Attached files
| file | filename |
|---|---|
| EX-32.2 - EXHIBIT 32.2 - Celcuity Inc. | tv487729_ex32-2.htm |
| EX-32.1 - EXHIBIT 32.1 - Celcuity Inc. | tv487729_ex32-1.htm |
| EX-31.2 - EXHIBIT 31.2 - Celcuity Inc. | tv487729_ex31-2.htm |
| EX-31.1 - EXHIBIT 31.1 - Celcuity Inc. | tv487729_ex31-1.htm |
| EX-23.1 - EXHIBIT 23.1 - Celcuity Inc. | tv487729_ex23-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _______________ to _______________
Commission File Number: 001-38207
Celcuity Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 82-2863566 |
|
(State or other jurisdiction of Incorporation or organization) |
(I.R.S. Employer Identification No.) |
| 16305 36th Avenue North, Suite 450 Minneapolis, MN |
55446 |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: (763) 392-0767
| Securities registered pursuant to Section 12(b) of the Act: | ||
| Title of Each Class | Name of Each Exchange on which Registered | |
| Common Stock, $0.001 par value per share | The Nasdaq Capital Market | |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ¨ Yes x No
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. ¨ Yes x No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. x Yes ¨ No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). x Yes ¨ No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or emerging growth company. See definition of “large accelerated filer,” “accelerated filer,”, “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.:
| Large accelerated filer ¨ | Accelerated filer ¨ | |
| Non-accelerated filer ¨ | (Do not check if a smaller reporting company) | Smaller reporting company x |
| Emerging growth company x |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act x
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). ¨ Yes x No
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the Registrant as of September 19, 2017 was approximately $54,831,578, based on a price per share of $9.50. The Registrant was privately held on June 30, 2017 (the last business day of the Registrant’s most recently completed second fiscal quarter), and as such, the Registrant has elected to use September 19, 2017 as the calculation date, which was the effective date of the registration statement for the Registrant’s initial public offering. The price per share listed above is equal to the initial public offering price of the Registrant’s common stock and the value of the Registrant’s voting and non-voting common equity listed above assumes conversion of the Company’s outstanding convertible promissory notes immediately following the Registrant’s initial public offering, which closed on September 22, 2017.
As of February 28, 2018, there were 10,106,464 shares of the Registrant’s common stock outstanding.
DOCUMENTS INCORPORATED IN PART BY REFERENCE
Portions of the Registrant’s definitive proxy statement relating to its 2018 Annual Meeting of Stockholders is incorporated by reference into Part III of this Annual Report on Form 10-K.
2017 Annual Report on Form 10-K
Table of Contents
| 2 |
Special Note Regarding Forward-Looking Statements
This Annual Report on Form 10-K contains forward-looking statements regarding us, our business prospects and our results of operations that are subject to certain risks and uncertainties posed by many factors and events that could cause our actual business, prospects and results of operations to differ materially from those that may be anticipated by such forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, those described in Part I, Item 1A. “Risk Factors” and elsewhere in this report. Readers are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date of this report. We expressly disclaim any intent or obligation to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise. Readers are urged to carefully review and consider the various disclosures made by us in this report and in our other reports filed with the Securities and Exchange Commission that advise interested parties of the risks and factors that may affect our business.
All statements, other than statements of historical facts, contained in this Annual Report on Form 10-K, including statements regarding our business, operations and financial performance and condition, as well as our plans, objectives and expectations for our business, operations and financial performance and condition, are forward-looking statements. In some cases, you can identify forward-looking statements by the following words: “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “might,” “target,” “ongoing,” “plan,” “potential,” “predict,” “project,” “should,” “will,” “would,” or the negative of these terms or other comparable terminology, although not all forward-looking statements contain these words. Forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause our results, levels of activity, performance or achievements to be materially different from the information expressed or implied by the forward-looking statements in this Annual Report on Form 10-K. Forward-looking statements may include, among other things, statements relating to:
| · | our plans to develop and commercialize our CELx platform and CELx tests for patients with cancer and our expectations regarding the various cancer sub-types our CELx tests will identify; |
| · | any perceived advantage of our CELx platform and CELx tests as compared to traditional molecular or other diagnostic tests, including without limitation, the ability of our platform and tests to help physicians treat their patients’ cancers or to identify new patient populations not diagnosable with currently available diagnostic tests; |
| · | our expected first-mover advantage in providing products to culture living tumor cells on a commercial scale, or the sustainability of our competitive advantages; |
| · | the size and growth potential of the markets for our CELx platform, and our ability to serve those markets; |
| · | the rate and degree of market acceptance, both in the United States and internationally, and clinical utility of our diagnostic platform and tests; |
| · | our ability to partner with and generate revenue from pharmaceutical partners and physicians, and the market opportunity for HER2 therapies and other CELx programs for our pharmaceutical partners as a result of our CELx platform; |
| · | the success of competing tests that are or may become available; |
| · | the ability of our CELx platform and tests to impact clinical trials by our pharmaceutical partners, such as streamlining approval from the U.S. Food and Drug Administration, or FDA of targeted therapeutics; |
| · | the success, cost and timing of our CELx platform development activities and planned clinical trials, as well as our reliance on collaboration with third parties to conduct our clinical trials; |
| · | our commercialization, marketing and manufacturing capabilities and strategy; |
| · | expectations regarding federal, state, and foreign regulatory requirements and developments, such as potential FDA regulation of our CELx platform and CELx tests, our operations, as well as our laboratory; |
| · | our plans with respect to pricing in the United States and internationally, and our ability to obtain reimbursement for CELx tests, including expectations as to our ability or the amount of time it will take to achieve successful reimbursement from third-party payors, such as commercial insurance companies and health maintenance organizations, and government insurance programs, such as Medicare and Medicaid; |
| · | our ability to obtain funding for our operations, including funding necessary to complete further development and commercialization of our CELx platform and CELx tests; |
| 3 |
| · | our expectations with respect to our facility needs; |
| · | our estimates regarding expenses, future revenue, capital requirements and needs for additional financing; |
| · | future agreements with third parties about the commercialization of our CELx diagnostic platform and tests; |
| · | our expectations regarding our ability to obtain and maintain intellectual property protection for CELx platform and approach; |
| · | our ability to attract and retain key scientific or management personnel; |
| · | our expectations regarding the period during which we qualify as an emerging growth company defined under the Jumpstart Our Business Startups Act of 2012, or the JOBS Act; |
| · | the requirements of being a public company; |
| · | our expectations regarding having our stock listed on The Nasdaq Capital Market; and |
| · | our anticipated use of the net proceeds from our initial public offering. |
You should read the matters described in Part I, Item 1A. “Risk Factors” and the other cautionary statements made in this Annual Report on Form 10-K. We cannot assure you that the forward-looking statements in this report will prove to be accurate and therefore you are encouraged not to place undue reliance on forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements we make. You are urged to carefully review and consider the various disclosures made by us in this report and in other filings with the SEC that advise of the risks and factors that may affect our business. Other than as required by law, we undertake no obligation to update or revise these forward-looking statements, even though our situation may change in the future. Our forward-looking statements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures or investments that we may make.
| 4 |
Overview
Unless otherwise provided in this Annual Report on Form 10-K, references to the “Company,” “we,” “us,” and “our” and similar references refer to Celcuity Inc., a Delaware corporation.
We are a cellular analysis company that is discovering new cancer sub-types and commercializing diagnostic tests designed to significantly improve the clinical outcomes of cancer patients treated with targeted therapies. Our proprietary CELx diagnostic platform is the only commercially ready technology we are aware of that uses a patient’s living tumor cells to identify the specific abnormal cellular process driving a patient’s cancer and the targeted therapy that best treats it. We believe our CELx platform provides two important improvements over traditional molecular diagnostics. First, molecular diagnostics can only provide a snapshot of the genetic mutations present in a patient’s tumor because they analyze dead cells. Using dead cells prevents molecular diagnostics from analyzing in real-time the dynamic cellular activities, known as cell signaling, that regulate cell proliferation or survival. Cancer can develop when certain cell signaling activity becomes abnormal. Since genetic mutations are often only weakly correlated to the cell signaling activity driving a patient’s cancer, a molecular diagnostic is prone to providing an incomplete diagnosis. CELx tests overcome this limitation by measuring real-time cell signaling activity in a patient’s living tumor cells. When a CELx test detects abnormal signaling activity, a more accurate diagnosis of the patient’s cancer driver is obtained. Second, molecular diagnostics can only estimate the probability of a patient’s potential drug response based on a statistical analysis of the drug’s clinical trial results. Instead of this indirect estimate of drug response, CELx tests directly measure the effectiveness of a targeted therapy in a patient’s living tumor cells. This enables physicians to confirm that the therapeutic matching the patient’s cancer driver is functional in the patient’s tumor cells before prescribing it, which significantly increases the likelihood of a positive clinical outcome. We have one U.S. patent, five pending U.S. patent applications, one pending PCT patent application, as well as numerous corresponding non-U.S. patent applications covering our diagnostic approach using cell signaling analysis in living patient cells to guide treatment of patients with targeted therapies.
Our first analytically validated and commercially ready test using our CELx platform, is our CELx HER2 Signaling Function Test, or CELx HSF Test. Our CELx HSF Test diagnoses two new sub-types of HER2-negative breast cancer that traditional molecular diagnostics cannot detect. Our internal studies show that approximately 20% of HER2-negative breast cancer patients have abnormal HER2 signaling activity similar to levels found in HER2+ breast cancer cells. As a result, these HER2-negative patients have undiagnosed HER2-driven breast cancer and would be likely to respond to the same anti-HER2 targeted therapies only HER2+ patients receive today. Our CELx HSF Test is targeting HER2-negative breast cancer patients receiving drug treatment. As a companion diagnostic, or CDx, our CELx HSF Test could potentially drive additional clinical usage of HER2 therapies to treat the patients our test identifies with abnormal HER2 signaling. We believe this creates financial motivation for pharmaceutical companies to partner with us. We also believe that having an analytically validated and commercially ready diagnostic platform is attractive to potential pharmaceutical company partners as it will enable them to use our diagnostic tests immediately after receiving the regulatory approvals required to sell their drug therapies to the newly identified patient populations identified by our CDx.
Once our CELx HSF Test was analytically validated and became commercially ready, we sought opportunities to collaborate with pharmaceutical companies that owned HER2 drugs. Our efforts resulted in a collaboration with Genentech, Inc., or Genentech and the National Surgical Adjuvant Breast and Bowel Project Foundation, or NSABP, to field a prospective clinical trial to evaluate the efficacy of Genentech’s HER2 targeted therapies in patients with these newly identified cancer sub-types. We expect interim results from this trial in late 2018 and final results six to nine months later. We believe a successful outcome of this collaboration will demonstrate the suitability of our CELx HSF Test as a CDx for HER2 therapies and support our activities to attract other pharmaceutical company partnerships.
| 5 |
In addition to the two new breast cancer sub-types our CELx HSF Test diagnoses, we discovered 14 new potential cancer sub-types in breast, lung, colon, ovarian, kidney, bladder and hematological cancers. Approved or investigational drugs are currently available to treat each of these new potential cancer sub-types. CELx tests for these additional cancer sub-types are in various stages of development, and we expect them to become commercially ready on a staggered basis over the next few years. The development process for these additional CELx tests will mirror the process used to develop our CELx HSF test. This process includes completion of internal animal, verification, training set, and validation studies. As new CELx tests become commercially ready, we expect to initiate collaborations with pharmaceutical companies to help them obtain new drug indications for the new cancer sub-types our tests identify. In addition, we will continue our research to identify additional new cancer sub-types and to develop the corresponding CELx tests to diagnose them.
The need for more complete cancer diagnoses is significant. The complexity and dynamic nature of cancer makes it difficult to determine the underlying cellular activity driving the disease. Molecular tests are used to identify genetic mutations and select targeted therapies, but the overall impact of those tests on patient outcomes has fallen far short of expectations, primarily due to two factors. First, molecular tests provide a static and limited genetic profile of a patient’s tumor, and, therefore, cannot measure dynamic disease activity. These tests rely on statistical correlations to diagnose patients, and when a genetic mutation is only weakly correlated to oncogenic-related cellular dysfunction, a high number of false positive diagnoses will result. With patient response rates to therapies targeting a genetic mutation typically less than 50%, and in some cases, only 10% to 20%, there is significant need for an alternative approach. Second, many cancers lack a genetic biomarker to guide treatment. For those patients, the cellular dysfunction responsible for their cancer goes undiagnosed, which means they are less likely to receive a potentially beneficial targeted therapy. Thus, current molecular tests have demonstrated only a limited ability to diagnose the specific cellular dysfunction that is driving most patients’ cancer.
Our CELx platform addresses the need for better cancer diagnostic tests using two complementary technologies that represent a significant departure from molecular-based analyses. Unlike molecular tests that use fixed or lysed (dead) cells and can only measure the static composition of a cell, our CELx platform measures real-time signaling activity in a patient’s live tumor cells. This enables us to: (1) identify the cellular signaling dysfunction driving a patient’s cancer; and (2) confirm whether the matching targeted therapy is functional in the patient’s cells. Our CELx tests are performed in our laboratory in Minneapolis, Minnesota that is certified under the Clinical Laboratory Improvement Amendments of 1988, or CLIA, and accredited by the College of American Pathologies, or CAP.
Our platform, comprised of our internally developed cell microenvironment and cell signaling quantification technologies, allows for more accurate diagnoses and the discovery of new cancer sub-types:
| · | Cell microenvironment. Culturing living tumor cells poses three primary challenges. First, there is typically only a small amount of patient tumor tissue available. Second, the tumor cells often die once they are removed from the tumor tissue. Third, tumor cells that do survive are difficult to maintain. Moreover, when conventional cell culture approaches are used it can often take more than two months to prepare a test sample and the success rate is typically less than 50%. Due to these challenges, the pharmaceutical industry principally relies on widely available immortalized or genetically modified cancer cell lines, which are easily maintained and proliferate indefinitely at predictable rates. While these properties are useful for drug discovery purposes, that usefulness has minimized incentives to transfer patient tumor cell culturing technologies to the clinical setting. Our proprietary cell microenvironment technologies were designed to overcome these challenges and provide a testable cell sample from a patient tumor specimen as small as 20 milligrams in 10 to 14 days for more than 90% of the patient tumor specimens we receive. |
| · | Dynamic cell signaling quantification. We analyze the signaling pathway activity of live patient tumor cells using a biosensor that converts the dynamic cellular response to pathway activators or pathway inhibitors to a measurable electrical signal in real-time. To determine the activity of a specific signaling pathway, an activating agent specific to a pathway receptor is used to turn on the pathway and a corresponding inhibitory agent specific to the pathway receptor is used to turn signaling off. Thus, our tests allow us to identify both the signaling pathway abnormalities driving a patient’s cancer and to confirm whether a matching targeted therapy may prove beneficial. |
We believe our CELx platform will fundamentally change the standard-of-care many cancer patients receive. Patients with the newly identified cancer sub-types we have discovered have oncogenic pathways that are signaling abnormally, and, we believe, may respond positively to a matching targeted therapy. By identifying patients with a new cancer sub-type, each CELx test will create, in effect, a proprietary patient population that molecular diagnostics cannot identify.
| 6 |
Our initial commercial strategy is to partner with pharmaceutical companies to provide companion diagnostics for the pharmaceutical partners’ existing or investigational targeted therapies. We expect such partnerships to involve collaboration on clinical trials, regulatory submissions, and commercialization activities. We will initiate activities to pursue partnerships as our CELx tests become commercially ready and can be matched with a potential partner’s targeted therapies. Our commercial-related efforts to date have focused on seeking partnerships for our CELx HSF test, which became commercially ready as a laboratory developed test after it was analytically validated in 2016. We expect to seek pharmaceutical partnerships for a variety of different targeted therapies in other solid tumor types as we are conducting our initial clinical trial with Genentech and the NSABP.
We believe our CELx CDx tests will expand the matching drug’s market size because they can facilitate approval of new drug indications that a pharmaceutical company would not otherwise be able to obtain.
We expect that successful pharmaceutical company partnerships will generate significant revenue from the sale of tests to identify patients eligible for clinical trials, from milestone payments, and, potentially, from royalties on the incremental drug revenues our tests enable. A key requirement for success of these partnerships will be clinical trial results that demonstrate the advantages of using a CELx test as a companion diagnostic. Once a new drug indication is received that requires use of our CDx to identify eligible patients, we will offer our tests directly to treating physicians and coordinate go-to-market strategies with our partner. This coordination of commercialization strategies will allow us to significantly leverage the sales, marketing and reimbursement resources of our pharmaceutical partner, unlike traditional molecular diagnostic companies.
Our Value Proposition
We believe we offer a clear and compelling value proposition to the key healthcare stakeholders:
| · | Patients & Providers—Improved patient outcomes. Our CELx tests provide a more accurate diagnosis of a patient’s cancer driver and an assessment of a matching targeted therapy’s effectiveness in blocking the cellular dysfunction. This will enable physicians to match more precisely the targeted therapy they use to treat their patients. We believe this will increase the percentage of patients responding to the drug, improving overall patient outcomes significantly. |
| · | Pharma—Increased revenue & optimized clinical trials. CELx tests can significantly increase the revenue potential for many existing targeted therapies by identifying entirely new pools of patients potentially responsive to their therapy. For some targeted therapies, we estimate a CELx test could double the number of patients approved to receive treatment, thus driving billions of dollars in incremental sales. Also, by providing more precise selection of patients, our CELx tests can increase the odds a clinical trial meets its trial endpoint, greatly enhancing the likelihood the drug will obtain FDA approval for a new indication. In addition, according to an ARK Invest publication dated August 2016, companion diagnostics that increase the response rates of a drug can reduce Phase 3 clinical trial size as much as ten-fold and costs as much as 60%. |
| · | Payors—Lower costs per responsive patient. By providing more precise cancer diagnoses and driving higher drug response rates, we will significantly reduce the money spent on drugs that do not benefit patients. Many targeted therapies cost more than $50,000 per treatment and only benefit a small fraction of patients receiving them. Calculating drug costs on a cost-per-responsive patient, and not just cost-per-treated patient, highlights the true cost of targeted therapies and the expense associated with low drug response rates. For instance, a $50,000 targeted therapy with a 30% response rate costs $167,000 per responsive patient; however, that same drug would only cost $83,000 per responsive patient if the response rate was 60%. |
Our Competitive Strengths
We have a number of key strengths that enhance our ability to achieve our mission and build a successful company:
| · | First mover. We are the first company that we are aware of to launch diagnostic tests that measure the signaling pathway activity in a patient’s live tumor cells, which we believe gives us a significant first mover advantage. |
| · | High barriers to entry. Our issued and pending patents, as well as our proprietary information and trade secrets, give us a strong intellectual property position that we believe creates a significant barrier to entry for potential competitors. |
| 7 |
| · | Broad range of applications for our platform. We can develop tests for a wide range of signaling pathways and a wide range of cancer types. This allows us to build a deep new product pipeline that creates multiple paths to build a large and profitable business. |
| · | Diverse revenue streams including pharma partnerships. We anticipate generating significant revenue from CDx pharmaceutical partners, including revenue from the sale of tests to identify patients eligible for clinical trials, milestone payments, and potentially, from royalties on the incremental drug revenues our tests enable. Our most significant revenue opportunity comes from ongoing sales of CELx tests to physicians during the commercialization stage of the CDx. |
| · | Strong senior leadership team. Our founders and senior leaders have a proven track record of success building, operating and selling several successful companies. We have deep and highly relevant and complementary diagnostic, scientific, product development, and commercialization experience that has enabled us to establish market leadership positions for the companies we previously led. |
Our Platform Advantages
Our unique and proprietary CELx functional cellular analysis technology represents a major shift from the diagnostic industry’s reliance on molecular profiling to characterize a patient’s cancer sub-type. Our goal is to leverage our technology to build a durable competitive advantage that enables us to improve outcomes for a significant percentage of cancer patients.
Our CELx platform advantages include:
| · | Powerful cancer sub-type discovery tool. We have already discovered 16 new potential cancer sub-types that are not currently diagnosed and treated with a matching targeted therapy. These sub-types are characterized by the dysfunctional signaling pathway activity our CELx tests identify. By identifying new cancer sub-types, we are creating new patient populations to which pharmaceutical companies can offer new and existing drug therapies. |
| · | Direct patient-specific assessment of disease status. Even though the response rates for many targeted therapeutics are low, for those patients who do respond, their outcomes can be improved significantly. The problem is matching the patient to the right drug. Our platform overcomes this problem by directly identifying whether an oncogenic signaling pathway is abnormally active in a patient’s cells. This provides the most complete assessment available today of the intracellular activity driving a patient’s cancer. Existing genomic tests typically can only provide a determination whether cancer is present and an assessment of molecular mutations that may or may not be associated with the patient’s cancer driver. |
| · | Direct measurement of matching drug effectiveness. An important advantage of the CELx platform is its ability to quantify the amount of signaling dysfunction that a matching targeted therapy can inhibit in an individual patient’s cancer cells. This allows us to evaluate whether there are inherent drug resistance mechanisms that would prevent the therapy from functioning in the patient’s tumor cells. Molecular tests cannot provide this evaluation. |
| · | Improved response rates. We believe a patient population will have a higher response rate to a matching targeted therapy when it is diagnosed with a CELx test than with a molecular biomarker. By first identifying whether dysfunctional signaling is present and then confirming that a matching targeted therapy can inhibit the dysfunction, a CELx test eliminates the two primary variables that confound patient response to targeted therapy signaling: the presence or absence of the disease and the drug not functioning as intended. A molecular test provides insight on neither of these variables in most cases. |
| · | Identify drug responsive proprietary patient cohorts. There are large numbers of cancer patients who lack a genetic biomarker to guide treatment. For these patients, the cellular dysfunction driving the cancer goes undiagnosed, thus excluding such patients from receiving a potentially beneficial targeted therapy. We believe our CELx tests will enable us to identify new proprietary patient populations not currently diagnosable with molecular tests and increase the number of patients likely to respond to a matching targeted therapy. Moreover, we will be the only partner a pharmaceutical company can work with to develop a CDx for a new indication of a targeted therapy addressing these new patient populations. By contrast, most molecular diagnostic tests are undifferentiated and have little proprietary value, which gives pharmaceutical companies a wide range of companies to select from when choosing a molecular-based CDx partner. |
| 8 |
| · | Streamlined FDA approval of targeted therapeutics. CELx tests will enable our pharmaceutical partners to enroll patients in their clinical trial with the same cellular dysfunction their targeted therapy is designed to inhibit. We believe this will improve patient response rates, increasing the likelihood the trial meets its endpoint target and thus the likelihood the drug receives FDA approval. Improved patient response rates would also help reduce the size, cost, and length of our partner’s clinical trials. |
Our Industry
According to the Centers for Disease Control and Prevention, or CDC, cancer was the second-leading cause of death in the United States in 2015, responsible for nearly one of every four deaths. There are many types of cancer treatment options, including surgery, radiation therapy, chemotherapy, immunotherapy, hormone therapy, stem cell transplant, and targeted therapy. Targeted therapies are drugs or other substances that block the growth and spread of cancer by interfering with specific molecular targets involved in the progression of cancer. Targeted therapies differ from standard chemotherapy drugs in that they are often cytostatic (block tumor cell proliferation) rather than cytotoxic (kill tumor cells). According to the National Cancer Institute, there are currently more than 80 approved targeted oncology therapies, some of which cost more than $100,000 per treatment course.
Diagnostic tests to detect single biomarkers are now widely used by pathologists to determine the molecular sub-type of a cancer. When a molecular biomarker test is used to support the choice of therapy to prescribe, it is often referred to as a “companion diagnostic.” Increasing numbers of targeted therapeutics are prescribed based on the results from a companion diagnostic test to detect the presence of a molecular biomarker. Only patients testing positive for the biomarker are eligible to receive the associated therapy.
Companion diagnostics are becoming increasingly important to the pharmaceutical industry. The use of companion diagnostics to better match patients to effective treatments positively impacts clinical outcomes and lowers expenditures on drugs that do not benefit patients. Stratifying the eligible patient population to include only likely responders is particularly important when the percentage of likely responders is only a fraction of the total cancer population. In these circumstances, narrowing the eligible patient population is often necessary to meet the clinical endpoint targets required to receive FDA drug approval.
Our Market Opportunities
CDx Development Opportunities
We believe there at least 50 different potential opportunities for our company to collaborate on CDx programs with pharmaceutical companies. Our ability to develop partnering relationships with these pharmaceutical companies will be predicated on a number of factors, including the size of the patient population our CELx test identifies, the remaining patent life of the matching targeted therapy, as well as the success or failure of clinical trials we have conducted with other pharmaceutical companies. Completing clinical trials requires, among other things, successful enrollment of patients, meeting trial endpoint goals, and completing the trial in a timely manner. The time to complete a clinical trial can vary widely depending on a number of factors, many of which will be specific to any particular clinical trial.
We believe the revenue opportunity per CDx program will be consistent with other development programs pharmaceutical companies support. In addition, the revenue for an individual CDx program would represent only a small fraction of the potential value the new drug indication our CDx could create for our pharmaceutical company partner. For some drugs, our tests could double the number of patients eligible for a targeted therapy. Our CELx HSF Test identified 20% of HER2-negative patients with abnormal HER2 signaling who may benefit from treatment with HER2 drugs. Fully deployed, this test could double the annual number of eligible patients to receive HER2 targeted therapies in the U.S. alone.
| 9 |
CELx Testing Opportunities
We expect to generate recurring CDx testing revenues once a CELx CDx-linked drug therapy is approved for patient use. On average, we believe that the lifetime value of providing the CDx test will significantly exceed the revenue generated from the CDx development program. We expect to offer each CELx test to patients at prices ranging from $4,000–$7,000, depending on the number of pathways evaluated. No tests directly comparable to the CELx tests are available today to offer reference points for pricing purposes. Pricing for several proprietary complex genomic tests, however, fall within this range and we believe this provides guidance on the amount insurance companies are willing to pay for highly informative tests that guide patient care.
CELx Technology Background
The Role of Cellular Signaling Pathways in Cancer
Cancer is a class of exceedingly complex and diverse diseases characterized by the development of abnormal cells that divide uncontrollably and can infiltrate and destroy normal body tissue and disrupt normal organ function. In normal cells, a series of biochemical activities, known as signal transduction, transmit biochemical signals through an interconnected network of signaling pathways to control cell proliferation and survival. Cancer arises when alterations occur in one or more of these signaling pathways and normal cell processes are disrupted, resulting in uncontrolled cell proliferation. These alterations are driven by a variety of cellular aberrations, including genetic mutations and dysfunctional signaling pathway mechanisms. Identifying the alteration driving an individual’s cancer is complicated by the immense complexity of these signal transduction processes and the practically unquantifiable number of pathway variables.
As recently as 20 years ago, most cancers were classified and subsequently treated solely on the basis of the anatomical location of the tumor in the body. Chemotherapies that kill rapidly dividing cells were widely used, but they had only limited efficacy for many patients and caused a wide range of dangerous side effects due to lack of discrimination for tumor tissue. As tools to identify molecular mutations became available, scientists began to uncover correlations between certain molecular mutations, cancer tissue type, and a patient’s prognosis. This fostered the development of molecularly targeted therapeutics that were designed to disrupt the specific cellular function of the drug target, typically abnormal signaling pathway activity, associated the molecular mutation. These targeted therapies greatly improved outcomes for some cancer patients and are a testament to the efficacy of targeted therapies when effectively prescribed. According to information published by the Journal of Clinical Oncology in July 2015, targeted therapies are oftentimes 10 to 20 times more expensive than chemotherapies.
In conjunction with the advent of targeted therapies, new molecular diagnostics were developed to help physicians refine the classification of a patient’s cancer into sub-types based on the presence of a specific molecular anomalies, such as genetic mutations or over-expressed proteins. Such mutations or over-expressed proteins are commonly referred to as “biomarkers” when they are used to diagnose a disease and evaluate treatment options. For instance, breast cancer diagnostic tests are performed to determine whether two protein biomarkers, human epidermal growth factor receptor 2 (HER2) or estrogen receptors (ER), are overexpressed in the cancer cells. The results of these tests are used to classify the patient’s cancer molecular sub-type and to guide selection of a corresponding targeted drug therapy.
The launch and on-going development of many new targeted therapies and the increasing use of companion molecular diagnostics to guide selection of the most appropriate therapy for each patient ushered in the era of so-called “precision medicine” in oncology. Advances in genomic and proteomic techniques and drug discovery enabled researchers to identify new drug targets, new molecular diagnostics, and drugs that would specifically bind to the target.
While the increased usage of targeted therapies has improved patient outcomes, there is increasing recognition that the promise of molecularly guided diagnoses and targeted treatment has fallen far short of expectations. This is generally due to the heterogeneous nature of these diseases from patient to patient and the challenge of identifying the specific cellular dysfunction driving a cancer patient’s tumor growth. No matter how sophisticated or detailed, a point-in-time molecular profile can only provide a snapshot of a tumor. As a result, the genetic mutations many current tests identify are often only weakly correlated to the abnormal signaling driving a patient’s cancer. This is because protein and gene profiling provide an incomplete assessment of the biochemical activity promoting cancer tumor growth. In fact, when dysfunctional, the activity of signaling pathway networks are, we believe, not possible to assess using current genetic analyses, despite the impressive investments in mapping the human genome and advancements in techniques to identify molecular mutations.
| 10 |
The combination of the heterogeneous nature of cancer and the weak correlation of abnormal signaling to many genetic mutations helps explain why the response rates for patients treated with many targeted therapies are often less than 50%, and in some cases as low as 10% to 20%. For a patient to respond to a targeted therapy designed to disrupt disease-related signaling activity, two factors must be present: (1) the patient’s diseased cells must have the same signaling pathway dysfunction the drug is designed to inhibit, and (2) the drug affects its targeted pathway as intended. Current state-of-the-art genomic tests use fixed (dead) cells, which limits them to evaluating the presence or concentration of a genetic mutation or protein. These tests cannot evaluate either dynamic signaling activity or whether a drug can affect that activity. When a patient’s genomic biomarker status does not represent underlying signaling pathway dysfunction, this can lead to selection of the wrong targeted therapy to treat the patient. Of particular interest to us are those patients with dysfunctional signaling who lack a corresponding biomarker; they are not currently eligible to receive any targeted therapy that treats their dysfunctional signaling.
To measure dynamic cellular activity, living patient tumor cells are required. Until our advancements, efforts to use living patient tumor cells have been limited by the lack of reliable methods to extract and culture cancer cells from patient tumors. These previously limited efforts reflect the emphasis amongst cancer researchers on creating stable cell lines for use to model cell function or to studies screen millions of test compounds in drug discovery programs. Pharmaceutical companies driving the commercial development of cell technologies work primarily with immortalized cells or cell lines genetically modified to express a target or mutation of interest. These cell lines consist of established cell cultures that proliferate indefinitely and very uniformly. They are used primarily because they provide a highly uniform response when tested with millions of small molecules in the search for potential new drugs, and because techniques to culture these cells are well known, their properties well understood, and other experimental results using them are available for comparison purposes. Conversely, live patient tumor cells are difficult to obtain, are only available in small quantities, and according to a 2014 article published by Science, the percentage of tumors that yield proliferative cells with conventional culturing methods has until now been well below 50%, which required months of culturing to obtain sufficient testable quantities of cells. For these reasons, researchers prefer paraffin-fixed tissue or cell lines over living tumor cells when studying disease processes or screening drug candidates. This lack of compelling rationale for pharmaceutical companies and academic institutions to work with live tumor cells for research purposes left the field of live tumor cell research in a relatively immature state.
Our CELx Platform
We have made significant investments in research and development to build the first commercially-ready cancer diagnostic platform that we are aware of that measures the signaling pathway activity in a patient’s living tumor cells. To measure dynamic cellular activity, we internally developed two distinct but complementary technologies, which now comprise our CELx platform:
| · | our proprietary cell microenvironment; and |
| · | our method to quantify dynamic patient cell signaling dysfunction. |
We utilize our CELx platform to create CELx tests that measure specific signaling pathway activity in various tumor types.
Cell microenvironment. Previous research has shown that cancer cells extracted from a patient’s tumor share the molecular features of the primary cancers from which they were derived and could provide an ex vivo (outside the patient) model of a patient’s tumor. The technology around tumor cell extraction from individual patients and culturing techniques, however, has largely remained undeveloped. For instance, no competing diagnostic tests use live patient tumor cells to measure dynamic cell signaling activity and studies on the topic have historically highlighted the challenges of deriving a viable patient tumor cell sample from an individual patient tumor specimen.
We have developed a cell microenvironment to extract and expand viable tumor cells from fresh human tumor tissue, which meets the three critical clinical parameters a patient-derived tumor cell sample would need to satisfy in order to meet the regulatory and clinical requirements for a diagnostic test measuring signaling activity:
| · | The patient cell sample tested must reflect the starting tumor’s composition. If samples do not reflect the original tumor’s composition, test results derived from that sample may not be representative of the patient’s tumor. Competing techniques largely rely upon use of irradiated non-tumor cells to foster cell proliferation, transformative “engineering” techniques or other un-natural manipulations of the cells to keep them alive. Because these competing approaches significantly increase the risk that the resulting patient cell test sample may not mirror the patient’s original tumor, we developed processes that only utilize tumor cells derived directly from the patient’s tumor specimen. |
| 11 |
| · | The sample must be available for testing in less than 21 days. Clinicians generally require test results in cases of complex diseases such as cancer within two to three weeks so they can begin treatment of their patient as soon as the initial symptoms are evaluated or a preliminary diagnosis is made. Competing techniques require two to six months to culture sufficient tumor cells for a test sample, making them unsuitable for use with a clinical diagnostic. To meet this time requirement, we developed processes that allow tumor cell proliferation outside the patient. |
| · | At least 90% of the tumor specimens obtained from a patient must yield testable samples. Clinicians will only order tests that require a patient specimen when they are highly likely to receive a test result. The challenges of increasing the cell sample yield from tumor tissue are well known and competing techniques are only able to obtain testable quantities of cells from less than 50% of patient tumor specimens. To meet this requirement, we developed processes that enhance cell survival when tumor cells are removed from live tissue. |
We believe our pioneering efforts have substantially advanced the technology of primary tumor cell culture technology. We have one issued U.S. patent, five pending U.S. patent applications, nine pending non-U.S. patent applications and one pending international PCT patent application, as well as significant proprietary know-how and trade secrets for the various cell sample preparation methods we have developed.
Dynamic patient cell signaling quantification. The second component of our CELx platform involves methods to quantify specific dynamic signal transduction events in patient derived tumor cells. The complexity of signal transduction processes is immense and the permutations of the pathway variables are practically unquantifiable. Current analytical methods to assess these variables use dead (fixed or lysed) cells. Point-in-time measurements are limited to assessment of the compositional status (e.g. mutation), concentration level (e.g. protein amount), or activation status (e.g. phosphorylation) of a finite number of signaling pathway components. A key insight underlying our technology was our observation that, no matter how sophisticated or detailed, a point-in-time molecular profile would only provide a snapshot. These methods could not provide a complete, dynamic assessment of the signaling activity driving a patient’s cancer. These point-in-time molecular analyses would, in many cases, only provide a weak correlation to the presence of the signaling pathway dysfunction driving a patient’s cancer. Instead, we concluded that a complete diagnosis of cancer and an assessment of a patient’s response to treating their disease requires measurement of the underlying activity of signaling pathways in live patient tumor cells.
To measure live real-time dynamic cell signaling activity, we utilize an impedance biosensor instrument. An impedance biosensor is an analytical platform that converts changes in cellular activity to a measurable electrical signal. We use the instrument to monitor dynamic changes in cell adhesion and morphology initiated by signal pathway activation or inhibition in live patient tumor cells. The instrument is comprised of a 96-well microplate with thin gold electrodes covering the bottom of each well. Wells employed with a selective extracellular matrix attach viable cells in a specific manner to the electrodes. The presence of viable cells on top of the electrodes affects the local ionic environment at the electrode/cell interface, leading to an increase in electrode impedance. To obtain a measurement, a small alternating current is applied across the electrode. When cells are added to the microplate wells and attach to the electrodes, they act as insulators increasing the impedance in each well.
As cells cover the electrodes, the current is impeded in a manner related to the number of cells and their adhesion properties. In addition, since cell signaling changes modulate a cell’s adhesion properties, the impedance biosensor detects and quantifies these changes. When cells are stimulated and change their function, the accompanying changes in cell adhesion thus alter the impedance that is measured.
To determine the activity of a specific signaling pathway, an activating agent specific to a pathway receptor is used to turn on the pathway and a corresponding inhibitory agent specific to the pathway receptor is used to turn signaling off. When signaling pathways are stimulated in this manner, adhesion molecules are effected and cause a change in the impedance measured in a well. By relying on the principle of detecting signaling pathway activity, we believe we can develop tests for a range of disease types and targeted therapies that affect various cellular pathways.
| 12 |
Data is recorded in real-time when a patient’s tumor cells are responding to activating or inhibitory agents and analyzed with the impedance biosensor. The output value is reported as the change in the electrical impedance measured. The change in impedance values is quantified over time and used to determine a Signaling Function Score.
New Product Development
We are leveraging our CELx technology to discover new cancer sub-types that a genomic test cannot detect. These new sub-types are characterized by the hyperactive signaling pathway our test identifies. These sub-types cannot be detected by genomic tests because they lack a corresponding molecular biomarker to identify it. We will translate our discoveries into diagnostic tests that incorporate the following primary steps:
| (1) | Extraction of proliferative tumor cells from patient biopsy. This step provides the patient tumor cells that we use to perform the test. |
| (2) | Activation of signaling pathway activity. This step determines whether the signaling pathway we are assessing is dysfunctional or not. |
| (3) | Inhibition of signaling pathway activity. This step determines whether the matching targeted therapy is functional in the patient’s tumor cells. If the drug can block a significant amount of the signaling dysfunction, this demonstrates lack of an inherent resistance mechanism in the patient’s tumor cells that would prevent the drug from functioning when prescribed to the patient. |
We confirmed that we can discover new cancer sub-types in 2015 with the discovery of two new breast cancer sub-types—HER2-/ER+ and HER2-/ER- breast cancer with abnormal HER2 signaling. We are now leveraging the expertise we gained while validating the resulting CELx HSF Test for breast cancer to guide our discovery of additional cancer sub-types.
We are currently conducting research to identify additional cancer sub-types in five solid tumor types. Our research studies to date have identified 14 potentially new breast, lung, ovarian, kidney, and bladder cancer sub-types that involve dysfunctional oncogenic signaling pathways. Multiple dysfunctional pathways were active in each of these tumor types. These studies confirm that the CELx platform can be a cancer sub-type discovery engine and that we can create a multi-pathway test to identify the specific driver in a patient’s tumor. We expect to eventually expand the tumor types we evaluate to include colon, head and neck, leukemia, esophageal, and gastric cancers.
We will seek to identify individual signaling pathways that may be driving at least 5% to 10% of the total cancers in each tissue area. Once we have characterized the prevalence of the different sub-types of signaling dysfunction in each tumor type and validated the tests for the different pathways, our plan will be to launch a corresponding CELx test. Eventually, each CELx test will analyze multiple pathways in a patient’s tumor to identify the specific pathway dysfunction driving a patient’s cancer. Testing multiple pathways will thus provide a systems view of the patient’s cancer using dynamic functional analysis. We believe this will result in more accurate diagnosis of a patient compared to molecular diagnostics that are using next generation sequencing to assess the status of multiple static biomarkers.
Clinical Trial Approach
A major component of our development and commercial activities is providing clinical data from interventional clinical trials using our CELx tests. Our clinical trial strategy is predicated on proving the correlation between our CELx Signaling Function Score and a patient’s clinical results. Once our first trial demonstrates that our CELx HER2 Test identifies patients responsive to HER2 targeted therapies, we expect pharmaceutical companies to partner with us to fund trials to evaluate new potential indications for their drugs with patients identified by one of our CELx tests. The trials will be designed to confirm that patients with abnormal pathway signaling obtain a superior clinical response to a therapy targeting that pathway than to the standard-of-care therapy they currently receive.
Each clinical trial would be structured as a prospective interventional study. The objective would be to confirm the relationship between the CELx Signaling Function Score generated by the CELx test and the study endpoint. Different primary endpoints will be used depending on the stage and type of cancer. For late-stage solid tumor trials, Time-to-Progression, or TTP, or Progression-Free Survival, or PFS, would likely be used, mirroring the endpoints used in the tested drug’s pivotal trial. For early stage solid tumor trials, pathological complete response, or pCR, would likely be used. The trials will either be single-arm or randomized two-arm.
| 13 |
We also expect to evaluate multiple CELx tests in the same trial when we have identified two or more cancer sub-types in the same tumor type (e.g., breast, ovarian, lung). Screened subjects would be assigned to a therapy arm that corresponds to the pathway found to be abnormal.
To obtain statistically significant results, a randomized two-arm trial is projected to require enrollment of approximately 120 to 150 patients for each drug evaluated, or between 60 to 75 patients in each arm. A single-arm trial would require 25 to 50 patients. We estimate each trial will require 18 to 27 months from the initiation of enrollment to completion of the follow-up period and that interim analysis will be performed in most trials with interim data available after 10 to 15 months.
For trials involving patients not currently eligible for a cancer drug that targets a certain pathway, we would first obtain a tissue specimen from each subject and perform the CELx test to identify subjects who have abnormal signaling. These patients would then be randomly assigned to either an arm that receives the current standard-of-care therapy or one that includes the current standard-of-care therapy plus the targeted therapy. All patients would be monitored until their disease progresses.
First Test—CELx HER2 Signaling Function Test
HER2+ breast cancer. Roughly 15% to 20% of breast cancer patients are diagnosed with HER2+ breast cancer when their tumor cells are found to have overexpressed or amplified levels of HER2. These patients are treated with anti-HER2 targeted therapies in combination with chemotherapies. Results from a number of clinical trial results for HER2 drugs reveal that only about 40% of HER2-positive patients respond to them. In addition, findings from several clinical trials have shown that a sub-set of HER2-negative patients benefit from therapies that target HER2. These results highlight the relatively weak correlation between HER2 receptor or gene amplification status and drug response.
Signaling activity status vs. HER2 receptor status. Based on this analysis, we concluded that measurement of HER2 signaling activity, rather than absolute HER2 levels, may more accurately diagnose HER2-driven breast cancer. This led to our successful studies in 2014 when we discovered abnormal HER2 signaling in HER2-negative breast cancer patient tissue. We concluded that this patient population provided an excellent opportunity to validate our hypothesis that signaling activity is more correlative to disease activity than receptor status. Measuring HER2-status not sufficient to diagnose all HER2 cancers. Despite the widely recognized role that a dysfunctional HER2-related signaling network plays in promoting breast cancer, only tests measuring a single reactant, HER2 protein, are performed in the clinic to diagnose it; we believe no diagnostic tests are available today that measure HER2 signaling activity within a patient’s breast tumor epithelial cells. This focus on measuring HER2 expression-levels reflects the widely-held view that measuring a patient’s HER2 status is sufficient to diagnose HER2-driven breast cancers. When only HER2 expression is measured, though, patients classified as HER2-negative but whose tumor cells have abnormal HER2 signaling are diagnosed as not having HER2-driven breast cancer, when, in fact, they do.
Diagnosing HER2 disease in HER2-negative patients with CELx HER2 Signaling Function Test. Since current genomic methods cannot identify the HER2-negative breast cancer patients who have the HER2-driven cancer, a new method was required. Such a method would need to analyze the HER2-signaling pathways (MAPK and PI3K) associated with HER2 cancers in a patient’s tumor cells. This is what our CELx HER2 Test is designed to do. Our test identifies patients whose HER2 status as determined by conventional techniques does not represent the correct diagnosis of their breast cancer at a functional level.
Improving outcomes, HER2 drug response rates, and lower cost per responding patient. Identifying these HER2-negative/HER2-signaling abnormal patients, and treating them with HER2 therapies, offers the potential to improve their clinical outcomes significantly. It is also likely that patients with abnormal HER2 signaling will respond at higher rates than HER2-positive patients. We believe that patients with abnormal HER signaling (e.g., those the CELx HSF Test diagnoses) are very likely to respond because they have the specific disease mechanism the HER2 therapies are designed to treat. This would result in a reduction in the cost of HER2 drugs per responsive patient for those the CELx HSF Test identifies compared to those identified with HER2 protein or gene status tests.
| 14 |
Clinical Studies
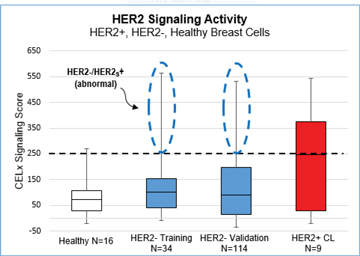
Prevalence Studies: 20% of HER2-Negative Patients Have Abnormal HER2-Signaling
To derive an initial estimate of the prevalence of abnormal HER2 signaling within the HER2-negative breast cancer population, we conducted a cell line survey, a training set study and a validation study using primary tumor cells. Live cell response to specific HER2 agonists (NRG1b and EGF) and antagonist (pertuzumab) was measured.
Key findings include:
| · | Cell Line Survey Study (N=19) |
| · | 4 of 9 HER2+ cell lines had HER2 signaling activity above 250 signaling units—these results helped establish an initial cut-off value; |
| · | Confirmed that normal HER2 signaling can occur in cells with overexpressed amounts of HER2; and |
| · | Confirmed that abnormal HER2 signaling can occur in cells with normal HER2 receptor levels. |
| · | Training Set Study (N=50). |
| · | 7 of 34 HER2-negative breast cancer patients (20.5%; 95% CI = 10%–37%) had tumor cells with HER2 signaling activity that was characterized as abnormally high and consistent with the HER2 signaling found in the upper 50% of the HER2+ cell lines; and |
| · | The 16 healthy breast specimens had a significantly lower average and standard deviation HER2 Signaling Scores than the HER2- and HER2+ breast cancer specimens. |
| · | Validation Study (N=114) |
| · | 27 of 114 patients (23.7%; 95% CI = 17%–32%) had tumor cells with HER2 signaling activity that was characterized as abnormally high and consistent with the HER2 signaling found in the upper 50% of the HER2+ cell lines. |
| · | The graph below presents the data from the Cell Line, Training Set and Validation Prevalence Studies in a Box-Whiskers plot format. |
| o | The dotted line at 250 represents the cut-off value for the CELx HSF Test. The cut-off value of 250 is equivalent to the median CELx Signaling Score recorded from the sample of HER2+ cell lines. |
| o | HER2-negative breast cancer patients with CELx Signaling Scores at or above 250 have abnormal HER2 signaling. |
| o | The circled portions of the plots for the HER2- Training Set and HER2 Validation Set results represent the specimens with Abnormal HER2-driven signaling. |
| 15 |
Xenograft Study: Abnormal HER2 Signaling Correlates to Drug Response Better than HER2 Status
We conducted a study in collaboration with the University of Minnesota using xenograft mouse models of human breast tumors to evaluate the relationship of HER2-driven signaling and response to lapatinib, a reversible dual-HER2 kinase inhibitor. Key findings:
| · | The HER2 signal inhibitor shrank a HER2-negative tumor with abnormal HER2 signaling; |
| · | The HER2 signal inhibitor did not affect the HER2+ tumor with normal HER2 signaling; and |
| · | Findings contradict HER2 receptor-based conclusions: |
| · | Lapatinib inhibition more correlative to HER2 signaling than HER2 receptor expression; and |
| · | HER2 signaling status independent of HER2 receptor expression. |
These findings support the hypothesis that HER2-negative breast cancer patients with abnormal HER2-driven signaling may benefit from treatment with anti-HER2 drugs.
A summary of results:
Xenograft Study Results
| Cell Line | ||||
| Parameter | HCC1954 | BT483 | ||
| HER2 Receptor Expression (IHC) | HER2+ (3+) | HER2- (0) | ||
| HER2 Signaling Status (CELx) | Normal | Abnormal | ||
| Lapatinib Inhibition (Xenograft) | 13% (p = 0.34) | 49% (p = 0.01) | ||
Study of Two HER2 Antibody Therapies, Trastuzumab and Pertuzumab, in HER2- and HER2+ Cells
We conducted this study to compare the effectiveness of two anti-HER2 antibodies in blocking HER2-driven signaling in HER2+ and HER2- cells. Tumor cells from 5 HER2- primary tumors and 4 HER2+ cell lines were obtained. Real-time live cell response to NRG1, a specific HER2/HER3 agonist, with or without pertuzumab, trastuzumab, or the combination of the two, was measured and quantified. All cell samples tested had comparable, and abnormal, levels of NRG1 activated HER2-driven signaling. Key findings:
| 16 |
| · | In each sample, the two mAb’s inhibited a higher percentage of signaling in combination than either mAb alone; no interference effects between the two mAb’s were detected; |
| · | Pertuzumab and trastuzumab alone were each more effective in the HER2- cell samples than in the HER2+ ones; and |
| · | Two HER2 mAb’s used to treat HER2+ breast cancer patients are as effective in blocking abnormal HER2-driven function ex vivo in HER2- primary cells as they are in HER2+ cell lines. |
Average % NRG1 Inhibition
| HER2 mAb | HER2+ Cell Lines |
HER2-(HER2S+) Primaries | ||
| Pertuzumab | 62% | 73% | ||
| Trastuzumab | 19% | 44% | ||
| T+P | 87% | 81% | ||
Study of Four HER2 Signal Inhibitors in HER2- and HER2+ Cells with Abnormal HER2 Signaling
Tumor cells from seven HER2-negative tumor specimens with abnormal HER2-driven signaling (HER2 S +) and the nine HER2-positive cell lines were obtained. Real time live cell response to NRG1, a specific HER2/HER3 agonist, with or without a HER2 targeted drug (pertuzumab, lapatinib, neratinib, afatinib) was measured and quantified. From these responses, the percentage inhibition of the HER2-driven signaling initiated by NRG1 by the HER2 drugs was determined. Key findings:
| · | Each of the HER2 drugs inhibited an average of at least 69% of the HER2-driven signaling activated by NRG1 stimulation in the HER2-negative primary cell samples; |
| · | The highest level of inhibition was found with the two irreversible covalent dual RTKi’s, afatinib and neratinib; and |
| · | All of the HER2 drugs inhibited a greater percentage of HER2-driven signaling in the HER2-negative primary tumor cells than in the HER2-positive cell lines. |
Average % NRG1 Inhibition
| HER2 Drugs | Mechanism of Action | Cell Lines (HER2+) |
Primaries (HER2-/HER2S+) | |||
| Pertuzumab | HER2 dimerization inhibitor | 46% | 78% | |||
| Lapatinib | Reversible Dual RTKi | 15% | 69% | |||
| Afatinib | Irreversible Covalent Dual RTKi | 47% | 93% | |||
| Neratinib | Irreversible Covalent Dual RTKi | 95% | 100% |
Analytical Validation Study
We conducted analytical validation studies in accordance with applicable FDA guidance and Clinical and Laboratory Standards Institute, or CLSI, standards to characterize the performance of the CELx HSF Test. CLSI standards define the test protocols that the FDA and CLIA require laboratories to use to characterize the performance of their diagnostic tests. The study results confirm that the CELx HSF Test has high analytical sensitivity and specificity. A summary of the results is provided below:
| 17 |
CELx HSF Test Analytical Study Results
| Performance Characteristics | Results | |
| Analytical Precision (Qualitative) | ||
| Analytical Sensitivity (95% CI) | 95.8%–100% (88/88) | |
| Analytical Specificity (95% CI) | 95.8%–100% (88/88) | |
| Detection Limits | ||
| Limit of Blank | 0.0020 cell attachment units | |
| Limit of Detection | 0.0099 cell attachment units | |
| Cut-Off Characterization | 250 signaling units | |
| Carry Over | 0% |
Clinical Trial (in process): CELx test FDA Approved for Use in Clinical Trials
On May 8, 2017, we entered into a non-exclusive Clinical Trial Agreement with NSABP to conduct a 54-patient single-arm interventional trial. Pursuant to the agreement, NSABP serves as the Sponsor and Principal Investigator of the trial and is responsible for, among other things, setting up clinical sites, enrolling patients, and managing clinical data. NSABP has contracted separately with Genentech to provide drugs for the study at no cost. We are performing the CELx HSF Test to select patients for the trial and are providing the funding for the trial’s patient-related costs. Completing this trial will require, among other things, successful enrollment of patients, meeting trial endpoint goals, and completing the trial in a timely manner. Based on our estimates of patient enrollment rates, we expect to obtain interim results in late 2018 and final results six to nine months later.
NSABP is one of the country’s premier clinical research cooperatives. Its members include many of the country’s leading medical centers and their investigators are amongst the most-respected in the breast cancer field. Genentech is one of the largest biopharmaceutical companies in the world and was the first company to launch a HER2 targeted therapy; their anti-HER2 targeted therapies have roughly 95% market share.
We submitted an Investigational Device Exemption, or IDE, application to the FDA to obtain approval to use our CELx HSF Test in a clinical trial setting. The IDE submission included validation test protocols and study reports, manufacturing process summaries, and relevant publications. The FDA approved our IDE in early 2017.
The goal is to demonstrate that patients who have an abnormal signaling pathway, as identified by our CELx test, respond to treatment with a matching targeted therapy. A synopsis of the trial protocol is provided below.
| 18 |
Clinical Trial Synopsis
| Objective | To evaluate the efficacy of neoadjuvant HER2 drug treatment in early stage HER2-/HER2S+ breast cancer patients |
Sites/Sponsor Endpoint Investigational (Single) Arm |
Multi-center in collaboration with NSABP and Genentech 54 HER2- early stage breast cancer (26 ER+/28ER-) Pathological complete response (ypT0/Tis ypN0) N=54 (HER2S+) |
Pursuant to our agreement with NSABP, the cost of the study is $2.65 million, subject to adjustment based on reimbursement for travel and similar expenses. Of this amount, we paid $300,000 upon entry into the agreement and $50,000 or one quarterly payment in 2017 and we will pay: (i) an aggregate of approximately $1.8 million as certain patient milestones are met, (ii) five quarterly payments of $50,000 in 2018 and ending in 2019, and (iii) a final payment of $250,000 upon completion of the study.
Our agreement with NSABP may be terminated by one or either party upon certain events, such as: (i) the FDA withdrawing its authorization and approval to perform the study, (ii) NSABP determining that the human and/or toxicology test results support termination of the study, (iii) either us or NSABP determining that an adverse reaction or side-effect of drugs administered in the study or a modification of the study’s protocol raises safety issues to support termination of the study, (iv) either party remaining in material breach of the agreement for a period of 30 days following notice of such breach, (v) us not performing the CELx HSF Tests or providing study kits, (vi) us failing to pay amounts owed to NSABP, and/or (vii) Genentech terminating its agreement with NSABP to supply drugs for the study or the drugs for the study no longer being manufactured or being available.
Commercialization Strategy
Our commercial activities will target three complementary groups at various phases of the development of our CELx tests.
| · | Pharmaceutical companies. For each CELx test we develop to diagnose a new cancer sub-type, we will identify the matching targeted therapies, either currently approved or in the investigational phase, and the manufacturer of those therapies. We will initiate discussions and seek to reach development agreements with each of these pharmaceutical companies when we have verified the prevalence of the cancer sub-type and completed successful animal studies. |
| · | Medical and surgical oncologists. We will initially target key opinion leaders, or KOLs, in each cancer type once we have completed the analytical validation of a CELx test. This will allow us to build awareness and credibility for the CELx test as we are generating clinical validation data. When a new drug indication is received that requires use of a CELx CDx to identify eligible patients, we will coordinate the pharmaceutical company’s go-to-market activities with our own. This coordination will allow us to significantly leverage the pharmaceutical company’s sales, marketing, and reimbursement, unlike traditional molecular diagnostic companies. |
| · | Payors. We will initiate pilot activities with payors for late stage patients during the clinical validation phase of a CELx test’s development. We will expand our payor efforts to include health economics analysis once we have clinical trial data available. When a new drug indication is received that requires use of a CELx CDx to identify eligible patients, we expect to coordinate the pharmaceutical company’s reimbursement activities with our own. |
| 19 |
Our CELx tests are laboratory developed tests and subject to regulation under CLIA. We completed the analytical validation of our first CELx test and received CLIA certification in 2016, at which time our CELx HSF Test was ready to sell commercially on a stand-alone basis to treating physicians. We expect to generate revenues from CELx tests performed in conjunction with the clinical trials a pharmaceutical company will field during the clinical phase of our partners’ drug approval process. We also expect that the agreements we enter into with the pharmaceutical companies partnering with us on these trials will include milestone payments at initiation and completion of trials and perhaps at various other negotiated points during the trials. We expect to generate revenue from the sale of CELx tests ordered by physicians either as stand-alone diagnostics or, upon the approval of our pharmaceutical company’s matching drug, as a CDx. A key requirement for success of these partnerships will be clinical trial results that demonstrate the advantages of using a CELx test as a companion diagnostic.
We intend to position our unique and highly differentiated tests as practice changing advancements in patient care. To inform key stakeholders of the value of our solution in order to drive adoption and reimbursement, we expect to employ the following diverse commercialization strategies over time:
| · | leverage our pharmaceutical partnership and their go-to-market initiatives for the drug our CDx is partnered with; |
| · | collaborate with oncology thought and KOLs and leading institutions on clinical research, publications, and product development; |
| · | build an experienced, oncology-focused sales force in the United States and international distribution channels that are supported by dedicated company personnel; |
| · | integrate into the everyday practice of clinicians through our medical affairs and client services efforts; |
| · | publish important medical and scientific data in peer-reviewed journals and present at major industry conferences, conduct clinical trials; and |
| · | work with patient advocacy groups, leading cancer philanthropic organizations, and medical societies to drive awareness of CELx tests and the importance of incorporating functional cellular analysis into cancer treatment. |
Through these efforts, we will seek to promote our CELx test’s unique capabilities throughout the oncology community—from patients, to the physicians treating them, to the third-party payors for these treatments and to biopharmaceutical companies developing new treatments—all with the goal of facilitating better-informed treatment decisions for the greatest number of patients.
Once results from our current clinical trial are available, we expect to launch our first test, the CELx HSF Test in two phases. During the first phase, we intend to target KOLs in the oncology field to build awareness of our CELx platform. These KOLs are primarily comprised of oncologists and surgeons practicing at major academic health systems who participate in clinical research and cancer research scientists. They often are early adopters of new technologies, particularly those involving new insights into disease mechanisms. We expect to gradually ramp up our efforts to the remainder of the medical oncologists once we have established our reputation within the KOL community.
Once a CELx test is launched to the broader market, we expect physicians, typically a medical or surgical oncologist, will order our tests either as stand-alone diagnostics or, upon the approval of a pharmaceutical company’s matching drug, as a CDx. The physician will prescribe a CELx test and coordinate provision of a patient specimen from a biopsy or surgical procedure. The fresh tissue would then be shipped overnight directly to our laboratory where we would use our proprietary methods to extract diseased cell samples from the patient’s tissue and perform the CELx tests ordered. Test results would typically be available in 10 to 14 days after receipt of the patient specimen. For each patient sample analyzed, a Signaling Function Score would be calculated quantitatively and converted into a final qualitative result: abnormal or normal. For patients found to have an abnormal signaling function, clinicians would use the results of the CELx test as a guide to select a targeted drug that inhibits the abnormal signaling activity identified.
United States
For our first tests, we will target the estimated 4,300 medical oncologists working in hospitals and cancer centers in the United States. We expect to hire domestic sales professionals with typically over 10 years of experience in clinical oncology sales working at leading biopharmaceutical or specialty reference laboratory companies.
In general, we intend to focus our initial sales efforts on building relationships with KOLs and researchers at leading academic research institutions to demonstrate the scientific credibility of our CELx tests. We also plan to build relationships in community oncology practice settings through leading physician networks and community hospitals and community based cancer centers. We will also attend national and regional clinical meetings focused on cancer treatment for our anti-cancer tests.
| 20 |
We believe the unique and important nature of the results our CELx tests provide, and their positioning as a CDx, will drive many medical oncologists to independently seek out our tests once they become aware of them. We believe this may allow us to achieve our market penetration goals with a sales force and marketing expenses significantly less costly than has been experienced by molecular diagnostic companies.
International
We believe we can serve the international market from our laboratory in Minnesota. We expect to establish an international presence using local distributors that sell to physicians and coordinate shipment of specimens to the United States. To serve international markets, we would expect to add dedicated regional managers located outside the United States to oversee our relationships at the local level.
Pricing and Reimbursement
The principal groups that we expect to pay us in the future for our CELx tests include:
| · | commercial third-party payors; |
| · | government payors, including Medicare and state Medicaid plans; |
| · | biopharmaceutical customers; |
| · | hospitals, cancer centers, and other institutions; and |
| · | patients. |
Adequate reimbursement will be an important factor in achieving broad clinical adoption of our CELx tests. At the same time, we believe broad clinical adoption will help drive favorable reimbursement decisions. To achieve broad reimbursement coverage with commercial third-party payors and government payors, including Medicare and Medicaid, we plan to demonstrate the economic and clinical value of our CELx tests to payors by employing a multi-pronged strategy:
| · | Set a high bar for analytical validation. We expect to present data on the reproducibility and precision of CELx tests at conferences and will seek to publish the results in peer-reviewed journals. |
| · | Meet the evidence standards necessary to be consistent with leading clinical guidelines. We believe inclusion in leading clinical practice guidelines plays a critical role in payers’ coverage decisions. We plan to conduct clinical validation and clinical utility studies that are consistent with the requirements of the widely recognized National Comprehensive Cancer Network clinical practice guidelines. |
| · | Execute an internal managed care policy and claims adjudication function as part of our core business operations. We plan to make obtaining adequate and widespread reimbursement a critical component of our business operations. We expect to hire a team of in-house claims processing and reimbursement specialists who will work with patients and payers to navigate the claims process and obtain maximum reimbursement. |
| · | Cultivate a network of KOLs. KOLs are able to influence clinical practice by publishing research and determining whether new tests should be integrated into practice guidelines. We expect to collaborate with KOLs early in the development process to ensure our clinical studies are designed and executed in a way that clearly demonstrates the benefits of our tests to physicians and payers.C. |
| · | Compile a growing library of peer-reviewed studies that demonstrate the test is effective. We will seek to publish peer-reviewed articles and review papers to help support our efforts to obtain widespread adoption and reimbursement of our CELx tests. In each disease area we pursue, we intend to conduct studies in order to develop similar supporting literature. |
| · | Reduce expenditures. We intend to build economic models to measure the financial benefits of using our CELx test in guiding patient treatment and minimizing the use of drugs that will not likely have a positive impact. We plan to use the data we gather through the use of these models as we meet with commercial third-party payors and government payors. |
| · | Commercial third-party payors and government payors are increasingly making significant efforts to contain healthcare costs. A major cost reduction opportunity is to reduce expenditures for drug courses that provide no patient benefit. Our technology will enable physicians to prescribe therapies that have significantly higher response rates than has been the case with targeted therapies to date. Since this will lower the drug cost per responsive patient, we believe widespread use of our CELx tests is consistent with payors goals of delivering health care more cost effectively. |
| 21 |
Our Competition
At present, we are not aware of any other companies that offer diagnostic tests that use a patient’s live tumor cells to identify the signaling pathway driving a patient’s cancer. There are several companies focused on developing genomic or proteomic analyses of a patient’s diseased cells. Initial efforts identified protein targets or genetic mutations, oftentimes referred to as “biomarkers,” that are associated with a disease process to enable development of drugs more closely tailored to specific patient populations.
As tools for human genome analysis have become less expensive, a number of companies have also recently launched more complex genomic test panels and gene expression signatures tests. These tests rely on a static measurement of molecular properties and mathematical analysis to identify statistically significant correlations between the selected molecular properties and a clinical condition or outcome of populations of patients with the “same” disease.
These genetic tests often have limited predictive success because they only identify some, but not all of, the molecular and cellular conditions required for a drug therapy to function in a patient. They may identify the presence of the genes associated with a disease but they cannot determine how the gene products function in the context of a particular individual.
Providers of genomic or proteomic tests includes diagnostic kit manufacturers, hospitals and independent laboratories. We do not plan to develop tests where a molecular biomarker can identify drug responsive patients, so our current tests will not compete directly against the tests provided by these other companies. The table below provides a summary of the points of differentiation between our signaling function analysis approach and the molecular approaches used by our potential competitors.
| 22 |
Current Molecular Methods vs. Celcuity’s Functional Cellular Analysis Platform
| Type of Cell Sample Used: | ||
| Dead tumor cells (fixed, lysed) |
Live tumor cells | |
Type of Analysis Performed |
Single point-in-time mutation(s) status or protein amount, or activation status | Quantify signaling pathway activity over 24-hour period |
| Relationship to disease driver | Correlative | Direct Cause |
| Disease driver evaluated | No. Only a single or small set of components of the cell are evaluated | Yes. The activity of the entire signaling pathway is assessed |
| Drug function evaluated | No. Cannot assess drug function with dead cells | Yes. Drug’s effect on signaling pathway activity in patient’s cells quantified |
| Companies | Foundation Medicine, Caris Life Sciences, NeoGenomics, LabCorp, Quest, Nanostring, Paradigm, Biocept, Exosome Diagnostics, Guardant Health, Roche Diagnostics, Qiagen, Myriad, Genomic Health | Celcuity |
We are not aware of any available tests directly comparable to the CELx tests. We expect to offer each CELx test to patients at list prices ranging from $4,000 to $7,000, depending on the number of pathways evaluated. List prices for several proprietary complex genomic tests fall within this range and we believe this provides guidance as to the pricing of highly informative tests that guide cancer patient care.
Intellectual Property
We believe one of our core competitive advantages is the strength of our intellectual property portfolio. We developed our CELx technology internally. We are seeking both U.S. and non-U.S. patents to protect our inventions. Our patent portfolio currently includes the following:
| 23 |
| Subject Matter | Patent/Application # | Status | Priority Date |
Expiration | ||||
| Methods of treating a cancer patient using cell signaling analysis | US 9,404,915 | U.S. granted; Europe and Japan pending | 6/12/2012 | 2033 | ||||
| Methods of treating a cancer patient using cell signaling analysis | US 15/192,280 | U.S. pending | 6/12/2012 | 2033 | ||||
| Methods of determining the functional status of a cellular pathway | US 15/179,119 | U.S. pending | 12/12/2013 | 2034 | ||||
| Methods of diagnosing a cancer patient using cell signaling analysis | US 15/533,897 | U.S., Australia, Canada, China, Europe, Japan, South Korea and New Zealand pending | 12/12/2014 | 2035 | ||||
| Methods of creating a patient cell microenvironment | PCT/US2016/057923 | PCT pending | 10/20/2015 | 2036 | ||||
| Methods of treating a cancer patient using cell signaling analysis | US 62/473,936 | U.S. pending | 3/20/2017 | 2038 | ||||
| Methods of treating a cancer patient using cell signaling analysis | US 62/587,572 | U.S. pending | 10/17/2017 | 2038 |
We understand we must develop and maintain protection on the proprietary aspects of our technologies in order to remain competitive. We rely on a combination of patents, copyrights, trademarks, trade secret and other intellectual property laws and confidentiality, material transfer agreements, invention assignment agreements and other contracts to protect our intellectual property rights.
We plan to develop names for new products and apply for trademarks and as appropriate secure trademark protection for them, including domain name registration, in relevant jurisdictions. We also have developed a number of proprietary methods, materials, processes, and techniques related to the preparation of patient samples and performance of the CELx test that we believe are most effectively protected as trade secrets rather than as patented subject matter.
Research and Development
We have made significant investments in research and development for our CELx platform. Our annual research and development expenses were approximately $3.1 million and $5.0 million for the years ended December 31, 2016 and 2017, respectively.
Principal Suppliers
We purchase commercially available reagents and instruments from a variety of suppliers. Our principal reagent suppliers include Bio-Techne Corporation, Selleck Chemicals, Sigma-Aldrich, and VWR International. Our principal instrument suppliers include Acea Biosciences, Integra Biosciences, Invitrogen, and Thermo Fisher Scientific. These items are purchased on a purchase order basis pursuant to the applicable supplier’s standard terms and conditions. The items purchased from these suppliers are standard products sold widely to the biotechnology industry. All items purchased are typically available within several days after an order is placed.
| 24 |
Government Regulation
CLIA and CMS
The Centers for Medicare & Medicaid Services, or CMS, an agency within the U.S. Department of Health and Human Services, regulates all clinical laboratory testing (except research) performed on humans in the U.S. through CLIA. All clinical laboratories that perform clinical lab services on human specimens for the purpose of providing information on the diagnosis, prevention or treatment of disease must receive CLIA certification. This covers approximately 175,000 laboratories as of 2017. Laboratories must obtain CLIA certification and demonstrate compliance with CLIA requirements as confirmed by an inspection by CMS. We received our CLIA certification in 2016. We also had our laboratory certified by the College of American Pathologies, or CAP, in 2016, an organization recognized by CMS as a third-party reviewer of clinical laboratories. Several states, including, among others, New York and California, require licensure of out-of-state labs that receive specimens from the state and compliance with the state’s individual laboratory regulations.
If our laboratory is out of compliance with CLIA requirements, we may be subject to sanctions such as suspension, limitation or revocation of our CLIA certificate, as well as directed plan of correction, state on-site monitoring, civil money penalties, civil injunctive suit or criminal penalties. We must maintain CLIA compliance and certification to be eligible to bill for services provided to Medicare and Medicaid beneficiaries. If we were to be found out of compliance with CLIA program requirements and subjected to sanction, our business could be harmed. Failure to comply with state licensure laws, if applicable, could subject us to additional sanctions imposed by state licensing authorities.
FDA
FDA approval or clearance is not currently required for CELx tests offered as a stand-alone laboratory developed test. If we are partnered with a drug company to launch a CELx test as a CDx for a new drug indication, we would be required to obtain a Pre-Market Approval, or PMA, in conjunction with the pharmaceutical company seeking a New Drug Approval for the matching therapy. Historically, the FDA has exercised enforcement discretion with respect to tests performed solely in a central laboratory, like the CELx tests, often referred to as Laboratory Developed Tests, or LDTs. The FDA has not required laboratories that furnish only LDTs to comply with the agency’s requirements for medical devices (e.g., establishment registration, device listing, quality systems regulations, pre-market clearance or pre-market approval, and post-market controls).
Although the FDA proposed regulations that would apply to LDTs, FDA recently decided that, at present, those regulations are not moving forward towards approval and implementation. In mid-2014, the FDA published a Draft Guidance Document describing a proposed approach for a regulatory framework for LDTs that would have resulted in most of the high-value LDT tests marketed today eventually being required to obtain 510(k) clearances or Pre-Market Approvals. If implemented, this regulatory framework would require most hospital clinical labs to abandon a number of tests it performs or to pursue regulatory clearances or approvals to perform them. These proposals met significant resistance from Congress, the hospital industry, and independent clinical laboratories. The FDA indicated in late 2016 that it does not intend to finalize the 2014 Draft Guidance Document at this time. However, FDA continues to discuss potential regulatory approaches to LDTs.
HIPAA and HITECH
Under the administrative simplification provisions of HIPAA, as amended by the HITECH Act, the U.S. Department of Health and Human Services, or HHS, issued regulations that establish uniform standards governing the conduct of certain electronic healthcare transactions and protecting the privacy and security of protected health information used or disclosed by healthcare providers and other covered entities. HIPAA includes the following primary sets of regulations: privacy regulations, security regulations, and standards for electronic transactions, which establish standards for certain healthcare transactions. The privacy and security regulations were extensively amended in 2013 to incorporate new requirements from the HITECH Act.
| 25 |
The privacy regulations cover the use and disclosure of protected health information by healthcare providers and other covered entities. They also set forth certain rights that an individual has with respect to his or her protected health information, including, but not limited to, the right to access or amend certain records containing protected health information, or to request restrictions on the use or disclosure of protected health information. The security regulations establish requirements for safeguarding the confidentiality, integrity, and availability of protected health information that is electronically transmitted or electronically stored. The HITECH Act, among other things, makes many of HIPAA’s privacy and security standards applicable to business associates of covered entities, and established certain protected health information security breach notification requirements. A covered entity must notify affected individual(s) and the HHS when there is a breach of unsecured protected health information. HIPAA also governs patient access to laboratory test reports. Effective October 6, 2014, individuals (or their personal representatives, as applicable), have the right to access test reports directly from clinical laboratories and to direct that copies of those test reports be transmitted to persons or entities designated by the individual.
These laws impose significant fines and other penalties for improper use or disclosure of protected health information. Additionally, to the extent that we submit electronic healthcare claims and payment transactions that do not comply with the electronic data transmission standards established under HIPAA and the HITECH Act, payments to us may be delayed or denied.
In addition to the federal privacy regulations, there are a number of state laws regarding the privacy and security of health information and personal data that are applicable to our operations. The HIPAA privacy and security regulations establish a uniform federal “floor” that covered entities and business associates must meet and do not supersede state laws that are more stringent or provide individuals with greater rights with respect to the privacy or security of, and access to, their records containing protected health information. The compliance requirements of these various state laws, including additional breach reporting requirements, and the penalties for violation vary widely and new privacy and security laws in this area are evolving. We believe that we have taken the steps required for us to comply with health information privacy and security statutes and regulations in all jurisdictions, both state and federal. However, we may not be able to maintain compliance in all jurisdictions where we do business. Failure to maintain compliance, or changes in state or federal laws regarding privacy or security, could result in civil and/or criminal penalties and could have a material adverse effect on our business.
Federal, State and Foreign Fraud and Abuse Laws
In the United States, there are various fraud and abuse laws with which we must comply and we are potentially subject to regulation by various federal, state and local authorities, including CMS, other divisions of the HHS (e.g., the Office of Inspector General), the U.S. Department of Justice, and individual U.S. Attorney offices within the Department of Justice, and state and local governments. We also may be subject to foreign fraud and abuse laws in connection with our international business activities.
In the United States, the federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce, or in return for patient referrals for, or purchasing, leasing, ordering, recommending or arranging for the purchase, lease or order of, any healthcare item or service reimbursable under a governmental payor program. Courts have stated that a financial arrangement may violate the Anti-Kickback Statute if any one purpose of the arrangement is to encourage patient referrals or other federal healthcare program business, regardless of whether there are other legitimate purposes for the arrangement. The definition of “remuneration” has been broadly interpreted to include anything of value, including gifts, discounts, credit arrangements, payments of cash, consulting fees, waivers of co-payments, ownership interests, and providing anything at less than its fair market value. Recognizing that the Anti-Kickback Statute is broad and may technically prohibit many innocuous or beneficial arrangements within the healthcare industry, the HHS issued a series of regulatory “safe harbors.” These safe harbor regulations set forth certain provisions, which, if met, will assure healthcare providers and other parties that they will not be prosecuted under the federal Anti-Kickback Statute. Although full compliance with these provisions protects against prosecution under the federal Anti-Kickback Statute, the failure of a transaction or arrangement to fit within a specific safe harbor does not necessarily mean that the transaction or arrangement is illegal or that prosecution under the federal Anti-Kickback Statute will be pursued. Many states also have anti-kickback statutes, some of which may apply to items or services reimbursed by any third-party payor, including commercial insurers.
| 26 |
In addition, federal false claims laws, including the federal civil False Claims Act, prohibit, among other things, any person or entity from knowingly presenting, or causing to be presented, a false claim for payment to, or approval by, the federal government or knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. As a result of a modification made by the Fraud Enforcement and Recovery Act of 2009, a claim includes “any request or demand” for money or property presented to the U.S. government. Recently, several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies’ marketing of the product for unapproved, and thus generally non-reimbursable, uses. The civil monetary penalties statute imposes penalties against any person or entity who, among other things, is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent.
HIPAA created additional federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud or to obtain, by means of false or fraudulent pretenses, representations or promises, any money or property owned by, or under the control or custody of, any healthcare benefit program, including private third-party payors and knowingly and willfully falsifying, concealing or covering up by trick, scheme or device, a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services.
In addition, various states have enacted false claim laws analogous to the federal False Claims Act, although many of these state laws apply where a claim is submitted to any third-party payor and not merely a governmental payor program. If our operations are found to be in violation of any of the federal or state healthcare laws described above or any other governmental regulations that apply to us, we may be subject to significant penalties, including without limitation, civil, criminal and/or administrative penalties, damages, fines, disgorgement, exclusion from participation in government programs, such as Medicare and Medicaid, injunctions, private “qui tam” actions brought by individual whistleblowers in the name of the government, or refusal to allow us to enter into government contracts, contractual damages, reputational harm, administrative burdens, diminished profits and future earnings, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
In Europe, various countries have adopted anti-bribery laws providing for severe consequences, in the form of criminal penalties and/or significant fines, for individuals and/or companies committing a bribery offence. Violations of these anti-bribery laws, or allegations of such violations, could have a negative impact on our business, results of operations and reputation. For instance, in the United Kingdom, under the Bribery Act 2010, which went into effect in July 2011, a bribery occurs when a person offers, gives or promises to give a financial or other advantage to induce or reward another individual to improperly perform certain functions or activities, including any function of a public nature. Bribery of foreign public officials also falls within the scope of the Bribery Act 2010. Under the new regime, an individual found in violation of the Bribery Act 2010 faces imprisonment of up to 10 years. In addition, the individual can be subject to an unlimited fine, as can commercial organizations for failure to prevent bribery.
Federal and State Physician Self-Referral Prohibitions
Under a federal law directed at “self-referral,” commonly known as the “Stark Law,” there are prohibitions, with certain exceptions, on referrals for certain designated health services, including laboratory services, that are covered by the Medicare and Medicaid programs by physicians who personally, or through a family member, have an investment or ownership interest in, or a compensation arrangement with, an entity performing the tests. The prohibition also extends to payment for any testing referred in violation of the Stark Law. A person who engages in a scheme to circumvent the Stark Law’s referral prohibition may be fined up to $100,000 for each such arrangement or scheme. In addition, any person who presents or causes to be presented a claim to the Medicare or Medicaid programs in violation of the Stark Law is subject to civil monetary penalties of up to $15,000 per bill submission, an assessment of up to three times the amount claimed and possible exclusion from participation in federal governmental payor programs. Bills submitted in violation of the Stark Law may not be paid by Medicare or Medicaid, and any person collecting any amounts with respect to any such prohibited bill is obligated to refund such amounts. Many states have comparable laws that are not limited to Medicare and Medicaid referrals.
| 27 |
Other Regulatory Requirements
Our operations do not currently use hazardous materials, but we do generate regulated medical waste in the normal course of performing our CELx tests. This subjects us to a variety of federal, state and local environmental and safety laws and regulations. Some of the regulations under the current regulatory structure provide for strict liability, holding a party potentially liable without regard to fault or negligence. We could be held liable for damages and fines as a result of our, or others’, business operations should contamination of the environment or individual exposure to hazardous substances occur. We cannot predict how changes in laws or development of new regulations will affect our business operations or the cost of compliance.
New Legislation and Regulations
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the testing, approval, manufacturing and marketing of products that are or will be regulated by the FDA or CMS. In addition to new legislation, CMS and FDA regulations and policies are often revised or interpreted by the agencies in ways that may significantly affect our business and our products. It is impossible to predict whether further legislative changes will be enacted or FDA or CMS regulations, guidance, policies or interpretations will be changed, or what the impact of such changes, if any, may be. The 2016 presidential election and change in administration make it even more difficult to predict if and how federal regulations may change and/or federal agencies might alter their positions.
Pharmaceutical Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any products for which we sell. Sales of any of our products will depend, in part, on the extent to which the costs of the products will be covered by third-party payors, including government health programs such as Medicare and Medicaid, commercial health insurers, managed care organizations or pharmaceutical companies. The process for determining whether a third-party payor will provide coverage for a test sometimes is separate from the process for setting the price of a drug product or for establishing the reimbursement rate that a payor will pay for the drug product. Third-party payors may limit coverage to specific testing products on an approved list, which might not include all of the tests available for a particular indication.
In order to obtain coverage and reimbursement for any product, we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the test. Whether or not we conduct such studies, our products may not be considered medically necessary or cost-effective. A third-party payor’s decision to provide coverage for a test does not imply that an adequate reimbursement rate will be approved. Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage, and adequate reimbursement, for the product. Third-party reimbursement may not be sufficient to enable us to maintain price levels high enough to realize an appropriate return on our investment in product development.
The containment of healthcare costs has become a priority of federal, state and foreign governments, and the prices of tests and drugs have been a focus in this effort. Third-party payors are increasingly challenging the prices charged for medical products and services, examining the medical necessity and reviewing the cost-effectiveness of testing products, drug products and medical services and questioning safety and efficacy. If these third-party payors do not consider our products to be cost-effective compared to other available tests, they may not cover our products or, if they do, the level of payment may not be sufficient to allow us to sell our products at a profit. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs to limit the growth of government-paid healthcare costs, including price controls and restrictions on reimbursement. Adoption of such controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for testing products or drugs that require use of our testing products and could adversely affect our net revenue and results.
Pricing and reimbursement schemes vary widely from country to country. Some countries may require the completion of additional studies that compare the cost-effectiveness of a particular test to currently available tests. The downward pressure on healthcare costs in general, particularly prescription drugs and testing products, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert competitive pressure that may reduce pricing within a country. Any country that has price controls or reimbursement limitations for testing products may not allow favorable reimbursement and pricing arrangements for any of our products.
| 28 |
Coverage policies, third-party reimbursement rates and test pricing regulation may change at any time. In particular, in the United States, the Affordable Care Act contains provisions that have the potential to substantially change healthcare delivery and financing, including impacting the profitability of testing and drugs. For example, the Affordable Care Act revised the methodology by which rebates owed by manufacturers for covered outpatient drugs are calculated under the Medicaid Drug Rebate Program, extended the Medicaid Drug Rebate Program to utilization of covered drugs dispensed to individuals enrolled in Medicaid managed care organizations and subjected manufacturers to new annual fees for certain branded prescription drugs. As the price of our test may be included in the reimbursement rates for certain drugs, this could significantly impact our pricing. Even if favorable coverage and reimbursement status is attained for one or more products, less favorable coverage policies and reimbursement rates may be implemented in the future. However, the proposed repeal of the Affordable Care Act and the uncertainty surrounding a potential replacement law make it even more difficult to predict the future for reimbursement and pricing of drugs and tests in the United States.
Corporate History
We were organized as a Minnesota limited liability company in 2011 and commenced operations in 2012. On September 15, 2017, we converted from a Minnesota limited liability company into a Delaware corporation and changed our name from Celcuity LLC to Celcuity Inc.
Employees and Labor Relations
As of December 31, 2017, we had 22 employees, each of which were full-time employees. None of our employees are currently covered by collective bargaining agreements and we believe that our relations with our employees are good.
| 29 |
Risk factors which could cause actual results to differ from our expectations and which could negatively impact our financial condition and results of operations are discussed below and elsewhere in this report. Additional risks and uncertainties not presently known to us or that are currently not believed to be significant to our business may also affect our actual results and could harm our business, financial condition and results of operations. If any of the risks or uncertainties described below or any additional risks and uncertainties actually occur, our business, results of operations and financial condition could be materially and adversely affected.
Risks Relating to Our Business
We have a limited operating history upon which you can evaluate us. We may never generate revenue or profit.
We are an early-stage biotechnology company that commenced activities in January 2012. We only have a limited operating history upon which you can evaluate us. Our business plan has not been tested. Since inception, we have had no revenue and have incurred significant operating losses. We have financed our operations primarily through private placements of common equity and convertible notes prior to our initial public offering, as well as proceeds from our initial public offering of common stock. To generate revenue and become and remain profitable, we must continue to develop and commercialize the CELx platform. To do so, we need to successfully complete our clinical trial collaboration with Genentech and the NSABP for our CELx HSF Test, continue to develop other CELx tests for other cancer sub-types and cultivate partnerships with pharmaceutical companies. We must also build operational and financial infrastructure to support commercial operations, train and manage employees, and market and sell our CELx tests (as a CDx and/or as a stand-alone test).
We may never succeed in any or all of these activities and, even if we do, we may never generate revenue that is sufficient to achieve profitability. We expect to continue to incur significant expenses and operating losses for the foreseeable future, and the net losses we incur may fluctuate significantly from quarter to quarter. Our failure to become and remain profitable would decrease the value of the company and could impair our ability to raise capital, maintain or expand our research and development efforts, expand our business, or continue our operations.
Our initial success is heavily dependent on the success of our CELx HSF Test.
Our business strategy is focused on attracting pharmaceutical company partnerships that provide revenue from the sale of CELx tests during clinical trials, from milestone payments during clinical trials, from sales of our CELx tests as companion diagnostics or stand-alone tests thereafter, and, potentially, from royalties on the incremental drug revenues our tests enable. Our ability to obtain such partnerships and generate such revenue depends in part on the ability of our CELx HSF Test to demonstrate the potential incremental opportunity available for pharmaceutical companies. We do not expect to receive interim results for our first prospective clinical trial for the CELx HSF Test until late 2018 and final results six to nine months later. Success of the HSF Test trial will depend on many factors, such as successful enrollment of patients, meeting trial endpoint goals, and completing the trial in a timely manner. Our ability to complete the trial could be delayed or prevented for several reasons that are out of our control, such as the FDA withdrawing its authorization and approval to perform the study, the NSABP determining that the human and/or toxicology test results do not support continuing the trial, or participants having adverse reactions or side-effects to the drugs administered in the study. If we are unable to demonstrate that the HSF Test is suitable as a CDx for the targeted therapy, we will likely not be able to generate future revenue from our CELx HSF Test and may not be able to attract other pharmaceutical companies to partner with us for the development and commercialization of other CELx tests. Further, potential pharmaceutical company partners may delay negotiating development agreements until results of the CELx HSF Test trial are available. Even if the ultimate outcome of our CELx HSF Test trial is positive, any delays could materially and adversely affect our business.
| 30 |
We may not be successful in finding pharmaceutical company partners for continuing development of additional CELx tests.
We intend to develop strategic partnerships with pharmaceutical companies for developing additional CELx tests. Many of the potential partners are global, multi-billion-dollar pharmaceutical companies with sophisticated research and development organizations and multiple priorities. We may not be successful in our efforts to establish such a strategic partnership or other alternative arrangements for our CELx tests because, among other things, our research and development pipeline may be insufficient, such tests may be deemed to be at too early of a stage of development for collaborative effort, or third parties may not view such tests as having the requisite potential to demonstrate efficacy. In addition, we may be restricted under collaboration agreements from entering into future agreements with other partners. Even if we are able to find suitable partners, we may not be successful in negotiating development agreements with such partners that provide revenue from the sale of our CELx tests to identify patients eligible for required clinical trials, milestone payments, and/or royalties on the incremental drug revenues that our tests enable. If we are unable to reach agreements with suitable strategic partners on a timely basis, on acceptable terms or at all, we may have to curtail the development of additional CELx tests, our expected revenue opportunities may be significantly smaller than expected and our business may fail.
While our CELx HSF Test is commercially ready, we have not attempted to market it to physicians or their patients as a stand-alone test and have no ability to determine this test or any of our other tests are currently commercially viable.
While our CELx HSF test has been analytically validated, is conducted in our CLIA certified and CAP accredited laboratory, and is currently ready for commercial use as a laboratory developed test, we have not attempted to market it to physicians or their patients. Furthermore, we have commenced only limited communications with KOLs to build awareness and credibility of our CELx diagnostic platform and CELx HSF Test. Accordingly, we have no ability to determine whether our CELx HSF Test, or any other future CELx test, will be commercially viable as a stand-alone test. We may never be successful in generating revenue from our CELx HSF Test or other CELx tests as stand-alone tests, and if we are unable to build pharmaceutical partnerships that enable us to market this and other tests as companion diagnostic tests, we may never generate any revenue and our business may fail.
Developing our CELx tests involves a lengthy and complex process that may not be successful.
Our CELx tests may take several years to develop from the time they are discovered to the time they are available for patient use, if ever. In order to develop additional CELx tests into commercially ready products, we need to successfully complete a variety of activities, including, among other, conducting substantial research and development, conducting extensive analytical testing, and maintaining our CLIA certified and CAP accredited laboratory. In addition, our business strategy is focused heavily on our CELx tests being sold as companion diagnostics. This will require obtaining and maintaining partnerships with pharmaceutical companies and successfully completing clinical studies that demonstrate the suitability of the applicable CELx test as a CDx for their targeted therapies.
These activities will require us to expend significant resources. Based on comparable companies in this industry, few research and development projects result in commercially viable products, and success in early clinical studies often is not replicated in later studies. At any point, we may abandon development of a product candidate for several reasons, such as a clinical validation study failing to demonstrate the prospectively defined endpoints of the study. We may also be required to expend considerable resources repeating clinical studies, which would adversely affect the timing for generating potential revenue from a new product and our ability to invest in other products in our pipeline.
Clinical trials are expensive and complex with uncertain outcomes, which may prevent or delay commercialization of our CELx tests.
For our CELx tests to become a CDx for a matching targeted therapy, we must conduct clinical trials to demonstrate that patients who have an abnormal signaling pathway, as identified by our CELx tests, respond to treatment with a matching targeted therapy. Clinical testing is expensive, difficult to design and implement, and can take many years to complete, and its outcome is inherently uncertain. As a company, we have no experience in conducting or participating in clinical trials. We cannot be certain that any future clinical trials will conclusively demonstrate that any CELx test is effective as a CDx. If our trials do not yield positive results, we may be unable to maintain the pharmaceutical company partnerships we build or find additional partners, we may not be able to successfully commercialize our CELx tests or generate any revenue, our business may fail, and you may lose part or all of your investment.
| 31 |
We cannot be certain that our existing clinical trial or future clinical trials, if any, will begin or be completed on time, if at all. We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent our ability to commercialize our CELx tests, such as:
| · | delays in reaching, or failure to reach, agreement on acceptable clinical trial contracts or clinical trial protocols with planned trial sites and/or strategic partners; |
| · | delay or failure in reaching agreement with the FDA or a comparable foreign regulatory authority on a trial design, in obtaining authorization from such authorities to commence the trial, and/or in complying with conditions or other requirements imposed by such regulatory authorities with respect to the trial; |
| · | delay or failure in recruiting and enrolling suitable subjects to participate in one or more clinical trials, or with such participants completing a trial or returning for follow-up during or after the trial; |
| · | clinical sites, investigators or other third-parties deviating from the trial protocol, failing to conduct the trial in accordance with regulatory and contractual requirements, and/or dropping out of a trial; |
| · | regulatory imposition of a clinical hold for any of our clinical trials, where a clinical hold in a trial in one indication would result in a clinical hold for clinical trials in other indications; and |
| · | changes in governmental regulations or administrative actions. |
Significant nonclinical or clinical trial delays could prevent us from maintaining and/or developing new pharmaceutical company partnerships. Delays could also shorten any periods during which we may have the exclusive right to commercialize our CELx tests or allow our competitors to bring products to market before we do. As such, any delays could impair our ability to successfully commercialize our CELx tests and may materially and adversely affect our business, financial condition, results of operations and prospects.
Even if our CELx tests achieve positive clinical trial results, they may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success.
If any of our potential CELx tests, including the CELx HSF Test, achieve positive clinical trial results, they may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success. For example, conventional genomic- or proteomic-based analyses are commonly used today to diagnose cancer and prescribe cancer medications, and physicians may continue to rely on these diagnostic tests instead of adopting the use of a CELx test. The degree of market acceptance of our CELx tests, will depend on a number of factors, including:
| · | their efficacy and other potential advantages compared to alternative diagnostic tests; |
| · | our ability to offer them for sale at competitive prices; |
| · | their convenience and ease of administration compared to alternative diagnostics; |
| · | the willingness of the target patient population to try new diagnostics and of physicians to initiate such diagnostics; |
| · | the strength of marketing and distribution support; |
| · | the availability of third-party coverage and adequate reimbursement for our diagnostic tests; and |
| · | our ability to partner with pharmaceutical companies to develop CDx programs for the new cancer sub-types we discover. |
If our CELx tests do not achieve an adequate level of acceptance, we may never generate significant product revenues and we may not become profitable.
| 32 |
Our business, operational and financial goals may not be attainable if the market opportunities for our CELx tests or our pharmaceutical company partners are smaller than we expect. Our internal research and estimates on market opportunities have not been verified by independent sources, and we have not independently verified market and industry data from third-parties that we have relied on.
The total market opportunities that we believe exist are based on a variety of assumptions and estimates, including the number of potential CDx programs we will be able to successfully pursue, the amount of potential milestone payments that we could receive in CDx programs, the number of patients we will test in clinical trials, the price we will be able to charge for our tests and the total annual number of cancer patients with undiagnosed abnormal cell signaling. In addition, we have relied on third-party publications, research, surveys and studies for information related to determining market opportunities, including without limitation, information on the number of cancer patients and those receiving various forms of treatment, the cost of drug therapy, the amount of revenue generated from various types of drug therapy, the objective response rates of drug therapies, the number of deaths caused by cancer and the expected growth in cancer drug therapy and diagnostic markets. Our internal research and estimates on market opportunities have not been verified by independent sources, and we have not independently verified market and industry data from third-parties that we have relied on. Any or all of our assumptions and/or estimates may prove to be incorrect for several reasons, such as inaccurate reports or information that we have relied on, potential patients or providers not being amenable to using our CELx platform for diagnostic testing or such patients becoming difficult to identify and access, limited reimbursement for companion diagnostics, pricing pressure due to availability of alternative diagnostic tests, or an inability of the CELx tests’ companion drugs to obtain the necessary regulatory approvals for new indications. If any or all of our assumptions and estimates prove inaccurate, we and our CDx pharmaceutical partners may not attain our business, operational and financial goals.
The expected selling price range of our CELx test is an estimate. We have not yet sold any such tests and the actual price we are able to charge may be substantially lower than our expected price range.
We have estimated the selling price range of our CELx test based on the pricing of other diagnostic tests currently available and assumptions regarding the efficacy and market acceptance of our tests. We have not yet sold our CELx tests and cannot be certain of the actual price we may be able to charge. The availability and price of our competitors’ products could limit the demand and the price we are able to charge. We may not achieve our business plan if acceptance is inhibited by price competition, if pharmaceutical companies refuse to pay our expected prices for CELx tests in clinical trials, if physicians are reluctant to switch from other diagnostic tests to our CELx tests or if physicians switch to other new products or choose to reserve our CELx tests for use in limited circumstances. Furthermore, reductions in the reimbursement rate of third-party payors have occurred and may occur in the future. Each of these factors could cause our selling price to be substantially lower than expected, and we may fail to obtain revenue or become profitable.
The insurance coverage and reimbursement status of new diagnostic products is uncertain. Failure to obtain or maintain adequate coverage and reimbursement for CELx tests could limit our ability to market those CELx tests and decrease our ability to generate revenue.
The availability and extent of reimbursement by governmental and private payors is essential for most patients to be able to afford expensive diagnostic tests and treatments. Sales of any of our potential CELx tests will depend substantially, both in the United States and internationally, on the extent to which the costs of our CELx tests will be paid by health maintenance, managed care, and similar healthcare management organizations, or reimbursed by government health administration authorities, private health coverage insurers and other third-party payors. Reimbursement by a payor may depend on a number of factors, including a payor’s determination that the CELx tests are:
| · | neither experimental nor investigational; |
| · | appropriate for the specific patient; |
| · | cost-effective; |
| · | supported by peer-reviewed publications; and |
| · | included in clinical practice guidelines. |
If reimbursement is not available, or is available only to a limited amount, we may not be able to successfully commercialize our CELx tests at expected levels, or potentially at all. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize a sufficient return on our research and development investment.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved diagnostic products. In the United States, the principal decisions about reimbursement for new diagnostic products and services are typically made by CMS. CMS decides whether and to what extent a new product or service will be covered and reimbursed under Medicare. Private payors tend to follow CMS to a substantial degree. As such, a significant portion of our potential revenue depends on CMS approving coverage and reimbursement of our CELx tests.
| 33 |
Outside the United States, international operations are generally subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis on cost-containment initiatives in Europe, Canada and other countries has and will continue to put pressure on the pricing and usage of diagnostic tests such as our potential CELx tests. In many countries, particularly the countries of the European Union, the prices of medical products are subject to varying price control mechanisms as part of national health systems. In these countries, pricing negotiations with governmental authorities can take considerable time. To obtain reimbursement or pricing approval in some countries, we may be required to demonstrate the cost-effectiveness of our CELx tests relative to other available diagnostic tests. The prices of products under such systems may be substantially lower than in the United States. Other countries allow companies to fix their own prices for products, but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our CELx tests. Accordingly, in markets outside the United States, the reimbursement for our potential CELx tests may be reduced compared with the United States and may be insufficient to generate commercially reasonable revenue and profit.
Moreover, increasing efforts by governmental and third-party payors, in the United States and internationally, to cap or reduce healthcare costs may cause such organizations to limit both coverage and level of reimbursement for new products approved and, as a result, they may not cover or provide adequate payment for our potential CELx tests. The downward pressure on healthcare costs in general, particularly prescription drugs and surgical procedures and other treatments, has become very intense. We expect to experience pricing pressures in connection with the sale of any CELx tests due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes.
We may encounter difficulties in commercializing and marketing our products.
In order to commercialize any CELx test, we must build marketing, sales, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and we may not be successful in doing so. For each CELx test we develop, we intend to pursue development agreements with the pharmaceutical companies that provide matching targeted therapies. Once we have completed the analytical validation of a CELx test, we plan to target key opinion leaders (KOLs) to build product awareness. Once we have clinical validation data available, we expect to expand our sales and marketing efforts to target the broader market, and coordinate our go-to-market activities with those of our partner pharmaceutical companies. These activities will be expensive and time consuming and will require significant attention of our executive officers to manage. Furthermore, there is no guarantee that any new drug indications will require our CELx tests as a CDx or that any pharmaceutical company will effectively coordinate sales and marketing activities with us. Any failure or delay in these activities would adversely impact the commercialization our CELx platform, and our business, financial condition, results of operations and prospects may be materially and adversely affected.
We may encounter difficulties in managing growth, which could disrupt our operations.
To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Due to our limited financial resources, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
We face significant competition from other diagnostic companies and our operating results will suffer if we fail to compete effectively.
The diagnostic testing related industry is intensely competitive. We have competitors both in the United States and abroad, including universities and other research institutions and providers of diagnostics that focus on developing genomic or proteomic analyses of a patient’s diseased cells or theranostic tests to predict specific patient responses to a drug therapy. Many of our competitors have substantially greater financial, technical and other resources, such as larger research and development staff and well-established marketing and sales forces. Our competitors may succeed in developing, acquiring or licensing, on an exclusive basis, products or services that are more effective or less costly than the CELx tests that we are currently developing or that we may develop. In addition, established medical technology, biotechnology and/or pharmaceutical companies may invest heavily to accelerate discovery and development of diagnostic tests that could make our CELx tests less competitive.
| 34 |
Our ability to compete successfully will depend largely on our ability to:
| · | discover and develop CELx tests for cancer sub-types that are superior to other products in the market; |
| · | demonstrate compelling advantages in the efficacy and convenience of our CELx tests on a cost competitive basis; |
| · | attract qualified scientific, product development and commercial personnel; |
| · | obtain and maintain patent and other proprietary protection as necessary for our CELx platform; |
| · | obtain required U.S. and international regulatory approvals; |
| · | successfully collaborate with research institutions and pharmaceutical companies in the discovery, development and commercialization of our current and future CELx tests; and |
| · | successfully expand our operations and build a sales force to support commercialization. |
We may not be able to compete effectively if we are unable accomplish one or more of these objectives.
If our sole laboratory facility becomes inoperable, we will be unable to perform our tests and our business will be harmed.
We do not have redundant laboratory facilities. We perform all of our diagnostic services in our laboratory located in Minneapolis, Minnesota. Our facility and the equipment we use to perform our tests would be costly to replace and could require substantial lead time to repair or replace. The facility may be harmed or rendered inoperable by physical damage from fire, floods, tornadoes, power loss, telecommunications failures, break-ins and similar events, which may render it difficult or impossible for us to perform our tests for some period of time. The inability to perform our tests may result in the loss of customers or harm our reputation, and we may be unable to regain those customers in the future. Although we possess insurance for damage to our property and the disruption of our business, this insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all.
In order to rely on a third party to perform our tests, we could only use another facility with established state licensure and CLIA accreditation under the scope of which our potential CELx tests could be performed following validation and other required procedures. We cannot assure you that we would be able to find another CLIA-certified facility willing to adopt CELx tests and comply with the required procedures, or that this laboratory would be willing to perform the tests for us on commercially reasonable terms.
We must hire and retain a qualified sales force.
Our ability to grow revenue for our CELx tests is dependent upon our ability to build an effective sales team. We do not currently have a dedicated sales force, and building one will be an expensive and time-consuming process. We face intense competition for qualified sales personnel and our inability to hire or retain an adequate number of sales representatives could limit our ability to maintain or expand our business and increase sales. Even if we are able to increase our sales force, our new sales personnel may not provide sufficient high quality service and attention to effectively market and sell our CELx platform. If we are unable to develop our marketing and sales networks or if our sales personnel do not perform as expected, we may be unable to maintain or grow our existing business and our business, financial condition, results of operations and prospects may be materially and adversely affected.
We will be dependent on our ability to attract and retain key personnel.
Our operations will be materially dependent upon the services of our officers and key employees, including Brian F. Sullivan, our Chief Executive Officer, and Dr. Lance G. Laing, our Chief Science Officer. Successful implementation of our business plan will also require the services of other consultants and additional personnel. We cannot assure you that we will be able to attract and retain such persons as employees, independent contractors, consultants or otherwise. If we are not able to attract individuals with the skills required for our business, or if we lose the services of either Mr. Sullivan or Dr. Laing, we may be unable successfully to implement our business plan.
| 35 |
Our inability to raise additional capital on acceptable terms in the future may limit our ability to develop and commercialize our CELx platform.
We may require additional capital to finance capital expenditures and operating expenses over the next several years as we launch our CELx platform and expand our infrastructure, commercial operations and research and development activities. We may seek to raise additional capital through equity offerings, debt financings, collaborations or licensing arrangements. Additional funding may not be available to us on acceptable terms, or at all. If we raise funds by issuing equity securities, dilution to our stockholders could result. Any equity securities issued may also provide for rights, preferences or privileges senior to those of holders of our existing securities. The incurrence of additional indebtedness or the issuance of certain equity securities could result in increased fixed payment obligations and could also include restrictive covenants, such as limitations on our ability to incur additional debt or issue additional equity, limitations on our ability to acquire or license intellectual property rights, and other operating restrictions that could adversely affect our ability to conduct our business. In the event that we enter into collaborations or licensing arrangements to raise capital, we may be required to accept unfavorable terms. If we are not able to secure additional funding when needed, we may have to delay, reduce the scope of or eliminate one or more research and development programs or selling and marketing initiatives. In addition, we may have to work with a partner on one or more of our products or market development programs, which could lower the economic value of those programs to our company.
Risks Related to Our Reliance on Third Parties
We will rely on collaboration with third parties to conduct our clinical trials, including the current trial involving the CELx HSF Test, and those third parties may not perform satisfactorily.
We will rely on third parties to conduct clinical trials for our CELx tests. For our CELx HSF Test, we are collaborating with Genentech and the NSABP to conduct a 54-patient single-arm interventional trial that is expected to obtain interim results in late 2018 and final results six to nine months later. We are funding the patient-related costs for this trial and Genentech is supplying the drugs. We will rely on NSABP to conduct our clinical trial of the CELx HSF Test, including setting up clinical sites, enrolling patients, and managing clinical data.
We expect to field additional clinical trials to evaluate new potential indications for drugs with patients identified by one of our new CELx tests. NSABP, other contract research organizations that we hire and/or pharmaceutical companies we partner with might not successfully carry out their contractual duties, meet expected deadlines, or conduct our planned clinical trials in accordance with regulatory requirements or our stated protocols. Any of them may also terminate their relationship with us for a variety of reasons. Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. If we need to enter into alternative arrangements, we may not be able to complete our clinical trials and may not be able to, or may be delayed in our efforts to, successfully commercialize our potential CELx tests.
The pharmaceutical companies that we partner with may not be successful in receiving regulatory approval for drug indications or may not commercialize their companion therapies for our expected CDx programs.
While we intend to provide our pharmaceutical company partners with new patient populations for such partners’ existing or investigational targeted therapies, there can be no assurances that such partners will be able to obtain regulatory approval for new indications to treat these patient populations or otherwise be successful in commercializing these new therapies. The pharmaceutical companies we partner with:
| · | may not meet clinical trial endpoint targets in evaluating efficacy of a targeted therapy in the patient population; |
| · | may encounter regulatory or production difficulties that could constrain the supply of the companion therapies; |
| · | may have difficulties gaining acceptance of the use of the companion therapies in the clinical community; |
| · | may not pursue commercialization of any companion therapies; |
| · | may elect not to continue or renew commercialization programs based on changes in their strategic focus or available funding, or external factors, such as an acquisition, that divert resources or create competing priorities; |
| · | may not commit sufficient resources to the marketing and distribution of such companion therapies; or |
| · | may terminate their relationship with us. |
| 36 |
Any of these factors could adversely affect our commercialization strategy, business, results of operations and financial condition.
Our instrument or reagent suppliers may fail to meet our quality requirements for the items we purchase or fail to provide a continuous supply of the items we utilize to perform our CELx tests.
We utilize highly specialized reagents and instruments to perform our CELx tests. We may be unable to find suitable replacement reagents and instruments on a timely basis, if at all. Interruption in the supply of these items or degradation in their quality could delay analytical and clinical studies, and/or render us unable to deliver CELx tests. This would interrupt sales and adversely affect our business, results of operations and financial condition.
Performance issues or price increases by our shipping carriers could adversely affect our business, results of operations and financial condition, and harm our reputation and ability to provide our CELx tests on a timely basis.
Expedited, reliable shipping is essential to our operations. Should our shipping carrier encounter delivery performance issues such as loss, damage or destruction of a sample, such occurrences may damage our reputation and lead to decreased demand for our services and increased cost and expense to our business. In addition, any significant increase in shipping rates could adversely affect our operating margins and results of operations. Similarly, strikes, severe weather, natural disasters or other service interruptions by delivery services we use would adversely affect our ability to receive and process patient samples on a timely basis. There are only a few providers of overnight nationwide transport services, and there can be no assurance that we will be able to maintain arrangements with providers on acceptable terms, if at all.
Risks Related to Government Regulation
Our CELx tests represent a novel approach to companion diagnostics, which could result in heightened regulatory scrutiny, delays in clinical development, or delays in our ability to commercialize any products.
Our unique and proprietary CELx technology is the first cancer diagnostic platform we are aware of that can detect the underlying signaling dysfunction driving a patient’s cancer. Because this is a novel approach to companion diagnostics, there can be no assurance as to the length of a clinical trial period, the number of patients the FDA or another applicable regulatory authority will require to be enrolled in the trials in order to establish the safety and efficacy of our CELx tests and the companion drugs, or that the data generated in these trials will be acceptable to the FDA or another applicable regulatory authority to support marketing approval of new indications for the companion drugs. This could delay or prohibit our clinical trials and/or commercialization of our CELx tests.
If the FDA were to begin regulating our tests, we could incur substantial costs and delays associated with trying to obtain premarket clearance or approval.
Most LDTs, are not currently subject to FDA regulation, although reagents, instruments, software or components provided by third parties and used to perform LDTs may be subject to regulation. We believe that the CELx tests are LDT’s, which is a term that describes tests that are designed and performed within a single laboratory. As a result, we believe the CELx tests are not currently subject to regulation by the FDA in accordance with the FDA’s current policy of exercising enforcement discretion regarding LDTs.
Historically, the FDA has not required laboratories that furnish only LDTs to comply with the agency’s requirements for medical devices (e.g., establishment registration, device listing, quality systems regulations, premarket clearance or premarket approval, and post-market controls). In mid-2014, the FDA published a Draft Guidance document describing a proposed approach for a regulatory framework for LDTs, but in late 2016, the FDA indicated it no longer intended to finalize the LDT Guidance Document at that time. It is not clear when or if the FDA will seek to alter the current LDT regulatory framework in the future. We cannot provide any assurance that FDA regulation, including premarket review, will not be required in the future for our tests, whether through additional guidance issued by the FDA, new enforcement policies adopted by the FDA or new legislation enacted by Congress. We cannot predict with certainty the timing or content of future legislation enacted or guidance issued regarding LDTs, or how it will affect our business.
| 37 |
If premarket review is required by the FDA at a future date or if we decide to voluntarily pursue FDA premarket review of our CELx tests, there can be no assurance that our CELx tests or any tests we may develop in the future will be cleared or approved by the FDA on a timely basis, if at all, nor can there be assurance that labeling claims will be consistent with our current claims or adequate to support continued adoption of and reimbursement for our CELx tests. If our CELx tests are allowed to remain on the market but there is uncertainty in the marketplace about our tests, if they are labeled investigational by the FDA, or if labeling claims the FDA allows us to make are more limited than we expect, reimbursement may be adversely affected and we may not be able to sell our CELx tests. Compliance with FDA regulations would increase the cost of conducting our business, and subject us to heightened regulation and scrutiny by the FDA and penalties for failure to comply with these requirements.
If we fail to obtain required federal and state laboratory licenses, we could lose the ability to perform our tests.
Clinical laboratory tests, including our CELx tests, are regulated under CLIA. CLIA is a federal law that regulates clinical laboratories that perform testing on specimens derived from humans for the purpose of providing information for the diagnosis, prevention or treatment of disease. CLIA regulations mandate specific standards for laboratories in the areas of personnel qualifications, administration, and participation in proficiency testing, patient test management and quality assurance. CLIA certification is also required in order for us to be eligible to bill state and federal healthcare programs, as well as many private third-party payers, for any tests we launch. We will also be required to maintain state licenses in certain states to conduct testing in our laboratories. While we currently have CLIA Certification for our Minnesota laboratory, failure to maintain this certification would adversely affect our ability to launch our CELx tests.
We generate medical waste and could face substantial liability if we violate laws with respect to the handling of medical waste.
We generate regulated medical waste in the normal course of performing our CELx tests. This subjects us to a variety of federal, state and local environmental and safety laws and regulations. Some of the regulations under the current regulatory structure provide for strict liability, holding a party potentially liable without regard to fault or negligence. We could be held liable for damages and fines as a result of our, or others’, business operations should contamination of the environment or individual exposure to hazardous substances occur related to our business. We cannot predict how changes in laws or development of new regulations will affect our business operations or the cost of compliance.
Failure to comply with the HIPAA security and privacy regulations may increase our operational costs.
A portion of the data that we obtain and handle for or on behalf of our clients is considered protected health information, or PHI, subject to the Health Insurance Portability and Accountability Act of 1996, or HIPAA. Under HIPAA and our contractual agreements with our HIPAA-covered entity health plan customers, we are considered a “business associate” to those customers, and are required to maintain the privacy and security of PHI in accordance with HIPAA and the terms of our business associate agreements with our clients, including by implementing HIPAA-required administrative, technical and physical safeguards. We are also required to maintain similar business associate agreements with our subcontractors that have access to PHI of our customers in rendering services to us or on our behalf. We will incur significant costs to establish and maintain these safeguards and, if additional safeguards are required to comply with HIPAA regulations or our clients’ requirements, our costs could increase further, which would negatively affect our operating results. Furthermore, we cannot guarantee that such safeguards have been and will continue to be adequate under applicable laws. If we have failed, or fail in the future, to maintain adequate safeguards, or we or our agents or subcontractors use or disclose PHI in a manner prohibited or not permitted by HIPAA, our subcontractor business associate agreements, or our business associate agreements with our customers, or if the privacy or security of PHI that we obtain and handle is otherwise compromised, we could be subject to significant liabilities and consequences.
| 38 |
We will also need to expend a considerable amount of resources complying with other federal, state and foreign laws and regulations. If we are unable to comply or have not complied with such laws, we could face substantial penalties or other adverse actions.
Our operations are subject, directly or indirectly, to other federal, state and foreign laws and regulations that are complex and their application to our specific products, services and relationships may not be clear and may be applied to our business in ways that we do not anticipate. Compliance with laws and regulations will require us to expend considerable resources implementing internal policies and procedures for compliance and ongoing monitoring, and will require significant attention of our management team. This will be challenging as an early-stage company with limited financial resources and human capital. These laws include, for example:
| · | Title XI of the Social Security Act, commonly referred to as the federal Anti-Kickback Statute, which prohibits the knowing and willful offer, payment, solicitation or receipt of remuneration, directly or indirectly, in cash or in kind, in return for or to reward the referral of patients or arranging for the referral of patients, or in return for the recommendation, arrangement, purchase, lease or order of items or services that are covered, in whole or in part, by a federal healthcare program such as Medicare or Medicaid; |
| · | The civil False Claims Act, that forbids the knowing submission or “causing the submission” of false or fraudulent information or the failure to disclose information in connection with the submission and payment of claims for reimbursement to Medicare, Medicaid, federal healthcare programs or private health plans; |
| · | The federal Physician Self-referral Law, commonly known as the Stark Law, which prohibits physicians from referring Medicare or Medicaid patients to providers of “designated health services” with whom the physician or a member of the physician’s immediate family has an ownership interest or compensation arrangement, unless a statutory or regulatory exception applies, and similar state equivalents that may apply regardless of payor; and |
| · | The U.S. Foreign Corrupt Practices Act of 1977, as amended, or FCPA, the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, and the USA PATRIOT Act, which among other things, prohibit companies and their employees, agents, third-party intermediaries, joint venture partners and collaborators from authorizing, promising, offering, or providing, directly or indirectly, improper payments or benefits to recipients in the public or private sector. |
Many states and foreign governments have adopted similar laws and regulations. Violations of law could subject us to civil or criminal penalties, monetary fines, disgorgement, individual imprisonment, contractual damages, reputational harm, diminished profits and future earnings and curtailment of our operations. We could also be required to change or terminate some portions of operations or business or could be disqualified from providing services to healthcare providers doing business with government programs.
New legislation and regulations could be passed that affect our operations and result in additional risks and/or costs to our business.
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the testing, approval, manufacturing and marketing of products that are or will be regulated by the FDA or CMS. In addition to new legislation, CMS and FDA regulations and policies are often revised or interpreted by the agencies in ways that may significantly affect our business and our products. It is impossible to predict whether further legislative changes will be enacted or FDA or CMS regulations, guidance, policies or interpretations will be changed, or what the impact of such changes, if any, may be. The 2016 presidential election and change in administration make it even more difficult to predict if and how federal regulations may change and/or federal agencies might alter their positions. Changes in laws and the development of new regulations could affect our business operations and/or the cost of compliance.
| 39 |
Risks Related to Intellectual Property
If we are unable to obtain and maintain intellectual property protection for our technology, or if the scope of the intellectual property protection obtained is not sufficiently broad, our competitors could develop and commercialize technology and diagnostic tests similar or identical to ours, and our ability to successfully commercialize our technology and diagnostic tests may be impaired.
Our ability to compete successfully will depend in part on our ability to obtain and enforce patent protection for our products, preserve our trade secrets and operate without infringing the proprietary rights of third parties. We have applied for patents that protect our technology. Our patent portfolio includes one U.S. patent, five pending U.S. patent applications, one pending international PCT patent application, and numerous corresponding non-U.S. patent applications. Each patent and patent application covers methods of use. However, we cannot assure you that our intellectual property position will not be challenged or that all patents for which we have applied will be granted. The validity and breadth of claims in patents involve complex legal and factual questions and, therefore, may be highly uncertain. Uncertainties and risks that we face include the following:
| · | our pending or future patent applications may not result in the issuance of patents; |
| · | the scope of any existing or future patent protection may not exclude competitors or provide competitive advantages to us; |
| · | our patents may not be held valid if subsequently challenged; |
| · | other parties may claim that our products and designs infringe the proprietary rights of others—even if we are successful in defending our patents and proprietary rights, the cost of such litigation may adversely affect our business; and |
| · | other parties may develop similar products, duplicate our products, or design around our patents. |
The patent prosecution process is expensive and time-consuming, and we may not be able to file, prosecute, maintain, enforce or license all necessary or desirable patent applications at a reasonable cost or in a timely manner, or in all jurisdictions. We may choose not to seek patent protection for certain innovations and may choose not to pursue patent protection in certain jurisdictions, and under the laws of certain jurisdictions, patents or other intellectual property rights may be unavailable or limited in scope. It is also possible that we will fail to identify patentable aspects of our discovery and nonclinical development output before it is too late to obtain patent protection.
The patent position of companies like ours is highly uncertain, involves complex legal and factual questions and has in recent years been the subject of much litigation. The U.S. Patent and Trademark Office, or U.S. PTO, has not established a consistent policy regarding the breadth of claims that it will allow in medical technology patents. In addition, the laws of foreign jurisdictions may not protect our rights to the same extent as the laws of the United States. For example, India and China do not allow patents for methods of treating the human body. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in our owned or licensed patents or pending patent applications, or that we were the first to file for patent protection of such inventions. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain. Our pending and future patent applications may not result in patents being issued that protect our technology or CELx tests, in whole or in part, or which effectively prevent others from commercializing competitive technologies and diagnostic tests. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection.
Moreover, we may be subject to a third-party pre-issuance submission of prior art to the U.S. PTO or patent offices in foreign jurisdictions, or become involved in opposition, derivation, reexamination, inter parties review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology and compete directly with us, without payment to us, or result in our inability to commercialize CELx platform without infringing third-party patent rights. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, it could dissuade companies from collaborating with us to develop or commercialize current or future CELx tests.
Even if our owned patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our owned patents by developing similar or alternative technologies or products in a non-infringing manner.
| 40 |
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our owned patents may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and product candidates, or limit the duration of the patent protection of our technology and potential diagnostic tests. Given the amount of time required for the development, testing and regulatory review of new diagnostic tests, patents protecting such tests might expire before or shortly after such candidates are commercialized. As a result, our owned patent portfolio may not provide us with sufficient rights to exclude others from commercializing diagnostic tests similar or identical to ours.
Patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted and may also affect patent litigation. The U.S. PTO recently developed new regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, only became effective on March 16, 2013. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business and financial condition. Depending on future actions by the U.S. Congress, the federal courts, and the U.S. PTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future. In addition, there may be patent law reforms in foreign jurisdictions that could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents in those foreign jurisdictions.
Third parties may initiate legal proceedings alleging that we are infringing their intellectual property rights, the outcome of which would be uncertain and could have a material adverse effect on the success of our business.
Our commercial success depends upon our ability, and the ability of our collaborators, to develop, manufacture, market and sell our CELx tests and use our proprietary technologies without infringing the proprietary rights of third parties. There is considerable intellectual property litigation in the medical technology, biotechnology and pharmaceutical industries. We may become party to, or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our CELx platform, including interference or derivation proceedings before the U.S. PTO and similar bodies in other jurisdictions. Third parties may assert infringement claims against us based on existing patents or patents that may be granted in the future.
If we are found to infringe a third party’s intellectual property rights, we could be required to obtain a license from such third party to continue developing and marketing our CELx platform and CELx tests. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. We could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees if we are found to have willfully infringed a patent. A finding of infringement could prevent us from commercializing our CELx platform or force us to cease some of our business operations, which could materially harm our business. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business.
We may be subject to claims by third parties asserting that our employees or we have misappropriated their intellectual property, or claiming ownership of what we regard as our own intellectual property.
Our current and future employees may have been previously employed at universities or other biotechnology, diagnostic technology or pharmaceutical companies, including our competitors or potential competitors and strategic partners. Although we try to ensure that our employees do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that these employees or we have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such employee’s former employer. Litigation may be necessary to defend against these claims.
| 41 |
In addition, while it is our policy to require our employees and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own. Our and their assignment agreements may not be self-executing or may be breached, and we may be forced to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of what we regard as our intellectual property.
If we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in prosecuting or defending against such claims, litigation could result in substantial costs and be a distraction to management.
Any lawsuits relating to infringement of intellectual property rights necessary to defend ourselves or enforce our rights will be costly and time consuming, and could be unsuccessful.
Because competition in our industry is intense, competitors may infringe or otherwise violate our issued patents, patents of our licensors or other intellectual property. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time consuming, and could distract our technical and management personnel from their normal responsibilities. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims against us alleging that we infringe their patents. In addition, in a patent infringement proceeding, a court may decide that a patent of ours is invalid or unenforceable, in whole or in part, construe the patent’s claims narrowly or refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation proceeding could put one or more of our patents at risk of being invalidated or interpreted narrowly. We may also elect to enter into license agreements in order to settle patent infringement claims or to resolve disputes prior to litigation, and any such license agreements may require us to pay royalties and other fees that could be significant. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure.
If we are not able to prevent disclosure of our trade secrets and other proprietary information, the value of our CELx platform could be significantly diminished.
We rely on trade secret protection to protect our interests in proprietary know-how and in processes for which patents are difficult to obtain or enforce. We may not be able to protect our trade secrets adequately. We have a policy of requiring our consultants, advisors and strategic partners to enter into confidentiality agreements and our employees to enter into invention, non-disclosure and non-compete agreements. However, no assurance can be given that we have entered into appropriate agreements with all parties that have had access to our trade secrets, know-how or other proprietary information. There is also no assurance that such agreements will provide meaningful protection of our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure of information. Furthermore, we cannot provide assurance that any of our employees, consultants, contract personnel, or strategic partners, either accidentally or through willful misconduct, will not cause serious damage to our programs and/or our strategy, for example by disclosing important trade secrets, know-how or proprietary information to our competitors. It is also possible that our trade secrets, know-how or other proprietary information could be obtained by third parties as a result of breaches of our physical or electronic security systems. Any disclosure of confidential data into the public domain or to third parties could allow our competitors to learn our trade secrets and use the information in competition against us. In addition, others may independently discover our trade secrets and proprietary information. Any action to enforce our rights is likely to be time consuming and expensive, and may ultimately be unsuccessful, or may result in a remedy that is not commercially valuable. These risks are accentuated in foreign countries where laws or law enforcement practices may not protect proprietary rights as fully as in the United States. Any unauthorized disclosure of our trade secrets or proprietary information could harm our competitive position.
| 42 |
Risks Relating to Our Common Stock
Our executive officers, directors and principal stockholders, if they choose to act together, have the ability to control all matters submitted to stockholders for approval.
Our executive officers and directors, combined with our stockholders who each own more than 5% of our outstanding common stock, in the aggregate, own approximately 50% of our outstanding capital stock. As a result, if these stockholders were to choose to act together, they would be able to control all matters submitted to our stockholders for approval, as well as our management and affairs. For example, these persons, if they choose to act together, would control the election of directors and approval of any merger, consolidation or sale of all or substantially all of our assets. This concentration of ownership control may:
| · | delay, defer or prevent a change in control; |
| · | make changes to our management and the board of directors challenging for other stockholders; or |
| · | impede a merger, consolidation, takeover or other business combination involving us that other stockholders may desire or may result in you obtaining a premium for your shares. |
Our internal control over financial reporting does not currently meet the standards required by Section 404 of the Sarbanes-Oxley Act, and failure to achieve and maintain effective internal control over financial reporting in accordance with Section 404 of the Sarbanes-Oxley Act could have a material adverse effect on our business and stock price.
We previously have not been required to maintain internal control over financial reporting in a manner that meets the standards of publicly traded companies required by Sections 404(a) or 404(b) of the Sarbanes-Oxley Act. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with GAAP. We are not currently in compliance with, and we cannot be certain when we will be able to implement the requirements of Section 404(a). We may encounter problems or delays in implementing any changes necessary to make a favorable assessment of our internal control over financial reporting. If we cannot favorably assess the effectiveness of our internal control over financial reporting, investors could lose confidence in our financial information and the price of our common stock could decline.
Additionally, the existence of any material weakness or significant deficiency would require management to devote significant time and incur significant expense to remediate any such material weaknesses or significant deficiencies and management may not be able to remediate any such material weaknesses or significant deficiencies in a timely manner. The existence of any material weakness in our internal control over financial reporting could also result in errors in our financial statements that could require us to restate our financial statements causing us to fail to meet our reporting obligations and cause stockholders to lose confidence in our reported financial information, all of which could materially and adversely affect us.
Provisions in our corporate charter documents and under Delaware law could make an acquisition of our company, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our certificate of incorporation and our bylaws may discourage, delay or prevent a merger, acquisition or other change in control of our company that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. In addition, because our board of directors will be responsible for appointing the members of our management team, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. Among other things, these provisions:
| · | allow the authorized number of our directors to be changed only by resolution of our board of directors; |
| · | limit the manner in which stockholders can remove directors from our board of directors; |
| · | establish advance notice requirements for stockholder proposals that can be acted on at stockholder meetings and nominations to our board of directors; |
| · | require that stockholder actions must be effected at a duly called stockholder meeting and prohibit actions by our stockholders by written consent; |
| · | limit who may call stockholder meetings; |
| · | authorize our board of directors to issue preferred stock without stockholder approval, which could be used to institute a “poison pill” that would work to dilute the stock ownership of a potential hostile acquirer, effectively preventing acquisitions that have not been approved by our board of directors; and |
| 43 |
| · | require the approval of the holders of at least two-thirds of the votes that all our stockholders would be entitled to cast to amend or repeal specified provisions of our certificate of incorporation or bylaws. |
Moreover, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
Any of these provisions of our charter documents or Delaware law could, under certain circumstances, depress the market price of our common stock.
The price of our common stock may be volatile and fluctuate substantially, which could result in substantial losses for purchasers of our common stock.
Our stock price may be extremely volatile. The stock market in general and the market for smaller medical technology companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, you may not be able to sell your common stock at or above the initial public offering price. The market price for our common stock may be influenced by many factors, including:
| · | the success of competitive products or technologies; |
| · | results of planned clinical trials of our CELx HSF Test or other CELx tests may develop in the future; |
| · | regulatory or legal developments in the United States and other countries; |
| · | developments or disputes concerning patent applications, issued patents or other proprietary rights; |
| · | the recruitment or departure of key personnel; |
| · | the level of expenses related to any of our CELx tests or clinical development programs; |
| · | actual or anticipated changes in estimates as to financial results, development timelines or recommendations by securities analysts; |
| · | operating results that fail to meet expectations of securities analysts that cover our company; |
| · | variations in our financial results or those of companies that are perceived to be similar to us; |
| · | changes in the structure of healthcare payment systems; |
| · | market conditions in the pharmaceutical, biotechnology and medical technology sectors; |
| · | sales of our stock by us, our insiders and our other stockholders; |
| · | general economic and market conditions; and |
| · | the other factors described in this “Risk Factors” section. |
We may be subject to securities litigation, which is expensive and could divert management attention.
Smaller medical technology companies like us often experience volatile stock prices, and companies that have experienced volatility in the market price of their stock have been subject to an increased incidence of securities class action litigation. We may be the target of this type of litigation in the future. Securities litigation against us could result in substantial costs and divert our management’s attention from other business concerns, which could seriously harm our business.
If securities or industry analysts do not publish research or reports about our business, or publish negative reports about our business, our stock price and trading volume could decline.
The trading market for our common stock depends in part on the research and reports that securities or industry analysts publish about us or our business. We do not have any control over these analysts. There can be no assurance that analysts will cover us or provide favorable coverage. If one or more of the analysts who cover us downgrade our stock or change their opinion of our stock, our stock price would likely decline. If one or more of these analysts cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause our stock price or trading volume to decline.
| 44 |
We are an “emerging growth company,” and the reduced disclosure requirements applicable to emerging growth companies may make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and may remain an emerging growth company for up to five years. For so long as we remain an emerging growth company, we are permitted and intend to rely on exemptions from certain disclosure requirements that are applicable to other public companies that are not emerging growth companies. These exemptions include:
| · | being permitted to provide only two years of audited financial statements, in addition to any required unaudited interim financial statements, with correspondingly reduced “Management’s Discussion and Analysis of Financial Condition and Results of Operations” disclosure; |
| · | not being required to comply with the auditor attestation requirements in the assessment of our internal control over financial reporting of Section 404(b) of the Sarbanes-Oxley Act; |
| · | not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements; |
| · | reduced disclosure obligations regarding executive compensation; and |
| · | exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
We have taken advantage of reduced reporting burdens in this report. We cannot predict whether investors will find our common stock less attractive if we rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
We incur increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives and corporate governance practices.
As a public company, and particularly after we are no longer an emerging growth company, we will incur significant legal, accounting and other expenses that we did not incur as a private company. The Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of The Nasdaq Capital Market and other applicable securities rules and regulations impose various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations have increased our ongoing legal and financial compliance costs and will make some activities more time-consuming and costly.
Pursuant to Section 404 of the Sarbanes-Oxley Act, or Section 404, we will first be required to furnish a report by our management on our internal control over financial reporting for the year ending December 31, 2018. As discussed above, if we cease to be an emerging growth company, we will be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm as required by Section 404(b). To achieve compliance with Section 404 within the prescribed period, we will be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that we will not be able to conclude, within the prescribed timeframe or at all, that our internal control over financial reporting is effective as required by Section 404. If we identify one or more material weaknesses, it could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
Since we do not anticipate paying any cash dividends on our capital stock in the foreseeable future, stock price appreciation, if any, will be your sole source of gain.
We currently intend to retain all of our future earnings, if any, to finance the growth and development of our business. In addition, the terms of any future debt agreements may preclude us from paying dividends. As a result, appreciation, if any, in the market price of our common stock will be your sole source of gain for the foreseeable future.
| 45 |
ITEM 1B. Unresolved Staff Comments
Not applicable
Facilities
We currently lease and occupy approximately 5,000 square feet in Minneapolis, Minnesota, which includes our clinical laboratory and offices. This lease expires in May 2018 and is renewable annually. In September 2017, the Company entered into an operating lease agreement for approximately 16,000 square feet in Minneapolis, Minnesota to accommodate expansion in research and development and general corporate office needs. The new lease commences and the Company will be moving to the facility in May 2018, in conjunction with the termination of the existing lease. The lease agreement extends through April 2021, with the right to extend the term for two periods of one year each and provides for monthly rent, real estate taxes and operating expenses. We believe that the new leased space is adequate to meet current and anticipated future requirements and that additional or substitute space will be available as needed to accommodate any expansions that our operations require.
From time to time we may be involved in disputes or litigation relating to claims arising out of our operations. We are not currently a party to any legal proceedings that could reasonably be expected to have a material adverse effect on our business, financial condition and results of operations.
ITEM 4. Mine Safety Disclosures
Not applicable.
ITEM 5. Market For Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Price Information
Our common stock has been listed on The Nasdaq Capital Market under the symbol “CELC” since September 20, 2017. Prior to that date, there was no public trading market for our common stock. Our common stock priced at $9.50 per share in our initial public offering on September 19, 2017. The following table sets forth for the periods indicated the high and low intra-day sale prices per share of our common stock as reported on The Nasdaq Capital Market:
| High | Low | |||||||
| Year Ended December 31, 2017 | ||||||||
| Third quarter from and after September 20, 2017 | $ | 15.55 | $ | 11.10 | ||||
| Fourth quarter | $ | 21.98 | $ | 11.68 | ||||
Holders
As of February 28, 2018, there were approximately 117 holders of record of our common stock. The actual number of holders of common stock is greater than this number of record holders and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers and nominees. The number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
| 46 |
Dividends
We have never declared or paid any cash dividends on our common stock. We currently intend to retain our future earnings, if any, to finance the operation and expansion of our business. We do not expect to pay cash dividends on our common stock in the foreseeable future. Payment of future cash dividends, if any, will be at the discretion of our board of directors after taking into account various factors, including our financial condition, operating results, current and anticipated cash needs, outstanding indebtedness and plans for expansion and restrictions imposed by lenders, if any.
Recent Sales of Unregistered Securities
In September 2017, upon the closing of our initial public offering, our then-outstanding convertible notes were automatically converted into an aggregate of 881,911 shares of common stock. These shares of common stock were issued pursuant to an exemption from the registration requirements of the Securities Act provided by Section 3(a)(9) or Section 4(a)(2) of the Securities Act.
Issuer Purchases of Equity Securities
None.
Equity Compensation Plan Information
The information required by this Item concerning equity compensation plans is incorporated herein by reference from Part III, Item 11 of this report.
Use of Proceeds from Registered Securities
On September 22, 2017, we issued and sold 2,760,000 shares of our common stock in the initial public offering at a public offering price of $9.50 per share, for aggregate gross proceeds of $26.2 million. All of the shares issued and sold in the initial public offering were registered under the Securities Act pursuant to a Registration Statement on Form S-1 (File No. 333-220128), which was declared effective by the SEC on September 19, 2017. Craig-Hallum Capital Group LLC acted as the sole manager for the offering. The offering terminated on September 22, 2017.
The net offering proceeds to us, after deducting underwriting discounts of approximately $1.8 million and offering expenses paid by us totaling approximately $1.1 million, were approximately $23.3 million. No offering expenses were paid directly or indirectly to any of our directors or officers (or their associates) or persons owning 10.0% or more of any class of our equity securities or to any other affiliates.
At December 31, 2017, the net proceeds from our initial public offering were held in a diversified portfolio of bank deposits, government money market funds, government securities (U.S. Treasury and U.S. government agency securities), and certificates of deposit. All investments are highly liquid, with liquidity and capital preservation being the primary investment objectives. There has been no material change in our planned uses of the net proceeds from those described in the Prospectus dated September 19, 2017.
ITEM 6. Selected Financial Data
The following tables present, as of the dates and for the years indicated, our selected historical financial data, as indicated therein. The statement of operations data for the years ended December 31, 2017 and 2016 and the balance sheet data as of December 31, 2017 and 2016 are derived from our audited financial statements that are included elsewhere in this report. Our historical results are not indicative of the results to be expected in the future.
You should read this information together with our financial statements and the related notes, as well as the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this report.
| 47 |
| Years Ended | ||||||||
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Statements of Operations Data: | ||||||||
| Operating expenses: | ||||||||
| Research and development | $ | 4,980,427 | $ | 3,064,762 | ||||
| General and administrative | 972,518 | 263,664 | ||||||
| Total operating expenses | 5,952,945 | 3,328,426 | ||||||
| Loss from operations | (5,952,945 | ) | (3,328,426 | ) | ||||
| Other income (expense) | ||||||||
| Interest expense | (451,664 | ) | - | |||||
| Interest income | 152,879 | 18,018 | ||||||
| Other income (expense), net | (298,785 | ) | 18,018 | |||||
| Net loss before income taxes | (6,251,730 | ) | (3,310,408 | ) | ||||
| Income tax benefits | - | - | ||||||
| Net loss | $ | (6,251,730 | ) | $ | (3,310,408 | ) | ||
| Net loss per share attributable to common stockholders, basic and diluted | $ | (0.84 | ) | $ | (0.52 | ) | ||
| Weighted average common shares outstanding used to compute net loss per share attributable to common stock, basic and diluted | 7,460,640 | 6,313,089 | ||||||
| As of December 31, | ||||||||
| 2017 | 2016 | |||||||
| Balance Sheet Data: | ||||||||
| Total assets | $ | 31,969,510 | $ | 6,056,977 | ||||
| Total current liabilities | 578,053 | 445,359 | ||||||
| Total stockholders’ equity | 31,391,457 | 5,611,618 | ||||||
| 48 |
ITEM 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of operations together in conjunction with our financial statements and the related notes included elsewhere in this report. Some of the information contained in this discussion and analysis or set forth elsewhere in this report, including information with respect to our plans and strategy for our business and expected financial results, includes forward-looking statements that involve risks and uncertainties. You should review the “Risk Factors” discussed in section 1A of Part I of this Form 10-K.
OVERVIEW
We are a cellular analysis company that is discovering new cancer sub-types and commercializing diagnostic tests designed to significantly improve the clinical outcomes of cancer patients treated with targeted therapies. Our proprietary CELx diagnostic platform is the only commercially ready technology we are aware of that uses a patient’s living tumor cells to identify the specific abnormal cellular process driving a patient’s cancer and the targeted therapy that best treats it. We believe our CELx platform provides two important improvements over traditional molecular diagnostics. First, molecular diagnostics can only provide a snapshot of the genetic mutations present in a patient’s tumor because they analyze dead cells. Using dead cells prevents molecular diagnostics from analyzing in real-time the dynamic cellular activities, known as cell signaling, that regulate cell proliferation or survival. Cancer can develop when certain cell signaling activity becomes abnormal. Since genetic mutations are often only weakly correlated to the cell signaling activity driving a patient’s cancer, a molecular diagnostic is prone to providing an incomplete diagnosis. CELx tests overcome this limitation by measuring real-time cell signaling activity in a patient’s living tumor cells. When a CELx test detects abnormal signaling activity, a more accurate diagnosis of the patient’s cancer driver is obtained. Second, molecular diagnostics can only estimate the probability of a patient’s potential drug response based on a statistical analysis of the drug’s clinical trial results. Instead of this indirect estimate of drug response, CELx tests directly measure the effectiveness of a targeted therapy in a patient’s living tumor cells. This enables physicians to confirm that the therapeutic matching the patient’s cancer driver is functional in the patient’s tumor cells before prescribing it, which significantly increases the likelihood of a positive clinical outcome.
Our first analytically validated and commercially ready test using our CELx platform, the CELx HSF Test, diagnoses two new sub-types of HER2-negative breast cancer that traditional molecular diagnostics cannot detect. Our internal studies show that approximately 20% of HER2-negative breast cancer patients have abnormal HER2 signaling activity similar to levels found in HER2+ breast cancer cells. As a result, these HER2-negative patients have undiagnosed HER2-driven breast cancer and would be likely to respond to the same anti-HER2 targeted therapies only HER2+ patients receive today. Our CELx HSF Test is targeting HER2-negative breast cancer patients receiving drug treatment. We are fielding a prospective clinical trial in collaboration with Genentech and the NSABP to evaluate the efficacy of Genentech’s HER2 targeted therapies in patients with these newly identified cancer sub-types. We expect interim results from this trial in late 2018 and final results six to nine months later.
In addition to our CELx tests for HER2-negative breast cancer, we are developing CELx tests to diagnose 14 new potential cancer sub-types we have discovered in breast, lung, colon, ovarian, kidney, bladder and hematological cancers. Approved or investigational drugs are currently available to treat these new potential cancer sub-types. We expect to launch these additional tests on a staggered basis over the next few years while continuing our research to identify additional new cancer sub-types.
On September 22, 2017, we completed our underwritten initial public offering, or IPO, of 2,760,000 shares of our common stock at a price to the public of $9.50 per share. The aggregate net proceeds received by the company from the offering were approximately $23.3 million, net of underwriting discounts and commissions and offering expenses. Shares of our common stock began trading on September 20, 2017 on The Nasdaq Capital Market under the symbol “CELC”.
| 49 |
Following the IPO, we entered into a lease agreement for building space to accommodate expansion in research and development and general corporate office needs. The new lease commences and we will be moving to the space in May 2018, in conjunction with the termination of our existing lease. The lease agreement extends through April 2021 and provides for monthly rent, real estate taxes and operating expenses.
We have not generated any revenue from sales to date, and we continue to incur significant research and development and other expenses related to our ongoing operations. As a result, we are not and have never been profitable and have incurred losses in each period since we began operations in 2012. For the year ended December 31, 2017 and 2016, we reported a net loss of approximately $6.3 million and $3.3 million, respectively. As of December 31, 2017, we had a combined accumulated deficit of approximately $12.6 million under Celcuity LLC and $2.0 million under Celcuity Inc. As of December 31, 2017, we had cash, cash equivalents, and investments of approximately $31.4 million.
RESULTS OF OPERATIONS
Components of Operating Results
Revenue
To date, we have not generated any revenue. Initially, our ability to generate revenue will depend primarily upon our ability to obtain partnership agreements with pharmaceutical companies to provide companion diagnostics for such pharmaceutical partners’ existing or investigational targeted therapies. We expect these partnerships to generate significant revenue from the sale of tests to identify patients eligible for clinical trials, from milestone payments, and, potentially, from royalties on the incremental drug revenues our tests enable. Once a new drug indication is received that requires use of our companion diagnostic to identify eligible patients, we expect to generate revenues from sales of tests to treating physicians.
Research and Development
Since our inception, we have primarily focused on research and development of our CELx platform, development and validation of our CELx HSF Test, and research related to the discovery of new cancer sub-types. Research and development expenses primarily include:
| · | employee-related expenses related to our research and development activities, including salaries, benefits, travel and stock-based compensation expenses; |
| · | laboratory supplies; |
| · | consulting fees paid to third parties; |
| · | clinical trial costs; |
| · | facilities expenses; and |
| · | legal costs associated with patent applications. |
Internal and external research and development costs are expensed as they are incurred. As we initiate clinical trials to evaluate efficacy of targeted therapies in cancer patients selected with one of our CELx tests, the proportion of research and development expenses allocated to external spending will grow at a faster rate than expenses allocated to internal expenses.
General and Administrative
General and administrative expenses consist primarily of salaries and related benefits, including stock-based compensation expense related to our executive, finance and support functions. Other general and administrative expenses include professional fees for auditing, tax, and legal services, insurance and travel expenses. We may incur additional fees for legal, accounting, insurance and other professional service fees associated with being a public company, which may increase further when we are no longer able to rely on the “emerging growth company” exemption we were afforded under the JOBS Act.
| 50 |
Sales and Marketing
Selling and marketing expenses consist primarily of professional and consulting fees related to these functions. To date, we have incurred immaterial sales and marketing expenses as we continue to focus primarily on the development of our CELx platform and corresponding CELx tests. We expect to begin to incur increased selling and marketing expenses in anticipation of the commercialization of our CELx HSF Test. These increased expenses are expected to include payroll-related costs as we add employees in the commercial departments, costs related to the initiation and operation of our sales and distribution network and marketing related costs.
Interest Expense
Interest expense primarily consists of the amortization of debt discount and debt financing costs related to the issuance of our unsecured convertible promissory notes that were converted to common stock upon our IPO.
Interest Income
Interest income consists of interest income earned on our cash, cash equivalents, and investment balances.
Results of Operations
Comparison of the Years Ended December 31, 2017 and 2016
| Years Ended | Increase | |||||||||||||||
| December 31, | (Decrease) | |||||||||||||||
| 2017 | 2016 | $ | % | |||||||||||||
| Statements of Operations Data: | ||||||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 4,980,427 | $ | 3,064,762 | $ | 1,915,665 | 63 | % | ||||||||
| General and administrative | 972,518 | 263,664 | 708,854 | 269 | ||||||||||||
| Total operating expenses | 5,952,945 | 3,328,426 | 2,624,519 | 79 | ||||||||||||
| Loss from operations | (5,952,945 | ) | (3,328,426 | ) | (2,624,519 | ) | 79 | % | ||||||||
| Other income (expense) | ||||||||||||||||
| Interest expense | (451,664 | ) | - | (451,664 | ) | n/a | ||||||||||
| Interest income | 152,879 | 18,018 | 134,861 | 748 | ||||||||||||
| Other income (expense), net | (298,785 | ) | 18,018 | (316,803 | ) | (1,758 | ) | |||||||||
| Net loss before income taxes | (6,251,730 | ) | (3,310,408 | ) | (2,941,322 | ) | 89 | |||||||||
| Income tax benefits | - | - | - | - | ||||||||||||
| Net loss | $ | (6,251,730 | ) | $ | (3,310,408 | ) | $ | (2,941,322 | ) | 89 | % | |||||
Research and Development
For the year ended December 31, 2017, our total research and development expenses increased $1.9 million, or 63%, to $4.98 million from $3.07 million for the prior year. The increase primarily resulted from a $1.32 million increase in compensation related expenses, including approximately $0.4 million of non-cash stock-based compensation, to support development of our CELx platform and validation studies of our CELx HSF Test. In addition, other research and development expenses increased $0.58 million due to legal expenses related to patent costs, start-up clinical trial costs, and business development activities.
| 51 |
Conducting a significant amount of research and development is central to our business model. We plan to increase our research and development expenses for the foreseeable future as we seek to discover new cancer sub-types and to develop and validate additional CELx tests to diagnose such sub-types. We also expect to incur increased expenses to support companion diagnostic business development activities with pharmaceutical companies as we develop additional CELx tests.
General and Administrative
For the year ended December 31, 2017, our total general and administrative expenses increased $0.7 million or 269% to $1.0 million from $0.3 million for the prior year. The increase primarily resulted from increases in compensation related expenses, including approximately $0.3 million of non-cash stock-based compensation, and other expenses associated with being a public company such as professional accounting and legal fees, and director and officer insurance.
We anticipate that our general and administrative expenses will increase in future periods, reflecting both increased costs in connection with the potential future commercialization of CELx tests, an expanding infrastructure, and increased professional fees associated with being a public reporting company.
To date, we have incurred immaterial sales and marketing expenses as we continue to focus primarily on the development of our CELx platform and corresponding CELx tests. We expect to begin to incur increased selling and marketing expenses in anticipation of the commercialization of our CELx HSF Test. These increased expenses are expected to include payroll-related costs as we add employees in the commercial departments, costs related to the initiation and operation of our sales and distribution network and marketing related costs.
Interest Expense
For the year ended December 31, 2017, interest expense increased $0.5 million, from zero for the prior year. The increase consisted of non-cash amortization of debt discount and debt financing costs and accrued interest related to the issuance of our unsecured convertible promissory notes.
Interest Income
For the year ended December 31, 2017, interest income increased by approximately $0.13 million, or 748% to $0.15 million from $0.02 million for the prior year. The increases resulted from interest earned on our cash, cash equivalents, and investments.
LIQUIDITY AND CAPITAL RESOURCES
Since our inception, we have incurred losses and cumulative negative cash flows from operations. Through December 31, 2017, we have raised capital of approximately $13.7 million and $7.5 million through private placements of common equity and unsecured convertible notes, respectively. On September 22, 2017, we also closed on the initial public offering of our common stock, which generated approximately $23.3 million of additional cash after taking into account underwriting discounts and commissions and offering expenses. Cash from these capital raising activities has been our primary source of funds for our operations since inception. As of December 31, 2017, our cash, cash equivalents, and investments were approximately $31.4 million, and we had a combined accumulated deficit of approximately $12.6 million under Celcuity LLC and $2.0 million under Celcuity Inc.
We expect that our research and development and general and administrative expenses will increase as we continue to develop our CELx platform and additional CELx tests, conduct research related to the discovery of new cancer sub-types, conduct clinical trials, and pursue other business development activities. We will also start to incur sales and marketing expenses as we commercialize our CELx HSF Test. We expect to use cash on hand to fund our research and development expenses, capital expenditures, working capital, sales and marketing expenses, and general corporate expenses, as well as for the increased costs associated with being a public company.
Based on our current business plan, we believe that our current cash on hand will provide sufficient cash to finance operations and pay obligations when due during at least the next 24 months.
| 52 |
We may seek to raise additional capital beyond the currently anticipated amount to expand our business, pursue strategic investments, and take advantage of financing or other opportunities that we believe to be in the best interests of the Company and our stockholders. Additional capital may be raised through the sale of common or preferred equity or convertible debt securities, entry into debt facilities or other third-party funding arrangements. The sale of equity and convertible debt securities may result in dilution to our stockholders and those securities may have rights senior to those of our common shares. Agreements entered into in connection with such capital raising activities could contain covenants that would restrict our operations or require us to relinquish certain rights. Additional capital may not be available on reasonable terms, or at all.
Cash Flows
The following table sets forth the primary sources and uses of cash for the years ending December 31.
| 2017 | 2016 | |||||||
| Net cash provided by (used in): | ||||||||
| Operating activities | $ | (4,947,982 | ) | $ | (2,888,288 | ) | ||
| Investing activities | (28,976,983 | ) | (40,903 | ) | ||||
| Financing activities | 30,708,406 | 3,718,299 | ||||||
| Net increase (decrease) in cash and cash equivalents | $ | (3,216,559 | ) | $ | 789,108 | |||
Operating Activities
Net cash used in operating activities was approximately $4.9 million for the year ended December 31, 2017 and consisted primarily of a net loss of approximately $6.3 million and approximately $0.1 million in working capital changes, adjusted for non-cash items of approximately $1.5 million. The approximately $0.1 million of working capital change was primarily due to approximately $0.2 million increase in prepaid insurance and deposits, offset by approximately $0.1 million increase in accounts payable and accrued expenses. Non-cash expense items of approximately $1.5 million consisted of depreciation of approximately $0.1 million, stock-based compensation expense of approximately $0.9 million and interest expense of approximately $0.5 million primarily related to amortization of debt discount and debt financing costs. The net cash used in operating activities was approximately $2.9 million for the year ended December 31, 2016 and consisted primarily of a net loss of approximately $3.3 million, adjusted for approximately $0.3 million of non-cash items, including depreciation of approximately $0.07 million and stock-based compensation expense of approximately $0.2 million.
Investing Activities
Net cash used in investing activities for the year ended December 31, 2017 was approximately $29.0 million and consisted of approximately $28.75 million of investments in certificates of deposit, government securities (U.S. Treasury Notes and U.S. government agency securities) and approximately $0.25 million in purchases of property and equipment. Net cash used in investing activities for the year ended December 31, 2016 was approximately $0.04 million and consisted of purchases of property and equipment.
| 53 |
Financing Activities
Net cash provided by financing activities for the year ended December 31, 2017 was approximately $30.7 million and reflects the net proceeds of approximately $7.5 million from the sale of unsecured convertible promissory notes and warrants to certain investors through a private placement, as well as the net proceeds of approximately $23.3 million from the sale of common stock in our initial public offering. Net cash provided by financing activities for the year ended December 31, 2016 was approximately $3.7 million and reflects the net proceeds from the sale of membership units of Celcuity LLC to certain investors.
OFF-BALANCE SHEET ARRANGEMENTS
We do not currently have any off-balance sheet arrangements as defined in Item 303(a)(4) of Regulation S-K.
RECENT ACCOUNTING PRONOUNCEMENTS
From time to time new accounting pronouncements are issued by the Financial Accounting Standards Board, or FASB, or other standard setting bodies and adopted by us as of the specified effective date. Unless otherwise discussed in Note 2 to our financial statements included elsewhere in this report, we believe that the impact of recently issued standards that are not yet effective will not have a material impact on our financial position or results of operations upon adoption.
CRITICAL ACCOUNTING POLICIES AND USE OF ESTIMATES
Our management’s discussion and analysis of financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or Generally Accepted Accounted Principles (“U.S. GAAP”). The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported expenses during the reporting periods. These items are monitored and analyzed by us for changes in facts and circumstances, and material changes in these estimates could occur in the future. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances; the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Changes in estimates are reflected in reported results for the period in which they become known. Actual results may differ materially from these estimates.
Our significant accounting policies are more fully described in Note 2 to our financial statements included in this report. Of our significant accounting policies, we believe that the following is the most critical:
Stock-Based Compensation
The Company recognizes compensation expense for employees based on an estimated grant date fair value using the Black-Scholes option-pricing method. The Company has elected to account for forfeitures as they occur.
The fair value of options granted to nonemployees is determined using the fair value of the service provided or the fair value of the option granted, whichever is more reliable. The fair value is measured at the value of the Company’s common shares at the earlier of the date that the commitment for performance by the counterparty has been reached or the counterparty’s performance is complete. Awards granted to nonemployees are remeasured to fair value at each period end date until vested and expensed on a straight-line basis over the vesting period.
The inputs for the Black-Scholes valuation model require management’s significant assumptions. Prior to the Company’s IPO, the common share price was determined by the Company’s board based on recent prices of common shares sold in private offerings prior to the IPO. Subsequent to the IPO, the common share price was determined by using the quoted price on the grant date. The risk-free interest rates were based on the rate for U.S. Treasury securities at the date of grant with maturity dates approximately equal to the expected life at the grant date. The expected life was based on the simplified method in accordance with the SEC Staff Accounting Bulletin Nos. 107 and 110. The expected volatility was estimated based on historical volatility information of peer companies that are publicly available.
| 54 |
All assumptions used to calculate the grant date fair value of nonemployee options are generally consistent with the assumptions used for options granted to employees, except the expected life is equal to the contractual term.
For grants of restricted stock, we record compensation expense based on the quoted fair value of the shares on the grant date over the requisite service period. Compensation expense for ESPP rights is recorded in line with each respective offering period.
ITEM 7A. Quantitative and Qualitative Disclosures About Market Risk
As a smaller reporting company, we are not required to provide disclosure pursuant to this item.
| 55 |
ITEM 8. Financial Statements and Supplementary Data
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of Celcuity Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Celcuity Inc. (the Company) as of December 31, 2017 and 2016, and the related statements of operations, changes in stockholders’ equity, and cash flows for each of the years in the two-year period ended December 31, 2017, and the related notes (collectively referred to as the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results of its operations and its cash flows for each of the years in the two-year period ended December 31, 2017, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Boulay PLLP
We have served as the Company’s auditor
since 2017.
Minneapolis, Minnesota
March 15, 2018
![]()
| 56 |
Balance Sheets
| December 31, 2017 | December 31, 2016 | |||||||
| Assets | ||||||||
| Current Assets: | ||||||||
| Cash and cash equivalents | $ | 2,639,789 | $ | 5,856,348 | ||||
| Investments | 21,556,857 | - | ||||||
| Restricted cash | 50,000 | 50,000 | ||||||
| Deposits | 27,726 | 5,717 | ||||||
| Prepaid assets | 209,708 | - | ||||||
| Total current assets | 24,484,080 | 5,912,065 | ||||||
| Property and equipment, net | 280,056 | 144,912 | ||||||
| Long term investments | 7,205,374 | - | ||||||
| Total Assets | $ | 31,969,510 | $ | 6,056,977 | ||||
| Liabilities and Stockholders’ Equity: | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable | $ | 71,913 | $ | 331,534 | ||||
| Accrued expenses | 506,140 | 113,825 | ||||||
| Total current liabilities | 578,053 | 445,359 | ||||||
| Total Liabilities | 578,053 | 445,359 | ||||||
| Commitments and contingencies | ||||||||
| Stockholders’ Equity: | ||||||||
| Members’ equity contributions: 0 and 6,440,139 shares issued and outstanding as of December 31, 2017 and December 31, 2016, respectively | - | 13,349,654 | ||||||
| Preferred stock, $0.001 par value: 5,000,000 and 0 shares authorized as of December 31, 2017 and December 31, 2016, respectively; 0 shares issued and outstanding as of December 31, 2017 and December 31, 2016 | - | - | ||||||
| Common stock, $0.001 par value: 45,000,000 and 0 shares authorized as of December 31, 2017 and December 31, 2016, respectively; 10,087,516 and 0 shares issued and outstanding as of December 31, 2017 and December 31, 2016, respectively | 10,087 | - | ||||||
| Additional paid-in capital | 33,388,597 | 593,365 | ||||||
| Accumulated deficit | (2,007,227 | ) | (8,331,401 | ) | ||||
| Total Stockholders’ Equity | 31,391,457 | 5,611,618 | ||||||
| Total Liabilities and Stockholders’ Equity | $ | 31,969,510 | $ | 6,056,977 | ||||
See accompanying notes to the financial statements
| 57 |
Statements of Operations
| Years Ended | ||||||||
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Operating expenses: | ||||||||
| Research and development | $ | 4,980,427 | $ | 3,064,762 | ||||
| General and administrative | 972,518 | 263,664 | ||||||
| Total operating expenses | 5,952,945 | 3,328,426 | ||||||
| Loss from operations | (5,952,945 | ) | (3,328,426 | ) | ||||
| Other income (expense) | ||||||||
| Interest expense | (451,664 | ) | - | |||||
| Interest income | 152,879 | 18,018 | ||||||
| Other income (expense), net | (298,785 | ) | 18,018 | |||||
| Net loss before income taxes | (6,251,730 | ) | (3,310,408 | ) | ||||
| Income tax benefits | - | - | ||||||
| Net loss | $ | (6,251,730 | ) | $ | (3,310,408 | ) | ||
| Net loss per unit, basic and diluted | $ | (0.84 | ) | $ | (0.52 | ) | ||
| Weighted average common shares outstanding, basic and diluted | 7,460,640 | 6,313,089 | ||||||
See accompanying notes to the financial statements
| 58 |
Statements of Changes in Stockholders’ Equity
| Member Contributions | Common Stock | Additional | Accumulated | |||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Paid-In Capital | Deficit | Total | ||||||||||||||||||||||
| Balance at December 31, 2015 | 5,891,181 | $ | 9,961,962 | - | $ | - | $ | 75,452 | $ | (5,020,993 | ) | $ | 5,016,421 | |||||||||||||||
| Member shares issued, net | 548,958 | 3,387,692 | - | - | 330,607 | - | 3,718,299 | |||||||||||||||||||||
| Stock-based compensation | - | - | - | - | 141,408 | - | 141,408 | |||||||||||||||||||||
| Non-employee stock-based compensation | - | - | - | - | 45,898 | - | 45,898 | |||||||||||||||||||||
| Net loss | - | - | - | - | - | (3,310,408 | ) | (3,310,408 | ) | |||||||||||||||||||
| Balance at December 31, 2016 | 6,440,139 | 13,349,654 | 593,365 | (8,331,401 | ) | 5,611,618 | ||||||||||||||||||||||
| Warrants issued - unsecured convertible promissory note holders | - | - | - | - | 776,717 | - | 776,717 | |||||||||||||||||||||
| Warrants issued - placement agent | - | - | - | - | 286,999 | - | 286,999 | |||||||||||||||||||||
| Corporate conversion from Celcuity LLC to Celcuity Inc. | - | - | - | - | (12,575,904 | ) | 12,575,904 | - | ||||||||||||||||||||
| Corporate conversion to common stock | (6,440,139 | ) | (13,349,654 | ) | 6,440,139 | 6,440 | 13,343,214 | - | - | |||||||||||||||||||
| Common stock issued in initial public offering, net of underwriter commission of $1,835,400, initial public offering costs of $1,133,553 and underwriter warrant of $784,111 | - | - | 2,760,000 | 2,760 | 22,464,226 | - | 22,466,986 | |||||||||||||||||||||
| Warrant issued - underwriter | - | - | - | - | 784,111 | - | 784,111 | |||||||||||||||||||||
| Conversion of unsecured convertible promissory note to common stock | - | - | 881,911 | 882 | 6,839,436 | - | 6,840,318 | |||||||||||||||||||||
| Exercise of common stock warrants | 216 | - | 2,052 | 2,052 | ||||||||||||||||||||||||
| Stock-based compensation | - | - | 5,250 | 5 | 677,366 | - | 677,371 | |||||||||||||||||||||
| Non-employee stock-based compensation | - | - | - | - | 197,015 | - | 197,015 | |||||||||||||||||||||
| Net loss | - | - | - | - | - | (6,251,730 | ) | (6,251,730 | ) | |||||||||||||||||||
| Balance at December 31, 2017 | - | $ | - | 10,087,516 | $ | 10,087 | $ | 33,388,597 | $ | (2,007,227 | ) | $ | 31,391,457 | |||||||||||||||
See accompanying notes to the financial statements
| 59 |
Statements of Cash Flows
| Years Ended | ||||||||
| December 31, | ||||||||
| 2017 | 2016 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (6,251,730 | ) | $ | (3,310,408 | ) | ||
| Adjustments to reconcile net loss to net cash used by operations: | ||||||||
| Depreciation | 104,704 | 73,059 | ||||||
| Stock-based compensation | 874,386 | 187,306 | ||||||
| Non-cash interest expense | 451,664 | - | ||||||
| Non-cash interest income | (25,097 | ) | - | |||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid assets and deposits | (231,717 | ) | - | |||||
| Accounts payable | (262,507 | ) | 69,779 | |||||
| Accrued expenses | 392,315 | 91,976 | ||||||
| Net cash used by operating activities | (4,947,982 | ) | (2,888,288 | ) | ||||
| Cash flows from investing activities: | ||||||||
| Purchases of investments | (28,737,135 | ) | - | |||||
| Purchases of property and equipment | (239,848 | ) | (40,903 | ) | ||||
| Net cash used by investing activities | (28,976,983 | ) | (40,903 | ) | ||||
| Cash flows from financing activities: | ||||||||
| Proceeds from sale of member shares, net of issuance costs | - | 3,718,299 | ||||||
| Proceeds from sale of convertible promissory notes | 7,493,330 | - | ||||||
| Proceeds from initial public offering of common stock | 24,109,650 | - | ||||||
| Proceeds from exercise of common stock warrants | 2,052 | - | ||||||
| Payments for debt issuance costs | (40,961 | ) | - | |||||
| Payments for initial public offering costs | (855,665 | ) | - | |||||
| Net cash provided by financing activities | 30,708,406 | 3,718,299 | ||||||
| Net change in cash and cash equivalents | (3,216,559 | ) | 789,108 | |||||
| Cash and cash equivalents: | ||||||||
| Beginning of period | 5,856,348 | 5,067,240 | ||||||
| End of period | $ | 2,639,789 | $ | 5,856,348 | ||||
| Non-cash financing activities: | ||||||||
| Debt issuance costs netted against proceeds from sale of convertible promissory notes | $ | 844,170 | $ | - | ||||
| Debt discount related to investor and agent warrants (Note 11) | 1,063,715 | - | ||||||
| Initial public offering costs included in accounts payable | 2,889 | - | ||||||
| Underwriter’s reimbursable offering costs netted against initial public offering proceeds | 275,000 | - | ||||||
See accompanying notes to the financial statements
| 60 |
NOTES TO FINANCIAL STATEMENTS
| 1. | ORGANIZATION |
Nature of Business
Celcuity Inc., a Delaware corporation (the “Company”), is a cellular analysis company that is discovering new cancer sub-types and commercializing diagnostic tests designed to significantly improve the response rates of cancer patients treated with targeted therapies. The Company’s proprietary CELx diagnostic platform is currently the only commercially ready technology the Company is aware of that uses a patient’s living tumor cells to evaluate the functional status of the cell signaling pathways associated with cancer. The CELx platform identifies the abnormal signaling activity driving a patient’s cancer and quantifies how effectively a targeted therapy can treat it. This enables physicians to select the therapeutic that precisely matches and inhibits a patient’s cellular dysfunction, which significantly increases the likelihood of a positive clinical outcome. The Company’s first analytically validated and commercially ready CELx test diagnoses two new sub-types of HER2-negative breast cancer. The Company is currently fielding a prospective clinical trial to evaluate the efficacy of HER2 targeted therapies in patients with these newly identified cancer sub-types. In addition to the CELx tests for HER2-negative breast cancer, the Company is developing CELx tests to diagnose 14 new potential cancer sub-types in breast, lung, colon, ovarian, kidney, bladder and hematological cancers. The Company expects to launch these additional tests on a staggered basis over the next few years. The Company was cofounded in 2012 by Brian Sullivan and Lance Laing and is based in Minnesota. The Company has not generated any revenues to date.
| 2. | SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
Basis of Presentation
The accompanying financial statements have been prepared in accordance with accounting principles generally accepted in the United States (“GAAP”) and pursuant to the rules and regulations of the SEC. Operating results for the year ended December 31, 2017 are not necessarily indicative of results to be expected for any future year.
On September 15, 2017, in relation to preparing for its initial public offering (“IPO”), Celcuity LLC filed a certificate of conversion, whereby Celcuity LLC effected a corporate conversion from a Minnesota limited liability company to a Delaware corporation and changed its name to Celcuity Inc. Pursuant to the corporate conversion, units of membership interest in the limited liability company were converted into shares of common stock of the corporation at a conversion ratio of 40 units for one share of common stock. As a result of the corporate conversion, accumulated deficit was reduced to zero on the date of the corporate conversion, and the corresponding amount was credited to additional paid-in capital. The corporate conversion was approved by members holding a majority of our outstanding units, and in connection with such conversion, the Company filed a certificate of incorporation and adopted bylaws. Pursuant to the Company’s certificate of incorporation, the Company is authorized to issue up to 45,000,000 shares of common stock $0.001 par value per share and 5,000,000 shares of preferred stock $0.001 par value per share. The Company determined that the corporate conversion is equivalent to a change in the Company’s capital structure. As such, all references in the audited financial statements to the number of shares and per-share amounts of member units are now presented as common stock and have been retroactively restated to reflect this conversion.
On September 22, 2017, the Company completed its IPO whereby it sold 2,760,000 shares of common stock at a public offering price of $9.50 per share. The aggregate net proceeds received by the Company from the offering were approximately $23.3 million, net of underwriting discounts and commissions of approximately $1.8 million and offering expenses of approximately $1.1 million. Upon the closing of the IPO, 10,082,050 shares of common stock were outstanding, which includes 881,911 shares of common stock as a result of the conversion of the Company’s Unsecured Convertible Promissory Notes (See Note 11). The shares began trading on September 20, 2017 on The Nasdaq Capital Market under the symbol “CELC”.
Accounting Estimates
Management uses estimates and assumptions in preparing these financial statements in accordance with U.S. GAAP. Those estimates and assumptions affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities, and the reported revenues and expenses. Actual results could differ from those estimates. Significant items subject to such estimates and assumptions include the valuation of stock-based compensation and warrants issued to investors, a placement agent and an underwriter, and prepaid or accrued clinical trial costs.
| 61 |
Cash and Cash Equivalents
The Company maintains its accounts primarily at one financial institution. At times throughout the year, the Company’s cash balances may exceed amounts insured by the Federal Deposit Insurance Corporation. At December 31, 2017 and December 31, 2016, the Company had $2,612,104 and $5,842,193, respectively, in money market funds and U.S. Treasury Bills that are considered cash equivalents.
Investments
The Company maintains its investments in certificates of deposit, U.S. governmental agency securities and U.S. treasury notes and has classified them as held-to-maturity at the time of purchase. Held-to-maturity securities are those securities in which the Company has the ability and intent to hold until maturity. Held-to maturity securities are recorded at amortized cost, adjusted for the amortization or accretion of premiums and discounts. Premiums and discounts are amortized or accreted over the life of the related held-to-maturity security using a straight-line method. At December 31, 2017 and 2016, the Company had $28,762,231 and $0, respectively, of investments.
Property and Equipment
Property and equipment are stated at cost. Depreciation is provided over estimated useful lives using the straight-line method. Maintenance and repairs are expensed as incurred; major improvements and betterments are capitalized.
Estimated useful lives of property and equipment are as follows for the major classes of assets:
| Estimated | ||
| Asset Description | Lives | |
| Furniture and Equipment | 4 | |
| Leasehold Improvements | 2-3 |
Long-Lived Assets
Long-lived assets, such as property and equipment, are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. If circumstances require a long-lived asset or asset group be tested for possible impairment, the Company first compares undiscounted cash flows expected to be generated by that asset or asset group to its carrying value. If the carrying value of the long-lived asset or asset group is not recoverable on an undiscounted cash flow basis, an impairment is recognized to the extent that the carrying value exceeds its fair value. Fair value is determined through various valuation techniques including discounted cash flow models, quoted market values, and third party independent appraisals, as considered necessary.
Comprehensive Loss
Comprehensive loss includes net loss as well as other changes in stockholders’ equity that result from transactions and economic events other than those with stockholders. For all periods presented, there was no difference between net loss and comprehensive loss.
Risks and Uncertainties
The Company is subject to risks common to companies in the development stage including, but not limited to, dependency on the clinical and commercial success of its diagnostic tests, ability to obtain regulatory approval of its diagnostic tests, the need for substantial additional financing to achieve its goals, uncertainty of broad adoption of its approved products, if any, by physicians and consumers, and significant competition.
Fair Value of Financial Instruments
The Company’s accounting for fair value measurements of assets and liabilities that are recognized or disclosed at fair value in the financial statements on a recurring or nonrecurring basis adheres to the Financial Accounting Standards Board (“FASB”) fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements) and the lowest priority to measurements involving significant unobservable inputs (Level 3 measurements). The three levels of the fair value hierarchy are as follows:
| · | Level 1 Inputs: Unadjusted quoted prices in active markets for identical assets or liabilities accessible to the Company at the measurement date. |
| · | Level 2 Inputs: Other than quoted prices included in Level 1 inputs that are observable for the asset or liability, either directly or indirectly, for substantially the full term of the asset or liability. |
| 62 |
| · | Level 3 Inputs: Unobservable inputs for the asset or liability used to measure fair value to the extent that observable inputs are not available, thereby allowing for situations in which there is little, if any, market activity for the asset or liability at measurement date. |
The level in the fair value hierarchy within which a fair measurement in its entirety falls is based on the lowest level input that is significant to the fair value measurement in its entirety.
The carrying values of cash equivalents, restricted cash, accounts payable, accrued expenses and other financial working capital items approximate fair value at December 31, 2017 and December 31, 2016, due to the short maturity nature of these items.
Income Taxes
The Company accounts for income taxes using the asset and liability method, as required by the accounting standard for income taxes. Under this method, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases, as well as net operating loss and tax credit carryforwards. Deferred taxes are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred taxes of a change in tax rates is recognized in results of operations in the period that includes the enactment date. The effects of any future changes in tax laws or rates have not been considered. The Company regularly reviews deferred tax assets to assess their potential realization and establish a valuation allowance for portions of such assets to reduce the carrying value if the Company does not consider it to be more likely than not that the deferred tax assets will be realized.
The Company recognizes the impact of an uncertain tax position in its financial statements if, in management’s judgment, the position is more-likely-than-not sustainable upon audit based on the position’s technical merits. This involves the identification of potential uncertain tax positions, the evaluation of applicable tax laws and an assessment of whether a liability for an uncertain tax position is necessary.
Stock-Based Compensation
The Company’s stock-based compensation consists of common stock options and restricted stock issued to certain employees and nonemployees of the Company and the Company’s Employee Stock Purchase Plan. The Company recognizes compensation expense for employees based on an estimated grant date fair value using the Black-Scholes option-pricing method. The Company has elected to account for forfeitures as they occur.
The fair value of options granted to nonemployees is determined using the fair value of the service provided or the fair value of the option granted, whichever is more reliable. The fair value is measured at the value of the Company’s common shares at the earlier of the date that the commitment for performance by the counterparty has been reached or the counterparty’s performance is complete. Awards granted to nonemployees are remeasured to fair value at each period end date until vested and expensed on a straight-line basis over the vesting period.
Research and Development
Research and development costs are expensed as incurred. Research and development costs amounted to $4,980,427 in 2017 and $3,064,762 in 2016.
Clinical Trial Costs
The Company records prepaid assets or accrued expenses for prepaid or estimated clinical trial costs conducted by third-party service providers, which include the conduct of preclinical studies and clinical trials. These costs are a significant component of the Company’s research and development expenses. The Company accrues for these costs based on factors such as estimates of the work completed and in accordance with agreements established with its third-party service providers under the service agreements. The Company makes significant judgments and estimates in determining the accrued liabilities balance in each reporting period. As actual costs become known, the Company adjusts its prepaid assets or accrued expenses. The Company has not experienced any material differences between accrued costs and actual costs incurred. However, the status and timing of actual services performed, number of patients enrolled and the rate of patient enrollments may vary from the Company’s estimates, resulting in an adjustment to expense in future periods. Changes in these estimates that result in material changes to the Company’s prepaid assets or accrued expenses could materially affect the Company’s results of operations.
Segment Data
The Company manages its operations as a single segment for the purposes of assessing performance and making operating decisions.
| 63 |
Application of New or Revised Accounting Standards
Pursuant to the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”), a company constituting an “emerging growth company” is, among other things, entitled to rely upon certain reduced reporting requirements. The Company is an emerging growth company, but has irrevocably elected not to take advantage of the extended transition period afforded by the JOBS Act for the implementation of new or revised accounting standards. As a result, the Company will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for public companies that are not emerging growth companies.
Recently Issued Accounting Pronouncements
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842), which provides guidance for accounting for leases. The new guidance requires companies to recognize the assets and liabilities for the rights and obligations created by leased assets, initially measured at the present value of the lease payments. The accounting guidance for lessors is largely unchanged. The ASU is effective for annual and interim periods beginning after December 15, 2018 with early adoption permitted. It is to be adopted using a modified retrospective approach. The Company is currently evaluating the impact that the adoption of this guidance will have on the Company’s financial statements.
| 3. | NET LOSS PER COMMON SHARE |
Basic and diluted net loss per common share is determined by dividing net loss attributable to common stockholders by the weighted-average common shares outstanding during the period. For all periods presented, the common shares underlying the options and warrants have been excluded from the calculation because their effect would be anti-dilutive. Therefore, the weighted-average shares outstanding used to calculate both basic and diluted loss per common shares are the same.
For the years ended December 31, 2017 and 2016, 64,410 and 55,283 options and 55,055 and 0 warrants, and 414 and 0 restricted stock share equivalents, respectively, have been excluded from the computations of diluted weighted-average shares outstanding.
| 4. | INVESTMENTS |
The following table summarizes the Company’s held-to-maturity investment securities at amortized cost as of December 31, 2017:
| Amortized Cost, as Adjusted | Gross Unrealized Holding Gains | Gross Unrealized Holding Losses | Estimated Fair Value | |||||||||||||
| Short-term investments: | ||||||||||||||||
| Certificates of Deposit | $ | 14,001,237 | $ | - | $ | 20,146 | $ | 13,981,091 | ||||||||
| Governmental Agency Securities | 5,945,314 | - | 18,101 | 5,927,213 | ||||||||||||
| U.S. Treasury Notes | 1,610,306 | - | 633 | 1,609,673 | ||||||||||||
| Total | $ | 21,556,857 | $ | - | $ | 38,880 | $ | 21,517,977 | ||||||||
| Amortized Cost, as Adjusted | Gross Unrealized Holding Gains | Gross Unrealized Holding Losses | Estimated Fair Value | |||||||||||||
| Long-term investments: | ||||||||||||||||
| Certificates of Deposit | $ | 4,165,000 | $ | - | $ | 21,481 | $ | 4,143,519 | ||||||||
| Governmental Agency Securities | 3,040,374 | - | 14,907 | 3,025,467 | ||||||||||||
| Total | $ | 7,205,374 | $ | - | $ | 36,388 | $ | 7,168,986 | ||||||||
The Company had no investments as of December 31, 2016. The Company’s held-to-maturity investments of $21,556,857 and $7,205,374 will mature in 2018 and 2019, respectively.
| 64 |
| 5. | PREPAID ASSETS |
Prepaid assets consisted of the following at December 31:
| 2017 | 2016 | |||||||
| Directors & Officers insurance | $ | 162,914 | $ | - | ||||
| Prepaid rent | 21,673 | - | ||||||
| Other | 25,121 | - | ||||||
| Total | $ | 209,708 | $ | - | ||||
| 6. | PROPERTY AND EQUIPMENT |
Property and equipment consisted of the following at December 31:
| 2017 | 2016 | |||||||
| Leasehold improvements | $ | 22,307 | $ | 22,307 | ||||
| Furniture and equipment | 517,868 | 278,020 | ||||||
| 540,175 | 300,327 | |||||||
| Less: Accumulated depreciation | (260,119 | ) | (155,415 | ) | ||||
| Total | $ | 280,056 | $ | 144,912 | ||||
Depreciation expense was $104,704 and $73,059 for the years ending December 31, 2017 and 2016, respectively.
| 7. | ACCRUED EXPENSES |
Accrued expenses consisted of the following at December 31:
| 2017 | 2016 | |||||||
| Accrued bonuses | $ | 389,802 | $ | 72,917 | ||||
| Accrued payroll | 61,829 | 40,341 | ||||||
| Other | 54,509 | 567 | ||||||
| Total | $ | 506,140 | $ | 113,825 | ||||
| 8. | COMMITMENTS |
Lease Obligation
The Company leases its corporate space in Minneapolis, Minnesota. At December 31, 2017, the Company had the following minimum commitments for payment of rentals which at inception had a non-cancellable term of more than one year:
| Operating | ||||
| Lease | ||||
| 2018 | $ | 141,456 | ||
| 2019 | 188,850 | |||
| 2020 | 193,338 | |||
| 2021 | 64,940 | |||
| Total | $ | 588,584 | ||
Annual rent expense for operating leases was $51,455 and $49,632 for 2017 and 2016, respectively. In connection with the corporate lease, the Company is required to maintain a $50,000 standby letter of credit. The standby letter of credit expires on July 31, 2018.
| 65 |
In September 2017, the Company entered into a non-cancelable operating lease agreement for building space to accommodate expansion in research and development and general corporate office needs. The new lease commences and the Company will be moving to the facility in May 2018, in conjunction with the termination of the existing lease. The new lease agreement extends through April 2021 and provides for monthly rent, real estate taxes and operating expenses.
Clinical Research Study
In May 2017, the Company entered into an agreement with a clinical research organization to conduct a clinical research study. The Company made a payment of $300,000 in June 2017 and $50,000 in November 2017 and is obligated to make payments of $200,000 and $50,000 in 2018 and 2019, respectively. Additional payments will be due as patients are enrolled in the study. The maximum amount of these additional payments is estimated to be approximately $2,040,000 over the course of the agreement.
| 9. | STOCKHOLDERS’ EQUITY |
On September 15, 2017, in connection with its IPO, Celcuity LLC filed a certificate of conversion, whereby Celcuity LLC effected a corporate conversion from a Minnesota limited liability company to a Delaware corporation and changed its name to Celcuity Inc. Pursuant to the conversion, units of membership interest in the limited liability company were converted into shares of common stock of the corporation at a conversion ratio of 40 units for one share of common stock. The Company had 257,604,208 member units issued and outstanding as of September 15, 2017. After giving effect to the corporate conversion, the number of common shares outstanding as of such date is 6,440,139. As a result of the corporate conversion, accumulated deficit was reduced to zero on the date of the corporate conversion, and the corresponding amount was credited to additional paid-in capital. The corporate conversion was approved by members holding a majority of our outstanding units, and in connection with such conversion, the Company filed a certificate of incorporation and adopted bylaws. Pursuant to the Company’s certificate of incorporation, the Company is authorized to issue up to 45,000,000 shares of common stock $0.001 par value per share and 5,000,000 shares of preferred stock $0.001 par value per share. All references in the audited financial statements to the number of shares and per-share amounts of common stock have been retroactively restated to reflect this conversion.
On September 22, 2017, the Company completed its IPO whereby it sold 2,760,000 shares of common stock at a public offering price of $9.50 per share. The aggregate net proceeds received by the Company from the offering were approximately $23.3 million, net of underwriting commissions of approximately $1.8 million and offering expenses of approximately $1.1 million. Upon the closing of the IPO, 10,082,050 shares of common stock were outstanding, which includes 881,911 shares of common stock issued as a result of the conversion of the Company’s Convertible Notes (See Note 11). The shares began trading on September 20, 2017 on The Nasdaq Capital Market under the symbol “CELC”.
At December 31, 2017 and 2016, the Company had common stock shares and member shares outstanding of 10,087,516 and 6,440,139, respectively.
Warrants
In connection with the 2016 private placement unit offering, the Company issued ten-year warrants to the placement agent of the private placement. The warrants allow the agent to purchase up to 55,249 common shares at $7.56 per share. The warrants are immediately exercisable and expire on January 14, 2026 and May 2, 2026. These warrants are equity classified and the fair value of $330,607 is reflected as additional paid-in capital.
In connection with the private offering of convertible notes (Note 11), the Company issued ten-year warrants to purchase 48,615 common shares at a price of $8.42 per share to the placement agent. In addition, the Company granted the convertible notes investors the right to receive a seven-year warrant to purchase 131,675 common shares at an exercise price that is equal to the conversion price of the notes (Note 11). With the completion of the IPO on September 22, 2017, these warrants were issued.
In connection with the IPO, the Company issued a five-year warrant to the underwriter. The warrant allows the underwriter to purchase up to 138,000 common shares at $10.45 per share. This warrant is immediately exercisable and expires on September 19, 2022. This warrant is equity classified and the fair value was $784,111 at the IPO offering date.
At December 31, 2017 and 2016, the Company had warrants to purchase 373,323 and 55,249 common shares outstanding, respectively, at a weighted average exercise price of $9.42 and $7.56, respectively. A total of 216 and 0 warrants were exercised in the years ending December 31, 2017 and 2016, respectively.
| 66 |
| 10. | STOCK-BASED COMPENSATION |
2012 Equity Incentive Plan
The 2012 Equity Incentive Plan, as amended, was adopted by the Company’s board and approved by the members of the Company on August 10, 2012. The Company reserved a maximum of 625,000 common shares available for issuance under the 2012 Equity Incentive Plan. The 2012 Equity Incentive Plan provides for share options, restricted share awards, performance share awards or share bonuses. The exercise price of each share option granted under the 2012 Equity Incentive Plan is not less than one hundred percent (100%) of the fair market value of one share on the date of grant. The maximum permitted term of options granted under the 2012 Equity Incentive Plan is ten years. The Company’s board has administered the plan and determined the provisions of incentive awards, including eligible recipients, number of shares subject to an incentive award, exercise price, vesting schedule, duration of an incentive award and other restrictions an incentive award may be subject to. The 2012 Equity Incentive Plan was fixed on September 6, 2017 and any new awards will be issued under the terms of the 2017 Stock Incentive Plan.
2017 Stock Incentive Plan
The 2017 Stock Incentive Plan, or the 2017 Plan, was adopted by the Company’s board on September 6, 2017 and became effective following the corporate conversion which took place on September 15, 2017. The Company reserved a maximum of 750,000 common shares available for issuance under the 2017 Plan. The number of shares reserved for issuance under the 2017 Plan will increase automatically on January 1, 2019 and each subsequent anniversary through January 1, 2027 by the number of shares equal to 1.0% of the aggregate number of outstanding shares of the Company’s common stock as of the immediately preceding December 31. However, the Company’s board may reduce the amount of the increase in any particular year. The maximum permitted term of options granted under the 2017 Plan is ten years. The 2017 Plan provides for share options, restricted stock awards, stock appreciation rights, restricted stock units, performance awards and stock bonuses. The exercise price of each share option granted under the 2017 Plan is not less than one hundred percent (100%) of the fair market. The 2017 Plan will generally be administered by the compensation committee of the Company’s board of directors and has the authority to interpret the plan, grant awards and make all other determinations necessary for the administration of the plan.
The Black-Scholes option-pricing model was used to estimate the fair value of equity-based awards with the following weighted-average assumptions at December 31:
| 2017 | 2016 | |||
| Risk-free interest rate | 1.89 - 2.40% | 2.00% | ||
| Expected volatility | 75.0% | 75.0% | ||
| Expected life (years) | 5.5 to 10.00 | 6.25 to 10.00 | ||
| Expected dividend yield | 0% | 0% |
The inputs for the Black-Scholes valuation model require management’s significant assumptions. Prior to the Company’s IPO, the common share price was determined by the Company’s board based on recent prices of common shares sold in private offerings prior to the IPO. Subsequent to the IPO, the common share price was determined by using the price on the grant date. The risk-free interest rates were based on the rate for U.S. Treasury securities at the date of grant with maturity dates approximately equal to the expected life at the grant date. The expected life was based on the simplified method in accordance with the SEC Staff Accounting Bulletin Nos. 107 and 110. The expected volatility was estimated based on historical volatility information of peer companies that are publicly available.
All assumptions used to calculate the grant date fair value of nonemployee options are generally consistent with the assumptions used for options granted to employees, except the expected life is equal to the contractual term. In the event the Company terminates any of its consulting agreements, the unvested options underlying the agreements would also be cancelled. Unvested nonemployee options are marked-to-market at each reporting period by using the price on the last trading date of the reporting period.
| 67 |
The following table summarizes the activity for all stock options outstanding at December 31 under the Plan:
| 2017 | 2016 | |||||||||||||||
| Shares | Weighted Average Exercise Price | Shares | Weighted Average Exercise Price | |||||||||||||
| Options outstanding at beginning of year | 302,088 | $ | 5.91 | 114,525 | $ | 3.07 | ||||||||||
| Granted | 233,630 | 9.78 | 190,063 | 7.60 | ||||||||||||
| Forfeited | (34,115 | ) | 7.87 | (2,500 | ) | 3.60 | ||||||||||
| Balance at December 31 | 501,603 | $ | 7.58 | 302,088 | $ | 5.91 | ||||||||||
| Options exercisable at December 31: | 201,705 | $ | 5.80 | 71,463 | $ | 2.91 | ||||||||||
| Weighted Average Grant Date Fair Value for Options Granted During the year: | $ | 6.35 | $ | 5.22 | ||||||||||||
The following table summarizes additional information about stock options outstanding and exercisable at December 31, 2017 under the Plan:
| Options Outstanding | Options Exercisable | |||||||||||||||||||||||||
| Options Outstanding | Weighted Average Remaining Contractual Life | Weighted Average Exercise Price | Aggregate Intrinsic Value | Options Exercisable | Weighted Average Exercise Price | Aggregate Intrinsic Value | ||||||||||||||||||||
| 501,603 | 8.56 | $ | 7.58 | $ | 5,702,716 | 201,705 | $ | 5.80 | $ | 2,652,723 | ||||||||||||||||
The Company recognized stock based compensation expense for stock options of $818,098 and $187,306 for the years ending December 31, 2017 and 2016, respectively.
A restricted stock award of 5,250 shares was granted to a member of the board of directors in 2017. The Company has 5,250 and 0 shares outstanding as of December 31, 2017 and 2016, respectively and 0 shares vested as of December 31, 2017 and 2016, respectively. The Company recognized stock based compensation expense for the restricted stock of $28,193 and $0 for the years ending December 31, 2017 and 2016, respectively.
The total remaining shares available for grant under the 2017 plan is 688,020.
Total unrecognized compensation cost related to stock options and restricted stock is estimated to be recognized as follows:
| 2018 | $ | 829,895 | ||
| 2019 | 412,803 | |||
| 2020 | 299,706 | |||
| 2021 | 116,950 | |||
| Total estimated compensation cost to be recognized | $ | 1,659,354 | ||
2017 Employee Stock Purchase Plan
The Company’s employee stock purchase plan, or ESPP, was adopted by the Company’s board on September 6, 2017, subject to stockholder approval at the next meeting of stockholders. If stockholders do not approve the ESPP, it will be terminated and all contributions returned to the participants without the purchase of any shares. The Company has reserved a total of 100,000 shares for issuance. The number of shares authorized and reserved for issuance under the ESPP will be automatically increased on the first day of each of the Company’s fiscal years beginning in 2019 by the number of shares equal to 0.5% of the total outstanding number of shares of common stock. However, the Company’s board may reduce the amount of the increase in any particular year. The ESPP provides participating employees with an opportunity to purchase shares of the Company’s common stock at a discount through payroll deductions. The plan is available to all employees unless they are employed for less than 20 hours per week or own 5% or more of the total combined voting power or value of the Company’s common stock. The plan is administered using overlapping 24 month offering periods, referred to as an Offering Period. Each Offering Period has four six-month purchase periods. A new Offering Period and purchase period begin every six months on May 1 and November 1 of each year. Participating employees may purchase common stock, on a voluntary after tax-basis, at a price equal to 85% of the fair market value of a share of common stock on either the offering date or the purchase date, whichever is lower. If the purchase date has a lower price, the employee will automatically be placed in the Offering Period beginning immediately after the purchase date. The Company recognized stock based compensation expense of $28,095 and $0 for the years ending December 31, 2017 and 2016, respectively.
| 68 |
The Company recognized total stock-based compensation, as follows for the years ended December 31:
| 2017 | 2016 | |||||||
| Stock-based compensation expense in operating expenses: | ||||||||
| Research and development | $ | 608,456 | $ | 187,306 | ||||
| General and administrative | 265,930 | - | ||||||
| Total | $ | 874,386 | $ | 187,306 | ||||
| 11. | UNSECURED CONVERTIBLE PROMISSORY NOTES |
In April and May of 2017, the Company issued to certain accredited investors convertible notes in the original principal amount of $5,750,000 and $2,587,500, respectively, for total principal of $8,337,500 (the “Convertible Notes”).
The Convertible Notes accrued interest at a rate of 1.25% per annum from date of issuance until December 31, 2018 on a non-compounding basis. All principal and interest was due on December 31, 2018. The IPO was considered a qualified financing, therefore the outstanding principal balance and all accrued interest under the Convertible Notes automatically converted into 881,911 shares of common stock pursuant to the terms of such notes. The conversion price of the Convertible Notes was equal to the price at which the equity securities were sold in the IPO, which was $9.50 per share.
In connection with the issuance of the Convertible Notes, the Company granted those investors the right to receive a seven-year warrant to purchase 131,675 common shares at an exercise price that is equal to the conversion price of the Convertible Notes. The gross proceeds of $8,337,500 was allocated $7,560,783 and $776,717 to the Convertible Notes and warrants, respectively, based on their relative fair value. The relative fair value of the warrants of $776,717 was recorded as debt discount and credited to additional paid-in capital. The resulting debt discount was amortized to interest expense using the effective interest method over the term of the Convertible Notes.
Cedar Point Capital, LLC (“Cedar”) served as the Company’s placement agent in connection with the placement of the Convertible Notes and earned a commission of approximately 10% of the original principal balance of such notes. Debt financing costs in the aggregate of $885,131 (not including the agent warrant discussed below), comprised primarily of the commission earned by Cedar, were amortized to interest expense using the effective interest method over the term of the Convertible Notes until converted. In addition to the commission earned by Cedar, the Company issued an agent’s ten-year warrant to purchase 48,615 common shares. The exercise price was $8.42 per share. The fair value of the agent’s warrant was $286,999 and is considered additional debt discount and was credited to additional paid-in capital. During the period beginning on January 1, 2017 and ending on September 22, 2017 (the date of the IPO closing), the Company amortized $411,375 of debt discount and financing costs to interest expense for these Convertible Notes.
| 12. | Income Taxes |
Following the conversion of Celcuity LLC to Celcuity Inc. on September 15, 2017, Celcuity Inc. will begin filing federal and state returns where required. No income tax benefit was recorded for the period September 16, 2017 through December 31, 2017, due to net losses and recognition of a valuation allowance. The following table presents a reconciliation of the tax expense computed at the statutory federal rate and the Company’s tax expense for the years ending December 31:
| 2017 |
2016 | |||||||
| Tax benefit at statutory federal rate | $ | (682,000 | ) | $ | - | |||
| State income tax benefit, net of federal tax effect | (130,000 | ) | - | |||||
| Change in valuation allowance on deferred tax assets | 897,000 | - | ||||||
| Other permanent items | 3,000 | - | ||||||
| Revaluation of deferred taxes due to U.S. Tax Reform | 366,000 | - | ||||||
| Change in tax status | (454,000 | ) | - | |||||
| Income tax benefits | $ | - | $ | - | ||||
| 69 |
Deferred income taxes reflect the net effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The Company’s deferred tax assets relate primarily to its net operating loss carryforwards and other balance sheet basis differences. In accordance with ASC 740, “Income Taxes,” the Company recorded a valuation allowance to fully offset the net deferred tax asset, because it is more likely than not that the Company will not realize future benefits associated with these deferred tax assets at December 31, 2017. The tax effects of temporary differences and carryforwards that give rise to significant portions of the deferred tax assets are as follows:
| 2017 | 2016 | |||||||
| Deferred tax assets (liabilities): | ||||||||
| Accrued expenses | $ | 112,000 | $ | - | ||||
| Share-based compensation | 325,000 | - | ||||||
| Property and equipment | 68,000 | - | ||||||
| Net operating losses | 392,000 | - | ||||||
| Valuation allowance | (897,000 | ) | - | |||||
| Net deferred tax assets (liabilities) | $ | - | $ | - | ||||
On December 22, 2017, the Tax Cuts and Jobs Act (the Act) was signed into United States tax law. As a result, certain provisions in this Act effect the deferred tax assets and liabilities and the deferred tax provision of the Company as of and for the year ended December 31, 2017. Among other provisions included in the Act, the US federal corporate tax rate is reduced from a graduated rate up to 35% to a flat rate of 21%. The Company has adjusted its deferred tax assets and liabilities at December 31, 2017 to reflect the Act’s reduction of corporate tax rates which are expected to be in effect in future years as the deferred tax assets and liabilities are realized. The effect of this adjustment has been recognized as an increase in the deferred provision for income taxes of $366,000.
At December 31, 2017, the Company had federal and state net operating loss carryforwards of approximately $1.36 million each. The net operating loss carryforwards will expire in the year ending December 31, 2037.
Under the provisions of Section 382 of the Internal Revenue Code of 1986, certain substantial changes in the Company’s ownership, including a sale of the Company, or significant changes in ownership due to sales of equity, may limit in the future the amount of net operating loss carryforwards available to offset future taxable income.
The Company recognizes uncertain tax positions in accordance with ASC 740 on the basis of evaluating whether it is more likely than not that the tax positions will be sustained upon examination by tax authorities. For those tax positions that meet the more-likely-than not recognition threshold, we recognize the largest amount of tax benefit that is more than 50 percent likely to be realized upon ultimate settlement. As of December 31, 2017 and 2016, the Company has no unrecognized tax benefits. There are no unrecognized tax benefits included on the balance sheet that would, if recognized, impact the effective tax rate. The Company does not anticipate there will be a significant change in unrecognized tax benefits within the next 12 months.
Prior to the conversion, Celcuity was a limited liability company and therefore was a disregarded legal entity for income tax purposes. Accordingly, no benefit for income taxes was recorded prior to the conversion.
For years before 2013, the Company is no longer subject to U.S. federal or state income tax examinations. The Company’s policy is to recognize interest and penalties related to uncertain tax positions as a component of general and administrative expenses. As of September 30, 2017, and December 31, 2016, the Company did not have any significant uncertain tax positions.
| 70 |
ITEM 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None
ITEM 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2017. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2017, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective.
Changes in Internal Control over Financial Reporting
There were no material changes in our internal control over financial reporting during the quarter ended December 31, 2017 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Management Report on Internal Control over Financial Reporting
This annual report does not include a report of management’s assessment regarding internal control over financial reporting or an attestation report of our registered independent public accounting firm due to a transition period established by rules of the SEC for newly public companies.
None.
ITEM 10. Directors, Executive Officers and Corporate Governance.
Directors
Brian F. Sullivan, age 56, is our co-Founder and has served as Chairman of the Board and Chief Executive Officer since we commenced operations in 2012. Mr. Sullivan has over 25 years of experience founding and building successful, high growth technology companies. He was Chairman and CEO of SterilMed, a medical device reprocessing company, from 2003, when he led an investment group to acquire a majority interest, until its sale to Ethicon Endo-Surgery Inc., a Johnson & Johnson company, for $330 million in 2011. Previously, he was co-founder and Chief Executive Officer of Recovery Engineering, a filtration company, which he took public and subsequently sold to Procter & Gamble for $265 million in 1999. Since 2003, Mr. Sullivan has served on the board of directors of Entegris, Inc., a publicly-held company. Mr. Sullivan has received four U.S. patents and has several pending. He graduated magna cum laude with distinction from Harvard College with an A.B. in economics. Among other attributes, skills, and qualifications, the board of directors believes Mr. Sullivan is uniquely qualified to serve as a director based on his extensive operational and business development experience, and his knowledge in building stockholder value, growing a company from inception and navigating significant corporate transactions and the public company process.
Lance G. Laing, Ph.D., age 56, is our co-Founder and has served as Chief Science Officer, Vice President, Secretary and Director since we commenced operations in 2012. Dr. Laing’s career spans more than 15 years in drug discovery research and technology development. He received his doctorate in biophysics and biochemistry from The Johns Hopkins University and completed a National Institutes of Health post-doctoral fellowship at Washington University Medical School. He has received 17 U.S. patents and has an additional 24 U.S. patents pending. His drug discovery research career began at Scriptgen/Anadys Pharmaceuticals (purchased by Novartis), where he worked under Professor Peter Kim, who became President of Merck Research. He also was Director of Chemistry and Bioapplications and Director of Detection Product Development for two companies that each developed instruments similar to those Celcuity uses to perform the CELx tests. His work at these two instrument companies gave him unique expertise and experience in developing a variety of patented applications for these instruments. Most recently, he served as an executive director for an international drug discovery and development company. Among other attributes, skills, and qualifications, the board of directors believes Dr. Laing is uniquely qualified to serve as a director based on his significant research, medical and scientific expertise.
| 71 |
Maureen Cronin, Ph.D., age 65, recently retired as Executive Director of Research Informatics at Celgene Corporation, where she served beginning 2012. Dr. Cronin’s career spans more than 25 years in biotechnology research and development and drug discovery research. She has served at seven biotechnology or molecular diagnostics startups, including service as a senior research executive at two leading oncology precision medicine diagnostics companies. From 2001 to 2010, she was Vice President of Research at Genomic Health, Inc. and from 2010 to 2012, she was Vice President of Research and Development at Foundation Medicine, Inc. Her technology research and development career began at Affymetrix (purchased by Thermo Fisher). She currently serves as a director of a privately-held company. She received her doctorate in physiology and pharmacology from University of California, San Diego as a Regents’ Fellow and completed a UCSD post-doctoral fellowship at the San Diego VA Hospital. She has been named an inventor on 17 U.S. and 11 European patents and has authored or coauthored more than 40 peer reviewed publications. Her work at diagnostics and drug development companies gives her unique expertise and experience in developing and delivering precision medicine tests. Among other attributes, skills, and qualifications, the board of directors believes Ms. Cronin is uniquely qualified to serve as a director based on her significant diagnostics research and applied scientific expertise.
David F. Dalvey, age 59, has served as a member of Celcuity’s board of directors since February 2014. He was designated to serve as a board member by certain institutional investors of Celcuity pursuant to an agreement entered into in connection with such investors’ purchase of equity units of the company. Mr. Dalvey has more than 30 years of experience in the fields of corporate finance and venture capital, working primarily with growth-oriented technology and life-science businesses. He has over 10 years of corporate finance advisory experience with two national investment banks, completing over 150 individual transactions. He has been the General Partner of Brightstone Venture Capital, a venture capital management company, since September 2000. Brightstone is a 25-year old venture capital management company that has raised and managed ten venture partnerships. Previously, he held management positions with R.J. Steichen and Company, an investment bank, from 1995 to 2000, The Food Fund LP, a venture capital firm, from 1992 to 1995 and Wessels, Arnold & Henderson, an investment bank, from 1987 to 1992. Mr. Dalvey served on the board of directors for Navarre Corporation (now Speed Commerce, Inc.) from 2009 until November 2012, on the board of managers for Blue Rock Market Neutral Fund, a mutual fund registered under the Investment Company Act of 1940 from 2000 to 2014 and on the board of directors for Digitiliti, Inc. from July 2011 until October 2012. Mr. Dalvey has significant operational exposure as a board director or advisor to many other public and privately held growth businesses and has served on these companies’ audit, strategic or governance committees, including companies such as HomeSpotter, Definity Health, AppTec Laboratories, CHF Solutions, BiteSquad, Agiliti, and Nature Vision. Mr. Dalvey received a B.S. in Business/Management Economics from University of Minnesota. Among other attributes, skills, and qualifications, the board of directors believes Mr. Dalvey is uniquely qualified to serve as a director based on his leadership experience in operating both public and private companies and his experience working in the investment community and with investment firms enable him to bring valuable insight and knowledge to our board of directors.
Richard J. Nigon, age 70, is currently Senior Vice President of Cedar Point Capital, LLC., a private company that raises capital for early stage companies, where he has served since 2007. Mr. Nigon has also been a board member for Tactile Systems Technology since September 2012 and Northern Technologies International Corp. since February 2010, including its non-executive Chairman of the board of directors since November 2012. Mr. Nigon also serves as a director of several private companies. Mr. Nigon previously served as a board member for Vascular Solutions, Inc. from November 2000 to February 2017, when it was acquired by Teleflex, Incorporated and as a board member for Virtual Radiologic Corporation from May 2007 until it was acquired in July 2010. From February 2001 until December 2006, Mr. Nigon was a Director of Equity Corporate Finance for Miller Johnson Steichen Kinnard, a privately held investment firm, which was acquired in December 2006 by Stifel Nicolaus, a brokerage and investment banking firm. After that acquisition, Mr. Nigon became a Managing Director of Private Placements of Stifel Nicolaus until May 2007. From February 2000 to February 2001, Mr. Nigon served as the Chief Financial Officer of Dantis, Inc., a web hosting company. Prior to joining Dantis, Mr. Nigon was employed by Ernst & Young LLP from 1970 to 2000, where he served as a partner from 1981 to 2000. While at Ernst & Young, Mr. Nigon served as the Director of Ernst & Young’s Twin Cities Entrepreneurial Services Group and was the coordinating partner on several publicly-traded companies in the consumer retailing and manufacturing sectors. We believe Mr. Nigon is qualified to serve on our board of directors because of his extensive public accounting and auditing experience, including particular experience with emerging growth companies. We also feel that he will bring to the board of directors a strong background in financial controls and reporting, financial management, financial analysis, SEC reporting requirements and mergers and acquisitions. His strategic planning expertise gained through his management and leadership roles at private investment firms also makes him well-suited to serve as a member of our board of directors.
| 72 |
Executive Officers
Information regarding our Chief Executive Officer, Brian F. Sullivan, and our Chief Science Officer, Lance G. Laing, PhD., is included above under the heading “Directors”.
Vicky Hahne, age 51, joined as our Chief Financial Officer in July 2017. She has more than 20 years of financial leadership experience, including the most recent 10 years in the healthcare industry. Prior to joining Celcuity, Ms. Hahne served as Controller of Respiratory Technologies Inc., a medical device manufacturer, from 2015 to 2017. While at Respiratory Technologies, she played a key role in the due diligence process to sell the company to Koninklijke Philips. In 2014, she served as Controller for Ability Network Inc., a healthcare information technology company. From 2007 to 2012, Ms. Hahne served as Controller of Sterilmed Inc., a medical device reprocessing company, where she was significantly involved in the sale of the company to Johnson & Johnson. Prior to these roles, Ms. Hahne held several senior financial positions at SimonDelivers Inc., including Chief Financial Officer. Ms. Hahne has extensive experience in early stage, high growth companies with responsibilities including financial controls and stewardship, financial analysis, mergers and acquisitions, building infrastructure and systems. She received a B.S. degree in Finance and Accounting from Northern State University and received her CPA certificate in 1990.
Other Senior Management
Laura Beggrow, age 55, joined as our Chief Commercial Officer in June 2016. She has more than 20 years of sales and marketing leadership experience in the pharmaceutical and molecular diagnostic industry, including service as a Chief Commercial Officer, VP Sales and Marketing and President. Between 2004 and 2013, Ms. Beggrow served in various sales and marketing leadership positions at one of the leading molecular diagnostic companies in the U.S., Genomic Health, Inc. During her tenure, her roles included V.P. U.S. Sales and Marketing and V.P. U.S. Sales, and she led successful launches of several key products. From 2013 to 2014, she served as President of Strand Diagnostics, Inc., where she provided commercial and operational leadership. From 2014 to 2015, she was Chief Commercial Officer of NantHealth, an early stage company developing a suite of molecular diagnostic products. Ms. Beggrow has a proven track record of successfully launching products, developing strategic product portfolio plans, and building market leading sales organizations. Prior to these roles, Ms. Beggrow served in various sales and sales management positions at Rhone-Poulenc Rorer, Genentech, Cell Therapeutics and Abbott Laboratories. She received a B.S. in Nursing from The Ohio State University.
Corporate Governance
Our board of directors has adopted a Code of Business Conduct and Ethics that applies to our directors, officers and employees. This code is available on the corporate governance section of our website (which is a subsection of the investor relations section of our website) at the following address: www.celcuity.com. We intend to disclose on our website any amendments or waivers to the Code that are required to be disclosed by SEC rules.
Additional information required by this Item 10 will be contained in our definitive proxy statement for our 2018 Annual Meeting of Stockholders (the “Definitive Proxy Statement”) and is incorporated herein by reference.
ITEM 11. Executive Compensation.
The information required by this Item 11 will be contained in the Definitive Proxy Statement and is incorporated herein by reference.
ITEM 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The information required by this Item 12 will be contained in the Definitive Proxy Statement and is incorporated herein by reference.
ITEM 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by this Item 13 will be contained in the Definitive Proxy Statement and is incorporated herein by reference.
ITEM 14. Principal Accounting Fees and Services.
The information required by this Item 14 will be contained in the Definitive Proxy Statement and is incorporated herein by reference.
| 73 |
ITEM 15. Exhibits, Financial Statement Schedules.
FINANCIAL STATEMENTS
None.
FINANCIAL STATEMENT SCHEDULES
None.
EXHIBITS
See Exhibit Index to Form 10-K immediately following the signature page hereto, which is incorporated herein by reference.
| 74 |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Dated: March 15, 2018 | CELCUITY INC. | ||
| By | /s/ Brian F. Sullivan | ||
| Brian F. Sullivan | |||
| Chairman and Chief Executive Officer | |||
| (Principal Executive Officer) | |||
| By | /s/ Vicky Hahne | ||
| Vicky Hahne | |||
| Chief Financial Officer | |||
| (Principal Financial and Accounting Officer) | |||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Each person whose signature appears below constitutes and appoints Brian F. Sullivan and Vicky Hahne as the undersigned’s true and lawful attorneys-in fact and agents, each acting alone, with full power of substitution and resubstitution, for the undersigned and in the undersigned’s name, place and stead, in any and all amendments to this Annual Report on Form 10-K and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granted unto said attorneys-in-fact and agents, each acting alone, full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as the undersigned might or could do in person, hereby ratifying and confirming all said attorneys-in-fact and agents, each acting alone, or his substitute or substitutes, may lawfully do or cause to be done by virtue thereof.
|
Signature |
Title |
Date | ||
| /s/ Brian F. Sullivan | Chief Executive Officer and Chief Financial Officer (Principal Executive Officer and Principal Accounting Officer) |
March 15, 2018 | ||
| Brian F. Sullivan | and Director (Chairman) | |||
| /s/ Lance G. Laing | Chief Science Officer, Vice President and Secretary, and | March 15, 2018 | ||
| Lance G. Laing | Director | |||
| /s/ Maureen Cronin | Director | March 15, 2018 | ||
| Maureen Cronin | ||||
| /s/ Dave F. Dalvey | Director | March 15, 2018 | ||
| Dave F. Dalvey | ||||
| /s/ Richard J. Nigon | Director | March 15, 2018 | ||
| Richard J. Nigon |
| 75 |
EXHIBIT INDEX
CELCUITY INC.
FORM 10-K
| 76 |
| * | Filed herewith. |
| ** | Furnished herewith. |
| + | Management contract or compensatory plan. |
| 77 |