Attached files
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| ☒ | ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
OR
| ☐ | TRANSITION REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number: 001-37406
CORINDUS VASCULAR ROBOTICS, INC.
(Exact name of Registrant as specified in its charter)
| Delaware | 30-0687898 | |||||
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |||||
309 Waverley Oaks Road, Suite 105, Waltham, MA 02452
(Address of principal executive offices)
| (508) 653-3335 | ||
| (Registrant’s telephone number, including area code) | ||
Securities registered pursuant to Section 12(b) of the Exchange Act:
| Title of each class | Name of each exchange on which registered | |
| Common Stock, $0.0001 par value per share | NYSE American |
Securities registered pursuant to Section 12(g) of the Exchange Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act.
Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☒ |
| Non-accelerated filer | ☐ | Smaller reporting company | ☐ |
| (Do not check if a smaller reporting company.) | Emerging growth company | ☐ | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 31(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
The aggregate market value of the common stock held by non-affiliates of the registrant, as of June 30, 2017 (the last business day of the registrant’s second quarter of fiscal 2017), was approximately $204,251,114. For purposes of this computation, all officers, directors, and 10% beneficial owners of the registrant are deemed to be affiliates. Such determination should not be deemed to be an admission that such officers, directors, or 10% beneficial owners are, in fact, affiliates of the registrant.
As of March 9, 2018, the registrant had outstanding 188,772,869 shares of common stock, $0.0001 par value, which is its only class of common stock.
DOCUMENTS INCORPORATED BY REFERENCE:
Portions of the registrant’s definitive Proxy Statement for its 2018 Annual Meeting of Stockholders, which is expected to be filed with the Securities and Exchange Commission no later than 120 days after the registrant’s fiscal year ended December 31, 2017, are incorporated by reference into Part III of this Annual Report on Form 10-K where indicated.
CORINDUS VASCULAR ROBOTICS, INC.
INDEX TO ANNUAL REPORT ON FORM 10-K
FOR THE YEAR ENDED DECEMBER 31, 2017
Table of Contents
CorPath® is a registered trademark of our company and technIQ™ is a trademark of our company. This Annual Report may also contain trademarks and trade names of other companies.
This Annual Report includes market and industry data that we obtained from periodic industry publications, third-party studies and surveys, government agency sources, filings of public companies in our industry, and internal company surveys. Industry publications and surveys generally state that the information contained therein has been obtained from sources believed to be reliable. Although we believe the foregoing industry and market data to be reliable at the date of the report, this information could prove to be inaccurate as a result of a variety of matters.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K and the information incorporated by reference in this Annual Report contain forward-looking statements that involve substantial risks and uncertainties. For example, statements regarding our operations, financial position, business strategy, product development, and other plans and objectives for future operations, and assumptions and predictions about future product development and demand, research and development, marketing, expenses and sales are all forward-looking statements. These statements may be found in the items of this Annual Report entitled “Business” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” as well as in this Annual Report generally. These statements are generally accompanied by words such as “intend,” “anticipate,” “believe,” “estimate,” “target,” “project,” “potential,” “continue,” “forecast,” “predict,” “plan,” “may,” “will,” “could,” “would,” “should,” “expect,” or the negative of such terms or other comparable terminology.
We have based these forward-looking statements on our current expectations and projections about future events. We believe that the assumptions and expectations reflected in such forward-looking statements are reasonable, based on information available to us on the date hereof, but we cannot assure you that these assumptions and expectations will prove to have been correct or that we will take any action that we may presently be planning. These forward-looking statements are inherently subject to known and unknown risks and uncertainties. Actual results or experience may differ materially from those expected or anticipated in the forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, research and product development uncertainties, regulatory policies and approval requirements, competition from other similar businesses, market and general economic factors, and the other risks discussed in Item 1A of this Annual Report. This discussion should be read in conjunction with the consolidated financial statements and notes thereto included in this Annual Report.
We have identified some of the important factors that could cause future events to differ from our current expectations and they are described in this Annual Report in the section entitled “Risk Factors” that you should review carefully. Please consider our forward-looking statements in light of those risks as you read this Annual Report. If one or more of these or other risks or uncertainties materialize, or if our underlying assumptions prove to be incorrect, actual results may vary materially from what we project. We do not undertake to update any forward-looking statements or to publicly announce the results of any revisions to any statements to reflect new information or future events or developments.
EXPLANATORY NOTE
Unless the context otherwise requires, the terms “Company,” “we,” “us,” or “our” refer to Corindus Vascular Robotics, Inc., a Delaware corporation, together with our subsidiaries, Corindus, Inc., a Delaware corporation, and Corindus Security Corporation, a Delaware corporation. Where appropriate, content related only to Corindus, Inc. is referenced as Corindus, Inc.
MARKET, INDUSTRY AND OTHER DATA
Unless otherwise indicated, information contained in this Report concerning our industry and the markets in which we operate, including information regarding our general expectations and market position, market opportunity and market size, is based on information from various sources and on assumptions that we have made based on such information and other similar sources and on our knowledge of the markets for our products. This information involves a number of assumptions and limitations, and you are cautioned not to give undue weight to such information. In addition, projections, assumptions and estimates of our future performance and the future performance of the industry in which we operate are necessarily subject to a high degree of uncertainty and risk due to a variety of factors and could differ materially from those expressed in the programs, assumptions and estimates made by third parties and by us.
1
Overview
We design, manufacture and sell precision vascular robotic-assisted systems for use in interventional vascular procedures (the “CorPath® System”). The CorPath System is the first medical device cleared by the U.S. Food and Drug Administration (“FDA”) to bring robotic-assisted precision to radial, coronary and peripheral procedures. During these procedures, the interventional cardiologist sits at a radiation-shielded workstation to advance interventional devices with millimeter-by-millimeter precision. The workstation allows the physician greater control and the freedom from wearing heavy lead protective equipment that can cause musculoskeletal injuries. The CorPath System brings robotic precision to radial and complex interventional procedures to help optimize clinical outcomes and minimize the costs associated with complications of improper stent placement with manual procedures. In October 2016, we received 510(k) clearance from the FDA for our CorPath GRX System, the second generation of the CorPath System. CorPath GRX significantly builds upon the CorPath 200 platform, adding a significant number of key upgrades that increase precision, improve workflow, and extend the capabilities and range of the procedures that can be performed robotically. These features include active guide management which enables control of the guide catheter along with robotic control of the guidewire and balloon or stent catheter, with one-millimeter advancement, from the control console. This precise positioning will enable physicians to adjust guide catheter position during procedures, and may expand use of CorPath to more complex cases. We began commercial shipment of the CorPath GRX System in late January 2017. While the CorPath GRX has been cleared for and we are targeting percutaneous coronary intervention procedures and peripheral vascular interventions, we believe our technology platform has the capability to be developed in the future for other segments of the vascular intervention market, including neurointerventional procedures and other more complex cardiac interventions such as structural heart procedures. As of December 31, 2017, we have installed 33 CorPath GRX Systems. Additionally, as of December 31, 2017, we shipped six CorPath GRX Systems that were accepted by a distributor. During 2017, the majority of our consumable revenues relate to the sale of CorPath GRX System cassettes and accessories.
Corporate History
Our Company was incorporated under the laws of the State of Nevada on May 4, 2011 under the name “Your Internet Defender Inc.” On August 12, 2014, we closed (the “Closing”) a reverse acquisition transaction (the “Acquisition”) in which we acquired Corindus, Inc. and Corindus Security Corporation as wholly owned subsidiaries. Immediately following the Closing, the business of Corindus, Inc. became our sole focus. We subsequently changed our name to Corindus Vascular Robotics, Inc. and increased our authorized capital stock to 260,000,000 shares (250,000,000 shares of common stock, $0.0001 par value per share, and 10,000,000 shares of preferred stock, $0.0001 par value per share). Effective June 28, 2016, our Company changed its state of incorporation from the State of Nevada to the State of Delaware.
On May 28, 2015, we completed a public offering by issuing 12,650,000 shares of our common stock at $3.80 per share in exchange for proceeds of $44,392,000 net of underwriting discounts, commissions and other offering costs. In connection with the public offering, our common stock was approved for listing on the NYSE American, where it commenced trading on May 29, 2015 under the symbol “CVRS.” Our Company’s common stock was previously traded on the OTCQB as provided by OTC Markets Group, Inc. under the symbol “CVRS.”
Percutaneous Coronary Intervention History and Development
PCI, sometimes known as coronary angioplasty, is a non-surgical technique used to open stenotic (narrowed or blocked) coronary arteries found in coronary artery disease. Coronary arteries supply the heart muscle with blood. PCI requires the use of a cardiac catheterization suite (sometimes called a cath lab) with special equipment, x-ray capability and trained personnel. Usually, access to the patient’s heart and major blood vessels is obtained percutaneously through the femoral artery in the groin area. The artery is punctured through the skin with a special needle. Under x-ray guidance, a guide catheter is introduced through the femoral artery up to the aorta (large artery from the heart) and then gently advanced into the blocked coronary artery. The catheter and its devices are passed through the inside of the artery into an area of coronary artery narrowing or blockage. At the leading tip of this catheter, several different devices (such as a balloon, stent or cutting device) can be deployed. A balloon is used to open the coronary artery and restore blood flow. Usually at that time, a stent (a mesh-like tube that holds open the artery) is placed to maintain good blood flow through the damaged area.
PCI is the single highest-volume vascular intervention, with more than 3.6 million procedures performed on a global basis annually according to J.P. Morgan’s 2016 Interventional Cardiology Market Model. PCI can be used to relieve or reduce angina, prevent heart attacks and alleviate congestive heart failure and allows some patients to avoid open heart surgery, which often involves an extensive procedure and a long rehabilitation period.
The first PCI procedure, then known as percutaneous transluminal coronary angioplasty, was performed in Zurich in September 1977 by Andreas Gruentzig, a Swiss radiologist. The early procedures had limited success due to risks associated with the use of large guide catheters that could easily rupture the vessel, no availability of guidewires and large balloon catheters with low burst pressure points. From 1977 to 1986, guide catheters, guidewires and balloon catheter technology were improved, with slimmer profiles and increased tolerance to higher inflation pressure. Stents, first introduced in 1986, are now used in most coronary interventions. The utility of stents has substantially increased procedural safety and success, thus significantly reducing the need for emergency coronary artery bypass surgery.
Occupational Hazards of Catheterization Labs
While there has been significant innovation in the devices and diagnostic tools used in interventional cardiology procedures, the way the manual procedures are generally performed by physicians has remained virtually unchanged since the first procedure by Dr. Gruentzig over 40 years ago. In order to perform the procedure, a physician stands by the patient who is lying on the cath lab table. The physician wears cumbersome and heavy protective apparel containing lead to block exposure to the ionizing radiation of x-rays used in the procedure and thereby combat its adverse effects. Already under bodily strain, the physician must deliver constant x-ray exposures to view the different vessels, which provides visual guidance for manual manipulation of interventional devices inside the patient’s heart. In addition to these physical demands, the current manual methods of performing PCI procedures make it difficult for physicians to visualize and estimate the length of the blocked lesion that requires the treatment, often leading to improper device selection and poor placement accuracy.
2
Interventional cardiologists who perform vascular interventional procedures face life-threatening risks from excessive radiation exposure, may suffer significant occupational hazards and must overcome procedural challenges when performing traditional coronary interventions. The chronic ionizing x-ray radiation exposure to the physician’s eyes associated with traditional PCI can cause posterior lens opacities, early cataracts and cancer malignancies. Orthopedic injuries from standing for long periods of time while wearing heavy radiation protection are also common, as are chronic pain complaints and missed physician workdays. In light of these risks, several professional societies and governmental agencies worldwide have called for reductions in radiation to improve catheterization laboratory safety.
Research shows that interventional cardiologists experience among the highest levels of radiation exposure of any medical professional, which leads to increased risk for cancer and cataract formation in addition to increased levels of orthopedic strain from the use of heavy protective garments required to block such exposure. In a study of 36 physicians (of which 28 were interventional cardiologists) with brain tumors potentially linked to radiation exposure over their careers, 86% had left-sided tumors, indicating a correlation with the physician’s position at the cath lab table. Additionally, in a survey of interventional cardiologists conducted by the Society for Cardiovascular Angiography and Interventions, 49.4% reported at least one orthopedic injury, 6.9% were required to limit their caseload due to radiation exposure, and 9.3% experienced health-related periods of absence. Many hospitals will not allow female interventional cardiologists to practice during pregnancy, while others require them to wear lead protective gear with twice the typical thickness to protect from radiation exposure.
We believe that the future of interventional procedures, where the physician sits inside the cath lab within a radiation-shielded interventional cockpit, will be greatly improved through the use of advanced robotic tools that provide (i) enhanced safety for the catheterization lab staff relative to radiation exposure, (ii) improved patient procedures through advanced precision, dexterity and visualization for the physician and (iii) an economically compelling solution for the hospital. We are pioneering the use of precision vascular robotics to achieve these goals and to improve the way that minimally invasive vascular interventions are performed.
Our Precision Robotics Systems
We design, manufacture and sell our CorPath System for use in radial and complex interventional vascular procedures to bring the precision and accuracy of the only FDA-cleared robotic platform to facilitate stent positioning for PCI procedures by allowing a physician to measure, manipulate and advance devices with robotic precision. Additionally, our CorPath System allows the physician to perform PCI procedures with a control console located within an interventional cockpit. While the CorPath GRX has been cleared for and we are targeting PCI procedures and the CorPath 200 has been cleared for an additional indication for peripheral vascular interventions, we believe our technology platform has the capability to be developed in the future for other segments of the vascular intervention market, including neurointerventional procedures and other more complex cardiac interventions such as structural heart procedures.
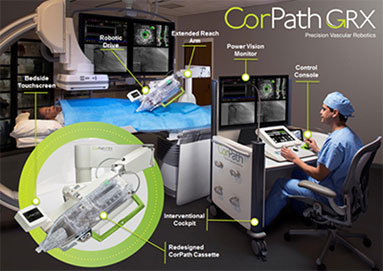
The CorPath GRX System enables the precise, robotic-assisted control of coronary guide catheters, guidewires and balloon/stent devices from the safety of a radiation-shielded interventional cockpit. The CorPath GRX System consists of two components: a Bedside Unit and an Interventional Cockpit. The radiation-shielded cockpit features a simple-to-use Control Console to precisely control the movement of guide catheters, guidewires, and balloon/stent catheters. Using joysticks and touch-screen controls, the physician is able to measure lengths of portions of anatomy to help in selecting the appropriate stent. At the bedside, the CorPath GRX System’s Robotic Drive and sterile, single-use cassette (“CorPath Cassette”) translate the physician’s commands into precise movements and manipulations of the coronary guide catheters, guidewires, stents and catheters. The CorPath GRX Cassette provides a single-use sterile interface with standard PCI catheters, guidewires and devices. The CorPath GRX System empowers physicians with precise sub-millimeter measurement and 1mm advancement accuracy. By optimizing stent selection and positioning, the CorPath GRX System enables the deliberate advancement of devices, provides the ability to hold the guidewire and balloon/stent in place during device deployment and helps to ensure that there are no unintended wire/device movements during the procedure.
The CorPath GRX System allows the interventional cardiologist to perform the procedure while seated in an ergonomic and comfortable position in a radiation-shielded cockpit positioned as close as a few feet away from the patient. Our radiation-shielded cockpit provides a reduction in radiation exposure for the primary operator as compared to levels found at the traditional table position for manual procedures. The cockpit allows the physician to control the procedure while seated outside of the radiation field without the need for heavy protective wear. The Percutaneous Robotically-Enhanced Coronary Intervention Study (the “PRECISE Study”) published in the Journal of the American College of Cardiology (the “JACC”), which we sponsored using the CorPath 200 System, demonstrated a 95.2% reduction in radiation exposure to the primary operator. The CorPath GRX System also provides physicians with enhanced visualization of the procedure through a high resolution widescreen monitor positioned at eye level in the cockpit. These improvements have the potential to reduce physician fatigue and could potentially extend a physician’s medical career. A photo of our CorPath GRX System appears below.
The CorPath GRX System
3
Overview of Industry and Market
Vascular Market
We developed vascular robotic technology to provide physicians with protection from the occupational hazards of the cath lab and to provide them with robotic precision while executing vascular procedures. Our initial indication for use of the CorPath System is for PCI and peripheral vascular procedures. We believe our technology can be applied to various vascular clinical applications and markets, including neurointerventional and structural heart, and we may decide to pursue these markets in the future.
Coronary Market (PCI)
Our current target market is all cardiac cath labs in the U.S. The IMV 2013 Cardiac Cath Lab Market summary report estimates that there are more than 3,250 cath lab rooms in the U.S. performing PCI procedures, which represents approximately 40% of the global market of more than 8,000 PCI cath lab rooms. According to the J.P. Morgan 2016 Interventional Cardiology Market report, there are over 3.6 million PCI procedures performed worldwide each year and approximately 940,000 procedures performed each year in the U.S. The portion of the U.S. cath lab rooms qualifying as customers likely to purchase our product is difficult to ascertain because potential customers are determined by our sales team on a case-by-case basis and is somewhat subjective based on the priorities of each individual facility. Cath lab patient volume has decreased over the past several years, leading to increased competition for patients.
Peripheral Vascular Market
According to Millenium Research Group’s 2013 report on U.S. Markets for Peripheral Devices, approximately 1.7 million peripheral vascular procedures are performed annually worldwide (approximately 40% of those in the U.S.) and the annual procedure volumes are expected to grow to over 2.3 million procedures worldwide by 2018. While some peripheral procedures are conducted in cath labs that also conduct PCI procedures, IMV’s 2012 Interventional Angiography Lab Market summary report estimates that there are over 3,500 non-PCI peripheral vascular labs worldwide which represent incremental CorPath System placement opportunities beyond PCI.
Neurointerventional Market
Medtech Insight’s 2011 report on U.S. Markets for Neurosurgical and Neurointerventional Surgical Products estimates that 395,000 neurointerventional procedures are performed each year, 160,000 in the U.S. and 235,000 internationally, and that the annual volume will grow to an estimated 720,000 worldwide procedures by 2018. The number of incremental, dedicated system worldwide sales opportunities exceeds 400 labs, with 40% in the U.S. and 60% outside the U.S. (“International”).
Structural Heart Market
The number of structural heart procedures has been growing and is expected to continue to grow significantly, with an estimated 40,000 worldwide procedures currently performed annually (25% U.S., 75% International) expected to grow to an estimated 120,000 annual structural heart procedures by the year 2018, according to a 2013 clinical report in the Journal of the American College of Cardiology.
Our Business Model
Our business model involves the launching of coronary robotic-assisted intervention programs which include the sale of a durable robotic system and a repeat consumable. After the program launch and the sale and installation of the CorPath System in a hospital cath lab, we provide customer support through training and sales of our CorPath Cassette, which provides a sterile interface with standard PCI guide catheters, guidewires and devices. The CorPath Cassette is consumed and replaced for each new patient procedure. The use of the CorPath Cassettes represents opportunity for recurring revenue for each PCI procedure using the CorPath System. We also sell service contracts providing various levels of ongoing service. Over time, we expect to have follow-on sales related to the CorPath System to offer and install robotic system upgrades with more features and new applications.
Our current product line is marketed and sold in the United States by our direct sales team that calls on interventional cardiologists, catheterization lab departments and executive administrators in hospitals across the U.S. to launch coronary robotic-assisted intervention programs and drive sales of our CorPath System and our CorPath Cassette. We employ two different types of sales representatives in the field. Our Regional Sales Managers (“RSMs”) operate as full line sales professionals responsible for selling CorPath Systems, Cassettes and all other associated disposable accessories. Our Clinical Specialists (“Clinicals”) focus on clinical education and training of physicians and their staff and selling CorPath Cassettes as well as associated disposable accessories.
The RSMs are responsible for identifying potential customers in the more than 3,250 cath lab rooms performing PCI procedures in the U.S. that desire to launch a coronary robotic-assisted intervention program and purchase a CorPath System. The RSMs may sell the CorPath System as a capital sale or through third-party financed leasing or rental programs. The RSMs are also responsible for selling service contracts for the CorPath System. The RSMs are supported by our marketing department, which provides them with leads and sales opportunities garnered through direct marketing activities at interventional cardiology conferences, online webinars, regional seminars and trade journal advertising. Our marketing department also provides the RSMs with the sales tools and marketing resources to help convey the value proposition of the CorPath System.
Our Clinicals focus their efforts on selling our CorPath Cassettes and other associated disposable accessories designed to maintain a sterile environment when using our products in a cath lab. They are responsible for increasing their account sales through new orders and repeat consumable sales within their specific accounts. The Clinicals build important relationships throughout the CorPath System installed base accounts, including with the interventional cardiologists, the cath lab technologists, nurses, cath lab directors, schedulers, purchasers and administrators. The Clinicals are responsible for ongoing training and development of the CorPath System installed base accounts to build successful CorPath robotic programs and expand system usage across physicians. The Clinicals are also responsible for ensuring purchase orders are obtained and that appropriate inventory levels are maintained on site.
4
Driving Utilization of the CorPath System
Following the launch of the coronary robotic-assisted intervention program and the initial sale of a CorPath System to a hospital, we provide enhanced training to the primary physicians and cath lab staff responsible for launching the program and then work to secure an increase in the number of cases performed over time. Subsequently, we expand training to the next group of physicians who use the system. We consistently focus our efforts to make sure that the system is well integrated into the customer’s everyday workflow within the cath lab. Dedicated sales and marketing efforts support awareness and use of the CorPath System. Utilization support comes both from encouraging the use of the system within customer accounts and by providing materials to educate general cardiologists and patients on the availability of the CorPath System at the customer site and in their geographical area.
The CorPath System uses a proprietary single-use sterile cassette, which is the source of recurring revenue as use of the CorPath System continues and increases. After a CorPath System is installed and initial training is complete, we provide ongoing support in order to increase customers’ familiarity with system features and benefits with the goal of increasing usage of the CorPath System.
Service Revenue
One year of customer support and warranty is included with the sale of each CorPath System. Thereafter, we sell service contracts under which we continue to provide support after the one-year period. We anticipate that service beyond the basic warranty will become an increasingly important additional source of revenue.
Our Growth Strategy
Our goal is to ensure that the coronary robotic-assisted intervention program and the use of the CorPath System becomes the standard of care for interventional procedures by providing unsurpassed protection for cath lab staff and being the leading precision robotic technology for patient procedures. We have identified key characteristics and criteria for customers that are imperative to implement a successful cardiovascular robotics program. We are meaningfully narrowing our target customers focus with the intent to be deeply integrated within our customer partnerships. We are working with selected customers around the country to establish CorPath System centers of excellence. These centers allow us to bring prospective customers to visit a hospital and cath lab that has previously installed a CorPath System. The robotic course visit will allow the prospective customer the opportunity to see the system installed and in use. It also provides the opportunity to discuss the benefits of the system with the hospital staff, including interventional cardiologists, technologists and administrators, and view the work flow of the system in a real life clinical setting. We have successfully conducted such courses at several sites around the country and we are continuing to establish these centers of excellence and collaborative relationships with key institutions.
We intend to establish our Company and technology as the brand that cares about and supports the physician and cath lab staff by leading the industry in providing solutions that address and remedy their occupational hazards. By promoting safety and providing awareness of occupational hazards in the cath lab and supporting education about solutions, we hope to become the preferred source for customers seeking to improve the safety of their operations.
A second prong of our growth strategy is to expand into new clinical segments. In addition to our objective to make the CorPath System the premier standard for PCI procedures, we may decide to pursue additional vascular interventional applications for our vascular robotic-assisted technology. Our closest adjacent opportunity is in peripheral vascular procedures performed by interventional cardiologists, vascular surgeons and interventional radiologists. The CorPath GRX System was cleared for use in peripheral vascular interventions in February 2018. These procedures treat vascular disease in non-coronary areas like the patient’s legs. These procedures are often quite lengthy and they expose physicians to x-ray radiation for extended periods of time. The peripheral vascular procedure market has been growing rapidly and is projected to grow at a compound annual growth rate, or CAGR, of 5.9% based on iData’s October 2013 Research Report: “US Markets for Peripheral Vascular Devices and Accessories.”
Further expansion into neuro-interventional procedures to treat stroke, brain aneurysms and other diseases of the head and neck could allow us to leverage precision robotic-assisted tools into these highly precise procedures which are very well reimbursed.
Another area of future growth is the emerging market of structural heart procedures. This market segment is experiencing rapid growth due to the advent of new catheter-delivered medical devices that are replacing open surgical procedures. One of the most prominent new devices in this market is the transcatheter aortic valve. The transcatheter aortic valve replacement (“TAVR”) procedure requires very complex integration of a variety of imaging modalities and precise deployment of the device. Our interventional cockpit and robotic-assisted control could potentially provide significant benefits to the execution of TAVR procedures.
Any of these potential applications would require additional clinical trials and various levels of research, engineering, software development, product development, system modifications and regulatory approvals.
An integral part of our growth strategy is to expand commercialization beyond the U.S. marketplace. Opportunities outside of the U.S. represent over 60% of the global procedure volume and are growing at a rate faster than the U.S. market. We intend to expand into and penetrate these new geographical international markets over time by leveraging our product development, clinical research and regulatory approvals gained in the U.S. Our initial international target markets include the Middle East, Northern Europe and Japan. We have a CE Mark for the CorPath System which permits us to commercially distribute these systems throughout the European Union. In February 2017, we announced the signing of a strategic distribution agreement with Japan Medicalnext Co., Ltd., a wholly-owned entity of MC Healthcare, Inc., (subsidiary of Mitsubishi Corporation) and prominent supplier of medical devices in Japan. Pursuant to the agreement, Japan Medicalnext became the exclusive distributor of our products in Japan. Japan is the third largest market for PCI, with an approximate annual volume of 250,000 procedures. We are working together with our distributor to secure Pharmaceutical and Medical Device Agency (“PMDA”) approval as the first step in preparing for the commercial launch of CorPath GRX in Japan, which we expect to occur in the first half of 2018.
Research and Development
We have built a leading Research and Development (“R&D”) organization comprised of mechanical, electrical, systems and software engineers with deep expertise in med-tech and robotics. We expect our investment in R&D to expand the capabilities of our robotic system – allowing for control and delivery of a wide range of interventional devices across the cardiovascular, peripheral and neurovascular spaces. In addition to scaling capabilities of our robotic platform, our technology roadmap includes development programs for tele-stenting, procedural automation, and artificial intelligence. Tele-stenting, or remote robotic treatment for PCI, will enable physicians to conduct procedures from virtually any location. Procedural automation will focus on automating techniques from the best physicians in the world – making them available to every hospital with a robotic PCI program. Our long-term vision is to enable autonomous navigation of medical devices for interventional procedures. Our artificial intelligence capabilities will leverage deep learning and algorithms to reduce variability of treatment and improve quality of care. Our research and development activities relating to tele-stenting, procedural automation, and other artificial intelligence capabilities are in early stages and we can provide no assurances that we will be able to successfully develop and commercialize this technology.
5
In addition to expanding the capabilities of the CorPath System, we will continue to invest in the design of system manufacturability improvements which we expect to result in a lower cost system and cassette. The engineering function will use Design for Manufacturability and Assembly (“DFMA”) processes in an effort to reduce costs. DFMA is the combination of two methodologies: Design for Manufacture, which means the design for ease of manufacture of the parts that will form a product, and Design for Assembly, which means the design of the product for ease of assembly. DFMA is used as the basis for concurrent engineering studies to provide guidance to the design team in simplifying the product structure to reduce manufacturing and assembly costs and to quantify improvements. DFMA is a component of lean manufacturing.
Research and development expense amounted to approximately $9.5 million, $10.3 million and $10.0 million for the years ended December 31, 2017, 2016 and 2015, respectively.
Clinical Studies
We are dedicated to continually advancing robotic-assisted PCI through the publication of clinical data supporting the CorPath System’s value and applicability. We are working with several leading institutions to conduct clinical research activities to further collect evidence regarding the applicability and benefits of robotic-assisted procedures. We are committed to collaboration with prominent interventional cardiologists to build evidence for the benefits of robotic-assisted PCI. We intend to continue to pursue opportunities to develop further evidence for the benefits of the CorPath GRX System in practice. An important component to making the CorPath GRX System the standard of care in the cath lab will be to demonstrate the clinical benefits and applicability of the CorPath GRX System and the advancement of robotic-assisted procedures.
First in Man Trial
In April 2011, the First in Man Trial for the CorPath Robotic-assisted PCI System was published in the Journal of the American College of Cardiologists. This clinical trial enrolled eight patients with coronary artery disease who required a PCI procedure at the Corbic Research Institute in Envigado, Colombia. All patients were treated for a single de novo coronary lesion up to 25mm in length located in a vessel 2.5-4.0 mm in diameter. The procedure was successfully completed in all eight patients utilizing the CorPath System to advance coronary guidewires and perform the intervention, and there were no reported device or procedure-related complications or major adverse events. Operator radiation exposure was 97% lower with the use of the CorPath System in comparison with levels found at the standard table position.
CorPath PRECISE Study
We sponsored the PRECISE Study aimed to evaluate the safety and effectiveness of the clinical and technical performance of the CorPath System in the delivery and manipulation of coronary guidewires and balloon/stent devices for use in PCI procedures. We sponsored the PRECISE Study under Investigational Device Exemption (“IDE”) approval from the FDA to obtain 510(k) clearance. The PRECISE Study was a prospective, single-arm, multi-center, non-randomized trial of the CorPath System. We enrolled 164 patients who were evaluated at nine clinical sites (eight in the U.S.). The PRECISE Study was conducted under Principal Investigators Dr. Giora Weisz, MD Associate Professor of Medicine at Columbia University Medical Center and Chairman of Cardiology, Shaare Zedek Medical Center, Jerusalem, Israel, and Dr. Joseph Carrozza, Chief of Cardiovascular Medicine at St. Elizabeth’s Medical Center in Boston. Physicians participating in the trial did not receive any direct financial compensation. Results of the PRECISE Study were published in the April 2013 issue of the Journal of the American College of Cardiology and reported a successful PCI completion with use of the CorPath System in 162 of the 164 cases. In each of the two cases in which the PCI procedure was not completed, the interventionalist left the CorPath cockpit to complete the procedure manually, resulting in an incomplete use of the CorPath System. The average radiation exposure to the cardiovascular interventionalist decreased by 95.2% in comparison with levels measured at the location where manual procedures are normally conducted during standard interventions. The overall rate of clinical procedure success was 97.6%, with 100% of patients achieving post-procedure stenosis of less than 30% (as evaluated by a Core Laboratory), and 97.6% of patients had an absence of Major Adverse Cardiac Events (“MACE”). The four MACE events that did arise in the PRECISE Study were cardiac enzyme elevations without symptoms. There were no device-related complications.
CorPath PRECISION Registry
In 2013, we initiated the PRECISION Registry, a multicenter post-market registry for the evaluation of the CorPath 200 System’s effectiveness in PCI procedures. PRECISION aimed to collect data on real-world use of the CorPath System. We were interested in learning about the patterns of the CorPath System’s use, safety and effectiveness from a broad registry participation perspective. Sixteen sites participated in the PRECISION registry. A subset of the data was presented as a Late Breaking Clinical Trial at SCAI 2017.
CorPath PRECISION GRX Registry
In 2017, we launched the PRECISION GRX registry, a multicenter post-market registry for the evaluation of the CorPath GRX System’s effectiveness in PCI procedures. PRECISION GRX aims to collect data on real-world use of the CorPath GRX System. With the addition of Active Guide Management we build on the data and experience gained from PRECISION with a focus on how adding guide catheter control impacts the overall outcome measures. There are currently 6 sites participating in the PRECISION GRX Registry, which is conducted under the leadership of Dr. Ehtisham Mahmud. Each site obtained approval to participate in the PRECISION GRX Registry from its hospital Institutional Review Board as part of the site’s regular clinical research approval process and other industry-standard protections are in place for patients enrolled in the post-market study. Data for the registry is being collected and monitored through industry-standard clinical research procedures.
6
Robotically-Assisted Peripheral Intervention for peripheral arterial Disease Study (RAPID)
The Robotically-Assisted Peripheral Intervention for peripheral arterial Disease (RAPID) Study to evaluate the safety and performance of the CorPath 200 System for use in percutaneous vascular interventions was completed in 2015. The RAPID trial was a single-arm, single center study conducted at the Medical University of Graz in Graz, Austria. The RAPID trial was led by Prof. Dr. Marianne Brodmann, MD, a leading researcher within the University’s Division of Angiology, in combination with Prof. Dr. Hannes Deutschmann, of the Medical University of Graz Department of Radiology, and study chairman, Dr. Ehtisham Mahmud, director, Sulpizio Cardiovascular Center-Medicine, UC San Diego. The trial was a prospective, single-arm, single-center study that enrolled 20 subjects to assess the safety and effectiveness of the CorPath System in recanalizing lower extremity arterial blockages during peripheral angioplasty procedures. Results of the RAPID Study were presented in a major cardiology conference. The PAD presentation (Rutherford Classification) was primarily severe (60%) or moderate (30%) claudication. A total of 29 lesions located in the superficial femoral (89.7%) or popliteal (10.3%) arteries were treated. Device technical success and clinical procedural success was 100%. Three minor procedure-related adverse events, all access site hematomas, were reported. There were no device-related complications. In 2016, the RAPID trial was published in JACC. The data was used to support FDA 510(k) clearance for the CorPath 200, therefore all devices used had to be FDA approved. This requirement limited robotic-assisted PVI to rapid exchange PTA balloons. In a subsequent study, RAPID II, femoropopliteal lesions were treated with robotic-assisted drug-coated balloons, allowing for a completely robotic-assisted PCI procedure. The results of this study are expected to be released later in 2018.
Staff Exposure to X-ray during PCI: CorPath vs. Manual: An Observational Study
The Staff Exposure to X-ray during PCI: CorPath vs. Manual: An Observational Study to compare cardiac catheterization staff and physician radiation exposure in robotic-assisted PCI with the CorPath 200 System vs. that in manual PCI was completed in 2015. This observational study was led by Dr. Paul T. Campbell, MD, a leading interventional cardiologist at Carolinas Medical Center – NorthEast, Concord, NC. The trial was a prospective, randomized, dual-arm, single-center study that enrolled 30 patients. This observational study showed a significant reduction in physician radiation exposure, and reduction in staff radiation exposure in the robotic-assisted PCI arm compared to the manual PCI arm.
Our Current Product Line
Our first product, the CorPath 200 System, brought the precision and accuracy of robotic technology to PCI procedures performed in an interventional cath lab. The CorPath 200 System is intended for use in the remote delivery and manipulation of coronary guidewires and rapid exchange balloon/stent catheters during PCI procedures and in the remote delivery and manipulation of guidewires and rapid exchange catheters during peripheral percutaneous vascular interventional procedures. The second generation CorPath GRX System is intended for use in the remote delivery and manipulation of guidewires and rapid exchange balloon/stent devices, and remote manipulation of guide catheters during percutaneous coronary intervention procedures and peripheral procedures. There is no contraindication for the use of either product in PCI procedures.
The CorPath System enables the precise, robotic-assisted control of coronary guidewires, guide catheters and balloon/stent devices from the safety of a radiation-shielded, ergonomic interventional cockpit. The CorPath System consists of two components: a bedside unit and an interventional cockpit. The radiation-shielded cockpit features a simple-to-use control console to precisely control the movement of guide catheters, guidewires and balloon/stent catheters. The bedside unit translates the physician’s commands into precise movements and manipulations of the coronary stents and catheters contained in a single-use cassette.
The CorPath Cassette provides a sterile interface with standard PCI guidewires and guide catheter devices and is replaced for each new patient procedure.
In July 2012, we received 510(k) clearance from the FDA for the CorPath 200 System and initiated a limited commercial launch in the U.S.
In October 2015, we received 510(k) clearance from the FDA for our robotic-assisted CorPath 200 System to be used during PCI procedures performed via radial access. The 510(k) clearance was based on results of a clinical trial conducted at Spectrum Health, Grand Rapids, Michigan, and St. Joseph’s Hospital Health Center, Syracuse, New York.
In March 2016, we received 510(k) clearance from the FDA for our robotic-assisted CorPath 200 System to be used during peripheral vascular interventions. The 510(k) clearance was based on results of a clinical trial conducted at Medical University of Graz in Graz, Austria.
In October 2016, we received 510(k) clearance from the FDA for our CorPath GRX, the second generation of our CorPath System. In February 2018, we received 510(k) clearance from the FDA for CorPath GRX to be used during peripheral vascular interventions. The CorPath GRX System is intended for use in the remote delivery and manipulation of guidewires and rapid exchange catheters, and remote manipulation of guide catheters during percutaneous coronary and vascular procedures.
In March 2018, we received 510(k) clearance from the FDA for the first automated robotic movement designed for the CorPath GRX platform. The software, named “Rotate on Retract” (RoR), is the first automated move in the technIQ™ Series. RoR allows the operator to quickly navigate to a targeted lesion by automatically rotating the guidewire upon joystick retraction. Over the next several years, we intend to focus on developing additional automated robotic movements to expand the technIQ™ Series.
While we are initially targeting PCI and peripheral vascular procedures, we believe our open platform technology has the capability to be developed in the future to address all segments of the vascular intervention market, including neurointerventional procedures and other more complex cardiac interventions such as structural heart procedures.
Products in Development
Our product is tailored to maximize penetration and adoption of our CorPath System technology while providing the best clinical outcomes to our customers and their patients. Our vision for the future is to provide physicians with a complete tool box to robotically perform any interventional procedure desired. We are seeking to expand our penetration within PCI to more complex cases. As we see robotics as the center of the lab, we will continue to integrate other technologies into our robotic system to enable a complete solution for physicians. In order to accomplish this goal, we may investigate proprietary devices, imaging integration and electronic medical record integration while continuing to optimize the workflow in the lab and the remote program we have launched.
7
Installed CorPath Systems and Backlog
As of December 31, 2017, there were 31 CorPath GRX Systems installed in hospitals across the U.S. and two installed at international locations. Physicians and their teams in these locations have received training and procedures are currently being performed. Currently these sites have between one to three primary physicians using the CorPath System. RSMs and Clinicals visit installed sites regularly to support current users and also to expand usage to new targeted users. As of December 31, 2017, we had orders for seven additional CorPath GRX Systems which we had not yet shipped or installed.
Intellectual Property
Our success depends, in part, on our ability to obtain patents, maintain trade secret protection and operate without infringing the proprietary rights of others. Our intellectual property (“IP”) portfolio covers aspects of our CorPath System and cassettes, as well as other technology that we have under development, and is one of the means by which we attempt to protect our competitive position. We rely primarily on a combination of know-how, trade secrets, patents, trademarks and contractual restrictions to protect our products and to maintain our competitive position. We are seeking other ways to protect our intellectual property through various legal mechanisms in relevant jurisdictions.
Our researchers and engineers work closely with our patent counsel to protect their inventions and intellectual property with patents issued around the world. We believe that we are building an extensive intellectual property portfolio to protect the fundamental scope of our technology, including our robotic technology, navigational methods, procedures, systems and consumable devices.
As of January 31, 2018, we currently own more than 70 patents with more than 50 pending patent applications. Of these, we had more than 30 issued U.S. patents with more than 30 pending U.S. patent applications and over 40 granted foreign patents and more than 20 pending foreign applications. The granted foreign patents are in France, Germany, Japan, Italy, Israel, the Netherlands and the United Kingdom. The pending applications are in China, Europe (through applications filed in the European Patent Office), India and Japan. Additionally, there are three Patent Cooperation Treaty applications pending. Our granted patents begin expiring in 2018, and continue to expire through 2030.
Our patents cover, among other things, technology related to robotic control of interventional devices, and the control of the CorPath System, including, but not limited to, the graphical and user interface, function and design of the CorPath Cassette, image-guided navigation for catheter-based interventions, measurement of the length of a structure, and radiation-protected work stations.
In addition to our existing patent coverage, we continue to invest in product development and new IP to further enhance the capabilities of the CorPath System for PCI and other vascular applications. Relative to our current and future portfolio, we believe it will be costly and technically difficult to reverse engineer our products.
We intend to actively protect our intellectual property with patents, trademarks, trade secrets or other legal avenues for the protection of intellectual property. We intend to aggressively prosecute, enforce and defend our patents, trademarks and proprietary technology. The loss, by expiration or otherwise, of any one patent may have a material effect on our business. Defense and enforcement of our IP rights can be expensive and time consuming, even if the outcome is favorable to us. It is possible that the patents issued or licensed to us will be successfully challenged. For example, a court may find that we are infringing on validly issued patents of third parties or that we may have to alter or discontinue the development of our products or pay licensing fees to take into account patent rights of third parties.
As we continue to develop proprietary intellectual property, we will expand our protection by applying for patents on future technologies. While we seek broad coverage under our patent applications, there is always a risk that an alteration to the process may provide sufficient basis for a competitor to avoid infringement claims. In addition, patents expire and we cannot provide any assurance that any patents will be issued from our pending application or that any potentially issued patents will adequately protect our intellectual property.
Sales and Marketing
We market, sell and support our products in the U.S. through our direct sales force of RSMs with support from our Clinicals who provide training and clinical support to our customers (collectively, our “Sales Team”). Our direct sales force is the primary distribution channel for CorPath System sales. Our Sales Team and headquarters-based marketing team work together to identify leads, evaluate clinical interest, navigate capital procurement processes and establish coronary robotic-assisted intervention programs, both in the United States and Internationally. Our sales and marketing program includes two important steps: selling CorPath Systems to the customer and then leveraging our installed base of systems to drive recurring sales of cassettes and service.
Sales targeting is based on segmentation to identify customers who are likely to purchase and utilize the CorPath System and customers who are likely to be influencers in their region which will help fuel further growth. All hospitals with cath lab rooms that perform PCI procedures are potential customers for a CorPath System. The portion of the approximately 3,250 cath lab rooms in the U.S. that will qualify as customers likely to purchase a CorPath System is difficult to ascertain because potential customers are determined by our sales team on a case-by-case basis and is somewhat subjective based on the priorities of each individual physician and hospital facility. We believe customers that are likely to purchase our product meet a critical criteria profile including (i) an awareness of the dangers faced by interventional cardiologists due to radiation and ergonomic issues in the cath lab, (ii) a practice volume large enough to economically support the CorPath System, (iii) hospital financial health that allows for the capital or operational expenditure for a CorPath System and (iv) regional competitiveness that demands the implementation of new technology.
Our sales effort begins with the interest of an influential physician; and, therefore, our marketing efforts are primarily directed toward interventional cardiologists. Our primary marketing objective is to raise awareness about the CorPath System and its features and benefits among our target customers.
Marketing awareness activities target two strategies:
| 1) | General awareness – build knowledge and understanding of the value that the CorPath System brings to the cardiology community, focused initially on awareness from interventional cardiologists; and | |
| 2) | Targeted awareness – using data analysis to identify a target segment of customers (hospitals and physicians) for additional marketing and sales focus. |
8
We also market and sell our products through certain distributor and partnership relationships. We also entered into a distributor relationship with Alliant Enterprises, LLC who assists us in working with U.S. Department of Veterans Affairs hospitals. In February 2017, we announced the signing of a strategic distribution agreement with Japan Medicalnext Co., Ltd., a wholly-owned entity of MC Healthcare, Inc., (subsidiary of Mitsubishi Corporation) and prominent supplier of medical devices in Japan. Pursuant to the agreement, Japan Medicalnext became the exclusive distributor of our products in Japan. We may continue to sell products through our former exclusive worldwide distributor, Philips Medical Systems Nederland B.V., although our distribution agreement entered into in 2010 has expired.
Clinical Benefits for Patients
Although more than 940,000 PCI procedures are performed annually in the U.S., interventionalists continue to face challenges of poorly selected or misplaced stents. Currently, PCI procedures are performed by interventional cardiologists who approximate lesion length using techniques of subjective visual estimation and tactile feel to position the stent. Published data from the Impact of Stent Deployment Procedural Factors on Long-Term Effectiveness and Safety of Sirolimus-Eluting Stents (“STLLR”) trial in 2008, a study designed to specifically examine PCI stent placement accuracy, showed that nearly 50% of coronary stent placements are not accurately positioned within the lesion using this technique. The clinical impact of longitudinal geographic miss includes complications such as re-occlusion that compels repeat intervention. The CorPath System presents a new option to interventional cardiologists with the potential to optimize clinical outcomes by providing enhanced visualization, precise anatomical measurement and improved control for optimal stent positioning. Using the CorPath System, physicians can (i) consistently measure the anatomy with sub-millimeter accuracy, helping them to choose the correct stent for each patient, (ii) move the guidewire straight into the vessel at the proper angle, potentially leading to a shortened procedure for the patient, (iii) view an enhanced, close-up view of the patient’s vessels and arteries for the entire procedure and (iv) hold the guidewire and balloon/stent in place during device deployment, helping to ensure no unintended wire/device movements during the procedure which could adversely affect the patient.
In addition, robotics can enable increased adoption of transradial access by eliminating the awkward working conditions and increased radiation exposure experienced by the physician. Data has shown that transradial access has significant benefits for patients including reduced incidence of procedural complications including major bleeding and vascular complications, reduced hospitalization, and an improved patient experience. Specifically, left transradial access has additional benefits for certain patients including those who have previously undergone coronary artery bypass grafting (CABG) surgery and those who are right-hand dominant Studies have demonstrated that combining robotics with transradial access can also increase the precision of stent positioning which may improve patient outcomes.
Physician Benefits
The cath lab is a hazardous work environment where interventional cardiologists are exposed to radiation on a daily basis. Physicians face two significant risks in the cath lab: damaging radiation exposure despite the use of heavy lead protective aprons and orthopedic strain due to wearing such protective garments while working in ergonomically compromising positions. The International Agency for Research on Cancer (part of the World Health Organization) and the U.S. Environmental Protection Agency independently recognize that ionizing radiation, such as x–rays, can cause cancer and have classified such radiation as a “known carcinogen.” The primary method recommended to partially protect oneself from radiation exposure in the cath lab environment entails wearing more than 20 pounds of lead while leaning over a patient’s table, which leads to interventionalist disc disease of the spine as well as knee, hip and neck injuries. Our CorPath System can limit these risks as evidenced by the results from our PRECISE Study, which demonstrated a 95.2% reduction in exposure to radiation obviating the need to wear lead during the procedure.
Hospital Benefits
Hospitals face increasing pressure to maintain or grow cath lab procedure volumes. By offering a differentiated service, such as robotic-assisted PCI, we can help a facility grow its business. As demonstrated with robotic surgery, hospitals that adopt and promote the technology can benefit in the form of additional patients and procedures.
Target Customers
The Interventional Cardiologist
The physician is a key decision maker in the evaluation and adoption of new technologies in the interventional cath lab. There are approximately 5,200 active interventional cardiologists in the United States, according to a 2013 article in the Catheterization and Cardiovascular Interventions journal, who are estimated to perform in the aggregate more than 940,000 PCI procedures per year. Interventional cardiologists tend to incorporate technology into their practice and are very focused on products that improve patient care and clinical outcomes. Additionally, interventional cardiologists experience unique occupational risk from their work environment, with the largest exposure to radiation of any medical professionals. To offset this risk, interventionalists wear heavy lead protection exposing them to a higher risk of orthopedic injuries and resulting pain.
The CorPath System allows physicians to measure anatomy with sub-millimeter accuracy and manipulate the interventional device in 1mm increments and with discrete rotational movements. The capability to accurately control and deliver treatment, using a guide catheter, guidewire and stent of their choice, allows physicians to optimize their PCI procedures and potentially provide better clinical outcomes for their patients. Specifically, the robotic precision can potentially minimize longitudinal geographic miss which has been demonstrated in the STLLR trial to correlate to a 2.3 times greater chance of needing to revascularize the target vessel in the first post-procedure year.
In addition, because physician safety is a growing concern (e.g., studies have shown an increased presence of left-sided brain tumors due to occupational radiation exposure), the ability of the CorPath System to reduce the level of occupational radiation will continue to be a key marketing message. The safety aspect of the device may be a key selling feature as more physicians become employed by healthcare groups which will need to address these concerns to avoid potential workers’ compensation claims and reduce insurance costs. Thus, messaging to physicians will focus on the ability of robotic-assisted PCI to improve procedures that can potentially lead to better clinical outcomes and the protection of physicians from radiation and orthopedic issues.
9
The Hospital Administrator
In this era of economic pressure, purchasing decisions by hospitals must be carefully evaluated to ensure an associated cost benefit. In the case of our products, hospital administrators must be convinced of both the clinical benefit and the economic benefit of having procedures performed using the CorPath System.
Cath lab patient volume has decreased over the past several years leading to increased competition for patients. Recent data has shown that sites that adopt robotic-assisted surgical procedures, such as prostatectomy, have been able to attract increased patient volumes. Similarly, by using the CorPath System to promote technological leadership in the field of advanced robotics, hospitals can more easily attract and retain physicians while also increasing patient volume.
Customers purchasing our elective Continuity Support program have access to our valuable CorPath Hospital Marketing Program. This broad-based program is a tool kit designed to assist our customer hospitals in launching their own CorPath Vascular Robotic Program using the CorPath System as a tool to market the hospital’s quality and commitment to patient care and innovation. The tool kit contains both the programmatic and content elements designed to (i) plan, initiate, and execute public relations and outreach campaigns, (ii) influence and change referral patterns to improve market share in the hospital’s catchment area, (iii) promote the benefits of our innovative robotic technology to hospital personnel and patients, and (iv) develop substantial community awareness of the technology and the physicians employing it.
Significant Customers
One customer, Japan Medicalnext Co., Ltd., accounted for 20% of revenues in 2017. Three customers, Central Circle Co., Philips and Northwest Texas Healthcare System accounted for 28%, 13%, and 12%, respectively, of revenues in 2016. Three customers, UCSD Medical Center, IA St. Luke’s Medical Center and Edward Heart Hospital accounted for 13%, 11%, and 10% of revenues in 2015.
Competition
We currently do not face any direct competition for robotic-assisted PCI as the CorPath System is the only FDA-cleared device for this indication. We have some indirect competition in regard to other interventional procedures. There are three companies that make vascular robotic systems for electrophysiology procedures; Hansen Medical, Catheter Precision and Stereotaxis. Hansen Medical, which was acquired by Auris Surgical Robotics in 2016, also has a system used for peripheral vascular procedures. Although Auris Surgical Robotics is not currently marketing or selling this system, they may become a direct competitor for those procedures. Our primary focus today is on converting customers from the traditional manual PCI procedure to the CorPath System PCI procedure.
The medical device industry, however, is very competitive and subject to significant technological changes. Our potential competitors may develop products, treatments or procedures that are similar, more advanced, safer or more effective than ours. We expect to face competition from many different sources with respect to our existing products and products that we may seek to develop or commercialize in the future.
Seasonality
Our CorPath System sales and purchase order cycle may typically take from 6 to 15 months due to the capital budgeting cycle and approval process at each hospital. Because it is a capital item, such a purchase generally requires the approval of senior management of hospitals, and sometimes their parent organizations, purchasing groups, and/or government bodies, as applicable. In addition, hospitals may delay or accelerate purchases of the CorPath System in conjunction with timing of their capital budget timelines. As a result, while it is difficult for us to precisely predict the exact timing of CorPath purchases, we believe that our sales may tend to be heaviest during the third month of each fiscal quarter and heavier in the fourth fiscal quarter than in the other quarters.
Timing of PCI procedures and changes in the PCI procedure market could directly affect the timing of the purchase of our products by hospitals. It is likely that adoption of our products will be more challenging in the third quarter of each year when new interventional fellows join the staff at several of our hospital customer sites. As they are untrained with respect to cath lab skills and patients’ cases, they may be devoted to their manual training techniques rather than use of the CorPath System. In the longer term, this risk should be mediated by the limited number of fellows programs relative to hospitals performing PCI procedures.
Financial Information by Geographic Area
Revenue from external customers located in:
| Years Ended December 31, | ||||||||||||||||||||||||
| 2017 | 2016 | 2015 | ||||||||||||||||||||||
| Amount | Percent | Amount | Percent | Amount | Percent | |||||||||||||||||||
| United States | $ | 6,694 | 69 | % | $ | 1,867 | 66 | % | $ | 2,683 | 98 | % | ||||||||||||
| Japan | 1,906 | 20 | % | — | — | — | — | |||||||||||||||||
| All others, combined | 1,050 | 11 | % | 975 | 34 | % | 46 | 2 | % | |||||||||||||||
| Total | $ | 9,650 | 100 | % | $ | 2,842 | 100 | % | $ | 2,729 | 100 | % | ||||||||||||
All of the Company’s long-lived assets are located in the United States.
Customer Service
Our goal is 100% customer satisfaction by consistently delivering superior customer experiences before, during, and after the sale. To achieve this goal, we maintain a headquarters-based customer support service team supplemented by our dedicated field-based Clinicals. Our customer support service team primarily handles all order processing for consumables to ensure that new orders arrive before inventories are depleted. We are committed to providing prompt service for repairs to equipment in order to keep customer uptime at maximum levels. Our Clinicals are field-based and at customer sites on a regular basis to support their needs including on-going training in and outside of the lab. All of our customer service representatives receive regular training so that they can effectively and efficiently field questions from current and prospective customers.
10
Our Return Policy: One Stent Program
Neither our equipment, once purchased and installed, nor our single-use cassettes, are returnable or refundable. We stand behind the quality of our products. We value frequent communication with and feedback from our customers in order to continue to improve our offerings and services.
By minimizing stent utilization, the use of the CorPath System has the potential to bring significant clinical, safety and financial benefits to a hospital. To demonstrate our commitment to the benefits of our robotic CorPath System, we offer our hospitals a unique, stent utilization efficiency program called the CorPath One Stent Program. For each eligible CorPath System procedure in which a second unplanned stent is used, we currently provide a credit to the hospital of $1,000 to be used toward the purchase of additional cassettes. These credits have not been significant to date.
Raw Materials for Our Products
We acquire all raw materials for our products from a group of third-party suppliers. These suppliers may be manufacturers of custom components or distributors of commodity, off-the-shelf, components. Whenever possible, secondary sources for the materials are identified and maintained on our Approved Supplier List. To be included on our Approved Supplier List, suppliers must pass the requirements of our documented Supplier Approval Process.
Availability of and Dependence upon Suppliers
We own all of the designs of all of the custom components used in our product. This allows us to source components which minimize risk of patent infringement or risk of sale to any other manufacturer. We are able to source components at any supplier that has the technical capability to manufacture them. Some of the items we use are off-the-shelf components which can be sourced on the open market and have very little risk in terms of supply and design change. We continually review our supply base for cost and delivery capacity and make adjustments as necessary.
Manufacturing of Our Products
The CorPath System and CorPath Cassettes are manufactured in accordance with the FDA’s Current Good Manufacturing Practices (“CGMPs”) for medical devices. Our product was initially cleared by the FDA in 2012 for commercial sale using the 510(k) process while our second generation CorPath GRX was cleared in 2016, and our Waltham, Massachusetts facility is registered with the FDA as the place of manufacture for both of these systems.
With the exception of our cockpit, which is manufactured by an outside source, all of our manufacturing is categorized as light assembly and is performed by trained personnel in our facility. The single-use cassette is manufactured in an International Organization for Standardization (“ISO”) Class 8 clean room. This room is monitored, controlled, and operated according to ISO Class 8 and associated FDA guidelines. Finished products are stored in our facility and shipped directly to the customer. No special environmental controls are required for the storage of our product.
Quality Control for Our Products
A quality assurance team establishes procedures for process control and tests products at various stages of the manufacturing process to ensure we meet product specifications and that our finished products are manufactured in compliance with FDA Quality System Regulations (“QSR”). We inspect incoming components and finished goods per established procedures. Prior to shipment of the product to customers, the quality assurance team reviews our manufacturing record, to ensure it meets established process control requirements and product specifications.
Our quality procedures are designed to meet current FDA regulations and ISO 13485 for compliance with CE Mark requirements. Our production requirements are established to meet product specifications cleared by the FDA and ensure safety of the patients and performance expected by the end users. Our quality system is routinely audited by an internal auditor team and annually assessed by BSI Group for Quality Management System (“QMS”) and CE certification. BSI Group is an independent entity, which assesses the compliance of the QMS to ISO 13485 and CE Mark requirements and, upon establishing compliance, provides CE certification (the “Notified Body”).
Government Regulation
U.S. Medical Device Regulation
Our products and operations in the U.S. are subject to extensive and rigorous regulation by the FDA. The FDA regulates the development, testing, manufacturing, labeling, storage, record-keeping, promotion, marketing, distribution and service of medical devices in the U.S. to ensure that medical products distributed domestically are safe and effective for their intended uses. Under the Federal Food, Drug, and Cosmetic Act (“FFDCA”), medical devices are classified into one of three classes (Class I, Class II or Class III), depending on the degree of risk associated with each medical device and the extent of regulatory controls needed to ensure the device’s safety and effectiveness. Our current products are Class II medical devices.
Class II medical devices are those that are subject to general controls and, frequently, additional special controls, such as premarket or specific labeling guidelines, as specified by the FDA. Class II devices also typically require premarket review and clearance by the FDA, which is accomplished through the submission of a 510(k) premarket notification before the device may be marketed in the U.S. As part of the 510(k) notification process, the FDA may require the following:
| ● | Development of comprehensive product description and indications for use. |
| ● | Completion of extensive preclinical tests and/or preclinical animal studies, all performed in accordance with the FDA’s Good Laboratory Practice (“GLP”) regulations. |
11
| ● | Comprehensive review of predicate devices and development of data supporting the new product’s substantial equivalence to one or more predicate devices. | |
| ● | If appropriate and required, certain types of clinical trials (IDE submission and approval may be required for conducting a clinical trial in the U.S.). |
Clinical trials involve use of the medical device on human subjects under the supervision of qualified investigators in accordance with current Good Clinical Practices (“GCPs”) which include the requirement that all research subjects provide their informed consent for their participation in any clinical study. A protocol with predefined end points, an appropriate sample size and pre-determined patient inclusion and exclusion criteria, is required before initiating and conducting clinical trial. The protocol is reviewed and approved by the participating hospital’s Institutional Review Board (“IRB”) before the clinical trial can be initiated at the site. Additionally, the IRB must monitor the study until complete. Any subsequent protocol amendments must be submitted and approved by the IRB.
| ● | Assuming successful completion of all required testing, a detailed 510(k) application is submitted to the FDA requesting clearance to market the product. The application includes all relevant data from pertinent preclinical and clinical trials, together with detailed information relating to the product’s manufacturing controls and proposed labeling, and other relevant documentation. | |
| ● | A 510(k) clearance letter from the FDA will authorize commercial marketing of the device for one or more specific indications for use. | |
| ● | After regulatory clearance, we are required to comply with a number of post-clearance requirements, including, but not limited to, complaint handling and Medical Device Reporting, trending and relevant corrective actions. Also, quality control and manufacturing procedures must continue to conform to QSR requirements. The FDA periodically inspects manufacturing facilities to assess compliance with QSRs, which imposes extensive procedural, substantive, and record keeping requirements on medical device manufacturers. In addition, changes to the manufacturing process are strictly regulated, and, depending on the change, validation activities may need to be performed. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with QSRs and types of regulatory controls. |
While not anticipated, future FDA inspections and Notified Body (in the EU) audits may identify compliance issues at our facilities that may potentially disrupt production or distribution, or require substantial resources to correct. In addition, discovery of previously unknown problems with a device or failure to comply with applicable requirements may result in restrictions on manufacturing and distribution of the device, including withdrawal/recall of the device from the market, or FDA-initiated or judicial action that could delay or prohibit further marketing. Newly identified safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and/or contraindications, and also may require the implementation of other risk management measures.
After a device receives FDA 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, requires a new 510(k) clearance or could require a premarket approval application (“PMA”). The FDA requires each manufacturer to make the determination of whether a modification requires a new 501(k) notification or PMA in the first instance, but the FDA can review any such decision. If the FDA disagrees with a manufacturer’s decision not to seek a new 510(k) clearance or PMA for a particular change, the FDA may retroactively require the manufacturer to seek 510(k) clearance or PMA. The FDA can also require the manufacturer to cease U.S. marketing and/or recall the modified device until additional 510(k) clearance or PMA approval is obtained.
The FDA and the Federal Trade Commission (“FTC”) also regulate the advertising claims of our products to ensure that the claims we make are consistent with our regulatory clearances, that there is scientific data to substantiate the claims and that our advertising is neither false nor misleading. In general, we may not promote or advertise our products for uses not within the scope of our intended use statement in our clearances or make unsupported safety and effectiveness claims. Many regulatory jurisdictions outside of the U.S. have similar regulations to which we would be subject. Our manufacturing processes are required to comply with the FDA’s cGMP requirements contained in its QSR and associated regulations and guidance. The QSR covers, among other things, the methods and documentation of the design, testing, production, processes, controls, quality assurance, labeling, packaging and shipping, installation and service of a company’s products. The QSR also requires maintenance of extensive records which demonstrate compliance with FDA regulation, the manufacturer’s own procedures, specifications and testing as well as distribution and post-market experience. Compliance with the QSR is necessary to receive FDA clearance or approval to market new products and is necessary for a manufacturer to be able to continue to market cleared or approved product offerings in the U.S. A company’s facilities, records, and manufacturing processes are subject to periodic scheduled or unscheduled inspections by the FDA, which may issue reports known as Forms FDA 483 or Notices of Inspectional Observations which list instances where the FDA inspector believes the manufacturer has failed to comply with applicable regulations and/or procedures. If the observations are sufficiently serious or the manufacturer fails to respond appropriately, the FDA may issue Warning Letters, which are notices of intended enforcement actions against the manufacturer; or Untitled Letters, which are used for less serious violations that may not rise to the level of regulatory significance. These enforcement actions could include legal actions, including fines and total shutdown of production facilities, seizure of product, prohibition on export or import and criminal prosecution. Such actions may have further indirect consequences for the manufacturer outside of the U.S., and may adversely affect the reputation of the manufacturer and the product. In the U.S., routine FDA inspections usually occur every two years, and may occur more often for cause.
We intend to submit 510(k) applications for our next generation devices and for any new indications for use of our existing products. The applications may rely upon published literature and/or the findings of safety and effectiveness based on pre-clinical or clinical studies conducted for an approved or cleared product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product or for new claims for the cleared product.
Foreign Medical Device Regulation
In order for us to market our products in other countries, we must comply with extensive safety and quality regulations in other countries. These regulations, including the requirements for approvals, clearance or grant of CE Certificates and Declaration of Conformity and the time required for regulatory review, vary from country to country. Failure to obtain regulatory approval, clearance or CE Certificates of Conformity (or equivalent) in any foreign country in which we plan to market our products, including obtaining PMDA approval in Japan, may harm our ability to generate revenue and harm our business.
12
The primary regulatory environment in Europe is that of the European Economic Area (the “EEA”), which is comprised of the 28 Member States of the European Union (“EU”), Iceland, Liechtenstein and Norway. In the EEA, our devices are required to comply with the Essential Requirements defined in Annex I to the EU Medical Devices Directive (applicable in the non-EU EEA Member States via the Agreement on the European Economic Area). We are also required to ensure compliance with the relevant quality system requirements defined in the Annexes to the Medical Devices Directive. Compliance with these requirements entitles us to affix the CE mark to our medical devices, without which they cannot be commercialized in EEA. To demonstrate compliance with the Essential Requirements defined in Annex I of the Medical Devices Directive to obtain the right to affix the CE mark to our medical devices, and thus be permitted to market our medical devices on the EEA market, we must undergo a conformity assessment procedure, which varies according to the type of medical device and its classification. With the exception of low risk medical devices (Class I devices with no measuring function and which are not sterile), in relation to which the manufacturer may issue a CE Declaration of Conformity based on a self-assessment of the conformity of its products with the Essential Requirements defined in the Medical Devices Directive, a conformity assessment procedure requires the intervention of an EU accredited organization. This is an organization designated by the competent authorities of an EEA country to conduct conformity assessments. Depending on the relevant conformity assessment procedure, the accredited organization may audit and examine products’ Technical File and/or the quality system for the manufacture, design and final inspection of our devices before issuing a CE Certificate of Conformity. This Certificate demonstrates substantive compliance with the relevant Essential Requirements laid down in Annex I of the Medical Devices Directive or the relevant quality system requirements defined in the Annexes to the Directive and constitutes the basis for manufacturers to issue their mandatory Declaration of Conformity. Companies compliant with ISO requirements such as “EN ISO 13485: 2012 Medical devices — Quality management systems — Requirements for regulatory purposes” benefit from a presumption of conformity with the relevant quality system requirements defined in the Annexes to the Medical Devices Directive. The Notified Body issues a CE Certificate of Conformity following successful completion of a conformity assessment procedure conducted in relation to the medical device and its manufacturer and their conformity with the Essential Requirements and quality system requirements. In 2016, we received CE Certificate of Conformity from our Notified Body permitting us to affix the CE mark and market our CorPath GRX System in the EEA. If we modify existing products or develop new products in the future, including new devices, it may be necessary to notify our Notified Body and go through a conformity assessment procedure before having the right to affix the CE mark to such products. We will be subject to regulatory audits, currently conducted annually, in order to maintain any CE Certificates of Conformity that have been issued by our Notified Body. We cannot be certain that we will be able to obtain CE Certificates of Conformity for new or modified products. We continually strive to maintain our quality system to comply with the regulatory requirements defined in the Medical Device Directive and EN ISO 13485 for the CE Certificate of Conformity that we have received. We will evaluate regulatory approval in other foreign countries on an opportunistic basis.
Third Party Coverage and Reimbursement
The U.S. government and health insurance companies together are responsible for hospital and physician reimbursement for virtually all covered interventional procedures. Governments and insurance companies generally reimburse hospitals and physicians for procedures considered medically necessary. The Centers for Medicare & Medicaid Services (“CMS”), administers the Medicare and Medicaid programs (the latter, along with applicable state governments). Many other third-party payors model their reimbursement methodologies after the Medicare program. As the single largest payor, this program has a significant impact on other payors’ payment systems.
Generally, reimbursement for professional services performed at a facility by physicians is reported under billing codes issued by the American Medical Association (“AMA”), known as Current Procedural Terminology (“CPT”) codes. Physician reimbursement under Medicare generally is based on a fee schedule and determined by the relative values of the professional service rendered. In addition, CMS and the National Center for Health Statistics (“NCHS”) are jointly responsible for overseeing changes and modifications to billing codes known as ICD-9-CM procedural codes used by hospitals to report inpatient procedures. For Medicare, CMS generally reimburses hospitals for services provided during an inpatient stay based on a prospective payment system that is determined by a classification system known as Medicare-Severity Diagnostic Related Groupings (“MS-DRGs”). MS-DRGs are assigned using a number of factors including the principal diagnosis, major procedures, discharged status, patient age and complicating secondary diagnoses among other things. Hospital outpatient services, reported by CPT codes, are assigned to clinically relevant Ambulatory Payment Classifications (“APCs”) used to determine the payment amount for services provided.
On October 1, 2008, CMS and NCHS issued a new family of ICD-9-CM procedure codes for “Robotically Assisted Procedures.” The purpose of the ICD-9-CM family of procedure codes is to gather data on robotic assisted surgical procedures. Effective October 1, 2014, ICD-9-CM procedure code 1743 was implemented for Percutaneous Robotic Assisted Procedure(s). A surgical procedure, completed with or without robotic assistance, continues to be assigned to the clinically relevant MS-DRG.
Governments and insurance companies carefully review and increasingly challenge the prices charged for medical products and surgical services. Reimbursement rates from private companies vary depending on the procedure performed, the third-party payor, contract terms, and other factors. Because both hospitals and physicians may receive the same reimbursement for their respective services, with or without robotics, regardless of actual costs incurred by the hospital or physician in furnishing the care, including for the specific products used in that procedure, hospitals and physicians may decide not to use our products if reimbursement amounts are insufficient to cover any additional costs incurred when purchasing our products.
Domestic institutions typically bill for the primary procedure that includes our products to various third-party payors, such as Medicare, Medicaid and other government programs and private insurance plans. Because our CorPath System has been cleared for commercial distribution in the U.S. by the FDA, coverage and reimbursement by payors are generally determined by the medical necessity of the primary procedure. While PCI procedures are typically reimbursed by third-party payors, currently, there is no incremental reimbursement provided for robotic-assisted PCI. Therefore, using the CorPath System and consumable cassettes without an incremental reimbursement will initially increase the up-front cost of the PCI procedure and the cath lab operation based on the cost of the CorPath System and also consumable cassettes. This lack of incremental reimbursement from third-party payors for procedures performed with our products, or lack of coverage by governmental and private payors’ policies of interventional procedures performed using our products, may make us unable to generate the revenues necessary to support our business.
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, “the PPACA”), was signed into law and makes changes significantly impacting healthcare providers, insurers, pharmaceutical and medical device manufacturers. One of the principal aims of the PPACA is to expand health insurance coverage to any Americans who are currently uninsured. The PPACA contains a number of provisions designed to generate the revenues necessary to fund this coverage expansion, including, but not limited to new fees or taxes on certain health-related industries, including medical device manufacturers. Beginning in 2013, medical device manufacturers are required to pay an excise tax (or sales tax) of 2.3% on certain U.S. medical device revenues. Under this provision, we have incurred an excise tax of approximately $104,000 cumulatively through December 31, 2015 which is reflected in our operating expenses. The excise tax was suspended for two years due to the operation of a provision in the Consolidated Appropriations Act of 2016, signed into law on December 18, 2015, but that two-year suspension period ended on December 31, 2017. In mid-January 2018, with the signing of P.L. 115-120, suspension of the medical device excise tax was extended for an additional two years ending on December 31, 2019 and made retroactive to January 1, 2018. At the present time, there is no guarantee that the excise tax will continue to be suspended by congressional action after this suspension ends, or that any provisions of the PPACA will continue to exist in their current form. Certain legislators are continuing their efforts to repeal the PPACA, although there is little clarity on how such a repeal would be implemented and what a PPACA replacement might look like. For the immediate future, there continues to be significant uncertainty regarding the health care, health care coverage and health care insurance markets.
13
In addition, other legislative changes have been proposed and adopted since the PPACA was enacted. On August 2, 2011, the Budget Control Act of 2011 was signed into law, which, among other things, creates the Joint Select Committee on Deficit Reduction to recommend proposals in spending reductions to Congress. The Joint Select Committee did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This included aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, which went into effect on April 1, 2013. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers.
Any regulatory or legislative developments in domestic markets that eliminate or reduce reimbursement rates for procedures performed using our products could harm our ability to sell our products or cause downward pressure on the prices of our products, either of which would affect our ability to generate the revenues necessary to support our business.
Properties
Our principal offices and manufacturing facilities are located at 309 Waverley Oaks Road, Suite 105, Waltham, Massachusetts 02452. In October 2012, Corindus, Inc. entered into a lease with Beaver Group, LLC for a term of approximately five years for 26,402 square feet of office and manufacturing space (the “Lease”). In October 2016, the Lease was extended for an additional three years ending on January 31, 2021 (“Extended Lease”). Over the term of the Extended Lease, we pay an average monthly cost of $53,000 of base rent, excluding common area fees, taxes and insurance. Our management believes that the leased premises are suitable and adequate to meet current needs.
Employees
As of March 9, 2018, we have 106 full-time employees. Additionally, from time to time, we hire temporary or contract employees. None of our employees are covered by a collective bargaining agreement and we are unaware of any union organizing efforts. We have never experienced a major work stoppage, strike or dispute. We consider our relationship with our employees to be good.
Subsidiaries
Our subsidiaries are Corindus, Inc., which is our operating company, and Corindus Security Corporation, which holds and invests the proceeds of the issuance of certain securities.
Corporate Information
We are a Delaware corporation. Our corporate headquarters and manufacturing facilities are located at 309 Waverley Oaks Road, Suite 105, Waltham, Massachusetts 02452. Our telephone number is 508-653-3335 and our fax number is 508-653-3355. We maintain a website at http://www.corindus.com.
Available Information
Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to such reports filed or furnished pursuant to section 13(a) or 15(d) of the Securities Exchange Act of 1934, as well as section 16 reports on Form 3, 4, or 5, are available free of charge on our Internet website at http://www.corindus.com as soon as is reasonably practicable after they are filed or furnished with the SEC. Our Code of Conduct and Ethics and the charters for the Audit Committee, the Nominating and Governance Committee and the Compensation and Management Development Committee of our Board of Directors are also available on our Internet website. The Code of Conduct and Ethics and charters are also available in print to any shareholder upon request. Requests for such documents should be directed to David Long, Chief Financial Officer, at 309 Waverley Oaks Road, Suite 105, Waltham, Massachusetts 02452. Our Internet website and the information contained on it or connected to it are not part of, nor incorporated by, reference into this Form 10-K. Our filings with the SEC are also available on the SEC’s website at http://www.sec.gov.
14
| ITEM 1A. | RISK FACTORS. |
Investing in our common stock or any other security that may be issued by us involves a high degree of risk. You should carefully consider the risks described below, together with all of the other information included in this Report, before making an investment decision. If any of the following risks actually occur, our business, financial condition or results of operations could suffer. In that case, the trading price of our shares of Common Stock could decline, and you may lose all or part of your investment. You should read the section entitled “Forward-Looking Statements” above for a discussion of what types of statements are forward-looking statements, as well as the significance of such statements in the context of this Report.
Risks Related to our Financial Position
Our net losses and significant cash used in operating activities may hinder our ability to continue as a going concern.
We have incurred losses since inception and have funded our cash flow deficits primarily through the issuance of capital stock and debt. As of December 31, 2017, we had an accumulated deficit of $180.8 million. As of December 31, 2017, we had cash and cash equivalents of $17.5 million and working capital of $16.4 million. We anticipate that these available resources and the additional financings will be sufficient to meet our cash requirements for at least the next 12 months from March 19, 2018. However, as we continue to incur losses and cash flow deficits, our transition to profitability is dependent upon achieving a level of revenues adequate to support our cost structure, but we will otherwise rely on additional capital funding until such time as that is achieved.
As such, we have evaluated whether or not our cash and cash equivalents on hand and the cash proceeds from the financing activities described above would be sufficient to sustain projected operating activities through March 19, 2019 as required by Accounting Standards Codification (ASC) 205-40 Disclosure of Uncertainties About an Entity’s Ability to Continue as a Going Concern. Based on our current forecasts of cash flow deficits, which are estimated to be approximately $37 million to $39 million per year, we will have cash through approximately May 2019.
Due to the inherent uncertainty in predicting revenues and certain variable costs, we have considered our ability to reduce cash flow deficits and have determined that if we do not achieve our revenue forecast, we would undertake the following activities to reduce our cash flow deficits:
●
Defer or limit some or all of its spending on capital equipment that we believe is necessary to reduce manufacturing costs of cassettes and CorPath systems to be used in marketing and training activities that were otherwise planned for 2018;
●
Eliminate or defer the 2018 discretionary bonus payouts for all bonus eligible employees, including executive management;
●
Reduce spending on travel, clinical programs and prototypes;
●
Eliminate planned headcount additions in marketing, sales, and manufacturing; and
●
Reduce external consulting resources who otherwise would be used to assist in installations of CorPath systems.
As a result, we believe our plans can be effectively implemented as all of the actions are within our control and will be finalized and will be able to be effectively implemented, if required.
We may never achieve profitability, and unless and until doing so, we intend to fund future operations through additional non-dilutive or dilutive financings. There can be no assurances, however, that additional funding will be available on terms acceptable to us, if at all.
Until we reach profitability and generate operating cash flows to grow the business, we will need to continue to raise additional funding. We may be unable to raise capital when needed, which would force us to delay, reduce or eliminate our product development programs, commercialization efforts and growth strategy.
We will need additional funding for establishing and expanding our sales and marketing infrastructure and for future product development and we may be unable to raise capital when needed or on attractive terms, which would force us to delay, reduce or eliminate our product development programs or commercialization efforts.
We have funded operations primarily through the issuance of capital stock and debt. On May 28, 2015, our Company completed a public offering by issuing 12,650,000 shares of our common stock at $3.80 per share in exchange for proceeds of $44.4 million, net of underwriting discounts, commissions and other offering costs. In March 2017, we completed a private placement of 68,055,700 shares of our common stock at $0.6616 per share in exchange for gross proceeds of approximately $45 million, before deducting offering expenses. As of December 31, 2017, we had approximately $17.5 million in cash and cash equivalents. On March 16, 2018, we closed on a private placement of convertible preferred stock for gross proceeds of $25 million. The preferred stock is convertible into an aggregate of 20,000,000 shares of common stock, and is entitled to receive non-compounding dividends in additional shares of preferred stock, at the rate of 12% per annum, subject to reduction in the event certain milestones are achieved. The preferred stock purchasers were also issued warrants to purchase an aggregate of 8,750,000 shares of common stock at an exercise price of $1.40 per share, exercisable either for cash or on a cashless basis. Also, on March 16, 2018, we completed a financing arrangement with two lenders which provides for borrowings of up to $26 million in the form of up to $23 million in term loans and up to a $3 million revolving line-of-credit through March 2022. As of March 16, 2018, we had $12 million in principal outstanding under the term loan facility and $0 in principal outstanding under the revolving loan facility. An additional $5.5 million in term loans may become available in the future provided we have achieved a specified gross profit milestone prior to January 1, 2019, and an additional $5.5 million may become available provided we receive net cash proceeds of $30 million from a future sale of our equity securities prior to July 1, 2019 and achieve a specified gross profit milestone prior to September 1, 2019 . Until such time that we achieve the specified criteria, the additional term loans are not available to us. We cannot assure you that we will achieve the gross profit or equity financing milestones that will trigger our ability to further draw the term loan facility. The revolving line-of-credit also has various clauses which restrict its availability and for which we currently do not meet such restrictions.
Should we raise additional funds by issuing equity securities, our stockholders will experience immediate dilution. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. Any additional debt or equity financing that we close may contain terms, such as liquidation and other preferences, which are not favorable to us or our stockholders. If we raise additional funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish valuable rights to our technologies, future revenue streams or product candidates or to grant licenses on terms that may not be favorable to us. If additional financing is not available when required or is not available on acceptable terms, we may be unable to fund expansion, successfully promote our brand name, develop or enhance our services, take advantage of business opportunities, or respond to competitive pressures or unanticipated requirements, any of which could seriously harm our business and reduce the value of your investment.
The terms of our term loan facility and revolving credit facility place restrictions on our ability to operate our business and on our financial flexibility, and we may be unable to achieve the milestones necessary for us to incur additional borrowings under the term loan facility or to satisfy the minimum revenue covenants.
On March 16, 2018, we completed a financing arrangement with two lenders which provides for borrowings of up to $26 million, in the form of up to $23 million in term loans and up to a $3 million revolving line of credit through March 2022. As of March 16, 2018, we had $12 million in principal outstanding under the term loan facility and $0 in principal outstanding under the revolving loan facility An additional $5.5 million in term loans may become available in the future provided we have achieved a specified gross profit milestone prior to January 1, 2019, and an additional $5.5 million may become available provided we receive net cash proceeds of $30 million from a future sale of our equity securities prior to July 1, 2019 and achieve a specified gross profit milestone prior to September 1, 2019 . Until such time that we achieve the specified criteria, the additional term loans are not available to us. We cannot assure you that we will achieve the gross profit or equity financing milestones that will trigger our ability to further draw the term loan facility. The revolving line-of-credit also has various clauses which restrict its availability and for which we currently do not meet such restrictions.
Both loan facilities include customary affirmative and negative covenants, which may make it difficult for us to run our business. Upon the earlier of the second advance under the term loan facility or the first advance under the revolving loan facility, we must also achieve minimum revenue on a monthly basis measured against a percentage of our Board of Directors approved projections for the applicable fiscal year. We cannot assure you that we will be able to achieve the minimum revenue requirements provided for in the loan facilities. Our failure to satisfy the revenue, or any other, covenant could result in an event of default under the loan facilities. Other events of default include, among other things, our failure to pay principal or interest due, a breach of other covenants under the loan facilities, our insolvency, a material adverse change, the occurrence of any default under certain other indebtedness in an amount greater than $250,000 and judgments against us in an amount greater than $250,000 individually or in the aggregate.
Changes in our effective tax rate may harm our results of operations.
A number of factors may harm our future effective tax rates including, but not limited to, the following:
| ● | the jurisdictions in which profits are determined to be earned and taxed; |
| ● | the resolution of issues arising from tax audits with various taxing authorities; |
| ● | change in valuation of our deferred tax assets and liabilities; |
| ● | increases in expenses not deductible for tax purposes; |
| ● | changes in available tax credits and deductions; |
| ● | changes in share-based compensation; and |
| ● | changes in tax laws or the interpretation of such tax laws and changes in generally accepted accounting principles. |
Because we have incurred losses to date, we have not recorded any income tax provision thus far. At December 31, 2017, we had U.S. federal and state net operating loss carryforwards of approximately $147.8 million and $91.6 million, respectively, that can be carried forward and offset against future taxable income. These federal net operating loss carryforwards will begin to expire in 2028 and the state net operating loss carryforwards will begin to expire in 2022.
Utilization of net operating losses may be subject to a substantial annual limitation due to the “change in ownership” provisions of the Internal Revenue Code of 1986, as amended, and similar state provisions. This limitation may result in the expiration of net operating losses before utilization. We have not yet determined whether any changes in ownership have triggered any such limitations. There can be no assurance that we will utilize the entire amount of our net operating loss carryforwards.
15
The recently passed comprehensive tax reform bill could adversely affect our business and financial condition.
On December 22, 2017, the President signed into law significant changes to federal income tax law passed by Congress. Among other things, the changes include a reduction of U.S. federal corporate income tax rates, significant new limitations on the deductibility of interest and net operating loss carryforwards, expansion of the expensing of capital expenditures, and the institution of a modified territorial system in place of the former “worldwide” system of taxation. Our net deferred tax assets and liabilities will be revalued at the newly enacted U.S. corporate rate, and the impact, if any, will be recognized in our tax expense in the year of enactment. We continue to examine the impact this tax reform legislation may have on our business. The impact of this tax reform is uncertain and could be adverse. We urge our stockholders to consult with their legal and tax advisors with respect to such legislation and the potential tax consequences of investing in our common stock.
Risks Related to Our Business and Industry
We are completely dependent on the success of our CorPath System, which has a limited commercial history. If our CorPath System fails to gain or loses market acceptance, our business will suffer.
During 2016, we received 510(k) clearance from the FDA for our CorPath GRX System, the second-generation of the CorPath System. We commercially introduced our CorPath GRX System in January 2017, and expect that sales of our CorPath GRX System will account for the majority of our revenue for the foreseeable future. Because of its recent commercial introduction, the CorPath GRX System has limited product and brand recognition. Demand for our original CorPath System had not increased as quickly as we expected and we do not know if we will be successful over the long term in generating increased demand for the use of our products. Failure of our CorPath GRX System to significantly penetrate current or new markets would negatively impact our business, financial condition and results of operations.
We operate in a competitive industry and if our competitors have products that are marketed more effectively or develop products, treatments or procedures that are similar, more advanced, safer or more effective than ours, our commercial opportunities will be reduced or eliminated and our business will be harmed.
Our potential competitors may develop products, treatments or procedures that are similar, more advanced, safer or more effective than ours. The medical device industry is very competitive and subject to significant technological and practice changes. We expect to face competition from many different sources with respect to our existing products and products that we may seek to develop or commercialize in the future.
Competing against large established competitors with significant resources may make establishing a market for any products that we develop difficult which would have a material adverse effect on our business. Our commercial opportunity could also be reduced or eliminated if our competitors develop and commercialize products, treatments or procedures that are safer, more effective, are more convenient or are less expensive than our existing products or any product that we may develop. Many of our potential competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we may have. Mergers and acquisitions in the medical device industry market may result in even more resources being concentrated among a smaller number of our potential competitors. Smaller and other early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These companies compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
If institutions or physicians are unable to obtain coverage and reimbursement from third-party payors for procedures using our products, or if reimbursement is insufficient to cover the costs of purchasing our products, we may be unable to generate sufficient sales to support our business.
In the United States, hospitals generally bill for the services performed with our products to various third-party payors, such as Medicare, Medicaid and other government programs and private insurance plans. Currently, there is no incremental reimbursement provided for robotic-assisted PCI. Therefore, using the CorPath System and consumable cassette without an incremental reimbursement will initially increase the up-front cost of the PCI procedure and the cath lab operation based on the cost of the CorPath System and also consumable cassettes. This lack of incremental reimbursement from third-party payors for procedures performed with our products, or lack of coverage by governmental and private payors’ policies of interventional procedures performed using our products, could deter hospitals from purchasing our products and may make us unable to generate the revenues necessary to support our business.
The commercial success of our products will depend upon the degree of market acceptance by hospitals and physicians. Should we not achieve market acceptance, we will not be able to generate the revenue necessary to support our business.
The CorPath System is a new technology that represents a fundamentally new way of performing PCI procedures; however, it competes with established treatment options for PCI procedures. Achieving physician, patient and third-party payor acceptance of the CorPath System as a preferred method of performing vascular procedures will be crucial to our success. If our products fail to achieve market acceptance, hospital customers will not purchase our products and we will not be able to generate the revenue necessary to support our business. We believe that acceptance by hospitals, physicians and third-party payors regarding the benefits of procedures performed using our products will be essential for acceptance of our products by patients. Physicians will not recommend the use of our products unless we can demonstrate that they produce results comparable or superior to existing PCI techniques. Even though we have proven the effectiveness of our products through clinical trials, physicians may elect not to use our products for any number of other reasons. For example, cardiologists may continue to recommend conventional PCI techniques simply because it is already widely accepted. In addition, physicians may be slow to adopt our products because of the perceived liability risks arising from the use of new products and the uncertainty of reimbursement from third-party payors, particularly in light of ongoing health care reform initiatives. We expect that there will be a learning process involved for physicians and their surgical teams to become proficient in the use of our products. Market acceptance could be delayed by the time required to complete this training. We may not be able to rapidly train physicians and their surgical teams in numbers sufficient to generate adequate demand for our products.
16
Development and awareness of our brand will largely depend upon our success in increasing our customer base. In order to attract and retain customers and to promote and maintain our brand in response to competitive pressures, management plans to significantly increase our sales and marketing budgets, particularly for our field sales force. If we are unable to economically promote or maintain our brand, our business, results of operations and financial condition could be severely harmed.
The successful use of our CorPath System depends in part on physician skill and experience. If we are unable to train physicians on the proper use of our system, we may experience a high risk of product liability.
The successful use of our CorPath System depends in part on the physician’s skill and experience. We train users on the proper techniques in using our system to achieve the intended outcome. Because of the acute nature of PCI procedure, we are unable to have a company representative attend cases using the CorPath System. As the number of users of our system increases, it is possible that the level of training that we are accustomed to providing will be insufficient and some physicians may not be willing to invest the time required to become properly trained with our procedure. We may find that physicians who are less skilled in the use of interventional devices will increasingly use the CorPath System, potentially leading to a higher rate of device failure, injury, negative publicity and an increased risk of product liability. We may be subject to claims against us even if the apparent injury is due to the actions of others. Any litigation that may occur based on physician error in the use of our products and our potential inability to train physicians to use our CorPath System may lead to inadequate demand for our products and have a material adverse impact on our business, financial condition and results of operations.
Our future success is dependent upon expanding our technology platform to other segments. Our potential inability to expand our technology platform beyond PCI may adversely affect our ability to increase our revenues.
Currently, our only users are interventional cardiologists in PCI procedures. We are dependent on our ability to expand our technology platform and sell our products to other vascular markets in the future, including neurointerventional and other more complex cardiac interventions such as structural heart. The techniques used in our procedure are similar to those used by not only our current user base of interventional cardiologists, but also to those used by other specialists who are generally trained in interventional techniques. Our revenue growth will depend on our ability to obtain approval to sell our CorPath System into these other markets and to sell our products to their physicians and their affiliated hospitals. In October 2015, we received 510(k) clearance from the FDA for our robotic-assisted CorPath System to be used during percutaneous coronary interventions performed via radial access, and on March 29, 2016, we received 510(k) clearance from the FDA for our robotic-assisted CorPath System for use in peripheral vascular interventions. In October 2016, we received 510(k) clearance from the FDA for the CorPath GRX, the second generation of our CorPath System and in February 2018, we received 510(k) clearance from the FDA for CorPath GRX to be used during peripheral vascular interventions. In March 2018, we received 510(k) clearance from the FDA for the first automated robotic movement designed for the CorPath GRX platform. Convincing physicians to dedicate the time and energy necessary for adequate training in the use of our system is challenging, and we cannot provide any assurance that we will be successful in these efforts. In addition, we do not have significant experience in selling our products to other specialists. They may require, among other things, additional clinical evidence supporting patient and physician benefits, training in a manner to which we are not accustomed or other resources that we do not readily have available or are not cost effective for us to provide. If we are unable to expand into other markets, growth of our sales will be limited and our revenue will be adversely impacted.
Decreasing cath lab patient volume and CorPath Cassette utilization could adversely affect our business, financial condition or results of operations.
Our current target market consists of the estimated 3,250 cath lab rooms in the U.S. that perform PCI procedures, which we estimate represents 40% of the global market of more than 8,000 PCI cath lab rooms. U.S. cath lab patient volume has decreased over the past several years, leading to increased competition for patients. If U.S. cath lab patient volume continues to decrease, it may become more difficult for us to grow revenue and increase market share and could adversely affect our business, financial condition or results of operations. In addition, revenue from the sale of our consumable cassettes to cath labs which already have a CorPath System installed is dependent on how often the systems are utilized. We sell CorPath Cassettes and accessories to end users and distributors on an as-needed basis. The revenue from the sale of these products is generally recorded when the items are shipped. If the utilization rate in cath labs decreases, our revenues, financial condition and results of operations will be adversely impacted.
Our marketing strategy is dependent on collaboration with physician “key opinion leaders.”
Our research and development efforts and our marketing strategy depend heavily on obtaining support, physician training assistance and collaboration from highly-regarded physicians at leading commercial and research hospitals, in the United States and abroad. If we are unable to gain and/or maintain such support, training services and collaboration, or if the reputation or standing of these physicians is impaired or otherwise adversely affected, our ability to market our products and, as a result, our financial condition, results of operations and cash flow, could be materially and adversely affected.
Revenue related to our CorPath Cassettes is derived predominately from customers using our CorPath GRX System. If we do not receive repeat orders of CorPath Cassettes for use with the CorPath GRX System from our customers, our revenue may not grow as expected.
For the fourth quarter and full year ended December 31, 2017, approximately 96% and 80%, respectively, of our unit volume of revenue generated from the sale of cassettes is from customers using our CorPath GRX system. Cassettes sales for our CorPath 200 first generation platform have declined since the launch of the CorPath GRX system as utilization for CorPath 200 has slowed down significantly and we expect this trend to continue. There are 33 CorPath GRX systems currently installed at customer sites. The CorPath Cassette is consumed and replaced during each procedure and represents an opportunity for recurring revenue for each procedure. If our customers do not purchase CorPath Cassettes for the CorPath GRX system in the future, our revenue would be adversely impacted.
Our products face the risk of technological obsolescence, which, if realized, could have a material adverse effect on our business.
The medical device industry is characterized by rapid and significant technological change. There can be no assurance that third parties will not succeed in developing or marketing technologies and products that are more effective than ours or that would render our technology and products obsolete or noncompetitive. Additionally, new, less invasive surgical procedures and medications could be developed that replace or reduce the importance of current procedures that use or could use our products. Accordingly, our success will depend in part upon our ability to respond quickly to medical and technological changes through the development of new products. Product development involves a high degree of risk, and we cannot assure you that our new product development efforts will result in any commercially successful products.
17
We may experience long and variable capital sales cycles and/or seasonality in our business, which may cause fluctuations in our financial results.
Our CorPath System may have a lengthy sales and purchase order cycle because it is a major capital item and such a purchase generally requires the approval of senior management of hospitals, their parent organizations, purchasing groups, and/or government bodies, as applicable. In addition, hospitals may delay or accelerate system purchases in conjunction with timing of their capital budget timelines. As a result, it is difficult for us to predict the length of capital sales cycles and, therefore, the exact timing of capital sales. We believe that our sales may tend to be heaviest during the third month of each fiscal quarter, and lighter in the third and first fiscal quarters and heavier in the fourth fiscal quarter. Timing of PCI procedures and changes in the PCI procedure market could directly affect the timing of the purchase of our products by hospitals.
The above factors may contribute to fluctuations in our quarterly operating results and it is possible that in some future quarters our operating results will fall below the expectations of securities analysts or investors. If that happens, the market price of our stock would likely decrease. These fluctuations, among other factors, also mean that you will not be able to rely upon our operating results in any particular period as an indication of future performance. In addition, the introduction of new products could adversely impact our sales cycle as customers take additional time to assess the benefits and costs of such products.
If defects are discovered in our products, we may incur additional unforeseen costs, hospitals may not purchase our products and our reputation may suffer.
Our products incorporate mechanical parts, electrical components, optical components and computer software, any of which can contain errors or failures, especially when first introduced. In addition, new products or enhancements may contain undetected errors or performance problems that, despite testing, are discovered only after commercial shipment. Because our products are designed to be used to perform complex medical procedures, we expect that our customers will have an increased sensitivity to such defects. We cannot provide any assurances that our products will not experience component aging, errors or performance problems in the future. If we experience flaws or performance problems, any of the following could occur:
| ● | delays in product shipments; |
| ● | loss of revenue; |
| ● | delay in market acceptance; |
| ● | diversion of our resources; |
| ● | damage to our reputation; |
| ● | product recalls; |
| ● | regulatory actions; |
| ● | increased service or warranty costs; or |
| ● | product liability claims. |
In the future, we may be subject to product liability and negligence claims relating to the use of our products that could be expensive, divert management’s attention and harm our business.
Our business exposes us to significant risks of product liability claims, which are inherent to the medical device industry, including product liability exposure related to the testing of our CorPath System in human clinical trials. Because our CorPath System is designed to be used in complex surgical procedures, defects could result in a number of complications, including serious personal injury or death. Claims could be brought against us if use or misuse of our CorPath System causes, or merely appears to have caused, personal injury or death. Product liability claims may be brought by individuals or by groups seeking to represent a class.
While we have and intend to maintain product liability insurance, our coverage may not be sufficient to cover claims that may be made against us and we may be unable to maintain such insurance. Additionally, we have entered into various agreements where we indemnify third parties for certain claims relating to our products. These indemnification obligations may require us to pay significant sums of money for claims that are covered by these indemnification obligations. We are not currently subject to any product liability claims; however, future product liability claims against us, regardless of their merit, may result in negative publicity about us that could ultimately harm our reputation and could have a material adverse effect on our business, financial condition, results of operations.
We may be subject to product recalls that could negatively affect our business.
We may be subject to product recalls, withdrawals or seizures if any of our products are believed to cause injury or are subject to serious malfunctions or if we are alleged to have violated governmental regulations in the manufacture, labeling, promotion, sale or distribution of our products. A recall, withdrawal or seizure of any of our products could materially and adversely affect consumer confidence in our brand and lead to decreased demand for our products. In addition, a recall, withdrawal or seizure of our products would require significant management attention, would likely result in substantial and unexpected expenditures and could materially and adversely affect our business, financial condition or results of operations.
Our business may be affected by unfavorable publicity or lack of consumer acceptance.
We are highly dependent upon consumer acceptance of the safety, efficacy and quality of our products. Consumer acceptance of a product can be significantly influenced by scientific research or findings, national media attention and other publicity about product use. A product may be received favorably resulting in high sales associated with that product that may not be sustainable as consumer preferences change. Future scientific research or publicity could be unfavorable to our industry or to any of our products and may not be consistent with earlier favorable research or publicity. A future research report or publicity that is perceived by our consumers as less than favorable or that may question earlier favorable research or publicity could have a material adverse effect on our ability to generate revenue. Adverse publicity in the form of published scientific research, statements by regulatory authorities or otherwise, whether or not accurate, that associates the use of our product with adverse effects, or that questions the benefits of our product or a similar product, or that claims that our products are ineffective or unsafe, could reduce market acceptance of our products and could result in decreased product demand and could have a material adverse effect on our business, reputation, financial condition or results of operations.
18
We could be subject to significant, uninsured liabilities.
In the future, we may not continue to maintain certain existing insurance coverage or adequate levels of coverage. Premiums for many types of insurance have increased significantly in recent years, and depending on market conditions and our circumstances, in the future, certain types of insurance such as directors’ and officers’ insurance or products liability insurance may not be available on acceptable terms or at all.
We may encounter manufacturing problems or delays that could result in lost revenue.
Manufacturing our products is a complex process. We may encounter difficulties in scaling up or maintaining production of our products, including:
| ● | problems involving production yields; |
| ● | quality control and assurance; |
| ● | component supply shortages; |
| ● | import or export restrictions on components, materials or technology; |
| ● | shortages of qualified personnel; and |
| ● | compliance with state and federal regulations. |
If demand for our products exceeds our manufacturing capacity, we could develop a substantial backlog of customer orders. If we are unable to maintain larger-scale manufacturing capabilities, our ability to generate revenues will be limited and our reputation in the marketplace could be damaged.
We depend on limited or single source suppliers and vendors for components and services used in the manufacture of our products, and the partial or complete loss of these suppliers or vendors could cause customer supply or production delays and a substantial loss of revenues.
We depend on limited or single source suppliers for certain key components and limited vendors for certain services used to manufacture our products, making us susceptible to quality issues, shortages and price changes. Any of these limited or single source suppliers or vendors could stop producing or supplying our components or stop performing services used to manufacture our products, cease operations or be acquired by, or enter into exclusive arrangements with, one or more potential competitors. As a result, these suppliers and vendors could stop providing components or services to us at commercially reasonable prices, or at all. Because there are a limited number of suppliers and vendors that manufacture the components and provide the services used to manufacture our products, it may be difficult to quickly identify alternate suppliers or vendors or to qualify alternative components or services on commercially reasonable terms, and our ability to satisfy customer demand may be adversely affected, which could result a substantial loss of revenue.
Disruption of critical information systems or material breaches in the security of our systems could harm our business, customer relations and financial condition.
Information technology helps us operate efficiently, interface with customers, maintain financial accuracy and efficiency and accurately produce our financial statements. If we do not allocate and effectively manage the resources necessary to build and sustain the proper technology infrastructure we could be subject to transaction errors, processing inefficiencies, the loss of customers, business disruptions or the loss of or damage to intellectual property through security breach. If our data management systems do not effectively collect, store, process and report relevant data for the operation of our business, whether due to equipment malfunction or constraints, software deficiencies or human error, our ability to effectively plan, forecast and execute our business plan and comply with applicable laws and regulations will be impaired, perhaps materially. As the use of digital technologies has increased, cyber incidents, including deliberate attacks and attempts to gain unauthorized access to computer systems and networks, have increased in frequency and sophistication. Despite the implementation of security measures, our internal computer systems are vulnerable to damage from such cyberattacks, including computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. Any such impairment could materially and adversely affect our financial condition, results of operations, cash flows and the timeliness with which we report our internal and external operating results.
Our business requires us to use and store personally identifiable information (“PII”) of our customers, employees and business partners. This may include names, addresses, phone numbers, email addresses, contact preferences, tax identification numbers and payment account information. We require user names and passwords in order to access our information technology systems. We also use encryption and authentication technologies to secure the transmission and storage of data. These security measures may be compromised as a result of third-party security breaches, employee error, malfeasance, faulty password management or other irregularity, and result in persons obtaining unauthorized access to our data or accounts. Third parties may attempt to fraudulently induce employees or customers into disclosing user names, passwords or other sensitive information, which may in turn be used to access our information technology systems.
We devote significant resources to network security, data encryption and other security measures to protect our systems and data, but these security measures cannot provide absolute security. We may experience a breach of our systems and may be unable to protect sensitive data. There can be no assurance that we will be successful in preventing cyber-attacks or successfully mitigating their effects. The costs to us to eliminate or alleviate network security problems, bugs, viruses, worms, malicious software programs and security vulnerabilities could be significant, and our efforts to address these problems may not be successful and could result in unexpected interruptions, delays, cessation of service and may harm our business operations. Moreover, if a computer security breach affects our systems or results in the unauthorized release of PII, our reputation and brand could be materially damaged and use of our products and services could decrease. As we grow our business outside of the U.S., we will be subject to additional, complex data security obligations. Compliance with these obligations may be costly and failure to comply may materially harm our business.
19
Failure to manage growth effectively could prevent us from achieving our goals.
Our growth strategy may impose a significant burden on our administrative and operational resources. Our ability to effectively manage growth depends on our ability to substantially expand the capabilities of our administrative and operational resources and to attract, train, manage and retain qualified management and other personnel. Our failure to successfully manage growth could result in our sales not increasing commensurately with capital investments. Our inability to successfully manage growth could materially adversely affect our business.
Any failure to adequately expand our direct sales force will impede our growth. If we are unable to attract, hire and retain qualified sales and management personnel, the commercial opportunity for our products may be diminished.
We expect to be substantially dependent on a direct sales force to attract new business and to manage customer relationships. We plan to expand our direct sales force and believe that there is significant competition for qualified, productive direct sales personnel with advanced sales skills and technical knowledge of our industry. Our ability to achieve significant growth in revenue in the future will depend, in large part, on our success in recruiting, training and retaining sufficient direct sales personnel. Recent hires and planned hires may not become as productive as expected and we may be unable to hire sufficient numbers of qualified individuals in the future in the markets where we do business. If we are unable to hire and develop sufficient numbers of productive sales personnel, our business prospects could suffer.
As of December 31, 2017, our sales force consisted of 11 and 12 regional sales managers and clinical specialists, respectively. We may not be able to attract, hire, train and retain qualified sales and sales management personnel. If we are not successful in our efforts to maintain and grow a qualified sales force, our ability to independently market and promote our products may be impaired. Even if we are able to effectively maintain a qualified sales force, our sales force may not be successful in commercializing our products.
To successfully market and sell our CorPath System internationally, we must address many issues with which we have little or no experience.
To date, we have primarily marketed our CorPath System domestically in the United States. Over the long term, we intend to grow our business internationally, and to do so we will need to attract distributors or expand our sales operations to effectively sell our CorPath System internationally. Distributors may not commit the necessary resources to market and sell our CorPath System in accordance with our expectations. If future distributors do not perform adequately, or we are unable to locate distributors for particular geographic areas, we may not realize expected long term international revenue growth. International sales are subject to a number of risks, including:
| ● | varying coverage and reimbursement processes and procedures; |
| ● | difficulties in staffing and managing foreign operations; |
| ● | reduced protection for intellectual property rights in some countries; |
| ● | export restrictions, trade regulations and foreign tax laws; |
| ● | fluctuating foreign currency exchange rates; |
| ● | foreign certification and regulatory requirements; |
| ● | lengthy payment cycles and difficulty in collecting accounts receivable; |
| ● | customs clearance and shipping delays; |
| ● | legal and regulatory requirements related to anti-bribery laws in foreign jurisdictions, as well as in relation to the U.S. Foreign Corrupt Practices Act (FCPA); |
| ● | political and economic instability; and |
| ● | preference for locally produced products. |
If one or more of these risks is realized, it could require us to dedicate significant resources to remedy the situation, our plan to expand internationally may fail and our financial performance may suffer as a result.
If we fail to attract and retain key personnel, or to retain our executive management team, we may be unable to successfully develop or commercialize our products.
Our success depends in part on our continued ability to attract, retain and motivate highly qualified managerial personnel. We are highly dependent upon our executive management team. The loss of the services of any one or more of the members of our executive management team could delay or prevent the successful completion of some of our development and commercialization objectives.
Recruiting and retaining qualified sales and marketing personnel is critical to our success. We may not be able to attract and retain these personnel on acceptable terms. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our development and commercialization strategy. Our consultants and advisors may also be employed by other companies and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could have a material adverse effect on our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include, but is not limited to, intentional or negligent failures to comply with FDA or U.S. health care laws and regulations or applicable laws, regulations, guidance or codes of conduct set by foreign governmental authorities or self-regulatory industry organizations, provide accurate information to any governmental authorities such as the FDA, comply with manufacturing standards we may establish, comply with federal and state healthcare fraud and abuse laws and regulations, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the medical device industry are subject to extensive laws, regulations, guidance and codes of conduct intended to prevent fraud, kickbacks, self-dealing and other abusive practices. It is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws, regulations, guidance or codes of conduct. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines and/or other sanctions.
20
Our business entails a significant risk of product liability and our ability to obtain sufficient insurance coverage could harm our business, financial condition, results of operations or prospects.
Our business exposes us to significant product liability risks inherent in the development, testing, manufacturing and marketing of medical device products. Product liability claims could lead to an inadequate demand for our products and have a material adverse impact on our business, financial condition and results of operations. Such claims could result in an investigation by certain regulatory authorities, such as the FDA or foreign regulatory authorities, of the safety and effectiveness of our products, our manufacturing processes and potentially a recall of our products or more serious enforcement action, or suspension or withdrawal of marketing clearances or approvals. Regardless of the merits or eventual outcome, liability claims may also result in decreased demand for our products, injury to our reputation, costs to defend related litigation, a diversion of management’s time and our resources, substantial monetary awards to clinical trial participants or patients and/or a decline in our stock price. We currently have product liability insurance that we believe is appropriate for our stage of commercialization. Any insurance we have or may obtain in the future may not provide sufficient coverage against potential liabilities. Furthermore, product liability insurance is becoming increasingly expensive. As a result, we may be unable to obtain sufficient insurance at a reasonable cost to protect us against losses caused by product liability claims that could have a material adverse effect on our business.
Our current operations are concentrated in one location and any events affecting this location may have material adverse consequences.
Our current operations are located in our facilities situated in Waltham. Any unplanned event, such as flood, fire, explosion, earthquake, extreme weather condition, medical epidemics, power shortage, telecommunication failure or other natural or manmade accidents or incidents that prevent us from fully utilizing the facilities, may have a material adverse effect on our ability to operate our business, particularly on a daily basis, and have significant negative consequences on our financial and operating conditions. Loss of access to these facilities may result in increased costs, delays in the development of our product candidates or interruption of our business operations. As part of our risk management policy, we maintain insurance coverage at levels that we believe are appropriate for our business. However, in the event of an accident or incident at these facilities, we cannot assure you that the amounts of insurance will be sufficient to satisfy any damages and losses. If our facilities are unable to operate because of an accident or incident or for any other reason, even for a short period of time, any or all of our research and development programs may be harmed. Any business interruption may have a material adverse effect on our business, financial position, results of operations and prospects.
Changes in accounting rules and regulations, or interpretations thereof, could result in unfavorable accounting charges or require us to change our compensation policies.
Accounting methods and policies for medical device companies, including policies governing revenue recognition, research and development and related expenses and accounting for stock-based compensation, are subject to review, interpretation and guidance from our independent registered accounting firm and relevant accounting authorities, including the SEC. Changes to accounting methods or policies, or interpretations thereof, may require us to reclassify, restate or otherwise change or revise our consolidated financial statements, including those contained in our Annual Reports on Form 10-K.
Software errors or other defects may be discovered in our products.
Our products incorporate many components, including sophisticated computer software. Complex software frequently contains errors, especially when first introduced. Because our products are designed to be used to perform complex procedures, we expect that physicians and hospitals will have an increased sensitivity to the potential for software defects. We cannot assure you that our software or other components will not experience errors or performance problems in the future. If we experience software errors or performance problems, we would likely also experience:
| ● | loss of revenue; | |
| ● | delay in market acceptance of our products; | |
| ● | damage to our reputation; | |
| ● | additional regulatory filings; | |
| ● | product recalls; | |
| ● | increased service or warranty costs; and/or | |
| ● |
product liability claims relating to the software defects. |
Risks Related to Intellectual Property
If we are unable to obtain and maintain protection for intellectual property relating to our technology and products, the value of our technology and products will be adversely affected.
Our success will depend in part on our ability to obtain and maintain protection for the intellectual property covering or incorporated into our technology and products. The patent situation in the field of medical devices involves complex legal and scientific questions. We rely upon patents, trade secret laws and confidentiality agreements to protect our technology and products. We may not be able to obtain patent rights relating to our technology or products and pending patent applications to which we have rights may not issue as patents or if issued, may not issue in a form that will be advantageous to us. Even if issued, any patents issued to us may be challenged, narrowed, invalidated, held to be unenforceable or circumvented. Changes in either patent laws or in interpretations of patent laws in the United States may diminish the value of our intellectual property or narrow the scope of our patent protection.
Trademarks and trademark protection of our products may not provide us with a meaningful competitive advantage.
We use trademarks on our products and believe that having distinctive marks is an important factor in marketing them. Distinctive marks may also be important for any additional products that we successfully develop and commercially market. If we are unable to establish name recognition based on our trademarks and trade names, we may not be able to compete effectively and our business may be adversely affected. Our trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. Any trademark litigation, including legal proceedings to seek to protect our trademarks, could be expensive, and it is possible that our efforts could be unsuccessful. We may not be able to protect our rights to these trademarks and trade names or may be forced to stop using these names.
21
We may in the future be a party to patent litigation and administrative proceedings that could be costly and could interfere with our ability to sell our CorPath System.
Litigation related to infringement and other intellectual property claims, with or without merit, is unpredictable, can be expensive and time consuming and can divert management’s attention from our core business. If we lose this kind of litigation, a court could require us to pay substantial damages, and prohibit us from using technologies essential to our CorPath System, any of which would have a material adverse effect on our business, results of operations and financial condition. If relevant patents are upheld as valid and enforceable and we are found to infringe, we could be prevented from selling our CorPath System unless we can obtain a license to use technology or ideas covered by such patents or are able to redesign our CorPath System to avoid infringement. We do not know whether any necessary licenses would be available to us on satisfactory terms, if at all, or whether we could redesign our CorPath System or processes to avoid infringement.
Competing products may also appear in other countries in which our patent coverage might not exist or be as strong. If we lose a foreign patent lawsuit, we could be prevented from marketing our CorPath System in one or more foreign countries.
Risks Related to Government Regulation
Recently enacted healthcare legislation reforming the U.S. healthcare system, as well as future reforms, may have a material adverse effect on our financial condition and results of operations.
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, the “PPACA”), was signed into law which makes changes that significantly impact healthcare providers, insurers, pharmaceutical and medical device manufacturers. One of the principal aims of the PPACA as currently enacted is to expand health insurance coverage to approximately 32,000,000 uninsured Americans. The consequences of these significant coverage expansions on the sales of our products are unknown and speculative at this point.
The PPACA contains a number of provisions designed to generate the revenues necessary to fund the coverage expansions, among other things. This includes new fees or taxes on certain health-related industries, including medical device manufacturers, regardless of whether the companies are profitable. Beginning in 2013, medical device manufacturers were required to pay an excise tax (or sales tax) of 2.3% of certain U.S. medical device revenues. Under this provision, we have paid an excise tax of approximately $0.1 million through December 31, 2015, which tax is reflected in our operating expenses. Though there are some exceptions to the excise tax, this excise tax applies to all or most of our products sold within the U.S. The excise tax was suspended due to the operation of a provision in the Consolidated Appropriations Act of 2016, signed into law on December 18, 2015 but that two-year suspension period ended on December 31, 2017. In mid-January 2018, with the signing of P.L. 115-120, suspension of the medical device excise tax was extended for an additional two years ending on December 31, 2019 and made retroactive to January 1, 2018. At the present time, there is no guarantee that the excise tax will continue to be suspended by congressional action after this two-year moratorium ends, or that any provisions of the PPACA will continue to exist in their current form. The PPACA also establishes a new Patient-Centered Outcomes Research Institute to oversee and identify priorities in comparative clinical effectiveness research in an effort to coordinate and develop such research; implements payment system reforms including a national pilot program on payment bundling to encourage hospitals, physicians, and other providers to improve the coordination, quality, and efficiency of certain healthcare services through bundled payment models; and creates an independent payment advisory board that will submit recommendations to reduce Medicare spending if projected Medicare spending exceeds a specified growth rate.
The PPACA provisions on comparative clinical effectiveness research also extend the initiatives of the American Recovery and Reinvestment Act of 2009, known as the stimulus package, which included $1.1 billion in funding to study the comparative effectiveness of health care treatments and strategies. This stimulus funding was designated for, among other things, conducting, supporting or reviewing research that compares and evaluates the risks and benefits, clinical outcomes, effectiveness and appropriateness of products. The PPACA appropriates additional funding to comparative clinical effectiveness research. Although Congress has indicated that this funding is intended to improve the quality of health care, it remains unclear how the research will impact current Medicare coverage and reimbursement or how new information will influence other third-party payor policies. The taxes imposed by the PPACA and the expansion in the government’s role in the U.S. healthcare industry may result in decreased profits to us, lower reimbursement by payors using our products, and/or reduced medical procedure volumes, all of which may adversely affect our business, financial condition and results of operations.
In addition, other legislative changes have been proposed and adopted since the PPACA was enacted. On August 2, 2011, the Budget Control Act of 2011 was signed into law, which, among other things, creates the Joint Select Committee on Deficit Reduction to recommend proposals in spending reductions to Congress. The Joint Select Committee did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This included aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, which went into effect on April 1, 2013. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Strong, partisan disagreement in Congress has prevented implementation of various PPACA provisions, and the Trump administration has made repeal of the PPACA a priority. Since its enactment, there have been judicial and Congressional challenges to certain aspects of the PPACA. Both Congress and President Trump have expressed their intention to repeal or repeal and replace the ACA, and as a result certain sections of the PPACA have not been fully implemented or were effectively repealed. The uncertainty around the future of the PPACA, and in particular the impact on reimbursement levels and the number of insured individuals, may lead to uncertainty or delay in the purchasing decisions of our customers, which may in turn negatively impact our product sales. If there are not adequate reimbursement levels, our business and results of operations could be adversely affected. One of the first executive orders of the Trump administration granted federal agencies broad powers to unwind regulations under the PPACA. For the immediate future, there is significant uncertainty regarding the health care, health care coverage and health care insurance market.
We expect that additional state and federal healthcare reform measures may be adopted in the future, any of which could have a material adverse effect on our industry generally and our ability to successfully commercialize our products or could limit or eliminate our spending on certain development projects.
22
The U.S. government has in the past considered, is currently considering and may in the future consider healthcare policies and proposals intended to curb rising healthcare costs, including those that could significantly affect both private and public reimbursement for healthcare services. State and local governments, as well as a number of foreign governments, are also considering or have adopted similar types of policies. Future significant changes in the healthcare systems in the United States or elsewhere, and current uncertainty about whether and how changes may be implemented, could have a negative impact on the demand for our products. We are unable to predict whether other healthcare policies, including policies stemming from legislation or regulations affecting our business, may be proposed or enacted in the future; what effect such policies would have on our business; or the effect ongoing uncertainty about these matters will have on the purchasing decisions of our customers.
We are subject to federal and state laws governing our business practices which, if violated, could result in substantial civil and criminal penalties. Additionally, challenges to or investigations of our practices could cause adverse publicity and be costly to respond to and could otherwise harm our business.
The Medicare and Medicaid anti-kickback laws, and similar state laws that may apply to items or services reimbursed by any third-party payor, including commercial insurers, prohibit payments or other remuneration that could be considered to induce hospitals, physicians or other potential purchasers of our products either to refer patients or to purchase, lease or order, or arrange for or recommend the purchase, lease or order of healthcare products or services for which payment may be made under federal and state healthcare programs, such as Medicare and Medicaid. Further, the recently enacted PPACA, among other things, amends the intent requirement of the federal anti-kickback and criminal health care fraud statutes. A person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. In addition, the PPACA provides that the government or a whistleblower may assert that a claim (including items or services resulting from a violation of the federal anti-kickback statute) constitutes a false or fraudulent claim for purposes of the false claims act. These laws may affect our sales, marketing and other contractual and promotional activities by limiting the kinds of financial arrangements we may have with hospitals, physicians or other potential purchasers of our products. They particularly impact how we structure our sales offerings, including discount practices, customer support, education and training programs, physician consulting and other service arrangements. These laws are broadly written, and it is often difficult to determine precisely how these laws will be applied to specific circumstances. Violating anti-kickback laws can result in civil and criminal penalties, which can be substantial and include exclusion from government healthcare programs for noncompliance. Even an unsuccessful challenge or investigation into our practices could cause adverse publicity, and be costly to defend, and thus could harm our business and results of operations.
The PPACA also imposes new reporting and disclosure requirements on device manufacturers for any “transfer of value” made or distributed to physicians and other healthcare providers. Such information must be made publicly available in a searchable format. In addition, device manufacturers will also be required to report and disclose any investment interests held by physicians and their immediate family members during the preceding calendar year. Failure to submit required information may result in civil monetary penalties of up to an aggregate of approximately $0.2 million per year (and up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests not reported in an annual submission. Device manufacturers were required to begin collecting data on August 1, 2013 and were required to submit reports to the Centers for Medicare & Medicaid Services (“CMS”) by March 31, 2014 and the 90th day of each subsequent calendar year. We submitted our report due March 31, 2014 in a timely manner and continue to be in compliance with this reporting requirement.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians, including the tracking and reporting of gifts, compensation and other remuneration to physicians. Certain states mandate implementation of corporate compliance programs to ensure compliance with these laws, impose restrictions on device manufacturer marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians. The shifting corporate compliance environment, and the need to build and maintain robust and expandable systems to comply with multiple jurisdictions with different compliance and/or reporting requirements, increases the possibility that we may be found out of compliance of one or more of the requirements, subjecting us to significant civil monetary penalties.
Compliance with complex foreign and U.S. laws and regulations that apply to our potential international operations increases our cost of doing business in international jurisdictions and could expose us or our employees to fines and penalties in the U.S. and/or abroad. These numerous and sometimes conflicting laws and regulations include U.S. laws such as the Foreign Corrupt Practices Act, and similar laws in foreign countries, such as the U.K. Bribery Act of 2010. Violations of these laws and regulations could result in fines, criminal sanctions against us, our officers or our employees, prohibitions on the conduct of our business and damage to our reputation. There can be no assurance that our policies and procedures are sufficient to ensure compliance with these laws or that our employees, contractors or agents will not violate our policies and procedures.
The Dodd-Frank Wall Street Reform and Consumer Protection Act requires us to track and disclose the source of certain metals used in manufacturing which may stem from minerals (so-called “conflict minerals”) which originate in the Democratic Republic of the Congo or adjoining regions. These metals include tantalum, tin, gold and tungsten. In most cases no acceptable alternative material exists which has the necessary properties. It is not possible to determine the source of the metals by analysis but instead a good faith description of the source of the intermediate components and raw materials must be obtained. The components which incorporate those metals may originate from many sources and we may purchase fabricated products from manufacturers who may have a long and difficult-to-trace supply chain. As the spot price of these materials varies, producers of the metal intermediates can be expected to change the mix of sources used, and components and assemblies which we buy may have a mix of sources as their origin. We do not believe these materials are present in the component parts that we use in our CorPath System, but there can be no assurance that these metals will not be included in our components and assemblies from time to time.
Our products are subject to a lengthy and uncertain domestic regulatory review process. If we do not obtain and maintain the necessary domestic regulatory authorizations, we will not be able to provide our products in the U.S.
Our products and operations are subject to extensive regulation in the U.S. by the FDA. The FDA regulates the development, bench and clinical testing, manufacturing, labeling, storage, record-keeping, promotion, marketing, sales, distribution and post-market support and reporting of medical devices in the U.S. to ensure that medical products distributed domestically are safe and effective for their intended uses. In order for us to market our Class II products for use in the U.S., we must first obtain clearance from the FDA pursuant to Section 510(k) of the Federal Food, Drug, and Cosmetic Act (“FFDCA”). In October 2015, we received 510(k) clearance from the FDA for our robotic-assisted CorPath System to be used during PCI procedures performed via radial access, and on March 29, 2016, we received 510(k) clearance from the FDA for our robotic-assisted CorPath System for use in peripheral vascular interventions. In October 2016, we received 510(k) clearance from the FDA for the CorPath GRX, the second generation of the CorPath System and in February 2018, we received 510(k) clearance from the FDA for CorPath GRX to be used during peripheral vascular interventions. In March 2018, we received 510k) clearance from the FDA for the first automated robotic movement designed for the CorPath GRX platform. Clearance under Section 510(k) requires demonstration that a new device is substantially equivalent to another device with 510(k) clearance or grandfathered (“pre-amendment”) status or to a device that was reclassified from Class III to Class II or Class I (those are referred to as predicate devices). If we significantly modify our products after they receive FDA clearance, or seek to market them for additional indications for use, the FDA may require us to submit a separate 510(k) or premarket approval application (“PMA”) for the modified product before we are permitted to market the products in the U.S. In addition, if we develop products in the future that are not considered to be substantially equivalent to a predicate device, we will be required to obtain FDA approval by submitting a PMA. The FDA may not act favorably or quickly in its review of our 510(k) or PMA submissions or we may encounter significant difficulties and costs in our efforts to obtain FDA clearance or approval, any of which could delay or preclude our sale of new products in the U.S. Furthermore, the FDA may request additional data or require us to conduct further testing, or compile more data, including clinical data and clinical studies, in support of a 510(k) submission.
23
Regulatory policy affecting our products can change at any time. The changes and their impact on our business cannot be accurately predicted. For example, in 2011, the FDA announced a Plan of Action to modernize and improve the FDA’s premarket review of medical devices, and has implemented, and continues to implement, reforms intended to streamline the premarket review process. In addition, as part of the Food and Drug Administration Safety and Innovation Act of 2012, Congress enacted several reforms through the Medical Device Regulatory Improvements and additional miscellaneous provisions which will further affect both pre- and post-approval medical device regulation. Most recently, FDA has been focusing on the cybersecurity risks associated with certain medical devices and encouraging device manufacturers to take a more proactive approach to assessing the cybersecurity risks of their devices both during development and on a periodic basis after the devices are in commercial distribution. These regulatory efforts could lead to new FDA requirements in the future or additional product liability or other litigation risks if one of our products is considered to be susceptible to third-party tampering. In August 2016, the FDA released its proposals for reforming long-standing procedures and requirements related to modifications to medical devices already on the market. In December 2016, Congress passed the 21st Century Cures Act, which makes multiple changes to the FDA’s rules for medical devices as well as for clinical trials, and Congress passed another large piece of legislation related to medical devices in August 2017,the Medical Device User Fee reauthorization package, that could have certain impacts on our business. Changes in the FDA 510(k) process could make clearance more difficult to obtain, increase delay, add uncertainty and have other significant adverse effects on our ability to obtain and maintain clearance for our products. The FDA may also, instead of accepting a 510(k) submission, require us to submit a PMA, which is typically a much more complex, lengthy and burdensome application than a 510(k) submission. To support a PMA, the FDA would likely require that we conduct one or more clinical studies to demonstrate that the device is safe and effective. In some cases such studies may be requested for a 510(k) as well. We may not be able to meet the requirements to obtain 510(k) clearance or PMA approval, in which case the FDA may not grant any necessary clearances or approvals. In addition, the FDA may place significant limitations upon the intended uses of our products as a condition to a 510(k) clearance or PMA approval. Product applications can also be denied or withdrawn due to failure to comply with regulatory requirements or the occurrence of unforeseen problems following clearance or approval. Any delays or failure to obtain FDA clearance or approval of new products we develop, any limitations imposed by the FDA on new product use or the costs of obtaining FDA clearance or approvals could have a material adverse effect on our business, financial condition and results of operations.
In order to conduct a clinical investigation involving human subjects for the purpose of demonstrating the safety and effectiveness of a medical device, a company must, among other things, apply for and obtain Institutional Review Board (‘IRB”) approval of the proposed investigation. In addition, if the clinical study involves a “significant risk” (as defined by the FDA) to human health, the sponsor of the investigation must also submit and obtain FDA approval of an Investigational Device Exemption (“IDE”) application. Our system product is considered a significant risk device requiring IDE approval prior to investigational use. We may not be able to obtain FDA and/or IRB approval to undertake clinical trials in the U.S. for any new devices we intend to market in the U.S. in the future. If we do obtain such approvals, we may not be able to conduct studies which comply with the IDE and other regulations governing clinical investigations or the data from any such trials may not support clearance or approval of the investigational device. Failure to obtain such approvals or to comply with such regulations could have a material adverse effect on our business, financial condition and results of operations. Certainty that clinical trials will meet desired endpoints, produce meaningful or useful data and be free of unexpected adverse effects, or that the FDA will accept the validity of foreign clinical study data cannot be assured, and such uncertainty could preclude or delay market clearance or authorizations resulting in significant financial costs and reduced revenue.
If we fail to obtain regulatory clearances in other countries for existing products or products under development, we will not be able to commercialize these products in those countries.
In order for us to market our products in other countries, we must comply with extensive safety and quality regulations in other countries regarding the quality, safety and efficacy of our products. These regulations, including the requirements for approvals, clearance or grant of Conformité Européenne (“CE”) Certificates of Conformity, PMDA approval in Japan, and the time required for regulatory review, vary from country to country. Failure to obtain regulatory approval, clearance or CE Certificates of Conformity (or equivalent) in any foreign country in which we plan to market our products may harm our ability to generate revenue and harm our business. Approval and CE marking procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval or CE Certificate of Conformity in other countries might differ from that required to obtain FDA clearance. The regulatory approval or CE marking process in other countries may include all of the risks detailed above regarding FDA clearance in the United States. Regulatory approval or the CE marking of a product in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval or a CE Certificate of Conformity in one country may negatively impact the regulatory process in others. Failure to obtain regulatory approval or a CE Certificate of Conformity in other countries or any delay or setback in obtaining such approval could have the same adverse effects described above regarding FDA clearance in the United States.
The primary regulatory environment in Europe is that of the European Economic Area (the “EEA”), which is comprised of the 28 Member States of the European Union (“EU”), Iceland, Liechtenstein and Norway. In the European Economic Area (EEA”), our devices are required to comply with the Essential Requirements laid down in Annex I to the Medical Devices Directive (applicable in the non-EU EEA Member States via the Agreement on the EEA). We are also required to ensure compliance with the relevant quality system requirements laid down in the Annexes to the Medical Devices Directive. Companies compliant with ISO requirements such as “EN ISO 13485: 2003 Medical devices—Quality management systems—Requirements for regulatory purposes” benefit from a presumption of conformity with the relevant Essential Requirements or the quality system requirements laid down in the Annexes to the Medical Devices Directive. Following successful completion of a conformity assessment procedure conducted in relation to the medical device and its manufacturer and their conformity with the Essential Requirements and quality system requirements, the Notified Body issues a CE Certificate of Conformity. This Certificate entitles the manufacturer to affix the CE mark to its medical devices after having prepared and signed a related CE Declaration of Conformity. We received a CE Certificate of Conformity for our CorPath GRX System in 2016. We cannot be certain that we will be successful in meeting and continuing to meet the requirements of the Medical Devices Directive in the EEA.
24
We may incur liability related to the off-label use or misleading advertising of our products.
The FDA regulates the promotional labeling for and the Federal Trade Commission (“FTC”) regulates the advertising of our products to ensure that the claims we make are consistent with our regulatory clearances, that there is scientific data to substantiate the claims and that our promotion and advertising is neither false nor misleading. The off-label marketing or false advertising of our products may harm our image in the marketplace, result in injuries that lead to product liability suits, which could be costly to our business, or result in costly investigations and sanctions from the FDA and other regulatory bodies if we are deemed to have engaged in off-label promotion or false advertising.
If we are found to have violated laws protecting the confidentiality of patient health information, we could be subject to civil or criminal penalties, which could increase our liabilities and harm our reputation or our business.
There are a number of federal and state laws in the United States protecting the confidentiality of certain patient health information, including patient records, and restricting the use and disclosure of that information. In particular, the U.S. Department of Health and Human Services promulgated patient privacy rules and security standards under the Health Insurance Portability and Accountability Act of 1996 and the HITECH Act of 2009 (collectively, “HIPAA”), which expanded the application of those rules and added breach notification requirements. These privacy rules protect medical records and other patient health information by limiting their use and disclosure. HIPAA also gives individuals the right to access, amend and seek accounting of their own health information and limits most use and disclosures of health information to the minimum amount reasonably necessary to accomplish the intended purpose of the use or disclosure. HIPAA does not apply to us, but it imposes significant restrictions on our customers and their ability to share patient information with us. Our failure to structure interactions with our physician customers in a HIPAA compliant manner may result in significant penalties to our customers and harm to our business.
Complying with FDA regulations is a complex process, and our failure to comply fully could subject us to significant enforcement actions.
Because our products are commercially distributed, numerous quality and post-market regulatory requirements apply, including the following:
| ● | continued compliance to the FDA Quality System Regulations (“QSR”), which requires manufacturers to follow elaborate design, testing, control, documentation and other quality assurance procedures during the development and manufacturing process, as well as to take into account newly emerging risks associated with a medical device such as cybersecurity vulnerabilities; |
| ● | labeling regulations; |
| ● | the FDA’s general prohibition against false or misleading statements in the labeling or promotion of products for unapproved uses; |
| ● | stringent complaint reporting and Medical Device Reporting regulations, which requires that manufacturers keep detailed records of investigations or complaints against their devices and to report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; |
| ● | adequate use of the Corrective and Preventive Actions process to identify and correct or prevent significant systemic failures of products or processes or in trends which suggest same; and |
| ● | the reporting of Corrections and Removals, which requires that manufacturers report to the FDA recalls and field corrective actions taken to reduce a risk to health or to remedy a violation of the FFDCA that may pose a risk to health. |
We are subject to inspection and marketing surveillance by the FDA to determine our compliance with regulatory requirements. If the FDA finds that we have failed to comply, it can institute a wide variety of regulatory or enforcement actions, ranging from inspectional observations (Form FDA 483) to a public Warning Letter to more severe civil and criminal sanctions including the seizure of our products and equipment or a ban on the import or export of our products. Our failure to comply with applicable requirements could lead to an enforcement action that may have an adverse effect on our financial condition and results of operations.
Any modification or change of medical devices cleared for market requires the manufacturer to make a determination whether the change is significant enough to require new 510(k) clearance. We have created labeling, advertising and user training for our CorPath System to describe specific procedures that we believe are fully within the scope of our existing 510(k) indications for use stated in our 510(k) clearances. We cannot assure that the FDA would agree that all such specific procedures are within the scope of the existing general clearance or that we have compiled adequate information to support the safety and efficacy of using the CorPath System for all such specific procedures.
If our manufacturing facilities do not continue to meet federal, state or other manufacturing standards, we may be required to temporarily cease all or part of our manufacturing operations, distribution of our products and/or recall our products which would result in significant product delivery delays and lost revenue.
Our manufacturing facilities are subject to periodic inspection by regulatory authorities and our operations will continue to be regulated and inspected by the FDA and other regulatory agencies for compliance with Current Good Manufacturing Practices (“CGMP”) requirements contained in the QSR and other regulatory requirements. For any CorPath Systems shipped internationally, we are also required to comply with the International Organization for Standardization (“ISO”) quality system standards as well as European Directives and norms in order to produce products for sale in the EU. In addition, many countries such as Canada and Japan have very specific additional regulatory requirements for quality assurance and manufacturing. If we fail to continue to comply with CGMP requirements, as well as ISO or other regulatory standards, we may be required to cease all or part of our operations until we comply with these regulations.
25
Risks Related to Our Common Stock
If our stock price declines, our common stock may be subject to delisting from the NYSE American.
Our common stock was approved for listing on the NYSE American and commenced trading on May 29, 2015 under the symbol “CVRS.” Our Company’s common stock was previously traded on the OTCQB as provided by OTC Markets Group, Inc. under the symbol “CVRS.” Our common stock is currently trading at a price of less than $2.00 per share. We currently meet the continued listing standards of NYSE American. However, we cannot assure you that we will be able to continue to comply with the standards that we are required to meet in order to maintain a listing of our common stock on the NYSE American. Our failure to continue to meet these requirements may result in our common stock being delisted from the NYSE American. If our common stock is delisted, this would, among other things, substantially impair our ability to raise additional funds and could result in a loss of institutional investor interest and fewer development opportunities for us.
The price of our common stock could be highly volatile due to a number of factors, which could lead to losses by investors and costly securities litigation.
We cannot predict the extent to which investor interest in our Company will result in an active trading market on the NYSE American or any other exchange that we may trade on in the future. The trading price of our common stock on the NYSE American has been highly volatile and is likely to continue to be highly volatile in response to a number of factors including, without limitation, the following:
| ● | fluctuations in price and volume due to investor speculation and other factors that may not be tied to our financial performance; |
| ● | performance by us in the execution of our business plan; |
| ● | financial viability; |
| ● | actual or anticipated variations in our operating results; |
| ● | announcements of developments by us or our competitors; |
| ● | market conditions in our industry; |
| ● | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures or capital commitments; |
| ● | adoption of new accounting standards affecting our industry; |
| ● | additions or departures of key personnel; |
| ● | introduction of new products by us or our competitors; |
| ● | sales of our common stock or other securities in the open market; |
| ● | regulatory developments in both the United States and foreign countries; |
| ● | performance of products sold and advertised by licensees in the marketplace; |
| ● | economic and other external factors; |
| ● | period-to-period fluctuations in financial results; and |
| ● | other events or factors, many of which are beyond our control. |
The stock market is subject to significant price and volume fluctuations. In the past, and several recent situations, following periods of volatility in the market price of a company’s securities, securities class action litigation has been initiated against such company. Litigation initiated against us, whether or not successful, could result in substantial costs and diversion of our management’s attention and resources, which could harm our business and financial condition.
If we are unable to successfully maintain our internal controls over financial reporting or if material weaknesses are discovered in our internal accounting procedures, the accuracy and timing of our financial reporting may be adversely affected, which may adversely affect investor confidence in us and, as a result, the value of our common stock.
Any failure to develop or maintain effective internal controls over financial reporting or difficulties encountered in maintaining or improving our internal controls over financial reporting could harm our operating results and prevent us from meeting our reporting obligations. Moreover, effective internal controls, particularly those related to revenue recognition, are necessary for us to produce reliable financial reports and are important to helping prevent financial fraud. If we cannot provide reliable financial reports or prevent fraud, our business and operating results could be harmed, investors could lose confidence in our reported financial information, and the trading price of our common stock could drop significantly. In addition, investors relying upon this misinformation could make an uninformed investment decision, and we could be subject to sanctions or investigations by the Commission or other regulatory authorities or to stockholder class action securities litigation.
Provisions in our certificate of incorporation and by-laws and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our certificate of incorporation and by-laws may discourage, delay or prevent a merger, acquisition or other change in control of us that stockholders may consider favorable, including transactions in which our common stockholders might otherwise receive a premium price for their shares. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. In addition, because our board of directors is responsible for appointing the members of our management team, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. Among other things, these provisions:
| ● | establish a classified board of directors such that not all members of the board are elected at one time; |
| ● | allow the authorized number of our directors to be changed only by resolution of our board of directors; |
| ● | limit the manner in which stockholders can remove directors from the board; |
| ● | establish advance notice requirements for stockholder proposals that can be acted on at stockholder meetings and for nominations to our board of directors; |
| ● | limit who may call stockholder meetings; |
| ● | prohibit actions by our stockholders by written consent; |
26
| ● | require that stockholder actions be effected at a duly called stockholders meeting; |
| ● | authorize our board of directors to issue preferred stock without stockholder approval, which could be used to institute a “poison pill” that would work to dilute the stock ownership of a potential hostile acquirer, effectively preventing acquisitions that have not been approved by our board of directors; and |
| ● | require the approval of the holders of at least 75% of the votes that all our stockholders would be entitled to cast to amend or repeal certain provisions of our certificate of incorporation or by-laws. |
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns 15% or more of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired 15% or more of our outstanding voting stock, unless the merger or combination is approved in a manner prescribed by the statute.
We may not have sufficient authorized and available shares if all outstanding stock options are exercised.
Our certificate of incorporation authorizes 250,000,000 shares of common stock. . As of February 28, 2018, we had 188,772,869 outstanding shares of common stock. There are 20,110,278 shares of common stock underlying outstanding stock options and restricted stock units and 355,028 shares of common stock underlying warrants. The weighted average exercise price of outstanding stock options is $1.27 per share, while the restricted stock units were granted at $1.09 per share, and the outstanding warrants have an exercise price of $1.41 per share. In addition, on March 16, 2018, we closed an equity financing pursuant to which we issued Series A preferred stock convertible into up to 20,000,000 shares of common stock at a conversion rate of $1.25 per share and warrants exercisable for 8,750,000 shares of common stock at an exercise price of $1.40 per share. The Shares of Series A preferred stock will also be entitled to receive non-compounding dividends in additional shares of preferred stock, at the rate of 12% per annum, subject to reduction in the event certain milestones are achieved. This dividend preferred stock is also convertible into shares of common stock at a conversion rate of $1.25 per share. If we are unable to increase the number of authorized shares of common stock by amendment to our certificate of incorporation, we may not be able to grant or sell additional shares of common stock in the future. Our limited available authorized common stock could hinder our ability to attract and retain employees with stock options and other equity incentives and could require us to pursue non-equity financing alternatives in the future, such as incurring significant debt, which may be unavailable on attractive terms or at all.
Insiders have substantial control over the outstanding shares of the Company’s common stock and could delay or prevent a change in corporate control, including a transaction in which the Company’s stockholders could sell or exchange their shares for a premium.
On March 9, 2018, our directors and executive officers beneficially own an aggregate of approximately 28% of our outstanding shares of common stock. As a result, our directors and executive officers, if acting together, may have the ability to affect the outcome of matters submitted to stockholders for approval, including the election and removal of directors, and any merger, consolidation or sale of all or substantially all of our assets. In addition, these persons acting together may have the ability to control our management and business affairs. Accordingly, this concentration of ownership may harm the value of our common stock by:
| ● | delaying, deferring or preventing a change in control; |
| ● | impeding a merger, consolidation, takeover or other business combination; or |
| ● | discouraging a potential acquirer from making an acquisition proposal or otherwise attempting to obtain control. |
We do not expect to pay dividends on our common stock and investors should not buy our common stock expecting to receive dividends.
We have never declared or paid any dividends on our common stock and we do not anticipate that we will declare or pay any dividends on our common stock in the foreseeable future. In addition, we are restricted from paying cash dividends on our common stock under our term loan and revolving loan facilities. Consequently, you will only realize an economic gain on your investment in our common stock if the price appreciates. You should not purchase our common stock expecting to receive cash dividends. Since we do not pay dividends on our common stock, and if we are not successful in establishing an orderly trading market for our shares, then you may not have any manner to liquidate or receive any payment on your investment. Therefore our failure to pay dividends on our common stock may cause you to not see any return on your investment even if we are successful in our business operations. In addition, because we do not pay dividends on our common stock we may have trouble raising additional funds which could affect our ability to expand our business operations.
We are likely to raise additional funds, finance acquisitions or develop strategic relationships by issuing capital stock.
We have financed our operations, and we expect to continue to finance our operations, make acquisitions and develop strategic relationships by issuing equity or convertible debt securities which could significantly reduce the percentage ownership of our existing stockholders. Furthermore, any newly issued securities could have rights, preferences and privileges senior to those of our existing common stock. Moreover, any issuances by us of equity securities may be at or below the prevailing market price of our common stock and in any event may have a dilutive impact on your ownership interest, which could cause the market price of our common stock to decline. We may also raise additional funds through the incurrence of debt, and the holders of any debt we may issue would have rights superior to your rights in the event we are not successful and are forced to seek the protection of the bankruptcy laws.
Securities analysts may not cover our common stock and this may have a negative impact on our common stock’s market price.
The future trading market for our common stock may depend on the research and reports that securities analysts publish about us or our business. We do not have any control over these analysts. We may face additional risks since we became a public company through an acquisition which, for accounting purposes, was treated as a reverse merger. There is no guarantee that securities analysts will cover our common stock and there may be little incentive to brokerage firms to recommend the purchase of our common stock. If securities analysts do not cover our common stock, the lack of research coverage may adversely affect our common stock’s market price, if any. If we are covered by securities analysts who downgrade our stock, our stock price would likely decline. If one or more of these analysts ceases to cover us or fails to publish regular reports on us, we could lose visibility in the financial markets, which could cause our stock price or trading volume to decline.
Our management devotes substantial time to our compliance with regulations and we incur significant costs as a public company.
As a public company, we incur significant legal, accounting and other expenses. In addition, the Sarbanes-Oxley Act and rules subsequently implemented by the SEC and the NYSE American have imposed various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other personnel devote a substantial amount of time to these compliance initiatives and we anticipate that such efforts will continue, particularly as we cease to be able to avail ourselves of the reduced disclosure requirements available to emerging growth companies. Moreover, these rules and regulations have increased our legal and financial compliance costs and will make some activities more time-consuming and costly.
27
In particular, the Sarbanes-Oxley Act and rules subsequently implemented by the Commission impose various requirements on public companies with respect to corporate governance practices. The Sarbanes-Oxley Act requires, among other things, that our management maintain adequate disclosure controls and procedures and internal control over financial reporting. In particular, we must perform system and process evaluation and testing of our internal control over financial reporting to allow management and, as applicable, our independent registered public accounting firm, to report on the effectiveness of our internal control over financial reporting.
We may become involved in securities class action litigation that could divert management’s attention and harm our business.
The stock market, in general, and the market for medical device companies, in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of those companies. These broad market and industry factors may materially harm the market price of our common stock, regardless of our operating performance. In the past, following periods of volatility in the market price of a particular company’s securities, securities class action litigation has often been brought against that company. We may become involved in this type of litigation in the future. Litigation often is expensive and diverts management’s attention and resources, which could materially harm our financial condition and results of operations.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS. |
None.
| ITEM 2. | PROPERTIES. |
Our principal offices and manufacturing facilities are located at 309 Waverley Oaks Road, Suite 105, Waltham, Massachusetts 02452. In October 2012, Corindus, Inc. entered into a lease with Beaver Group, LLC for a term of approximately five years for 26,402 square feet of office and manufacturing space (the “Lease”). In October 2016, the Lease term was extended for an additional three years ending on January 31, 2021 (“Extended Lease”). Over the term of the Extended Lease, we pay an average monthly cost of $53 thousand of base rent, excluding common area fees, taxes and insurance. Our management believes that the leased premises are suitable and adequate to meet current needs.
| ITEM 3. | LEGAL PROCEEDINGS. |
From time to time, we may become involved in various lawsuits and legal proceedings that arise in the ordinary course of business. Litigation is subject to inherent uncertainties and an adverse result in these or other matters may arise from time to time that may harm our business. We are currently not aware of any such legal proceedings or claims that we believe will have a material adverse effect on our business, financial condition or operating results.
| ITEM 4. | MINE SAFETY DISCLOSURES. |
Not applicable.
28
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES. |
Market Information
Since May 29, 2015, our common stock has been listed on the NYSE American under the symbol “CVRS.”
The following table sets forth the high and low sales prices of our common stock as reported by the NYSE American for each quarter in the fiscal years ended December 31, 2017 and 2016:
| High | Low | |||||||
| 2017 | ||||||||
| Fourth Quarter | $ | 1.60 | $ | 0.66 | ||||
| Third Quarter | $ | 2.25 | $ | 1.35 | ||||
| Second Quarter | $ | 2.03 | $ | 0.98 | ||||
| First Quarter | $ | 1.75 | $ | 0.40 | ||||
| 2016 | ||||||||
| Fourth Quarter | $ | 1.23 | $ | 0.56 | ||||
| Third Quarter | $ | 1.80 | $ | 1.04 | ||||
| Second Quarter | $ | 1.71 | $ | 0.87 | ||||
| First Quarter | $ | 3.28 | $ | 0.73 | ||||
On March 9, 2018, the closing price of a share of our common stock on the NYSE American was $1.28.
Stockholders
As of March 9, 2018, there were 188,772,869 shares of common stock outstanding, which were held by approximately 109 record holders.
Dividends
We have never declared or paid any cash dividend. We do not anticipate that we will declare or pay any dividends in the foreseeable future. Our current policy is to retain earnings, if any, to fund operations, and the development and growth of our business. Any future determination to pay cash dividends will be at the discretion of our Board of Directors and will be dependent upon our financial condition, operating results, capital requirements, applicable contractual restrictions, restrictions in our organizational documents, and any other factors deemed relevant by our Board of Directors. In addition, we are restricted from paying cash dividends under our term loan and revolving loan facilities.
Comparative Stock Performance
The following line graph compares cumulative total shareholder return for the period beginning August 18, 2014 and ended December 31, 2017 for (i) our common stock; (ii) NYSE Medical Equipment (U.S. Companies); (iii) NASDAQ Stock Market (U.S. Companies); (iv) NASDAQ Medical Equipment, and (v) NYSE American Stock Market. The graph assumes $100 invested on August 18, 2014 and includes reinvestment of dividends. Measurement points are August 18, 2014 and the last trading day of the fiscal years ended December 31, 2017, 2016, 2015, and 2014. The stock price performance on the following graph is not necessarily indicative of future stock price performance.
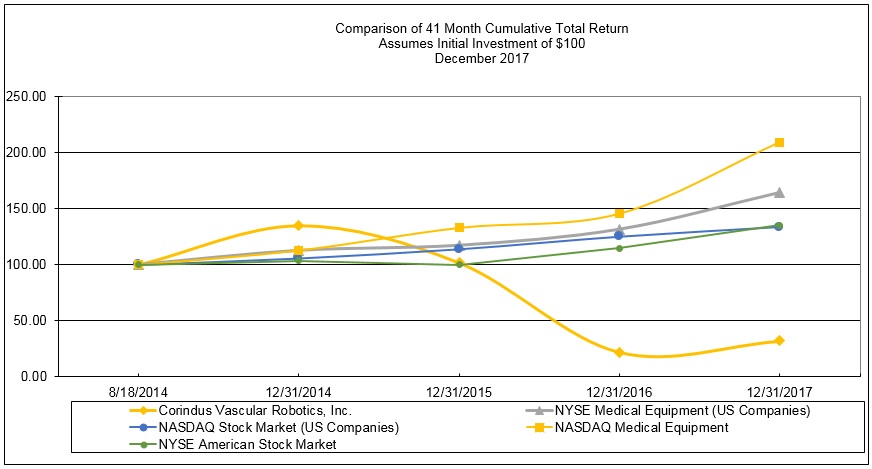
29
The information under the caption “Comparative Stock Performance” above is not deemed to be “filed” as part of this Annual Report, and is not subject to the liability provisions of Section 18 of the Securities Exchange Act of 1934. Such information will not be deemed to be incorporated by reference into any filing we make under the Securities Act of 1933 unless we explicitly incorporate it into such a filing at the time.
Unregistered Sales of Securities
On October 11, 2017, a warrant to purchase 4,728,191 shares of Common Stock held by Koninklijke Philips NV (“Philips”) was automatically exercised on a net exercise basis in connection with its expiration. Upon expiration of the warrant, Philips paid the exercise price of $1.06 per share through the Company’s withholding of 3,334,586 of the warrant shares to pay the exercise price and issuing 1,393,605 shares of Common Stock to Philips. The shares were issued in reliance on an exemption from registration under Section 4(a)(2) of the Securities Act of 1933, as amended.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
During the fourth quarter of the year ended December 31, 2017, neither we nor any “affiliated purchaser,” as that term is defined in Rule 10b-18(a)(3) under the Exchange Act, repurchased any of our registered equity securities.
| ITEM 6. | SELECTED FINANCIAL DATA. |
The selected financial data set forth below for the years ended December 31, 2017, 2016, 2015, 2014 and 2013 and historical balance sheet data as of December 31, 2017, 2016, 2015, 2014 and 2013, have been derived from our consolidated financial statements. The selected consolidated financial data included below should be read in conjunction with the consolidated financial statements (and notes thereon) and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” included in Item 7.
| Fiscal Year Ended December 31, | ||||||||||||||||||||
| 2017 | 2016 | 2015 | 2014(1) | 2013(1) | ||||||||||||||||
| Consolidated Statements of Operations Data: | (in thousands, except share and per share amounts) | |||||||||||||||||||
| Revenue | $ | 9,650 | $ | 2,842 | $ | 2,729 | $ | 2,983 | $ | 896 | ||||||||||
| Cost of goods sold | 9,265 | 5,042 | 3,724 | 4,904 | 2,430 | |||||||||||||||
| Gross (loss) profit | 385 | (2,200 | ) | (995 | ) | (1,921 | ) | (1,534 | ) | |||||||||||
| Operating expenses: | ||||||||||||||||||||
| Research and development | 9,517 | 10,313 | 10,033 | 6,607 | 4,793 | |||||||||||||||
| Selling, general and administrative | 24,777 | 19,564 | 16,143 | 13,002 | 8,221 | |||||||||||||||
| Restructuring charges | — | — | — | 175 | — | |||||||||||||||
| Total operating expenses | 34,294 | 29,877 | 26,176 | 19,784 | 13,014 | |||||||||||||||
| Operating loss | (33,909 | ) | (32,077 | ) | (27,171 | ) | (21,705 | ) | (14,548 | ) | ||||||||||
| Other income (expense): | ||||||||||||||||||||
| Warrant revaluation | — | — | — | (2,421 | ) | (171 | ) | |||||||||||||
| Interest and other income (expense) | (214 | ) | (1,001 | ) | (1,592 | ) | (415 | ) | 28 | |||||||||||
| Total other expense, net | (214 | ) | (1,001 | ) | (1,592 | ) | (2,836 | ) | (143 | ) | ||||||||||
| Net loss | $ | (34,123 | ) | $ | (33,078 | ) | $ | (28,763 | ) | $ | (24,541 | ) | $ | (14,691 | ) | |||||
| Net loss per share, basic and diluted | $ | (0.20 | ) | $ | (0.28 | ) | $ | (0.25 | ) | $ | (0.29 | ) | $ | (0.20 | ) | |||||
| Weighted average number of common shares outstanding | 173,925,450 | 119,019,700 | 113,254,925 | 84,990,198 | 73,360,259 | |||||||||||||||
| (1) | On August 12, 2014, Your Internet Defender Inc. (“Your Internet Defender”), closed a reverse acquisition transaction in which it acquired Corindus, Inc. (“Corindus”) and Corindus Security Corporation as wholly owned subsidiaries (the “Acquisition”). The Acquisition was accounted as a reverse acquisition pursuant to which Corindus was considered the acquiring entity for accounting purposes. As such, Corindus’ historical results of operations replace Your Internet Defender’s historical results of operations for all periods prior to the Acquisition. The results of operations of both companies are included in our consolidated financial statements for all periods after the completion of the Acquisition. |
| As of December 31, | ||||||||||||||||||||
| 2017 | 2016 | 2015 | 2014 | 2013 | ||||||||||||||||
| Consolidated Balance Sheet Data: | (in thousands) | |||||||||||||||||||
| Cash, cash equivalents and marketable securities | $ | 17,458 | $ | 9,183 | $ | 42,666 | $ | 28,526 | $ | 9,845 | ||||||||||
| Total assets | $ | 24,566 | $ | 13,013 | $ | 47,139 | $ | 32,836 | $ | 14,768 | ||||||||||
| Long-term debt, net of current portion | $ | — | $ | — | $ | 3,673 | $ | 7,594 | $ | — | ||||||||||
| Long-term capital lease obligation, net of current portion | $ | 102 | $ | — | $ | — | $ | — | $ | — | ||||||||||
| Total liabilities | $ | 7,051 | $ | 8,943 | $ | 11,292 | $ | 13,054 | $ | 4,728 | ||||||||||
| Working capital | $ | 16,429 | $ | 3,048 | $ | 37,993 | $ | 26,231 | $ | 11,387 | ||||||||||
| Accumulated deficit | $ | (180,841 | ) | $ | (146,718 | ) | $ | (113,640 | ) | $ | (84,877 | ) | $ | (60,336 | ) | |||||
| Total stockholders’ (deficit) equity | $ | 17,515 | $ | 4,070 | $ | 35,847 | $ | 19,782 | $ | (10,040 | ) | |||||||||
30
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS. |
The following discussion of our financial condition and results of operations should be read in conjunction with our financial statements and related notes appearing elsewhere in this Annual Report on Form 10-K. The following discussion contains forward-looking statements that reflect our plans, estimates and beliefs. Our actual results may differ materially from those discussed in the forward-looking statements. Factors that might cause future results to differ materially from those projected in the forward-looking statements include, but are not limited to, those discussed in “Risk Factors” and elsewhere in this Annual Report on Form 10-K. See also “Cautionary Note Regarding Forward-Looking Statements.”
Overview
We design, manufacture and sell precision vascular robotic-assisted systems for use in interventional vascular procedures (the “CorPath® System”). The CorPath System is the first medical device cleared by the FDA to bring robotic-assisted precision to radial, coronary and peripheral procedures. During these procedures, the interventional cardiologist sits at a radiation-shielded workstation to advance interventional devices with millimeter-by-millimeter precision. The workstation allows the physician greater control and the freedom from wearing heavy lead protective equipment that causes musculoskeletal injuries. The CorPath System brings robotic precision to radial and complex interventional procedures to help optimize clinical outcomes and minimize the costs associated with complications of improper stent placement with manual procedures. In October 2016, we received 510(k) clearance from the FDA for our CorPath GRX System, the second generation of the CorPath System. CorPath GRX significantly builds upon the CorPath 200 platform, the first generation of the CorPath System, adding a significant number of key upgrades that increase precision, improve workflow, and extend the capabilities and range of the procedures that can be performed robotically. These features include active guide management which enables control of the guide catheter along with robotic control of the guidewire and balloon or stent catheter, with one-millimeter advancement, from the control console. This precise positioning will enable physicians to adjust guide catheter position during procedures and may expand use of CorPath to more complex cases. We began commercial shipment of the CorPath GRX System in late January 2017. While the CorPath GRX has been cleared for and we are targeting percutaneous coronary intervention procedures and peripheral vascular interventions, we believe our technology platform has the capability to be developed in the future for other segments of the vascular intervention market, including neurointerventional and other more complex cardiac interventions such as structural heart procedures. As of December 31, 2017, we have installed 33 CorPath GRX Systems. Additionally, as of December 31, 2017, we shipped six CorPath GRX Systems that were accepted by a distributor. During 2017, the majority of our consumable revenues relate to the CorPath GRX System.
On March 16, 2018, we closed on a private placement of convertible preferred stock for gross proceeds of $25 million. The preferred stock is convertible into an aggregate of 20,000,000 shares of common stock, and is entitled to receive non-compounding dividends in additional shares of preferred stock, at the rate of 12% per annum, subject to reduction in the event certain milestones are achieved. The preferred stock purchasers were also issued warrants to purchase an aggregate of 8,750,000 shares of common stock at an exercise price of $1.40 per share, exercisable either for cash or on a cashless basis.
On March 16, 2018, we completed a financing arrangement with two lenders which provides for borrowings of up to $26 million in the form of up to $23 million in term loans and up to a $3 million revolving line-of-credit through March 2022. As of March 16, 2018, we had $12 million in principal outstanding under the term loan facility and $0 in principal outstanding under the revolving loan facility An additional $5.5 million in term loans may become available in the future provided we have achieved a specified gross profit milestone prior to January 1, 2019, and an additional $5.5 million may become available provided we receive net cash proceeds of $30 million from a future sale of our equity securities prior to July 1, 2019 and achieve a specified gross profit milestone prior to September 1, 2019 . Until such time that we achieve the specified criteria, the additional term loans are not available to us. We cannot assure you that we will achieve the gross profit or equity financing milestones that will trigger our ability to further draw the term loan facility. The revolving line-of-credit also has various clauses which restrict its availability and for which we currently do not meet such restrictions. The outstanding term loans bear interest at a floating rate per annum equal to the greater of (i) 8.83% and (ii) the sum of (a) the one month ICE Benchmark LIBOR based on U.S. Dollar deposits, plus (b) 7.25%. The outstanding principal under the revolving line bears interest at a floating rate per annum equal to the greater of (i) 5.0% and (ii) the sum of (a) the “prime rate,” as reported in the Wall Street Journal, plus (b) 0.5%, which interest is payable monthly. Both loan facilities are secured by substantially all of our personal property other than our intellectual property. Both loan facilities include customary affirmative and negative covenants. Upon the earlier of the second advance under the term loan facility or the first advance under the revolving loan facility, we must also achieve minimum revenue on a monthly basis measured against a percentage of our Board of Directors-approved projections for the applicable fiscal year. Our failure to satisfy the revenue, or any other, covenant could result in an event of default under the loan facilities. Both loan facilities also include other events of default, the occurrence and continuation of which could cause interest to be charged at the rate that is otherwise applicable plus 5.0% and would provide the collateral agent under the term loan facility or the lender under the revolving loan facility, as applicable, with the right to exercise remedies against us and the collateral securing the loan facilities. These events of default include, among other things, any failure by us to pay principal or interest due under the loan facilities, a breach of certain covenants under the loan facilities, our insolvency, a material adverse change, the occurrence of any default under certain other indebtedness in an amount greater than $0.25 million, one or more judgments against us in an amount greater than $0.25 million individually or in the aggregate, and any default under the other loan facility.
Critical Accounting Policies
Our discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments, including those related to revenue recognition, inventory valuation, assumptions used in the valuation of stock-based awards, and valuation allowances against deferred income tax assets. We base our estimates on historical experience, known trends and events and various other factors that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
We believe that several accounting policies are important to understanding our historical and future performance. We refer to these policies as “critical” because these specific areas generally require us to make judgments and estimates about matters that are uncertain at the time we make the estimate. We use the best information available to us to make our judgments and estimates; however, actual results may be different. We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our financial statements. It is important that the discussion of our operating results that follows be read in conjunction with the critical accounting policies discussed below.
Revenue Recognition
The CorPath System is a capital medical device used by hospitals and surgical centers to perform heart catheterizations. Use of the CorPath System requires a sterile, single-use cassette (the “CorPath Cassette”), which are sold separately, for each procedure. Products are sold to customers with no rights of return. We recognize all revenue on the sale of products when the following criteria are met:
| ● | Persuasive evidence of an arrangement exists | |
| ● | The price to the buyer is fixed or determinable | |
| ● | Collectability is reasonably assured | |
| ● | Risk of loss transfers and the product is delivered. |
In each arrangement, we are responsible for installation of the CorPath System and initial user training, which services are deemed essential to the functionality of the system. Therefore, we recognize system revenue when the CorPath System is delivered, installed and accepted by the end user customer.
Each CorPath System is sold with a standard one-year warranty, which provides that the CorPath System will function as intended, and during that one-year period, we will either replace the product or a portion thereof or provide the necessary repair service during our normal service hours. We accrue for the estimated costs of the warranty once the CorPath System revenue is recognized.
We generally enter into multiple element arrangements, which include the sale of a CorPath System with an initial order of CorPath Cassettes, and may include either a basic service plan or a premium service plan. The basic service plan provides for an extended warranty period and the premium service plan provides for the extended warranty as well as component upgrades, when and if they become available during the service period. Deliverables, which are accounted for as separate units of accounting under multiple-element arrangements include: (a) the CorPath System, including installation and initial training, which are subject to customer acceptance and (b) the initial shipment of CorPath Cassettes to the customer, and may include either (c) an extended warranty or (d) component upgrades.
31
We recognize revenue on multiple-element arrangements in accordance with Accounting Standards Update (“ASU”) ASU 2009-13, Revenue Recognition (Topic 605): Multiple Deliverable Revenue Arrangements, based on the estimated selling price of each element. In accordance with ASU 2009-13, we use vendor-specific objective evidence (“VSOE”), if available, to determine the selling price of each element. If VSOE is not available, we use third-party evidence (“TPE”) to determine the selling price. If TPE is not available, we use our best estimate to develop the estimated selling price (“BESP”). We use BESP to determine the selling price of our systems as well as the basic and premium service plans. BESP is determined based on estimated costs plus a reasonable margin, and has generally been consistent with the price charged to the customer for such products and services. The determination of BESP also considers the price of the service plans charged to customers when such services are sold separately in subsequent transactions. We also use BESP to determine the selling price of the initial order of cassettes, which considers the price at which we charge customers when the cassettes are sold separately.
Revenue related to basic and premium service plans is recognized on a straight-line basis over the life of the service contract. Revenue from cassettes and accessories is recorded upon delivery and services provided by us outside of a basic or premium service contract is recognized as the services are provided. If a revenue arrangement contains an undelivered element, such as an unspecified upgrade, revenues are deferred until delivery is complete.
Income Taxes
We account for income taxes using the liability method, whereby deferred tax asset and liability account balances are determined based on differences between financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates that will be in effect when the differences are expected to reverse. We have provided a valuation allowance to reduce deferred tax assets to amounts that are realizable based on uncertainty of future taxable income.
We account for uncertain tax positions using a “more-likely-than-not” threshold for recognizing and resolving uncertain tax positions. The evaluation of uncertain tax positions is based on factors including, but not limited to, changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, the effective settlement of matters subject to audit, new audit activity and changes in facts or circumstances related to a tax position. Corindus has not had an uncertain tax position to date.
Stock-Based Compensation
We recognize compensation costs resulting from the issuance of stock-based awards to employees as an expense in the consolidated statements of operations over the requisite service or performance period based on a measurement of fair value for each stock award. The awards issued to date have primarily been stock options with service-based vesting periods over two or four years, restricted stock units with service-based vesting periods of one year, and shares of unrestricted common stock. During 2017, certain shares of unrestricted common stock were granted to a non-employee director of the Company which were fully vested upon grant. Accordingly, compensation expense related to unrestricted stock was recognized on the date of the grant. Stock-based compensation is charged to the respective line items in our statements of operations to which the employee’s services are classified. Compensation costs on awards with both performance and service-based vesting are recorded when it becomes probable that the performance condition and requisite service will be met. Compensation costs associated with stock-based awards to non-employees are measured at fair value on the date of grant and re-measured at the fair value on the date the awards vest and for those awards that have not vested at the end of each reporting period. We use the Black-Scholes-Merton option pricing model (“Black-Scholes Model”) to determine the fair value of the awards. The key assumptions in the Black-Scholes Model include an estimate of the volatility of our stock, the risk-free interest rate, and the expected period the stock option will be exercised over and a key assumption in stock-based compensation expense recognition is the estimated forfeiture rate.
The fair value of our common stock is based on the trading of our stock on the OTCQB and NYSE American, as applicable. We utilize quoted market prices to calculate the fair value of stock-based awards.
Inventories
Inventories are valued at the lower of cost or net realizable value using the first-in, first-out (FIFO) method. We routinely monitor the recoverability of our inventories and record the lower of cost or market reserves, or reserves for excess and obsolete inventories, as required. We also monitor the utilization of our production facility and we record the costs of under-utilization of the production facility directly to cost of revenue.
Components of Results of Operations
The following is a description of what comprises each of our significant statement of operations captions:
Revenues
We generate our revenues primarily from the sale of the CorPath System, CorPath Cassettes, accessories and service contracts.
Cost of Revenue
Cost of revenue represents the cost of materials for the CorPath System, CorPath Cassettes and accessories, service labor and labor and overhead of production facilities.
Research and Development
Research and development expenses consist primarily of salaries and stock based compensation for our research and development, clinical and regulatory employees, and certain operating costs related to research and development and third party contractor costs.
32
Selling, General and Administrative
Selling, general and administrative expenses consist primarily of salaries and stock based compensation for our executives and our marketing, finance, legal, human resource, and other administrative employees as well as salaries and commissions of our internal sales force. Selling, general and administrative expenses also include marketing program costs and outside consulting, legal and accounting services, and facilities and other supporting overhead costs. We also included shipping costs for CorPath Systems and CorPath Cassettes and Accessories in selling, general and administrative expense.
Interest and Other Expense, net
Interest and other expense, net represents interest expense on borrowings under the Company’s Loan and Security Agreement (as defined below) and capital lease obligation, accretion of discounts and amortization of premiums on available-for-sale marketable securities, and interest income.
Results of Operations
Discussion of Year Ended December 31, 2017 compared to Year Ended December 31, 2016:
| Year Ended December 31, | ||||||||||||
| 2017 | 2016 | Change | ||||||||||
| (In thousands) | ||||||||||||
| Revenue | $ | 9,650 | $ | 2,842 | $ | 6,808 | ||||||
| Cost of revenue | 9,265 | 5,042 | 4,223 | |||||||||
| Gross profit (loss) | 385 | (2,200 | ) | 2,585 | ||||||||
| Operating expenses: | ||||||||||||
| Research and development | 9,517 | 10,313 | (796 | ) | ||||||||
| Selling, general and administrative | 24,777 | 19,564 | 5,213 | |||||||||
| Total operating expenses | 34,294 | 29,877 | 4,417 | |||||||||
| Operating loss | (33,909 | ) | (32,077 | ) | (1,832 | ) | ||||||
| Interest and other expense, net | (214 | ) | (1,001 | ) | 787 | |||||||
| Net loss | $ | (34,123 | ) | $ | (33,078 | ) | $ | (1,045 | ) | |||
Revenue
Revenue increased to $9.7 million for the year ended December 31, 2017 from $2.8 million for the year ended December 31, 2016. This revenue increase was due primarily to the increase in CorPath Systems and capital upgrades installed during 2017.
Our revenue associated with CorPath Systems increased to $6.9 million for the year ended December 31, 2017 from $1.5 million for the year ended December 31, 2016. We installed 17 and seven new CorPath Systems during the years ended December 31, 2017 and 2016, respectively. Additionally, CorPath System revenues included revenues related to six CorPath Systems shipped and accepted by a distributor during the year ended December 31, 2017. For the year ended December 31, 2017, CorPath Systems revenue included $0.7 million of previously deferred CorPath System revenue where the customer arrangements included a previously undelivered item that was completed in 2017. Our average selling price associated with CorPath Systems for which revenue was recognized during the year ended December 31, 2017 when compared to CorPath Systems for which revenue was recognized during the year ended December 31, 2016 decreased by 3%. We have experienced, and we expect to continue to experience, some unevenness in the number and trend of CorPath Systems sold and the average selling price of CorPath Systems sold on a quarterly basis given the early stage of commercialization of our product and market acceptance along with the continued development of a dedicated and consistent sales force.
With the launch of our second generation CorPath GRX System in January 2017, revenue also includes upgrade revenues for existing customers of CorPath 200 Systems who purchased capital upgrades to the CorPath GRX System. Revenues associated with capital upgrades during the year ended December 31, 2017 totaled $1.2 million and there were no such items during the year ended December 31, 2016.
Our revenue for CorPath Cassettes and accessories, which represents our sale of consumables, increased to $0.9 million for the year ended December 31, 2017 as compared to $0.6 million for the year ended December 31, 2016. The volume and average price of our CorPath Cassettes increased by 482 units and decreased by 11.3%, respectively, from the year ended December 31, 2017 to the year ended December 31, 2016.
Our revenue associated with services performed totaled $0.7 million for both the year ended December 31, 2017 and 2016. We have experienced, and expect to continue to experience, fluctuations in our service revenues based upon whether customers elect to purchase service contracts with their CorPath Systems.
Given the relatively small number of customers due to the early stage of the commercialization and the higher price of the CorPath System relative to consumables, customers that purchase a CorPath System in a specific period tend to make up a significant percentage of revenue in that period.
Cost of Revenue
Cost of revenue increased to $9.3 million for the year ended December 31, 2017 from $5.0 million for the year ended December 31, 2016. Cost of revenue for both the years ended December 31, 2017 and 2016 included the effect of under-utilization of our production facilities. For the year ended December 31, 2017, cost of revenue included the cost of multiple CorPath GRX System upgrades that were installed pursuant to pre-existing and new contractual arrangements, and the costs associated with 33 Systems installed or delivered during 2017, as well as our recording a lower of cost or net realizable value reserve of $268 thousand for our CorPath GRX Cassettes due to the higher material and production costs associated with the early stage of our production of these items.
33
Cost of revenue represents the cost of materials for the CorPath System and CorPath Cassettes, as well as labor and overhead at our production facility. At current volumes, our cost to manufacture the CorPath GRX System and CorPath GRX Cassettes is approximately $0.2 million and $2 thousand, respectively. We expect these costs to decrease as we obtain economies of scale with respect to purchasing and production and continue to incorporate design enhancements.
Gross Profit (Loss)
Our gross margin increased to a gross profit of $0.4 million for the year ended December 31, 2017 from a gross loss of $2.2 million for the year ended December 31, 2016 based on the changes in revenue and cost of revenues as discussed above. For the year ended December 31, 2017, we have not generated enough sales volume of CorPath Systems and CorPath Cassettes to significantly offset our manufacturing costs, including the effect of the under-utilization of our production facility, a portion of which represents excess manufacturing capacity, and we have therefore generated minimal gross profit.
Research and Development
Research and development expenses decreased to $9.5 million for the year ended December 31, 2017 from $10.3 million for the year ended December 31, 2016 primarily due to reduced purchases of prototype materials of $1.0 million and reduced spending for consulting services of $0.3 million for the year ended December 31, 2017. These reductions were offset by increased compensation expense of $0.6 million primarily related to employees hired subsequent to December 31, 2016.
Selling, General and Administrative
Selling, general and administrative expenses increased to $24.8 million for the year ended December 31, 2017 from $19.6 million for the year ended December 31, 2016 primarily due to increased compensation expense of $3.9 million primarily for sales-related employees hired subsequent to December 31, 2016, incremental stock-based compensation expense of $0.4 million associated with stock options granted in 2016 and 2017, and increased travel-related expenses of $0.6 million.
Interest and Other Expense, net
Interest and other expense decreased to $0.2 million for the year ended December 31, 2017 from $1.0 million for the year ended December 31, 2016. The interest and other expense for both periods was primarily the result of interest expense on borrowings under the Company’s Loan and Security Agreement (as defined below). The decrease in interest and other expense for the year ended December 31, 2017 as compared to the prior year period is primarily due to lower interest expense as a result of a reduction in our overall debt balance as a result of contractual principal payments made during the past year whereby the debt balance was fully repaid in 2017.
Income Taxes
We have not recorded any benefit related to operating losses due to uncertainty about future taxable income.
Discussion of Year Ended December 31, 2016 compared to Year Ended December 31, 2015:
| Year Ended December 31, | ||||||||||||
| 2016 | 2015 | Change | ||||||||||
| (In thousands) | ||||||||||||
| Revenue | $ | 2,842 | $ | 2,729 | $ | 113 | ||||||
| Cost of revenue | 5,042 | 3,724 | 1,318 | |||||||||
| Gross loss | (2,200 | ) | (995 | ) | (1,205 | ) | ||||||
| Operating expenses: | ||||||||||||
| Research and development | 10,313 | 10,033 | 280 | |||||||||
| Selling, general and administrative | 19,564 | 16,143 | 3,421 | |||||||||
| Total operating expenses | 29,877 | 26,176 | 3,701 | |||||||||
| Operating loss | (32,077 | ) | (27,171 | ) | (4,906 | ) | ||||||
| Interest and other expense, net | (1,001 | ) | (1,592 | ) | 591 | |||||||
| Net loss | $ | (33,078 | ) | $ | (28,763 | ) | $ | (4,315 | ) | |||
Revenue
Revenue increased to $2.8 million for the year ended December 31, 2016 from $2.7 million for the year ended December 31, 2015 due primarily to an increase in service revenue.
Our revenue associated with CorPath System sales decreased to $1.5 million for the year ended December 31, 2016 from $1.8 million for the year ended December 31, 2015. We sold seven and eight CorPath Systems during the years ended December 31, 2016 and 2015, respectively, and our average selling price increased by 51% for the year ended December 31, 2016 compared to the year ended December 31, 2015. The decline in CorPath System revenue for 2016 despite the increase in the sales price related to the deferral of revenue on two systems in 2016 due to undelivered elements for specified upgrade rights. We have experienced, and we expect to continue to experience, some unevenness in the number and trend of units sold and the average selling price of units sold on a quarterly basis given the early stage of commercialization of our product and market acceptance along with the continued development of a dedicated and consistent sales force. In 2016, we sold two systems in the first quarter, zero in the second quarter, one in the third quarter, and four in the fourth quarter. Additionally, we expect variability in the sales of our consumables until our product receives wider market acceptance.
Our revenue from CorPath Cassettes and accessories, which represent our sale of consumables, decreased to $0.6 million for the year ended December 31, 2016 from $0.7 million for the year ended December 31, 2015 due to a decline in volume. The volume and average price of our CorPath Cassettes decreased by 65 units and increased by 0.6%, respectively, for the year ended December 31, 2016 compared to the year ended December 31, 2015. Revenues under our CUPs represented 21.9% and 36.8% for the year ended December 31, 2016 and 2015, respectively, of our total revenues for the sale of consumables.
Our revenue associated with services performed increased to $0.7 million for the year ended December 31, 2016 from $0.3 million for the year ended December 31, 2015. The increase relates to revenue recognized under the terms of outstanding service agreements and an increase in service projects performed for the year ended December 31, 2016. We have experienced, and expect to continue to experience, fluctuations in our service revenues based upon whether or not customers elect to purchase service contracts with their CorPath Systems and the related deferral of any expected services to be performed under such arrangements.
Given the relatively small number of customers due to the early stage of the Company’s commercialization and the price of the CorPath System relative to consumables, customers that purchase a system in a specific period tend to make up a significant percentage of revenue in that period.
Cost of Revenue
Cost of revenue increased to approximately $5.0 million for the year ended December 31, 2016 from approximately $3.7 million for the year ended December 31, 2015. Cost of revenues for both the year ended December 31, 2016 and December 31, 2015 included the effect of under-utilization of our production facilities.
Cost of revenue represents the cost of materials for the CorPath System and CorPath Cassettes, as well as labor and overhead at our production facility.
34
Gross Loss
Gross loss increased to approximately $2.2 million for the year ended December 31, 2016 from approximately $1.0 million for the year ended December 31, 2015 based on the changes in revenue and cost of revenue as discussed above. We have not generated enough sales volume of CorPath Systems to offset our manufacturing costs including the effect of the under-utilization of our production facility and have generated a gross loss.
Research and Development
Research and development expenses increased to approximately $10.3 million for the year ended December 31, 2016 from approximately $10.0 million for the year ended December 31, 2015. This increase of $0.3 million is primarily due to increased compensation expense of $0.6 million and incremental stock-based compensation of $0.1 million for the year ended December 31, 2016, partially offset by decreases in purchasing of prototype material of $0.3 million and reduced spending on consultants and subcontractors of $0.2 million.
Selling, General and Administrative
Selling, general and administrative expenses increased to approximately $19.6 million for the year ended December 31, 2016 from approximately $16.1 million for the year ended December 31, 2015. This increase of $3.5 million is primarily due to incremental stock-based compensation expense of $1.8 million, increased sales salaries and commissions of $1.1 million, incremental costs of $0.7 associated with the transition of the Company’s former Chief Executive Officer that was completed in March 2016, and increased travel expenses of $0.4 million, partially offset by a reduction in marketing program costs of $0.4 million and recruiting costs of $0.2 million.
Interest and Other Expense, net
Other expense, net, decreased to approximately $1.0 million for the year ended December 31, 2016 from approximately $1.6 million for the year ended December 31, 2015. Other expense for both the year ended December 31, 2016 and December 31, 2015 was primarily the result of interest expense on borrowings under our Loan and Security Agreement (as defined below). The decrease in other expense for the year ended December 31, 2016 as compared to the year ended December 31, 2015 is primarily due to lower interest expense as a result of a reduction in our overall debt balance through contractual principal payments over the past year.
Income Taxes
We have not recorded any benefit related to operating losses due to uncertainty about future taxable income.
Liquidity and Capital Resources
On March 16, 2018, we closed on a private placement of convertible preferred stock for gross proceeds of $25 million. The preferred stock is convertible into an aggregate of 20,000,000 shares of common stock, and is entitled to receive non-compounding dividends in additional shares of preferred stock, at the rate of 12% per annum, subject to reduction in the event certain milestones are achieved. The preferred stock purchasers were also issued warrants to purchase an aggregate of 8,750,000 shares of common stock at an exercise price of $1.40 per share, exercisable either for cash or on a cashless basis.
On March 16, 2018, we completed a financing arrangement with two lenders which provides for borrowings of up to $26 million in the form of up to $23 million in term loans and up to a $3 million revolving line-of-credit through March 2022. As of March 16, 2018, we had $12 million in principal outstanding under the term loan facility and $0 in principal outstanding under the revolving loan facility. An additional $5.5 million in term loans may become available in the future provided we have achieved a specified gross profit milestone prior to January 1, 2019, and an additional $5.5 million may become available provided we receive net cash proceeds of $30 million from a future sale of our equity securities prior to July 1, 2019 and achieve a specified gross profit milestone prior to September 1, 2019 . Until such time that we achieve the specified criteria, the additional term loans are not available to us. We cannot assure you that we will achieve the gross profit or equity financing milestones that will trigger our ability to further draw the term loan facility. The revolving line-of-credit also has various clauses which restrict its availability and for which we currently do not meet such restrictions. The outstanding term loans bear interest at a floating rate per annum equal to the greater of (i) 8.83% and (ii) the sum of (a) the one month ICE Benchmark LIBOR based on U.S. Dollar deposits, plus (b) 7.25%. The outstanding principal under the revolving line bears interest at a floating rate per annum equal to the greater of (i) 5.0% and (ii) the sum of (a) the “prime rate,” as reported in the Wall Street Journal, plus (b) 0.5%, which interest is payable monthly. Both loan facilities are secured by substantially all of our personal property other than our intellectual property. Both loan facilities include customary affirmative and negative covenants. Upon the earlier of the second advance under the term loan facility or the first advance under the revolving loan facility, we must also achieve minimum revenue on a monthly basis measured against a percentage of our Board of Directors-approved projections for the applicable fiscal year. Our failure to satisfy the revenue, or any other, covenant could result in an event of default under the loan facilities. Both loan facilities also include other events of default, the occurrence and continuation of which could cause interest to be charged at the rate that is otherwise applicable plus 5.0% and would provide the collateral agent under the term loan facility or the lender under the revolving loan facility, as applicable, with the right to exercise remedies against us and the collateral securing the loan facilities. These events of default include, among other things, any failure by us to pay principal or interest due under the loan facilities, a breach of certain covenants under the loan facilities, our insolvency, a material adverse change, the occurrence of any default under certain other indebtedness in an amount greater than $0.25 million, one or more judgments against us in an amount greater than $0.25 million individually or in the aggregate, and any default under the other loan facility.
We have incurred losses since inception and have funded our cash flow deficits primarily through the issuance of capital stock and debt. As of December 31, 2017, we had an accumulated deficit of $180.8 million. As of December 31, 2017, we had cash and cash equivalents of $17.5 million and working capital of $16.4 million. We anticipate that these available resources and the additional financings discussed above will be sufficient to meet our cash requirements for at least the next 12 months from March 19, 2018. However, as we continue to incur losses and cash flow deficits, our transition to profitability is dependent upon achieving a level of revenues adequate to support our cost structure, but we will otherwise rely on additional capital funding until such time as that is achieved.
As such, we have evaluated whether or not our cash and cash equivalents on hand and the cash proceeds from the financing activities described above would be sufficient to sustain projected operating activities through March 19, 2019 as required by Accounting Standards Codification (ASC) 205-40 Disclosure of Uncertainties About an Entity’s Ability to Continue as a Going Concern. Based on our current forecasts of cash flow deficits, which are estimated to be approximately $37 million to $39 million per year, we will have cash through approximately May 2019.
Due to the inherent uncertainty in predicting revenues and certain variable costs, we have considered our ability to reduce cash flow deficits and have determined that if we do not achieve our revenue forecast, we would undertake the following activities to reduce our cash flow deficits:
●
Defer or limit some or all of its spending on capital equipment that we believe is necessary to reduce manufacturing costs of cassettes and CorPath systems to be used in marketing and training activities that were otherwise planned for 2018;
●
Eliminate or defer the 2018 discretionary bonus payouts for all bonus eligible employees, including executive management;
●
Reduce spending on travel, clinical programs and prototypes;
●
Eliminate planned headcount additions in marketing, sales, and manufacturing; and
●
Reduce external consulting resources who otherwise would be used to assist in installations of CorPath systems.
As a result, we believe our plans can be effectively implemented as all of the actions are within our control and will be finalized and will be able to be effectively implemented, if required.
We may never achieve profitability, and unless and until doing so, we intend to fund future operations through additional non-dilutive or dilutive financings. There can be no assurances, however, that additional funding will be available on terms acceptable to us, if at all.
35
On June 11, 2014, the Company entered into a Loan and Security Agreement pursuant to which the lender agreed to make available to the Company $10 million in two separate $5 million loans under secured promissory notes (the “Loan and Security Agreement”). The initial note was made on June 11, 2014 in an aggregate principal amount equal to $5 million (the “Initial Promissory Note”) and was repayable in equal monthly installments of principal and interest over 27 months beginning on July 1, 2015. On December 31, 2014, the Company borrowed the additional $5 million (the “Second Promissory Note”) under the Loan and Security Agreement. The Second Promissory note was also repayable in equal monthly installments of principal and interest over 27 months beginning on July 1, 2015.
All amounts due under the Loan and Security Agreements were fully repaid on October 2, 2017.
In summary, our cash flows were:
| Years Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| (In thousands) | ||||||||||||
| Net cash used in operating activities | $ | (32,358 | ) | $ | (27,594 | ) | $ | (27,856 | ) | |||
| Net cash provided by (used in) investing activities | $ | (174 | ) | $ | 20,087 | $ | (20,793 | ) | ||||
| Net cash provided by (used in) financing activities | $ | 40,807 | $ | (5,452 | ) | $ | 42,265 | |||||
Operating Activities
Cash used in operating activities was $32.4 million for the year ended December 31, 2017 compared to $27.6 million for the year ended December 31, 2016. The $4.8 million increase in the use of cash was due primarily to the changes in working capital.
Cash used in operating activities was $27.6 million for the year ended December 31, 2016 compared to $27.9 million for the year ended December 31, 2015. The $0.3 million decrease in the use of cash was due primarily to the changes in working capital.
Cash used for operating activities was primarily comprised of research and development activities related to the CorPath GRX and selling activities related to both the CorPath 200 and CorPath GRX in addition to general and administrative costs required to operate the Company.
Investing Activities
Cash used in investing activities was $0.2 million for the year ended December 31, 2017 compared to $20.1 million provided by investing activities for the year ended December 31, 2016. The change was primarily due to maturities of available-for-sale securities during 2016 with no similar activity during 2017.
Cash provided by investing activities was $20.1 million for the year ended December 31, 2016 compared to $20.8 million used in investing activities for the year ended December 31, 2015. The cash generated from investing activities during 2016 was primarily the maturity of $20.6 million of available-for-sale securities while the use of funds in investing activities during 2015 was primarily due to purchases of available-for sale securities of $22.8 million from the proceeds of our May 2015 public offering.
Financing Activities
Cash provided by financing activities for the year ended December 31, 2017, totaled $40.8 million and was primarily due to proceeds from the issuance of common stock in a private placement offset by contractual payments on long-term debt.
Cash used in financing activities for the year ended December 31, 2016 totaled $5.5 million and was due to contractual payments on long-term debt, payment for common stock repurchase and retirement and the payment of withholding taxes on stock option exercises.
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements (as that term is defined in Item 303(a)(4)(ii) of Regulation S-K) as of December 31, 2017 that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources.
Contractual Obligations
The following table summarizes our contractual cash obligations at December 31, 2017 and the effect such obligations are expected to have on our liquidity and cash flow in future periods:
|
Payments Due By Period (In thousands) |
||||||||||||||||||||
| Total | Less than 1 Year |
1 - 3 Years |
3 - 5 Years |
More than 5 Years |
||||||||||||||||
| Capital lease liability | $ | 182 | $ | 67 | $ | 115 | $ | — | $ | — | ||||||||||
| Building lease liability | 1,945 | 577 | 1,313 | 55 | — | |||||||||||||||
| $ | 2,127 | $ | 644 | $ | 1,428 | $ | 55 | $ | — | |||||||||||
36
Recently Issued Accounting Standards
In May 2014, August 2015, April 2016 and May 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2014-09, Revenue from Contracts with Customers, ASU 2015-14, Revenue from Contracts with Customers, Deferral of the Effective Date, ASU 2016-10, Revenue from Contracts with Customers, Identifying Performance Obligations and Licensing, and ASU 2016-12, Revenue from Contracts with Customers, Narrow-Scope Improvements and Practical Expedients, respectively, (collectively referred to as “Topic 606”). Topic 606 outlines a single comprehensive model for entities to use in accounting for revenue arising from contracts with customers, and supersedes current revenue recognition guidance, including industry-specific guidance. It also requires entities to disclose both quantitative and qualitative information that enable financial statements users to understand the nature, amount, timing, and uncertainty of revenue and cash flows arising from contracts with customers. The amendments in these ASUs are effective for fiscal years, and interim periods within those years, beginning after December 15, 2017. The two permitted transition methods under the new standard are the full retrospective method, in which case the standard would be applied to each prior reporting period presented and the cumulative effect of applying the standard would be recognized at the earliest period shown, or the modified retrospective method, in which case the cumulative effect of applying the standard would be recognized at the date of initial application. We will adopt the guidance on January 1, 2018 and will apply the modified retrospective transition approach only to contracts that have undelivered performance obligations as of this date. We performed a review of all revenue streams to identify any differences in the timing, measurement or presentation of revenue recognition. We have an installed base of 62 CorPath Systems as of December 31, 2017. We assessed all of these systems, and determined that a large number of those installations will not be impacted by the implementation of Topic 606 since there will be no future performance obligations under those arrangements upon adoption. As a result, we identified a limited number of contracts for which there will be future performance obligations as of the adoption date. Pertaining to these contracts, the adoption of Topic 606 will result in certain components of services being unbundled from previously bundled arrangements, and result in an acceleration of the timing of revenue recognition related to our services. The new standard requires revenues to be estimated and recognized upon transfer of the promised goods and services, and we determined that the accelerated recognition of service revenues will result in a cumulative adjustment to revenues as of the adoption date. Adoption of the new standard will result in the capitalization of certain costs to obtain service contracts and the capitalization of such costs will result in an adjustment to capitalized expenses as of the adoption date. We are in the process of finalizing the overall impact of the adoption of this standard on our fourth quarter sales and will finalize in the first quarter of 2018. Our calculations indicate that such amounts may not be material to our results of operations.
In February 2016, the FASB issued ASU 2016-02, Leases (Topic 842), which amends leasing accounting requirements. The new standard requires lessee recognition on the balance sheet of a right-of-use asset and a lease liability, initially measured at the present value of the lease payments. It further requires recognition in the income statement of a single lease cost, calculated so that the cost of the lease is allocated over the lease term on a generally straight-line basis. Finally, it requires classification of all cash payments within operating activities in the statement of cash flows. It is effective for fiscal years commencing after December 15, 2018 and early adoption is permitted. We are currently evaluating the impact of this standard on our consolidated financial statements.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows (Topic 230), Classification of Certain Cash Receipts and Cash Payments, which reduces diversity in how certain cash receipts and cash payments are presented and classified in the Consolidated Statements of Cash Flows. It is effective for fiscal years, and interim periods within those years, beginning after December 15, 2017 and will be required to be applied retrospectively, with early adoption permitted. We are currently evaluating the impact of this update on its consolidated financial statements and related disclosures.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK. |
We are exposed to market risk related to changes in interest rates. We had cash and cash equivalents of $17.5 million as of December 31, 2017. The cash and cash equivalents as of December 31, 2017 consists of cash in bank deposits and money market funds. Our primary exposure to market risk is interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates, particularly because our investment strategy is primarily in short term securities. The effect of an immediate hypothetical change in variable interest rates would not have a material effect on our consolidated financial statements.
We have generated limited net revenue from operations to date and depend on funds raised through other sources. One possible source of funding is through further equity offerings. Our ability to raise funds in this manner depends upon capital market forces affecting our stock price.
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA. |
The consolidated financial statements, together with the report of our independent registered public accounting firm, Ernst & Young LLP, appear at page F-1 through F-24 of this Annual Report on Form 10-K.
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE. |
None.
| ITEM 9A. | CONTROLS AND PROCEDURES. |
Evaluation of Disclosure Controls and Procedures
As required by Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), we carried out an evaluation under the supervision and with the participation of our senior management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures as of December 31, 2017. Based upon their evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures are effective to ensure that material information relating to the Company required to be disclosed by the Company in reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in Securities and Exchange Commission rules and forms and to ensure that such information is accumulated and communicated to senior management, including our Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, our management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurances of achieving the desired control objectives, and management necessarily was required to apply its judgment in designing and evaluating the controls and procedures.
37
Internal Control over Financial Reporting
Management’s Report on Internal Controls over Financial Reporting
Our senior management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rules 13a-15(f) and 15d-15(f) promulgated under the Exchange Act as a process designed by, or under the supervision of, our principal executive and principal financial officers, or persons performing similar functions, and effected by our board of directors, senior management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. GAAP, and includes policies and procedures that:
| ● | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; | |
| ● | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and | |
| ● | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the consolidated financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. We continue to review our internal control over financial reporting, and may from time to time make changes aimed at enhancing their effectiveness and to ensure that our systems evolve with our business.
Under the supervision and with the participation of senior management, including the Chief Executive Officer and Chief Financial Officer, the Company conducted an evaluation of the effectiveness of the Company’s internal control over financial reporting based on the framework in “Internal Control — Integrated Framework (2013)” issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on this assessment, our senior management has concluded that the internal control over financial reporting was effective as of December 31, 2017.
The effectiveness of our internal control over financial reporting as of December 31, 2017 has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated in their report which we include herein.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting during the quarter ended December 31, 2017 that have materially affected, or that are reasonably likely to materially affect, our internal control over financial reporting.
| 38 |
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of
Corindus Vascular Robotics, Inc.
Opinion on Internal Control over Financial Reporting
We have audited Corindus Vascular Robotics, Inc.’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Corindus Vascular Robotics, Inc. (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of Corindus Vascular Robotics, Inc. as of December 31, 2017 and 2016, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2017, and the related notes of the Company and our report dated March 19, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Controls over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Boston, Massachusetts
March 19, 2018
39
| ITEM 9B. | OTHER INFORMATION. |
None.
40
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS, AND CORPORATE GOVERNANCE. |
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Management and Corporate Governance,” “ Section 16(A) Beneficial Ownership Reporting Compliance,” and “Code of Conduct and Ethics” in the Company’s Proxy Statement for the 2018 Annual Meeting of Stockholders.
| ITEM 11. | EXECUTIVE COMPENSATION. |
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Executive Officer and Director Compensation,” “Compensation Discussion and Analysis,” “Pay Ratio Disclosure,” “Management and Corporate Governance,” and “Compensation Committee Report” in the Company’s Proxy Statement for the 2018 Annual Meeting of Stockholders.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS. |
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Equity Compensation Plan Information” and “Security Ownership of Certain Beneficial Owners and Management” to the definitive Proxy Statement for the 2018 Annual Meeting of Stockholders.
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS AND DIRECTOR INDEPENDENCE. |
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Certain Relationships and Related Person Transactions” and “Management and Corporate Governance” in the Company’s Proxy Statement for the 2018 Annual Meeting of Stockholders.
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES. |
The response to this item is incorporated by reference from the discussion responsive thereto under the captions “Proposal 6: Ratification of Independent Registered Public Accounting Firm” in the Company’s Proxy Statement for the 2018 Annual Meeting of Stockholders.
| 41 |
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES. |
| (a) | Financial Statements and Financial Statements Schedules |
| (1) | Financial Statements are listed in the Consolidated Financial Statements Contents on page F-1 of this Annual Report. | |
| (2) | No financial statement schedules are included because such schedules are not applicable, are not required, or because required information is included in the consolidated financial statements or notes thereto. | |
| (3) | Exhibits. |
| 42 |
___________
| * | Filed herewith. |
| ** | This certification is being furnished and shall not be deemed “filed” with the SEC for purposes of Section 18 of the Exchange Act, or otherwise subject to the liability of that section, and shall not be deemed to be incorporated by reference into any filing under the Securities Act or the Exchange Act, except to the extent that the Registrant specifically incorporates it by reference. |
| † | Portions of this exhibit have been omitted pursuant to a request for confidential treatment. Omitted material has been separately filed with the Securities and Exchange Commission. |
| # | Management contract or compensatory plan or arrangement. |
43
| ITEM 16. | FORM 10-K SUMMARY |
Not applicable.
44
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Company has duly caused this Report to be signed on its behalf by the undersigned thereunto duly authorized.
DATE: March 19, 2018
| CORINDUS VASCULAR ROBOTICS, INC. | ||
| By: | /s/ Mark J. Toland | |
| Mark J. Toland | ||
| Chief Executive Officer and President | ||
| Principal Executive Officer | ||
| By: | /s/ David W. Long | |
| David W. Long | ||
| Chief Financial Officer and Senior Vice President | ||
| Principal Financial and Accounting Officer | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
| Signature | Capacity | Date | ||
| /s/ Mark J. Toland | Chief Executive Officer, President and Director | March 19, 2018 | ||
| Mark J. Toland | (Principal Executive Officer) | |||
| /s/ David W. Long | Chief Financial Officer, Senior Vice President, | March 19, 2018 | ||
| David W. Long | Treasurer and Secretary (Principal Financial and Accounting Officer) | |||
| /s/ Jeffrey C. Lightcap | Chairman | March 19, 2018 | ||
| Jeffrey C. Lightcap | ||||
| /s/ Jeffrey Gold | Director | March 19, 2018 | ||
| Jeffrey Gold | ||||
| /s/ Campbell Rogers | Director | March 19, 2018 | ||
| Campbell Rogers | ||||
| /s/ Louis A. Cannon | Director | March 19, 2018 | ||
| Louis A. Cannon | ||||
| /s/ Nathan R. Harrington | Director | March 19, 2018 | ||
| Nathan R. Harrington |
45
F- 1
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of
Corindus Vascular Robotics, Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Corindus Vascular Robotics, Inc. (the Company) as of December 31, 2017 and 2016, the related consolidated statements of operations and comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2017, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control-Integrated framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated March 19, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2010.
Boston, Massachusetts
March 19, 2018
F- 2
| CORINDUS VASCULAR ROBOTICS, INC. | |||||
| CONSOLIDATED BALANCE SHEETS | |||||
| (In thousands, except share and per share amounts) | |||||
| December 31, | December 31, | |||||||
| 2017 | 2016 | |||||||
| Assets | ||||||||
| Current Assets: | ||||||||
| Cash and cash equivalents | $ | 17,458 | $ | 9,183 | ||||
| Accounts receivable | 2,863 | 384 | ||||||
| Due from related party | — | 250 | ||||||
| Inventories, net | 2,103 | 1,545 | ||||||
| Prepaid expenses and other current assets | 539 | 448 | ||||||
| Total current assets | 22,963 | 11,810 | ||||||
| Property and equipment, net | 1,452 | 982 | ||||||
| Deposits and other assets | 151 | 221 | ||||||
| Total assets | $ | 24,566 | $ | 13,013 | ||||
| Liabilities and stockholders’ equity | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable | $ | 2,416 | $ | 2,463 | ||||
| Accrued expenses | 3,637 | 1,794 | ||||||
| Customer deposits | 93 | — | ||||||
| Deferred revenue | 339 | 750 | ||||||
| Current portion of capital lease obligation | 49 | — | ||||||
| Current portion of long-term debt | — | 3,755 | ||||||
| Total current liabilities | 6,534 | 8,762 | ||||||
| Long-term Liabilities | ||||||||
| Deferred revenue, net of current portion | 342 | 129 | ||||||
| Long-term capital lease obligation, net of current portion | 102 | — | ||||||
| Other liabilities | 73 | 52 | ||||||
| Total long-term liabilities | 517 | 181 | ||||||
| Total liabilities | 7,051 | 8,943 | ||||||
| Commitments and Contingencies (Note 12) | ||||||||
| Stockholders’ equity: | ||||||||
| Preferred stock, $0.0001 par value; 10,000,000 shares authorized; none issued and outstanding | — | — | ||||||
| Common stock, $0.0001 par value; 250,000,000 shares authorized; 188,764,851 shares at December 31, 2017 and 119,025,221 shares at December 31, 2016 issued and outstanding | 19 | 12 | ||||||
| Additional paid-in capital | 198,337 | 150,776 | ||||||
| Accumulated deficit | (180,841 | ) | (146,718 | ) | ||||
| Total stockholders’ equity | 17,515 | 4,070 | ||||||
| Total liabilities and stockholders’ equity | $ | 24,566 | $ | 13,013 | ||||
The accompanying notes are an integral part of the consolidated financial statements.
F- 3
| CORINDUS VASCULAR ROBOTICS, INC. | |||||||||||
| CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS | |||||||||||
| (In thousands, except share and per share amounts) | |||||||||||
| Year Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Revenue | $ | 9,650 | $ | 2,842 | $ | 2,729 | ||||||
| Cost of revenue | 9,265 | 5,042 | 3,724 | |||||||||
| Gross profit (loss) | 385 | (2,200 | ) | (995 | ) | |||||||
| Operating expenses: | ||||||||||||
| Research and development | 9,517 | 10,313 | 10,033 | |||||||||
| Selling, general and administrative | 24,777 | 19,564 | 16,143 | |||||||||
| Total operating expense | 34,294 | 29,877 | 26,176 | |||||||||
| Operating loss | (33,909 | ) | (32,077 | ) | (27,171 | ) | ||||||
| Interest and other expense, net | (214 | ) | (1,001 | ) | (1,592 | ) | ||||||
| Net loss | $ | (34,123 | ) | $ | (33,078 | ) | $ | (28,763 | ) | |||
| Net loss per share--basic and diluted | $ | (0.20 | ) | $ | (0.28 | ) | $ | (0.25 | ) | |||
|
Weighted-average common shares used in computing net loss
per share--basic and diluted |
173,925,450 | 119,019,700 | 113,254,925 | |||||||||
| Other comprehensive loss: | ||||||||||||
| Net loss | $ | (34,123 | ) | $ | (33,078 | ) | $ | (28,763 | ) | |||
| Unrealized gain (loss) on marketable securities | — | 14 | (14 | ) | ||||||||
| Comprehensive loss | $ | (34,123 | ) | $ | (33,064 | ) | $ | (28,777 | ) | |||
The accompanying notes are an integral part of the consolidated financial statements.
F- 4
| CORINDUS VASCULAR ROBOTICS, INC. | |||||||||||||||||||||||||
| CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY | |||||||||||||||||||||||||
| (In thousands, except share and per share amounts) | |||||||||||||||||||||||||
| Additional | ||||||||||||||||||||||||
| Common Stock, $0.0001 Par Value | Paid-in | Accumulated Other | Accumulated | |||||||||||||||||||||
| Shares | Amount | Capital | Comprehensive Loss | Deficit | Total | |||||||||||||||||||
| Balance at December 31, 2014 | 105,883,157 | $ | 11 | $ | 104,648 | $ | — | $ | (84,877 | ) | $ | 19,782 | ||||||||||||
| Stock-based compensation expense | — | — | 505 | — | — | 505 | ||||||||||||||||||
| Issuance of common stock in connection with public offering of common stock, net of issuance costs of $794 | 12,650,000 | 1 | 44,391 | — | — | 44,392 | ||||||||||||||||||
| Issuance of common stock upon exercise of stock options | 340,345 | — | 76 | — | — | 76 | ||||||||||||||||||
| Common stock withheld to pay statutory minimum withholding taxes on exercise of stock options | (41,061 | ) | — | (131 | ) | — | — | (131 | ) | |||||||||||||||
| Change in fair value of marketable securities | — | — | — | (14 | ) | — | (14 | ) | ||||||||||||||||
| Net loss | — | — | — | — | (28,763 | ) | (28,763 | ) | ||||||||||||||||
| Balance at December 31, 2015 | 118,832,441 | 12 | 149,489 | (14 | ) | (113,640 | ) | 35,847 | ||||||||||||||||
| Stock-based compensation expense | — | — | 2,366 | — | — | 2,366 | ||||||||||||||||||
| Issuance of common stock upon exercise of stock options | 848,297 | — | (338 | ) | — | — | (338 | ) | ||||||||||||||||
| Issuance of common stock upon exercise of warrants | 93,325 | — | — | — | — | — | ||||||||||||||||||
| Common stock repurchase and retirement | (748,842 | ) | — | (741 | ) | — | — | (741 | ) | |||||||||||||||
| Change in fair value of marketable securities | — | — | — | 14 | — | 14 | ||||||||||||||||||
| Net loss | — | — | — | — | (33,078 | ) | (33,078 | ) | ||||||||||||||||
| Balance at December 31, 2016 | 119,025,221 | 12 | 150,776 | — | (146,718 | ) | 4,070 | |||||||||||||||||
| Stock-based compensation expense | — | — | 2,892 | — | — | 2,892 | ||||||||||||||||||
| Issuance of common stock in connection with private placement of common stock, net of issuance costs of $415 | 68,055,700 | 7 | 44,604 | — | — | 44,611 | ||||||||||||||||||
| Issuance of common stock upon exercise of stock options | 261,670 | — | 65 | — | — | 65 | ||||||||||||||||||
| Issuance of common stock upon exercise of warrants | 1,393,605 | — | — | — | — | — | ||||||||||||||||||
| Issuance of common stock upon vesting of restricted stock units | 26,362 | — | — | — | — | — | ||||||||||||||||||
| Issuance of unrestricted common stock | 2,293 | — | — | — | — | — | ||||||||||||||||||
| Net loss | — | — | — | — | (34,123 | ) | (34,123 | ) | ||||||||||||||||
| Balance at December 31, 2017 | 188,764,851 | $ | 19 | $ | 198,337 | $ | — | $ | (180,841 | ) | $ | 17,515 | ||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F- 5
| CORINDUS VASCULAR ROBOTICS, INC. | |||||||||||
| CONSOLIDATED STATEMENTS OF CASH FLOWS | |||||||||||
| (In thousands) | |||||||||||
| Year Ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Operating activities | ||||||||||||
| Net loss | $ | (34,123 | ) | $ | (33,078 | ) | $ | (28,763 | ) | |||
| Adjustments to reconcile net loss to net cash flows used in operating activities: | ||||||||||||
| Loss on disposal of fixed assets | 28 | 81 | — | |||||||||
| Impairment of property and equipment | — | 125 | — | |||||||||
| Depreciation and amortization | 723 | 725 | 706 | |||||||||
| Stock-based compensation expense | 2,892 | 2,366 | 505 | |||||||||
| Write down of inventories | 268 | — | — | |||||||||
| Accretion of interest expense | 101 | 422 | 625 | |||||||||
| Accretion of available-for-sale securities | — | (15 | ) | (13 | ) | |||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Accounts receivable | (2,479 | ) | 494 | (404 | ) | |||||||
| Due from related party | 250 | (250 | ) | — | ||||||||
| Prepaid expenses and other current assets | (91 | ) | 143 | (17 | ) | |||||||
| Deferred inventory costs | — | — | 102 | |||||||||
| Inventories | (1,638 | ) | (216 | ) | (346 | ) | ||||||
| Deposits and other assets | (1 | ) | 7 | 56 | ||||||||
| Accounts payable, accrued expenses and other liabilities | 1,817 | 1,530 | (380 | ) | ||||||||
| Customer deposits | 93 | — | — | |||||||||
| Deferred revenue | (198 | ) | 72 | 73 | ||||||||
| Net cash used in operating activities | (32,358 | ) | (27,594 | ) | (27,856 | ) | ||||||
| Investing activities | ||||||||||||
| Purchases of available-for-sale securities | — | — | (22,766 | ) | ||||||||
| Maturities of available-for-sale securities | — | 20,553 | 2,241 | |||||||||
| Collection of notes receivable | 71 | 65 | — | |||||||||
| Purchase of property and equipment | (245 | ) | (531 | ) | (268 | ) | ||||||
| Net cash provided by (used in) investing activities | (174 | ) | 20,087 | (20,793 | ) | |||||||
| Financing activities | ||||||||||||
| Proceeds from issuance of common stock, net of offering costs | 44,611 | — | 44,392 | |||||||||
|
Proceeds from issuance of long term debt and warrants, net of deferred
financing costs and discounts |
— | — | (50 | ) | ||||||||
| Proceeds from exercise of stock options | 65 | 72 | 76 | |||||||||
| Payment for common stock repurchase and retirement | — | (741 | ) | — | ||||||||
| Payments for withholding taxes on stock option exercises | — | (410 | ) | (131 | ) | |||||||
| Payments on capital lease obligation | (13 | ) | — | — | ||||||||
| Payments on debt | (3,856 | ) | (4,373 | ) | (2,022 | ) | ||||||
| Net cash provided by (used in) financing activities | 40,807 | (5,452 | ) | 42,265 | ||||||||
| Net increase (decrease) in cash and cash equivalents | 8,275 | (12,959 | ) | (6,384 | ) | |||||||
| Cash and cash equivalents at beginning of period | 9,183 | 22,142 | 28,526 | |||||||||
| Cash and cash equivalents at end of period | $ | 17,458 | $ | 9,183 | $ | 22,142 | ||||||
| Supplemental Cash Flow Information | ||||||||||||
| Transfer from inventories to property and equipment in the field | $ | 812 | $ | — | $ | 587 | ||||||
| Interest paid | $ | 157 | $ | 652 | $ | 976 | ||||||
| Assets acquired under capital lease | $ | 164 | $ | — | $ | — | ||||||
| The accompanying notes are an integral part of the consolidated financial statements. | ||||||||||||
F- 6
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
| 1. | Nature of Operations |
The Company
Corindus Vascular Robotics, Inc. (the “Company”), formerly named Your Internet Defender, Inc. (“YIDI”), acquired Corindus, Inc., a privately-held company, in a reverse acquisition on August 12, 2014 (the “Acquisition”). The Company was previously a Nevada corporation, but effective June 28, 2016, the Company changed its state of incorporation from the State of Nevada to the State of Delaware. The Company’s corporate headquarters and research and development facility are in Waltham, Massachusetts and the Company is engaged in the design, manufacture and sale of precision vascular robotic-assisted systems (“CorPath System”) for use in interventional vascular procedures.
Since its inception on March 21, 2002, the Company has devoted its efforts principally to research and development, business development activities and raising capital. In July 2012, the Company received 510(k) clearance from the United States Food and Drug Administration (“FDA”) for the CorPath 200 System and initiated a limited commercial launch in the U.S. In 2013, the Company moved into the growth stage, investing in sales and marketing in order to build the customer base. While the Company’s device is initially cleared for and targeting percutaneous coronary intervention (“PCI”) and peripheral vascular procedures, the Company believes its technology platform has the capability to be developed in the future for other segments of the vascular intervention market, including neurointerventional and other more complex cardiac interventions such as structural heart procedures.
In October 2015, the Company received 510(k) clearance from the FDA for its robotic-assisted CorPath 200 System to be used during PCI procedures performed via radial access. The 510(k) clearance was based on results of a clinical trial conducted at Spectrum Health, Grand Rapids, Michigan, and St. Joseph’s Hospital Health Center, Syracuse, New York.
In March 2016, the Company received 510(k) clearance from the FDA for its robotic-assisted CorPath 200 System for use in peripheral vascular interventions. The 510(k) clearance for peripheral intervention was based on results of a clinical trial conducted at Medical University of Graz in Graz, Austria.
In October 2016, the Company received 510(k) clearance from the FDA for its CorPath GRX, the second generation of its CorPath System and in February 2018, the Company received 510(k) clearance from the FDA for CorPath GRX to be used during peripheral vascular interventions. In March 2018, the Company received 510(k) clearance from the FDA for the first automated robotic movement designed for the CorPath GRX platform. The CorPath GRX System is intended for use in the remote delivery and manipulation of guidewires and rapid exchange catheters, and remote manipulation of guide catheters during percutaneous coronary and vascular procedures. The Company began commercial shipment of the CorPath GRX in late January 2017.
The Company’s future capital requirements will depend upon many factors, including progress with developing, manufacturing and marketing its technologies, the time and costs involved in preparing, filing, prosecuting, maintaining and enforcing patent claims and other proprietary rights, its ability to establish collaborative arrangements, marketing activities and competing technological and market developments, including regulatory changes affecting medical procedure reimbursement, and overall economic conditions in the Company’s target markets.
Liquidity
On March 16, 2018, the Company closed on a private placement of convertible preferred stock for gross proceeds of $25,000. The preferred stock is convertible into an aggregate of 20,000,000 shares of common stock, and is entitled to receive non-compounding dividends in additional shares of preferred stock, at the rate of 12% per annum, subject to reduction in the event certain milestones are achieved. The preferred stock purchasers were also issued warrants to purchase an aggregate of 8,750,000 shares of common stock at an exercise price of $1.40 per share, exercisable either for cash or on a cashless basis.
On March 16, 2018, the Company completed a financing arrangement with two lenders which provides for borrowings of up to $26 million in the form of up to $23 million in term loans and up to a $3 million revolving line-of-credit through March 2022. As of March 16, 2018, the Company had $12 million in principal outstanding under the term loan facility and $0 in principal outstanding under the revolving loan facility. An additional $5.5 million in term loans may become available in the future provided the Company has achieved a specified gross profit milestone prior to January 1, 2019, and an additional $5.5 million may become available provided the Company receives net cash proceeds of $30 million from a future sale of the Company's equity securities prior to July 1, 2019 and achieves a specified gross profit milestone prior to September 1, 2019. Until such time that the Company achieves the specified criteria, the additional term loans are not available to the Company. The Company cannot assure you that the Company will achieve the gross profit or equity financing milestones that will trigger the Company's ability to further draw the term loan facility. The revolving line-of-credit also has various clauses which restrict its availability and for which the Company currently does not meet such restrictions. The outstanding term loans bear interest at a floating rate per annum equal to the greater of (i) 8.83% and (ii) the sum of (a) the one month ICE Benchmark LIBOR based on U.S. Dollar deposits, plus (b) 7.25%. The outstanding principal under the revolving line bears interest at a floating rate per annum equal to the greater of (i) 5.0% and (ii) the sum of (a) the “prime rate,” as reported in the Wall Street Journal, plus (b) 0.5%, which interest is payable monthly. Both loan facilities are secured by substantially all of the Company's personal property other than the Company's intellectual property. Both loan facilities include customary affirmative and negative covenants. Upon the earlier of the second advance under the term loan facility or the first advance under the revolving loan facility, the Company must also achieve minimum revenue on a monthly basis measured against a percentage of the Company's Board of Directors-approved projections for the applicable fiscal year. The Company's failure to satisfy the revenue, or any other, covenant could result in an event of default under the loan facilities. Both loan facilities also include other events of default, the occurrence and continuation of which could cause interest to be charged at the rate that is otherwise applicable plus 5.0% and would provide the collateral agent under the term loan facility or the lender under the revolving loan facility, as applicable, with the right to exercise remedies against the Company and the collateral securing the loan facilities. These events of default include, among other things, any failure by the Company to pay principal or interest due under the loan facilities, a breach of certain covenants under the loan facilities, the Company's insolvency, a material adverse change, the occurrence of any default under certain other indebtedness in an amount greater than $0.25 million, one or more judgments against the Company in an amount greater than $0.25 million individually or in the aggregate, and any default under the other loan facility.
F- 7
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
The Company has incurred losses since inception and has funded its cash flow deficits primarily through the issuance of capital stock and debt. As of December 31, 2017, the Company had an accumulated deficit of $180,841. As of December 31, 2017, the Company had cash and cash equivalents of $17,458 and working capital of $16,429. The Company anticipates that these available resources and the additional financings discussed above will be sufficient to meet the Company’s cash requirements for at least the next 12 months from March 19, 2018. However, as the Company continues to incur losses and cash flow deficits, its transition to profitability is dependent upon achieving a level of revenues adequate to support its cost structure, but the Company will otherwise rely on additional capital funding until such time as that is achieved.
As such, the Company has evaluated whether or not its cash and cash equivalents on hand and the cash proceeds from the financing activities described above would be sufficient to sustain projected operating activities through March 19, 2019 as required by Accounting Standards Codification (ASC) 205-40 Disclosure of Uncertainties About an Entity’s Ability to Continue as a Going Concern. Based on the Company’s current forecasts of annual cash flow deficits, which are estimated to be approximately $37,000 to $39,000 per year, the Company will have cash through approximately May 2019.
Due to the inherent uncertainty in predicting revenues and certain variable costs, management has considered its ability to reduce cash flow deficits and has determined that if it does not achieve its revenue forecast, it would undertake the following activities to reduce its cash flow deficits:
●
Defer or limit some or all of its spending on capital equipment that management believes is necessary to reduce manufacturing costs of cassettes and CorPath systems to be used in marketing and training activities that were otherwise planned for 2018;
●
Eliminate or defer the 2018 discretionary bonus payouts for all bonus eligible employees, including executive management;
●
Reduce spending on travel, clinical programs and prototypes;
●
Eliminate planned headcount additions in marketing, sales, and manufacturing; and
●
Reduce external consulting resources who otherwise would be used to assist in installations of CorPath systems.
As a result, management believes its plans can be effectively implemented as all of the actions are within the Company’s control and will be finalized and will be able to be effectively implemented, if required.
The Company may never achieve profitability, and unless and until doing so, the Company intends to fund future operations through additional non-dilutive or dilutive financings. There can be no assurances, however, that additional funding will be available on terms acceptable to the Company, if at all.
| 2. | Significant Accounting Policies |
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries, Corindus, Inc. and Corindus Security Corporation. All intercompany transactions and balances have been eliminated in consolidation. The functional currency of both wholly-owned subsidiaries is the U.S. dollar and, therefore, the Company has not recorded any currency translation adjustments.
In the fourth quarter of 2014, the Company participated in the formation of a not-for-profit, which was established to generate awareness of the health risks linked to the use of fluoroscopy in hospital catheterization. As of December 31, 2017, the Company’s Chief Executive Officer and one of its senior executives represented two of the four voting members of the board of directors of the entity. As a result, under the voting model used for the consolidation of related parties which are controlled by a company, the Company has consolidated the financial statements of the entity, and recognized expenses of $36, $123, and $386 for the years ended December 31, 2017, 2016 and 2015, respectively. The entity had assets and liabilities of $15 and $7, respectively, on its balance sheet at December 31, 2017 and had both assets and liabilities of $23 on its balance sheet at December 31, 2016.
Segment Information
The Company operates in one business segment, which is the development, marketing and sale of robotic-assisted vascular intervention devices. Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker in making decisions regarding resource allocation and assessing performance. To date, the chief operating decision maker has made such decisions and assessed performance at the company level, as one segment. The Company’s chief operating decision maker is the Chief Executive Officer.
Revenues from domestic customers were $6,694, $1,867 and $2,683 for the years ending December 31, 2017, 2016 and 2015, respectively. Revenues from international customers in Japan, Dubai, Israel, and Kuwait, were $2,956, $975 and $46 for the years ending December 31, 2017, 2016 and 2015, respectively.
F- 8
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
Use of Estimates
The process of preparing financial statements in conformity with accounting principles generally accepted in the United States (“U.S. GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of assets and liabilities at the date of the financial statements. Such management estimates include those relating to revenue recognition, inventory valuation, assumptions used in the valuation of stock-based awards, and valuation allowances against deferred income tax assets. Actual results could differ from those estimates.
Cash Equivalents
The Company considers highly liquid short-term investments, which consist of money market funds, with original maturity dates of three months or less at the date of purchase, to be cash equivalents. From time to time, the Company’s cash balances may exceed federal deposit insurance limits.
Marketable Securities
The Company determines the appropriate classification of marketable securities at the time of purchase and reevaluates such designation at each balance sheet date. The Company had classified all of its marketable securities during 2016 “available-for-sale” pursuant to ASC 320, Investments – Debt and Equity Securities. The Company records available-for-sale securities at fair value, with the unrealized gains and losses included in accumulated other comprehensive loss in stockholders’ equity.
The Company adjusts the cost of available-for-sale debt securities for amortization of premiums and accretion of discounts to maturity. The Company includes such amortization and accretion in interest and other expense. The cost of securities sold is based on the specific identification method. The Company includes interest income on securities classified as available-for-sale in interest and other expense.
The Company reviews marketable securities for other-than-temporary impairment whenever the fair value of a marketable security is less than the amortized cost and evidence indicates that a marketable security’s carrying amount is not recoverable within a reasonable period of time. Other-than-temporary impairments of investments are recognized in the consolidated statements of operations if the Company has experienced a credit loss, has the intent to sell the marketable security, or if it is more likely than not that the Company will be required to sell the marketable security before recovery of the amortized cost basis.
During 2016, the activity in the Company’s accumulated other comprehensive loss was composed solely of activity related to the Company’s available-for-sale securities. There were no realized gains or losses recognized on the maturity of available-for-sale securities during the year ended December 31, 2016, and as a result, the Company did not reclassify any amount out of accumulated other comprehensive loss during the year.
Fair Value Measurements
In accordance with ASC 820, Fair Value Measurements and Disclosures, the Company generally defines fair value as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date (exit price). The Company uses a three-tier fair value hierarchy, which classifies the inputs used in measuring fair values. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements), and the lowest priority to unobservable inputs (Level 3 measurements). The three levels of the fair value hierarchy are described below:
| ● | Level 1 – inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities that the reporting entity has the ability to access at the measurement date. |
| ● | Level 2 – inputs are inputs other than quoted prices included within Level 1 that are observable for the asset or liability, either directly or indirectly. If the asset or liability has a specified (contractual) term, a Level 2 input must be observable for substantially the full term of the asset or liability. |
F- 9
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
| ● | Level 3 – inputs are unobservable inputs for the asset or liability in which there is little, if any, market activity for the asset or liability at the measurement date. |
At both December 31, 2017 and 2016, the Company had only one asset, cash equivalents, that was measured at fair value on a recurring basis. The Company had cash equivalents totaling approximately $0 and $164 at December 31, 2017 and 2016, respectively, which were valued based on Level l inputs.
The Company’s financial instruments of deposits and notes receivable are carried at cost and approximate their fair values given the liquid nature of such items. The fair value of the Company’s capital lease obligation approximates its carrying value at December 31, 2017 due to its recent negotiation with lessor. The fair value of the Company’s long-term debt amounted to $3,759 at December 31, 2016 based on discounted cash flow analysis, which includes Level 3 inputs and fair value approximates recorded amounts.
Concentrations of Credit Risk and Significant Customers
The Company had the following customers that accounted for greater than 10% of its revenues for the years ended December 31, 2017, 2016 and 2015, respectively:
| For the Year ended December 31, | |||||||||||||
| Customer | 2017 | 2016 | 2015 | ||||||||||
| A | 20 | % | — | — | |||||||||
| B | 1 | % | 28 | % | — | ||||||||
| C | — | 13 | % | 5 | % | ||||||||
| D | — | 12 | % | — | |||||||||
| E | 1 | % | 1 | % | 13 | % | |||||||
| F | — | — | 11 | % | |||||||||
| G | — | 1 | % | 10 | % | ||||||||
The Company had five other customers that together accounted for 84% of the Company’s accounts receivable balance at December 31, 2017, but none of these customers exceeded 10% of its revenues in 2017. Given the current revenue levels, in a period in which the Company sells a system, that customer is likely to represent a significant customer.
The Company had four other customers that together accounted for 73% of the Company’s accounts receivable balance at December 31, 2016, but none of these customers exceeded 10% of its revenues in 2016.
Off-Balance Sheet Arrangements
The Company has no significant off-balance sheet risk such as foreign exchange contracts, option contracts, or other hedging arrangements.
Allowance for Doubtful Accounts
The Company evaluates the collectability of accounts receivable on a regular basis. The allowance for doubtful accounts, if any, is based upon various factors including the financial condition and payment history of customers, an overall review of collections experience on other accounts and economic factors or events expected to affect future collections experience. The Company’s accounts receivable consist primarily of amounts due from large, well-capitalized customers and while the Company reviews their creditworthiness, collectability is generally not an issue. The Company records an allowance for doubtful accounts, when necessary, based on the potential for collectability issues within the customer base. The Company’s allowance for doubtful accounts was $0 at December 31, 2017 and 2016.
F- 10
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
Product Warranty
Customers are permitted to return defective products under the Company’s standard product warranty program. For CorPath Systems, the Company’s standard one-year warranty provides for the repair of any product that malfunctions. Return and replacement can only occur if a material breach of the warranty remains uncured for 30 days. A roll-forward of the Company’s warranty liability is as follows:
| Balance at December 31, 2015 | $ | 68 | ||
| Provision for warranty obligations | 67 | |||
| Settlements | (78 | ) | ||
| Balance at December 31, 2016 | 57 | |||
| Provision for warranty obligations | 352 | |||
| Settlements | (113 | ) | ||
| Balance at December 31, 2017 | $ | 296 |
Inventories
Inventories are valued at the lower of cost or net realizable value using the first-in, first-out (FIFO) method. The Company routinely monitors the recoverability of its inventory and records the lower of cost or market reserves based on current selling prices and reserves for excess and obsolete inventory based on historical and forecasted usage, as required. Scrap and excess manufacturing costs are charged to cost of revenue as incurred and not capitalized as part of inventories. The Company only capitalizes pre-launch inventories when purchased for commercial use and it deems regulatory approval to be probable.
Property and Equipment
Property and equipment is carried at cost. Major items and betterments are capitalized; maintenance and repairs are charged to expense as incurred. The Company capitalizes certain costs incurred in connection with developing or obtaining internal-use software. Software costs that do not meet capitalization criteria are expensed as incurred. Demonstration equipment represents internally manufactured capital equipment that is used on-site at trade shows and at customer locations to demonstrate the CorPath System. Field equipment represents internally manufactured capital equipment placed at customer locations under programs that involve the customer’s agreement to provide their facility as a training/showsite for other potential customers while purchasing cassettes for their cases performed. As of December 31, 2017, the Company had placed three CorPath GRX field equipment units and one CorPath GRX for a customer’s evaluation purposes.
Depreciation on the demonstration equipment is charged to selling, general and administrative and the depreciation on the field equipment is charged to cost of revenue. Depreciation is computed under the straight-line method over the estimated useful lives of the respective assets.
Depreciation is provided over the following estimated asset lives:
| Machinery and equipment | 5 years |
| Computer equipment | 3 years |
| Office furniture and equipment | 5 years |
| Leasehold improvements | Shorter of life of lease or useful life |
| Vendor tooling | 1.5 - 3 years, based on planned usage |
| Software | 4 years |
| Demonstration equipment | 3 years |
| Field equipment | 3 years |
F- 11
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
Impairment of Long-Lived Assets
The Company’s long-lived assets principally consist of property and equipment. The Company continually monitors events and changes in circumstances that could indicate carrying amounts of long-lived assets may not be recoverable. An impairment loss is recognized when expected cash flows are less than an asset’s carrying value. Accordingly, when indicators of impairment are present, the Company evaluates the carrying value of such assets in relation to the operating performance and estimated future undiscounted cash flows of the underlying assets. The Company’s policy is to record an impairment loss when it is determined that the carrying amount of the asset may not be recoverable. During 2016, the undiscounted estimated cash flows from certain equipment placed at customer locations was less than the related equipment’s carrying value. As such, the Company recorded an impairment charge of $125 based on the difference between the estimated fair value of the equipment and its carrying value. The impairment charge of $88 and $37 is recorded within cost of revenues and selling, general and administrative, respectively, in the accompanying 2016 consolidated statement of operations. There were no other impairment charges or indicators of impairment for the years ended December 31, 2017, 2016 or 2015.
Comprehensive Loss
Comprehensive loss is comprised of net loss and changes in the unrealized gains and losses on marketable securities. Accumulated other comprehensive loss, a component of stockholders’ equity, is comprised of the cumulative unrealized gains and/or losses from the change in fair market value of the Company’s marketable securities. Accumulated other comprehensive loss was $0 as of both December 31, 2017 and 2016.
Revenue Recognition
The CorPath System is a capital medical device used by hospitals and surgical centers to perform heart catheterizations. Use of the CorPath System requires a sterile, single-use cassette (the “CorPath Cassette”), which is sold separately, for each procedure. Products are sold to customers with no rights of return. The Company recognizes revenue on the sale of products when the following criteria are met:
| ● | Persuasive evidence of an arrangement exists |
| ● | The price to the buyer is fixed or determinable |
| ● | Collectability is reasonably assured |
| ● | Risk of loss transfers and the product is delivered |
In each arrangement, the Company is responsible for installation of the CorPath System and initial user training, which services are deemed essential to the functionality of the system. Therefore, the Company recognizes system revenue when the CorPath System is delivered and installed, and accepted by the end user customer.
Each CorPath System is sold with a standard one year warranty, which provides that the CorPath System will function as intended and during that one year period, the Company will either replace the product or a portion thereof or provide the necessary repair service during the Company’s normal service hours. The Company accrues for the estimated costs of the warranty once the CorPath System is installed.
The Company generally enters into multiple element arrangements, which include the sale of a CorPath System with an initial order of CorPath Cassettes, and may include either a basic service plan or a premium service plan. The basic service plan provides for an extended warranty period and the premium service plan provides for the extended warranty as well as component upgrades, when and if they become available during the service period. Deliverables, which are accounted for as separate units of accounting under multiple-element arrangements include: (a) the CorPath System, including installation and initial training, which are subject to customer acceptance and (b) the initial shipment of CorPath Cassettes to the customer, and may include either (c) an extended warranty or (d) component upgrades.
The Company recognizes revenue on multiple-element arrangements in accordance with ASU 2009-13, Revenue Recognition (Topic 605): Multiple Deliverable Revenue Arrangements, based on the estimated selling price of each element. In accordance with ASU 2009-13, the Company uses vendor-specific objective evidence (“VSOE”), if available, to determine the selling price of each element. If VSOE is not available, the Company uses third-party evidence (“TPE”) to determine the selling price. If TPE is not available, the Company uses its best estimate to develop the estimated selling price (“BESP”). The Company uses BESP to determine the selling price of its systems as well as the basic and premium service plans. BESP is determined based on estimated costs plus a reasonable margin, and has generally been consistent with the price charged to the customer for such products and services. The determination of BESP also considers the price of the service plans charged to customers when such services are sold separately in subsequent transactions. The Company also uses BESP to determine the selling price of the initial order of cassettes, which considers the price at which it charges its customers when the cassettes are sold separately.
F- 12
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
Revenue related to basic and premium service plans is recognized on a straight-line basis over the life of the service contract. Revenue from cassettes and accessories is recorded upon delivery and services provided by the Company outside of a basic or premium service contract are recognized as the services are provided. If a revenue arrangement contains an undelivered element, such as a specified upgrade, revenues are deferred until delivery is complete.
There are no performance, cancellation, termination, and refund-type provisions under the Company’s multiple element arrangements.
At times, the Company may provide products to customers in exchange for future purchases of products and provision of services. The costs of any such products are included in cost of revenue in the accompanying consolidated statements of operations.
The Company also uses a One-Stent Program to demonstrate its confidence in the CorPath System’s ability to help accurately measure anatomy and precisely place only one stent per lesion. The Company provides eligible customers registered under the program a $1 credit against future CorPath Cassette purchases for a qualifying CorPath PCI procedure which uses more than one stent per lesion. The estimated cost of honoring the potential obligation under the program is recorded as a reduction of revenue at the time of shipment. These costs have not been significant to date.
The Company records shipping and handling costs as a selling expense in the period incurred, and records payments from customers for shipping costs as a reduction of selling expenses. Such amounts have not been material in the periods presented.
Research and Development
Costs for research and development are expensed as incurred. Research and development expense consists primarily of salaries, salary-related expenses and costs of contractors and materials.
Income Taxes
The Company accounts for income taxes using the liability method, whereby deferred tax asset and liability account balances are determined based on differences between financial reporting and tax basis of assets and liabilities and are measured using the enacted tax rates that will be in effect when the differences are expected to reverse. The Company provides a valuation allowance, if necessary, to reduce deferred tax assets to amounts that are realizable.
The Company accounts for uncertain tax positions using a “more-likely-than-not” threshold for recognizing and resolving uncertain tax positions. The evaluation of uncertain tax positions is based on factors including, but not limited to, changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, the effective settlement of matters subject to audit, new audit activity and changes in facts or circumstances related to a tax position. The Company evaluates these tax positions on an annual basis. The Company also accrues for potential interest and penalties related to unrecognized tax benefits in income tax expense.
F- 13
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
The Company recognizes the tax benefit of tax positions to the extent that the benefit will more likely than not be realized. The determination as to whether the tax benefit will more likely than not be realized is based upon the technical merits of the tax position as well as consideration of the available facts and circumstances.
Stock-Based Compensation
The Company recognizes compensation costs resulting from the issuance of service stock-based awards to employees and directors as an expense in the consolidated statements of operations over the requisite service period based on a measurement of fair value for each stock award. The awards issued to date have primarily been stock options with service-based vesting periods over two or four years, restricted stock units with service-based vesting periods of one year, and shares of unrestricted common stock. During 2017, certain shares of unrestricted common stock were granted to a non-employee director of the Company which were fully vested upon grant. Accordingly, compensation expense related to unrestricted stock was recognized on the date of the grant. During 2016, the Company issued certain stock-based awards that contain both performance and service-based vesting conditions which vest over periods of up to 25 months. The Company records expense on these awards when it becomes probable that the performance condition and requisite service will be met. The Company recognizes compensation costs resulting from the issuance of stock-based awards to non-employees as an expense in the consolidated statements of operations over the service period based on a measurement of fair value for each stock award at each performance date and period end.
Prior to the Company’s initial public offering, the fair value of the Common Stock was obtained from quoted market prices on the OTCQB as provided by OTC Market Groups, Inc. In connection with the public offering in May 2015, the Company’s Common Stock was approved for listing on the NYSE American, where it commenced trading under the symbol “CVRS”. As such, subsequent to this, the Company utilizes quoted market prices to calculate fair value of stock-based awards.
The following assumptions were used to estimate the fair value of stock options granted using the Black-Scholes-Merton option-pricing model (“Black-Scholes Model”).
| For the Year ended December 31, | ||||||
| 2017 | 2016 | 2015 | ||||
| Risk-free interest rate | 1.87-2.38% | 1.27-2.45% | 1.54-1.97% | |||
| Expected term in years | 6.00-10.00 | 6.08-10.00 | 6.08 | |||
| Expected volatility | 55-67% | 48-64% | 50% | |||
| Expected dividend yield | 0% | 0% | 0% | |||
The risk-free interest rate assumption is based upon observed U.S. government treasury interest rates with a term that is consistent with the expected term of the Company’s employee stock options. The expected term is based on the average of the vesting period and contractual term of the Company’s options given the lack of historical data available. The Company does not pay a dividend, and is not expected to pay a dividend in the foreseeable future.
Due to a lack of a public market for the Company’s Common Stock for an extended period of time, the Company utilized comparable public companies’ volatility rates as a proxy of its expected volatility for purposes of the Black-Scholes Model. Stock-based compensation expense is recorded net of estimated forfeitures and is adjusted periodically for actual forfeitures. The Company uses historical data to estimate forfeiture rates. For the years ended December 31, 2017, 2016 and 2015, forfeitures were estimated to be 5.0% each year.
F- 14
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
Recent Accounting Pronouncements
In May 2014, August 2015, April 2016 and May 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2014-09, Revenue from Contracts with Customers, ASU 2015-14, Revenue from Contracts with Customers, Deferral of the Effective Date, ASU 2016-10, Revenue from Contracts with Customers, Identifying Performance Obligations and Licensing, and ASU 2016-12, Revenue from Contracts with Customers, Narrow-Scope Improvements and Practical Expedients, respectively, (collectively referred to as “Topic 606”). Topic 606 outlines a single comprehensive model for entities to use in accounting for revenue arising from contracts with customers, and supersedes current revenue recognition guidance, including industry-specific guidance. It also requires entities to disclose both quantitative and qualitative information that enable financial statements users to understand the nature, amount, timing, and uncertainty of revenue and cash flows arising from contracts with customers. The amendments in these ASUs are effective for fiscal years, and interim periods within those years, beginning after December 15, 2017. The two permitted transition methods under the new standard are the full retrospective method, in which case the standard would be applied to each prior reporting period presented and the cumulative effect of applying the standard would be recognized at the earliest period shown, or the modified retrospective method, in which case the cumulative effect of applying the standard would be recognized at the date of initial application. The Company will adopt the guidance on January 1, 2018 and will apply the modified retrospective transition approach only to contracts that have undelivered performance obligations as of this date. The Company performed a review of all revenue streams to identify any differences in the timing, measurement or presentation of revenue recognition. The Company has an installed base of 62 CorPath Systems as of December 31, 2017. The Company assessed all of these systems, and determined that a large number of those installations will not be impacted by the implementation of Topic 606 since there will be no future performance obligations under those arrangements upon adoption. As a result, the Company identified a limited number of contracts for which there will be future performance obligations as of the adoption date. Pertaining to these contracts, the adoption of Topic 606 will result in certain components of services being unbundled from previously bundled arrangements, and result in an acceleration of the timing of revenue recognition related to the Company’s services. The new standard requires revenues to be estimated and recognized upon transfer of the promised goods and services, and the Company determined that the accelerated recognition of service revenues will result in a cumulative adjustment to revenues as of the adoption date. Adoption of the new standard will result in the capitalization of certain costs to obtain service contracts and the capitalization of such costs will result in an adjustment to capitalized expenses as of the adoption date. The Company is in the process of finalizing the overall impact of the adoption of this standard on its fourth quarter sales and will finalize in the first quarter of 2018. The Company’s calculations indicate that such amounts may not be material to the Company’s results of operations.
In February 2016, the FASB issued ASU 2016-02, Leases (Topic 842), which amends leasing accounting requirements. The new standard requires lessee recognition on the balance sheet of a right-of-use asset and a lease liability, initially measured at the present value of the lease payments. It further requires recognition in the income statement of a single lease cost, calculated so that the cost of the lease is allocated over the lease term generally on a straight-line basis. Finally, it requires classification of all cash payments within operating activities in the statement of cash flows. It is effective for fiscal years commencing after December 15, 2018 and early adoption is permitted. The Company is currently evaluating the impact of this standard on its consolidated financial statements.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows (Topic 230), Classification of Certain Cash Receipts and Cash Payments, which reduces diversity in how certain cash receipts and cash payments are presented and classified in the Consolidated Statements of Cash Flows. It is effective for fiscal years, and interim periods within those years, beginning after December 15, 2017 and will be required to be applied retrospectively, with early adoption permitted. The Company is currently evaluating the impact of this update on its consolidated financial statements and related disclosures.
In May 2017, the FASB issued ASU 2017-09, Compensation—Stock Compensation (Topic 718) - Scope of Modification Accounting, which clarifies when to account for a change to the terms or conditions of a share-based payment award as a modification. Under the new guidance, modification accounting is required only if the fair value, the vesting conditions, or the classification of the award (as equity or liability) changes as a result of the change in terms or conditions. It is effective prospectively for annual periods beginning on or after December 15, 2017, with early adoption permitted. The Company is currently evaluating the impact of this update on its consolidated financial statements and related disclosures.
F- 15
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
| 3. | Inventories |
The Company’s inventories are valued at the lower of cost or net realizable value using the FIFO method and consist of the following:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Raw material | $ | 945 | $ | 578 | ||||
| Work in progress | 310 | 163 | ||||||
| Finished goods | 848 | 804 | ||||||
| Total | $ | 2,103 | $ | 1,545 | ||||
The Company wrote down inventories by $268 during the year ended December 31, 2017 to properly state amounts at the lower of cost or net realizable value.
| 4. | Property and Equipment |
Property and equipment are stated at cost and are being depreciated using the straight-line basis over the assets’ estimated useful lives. Depreciation and amortization expense was $723, $725 and $706 for the fiscal years 2017, 2016 and 2015, respectively. Property and equipment consist of the following:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Machinery and equipment | $ | 546 | $ | 561 | ||||
| Computer equipment | 147 | 136 | ||||||
| Office furniture and equipment | 320 | 267 | ||||||
| Leasehold improvements | 62 | 45 | ||||||
| Vendor tooling | 898 | 942 | ||||||
| Software | 658 | 554 | ||||||
| Demonstration equipment | 593 | 717 | ||||||
| Field equipment | 819 | 1,004 | ||||||
| Construction in progress | — | 29 | ||||||
| 4,043 | 4,255 | |||||||
| Less accumulated depreciation and amortization | (2,591 | ) | (3,273 | ) | ||||
| Property and equipment net | $ | 1,452 | $ | 982 | ||||
| 5. | Accrued Expenses |
Accrued expenses consist of the following:
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Payroll and benefits | $ | 1,764 | $ | 751 | ||||
| Professional and consultant fees | 579 | 368 | ||||||
| Commissions | 542 | 433 | ||||||
| Warranty | 296 | 57 | ||||||
| Travel expense | 242 | 43 | ||||||
| Sales tax payable | 83 | 41 | ||||||
| Interest | — | 33 | ||||||
| Other | 131 | 68 | ||||||
| Total | $ | 3,637 | $ | 1,794 | ||||
F- 16
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
| 6. | Long-Term Debt |
On June 11, 2014, the Company entered into a Loan and Security Agreement pursuant to which the lender agreed to make available to the Company $10,000 in two separate $5,000 loans under secured promissory notes. The initial note was made on June 11, 2014 in an aggregate principal amount equal to $5,000 (the “Initial Promissory Note”) and was repayable in equal monthly installments of principal and interest over 27 months beginning on July 1, 2015. On December 31, 2014, the Company borrowed the additional $5,000 (the “Second Promissory Note”) under the Loan and Security Agreement. The Second Promissory note was also repayable in equal monthly installments of principal and interest over 27 months beginning on July 1, 2015.
In connection with the Initial Promissory Note, the Company issued the lender warrants to purchase 177,514 shares of the Company’s Common Stock at an exercise price of $1.41 per share. The fair value of the warrant issued to the lender was determined to be $230 at the date of issuance, and was recorded as a discount on the debt. Additionally, in connection with the Second Promissory Note, the Company issued the lender warrants to purchase 177,514 shares of the Company’s Common Stock at an exercise price of $1.41 per share. The fair value of the warrant issued to the lender was determined to be $619 at the date of issuance, and was recorded as a discount on the debt. The Company amortized the debt discount to interest expense over the term of the debt using the effective interest method.
All amounts due under the Loan and Security Agreements were fully repaid on October 2, 2017.
| 7. | Income Taxes |
There was no federal or state provision for income taxes for the years ended December 31, 2017, 2016 or 2015 due to the Company’s operating losses and a full valuation allowance on deferred income tax assets for all periods since inception. All of the Company’s loss before provision for income taxes is attributable to its United States operations.
The Company’s effective income tax rate differs from the statutory federal income tax rate as follows:
| Years ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Statutory U.S. federal rate | 34.0 | % | 34.0 | % | 34.0 | % | ||||||
| State income tax | 2.6 | 1.7 | 3.7 | |||||||||
| Permanent items | 0.3 | (0.1 | ) | (0.4 | ) | |||||||
| Other | 1.0 | — | (0.5 | ) | ||||||||
| Change in state tax rate | 0.1 | (0.6 | ) | 0.7 | ||||||||
| Deferred federal rate reduction (Effect of US tax reform) | (58.9 | ) | — | — | ||||||||
| Federal R&D credit | 1.0 | 1.1 | 1.3 | |||||||||
| State R&D and other credits | 0.5 | 0.5 | 0.5 | |||||||||
| Change in valuation allowance | 19.4 | (36.6 | ) | (39.3 | ) | |||||||
| Total expense (benefit) | -% | -% | -% | |||||||||
F- 17
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets and the related valuation allowance were as follows:
| Years ended December 31, | ||||||||
| 2017 | 2016 | |||||||
| Deferred income tax assets: | ||||||||
| Operating loss carryforwards | $ | 35,950 | $ | 42,130 | ||||
| Start-up expenditures | 1,404 | 2,351 | ||||||
| Property and equipment | 40 | 77 | ||||||
| Stock-based compensation expense | 1,420 | 1,197 | ||||||
| Research and development credit carryforwards | 2,928 | 2,258 | ||||||
| Accrued expenses and other | 628 | 695 | ||||||
| Total deferred income tax assets | 42,370 | 48,708 | ||||||
| Valuation allowance | (42,370 | ) | (48,708 | ) | ||||
| Net deferred income tax assets | $ | — | $ | — | ||||
On December 22, 2017, the SEC staff issued Staff Accounting Bulletin No. 118, Income Tax Accounting Implications of the Tax Cuts and Jobs Act (“SAB 118”), which allows the recording of provisional amounts during a measurement period not to extend beyond one year of the enactment date. In accordance with SAB 118, the Company has determined that its deferred tax asset value and associated valuation allowance reduction of $20,104 is a provisional amount and a reasonable estimate at December 31, 2017. The final impact may differ from this provisional amount due to, among other things, changes in interpretations and assumptions the Company has made thus far and the issuance of additional regulatory or other guidance. The Company expects to complete the final impact within the measurement period.
The Company has provided a full valuation allowance against the deferred income tax assets, since it has a history of losses, which are all attributable to the U.S. and currently does not have enough positive evidence required under U.S. GAAP to reverse its valuation allowance. Management does not believe it is more likely than not that its deferred tax assets relating to the loss carryforwards and other temporary differences will be realized in the future. For the year ended December 31, 2016, the valuation allowance increased by $12,121, resulting principally from increased operating loss carryforward. For the year ended December 31, 2017, the valuation allowance decreased by $6,338, resulting principally from the change in deferred federal tax rate from 34% to 21% due to US tax reform.
At December 31, 2017, the Company had U.S. federal and state net operating loss carryforwards of approximately $147,795 and $91,631, respectively, that can be carried forward and offset against future taxable income. The Federal net operating loss carryforwards will begin to expire in 2028 and the state NOL carryforwards expire in various amounts, but none before 2022.
Effective as of January 1, 2017, the Company adopted a change in accounting policy in accordance with ASU 2016-09 to account for excess tax benefits and tax deficiencies as income tax expense or benefit, treated as discrete items in the reporting period in which they occur, and to recognize previously unrecognized deferred tax assets that arose directly from (or the use of which was postponed by) tax deductions related to equity compensation in excess of compensation recognized for financial reporting. The recognition of the federal and state excess tax benefit net operating losses increased the net operating loss deferred tax asset by $438. No prior periods were restated as a result of this change in accounting policy as the Company maintains a valuation allowance against its deferred tax assets, which also increased by $438 after adoption.
The Company also had federal and state tax credits of approximately $1,935 and $1,256 at December 31, 2017, respectively, which may be used to offset future tax liabilities. These tax credit carryforwards will expire at various times beginning in 2029 for federal purposes and 2018 for state purposes.
F- 18
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
Utilization of net operating losses and tax credit carryforwards may be subject to a substantial annual limitation due to the “change in ownership” provisions of the Internal Revenue Code of 1986, and similar state provisions. The annual limitation may result in the expiration of net operating losses before utilization. Through December 31, 2017, the Company has completed several financings since its inception which it believes has not resulted in any changes in ownership as defined by Sections 382 and 383 of the Internal Revenue Code. If the financings caused an “ownership change”, generally defined as a greater than 50 percent change (by value) in its equity ownership over a three-year period, the Company’s attributes may be subject to an annual limitation. Subsequent ownership changes may further affect the limitation in future years.
Significant judgment is required in evaluating the Company’s tax positions and in determining the Company’s provision for income taxes. In the ordinary course of business, there are many transactions and calculations for which the ultimate tax determination is uncertain. As of December 31, 2017, the Company was not under audit in any tax jurisdiction. The U.S. statute of limitations will remain open to examination by the tax authorities until the utilization of net operating loss carryforwards. The Company accrues interest and penalties, if any, related to unrecognized tax benefits in income tax expense.
| 8. | Stockholders’ Equity |
The Company is authorized to issue 250,000,000 shares of common stock. Holders of Common Stock are entitled to vote on all matters and are entitled to the number of votes equal to the number of common shares held. Holders of Common Stock shall be entitled to receive dividends when and if declared by the Board of Directors. No dividends have been declared to date. In certain events, including the liquidation, dissolution or winding up of the Company, the remaining assets of the Company shall be distributed ratably among the holders of Common Stock.
The Company is authorized to issue 10,000,000 shares of preferred stock. As of December 31, 2017 and 2016, the Company had no shares of preferred stock issued or outstanding.
At December 31, 2017, there were 18,613,366 shares of common stock reserved for the potential exercise of warrants (355,028), stock options (18,215,932), and restricted stock units (42,406) and 3,752,598 shares that are available for grant under the 2014 Stock Award Plan.
| 9. | Stock-Based Compensation |
In connection with the Acquisition, Corindus exchanged options to purchase shares of its Common Stock for YIDI’s options to purchase shares of YIDI’s Common Stock (the “Replacement Plan Options”). The 2014 Stock Award Plan is the replacement plan for options previously awarded under the Corindus, Inc. 2006 Umbrella Option Plan and the Corindus, Inc. 2008 Stock Incentive Plan and is the plan under which all future Company options will be issued. The 2014 Stock Award Plan was limited to award issuances which in the aggregate could not exceed 9,035,016 shares, all of which shares will be used for the issuance of the Company stock-based awards, including options to purchase common stock, restricted stock and restricted stock units. Replacement Plan Options are exercisable for up to ten years from the date of original vesting commencement date of the options.
On April 30, 2015, the Company’s Board of Directors and shareholders owning a majority of the Company’s outstanding shares of common stock approved an increase in the authorized shares of common stock under the 2014 Stock Award Plan from 9,035,016 shares to 18,661,856 shares. On June 22, 2017, the stockholders of the Company approved an amendment to the Company’s Amended and Restated 2014 Stock Award Plan to increase the number of shares of common stock available for issuance under the plan by 4,038,144 shares, from 18,661,856 shares to 22,700,000 shares (the “Plan Amendment”). The Plan Amendment was previously adopted by the Company’s Board of Directors, subject to stockholder approval, and became effective upon the receipt of stockholder approval at the Company’s Annual Meeting.
F- 19
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
Awards of 2,293 shares of unrestricted common stock granted to a non-employee director in 2017 were valued at the closing price of the Company’s common stock at the date of grant and were fully vested on the date of grant. Stock-based compensation awards of 68,768 shares of restricted stock units granted to non-employee directors in 2017. Upon vesting, the Company issues shares of common stock for its restricted stock units. The shares issues have a required holding period of 36 months from the date of grant of the restricted stock units. As a result, the Company has valued the restricted stock units based on the closing price of the Company’s common stock on the date of grant less a discount for lack of marketability during the holding period of 20%. The 20% discount was calculated based on various studies quantifying discounts for lack of marketability with similar metrics to the Company. The factors considered in the discount for lack of marketability analysis include market value of equity, holding period, total revenue, total assets, volatility, total shareholders’ equity, and net income. Restricted stock studies are a source of public information on the discount for lack of marketability as the focus of these studies is the purchase of restricted securities. Restricted securities are shares issued and sold by a publicly traded company without prior registration with the Securities and Exchange Commission. Because of the restriction on the marketability of the securities, companies purchase the securities at prices lower than the price of a registered security of the same company. The difference between the two prices represents the discount for the lack of marketability. The related compensation expense is being amortized over the twelve month vesting period. Compensation costs recognized related to these awards totaled $27 in 2017 and was included in selling, general and administrative in the accompanying consolidated statement of operations, with remaining unrecognized compensation expense of $33 to be recognized over the remaining vesting period of approximately 6.6 months. Restricted stock units of 26,362 vested and were issued during 2017 leaving 42,406 to be issued upon vesting in 2018.
A summary of the activity under the Company’s stock option plans is as follows:
| Options | Weighted Average Exercise Price |
Weighted Average Remaining Contractual Term/Years |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding at December 31, 2016 | 17,523,072 | $ | 1.30 | 8.34 | $ | 511 | ||||||||||
| Granted | 2,020,933 | $ | 1.44 | |||||||||||||
| Exercised | (261,670 | ) | $ | 0.25 | ||||||||||||
| Canceled | (1,066,403 | ) | $ | 2.05 | ||||||||||||
| Outstanding at December 31, 2017 | 18,215,932 | $ | 1.29 | 7.64 | $ | 1,830 | ||||||||||
| Exercisable at December 31, 2017 | 9,807,397 | $ | 1.22 | 6.82 | $ | 1,624 | ||||||||||
| Vested and expected to vest at December 31, 2017 | 17,795,505 | $ | 1.29 | 7.62 | $ | 1,824 | ||||||||||
The fair value of employee options is estimated on the date of each grant using the Black-Scholes Model. The weighted-average grant date fair value of options granted during the year ended December 31, 2017, 2016 and 2015 were $0.81, $0.65 and $1.80, respectively. As of December 31, 2017, there was approximately $5,659 of unrecognized compensation cost related to non-vested stock-based compensation arrangements under the 2014 Stock Award Plan. That cost was expected to be recognized over a weighted-average period of 2.51 years.
The total intrinsic value of options exercised in 2017 was $251.
Stock-based compensation expense was allocated based on the employees’ function as follows:
| Years ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Research and development | $ | 258 | $ | 170 | $ | 74 | ||||||
| Selling, general and administrative | 2,634 | 2,196 | 431 | |||||||||
| $ | 2,892 | $ | 2,366 | $ | 505 | |||||||
F- 20
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
| 10. | Warrants to Purchase Common Stock |
The Company issued warrants to purchase 355,028 shares of the Company’s common stock at $1.41 per share in connection with its outstanding borrowing arrangement as described in Note 6.
The table below is a roll-forward of the Company’s warrant activity for the year ended December 31, 2017:
| Number of Warrants |
Weighted- Average Exercise Price |
||||||||
| Outstanding at December 31, 2016 | 5,083,219 | $ | 1.08 | ||||||
| Granted | — | — | |||||||
| Exercised | (4,728,191 | ) | 1.06 | ||||||
| Expired | — | — | |||||||
| Outstanding at December 31, 2017 | 355,028 | $ | 1.41 | ||||||
On October 11, 2017, a warrant to purchase 4,728,191 shares of Common Stock held by Koninklijke Philips NV (“Philips”) was automatically exercised on a net exercise basis in connection with its expiration. Upon expiration of the warrant, Philips paid the exercise price of $1.06 per share through the Company’s withholding of 3,334,586 of the warrant shares to pay the exercise price and issuing 1,393,605 shares of Common Stock to Philips. The shares were issued in reliance on an exemption from registration under Section 4(a)(2) of the Securities Act of 1933, as amended.
The Company has 355,028 warrants outstanding at December 31, 2017 with an exercise price of $1.41 and an expiration date of May 28, 2020.
| 11. | Related Party Transactions |
Philips Medical Systems, Nederland B.V.
On January 21, 2011, the Company entered into a distributor agreement with Philips appointing Philips to be the sole distributor for the promotion and sale of the Company’s CorPath System. The agreement was terminated on August 7, 2014. The Company may continue to sell CorPath Systems through Philips on a sale by sale basis under a non-exclusive arrangement under mutually agreeable terms, which may include a continued level of discounted pricing, until such time the Company either executes a new distribution arrangement with Philips or the Company no longer does business with Philips.
There were no revenues from or shipments to Philips during the year ended December 31, 2017. For the years ended December 31, 2016 and 2015, the Company recorded revenues of $375 and $125, respectively, from shipments to Philips under the distributor agreement. There were no amounts outstanding from Philips at December 31, 2017 resulting from selling activity under the agreement. At December 31, 2016, Philips owed the Company $250 resulting from selling activity under the agreement.
On October 11, 2017, a warrant to purchase 4,728,191 shares of Common Stock held by Philips was automatically exercised on a net exercise basis in connection with its expiration. Upon expiration of the warrant, Philips paid the exercise price of $1.06 per share through the Company’s withholding of 3,334,586 of the warrant shares to pay the exercise price and issuing 1,393,605 shares of Common Stock to Philips. The shares were issued in reliance on an exemption from registration under Section 4(a)(2) of the Securities Act of 1933, as amended.
Shareholder Loans
On June 14, 2010, the Company loaned funds to certain stockholders of the Company for tax payments to be made to the Israel Tax Authority in connection with a tax ruling related to a reorganization that took place in 2008 and the Company received non-interest bearing notes receivable, which documented such loans. The total amount of notes receivable issued was $145. The notes receivable were repayable upon the disposition of the Company’s common stock by the existing shareholder. Notes receivable amounted to $71 at December 31, 2016 and was included in prepaid expenses and other current assets in the accompanying consolidated balance sheet. The notes receivable were collected during 2017.
F- 21
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
Private Placement
On March 15, 2017, the Company closed on a private placement for the sale of an aggregate of 68,055,700 shares of its common stock at $0.6616 per share, for an aggregate purchase price of approximately $45,026 before deducting offering expenses of approximately $415. The financing round involved a syndicate of top-tier healthcare investors. Existing key investors who participated in this financing included HealthCor Partners Management, the Company’s largest shareholder, Royal Philips, an affiliate of Philips, and Energy Capital. New investors whose participation in this financing resulted in beneficial ownership of greater than 5% of the Company’s common stock included Consonance Capital and Hudson Executive Capital, L.P.
| 12. | Commitments and Contingencies |
The Company has an operating lease for approximately 26,402 square feet at its corporate headquarters and manufacturing plant in Waltham, Massachusetts, which expires in January 2021. The lease terms include escalating rent payments over the life of the lease and rent expense is recognized over the life of the lease on a straight-line basis. The difference between the amount expensed and actual rent payments is recorded as deferred rent included within other liabilities in the consolidated balance sheets. In connection with the lease, the Company is required to maintain a security deposit with its landlord. The security deposit is approximately $134 at December 31, 2017 and 2016, and is included in deposits and other assets in the accompanying consolidated balance sheets. The Company has a capital lease covering office furniture and carpeting, which expires in November 2020.
Total rent expense was $661, $597, and $574 for the years end December 31, 2017, 2016 and 2015, respectively. At December 31, 2017, the Company’s future minimum lease payments are indicated below:
|
For the Year Ended December 31, |
Operating |
Capital |
|||||||
| 2018 | $ | 577 | $ | 67 | |||||
| 2019 | 648 | 67 | |||||||
| 2020 | 665 | 48 | |||||||
| 2021 | 55 | — | |||||||
| $ | 1,945 | 182 | |||||||
| Less amount representing interest | 31 | ||||||||
| Minimum lease payments | 151 | ||||||||
| Less current portion of obligation under capital lease | 49 | ||||||||
| Long-term obligation under capital lease | $ | 102 | |||||||
Assets held under the capital lease arrangement and the related accumulated amortization totaled $164 and $6, respectively, at December 31, 2017. Amortization of property under the capital lease is included in depreciation and amortization in the accompanying financial statements.
F- 22
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
| 13. | Net Loss per Share |
Basic net loss per share is computed by dividing net loss by the weighted average shares of common stock outstanding for each period. Diluted net loss per share is the same as basic net loss per share since the Company has net losses for each period presented. The following common stock equivalents were excluded from the calculation of diluted net loss per share for the periods indicated because including them would have had an anti-dilutive effect:
| For the Year ended December 31, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Options to purchase common stock | 18,215,932 | 17,523,072 | 8,778,503 | |||||||||
| Warrants to purchase common stock | 355,028 | 5,083,219 | 5,207,379 | |||||||||
| Restricted stock units | 42,406 | — | — | |||||||||
| Total | 18,613,366 | 22,606,291 | 13,985,882 | |||||||||
| 14. | 401(k) Plan |
The Company has a tax-qualified employee savings and retirement 401(k) plan, covering all qualified employees. Participants may elect a salary deferral up to the statutorily prescribed annual limit for tax-deferred contributions. The Company matches 100% of the participant’s first 3% of eligible contributions plus 50% of the participant’s next 2% of contributions. Amounts expensed related to this plan totaled $392, $254 and $215 in 2017, 2016 and 2015, respectively.
| 15. | Selected Quarterly Financial Data (Unaudited) |
The following table presents unaudited operating results for each of the Company’s quarters in the years ended December 31, 2017 and 2016:
| Fiscal Year 2017 Quarters | ||||||||||||||||||||
| First | Second | Third | Fourth | Year | ||||||||||||||||
| Revenue | $ | 777 | $ | 2,258 | $ | 2,425 | $ | 4,190 | $ | 9,650 | ||||||||||
| Cost of revenue | 1,892 | 2,200 | 2,183 | 2,990 | 9,265 | |||||||||||||||
| Gross profit (loss) | (1,115 | ) | 58 | 242 | 1,200 | 385 | ||||||||||||||
| Operating expenses | 8,636 | 8,395 | 8,081 | 9,182 | 34,294 | |||||||||||||||
| Operating loss | (9,751 | ) | (8,337 | ) | (7,839 | ) | (7,982 | ) | (33,909 | ) | ||||||||||
| Total other expense, net | (134 | ) | (77 | ) | (14 | ) | 11 | (214 | ) | |||||||||||
| Net loss | $ | (9,885 | ) | $ | (8,414 | ) | $ | (7,853 | ) | $ | (7,971 | ) | $ | (34,123 | ) | |||||
| Net loss per share – basic and diluted | $ | (0.07 | ) | $ | (0.04 | ) | $ | (0.04 | ) | $ | (0.04 | ) | $ | (0.20 | ) | |||||
| Fiscal Year 2016 Quarters | ||||||||||||||||||||
| First | Second | Third | Fourth | Year | ||||||||||||||||
| Revenue | $ | 1,108 | $ | 508 | $ | 688 | $ | 538 | $ | 2,842 | ||||||||||
| Cost of revenue | 1,078 | 1,114 | 1,220 | 1,630 | 5,042 | |||||||||||||||
| Gross profit (loss) | 30 | (606 | ) | (532 | ) | (1,092 | ) | (2,200 | ) | |||||||||||
| Operating expenses | 7,275 | 6,772 | 7,313 | 8,517 | 29,877 | |||||||||||||||
| Operating loss | (7,245 | ) | (7,378 | ) | (7,845 | ) | (9,609 | ) | (32,077 | ) | ||||||||||
| Total other expense, net | (382 | ) | (216 | ) | (221 | ) | (182 | ) | (1,001 | ) | ||||||||||
| Net loss | $ | (7,627 | ) | $ | (7,594 | ) | $ | (8,066 | ) | $ | (9,791 | ) | $ | (33,078 | ) | |||||
| Net loss per share – basic and diluted | $ | (0.06 | ) | $ | (0.06 | ) | $ | (0.07 | ) | $ | (0.08 | ) | $ | (0.28 | ) | |||||
Note: Quarterly net loss per share amounts may not sum to net loss per share for the year due to rounding.
F- 23
Corindus Vascular Robotics, Inc.
Notes to Consolidated Financial Statements
In thousands, except share and per share amounts
| 16. | Subsequent Events |
On March 16, 2018, the Company closed on a private placement of convertible preferred stock for gross proceeds of $25,000. The preferred stock is convertible into an aggregate of 20,000,000 shares of common stock, and is entitled to receive non-compounding dividends in additional shares of preferred stock, at the rate of 12% per annum, subject to reduction in the event certain milestones are achieved. The preferred stock purchasers were also issued warrants to purchase an aggregate of 8,750,000 shares of common stock at an exercise price of $1.40 per share, exercisable either for cash or on a cashless basis.
On March 16, 2018, the Company completed a financing arrangement with two lenders which provides for borrowings of up to $26 million in the form of up to $23 million in term loans and up to a $3 million revolving line-of-credit through March 2022. As of March 16, 2018, the Company had $12 million in principal outstanding under the term loan facility and $0 in principal outstanding under the revolving loan facility. An additional $5.5 million in term loans may become available in the future provided the Company has achieved a specified gross profit milestone prior to January 1, 2019, and an additional $5.5 million may become available provided the Company receives net cash proceeds of $30 million from a future sale of the Company's equity securities prior to July 1, 2019 and achieves a specified gross profit milestone prior to September 1, 2019. Until such time that the Company achieves the specified criteria, the additional term loans are not available to the Company. The Company cannot assure you that the Company will achieve the gross profit or equity financing milestones that will trigger the Company's ability to further draw the term loan facility. The revolving line-of-credit also has various clauses which restrict its availability and for which the Company currently does not meet such restrictions. The outstanding term loans bear interest at a floating rate per annum equal to the greater of (i) 8.83% and (ii) the sum of (a) the one month ICE Benchmark LIBOR based on U.S. Dollar deposits, plus (b) 7.25%. The outstanding principal under the revolving line bears interest at a floating rate per annum equal to the greater of (i) 5.0% and (ii) the sum of (a) the “prime rate,” as reported in the Wall Street Journal, plus (b) 0.5%, which interest is payable monthly. Both loan facilities are secured by substantially all of the Company's personal property other than the Company's intellectual property. Both loan facilities include customary affirmative and negative covenants. Upon the earlier of the second advance under the term loan facility or the first advance under the revolving loan facility, the Company must also achieve minimum revenue on a monthly basis measured against a percentage of the Company's Board of Directors-approved projections for the applicable fiscal year. The Company's failure to satisfy the revenue, or any other, covenant could result in an event of default under the loan facilities. Both loan facilities also include other events of default, the occurrence and continuation of which could cause interest to be charged at the rate that is otherwise applicable plus 5.0% and would provide the collateral agent under the term loan facility or the lender under the revolving loan facility, as applicable, with the right to exercise remedies against the Company and the collateral securing the loan facilities. These events of default include, among other things, any failure by the Company to pay principal or interest due under the loan facilities, a breach of certain covenants under the loan facilities, the Company's insolvency, a material adverse change, the occurrence of any default under certain other indebtedness in an amount greater than $0.25 million, one or more judgments against the Company in an amount greater than $0.25 million individually or in the aggregate, and any default under the other loan facility.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
F- 24