Attached files
| file | filename |
|---|---|
| EX-32.1 - EX-32.1 - Presbia PLC | lens-ex321_7.htm |
| EX-31.2 - EX-31.2 - Presbia PLC | lens-ex312_6.htm |
| EX-31.1 - EX-31.1 - Presbia PLC | lens-ex311_8.htm |
| EX-23.1 - EX-23.1 - Presbia PLC | lens-ex231_10.htm |
| EX-21.1 - EX-21.1 - Presbia PLC | lens-ex211_9.htm |
| EX-10.28 - EX-10.28 - Presbia PLC | lens-ex1028_756.htm |
| EX-10.27 - EX-10.27 - Presbia PLC | lens-ex1027_755.htm |
| EX-10.26 - EX-10.26 - Presbia PLC | lens-ex1026_798.htm |
Ch
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
(Mark One)
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
or
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission File Number 001-36824
PRESBIA PLC
(Exact name of registrant as specified in its charter)
|
Ireland |
98-1162329 |
|
(State or other jurisdiction of incorporation or organization) |
(IRS Employer Identification No.) |
Suite 7, Sandyford Office Centre, 17 Corrig Road, Sandyford
Dublin 18 Ireland
(Address of principal executive offices, including zip code)
Registrant’s Telephone Number, Including Area Code:
+353 (1) 551 1487
Securities registered pursuant to Section 12(b) of the Act:
|
Ordinary Shares, $0.001 Par Value |
The NASDAQ Capital Market |
|
(Title of each class) |
(Name of each exchange on which registered) |
Securities registered pursuant to Section 12(g) of the Act: NONE
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). ☒ Yes ☐ No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in 12b-2 of the Exchange Act.
|
Large accelerated filer |
☐ |
Accelerated filer |
☐ |
|
Non-accelerated filer |
☐ |
Smaller reporting company |
☒ |
|
|
|
Emerging growth company |
☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☒
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the Registrant’s ordinary shares (the only common equity of the Registrant) held by non-affiliates for the last business day of the Registrant’s most recently completed second fiscal quarter: $10,932,997.
As of March 30, 2018, there were 17,121,857 ordinary shares outstanding.
PRESBIA PLC
2017 ANNUAL REPORT ON FORM 10-K
TABLE OF CONTENTS
|
|
|
Page |
|
|
|
|
|
Item 1. |
2 |
|
|
Item 1A. |
26 |
|
|
Item 1B. |
53 |
|
|
Item 2. |
54 |
|
|
Item 3. |
54 |
|
|
Item 4. |
54 |
|
|
|
|
|
|
|
|
|
|
Item 5. |
55 |
|
|
Item 6. |
55 |
|
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
56 |
|
Item 7A. |
64 |
|
|
Item 8. |
65 |
|
|
Item 9. |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
86 |
|
Item 9A. |
86 |
|
|
Item 9B. |
87 |
|
|
|
|
|
|
|
|
|
|
Item 10. |
88 |
|
|
Item 11. |
92 |
|
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
99 |
|
Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
101 |
|
Item 14. |
103 |
|
|
|
|
|
|
|
|
|
|
Item 15. |
104 |
|
|
Item 16. |
104 |
|
|
105 |
||
Cautionary Note Regarding Forward-Looking Statements
This Annual Report on Form 10-K contains forward-looking statements, within the meaning of the Private Securities Litigation Reform Act of 1995, that involve substantial risks and uncertainties. The forward-looking statements are contained principally in the sections of this Annual Report on Form 10-K titled “Business,” “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” but are also contained elsewhere in this Annual Report on Form 10-K. In some cases, you can identify forward-looking statements by the words “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “might,” “objective,” “ongoing,” “plan,” “predict,” “project,” “potential,” “should,” “will,” or “would,” and or the negative of these terms, or other comparable terminology intended to identify statements about the future. These statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to be materially different from the information expressed or implied by these forward-looking statements. Although we believe that we have a reasonable basis for each forward-looking statement contained in this Annual Report on Form 10-K, we caution you that these statements are based on a combination of facts and factors currently known by us and our expectations of the future, about which we cannot be certain.
The forward-looking statements in this Annual Report on Form 10-K include, among other things, statements about:
|
|
• |
the timing, progress and results of our U.S. staged pivotal clinical trial and our regulatory submissions; |
|
|
• |
our ability to advance our microlens and successfully complete our U.S. staged pivotal clinical trial; |
|
|
• |
our ability to obtain pre-market approvals; |
|
|
• |
the commercialization of our microlens outside the U.S.; |
|
|
• |
our ability to continue as a going concern; |
|
|
• |
our anticipated cash needs and our needs for additional financing; |
|
|
• |
the implementation of our business model, strategic plans for our business, products and technology; |
|
|
• |
the scope of protection we are able to establish and maintain for intellectual property rights covering our products |
|
|
• |
estimates of our expenses, future revenues, capital requirements and our needs for additional financing; |
|
|
• |
the timing or likelihood of regulatory filings and approvals; |
|
|
• |
our financial performance; and |
|
|
• |
developments relating to our competitors and our industry. |
You should refer to “Part I, Item 1A. Risk Factors” of this Annual Report on Form 10-K for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Annual Report on Form 10-K will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame or at all. We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
You should read this Annual Report on Form 10-K and the documents that we reference in this Annual Report on Form 10-K and have filed as exhibits to this Annual Report on Form 10-K completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
Industry and Market Data
We obtained the industry and market data in this Annual Report on Form 10-K from our own research as well as from industry and general publications and surveys and studies conducted by third parties. Industry and general publications, studies and surveys generally state that the information contained therein has been obtained from sources believed to be reliable. These third parties may, in the future, alter the manner in which they conduct surveys and studies regarding the markets in which we operate our business. As a result, you should carefully consider the inherent risks and uncertainties associated with the industry and market data contained in this Annual Report on Form 10-K, including those discussed in “Part I, Item 1A. Risk Factors.”
1
This Annual Report on Form 10-K includes trademarks, service marks and trade names owned by us or other companies. All trademarks, service marks and trade names included in this Annual Report on Form 10-K are the property of their respective owners. Our principal trademark or trade name that we use is PresbiaTM.
In January 2015, we completed a series of corporate reorganization transactions described in “Item 1. Business—Corporate History and Information,” which we refer to herein as the Reorganization Transactions. Unless we state otherwise, the terms “we,” “us,” “our,” “Presbia” and the “Company” refer to Presbia PLC and its consolidated subsidiaries after giving effect to the Reorganization Transactions. Prior to the completion of the Reorganization Transactions, the foregoing terms refer to the entities that became the consolidated subsidiaries of Presbia PLC upon consummation of the Reorganization Transactions.
Overview
We are an ophthalmic device company which has developed and is in the process of obtaining FDA approval for a proprietary optical lens implant for treating presbyopia, the age-related loss of the ability to focus on near objects. Our lens, which we refer to as our microlens, is a miniature lens designed to be surgically implanted in a patient’s eye to improve that patient’s ability to see objects at close distances. Our current strategy is to continue to commercialize our microlens in certain strategic countries where we currently have marketing approval and to obtain FDA approval for our microlens in the United States. Our goal is to become a leading provider of corneal inlay presbyopia-correcting treatment worldwide.
According to Market Scope, an ophthalmic market research organization, presbyopia is a common vision disorder that affects approximately 1.9 billion people worldwide. Presbyopia is associated with the inability of the eye’s natural lens to change shape, or accommodate, in order to see clearly objects in the near and middle distance ranges. According to Market Scope, the worldwide presbyopic population is expected to grow to approximately 2.1 billion by 2021. We believe that the optimum market for our microlens is the global emmetropic presbyopic market, which as an example, in the U.S., is estimated at 38% of the total U.S. presbyopic market, estimated at 119 million presbyopes. According to Market Scope, spending on devices, equipment and procedure fees for presbyopia-correcting surgery is expected to increase from approximately $476 million in 2015 to approximately $906 million in 2020 at the manufacturer level. We do not currently have marketing approval in many jurisdictions included in the foregoing global data, which jurisdictions collectively represent a majority of the worldwide presbyopic population, including the United States.
We believe that our solution offers each of the following benefits:
|
|
• |
our solution is intended primarily for emmetropic-presbyopes, or those individuals who suffer from presbyopia but do not have any other visual disorder, and it has also been used effectively after laser procedures. |
|
|
• |
our solution is also intended for plano-presbyopes, those who have had prior lens replacement procedures, and currently seek treatment for presbyopia. |
|
|
• |
our solution is minimally invasive; our microlens can be implanted and removed in simple, surgical procedures. |
|
|
• |
our solution offers significant near vision improvement with little or no loss of binocular distance visual acuity (the ability to see distant objects with both eyes without prescription enhancement) and minimal risk of adverse side effects. |
|
|
• |
our solution offers a wide range of corrective power, from +1.5 diopters to +3.5 diopters, in 0.25 diopter increments (a diopter is a unit of measurement of the optical power of a lens). |
|
|
• |
once implanted, our microlens is invisible to the naked eye. |
In addition, our microlens and the procedure to implant our microlens are not currently reimbursed through private or governmental third-party payors in any country, nor do we anticipate that they will be reimbursable in the foreseeable future. The commercialization of our microlens depends on a prospective patient’s ability to cover the costs of our microlens and the implantation procedure. We believe that a substantial portion of presbyopes worldwide do not have the financial means to cover the costs of our microlens, but we believe that a direct patient pay model enables medical providers to avoid pricing pressure from private or governmental third-party payors.
Our microlens procedure is performed using a 150 kilohertz or greater frequency femtosecond laser, which is a laser that is currently used in certain LASIK surgeries, cataract surgeries and cornea replacement surgeries. In commercializing our solution, we intend to target those markets with a well-established presence of ophthalmic clinics equipped with femtosecond lasers. We believe that the existing infrastructure in most such ophthalmic clinics is sufficient to make our solution an attractive opportunity for such ophthalmic
2
clinics and our commercialization strategy includes working closely with such ophthalmic clinics to train and qualify ophthalmic surgeons on the use of our solution.
Through our European Union CE Mark, we are generally authorized to market our microlens throughout the European Economic Area (all European Union member states plus Iceland, Liechtenstein and Norway), or “EEA”, and, through mutual recognition agreements, in Switzerland. We currently market our microlens in certain strategic EEA countries as well as certain strategic countries outside of the EEA in which we possess marketing approval, such as South Korea. Through February 2018, we have shipped approximately 2,200 of our microlenses to commercial partners outside of the United States, of which ophthalmic surgeons have implanted approximately 1,300. For geographic information regarding our revenues and long-lived assets, please see Note 11 to our audited financial statements included elsewhere in this Annual Report on Form 10-K.
We are presently seeking marketing approval in the United States. In order to commercialize our microlens in the United States, we must first obtain a premarket approval, or “PMA”, from the U.S. Food and Drug Administration, or the “FDA”. In December 2013, we received approval to commence a staged pivotal clinical trial as part of the FDA approval process. Beginning in May 2014, we enrolled a total of seventy-five (75) subjects at six (6) investigational sites in the U.S. and each subject underwent insertion of our microlens in the non-dominant eye. Based on nine-month data on fifty-two (52) subjects, in January 2015, we submitted an interim safety report as a supplement to our investigational device exemption, or IDE, to the FDA. In February 2015, we received approval from the FDA to commence second stage enrollment in this trial. In September 2015, we completed enrollment of the second stage study of 346 subjects at up to five (5) additional investigational sites. This trial is necessary in order to obtain clinical data to provide the primary support for a safety and effectiveness evaluation to support a PMA for commercial distribution in the U.S.. Data on a minimum of 300 subjects with 24-month data will be submitted as part of the PMA, and all subjects will be followed for three (3) years following implantation. We are pursuing a modular PMA submission strategy whereby we submitted to the FDA information regarding biocompatibility in the second quarter of 2016. We submitted to the FDA the second and third PMA modules in the first quarter of 2017, which contains information regarding preclinical testing, engineering and manufacturing. We are targeting submission to the FDA of our final PMA module, containing 24-month data on 300 subjects, in the second quarter of 2018. We are targeting PMA approval of our microlens in the fourth quarter of 2018. We are also targeting submission to the FDA of a final report with 36-month data on these 300 subjects in the fourth quarter of 2018.
The Eye and Vision Problems
The human eye is a specialized sensory organ capable of receiving visual images and transmitting them to the visual center in the brain. Among the main parts of the eye are the cornea, the iris, the lens and the retina. The cornea is the clear window in the front of the eye through which light first passes. The interior surface of the cornea is lined with a single layer of flat, tile-like endothelial cells, whose function is to maintain the transparency of the cornea. The iris is a pigmented muscular curtain located behind the cornea that opens and closes to regulate the amount of light entering the eye through the pupil, an opening at the center of the iris. The lens, known in medical terminology as the “crystalline lens,” is a clear structure located behind the iris that changes shape, or accommodates, to focus light on the back of the eye. The retina is a layer of nerve tissue in the back of the eye that senses the light image and transmits it to the brain via the optic nerve. The figure below illustrates certain elements of the basic anatomy of the human eye.

3
The eye may be affected by common visual disorders, disease or trauma. A normal, well-functioning eye receives images of objects at varying distances and focuses the images on the retina. Refractive errors (including myopia, hyperopia, presbyopia and astigmatism, each described below) occur when the eye cannot properly focus an image on the retina. In addition to presbyopia, common vision problems include:
|
|
• |
myopia, or nearsightedness, which occurs when the eye’s lens focuses images in front of the retina; |
|
|
• |
hyperopia, or farsightedness, which occurs when the eye’s lens focuses images behind the plane of the retina; |
|
|
• |
astigmatism, an optical defect in which vision is blurred due to an oval-shaped cornea or, in some cases, an oval-shaped natural lens, producing a distorted image on the retina. Astigmatism may accompany myopia or hyperopia; and |
|
|
• |
cataracts, a clouding of the lens, which worsens with time and gradually occludes incoming light images. |
Cataracts are mostly age-related, while myopia, hyperopia and astigmatism are not. The most common surgical treatment for myopia, hyperopia and astigmatism is LASIK surgery, in which the surface of the cornea is carefully mapped and then a computerized optical laser uses this mapping to reshape the surface of the cornea by ablation to permit proper focusing. Cataracts are most often treated by surgically removing the affected lens and replacing it with a monofocal or multi-focal IOL.
Presbyopia is an age-related refractive disorder that generally begins to develop when a person reaches the age of 40. The disorder may go unnoticed for several years after its initial onset and can worsen with age. The first symptoms of presbyopia are typically experienced when a person begins to have difficulty reading fine print. Presbyopia is associated with a loss of lens “elasticity,” the ability of the lens to change shape in order to focus incoming light on the retina from objects in near and middle distance ranges. Elasticity is slowly lost as people age, resulting in a slow decrease in the ability of the eye to focus on nearby objects. Presbyopia is a natural part of aging and affects substantially all people at some point in their adult lives.
Presbyopia Market
According to Market Scope, presbyopia currently affects approximately 1.9 billion people worldwide, or approximately 25% of the global population. According to Market Scope, the worldwide presbyopic population is expected to grow to approximately 2.1 billion people by the end of 2021. The global market opportunity for surgical treatment of presbyopia is large and growing due to the aging of the population. Globally, the median age is projected to increase from 29 years in 2011 to 38 years by 2050. Consistent with the expected growth in the worldwide presbyopic population, according to Market Scope, the annual number of presbyopia-correcting surgeries performed globally is expected to increase from approximately 712,000 procedures in 2015 to approximately 1.2 million procedures by 2020. According to Market Scope, corneal inlays are projected to be the fastest growing segment of this market and are expected to grow from approximately 18,000 procedures in 2014 to approximately 204,000 procedures in 2019. In addition, according to Market Scope, spending on devices, equipment and procedure fees for presbyopia-correcting surgery is expected to increase from approximately $476 million in 2015 to approximately $906 million in 2020 at the manufacturer level. We believe that the optimum market for our microlens is the emmetropic presbyopic market, which, as an example, in the U.S., is estimated at 38% of the total U.S. presbyopic population. We do not have marketing approval in many jurisdictions included in the foregoing global data, which jurisdictions collectively represent a majority of the worldwide presbyopic population. We have marketing approval in a number of strategic countries that we are targeting for commercialization and we are actively seeking marketing approval in the United States.
Approaches for Treating Presbyopia
Although reading glasses and contact lenses have historically been, and remain, the most common solution for presbyopia, there are significant drawbacks associated with these non-surgical approaches. Eyeglasses can easily be lost, misplaced, broken or scratched and require frequent cleaning. Also, many people wish to avoid the inconvenience of keeping reading glasses close at hand. Contact lenses require daily insertion, removal and maintenance, which can be problematic for an increasingly mobile population and for people living and working in dusty environments or in unsanitary conditions.
There are presently four surgical correction categories for treating presbyopia:
Monovision. Monovision treatments correct one eye, typically the dominant eye, for distance vision and correct the other, non-dominant eye for near vision. While monovision may be accomplished through the use of glasses with two different lenses with varying thickness, that approach can cause bothersome symptoms when a person looks through the edges of the glasses. A more typical approach to monovision is the use of two different contact lenses. A more permanent monovision approach is to undergo laser or IOL-based refractive surgeries adapted for presbyopia correction. A significant drawback of monovision surgical treatments is the complexity of achieving additional correction, if vision further deteriorates. Additional drawbacks include occasional patient adaptation issues, whereby patients have difficulty adjusting to the monovision arrangement and suffer from blurring of vision, difficulty driving at night and loss of stereopsis, or the ability to focus upon an object with both eyes and create a single stereoscopic image.
4
Multifocal. Multifocal approaches are designed to provide both distance and near focus at the same time in each eye. Generally, both depth perception, or the ability to judge the distance of an object, and contrast sensitivity, or the ability to detect detail having subtle color gradations, are generally improved when two eyes can focus on an object. In addition, these approaches are intended to be improvements over constantly taking glasses off and putting them back on (possibly by wearing glasses around the neck) or by using bifocal or varifocal glasses or contact lenses, in which the eye is trained to look through the top part for distance vision and the bottom part for near vision. Multifocal effects can be achieved by lens replacement, including multifocal IOLs (IOLs with different zones of varying power), or through the creation of a multifocal cornea using laser refractive surgery (to create two or more refractive zones on the central cornea) or intrastromal ablation (laser used to make small changes in the thickness of the cornea). As with monovision, a significant drawback of these multifocal approaches is the complexity of achieving additional correction, if vision further deteriorates. In addition, some patients may experience halos, or rings around lights, at night, and it may also take time for multifocal patients to adapt to the different focal areas.
Restoring Accommodation. Accommodating approaches generally attempt to replace the natural lens with an accommodating IOL, which is an artificial lens that is designed to mimic the movement of the natural crystalline lens of the eye. All IOL-based surgeries are susceptible to opacification, or clouding, of the lens capsule, which is the part of the natural lens covering that remains after surgery, decreasing vision and requiring a laser procedure to cut a hole in the clouded back lining of the lens capsule to allow light to pass through the membrane to the retina. Accommodating IOLs are also subject to certain other complications pertaining to the shrinkage, closure or clouding of the capsule that can reduce the mode of action of the accommodating IOLs, rendering them less effective. Other less common accommodating techniques include lens softening and scleral relaxation techniques, which are designed to improve near vision by restoring the function of the eye’s own accommodative system. Lens-softening techniques use pharmaceuticals or lasers to soften or change the structure of the natural crystalline lens, allowing it to flex better to increase accommodation. Scleral relaxation techniques use implants in the sclera of the eye to increase the eye’s ability to focus at near distances. To date, these procedures have had little documented success.
Corneal Inlays. Corneal inlays include miniature surgically implanted lenses (such as our microlens), optical devices inserted into the cornea to reshape the front surface of the eye, and small implants to reduce the size of the opening into the eye to reset the angle of the light rays entering the eye and reduce both the number of rays and the light scatter, each of which is designed to improve near vision.
Our Solution
We have designed our microlens to address certain limitations of other surgical approaches to treat presbyopia. The critical aspects of our solution include:
|
|
• |
Effective Treatment Option for Emmetropic Presbyopes. The largest sub-group of the presbyopic population is emmetropic presbyopes, or those individuals without significant refractive error who suffer from presbyopia. Emmetropic presbyopes account for approximately 38% of the total presbyopic population. We believe that ophthalmologists are generally reluctant to recommend a LASIK or IOL procedure as a solution for an emmetropic presbyope given the inherent risks and visual compromises of such procedures. Because our procedure does not involve the removal of the natural lens, the reshaping of the cornea or the removal of corneal tissue, we believe that ophthalmologists will be more likely to recommend our microlens as a solution for emmetropic presbyopes than a LASIK or IOL procedure. |
|
|
• |
Complementary Solution. While our solution is intended primarily for emmetropic-presbyopes, or those individuals who suffer from presbyopia but do not have any other visual disorder, it has also been used effectively on plano-presbyopes after laser procedures or lens replacement procedures for treatment of presbyopia. |
|
|
• |
Minimally Invasive. Our microlens is implanted in a pocket in the cornea created with a femtosecond laser. The pocket seals itself within a few days, holding the lens in place. The procedure does not require the reshaping of the cornea and no corneal tissue is removed. Moreover, the nature of our solution permits normal nutrient flow to the cornea, enabling corneal metabolism. As a result, there is less potential for dry-eye symptoms and less damage to the collagen fibers that support corneal shape and structure. |
|
|
• |
Correction Options. The range of optical power corrections available in our microlens allows the ophthalmic surgeon to choose the correction most appropriate for the patient’s specific near vision requirements, as opposed to a unilateral “one size fits all” approach. |
|
|
• |
Invisible. The clear nature of our microlens renders it invisible to the naked eye which we believe will make it appealing to patients. |
|
|
• |
Does Not Hinder Certain Other Procedures. Our microlens does not hinder examination of the retina and other structures in the eye necessary to diagnose other ocular health disorders. |
5
|
|
generally begin immediately after the surgery, with the immediate common side effects of such a procedure generally being mild eye dryness and irritation, transient elevated intraocular pressure due to standard post-surgery medication regimen, corneal haze (the activation of inflammatory cells in connection with surgery), transient light sensitivity (an abnormal occurrence of photosensitivity associated with the femtosecond laser) and certain visual symptoms, such as halos or glare. |
We believe that surgical treatment for presbyopia represents a large new market opportunity for ophthalmic surgeons. The market for traditional surgical ophthalmic treatments, such as LASIK for myopia, hyperopia and astigmatism, and traditional monofocal IOLs for cataracts, is highly mature. Our procedure utilizes the femtosecond laser currently used for certain LASIK surgeries, cataract surgeries and cornea replacement surgeries. We believe that many ophthalmic clinics equipped with such lasers are not operating at full capacity, and we hope to utilize such untapped capacity. Our procedure would allow these ophthalmic clinics and ophthalmic surgeons to introduce a new treatment modality using their existing laser equipment, adding incremental revenue without the need for significant new capital commitments.
We believe demand is likely to be fueled further by the ever-evolving, near-vision needs resulting from the increasing reliance on smart phones, tablets and other technological advances requiring good near vision.
Our Technology
Our microlens is a disc shaped lens that has a refractive zone in the periphery designed to improve near vision problems associated with presbyopia. The two figures below illustrate the design of our microlens.
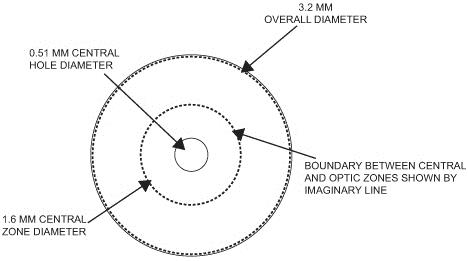
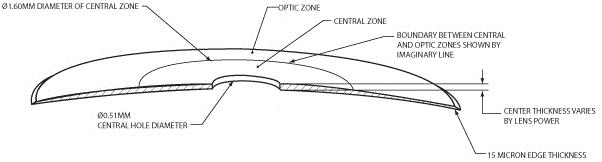
Our microlens is 3.2 millimeters (mm) in diameter, has an edge thickness of 0.015 mm and has a center thickness that ranges from approximately 0.03 mm to 0.05 mm (depending on the lens power). Once implanted, it is invisible to the naked eye. Our microlens is made of a hydrophilic acrylic material, similar to the kind that has been used to make IOLs for over 20 years. Our microlens is designed to be removable. In addition, our microlens is designed to reduce the risk of permanent corneal tissue loss and is designed to be biocompatible with the cornea, allowing for corneal metabolism, which is essential to the health and normal functioning of the cornea.
6
Ocular dominance is the tendency to prefer visual input from one eye to the other. Our microlens is implanted in a patient’s non-dominant eye to minimize impact to binocular uncorrected distance vision. Through implantation in the patient’s non-dominant eye, our solution seeks to exploit the brain’s ability to perceptually suppress central vision in one eye when the two eyes are receiving disparate stimuli and focus on the clearer images while ignoring the blurrier images. Prior to implantation, we require patients to wear a contact lens for near vision correction in their non-dominant eye for a minimum of five to seven days before insertion of our microlens in order to assess whether or not the patient is able to adapt to the change in the visual system. Not all prospective patients are able to adapt to the change in the visual system. Based on feedback that we have received from surgeons to date, we believe that approximately 40% of prospective patients who underwent monofocal contact lens correction in their non-dominant eye were unable to adapt to the change in the visual system and approximately 25% of prospective patients who underwent multifocal contact lens correction in their non-dominant eye were unable to adapt to the change in the visual system.
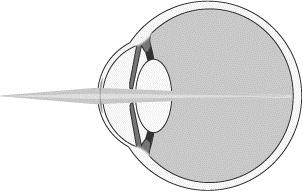
To improve near vision, as shown below, the refractive peripheral portion of our microlens is designed to help focus light from near objects (darker shaded light) onto the retina.
Near Vision with Microlens
To improve maximum distance vision, the central refractive portion of our microlens is designed to permit light from distant objects to pass through the cornea and lens and focus on the retina (lighter shaded light shown below). The refractive peripheral portion of the lens causes some distant light rays to focus in front of the retina, instead of on it (darker shaded light shown below). However, when the brain receives dual visual stimulus from the corrected non-dominant eye, as well as the uncorrected dominant eye, it is able to correctly combine the information into an image for the patient.
Distance Vision with Microlens
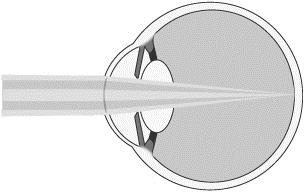
Insertion of our Microlens
Our microlens is surgically implanted in an outpatient setting. The procedure, requiring only topical anesthesia, typically takes a trained ophthalmic surgeon approximately 10 minutes. The procedure and equipment needed to create a corneal pocket to insert the microlens are similar to those currently used in LASIK procedures. In addition to our microlens inserter, we believe that the existing infrastructure in most ophthalmic clinics equipped with femtosecond lasers and existing surgical tools in ophthalmic surgery clinics is sufficient for our procedure.
7
The ophthalmic surgeon starts the procedure by making a mark on the cornea at the center of the visual axes in order to determine the most appropriate location of the corneal pocket as well as the microlens placement and alignment once in place. Then, using a 150 kilohertz or greater frequency femtosecond laser, the ophthalmic surgeon creates a pocket, approximately four to 5.5 mm in diameter, in the cornea of the patient’s non-dominant eye. Using a microlens inserter, the ophthalmic surgeon then inserts our microlens into the corneal pocket. Finally, the ophthalmic surgeon centers and checks the position of the implanted microlens before completing the surgery. The corneal pocket automatically seals itself within a few days, holding the microlens in place at the center of the eye’s visual axis.
We have designed our microlens and procedure to be removable in a minimally invasive manner in the event that a patient wishes to have a stronger prescription microlens implanted. This may occur if a patient’s presbyopia significantly progresses over time or in the event that a patient wishes to have the microlens removed for any other reason, including if the patient is uncomfortable with the results, if neural adaptation is not achieved, or if technological advances produce alternative solutions in the future. In the United States, our IDE does not permit replacement of a microlens in the event that a patient’s microlens is removed after implantation. Also, in the United States, our IDE requires any removal of the microlens to be reported as an adverse event.
The procedure to remove our microlens may take place in an outpatient setting, using only topical anesthesia. The removal procedure consists of opening the corneal pocket entry point and, using a fluid to lubricate the pocket of the lens, sliding the lens from the corneal pocket. This procedure typically takes a trained ophthalmic surgeon approximately 10 minutes. A new microlens can be immediately inserted into a patient’s existing corneal pocket.
Through February 2018, we have shipped approximately 2,200 of our microlenses to commercial partners outside of the United States, of which ophthalmic surgeons have implanted approximately 1,300.
Clinical Studies
We have completed a multicenter clinical study outside the United States. In addition, several third parties have conducted limited studies of our microlens. These studies are summarized below.
Evaluation Conducted Outside of the United States
In early 2012, we completed a 12-month multicenter, post-market evaluation in Italy and Greece of our microlens in presbyopic patients between the ages of 45 and 60 to evaluate the safety and effectiveness of our microlens. We designed, and oversaw the implementation of, the protocol for this evaluation, which was conducted at our request by a surgeon at the Vardinoyannion Eye Institute at the University of Crete in Crete, Greece and by a surgeon at Prato Hospital in Prato, Italy. The 12-month data for the 70 patients who completed the study demonstrated successful patient outcomes and a low rate of post-operative adverse events. The average UCVA-near in the operated eye pre-surgery for those 70 patients was 20/110 and 99% of those patients started the study with UCVA-near in the operated eye of 20/50 or worse. Key effectiveness findings from this evaluation included the following:
|
|
• |
the average UCVA-near in the operated eye for such patients post-surgery was 20/27, 99% of such patients completed the study with 20/40 or better UCVA-near in the operated eye and 70% of such patients completed the study with 20/25 or better UCVA-near in the operated eye (see Figure 1 below); |
|
|
• |
although there was a slight loss in uncorrected distance visual acuity (the ability to see distant objects without prescription enhancement), or UCVA-distance, in the operated eye (see Figure 4 below), there was no significant change in binocular UCVA-distance (UCVA-distance when using both eyes) from before treatment to after treatment in this study (see Figure 2 below); and |
|
|
• |
there was no significant change in best corrected distance visual acuity (distance vision using prescription enhancement), or BCVA-distance, in the operated eye after 12 months (see Figure 3 below). |
A Snellen chart is an eye chart used by eye care professionals and others to measure visual acuity. It usually consists of letters printed in lines of decreasing size which a person is asked to read at a fixed distance. 20/20 is a term used to define normal visual acuity, which relates to the Snellen chart. The first number denotes a certain distance, and the second number denotes the distance at which a person with normal visual acuity could read clearly those letters that the subject of the assessment can read clearly at the distance denoted in the first number. The standard distance for testing distance visual acuity is 20 feet. Thus, with respect to distance vision, if an individual has 20/100 vision, it means that a person with normal distance vision can read at 100 feet what the patient can only read at 20 feet (poor distance vision). 20/10 vision, on the other hand, would mean the individual has better than normal distance vision, being able to read at 20 feet what a person with normal distance vision could only read at 10 feet. With respect to near vision, the 20/20 nomenclature is used with the distances in the first number and the second number scaled to the distance used in the study. Thus, an individual with 20/20 near vision means the patient can read clearly those letters at the distance tested (usually 40 centimeters (cm) in the United States and 33 cm outside of the United States) that a person with normal near visual acuity could read clearly at that
8
distance. In our post-market evaluation, we tested visual acuity using an Early Treatment Diabetic Retinopathy Chart, or ETDRS, Snellen chart; the distance used to test distance visual acuity was 20 feet and the distance used to test near visual acuity was 33 cm.
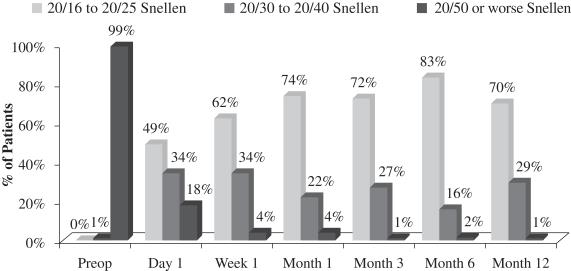
An important measurement is the number of patients who reach better visual acuity levels, or visual correction, after treatment. Before surgery, the 70 patients who completed the study had an average UCVA-near in the eye to be operated on of 20/110 and 99% of such patients started the study with UCVA-near measurements of 20/50 or worse in that eye. After treatment with our microlens, such patients had an average UCVA-near in the operated eye of 20/27 and 99% of such patients achieved UCVA-near measurements of 20/40 or better in the operated eye and 70% of such patients achieved UCVA-near measurements of 20/25 or better in the operated eye. The following chart summarizes the positive UCVA-near results in this post-market evaluation:
Figure 1
Uncorrected Near Visual Acuity Operated Eye (33 cm chart)
(N=70 at Month 12)
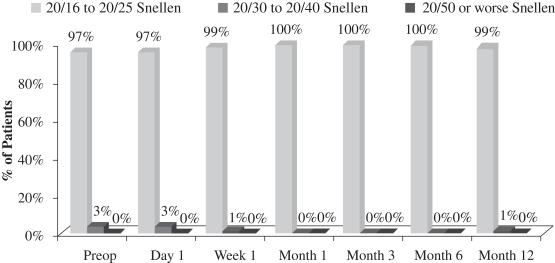
Another important measurement is the number of patients who maintain binocular UCVA-distance levels post-treatment. There was no significant change in binocular UCVA-distance from before treatment to after treatment in this study. This stability in binocular vision is important because it indicates that patients in the study did not experience a significant compromise in binocular UCVA-distance as a result of the insertion of our microlens, meaning that their normal binocular far vision was not compromised. The following chart summarizes the binocular UCVA-distance findings in this post-market evaluation:
9
Uncorrected Binocular Distance Visual Acuity
(N=70 at Month 12)
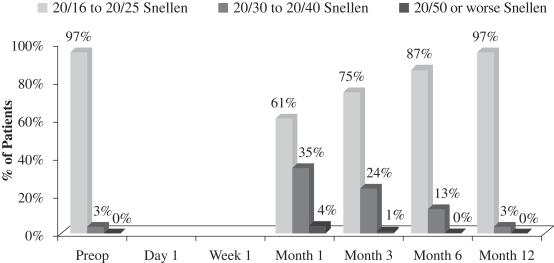
In addition, patients in this study experienced no significant change in BCVA-distance in the operated eye at 12 months post-implantation, which indicates that there was no compromise in the operated eye’s optical system at 12 months. The following chart summarizes BCVA-distance findings with respect to the patient’s operated eye in this post-market evaluation:
Figure 3
Best Corrected Distance Vision Acuity Operated Eye
(N=70 at Month 12)
10
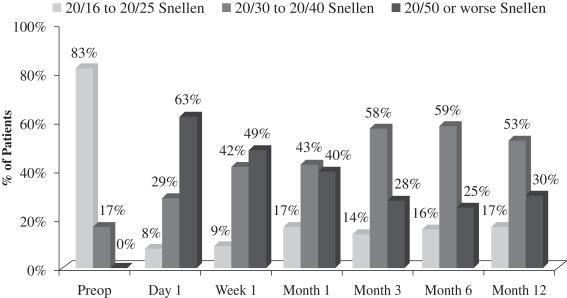
There was a slight loss of UCVA-distance in the operated eye in the study population. Before surgery, 83% of patients achieved UCVA-distance in the operated eye of 20/16 to 20/25 and 17% achieved UCVA-distance in the operated eye of 20/30 to 20/40. After treatment with our microlens, 17% of patients achieved UCVA-distance in the operated eye of 20/16 to 20/25, 53% achieved UCVA-distance in the operated eye of 20/30 to 20/40, and 30% achieved UCVA-distance in the operated eye of 20/50 or worse. Although, as mentioned above, there was no significant change in binocular UCVA-distance from before treatment to after treatment in this study, far distance vision in the operated eye is also important, particularly as it relates to overall patient satisfaction. In the study population, 78% of patients who responded reported that they perceived their UCVA-distance in the operated eye as “excellent” to “good.” This generally correlates to the data at month 12, where 70% of patients achieved 20/40 vision or better in the operated eye. The remaining 30% of patients achieved UCVA-distance in the operated eye of 20/50 or worse, and consistent with such results, 20% of patients who responded reported that they perceived their UCVA-distance in the operated eye as “fair.” One patient who responded considered his outcome with respect to UCVA-distance in the operated eye as “poor.” The following chart summarizes UCVA-distance findings with respect to the patient’s operated eye in this post-market evaluation:
Figure 4
Uncorrected Distance Visual Acuity Operated Eye
(N=70 at Month 12)
There are several possible explanations for the loss of distance visual acuity in the operated eye, including, but not limited to the following:
|
|
• |
the time required for neural adaptation, or the time it takes the brain to adapt to the change in the visual system; |
|
|
• |
improper patient selection, or the selection of patients who are intolerant of monovision, impatient or not willing to wait for the neural adaptation time period; and |
|
|
• |
improper lens power selection, meaning the patient is difficult to refract. |
11
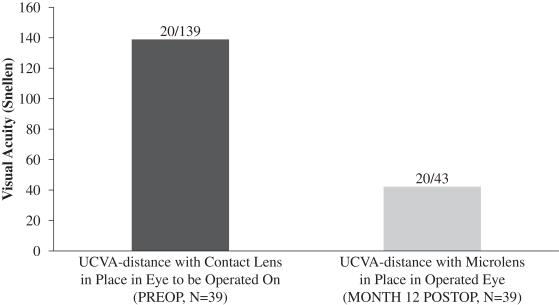
A subset of 39 patients enrolled in this evaluation underwent a monovision simulation, whereby pre-surgery UCVA-distance in the eye to be operated on with near vision contact lens correction in place was compared to UCVA-distance in the operated eye following implantation of our microlens. The purpose of this simulation was to evaluate whether UCVA-distance in the operated eye following implantation of our microlens is better than pre-surgery UCVA-distance in such eye with the use of a near vision correction contact lens. These patients were found to have an average UCVA-distance in the operated eye of 20/43 12 months post-surgery, while the same patients pre-surgery had an average UCVA-distance in the eye with near vision contact lens correction of 20/139. We believe that this result is due to the nature of the design of our microlens which is intended to maintain distance vision in the operated eye to the greatest extent possible. The central portion of our microlens is designed to allow for light from distant objects to enter the eye and focus on the retina, and the retina ultimately transmits that image to the brain. Figure 5 below illustrates the findings in this evaluation with respect to post-surgery UCVA-distance in the operated eye compared to pre-surgery UCVA-distance in the eye to be operated upon with near vision contact lens correction in place:
Figure 5
Overall, patient satisfaction with the procedure was generally high. 97% of patients who responded reported “excellent” or “good” perception of UCVA-near, and 97% of patients who responded reported “excellent” or “good” perception of binocular UCVA-distance. 75% of patients who responded reported no use of glasses for near tasks while the remaining 25% reported use of glasses less than 50% of the time. 78% of patients who responded indicated that they used glasses for near tasks more than 50% of the time prior to implantation.
Key safety findings from the evaluation over a 12-month period included the following:
|
|
• |
low rate of post-operative adverse events; |
|
|
• |
one patient complained one week after implantation of significant halos and glare when driving at night and requested removal of the microlens (the lens was removed one month post-surgery); |
|
|
• |
one case of transient light sensitivity syndrome was reported: this represents an abnormal occurrence of photosensitivity associated with the femtosecond laser, which resolved after application of a topical steroid regimen; |
|
|
• |
one case of epithelial ingrowth was reported: this represents an abnormal growth of corneal epithelium in an area where it does not belong, associated with the femtosecond laser, which resolved after the ingrowth was surgically cleared; and |
|
|
• |
four cases of transient stromal haze were reported: these cases involved the activation of inflammatory cells in connection with surgery, which resolved after application of a topical steroid regimen. |
There was no significant change in:
|
|
• |
intraocular pressure, or the fluid pressure in the eye; |
|
|
• |
endothelial cell density, or the tissue layer undersurface of the cornea and which regulates corneal water content; |
12
|
|
• |
binocular contrast sensitivity. |
We continue to evaluate our microlens through clinical studies and marketing and post-marketing evaluations in connection with regulatory requirements and our commercialization efforts. In addition, through February 2018, we have shipped approximately 2,200 of our microlenses to commercial partners outside of the United States, of which ophthalmic surgeons have implanted approximately 1,300.
Third Party Studies
Our microlens has been the subject of certain third party studies that have been conducted to assess the effectiveness and safety of our microlens. We did not commission these studies or design, review or oversee the implementation of their protocols (although we paid the annual fees of the institutional review board, or “IRB”, reviewing one such study in Japan), and we have limited information with respect to these studies. These studies have reported certain adverse effects relating to the safety and effectiveness of our microlens. In connection with the findings in certain of such studies and observations of other surgeons regarding our procedure, we have undertaken certain investigative actions as part of our ongoing risk mitigation efforts. See “Risk Factors—Risks Related to Our Business—If concerns regarding side effects from presbyopia-correcting surgery generally, or our products specifically, develop, including as a result of third-party studies and publications, our business, results of operations and financial condition will be materially and adversely affected.”
U.S. Staged Pivotal Clinical Trial
In May 2014, we began a staged pivotal clinical trial to seek marketing approval for our microlens in the United States. See “Clinical Development and Commercialization Targets” below for a description of this study. For a description of adverse events to date in this study, see “Risk Factors—Risks Related to Our Business—If concerns regarding side effects from presbyopia-correcting surgery generally, or our products specifically, develop, including as a result of third-party studies and publications, our business, results of operations and financial condition will be materially and adversely affected.”
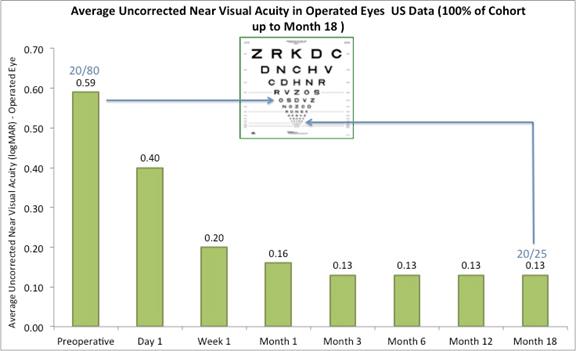
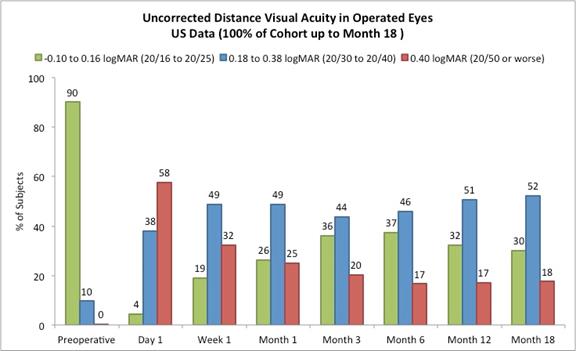
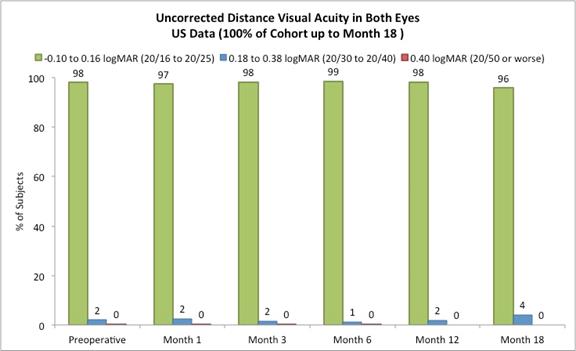
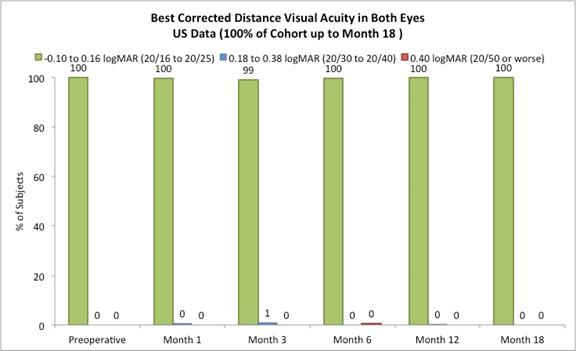
On May 23, 2017, we reported the following data from our U.S. staged pivotal clinical trial. Through May 19, 2017, 421 subjects had undergone insertion of the Company’s microlens during the staged pivotal clinic trial that the Company is performing to obtain the clinical data required to enable the Company to obtain pre-market approval from the FDA. Data (representing 100% of the study cohort at month 18 and excluding subjects who had the Microlens explanted or those that did not return for scheduled trial visits and considered lost to follow-up) made available to the Company in May 2017 indicates that:
|
|
• |
Subjects gained an average of 5 lines of uncorrected near visual acuity (the ability to see close objects without prescription enhancement) in treated eyes (Figure A). |
|
|
• |
Approximately 82% of subjects achieved 20/40 or better uncorrected distance vision in treated eyes (Figure B) and there was little to no change in binocular uncorrected distance vision from preoperative values (Figure C), and |
|
|
• |
Approximately 98% of subjects achieved 20/40 or better best corrected distance vision in the treated eyes (Figure D) and there was little to no change in binocular best corrected distance vision from preoperative values (Figure E). (Presbia Flexivue MicrolensTM is designed to take advantage of binocular vision. Most patients have the ability to fuse images in the brain from each eye to ensure the best image. This process is known as “neuroadaptation.”). |
13
The following chart summarizes the uncorrected near visual acuity in the treated eyes:
Figure A
The following chart summarizes the uncorrected distance vision in the operated eyes:
Figure B
14
The following chart summarizes the binocular uncorrected distance visual acuity:
Figure C
The following chart summarizes the best corrected distance vision in the operated eyes:
Figure D
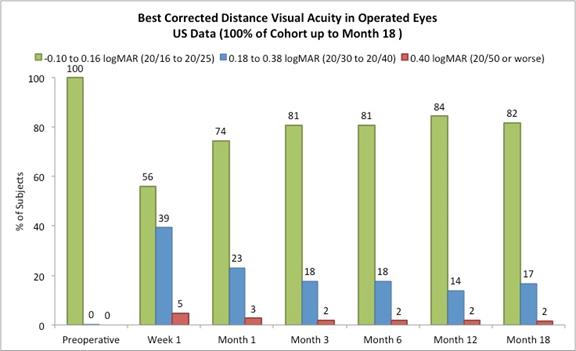
15
The following chart summarizes the binocular best corrected distance vision:
Figure E
Notwithstanding these results, we cannot assure you when or whether the Company will obtain pre-market approval, or what expenditures the Company will incur whether or not we obtain such approval, given the many significant risks associated with seeking such an approval from the FDA. Furthermore, certain adverse events have been reported as part of the on-going staged pivotal clinical trial. For a discussion of previously reported adverse events, please see the risk factors, including the risk factor titled “If concerns regarding side effects from presbyopia correction surgery generally, or our products specifically, develop, including as a result of third-party studies and publications, our business, results of operations and financial condition will be materially and adversely affected.”, in the Company’s annual report on Form 10-K for the year ended December 31, 2017.
The Company requires PMA approval in order to market its products in the United States.
To date, 100% of the subjects have passed through the 24-month post-operative visit. We are in the process of analyzing this data and preparing to submit to the FDA. We are targeting submission to the FDA of our final PMA module, containing 24-month data, in the second quarter of 2018. We do not expect to disclose any additional clinical data until we have received FDA approval or feedback on our fourth module PMA submission.
Clinical Development and Commercialization Targets
In December 2013, we received approval from the FDA to commence a staged pivotal clinical trial of our microlens in the United States. This clinical trial is a prospective, non-randomized, unmasked, multicenter clinical investigation. Beginning in May 2014, we enrolled a total of 75 subjects at six investigational sites in the United States. Beginning in June 2014, each of these subjects underwent insertion of our microlens in the subject’s non-dominant eye. Based on six-month data on 52 of these subjects, in January 2015, we submitted to the FDA an interim safety report as a supplement to our IDE. In February 2015, we received approval from the FDA to commence second stage enrollment in this trial. Through February 15, 2018, 346 subjects have undergone insertion of our microlens in the second stage of this study. All subjects will be followed for three years following implantation. Subjects from outside the United States are not enrolled in this study. The primary endpoint is UCVA-near at 24 months post- implantation, together with safety objectives such as a low rate of ocular adverse events, endothelial cell loss over time in the operated eye, and an assessment of BCVA-distance and contrast sensitivity in the operated eye (the visual ability, with distance vision correction in place, to see objects that may not be outlined clearly or that do not stand out from their background). Although our microlens is designed to be removable, our IDE requires any removal to be reported as an adverse event. We are pursuing a modular PMA submission strategy whereby we submitted to the FDA information regarding biocompatibility in the second quarter of 2016. We submitted to the FDA the second and third PMA modules in the first quarter of 2017, which contains information regarding preclinical testing, engineering and manufacturing. We are targeting submission to the FDA of our final PMA module, containing 24-month data, in the second quarter of 2018. We are targeting PMA approval of our microlens in the fourth quarter of 2018. We are also targeting submission to the FDA of
16
a final report with 36-month data on all available subjects in the fourth quarter of 2018. These milestones could be delayed by further interactions with the FDA or by a variety of other factors, including the final design of the study that is approved by the FDA, and are subject to risks and uncertainties. There can be no assurance that the FDA will grant our PMA approval or, if granted, that it will be granted in accordance with our anticipated time schedule. In addition, the FDA may require us to conduct post-approval studies as a condition of approval.
Outside of the United States, we plan to focus our commercialization and ongoing clinical trials in Germany and South Korea.
Strategy
Our goal is to become the leading provider of corneal lens implants for patients with presbyopia, which includes obtaining regulatory approval of the Flexivue Microlens from the FDA and, through our European CE Mark, selective commercialization and ongoing clinical trials in Germany and South Korea.
U.S. Staged Pivotal Clinical Trial
Gaining approval to market our products in the United States is a critical element in our strategy. In order to obtain such approval, we must obtain a PMA from the FDA. We cannot assure you when or whether we will obtain such an approval, or what expenditures we will incur whether or not we obtain such approval, given the many significant risks associated with seeking such an approval from the FDA.
International Commercialization
By affixing the Conformité Européene mark, or European Union CE Mark, to the products, we are authorized to market the Flexivue microlens throughout the EEA and, through mutual recognition agreements, in Switzerland (certain EEA countries also require additional in-country registration).
On December 10, 2017, our board of directors approved the re-ordering of the Company’s operational priorities, focusing its resources on FDA approval of its microlens and ongoing clinical trials and commercialization efforts in Germany and South Korea. These actions reduce the pre-FDA approval marketing, manufacturing and engineering expenses associated with the post-FDA approval U.S. commercial launch of our microlens.
Sales & Marketing
We intend to utilize a direct selling structure unless a country requires us to sell through a distributor, agent or we determine that a distributor/agent will offer us a more effective path to commercialization. We have a focused team consisting of our European commercial vice president, our clinical services director, clinical application specialists and surgical trainers. Our microlens and the procedure to implant our microlens are not currently reimbursed through private or governmental third-party payors in any country, nor do we anticipate that our microlens and the procedure to implant our microlens will be reimbursable through private or governmental third-party payors in the foreseeable future. Although the commercialization of our microlens depends on a prospective patient’s ability to cover the costs of our microlens and the implantation procedure and we believe that a substantial portion of presbyopes worldwide do not have the financial means to cover the costs of our microlens, we believe that a direct patient-pay model enables medical providers to avoid pricing pressure from private or governmental third-party payors. We do not have control over the prices that medical providers charge patients for our microlens and the implantation procedure.
Research and Development
We remain focused on advancing our technology and continuing to improve our microlens. We maintain an active internal research and development process, which also includes clinical activities and regulatory affairs. We expended $7.1 million and $5.5 million for research and development during the years ended December 31, 2017 and December 31, 2016, respectively.
Intellectual Property
Our commercial success depends, in part, on our ability to obtain and maintain proprietary protection for our products, technologies and other know-how, to operate without infringing the proprietary rights of others and to prevent others from infringing on our proprietary rights. We strive to protect our investment in the research, development, manufacturing and marketing of our products through the use of patents, trademarks, copyrights and trade secrets, as well as customary confidentiality and other contractual protections. We own intellectual property related to refractive powered inlays to treat presbyopia. We believe that our intellectual property portfolio, specifically the patents therein, also gives us the ability to expand into broader vision correction solutions if we so
17
decide. However, the extent to which our intellectual property will provide us with protection and enable us to commercialize our proprietary technology without interference from others is subject to numerous risks. See “Risk Factors—Risk Relating to Our Intellectual Property.”
Patents
We currently own six issued U.S. patents, and four pending U.S. patent applications, all of which we consider material to our business. Three of our existing patents relate to our microlens inserter and corresponding methods of use; these patents expire in 2030 or 2031. Two additional patents relate to the method and apparatus to package and transport our microlens, making it readily accessible; these patents expire in 2030. The other patent relates to a method that can be used by a laser to cut a pocket in the cornea and insert the microlens; this patent expires in 2028. Our four pending applications relate to a method and apparatus to package and transport our microlens, to an extension of the method for laser cutting a corneal pocket to insert our microlens, to the apparatus and method to use a preloaded inserter to insert our microlens, and a system for inserting our intracorneal lens.
Additionally, we have a total of 33 foreign patent applications, 27 of which are still pending in Australia, Brazil, Canada, China, Europe, Israel, Hong Kong, India, Japan, Korea and Russia. The foreign applications correspond to the content of the six issued U.S. patents. We currently own a patent in Canada, which corresponds to the U.S. patent which covers the method and apparatus to insert our microlens, and we have granted applications in Israel and China which correspond to the same U.S. patent. We also own patents in Japan and Australia, which correspond to the U.S. patent which covers the microlens inserter.
Our patents and patent applications may allow us to exclude others from practicing our proprietary inventions and may provide us with an opportunity to obtain royalties or cross-licenses of intellectual property from other manufacturers. Because we have limited knowledge of the research and development efforts and strategic plans of our competitors, we can only estimate the value of our patents and patent applications. Competitors may be able to design products and/or processes that avoid infringing our patent portfolio as it may exist from time to time. We may choose to abandon any of our issued or pending U.S. and/or foreign patents and/or patent applications that no longer match our commercialization and IP strategy.
Trademarks
Worldwide, we have several registered trademarks and pending trademark applications that we consider to be important to our business. The scope and duration of trademark protection varies widely throughout the world. In some countries, trademark protection continues only as long as the mark is used. Other countries require registration of trademarks and the payment of registration fees. Trademark registrations are generally for fixed but renewable terms.
Confidentiality Agreements
We protect our proprietary technology, in part, through confidentiality and nondisclosure agreements with employees, consultants and other parties. Our confidentiality agreements with employees and consultants generally contain standard provisions requiring those individuals to assign to Presbia, without additional consideration, inventions conceived or reduced to practice by them while employed or retained by Presbia, subject to customary exceptions.
Competition
The medical device industry in general, and the ophthalmic medical device market in particular, are highly competitive, subject to rapid technological change and significantly affected by new product introductions and market activities of other participants. Our currently marketed products are, and any future products we commercialize will be, subject to intense competition.
We expect to compete against companies that are developing corneal inlay surgical solutions for presbyopia, such as the KAMRA lens owned by Sightlife Surgical Inc., which acquired the product from AcuFocus, Inc. in March 2018. The KAMRA corneal inlay attempts to use small aperture optics to reduce distortion by eliminating peripheral light rays and limiting the width of diverging light rays. The KAMRA completed pivotal clinical trials in the U.S. and received FDA approval in April 2015. LensGen, Inc. is a newer company with little publicly available information regarding its intraocular lens which is being designed to harness fluidics and displacement to manipulate curvature to better capture light. ReVision Optics, Inc., which developed a corneal inlay solution and received FDA approval in June 2016, discontinued commercial activity in February 2018. Refocus Group, Inc. has developed the VisAbility Micro-Insert System, a procedure performed on both eyes and consisting of micro implants inserted just below the surface of the eye. Refocus Group, Inc. submitted its final module to the FDA for pre-market approval of the VisAbility Micro-Insert System in December 2017. The KAMRA corneal inlay, like the Presbia microlens, has marketing approval in certain jurisdictions outside the United States, including the EEA, and can be expected to compete with Presbia in such jurisdictions. However, we believe our microlens is less cosmetically conspicuous than the KAMRA and offers a wider range of optical power corrections.
We expect to compete against companies that offer alternative surgical treatment methodologies, including monovision, multifocal and accommodating approaches, and companies that promote reading glasses and/or contact lenses as approaches for responding to
18
presbyopia. At any time, our known competitors and other potential market entrants may develop new devices or treatment alternatives that may compete directly with our products. In addition, they may gain a market advantage by developing and patenting competitive products or processes earlier than we can or by obtaining regulatory approvals/clearances or market registrations more rapidly than we can.
Certain of our current and potential competitors may have significantly greater financial, technical, marketing and other resources than we do and may be able to devote greater resources to the development, regulatory approval, promotion, sale and support of their products. Our competitors may also have more extensive customer bases and broader customer relationships than we do, including relationships with our potential customers. In addition, many of these companies have longer operating histories and greater brand recognition than we do. Because of the size of the presbyopia market and the high growth profile of that market, we anticipate that companies will dedicate significant resources to developing competing products. We believe that the principal competitive factors in our market include:
|
|
• |
improved outcomes for patients and other product quality issues; |
|
|
• |
product innovation; |
|
|
• |
acceptance by ophthalmic surgeons; |
|
|
• |
ease of use and reliability; |
|
|
• |
regulatory status and speed to market; |
|
|
• |
product price and procedure price; and |
|
|
• |
reputation for technical leadership. |
We cannot assure you that we will be able to compete effectively against our competitors in regard to any one or all of these factors.
Manufacturing
Our microlens is manufactured using hydrophilic acrylic material that has been utilized in the lens manufacturing market for the last 20 years. This material is well known and has an established safety profile. High precision lathing machines are used to generate sub-micro level accuracy of convex/concave radii. Like other traditional IOL manufacturing processes, the manufacturing of the microlens is divided into a dry and a wet process.
We currently have certified ISO Class 8 cleanroom manufacturing capacity in Irvine, California. Items manufactured in this facility to date have been used solely for the current IDE. We believe the facility is scalable to meet future U.S. and out of the U.S., or “OUS”, demand with the Irvine facility having received all applicable regulatory registrations, approvals and certifications in December 2016, allowing us to manufacture CE marked products at this site. Our U.S. facility received regulatory approval from the State of California to manufacture our microlens for our U.S. staged pivotal trial, and during 2014 and 2015, this facility provided all of the required lenses that were used in the treatment phase for our ongoing IDE. Additionally, our U.S. facility has demonstrated conformity with the Essential Requirements under Annex I of Directive 93/42/EEC, referred herein as the EU Medical Device Directive, with respect to the manufacture of our microlens for sale in the EEA and FDA Quality System Regulation 21 CFR Part 820, current good manufacturing practices (“CGMPs”).
We believe that our current manufacturing arrangements are sufficient to support our foreseeable manufacturing needs. Product manufactured at the Irvine, California site includes lens manufacturing, final assembly, and packaging. Sterilization for the Irvine manufactured product is conducted at an FDA registered and ISO 13485 certified full service medical device contract manufacturer. Inventory of our microlens is housed at our facilities in Ireland as well as in Irvine, CA.
Sources and Availability of Raw Materials
We use a wide range of raw materials in the production of our products, with most of the raw materials and components being purchased from external suppliers. The hydrophilic acrylic material used to manufacture our microlens is supplied to us by a single supplier located in the United Kingdom. We would be required to obtain approval from the FDA in the event that we wished to use different material or similar material from a different supplier with respect to any products to be offered and sold in the United States. Although we do not have a guaranteed supply commitment from our sole supplier of such hydrophilic acrylic material, we believe that such supplier will be able to sufficiently meet our currently anticipated supply needs. Additionally, we do not currently have any long-term agreements in place for the supply of any other raw materials used in manufacturing of the microlens, however, all other materials are currently readily available from a number of suppliers, both in the United States and abroad.
19
Our medical device products are subject to extensive regulation by the FDA and various other U.S. federal, state and non-U.S. governmental authorities, such as the competent authorities of the countries of the EEA. Government regulation of medical devices is meant to assure their safety and effectiveness, and includes regulation of, among other things:
|
|
• |
design, development and manufacturing; |
|
|
• |
testing, labeling, content and language of instructions for use and storage; |
|
|
• |
clinical trials; |
|
|
• |
product safety; |
|
|
• |
marketing, sales and distribution; |
|
|
• |
regulatory clearances and approvals, including premarket clearance and approval; |
|
|
• |
conformity assessment procedures; |
|
|
• |
product traceability and record keeping procedures; |
|
|
• |
advertising and promotion; |
|
|
• |
product complaints, complaint reporting, recalls and field safety corrective actions; |
|
|
• |
post-market surveillance, including reporting of deaths or serious injuries and malfunctions that, if they were to recur, could lead to death or serious injury; |
|
|
• |
post-market studies; and |
|
|
• |
product import and export. |
To market and sell our products in any country, we must first seek and obtain regulatory approvals, certifications or registrations and comply with the laws and regulations of that country. These laws and regulations, including the requirements for approvals, certifications or registrations and the time required for regulatory review, vary from country to country. Obtaining and maintaining regulatory approvals, certifications and/or registrations are expensive, and we cannot be certain that we will receive regulatory approvals, certifications and/or registrations in any country for which we have yet to receive such approvals, certifications and/or registrations or that we will be able to maintain any regulatory approvals, certifications or registrations that we currently possess in any country. If we fail to obtain or maintain regulatory approvals, certifications or registrations in any country in which we currently market or plan to market our products or if we fail to comply with all applicable regulatory laws, rules and regulations, our ability to sell our products could be jeopardized and we could be subject to enforcement actions. See “Part I, Item 1a. Risk Factors—Risks Related to Regulatory Requirements” for a discussion of the risks and uncertainties that apply to Presbia in connection with government regulation of its products.
Regulatory Requirements in the United States
Under the U.S. Food, Drug and Cosmetic Act, or the FD&C Act, manufacturers of medical devices must comply with extensive regulation relating to the issues described above, including regulations governing the design, testing, manufacturing, packaging, quality, servicing and marketing of medical products. Our immediate focus is upon the steps that we must take before our products can be marketed and sold in the United States.
FDA’s Premarket Clearance and Approval Requirements
Unless an exemption applies, each medical device that is distributed commercially in the United States requires either prior 510(k) clearance or prior approval of a PMA application from the FDA. The FDA classifies medical devices into one of three classes. Devices deemed to pose low to moderate risk are placed in either Class I or II, which, absent an exemption, requires the manufacturer to submit to the FDA a premarket notification requesting permission for commercial distribution. This process is known as 510(k) clearance. Some low risk devices are exempt from this requirement. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a previously cleared 510(k) device, are placed in Class III, requiring approval of a PMA application. Our Class III products/devices require prior approval of a PMA application from the FDA. Both premarket clearance and PMA applications are subject to the payment of user fees, paid at the time of submission for FDA review. The FDA can also impose restrictions on the sale, distribution or use of devices at the time of their clearance or approval, or subsequent to marketing.
20
A PMA application must be submitted to the FDA if, as is the case with the microlens, the device cannot be cleared through the 510(k) process. The PMA application process is generally more costly and time consuming than the 510(k) process and requires proof of the safety and effectiveness of the device to the FDA’s regulations. Accordingly, a PMA application must be supported by extensive data including, but not limited to, technical information regarding device design and development, pre-clinical and clinical trials, data and labeling to support the FDA’s determination that the device is safe and effective for its intended use. After a PMA application is complete, the FDA will accept the application and begin an in-depth review of the submitted information. By statute, the FDA has 180 days to review the “accepted application,” although, generally, review of the application takes between one and three years, and may take significantly longer. During this review period, the FDA may request additional information and/or clarification of information already provided. Also, during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. In addition, the FDA will conduct a preapproval inspection of the manufacturing facility to ensure compliance with its Quality System Regulations, or QSRs, which impose elaborate design development, testing, control, documentation and other quality assurance procedures in the design and manufacturing process. The FDA may approve a PMA application with post-approval conditions intended to ensure the safety and effectiveness of the device, including, among other things, restrictions on labeling, promotion, sale and distribution and collection of long-term follow-up data from patients in the clinical study that supported approval. Failure to comply with the conditions of approval can result in materially adverse enforcement actions, including the loss or withdrawal of the approval. New PMA applications or PMA application supplements are required for significant modifications to the manufacturing process, as well as for modifications that affect the safety or effectiveness of the device, including, for example, certain types of modifications to the device’s indication for use, manufacturing process, labeling and design. PMA supplements often require submission of the same type of information as a PMA application, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA application, and may not require as extensive clinical data or the convening of an advisory panel.
Our microlens, as an implanted device, cannot be marketed and sold in the United States without PMA approval. We anticipate that other products that we may develop in the future, as well as modifications to our existing products, will also be associated with the implantation process and thus in all likelihood will be subject to PMA approval rather than 510(k) clearance.
IDE Applications
A clinical trial is almost always required to support a PMA application. In the United States, absent certain limited exceptions, human clinical trials intended to support product clearance or approval require an IDE application. Some types of studies deemed to present “non-significant risk” are deemed to have an approved IDE once certain requirements are addressed and IRB approval is obtained. If the device presents a “significant risk” to human health, as defined by the FDA, the sponsor must submit an IDE application to the FDA and obtain IDE approval prior to commencing the human clinical trials. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to evaluate the device in humans and that the testing protocol is scientifically sound. The IDE application must be approved in advance by the FDA for a specified number of subjects, unless the product is deemed a non-significant risk device and eligible for more abbreviated IDE requirements.
In December 2013, we received approval of our IDE from the FDA to begin a staged pivotal clinical trial of our microlens in the United States.
Clinical Trials
Clinical trials for a Class III device may begin once the IDE application is approved by the FDA and the responsible IRBs at the clinical trial sites. There can be no assurance that submission of an IDE will result in the ability to commence clinical trials. Additionally, after a trial begins, the FDA may place it on hold or terminate it if, among other reasons, it concludes that the clinical subjects are exposed to unacceptable health risks that outweigh the benefits of participation in the study. During a study, sponsors are required to comply with the FDA’s IDE requirements for investigator selection, trial monitoring, reporting and record-keeping and with prohibitions on promoting investigational devices or making safety or effectiveness claims for them. Sponsors are also responsible for the appropriate labeling and distribution of investigational devices.
We began enrollment for our U.S. staged pivotal clinical trial in May 2014 and began treating patients in June 2014. Initially, 75 subjects underwent insertion of our microlens at six investigational sites in the first stage of this study. Based on six-month data on 52 of these subjects, in January 2015, we submitted an interim safety report to the FDA along with a supplement to our IDE requesting approval to begin second stage enrollment. In February 2015, we received approval from the FDA to commence second stage enrollment in this trial. We were authorized to enroll up to an additional 337 subjects at up to nine additional investigational sites. The clinical trial that we have commenced for our microlens is expected to extend at least through 2018. We do not anticipate receiving the approval for our microlens before the fourth quarter of 2018.
21
Our clinical trials must be conducted in accordance with U.S. federal and state regulations concerning human subject protection, including informed consent and healthcare privacy. The investigators must obtain patient informed consents, rigorously follow the investigational plan and study protocol, control the disposition of investigational devices and comply with all reporting and record-keeping requirements.
In addition, the FDA’s grant of permission to proceed with clinical testing does not constitute a binding commitment that the FDA will consider our study design adequate to support PMA approval. In addition, there can be no assurance that the data that we generate during a clinical study will meet chosen safety and effectiveness endpoints or otherwise produce results that will lead the FDA to grant marketing approval.
Pervasive and Continuing FDA Regulation
After a device is placed on the market, regardless of its classification or premarket pathway, numerous regulatory requirements apply. These include, but are not limited to:
|
|
• |
establishment registration and device listings with the FDA, which helps facilitate FDA inspections and other regulatory action; |
|
|
• |
QSRs, which requires manufacturers, including third-party manufacturers, to follow stringent design, testing, process control, documentation and other quality assurance procedures during all aspects of the development and manufacturing process; |
|
|
• |
labeling control and advertising regulations, which prohibit the promotion of products for uncleared or unapproved, or off-label, uses or indications, and impose other restrictions on labeling; |
|
|
• |
approval or clearance of product modifications that could significantly affect safety or efficacy or that would constitute a major change in intended use; |
|
|
• |
medical device reporting regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; |
|
|
• |
corrections and removal reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health. In addition, the FDA may order a mandatory recall if there is a reasonable probability that the device would cause serious adverse health consequences or death; and |
|
|
• |
post-approval restrictions or conditions, including requirements to conduct post-market surveillance studies to establish continued safety data. |
The FDA has broad post-market and regulatory enforcement powers. We will be required to register with the FDA as a medical device manufacturer. As such, our manufacturing facilities will be subject to FDA inspections for compliance with QSRs. These regulations will require that we manufacture our products and maintain our documents in a prescribed manner with respect to design, manufacturing, testing and quality control activities. As a medical device manufacturer, we will also be required to comply with FDA requirements regarding the reporting of adverse events associated with the use of our medical devices, as well as product malfunctions that would likely cause or contribute to death or serious injury if the malfunction were to recur. FDA regulations also govern product labeling and prohibit a manufacturer from marketing a medical device for unapproved applications. The FDA may conduct unannounced inspections to determine compliance with the QSR and other regulations, and these inspections may include the manufacturing facilities of subcontractors. Failure by us or our suppliers to comply with applicable regulatory requirements can result in enforcement actions by the FDA or other regulatory authorities, which may result in sanctions and related consequences including, but not limited to:
|
|
• |
untitled letters or warning letters; |
|
|
• |
fines, injunctions, consent decrees and civil penalties; |
|
|
• |
recall, detention or seizure of our products; |
|
|
• |
operating restrictions, partial suspension or total shutdown of production; |
|
|
• |
refusal of or delay in granting our requests for premarket approval or clearances of new products or modified products; |
|
|
• |
once we have received premarket approvals or clearances, withdrawing those approvals or clearances that are already granted; |
|
|
• |
refusal to grant export approval for our products; |
22
|
|
• |
unanticipated expenditures to address or defend such actions. |
Regulatory Requirements Outside of the United States
Sales of medical devices outside the United States are subject to non-U.S. regulatory requirements that vary widely from country to country. These laws and regulations range from simple product registration requirements in some countries to complex clearance and production controls in others. As a result, the processes and time periods required to obtain foreign marketing approval may be longer or shorter than those necessary to obtain FDA market approval. These differences may affect the efficiency and timeliness of international market introduction of our products.
In order to be placed on the market within the EEA, medical devices must meet the essential requirements set out in the relevant medical device legislation. The principal legislation regulating general medical devices in the EEA is Directive 93/42/EEC, referred to herein as the EU Medical Devices Directive. Where a medical device meets the essential requirements set out in the EU Medical Devices Directive and complies with the appropriate conformity assessment procedure, based on the classification of the medical device, a declaration or certificate of conformity will issue and a CE Mark may then be affixed to the product. Once a CE Mark has been affixed to the medical device, it may then be placed on the market in any country within the EEA and, through mutual recognition agreements, in Switzerland (subject to certain localized registration and language requirements).
In February 2010, we received a CE certificate of conformity from our notified body (a private organization designated by the competent authorities of the EEA to conduct conformity assessments and verify the conformity of manufacturers and their medical devices with Essential Requirements laid down in Annex I of the EU Medical Devices Directive) for our microlens allowing the CE mark to be affixed to our microlens, permitting our microlens to be placed on the market within any state in the EEA and, through mutual recognition agreements, Switzerland (subject to certain localized registration and language requirements). In May 2013, we issued a declaration of conformity for our microlens inserter allowing the CE Mark to be affixed to our microlens inserter. We have also obtained an ISO 13485 quality system certification, which confirms that our medical device manufacturing quality management system is compliant with globally recognized standards set forth by the International Organization for Standardization. We are required to keep up-to-date and remain compliant with the most recently issued standards. In order to maintain our certificate of conformity and CE Mark, we must continue to comply with the EU Medical Devices Directive and pass annual facilities audit inspections by an inspection agency of the EEA to ISO 13485 standards. In addition, a notified body or other competent authority in an EEA country may perform post-marketing audits on our products and premises from time to time. Failure to comply with such requests in a timely manner, and any adverse findings in any such audit, could result in the withdrawal of our certificate of conformity and our CE Mark, and the recall or withdrawal of our products from the EEA market. Each certificate of conformity may be valid for a maximum of five years but would typically be valid for three years. Our existing certificate of conformity for our microlens is valid until March 2019, and our CE Mark is valid until June 2018. At the end of each period of validity, we are required to apply to the notified body for a renewal of our certificate of conformity. There may be delays in the renewal of our certificate of conformity and the notified body may require modifications to our products or to the related technical files before it agrees to issue a new certificate of conformity.
On May 5, 2017, the European Commission officially published the Medical Device Regulation (MDR 2017/745) and came into force on May 25, 2017. Manufacturers of currently approved medical devices will have a transition time of three years until May 26th 2020 to meet the requirements of the MDR. For some manufacturers the new MDR provides an additional time after the date of application allowing them to place new products for a maximum of 4 more years on the market. Additional requirements will apply for this extended transition period.

In addition, we have obtained marketing authorization for our microlens and microlens inserter in certain countries outside of the EEA, including certain countries in which our microlens is currently commercially available. We are subject to the regulatory laws and regulations of each such country in order to maintain our marketing authorization. In addition, we will be subject to the regulatory
23
laws and regulations of any additional country in which we obtain marketing approval to maintain such approval. These regulatory laws are complex and vary from country to country. Failure to comply with applicable laws and regulations could jeopardize our ability to sell our products and result in a variety of enforcement actions, all of which would negatively impact our business, results of operations and financial condition.
Corruption Laws
The U.S. Foreign Corrupt Practices Act and similar foreign anti-corruption laws generally prohibit companies and their intermediaries from making improper payments or providing anything of value to improperly influence foreign government officials for the purpose of obtaining or retaining business, or obtaining an unfair advantage. In recent years, there has been a substantial increase in the global enforcement of anti-corruption laws. Our ongoing non-U.S. operations and our expansion into additional countries outside the United States, including in developing countries, could increase the risk of such violations. Violations of these laws may result in severe criminal or civil sanctions, could disrupt our business, and could adversely affect our reputation, business and results of operations or financial condition.
Environmental Matters
Our activities currently require the controlled use of potentially harmful biological materials and hazardous materials and chemicals. We are subject to U.S. federal, state and local and non-U.S. environmental and pollution control laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. We believe that our operations comply in all material respects with applicable environmental laws and regulations in each country where we conduct business or have operations. We do not expect compliance with these laws to affect materially our capital expenditures, earnings or competitive position. We have no plans to invest in material capital expenditures for environmental control facilities for the remainder of our current fiscal year or for the next fiscal year. We are not aware of any pending actions, litigation or significant financial obligations arising from current or past environmental practices that are likely to have a material adverse impact on our financial position. However, environmental problems relating to our properties could develop in the future, and such problems could require significant expenditures. In addition, we cannot predict changes in environmental legislation or regulations that may be adopted or enacted in the future and that may adversely affect us.
Employees
As of December 31, 2017, we had 24 employees, one of whom holds a doctorate in optical physics and physiological optics degree, and three of whom hold other advanced degrees. We have no collective bargaining agreements with our employees and we have not experienced any work stoppages. We believe that our relations with our employees are good.
Facilities
Our operations are currently conducted at three leased facilities. We lease two locations with an aggregate of approximately 17,500 square feet of office, laboratory and manufacturing space in Irvine, California, of which one facility consisting of approximately 8,500 square feet is vacant and is available for subtenant lease. We lease 610 square feet of office and warehouse space in Dublin, Ireland. Our corporate headquarters is currently located at our Dublin location.
We believe that our existing facilities are adequate for our current needs. When our leases expire, we may seek to renew our leases or look for additional or alternate space for our operations. We believe that suitable additional or alternative space will be available in the future on commercially reasonable terms.
Corporate History and Information
In February 2015, Presbia PLC consummated its initial public offering of ordinary shares. Prior to our initial public offering, we effected a series of reorganization transactions described below.
Presbia Holdings was organized in the Cayman Islands in 2007 as an exempted company with limited liability. In 2009, Presbia Holdings acquired Visitome, Inc., a California corporation and the developer of our corneal inlay technology.
In October 2013, we completed a restructuring which involved the establishment of our interim holding company, Presbia Ireland, Limited, that directly or indirectly owns 100% of our business, assets and subsidiaries. Presbia Ireland, Limited is organized under the laws of Ireland as a private limited company. At the time of the restructuring, Presbia Ireland, Limited was wholly-owned by Presbia Holdings and certain intercompany debt was owed to Presbia Holdings by certain of its other subsidiaries. As part of the restructuring, approximately $12.2 million of such outstanding intercompany debt owed to Presbia Holdings was converted to equity of such subsidiaries. We refer to this transaction as the 2013 Restructuring.
24
In November 2014, Presbia Holdings converted additional indebtedness owed to Presbia Holdings by certain subsidiaries of Presbia Ireland, Limited at that time to equity. In this transaction, approximately $23.5 million of outstanding intercompany debt owed to Presbia Holdings was converted to equity of such subsidiaries. We refer to this transaction as the 2014 Debt Conversion.
In January 2015, Presbia Holdings converted all the remaining indebtedness owed by a subsidiary of Presbia Ireland, Limited at that time to equity. In this transaction, approximately $1.6 million of outstanding intercompany debt owed to Presbia Holdings was converted to equity of such subsidiary. We refer to this transaction as the 2015 Debt Conversion. In addition, immediately following the 2015 Debt Conversion, Presbia Holdings contributed all the share capital in issue in Presbia Ireland, Limited to Presbia PLC, an Irish incorporated public limited company formed in February 2014 for the purpose of consummating our initial public offering, in exchange for 9,166,667 ordinary shares of Presbia PLC. We refer to this transaction as the 2015 Capital Contribution. Presbia PLC previously issued 40,000 ordinary shares to Presbia Holdings upon its formation, in order to satisfy statutory requirements for the incorporation of all Irish public limited companies, which were re-designated as deferred shares under our memorandum and articles of association prior to the consummation of our initial public offering. We refer to the 2014 Debt Conversion, the 2015 Debt Conversion and the 2015 Capital Contribution, collectively, as the 2014-2015 Restructuring.
We refer to the 2013 Restructuring, the formation and initial capitalization of Presbia PLC, and the 2014-2015 Restructuring, collectively, as the Reorganization Transactions.
In August 2015, Presbia Holdings distributed the 9,166,667 ordinary shares of Presbia PLC, referred to herein as the 2015 Capital Contribution and an additional 500,000 ordinary shares acquired from the initial public offering for an aggregate of 9,666,667 ordinary shares, to its ordinary shareholders. Presbia Holdings was liquidated in November 2015.
Our corporate structure is set forth below.
Our principal executive offices are located at Suite 7, Sandyford Office Centre, 17 Corrig Road, Sandyford, Dublin 18 Ireland, and our telephone number is +353 (1) 551 1487.
Our website address is http://www.presbia.com. The information in, or that can be accessed through, our website is not part of this Annual Report on Form 10-K. Our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K and amendments to those reports are available, free of charge, on or through our website as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities Exchange Commission, or the SEC. The public may read and copy
25
any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, D.C. 20549. Information on the operation of the Public Reference Room can be obtained by calling 1-800-SEC-0330. The SEC maintains an Internet site that contains reports, proxy and information statements and other information regarding our filings at www.sec.gov.
We are providing the following cautionary discussion of risk factors, uncertainties and assumptions that we believe are relevant to our business. These are factors that, individually or in the aggregate, we think could cause our actual results to differ materially from expected and historical results and our forward-looking statements. We note these factors for investors as permitted by Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act, and Section 27A of the Securities Act of 1933, as amended, or the Securities Act. You should understand that it is not possible to predict or identify all such factors. Consequently, you should not consider this section to be a complete discussion of all potential risks or uncertainties that may substantially impact our business. Moreover, we operate in a competitive and rapidly changing environment. New factors emerge from time to time and it is not possible to predict the impact of all of these factors on our business, financial condition or results of operations.
Risks Related to Our Business
We anticipate that we will continue to incur significant losses for the foreseeable future and, if we are unable to achieve and sustain profitability, the market value of our ordinary shares will likely decline.
We are an ophthalmic device company with a limited operating history. We do not possess the regulatory approvals necessary to market our products in the United States, and we continue to incur significant research and development, sales and marketing and general and administrative expenses related to our operations. We are not profitable and have incurred losses in each year since our formation. Our net losses for the years ended December 31, 2017 and 2016 were $17.0 million and $15.8 million, respectively. As of December 31, 2017, we had an accumulated deficit of $88.3 million.
We expect to continue to incur significant losses for the foreseeable future. We expect that these losses and our cash needs will increase in the near term as we continue to conduct our staged pivotal clinical trial in the United States, seek marketing approval in other countries, and commercialize our products in those non-U.S. markets where we are permitted to sell our microlens. We may never achieve profitability, and unless and until we do, we will need to continue to raise capital.
We expect to finance future cash needs through public or private equity offerings, debt financings or corporate collaborations and licensing arrangements. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available, we may be required to delay, reduce the scope of, or curtail, our operations. To the extent that we raise additional funds by issuing equity securities, our shareholders will experience dilution, and debt financing, if available, may involve restrictive covenants. We may not be able to enter into collaborations that we seek to establish. To the extent that we raise additional funds through collaborations and licensing arrangements, it may be necessary to relinquish some rights to our technologies or our product candidates or grant licenses on terms that may not be favorable to us. We may seek to access the public or private capital markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time.
We expect to incur substantial expenses in our pursuit of regulatory approval in the United States and can provide no assurances that we will obtain the necessary approvals from the FDA to market our products in the United States.
The United States is a key market for commercialization of our microlens. Before we can market our products in the United States, we must conduct and successfully complete extensive clinical trials and then receive PMA from the FDA. The earliest that we can reasonably expect to receive a PMA for our microlens is in the fourth quarter of 2018, and it is possible that none of our existing products or any products we may seek to develop in the future will ever obtain a PMA. Furthermore, even if we were to obtain a PMA, neither approval by the FDA nor our existing CE Mark ensures approval by regulatory authorities in other countries or jurisdictions that we are targeting for commercialization of our microlens, and approval by one regulatory authority does not ensure approval by regulatory authorities in other countries or by the FDA.
The time required to obtain approval by the FDA and comparable non-U.S. regulatory authorities is unpredictable and depends upon numerous factors, including the substantial discretion of such regulatory authorities. In addition, approval policies, regulations or the type and amount of preclinical and clinical data necessary to gain approval may change during the course of a product’s life-cycle and may vary among jurisdictions. We will be required to undertake and complete certain studies to generate data required to support submissions to the FDA and certain other regulatory authorities, which studies may require additional capital and time. If we do not receive or maintain regulatory approvals for our products in the United States and other jurisdictions that we target for commercialization of our products, we will not be able to successfully commercialize our products, which would substantially impair our ability to generate revenues and materially harm our business, results of operations and financial condition.
26
Based on our current plan, we believe we will need additional capital to support our operations.
Based on our current business plan, we believe that our cash and cash equivalents at December 31, 2017 will not be sufficient to meet our anticipated cash requirements during the twelve-month period subsequent to the issuance of the financial statements included in this Annual Report on Form 10-K. Our current commercialization and clinical trial strategy is targeted to countries where we believe we can establish the market for our microlens. This commercialization and clinical trial strategy will undergo continual prioritization and in the future we may adjust our commercialization efforts to preserve our existing cash or realize better results than anticipated which could have a positive impact on cash. Our U.S. pivotal clinical trial and planned FDA approval is our highest priority. We need to raise additional capital to fund our operations. We may raise additional capital through equity offerings, debt financings, collaborations and/or licensing arrangements. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available, we may be required to delay, reduce the scope of, or curtail, our operations. To the extent that we raise additional funds by issuing equity securities, our shareholders will experience dilution, and debt financing or other preferred equity instruments, if available, may involve restrictive covenants.
There is substantial doubt about our ability to continue as a going concern.
Our recurring losses from operations raise substantial doubt about our ability to continue as a going concern, and as a result, our independent registered public accounting firm included an explanatory paragraph in its report on our financial statements as of and for the year ended December 31, 2017 with respect to this uncertainty. This going concern uncertainty, and any future going concern uncertainty, could materially limit our ability to raise additional capital. We have incurred significant losses since our inception and have never been profitable, and it is possible we will never achieve profitability. We are an ophthalmic device company with a limited operating history. We do not possess the regulatory approvals necessary to market our products in the United States, and we continue to incur significant research and development, sales and marketing and general and administrative expenses related to our operations. As a result, we have not generated any significant revenue from operations since inception, and we have incurred substantial net losses to date. Our net losses for the years ended December 31, 2017 and 2016 were $17.0 million and $15.8 million, respectively. As of December 31, 2017, we had an accumulated deficit of $88.3 million. Meaningful revenues will likely not be available until, and unless, our microlens is approved by the FDA and successfully commercialized. The perception that we may not be able to continue as a going concern may cause potential partners or investors to choose not to deal with us due to concerns about our ability to meet our contractual and financial obligations.
Our microlens is currently our sole product and we are highly dependent on the successful marketing and sales of this product. There is no assurance that we will be able to develop any additional products.
Our microlens is currently our sole product focus. We may fail to successfully commercialize our product. Successfully commercializing medical devices such as our microlens is a complex and uncertain process, dependent on the efforts of management, distributors, outside consultants and general economic conditions, among other factors. Any factors that adversely impact the commercialization of our microlens including, but not limited to, the delay or denial of regulatory approvals that we seek, competition or acceptance in the marketplace, will have a negative impact on our business, results of operations and financial condition. We cannot assure you that we will be successful in developing or commercializing any potential enhancements to our microlens or any other products. Our inability to successfully commercialize our current products and/or successfully develop and commercialize additional products or any enhancements to our products which we may develop would have a material adverse effect on our business, results of operations and financial condition.
Our U.S. staged pivotal clinical trial may be delayed, suspended or terminated, which could delay or prohibit us from obtaining regulatory approvals or make obtaining such regulatory approvals more costly.
In February 2015, we received approval from the FDA to commence second stage enrollment in our U.S. staged pivotal clinical trial. By September 2015 we had completed the second stage enrollment in our U.S. staged pivotal clinical trial. However, delays in the completion of clinical testing could significantly affect our product development costs. The completion of clinical trials can be delayed for a number of reasons, including delays related to:
|
|
• |
unexpected adverse effects experienced by patients in a clinical trial; and |
|
|
• |
retaining patients who have initiated a clinical trial, but may withdraw due to treatment protocol, adverse effects from the therapy, lack of efficacy from the treatment or personal issues or who may not return for a sufficient number of post-operative visits. Clinical trials may also be delayed, suspended or terminated as a result of ambiguous or negative interim results, or results that are inconsistent with earlier results. In addition, a clinical trial may be suspended or terminated by us, the FDA or other regulatory authorities due to a number of factors, including: |
27
|
|
• |
failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
|
|
• |
inspection of the clinical trial operations, trial sites or manufacturing sites by the FDA or other regulatory authorities, resulting in the imposition of a clinical hold; |
|
|
• |
unforeseen safety issues or any determination that a clinical trial presents unacceptable health risks; and |
|
|
• |
lack of adequate funding to continue the clinical trial, including the incurrence of unforeseen costs due to enrollment delays, requirements to conduct additional clinical trials or increased expenses associated with the services of our contract research organizations, or “CROs”, and other third parties. |
Our product development costs will increase if we experience delays in testing or if we need to perform more or larger clinical trials than planned. Additionally, changes in regulatory requirements and policies may occur in any jurisdiction and we may need to amend clinical trial protocols to reflect these changes. Amendments may require us to resubmit our clinical trial protocols to independent ethical committees, known as IRBs, for reexamination, which may impact the costs, timing or successful completion of a clinical trial. In addition, IRBs or other regulatory authorities may order the temporary discontinuation or termination of our clinical trials at any time if they believe that the clinical trials are not being conducted in accordance with applicable law or regulatory requirements, including if they present an unacceptable safety risk to patients. If we experience delays in completion of, or if we, the FDA or other regulatory authorities, an IRB or other reviewing entities, or any of our clinical trial sites suspend or terminate our U.S. staged pivotal trial, the commercial prospects for our products may be harmed and our ability to generate revenues will be delayed. In addition, many of the factors that cause, or lead to, termination or suspension of, or a delay in the commencement or completion of, clinical trials may also ultimately lead to the denial of regulatory approval of a product. Also, if our U.S. staged pivotal trial is delayed, our competitors may be able to bring products to market before we do or further entrench their products in the market, and the commercial viability of our product candidates could be significantly reduced. For example, the KAMRA received FDA approval in April 2015 and Revision Optics, Inc. received FDA approval in June 2016 for its corneal inlay. In February 2018, Revision Optics ceased business operations. In March 2018, AcuFocus sold its AcuFocus KAMRA corneal inlay designed for surgically treating presbyopia to SightLife Surgical, a private company, for an undisclosed amount.
If concerns regarding side effects from surgical procedures to improve near vision generally, or our products specifically, develop, including as a result of third-party studies and publications, our business, results of operations and financial condition will be materially and adversely affected.
Concerns about potential side effects and long-term results may negatively impact market acceptance of surgical procedures to improve near vision generally or our products specifically, result in potential liability for us and prevent us from growing our business. Any undesirable side effects that may be discovered in our clinical trials or evaluations or in any third party studies or evaluations as part of our post marketing vigilance obligations could delay or prevent regulatory approval, including FDA approval, could prevent us from maintaining our existing regulatory approvals or our CE mark, or limit marketability of our products.
In early 2012, we completed a 12-month multicenter, post-market evaluation in Italy and Greece of our microlens in presbyopic patients between the ages of 45 and 60. The 12-month data for 70 patients indicated certain post-operative adverse events, including: one removal of the microlens, as a result of a patient’s complaints of significant halos and glare when driving at night; one case of transient light sensitivity syndrome (an abnormal occurrence of photosensitivity associated with the femtosecond laser); one case of epithelial ingrowth (an abnormal growth of corneal epithelium in an area where it does not belong, associated with the femtosecond laser); and four cases of transient stromal haze (the activation of inflammatory cells in connection with surgery). In addition, certain patients experienced a slight loss in uncorrected visual acuity-distance, or UCVA-distance, which is distance vision in the operated eye without prescription enhancement. For further information regarding this post-market evaluation, see “Part I, Item 1. Business—Our Solution—Evaluation Conducted Outside of the United States.”
In addition, our microlens has been the subject of certain third party studies that have been conducted to assess the effectiveness and safety of our microlens. We did not commission these studies or design, review or oversee the implementation of their protocols, and we have limited information with respect to these studies. These studies have reported certain adverse effects relating to the safety and effectiveness of our microlens and microlens inserter. With respect to the below referenced third-party study conducted in Japan, we paid the annual fees of the IRB which reviews the study’s protocol. The types of adverse events observed in these third-party studies include:
|
|
• |
Italy - A third-party study conducted in Italy in 81 patients in 2011 indicated the following adverse events: removal of our microlens within 12 months of implantation due to patient-reported reduction in distance vision and the presence of halos. |
|
|
• |
Japan - A third-party study conducted in Japan in 38 patients from 2012 to April 2014 reported the following adverse events: inlay defect due epithelial ingrowth microlens removal due to halos and glare; meibomianitis; moderate foreign debris; keratic precipitates; superficial punctate keratitis; vertical gas bubbles surgeon performing the testing initially reported such foreign debris to be metallic. |
28
|
|
• |
Russia - Third-party study was conducted in 2012 in Russia in ten patients. This study reported the following adverse events: a decrease in UCVA-distance and BCVA-distance due to night glare; removal of our microlens and minimal debris. |
We are aware of certain adverse events observed in the commercial setting. Between 2015 and 2017, three adverse events were reported. In a commercial setting in Ireland, two cases of microbial infection were reported. Neither case resulted in explantation of the microlens with the issue resolving with topical medications. After an extensive investigation conducted by Presbia, it was determined that the two incidents were not associated with the microlens and the HPRA (Health Products Regulatory Authority), the Irish competent authority, closed the two cases. A third incident was reported involving suspected corneal ectasia, which resolved with explantation of the microlens. If our microlens or microlens inserter or any other equipment supplied by us are ultimately determined to produce undesirable side effects, including posing a health risk through the deposit of foreign particles in a patient’s eye, such determination could result in the suspension of our U.S. staged pivotal clinical trial, delay, make it more difficult and expensive for us to receive and/or prevent us from receiving, or prevent us from maintaining, regulatory approvals, including FDA approval or our CE mark, limit marketability of our products and subject us to lawsuits or claims.
Adverse findings in post-marketing vigilance or regulatory audits could subject us to suspension or withdrawal of our certificates of conformity, mandatory product recalls and significant legal liability, which would materially and adversely affect our business, results of operations and financial condition.
In February 2010, we received a CE certificate of conformity from our notified body (a private organization designated by the competent authorities of the EEA, to conduct conformity assessments and verify the conformity of manufacturers and their medical devices with the Essential Requirements of the EU Medical Devices Directive ) for our microlens allowing the CE Mark to be affixed to our microlens, permitting our microlens to be placed on the market within any state in the EEA and, through mutual recognition agreements, Switzerland (subject to certain localized registration and language requirements). Manufacturers of medical devices in the EEA are required to implement post-marketing vigilance procedures with respect to their CE Marked medical devices in accordance with the rules governing the Medical Device Vigilance System provided for in European Commission’s MEDDEV 12.12/1. Such post-marketing vigilance procedures include surveillance of patient and user complaints and alleged incidents associated with the use of CE Marked medical devices. MEDDEV 12.12/1 defines incidents as any malfunction or deterioration in the characteristics and/or performance of a device, as well as any inadequacy in the labeling or the instructions for use which, directly or indirectly, might lead to or might have led to the death of a patient, or user or of other persons or to a serious deterioration in their state of health. When a medical device is suspected to be a contributory cause of an incident that led or might have led to death of or the serious deterioration of the health of a patient, or user or of other person, its manufacturer or authorized representative in the European Union must report it to the competent authority in whose territory the incident occurred. Incidents must be reported as soon as possible, and in some cases immediately, after the manufacturer becomes aware of the incident. In addition to reporting the incident, the manufacturer must investigate the incident and take any corrective action required, including Field Safety Corrective Actions (“FSCAs”). The manufacturer’s investigation is monitored by the competent authority, which may intervene, or initiate an independent investigation if considered appropriate. The required corrective action depends on the seriousness of the incident, and varies from the issuance of advisory notices to the implementation of product recalls. FSCAs must be reported by the manufacturer or its authorized representative to the competent authorities of the countries affected by the FSCA. Customers and/or the end users of the medical device must also be notified. Incidents not requiring notification to the competent authorities must be documented, reviewed, investigated and analyzed on a regular basis by the manufacturer to determine whether trending conclusions can be made concerning the safety or performance of the medical device and whether actions must be taken in relation to the continued marketing of medical devices currently on the market. We expect to incur ongoing costs to comply with these post-market vigilance obligations in EEA markets for so long as we continue to market and sell products in those markets. Moreover, any patient or user complaints and/or adverse events discovered during such post-market vigilance could subject us to suspension or withdrawal of our CE certificates of conformity or CE mark, mandatory product recalls and significant legal liability, which would materially and adversely affect our business, results of operation and financial condition. In addition, a notified body or competent authority in an EEA country may perform post-marketing audits on our products and premises from time to time. Failure to comply with such requests in a timely manner, and any adverse findings in any such audit, could subject us to suspension or withdrawal of our CE certificates of conformity or CE mark, mandatory product recalls and significant legal liability, which would materially and adversely affect our business, results of operations and financial condition.
29
We were previously subject to certain legal proceedings relating to the ownership of certain assets, including intellectual property. As demonstrated by such proceedings, future claims regarding intellectual property may be costly and time consuming to defend and future claims may delay or prevent the development and commercialization of our products or place our patent portfolio and other proprietary rights at risk.
In June 2008, Biovision AG, a Swiss company, was liquidated in bankruptcy in Switzerland. Vladimir Feingold, formerly one of our directors, a consultant to us, and our former Chief Technology Officer, was a minority owner of Biovision AG and served as its President and Chief Executive Officer. During the bankruptcy auction in Switzerland, Thomke Invest AG, or Thomke, purchased certain assets of Biovision AG with the stated intention of transferring those assets to Biovision Technology AG, which became Neoptics AG. We subsequently acquired assets from Neoptics AG in the third quarter of 2016. Disputes arose as to the scope of the purchased assets, the propriety of the Swiss auction, and what persons or entities had superior rights with respect to certain property, data, know-how, processes, and technology relating to a specialized surgically implanted lens to treat presbyopia, which we refer to collectively as the Disputed Assets.
Two competing lawsuits were filed in July 2008. The first lawsuit, which we refer to as the Thomke Action, was filed in the Superior Court of the State of California, County of Orange, by Thomke against Mr. Feingold, Biovision AG, our Visitome, Inc. subsidiary, Zohar Loshitzer, one of our directors, Orchard Capital Corporation, which is owned by Richard Ressler (one of our directors and our controlling shareholder), and Swiss Investment Corporation. In its complaint, Thomke alleged, among other things, wrongful possession of personal property and conversion of the Disputed Assets. The second lawsuit, which we refer to as the Visitome Action, was filed on the same day in the same court by our Visitome, Inc. subsidiary seeking a declaration of rights with respect to the Disputed Assets. In November 2012, Swiss Investment Corporation and Mr. Feingold filed a lawsuit in Switzerland to invalidate certain orders issued by the Swiss Insolvency Office with respect to the Disputed Assets.
The Thomke Action and the Visitome Action were eventually consolidated. On December 12, 2012, the parties to the various lawsuits and certain of their affiliates entered into a settlement agreement to settle the three lawsuits. The parties to the settlement agreement included certain of our affiliates, including Mr. Feingold, Mr. Loshitzer, Visitome, Inc., PresbiBio, LLC, Presbia Holdings and Orchard Capital Corporation. We subsequently acquired assets from Neoptics AG in the third quarter of 2016.
We cannot guarantee that we will not be subject to future claims regarding intellectual property. If successful, any such claims could place our patent portfolio and other proprietary rights at risk, which would have a material and adverse effect on our business, results of operations and financial condition. Even if such claims are not successful, they could be costly and time consuming to defend and they could delay or prevent the development and commercialization of our products.
We have a limited operating history and may face difficulties encountered by early stage companies in new and rapidly evolving markets.
We concluded our initial public offering (“IPO”) in January 2015 and have a limited operating history. In assessing our future prospects, you should consider the risks and difficulties frequently encountered by early stage companies in new and rapidly evolving markets, particularly companies engaged in the development and sales of medical devices. These risks include our ability to:
|
|
• |
manage expectations during the lengthy process of obtaining PMA approval from the FDA; |
|
|
• |
establish and increase awareness of our brand and strengthen customer loyalty; |
|
|
• |
grow our business in targeted markets outside of the United States while awaiting FDA approval; |
|
|
• |
implement and successfully execute our commercialization strategy; |
|
|
• |
respond effectively to competitive pressures and developments; |
|
|
• |
continue to develop and enhance our products in development; |
|
|
• |
obtain and publish sufficient clinical data to reduce the uncertainty surrounding clinical acceptance of our microlens; |
|
|
• |
obtain regulatory approval to commercialize our products and, when and if approved, enhance those products; |
|
|
• |
maintain compliance with all applicable regulatory statutes and regulations; |
|
|
• |
expand our global presence; |
|
|
• |
perform clinical research and trials on our existing products and future product candidates; |
|
|
• |
attract, retain and motivate qualified personnel; and |
|
|
• |
raise additional capital, on favorable or acceptable terms, if at all. |
30
As a result of these or other risks, our business strategy might not be successful.
We are engaged in an intensely competitive business with competitors that may enjoy significant competitive advantages over us and if we are unable to compete successfully against our existing or potential competitors, our sales and operating results may be negatively affected and we may not grow.
The market for surgical presbyopia correction is intensely competitive, both in and outside of the U.S., and competition may increase. In addition to our company, there are several other companies who have developed or are currently developing competing corneal inlay or other micro implant solutions—SightLife Surgical, Inc., LensGen Inc. and Refocus Group, Inc.. The KAMRA, purchased by SightLife Surgical Inc. from AcuFocus in March 2018, completed pivotal clinical trials in the U.S. and received FDA approval in April 2015. Refocus Group, Inc. submitted its final module to the FDA for pre-market approval of the VisAbility Micro-Insert System in December 2017. Other non-corneal inlay procedures also offer solutions to presbyopia, including: monovision approaches (whereby one eye, typically the dominant eye, is corrected for distance vision and the other eye is corrected for near vision using glasses, contact lenses or surgical procedures); multifocal approaches (whereby both a distance focus and a near focus are provided at the same time in each eye using glasses, contact lenses, surgically implanted artificial lenses or laser surgery); and accommodating approaches (whereby surgically implanted artificial lenses are designed to mimic the movement of the natural crystalline lens of the eye or techniques are used to attempt to restore the function of the eye’s own accommodative system). Certain companies enjoy competitive advantages over us, including, but not limited to: significantly greater name recognition; established relations with healthcare professionals and customers; established distribution networks; additional lines of products; greater experience in conducting research and development, manufacturing, clinical trials, obtaining regulatory approval for products, and marketing approved or CE marked products; greater financial and human resources for product development, sales and marketing, and patent litigation; and earlier commencement and successful completion of U.S. pivotal clinical trials. To compete in this market requires an ongoing, extensive search for technological innovation and the ability to respond to rapid technological change. It also requires, among other things, the ability to effectively discover, develop, test and obtain regulatory approvals for products, complete conformity assessment and CE mark products, as well as the ability to effectively commercialize, market and promote approved products, including communicating the effectiveness, safety and value of products to actual and prospective patients and medical professionals. A better-financed or lower-cost provider of corneal inlay surgical solutions or a competing vision treatment could take market share away from us or force us to lower product prices, causing our revenues and results of operations to decline materially.
If our microlens is approved by FDA but fails to achieve an adequate level of acceptance by ophthalmic surgeons, we will not be able to generate sufficient revenue to become or remain profitable. Ophthalmic surgeons play a significant role in determining the course of treatment and, ultimately, the type of products that will be used to treat a patient for presbyopia. If our microlens fails to receive an adequate level of market acceptance from ophthalmic surgeons, clinical staff and the broader ophthalmic community (including optometrists) and we do not convince ophthalmic surgeons and other ophthalmic stakeholders that our products are attractive alternatives to our competitors’ products as well as a complementary solution to other existing vision correction procedures, we will never generate sufficient revenue to become or remain profitable. Market adoption of corneal inlays by surgeons in the U.S. market has been much slower than anticipated. For example, Revision Optics, a former competitor of ours which received FDA approval for its Raindrop Near Vision Inlay in June 2016, experienced challenges in driving market adoption of its inlay and closed its business operations in February 2018. Obtaining market acceptance for corneal inlays such as ours, which require a significant number of patient follow-up visits, is challenging. As a result, it will be important for us to effectively market our products to ophthalmic surgeons, clinical staff and the broader ophthalmic community (including optometrists). Acceptance of our products depends on educating ophthalmic surgeons and their clinical staff as to the distinctive characteristics, perceived clinical benefits, safety and cost effectiveness of our products as compared to our competitors’ products as well as the utility of our microlens to be used as a complementary procedure to existing surgical treatments for visual problems. It also depends on training ophthalmic surgeons and their clinical staff in the proper application of our products. To date, we have deployed limited resources to market our products in certain targeted jurisdictions outside the U.S. If we are not successful in appropriately convincing ophthalmic surgeons and their clinical staff of the merits of our products or educating them on the use of our products, they may not use our products and we will be unable to fully commercialize our products or reach profitability. Ophthalmic surgeons and/or their clinical staff may be hesitant to change their medical treatment practices for the following reasons, among others:
|
|
• |
lack of experience with our products and concerns regarding potential side effects; |
|
|
• |
prior negative experience with competitors’ inlay products and surgeons’ concerns that our products may lead to similar negative patient outcomes; |
|
|
• |
lack of clinical data currently available to support the safety and effectiveness of our products; |
|
|
• |
existing relationships with competitors and distributors that sell their products; |
|
|
• |
lack or perceived lack of evidence supporting additional patient benefits; |
31
|
|
• |
the time commitment that may be required for training. |
In addition, we believe recommendations and support of our products by influential ophthalmic surgeons are important for market acceptance and adoption. If we do not receive support from such ophthalmic surgeons or long term data does not show the benefits of using our products, ophthalmic surgeons may not use our products. In such circumstances, we may not be able to grow our revenues or achieve profitability.
If we are unable to train ophthalmic surgeons and their clinical staff on the safe and appropriate use of our products, we may be unable to achieve revenue growth or profitability.
An important part of our sales process includes the ability to train ophthalmic surgeons and their clinical staff on the safe and appropriate use of our products. We have very limited experience in training and retaining qualified independent ophthalmic surgeons to perform presbyopia correction surgery using our products. If we are unable to attract ophthalmic surgeons to our training programs, we may be unable to achieve growth or profitability.
There is a learning process involved in ophthalmic surgeons and their clinical staff becoming proficient in the use of our products. It is critical to the success of our commercialization efforts to train a sufficient number of ophthalmic surgeons and to provide them with adequate instruction in the use of our microlens. This training process may take longer than expected and may therefore affect our ability to increase sales. Following completion of training, we expect to rely on the trained ophthalmic surgeons to appropriately advocate the benefits of our products in the broader marketplace. Convincing ophthalmic surgeons to dedicate the time and energy necessary for adequate training is challenging, and we cannot assure you we will be successful in these efforts. If ophthalmic surgeons and their clinical staff are not properly trained, they may misuse or ineffectively use our products. Such uses may result in unsatisfactory patient outcomes, patient injury, negative publicity or lawsuits against us, any of which would have a material adverse effect on our business, results of operations and financial condition.
Our reliance on a limited number of third-party suppliers for our microlens could harm our ability to meet demand for our products in a timely and cost effective manner.
We have manufacturing capacity in Irvine, California, but items manufactured in that facility to date have been produced solely for IDE use in the U.S. We believe the facility is also scalable to meet future U.S. and out of the U.S., or OUS, demand once it has received all applicable regulatory registrations, approvals and certifications. In December 2016, we received approval to manufacture CE mark products in Irvine. Our U.S. facility received regulatory approval from the State of California to manufacture our microlens for our U.S. staged pivotal trial and during 2014 and 2015 provided all of the required lenses that were used in the treatment phase for 421 patients. Also, our U.S. facility has demonstrated conformity with the Essential Requirements of the EU Medical Devices Directive with respect to the manufacture of our microlens for sale in the EEA, including all applicable quality assurance requirements. We may continue to utilize our existing Israeli supplier for products sold outside of the U.S., including in the EEA, if OUS demand exceeds our internal manufacturing capacity. Given the location of our Israeli supplier, the supply of our microlens could be disrupted if events were to occur in the Middle East that resulted in social, political, economic or military instability. Our supply agreement with this supplier expired in January 2017 and going forward we may continue to utilize this supplier and would order products on a purchase order basis. Whether we manufacture our microlens at our California facility, order products from our existing Israeli supplier or identify one or more alternate suppliers, we cannot assure you that we will be able to obtain sufficient quantities of our microlens in the future, which could have a material adverse effect on our business, results of operations and financial condition.
For us to be successful, our suppliers must be able to provide us with products in desired quantities, in compliance with regulatory requirements, in accordance with agreed-upon detailed specifications, at acceptable costs and on a timely basis. Reliance on third party suppliers entails risks to which we would not be subject if we manufactured all of our products ourselves, including reliance on the third parties for regulatory compliance and quality assurance, the possibility that products will not be delivered on a timely basis, the possibility of increases in pricing for our products, the possibility of breach of the applicable manufacturing agreement by third parties and the possibility of termination or non-renewal of the agreement by third parties. If any of these risks materialize, it could significantly increase our costs and impact our ability to meet demand for our products and could have a material adverse effect on our business, results of operations and financial condition. If we are unable to satisfy commercial demand for our products in a timely manner, our ability to generate revenue would be impaired, market acceptance of our products could be adversely affected and customers may instead purchase or use our competitors’ products. Securing a replacement supplier could be difficult, time-consuming and expensive.
There are a limited number of suppliers and third-party manufacturers that operate under the FDA’s current Good Manufacturing Practices, or cGMP, maintain certifications of the International Standards Organization, or ISO, that are recognized as harmonized
32
standards in the EEA, and have the necessary expertise and capacity to manufacture our products. As a result, if it were necessary to terminate our relationship with our existing suppliers, it may be difficult for us to locate another supplier that could promptly fulfill our anticipated future needs. If we are unable to arrange for third-party manufacturing of our products, or are unable to do so on commercially reasonable terms, our sales may be materially and adversely affected.
We rely on a single third-party supplier to supply the raw material used to manufacture our microlens.
The hydrophilic acrylic material used to manufacture our microlens is supplied to us by a single supplier located in the United Kingdom. We do not have a guaranteed supply commitment from this supplier. Although we believe that such supplier will be able to sufficiently meet our currently anticipated supply needs, we cannot assure you that we will be able to obtain sufficient quantities of the hydrophilic acrylic material in the future, which could have a material adverse effect on our business, results of operations and financial condition. In addition, we would be required to obtain approval from the FDA in the event that we wished to use different material or similar material from a different supplier with respect to any products to be offered and sold in the United States.
The global nature of our business may result in fluctuations and declines in our sales and profits.
Our products are currently available in several countries outside of the U.S. Because we have a CE Mark for our microlens, we have the ability presently to market that product within the EEA, and, through mutual recognition agreements, in Switzerland. For the foreseeable future, pending receipt of the necessary FDA approvals to market our products in the U.S., we expect that sales outside of the U.S. will represent 100% of our revenues. We may be exposed to transaction risk because some of our sales and expenses will be incurred in a different currency than the local currency. To date, we have not attempted to offset our exposure to this risk by investing in derivatives or engaging in other hedging transactions.
Economic, social and political conditions, laws, practices and local customs vary widely among the countries in which we sell our products. Our operations outside of the U.S. face a number of risks and potential costs, enjoy less stringent protection of intellectual property and face economic, political and social uncertainty in some countries, especially in emerging markets. We have limited experience developing and manufacturing our products to comply with the commercial and legal requirements of markets outside of the U.S. Our success in markets outside of the U.S. will depend, in part, on our ability to manufacture products that meet applicable regulatory and commercial requirements, our ability to enforce contractual commitments and our ability to develop and implement policies and strategies that are effective in anticipating and managing these and other risks in the countries where we do business. Such risks may have a material adverse effect on our operations in any particular country and on our business as a whole. Inflation in emerging markets also may make our products more expensive there and increase the credit risks to which we will be exposed.
If we do not successfully implement our commercialization strategy, our business, results of operations and financial condition will be adversely affected.
We have developed our commercialization strategy based on assumptions about the presbyopia market that might prove to be wrong. We believe that various demographics and industry-specific trends, including adults noticing the onset of presbyopia as they reach their forties, the demands upon our eyes resulting from the increased use of electronic devices and increasing acceptance of eye surgeries as alternatives to reading glasses and contact lenses, will help drive growth in our market and our business, but these demographics and trends are uncertain. Actual demand for our products could differ materially from projected demand if our assumptions regarding these factors prove to be incorrect or do not materialize, or if alternative treatments to those offered by our products gain widespread acceptance.
We may not be able to successfully implement our commercialization strategy. To implement our commercialization strategy of initially dealing directly with ophthalmic clinics, we must, among other things, appropriately educate the decision-makers within these organizations regarding the advantages of our products and processes, train professionals working in those centers on how to use our products, enter into commercially reasonable agreements with those centers and engage in careful follow-up to capture relevant experience and demonstrate our goal to partner with our laser center customers. Our strategy of focusing exclusively on the presbyopia market may limit our ability to grow. Moreover, even if we successfully implement our commercialization strategy, our operating results may not improve or may decline. We may decide to alter or discontinue aspects of our commercialization strategy and may adopt different strategies due to business or competitive factors not currently foreseen, such as new medical technologies that would make our products obsolete. Any failure to implement our business strategy may materially and adversely affect our business, results of operations and financial condition.
33
If the market does not accept and endorse presbyopia correction surgery, we will not be able to successfully execute our business plan.
We believe that our profitability and our ability to expand depend to a large extent on the acceptance of vision correction surgeries in general, as well as presbyopia correction surgery specifically, as a safe and effective treatment option. Even if we obtain FDA and other required regulatory approvals, if presbyopia correction surgery does not gain broad market acceptance, our opportunity to achieve profitability and sustained growth will be severely limited. We cannot assure you that presbyopia correction surgery will be accepted widely, if at all, by ophthalmic surgeons, ophthalmologists, optometrists or the general population as an alternative to existing or future methods of treating presbyopia or other refractive vision disorders. Market acceptance depends on a number of factors, including but not limited to:
|
|
• |
the efficacy and safety of our products as demonstrated in clinical trials, as well as by actual usage in jurisdictions where our products are authorized for marketing and sale; |
|
|
• |
the clinical indications and intended purpose for which our products are approved and/or CE marked if and when approvals are granted and CE marks affixed following the completion of the conformity assessment; |
|
|
• |
acceptance by ophthalmic surgeons, ophthalmologists, optometrists and ophthalmic centers; |
|
|
• |
third-party publications reporting findings with respect to the efficacy and safety of our products; |
|
|
• |
the potential and demonstrable advantages and disadvantages of our products and of competitive products and processes; |
|
|
• |
relative convenience and ease of administration; |
|
|
• |
the tolerance of our products by patients, including prevalence and severity of side effects; and |
|
|
• |
the effectiveness of our sales and marketing efforts. |
Any factor that adversely impacts market acceptance of presbyopia correction surgery will have a negative impact on our business, results of operations and financial condition.
We do not anticipate that our microlens and the procedure to implant our microlens will be reimbursable through private or governmental third-party payors, which could limit market acceptance.
Our microlens and the procedure to implant our microlens are not currently reimbursable through private or governmental third-party payors in any country. In addition, we do not anticipate that our microlens and the procedure to implant our microlens will be reimbursable through private or governmental third-party payors in the foreseeable future. The commercialization of our microlens depends on prospective patients’ ability to cover the costs of our microlens and the implantation procedure. We believe that a substantial portion of presbyopes worldwide do not have the financial means to cover the costs of our microlens. A general regional or worldwide economic downturn could negatively impact demand for our microlens. In the event that medically eligible patients deem the costs of our procedure to be prohibitively high or consider alternative treatment options to be more affordable, our business, results of operations and financial condition would be negatively impacted.
Our ability and the ability of our subsidiaries to use net operating loss carryforwards and certain other tax attributes may be limited.
Our ability and the ability of our subsidiaries to utilize U.S. federal net operating loss carryforwards and federal tax credits may be limited under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, or the Code. The limitations apply if an “ownership change,” as defined by Section 382, occurs. Generally, an ownership change occurs if the percentage of the value of the stock that is owned by one or more direct or indirect “five percent shareholders” increases by more than 50 percentage points over their lowest ownership percentage at any time during the applicable testing period (typically three years). The ownership change of a parent entity may result in the ownership change of a subsidiary. If we or any of our subsidiaries have experienced an “ownership change” at any time since formation, that corporation may already be subject to limitations on the ability to utilize existing net operating losses and other tax attributes to offset taxable income. In addition, future changes in our stock ownership, which may be outside of our control, may trigger an “ownership change” and, consequently, Section 382 and 383 limitations. As a result, if we or our subsidiaries earn net taxable income, the ability to use pre-change net operating loss carryforwards and other tax attributes to offset U.S. federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us or our subsidiaries.
Our ability to utilize net operating losses to offset future taxable income under Irish tax regulations may be limited due to the historical tax losses incurred by our Irish trading entity, Presbia Ireland Limited, have been on a pre-trading basis. Under the Irish rules, only such losses that occur within a three year period prior to commencing trading can be utilized to offset future trading taxable income.
34
Additionally, Presbia PLC has been categorized as a holding company under Irish tax regulations and, as such, losses incurred by Presbia PLC are not deductible for tax purposes and do not result in tax loss carryforward against future taxable income.
We may not be able to achieve a competitive worldwide effective corporate tax rate.
We cannot give any assurance as to what our effective tax rate will be, because of, among other things, uncertainty regarding the geographic mix of any income we generate and the tax policies of the jurisdictions where we operate. Our actual effective tax rate may vary from our expectation and that variance may be material. Additionally, the tax laws of Ireland and other jurisdictions could change in the future, and such changes could cause a material change in our effective tax rate, which may negatively impact our business, results of operations and financial condition.
Presbia PLC and Presbia Ireland, Limited are incorporated in and resident for tax purposes in Ireland. Accordingly, they are subject to Irish corporation tax on their worldwide income and gains. The current rates of Irish corporation tax are 12.5% for certain trading income, 25% for all other income, and 33% for capital gains. It is anticipated that we will be subject to the lower rate of Irish corporation tax applicable to our trading income (currently 12.5%) on the basis that we will be carrying on a trade in Ireland for Irish tax purposes. However, we cannot guarantee that our activities in Ireland will be sufficient to qualify for trading status in respect of all or any portion of our income. There is no comprehensive definition of what constitutes “trading” for Irish tax purposes, and whether or not a company is carrying on a trade in Ireland for Irish tax purposes is determined on the facts of each individual case. Consequently, we cannot assure you that the Irish Revenue (Tax) authorities would accept our trading status for Irish tax purposes in respect of all or any portion of our income. If it is determined that we are not in fact carrying on a trade in Ireland for Irish tax purposes, our income in Ireland could be subject to a 25% tax rate.
Changes in tax laws or tax rulings in the U.S. and abroad could have a significant adverse impact on our effective tax rate.
On December 22, 2017, the Tax Cuts and Jobs Act (“TCJA”), was enacted into law. The TCJA makes significant changes to the U.S. taxation of our domestic and international operations. The TCJA contains a number of provisions that may adversely impact our effective tax rate or operating cash flows going forward, including:
|
|
• |
The limitation on the amount of interest expense deduction available to our U.S. subsidiaries to the extent we are unable to absorb any unused interest deductions over time; |
|
|
• |
The “Base Erosion Anti-Abuse Tax”, which requires our U.S. subsidiaries to make an alternative determination of taxable income without regard to tax deductions for certain payments to affiliates; |
|
|
• |
Provisions that may deny deductions for certain payments made by our U.S. subsidiaries to non-U.S. affiliates to the extent such payments are classified as “hybrid payments”; and |
|
|
• |
The one-time transition tax (i.e. toll charge) on the pre-2018 earnings of certain non-U.S. subsidiaries. The tax is payable over eight years, but is not dependent on our future earnings and therefore may have an adverse impact on our future operating cash flow. |
Many countries in Europe, as well as a number of other countries and organizations, have recently proposed or recommended changes to existing tax laws which could impact our effective tax rate or future tax obligations. The Organization for Economic Cooperation and Development has been working on a Base Erosion and Profit Sharing Project, and is expected to continue to issue guidelines and proposals that may change various aspects of the existing framework under which our tax obligations are determined in many of the countries in which we do business. The European Commission has conducted investigations in multiple countries focusing on whether local country tax rulings or tax legislation provides preferential tax treatment that violates European Union state aid rules. If the Company’s effective tax rates were to increase, or if the ultimate determination of the Company’s taxes owed is for an amount in excess of amounts previously accrued, the Company’s operating results, cash flows, and financial condition could be adversely affected.
We would be adversely affected if, either based on current law or in the event of a change in law, the Internal Revenue Service (“IRS”) did not agree that Presbia is a foreign corporation for U.S. federal tax purposes.
We believe that, under current law, we are treated as a foreign corporation for U.S. federal income tax purposes. However, changes to the inversion rules in Section 7874 of the Code or the U.S. Treasury Regulations promulgated thereunder or other U.S. Internal Revenue Service, or IRS, or U.S. Treasury Department guidance could adversely affect our status as a foreign corporation for U.S. federal income tax purposes, and any such changes could have prospective or retroactive application to us and/or our respective shareholders and affiliates. Most recently, the U.S. Treasury Department issued Notice 2014-52, which applies stricter “anti-inversion” rules to inversion transactions occurring on or after September 22, 2014. Although the Notice in its current form would not affect our status as a foreign corporation, the U.S. Congress may enact legislation in the future to change the inversion rules, possibly
35
retroactively. In addition, recent legislative proposals have aimed to expand the scope of U.S. corporate tax residence, and such legislation, if passed, could have a material and adverse effect on us.
In addition, the TCJA significantly changes U.S. tax law by, among other things, lowering the corporate income tax rate from a maximum of 35% to a flat 21%, implementing a modified territorial tax system, imposing a one-time transition tax on deemed repatriated earnings of foreign subsidiaries and changing the rules which determine whether a U.S. person is a U.S. shareholder of a controlled foreign corporation, or CFC, for 2017 and onwards. Notwithstanding the reduction in the corporate income tax rate, the overall impact of the new federal tax law is uncertain and our business and financial condition could be adversely affected. In addition, it is uncertain if and to what extent various states will conform to the newly enacted federal tax law. The impact of this tax reform on holders of our ordinary shares is also uncertain and could be adverse.
We depend on key employees, the loss of which could substantially damage our business and our ability to compete.
We depend on the continued service of our executive officers and other key employees. The loss of a key employee could hurt our business. Our executive officers are employees at will and are not subject to a non-compete obligation. We could be particularly damaged if any of our executive officers or any other key employee or employees went to work for our competitors. Our future success depends on our ability to identify, attract, train, motivate and retain other highly skilled personnel. Failure to do so may adversely affect our results. We do not maintain insurance policies to cover the cost of replacing the services of any of our key employees who may unexpectedly die or become disabled.
We may seek to grow our business through acquisitions of or investments in new or complementary businesses, products or technologies, and the failure to manage any acquisitions or investments, or the failure to integrate them with our existing business, could have a material adverse effect on us.
From time to time, we expect to consider opportunities to acquire or make investments in other technologies, products and businesses that may enhance our capabilities, complement our current products or expand the breadth of our markets or customer base. For example, in August 2016, we entered into an asset purchase agreement with Neoptics AG pursuant to which we acquired certain assets from Neoptics including patents, pending patents, specified trademarks, equipment, inventory, technical documents and other related documents. Potential and completed acquisitions and strategic investments involve numerous risks, including, but not limited to:
|
|
• |
problems assimilating the purchased technologies, products or business operations; |
|
|
• |
maintaining uniform standards, procedures, controls and policies; |
|
|
• |
unanticipated costs associated with acquisitions; |
|
|
• |
diversion of management’s attention from our core business; |
|
|
• |
adverse effects on existing business relationships with suppliers; |
|
|
• |
risks associated with entering new markets in which we have limited or no experience; |
|
|
• |
potential loss of key employees of acquired businesses; and |
|
|
• |
increased legal and accounting compliance costs. |
We have no current commitments or intentions with respect to any acquisition or investment. We do not know if we will be able to identify suitable acquisitions, complete any such acquisitions on favorable terms or at all, successfully integrate any acquired business, product or technology into our business or retain any key personnel, suppliers or distributors. Our ability to grow through acquisitions successfully depends upon our ability to identify, negotiate, complete and integrate suitable target businesses and to obtain any necessary financing. These efforts could be expensive and time-consuming, and may disrupt our ongoing business and prevent management from focusing on our operations. If we are unable to integrate any acquired businesses, products or technologies effectively, our business, results of operations and financial condition would be materially and adversely affected.
36
We may need to increase the size of our organization, and we may experience difficulties in managing growth.
As of December 31, 2017, we had 24 employees. Whether or not we grow by acquisition or internal growth, we expect that it will be necessary to expand our managerial, operational, financial and other resources in order to manage our operations and clinical trials, continue our development activities and fully commercialize our products. Our systems currently in place may not be adequate to support this future growth. Our need to effectively execute our business strategy requires that we:
|
|
• |
manage our clinical trials effectively; |
|
|
• |
provide substantial clinical, training, sales and operational support to ophthalmic centers at the time that we enter into contractual relationships with them and provide ongoing support even after the centers are fully trained; |
|
|
• |
manage our internal development efforts effectively; |
|
|
• |
continue to improve our operational, financial and management controls, reporting systems and procedures; and |
|
|
• |
identify, recruit, maintain, motivate and integrate additional employees. |
If we are unable to expand our managerial, operational, financial, and other resources to the extent required to manage our development and commercialization activities, our business, results of operations and financial condition would be materially and adversely affected.
We may be subject to costly product liability claims related to our clinical trials and products and, if we are unable to obtain adequate insurance or are required to pay for liabilities resulting from a claim excluded from, or beyond the limits of, our insurance coverage, a material liability claim could adversely affect our financial condition.
We face the risk that the use or misuse of our products may result in adverse side effects to patients in our clinical trials. We face even greater risks in connection with the commercialization of our products, including our current sales outside of the U.S. Although we maintain product liability insurance and request that ophthalmic clinics and hospitals offering our products, and the physicians at such facilities, maintain product liability insurance, any such insurance coverage may be insufficient to reimburse us for any expenses or losses we may suffer, and we may be required to increase our product liability insurance coverage for trials that we initiate in the future. We do not know whether we will be able to continue to obtain product liability coverage and obtain expanded coverage if we require it, on acceptable terms, or at all. We may not have sufficient resources to pay for any liabilities resulting from a claim excluded from, or beyond the limits of, our insurance coverage. To the extent that we provide indemnities in favor of third parties under our agreements with them, there is also a risk that these third parties could incur liability and bring a claim under such indemnities. An individual may bring a product liability claim against us alleging that one of our products caused an injury or is found to be unsuitable for consumer use. Any product liability claim brought against us, with or without merit, could result in:
|
|
• |
withdrawal of clinical trial volunteers, investigators, patients or trial sites; |
|
|
• |
difficulties in commercializing our products; |
|
|
• |
decreased demand for our products; |
|
|
• |
regulatory investigations that could require costly recalls or product modifications; |
|
|
• |
loss of revenues; |
|
|
• |
substantial costs of litigation; |
|
|
• |
liabilities that substantially exceed our product liability insurance, which we would then be required to pay ourselves; |
|
|
• |
an increase in our product liability insurance rates or the inability to maintain insurance coverage in the future on acceptable terms, if at all; |
|
|
• |
the diversion of management’s attention from our business; and |
|
|
• |
damage to our reputation and the reputation of our products. |
Product liability claims may subject us to the foregoing and other risks, which could have a material adverse effect on our business, results of operations and financial condition.
If we use biological and hazardous materials in a manner that causes injury or violates applicable laws or regulations, we could be liable for damages.
Our activities currently require the controlled use of potentially harmful biological materials and hazardous materials and chemicals. We cannot eliminate the risk of accidental contamination or injury to employees or third parties from the use, storage, handling or
37
disposal of these materials. In the event of contamination or injury, we could be held liable for any resulting damages, and any liability could exceed our resources or any applicable insurance coverage we may have. Additionally, we are subject to, on an ongoing basis, a variety of U.S. federal, state and non-U.S. environmental and pollution control laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. The cost of compliance with these laws and regulations may become increasingly significant and could have a material adverse effect on our business, results of operations and financial condition. In the event of an accident or if we otherwise fail to comply with applicable regulations, we could lose our permits or approvals or be held liable for damages or penalized with fines.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with FDA or other governmental regulations, to provide accurate information to the FDA or other governmental authorities, to comply with applicable privacy laws, to comply with manufacturing standards we have established, to adequately monitor clinical investigation sites, or to report financial information or data accurately. Employee misconduct could involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Our sales volumes and our operating results may fluctuate from quarter to quarter, which may make our performance more difficult to understand and may make our future performance more difficult to predict.
We may experience meaningful variability in our sales and operating expenses among quarters, as well as within each quarter, as a result of a number of factors, including, among other things:
|
|
• |
the timing of or failure to obtain regulatory approvals or clearances for products; |
|
|
• |
the number of products sold in the quarter; |
|
|
• |
the demand for, and pricing of, our products and the products of our competitors; |
|
|
• |
costs, benefits and timing of new product introductions; |
|
|
• |
increased competition; |
|
|
• |
the availability and cost of components and materials; |
|
|
• |
the number of selling days in the quarter; and |
|
|
• |
impairment and other special charges. |
Such quarterly fluctuations may make it difficult to understand our performance and predict our future performance.
If we experience material weaknesses in the future or otherwise fail to maintain an effective system of internal controls in the future, we may not be able to accurately report our financial condition or results of operations which may adversely affect investor confidence in us and, as a result, the value of our ordinary shares.
As a result of becoming a public company, we are required, under Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, to establish adequate internal control over financial reporting and disclosure controls and procedures and to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting. This assessment must include disclosure of any material weaknesses identified by our management in our internal control over financial reporting. A material weakness is a deficiency or combination of deficiencies in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of a company’s annual and interim financial statements will not be detected or prevented on a timely basis.
The effectiveness of our controls and procedures may be limited by a variety of factors, including:
|
|
• |
faulty human judgment and simple errors, omissions or mistakes; |
|
|
• |
fraudulent action of an individual or collusion of two or more people; |
38
|
|
• |
inappropriate management override of procedures; and |
|
|
• |
the possibility that any enhancements to controls and procedures may still not be adequate to assure timely and accurate financial control. |
When we cease to be an “emerging growth company” and a “smaller reporting company” under the U.S. federal securities laws, our auditors will be required to express an opinion on the effectiveness of our internal controls. If we are unable to confirm that our internal control over financial reporting is effective, or if our auditors are unable to express an opinion on the effectiveness of our internal controls, we could lose investor confidence in the accuracy and completeness of our financial reports, which could cause the price of our ordinary shares to decline.
We are incurring significant costs as a result of being a public company, which may adversely affect our operating results and financial condition.
We are incurring costs associated with corporate governance requirements, including requirements under the Sarbanes-Oxley Act, as well as rules implemented by the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010, or the Dodd-Frank Act, the SEC, and the NASDAQ listing rules. These rules and regulations are expected to increase our accounting, legal and financial compliance costs and make some activities more time-consuming and costly. In addition, we are incurring additional costs associated with our public company reporting requirements and we expect those costs to increase in the future. As a public company, it is more expensive for us to maintain directors’ and officers’ liability insurance and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified persons to serve on our board of directors, or our Board, committees of our Board, or as executive officers. We cannot predict or estimate the amount of additional costs we may incur or the timing of such costs.
If we experience significant disruptions or security breaches in our information technology systems, our business may be adversely affected.
We depend on our information technology systems for the efficient functioning of our business, including accounting, data storage, compliance, purchasing and inventory management. Although we attempt to mitigate interruptions, we may experience difficulties in implementing certain upgrades, which would impact our business operations, or experience difficulties in operating our business during the upgrade, either of which could disrupt our operations, including our ability to timely ship and track product orders, project inventory requirements, manage our supply chain and otherwise adequately service our customers. In the event we experience significant disruptions as a result of the implementation of our information technology systems, we may not be able to repair our systems in an efficient and timely manner. Furthermore, despite the implementation of security measures, our information technology systems and those of our clinical research organizations, contract manufacturers and other contractors and consultants are vulnerable to damage from cyber-attacks, malicious intrusion, computer viruses, unauthorized access, loss of data privacy, natural disasters, terrorism, war and telecommunication, electrical failures or other significant disruption. Accordingly, such events may disrupt or reduce the efficiency of our entire operation, and result in a loss or damage to our data or inappropriate disclosure of confidential or proprietary information, and have a material adverse effect on our results of operations and cash flows.
Fluctuations in insurance cost and availability could adversely affect our profitability or our risk management profile.
We hold a number of insurance policies, including product liability insurance, directors’ and officers’ liability insurance, general liability insurance, property insurance and workers’ compensation insurance. If the costs of maintaining adequate insurance coverage increase significantly in the future, our operating results could be materially and adversely affected. Likewise, if any of our current providers should no longer be able to provide coverage to us, we may not be able to find another provider that provides comparable coverage for comparable costs, which could impact our coverage and materially and adversely affect our operating results.
Risks Related to our Regulatory Requirements
Our products are subject to extensive governmental regulation both in the U.S. and in other countries, and our failure to comply with applicable requirements could cause our business to suffer.
Our products are subject to extensive regulation by the FDA and various other U.S. federal, state and non-U.S. governmental authorities, such as the competent authorities and notified bodies of the countries of the EEA and other countries in which we currently have marketing approval and/or conduct operations. Government regulation of medical devices is meant to assure their safety and effectiveness, and includes regulation of, among other things:
|
|
• |
design, development and manufacturing; |
|
|
• |
testing, labeling, content and language of instructions for use and storage; |
39
|
|
• |
clinical trials; |
|
|
• |
product safety; |
|
|
• |
marketing, sales and distribution; |
|
|
• |
regulatory approvals and clearances, including premarket approval and clearance; |
|
|
• |
conformity assessment procedures and CE marking; |
|
|
• |
product traceability and record keeping procedures; |
|
|
• |
advertising and promotion; |
|
|
• |
product complaints, complaint reporting, recalls and field safety corrective actions; |
|
|
• |
post-market surveillance, including reporting of deaths or serious injuries and malfunctions that, if they were to recur, could lead to death or serious injury or reports of events that led or might have led to death of or the serious deterioration of the health of a patient, or user or of other person; |
|
|
• |
post-market studies; and |
|
|
• |
product import and export. |
Failure to comply with applicable laws and regulations could jeopardize our ability to sell our products and result in enforcement actions such as:
|
|
• |
delays in the introduction of products into the market; |
|
|
• |
warning letters; |
|
|
• |
injunctions; |
|
|
• |
inspections and audits; |
|
|
• |
fines and other civil penalties; |
|
|
• |
termination of distribution; |
|
|
• |
recalls or seizures of products; |
|
|
• |
total or partial suspension of production; |
|
|
• |
refusal of the FDA or other regulators to grant necessary approvals or clearances; |
|
|
• |
withdrawals or suspensions of then current approvals or clearances, resulting in prohibitions on sales of our products; |
|
|
• |
withdrawal or suspension of the CE certificates of conformity granted by the notified body or delay in obtaining these certificates; |
|
|
• |
withdrawal of the EC declarations of conformity; and/or |
|
|
• |
in the most serious cases, criminal penalties. |
Any of these sanctions could result in higher than anticipated costs or lower than anticipated sales and have a material adverse effect on our reputation, business, results of operations and financial condition.
We are subject to complex regulations which have tended to become more stringent over time. Regulatory changes could result in restrictions on our ability to carry on or expand our operations, higher than anticipated costs or lower than anticipated sales.
Our current products are Class III devices under the U.S. Food, Drug and Cosmetic Act, or FDCA, and thus subject to more stringent regulatory controls than other medical devices. Before we can market or sell our microlens in the U.S., we must obtain approval of a PMA application from the FDA. Our IDE enables us to use our microlens in clinical studies in order to begin to collect safety and effectiveness data for the PMA application. In the PMA approval process, the FDA must determine that a proposed device is safe and effective for its intended use based, in part, on extensive data, including, but not limited to, technical, pre-clinical, clinical trial, manufacturing and labeling data. The PMA process is typically required for devices that are deemed to pose the greatest risk, such as implantable devices, as well as life-sustaining and life-supporting devices. The process of obtaining a PMA generally takes from one to four years, or even longer, from the time the application is submitted to the FDA until an approval is obtained. We do not expect to receive our PMA any earlier than in the fourth quarter of 2018.
40
Future products that we may develop, as well as material modifications to our existing products, will require a PMA Supplement, new Clinical Data and/or a new PMA. If the FDA requires us to go through a lengthier, more rigorous examination for future products or modifications to existing products than we had expected, our product introductions or modifications could be delayed or canceled, which could cause our sales to decline. Outcomes under the PMA process are difficult to predict, as are the time and expense associated with that process. Further, even if any of our future products do not require a PMA, we cannot assure you that we will be able to obtain clearances under Section 510(k) of the FDCA, or 510(k) clearances, which is a less onerous approval process than the PMA process, with respect to those products.
The FDA can delay, limit or deny approval or clearance of a device for many reasons, including but not limited to:
|
|
• |
our inability to demonstrate to the FDA’s satisfaction that our products are safe and effective for their intended uses; |
|
|
• |
the data from our pre-clinical studies and clinical trials may be insufficient to support clearance or approval, where required; and |
|
|
• |
the manufacturing process or facilities we use may not meet applicable requirements. |
Significant delays in receiving approval or clearance, or the failure to receive approval or clearance for our products, would adversely affect our ability to generate revenues and negatively impact our business, results of operations and financial condition.
In addition, the FDA may change its approval and clearance policies, adopt additional regulations or revise existing regulations, or take other actions that may prevent or delay approval or clearance of our products under development or impact our ability to modify any products that may be approved or cleared. For example, in 2011, the FDA announced a Plan of Action to modernize and improve the FDA’s premarket review of medical devices, and has implemented, and continues to implement, reforms intended to streamline the premarket review process. In addition, as part of the U.S. Food and Drug Administration Safety and Innovation Act of 2012, or FDASIA, the U.S. Congress enacted several reforms entitled “Medical Device Regulatory Improvements” and additional miscellaneous provisions which will further affect medical device regulation both pre- and post-approval. Any change in the laws or regulations that govern the approval and clearance processes relating to our current and future products could make it more difficult and costly to obtain approval or clearance for new products, or to produce, market and distribute existing products.
Any delay in, or failure to receive or maintain, approval or clearance for our products under development could prevent us from generating revenue in the U.S from these products or achieving profitability. Additionally, the FDA and other regulatory authorities have broad enforcement powers. Regulatory enforcement or inquiries, or other increased scrutiny on us, could dissuade some ophthalmic surgeons from using our products and adversely affect our reputation and the perceived safety and effectiveness of our products.
In addition, even if we obtain the proper regulatory approval or clearance to market a product, the FDA has the power to require us to conduct post-market surveillance studies, which are designed to identify adverse events, device malfunctions or complaints from patients implanted with the device during a specified period after the commencement of commercial use in the U.S. The FDA may also require us to conduct post-approval studies to further monitor the safety and/or effectiveness of our products. Failure to conduct required surveillance or studies in a timely manner could result in the revocation of the PMA approval or 510(k) clearance for the product that is subject to such a requirement and could also result in the recall or withdrawal of the product, which would prevent us from generating sales from that product in the U.S.
In order to be placed on the market within the EEA, medical devices must meet the Essential Requirements set out in Annex I of the EU Medical Devices Directive such that we can affix the CE Mark to our products. The principal legislation regulating general medical devices in the EEA is the EU Medical Devices Directive. In the case of low risk (Class I) medical devices, such as our microlens inserter, the manufacturer may self-certify conformity with the EU Medical Devices Directive by issuing an EC declaration of conformity. In the case of medium to high risk (Class IIa, IIb and III) medical devices, including our microlens which is a Class IIb medical device, the CE certificate of conformity issues from a notified body. Where a medical device meets the Essential Requirements set out in the EU Medical Devices Directive and complies with the appropriate conformity assessment procedure, based on the classification of the medical device, an EC declaration or CE certificate of conformity will issue and a CE Mark may then be affixed to the product. Once a CE Mark has been affixed to the medical device, it may then be placed on the market in any country within the EEA and, through mutual recognition agreements, Switzerland (subject to certain localized registration and language requirements).
In February 2010, we received a CE certificate of conformity from our notified body for our microlens allowing the CE Mark to be affixed to our microlens. In May 2013, we issued an EC declaration of conformity for our microlens inserter allowing the CE Mark to be affixed to our microlens inserter. We have also obtained an ISO 13485 quality system certification, which confirms that our medical device manufacturing quality management system is compliant with globally recognized standards set forth by the International Organization for Standardization. We are required to keep up-to-date and remain compliant with the most recently issued
41
standards. In order to maintain our CE certificate and EC declaration of conformity and CE Mark, we must continue to comply with the EU Medical Devices Directive and pass annual facilities audit inspections by an inspection agency of the EEA to ISO 13485 standards. In addition, a notified body or other competent authority in an EEA country may perform post-marketing audits on our products and premises from time to time. Failure to comply with such requests in a timely manner, and any adverse findings in any such audit, could result in the withdrawal of our CE certificate and EC declaration of conformity and our CE Mark, and the suspension, recall or withdrawal of our products from the EEA market. Each certificate of conformity may be valid for a maximum of five years but would typically be valid for three years. Our existing CE certificate of conformity for our microlens is valid until November 2019. At the end of each period of validity, we are required to apply to the notified body for a renewal of our CE certificate of conformity. There may be delays in the renewal of our CE certificate of conformity and the notified body may require modifications to our products or to the related technical files before it agrees to issue a new certificate of conformity. We may face difficulties or delays in renewing our existing CE certificate of conformity in light of the new EU medical devices regulations.
On September 26, 2012, the European Commission adopted a package of legislative proposals designed to replace the existing regulatory framework for medical devices in the EEA. On June 14, 2016, the agreed texts of the new medical devices and in vitro diagnostic medical devices regulations (“Regulations”) were finally published. The Regulations still have to be formally adopted by the Council and the Parliament as part of the EEA legislative process. If and when adopted, the proposed new legislation may prevent or delay the EEA approval clearance or CE marking of any future products we may develop or impact our ability to modify currently CE marked products on a timely basis.
On May 5, 2017, the European Commission officially published the Medical Device Regulation (MDR 2017/745) and came into force on May 25, 2017. Manufacturers of currently approved medical devices will have a transition time of three years until May 26th 2020 to meet the requirements of the MDR. For some manufacturers the new MDR provides an additional time after the date of application allowing them to place new products for a maximum of 4 more years on the market. Additional requirements will apply for this extended transition period.

The U.S., in which we are seeking marketing approval, those countries which recognize our CE mark, and those other countries in which we have marketing approval, collectively, only represent a portion of the worldwide presbyopic population. To market and sell our products in other countries, including those countries that may represent a substantial portion of the worldwide presbyopic population, we must seek and obtain regulatory approvals, certifications and/or registrations and comply with the laws and regulations of those countries. These laws and regulations, including the requirements for approvals, certifications and/or registrations and the time required for regulatory review, vary from country to country. Obtaining and maintaining regulatory approvals, certifications and/or registrations are expensive, and we cannot be certain that we will receive regulatory approvals, certifications and/or registrations in any country for which we have yet to receive such approvals, certifications and/or registrations or that we will be able to maintain any regulatory approvals, certifications and/or registrations that we currently possess. If we fail to obtain or maintain regulatory approvals, certifications and/or registrations in any country in which we plan to market our products, our ability to generate revenue will be harmed.
Failure to comply with applicable laws and regulations could jeopardize our ability to sell our products and result in a variety of enforcement actions, all of which would negatively impact our business, results of operations and financial condition.
Modifications to our products may require new premarket approvals or notified assessments or may require us to cease marketing or recall the modified products until approvals are obtained.
Any modification to a PMA-approved device that could significantly affect its safety or effectiveness, including significant design and manufacturing changes, or that would constitute a major change in its intended use, design or manufacture, may require approval of a PMA Supplement, new Clinical Data, and/or a new PMA. The FDA requires every manufacturer to make this determination in the first instance, but the FDA may review any manufacturer’s decision. The FDA may not agree with our decisions regarding whether new approvals are necessary. If the FDA disagrees with any determination that we may make in the future and requires us to seek new PMA approvals for modifications to any previously approved or cleared products for which we have concluded that new approvals are
42
unnecessary, we may be required to cease marketing or distribution of our products or to recall the modified product until we obtain approval, and we may be subject to significant regulatory fines or penalties.
In the EEA, we are required to inform the notified body that carried out the conformity assessment of the medical devices we market or sell in the EEA of any substantial changes to the CE marked device or our quality system such as changes to our devices which could affect compliance with the Essential Requirements set forth in the EU Medical Devices Directive or the indications and/or contraindications and/or warnings determined by the manufacturer to be appropriate to ensure the clinical performance of the device. These substantial changes may require further conformity assessment by a notified body and variation to any existing CE certificate of conformity. If the assessment is favorable, the notified body will issue a new CE certificate of conformity or an addendum to the existing CE certificate of conformity attesting compliance with the Essential Requirements set forth in the EU Medical Devices Directive. If it is not, we may not be able to continue to market and sell the product in the EEA.
We may fail to obtain or maintain regulatory approvals or complete conformity assessment and CE marking to market our products in countries outside of the U.S.
We market our products in certain countries outside of the U.S. and intend to expand our non-U.S. marketing. Each jurisdiction that we target for commercialization of our products requires regulatory approvals and compliance with numerous and sometimes varying regulatory requirements. In addition to the countries in which we currently have marketing approval or CE marking, from time to time we may seek regulatory approval or clearance to market our products in other jurisdictions. The approval procedures vary among countries and may involve requirements for additional testing, and the time required to obtain approval may differ from country to country and from that required to obtain clearance or approval in the U.S. and the necessary CE certificates and EC declarations of conformity in the EEA countries.
Approval or clearance in the U.S. and/or a CE certificate of conformity or CE marking in the EEA countries does not ensure approval or certification by regulatory authorities in other countries or jurisdictions, and approval or certification by one regulatory authority does not ensure approval or certification by regulatory authorities in other countries or by the FDA. Any non-U.S. regulatory approval or certification process may include similar risks associated with obtaining FDA clearance or approval. In addition, some countries only approve or certify a product for a certain period of time, in which case we will be required to re-approve or re-certify our products in a timely manner prior to the expiration of our prior approval or certification. We may not obtain regulatory approvals that we seek on a timely basis, if at all. We may not be able to file for regulatory approvals or certifications and may not receive or maintain necessary approvals to commercialize our products in any market. If we fail to receive or maintain necessary approvals or certifications to commercialize our products in any non-U.S. jurisdiction on a timely basis, or at all, or if we fail to have our products re-approved or re-certified, our business, results of operations and financial condition could be materially and adversely affected.
If we or our suppliers fail to comply with ongoing EEA and FDA or other regulatory authority requirements, or if we experience unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market.
Any product for which we obtain approval clearance or complete conformity assessment and CE mark, and the manufacturing processes, reporting requirements, post-approval clinical data and promotional activities for such product, will be subject to continued regulatory review, oversight and periodic inspections by the FDA and other U.S. and non-U.S. regulatory authorities. In particular, we and our third-party suppliers will be required to comply with the FDA’s Quality System Regulation, or QSR. In EEA countries, compliance with harmonized standards is also recommended as this is often interpreted as a presumption of conformity with the relevant Essential Requirements set forth in Annex I to the EU Medical Devices Directive. These FDA regulations and EU standards cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of our products. Compliance with applicable regulatory requirements is subject to continual review and is monitored rigorously through periodic inspections by the FDA. Compliance with harmonized standards in the EEA is also subject to regular review through the conduct of inspections by notified bodies or other certification bodies. If we, or our suppliers, fail to adhere to QSR requirements in the U.S. or other harmonized standards in the EEA, this could delay production of our products and lead to fines, difficulties in obtaining regulatory clearances and CE certificates of conformity and CE marking, recalls, enforcement actions, including injunctive relief or consent decrees, or other consequences, which could, in turn, have a material adverse effect on our business, financial condition or results of operations.
In addition, the FDA audits compliance with the QSR through periodic announced and unannounced inspections of manufacturing and other facilities. The failure by our company or any of our suppliers to comply with applicable statutes and regulations administered by the FDA, or the failure to timely and adequately respond to any adverse inspectional observations or product safety issues, could result in any of the following enforcement actions:
|
|
• |
untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
|
|
• |
unanticipated expenditures to address or defend such actions; |
43
|
|
• |
customer notifications or repair, replacement, refunds, recalls, detention or seizure of our products; |
|
|
• |
operating restrictions or partial suspension or total shutdown of production; |
|
|
• |
refusal of or delaying our requests for PMA approval of new products or modified products; |
|
|
• |
withdrawing PMA approvals that have already been granted; |
|
|
• |
refusal to grant export approval for our products; and |
|
|
• |
criminal prosecution. |
Any of these sanctions could have a material adverse effect on our reputation, business, results of operations and financial condition. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with all applicable regulatory requirements, which could result in our failure to produce our products on a timely basis and in the required quantities, if at all.
Outside the EEA and the U.S., our products and operations are required to comply with standards set by the applicable regulatory authorities in each jurisdiction that we target for commercialization of our products, and those standards, types of evaluation and scope of review differ among such regulatory authorities. We intend to comply with the standards enforced by such regulatory authorities as needed to commercialize our products. If we fail to comply with any of these standards adequately, a regulatory authority may take adverse actions similar to those within the power of a notified body or competent authority or the FDA. Any such action may harm our reputation and business, and could have a material adverse effect on our business, results of operations and financial condition.
If our products, or the malfunction of our products, cause or contribute to a serious injury or a death, we will be subject to medical device reporting regulations, which can result in voluntary corrective actions or agency or regulatory authority enforcement actions.
Under the FDA medical device reporting regulations, medical device manufacturers are required to report to the FDA information that a device has or may have caused or contributed to a serious injury or death or has malfunctioned in a way that would likely cause or contribute to serious injury or death if the malfunction of the device or a similar device were to recur. When a medical device is suspected to be a contributory cause of an incident event that led or might have led to death of or the serious deterioration of the health of a patient, or user or of the other person, all manufacturers placing such medical devices on the market in the EEA are legally bound to report those events to the competent authority in whose jurisdiction the incident occurred. Were this to happen to us, the relevant competent authority would file an initial report, and there would then be a further inspection or assessment if there were particular issues. This would be carried out either by the competent authority or it could require that the notified body carry out the inspection or assessment.
Any such adverse event involving our products could result in future voluntary corrective actions, such as recalls, modification exchange and/or destruction of devices, or customer notifications, or agency or competent authority action, such as inspection or enforcement action. Any corrective action, whether voluntary or involuntary, will require the dedication of our time and capital, distract management from operating our business and may harm our business, results of operations and financial condition.
In the EEA, we must comply with the EU Medical Device Vigilance System. Under this system, incidents must be reported to the relevant authorities of the EEA countries, and manufacturers may be required to take Field Safety Corrective Actions, or FSCAs, to reduce a risk of death or serious deterioration in the state of health associated with the use of a medical device that is already placed on the market. An incident is defined as any malfunction or deterioration in the characteristics and/or performance of a device, as well as any inadequacy in the labeling or the instructions for use which, directly or indirectly, might lead to or might have led to the death of a patient or user or of other persons or to a serious deterioration in their state of health. An FSCA may include the recall, modification, exchange, destruction or retrofitting of the device. FSCAs must be communicated by the manufacturer or its European Authorized Representative to its customers and/or to the end users of the device through Field Safety Notices.
Our products may in the future be subject to product recalls. A recall of our products, either voluntarily or at the direction of governmental authorities, or the discovery of serious safety issues with our products, could have a significant adverse impact on us.
Governmental authorities, including the FDA and competent authorities of the EEA member states, have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture. In the case of the FDA, the authority to require a recall must be based on an FDA finding that there is reasonable probability that the device would cause serious injury or death. In addition, non-U.S. governmental authorities have the authority to require the recall of our products in the event of material deficiencies or defects in design or manufacture or to reduce the risk of death or serious deterioration in the state of health of patients, users or other persons. Manufacturers may, under their own initiative, recall a product if any material deficiency in a device is found. A government-mandated or voluntary recall by us could occur as a result of an unacceptable risk to health, product failures,
44
malfunctions, manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls of any of our products would divert managerial and financial resources and would have an adverse effect on our reputation, results of operations and financial condition, which could impair our ability to produce our products in a cost-effective and timely manner in order to meet our customers’ demands. We may also be subject to liability claims, be required to bear other costs, or take other actions that may have a negative impact on our future sales and our ability to reach profitability.
We rely on third parties to conduct our clinical trials and assist us with pre-clinical development. If these third parties do not perform as contractually required or expected, we may not be able to obtain regulatory clearance or approval for, or commercialize, our products.
We rely on third parties, including contract research organizations, medical institutions, clinical investigators and contract laboratories, to conduct our clinical trials and to assist in the preparation of our PMA submissions. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if these third parties need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our pre-clinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory clearance or approval for, or successfully commercialize, our products on a timely basis, if at all, and our business, operating results and prospects may be materially and adversely affected. Furthermore, our third-party clinical trial investigators may be delayed in conducting our clinical trials for reasons outside of their control. We had previously engaged a contract research organization to monitor the clinical investigation sites, but terminated the engagement in January 2016 and are now relying on our own internal staff to monitor the clinical investigation sites. As stated above, we may determine to outsource this function again in the future.
The results of our clinical trials may not support our product claims or may result in the discovery of adverse side effects.
Our ongoing research and development, pre-clinical testing, clinical trial and post-market evaluation activities will be subject to extensive regulation and review by numerous governmental authorities, both in and outside of the U.S. We are currently conducting a pivotal clinical trial under our IDE for our microlens, to gather information about these products’ safety, efficacy or optimal use. In the future we may conduct clinical trials to support approval of new products. All such clinical studies must be conducted in compliance with applicable regulations or the applicable regulatory authorities may take enforcement action. The data collected from these clinical studies may ultimately be used to support market clearance for these products. Even if our clinical trials are completed as planned, we cannot be certain that their results will support our product claims or that the applicable regulatory authorities and notified bodies will agree with our conclusions regarding them. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that later trials will replicate the results of prior trials and pre-clinical studies. The clinical trial process may fail to demonstrate that our products are safe and effective for the proposed indicated uses, which could cause us to abandon a product and may delay development of others. Any delay or termination of our clinical trials will delay the filing of our product submissions and, ultimately, our ability to commercialize our products and generate revenues. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of the product’s profile.
We may be subject to enforcement action if we engage in improper marketing or promotion of our products.
The marketing and promotion of our products is subject to EEA Member States laws implementing the EU Medical Devices Directive, Directive 2006/114/EC concerning misleading and comparative advertising, and Directive 2005/29/EC on unfair commercial practices, as well as other EEA Member State legislation governing the advertising and promotion of medical devices. In addition, we are subject to EU and national self-regulatory Codes of Conduct. These laws and Codes of Conduct may limit or restrict the advertising and promotion of our products to the general public and may impose limitations on our promotional activities with healthcare professionals.
Further, once our products are approved, our promotional materials and training methods must comply with FDA and other applicable laws and regulations, including the prohibition of the promotion of unapproved, or off-label, use. If the FDA determines that our promotional materials or training constitutes promotion of an off-label use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine or criminal penalties. It is also possible that other U.S. federal, state or non-U.S. enforcement authorities might take action if they consider our promotional or training materials to constitute promotion of an off-label use. In that event, our reputation could be damaged and adoption of the products could be impaired. In addition, the off-label use of our products may increase the risk of product liability claims, which are expensive to defend and could divert our management’s attention, result in substantial damage awards against us, and harm our reputation.
The provision of benefits or advantages to physicians in order to induce or encourage the prescription, recommendation, endorsement, purchase, supply, order or use of medical devices is prohibited in the EEA. The provision of benefits or advantages to physicians is
45
also governed by the national anti-bribery laws of the member states of the EEA. One such example is the UK Bribery Act 2010. Infringement of these laws could result in substantial fines and imprisonment.
Payments made to physicians in certain EEA Member States must be publically disclosed. Moreover, agreements with physicians must often be the subject of prior notification and approval by the physician’s employer, his/her competent professional organization and/or the competent authorities of the individual EEA Member States. These requirements are provided in the national laws, industry codes, or professional codes of conduct, applicable in the EEA Member States.
Failure to comply with these requirements could result in reputational risk, public reprimands, administrative penalties, fines or imprisonment.
Regulatory healthcare reforms may make it more difficult and costly for us to obtain regulatory approval or clearance of our products or complete conformity assessment and CE marking to produce, market and distribute our products after approval or clearance is obtained.
FDA regulations and guidance and EEA laws and regulations and guidance are often revised or reinterpreted by the FDA and EEA competent authorities in ways that may significantly affect our business and our products. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of our products. Delays in receipt of, or failure to receive, regulatory approvals or clearances for our products would have a material adverse effect on our business, results of operations and financial condition.
Risks Related to Our Intellectual Property
We may become subject to third parties’ claims alleging infringement of their patents and proprietary rights or seeking to invalidate our patents or proprietary rights, or we may need to become involved in lawsuits to protect or enforce our patent portfolio, which could be costly, time consuming, delay or prevent the development and commercialization of our products, or put our patent portfolio and other proprietary rights at risk.
Litigation relating to infringement or misappropriation of patent and other intellectual property rights in the medical device industry is common. For example, we were previously a party to legal proceedings relating to the ownership of certain assets, including intellectual property. See “Risks Related to Our Business—we were previously subject to certain legal proceedings relating to the ownership of certain assets, including intellectual property. As demonstrated by such proceedings, future claims regarding intellectual property may be costly and time consuming to defend and future claims may delay or prevent the development and commercialization of our products or place our patent portfolio and other proprietary rights at risk.” We may be subject to third-party claims in the future that would cause us to incur substantial expenses and which, if successful, could cause us to pay substantial damages. These damages potentially include increased damages and attorneys’ fees if we are found to have infringed such rights willfully. Further, if a patent infringement suit is brought against us, our research, development, manufacturing or sales activities relating to the product that is the subject of the suit may be delayed or terminated. As a result of patent infringement claims, or in order to avoid potential infringement claims, we may choose to seek, or be required to seek, a license from the claimant, which would be likely to include a requirement to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if a license can be obtained on acceptable terms, the rights may be nonexclusive, which would give our competitors access to the same intellectual property rights. If we are unable to enter into a license on acceptable terms, we could be prevented from commercializing one or more of our products, or forced to modify such products, or to cease some aspect of our business operations, which could harm our business significantly.
U.S. and non-U.S. issued patents and pending patent applications controlled by third parties may relate to areas in which we are developing products. In such an instance, because all issued patents are entitled to a presumption of validity in many countries, including the U.S. and many European countries, issued patents held by others that claim our products or technology may limit our freedom to operate unless and until those patents expire or are declared invalid or unenforceable in a court of applicable jurisdiction, if we do not obtain a license or other right to practice the claimed inventions. Pending patent applications controlled by third parties may result in additional issued patents claiming our products and technology. In addition, the publication of patent applications occurs with a certain delay after the date of filing, so we may not be aware of all relevant patent applications of third parties at a given point in time. Further, publication of discoveries in the scientific or patent literature often lags behind actual discoveries, so we may not be able to determine whether inventions claimed in patent applications of third parties have been made before or after the date on which inventions claimed in our patent applications and patents have been made. If third parties prepare and file patent applications in the U.S. that also claim technology or therapeutics claimed by our patent applications or patents, we may have to participate in interference proceedings in the U.S. Patent and Trademark Office, or USPTO, to determine the priority of invention. An unfavorable outcome could require us to attempt to license rights from the prevailing party, or to cease using the related technology or developing or commercializing the related product candidate. We may also become involved in opposition proceedings in the European Patent Office regarding our intellectual property rights with respect to our products and technology.
46
Competitors may infringe our patent rights, or misappropriate or violate our other intellectual property rights. To counter infringement or unauthorized use, we may find it necessary to file infringement or other claims to protect our intellectual property rights. In addition, in any infringement proceeding brought by us against a third party to enforce our rights, a court may decide that a patent of ours is invalid or unenforceable, or may refuse to stop the other party from using the technology at issue on the basis that our patent does not cover the technology in question. An adverse result in any such litigation proceeding could put our patent protections at risk of being invalidated or interpreted narrowly, which could open us up to additional competition and have a material adverse effect on our business.
The cost to us of any patent litigation or other proceedings, such as interference proceedings, which are meant to determine who first invented any of the claims covered by the patent, even if resolved in our favor, could be substantial. Such litigation or proceedings could substantially increase our operating losses and reduce our resources available for development activities. We may not have sufficient financial or other resources to adequately conduct such litigation or proceedings. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than us because of their substantially greater financial resources. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments, and, if securities analysts or investors perceive these results to be negative, there could be a substantial adverse effect on the price of our ordinary shares. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Patent litigation and other proceedings may also require significant time and attention of management and technical staff, which may materially and adversely impact our financial position and results of operations. Furthermore, because of the substantial amount of discovery required in connection with most intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
Our proprietary rights may not adequately protect our technologies and product candidates. If we are unable to protect our product candidates and our intellectual property rights, our position in the market may be materially and adversely affected.
Our commercial success may depend on our ability to obtain patents and maintain adequate protection for our technologies, intellectual property and product candidates in the U.S. and other countries. Our patent portfolio consists of certain U.S. patents, patents issued in other jurisdictions and patent applications in the U.S. and other jurisdictions relating to our technologies. There is no guarantee that any of our patent applications will result in issued patents, or that any patents, if issued, will include claims that are sufficiently broad to cover our existing products or products in development, or to provide meaningful protection from our competitors. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our proprietary technologies and future products are covered by valid and enforceable patents or are effectively maintained as trade secrets within our organization. If third parties disclose or misappropriate our proprietary rights, it may materially and adversely impact our position in the market.
We have applied for patents covering both our technologies and the products we are developing. We may fail to apply for patents on important technologies or products in development in a timely fashion, or at all. Our existing patents and any future patents we obtain may not be sufficiently broad to prevent others from using our technologies or from developing competing products and technologies. Moreover, the patent positions of many medical device companies are highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. As a result, the validity and enforceability of our patent portfolio cannot be predicted with certainty. In addition, we cannot guarantee you that:
|
|
• |
we were the first to make the inventions covered by our issued patents and our pending patent applications; |
|
|
• |
we were the first to file patent applications for these inventions; |
|
|
• |
others will not independently develop similar or alternative technologies or duplicate any of our technologies by inventing around our claims; |
|
|
• |
a third party will not challenge our proprietary rights, and, if challenged, that a court will hold that our existing or future patents are valid and enforceable; |
|
|
• |
any patents issued to us will cover our products as ultimately developed, or provide us with any competitive advantages; |
|
|
• |
we will develop additional proprietary technologies that are patentable; or |
|
|
• |
the patents of others will not have a material adverse effect on our business. |
In addition, there are numerous recent changes to the patent laws and proposed changes to the rules of the USPTO which may have a significant impact on our ability to protect our technology and enforce our intellectual property rights. For example, on September 16, 2011, U.S. President Obama signed the America Invents Act which codifies several significant changes to the U.S. patent laws, including, among other things, changing from a “first to invent” to a “first inventor to file” system, limiting where a patentee may file a patent suit, requiring the apportionment of patent damages, eventually eliminating interference proceedings while maintaining
47
derivation actions, and creating a post-grant opposition process to challenge patents after they have issued. The effects of these changes are currently uncertain as the USPTO must still implement various regulations, and the courts have yet to address many of these provisions in the context of a dispute.
Restrictions on our patent rights relating to our products may limit our ability to prevent third parties from competing against us.
Our success will depend, in part, on our ability to obtain and maintain patent protection for our products, preserve our trade secrets, prevent third parties from infringing upon our proprietary rights and operate without infringing upon the proprietary rights of others. We cannot be certain that the claims in our patent applications to inventions covering our current or future products will be considered patentable by the USPTO and courts in the U.S. or by the patent offices and courts in countries outside of the U.S.
We have filed a method-of-use patent application and may file additional method-of-use patent applications in the future. This type of patent protects the use of the product only for the specified method and does not prevent a competitor from making and marketing a product that is identical to our product for an indication that is outside the scope of the patented method. Moreover, even if these competitors do not actively promote their product for our targeted indication, ophthalmic surgeons and ophthalmologists may use these products “off-label.” Although off-label use may infringe or contribute to the infringement of method-of-use patents, the practice is difficult to prevent or prosecute.
Patent applications in the U.S. and most other countries are confidential for a period of time until they are published, and publication of discoveries in scientific or patent literature typically lags actual discoveries by several months or more. As a result, we cannot be certain that we and the inventors of the issued patents and applications that we may in-license were the first to conceive of the inventions covered by such patents and pending patent applications or that we and those inventors were the first to file patent applications covering such inventions. Also, patent protection may lapse before we manage to obtain commercial value from patents that we may obtain, which might result in increased competition and materially and adversely affect our position in the market.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on our products and technologies throughout the world would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, but where enforcement is not as strong as that in the U.S. These products may compete with our future products in jurisdictions where we do not have any issued patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in jurisdictions outside of the U.S. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, which could make it difficult for us to stop the infringement of any patent issued to us or the marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in jurisdictions outside of the U.S. could result in substantial cost and divert our efforts and attention from other aspects of our business.
Obtaining and maintaining our patents depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors may be able to enter the market earlier than would otherwise have been the case.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our registered or unregistered trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names, which we need to build name recognition by potential partners or customers in our markets of interest. Over the long term, if we are unable to establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively and our business may be materially and adversely affected.
48
We may be subject to claims that we or our employees or consultants have wrongfully used or disclosed alleged trade secrets of our employees’ or consultants’ former employers or their clients. These claims may be costly to defend and, if we do not successfully do so, we may be required to pay monetary damages and may lose valuable intellectual property rights or personnel.
Although no claims against us are currently pending, we may be subject to claims that our employees or our company have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of the former employers of our employees. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. A loss of key research personnel or their work product could hamper our ability to commercialize, or prevent us from commercializing, our products, which could severely harm our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a significant distraction to management.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for some of our technology and products, we will also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position. We seek to protect our trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, contract manufacturers, consultants and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants that obligate them to assign their inventions to us. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the U.S. are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
Risks Related to Ownership of Our Ordinary Shares
If we or any of our subsidiaries were to constitute a passive foreign investment company, or PFIC, for U.S. federal income tax purposes, then U.S. Holders could be subject to adverse U.S. federal income tax consequences.
A PFIC generally is a foreign corporation if either at least (i) 75% of its gross income is “passive income” or (ii) 50% of the gross value of its assets is attributable to assets that produce, or are held for the production of, passive income. We believe that we were not a PFIC in prior taxable years, and based on current business plans and financial expectations, we expect that we will not be a PFIC for the current taxable year. However, PFIC classification is fundamentally factual in nature, generally cannot be determined until the close of the taxable year in question, and is determined annually. Additionally, the analysis depends, in part, on the application of complex U.S. federal income tax rules, which are subject to differing interpretations. Consequently, there can be no assurance that we have never been, are not, and will not become a PFIC for any taxable year during which U.S. Holders hold ordinary shares. If we are a PFIC in any taxable year in which you hold shares and you are a U.S. Holder, we always will be a PFIC with respect to your stock ownership. If we are a PFIC and you are a U.S. Holder and do not make a Qualified Electing Fund election, or QEF election, with respect to us or a “mark-to-market” election with respect to our ordinary shares, you will be subject to adverse tax consequences, including deferred tax and interest charges with respect to certain distributions on our ordinary shares, any gain realized on a disposition of our ordinary shares and certain other events. The effect of these adverse tax consequences could be materially adverse to you. We do not believe that any of our subsidiaries will be PFICs in the current taxable year or foreseeable future taxable years based on their current and projected assets and income; however, there can be no assurance that our subsidiaries have never been, are not, and will not become PFICs for any taxable year during which U.S. Holders hold ordinary shares. In addition, we may form or acquire a subsidiary that is a PFIC in the future. In such event, U.S. Holders will also need to make the QEF election with respect to each such subsidiary in order to avoid the adverse tax consequences described above.
An active, liquid and orderly trading market for our ordinary shares may not develop and you may not be able to resell your shares at or above the price that you paid for them.
Prior to our initial public offering in January 2015, there was no public market for our ordinary shares. In January 2015, our ordinary shares were listed on the NASDAQ Global Market. On February 12, 2018, the Company decided to transfer the listing of its ordinary shares from the NASDAQ Global Market to the NASDAQ Capital Market, which requires the Company to maintain at least $2.5 million of stockholders’ equity. Subsequently, the Company’s ordinary shares were transferred from The NASDAQ Global Market to The NASDAQ Capital Market effective at the opening of business on February 14, 2018. There can be no assurances that we will maintain compliance with applicable NASDAQ Capital Market listing requirements. Failure to maintain compliance with applicable NASDAQ listing requirements could result in the delisting of our shares from trading on the Nasdaq system, which could have a material adverse effect on the trading price, volume and marketability of our common stock.
49
Although our ordinary shares are listed on the NASDAQ Capital Market, an active, liquid, and orderly trading market for our shares may never develop or be sustained. If an active market for our ordinary shares does not continue to develop or is not sustained, it may be difficult for investors in our ordinary shares to sell shares without depressing the market price for the shares or to sell the shares at all.
Our share price may be volatile, and you may not be able to resell your shares at or above the price that you paid for them.
Since our initial public offering, the trading price of our ordinary shares has been volatile, and it is likely that the trading price of our ordinary shares will continue to be volatile. As a result of this volatility, investors may not be able to sell their ordinary shares at or above the price paid for the shares. The market price for our ordinary shares may be influenced by many factors, including:
|
|
• |
announcements regarding the timing, progress or results of our clinical trials, post-market evaluation studies, research and development programs and commercialization efforts; |
|
|
• |
fluctuations in our quarterly financial results or the quarterly financial results of companies perceived to be similar to us; |
|
|
• |
actual or anticipated fluctuations in our key operating metrics, financial condition and operating results; |
|
|
• |
third-party publications reporting findings with respect to the effectiveness and safety of our products; |
|
|
• |
difficulties in establishing relationships with ophthalmic clinics; |
|
|
• |
actual or anticipated changes in our growth rate; |
|
|
• |
announcements of technological innovations or new offerings by us or our competitors; |
|
|
• |
our announcement of actual results for a fiscal period that are worse than projected or expected or our announcement of revenue or earnings guidance that is lower than expected; |
|
|
• |
changes in estimates of our financial results or recommendations by securities analysts; |
|
|
• |
failure of any of our products to achieve or maintain market acceptance; |
|
|
• |
changes in market valuations of similar companies; |
|
|
• |
success of competitive products or services; |
|
|
• |
changes in our capital structure, such as future issuances of securities or the incurrence of debt; |
|
|
• |
announcements by us or our competitors of significant products or services, contracts, acquisitions or strategic alliances; |
|
|
• |
regulatory developments in the United States or other countries; |
|
|
• |
actual or threatened litigation involving us or our industry; |
|
|
• |
additions or departures of key personnel; |
|
|
• |
share price and volume fluctuations attributable to inconsistent trading volume levels of our shares; |
|
|
• |
further issuances of ordinary shares by us; |
|
|
• |
sales of ordinary shares by our shareholders; |
|
|
• |
repurchases or redemptions of ordinary shares; and |
|
|
• |
changes in general economic, industry and market conditions. |
In addition, the stock market in general, and the market for medical device companies in particular, has experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. Securities class action litigation has often been instituted against companies following periods of volatility in the overall market and in the market price of a company’s securities. Any such litigation, if instituted against us, could result in very substantial costs, divert our management’s attention and resources, and harm our business, operating results and financial condition.
A sale of a substantial number of our ordinary shares into the public market could cause the market price of our ordinary shares to drop significantly, even if our business is doing well.
As of March 30, 2018, we had 17,121,857 ordinary shares issued and outstanding, of which 10,479,449 shares, or 61.2% of our total outstanding shares, are held by Richard Ressler and his affiliates, our controlling shareholder.
50
If our controlling shareholder, sells, or indicates an intention to sell, or if our controlling shareholder distributes our shares to its equity holders and those equity holders sell or indicate an intention to sell, substantial amounts of our ordinary shares in the public market, the trading price of our ordinary shares could decline. The perception in the market that these sales may occur could also cause the trading price of our ordinary shares to decline.
Our controlling shareholder is entitled to rights with respect to the registration of the ordinary shares that it holds under the Securities Act. See “Part II, Item 13. Certain Relationships and Related Party Transactions—Registration Rights Agreement.” Registration of these shares under the Securities Act would result in the shares becoming freely tradable without restriction under the Securities Act, except for shares purchased by affiliates.
In, 2015, we filed a registration statement on Form S-8 registering the issuance of 1.8 million ordinary shares subject to options or other equity awards issued or reserved for issuance under our Presbia Incentive Plan. In 2016, we filed a registration statement on Form S-8 registering the issuance of an additional 400,000 ordinary shares subject to options or other equity awards issued or reserved for issuance under our Presbia Incentive Plan. Shares registered under this registration statement on Form S-8 are available for sale in the public market subject to applicable vesting arrangements and the exercise of options, and, in the case of our affiliates, the restrictions of Rule 144. If these additional ordinary shares are sold, or if it is perceived that they will be sold, in the public market, the trading price of our ordinary shares could decline.
In addition, our controlling shareholder, our directors and our executive officers may establish programmed selling plans under Rule 10b5-1 of the Exchange Act with respect to shares that they hold or thereafter acquire, for the purpose of effecting sales of our ordinary shares. Any sales of ordinary shares by these shareholders, or the perception that those sales may occur, including the entry into such programmed selling plans, could have a material adverse effect on the trading price of our ordinary shares.
If securities or industry analysts do not publish or cease publishing research or reports about us, our business, or our market, or if they change their recommendations regarding our shares adversely, our share price and trading volume could decline.
The trading market for our ordinary shares will be influenced by the research and reports that industry or securities analysts may publish about us, our business, our market or our competitors. Securities and industry analysts do not currently, and may never, publish research on us. If no securities or industry analysts commence coverage of our company, our share price and trading volume would likely be negatively impacted. If any of the analysts who may cover us change their recommendation regarding our shares adversely, or provide more favorable relative recommendations about our competitors, our share price would likely decline. If any of the analysts who may cover us were to cease coverage or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause our share price or trading volume to decline.
Our controlling shareholder has substantial control over us and beneficially owns a majority of our issued ordinary shares, which could delay or prevent a change in corporate control.
Richard Ressler, one of our directors and his affiliates, hold a majority of our issued ordinary shares. As of March 30, 2018, Mr. Ressler effectively controlled approximately 61.2% of the 17,121,857 shares outstanding. As a result, our controlling shareholder has the ability to control the outcome of matters submitted to our shareholders for approval, including the election of directors and any sale, merger, consolidation or sale of all or substantially all of our assets. In addition, our controlling shareholder has the ability to control or influence our management and our affairs. The concentration of voting power in our controlling shareholder may have an adverse effect on our share price. Furthermore, on December 14, 2017, we concluded a services agreement with OCV Management, LLC, a related party co-founded by Mr. Ressler and Mark Yung, our Chairman and CEO, for the purpose of providing management services to us.
We are an “emerging growth company” and we cannot be certain if the reduced disclosure requirements applicable to emerging growth companies will make our ordinary shares less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act, and we are taking advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies,” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes Oxley Act for an extended period of time, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We may take advantage of these exemptions until we are no longer an “emerging growth company.” We will remain an “emerging growth company” until December 31, 2020, although if the market value of our ordinary shares that are held by non-affiliates exceeds $700 million as of any June 30 before that time and in certain other circumstances, we would cease to be an “emerging growth company” as of the following December 31. We cannot predict if investors will find our ordinary shares less attractive because we may rely on these exemptions. If some investors find our ordinary shares less attractive as a result, there may be a less active trading market for our ordinary shares.
51
We are a “controlled company” under the NASDAQ listing rules, and as such we are entitled to exemption from certain NASDAQ corporate governance standards, and you may not have the same protections afforded to shareholders of companies that are subject to all NASDAQ corporate governance requirements.
Richard Ressler and his affiliates, including Mark Yung, our Chairman and CEO, hold a majority of the voting power of our issued ordinary shares. As a result, we are a “controlled company” within the meaning of the corporate governance rules of NASDAQ. Under these rules, a controlled company may elect not to comply with certain corporate governance requirements, including: the requirement that we have a compensation committee that is composed entirely of independent directors; the requirement that we have a nominating/corporate governance committee that is composed entirely of independent directors; and the requirement that a majority of the members of our Board be independent directors. We are utilizing and intend to continue to utilize some or all of those exemptions. Accordingly, you will not be similarly situated to shareholders of companies that are subject to all of the corporate governance requirements of NASDAQ. Our status as a controlled company could make our ordinary shares less attractive to some investors or otherwise harm our stock price.
We do not currently intend to pay dividends on our ordinary shares and, consequently, your ability to achieve a return on your investment will depend on appreciation in the price of our ordinary shares.
We have never declared or paid any cash dividends on our ordinary shares and do not intend to do so for the foreseeable future. We currently intend to retain all available funds and any future earnings to support the operation of, and to finance the growth and development of, our business. Any future determination to declare cash dividends will be made at the discretion of our Board, subject to compliance with applicable laws (including the Irish Companies Acts (which we refer to herein as the “Companies Act”), which require Irish companies to have “profits available for distribution” before they can pay dividends) and covenants under credit facilities, which may restrict or limit our ability to pay dividends and will depend on our financial condition, operating results, capital requirements, general business conditions and other factors that our Board may deem relevant. As a result, any return to shareholders will be limited to the appreciation, if any, of their ordinary shares.
Provisions contained in our articles of association, as well as provisions of Irish law, could impair a takeover attempt.
Our articles of association and certain provisions of the Companies Acts contain provisions that could have the effect of delaying or preventing changes in control or changes in our management without the consent of our Board.
There are a number of methods for acquiring an Irish public limited company, including a court-approved scheme of arrangement under the Companies Acts, through a tender offer by a third party under the Irish Takeover Panel Act 1997 (as amended) and the takeover rules made thereunder, which we refer to herein as the “Irish Takeover Rules,” and by way of a merger with a company incorporated in the EEA under the European Communities (Cross-Border Mergers) Regulations 2008 (as amended). Each method requires shareholder approval or acceptance and different thresholds apply. The Irish Takeover Rules will govern a takeover or attempted takeover of our company by means of a court-approved scheme of arrangement, a tender offer or a cross-border merger. These Rules contain detailed provisions for takeovers including as to disclosure, dealing and timetable. The Irish Takeover Rules could discourage an investor from acquiring 30% or more of the outstanding ordinary shares of our company unless such investor were prepared to make a bid to acquire all outstanding ordinary shares.
Our Board may be limited by the Irish Takeover Rules in its ability to defend an unsolicited takeover attempt.
Under the Irish Takeover Rules, we will not be permitted to take certain actions that might “frustrate” an offer for our ordinary shares once our Board has received an offer, or has reason to believe an offer is or may be imminent, without the approval of more than 50% of shareholders entitled to vote at a general meeting of our shareholders and/or the consent of the Irish Takeover Panel. This could limit the ability of our Board to take defensive actions even if it believes that such defensive actions would be in the best interests of our company.
Irish law differs from the laws in effect in the U.S. and may afford less protection to holders of our securities.
It may not be possible to enforce court judgments obtained in the U.S. against us in Ireland based on the civil liability provisions of the U.S. federal or state securities laws. In addition, there is some uncertainty as to whether the courts of Ireland would recognize or enforce judgments of U.S. courts obtained against us or our directors or officers based on the civil liabilities provisions of the U.S. federal or state securities laws or hear actions against us or those persons based on those laws. We have been advised that the U.S. currently does not have a treaty with Ireland providing for the reciprocal recognition and enforcement of judgments in civil and commercial matters. Therefore, a final judgment for the payment of money rendered by any U.S. federal or state court based on civil liability, whether or not based solely on U.S. federal or state securities laws, would not automatically be enforceable in Ireland.
52
As an Irish company, we are governed by the Companies Acts, which differ in some material respects from laws generally applicable to U.S. corporations and shareholders, including, among others, differences relating to interested director and officer transactions and shareholder lawsuits. Likewise, the duties of directors and officers of an Irish company generally are owed to the company only. Shareholders of Irish companies generally do not have a personal right of action against directors or other officers of the company and may exercise such rights of action on behalf of the company only in limited circumstances. Accordingly, holders of our ordinary shares may have more difficulty protecting their interests than would holders of shares of a corporation incorporated in a jurisdiction of the U.S.
The rights of our shareholders may differ from the rights typically offered to shareholders of a U.S. corporation and these differences may make our ordinary shares less attractive to investors.
We are incorporated under Irish law and, therefore, certain of the rights of holders of our shares are governed by Irish law, including the provisions of the Companies Acts, and by our memorandum and articles of association. These rights differ in certain respects from the rights of shareholders in typical U.S. corporations and these differences may make our ordinary shares less attractive to investors. The principal differences include the following:
|
|
• |
under Irish law, dividends may only be declared if we have, on an individual entity basis, profits available for distribution, within the meaning of the Companies Acts; |
|
|
• |
under Irish law, each shareholder generally has preemptive rights to subscribe on a proportionate basis to any issuance of shares for cash. Under U.S. law, shareholders generally do not have preemptive rights unless specifically granted in the certificate of incorporation or otherwise. Pre-emption rights may be disapplied under Irish law for a maximum renewable five-year period by Irish companies by way of a provision in their articles of association or special resolution of their shareholders, which is an option we have availed ourselves of in our articles of association prior to the consummation of our initial public offering; |
|
|
• |
under Irish law, certain matters require the approval of holders of 75% of the votes cast at a general meeting of our shareholders, including amendments to our memorandum and articles of association. This may make it more difficult for us to complete certain types of corporate actions deemed advisable by our Board. Under U.S. law, generally only majority shareholder approval is required to amend the certificate of incorporation or to approve other significant transactions. There is no requirement under Irish law for the shareholder approval of transactions generally; |
|
|
• |
under Irish law, a bidder seeking to acquire us would need, on a tender offer, to receive shareholder acceptance in respect of 80% of our outstanding shares in order to effect a compulsory acquisition of the remaining outstanding shares. If this 80% threshold is not achieved in the offer, under Irish law, the bidder cannot complete a “second step merger” to obtain 100% control of us. Accordingly, acceptance of an offer by 80% of our outstanding shares will likely be a compulsory acquisition of the non-accepting shares or a condition in a tender offer to acquire us, not 50% as is more common in tender offers for corporations organized under U.S. law; and |
|
|
• |
under Irish law, shareholders may be required to disclose information regarding their equity interests upon our request, and the failure to provide the required information could result in the loss or restriction of rights attaching to the shares, including prohibitions on the transfer of the shares, as well as restrictions on voting, dividends and other payments. Comparable provisions generally do not exist under U.S. law. |
A future transfer of your ordinary shares, other than one effected by means of the transfer of book entry interests in DTC, may be subject to Irish stamp duty.
Transfers of ordinary shares effected by means of the transfer of book entry interests in the Depository Trust Company, or “DTC,” should not be subject to Irish stamp duty. It is anticipated that the majority of ordinary shares will be traded through DTC through brokers who hold such ordinary shares on behalf of customers through DTC. This exemption should be available because our ordinary shares will be traded on a recognized stock exchange in the U.S. However, if you hold your ordinary shares as of record rather than beneficially through DTC or through a broker that holds your ordinary shares through DTC, any transfer of your ordinary shares could be subject to Irish stamp duty (currently at the rate of 1% of the higher of the price paid or the market value of the ordinary shares acquired). Payment of Irish stamp duty is generally a legal obligation of the transferee. The potential for stamp duty to arise could adversely affect the price of our ordinary shares.
Item 1B. Unresolved Staff Comments.
None.
53
Our operations are currently conducted at three leased facilities. We lease one location with an aggregate of approximately 9,000 square feet of office, laboratory and manufacturing space in Irvine, California. The lease expires in May 2022. In addition, we lease approximately 610 square feet of office and storage space in Dublin, Ireland. Our corporate headquarters are currently located at our Dublin location. In December 2017, we vacated excess office space in Irvine, California consisting of approximately 8,500 square feet in December 2017. We are actively seeking to sublease this space.
We believe that our current facilities are suitable and adequate to meet our current needs.
We are not aware of any pending or threatened legal proceeding against us that could have a material adverse effect on our business, operating results or financial condition. However, the medical device industry is characterized by frequent claims and litigation, including claims regarding patent and other intellectual property rights as well as improper hiring practices. As a result, we may be involved in various legal proceedings from time to time.
Item 4. Mine Safety Disclosures.
Not applicable.
54
Item 5. Market for Registrant’s Common Equity, Related Shareholder Matters and Issuer Purchases of Equity Securities.
Market Information for Ordinary Shares
On February 12, 2018, the Company decided to transfer the listing of its ordinary shares from the NASDAQ Global Market to the NASDAQ Capital Market. The Company's common stock continues to trade under the symbol "LENS". Prior to such time, our ordinary shares traded on The NASDAQ Global Market under the symbol “LENS.”. The following table sets forth the high and low sales prices per share of our ordinary shares as reported on The NASDAQ Global Market for the period indicated.
|
|
|
Year Ended December 31, 2017 |
|
|
Year Ended December 31, 2016 |
|
||||||||||
|
|
|
High |
|
|
Low |
|
|
High |
|
|
Low |
|
||||
|
Dollars per share |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
First quarter |
|
$ |
3.74 |
|
|
$ |
3.06 |
|
|
$ |
6.00 |
|
|
$ |
2.94 |
|
|
Second quarter |
|
$ |
3.24 |
|
|
$ |
2.20 |
|
|
$ |
4.99 |
|
|
$ |
3.45 |
|
|
Third quarter |
|
$ |
4.54 |
|
|
$ |
2.03 |
|
|
$ |
5.50 |
|
|
$ |
3.79 |
|
|
Fourth quarter |
|
$ |
5.35 |
|
|
$ |
1.93 |
|
|
$ |
4.97 |
|
|
$ |
3.25 |
|
The last reported sales price of our ordinary shares as reported on The NASDAQ Capital Market on March 15, 2018 was $3.06 per share.
Shareholders
As of March 30, 2018, there were twenty-one registered holders of record of our ordinary shares. This number does not reflect the beneficial holders of our ordinary shares who hold shares in street name through brokerage accounts or other nominees.
Dividend Policy
We have never declared or paid any cash dividends on our ordinary shares. We currently intend to retain all available funds and any future earnings to support our operations and finance the growth and development of our business. We do not intend to pay cash dividends on our ordinary shares for the foreseeable future. Any future determination relating to our dividend policy will be made at the discretion of our Board and will depend on, among other factors, our results of operations, financial condition, capital requirements, contractual restrictions, business prospects and other factors our Board may deem relevant.
Item 6. Selected Financial Data.
Under SEC rules and regulations, as a smaller reporting company, we are not required to provide the information otherwise required by this item.
55
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and the related notes appearing elsewhere in this Annual Report on Form 10-K. Some of the information contained in this discussion and analysis or set forth elsewhere in this Annual Report on Form 10-K, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. You should read “Cautionary Note Regarding Forward-Looking Statements” and “Part I, Item 1A. Risk Factors” of this Annual Report on Form 10-K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Overview
We are an ophthalmic device company which has developed and is currently marketing a proprietary optical lens implant for treating presbyopia, the age-related loss of the ability to focus on near objects. Our microlens is a miniature lens designed to be surgically implanted in a patient’s eye to improve that patient’s ability to see objects at close distances. Our current strategy is to continue to commercialize and pursue clinical trial efforts for our microlens in Germany and South Korea and to continue to seek to obtain FDA approval in the United States. Our goal is to become a leading provider of corneal inlay presbyopia-correcting treatment worldwide.
Although reading glasses and contact lenses have historically been, and remain, the most common solution for presbyopia, there are significant drawbacks associated with these approaches, as well as with alternative surgical approaches. We believe that our microlens provides an alternative solution to those presbyopic individuals who desire greater freedom from glasses and wish to avoid the daily maintenance and other complications of contact lenses. We believe that our microlens can be both an effective standalone solution for presbyopia and an effective complementary solution that can be used in conjunction with certain other surgical approaches that are used to treat vision disorders other than presbyopia.
We are an ophthalmic device company with a limited operating history. We are not profitable and have incurred losses in each year since our formation. We have reported recurring net losses and negative cash flow from operating activities since inception and, as of December 31, 2017, we had an accumulated deficit of $88.3 million. We expect to continue to incur significant losses for the foreseeable future.
Factors Affecting our Industry
The medical device industry in general, and the ophthalmic medical device market in particular, are highly competitive. In order for us to succeed in this market as a development stage company, we must:
|
|
• |
incur substantial expenditures to obtain regulatory approvals necessary to commence marketing our products in particular jurisdictions; |
|
|
• |
develop a commercialization strategy that is responsive both to the needs of ophthalmic clinics and ophthalmic surgeons and to our own requirements and limitations; |
|
|
• |
invest in our future by continuing to advance our technology and improve our microlens and other auxiliary products; |
|
|
• |
focus on, and respond to, the feedback we receive in post-operative situations and comply with various post-marketing reporting obligations; and |
|
|
• |
invest in our infrastructure, to assure that if we are successful in gaining necessary regulatory approvals, commercializing our products and advancing our technology, we will be able to grow our business accordingly. |
We expect to compete against companies that develop corneal inlay surgical solutions for presbyopia, companies that offer alternative surgical methodologies for the treatment of presbyopia, including monovision, multifocal and accommodating approaches, and companies that promote reading glasses and/or contact lenses as approaches for treating presbyopia. At any time, our known competitors and other potential market entrants, many of which have greater resources and experience in the ophthalmic medical device market than we have, may develop new devices or treatment alternatives that may compete directly with our products. In addition, they may gain a market advantage by developing and patenting competitive products or processes earlier than we can or by obtaining regulatory approvals/clearances or market registrations more rapidly than we can.
The competitive nature of the market, the high degree of government regulation, the importance of technological innovation and the significance that most people place on near vision combine to elevate the risks facing any development stage company seeking to enter our market.
56
Factors Affecting our Revenues
We believe that the principal factors affecting our revenues will include:
|
|
• |
our ability to obtain pre-market approval for our microlens in the United States and, if we are able to obtain that approval, the time that it will take us to obtain that approval and the associated expenses; |
|
|
• |
our ability to obtain necessary regulatory approvals in other jurisdictions that we target to commercialize our products, and, if we are able to obtain those approvals, the time that it will take us to obtain those approvals and the associated expenses; |
|
|
• |
the growth in the worldwide presbyopic population and the increasing needs of significant elements of that population to view small print on a variety of electronic devices; |
|
|
• |
our ability to maintain the regulatory approvals that we currently possess and may acquire in the future and the associated expense; |
|
|
• |
our ability to obtain commercialization commitments from ophthalmic clinics and individual surgeons in the jurisdictions in which we are authorized to market our products; |
|
|
• |
our ability to gain acceptance by ophthalmic surgeons, to train those ophthalmic surgeons and to cause those ophthalmic surgeons to train other ophthalmic surgeons; and |
|
|
• |
the effects that our competitors will have on us, in terms of our ability to meet price competition, to respond to product announcements and developments by others and to respond to other developments in the market. |
Factors Affecting Our Expenses
Our expenses are principally driven by the following factors:
|
|
• |
Cost of goods sold. At present, our cost of goods sold relates principally to amounts that we previously paid to our microlens supplier in Israel and our OEM microlens inserter supplier in the United States. Although we have developed manufacturing capacity in our California facility, we have used all output from that facility for evaluation purposes and for clinical trials. We believe the facility is also scalable to meet future U.S. and outside of the U.S., or OUS, demand once it has received all applicable regulatory registrations, approvals and certifications. Until there is an alternative use for that facility or we receive those approvals, the direct manufacturing costs that we incur in producing products in the United States will be included within research and development expenses. When and if we receive those approvals and manufacture products in the United States for sale in the United States, our cost of goods sold will also include raw material costs, labor expenses and other expenses that we incur to manufacture our products. We do not expect to increase production at our California facility significantly beyond what has been required for the clinical trials unless and until we receive approval of our PMA application from the FDA. When and if we ourselves manufacture products for sale, our costs of goods sold will also be directly impacted by: |
|
|
• |
the number of employees that will be engaged in manufacturing and the wages and benefits, including stock-based compensation, that we will pay to those employees; |
|
|
• |
to the extent we invest in fixed assets associated with manufacturing activities, the depreciation expenses associated with those fixed assets; |
|
|
• |
the costs we incur to purchase raw materials; |
|
|
• |
inventory write-downs for excess or obsolete inventory; |
|
|
• |
the costs of non-production materials; and |
|
|
• |
lease expenses associated with our production facilities. |
|
|
• |
Research and development. Our research and development expenses consist of the expenses we incur to develop our products, to pursue patent and trademark protection, to respond to technological challenges, to conduct clinical trials and post-market evaluation studies and to pursue governmental approvals. We expect to continue to expense all research and development costs as they are incurred with the exception of capital expenditures that would have alternative uses. Our research and development expenses consist of employee salaries and related benefits, including stock-based compensation, third-party contract costs relating to research, manufacturing, preclinical studies, clinical trial activities and post-market evaluation studies, and allocated facility costs. We may not succeed in achieving certain marketing approvals that we seek for our products. The probability of success of each product may be affected by numerous factors, including preclinical data, clinical data, post-market and third-party evaluation studies, competition, manufacturing capability and commercial viability. |
57
As of December 31, 2017, we had seven employees directly engaged in research and development for at least a portion of their responsibilities. We also engage outside advisors and counsel to assist in development projects and in prosecuting patent and trademark applications. Our research and development expenses will be directly impacted by:
|
|
• |
the number of employees that will be engaged in research and development and the wages and benefits, including stock-based compensation, that we will pay to those employees; |
|
|
• |
the extent to which we will rely on outside sources to provide research and development assistance and the fees charged to us for those services; |
|
|
• |
the extent to which we pursue post-market evaluation studies, our ability to sign-up patients for those studies and retain patients in those studies, the outcomes arising from those studies and the regulatory responses to those studies; |
|
|
• |
the results of third-party evaluation studies; |
|
|
• |
the size and geographical scope of the patent and trademark portfolio and the maintenance fees required to maintain that portfolio. |
|
|
• |
Sales and marketing. Consistent with the re-ordering of our operational priorities initiated in December 2017, we are focusing our resources on ongoing commercialization and clinical trial efforts in Germany and South Korea. Our commercialization strategy involves engaging ophthalmic clinics to ultimately sponsor our products after gaining confidence in our products and processes. We will train the staff of these centers in practice integration, support patient recruitment with direct response advertising campaigns, surgical performance, patient management and post-operative reporting, enabling the centers to perform a substantial portion of the commercialization process on their own. If we are successful in implementing this strategy, our principal expenses will be in furnishing training teams to ophthalmic clinics and then arranging for a smaller Presbia team to remain available to the center once the center is able to perform the necessary skills on its own. We have incurred, and will continue to incur, expenses in connection with conferences, seminars and trade shows that we attend and/or sponsor. Our sales and marketing expenses will be directly impacted by: |
|
|
• |
the volume of our revenues; |
|
|
• |
the number of countries in which we obtain authorization to market our products and the associated regulations; |
|
|
• |
the number of ophthalmic clinics that will be willing to partner with us; |
|
|
• |
the extent to which we are successful in training ophthalmic surgeons to train other ophthalmic surgeons and their clinical staff; |
|
|
• |
the extent to which we will be required to develop a distributorship network in countries that mandate that approach; |
|
|
• |
the number of employees that will be engaged in sales and marketing and the wages and benefits, including stock-based compensation, that we will pay to those employees; |
|
|
• |
the extent to which we identify advertising opportunities that we believe are likely to produce revenue growth; and |
|
|
• |
the extent to which we continue to participate in conferences, seminars and similar opportunities. |
|
|
• |
General and administrative expenses. Our general and administrative expenses consist of finance, human resources, purchasing and information technology services, other administrative services, foreign exchange costs and expenses associated with planning for and implementing the Reorganization Transactions. To date, our general and administrative expenses have been our largest single cost element, reflecting our approach of concentrating our own efforts on research and development and contracting with third-parties, principally affiliated entities and outside professionals, to provide administrative services to us. Over time, we expect to build our own infrastructure and perform more of these services in-house, in which case our general and administrative expenses will relate more to our own payroll than to the amount that we pay to third-parties. Furthermore, we are incurring additional expenses as a result of operating as a public company, including costs to comply with the rules and regulations applicable to companies listed on NASDAQ Capital Market and costs related to compliance and reporting obligations pursuant to the rules and regulations of the SEC. In addition, as a public company, we are incurring increased expenses related to additional insurance, investor relations and other increases related to needs for additional human resources and professional services. Our general and administrative expenses will be directly impacted by: |
|
|
• |
the extent to which we purchase services from third-parties; |
|
|
• |
the costs we incur to build an infrastructure capable of performing services in-house; |
58
|
|
• |
the number of employees that will be engaged in general and administrative functions and the wages and benefits, including stock-based compensation, that we will pay to those employees; |
|
|
• |
the geographical breadth of our company; and |
|
|
• |
the extent to which costs associated with being a public company increase over time. |
Critical Accounting Polices and Estimates
Our consolidated financial statements are prepared in accordance with generally accepted accounting principles in the United States, or GAAP. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, costs and expenses and related disclosures. We have based and will base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. In many instances, we could have reasonably used different accounting estimates, and in other instances changes in the accounting estimates are reasonably likely to occur from period to period. Accordingly, actual results could differ significantly from the estimates made by our management. To the extent that there are material differences between these estimates and actual results, our future financial statement presentation, financial condition, results of operations and cash flows will be affected. We believe that the accounting policies discussed below are critical to understanding our historical and future performance, as these policies relate to the more significant areas involving management’s judgments and estimates.
Emerging Growth Company Status
The Jumpstart Our Business Startups Act of 2012, or the JOBS Act, permits an “emerging growth company” such as us to take advantage of an extended transition period to comply with new or revised accounting standards applicable to public companies. We have chosen to “opt out” of this provision and, as a result, we will comply with new or revised accounting standards as required when they are adopted. This decision to opt out of the extended transition period under the JOBS Act is irrevocable.
Revenue Recognition
Prior to 2018, we recognized revenue when there is persuasive evidence that an arrangement exists with our customers, selling prices are fixed or determinable, title or risk of loss has passed, and collection is reasonably assured. Revenue was recognized upon shipment and payments are either received in advance, or net 30 days for lenses or net 14 days for accessories. Distributor arrangements included certain perfunctory acceptance provisions and a one-year warranty, from the date of shipment, that products are free from defects in material workmanship. Under such provisions customers may reject shipments via written notifications ranging from 14-45 days or exchange defective product under warranty for the same non-defective product. We have not had any significant rejected shipments or warranty claims. We did not grant price concessions to our distributors.
In 2012, we changed our commercialization strategy from exclusively using distributors to also targeting refractive laser centers equipped with femtosecond lasers, except in countries that require the use of distributors or sales representatives. In December 2017, in light our re-ordering of operational priorities, we have focused our commercialization and clinical trial efforts outside of the U.S. in Germany and South Korea, including established ophthalmic clinics in those countries. We recognize revenue from laser centers and ophthalmic clinics based upon an analysis of the terms of each customer arrangement and upon determination that persuasive evidence of an arrangement exists, selling prices are fixed or determinable, title or risk of loss has passed, and collection is reasonably assured. Revenues from laser centers and ophthalmic clinics during the years ended December 31, 2017 and 2016 were not material.
Beginning in 2018, we will recognize revenue based on a new revenue recognition standard which focuses on an analysis of the goods and services provided to our customers in the ordinary course. We will be required to analyze all commercial agreements to identify (i) contracts and arrangements with customers that meet the criteria for revenue recognition under the new standard (ii) identify all performance obligations under the contractual arrangement, such as the delivery of our Microlens to our customers and any other deliverable(s) as defined in a commercial arrangement (iii) the determination of the transaction price and the allocation of such pricing over the defined contractual obligations in the contract with the customer and (iv) the recognition of revenue once the customer has assumed control over the contractual obligations. Control refers to the ability on the part of the customer to obtain substantially all the benefits of possession of the delivered product or service. If revenue transactions do not qualify as revenue under the new standard, then the previous revenue standard or other GAAP standards, as appropriate, will be applied.
Stock-Based Compensation
We record share-based compensation in the consolidated statements of operations and comprehensive loss as expense, based on the estimated grant date fair value of our share-based awards, whereby such fair values are amortized to expense generally over the requisite service period. Our share-based awards are currently comprised of common stock options, restricted share awards and
59
restricted stock units granted under our various plans. The fair value of our common stock options is generally estimated on the grant date using the Black-Scholes-Merton, or BSM, option-pricing formula. While utilizing the BSM model meets established requirements, the estimated fair values generated by the model may not be indicative of the actual fair values of our share-based awards as it does not consider certain factors important to those awards to employees and non-employees, such as continued employment and periodic vesting requirements as well as limited transferability. The determination of the fair value of share-based awards utilizing the BSM model is affected by our stock price and a number of assumptions, including the expected term, expected volatility, risk-free interest rate and expected dividend yields. The expected term of our stock options for those options granted to employees is generally estimated using the simplified method, as permitted by guidance issued by the SEC. For those options granted to non-employees, the expected term of the options is the remaining contractual term. The expected volatility is based on the historical volatility of our stock price. Due to our limited stock price history, we relied upon the historical volatilities using a peer group average. The risk-free interest rate assumption is based on the U.S. Treasury interest rates appropriate for the expected term of our stock options and stock purchase rights. The fair value of our restricted stock awards is based on the closing market price of our common stock on the date of grant. If factors change and we employ different assumptions, share-based compensation expense may differ significantly from what we have recorded in the past. If there are any modifications or cancellations of the underlying unvested share-based awards, we may be required to accelerate, increase or cancel any remaining unearned share-based compensation expense.
Restricted stock units (“RSU’s”) awarded to employees vest when the closing price of our Ordinary shares closes above pre-defined price ranges beginning at $10 per share and remains above that threshold price for a period of 20 trading days. Additional vesting occurs if the closing price exceeds $15 per share and increases in increments of $5 per share to a maximum price of $30 per share. The presence of a market condition, such as the movement in our stock price across several pricing thresholds, requires that we utilize a Monte Carlo Simulation (“MCS”) approach in determining the fair value of an RSU upon grant. This methodology calculates the separate probabilities that our Ordinary share price will achieve and sustain the price levels between $10 and $30 per share including the intermediate price points. The aggregate fair value is determined by weighting the respective probabilities by its respective price level. The MCS requires the following inputs (i) the price volatility of our trading shares and (ii) the closing price of our Ordinary shares as of the grant date. The aggregate fair value is recognized as expense over the derived service period, on a straight-line basis subject to an adjustment for estimated forfeitures, as determined by the MCS model. This methodology, which considers the separate probabilities that each of the price thresholds or market conditions will be achieved under the RSU Plan guidelines.
Research and Development Expenses
We recognize research and development expenses as they are incurred. With respect to capital expenditures for property and equipment used in conducting research and development activities, these costs are generally capitalized on the balance sheet as part of property and equipment and depreciated over their useful lives to research and development expense provided these assets have future alternative uses.
During 2017 and 2016, we incurred costs in connection with the FDA staged-enrollment pivotal clinical trial, which is expected to continue into 2018 or possibly further. We incurred costs for patient recruiting, acquisition of clinical test equipment to be used in the trial, outside experts to read and interpret the results of the studies, third party costs to monitor the investigational sites and perform data collection activities and surgeon and patient fees in connection with surgical procedures and follow-up visits. Our policy with respect to the recognition of these expenses is to record such expenses as research and development expense in the period in which the services are provided. We will evaluate the purchases of clinical test equipment, on a case by case basis, to determine if there exists an alternative use for the equipment following the clinical trial. In the event we determine that there is no alternative use for the test equipment, then that cost will be expensed as part of research and development expense in the period in which we take title to the equipment from the supplier.
Impairment of Long-Lived Assets
We review the recoverability of long-lived and finite-lived intangible assets when circumstances indicate that the carrying amount of assets might not be recoverable. This evaluation compares the carrying value of the long-lived asset to the undiscounted cash flow projections associated with an asset or group of assets. In the event undiscounted cash flow projections indicate impairment, we would record an impairment loss on the statements of operations and comprehensive loss in the period in which the impairment occurred and adjust the carrying value of the asset or group of assets to its fair value.
Income Taxes
Deferred income tax assets and liabilities are recorded for the expected future tax consequences of temporary differences between the financial statement carrying amounts and the income tax basis of assets and liabilities. A valuation allowance is recorded against all of our net deferred tax asset balance due to uncertainties related to the realizability of our deferred tax assets as a result of our history of operating losses.
60
In accordance with generally accepted accounting principles, we identify operating segments as components or elements of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision-maker in making decisions regarding resource allocation and assessing performance. To date, the Company has viewed its operations and manages its business as one segment.
Results of Operations
Comparison of the Years Ended December 31, 2017 and 2016 (in thousands)
|
|
|
Year Ended December 31, |
|
|
Change |
|
||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
$000's |
|
|
% |
|
||||
|
Revenues |
|
$ |
14 |
|
|
$ |
14 |
|
|
$ |
— |
|
|
|
0 |
% |
|
Cost of goods sold |
|
|
97 |
|
|
|
279 |
|
|
|
(182 |
) |
|
|
(65 |
%) |
|
Gross loss |
|
|
(83 |
) |
|
|
(265 |
) |
|
|
182 |
|
|
|
(69 |
%) |
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
7,074 |
|
|
|
5,541 |
|
|
|
1,533 |
|
|
|
28 |
% |
|
Sales and marketing |
|
|
3,278 |
|
|
|
3,103 |
|
|
|
175 |
|
|
|
6 |
% |
|
General and administrative |
|
|
6,280 |
|
|
|
6,750 |
|
|
|
(470 |
) |
|
|
(7 |
%) |
|
Total operating expenses |
|
|
16,632 |
|
|
|
15,394 |
|
|
|
1,238 |
|
|
|
8 |
% |
|
Operating loss |
|
|
(16,715 |
) |
|
|
(15,659 |
) |
|
|
(1,056 |
) |
|
|
7 |
% |
|
Interest expense, net |
|
|
37 |
|
|
|
20 |
|
|
|
17 |
|
|
|
85 |
% |
|
Other expense |
|
|
1 |
|
|
|
2 |
|
|
|
(1 |
) |
|
|
(50 |
%) |
|
Loss before income tax provision |
|
|
(16,753 |
) |
|
|
(15,681 |
) |
|
|
(1,072 |
) |
|
|
7 |
% |
|
Income tax provision |
|
|
245 |
|
|
|
76 |
|
|
|
169 |
|
|
|
222 |
% |
|
Net loss |
|
$ |
(16,998 |
) |
|
$ |
(15,757 |
) |
|
$ |
(1,241 |
) |
|
|
8 |
% |
Revenue
Revenue for the years ended December 31, 2017 and 2016 was $14,000. Revenues were insignificant in both years reflecting a longer lead time to achieve significant volumes in selected markets. Until such time that we can be assured that our commercialization strategy, marketing activities, and execution plans are validated in selected key markets, including the U.S., we will commercialize in limited markets consistent with the re-ordering of our operational priorities initiated in December 2017.
Cost of Goods Sold
Cost of goods sold was $97,000 in the year ended December 31, 2017 as compared to $279,000 in the year ended December 31, 2016. During fiscal year 2017, cost of goods sold primarily consisted of $90,000 for provision of inventory obsolescence. During the year ended December 31, 2016, cost of goods sold primarily consisted of $278,000 for provision of inventory obsolescence.
Research and Development
Research and development expense increased by $1.5 million, or 28%, for the year ended December 31, 2017 as compared to 2016. The increase in research and development spend is primarily due to: (i) a $427,000 of cost increase related to our U.S. staged pivotal clinical trials consisting of costs incurred for patient compliance activities under both Phases I and II and the costs of developing subsequent PMA modules for FDA submission; (ii) manufacturing costs, which were higher by $101,000 due to an increase in expenses related to our pilot manufacturing facility used to provide the microlens used in the clinical trials and subsequent approval for OUS commercialization; (iii) increased engineering costs of $359,000 related to the currently discontinued development of our disposable inserter; (iv) a $259,000 cost increase related to regulatory and quality assurance personnel; and (v) other compensation related costs, which were higher by $427,000 due to additional clinical personnel costs.
Sales and Marketing
Sales and marketing expense increased by $0.2 million, or 6%, for the year ended December 31, 2017 as compared to the year ended December 31, 2016. We are not authorized to sell our microlens in the United States until we receive FDA approval to do so; therefore, our sales and marketing activities are limited to developing the markets outside the United States. During the year ended December 31, 2017, (i) we incurred lower costs for sales and marketing personnel of $307,000, a decrease of 16%; (ii) lower travel expenses related to supporting clients outside the United States of $114,000, an decrease of 32% relative to the prior year; (iii) and an
61
increase in other marketing costs of $597,000, representing an increase of 78% versus the prior year, primarily related to commercialization and clinical trial efforts in Germany and South Korea. We expect sales and marketing costs to decrease in 2018 as we focus our resources on FDA approval as well as ongoing clinical and commercial efforts in Germany and South Korea.
General and Administrative
General and administrative expenses declined by $0.5 million, or 7%, in the year ended December 31, 2017 as compared to the year ended December 31, 2016. General and administrative expenses decreased primarily due to a reduction of stock-based compensation of $558,000. For additional information on 2017 movements, see discussion under Fourth Quarter 2017 Adjustments below.
Interest and Other Income and Expense
Interest and other expense for the year ended December 31, 2017 was $37,000, or $17,000 higher than the year ended December 31, 2016. The change is due primarily to an increase in interest expense related to the Neoptics asset purchase. For additional information, see discussion above under Note 5 to our Consolidated Financial Statements (Note Payable).
Net Loss
Our net loss of $17.0 million for the year ended December 31, 2017 was $1.2 million higher, or 8% higher, than the loss in the corresponding period of 2016 of $15.8 million, due primarily to higher research and development expenses of $1.5 million and higher sales and marketing costs of $0.2 million, partially offset by lower general and administrative costs of $0.5 million, as previously discussed. We expect that losses will continue through 2018 and further, due to anticipated costs related to our U.S. staged pivotal clinical trial and ongoing costs required to develop the market outside of the United States for our microlens.
Fourth Quarter 2017 Adjustments
In December 2017, we announced a reorganization which resulted in a reduction of 15 employees, or 38.5% of total headcount, which affected payroll, benefits, severance and incentive pay expenses; reduced need for office space which resulted in vacant office space and termination costs; and we recognized a tax reserve of $238,000 for unpaid withholding taxes due to Irish tax authorities. The total amount of fourth quarter adjustments is a reduction in operating expense of $1,078,000 and an increase of tax provision expense of $238,000.
|
|
• |
Compensation Related - ($465,000) |
In December 2017, 15 employees were terminated as we reprioritized our efforts towards achieving FDA clinical trials approval of our microlens by the fourth quarter of 2018. Post-employment costs were approximately $332,000 consisting of severance costs to be paid out primarily over a six-month period ending June 2018 and a reversal of $797,000 in accrued incentive compensation costs for amounts that were accrued in 2017 but are not payable as of December 31, 2017.
|
|
• |
Facilities Related - $357,000 |
Also, in December 2017, we vacated one of our Irvine, California facilities and consolidated our operations into another facility also located in Irvine. As of December 31, 2017, the remaining lease term of the vacated facility, of approximately 8,500 square feet, was 33 months through September 30, 2020. We recorded a current liability of $222,000 representing the fair value of the remaining lease obligation as part of deferred rent on the Consolidated Balance Sheet as of December 31, 2017. The determination of the fair value was inclusive of reasonable prospective sublease assumptions, which may, or may not, materialize during the remaining term of the lease. These assumptions are reviewed and updated quarterly over the remaining term of the lease with possible adjustments to the accrual and corresponding increases or decreases to rent expense. In addition, we recorded write-downs of leasehold expenses, furniture and fixtures of an aggregate of $89,000 and accrued expense of $8,000 for unneeded telecom facilities. We also wrote down to net realizable value certain engineering tools, molds and fixtures related to discontinued engineering programs in the amount $52,000.
|
|
• |
Stock-Based Compensation Related – ($970,000) |
Equity awards granted by us do not include provisions to extend the terms and conditions related to vesting or exercising an award beyond the date of termination of the optionee (or holder), with the exception of stock options grant that contain a provision that allows the holder to exercise vested options (vesting must have occurred on or prior to termination date) for a period of 90 days following termination. In December 2017, we extended the terms and conditions for all equity grants for all terminated employees providing at least a one-year extension past the termination date for all vested stock options, and, in certain cases, the acceleration of
62
unvested options or restricted share awards. For those terminated employees with RSU grants based on a market condition, we granted a one-year extension to vest according to the same market conditions as defined in the original grants. According to U.S. GAAP, when a modification of an award occurs, the prior periods’ expense recognition associated with all unvested awards is reversed in the period of the modification on the basis that the old award is being exchanged for a new award. This resulted in a reduction of stock-based compensation expense of $1,350,000. We then compared the relative fair values of the old award with the terms and conditions of the modified award immediately before and after the date of modification and recorded the increase in fair value as stock-based compensation expense in the amount of $380,000, also in the same period of the date of modifications, resulting in a net reduction of stock-based compensation expense of $970,000.
|
|
• |
Irish Taxes - $238,000 |
At December 31, 2017, we established a reserve in the amount of $238,000 for a tax matter in Ireland in which we expect to make payments to the Irish tax authorities for past due amounts originating in 2015 and through 2017. These amounts consist of unpaid income taxes, interest and penalties on compensation paid to members of board of directors for their services while in Ireland. We believe that we are jointly liable along with the board members to make these payments and that recording a reserve as of December 31, 2017 is appropriate.
Liquidity and Capital Resources
On February 3, 2015, we closed the initial public offering of our ordinary shares. We sold a total of 4,166,667 ordinary shares in the offering at a public offering price of $10.00 per share. The aggregate proceeds from our initial public offering was $41.7 million, and we received net proceeds of approximately $36.8 million from the offering, after deducting $4.9 million of underwriting discounts and commissions and estimated offering expenses payable by us. Subsequently in March 2017, we closed a rights offering, pursuant to which we raised approximately $10.8 million in gross proceeds through the sale of 3,611,764 of its ordinary shares at a subscription price of $3.00 per whole share.
Our primary uses of cash are to fund operating expenses, primarily general and administrative expenditures and research and development expenditures. Cash used to fund operating expenses is impacted by the timing of when we pay these expenses, as reflected in the change in our outstanding accounts payable and accrued expenses.
Our future capital requirements are difficult to forecast and will depend on many factors, including:
|
|
• |
the progress, timing and completion of clinical trials for our products; |
|
|
• |
the number and characteristics of products that we pursue; |
|
|
• |
the progress, costs and results of our clinical trials; |
|
|
• |
the outcome, timing and cost of regulatory approvals; |
|
|
• |
delays that may be caused by changing regulatory requirements; |
|
|
• |
timing and amount of revenue resulting from sales of our microlens outside the U.S.; and |
|
|
• |
timing and investment in our commercialization efforts outside the U.S. |
The following table summarizes our cash flows for the periods indicated (in thousands):
|
|
|
Year Ended December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Net cash used in operating activities |
|
$ |
(13,971 |
) |
|
$ |
(13,407 |
) |
|
Net cash used in investing activities |
|
$ |
(198 |
) |
|
$ |
(285 |
) |
|
Net cash provided by (used in) financing activities |
|
$ |
10,024 |
|
|
$ |
(682 |
) |
At December 31, 2017, we had an accumulated deficit of approximately $88.3 million and we expect to incur additional operating losses through 2018, and further. As we continue to incur losses, our transition to profitability will depend on the successful development, approval and commercialization of our microlens. We may never achieve profitability, and unless and until we do, we will need to continue to raise additional capital. Based on our current business plan, we believe that our cash and cash equivalents at December 31, 2017, including the second quarter of 2018 financing will not be sufficient to meet our anticipated cash requirements during the twelve-month period subsequent to the issuance of the financial statements included in this Annual Report on Form 10-K. We need to raise additional capital to fund our operations. We may raise additional capital through equity offerings, debt financings, collaborations and/or licensing arrangements. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available on acceptable terms, we may be required to delay, reduce the scope of, or
63
curtail, our operations. To the extent that we raise additional funds by issuing equity securities, our shareholders will experience dilution, and debt financing, if available, may involve restrictive covenants.
Off-Balance Sheet Arrangements
We have not entered into any off-balance sheet arrangements and do not have any holdings in variable interest entities.
Recent Accounting Pronouncements
Refer to Note 2 of Notes to the Consolidated Financial Statements, included in Item 8 of this report, for discussion of recent accounting pronouncements.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
Under SEC rules and regulations, as a smaller reporting company, we are not required to provide the information otherwise required by this item.
64
Item 8. Financial Statements and Supplementary Data.
INDEX TO FINANCIAL STATEMENTS
|
|
Page |
|
PRESBIA PLC |
|
|
Consolidated Financial Statements: |
|
|
|
|
|
Report of Independent Registered Public Accounting Firm - Squar Milner LLP |
66 |
|
Consolidated Balance Sheets as of December 31, 2017 and 2016 |
67 |
|
68 |
|
|
69 |
|
|
Consolidated Statements of Cash Flows for the Years Ended December 31, 2017 and 2016 |
70 |
|
71 |
65
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and the Board of Directors of Presbia PLC
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Presbia PLC and its subsidiaries (the Company) as of December 31, 2017 and 2016, the related consolidated statements of operations and comprehensive loss, changes in shareholders’ equity and cash flows for the years then ended, and the related notes to the consolidated financial statements (collectively, the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2017 and 2016, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern Uncertainty
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has a history of significant recurring losses from operations through December 31, 2017, and does not have sufficient working capital at December 31, 2017 to fund its planned operations during the twelve-month period subsequent to the issuance of these financial statements. This raises substantial doubt about the Company's ability to continue as a going concern. Management's plans in regard to these matters also are described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ SQUAR MILNER LLP
We have served as the Company's auditor since 2015.
San Diego, California
March 30, 2018
66
CONSOLIDATED FINANCIAL STATEMENTS
PRESBIA PLC
(in thousands, except shares and par value amount)
|
|
|
December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Assets |
|
|
|
|
|
|
|
|
|
Current assets |
|
|
|
|
|
|
|
|
|
Cash |
|
$ |
3,236 |
|
|
$ |
7,333 |
|
|
Accounts receivable |
|
|
8 |
|
|
|
3 |
|
|
Inventory |
|
|
127 |
|
|
|
302 |
|
|
Prepaid expenses and other current assets |
|
|
108 |
|
|
|
392 |
|
|
Total current assets |
|
|
3,479 |
|
|
|
8,030 |
|
|
Property and equipment, net |
|
|
477 |
|
|
|
727 |
|
|
Intangible assets, net |
|
|
1,479 |
|
|
|
1,494 |
|
|
Other assets |
|
|
127 |
|
|
|
126 |
|
|
Total assets |
|
$ |
5,562 |
|
|
$ |
10,377 |
|
|
Liabilities and shareholders’ equity |
|
|
|
|
|
|
|
|
|
Current liabilities |
|
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
628 |
|
|
$ |
399 |
|
|
Due to related parties |
|
|
23 |
|
|
|
18 |
|
|
Note payable, current portion |
|
|
480 |
|
|
|
490 |
|
|
Other current liabilities |
|
|
1,151 |
|
|
|
634 |
|
|
Total current liabilities |
|
|
2,282 |
|
|
|
1,541 |
|
|
Note payable, net of current portion |
|
|
— |
|
|
|
369 |
|
|
Deferred rent |
|
|
107 |
|
|
|
28 |
|
|
Due to related parties, net of current portion |
|
|
12 |
|
|
|
— |
|
|
Other liabilities, net of current portion |
|
|
30 |
|
|
|
— |
|
|
Total liabilities |
|
|
2,431 |
|
|
|
1,938 |
|
|
Commitments and contingencies (note 12) |
|
|
|
|
|
|
|
|
|
Shareholders' equity |
|
|
|
|
|
|
|
|
|
Ordinary Shares |
|
|
|
|
|
|
|
|
|
$0.001 par value, 350,000,000 shares authorized; 17,121,857 and 13,420,927 shares issued and outstanding at December 31, 2017 and 2016, respectively |
|
|
17 |
|
|
|
13 |
|
|
Deferred Ordinary Shares |
|
|
|
|
|
|
|
|
|
€ 1.00 (US$1.35) par value, 39,994 shares authorized, issued and outstanding at December 31, 2017 and 2016 |
|
|
54 |
|
|
|
54 |
|
|
Preferred Shares |
|
|
|
|
|
|
|
|
|
$0.001 par value, 50,000,000 shares authorized; -0- shares issued and outstanding at December 31, 2017 and 2016 |
|
|
— |
|
|
|
— |
|
|
Additional paid-in capital |
|
|
91,362 |
|
|
|
79,676 |
|
|
Accumulated deficit |
|
|
(88,302 |
) |
|
|
(71,304 |
) |
|
Total shareholders' equity |
|
|
3,131 |
|
|
|
8,439 |
|
|
Total liabilities and shareholders’ equity |
|
$ |
5,562 |
|
|
$ |
10,377 |
|
The accompanying notes are an integral part of these consolidated financial statements
67
Consolidated Statements of Operations and Comprehensive Loss
(in thousands, except share and per share data)
|
|
|
Year Ended December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Revenues |
|
$ |
14 |
|
|
$ |
14 |
|
|
Cost of goods sold |
|
|
97 |
|
|
|
279 |
|
|
Gross loss |
|
|
(83 |
) |
|
|
(265 |
) |
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
Research and development |
|
|
7,074 |
|
|
|
5,541 |
|
|
Sales and marketing |
|
|
3,278 |
|
|
|
3,103 |
|
|
General and administrative |
|
|
6,280 |
|
|
|
6,750 |
|
|
Total operating expenses |
|
|
16,632 |
|
|
|
15,394 |
|
|
Operating loss |
|
|
(16,715 |
) |
|
|
(15,659 |
) |
|
Interest expense, net |
|
|
37 |
|
|
|
20 |
|
|
Other expense |
|
|
1 |
|
|
|
2 |
|
|
Loss before income tax provision |
|
|
(16,753 |
) |
|
|
(15,681 |
) |
|
Income tax provision |
|
|
245 |
|
|
|
76 |
|
|
Net loss |
|
$ |
(16,998 |
) |
|
$ |
(15,757 |
) |
|
Other comprehensive loss |
|
|
— |
|
|
|
— |
|
|
Total comprehensive loss |
|
$ |
(16,998 |
) |
|
$ |
(15,757 |
) |
|
Net loss per share - basic and diluted |
|
$ |
(1.04 |
) |
|
$ |
(1.18 |
) |
|
Weighted average, shares outstanding - basic and diluted |
|
|
16,298,505 |
|
|
|
13,337,560 |
|
The accompanying notes are an integral part of these consolidated financial statements
68
Consolidated Statements of Changes in Shareholders’ Equity
(in thousands, except share data)
|
|
|
Ordinary Shares |
|
|
Deferred Shares |
|
|
Additional Paid-In |
|
|
Accumulated |
|
|
|
|
|
||||||||||||
|
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Total |
|
|||||||
|
Balance, December 31, 2015 |
|
|
13,355,477 |
|
|
|
13 |
|
|
|
39,994 |
|
|
|
54 |
|
|
|
77,505 |
|
|
|
(55,547 |
) |
|
|
22,025 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
2,171 |
|
|
|
— |
|
|
|
2,171 |
|
|
Issuance of restricted shares |
|
|
65,450 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(15,757 |
) |
|
|
(15,757 |
) |
|
Balance, December 31, 2016 |
|
|
13,420,927 |
|
|
|
13 |
|
|
|
39,994 |
|
|
|
54 |
|
|
|
79,676 |
|
|
|
(71,304 |
) |
|
|
8,439 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from issuance of ordinary shares from rights offering, net of costs |
|
|
3,611,764 |
|
|
|
4 |
|
|
|
— |
|
|
|
— |
|
|
|
10,530 |
|
|
|
— |
|
|
|
10,534 |
|
|
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
1,156 |
|
|
|
— |
|
|
|
1,156 |
|
|
Issuance of restricted shares |
|
|
89,166 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(16,998 |
) |
|
|
(16,998 |
) |
|
Balance, December 31, 2017 |
|
|
17,121,857 |
|
|
$ |
17 |
|
|
|
39,994 |
|
|
$ |
54 |
|
|
$ |
91,362 |
|
|
|
(88,302 |
) |
|
|
3,131 |
|
The accompanying notes are an integral part of these consolidated financial statements
69
Consolidated Statements of Cash Flows
(in thousands)
|
|
|
Year Ended |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Cash flow from operating activities: |
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(16,998 |
) |
|
$ |
(15,757 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
477 |
|
|
|
253 |
|
|
Inventory provisions |
|
|
90 |
|
|
|
278 |
|
|
Loss on disposal of fixed assets |
|
|
1 |
|
|
|
4 |
|
|
Stock-based compensation |
|
|
1,168 |
|
|
|
2,171 |
|
|
Imputed interest expense |
|
|
83 |
|
|
|
37 |
|
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
Accounts receivable |
|
|
(5 |
) |
|
|
113 |
|
|
Inventory |
|
|
85 |
|
|
|
(149 |
) |
|
Prepaid expenses and other current assets |
|
|
286 |
|
|
|
16 |
|
|
Other assets |
|
|
(1 |
) |
|
|
(98 |
) |
|
Accounts payable and other current liabilities |
|
|
528 |
|
|
|
(305 |
) |
|
Income taxes payable |
|
|
231 |
|
|
|
8 |
|
|
Deferred rent |
|
|
79 |
|
|
|
59 |
|
|
Due to related parties |
|
|
5 |
|
|
|
(37 |
) |
|
Net cash used in operating activities |
|
|
(13,971 |
) |
|
|
(13,407 |
) |
|
Cash flow from investing activities: |
|
|
|
|
|
|
|
|
|
Purchase of intangible assets |
|
|
(125 |
) |
|
|
(145 |
) |
|
Purchases of property and equipment |
|
|
(73 |
) |
|
|
(140 |
) |
|
Net cash used in investing activities |
|
|
(198 |
) |
|
|
(285 |
) |
|
Cash flow from financing activities: |
|
|
|
|
|
|
|
|
|
Proceeds from issuance of ordinary shares from rights offering, net of costs |
|
|
10,534 |
|
|
|
— |
|
|
Payment of note payable to Neoptics A.G. |
|
|
(510 |
) |
|
|
(516 |
) |
|
Deferred offering costs |
|
|
— |
|
|
|
(167 |
) |
|
Proceeds from sale of equipment |
|
|
— |
|
|
|
1 |
|
|
Net cash (used in) provided by financing activities |
|
|
10,024 |
|
|
|
(682 |
) |
|
Net decrease in cash |
|
|
(4,145 |
) |
|
|
(14,374 |
) |
|
Effect of exchange rate on cash |
|
|
48 |
|
|
|
(42 |
) |
|
Cash balance at beginning of period |
|
|
7,333 |
|
|
|
21,749 |
|
|
Cash balance at end of period |
|
$ |
3,236 |
|
|
$ |
7,333 |
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental disclosure of cash flow information: |
|
|
|
|
|
|
|
|
|
Cash paid for income taxes |
|
$ |
19 |
|
|
$ |
50 |
|
|
|
|
|
|
|
|
|
|
|
|
Supplemental disclosures of non-cash investing and financing activities: |
|
|
|
|
|
|
|
|
|
Capitalized intangible asset costs included in accounts payable |
|
$ |
15 |
|
|
$ |
4 |
|
|
Deferred offering costs included in accounts payable and other current liabilities |
|
$ |
— |
|
|
$ |
91 |
|
The accompanying notes are an integral part of these consolidated financial statements
70
Notes to the Consolidated Financial Statements
(1) Basis of Presentation and Description of the Business
Presbia PLC (the “Company” or “Presbia PLC”), an Irish public limited company, was formed on February 6, 2014 through the issuance of 40,000 shares of €1.00 each for 40,000 Euro (approximately $54,000) for the purpose of consummating an initial public offering (“IPO”) of its ordinary shares. Presbia PLC’s ultimate controlling shareholder, Presbia Holdings (the “Parent”), was organized in the Cayman Islands in 2007 as an exempted company with limited liability. On October 21, 2013, the Parent completed a restructuring (the “2013 Restructuring”) which involved the formation on September 13, 2013 of an interim holding company, Presbia Ireland, Limited, and the contribution by the Parent of 100% of its direct and indirect ownership interests in its business, assets and subsidiaries to Presbia Ireland, Limited.
On November 30, 2014, Presbia Holdings converted all the remaining indebtedness owed to Presbia Holdings by certain subsidiaries of Presbia Ireland, Limited at that time to equity (“2014 Debt Conversion”). In the 2014 Debt Conversion, approximately $23.5 million of outstanding intercompany debt owed to Presbia Holdings was converted to equity of Presbia Ireland, Limited. On January 14, 2015, Presbia Holdings converted all the remaining indebtedness owed to Presbia Holdings by a subsidiary of Presbia Ireland, Limited at that time to equity (the “2015 Debt Conversion”) in the amount of approximately $1.6 million.
On January 14, 2015, the Parent contributed all the share capital in issue in Presbia Ireland, Limited to Presbia PLC (the “2015 Capital Contribution”) in exchange for 9,166,667 ordinary shares of US$0.01 each of Presbia PLC. The aggregate 9,166,667 ordinary shares of Presbia PLC have been reflected as issued and outstanding as of the earliest date of the periods presented for purposes of computation of basic and diluted net loss per share.
On February 3, 2015, Presbia PLC completed its initial public offering (“IPO”) of 4,166,667 of its ordinary shares at a price to the public of $10.00 per ordinary share and commenced trading on The NASDAQ Global Market under the symbol LENS. The Parent acquired 500,000 ordinary shares from the public offering. The net proceeds from the IPO consisted of aggregate gross proceeds of approximately $41.7 million less underwriting discounts and commissions of approximately $2.9 million and other issuance costs of approximately $2.0 million resulting in net proceeds of approximately $36.8 million.
On August 3, 2015, Presbia Holdings completed the distribution of its holdings of 9,666,667 ordinary shares of Presbia PLC to its ordinary shareholders for the purpose of dissolving Presbia Holdings on November 25, 2015 and ceasing to be the Parent of Presbia PLC.
The accompanying consolidated financial statements have been derived from the historical cost basis of the assets and liabilities, financial condition and cash flows of Presbia PLC, Presbia Ireland, Limited, organized in Ireland, Presbia Investments organized in the Cayman Islands and Presbia USA, Inc., and OPL, LLC. Presbia USA, Inc. and OPL, LLC are both entities organized in the United States, and include Presbia USA, Inc.’s subsidiaries, Visitome, Inc. and PresbiBio, LLC, both organized in the United States, and OPL, LLC’s direct and indirect subsidiaries, PIP Holdings, C.V and Presbia Cooperatief U.A., both organized in the Netherlands, and PresbiOptical LLC, organized in the United States (collectively, including Presbia PLC, the “Company”). The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America. The Company’s fiscal year ends on December 31. The entities presented in the consolidated financial statements have been under common control during the periods presented. All intercompany accounts have been eliminated in consolidation. All amounts are shown in U.S. dollars.
The Company, which began to be formed on June 29, 2007, has developed and is marketing a surgical solution to the age-related vision condition known as presbyopia. The Company’s primary objectives have been twofold: (1) achieve commercial success in those parts of the world where the Company’s surgical procedure has been approved by regulatory agencies and (2) successfully apply to the United States Food and Drug Administration (the “FDA”) for the authorization to market and manufacture the surgical procedure in the United States. Other than the FDA clinical trials that begun in 2014 and will continue to at least 2018, this procedure is currently not authorized by the FDA and may not be manufactured, sold, distributed or surgically performed on any individual in the United States. The Company’s principal revenue generating activity is the sale of the Company’s microlens to approved ophthalmologist physicians who, in turn, perform the surgical procedure on their patient base. Presbia is authorized to sell, market and perform this procedure in 40 countries through its wholly-owned Dutch subsidiary, Presbia Cooperatief U.A. Activities to-date have consisted primarily of research and development and expansion of its commercial activities in selected markets outside the United States.
71
At December 31, 2017, the Company has an accumulated deficit of $88.3 million and it expects to incur additional operating losses through 2018, and possibly further. As it continues to incur losses, the Company’s transition to profitability will depend on the successful development, approval and commercialization of its microlens. The Company may never achieve profitability, and unless and until it does, it will need to continue to raise additional capital. Based on the Company’s current business plan, management believes that its cash and cash equivalents at December 31, 2017, will not be sufficient to meet its anticipated cash requirements during the twelve-month period subsequent to the issuance of the financial statements included in this Annual Report on Form 10-K. These factors raise substantial doubt about the Company’s ability to continue as a going concern within one year from the date this Annual Report on Form 10-K is filed with the U.S. Securities and Exchange Commission (SEC). The Company’s current commercialization strategy is targeted to countries where the Company can establish the market for its technology. This commercialization strategy will undergo continual prioritization and in the future the Company may adjust its commercialization efforts to preserve its existing cash or realize better results than anticipated which could have a positive impact on cash. The Company’s U.S. pivotal clinical trial and planned FDA approval is its highest priority. The Company needs to raise additional capital to fund its operations. It may raise additional capital through equity offerings, debt financings, collaborations and/or licensing arrangements. Additional funds may not be available when the Company needs them on terms that are acceptable to the Company, or at all. If adequate funds are not available on acceptable terms, the Company may be required to delay, reduce the scope of, or curtail, its operations. To the extent that the Company raises additional funds by issuing equity securities, its shareholders will experience dilution, and debt financing, if available, may involve restrictive covenants.
(2) Summary of Significant Accounting Policies
Use of Estimates
The preparation of the Company’s consolidated financial statements in conformity with accounting principles generally accepted in the United States of America requires that management makes estimates and assumptions that are included in the consolidated financial statements and accompanying notes. The actual results may differ from those estimates.
Segment Information
In accordance with Accounting Standard Codification (“ASC”) 280-10-50, Segment Reporting, operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision-maker in making decisions regarding resource allocation and assessing performance. Since 2013, the Company has viewed and managed its operations and business as one segment through its wholly-owned Dutch subsidiary, Presbia Cooperatief U.A. and its wholly-owned Irish entity, Presbia Ireland, Limited, operating primarily in South Korea, Australia, the Netherlands, Ireland, Canada and Germany.
Foreign Currency
The functional currency of subsidiaries outside the United States of America is the U.S. Dollar. Transactions in foreign currencies during the year are re-measured at rates of exchange on the dates of the transactions. Gains and losses related to re-measurement of items arising through operating activities are accounted for in the statement of operations and comprehensive loss and included in general and administrative expense. Aggregate foreign exchange loss was $59,000 for the year ended December 31, 2017, and aggregate foreign exchange gain was $1,000 for the year ended December 31, 2016.
Reclassifications
Certain prior year balances within the consolidated financial statements have been classified to conform to current year presentation.
Concentrations of Credit Risk
Cash is generally deposited in demand deposit accounts that, from time to time, may exceed insurable limits. The Company has not experienced any losses from its deposits of cash.
Fair Value of Financial Instruments
The carrying values of certain of the Company’s financial instruments, such as prepaid expenses, accounts payable and accrued expenses, approximate fair value due to their short maturities. The Company’s debt obligation approximates fair value as a result of imputed interest which approximates prevailing market rates. Amounts payable to related parties, including the payable due to the Parent, which has no fixed maturity or expiration date, do not have readily determinable fair values.
72
The Company considers highly liquid investments with original maturities less than 90 days to be cash equivalents. As of December 31, 2017 and 2016, the Company had no such short-term investment instruments and maintained its cash in bank demand deposit accounts.
Inventory
The Company accounts for inventory at the lower of market or cost. Inventory is stated at weighted average cost, which is determined by applying the current average cost to the ending inventory. Inventory consists of lenses and lens inserters and other accessories used by physicians in the surgical process associated with the lenses. The Company maintains serialized records of all lenses, including the five-year expiration date of each lens, after which the lens cannot be sold. The Company considers the expiration dates of lenses, in addition to comparing the carrying amount of inventory to expected demand, and will write-down inventory for amounts determined to be excess or obsolete.
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation and amortization. Depreciation of property and equipment is computed using the straight-line method over their estimated useful lives of three to ten years. Leasehold improvements are amortized over the lesser of the useful life of the asset or the remaining term of the lease. Repairs and maintenance of property and equipment are expensed as incurred. Upon retirement or sale, the original cost and accumulated depreciation are removed and any gain or loss is recognized in the consolidated statements of operations and comprehensive loss.
Impairment of Long-Lived Assets
The Company reviews the recoverability of long-lived and finite-lived intangible assets when circumstances indicate that the carrying amount of assets might not be recoverable. This evaluation is based on various analyses, including undiscounted cash flow projections. In the event undiscounted cash flow projections indicate impairment, the Company would record an impairment based on the fair value of the assets at the date of the impairment.
Intangible Assets
On August 2, 2016, Presbia Ireland, Limited, a wholly-owned subsidiary of the Company, entered into an asset purchase agreement (the “Agreement”) with Neoptics AG (“Neoptics”) pursuant to which the Company acquired certain assets from Neoptics for an aggregate purchase price of 1.5 million Swiss Francs (approximately $1.5 million based on the exchange rate between the Swiss Franc and the U.S. Dollar on August 2, 2016) payable in three equal installments on each of August 2, 2016, December 31, 2017 and December 31, 2018. The assets acquired include patents, pending patents, and other immaterial assets. Effective August 2, 2016, the Company became responsible for all expenses associated with such assets. Pursuant to the terms of the Agreement, the acquisition closed on August 2, 2016. The Company included a reclassification of $516,000 from cash flow from investing activities to cash flow from financing activities in its Consolidated Statements of Cash Flows for the year ended December 31, 2016. This reclassification is related to its payment of the first installment to Neoptics and conforms to current year presentation.
Substantially all of the fair value of the gross assets acquired is concentrated in the acquired intellectual property rights or patents and not in the trademarks, documents or electronic data obtained from the Seller. The Company determined the intellectual property right or patents were akin to inputs within the scope of ASC 805 “Business Combinations” and that no substantive processes were acquired in the assets acquisition. Therefore, the Company applied the asset acquisition accounting guidance in ASC 805-50-30 “Initial Measurement of Acquisition of Assets Rather than a Business.”
The Company paid $0.6 million in cash in exchange for these patents and patent applications in the year ended December 31, 2017. The useful life of the patents was determined to be 12 years and 4 months and will be amortized through November 25, 2028. The Company recorded amortization expense of approximately $140,000 with net carrying amount of approximately $1,477,000 as of December 31, 2017. At December 31, 2017, a note payable of $480,000 is recorded on the consolidated Balance Sheet in connection with the final payment due by December 31, 2018. For fiscal years 2018 to 2022, approximately $135,000 will be recorded annually as amortization expense for the Neoptics assets.
Costs incurred to develop software for the Company’s website are capitalized and amortized over the estimated useful life of the software and are included in intangible assets. Costs related to design or maintenance of website development is expensed as incurred. For the years ended December 31, 2017 and 2016, the Company capitalized $0 associated with website development. Amortization expense of $15,000 was recorded in each of the years ended December 31, 2017 and 2016.
73
Comprehensive income or loss is defined as a change in equity of a company attributable to all transactions excluding those transactions resulting from investment with owners and distributions to owners. There were no differences between net loss and comprehensive loss in the years ended December 31, 2017 and 2016.
Revenue Recognition
The Company recognizes revenue when there is persuasive evidence that an arrangement exists with the customer, selling prices are fixed or determinable, title or risk of loss has passed, and collection is reasonably assured. Revenue is recognized upon shipment and payments are either received in advance, or net 30 days. In the years ended December 31, 2017 and 2016 there were four and three customers, respectively, that represented 100% of total sales recognized for each year. As of December 31, 2017, the Company was not authorized to manufacture or sell any of its products or services within the United States and, as a result, all of the Company’s revenues are derived from foreign customers. The Company recognized revenues of $14,000 from foreign ophthalmic clinics during the years ended December 31, 2017 and 2016.
Clinical Trials
In February 2015, the Company received approval from the FDA to commence enrollment of the second stage of its U.S. staged pivotal clinical trial of its Microlens involving the implantation of presbyopic patients. Enrollment for the second stage was completed in September 2015. The first staged-enrollment commenced during the first quarter of 2014, and during these trials, the Company incurred costs for patient recruiting, acquisition of clinical test equipment to be used in the trials, outside experts to read and interpret the results of the studies, third party costs to monitor the investigational sites and perform data collection activities and surgeon and patient fees in connection with the surgical procedures and follow-up visits. The Company’s policy with respect to the recognition of these costs is to record such costs as research and development expense in the consolidated statements of operations and comprehensive loss in the period in which the services are provided. The Company will evaluate the purchases of clinical test equipment, on a case by case basis, to determine if there exists an alternative use for the equipment following the clinical trials. In the event that the Company determines that there is no alternative use for the test equipment, that cost will be expensed as part of research and development expense.
Stock-Based Compensation
The compensation cost of stock-based awards granted to employees is measured at grant date, based on the estimated fair value of the award. The Company estimates the fair value of stock options using a Black-Scholes option pricing model. Additionally, the fair value of restricted stock units is determined using a Monte Carlo Simulation, or “MCS”, methodology, which considers the separate probabilities that of the price thresholds or market conditions will be achieved under the Restricted Stock Units Plan guidelines. Compensation expense for options granted to non-employees is determined as the fair value of consideration received or the fair value of the equity instruments issued, whichever is more reliably measured. Stock-based compensation costs are expensed on a straight-line basis over the service period. The fair value of awards granted to non-employees is remeasured each period until the related service is complete or there exists a significant disincentive not to perform the required services. Stock-based compensation costs are reflected in the accompanying statements of operations and comprehensive loss based upon the underlying employees’ roles within the Company.
Deferred Offering Costs
Generally, costs incurred such as legal, accounting and other professional fees including printing costs in the course of preparing for a capital raise are deferred on the balance sheet as deferred offering costs, which are eventually reclassified when the capital raise is concluded as a reduction of the proceeds of the capital raise as part of additional paid in capital. On December 5, 2016, the Company filed with the SEC a Form S-1 Registration Statement under the Securities Act of 1933 for the purpose of distributing to holders of the Company’s ordinary shares, non-transferable and non-tradeable subscription rights to purchase ordinary shares (“rights offering”). During the years ended December 31, 2017 and 2016, the Company incurred approximately $0.3 million and $0.2 million related to its rights offering, respectively, which was completed on March 8, 2017. Upon completion of the rights offering, the Company netted approximately $0.3 million in offering costs against the gross proceeds in shareholders’ equity as of December 31, 2017.
Income Taxes
As described in Note 1, the Parent completed a restructuring on October 21, 2013. As a result, some of the entities are no longer pass-through entities or were restructured as taxable entities. Provisions for federal, foreign, state, and local income taxes are calculated on pre-tax income based on current tax law and include the cumulative effect of any changes in tax rates from those used previously in determining deferred tax assets and liabilities. Deferred income tax assets and liabilities are recorded for the expected future tax
74
consequences of temporary differences between the financial statement carrying amounts and the income tax basis of assets and liabilities. A valuation allowance is recorded to reduce net deferred income tax assets to amounts that are more likely than not to be realized.
Advertising Costs
The Company incurs direct response advertising expense outside the United States in order to create awareness of the Company’s Microlens solution to presbyopia. The Company’s policy with respect to direct response advertising is to defer costs related to media and setup costs and amortize those costs to the consolidated statements of operations and comprehensive loss in the period in which the related revenue is recognized. Due to the early stages of market development and the uncertainty around the impact of advertising campaigns to-date, the Company expensed approximately $395,000, and $198,000 as part of sales and marketing expense in the consolidated statements of operations and comprehensive loss for the years ended December 31, 2017 and 2016, respectively.
Risks and Uncertainties
The Company’s product requires the approval of the FDA and regulatory agencies in the countries where the Company operates or expects to establish operations in the future. There is no assurance that the Company’s products will receive or maintain the necessary approvals to begin or continue operations. If the approvals are denied, delayed or withdrawn, there may be a material adverse impact on the Company’s results of operations and related cash flows.
Recently Adopted Accounting Standards
In November 2015, the FASB issued ASU 2015-17, Balance Sheet Classification of Deferred Taxes. ASU 2015-17 requires all deferred tax assets and liabilities to be classified as non-current on the balance sheet. This amendment simplifies the presentation of deferred income taxes. ASU 2015-17 became effective January 1, 2017 and did not have a material impact on our consolidated financial statements.
In March 2016, the Financial Accounting Standards Board issued ASU 2016-09 Compensation - Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting. ASU 2016-09 modifies multiple provisions intended to simplify various aspects of accounting for share-based payments including income tax consequences, accounting for forfeitures, classification of awards as either equity or liabilities, and classification on the statement of cash flows. ASU 2016-09 became effective January 1, 2017 and did not have a material impact on our consolidated financial statements.
Future Accounting Pronouncements
In May 2014, the FASB issued ASU 2014-09, Revenue from Contracts with Customers (Topic 606). The new standard is based on the principle that revenue should be recognized to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. ASU 2014-09 will be effective for the Company in the first quarter of 2018 and allows for full retrospective or a modified retrospective adoption approach. The Company does not believe that there is a material adjustment on the basis of either a full retrospective or the modified retrospective restatements of 2016 and 2017.
In February 2016, the FASB issued ASU 2016-02, Leases (Topic 842). The new standard requires lessees to recognize most leases on their balance sheets as lease liabilities with corresponding right-of-use assets and eliminates certain real estate-specific provisions. ASU 2016-02 will be effective for the Company in the first quarter of 2019. ASU 2016-02 will be adopted on a modified retrospective transition basis for leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements. Although the Company is currently evaluating the full impact of ASU 2016-02 on its consolidated financial statements, upon adoption of ASU 2016-2 the Company anticipates that its operating leases will be recognized on the balance sheet as a lease liability if certain criteria is met, with a corresponding right of use asset.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows, which clarifies the classification of certain cash receipts and payments. The specific cash flow issues addressed by ASU 2016-15, with the objective of reducing the existing diversity in practice, are as follows: (1) Debt prepayment or debt extinguishment costs; (2) Settlement of zero-coupon debt instruments or other debt instruments with insignificant coupon interest rates; (3) Contingent consideration payments made after a business combination; (4) Proceeds from the settlement of insurance claims; (5) Proceeds from the settlement of corporate-owned life insurance policies; (6) Distributions received from equity method investees; (7) Beneficial interest in securitization transactions; and (8) Separately identifiable cash flows and application of the predominance in principle. ASU 2016-15 will be effective for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2017. The Company adopted this new standard in January 2018 and it did not have a material impact on its consolidated financial statements.
75
In October 2016, the FASB issued Accounting Standards Update (ASU) No. 2016-16, Income Taxes (Topic 740): Intra-Entity Transfers of Assets Other than Inventory. This update requires entities to recognize the income tax consequences of an intra-entity transfer of an asset other than inventory when the transfer occurs. This update is effective for fiscal years beginning after December 15, 2017, including interim periods within those fiscal years. Early adoption is permitted as of the beginning of a fiscal year. The new standard must be adopted using a modified retrospective transition method, which is a cumulative-effective adjustment to retained earnings as of the beginning of the first effective reporting period. The Company adopted this new standard in January 2018 and it did not have a material impact on its consolidated financial statements.
In May 2017, the FASB issued ASU 2017-09, Compensation - Stock Compensation (Topic 718): Scope of Modification Accounting, which provides guidance about which changes to the terms or conditions of a share-based payment award require an entity to apply modification accounting in Topic 718. This update is effective for annual reporting periods beginning after December 15, 2017. Early adoption is permitted. The Company does not anticipate that the adoption of this ASU will have a material impact on its consolidated financial statements.
In February 2018, the FASB issued Accounting Standards Update (“ASU”) No. 2018-02, Income Statement—Reporting Comprehensive Income (Topic 220)—Reclassification of Certain Tax Effects from Accumulated Other Comprehensive Income. This update was issued to address the income tax accounting treatment of the stranded tax effects within other comprehensive income due to the prohibition of backward tracing due to an income tax rate change that was initially recorded in other comprehensive income. This issue came about from the enactment of the Tax Cuts and Jobs Act on December 22, 2017, which changed the Company’s income tax rate from 35% to 21%. The ASU changed current accounting whereby an entity may elect to reclassify the stranded tax effect from accumulated other comprehensive income to retained earnings. The ASU is effective for periods beginning after December 15, 2018, although early adoption is permitted. The Company does not anticipate that the adoption of this ASU will have a material impact on its consolidated financial statements.
(3) Inventory
The Company maintains a serial number tracking system that measures shelf life such that no lens that has aged beyond four years shall be delivered to a customer. During the years ended December 31, 2017 and 2016, inventory write-downs reflecting excess quantities on hand were recognized in the amount of $90,000 and $278,000, respectively, based on the age of the lens inventory, inserters, accessories and the refractive mix of the inventory. Finished goods inventory consists of the following (in thousands):
|
|
|
December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Lenses |
|
$ |
55 |
|
|
$ |
175 |
|
|
Accessories |
|
|
72 |
|
|
|
127 |
|
|
Inventory |
|
$ |
127 |
|
|
$ |
302 |
|
(4) Property and Equipment, net
Property and equipment, net consist of the following (in thousands):
|
|
|
December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Office equipment and computers |
|
$ |
79 |
|
|
$ |
78 |
|
|
Leasehold improvements |
|
|
214 |
|
|
|
168 |
|
|
Production equipment and facilities |
|
|
802 |
|
|
|
788 |
|
|
Software |
|
|
62 |
|
|
|
62 |
|
|
Furniture and vehicles |
|
|
237 |
|
|
|
227 |
|
|
|
|
|
1,394 |
|
|
|
1,323 |
|
|
Less: accumulated depreciation |
|
|
(917 |
) |
|
|
(596 |
) |
|
Property and equipment, net |
|
$ |
477 |
|
|
$ |
727 |
|
Depreciation and amortization expense related to property and equipment for the years ended December 31, 2017 and 2016 was $322,000 and $186,000, respectively and classified as part of operating expenses in the Consolidated Statements of Operations and Comprehensive Loss for all periods. In connection with the closure of an office facility in December 2017, approximately $67,000 of unrecognized depreciation for furniture and leasehold improvements and $52,000 of engineering tools, molds and fixtures were recorded in the Consolidated Statements of Operations and Comprehensive Loss for the period ended December 31, 2017 (see Note 14).
76
(5) Note Payable
Note payable is summarized as follows (in thousands):
|
|
|
December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Noninterest bearing note payable to Neoptics A.G. |
|
$ |
518 |
|
|
$ |
981 |
|
|
Less present value discount |
|
|
(38 |
) |
|
|
(122 |
) |
|
Note payable, net |
|
|
480 |
|
|
|
859 |
|
|
Less current portion |
|
|
480 |
|
|
|
490 |
|
|
Note payable, long term |
|
$ |
— |
|
|
$ |
369 |
|
Presbia Ireland, Limited, a wholly-owned subsidiary of the Company, entered into an asset purchase agreement with Neoptics AG pursuant to which the Company acquired certain assets from Neoptics for an aggregate purchase price of 1.5 million Swiss Francs (approximately $1.5 million based on the exchange rate between the Swiss Franc and US Dollar on August 2, 2016), bearing no interest and payable in three equal installments on each of August 2, 2016, December 31, 2017 and December 31, 2018. The Company recorded the intangible asset and a loan discount using a rate of 9%. The remaining liability is recorded in the Company’s balance sheet, net of unamortized discount of $38,000 at December 31, 2017. The discount on the note payable is being amortized to imputed interest expense over the life of the note.
(6) Other Current Liabilities
Other current liabilities consist of the following as of the dates set forth below (in thousands):
|
|
|
December 31, |
|
||||||||||
|
|
|
2017 |
|
|
2016 |
|
|||||||
|
Accrued payroll and severance related |
|
$ |
352 |
|
|
|
392 |
|
|||||
|
Accrued professional fees |
|
|
109 |
|
|
|
71 |
|
|||||
|
Accrued clinical expense |
|
|
125 |
|
|
|
— |
|
|||||
|
Accrued deferred rent expense |
|
|
236 |
|
|
|
52 |
|
|||||
|
Accrued other expense |
|
|
46 |
|
|
|
67 |
|
|||||
|
Income tax payable |
|
|
283 |
|
|
|
52 |
|
|||||
|
|
|
$ |
1,151 |
|
|
$ |
634 |
|
|||||
Other current liabilities for the year ended December 31, 2016 includes a reclassification of $52,000 related to the current portion of deferred rent, to conform to the current year presentation.
(7) Loss per Share
Basic net loss per ordinary share is calculated by dividing net loss allocated to ordinary shareholders by the weighted average number of ordinary shares outstanding during the reporting period, less unvested restricted stock awards. Diluted net loss allocated to ordinary shareholders per share is calculated based on the weighted average number of ordinary shares and dilutive potential ordinary shares outstanding during the period. Dilutive potential ordinary shares consist of the shares issuable upon the exercise of options and upon the vesting of restricted shares under the treasury stock method. In net loss periods, basic and diluted net loss per share are identical since the effect of potential ordinary shares is anti-dilutive and therefore excluded.
Basic and diluted net loss per share were calculated as follows (in thousands, except share and per share data):
|
|
|
December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Net Loss |
|
$ |
(16,998 |
) |
|
$ |
(15,757 |
) |
|
Net income allocated to participating securities |
|
|
— |
|
|
|
— |
|
|
Net loss allocated to common shareholders |
|
$ |
(16,998 |
) |
|
$ |
(15,757 |
) |
|
Weighted average shares outstanding - basic and diluted |
|
|
16,298,505 |
|
|
|
13,337,560 |
|
|
Basic and diluted net loss per share |
|
$ |
(1.04 |
) |
|
$ |
(1.18 |
) |
77
Antidilutive securities, which consist of options and restricted shares that are not included in the diluted net loss per share calculation, consisted of an aggregate of 1,037,964 and 1,082,916 weighted average shares for the fiscal years ended December 31, 2017 and 2016, respectively.
(8) Shareholders’ Equity
Ordinary Shares
As of December 31, 2017, our authorized share capital includes $400,000 and €40,000 divided into 350,000,000 ordinary shares of $0.001 each, which we refer to as the ordinary shares.
The rights and preferences of the ordinary shares are as follows: (i) subject to the right of the Company to set record dates for the purposes of determining the identity of shareholders entitled to notice of and/or to vote at a general meeting, the right to attend and speak at any general meeting of the Company and to exercise one vote per ordinary share held at any general meeting of the Company; (ii) the right to participate pro rata in all dividends declared by the Company; and (iii) the right, in the event of the Company’s winding up, to participate pro rata in the total assets of the Company. The rights attaching to the ordinary shares may be subject to the terms of issue of any series or class of preferred shares allotted by the Company’s board of directors from time to time in accordance with our articles of association.
Deferred Ordinary Shares
Capital related to Presbia PLC at December 31, 2014 consisted of ordinary shares of €1.00 each (“Euro Ordinary Shares”) of $54,000, €1.00 par value, 40,000 authorized and issued with effective date of formation of February 6, 2014 (paid-in on January 10, 2014 with $1.35: €1.00), being $54,000 in aggregate. On January 14, 2015 the Company amended its memorandum and articles of association, resulting in the re-designation of the 40,000 Euro Ordinary Shares as deferred ordinary shares (“Deferred Ordinary Shares”). In August 2015, the Company cancelled 6 Deferred Ordinary Shares pursuant to the distribution of assets held by Presbia Holdings to its ordinary shareholders and 39,994 Deferred Ordinary Shares held by Presbia Holdings were transferred to Richard Ressler.
Our articles of association provide that the holders of Deferred Ordinary Shares shall not be entitled to receive notice of, nor attend, speak or vote at, any general meeting of the Company and the holders of Deferred Ordinary Shares shall not be entitled to receive any dividend or distribution declared, made or paid or any return of capital or to any further or other right of participation in the assets of the Company. On a winding up of, or other return of capital (other than on a redemption of any class of shares in the capital of the Company) by the Company, the holders of Deferred Ordinary Shares are entitled to participate in such return of capital or winding up, such entitlement to be limited to the repayment of the amount paid up or credited as paid up on such Deferred Ordinary Shares, which shall be paid only after the holders of ordinary shares have received payment in respect of such amount as is paid up or credited as paid up on those ordinary shares held by them at that time, plus the payment in cash of $5,000,000 on each such ordinary share. The rights attaching to the Deferred Ordinary Shares may be subject to the terms of issue of any series or class of preferred shares allotted by the Company’s board of directors from time to time in accordance with our articles of association.
Preferred Shares
Our authorized share capital also includes 50,000,000 preferred shares of $0.001 each, which we refer to as preferred shares. As of December 31, 2017, no preferred shares have been issued.
Our articles of association authorize our Board, without shareholder approval, to determine the terms of the preferred shares that may be issued by us. Our Board is authorized, without obtaining any shareholder vote or consent, unless expressly provided by the terms of that class or series of class of shares, to provide from time to time for the issuance of ordinary shares or other classes or series of shares and to establish the characteristics of each such other class or series, including the number of shares, designations, relative voting rights, dividend rights, liquidation and other rights, redemption, repurchase or exchange rights and any other preferences and relative, participating, optional or other rights and limitations not inconsistent with applicable law.
Shares Issuable Pursuant to Services Agreement with OCV Management, LLC
On December 14, 2017, the Company concluded a services agreement with OCV Management, LLC for the purpose of providing management services for the Company for the period commencing December 14, 2017 until terminated upon thirty (30) days’ prior written notice by either of the parties. OCV Management, LLC will receive an annual fee of $250,000, payable annually in arrears. The obligation will be settled in the form of ordinary shares issued by the Company in a private offering. For 2018, the number of shares to be issued will be determined by dividing $250,000 by the closing price on January 2, 2018 and the shares will be issued on January 2, 2019. The Company recorded a non-current liability and a corresponding stock-based compensation expense of $12,000 as of December 31, 2017 in relation to services provided in 2017.
78
(9) Equity Based Awards
Presbia Incentive Plan
On January 14, 2015, the Company approved a compensation incentive plan (the “Presbia Incentive Plan”). The Presbia Incentive Plan permits the Company to grant awards of options, restricted shares, share appreciation rights, restricted stock units, performance shares, performance share units, dividend equivalent rights in respect of awards and other share-based and cash-based awards, including annual and long-term cash incentive awards. A total of 2,200,000 ordinary shares were authorized for issuance under the Presbia Incentive Plan of which approximately 435,141 were available on December 31, 2017 for future grants and awards. The exercise price of each option award shall be determined by the Board of Directors (or a committee thereof) at the date of grant in accordance with the terms of the Presbia Incentive Plan awards, and generally vest 20% annually over a five-year period and expire no later than 10 years from the grant date. The Presbia Incentive Plan terminates on January 14, 2025, unless terminated earlier by the board of directors. Awards under the Presbia Incentive Plan may be granted to employees, directors, consultants and other persons who perform services for the Company or a subsidiary of the Company.
The following table shows share-based compensation expense based upon all equity awards issued by Presbia PLC included in the Consolidated Statements of Operations and Comprehensive Loss for the years ended December 31, 2017 and 2016.
|
|
|
Year Ended December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Research and development |
|
$ |
251 |
|
|
$ |
259 |
|
|
General and administrative |
|
|
951 |
|
|
|
1,549 |
|
|
Sales and marketing |
|
|
(34 |
) |
|
|
363 |
|
|
|
|
$ |
1,168 |
|
|
$ |
2,171 |
|
Following the December 2017 reorganization in which 15 employees were terminated, approximately $970,000 was recorded as a reduction to share-based compensation expense in connection with the extension of vesting and exercise periods related to equity awards for these employees (see Note 14).
Options
The following table sets forth the Company’s option activity for the year ended December 31, 2017:
|
|
|
Number of Presbia PLC Shares |
|
|
Weighted Average Exercise Price Per Share |
|
|
Aggregate Intrinsic Value |
|
|||
|
Balance, January 1, 2017 |
|
|
1,050,000 |
|
|
$ |
9.77 |
|
|
|
— |
|
|
Granted |
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
Exercised |
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
Forfeited/cancelled/expired |
|
|
(200,900 |
) |
|
$ |
9.74 |
|
|
|
— |
|
|
Balance, December 31, 2017 |
|
|
849,100 |
|
|
$ |
9.77 |
|
|
|
— |
|
|
Vested, December 31, 2017 |
|
|
590,800 |
|
|
$ |
9.80 |
|
|
|
— |
|
|
Not Vested, December 31, 2017 |
|
|
258,300 |
|
|
$ |
9.72 |
|
|
|
— |
|
|
Exercisable, December 31, 2017 |
|
|
590,800 |
|
|
$ |
9.80 |
|
|
|
— |
|
79
The Company utilizes the Black-Scholes valuation model for estimating the fair value of granted stock options with the following assumptions in addition to the closing price of the Company’s ordinary shares on the date of the grant: (i) the Company estimates the expected term of the option utilizing the simplified method because of its limited history of option exercise activity and its options meet the criteria of a "plain-vanilla" option as defined by the Securities Exchange Commission (ii) due to its limited stock price volatility history, the Company uses a peer group average as permitted under ASC 718 consistent with the expected term of the stock option at the time of the grant and (iii) applies a risk-free interest rate based on the U.S. Treasury securities yield consistent with the expected term of the option at the time of the grant. The simplified method calculates the expected term as the average of the weighted average vesting period and contractual terms of the award.
For those options granted to employees, stock-based compensation expense is based upon the fair value of the option as of the grant-date and attributed to future reporting periods on a straight-line basis over the vesting period, or the requisite service period. A 3% forfeiture rate assumption was applied, which reduced the amount of expense recognized each period anticipating that a portion of all options granted would, more likely than not, be cancelled prior to the dates of its vesting periods. The forfeiture rate is subject to review and may be adjusted based upon experience. The Company did not issue employee options during the years ended December 31, 2017 and 2016.
Non-Employee Options
During the years ended December 31, 2017 and 2016, the Company did not grant options to non-employee consultants and medical advisors. In contrast to the determination of the fair value of options granted to employees, which are determined based upon the grant-date assumptions and applying the Black-Scholes model, the fair values for non-employee options and the related stock-based compensation expense are remeasured each financial reporting period based upon the assumptions applicable on the dates in which the financial statements are prepared, which are disclosed in the following table:
|
|
|
Year Ended December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Stock price per share |
|
$1.31 - $2.83 |
|
|
$2.50 - $3.49 |
|
||
|
Expected term |
|
7.5 - 8.3 Yrs. |
|
|
8.2 - 9.3 Yrs. |
|
||
|
Volatility |
|
69.3% - 85.8% |
|
|
66.1% - 92.6% |
|
||
|
Dividends |
|
|
— |
|
|
|
— |
|
|
Risk-free rate |
|
2.2% - 2.4% |
|
|
1.4% - 2.5% |
|
||
Because the performance criteria of these grants is based solely upon a requisite service period, but are subject to forfeiture if the service conditions are not met, stock-based compensation expense is determined by a straight-line attribution of the remeasured expense (mark-to-market) over the requisite service period subject to a forfeiture rate of 3%.
Restricted Shares
Consistent with the Company’s director compensation policy, during the years ended December 31, 2017 and 2016, the Company’s board of directors approved the grant of 89,166 and 65,450 restricted ordinary shares of the Company, with a grant date weighted average fair value of $3.58 and $3.50, and one-year and three-year vesting periods, respectively.
The following table sets forth the Company’s restricted share activity for the year ended December 31, 2017:
|
|
|
Unvested Number of Shares |
|
|
Weighted Average Fair Value per Share |
|
||
|
Balance, December 31, 2016 |
|
|
81,682 |
|
|
$ |
3.50 |
|
|
Granted |
|
|
89,166 |
|
|
$ |
3.58 |
|
|
Vested |
|
|
(36,481 |
) |
|
$ |
2.96 |
|
|
Forfeited/cancelled |
|
|
— |
|
|
|
— |
|
|
Unvested, December 31, 2017 |
|
|
134,367 |
|
|
$ |
3.78 |
|
80
During the year ended December 31, 2017, the Board of Directors approved the award of 97,500 restricted stock units (“RSU” or “RSU’s” or “RSU Plan”), respectively, to officers and employees in accordance with the guidelines provided by the Presbia Incentive Plan, which includes a provision that the recipient must be employed as a condition of vesting. The Presbia RSU Plan authorizes the issuance of 20% of each recipient’s total RSU award for the first occurrence that the closing price of the Company’s ordinary shares exceed, for a period of 20 consecutive business days, price thresholds of $10.00, $15.00, $20.00, $25.00 and $30.00, respectively. The RSU Plan also provides for a one-year “wait” or service period prior to any vesting permitted under the plan. The RSU Plan has a seven-year expiration period following the date of the grant.
Fair value of the RSU’s awarded was determined using a MCS methodology, which considers the separate probabilities that each of the price thresholds or market conditions will be achieved under the RSU Plan guidelines. Each probability is weighted by its respective price threshold, or its intrinsic value, which provides the basis for an aggregate fair value. The Company used the following key inputs in determining the fair value using the MCS model: (i) the volatility of the entity’s common stock and (ii) the closing price of the entity’s stock as of the measurement date of the RSU award. In accordance with GAAP, the Company recognizes as stock-based compensation expense, using a straight-line attribution method, the aggregate fair value over future periods based upon the respective derived service periods and fair values for each of the price thresholds as provided by the MCS model. A 3% forfeiture rate was applied to account for future cancellations and forfeitures. During the years ended December 31, 2017 and 2016 approximately $302,000 and $467,000, respectively, was recorded as stock-based compensation related to the RSU Plan.
During the year ended December 31, 2017, 60,000 restricted stock unit were granted to new employees and 37,500 restricted stock units were granted to existing employees.
The following table sets forth the Company’s RSU activity for the year ended December 31, 2017:
|
|
|
Unvested Number of |
|
|
Weighted Average Fair |
|
||
|
|
|
Shares |
|
|
Value per Share |
|
||
|
Balance, December 31, 2016 |
|
|
719,000 |
|
|
$ |
3.10 |
|
|
Granted |
|
|
97,500 |
|
|
$ |
1.92 |
|
|
Vested |
|
|
— |
|
|
$ |
— |
|
|
Forfeited/cancelled |
|
|
(77,500 |
) |
|
$ |
2.90 |
|
|
Unvested, December 31, 2017 |
|
|
739,000 |
|
|
$ |
2.09 |
|
As of December 31, 2017, 739,000 RSU’s were outstanding with none vested.
Unrecognized Share-based Compensation
As of December 31, 2017 and 2016, there were $0.5 million and $3.4 million, respectively, of unrecognized compensation expense related to employee and non-employee options of the Company, which collectively is expected to be recognized by the Company over the weighted average vesting period of 1.5 and 2.0 years, respectively. Unrecognized compensation expense for the same periods related to restricted shares was $0.2 million and $0.3 million, respectively, and is expected to be recognized over the weighted average vesting periods of 1.6 and 2.6 years, respectively. As of December 31, 2017, there was approximately $7.3 million of unrecognized compensation expense with respect to the RSU’s over a weighted average remaining derived service period of 2.0 years.
(10) Related Party Transactions
The following table sets forth the amounts due to related parties reflected in the accompanying consolidated balance sheets (in thousands):
|
|
|
Year Ended December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Payable to related parties-current: |
|
|
|
|
|
|
|
|
|
Management services provided by related parties |
|
$ |
35 |
|
|
$ |
18 |
|
|
Less current portion |
|
$ |
23 |
|
|
$ |
18 |
|
|
Management services provided by related parties, long term |
|
$ |
12 |
|
|
$ |
— |
|
81
Since 2011, Orchard Capital Corporation (“Orchard”), which is wholly-owned by Mr. Ressler, has provided financial analysis and bookkeeping, accounting, legal, compliance and directorial services to the Company pursuant to a Services Agreement. During the years ended December 31, 2017, and 2016, the Company recognized general and administrative expense of $14,000 and $14,000, respectively, for services invoiced by Orchard. As of December 31, 2017 and 2016, amounts due to Orchard for management and accounting services amounted to $3,000 and $4,000, respectively.
Commencing with the second quarter of 2013, the Company has received human resources management services, payroll services, IT support and risk management services from CIM Group, an entity in which Mr. Ressler has a controlling interest. The Company has incurred charges of $37,000 and $73,000 payable to CIM Group for such services for the years ended December 31, 2017 and 2016, respectively. As of December 31, 2017 and 2016, amounts due to CIM Group for human resources, payroll, information technology and legal services amounted to $19,000 and $14,000, respectively.
On December 14, 2017, the Company concluded a services agreement with OCV Management, LLC, a related party co-founded by Mr. Ressler and Mark Yung, for the purpose of providing management services for the Company for the period commencing December 14, 2017 until terminated upon thirty (30) days’ prior written notice by either of the parties. OCV Management, LLC will receive an annual fee of $250,000, payable annually in arrears. The obligation will be settled in the form of ordinary shares issued by the Company in a private offering. For 2018, the number of shares to be issued will be determined by dividing $250,000 by the closing price on January 2, 2018 and the shares will be issued on January 2, 2019. The Company recorded a non-current liability of $12,000 as of December 31, 2017 in relation to services provided in 2017 (see Note 15).
(11) Operating Segments and Geographic Information
The Company operates in one operating segment, the restoration of clear vision caused by presbyopia. The Company provides the refractive lens for patient surgeries and accessories for procedures performed exclusively outside the United States. Revenue originating in the United States is limited to intercompany transactions that do not result in any revenue generating activities to any individual or physician in the United States, and these amounts are eliminated upon consolidation. The operating losses in the United States result primarily from research and development and general and administrative costs while the operating losses in the foreign operations result primarily from sales and marketing costs and an allocation of general and administrative costs to foreign operations.
Revenues from external customers to individual countries are allocated based on the location of the customer. For the periods presented, there was no more than one customer in each individual country.
The following table sets forth the Company’s revenues generated from external customers located in foreign countries and long-lived assets by area (in thousands):
|
|
|
Year Ended December 31, |
|
|||||
|
Revenues |
|
2017 |
|
|
2016 |
|
||
|
Australia |
|
$ |
5 |
|
|
$ |
— |
|
|
New Zealand |
|
|
1 |
|
|
|
— |
|
|
Canada |
|
|
— |
|
|
|
1 |
|
|
Ireland |
|
|
— |
|
|
|
14 |
|
|
Italy |
|
|
3 |
|
|
|
— |
|
|
The Netherlands |
|
|
— |
|
|
|
(1 |
) |
|
Germany |
|
|
5 |
|
|
|
— |
|
|
Total |
|
$ |
14 |
|
|
$ |
14 |
|
|
|
|
Year Ended December 31, |
|
|||||
|
Long-lived Assets |
|
2017 |
|
|
2016 |
|
||
|
U.S. |
|
|
453 |
|
|
|
697 |
|
|
Foreign |
|
|
24 |
|
|
|
30 |
|
|
Total |
|
$ |
477 |
|
|
$ |
727 |
|
In the years ended December 31, 2017 and 2016 there were four and three customers, respectively, that represented 100% of total sales recognized for each year. As of December 31, 2017, the Company was not authorized to manufacture or sell any of its products or services within the United States and, as a result, all of the Company’s revenues are derived from foreign customers.
82
With respect to suppliers for the microlens, the Company has a five-year supplier agreement that expired in January 2017 with a lens manufacturer in Israel from which the Company received 100% of its lens supply for use in commercial activities outside the United States. The Company also has its own manufacturing facility in Irvine, California that we believe can be scaled to meet excessive lens demand or a disruption of supply sourced from outside the United States.
(12) Commitments and Contingencies
Facility Leases
In May 2012, the Company entered into a five-year non-cancelable lease for office and manufacturing space in Irvine, California that was renewed on June 1, 2016 and provided for an extension of five consecutive years starting from the original lease expiration date of May 31, 2017 through May 31, 2022. The Company maintains a one-year lease (which is now month to month) in Dublin, Ireland that commenced on December 1, 2013, and, in February 2018, it terminated a 30-month lease in Amsterdam, the Netherlands, that commenced on January 1, 2016. This termination was based on a mutual agreement with the landlord. The Company also maintains a four-year lease for office space in Irvine, California, that commenced on August 1, 2016 and will expire in September 2020. Aggregate rent expense for the years ended December 31, 2017 and 2016 was $867,000 and $388,000, respectively. The following table shows the annual base rental cost over the term of the leases (in thousands).
|
Years Ended December 31, |
|
|
|
Obligations Under Facility Leases |
|
|
|
2018 |
|
|
|
$ |
580 |
|
|
2019 |
|
|
|
|
569 |
|
|
2020 |
|
|
|
|
494 |
|
|
2021 |
|
|
|
|
283 |
|
|
2022 |
|
|
|
|
120 |
|
|
Thereafter |
|
|
|
|
— |
|
|
Total |
|
|
|
$ |
2,046 |
|
Contingencies
From time to time, the Company may be subject to legal proceedings and claims arising in the ordinary course of business. Management does not believe that the outcome of any of these matters will have a material effect on the Company’s consolidated financial statements.
(13) Income Taxes
The provision for income taxes consists of the following (in thousands):
|
|
|
Year Ended December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Current provision: |
|
|
|
|
|
|
|
|
|
Federal |
|
$ |
— |
|
|
$ |
— |
|
|
State |
|
|
2 |
|
|
|
4 |
|
|
Foreign |
|
|
243 |
|
|
|
72 |
|
|
Total current |
|
$ |
245 |
|
|
$ |
76 |
|
|
Deferred provision |
|
|
|
|
|
|
|
|
|
Federal |
|
|
(3,335 |
) |
|
|
(255 |
) |
|
State |
|
|
— |
|
|
|
— |
|
|
Foreign |
|
|
1,065 |
|
|
|
(1,228 |
) |
|
Total deferred |
|
|
(2,270 |
) |
|
|
(1,483 |
) |
|
Valuation allowance |
|
|
2,270 |
|
|
|
1,483 |
|
|
Total deferred |
|
|
— |
|
|
|
— |
|
|
Total income tax provision |
|
$ |
245 |
|
|
$ |
76 |
|
83
A reconciliation of the federal statutory rate to the effective rate is as follows:
|
|
|
Year Ended December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Expected income tax benefit at federal statutory rate |
|
|
35 |
% |
|
|
35 |
% |
|
State tax provision, net of federal benefit |
|
|
(0 |
%) |
|
|
0 |
% |
|
Foreign rate differential |
|
|
(15 |
%) |
|
|
(25 |
%) |
|
Nondeductible expenses |
|
|
(0 |
%) |
|
|
0 |
% |
|
Foreign Tax |
|
|
(1 |
%) |
|
|
0 |
% |
|
Change in valuation allowance |
|
|
(4 |
%) |
|
|
(10 |
%) |
|
Effect of Change in Tax Rate |
|
|
(9 |
%) |
|
|
0 |
% |
|
Other |
|
|
(7 |
%) |
|
|
0 |
% |
|
Income tax provision |
|
|
(1.4 |
%) |
|
|
(0.0 |
%) |
The components of the Company’s deferred tax assets are summarized as follows (in thousands):
|
|
|
Year Ended |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Deferred tax assets: |
|
|
|
|
|
|
|
|
|
Net operating loss carryforwards |
|
$ |
5,019 |
|
|
$ |
4,444 |
|
|
Accrued expenses |
|
|
31 |
|
|
|
195 |
|
|
Stock based compensation |
|
|
0 |
|
|
|
(11 |
) |
|
Deferred tax assets |
|
|
5,050 |
|
|
|
4,628 |
|
|
Valuation allowance |
|
|
(5,033 |
) |
|
|
(4,559 |
) |
|
Net deferred tax assets |
|
|
17 |
|
|
|
69 |
|
|
Deferred tax liabilities: |
|
|
|
|
|
|
|
|
|
Depreciation |
|
|
(17 |
) |
|
|
(69 |
) |
|
Total deferred tax liabilities |
|
|
(17 |
) |
|
|
(69 |
) |
|
Net deferred tax asset |
|
$ |
— |
|
|
$ |
— |
|
The valuation allowance has been established to offset the Company’s net deferred tax assets, as realization of such assets is not considered to be more likely than not due to the Company’s history of losses and uncertainties regarding the Company’s ability to generate future taxable income sufficient to realize the benefit of these deferred tax assets.
The Company has U.S. Federal net operating loss (“NOL”) carryforwards of approximately $11.1 million and $2.0 million for December 31, 2017 and 2016 respectively, subject to potential limitations pursuant to Internal Revenue Code section 382 as discussed below. The federal NOL carryforwards will begin to expire in 2033, unless previously utilized. The Company has NOL carryforwards in Ireland of $21.5 million and $21.2 million for December 31, 2017 and 2016, respectively. Under Irish rules, only such losses that occur within a three year period prior to commencing trading can be utilized to offset future trading taxable income.
Pursuant to Sections 382 of the Internal Revenue Code (the “Code”), annual use of the Company’s NOL carryforwards may be limited in the event a cumulative change in ownership of 50% of certain shareholders occurs within a three year period. An ownership change may limit the amount of NOL carryforwards that can be utilized annually to offset future taxable income and tax. In general, an “ownership change” as defined by Section 382 of the Code results from a transaction or series of transactions over a three-year period resulting in an ownership change of more than 50 percentage points of the outstanding stock of a company by certain shareholders.
As of December 31, 2017, the Company has provided a liability of $199,000 for unrecognized tax benefits relating to a foreign income tax matter. This amount is expected to reduce our effective income tax rate if recognized in future reporting periods.
The Company does not anticipate a significant increase in the unrecognized tax benefits over the next 12 months. The Company’s policy is to recognize interest expense and penalties related to income tax matters as a component of the income tax provision. As of December 31, 2017 and December 31, 2016, the Company has $39,000 and $0, respectively, of tax related accrued interest and penalties on its balance sheet or on its statement of operations.
A reconciliation of gross unrecognized tax benefits is as follows:
|
|
|
|
|
Year Ended December 31, |
||||
|
|
|
|
|
2017 |
|
|
2016 |
|
|
Balance at the beginning of the year |
|
|
|
— |
|
|
— |
|
84
|
|
|
|
199,000 |
|
|
— |
|
|
|
Balance at the end of the year |
|
|
$ |
199,000 |
|
$ |
— |
|
Due to net operating loss carryovers, the U.S. federal and state returns are open to examination by the Internal Revenue Service and state jurisdictions for years 2014 through 2017. The foreign income tax returns are open to examination for the years 2013 through 2017.
On December 22, 2017, the President signed the Tax Cuts and Jobs Act (the 2017 Act) into law. The Act will have pervasive financial reporting implications for all companies with US operations, including reduction of the U.S. federal corporate tax rate from 35 percent to 21 percent. We reviewed and incorporated the new tax bill implications through 2017 financial statements. We remeasured the deferred taxes at new corporation rate of 21%, which reduced the net deferred tax assets, before valuation allowance, by $1,567,000. Due to full valuation allowance, the change in deferred taxes was fully offset by the change in valuation allowance. The 2017 Act has no significant impact on the 2017 financial statements.
On December 22, 2017, Staff Accounting Bulletin No. 118, or SAB 118, was issued to address the application of GAAP in situations when a registrant does not have the necessary information available, prepared, or analyzed (including computations) in reasonable detail to complete the accounting for certain income tax effects of the 2017 Act. During the measurement period, impacts of the law are expected to be recorded at the time a reasonable estimate for all or a portion of the effects can be made, and provisional amounts can be recognized and adjusted as information becomes available, prepared, or analyzed. Any subsequent adjustment to these amounts will be recorded to current tax expense in 2018 when the analysis is complete.
(14) Fourth Quarter 2017 Adjustments
In December 2017, the Company announced a reorganization which resulted in a reduction of 15 employees, or 38.5% of total headcount, which affected payroll, benefits, severance and incentive pay expenses; reduced need for office space which resulted in vacant office space and termination costs; and adjustments to stock-based compensation expense related to modification of equity awards for terminated employees. In addition, the Company is recognizing a tax reserve of $238,000 for unpaid withholding taxes due to Irish tax authorities. The total amount of fourth quarter adjustments is a reduction in operating expense of $1,078,000 and an increase of tax provision expense of $238,000.
|
|
• |
Compensation Related - ($465,000) |
In December 2017, 15 employees were terminated as the Company reprioritized its efforts towards achieving FDA clinical trials approval of its Microlens by the fourth quarter of 2018. Post-employment costs were approximately $332,000 consisting of severance costs to be paid out primarily over a six-month period ending June 2018 and a reversal of $797,000 in accrued incentive compensation costs for amounts that were accrued in 2017 but are not payable as of December 31, 2017.
|
|
• |
Facilities Related - $357,000 |
Also, in December 2017, the Company vacated one of its Irvine, California facilities and consolidated its operations into another facility also located in Irvine. As of December 31, 2017, the remaining lease term of the vacated facility, of approximately 8,500 square feet, was 33 months through September 30, 2020. The Company recorded a current liability of $222,000 representing the fair value of the remaining lease obligation as part of the short-term portion of deferred rent on the Consolidated Balance Sheet as of December 31, 2017 (see note 6). The determination of the fair value was inclusive of reasonable prospective sublease assumptions, which may, or may not, materialize during the remaining term of the lease. These assumptions are reviewed and updated quarterly over the remaining term of the lease with possible adjustments to the accrual and corresponding increases or decreases to rent expense. In addition, the Company recorded write-downs of leasehold expenses, furniture and fixtures of an aggregate of $89,000 and accrued expense of $8,000 for unneeded telecom facilities. The Company also wrote down to net realizable value certain engineering tools, molds and fixtures related to discontinued engineering programs in the amount $52,000.
|
|
• |
Stock-Based Compensation Related – ($970,000) |
Equity awards granted by the Company do not include provisions to extend the terms and conditions related to vesting or exercising an award beyond the date of termination of the optionee (or holder), with the exception of stock option grants that contain a provision that allows the holder to exercise vested options (vesting must have occurred on or prior to termination date) for a period of 90 days following termination. On the date of reorganization, the Company extended the terms and conditions for all equity grants for all terminated employees providing at least a one-year extension past the termination date for all vested stock options (Type I modification as prescribed by ASC 718), and, in certain cases, the acceleration of unvested options or restricted share awards (Type III modification as prescribed by ASC 718). For those terminated employees with RSU grants based on a market condition, the Company
85
granted a one-year extension to vest according to the same market conditions as defined in the original grants. Generally, when a modification of an award occurs, the prior periods’ expense recognition associated with all unvested awards is reversed in the period of the modification on the basis that the old award is being exchanged for a new award. This resulted in a reduction of stock-based compensation expense of $1,350,000. The Company then compared the relative fair values of the old award with the terms and conditions of the modified award immediately before and after the date of modification and recorded the increase in fair value as stock-based compensation expense in the amount of $380,000, also in the same period of the date of modifications, resulting in a net reduction of stock-based compensation expense of $970,000.
|
|
• |
Irish Taxes - $238,000 |
Under Irish law non-executive Directors of Irish companies, regardless of the tax residency of such Directors, are subject to Irish income tax related to meetings held in Ireland. The Company is also jointly liable for the Irish taxes. As of December 31, 2017, the Company has not paid or arranged to be paid approximately $238,000 in withholding of income taxes required under Irish law including potential penalties and interest costs associated with the remuneration of certain board of director members with tax residencies outside of Ireland for the fiscal years 2015 to 2017. The estimated amount due for 2017 is $70,000, which the Company intends to recover from directors. The Company is working with directs to recover amounts related to years prior to 2017. As of December 31, 2017, the Company recognized a reserve of $238,000 under FIN 48, Accounting for Uncertainty of Income Taxes. The Company has not established a dialogue with Irish Revenue and cannot be certain as to the outcome of this issue.
(15) Subsequent Events
On December 14, 2017, in connection with the December 2017 reorganization, the Company concluded a management services agreement with OCV Management, LLC, a related party (see Note 10), in which the consideration to be provided to OCV for its services would be in ordinary shares equivalent in value to an annual rate of $250,000 per year and the number of ordinary shares to be determined by the closing price of the Company’s ordinary shares as of January 2, 2018. The closing price on January 2, 2018 was $3.29 indicating that the number of ordinary shares required to settle the annual obligation in January 2019 is 75,988.
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures.
Our management, with the participation of our Chief Executive Officer and Chief Accounting Officer, has evaluated the effectiveness of our disclosure controls and procedures (as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended) as of December 31, 2017. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost benefit relationship of possible controls and procedures. Based on such evaluation, our Chief Executive Officer and Chief Accounting Officer have concluded that, as of December 31, 2017, our disclosure controls and procedures were effective.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) and Rule 15d-15(f) of the Exchange Act. Our internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our internal control over financial reporting includes those policies and procedures that:
|
|
(i) |
pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; |
|
|
(ii) |
provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and |
|
|
(iii) |
provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements. |
86
The effectiveness of any system of internal control over financial reporting, including ours, is subject to inherent limitations, including the exercise of judgment in designing, implementing, operating, and evaluating the controls and procedures, and the inability to eliminate misconduct completely. Accordingly, any system of internal control over financial reporting, including ours, no matter how well designed and operated, can only provide reasonable, not absolute, assurances. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies or procedures may deteriorate. Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2017. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework (2013). Based on our assessment using those criteria, our management, including the Company’s principal executive officer and principal financial officer, has concluded that our internal control over financial reporting was effective as of December 31, 2017.
Changes in Internal Control Over Financial Reporting
There was no change in our internal control over financial reporting identified in connection with the evaluation required by Rule 13a-15(d) and 15d-15(d) of the Exchange Act that occurred during the quarter ended December 31, 2017 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
None.
87
Item 10. Directors, Executive Officers and Corporate Governance.
The following table sets forth information regarding our executive officers and directors as of March 30, 2018.
|
NAME |
|
AGE |
|
POSITION(S) |
|
Mark Yung(2)(3) |
|
44 |
|
Chief Executive Officer and Executive Chairman of the Board |
|
Richard Fogarty |
|
67 |
|
Chief Accounting Officer, Vice President, Finance and Secretary |
|
Casey Lind |
|
55 |
|
Chief Operating Officer |
|
Zohar Loshitzer |
|
60 |
|
Director |
|
Gerald Farrell(1) |
|
58 |
|
Director |
|
Gerd U. Auffarth(1)(2)(3) |
|
53 |
|
Director |
|
Richard Ressler(2)(3) |
|
59 |
|
Director |
|
Robert J. Cresci(1)(2)(3) |
|
74 |
|
Director |
|
(1) |
Member of our audit committee. |
|
(2) |
Member of our nominating and corporate governance committee. |
|
(3) |
Member of our compensation committee. |
Mark Yung was elected as Executive Chairman of the board and Chief Executive Officer of Presbia PLC on December 11, 2017. Mr. Yung is co-founder and Managing Principal of OCV Management, LLC (“OCV”), an investor, owner and operator of technology and life science companies based in Los Angeles, which was founded in 2016 and is majority-owned by Richard Ressler (one of our directors and the beneficial owner of the majority of the ordinary shares of our company). Previously, Mr. Yung was a Managing Director at Orchard Capital Corp., a firm he joined in 2006. Through his affiliation with OCV and Orchard Capital, Mr. Yung has and continues to serve in various senior capacities including, among others, as Chairman and CEO of Presbia PLC (NASDAQ: LENS), an ophthalmic device company, as Executive Chairman of the board of directors of Environmental Solutions Worldwide, Inc., a clean technology company focused on the reduction of diesel emissions, as Chief Financial Officer and director of Polymer Plainfield Holdings, Inc., an OEM automotive supplier with operations in the United States, Canada, Mexico and the Caribbean, as Chairman of the Board of Vantage Surgical Systems, Inc., and as director and/or officer of Coreolis Holdings and Tradewinds Holdings. Prior to joining Orchard Capital Corp., Mr. Yung was a Senior Vice President in the Corporate Strategy and Merger and Acquisitions groups of Citigroup in New York and ABN AMRO in Amsterdam, Netherlands. Prior to his corporate strategy roles, Mr. Yung was an investment professional at JPMorgan Partners (“JPMP”). At JPMP, Mr. Yung focused on venture capital, growth equity and buyout transactions in Latin America and served as board member for various emerging companies in the region. Mr. Yung began his career in 1996 at Chase Securities, Inc., focusing on leveraged finance for cross-border buyouts and privatizations. Mr. Yung is an Independent Director of PacWest Bancorp, Inc. (NASDAQ “PACW”). Mr. Yung holds a B.A. from Cornell University and an MBA from INSEAD.
Richard Fogarty has served as Chief Accounting Officer of Presbia PLC since January 2018. He previously served as the Chief Accounting Officer of Presbia PLC and the Vice President, Finance of Presbia PLC from February 2014 to June 2016. From February 2010 to August 2013, Mr. Fogarty was Vice President, Finance and Administration and Chief Financial Officer for Plainfield Precision, Inc., a provider of contract manufacturing services to the automotive and medical device industries. Mr. Fogarty held corporate controller and corporate finance positions in publicly-held companies Collectors Universe, Inc., which provides authentication and grading services for high-value collectibles, from March 2006 to February 2010, and Impco Technologies, Inc. (now Fuel Systems Solutions, Inc.), which designs, manufactures and supplies alternative fuel products and systems, from November 2002 to March 2006. Mr. Fogarty holds an M.B.A. from Fairleigh Dickinson University and a B.S. degree from Union College. Mr. Fogarty is a Certified Management Accountant.
Casey Lind has served as Chief Operating Officer since February 2018. Prior to joining Presbia, from October 2017 to February 2018, Ms. Lind was the managing member of Pathfinder Global Way, LLC, a private consulting company. Prior to that, from 1998 to June 2017, Ms. Lind worked for Alcon, a Novartis (NYSE: NVS) company, where she held positions of increasing responsibilities, including leading the early Surgical Glaucoma and Drug Delivery R&D teams and most recently as a Project Head for the Retina group. Ms. Lind has numerous issued patents focused on drug delivery, injection control, MEMS based posterior segment drainage and flow, and manufacturing processes for phase transition drug formulations. Ms. Lind has an active role at both the national and local level in Ophthalmic World Leaders (OWL). She is an OWL Champion Member and contributes at the national level on the Executive Membership Committee, and locally, by actively planning and moderating Southern California OWL chapter meetings. Ms. Lind is a graduate of Iowa State University, with a degree in Business, and received her MBA from Webster University.
Zohar Loshitzer has served as a director of Presbia PLC since February 2014. He served as a director of Presbia Holdings from May 2007 until its liquidation in 2015. He served as the Chief Executive Officer of Presbia PLC from February 2014 to October 2014,
88
served as the President of Presbia PLC from February 2014 to January 2015, served as Chief Business Development Officer from January 2015 to September 2015. Since 2016, Mr. Loshitzer has served as principal at OCV. Since January 2005, Mr. Loshitzer has served as a principal at Orchard Capital. OCV and Orchard Capital are majority owned by Richard Ressler (one of our directors and the beneficial owner of the majority of the ordinary shares of our company), where he supports the portfolio companies of Orchard Capital by designing operational efficiencies and cost reductions, and he has served since August 2000 as the President and Chief Executive Officer of Universal Telecom Services, Inc., a provider of telecommunications services and solutions to emerging markets. He has served as Executive Vice President of Corporate Strategy of j2 Global since June 2001 and from July 1997 through June 2001 he served as the Chief Information Officer at j2 Global. Mr. Loshitzer was the founder and President of MTP Consulting, Inc., a business consulting firm, from January 2011 to August 2013, and he was the founder and President of Imali, Inc., another business consulting firm, from January 2007 to December 2010. Since 1995, he has been a Managing Director at Orchard Telecom, a provider of telecommunications products. He previously served as a consultant to MAI Systems Corporation, a provider of information technology solutions, and as a General Manager and Managing Director at Life Alert Emergency Response, Inc., a provider of security services for the elderly, which Mr. Loshitzer co-founded. He was a director of the publicly-traded OCATA Therapeutics (formerly Advanced Cell Technology Inc.) from December 2011 to February 2016, when the company was sold to a Japanese private company. He was also a director of publicly-traded Environmental Solutions Worldwide, Inc. from January 2011 to December 2015. He graduated from Tel Aviv University with a degree in Electronics Engineering.
Mr. Loshitzer has been chosen to serve on our Board as a result of his finance and business management knowledge and experience and his investment experience in start-ups and early stage financings.
Dr. Gerald Farrell has served as a director of Presbia PLC since January 2016. Dr. Farrell has been involved in the pharmaceutical industry for more than 25 years, and worked for Eli Lilly Company Limited in Ireland and the United Kingdom for the majority of his successful career. Dr. Farrell’s tenure at Eli Lilly included positions of Managing Director, leading the commercial sales and marketing in Ireland, Director Strategy, Commercial and Business in the UK accountable for long-term strategic planning and business operations as well as other positions related to compliance, production, sales, and planning. Dr. Farrell earned a Ph.D. Plant Biotechnology from University College, Cork, and a BSc Honors – Plant Science and Higher Diploma Education with Honors from University College, Galway. Dr. Farrell serves on the Board for UCD Michael Smurfit Graduate Business School, serves as a director, and previously served as president, for Irish Pharmaceutical Healthcare Association, and serves as a council member for the Dublin Chamber of Commerce. Previously, from 2003 until 2014, Dr. Farrell served as a director of Eli Lilly and Company (Ireland) Ltd.
Dr. Farrell was chosen to serve on our Board as a result of his deep experience and leadership in the pharmaceutical industry developed over the course of an over 25-year career with Eli Lilly.
Prof. Dr. Gerd U. Auffarth has served as a director of Presbia PLC since August 2015 and Dr. Auffarth has also served as the Director and Chairman of the Department of Ophthalmology at the Ruprecht-Karls-University of Heidelberg, Germany since April 2011. Since August 2006, Dr. Auffarth has been the Director of the International Vision Correction Research Centre, a research center focusing on cataract and refractive surgery, laboratory studies, and ocular pharmacology. Since June 2012, he has been the Director of The David J Apple International Laboratory for Ophthalmic Pathology, which provides analyses on ophthalmic devices. Since August 2006 he has served as the Founder and Chief Executive Officer of' Steinbeis Technology Transfer Research Company IVCRC, which carries out research with companies, including start-up companies, in the ophthalmic medical device business in cooperation with the University Eye Clinic of Heidelberg. Since March 2012, he has served as the President of the German Society for Intraocular Lens Implantation and Refractive Surgery, and he has served on the board of this society since March 1999. Since September 2012, he has served as a member of the board of the German Ophthalmological Society (DOG), and since September 2005, he has served as an emeritus member of the board of the European Society for Cataract and Refractive Surgeons (ESCRS). He received an M.D. degree from the RWTH Aachen Medical School, Aachen University of Technology, Germany, and a Professorship in Ophthalmology and Ocular Pathology of the Ruprecht-Karls-University of Heidelberg, Germany.
Dr. Auffarth was chosen to serve on our Board due to his expertise in ophthalmic matters.
Richard S. Ressler has served as a director of Presbia PLC since January 2015. Mr. Ressler served as an officer and director of Presbia Holdings from May 2007 until its liquidation in 2015. Mr. Ressler is the founder and President of Orchard Capital Corp. ("Orchard Capital"), a firm through which Mr. Ressler oversees companies in which Orchard Capital or its affiliates invest. Through his affiliation with Orchard Capital, Mr. Ressler serves in various senior capacities with, among others, CIM Group, LLC (together with its controlled affiliates, "CIM"), a vertically-integrated owner and operator of real assets, Orchard First Source Asset Management (together with its controlled affiliates, "OFSAM"), a full-service provider of capital and leveraged finance solutions to U.S. corporations, and OCV Management, LLC (“OCV”), an investor, owner and operator of technology companies. Mr. Ressler also serves as a board member for various public and private companies in which Orchard Capital or its affiliates invest, including as chairman of j2 Global, Inc. (NASDAQ "JCOM"), and chairman of CIM Commercial Trust Corporation (NASDAQ “CMCT”). Mr. Ressler served as Chairman and CEO of JCOM from 1997 to 2000 and, through an agreement with Orchard Capital, currently serves
89
as its non-executive Chairman. Mr. Ressler has served as a director of LENS since January 2015 and as chairman of CMCT since 2014. Mr. Ressler co-founded CIM in 1994 and, through an agreement with Orchard Capital, chairs its executive, investment, allocation and asset management committees and serves on its credit committee. CIM Investment Advisors, LLC, an affiliate of CIM, is registered with the United States Securities and Exchange Commission as a registered investment adviser. Mr. Ressler co-founded the predecessor of OFSAM in 2001 and, through an agreement with Orchard Capital, chairs its executive committee. OFS Capital Management, LLC, an affiliate of OFSAM, is registered with the United States Securities and Exchange Commission as a registered investment adviser. Mr. Ressler co-founded OCV in 2016 and, through an agreement with Orchard Capital, chairs its executive committee. OCV is a relying adviser of OFS Capital Management, LLC. Prior to founding Orchard Capital, from 1988 until 1994, Mr. Ressler served as Vice Chairman of Brooke Group Limited, the predecessor of Vector Group, Ltd. (NYSE "VGR") and served in various executive capacities at VGR and its subsidiaries. Prior to VGR, Mr. Ressler was with Drexel Burnham Lambert, Inc., where he focused on merger and acquisition transactions and the financing needs of middle-market companies. Mr. Ressler began his career in 1983 with Cravath, Swaine and Moore, working on public offerings, private placements, and merger and acquisition transactions. Mr. Ressler holds a B.A. from Brown University, and J.D. and M.B.A. degrees from Columbia University.
Mr. Ressler has been chosen to serve on our Board as a result of his extensive experience with, and knowledge of, business management and finance.
Robert J. Cresci has served as a director of Presbia PLC since March 2015. Robert J. Cresci has been a managing director of Pecks Management Partners Ltd., an investment management firm, since 1990. Mr. Cresci currently serves on the boards of j2 Global, Inc., Luminex Corporation, OFS Capital Corporation, CIM Commercial Trust Corporation. Mr. Cresci previously served on the board of Continucare Corporation until 2011 and the board of Sepracor, Inc. until 2009. By virtue of his time with Pecks Management Partners and the other business entities mentioned, Mr. Cresci brings to our board of directors his broad expertise and experience in accounting issues, and public company matters. Mr. Cresci holds an undergraduate degree in Engineering from the United States Military Academy at West Point and holds a M.B.A. in Finance from the Columbia University Graduate School of Business.
Mr. Cresci was chosen to serve on our Board as a result of his broad experience in accounting and investment management developed during his tenure at Pecks Management Partners and his previous experience serving on the boards of other public companies.
Each of our executive officers and directors also serves as an executive officer and/or director of our Presbia USA, Inc. subsidiary. Certain of our executive officers and directors are also officers and/or directors of other subsidiaries of our company.
There are no family relationships among any of our directors or executive officers.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires persons who own more than ten percent of a registered class of our equity securities and our directors and executive officers to file with the SEC initial reports of ownership and reports in changes in ownership of any Presbia PLC equity securities. To our knowledge, based solely on a review of the copies of such reports furnished to us, and written representations that no other reports were required during the fiscal year ended December 31, 2017, all reports required to be filed under Section 16(a) were filed on a timely basis, except as set forth below.
In connection with the exercise of his subscription rights, Robert J. Cresci had a Form 4 filing due on March 10, 2017. Such Form 4 was filed on March 13, 2017.
In connection with the grant of restricted ordinary shares, Ralph H. Thurman had a Form 4 filing due on March 15, 2017. Such Form 4 was filed on March 22, 2017.
In connection with the grant of restricted ordinary shares, Gerald Farrell had a Form 4 filing due on March 15, 2017. Such Form 4 was filed on March 22, 2017.
In connection with the grant of restricted ordinary shares, Robert J. Cresci had a Form 4 filing due on March 15, 2017. Such Form 4 was filed on March 22, 2017.
In connection with the grant of restricted ordinary shares, Vladimir Feingold had a Form 4 filing due on March 15, 2017. Such Form 4 was filed on March 22, 2017.
In connection with the grant of restricted ordinary shares, Dr. Gerd Auffarth had a Form 4 filing due on March 15, 2017. Such Form 4 was filed on March 22, 2017.
90
In connection with the grant of restricted ordinary shares, Richard Ressler had a Form 4 filing due on March 15, 2017. Such Form 4 was filed on March 22, 2017.
In connection with the exercise of his subscription rights, Todd Cooper had a Form 4 filing due on March 10, 2017. Such Form 4 was filed on March 22, 2017.
In connection with the exercise of subscription rights and the grant of restricted ordinary shares, Zohar Loshitzer had a Form 4 filing due on March 10, 2017 and March 15, 2017. Such Form 4 was filed on April 13, 2017.
Code of Business Conduct and Ethics
We have adopted a code of business conduct and ethics that applies to all of our employees, officers and Board members, including those officers responsible for financial reporting. Our code of business conduct and ethics is available on our website (http://www.presbia.com). We expect that any amendments to the code, or any waivers of its requirements, will be disclosed on our website.
Board Composition and Committees
Richard Ressler and his affiliates control a majority of our issued and outstanding ordinary shares. As a result, we are a “controlled company” within the meaning of NASDAQ listing rules, and thus, we are exempt from the following NASDAQ requirements:
|
|
• |
the requirement that a majority of our Board consist of independent directors; |
|
|
• |
the requirement that director nominees be selected, or recommended for the Board’s selection, either by independent directors constituting a majority of the Board’s independent directors in a vote in which only independent directors participate, or by a nominating committee comprised solely of independent directors; and |
|
|
• |
the requirement that we have a compensation committee that is composed entirely of independent directors with a written charter addressing the committee’s purposes and responsibilities. |
We rely on these exemptions. As a result, we do not have a majority of independent directors and we do not have a compensation committee or nominating and corporate governance committee consisting entirely of independent directors. Accordingly, our investors do not have the same protections afforded to shareholders of companies that are subject to all of NASDAQ’s corporate governance requirements.
Audit Committee. The audit committee of our Board consists of Drs. Farrell and Auffarth and Mr. Cresci, and Mr. Cresci serves as the chairman of this committee. The audit committee assists our Board in its oversight responsibilities relating to the integrity of our financial statements, the qualifications, independence, compensation and performance of our independent auditors, our systems of internal accounting and financial controls, the performance of our internal audit function, the compliance of our company with legal and regulatory requirements and compliance with our company’s Code of Business Conduct and Ethics.
Our audit committee members must satisfy both NASDAQ and SEC independence criteria. Under the NASDAQ listing rules, a director will only qualify as an “independent director” if (i) the director is not disqualified under certain objective tests established by the NASDAQ listing rules and (ii) in the opinion of the issuer’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. To be considered independent for purposes of the SEC’s rules, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the board of directors, or any other board committee: (1) accept, directly or indirectly, any consulting, advisory, or other compensatory fee from the listed company or any of its subsidiaries or (2) be an affiliated person of the listed company or any of its subsidiaries.
Our Board has determined that all members of the audit committee meet both the SEC and the NASDAQ definitions of an independent director with respect to their service on our audit committee. In making this determination, our Board considered the relationships that Drs. Farrell and Auffarth and Mr. Cresci have with our company and all other facts and circumstances our Board deemed relevant in determining their independence. Our Board has determined that Mr. Cresci qualifies as an “audit committee financial expert” under SEC rules and regulations.
Compensation Committee. The compensation committee of our Board consists of Dr. Auffarth and Messrs. Ressler, Cresci and Yung, and Mr. Yung serves as the chairman of this committee. The primary purpose of the compensation committee of our Board is to (i) facilitate our Board’s discharge of its responsibilities relating to the evaluation and compensation of our executives, (ii) oversee the administration of our compensation plans, including the Presbia Incentive Plan, (iii) review and determine Board member
91
compensation and (iv) prepare any report on executive compensation required by the rules and regulations of the SEC and the listing rules of NASDAQ.
Nominating and Corporate Governance Committee. The nominating and corporate governance committee of our Board consists of Dr. Auffarth and Messrs. Yung, Cresci and Ressler, and Mr. Yung serves as the chairman of this committee. The primary purpose of our nominating and corporate governance committee is to (i) review the qualifications of, and recommend to our Board, proposed nominees for election to our Board, consistent with criteria approved by our Board, (ii) select, or recommend that our Board select, the director nominees for the next annual meeting of shareholders, (iii) develop, evaluate and recommend to our Board corporate governance practices applicable to our company and (iv) lead our Board in its annual review of the Board and management.
Our Board has adopted written charters under which the audit committee, compensation committee and nominating and corporate governance committee operate. A copy of each of these charters, which satisfy the applicable standards and rules of the SEC and NASDAQ, is available on our website (http://www.presbia.com).
Board Diversity
Our nominating and corporate governance committee is responsible for reviewing with the Board, on an annual basis, the appropriate characteristics, skills and experience required for the Board as a whole and its individual members. In evaluating the suitability of individual candidates (both new candidates and current members), the nominating and corporate governance committee, in recommending candidates for election, and the Board, in approving (and, in the case of vacancies, appointing) such candidates, will take into account many factors, including the following:
|
|
• |
diversity of personal and professional background, perspective and experience; |
|
|
• |
personal and professional integrity, ethics and values; |
|
|
• |
experience in corporate management, operations or finance, such as serving as an officer or former officer of a publicly-traded company, and a general understanding of marketing, finance and other elements relevant to the success of a publicly-traded company in today’s business environment; |
|
|
• |
experience relevant to our industry and with relevant social policy concerns; |
|
|
• |
experience as a board member or executive officer of another publicly-traded company; |
|
|
• |
relevant academic expertise or other proficiency in an area of our operations; |
|
|
• |
practical and mature business judgment, including ability to make independent analytical inquiries; |
|
|
• |
promotion of a diversity of business or career experience relevant to the success of our company; and |
|
|
• |
any other relevant qualifications, attributes or skills. |
Our Board intends to evaluate each individual in the context of the Board as a whole, with the objective of assembling a group that can best maximize the success of the business and represent shareholder interests through the exercise of sound judgment using its diversity of experience in these various areas.
Item 11. Executive Compensation.
This section discusses the material components of the executive compensation program for our named executive officers who are identified in the Summary Compensation Table below. This discussion may contain forward-looking statements that are based on our current plans, considerations, expectations and determinations regarding future compensation programs. Actual compensation programs that we adopt following the completion of this offering may differ materially from the currently planned programs summarized in this discussion.
Summary Compensation Table
The following table shows the compensation awarded to or earned by our principal executive officer, and all individuals who served as principal executive officer at any time during the fiscal year ended December 31, 2017, our two other most highly compensated executive officers who were serving as executive officers as of December 31, 2017 and up to two additional individuals for whom disclosure would have been provided but for the fact that the individual was not serving as an executive officer as of December 31, 2017. The persons listed in the following table are referred to herein as the “named executive officers.”
92
|
NAME AND PRINCIPLE POSITION |
|
YEAR |
|
|
SALARY ($) |
|
|
|
BONUS ($) |
|
|
STOCK AWARDS ($) |
|
|
|
RESTRICTED STOCK UNITS ($) |
|
|
|
NON-EQUITY INCENTIVE PLAN COMPENSA- TION ($) |
|
|
NON- EQUITY INCENTIVE PLAN COMPENSA- TION ($) |
|
|
NON-QUALI- FIED DEFERRED COMPENSA- TION EARNINGS ($) |
|
|
TOTAL ($) |
|
||||||||
|
Todd Cooper |
|
2017 |
(1) |
|
|
446,705 |
|
|
|
|
46,800 |
|
|
|
— |
|
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
493,505 |
|
|
Chief Executive Officer and President (January 2015 – December 2017) |
|
2016 |
|
|
|
412,917 |
|
|
|
|
40,000 |
|
|
|
— |
|
|
|
|
221,568 |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
674,485 |
|
|
Ralph Thurman |
|
2017 |
(2) |
|
|
144,437 |
|
|
|
|
— |
|
|
|
45,633 |
|
|
(3) |
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
190,070 |
|
|
Chairman (January 2015 - December 2017) |
|
2016 |
|
|
|
130,000 |
|
|
|
|
— |
|
|
|
39,998 |
|
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
169,998 |
|
|
Vladimir Feingold |
|
2017 |
(4) |
|
|
456,101 |
|
|
|
|
20,472 |
|
|
|
38,978 |
|
|
(5) |
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
515,551 |
|
|
Chief Technology Officer (June 2007 – January 2017) |
|
2016 |
|
|
|
323,678 |
|
|
|
|
15,450 |
|
|
|
— |
|
|
|
|
177,254 |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
516,382 |
|
|
Jarett Fenton |
|
2017 |
(6) |
|
|
232,125 |
|
|
|
|
14,063 |
|
|
|
— |
|
|
|
|
— |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
246,188 |
|
|
Chief Financial Officer (June 2016 – January 2018) |
|
2016 |
|
|
|
122,741 |
|
|
|
|
— |
|
|
|
— |
|
|
|
|
393,990 |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
516,731 |
|
|
(1) |
In December 2017, Mr. Cooper resigned as an officer of Presbia PLC and its subsidiaries. Mr. Cooper was provided with an extension through three years after termination date of the exercise period for his stock options and of the vesting period for his restricted stock units. In addition, Mr. Cooper was entitled to receive continuation of his base salary for six months, reimbursement of COBRA payments for six months. On April 28, 2016, Mr. Cooper was awarded 75,000 restricted stock units. |
|
(2) |
In December 2017, Mr. Thurman resigned as an officer and director of Presbia PLC and its subsidiaries. Mr. Thurman was provided with an extension through three years after termination date of the exercise period for his stock options and of accelerated vesting of his restricted share awards. |
|
(3) |
Mr. Thurman was awarded 12,738 restricted share awards in March 2017. The amount reported represents the aggregate grant date fair value calculated in accordance with FASB ASC 718. Information concerning the assumptions used to calculate these amounts is set forth in note 9 of audited consolidated financial statements presented elsewhere in this Annual Report on Form 10-K. |
|
(4) |
Mr. Feingold resigned from his position as Chief Technology Officer effective January 13, 2017 and transitioned to become a consultant. Mr. Feingold continued to serve as member of the Company’s board of directors until February 16, 2018, when he resigned from the board of the directors and its subsidiaries. Following his resignation from the board, Mr. Feingold has continued to provide consulting services to the Company. |
|
(5) |
Mr. Feingold was awarded 12,738 restricted share awards in March 2017. The amount reported represents the aggregate grant date fair value calculated in accordance with FASB ASC 718. Information concerning the assumptions used to calculate these amounts is set forth in note 9 of audited consolidated financial statements presented elsewhere in this Annual Report on Form 10-K. On April 28, 2016, Mr. Feingold was awarded 60,000 restricted stock units. |
|
(6) |
Mr. Fenton joined the Company as our Chief Financial Officer effective June 16, 2016 and resigned effective January 5, 2018. Mr. Fenton was provided with an extension through one year after termination date of the vesting period for his restricted stock units. On August 05, 2016, Mr. Fenton was awarded 100,000 restricted stock units. |
Cash Bonus Program
Our Board has established for 2017, and expects to continue in subsequent years, a cash bonus program for our employees. Pursuant to this program, employees will be eligible to receive an annual cash target bonus based on a specified percentage of the employee’s salary, which bonus will be earned upon the achievement of certain specified individual and corporate milestones.
Equity Awards
Presbia PLC
In 2016, we approved granted restricted stock units, or RSUs, to our named executive officers as follows:
|
|
• |
to Vladimir Feingold, in April 2016, 60,000 RSUs; |
|
|
• |
to Todd Cooper, in April 2016, 75,000 RSUs; and |
|
|
• |
to Jarrett Fenton, in August 2016, 100,000 RSUs. |
The Company intends to settle the vested RSUs in ordinary shares. The RSUs will vest in increments of 20% if certain specified stock price thresholds are achieved and maintained for twenty consecutive trading days; provided, however, that in no event shall such ordinary shares vest on a date prior to first annual anniversary of the date of grant or after the recipient’s termination of employment or service with the Company. The RSUs have a seven-year term and will vest in full upon a change of control, as defined in the RSU agreement. If dividends are distributed to shareholders, a number of dividend equivalent units will be credited to outstanding RSUs equal to the dollar amount of dividends that would have been paid with respect to the RSUs as of the dividend record date had they
93
been ordinary shares, divided by the closing price of an ordinary share on that date. Dividend equivalent units vest at the same times as the RSUs to which they relate vest.
We expect from time to time to make equity grants under the Presbia Incentive Plan to our employees to align the interests of our employees with our company.
Outstanding Equity Awards at Fiscal-End
The following table sets forth information regarding holdings by our named executive officers, as of December 31, 2017, of unexercised stock options, outstanding restricted stock awards and restricted stock units granted by Presbia PLC.
|
|
|
OPTIONS AWARDS(1) |
|
STOCK AWARDS |
|
||||||||||||||||||||||||||||||||
|
NAME |
|
NUMBER OF ORDINARY SHARES UNDERLYING UNEXERCISED OPTIONS (#) EXERCISABLE |
|
|
|
NUMBER OF ORDINARY SHARES UNDERLYING UNEXERCISED OPTIONS (#) UNEXERCIS- ABLE |
|
|
|
OPTION EXERCISE PRICE ($) |
|
|
OPTION EXPIRATION DATE |
|
NUMBER OF SHARES OR UNITS OF STOCK THAT HAVE NOT VESTED (#) |
|
|
|
MARKET VALUE OF SHARES OF UNITS OF STOCK THAT HAVE NOT VESTED ($) |
|
|
|
|
|
EQUITY INCENTIVE PLAN AWARDS: NUMBER OF UNEARNED SHARES, UNITS OR OTHER RIGHTS THAT HAVE NOT VESTED (#) |
|
|
|
EQUITY INCENTIVE PLAN AWARDS: MARKET OR PAYOUT VALUE OF UNEARNED SHARES, UNITS OR OTHER RIGHTS THAT HAVE NOT VESTED ($) |
|
|||||||
|
Todd Cooper |
|
|
180,000 |
|
|
|
|
90,000 |
|
(2) |
|
$ |
10.00 |
|
|
12/10/2020 |
|
|
75,000 |
|
|
|
|
45,000 |
|
|
(5 |
) |
|
|
75,000 |
|
|
|
|
45,000 |
|
|
Ralph Thurman |
|
|
250,000 |
|
(3) |
|
|
— |
|
(3) |
|
$ |
10.00 |
|
|
12/10/2020 |
|
|
— |
|
|
|
|
— |
|
|
|
|
|
|
— |
|
|
|
|
— |
|
|
Vladimir Feingold |
|
|
40,000 |
|
|
|
|
60,000 |
|
(4) |
|
$ |
10.00 |
|
|
1/28/2025 |
|
|
60,000 |
|
|
|
|
162,000 |
|
|
(5 |
) |
|
|
60,000 |
|
|
|
|
162,000 |
|
|
Jarett Fenton |
|
|
— |
|
|
|
|
— |
|
|
|
|
|
|
|
|
|
|
100,000 |
|
|
|
|
394,000 |
|
|
(5 |
) |
|
|
100,000 |
|
|
|
|
394,000 |
|
|
(1) |
There were no option exercises by any of our executive officers during the year ended December 31, 2017. |
|
(2) |
Such shares vested on January 28, 2018. Mr. Cooper was provided with an extension through three years after termination date of the exercise period for his stock options and of the vesting period for his restricted stock units. |
|
(3) |
Mr. Thurman was provided with an extension through three years after termination date of the exercise period for his stock options. |
|
(4) |
Until the end of the Initial Term and any renewal Terms of Mr. Feingold’s consulting agreement, all Restricted Share Units granted to Mr. Feingold on April 28, 2016 and all Stock Options granted to him on January 28, 2015 shall continue to vest in accordance with their respective agreements and the Company's Incentive Plan. |
|
(5) |
Restricted stock units issued in the year December 31, 2017. Fair value of restricted stock units is determined using a Monte Carlo Simulation (see note 9 for additional details). |
Retirement Benefits
We do not maintain, and during 2017 and 2016 did not maintain, any tax-qualified or non-qualified plans that provide for the payment of retirement benefits or benefits paid primarily following retirement to any of our named executive officers.
Agreements with Named Executive Officers
Each of our named executive officers is an employee at will. Other than as described below, we are not party to employment agreements with any of our named executive officers.
We entered into a letter agreement, which includes a severance arrangement, with Mr. Cooper, our President and Chief Executive Officer. The agreement provides that Mr. Cooper’s annual base salary shall be $400,000, with a target annual bonus of 50% of base pay based on agreed to objectives of our Board. Also, pursuant to this letter agreement, in connection with our initial public offering, we issued to Mr. Cooper an option to purchase 450,000 of our ordinary shares at an exercise price of $10.00 per share. In addition, if Mr. Cooper is terminated without cause or if he resigns because his base salary is unilaterally reduced or his title is diminished, he will be entitled to six months base pay, six months of reimbursement of certain medical benefits and vesting of stock options that would otherwise vest during that six-month period.
In further of our reprioritization plan, effective December 10, 2017, Mr. Cooper resigned as chief executive officer and a member of the board of directors and Mr. Thurman resigned as Executive Chairman. We entered into a separation agreement and general release with each of Mr. Cooper and Mr. Thurman pursuant to which each of Mr. Cooper and Mr. Thurman received an extension through three years after termination date of the exercise period for the stock options and of the vesting period for the restricted stock units held by each of them, In addition, pursuant to the terms of the letter agreement described above, Mr. Cooper will receive continuation
94
of his base salary for six months, reimbursement of COBRA payments for six months and the vesting of his stock options will be accelerated by six months.
In connection with Mr. Feingold’s resignation in January 2017, we entered into a Separation and General Release Agreement and a Consulting Agreement with Mr. Feingold. The Separation and General Release Agreement provides for, among other things, (i) the payment of his 2016 annual bonus in the range of $20,472 to $40,944 depending on the Board’s determination of the achievement of certain 2016 personal development and corporate goals, (ii) January 13, 2017 as the last date of employment, (iii) general release by Mr. Feingold and (iv) a consulting agreement.
Pursuant to the terms of Mr. Fenton’s offer letter he is eligible to receive an annual base salary of $225,000, with a target annual bonus of 25% of base salary. In furtherance of our reprioritization plan, effective January 5, 2018, Mr. Fenton’s employment terminated. We entered into a release agreement with Mr. Fenton pursuant to which Mr. Fenton received an extension through one year after his termination date of the vesting period for the restricted stock units held by him.
Presbia Incentive Plan
In January 2015, prior to the consummation of our initial public offering, we adopted a stock plan, which we refer to as the Presbia Incentive Plan. Unless sooner terminated by the Board, the Presbia Incentive Plan will expire 10 years after its adoption. On June 15, 2016, our Board of Directors approved an amendment (the “Plan Amendment”) to the Presbia Incentive Plan (as amended by the Plan Amendment, the “Amended Plan”), subject to shareholder approval at the Annual Meeting, which, among other things, increases the number of shares reserved for issuance under the Amended Plan by 400,000 ordinary shares and sets forth certain limits on the number of shares underlying options and stock appreciation rights, or SARs, granted to an individual in a given fiscal year so that options and SARs granted under the Plan may qualify as exempt performance-based compensation under Section 162(m) of the U.S. Internal Revenue Code of 1986, as amended (“Section 162(m)”), which otherwise generally disallows the corporate tax deduction for compensation paid in excess of $1 million annually to each of the chief executive officer and the other named executive officers of publicly-held companies. On August 4, 2016, the shareholders approved the Plan Amendment.
The Presbia Incentive Plan permits us to grant awards of stock options, restricted shares, stock appreciation rights, restricted stock units, performance shares, performance share units, dividend equivalent rights in respect of awards and other share-based and cash-based awards, including annual and long-term cash incentive awards. Awards under the Presbia Incentive Plan may be granted to employees, directors, consultants and other persons who perform services for our company or a subsidiary of our Company.
A total of 2,200,000 of our ordinary shares are authorized for issuance under the Presbia Incentive Plan. For purposes of calculating the number of shares available under the Presbia Incentive Plan, shares covered by forfeited, terminated, or cancelled awards are available for future awards under the Presbia Incentive Plan, as are shares that are surrendered or withheld from any award to satisfy tax withholding obligations or the exercise price of an award or that are tendered by an award recipient to pay the exercise price of any awards. Such shares may be authorized but unissued shares or authorized and issued shares held in our treasury or acquired by our Company for purposes of the Presbia Incentive Plan.
The Presbia Incentive Plan is administered by the Board’s compensation committee. The compensation committee has the authority to:
|
|
• |
determine which individuals shall be granted awards and the provisions of award agreements; |
|
|
• |
interpret the Presbia Incentive Plan and award agreements; |
|
|
• |
prescribe, amend and rescind rules and regulations, if any, relating to the Presbia Incentive Plan; |
|
|
• |
make all determinations necessary or advisable for the administration of the Presbia Incentive Plan; and |
|
|
• |
correct any defect, supply any omission and reconcile any inconsistency in the Presbia Incentive Plan or any award agreement. |
Payments to our Company upon the grant, exercise or payment of an award may be made in such form as our compensation committee determines, including cash, ordinary shares, net share exercise, other securities, other awards or other property.
Options granted pursuant to the Presbia Incentive Plan will have an exercise price that is not less than 100% of the fair market value of the shares subject to the option on the date of grant and a term of not more than 10 years from the date of grant. In general, unless an award agreement specifies otherwise, options will become exercisable with respect to 20% of the shares subject thereto on each of the first five anniversaries of the date of grant. However, each option will become fully exercisable upon a “change in control” of our company (as defined in the Presbia Incentive Plan), unless the Board determines that the optionee has been offered substantially identical replacement options and a comparable position at the acquiring company. In general, upon an optionee’s termination of employment, any then exercisable options held by the optionee may be exercised for a period of three months following such
95
termination (one year in the case of death), but in no event beyond the stated expiration date of such options; provided that all options shall immediately terminate upon termination of an optionee’s employment for cause.
Restricted shares granted pursuant to the Presbia Incentive Plan may not be sold, assigned or otherwise transferred during the restricted period determined by our compensation committee at grant. Except as otherwise determined by our compensation committee, upon a recipient’s termination of employment prior to the expiration of the applicable restricted period, all shares for which the restricted period has not lapsed shall be forfeited and reacquired by us at no cost (or for nil consideration). Our compensation committee may accelerate the vesting of all or any restricted shares at any time on such terms as it shall determine by cancelling the outstanding restrictions to which such shares are subject prior to the expiration of the restricted period of such shares. In addition, all restricted shares will become fully vested, and the restrictions to which shares are subject shall lapse, upon a “change in control” of our company (as defined in the Presbia Incentive Plan) unless the Board determines that the recipient has been offered substantially identical replacement restricted shares and a comparable position at the acquiring company. During the restricted period, the recipient shall possess all incidents of ownership of the restricted shares, including the right to receive dividends on and vote such shares; provided that, unless otherwise set forth in an award agreement, any cash or share dividends with respect to restricted shares shall be withheld by us for the recipient’s account and shall be subject to the same restrictions as the corresponding restricted shares to which such dividends relate.
Share appreciation rights granted pursuant to the Presbia Incentive Plan will confer the right to receive, for each ordinary share with respect to which the share appreciation right is exercised, an amount equal to (i) the excess of the fair market value of an ordinary share on the date of exercise over (ii) the base price of the share appreciation right. The base price of share appreciation rights will not be less than 100% of the fair market value of the ordinary shares subject to the share appreciation right on the date of grant. Share appreciation rights will become exercisable at such time or times as our compensation committee shall determine. Payment upon exercise of a share appreciation right may be made in cash or in our ordinary shares or both, as determined by our compensation committee.
Restricted stock units granted pursuant to the Presbia Incentive Plan will be subject to such terms as the compensation committee may determine. At the time of grant, our compensation committee will specify the date or dates on which restricted stock units will vest and the conditions to vesting and will specify the date on which ordinary shares will be transferred to a recipient in respect of vested restricted stock units (which date may be later than the vesting date or dates of such award). Except as otherwise determined by our compensation committee, upon a recipient’s termination of employment, restricted stock units that have not vested shall be forfeited and cancelled (or reacquired by us for nil consideration). Our compensation committee may at any time accelerate the vesting dates of all or any restricted stock units or waive or amend any conditions of such awards.
Our compensation committee may grant performance shares in the form of actual ordinary shares or performance share units having a value equal to an identical number of ordinary shares, in such amounts and subject to such terms as the compensation committee may determine. The performance conditions and the length of the performance period applicable to performance shares and performance share unit awards shall be determined by our compensation committee. In addition, our compensation committee shall determine whether performance share units will be paid in cash, ordinary shares or a combination of both.
Our compensation committee may award other types of share-based or cash-based awards under the Presbia Incentive Plan in such amounts and subject to such terms and conditions as our compensation committee may determine. Such awards may entail the transfer of actual ordinary shares or payment in cash or otherwise of amounts based on the value of our ordinary shares or the payment of cash pursuant to annual and long-term incentive awards approved by our compensation committee that may or may not be based on the value of our ordinary shares.
No employee may receive options or SARs with respect to more than 400,000 of our ordinary shares in the aggregate in any fiscal year. With respect to other awards under the Presbia Incentive Plan intended to be exempt from the deduction limitation under Section 162(m) for performance-based compensation (“Section 162(m) Awards”), no employee may be granted in any one fiscal year of the Company (a) restricted shares or restricted share units with respect to more than 400,000 ordinary shares each; and (b) performance shares, performance share units or other share-based awards that are denominated in ordinary shares with respect to more than 400,000 ordinary shares each. The maximum dollar value payable to any employee in any one fiscal year of the Company with respect to performance shares, performance share units or other share- or cash-based awards that may be settled in cash or other property (other than ordinary shares) is $1 million. These ordinary share limitations are subject to customary adjustments for stock splits, stock dividends or similar transactions.
Section 162(m) places a limit of $1 million on the amount our company may deduct in any one year for compensation paid to a “covered employee,” which is defined by Section 162(m) to mean any person who as of the last day of the fiscal year is the chief executive officer or one of our three highest compensated executive officers other than our principal financial officer. There is, however, an exception to this limit on deductibility for compensation that satisfies certain conditions for “qualified performance-based compensation” set forth under Section 162(m). One of the conditions requires shareholder approval every five years of the material
96
terms of the performance goals of the plan under which the compensation will be paid. Under the Amended Plan, the business criteria on which performance goals for awards that are intended to satisfy the conditions for deductibility under Section 162(m) as “performance-based compensation” will be selected from among the following, which may be applied to our company as a whole, or to an individual recipient, or to a department, unit, division or function within the company or an affiliate, and they may apply on a pre- or post-tax basis, either alone or relative to the performance of other businesses or individuals (including industry or general market indices): (i) net earnings or net income (before or after taxes); (ii) earnings growth; (iii) earnings per share (including, but not limited to, growth in diluted earnings per share from continuing operations); (iv) net sales (including, but not limited to, net sales growth); (v) gross profits or net operating profit; (vi) return on assets, return on equity, return on capital or return on sales; (vii) cash flow (including, but not limited to, operating cash flow, free cash flow, cash flow return on capital and statutory cash measures); (viii) revenue growth; (ix) earnings before or after taxes, interest, depreciation, and/or amortization; (x) productivity ratios; (xi) ordinary share price (including, but not limited to, growth measures), (xii) total stockholder return; (xiii) expense targets; (xiv) gross or operating margins, earnings before or after taxes, interest, depreciation, and/or amortization margins; (xv) operating efficiency; (xvi) customer satisfaction or increase in the number of customers; (xvii) division working capital turnover; (xviii) strategic business or operational criteria consisting of one or more objectives based on meeting specified goals relating to (A) acquisitions or divestitures, (B) business expansion, (C) cost targets, (D) diversity and inclusion, (E) efficiency, (F) management of employment practices and employee benefits, (G) market penetration, (H) product quality and quality audit scores, (I) reductions in errors and omissions, (J) reductions in lost business, (K) supervision of litigation and information technology, or (L) sustainability; (xix) market share; (xx) cost reductions; (xxi) working capital targets; (xxii) sales backlog; (xxiii) net debt and (xxiv) economic value added. Prior to payment or settlement of any Section 162(m) Award, our compensation committee must certify in writing that the performance goals and all other material terms applicable to such award were in fact satisfied. At the time of certification, the compensation committee will also determine the amount of compensation payable as a result of the attainment of such performance goals.
The compensation committee has no discretion to waive all or part of the performance goals applicable to a Section 162(m) Award, except in the event of a change of control, or in the event of the death or disability of an employee. Notwithstanding the foregoing, the compensation committee has the discretion to reduce the amount of any Section 162(m) Award based on such factors as determined by the compensation committee, including, without limitation, a determination that a reduction is appropriate in light of pay practices of competitors, the performance of the Company, a subsidiary, or an employee relative to the performance of competitors, or performance with respect to the Company’s strategic goals.
The Board may amend the Presbia Incentive Plan at any time, but no amendment may materially alter or adversely impair rights and obligations under previously granted awards without consent. Amendments to the Presbia Incentive Plan require shareholder approval to the extent required by applicable laws, regulations or rules.
This description is not complete. For more information, we refer you to the full text of the Presbia Incentive Plan, which we filed as an exhibit to the 2014 Annual Report on Form 10-K, and the Plan Amendment, which we filed as an exhibit to our Quarterly Report on Form 10-Q for the quarter ended June 30, 2016.
Securities Laws and U.S. Federal Income Taxes.
The Presbia Incentive Plan is designed to comply with various U.S. federal securities and tax laws as follows:
|
|
• |
Securities Laws. The Presbia Incentive Plan is intended to conform to all provisions of the Securities Act and the Exchange Act and any and all regulations and rules promulgated by the SEC thereunder, including without limitation, Rule 16b-3. The Presbia Incentive Plan will be administered, and options will be granted and may be exercised, only in such a manner as to conform to such laws, rules and regulations. |
|
|
• |
Section 162(m) of the Code. In general, under Section 162(m) of the Code, income tax deductions of publicly held corporations may be limited to the extent total compensation for certain executive officers exceeds $1,000,000 in any taxable year of the corporation. However, under Section 162(m), the deduction limit does not apply to certain “performance-based compensation” established by an independent compensation committee that is adequately disclosed to and approved by shareholders. Under a Section 162(m) transition rule for compensation plans of corporations that are privately held and that become publicly held in an initial public offering, the Presbia Incentive Plan will not be subject to Section 162(m) until a specified transition date, which is the earliest of: |
|
|
• |
the date on which the Presbia Incentive Plan is materially modified; |
|
|
• |
the date on which all of the ordinary shares reserved for issuance and other compensation allocated under the Presbia Incentive Plan are issued; |
97
|
|
• |
the date of the first meeting of our shareholders at which members of our Board are to be elected that occurs after the close of the third calendar year following the calendar year in which our initial public offering occurred. |
Prior to the transition date, the deduction limitation under Section 162(m) of the Code will not apply to compensation received pursuant to rights or awards granted under the Presbia Incentive Plan.
After the transition date, rights or awards granted under the Presbia Incentive Plan, other than compensation received pursuant to options and stock appreciation rights or the vesting of restricted shares granted prior to the transition date, will not qualify as “performance-based compensation” for purposes of Section 162(m), unless such rights or awards are granted or vest upon pre-established objective performance goals, the material terms of which are disclosed to and approved by our shareholders.
Director Compensation
For information regarding the compensation that we paid to our named executive officers who serve on the Board of Presbia PLC, Messrs. Thurman, Cooper, and Feingold, see “Summary Compensation Table.”
At present, non-employee directors of Presbia PLC are compensated as follows: (i) an annual board cash fee of $60,000; (ii) a cash fee of $2,500 for each board or committee meeting attended in person in Ireland; and (iii) reimbursement for out-of-pocket expenses incurred in connection with attending Board and committee meetings. In addition, we intend to grant new directors a one-time restricted share award valued at $80,000 in connection with their appointment to our Board, which shares will vest ratably over a five year vesting period, and we intend to grant our non-employee directors an annual restricted share award valued at $40,000 for their continued service on our Board, which shares vested ratably over a one and five year vesting periods for the years ended December 31, 2017 and 2016, respectively. In January 2017, we entered into a consulting agreement with Mr. Feingold, which provides for, among other things, (i) an initial one-year term, which shall automatically renew for successive one-year terms, unless terminated in accordance with the provisions of the Consulting Agreement, (ii) a $35,000 monthly consulting fee for the services specified in the applicable statement of work, including services related to regulatory and IP-related matters, (iii) termination by the Company upon sixty days’ notice and termination by Mr. Feingold upon thirty days’ notice, and (iv) the payment of the balance of the consulting fees for the initial term (as defined in the Consulting Agreement), if the Company terminates the Consulting Agreement without cause (as defined in the Consulting Agreement) during the initial term.
Director Equity Awards
Presbia PLC
On March 14, 2017, we granted 12,738 restricted ordinary shares to each of the following board members: Gerald Farrell, Gerd Auffarth, Richard Ressler, Robert J. Cresci and Zohar Loshitzer. The restricted shares fully vested at March 14, 2018, one year after the grant date.
98
The following table summarizes the annual compensation for our non- employee directors during the year ended December 31, 2017.
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
NAME |
|
Fees Earned or Paid in Cash ($) |
|
|
Stock Awards ($)(1) |
|
|
Option Awards ($)(2) |
|
|
Non-Equity Incentive Plan Compensation ($) |
|
|
Change in Pension Value and Nonqualified Deferred Compensation Earnings |
|
|
All Other Compensation ($) |
|
|
Total ($) |
|
|||||||
|
Gerald Farrell |
|
$ |
15,000 |
|
|
$ |
38,978 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
$ |
53,978 |
|
|
Gerd U. Auffarth |
|
|
67,500 |
|
|
|
38,978 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
106,478 |
|
|
Richard Ressler |
|
|
— |
|
|
|
38,978 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
38,978 |
|
|
Robert J. Cresci |
|
|
62,500 |
|
|
|
38,978 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
101,478 |
|
|
Zohar Loshitzer |
|
|
7,500 |
|
|
|
38,978 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
46,478 |
|
|
(1) |
This column reflects grant date fair value assigned to restricted stock awards (RSA's) on the basis of closing price of Company's ordinary shares on the date of the award. Such restricted shares will become vested one year after the grant date. |
|
(2) |
Amounts in the Option Award column reflect grant date fair value of options granted determined in accordance with ASC 718. Information concerning the assumptions used to calculate this value is set forth in Note 9 to the consolidated financial statements presented elsewhere in this Annual Report on Form 10-K. |
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The following table sets forth certain information as of March 30, 2018 with respect to the beneficial ownership of our ordinary shares by:
|
|
• |
each of our named executive officers and directors; |
|
|
• |
all of our executive officers and directors as a group; and |
|
|
• |
each person or group of affiliated persons who is known by us to beneficially own more than 5% of our outstanding shares. |
The amounts and percentages of shares beneficially owned are reported on the basis of regulations of the SEC governing the determination of beneficial ownership of securities. Under the SEC’s rules, a person is deemed to be a beneficial owner of a security if that person has or shares voting power, which includes the power to vote or to direct the voting of such security, or investment power, which includes the power to dispose of or to direct the disposition of such security. Unless otherwise indicated below, each beneficial owner named in the table below has sole voting and sole investment power with respect to all shares beneficially owned, subject to community property laws where applicable.
Unless otherwise indicated, the address of each person listed below is c/o Presbia PLC, Suite 7, Sandyford Office Centre, 17 Corrig Road, Sandyford, Dublin 18 Ireland.
99
|
|
|
ORDINARY SHARES BENEFICIALLY OWNED |
|
||||||
|
NAME OF BENEFICIAL OWNER |
|
NUMBER |
|
|
|
PERCENT |
|
||
|
5% Holders |
|
|
|
|
|
|
|
|
|
|
FMR, LLC |
|
|
2,568,278 |
|
(1) |
|
|
14.9 |
% |
|
245 Summer Street, Boston, MA 02210 |
|
|
|
|
|
|
|
|
|
|
Directors and Officers |
|
|
|
|
|
|
|
|
|
|
Todd Cooper |
|
|
272,514 |
|
(2) |
|
|
1.6 |
% |
|
Zohar Loshitzer |
|
|
427,960 |
|
(3) |
|
|
2.5 |
% |
|
Vladimir Feingold |
|
|
1,421,270 |
|
(4) |
|
|
8.3 |
% |
|
Richard Ressler |
|
|
10,494,048 |
|
(5) |
|
|
61.6 |
% |
|
Gerd Auffarth |
|
|
38,464 |
|
(6) |
|
* |
|
|
|
Ralph Thurman |
|
|
309,664 |
|
(7) |
|
|
1.8 |
% |
|
Robert Cresci |
|
|
96,128 |
|
(8) |
|
* |
|
|
|
Gerald Farrell |
|
|
59,664 |
|
(9) |
|
* |
|
|
|
Jarett Fenton |
|
|
- |
|
(10) |
|
* |
|
|
|
Executive officers and directors as a group (9 persons) |
|
|
13,119,712 |
|
(11) |
|
|
74.7 |
% |
|
* |
Less than one percent. |
|
(1) |
Based solely on information set forth in a Schedule 13G/A filed with the SEC on February 13 2018 by FMR LLC ("FMR") reporting that FMR: (i) beneficially owned 2,568,278 shares; (ii) had the sole power to dispose or direct the disposition of 2,568,278 shares; and (iii) had the sole power to vote or to direct the vote of 821,590 shares. In addition, the Schedule 13G/A shows that the following person s or entities beneficially own certain of the shares reported: Abigail P. Johnson and Fidelity Growth Company Fund. The Schedule 13G/A also shows that Abigail P. Johns is a Director, the Chairman and Chief Executive Officer and of FMR. Members of the Johnson family, including Abigail P. Johnson, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR, representing 49% of the voting power of FMR. The Johnson family group and all other Series B shareholders have entered into a shareholders' voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders' voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR. Neither FMR nor Abigail P. Johnson has the sole power to vote or direct the voting of the shares owned directly by the various investment companies registered under the Investment Company Act (the "Fidelity Funds"), advised by Fidelity Management & Research Company, a wholly-owned subsidiary of FMR, which power resides with the Fidelity Funds' Board of Trustees. Fidelity Management & Research Company carries out the voting of the shares under written guidelines established by the Fidelity Funds' Board of Trustees. |
|
(2) |
Includes 180,000 ordinary shares covered by options vested on January 28, 2017. Excludes 270,000 options, none of which are exercisable within 60 days of March 30, 2018. |
|
(3) |
Includes 40,000 ordinary shares covered by options vested on January 28, 2017. Excludes 60,000 options, none of which are exercisable within 60 days of March 30, 2018. Includes 20,985 restricted shares held by Mr. Loshitzer as of March 30, 2018. |
|
(4) |
Includes 40,000 ordinary shares covered by options vested on January 28, 2017. Excludes 60,000 options, none of which are exercisable within 60 days of March 30, 2018. Includes 12,738 restricted shares held by Mr. Feingold as of March 30, 2018. |
|
(5) |
Includes 10,000 ordinary shares covered by options vested on January 28, 2017. Includes 20,985 restricted shares held by Mr. Ressler as of March 30, 2018. |
|
(6) |
Includes 32,004 restricted shares held by Dr. Auffarth as of March 30, 2018. |
|
(7) |
Includes 250,000 ordinary shares covered by options vested on January 28, 2017, and 20,985 restricted shares held by Mr. Thurman as of March 30, 2018. |
|
(8) |
Includes 30,255 restricted shares held by Mr. Cresci as of March 30, 2018. |
|
(9) |
Includes 36,953 restricted shares held by Dr. Farrell as of March 30, 2018. |
|
(10) |
No shares are beneficially owned by Mr. Fenton as of March 30, 2018. |
|
(11) |
Includes 520,000 ordinary shares covered by options, and 174,905 restricted shares held as of March 30, 2018. Excludes 390,000 options, none of which are exercisable within 60 days of March 30, 2018. |
100
Securities Authorized for Issuance Under Equity Compensation Plans
The following table provides information as of December 31, 2017 regarding shares of our common stock that may be issued under the Presbia Incentive Plan (the “Plan”).
|
|
|
Equity Compensation Plan Information |
|
||||||||||
|
|
|
Number of securities to be issued upon exercise of outstanding options and rights(1) |
|
Weighted Average exercise price of outstanding options and rights |
|
Number of securities remaining available for future issuance under equity compensation plan (excluding securities referenced in column (a)) |
|
||||||
|
Equity compensation plans approved by security holders |
|
|
1,722,467 |
|
|
$ |
6.20 |
|
|
|
435,141 |
|
|
|
Equity compensation plans not approved by security holders |
|
— |
|
|
— |
|
|
— |
|
|
|||
|
Total |
|
|
1,722,467 |
|
|
|
6.20 |
|
|
|
435,141 |
|
|
(1) Does not include restricted shares of 176,759, issued as of December 31, 2017, as such shares are already reflected in our outstanding shares.
Item 13. Certain Relationships and Related Transactions and Director Independence.
The following is a description of transactions since January 1, 2016 to which we have been a party, in which the amount involved exceeds $120,000, and in which any of our directors, executive officers or holders of more than 5% of our share capital, or an affiliate or immediate family member thereof, had or will have a direct or indirect material interest.
Participation in Rights Offering
In the first quarter of 2017 we conducted a rights offering, pursuant to which we distributed to holders of our ordinary shares, at no charge, non-transferable and non-tradeable subscription rights to purchase ordinary shares. Each shareholder received one subscription right for each share of our ordinary shares owned at 5:00 PM Eastern on February 6, 2017, the record date for this rights offering. Each subscription right entitled its holder to purchase 0.335297256 ordinary shares at a subscription price of $3.00 per whole share, which we refer to as the “basic subscription right.” The holders also received an oversubscription privilege, pursuant to which a holder was entitled to purchase a portion of the unsubscribed ordinary shares at the subscription price, subject to proration if such holder had exercised its basic subscription rights in full, and other shareholders did not fully exercise their basic subscription rights. Each subscription right consists of a basic subscription right and an over-subscription privilege, which we refer to as the “subscription right.” The following officers and directors of Presbia purchased shares in the rights offering: (i) Richard Ressler directly and through certain entities controlled by him purchased an aggregate of 2,628,222 shares, (ii) Ralph Thurman purchased 5,543 shares, (iii) Mr. Loshitzer purchased 91,959 shares, (iv) Dr. Auffarth purchased 6,460 shares, (v) Mr. Farrell purchased 17,111 shares and (vi) Mr. Cresci purchased 55,873 shares.
Transactions with Orchard Capital and its Affiliates
Orchard Capital has provided financial analysis and bookkeeping, accounting, legal, compliance and directorial services to Presbia since January 2011 pursuant to a Services Agreement. Such agreement will remain in effect until terminated by either party thereto upon 30 days’ notice. Orchard Capital invoices us quarterly for such services at cost. During the years ended December 31, 2017 and 2016, we recognized general and administrative expenses of $14,000 and $14,000 for services invoiced by Orchard Capital. As of December 31, 2017 and 2016, $3,000 and $4,000 was due to Orchard Capital for management and accounting services, respectively.
Also, commencing during the second quarter of 2013, we have received human resources management services, payroll services, IT support and risk management services from CIM Group. We have incurred charges of $37,000 and $73,000 payable to CIM Group for such services for the years ended December 31, 2017 and 2016, respectively. As of December 31, 2017 and 2016, amounts due to CIM Group for human resources, payroll, information technology and legal services amounted to $19,000 and $14,000, respectively.
On December 14, 2017, the Company concluded a services agreement with OCV Management, LLC, a related party co-founded by Mr. Ressler and Mark Yung, for the purpose of providing management services for the Company for the period commencing December 14, 2017 until terminated upon thirty (30) days’ prior written notice by either of the parties. OCV Management, LLC will receive an annual fee of $250,000, payable annually in arrears. The obligation will be settled in the form of ordinary shares issued by the Company in a private offering. For 2018, the number of shares to be issued will be determined by dividing $250,000 by the closing
101
price on January 2, 2018 and the shares will be issued on January 2, 2019. The Company recorded a non-current liability of $12,000 as of December 31, 2017 in relation to services provided in 2017.
For information regarding the relationship of Richard Ressler (one of our directors and the beneficial owner of the majority of the ordinary shares of our company) and Mark Yung (our Executive Chairman of the board and Chief Executive Officer) with Orchard Capital, CIM Group and OCV, see “Part III, Item 10. Directors, Executive Officers and Corporate Governance.”
Registration Rights Agreement
Presbia Holdings, our former controlling shareholder, and certain of its transferees, had rights to cause our Company to register their ordinary shares, including any ordinary shares that Presbia Holdings transfers to its equity owners, under the Securities Act. In August 2015, in connection with the dissolution of Presbia Holdings, Presbia Holdings transferred all rights under the Registration Rights Agreement to Richard Ressler, one of our board members, and his affiliates. These rights are provided under the terms of a registration rights agreement between us and Presbia Holdings and includes demand registration rights and piggyback registration rights. These registration rights are assignable, subject to certain conditions, including that the assignee be bound by the terms and conditions of the registration rights agreement. To the extent permitted by applicable law, we will pay, or if not permitted by applicable law, we will cause one of our non-Irish subsidiaries to pay, all registration expenses in connection with registrations under this agreement.
Demand registration rights
Under the terms of the registration rights agreement, at any time beyond six months after the consummation of our initial public offering, we are required, upon the written request of the holders of the shares that are entitled to rights under the registration rights agreement, to use our best efforts to register all or a portion of these shares for public resale. We are not required to effect a registration pursuant to this provision of the registration rights agreement (i) if the shares requested to be registered do not represent (a) at least 10% of the shares that are entitled to registration rights under the agreement or (b) an anticipated aggregate public offering price of at least $10 million; or (ii) during the period starting with the date 30 days prior to our good faith estimate of the date of filing of, and ending on a date 180 days following the effective date of, any company-initiated registration under the Securities Act. If such a registration is to be an underwritten offering, then the holders’ registration rights are conditioned upon the holders’ participation in that underwriting. We may defer the filing of a registration statement once during any 12-month period for a period of not more than ninety days, if we provide written notice stating that in the good faith judgment of our Board, a disadvantageous condition exists, including the existence of certain material transactions or financings, the unavailability of any required financial statements, or the possession by our company of any material information which would not be in the best interests of our company to disclose.
Piggyback registration rights
In addition, the holders are entitled to piggyback registration rights. If we register any of our securities for our own account, the holders of these shares are entitled to include their shares in the registration. If such registration is to be an underwritten offering, then the holders’ registration rights are conditioned on such holders’ participation in that underwriting.
Director and Executive Officer Compensation
See “Item 11. Executive Compensation” for information regarding compensation of our directors and executive officers.
Indemnification Agreements and Directors’ and Officers’ Liability Insurance
We have entered into indemnification agreements with each of our directors and executive officers. Also, our Presbia USA, Inc. subsidiary has entered into an indemnification agreement with each of our executive officers and directors (each of our executive officers and directors is also an officer and/or director of our Presbia USA, Inc. subsidiary). These agreements, among other things, require us to indemnify an indemnitee to the fullest extent permitted by applicable law, including indemnification of expenses such as attorneys’ fees, judgments, fines and settlement amounts incurred by the indemnitee in any action or proceeding, including any action or proceeding by us or in our right, arising out of the person’s services as a director or executive officer. We also maintain directors’ and officers’ liability insurance for our directors and officers.
Policies and Procedures for Related Party Transactions
Our Board has adopted a written related person transaction policy to set forth the policies and procedures for the review and approval or ratification of related person transactions. This policy covers, with certain exceptions, any transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships in which we were or are to be a participant, where the amount involved exceeds $120,000 and a related person had, has or will have a direct or indirect material interest, including, without
102
limitation, purchases of goods or services by or from the related person or entities in which the related person has a material interest, indebtedness, guarantees of indebtedness and employment by us of a related person.
As provided by our audit committee charter, our audit committee is responsible for reviewing and approving in advance the related party transactions covered by our Company’s related transaction policies and procedures.
Richard Ressler and his affiliates control a majority of our issued and outstanding ordinary shares. As a result, we are a “controlled company” within the meaning of NASDAQ listing rules, and thus, we are exempt from the requirement that a majority of our Board consist of independent directors
The Company is currently managed by a six-member Board. Each of Dr. Auffarth, Mr. Cresci and Mr. Farrell is “independent” as that term is defined under the rules of The NASDAQ Stock Market.
Item 14. Principal Accountant Fees and Services.
The following tables present fees for professional services provided Squar Milner LLP for the years ended December 31, 2017 and 2016 (amounts in thousands):
|
|
|
Year Ended December 31, |
|
|||||
|
Squar Milner LLP |
|
2017 |
|
|
2016 |
|
||
|
Audit fees |
|
$ |
114 |
|
|
$ |
114 |
|
|
Audit-related fees |
|
|
9 |
|
|
|
20 |
|
|
Tax fees |
|
|
— |
|
|
|
— |
|
|
Total |
|
$ |
123 |
|
|
$ |
134 |
|
Audit fees. Audit fees consist of the aggregate fees billed for professional services rendered for (i) the audit of our annual financial statements and (ii) the reviews of the financial statements included in the Quarterly Reports on Form 10-Q.
Audit-Related fees. Audit-related fees are for assurance and related services that are reasonably related to the audit and reviews of our financial statements, exclusive of the fees disclosed as audit fees above.
Tax Fees. Tax fees consisted of professional services rendered for tax compliance, tax advice and tax planning. The services for the fees disclosed under this category include tax return preparation and technical tax advice.
Consistent with SEC policies regarding auditor independence and the audit committee’s charter, the audit committee has responsibility for engaging, setting compensation for and reviewing the performance of the independent registered public accounting firm.
For the years ended December 31, 2017 and 2016, the Audit Committee was responsible for appointing, setting compensation and overseeing the work of the independent auditors. The Audit Committee has established a policy regarding pre-approval of all auditing services and the terms thereof and non-audit services (other than non-audit services prohibited under Section 10A(g) of the Exchange Act or the applicable rules of the SEC or the Public Company Accounting Oversight Board) to be provided to us by the independent auditor. However, the pre-approval requirement may be waived with respect to the provision of non-audit services for us if the “de minimus” provisions of Section 10A(i)(1)(B) of the Exchange Act are satisfied.
The Audit Committee has considered whether the provision of other fees as described above is compatible with maintaining Squar Milner’s independence and has determined that such services for fiscal years ending in December 31, 2017 and 2016 were compatible. All such services were approved by the Audit Committee pursuant to Rule 2-01 of Regulation S-X under the Exchange Act to the extent that rule was applicable.
The Audit Committee is responsible for reviewing and discussing the audited financial statements with management, discussing with the independent registered public accountants the matters required in Auditing Standards No. 16, receiving written disclosures from the independent registered public accountants required by the applicable requirements of the Public Company Accounting Oversight Board regarding the independent registered public accountants’ communications with the Audit Committee concerning independence and discussing with the independent registered public accountants their independence, and recommending to the Board of Directors that the audit financial statements be included in our annual report on Form 10-K.
103
Item 15. Exhibits and Financial Statement Schedules.
(a)(1) Financial Statements. The financial statements filed as part of this report are listed on the Index to the Consolidated Financial Statements in “Part II, Item 8 Financial Statements and Supplementary Data.”
(a)(2) Financial Statement Schedules. These schedules have been omitted because they are not applicable or not required, or the information is included in the Consolidated Financial Statements or Notes thereto.
(a)(3) Exhibits required by Item 601 of Regulation S-K. The information required by this Section (a)(3) of Item 15 is set forth on the exhibit index that follows the signatures page of this Annual Report on Form 10-K.
None.
104
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
Date: March 30, 2018 |
PRESBIA PLC |
|
|
|
|
|
|
|
By: |
/s/ Mark Yung |
|
|
|
Mark Yung |
|
|
|
President and Chief Executive Officer |
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Todd Cooper and Richard Fogarty, jointly and severally, his attorneys-in-fact, each with the power of substitution, for him in any and all capacities, to sign any amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact, or his substitute or substitutes, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
|
|
Name |
|
|
Title |
|
|
Date |
|
|
|
|
|
||||||
|
/s/ Mark Yung Mark Yung |
President, Chief Executive Officer and Director |
March 30, 2018 |
||||||
|
|
|
|
||||||
|
/s/ Richard Fogarty Richard Fogarty |
Chief Accounting Officer, Vice President, Finance and Secretary (Principal Financial and Accounting Officer) |
March 30, 2018 |
||||||
|
|
|
|
||||||
|
/s/ Zohar Loshitzer Zohar Loshitzer |
Director |
March 30, 2018 |
||||||
|
|
|
|
||||||
|
/s/ Richard Ressler Richard Ressler |
Director |
March 30, 2018 |
||||||
|
|
|
|
||||||
|
/s/ Robert Cresci Robert Cresci |
Director |
March 30, 2018 |
||||||
|
|
|
|
||||||
|
/s/ Gerd U. Auffarth Gerd U. Auffarth |
Director |
March 30, 2018 |
||||||
|
|
|
|
||||||
|
/s/ Gerald Farrell Gerald Farrell |
Director |
March 30, 2018 |
||||||
105
106
|
Exhibit No. |
|
Description of Exhibit |
|
|
||
|
|
|
|
|
101.INS+ |
|
XBRL Instance |
|
|
|
|
|
101.SCH+ |
|
XBRL Taxonomy Extension Schema |
|
|
|
|
|
101.CAL+ |
|
XBRL Taxonomy Extension Calculation |
|
|
|
|
|
101.DEF+ |
|
XBRL Taxonomy Extension Definition |
|
|
|
|
|
101.LAB+ |
|
XBRL Taxonomy Extension Label |
|
|
|
|
|
101.PRE+ |
|
XBRL Taxonomy Extension Presentation |
|
|
|
|
|
# |
Indicates management contract or compensatory plan. |
|
+ |
Indicates filed herewith. |
|
(1) |
Previously filed as an exhibit to the registrant’s Registration Statement on Form S-1 (File No. 333-194713) filed with the Securities and Exchange Commission on January 23, 2015 and incorporated herein by reference. |
|
(2) |
Previously filed as an exhibit to the registrant’s Registration Statement on Form S-1 (File No. 333-194713) filed with the Securities and Exchange Commission on October 9, 2014 and incorporated herein by reference. |
|
(3) |
Previously filed as an exhibit to the registrant’s Registration Statement on Form S-1 (File No. 333-194713) filed with the Securities and Exchange Commission on January 12, 2015 and incorporated herein by reference. |
|
(4) |
Previously filed as an exhibit to the registrant’s Registration Statement on Form S-1 (File No. 333-194713) filed with the Securities and Exchange Commission on January 15, 2015 and incorporated herein by reference. |
|
(5) |
Previously filed as an exhibit to the registrant’s Annual Report on Form 10-K for the year ended December 31, 2014, filed with the Securities and Exchange Commission on March 31, 2015 and incorporated herein by reference. |
|
(6) |
Previously filed as an exhibit to the registrant’s Current Report on Form 8-K, filed with the Securities and Exchange Commission on October 2, 2015 and incorporated herein by reference. |
|
(7) |
Previously filed as an exhibit to the registrant’s Current Report on Form 8-K, filed with the Securities and Exchange Commission on May 2, 2016 and incorporated herein by reference. |
|
(8) |
Previously filed as an exhibit to the registrant’s Quarterly Report on Form 10-Q for the period ended June 30, 2016, filed with the Securities and Exchange Commission on August 12, 2016 and incorporated herein by reference. |
|
(9) |
Previously filed as an exhibit to the registrant’s Current Report on Form 8-K, filed with the Securities and Exchange Commission on January 18, 2017. |
|
(10) |
Previously filed as an exhibit to the registrant’s Current Report on Form 8-K, filed with the Securities and Exchange Commission on December 14, 2017. |
|
(11) |
Previously filed as an exhibit to the registrant’s Current Report on Form 8-K, filed with the Securities and Exchange Commission on January 24, 2018. |
|
(12) |
Previously filed as an exhibit to the registrant’s Current Report on Form 8-K, filed with the Securities and Exchange Commission on February 14, 2018. |
107