Attached files
| file | filename |
|---|---|
| EX-32.1 - EX-32.1 - Voyager Therapeutics, Inc. | vygr-20170930ex3217e18eb.htm |
| EX-31.2 - EX-31.2 - Voyager Therapeutics, Inc. | vygr-20170930ex31293915d.htm |
| EX-31.1 - EX-31.1 - Voyager Therapeutics, Inc. | vygr-20170930ex311002dfe.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-Q
|
☒ |
QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934. |
For the quarterly period ended September 30, 2017.
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934. |
For the transition period from ______ to ______
Commission file number: 001-37625
Voyager Therapeutics, Inc.
(Exact name of Registrant as specified in its charter)
|
Delaware |
|
46-3003182 |
|
(State or other jurisdiction of |
|
(I.R.S. Employer |
|
|
|
|
|
75 Sidney Street, |
|
02139 |
|
(Address of principal executive offices) |
|
(Zip Code) |
(857) 259-5340
(Registrant’s telephone number, including area code)
Not Applicable
(Former name, former address and former fiscal year, if changed since last report)
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer |
☐ |
|
Accelerated filer |
☒ |
|||||
|
Non-accelerated filer |
☐ |
(Do not check if a smaller reporting company) |
Smaller reporting company |
☐ |
|||||
|
|
|
|
Emerging growth company |
☒ |
|||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The number of outstanding shares of the registrant’s common stock, par value $0.001 per share, as of October 30, 2017 was 26,949,523.
Forward-Looking Statements
This Quarterly Report on Form 10-Q contains forward-looking statements that involve substantial risks and uncertainties. All statements other than statements of historical facts contained in this Quarterly Report on Form 10-Q, including statements regarding our strategy, future operations, future financial position, future revenue, projected costs, prospects, plans, objectives of management and expected market growth, are forward-looking statements. These statements involve known and unknown risks, uncertainties and other important factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements.
The words “anticipate,” “believe,” “estimate,” “expect,” “intend,” “may,” “might,” “plan,” “predict,” “project,” “target,” “potential,” “goals,” “will,” “would,” “could,” “should,” “continue,” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. These forward-looking statements include, among other things, statements about:
|
· |
our plans to develop and commercialize our product candidates based on adeno-associated virus, or AAV, gene therapy; |
|
· |
our ability to identify and optimize product candidates and novel AAV gene therapy capsids; |
|
· |
our ongoing and planned clinical trials, including our ability to continue to advance VY-AADC through the current Phase 1b clinical trial and into a planned Phase 2-3 clinical program as a treatment for advanced Parkinson’s disease, and our preclinical development efforts and studies; |
|
· |
the timing of and our ability to submit applications for, and obtain and maintain regulatory approvals for, our product candidates, including our ability to file Investigational New Drug applications, or INDs, for our lead candidate in our program for the treatment of a monogenic form of amyotrophic lateral sclerosis, VY-HTT01 for the treatment of Huntington’s disease, and VY-FXN01 for the treatment of Friederich’s ataxia; |
|
· |
our estimates regarding expenses, future revenues, capital requirements, and needs for additional financing; |
|
· |
our ability to continue to develop our product engine; |
|
· |
our ability to develop a manufacturing capability compliant with current good manufacturing practices for our product candidates; |
|
· |
our ability to access, develop, and obtain regulatory clearance for devices to deliver our AAV gene therapies to critical targets of neurological disease; |
|
· |
our intellectual property position and our ability to obtain and maintain intellectual property protection for our proprietary assets; |
|
· |
our estimates regarding the size of the potential markets for our product candidates and our ability to serve those markets; |
|
· |
the rate and degree of market acceptance of our product candidates for any indication once approved; |
|
· |
the possibility and timing of Sanofi Genzyme’s exercise of their options to the programs identified in our Collaboration Agreement; |
|
· |
our plans and ability to raise additional capital, including through equity offerings, debt financings, collaborations, strategic alliances, and licensing arrangements; |
|
· |
our competitive position and the success of competing products that are or become available for the indications that we are pursuing; |
|
· |
the impact of government laws and regulations including in the United States, the European Union, and other important geographies such as Japan; |
2
|
· |
our ability to sustain consistency with recently announced results from our ongoing Phase 1b clinical trial in future clinical trials; and |
|
· |
our ability to enter into future collaborations, strategic alliances, or licensing arrangements. |
These forward-looking statements are only predictions and we may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements. You should not place undue reliance on our forward-looking statements. Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements we make. We have based these forward-looking statements largely on our current expectations and projections about future events and trends that we believe may affect our business, financial condition and operating results. We have included important factors in the cautionary statements included in this Quarterly Report on Form 10-Q, particularly in Part II, Item 1A. Risk Factors that could cause actual future results or events to differ materially from the forward-looking statements that we make. Our forward-looking statements do not reflect the potential impact of any future acquisitions, mergers, dispositions, joint ventures or investments we may make.
You should read this Quarterly Report on Form 10-Q and the documents that we have filed as exhibits to the Quarterly Report on Form 10-Q with the understanding that our actual future results may be materially different from what we expect. We do not assume any obligation to update any forward-looking statements whether as a result of new information, future events or otherwise, except as required by applicable law.
3
FORM 10-Q
TABLE OF CONTENTS
4
Voyager Therapeutics, Inc.
Condensed Consolidated Balance Sheets
(amounts in thousands, except share and per share data)
(unaudited)
|
|
|
September 30, |
|
December 31, |
|
||
|
|
|
2017 |
|
2016 |
|
||
|
Assets |
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
48,201 |
|
$ |
36,641 |
|
|
Marketable securities, current |
|
|
77,386 |
|
|
137,777 |
|
|
Prepaid expenses and other current assets |
|
|
2,604 |
|
|
4,368 |
|
|
Total current assets |
|
|
128,191 |
|
|
178,786 |
|
|
Property and equipment, net |
|
|
10,558 |
|
|
7,893 |
|
|
Deposits and other non-current assets |
|
|
1,206 |
|
|
1,527 |
|
|
Marketable securities, non-current |
|
|
960 |
|
|
1,360 |
|
|
Total assets |
|
$ |
140,915 |
|
$ |
189,566 |
|
|
Liabilities and stockholders’ equity |
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
911 |
|
$ |
550 |
|
|
Accrued expenses |
|
|
10,493 |
|
|
6,488 |
|
|
Deferred revenue, current portion |
|
|
9,537 |
|
|
6,764 |
|
|
Total current liabilities |
|
|
20,941 |
|
|
13,802 |
|
|
Deferred rent |
|
|
5,385 |
|
|
4,999 |
|
|
Deferred revenue, net of current portion |
|
|
28,368 |
|
|
34,818 |
|
|
Other non-current liabilities |
|
|
1,015 |
|
|
25 |
|
|
Total liabilities |
|
|
55,709 |
|
|
53,644 |
|
|
Commitments and contingencies (see note 6) |
|
|
|
|
|
|
|
|
Stockholders’ equity: |
|
|
|
|
|
|
|
|
Preferred stock $0.001 par value: 5,000,000 shares authorized at September 30, 2017 and December 31, 2016; no shares issued and outstanding at September 30, 2017 and December 31, 2016 |
|
|
— |
|
|
— |
|
|
Common stock, $0.001 par value: 120,000,000 shares authorized at September 30, 2017 and December 31, 2016; 26,250,327 and 25,597,912 shares issued and outstanding at September 30, 2017 and December 31, 2016, respectively |
|
|
26 |
|
|
26 |
|
|
Additional paid-in capital |
|
|
234,519 |
|
|
225,963 |
|
|
Accumulated other comprehensive loss |
|
|
(455) |
|
|
(52) |
|
|
Accumulated deficit |
|
|
(148,884) |
|
|
(90,015) |
|
|
Total stockholders’ equity |
|
|
85,206 |
|
|
135,922 |
|
|
Total liabilities and stockholders’ equity |
|
$ |
140,915 |
|
$ |
189,566 |
|
The accompanying notes are an integral part of these condensed consolidated financial statements.
5
Voyager Therapeutics, Inc.
Condensed Consolidated Statements of Operations and Comprehensive Loss
(amounts in thousands, except share and per share data)
(unaudited)
|
|
|
Three Months Ended |
|
Nine Months Ended |
|
||||||||
|
|
|
September 30, |
|
September 30, |
|
||||||||
|
|
|
2017 |
|
2016 |
|
2017 |
|
2016 |
|
||||
|
Collaboration revenue |
|
$ |
1,148 |
|
$ |
3,308 |
|
$ |
3,790 |
|
$ |
11,858 |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
19,561 |
|
|
10,309 |
|
|
48,933 |
|
|
29,526 |
|
|
General and administrative |
|
|
4,942 |
|
|
3,370 |
|
|
14,372 |
|
|
9,789 |
|
|
Total operating expenses |
|
|
24,503 |
|
|
13,679 |
|
|
63,305 |
|
|
39,315 |
|
|
Operating loss |
|
|
(23,355) |
|
|
(10,371) |
|
|
(59,515) |
|
|
(27,457) |
|
|
Other income: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income, net |
|
|
291 |
|
|
1,171 |
|
|
799 |
|
|
2,069 |
|
|
Other expense, net |
|
|
(282) |
|
|
(99) |
|
|
(184) |
|
|
(435) |
|
|
Total other income |
|
|
9 |
|
|
1,072 |
|
|
615 |
|
|
1,634 |
|
|
Loss before income taxes |
|
|
(23,346) |
|
|
(9,299) |
|
|
(58,900) |
|
|
(25,823) |
|
|
Income tax benefit |
|
|
— |
|
|
303 |
|
|
31 |
|
|
303 |
|
|
Net loss |
|
$ |
(23,346) |
|
$ |
(8,996) |
|
$ |
(58,869) |
|
$ |
(25,520) |
|
|
Other comprehensive (loss) income |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Unrealized (loss) gain on available-for-sale-securities, net of income tax (benefit) provision of ($53) for the three months ended September 30, 2017, and $570 for the three and nine months ended September 30, 2016 |
|
|
(485) |
|
|
570 |
|
|
(403) |
|
|
881 |
|
|
Total other comprehensive (loss) income |
|
|
(485) |
|
|
570 |
|
|
(403) |
|
|
881 |
|
|
Comprehensive loss |
|
$ |
(23,831) |
|
$ |
(8,426) |
|
$ |
(59,272) |
|
$ |
(24,639) |
|
|
Net loss per share, basic and diluted |
|
$ |
(0.89) |
|
$ |
(0.35) |
|
$ |
(2.27) |
|
$ |
(1.01) |
|
|
Weighted-average common shares outstanding, basic and diluted |
|
|
26,164,527 |
|
|
25,374,381 |
|
|
25,968,849 |
|
|
25,227,058 |
|
The accompanying notes are an integral part of these condensed consolidated financial statements.
6
Voyager Therapeutics, Inc.
Condensed Consolidated Statements of Cash Flows
(amounts in thousands)
(unaudited)
|
|
|
Nine Months Ended |
|
||||
|
|
|
September 30, |
|
||||
|
|
|
2017 |
|
2016 |
|
||
|
Cash flow from operating activities |
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(58,869) |
|
$ |
(25,520) |
|
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
Stock-based compensation expense |
|
|
6,945 |
|
|
4,528 |
|
|
Depreciation |
|
|
1,159 |
|
|
448 |
|
|
Amortization of premiums and discounts on marketable securities |
|
|
197 |
|
|
571 |
|
|
In-kind research and development expenses |
|
|
113 |
|
|
989 |
|
|
Other non-cash items |
|
|
139 |
|
|
(409) |
|
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
Prepaid expenses and other current assets |
|
|
1,764 |
|
|
(311) |
|
|
Other non-current assets |
|
|
— |
|
|
57 |
|
|
Deferred revenue |
|
|
(3,790) |
|
|
(12,219) |
|
|
Accounts payable |
|
|
361 |
|
|
175 |
|
|
Accrued expenses |
|
|
3,950 |
|
|
2,301 |
|
|
Other non-current liabilities |
|
|
1,000 |
|
|
— |
|
|
Lease incentive benefit |
|
|
515 |
|
|
— |
|
|
Net cash used in operating activities |
|
|
(46,516) |
|
|
(29,390) |
|
|
Cash flow from investing activities |
|
|
|
|
|
|
|
|
Purchases of property and equipment |
|
|
(3,824) |
|
|
(1,160) |
|
|
Change in restricted cash |
|
|
— |
|
|
(471) |
|
|
Purchases of marketable securities and warrants |
|
|
(49,856) |
|
|
(66,627) |
|
|
Proceeds from maturities of marketable securities |
|
|
110,100 |
|
|
129,700 |
|
|
Net cash provided by investing activities |
|
|
56,420 |
|
|
61,442 |
|
|
Cash flow from financing activities |
|
|
|
|
|
|
|
|
Proceeds from the exercise of stock options |
|
|
1,656 |
|
|
146 |
|
|
Net cash provided by financing activities |
|
|
1,656 |
|
|
146 |
|
|
Net increase in cash and cash equivalents |
|
|
11,560 |
|
|
32,198 |
|
|
Cash and cash equivalents, beginning of period |
|
|
36,641 |
|
|
31,309 |
|
|
Cash and cash equivalents, end of period |
|
$ |
48,201 |
|
$ |
63,507 |
|
The accompanying notes are an integral part of these condensed consolidated financial statements.
7
VOYAGER THERAPEUTICS, INC.
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
1. Nature of business
Voyager Therapeutics, Inc. (“the Company”) is a clinical-stage gene therapy company focused on developing life-changing treatments for patients suffering from severe neurological diseases. The Company focuses on neurological diseases where it believes an adeno associated virus (“AAV”) gene therapy approach that either increases or decreases the production of a specific protein can slow or reduce the symptoms experienced by patients, and therefore have a clinically meaningful impact. The Company has created a product engine that enables it to engineer, optimize, manufacture, and deliver its AAV-based gene therapies that have the potential to provide durable efficacy following a single administration. The Company’s pipeline consists of six programs including advanced Parkinson's disease; a monogenic form of amyotrophic lateral sclerosis; Huntington's disease; Friedreich's ataxia; frontotemporal dementia / Alzheimer’s disease; and severe, chronic pain. Additionally, the Company is working to identify and optimize novel AAV capsids.
The Company is devoting substantially all of its efforts to product research and development, activities related to its product engine, novel AAV capsid identification and optimization, and raising capital. The Company is subject to risks common to companies in the biotechnology and gene therapy industry, including but not limited to, risks of failure of pre-clinical studies and clinical trials, the need to obtain marketing approval for its drug product candidates, the need to successfully commercialize and gain market acceptance of its drug product candidates, dependence on key personnel, protection of proprietary technology, compliance with government regulations, development by competitors of technological innovations, and ability to transition from pilot scale manufacturing to large scale commercial production.
In February 2015, the Company entered into an agreement with Sanofi Genzyme (“Collaboration Agreement”), which included a non-refundable upfront payment of $65.0 million. In addition, contemporaneous with entering into the Collaboration Agreement, Sanofi Genzyme entered into a Series B Stock Purchase Agreement, under which Sanofi Genzyme purchased 10,000,000 shares of Series B Preferred Stock for $30.0 million.
Through September 30, 2017, the Company has raised capital primarily from sales of convertible preferred stock and common stock, including its initial public offering of its common stock (“IPO”), and proceeds from the Collaboration Agreement. The Company operates in a net loss position and has recorded a net loss of $58.9 million and $25.5 million for the nine months ended September 30, 2017 and 2016, respectively. As of September 30, 2017, the Company has recognized an accumulated deficit of $148.9 million. The Company believes that its cash, cash equivalents, and marketable debt securities of $125.6 million as of September 30, 2017 is sufficient to fund its current operating plan into 2019. There can be no assurance, however, that the current operating plan will be achieved in the timeframe anticipated by the Company or will not change, that its cash resources will fund the Company’s operating plan for the period anticipated by the Company, or that if the Company needs additional funding, such funding will be available on terms acceptable to the Company, or at all.
2. Summary of significant accounting policies and basis of presentation
Basis of Presentation
The accompanying unaudited condensed consolidated financial statements of the Company have been prepared in accordance with accounting principles generally accepted in the United States (“GAAP”) for interim financial reporting and as required by Regulation S-X, Rule 10-01. Accordingly, they do not include all of the information and footnotes required by GAAP for complete financial statements. For further information, refer to the consolidated financial statements and footnotes included in the Company’s Annual Report on Form 10-K for the year ended December 31, 2016 as filed with the Securities and Exchange Commission (“SEC”). These interim condensed consolidated financial statements, in the opinion of management, reflect all normal recurring adjustments necessary for a fair presentation of the Company’s financial position and results of operations for the periods presented. Any reference in these notes to applicable guidance is meant to refer to the authoritative United States generally accepted accounting
8
principles as found in the Accounting Standards Codification (“ASC”) and Accounting Standards Updates (“ASU”) of the Financial Accounting Standards Board (“FASB”).
Principles of Consolidation
The unaudited interim consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries as disclosed in Note 2, Summary of Significant Accounting Policies, within the “Notes to Consolidated Financial Statements” accompanying its Annual Report on Form 10-K for the fiscal year ended December 31, 2016. Intercompany accounts and transactions have been eliminated.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. On an ongoing basis, the Company’s management evaluates its estimates, which include, but are not limited to, estimates related to revenue recognition, accrued expenses, stock‑based compensation expense, income taxes, and the fair value of common stock. The Company bases its estimates on historical experience and other market specific or other relevant assumptions that it believes to be reasonable under the circumstances. Actual results may differ from those estimates or assumptions.
Recent Accounting Pronouncements
In May 2014, the FASB issued ASU No. 2014-09, Revenue from Contracts with Customers (Topic 606) (“ASU 2014-09”). Subsequently, the FASB also issued ASU 2015-14, Revenue from Contracts with Customers (Topic 606), which adjusted the effective date of ASU 2014-09; ASU No. 2016-08, Revenue from Contracts with Customers (Topic 606): Principal versus Agent Considerations (Reporting Revenue Gross versus Net), which amends the principal-versus-agent implementation guidance and illustrations in ASU 2014-09; ASU No. 2016-10, Revenue from Contracts with Customers (Topic 606): Identifying Performance Obligations and Licensing, which clarifies identifying performance obligation and licensing implementation guidance and illustrations in ASU 2014-09; and ASU No. 2016-12, Revenue from Contracts with Customers (Topic 606): Narrow-Scope Improvements and Practical Expedients, which addresses implementation issues and is intended to reduce the cost and complexity of applying the new revenue standard in ASU 2014-09 (collectively, the “Revenue ASUs”).
The Revenue ASUs provide an accounting standard for a single comprehensive model for use in accounting for revenue arising from contracts with customers and supersedes most current revenue recognition guidance. The accounting standard is effective for interim and annual periods beginning after December 15, 2017, with an option to early adopt for interim and annual periods beginning after December 15, 2016. The guidance permits two methods of adoption: retrospectively to each prior reporting period presented (the full retrospective method), or retrospectively with the cumulative effect of initially applying the guidance recognized at the date of initial application (the modified retrospective method). The Company will adopt the Revenue ASUs effective January 1, 2018 and expects to utilize the modified retrospective methodology. As of September 30, 2017, revenue is generated exclusively from the Company’s collaboration arrangement with Sanofi Genzyme. The Company is currently evaluating the potential impact that the Revenue ASUs will have on its financial position and results of operations as it relates to this single arrangement. The adoption of the Revenue ASUs is expected to have a significant impact on the Company’s notes to consolidated financial statements and its internal controls over financial reporting.
In February 2016, the FASB issued ASU 2016-02, Leases (“ASC 842”), which sets out the principles for the recognition, measurement, presentation and disclosure of leases for both parties to a contract (i.e., lessees and lessors). The standard requires lessees to apply a dual approach, classifying leases as either finance or operating leases based on the principle of whether or not the lease is effectively a financed purchase by the lessee. This classification will determine whether lease expense is recognized based on an effective interest method or on a straight-line basis over the term of the lease. A lessee is also required to record a right-of-use asset and a lease liability for all leases with a term of greater than 12 months regardless of their classification. Leases with a term of 12 months or less will be accounted for similarly to existing guidance for operating leases today. ASC 842 supersedes the previous leases standard, ASC 840
9
Leases. The standard will be effective on January 1, 2019, with early adoption permitted. The Company is in the process of evaluating the impact that this new guidance will have on its consolidated financial statements.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows: Classification of Certain Cash Receipts and Cash Payments. The new standard clarifies certain aspects of the statement of cash flows, including the classification of contingent consideration payments made after a business combination and several other clarifications not currently applicable to the Company. The new standard also clarifies that an entity should determine each separately identifiable source or use within the cash receipts and cash payments on the basis of the nature of the underlying cash flows. In situations in which cash receipts and payments have aspects of more than one class of cash flows and cannot be separated by source or use, the appropriate classification should depend on the activity that is likely to be the predominant source or use of cash flows for the item. The new standard will be effective for the Company on January 1, 2018. The adoption of this standard is not expected to have a material impact on the Company’s consolidated statement of cash flows upon adoption.
In November 2016, the FASB issued ASU 2016-18, Statement of Cash Flows: Restricted Cash (“ASU 2016-18”). The amendments in this update require that amounts generally described as restricted cash and restricted cash equivalents be included within cash and cash equivalents when reconciling the beginning-of-period and end-of-period total amounts shown on the statement of cash flows. ASU 2016-18 will be effective January 1, 2018, with early adoption permitted. The Company expects the adoption to impact its consolidated statement of cash flows as, upon adoption, it will include the Company’s restricted cash balance in the cash and cash equivalents reconciliation of operating, investing and financing activities.
3. Fair Value Measurements
Assets and liabilities measured at fair value on a recurring basis as of September 30, 2017 and December 31, 2016 are as follows:
|
|
|
|
|
|
Quoted Prices |
|
Significant |
|
|
|
|
||
|
|
|
|
|
|
in Active |
|
Other |
|
Significant |
|
|||
|
|
|
|
|
|
Markets for |
|
Observable |
|
Unobservable |
|
|||
|
|
|
|
|
|
Identical Assets |
|
Inputs |
|
Inputs |
|
|||
|
Assets |
|
Total |
|
(Level 1) |
|
(Level 2) |
|
(Level 3) |
|
||||
|
September 30, 2017 |
|
(in thousands) |
|
||||||||||
|
Money market funds included in cash and cash equivalents |
|
$ |
47,246 |
|
$ |
47,246 |
|
$ |
— |
|
$ |
— |
|
|
Marketable securities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
U.S. Treasury notes |
|
|
67,407 |
|
|
67,407 |
|
|
— |
|
|
— |
|
|
U.S. Government agency securities |
|
|
9,979 |
|
|
— |
|
|
9,979 |
|
|
— |
|
|
Equity securities |
|
|
960 |
|
|
960 |
|
|
— |
|
|
— |
|
|
Total marketable securities |
|
$ |
78,346 |
|
$ |
68,367 |
|
$ |
9,979 |
|
$ |
— |
|
|
Warrants to purchase equity securities |
|
|
472 |
|
|
— |
|
|
472 |
|
|
— |
|
|
Total |
|
$ |
126,064 |
|
$ |
115,613 |
|
$ |
10,451 |
|
$ |
— |
|
|
December 31, 2016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds included in cash and cash equivalents |
|
$ |
36,003 |
|
$ |
36,003 |
|
$ |
— |
|
$ |
— |
|
|
Marketable securities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
U.S. Treasury notes |
|
|
130,173 |
|
|
130,173 |
|
|
— |
|
|
— |
|
|
U.S. Government agency securities |
|
|
7,604 |
|
|
— |
|
|
7,604 |
|
|
— |
|
|
Equity securities |
|
|
1,360 |
|
|
1,360 |
|
|
— |
|
|
— |
|
|
Total marketable securities |
|
$ |
139,137 |
|
$ |
131,533 |
|
$ |
7,604 |
|
$ |
— |
|
|
Warrants to purchase equity securities |
|
|
792 |
|
|
— |
|
|
792 |
|
|
— |
|
|
Total |
|
$ |
175,932 |
|
$ |
167,536 |
|
$ |
8,396 |
|
$ |
— |
|
10
The Company measures the fair value of money market funds, U.S. Treasury notes, and equity securities based on quoted prices in active markets for identical securities. The Level 2 debt securities include U.S. Government agency securities that are valued either based on recent trades of securities in inactive markets or based on quoted market prices of similar instruments and other significant inputs derived from or corroborated by observable market data. The Level 2 equity securities include warrants to purchase equity securities that are valued using the Black-Scholes model. The Black-Scholes option pricing model requires inputs based on certain assumptions, including (a) the expected stock price volatility, (b) the calculation of expected term of the awards, (c) the risk-free interest rate, and (d) expected dividends. The assumptions utilized to value the warrants to purchase equity securities as of September 30, 2017 and December 31, 2016 are as follows:
|
|
|
As of September 30, |
|
As of December 31, |
|
||
|
|
|
2017 |
|
2016 |
|
||
|
Risk-free interest rate |
|
1.8 |
% |
|
1.8 |
% |
|
|
Expected dividend yield |
|
— |
% |
|
— |
% |
|
|
Expected term (in years) |
|
3.9 |
|
|
4.7 |
|
|
|
Expected volatility |
|
100.1 |
% |
|
97.5 |
% |
|
The expected volatility is based on the historic volatility for the equity securities underlying the warrants and is calculated based on a period of time commensurate with the expected term assumption. The expected term is based on the remaining contractual life of the warrants on each measurement date. The risk-free interest rate is based on a treasury instrument whose term is consistent with the expected term of the warrants. The expected dividend yield is assumed to be zero as the entity that issued the warrants has never paid and has not indicated any intention to pay dividends.
4. Cash, Cash Equivalents, and Available for Sale Marketable Securities
Cash equivalents include all highly liquid investments maturing within 90 days from the date of purchase. Investments consist of securities with original maturities greater than 90 days when purchased. The Company classifies these investments as available-for-sale and records them at fair value in the accompanying condensed consolidated balance sheets. Unrealized gains or losses are included in accumulated other comprehensive income (loss). Premiums or discounts from par value are amortized to investment income over the life of the underlying investment.
The Company classifies marketable securities as available‑for‑sale. Marketable debt securities with a maturity date within the next year are classified as current assets. Marketable debt securities with a maturity date greater than one year and marketable equity securities are classified as non‑current where the Company has the intent and ability to hold these securities for at least the next 12 months. Available‑for‑sale debt securities are maintained by an investment manager and consist of U.S. Treasury notes and U.S. Government agency securities. During the third quarter in 2016, the Company invested in a supplier and received common stock and warrants to purchase common stock in that entity. The common stock is included in non-current marketable securities and the warrants are included in non-current assets.
All available‑for‑sale securities are carried at fair value with the unrealized gains and losses included in other comprehensive income (loss) as a component of stockholders’ equity until realized. Any premium or discount arising at purchase is amortized and/or accreted to interest income and/or expense. Realized gains and losses are determined using the specific identification method and are included in other income (expense). If any adjustment to fair value reflects a decline in value of the investment, the Company considers all available evidence to evaluate the extent to which the decline is “other‑than‑temporary” and, if so, recognizes the loss through a charge to the Company’s statement of operations and comprehensive loss. No other than temporary losses have been recognized.
11
Cash, cash equivalents, and marketable securities included the following at September 30, 2017 and December 31, 2016:
|
|
|
Amortized |
|
Unrealized |
|
Unrealized |
|
Fair |
|
||||
|
|
|
Cost |
|
Gains |
|
Losses |
|
Value |
|
||||
|
|
|
(in thousands) |
|
||||||||||
|
As of September 30, 2017 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds included in cash and cash equivalents |
|
$ |
47,246 |
|
$ |
— |
|
$ |
— |
|
$ |
47,246 |
|
|
Marketable securities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
U.S. Treasury notes |
|
|
67,421 |
|
|
— |
|
|
14 |
|
|
67,407 |
|
|
U.S. Government agency securities |
|
|
9,978 |
|
|
1 |
|
|
— |
|
|
9,979 |
|
|
Total debt securities |
|
$ |
77,399 |
|
$ |
1 |
|
$ |
14 |
|
$ |
77,386 |
|
|
Equity securities |
|
|
1,220 |
|
|
— |
|
|
260 |
|
|
960 |
|
|
Total marketable securities |
|
$ |
78,619 |
|
$ |
1 |
|
$ |
274 |
|
$ |
78,346 |
|
|
Total money market funds and marketable securities |
|
$ |
125,865 |
|
$ |
1 |
|
$ |
274 |
|
$ |
125,592 |
|
|
As of December 31, 2016 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds included in cash and cash equivalents |
|
$ |
36,003 |
|
$ |
— |
|
$ |
— |
|
$ |
36,003 |
|
|
Marketable securities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
U.S. Treasury notes |
|
|
130,237 |
|
|
2 |
|
|
66 |
|
|
130,173 |
|
|
U.S. Government agency securities |
|
|
7,604 |
|
|
— |
|
|
— |
|
|
7,604 |
|
|
Total debt securities |
|
$ |
137,841 |
|
$ |
2 |
|
$ |
66 |
|
$ |
137,777 |
|
|
Equity securities |
|
|
1,220 |
|
|
140 |
|
|
— |
|
|
1,360 |
|
|
Total marketable securities |
|
$ |
139,061 |
|
$ |
142 |
|
$ |
66 |
|
$ |
139,137 |
|
|
Total money market funds and marketable securities |
|
$ |
175,064 |
|
$ |
142 |
|
$ |
66 |
|
$ |
175,140 |
|
The estimated fair value of the Company’s debt securities balance at September 30, 2017, by contractual maturity, is as follows:
|
Due in one year or less |
|
$ |
77,386 |
|
5. Accrued Expenses
Accrued expenses as of September 30, 2017 and December 31, 2016 consist of the following:
|
|
|
As of September 30, |
|
As of December 31, |
|
||
|
|
|
2017 |
|
2016 |
|
||
|
|
|
(in thousands) |
|
||||
|
External research and development costs |
|
$ |
6,447 |
|
$ |
2,384 |
|
|
Employee compensation costs |
|
|
2,486 |
|
|
2,399 |
|
|
Professional services |
|
|
1,020 |
|
|
698 |
|
|
Accrued goods and services |
|
|
309 |
|
|
842 |
|
|
Patent costs |
|
|
180 |
|
|
89 |
|
|
Other |
|
|
51 |
|
|
76 |
|
|
Total |
|
$ |
10,493 |
|
$ |
6,488 |
|
6. Commitments and Contingencies
Operating Leases
During March 2014, the Company entered into an agreement to lease its facility located at 75 Sidney Street under a non-cancelable operating lease that would expire on December 15, 2019. The lease contains escalating rent clauses which require higher rent payments in future years. The Company expenses rent on a straight-line basis over the term of the lease, including any rent-free periods.
12
In December 2015, the Company executed an amendment to extend the 75 Sidney Street lease and executed an agreement to lease the 64 Sidney Street facility until December 31, 2024. The additional facility includes laboratory and office space, and was ready for occupancy in early 2017.
The Company received leasehold improvement incentives from the landlord totaling $1.3 million and $3.5 million for 75 Sidney Street and 64 Sidney Street, respectively. The Company recorded these incentives as a component of deferred rent and is amortizing these incentives as a reduction of rent expense over the life of the lease. The leasehold improvements have been recorded as fixed assets.
The following table summarizes the Company’s significant contractual obligations as of payment due date by period at September 30, 2017, related to the amended 75 Sidney Street lease and the 64 Sidney Street lease through December 2024:
|
|
|
Total Minimum |
|
|
|
|
|
Lease Payments |
|
|
|
|
|
(in thousands) |
|
|
|
2017 |
|
$ |
806 |
|
|
2018 |
|
|
3,290 |
|
|
2019 |
|
|
3,382 |
|
|
2020 |
|
|
3,762 |
|
|
2021 |
|
|
3,868 |
|
|
2022+ |
|
|
12,273 |
|
|
|
|
$ |
27,381 |
|
Significant Agreements
Sanofi Genzyme Collaboration Agreement
Summary of Agreement
In February 2015, the Company entered into the Collaboration Agreement with Sanofi Genzyme, which included a non‑refundable upfront payment of $65.0 million. In addition, contemporaneously with entering into the Collaboration Agreement, Sanofi Genzyme entered into a Series B Stock Purchase Agreement, under which Sanofi Genzyme purchased 10,000,000 shares of Series B Preferred Stock for $30.0 million. The fair value of the Series B Preferred Stock at the time of issuance was approximately $25.0 million. The $5.0 million premium over the fair value is accounted for as additional consideration under the Collaboration Agreement.
Under the Collaboration Agreement, the Company granted Sanofi Genzyme an exclusive option to license, develop and commercialize (i) ex‑U.S. rights to the following programs, which are referred to as Split Territory Programs; VY‑AADC (“Parkinson’s Program”), VY‑FXN01 (“Friedreich’s Ataxia Program”), a future program to be designated by Sanofi Genzyme (“Future Program”), and VY‑HTT01 (“Huntington’s Program”) with an incremental option to co‑commercialize VY‑HTT01 in the United States and (ii) worldwide rights to VY‑SMN101 (“Spinal Muscular Atrophy Program”). Sanofi Genzyme’s option for the Split Territory Programs and the Spinal Muscular Atrophy Program is triggered following the completion of the first proof of principle human clinical study (“POP Study”), on a program-by-program basis.
Prior to any option exercise by Sanofi Genzyme, the Company will collaborate with Sanofi Genzyme in the development of products under each Split Territory Program and the Spinal Muscular Atrophy Program pursuant to a written development plan and under the guidance of an Alliance Joint Steering Committee (“AJSC”), comprised of an equal number of employees from the Company and Sanofi Genzyme.
The Company is required to use commercially reasonable efforts to develop products under each Split Territory Program and the Spinal Muscular Atrophy Program through the completion of the applicable POP Study. During the development of these joint programs, the activities are guided by a Development Advisory Committee (“DAC”). The
13
DAC may elect to utilize certain Sanofi Genzyme technology relating to the Parkinson’s Program, the Huntington’s Program, or generally with the manufacture of Split Territory Program products.
The Company is solely responsible for all costs incurred in connection with the development of the Split Territory Programs and the Spinal Muscular Atrophy Program products prior to the exercise of an option by Sanofi Genzyme except that, at the Company’s request and upon mutual agreement, Sanofi Genzyme will provide “in‑kind” services valued at up to $5.0 million. As of September 30, 2017, the Company has received in-kind services from Sanofi Genzyme valued at $3.6 million.
Other than the Parkinson’s Program (for which a POP Study has already commenced), if the Company does not initiate a POP Study for a given Split Territory Program by December 31, 2026 (or for the Future Program by the tenth anniversary of the date the Future Program is nominated by Sanofi Genzyme), and Sanofi Genzyme has not terminated the Collaboration Agreement with respect to the collaboration program, then Sanofi Genzyme shall be entitled, as its sole and exclusive remedy, to a credit of $10.0 million for each such program against other milestone or royalty payments payable by Sanofi Genzyme under the Collaboration Agreement. However, if the POP Study is not initiated due to a regulatory delay or a force majeure event, such time period shall be extended for so long as such delay continues.
Under the Collaboration Agreement, Sanofi Genzyme is not required to pay an option exercise payment to exercise its option regarding the Parkinson’s Program. Sanofi Genzyme is required to pay $20.0 million for the exercise of each Split Territory Program option other than the Parkinson’s Program and $30.0 million for the exercise of the Spinal Muscular Atrophy Program option.
Upon Sanofi Genzyme’s exercise of its option to license a given product in a Split Territory Program (“Split Territory Licensed Product”), the Company will have sole responsibility for the development of such Split Territory Licensed Product in the United States and Sanofi Genzyme shall have sole responsibility for development of such Split Territory Licensed Product in the rest of the world. The Company and Sanofi Genzyme will have shared responsibility for execution of ongoing development of such Split Territory Licensed Product that is not specific to either territory, including costs associated therewith. The Company is responsible for all commercialization activities relating to Split Territory Licensed Products in the United States, including all of the associated costs. Sanofi Genzyme is responsible for all commercialization activities relating to the Split Territory Licensed Products in the rest of the world, including all of the associated costs. If Sanofi Genzyme exercises its co‑commercialization rights for the Huntington’s Program product (“Huntington’s Licensed Product”), Sanofi Genzyme will be the lead party responsible for all commercialization activities related to such product in the United States.
Upon exercise of the option, Sanofi Genzyme shall have the sole right to develop the licensed product from the Spinal Muscular Atrophy Program (“Spinal Muscular Atrophy Licensed Product”) worldwide. Sanofi Genzyme shall be responsible for all of the development costs that occur after the option exercise date for the Spinal Muscular Atrophy Program. Sanofi Genzyme is also responsible for commercialization activities relating to the Spinal Muscular Atrophy Licensed Product worldwide. In November 2016, the Company and Sanofi Genzyme elected to deprioritize the Spinal Muscular Atrophy Program due to, among other things, the progress made in other preclinical programs and the evolving competitive landscape.
Sanofi Genzyme is required to pay the Company for specified regulatory and commercial milestones, if achieved, up to $645.0 million across all programs. The regulatory approval milestones are payable upon either regulatory approval in the United States or regulatory and reimbursement approval in the European Union and range from $40.0 million to $50.0 million per milestone, with an aggregate total of $265.0 million. The commercial milestones are payable upon achievement of specified annual net sales in each program and range from $50.0 million to $100.0 million per milestone, with an aggregate total of $380.0 million.
In addition, to the extent any Split Territory Licensed Products or the Spinal Muscular Atrophy Licensed Product are commercialized, the Company is entitled to tiered royalty payments ranging from the mid‑single digits to mid‑teens based on a percentage of net sales by Sanofi Genzyme. Sanofi Genzyme is entitled to receive tiered royalty payments related to sales of Split Territory Licensed Product ranging from the low‑single digits to mid‑single digits based on a percentage of net sales by the Company depending on whether the Company uses Sanofi Genzyme technology in the Split Territory Licensed Product. If Sanofi Genzyme elects to co‑commercialize the Huntington’s
14
Licensed Product in the United States, the Company and Sanofi Genzyme will share in any profits or losses from sales of such product.
The Collaboration Agreement will continue in effect until the later of (i) the expiration of the last to expire of the option rights and (ii) the expiration of all payment obligations unless sooner terminated by the Company or Sanofi Genzyme. The Company and Sanofi Genzyme have customary termination rights including the right to terminate for an uncured material breach of the agreement committed by the other party and Sanofi Genzyme has the right to terminate for convenience.
In October 2017, the Company announced that Sanofi Genzyme decided not to exercise its option for the ex-U.S. rights to the Parkinson’s Program. Therefore, the Company now maintains global rights to VY-AADC. If the Company uses certain Sanofi Genzyme technology in VY-AADC, Sanofi Genzyme is entitled to receive low-single digit royalty payments based on a percentage of net sales by the Company and the Company may be obligated to make certain regulatory milestone payments to a third-party licensor. As a result of Sanofi Genzyme’s decision, the Company is no longer entitled to receive $45 million and $60 million of regulatory and commercial milestone payments, respectively, related to the Parkinson’s Program.
Accounting Analysis
The Collaboration Agreement includes the following deliverables: (i) research and development services for each of the Split Territory License Programs and the Spinal Muscular Atrophy Program, (ii) participation in the AJSC, (iii) participation in the DAC and (iv) the option to obtain a development and commercial license in the Parkinson’s Program and related deliverables. The Company has determined that the option to obtain a development and commercial license in the Parkinson’s Program is not a substantive option for accounting purposes, primarily because there is no additional option exercise payment payable by Sanofi Genzyme at the time the option is exercised. Therefore, the option to obtain a license and other obligations of the Company that are contingent upon exercise of the option are considered deliverables at the inception of the arrangement. The options in the other Split Territory Programs and the Spinal Muscular Atrophy Program are considered substantive as there are substantial option exercise payments payable by Sanofi Genzyme upon exercise. In addition, as a result of the uncertainties related to the discovery, research, development and commercialization activities, the Company is at risk with regard to whether Sanofi Genzyme will exercise the options. Moreover, the substantive options are not priced at a significant incremental discount. Accordingly, the substantive options are not considered deliverables at the inception of the arrangement and the associated option exercise payments are not included in allocable arrangement consideration. The Company has also determined that any obligations which are contingent upon the exercise of a substantive option are not considered deliverables at the outset of the arrangement, as these deliverables are contingent upon the exercise of the options. In addition, any option exercise payments associated with the substantive options are not included in the allocable arrangement consideration.
The Company has concluded that each of the deliverables identified at the inception of the arrangement has standalone value from the other undelivered elements. Additionally, the Collaboration Agreement does not include return rights related to the initial collaboration term. Accordingly, each deliverable qualifies as a separate unit of accounting.
The Company identified $79.3 million of allocable arrangement consideration consisting of the $65.0 million upfront fee, the $5.0 million premium paid in excess of fair value of the Series B Preferred Stock and $9.3 million of Sanofi Genzyme “in‑kind” and other funding.
The Company has allocated the allocable arrangement consideration based on the relative selling price of each unit of accounting. For all units of accounting, the Company determined the selling price using the best estimate of selling price (“BESP”). The Company determined the BESP for the service related deliverable for the research and development activities based on internal estimates of the costs to perform the services, including expected internal expenses and expenses with third parties for services and supplies, marked up to include a reasonable profit margin and adjusted for the scope of the potential license. Significant inputs used to determine the total expense of the research and development activities include, the length of time required and the number and costs of various studies that will be performed to complete the applicable POP Study. The BESP for the AJSC and DAC have been estimated based on the costs incurred to participate in the committees, marked up to include a reasonable profit margin. The BESP for the
15
license option was determined based on the estimated value of the license and related deliverables adjusted for the estimated probability that the option would be exercised by Sanofi Genzyme.
Based on the relative selling price allocation, the allocable arrangement consideration was allocated as follows:
|
|
|
|
|
|
|
Unit of Accounting |
|
Amount |
|
|
|
|
|
(in thousands) |
|
|
|
Research and Development Services for: |
|
|
|
|
|
Huntington’s Program |
|
$ |
15,662 |
|
|
Parkinson’s Program |
|
|
6,648 |
|
|
Friedreich’s Ataxia Program |
|
|
16,315 |
|
|
Spinal Muscular Atrophy Program |
|
|
32,050 |
|
|
Future Program |
|
|
2,464 |
|
|
Committee Obligations: |
|
|
|
|
|
AJSC |
|
|
147 |
|
|
DAC |
|
|
227 |
|
|
License Option and related deliverables |
|
|
5,743 |
|
|
Total |
|
$ |
79,256 |
|
The Company recognizes the amounts associated with research and development services on a straight-line basis over the estimated period of service as there is no discernable pattern or objective measure of performance for the services. Similarly, the Company recognizes the amount associated with the committee obligations on a straight-line basis over the period of service consistent with the expected pattern of performance. The amount allocated to the license option is being deferred until the point in time in which Sanofi Genzyme is required to make a decision. In October 2017, Sanofi Genzyme elected not to exercise the option. The amount allocated to the Parkinson’s Program license option and related deliverables unit of accounting will be recognized as revenue at the time of the decision.
During 2016, the Company reassessed the estimated period of performance for each of its units of accounting and determined that the estimated period would be extended for two units of accounting. Additionally, the Company and Sanofi Genzyme agreed to deprioritize the development of the Spinal Muscular Atrophy Licensed Product and reduce the estimates related to the amount of “in-kind” services that would be provided by Sanofi Genzyme. These adjustments were made on a prospective basis and resulted in decreases in revenue recognized by $2.4 million per quarter.
During the first two quarters of 2017, the Company reassessed the estimated period of performance for each of its units of accounting and, based on the current facts and circumstances, determined that the estimated period would be extended for three units of accounting. These adjustments were made on a prospective basis and resulted in a decrease in revenue recognized by $0.5 million per quarter.
The Company has evaluated all of the milestones that may be received in connection with each Split Territory Licensed Product and the Spinal Muscular Atrophy Licensed Product. In evaluating if a milestone is substantive, the Company assesses whether: (i) the consideration is commensurate with either the Company’s performance to achieve the milestone or the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the Company performance to achieve the milestone, (ii) the consideration relates solely to past performance and (iii) the consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. All regulatory milestones are considered substantive on the basis of the contingent nature of the milestone, specifically reviewing factors such as the scientific, clinical, regulatory, and other risks that must be overcome to achieve the milestone as well as the level of effort and investment required. Accordingly, such amounts will be recognized as revenue in full in the period in which the associated milestone is achieved, assuming all other revenue recognition criteria are met. All commercial milestones will be accounted for in the same manner as royalties and recorded as revenue upon achievement of the milestone, assuming all other revenue recognition criteria are met.
During the three and nine months ended September 30, 2017, the Company recognized $1.1 million and $3.8 million, respectively, of revenue associated with its collaboration with Sanofi Genzyme related to research and development services performed during the periods. As of September 30, 2017, there is $37.9 million of deferred
16
revenue related to the Collaboration Agreement, which is classified as either current or noncurrent in the accompanying consolidated balance sheet based on the period the consideration is expected to be recognized.
Costs incurred relating to the programs that Sanofi Genzyme has the option to license under the Collaboration Agreement consist of internal and external research and development costs, which primarily include: salaries and benefits, lab supplies and preclinical research studies. The Company does not separately track or segregate the amount of costs incurred under the Collaboration Agreement. These costs are included in research and development expenses in the Company’s statement of operations during the three and nine months ended September 30, 2017. The Company estimates that the majority of research and development expense during the period relate to programs for which Sanofi Genzyme has an option right.
MRI Interventions License and Securities Purchase Agreements
In September 2016, the Company entered into securities purchase and license agreements with MRI Interventions, Inc. (“MRIC”). MRIC is the primary supplier of the ClearPoint System, which is being used by the Company in ongoing development and clinical trials. Under a securities purchase agreement, the Company paid $2.0 million for shares of MRIC common stock and a warrant to purchase additional shares of MRIC common stock. The Company also entered into a license agreement with MRIC that provided for certain rights to MRIC technology and for MRIC to transfer the rights and know-how to manufacture the ClearPoint System to enable the Company to utilize an alternative supplier for the ClearPoint System for use in the Company’s development and clinical trials.
During the three months ended March 31, 2017, the Company terminated the license agreement with MRIC, and all prior and future commitments and obligations under such agreement became null and void. As of September 30, 2017 the Company continued to hold the common stock and warrants to purchase additional shares of common stock as an available-for-sale security and non-current asset, respectively.
Other Agreements
During September 2016, the Company entered into a research and development funding arrangement with a non-profit organization that provides up to $4.0 million in funding upon the achievement of clinical and development milestones. The agreement provides that the Company repay amounts received under certain circumstances including termination of the agreement, and to pay an amount up to 2.6 times the funding received upon successful development and commercialization of any products developed. During the three months ended March 31, 2017, the Company earned a milestone payment of $1.0 million. No milestone payments were earned in the three months ended September 30, 2017. The Company has evaluated the arrangement and has concluded that it represents a research and development financing arrangement as it is probable that the Company will repay amounts received under the arrangement. As a result, the $1.0 million earned through September 30, 2017 is recorded as a non-current liability in the accompanying balance sheet.
Litigation
The Company is not a party to any material legal matters and does not have contingency reserves established for any litigation liabilities as of September 30, 2017 or December 31, 2016.
7. Redeemable Convertible Preferred Stock
The Company has authorized preferred stock amounting to 5,000,000 shares as of September 30, 2017 and December 31, 2016. The authorized preferred stock was classified under stockholders’ equity at September 30, 2017 and December 31, 2016.
17
8. Stock‑Based Compensation
2015 Stock Option Plan
In October 2015, the Company’s board of directors and stockholders approved the 2015 Stock Option and Incentive Plan (“2015 Stock Option Plan”) which became effective upon the completion of the IPO. The 2015 Stock Option Plan provides the Company with the flexibility to use various equity-based incentive and other awards as compensation tools to motivate its workforce. These tools include stock options, stock appreciation rights, restricted stock, restricted stock units, unrestricted stock, performance share awards and cash-based awards. The 2015 Stock Option Plan replaced the Voyager Therapeutics, Inc. 2014 Stock Option and Grant Plan (“2014 Plan”). Any options or awards outstanding under the 2014 Plan remained outstanding and effective. The number of shares initially reserved for issuance under the 2015 Stock Option Plan is the sum of (i) 1,311,812 shares of common stock and (ii) the number of shares under the 2014 Plan that are not needed to fulfill the Company’s obligations for awards issued under the 2014 Plan as a result of forfeiture, expiration, cancellation, termination or net issuances of awards thereunder. The number of shares of common stock that may be issued under the 2015 Stock Option Plan is also subject to increase on the first day of each fiscal year by up to 4% of the Company’s issued and outstanding shares of common stock on the immediately preceding December 31.
Effective January 1, 2016 and 2017, an additional 1,069,971 and 1,070,635 shares, respectively, were added to the Company’s 2015 Stock Option Plan for future issuance. As of September 30, 2017, there were 1,628,505 shares of common stock available for future award grants under the 2015 Stock Option Plan. During the three and nine months ended September 30, 2017, the Company issued a total of 265,500 and 1,521,000 stock options, respectively, to employees and directors under the 2015 Stock Option Plan. During the three and nine months ended September 30, 2017, the Company issued a total of 20,000 stock options to non-employees under the 2015 Stock Option Plan.
The terms of stock awards agreements, including vesting requirements, are determined by the Board of Directors and are subject to the provisions of the 2015 Stock Option Plan.
2014 Stock Option and Grant Plan
In January 2014, the Company adopted the 2014 Plan, under which it may grant incentive stock options, non‑qualified stock options, restricted stock awards, unrestricted stock awards, or restricted stock units to purchase up to 823,529 shares of common stock to employees, officers, directors and consultants of the Company.
In April 2014, the Company amended the 2014 Plan to allow for the issuance of up to 1,411,764 shares of common stock. In August 2014, April 2015, August 2015, and October 2015, the Company further amended the 2014 Plan to allow for the issuance of up to 2,000,000, 2,047,058, 2,669,411, and 2,998,823 shares of common stock, respectively. During 2014 the Company issued only restricted stock awards under the 2014 Plan and during 2015 the Company only granted stock options.
Founder Awards
In January 2014, the Company issued 1,188,233 shares of restricted stock to its founders at an original issuance price of $0.0425 per share. Of the total restricted shares awarded to the founders, 835,292 shares generally vest over one to four years, based on each founder’s continued service to the Company in varying capacity as a Scientific Advisory Board member, consultant, director, officer or employee, as set forth in each grantee’s individual restricted stock purchase agreement. The remaining 352,941 of the shares issued begin vesting upon the achievement of certain performance objectives as well as continued service to the Company, as set forth in the agreements.
These performance conditions are tied to certain milestone events specific to the Company’s corporate goals, including but not limited to preclinical and clinical development milestones related to the Company’s product candidates. Stock‑based compensation expense associated with these performance‑based awards is recognized when the achievement of the performance condition is considered probable, using management’s best estimates. During 2016, management determined that the achievement of the performance milestone for one of the three performance‑based awards had become probable and began recognizing stock-based compensation accordingly. The Company recorded $0.6 million
18
and $1.5 million in stock-based compensation expense related to this award during the three and nine months ended September 30, 2017, respectively. The Company recorded $0.2 million and $1.0 million in stock-based compensation expense related to this award during the three and nine months ended September 30, 2016, respectively. No stock‑based compensation expense was recorded for the remaining two founders’ awards with performance-based vesting as of September 30, 2017 as the performance-based milestones related to these awards were not probable.
2015 Employee Stock Purchase Plan
In October 2015, the Company’s board of directors and stockholders approved the 2015 Employee Stock Purchase Plan (“2015 ESPP”). Under the 2015 ESPP, all full-time employees of the Company are eligible to purchase common stock of the Company twice per year, at the end of each six-month payment period. During each payment period, eligible employees who so elect, may authorize payroll deductions in an amount of 1% to 10% (whole percentages only) of the employee’s base pay for each payroll period. At the end of each payment period, the accumulated deductions are used to purchase shares of common stock from the Company at a discount. A total of 262,362 shares of common stock were initially authorized for issuance under this plan. The 2015 ESPP became effective upon the completion of the IPO. The number of shares of common stock that may be issued under the 2015 ESPP is also subject to increase on the first day of each fiscal year by up to 1% of the Company’s issued and outstanding shares of common stock on the immediately preceding December 31. The 2015 ESPP became effective upon the completion of the IPO. Effective January 1, 2016 and 2017, 267,492 and 267,658 shares of common stock, respectively, were added to the 2015 ESPP.
Stock‑Based Compensation Expense
Total compensation cost recognized for all stock‑based compensation awards in the statements of operations and comprehensive loss is as follows:
|
|
|
Three Months Ended |
|
Nine Months Ended |
|
||||||||
|
|
|
September 30, |
|
September 30, |
|
||||||||
|
|
|
2017 |
|
2016 |
|
2017 |
|
2016 |
|
||||
|
|
|
(in thousands) |
|
||||||||||
|
Research and development |
|
$ |
2,231 |
|
$ |
1,378 |
|
$ |
4,107 |
|
$ |
3,425 |
|
|
General and administrative |
|
|
1,010 |
|
|
411 |
|
|
2,838 |
|
|
1,103 |
|
|
Total stock compensation expense |
|
$ |
3,241 |
|
$ |
1,789 |
|
$ |
6,945 |
|
$ |
4,528 |
|
Restricted Stock
A summary of the status of and changes in unvested restricted stock unit activity under the Company’s equity award plan for the nine months ended September 30, 2017 was as follows:
|
|
|
|
|
Weighted |
|
|
|
|
|
|
|
Average |
|
|
|
|
|
|
|
Grant Date |
|
|
|
|
|
|
|
Fair Value |
|
|
|
|
|
Shares |
|
Per Share |
|
|
|
Unvested restricted common stock as of December 31, 2016 |
|
1,167,984 |
|
$ |
0.76 |
|
|
Issued |
|
— |
|
|
— |
|
|
Vested |
|
(455,634) |
|
$ |
0.76 |
|
|
Repurchased |
|
(36,202) |
|
$ |
0.67 |
|
|
Unvested restricted common stock as of September 30, 2017 |
|
676,148 |
|
$ |
0.72 |
|
The expense related to awards granted to employees and non-employees was $0.1 million and $1.6 million, respectively, for the three months ended September 30, 2017, and $0.4 million and $2.3 million for the nine months ended September 30, 2017, respectively. The expense related to awards granted to employees and non-employees was $0.1 million and $0.7 million, respectively, for the three months ended September 30, 2016, and $0.4 million and $1.9 million for the nine months ended September 30, 2016, respectively.
19
As of September 30, 2017, the Company had unrecognized stock-based compensation expense related to its unvested restricted stock unit awards of $8.1 million, which is expected to be recognized over the remaining average vesting period of 0.6 years.
Stock Options
The following is a summary of stock option activity for the nine months ended September 30, 2017:
|
|
|
|
|
Weighted |
|
Remaining |
|
Aggregate |
|
||
|
|
|
|
|
Average |
|
Contractual |
|
Intrinsic |
|
||
|
|
|
|
|
Exercise |
|
Life |
|
Value |
|
||
|
|
|
Shares |
|
Price |
|
(in years) |
|
(in thousands) |
|
||
|
Outstanding at December 31, 2016 |
|
1,871,237 |
|
$ |
10.21 |
|
|
|
|
|
|
|
Granted |
|
1,541,000 |
|
$ |
12.16 |
|
|
|
|
|
|
|
Exercised |
|
(130,129) |
|
$ |
8.42 |
|
|
|
|
|
|
|
Cancelled or forfeited |
|
(237,494) |
|
$ |
10.63 |
|
|
|
|
|
|
|
Outstanding at September 30, 2017 |
|
3,044,614 |
|
$ |
11.23 |
|
8.8 |
|
$ |
28,542 |
|
|
Exercisable at September 30, 2017 |
|
827,081 |
|
$ |
10.34 |
|
8.2 |
|
$ |
8,499 |
|
|
Vested and expected to vest at September 30, 2017 |
|
3,044,614 |
|
$ |
11.23 |
|
8.8 |
|
$ |
28,542 |
|
Using the Black‑Scholes option pricing model, the weighted average fair value of options granted to employees and directors during the three and nine months ended September 30, 2017 was $9.43 and $7.88 per share, respectively. The expense related to options granted to employees and directors was $1.4 million and $3.8 million for the three and nine months ended September 30, 2017, respectively. The weighted average fair value of options granted to employees and directors during the three and nine months ended September 30, 2016 was $8.98 and $7.62 per share, respectively. The expense related to awards granted to employees and directors was $0.9 million and $2.2 million for the three and nine months ended September 30, 2016, respectively.
The fair value of each option issued to employees and directors was estimated at the date of grant using the Black‑Scholes option pricing model with the following weighted‑average assumptions:
|
|
|
Three Months Ended |
|
Nine Months Ended |
|
||||||||
|
|
|
September 30, |
|
September 30, |
|
||||||||
|
|
|
2017 |
|
2016 |
|
2017 |
|
2016 |
|
||||
|
Risk-free interest rate |
|
1.8 |
% |
|
1.2 |
% |
|
2.0 |
% |
|
1.5 |
% |
|
|
Expected dividend yield |
|
— |
% |
|
— |
% |
|
— |
% |
|
— |
% |
|
|
Expected term (in years) |
|
6.0 |
|
|
6.0 |
|
|
6.0 |
|
|
6.0 |
|
|
|
Expected volatility |
|
72.9 |
% |
|
73.2 |
% |
|
73.8 |
% |
|
73.1 |
% |
|
Using the Black-Scholes option pricing model, the weighted average grant date fair value of options granted to non‑employees during the nine months ended September 30, 2017 was $6.88 per share. Unvested options granted to non‑employees are revalued at each measurement period until fully vested. The expense related to stock option awards granted to non‑employees was $0.1 million and $0.3 million for the three and nine months ended September 30, 2017, respectively. The weighted average grant date fair value of options granted to non-employees during the nine months ended September 30, 2016 was $9.21 per share. The expense related to awards granted to non-employees was $0.1 million and $0.1 the three and nine months ended September 30, 2016, respectively.
20
The fair value of each option issued to non‑employees was estimated at each vesting and reporting date using the Black‑Scholes option pricing model. The reporting date fair value was determined using the following weighted‑average assumptions:
|
|
|
As of September 30, |
|
||||
|
|
|
2017 |
|
2016 |
|
||
|
Risk-free interest rate |
|
2.2 |
% |
|
1.9 |
% |
|
|
Expected dividend yield |
|
— |
% |
|
— |
% |
|
|
Expected term (in years) |
|
8.7 |
|
|
8.9 |
|
|
|
Expected volatility |
|
77.5 |
% |
|
84.0 |
% |
|
As of September 30, 2017, the Company had unrecognized stock-based compensation expense related to its unvested stock options of $15.5 million which is expected to be recognized over the remaining weighted average vesting period of 2.8 years.
For the three and nine months ended September 30, 2017 and 2016, expected volatility was estimated using the historical volatility of the common stock of a peer group of publicly-traded companies that are similarly situated to the Company, blended with historical volatility of the Company’s own stock price.
9. Income taxes
Deferred tax assets and deferred tax liabilities are recognized based on temporary differences between the financial reporting and tax basis of assets and liabilities using statutory rates. A valuation allowance is recorded against deferred tax assets if it is more likely than not that some or all of the deferred tax assets will not be realized. Due to the uncertainty surrounding the realization of the favorable tax attributes in future tax returns, the Company has recorded a full valuation allowance against the Company’s otherwise recognizable net deferred tax assets.
For the three months ended September 30, 2017, the Company recognized a tax benefit in other comprehensive loss of $0.1 million related to the unrealized loss on available-for-sale securities. No income tax benefit or provision has been recognized in other comprehensive loss for the nine months ended September 30, 2017. For the three and nine months ended September 30, 2016, the Company recognized an income tax benefit of $0.3 million, and tax expense in other comprehensive income of $0.6 million related to the unrealized gain on available-for-sale securities. As of September 30, 2016, the Company recorded an accrued income tax provision of $0.3 million related to this tax benefit included within accrued expenses and other current liabilities in the condensed consolidated balance sheet, which is expected to be generated from continuing operations.
10. Net Loss Per Share
Basic net loss per share is calculated by dividing the net loss by the weighted-average number of shares of common stock outstanding during the period, without consideration for potentially dilutive securities. Diluted net loss per share is computed by dividing the net loss by the weighted-average number of shares of common stock and potentially dilutive securities outstanding for the period determined using the treasury-stock and if-converted methods.
For purposes of the diluted net loss per share calculation, unvested restricted common stock and outstanding stock options are considered to be potentially dilutive securities, but are excluded from the calculation of diluted net loss per share because their effect would be anti-dilutive and therefore, basic and diluted net loss per share were the same for all periods presented.
21
The following table sets forth the outstanding potentially dilutive securities that have been excluded in the calculation of diluted net loss per share because to include them would be anti-dilutive:
|
|
|
As of September 30, |
|
||
|
|
|
2017 |
|
2016 |
|
|
Unvested restricted common stock |
|
676,148 |
|
1,300,837 |
|
|
Outstanding stock options |
|
3,044,614 |
|
1,862,619 |
|
|
Total |
|
3,720,762 |
|
3,163,456 |
|
11. Related‑Party Transactions
During the three and nine months ended September 30, 2017 and 2016, the Company received consulting and management services from one of its investors. The total amount of consulting and management services provided by this investor was immaterial during the three and nine months ended September 30, 2017.
During the three and nine months ended September 30, 2017, the Company recognized $1.1 million and $3.8 million, respectively, of revenue associated with the Collaboration Agreement related to research and development services provided during this period. During the three and nine months ended September 30, 2016, the Company recognized $3.3 million and $11.9 million, respectively, of revenue associated with the Collaboration Agreement related to research and development services provided during the period. The Company also recognized $0.1 million of expense during the three and nine months ended September 30, 2017 related to “in‑kind” services provided by Sanofi Genzyme associated with the Collaboration Agreement. No amounts were recognized related to “in-kind” services provide by Sanofi Genzyme during the three months ended September 30, 2017. The Company recognized $0.2 million and $1.0 million of expense during the three and nine months ended September 30, 2016, respectively, related to “in-kind” services provided by Sanofi Genzyme under the Collaboration Agreement.
12. Subsequent Events
In October 2017, the Company announced that Sanofi Genzyme decided not to exercise its option for the ex-U.S. rights to the Parkinson’s Program. Therefore, the Company now maintains global rights to VY-AADC. If the Company uses certain Sanofi Genzyme technology in VY-AADC, Sanofi Genzyme is entitled to receive low-single digit royalty payments based on a percentage of net sales by the Company and the Company may be obligated to make certain regulatory milestone payments to a third-party licensor. As a result of Sanofi Genzyme’s decision, the Company is no longer entitled to receive the regulatory and commercial milestone payments related to the Parkinson’s Program.
The amount of the arrangement consideration allocated to the Parkinson’s Program License Option and related deliverables was included in current deferred revenue as of September 30, 2017 and will be recognized as revenue at the time of their election not to exercise the option.
ITEM 2.MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our unaudited condensed consolidated financial statements and related notes appearing elsewhere in this Quarterly Report on Form 10-Q, our unaudited condensed consolidated financial statements and related notes in our Quarterly Report on Form 10-Q, which was filed with the Securities and Exchange Commission, or SEC, on August 8, 2017, our unaudited condensed consolidated financial statements and related notes in our Quarterly Report on Form 10-Q, which was filed with the SEC on May 9, 2017, and the audited financial information and the notes thereto included in our Annual Report on Form 10-K for the year ended December 31, 2016, which was filed with the SEC on March 15, 2017.
Our actual results and timing of certain events may differ materially from the results discussed, projected, anticipated, or indicated in any forward-looking statements. We caution you that forward-looking statements are not
22
guarantees of future performance and that our actual results of operations, financial condition and liquidity, and the development of the industry in which we operate may differ materially from the forward-looking statements contained in this Quarterly Report on Form 10-Q. In addition, even if our results of operations, financial condition and liquidity, and the development of the industry in which we operate are consistent with the forward-looking statements contained in this Quarterly Report on Form 10-Q, they may not be predictive of results or developments in future periods.
The following information and any forward-looking statements should be considered in light of factors discussed elsewhere in the Quarterly Report on Form 10-Q, including those risks identified under Part II, Item 1A. Risk Factors.
These forward-looking statements are made under the safe harbor provisions of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act. These statements are neither promises nor guarantees. We caution readers not to place undue reliance on any forward-looking statements made by us, which speak only as of the date they are made. We disclaim any obligation, except as specifically required by law and the rules of the SEC, to publicly update or revise any such statements to reflect any change in our expectations or in events, conditions or circumstances on which any such statements may be based, or that may affect the likelihood that actual results will differ from those set forth in the forward-looking statements.
Overview
We are a clinical-stage gene therapy company focused on developing life-changing treatments for patients suffering from severe neurological diseases. We focus on neurological diseases where we believe an adeno-associated virus, or AAV, gene therapy approach that either increases or decreases the production of a specific protein can slow or reduce the symptoms experienced by patients, and therefore have a clinically meaningful impact. We have built a product engine, that we believe positions us to be the leading company at the intersection of AAV gene therapy and severe neurological disease. Our product engine enables us to engineer, optimize, manufacture and deliver our AAV-based gene therapies that have the potential to provide durable efficacy following a single administration. Additionally, we are working to identify and optimize novel AAV capsids, which are the outer viral protein shells that enclose the genetic material of the virus. Our team of experts in the fields of AAV gene therapy and neuroscience first identifies and selects severe neurological diseases that are well-suited for treatment using AAV gene therapy. We then engineer and optimize AAV vectors for delivery of the payload to the targeted tissue or cells. Our manufacturing process employs an established system that we believe will enable production of high quality AAV vectors at commercial-scale. Finally, we leverage established routes of administration and advances in dosing techniques to optimize delivery of our AAV gene therapies directly to discrete regions of the brain, more broadly to the spinal cord region, and to target cells that are critical to the disease of interest.
23
Our pipeline of gene therapy programs is summarized in the table below:
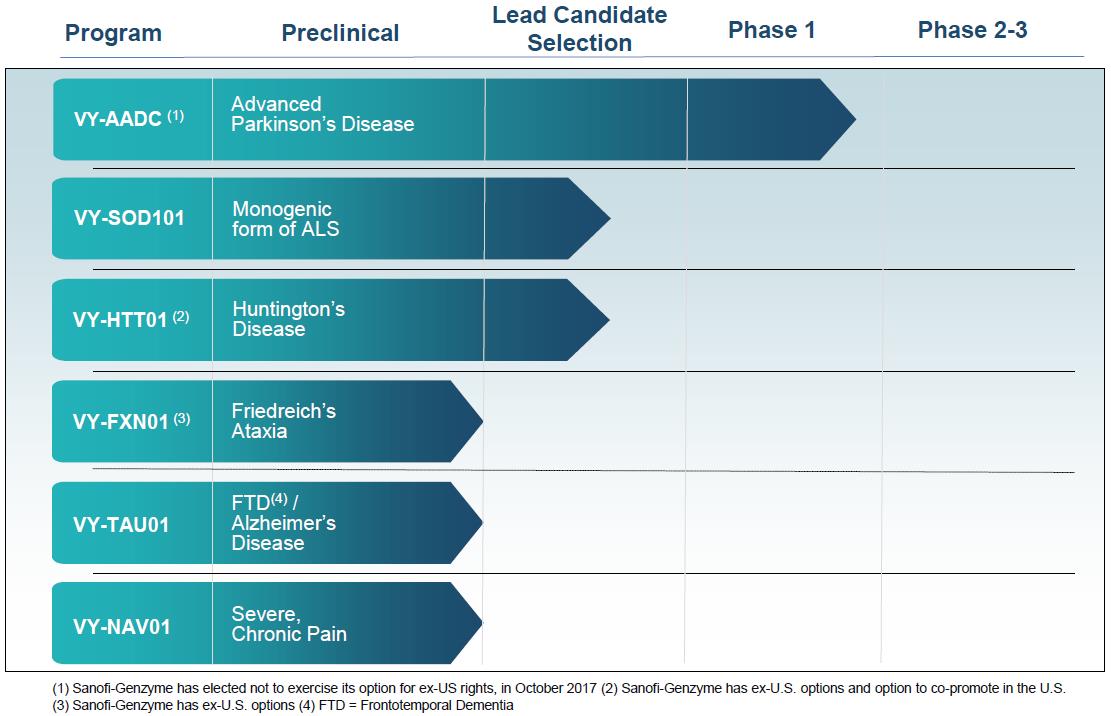
In November 2016, we and Sanofi Genzyme elected to deprioritize the development of VY-SMN101 for spinal muscular atrophy due to, among other things, the progress made in our other preclinical programs and the evolving competitive landscape.
In September 2017, we announced interim results from our ongoing, open-label Phase 1b clinical trial of VY-AADC01 for the treatment of advanced Parkinson’s disease. The trial includes 15 patients with advanced Parkinson’s disease and disabling motor fluctuations, treated with a single administration of VY-AADC01 to assess the safety and distribution of ascending doses of VY-AADC01 administered under magnetic resonance imaging, or MRI, guidance to the putamen, a region of the brain associated with impaired motor function in Parkinson’s disease. Secondary objectives include assessment of aromatic L-amino acid decarboxylase, or AADC, expression and activity in the putamen measured by F-Dopa positron emission tomography, which we refer to as a F-Dopa PET scan, that reflects the capacity to convert levodopa to dopamine. Other secondary measures include assessments of motor function and activities of daily living, as measured by the Unified Parkinson’s Disease Rating Scale, which we refer to as UPDRS-III and UPDRS-II, respectively; quality of life; and a patient-completed (Hauser) diary. These interim results included data from all 15 patients treated in Cohorts 1, 2 and 3 (five patients in each Cohort) including data from patients in Cohort 1 at 24 months, Cohort 2 at 12 months and Cohort 3 at six months. Cohort 1 patients received a single administration of VY-AADC01 at a concentration of 8.3×1011 vector genomes per milliliter, or vg/ml, using an infusion volume of up to 450 µL per putamen, or up to 900 µL per patient, for a total dose of 7.5×1011 vector genomes. Cohort 2 patients received a single administration of VY-AADC01 at a concentration of 8.3×1011 vg/ml, using an infusion volume of up to 900 µL per putamen, or up to 1,800 µL per patient, for a total dose of 1.5 × 1012 vector genomes. Cohort 3 patients received similar infusion volumes (up to 900 µL per putamen) of VY-AADC01 to Cohort 2 but at three-fold higher vector genome concentrations for a total dose of up to 4.5×1012.
24
In this trial:
|
· |
The use of real-time, MRI-guided delivery and increasing infusion volumes resulted in progressively greater coverage of the putamen, from 21% mean coverage of the volume of the putamen with VY-AADC01 in Cohort 1, 34% mean coverage in Cohort 2, and 42% mean coverage in Cohort 3. |
|
· |
VY-AADC01 treatment resulted in a 13% increase, a 56% increase, and a 79% increase in mean putaminal AADC enzyme activity in Cohort 1, 2, and 3, respectively, at six months relative to baseline as measured by F-Dopa PET scan. Coverage of the putamen and AADC enzyme activity were highly correlated (r=0.84, p=0.0002). |
|
· |
VY-AADC01 treatment resulted in reduced daily doses of oral levodopa and related medications to achieve optimal motor control, suggesting a greater capacity for patients to make more dopamine but with less need for oral levodopa. Patients’ Parkinson’s medications were reduced by a mean of 208 mg (14%), 553 mg (34%) and 618 mg (42%) for Cohorts 1, 2 and 3, respectively, at six months compared with baseline. |
|
· |
Treatment with VY-AADC01 resulted in dose-dependent (Cohorts 2 and 3 versus Cohort 1) and time-dependent clinically meaningful improvements in the patients’ motor function, as reflected in the amount of time patients self-reported that they did not experience troublesome dyskinesia, which we refer to as diary on-time. |
|
· |
Scores for UPDRS-III, the physician-rated motor examination, also improved while patients were on their medication in a dose-related manner (Cohorts 2 and 3 versus Cohort 1). |
|
· |
VY-AADC01 reduced patients’ periods of reduced mobility and stiffness, which we refer to as diary off-time, as self-reported in their diaries. |
|
· |
VY-AADC01 resulted in dose-dependent and time-dependent improvements in patients’ activities of daily living as measured by reductions in the UPDRS-II off medication score, including a change in mean score from baseline of -2.4 for Cohort 2 at 6 months compared with a change in score from baseline of -3.6 for Cohort 3 at 6 months. |
|
· |
VY-AADC01 infusion was successfully completed in all 15 patients and infusions of VY-AADC01 have been well-tolerated with no vector-related serious adverse events, which we refer to as SAEs. Fourteen of the 15 patients were discharged from the hospital within two days following surgery. As previously reported, one patient experienced two SAEs: a pulmonary embolism or blood clot in the lungs, and related heart arrhythmia or irregular heartbeat. The patient was treated with an anti-coagulant and symptoms associated with the SAEs have completely resolved. Investigators determined that this was most likely related to immobility during the administration and subsequent formation of a blood clot in the lower extremity. Consequently, deep vein thrombosis prophylaxis was added to the protocol and no subsequent events have been observed. |
Additionally, in September 2017, we announced that investigators recently completed dosing three additional patients in a separate Phase 1 clinical trial designed to optimize the intracranial delivery of VY-AADC01. We have subsequently completed doesing of a fourth patient. This Phase 1 trial explores a posterior, or back of the head, delivery approach, compared to Cohorts 1 through 3 from the ongoing Phase 1b clinical trial that used a transfrontal, or top of the head, delivery approach into the putamen. A posterior approach better aligns the infusion of VY-AADC01 with the anatomical structure of the putamen to potentially reduce the total procedure time and increase the total coverage of the putamen.
25
Administration of VY-AADC01 with this posterior approach was well-tolerated by the four patients dosed since the start of the trial. No serious adverse events were reported, and patients were discharged from the hospital the day after surgery. The posterior approach resulted in greater average putaminal coverage (approximately 50%) and reduced average administration times compared with the transfrontal approach of Cohorts 1 through 3 in the Phase 1b clinical trial. We expect to enroll more patients in this trial prior to the start of a pivotal Phase 2-3 program for advanced Parkinson’s disease.
We remain on track with our plans to begin a pivotal Phase 2-3 clinical program for advanced Parkinson’s disease in late 2017 and dose the first patient during the second quarter of 2018. We continue to follow patients from Cohorts 1 through 3 in the Phase 1b clinical trial of VY-AADC01 and patients in the Phase 1 posterior trajectory trial, and plan to report updated results from these trials during the first quarter of 2018, prior to the start of patient enrollment in the Phase 2-3 program. We plan to use VY-AADC02 in our Phase 2-3 clinical program as opposed to VY-AADC01. VY-AADC02 uses the same vector as VY-AADC01 but is manufactured using our baculovirus/Sf9 system as opposed to in HEK 293 cells, which are used to manufacture VY-AADC01. Before dosing the first patient in the Phase 2-3 program, we will need to file an IND for the baculovirus/Sf9 material for VY-AADC and gain clearance of that IND. Our Phase 2-3 pivotal program design provides for a single Phase 2 and a single Phase 3 clinical trial, conducted in staggered parallel and focused on key aspects of motor function measured over a sufficient period of time to detect a meaningful and durable benefit versus placebo. We expect that the Phase 2 trial will inform us early on if we properly blinded the study with placebo surgery and if we obtained sufficient coverage of the putamen with the increased number of surgical sites from the Phase 1 trial. Achieving both of these objectives will allow us to begin enrolling the Phase 3 trial in staggered parallel while the Phase 2 trial continues blinded follow-up.
We expect that the Phase 2 trial will enroll approximately 30 to 42 patients at about 16 trial sites, consisting of eight surgical sites and eight corresponding patient-referral sites. The surgical sites will perform the infusions. The clinical sites are the nearby hospitals and academic institutions from where eligible patients treated by movement disorder specialists will be referred and followed. We expect that the primary endpoint of both trials will likely be self-reported diary on-time without troublesome dyskinesia. Based on our estimates of patient enrollment and a 12-month blinded treatment period, we expect top-line data from the Phase 2 portion of the pivotal program in the second half of 2020. Shortly after dosing the patients in the Phase 2 trial and receiving their baseline PET images, we expect to be able to begin enrollment in the Phase 3 trial. We currently anticipate that enrollment in the Phase 3 trial will begin during the first half of 2019. We expect that the Phase 3 trial will enroll approximately 100 to 120 patients and will include approximately 30 trial sites, consisting of approximately 10 surgical sites and 20 clinical referral sites. Based on our estimates of patient enrollment and a 12-month blinded treatment period, we anticipate top-line data from the Phase 3 trial during the first half of 2022. Pending discussions with regulatory authorities and depending upon actual enrollment timelines, we believe a large enough treatment effect observed in the Phase 2 trial could support applications for marketing approval in the United States and Europe during the first half of 2021, with the ongoing Phase 3 trial acting as a supportive confirmatory trial. Alternatively, we believe that favorable results from both the Phase 2 and Phase 3 trials could support applications for marketing approval in the United States and Europe in mid-2022.
In October 2017, we announced that Sanofi Genzyme decided not to exercise its option for the ex-U.S. rights to the Parkinson’s Program. Therefore, we now maintain global rights to VY-AADC. If we use certain Sanofi Genzyme technology in VY-AADC, Sanofi Genzyme is entitled to receive low-single digit royalty payments based on a percentage of net sales by us and we may be obligated to make certain regulatory milestone payments to a third-party licensor. As a result of Sanofi Genzyme’s decision, we are no longer entitled to receive the regulatory and commercial milestone payments related to the Parkinson’s Program.
Earlier in the year, we selected VY-SOD101 as a clinical candidate for the treatment of amyotrophic lateral sclerosis, or ALS, caused by mutations in the superoxide dismutase 1 gene, or SOD1. During the third quarter, we conducted preclinical studies with additional routes of administration in large animal models, including studies with our novel AAV capsids. We now plan to further investigate these additional routes of administration and novel capsids in our ALS program before filing an IND application, previously planned for late 2017 or early 2018. These studies, together with the ongoing efforts on the Huntington’s disease and Friedreich’s ataxia programs are expected to support two IND filings from these programs during 2019.
26
Since our inception, our operations have focused on building our team, business planning, raising capital, establishing our intellectual property portfolio, determining which neurological diseases to pursue, advancing our product engine including delivery and manufacturing, and conducting preclinical studies and clinical trials. We do not have any product candidates approved for sale and have not generated any revenue from product sales.
We have incurred significant operating losses since our inception. Our net losses were $58.9 million for the nine months ended September 30, 2017. As of September 30, 2017, we had an accumulated deficit of $148.9 million. We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate that our expenses will increase significantly in connection with our ongoing activities, if, and as, we:
|
· |
continue investing in our product engine to optimize vector engineering, manufacturing and dosing and delivery techniques; |
|
· |
continue to advance our clinical candidate, VY-AADC01, through the current Phase 1b clinical trial and advance VY-AADC02 into later stage trials; |
|
· |
initiate additional preclinical studies and clinical trials for, and continue research and development of, our other programs; |
|
· |
continue our process research and development activities, as well as establish our research-grade and commercial manufacturing capabilities; |
|
· |
identify additional neurological diseases for treatment with our AAV gene therapies and develop additional programs or product candidates; |
|
· |
work to identify and optimize novel AAV capsids; |
|
· |
seek marketing and regulatory approvals for VY-AADC or other product candidates or devices that arise from our programs that successfully complete clinical development; |
|
· |
maintain, expand and protect our intellectual property portfolio; |
|
· |
identify, acquire or in-license other product candidates and technologies; |
|
· |
develop a sales, marketing and distribution infrastructure to commercialize any product candidates for which we may obtain marketing approval; |
|
· |
expand our operational, financial and management systems and personnel, including personnel to support our clinical development, manufacturing and commercialization efforts and our operations as a public company; |
|
· |
increase our product liability and clinical trial insurance coverage as we expand our clinical trials and commercialization efforts; and |
|
· |
continue to operate as a public company. |
27
Financial Operations Overview
Collaboration Revenue
To date, we have not generated any revenue from product sales and do not expect to generate any revenue from product sales for the foreseeable future. For the nine months ended September 30, 2017, we recognized $3.8 million of collaboration revenue from our collaboration with Sanofi Genzyme, which we refer to as the Sanofi Genzyme Collaboration.
For the foreseeable future, we expect substantially all of our revenue will be generated from the Sanofi Genzyme Collaboration and any other strategic relationships we may enter into. If our development efforts are successful, we may also generate revenue from product sales.
Expenses
Research and Development Expenses
Research and development expenses consist primarily of costs incurred for our research activities, including our program discovery efforts, and the development of our programs and product engine, which include:
|
· |
employee-related expenses including salaries, benefits, payroll taxes, travel, and stock-based compensation expense; |
|
· |
costs of funding research performed by third parties that conduct research and development, clinical and preclinical activities, manufacturing, device development, and production on our behalf; |
|
· |
the cost of purchasing lab supplies and non-capital equipment used in designing, developing, and manufacturing preclinical study materials; |
|
· |
expenses relating to regulatory activities, including any filing fees paid to the FDA in connection with our product candidates; |
|
· |
the cost of consulting services by external experts and related fees; |
|
· |
facility costs including rent, depreciation, and maintenance expenses; and |
|
· |
fees for maintaining licenses under our third‑party licensing agreements. |
Research and development costs are expensed as incurred. Costs for certain activities such as manufacturing, preclinical studies, and clinical trials, are generally recognized based on an evaluation of the progress to completion of specific tasks using information and data provided to us by our vendors and collaborators.
At this time, we cannot reasonably estimate or know the nature, timing, and estimated costs of the efforts that will be necessary to complete the development of our product candidates. We are also unable to predict when, if ever, material net cash inflows will commence from sales of our product candidates due to the numerous risks and uncertainties associated with developing such product candidates, including the uncertainty of:
|
· |
the scope, progress, results, and costs of product discovery, preclinical studies, and clinical trials for our product candidates; |
|
· |
establishing an appropriate safety profile and demonstrating efficacy for a particular indication; |
28
|
· |
establishing commercial manufacturing capabilities or making arrangements with third‑party manufacturers; |
|
· |
timing, receipt, and terms of marketing approvals from applicable regulatory authorities; |
|
· |
commercializing our product candidates, if and when approved, whether alone or in collaboration with others; |
|
· |
obtaining and maintaining patent and trade secret protection and regulatory exclusivity for our product candidates; |
|
· |
the potential and perceived advantages of our product candidates over alternative treatments; |
|
· |
achieving an adequate level of market acceptance of our product candidates as a viable treatment option; |
|
· |
continued acceptable safety profiles of the products following approval; and |
|
· |
retention of key research and development personnel. |
A change in the outcome of any of these variables with respect to the development of any of our product candidates would significantly change the costs, timing, and viability associated with the development of that product candidate.
Research and development activities are central to our business model. We expect research and development costs to increase significantly for the foreseeable future as our development programs progress, including the continuation of VY-AADC through the current Phase 1b clinical trial and into later-stage clinical trials as a treatment for advanced Parkinson’s disease, and our preclinical development efforts and studies. There are numerous factors associated with the successful commercialization of any of our product candidates, including future trial design and various regulatory requirements, many of which cannot be determined with accuracy at this time based on our stage of development. Additionally, future commercial, competitive, and regulatory factors beyond our control will impact our clinical development programs and plans.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and other related costs, including stock‑based compensation for personnel in executive, finance, accounting, business development, legal, and human resource functions. Other significant costs include corporate facility costs not otherwise included in research and development expenses, legal fees related to patent and corporate matters, and fees for accounting and consulting services.
We anticipate that our general and administrative expenses will increase in the future to support continued research and development activities, including the continuation of VY-AADC through the current Phase 1b clinical trial and into later-stage clinical trials as a treatment for advanced Parkinson’s disease, and our preclinical development efforts and studies. These increases will likely include increased costs related to the hiring of additional personnel and fees paid to outside consultants. We also anticipate increased expenses, in comparison to general and administrative expenses of the prior year, resulting from costs associated with being a public company including audit, legal, regulatory, and tax‑related services, director and officer insurance premiums, and investor relations costs.
Critical Accounting Policies and Estimates
We have prepared our financial statements in accordance with U.S. generally accepted accounting principles. Our preparation of these financial statements requires us to make estimates, assumptions, and judgments that affect the reported amounts of assets, liabilities, expenses, and related disclosures at the date of the financial statements, as well as
29
revenue and expenses recorded during the reporting periods. We evaluate our estimates and judgments on an ongoing basis. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results could therefore differ materially from these estimates under different assumptions or conditions.
There have been no material changes to our critical accounting policies from those described in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included in our Annual Report on Form 10-K for the year ended December 31, 2016, which was filed with the SEC on March 15, 2017.
Results of Operations
Comparison of the three months ended September 30, 2017 and 2016
The following table summarizes our results of operations for the three months ended September 30, 2017 and 2016, together with the changes in those items in dollars:
|
|
|
Three Months Ended |
|
|
|
|||||
|
|
|
September 30, |
|
|
|
|||||
|
|
|
2017 |
|
2016 |
|
Change |
|
|||
|
|
|
(in thousands) |
|
|||||||
|
Collaboration revenue |
|
$ |
1,148 |
|
$ |
3,308 |
|
$ |
(2,160) |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
19,561 |
|
|
10,309 |
|
|
9,252 |
|
|
General and administrative |
|
|
4,942 |
|
|
3,370 |
|
|
1,572 |
|
|
Total operating expenses |
|
|
24,503 |
|
|
13,679 |
|
|
10,824 |
|
|
Other income: |
|
|
|
|
|
|
|
|
|
|
|
Interest income, net |
|
|
291 |
|
|
1,171 |
|
|
(880) |
|
|
Other expense, net |
|
|
(282) |
|
|
(99) |
|
|
(183) |
|
|
Total other income |
|
|
9 |
|
|
1,072 |
|
|
(1,063) |
|
|
Loss before income taxes |
|
|
(23,346) |
|
|
(9,299) |
|
|
(14,047) |
|
|
Income tax benefit |
|
|
— |
|
|
303 |
|
|
(303) |
|
|
Net loss |
|
$ |
(23,346) |
|
$ |
(8,996) |
|
$ |
(14,350) |
|
Collaboration Revenue
Collaboration revenue was $1.1 million and $3.3 million for the three months ended September 30, 2017 and 2016, respectively, all of which related to the Sanofi Genzyme Collaboration for research and development services for various programs under our collaboration agreement with Sanofi Genzyme, which we refer to as the Collaboration Agreement. The decrease in collaboration revenue is largely related to changes in our estimated period of performance for reaching proof of principle for certain programs under the Collaboration Agreement.
During 2016 we reassessed the estimated period of performance for each of the units of accounting and determined that the estimated period would be extended for two units of accounting. Additionally, we and Sanofi Genzyme agreed to deprioritize the development of a product candidate for spinal muscular atrophy and reduce the estimates related to the amount of “in-kind” services that would be provided by Sanofi Genzyme. These adjustments were made on a prospective basis and resulted in decreases in revenue recognized by $2.4 million per quarter.
During the first two quarters of 2017, we reassessed the estimated period of performance for each of the units of accounting and, based on the current facts and circumstances, determined that the estimated period would be extended for three units of accounting. These adjustments were made on a prospective basis and resulted in a decrease in revenue recognized by $0.5 million per quarter.
30
Research and Development Expense
Research and development expense increased by $9.3 million from $10.3 million for the three months ended September 30, 2016, to $19.6 million for the three months ended September 30, 2017. The following table summarizes our research and development expenses for the three months ended September 30, 2017 and 2016:
|
|
|
Three Months Ended |
|
|
|
|||||
|
|
|
September 30, |
|
|
|
|||||
|
|
|
2017 |
|
2016 |
|
Change |
|
|||
|
|
|
(in thousands) |
|
|||||||
|
External research and development expenses |
|
$ |
12,538 |
|
$ |
4,238 |
|
$ |
8,300 |
|
|
Employee and contractor related expenses |
|
|
5,648 |
|
|
3,961 |
|
|
1,687 |
|
|
Facility and other expenses |
|
|
1,315 |
|
|
1,597 |
|
|
(282) |
|
|
License fees |
|
|
60 |
|
|
513 |
|
|
(453) |
|
|
Total research and development expenses |
|
$ |
19,561 |
|
$ |
10,309 |
|
$ |
9,252 |
|
The increase in research and development expense for the three months ended September 30, 2017 was primarily attributable to the following:
|
· |
approximately $8.3 million for increased costs of funding research performed by third parties that conduct research and development, preclinical and clinical activities, and manufacturing and production preparation activities on our behalf, and increased purchases of lab supplies and non‑capital equipment used in designing, developing, and manufacturing preclinical study materials. These increases were offset by a reduction in in‑kind research and development services incurred by Genzyme and provided to us under the Genzyme Collaboration; |
|
· |
approximately $1.7 million for increases in employee compensation cost due to increases in headcount and stock-based compensation; |
|
· |
offset by approximately $0.5 million for decreases in intellectual property and license fees; and |
|
· |
further offset by approximately $0.3 million for decreases in facilities and other costs including rent, depreciation, maintenance, and other expenses. |
General and Administrative Expense
General and administrative expense increased by $1.5 million from $3.4 million for the three months ended September 30, 2016 to $4.9 million for the three months ended September 30, 2017. The increase in general and administrative expense was primarily attributable to the following:
|
· |
approximately $1.0 million for increases in employee compensation cost due to increases in headcount and stock-based compensation; |
|
· |
approximately $0.5 million for increases in legal and maintenance costs related to our intellectual property; and |
|
· |
approximately $0.3 million for increases in facilities and other costs including rent, depreciation, maintenance, and other expenses. |
31
Other Income, net
Interest income of approximately $0.3 million related to our marketable securities balances was recognized during the three months ended September 30, 2017. The balance was offset by a $0.3 million loss related to the mark-to-market adjustments recognized on the fair value of warrants to purchase equity securities. Investment income of approximately $1.1 million was recognized during the three months ended September 30, 2016 including $0.8 million related mark-to-market gain recognized on the fair value of warrants to purchase equity securities, and higher interest income due to increased marketable securities balances resulting from our underwritten initial public offering in November 2015.
Comparison of the nine months ended September 30, 2017 and 2016
The following table summarizes our results of operations for the nine months ended September 30, 2017 and 2016, together with the changes in those items in dollars:
|
|
|
Nine Months Ended |
|
|
|
|||||
|
|
|
September 30, |
|
|
|
|||||
|
|
|
2017 |
|
2016 |
|
Change |
|
|||
|
|
|
(in thousands) |
|
|||||||
|
Collaboration revenue |
|
$ |
3,790 |
|
$ |
11,858 |
|
$ |
(8,068) |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
48,933 |
|
|
29,526 |
|
|
19,407 |
|
|
General and administrative |
|
|
14,372 |
|
|
9,789 |
|
|
4,583 |
|
|
Total operating expenses |
|
|
63,305 |
|
|
39,315 |
|
|
23,990 |
|
|
Other income: |
|
|
|
|
|
|
|
|
|
|
|
Interest income, net |
|
|
799 |
|
|
2,069 |
|
|
(1,270) |
|
|
Other expense, net |
|
|
(184) |
|
|
(435) |
|
|
251 |
|
|
Total other income |
|
|
615 |
|
|
1,634 |
|
|
(1,019) |
|
|
Loss before income taxes |
|
|
(58,900) |
|
|
(25,823) |
|
|
(33,077) |
|
|
Income tax benefit |
|
|
31 |
|
|
303 |
|
|
(272) |
|
|
Net loss |
|
$ |
(58,869) |
|
$ |
(25,520) |
|
$ |
(33,349) |
|
Collaboration Revenue
Collaboration revenue was $3.8 million and $11.9 million for the nine months ended September 30, 2017 and 2016, respectively, all of which related to the Sanofi Genzyme Collaboration in recognition of amounts allocated to research and development services for various programs under the Collaboration Agreement. The decrease in collaboration revenue is largely related to changes in our estimated period of performance for reaching proof of principle for certain programs under the Collaboration Agreement.
During 2016, we reassessed the estimated period of performance for each of the units of accounting and determined that the estimated period would be extended for two units of accounting. Additionally, we and Sanofi Genzyme agreed to deprioritize the development of a product candidate for spinal muscular atrophy and reduce the estimates related to the amount of “in-kind” services that would be provided by Sanofi Genzyme. These adjustments were made on a prospective basis and resulted in decreases in revenue recognized by $2.4 million per quarter.
During the first two quarters of 2017, we reassessed the estimated period of performance for each of the units of accounting and, based on the current facts and circumstances, determined that the estimated period would be extended for three units of accounting. These adjustments were made on a prospective basis and resulted in a decrease in revenue recognized by $0.5 million per quarter.
32
Research and Development Expense
Research and development expense increased by $19.4 million from $29.5 million for the nine months ended September 30, 2016, to $48.9 million for the nine months ended September 30, 2017. The following table summarizes our research and development expenses, for the nine months ended September 30, 2017 and 2016:
|
|
|
Nine Months Ended |
|
|
|
|||||
|
|
|
September 30, |
|
|
|
|||||
|
|
|
2017 |
|
2016 |
|
Change |
|
|||
|
|
|
(in thousands) |
|
|||||||
|
External research and development expenses |
|
$ |
27,882 |
|
$ |
14,200 |
|
$ |
13,682 |
|
|
Employee and contractor related expenses |
|
|
15,657 |
|
|
11,162 |
|
|
4,495 |
|
|
Facility and other expenses |
|
|
5,005 |
|
|
3,527 |
|
|
1,478 |
|
|
License fees |
|
|
389 |
|
|
637 |
|
|
(248) |
|
|
Total research and development expenses |
|
$ |
48,933 |
|
$ |
29,526 |
|
$ |
19,407 |
|
The increase in research and development expense for the nine months ended September 30, 2017 was primarily attributable to the following:
|
· |
approximately $13.6 million for increased costs of funding research performed by third parties that conduct research and development, preclinical and clinical activities, and manufacturing and production preparation activities on our behalf, and increased purchases of lab supplies and non‑capital equipment used in designing, developing, and manufacturing preclinical study materials, and an additional expense of approximately $0.1 million attributable to in‑kind research and development services incurred by Genzyme and provided to us under the Genzyme Collaboration; |
|
· |
approximately $4.5 million for increases in employee compensation cost due to increases in headcount and stock-based compensation; |
|
· |
approximately $1.5 million for increases in facilities and other costs including rent, depreciation, maintenance and other expenses; and |
|
· |
offset by approximately $0.2 million for decreases in license fees. |
General and Administrative Expense
General and administrative expense increased by $4.6 million from $9.8 million for the nine months ended September 30, 2016 to $14.4 million for the nine months ended September 30, 2017. The increase in general and administrative expense was primarily attributable to the following:
|
· |
approximately $2.7 million for increases in employee compensation cost due to increases in headcount and stock-based compensation; |
|
· |
approximately $0.8 million for increases in facilities and other costs including rent, depreciation, and maintenance; |
|
· |
approximately $0.8 million for increases in legal and maintenance costs related to our intellectual property; and |
|
· |
approximately $0.2 million for increases in other legal and professional fees. |
33
Other Income, net
Interest income of approximately $0.8 million related to our marketable securities balances was recognized during the nine months ended September 30, 2017. The balance was offset by a $0.2 million loss related to the mark-to-market adjustments recognized on the fair value of warrants to purchase equity securities. Investment income of approximately $1.6 million was recognized during the nine months ended September 30, 2016 including $0.8 million related mark-to-market gain recognized on the fair value of warrants to purchase equity securities, and higher interest income due to increased marketable securities balances resulting from our underwritten initial public offering in November 2015.
Liquidity and Capital Resources
Sources of Liquidity
We have raised capital primarily through proceeds from private placements of our redeemable convertible preferred stock and convertible promissory notes, sales of our common stock, including our initial public offering, and proceeds associated with an up front payment from the Sanofi-Genzyme Collaboration of $65.0 million.
As of September 30, 2017, we had cash, cash equivalents, and marketable debt securities of $125.6 million.
Cash Flows
The following table provides information regarding our cash flows for the nine months ended September 30, 2017 and 2016:
|
|
|
Nine Months Ended |
|
||||
|
|
|
September 30, |
|
||||
|
|
|
2017 |
|
2016 |
|
||
|
|
|
(in thousands) |
|||||
|
Net cash (used in) provided by: |
|
|
|
|
|
|
|
|
Operating activities |
|
$ |
(46,516) |
|
$ |
(29,390) |
|
|
Investing activities |
|
|
56,420 |
|
|
61,442 |
|
|
Financing activities |
|
|
1,656 |
|
|
146 |
|
|
Net increase in cash and cash equivalents |
|
$ |
11,560 |
|
$ |
32,198 |
|
Net Cash Used in Operating Activities
Net cash used in operating activities was $46.5 million during the nine months ended September 30, 2017 compared to $29.4 million during the nine months ended September 30, 2016. This increase in cash used in operating activities was primarily due to increased research and development activities as we advanced our programs, as well as higher general and administrative expenses including employee-related costs and legal fees to support our growing business.
Net Cash Provided by Investing Activities
Net cash provided by investing activities was $56.4 million during the nine months ended September 30, 2017 compared to $61.4 million during the nine months ended September 30, 2016. The decrease in cash provided by investing activities for the nine months ended September 30, 2017 was primarily due to proceeds from maturities of marketable securities of $110.1 million, offset by $49.9 million for purchases of marketable securities and $3.8 million for purchases of property and equipment. The cash provided by investing activities for the nine months ended September 30, 2016 was primarily due to proceeds from maturities of marketable securities of $129.7 million, offset by $66.6 million for purchases of marketable securities and $1.1 million for purchases of property and equipment.
34
Net Cash Provided by Financing Activities
Net cash provided by financing activities was $1.7 million and $0.1 million during the nine months ended September 30, 2017 and 2016, respectively, primarily from proceeds of exercises of stock options and purchases of common stock under our 2015 Employee Stock Purchase Plan.
Funding Requirements
We expect our expenses to increase in connection with our ongoing activities, particularly as we continue the research and development of, continue or initiate clinical trials of, and seek marketing approval for, our product candidates. In addition, if we obtain marketing approval for any of our product candidates, we expect to incur significant commercialization expenses related to program sales, marketing, manufacturing, and distribution to the extent that such sales, marketing, and distribution are not the responsibility of potential collaborators. Furthermore, we expect to incur additional costs associated with operating as a public company during 2017 and future years. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce, or eliminate our research and development programs or future commercialization efforts.
Based upon our current operating plan, we expect our existing cash, cash equivalents, and marketable securities will enable us to fund our operating expenses and capital expenditure requirements into 2019. Our future capital requirements will depend on many factors, including:
|
· |
the scope, progress, results and costs of product discovery, preclinical studies and clinical trials for our product candidates; |
|
· |
the scope, progress, results, costs, prioritization and number of our research and development programs; |
|
· |
the costs, timing and outcome of regulatory review of our product candidates; |
|
· |
our ability to establish and maintain collaboration, distribution, or other marketing arrangements for our product candidates on favorable terms, if at all; |
|
· |
the achievement of milestones or occurrence of other developments that trigger payments under the Sanofi Genzyme Collaboration and any other collaboration agreements to which we become a party; |
|
· |
the ability of our collaboration partners to exercise options to extend research and development programs; |
|
· |
the extent to which we are obligated to reimburse, or entitled to reimbursement of, preclinical development and clinical trial costs under collaboration agreements, if any; |
|
· |
the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending intellectual property-related claims; |
|
· |
the extent to which we acquire or in-license other product candidates and technologies or acquire or invest in other businesses, such as our investment in MRI Interventions, Inc.; |
|
· |
the costs related to evaluating possible alternative devices that may be useful in the delivery of our product candidates; |
|
· |
the costs of securing manufacturing arrangements for commercial production; |
|
· |
the costs of establishing or contracting for manufacturing, sales, marketing, distribution and other commercialization capabilities if we obtain regulatory approvals to market our product candidates; and |
|
· |
the level of product sales from any product candidates for which we obtain marketing approval in the future. |
35
Identifying potential product candidates and conducting preclinical studies and clinical trials is a time‑consuming, expensive and uncertain process that takes many years to complete. We may never generate the necessary data or results required to obtain marketing approval and achieve product sales. In addition, our product candidates, if approved, may not achieve commercial success. Our product revenues, if any, and any commercial milestone or royalty payments under our collaboration agreements, will be derived from sales of gene therapies that we do not expect to be commercially available for many years, if at all. Accordingly, we will need to continue to rely on additional financing to achieve our business objectives. Adequate additional financing may not be available to us on acceptable terms, or at all. In addition, we may seek additional capital due to favorable market conditions or strategic considerations, even if we believe we have sufficient funds for our current or future operating plans.
Until such time, if ever, as we can generate product revenues sufficient to achieve profitability, we expect to finance our cash needs through a combination of equity offerings, debt financings, collaborations, strategic alliances, and licensing arrangements. We do not have any committed external source of funds other than the amounts we are entitled to receive from Sanofi Genzyme for potential option exercises, the achievement of specified regulatory and commercial milestones, and royalty payments under the Sanofi Genzyme Collaboration. To the extent that we raise additional capital through the sale of equity or equity-linked securities, including convertible debt, our stockholders’ ownership interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect our existing stockholders’ rights as holders of our common stock. Debt financing and preferred equity financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, obtaining additional capital, acquiring or divesting businesses, making capital expenditures or declaring dividends.
If we raise additional funds through collaborations, strategic alliances, or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market products or product candidates that we would otherwise prefer to develop and market ourselves.
Contractual Obligations
The following table summarizes our significant contractual obligations as of payment due date by period at September 30, 2017:
|
|
|
|
|
|
Less Than |
|
|
|
|
|
|
|
More than |
|
||
|
|
|
Total |
|
1 Year |
|
1 to 3 Years |
|
3 to 5 Years |
|
5 Years |
|
|||||
|
Operating lease commitments(1) |
|
$ |
27,381 |
|
$ |
806 |
|
$ |
6,672 |
|
$ |
7,630 |
|
$ |
12,273 |
|
|
(1) |
We lease office space at 75 Sidney Street and 64 Sidney Street in Cambridge, Massachusetts under non‑cancelable operating leases that expire in December 2024. |
In December 2015, we executed an amendment to extend the lease for 75 Sidney Street in Cambridge, Massachusetts and executed a new agreement to lease 64 Sidney Street in Cambridge, Massachusetts, both terms going through December 2024.
We enter into agreements in the normal course of business with clinical research organizations, contract manufacturing organizations, and institutions to license intellectual property. We have not included these future payments in the table of contractual obligations above since the contracts are cancelable at any time by us, generally upon 30 to 90 days prior written notice.
Our agreements to license intellectual property include potential milestone payments that are dependent upon the development of products using the intellectual property licensed under the agreements and contingent upon the achievement of clinical trial results or regulatory approval milestones. Under the terms of one agreement, we licensed intellectual property that may be used in the development of therapies for three disease indications. Under the agreement, we would be obligated to pay milestone payments that are contingent upon clinical trial results and regulatory approval
36
of $5.0 million per disease indication, or up to $20.0 million in total. We are also required to pay annual maintenance fees to maintain the licenses. During the nine months ended September 30, 2017, we have paid $1.0 million to exercise the license to three indications under this agreement and an additional $90.0 thousand for maintenance fees.
In September 2016, we entered into securities purchase and license agreements with MRI Interventions, Inc., or MRIC. Under a securities purchase agreement, we paid $2.0 million for shares of MRIC common stock and a warrant to purchase additional shares of MRIC common stock. We also entered into a license agreement with MRIC that provided for certain rights to MRIC technology and for MRIC to transfer the rights and know-how to manufacture the ClearPoint System to enable us to utilize an alternative supplier for the ClearPoint System for use in our development and clinical trials. Additionally, we committed to purchase $1.0 million of the common stock of MRIC, at MRIC’s option upon the transfer of the manufacturing technology know-how for the ClearPoint System to the alternate manufacturer.
During the three months ended March 31, 2017, we terminated the license agreement with MRIC, and all prior and future commitments and obligations under such agreement became null and void. As of September 30, 2017, we continued to hold the common stock and warrants to purchase additional shares of common stock as an available-for-sale security and non-current asset, respectively.
During September 2016, we entered into a research and development funding arrangement with a non-profit organization that provides up to $4.0 million in funding upon the achievement of clinical and development milestones. The agreement provides that we repay amounts received under certain circumstances including termination of the agreement, and to pay an amount up to 2.6 times the funding received upon successful development and commercialization of any products developed. During the three months ended March 31, 2017, we earned a milestone payment of $1.0 million. No other milestone amounts have been earned under the agreement. We have evaluated the arrangement and have concluded that it represents a research and development financing arrangement as it is probable that we will repay amounts received under the arrangement. As a result, the $1.0 million earned to date is recorded as a non-current liability in the accompanying balance sheet.
Other than the above, there were no material changes to our contractual obligations and commitments described under Management’s Discussion and Analysis of Financial Condition and Results of Operations in our Annual Report on Form 10-K for the fiscal year ended December 31, 2016, as filed with the SEC on March 15, 2017.
Off‑Balance Sheet Arrangements
We did not have, during the periods presented, and we do not currently have, any off‑balance sheet arrangements, as defined under applicable SEC rules.
JOBS Act
In April 2012, the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, was enacted. Section 107 of the JOBS Act provides that an emerging growth company, or EGC, can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. Thus, an EGC can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies.
Subject to certain conditions, as an EGC, we intend to rely on certain of these exemptions, including without limitation, (i) providing an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404(b) of the Sarbanes‑Oxley Act and (ii) complying with any requirement that may be adopted by the Public Company Accounting Oversight Board, regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements, known as the auditor discussion and analysis. We will remain an EGC until the earlier of (i) the last day of the fiscal year in which we have total annual gross revenues of $1.0 billion or more; (ii) December 31, 2020; (iii) the date on which we have issued more than $1.0 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
37
ITEM 3. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are exposed to market risk related to changes in interest rates. We have policies requiring us to invest in high-quality issuers, limit our exposure to any individual issuer, and ensure adequate liquidity. Our primary exposure to market risk is interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates, particularly because our investments, including cash equivalents, are in the form of money market fund and marketable securities and are invested in U.S. Treasury obligations. Due to the short-term duration of our investment portfolio and the low risk profile of our investments, an immediate 100 basis point change in interest rates would not have had a material effect on the fair market value of our portfolio.
We are not currently exposed to market risk related to changes in foreign currency exchange rates; however, we may contract with vendors that are located in Asia and Europe in the future and may be subject to fluctuations in foreign currency rates at that time.
Inflation generally affects us by increasing our cost of labor and clinical trial costs. We do not believe that inflation had a material effect on our business, financial condition or results of operations during the nine months ended September 30, 2017 and 2016, respectively.
ITEM 4. CONTROLS AND PROCEDURES
Management’s Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures (as defined in Rules 13a-15(e) or 15d-15(e) under the Exchange Act) that are designed to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act is (1) recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms and (2) accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosure.
As of September 30, 2017, our management, with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this report. In designing and evaluating our disclosure controls and procedures, our management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Our principal executive officer and principal financial officer have concluded based upon the evaluation described above that, as of September 30, 2017, our disclosure controls and procedures were effective at the reasonable assurance level in accomplishing the goals described above.
We continue to review and document our disclosure controls and procedures, including our internal controls and procedures for financial reporting, and may from time to time make changes aimed at enhancing their effectiveness and to ensure that our systems evolve with our business.
Changes in Internal Control over Financial Reporting
During the three and nine months ended September 30, 2017, there have been no changes in our internal control over financial reporting, as such term is defined in Rules 13a-15(f) and 15(d)-15(f) promulgated under the Exchange Act, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
As of the date of this Quarterly Report on Form 10-Q, we were not party to any material legal matters or claims. In the future, we may become party to legal matters and claims arising in the ordinary course of business, the resolution of which we do not anticipate would have a material adverse impact on our financial position, results of operations, or cash flows.
38
An investment in shares of our common stock involves a high degree of risk. The risk factors below are the only ones that have changed materially since disclosed in Part I, Item 1A of our Annual Report on Form 10-K for the year ended December 31, 2016, filed with the SEC on March 15, 2017. You should carefully consider the risk factors below as well as the other risk factors included in our Annual Report on Form 10-K for the year ended December 31, 2016, filed with the SEC on March 15, 2017, together with the other information appearing elsewhere in this Quarterly Report on Form 10-Q, including our condensed consolidated financial statements and related notes hereto, our unaudited condensed consolidated financial statements and related notes in our Quarterly Report on Form 10-Q, which was filed with the SEC, on August 8, 2017, or our unaudited condensed consolidated financial statements and related notes in our Quarterly Report on Form 10-Q, which was filed with the SEC on May 9, 2017, before deciding to invest in our common stock. The occurrence of any of these risks could have a material adverse effect on our business, financial condition, results of operations and future growth prospects. In these circumstances, the market price of our common stock could decline, and you may lose all or part of your investment.
Risks Related to Our Financial Position and Need for Capital
We have incurred significant losses since inception and anticipate that we will continue to incur losses for the foreseeable future and may never achieve or maintain profitability.
We are a clinical-stage biotechnology company with a limited operating history, and have not yet generated revenues from the sales of our product candidates. Investment in biotechnology companies is highly speculative because it entails substantial upfront capital expenditures and significant risk that the product candidate will fail to obtain regulatory approval or become commercially viable. We have not yet demonstrated the ability to complete any clinical trials of our product candidates, obtain marketing approvals, manufacture a commercial‑scale product or conduct sales and marketing activities necessary for successful commercialization. We continue to incur significant expenses related to research and development, and other operations to commercialize our product candidates. As a result, we are not and have never been profitable and have incurred significant operating losses since our inception. Our losses were $58.9 million and $25.5 million for the nine months ended September 30, 2017 and 2016, respectively. As of September 30, 2017, we had an accumulated deficit of $148.9 million.
We historically have financed our operations primarily through private placements of our redeemable convertible preferred stock, public offerings of our common stock, and our recent collaboration agreement with Sanofi Genzyme. On November 16, 2015, we closed our Initial Public Offering, or IPO, whereby we sold 5,750,000 shares of common stock at a public offering price of $14.00 per share, including 750,000 shares of common stock issued upon the full exercise by the underwriters of their option to purchase additional shares, resulting in net proceeds to us of $72.9 million after deducting underwriting discounts and commissions and offering expenses payable by us. To date, we have devoted substantially all of our financial resources to building our team, business planning, raising capital, establishing our intellectual property portfolio, determining which neurological diseases to pursue, advancing our product engine including delivery and manufacturing and conducting preclinical studies and clinical trials. We expect that it could be several years, if ever, before we have a commercialized product candidate. We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. The net losses we incur may fluctuate significantly from quarter to quarter. We anticipate that our expenses will increase substantially if, and as, we:
|
· |
continue investing in our product engine to optimize vector engineering, manufacturing and dosing and delivery techniques; |
|
· |
continue to advance our clinical candidate, VY-AADC01, through the current Phase 1b clinical trial and advance VY-AADC02 into later stage trials; |
|
· |
initiate additional preclinical studies and clinical trials for, and continue research and development of, our other programs; |
|
· |
continue our process research and development activities, as well as establish our research‑grade and commercial manufacturing capabilities; |
39
|
· |
identify additional neurological diseases for treatment with our adeno-associated virus, or AAV, gene therapies and develop additional programs or product candidates; |
|
· |
work to identify and optimize novel AAV capsids; |
|
· |
develop and obtain regulatory clearance for devices to deliver our AAV gene therapies; |
|
· |
seek marketing and regulatory approvals for VY-AADC or other product candidates or devices that arise from our programs that successfully complete clinical development; |
|
· |
maintain, expand and protect our intellectual property portfolio; |
|
· |
identify, acquire or in-license other product candidates and technologies; |
|
· |
develop a sales, marketing and distribution infrastructure to commercialize any product candidates for which we may obtain marketing approval; |
|
· |
expand our operational, financial and management systems and personnel, including personnel to support our clinical development, manufacturing and commercialization efforts and our operations as a public company; |
|
· |
increase our product liability and clinical trial insurance coverage as we expand our clinical trials and commercialization efforts; and |
|
· |
continue to operate as a public company. |
Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. Our expenses will increase if:
|
· |
we are required by the U.S. Food and Drug Administration, or FDA, or the European Medicines Agency, or EMA, or other regulatory agencies to perform trials or studies in addition to those currently expected; |
|
· |
there are any delays in receipt of regulatory clearance to begin our planned clinical programs; or |
|
· |
there are any delays in enrollment of patients in or completing our clinical trials or the development of our product candidates. |
To become and remain profitable, we must develop and eventually commercialize product candidates with significant market potential, which will require us to be successful in a range of challenging activities. These activities can include completing preclinical studies and clinical trials of our product candidates; obtaining marketing approval for these product candidates; manufacturing at commercial scale; marketing and selling those products that are approved; satisfying any post‑marketing requirements and achieving an adequate level of market acceptance of and obtaining and maintaining adequate coverage and reimbursement from third-party payors for such products; and protecting our rights to our intellectual property portfolio. We may never succeed in any or all of these activities and, even if we do, we may never generate revenues that are significant or large enough to achieve profitability. If we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would decrease the value of our company and could impair our ability to raise capital, maintain our research and development efforts, expand our business or continue our operations. A decline in the value of our company also could cause our stockholders to lose all or part of their investment.
We will need to raise additional funding, which may not be available on acceptable terms, or at all. Failure to obtain this necessary capital when needed may force us to delay, limit or terminate certain of our product development efforts or other operations.
We expect our expenses to increase in connection with our ongoing and planned activities, particularly as we continue the research and development of, initiate further clinical trials of and seek marketing approval for, our product candidates. In addition, if we obtain marketing approval for any of our product candidates, we expect to incur significant
40
expenses related to product sales, medical affairs, marketing, manufacturing and distribution. Since the completion of our IPO on November 16, 2015, we have also incurred costs associated with operating as a public company. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. If we are unable to raise capital or enter into business development transactions when needed or on acceptable terms, we could be forced to delay, reduce or eliminate certain of our research and development programs or any future commercialization efforts.
Our operations have consumed significant amounts of cash since inception. As of September 30, 2017, our cash, cash equivalents, and marketable debt securities were $125.6 million. Based upon our current operating plan, we expect that our existing cash, cash equivalents, and marketable securities will enable us to fund our operating expenses and capital expenditure requirements into 2019.
Our future capital requirements will depend on many factors, including:
|
· |
the scope, progress, results, and costs of product discovery, preclinical studies, and clinical trials for our product candidates; |
|
· |
the scope, progress, results, costs, prioritization, and number of our research and development programs; |
|
· |
the costs, timing, and outcome of regulatory review of our product candidates; |
|
· |
our ability to establish and maintain collaboration, distribution, or other marketing arrangements for our product candidates on favorable terms, if at all; |
|
· |
the achievement of milestones or occurrence of other developments that trigger payments under the collaboration agreement with Sanofi Genzyme and any other collaboration agreements to which we become a party; |
|
· |
the ability of our collaboration partners to exercise options to extend research and development programs; |
|
· |
the extent to which we are obligated to reimburse, or entitled to reimbursement of, preclinical development and clinical trial costs under collaboration agreements, if any; |
|
· |
the costs and timing of preparing, filing, and prosecuting patent applications, maintaining and enforcing our intellectual property rights, and defending intellectual property‑related claims; |
|
· |
the extent to which we acquire or in‑license other product candidates and technologies, or acquire or invest in other businesses, such as our investment in MRI Interventions, Inc.; |
|
· |
the costs related to evaluating possible alternative devices that may be useful in the delivery of our product candidates; |
|
· |
the costs of securing manufacturing arrangements for commercial production; |
|
· |
the level of product sales from any product candidates for which we obtain marketing approval in the future; and |
|
· |
the costs of establishing or contracting for manufacturing, sales, marketing, distribution, and other commercialization capabilities if we obtain regulatory approvals to market our product candidates. |
Identifying potential product candidates and conducting preclinical studies and clinical trials is a time‑consuming, expensive, and uncertain process that takes years to complete. We may never generate the necessary data or results required to obtain marketing approval and achieve product sales. In addition, our product candidates, if approved, may not achieve commercial success. Our product revenues, if any, and any commercial milestones or royalty payments under our collaboration agreements, will be derived from sales of products that may not be commercially available for many years, if at all. Accordingly, we will need to continue to rely on additional financing and business development to achieve our business objectives. Adequate additional financing or business development transactions may not be available to us on acceptable terms, or at all. In addition, we may seek additional capital due to favorable
41
market conditions or strategic considerations, even if we believe we have sufficient funds for our current or future operating plans.
Raising additional capital may cause dilution to our stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
Until such time, if ever, as we can generate product revenues sufficient to achieve profitability, we expect to finance our cash needs through equity offerings, debt financings, collaborations, strategic alliances, licensing arrangements and marketing and distribution arrangements. We do not have any committed external source of funds other than the amounts we are entitled to receive from Sanofi Genzyme for potential option exercises, the achievement of specified regulatory and commercial milestones, and royalty payments under our collaboration. To the extent that we raise additional capital through the sale of equity or equity-linked securities, including convertible debt, our stockholders’ ownership interests will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect our existing stockholders’ rights as holders of our common stock. Debt financing and preferred equity financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, obtaining additional capital, acquiring or divesting businesses, making capital expenditures or declaring dividends. Our issuance of additional securities, whether equity or debt, or the possibility of such issuance, may cause the market price of our common stock to decline. Further, our existing stockholders may not agree with the terms of such financings.
If we raise additional funds through collaborations, strategic alliances, licensing arrangements or marketing and distribution arrangements, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, or terminate our product development or future commercialization efforts or grant rights to develop and market products or product candidates that we would otherwise prefer to develop and market ourselves.
Risks Related to Third Parties
To date, all of our revenue has been derived from our collaboration with Sanofi Genzyme, and if this collaboration agreement were to be terminated, our business financial condition, results of operations and prospects would be harmed.
In February 2015, we entered into a collaboration agreement with Sanofi Genzyme to leverage our combined expertise and assets in gene therapy for neurological diseases. Under the agreement, we received an upfront commitment of approximately $100.0 million. Pursuant to the agreement, we granted Sanofi Genzyme an exclusive option to license, develop and commercialize (i) ex‑U.S. rights to our advanced Parkinson’s disease, Friedreich’s ataxia and Huntington’s disease programs and a future program, or the Split Territory Programs, with an incremental option to co‑commercialize the product candidate from our Huntington’s disease program in the United States and (ii) worldwide rights to our spinal muscular atrophy program. If Sanofi Genzyme exercises an option for a Split Territory Program, except for our advanced Parkinson’s disease program, it is required to make an option exercise payment to us. Furthermore, Sanofi Genzyme shall pay up to $645.0 million in the aggregate upon the achievement of specified regulatory and commercial milestones and will pay us tiered royalty payments based on a percentage of net sales of product candidates from the programs for which it is exercised its option, or the Optioned Programs.
Following Sanofi Genzyme’s exercise of an option for a program, Sanofi Genzyme will have sole responsibility for the development and commercialization of the product candidates from such program in the applicable territory. Sanofi Genzyme will have the sole discretion to determine and direct its efforts and resources, including the ability to discontinue all efforts and resources, it applies to the development and, if approval is obtained, commercialization and marketing of the product candidates covered by the Optioned Programs in the applicable territories. Sanofi Genzyme may not be effective in obtaining approvals for the product candidates developed from the Optioned Programs or in marketing, or arranging for necessary supply, manufacturing or distribution relationships for, any approved products. Furthermore, Sanofi Genzyme may change its strategic focus or pursue alternative technologies in a manner that results in reduced, delayed or no revenue to us. Sanofi Genzyme has a variety of marketed products and product candidates
42
under collaboration with other companies, including some of our competitors, and its own corporate objectives may not be consistent with our best interests. If Sanofi Genzyme fails to develop, obtain regulatory approval for or ultimately commercialize any product candidate from the Optioned Programs in the applicable territories, or if Sanofi Genzyme terminates our collaboration, our business, financial condition, results of operations and prospects would be harmed. In October 2017, Sanofi Genzyme notified us that it decided not to exercise its option for the ex-U.S. rights to VY-AADC and terminated the portion of its collaboration with us concerning Parkinson’s disease.
In addition, any dispute or litigation proceedings we may have with Sanofi Genzyme in the future could delay development programs, create uncertainty as to ownership of or access to intellectual property rights, distract management from other business activities and generate substantial expense. Finally, we also may not be able to seek and obtain a viable, alternative collaborator to partner with for the development and commercialization of the Split Territory Programs or the Optioned Programs.
Risks Related to the Development and Regulatory Approval of Our Product Candidates
Our AAV gene therapy product candidates are based on a novel technology, which makes it difficult to predict the time and cost of development and of subsequently obtaining regulatory approval, if at all. Very few gene therapy products have been approved by U.S. and European regulatory authorities.
We have concentrated our research and development efforts to date on our product engine, identifying our initial targeted disease indications, and our initial product candidates. Our future success depends on our successful development of viable AAV gene therapy product candidates. Currently, only one of our product candidates, VY‑AADC01, is in clinical development, and the remainder of our product candidates are in preclinical development. We cannot accurately predict when or if any of our product candidates will prove effective or safe in humans or whether these product candidates will receive marketing approval. There can be no assurance that we will not experience problems or delays in developing our product candidates and that such problems or delays will not cause unanticipated costs, or that any such development problems can be solved. We also may experience unanticipated problems or delays in expanding our manufacturing capacity.
The clinical trial requirements of the FDA, the EMA and other regulatory authorities and the criteria these regulators use to determine the safety and efficacy of a product candidate vary substantially according to the type, complexity, novelty and intended use and market of the product candidate. The regulatory approval process for novel product candidates such as gene therapies can be more expensive and take longer than for other, better known or more extensively studied product candidates. Until August 2017, the FDA had never approved a gene therapy product. Since that time, it has approved Kymriah by Novartis International AG, for pediatric and young adult patients with a form of acute lymphoblastic leukemia, and Yescarta by Kite Pharma, Inc., for adult patients with certain forms of non-Hodgkin lymphoma. Similarly, only two gene therapy products, Glybera by uniQure N.V. and Strimvelis by GlaxoSmithKline PLC, have received marketing authorization from the European Commission.
It is difficult to determine how long it will take or how much it will cost to obtain regulatory approvals for our product candidates in either the United States or the European Union or how long it will take to commercialize our product candidates. The few regulatory approvals to date may not be indicative of what the FDA, European Commission, or other regulatory authorities may require for approval or of whether different or additional preclinical studies or clinical trials may be required to support regulatory approval in a particular jurisdiction. Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a potential product candidate to market could decrease our ability to generate sufficient product revenue, and our business, financial condition, results of operations and prospects may be harmed.
The doses and coverage of the putamen being employed in the ongoing VY-AADC01 Phase 1b clinical trial are higher than those used in prior trials. Such dose and coverage levels may need to be further optimized or we may not generate sufficient clinical data in a placebo-controlled trial to achieve market authorization.
The clinical trial results of some of our collaborators have been negatively affected by factors that had not been fully anticipated. For example, the magnitude of the clinical responses seen in the Phase 1 clinical trial of VY-AADC01 conducted by University of California San Francisco, or UCSF, were similar to placebo effects observed in previous
43
surgical therapies for Parkinson’s disease. As a result, we are unable to rely on the results of this Phase 1 trial for an indication of the efficacy of treatment with VY-AADC01. We believe that there is a need to optimize the dose and volume of infusion of VY-AADC01 to substantially increase the coverage of the putamen, the region of the brain targeted by VY-AADC01, to achieve a clinical benefit. However, we can provide no assurances that we will be able to optimize these parameters and thereby achieve sufficient coverage of the putamen to achieve a clinical benefit.
The ongoing Phase 1b clinical trial of VY-AADC01 incorporates several design features in an attempt to increase the area of the putamen, particularly the posterior putamen, which receives VY-AADC01 treatment. We are employing larger infusion volumes and higher doses of VY-AADC01, and we are using the ClearPoint System to provide real time, intra operative MRI assistance to the physician surgically administering VY-AADC01 to the patient.
Additionally, we have initiated a separate Phase 1 clinical trial to explore a posterior, or back of the head, delivery approach of VY-AADC01, compared with the transfrontal, or top of the head, delivery approach employed in our ongoing Phase 1b clinical trial.
Due to the nature of the techniques being used in the Phase 1b clinical trial and the numerous variables that can be changed, it is possible that the data generated from this trial may not provide evidence of clinical benefit. For example, physicians may use cannulas, which are small tubes, of differing lengths in the infusion procedure, or may use differing infusion speeds or infusion angles. These differences could affect the dose of VY-AADC01 that ultimately reaches the putamen, leading to highly variable results. Similarly, our limited experience to date with the posterior delivery approach we are testing in our Phase 1 clinical trial, coupled with that trial’s small size, may not generate sufficient data for us to determine whether a posterior approach would provide clinically superior outcomes to the transfrontal approach.
Furthermore, we plan to use VY-AADC02 in our later-stage clinical trials as opposed to VY-AADC01. VY-AADC02 uses the same vector as VY-AADC01 but is manufactured using our baculovirus/Sf9 system as opposed to in HEK 293 cells, which are used to manufacture VY-AADC01. Based on our discussions with the FDA, we believe that we have a good understanding of what in vitro testing and preclinical evaluation are required, which we will need to conduct to demonstrate comparability between the current version and the new version. Although we believe that VY-AADC02 will be similar or comparable to VY-AADC01, we can provide no assurances that we will be able to successfully complete the necessary in vitro testing and preclinical studies or that the results of such studies and tests will demonstrate the comparability between VY-AADC01 and VY-AADC02.
Risks Related to Ownership of Our Common Stock
The price of our common stock may be volatile and fluctuate substantially, which could result in substantial losses for holders of our common stock.
The price of our common stock may be volatile and fluctuate substantially. The stock market in general and the market for biopharmaceutical companies in particular have experienced extreme volatility that have often been unrelated to the operating performance of particular companies. The market price for our common stock may be influenced by many factors, including:
|
· |
our success in commercializing any product candidates for which we obtain marketing approval; |
|
· |
regulatory action and results of clinical trials of our product candidates or those of our competitors; |
|
· |
the success of competitive products or technologies; |
|
· |
the results of clinical trials of our product candidates; |
|
· |
the results of clinical trials of product candidates of our competitors; |
|
· |
the commencement, termination, and success of our collaborations; |
|
· |
regulatory or legal developments in the United States and other countries; |
44
|
· |
developments or disputes concerning patent applications, issued patents or other proprietary rights; |
|
· |
the recruitment or departure of key personnel; |
|
· |
the level of expenses related to any of our product candidates or clinical development programs; |
|
· |
the results of our efforts to discover, develop, acquire or in-license additional product candidates or technologies, the cost of commercializing such product candidates, and the cost of development of any such product candidates or technologies; |
|
· |
actual or anticipated changes in estimates as to financial results, development timelines or recommendations by securities analysts; |
|
· |
variations in our financial results or those of companies that are perceived to be similar to us; |
|
· |
the ability to secure third party reimbursement for our product candidates; |
|
· |
changes in the structure of healthcare payment systems; |
|
· |
market conditions in the pharmaceutical and biotechnology sectors; |
|
· |
general economic, industry and market conditions; and |
|
· |
the other factors described in the section titled “Risk Factors” and elsewhere in this Quarterly Report on Form 10‑Q. |
If our operating results fall below the expectations of investors or securities analysts for a given period, the price of our common stock could decline substantially. Furthermore, any fluctuations in our operating results from period to period may, in turn, cause the price of our stock to fluctuate substantially. We believe that such comparisons of our financial results are not necessarily meaningful and should not be relied upon as an indication of our future performance.
In the past, following periods of volatility in the market price of a company’s securities, securities class‑action litigation often has been instituted against that company. We also may face securities class-action litigation if we cannot obtain regulatory approvals for or if we otherwise fail to commercialize our product candidates. Such litigation, if instituted against us, could cause us to incur substantial costs to defend such claims and divert management’s attention and resources, which could seriously harm our business, financial condition, results of operations and prospects.
ITEM 2.UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS
We have no unregistered sales of securities for the three and nine months ended September 30, 2017.
The exhibits filed or furnished as part of this Quarterly Report are set forth on the Exhibit Index, which is incorporated herein by reference.
45
INDEX TO EXHIBITS
|
|
|
|
|
Incorporated by Reference to: |
||||||
|
Exhibit |
|
Description |
|
Form or |
|
Exhibit |
|
Filing |
|
SEC File |
|
10.1 |
|
Employment Agreement, effective September 11, 2017, between the Company and Matthew Ottmer |
|
8-K |
|
10.1 |
|
9/18/2017 |
|
001-37625 |
|
31.1 |
|
Certification of Chief Executive Officer pursuant to Exchange Act Rules 13a-14 or 15d-14. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.2 |
|
Certification of Chief Financial Officer pursuant to Exchange Act Rules 13a-14 or 15d-14. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
32.1+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.INS |
|
XBRL Instance Document. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.SCH |
|
XBRL Taxonomy Extension Schema Document. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.CAL |
|
XBRL Taxonomy Extension Calculation Document. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.LAB |
|
XBRL Taxonomy Extension Definition Linkbase Document. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.PRE |
|
XBRL Taxonomy Extension Labels Linkbase Document. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.DEF |
|
XBRL Taxonomy Extension Presentation Link Document. |
|
|
|
|
|
|
|
|
+ The certification furnished in Exhibit 32.1 hereto is deemed to be furnished with this Quarterly Report on Form 10-Q and will not be deemed to be “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended, except to the extent that the Registrant specifically incorporates it by reference.
46
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
Date: November 2, 2017 |
VOYAGER THERAPEUTICS, INC. |
||
|
By: |
/s/ Steven M. Paul, M.D. |
||
|
Steven M. Paul, M.D. |
|||
|
President, Chief Executive Officer and Director |
|||
|
(Principal Executive Officer) |
|||
|
By: |
/s/ Jane Henderson |
||
|
Jane Henderson |
|||
|
Chief Financial Officer and Senior Vice President of Corporate Development |
|||
|
(Principal Financial and Accounting Officer) |
|||
47