Attached files
| file | filename |
|---|---|
| EX-32.1 - EX-32.1 - Vanda Pharmaceuticals Inc. | d474152dex321.htm |
| EX-31.2 - EX-31.2 - Vanda Pharmaceuticals Inc. | d474152dex312.htm |
| EX-31.1 - EX-31.1 - Vanda Pharmaceuticals Inc. | d474152dex311.htm |
| EX-23.1 - EX-23.1 - Vanda Pharmaceuticals Inc. | d474152dex231.htm |
| EX-21.1 - EX-21.1 - Vanda Pharmaceuticals Inc. | d474152dex211.htm |
| EX-10.46 - EX-10.46 - Vanda Pharmaceuticals Inc. | d474152dex1046.htm |
| EX-10.45 - EX-10.45 - Vanda Pharmaceuticals Inc. | d474152dex1045.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE |
| SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2016
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE |
| SECURITIES EXCHANGE ACT OF 1934 |
Commission File No. 001-34186
VANDA PHARMACEUTICALS INC.
(Exact name of registrant as specified in its charter)
| Delaware | 03-0491827 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
2200 Pennsylvania Avenue NW, Suite 300 E
Washington D.C. 20037
(202) 734-3400
(Address and telephone number, including area code, of registrant’s principal executive offices)
Securities registered pursuant to Section 12(b) of the Exchange Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common Stock, par value $0.001 | The Nasdaq Stock Market LLC | |
| (NASDAQ Global Market) | ||
| Rights to Purchase Series A Junior Participating Preferred Stock | The Nasdaq Stock Market LLC (NASDAQ Global Market) |
Securities registered pursuant to Section 12(g) of the Exchange Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer | ☐ | Accelerated filer | ☒ | |||
| Non-accelerated filer | ☐ (Do not check if a smaller reporting company) | Smaller reporting company | ☐ | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Securities Exchange Act of 1934). Yes ☐ No ☒
As of June 30, 2016, the last business day of the registrant’s last completed second quarter, the aggregate market value of the Common Stock held by non-affiliates of the registrant was approximately $473.6 million based on the closing price of the registrant’s Common Stock, as reported by the NASDAQ Global Market, on such date. Shares of Common Stock held by each executive officer and director and stockholders known by the registrant to own 10% or more of the outstanding stock based on public filings and other information known to the registrant have been excluded since such persons may be deemed affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
The number of shares of the registrant’s Common Stock, par value $0.001 per share, outstanding as of February 6, 2017 was 44,346,431.
The exhibit index as required by Item 601(a) of Regulation S-K is included in Item 15 of Part IV of this report.
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the registrant’s proxy statement with respect to the registrant’s 2017 Annual Meeting of Stockholders, which is to be filed pursuant to Regulation 14A within 120 days after the end of the registrant’s fiscal year ended December 31, 2016, are incorporated by reference into Part III of this Form 10-K.
Table of Contents
Vanda Pharmaceuticals Inc.
Form 10-K
Table of Contents
PART I
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
Various statements throughout this report are “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. Forward-looking statements may appear throughout this report. Words such as, but not limited to, “believe,” “expect,” “anticipate,” “estimate,” “intend,” “plan,” “project,” “target,” “goal,” “likely,” “will,” “would,” and “could,” or the negative of these terms and similar expressions or words, identify forward-looking statements. Forward-looking statements are based upon current expectations that involve risks, changes in circumstances, assumptions and uncertainties. Important factors that could cause actual results to differ materially from those reflected in our forward-looking statements include, among others:
| • | the ability of Vanda Pharmaceuticals Inc. (we, our or Vanda) to continue to commercialize HETLIOZ® (tasimelteon) for the treatment of Non-24-Hour Sleep-Wake Disorder (Non-24) in the U.S. and Europe; |
| • | uncertainty as to the market awareness of Non-24 and the market acceptance of HETLIOZ®; |
| • | our ability to generate U.S. sales of Fanapt® (iloperidone) for the treatment of schizophrenia; |
| • | the timing and costs of continuing to build a sales and marketing, supply chain, distribution, pharmacovigilance, compliance and safety infrastructure to promote Fanapt® in the U.S.; |
| • | our dependence on third-party manufacturers to manufacture HETLIOZ® and Fanapt® in sufficient quantities and quality; |
| • | the regulatory status of Fanapt® in Europe; |
| • | our ability to successfully commercialize HETLIOZ® and Fanapt® outside of the U.S.; |
| • | our ability to prepare, file, prosecute, defend and enforce any patent claims and other intellectual property rights; |
| • | a loss of rights to develop and commercialize our products under our license agreements; |
| • | the ability to obtain and maintain regulatory approval of our products, and the labeling for any approved products; |
| • | the timing and success of preclinical studies and clinical trials conducted by us and our development partners; |
| • | a failure of our products to be demonstrably safe and effective; |
| • | the size and growth of the potential markets for our products and the ability to serve those markets; |
| • | our expectations regarding trends with respect to our revenues, costs, expenses and liabilities; |
| • | the scope, progress, expansion, and costs of developing and commercializing our products; |
| • | our failure to identify or obtain rights to new products; |
| • | a loss of any of our key scientists or management personnel; |
| • | limitations on our ability to utilize some of all of our prior net operating losses and orphan drug and research and development credits; |
| • | the cost and effects of litigation; |
| • | our ability to obtain the capital necessary to fund our research and development or commercial activities; |
| • | losses incurred from product liability claims made against us; and |
| • | use of our existing cash, cash equivalents and marketable securities. |
All written and verbal forward-looking statements attributable to us or any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section. We caution investors not to rely too heavily on the forward-looking statements we make or that are made on our behalf. We undertake no obligation, and specifically decline any obligation, to update or revise publicly any forward-looking statements, whether as a result of new information, future events or otherwise.
We encourage you to read Management’s Discussion and Analysis of our Financial Condition and Results of Operations and our consolidated financial statements contained in this annual report on Form 10-K. We also encourage you to read Item 1A of Part I of this annual report on Form 10-K, entitled Risk Factors, which contains a more complete discussion of the risks and uncertainties associated with our business. In addition to the risks described above and in Item 1A of this report, other unknown or unpredictable factors also could affect our results. Therefore, the information in this report should be read together with other reports and documents that we file with the Securities and Exchange Commission from time to time, including on Form 10-Q and Form 8-K, which may supplement, modify, supersede or update those risk factors. As a result of these factors, we cannot assure you that the forward-looking statements in this report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all.
1
Table of Contents
Overview
Vanda Pharmaceuticals Inc. (we or Vanda ) is a global biopharmaceutical company focused on the development and commercialization of innovative therapies to address high unmet medical needs and improve the lives of patients. Vanda commenced its operations in 2003 and our product portfolio includes:
| • | HETLIOZ® (tasimelteon), a product for the treatment of Non-24-Hour Sleep-Wake Disorder (Non-24), was approved by the U.S. Food and Drug Administration (FDA) in January 2014 and launched commercially in the U.S. in April 2014. In July 2015, the European Commission (EC) granted centralized marketing authorization with unified labeling for HETLIOZ® for the treatment of Non-24 in totally blind adults. HETLIOZ® was commercially launched in Germany in August 2016. HETLIOZ® has potential utility in a number of other circadian rhythm disorders and is presently in clinical development for the treatment of Pediatric Non-24, Jet Lag Disorder and Smith-Magenis Syndrome (SMS). |
| • | Fanapt® (iloperidone), a product for the treatment of schizophrenia, the oral formulation of which was approved by the FDA in May 2009 and launched commercially in the U.S. by Novartis Pharma AG (Novartis) in January of 2010. Novartis transferred all the U.S. and Canadian commercial rights to the Fanapt® franchise to us on December 31, 2014. Additionally, our distribution partners launched Fanapt® in Israel and Mexico in 2014. Fanapt® has potential utility in a number of other disorders. An assessment of new Fanapt® clinical opportunities is ongoing. |
| • | Tradipitant (VLY-686), a small molecule neurokinin-1 receptor (NK-1R) antagonist, which is presently in clinical development for the treatment of chronic pruritus in atopic dermatitis and gastroparesis. |
| • | Trichostatin A, a small molecule histone deacetylase (HDAC) inhibitor which is in development for the treatment of hematologic malignancies. |
| • | AQW051, a Phase II alpha-7 nicotinic acetylcholine receptor partial agonist. |
Since we began operations in March 2003, we have devoted substantially all of our resources to the in-licensing, clinical development and commercialization of our products. Our ability to generate meaningful product sales and achieve profitability largely depends on our ability to successfully commercialize HETLIOZ® and Fanapt® in the U.S. and Europe alone or with others, to complete the development of our products, and to obtain the regulatory approvals for and to manufacture, market and sell our products. The results of our operations will vary significantly from year-to-year and quarter-to-quarter and depend on a number of factors, including risks related to our business, risks related to our industry, and other risks which are detailed in Item 1A of Part I entitled Risk Factors and Item 7 of Part II entitled Management’s Discussion and Analysis of Financial Condition and Results of Operations of this annual report on Form 10-K.
Our activities will necessitate significant uses of working capital in 2017 and beyond. We are currently concentrating our efforts on selling HETLIOZ® and Fanapt® in the U.S. and our continued commercialization of HETLIOZ® in Europe. Additionally, we continue to pursue market approval of HETLIOZ® in other regions and Fanapt® in Europe and other regions. We will continue to work with our distribution partners who launched Fanapt® in Mexico and Israel. We see opportunities to grow our commercial products through life cycle management strategies that include the addition of new indications and formulations. We have built a research and development organization that includes extensive expertise in the scientific disciplines of pharmacogenetics and pharmacogenomics. We operate cross-functionally and are led by an experienced research and development management team. Our pipeline includes novel programs that could address largely unmet medical needs.
Our founder and Chief Executive Officer, Mihael H. Polymeropoulos, M.D., started Vanda’s operations in early 2003 after establishing and leading the Pharmacogenetics Department at Novartis. In acquiring and developing our products, we have relied upon our deep expertise in the scientific disciplines of pharmacogenetics and pharmacogenomics. These scientific disciplines examine both genetic variations among people that influence response to a particular drug, and the multiple pathways through which drugs affect people.
Our Strategy
Our goal is to create a leading global biopharmaceutical company focused on developing and commercializing innovative therapies addressing high unmet medical needs through the application of our drug development expertise and our pharmacogenetics and pharmacogenomics expertise. The key elements of our strategy to accomplish this goal are to:
| • | Maximize the commercial success of HETLIOZ® and Fanapt®; |
| • | Enter into strategic partnerships to supplement our capabilities and to extend our commercial reach; |
| • | Pursue the clinical development and regulatory approval of our products; |
| • | Apply our pharmacogenetics and pharmacogenomics expertise to differentiate our products; and |
| • | Expand our product portfolio through the identification and acquisition of additional products. |
2
Table of Contents
Products
We have the following products on the market or under regulatory review:
| Product |
Indication |
Geography |
Select Historical Milestones | |||
| HETLIOZ® (tasimelteon) |
Non-24 | United States | FDA approval in January 2014; Commercial launch in April 2014 | |||
| Europe | EC approval in July 2015; Commercial launch in Germany in August 2016 | |||||
| Fanapt® (Oral) (iloperidone) |
Schizophrenia | United States | FDA approval in May 2009; Commercial launch in January 2010; U.S. and Canada rights sublicensed to Novartis in October 2009 and reacquired by Vanda in December 2014; Long term maintenance sNDA approval in May 2016 | |||
| Fanaptum® (Oral) (iloperidone) |
Europe | EMA accepted for evaluation our MAA in December 2015 | ||||
| Mexico | Market approval in October 2013; Commercial launch in the fourth quarter of 2014 by our local distribution partner, Probiomed S.A. de C.V. | |||||
| Israel | Market approval August 2012; Commercial launch in the fourth quarter of 2014 by our local distribution partner, Megapharm Ltd. | |||||
We have the following products in clinical development:
| Product |
Target Indication |
Select Historical Milestones | ||
| HETLIOZ® (tasimelteon) |
Pediatric Non-24 | Initiated a liquid formulation pharmacokinetic study in the fourth quarter of 2016 | ||
| SMS | Initiated a placebo controlled study in the fourth quarter of 2016 | |||
| Jet Lag Disorder | Initiated a placebo controlled study in the fourth quarter of 2016 | |||
| Fanapt® (Oral) (iloperidone) |
Schizophrenia | Long-acting injectable under evaluation; | ||
| Other Disorders | Potential indications are under evaluation including bipolar depression, major depressive disorder and post-traumatic stress disorder – nightmares | |||
| Tradipitant (VLY-686) | Pruritus in patients with Atopic Dermatitis | Initiated a placebo controlled pruritus clinical study in the second quarter of 2016 | ||
| Gastroparesis | Initiated a placebo controlled study in fourth quarter of 2016 | |||
| Trichostatin A | Oncology | In development for hematologic malignancies | ||
| AQW051 | CNS Disorders | Potential indications are under strategic evaluation including cognitive impairment | ||
3
Table of Contents
HETLIOZ®
Commercial opportunity: Non-24
In January 2014, HETLIOZ® was approved in the U.S. for the treatment of Non-24. Non-24 is a serious, rare and chronic circadian rhythm disorder characterized by the inability to entrain (synchronize) the master body clock with the 24-hour day-night cycle. HETLIOZ® is the first FDA approved treatment for Non-24. HETLIOZ® is a melatonin agonist of the human MT1 and MT2 receptors, with greater specificity for MT2. These receptors are thought to be involved in the control of circadian rhythms. HETLIOZ® is believed to reset the master body clock in the suprachiasmatic nucleus (SCN), located in the hypothalamus, resulting in the entrainment and alignment of the body’s melatonin and cortisol rhythms to the 24-hour day-night cycle. HETLIOZ® was launched commercially in the U.S. in April 2014. In addition, in July 2015, the EC granted centralized marketing authorization with unified labeling for HETLIOZ® for the treatment of Non-24 in totally blind adults and included post-marketing commitments related to a pediatric investigation plan. This authorization is valid in the 28 countries that are members of the European Union, as well as European Economic Area members Iceland, Liechtenstein and Norway. HETLIOZ® was launched commercially in Germany in August 2016.
In January 2010, the FDA granted orphan drug designation status for HETLIOZ® in Non-24 in blind individuals. The FDA grants orphan drug designation to drugs that may provide significant therapeutic advantage over existing treatments and target conditions affecting 200,000 or fewer U.S. patients per year. Orphan drug designation provides potential financial and regulatory incentives, including study design assistance, tax credits, waiver of FDA user fees, and up to seven years of market exclusivity upon marketing approval. In February 2011, the European Medicines Agency (EMA) designated HETLIOZ® as an orphan medicinal product for the same indication.
Non-24 is a serious, rare and chronic circadian rhythm disorder characterized by the inability to synchronize the master body clock with the 24-hour day-night cycle. Non-24 affects a majority of totally blind individuals, or approximately 80,000 people in the U.S. Non-24 occurs almost entirely in individuals who lack the light sensitivity necessary to synchronize the master body clock in the brain with the 24-hour day-night cycle. Most people have a master body clock that naturally runs longer than 24-hours and light is the primary environmental cue that resets it to 24 hours each day. Individuals with Non-24 have a master body clock that is not reset, and continually delays, resulting in prolonged periods of misalignment between their circadian rhythms and the 24-hour day-night cycle, including the timing of melatonin and cortisol secretion. As a result of this misalignment, Non-24 is associated with significant disruption of the sleep-wake cycle and impairments in social and occupational functioning, and marked subjective distress. Individuals with Non-24 cycle in-and out-of phase and suffer from disrupted nighttime sleep patterns and/or excessive daytime sleepiness.
While there are no FDA or EC approved treatments for Non-24 other than HETLIOZ®, there are a number of drugs approved and prescribed for patients with sleep disorders. The most commonly prescribed drugs are hypnotics. See Competition below for a discussion of commonly prescribed drugs for patients with sleep disorders.
Therapeutic opportunity: Circadian Rhythm Sleep Disorders
Sleep disorders are segmented into three major categories: primary insomnia, secondary insomnia and circadian rhythm sleep disorders (CRSDs). Insomnia is a symptom complex that comprises difficulty falling asleep or staying asleep, or non-refreshing sleep, in combination with daytime dysfunction or distress. The symptom complex can be an independent disorder (primary insomnia) or be a result of another condition such as depression or anxiety (secondary insomnia). CRSDs result from a misalignment of the sleep/wake cycle and an individual’s daily activities or lifestyle. The circadian rhythm is the rhythmic output of the human biological clock and is governed by the hormones melatonin and cortisol. Both the timing of behavioral events (activity, sleep, and social interactions) and the environmental light/dark cycle result in a sleep/wake cycle that follows the circadian rhythm. Examples of CRSDs include transient disorders such as jet lag and chronic disorders such as delayed sleep phase disorder, shift work sleep disorder and Non-24. We are planning to develop HETLIOZ® for the treatment of pediatric Non-24. We initiated a pediatric liquid formulation pharmacokinetic study in the fourth quarter of 2016.
We initiated an open label interventional study in patients with SMS in the fourth quarter of 2015 and a SMS placebo controlled study in the fourth quarter of 2016. Enrollment in these studies is ongoing. SMS is a rare genetic disorder caused by a deletion on chromosome 17. The U.S. National Institute of Health estimates that SMS affects approximately one in 20,000 births in the U.S.
We initiated an observational study in Jet Lag Disorder in the fourth quarter of 2015, which has been extended. We initiated a placebo controlled study in the fourth quarter of 2016.
Fanapt®
Commercial Opportunity: Schizophrenia
Fanapt® is a product for the treatment of schizophrenia. In May 2009, the FDA granted U.S. marketing approval of Fanapt® for the acute treatment of schizophrenia in adults. In October 2009, we entered into an amended and restated sublicense agreement with Novartis. We had originally entered into a sublicense agreement with Novartis in June 2004 pursuant to which we obtained certain worldwide exclusive licenses from Novartis relating to Fanapt®. Pursuant to the amended and restated sublicense agreement, Novartis
4
Table of Contents
had exclusive commercialization rights to all formulations of Fanapt® in the U.S. and Canada. In January 2010, Novartis launched Fanapt® in the U.S. On December 31, 2014, Novartis transferred all the U.S. and Canadian commercial rights to the Fanapt® franchise to Vanda as part of the Settlement Agreement. See Note 3, Settlement Agreement with Novartis, to the consolidated financial statements included in Part II of this annual report on Form 10-K for additional information. In June 2015, we announced positive results from REPRIEVE, a Phase III long-term maintenance study that was conducted by Novartis. In May 2016, the FDA approved a supplemental New Drug Application (sNDA) for Fanapt® for the maintenance treatment of schizophrenia in adults.
We continue to explore the regulatory path and commercial opportunity for Fanapt® oral formulation outside of the U.S. In December 2012, the EMA’s Committee for Medicinal Products for Human Use (CHMP) issued a negative opinion recommending against approval of Fanaptum® (oral iloperidone tablets) for the treatment of schizophrenia in adult patients in the European Union. The CHMP was of the opinion that the benefits of Fanaptum® did not outweigh its risks and recommended against marketing authorization. We initiated an appeal of this opinion and requested a re-examination of the decision by the CHMP, but withdrew our Marketing Authorization Application (MAA) in the first quarter of 2013 because the additional clinical data requested by the CHMP would not have been available in the timeframe allowed by the EMA’s Centralized Procedure. In December 2015, we refiled a MAA with the EMA for Fanaptum® which included the results from the REPRIEVE study.
We have entered into agreements with the following partners for the commercialization of Fanapt® in the countries set forth below:
| Country |
Partner | Market Approval Date | ||
| Mexico |
Probiomed S.A. de C.V. | October 2013 | ||
| Israel |
Megapharm Ltd. | August 2012 |
Schizophrenia is a chronic, debilitating mental disorder characterized by hallucinations, delusions, racing thoughts and other psychotic symptoms (collectively referred to as “positive symptoms”), as well as moodiness, anhedonia (inability to feel pleasure), loss of interest, eating disturbances and withdrawal (collectively referred to as “negative symptoms”), and attention and memory deficits (collectively referred to as “cognitive symptoms”). Schizophrenia develops in late adolescence or early adulthood in approximately 1% of the world’s population. Most schizophrenia patients today are treated with drugs known as “atypical” antipsychotics, which were first approved in the U.S. in the late 1980s. These antipsychotics have been named “atypical” for their ability to treat a broader range of negative symptoms than the first-generation “typical” antipsychotics, which were introduced in the 1950s and are now generic. Atypical antipsychotics are generally regarded as having improved side effect profiles and efficacy relative to typical antipsychotics. See Competition below for a discussion of commonly prescribed atypical antipsychotics in addition to Fanapt®.
Pursuant to the Settlement Agreement with Novartis, we reacquired the U.S. and Canadian rights to the long-acting injectable (depot) formulation of Fanapt®. We are evaluating the commercial opportunity around the depot formulation.
Therapeutic opportunity: Other
We are currently in the process of evaluating potential indications, including bipolar depression, major depressive disorder and post-traumatic stress disorder – nightmares.
Tradipitant (VLY-686)
Tradipitant is a small molecule NK-1R antagonist that we licensed from Eli Lilly and Company (Lilly) in April 2012. NK-1R antagonists have been evaluated in a number of indications including chemotherapy-induced nausea and vomiting (CINV), post-operative nausea and vomiting (PONV), gastroparesis, alcohol dependence, anxiety, depression and chronic pruritus associated with atopic dermatitis.
We commenced a Phase II clinical study of tradipitant in the treatment of chronic pruritus in patients with atopic dermatitis in 2014. Results from this study, which were announced in March 2015, showed no significant difference from placebo on the pre-specified primary endpoint. Vanda believes this proof of concept study was informative, in that through subsequent analyses, it revealed statistically significant and clinically meaningful responses across multiple outcomes evaluated in individuals with higher blood plasma levels of tradipitant at the time of their pruritus assessments. We initiated a placebo controlled pruritus proof of concept study in the second quarter of 2016.
We initiated a placebo controlled Phase II clinical study of tradipitant in the treatment of gastroparesis in the fourth quarter of 2016.
Trichostatin A
Trichostatin A is a small molecule HDAC inhibitor with potential use as a treatment for several oncology indications. We plan to file an IND application in 2017.
5
Table of Contents
AQW051
AQW051 is a Phase II alpha-7 nicotinic acetylcholine receptor partial agonist that we licensed from Novartis on December 31, 2014 pursuant to the Settlement Agreement. We are evaluating potential indications, including cognitive impairment.
License Agreements
Our rights to develop and commercialize our products are subject to the terms and conditions of licenses granted to us by other pharmaceutical companies.
HETLIOZ®
In February 2004, we entered into a license agreement with Bristol-Myers Squibb Company (BMS) under which we received an exclusive worldwide license under certain patents and patent applications, and other licenses to intellectual property, to develop and commercialize HETLIOZ®. In partial consideration for the license, we paid BMS an initial license fee of $0.5 million. We made developmental milestone payments to BMS totaling $12.0 million under the license agreement, including an $8.0 million milestone payment in the first quarter of 2014 as a result of the FDA’s approval of our HETLIOZ® NDA. The $8.0 million milestone payment was capitalized as an intangible asset and is being amortized over the expected HETLIOZ® patent life in the U.S. We are obligated to make a future milestone payment to BMS of $25.0 million in the event that cumulative worldwide sales of HETLIOZ® reach $250.0 million, which we expect to reach by the end of 2018. Additionally, we are obligated to make royalty payments on HETLIOZ® net sales to BMS in any territory where we commercialize HETLIOZ® for a period equal to the greater of 10 years post the first commercial sale in the territory or the expiry of the new chemical entity patent in that territory. During the period prior to the expiry of the new chemical entity patent in a territory, we are obligated to pay a 10% royalty on net sales in that territory. The royalty rate is decreased by half for countries in which no new chemical entity patent existed or for the remainder of the 10 years after the expiry of the new chemical entity patent. We are also obligated under the license agreement to pay BMS a percentage of any sublicense fees, upfront payments and milestone and other payments (excluding royalties) that we receive from a third party in connection with any sublicensing arrangement, at a rate which is in the mid-twenties. We have agreed with BMS in our license agreement for HETLIOZ® to use our commercially reasonable efforts to develop and commercialize HETLIOZ®.
Either party may terminate the HETLIOZ® license agreement under certain circumstances, including a material breach of the agreement by the other. In the event we terminate our license, or if BMS terminates our license due to our breach, all rights licensed and developed by us under this agreement will revert or otherwise be licensed back to BMS on an exclusive basis.
Fanapt®
Pursuant to the terms of the Settlement Agreement with Novartis, Novartis transferred all U.S. and Canadian rights in the Fanapt® franchise to Vanda on December 31, 2014.
A predecessor company of Sanofi, Hoechst Marion Roussel, Inc. (HMRI), discovered Fanapt® and completed early clinical work on the compound. In 1996, HMRI licensed its rights to the Fanapt® patents and patent applications to Titan Pharmaceuticals, Inc. (Titan) on an exclusive basis. In 1997, soon after it had acquired its rights, Titan sublicensed its rights to Fanapt® on an exclusive basis to Novartis. In June 2004, we acquired exclusive worldwide rights to these patents and patent applications as well as certain Novartis patents and patent applications to develop and commercialize Fanapt® through a sublicense agreement with Novartis. In partial consideration for this sublicense, we paid Novartis an initial license fee of $0.5 million and were obligated to make future milestone payments to Novartis of less than $100.0 million in the aggregate (the majority of which were tied to sales milestones), as well as royalty payments to Novartis at a rate which, as a percentage of net sales, was in the mid-twenties. As a result of the FDA’s approval of the NDA for Fanapt® in May 2009, we met a milestone under the sublicense agreement, which required us to make a payment of $12.0 million to Novartis.
In October 2009, we entered into an amended and restated sublicense agreement with Novartis, which amended and restated the June 2004 sublicense agreement. Pursuant to the amended and restated sublicense agreement, Novartis had exclusive commercialization rights to all formulations of Fanapt® in the U.S. and Canada. Novartis began selling Fanapt® in the U.S. during the first quarter of 2010. Novartis was responsible for the further clinical development activities in the U.S. and Canada. Pursuant to the amended and restated sublicense agreement, we received an upfront payment of $200.0 million and were eligible for additional payments totaling up to $265.0 million upon Novartis’ achievement of certain commercial and development milestones for Fanapt® in the U.S. and Canada. We also received royalties, which, as a percentage of net sales, were in the low double-digits, on net sales of Fanapt® in the U.S. and Canada. We retained exclusive rights to Fanapt® outside the U.S. and Canada and are obligated to make royalty payments to Sanofi S.A. on Fanapt® sales outside the U.S. and Canada.
Pursuant to the terms of the Settlement Agreement, Novartis transferred all U.S. and Canadian rights in the Fanapt® franchise to us on December 31, 2014. We are obligated to make royalty payments to Sanofi, S.A. (Sanofi) and Titan, at a percentage rate equal to 23% on annual U.S. net sales of Fanapt® up to $200.0 million, and at a percentage rate in the mid-twenties on sales over $200.0 million through November 2016. See Note 3, Settlement Agreement with Novartis, to the consolidated financial statements included in Part II of this annual report on Form 10-K for additional information. In February 2016, we amended the agreement with Sanofi and Titan to remove Titan as the entity through which royalty payments from us are directed to Sanofi following the expiration of the new
6
Table of Contents
chemical entity (NCE) patent for Fanapt® in the U.S. on November 15, 2016. Under the amended agreement, we will pay directly to Sanofi a fixed royalty of 3% of net sales from November 16, 2016 through December 31, 2019 related to manufacturing know-how. We made a $2.0 million payment during the year ended December 31, 2016 that applied to this 3% manufacturing know-how royalty and will make additional royalty payments only to the extent that our cumulative royalty obligations during this period exceed the amount of the pre-payment. No further royalties on manufacturing know-how are payable by us after December 31, 2019. This amended agreement does not alter Titan’s obligation under the License Agreement to make royalty payments to Sanofi prior to November 16, 2016 or our obligations under the Sublicense Agreement to pay Sanofi a fixed royalty on Fanapt net sales equal up to 6% on Sanofi know-how not related to manufacturing under certain conditions for a period of up to 10 years in markets where the NCE patent has expired or was not issued.
Tradipitant (VLY-686)
In April 2012, we entered into a license agreement with Lilly pursuant to which we acquired an exclusive worldwide license under certain patents and patent applications, and other licenses to intellectual property, to develop and commercialize an NK-1R antagonist, tradipitant, for all human indications.
Pursuant to the agreement, we paid Lilly an initial license fee of $1.0 million and we will be responsible for all development costs for tradipitant. Lilly is also eligible to receive additional payments based upon achievement of specified development and commercialization milestones as well as tiered-royalties on net sales at percentage rates up to the low double digits. These milestones include $4.0 million for pre-NDA approval milestones and up to $95.0 million for future regulatory approval and sales milestones. We have agreed to use commercially reasonable efforts to develop and commercialize tradipitant.
Either party may terminate the agreement under certain circumstances, including a material breach of the agreement by the other. In the event that we terminate the agreement, or if Lilly terminates the agreement due to our breach or for certain other reasons set forth in the agreement, all rights licensed and developed by us under the agreement will revert or otherwise be licensed back to Lilly on an exclusive basis, subject to payment by Lilly to us of a royalty on net sales of products that contain tradipitant.
AQW051
In December 2014, we entered into a license agreement with Novartis pursuant to which we acquired an exclusive worldwide license under certain patents and patent applications, and other licenses to intellectual property, to develop and commercialize an alpha-7 nicotinic acetylcholine receptor partial agonist, AQW051, for all human indications.
Pursuant to the agreement, we will be responsible for all development costs for AQW051. Novartis is eligible to receive tiered royalties on net sales at percentage rates up to the low double digits. We have agreed to use commercially reasonable efforts to develop and commercialize AQW051.
Either party may terminate the agreement under certain circumstances, including a material breach of the agreement by the other. In the event that we terminate the agreement, or if Novartis terminates the agreement due to our breach or for certain other reasons set forth in the agreement, all rights licensed and developed by us under the agreement will revert or otherwise be licensed back to Novartis on an exclusive basis, subject to payment by Novartis to us of a royalty on net sales of products that contain AQW051.
Government Regulation
Government authorities in the U.S., at the federal, state and local level, as well as foreign countries and local foreign governments, regulate the research, development, testing, manufacture, labeling, promotion, advertising, distribution, sampling, marketing, import and export of our products. Other than HETLIOZ® in the U.S. and the European Union and Fanapt® in the U.S., Israel and Mexico, all of our products will require regulatory approval by government agencies prior to commercialization. In particular, human pharmaceutical products are subject to rigorous pre-clinical and clinical trials and other approval procedures of the FDA and similar regulatory authorities in foreign countries. The process of obtaining these approvals and the subsequent compliance with appropriate domestic and foreign laws, rules and regulations require the expenditure of significant time and human and financial resources.
United States government regulation
FDA approval process
In the U.S., the FDA regulates drugs under the Federal Food, Drug and Cosmetic Act, as amended, and implements regulations. If we fail to comply with the applicable requirements at any time during the product development process, approval process, or after approval, we may become subject to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawals of approvals, clinical holds, warning letters, product recalls, product seizures, total or partial suspension of our operations, injunctions, fines, civil penalties or criminal prosecution. Any such sanction could have a material adverse effect on our business.
7
Table of Contents
The steps required before a drug may be marketed in the U.S. include:
| • | pre-clinical laboratory tests, animal studies and formulation studies under Current Good Laboratory Practices (cGLP); |
| • | submission to the FDA of an investigational new drug application (IND), which must become effective before human clinical trials may begin; |
| • | execution of adequate and well-controlled clinical trials to establish the safety and efficacy of the drug for each indication for which approval is sought; |
| • | submission to the FDA of an NDA; |
| • | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with Current Good Manufacturing Practices (cGMP); and |
| • | FDA review and approval of the NDA. |
Pre-clinical studies generally are conducted in laboratory animals to evaluate the potential safety and activity of a drug. Violation of the FDA’s cGLP regulations can, in some cases, lead to invalidation of the studies, requiring these studies to be replicated. In the U.S., drug developers submit the results of pre-clinical trials, together with manufacturing information and analytical and stability data, to the FDA as part of the IND, which must become effective before clinical trials can begin in the U.S. An IND becomes effective 30 days after receipt by the FDA unless before that time the FDA raises concerns or questions about issues such as the proposed clinical trials outlined in the IND. In that case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. If these concerns or questions are unresolved, the FDA may not allow the clinical trials to commence.
Pilot studies generally are conducted in a limited patient population, approximately three to 25 subjects, to determine whether the drug warrants further clinical trials based on preliminary indications of efficacy. These pilot studies may be performed in the U.S. after an IND has become effective or outside of the U.S. prior to the filing of an IND in the U.S. in accordance with applicable government regulations and institutional procedures.
Clinical trials involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in assessing the safety and the effectiveness of the drug. Each protocol must be submitted to the FDA as part of the IND prior to beginning the trial.
Typically, clinical evaluation involves a time-consuming and costly three-Phase sequential process, but the phases may overlap. Each trial must be reviewed, approved and conducted under the auspices of an independent Institutional Review Board, and each trial must include the patient’s informed consent.
| • | Phase I: refers typically to closely-monitored clinical trials and includes the initial introduction of an investigational new drug into human patients or healthy volunteer subjects. Phase I trials are designed to determine the safety, metabolism and pharmacologic actions of a drug in humans, the potential side effects associated with increasing drug doses and, if possible, to gain early evidence of the drug’s effectiveness. Phase I trials also include the study of structure-activity relationships and mechanism of action in humans, as well as studies in which investigational new drugs are used as research tools to explore biological phenomena or disease processes. During Phase I trials, sufficient information about a drug’s pharmacokinetics and pharmacological effects should be obtained to permit the design of well-controlled, scientifically valid Phase II studies. The total number of subjects and patients included in Phase I trials varies, but is generally in the range of 20 to 80 people. |
| • | Phase II: refers to controlled clinical trials conducted to evaluate appropriate dosage and the effectiveness of a drug for a particular indication or indications in patients with a disease or condition under study and to determine the common short-term side effects and risks associated with the drug. These trials are typically well-controlled, closely monitored and conducted in a relatively small number of patients, usually involving no more than several hundred subjects. |
| • | Phase III: refers to expanded controlled and uncontrolled clinical trials. These trials are performed after preliminary evidence suggesting effectiveness of a drug has been obtained. Phase III trials are intended to gather additional information about the effectiveness and safety that is needed to evaluate the overall benefit-risk relationship of the drug and to provide an adequate basis for physician labeling. Phase III trials usually include several hundred to several thousand subjects. |
Phase I, II and III testing may not be completed successfully within any specified time period, if at all. The FDA closely monitors the progress of each of the three phases of clinical trials that are conducted in the U.S. and may, at its discretion, reevaluate, alter, suspend or terminate the testing based upon the data accumulated to that point and the FDA’s assessment of the risk/benefit ratio to the patient. A clinical program is designed after assessing the causes of the disease, the mechanism of action of the active pharmaceutical ingredient of the drug and all clinical and pre-clinical data of previous trials performed. Typically, the trial design protocols and efficacy endpoints are established in consultation with the FDA. Upon request through a special protocol assessment, the FDA can also provide specific guidance on the acceptability of protocol design for clinical trials. The FDA, we or our partners may suspend or terminate clinical trials at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health risk. The FDA can also request additional clinical trials be conducted as a condition to drug approval. During all clinical trials, physicians monitor the patients to determine effectiveness and to observe and report any reactions or other safety risks that may result from use of the drug.
8
Table of Contents
Assuming successful completion of the required clinical trials, drug developers submit the results of pre-clinical studies and clinical trials, together with other detailed information including information on the manufacture and composition of the drug, to the FDA, in the form of an NDA, requesting approval to market the drug for one or more indications. In most cases, the NDA must be accompanied by a substantial user fee. The FDA reviews an NDA to determine, among other things, whether a drug is safe and effective for its intended use.
Before approving an NDA, the FDA will inspect the facility or facilities where the drug is manufactured. The FDA will not approve the application unless cGMP compliance is satisfactory. The FDA will issue an approval letter if it determines that the application, manufacturing process and manufacturing facilities are acceptable. If the FDA determines that the NDA, manufacturing process or manufacturing facilities are not acceptable, it will issue a complete response letter (CRL), in which it will outline the deficiencies in the submission and will often request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA may ultimately decide that the NDA does not satisfy the regulatory criteria for approval and refuse to approve the NDA.
The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete. The FDA may not grant approval on a timely basis, or at all. We or our partners may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us or our partners from marketing our products. Furthermore, the FDA may prevent a drug developer from marketing a drug under a label for its desired indications or place other conditions on distribution as a condition of any approvals, which may impair commercialization of the drug. After approval, some types of changes to the approved drug, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval. Similar regulatory procedures must also be complied within countries outside the U.S.
If the FDA approves the NDA, the drug becomes available for physicians to prescribe in the U.S. After approval of our products, we have to comply with a number of post-approval requirements, including delivering periodic reports to the FDA, submitting descriptions of any adverse reactions reported, and complying with drug sampling and distribution requirements. We and our partners also are required to provide updated safety and efficacy information and to comply with requirements concerning advertising and promotional labeling. Also, our quality control and manufacturing procedures must continue to conform to cGMP after approval. Drug manufacturers and their subcontractors are required to register their facilities and are subject to periodic unannounced inspections by the FDA to assess compliance with cGMP which imposes certain procedural and documentation requirements relating to quality assurance and quality control. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance. The FDA may require post market testing and surveillance to monitor the drug’s safety or efficacy, including additional studies, known as Phase IV trials, to evaluate long-term effects.
In addition to studies requested by the FDA after approval, we or our partners may have to conduct other trials and studies to explore use of the approved product for treatment of new indications, which require FDA approval. The purpose of these trials and studies is to broaden the application and use of the product and its acceptance in the medical community.
We use, and will continue to use, third-party manufacturers to produce our products in clinical and commercial quantities. Future FDA inspections may identify compliance issues at our facilities or at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of problems with a product or the failure to comply with requirements may result in restrictions on a product, manufacturer or holder of an approved NDA, including withdrawal or recall of the product from the market or other voluntary or FDA-initiated action that could delay further marketing. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications.
In September 2007, the Food and Drug Administration Amendments Act (FDAAA), was enacted into law, amending the U.S. Federal Food, Drug, and Cosmetic Act and the Public Health Service Act. The FDAAA made a number of substantive and incremental changes to the review and approval processes in ways that could make it more difficult or costly to obtain approval for new pharmaceutical products, or to produce, market and distribute existing pharmaceutical products. Most significantly, the law changed the FDA’s handling of postmarked drug product safety issues by giving the FDA authority to require post approval studies or clinical trials, to request that safety information be provided in labeling, or to require an NDA applicant to submit and execute a Risk Evaluation and Mitigation Strategy (REMS).
The FDAAA made certain changes to the user fee provisions to permit the use of user fee revenue to fund the FDA’s drug product safety activities and the review of Direct-to-Consumer advertisements. The Food and Drug Administration Safety and Innovation Act of 2012, which became effective in October 2012, reauthorized the authority of the FDA to collect user fees to fund the FDA’s review activities.
In addition, new government requirements may be established that could delay or prevent regulatory approval of our products under development.
9
Table of Contents
The Hatch-Waxman Act
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent with claims that cover the applicant’s drug. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn be cited by potential competitors in support of approval of an abbreviated new drug application (ANDA). An ANDA provides for marketing of a drug that has the same active ingredients in the same strengths and dosage form as the listed drug and has been shown through bioequivalence testing to be therapeutically equivalent to the listed drug. ANDA applicants are not required to conduct or submit results of pre-clinical or clinical tests to prove the safety or effectiveness of their drug, other than the requirement for bioequivalence testing. Drugs approved in this way are commonly referred to as “generic equivalents” to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA applicant is required to certify to the FDA concerning any patents listed for the approved drug in the FDA’s Orange Book. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new drug. A certification that the new drug will not infringe the already approved drug’s listed patents or that such patents are invalid is called a Paragraph IV certification. If the applicant does not challenge the listed patents, the ANDA application will not be approved until all the listed patents claiming the referenced drug have expired.
If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the ANDA applicant.
The ANDA application also will not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced drug has expired. The U.S. Drug Price Competition and Patent Term Restoration Act of 1984, more commonly known as the “Hatch-Waxman Act,” provides a period of five years following approval of a drug containing no previously approved active ingredients, during which ANDAs for generic versions of those drugs cannot be submitted unless the submission contains a Paragraph IV challenge to a listed patent, in which case the submission may be made four years following the original drug approval. Federal law provides for a period of three years of exclusivity following approval of a listed drug that contains previously approved active ingredients but is approved in a new dosage form, route of administration or combination, or for a new use, the approval of which was required to be supported by new clinical trials conducted by or for the sponsor, during which FDA cannot grant effective approval of an ANDA based on that listed drug.
Foreign regulation
Whether or not we or our partners obtain FDA approval for a product, we must obtain approval by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement also vary greatly from country to country. Although governed by the applicable country, clinical trials conducted outside of the U.S. typically are administered with the three-Phase sequential process that is discussed above under “United States government regulation.” However, the foreign equivalent of an IND is not a prerequisite to performing pilot studies or Phase I clinical trials.
Under European Union regulatory systems, we may submit MAAs either under a centralized or decentralized procedure. The centralized procedure, which is available for drugs produced by biotechnology or which are highly innovative, provides for the grant of a single marketing authorization that is valid for all European Union member states. This authorization is a marketing authorization approval. The decentralized procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the applications and assessment report, each member state must decide whether to recognize approval. This procedure is referred to as the mutual recognition procedure.
In addition, regulatory approval of prices is required in most countries other than the U.S. We face the risk that the resulting prices would be insufficient to generate an acceptable return to us or our partners.
Patents and proprietary rights; Hatch-Waxman protection
We and our partners will be able to protect our products from unauthorized use by third parties only to the extent that our products are covered by valid and enforceable patents, either licensed in from third parties or generated internally, that give us or our partners sufficient proprietary rights. Accordingly, patents and other proprietary rights are essential elements of our business.
HETLIOZ®, Fanapt®, tradipitant and AQW051 are covered by new chemical entity and other patents and patent applications. The patents cover the active pharmaceutical ingredient and provide patent protection for all formulations containing these active
10
Table of Contents
pharmaceutical ingredients. For more on these license and sublicense arrangements, see License Agreements above. In addition, we have generated our own intellectual property, and filed patent applications covering this intellectual property, for HETLIOZ® and Fanapt.
The table below is a summary of FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations (Orange Book) listed patents for our commercial products. Members of these patent families are also issued or pending in a number of major market territories, such as Europe and Japan.
| Number | Type | |||||
| HETLIOZ® |
US 5,856,529 | NCE | ||||
| US 8,785,492 | Method of treatment | |||||
| US 9,060,995 | Method of treatment | |||||
| US 9,539,234 | Method of treatment | |||||
| US 9,549,913 | Method of treatment | |||||
| Fanapt® |
US 8,586,610 | Method of treatment | ||||
| US 8,652,776 | Method of treatment | |||||
| US 8,999,638 | Method of treatment | |||||
| US 9,072,742 | Method of treatment | |||||
| US 9,074,254 | Method of treatment | |||||
| US 9,074,255 | Method of treatment | |||||
| US 9,074,256 | Method of treatment | |||||
| US 9,138,432 | Method of treatment | |||||
| US 9,157,121 | Method of treatment | |||||
HETLIOZ®
Our rights to the new chemical entity patent covering HETLIOZ® and related intellectual property have been acquired through a license with BMS. HETLIOZ® and its formulations, genetic markers and uses are covered by a total of 14 patent and patent application families worldwide. The primary new chemical entity patent covering HETLIOZ® expires normally in 2017 in the U.S. and in most European markets. The “Hatch-Waxman Act” provides for an extension of new chemical entity patents for a period of up to five years following the expiration of the patent covering that compound to compensate for time spent in development. We believe that HETLIOZ® will meet the various criteria of the Hatch-Waxman Act and will receive five additional years of patent protection in the U.S., which would extend its new chemical entity patent protection in the U.S. until 2022. An application for the five year patent term extension has been filed and is being processed by the U.S. Patent and Trademark Office. In July 2014, a new method of use patent was issued to us by the U.S. Patent and Trademark Office for HETLIOZ® in the treatment of Non-24. This method of use patent is expected to expire in 2033, potentially further extending the exclusivity protection of HETLIOZ®. In June 2015, an additional method of use patent was issued to us by the U.S. Patent and Trademark Office for HETLIOZ®. This method of use patent is also expected to expire in 2033. Both the new chemical entity patent and the method of use patents are listed in the FDA’s Orange Book.
In Europe, the law provides for ten years of data exclusivity (with the potential for an additional year if the drug is developed for a significant new indication). As such, in Europe, data exclusivity will protect HETLIOZ® for at least ten years from approval. A completed Pediatric Investigation Plan could further extend this exclusivity for two years in an orphan indication, for a total of 12 years of exclusivity. It is also possible that the term of the new chemical entity patent in Europe could be extended by issuance of a supplementary protection certificate (SPC). The European Patent Office has granted our patent application directed to the 20 mg/day dose. This patent will expire normally in 2027. Patent applications directed to the treatment of Non-24, if granted, would provide exclusivity in Europe for this indication until at least 2033.
Outside the U.S. and Europe, data exclusivity will protect HETLIOZ® from generic competition for varying numbers of years depending on the country.
Additional patent applications directed to specific sleep disorders and to methods of treating patients with HETLIOZ®, if issued, would provide exclusivity for such indications and methods of treatment, potentially extending the effective patent protection period in the U.S., Europe, and other major markets.
Fanapt®
The new chemical entity patent for Fanapt®, which expired in 2016, is owned by Sanofi, and other patents and patent applications relating to Fanapt® previously owned by Novartis are now owned by Vanda. We originally obtained exclusive worldwide rights to develop and commercialize the products covered by these patents through license and sublicense arrangements. Then, pursuant to an amended sublicense agreement with Novartis, Novartis retained exclusive commercialization rights to all formulations of Fanapt® in the U.S. and Canada. However, as of December 2014, pursuant to an asset transfer agreement, we acquired all rights in Fanapt®, including in the U.S. and Canada.
Fanapt® and its metabolites, formulations, genetic markers and uses are covered by a total of 17 patent and patent application families in the U.S., Europe, and other markets. The primary new chemical entity patent covering Fanapt® expired in November 2016
11
Table of Contents
in the U.S. and expired in 2010 in major markets outside the U.S. In November 2013, a patent directed to a method of treating patients with Fanapt® based on genotype was issued to us by the U.S. Patent and Trademark Office. This patent, which was listed in the FDA’s Orange Book in January 2015, is set to expire in 2027, potentially further extending the exclusivity protection of Fanapt®. Additional method of treatment patents were issued and listed in the Orange Book with the latest expected expiry in December 2031. See Note 18, Legal Matters, to the consolidated financial statements included in Part II of this annual report on Form 10-K for additional information.
In Europe, the law provides for ten years of data exclusivity (with the potential for an additional year if the drug is developed for a significant new indication). No generic versions of Fanapt® would be permitted to be marketed or sold during this 10-year (or 11-year) period in most European countries. Consequently, we expect our rights to commercialize Fanapt® will be exclusive for at least 10 years from approval in Europe. Outside the U.S. and Europe, data exclusivity will protect Fanapt® from generic competition for varying numbers of years depending upon the country. Several other patent applications covering metabolites, uses, formulations and genetic markers relating to Fanapt® extend beyond 2020. The patent family for the microsphere depot formulation of Fanapt® expires in 2024 in the U.S. and 2022 in most of the major markets in Europe. The patent family for the aqueous microcrystals depot formulation of Fanapt® expires in 2023 in the U.S. and in most of the major markets in Europe.
Tradipitant
Lilly owns a new chemical entity patent as well as patent applications directed to polymorphic forms of, and methods of making tradipitant. Thus, tradipitant is covered by a total of three patent and patent application families worldwide, which have been licensed to us. The new chemical entity patent covering tradipitant expires in 2023, except in the U.S., where it expires normally in 2024 subject to any extension that may be received under Hatch-Waxman. We have filed additional patent applications based on discoveries made during recent studies with tradipitant.
AQW051
Novartis owns a new chemical entity patent as well as patent applications directed to methods of using AQW051, AQW051 formulations, and combinations of AQW051 with other active pharmaceutical ingredients. The new chemical entity patent expires normally in 2023 in the U.S., Europe, and other markets.
Trichostatin A
Trichostatin A is a small molecule HDAC inhibitor with potential use as a treatment for several oncology indications. We have pending patent applications covering the use of Trichostatin A and plan on filing additional applications based on discoveries made throughout the development plan of this molecule.
Other Patents
Aside from the new chemical entity patents and other in-licensed patents relating to Fanapt®, HETLIOZ®, tradipitant, and AQW051, we have numerous patent and patent application families, most of which have been filed in key markets including the U.S., relating to our products and development compounds. In addition, we have several other patent application families relating to drugs not presently in clinical studies. The claims in these various patents and patent applications are directed to compositions of matter, including claims covering other products, pharmaceutical compositions and methods of use.
Proprietary Know-how
For proprietary know-how that is not appropriate for patent protection, processes for which patents are difficult to enforce and any other elements of our discovery process that involve proprietary know-how and technology that are not covered by patent applications, we generally rely on trade secret protection and confidentiality agreements to protect our interests. We require all of our employees, consultants and advisors to enter into confidentiality agreements. Where it is necessary to share our proprietary information or data with outside parties, our policy is to make available only that information and data required to accomplish the desired purpose and only pursuant to a duty of confidentiality on the part of those parties.
Third-Party Reimbursement and Pricing Controls
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010 (together, PPACA), has changed and is expected to further significantly change the way healthcare is financed by both governmental and private insurers. The provisions of PPACA became effective over various periods from 2010 through 2014. We cannot predict the complete impact of PPACA on pharmaceutical companies because many of PPACA’s reforms require the promulgation of detailed regulations to implement the statutory provisions, which has not yet occurred. While we cannot predict the complete impact on federal reimbursement policies this law will have in general or specifically on any product we commercialize, PPACA may result in downward pressure on pharmaceutical reimbursement, which could negatively affect market acceptance of new products. The rebates, discounts, taxes and other costs resulting from PPACA may have a significant effect on our profitability in the future. In addition, potential reductions of the per capita rate of growth in Medicare spending under PPACA, could potentially limit access to certain treatments or mandate price controls for our products. Moreover, although the United States Supreme Court has upheld the constitutionality of most of PPACA, some states have indicated that they intend not to implement certain sections of PPACA, and some members of the U.S. Congress are still working to repeal PPACA. We cannot predict whether these challenges will continue or other proposals will be made or adopted, or what impact these efforts may have on us or our partners.
12
Table of Contents
In the U.S. and elsewhere, sales of pharmaceutical products depend in significant part on the availability of reimbursement to the consumer from third-party payors, such as government and private insurance plans. Third-party payors are increasingly challenging the prices charged for medical products and services. It will be time consuming and expensive for us or our partners to go through the process of seeking reimbursement from Medicare and private payors. Our products may not be considered cost-effective, and coverage and reimbursement may not be available or sufficient to allow us or our partners to sell our compounds on a competitive and profitable basis. The passage of the Medicare Prescription Drug and Modernization Act of 2003 imposes additional requirements for the distribution and pricing of prescription drugs which may affect the marketing of our products.
In many foreign markets, including the countries in the European Union and Japan, pricing of pharmaceutical products is subject to governmental control. In the U.S., there have been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental pricing control. While we cannot predict whether such legislative or regulatory proposals will be adopted, the adoption of such proposals could have a material adverse effect on our business, financial condition and profitability.
Marketing and Sales
HETLIOZ® was approved in the U.S. for the treatment of Non-24 in January 2014 and commercially launched in the U.S. in April 2014. Additionally, HETLIOZ® was approved in the Europe Union for the treatment of Non-24 in July 2015. We commercially launched HETLIOZ® in Germany in the third quarter of 2016.
Given the range of potential indications for HETLIOZ®, we may pursue one or more partnerships for the development and commercialization of HETLIOZ® worldwide.
Fanapt® was approved in the U.S. for the treatment of schizophrenia in May 2009 and commercially launched in the U.S. in January 2010. In October 2009, we entered into an amended and restated sublicense agreement with Novartis pursuant to which Novartis has exclusive commercialization rights to all formulations of Fanapt® in the U.S. and Canada. Novartis began selling Fanapt® in the U.S. during the first quarter of 2010. Pursuant to the terms of the Settlement Agreement with Novartis, Novartis transferred all U.S. and Canadian rights in the Fanapt® franchise to Vanda on December 31, 2014.
Fanapt® was launched in Israel and Mexico by our distribution partners in 2014. We continue to explore the regulatory path and commercial opportunity for Fanapt® oral formulation in other regions.
Manufacturing
We currently utilize a virtual supply manufacturing and distribution chain in which we do not have our own facilities to manufacture commercial or clinical trial supplies of drugs and we do not have our own distribution facilities. Additionally, we do not intend to develop such facilities for any product in the near future. Instead, we contract with third parties for the manufacture, warehousing, order management, billing and collection and distribution of our products and product candidates.
We expect to continue to rely solely on third-party manufacturers to manufacture drug substance and final drug products for both clinical development and commercial sale. However, there are numerous factors that could cause interruptions in the supply of our products, including regulatory reviews, changes in our sources for manufacturing, disputes with a manufacturer, or financial instability of manufacturers, all of which could negatively impact our operation and our financial results.
In January 2014, we entered into a manufacturing agreement with Patheon Pharmaceuticals Inc. (Patheon) for the manufacture of commercial supplies of HETLIOZ® 20 mg capsules at Patheon’s Cincinnati, Ohio manufacturing site. Under the HETLIOZ® manufacturing agreement, we are responsible for supplying the active pharmaceutical ingredient for HETLIOZ® to Patheon and have agreed to certain minimum yearly order requirements. Patheon is responsible for manufacturing the HETLIOZ® 20 mg capsules, conducting quality control and stability testing, and packaging the HETLIOZ® capsules. The HETLIOZ® manufacturing agreement has an initial term of five years and will automatically renew after the initial term for successive terms of one year each, unless either party gives notice of its intention to terminate the agreement at least twelve months prior to the end of the then current term. Either party may terminate the HETLIOZ® manufacturing agreement under certain circumstances upon specified written notice to the other party.
As part of the Settlement Agreement, we assumed Novartis’ manufacturing agreement with Patheon for the manufacture of commercial supplies of Fanapt® In May 2016, we entered into a new manufacturing agreement with Patheon for the manufacture of commercial supplies of Fanapt® 1, 2, 4, 6, 8, 10 and 12 mg tablets at Patheon’s Mississauga, Ontario, Canada manufacturing site. Under the Fanapt® manufacturing agreement, we are responsible for sourcing the supply of the active pharmaceutical ingredient (iloperidone), and have agreed to order from Patheon at least 70% of the total yearly requirement of new units of Fanapt® tables for the U.S. and other specified countries each year for the term of the agreement. The Fanapt® manufacturing agreement has an initial term of five years and will automatically renew after the initial term for successive terms of one year each, unless either party gives notice of its intention to terminate the agreement at least twelve months prior to the end of the then current term. Either party may terminate the Fanapt® manufacturing agreement under certain circumstances upon specified written notice to the other party.
13
Table of Contents
Research and Development
We have built a research and development organization that includes extensive expertise in the scientific disciplines of pharmacogenetics and pharmacogenomics. We operate cross-functionally and are led by an experienced research and development management team. We use rigorous project management techniques to assist us in making disciplined strategic research and development program decisions and to help limit the risk profile of our product pipeline. We also access relevant market information and key opinion leaders in creating target product profiles and, when appropriate, as we advance our programs towards commercialization. We engage third parties to conduct portions of our preclinical research. In addition, we utilize multiple clinical sites to conduct our clinical trials; however, we are not substantially dependent upon any one of these sites for our clinical trials nor do any of them conduct a major portion of our clinical trials.
Research and development expenses amounted to $29.2 million, $29.1 million and $19.2 million for the years ended December 31, 2016, 2015 and 2014, respectively.
Major Customers
Our revenues are generated from product sales and are concentrated with specialty pharmacies and wholesalers. There were five major customers that each accounted for more than 10% of total revenues and, as a group, represented 86% of total revenues for the year ended December 31, 2016.
Competition
The pharmaceutical industry and the central nervous system segment of that industry, in particular, is highly competitive and includes a number of established large and mid-sized companies with greater financial, technical and personnel resources than we have and significantly greater commercial infrastructures than we have. Our market segment also includes several smaller emerging companies whose activities are directly focused on our target markets and areas of expertise. Our products, once approved for commercial use, will compete with numerous therapeutic treatments offered by these competitors. While we believe that our products will have certain favorable features, existing and new treatments may also possess advantages. Additionally, the development of other drug technologies and methods of disease prevention are occurring at a rapid pace. These developments may render our products or technologies obsolete or noncompetitive.
We believe the primary competitors for HETLIOZ® and Fanapt® are as follows:
| • | For HETLIOZ® in the treatment of Non-24, there are no FDA approved direct competitors. Sedative-Hypnotic treatments for certain sleep related disorders include, Ambien® (zolpidem) by Sanofi (including Ambien CR®), Lunesta® (eszopiclone) by Sunovion Pharmaceuticals Inc., Sonata® (zaleplon) by Pfizer Inc., Rozerem® (ramelteon) by Takeda Pharmaceuticals Company Limited, Silenor® (doxepin) by Pernix Therapeutics, Belsomra® (suvorexant) by Merck & Co., Inc., generic products such as zolpidem, trazodone and doxepin, and over-the-counter remedies such as Benadryl® and Tylenol PM®. The class of melatonin agonists includes Rozerem® (ramelteon) by Takeda Pharmaceuticals Company Limited, Valdoxan® (agemelatine) by Servier, Circadin® (long-acting melatonin) by Neurim Pharmaceuticals and the food supplement melatonin. Shift work and excessive sleepiness disorder treatments include Nuvigil® (armodafinil) and Provigil® (modafinil) both by Teva Pharmaceutical Industries Ltd. |
| • | For Fanapt® in the treatment of schizophrenia, the atypical antipsychotics competitors are Risperdal® (risperidone), including the depot formulation Risperdal® Consta® and Invega® (paliperidone), including the depot formulation Invega® Sustenna®, each by Ortho-McNeil-Janssen Pharmaceuticals, Inc., Zyprexa® (olanzapine), including the depot formulation Zyprexa® Relprevv™, each by Eli Lilly and Company, Seroquel® and Seroquel XR (quetiapine) by AstraZeneca PLC, Abilify® (aripiprazole) by BMS/Otsuka America Pharmaceutical Inc., Abilify® Maintena® (the depot formulation of Abilify®) by Lundbeck/Otsuka America Pharmaceutical Inc., Geodon® (ziprasidone) by Pfizer Inc., Saphris® (asenapine) by Actavis plc, Latuda® (lurasidone) by Sunovion Pharmaceuticals Inc., Rexulti® (brexpiprazole) by Lundbeck/Otsuka America Pharmaceutical, Inc., Aristada™ (aripiprazole lauroxil) extended-release injectible suspension by Alkermes, Inc., Vraylar™ (cariprazine) by Teva Pharmaceutical Industries Ltd., and generic clozapine, as well as the typical antipsychotics haloperidol, chlorpromazine, thioridazine, and sulpiride (all of which are generic). |
Our ability to compete successfully will depend in part on our ability to utilize our pharmacogenetics and pharmacogenomics and drug development expertise to identify, develop, secure rights to and obtain regulatory approvals for promising pharmaceutical products before others are able to develop competitive products. Our ability to compete successfully will also depend on our ability to attract and retain skilled and experienced personnel. Additionally, our ability to compete may be affected because insurers and other third-party payors in some cases seek to encourage the use of cheaper, generic products, which could make our products less attractive.
14
Table of Contents
Employees
We had 142 full-time employees as of December 31, 2016, compared with 118 employees as of December 31, 2015. None of our employees are represented by a labor union. We have not experienced any work stoppages and consider our employee relations to be good.
Corporate Information
We were incorporated in Delaware in 2002. Our principal executive offices are located at 2200 Pennsylvania Avenue NW, Suite 300E, Washington D.C. 20037, and our telephone number is (202) 734-3400. Our website address is www.vandapharma.com and the information contained in, or that can be accessed through, our website is not part of this annual report and should not be considered part of this annual report.
Available Information
We file annual, quarterly, and current reports, proxy statements, and other documents with the Securities and Exchange Commission (SEC) under the Securities Exchange Act of 1934 (the Exchange Act). The public may read and copy any materials that we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, D.C. 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. Also, the SEC maintains an internet website at www.sec.gov that contains reports, proxy and information statements, and other information regarding issuers, including us, that file electronically with the SEC.
We also make available free of charge on our Internet website at www.vandapharma.com our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and, if applicable, amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC.
Our code of ethics, other corporate policies and procedures, and the charters of our Audit Committee, Compensation Committee and Nominating/Corporate Governance Committee are available through our Internet website at www.vandapharma.com.
| ITEM 1A. | RISK FACTORS |
Our business, financial condition and operating results can be affected by a number of factors, whether current known or unknown, including but not limited to those described below, any one or more of which could, directly or indirectly, cause our actual operating results and financial condition to vary materially from past, or anticipated future, operating results and financial condition. Any of these factors, in whole or in part, could materially and adversely affect our business, financial condition, operating results and the price of our common stock.
The following discussion of risk factors contains forward-looking statements. These risk factors may be important to understanding any statement in this annual report on Form 10-K or elsewhere. The following information should be read in conjunction with the consolidated financial statements and related notes in Part I, Item 1, Financial Statements and Part I, Item 2, Management’s, Discussion and Analysis of Financial Condition and Results of Operations.
Because of the following factors, as well as other factors affecting our financial condition and operating results, past financial performance should not be considered to be a reliable indicator of future performance, and investors should not use historical trends to anticipate results or trends in future periods.
Risks related to our business and industry
We are dependent on the commercial success of HETLIOZ® and Fanapt®.
Our future success is currently substantially dependent upon the commercial success of HETLIOZ® for the treatment of Non-24-Hour Sleep-Wake Disorder (Non-24) and Fanapt® for the treatment of schizophrenia.
In January 2014, the FDA approved our New Drug Application (NDA) for HETLIOZ® for the treatment of Non-24 and in April 2014, we commenced the U.S. commercial launch of HETLIOZ®. In July 2015, the European Commission (EC) granted centralized marketing authorization with unified labeling for HETLIOZ® for the treatment of Non-24 in blind adults, and in August 2016 we commenced the commercial launch of HETLIOZ® in Germany. This authorization is valid in the 28 countries that are members of the European Union, as well as European Economic Area members Iceland, Liechtenstein and Norway.
In the first quarter of 2015, we acquired the U.S. commercial rights to Fanapt®, and began selling, marketing and distributing Fanapt® in the U.S.
Our ability to generate significant product revenue from sales of HETLIOZ® and Fanapt®, both in the U.S. and abroad, in the near term will depend on, among other things, our ability to:
| • | defend our patents and intellectual property from generic competition; |
| • | maintain commercial manufacturing arrangements with third-party manufacturers; |
15
Table of Contents
| • | produce, through a validated process, sufficiently large quantities of inventory of our products to meet demand; |
| • | continue to maintain and grow a wide variety of internal sales, distribution and marketing capabilities sufficient to sustain growth in sales of our products; |
| • | gain broad acceptance of our products from physicians, health care payors, patients, pharmacists and the medical community; |
| • | properly price and obtain adequate coverage and reimbursement of these products by governmental authorities, private health insurers, managed care organizations and other third-party payors; |
| • | maintain compliance with ongoing FDA labeling, packaging, storage, advertising, promotion, recordkeeping, safety and other post-market requirements; |
| • | obtain approval from the FDA to expand the labeling of our approved products for additional indications; |
| • | obtain regulatory approval for HETLIOZ® or Fanapt® in additional countries; |
| • | adequately protect against and effectively respond to any claims by holders of patents and other intellectual property rights that our products infringe their rights; and |
| • | adequately protect against and effectively respond to any unanticipated adverse effects or unfavorable publicity that develops in respect to our products, as well as the emergence of new or existing competitive products, which may be proven to be more clinically effective and cost-effective. |
We expect to continue to incur significant expenses and to utilize a substantial portion of our cash resources as we continue the commercialization of HETLIOZ® and Fanapt®, evaluate foreign market opportunities for HETLIOZ® and Fanapt® and continue to grow our operational capabilities, both domestically and abroad. This activity represents a significant investment in the commercial success of HETLIOZ® and Fanapt®, which is uncertain.
If our continued commercial efforts are not successful with respect to HETLIOZ® and Fanapt® in the U.S., Europe or other jurisdictions in which these products may be approved for sale, our ability to generate increased product sales revenue may be jeopardized and, consequently, our business may be seriously harmed.
The cost of growing and maintaining a sales, marketing and distribution organization may exceed its cost effectiveness. If we fail to continue to develop sales, marketing and distribution capabilities, if sales efforts are not effective or if costs of developing sales, marketing and distribution capabilities exceed their cost effectiveness, our business, results of operations and financial condition could be materially adversely affected.
Continued growth of HETLIOZ® and Fanapt® may be slow or limited for a variety of reasons including competing products or unanticipated safety issues. If either HETLIOZ® or Fanapt® is not successful in gaining broad commercial acceptance, our business would be harmed.
Any increase in sales of HETLIOZ® and Fanapt® will be dependent on several factors, including our ability to educate physicians and to increase physician awareness of the benefits and cost-effectiveness of our products relative to competing products. The degree of further market acceptance of any of our products or market acceptance of approved product candidates among physicians, patients, health care payors and the medical community will depend on a number of factors, including:
| • | acceptable evidence of safety and efficacy; |
| • | relative convenience and ease of administration; |
| • | the prevalence and severity of any adverse side effects; |
| • | availability of alternative treatments; and |
| • | pricing and cost effectiveness. |
In addition, HETLIOZ® and Fanapt® are subject to continual review by the FDA, and we cannot assure that newly discovered or reported safety issues will not arise. With the use of any newly marketed drug by a wider patient population, serious adverse events may occur from time to time that initially do not appear to relate to the drug itself. Any safety issues could cause us to suspend or cease marketing of our approved products, cause us to modify how we market our approved products, subject us to substantial liabilities and adversely affect our revenues and financial condition. In the event of a withdrawal of either HETLIOZ® or Fanapt® from the market, our revenues would decline significantly and our business would be seriously harmed.
We may enter into third party collaborations from time to time in order to commercialize our products. If we are unable to identify or enter into an agreement with any material third-party collaborator, if our collaborations with any such third-party are not commercially successful or if our agreement with any such third-party is terminated or allowed to expire, we could be adversely affected financially or our business reputation could be harmed.
16
Table of Contents
Our business strategy includes entering into collaborations with corporate collaborators for the commercialization of HETLIOZ®, Fanapt® and our other products. Areas in which we may potentially enter into third-party collaboration arrangements include joint sales and marketing arrangements for sales and marketing in certain European Union countries and elsewhere outside of the U.S., and future product development arrangements. If we are unable to identify or enter into an agreement with any material third-party collaborator, this could result in an adverse effect on our business, results of operations or financial condition. Any arrangements we do enter into may not be scientifically or commercially successful. The termination of any of these arrangements might adversely affect our ability to develop, commercialize and market our products.
The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Our collaborators will have significant discretion in determining the efforts and resources that they will apply to these collaborations. We expect that the risks which we face in connection with these future collaborations will include the following:
| • | our collaboration agreements are expected to be for fixed terms and subject to termination under various circumstances, including, in many cases, on short notice without cause; |
| • | our collaborators may develop and commercialize, either alone or with others, products and services that are similar to or competitive with our products which are the subject of their collaboration with us; and |
| • | our collaborators may change the focus of their commercialization efforts. |
In recent years there have been a significant number of mergers and consolidations in the pharmaceutical and biotechnology industries, some of which have resulted in the participant companies reevaluating and shifting the focus of their business following the completion of these transactions. The ability of our products to reach their potential could be limited if any of our future collaborators decreases or fails to increase spending relating to such products.
Collaborations with pharmaceutical companies and other third-parties often are terminated or allowed to expire by the other party. With respect to our future collaborations, any such termination or expiration could adversely affect us financially as well as harm our business reputation.
Even after we or our partners obtain regulatory approvals of a product, acceptance of the product in the marketplace is uncertain and failure to achieve commercial acceptance will prevent or delay our ability to generate significant revenue from such product.
Even after obtaining regulatory approvals for the sale of our products, the commercial success of these products will depend, among other things, on their acceptance by physicians, patients, third-party payors and other members of the medical community as therapeutic and cost-effective alternatives to competing products and treatments. The degree of market acceptance of any product will depend on a number of factors, including the demonstration of its safety and efficacy, its cost-effectiveness, its potential advantages over other therapies, the reimbursement policies of government and third-party payors with respect to such product, our ability to attract and maintain corporate partners, including pharmaceutical companies, to assist in commercializing our products, receipt of regulatory clearance of marketing claims for the uses that we or our partners are developing and the effectiveness of our and our partners’ marketing and distribution capabilities. If our approved products fail to gain market acceptance or do not become widely accepted by physicians, patients, third-party payors and other members of the medical community, it is unlikely that we will ever become profitable on a sustained basis or achieve significant revenues.
We rely and will continue to rely on outsourcing arrangements for many of our activities, including clinical development and supply of HETLIOZ®, Fanapt® and our other products.
As of December 31, 2016, we had 142 full-time employees. We rely on outsourcing arrangements for a significant portion of our activities, including distribution, clinical research and development, data collection and analysis and manufacturing, as well as for certain functions as a public company. We may have limited control over these third parties and we cannot guarantee that they will perform their obligations in an effective and timely manner.
Disruptions to our HETLIOZ® or Fanapt® supply chains could materially affect our ability to successfully commercialize HETLIOZ® or Fanapt®, thereby reducing our future earnings and prospects.
A loss or disruption with any one of our manufacturers or suppliers could disrupt the supply of HETLIOZ® or Fanapt®, possibly for a significant time period, and we may not have sufficient inventories to maintain supply before the manufacturer or supplier could be replaced or the disruption is resolved. In addition, marketed drugs and their contract manufacturing organizations are subject to continual review, including review and approval of their manufacturing facilities and the manufacturing processes, which can result in delays in the regulatory approval process and/or commercialization. Introducing a replacement or backup manufacturer or supplier for HETLIOZ® or Fanapt® requires a lengthy regulatory and commercial process and there can be no guarantee that we could obtain necessary regulatory approvals in a timely fashion or at all. In addition, it is difficult to identify and select qualified suppliers and manufacturers with the necessary technical capabilities, and establishing new supply and manufacturing sources involves a lengthy and technical engineering process.
Failure to comply with government regulations regarding the sale and marketing of our products could harm our business.
17
Table of Contents
Our business is subject to extensive government regulation and oversight. As a result, we may become subject to governmental actions which could materially and adversely affect our business, results of operations and financial condition, certain of which are described below.
Pharmaceutical Pricing and Reimbursement
In U.S. markets, our ability and that of our partners to commercialize our products successfully, and to attract commercialization partners for our products, depends in significant part on the availability of adequate financial coverage and reimbursement from third-party payors, including, in the U.S., governmental payors such as the Medicare and Medicaid programs, managed care organizations, and private health insurers.
We participate in the Medicaid Drug Rebate Program for both HETLIOZ® and Fanapt®. Under the Medicaid Drug Rebate Program, we are required to pay a rebate to each state Medicaid program for our covered outpatient drugs that are dispensed to Medicaid beneficiaries and paid for by a state Medicaid program as a condition of having federal funds being made available to the states for our drugs under Medicaid and Medicare Part B. Those rebates are based on pricing data that are reported by us on a monthly and quarterly basis to the Centers for Medicare & Medicaid Services (CMS). Federal law requires that any company that participates in the Medicaid Drug Rebate Program also participate in the Public Health Service’s 340B drug pricing discount program (the 340B program), in order for federal funds to be available for the manufacturer’s drugs under Medicaid and Medicare Part B. The 340B program requires participating manufacturers to agree to charge statutorily-defined covered entities no more than the 340B “ceiling price” for the manufacturer’s covered outpatient drugs. The ceiling price can represent a significant discount and is based on the pricing data reporting to the Medicaid Drug Rebate Program.
PPACA expanded the 340B program to include additional entity types: certain free-standing cancer hospitals, critical access hospitals, rural referral centers and sole community hospitals, each as defined by PPACA. PPACA exempts drugs designated under section 526 of the FDC Act as “orphan drugs” from the ceiling price requirements for these newly-eligible entities.
PPACA also obligates HRSA to create regulations and processes to improve the integrity of the 340B program and to update the agreement that manufacturers must sign to participate in the 340B program. HRSA issued a final regulation in January of 2017 regarding the calculation of the 340B ceiling price and the imposition of civil monetary penalties on manufacturers that knowingly and intentionally overcharge covered entities. The final regulation regarding the 340B program include the requirement that a manufacturer calculate the 340B ceiling price on a quarterly basis, the requirement that a manufacturer charge $0.01 per unit of measure if the 340B ceiling price calculation results in a ceiling price that equals zero (penny pricing), the methodology manufacturers must use when estimating the ceiling price for a new covered outpatient drug, an explanation of how a civil monetary penalty (CMP) would be imposed on a manufacturer that knowingly and intentionally overcharges a covered entity; and an explanation of what would constitute an instance of overcharging to trigger a CMP. This final regulation becomes effective March 6, 2017, but recognizing such date is in the middle of a quarter, HRSA plans to begin enforcing the requirements of this final rule at the start of the next quarter, which begins April 1, 2017. HRSA recently issued a proposed regulation regarding an administrative dispute resolution process for the 340B program. Any final regulations and guidance could affect our obligations under the 340B program in ways we cannot anticipate. In addition, legislation may be introduced that, if passed, would further expand the 340B program to additional covered entities or otherwise expand the 340B program.
Federal law also requires that for a drug manufacturer’s products to be eligible for payment with federal funds under the Medicaid and Medicare Part B programs and to be purchased by certain federal agencies and grantees, the manufacturer must participate in the Department of Veterans Affairs Federal Supply Schedule (FSS), pricing program, established by Section 603 of the Veterans Health Care Act of 1992. Manufacturers that participate in the FSS pricing program must list their covered (innovator) drugs on an FSS contract and charge no more than Federal Ceiling Price (FCP), to the Department of Veterans Affairs, Department of Defense, Public Health Service, and Coast Guard when those agencies purchase from the FSS contract or a depot contract. FCP is calculated based on non-federal average manufacturer price data, which manufacturers must submit quarterly and annually. In addition, because our products are available in the retail and specialty pharmacy setting, we are required to provide rebates to the Department of Defense for prescriptions dispensed to Tricare beneficiaries from Tricare retail network pharmacies under the Tricare Retail Refund Program. These programs obligate the manufacturer to pay rebates and offer its drugs at certain prices to certain federal purchasers. To the extent we choose to participate in these government healthcare programs for our current and future products, these and other requirements may affect our ability to profitably sell any product for which we obtain marketing approval.
Pricing and rebate calculations vary among products and programs. The calculations are complex and will often be subject to interpretation by us, governmental or regulatory agencies and the courts. If we become aware that our reporting of pricing data for a prior quarter was incorrect, we will be obligated to resubmit the corrected data. For the Medicaid Drug Rebate Program, corrected data must be submitted for a period not to exceed 12 quarters from the quarter in which the data originally were due. Such restatements and recalculations increase our costs for complying with the laws and regulations governing the Medicaid Drug Rebate Program and other governmental pricing programs.
We may be liable for errors associated with our submission of pricing data. If we are found to have knowingly submitted false pricing data to the Medicaid program or the FSS pricing program, we may be liable for civil monetary penalties in the amount of up to $100,000 per item of false information. Our failure to submit pricing data to the Medicaid program or the FSS pricing program on a timely basis could result in a civil monetary penalty of $10,000 per day for each day the information is late. Such failure also could be grounds for CMS to terminate our Medicaid drug rebate agreement, which is the agreement under which we would participate in the Medicaid Drug Rebate Program. In the event that CMS terminates our rebate agreement, federal payments may not be available under Medicaid or Medicare Part B for our covered outpatient drugs. We cannot assure you that our submissions will not be found to be incomplete or incorrect.
18
Table of Contents
Third-party payors decide which drugs they will pay for and establish reimbursement and co-pay levels. Third-party payors are increasingly challenging the prices charged for medical products and services and examining their cost effectiveness, in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost effectiveness of our products. Even with such studies, any of our products that are commercialized may be considered less safe, less effective or less cost-effective than other products, and third-party payors may not provide coverage and reimbursement, in whole or in part, for our products.
Political, economic and regulatory influences are subjecting the healthcare industry in the U.S. to fundamental changes. There have been, and we expect there will continue to be, legislative and regulatory proposals to change the healthcare system and reimbursement systems in ways that could impact our ability and that of our partners to profitably sell commercialized products.
Payors also are increasingly considering new metrics as the basis for reimbursement rates, such as average sales price, average manufacturer price and actual acquisition cost. It is difficult to project the impact of these evolving reimbursement mechanics on the willingness of payors to cover any of our products that are commercialized.
In addition, we anticipate that a significant portion of our or our partners’ revenue from sales of commercialized products will be obtained through government payors, including Medicaid, and any failure to qualify for reimbursement for products we are able to commercialize under those programs would have a material adverse effect on revenues and royalties from sales of such products.
Interactions with Healthcare Providers
Healthcare providers, physicians and others often play a primary role in the recommendation and prescription of pharmaceutical products. Manufacturers of branded prescription drugs are subject to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which manufacturers market, sell and distribute the products for which they obtain marketing approval. Some of the laws and regulations that may affect our ability to operate are described below.
Anti-Kickback Laws
The federal Anti-Kickback Statute prohibits, among other things, persons from knowingly and willfully offering, paying, soliciting, or receiving remuneration, directly or indirectly, in cash or in kind, to induce or reward the purchasing, leasing, ordering or arranging for the purchase, lease, or order of any health care item or service reimbursable under federal healthcare programs such as Medicare and Medicaid. The term “remuneration” has been broadly interpreted to include anything of value, and the government can establish a violation of the Anti-Kickback Statute without proving that a person or entity had actual knowledge of the law or specific intent to violate it. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. There are a number of statutory exceptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, however, the exceptions and safe harbors are drawn narrowly. Failure to meet all of the requirements of a particular statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti-Kickback Statute, but the legality of the arrangement will be evaluated on a case-by-case basis based on the totality of the facts and circumstances. A number of states also have anti-kickback laws that establish similar prohibitions that may apply to items or services reimbursed by government programs, as well as any third-party payors, including commercial payors.
False Claims Act
The federal civil False Claims Act prohibits, among other things, individuals or entities from knowingly presenting, or causing to be presented false or fraudulent claims for payment of government funds and knowingly making, or causing to be made or used, a false record or statement to get a false claim paid. Certain marketing practices may implicate the federal civil False Claims Act, including promotion of pharmaceutical products for unapproved uses, providing free product to customers with the expectation that the customer would bill federal programs for the product, or inflating prices report to private price publication services used to set drug reimbursement rates under federal healthcare programs. In addition, PPACA amended the Social Security Act to provide that a claim including items or services resulting from a violation of the Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act. Actions under the False Claims Act may be brought by the government or as a qui tam action by a private individual in the name of the government. False Claims Act liability is potentially significant in the healthcare industry because the statute provides for treble damages and mandatory penalties of $5,500 to $11,000 per false claim or statement, which will increase to a range of $10,781 to $21,563 for violations after November 2, 2015, and assessed after August 1, 2016. Because of the potential for large monetary exposure, healthcare companies often resolve allegations without admissions of liability for significant and sometimes material amounts to avoid the uncertainty of treble damages and per claim penalties that may awarded in litigation proceedings. They may be required, however, to enter into corporate integrity agreements with the government, which may impose substantial costs on companies to ensure compliance. Pharmaceutical companies also are subject to other federal false claim laws, including laws that impose criminal penalties, including imprisonment and criminal fines, for making or presenting a false or fictitious or fraudulent claim to the federal government.
19
Table of Contents
Health Insurance Portability and Accountability Act
The federal Health Insurance Portability and Accountability Act of 1996 (HIPAA), created federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services.
Physician Payment Sunshine Act
The federal Physician Payment Sunshine Act requires manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program to report annually (with certain exceptions) to CMS, information related to payments or other “transfers of value” made to physicians and teaching hospitals, and requires applicable manufacturers and group purchasing organizations to report annually to CMS ownership and investment interests held by physicians and their immediate family members and payments or other ‘‘transfers of value’’ to such physician owners.
Analogous State and Foreign Laws
The majority of states also have statutes or regulations similar to the federal laws described above, including state anti-kickback and false claims laws. These state laws apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. In addition, a number of states require pharmaceutical companies to report expenses relating to the marketing and promotion of pharmaceutical products and to report gifts and payments to individual physicians in the states. Other states restrict when pharmaceutical companies may provide meals to prescribers or engage in other marketing related activities, or require pharmaceutical companies to implement compliance programs or marketing codes of conduct.
Outside the U.S., we are subject to similar regulations in those countries where we market and sell products. In some foreign countries, including major markets in the European Union and Japan, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take nine to twelve months or longer after the receipt of regulatory marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product to other available therapies. Our business could be materially harmed if reimbursement of our products is unavailable or limited in scope or amount or if pricing is set at unsatisfactory levels.
Additionally, drug prices are under significant scrutiny, and along with other health care costs, continue to be subject to intense political and societal pressures, which we anticipate will continue and escalate, including on a global basis. As a result, our business and reputation may be harmed, our stock price may be adversely impacted and experience periods of volatility, and our results of operations may be adversely impacted.
Foreign Corrupt Practices Act
The U.S. Foreign Corrupt Practices Act (FCPA), prohibits U.S. corporations and their representatives and intermediaries from offering, promising, authorizing or making payments to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business abroad. The scope of the FCPA includes interactions with certain healthcare professionals in many countries. Other countries have enacted similar anti-corruption laws and/or regulations.
Efforts to ensure that business activities and business arrangements comply with applicable healthcare laws and regulations can be costly for manufacturers of branded prescription products. If a manufacturer’s operations, including activities conducted by its sales team, are found to be in violation of any of these laws or any other governmental regulations that apply to the company, the company may be subject to significant civil, criminal and administrative sanctions, including imprisonment, monetary penalties, damages, fines, exclusion from participation in federal healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of operations.
We intend to seek regulatory approvals for our products in additional foreign jurisdictions, but we may not obtain any such approvals.
We intend to market our products, alone or with others, in foreign jurisdictions. In order to market our products in foreign jurisdictions, we or our partners may be required to obtain separate regulatory approvals and to comply with numerous and varying regulatory requirements. The approval procedure varies among countries and jurisdictions and can involve additional trials, and the time required to obtain approval may differ from that required to obtain FDA approval. Additionally, the foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. For all of these reasons, we or our partners may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory
20
Table of Contents
authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or jurisdictions or by the FDA. We or our partners may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market. The failure to obtain these approvals could harm our business materially.
We rely on a limited number of specialty pharmacies for distribution of HETLIOZ® in the U.S., and the loss of one or more of these specialty pharmacies or their failure to distribute HETLIOZ® effectively would materially harm our business.
HETLIOZ® is only available for distribution through a limited number of specialty pharmacies in the U.S. A specialty pharmacy is a pharmacy that specializes in the dispensing of medications for complex or chronic conditions, which often require a high level of patient education and ongoing management. The use of specialty pharmacies involves certain risks, including, but not limited to, risks that these specialty pharmacies will:
| • | not provide us accurate or timely information regarding their inventories, the number of patients who are using HETLIOZ® or complaints about HETLIOZ®; |
| • | reduce their efforts or discontinue to sell or support or otherwise not effectively sell or support HETLIOZ®; |
| • | not devote the resources necessary to sell HETLIOZ® in the volumes and within the time frames that we expect; |
| • | be unable to satisfy financial obligations to us or others; or |
| • | cease operations. |
In addition, if one or more of our specialty pharmacies do not fulfill their contractual obligations to us, or refuse or fail to adequately serve patients, or their agreements are terminated without adequate notice, shipments of HETLIOZ®, and associated revenues, would be adversely affected. We expect that it would take a significant amount of time if we were required to replace one or more of our specialty pharmacies.
Our revenues from Fanapt® are substantially dependent on sales through a limited number of wholesalers, and such revenues may fluctuate from quarter to quarter.
We sell Fanapt® primarily through a limited number of pharmaceutical wholesalers in the U.S. The use of pharmaceutical wholesalers involves certain risks, including, but not limited to, risks that these pharmaceutical wholesalers will:
| • | not provide us accurate or timely information regarding their inventories, demand from wholesaler customers buying Fanapt® or complaints about Fanapt®; |
| • | reduce their efforts or discontinue to sell or support or otherwise not effectively sell or support Fanapt®; |
| • | not devote the resources necessary to sell Fanapt® in the volumes and within the time frames that we expect; |
| • | be unable to satisfy financial obligations to us or others; or |
| • | cease operations. |
Additionally, our reliance on a small number of wholesalers could cause revenues to fluctuate from quarter to quarter based on the buying patterns of these wholesalers. In addition, if any of these wholesalers fails to pay on a timely basis or at all, our business, financial condition and results of operations could be materially adversely affected.
We face substantial competition, which may result in others developing or commercializing products before or more successfully than we do.
Our future success will depend on our or our partners’ ability to demonstrate and maintain a competitive advantage with respect to our products and our ability to identify and develop additional products. Large, fully integrated pharmaceutical companies, either alone or together with collaborative partners, have substantially greater financial resources and have significantly greater experience than we do in:
| • | developing products; |
| • | undertaking pre-clinical testing and clinical trials; |
| • | obtaining FDA and other regulatory approvals of products; and |
| • | manufacturing, marketing and selling products. |
These companies may invest heavily and quickly to discover and develop novel products that could make our products obsolete. Accordingly, our competitors may succeed in obtaining patent protection, receiving FDA or foreign regulatory approval or commercializing superior products or other competing products before we do. Technological developments or the FDA or foreign regulatory approval of new therapeutic indications for existing products may make our products obsolete or may make them more difficult to market successfully, any of which could have a material adverse effect on our business, results of operations and financial condition.
21
Table of Contents
Our products, if successfully developed and approved for commercial sale, will compete with a number of drugs and therapies currently manufactured and marketed by major pharmaceutical and other biotechnology companies. Our products may also compete with new products currently under development by others or with products which may cost less than our products. Physicians, patients, third party payors and the medical community may not accept or utilize any of our products that may be approved. If HETLIOZ®, Fanapt® and our other products, if and when approved, do not achieve significant market acceptance, our business, results of operations and financial condition would be materially adversely affected. We believe the primary competitors for HETLIOZ® and Fanapt® are as follows:
| • | For HETLIOZ® in the treatment of Non-24, there are no FDA approved direct competitors. Sedative-Hypnotic treatments for certain sleep related disorders include, Ambien® (zolpidem) by Sanofi (including Ambien CR®), Lunesta® (eszopiclone) by Sunovion Pharmaceuticals Inc., Sonata® (zaleplon) by Pfizer Inc., Rozerem® (ramelteon) by Takeda Pharmaceuticals Company Limited, Silenor® (doxepin) by Pernix Therapeutics, Belsomra® (suvorexant) by Merck & Co., Inc., generic products such as zolpidem, trazodone and doxepin, and over-the-counter remedies such as Benadryl® and Tylenol PM®. The class of melatonin agonists includes Rozerem® (ramelteon) by Takeda Pharmaceuticals Company Limited, Valdoxan® (agemelatine) by Servier, Circadin® (long-acting melatonin) by Neurim Pharmaceuticals and the food supplement melatonin. Shift work and excessive sleepiness disorder treatments include Nuvigil® (armodafinil) and Provigil® (modafinil) both by Teva Pharmaceutical Industries Ltd. |
| • | For Fanapt® in the treatment of schizophrenia, the atypical antipsychotics competitors are Risperdal® (risperidone), including the depot formulation Risperdal® Consta® and Invega® (paliperidone), including the depot formulation Invega® Sustenna®, each by Ortho-McNeil-Janssen Pharmaceuticals, Inc., Zyprexa® (olanzapine), including the depot formulation Zyprexa® Relprevv™, each by Eli Lilly and Company, Seroquel® and Seroquel XR (quetiapine) by AstraZeneca PLC, Abilify® (aripiprazole) by BMS/Otsuka America Pharmaceutical Inc., Abilify® Maintena® (the depot formulation of Abilify®) by Lundbeck/Otsuka America Pharmaceutical Inc., Geodon® (ziprasidone) by Pfizer Inc., Saphris® (asenapine) by Actavis plc, Latuda® (lurasidone) by Sunovion Pharmaceuticals Inc., Rexulti® (brexpiprazole) by Lundbeck/Otsuka America Pharmaceutical, Inc., Aristada™ (aripiprazole lauroxil) extended-release injectible suspension by Alkermes, Inc., Vraylar™ (cariprazine) by Teva Pharmaceutical Industries Ltd., and generic clozapine, as well as the typical antipsychotics haloperidol, chlorpromazine, thioridazine, and sulpiride (all of which are generic). |
Additionally, we may face competition from newly developed generic products. Under the Hatch-Waxman Act newly approved drugs and indications may benefit from a statutory period of non-patent marketing exclusivity. The Hatch-Waxman Act seeks to stimulate competition by providing incentives to generic pharmaceutical manufacturers to introduce non-infringing forms of patented pharmaceutical products and to challenge patents on branded pharmaceutical products. If we are unsuccessful at challenging an Abbreviated New Drug Application (ANDA), filed pursuant to the Hatch-Waxman Act, cheaper generic versions of our products, which may be favored by insurers and third-party payors, may be launched commercially, which would significantly harm our business.
In June 2014, we filed suit against Roxane Laboratories, Inc. (Roxane) in the U.S. District Court for the District of Delaware (the Delaware District Court). The suit seeks an adjudication that Roxane has infringed one or more claims of our U.S. Patent No. 8,586,610 (the ‘610 Patent) by submitting to the U.S. Food and Drug Administration (the FDA) an ANDA for a generic version of Fanapt® prior to the expiration of the ‘610 Patent in November 2027. In addition, pursuant to the Settlement Agreement with Novartis, we assumed Novartis’ patent infringement action against Roxane in the Delaware District Court. That suit alleges that Roxane has infringed one or more claims of U.S Patent RE39198 (the ‘198 Patent), which is licensed exclusively to us, by filing an ANDA for a generic version of Fanapt® prior to the expiration of the ‘198 Patent in November 2016. These two cases against Roxane were consolidated by agreement of the parties and were tried together in a five-day bench trial that concluded on March 4, 2016. On August 25, 2016, the Delaware District Court ruled in our favor, finding that Roxane’s ANDA product infringed the asserted claims of the ‘610 Patent and the ‘198 Patent. The Delaware District Court ruled that we are entitled to a permanent injunction against Roxane enjoining Roxane from infringing the ‘610 Patent, including the manufacture, use, sale, offer to sell, sale, distribution or importation of any generic iloperidone product described in the ‘610 Patent ANDA until the expiration of the ‘610 Patent in November 2027. If we obtain pediatric exclusivity, the injunction against Roxane would be extended until May 2028 under the Delaware District Court’s order. On September 23, 2016, Roxane filed a notice of appeal with the Federal Circuit Court of Appeals. Roxane filed its opening appellate brief on February 7, 2017.
In 2015, we filed six separate patent infringement lawsuits in the Delaware District Court against Roxane, Inventia Healthcare Pvt. Ltd. (Inventia), Lupin Ltd. and Lupin Pharmaceuticals, Inc. (Lupin), Taro Pharmaceuticals USA, Inc. and Taro Pharmaceutical Industries, Ltd. (Taro), and Apotex Inc. and Apotex Corp., (collectively, the Defendants). The lawsuits each seek an adjudication that the respective Defendants infringed one or more claims of the ‘610 Patent and/or our U.S. Patent No. 9,138,432 (the ‘432 Patent) by submitting to the FDA an ANDA for a generic version of Fanapt® prior to the expiration of the ‘610 Patent in November 2027 or the ‘432 Patent in September 2025. The Defendants have denied infringement and counterclaimed for declaratory judgment of invalidity
22
Table of Contents
and noninfringement of the ‘610 Patent and the ‘432 Patent. A trial on these matters was scheduled to begin on May 15, 2017. On December 15, 2016, following a submission by the parties that it may be appropriate to remove the May 15, 2017 trial from the Delaware District Court’s calendar pending the Federal Circuit’s decision in Roxane’s appeal of the Delaware District Court’s August 25, 2016 ruling finding that Roxane’s proposed products infringed the asserted claims of the ‘610 Patent, the Delaware District Court removed the May 15, 2017 trial from the calendar and adopted a new schedule for discovery. The parties agreed that within 14 days after any decision on the merits in the Roxane appeal, the parties will submit to the Delaware District Court a status report and request a schedule for trial. We entered into a confidential stipulation with Inventia regarding any potential launch of Inventia’s generic ANDA product. We also entered into a confidential stipulation with Lupin regarding any potential launch of Lupin’s generic ANDA product.
Lupin filed counter claims for declaratory judgment of invalidity and noninfringement of seven of our method of treatment patents that are listed in the Approved Drug Products with Therapeutic Equivalence Evaluations (the Orange Book) related to Fanapt® (such seven patents, the Method of Treatment Patents). We have not sued Lupin for infringing the Method of Treatment Patents. On October 13, 2016, we, along with Lupin, filed a Stipulation of Dismissal in the Delaware District Court pursuant to which Lupin’s counterclaims relating to the Method of Treatment Patents were dismissed without prejudice in recognition of an agreement reached between Lupin and us by which we would not assert those patents against Lupin absent certain changes in Lupin’s proposed prescribing information for its iloperidone tablets.
On October 24, 2016, we entered into a License Agreement with Taro to resolve our patent litigation against Taro regarding Taro’s ANDA seeking approval of its generic version of Fanapt® (the Taro License Agreement). Under the Taro License Agreement, we granted Taro a non-exclusive license to manufacture and commercialize Taro’s version of Fanapt® in the U.S. effective November 2, 2027, unless prior to that date we obtain pediatric exclusivity for Fanapt®, in which case, the license will be effective May 2, 2028. Taro may enter the market earlier under certain limited circumstances. The Taro License Agreement, which is subject to review by the U.S. Federal Trade Commission (FTC) and the U.S. Department of Justice (DOJ), provides for a full settlement and release by us and Taro of all claims that are the subject of the litigation.
On December 7, 2016, we entered into a License Agreement with Apotex to resolve our patent litigation against Apotex regarding Apotex’s ANDA seeking approval of its generic version of Fanapt® (the Apotex License Agreement). Under the Apotex License Agreement, we granted Apotex a non-exclusive license to manufacture and commercialize Apotex’s version of Fanapt® in the U.S. effective November 2, 2027, unless prior to that date we obtain pediatric exclusivity for Fanapt®, in which case, the license will be effective May 2, 2028. Apotex may enter the market earlier under certain limited circumstances. The Apotex License Agreement, which is subject to review by the FTC and the DOJ, provides for a full settlement and release by us and Apotex of all claims that are the subject of the litigation.
On February 26, 2016, Roxane filed suit against us in the U.S. District Court for the Southern District of Ohio (Ohio District Court). The suit seeks a declaratory judgment of invalidity and noninfringement of the Method of Treatment Patents. We have not sued Roxane for infringing the Method of Treatment Patents. We filed a motion to dismiss this lawsuit for lack of personal jurisdiction or to transfer the lawsuit to the Delaware District Court. On December 20, 2016, the Ohio District Court ruled in our favor, dismissing Roxane’s suit without prejudice for lack of personal jurisdiction.
On February 26, 2016, Roxane filed a Petition for Inter Partes Review (IPR) of the ‘432 Patent with the Patent Trials and Appeals Board (PTAB) of the U.S. Patent and Trademark Office. We filed a Preliminary Response on June 7, 2016, and on August 30, 2016, the PTAB denied the request by Roxane to institute an IPR of the ‘432 Patent. On September 29, 2016, Roxane filed a Petition for Rehearing with the PTAB, and on October 13, 2016, we filed a Response to Roxane’s Petition. On November 4, 2016, the PTAB denied Roxane’s Petition for Rehearing.
FDA and foreign regulatory approval of our products is uncertain.
The research, testing, manufacturing and marketing of products such as those that we have developed or that we or our partners are developing are subject to extensive regulation by federal, state and local government authorities, including the FDA, as well as foreign regulatory authorities in jurisdictions in which we seek approval. To obtain regulatory approval of such products, we or our partners must demonstrate to the satisfaction of the applicable regulatory agency that, among other things, the product is safe and effective for its intended use. In addition, we or our partners must show that the manufacturing facilities used to produce such products are in compliance with current Good Manufacturing Practices regulations (cGMP).
The process of obtaining FDA and other required regulatory approvals and clearances can take many years and will require us and our partners, as applicable, to expend substantial time and capital. Despite the time and expense expended, regulatory approval is never guaranteed. The number of pre-clinical and clinical trials that will be required for FDA or foreign regulatory approval varies depending on the product, the disease or condition that the product is in development for, and the requirements applicable to that particular product. The FDA or applicable foreign regulatory agency can delay, limit or deny approval of a product for many reasons, including that:
| • | a product may not be shown to be safe or effective; |
23
Table of Contents
| • | the FDA or foreign agency may interpret data from pre-clinical and clinical trials in different ways than we or our partners do; |
| • | the FDA or foreign agency may not approve our or our partners’ manufacturing processes or facilities; |
| • | a product may not be approved for all the indications we or our partners request; |
| • | the FDA or foreign agency may change its approval policies or adopt new regulations; |
| • | the FDA or foreign agency may not meet, or may extend, the Prescription Drug User Fee Act (PDUFA-V) date or its foreign equivalent with respect to a particular NDA or foreign application; and |
| • | the FDA or foreign agency may not agree with our or our partners’ regulatory approval strategies or components of the regulatory filings, such as clinical trial designs. |
For example, if certain of our or our partners’ methods for analyzing trial data are not accepted by the FDA or the applicable foreign agency, we or our partners may fail to obtain regulatory approval for our products.
Any delay or failure to obtain regulatory approvals for our products will result in increased costs, could diminish competitive advantages that we may attain and would adversely affect the marketing and sale of our products. Other than HETLIOZ® in the U.S. and the 31 countries in Europe covered by the centralized marketing authorization by the EC, and Fanapt® in the U.S., Mexico and Israel, we have not received regulatory approval to market any of our products in any jurisdiction.
Even following regulatory approval of our products, the FDA or the applicable foreign agency may impose limitations on the indicated uses for which such products may be marketed, subsequently withdraw approval or take other actions against us, our partners or such products that are adverse to our business. The FDA and foreign agencies generally approve drugs for particular indications. An approval for a more limited indication reduces the size of the potential market for the product. Product approvals, once granted, may be withdrawn or modified if problems occur after initial marketing.
We and our partners also are subject to numerous federal, state, local and foreign laws, regulations and recommendations relating to safe working conditions, laboratory and manufacturing practices, the environment and the use and disposal of hazardous substances used in connection with discovery, research and development work. In addition, we cannot predict the extent to which new governmental regulations might significantly impede the discovery, development, production and marketing of our products. We or our partners may be required to incur significant costs to comply with current or future laws or regulations, and we may be adversely affected by the cost of such compliance or the inability to comply with such laws or regulations.
If our products are determined to be unsafe or ineffective in humans, whether commercially or in clinical trials, our business will be materially harmed.
Despite the FDA’s approval of the NDA for HETLIOZ® in January 2014 and the NDA for Fanapt® in May 2009, the EC’s grant of the centralized marketing authorization for HETLIOZ® in July 2015, and the positive results of our completed trials for HETLIOZ® and Fanapt®, we are uncertain whether either of these products will ultimately prove to be effective and safe in humans long term and in all uses. Frequently, products that have shown promising results in clinical trials have suffered significant setbacks in later clinical trials or even after they are approved for commercial sale. Future uses of our products, whether in clinical trials or commercially, may reveal that the product is ineffective, unacceptably toxic, has other undesirable side effects, is difficult to manufacture on a large scale, is uneconomical, infringes on proprietary rights of another party or is otherwise not fit for further use. If our products are determined to be unsafe or ineffective in humans, our business will be materially harmed.
Clinical trials for our products are expensive and their outcomes are uncertain. Any failure or delay in completing clinical trials for our products could severely harm our business.
Pre-clinical studies and clinical trials required to demonstrate the safety and efficacy of our products are time-consuming and expensive and together take several years to complete. Before obtaining regulatory approvals for the commercial sale of any of our products, we or our partners must demonstrate through preclinical testing and clinical trials that such product is safe and effective for use in humans. We have incurred, and we will continue to incur, substantial expense for, and devote a significant amount of time to, preclinical testing and clinical trials.
Historically, the results from preclinical testing and early clinical trials often have not predicted results of later clinical trials. A number of new drugs have shown promising results in clinical trials, but subsequently failed to establish sufficient safety and efficacy data to obtain necessary regulatory approvals. Clinical trials conducted by us, by our partners or by third parties on our or our partners’ behalf may not demonstrate sufficient safety and efficacy to obtain the requisite regulatory approvals for our products. Regulatory authorities may not permit us or our partners to undertake any additional clinical trials for our products, may force us to stop any ongoing clinical trials and it may be difficult to design efficacy studies for our products in new indications.
Clinical development efforts performed by us or our partners may not be successfully completed. Completion of clinical trials may take several years or more. The length of time can vary substantially with the type, complexity, novelty and intended use of the products and the size of the prospective patient population. The commencement and rate of completion of clinical trials for our products may be delayed by many factors, including:
| • | the inability to manufacture or obtain from third parties materials sufficient for use in pre-clinical studies and clinical trials; |
| • | delays in beginning a clinical trial; |
24
Table of Contents
| • | delays in patient enrollment and variability in the number and types of patients available for clinical trials; |
| • | difficulty in maintaining contact with patients after treatment, resulting in incomplete data; |
| • | poor effectiveness of our products during clinical trials; |
| • | unforeseen safety issues or side effects; and |
| • | governmental or regulatory delays and changes in regulatory requirements and guidelines. |
If we or our partners fail to complete successfully one or more clinical trials for our products, we or they may not receive the regulatory approvals needed to market that product. Therefore, any failure or delay in commencing or completing these clinical trials would harm our business materially.
Our products may cause undesirable side effects or have other properties that could delay, prevent or result in the revocation of their regulatory approval or limit their marketability.
Undesirable side effects caused by our products could interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by the FDA or other regulatory authorities for any or all targeted indications, and in turn prevent us or our partners from commercializing or continuing the commercialization of such products and generating revenues from their sale. We will continue to assess the side effect profile of our products in ongoing clinical development programs. However, we cannot predict whether the commercial use of our approved products (or our products in development, if and when they are approved for commercial use) will produce undesirable or unintended side effects that have not been evident in the use of, or in clinical trials conducted for, such products to date. Additionally, incidents of product misuse may occur. These events, among others, could result in product recalls, product liability actions or withdrawals or additional regulatory controls, all of which could have a material adverse effect on our business, results of operations and financial condition.
In addition, if after receiving marketing approval of a product, we, our partners or others identify undesirable side effects caused by such product, we or our partners could face one or more of the following:
| • | regulatory authorities may require the addition of labeling statements, such as a “black box” warning or a contraindication; |
| • | regulatory authorities may withdraw their approval of the product; |
| • | we or our partners may be required to change the way the product is administered, conduct additional clinical trials or change the labeling of the product; and |
| • | our, our partner’s or the product’s reputation may suffer. |
Any of these events could prevent us or our partners from achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing the product, which in turn could delay or prevent us from generating significant revenues from its sale.
We have a history of operating losses, anticipate future losses and may never become profitable on a sustained basis.
We have been engaged in identifying and developing products since March 2003, which has required, and will continue to require, significant research and development expenditures. The continued commercialization of HETLIOZ® and Fanapt® will require substantial additional expenditures.
As of December 31, 2016, we had an accumulated deficit of $345.9 million and we cannot estimate with precision the extent of our future losses. In April 2014, we commercially launched HETLIOZ® in the U.S. for the treatment of Non-24 and in August 2016 we commercially launched HETLIOZ® in Germany for the treatment of Non-24. We are currently evaluating the commercial opportunity for HETLIOZ® in the rest of Europe. In the fourth quarter of 2014, we acquired all further rights to Fanapt® from Novartis. The continued commercialization of HETLIOZ® and Fanapt® will require substantial additional expenditures. In addition, we may not succeed in commercializing HETLIOZ®, Fanapt® or any other products. Novartis launched Fanapt® in the U.S. in the first quarter of 2010 and we began selling Fanapt® on our own in the first quarter of 2015. We may not succeed in gaining additional market acceptance of Fanapt® in the U.S. and we may not succeed in commercializing HETLIOZ® or Fanapt® outside of the U.S. We may not be profitable even if our products are successfully commercialized. We may be unable to fully develop, obtain regulatory approval for, commercialize, manufacture, market, sell and derive revenue from our products in the timeframes we project, if at all, and our inability to do so would materially and adversely impact the market price of our common stock and our ability to raise capital and continue operations.
25
Table of Contents
There can be no assurance that we will achieve sustained profitability. Our ability to achieve sustained profitability in the future depends, in part, upon:
| • | our ability to obtain and maintain regulatory approval for our products, particularly HETLIOZ® for the treatment of Non-24, both in the U.S. and in foreign countries; |
| • | our ability to successfully commercialize HETLIOZ® in the U.S., Europe and other jurisdictions in which HETLIOZ® may receive regulatory approval, if any; |
| • | our ability to successfully raise awareness regarding Non-24 in the medical and patient communities; |
| • | our ability to successfully market and sell Fanapt® in the U.S. and our or our partners’ ability to successfully market and sell Fanapt® in Israel, Mexico and other jurisdictions in which we may receive regulatory approval, if any; |
| • | our ability to enter into and maintain agreements to develop and commercialize our products; |
| • | our and our partners’ ability to develop, have manufactured and market our products; |
| • | our and our partners’ ability to obtain adequate reimbursement coverage for our products from insurance companies, government programs and other third party payors; and |
| • | our ability to obtain additional research and development funding from collaborative partners or funding for our products. |
In addition, the amount we spend will impact our profitability. Our spending will depend, in part, upon:
| • | the costs of our marketing or awareness campaigns; |
| • | the progress of our research and development programs for our products, including clinical trials; |
| • | the time and expense that will be required to pursue FDA and/or foreign regulatory approvals for our products and whether such approvals are obtained on a timely basis, if at all; |
| • | the time and expense required to prosecute, enforce and/or challenge patent and other intellectual property rights; |
| • | the cost of operating and maintaining development and research facilities; |
| • | the cost of third party manufacturers; |
| • | the number of additional products we pursue; |
| • | how competing technological and market developments affect our products; |
| • | the cost of possible acquisitions of technologies, products, product rights or companies; |
| • | the cost of obtaining licenses to use technology owned by others for proprietary products and otherwise; |
| • | the costs and effects of potential litigation; and |
| • | the costs associated with recruiting and compensating a highly skilled workforce in an environment where competition for such employees may be intense. |
We may not achieve all or any of these goals and, thus, we cannot provide assurances that we will ever be profitable on a sustained basis or achieve significant revenues. Even if we do achieve some or all of these goals, we may not achieve significant or sustained commercial success.
Our ability to use net operating loss carryforwards and tax credit carryforwards to offset future taxable income may be limited as a result of transactions involving our common stock.
In general, under Section 382 of the Internal Revenue Code of 1986, as amended (IRC), a corporation that undergoes an “ownership change” is subject to limitations on its ability to utilize its pre-change net operating losses (NOLs) and certain other tax assets (tax attributes) to offset future taxable income. In general, an ownership change occurs if the aggregate stock ownership of certain stockholders increases by more than 50 percentage points over such stockholders’ lowest percentage ownership during the testing period (generally three years). Transactions involving our common stock, even those outside our control, such as purchases or sales by investors, within the testing period could result in an ownership change. A limitation on our ability to utilize some or all of our NOLs or credits could have a material adverse effect on our results of operations and cash flows. Ownership changes did occur as of December 31, 2008 and December 31, 2014. Our management determined that there was sufficient Built-In-Gain as of December 31, 2008 to offset the IRC Section 382 limitation generated by the ownership change. Our management believes that there was sufficient Built-In-Gain as of December 31, 2014 to offset the IRC Section 382 limitation generated by the ownership change. Any future ownership changes may cause our existing tax attributes to have additional limitations.
26
Table of Contents
If we fail to obtain the capital necessary to fund our research and development activities and commercialization efforts, we may be unable to continue operations or we may be forced to share our rights to commercialize our products with third parties on terms that may not be attractive to us.
Our activities will necessitate significant uses of working capital throughout 2017 and beyond. It is uncertain whether our existing funds will be sufficient to meet our operating needs. As of December 31, 2016, our total cash and cash equivalents and marketable securities were $141.3 million. Our long term capital requirements are expected to depend on many factors, including, among others:
| • | our ability to successfully commercialize HETLIOZ® and Fanapt® globally; |
| • | outcomes of ongoing and potential patent litigation; |
| • | costs of developing and maintaining sales, marketing and distribution channels and our ability to sell our products; |
| • | market acceptance of our products; |
| • | costs involved in establishing manufacturing capabilities for commercial quantities of our products; |
| • | the number of potential formulations and products in development; |
| • | progress with pre-clinical studies and clinical trials; |
| • | time and costs involved in obtaining regulatory (including FDA) approval; |
| • | costs involved in preparing, filing, prosecuting, maintaining and enforcing patent, trademark and other intellectual property claims; |
| • | competing technological and market developments; |
| • | costs for recruiting and retaining employees and consultants; |
| • | costs for training physicians; and |
| • | legal, accounting, insurance and other professional and business related costs. |
As a result, we may need to raise additional capital to fund our anticipated operating expenses and execute on our business plans. In our capital-raising efforts, we may seek to sell debt securities or additional equity securities, obtain a bank credit facility, or enter into partnerships or other collaboration agreements. The sale of additional equity or debt securities, if convertible, could result in dilution to our stockholders and may also result in a lower price for our common stock. The incurrence of indebtedness would result in increased fixed obligations and could also result in covenants that could restrict our operations. However, we may not be able to raise additional funds on acceptable terms, or at all. If we are unable to secure sufficient capital to fund our planned activities, we may not be able to continue operations, or we may have to enter into partnerships or other collaboration agreements that could require us to share commercial rights to our products to a greater extent or at earlier stages in the drug development process than is currently intended. These partnerships or collaborations, if consummated prior to proof-of-efficacy or safety of a given product, could impair our ability to realize value from that product. If additional financing is not available when required or is not available on acceptable terms, we may be unable to fund our operations and planned growth, develop or enhance our technologies or products, take advantage of business opportunities or respond to competitive market pressures, any of which would materially harm our business, financial condition and results of operations.
If our contract research organizations do not successfully carry out their duties or if we lose our relationships with contract research organizations, our drug development efforts could be delayed.
Our arrangements with contract research organizations are critical to our success in bringing our products to the market and promoting such marketed products profitably. We are dependent on contract research organizations, third-party vendors and investigators for pre-clinical testing and clinical trials related to our drug discovery and development efforts and we will likely continue to depend on them to assist in our future discovery and development efforts. These parties are not our employees and we cannot control the amount or timing of resources that they devote to our programs. As such, they may not complete activities on schedule or may not conduct our clinical trials in accordance with regulatory requirements or our stated protocols. The parties with which we contract for execution of our clinical trials play a significant role in the conduct of the trials and the subsequent collection and analysis of data. If they fail to devote sufficient time and resources to our drug development programs or if their performance is substandard, it will delay the development, approval and commercialization of our products. Moreover, these parties may also have relationships with other commercial entities, some of which may compete with us. If they assist our competitors, it could harm our competitive position.
Our contract research organizations could merge with or be acquired by other companies or experience financial or other setbacks unrelated to our collaboration that could, nevertheless, materially adversely affect our business, results of operations and financial condition.
If we lose our relationship with any one or more of these parties, we could experience a significant delay in both identifying another comparable provider and then contracting for its services. We may be unable to retain an alternative provider on reasonable terms, if at all. Even if we locate an alternative provider, it is likely that this provider may need additional time to respond to our needs and may not provide the same type or level of service as the original provider. In addition, any provider that we retain will be subject to current Good Laboratory Practices, and similar foreign standards and we do not have control over compliance with these regulations by these providers. Consequently, if these practices and standards are not adhered to by these providers, the development and commercialization of our products could be delayed.
27
Table of Contents
We rely on a limited number of third party manufacturers to formulate and manufacture our products and our business will be seriously harmed if these manufacturers are not able to satisfy our demand and alternative sources are not available.
Our expertise is primarily in the research and development and pre-clinical and clinical trial phases of product development. We do not have an in-house manufacturing capability and depend completely on a small number of third-party manufacturers and active pharmaceutical ingredient formulators for the manufacture of our products. Therefore, we are dependent on third parties for our formulation development and manufacturing of our products. This may expose us to the risk of not being able to directly oversee the production and quality of the manufacturing process and provide ample commercial supplies to successfully launch and maintain the marketing of our products. Furthermore, these third party contractors, whether foreign or domestic, may experience regulatory compliance difficulty, mechanical shut downs, employee strikes, or other unforeseeable events that may delay or limit production. Our inability to adequately establish, supervise and conduct (either ourselves or through third parties) all aspects of the formulation and manufacturing processes would have a material adverse effect on our ability to develop and commercialize our products.
In January 2014, we entered into a manufacturing agreement with Patheon Pharmaceuticals Inc. (Patheon) for the manufacture of commercial supplies of HETLIOZ® 20 mg capsules. In May 2016, we entered into a manufacturing agreement with Patheon for the manufacture of commercial supplies of Fanapt® capsules. We do not have exclusive long-term agreements with any other third party manufacturers of our products. If Patheon, or any other third party manufacturer, is unable or unwilling to perform its obligations under our manufacturing agreements for any reason, we may not be able to locate alternative acceptable manufacturers or formulators or enter into favorable agreements with them. Any inability to acquire sufficient quantities of our products in a timely manner from these third parties could adversely affect sales of our products, delay clinical trials and prevent us from developing our products in a cost-effective manner or on a timely basis. In addition, manufacturers of our products are subject to cGMP and similar foreign standards and we do not have control over compliance with these regulations by our manufacturers. If one of our contract manufacturers fails to maintain compliance, the production of our products could be interrupted, resulting in delays and additional costs. In addition, if the facilities of such manufacturers do not pass a pre-approval or post-approval plant inspection, the FDA will not grant approval and may institute restrictions on the marketing or sale of our products.
Our manufacturing strategy presents the following additional risks:
| • | because most of our third-party manufacturers and formulators are located outside of the U.S., there may be difficulties in importing our products or their components into the U.S. as a result of, among other things, FDA import inspections, incomplete or inaccurate import documentation or defective packaging; and |
| • | because of the complex nature of our products, our manufacturers may not be able to successfully manufacture our products in a cost-effective and/or timely manner. |
Materials necessary to manufacture our products may not be available on commercially reasonable terms, or at all, which may delay the development, regulatory approval and commercialization of our products.
We and our partners rely on manufacturers to purchase from third-party suppliers the materials necessary to produce our products for clinical trials and commercialization. Suppliers may not sell these materials to such manufacturers at the times we or our partners need them or on commercially reasonable terms. We do not have any control over the process or timing of the acquisition of these materials by these manufacturers. Moreover, we currently do not have any agreements for the commercial production of these materials. If the manufacturers are unable to obtain these materials for our or our partners’ clinical trials, product testing, potential regulatory approval of our products and commercial scale manufacturing could be delayed, significantly affecting our and our partners’ ability to further develop and commercialize our products. If we, our manufacturers or our partners, as applicable, are unable to purchase these materials for our products, there would be a shortage in supply or the commercial launch of such products would be delayed, which would materially and adversely affect our or our partners’ ability to generate revenues from the sale of such products.
If we cannot identify, or enter into licensing arrangements for, new products, our ability to develop a diverse product portfolio will be limited.
A component of our business strategy is acquiring rights to develop and commercialize products discovered or developed by other pharmaceutical and biotechnology companies for which we may find effective uses and markets through our unique pharmacogenetics and pharmacogenomics expertise for the treatment of central nervous system disorders. Competition for the acquisition of these products is intense. If we are not able to identify opportunities to acquire rights to commercialize additional products, we may not be able to develop a diverse portfolio of products and our business may be harmed. Additionally, it may take substantial human and financial resources to secure commercial rights to promising products. Moreover, if other firms develop pharmacogenetics and pharmacogenomics capabilities, we may face increased competition in identifying and acquiring additional products.
We may not be successful in the development of products for our own account.
In addition to our business strategy of acquiring rights to develop and commercialize products, we may develop products for our own account by applying our technologies to off-patent drugs as well as developing our own proprietary molecules. Because we will be funding the development of such programs, there is a risk that we may not be able to continue to fund all such programs to
28
Table of Contents
completion or to provide the support necessary to perform the clinical trials, obtain regulatory approvals or market any approved products. We expect the development of products for our own account to consume substantial resources. If we are able to develop commercial products on our own, the risks associated with these programs may be greater than those associated with our programs with collaborative partners.
If we lose key scientists or management personnel, or if we fail to recruit additional highly skilled personnel, it will impair our ability to identify, develop and commercialize products.
We are highly dependent on principal members of our management team and scientific staff, including our Chief Executive Officer, Mihael H. Polymeropoulos, M.D. These executives each have significant pharmaceutical industry experience. The loss of any such executives, including Dr. Polymeropoulos, or any other principal member of our management team or scientific staff, would impair our ability to identify, develop and market new products. Our management and other employees may voluntarily terminate their employment with us at any time. The loss of the services of these or other key personnel, or the inability to attract and retain additional qualified personnel, could result in delays to development or approval, loss of sales and diversion of management resources. In addition, we depend on our ability to attract and retain other highly skilled personnel, including research scientists. Competition for qualified personnel is intense, and the process of hiring and integrating such qualified personnel is often lengthy. We may be unable to recruit such personnel on a timely basis, if at all, which would negatively impact our development and commercialization programs.
Additionally, we do not currently maintain “key person” life insurance on the lives of our executives or any of our employees. This lack of insurance means that we may not have adequate compensation for the loss of the services of these individuals.
Product liability lawsuits could divert our resources, result in substantial liabilities and reduce the commercial potential of our products.
The risk that we may be sued on product liability claims is inherent in the development and sale of pharmaceutical products. For example, we face a risk of product liability exposure related to the testing of our products in clinical trials and will face even greater risks upon commercialization by us or our partners of our products. We believe that we may be at a greater risk of product liability claims relative to other pharmaceutical companies because our products are intended to treat central nervous system disorders, and it is possible that we may be held liable for the behavior and actions of patients who use our products. These lawsuits may divert our management from pursuing our business strategy and may be costly to defend. In addition, if we are held liable in any of these lawsuits, we may incur substantial liabilities and we or our partners may be forced to limit or forego further commercialization of one or more of our products. Although we maintain product liability insurance, our aggregate coverage limit under this insurance is $25.0 million, and while we believe this amount of insurance is sufficient to cover our product liability exposure, these limits may not be high enough to fully cover potential liabilities. As our development activities and commercialization efforts progress and we and our partners sell our products, this coverage may be inadequate, we may be unable to obtain adequate coverage at an acceptable cost or we may be unable to get adequate coverage at all or our insurer may disclaim coverage as to a future claim. This could prevent the commercialization or limit the commercial potential of our products. Even if we are able to maintain insurance that we believe is adequate, our results of operations and financial condition may be materially adversely affected by a product liability claim. Uncertainties resulting from the initiation and continuation of products liability litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Product liability litigation and other related proceedings may also require significant management time.
European Union Member States tend to impose strict price controls, which may delay or prevent the further commercial launch or impede the commercial success of HETLIOZ® in Europe and adversely affect our future results of operations.
In the European Union, prescription drug pricing and reimbursement are subject to governmental control and reimbursement mechanisms used by private and public health insurers in the European Union vary by Member State. For the public systems, reimbursement is determined by guidelines established by the legislature or responsible national authority. As elsewhere, inclusion in reimbursement catalogues focuses on the medical usefulness, need, quality and economic benefits to patients and the health care system. Acceptance for reimbursement comes with cost, use and often volume restrictions, which can vary by Member State. Although we have received marketing authorization for HETLIOZ® from the EC, pricing negotiations with governmental authorities may take a considerable amount of time in those Member States that impose price controls. For example, we launched HETLIOZ® commercially in Germany in August 2016, but we have not yet concluded our pricing negotiations with German authorities. In addition, to obtain reimbursement or pricing approval for HETLIOZ® in some Member States, we may be required to conduct a clinical trial that compares the cost-effectiveness of HETLIOZ®, to other available therapies.
Some Member States require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some Member States, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we may be subject to lengthy price regulations that delay or prevent the commercial launch of HETLIOZ® in a particular Member State and negatively impact the revenues that are generated from the sale of HETLIOZ® in that country. If reimbursement of HETLIOZ® is unavailable or limited in scope or amount, or if pricing for HETLIOZ® is set at unsatisfactory levels or takes too long to establish, or if there is competition from lower priced cross-border sales, our results of operations will be negatively affected.
29
Table of Contents
We may not be able to effectively market and sell our future products, if approved, in the U.S.
We plan on building our sales and marketing capabilities in the U.S. to commercialize future products, if approved. We expect to build such capabilities by investing significant amounts of financial and management resources. Furthermore, the cost of establishing and maintaining marketing and sales capabilities may not be justifiable in light of the revenues generated by any future products.
If we are unable to establish and maintain adequate sales and marketing capabilities for future products or are unable to do so in a timely manner, we may not be able to generate product revenues from these products which may prevent us from reaching or maintaining profitability.
Legislative or regulatory reform of the healthcare system in the U.S. may affect our ability to sell our products profitably.
PPACA substantially changes the way healthcare is financed by both governmental and private insurers, and significantly impacts the pharmaceutical industry. PPACA contains a number of provisions that are expected to impact our business and operations, in some cases in ways we cannot currently predict. Changes that may affect our business if we or our partners commercialize our products in the future include those governing enrollment in federal healthcare programs, reimbursement changes, rules regarding prescription drug benefits under the health insurance exchanges, and fraud and abuse and enforcement. In addition, continued implementation of PPACA may result in the expansion of new programs such as Medicare payment for performance initiatives, and may impact existing government healthcare programs, such as by improving the physician quality reporting system and feedback program.
Additional provisions of PPACA may negatively affect our revenues from products that we or our partners commercialize in the future. For example, as part of PPACA’s provisions closing a coverage gap that currently exists in the Medicare Part D prescription drug program, manufacturers of branded prescription drugs are required to provide a 50% discount on branded prescription drugs dispensed to beneficiaries within this coverage gap. Medicare Part D is a prescription drug benefit available to all Medicare beneficiaries. It is a voluntary benefit that is implemented through private plans under contractual arrangements with the federal government. Similar to pharmaceutical coverage through private health insurance, Part D plans negotiate discounts from drug manufacturers and pass on some of those savings to Medicare beneficiaries. PPACA also makes changes to the Medicaid Drug Rebate Program, discussed in more detail below, including increasing the minimum rebate from 15.1% to 23.1% of the average manufacturer price for most innovator products. On February 1, 2016, CMS, the federal agency that administers the Medicare and Medicaid programs, issued final regulations to implement the changes to the Medicaid Drug Rebate Program under PPACA. These regulations became effective on April 1, 2016.
Many of PPACA’s most significant reforms did not take effect until 2014 or thereafter, and the resulting new programs and requirements will continue to evolve in the next few years. Some states have chosen not to expand their Medicaid programs by raising the income limit to 133% of the federal poverty level. In part because not all states have expanded their Medicaid programs, it is unclear whether there will be more uninsured patients than anticipated when Congress passed PPACA. For each state that has opted not to expand its Medicaid program, there will be fewer insured patients overall. An increase in the proportion of uninsured patients who are prescribed products resulting from our proprietary or partnered programs could impact the future sales of any products that are commercialized in the future and our business and results of operations.
Further, in September 2007, the Food and Drug Administration Amendments Act of 2007 was enacted giving the FDA enhanced post-marketing authority including the authority to require post-marketing studies and clinical trials, labeling changes based on new safety information and compliance with REMS approved by the FDA. The FDA’s exercise of this authority could result in delays or increased costs during product development, clinical trials and regulatory review, increased costs to ensure compliance with post-approval regulatory requirements and potential restrictions on the sale and/or distribution of approved products.
In addition, other legislative changes have been proposed and adopted in the U.S. since PPACA was enacted. On August 2, 2011, the Budget Control Act of 2011 among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013, which will remain in effect until 2024 unless additional congressional action is taken. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, increased the statute of limitations period for the government to recover overpayments to providers from three to five years. We expect that additional federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, and in turn could significantly reduce the projected value of certain development projects and reduce our profitability.
More recently, the new presidential administration and the U.S. Congress have indicated that they may seek to replace PPACA and related legislation with new healthcare legislation. There is uncertainty with respect to the impact these potential changes may have, if any, and any changes will likely take time to unfold, and could have an impact on coverage and reimbursement for healthcare items and services covered by plans that were authorized by PPACA. However, we cannot predict the ultimate content, timing or effect of any healthcare reform legislation or the impact of potential legislation on us.
30
Table of Contents
We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our products once approved or additional pricing pressures, and may adversely affect our operating results.
Future transactions may harm our business or the market price of our stock.
We regularly review potential transactions related to technologies, products or product rights and businesses complementary to our business. These transactions could include:
| • | mergers; |
| • | acquisitions; |
| • | strategic alliances; |
| • | licensing agreements; and |
| • | co-promotion and similar agreements. |
We may choose to enter into one or more of these transactions at any time, which may cause substantial fluctuations in the market price of our stock. Moreover, depending upon the nature of any transaction, we may experience a charge to earnings, which could also materially adversely affect our results of operations and could harm the market price of our stock.
We may undertake strategic acquisitions in the future, and difficulties integrating such acquisitions could damage our ability to achieve or sustain profitability.
Although we have no experience in acquiring businesses, we may acquire businesses or assets that complement or augment our existing business. If we acquire businesses with promising products or technologies, we may not be able to realize the benefit of acquiring such businesses if we are unable to move one or more products through preclinical and/or clinical development to regulatory approval and commercialization. Integrating any newly acquired businesses or technologies could be expensive and time-consuming, resulting in the diversion of resources from our current business. We may not be able to integrate any acquired business successfully. We cannot assure you that, following an acquisition, we will achieve revenues, specific net income or loss levels that justify the acquisition or that the acquisition will result in increased earnings, or reduced losses, for the combined company in any future period. Moreover, we may need to raise additional funds through public or private debt or equity financing to acquire any businesses, which would result in dilution for stockholders or the incurrence of indebtedness and may not be available on terms which would otherwise be acceptable to us. We may not be able to operate acquired businesses profitably or otherwise implement our growth strategy successfully.
Our operating results may fluctuate significantly due to a number of factors which make our future results difficult to predict and could cause our operating results to fall below expectations or our guidance.
Our operating results will continue to be subject to fluctuations. The revenues we generate and our operating results will be affected by numerous factors, including:
| • | product sales; |
| • | cost of product sales; |
| • | marketing and other expenses; |
| • | manufacturing or supply issues; |
| • | the timing and amount of royalties or milestone payments; |
| • | our addition or termination of development programs; |
| • | variations in the level of expenses related to our products or future development programs; |
| • | regulatory developments affecting our products or those of our competitors; our execution of collaborative, licensing or other arrangements, and the timing of payments we may make or receive under these arrangements; |
| • | any intellectual property infringement or other lawsuit in which we may become involved; and |
| • | the timing and recognition of stock-based compensation expense. |
If our operating results fall below the expectations of investors or securities analysts or below any guidance we may provide, the price of our common stock could decline substantially. Furthermore, any fluctuations in our operating results may, in turn, cause the price of our stock to fluctuate substantially. We believe that comparisons of our financial results are not necessarily meaningful and should not be relied upon as an indication of our future performance.
31
Table of Contents
We are increasingly dependent on information technology systems, infrastructure and data.
We are increasingly dependent upon information technology systems, infrastructure and data. Our computer systems may be vulnerable to service interruption or destruction, malicious intrusion and random attack. Security breaches pose a risk that sensitive data, including intellectual property, trade secrets or personal information may be exposed to unauthorized persons or to the public. Cyber-attacks are increasing in their frequency, sophistication and intensity, and have become increasingly difficult to detect. Cyber-attacks could include the deployment of harmful malware, denial-of service, social engineering and other means to affect service reliability and threaten data confidentiality, integrity and availability. Our key business partners face similar risks, and a security breach of their systems could adversely affect our security posture. While we continue to invest data protection and information technology, there can be no assurance that our efforts will prevent service interruptions, or identify breaches in our systems, that could adversely affect our business and operations and/or result in the loss of critical or sensitive information, which could result in financial, legal, business or reputational harm.
Risks related to intellectual property and other legal matters
Our rights to develop and commercialize our products are subject in part to the terms and conditions of licenses or sublicenses granted to us by other pharmaceutical companies.
HETLIOZ® is based in part on patents that we have licensed on an exclusive basis and other intellectual property licensed from Bristol-Myers Squibb Company (BMS). BMS holds certain rights with respect to HETLIOZ® in the license agreement. Either party may terminate the license agreement under certain circumstances, including a material breach of the agreement by the other. In the event we terminate our license, or if BMS terminates our license due to our breach, all rights to HETLIOZ® (including any intellectual property we develop with respect to HETLIOZ®) will revert or otherwise be licensed back to BMS on an exclusive basis. Any termination or reversion of our rights to develop or commercialize HETLIOZ®, including any reacquisition by BMS of our rights, would have a material adverse effect on our business.
Fanapt® is based in part on patents and other intellectual property owned by Sanofi. Titan Pharmaceuticals, Inc. (Titan) holds an exclusive license from Sanofi to the intellectual property owned by Sanofi, and Titan has sublicensed its rights under such license on an exclusive basis to Novartis. We acquired exclusive rights to this and other intellectual property through a further sublicense from Novartis. The sublicense with Novartis was amended and restated in October of 2009 to provide Novartis with exclusive rights to commercialize Fanapt® in the U.S. and Canada. We retained exclusive rights to Fanapt® outside the U.S. and Canada. We acquired all of Novartis’ rights to Fanapt® in the fourth quarter of 2014 pursuant to an asset transfer agreement and related agreements with Novartis. We may lose our rights to develop and commercialize Fanapt® if we fail to comply with certain requirements in the Titan license agreement regarding our financial condition, or if we fail to comply with certain diligence obligations regarding our development or commercialization activities. Our loss of rights in Fanapt® would have a material adverse effect on our business, financial condition and results of operations.
Tradipitant is based in part on patents that we have licensed on an exclusive basis and other intellectual property licensed from Eli Lilly and Company (Lilly). Lilly may terminate our license if we fail to use our commercially reasonable efforts to develop and commercialize tradipitant or if we materially breach the agreement and fail to cure that breach. In the event that we terminate our license, or if Lilly terminates our license for the reasons stated above, all of our rights to tradipitant (including any intellectual property we develop with respect to tradipitant) will revert back to Lilly, subject to payment by Lilly to us of a royalty on net sales of products that contain tradipitant.
AQW051, to which we acquired rights from Novartis in the fourth quarter of 2014, is based on patents and other intellectual property that we have licensed on an exclusive basis from Novartis. Novartis may terminate our license if we materially breach the agreement, which includes an obligation to use commercially reasonable efforts to develop and commercialize AQW051, and fail to cure that breach. In the event that Novartis terminates our license for the reasons stated above, all of our rights to AQW051 (including any intellectual property we develop with respect to AQW051) will revert back to Novartis without compensation.
If our efforts to protect the proprietary nature of the intellectual property related to our products are not adequate, we may not be able to compete effectively in our markets.
Method of treatment patents protect the use of a product for the method specified in the patent claims. This type of patent does not prevent a competitor from making and marketing a product that is identical to our product for a use that is outside the scope of the patented method. Moreover, even if competitors do not actively promote their product for our patented methods, physicians may prescribe these products “off-label.” Although off-label prescriptions may infringe or contribute to the infringement of method of treatment patents, such infringement may be difficult to prevent.
Our patents and patent applications may be challenged or fail to result in issued patents and our existing or future patents may be too narrow to prevent third parties from developing or designing around these patents. In addition, we generally rely on trade secret protection and confidentiality agreements to protect certain proprietary know-how that is not patentable, for processes for which patents are difficult to enforce and for any other elements of our drug development processes that involve proprietary know-how, information and technology that is not covered by patent applications. While we require all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information and technology to enter into confidentiality
32
Table of Contents
agreements, we cannot be certain that this know-how, information and technology will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. Further, the laws of some foreign countries do not protect proprietary rights to the same extent as the laws of the U.S. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the U.S. and abroad. If we are unable to protect or defend the intellectual property related to our technologies, we will not be able to establish or maintain a competitive advantage in our market.
If we do not obtain protection under the Hatch-Waxman Act and similar foreign legislation to extend our patents and to obtain market exclusivity for our products, our business will be harmed.
The Hatch-Waxman Act provides for an extension of patent term for drugs for a period of up to five years to compensate for time spent in development. Assuming we gain a five-year patent term extension for HETLIOZ®, and that we continue to have rights under our license agreement with respect to this product, we would have exclusive rights to the HETLIOZ® U.S. new chemical entity patent (the primary patent covering the product as a new composition of matter) until 2022. We also own HETLIOZ® U.S. method of treatment patents (directed to the approved method of treatment as described in the HETLIOZ® label approved by the FDA). These patents expire normally in 2033. The Fanapt® U.S. new chemical entity patent received the full five-year patent term extension under the Hatch-Waxman Act and so this patent in the U.S. expired in November 2016. In November 2013, a patent directed to a method of treating patients with Fanapt® based on genotype was issued to us by the U.S. Patent and Trademark Office. This patent, which was listed in the FDA’s Orange Book in January 2015, is set to expire in 2027. Please see the risk factor entitled “We face substantial competition, which may result in others developing or commercializing products before or more successfully than we do,” and Part I, Item 3, Legal Proceedings, of this annual report on Form 10-K for additional information. See also Note 18, Legal Matters, to the consolidated financial statements included in Part II of this annual report on Form 10-K for additional information. Eight additional U.S. patents directed to methods of treating patients with Fanapt®, which are set to expire between 2025 and 2031, were issued to us in 2014 and 2015.
A directive in the European Union provides that companies that receive regulatory approval for a new medicinal product will have a 10-year period of market exclusivity for that product (with the possibility of a further one-year extension), beginning on the date of such European regulatory approval, regardless of when the European new chemical entity patent covering such product expires. A generic version of the approved drug may not be marketed or sold in Europe during such market exclusivity period. This directive is of material importance with respect to Fanapt®, since the European new chemical entity patent for Fanapt® has expired.
Assuming we gain a five-year patent term restoration for tradipitant, and that we continue to have rights under our license agreement with respect to this product, we would have exclusive rights to tradipitant’ s U.S. new chemical entity patent until 2029. Assuming we gain a five-year patent term restoration for AQW051, and that we continue to have rights under our license agreement with respect to this product, we would have exclusive rights to AQW051’s U.S. new chemical entity patent until 2028.
However, there is no assurance that we will receive the extensions of our patents or other exclusive rights available under the Hatch-Waxman Act or similar foreign legislation. If we fail to receive such extensions or exclusive rights, our or our partners’ ability to prevent competitors from manufacturing, marketing and selling generic versions of our products will be materially impaired.
Litigation or third-party claims of intellectual property infringement could require us to divert resources and may prevent or delay our drug discovery and development efforts.
Our commercial success depends in part on our not infringing the patents and proprietary rights of third parties. Third parties may assert that we are employing their proprietary technology without authorization. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents.
Furthermore, parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to develop and commercialize one or more of our products. Defense of these claims, regardless of their merit, would divert substantial financial and employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, obtain one or more licenses from third parties or pay royalties. In addition, even in the absence of litigation, we may need to obtain additional licenses from third parties to advance our research or allow commercialization of our products. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In that event, we would be unable to develop and commercialize further one or more of our products.
In addition, in the future we could be required to initiate litigation to enforce our proprietary rights against infringement by third parties. Prosecution of these claims to enforce our rights against others could divert substantial financial and employee resources from our business. If we fail to enforce our proprietary rights against others, our business will be harmed.
As described elsewhere in these risk factors and in Part I, Item 3, Legal Proceedings, of this annual report on Form 10-K, we have initiated lawsuits to enforce our patent rights against certain generic pharmaceutical companies.
33
Table of Contents
Risks related to our common stock
Our stock price has been highly volatile and may be volatile in the future, and purchasers of our common stock could incur substantial losses.
The realization of any of the risks described in these risk factors or other unforeseen risks could have a dramatic and adverse effect on the market price of our common stock. Between January 1, 2016 and December 31, 2016, the high and low sale prices of our common stock as reported on The NASDAQ Global Market varied between $6.91 and $18.00. Additionally, market prices for securities of biotechnology and pharmaceutical companies, including ours, have historically been very volatile. The market for these securities has from time to time experienced significant price and volume fluctuations for reasons that were unrelated to the operating performance of any one company.
The following factors, in addition to the other risk factors described in this section, may also have a significant impact on the market price of our common stock:
| • | our or our partners’ ability to successfully commercialize our products; |
| • | our ability to successfully execute our commercialization strategies; |
| • | publicity regarding actual or potential testing or trial results relating to products under development by us or our competitors; |
| • | the outcome of regulatory review relating to products under development by us or our competitors; |
| • | regulatory developments in the U.S. and foreign countries; |
| • | developments concerning any collaboration or other strategic transaction we may undertake; |
| • | publicity regarding actual or potential litigation involving us; |
| • | announcements of patent issuances or denials, technological innovations or new commercial products by us or our competitors; |
| • | termination or delay of development or commercialization program(s) by our partners; |
| • | safety issues with our products or those of our competitors; |
| • | announcements of technological innovations or new therapeutic products or methods by us or others; |
| • | actual or anticipated variations in our quarterly operating results; |
| • | changes in estimates of our financial results or recommendations by securities analysts or failure to meet such financial expectations; |
| • | changes in government regulations or policies; |
| • | changes in patent legislation or patent decisions or adverse changes to patent law; |
| • | additions or departures of key personnel or members of our board of directors; |
| • | the publication of negative research or articles about our company, our business or our products by industry analysts or others; |
| • | market rumors or press reports; |
| • | publicity regarding actual or potential transactions involving us; and |
| • | economic, political and other external factors beyond our control. |
We have been and may in the future be subject to litigation, which could harm our stock price, business, results of operations and financial condition.
We have been the subject of litigation in the past and may be subject to litigation in the future. In the past, following periods of volatility in the market price of their stock, many companies, including us, have been the subjects of securities class action litigation. Any such litigation can result in substantial costs and diversion of management’s attention and resources and could harm our stock price, business results of operations and financial condition. As a result of these factors, holders of our common stock might be unable to sell their shares at or above the price they paid for such shares.
If there are substantial sales of our common stock, our stock price could decline.
A small number of institutional investors and private equity funds hold a significant number of shares of our common stock. Sales by these stockholders of a substantial number of shares, or the expectation of such sales, could cause a significant reduction in the market price of our common stock.
In addition to our outstanding common stock, as of December 31, 2016 there were a total of 6,686,764 shares of our common stock that we have registered and that we are obligated to issue upon the exercise of currently outstanding options and settlement of
34
Table of Contents
restricted stock unit awards granted under our 2006 and 2016 Equity Incentive Plans. Upon the exercise of these options or settlement of the shares underlying these restricted stock units, as the case may be, in accordance with their respective terms, these shares may be resold freely, subject to restrictions imposed on our affiliates under Rule 144. If significant sales of these shares occur in short periods of time, these sales could reduce the market price of our common stock. Any reduction in the trading price of our common stock could impede our ability to raise capital on attractive terms, if at all.
If we fail to maintain the requirements for continued listing on The NASDAQ Global Market, our common stock could be delisted from trading, which would adversely affect the liquidity of our common stock and our ability to raise additional capital.
Our common stock is currently listed for quotation on The NASDAQ Global Market. We are required to meet specified listing criteria in order to maintain our listing on The NASDAQ Global Market. If we fail to satisfy The NASDAQ Global Market’s continued listing requirements, our common stock could be delisted from The NASDAQ Global Market, in which case we may transfer to The NASDAQ Capital Market, which generally has lower financial requirements for initial listing or, if we fail to meet its listing requirements, the over-the-counter bulletin board. Any potential delisting of our common stock from The NASDAQ Global Market would make it more difficult for our stockholders to sell our stock in the public market and would likely result in decreased liquidity and increased volatility for our common stock.
If securities or industry analysts do not publish research or reports or publish unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. We currently have research coverage by securities and industry analysts. If one or more of the analysts who covers us downgrades our stock, our stock price would likely decline. If one or more of these analysts ceases coverage of our Company or fails to regularly publish reports on us, interest in the purchase of our stock could decrease, which could cause our stock price or trading volume to decline.
You may experience future dilution as a result of future equity offerings.
In order to raise additional capital, we may in the future offer additional shares of our common stock or other securities convertible into or exchangeable for our common stock at prices that may not be the same as the price per share in previous offerings. We may sell shares or other securities in any other offering at a price per share that is less than the price per share paid by investors in previous offerings, and investors purchasing shares or other securities in the future could have rights superior to existing stockholders. The price per share at which we sell additional shares of our common stock, or securities convertible or exchangeable into common stock, in future transactions may be higher or lower than the price per share paid by investors.
Our business could be negatively affected as a result of the actions of activist stockholders.
Proxy contests have been waged against many companies in the biopharmaceutical industry, including us, over the last several years. If faced with a proxy contest or other type of shareholder activism, we may not be able to respond successfully to the contest or dispute, which would be disruptive to our business. Even if we are successful, our business could be adversely affected by a proxy contest or shareholder dispute involving us or our partners because:
| • | responding to proxy contests and other actions by activist stockholders can be costly and time-consuming, disrupting operations and diverting the attention of management and employees; |
| • | perceived uncertainties as to future direction may result in the loss of potential acquisitions, collaborations or in-licensing opportunities, and may make it more difficult to attract and retain qualified personnel and business partners; and |
| • | if individuals are elected to a board of directors with a specific agenda, it may adversely affect our ability to effectively and timely implement our strategic plan and create additional value for our stockholders. |
These actions could cause our stock price to experience periods of volatility.
Anti-takeover provisions in our charter and bylaws and under Delaware law, and our rights plan could prevent or delay a change in control of our company.
We are a Delaware corporation and the anti-takeover provisions of Section 203 of the Delaware General Corporation Law may discourage, delay or prevent a change in control by prohibiting us from engaging in a business combination with an interested stockholder for a period of three years after the person becomes an interested stockholder, even if a change of control would be beneficial to our existing stockholders. In addition, our amended and restated certificate of incorporation and bylaws may discourage, delay or prevent a change in our management or control over us that stockholders may consider favorable. Our amended and restated certificate of incorporation and bylaws:
| • | authorize the issuance of “blank check” preferred stock that could be issued by our board of directors to thwart a takeover attempt; |
35
Table of Contents
| • | do not provide for cumulative voting in the election of directors, which would allow holders of less than a majority of the stock to elect some directors; |
| • | establish a classified board of directors, as a result of which the successors to the directors whose terms have expired will be elected to serve from the time of election and qualification until the third annual meeting following their election; |
| • | require that directors only be removed from office for cause; |
| • | provide that vacancies on the board of directors, including newly-created directorships, may be filled only by a majority vote of directors then in office; |
| • | limit who may call special meetings of stockholders; |
| • | prohibit stockholder action by written consent, requiring all actions to be taken at a meeting of the stockholders; and |
| • | establish advance notice requirements for nominating candidates for election to the board of directors or for proposing matters that can be acted upon by stockholders at stockholder meetings. |
Moreover, in September 2008, our board of directors adopted a rights agreement that unless renewed expires in September 2018, the provisions of which could result in significant dilution of the proportionate ownership of a potential acquirer and, accordingly, could discourage, delay or prevent a change in our management or control over us.
Prolonged economic uncertainties or downturns, as well as unstable market, credit and financial conditions, may exacerbate certain risks affecting our business and have serious adverse consequences on our business.
The global economic downturn and market instability has made the business climate more volatile and more costly. These economic conditions, and uncertainty as to the general direction of the macroeconomic environment, are beyond our control and may make any necessary debt or equity financing more difficult, more costly, and more dilutive. While we believe we have adequate capital resources to meet current working capital and capital expenditure requirements, a lingering economic downturn or significant increase in our expenses could require additional financing on less than attractive rates or on terms that are excessively dilutive to existing stockholders. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our stock price and could require us to delay or abandon clinical development plans.
Sales of our products will be dependent, in large part, on reimbursement from government health administration authorities, private health insurers, distribution partners and other organizations. As a result of negative trends in the general economy in the U.S. or other jurisdictions in which we may do business, these organizations may be unable to satisfy their reimbursement obligations or may delay payment. In addition, federal and state health authorities may reduce Medicare and Medicaid reimbursements, and private insurers may increase their scrutiny of claims. A reduction in the availability or extent of reimbursement could negatively affect our or our partners’ product sales and revenue.
In addition, we rely on third parties for several important aspects of our business. For example, we use third parties for sales, distribution, medical affairs and clinical research, and we rely upon several single source providers of raw materials and contract manufacturers for the manufacture of our products. During challenging and uncertain economic times and in tight credit markets, there may be a disruption or delay in the performance of our third party contractors, suppliers or partners. If such third parties are unable to satisfy their commitments to us, our business and results of operations would be adversely affected.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
Not applicable.
| ITEM 2. | PROPERTIES |
Our headquarters office consists of a total of 40,188 square feet of office space located at 2200 Pennsylvania Avenue, N.W. in Washington, D.C. under operating leases and subleases that expire in 2026 and are subject to renewal options. In addition, we have 2,880 square feet of office space for our European headquarters in London under an operating lease that has a lease term ending in 2021 and is subject to a renewal option, and 1,249 square feet of office space in Berlin under a short-term operating lease. We believe that these facilities are suitable and adequate to meet our anticipated near-term needs. We anticipate that following the expiration of the leases, additional or alternative space will be available at commercially reasonable terms.
| ITEM 3. | LEGAL PROCEEDINGS |
In June 2014, we filed suit against Roxane Laboratories, Inc. (Roxane) in the U.S. District Court for the District of Delaware (the Delaware District Court). The suit seeks an adjudication that Roxane has infringed one or more claims of our U.S. Patent
36
Table of Contents
No. 8,586,610 (the ‘610 Patent) by submitting to the U.S. Food and Drug Administration (the FDA) an Abbreviated New Drug Application (ANDA) for a generic version of Fanapt® prior to the expiration of the ‘610 Patent in November 2027. In addition, pursuant to the settlement agreement with Novartis Pharma AG (Novartis), we assumed Novartis’ patent infringement action against Roxane in the Delaware District Court. That suit alleges that Roxane has infringed one or more claims of U.S. Patent RE39198 (the ‘198 Patent), which is licensed exclusively to us, by filing an ANDA for a generic version of Fanapt® prior to the expiration of the ‘198 Patent in November 2016. These two cases against Roxane were consolidated by agreement of the parties and were tried together in a five-day bench trial that concluded on March 4, 2016. On August 25, 2016, the Delaware District Court ruled in our favor, finding that Roxane’s ANDA product infringed the asserted claims of the ‘610 Patent and the ‘198 Patent. The Delaware District Court ruled that we are entitled to a permanent injunction against Roxane enjoining Roxane from infringing the ‘610 Patent, including the manufacture, use, sale, offer to sell, sale, distribution or importation of any generic iloperidone product described in the ‘610 Patent ANDA until the expiration of the ‘610 Patent in November 2027. If we obtain pediatric exclusivity, the injunction against Roxane would be extended until May 2028 under the Delaware District Court’s order. On September 23, 2016, Roxane filed a notice of appeal with the Federal Circuit Court of Appeals. Roxane filed its opening appellate brief on February 7, 2017.
In 2015, we filed six separate patent infringement lawsuits in the Delaware District Court against Roxane, Inventia Healthcare Pvt. Ltd. (Inventia), Lupin Ltd. and Lupin Pharmaceuticals, Inc. (Lupin), Taro Pharmaceuticals USA, Inc. and Taro Pharmaceutical Industries, Ltd. (Taro), and Apotex Inc. and Apotex Corp., (collectively, the Defendants). The lawsuits each seek an adjudication that the respective Defendants infringed one or more claims of the ‘610 Patent and/or our U.S. Patent No. 9,138,432 (the ‘432 Patent) by submitting to the FDA an ANDA for a generic version of Fanapt® prior to the expiration of the ‘610 Patent in November 2027 or the ‘432 Patent in September 2025. The Defendants have denied infringement and counterclaimed for declaratory judgment of invalidity and noninfringement of the ‘610 Patent and the ‘432 Patent. A trial on these matters was scheduled to begin on May 15, 2017. On December 15, 2016, following a submission by the parties that it may be appropriate to remove the May 15, 2017 trial from the Delaware District Court’s calendar pending the Federal Circuit’s decision in Roxane’s appeal of the Delaware District Court’s August 25, 2016 ruling finding that Roxane’s proposed products infringed the asserted claims of the ‘610 Patent, the Delaware District Court removed the May 15, 2017 trial from the calendar and adopted a new schedule for discovery. The parties agreed that within 14 days after any decision on the merits in the Roxane appeal, the parties will submit to the Delaware District Court a status report and request a schedule for trial. We entered into a confidential stipulation with Inventia regarding any potential launch of Inventia’s generic ANDA product. We also entered into a confidential stipulation with Lupin regarding any potential launch of Lupin’s generic ANDA product.
Lupin filed counter claims for declaratory judgment of invalidity and noninfringement of seven of our method of treatment patents that are listed in the Approved Drug Products with Therapeutic Equivalence Evaluations (the Orange Book) related to Fanapt® (such seven patents, the Method of Treatment Patents). We have not sued Lupin for infringing the Method of Treatment Patents. On October 13, 2016, we, along with Lupin, filed a Stipulation of Dismissal in the Delaware District Court pursuant to which Lupin’s counterclaims relating to the Method of Treatment Patents were dismissed without prejudice in recognition of an agreement reached between Lupin and us by which we would not assert those patents against Lupin absent certain changes in Lupin’s proposed prescribing information for its iloperidone tablets.
On October 24, 2016, we entered into a License Agreement with Taro to resolve our patent litigation against Taro regarding Taro’s ANDA seeking approval of its generic version of Fanapt® (the Taro License Agreement). Under the Taro License Agreement, we granted Taro a non-exclusive license to manufacture and commercialize Taro’s version of Fanapt® in the U.S. effective November 2, 2027, unless prior to that date we obtain pediatric exclusivity for Fanapt®, in which case, the license will be effective May 2, 2028. Taro may enter the market earlier under certain limited circumstances. The Taro License Agreement, which is subject to review by the U.S. Federal Trade Commission (FTC) and the U.S. Department of Justice (DOJ), provides for a full settlement and release by us and Taro of all claims that are the subject of the litigation.
On December 7, 2016, we entered into a License Agreement with Apotex to resolve our patent litigation against Apotex regarding Apotex’s ANDA seeking approval of its generic version of Fanapt® (the Apotex License Agreement). Under the Apotex License Agreement, we granted Apotex a non-exclusive license to manufacture and commercialize Apotex’s version of Fanapt® in the U.S. effective November 2, 2027, unless prior to that date we obtain pediatric exclusivity for Fanapt®, in which case, the license will be effective May 2, 2028. Apotex may enter the market earlier under certain limited circumstances. The Apotex License Agreement, which is subject to review by the FTC and the DOJ, provides for a full settlement and release by us and Apotex of all claims that are the subject of the litigation.
On February 26, 2016, Roxane filed suit against us in the U.S. District Court for the Southern District of Ohio (Ohio District Court). The suit seeks a declaratory judgment of invalidity and noninfringement of the Method of Treatment Patents. We have not sued Roxane for infringing the Method of Treatment Patents. We filed a motion to dismiss this lawsuit for lack of personal jurisdiction or to transfer the lawsuit to the Delaware District Court. On December 20, 2016, the Ohio District Court ruled in our favor, dismissing Roxane’s suit without prejudice for lack of personal jurisdiction.
37
Table of Contents
On February 26, 2016, Roxane filed a Petition for Inter Partes Review (IPR) of the ‘432 Patent with the Patent Trials and Appeals Board (PTAB) of the United States Patent and Trademark Office. We filed a Preliminary Response on June 7, 2016, and on August 30, 2016, the PTAB denied the request by Roxane to institute an IPR of the ‘432 Patent. On September 29, 2016, Roxane filed a Petition for Rehearing with the PTAB, and on October 13, 2016, we filed a Response to Roxane’s Petition. On November 4, 2016, the PTAB denied Roxane’s Petition for Rehearing.
| ITEM 4. | MINE SAFETY DISCLOSURES |
Not applicable.
PART II
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Our common stock is quoted on The NASDAQ Global Market under the symbol “VNDA.” The following table sets forth, for the periods indicated, the range of high and low sale prices of our common stock as reported on The NASDAQ Global Market:
| Year Ended December 31, 2016 | High | Low | ||||||
| First quarter |
$ | 9.58 | $ | 6.91 | ||||
| Second quarter |
11.40 | 8.02 | ||||||
| Third quarter |
18.00 | 10.81 | ||||||
| Fourth quarter |
17.65 | 13.55 | ||||||
| Year Ended December 31, 2015 | High | Low | ||||||
| First quarter |
$ | 15.00 | $ | 8.80 | ||||
| Second quarter |
13.92 | 8.92 | ||||||
| Third quarter |
14.50 | 10.57 | ||||||
| Fourth quarter |
12.28 | 8.00 | ||||||
As of February 6, 2017, there were 9 holders of record of our common stock. The number of holders of record of our common stock does not reflect the number of beneficial holders whose shares are held by depositors, brokers or other nominees.
Dividends
We have not paid dividends to our stockholders (other than a dividend of preferred share purchase rights which was declared in September 2008) since our inception and do not plan to pay dividends in the foreseeable future. We currently intend to retain earnings, if any, to finance our growth.
38
Table of Contents
Market Price of and Dividends on the Registrant’s Common Equity and Related Stockholder Matters
The following graph shows the cumulative five-year total return on our common stock relative to the cumulative total returns of the NASDAQ Composite Index and the NASDAQ Biotechnology Index. An investment of $100 (with reinvestment of dividends) is assumed to have been made in our common stock and in each of the indexes on December 31, 2011 and its relative performance is tracked through December 31, 2016. The comparisons in the table are required by the Securities and Exchange Commission (SEC) and are not intended to forecast or be indicative of possible future performance of our common stock. We have not paid dividends to our stockholders since the inception (other than a dividend of preferred share purchase rights which was declared in September 2008) and do not plan to pay dividends in the foreseeable future. The following graph and related information is being furnished solely to accompany this annual report on Form 10-K pursuant to Item 201(e) of Regulation S-K and shall not be deemed “soliciting materials” or to be “filed” with the SEC (other than as provided in Item 201), nor shall such information be incorporated by reference into any of our filings under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date hereof, and irrespective of any general incorporation language in any such filing.
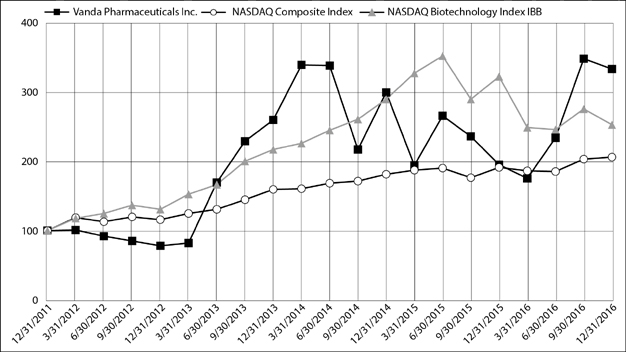
Securities Authorized for Issuance under Equity Incentive Plans
Information regarding securities authorized for issuance under equity incentive plans will be contained in our Proxy Statement for the 2017 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2016, under the captions “Equity Compensation Plan Information” and “Security Ownership of Certain Beneficial Owners and Management” and is incorporated herein by reference pursuant to General Instruction G(3) to Form 10-K.
| ITEM 6. | SELECTED CONSOLIDATED FINANCIAL DATA |
The consolidated statements of operations data for the years ended December 31, 2016, 2015 and 2014 and the consolidated balance sheet data as of December 31, 2016 and 2015 are each derived from our audited consolidated financial statements included in this annual report on Form 10-K. The consolidated statements of operations data for the years ended December 31, 2013 and 2012, and the consolidated balance sheet data as of December 31, 2014, 2013 and 2012 are each derived from our audited consolidated financial statements not included herein. Our historical results for any prior period are not necessarily indicative of results to be expected in any future period.
39
Table of Contents
The following data should be read together with our consolidated financial statements and accompanying notes and the section entitled Management’s Discussion and Analysis of Financial Condition and Results of Operations included in this annual report on Form 10-K.
| Year Ended December 31, | ||||||||||||||||||||
| (in thousands, except for share and per share amounts) | 2016 | 2015 | 2014 (1) | 2013 | 2012 | |||||||||||||||
| Statements of Operations Data |
||||||||||||||||||||
| Total revenues |
$ | 146,017 | $ | 109,925 | $ | 50,157 | $ | 33,879 | $ | 32,727 | ||||||||||
| Operating expenses: |
||||||||||||||||||||
| Cost of goods sold, excluding amortization |
24,712 | 23,462 | 1,583 | — | 129 | |||||||||||||||
| Research and development |
29,156 | 29,145 | 19,230 | 28,502 | 45,764 | |||||||||||||||
| Selling, general and administrative |
99,787 | 84,531 | 84,644 | 25,082 | 14,517 | |||||||||||||||
| Intangible asset amortization |
10,933 | 12,972 | 2,254 | 1,495 | 1,495 | |||||||||||||||
| Gain on arbitration settlement |
— | — | (77,616 | ) | — | — | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating expenses |
164,588 | 150,110 | 30,095 | 55,079 | 61,905 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Income (loss) from operations |
(18,571 | ) | (40,185 | ) | 20,062 | (21,200 | ) | (29,178 | ) | |||||||||||
| Other income |
665 | 320 | 124 | 145 | 561 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Income (loss) before income taxes |
(17,906 | ) | (39,865 | ) | 20,186 | (21,055 | ) | (28,617 | ) | |||||||||||
| Provision for income taxes |
104 | — | — | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net income (loss) |
$ | (18,010 | ) | $ | (39,865 | ) | $ | 20,186 | $ | (21,055 | ) | $ | (28,617 | ) | ||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net income (loss) per share: |
||||||||||||||||||||
| Basic |
$ | (0.41 | ) | $ | (0.94 | ) | $ | 0.58 | $ | (0.69 | ) | $ | (1.01 | ) | ||||||
| Diluted |
$ | (0.41 | ) | $ | (0.94 | ) | $ | 0.55 | $ | (0.69 | ) | $ | (1.01 | ) | ||||||
| Weighted average shares outstanding: |
||||||||||||||||||||
| Basic |
43,449,441 | 42,250,254 | 34,774,163 | 30,351,353 | 28,228,409 | |||||||||||||||
| Diluted |
43,449,441 | 42,250,254 | 36,686,723 | 30,351,353 | 28,228,409 | |||||||||||||||
| December 31, | ||||||||||||||||||||
| 2016 | 2015 | 2014 | 2013 | 2012 | ||||||||||||||||
| Balance Sheet Data |
||||||||||||||||||||
| Cash and cash equivalents |
$ | 40,426 | $ | 50,843 | $ | 60,901 | $ | 64,764 | $ | 88,772 | ||||||||||
| Marketable securities |
100,914 | 92,337 | 68,921 | 65,586 | 31,631 | |||||||||||||||
| Working capital |
123,855 | 115,230 | 133,944 | 102,763 | 93,705 | |||||||||||||||
| Total assets |
210,374 | 213,050 | 171,704 | 143,349 | 135,448 | |||||||||||||||
| Total liabilities |
79,044 | 80,023 | 10,887 | 99,225 | 125,543 | |||||||||||||||
| Accumulated deficit |
(345,859 | ) | (327,849 | ) | (287,984 | ) | (308,170 | ) | (287,115 | ) | ||||||||||
| Total stockholders’ equity |
131,330 | 133,027 | 160,817 | 44,124 | 9,905 | |||||||||||||||
| (1) | Net income for the year ended December 31, 2014 includes a gain on arbitration settlement of $77.6 million, or $2.23 and $2.12 per basic and diluted share, respectively. |
40
Table of Contents
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
You should read the following discussion and analysis of our financial condition and results of operations together with Selected Consolidated Financial Data and our consolidated financial statements and related notes appearing in this annual report on Form 10-K. Some of the information contained in this discussion and analysis or set forth elsewhere in this annual report on Form 10-K include historical information and other information with respect to our plans and strategy for our business and contain forward-looking statements that involve risks, uncertainties and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of certain factors, including but not limited to those set forth under the “Risk Factors” section of this report and elsewhere in this annual report on Form 10-K.
Overview
Vanda Pharmaceuticals Inc. (we, our or Vanda) is a global biopharmaceutical company focused on the development and commercialization of innovative therapies to address high unmet medical needs and improve the lives of patients. We commenced operations in 2003 and our product portfolio includes:
| • | HETLIOZ® (tasimelteon), a product for the treatment of Non-24-Hour Sleep-Wake Disorder (Non-24), was approved by the U.S. Food and Drug Administration (FDA) in January 2014 and launched commercially in the U.S. in April 2014. In July 2015, the European Commission (EC) granted centralized marketing authorization with unified labeling for HETLIOZ® for the treatment of Non-24 in totally blind adults. HETLIOZ® was commercially launched in Germany in August 2016. HETLIOZ® has potential utility in a number of other circadian rhythm disorders and is presently in clinical development for the treatment of Pediatric Non-24, Jet Lag Disorder and Smith-Magenis Syndrome (SMS). |
| • | Fanapt® (iloperidone), a product for the treatment of schizophrenia, the oral formulation of which was approved by the FDA in May 2009 and launched commercially in the U.S. by Novartis Pharma AG (Novartis) in January of 2010. Novartis transferred all the U.S. and Canadian commercial rights to the Fanapt® franchise to us on December 31, 2014. Additionally, our distribution partners launched Fanapt® in Israel and Mexico in 2014. Fanapt® has potential utility in a number of other disorders. An assessment of new Fanapt® clinical opportunities is ongoing. |
| • | Tradipitant (VLY-686), a small molecule neurokinin-1 receptor (NK-1R) antagonist, which is presently in clinical development for the treatment of chronic pruritus in atopic dermatitis and gastroparesis. |
| • | Trichostatin A, a small molecule histone deacetylase (HDAC) inhibitor. |
| • | AQW051, a Phase II alpha-7 nicotinic acetylcholine receptor partial agonist. |
Operational Highlights
HETLIOZ®
| • | Enrollment of patients for a Jet Lag Disorder clinical study is ongoing. Results are expected in the second half of 2017. |
| • | Enrollment in the Smith-Magenis Syndrome (SMS) clinical study is ongoing with results expected in 2018. |
| • | A pharmacokinetic study of the HETLIOZ® pediatric formulation is enrolling with results expected in 2018. |
Fanapt®
| • | An expansion of the Fanapt® U.S. field sales team is expected to be completed during the first quarter of 2017. |
| • | The Marketing Authorization Application (MAA) for oral Fanaptum® tablets is under evaluation by the European Medicines Agency for the treatment of schizophrenia in adults. A decision on the Fanaptum® MAA is expected during the second half of 2017. |
| • | An assessment of new Fanapt® clinical opportunities is ongoing. |
Tradipitant
| • | Enrollment in a tradipitant clinical study for the treatment of chronic pruritus in patients with atopic dermatitis is approaching completion. Results are expected in mid 2017. |
| • | A tradipitant clinical study for the treatment of gastroparesis began enrolling patients in the fourth quarter of 2016. Results are expected in the fourth quarter of 2017. |
41
Table of Contents
Trichostatin A
| • | Vanda expects to submit an investigational new drug application to the FDA in mid 2017 for trichostatin A, which will seek clearance to begin a clinical study of hematologic malignancies. |
Since we began operations in March 2003, we have devoted substantially all of our resources to the in-licensing, clinical development and commercialization of our products. Our ability to generate meaningful product sales and achieve profitability largely depends on our ability to successfully commercialize HETLIOZ® and Fanapt® in the U.S. and Europe, on our ability, alone or with others, to complete the development of our products, and to obtain the regulatory approvals for and to manufacture, market and sell our products. The results of our operations will vary significantly from year-to-year and quarter-to-quarter and depend on a number of factors, including risks related to our business, risks related to our industry, and other risks which are detailed in Risk Factors reported in Item 1A of Part I of this annual report on Form 10-K.
As described in Part I, Item 3, Legal Proceedings, of this annual report on Form 10-K, we have initiated lawsuits to enforce our patent rights against certain generic pharmaceutical companies.
Critical Accounting Policies
The preparation of our consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of our financial statements, as well as the reported revenues and expenses during the reported periods. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
A summary of our significant accounting policies appears in the notes to our audited consolidated financial statements for the year ended December 31, 2016 included in this annual report on Form 10-K. However, we believe that the following accounting policies are important to understanding and evaluating our reported financial results, and we have accordingly included them in this discussion.
Inventory. Inventory, which is recorded at the lower of cost or net realizable value, includes the cost of third-party manufacturing and other direct and indirect costs and is valued using the first-in, first-out method. We capitalize inventory costs associated with our products upon regulatory approval when, based on management’s judgment, future commercialization is considered probable and the future economic benefit is expected to be realized; otherwise, such costs are expensed as research and development. Inventory is evaluated for impairment by consideration of factors such as lower of cost or net realizable value, obsolescence or expiry.
Net Product Sales. Our net product sales consist of sales of HETLIOZ® and sales of Fanapt®. We apply the revenue recognition guidance in accordance with Financial Accounting Standards Board Accounting Standards Codification (ASC) Subtopic 605-15, Revenue Recognition—Products. We recognize revenue from product sales when there is persuasive evidence that an arrangement exists, title to product and associated risk of loss has passed to the customer, the price is fixed or determinable, collectability is reasonably assured and we have no further performance obligations.
HETLIOZ® is available in the U.S. for distribution through a limited number of specialty pharmacies, and is not available in retail pharmacies. Fanapt® is available in the U.S. for distribution through a limited number of wholesalers and is available in retail pharmacies. We invoice and record revenue when our customers, specialty pharmacies and wholesalers, receive product from the third-party logistics warehouse. Revenues and accounts receivable are concentrated with these customers.
We have entered into distribution agreements with Probiomed S.A. de C.V. (Probiomed) for the commercialization of Fanapt® in Mexico and Megapharm Ltd. for the commercialization of Fanapt® in Israel.
42
Table of Contents
Product Sales Discounts and Allowances. Product sales are recorded net of applicable discounts, chargebacks, rebates, co-pay assistance, service fees and product returns that are applicable for various government and commercial payors. Reserves established for discounts and returns are classified as reductions of accounts receivable if the amount is payable to direct customers, with the exception of service fees. Service fees are classified as a liability. Reserves established for chargebacks, rebates or co-pay assistance are classified as a liability if the amount is payable to a party other than customers. We currently record sales allowances for the following:
Prompt-pay: Specialty pharmacies and wholesalers are offered discounts for prompt payment. We expect that the specialty pharmacies and wholesalers will earn prompt payment discounts and, therefore, deduct the full amount of these discounts from total product sales when revenues are recognized.
Rebates: Allowances for rebates include mandated and supplemental discounts under the Medicaid Drug Rebate Program as well as contracted rebate programs with other payors. Rebate amounts owed after the final dispensing of the product to a benefit plan participant are based upon contractual agreements or legal requirements with public sector benefit providers, such as Medicaid. The allowance for rebates is based on statutory or contracted discount rates and expected utilization. Estimates for the expected utilization of rebates are based on historical activity and, where available, actual and pending prescriptions for which we have validated the insurance benefits. Rebates are generally invoiced and paid in arrears, such that the accrual balance consists of an estimate of the amount expected to be incurred for the current quarter’s activity, plus an accrual balance for known prior quarter’s unpaid rebates. If actual future invoicing varies from estimates, we may need to adjust accruals, which would affect net revenue in the period of adjustment.
Chargebacks: Chargebacks are discounts that occur when contracted customers purchase directly from specialty pharmacies and wholesalers. Contracted customers, which currently consist primarily of Public Health Service institutions, non-profit clinics, and Federal government entities purchasing via the Federal Supply Schedule, generally purchase the product at a discounted price. The specialty pharmacy or wholesaler, in turn, charges back the difference between the price initially paid by the specialty pharmacy or wholesaler and the discounted price paid to the specialty pharmacy or wholesaler by the contracted customer. The allowance for chargebacks is based on historical activity and, where available, actual and pending prescriptions for which we have validated the insurance benefits.
Medicare Part D Coverage Gap: Medicare Part D prescription drug benefit mandates manufacturers to fund approximately 50% of the Medicare Part D insurance coverage gap for prescription drugs sold to eligible patients. Estimates for expected Medicare Part D coverage gap are based in part on historical activity and, where available, actual and pending prescriptions for which we have validated the insurance benefits. Funding of the coverage gap is generally invoiced and paid in arrears so that the accrual balance consists of an estimate of the amount expected to be incurred for the current quarter’s activity, plus an accrual balance for known prior quarter activity. If actual future funding varies from estimates, we may need to adjust accruals, which would affect net sales in the period of adjustment.
Service Fees: We also incur specialty pharmacy and wholesaler fees for services and their data. These fees are based on contracted terms and are known amounts. We accrue service fees at the time of revenue recognition, resulting in a reduction of product sales and the recognition of an accrued liability, unless it receives an identifiable and separate benefit for the consideration and it can reasonably estimate the fair value of the benefit received. In which case, service fees are recorded as selling, general and administrative expense.
Co-payment Assistance: Patients who have commercial insurance and meet certain eligibility requirements may receive co-payment assistance. Co-pay assistance utilization is based on information provided by our third-party administrator. The allowance for co-pay assistance is based on actual sales and an estimate for pending sales based on either historical activity or pending sales for which we have validated the insurance benefits.
Product Returns: Consistent with industry practice, we generally offer direct customers a limited right to return as defined within our returns policy. We consider several factors in the estimation process, including historical return activity, expiration dates of product shipped to specialty pharmacies, inventory levels within the distribution channel, product shelf life, prescription trends and other relevant factors.
The following table summarizes sales discounts and allowance activity as of and for the years ended December 31, 2016, 2015 and 2014:
43
Table of Contents
| (in thousands) | Rebates & Chargebacks |
Discounts, Returns and Other |
Total | |||||||||
| Balance at December 31, 2013 |
$ | — | $ | — | $ | — | ||||||
| Provision related to current period sales |
419 | 720 | 1,139 | |||||||||
| Credits/payments made |
(51 | ) | (452 | ) | (503 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Balance at December 31, 2014 |
368 | 268 | 636 | |||||||||
| Provision related to current period sales |
57,424 | 17,940 | 75,364 | |||||||||
| Adjustments for prior period sales |
(114 | ) | (25 | ) | (139 | ) | ||||||
| Credits/payments made |
(24,255 | ) | (14,626 | ) | (38,881 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Balance at December 31, 2015 |
33,423 | 3,557 | 36,980 | |||||||||
| Provision related to current period sales |
56,133 | 19,451 | 75,584 | |||||||||
| Adjustments for prior period sales |
(1,842 | ) | 790 | (1,052 | ) | |||||||
| Credits/payments made |
(56,512 | ) | (17,340 | ) | (73,852 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Balance at December 31, 2016 |
$ | 31,202 | $ | 6,458 | $ | 37,660 | ||||||
|
|
|
|
|
|
|
|||||||
The provision for rebates and chargebacks of $56.1 million and $57.4 million for the years ended December 31, 2016 and 2015, respectively, primarily represents Medicaid rebates and contracted rebate programs applicable to sales of Fanapt®. The provision for discounts, returns and other of $19.5 million and $17.9 for the years ended December 31, 2016 and 2015, respectively, primarily represents wholesaler distribution fees applicable to sales of Fanapt® and prompt pay discounts applicable to the sales of both HETLIOZ® and Fanapt®.
License revenue. Our license revenues in 2014 were derived from the amended and restated sublicense agreement with Novartis and include an upfront payment and future milestone and royalty payments. Pursuant to the amended and restated sublicense agreement, Novartis had the right to commercialize and develop Fanapt® in the U.S. and Canada. Under the amended and restated sublicense agreement, we received an upfront payment of $200.0 million. Revenue related to the upfront payment was recognized ratably from the date the amended and restated sublicense agreement became effective (November 2009) through the expected duration of the Novartis commercialization of Fanapt® in the U.S. which was estimated to be through the expiry of the Fanapt® composition of patent, including a granted Hatch-Waxman extension (November 2016). In connection with the Settlement Agreement, we recognized the remaining deferred revenue as of December 31, 2014 as part of the gain on arbitration settlement. See Note 3, Settlement Agreement with Novartis, to the consolidated financial statements included in Part II of this annual report on Form 10-K for additional information.
Stock-based compensation. We use the Black-Scholes-Merton option pricing model to determine the fair value of stock options. The determination of the fair value of stock options on the date of grant using an option pricing model is affected by our stock price as well as assumptions regarding a number of complex and subjective variables. These variables include the expected stock price volatility over the expected term of the awards, actual and projected employee stock option exercise behaviors, risk-free interest rate and expected dividends. Expected volatility rates are based on the historical volatility of our publicly traded common stock and other factors. The risk-free interest rates are based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of the grant. We have not paid dividends to our stockholders since our inception (other than a dividend of preferred share purchase rights which was declared in September 2008) and do not plan to pay dividends in the foreseeable future. Stock-based compensation expense is also affected by the expected forfeiture rate for the respective option grants. If our estimates of the fair value of these equity instruments or expected forfeitures are too high or too low, it would have the effect of overstating or understating expenses.
Research and development expenses. Research and development expenses consist primarily of fees for services provided by third parties in connection with the clinical trials, costs of contract manufacturing services, milestone payments made under licensing agreements prior to regulatory approval, costs of materials used in clinical trials and research and development, costs for regulatory consultants and filings, depreciation of capital resources used to develop products, related facilities costs, and salaries, other employee-related costs and stock-based compensation for research and development personnel. We expense research and development costs as they are incurred for products in the development stage, including manufacturing costs and milestone payments made under license agreements prior to FDA approval. Upon and subsequent to FDA approval, manufacturing and milestone payments made under license agreements are capitalized. Milestone payments are accrued when it is deemed probable that the milestone event will be achieved. Costs related to the acquisition of intellectual property are expensed as incurred if the underlying technology is developed in connection with our research and development efforts and has no alternative future use.
44
Table of Contents
Selling, general and administrative expenses. Selling, general and administrative expenses consist primarily of salaries, other related costs for personnel, including stock-based compensation, related to executive, finance, accounting, information technology, marketing, medical affairs and human resource functions. Other costs include facility costs not otherwise included in research and development expenses and fees for marketing, medical affairs, legal, accounting and other professional services. Selling, general and administrative expenses also include third party expenses incurred to support sales, business development, and other business activities. Additionally, selling, general and administrative expenses included our estimate for the annual Patient Protection and Affordable Care fee.
Income taxes. On a periodic basis, we evaluate the realizability of our deferred tax assets and liabilities and will adjust such amounts in light of changing facts and circumstances, including but not limited to future projections of taxable income, the reversal of deferred tax liabilities, tax legislation, rulings by relevant tax authorities and tax planning strategies. Settlement of filing positions that may be challenged by tax authorities could impact our income taxes in the year of resolution.
In assessing the realizability of deferred tax assets, we consider whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the period in which those temporary differences becomes deductible or the net operating losses (NOLs) and credit carryforwards can be utilized. When considering the reversal of the valuation allowance, we consider the level of past and future taxable income, the reversal of deferred tax liabilities, the utilization of the carryforwards and other factors. Revisions to the estimated net realizable value of the deferred tax asset could cause our provision for income taxes to vary significantly from period to period.
Intangible Assets
The following is a summary of our intangible assets as of December 31, 2016:
| December 31, 2016 | ||||||||||||||||
| (in thousands) | Estimated Useful Life (Years) |
Gross Carrying Amount |
Accumulated Amortization |
Net Carrying Amount |
||||||||||||
| HETLIOZ® |
January 2033 | $ | 33,000 | $ | 5,181 | $ | 27,819 | |||||||||
| Fanapt® |
November 2016 | 27,941 | 27,941 | — | ||||||||||||
|
|
|
|
|
|
|
|||||||||||
| $ | 60,941 | $ | 33,122 | $ | 27,819 | |||||||||||
|
|
|
|
|
|
|
|||||||||||
HETLIOZ®. In January 2014, we announced that the FDA had approved the NDA for HETLIOZ®. As a result of this approval, we met a milestone under its license agreement with Bristol-Myers Squibb (BMS) that required us to make a license payment of $8.0 million to BMS. The $8.0 million is being amortized on a straight-line basis over the remaining life of the U.S. patent for HETLIOZ®, which prior to June 2014, we expected to last until December 2022. In June 2014, we received a notice of allowance from the U.S. Patent and Trademark Office for a patent covering the method of use of HETLIOZ®. The patent expires in January 2033, thereby potentially extending the exclusivity protection in the U.S. beyond the composition of matter patent. As a result of the patent allowance, we extended the estimated useful life of the U.S. patent for HETLIOZ® from December 2022 to January 2033.
We are obligated to make a future milestone payment to BMS of $25.0 million in the event that cumulative worldwide sales of HETLIOZ® reach $250.0 million. The likelihood of achieving the milestone and the related milestone obligation was determined to be probable during the year ended December 31, 2015. As a result, the future obligation of $25.0 million was recorded as a non-current liability as of December 31, 2016 and 2015. The $25.0 million was determined to be additional consideration for the acquisition of the HETLIOZ® intangible asset, which was created upon FDA approval on January 31, 2014. The actual payment of the $25.0 million will occur once the $250.0 million in cumulative worldwide sales of HETLIOZ® is realized, which we expect to occur by the end of 2018. The intangible asset of $25.0 million is being amortized on a straight-line basis over the remaining life of the U.S. patent for HETLIOZ®, which is expected to be January 2033. Amortization of intangible assets relating to HETLIOZ® amounted to $1.7 million, $2.9 million and $0.6 million for the years ended December 31, 2016, 2015 and 2014, respectively. In future periods we expect annual amortization of capitalized intangible asset costs relating to HETLIOZ® will amount to $1.7 million until the expiration of the patent in 2033.
Fanapt®. In 2009, we announced that the FDA had approved the NDA for Fanapt®. As a result of this approval, we met a milestone under our original sublicense agreement with Novartis that required us to make a license payment of $12.0 million to Novartis. The $12.0 million has been amortized on a straight-line basis over the remaining life of the U.S. composition of matter patent for Fanapt® to November 2016.
Pursuant to the Settlement Agreement, Novartis transferred all U.S. and Canadian rights in the Fanapt® franchise to us. As a result, we recognized an intangible asset of $15.9 million on December 31, 2014 related to the reacquired rights to Fanapt®, which has been amortized on a straight-line basis through November 2016. The useful life estimation for the Fanapt® intangible asset was based on the market participant methodology prescribed by ASC 805, and therefore does not reflect the impact of additional Fanapt® patents solely owned by us with varying expiration dates, the latest of which is December 2031. Amortization of intangible assets relating to Fanapt® was completed in November 2016 and amounted to $9.2 million, $10.1 million and $1.7 million for the years ended December 31, 2016, 2015 and 2014, respectively.
45
Table of Contents
Intangible assets are amortized over their estimated useful economic life using the straight line method. Total amortization expense was $10.9 million, $13.0 million and $2.3 million for the years ended December 31, 2016, 2015 and 2014, respectively.
The following is a summary of the future intangible asset amortization schedule as of December 31, 2016:
| (in thousands) | Total | 2017 | 2018 | 2019 | 2020 | 2021 | Thereafter | |||||||||||||||||||||
| HETLIOZ® |
$ | 27,819 | $ | 1,721 | $ | 1,721 | $ | 1,721 | $ | 1,721 | $ | 1,721 | $ | 19,214 | ||||||||||||||
Recent Accounting Pronouncements
See Note 2, Summary of Significant Accounting Policies, to the consolidated financial statements included in Part II of this annual report on Form 10-K for information on recent accounting pronouncements.
Results of Operations
We anticipate that our results of operations will fluctuate for the foreseeable future due to several factors, including our and our partners’ ability to successfully commercialize our products, any possible payments made or received pursuant to license or collaboration agreements, progress of our research and development efforts, the timing and outcome of clinical trials and related possible regulatory approvals. Our limited operating history makes predictions of future operations difficult. Since our inception, we have incurred significant losses resulting in an accumulated deficit of $345.9 million as of December 31, 2016. Our total stockholders’ equity was $131.3 million as of December 31, 2016.
Year ended December 31, 2016 compared to year ended December 31, 2015
Revenues. Total revenues increased by $36.1 million, or 33%, to $146.0 million for the year ended December 31, 2016 compared to $109.9 million for the year ended December 31, 2015. During the years ended December 31, 2016 and 2015, revenues consisted of the following:
| Year Ended December 31, | ||||||||||||||||
| (in thousands) | 2016 | 2015 | Net Change | Percent | ||||||||||||
| HETLIOZ® product sales, net |
$ | 71,671 | $ | 44,302 | $ | 27,369 | 62 | % | ||||||||
| Fanapt® product sales, net |
74,346 | 65,623 | 8,723 | 13 | % | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| $ | 146,017 | $ | 109,925 | $ | 36,092 | 33 | % | |||||||||
|
|
|
|
|
|
|
|||||||||||
HETLIOZ® product sales increased by $27.4 million, or 62%, to $71.7 million for the year ended December 31, 2016 compared to $44.3 million for the year ended December 31, 2015.
Fanapt® product sales increased by $8.7 million, or 13%, to $74.3 million for the year ended December 31, 2016 compared to $65.6 million for the year ended December 31, 2015.
Cost of goods sold. Cost of goods sold was $24.7 million for the year ended December 31, 2016, compared to $23.5 million for the year ended December 31, 2015. Cost of goods sold includes third party manufacturing costs of product sold, third party royalty costs and distribution and other costs. Third party royalty costs are 10% of net U.S. sales of HETLIOZ®. Third party royalty costs were 23% of net U.S. sales of Fanapt® through November 15, 2016 and 9% thereafter.
HETLIOZ® cost of goods sold as a percentage of HETLIOZ® revenue depends upon our cost to manufacture inventory at normalized production levels with our third party manufacturers. We expect that, in the future, total HETLIOZ® manufacturing costs included in cost of goods sold will be less than 2% of our net HETLIOZ® product sales.
Fanapt® work-in-process inventory and finished goods inventory acquired from Novartis as part of the acquisition of the Fanapt® business were recorded at fair value. The fair value of the inventory acquired from Novartis represents a higher cost than if new work-in-process inventory and finished goods inventory was manufactured at this time. We expect that, in the future, total U.S. Fanapt® manufacturing costs included in cost of goods sold will be less than 4% of our net U.S. Fanapt® product sales.
Research and development expenses. Research and development expenses were $29.2 and $29.1 million for the years ended December 31, 2016 and 2015, respectively. Increased clinical trial expenses associated with the HETLIOZ® Jet Lag Disorder and SMS programs and the tradipitant chronic pruritus in atopic dermatitis program that were incurred for the year ended December 31, 2016 were offset by the close out of Fanapt® clinical trial expenses transitioned to us as part of the Settlement Agreement with Novartis and regulatory expenses related to our sNDA filing incurred during the year ended December 31, 2015. The following table summarizes the costs of our product development initiatives for the year ended December 31, 2016 and 2015.
46
Table of Contents
| Year Ended December 31, |
||||||||
| (in thousands) | 2016 | 2015 | ||||||
| Direct project costs (1) |
||||||||
| HETLIOZ® |
$ | 12,658 | $ | 10,444 | ||||
| Fanapt® |
2,598 | 8,501 | ||||||
| Tradipitant |
7,010 | 4,006 | ||||||
| Trichostatin A |
2,218 | 1,681 | ||||||
|
|
|
|
|
|||||
| 24,484 | 24,632 | |||||||
|
|
|
|
|
|||||
| Indirect project costs (1) |
||||||||
| Stock-based compensation |
2,087 | 2,269 | ||||||
| Other indirect overhead |
2,585 | 2,244 | ||||||
|
|
|
|
|
|||||
| 4,672 | 4,513 | |||||||
|
|
|
|
|
|||||
| Total research & development expense |
$ | 29,156 | $ | 29,145 | ||||
|
|
|
|
|
|||||
| (1) | We record direct costs, including personnel costs and related benefits, on a project-by-project basis. Many of our research and development costs are not attributable to any individual project because we share resources across several development projects. We record indirect costs that support a number of our research and development activities in the aggregate, including stock-based compensation expense. |
We expect to incur significant research and development expenses as we continue to develop our products. In addition, we expect to incur licensing costs in the future that could be substantial, as we continue our efforts to develop our products.
Selling, general and administrative expenses. Selling, general and administrative expenses increased by $15.3 million, or 18%, to $99.8 million for the year ended December 31, 2016, compared with $84.5 million for the year ended December 31, 2015. The increase was primarily the result of marketing and sales efforts around Fanapt® in the U.S. and HETLIOZ® in Europe, an increase in the number of employees, including the hiring of new members of the executive management team at the end of 2015, as well as increased legal fees associated with ongoing patent litigation.
Intangible asset amortization. Intangible asset amortization decreased by $2.1 million, or 16 %, to $10.9 million for year ended December 31, 2016 compared to $13.0 million for the year ended December 31, 2015. The likelihood of achieving a future milestone obligation that becomes payable to BMS when cumulative sales of HETLIOZ® equal $250.0 million was determined to be probable in the first quarter of 2015 resulting in an increase in capitalized intangible assets of $25.0 million. As a result, intangible asset amortization relating to HETLIOZ® for the year ended December 31, 2015 had included additional amortization of $1.2 million for a catch-up adjustment to retroactively record cumulative amortization from February 1 to December 31, 2014 relating to the capitalized intangible asset of $25.0 million. We expect that annual amortization of capitalized intangible asset costs relating to HETLIOZ® will amount to $1.7 million in future years until the expiration of the patent in 2033.
Amortization of intangible assets relating to Fanapt® was completed in November 2016 and amounted to $9.2 million for the year ended December 31, 2016, compared to $10.1 million for the year ended December 31, 2015. Pursuant to the terms of the Settlement Agreement with Novartis, Novartis transferred all U.S. and Canadian rights in the Fanapt® franchise to us on December 31, 2014 resulting in an increase in capitalized intangible assets of $15.9 million that has been amortized until November 2016. The useful life estimation for the Fanapt® intangible asset was based on the market participant methodology prescribed by ASC 805, and therefore does not reflect the impact of additional Fanapt® patents solely owned by us with varying expiration dates, the latest of which is December 2031.
Provision for income taxes. The provision for income taxes was $0.1 million and zero for the years ended December 31, 2016 and 2015, respectively. The tax provision for the year ended December 31, 2016 is attributable to activities at our foreign subsidiaries and state income taxes. The tax benefit relating to the loss before income taxes for the years ended December 31, 2016 and 2015 in the U.S. was fully offset by a tax valuation allowance resulting from our assessment that it is more likely than not that our deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the period in which NOLs and credit carryforwards can be utilized.
47
Table of Contents
Year ended December 31, 2015 compared to year ended December 31, 2014
Revenues. Total revenues increased by $59.8 million, or 119%, to $109.9 million for the year ended December 31, 2015 compared to $50.2 million for the year ended December 31, 2014. During the years ended December 31, 2015 and 2014, revenues consisted of the following:
| Year Ended December 31, | ||||||||||||||||
| (in thousands) | 2015 | 2014 | Net Change | Percent | ||||||||||||
| HETLIOZ® product sales, net |
$ | 44,302 | $ | 12,802 | $ | 31,500 | 246 | % | ||||||||
| Fanapt® product sales, net |
65,623 | 107 | 65,516 | (1 | ) | |||||||||||
| Fanapt® royalty revenue |
— | 6,502 | (6,502 | ) | -100 | % | ||||||||||
| Fanapt® licensing agreement |
— | 30,746 | (30,746 | ) | -100 | % | ||||||||||
|
|
|
|
|
|
|
|||||||||||
| $ | 109,925 | $ | 50,157 | $ | 59,768 | 119 | % | |||||||||
|
|
|
|
|
|
|
|||||||||||
| (1) | Percentage change not meaningful. |
HETLIOZ® was commercially launched in the U.S. in April 2014. Pursuant to the terms of the Settlement Agreement, Novartis transferred all U.S. and Canadian rights in the Fanapt® franchise to us in December 2014. We began selling Fanapt® commercially in the U.S. in January 2015. Fanapt® royalty revenue for the year ended December 31, 2014 represented amounts due from Novartis based on quarterly U.S. sales of Fanapt® by Novartis, and Fanapt® license revenue for the year ended December 31, 2014 represented amortization of deferred revenue from the $200.0 million up-front license fee received from Novartis. Pursuant to the Settlement Agreement, royalties from Novartis ceased, and the remaining balance of the deferred revenue as of December 31, 2014 related to the up-front license fee was recognized as part of gain on arbitration settlement in the consolidated statement of operations for the year ended December 31, 2014.
Cost of goods sold. Cost of goods sold was $23.5 million for the year ended December 31, 2015, compared to $1.6 million for the year ended December 31, 2014. HETLIOZ® was commercially launched in the U.S. in April 2014, and we began selling Fanapt® commercially in the U.S. in January 2015. Cost of goods sold includes third party manufacturing costs of product sold, third party royalty costs and distribution and other costs. Third party royalty costs are 10% of net U.S. sales of HETLIOZ® and 23% of net U.S. sales of Fanapt®.
HETLIOZ® cost of goods sold as a percentage of HETLIOZ® revenue depends upon our cost to manufacture inventory at normalized production levels with our third party manufacturers. We expect that, in the future, total HETLIOZ® manufacturing costs included in cost of goods sold will be less than 2% of our net HETLIOZ® product sales.
Fanapt® work-in-process inventory and finished goods inventory acquired from Novartis as part of the acquisition of the Fanapt® business were recorded at fair value. The fair value of the inventory acquired from Novartis represents a higher cost than if new work-in-process inventory and finished goods inventory was manufactured at this time. We expect that, in the future, total U.S. Fanapt® manufacturing costs included in cost of goods sold will be less than 4% of our net U.S. Fanapt® product sales.
Research and development expenses. Research and development expenses increased by $9.9 million, or 52%, to $29.1 million for year ended December 31, 2015 compared to $19.2 million for the year ended December 31, 2014. The increase is primarily the result of the close out of Fanapt® clinical trial expenses transitioned to us as part of the Settlement Agreement and regulatory expenses related to our sNDA and EMA filings. The following table summarizes the costs of our product development initiatives for the years ended December 31, 2015 and 2014.
48
Table of Contents
| Year Ended December 31, |
||||||||
| (in thousands) | 2015 | 2014 | ||||||
| Direct project costs (1) |
||||||||
| HETLIOZ® |
$ | 10,444 | $ | 12,478 | ||||
| Fanapt® |
8,501 | 160 | ||||||
| Tradipitant |
4,006 | 2,303 | ||||||
| Trichostatin A |
1,681 | 335 | ||||||
|
|
|
|
|
|||||
| 24,632 | 15,276 | |||||||
|
|
|
|
|
|||||
| Indirect project costs (1) |
||||||||
| Stock-based compensation |
2,269 | 1,933 | ||||||
| Other indirect overhead |
2,244 | 2,021 | ||||||
|
|
|
|
|
|||||
| 4,513 | 3,954 | |||||||
|
|
|
|
|
|||||
| Total research & development expense |
$ | 29,145 | $ | 19,230 | ||||
|
|
|
|
|
|||||
| (1) | We record direct costs, including personnel costs and related benefits, on a project-by-project basis. Many of our research and development costs are not attributable to any individual project because we share resources across several development projects. We record indirect costs that support a number of our research and development activities in the aggregate, including stock-based compensation expense. |
We expect to incur significant research and development expenses as we continue to develop our products. In addition, we expect to incur licensing costs in the future that could be substantial, as we continue our efforts to develop our products.
Selling, general and administrative expenses. Selling, general and administrative expenses were $84.5 million for the year ended December 31, 2015, relatively unchanged from $84.6 million for the year ended December 31, 2014. We incurred costs associated with our Non-24 Disease Awareness campaign and HETLIOZ® branded advertising campaign during the year ended December 31, 2014. During the year ended December 31, 2015, we continued to incur costs associated with these campaigns but at reduced amounts when compared with the year ended December 31, 2014. The decrease associated with these campaigns was partially offset by marketing and sales efforts around both HETLIOZ® and Fanapt® in the U.S., an increase in the number of employees during the year ended December 31, 2015, as well as increased legal fees associated with ongoing patent litigation. Stock-based compensation expense associated with selling, general and administrative expense increased by $1.8 million, or 46%, to $5.7 million, for year ended December 31, 2015 compared to $3.9 million for the year ended December 31, 2014. The increase in expense was primarily the result of an increase in the number of employees during 2015 due to the hiring of new members of the executive management team, the field-based sales team and the medical affairs team.
Intangible asset amortization. Intangible asset amortization increased by $10.7 million to $13.0 million for year ended December 31, 2015 compared to $2.3 million for the year ended December 31, 2014. The increase reflects additional amortization of $8.3 million relating to Fanapt®. Pursuant to the terms of the Settlement Agreement, Novartis transferred all U.S. and Canadian rights in the Fanapt® franchise to us in December 2014 resulting in an increase in capitalized intangible assets of $15.9 million that is being amortized until November 2016. The increase also reflects additional amortization of $2.4 million relating to HETLIOZ®. The likelihood of achieving a future milestone obligation that becomes payable to BMS when cumulative sales of HETLIOZ® equal $250.0 million was determined to be probable in the first quarter of 2015 resulting in an increase in capitalized intangible assets of $25.0 million and a corresponding increase in accrued non-current liabilities. The additional amortization relating to HETLIOZ® in 2015 includes a catch-up adjustment of $1.2 million to retroactively record cumulative amortization from February 1, 2014 to December 31, 2014. We expect that annual amortization of capitalized intangible asset costs relating to HETLIOZ® will amount to $1.7 million in future years until the expiration of the patent in 2033.
Gain on arbitration settlement. Pursuant to the Settlement Agreement with Novartis, we recorded a gain of $77.6 million for the year ended December 31, 2014.
Provision (benefit) for income taxes. The tax provision (benefit) for the years ended December 31, 2015 and 2014 was fully offset by a tax valuation allowance resulting from our assessment that it is more likely than not that our deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the period in which NOLs and credit carryforwards can be utilized.
Liquidity and Capital Resources
As of December 31, 2016, our total cash and cash equivalents and marketable securities were $141.3 million compared to $143.2 million at December 31, 2015. Our cash and cash equivalents are deposits in operating accounts and highly liquid investments
49
Table of Contents
with an original maturity of 90 days or less at date of purchase and consist of investments in money market funds with commercial banks and financial institutions, and commercial paper of high-quality corporate issuers. Our marketable securities consist of investments in government sponsored enterprises and commercial paper.
Our liquidity resources as of December 31, 2016 and December 31, 2015 are summarized as follows:
| (in thousands) | December 31, 2016 |
December 31, 2015 |
||||||
| Cash and cash equivalents |
$ | 40,426 | $ | 50,843 | ||||
| Marketable securities: |
||||||||
| U.S. Treasury and government agencies |
50,647 | 44,057 | ||||||
| Corporate debt |
50,267 | 48,280 | ||||||
|
|
|
|
|
|||||
| Total marketable securities |
100,914 | 92,337 | ||||||
|
|
|
|
|
|||||
| Total cash and cash equivalents |
$ | 141,340 | $ | 143,180 | ||||
|
|
|
|
|
|||||
As of December 31, 2016, we maintained all of our cash and cash equivalents in three financial institutions. Deposits held with these institutions may exceed the amount of insurance provided on such deposits, but we do not anticipate any losses with respect to such deposits.
We expect to incur substantial costs and expenses throughout 2017 and beyond in connection with our U.S. commercial activities for HETLIOZ® and Fanapt®, including Medicaid rebates, the European commercial launch activities for HETLIOZ® and a probable future milestone payment of $25.0 million to BMS in the event cumulative worldwide sales of HETLIOZ® reach $250.0 million. During this time, we will evaluate the commercial opportunity for Fanapt® in Europe, assuming EMA approval. Additionally, we continue to pursue market approval of HETLIOZ® and Fanapt® in other regions. Because of the uncertainties discussed above, the costs to advance our research and development projects and the U.S. commercial activities for HETLIOZ® and Fanapt® are difficult to estimate and may vary significantly. Management believes that our existing funds will be sufficient to meet our operating plans for the foreseeable future. Our future capital requirements and the adequacy of our available funds will depend on many factors, primarily including our ability to generate revenue, the scope and costs of our commercial, manufacturing and process development activities and the magnitude of our discovery, preclinical and clinical development programs.
We may need or desire to obtain additional capital to finance our operations through debt, equity or alternative financing arrangements. We may also seek capital through collaborations or partnerships with other companies. The issuance of debt could require us to grant liens on certain of our assets that may limit our flexibility and debt securities may be convertible into common stock. If we raise additional capital by issuing equity securities, the terms and prices for these financings may be much more favorable to the new investors than the terms obtained by our existing stockholders. These financings also may significantly dilute the ownership of our existing stockholders. If we are unable to obtain additional financing, we may be required to reduce the scope of our future activities which could harm our business, financial condition and operating results. There can be no assurance that any additional financing required in the future will be available on acceptable terms, if at all.
50
Table of Contents
Cash flow
The following table summarizes our net cash flows from operating, investing and financing activities for the years ended December 31, 2016, 2015 and 2014:
| Year Ended December 31, | ||||||||||||
| (in thousands) | 2016 | 2015 | 2014 | |||||||||
| Net cash provided by (used in): |
||||||||||||
| Operating activities: |
||||||||||||
| Net income (loss) |
$ | (18,010 | ) | $ | (39,865 | ) | $ | 20,186 | ||||
| Deferred licensing revenues |
— | — | (30,572 | ) | ||||||||
| Gain on arbitration settlement with Novartis Pharma AG |
— | — | (77,616 | ) | ||||||||
| Other non-cash charges |
21,015 | 22,675 | 9,075 | |||||||||
| Net change in operating assets and liabilities |
(11,108 | ) | 29,639 | (2,627 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Operating activities |
(8,103 | ) | 12,449 | (81,554 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Investing activities: |
||||||||||||
| Net purchases of marketable securities |
(8,618 | ) | (24,071 | ) | (3,513 | ) | ||||||
| Other |
(1,453 | ) | (2,527 | ) | (8,524 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Investing activities |
(10,071 | ) | (26,598 | ) | (12,037 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Financing activities |
||||||||||||
| Net proceeds from common stock offerings |
— | — | 87,313 | |||||||||
| Proceeds from exercise of employee stock options and other |
7,751 | 4,091 | 2,415 | |||||||||
|
|
|
|
|
|
|
|||||||
| Financing activities |
7,751 | 4,091 | 89,728 | |||||||||
|
|
|
|
|
|
|
|||||||
| Effect of foreign currencies on cash and cash equivalents |
6 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net decrease in cash and cash equivalents |
$ | (10,417 | ) | $ | (10,058 | ) | $ | (3,863 | ) | |||
|
|
|
|
|
|
|
|||||||
Year ended December 31, 2016 compared to year ended December 31, 2015
Net cash used in operating activities was $8.1 million for the year ended December 31, 2016, a decrease of $20.6 million compared with net cash provided of $12.4 million for the year ended December 31, 2015. The decrease reflects a net reduction of $40.7 million from the net change in operating assets and liabilities, including a decrease in accrued government and other rebates of $36.5 million primarily from sales allowances relating to our initial sales of Fanapt® in 2015 and a decrease in accounts payable and accrued liabilities of $8.9 million, partly offset by a net decrease of $8.4 million in accounts receivable primarily relating to initial U.S. sales of Fanapt® in 2015. The effect of the net changes in operating assets and liabilities was partially offset by a reduction in the net loss of $21.9 million.
Year ended December 31, 2015 compared to year ended December 31, 2014
Net cash provided by operating activities was $12.4 million for the year ended December 31, 2015, an increase of $94.0 million from net cash used in operating activities of $81.6 million for the year ended December 31, 2014. The increase resulted from a reduction in the net loss of $17.6 million, excluding the non-cash gain of $77.6 million on arbitration settlement recognized in the year ended December 31, 2014, non-cash amortization of deferred licensing revenue of $30.6 million relating to Fanapt® in the year ended December 31, 2014, and an increase of $13.6 million in other non-cash charges primarily from an increase the amortization of intangible assets relating to Fanapt®. In addition, the increase in net cash provided by operating activities reflects a net increase in accrued government and other rebates of $34.6 million primarily for sales allowances relating to initial sales of Fanapt® in 2015 and an increase in accounts payable and accrued liabilities of $8.4 million. The increase in net cash provided by operating activities was partly offset by a net increase of $11.1 million in accounts receivable resulting from U.S. sales of Fanapt® which we commenced in 2015.
Off-balance sheet arrangements
We have no off-balance sheet arrangements, as defined in Item 303(a)(4) of the Securities and Exchange Commission’s Regulation S-K.
51
Table of Contents
Contractual obligations and commitments
The following is a summary of our non-cancellable long-term contractual cash obligations as of December 31, 2016:
| Cash payments due by year (1) (2) | ||||||||||||||||||||||||||||
| (in thousands) | Total | 2017 | 2018 | 2019 | 2010 | 2021 | Thereafter | |||||||||||||||||||||
| Operating leases |
$ | 21,623 | $ | 1,981 | $ | 2,216 | $ | 2,271 | $ | 2,327 | $ | 2,170 | $ | 10,658 | ||||||||||||||
| Milestone obligation (3) |
25,000 | — | 25,000 | — | — | — | — | |||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| $ | 46,623 | $ | 1,981 | $ | 27,216 | $ | 2,271 | $ | 2,327 | $ | 2,170 | $ | 10,658 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| (1) | This table does not include various agreements that we have entered into for services with third party vendors, including agreements to conduct clinical trials, to manufacture products, and for consulting and other contracted services due to the cancelable nature of the services. We accrued the costs of these agreements based on estimates of work completed to date. Additionally, this table does not include rebates, chargebacks or discounts recorded as liabilities at the time that product sales are recognized as revenue. |
| (2) | This table does not include potential future milestone obligations under our license agreement with Eli Lilly for the exclusive rights to develop and commercialize tradipitant where we could be obligated to make future milestone payments of up to $4.0 million for pre-NDA approval milestones and up to $95.0 million for future regulatory approval and sales milestones. |
| (3) | This table includes a probable future milestone obligation under our license agreement with BMS, where we will be obligated to make a future milestone payment of $25.0 million in the event cumulative worldwide sales of HETLIOZ® reach $250.0 million. This probable obligation has been accrued as a non-current liability in our consolidated balance sheet as of December 31, 2016. |
Operating leases
Commitments relating to operating leases represent the minimum annual future payments under operating leases and subleases for a total of 40,188 square feet of office space for our headquarters office at 2200 Pennsylvania Avenue, N.W. in Washington, D.C. that expire in 2026, the operating lease for 2,880 square feet of office space for our European headquarters in London that has a noncancellable lease term ending in 2021, and 1,249 square feet of office space in Berlin under a short-term operating lease.
| ITEM 7A. | QUALITATIVE AND QUANTITATIVE DISCLOSURES ABOUT MARKET RISK |
Interest rate risks
Our exposure to market risk is currently confined to our cash and cash equivalents, marketable securities and restricted cash. We currently do not hedge interest rate exposure. We have not used derivative financial instruments for speculation or trading purposes. Because of the short-term maturities of our cash and cash equivalents and marketable securities, we do not believe that an increase in market rates would have any significant impact on the realized value of our investments.
Concentrations of credit risk
We deposit our cash with financial institutions that we consider to be of high credit quality and purchase marketable securities which are generally investment grade, liquid, short-term fixed income securities and money-market instruments denominated in U.S. dollars. Our marketable securities consist of certificates of deposit, commercial paper, corporate notes and U.S. government agency notes.
Revenues and accounts receivable are concentrated with specialty pharmacies and wholesalers. There were five major customers that each accounted for more than 10% of total revenues and, as a group, represented 86% of total revenues for the year ended December 31, 2016. There were five major customers that each accounted for more than 10% of accounts receivable and, as a group, represented 92% of total accounts receivable at December 31, 2016. We mitigate our credit risk relating to accounts receivable from customers by performing ongoing credit evaluations.
Foreign currency risk
We are exposed to risks related to changes in foreign currency exchange rates relating to our foreign operations. The functional currency of our international subsidiaries is the local currency. We are exposed to foreign currency risk to the extent that we enter into transactions denominated in currencies other than our subsidiaries’ respective functional currencies. We are also exposed to unfavorable fluctuations of the U.S. dollar, which is our reporting currency, against the currencies of our operating subsidiaries when their respective financial statements are translated into U.S. dollars for inclusion in our consolidated financial statements. We do not currently hedge our foreign currency exchange rate risk. Foreign currency has not had a material impact on our results of operations.
52
Table of Contents
Effects of inflation
Inflation has not had a material impact on our results of operations.
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
The consolidated financial statements and related financial statement schedules required to be filed are listed in the Index to Consolidated Financial Statements and are incorporated in Item 15 of Part IV of this annual report on Form 10-K.
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
| ITEM 9A. | CONTROLS AND PROCEDURES |
Conclusion Regarding the Effectiveness of Disclosure Controls and Procedures
Under the supervision and with the participation of our management, including the Chief Executive Officer and Chief Financial Officer, we evaluated the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities and Exchange Act of 1934 (Exchange Act)) as of December 31, 2016. Based upon that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures are effective as of December 31, 2016, the end of the period covered by this annual report on Form 10-K, to ensure that the information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosures.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining an adequate system of internal control over financial reporting, as defined in the Exchange Act Rule 13a-15(f). Management conducted an assessment of our internal control over financial reporting based on the original framework established in 2013 by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control — Integrated Framework. Based on the assessment, management concluded that, as of December 31, 2016, our internal control over financial reporting was effective. The effectiveness of our internal control over financial reporting as of December 31, 2016 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report included in this annual report on Form 10-K.
Changes in Internal Control over Financial Reporting
There has been no change in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) during the fourth quarter of 2016 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting, other than additional controls related to the consolidation of our foreign subsidiaries.
| ITEM 9B. | OTHER INFORMATION |
None.
PART III
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
Information required under this item will be contained in our Proxy Statement for the 2017 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2016, under the captions “Election of Directors,” “Executive Officers,” “Corporate Governance,” and “Section 16(a) Beneficial Ownership Reporting Compliance” and is incorporated herein by reference pursuant to General Instruction G(3) to Form 10-K.
| ITEM 11. | EXECUTIVE COMPENSATION |
Information required under this item will be contained in our Proxy Statement for the 2017 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2016, under the captions “Corporate Governance” and “Executive Compensation,” and is incorporated herein by reference pursuant to General Instruction G(3) to Form 10-K, except that information required by Item 407(e)(5) of Regulation S-K will be deemed furnished in this Form 10-K and will not be deemed incorporated by reference into any filing under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, except to the extent that we specifically incorporate it by reference into such filing.
53
Table of Contents
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
Information required under this item will be contained in our Proxy Statement for the 2017 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2016, under the captions “Equity Compensation Plan Information” and “Security Ownership of Certain Beneficial Owners and Management” and is incorporated herein by reference pursuant to General Instruction G(3) to Form 10-K.
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
Information required under this item will be contained in our Proxy Statement for the 2017 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2016, under the caption “Corporate Governance” and is incorporated herein by reference pursuant to General Instruction G(3) to Form 10-K.
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
Information required under this item will be contained in our Proxy Statement for the 2017 Annual Meeting of Stockholders to be filed with the SEC within 120 days after the end of the fiscal year ended December 31, 2016, under the caption “Ratification of Selection of Independent Registered Public Accounting Firm” and is incorporated herein by reference pursuant to General Instruction G (3) to Form 10-K.
PART IV
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENTS SCHEDULES |
The consolidated financial statements filed as part of this annual report on Form 10-K are listed in the Index to Consolidated Financial Statements. Certain schedules are omitted because they are not applicable, or not required, or because the required information is included in the consolidated financial statements or notes thereto. The Exhibits are listed in the Exhibit Index.
54
Table of Contents
Pursuant to the requirements of Section 13 and 15(d) of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this annual report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized.
| Vanda Pharmaceuticals Inc. | ||||||
| February 17, 2017 | By: | /s/ Mihael H. Polymeropoulos, M.D. | ||||
| Mihael H. Polymeropoulos, M.D. | ||||||
| President and Chief Executive Officer | ||||||
Pursuant to the requirements of the Securities Act of 1934, this annual report on Form 10-K has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Name |
Title |
Date | ||
| /s/ Mihael H. Polymeropoulos, M.D. Mihael H. Polymeropoulos, M.D. |
President and Chief Executive Officer and Director (principal executive officer) | February 17, 2017 | ||
| /s/ James P. Kelly James P. Kelly |
Senior Vice President, Chief Financial Officer and Treasurer (principal financial officer and principal accounting officer) | February 17, 2017 | ||
| /s/ H. Thomas Watkins H. Thomas Watkins |
Chairman of the Board and Director |
February 17, 2017 | ||
| /s/ Kenneth M. Bate Kenneth M. Bate |
Director | February 17, 2017 | ||
| /s/ Michael Cola Michael Cola |
Director | February 17, 2017 | ||
| /s/ Richard W. Dugan Richard W. Dugan |
Director | February 17, 2017 | ||
| /s/ Vincent J. Milano Vincent J. Milano |
Director | February 17, 2017 | ||
55
Table of Contents
Vanda Pharmaceuticals Inc.
Index to Consolidated Financial Statements
56
Table of Contents
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Vanda Pharmaceuticals Inc.
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations, of comprehensive income (loss), of changes in stockholders’ equity, and of cash flows present fairly, in all material respects, the financial position of Vanda Pharmaceuticals Inc. and its subsidiaries at December 31, 2016 and 2015, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2016 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2016, based on criteria established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for these financial statements, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in Management’s Report on Internal Control over Financial Reporting appearing under item 9A. Our responsibility is to express opinions on these financial statements and on the Company’s internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
Baltimore, Maryland
February 17, 2017
57
Table of Contents
VANDA PHARMACEUTICALS INC.
| (in thousands, except for share and per share amounts) | December 31, 2016 |
December 31, 2015 |
||||||
| ASSETS |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 40,426 | $ | 50,843 | ||||
| Marketable securities |
100,914 | 92,337 | ||||||
| Accounts receivable, net |
20,268 | 16,331 | ||||||
| Inventory |
779 | 1,294 | ||||||
| Prepaid expenses and other current assets |
11,788 | 5,742 | ||||||
|
|
|
|
|
|||||
| Total current assets |
174,175 | 166,547 | ||||||
| Property and equipment, net |
5,015 | 4,570 | ||||||
| Intangible assets, net |
27,819 | 38,752 | ||||||
| Non-current inventory and other |
3,365 | 3,181 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 210,374 | $ | 213,050 | ||||
|
|
|
|
|
|||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY |
||||||||
| Current liabilities: |
||||||||
| Accounts payable and accrued liabilities |
$ | 16,196 | $ | 15,767 | ||||
| Accrued government and other rebates |
34,124 | 35,550 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
50,320 | 51,317 | ||||||
| Milestone obligation under license agreement |
25,000 | 25,000 | ||||||
| Other non-current liabilities |
3,724 | 3,706 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
79,044 | 80,023 | ||||||
|
|
|
|
|
|||||
| Commitments and contingencies (Notes 13 and 18) |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.001 par value; 20,000,000 shares authorized, and no shares issued or outstanding |
— | — | ||||||
| Common stock, $0.001 par value; 150,000,000 shares authorized; 44,000,614 and 42,815,291 shares issued and outstanding at December 31, 2016 and December 31, 2015, respectively |
44 | 43 | ||||||
| Additional paid-in capital |
477,087 | 460,794 | ||||||
| Accumulated other comprehensive income |
58 | 39 | ||||||
| Accumulated deficit |
(345,859 | ) | (327,849 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
131,330 | 133,027 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 210,374 | $ | 213,050 | ||||
|
|
|
|
|
|||||
The accompanying notes are an integral part of these consolidated financial statements.
58
Table of Contents
VANDA PHARMACEUTICALS INC.
CONSOLIDATED STATEMENTS OF OPERATIONS
| (in thousands, except for share and | Year Ended December 31, | |||||||||||
| per share amounts) | 2016 | 2015 | 2014 | |||||||||
| Revenues: |
||||||||||||
| Net product sales |
$ | 146,017 | $ | 109,925 | $ | 12,909 | ||||||
| Royalty revenue |
— | — | 6,502 | |||||||||
| Licensing agreement |
— | — | 30,746 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total revenues |
146,017 | 109,925 | 50,157 | |||||||||
| Operating expenses: |
||||||||||||
| Cost of goods sold, excluding amortization |
24,712 | 23,462 | 1,583 | |||||||||
| Research and development |
29,156 | 29,145 | 19,230 | |||||||||
| Selling, general and administrative |
99,787 | 84,531 | 84,644 | |||||||||
| Intangible asset amortization |
10,933 | 12,972 | 2,254 | |||||||||
| Gain on arbitration settlement |
— | — | (77,616 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
164,588 | 150,110 | 30,095 | |||||||||
|
|
|
|
|
|
|
|||||||
| Income (loss) from operations |
(18,571 | ) | (40,185 | ) | 20,062 | |||||||
| Other income |
665 | 320 | 124 | |||||||||
|
|
|
|
|
|
|
|||||||
| Income (loss) before income taxes |
(17,906 | ) | (39,865 | ) | 20,186 | |||||||
| Provision for income taxes |
104 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net income (loss) |
$ | (18,010 | ) | $ | (39,865 | ) | $ | 20,186 | ||||
|
|
|
|
|
|
|
|||||||
| Net income (loss) per share: |
||||||||||||
| Basic |
$ | (0.41 | ) | $ | (0.94 | ) | $ | 0.58 | ||||
|
|
|
|
|
|
|
|||||||
| Diluted |
$ | (0.41 | ) | $ | (0.94 | ) | $ | 0.55 | ||||
|
|
|
|
|
|
|
|||||||
| Weighted average shares outstanding: |
||||||||||||
| Basic |
43,449,441 | 42,250,254 | 34,774,163 | |||||||||
|
|
|
|
|
|
|
|||||||
| Diluted |
43,449,441 | 42,250,254 | 36,686,723 | |||||||||
|
|
|
|
|
|
|
|||||||
The accompanying notes are an integral part of these consolidated financial statements.
59
Table of Contents
VANDA PHARMACEUTICALS INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME (LOSS)
| Year Ended December 31, | ||||||||||||
| (in thousands) | 2016 | 2015 | 2014 | |||||||||
| Net income (loss) |
$ | (18,010 | ) | $ | (39,865 | ) | $ | 20,186 | ||||
|
|
|
|
|
|
|
|||||||
| Other comprehensive income (loss): |
||||||||||||
| Net foreign currency translation loss |
(1 | ) | — | — | ||||||||
| Change in net unrealized gain (loss) on marketable securities |
20 | 23 | (5 | ) | ||||||||
| Tax provision on other comprehensive income (loss) |
— | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Other comprehensive income (loss), net of tax |
19 | 23 | (5 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Comprehensive income (loss) |
$ | (17,991 | ) | $ | (39,842 | ) | $ | 20,181 | ||||
|
|
|
|
|
|
|
|||||||
The accompanying notes are an integral part of these consolidated financial statements.
60
Table of Contents
VANDA PHARMACEUTICALS INC.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
| Common Stock | Additional Paid-in Capital |
O ther Comprehensive Income (Loss) |
Accumulated Deficit |
|||||||||||||||||||||
| (in thousands, except for share amounts) | Shares | Par Value | Total | |||||||||||||||||||||
| Balances at December 31, 2013 |
33,338,543 | $ | 33 | $ | 352,240 | $ | 21 | $ | (308,170 | ) | $ | 44,124 | ||||||||||||
| Net proceeds from public offering of common stock |
5,750,000 | 5 | 62,308 | — | — | 62,313 | ||||||||||||||||||
| Issuance of common stock to Novartis Pharma AG |
1,808,973 | 2 | 25,903 | — | — | 25,905 | ||||||||||||||||||
| Issuance of common stock from the exercise of stock options and settlement of restricted stock units |
621,231 | 1 | 2,851 | — | — | 2,852 | ||||||||||||||||||
| Shares withheld upon settlement of restricted stock units |
(32,386 | ) | — | (436 | ) | — | — | (436 | ) | |||||||||||||||
| Stock-based compensation expense |
— | — | 5,878 | — | — | 5,878 | ||||||||||||||||||
| Net income |
— | — | — | — | 20,186 | 20,186 | ||||||||||||||||||
| Other comprehensive loss, net of tax |
— | — | — | (5 | ) | — | (5 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balances at December 31, 2014 |
41,486,361 | 41 | 448,744 | 16 | (287,984 | ) | 160,817 | |||||||||||||||||
| Issuance of common stock from the exercise of stock options and settlement of restricted stock units |
1,353,877 | 2 | 4,372 | — | — | 4,374 | ||||||||||||||||||
| Shares withheld upon settlement of equity awards |
(24,947 | ) | — | (283 | ) | — | — | (283 | ) | |||||||||||||||
| Stock-based compensation expense |
— | — | 7,961 | — | — | 7,961 | ||||||||||||||||||
| Net loss |
— | — | — | — | (39,865 | ) | (39,865 | ) | ||||||||||||||||
| Other comprehensive income, net of tax |
— | — | — | 23 | — | 23 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balances at December 31, 2015 |
42,815,291 | 43 | 460,794 | 39 | (327,849 | ) | 133,027 | |||||||||||||||||
| Issuance of common stock from the exercise of stock options and settlement of restricted stock units |
1,185,323 | 1 | 7,750 | — | — | 7,751 | ||||||||||||||||||
| Stock-based compensation expense |
— | — | 8,543 | — | — | 8,543 | ||||||||||||||||||
| Net loss |
— | — | — | — | (18,010 | ) | (18,010 | ) | ||||||||||||||||
| Other comprehensive income, net of tax |
— | — | — | 19 | — | 19 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balances at December 31, 2016 |
44,000,614 | $ | 44 | $ | 477,087 | $ | 58 | $ | (345,859 | ) | $ | 131,330 | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
61
Table of Contents
VANDA PHARMACEUTICALS INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
| Year Ended December 31, | ||||||||||||
| (in thousands) | 2016 | 2015 | 2014 | |||||||||
| Cash flows from operating activities |
||||||||||||
| Net income (loss) |
$ | (18,010 | ) | $ | (39,865 | ) | $ | 20,186 | ||||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: |
||||||||||||
| Depreciation of property and equipment |
935 | 582 | 530 | |||||||||
| Stock-based compensation |
8,543 | 7,961 | 5,878 | |||||||||
| Amortization of discounts and premiums on marketable securities |
62 | 677 | 174 | |||||||||
| Intangible asset amortization |
10,933 | 12,972 | 2,254 | |||||||||
| Deferred income taxes |
(104 | ) | — | — | ||||||||
| Deferred licensing revenues |
— | — | (30,572 | ) | ||||||||
| Gain on arbitration settlement with Novartis Pharma AG |
— | — | (77,616 | ) | ||||||||
| Other non-cash adjustments, net |
646 | 483 | 239 | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Accounts receivable |
(4,298 | ) | (12,677 | ) | (1,623 | ) | ||||||
| Prepaid expenses and other assets |
(6,159 | ) | (2,558 | ) | (318 | ) | ||||||
| Inventory |
200 | 387 | (2,210 | ) | ||||||||
| Accounts payable and accrued liabilities |
575 | 9,432 | 1,029 | |||||||||
| Accrued government and other rebates |
(1,426 | ) | 35,055 | 495 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) operating activities |
(8,103 | ) | 12,449 | (81,554 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from investing activities |
||||||||||||
| Acquisition of intangible assets |
— | — | (8,000 | ) | ||||||||
| Purchases of property and equipment |
(1,407 | ) | (2,527 | ) | (769 | ) | ||||||
| Purchases of marketable securities |
(165,405 | ) | (193,111 | ) | (93,343 | ) | ||||||
| Proceeds from sale of marketable securities |
— | 999 | 8,948 | |||||||||
| Maturities of marketable securities |
156,787 | 168,041 | 80,882 | |||||||||
| Change in restricted cash |
(46 | ) | — | 245 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in investing activities |
(10,071 | ) | (26,598 | ) | (12,037 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from financing activities |
||||||||||||
| Net proceeds from public offering of common stock |
— | — | 62,313 | |||||||||
| Net proceeds from offering common stock to Novartis Pharma AG |
— | — | 25,000 | |||||||||
| Proceeds from exercise of employee stock options |
7,751 | 4,374 | 2,851 | |||||||||
| Tax obligations paid in connection with settlement of restricted stock units |
— | (283 | ) | (436 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
7,751 | 4,091 | 89,728 | |||||||||
|
|
|
|
|
|
|
|||||||
| Effect of foreign currencies on cash and cash equivalents |
6 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net decrease in cash and cash equivalents |
(10,417 | ) | (10,058 | ) | (3,863 | ) | ||||||
| Cash and cash equivalents |
||||||||||||
| Beginning of year |
50,843 | 60,901 | 64,764 | |||||||||
|
|
|
|
|
|
|
|||||||
| End of year |
$ | 40,426 | $ | 50,843 | $ | 60,901 | ||||||
|
|
|
|
|
|
|
|||||||
The accompanying notes are an integral part of these consolidated financial statements.
62
Table of Contents
VANDA PHARMACEUTICALS INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Business Organization and Presentation
Business organization
Vanda Pharmaceuticals Inc. (the Company) is a global biopharmaceutical company focused on the development and commercialization of innovative therapies to address high unmet medical needs and improve the lives of patients. The Company commenced its operations in 2003 and operates in one reporting segment. The Company’s portfolio includes the following products:
| • | HETLIOZ® (tasimelteon), a product for the treatment of Non-24-Hour Sleep-Wake Disorder (Non-24), was approved by the U.S. Food and Drug Administration (FDA) in January 2014 and launched commercially in the U.S. in April 2014. In July 2015, the European Commission (EC) granted centralized marketing authorization with unified labeling for HETLIOZ® for the treatment of Non-24 in totally blind adults. HETLIOZ® was commercially launched in Germany in August 2016. HETLIOZ® has potential utility in a number of other circadian rhythm disorders and is presently in clinical development for the treatment of Pediatric Non-24, Jet Lag Disorder and Smith-Magenis Syndrome (SMS). |
| • | Fanapt® (iloperidone), a product for the treatment of schizophrenia, the oral formulation of which was approved by the FDA in May 2009 and launched commercially in the U.S. by Novartis Pharma AG (Novartis) in January of 2010. Novartis transferred all the U.S. and Canadian commercial rights to the Fanapt® franchise to the Company on December 31, 2014. Additionally, the Company’s distribution partners launched Fanapt® in Israel and Mexico in 2014. Fanapt® has potential utility in a number of other disorders. An assessment of new Fanapt® clinical opportunities is ongoing. |
| • | Tradipitant (VLY-686), a small molecule neurokinin-1 receptor (NK-1R) antagonist, which is presently in clinical development for the treatment of chronic pruritus in atopic dermatitis and gastroparesis. |
| • | Trichostatin A, a small molecule histone deacetylase (HDAC) inhibitor. |
| • | AQW051, a Phase II alpha-7 nicotinic acetylcholine receptor partial agonist. |
Basis of presentation
The accompanying consolidated financial statements includes the accounts of Vanda Pharmaceuticals Inc. and its wholly-owned subsidiaries and have been prepared in accordance with accounting principles generally accepted in the United States of America. All intercompany accounts and transactions have been eliminated in consolidation.
2. Summary of Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates that affect the reported amounts of assets and liabilities at the date of the financial statements, disclosure of contingent assets and liabilities, and the reported amounts of revenue and expenses during the reporting period. The Company has estimated its annual fees for Fanapt® under the Patient Protection and Affordable Care Act of 2010, as amended by the Health Care and Education Reconciliation Act of 2010, however, the amount of the estimated liability could increase, but the range of this increase is not reasonably estimable at this time. Management continually re-evaluates its estimates, judgments and assumptions, and management’s evaluation could change. Actual results could differ from those estimates.
Cash and Cash Equivalents
For purposes of the consolidated balance sheets and consolidated statements of cash flows, cash equivalents represent highly-liquid investments with a maturity date of three months or less at the date of purchase. Restricted cash of $0.8 million relating primarily to leases for office space is included in other current and other non-current assets at December 31, 2016 and 2015.
Marketable Securities
The Company classifies all of its marketable securities as available-for-sale securities. The Company’s investment policy requires the selection of high-quality issuers, with bond ratings of AAA to A1+/P1. Available-for-sale securities are carried at fair market value, with unrealized gains and losses reported as a component of stockholders’ equity in accumulated other comprehensive income/loss. Interest and dividend income is recorded when earned and included in interest income. Premiums and discounts on marketable securities are amortized and accreted, respectively, to maturity and included in interest income. The Company uses the specific identification method in computing realized gains and losses on the sale of investments, which would be included in the consolidated statements of operations when generated. Marketable securities with a maturity of more than one year as of the balance sheet date and which the Company does not intend to sell within the next twelve months are classified as non-current. All other marketable securities are classified as current.
63
Table of Contents
Inventory
Inventory, which is recorded at the lower of cost or net realizable value, includes the cost of third-party manufacturing and other direct and indirect costs and is valued using the first-in, first-out method. The Company capitalizes inventory costs associated with its products upon regulatory approval when, based on management’s judgment, future commercialization is considered probable and the future economic benefit is expected to be realized; otherwise, such costs are expensed as research and development. Inventory is evaluated for impairment by consideration of factors such as lower of cost or net realizable value, obsolescence or expiry. Inventory not expected to be consumed within 12 months following the balance sheet date are classified as non-current.
Intangible Assets
Costs incurred for products not yet approved by the FDA and for which no alternative future use exists are recorded as expense. In the event a product has been approved by the FDA or an alternative future use exists for a product, patent and license costs are capitalized and amortized over the expected patent life of the related product. Milestone payments to the Company’s partners are recognized when it is deemed probable that the milestone event will occur.
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation. The costs of leasehold improvements funded by or reimbursed by the lessor are capitalized and amortized as leasehold improvements along with a corresponding deferred rent liability. Depreciation of most property and equipment is provided on a straight-line basis over the estimated useful lives of the assets. Leasehold improvements are amortized using a straight-line basis over the lesser of the estimated useful lives of the assets or the terms of the related leases. The costs of additions and improvements are capitalized, and repairs and maintenance costs are charged to operations in the period incurred. Upon retirement or disposition of property and equipment, the cost and accumulated depreciation are removed from the accounts and any resulting gain or loss is reflected in the statement of operations for that period.
Accounts Payable and Accrued Liabilities
The Company’s management is required to estimate accrued liabilities as part of the process of preparing financial statements. The estimation of accrued liabilities involves identifying services that have been performed on the Company’s behalf, and then estimating the level of service performed and the associated cost incurred for such services as of each balance sheet date in the financial statements. Accrued liabilities include professional service fees, such as lawyers and accountants, contract service fees, such as those under contracts with clinical monitors, data management organizations and investigators in conjunction with clinical trials, fees to contract manufacturers in conjunction with the production of clinical materials, and fees for marketing and other commercialization activities. Pursuant to management’s assessment of the services that have been performed on clinical trials and other contracts, the Company recognizes these expenses as the services are provided. Such management assessments include, but are not limited to: (i) an evaluation by the project manager of the work that has been completed during the period, (ii) measurement of progress prepared internally and/or provided by the third-party service provider, (iii) analyses of data that justify the progress, and (iv) management’s judgment. In the event that the Company does not identify certain costs that have begun to be incurred or the Company under- or over-estimates the level of services performed or the costs of such services, the Company’s reported expenses for such period would be too low or too high.
Net Product Sales
The Company’s net product sales consist of sales of HETLIOZ® and, beginning in 2015, sales of Fanapt®. Net sales by product for the years ended December 31, 2016, 2015 and 2014 were as follows:
| Year Ended December 31, | ||||||||||||
| (in thousands) | 2016 | 2015 | 2014 | |||||||||
| HETLIOZ® product sales, net |
$ | 71,671 | $ | 44,302 | $ | 12,802 | ||||||
| Fanapt® product sales, net |
74,346 | 65,623 | 107 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 146,017 | $ | 109,925 | $ | 12,909 | |||||||
|
|
|
|
|
|
|
|||||||
The Company applies the revenue recognition guidance in accordance with Financial Accounting Standards Board (FASB) Accounting Standards Codification (ASC) Subtopic 605-15, Revenue Recognition—Products. The Company recognizes revenue from product sales when there is persuasive evidence that an arrangement exists, title to product and associated risk of loss has passed to the customer, the price is fixed or determinable, collectability is reasonably assured and the Company has no further performance obligations.
64
Table of Contents
Product Sales Discounts and Allowances
The Company’s product sales are recorded net of applicable discounts, chargebacks, rebates, co-pay assistance, service fees and product returns that are applicable for various government and commercial payors. Reserves established for discounts and returns are classified as reductions of accounts receivable if the amount is payable to direct customers, with the exception of service fees. Service fees are classified as a liability. Reserves established for chargebacks, rebates or co-pay assistance are classified as a liability if the amount is payable to a party other than customers. The Company currently records sales allowances for the following:
Rebates: Allowances for rebates include mandated and supplemental discounts under the Medicaid Drug Rebate Program as well as contracted rebate programs with other payors. Rebate amounts owed after the final dispensing of the product to a benefit plan participant are based upon contractual agreements or legal requirements with public sector benefit providers, such as Medicaid. The allowance for rebates is based on statutory or contracted discount rates and expected utilization. Estimates for the expected utilization of rebates are based on historical activity and, where available, actual and pending prescriptions for which the Company has validated the insurance benefits. Rebates are generally invoiced and paid in arrears, such that the accrual balance consists of an estimate of the amount expected to be incurred for the current quarter’s activity, plus an accrual balance for known prior quarter’s unpaid rebates. If actual future invoicing varies from estimates, the Company may need to adjust accruals, which would affect net revenue in the period of adjustment.
Chargebacks: Chargebacks are discounts that occur when contracted customers purchase directly from specialty pharmacies and wholesalers. Contracted customers, which currently consist primarily of Public Health Service institutions, non-profit clinics, and Federal government entities purchasing via the Federal Supply Schedule, generally purchase the product at a discounted price. The specialty pharmacy or wholesaler, in turn, charges back the difference between the price initially paid by the specialty pharmacy or wholesaler and the discounted price paid to the specialty pharmacy or wholesaler by the contracted customer. The allowance for chargebacks is based on historical activity and, where available, actual and pending prescriptions for which the Company has validated the insurance benefits.
Medicare Part D Coverage Gap: Medicare Part D prescription drug benefit mandates manufacturers to fund approximately 50% of the Medicare Part D insurance coverage gap for prescription drugs sold to eligible patients. Estimates for expected Medicare Part D coverage gap are based in part on historical activity and, where available, actual and pending prescriptions for which the Company has validated the insurance benefits. Funding of the coverage gap is generally invoiced and paid in arrears so that the accrual balance consists of an estimate of the amount expected to be incurred for the current quarter’s activity, plus an accrual balance for known prior quarter activity. If actual future funding varies from estimates, the Company may need to adjust accruals, which would affect net revenue in the period of adjustment.
Service Fees: The Company also incurs specialty pharmacy and wholesaler fees for services and their data. These fees are based on contracted terms and are known amounts. The Company accrues service fees at the time of revenue recognition, resulting in a reduction of product sales and the recognition of an accrued liability, unless it receives an identifiable and separate benefit for the consideration and it can reasonably estimate the fair value of the benefit received. In which case, service fees are recorded as selling, general and administrative expense.
Co-payment Assistance: Patients who have commercial insurance and meet certain eligibility requirements may receive co-payment assistance. Co-pay assistance utilization is based on information provided by the Company’s third-party administrator. The allowance for co-pay assistance is based on actual sales and an estimate for pending sales based on either historical activity or pending sales for which the Company has validated the insurance benefits.
Prompt-pay: Specialty pharmacies and wholesalers are offered discounts for prompt payment. The Company expects that the specialty pharmacies and wholesalers will earn prompt payment discounts and, therefore, deducts the full amount of these discounts from total product sales when revenues are recognized.
Product Returns: Consistent with industry practice, the Company generally offers direct customers a limited right to return as defined within the Company’s returns policy. The Company considers several factors in the estimation process, including historical return activity, expiration dates of product shipped to specialty pharmacies, inventory levels within the distribution channel, product shelf life, prescription trends and other relevant factors. The following table summarizes activity for product returns as of and for the years ended December 31, 2016, 2015 and 2014:
| (in thousands) |
||||
| Balance at December 31, 2013 |
$ | — | ||
| Additions |
85 | |||
|
|
|
|||
| Balance at December 31, 2014 |
85 | |||
| Additions |
986 | |||
| Credits/payments |
(12 | ) | ||
|
|
|
|||
| Balance at December 31, 2015 |
1,059 | |||
| Additions |
2,507 | |||
| Credits/payments |
(486 | ) | ||
|
|
|
|||
| Balance at December 31, 2016 |
$ | 3,080 | ||
|
|
|
65
Table of Contents
License Revenue
The Company’s license and royalty revenue for the year ended December 31, 2014 was derived from the amended and restated sublicense agreement with Novartis. Pursuant to the amended and restated sublicense agreement, Novartis had the right to commercialize and develop Fanapt® in the U.S. and Canada. Under the amended and restated sublicense agreement, the Company received an upfront payment of $200.0 million. Revenue related to the upfront payment was recognized ratably from the date the amended and restated sublicense agreement became effective (November 2009) through the expected duration of the Novartis commercialization of Fanapt® in the U.S. which was estimated to be through the expiry of the Fanapt® composition of patent, including a granted Hatch-Waxman extension (November 2016). In connection with the Settlement Agreement with Novartis, the Company recognized the remaining deferred revenue as of December 31, 2014 as part of the gain on arbitration settlement. See Note 3, Settlement Agreement with Novartis, for additional information.
Major Customers
HETLIOZ® is only available in the U.S. for distribution through a limited number of specialty pharmacies, and is not available in retail pharmacies. Fanapt® is available in the U.S. for distribution through a limited number of wholesalers and is available in retail pharmacies. The Company invoices and records revenue when its customers, specialty pharmacies and wholesalers, receive product from the third-party logistics warehouse. Revenues and accounts receivable are concentrated with these customers. The following table presents each major customer that represented more than 10% of total revenues for the years ended December 31, 2016, 2015 and 2014:
| Year Ended December 31, | ||||||||||||
| Percent of Total Revenues | 2016 | 2015 | 2014 | |||||||||
| Distributor A |
23 | % | 14 | % | — | |||||||
| Distributor B |
16 | % | 18 | % | — | |||||||
| Distributor C |
16 | % | 19 | % | — | |||||||
| Distributor D |
16 | % | 14 | % | — | |||||||
| Distributor E |
15 | % | 17 | % | — | |||||||
| Distributor F |
— | 12 | % | — | ||||||||
| Novartis royalty revenue |
— | — | 13 | % | ||||||||
| Novartis license agreement |
— | — | 61 | % | ||||||||
The following table presents each major customer that represented more than 10% of accounts receivable, net, as of December 31, 2016 and 2015:
| December 31, | ||||||||
| Percent of Accounts Receivable, Net | 2016 | 2015 | ||||||
| Distributor A |
22 | % | 12 | % | ||||
| Distributor B |
19 | % | 24 | % | ||||
| Distributor C |
15 | % | 17 | % | ||||
| Distributor D |
11 | % | 12 | % | ||||
| Distributor E |
25 | % | 22 | % | ||||
Cost of Goods Sold
Cost of goods sold includes royalties payable, the cost of inventory sold, manufacturing and supply chain costs and product shipping and handling costs related to U.S. sales of HETLIOZ® and sales of Fanapt® to the Company’s distribution partners.
Research and Development Expenses
Research and development expenses consist primarily of fees for services provided by third parties in connection with the clinical trials, costs of contract manufacturing services, milestone payments, costs of materials used in clinical trials and research and development, costs for regulatory consultants and filings, depreciation of capital resources used to develop products, related facilities costs, and salaries, other employee-related costs and stock-based compensation for research and development personnel. The Company expenses research and development costs as they are incurred for products in the development stage, including manufacturing costs and milestone payments made under license agreements prior to FDA approval. Upon and subsequent to FDA approval, manufacturing and milestone payments related to license agreements are capitalized. Milestone payments are accrued when it is deemed probable that the milestone event will be achieved. Costs related to the acquisition of intellectual property are expensed as incurred if the underlying technology is developed in connection with the Company’s research and development efforts and has no alternative future use.
66
Table of Contents
Selling, General and Administrative Expenses
Selling, general and administrative expenses consist of salaries, stock-based compensation, facilities and third party expenses. Selling, general and administrative expenses are associated with the activities of the executive, finance, accounting, information technology, business development, commercial support, trade and distribution, sales, marketing, legal, medical affairs and human resource functions. Additionally, selling, general and administrative expenses included an estimate for the annual Patient Protection and Affordable Care fee.
Stock-Based Compensation
Compensation costs for all stock-based awards to employees and directors are measured based on the grant date fair value of those awards and recognized over the period during which the employee or director is required to perform service in exchange for the award. The fair value of stock options granted and restricted stock units (RSUs) awarded are amortized using the straight-line method. As stock-based compensation expense recognized in the consolidated statements of operations is based on awards ultimately expected to vest, it has been reduced for estimated forfeitures. Forfeitures are required to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates
Advertising Expense
The Company expenses the costs of advertising, including branded promotional expenses, as incurred. Branded advertising expenses, recorded in selling, general and administrative expenses, were $1.4 million, $3.4 million and $5.0 million for the years ended December 31, 2016, 2015 and 2014, respectively.
Foreign Currency
The reporting currency of the Company is the U.S. dollar. The functional currency of the Company’s international subsidiaries is the local currency. Assets and liabilities, including inter-company balances for which settlement is anticipated in the foreseeable future, denominated in foreign currencies are translated at exchange rates in effect at the balance sheet date. Foreign currency equity balances are translated at historical rates. Revenues and expenses denominated in foreign currencies are translated at average exchange rates for the respective periods. Foreign currency translation adjustments are recorded in accumulated other comprehensive income.
Transactions denominated in currencies other than subsidiaries’ functional currencies are recorded based on exchange rates at the time such transactions arise. Changes in exchange rates with respect to amounts recorded in the consolidated balance sheets related to these items will result in unrealized foreign currency transaction gains and losses based upon period-end exchange rates. The Company also records realized foreign currency transaction gains and losses upon settlement of the transactions. Foreign currency transaction gains and losses are included in other income and amounted to a loss of $0.2 million for the year ended December 31, 2016. Foreign currency transaction gains and losses were not material for the years ended December 31, 2015 and 2014.
Income Taxes
The Company accounts for income taxes in accordance with the authoritative guidance on accounting for income taxes, which requires companies to account for deferred income taxes using the asset and liability method. Under the asset and liability method, current income tax expense or benefit is the amount of income taxes expected to be payable or refundable for the current year. A deferred income tax asset or liability is recognized for future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and tax credits and loss carryforwards. Deferred tax assets are reduced by a valuation allowance when, in the opinion of management, it is more likely than not that some portion or all of the deferred tax assets will not be realized. Tax rate changes are reflected in income during the period such changes are enacted. Changes in ownership may limit the amount of NOL carryforwards that can be utilized in the future to offset taxable income.
Non-Cash Investing and Financing Activities
Purchases of property and equipment accrued in current liabilities amounted to $0.2 million for each of the years ended December 31, 2016 and 2015 and zero for the year ended December 31, 2014. The acquisition of an intangible asset relating to HETLIOZ® accrued in non-current liabilities amounted to $25.0 million for the year ended December 31, 2015. The reacquired right relating to Fanapt® resulted in an intangible asset of $15.9 million, inventories of $3.0 million and a prepaid asset of $0.1 million for the year ended December 31, 2014.
Certain Risks and Uncertainties
The Company’s products under development require approval from the FDA or other international regulatory agencies prior to commercial sales. There can be no assurance the products will receive the necessary clearance. If the Company is denied clearance or clearance is delayed, it may have a material adverse impact on the Company.
67
Table of Contents
The Company’s products are concentrated in rapidly-changing, highly-competitive markets, which are characterized by rapid technological advances, changes in customer requirements and evolving regulatory requirements and industry standards. Any failure by the Company to anticipate or to respond adequately to technological developments in its industry, changes in customer requirements or changes in regulatory requirements or industry standards or any significant delays in the development or introduction of products or services could have a material adverse effect on the Company’s business, operating results and future cash flows.
The Company depends on single source suppliers for critical raw materials for manufacturing, as well as other components required for the administration of its products. The loss of these suppliers could delay the clinical trials or prevent or delay commercialization of the products.
Concentrations of Credit Risk
Financial instruments, which potentially subject the Company to significant concentrations of credit risk, consist primarily of cash, cash equivalents and marketable securities. The Company places its cash, cash equivalents and marketable securities with highly-rated financial institutions. At December 31, 2016, the Company maintained all of its cash, cash equivalents and marketable securities in three financial institutions. Deposits held with these institutions may exceed the amount of insurance provided on such deposits. Generally, these deposits may be redeemed upon demand, and the Company believes there is minimal risk of losses on such balances.
Segment and Geographic Information
The Company operates in one reporting segment and, accordingly, no segment disclosures are presented herein. HETLIOZ® was commercially launched in Germany in August 2016. Foreign sales were not material for the year ended December 31, 2016.
Recent Accounting Pronouncements
In November 2016 the FASB issued Accounting Standards Update (ASU) 2016-18, Restricted Cash. The new standard requires that a statement of cash flows explain the change during the period in the total of cash, cash equivalents and amounts generally described as restricted cash or restricted cash equivalents. Therefore, amounts generally described as restricted cash and restricted cash equivalents should be included with cash and cash equivalents when reconciling the beginning-of-period and end-of-period total amounts shown on the statement of cash flows. The standard is effective for annually reporting periods beginning after December 15, 2017, and interim periods within annual periods beginning after December 15, 2017. Early adoption is permitted. The Company is evaluating this standard to determine if adoption will have a material impact on the Company’s consolidated financial statements.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows – Classification of Certain Cash Receipts and Cash Payments, to clarify guidance on the classification of certain cash receipts and cash payments in the statement of cash flow. The standard is effective for annual reporting periods beginning after December 15, 2017, and interim periods within annual periods beginning after December 15, 2017. Early adoption is permitted. Adoption of this new standard is not expected to have a material impact on the Company’s consolidated financial statements.
In June 2016, the FASB issued ASU 2016-13, Financial Instruments – Credit Losses, related to the measurement of credit losses on financial instruments. The standard will require the use of an “expected loss” model for instruments measured at amortized cost. The standard is effective for years beginning after December 15, 2019, and interim periods within annual periods beginning after December 15, 2019. The Company is evaluating this standard to determine if adoption will have a material impact on the Company’s consolidated financial statements.
In March 2016, the FASB issued ASU 2016-09, Improvements to Employee Share-Based Payment Accounting, to simplify various aspects related to how share-based payments are accounted for and presented in the financial statements. The ASU provides that all of the tax effects related to share-based payments are recorded as part of the provision for income taxes, allows entities to withhold an amount up to the employees’ maximum individual tax rate in the relevant jurisdiction, allows entities to estimate the effect of forfeitures or recognized forfeitures when they occur, and other improvements to the accounting for share-based awards. The new standard is effective for annual periods beginning after December 15, 2016, and interim periods within annual periods beginning after December 15, 2016. Early adoption is permitted. Adoption of this new standard is not expected to have a material impact on the Company’s consolidated financial statements.
In February 2016, the FASB issued ASU 2016-02, Leases. The new standard requires that lessees will need to recognize a right-of-use asset and a lease liability for virtually all of their leases (other than leases that meet the definition of a short-term lease). The liability will be equal to the present value of lease payments. The asset will be based on the liability subject to certain adjustments. For income statement purposes, the FASB retained a dual model, requiring leases to be classified as either operating or finance. Operating leases will result in straight-line expense (similar to current operating leases) while finance leases will result in a front-loaded expense pattern (similar to current capital leases). The new standard is effective for annual periods ending after December 15, 2018, and interim periods within annual periods beginning after December 15, 2018. Early adoption is permitted. The Company is evaluating this standard to determine if adoption will have a material impact on the Company’s consolidated financial statements.
68
Table of Contents
In July 2015, the FASB issued ASU 2015-11, Simplifying the Measurement of Inventory, dealing with changes to the subsequent measurement of inventory. Currently, an entity is required to measure its inventory at the lower of cost or market, whereby market can be replacement cost, net realizable value, or net realizable value less an approximately normal profit margin. The changes require that inventory be measured at the lower of cost and net realizable value, thereby eliminating the use of the other two market methodologies. Net realizable value is defined as the estimated selling prices in the ordinary course of business less reasonably predictable costs of completion, disposal, and transportation. The new standard is effective for periods beginning after December 15, 2016. The Company adopted this new standard in the second quarter of 2016, and adoption did not have a material impact on the Company’s consolidated financial statements.
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements – Going Concern. The new standard requires management of public and private companies to evaluate whether there is substantial doubt about the entity’s ability to continue as a going concern and, if so, disclose that fact. Management will also be required to evaluate and disclose whether its plans alleviate that doubt. The new standard is effective for annual periods ending after December 15, 2016, and interim periods within annual periods beginning after December 15, 2016. The Company adopted this new standard for the year ended December 31, 2016, and adoption did not have a material impact on the Company’s consolidated financial statements.
In May 2014, the FASB issued ASU 2014-09, Revenue from Contracts with Customers. This new standard requires companies to recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which a company expects to be entitled in exchange for those goods or services. Under the new standard, revenue is recognized when a customer obtains control of a good or service. The standard allows for two transition methods—entities can either apply the new standard (i) retrospectively to each prior reporting period presented, or (ii) retrospectively with the cumulative effect of initially applying the standard recognized at the date of initial adoption. In July 2015, the FASB issued ASU 2015-14, Revenue from Contracts with Customers, which defers the effective date by one year to December 15, 2017 for fiscal years, and interim periods within those fiscal years, beginning after that date. Early adoption of the standard is permitted, but not before the original effective date of December 15, 2016. In March 2016, the FASB issued ASU 2016-08 Revenue from Contracts with Customers, Principal versus Agent Considerations (Reporting Revenue versus Net), in April 2016, the FASB issued ASU 2016-10, Revenue from Contracts with Customers, identifying Performance Obligations and Licensing, and in May 2016, the FASB issued ASU 2016-12, Revenue from Contracts with Customers, Narrow-Scope Improvements and Practical Expedients, which provide additional clarification on certain topics addressed in ASU 2014-09. ASU 2016-08, ASU 2016-10, and ASU 2016-12 follow the same implementation guidelines as ASU 2014-09 and ASU 2015-14. The initial analysis identifying areas that will be impacted by the new guidance is substantially complete, and the Company is currently analyzing the potential impacts to the consolidated financial statements and related disclosures. Revenue from the Company’s product sales is expected to remain substantially unchanged.
3. Settlement Agreement with Novartis
In May 2014, the Company commenced arbitration proceedings with Novartis relating to the license of Fanapt® (the Fanapt® Arbitration). In December 2014, the Company entered into a settlement agreement with Novartis and certain of its affiliates (the Settlement Agreement). Pursuant to the terms of the Settlement Agreement, the Company and Novartis dismissed the Fanapt® Arbitration and released each other from any related claims. In addition, in connection with the Settlement Agreement, Novartis (i) transferred all U.S. and Canadian rights in the Fanapt® franchise to the Company, (ii) purchased $25.0 million of the Company’s common stock at a price per share equal to $13.82, and (iii) granted to the Company an exclusive worldwide license to AQW051, a Phase II alpha-7 nicotinic acetylcholine receptor partial agonist.
Pursuant to the stock purchase agreement entered into as part of the Settlement Agreement, Novartis purchased $25.0 million of the Company’s common stock. The Company issued to Novartis an aggregate of 1,808,973 shares at $13.82 per share, which per share represented a 10% premium to the average closing prices of the Company’s common stock for the ten trading days prior to December 22, 2014. The Company recorded a loss of $0.9 million as part of gain on arbitration settlement in the consolidated statement of operations for the year ended December 31, 2014 related to the issuance of stock, which was valued using the Company’s closing stock price on December 31, 2014, the effective date of the transaction.
In connection with the Settlement Agreement, the Company received an exclusive worldwide license under certain patents and patent applications, and other licenses to intellectual property, to develop and commercialize AQW051. Under the AQW051 license agreement, the Company is obligated to use its commercially reasonable efforts to develop and commercialize AQW051 and is responsible for all development costs under the AQW051 license agreement. Novartis is eligible to receive tiered-royalties on net sales at percentage rates up to the mid-teens. The Company evaluated AQW051 and determined that the asset is both incomplete and has substance. However, given the early stage of AQW051 and the future costs of development, no transaction value was allocated to this asset.
The Company accounted for the Settlement Agreement in accordance with the provisions of ASC Subtopic 805, Business Combinations (ASC 805). Under the provisions of ASC 805, the acquisition date for a business is the date on which the company obtains control of the acquiree. The Company obtained control on December 31, 2014, the effective date of the Settlement Agreement.
69
Table of Contents
The following summarizes the fair value of consideration exchanged as part of the Settlement Agreement:
| (in thousands) | ||||
| Equity issued |
$ | 25,904 | ||
| Cash received |
(25,000 | ) | ||
| Settlement of pre-existing non-contractual relationship |
18,087 | |||
|
|
|
|||
| $ | 18,991 | |||
|
|
|
|||
Assets acquired and recorded at fair value as of December 31, 2014 were as follows:
| (in thousands) | ||||
| Inventory |
$ | 2,960 | ||
| Intangible—Re-acquired right |
15,940 | |||
| Prepaid services |
91 | |||
|
|
|
|||
| $ | 18,991 | |||
|
|
|
|||
The Company recorded the reacquired right as an intangible asset as of December 31, 2014 and has amortized the reacquired right on a straight-line basis through November 2016.
Due to the effective date of the Settlement Agreement being December 31, 2014, the Company did not recognize any revenue or operating expenses related to U.S. or Canadian commercial sales of Fanapt® in the consolidated statement of operations for the year ended December 31, 2014.
In connection with the Settlement Agreement, the Company and Novartis terminated the 2009 Amended Sublicense Agreement (the 2009 Agreement). Given the termination of this pre-existing contractual relationship and that there is no further obligation under the 2009 Agreement, the Company recognized a gain of $59.5 million, representing the remaining deferred revenue related to the $200.0 million upfront payment received from Novartis under the 2009 Agreement. This amount was included in gain on arbitration settlement in the consolidated statement of operations in the fourth quarter of 2014.
The Settlement Agreement provided for a mutual release of claims and dismissed the Fanapt® Arbitration, which effectively settled a pre-existing non-contractual relationship. As a result, the Company recorded an $18.1 million gain on the settlement of arbitration, which represented the value of a potential future arbitration outcome. This amount was valued based on a probability weighted scenario analysis that took into consideration the probability of each potential future alternative outcomes of the arbitration between the parties. This amount is included in gain on arbitration settlement in the consolidated statement of operations for the year ended December 31, 2014.
4. Earnings per Share
Basic earnings per share (EPS) is calculated by dividing the net income (loss) by the weighted average number of shares of common stock outstanding. Diluted EPS is computed by dividing the net income (loss) by the weighted average number of shares of common stock outstanding, plus potential outstanding common stock for the period. Potential outstanding common stock includes stock options and shares underlying RSUs, but only to the extent that their inclusion is dilutive.
70
Table of Contents
The following table presents the calculation of basic and diluted net income (loss) per share of common stock for the years ended December 31, 2016, 2015, and 2014:
| Year Ended December 31, | ||||||||||||
| (in thousands, except for share and per share amounts) | 2016 | 2015 | 2014 | |||||||||
| Numerator: |
||||||||||||
| Net income (loss) |
$ | (18,010 | ) | $ | (39,865 | ) | $ | 20,186 | ||||
|
|
|
|
|
|
|
|||||||
| Denominator: |
||||||||||||
| Weighted average shares outstanding: Basic |
43,449,441 | 42,250,254 | 34,774,163 | |||||||||
| Effect of dilutive securities |
— | — | 1,912,560 | |||||||||
|
|
|
|
|
|
|
|||||||
| Weighted average shares outstanding: Diluted |
43,449,441 | 42,250,254 | 36,686,723 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net income (loss) per share, basic and diluted: |
||||||||||||
| Basic |
$ | (0.41 | ) | $ | (0.94 | ) | $ | 0.58 | ||||
|
|
|
|
|
|
|
|||||||
| Diluted |
$ | (0.41 | ) | $ | (0.94 | ) | $ | 0.55 | ||||
|
|
|
|
|
|
|
|||||||
| Antidilutive securities excluded from calculations of diluted net income (loss) per share |
4,943,797 | 5,660,199 | 3,524,656 | |||||||||
|
|
|
|
|
|
|
|||||||
The Company incurred a net loss for each of the years ended December 31, 2016 and 2015 causing inclusion of any potentially dilutive securities to have an anti-dilutive effect, resulting in dilutive loss per share and basic loss per share attributable to common stockholders being equivalent.
5. Marketable Securities
The following is a summary of the Company’s available-for-sale marketable securities as of December 31, 2016, all of which have contract maturities of less than one year:
| December 31, 2016 (in thousands) |
Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Fair Market Value |
||||||||||||
| U.S. Treasury and government agencies |
$ | 50,661 | $ | 3 | $ | (17 | ) | $ | 50,647 | |||||||
| Corporate debt |
50,194 | 89 | (16 | ) | 50,267 | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 100,855 | $ | 92 | $ | (33 | ) | $ | 100,914 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The following is a summary of the Company’s available-for-sale marketable securities as of December 31, 2015:
| December 31, 2015 (in thousands) |
Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Fair Market Value |
||||||||||||
| U.S. Treasury and government agencies |
$ | 44,059 | $ | 6 | $ | (8 | ) | $ | 44,057 | |||||||
| Corporate debt |
48,239 | 46 | (5 | ) | 48,280 | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 92,298 | $ | 52 | $ | (13 | ) | $ | 92,337 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
6. Fair Value Measurements
Authoritative guidance establishes a three-tier fair value hierarchy, which prioritizes the inputs used in measuring fair value. These tiers include:
| • | Level 1 — defined as observable inputs such as quoted prices in active markets |
| • | Level 2 — defined as inputs other than quoted prices in active markets that are either directly or indirectly observable |
| • | Level 3 — defined as unobservable inputs in which little or no market data exists, therefore requiring an entity to develop its own assumptions |
Marketable securities classified in Level 1 and Level 2 as of December 31, 2016 and 2015 consist of available-for-sale marketable securities. The valuation of Level 1 instruments is determined using a market approach, and is based upon unadjusted quoted prices for identical assets in active markets. The valuation of investments classified in Level 2 also is determined using a market approach based upon quoted prices for similar assets in active markets, or other inputs that are observable for substantially the full term of the financial instrument. Level 2 securities include certificates of deposit, commercial paper and corporate notes that use as their basis readily observable market parameters. The Company did not transfer any assets between Level 2 and Level 1 during the years ended December 31, 2016 and 2015.
71
Table of Contents
The Company held certain assets that are required to be measured at fair value on a recurring basis as of December 31, 2016, as follows:
| Fair Value Measurement as of December 31, 2016 Using | ||||||||||||||||
| (in thousands) | December 31, 2016 |
Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
||||||||||||
|
Available-for-sale securities: |
||||||||||||||||
| U.S. Treasury and government agencies |
$ | 50,647 | $ | 50,647 | $ | — | $ | — | ||||||||
| Corporate debt |
50,267 | — | 50,267 | |||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 100,914 | $ | 50,647 | $ | 50,267 | $ | — | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The Company held certain assets that are required to be measured at fair value on a recurring basis as of December 31, 2015, as follows:
| Fair Value Measurement as of December 31, 2015 Using | ||||||||||||||||
| (in thousands) | December 31, 2015 |
Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
||||||||||||
|
Available-for-sale securities: |
||||||||||||||||
| U.S. Treasury and government agencies |
$ | 44,057 | $ | 44,057 | $ | — | $ | — | ||||||||
| Corporate debt |
48,280 | — | 48,280 | |||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 92,337 | $ | 44,057 | $ | 48,280 | $ | — | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
The Company also has financial assets and liabilities, not required to be measured at fair value on a recurring basis, which primarily consist of cash and cash equivalents, accounts receivable, restricted cash, accounts payable and accrued liabilities, the carrying value of which materially approximate their fair values.
7. Inventory
The Company evaluates expiry risk by evaluating current and future product demand relative to product shelf life. The Company builds demand forecasts by considering factors such as, but not limited to, overall market potential, market share, market acceptance and patient usage. Inventory levels are evaluated for the amount of inventory that would be sold within one year. At certain times, the level of inventory can exceed the forecasted level of cost of goods sold for the next twelve months. The Company classifies the estimate of such inventory as non-current. Inventory consisted of the following as of December 31, 2016 and 2015:
| December 31, | ||||||||
| (in thousands) | 2016 | 2015 | ||||||
| Current assets |
||||||||
|
Work-in-process |
$ | 17 | $ | — | ||||
| Finished goods |
762 | 1,294 | ||||||
|
|
|
|
|
|||||
| $ | 779 | $ | 1,294 | |||||
|
|
|
|
|
|||||
| Non-Current assets |
||||||||
| Raw materials |
$ | 127 | $ | 127 | ||||
|
Work-in-process |
2,225 | 2,369 | ||||||
| Finished goods |
83 | — | ||||||
|
|
|
|
|
|||||
| $ | 2,435 | $ | 2,496 | |||||
|
|
|
|
|
|||||
72
Table of Contents
8. Prepaid Expenses and Other Current Assets
The following is a summary of the Company’s prepaid expenses and other current assets as of December 31, 2016 and 2015:
| December 31, | ||||||||
| (in thousands) | 2016 | 2015 | ||||||
| Research and development expenses |
$ | 2,397 | $ | 2,948 | ||||
| Consulting and other professional fees |
6,051 | 1,815 | ||||||
| Prepaid royalties |
1,761 | — | ||||||
| Other |
1,579 | 979 | ||||||
|
|
|
|
|
|||||
| $ | 11,788 | $ | 5,742 | |||||
|
|
|
|
|
|||||
9. Property and Equipment
The following is a summary of the Company’s property and equipment, at cost, as of December 31, 2016 and 2015:
| Estimated Useful Life (Years) |
December 31, |
|||||||||||
| (in thousands) | 2016 | 2015 | ||||||||||
| Computer and other equipment |
3 | $ | 2,426 | $ | 2,046 | |||||||
| Furniture and fixtures |
5 - 7 | 1,412 | 1,318 | |||||||||
| Leasehold improvements |
5 -11 | 4,408 | 3,519 | |||||||||
|
|
|
|
|
|||||||||
| 8,246 | 6,883 | |||||||||||
| Accumulated depreciation and amortization |
|
(3,231 | ) | (2,313 | ) | |||||||
|
|
|
|
|
|||||||||
| $ | 5,015 | $ | 4,570 | |||||||||
|
|
|
|
|
|||||||||
Depreciation expense was $0.9 million, $0.6 million and $0.5 million for the years ended December 31, 2016, 2015 and 2014, respectively.
10. Intangible Assets
The following is a summary of the Company’s intangible assets as of December 31, 2016:
| December 31, 2016 | ||||||||||||||||
| (in thousands) | Estimated Useful Life (Years) |
Gross Carrying Amount |
Accumulated Amortization |
Net Carrying Amount |
||||||||||||
| HETLIOZ® |
January 2033 | $ | 33,000 | $ | 5,181 | $ | 27,819 | |||||||||
| Fanapt® |
November 2016 | 27,941 | 27,941 | — | ||||||||||||
|
|
|
|
|
|
|
|||||||||||
| $ | 60,941 | $ | 33,122 | $ | 27,819 | |||||||||||
|
|
|
|
|
|
|
|||||||||||
The following is a summary of the Company’s intangible assets as of December 31, 2015:
| December 31, 2015 | ||||||||||||||||
| (in thousands) | Estimated Useful Life (Years) |
Gross Carrying Amount |
Accumulated Amortization |
Net Carrying Amount |
||||||||||||
| HETLIOZ® |
January 2033 | $ | 33,000 | $ | 3,460 | $ | 29,540 | |||||||||
| Fanapt® |
November 2016 | 27,941 | 18,729 | 9,212 | ||||||||||||
|
|
|
|
|
|
|
|||||||||||
| $ | 60,941 | $ | 22,189 | $ | 38,752 | |||||||||||
|
|
|
|
|
|
|
|||||||||||
HETLIOZ®. In January 2014, the Company announced that the FDA had approved the NDA for HETLIOZ®. As a result of this approval, the Company met a milestone under its license agreement with Bristol-Myers Squibb (BMS) that required the Company to make a license payment of $8.0 million to BMS. The $8.0 million is being amortized on a straight-line basis over the remaining life of the U.S. patent for HETLIOZ®, which prior to June 2014, the Company expected to last until December 2022. In June 2014, the Company received a notice of allowance from the U.S. Patent and Trademark Office for a patent covering the method of use of HETLIOZ®. The patent expires in January 2033, thereby potentially extending the exclusivity protection in the U.S. beyond the composition of matter patent. As a result of the patent allowance, the Company extended the estimated useful life of the U.S. patent for HETLIOZ® from December 2022 to January 2033.
73
Table of Contents
The Company is obligated to make a future milestone payment to BMS of $25.0 million in the event that cumulative worldwide sales of HETLIOZ® reach $250.0 million. The likelihood of achieving the milestone and the related milestone obligation was determined to be probable during the year ended December 31, 2015. As a result, the future obligation of $25.0 million was recorded as a non-current liability as of December 31, 2016 and 2015. The $25.0 million was determined to be additional consideration for the acquisition of the HETLIOZ® intangible asset, which was created upon FDA approval on January 31, 2014. The actual payment of the $25.0 million will occur once the $250.0 million in cumulative worldwide sales of HETLIOZ® is realized, which the Company expects to occur by the end of 2018. The intangible asset of $25.0 million is being amortized on a straight-line basis over the remaining life of the U.S. patent for HETLIOZ®, which is expected to be January 2033. Amortization of intangible assets relating to HETLIOZ® amounted to $1.7 million, $2.9 million and $0.6 million for the years ended December 31, 2016, 2015 and 2014, respectively. In future periods the Company expects annual amortization of capitalized intangible asset costs relating to HETLIOZ® will amount to $1.7 million until the expiration of the patent in 2033.
Fanapt®. In 2009, the Company announced that the FDA had approved the NDA for Fanapt®. As a result of this approval, the Company met a milestone under its original sublicense agreement with Novartis that required the Company to make a license payment of $12.0 million to Novartis. The $12.0 million has been amortized on a straight-line basis over the remaining life of the U.S. composition of matter patent for Fanapt® to November 2016.
Pursuant to the Settlement Agreement, Novartis transferred all U.S. and Canadian rights in the Fanapt® franchise to the Company. As a result, the Company recognized an intangible asset of $15.9 million on December 31, 2014 related to the reacquired rights to Fanapt®, which has been amortized on a straight-line basis through November 2016. The useful life estimation for the Fanapt® intangible asset was based on the market participant methodology prescribed by ASC 805, and therefore does not reflect the impact of additional Fanapt® patents solely owned by the Company with varying expiration dates, the latest of which is December 2031. Amortization of intangible assets relating to Fanapt® was completed in November 2016 and amounted to $9.2 million, $10.1 million and $1.7 million for the years ended December 31, 2016, 2015 and 2014, respectively.
Intangible assets are amortized over their estimated useful economic life using the straight line method. Total amortization expense was $10.9 million, $13.0 million and $2.3 million for the years ended December 31, 2016, 2015 and 2014, respectively.
The following is a summary of the future intangible asset amortization schedule as of December 31, 2016:
| (in thousands) | Total | 2017 | 2018 | 2019 | 2020 | 2021 | Thereafter | |||||||||||||||||||||
| HETLIOZ® |
$ | 27,819 | $ | 1,721 | $ | 1,721 | $ | 1,721 | $ | 1,721 | $ | 1,721 | $ | 19,214 | ||||||||||||||
11. Accounts Payable and Accrued Liabilities
The following is a summary of the Company’s accounts payable and accrued liabilities as of December 31, 2016 and 2015:
| December 31, | ||||||||
| (in thousands) | 2016 | 2015 | ||||||
| Research and development expenses |
$ | 3,024 | $ | 3,199 | ||||
| Consulting and other professional fees |
3,192 | 5,088 | ||||||
| Compensation and employee benefits |
4,291 | 468 | ||||||
| Royalties payable |
4,555 | 5,328 | ||||||
| Other |
1,134 | 1,684 | ||||||
|
|
|
|
|
|||||
| $ | 16,196 | $ | 15,767 | |||||
|
|
|
|
|
|||||
12. Deferred Licensing Revenue
The following is a summary of changes in deferred licensing revenue for the year ended December 31, 2014:
| (in thousands) | Year Ended December 31, 2014 |
|||
| Balance December 31, 2013 |
$ | 90,275 | ||
| Licensing revenue recognized |
(30,746 | ) | ||
| Recognized as part of gain on arbitration settlement |
(59,529 | ) | ||
|
|
|
|||
| Balance December 31, 2014 |
$ | — | ||
|
|
|
|||
During the year ended December 31, 2014, the Company recognized revenue of $30.7 million related to the amended and restated sublicense agreement with Novartis entered into in 2009. In connection with the Settlement Agreement, the Company
recognized the remaining deferred revenue balance of $59.5 million during year ended December 31, 2014, as part of the gain on arbitration settlement. See Note 3, Settlement Agreement with Novartis, for additional information.
74
Table of Contents
13. Commitments and Contingencies
Operating Leases
Commitments relating to operating leases represent the minimum annual future payments under operating leases and subleases for a total of 40,188 square feet of office space for the Company’s headquarters at 2200 Pennsylvania Avenue, N.W. in Washington, D.C. that expire in 2026, the operating lease for 2,880 square feet of office space for the Company’s European headquarters in London that has a noncancellable lease term ending in 2021, and 1,249 square feet of office space in Berlin under a short-term operating lease. The following is a summary of the minimum annual future payments under operating leases and subleases for office space as of December 31, 2016:
| Cash payments due by year | ||||||||||||||||||||||||||||
| (in thousands) | Total | 2017 | 2018 | 2019 | 2010 | 2021 | Thereafter | |||||||||||||||||||||
| Operating leases |
$ | 21,623 | $ | 1,981 | $ | 2,216 | $ | 2,271 | $ | 2,327 | $ | 2,170 | $ | 10,658 | ||||||||||||||
In 2011, the Company entered into an operating lease for its headquarters at 2200 Pennsylvania Avenue, N.W. in Washington, D.C. A lease amendment in 2014 increased the office space under lease to 30,260 square feet, and a lease amendment in June 2016 extended the lease term from April 2023 to September 2026. Subject to the prior rights of other tenants, the Company has the right to renew the lease for five years following its expiration. The Company has the right to sublease or assign all or a portion of the premises, subject to standard conditions. The lease may be terminated early by the Company or the landlord under certain circumstances.
In June 2016, the Company entered into a sublease under which the Company will lease 9,928 square feet of office space for its headquarters at 2200 Pennsylvania Avenue, N.W. in Washington, D.C. The sublease term begins in January 2017 and ends in July 2026, but may be terminated earlier by either party under certain circumstances. The Company has the right to sublease or assign all or a portion of the premises, subject to standard conditions.
Rent expense under operating leases and subleases, was $2.5 million, $1.9 million and $1.7 million for the years ended December 31, 2016, 2015 and 2014, respectively.
Guarantees and Indemnifications
The Company has entered into a number of standard intellectual property indemnification agreements in the ordinary course of its business. Pursuant to these agreements, the Company indemnifies, holds harmless, and agrees to reimburse the indemnified party for losses suffered or incurred by the indemnified party, generally the Company’s business partners or customers, in connection with any U.S. patent or any copyright or other intellectual property infringement claim by any third party with respect to the Company’s products. The term of these indemnification agreements is generally perpetual from the date of execution of the agreement. The maximum potential amount of future payments the Company could be required to make under these indemnification agreements is unlimited. Since inception, the Company has not incurred costs to defend lawsuits or settle claims related to these indemnification agreements. The Company also indemnifies its officers and directors for certain events or occurrences, subject to certain conditions.
License Agreements
The Company’s rights to develop and commercialize its products are subject to the terms and conditions of licenses granted to the Company by other pharmaceutical companies.
HETLIOZ®. In February 2004, the Company entered into a license agreement with BMS under which it received an exclusive worldwide license under certain patents and patent applications, and other licenses to intellectual property, to develop and commercialize HETLIOZ®. In partial consideration for the license, the Company paid BMS an initial license fee of $0.5 million. The Company made a milestone payment to BMS of $1.0 million under the license agreement in 2006 relating to the initiation of its first Phase III clinical trial for HETLIOZ®. As a result of the FDA acceptance of the Company’s NDA for HETLIOZ® for the treatment of Non-24 in July 2013, the Company incurred a $3.0 million milestone obligation under the license agreement with BMS. As a result of the FDA’s approval of the HETLIOZ® NDA in January 2014, the Company incurred an $8.0 million milestone obligation in the first quarter of 2014 under the same license agreement that was capitalized as an intangible asset and is being amortized over the expected HETLIOZ® patent life in the U.S. The Company is obligated to make a future milestone payment to BMS of $25.0 million in the event that cumulative worldwide sales of HETLIOZ® reach $250.0 million. During the first quarter of 2015, the likelihood of achieving the milestone and the related milestone obligation was determined to be probable. As such, the $25.0 million milestone obligation was capitalized as an intangible asset and is being amortized over the expected HETLIOZ® patent life in the U.S. The actual payment of the $25.0 million will occur once the $250.0 million in cumulative worldwide sales of HETLIOZ® is realized, which is expected to be in 2018. Additionally, the Company is obligated to make royalty payments on HETLIOZ® net sales to BMS in any territory where the
75
Table of Contents
Company commercializes HETLIOZ® for a period equal to the greater of 10 years following the first commercial sale in the territory or the expiry of the new chemical entity patent in that territory. During the period prior to the expiry of the new chemical entity patent in a territory, the Company is obligated to pay a 10% royalty on net sales in that territory. The royalty rate is decreased by half for countries in which no new chemical entity patent existed or for the remainder of the 10 years after the expiry of the new chemical entity patent. The Company is also obligated under the license agreement to pay BMS a percentage of any sublicense fees, upfront payments and milestone and other payments (excluding royalties) that it receives from a third party in connection with any sublicensing arrangement, at a rate which is in the mid-twenties. The Company has agreed with BMS in the license agreement for HETLIOZ® to use its commercially reasonable efforts to develop and commercialize HETLIOZ®.
The license agreement was amended in April 2013 to add a process that would allow BMS to waive the right to develop and commercialize HETLIOZ® in those countries not covered by a development and commercialization agreement. Subsequent to the execution of the April 2013 amendment, BMS provided the Company with formal written notice that it irrevocably waived the option to exercise the right to reacquire any or all rights to any product (as defined in the license agreement) containing HETLIOZ®, or to develop or commercialize any such product, in the countries not covered by a development and commercialization agreement.
Either party may terminate the HETLIOZ® license agreement under certain circumstances, including a material breach of the agreement by the other. In the event the Company terminates the license, or if BMS terminates the license due to the Company’s breach, all rights licensed and developed by the Company under the license agreement will revert or otherwise be licensed back to BMS on an exclusive basis.
Fanapt®. A predecessor company of Sanofi, Hoechst Marion Roussel, Inc. (HMRI) discovered Fanapt® and completed early clinical work on the product. In 1996, following a review of its product portfolio, HMRI licensed its rights to the Fanapt® patents and patent applications to Titan Pharmaceuticals, Inc. (Titan) on an exclusive basis. In 1997, soon after it had acquired its rights, Titan sublicensed its rights to Fanapt® on an exclusive basis to Novartis. In June 2004, the Company acquired exclusive worldwide rights to these patents and patent applications, as well as certain Novartis patents and patent applications to develop and commercialize Fanapt®, through a sublicense agreement with Novartis. In October 2009, subsequent to the FDA’s approval of the NDA for Fanapt®, the Company entered into an amended and restated sublicense agreement with Novartis, which amended and restated the June 2004 sublicense agreement. Pursuant to the amended and restated sublicense agreement, Novartis had exclusive commercialization rights to all formulations of Fanapt® in the U.S. and Canada. Novartis began selling Fanapt® in the U.S. during the first quarter of 2010. Novartis was responsible for the further clinical development activities in the U.S. and Canada. The Company also received royalties equal to 10% of net sales of Fanapt® in the U.S. and Canada up to December 31, 2014. The Company retained exclusive rights to Fanapt® outside the U.S. and Canada and was obligated to make royalty payments to Sanofi S.A. (Sanofi) on Fanapt® sales outside the U.S. and Canada.
Pursuant to the terms of the Settlement Agreement with Novartis, Novartis transferred all U.S. and Canadian rights in the Fanapt® franchise to the Company on December 31, 2014. The Company is obligated to make royalty payments to Sanofi and Titan, at a percentage rate equal to 23% on annual U.S. net sales of Fanapt® up to $200.0 million, and at a percentage rate in the mid-twenties on sales over $200.0 million through November 2016. In February 2016, the Company amended the agreement with Sanofi and Titan to remove Titan as the entity through which royalty payments from the Company are directed to Sanofi following the expiration of the new chemical entity (NCE) patent for Fanapt® in the U.S. on November 15, 2016. Under the amended agreement, the Company will pay directly to Sanofi a fixed royalty of 3% of net sales from November 16, 2016 through December 31, 2019 related to manufacturing know-how. The Company made a $2.0 million payment during the year ended December 31, 2016 that applied to this 3% manufacturing know-how royalty and will make additional royalty payments only to the extent that the Company’s cumulative royalty obligations during this period exceed the amount of the pre-payment. No further royalties on manufacturing know-how are payable by the Company after December 31, 2019. This amended agreement does not alter Titan’s obligation under the license agreement to make royalty payments to Sanofi prior to November 16, 2016 or the Company’s obligations under the sublicense agreement to pay Sanofi a fixed royalty on Fanapt® net sales equal up to 6% on Sanofi know-how not related to manufacturing under certain conditions for a period of up to 10 years in markets where the NCE patent has expired or was not issued.
The Company has entered into distribution agreements with Probiomed S.A. de C.V. for the commercialization of Fanapt® in Mexico and Megapharm Ltd. for the commercialization of Fanapt® in Israel.
Tradipitant. In April 2012, the Company entered into a license agreement with Eli Lilly and Company (Lilly) pursuant to which the Company acquired an exclusive worldwide license under certain patents and patent applications, and other licenses to intellectual property, to develop and commercialize an NK-1R antagonist, tradipitant, for all human indications. The patent describing tradipitant as a new chemical entity expires in April 2023, except in the U.S., where it expires in June 2024 absent any applicable patent term adjustments. Pursuant to the license agreement, the Company paid Lilly an initial license fee of $1.0 million and will be responsible for all development costs. The initial license fee was recognized as research and development expense in the consolidated statement of operations for the year ended December 31, 2012. Lilly is also eligible to receive additional payments based upon achievement of specified development and commercialization milestones as well as tiered-royalties on net sales at percentage rates up to the low
76
Table of Contents
double digits. These milestones include $4.0 million for pre-NDA approval milestones and up to $95.0 million for future regulatory approval and sales milestones. The Company is obligated to use its commercially reasonable efforts to develop and commercialize tradipitant. Either party may terminate the license agreement under certain circumstances, including a material breach of the license agreement by the other. In the event that the Company terminates the license agreement, or if Lilly terminates due to the Company’s breach or for certain other reasons set forth in the license agreement, all rights licensed and developed by the Company under the license agreement will revert or otherwise be licensed back to Lilly on an exclusive basis, subject to payment by Lilly to the Company of a royalty on net sales of products that contain tradipitant.
AQW051. In connection with the settlement agreement with Novartis relating to Fanapt®, the Company received an exclusive worldwide license under certain patents and patent applications, and other licenses to intellectual property, to develop and commercialize AQW051, a Phase II alpha-7 nicotinic acetylcholine receptor partial agonist. Pursuant to the license agreement, the Company is obligated to use its commercially reasonable efforts to develop and commercialize AQW051 and is responsible for all development costs under the AQW051 license agreement. The Company has no milestone obligations; however, Novartis is eligible to receive tiered-royalties on net sales at percentage rates up to the mid-teens.
Research and Development and Marketing Agreements
In the course of its business, the Company regularly enters into agreements with clinical organizations to provide services relating to clinical development and clinical manufacturing activities under fee service arrangements. The Company’s current agreements for clinical services may be terminated on generally 60 days’ notice without incurring additional charges, other than charges for work completed but not paid for through the effective date of termination and other costs incurred by the Company’s contractors in closing out work in progress as of the effective date of termination.
14. Income Taxes
The Company recorded total tax expense of $0.1 million on consolidated pretax loss of $17.9 million, consisting of $18.1 million of pretax loss in the U.S. and $0.2 million of pretax income from foreign subsidiaries. The following is a summary of the provision (benefit) for income taxes for the years ended December 31, 2016, 2015 and 2014:
| Year Ended December 31, | ||||||||||||
| (in thousands) | 2016 | 2015 | 2014 | |||||||||
| Current: |
||||||||||||
| Federal |
$ | — | $ | — | $ | — | ||||||
| State |
66 | — | — | |||||||||
| Foreign |
142 | — | — | |||||||||
| Deferred: |
||||||||||||
| Federal |
— | — | — | |||||||||
| State |
— | — | — | |||||||||
| Foreign |
(104 | ) | — | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Provision for income taxes |
$ | 104 | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
Deferred tax assets are reduced by a tax valuation allowance when, in the opinion of management, it is more likely than not that some portion or all of the deferred tax assets will not be realized. The fact that the Company has historically generated pretax losses in the U.S. serves as strong evidence that it is more likely than not that deferred tax assets in the U.S. will not be realized in the future. Therefore, the Company had a full tax valuation allowance against all deferred tax assets in the U.S. as of December 31, 2016 and 2015. As a result of the tax valuation allowance against deferred tax assets in the U.S., there was no benefit for income taxes for the years ended December 31, 2016 and 2015 and there was no provision for income taxes for the years ended December 31, 2015 and 2014.
77
Table of Contents
The following is reconciliation between the federal statutory tax rate and the Company’s effective tax rate for the years ended December 31, 2016, 2015 and 2014:
| Year Ended December 31, | ||||||||||||
| 2016 | 2015 | 2014 | ||||||||||
| Federal tax at statutory rate |
35.0 | % | 35.0 | % | 34.0 | % | ||||||
| State taxes |
0.8 | % | -0.1 | % | 7.2 | % | ||||||
| Change in valuation allowance |
-38.4 | % | -25.4 | % | -59.7 | % | ||||||
| Research and development credit |
3.8 | % | 1.5 | % | 1.3 | % | ||||||
| Orphan drug credit |
7.6 | % | 1.6 | % | 8.5 | % | ||||||
| Section 162(m) limitation |
0.0 | % | -5.7 | % | 1.1 | % | ||||||
| Tax rate change |
3.9 | % | -0.3 | % | 4.8 | % | ||||||
| Change in state NOLs |
0.0 | % | -1.4 | % | 0.0 | % | ||||||
| Non-deductible stock-based compensation |
-12.5 | % | -5.1 | % | 0.0 | % | ||||||
| Other non-deductible items |
-0.8 | % | -0.1 | % | 2.8 | % | ||||||
|
|
|
|
|
|
|
|||||||
| Effective tax rate |
-0.6 | % | 0.0 | % | 0.0 | % | ||||||
|
|
|
|
|
|
|
|||||||
The following is a summary of the components of the Company’s deferred tax assets, net, and the related tax valuation allowance as of December 31, 2016 and 2015:
| December 31, | ||||||||
| (in thousands) | 2016 | 2015 | ||||||
| Deferred tax assets: |
||||||||
| Net operating loss carryforwards |
$ | 84,177 | $ | 83,686 | ||||
| Stock-based compensation |
12,443 | 12,919 | ||||||
| Accrued and deferred expenses |
2,558 | 1,443 | ||||||
| Research and development and orphan drug credit carryforwards |
41,104 | 38,333 | ||||||
| Intangible assets |
5,477 | 2,954 | ||||||
| Other |
1,019 | 802 | ||||||
|
|
|
|
|
|||||
| Total deferred tax assets |
146,778 | 140,137 | ||||||
| Deferred tax liabilities: |
||||||||
| Other |
(666 | ) | (1,100 | ) | ||||
|
|
|
|
|
|||||
| Total deferred tax liabilities |
(666 | ) | (1,100 | ) | ||||
| Deferred tax assets, net |
146,112 | 139,037 | ||||||
| Valuation allowance |
(146,012 | ) | (139,037 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax assets |
$ | 100 | $ | — | ||||
|
|
|
|
|
|||||
The Company’s net deferred tax asset of $0.1 million as of December 31, 2016 was included as a component of non-current inventory and other in the consolidated balance sheet.
The following is a summary of changes in the Company’s tax valuation allowance for the years ended December 31, 2016, 2015 and 2014:
| (in thousands) | Balance at Beginning of Year |
Additions | Reductions | Balance at End of Year |
||||||||||||
| Year Ended: |
||||||||||||||||
| December 31, 2016 |
$ | 139,037 | $ | 11,031 | $ | (4,056 | ) | $ | 146,012 | |||||||
| December 31, 2015 |
128,890 | 17,002 | (6,855 | ) | 139,037 | |||||||||||
| December 31, 2014 |
141,150 | 27,893 | (40,153 | ) | 128,890 | |||||||||||
As of December 31, 2016, the Company had federal and state net operating loss (NOL) carryforwards of $228.0 million, including $8.7 million of gross excess windfall benefits generated from stock-based compensation from which the tax benefit would be recorded to Additional Paid-in Capital if realized. As of December 31, 2016, the Company also had research and development credits of $7.5 million and orphan drug carryforward credits of $33.7 million. These NOL carryforwards and credits will begin to expire in 2028 and 2024, respectively.
Because the Company has generated NOLs from inception through December, 31, 2016, all income tax returns filed by the Company are open to examination by tax jurisdictions. As of December 31, 2016, the Company’s income tax returns had not been under examination by any federal or state tax jurisdictions. As of December 31, 2016 and 2015, the Company had no uncertain tax positions.
78
Table of Contents
Certain tax attributes of the Company, including NOLs and credits, are potentially subject to a limitation should an ownership change as defined under the Internal Revenue Code of 1986, as amended (IRC), Section 382, occur. The limitations resulting from a change in ownership could affect the Company’s ability to use NOLs and credit carryforward (tax attributes). Ownership changes did occur as of December 31, 2014 and December 31, 2008. However, the Company believes that it had sufficient Built-In-Gain to offset the IRC Section 382 limitation generated by the ownership changes. Any future ownership changes may cause the Company’s existing tax attributes to have additional limitations. Additionally, the Company maintains a valuation allowance on its tax attributes, therefore, any IRC Section 382 limitation would not have a material impact on the Company’s provision for income taxes as of December 31, 2016.
15. Public Offering of Common Stock
In October 2014, the Company completed a public offering of 5,750,000 shares of common stock at a price to the public of $11.60 per share. Net cash proceeds from the public offering were $62.3 million, after deducting the underwriting discounts and commissions and offering expenses.
16. Equity Incentive Plans
As of December 31, 2016, there were 6,686,764 shares that were subject to outstanding options and RSUs under the 2006 Equity Incentive Plan (the 2006 Plan) and the 2016 Equity Incentive Plan (the 2016 Plan) (collectively, the Plans). The 2006 Plan expired by its terms on April 12, 2016. Outstanding options and RSUs under the 2006 Plan remain in effect and the terms of the 2006 Plan continue to apply, but no additional awards can be granted under the 2006 Plan. On June 16, 2016, the Company’s stockholders approved the 2016 Plan. There are 2,000,000 shares of common stock reserved for issuance under the 2016 Plan, of which 1,822,500 shares remained available for future grant as of December 31, 2016.
Stock Options
The Company has granted option awards under the Plans with service conditions (service option awards) that are subject to terms and conditions established by the compensation committee of the board of directors. Service option awards have 10-year contractual terms and all service option awards granted prior to December 31, 2006, service option awards granted to new employees, and certain service option awards granted to existing employees vest and become exercisable on the first anniversary of the grant date with respect to the 25% of the shares subject to service option awards. The remaining 75% of the shares subject to the service option awards vest and become exercisable monthly in equal installments thereafter over three years. Certain service option awards granted to existing employees after December 31, 2006 vest and become exercisable monthly in equal installments over four years. The initial service option awards granted to directors upon their election vest and become exercisable in equal monthly installments over a period of four years, while the subsequent annual service option awards granted to directors vest and become exercisable in equal monthly installments over a period of one year. Certain service option awards to executives and directors provide for accelerated vesting if there is a change in control of the Company. Certain service option awards to employees and executives provide for accelerated vesting if the respective employee’s or executive’s service is terminated by the Company for any reason other than cause or permanent disability. As of December 31, 2016, $8.9 million of unrecognized compensation costs related to unvested service option awards are expected to be recognized over a weighted average period of 1.2 years. No option awards are classified as a liability as of December 31, 2016.
The Company’s equity incentive plan, the Second Amended and Restated Management Equity Plan (the 2004 Plan), expired by its terms in 2014 and no additional options will be granted under the 2004 Plan. There were no shares subject to outstanding options granted under the 2004 Plan as of December 31, 2016 and 2015. The following is a summary of option activity for the 2004 Plan for the years ended December 31, 2015 and 2014:
| 2004 Plan (in thousands, except for share and per share amounts) |
Number of Shares |
Weighted Average Exercise Price at Grant Date |
Weighted Average Remaining Term (Years) |
Aggregate Intrinsic Value |
||||||||||||
| Outstanding at December 31, 2013 |
670,744 | $ | 1.79 | 1.78 | $ | 7,124 | ||||||||||
| Exercised |
(17,934 | ) | 3.57 | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2014 |
652,810 | 1.74 | 0.78 | 8,212 | ||||||||||||
| Exercised |
(652,810 | ) | 1.74 | 6,129 | ||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2015 |
— | |||||||||||||||
|
|
|
|||||||||||||||
79
Table of Contents
The following is a summary of option activity for the 2006 Plan and the 2016 Plan for the years ended December 31, 2016, 2015, and 2014:
| 2006 and 2016 Plans (in thousands, except for share and per share amounts) |
Number of Shares |
Weighted Average Exercise Price at Grant Date |
Weighted Average Remaining Term (Years) |
Aggregate Intrinsic Value |
||||||||||||
| Outstanding at December 31, 2013 |
5,533,618 | $ | 10.98 | 6.93 | $ | 21,264 | ||||||||||
| Granted |
1,324,337 | 12.17 | ||||||||||||||
| Forfeited |
(237,108 | ) | 8.35 | |||||||||||||
| Exercised |
(393,735 | ) | 7.08 | 2,923 | ||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2014 |
6,227,112 | 11.58 | 6.71 | 28,523 | ||||||||||||
| Granted |
1,056,500 | 11.74 | ||||||||||||||
| Forfeited |
(496,854 | ) | 10.75 | |||||||||||||
| Expired |
(64,336 | ) | 25.69 | |||||||||||||
| Exercised |
(469,974 | ) | 7.02 | 2,594 | ||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2015 |
6,252,448 | 11.87 | 6.16 | 7,498 | ||||||||||||
| Granted |
866,011 | 8.43 | ||||||||||||||
| Forfeited |
(392,700 | ) | 11.23 | |||||||||||||
| Expired |
(279,766 | ) | 17.38 | |||||||||||||
| Exercised |
(897,657 | ) | 8.63 | 4,264 | ||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2016 |
5,548,336 | 11.62 | 5.58 | 32,453 | ||||||||||||
|
|
|
|||||||||||||||
| Exercisable at December 31, 2016 |
3,983,012 | 12.07 | 4.45 | 23,924 | ||||||||||||
|
|
|
|||||||||||||||
| Vested and expected to vest at December 31, 2016 |
5,450,280 | 11.66 | 5.52 | 31,850 | ||||||||||||
|
|
|
|||||||||||||||
The weighted average grant-date fair value of options granted was $4.53, $6.59 and $6.99 per share for the years ended December 31, 2016, 2015 and 2014, respectively. Proceeds from the exercise of stock options amounted to $7.8 million, $4.4 million and $2.9 million for the years ended December 31, 2016, 2015 and 2014, respectively.
Restricted Stock Units
An RSU is a stock award that entitles the holder to receive shares of the Company’s common stock as the award vests. The fair value of each RSU is based on the closing price of the Company’s stock on the date of grant. The Company has granted RSUs under the Plans with service conditions (service RSUs) that vest in four equal annual installments provided that the employee remains employed with the Company. As of December 31, 2016, $8.2 million of unrecognized compensation costs related to unvested service RSUs are expected to be recognized over a weighted average period of 1.8 years. No RSUs are classified as a liability as of December 31, 2016.
80
Table of Contents
The following is a summary of RSU activity for the 2006 Plan and the 2016 Plan for the years ended December 31, 2016, 2015, and 2014:
| RSUs | Number of Shares Underlying RSUs |
Weighted Average Grant Date Fair Value |
||||||
| Unvested at December 31, 2013 |
883,690 | $ | 7.70 | |||||
| Granted |
436,115 | 12.28 | ||||||
| Forfeited |
(84,282 | ) | 6.75 | |||||
| Vested |
(209,562 | ) | 6.67 | |||||
|
|
|
|||||||
| Unvested at December 31, 2014 |
1,025,961 | 9.94 | ||||||
| Granted |
417,000 | 11.51 | ||||||
| Forfeited |
(189,187 | ) | 10.60 | |||||
| Vested |
(231,093 | ) | 7.96 | |||||
|
|
|
|||||||
| Unvested at December 31, 2015 |
1,022,681 | 10.90 | ||||||
| Granted |
657,742 | 8.71 | ||||||
| Forfeited |
(254,329 | ) | 10.38 | |||||
| Vested |
(287,666 | ) | 9.65 | |||||
|
|
|
|||||||
| Unvested at December 31, 2016 |
1,138,428 | 10.07 | ||||||
|
|
|
|||||||
The grant date fair value for the 287,666 shares underlying RSUs that vested during the year ended December 31, 2016 was $2.8 million.
Stock-Based Compensation Expense
Stock-based compensation expense recognized for the years ended December 31, 2016, 2015 and 2014 was allocated as follows:
| Year Ended December 31, | ||||||||||||
| (in thousands) | 2016 | 2015 | 2014 | |||||||||
| Research and development |
$ | 2,087 | $ | 2,269 | $ | 1,933 | ||||||
| Selling, general and administrative |
6,456 | 5,692 | 3,945 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 8,543 | $ | 7,961 | $ | 5,878 | |||||||
|
|
|
|
|
|
|
|||||||
The fair value of each option award is estimated on the date of grant using the Black-Scholes-Merton option pricing model that uses the assumptions noted in the following table. Expected volatility rates are based on the historical volatility of the Company’s publicly traded common stock. The risk-free interest rates are based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of the grant. The Company has not paid dividends to its stockholders since its inception (other than a dividend of preferred share purchase rights, which was declared in September 2008) and does not plan to pay dividends in the foreseeable future. Assumptions used in the Black-Scholes-Merton option pricing model for employee and director stock options granted during the years ended December 31, 2016, 2015 and 2014 were as follows:
| Year Ended December 31, | ||||||||||||
| 2016 | 2015 | 2014 | ||||||||||
| Expected dividend yield |
0 | % | 0 | % | 0 | % | ||||||
| Weighted average expected volatility |
57 | % | 60 | % | 62 | % | ||||||
| Weighted average expected term (years) |
6.08 | 6.00 | 5.90 | |||||||||
| Weighted average risk-free rate |
1.37 | % | 1.67 | % | 1.73 | % | ||||||
17. Employee Benefit Plan
The Company has a defined contribution plan under IRC Section 401(k). This plan covers substantially all employees who meet minimum age and service requirements and allows participants to defer a portion of their annual compensation on a pre-tax basis. Currently, the Company matches 50 percent up to the first six percent of employee contributions. All matching contributions have been paid by the Company. The Company match vests over a four-year period and amounted to $0.4 million, $0.3 million and $0.2 million for the years ended December 31, 2016, 2015 and 2014, respectively.
81
Table of Contents
18. Legal Matters
In June 2014, the Company filed suit against Roxane Laboratories, Inc. (Roxane) in the U.S. District Court for the District of Delaware (the Delaware District Court). The suit seeks an adjudication that Roxane has infringed one or more claims of the Company’s U.S. Patent No. 8,586,610 (the ‘610 Patent) by submitting to the FDA an Abbreviated New Drug Application (ANDA) for a generic version of Fanapt® prior to the expiration of the ‘610 Patent in November 2027. In addition, pursuant to the settlement agreement with Novartis, the Company assumed Novartis’ patent infringement action against Roxane in the Delaware District Court. That suit alleges that Roxane has infringed one or more claims of U.S. Patent RE39198 (the ‘198 Patent), which is licensed exclusively to the Company, by filing an ANDA for a generic version of Fanapt® prior to the expiration of the ‘198 Patent in November 2016. These two cases against Roxane were consolidated by agreement of the parties and were tried together in a five-day bench trial that concluded on March 4, 2016. On August 25, 2016, the Delaware District Court ruled in favor of the Company, finding that Roxane’s ANDA product infringed the asserted claims of the ‘610 Patent and the ‘198 Patent. The Delaware District Court ruled that the Company is entitled to a permanent injunction against Roxane enjoining Roxane from infringing the ‘610 Patent, including the manufacture, use, sale, offer to sell, sale, distribution or importation of any generic iloperidone product described in the ‘610 Patent ANDA until the expiration of the ‘610 Patent in November 2027. If the Company obtains pediatric exclusivity, the injunction against Roxane would be extended until May 2028 under the Delaware District Court’s order. On September 23, 2016, Roxane filed a notice of appeal with the Federal Circuit Court of Appeals. Roxane filed its opening appellate brief on February 7, 2017.
In 2015, the Company filed six separate patent infringement lawsuits in the Delaware District Court against Roxane, Inventia Healthcare Pvt. Ltd. (Inventia), Lupin Ltd. and Lupin Pharmaceuticals, Inc. (Lupin), Taro Pharmaceuticals USA, Inc. and Taro Pharmaceutical Industries, Ltd. (Taro), and Apotex Inc. and Apotex Corp., (collectively, the Defendants). The lawsuits each seek an adjudication that the respective Defendants infringed one or more claims of the ‘610 Patent and/or the Company’s U.S. Patent No. 9,138,432 (the ‘432 Patent) by submitting to the FDA an ANDA for a generic version of Fanapt® prior to the expiration of the ‘610 Patent in November 2027 or the ‘432 Patent in September 2025. The Defendants have denied infringement and counterclaimed for declaratory judgment of invalidity and noninfringement of the ‘610 Patent and the ‘432 Patent. A trial on these matters was scheduled to begin on May 15, 2017. On December 15, 2016, following a submission by the parties that it may be appropriate to remove the May 15, 2017 trial from the Delaware District Court’s calendar pending the Federal Circuit’s decision in Roxane’s appeal of the Delaware District Court’s August 25, 2016 ruling finding that Roxane’s proposed products infringed the asserted claims of the ‘610 Patent, the Delaware District Court removed the May 15, 2017 trial from the calendar and adopted a new schedule for discovery. The parties agreed that within 14 days after any decision on the merits in the Roxane appeal, the parties will submit to the Delaware District Court a status report and request a schedule for trial. The Company entered into a confidential stipulation with Inventia regarding any potential launch of Inventia’s generic ANDA product. The Company also entered into a confidential stipulation with Lupin regarding any potential launch of Lupin’s generic ANDA product.
Lupin filed counter claims for declaratory judgment of invalidity and noninfringement of seven of the Company’s method of treatment patents that are listed in the Approved Drug Products with Therapeutic Equivalence Evaluations (the Orange Book) related to Fanapt® (such seven patents, the Method of Treatment Patents). The Company has not sued Lupin for infringing the Method of Treatment Patents. On October 13, 2016, the Company and Lupin filed a Stipulation of Dismissal in the Delaware District Court pursuant to which Lupin’s counterclaims relating to the Method of Treatment Patents were dismissed without prejudice in recognition of an agreement reached between the parties by which the Company would not assert those patents against Lupin absent certain changes in Lupin’s proposed prescribing information for its iloperidone tablets.
On October 24, 2016, the Company entered into a License Agreement with Taro to resolve the Company’s patent litigation against Taro regarding Taro’s ANDA seeking approval of its generic version of Fanapt® (the Taro License Agreement). Under the Taro License Agreement, the Company granted Taro a non-exclusive license to manufacture and commercialize Taro’s version of Fanapt® in the U.S. effective November 2, 2027, unless prior to that date the Company obtains pediatric exclusivity for Fanapt®, in which case, the license will be effective May 2, 2028. Taro may enter the market earlier under certain limited circumstances. The Taro License Agreement, which is subject to review by the U.S. Federal Trade Commission (FTC) and the U.S. Department of Justice (DOJ), provides for a full settlement and release by the Company and Taro of all claims that are the subject of the litigation.
On December 7, 2016, the Company entered into a License Agreement with Apotex to resolve the Company’s patent litigation against Apotex regarding Apotex’s ANDA seeking approval of its generic version of Fanapt® (the Apotex License Agreement). Under the Apotex License Agreement, the Company granted Apotex a non-exclusive license to manufacture and commercialize Apotex’s version of Fanapt® in the U.S. effective November 2, 2027, unless prior to that date the Company obtains pediatric exclusivity for Fanapt®, in which case, the license will be effective May 2, 2028. Apotex may enter the market earlier under certain limited circumstances. The Apotex License Agreement, which is subject to review by the FTC and the DOJ, provides for a full settlement and release by the Company and Apotex of all claims that are the subject of the litigation.
82
Table of Contents
On February 26, 2016, Roxane filed suit against the Company in the U.S. District Court for the Southern District of Ohio. The suit seeks a declaratory judgment of invalidity and noninfringement of the Method of Treatment Patents. The Company has not sued Roxane for infringing the Method of Treatment Patents. The Company filed a motion to dismiss this lawsuit for lack of personal jurisdiction or to transfer the lawsuit to the Delaware District Court. On December 20, 2016, the Ohio District Court ruled in our favor, dismissing Roxane’s suit without prejudice for lack of personal jurisdiction.
On February 26, 2016, Roxane filed a Petition for Inter Partes Review (IPR) of the ‘432 Patent with the Patent Trials and Appeals Board (PTAB) of the United States Patent and Trademark Office. The Company filed a Preliminary Response on June 7, 2016, and on August 30, 2016 the PTAB denied the request by Roxane to institute an IPR of the ‘432 Patent. On September 29, 2016, Roxane filed a Petition for Rehearing with the PTAB, and on October 13, 2016 the Company filed a Response to Roxane’s Petition. On November 4, 2016, the PTAB denied Roxane’s Petition for Rehearing.
19. Quarterly Financial Data (Unaudited)
The following is a summary of quarterly financial data for the years ended December 31, 2016 and 2015:
| (in thousands, except for per share amounts) | First Quarter |
Second Quarter |
Third Quarter |
Fourth Quarter |
||||||||||||
| Year Ended December 31, 2016 | ||||||||||||||||
| Revenue |
$ | 33,262 | $ | 36,029 | $ | 38,482 | $ | 38,244 | ||||||||
| Gross profit (1) |
24,363 | 26,593 | 28,549 | 30,867 | ||||||||||||
| Loss from operations |
(12,475 | ) | (4,789 | ) | (653 | ) | (654 | ) | ||||||||
| Net loss |
(12,358 | ) | (4,618 | ) | (430 | ) | (604 | ) | ||||||||
| Net loss per share, basic and diluted |
$ | (0.29 | ) | $ | (0.11 | ) | $ | (0.01 | ) | $ | (0.01 | ) | ||||
| Year Ended December 31, 2015 | ||||||||||||||||
| Revenue |
$ | 22,150 | $ | 27,582 | $ | 28,344 | $ | 31,849 | ||||||||
| Gross profit (1) |
12,991 | 18,874 | 18,891 | 22,735 | ||||||||||||
| Loss from operations |
(10,293 | ) | (5,458 | ) | (9,541 | ) | (14,893 | ) | ||||||||
| Net loss |
(10,221 | ) | (5,386 | ) | (9,461 | ) | (14,797 | ) | ||||||||
| Net loss per share, basic and diluted |
$ | (0.24 | ) | $ | (0.13 | ) | $ | (0.22 | ) | $ | (0.35 | ) | ||||
| (1) | Gross profit includes revenue less cost of goods sold, excluding amortization and intangible asset amortization. |
83
Table of Contents
EXHIBIT INDEX
| Exhibit |
Description | |
| 3.1 | Form of Amended and Restated Certificate of Incorporation of the registrant (filed as Exhibit 3.8 to Amendment No. 2 to the registrant’s registration statement on Form S-1 (File No. 333-130759) on March 17, 2006 and incorporated herein by reference). | |
| 3.2 | Form of Certificate of Designation of Series A Junior Participating Preferred Stock (filed as Exhibit 3.10 to the registrant’s current report on Form 8-K (File No. 001-34186) on September 25, 2008 and incorporated herein by reference). | |
| 3.3 | Fourth Amended and Restated Bylaws of the registrant, as amended and restated on December 17, 2015 (filed as Exhibit 3.1 to the registrant’s current report on Form 8-K (File No. 001-34186) on December 21, 2015 and incorporated herein by reference). | |
| 4.1 | Specimen certificate representing the common stock of the registrant (filed as Exhibit 4.4 to Amendment No. 2 to the registrant’s registration statement on Form S-1 (File No. 333-130759) on March 17, 2006, and incorporated herein by reference). | |
| 4.2 | Rights Agreement, dated as of September 25, 2008, by and between the registrant and American Stock Transfer & Trust Company, LLC, as Rights Agent (filed as Exhibit 4.5 to the registrant’s current report on Form 8-K (File No. 001-34186) on September 25, 2008 and incorporated herein by reference). | |
| 4.3 | Amendment to Rights Agreement, dated as of December 22, 2009, by and between the registrant and American Stock Transfer & Trust Company, LLC, as Rights Agent (filed as Exhibit 4.6 to the registrant’s current report on Form 8-K (File No. 001-34186) on December 22, 2009 and incorporated herein by reference). | |
| 10.1† | Registrant’s Second Amended and Restated Management Equity Plan (filed as Exhibit 10.1 to the registrant’s registration statement on Form S-1 (File No. 333-130759) on December 29, 2005 and incorporated herein by reference). | |
| 10.2# | Amended and Restated License, Development and Commercialization Agreement, dated July 24, 2005, by and between Bristol-Myers Squibb Company and the registrant (relating to HETLIOZ®) (filed as Exhibit 10.3 to Amendment No. 1 to the registrant’s registration Statement on Form S-1 (File No. 333-130759) on February 16, 2006 and incorporated herein by reference). | |
| 10.3 | Summary Plan Description provided for the registrant’s 401(k) Profit Sharing Plan & Trust (filed as Exhibit 10.10 to the registrant’s registration statement on Form S-1 (File No. 333-130759) on December 29, 2005 and incorporated herein by reference). | |
| 10.4 | Form of Indemnification Agreement entered into by directors and executive officers (filed as Exhibit 10.11 to the registrant’s registration statement on Form S-1 (File No. 333-130759) on December 29, 2005 and incorporated herein by reference). | |
| 10.5† | 2006 Equity Incentive Plan, as amended (filed as Exhibit 10.38 to the registrant’s current report on Form 8-K (File No. 001-34186) on March 7, 2010 and incorporated herein by reference). | |
| 10.6 | Form of Tax Indemnity Agreement (filed as Exhibit 10.20 to the registrant’s quarterly report on Form 10-Q (File No. 000-51863) on August 8, 2007 and incorporated herein by reference). | |
| 10.7† | Amended and Restated Employment Agreement, dated December 16, 2008, by and between Mihael H. Polymeropoulos and the registrant (filed as Exhibit 10.34 to the registrant’s quarterly report on Form 10-Q (File No. 001-34186) on August 10, 2009 and incorporated herein by reference). | |
| 10.8† | Employment Agreement, dated December 13, 2010, by and between James Kelly and the registrant (filed as Exhibit 10.38 to the registrant’s annual report on Form 10-K (File No. 001-34186) on March 10, 2011 and incorporated herein by reference). | |
84
Table of Contents
| Exhibit |
Description | |
| 10.9† | Amendment to Amended and Restated Employment Agreement, dated December 16, 2010, by and between Mihael H. Polymeropoulos and the registrant (filed as Exhibit 10.39 to the registrant’s annual report on Form 10-K (File No. 001-34186) on March 10, 2011 and incorporated herein by reference). | |
| 10.10 | Amended and Restated Tax Indemnity Agreement, dated December 16, 2010, by and between Mihael H. Polymeropoulos and the registrant (filed as Exhibit 10.41 to the registrant’s annual report on Form 10-K (File No. 001-34186) on March 10, 2011 and incorporated herein by reference). | |
| 10.11 | Lease, effective as of July 25, 2011, by and between Square 54 Office Owner LLC and the registrant (filed as Exhibit 10.42 to the registrant’s quarterly report on Form 10-Q (File No. 001-34186) on November 7, 2011 and incorporated herein by reference). | |
| 10.12† | Form of Notice of Stock Option Grant and Stock Option Agreement under 2006 Equity Incentive Plan 2011 (filed as Exhibit 10.44 to the registrant’s annual report on Form 10-K (File No. 001-34186) on March 9, 2012 and incorporated herein by reference). | |
| 10.13† | Form of Restricted Stock Unit Award Agreement under 2006 Equity Incentive Plan 2011 (filed as Exhibit 10.45 to the registrant’s annual report on Form 10-K (File No. 001-34186) on March 9, 2012 and incorporated herein by reference). | |
| 10.14 | Amendment to Amended and Restated License, Development and Commercialization Agreement, dated as of April 15, 2010, by and between Bristol-Myers Squibb Company and the registrant (filed as Exhibit 10.38 to the registrant’s current report on Form 8-K (File No. 001-34186) on April 19, 2010 and incorporated herein by reference). | |
| 10.15 | Amendment to Amended and Restated License, Development and Commercialization Agreement, dated as of May 24, 2012, by and between Bristol-Myers Squibb Company and the registrant (filed as Exhibit 10.46 to the registrant’s current report on Form 8-K (File No. 001-34186) on May 30, 2012 and incorporated herein by reference). | |
| 10.16# | License, Development and Commercialization Agreement, dated as of April 12, 2012, by and between Eli Lilly and Company and the registrant (filed as Exhibit 10.48 to the registrant’s quarterly report on Form 10-Q (File No. 001-34186) on August 3, 2012 and incorporated herein by reference). | |
| 10.17 | Amendment to Amended and Restated License, Development and Commercialization Agreement, dated as of April 25, 2013, by and between Bristol-Myers Squibb Company and the registrant (filed as Exhibit 10.50 to the registrant’s current report on Form 8-K (File No. 001-34186) on April 29, 2013 and incorporated herein by reference). | |
| 10.18† | Employment Agreement, dated as of April 15, 2013, by and between Paolo Baroldi and the registrant (filed as Exhibit 10.51 to the registrant’s quarterly report on Form 10-Q (File No. 001-34186) on July 31, 2013 and incorporated herein by reference). | |
| 10.19# | Manufacturing Agreement, dated January 24, 2014, by and between Patheon Pharmaceuticals Inc. and the registrant (relating to HETLIOZ®) (filed as Exhibit 10.53 to the registrant’s quarterly report on Form 10-Q (File No. 001-34186) on May 8, 2014 and incorporated herein by reference). | |
| 10.20 | Amendment to Lease Agreement, dated March 18, 2014, by and between Square 54 Office Owner LLC and the registrant (filed as Exhibit 10.54 to the registrant’s quarterly report on Form 10-Q (File No. 001-34186) on May 8, 2014 and incorporated herein by reference). | |
| 10.21 | Settlement Agreement and Mutual General Release, dated December 22, 2014, by and among Novartis Pharma AG and the registrant (filed as Exhibit 10.55 to the registrant’s annual report on Form 10-K (File No. 001-34186) on March 13, 2015 and incorporated herein by reference). | |
| 10.22# | Asset Transfer Agreement, dated December 22, 2014, by and among Novartis Pharma AG, Novartis AG and the registrant (relating to Fanapt®) (filed as Exhibit 10.56 to the registrant’s annual report on Form 10-K/A (File No. 001-34186) on June 10, 2015 and incorporated herein by reference). | |
85
Table of Contents
| Exhibit |
Description | |
| 10.23# | Sublicense Agreement, dated November 20, 1997, by and between Titan Pharmaceuticals, Inc. and Novartis Pharma AG (filed as Exhibit 10.30 to Titan Pharmaceutical Inc.’s registration statement on Form S-3 (File No. 333-42367) on December 16, 1997 and incorporated herein by reference). | |
| 10.24# | Amendment No. 1 to Sublicense Agreement by and between Titan Pharmaceuticals, Inc. and Novartis Pharma AG, dated November 30, 1998 (filed as Exhibit 10.58 to the registrant’s annual report on Form 10-K for the year ended December 31, 2014 and incorporated herein by reference). | |
| 10.25# | Amendment No. 2 to Sublicense Agreement, dated April 10, 2001, by and between Titan Pharmaceuticals, Inc. and Novartis Pharma AG (filed as Exhibit 10.59 to the registrant’s annual report on Form 10-K/A (File No. 001-34186) on June 10, 2015 and incorporated herein by reference) | |
| 10.26# | Amendment No. 3 to Sublicense Agreement, dated June 4, 2004, by and between Titan Pharmaceuticals, Inc. and Novartis Pharma AG (filed as Exhibit 10.60 to the registrant’s annual report on Form 10-K (File No. 001-34186) on March 13, 2015 and incorporated herein by reference). | |
| 10.27 | Stock Purchase Agreement, dated December 22, 2014, by and between Novartis AG and the registrant (filed as Exhibit 10.61 to the registrant’s annual report on Form 10-K (File No. 001-34186) on March 13, 2015 and incorporated herein by reference). | |
| 10.28# | License Agreement, dated December 22, 2014, by and between Novartis Pharma AG and the registrant (relating to AQW051) (filed as Exhibit 10.62 to the registrant’s annual report on Form 10-K (File No. 001-34186) on March 13, 2015 and incorporated herein by reference). | |
| 10.29† | Employment Agreement, dated April 20, 2015, by and between Thomas E. Gibbs, Senior Vice President and Chief Commercial Officer, and the registrant (filed as Exhibit 10.65 to the registrant’s quarterly report on Form 10-Q (File No. 001-34186) on July 31, 2015 and incorporated herein by reference). | |
| 10.30† | Employment Agreement, dated September 3, 2015, by and between Gian Piero Reverberi, Senior Vice President and General Manager Europe, and the registrant (filed as Exhibit 10.64 to the registrant’s quarterly report on Form 10-Q (File No. 001-34186) on November 4, 2015 and incorporated herein by reference). | |
| 10.31† | Employment Agreement, dated September 14, 2015, by and between Richard L. Gulino, Senior Vice President, General Counsel and Secretary, and the registrant (filed as Exhibit 10.65 to the registrant’s quarterly report on Form 10-Q (File No. 001-34186) on November 4, 2015 and incorporated herein by reference). | |
| 10.32 | Agreement, dated February 2, 2016, by and among Titan Pharmaceuticals, Inc., Aventisub LLC, the successor-in-interest to Aventisub II Inc. Sanofi-Aventis and the registrant (filed as Exhibit 10.1 to the registrant’s current report on Form 8-K (File No. 001-34186) on February 4, 2016 and incorporated herein by reference). | |
| 10.33† | UK Sub Plan under the 2006 Equity Incentive Plan (filed as Exhibit 10.33 to the registrant’s annual report on Form 10-K (File No. 001-34186) on February 12, 2016 and incorporated herein by reference). | |
| 10.34† | Form of Stock Option Grant and Stock Option Agreement under the UK Sub Plan (filed as Exhibit 10.34 to the registrant’s annual report on Form 10-K (File No. 001-34186) on February 12, 2016 and incorporated herein by reference). | |
| 10.35† | Form of Restricted Stock Unit Award Agreement under the UK Sub Plan (filed as Exhibit 10.35 to the registrant’s annual report on Form 10-K (File No. 001-34186) on February 12, 2016 and incorporated herein by reference). | |
| 10.36† | Vanda Pharmaceuticals Inc. 2016 Equity Incentive Plan, effective as of June 16, 2016 (filed as Exhibit 10.36 to the registrant’s registration statement on Form S-8 (File No. 333-212255) on June 27, 2016 and incorporated herein by reference). | |
| 10.37† | Form of Notice of Stock Option Grant and Stock Option Agreement under 2016 Equity Incentive Plan (filed as Exhibit 10.37 to the registrant’s registration statement on Form S-8 (File No. 333-212255) on June 27, 2016 and incorporated herein by reference). | |
86
Table of Contents
| Exhibit |
Description | |
| 10.38† | Form of Restricted Stock Unit Award Agreement under 2016 Equity Incentive Plan (filed as Exhibit 10.38 to the registrant’s registration statement on Form S-8 (File No. 333-212255) on June 27, 2016 and incorporated herein by reference). | |
| 10.39† | UK Sub Plan under the 2016 Equity Incentive Plan (filed as Exhibit 10.39 to the registrant’s registration statement on Form S-8 (File No. 333-212255) on June 27, 2016 and incorporated herein by reference). | |
| 10.40† | Form of Stock Option Grant and Stock Option Agreement under the UK Sub Plan under the 2016 Equity Incentive Plan (filed as Exhibit 10.40 to the registrant’s registration statement on Form S-8 (File No. 333-212255) on June 27, 2016 and incorporated herein by reference). | |
| 10.41† | Form of Restricted Stock Unit Award Agreement under the UK Sub Plan under the 2016 Equity Incentive Plan (filed as Exhibit 10.41 to the registrant’s registration statement on Form S-8 (File No. 333-212255) on June 27, 2016 and incorporated herein by reference). | |
| 10.42# | Manufacturing Agreement, dated May 6, 2016, by and between Patheon Pharmaceuticals Inc. and the registrant (relating to Fanapt®) (filed as Exhibit 10.42 to the registrant’s quarterly report on Form 10-Q (File No. 001-34186) on July 31, 2016 and incorporated herein by reference). | |
| 10.43 | Second Amendment to Lease Agreement, dated June 20, 2016, by and between Square 54 Office Owner LLC and the registrant (filed as Exhibit 10.43 to the registrant’s quarterly report on Form 10-Q (File No. 001-34186) on July 31, 2016 and incorporated herein by reference). | |
| 10.44 | Sublease Agreement, dated June 22, 2016, by and between Hunton & Williams LLP and the registrant (filed as Exhibit 10.44 to the registrant’s quarterly report on Form 10-Q (File No. 001-34186) on July 31, 2016 and incorporated herein by reference). | |
| 10.45‡* | License Agreement, dated October 24, 2016, by and among Taro Pharmaceuticals USA, Inc., Taro Pharmaceuticals Industries Ltd. and the registrant. | |
| 10.46‡* | License Agreement, dated December 7, 2016, by and between Apotex, Inc. and the registrant. | |
| 18.1 | Preferability Letter of Independent Public Accounting Firm, dated May 8, 2014 (filed as Exhibit 18.1 to the registrant’s quarterly report on Form 10-Q (File No. 001-34186) on May 8, 2014 and incorporated herein by reference). | |
| 21.1* | List of Subsidiaries. | |
| 23.1* | Consent of PricewaterhouseCoopers LLP, Independent Registered Public Accounting Firm. | |
| 31.1* | Certification of the Chief Executive Officer, as required by Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2* | Certification of the Chief Financial Officer as required by Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1* | Certification of the Chief Executive Officer and Chief Financial Officer as required by Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 101* | The following financial information from this annual report on Form 10-K for the fiscal year ended December 31, 2016, formatted in XBRL (eXtensible Business Reporting Language) and furnished electronically herewith: (i) Consolidated Balance Sheets as of December 31, 2016 and 2015; (ii) Consolidated Statements of Operations for the years ended December 31, 2016, 2015 and 2014; (iii) Consolidated Statements of Comprehensive Income (Loss) for the years ended December 31, 2016, 2015 and 2014; (iv) Consolidated Statements of Changes in Stockholders’ Equity for the years ended December 31, 2016, 2015 and 2014; (v) Consolidated Statements of Cash Flows for the years ended December 31, 2016, 2015 and 2014; and (vi) Notes to the Consolidated Financial Statements. | |
| † | Indicates management contract or compensatory plan. |
| # | Confidential treatment has been granted with respect to certain provisions of this exhibit. |
| ‡ | Portions of this exhibit (indicated by asterisks) have been omitted pursuant to a request for confidential treatment. The omitted portions of this exhibit have been filed with the Securities and Exchange Commission. |
| * | Filed herewith. |
87