December 31, 2016, respectively | - | - | ||||||
| Common stock, $0.006 par value; 180,000,000 shares authorized, 10,521,728 and 8,977,138 shares issued and outstanding as of December 31, 2017 and 2016, respectively | 63,127 | 53,863 | ||||||
| Additional paid-in capital | 8,413,823 | 5,115,983 | ||||||
| Accumulated deficit | (8,028,707 | ) | (1,951,082 | ) | ||||
| Total stockholders’ equity | 448,243 | 3,218,764 | ||||||
| Total liabilities and stockholders’ equity | $ | 6,183,746 | $ | 6,186,433 | ||||
The accompanying footnotes are an integral part of these consolidated financial statements.
| F-3 |
MARINA BIOTECH, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
| For
the Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Operating expenses | ||||||||
| Research and development | 907,493 | 132,513 | ||||||
| General and administrative | 4,732,221 | 576,028 | ||||||
| Amortization | 450,903 | 49,189 | ||||||
| Total operating expenses | 6,090,617 | 757,730 | ||||||
| Loss from operations | (6,090,617 | ) | (757,730 | ) | ||||
| Other expense | ||||||||
| Interest expense | (78,890 | ) | (3,513 | ) | ||||
| Change in fair value liability of warrants | - | (75,100 | ) | |||||
| Amortization of debt discount | (49,041 | ) | - | |||||
| (127,931 | ) | (78,613 | ) | |||||
| Loss before provision for income taxes | (6,218,548 | ) | (836,343 | ) | ||||
| Provision for income taxes | 800 | 800 | ||||||
| Net loss | $ | (6,219,348 | ) | $ | (837,143 | ) | ||
| Net loss per share – basic and diluted | $ | (0.63 | ) | $ | (0.02 | ) | ||
| Weighted average shares outstanding | 9,836,109 | 47,431,096 | ||||||
The accompanying footnotes are an integral part of these consolidated financial statements.
| F-4 |
MARINA BIOTECH, INC.
CONSOLIDATED STATEMENTS OF SHAREHOLDERS’ DEFICIENCY
| Additional | ||||||||||||||||||||
| Common Stock | Paid-in | Accumulated | ||||||||||||||||||
| Number | Par Value | Capital | Deficit | Total | ||||||||||||||||
| Balance, December 31, 2015 | 5,839,283 | $ | 35,036 | $ | 1,169,088 | $ | (1,113,939 | ) | $ | 90,185 | ||||||||||
| Issuance of warrants | - | – | 36,470 | – | 36,470 | |||||||||||||||
| Capital contribution from related party | - | – | 257,252 | – | 257,252 | |||||||||||||||
| Effect of reverse acquisition on November 15, 2016: | ||||||||||||||||||||
| Adjustment for reverse merger | 3,137,855 | 18,827 | 3,653,173 | – | 3,672,000 | |||||||||||||||
| Net loss | – | – | – | (837,143 | ) | (837,143 | ) | |||||||||||||
| Balance, December 31, 2016 | 8,977,138 | $ | 53,863 | $ | 5,115,983 | $ | (1,951,082 | ) | $ | 3,218,764 | ||||||||||
| Modified retrospective adjustment of adoption of new accounting standard (Note 1) | $ | 141,723 | 141,723 | |||||||||||||||||
| Sale of common stock to related party | 86,207 | 517 | 249,483 | – | 250,000 | |||||||||||||||
| Common stock issued for services | 530,058 | 3,180 | 1,400,977 | – | 1,404,157 | |||||||||||||||
| Common stock issued to settle accounts payable and accrued liabilities | 622,296 | 3,734 | 972,980 | – | 976,714 | |||||||||||||||
| Return of common stock against settlement of note receivable | (8,725 | ) | (52 | ) | (31,352 | ) | – | (31,404 | ) | |||||||||||
| Restricted stock issued to officers | 70,000 | 420 | 245,580 | – | 246,000 | |||||||||||||||
| Share based compensation | – | – | 178,784 | – | 178,784 | |||||||||||||||
| Warrants issued with convertible debt as loan fee | – | – | 112,210 | – | 112,210 | |||||||||||||||
| Exercise of warrants for common stock | 60,944 | 366 | 170,277 | – | 170,643 | |||||||||||||||
| Conversion of Series C Preferred to common stock | 180,000 | 1,080 | (1,080 | ) | - | |||||||||||||||
| Effects of rounding due to reverse split | 3,360 | 19 | (19 | ) | – | - | ||||||||||||||
| Net loss | – | – | – | (6,219,348 | ) | (6,219,348 | ) | |||||||||||||
| Balance, December 31, 2017 | 10,521,278 | $ | 63,127 | $ | 8,413,823 | $ | (8,028,707 | ) | $ | 448,243 | ||||||||||
The accompanying footnotes are an integral part of these consolidated financial statements.
| F-5 |
MARINA BIOTECH, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| For the Years Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Cash Flows Used in Operating Activities: | ||||||||
| Net loss | $ | (6,219,348 | ) | $ | (837,143 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Share based compensation | 424,784 | - | ||||||
| Common shares issued to third party for services | 1,404,157 | - | ||||||
| Warrants issued for services | - | 36,470 | ||||||
| Amortization of intangibles | 450,903 | 49,189 | ||||||
| Change in fair value liabilities for price adjustable warrants | - | 75,100 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other assets | 161,164 | (87,814 | ) | |||||
| Accounts payable | 425,339 | (876,044 | ) | |||||
| Accrued expenses | 837,714 | 1,130,757 | ||||||
| Due to related party | 1,205,561 | 23,641 | ||||||
| Other liabilities) | - | (36,470 | ) | |||||
| Net Cash Used in Operating Activities | (1,260,685 | ) | (522,314 | ) | ||||
| Cash Flows (Used in) Provided by Investing Activities: | ||||||||
| Net cash acquired in reverse acquisition | - | 5,867 | ||||||
| Purchase of intangible assets | (375,000 | ) | - | |||||
| Net Cash (Used in) Provided by Investing Activities | (375,000 | ) | 5,867 | |||||
| Cash Flows from Financing Activities: | ||||||||
| Proceeds from sale of common stock to related party | 250,000 | - | ||||||
| Proceeds from notes payable due to related party | 500,000 | - | ||||||
| Proceeds from convertible notes | - | 124,973 | ||||||
| Proceeds from convertible notes due to related parties, net | 790,073 | 234,973 | ||||||
| Proceeds from exercise of warrants for common stock | 170,643 | - | ||||||
| Payments for notes payable | (24,000 | ) | - | |||||
| Loan fees | (50,000 | ) | - | |||||
| Net Cash Provided by Financing Activities | 1,636,716 | 359,946 | ||||||
| Net increase (decrease) in cash | 1,031 | (156,501 | ) | |||||
| Cash – Beginning of Period | 105,347 | 261,848 | ||||||
| Cash - End of Period | $ | 106,378 | $ | 105,347 | ||||
| Supplementary Cash Flow Information: | ||||||||
| Interest paid | $ | - | $ | - | ||||
| Income taxes paid | $ | 800 | $ | 800 | ||||
| Non-cash Investing and Financing Activities: | ||||||||
| Issuance of warrants for services | $ | - | $ | 36,470 | ||||
| Common stock issued in reverse acquisition | $ | - | $ | 3,672,000 | ||||
| Contributed capital | $ | - | $ | 257,252 | ||||
| Common stock issued for accounts payable | $ | 976,714 | $ | - | ||||
| Return of common stock for other assets | $ | 31,404 | $ | - | ||||
| Adjustment to goodwill for change in value of pre-acquisition accounts payable | $ | 55,247 | $ | - | ||||
| Assumption of liabilities for acquisition of assets | $ | 320,000 | $ | - | ||||
| Warrants issued with convertible debt as loan fee | $ | 112,210 | $ | - | ||||
| Non-cash retrospective adjustment (Note 1) | $ | 141,723 | $ | - | ||||
The accompanying footnotes are an integral part of these consolidated financial statements.
| F-6 |
MARINA BIOTECH, INC.
Notes to Consolidated Financial Statements
FOR THE YEAR ENDED dECEMBER 31, 2017
Note 1 – organization and business operations
Reverse Merger with IThenaPharma
On November 15, 2016, Marina entered into, and consummated the transactions contemplated by, an Agreement and Plan of Merger between and among IthenaPharma, Inc., a Delaware corporation (“IThena”), IThena Acquisition Corporation, a Delaware corporation and wholly-owned subsidiary of Marina (“Merger Sub”), and Vuong Trieu as the IThena representative (the “Merger Agreement”), pursuant to which IThena merged into Merger Sub (the “Merger”). For a more detailed discussion on the reverse merger, refer to Note 3 – Intangible Assets below.
IThena is a developer of personalized therapies for combined pain/hypertension through its proprietary Fixed Dose Combination (“FDC”) technology and point of care Therapeutic Drug Monitoring (“TDM”). Through the combination of these technologies, IThenaPharma is looking to deliver therapies with improved compliance and personalized dosing. IThena’s lead products are the celecoxib FDCs which include IT-102 and IT-103, fixed dose combinations of celecoxib and lisinopril and celecoxib and olmesartan, respectively. IT-102 and IT-103 are being developed as celecoxib without the drug induced edema associated with celecoxib alone. IT-102 and IT-103 are being developed initially for combined arthritis / hypertension and subsequently for treatment of pain, or cancer, or other indications requiring high doses of celecoxib.
Business Operations
Marina Biotech, Inc. and its wholly-owned subsidiaries, MDRNA Research, Inc., Cequent Pharmaceuticals, Inc., Atossa Healthcare, Inc., and IthenaPharma, Inc. (collectively “Marina,” “we,” “our,” or “us”) is a fully integrated, commercial stage biopharmaceutical company delivering proprietary drug therapeutics for significant unmet medical needs in the U.S., Europe and certain additional international markets. Its portfolio of products currently focuses on FDCs in hypertension, arthritis, pain and oncology allowing for innovative solutions to such unmet medical needs. Its mission is to provide effective and patient centric treatment for hypertension – including resistant hypertension. In this connection, we acquired from Symplmed and its wholly-owned subsidiary, Symplmed Technologies, LLC, certain of the intellectual property assets related to the patented technology platform known as DyrctAxess, also called Total Care, that offers enhanced efficiency, control and information to empower patients, physicians and manufacturers to help achieve optimal care.
In doing so, we have created a universal platform for the effective treatment of hypertension as well as for the distribution of FDC hypertensive drugs, such as our FDA-approved product Prestalia, and the other products in our pipeline, devices for therapeutic drug monitoring, blood pressure, and other cardiac monitors, as well as services such as counseling and prescription reminders.
We currently have one commercial and three clinical development programs underway: (i) Prestalia®, a single-pill FDC of perindopril, an angiotensin-converting-enzyme (“ACE”) inhibitor and amlodipine, a calcium channel blocker (“CCB”), which has been approved by the U.S. Food and Drug Administration (“FDA”) and is actively marketed in the U.S.; (ii) our next generation celecoxib program drug candidates for the treatment of acute and chronic pain, IT-102 and IT-103, each of which is an FDC of celecoxib, a COX-2 selective nonsteroidal anti-inflammatory drug (“NSAID”) and either lisinopril (IT-102) or olmesartan (IT-103) – both Lisinopril and olmesartan are antihypertension drugs; (iii) CEQ508, an oral delivery of small interfering RNA (“siRNA”) against beta-catenin, combined with IT-102 to suppress polyps in the precancerous syndrome and orphan indication Familial Adenomatous Polyposis (“FAP”); and (iv) CEQ508 combined with IT-103 to treat Colorectal Cancer.
Our current focus is primarily on the commercialization of Prestalia. The development of IT-102 and IT-103 will be achieved through partnering with other entities or raising additional funds to advance the development of the products. We believe that by combining a COX-2 inhibitor with an antihypertensive in a single FDC oral tablet, IT-102 and IT-103 will each offer improved safety profiles as compared to currently available and previously marketed COX-2 inhibitors as well as address patients with chronic pain who are commonly taking antihypertension drugs concurrently. We further believe that the current opioid addiction epidemic in the U.S. has been driven in part by the withdrawal from the market of certain COX-2 inhibitors due to their associated risk of cardiovascular-related adverse events. We plan to license or divest our other assets since they no longer align with our focus on the treatment of hypertension.
We intend to create value through the continued commercialization of our FDA-approved product, Prestalia, while moving our FDC development programs forward to further strengthen our commercial presence. We intend to retain ownership and control of all of our product candidates, but in the interest of accelerated growth and market penetration, we will also consider partnerships with pharmaceutical or biotechnology companies in order to reduce time to market and to balance development risks, both clinically and financially.
| F-7 |
As our strategy is to be a fully integrated biopharmaceutical company, we will drive a primary corporate focus on revenue generation through our commercial assets, with a secondary focus on advancing our FDC pipeline to further enhance our commercial presence.
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”). The summary of significant accounting policies presented below is designed to assist in understanding the Company’s financial statements. Such financial statements and accompanying notes are the representations of Company’s management, who is responsible for their integrity and objectivity.
Principles of Consolidation
The consolidated financial statements include the accounts of IThenaPharma Inc. and Marina Biotech, Inc. and the wholly-owned subsidiaries, Cequent, MDRNA, and Atossa, and eliminate any inter-company balances and transactions.
Reverse Stock Split
On August 1, 2017, we filed a Certificate of Amendment of our Amended and Restated Certificate of Incorporation to effect a one-for-ten reverse split of our issued and outstanding shares of common stock. Our common stock commenced trading on the OTCQB tier of the OTC Markets on a split-adjusted basis on August 3, 2017. Unless indicated otherwise, all share and per share information included in these financial statements give effect to the reverse split.
Going Concern and Management’s Liquidity Plans
The accompanying consolidated financial statements have been prepared on the basis that we will continue as a going concern, which contemplates realization of assets and the satisfaction of liabilities in the normal course of business. At December 31, 2017, we had a significant accumulated deficit of approximately $8 million and a negative working capital of approximately $6 million. Our operating activities consume the majority of our cash resources. We anticipate that we will continue to incur operating losses as we execute our commercialization and research and development plans, as well as strategic and business development initiatives. In addition, we have had and will continue to have negative cash flows from operations, at least into the near future. We have previously funded, and plan to continue funding, our losses primarily through the sale of common and preferred stock, combined with or without warrants, the sale of notes, revenue provided from our license agreements and, to a lesser extent, equipment financing facilities and secured loans. However, we cannot be certain that we will be able to obtain such funds required for our operations at terms acceptable to us or at all.
On April 16, 2018, we held the initial closing of our private placement of shares of our Series E Convertible Preferred Stock, and warrants to purchase shares of our common stock, as a result of which we raised over $10 million in net proceeds to our company. For our assessment as of December 31, 2017, we have considered the amount raised and we will continue to reassess our ability to address the going concern.
| F-8 |
The accompanying consolidated financial statements do not include any adjustments relating to the recoverability of and classification of assets carrying amounts or the amount and classification of liabilities that might result should the Company be unable to continue as a going concern.
Use of Estimates
The preparation of the accompanying consolidated financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reported period. Significant areas requiring the use of management estimates include valuation allowance for deferred income tax assets and fair value of financial instruments. Actual results could differ materially from such estimates under different assumptions or circumstances.
Cash and Cash Equivalents
The Company considers all highly liquid investments with maturities of three months or less at the time of purchase to be cash equivalents. There are no cash equivalents as of December 31, 2017 or 2016.
The Company deposits its cash with major financial institutions and may at times exceed the federally insured limit. At December 31, 2017 and 2016, the Company had no cash balances in excess of the federal insurance limit.
Goodwill
The Company periodically reviews the carrying value of intangible assets not subject to amortization, including goodwill, to determine whether impairment may exist. Goodwill and certain intangible assets are assessed annually, or when certain triggering events occur, for impairment using fair value measurement techniques. These events could include a significant change in the business climate, legal factors, a decline in operating performance, competition, sale or disposition of a significant portion of the business, or other factors. Specifically, goodwill impairment is determined using a two-step process. The first step of the goodwill impairment test is used to identify potential impairment by comparing the fair value of a reporting unit with its carrying amount, including goodwill. The Company uses level 3 inputs and a discounted cash flow methodology to estimate the fair value of a reporting unit. A discounted cash flow analysis requires one to make various judgmental assumptions including assumptions about future cash flows, growth rates, and discount rates. The assumptions about future cash flows and growth rates are based on the Company’s budget and long-term plans. Discount rate assumptions are based on an assessment of the risk inherent in the respective reporting units. If the fair value of a reporting unit exceeds its carrying amount, goodwill of the reporting unit is considered not impaired and the second step of the impairment test is unnecessary. If the carrying amount of a reporting unit exceeds its fair value, the second step of the goodwill impairment test is performed to measure the amount of impairment loss, if any. The second step of the goodwill impairment test compares the implied fair value of the reporting unit’s goodwill with the carrying amount of that goodwill. If the carrying amount of the reporting unit’s goodwill exceeds the implied fair value of that goodwill, an impairment loss is recognized in an amount equal to that excess. The implied fair value of goodwill is determined in the same manner as the amount of goodwill recognized in a business combination. That is, the fair value of the reporting unit is allocated to all of the assets and liabilities of that unit (including any unrecognized intangible assets) as if the reporting unit had been acquired in a business combination and the fair value of the reporting unit was the purchase price paid to acquire the reporting unit.
The Company did not record an impairment loss on goodwill for the years ended December 31, 2017 or 2016.
| F-9 |
Fair Value of Financial Instruments
We consider the fair value of cash, accounts payable, due to related parties, notes payable, notes payable to related parties, convertible notes payable and accrued liabilities not to be materially different from their carrying value. These financial instruments have short-term maturities. We follow authoritative guidance with respect to fair value reporting issued by the Financial Accounting Standards Board (“FASB”) for financial assets and liabilities, which defines fair value, provides guidance for measuring fair value and requires certain disclosures. The guidance does not apply to measurements related to share-based payments. The guidance discusses valuation techniques, such as the market approach (comparable market prices), the income approach (present value of future income or cash flow), and the cost approach (cost to replace the service capacity of an asset or replacement cost). The guidance establishes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three broad levels. The following is a brief description of those three levels:
| Level 1: | Observable inputs such as quoted prices (unadjusted) in active markets for identical assets or liabilities. |
| Level 2: | Inputs other than quoted prices that are observable for the asset or liability, either directly or indirectly. These include quoted prices for similar assets or liabilities in active markets and quoted prices for identical or similar assets or liabilities in markets that are not active. |
| Level 3: | Unobservable inputs in which little or no market data exists, therefore developed using estimates and assumptions developed by us, which reflect those that a market participant would use. |
Our cash is subject to fair value measurement and is determined by Level 1 inputs. We measure the liability for committed stock issuances with a fixed share number using Level 1 inputs. We measured the liability for price adjustable warrants and certain features embedded in notes, using the probability adjusted Black-Scholes option pricing model (“Black-Scholes”), which management has determined approximates values using more complex methods, using Level 3 inputs. There were no liabilities measured at fair value as of December 31, 2017 (see Recent Accounting Pronouncements below). The following tables summarize our liabilities measured at fair value on a recurring basis as of December 31, 2016:
Balance at December 31, 2016 | Level
1 Quoted prices in active markets for identical assets | Level
2 Significant other observable inputs | Level
3 Significant unobservable inputs | |||||||||||||
| Liabilities: | ||||||||||||||||
| Fair value liability for price adjustable warrants | $ | 141,723 | $ | - | $ | - | $ | 141,723 | ||||||||
| Total liabilities at fair value | $ | 141,723 | $ | - | $ | - | $ | 141,723 | ||||||||
The following presents activity of the fair value liability of price adjustable warrants determined by Level 3 inputs for the period ended December 31, 2017:
| Fair
value liability for price adjustable warrants |
||||
| Balance at December 31, 2016 | $ | 141,723 | ||
| Reclassification of derivative liability due to adoption of ASU 2017-11 (See Recent Accounting Pronouncements below) | (141,723 | ) | ||
| Balance at December 31, 2017 | $ | - | ||
| F-10 |
Impairment of Long-Lived Assets
We review all of our long-lived assets for impairment indicators throughout the year and perform detailed testing whenever impairment indicators are present. In addition, we perform detailed impairment testing for indefinite-lived intangible assets, at least annually, at December 31. When necessary, we record charges for impairments. Specifically:
| ● | For finite-lived intangible assets, such as developed technology rights, and for other long-lived assets, we compare the undiscounted amount of the projected cash flows associated with the asset, or asset group, to the carrying amount. If the carrying amount is found to be greater, we record an impairment loss for the excess of book value over fair value. In addition, in all cases of an impairment review, we re-evaluate the remaining useful lives of the assets and modify them, as appropriate; and |
| ● | For indefinite-lived intangible assets, such as acquired in-process R&D assets, each year and whenever impairment indicators are present, we determine the fair value of the asset and record an impairment loss for the excess of book value over fair value, if any. |
Management determined that no impairment indicators were present and that no impairment charges were necessary as of December 31, 2017 or 2016.
Revenue Recognition
Revenue will be recognized when persuasive evidence that an arrangement exists, delivery has occurred, collectability is reasonably assured, and fees are fixed or determinable. Deferred revenue expected to be recognized within the next 12 months will be classified as current. Substantially, a majority of our revenues is planned to be generated from the sales of product. We plan to launch Prestalia, our FDA approved product. We also expect some of our revenue will be generated from licensing arrangements that do not involve multiple deliverables and have no ongoing influence, control or R&D obligations. Our license arrangements may include upfront non-refundable payments, development milestone payments, patent-based or product sale royalties, and commercial sales, all of which are treated as separate units of accounting. In addition, we may receive revenues from sub-licensing arrangements. For each separate unit of accounting, we will determine that the delivered items have value to the other party on a stand-alone basis, we will have objective and reliable evidence of fair value using available internal evidence for the undelivered item(s) and our arrangements generally do not contain a general right of return relative to the delivered item.
Product Sales
Revenue from our product sales will be recognized when title and risk of loss have transferred to our customer, and the following additional criteria are met:
| (1) | appropriate evidence of a binding arrangement exists with our customer; |
| (2) | price is substantially fixed or determinable; |
| (3) | collection from our customer is reasonably assured; |
| (4) | our customer’s obligation to pay us is not contingent on resale of the product; |
| (5) | we do not have significant continued performance obligations to our customer; and |
| (6) | we have a reasonable basis to estimate returns. |
Our gross revenue will be reduced by our gross-to-net, or GTN, estimates each period, resulting in our reported “product sales, net.” We will defer revenue recognition in full if these estimates are not reasonably determinable at the time of sale. These estimates will be based upon information received from external sources (such as written or oral information obtained from our customers with respect to their period-end inventory levels and their sales to end-users during the period), in combination with management’s informed judgments. Due to the inherent uncertainty of estimates, the actual amount we incur may be materially different than our GTN estimates, and require prospective revenue adjustments in periods after the initial sale was recorded.
Our GTN estimates may be comprised of the following categories:
| ● | Product Returns Allowances: Our customers may be permitted to return purchased product beginning at its expiration date, and within six months thereafter. Returned product is generally destroyed and not resold. Returns outside of the above referenced criteria for expiry. | |
| ● | Prompt Pay Discounts: Discounts for prompt payment will be estimated at the time of sale, based on our future eligible customers’ prompt payment history and the contractual discount percentage. | |
| ● | Medicaid Rebates: Our products may be subject to state government-managed Medicaid programs, whereby rebates are issued to participating state governments. These rebates arise when a patient treated with our product is covered under Medicaid, resulting in a discounted price for our product under the applicable Medicaid program. Our Medicaid rebate accrual calculations will require us to project the magnitude of our sales, by state, that will be subject to these rebates. There could be a significant time lag in us receiving rebate notices from each state (generally several months or longer after our sale is recognized). Our estimates will be based on our historical claim levels by state, as supplemented by management’s judgment. |
Revenue from licensing arrangements will be recorded when earned based on the specific terms of the contracts. Upfront non-refundable payments, where we are not providing any continuing services as in the case of a license to our IP, are recognized when the license becomes available to the other party.
Milestone payments typically represent nonrefundable payments to be received in conjunction with the uncertain achievement of a specific event identified in the contract, such as initiation or completion of specified development activities or specific regulatory actions such as the filing of an Investigational New Drug Application (“IND”). We believe a milestone payment represent the culmination of a distinct earnings process when it is not associated with ongoing research, development or other performance on our part and it is substantive in nature. We recognize such milestone payments as revenue when it becomes due and collection is reasonably assured.
Royalty and earn-out payment revenues will generally be recognized upon commercial product sales by the licensee as reported by the licensee.
| F-11 |
Stock-based Compensation
We use Black-Scholes for the valuation of stock-based awards. Stock-based compensation expense is based on the value of the portion of the stock-based award that will vest during the period, adjusted for expected forfeitures. The estimation of stock-based awards that will ultimately vest requires judgment, and to the extent actual or updated results differ from our current estimates, such amounts will be recorded in the period the estimates are revised. Black-Scholes requires the input of highly subjective assumptions, and other reasonable assumptions could provide differing results. Our determination of the fair value of stock-based awards on the date of grant using an option pricing model is affected by our stock price as well as assumptions regarding a number of highly complex and subjective variables. These variables include, but are not limited to, the expected life of the award and expected stock price volatility over the term of the award. Stock-based compensation expense is recognized immediately for immediately-vested portions of the grant, with the remaining portions recognized on a straight-line basis over the applicable vesting periods based on the fair value of such stock-based awards on the grant date. Forfeiture rates have been estimated based on historical rates and compensation expense is adjusted for general forfeiture rates in each period. Beginning in September 2014, Marina did not use historical forfeiture rates and did not apply a forfeiture rate as the historical forfeiture rate was not believed to be a reasonable estimate of the probability that the outstanding awards would be exercised in the future. Given the specific terms of the awards and the recipient population, we expect these options will all be exercised in the future.
Non-employee stock compensation expense is recognized immediately for immediately-vested portions of a grant, with the remaining portions recognized on a straight-line basis over the applicable vesting periods. At the end of each financial reporting period prior to vesting, the value of the unvested stock options, as calculated using Black-Scholes, is re-measured using the fair value of our common stock, and the stock-based compensation recognized during the period is adjusted accordingly.
Net Income (Loss) per Common Share
Basic net income (loss) per common share (after giving effect of the one for ten reverse stock split) is computed by dividing the net income (loss) by the weighted average number of common shares outstanding during the period, excluding any unvested restricted stock awards. Diluted net income (loss) per share includes the effect of common stock equivalents (stock options, unvested restricted stock, and warrants) when, under either the treasury or if-converted method, such inclusion in the computation would be dilutive. Net income (loss) is adjusted for the dilutive effect of the change in fair value liability for price adjustable warrants, if applicable. The following number of shares have been excluded from diluted net income (loss) since such inclusion would be anti-dilutive:
| Year ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Stock options outstanding | 745,707 | 168,811 | ||||||
| Warrants | 2,559,612 | 2,446,678 | ||||||
| Shares to be issued upon conversion of notes payable | 319,617 | 89,286 | ||||||
| Total | 3,624,936 | 2,704,775 | ||||||
Income Taxes
Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets, including tax loss and credit carry forwards, and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date.
The Company accounts for income taxes using the asset and liability method to compute the differences between the tax basis of assets and liabilities and the related financial amounts, using currently enacted tax rates.
Reclassification of Prior Year Presentation
Certain prior year amounts have been reclassified for consistency with the current year presentation. These reclassifications had no effect on the reported results of operations or cash flows.
| F-12 |
Under the provisions of ASC 740, Income Taxes (“ASC 740”), the Company recognizes the financial statement benefit of a tax position only after determining that the relevant tax authority would more likely than not sustain the position following an audit. For tax positions meeting the more- likely-than-not threshold, the amount recognized in the financial statements is the largest benefit that has a greater than 50 percent likelihood of being realized upon ultimate settlement with the relevant tax authority. The Company did not have any unrecognized tax benefits and there was no effect on the Company’s financial condition or results of operations as a result of adopting the provisions of ASC 740.
The Company recognizes a tax benefit only if it is more likely than not that the position is sustainable, based solely on its technical merits and considerations of the relevant taxing authorities; administrative practice and precedents. The Company completed its analysis of uncertain tax positions in accordance with applicable accounting guidance and determined no amounts were required to be recognized in the financial statements at December 31, 2017 and 2016.
Recent Accounting Pronouncements
In July 2017, the Financial Accounting Standards Board issued Accounting Standads Update (“ASU”) 2017-11, Earnings Per Share (Topic 260), Distinguishing Liabilities from Equity (Topic 480), Derivatives and Hedging (Topic 815) – I. Accounting for Certain Financial Instruments with Down Round Features and II. Replacement of the Indefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Noncontrolling Interests with a Scope Exception (“ASU 2017-11”), which addresses the complexity of accounting for certain financial instruments with down round features and addresses the difficulty of navigating Topic 480 because of the existence of extensive pending content in the ASC as a result of the indefinite deferral of accounting requirements about mandatorily redeemable financial instruments of certain nonpublic entities and certain mandatorily redeemable noncontrolling interests. This update applies to all entities that issue financial instruments that include down round features and entities that present earnings per share in accordance with Topic 260. This guidance is effective for financial statements issued for fiscal years beginning after December 15, 2018, and interim periods within those fiscal years. Early adoption is permitted, including adoption in an interim period. If early adopted in an interim period, any adjustments should be reflected as of the beginning of the fiscal year that includes that interim period. In November 2017, the Company adopted the provisions relating to the issuance of financial statements that include down round provisions utilizing the modified retrospective approach. See Fair Value of Accounting Instruments above for the impact of the adoption on the consolidated financial statements.
In May 2017, the FASB issued ASU 2017-09, Compensation – Stock Compensation (Topic 718) – Scope of Modification Accounting (“ASU 2017-09”), which provides guidance about which changes to the terms or conditions of a stock-based payment award require an entity to apply modification accounting in Topic 718. This update applies to any entity that changes the terms or conditions of a stock-based payment award. This guidance is effective for financial statements issued for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years. The Company does not currently expect ASU 2017-09 to have a material effect on its financial statements and disclosures upon adoption.
In January 2017, the FASB issued ASU No. 2017-04, “Intangibles—Goodwill and Other (Topic 350): Simplifying the Test for Goodwill Impairment” (ASU 2017-04)”. Under the amendments, an entity should perform its annual or interim goodwill impairment test by comparing the fair value of a reporting unit with its carrying amount. An entity should recognize an impairment charge for the amount by which the carrying amount exceeds the reporting unit’s fair value, but the loss recognized should not exceed the total amount of goodwill allocated to that reporting unit. The FASB also eliminated the requirements for any reporting unit with a zero or negative carrying amount to perform a qualitative assessment, and if it fails that qualitative test, to perform Step 2 of the goodwill impairment test. The same impairment test will therefore apply to all reporting units, and an entity will be required to disclose the amount of goodwill allocated to each reporting unit with a zero or negative carrying amount of net assets. SEC filers are required to adopt the new standard for annual or any interim goodwill impairment tests in fiscal years beginning after December 15, 2019. Early adoption is permitted for interim or annual goodwill impairment tests performed on testing dates on or after January 1, 2017. The Company does not currently expect ASU 2017-04 to have a material effect on its financial statements and disclosures upon adoption.
| F-13 |
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows – Classification of Certain Cash Receipts and Cash Payments (Topic 230) (“ASU 2016-15”), which addresses eight specific cash flow issues with the objective of reducing the existing diversity in practice. This update applies to all entities that are required to present a statement of cash flows under Topic 230. This guidance is effective for financial statements issued for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years. The Company does not currently expect ASU 2016-15 to have a material effect on its financial statements and disclosures upon adoption.
In March 2016, FASB issued (“ASU”) 2016-09, Compensation – Stock Compensation – Improvements to Employee Shared-Based Payment Accounting (“ASU 2016-09”). This guidance is intended to simplify the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities and classification on the statement of cash flows. The amendments in this update are effective for public business entities for financial statements issued for annual periods beginning after December 15, 2016, including interim periods within those annual periods. For all other entities, the amendments are effective for annual periods beginning after December 15, 2017, and interim periods within annual periods beginning after December 15, 2018. Early adoption is permitted for any entity in any interim or annual periods. The Company is currently assessing the impact of this ASU on its financial statements.
In February 2016, the FASB issued Accounting Standards Update, Leases (“ASU 2016-02”),. This update requires lessees to recognize at the lease commencement date a lease liability which is the lessee’s obligation to make lease payments arising from a lease, measured on a discounted basis, and right-of-use assets, which is an asset that represents the lessee’s right to use, or control the use of, a specified asset for the lease term. Lessees will no longer be provided with a source of off-balance sheet financing. This update is effective for financial statements issued for annual periods beginning after December 15, 2019, including interim periods within those fiscal years. Early adoption is permitted. Lessees and lessors must apply a modified retrospective transition approach for leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements. The modified retrospective approach would not require any transition accounting for leases that expired before the earliest comparative period presented. Applying a full retrospective transition approach is not allowed. The Company does not expect the adoption of this standard to have a material effect on its financial statements.
In January 2016, FASB ASU 2016-01, Recognition and Measurement of Financial Assets and Financial Liabilities (“ASU 2016-01”). The Update intends to enhance the reporting model for financial instruments to provide users of financial instruments with more decision-useful information and addresses certain aspects of the recognition, measurement, presentation, and disclosure of financial instruments. This new standard affects all entities that hold financial assets or owe financial liabilities. Entities should apply the amendments as a cumulative-effect adjustment to the balance sheet as of the beginning of the fiscal year of adoption. The amendments related to equity securities without readily determinable fair values, including disclosure requirement, should be applied prospectively to equity investments that exist as of the date of adoption of the update. ASU 2016-01 is effective for public business entities for fiscal years beginning after December 15, 2017, including interim periods within those fiscal years. For all other entities, including not-for-profit entities and employee benefit plans within the scope of ASC Topics 960 through 965 on plan accounting, ASC 2016-01 is effective for fiscal years beginning after December 15, 2018, and interim periods within fiscal years beginning after December 15, 2019. All entities that are not public business entities may adopt the ASC 2016-01 earlier as of the fiscal years beginning after December 15, 2017, including interim periods within those fiscal years. The adoption of this guidance is not expected to have a material impact on the Company’s financial position, results of operations or cash flows.
In May 2014, FASB issued changes to the recognition of revenue from contracts with customers. These changes created a comprehensive framework for all entities in all industries to apply in the determination of when to recognize revenue, and, therefore, supersede virtually all existing revenue recognition requirements and guidance. This framework is expected to result in less complex guidance in application while providing a consistent and comparable methodology for revenue recognition. The core principle of the guidance is that an entity should recognize revenue to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. To achieve this principle, an entity should apply the following steps: (i) identify the contract(s) with a customer, (ii) identify the performance obligations in the contract(s), (iii) determine the transaction price, (iv) allocate the transaction price to the performance obligations in the contract(s), and (v) recognize revenue when, or as, the entity satisfies a performance obligation. In August 2015, the FASB deferred the effective date by one year, making these changes effective for the Company on January 1, 2018. The Company does not expect the adoption of this standard to have a material effect on its financial statements.
| F-14 |
Subsequent Event Policy
Management has evaluated all activity since December 31, 2017, through the date the financial statements were issued and has concluded that no subsequent events have occurred that would require recognition in the Financial Statements or disclosure in the Notes to the Financial Statements, other than those described in Note 11.
Note 3 – Intangible Assets
Reverse Merger with IThenaPharma
On November 15, 2016, we entered into, and consummated the transactions contemplated by, an Agreement and Plan of Merger between and among Marina Biotech, Inc., IThenaPharma Inc., a Delaware corporation (“IThena”), IThena Acquisition Corporation, a Delaware corporation and wholly-owned subsidiary of Marina (“Merger Sub”), and Vuong Trieu as the IThena representative (the “Merger Agreement”), pursuant to which IThena merged into Merger Sub (the “Merger”). Upon completion of the Merger and subject to the applicable provisions of the Merger Agreement, Merger Sub has ceased to exist and IThena continued as the surviving corporation of the Merger and as a wholly-owned subsidiary of Marina. As consideration for the Merger, Marina issued to the former shareholders of IThena 5,839,283 shares of the Company’s common stock, representing approximately 65% of the issued and outstanding shares of Marina’s common stock following the completion of the Merger. Outstanding warrants to purchase 30,000 shares of common stock of IThena were converted into warrants to purchase common stock of Marina. In addition, Marina appointed Vuong Trieu, the president of IThena, as the Chairman of the Board of Directors of Marina, effective November 15, 2016. Dr. Trieu, in his capacity as the IThena representative, later appointed Philippe P. Calais, Ph.D., as a member of the Board of Directors of Marina effective December 8, 2016, pursuant to the rights granted to the former shareholders of IThena in the Merger Agreement.
As the former shareholders of IThena control greater than 50% of the Company subsequent to the Merger, for accounting purposes, the Merger was treated as a “reverse acquisition” and IThena is considered the accounting acquirer. IThena accounted for the acquisition of Marina under the purchase accounting method following completion. Accordingly, IThena’s historical results of operations replace Marina’s historical results of operations for all periods prior to the Merger, and for all periods following the Merger, the results of operations of both companies are included. As a result of the Merger, while we have presented the results for the year ended December 31, 2017; the results for the year ended 2016 reflect only the results of IThena.
The purchase price of approximately $3.7 million represents the consideration in the reverse merger transaction and is calculated based on the number of shares of common stock of the combined company that Marina stockholders owned as of the closing of the transaction and the fair value of assets and liabilities assumed by IThena.
The number of shares of common stock Marina issued to IThena stockholders is calculated pursuant to the terms of the Merger Agreement based on Marina common stock outstanding as of November 15, 2016, as follows:
| Shares of Marina common stock outstanding as of November 15, 2016 | 3,137,855 | |||
| Divided by the percentage of Marina ownership of combined company | 35 | % | ||
| Adjusted total shares of common stock of combined company | 8,977,138 | |||
| Multiplied by the assumed percentage of IThena ownership of combined company | 65 | % | ||
| Shares of Marina common stock issued to IThena upon closing of transaction | 5,839,283 |
The application of the acquisition method of accounting is dependent upon certain valuations and other studies that have yet to be completed. The purchase price allocation will remain preliminary until IThena management determines the fair values of assets acquired and liabilities assumed. The final determination of the purchase price allocation is anticipated to be completed as soon as practicable after completion of the transaction and will be based on the fair values of the assets acquired and liabilities assumed as of the transaction closing date. The final amounts allocated to assets acquired and liabilities assumed could differ significantly from the amounts presented.
| F-15 |
The purchase price as of December 31, 2017 has been allocated based on a preliminary estimate of the fair value of assets acquired and liabilities assumed:
| Assets and Liabilities Acquired: | ||||
| Cash | $ | 5,867 | ||
| Net current liabilities assumed (excluding cash) | (1,871,725 | ) | ||
| Identifiable intangible assets | 2,361,066 | |||
| Debt | (326,037 | ) | ||
| Net assets acquired | 169,171 | |||
| Goodwill | 3,502,829 | |||
| Purchase price | $ | 3,672,000 |
The above estimated purchase price allocation and goodwill valuation reflects changes in fair value determinations of $55,247 for the year ended December 31, 2017 and approximately $1,238,000 since the Merger date. As part of the Merger, the Company allocated $3,502,829 to goodwill. Additionally, a substantial portion of the assets acquired were allocated to identifiable intangible assets. The fair value of the identifiable intangible asset is determined primarily using the “income approach,” which requires a forecast of all the expected future cash flows.
The Company used the discounted cash flow method for the impairment testing as of December 31, 2017. The Company performed discounted cash flow analysis projected over 5 years to estimate the fair value of the reporting unit, using management’s best judgement as to revenue growth rates and expense projections. This analysis indicated cash flows (and discounted cash flows) greater than the $3.5 million book value of goodwill. The Company determined these were no indicators of impairment in goodwill for the Company as of December 31, 2017.
On November 15, 2016, Marina agreed to issue to Novosom Verwaltungs GmbH (“Novosom”) 150,000 shares of common stock upon the closing of the Merger in consideration of Novosom’s agreement that the consummation of the Merger would not constitute a “Liquidity Event” under that certain Asset Purchase Agreement dated as of July 27, 2010 between and among Marina, Novosom and Steffen Panzner, Ph.D., and thus that no additional consideration under such agreement would be due to Novosom as a result of the consummation of the Merger.
In July 2016, Marina pledged to issue common stock valued at approximately $15,000 to Novosom for the portion due under our July 2010 Asset Purchase Agreement with Novosom, related to Marina’s license agreement with an undisclosed licensee that grants such licensee rights to use Marina’s technology and intellectual property to develop and commercialize products combining certain molecules with Marina’s liposomal delivery technology known as NOV582. In November 2016, we issued 11,905 shares with a value of approximately $15,000 to Novosom as the equity component owed under our July 2016 license agreement.
Acquisition of Assets from Symplmed
In June 2017, we entered into an Asset Purchase Agreement (the “Purchase Agreement”) with Symplmed Pharmaceuticals LLC (“Symplmed”) pursuant to which we purchased from Symplmed, for aggregate consideration of approximately $620,000(consisting of $300,000 in cash plus the assumption of certain liabilities of Symplmed in the amount of approximately $320,000), Symplmed’s assets relating to a single-pill FDC of perindopril arginine and amlodipine besylate known as Prestalia® (“Prestalia”), that has been approved by the FDA for the treatment of hypertension. In addition, as part of the transactions contemplated by the Purchase Agreement: (i) Symplmed agreed to transfer to us, not later than 150 days following the closing date, the New Drug Applications for the approval of Prestalia as a new drug by the FDA; and (ii) Symplmed assigned to us all of its rights and obligations under that certain Amended and Restated License and Commercialization Agreement by and between Symplmed and Les Laboratoires Servier (“Servier”) dated January 11, 2012, pursuant to which Symplmed has an exclusive license from Servier to manufacture, have manufactured, develop, promote, market, distribute and sell Prestalia in the U.S. (and its territories and possessions) in consideration of regulatory and sales-based milestone payments and royalty payments based on net sales. Management has determined that this acquisition was deemed an asset purchase under FASB ASC 805.
The purchase price of $620,000 has been allocated based on a preliminary estimate of the fair value of the assets acquired and is included in intangible assets as of December 31, 2017 and is subject to change.
| F-16 |
Further, we entered into an offer letter to hire our current Chief Commercial Officer, who was the President and Chief Executive Officer of Symplmed, which appointment became effective on June 22, 2017. We also agreed in such offer letter to issue 60,000 restricted shares of our common stock under our 2014 Long-Term Incentive Plan to our Chief Commercial Officer, with all of such shares vesting on the six (6) month anniversary of the date of grant. These shares were fully vested on December 31, 2017.
In furtherance of the acquisition and commercialization of Prestalia, on July 21, 2017 we acquired from Symplmed and its wholly-owned subsidiary, Symplmed Technologies, LLC, certain of the intellectual property assets related to the patented technology platform known as DyrctAxess, also known as Total Care, that offers enhanced efficiency, control and information to empower patients, physicians and manufacturers to help achieve optimal care for $75,000 in cash.
The following table summarizes the estimated fair value of the identifiable intangible assets acquired, their useful life, and method of amortization:
| Estimated
Fair Value | Estimated
Useful Life (Years) | Annual
Amortization Expense | ||||||||||
| Intangible asset from Merger | $ | 2,361,066 | 6.0 | $ | 393,511 | |||||||
| Intangible asset - Prestalia | 620,000 | 6.6 | 94,177 | |||||||||
| Intangible asset – DyrctAxess | 75,000 | 14.0 | 5,357 | |||||||||
| Total | $ | 3,056,066 | $ | 493,045 | ||||||||
The net intangible asset was $2,555,974, net of accumulated amortization of $500,092, as of December 31, 2017. Amortization expense was $450,903 and $49,189 for the year ended December 31, 2017 and 2016, respectively.
Note 4 - Related Party Transactions
Due to Related Party
The Company and other related entities have a commonality of ownership and/or management control, and as a result, the reported operating results and /or financial position of the Company could significantly differ from what would have been obtained if such entities were autonomous.
The Company has a Master Services Agreement (“MSA”) with a related party that is partly-owned by the Company’s Executive Chairman, Autotelic Inc., effective November 15, 2016. Autotelic Inc. currently owns less than 5% of the Company. The MSA states that Autotelic Inc. will provide business functions and services to the Company and allows Autotelic Inc. to charge the Company for these expenses paid on its behalf. The MSA includes personnel costs allocated based on amount of time incurred and other services such as consultant fees, clinical studies, conferences and other operating expenses incurred on behalf of the Company.
During the period commencing November 15, 2016 (the “Effective Date”) and ending on the date that the Company has completed an equity offering of either common or preferred stock in which the gross proceeds therefrom is no less than $10 million (the “Equity Financing Date”), the Company shall pay Autotelic the following compensation: cash in an amount equal to the actual labor cost (paid on a monthly basis), plus 100% markup in warrants for shares of the Company’s common stock with a strike price equal to the fair market value of the Company’s common stock at the time said warrants are issued. The Company shall also pay Autotelic for the services provided by third party contractors plus 20% mark up. The warrant price per share will be calculated based on the Black-Scholes model.
After the Equity Financing Date, the Company shall pay Autotelic Inc. a cash amount equal to the actual labor cost plus 100% mark up of provided services and 20% mark up of provided services by third party contractors or material used in connection with the performance of the contracts, including but not limited to clinical trial, non-clinical trial, Contract Manufacturing Organizations (“CMO”), FDA regulatory process, Contract Research Organizations (“CRO”) and Chemistry and Manufacturing Controls (“CMC”).
| F-17 |
In accordance with the MSA, Autotelic Inc. billed the Company for personnel and service expenses Autotelic Inc. incurred on behalf of the Company. For the year ended December 31, 2017 and 2016, Autotelic Inc. billed a total of $791,889 and $344,563, including personnel costs of $558,098 and $166,550, respectively. An unpaid balance of $730,629 is included in due to related party in the accompanying balance sheet as of December 31, 2017. The Company agreed to issue warrants at a future date for the remaining balance due of $605,889, which is included in due to related parties as of December 31, 2017.
In connection with a private placement of our Series E Convertible Preferred Stock (Preferred Shares), Autotelic agreed to convert the entire unpaid balance due to it as of March 31, 2018 into shares of Series E Convertible Preferred Stock and warrants in full and complete satisfaction of the unpaid balance. Also see Note 11 – Subsequent Events below.
Note 5 – Notes Payable
Following is a breakdown of notes payable as of December 31, 2017
| 2017 | 2016 | |||||||
| Notes payable | $ | 97,523 | $ | 121,523 | ||||
| Convertible notes payable | 346,700 | 314,475 | ||||||
| Total notes payable | $ | 444,223 | $ | 435,998 | ||||
| Notes payable – related parties | 93,662 | $ | - | |||||
| Convertible notes payable – related parties (net of debt discount of $113,170) | 1,368,378 | 250,000 | ||||||
| Total notes payable – related parties | $ | 1,462,040 | $ | 250,000 | ||||
Note Payable – Service Provider
In December 2016, we entered into an Agreement and Promissory Note with a law firm for past services performed totaling $121,523. The note calls for monthly payments of $6,000 per month, beginning with an initial payment on March 31, 2017. The note is unsecured and non-interest bearing. The note will be considered paid in full if the Company pays $100,000 by December 31, 2017, which was not paid. The balance due on the note was $121,523 and $97,523 as of December 31, 2017 and 2016, respectively.
Note Purchase Agreement and Amendment
In June 2016, Marina entered into a Note Purchase Agreement (the “Purchase Agreement”) with certain investors (the “Purchasers”), pursuant to which Marina issued to the Purchasers unsecured promissory notes in the aggregate principal amount of $300,000 (the “Notes”). Interest accrued on the unpaid principal balance of the Notes at the rate of 12% per annum beginning on September 20, 2016. The Notes were due and payable on June 20, 2017.
In July 2017, we entered into an amendment agreement (the “Amendment Agreement”) with respect to those Notes and the warrants to purchase shares of our common stock that are currently held by the Purchasers and that were originally issued pursuant to a certain Note and Warrant Purchase Agreement dated as of February 10, 2012 by and among Marina, MDRNA Research, Inc., Cequent Pharmaceuticals, Inc. and the purchasers identified on the signature pages thereto (as amended from time to time), to, among other things, extend the maturity date of the Notes to December 31, 2017 and to extend the price protection applicable to certain of the warrants held by the Purchasers with respect to dilutive offerings afforded thereunder to February 10, 2020. Refer to our Form 10-Q for the six months ended June 30, 2017 for a more detailed discussion and additional terms for these Notes.
The unpaid principal balance of the Notes, together with accrued and unpaid interest thereon, converted into shares of Series E Convertible Preferred Stock and warrants to purchase shares of common stock upon the Initial Closing of the Private Placement. As a result of the conversion of the Notes, all obligations of the Company to the Purchasers thereunder have been satisfied and the Notes are no longer outstanding. Also see Note 11 – Subsequent Events below.
As of December 31, 2017 and 2016, the accrued interest expense on the Notes amounted to $46,700 and $14,475, respectively, with a total balance of principal and interest of $346,700 and $314,475, respectively.
Bridge Note Financing
In June 2017, we issued convertible promissory notes (the “Notes”) in the aggregate principal amount of $400,000 to 10 investors pursuant to a Note Purchase Agreement (the “Note Purchase Agreement”) that we entered into with such investors. The Notes bear interest at a rate of five percent (5%) per annum and are due and payable at any time on or after the earlier of (i) June 1, 2018 and (ii) the occurrence of an event of default (as defined in the Note Purchase Agreement). Our Executive Chairman and our Chief Science Officer were each investors in the Notes.
| F-18 |
Upon written notice delivered to us by the holders of a majority in interest of the aggregate principal amount of Notes that are outstanding at the time of such calculation (the “Majority Holders”) not more than five (5) days following the maturity date of the Notes, the Majority Holders shall have the right, but not the obligation, on behalf of themselves and all other holders of Notes, upon written notice delivered to us, to elect to convert the entire unpaid principal amount of all, but not less than all, of the Notes and the accrued and unpaid interest thereon into such number of shares of our common stock as is equal to, with respect to each Note: (x) the entire unpaid principal amount of such Note and the accrued and unpaid interest thereon on the date of the delivery of such notice by (y) $3.50.
The unpaid principal balance of the Notes, together with accrued and unpaid interest thereon, converted into shares of Series E Convertible Preferred Stock and warrants to purchase shares of common stock upon the Initial Closing of the Private Placement. As a result of the conversion of the Notes, all obligations of the Company to the holders of the Notes thereunder have been satisfied and the Notes are no longer outstanding. Also see Note 11 – Subsequent Events below.
As of December 31, 2017, the accrued interest expense on the Notes amounted to $11,365, with a total balance of principal and interest of $411,365 and is included in convertible notes payable – related parties on the accompanying balance sheet.
Convertible Notes Payable
In July 2016, IThena issued convertible promissory notes with an aggregate principal balance of $50,000 to certain related-party investors. Borrowings under each of these convertible notes bore interest at 3% per annum and these notes mature on June 30, 2018. Upon the completion of certain funding events, IThena had the right to convert the outstanding principal amount of these notes into shares of the IThena’s common stock. The notes were assumed by Autotelic Inc. on November 15, 2016 as part of its acquisition of the technology asset (IT-101).
On November 22, 2017, the Company issued a convertible promissory note with a related party for $500,000, with annual interest at 8%, maturing on March 31, 2018, and convertible at the price equal to any financing transaction involving the sale by the Company of its equity securities yielding aggregate gross proceeds to the Company of not less than $5 million. Total principal and interest was $504,274 as of December 31, 2017 and is included in convertible notes to related parties on the accompanying balance sheet. The note included warrants to purchase 66,667 shares of the Company’s common stock, with a 5-year term and an exercise price of $0.75. The note included a debt discount of $162,210 consisting of loan costs of $50,000 and the fair value of the warrants of $112,210. Total amortization of this debt discount was $49,041 for the year ended December 31, 2017, with a remaining unamortized value of $113,169. The net balance of the note of $391,105 is included in convertible notes to related parties on the accompanying balance sheet at December 31, 2017.
The unpaid principal balance of the related party note, together with accrued and unpaid interest thereon, automatically converted into shares of Series E Convertible Preferred Stock and Warrants to purchase shares of common stock upon the Initial Closing of the Private Placement. As a result of the conversion of the related party note, all obligations of the Company to the holder under such note have been satisfied and the note is no longer outstanding. Also see Note 11 – Subsequent Events below.
Convertible Notes Payable, Dr. Trieu
In connection with the Merger, Marina entered into a Line Letter dated November 15, 2016 with Dr. Trieu, our Executive Chairman, for an unsecured line of credit in an amount not to exceed $540,000 , to be used for current operating expenses. Dr. Trieu has advanced the full $540,000 under the Line Letter as of December 31, 2017 ($250,000 as of December 31, 2016). Accrued interest on the Line Letter was $25,836 and $0 as of December 31, 2017 and December 31, 2016, respectively, and is included in convertible notes payable to related parties on the accompanying balance sheets. The line of credit is currently convertible at any time into shares of the Company’s common stock at a price of $1.77 per share.
The unpaid principal balance of the line of credit, together with accrued and unpaid interest thereon, converted into shares of Series E Convertible Preferred Stock and Warrants to purchase shares of common stock upon the Initial Closing of the Private Placement. As a result of the conversion of the line of credit, all obligations of the Company to Dr. Trieu thereunder have been satisfied and the line of credit is no longer outstanding. Also see Note 11 – Subsequent Events below.
Line Letter with Autotelic Inc.
On April 4, 2017, the Company entered into a Line Letter with Autotelic Inc for an unsecured line of credit in an amount not to exceed $500,000, to be used for current operating expenses. Autotelic Inc. is. a stockholder of IThenaPharma that became the holder of 525,535 shares of Marina common stock as a result of the Merger, and an entity of which Dr. Trieu serves as Chairman of the Board. Autotelic Inc. was to consider requests for advances under the Line Letter until September 1, 2017. Autotelic Inc. shall have the right at any time for any reason in its sole and absolute discretion to terminate the line of credit available under the Line Letter or to reduce the maximum amount available thereunder without notice . Advances made under the Line Letter bear interest at the rate of five percent (5%) per annum, are evidenced by the Demand Promissory Note issued to Autotelic Inc., and are due and payable upon demand by Autotelic, Inc. Autotelic Inc. advanced funds after September 1, 2017 but is no longer considering additional requests for advances as of December 31, 2017.
| F-19 |
The balance under the line was $93,662, including accrued interest of $2,847 as of December 31, 2017 and is included in notes to related parties on the accompanying balance sheet. Since this line was not extended, no further funds are available under this line of credit.
The unpaid principal balance of the line of credit, together with accrued and unpaid interest thereon, converted into shares of Series E Convertible Preferred Stock and warrants to purchase shares of common stock upon the Initial Closing of the Private Placement. As a result of the conversion of the line of credit, all obligations of the Company to Autotelic Inc. thereunder have been satisfied and the line of credit is no longer outstanding. Also see Note 11 – Subsequent Events below.
Note 6 – Stockholders’ Equity
Preferred Stock
Marina designated 1,000 shares as Series B Preferred Stock (“Series B Preferred”) and 90,000 shares as Series A Junior Participating Preferred Stock (“Series A Preferred”). No shares of Series B Preferred or Series A Preferred are outstanding. In March 2014, Marina designated 1,200 shares as Series C Convertible Preferred Stock (“Series C Preferred”). In August 2015, Marina designated 220 shares as Series D Convertible Preferred Stock (“Series D Preferred”). In April 2018, Marina designated 3,500 shares of Series E Convertible Preferred Stock,
Series C Preferred
Each share of Series C Preferred has a stated value of $5,000 per share, has a $5,100 liquidation preference per share, has voting rights of 666.67 votes per share, and is convertible into shares of common stock at a conversion price of $7.50 per share. In June 2015, an investor converted 90 shares of Series C Preferred into 60,000 shares of common stock with a value of $5.40 per share. In November 2015, an investor converted an additional 90 shares of Series C Preferred into 60,000 shares of common stock with a value of $3.10 per share. On September 15, 2017, an investor converted 270 shares of Series C Preferred stock into 180,000 shares of our common stock.
Series D Preferred
In August 2015, Marina entered into a Securities Purchase Agreement with certain investors pursuant to which Marina sold 220 shares of Series D Preferred and warrants to purchase up to 344,000 shares of Marina’s common stock at an initial exercise price of $4.00 per share before August 2021, for an aggregate purchase price of $1.1 million. Each share of Series D Preferred has a stated value of $5,000 per share, has a liquidation preference of $300 per share, has voting rights of 1,250 votes per share and is convertible into shares of common stock at a conversion price of $4.00 per share. The Series D Preferred is initially convertible into an aggregate of 275,000 shares of Marina’s common stock, subject to certain limitations and adjustments, has a 5% stated dividend rate, is not redeemable and has voting rights on an as-converted basis. In November 2015, an investor converted 50 shares of Series D Preferred into 62,500 shares of common stock. In February 2016, an investor converted 110 shares of Series D Preferred into 137,500 shares of common stock.
Series E Convertible Preferred Shares
In connection with a private placement offering, we recently created a new Series E Convertible Preferred Stock (Preferred Shares). Also see Note 11 – Subsequent Events below.
Each share of Series E Convertible Preferred Stock has a stated value of $5,000 per share and is convertible into shares of common stock at a conversion price of $0.50 per share. In connection with the private placement offering we issued 2,334 Preferred Shares.
Common Stock
Our common stock currently trades on the OTCQB tier of the OTC Markets.
Stock Issuances
In February 2017, we entered into two privately negotiated transactions pursuant to which we issued an aggregate of 615,368 shares of our common stock for an effective price per share of $1.55 to settle aggregate liabilities of approximately $956,000, which had been reflected in accrued expenses as of December 31, 2016.
In February 2017, we issued 30,000 shares of our common stock with a fair value of $54,000 ($1.80 per share) to a consultant providing investment advisory services.
In February 2017, we issued 10,000 restricted shares of our common stock with a fair value of $1.80 per share to our CEO for services and were fully vested by December 31, 2017.
In February 2017, we entered into a Stock Purchase Agreement with LipoMedics, a related party, pursuant to which we issued to LipoMedics an aggregate of 86,207 shares of our common stock for a total purchase price of $250,000.
| F-20 |
In March 2017, we entered into a Settlement Agreement, whereby a note receivable for $45,000 was settled with a cash payment by the note holder to the Company of $14,049, the surrender of 6,000 warrants, and the surrender of 8,725 shares of common stock held by the noteholder with a fair value of $31,404, which were cancelled effective March 31, 2017.
In April 2017, the Company entered into a Compromise and Release Agreement to settle $36,047 due to a service provider for $15,957 in cash and $20,090 of the Company’s common stock at $2.90 per share (for a total issuance of 6,928 shares). The Company issued 6,928 shares to the service provider in May 2017.
In May 2017, the holders of warrants purchased 60,944 shares of our common stock at an exercise price of $2.80 per share exercised such warrants, yielding aggregate gross proceeds to us of $170,643.
In June 2017, we entered into an offer letter to hire our current Chief Commercial Officer, who was the President and Chief Executive Officer of Symplmed, which appointment became effective on June 22, 2017. We also agreed in such offer letter to issue 60,000 restricted shares of our common stock under our 2014 Long-Term Incentive Plan to our Chief Commercial Officer, with a fair value of $228,000 ($3.80 per share), with all of such shares to vest on the six (6) month anniversary of the date of grant. These shares were issued in June 2017 and were fully vested as of December 31, 2017.
In August 2017, in connection with the reverse split, we issued 3,360 shares of common stock due to rounding at exchange and participant levels.
In September 2017, an investor converted 270 shares of Series C Preferred stock into 180,000 shares of our common stock on a cashless basis.
In September 2017, we entered into an engagement letter with a financial advisor pursuant to which, among other things, we agreed to issue to such financial advisor, in partial consideration of the services to be rendered under the engagement letter, an aggregate of 500,058 shares of our common stock. The shares were issued in November 2017 with an aggregate fair value of approximately $1,350,157.
Warrants
As of December 31, 2017, there were 2,559,612 warrants outstanding, with a weighted average exercise price of $4.28 per share, and annual expirations as follows:
| Expiring in 2018 | 11,383 | |||
| Expiring in 2019 | 600,000 | |||
| Expiring in 2020 | 1,189,079 | |||
| Expiring in 2021 | 343,750 | |||
| Expiring in 2022 | 66,667 | |||
| Expiring thereafter | 348,733 | |||
| 2,559,612 |
On May 21, 2017, the holders of warrants, to purchase 60,944 shares of our common stock at an exercise price of $2.80 per share, exercised such warrants, yielding aggregate gross proceeds to us of $170,643.
On November 22, 2017, we issued warrants to purchase 66,667 shares of our common stock to a consultant in connection with a related party convertible note issued in November 2017 at an exercise price of $0.75 and a term of five years with an aggregate fair value of $112,210.
A total of 149,111 warrants expired in May 2017.
On April 13, 2018, in connection with a private placement of our Series E Convertible Preferred Stock, we issued 22,665,525 warrants. Also see Note 11 – Subsequent events.
| F-21 |
Note 7 — Stock Incentive Plans
Stock Options
Stock option activity was as follows:
| Options Outstanding | ||||||||
| Shares | Weighted Average Exercise Price |
|||||||
| Outstanding, January 1, 2016 | - | $ | - | |||||
| Options acquired in reverse merger | 168,811 | 36.80 | ||||||
| Outstanding, December 31, 2016 | 168,811 | $ | 36.80 | |||||
| Options granted | 658,057 | 1.93 | ||||||
| Options expired and forfeited | (81,161 | ) | 1.14 | |||||
| Outstanding, December 31, 2017 | 745,707 | 8.84 | ||||||
| Exercisable, December 31, 2017 | 139,673 | $ | 3.85 | |||||
The following table summarizes additional information on Marina’s stock options outstanding at December 31, 2017:
| Options Outstanding | Options Exercisable | |||||||||||||||||||||
| Range
of Exercise Prices | Number Outstanding | Weighted- Average Remaining Contractual Life (Years) | Weighted Average Exercise Price | Number Exercisable | Weighted Average Exercise Price | |||||||||||||||||
| $ | 1.00 | 14,000 | 3.88 | $ | 1.00 | 14,000 | $ | 1.00 | ||||||||||||||
| $ | 1.70 – $1.80 | 534,007 | 9.23 | 1.79 | 47,973 | 1.75 | ||||||||||||||||
| $ | 2.60 – $8.20 | 168,400 | 4.12 | 3.15 | 48,400 | 4.62 | ||||||||||||||||
| $ | 10.70 – $22.00 | 25,050 | 1.73 | 10.81 | 25,050 | 10.81 | ||||||||||||||||
| $ | 476.00 - $876.00 | 2,100 | .44 | 676.00 | 2,100 | 676.00 | ||||||||||||||||
| $ | 1276.00 - $2076.00 | 2,150 | .44 | 1,582.98 | 2,150 | 1,582.98 | ||||||||||||||||
| Totals | 745,707 | 7.68 | $ | 8.84 | 139,673 | $ | 3.85 | |||||||||||||||
Weighted-Average Exercisable Remaining Contractual Life (Years) 3.95
In January 2017, the Company granted a total of 48,600 stock options to directors and officers for services. One-half of the options vest immediately and one-half of the options vest on the one-year anniversary of the grant date. The options have an exercise price of $1.70 and a five-year term. During the year ended December 31, 2017, a total of 4,050 of these options were forfeited, and 4,050 options were extended to January 2022.
In February 2017, the Company granted a total of 16,000 stock options to key employees for services. The options vest on the one-year anniversary of the grant date, have an exercise price of $1.80, and have a five-year term.
In October 2017, we appointed our Chief Financial Officer. In connection with this appointment, we granted the officer options to purchase up to 60,000 shares of our common stock under our 2014 Long-Term Incentive Plan, with all of such options vesting and becoming exercisable on the one-year anniversary of the grant date. The options have a 5-year term and an exercise price of $2.70.
On October 19, 2017, we appointed our Chief Legal Officer. In connection with this appointment, we granted the officer options to purchase up to 60,000 shares of our common stock under our 2014 Long-Term Incentive Plan, with all of such options vesting and becoming exercisable on the one-year anniversary of the grant date. The options have a 5-year term and an exercise price of $2.40.
On November 22, 2017, the Company granted a total of 473,457 stock options to a related party noteholder in connections with the issuance of a convertible note. The options vest 5% each quarter over 5 years, have an exercise price equal to the Company’s stock price on the date of vesting, and have a ten-year term.
| F-22 |
At December 31, 2017, we had approximately $949,500 of total unrecognized compensation expense related to unvested stock options. Total expense related to stock options was $178,784 for the year ended December 31, 2017.
At December 31, 2017, the intrinsic value of options outstanding or exercisable was $9,100 as there were 14,000 options outstanding with an exercise price less than $1.65, the per share closing market price of our common stock at that date.
Note 8 — Intellectual Property and Collaborative Agreements
Novosom Agreements
In July 2010, Marina entered into an agreement pursuant to which Marina acquired intellectual property for Novosom’s SMARTICLES-based liposomal delivery system. In February 2016, Marina issued Novosom 20,548 shares of common stock valued at approximately $58,000 as additional consideration under such agreement.
In March 2016, Marina entered into a license agreement covering certain of Marina’s platforms for the delivery of an undisclosed genome editing technology. Under the terms of the agreement, Marina received an upfront license fee of $250,000 and could receive up to $40 million in success-based milestones. In April 2016, Marina issued Novosom 47,468 shares of common stock valued at approximately $75,000 for amounts due under this agreement.
In July 2016, Marina entered into a license agreement with an undisclosed licensee that grants such licensee rights to use Marina’s technology and intellectual property to develop and commercialize products combining certain molecules with Marina’s liposomal delivery technology known as NOV582. Under the terms of this agreement, the licensee agreed to pay to us an upfront license fee in the amount of $350,000 (to be paid in installments through the end of 2017), along with milestone payments on a per-licensed-product basis and royalty payments in the low single digit percentages. As of September 30, 2016, Marina had received $50,000 per the terms of this license agreement. In November 2016, we issued 11,905 shares with a value of $15,000 to Novosom as the equity component owed under Marina’s July 2016 license agreement.
Arrangements with LipoMedics
In February 2017, we entered into a License Agreement (the “License Agreement”) with LipoMedics, pursuant to which, among other things, we provided to LipoMedics a license to our SMARTICLES platform for further development of Lipomedics’s proprietary phospholipid nanoparticles that can deliver protein, small molecule drugs, and peptides. These are not currently being developed at Marina and Marina has no IP around these products. On the same date, we also entered into a Stock Purchase Agreement with LipoMedics pursuant to which we issued to LipoMedics an aggregate of 86,207 shares of our common stock for a total purchase price of $250,000.
Under the terms of the License Agreement, we could receive up to $90 million in success-based milestones based on commercial sales of licensed products. In addition, if LipoMedics determines to pursue further development and commercialization of products under the License Agreement, LipoMedics agreed, in connection therewith, to purchase shares of our common stock for an aggregate purchase price of $500,000, with the purchase price for each share of common stock being the greater of $2.90 or the volume weighted average price of our common stock for the thirty (30) trading days immediately preceding the date on which LipoMedics notifies us that it intends to pursue further development or commercialization of a licensed product.
If LipoMedics breaches the License Agreement, we shall have the right to terminate the License Agreement effective sixty (60) days following delivery of written notice to LipoMedics specifying the breach, if LipoMedics fails to cure such material breach within such sixty (60) day period. LipoMedics may terminate the License Agreement by giving thirty (30) days’ prior written notice to us.
Vuong Trieu, Ph.D., our Executive Chairman, is the Chairman of the Board and Chief Operating Officer of LipoMedics.
| F-23 |
In consideration Lipomedics agreed to the following fee schedule: 1) Evaluations License Fee. Simultaneous with the execution and delivery of the License Agreement, Lipomedics shall enter into a Stock Purchase Agreement in form and substance reasonably acceptable to Marina and Lipomedics, pursuant to which Marina will sell to Lipomedics shares of the common stock of Marina for an aggregate purchase price of $0.25 million, with the purchase price for each share of Marina common stock being $2.90. 2) Commercial License Fee. Unless the License Agreement is earlier terminated, within thirty (30) days following Lipomedics’s delivery of an Evaluation Notice advising that it intends to pursue, or cause to be pursued, further development and commercialization of Licensed Products. 3) For up to and including three Licensed Products, Lipomedics shall pay to Marina a milestone of $10 million upon reaching Commercial Sales in the Territory in any given twelve month period equal to or greater than $500 million for a given Licensed Product and of $20 million upon reaching Commercial Sales in any given twelve month period equal to or greater than $1 million for such Licensed Product, such payments to be made within thirty (30) days following the month in which such Commercial Sale targets are met.
Arrangements with Oncotelic Inc.
In July 2017, we entered into a License Agreement (the “License Agreement”) with Oncotelic, Inc. (“Oncotelic”) pursuant to which, among other things, we provided to Oncotelic a license to our SMARTICLES platform for the delivery of antisense DNA therapeutics, as well as a license to our conformationally restricted nucleotide (“CRN”) technology with respect to TGF-Beta. [Under the terms of the License Agreement, Oncotelic also agreed to purchase 49,019 shares of our common stock for an aggregate purchase price of $0.25 million ($5.10 per share), with such purchase and sale to be made pursuant to a Stock Purchase Agreement to be entered into between us and Oncotelic within thirty (30) days following the date of the License Agreement. As of the date of this filing, such stock purchase has not occurred and we are in discussion with Oncotelic to complete the purchase.
Under the terms of the License Agreement, we could receive up to $90 million in success-based milestones based on commercial sales of licensed products. In addition, if Oncotelic determines to pursue further development and commercialization of products under the License Agreement, Oncotelic agreed, in connection therewith, to purchase shares of our common stock for an aggregate purchase price of $0.5 million, with the purchase price for each share of common stock being the greater of $5.10 or the volume weighted average price of our common stock for the thirty (30) trading days immediately preceding the date on which Oncotelic notifies us that it intends to pursue further development or commercialization of a licensed product.
If Oncotelic breaches the License Agreement, we shall have the right to terminate the License Agreement effective sixty (60) days following delivery of written notice to Oncotelic specifying the breach, if Oncotelic fails to cure such material breach within such sixty (60) day period. Oncotelic may terminate the License Agreement by giving thirty (30) days’ prior written notice to us.
Dr. Trieu, our Executive Chairman, is the principal stockholder and Chief Executive Officer of Oncotelic.
Sale of DiLA 2 Assets
On March 16, 2018, Marina entered into an Licensing Agreement with Agenovir Corporation, whereby Marina granted Agenovir exclusive rights to the company’s DiLA2 delivery system in exchange for an upfront payment of $200,000 and further potential future consideration dependent upon event and sales-based milestones. Under the terms of the agreement, Marina has agreed to assign ownership of the intellectual property associated with the DiLA2 delivery system to Agenovir Corporation.
Asset Purchase Agreement
In July 2017, Marina entered into an Asset Purchase Agreement with Symplmed Pharmaceuticals LLC and its wholly-owned subsidiary Symplmed Technologies, LLC pursuant to which the Company purchased from the Sellers, for an aggregate purchase price of $75,000 in cash, certain specified assets of the Sellers relating to the Sellers’ patented technology platform known as DyrctAxess that offers enhanced efficiency, control and information to empower patients, physicians and manufacturers to help achieve optimal care (see Note 3).
| F-24 |
Note 9 – Commitments and Contingencies
Amendment to Agreement with Windlas Healthcare Private Limited
On August 17, 2017, we entered into an amendment (the “Amendment”) of that certain Pharmaceutical Development Agreement dated as of March 30, 2017 by and between Windlas Healthcare Private Limited (“Windlas”) and our company (the “Development Agreement”), relating to the development by Windlas of certain pharmaceutical products to be used for conducting clinical trials or for regulatory submissions, as more fully described therein. Pursuant to the Amendment, we and Windlas agreed to amend the Development Agreement to reflect our agreement to issue to Windlas, and Windlas’ agreement to accept from us, in lieu of cash payments with respect to forty percent (40%) of the total amount reflected on invoices sent from time to time by Windlas to us, shares of our common stock having an aggregate value equal to forty percent (40%) of such invoiced amount (with the remaining portion of the invoiced amount being paid in cash). The maximum value of common stock that may be issued to Windlas pursuant to the Development Agreement (as modified by the Amendment) is $2 million. The parties also agreed that the foregoing payment arrangement would apply to any Contract Manufacturing and Supply Agreement (or similar agreement) relating to the manufacturing of commercial batches of the products covered by the Development Agreement that may be entered into between the parties.
Litigation
Because of the nature of the Company’s activities, the Company is subject to claims and/or threatened legal actions, which arise out of the normal course of business. Other than the disclosure below, as of the date of this filing, the Company is not aware of any pending lawsuits against the Company, its officers or directors.
The Company has been named on a complaint filed in New York State as a defendant in the matter entitled Vaya Pharma, Inc. v. Symplmed Technologies, Inc., Symplmed Pharmaceuticals, Inc., Erik Emerson and Marina Biotech, Inc. While this complaint has been filed in the Supreme Court of the State of New York, the Company has not been legally served. The complaint alleges, in relevant part, that: (i) the sale by Symplmed Pharmaceuticals, Inc. of its assets related to its Prestalia product, and the sale by Symplmed Technologies, Inc. of its assets related to its DyrctAxess platform, should be set aside pursuant to New York law as they were consummated without fair consideration to the sellers (the “Symplmed Defendants”), and thereby had the effect of fraudulently depriving the creditors of the Symplmed Defendants, including Vaya Pharma, Inc., of funds that could have been used to pay their debts; and (ii) the Company is liable, as successor, for any and all claims by Vaya Pharma, Inc. against the Symplmed Defendants, though pursuant to the agreement the Company is only contractually responsible for liabilities that accrue after the parties entered into the agreement for Prestalia and any liabilities that existed prior to the agreement are contractually held by Symplmed. If and when the Company is legally served, it is the intention of the Company to dispute jurisdiction, the sufficiency of the pleading and the claims set forth in this complaint, and to defend this matter, vigorously. However, due to the inherent uncertainties of litigation, the ultimate outcome of this matter is uncertain. The Company reached a tentative settlement agreement and, although such settlement has not been finalized, the Company accrued a total provision of $250,000 for this complaint, which is included in accrued expenses on the accompanying consolidated balance sheet.
Our Prestalia product is currently involved in a paragraph IV challenge regarding patents issued to perindopril arginine. This challenge, which is currently pending in the United States District Court for the District of Delaware (No. 1:17-cv-00276), is captioned Apotex Inc. and Apotex Corp. v. Symplmed Pharmaceuticals, LLC and Les Laboratoires Servier. The challengers (Apotex Inc. and Apotex Corp. (“Apotex”)) have filed an Abbreviated New Drug Application seeking FDA approval to market a generic version of Prestalia and included a Paragraph (IV) certification. In the litigation, Apotex seeks a declaratory judgment that no valid claims of the two patents Symplmed listed in the FDA Orange Book as having claims covering Prestalia, U.S. Patent No. 6,696,481 and 7,846,961, will be infringed by the Apotex proposed generic version of Prestalia and that the claims of those patents are invalid. The challenge is designed to provide Apotex with an opportunity to enter the market with a generic version of Prestalia, ahead of the expiration of the patents with claims covering that product. Apotex entered into negotiations with Symplmed Pharmaceuticals, LLC (which entity sold its assets relating to Prestalia to us in June 2017, including its License and Commercialization Agreement with Les Laboratories Servier) and Les Laboratories Servier (which entity owns or controls intellectual property rights relating to pharmaceutical products containing as an active pharmaceutical ingredient perindopril in combination with other active pharmaceutical ingredients, which rights have been licensed to Symplmed Pharmaceuticals) to resolve the challenge in the second quarter of 2017, and such parties, along with us, have come to a general agreement on terms that will result in a delay to the challengers’ ability to enter the market with a generic version of Prestalia, while still providing the challenger with the right to enter the market prior to the expiration of the patent covering such product. The term sheet memorializing such terms is pending execution in a final settlement agreement. In the meantime, the District Court has entered an order extending the time for the defendants to respond to Apotex’s Complaint until January 9, 2018 .
We do not anticipate that the proposed settlement terms – which would provide us with the ability to exclusively market Prestalia for a specified period of time without incurring the risks or costs of litigation – will have a material adverse impact on our long term business as a whole. We hold this expectation because it is our intention that Prestalia will be a building block toward the development of our company into a larger organization, which we hope will include the commercial launch by us of Prestalia and the expansion of our commercial capabilities in connection with such activities, followed by the launch of products that we license for commercialization purposes from our partners and the commercial launch of products that we develop internally. We anticipate that the aforementioned steps to expand and strengthen our commercial capabilities and diversify our product offerings so as to generate meaningful revenues in excess of those revenues generated by sales of Prestalia will occur during our exclusivity period with respect to Prestalia (i.e., prior to the date on which Apotex enters the market with a generic version of Prestalia). If we are able to achieve this growth, the overall economic impact that would result to us even if Apotex markets a generic version of Prestalia and captures a significant percentage of the market for that drug following our exclusivity period would be reduced.
However, there can be no assurance that the paragraph IV challenge will be resolved without litigation or resolved on the terms currently proposed. There also cannot be any assurance that our plans to commercialize Prestalia (or any of the products that it may license from its partners or that it may develop internally) and expand our product offerings and the revenue generated therefrom so as to lessen our reliance on sales of Prestalia will be achieved. Any failure of our expectations regarding the resolution of the paragraph IV challenge and the expansion of our commercial activities (and the revenues to be derived therefrom) could have a material adverse effect on us, our prospects and our results of operations.
NOTE 10 - INCOME TAXES
We have identified our federal and California state tax returns as “major” tax jurisdictions. The periods our income tax returns are subject to examination for these jurisdictions are 2014 through 2016. We believe our income tax filing positions and deductions will be sustained on audit, and we do not anticipate any adjustments that would result in a material change to our financial position. Therefore, no liabilities for uncertain income tax positions have been recorded.
At December 31, 2017, we had available net operating loss carry-forwards for federal income tax reporting purposes of approximately $323 million and had available tax credit carry-forwards for federal tax reporting purposes of approximately $10.6 million, which are available to offset future taxable income. Portions of these carry-forwards will expire through 2037 if not otherwise utilized. We have not performed a formal analysis, but we believe our ability to use such net operating losses and tax credit carry-forwards is subject to annual limitations due to change of control provisions under Sections 382 and 383 of the Internal Revenue Code, which significantly impacts our ability to realize these deferred tax assets.
| F-25 |
Our net deferred tax assets, liabilities and valuation allowance are as follows:
| Year Ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryforwards (a) | $ | 2,075,529 | $ | 892,107 | ||||
| Tax credit carryforwards (a) | - | |||||||
| Depreciation and amortization | 3,107,094 | 3,414,971 | ||||||
| Share based compensation | 297,893 | - | ||||||
| Other | 389,146 | 533,034 | ||||||
| Total deferred tax assets | 5,869,662 | 4,840,112 | ||||||
| Valuation allowance | (5,354,880 | ) | (3,918,791 | ) | ||||
| Net deferred tax assets | 514,782 | 921,321 | ||||||
| Deferred tax liabilities: | ||||||||
| Intangible assets | (514,782 | ) | (921,321 | ) | ||||
| Net deferred tax liabilities | $ | - | $ | - | ||||
(a) reflects estimated change of control limitation under Section 382 and 383 of the Internal Revenue Code as of December 31, 2016 due to reverse merger on November 15, 2016.
We record a valuation allowance in the full amount of our net deferred tax assets since realization of such tax benefits has been determined by our management to be less likely than not. The valuation allowance increased $1,436,089 and $3,444,024 during 2017 and 2016, respectively.
In 2017 and 2016, there was income tax expense of $800 and $800, respectively, due to IThena’s income tax due the state of California.
As of the date of this filing, the Company has not filed its 2016 federal and state corporate income tax returns. The Company expects to file these documents as soon as practicable.
The Tax Cuts and Jobs Act (the Act) was enacted on December 22, 2017. The Act reduces the US federal corporate tax rate from 35% to 21% and will require the Company to re-measure certain deferred tax assets and liabilities based on the rates at which they are anticipated to reverse in the future, which is generally 21%. The Company adopted the new rate as it relates to the calculations, of deferred tax amounts as of December 31, 2017.
Note 11 - Subsequent Events
Except for the events discussed below, there were no subsequent events that required recognition or disclosure. The Company evaluated subsequent events through the date the financial statements were issued and filed with the Securities and Exchange Commission.
On March 16, 2018, Marina entered into an Licensing Agreement with Agenovir Corporation, whereby Marina granted Agenovir exclusive rights to the company’s DiLA2 delivery system in exchange for an upfront payment of $200,000 and further potential future consideration dependent upon event and sales-based milestones. Under the terms of the agreement, Marina has agreed to assign ownership of the intellectual property associated with the DiLA2 delivery system to Agenovir Corporation.
Series E Convertible Preferred Share Private Placement Offering
On April 16, 2018, we entered into Subscription Agreements (the “Purchase Agreements”) with certain accredited investors and conducted a closing pursuant to which we sold 2,334 shares of our Series E convertible preferred stock (the “Preferred Stock”), at a purchase price of $5,000 per share of Preferred Stock. Each share of Preferred Stock is initially convertible into shares of our common stock (the “Common Stock”), at a conversion price of $0.50 per share of Common Stock. In addition, each investor received a 5-year warrant (the “Warrants”, and collectively with the Preferred Stock, the “Securities”) to purchase 0.75 shares of Common Stock for each share of Common Stock issuable upon the conversion of the Preferred Stock purchased by such investor at an exercise price equal to $0.55 per share of Common Stock, subject to adjustment thereunder. The closing is the initial closing (the “Initial Closing”) of our private placement (the “Private Placement”) of up to $10,000,000 of Securities, which amount may be increased to $15,000,000 at the discretion of our company and its placement agent in the Private Placement (the “Maximum Offering Amount”). The Preferred Stock has voting rights, dividend rights, liquidation preferences, conversion rights and anti-dilution rights as described in the Certificate of Designation of Preferences, Rights and Limitations of the Preferred Stock. The Warrants have full-ratchet anti-dilution protection, are exercisable for a period of five years following the final closing of the Private Placement and contain customary exercise limitations.
We received total gross proceeds of approximately $11.7 million from the Initial Closing, prior to deducting placement agent fees and estimated expenses payable by us associated with the Initial Closing. We intend intends to use the proceeds of the Private Placement for funding our commercial operations to the sale and promotion of our Prestalia® product, working capital needs, capital expenditures, the repayment of certain liabilities and other general corporate purposes. Pursuant to the Purchase Agreements, we may periodically conduct additional closings until we have sold the Maximum Offering Amount.
| F-26 |