Attached files
| file | filename |
|---|---|
| EX-32.1 - EXHIBIT 32.1 - Orgenesis Inc. | exhibit32-1.htm |
| EX-31.2 - EXHIBIT 31.2 - Orgenesis Inc. | exhibit31-2.htm |
| EX-31.1 - EXHIBIT 31.1 - Orgenesis Inc. | exhibit31-1.htm |
| EX-32.2 - EXHIBIT 32.2 - Orgenesis Inc. | exhibit32-2.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
[X] ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended: November 30, 2015
or
[ ] TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from __________________________ to __________________________
Commission file number 000-54329
![]()
ORGENESIS INC.
((Exact
name of registrant as specified in its charter)
| Nevada | 98-0583166 |
| State or other jurisdiction | (I.R.S. Employer |
| of incorporation or organization | Identification No.) |
20271 Goldenrod Lane, Germantown, MD 20876
((Address of principal executive offices) (Zip Code)
Registrant’s telephone number, including area code: (480) 659-6404
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to section 12(g) of the Act:
Common Stock, par value $0.0001 per share
(Title of class)
Indicate by check mark if the registrant is a well-known
seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes [
] No [X]
Indicate by check mark if the registrant is not required to
file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes [
] No [X]
Indicate by check mark whether the registrant (1) has filed all
reports required to be filed by Section 13 or 15(d) of the Securities Exchange
Act of 1934 during the preceding 12 months (or for such shorter period that the
registrant was required to file such reports), and (2) has been subject to such
filing requirements for the past 90 days.
Yes
[X] No [ ]
Indicate by check mark whether the registrant has submitted
electronically and posted on its corporate Web site, if any, every Interactive
Data File required to be submitted and posted pursuant to Rule 405 of Regulation
S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such
shorter period that the registrant was required to submit and post such files).
Yes
[X] No [ ]
0
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. [X]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer [ ] | Accelerated filer [ ] |
| Non-accelerated filer [ ] | |
| (Do not check if a smaller reporting company) | Smaller reporting company [X] |
Indicate by check mark whether the registrant is a shell
company (as defined in Rule 12b-2 of the Act).
Yes [ ]
No [X]
The registrant had 108,932,129 shares of common stock outstanding as of February 26, 2016. The aggregate market value of the voting and non-voting common stock held by non-affiliates of the registrant as of May 29, 2015 was $16,961,572 as computed by reference to the closing price of such common stock on the OTCQB on such date.
DOCUMENTS INCORPORATED BY REFERENCE
The registrant intends to file a definitive proxy statement pursuant to Regulation 14A in connection with its 2016 Annual Meeting of Stockholders within 120 days after the close of the fiscal year covered by this Form 10-K. Portions of such proxy statement are incorporated by reference into Items 10, 11, 12, 13 and 14 of Part III of this report.
1
ORGENESIS INC.
2015 FORM 10-K ANNUAL REPORT
TABLE OF CONTENTS
2
FORWARD-LOOKING STATEMENTS
CAUTIONARY STATEMENT FOR PURPOSES OF THE "SAFE HARBOR" PROVISIONS OF THE PRIVATE SECURITIES LITIGATION REFORM ACT OF 1995
The following discussion should be read in conjunction with the financial statements and related notes contained elsewhere in this Form 10-K. Certain statements made in this discussion are "forward-looking statements" within the meaning of The Private Securities Litigation Reform Act of 1995. Forward-looking statements are projections in respect of future events or financial performance. In some cases, you can identify forward-looking statements by terminology such as “may”, “should”, “expects”, “plans”, “anticipates”, “believes”, “estimates”, “predicts”, “potential” or “continue” or the negative of these terms or other comparable terminology. Forward-looking statements made in an annual report on Form 10-K include statements about our:
| • |
ability to obtain sufficient capital or strategic business arrangements to fund our operations and realize our business plan; |
| • |
ability to grow the business of MaSTherCell, which we recently acquired, our Contract Development and Manufacturing Organization (“CDMO”) business; |
| • |
belief as to whether a meaningful and profitable global market can be established for our CDMO business for cell therapy; |
| • |
intention to develop to the clinical stage a new technology to transdifferentiate liver cells into functional insulin-producing cells, thus enabling normal glucose regulated insulin secretion, via cell therapy; |
| • |
belief that our treatment seems to be safer than other options; |
| • |
belief that one of our principal competitive advantages is our cell transdifferentiation technology being developed by our Israeli Subsidiary; |
| • |
expectations regarding our Israeli Subsidiary’s ability to obtain and maintain intellectual property protection for our technology and therapies; |
| • |
ability to commercialize products in light of the intellectual property rights of others; |
| • |
ability to obtain funding for operations, including funding necessary to prepare for clinical trials and to complete such clinical trials; |
| • |
future agreements with third parties in connection with the commercialization of our technologies; |
| • |
size and growth potential of the markets for our product candidates, and our ability to serve those markets; |
| • |
regulatory developments in the United States and foreign countries; |
| • |
ability to contract with third-party suppliers and manufacturers and their ability to perform adequately; |
| • |
plans to integrate and support our manufacturing facilities in Belgium; |
| • |
success as it is compared to competing therapies that are or may become available; |
| • |
ability to attract and retain key scientific or management personnel and to expand our management team; |
| • |
accuracy of estimates regarding expenses, future revenue, capital requirements, profitability, and needs for additional financing; |
| • |
belief that Diabetes Mellitus will be one of the most challenging health problems in the 21st century and will have staggering health, societal and economic impact; |
| • |
need to raise additional funds on an immediate basis which may not be available on acceptable terms or at all; |
| • |
research facility in Israel and the surrounding Middle East political situation which may materially adversely affect our Israeli Subsidiary’s operations and personnel; |
| • |
relationship with with Tel Hashomer - Medical Research, Infrastructure and Services Ltd. (“THM”) and the risk that THM may cancel the License Agreement; |
| • |
expenditures not resulting in commercially successful products; and |
| • |
extensive industry regulation, and how that will continue to have a significant impact on our business, especially our product development, manufacturing and distribution capabilities. |
These statements are only predictions and involve known and unknown risks, uncertainties and other factors, including the risks in the section entitled “Risk Factors” set forth in this Annual Report on Form 10-K for the year ended November 30, 2015, any of which may cause our company’s or our industry’s actual results, levels of activity, performance or achievements to be materially different from any future results, levels of activity, performance or achievements expressed or implied by these forward-looking statements. These risks may cause the Company’s or its industry’s actual results, levels of activity or performance to be materially different from any future results, levels of activity or performance expressed or implied by these forward looking statements.
3
Although the Company believes that the expectations reflected in the forward-looking statements are reasonable, it cannot guarantee future results, levels of activity or performance. Moreover, neither the company nor any other person assumes responsibility for the accuracy and completeness of these forward-looking statements. The company is under no duty to update any forward-looking statements after the date of this report to conform these statements to actual results.
4
PART I
ITEM 1. BUSINESS
As used in this annual report on Form 10-K and unless otherwise indicated, the terms “we,” “us”, "our", “Orgenesis” or the “Company” refer to Orgenesis Inc. and its wholly-owned Subsidiaries, Orgenesis Ltd. (the “Israeli Subsidiary”), Orgenesis SPRL (the “Belgian Subsidiary”), Orgenesis Maryland, Inc. (the “U.S. Subsidiary”) and MaSTherCell SA (“MaSTherCell”), our Belgian-based subsidiary. Unless otherwise specified, all dollar amounts are expressed in United States dollars.
Corporate Overview
We are among the first of a new breed of regenerative therapy companies with expertise and unique experience in cell therapy development and manufacturing. We are building a fully-integrated biopharmaceutical company focused not only on developing our trans-differentiation technologies for diabetes and vertically integrating manufacturing that can optimize our abilities to scale-up our technologies for clinical trials and eventual commercialization, but also do the same for the technologies of other cell therapy markets in such areas as cell-based cancer immunotherapies and neurodegenerative diseases. This integrated approach supports our business philosophy of bringing to market significant life-improving medical treatments.
Our cell therapy technology derives from published work of Prof. Sarah Ferber, our Chief Science Officer and a researcher at Tel Hashomer Medical Center, a leading medical hospital and research center in Israel (“THM”), who established a proof of concept that demonstrates the capacity to induce a shift in the developmental fate of cells from the liver and transdifferentiating (converting) them into “pancreatic beta cell-like” insulin-producing cells. Furthermore, those cells were found to be resistant to autoimmune attack and to produce insulin in a glucose-sensitive manner in relevant animal models. Our development activities with respect to cell-derived and related therapies, which are conducted through the Israeli Subsidiary, have, to date, been limited to laboratory and preclinical testing. Our development plan calls for conducting additional preclinical safety and efficacy studies with respect to diabetes and other potential indications.
Our Belgian-based subsidiary, MaSTherCell, is a contract development manufacturing organization, or CDMO, specialized in cell therapy development for advanced medicinal products. In the last decade, cell therapy medicinal products have gained significant importance, particularly in the fields of ex-vivo gene therapy, immunotherapy and regenerative medicine. While academic and industrial research has led scientific development in the sector, industrialization and manufacturing expertise remains insufficient. MaSTherCell plans to fill this need by providing two types of services to its customers: (i) process and assay development services and (ii) Good Manufacturing Practices (GMP) contract manufacturing services. These services offer a double advantage to MaSTherCell's customers. First, customers can continue focusing their financial and human resources on their product/therapy, while relying on a trusted source for their process development/production. Second, it allows customers to profit from MaSTherCell's expertise in cell therapy manufacturing and all related aspects.
We intend to leverage the expertise and experience of MaSTherCell, our subsidiary, in cell process development and manufacturing capability, to build a fully integrated bio-pharmaceutical company in the cell therapy development and manufacturing area.
We need to raise significant capital in order to realize our business plan. See “Risk Factors”.
We were incorporated in the state of Nevada on June 5, 2008, under the name Business Outsourcing Services, Inc. Effective August 31, 2011, we completed a merger with our subsidiary, Orgenesis Inc., a Nevada corporation which was incorporated solely to effect a change in our name. As a result, we changed our name from “Business Outsourcing Services, Inc.” to “Orgenesis Inc.” Our common stock is currently listed on the OTC Market, QB tier, under the symbol “ORGS”.
5
Cell Therapy and Regenerative Medicine Field
Regenerative medicine is generally the process of replacing or regenerating human cells, tissues or organs to restore normal function. Our business model is focused on two of these areas. First, through our wholly-owned CDMO subsidiary, MaSTherCell, we are afforded a unique and fundamental base platform of experience and expertise with a multitude of cell types in development. MaSTherCell is strategically positioning us in a way that allows us to participate in the cell therapy field on multiple levels as the cell therapy industry evolves. Our goal is to nurture our reputation as a premier service provider in the regenerative medicine industry by continuing to leverage the experience and expertise of MaSTherCell as a recognized leader of cell therapy manufacturing and development. Second, on our clinical development side, through our Israeli Subsidiary, our goal is to advance a unique product that combines cell-based therapy and regenerative medicine, Autologous Insulin Producing (“AIP”) cells, into clinical development. AIP cells utilize the technology of ‘cellular trans-differentiation’ to transform an autologous adult liver cell into an adult, fully functional and physiologically glucose-responsive pancreatic-like insulin producing cell. Treatment with AIP cells is expected to provide Type 1 Diabetes patients with long-term insulin independence. Because the AIP cells are autologous, this benefit should be achieved and maintained without the need for concomitant immunosuppressive therapy.
All living complex organisms start as a single cell that replicates, differentiates (matures) and perpetuates in an adult organism throughout its lifetime. Cell therapy is the prevention or treatment of human disease by the administration of cells that have been selected, multiplied and pharmacologically treated or altered outside the body (ex vivo). To date, the most common type of cell therapy has been the replacement of mature, functioning cells through blood and platelet transfusions. Since the 1970s, first bone marrow and then blood and umbilical cord-derived stem cells have been used to restore bone marrow, as well as blood and immune system cells damaged by the chemotherapy and radiation that are used to treat many cancers. These types of cell therapies are standard of practice world-wide and are typically reimbursed by insurance.
Within the field of cell therapy, research and development using stem cells to treat a host of diseases and conditions has greatly expanded. Stem cells (in either embryonic or adult forms) are primitive and undifferentiated cells that have the unique ability to transform into or otherwise affect many different cells, such as white blood cells, nerve cells or heart muscle cells. Our cell therapy development efforts do not use stem cells, but rather are focused on the use of fully mature, adult cells; for our purposes in the treatment of diabetes, our cells are derived from the liver or other adult tissue and are transdifferentiated to become adult AIP cells.
There are two general classes of cell therapies: Patient Specific Cell Therapies (PSCTs) and Off-the-Shelf Cell Therapies (OSCTs). In PSCTs, cells collected from a person (donor) are transplanted into, or used to develop a treatment for a patient (recipient) with or without modification. In cases where the donor and the recipient are the same individual, these procedures are referred to as “autologous”. In cases in which the donor and the recipient are not the same individual, these procedures are referred to as “allogeneic.” A notable form of autologous PSCT involves the use of autologous cells to create vaccines directed against tumor cells in the body which has been demonstrated to be effective and safe in clinical trials. Our treatment for diabetes focuses on PSCTs using autologous cells. Autologous cells offer a low likelihood of rejection by the patient and we believe the long-term benefits of these PSCTs can best be achieved with an autologous product.
Various cell therapies are in clinical development for an array of human diseases, including autoimmune, oncologic, neurologic and orthopedic diseases, among other indications. Orgenesis, as well as other companies, are developing cell therapies that are designed to address cancers, ischemic repair and immune modulation. While no assurances can be given regarding future medical developments, we believe that the field of cell therapy holds the promise to better the human experience and minimize or ameliorate the pain and suffering from many common diseases and/or from the process of aging.
6
Diabetes Mellitus (DM), or simply diabetes, is a metabolic disorder usually caused by a combination of hereditary and environmental factors, and results in abnormally high blood sugar levels (hyperglycemia). Diabetes occurs as a result of impaired insulin production by the pancreatic islet cells. The most common types of the disease are Type-1 Diabetes (T1D) and Type-2 Diabetes (T2D). In T1D, the onset of the disease follows an autoimmune attack of β-cells that severely reduces β-cell mass. T1D usually has an early onset and is sometimes also called juvenile diabetes. In T2D, the pathogenesis involves insulin resistance, insulin deficiency and enhanced gluconeogenesis, while late progression stages eventually leads to β-cell failure and a significant reduction in β-cell function and mass. T2D often occurs later in life and is sometimes called adult onset diabetes. Both T1D and late-stage T2D result in marked hypoinsulinemia, reduction in β-cell function and mass and lead to severe secondary complications, such as myocardial infarcts, limb amputations, neuropathies and nephropathies and even death. In both cases, patients become insulin-dependent, requiring either multiple insulin injections per day or reliance on an insulin pump.
We believe that diabetes will be one of the most challenging health problems in the 21st century, and will have a staggering health, societal, and economic impact. Diabetes is currently the fourth or fifth leading cause of death in most developed countries. There also is substantial evidence that it is an epidemic in many developing and newly industrialized nations.
Threats from Pancreas Islet Transplantation and Cell Therapies
For some patients with severe and difficult to control diabetes (hypoglycemic unawareness), islet transplants are considered. Pancreatic islets are the cells in the pancreas that produce insulin. Physicians use enzymes to isolate the islets from the pancreas of a deceased donor. Because the islets are fragile, transplantation must occur soon after they are removed. Typically, a patient receives at least 10,000 islet “equivalents” per kilogram of body weight, extracted from pancreases obtained from different donors. Patients often require two separate transplants to achieve insulin independence.
Transplants are often performed by an interventional radiologist, who uses x-rays and ultrasound to guide placement of a catheter - a small plastic tube - through the upper abdomen and into the portal vein of the liver. The islets are then infused slowly through the catheter into the liver. The patient receives a local anesthetic and a sedative. In some cases, a surgeon may perform the transplant through a small incision, using general anesthesia.
Because the islets are obtained from cadavers that are unrelated to the patient, the patient needs to be treated with drugs that inhibit the immune response so that the patient doesn’t reject the transplant. In the early days of islet transplantation, the drugs were so powerful that they actually were toxic to the islets; improvements in the procedure are widely used and are now referred to as the Edomonton Protocol.
Studies and Reports
Since reporting their findings in the June 2000 issue of the New England Journal of Medicine, researchers at the University of Alberta in Edmonton, Canada, have continued to use and refine Edmonton Protocol to transplant pancreatic islets into selected patients with T1D that is difficult to control.
In 2005, the researchers published 5-year follow-up results for 65 patients who received transplants at their center and reported that about 10 percent of the patients remained free of the need for insulin injections at 5-year follow-up. Most recipients returned to using insulin because the transplanted islets lost their ability to function over time, potentially due to the immune suppression protocol, which prevents the immune rejection of the implanted cells. The researchers noted, however, that many transplant recipients were able to reduce their need for insulin, achieve better glucose stability, and reduce problems with hypoglycemia, also called low blood sugar level.
In its 2006 annual report, the Collaborative Islet Transplant Registry, which is funded by the National Institute of Diabetes and Digestive and Kidney Diseases, presented data from 23 islet transplant programs on 225 patients who received islet transplants between 1999 and 2005. According to the report, nearly two-thirds of recipients achieved “insulin independence” - defined as being able to stop insulin injections for at least 14 days - during the year following transplantation. However, other data from the report showed that insulin independence is difficult to maintain over time. Six months after their last infusion of islets, more than half of recipients were free of the need for insulin injections, but at 2-year follow-up, the proportion dropped to about one-third of recipients. The report described other benefits of islet transplantation, including reduced need for insulin among recipients who still needed insulin, improved blood glucose control, and greatly reduced risk of episodes of severe hypoglycemia.
7
In a 2006 report of the Immune Tolerance Network’s international islet transplantation study, researchers emphasized the value of transplantation in reversing a condition known as hypoglycemia unawareness. People with hypoglycemia unawareness are vulnerable to dangerous episodes of severe hypoglycemia because they are not able to recognize that their blood glucose levels are too low. The study showed that even partial islet function after transplant can eliminate hypoglycemia unawareness.
Pancreatic islet transplantation (cadaver donors) is an allogeneic transplant, and, as in all allogeneic transplantations, there is a risk for graft rejection and patients must receive lifelong immune suppressants. Though this technology has shown good results clinically, there are several setbacks, such as patients being sensitive to recurrent T1D autoimmune attacks and a shortage in tissues available for islet cells transplantation.
Our Cell Therapy Business
We are developing and bringing to the clinical stage a technology that is based on the published work of Prof. Sarah Ferber, our Chief Science Officer and a researcher at THM, who established a proof of concept that demonstrates the capacity to induce a shift in the developmental fate of cells from the liver and differentiating (converting) them into “pancreatic beta cell-like” insulin-producing cells. Furthermore, those cells were found to be resistant to the autoimmune attack and to produce insulin in a glucose-sensitive manner.
We intend to grow our cell therapy business by furthering this technology to the clinical stage. We intend to devote significant resources to process development and manufacturing in order to optimize the safety and efficacy of our future product candidates, as well as our cost of goods and time to market. Our goal is to carefully manage our fixed cost structure, maximize optionality, and drive long-term cost of goods as low as possible. We believe that operating our own manufacturing facility will provide the Company with enhanced control of material supply for both clinical trials and the commercial market, will enable the more rapid implementation of process changes, and will allow for better long-term margins.
The License Agreement
On February 2, 2012, our Israeli Subsidiary entered into a licensing agreement with THM pursuant to which the Israeli Subsidiary was granted a worldwide royalty bearing and exclusive license to certain information regarding a molecular and cellular approach directed at converting liver cells into functional insulin producing cells as a treatment for diabetes (the “License Agreement”). By using therapeutic agents (i.e., PDX-1, and additional pancreatic transcription factors in an adenovirus-vector) that efficiently convert a sub-population of liver cells into pancreatic islets phenotype and function, this approach allows the diabetic patient to be the donor of his own therapeutic tissue. We believe that this provides major competitive advantage the cell transformation technology our Israeli Subsidiary is developing. Based on the licensed knowhow and patents, it is our intention, through the Israeli Subsidiary, to develop to the clinical stage a new technology for regeneration of functional insulin-producing cells, thus enabling normal glucose regulated insulin secretion, via cell therapy. By using therapeutic agents (i.e., PDX-1, and additional pancreatic transcription factors in an adenovirus-vector) that efficiently convert a sub-population of liver cells into pancreatic islets phenotype and function, this approach allows the diabetic patient to be the donor of his own therapeutic tissue.
As consideration for the license under the License Agreement, the Israeli Subsidiary has agreed to pay the following to THM:
| 1) |
A royalty of 3.5% of net sales; | |
| 2) |
16% of all sublicensing fees received; | |
| 3) |
An annual license fee of $15,000, which commenced on January 1, 2012 and is due once every year thereafter (the “Annual Fee”). The Annual Fee is non-refundable, but it shall be credited each year due, against the royalty noted above, to the extent that such are payable, during that year; and | |
| 4) |
Milestone payments as follows: |
8
| a) |
$50,000 on the date of initiation of phase I clinical trials in human subjects; | |
| b) |
$50,000 on the date of initiation of phase II clinical trials in human subjects; | |
| c) |
$150,000 on the date of initiation of phase III clinical trials in human subjects; | |
| d) |
$750,000 on the date of initiation of issuance of an approval for marketing of the first product by the FDA; and | |
| e) |
$2,000,000, when worldwide net sales of products have reached the amount of $150,000,000 for the first time, (The “Sales Milestone”). |
As of November 30, 2015, the Israeli Subsidiary has not reached any of these milestones.
In the event of an acquisition of all of the issued and outstanding share capital of the Israeli Subsidiary or of the Company and/or consolidation of the Israeli Subsidiary or the Company into or with another corporation (“Exit”), under the License Agreement, THM is entitled to elect, at its sole option, whether to receive from the Company a one-time payment based, as applicable, on the value at the time of the Exit of either 5,563,809 shares of common stock of the Company or the value of 1,000 ordinary shares of the Israeli Subsidiary at the time of the Exit. If THM elects to receive the consideration as a result of an Exit, the royalty payments will cease.
If THM elects to not receive any consideration as a result of an Exit, THM is entitled under the License Agreement to continue to receive all the rights and consideration it is entitled to pursuant to the License Agreement (including, without limitation, the exercise of the rights pursuant to future Exit events), and any agreement relating to an Exit event shall be subject to the surviving entity’s and/or the purchaser’s undertaking towards THM to perform all of the Israeli Subsidiary's obligations pursuant to the License Agreement.
The Israeli Subsidiary agreed to submit to THM a commercially reasonable plan which shall include all research and development activities as required for the development and manufacture of the products, including preclinical and clinical activities until an FDA or any other equivalent regulatory authority’s approval for marketing and including all regulatory procedures required to obtain such approval for each product candidate (a “Development Plan”), within 18 months from the date of the License Agreement. Under the License agreement, the Israeli Subsidiary undertook to develop, manufacture, sell and market the products pursuant to the milestones and time-frame schedule specified in the Development Plan. The Israeli Subsidiary submitted the Development Plan in May 2014.
Under the License Agreement, THM is entitled to terminate the License Agreement in each of the following events:
| • | The Israeli Subsidiary materially changes its business. |
|
• |
The Israeli Subsidiary breaches any of its material obligations under the License Agreement, provided that THM has provided the Israeli Subsidiary with written notice of such material breach and THM’s intention to terminate, and the Israeli Subsidiary has not cured such breach within 180 days of receiving such written notice from THM. The Israeli Subsidiary's failure to comply with sections relating to the following are deemed to be a material breach of the License Agreement: |
| o | granting of sublicenses; | |
| o | confidentiality provisions; | |
| o | performance of payments to the Licensor; or | |
| o | indemnity and insurance. |
| • |
The Israeli Subsidiary breaches any of its obligations thereunder other than material breaches, and such breach remains uncured for 200 days after written notice from THM. |
| • |
The Israeli Subsidiary becomes insolvent; file a petition or have a petition filed against it, under any laws relating to insolvency; enter into any voluntary arrangement for the benefit of our creditors; or appoint or have appointed on our behalf a receiver, liquidator or trustee of any of its property or assets, under any laws relating to insolvency; and such petition, arrangement or appointment is not dismissed or vacated within 90 days. |
| • |
The Israeli Subsidiary has ceased to carry on our business for a period of more than 60 days. |
9
|
• |
The Israeli Subsidiary challenges, or causes any third party to challenge, the intellectual property rights or other rights of THM to the licensed information anywhere in the world. |
The Israeli Subsidiary may terminate the License Agreement and return the licensed information to THM only in the following events:
| • |
the development and/or manufacture of the licensed information is not successful according to the scientific criteria acceptable in the relevant field of the invention; |
| • |
if the registration and/or defense of a patent is not successful, in any country for reasons not dependent upon the Israeli Subsidiary; |
| • |
the development and/or manufacture of the licensed information is not approved by the proper regulation procedures as mandated under the relevant laws for reasons not dependent upon the Israeli Subsidiary; or |
| • |
an external specialist in the field of the product(s) determined in a reasoned and explained written opinion that there is insufficient market demand for the products and such written opinion was provided to THM. |
On March 22, 2012, the Israeli Subsidiary entered into a research service agreement with THM pursuant to which THM is to perform a study at the facilities and use the equipment and personnel of the Chaim Sheba Medical Center (the “Hospital”), for the consideration of approximately $74,000 for a year. On each of May 2013 and 2014, the Israeli Subsidiary renewed the research agreement for an annual consideration of approximately $92,000 and approximately $114,000, respectively.
We intend to advance our cell therapy business by furthering this technology to a clinical stage. We intend to devote significant resources to process development and manufacturing in order to optimize the safety and efficacy of our future product candidates, as well as our cost of goods and time to market. Our goal is to carefully manage our fixed cost structure, maximize optionality, and drive long-term cost of goods as low as possible. We believe that operating our own manufacturing facility will provide the Company with enhanced control of material supply for both clinical trials and the commercial market, will enable the more rapid implementation of process changes, and will allow for better long-term margins.
Toward this goal, we are working to advance a unique product that combines cell-based therapy and regenerative medicine, (AIP) cells, into clinical development. AIP cells utilize the technology of ‘cellular trans-differentiation’ to transform an autologous adult liver cell into an adult, fully functional and physiologically glucose-responsive pancreatic-like insulin producing cell. Treatment with AIP cells is expected to provide Diabetes patients with long-term insulin independence. Because the AIP cells are autologous, this benefit should be achieved and maintained without the need for concomitant immunosuppressive therapy. The procedure to generate AIP cells begins with liver tissue accessed via needle biopsy from a patient. The liver tissue is then sent to a central facility where biopsied liver cells are isolated, expanded and trans-differentiated into AIP cells. The final product is a solution of AIP cells, which are packaged in an infusion bag and sent back to the patient’s treating physician where the cells are transplanted back into the patient’s liver via portal vein infusion. The entire process, from biopsy to transplantation, is expected to take 5-6 weeks.
10
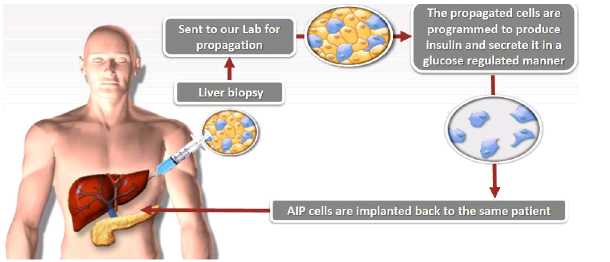
Unique benefits of AIP cells
We believe that our singular focus on the acquisition, development, and commercialization of AIP cells may have many and meaningful benefits over other technologies, including:
| • | Physiologically glucose-responsive insulin production within one week of AIP cell transplantation; |
| • | Insulin-independence within one month; |
| • | Single course of therapy (~10-year insulin-independence); |
| • | No need for concomitant immunosuppressive therapy; |
| • | Return to (near) normal quality of life for patients; |
| • | Single liver biopsy supplies unlimited source of therapeutic tissue (bio-banking for future use if needed); |
| • | Highly controlled and tightly closed GMP systems; and |
| • | Quality Control of final product upon release and distribution |
We are aware of no other company focused on development of AIP cells based on transdifferentiatation. The pharmaceutical industry is fragmented and it is a competitive market. We compete with many pharmaceutical companies, both large and small and there may be technologies in development of which we are not aware.
Marketing
Our plan is to market and sell AIP cellular therapy as a stand-alone product, and to provide supporting education and services to treating physicians and the healthcare providers that support them. In addition, we expect to provide appropriate and supportive services to the distribution networks that make our product available to treating physicians and facilities. Once marketing authorization is granted, we plan to market our product in the North American, European and Asian regions.
As part of our long-term strategy, we will consider clinical development and commercialization collaborations and/or partnerships with international companies involved in the diabetes therapeutic area. Currently, leading companies in this field include Novo Nordisk, Takeda Pharmaceutical, Eli Lilly, GlaxoSmithKline, Sanofi Aventis and Merck.
Future Product Candidates
Currently, liver cells are best suited for generating AIP cells. Future products may involve the use of cell types other than liver that are more easily accessible from the diabetic patient or from unrelated donors. Additionally, other adult cells (i.e. fibroblasts) may be studied for trans-differentiation into functional cells in diseases other than insulin-dependent disorders (i.e. neurodegenerative).
11
Competition
Insulin therapy is used for Insulin-Dependent Diabetes Mellitus (IDDM) patients who are not controlled with oral medications, but this therapy has well-known and well-characterized disadvantages. Weight gain is a common side effect of insulin therapy, which is a risk factor for cardiovascular disease. Injection of insulin causes pain and inconvenience for patients. Patient compliance and inconvenience of self-administering multiple daily insulin injections is also considered a disadvantage of this therapy. The most serious adverse effect of insulin therapy is hypoglycemia.
The global diabetes market comprising the insulin, insulin analogues and other anti-diabetic drugs has been evolving rapidly. Today’s overall diabetes market is dominated by a handful of participants such as Novo Nordisk A/S, Eli Lilly and Company, Sanofi-Aventis, Takeda Pharmaceutical Company Limited, Pfizer Inc., Merck KgaA, and Bayer AG.
Collaboration Agreements
We have embarked on a strategy of collaborative arrangements with strategically situated third parties around the world. We believe that these parties have the expertise, experience and strategic location to advance our clinical development business.
Korea
On April 7, 2015, we executed a non-binding memorandum of understanding with Global Stem Cell & Regenerative Medicine Acceleration Center of Korea in order to collaborate research in stem cell and regenerative medicine. Both parties will absorb their own expenses related to the memorandum of understanding. The memorandum of understanding is for three years unless one of the parties provides written notice terminating the memorandum of understanding.
On March 25, 2015 the Israeli Subsidiary executed a non-binding memorandum of understanding with CureCell Co., Ltd. of Korea (“CureCell”) as an initial step for finalizing a joint venture to develop and bring to market Orgenesis' AIP cell therapy product in Korea. The parties jointly aim to raise all the available funds from both public and private sectors. The memorandum of understanding is intended to be finalized by a definitive agreement and will be in effect until we sign a formal agreement with CureCell or either party withdraws their collaboration.
Russia
On November 12, 2015, our Israeli Subsidiary entered into a collaboration agreement with Biosequel LLC, a company incorporated and existing under the laws of Russia (“Biosequel”) to collaborate, in carrying out clinical trials and eventually marketing the Company’s products in Russia, Belarus and Kazakhstan. The collaboration is divided into two stages, with the first focused on obtaining the requisite regulatory approvals for conducting clinical trials, as well as performing all clinical and other testing required for market authorization in the defined territory. The second stage will focus on marketing the products in the territory and will be subject to obtaining requisite approvals for such marketing. Biosequel will fund the costs for the first stage, which is expected to last for five or more years, but such stage may terminate earlier if the necessary regulatory approvals for commencement of clinical trials are not obtained by the second anniversary of the agreement or if the malting approvals are not obtained with 48 months following the commencement of the clinical trials. The collaboration agreement is also terminable under certain limited conditions relating to a party’s insolvency or bankruptcy related event or breach of a material term of the agreement and force majeure events or upon the termination of the THM License Agreement.
China, China, Hong Kong SAR and Macau SAR
On February 18, 2016, our Israeli Subsidiary entered into a Collaboration Agreement (the “Collaboration Agreement) with Grand China Energy Group Limited with headquarters in Beijing, China (“Grand China”) to collaborate in carrying out clinical trials and marketing the Company’s autologous insulin producing cell therapy product in the Peoples Republic of China, Hong Kong and Macau, based on achieving certain pre-market development milestones that include Grand China obtaining the requisite regulatory approvals for commercialization of our AIP cells, including performing all clinical and other testing required for market authorization in each jurisdiction in the territory. Upon achieving the pre-market development milestones by Grand China, the parties will collaborate on marketing the products in the territory. Grand China will bear all costs associated with the pre-marketing development efforts in the territory, which is expected to last for approximately four years. Subject to the completion of the pre-marketing development milestones, our Israeli Subsidiary has agreed to grant to Grand China, or a fully owned subsidiary thereof, under a separate sub-license agreement, an exclusive sub-license to the intellectual property underlying the solely for commercialization of the Company’s products in each such jurisdiction in the territory where all of the pre-marketing development required to commercialize the AIP cells have been successfully completed by Grand China. Grand China has agreed to pay annual license fees, ongoing royalties based on net sales generated by Grand China and its sublicensees, milestone payments and sublicense fees.
12
Research and Development Expenditures
We incurred $1,860 thousand in research and development expenditures in the last fiscal year ended November 30, 2015, of which $793 thousand was covered by grant funding (See Item 8). We intend to dedicate most of our capital to research and development with no expectation of revenue from product sales in the foreseeable future.
Contract Development and Manufacturing Businesss
Acquisition of MaSTherCell
We acquired MaSTherCell in November 2014 pursuant to a share purchase agreement with MaSTherCell’s shareholders dated as of November 12, 2014, as subsequently amended (the “SEA”). Under the SEA, as amended in November 2015, we agreed to remit to MaSTherCell, by way of an equity investment, EUR 3.8 million by November 30, 2015 (the “Initial Investment”), to be followed by a subsequent equity investment by December 31, 2015 in MaSTherCell of EUR 1.2 million. By agreement with the MaSTherCell shareholders, we remitted in December 2015, the sum of EUR 3.8 million or $4,103,288, in compliance with our obligations as required under the SEA. The right of the former MaSTherCell shareholders to unwind the merger with our Company terminated upon the such investment. Additionally, in connection with the equity investment, on December 10, 2015 we agreed to invest an additional EUR 2.2 million in MaSTherCell equity in addition to the Initial Investment, which additional amount becomes due upon the request of the MaSTherCell board of directors, of whom Company directors/officers currently represent a majority.
In connection with the above agreements, we granted to certain former MaSTherCell shareholders, who currently hold approximately 12% of the Company’s outstanding common stock, the first right to negotiate the terms of the sale of MaSTherCell, should the Company decide at a future date to sell its shares in MaSTherCell or otherwise sell equity interests in MaSTherCell (the “Sale Event”), on an exclusive basis, for the first thirty days following our delivery to such shareholders of notice of such intention. We agreed to accept the offer of such shareholders resulting from the Sale Event negotiations, unless our board of directors determines that a materially superior offer may be available to us if the Sale Event were open to other parties, in which case we are entitled to negotiate the Sale Event with unrelated third parties.
Our Plans for MaSTherCell
We are conducting our CDMO business thorugh MaSTherCell. Subject to raising additional working capital, we intend to devote significant resources to process development and manufacturing in order to optimize the safety and efficacy of our future product candidates for our customers, as well as our cost of goods and time to market. Our goal is to carefully manage our fixed cost structure, maximize optionality, and drive long-term cost of goods as low as possible. We believe that operating our own manufacturing facility provides us with enhanced control of material supply for both clinical trials and the commercial market, will enable the more rapid implementation of process changes, and will allow for better long-term margins.
MaSTherCell's target customers are primarily cell therapy companies that are in pre- or early-stage clinical trials. This stems from the finding that these companies' processes have to be set up right from start in order for them to obtain approved products that have the simplest possible process and with the lowest possible cost of goods sold (COGS). Therefore, MaSTherCell's strategy is to build long term relationships with its customers in order to help them bring highly potent cell therapy products faster to the market and in cost-effective ways.
13
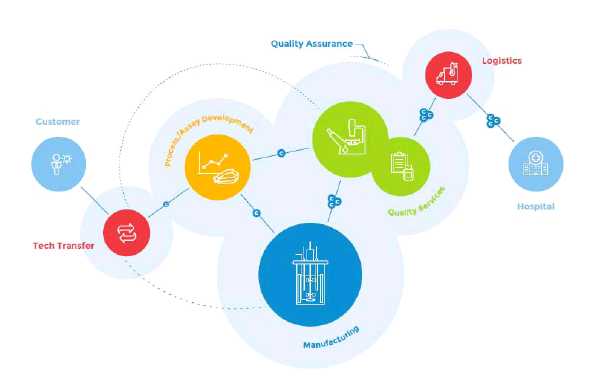
To provide these services MaSTherCell relies on a team of dedicated experts both from academic and industry backgrounds. It operates through state-of-the-art facilities located just 40 minutes from Brussels, which have received the final cGMP manufacturing authorization from the Belgian Drug Agency (AFMPS) in September 2013.
Competition in the CDMO Field
MaSTherCell competes with a number of companies both directly and indirectly. Key competitors include the following CDMOs: Lonza Group Ltd, Progenitor Cell Therapy (PCT) LLC, Pharmacell BV, WuxiAppTec (WuXi PharmaTech (Cayman) Inc.), Cognate Bioservices Inc., Apceth GmbH & Co. KG, Eufets GmbH, Fraunhofer Gesellschaft, Cellforcure SASU, Cell Therapy Catapult Limited and Molmed S.p.A. MaSTherCell's services differ from these companies in two major aspects:
| • |
Quality and expertise of its services: Clients identify the excellence of its facility, quality system, and people as a major differentiating point compared to competitors; and |
| • |
Flexible and tailored approach: MaSTherCell's philosophy is to build a true partnership with its clients and adapt itself to the clients’ needs, which entails no “off-the-shelf process” nor in-house technology platform, but a dedicated person in plant (of client), joint steering committees on each project and dedicated project managers. |
Neither of these differentiating points results in a price premium compared to other CMO’s as MaSTherCell operates with a lean organization focused solely on cell therapy.
Finally, MaSTherCell is the only CDMO located in Belgium which logistically offers an ideal location given the high concentration of companies active in cell therapy (potential clients and companies with complementary know-how, products and services).
Subsidiaries and Grant Funding
Subsidiaries
14
In addition to our Israeli Subsidiary and MaSTherCell, we have the following subsidiaries:
On July 31, 2013, we incorporated a wholly-owned subsidiary in Maryland, Orgenesis Maryland Inc., or the U.S. Subsidiary, which was formed as the U.S. center for research and development and manufacturing scale-up for our technology. The U.S. Subsidiary received a grant from TEDCO which is being used for pre-clinical research and will oversee initiation and conduct of our Phase 1 clinical trial program. The TEDCO grant is further discussed below.
On October 11, 2013, Orgenesis Ltd. incorporated a wholly-owned subsidiary in Belgium, Orgenesis SPRL, our Belgian Subsidiary, which is engaged in development and manufacturing activities together with clinical development studies in Europe. The incorporation of Orgenesis SPRL followed a strategic decision in May 2013 to work with Pall Life Science Belgium BVBA (formerly ATMI BVBA), a Belgian company, to supply disposable bioreactors as the major component in our product manufacturing. In addition, we made another strategic decision in September 2013 to work with Masthercell SPRL, which we subsequently acquired, in order to develop a manufacturing process and to manufacture our product. Both companies are located in Belgium.
A breakdown of our various subsidiaries is as follows:
| Entity | Percentage of | Location |
| Ownership | ||
| Orgenesis Ltd. | 100% | Israel |
| Orgenesis Maryland Inc. | 100% | United States of America |
| Orgenesis SPRL (1) | 95% | Belgium |
| Cell Therapy Holding SA(2) | 100% | Belgium |
| MaSTherCell SA | 100% | Belgium |
| (1) |
Orgenesis Ltd. Owns 5% of Orgenesis SPRL. |
| (2) |
Cell Therapy Holding SA is a financial holding company of MaSTherCell SA, but has no operations. |
Grant Funding
Maryland Technology Development Corporation (“TEDCO”).
On June 30, 2014, the Company’s U.S. Subsidiary entered into a grant agreement with Maryland Technology Development Corporation (“TEDCO”). TEDCO was created by the Maryland State Legislature in 1998 to facilitate the transfer and commercialization of technology from Maryland’s research universities and federal labs into the marketplace and to assist in the creation and growth of technology based businesses in all regions of the State. TEDCO is an independent organization that strives to be Maryland’s lead source for entrepreneurial business assistance and seed funding for the development of startup companies in Maryland’s innovation economy. TEDCO administers the Maryland Stem Cell Research Fund to promote State funded stem cell research and cures through financial assistance to public and private entities within the State. Under the agreement, TEDCO has agreed to give the U.S Subsidiary an amount not to exceed $406,431 (the “Grant”). The Grant will be used solely to finance the costs to conduct the research project entitled “Autologous Insulin Producing (AIP) Cells for Diabetes” during a period of two years. On July 22, 2014, the U.S Subsidiary received an advance payment of $203,000 under the grant. Through November 30, 2015, the company spent the full amount of the grant. On September 21, 2015 the U.S Subsidiary received the second advance payment in amount of $203,000.
Department De La Gestion Financiere Direction De L’analyse Financiere (“DGO6”)
On November 17, 2014, our Belgian Subsidiary received the formal approval from the Walloon Region, Belgium (Service Public of Wallonia, DGO6) for a €2.015 million ($2.4 million) support program for the research and development of a potential cure for Type 1 Diabetes. The financial support is composed of a €1,085 thousand (70% of budgeted costs) grant for the industrial research part of the research program and a further recoverable advance of €930 thousand (60% of budgeted costs) of the experimental development part of the research program. The grants will be paid to the Company over a period of approximately 3 years. The grants are subject to certain conditions with respect to our work in the Walloon Region.
15
In addition, the DGO6 is also entitled to a royalty upon revenue being generated from any commercial application of the technology. On December 9 and 16, 2014, the Belgian Subsidiary received €651 thousand and €558 thousand under the grant, respectively. Up through November 30, 2015, an amount of $1.4 million (€1.1 million) was recorded as deduction of research and development expenses and an amount of $114 thousand was recorded as advance payments on account of grant. On March 20, 2012, MaSTherCell had been granted an investment grant from the DGO6 for an amount of €1,421 thousand. This grant is related to the investment in the production facility with a coverage of 32% of the investment planned. A first payment of €568 thousand was received in August 2013. The remaining part is expected to be paid by the end of fiscal 2016.
Israel-U.S. Binational Industrial Research and Development Foundation (“BIRD”)
On September 9, 2015, the Israeli Subsidiary entered into a pharma Cooperation and Project Funding Agreement (CPFA) with BIRD and Pall Corporation, a U.S. company. BIRD will give a conditional grant of $400 thousand each (according to terms defined in the agreement), for a joint research and development project for the use Autologous Insulin Producing (AIP) Cells for the Treatment of Diabetes (the “Project”). The Project started on March 1, 2015. Upon the conclusion of product development, the grant shall be repaid at the rate of 5% of gross sales. The grant will be used solely to finance the costs to conduct the research of the project during a period of 18 months starting on March 1, 2015.
Up through November 30, 2015, an amount of $153 thousand was recorded as deduction of research and development expenses and receivable on account of grant. On September 21, 2015, the Israeli Subsidiary received $100 thousand under the grant.
Intellectual Property
We will be able to protect our technology and products from unauthorized use by third parties only to the extent it is covered by valid and enforceable patents or is effectively maintained as trade secrets. Patents and other proprietary rights are thus an essential element of our business.
Our success will depend in part on our ability to obtain and maintain proprietary protection for our product candidates, technology, and know-how, to operate without infringing on the proprietary rights of others, and to prevent others from infringing it proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, filing U.S. and foreign patent applications related to our proprietary technology, inventions, and improvements that are important to the development of our business. We also rely on trade secrets, know-how, continuing technological innovation, and in-licensing opportunities to develop and maintain our proprietary position.
We own or have exclusive rights to three (3) United States and seven (7) foreign issued patents and allowed patent applications, three (3) pending applications in the United States, twelve (12) pending applications in foreign jurisdictions: Europe, Australia, Brazil, Canada, China, Columbia, Eurasia, Israel, Japan, South Korea, Mexico, and Singapore, and one (1) international PCT patent application, relating to the transdifferentiation of cells (including hepatic cells) to cells having pancreatic β-cell phenotype and function, and their use in the treatment of degenerative pancreatic disorders including diabetes, pancreatic cancer, and pancreatitis.
Granted United States patents which are directed to methods of making transdifferentiated cells will expire between 2021 and 2023, excluding any patent term extensions that might be available following the grant of marketing authorizations. Granted patents outside of the United States directed to making transdifferentiated cells and their uses will expire between 2020 and 2024. We have pending patent applications for methods of making our product, the product itself, and methods of using the product that, if issued, would expire in the United States and in countries outside of the United States between 2034 and 2035, excluding any patent term adjustment that might be available following the grant of the patent and any patent term extensions that might be available following the grant of marketing authorizations. These pending patent applications are directed to the following specific compositions and methods: a method of producing a transdifferentiated population of cells, a population of transdifferentiated cells, a method of treating a degenerative pancreatic disorder in a subject in need, a method of isolating a population of cells that have an enriched capacity for transcription factor induced transdifferentiation, an isolated population of cells having enriched transdifferentiation capacity, a method of increasing transdifferentiation efficiency in a population of cells, a population of liver cells enriched for cells predisposed to transdifferentiation, and a method of manufacturing a population of human insulin producing cells and the population of cells produced by the recited manufacturing method.
16
Government Regulation
We have not sought approval from the FDA for the AIP cells. Among all forms of cell therapy modalities, we believe that autologous cell replacement therapy seems to be of the highest benefit. We believe that it seems to be safer than other options as it does not alter the host genome but only alters the set of expressed epigenetic information that seems to be highly specific to the reprogramming protocol. It provides an abundant source of therapeutic tissue, which is not rejected by the patient and does not have to be treated by immune suppressants. It is highly ethical since no human organ donations or embryo-derived cells are needed. The proposed therapeutic approach does not require cell bio-banking at birth, which is both expensive and cannot be used for patients born prior to 2000.
Within the last decade, many studies published in leading scientific journal confirmed the capacity of reprogramming adult cells from many of our mature organs to either alternate organs or to “stem like cells”. The most widely used autologous cell replacement protocol is the one used for autologous implantation of bone marrow stem cells. This protocol is widely used in patients undergoing a massive chemotherapy session that destroys their bone marrow cells. However, the stem cells used for cancer patients delineated above do not require extensive manipulation and is regarded by FDA as “minimally manipulated”.
An additional autologous cell therapy approach already used in man is autologous chondrocyte implantation (ACI). In the United States, Genzyme Corporation provides the only FDA approved ACI treatment: Carticel. The Carticel treatment is designated for young, healthy patients with medium to large sized damage to cartilage. During an initial procedure, the patient’s own chondrocytes are removed arthroscopically from a non-load-bearing area from either the intercondylar notch or the superior ridge of the medial or lateral femoral condyles.
To aid us in our efforts to achieve the highest level of compliance with FDA requirements, we have looked to hire experts in the field of pharmaceutical compliance.
Regulatory Process in the United States
Our product is subject to regulation as a biological product under the Public Health Service Act and the Food, Drug and Cosmetic Act. FDA generally requires the following steps for pre-market approval or licensure of a new biological product:
| • |
Pre-clinical laboratory and animal tests conducted in compliance with the Good Laboratory Practice, or GLP, requirements to assess a drug’s biological activity and to identify potential safety problems, and to characterize and document the product’s chemistry, manufacturing controls, formulation, and stability; |
| • |
Submission to FDA of an Investigational New Drug, or IND application, which must become effective before clinical testing in humans can begin; |
| • |
Obtaining approval of Institutional Review Boards, or IRBs, of research institutions or other clinical sites to introduce the biologic drug candidate into humans in clinical trials; |
| • |
Conducting adequate and well-controlled human clinical trials to establish the safety and efficacy of the product for its intended indication conducted in compliance with Good Clinical Practice, or GCP requirements; |
| • |
Compliance with current Good Manufacturing Practices, or cGMP regulations and standards; |
| • |
Submission to FDA of a Biologics License Application, or BLA, for marketing that includes adequate results of pre-clinical testing and clinical trials; |
| • |
FDA reviews the marketing application in order to determine, among other things, whether the product is safe, effective and potent for its intended uses; and |
| • |
Obtaining FDA approval of the BLA, including inspection and approval of the product manufacturing facility as compliant with cGMP requirements, prior to any commercial sale or shipment of the pharmaceutical agent. FDA may also require post marketing testing and surveillance of approved products, or place other conditions on the approvals. |
17
Regulatory Process in Europe
The European Union (“EU”) has approved a regulation specific to cell and tissue therapy product, the Advanced Therapy Medicinal Product (ATMP) regulation. For products such as our AIP that are regulated as an ATMP, the EU Directive requires:
| • |
Compliance with current Good Manufacturing Practices, or cGMP regulations and standards, pre-clinical laboratory and animal testing; |
| • |
Filing a Clinical Trial Application (CTA) with the various member states or a centralized procedure; Voluntary Harmonization Procedure (VHP), a procedure which makes it possible to obtain a coordinated assessment of an application for a clinical trial that is to take place in several European countries; |
| • |
Obtaining approval of Ethic Committees of research institutions or other clinical sites to introduce the AIP into humans in clinical trials; |
| • |
Adequate and well-controlled clinical trials to establish the safety and efficacy of the product for its intended use; and |
| • |
Submission to EMEA for a Marketing Authorization (MA); Review and approval of the MAA (Marketing Authorization Application). |
Clinical trials
Typically, both in the U.S. and the European Union, clinical testing involves a three-phase process, although the phases may overlap. In Phase I, clinical trials are conducted with a small number of healthy volunteers or patients and are designed to provide information about product safety and to evaluate the pattern of drug distribution and metabolism within the body. In Phase II, clinical trials are conducted with groups of patients afflicted with a specific disease in order to determine preliminary efficacy, optimal dosages and expanded evidence of safety. In some cases, an initial trial is conducted in diseased patients to assess both preliminary efficacy and preliminary safety and patterns of drug metabolism and distribution, in which case it is referred to as a Phase I/II trial. Phase III clinical trials are generally large-scale, multi-center, comparative trials conducted with patients afflicted with a target disease in order to provide statistically valid proof of efficacy, as well as safety and potency. In some circumstances, FDA or EMA may require Phase IV or post-marketing trials if it feels that additional information needs to be collected about the drug after it is on the market. During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data and clinical trial investigators. An agency may, at its discretion, re-evaluate, alter, suspend, or terminate the testing based upon the data that have been accumulated to that point and its assessment of the risk/benefit ratio to the patient. Monitoring all aspects of the study to minimize risks is a continuing process. All adverse events must be reported to the FDA or EMA.
Employees
As of November 30, 2015, we had 41 full-time employees, including the employees of our subsidiaries. Most of our senior management and professional employees have had prior experience in pharmaceutical or biotechnology companies. None of our employees is covered by collective bargaining agreements. We believe that our relations with our employees are good.
Corporate and Available Information
Our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and all amendments to those reports are made available free of charge though our Internet website (http://www.orgenesis.com) as soon as practicable after such material is electronically filed with, or furnished to, the Securities and Exchange Commission. Except as otherwise stated in these documents, the information contained on our website or available by hyperlink from our website is not incorporated by reference into this report or any other documents we file, with or furnish to, the Securities and Exchange Commission.
18
ITEM 1A. RISK FACTORS
An investment in our common stock involves a number of very significant risks. You should carefully consider the following risks and uncertainties in addition to other information in this report in evaluating our company and its business before purchasing shares of our common stock. Our business, operating results and financial condition could be seriously harmed due to any of the following risks. You could lose all or part of your investment due to any of these risks.
Risks Related to Our Financial Condition and Liquidity
We are at an early stage of development, making our future viability, going concern, and success uncertain.
Our business is at an early stage of development. We are subject to all of the business risks associated with an early stage enterprise. We are still in the process of conducting laboratory research to determine potential product candidates for our cell therapy technology, and have not yet identified a lead product candidate or begun clinical trials with respect to any product candidates. The success of our business will depend on many factors including, without limitation, our ability to obtain additional capital as and when needed, identify a viable, lead product candidate, successfully complete preclinical testing and clinical trials, obtain marketing approvals and establish sales and marketing capacity. We may not be able to develop any products, initiate or complete clinical trials for any product candidates, obtain requisite regulatory approvals or commercialize any products. Any potential products may prove to have undesirable and unintended side effects or other characteristics adversely affecting their safety, efficacy or cost-effectiveness that could prevent or limit their use. Any potential product incorporating our technology may fail to provide the intended therapeutic benefits or to achieve therapeutic benefits equal to or better than the standard of treatment at the time of testing or production. As a result of our early stage of development, the future viability, going concern, and success of our company is uncertain.
If we fail to obtain the capital necessary to fund our operations, our financial results, financial condition and ability to continue as a going concern will be adversely affected and we will have to delay, reduce the scope of or terminate some or all of our research and development programs and may be forced to cease operations.
Our consolidated financial statements for the year ended November 30, 2015 have been prepared assuming that we will continue as a going concern. Our recurring losses from operations and our stockholders’ deficit raise substantial doubt about our ability to continue as a going concern. As a result, our independent registered public accounting firm included an explanatory paragraph in its report on our consolidated financial statements for the year ended November 30, 2015 with respect to this uncertainty. We do not anticipate generating meaningful revenue in the foreseeable future and we will need to raise additional capital to fund our operating requirements and continue as a going concern. In addition, the perception that we may not be able to continue as a going concern may cause others to choose not to deal with us due to concerns about our ability to meet our contractual obligations and may adversely affect our ability to raise additional capital.
Based upon our current plan of operations, and assuming we raise capital as and when needed, we anticipate spending approximately $5.7 million on manufacturing and scale-up activities during the 12 months ending November 30, 2016. Our capital requirements will depend on many factors, including:
| • |
The number and type of product candidates that we pursue; |
| • |
The progress, timing, scope, number and complexity of preclinical studies and clinical trials that we undertake; |
| • |
The timing and cost involved in obtaining FDA and other regulatory approvals; |
| • |
Costs related to maintaining, expanding and enforcing our intellectual property portfolio; |
| • |
Whether we enter into joint venture, licensing, collaboration or other strategic transactions involving funding or otherwise relating to research and development, manufacturing or marketing activities, and the scope and terms of any such arrangements; and |
| • |
The time and cost necessary to launch and successfully commercialize our product candidates, if approved. |
These factors raise substantial doubt about the Company’s ability to continue as a going concern.
19
We need to raise additional capital through equity or debt financing and collaborative arrangements, or some combination thereof. Substantially all of our operating capital requirements since inception have been provided by existing investors, on an as needed basis. Additional capital may not be available on acceptable terms, or at all. If we raise capital through the sale of equity-based securities, dilution to our then-existing equity investors would result. If we obtain capital through the incurrence of debt, we would likely become subject to covenants restricting our business activities, and holders of debt instruments would have rights and privileges senior to those of our equity investors. In addition, servicing the interest and repayment obligations under borrowings would divert funds that would otherwise be available to support research and development, clinical or commercialization activities. If we obtain capital through collaborative arrangements, these arrangements could require us to relinquish some rights to our technologies or product candidates and we may become dependent on third parties.
If we are unable to obtain adequate financing on a timely basis, we may be required to delay, reduce the scope of or eliminate one or more of our planned research and development programs and otherwise limit or cease our operations.
We have incurred losses since inception and may never achieve or sustain profitability.
We have incurred significant operating losses. We expect to incur operating losses for the foreseeable future. We had an accumulated deficit of $20.6 million as of November 30, 2015. Currently, our revenues are not substantial enough to cover our operating expenses. The extent of future operating losses is highly uncertain, and we may never achieve or sustain profitability
Risk Related to our CDMO Business
We need additional financing to grow our CDMO operations; if we are unable to raise additional capital, as and when needed, or on acceptable terms, we may be forced to delay, reduce or eliminate the expansion of our contract development and manufacturing operations.
The current operating plan of our subsidiary, MaSTherCell, requires additional capital to fund, among other things, the operation, enhancement and expansion of its operations to support its customers. The amount and timing of our future capital requirements also will likely depend on many other factors, including:
| • |
the cost of expansion of our contract development and manufacturing operations, including but not limited to, the costs of expanded facilities, equipment costs, engineering and innovation initiatives and personnel; and |
| • |
the opportunity to produce therapies in commercial phases for a customer which will require large production units. |
We have committed to remit to MaSTherCell, as an equity investment, an additional EUR 2.2 milllion, as needed by MaSTherCell. We will need to raise additional capital in order to fund MaSTherCell. Currently, we have no commitments for our working capital needs and we may ultimately be unable to raise capital on terms that are acceptable to us, if at all. Our inability to obtain necessary capital or financing to fund our future operating needs could adversely affect our business, results of operations and financial condition.
MaSTherCell has incurred substantial losses and negative cash flow from operations in the past, and expects to continue to incur losses and negative cash flow for the foreseeable future.
MaSTherCell has a limited operating history, limited capital, and limited sources of revenue. Since its inception in 2011 through November 30, 2015, the revenues generated have not been sufficient to cover costs attributable to that business. Based upon current plans, it is expected that MaSTherCell will reach a positive EBITDA in 2016, although our we will incur operating losses in future periods. This will happen because there are expenses associated with the development, marketing, and sales of our services. As a result, we may not generate significant revenues in the future. Failure to generate significant revenues in near future may cause us to suspend or cease activities. Our ability to achieve and maintain profitability and positive cash flow is dependent upon our ability to generate revenues, manage expenses, and compete successfully with our direct and indirect competitors.
A significant global market for our third party manufacturing services at MaSTherCell may never emerge.
20
At MaSTherCell, the current market and our existing contracts principally consist of providing consulting and manufacturing of cell and tissue-based therapeutic products in clinical trials. The number of people who may use cell or tissue-based therapies and thus the demand for stem cell processing services is difficult to forecast. If cell therapies under development by our customers to treat disease are not proven effective, demonstrate unacceptable risks or side effects or, where required, fail to receive regulatory approval, our business will be significantly impaired. While the therapeutic application of cells to treat serious diseases is currently being explored by a number of companies, to date there are only a handful of approved products in the United States, Asia and in Europe. Ultimately, our success in developing our contract development and manufacturing business depends on the development and growth of a broad and profitable global market for cell- and tissue-based therapies and services and our ability to capture a share of this market through MaSTherCell.
MaSTherCell's revenues may vary dramatically from period to period making it difficult to forecast future results.
MaSTherCell recorded revenues of $2.9 million for the year ended November 30, 2015. The nature and duration of MaSTherCell's contracts with customers often involve regular renegotiation of the scope, level and price of the services we are providing. If our customers reduce the level of their spending on research and development or marketing or are unsuccessful in attaining or retaining product sales due to market conditions, reimbursement issues or other factors, our results of operations may be materially impacted. In addition, other factors, including the rate of enrollment for clinical studies, will directly impact the level and timing of the products and services we deliver. As such, the levels of our revenues and profitability can fluctuate significantly from one period to another and it can be difficult to forecast the level of future revenues with any certainty.
The loss of one or more of MaSTherCell’s major clients or a decline in demand from one or more of these clients could harm MaSTherCell’s business.
MaSTherCell has a limited number of major clients that together account for a large percentage of the total revenues earned. There can be no assurance that such clients will continue to use MaSTherCell’s services at the same level or at all. A reduction or delay in the use of MaSTherCell’s services, including reductions or delays due to market, economic or competitive conditions, could have a material adverse effect on MaSTherCell’s business, operating results and financial condition.
MaSTherCell has a finite manufacturing capacity, which could inhibit the long-term growth prospects of this business.
MaSTherCell currently provides services and produces materials for clinical trials at its existing manufacturing facilities in Gosselies (Belgium), which it has designed and operated to be compliant with cGMP requirements. While we believe these facilities provide it with sufficient capacity to meet expected near term demand, it is possible that the demand for its services and products could exceed its existing manufacturing capacity. It may become necessary or desirable for it to expand its manufacturing capabilities for cell therapy services and products in the future which may require it to invest significant amounts of capital and to obtain regulatory approvals. In this regard, we are reviewing opportunities for expansion to both commercial level and international manufacturing capabilities. If we are unable to meet rising demand for products and services on a timely basis or unable to maintain cGMP compliance standards, then it is likely that our clients and potential clients will elect to obtain the products and services from competitors, which could materially and adversely affect the level of our revenues and our prospects for growth.
MaSTherCell’s business is subject to risks associated with a single manufacturing facility.
MaSTherCell’s contract manufacturing services are dependent upon a single facility located in Gosselies (Belgium). A catastrophic loss of the use of all or a portion of MaSTherCell’s manufacturing facility due to accident, fire, explosion, labor issues, weather conditions, other natural disaster or otherwise, whether short or long-term, could have a material adverse effect on MaSTherCell’s customer relationships and financial results.
21
If MaSTherCell loses electrical power at its manufacturing facility, its business operations may be adversely affected.
MaSTherCell owns a back-up generator allowing it to provide for its manufacturing power consumption needs for a few hours. However, if MaSTherCell loses electrical power at its manufacturing facility for more than a few hours, MaSTherCell would be unable to continue its manufacturing operations for an extended period of time because MaSTherCell does not own any other back-up power source large enough to provide for its manufacturing power consumption needs. Additionally, MaSTherCell does not have an alternative manufacturing location. Therefore, a significant disruption in MaSTherCell’s manufacturing operations could materially and adversely affect its business operations during an extended period of power outage.
We have a limited marketing staff and budget for our MaSTherCell operations, which could limit our ability to grow this business.
The degree of market acceptance of our products and services depends upon a number of factors, including the strength of our sales and marketing support. If our marketing is not effective, our ability to generate revenues could be significantly impaired. The newness of the industry and capital constraints provide challenges to our marketing and sales activities at MaSTherCell, and the failure to attract a sufficient base of customers will affect our ability to increase our revenues and operate profitably.
The logistics associated with the distribution of materials produced by MaSTherCell for third parties and for us are significant, complex and expensive and may negatively impact our ability to generate and meet future demand for our products and improve profitability.
Current cell therapy products and product candidates, have a limited shelf life, in certain instances limited to less than 12 hours. Thus, it is necessary to minimize the amount of time between when the cell product is extracted from a patient, arrives at our facility for processing, and is returned for infusion in the patient.
To do so, we need our cell therapy facilities to be located in major population centers in which patients are likely to be located and within close proximity of major airports. In the future, it may be necessary to build new facilities, which would require a significant commitment of capital and may not then be available to us. Even if we are able to establish such new facilities, we may experience challenges in ensuring that they are compliant with cGMP standards, EMEA requirements, and/or applicable state or local regulations. We cannot be certain that we would be able to recoup the costs of establishing a facility in a given market. Given these risks, we could choose not to expand our cell processing and manufacturing services into new geographic markets which will limit our future growth prospects.
Product liability and uninsured risks may adversely affect MaSTherCell’s continuing operations and damage its reputation.
MaSTherCell operates in an industry susceptible to significant product liability claims. MaSTherCell may be liable if it manufactures any product that causes injury, illness, or death. In addition, product liability claims may be brought against MaSTherCell’s clients, in which case MaSTherCell’s clients or others may seek contribution from MaSTherCell if they incur any loss or expenses related to such claims. These claims may be brought by individuals seeking relief or by groups seeking to represent a class. The defense of such claims may be costly and time-consuming, and could divert the attention of MaSTherCell’s management and technical personnel.
A breakdown or breach of MaSTherCell’s information technology systems could subject MaSTherCell to liability or interrupt the operation of its business.
MaSTherCell relies upon its information technology systems and infrastructure for its business. The size and complexity of MaSTherCell’s computer systems make it potentially vulnerable to breakdown and unauthorized intrusion. MaSTherCell could also experience a business interruption, theft of confidential information, or reputational damage from industrial espionage attacks, malware or other cyber attacks, which may compromise MaSTherCell’s system infrastructure or lead to data leakage, either internally or at MaSTherCell’s third-party providers.
22
Similarly, data privacy breaches by those who access MaSTherCell’s systems may pose a risk that sensitive data, including intellectual property, trade secrets or personal information belonging to MaSTherCell or its employees, clients or other business partners, may be exposed to unauthorized persons or to the public. There can be no assurance that MaSTherCell’s efforts to protect its data and information technology systems will prevent breakdowns or breaches in MaSTherCell’s systems that could adversely affect its business and result in financial and reputational harm to MaSTherCell.
We face competition from established as well as other emerging companies, which could divert clients to our competitors, result in pricing pressure and significantly reduce our revenue.
We expect existing competitors and new entrants to CDMO market to constantly revise and improve their business models in response to challenges from competing businesses, including ours. Some of our competitors and potential competitors have significantly greater resources than we do. Increased competition may result in pricing pressure for us in terms of the prices we are able to negotiate to receive from a client. If we cannot compete successfully against our competitors, our ability to grow our business and achieve profitability could be impaired.
Risks Relating to the Biopharmaceutical Business
THM is entitled to cancel the License Agreement.
Pursuant to the terms of the License Agreement with THM, the Israeli Subsidiary must develop, manufacture, sell and market the products pursuant to the milestones and time schedule specified in the Development Plan. In the event the Israeli Subsidiary fails to fulfill the terms of the Development Plan, THM shall be entitled to terminate the License Agreement by providing the Israeli Subsidiary with written notice of such a breach and if the Israeli Subsidiary does not cure such breach within one year of receiving the notice. If THM cancels the License Agreement, our business may be materially adversely affected. THM may also terminate the License Agreement if the Israeli Subsidiary breaches an obligation contained in the License Agreement and does not cure it within 180 days of receiving notice of the breach. Any termination or cancellation of the License Agreement is likely to materially adversely affect our business and prospects.
If we are unable to successfully acquire, develop or commercialize new products, our operating results will suffer.
Our future results of operations will depend to a significant extent upon our ability to successfully develop and commercialize our technology and businesses in a timely manner. There are numerous difficulties in developing and commercializing new technologies and products, including:
| • |
successfully achieving major developmental steps required to bring the product to a clinical testing stage and clinical testing may not be positive; |
| • |
developing, testing and manufacturing products in compliance with regulatory standards in a timely manner; |
| • |
the failure to receive requisite regulatory approvals for such products in a timely manner or at all; |
| • |
developing and commercializing a new product is time consuming, costly and subject to numerous factors, including legal actions brought by our competitors, that may delay or prevent the development and commercialization of our product; |
| • |
incomplete, unconvincing or equivocal clinical trials data; |
| • |
experiencing delays or unanticipated costs; |
| • |
significant and unpredictable changes in the payer landscape, coverage and reimbursement for our future product; |
| • |
experiencing delays as a result of limited resources at the U.S. Food and Drug Administration (“FDA”) or other regulatory agencies; and |
| • |
changing review and approval policies and standards at the FDA and other regulatory agencies. |
As a result of these and other difficulties, products in development by us may or may not receive timely regulatory approvals, or approvals at all, necessary for marketing by us or other third-party partners. If any of our future products are not approved in a timely fashion or, when acquired or developed and approved, cannot be successfully manufactured, commercialized or reimbursed, our operating results could be adversely affected. We cannot guarantee that any investment we make in developing product will be recouped, even if we are successful in commercializing these products.
23
Our research and development programs are based on novel technologies and are inherently risky.
We are subject to the risks of failure inherent in the development of products based on new technologies. The novel nature of our cell therapy technology creates significant challenges with respect to product development and optimization, manufacturing, government regulation and approval, third-party reimbursement and market acceptance. For example, the FDA has relatively limited experience with the development and regulation of cell therapy products and, therefore, the pathway to marketing approval for our cell therapy product candidates may accordingly be more complex, lengthy and uncertain than for a more conventional product candidate. The indications of use for which we choose to pursue development may have clinical effectiveness endpoints that have not previously been reviewed or validated by the FDA, which may complicate or delay our effort to ultimately obtain FDA approval. Our efforts to overcome these challenges may not prove successful, and any product candidate we seek to develop may not be successfully developed or commercialized.
Extensive industry regulation has had, and will continue to have, a significant impact on our business, especially our product development, manufacturing and distribution capabilities.
All pharmaceutical companies are subject to extensive, complex, costly and evolving government regulation. For the U.S., this is principally administered by the FDA and to a lesser extent by the Drug Enforcement Administration (“DEA”) and state government agencies, as well as by varying regulatory agencies in foreign countries where products or product candidates are being manufactured and/or marketed. The Federal Food, Drug and Cosmetic Act, the Controlled Substances Act and other federal statutes and regulations, and similar foreign statutes and regulations, govern or influence the testing, manufacturing, packing, labeling, storing, record keeping, safety, approval, advertising, promotion, sale and distribution of our future products.
Under these regulations, we may become subject to periodic inspection of our facilities, procedures and operations and/or the testing of our future products by the FDA, the DEA and other authorities, which conduct periodic inspections to confirm that we are in compliance with all applicable regulations. In addition, the FDA and foreign regulatory agencies conduct pre-approval and post-approval reviews and plant inspections to determine whether our systems and processes are in compliance with current good manufacturing practice (“cGMP”) and other regulations. Following such inspections, the FDA or other agency may issue observations, notices, citations and/or warning letters that could cause us to modify certain activities identified during the inspection. FDA guidelines specify that a warning letter is issued only for violations of “regulatory significance” for which the failure to adequately and promptly achieve correction may be expected to result in an enforcement action. We may also be required to report adverse events associated with our future products to FDA and other regulatory authorities. Unexpected or serious health or safety concerns would result in labeling changes, recalls, market withdrawals or other regulatory actions.
The range of possible sanctions includes, among others, FDA issuance of adverse publicity, product recalls or seizures, fines, total or partial suspension of production and/or distribution, suspension of the FDA’s review of product applications, enforcement actions, injunctions, and civil or criminal prosecution. Any such sanctions, if imposed, could have a material adverse effect on our business, operating results, financial condition and cash flows. Under certain circumstances, the FDA also has the authority to revoke previously granted drug approvals. Similar sanctions as detailed above may be available to the FDA under a consent decree, depending upon the actual terms of such decree. If internal compliance programs do not meet regulatory agency standards or if compliance is deemed deficient in any significant way, it could materially harm our business.
For Europe, the European Medicines Agency (“EMEA”) will regulate our future products. Regulatory approval by the EMEA will be subject to the evaluation of data relating to the quality, efficacy and safety of our future products for its proposed use. The time taken to obtain regulatory approval varies between countries. Different regulators may impose their own requirements and may refuse to grant, or may require additional data before granting, an approval, notwithstanding that regulatory approval may have been granted by other regulators. Regulatory approval may be delayed, limited or denied for a number of reasons, including insufficient clinical data, the product not meeting safety or efficacy requirements or any relevant manufacturing processes or facilities not meeting applicable requirements.
24
Further trials and other costly and time-consuming assessments of the product may be required to obtain or maintain regulatory approval. Medicinal products are generally subject to lengthy and rigorous pre-clinical and clinical trials and other extensive, costly and time-consuming procedures mandated by regulatory authorities. We may be required to conduct additional trials beyond those currently planned, which could require significant time and expense.
We have never generated any revenue from product sales and our ability to generate revenue from product sales and become profitable depends significantly on our success in a number of factors.
We have no products approved for commercial sale, have not generated any revenue from product sales, and do not anticipate generating any revenue from product sales until sometime after we have received regulatory approval for the commercial sale of a product candidate. Our ability to generate revenue and achieve profitability depends significantly on our success in many factors, including:
| • |
completing research regarding, and nonclinical and clinical development of, our product candidates; |
| • |
obtaining regulatory approvals and marketing authorizations for product candidates for which we complete clinical studies; |
| • |
developing a sustainable and scalable manufacturing process for our product candidates, including establishing and maintaining commercially viable supply relationships with third parties and establishing our own manufacturing capabilities and infrastructure; |
| • |
launching and commercializing product candidates for which we obtain regulatory approvals and marketing authorizations, either directly or with a collaborator or distributor; |
| • |
obtaining market acceptance of our product candidates as viable treatment options; |
| • |
addressing any competing technological and market developments; |
| • |
identifying, assessing, acquiring and/or developing new product candidates; |
| • |
negotiating favorable terms in any collaboration, licensing, or other arrangements into which we may enter; |
| • |
maintaining, protecting, and expanding our portfolio of intellectual property rights, including patents, trade secrets, and know-how; and |
| • |
attracting, hiring, and retaining qualified personnel. |
Even if one or more of the product candidates that we develop is approved for commercial sale, we anticipate incurring significant costs associated with commercializing any approved product candidate. Our expenses could increase beyond expectations if we are required by the U.S. Food and Drug Administration, or the FDA, or other regulatory agencies, domestic or foreign, to change our manufacturing processes or assays, or to perform clinical, nonclinical, or other types of studies in addition to those that we currently anticipate. If we are successful in obtaining regulatory approvals to market one or more of our product candidates, our revenue will be dependent, in part, upon the size of the markets in the territories for which we gain regulatory approval, the accepted price for the product, the ability to get reimbursement at any price, and whether we own the commercial rights for that territory. If the number of our addressable disease patients is not as significant as we estimate, the indication approved by regulatory authorities is narrower than we expect, or the reasonably accepted population for treatment is narrowed by competition, physician choice or treatment guidelines, we may not generate significant revenue from sales of such products, even if approved. If we are not able to generate revenue from the sale of any approved products, we may never become profitable.
We have concentrated our research and development efforts on technology using cell-based therapy, and our future success is highly dependent on the successful development of that technology for diabetes.
We have developed a technology that demonstrates the capacity to induce a shift in the developmental fate of cells from the liver and differentiating (converting) them into “pancreatic beta cell-like” insulin-producing cells for patients with diabetes. Based on licensed knowhow and patents, our intention is to develop our technology to the clinical stage for regeneration of functional insulin-producing cells, thus enabling normal glucose regulated insulin secretion, via cell therapy. By using therapeutic agents (i.e., PDX-1, and additional pancreatic transcription factors in an adenovirus-vector) that efficiently convert a sub-population of liver cells into pancreatic islets phenotype and function, this approach allows the diabetic patient to be the donor of his own therapeutic tissue and to start producing his/her own insulin in a glucose-responsive manner, thereby eliminating the need for insulin injections. Because this is a new approach to treating diabetes, developing and commercializing our product candidates subjects us to a number of challenges, including:
25
| • |
obtaining regulatory approval from the FDA and other regulatory authorities that have very limited experience with the commercial development of our technology for diabetes; |
| • |
developing and deploying consistent and reliable processes for engineering a patient’s liver cells ex vivo and infusing the engineered cells back into the patient; |
| • |
developing processes for the safe administration of these products, including long-term follow-up for all patients who receive our products; |
| • |
sourcing clinical and, if approved, commercial supplies for the materials used to manufacture and process our products; |
| • |
developing a manufacturing process and distribution network with a cost of goods that allows for an attractive return on investment; and |
| • |
establishing sales and marketing capabilities after obtaining any regulatory approval to gain market acceptance; |
When we are able to commence our clinical trials, we may not be able to conduct our trials on the timelines we expect.
Clinical testing is expensive, time consuming, and subject to uncertainty. We cannot guarantee that any clinical studies will be conducted as planned or completed on schedule, if at all. We expect that our early clinical work will help support the filing with the FDA of an IND for our product in 2016. However, we cannot be sure that we will be able to submit an IND in this time-frame, and we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin. Moreover, even if these trials begin, issues may arise that could suspend or terminate such clinical trials. A failure of one or more clinical studies can occur at any stage of testing, and our future clinical studies may not be successful. Events that may prevent successful or timely completion of clinical development include:
| • |
the inability to generate sufficient preclinical or other in vivo or in vitro data to support the initiation of clinical studies; |
| • |
delays in reaching a consensus with regulatory agencies on study design; |
| • |
the FDA not allowing us to use the clinical trial data from a research institution to support an IND if we cannot demonstrate the comparability of our product candidates with the product candidate used by the relevant research institution in its clinical studies; |
| • |
delays in obtaining required Institutional Review Board, or IRB, approval at each clinical study site; |
| • |
imposition of a temporary or permanent clinical hold by regulatory agencies for a number of reasons, including after review of an IND application or amendment, or equivalent application or amendment; as a result of a new safety finding that presents unreasonable risk to clinical trial participants; a negative finding from an inspection of our clinical study operations or study sites; developments on trials conducted by competitors for related technology that raises FDA concerns about risk to patients of the technology broadly; or if FDA finds that the investigational protocol or plan is clearly deficient to meet its stated objectives; |
| • |
delays in recruiting suitable patients to participate in our clinical studies; |
| • |
difficulty collaborating with patient groups and investigators; |
| • |
failure to perform in accordance with the FDA’s current good clinical practices, or cGCPs, requirements, or applicable regulatory guidelines in other countries; |
| • |
delays in having patients complete participation in a study or return for post-treatment follow-up; |
| • |
patients dropping out of a study; |
| • |
occurrence of adverse events associated with the product candidate that are viewed to outweigh its potential benefits; |
26
| • |
changes in regulatory requirements and guidance that require amending or submitting new clinical protocols; |
| • |
changes in the standard of care on which a clinical development plan was based, which may require new or additional trials; |
| • |
the cost of clinical studies of our product candidates being greater than we anticipate; |
| • |
clinical studies of our product candidates producing negative or inconclusive results, which may result in our deciding, or regulators requiring us, to conduct additional clinical studies or abandon product development programs; and |
| • |
delays in manufacturing, testing, releasing, validating, or importing/exporting sufficient stable quantities of our product candidates for use in clinical studies or the inability to do any of the foregoing. |
Any inability to successfully complete preclinical and clinical development could result in additional costs to us or impair our ability to generate revenue. In addition, if we make manufacturing or formulation changes to our product candidates, we may be required to or we may elect to conduct additional studies to bridge our modified product candidates to earlier versions. Clinical study delays could also shorten any periods during which our products have patent protection and may allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates and may harm our business and results of operations.
Our clinical trial results may also not support approval, whether accelerated approval, conditional marketing authorizations, or regular approval. The results of preclinical and clinical studies may not be predictive of the results of later-stage clinical trials, and product candidates in later stages of clinical trials may fail to show the desired safety and efficacy despite having progressed through preclinical studies and initial clinical trials. In addition, our product candidates could fail to receive regulatory approval for many reasons, including the following:
| • |
the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; |
| • |
the population studied in the clinical program may not be sufficiently broad or representative to assure safety in the full population for which we seek approval; |
| • |
we may be unable to demonstrate that our product candidates’ risk-benefit ratios for their proposed indications are acceptable; |
| • |
the results of clinical trials may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval; |
| • |
we may be unable to demonstrate that the clinical and other benefits of our product candidates outweigh their safety risks; |
| • |
the FDA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials; |
| • |
the data collected from clinical trials of our product candidates may not be sufficient to the satisfaction of the FDA or comparable foreign regulatory authorities to obtain regulatory approval in the United States or elsewhere; |
| • |
the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes, our own manufacturing facilities, or our third-party manufacturers’ facilities with which we contract for clinical and commercial supplies; and |
| • |
the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
Further, failure to obtain approval for any of the above reasons may be made more likely by the fact that the FDA and other regulatory authorities have very limited experience with commercial development of our cell therapy for the treatment of Type 1 Diabetes.
Our product candidates may cause undesirable side effects or have other properties that could halt their clinical development, prevent their regulatory approval, limit their commercial potential, or result in significant negative consequences.
As with most biological drug products, use of our product candidates could be associated with side effects or adverse events which can vary in severity from minor reactions to death and in frequency from infrequent to prevalent. Any of these occurrences may materially and adversely harm our business, financial condition and prospects.
27
Research and development of biopharmaceutical products is inherently risky.
We may not be successful in our efforts to use and enhance our technology platform to create a pipeline of product candidates and develop commercially successful products, or we may expend our limited resources on programs that do not yield a successful product candidate and fail to capitalize on product candidates or diseases that may be more profitable or for which there is a greater likelihood of success. If we fail to develop additional product candidates, our commercial opportunity will be limited. Even if we are successful in continuing to build our pipeline, obtaining regulatory approvals and commercializing additional product candidates will require substantial additional funding beyond the net proceeds of this offering and are prone to the risks of failure inherent in medical product development. Investment in biopharmaceutical product development involves significant risk that any potential product candidate will fail to demonstrate adequate efficacy or an acceptable safety profile, gain regulatory approval, and become commercially viable. We cannot provide you any assurance that we will be able to successfully advance any of these additional product candidates through the development process. Our research programs may initially show promise in identifying potential product candidates, yet fail to yield product candidates for clinical development or commercialization for many reasons, including the following:
| • |
our platform may not be successful in identifying additional product candidates; |
| • |
we may not be able or willing to assemble sufficient resources to acquire or discover additional product candidates; |
| • |
our product candidates may not succeed in preclinical or clinical testing; |
| • |
a product candidate may on further study be shown to have harmful side effects or other characteristics that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory criteria; |
| • |
competitors may develop alternatives that render our product candidates obsolete or less attractive; |
| • |
product candidates we develop may nevertheless be covered by third parties’ patents or other exclusive rights; |
| • |
the market for a product candidate may change during our program so that the continued development of that product candidate is no longer reasonable; |
| • |
a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and |
| • |
a product candidate may not be accepted as safe and effective by patients, the medical community or third- party payers, if applicable. |
If any of these events occur, we may be forced to abandon our development efforts for a program or programs, or we may not be able to identify, discover, develop, or commercialize additional product candidates, which would have a material adverse effect on our business and could potentially cause us to cease operations.
Our product candidates are biologics and the manufacture of our product candidates is complex and we may encounter difficulties in production, particularly with respect to process development or scaling-out of our manufacturing capabilities.
If we encounter such difficulties, our ability to provide supply of our product candidates for clinical trials or our products for patients, if approved, could be delayed or stopped, or we may be unable to maintain a commercially viable cost structure. Our product candidates are biologics and the process of manufacturing our products is complex, highly regulated and subject to multiple risks. The manufacture of our product candidates involves complex processes, including the biopsy of tissue from a patient’s liver, propagation of the patient’s liver cells from that liver tissue to obtain the desired dose, trans-differentiating those cells into insulin-producing cells ex vivo and ultimately infusing the cells back into a patient’s body. As a result of the complexities, the cost to manufacture biologics is generally higher than traditional small molecule chemical compounds, and the manufacturing process is less reliable and is more difficult to reproduce. Our manufacturing process will be susceptible to product loss or failure due to logistical issues associated with the collection of liver cells, or starting material, from the patient, shipping such material to the manufacturing site, shipping the final product back to the patient, and infusing the patient with the product, manufacturing issues associated with the differences in patient starting materials, interruptions in the manufacturing process, contamination, equipment or reagent failure, improper installation or operation of equipment, vendor or operator error, inconsistency in cell growth, and variability in product characteristics. Even minor deviations from normal manufacturing processes could result in reduced production yields, product defects, and other supply disruptions. If for any reason we lose a patient’s starting material or later-developed product at any point in the process, the manufacturing process for that patient will need to be restarted and the resulting delay may adversely affect that patient’s outcome. If microbial, viral, or other contaminations are discovered in our product candidates or in the manufacturing facilities in which our product candidates are made, such manufacturing facilities may need to be closed for an extended period of time to investigate and remedy the contamination. Because our product candidates are manufactured for each particular patient, we will be required to maintain a chain of identity with respect to materials as they move from the patient to the manufacturing facility, through the manufacturing process, and back to the patient. Maintaining such a chain of identity is difficult and complex, and failure to do so could result in adverse patient outcomes, loss of product, or regulatory action including withdrawal of our products from the market. Further, as product candidates are developed through preclinical to late stage clinical trials towards approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods, are altered along the way in an effort to optimize processes and results. Such changes carry the risk that they will not achieve these intended objectives, and any of these changes could cause our product candidates to perform differently and affect the results of planned clinical trials or other future clinical trials.
28
Although we are working to develop commercially viable processes, doing so is a difficult and uncertain task, and there are risks associated with scaling to the level required for advanced clinical trials or commercialization, including, among others, cost overruns, potential problems with process scale-out, process reproducibility, stability issues, lot consistency, and timely availability of reagents or raw materials. We may ultimately be unable to reduce the cost of goods for our product candidates to levels that will allow for an attractive return on investment if and when those product candidates are commercialized.
We expect that continued development of our manufacturing facility via MaSTherCell will provide us with enhanced control of material supply for both clinical trials and the commercial market, enable the more rapid implementation of process changes, and allow for better long-term margins. We may establish multiple manufacturing facilities as we expand our commercial footprint to multiple geographies, which may lead to regulatory delays or prove costly. Even if we are successful, our manufacturing capabilities could be affected by cost-overruns, unexpected delays, equipment failures, labor shortages, natural disasters, power failures and numerous other factors that could prevent us from realizing the intended benefits of our manufacturing strategy and have a material adverse effect on our business.
In addition, the manufacturing process for any products that we may develop is subject to FDA and foreign regulatory authority approval process, and we will need to contract with manufacturers who can meet all applicable FDA and foreign regulatory authority requirements on an ongoing basis. If we or our CMOs are unable to reliably produce products to specifications acceptable to the FDA or other regulatory authorities, we may not obtain or maintain the approvals we need to commercialize such products. Even if we obtain regulatory approval for any of our product candidates, there is no assurance that either we or our CMOs will be able to manufacture the approved product to specifications acceptable to the FDA or other regulatory authorities, to produce it in sufficient quantities to meet the requirements for the potential launch of the product, or to meet potential future demand. Any of these challenges could delay completion of clinical trials, require bridging clinical trials or the repetition of one or more clinical trials, increase clinical trial costs, delay approval of our product candidate, impair commercialization efforts, increase our cost of goods, and have an adverse effect on our business, financial condition, results of operations and growth prospects.
On July 3, 2014, our Belgian Subsidiary entered into a service agreement with MaSTherCell, pursuant to which MaSTherCell will function as our CMO and conduct certain clinical tests related to diabetes treatment research. The term of the service agreement will run until all work is completed (or by either party providing 30 days’ written notice of termination) in order to develop a manufacturing process and to manufacture our product. While we anticipate that MaSTherCell will be able to sufficiently support our needs as a CMO, we may need to find other CMOs to meet our clinical and manufacturing needs, of which there are a limited number of third-party manufacturers. This exposes us to the following risks:
29
| • |
We may be unable to identify manufacturers on acceptable terms or at all because the number of potential manufacturers is limited and the FDA must approve any manufacturers. This approval would require new testing and good manufacturing practices compliance inspections by FDA. In addition, a new manufacturer would have to be educated in, or develop substantially equivalent processes for, production of our products; |
| • |
Other manufacturers may have little or no experience with autologous cell products, which are products made from a patient’s own cells, and therefore may require a significant amount of support from us in order to implement and maintain the infrastructure and processes required to manufacture our product candidates; |
| • |
Our third-party manufacturers might be unable to timely manufacture our product or produce the quantity and quality required to meet our clinical and commercial needs, if any; |
| • |
Contract manufacturers may not be able to execute our manufacturing procedures and other logistical support requirements appropriately; |
| • |
Our future contract manufacturers may not perform as agreed, may not devote sufficient resources to our products, or may not remain in the contract manufacturing business for the time required to supply our clinical trials or to successfully produce, store, and distribute our products; |
| • |
Manufacturers are subject to ongoing periodic unannounced inspection by the FDA and corresponding state agencies to ensure strict compliance with current good manufacturing practices, or cGMP, and other government regulations and corresponding foreign standards. We do not have control over third-party manufacturers’ compliance with these regulations and standards; |
| • |
We may not own, or may have to share, the intellectual property rights to any improvements made by our third-party manufacturers in the manufacturing process for our products; |
| • |
Our third-party manufacturers could breach or terminate their agreement with us; |
| • |
Raw materials and components used in the manufacturing process, particularly those for which we have no other source or supplier, may not be available or may not be suitable or acceptable for use due to material or component defects; |
| • |
Our contract manufacturers and critical reagent suppliers may be subject to inclement weather, as well as natural or man-made disasters; or |
| • |
Our contract manufacturers may have unacceptable or inconsistent product quality success rates and yields. |
Each of these risks could delay or prevent the completion of our clinical trials or the approval of any of our product candidates by the FDA, result in higher costs or adversely impact commercialization of our product candidates. In addition, we will rely on third parties to perform certain specification tests on our product candidates prior to delivery to patients. If these tests are not appropriately done and test data are not reliable, patients could be put at risk of serious harm and the FDA could place significant restrictions on our company until deficiencies are remedied.
The manufacture of biological drug products is complex and requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of biologic products often encounter difficulties in production, particularly in scaling up or out, validating the production process, and assuring high reliability of the manufacturing process (including the absence of contamination). These problems include logistics and shipping, difficulties with production costs and yields, quality control, including stability of the product, product testing, operator error, availability of qualified personnel, as well as compliance with strictly enforced federal, state and foreign regulations. Furthermore, if contaminants are discovered in our supply of our product candidates or in the manufacturing facilities, such manufacturing facilities may need to be closed for an extended period of time to investigate and remedy the contamination. We cannot assure you that any stability failures or other issues relating to the manufacture of our product candidates will not occur in the future. Additionally, our manufacturers may experience manufacturing difficulties due to resource constraints or as a result of labor disputes or unstable political environments. If our manufacturers were to encounter any of these difficulties, or otherwise fail to comply with their contractual obligations, our ability to provide our product candidate to patients in clinical trials would be jeopardized. Any delay or interruption in the supply of clinical trial supplies could delay the completion of clinical trials, increase the costs associated with maintaining clinical trial programs and, depending upon the period of delay, require us to begin new clinical trials at additional expense or terminate clinical trials completely.
Cell-based therapies rely on the availability of reagents, specialized equipment, and other specialty materials, which may not be available to us on acceptable terms or at all. For some of these reagents, equipment, and materials, we rely or may rely on sole source vendors or a limited number of vendors, which could impair our ability to manufacture and supply our products.
30
Manufacturing our product candidates will require many reagents, which are substances used in our manufacturing processes to bring about chemical or biological reactions, and other specialty materials and equipment, some of which are manufactured or supplied by small companies with limited resources and experience to support commercial biologics production. We currently depend on a limited number of vendors for certain materials and equipment used in the manufacture of our product candidates. Some of these suppliers may not have the capacity to support commercial products manufactured under cGMP by biopharmaceutical firms or may otherwise be ill-equipped to support our needs. We also do not have supply contracts with many of these suppliers and may not be able to obtain supply contracts with them on acceptable terms or at all. Accordingly, we may experience delays in receiving key materials and equipment to support clinical or commercial manufacturing.
For some of these reagents, equipment, and materials, we rely and may in the future rely on sole source vendors or a limited number of vendors. An inability to continue to source product from any of these suppliers, which could be due to regulatory actions or requirements affecting the supplier, adverse financial or other strategic developments experienced by a supplier, labor disputes or shortages, unexpected demands, or quality issues, could adversely affect our ability to satisfy demand for our product candidates, which could adversely and materially affect our product sales and operating results or our ability to conduct clinical trials, either of which could significantly harm our business.
As we continue to develop and scale our manufacturing process, we expect that we will need to obtain rights to and supplies of certain materials and equipment to be used as part of that process. We may not be able to obtain rights to such materials on commercially reasonable terms, or at all, and if we are unable to alter our process in a commercially viable manner to avoid the use of such materials or find a suitable substitute, it would have a material adverse effect on our business.
We currently have no marketing and sales organization and have no experience in marketing products. If we are unable to establish marketing and sales capabilities or enter into agreements with third parties to market and sell our product candidates, we may not be able to generate product revenue.
We currently have no sales, marketing, or commercial product distribution capabilities and have no experience in marketing products. We intend to develop an in-house marketing organization and sales force, which will require significant capital expenditures, management resources, and time. We will have to compete with other pharmaceutical and biotechnology companies to recruit, hire, train, and retain marketing and sales personnel. If we are unable or decide not to establish internal sales, marketing and commercial distribution capabilities for any or all products we develop, we will likely pursue collaborative arrangements regarding the sales and marketing of our products. However, there can be no assurance that we will be able to establish or maintain such collaborative arrangements, or if we are able to do so, that they will have effective sales forces. Any revenue we receive will depend upon the efforts of such third parties, which may not be successful. We may have little or no control over the marketing and sales efforts of such third parties, and our revenue from product sales may be lower than if we had commercialized our product candidates ourselves. We also face competition in our search for third parties to assist us with the sales and marketing efforts of our product candidates.
There can be no assurance that we will be able to develop in-house sales and commercial distribution capabilities or establish or maintain relationships with third-party collaborators to successfully commercialize any product in the United States or overseas, and as a result, we may not be able to generate product revenue.
A variety of risks associated with operating our business internationally could materially adversely affect our business. We plan to seek regulatory approval of our product candidates outside of the United States and, accordingly, we expect that we, and any potential collaborators in those jurisdictions, will be subject to additional risks related to operating in foreign countries, including:
| • |
differing regulatory requirements in foreign countries; |
| • |
unexpected changes in tariffs, trade barriers, price and exchange controls, and other regulatory requirements; |
31
| • |
economic weakness, including inflation, or political instability in particular foreign economies and markets; |
| • |
compliance with tax, employment, immigration, and labor laws for employees living or traveling abroad; |
| • |
foreign taxes, including withholding of payroll taxes; |
| • |
foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; |
| • |
difficulties staffing and managing foreign operations; |
| • |
workforce uncertainty in countries where labor unrest is more common than in the United States; |
| • |
potential liability under the Foreign Corrupt Practices Act of 1977 or comparable foreign laws; |
| • |
challenges enforcing our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States; |
| • |
production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
| • |
business interruptions resulting from geo-political actions, including war and terrorism. |
These and other risks associated with our planned international operations may materially adversely affect our ability to attain or maintain profitable operations.
We face significant competition from other biotechnology and pharmaceutical companies, and our operating results will suffer if we fail to compete effectively.
The biopharmaceutical industry, and the rapidly evolving market for developing cell-based therapies in particular, is characterized by intense competition and rapid innovation. Our competitors may be able to develop other compounds or drugs that are able to achieve similar or better results. Our potential competitors include major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical companies, universities, and other research institutions. Many of our competitors have substantially greater financial, technical and other resources, such as larger research and development staff and experienced marketing and manufacturing organizations as well as established sales forces. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated in our competitors. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our competitors, either alone or with collaborative partners, may succeed in developing, acquiring or licensing on an exclusive basis drug or biologic products that are more effective, safer, more easily commercialized, or less costly than our product candidates or may develop proprietary technologies or secure patent protection that we may need for the development of our technologies and products.
Specifically, we face significant competition from companies in the insulin therapy market. Insulin therapy is widely used for Insulin-Dependent Diabetes Mellitus (IDDM) patients who are not controlled with oral medications. The global diabetes market comprising the insulin, insulin analogues and other anti-diabetic drugs has been evolving rapidly. A look at the diabetes market reveals that it is dominated by a handful of participants such as Novo Nordisk A/S, Eli Lilly and Company, Sanofi-Aventis, Takeda Pharmaceutical Company Limited, Pfizer Inc., Merck KgaA, and Bayer AG. Even if we obtain regulatory approval of our product candidates, we may not be the first to market and that may affect the price or demand for our product candidates. Additionally, the availability and price of our competitors’ products could limit the demand and the price we are able to charge for our product candidates. We may not be able to implement our business plan if the acceptance of our product candidates is inhibited by price competition or the reluctance of physicians to switch from existing methods of treatment to our product candidates, or if physicians switch to other new drug or biologic products or choose to reserve our product candidates for use in limited circumstances. Additionally, a competitor could obtain orphan product exclusivity from the FDA with respect to such competitor’s product. If such competitor product is determined to be the same product as one of our product candidates, that may prevent us from obtaining approval from the FDA for such product candidate for the same indication for seven years, except in limited circumstances.
We are highly dependent on our key personnel, and if we are not successful in attracting, motivating and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
32
Our ability to compete in the highly competitive biotechnology and pharmaceutical industries depends upon our ability to attract, motivate and retain highly qualified managerial, scientific and medical personnel. We are highly dependent on our management, particularly our chief science officer, Prof. Sarah Ferber, our chief executive officer, Vered Caplan and the chief executive officer of our U.S. Subsidiary, Scott Carmer. The loss of the services of any of our executive officers, other key employees, and other scientific and medical advisors, and our inability to find suitable replacements, could result in delays in product development and harm our business. Competition for skilled personnel is intense and the turnover rate can be high, which may limit our ability to hire and retain highly qualified personnel on acceptable terms or at all.
To induce valuable employees to remain at our company, in addition to salary and cash incentives, we have provided stock option grants that vest over time. The value to employees of these equity grants that vest over time may be significantly affected by movements in our stock price that are beyond our control, and may at any time be insufficient to counteract more lucrative offers from other companies. Although we have employment agreements with our key employees, these employment agreements provide for at-will employment, which means that any of our employees could leave our employment at any time, with or without notice. We do not maintain “key man” insurance policies on the lives of all of these individuals or the lives of any of our other employees.
Risks Related to our Company and Business Generally
Our success will depend, in large part, on strategic collaborations with third parties to develop and commercialize product candidates, and we may not have control over a number of key elements relating to the development and commercialization of any such product candidate.
A key aspect of our strategy is to seek collaboration with a partner, such as a large pharmaceutical organization, that is willing to further develop and commercialize a selected product candidate. To date, we have not entered into any such collaborative arrangement.
By entering into any such strategic collaboration, we may rely on our partner for financial resources and for development, regulatory and commercialization expertise. Our partner may fail to develop or effectively commercialize our product candidate because they:
| • |
do not have sufficient resources or decide not to devote the necessary resources due to internal constraints such as limited cash or human resources; |
| • |
decide to pursue a competitive potential product developed outside of the collaboration; |
| • |
cannot obtain the necessary regulatory approvals; |
| • |
determine that the market opportunity is not attractive; or |
| • |
cannot manufacture or obtain the necessary materials in sufficient quantities from multiple sources or at a reasonable cost. |
We may not be able to enter into a collaboration on acceptable terms, if at all. We face competition in our search for partners from other organizations worldwide, many of whom are larger and are able to offer more attractive deals in terms of financial commitments, contribution of human resources, or development, manufacturing, regulatory or commercial expertise and support.
If we are not successful in attracting a partner and entering into a collaboration on acceptable terms, we may not be able to complete development of or commercialize any product candidate. In such event, our ability to generate revenues and achieve or sustain profitability would be significantly hindered and we may not be able to continue operations as proposed, requiring us to modify our business plan, curtail various aspects of our operations or cease operations.
Third parties to whom we may license or transfer development and commercialization rights for products covered by intellectual property rights may not be successful in their efforts, and as a result, we may not receive future royalty or other milestone payments relating to those products or rights.
We will need to grow the size and capabilities of our organization, and we may experience difficulties in managing this growth.
33
As of November 30, 2015, we had 41 full-time employees. As our development and commercialization plans and strategies develop, we must add a significant number of additional managerial, operational, sales, marketing, financial, and other personnel. Future growth will impose significant added responsibilities on members of management, including:
| • |
identifying, recruiting, integrating, maintaining, and motivating additional employees; |
| • |
managing our internal development efforts effectively, including the clinical and FDA review process for our product candidates, while complying with our contractual obligations to contractors and other third parties; and |
| • |
improving our operational, financial and management controls, reporting systems, and procedures. |
Our future financial performance and our ability to commercialize our product candidates will depend, in part, on our ability to effectively manage any future growth, and our management may also have to divert a disproportionate amount of its attention away from day-to-day activities in order to devote a substantial amount of time to managing these growth activities. Our efforts to manage our growth are complicated by the fact that all of our executive officers other than our chief executive officer have joined us since August 2014. This lack of long-term experience working together may adversely impact our senior management team’s ability to effectively manage our business and growth.
We currently rely, and for the foreseeable future will continue to rely, in substantial part on certain independent organizations, advisors and consultants to provide certain services. There can be no assurance that the services of these independent organizations, advisors and consultants will continue to be available to us on a timely basis when needed, or that we can find qualified replacements. In addition, if we are unable to effectively manage our outsourced activities or if the quality or accuracy of the services provided by consultants is compromised for any reason, our clinical trials may be extended, delayed, or terminated, and we may not be able to obtain regulatory approval of our product candidates or otherwise advance our business. There can be no assurance that we will be able to manage our existing consultants or find other competent outside contractors and consultants on economically reasonable terms, if at all. If we are not able to effectively expand our organization by hiring new employees and expanding our groups of consultants and contractors, we may not be able to successfully implement the tasks necessary to further develop and commercialize our product candidates and, accordingly, may not achieve our research, development, and commercialization goals.
We have entered into collaborations and may form or seek collaborations or strategic alliances or enter into additional licensing arrangements in the future, and we may not realize the benefits of such alliances or licensing arrangements.
We may form or seek strategic alliances, create joint ventures or collaborations, or enter into additional licensing arrangements with third parties that we believe will complement or augment our development and commercialization efforts with respect to our product candidates and any future product candidates that we may develop. Any of these relationships may require us to incur non-recurring and other charges, increase our near and long-term expenditures, issue securities that dilute our existing stockholders, or disrupt our management and business. In addition, we face significant competition in seeking appropriate strategic partners and the negotiation process is time-consuming and complex. Moreover, we may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for our product candidates because they may be deemed to be at too early of a stage of development for collaborative effort and third parties may not view our product candidates as having the requisite potential to demonstrate safety and efficacy. Further, collaborations involving our product candidates, such as our collaborations with third-party research institutions, are subject to numerous risks, which may include the following:
| • |
collaborators have significant discretion in determining the efforts and resources that they will apply to a collaboration; |
| • |
collaborators may not pursue development and commercialization of our product candidates or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in their strategic focus due to the acquisition of competitive products, availability of funding, or other external factors, such as a business combination that diverts resources or creates competing priorities; |
34
| • |
collaborators may delay clinical trials, provide insufficient funding for a clinical trial, stop a clinical trial, abandon a product candidate, repeat or conduct new clinical trials, or require a new formulation of a product candidate for clinical testing; |
| • |
collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products or product candidates; |
| • |
a collaborator with marketing and distribution rights to one or more products may not commit sufficient resources to their marketing and distribution; |
| • |
collaborators may not properly maintain or defend our intellectual property rights or may use our intellectual property or proprietary information in a way that gives rise to actual or threatened litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential liability; |
| • |
disputes may arise between us and a collaborator that cause the delay or termination of the research, development or commercialization of our product candidates, or that result in costly litigation or arbitration that diverts management attention and resources; |
| • |
collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates; and |
| • |
collaborators may own or co-own intellectual property covering our products that results from our collaborating with them, and in such cases, we would not have the exclusive right to commercialize such intellectual property. |
As a result, if we enter into collaboration agreements and strategic partnerships or license our products or businesses, we may not be able to realize the benefit of such transactions if we are unable to successfully integrate them with our existing operations and company culture, which could delay our timelines or otherwise adversely affect our business. We also cannot be certain that, following a strategic transaction or license, we will achieve the revenue or specific net income that justifies such transaction. Any delays in entering into new collaborations or strategic partnership agreements related to our product candidates could delay the development and commercialization of our product candidates in certain geographies for certain indications, which would harm our business prospects, financial condition, and results of operations.
If we engage in future acquisitions or strategic partnerships, this may increase our capital requirements, dilute our stockholders, cause us to incur debt or assume contingent liabilities, and subject us to other risks.
We may evaluate various acquisitions and strategic partnerships, including licensing or acquiring complementary products, intellectual property rights, technologies, or businesses. Any potential acquisitions or strategic partnerships may entail numerous risks, including:
| • |
increased operating expenses and cash requirements; |
| • |
the assumption of additional indebtedness or contingent liabilities; |
| • |
the issuance of our equity securities; |
| • |
assimilation of operations, intellectual property and products of an acquired company, including difficulties associated with integrating new personnel; |
| • |
the diversion of our management’s attention from our existing product programs and initiatives in pursuing such a strategic merger or acquisition; |
| • |
retention of key employees, the loss of key personnel, and uncertainties in our ability to maintain key business relationships; |
| • |
risks and uncertainties associated with the other party to such a transaction, including the prospects of that party and their existing products or product candidates and regulatory approvals; and |
35
| • |
our inability to generate revenue from acquired technology and/or products sufficient to meet our objectives in undertaking the acquisition or even to offset the associated acquisition and maintenance costs. |
In addition, if we undertake acquisitions, we may issue dilutive securities, assume or incur debt obligations, incur large one-time expenses and acquire intangible assets that could result in significant future amortization expense. Moreover, we may not be able to locate suitable acquisition opportunities and this inability could impair our ability to grow or obtain access to technology or products that may be important to the development of our business.
Our success depends on our ability to protect our intellectual property and our proprietary technologies.
Our commercial success depends in part on our ability to obtain and maintain patent protection and trade secret protection for our product candidates, proprietary technologies, and their uses as well as our ability to operate without infringing upon the proprietary rights of others. We can provide no assurance that our patent applications or those of our licensors will result in additional patents being issued or that issued patents will afford sufficient protection against competitors with similar technologies, nor can there be any assurance that the patents issued will not be infringed, designed around or invalidated by third parties. Even issued patents may later be found unenforceable or may be modified or revoked in proceedings instituted by third parties before various patent offices or in courts. The degree of future protection for our proprietary rights is uncertain. Only limited protection may be available and may not adequately protect our rights or permit us to gain or keep any competitive advantage. Composition-of-matter patents on the biological or chemical active pharmaceutical ingredients are generally considered to offer the strongest protection of intellectual property and provide the broadest scope of patent protection for pharmaceutical products, as such patents provide protection without regard to any method of use or any method of manufacturing. While we have an issued patent in the United States with a claim for a composition directed to a vector comprising a promoter linked to a a pancreatic and duodenal homeobox 1 (PDX-1) polypeptide, and a carrier, we cannot be certain that the claim in our issued patent will not be found invalid or unenforceable if challenged. We cannot be certain that the claims in our issued United States methods of use patents will not be found invalid or unenforceable if challenged. We cannot be certain that the pending applications covering composition-of-matter of our transdifferentiated cell populations will be considered patentable by the United States Patent and Trademark Office (USPTO), and courts in the United States or by the patent offices and courts in foreign countries, nor can we be certain that the claims in our issued patents will not be found invalid or unenforceable if challenged. Even if our patent applications covering populations of transdifferentiated cells issue as patents, the patents protect a specific transdifferentiated cell product and may not be enforced against competitors making and marketing a product that has the same activity. Method-of-use patents protect the use of a product for the specified method or for treatment of a particular indication. This type of patents may not be enforced against competitors making and marketing a product that has cells that may provide the same activity but is used for a method not included in the patent. Moreover, even if competitors do not actively promote their product for our targeted indications, physicians may prescribe these products “off-label.” Although off-label prescriptions may infringe or contribute to the infringement of method-of-use patents, the practice is common and such infringement is difficult to prevent or prosecute.
Our issued patent having a claim for a composition comprising a vector comprising a promoter linked to PDX-1 is expected to expire in the United States in 2021. Our additional patents to methods of use of the vector, and pending patent applications to transdifferentiated cell populations made by a process of contacting non-pancreatic β-cells with PDX-1, NeuroD1 or Pax-4, and MafA and their use to treat various indications are expected to expire at various times that range from 2023 (for issued United States patents) to potentially 2035 (for pending patent applications if patents were to issue on the pending applications filed thereon).
The patent application process is subject to numerous risks and uncertainties, and there can be no assurance that we or any of our future development partners will be successful in protecting our product candidates by obtaining and defending patents. These risks and uncertainties include the following:
| • | the USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case; |
36
| • |
patent applications may not result in any patents being issued; |
| • |
patents that may be issued or in-licensed may be challenged, invalidated, modified, revoked, circumvented, found to be unenforceable or otherwise may not provide any competitive advantage; |
| • |
our competitors, many of whom have substantially greater resources and many of whom have made significant investments in competing technologies, may seek or may have already obtained patents that will limit, interfere with or eliminate our ability to make, use, and sell our potential product candidates; |
| • |
there may be significant pressure on the U.S. government and international governmental bodies to limit the scope of patent protection both inside and outside the United States for disease treatments that prove successful, as a matter of public policy regarding worldwide health concerns; and |
| • |
countries other than the United States may have patent laws less favorable to patentees than those upheld by U.S. courts, allowing foreign competitors a better opportunity to create, develop and market competing product candidates. |
In addition, we rely on the protection of our trade secrets and proprietary know-how. Although we have taken steps to protect our trade secrets and unpatented know-how, including entering into confidentiality agreements with third parties, and confidential information and inventions agreements with employees, consultants and advisors, we cannot provide any assurances that all such agreements have been duly executed, and third parties may still obtain this information or may come upon this or similar information independently. Additionally, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating its trade secrets. If any of these events occurs or if we otherwise lose protection for our trade secrets or proprietary know-how, our business may be harmed.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize any products. For example, we may be sued if our product candidates cause or are perceived to cause injury or are found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| • | decreased demand for our products; |
| • | injury to our reputation; |
| • | withdrawal of clinical trial participants and inability to continue clinical trials; |
| • | initiation of investigations by regulators; |
| • | costs to defend the +related litigation; |
| • | a diversion of management’s time and our resources; |
| • | substantial monetary awards to trial participants or patients; |
| • | product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| • | loss of revenue; |
| • | exhaustion of any available insurance and our capital resources; |
| • | the inability to commercialize any product candidate; and |
| • | a decline in our share price. |
Because our products have not reached clinical or commercial stage, we do not currently carry clinical trial or product liability insurance. In the future, our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of products we develop, alone or with collaborators. Such insurance policies may also have various exclusions, and we may be subject to a product liability claim for which we have no coverage.
37
Because some of our directors and officers are not residents of the United States, investors may find it difficult to enforce, within the United States, any judgments obtained against some of our directors and officers.
Some of our directors and officers are not residents of the United States, and all or a substantial portion of their assets is located outside the United States. As a result, it may be difficult for investors to enforce within the United States any judgments obtained against some of our directors and officers, including judgments predicated upon the civil liability provisions of the securities laws of the United States or any state thereof.
Risks Relating to Our Common Stock
If we issue additional shares in the future, it will result in the dilution of our existing stockholders.
Our articles of incorporation authorize the issuance of up to 1,750,000,000 shares of our common stock with a par value of $0.0001 per share. Our board of directors may choose to issue some or all of such shares to acquire one or more companies or products and to fund our overhead and general operating requirements. The issuance of any such shares will reduce the book value per share and may contribute to a reduction in the market price of the outstanding shares of our common stock. If we issue any such additional shares, such issuance will reduce the proportionate ownership and voting power of all current stockholders. Further, such issuance may result in a change of control of our company.
Trading of our stock is restricted by the Securities Exchange Commission’s penny stock regulations, which may limit a stockholder’s ability to buy and sell our common stock.
The Securities and Exchange Commission has adopted regulations which generally define “penny stock” to be any equity security that has a market price (as defined) less than $5.00 per share or an exercise price of less than $5.00 per share, subject to certain exceptions. Our securities are covered by the penny stock rules, which impose additional sales practice requirements on broker-dealers who sell to persons other than established customers and “accredited investors”. The term “accredited investor” refers generally to institutions with assets in excess of $5,000,000 or individuals with a net worth in excess of $1,000,000 or annual income exceeding $200,000 or $300,000 jointly with their spouse. The penny stock rules require a broker-dealer, prior to a transaction in a penny stock not otherwise exempt from the rules, to deliver a standardized risk disclosure document in a form prepared by the Securities and Exchange Commission, which provides information about penny stocks and the nature and level of risks in the penny stock market. The broker-dealer also must provide the customer with current bid and offer quotations for the penny stock, the compensation of the broker-dealer and its salesperson in the transaction and monthly account statements showing the market value of each penny stock held in the customer’s account. The bid and offer quotations, and the broker-dealer and salesperson compensation information, must be given to the customer orally or in writing prior to effecting the transaction and must be given to the customer in writing before or with the customer’s confirmation. In addition, the penny stock rules require that prior to a transaction in a penny stock not otherwise exempt from these rules, the broker-dealer must make a special written determination that the penny stock is a suitable investment for the purchaser and receive the purchaser’s written agreement to the transaction. These disclosure requirements may have the effect of reducing the level of trading activity in the secondary market for the stock that is subject to these penny stock rules. Consequently, these penny stock rules may affect the ability of broker-dealers to trade our securities. We believe that the penny stock rules discourage investor interest in and limit the marketability of our common stock.
FINRA sales practice requirements may also limit a stockholder’s ability to buy and sell our stock.
In addition to the “penny stock” rules described above, the Financial Industry Regulatory Authority (“FINRA”) has adopted rules that require that in recommending an investment to a customer, a broker-dealer must have reasonable grounds for believing that the investment is suitable for that customer. Prior to recommending speculative low priced securities to their non-institutional customers, broker-dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status, investment objectives and other information. Under interpretations of these rules, FINRA believes that there is a high probability that speculative low priced securities will not be suitable for at least some customers. FINRA requirements make it more difficult for broker-dealers to recommend that their customers buy our common stock, which may limit your ability to buy and sell our stock and have an adverse effect on the market for our stock.
38
Our common stock is illiquid and the price of our common stock may be negatively impacted by factors that are unrelated to our operations.
Although our common stock is currently listed for quotation on the QB, there is no market for our common stock. Even when a market is established and trading begins, trading through the OTCQB is frequently thin and highly volatile. There is no assurance that a sufficient market will develop in our stock, in which case it could be difficult for stockholders to sell their stock. The market price of our common stock could fluctuate substantially due to a variety of factors, including market perception of our ability to achieve our planned growth, quarterly operating results of our competitors, trading volume in our common stock, changes in general conditions in the economy and the financial markets or other developments affecting our competitors or us. In addition, the stock market is subject to extreme price and volume fluctuations. This volatility has had a significant effect on the market price of securities issued by many companies for reasons unrelated to their operating performance and could have the same effect on our common stock.
We do not intend to pay dividends on any investment in the shares of stock of our company.
We have never paid any cash dividends, and currently do not intend to pay any dividends for the foreseeable future. Because we do not intend to declare dividends, any gain on an investment in our company will need to come through an increase in the stock’s price. This may never happen and investors may lose all of their investment in our company.
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not Applicable.
ITEM 2. PROPERTIES
We do not own any real property. A description of our properties is as follows:
| Entity | Property Description | |
| These are the principal offices | ||
| Orgenesis Inc./Orgenesis Maryland Inc. | • |
Located at 20271 Goldenrod Lane, Germantown, MD 20876. |
| • |
Occupy office space at the Germantown Innovation Center. | |
| • |
Cost is $200 per month on a month-to-month contract. | |
| Lab and office : | ||
| Orgenesis Ltd. | • |
Located at HaMazmera St 9, Ness Ziona, Israel. |
| • |
Monthly costs are 10 thousand ILS | |
| • |
Lease agreement expires 2018 | |
| Orgenesis SPRL (1) | No physical office* | |
| Cell Therapy Holding SA | No physical office* | |
| MaSTherCell SA | All activities located in Gosselies, Belgium, in the I-Tech Incubator. Property consists of: | |
| • |
Operational production area represent +/- 1,200 m² | |
| • |
Monthly costs are approximately €7,500 | |
| • |
Lease agreement expires 2027 |
|
| • |
The new production area will be build during 2016 and will be opertational in January 2017. |
|
| • |
Office area is approximatively 600 m² |
|
| • |
Monthly cost is approximately €6,000 |
|
39
*Any functions reside on the offices MaSTherCell.
We believe that our facilities are generally in good condition and suitable to carry on our business. We also believe that, if required, suitable alternative or additional space will be available to us on commercially reasonable terms.
ITEM 3. LEGAL PROCEEDINGS
We are not involved in any pending legal proceedings that we anticipate would result in a material adverse effect on our business or operations.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
40
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market information
Our common stock is quoted on the OTCQB under the symbol “ORGS”. Set forth below are the range of high and low bid quotations for the period indicated as reported by the OTC Markets Group for the periods provided. The market quotations reflect inter-dealer prices, without retail mark-up, mark-down or commissions and may not necessarily represent actual transactions.
|
Quarter Ended |
High | Low |
|
Year Ended November 30, 2015 |
||
|
Fourth Quarter |
$ 0.30 | $ 0.48 |
|
Third Quarter |
$ 0.33 | $ 0.55 |
|
Second Quarter |
$ 0.50 | $ 0.73 |
|
First Quarter |
$ 0.38 | $ 0.69 |
|
Year Ended November 30, 2014 |
||
|
Fourth Quarter |
$ 0.90 | $ 0.46 |
|
Third Quarter |
$ 1.00 | $ 0.34 |
|
Second Quarter |
$ 0.78 | $ 0.44 |
|
First Quarter |
$ 0.80 | $ 0.41 |
As of February 26, 2016, there were 54 holders of record of our common stock. A significant number of shares of our common stock are held in either nominee name or street name brokerage accounts, and consequently, we are unable to determine the number of beneficial owners of our stock.
Dividend Policy
We have paid no dividends on our common stock and do not expect to pay cash dividends in the foreseeable future. We plan to retain all earnings to provide funds for the operations of our company. In the future, our Board of Directors will decide whether to declare and pay dividends based upon our earnings, financial condition, capital requirements, and other factors that our Board of Directors may consider relevant. We are not under any contractual restriction as to present or future ability to pay dividends.
Unregistered Sales of Equity Securities
There were no unregistered sales of securities during the three months ended November 30, 2015
Issuer Purchases of Equity Securities
We do not have a stock repurchase program for our common stock and have not otherwise purchased any shares of our common stock.
ITEM 6. SELECTED FINANCIAL DATA
Not applicable.
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND
41
RESULTS OF OPERATIONS
Overview
This subsection of MD&A provides an overview of the important factors that management focuses on in evaluating our businesses, financial condition and operating performance, our overall business strategy and our financial results for the periods covered.
We are building a fully-integrated biopharmaceutical company focused not only on developing our trans-differentiation technologies for Diabetes and vertically integrating manufacturing that can optimize our abilities to scale-up our technologies for clinical trials and eventual commercialization, but also do the same for the technologies of other cell therapy markets.
We are focused on developing our cell therapy technologies to market with respect to diabetes as well as other indications. Our development plan calls for conducting additional preclinical safety and efficacy studies with respect to diabetes and other potential indications. Our Belgian based subsidiary, MaSTherCell, is a contract development manufacturing organization, or CDMO, specialized in cell therapy development for advanced medicinal products. In the last decade, cell therapy medicinal products have gained significant importance, particularly in the fields of ex-vivo gene therapy, immunotherapy and regenerative medicine. While academic and industrial research has led scientific development in the sector, industrialization and manufacturing expertise remains insufficient. MaSTherCell plans to fill this need by providing two types of services to its customers: (i) process and assay development services and (ii) Good Manufacturing Practices (GMP) contract manufacturing services.
As noted below, fiscal 2015 is the first year in which we recorded revenues of approximately $3 million attributable to our subsidiary, MaSTherCell, which we acquired in March 2015. As of November 30, 2015, MaSTherCell had a backlog of approximately €7.5 million, or approximately $7.9 million, consisting of purchase orders/service contracts for production activities that MaSTherCell expects to fulfill into fiscal year 2016.
Results of Operations
Comparison of the Year Ended November 30, 2015 and the Year Ended November 30, 2014
Revenue
Following the MaSTherCell acquisition which closed in March 2015, this is the first year that the Company recognizes revenues from MaSTherCell’s operations. MaSTherCell bills for services linked to cell process development and cell manufacturing services based on individual contracts in accordance with ASC 605, Revenue Recognition. Cell manufacturing services are generally distinct arrangements whereby MaSTherCell is paid for time and materials or for fixed monthly amounts. The Company also recognizes revenue via the sale of consumables to customers that are incidental to the services provided as foreseen in the clinical services contracts. On a monthly basis, the Company bills customers for reimbursable expenses and immediately recognizes these billings in revenue, as the revenue is deemed earned.
For the year ended November 30, 2015, our total revenues were approximately $3 million, as opposed to none for the corresponding period in 2014, respectively. The increase in revenue is attributable to our acquisition of MaSTherCell and the revenues they recognize from services and sales of consumables.
Expenses
The Company’s expenses for the year ended November 30, 2015 are summarized as follows in comparison to its expenses for the year ended November 30, 2014:
|
|
Year Ended November 30, | |||||
|
|
2015 | 2014 | ||||
|
|
(in thousands) | |||||
|
Revenues |
$ | 2,974 | $ | - | ||
|
Cost of sales |
3,880 | - | ||||
|
Research and development expenses, net |
1,067 | 1,549 | ||||
|
Amortization of intangible assets |
1,203 | - | ||||
|
General and administration expenses |
4,035 | 3,028 | ||||
|
Financial expenses (income), net |
(1,850 | ) | 927 | |||
|
Loss before income taxes |
$ | 5,361 | $ | 5,504 | ||
42
Research and Development Expenses
|
|
Year Ended November 30, | |||||
|
|
(in thousands) | |||||
|
|
2015 | 2014 | ||||
|
Salaries and related expenses |
$ | 550 | 778 | |||
|
Stock-based compensation |
129 | 766 | ||||
|
Professional fees and consulting services |
476 | 126 | ||||
|
Lab expenses |
468 | 609 | ||||
|
Other research and development expenses |
237 | 199 | ||||
|
Less – grant |
(793 | ) | (929 | ) | ||
|
Total |
$ | 1,067 | $ | 1,549 | ||
The decrease in salaries and related expenses and in stock-based compensation in the year ended November 30, 2015, compared to 2014, is primarily due to lower compensation expense for certain executives that are no longer employed by the Company. This was offset by expenses due to the employment of a new development director for the cell therapy business. The decrease in grant income is due to a decrease of $266 thousand on the DGO6 project, mainly due to less work performed under grants approved from DGO6 in our Belgian Subsidiary and due to changes in the exchange rate currency. This was offset by grant income of $153 thousand due to work performed under the grant approved from BIRD.
Selling, General and Administrative Expenses
|
|
Year Ended November 30, | |||||
|
|
(in thousands) | |||||
|
|
2015 | 2014 | ||||
|
Salaries and related expenses |
$ | 874 | $ | 330 | ||
|
Stock-based compensation |
674 | 1,721 | ||||
|
Accounting and legal fees |
633 | 476 | ||||
|
Professional fees |
1,045 | 213 | ||||
|
Rent and related expenses |
291 | - | ||||
|
Business development |
326 | 144 | ||||
|
Other general and administrative expenses |
192 | 144 | ||||
|
Total |
$ | 4,035 | $ | 3,028 | ||
The increase in salaries and related expenses for the year ended November 30, 2015, compared to 2014 is due to the increase in the number of employees following the acquisition of MaSTherCell. In addition, there was an increase in legal and accounting fees due to the various material transactions that occurred for certain collaboration agreements and as a result of the acquisition of MaSTherCell. The rent and related expenses for the year ended November 30, 2015 arise from the offices of MaSTherCell. The increase in professional fees for the year ended November 30, 2015 is due to a reduction in the reliance on outside professionals as compared to the same period last year.
Financial Expenses (Income), net
|
|
Year Ended November 30, | |||||
|
|
(in thousands) | |||||
|
|
2015 | 2014 | ||||
|
Decrease in fair value of warrants and financial liabilities measured at fair value |
$ | (2,596 | ) | $ | (180 | ) |
|
Interest expense on convertible loans |
726 | 691 | ||||
|
Funding fees |
135 | |||||
|
Foreign exchange loss, net |
50 | 10 | ||||
|
Issuance of warrants as induced conversion |
260 | |||||
|
Other expenses |
(30 | ) | 11 | |||
|
Total |
$ | (1,850 | ) | $ | 927 | |
43
The increase in financial income for the year ended November 30, 2015 compared to 2014 is mainly attributable to a decrease in the fair value of warrants, embedded derivative and convertible bonds, which is a non-cash metric that is based on the Company’s share price as of the measurement date and reflects the issuance of beneficial warrants that were granted during 2015. Due to a decrease in the Company’s shares price during the period, there was a resultant income impact.
Liquidity and Financial Condition
Working Capital Deficiency
|
|
As of November 30, | |||||
|
|
(in thousands) | |||||
|
|
2015 | 2014 | ||||
|
Current assets |
$ | 8,206 | $ | 2,230 | ||
|
Current liabilities |
16,476 | 4,663 | ||||
|
Working capital deficiency |
$ | (8,270 | ) | $ | (2,433 | ) |
The increase in current assets is mainly due to an increase of $3 million in cash and cash equivalents following the capital raise and an increase of $3.4 million in accounts receivable, inventory, prepaid expenses and other receivables, as well as grants receivable due to the acquisition of MaSTherCell.
Cash Flows
|
|
For the Year Ended November 30, | |||||
|
|
(in thousands) | |||||
|
|
2015 | 2014 | ||||
|
Net loss |
$ | (5,261 | ) | $ | (5,504 | ) |
|
|
||||||
|
Net cash used in operating activities |
(2,706 | ) | (1,429 | ) | ||
|
Net cash used in investing activities |
(932 | ) | 2 | |||
|
Net cash provided by financing activities |
6,666 | 2,737 | ||||
|
Increase (decrease) in cash and cash equivalents |
$ | 3,028 | $ | 1,310 | ||
Our cash and cash equivalents balance increased to $4.2 million at November 30, 2015 from $1.3 million at November 30, 2014. The increase in cash and cash equivalents during the period was primarily due to increase in financing activities of $4 million that was offset by increase in cash flows used in operating activities in an amount of $1.3 million and by increase in cash used in investing activities of $0.9 million.
Net cash used in operations of approximately $2.7 million for the year ended November 30, 2015 was mainly due to increases in accounts receivable of $0.7 million and a change in the fair value of convertible bonds and warrants of $2.6 million, but was offset by decrease of $1.1 million in prepaid expenses and other accounts receivable.
Net cash provided by financing activities for the year ended November 30, 2015 was $6.7 million, compared to $2.7 million for the same period last year. The increase of $4 million was mainly due to an increase in the amount of $3.3 million from the issuance of shares and warrants and proceeds from issuance of loans to MaSTherCell totaling $3.9 million, which was offset by repayment of short and long term loans in amount of $2.4 million, and by a decrease in the issuance convertible loans in amount of $0.7 million.
We need to raise additional operating capital on an immediate basis. Management believes that current cash resources, together with the credit line facilities referred to below under “Private Placement in Fiscal year 2016”, will not allow us to meet current working capital requirements through fiscal 2016. Without additional sources of cash and/or the deferral, reduction, or elimination of significant planned expenditures, the Company will not have the cash resources to remain as a going concern thereafter.
The factors that can impact our ability to continue to fund our operating needs through fiscal 2016 include, but are not limited to:
- Our ability to expand revenue volume at MaSTherCell, which is highly dependent on finite manufacturing facilities;
- Our ability to maintain manufacturing costs at MaSTherCell as expected; and
- Our continued need to reduce our cost structure while simultaneously expanding the breadth of our business, enhancing our technical capabilities, and pursing new business opportunities.
If we cannot effectively manage these factors, including closing new revenue opportunities from existing and new customers for our CDMO business, we will need to raise additional capital to support our business. Except for the credit facility discussed below, we have no commitments for any such funding, and there are no assurances that such additional sources of liquidity can be obtained on terms acceptable to the Company, or at all. If the Company is unable to obtain adequate financing or financing on terms satisfactory to the Company, the Company will not have the cash resources to continue as a going concern.
While we will actively seek to identify sources of liquidity to, there are no assurances that such additional sources of liquidity can be obtained on terms acceptable to us on a commercially reasonable basis, or at all. These factors raise substantial doubt about our ability to continue as a going concern. Furthermore, our “going concern” may make it more difficult for us to raise funds.
44
Corporate Developments in Fiscal Year 2015
Since the commencement of the year through November 30, 2015, we experienced the following corporate developments:
Department De La Gestion Financiere Direction De L’analyse Financiere (“DGO6”)
On November 17, 2014, the Company’s Belgian Subsidiary received the formal approval from the Walloon Region, Belgium (Service Public of Wallonia, DGO6) for a €2.015 million ($2.4 million) support program for the research and development of a potential cure for Type 1 Diabetes. The Financial support is composed of a €1,085 thousand (70% of budgeted costs) grant for the industrial research part of the research program and a further recoverable advance of €930 thousand (60% of budgeted costs) of the experimental development part of the research program. The grants will be paid to the company over a period of approximately 3 years. The grants are subject to certain conditions with respect to the Company’s work in the Walloon Region, and contain a repayment provision upon attaining a favorable outcome. In addition, the DGO6 is also entitled to a royalty upon revenue being generated from any commercial application of the technology. On December 9 and 16, 2014, the Company received €651 thousand and €558 thousand under the grant, respectively.
Israel-U.S. Binational Industrial Research and Development Foundation (“BIRD”)
On September 9, 2015, the Israeli Subsidiary entered into a pharma Cooperation and Project Funding Agreement (CPFA) with BIRD and Pall Corporation, a U.S. company. BIRD will give a conditional grant of $400 thousand each (according to terms defined in the agreement), for a joint research and development project for the use Autologous Insulin Producing (AIP) Cells for the Treatment of Diabetes (the “Project”). The Project started on March 1, 2015. Upon the conclusion of product development, the grant shall be repaid at the rate of 5% of gross sales. The grant will be used solely to finance the costs to conduct the research of the project during a period of 18 months starting on March 1, 2015. On September 21, 2015, the Israeli Subsidiary received $100 thousand under the grant.
Nine Investments Limited Convertible loan agreement
On December 31, 2014, the Company executed an amendment to convertible loan agreement with Nine Investments Limited to extend the due date of the loan of $1,500 thousand from December 31, 2014 to January 31, 2015. As of the date of this report, the Company has not finalized the terms and revised maturity date of this loan, although it believes it will be successful in extending the agreement upon mutually agreeable terms as soon as practicable.
Share Exchange Agreement with MaSTherCell SA
On November 12, 2015, the Company and MaSTherCell and each of the shareholders of MaSTherCell (the “MaSTherCell Shareholders”), entered into an amendment (“Amendment No. 2”) to the Share Exchange Agreement. Under Amendment No. 2, the conditions under which the MaSTherCell Shareholders under the original agreement could unwind the transaction was extended to November 30, 2015. Under Amendment No. 2, the Company agreed to remit to MaSTherCell, by way of an equity investment, the sum of EUR 3.8 million by November 30, 2015 (the “Initial Investment”), to be followed by a subsequent equity investment by December 31, 2015 in MaSTherCell of EUR 1.2 million. These equity investments were to be made out of the proceeds from equity or equity-linked investments and/or credit facilities that the Company was required to have in place on or before November 30, 2015 in the aggregate amount of at least $10 million (the “Post Closing Financing”).
On December 10, 2015, we remitted to MaSTherCell, SA (“MaSTherCell”) by way of an equity investment, the sum of EUR 3.8 million, or $4,103,288, representing the “initial Investment” , in compliance with our obligations as required under Amendment No.2. We also agreed to invest an additional EUR 2.2 million in MaSTherCell equity in addition to the Initial Investment, which additional amount becomes due upon the request of the MaSTherCell board of directors, of whom Company directors/officers currently represent a majority. The additional EUR 2.2 million represents the sum total of our funding obligation to MaSTherCell.
Private Placement
On various dates from October 27, 2015 up through November 30, 2015, the Company entered into definitive agreements with accredited investors relating to a private placement (the “Private Placement”) of (i) 8,083,416 shares of the Company’s common stock and (ii) three year warrants to purchase up to an additional 8,083,416 shares of the Company’s Common Stock at a per share exercise price of $0.52. The purchased securities were issued pursuant to subscription agreements between the Company and the purchasers for aggregate proceeds to the Company of $4,203,000.
45
In connection therewith, on December 10 2015, we also entered into securities purchase agreements with two accredited investors pursuant to which these lenders (“Lenders”) furnished to us access to a $5.0 million credit line (collectively, the “Credit Facility Agreements”). Pursuant to the Credit Facility Agreements, upon request we are entitled to receive $500,000 or such lesser amount as may then be available under the credit facility (the “Advance Amount”), pro-rata from the credit providers under the Credit Facility Agreements, in consideration of which, we will issue to such persons, promissory notes for the amount advanced (each a “Credit Note”). Orgenesis may draw down on the credit facility as needed until the entire $5.0 million is exhausted. Unless extended by mutual arrangement, the credit facility terminates on the earlier to occur of (i) November 30, 2016 and (ii) such time as we shall have raised in excess of $10 million in an equity investment. In consideration of the funding commitment under the Credit Facility Agreements, we issued to these Lenders warrants to purchase up to an aggregate of 2,358,491 shares of the Company’s Common Stock at a per share exercise price of $0.53 per share (the “Commitment Warrants”). The Commitment Warrants become first exercisable upon the scheduled expiration or termination of the credit facility through the third anniversary thereof; provided, that the Commitment Warrants issued to a Lender are subject to cancellation if for whatever reason a funding request by such Lender is not honored. Additionally, upon the issuance of Credit Notes, the Lender is entitled to three year warrants (“Drawdown Warrants”) to purchase additional shares of the Company’s Common Stock in an amount equal to the quotient of: 0.50 X Advance Amount / $0.53. If the entire $5,000,000 were drawn down by the Company, it would issue to the Lenders a total of 4,716,980 Drawdown Warrants.
All Credit Notes that may be issued under the Credit Facility mature on November 30, 2016. Interest on the outstanding principal amount of the Credit Notes accrues at a per annum rate of 12%, payable at maturity or upon an event of default. The Credit Notes contain customary events of default for transactions of this nature. Upon an event of default, the Lender has the right to require the Company to prepay the outstanding principal amount of the Credit Notes plus all accrued and unpaid interest. In addition, the Lender may require prepayment of the Notes at par in connection with certain major transactions and the occurrence of certain other triggering events.
Going Concern
The audited consolidated financial statements contained in this report have been prepared assuming that the Company will continue as a going concern. The Company has net losses for the period from inception (June 5, 2008) through November 30, 2015 of $20.6 million, as well as negative cash flows from operating activities. Company's management estimates that the cash and cash equivalents balance as of November 30, 2015 of $4.2 million that was used for further investment in MaSTherCell, is not sufficient to fund the Company’s other Subsidiary operational and clinical development activities for the twelve months following November 30, 2015. These factors raise substantial doubt about the Company's ability to continue as a going concern. Management is in the process of evaluating various financing alternatives for operations, as the Company will need to finance future research and development activities and general and administrative expenses through fund raising in the public or private equity markets.
Management believes that it will be able to secure the necessary financing as a result of ongoing financing discussions with third party investors and existing shareholders. However, there is no assurance that the Company will be successful with those initiatives.
The consolidated financial statements do not include any adjustments that may be necessary should the Company be unable to continue as a going concern. The Company’s continuation as a going concern is dependent on its ability to obtain additional financing as may be required and ultimately to attain profitability. If the Company raises additional funds through the issuance of equity, the percentage ownership of current shareholders could be reduced, and such securities might have rights, preferences or privileges senior to its common stock. Additional financing may not be available upon acceptable terms, or at all. If adequate funds are not available or are not available on acceptable terms, the Company may not be able to take advantage of prospective business endeavors or opportunities, which could significantly and materially restrict its future plans for developing its business and achieving commercial revenues. If the Company is unable to obtain the necessary capital, the Company may have to cease operations.
We expect that our operating expenses will increase over the next twelve months to continue our development activities. As of the date of this filing, we had cash and cash equivalents of approximately $780,000. We do not expect to raise capital through debt financing from traditional lending sources since we are not currently producing revenue and cannot assure a lender that we will be able to successfully achieve commercial revenues from the development of our technology. Therefore, we only expect to raise money through equity financing via the sale of our common stock. If we cannot raise the money that we need in order to continue to operate our business, we will be forced to delay, scale back or eliminate some or all of our proposed operations. If any of these were to occur, there is a substantial risk that our business would fail. If we are unsuccessful in raising additional financing, we may need to curtail, discontinue or cease operations.
During 2015 and 2014, we have received certain grant funding and have relied and expect to continue to rely on such funding to further our clinical development in the future.
On June 30, 2014, our U.S. Subsidiary entered into a grant agreement with TEDCO. Under the agreement, TEDCO has agreed to give us an amount not to exceed $406,431 to be used solely to finance the costs to conduct the research project entitled “Autologous Insulin Producing (AIP) Cells for Diabetes” during a period of two years. On July 22, 2014, our U.S. Subsidiary received an advance payment of $203,215 on account of the grant. Through November 30, 2014, an amount of $118,305 out of the $203,215 was spent.
On November 17, 2014, our Belgian Subsidiary received the formal approval from the Walloon Region, Belgium (Service Public of Wallonia, DGO6) for a €2.015 million ($2.4 million) support program for the research and development of a potential cure for Type 1 Diabetes. The Financial support is composed of a 1,085 thousand Euros (70% of budgeted costs) grant for the industrial research part of the research program and a further recoverable advance of €930 thousand (60% of budgeted costs) of the experimental development part of the research program. The grants will be paid to us over a period of approximately 3 years. The grants are subject to certain conditions with respect to our work in the Walloon Region. On December 9 and 16, 2014, we received €651 thousand and €558 thousand under the grant, respectively, to have relied on grant funding in 2014 may need to raise additional funds in the future that may not be available on acceptable terms or at all.
46
On September 9, 2015, the Israeli Subsidiary entered into a pharma Cooperation and Project Funding Agreement (CPFA) with BIRD and Pall Corporation, a U.S. company. BIRD will give a conditional grant of $400 thousand each (according to terms defined in the agreement), for a joint research and development project for the use Autologous Insulin Producing (AIP) Cells for the Treatment of Diabetes (the “Project”). The Project started on March 1, 2015. Upon the conclusion of product development, the grant shall be repaid at the rate of 5% of gross sales. The grant will be used solely to finance the costs to conduct the research of the project during a period of 18 months starting on March 1, 2015. On September 21, 2015, the Israeli Subsidiary received $100 thousand under the grant.
Cash Requirements
The Company’s plan of operation over the next 12 months is to:
| • | initiate regulatory activities in Europe and the United States; |
| • | locate suitable facility on the U.S. for tech transfer and manufacturing scale-up; |
| • | purchase equipment needed for its cell production process; |
| • | hire key personnel including in GMP implementation and general and administrative; |
| • | collaborate with clinical centers and regulators to carry out clinical studies and clinical safety testing; |
| • | identify optional technologies for scale up of the cells production process; and |
| • | initialize efforts to validate the manufacturing process. |
The Company estimates its operating capital needs for the next 12 months as of November 30, 2015 to be as follows (in thousands):
|
GMP process development and validation |
$ | 2,200 | |
|
Scale-up of Manufacturing |
3,500 | ||
|
General and administrative |
1,300 | ||
|
Working capital |
3,000 | ||
|
Total |
$ | 10,000 |
The above amounts do not include the additional EUR 2.2 million per Amendment No. 2 under the share exchange agreement with MaSTherCell shareholders that becomes due upon the request of the MaSTherCell board of directors, of whom Company directors/officers currently represent a majority
Future Financing
The Company will require additional funds to implement the Company’s growth strategy for its business. In addition, while the Company has received various grants that have enabled the company to fund its clinical developments, these funds are largely restricted for use for other corporate operational and working capital purposes. Therefore, the Company will need to raise additional capital to both supplement the Company’s clinical developments that are not covered by any grant funding and to cover the Company’s operational expenses. These funds may be raised through equity financing, debt financing, or other sources, which may result in further dilution in the equity ownership of the Company’s shares. There can be no assurance that additional financing will be available to the company when needed or, if available, that it can be obtained on commercially reasonable terms. If the Company is not able to obtain the additional financing on a timely basis should it be required, or generate significant material revenues from operations, the Company will not be able to meet its other obligations as they become due and will be forced to scale down or perhaps even cease the Company’s operations.
47
Off-Balance Sheet Arrangements
The Company has no off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on the Company’s financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that is material to stockholders.
Critical Accounting Policies and Estimates
Our significant accounting policies are more fully described in the notes to our consolidated financial statements included herein for the fiscal year ended November 30, 2015. We believe that the accounting policies below are critical for one to fully understand and evaluate our financial condition and results of operations.
Business Combination
We allocated the purchase price of business we acquired to the tangible and intangible assets acquired and liabilities assumed based upon their estimated fair values on the acquisition date. Any excess of the purchase price over the fair value of the net assets acquired is recorded as goodwill. Acquired in-process backlog, customer relations, brand name and know how are recognized at fair value. The purchase price allocation process requires from us to make significant estimates and assumptions, especially at the acquisition date with respect to intangible assets. Direct transaction costs associated with the business combination are expensed as incurred. The allocation of the consideration transferred in certain cases may be subject to revision based on the final determination of fair values during the measurement period, which may be up to one year from the acquisition date. We included the results of operations of the business that we acquired in the consolidated results prospectively from the date of acquisition.
Research and Development, net
Research and development expenses include costs directly attributable to the conduct of research and development programs, including the cost of salaries, stock-based compensation expenses, payroll taxes and other employees' benefits, lab expenses, consumable equipment and consulting fees. All costs associated with research and developments are expensed as incurred. Participation from government departments and from research foundations for development of approved projects is recognized as a reduction of expense as the related costs are incurred.
Intangible Assets
Intangible assets are recorded at acquisition cost less accumulated amortization and impairment. Definite lived intangible assets are amortized over their estimated useful life using the straight-line method, which is determined by identifying the period over which the cash flows from the asset are expected to be generated.
Goodwill
Goodwill represents the excess of the purchase price of acquired businesse over the estimated fair value of the identifiable net assets acquired. Goodwill is not amortized but is tested for impairment at least annually (at November 30), at the reporting unit level or more frequently if events or changes in circumstances indicate that the asset might be impaired. The goodwill impairment test is applied by performing a qualitative assessment before calculating the fair value of the reporting unit. If, on the basis of qualitative factors, it is considered not more likely than not that the fair value of the reporting unit is less than the carrying amount, further testing of goodwill for impairment would not be required. Otherwise, goodwill impairment is tested using a two-step approach.
Fair Value Measurement
The fair value measurement guidance clarifies that fair value is an exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use in the valuation of an asset or liability. It establishes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements) and the lowest priority to unobservable inputs (Level 3 measurements). The three levels of the fair value hierarchy under the fair value measurement guidance are described below:
48
| • |
Level 1: Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs. |
| • |
Level 2: Observable inputs that are based on inputs not quoted on active markets, but corroborated by market data. |
| • |
Level 3: Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs. |
Embedded Derivatives
We entered into convertible debentures agreements in which a derivative instrument is “embedded”. Embedded derivative is separated from the host contract and carried at fair value when (1) the embedded derivative possesses economic characteristics that are not clearly and closely related to the economic characteristics of the host contract and (2) a separate, stand-alone instrument with the same terms would qualify as a derivative instrument. The derivative is measured both initially and in subsequent periods at fair value, with changes in fair value charged to finance expenses, net. As to embedded derivatives arising from the issuance of convertible debentures, see Note 13.
Volatility in Stock-Based Compensation
The volatility is based on historical volatilities of companies in comparable stages as well as the historical volatility of companies in the industry and, by statistical analysis of the daily share-pricing model. The volatility of stock-based compensation granted after November 30, 2013 is based on historical volatility of the Company for the last two years.
Warrants and Price Protection Mechanism Derivative Classified as a Liability
Warrants that entitle the holder to down-round protection (through ratchet and anti-dilution provisions) and price protection mechanism derivatives in respect of shares entitled to down-round protection are classified as liabilities on the balance sheet. The liability is measured both initially and in subsequent periods at fair value, with changes in fair value charged to finance expenses, net.
The fair value of the warrants and the price protection mechanism derivatives are determining by using a Monte Carlo type model based on a risk neutral approach. The model takes as an input the estimated future dates when new capital will be raised, and builds a multi-step dynamic model. The first step is to model the risk neutral distribution of the share value on the new issue dates, then for each path to use the Black-Scholes model to estimate the value of the warrants and the price protection mechanism derivatives on the last issue date including all the changes in exercise price and quantity along this path. The significant unobservable input used in the fair value measurement is the future expected issue dates. bSignificant delay in this input would result a higher fair value measurement.
Impairment of Long-lived Assets
We are reviewing the property and equipment, intangible assets subject to amortization and other long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset class may not be recoverable. Indicators of potential impairment include: an adverse change in legal factors or in the business climate that could affect the value of the asset; an adverse change in the extent or manner in which the asset is used or is expected to be used, or in its physical condition; and current or forecasted operating or cash flow losses that demonstrate continuing losses associated with the use of the asset. If indicators of impairment are present, the asset is tested for recoverability by comparing the carrying value of the asset to the related estimated undiscounted future cash flows expected to be derived from the asset. If the expected cash flows are less than the carrying value of the asset, then the asset is considered to be impaired and its carrying value is written down to fair value, based on the related estimated discounted cash flows. There were no impairment charges in 2015.
49
Revenue Recognition
We recognize the revenue for services linked to cell process development and cell manufacturing services based on individual contracts in accordance with ASC 605, Revenue Recognition, when the following criteria have been met: persuasive evidence of an arrangement exists; delivery has occurred or services have been provided; the seller’s price to the buyer is fixed or determinable and collectability is reasonably assured. We determine that persuasive evidence of an arrangement exists based on written contracts that define the terms of the arrangements. In addition, we determine that services have been delivered in accordance with the arrangement. We assess whether the fee is fixed or determinable based on the payment terms associated with the transaction and whether the sales price is subject to refund or adjustment. Service revenues are recognized as the services are provided.
We are assessing cash collectability based on a number of factors, including past collection history with the client and the client's creditworthiness. If we determine that collectability is not reasonably assured, we defer revenue recognition until collectability becomes reasonably assured, which is generally upon receipt of the cash. Our arrangements are generally non-cancellable, though clients typically have the right to terminate their agreement for cause if we materially fails to perform. Cell manufacturing services are generally distinct arrangements whereby we are paid for time and materials or for fixed monthly amounts. Revenue is recognized when efforts are expended or contractual terms have been met.
For service agreement contracts we deliver services by executing more than one act, we recognised revenue based on the proportional performance method. Under this method, the costs are recognised in the income statement as incurred and the revenue recognized will be a proportion of the total contract consistent with the costs proportion of total costs. Any amounts invoiced to clients as a result of contractual terms are recognized as deferred income to the extent it exceeds the performance completed.
We also incur revenue corresponding to invoicing to customers of some consumables which are incidental to the services provided as foreseen in the clinical services contracts. We bill customers for reimbursable expenses and immediately recognize these billings in revenue, as the revenue is deemed earned.
Recently Adopted Accounting Pronouncements
See Note 2 of the annual financial statements for our Recently Adopted Accounting Pronouncments and Recently Issuued Accounting Pronouncements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The information called for by Item 8 is included following the "Index to Financial Statements" on page F-1 contained in this annual report on Form 10-K.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
The Company maintains disclosure controls and procedures that are designed to ensure that information required to be disclosed in the Company’s reports filed under the Securities Exchange Act of 1934, as amended, is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to the Company’s management, including the Company’s interim president and chief executive officer (who is the Company’s principal executive officer) and the Company’s chief financial officer, treasurer, and secretary (who is the Company’s principal financial officer and principal accounting officer) to allow for timely decisions regarding required disclosure. In designing and evaluating the Company’s disclosure controls and procedures, the Company’s management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and the Company’s management is required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures. The ineffectiveness of the Company’s disclosure controls and procedures was due to material weaknesses identified in the Company’s internal control over financial reporting, described below.
50
Management’s Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over the Company’s financial reporting. In order to evaluate the effectiveness of internal control over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act of 2002. Our management, with the participation of the Company’s principal executive officer and principal financial officer has conducted an assessment, including testing, using the criteria in Internal Control - Integrated Framework, issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) (2013). Our system of internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. This assessment included review of the documentation of controls, evaluation of the design effectiveness of controls, testing of the operating effectiveness of controls and a conclusion on this evaluation. Based on this evaluation, the Company’s management concluded its internal control over financial reporting was not effective as of November 30, 2015. The ineffectiveness of the Company’s internal controls over financial reporting was due to the following material weaknesses which are indicative of many small companies with limited staff:
| (i) |
inadequate segregation of duties consistent with control objectives; | |
| (ii) |
an ineffective internal audit function or risk assessment function for which such functions are important to the monitoring or risk assessment component of internal control; and | |
| (iii) |
ineffective controls over period end financial disclosure and reporting processes. |
Our management believes the weaknesses identified above have not had any material effect on our financial statements. However, we are currently reviewing our disclosure controls and procedures related to these material weaknesses and expect to implement changes as soon as practicable, including identifying specific areas within our governance, accounting and financial reporting processes to add adequate resources to remediate these material weaknesses.
Our management will continue to monitor and evaluate the effectiveness of our internal controls and procedures and our internal controls over financial reporting on an ongoing basis and is committed to taking further action and implementing additional enhancements or improvements, as necessary and as funds allow.
Because of its inherent limitations, internal controls over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting during the quarter ended November 30, 2015 that have materially affected, or are reasonably likely to materially affect our internal control over financial reporting.
51
Management’s Remediation Plan
Subject to raising additional working capital, we plan to take steps to enhance and improve the design of our internal control over financial reporting. During the period covered by this annual report on Form 10-K, we have not been able to remediate the material weaknesses identified above. To remediate such weaknesses, we plan to implement the following changes in the next fiscal year as our capital resources allow:
| (i) |
appoint additional qualified personnel to address inadequate segregation of duties and ineffective risk management and implement modifications to our financial controls to address such inadequacies; and |
| (ii) |
adopt sufficient written policies and procedures for accounting and financial reporting. |
The remediation efforts set out in (i) is largely dependent upon our company securing additional financing to cover the costs of hiring the requisite personnel and implementing the changes required. If we are unsuccessful in securing such funds, remediation efforts may be delayed. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues, if any, within our company have been detected. These inherent limitations include the realities that judgments in decision-making can be faulty and that breakdowns can occur because of simple error or mistake.
Management believes that despite our material weaknesses set forth above, our financial statements for the year ended November 30, 2015 are fairly stated, in all material respects, in accordance with US GAAP.
ITEM 9B. OTHER INFORMATION
None.
52
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this Item is incorporated by reference to the Company's definitive proxy statement for the 2016 annual meeting of stockholders.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this Item is incorporated by reference to the Company's definitive proxy statement for the 2016 annual meeting of stockholders.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this Item is incorporated by reference to the Company's definitive proxy statement for the 2016 annual meeting of stockholders.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTORS INDEPENDENCE
The information required by this Item is incorporated by reference to the Company's definitive proxy statement for the 2016 annual meeting of stockholders.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by this Item is incorporated by reference to the Company's definitive proxy statement for the 2016 annual meeting of stockholders.
53
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
Exhibits required by Regulation S-K
|
No. |
Description |
|
3.1 |
Articles of Incorporation (incorporated by reference to an exhibit to a registration statement on Form S-1 filed on April 2, 2009) |
|
3.2 |
Certificate of Change (incorporated by reference to an exhibit to a current report on Form 8-K filed on September 2, 2011) |
|
3.3 |
Articles of Merger (incorporated by reference to an exhibit to a current report on Form 8-K filed on September 2, 2011) |
|
3.4 |
Certificate of Amendment to Articles of Incorporation (incorporated by reference to an exhibit to a current report on Form 8-K filed on September 21, 2011) |
|
3.5 |
Certificate of Correction dated February 27, 2012 (incorporated by reference to an exhibit to a current report on Form 8-K/A filed on March 16, 2012) |
| 3.6 |
Amended and Restated Bylaws effective sa of March 2, 2015 (incorporated by reference to an exhibit to a current report on Form 8-K filed on September 21, 2011) |
|
10.3 |
Investment Agreement dated December 13, 2013 with Kodiak Capital Group, LLC (incorporated by reference to our current report on Form 8-K filed on December 16, 2013) |
|
10.4 |
Registration Rights Agreement dated December 13, 2013 with Kodiak Capital Group, LLC (incorporated by reference to our current report on Form 8-K filed on December 16, 2013) |
|
10.5 |
Form of subscription agreement (incorporated by reference to our current report on Form 8-K filed on March 4, 2014) |
|
10.6 |
Form of warrant (incorporated by reference to our current report on Form 8-K filed on March 4, 2014) |
|
10.7 |
Consulting Agreement dated April 3, 2014 with Aspen Agency Limited (incorporated by reference to our current report on Form 8-K filed on April 7, 2014) |
|
10.8 |
Stock Option Agreement dated April 3, 2014 with Aspen Agency Limited (incorporated by reference to our current report on Form 8-K filed on April 7, 2014) |
|
10.9 |
Personal Employment Agreement dated April 16, 2014 by and between Orgenesis Ltd. and Joseph Tenne (incorporated by reference to our current report on Form 8-K filed on April 16, 2014)+ |
|
10.10 |
Form of subscription agreement with form of warrant (incorporated by reference to our current report on Form 8-K filed on April 28, 2014) |
|
10.11 |
Convertible Loan Agreement dated May 29, 2014 with Nine Investments Limited (incorporated by reference to our current report on Form 8-K filed on May 30, 2014) |
|
10.12 |
Services Agreement between Orgenesis SPRL and MaSTherCell SA dated July 3, 2014 incorporated by reference to our current report on Form 8-K filed on July 7, 2014) |
|
10.13 |
Financial Consulting Agreement dated August 1, 2014 with Eventus Consulting, P.C., (incorporated by reference to our current report on Form 8-K filed on August 5, 2014) |
|
10.14 |
Personal Employment Agreement dated August 1, 2014 by and between Orgenesis, Inc. and Neil Reithinger (incorporated by reference to our current report on Form 8-K filed on August 5, 2014)+ |
|
10.15 |
Personal Employment Agreement dated as of July 23, 2014 by and between Orgenesis Maryland Inc. and Scott Carmer (incorporated by reference to our current report on Form 8-K filed on August 6, 2014)+ |
|
10.16 |
Personal Employment Agreement dated August 22, 2014 by and between Orgenesis Ltd. and Vered Caplan (incorporated by reference to our current report on Form 8-K filed on August 25, 2014)+ |
|
10.17 |
Share Exchange Agreement dated November 6, 2014 with MaSTherCell SA and Cell Therapy Holding SA (collectively “MaSTherCell”) and each of the shareholders of MaSTherCell (incorporated by reference to our current report on Form 8-K filed on November 10, 2014) |
54
|
No. |
Description |
|
10.18 |
Addendum 1 to Share Exchange Agreement dated March 2, 2015 with MaSTherCell SA, Cell Therapy Holding SA and their shareholders (incorporated by reference to the Company’s current report on Form 8-K filed on March 5, 2015) |
|
10.19 |
Escrow Agreement dated February 27, 2015 with the shareholders of MaSTherCell SA and Cell Therapy Holding SA and bondholders of MaSTherCell SA and Securities Transfer Corporation (incorporated by reference to the Company’s current report on Form 8-K filed on March 5, 2015) |
Addendum No. 2 to Share Exchange Agreement dated November 12, 2015 with MaSTherCell SA, Cell Therapy Holding SA and their shareholders (incorporated by reference to the Company’s current report on Form 8-K filed on November 13, 2015) |
|
|
10.20 |
Orgenesis Inc. Board of Advisors Consulting Agreement dated March 16, 2015 (incorporated by reference to the Company’s current report on Form 8-K filed on March 17, 2015) |
|
21.1 |
List of Subsidiaries of Orgenesis Inc |
|
99.1 |
Global Share Incentive Plan (2012) (incorporated by reference to our current report on Form 8K filed on May 31, 2012) |
|
99.2 |
Appendix – Israeli Taxpayers Global Share Incentive Plan (incorporated by reference to our current report on Form 8K filed on May 31, 2012) |
|
99.3 |
Audit Committee Charter (incorporated by reference to our current report on Form 8K filed on January 15, 2013) |
|
99.4 |
Compensation Committee Charter (incorporated by reference to our current report on Form 8K filed on January 15, 2013) |
|
101* |
Interactive Data Files pursuant to Rule 405 of Regulation ST. |
*Filed herewith
+ Management Agreement
55
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
ORGENESIS INC.
By: /s/ Vered
Caplan
Vered
Caplan
President, Chief Executive Officer and Chairperson
of the Board
(Principal Executive Officer)
Date: February 29, 2016
By: /s/ Neil
Reithinger
Neil
Reithinger
Chief Financial Officer, Treasurer and Secretary
(Principal
Accounting Officer)
Date: February 29, 2016
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
By: /s/ Guy
Yachin
Guy
Yachin
Director
Date: February 29, 2016
By: /s/ David
Sidransky
David
Sidransky
Director
Date: February 29, 2016
By: /s/ Yaron
Adler
Yaron
Adler
Director
Date: February 29, 2016
By: /s/ Etti
Hanochi
Etti
Hanochi
Director
Date: February 29, 2016
By: /s/ Chris
Buyse
Chris
Buyse
Director
Date: February 29, 2016
By: /s/ Hugues
Bultot
Hugues
Bultot
Director
Date: February 29, 2016
56
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
ORGENESIS INC.
CONSOLIDATED FINANCIAL STATEMENTS
AS OF NOVEMBER 30, 2015
TABLE OF CONTENTS

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders of
ORGENESIS INC.
We have audited the accompanying consolidated balance sheets of Orgenesis Inc. and its subsidiaries (the “Company”) as of November 30, 2015 and 2014, and the related consolidated statements of comprehensive loss, changes in capital deficiency and cash flows for each of the years then ended. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company at November 30, 2015 and 2014, and the results of their comprehensive loss, changes in capital deficiency and cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
As discussed in Note 1b to the financial statements, the Company has recurring losses for the period from inception through November 30, 2015 and presently the Company does not have sufficient cash and other resources to meet its requirements in the following twelve months. These factors raise substantial doubt as to the Company's ability to continue as a going concern. The accompanying financial statements have been prepared assuming that the Company will continue as a going concern and do not include any adjustments that might result from the outcome of this uncertainty.
| Tel-Aviv, Israel | Kesselman & Kesselman |
| February 29, 2016 | Certified Public Accountants (Isr.) |
| A member firm of PricewaterhouseCoopers International Limited |
Kesselman & Kesselman, Trade Tower, 25 Hamered Street,
Tel-Aviv 6812508, Israel,
P.O Box 50005 Tel-Aviv 6150001
Telephone: +972 -3- 7954555, Fax:+972 -3- 7954556, www.pwc.com/il
ORGENESIS INC.
CONSOLIDATED BALANCE SHEETS
(U.S. Dollars in thousands)
|
|
November 30, |
|||||
|
|
2015 |
|
|
2014 |
||
|
Assets |
||||||
|
CURRENT ASSETS: |
||||||
|
Cash and cash equivalents |
$ | 4,168 | $ | 1,314 | ||
|
Accounts receivable |
1,173 | - | ||||
|
Prepaid expenses and other receivables |
1,118 | 105 | ||||
|
Grants receivable |
1,446 | 811 | ||||
|
Inventory |
301 | |||||
|
Total current assets |
8,206 | 2,230 | ||||
|
NON CURRENT ASSETS: |
||||||
|
Property and equipment, net |
4,296 | 13 | ||||
|
Restricted cash |
5 | - | ||||
|
Intangible assets, net |
16,653 | - | ||||
|
Goodwill |
9,535 | - | ||||
|
Other assets |
53 | 6 | ||||
|
Total non current assets |
30,542 | 19 | ||||
|
TOTAL ASSETS |
38,748 | 2,249 | ||||
|
Liabilities net of capital deficiency |
||||||
|
CURRENT LIABILITIES: |
||||||
|
Accounts payable |
3,475 | 1,084 | ||||
|
Accrued expenses |
816 | 375 | ||||
|
Employee and related payables |
1,348 | 626 | ||||
|
Related parties |
42 | 42 | ||||
|
Advance payments on account of grant |
307 | 85 | ||||
|
Short-term loans and current maturities of long term loans |
2,829 | 14 | ||||
|
Deferred income |
1,216 | - | ||||
|
Convertible loans |
3,022 | 2,437 | ||||
|
Convertible bonds |
1,888 | - | ||||
|
Price protection derivative |
1,533 | - | ||||
|
TOTAL CURRENT LIABILITIES |
16,476 | 4,663 | ||||
|
|
||||||
|
LONG-TERM LIABILITIES: |
||||||
|
Loans payable |
2,540 | - | ||||
|
Warrants |
1,382 | 560 | ||||
|
Retirement benefits obligation |
5 | 5 | ||||
|
Deferred taxes |
3,327 | - | ||||
|
TOTAL LONG-TERM LIABILITIES |
7,254 | 565 | ||||
|
TOTAL LIABILITIES |
23,730 | 5,228 | ||||
|
COMMITMENTS |
||||||
|
REDEEMABLE COMMON STOCK |
21,458 | |||||
|
CAPITAL DEFICIENCY: |
||||||
|
Common stock of
$0.0001 par value, 1,750,000,000 shares
authorized, |
6 | 6 | ||||
|
Additional paid-in capital |
14,229 | 13,152 | ||||
|
Receipts on account of shares to be allotted |
1,251 | 60 | ||||
|
Accumulated other comprehensive loss |
(1,286 | ) | (18 | ) | ||
|
Accumulated deficit |
(20,640 | ) | (16,179 | ) | ||
|
TOTAL CAPITAL DEFICIENCY |
(6,440 | ) | (2,979 | ) | ||
|
TOTAL LIABILITIES NET OF CAPITAL DEFICIENCY |
$ | 38,748 | $ | 2,249 | ||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
ORGENESIS INC.
CONSOLIDATED STATEMENTS OF
COMPREHENSIVE LOSS
(U.S. Dollars in thousands, except share and per
share amounts)
|
|
For the Year Ended | |||||
|
|
November 30, | |||||
|
|
2015 | 2014 | ||||
|
REVENUES |
$ | 2,974 | $ | - | ||
|
COST OF REVENUES |
3,880 | |||||
|
GROSS LOSS |
906 | - | ||||
|
|
||||||
|
RESEARCH AND DEVELOPMENT EXPENSES, net |
1,067 | 1,549 | ||||
|
AMORTIZATION OF INTANGIBLE ASSETS |
1,203 | - | ||||
|
SELLING, GENERAL AND ADMINISTRATIVE EXPENSES |
4,035 | 3,028 | ||||
|
OPERATING LOSS |
7,211 | 4,577 | ||||
|
FINANCIAL EXPENSES (INCOME), net |
(1,850 | ) | 927 | |||
|
LOSS BEFORE INCOME TAXES |
5,361 | 5,504 | ||||
|
INCOME TAX BENEFIT |
(900 | ) | ||||
|
NET LOSS |
$ | 4,461 | $ | 5,504 | ||
|
|
||||||
|
LOSS PER SHARE: |
||||||
|
Basic |
$ | 0.08 | $ | 0.10 | ||
|
Diluted |
$ | 0.11 | $ | 0.11 | ||
|
WEIGHTED AVERAGE NUMBER OF SHARES USED IN COMPUTATION OF BASIC AND DILUTED LOSS PER SHARE: |
||||||
|
Basic |
55,798,416 | 54,162,596 | ||||
|
Diluted |
56,920,912 | 54,721,969 | ||||
|
|
||||||
|
OTHER COMPREHENSIVE LOSS - |
||||||
|
Net loss |
$ | 4,461 | $ | 5,504 | ||
|
Translation adjustments |
1,268 | 18 | ||||
|
TOTAL COMPREHENSIVE LOSS |
$ | 5,729 | $ | 5,522 | ||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
ORGENESIS INC.
CONSOLIDATED
STATEMENTS OF CHANGES IN CAPITAL
DEFICIENCY
(U.S. Dollars in
thousands, except share amounts)
|
|
Common Stock | Additional | Receipts on | Accumulated | |||||||||||||||||
|
|
Par | Paid-in | Account of | Other | Accumulated | ||||||||||||||||
|
|
Number | Value | Capital | Share to be Allotted | Comprehensive Loss | Deficit | Total | ||||||||||||||
|
BALANCE AT DECEMBER 1, 2013 |
51,144,621 | $ | 5 | $ | 8,635 | $ | - | $ | - | $ | (10,675 | ) | $ | (2,035 | ) | ||||||
|
Changes during the Year ended November 30, 2014 |
|||||||||||||||||||||
|
Stock-based compensation to employees and directors |
1,200 | 1,200 | |||||||||||||||||||
|
Stock-based compensation to service providers |
913,333 | 1,287 | 1,287 | ||||||||||||||||||
|
Issuances of shares and warrants |
2,479,628 | 1,214 | 60 | 1,274 | |||||||||||||||||
|
Conversions of convertible loans into shares and warrants |
713,023 | 1 | 630 | 631 | |||||||||||||||||
|
Proceeds from exercise of stock options |
623,806 | 1 | 1 | ||||||||||||||||||
|
Exercise of warrants into shares and warrants |
96,154 | 50 | 50 | ||||||||||||||||||
|
Beneficial conversion feature of convertible loans |
135 | 135 | |||||||||||||||||||
|
Comprehensive loss for the year |
(18 | ) | (5,504 | ) | (5,522 | ) | |||||||||||||||
|
BALANCE AT NOVEMBER 30, 2014 |
55,970,565 | $ | 6 | $ | 13,152 | $ | 60 | $ | (18 | ) | $ | (16,179 | ) | $ | (2,979 | ) | |||||
|
Changes during the Year ended November 30, 2015 |
|||||||||||||||||||||
|
Stock-based compensation to employees and directors |
713 | 713 | |||||||||||||||||||
|
Stock-based compensation to service providers |
90 | 90 | |||||||||||||||||||
|
Warrants issued to credit providers |
208 | 208 | |||||||||||||||||||
|
Issuances of shares |
115,385 | 60 | (60 | ) | |||||||||||||||||
|
Shares cancellation |
(250,000 | ) | |||||||||||||||||||
|
Receipts on account of shares to be issued |
6 | 1,251 | 1,257 | ||||||||||||||||||
|
Comprehensive loss for the year |
(1,268 | ) | (4,461 | ) | (5,729 | ) | |||||||||||||||
|
BALANCE AT NOVEMBER 30, 2015 |
55,835,950 | $ | 6 | $ | 14,229 | $ | 1,251 | $ | (1,286 | ) | $ | (20,640 | ) | $ | (6,440 | ) | |||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
ORGENESIS INC.
CONSOLIDATED STATEMENTS OF CASH
FLOWS
(U.S. Dollars in thousands)
|
|
Year Ended November 30, | |||||
|
|
2015 | 2014 | ||||
|
CASH FLOWS FROM OPERATING ACTIVITIES: |
||||||
|
Net loss |
$ | (4,461 | ) | $ | (5,504 | ) |
|
Adjustments required to reconcile net loss to net cash used in operating activities: |
||||||
|
Stock-based compensation |
803 | 2,487 | ||||
|
Depreciation and amortization expenses |
1,991 | 5 | ||||
|
Change in fair value of warrants and embedded derivatives |
(1,375 | ) | (180 | ) | ||
|
Change in fair value of convertible bonds |
(1,221 | ) | ||||
|
Interest expense accrued on loans and convertible loans |
502 | 1,086 | ||||
|
Changes in operating assets and liabilities: |
||||||
|
Increase in accounts receivable |
(731 | ) | ||||
|
Increase in inventory |
(87 | ) | ||||
|
Increase in other assets |
(22 | ) | ||||
|
Increase in prepaid expenses and other accounts receivable |
(1,083 | ) | (74 | ) | ||
|
Increase in accounts payable |
1,497 | 1,016 | ||||
|
Increase (decrease) in accrued expenses |
538 | (11 | ) | |||
|
Increase in employee and related payables |
353 | 472 | ||||
|
Increase in deferred income |
1,039 | |||||
|
Increase (decrease) in advance payments and receivables on account of grant |
451 | (726 | ) | |||
|
Decrease in deferred taxes |
(900 | ) | ||||
|
Net cash used in operating activities |
(2,706 | ) | (1,429 | ) | ||
|
CASH FLOWS FROM INVESTING ACTIVITIES: |
||||||
|
Purchase of property and equipment |
(982 | ) | (5 | ) | ||
|
Restricted cash |
(5 | ) | ||||
|
Acquisition of MaSTherCell, net of cash acquired, see note 3 |
305 | |||||
|
Short term investments and deposits |
(250 | ) | 10 | |||
|
Amounts funded in respect of retirement benefits obligation |
(3 | ) | ||||
|
Net cash provided by (used in) investing activities |
(932 | ) | 2 | |||
|
CASH FLOWS FROM FINANCING ACTIVITIES: |
||||||
|
Short-term line of credit |
(14 | ) | 14 | |||
|
Proceeds from issuance of shares and warrants |
4,203 | 922 | ||||
|
Proceeds from exercise of stock options |
1 | |||||
|
Proceeds from issuance of loans payable |
3,946 | |||||
|
Proceeds from exercise of warrants into shares and warrants |
50 | |||||
|
Repayment of short and long-term debt |
(2,419 | ) | ||||
|
Proceeds from issuance of convertible loans |
950 | 1,750 | ||||
|
Net cash provided by financing activities |
6,666 | 2,737 | ||||
|
NET CHANGE IN CASH AND CASH EQUIVALENTS |
3,028 | 1,310 | ||||
|
EFFECT OF EXCHANGE RATE CHANGES ON CASH AND CASH EQUIVALENTS |
(174 | ) | (47 | ) | ||
|
CASH AND CASH EQUIVALENTS AT BEGINNING OF YEAR |
1,314 | 51 | ||||
|
CASH AND CASH EQUIVALENTS AT END OF YEAR |
$ | 4,168 | $ | 1,314 | ||
|
|
||||||
|
SUPPLEMENTAL NON-CASH FINANCING ACTIVITY |
||||||
|
Conversion of loans (including accrued interest) to common stock and warrants |
$ | 631 | ||||
|
Common stock issued for rended services |
$ | 37 | ||||
|
Warrants to be issued to credit providers |
$ | 208 | ||||
|
SUPPLEMENTAL INFORMATION ON INTEREST PAID IN CASH |
$ | 125 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
NOTE 1 – DESCRIPTION OF BUSINESS
a. General
Orgenesis Inc. (“the Company”) was incorporated in the state of Nevada on June 5, 2008. The Company is developing a technology that demonstrates the capacity to induce a shift in the developmental fate of cells from the liver and differentiating (converting) them into “pancreatic beta cell-like” insulin producing cells for patients with Type 1 Diabetes.
On October 11, 2011, the Company incorporated a wholly owned subsidiary in Israel, Orgenesis Ltd. (the “Israeli Subsidiary”), which is engaged in research and development. On February 2, 2012, the Israeli Subsidiary entered into an agreement with Tel Hashomer Medical Research (“THM”), Infrastructure and Services Ltd (the “Licensor”). The Israeli Subsidiary was granted a worldwide, royalty bearing, exclusive license to transdifferentiation of cells to insulin producing cells, including the population of insulin producing cells, methods of making this population, and methods of using this population of cells for cell therapy or diabetes treatment developed by Dr. Sarah Ferber of THM.
On July 31, 2013, the Company incorporated a wholly owned subsidiary in Maryland, Orgenesis Maryland Inc. (the “U.S. Subsidiary”), which is engaged in research and development.
On October 11, 2013, the Company incorporated a wholly owned subsidiary in Belgium, Orgenesis SPRL (the “Belgian Subsidiary”), which is engaged in development and manufacturing activities together with clinical development studies in Europe, and later on to be the Company’s center for activities in Europe.
As discussed in Note 3, on March 2, 2015, the Company completed the acquisition of MaSTherCell SA and Cell Therapy Holding SA (collectively “MaSTherCell”). MaSTherCell is a Contract Development and Manufacturing Organization (CDMO) specializing in cell therapy development for advanced medicinal products. Cell therapy is the prevention or treatment of human disease by the administration of cells that have been selected, multiplied and pharmacologically treated or altered outside the body (ex vivo). MaSTherCell's CDMO activity is operated as a separate reporting segment (See Note 4).
As used in this report and unless otherwise indicated, the term “Company” refers to Orgenesis Inc. and its wholly-owned subsidiaries (“Subsidiaries”). Unless otherwise specified, all dollar amounts are expressed in United States dollars.
b. Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. The Company has net losses for the period from inception (June 5, 2008) through November 30, 2015 of $20.6 million, as well as negative cash flows from operating activities. Presently, the Company does not have sufficient cash resources to meet its plans in the twelve months following November 30, 2015. These factors raise substantial doubt about the Company's ability to continue as a going concern. Management is in the process of evaluating various financing alternatives for operations, as the Company will need to finance future research and development activities and general and administrative expenses through fund raising in the public or private equity markets.
The consolidated financial statements do not include any adjustments that may be necessary should the Company be unable to continue as a going concern. The Company’s continuation as a going concern is dependent on its ability to obtain additional financing as may be required and ultimately to attain profitability. If the Company raises additional funds through the issuance of equity, the percentage ownership of current shareholders could be reduced, and such securities might have rights, preferences or privileges senior to its common stock. Additional financing may not be available upon acceptable terms, or at all. If adequate funds are not available or are not available on acceptable terms, the Company may not be able to take advantage of prospective business endeavors or opportunities, which could significantly and materially restrict its future plans for developing its business and achieving commercial revenues. If the Company is unable to obtain the necessary capital, the Company may have to cease operations.
F-7
On December 7, 2015, and December 21, 2015, the Company entered into definitive agreements with accredited investors relating to a private placement (See Note 18(a)).
NOTE 2- SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
The accounting policies adopted are generally consistent with those of the previous financial year. Due to the acquisition of MaSTherCell, new accounting policies had to be adopted.
a. Use of Estimates in the Preparation of Financial Statements
The preparation of the consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the financial statement date and the reported expenses during the reporting periods. Actual results could differ from those estimates. As applicable to these consolidated financial statements, the most significant estimates and assumptions relate to the valuation of stock based compensation, valuation of financial instruments measured at fair value and valuation of intangible assets.
b. Business Combination
The Company allocates the purchase price of an acquired business to the tangible and intangible assets acquired and liabilities assumed based upon their estimated fair values on the acquisition date. Any excess of the purchase price over the fair value of the net assets acquired is recorded as goodwill. Acquired in-process backlog, customer relations, brand name and know how are recognized at fair value. The purchase price allocation process requires management to make significant estimates and assumptions, especially at the acquisition date with respect to intangible assets. Direct transaction costs associated with the business combination are expensed as incurred. The allocation of the consideration transferred in certain cases may be subject to revision based on the final determination of fair values during the measurement period, which may be up to one year from the acquisition date. The Company includes the results of operations of the business that it has acquired in its consolidated results prospectively from the date of acquisition.
c. Cash equivalents
The Company considers all short term, highly liquid investments, which include short term bank deposits with original maturities of three months or less from the date of purchase, that are not restricted as to withdrawal or use and are readily convertible to known amounts of cash, to be cash equivalents.
d. Restricted Cash
The company has restricted cash deposited as a guarantee for the use of the Company's credit card. The Company classifies these amounts as a non-current asset since the Company expects to continue the use of the credit card.
e. Research and Development, net
Research and development expenses include costs directly attributable to the conduct of research and development programs, including the cost of salaries, stock-based compensation expenses, payroll taxes and other employees' benefits, lab expenses, consumable equipment and consulting fees. All costs associated with research and developments are expensed as incurred. Participation from government departments and from research foundations for development of approved projects is recognized as a reduction of expense as the related costs are incurred.
f. Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly owned Subsidiaries. All intercompany transactions and balances have been eliminated in consolidation.
g. Functional Currency
F-8
The currency of the primary economic environment in which the operations of the Company and part of its Subsidiaries are conducted is in U.S. dollars (“$” or “dollar”). The functional currency of the Belgian Subsidiaries is the Euro (“€” or “Euro”). Most of the Company’s expenses are incurred in dollars and the source of the Company’s financing has been provided in dollars. Thus, the functional currency of the Company and its Subsidiaries is the dollar. Transactions and balances originally denominated in dollars are presented at their original amounts. Balances in foreign currencies are translated into dollars using historical and current exchange rates for nonmonetary and monetary balances, respectively. For foreign transactions and other items reflected in the statements of operations, the following exchange rates are used: (1) for transactions – exchange rates at transaction dates or average rates and (2) for other items (derived from nonmonetary balance sheet items such as depreciation) – historical exchange rates. The resulting transaction gains or losses are recorded as financial income or expenses. The financial statements of the Belgian Subsidiaries are included in the consolidated financial statements, translated into U.S. dollars. Assets and liabilities are translated at year-end exchange rates, while revenues and expenses are translated at yearly average exchange rates during the year. Differences resulting from translation of assets and liabilities are presented as other comprehensive income.
h. Inventory
Inventory is stated at the lower of cost or net realizable value with cost determined under the first-in-first-out (FIFO) cost method. The entire balance of inventory at November 30, 2015, consists of raw material.
i. Property and Equipment
Property and equipment are recorded at cost and depreciated by the straight-line method over the estimated useful lives of the related assets.
Annual rates of depreciation are presented in the table below:
|
|
Weighted Average |
|
|
Useful Life (Years) |
|
Production facility |
10 |
|
Laboratory equipment |
5 |
|
Office equipment and computers |
3-5 |
j. Intangible Assets
Intangible assets and their useful lives are as follows:
|
|
Weighted Average | Amortization Recorded at |
|
|
Useful Life (Years) | Comprehensive Loss Line Item |
|
Backlog |
1.75 | Cost of revenues |
|
Customer Relationships |
7.75 | Amortization of intangible assets |
|
Brand |
9.75 | Amortization of intangible assets |
|
Know-How |
11.75 | Amortization of intangible assets |
Intangible assets are recorded at acquisition cost less accumulated amortization and impairment. Definite lived intangible assets are amortized over their estimated useful life using the straight-line method, which is determined by identifying the period over which the cash flows from the asset are expected to be generated.
k. Goodwill
Goodwill represents the excess of the purchase price of acquired business over the estimated fair value of the identifiable net assets acquired. Goodwill is not amortized but is tested for impairment at least annually (at November 30), at the reporting unit level or more frequently if events or changes in circumstances indicate that the asset might be impaired. The goodwill impairment test is applied by performing a qualitative assessment before calculating the fair value of the reporting unit. If, on the basis of qualitative factors, it is considered not more likely than not that the fair value of the reporting unit is less than the carrying amount, further testing of goodwill for impairment would not be required. Otherwise, goodwill impairment is tested using a two-step approach.
F-9
The first step involves comparing the fair value of the reporting unit to its carrying amount. If the fair value of the reporting unit is determined to be greater than its carrying amount, there is no impairment. If the reporting unit’s carrying amount is determined to be greater than the fair value, the second step must be completed to measure the amount of impairment, if any. The second step involves calculating the implied fair value of goodwill by deducting the fair value of all tangible and intangible assets, excluding goodwill, of the reporting unit from the fair value of the reporting unit as determined in step one. The implied fair value of the goodwill in this step is compared to the carrying value of goodwill. If the implied fair value of the goodwill is less than the carrying value of the goodwill, an impairment loss equivalent to the difference is recorded.
l. Impairment of Long-lived Assets
The Company reviews its property and equipment, intangible assets subject to amortization and other long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset class may not be recoverable. Indicators of potential impairment include: an adverse change in legal factors or in the business climate that could affect the value of the asset; an adverse change in the extent or manner in which the asset is used or is expected to be used, or in its physical condition; and current or forecasted operating or cash flow losses that demonstrate continuing losses associated with the use of the asset. If indicators of impairment are present, the asset is tested for recoverability by comparing the carrying value of the asset to the related estimated undiscounted future cash flows expected to be derived from the asset. If the expected cash flows are less than the carrying value of the asset, then the asset is considered to be impaired and its carrying value is written down to fair value, based on the related estimated discounted cash flows. There were no impairment charges in 2015.
m. Revenue Recognition
The Company recognizes revenue for services linked to cell process development and cell manufacturing services based on individual contracts in accordance with ASC 605, Revenue Recognition, when the following criteria have been met: persuasive evidence of an arrangement exists; delivery has occurred or services have been provided; the price is fixed or determinable and collectability is reasonably assured. The Company determines that persuasive evidence of an arrangement exists based on written contracts that define the terms of the arrangements. In addition, the Company determines that services have been delivered in accordance with the arrangement. The Company assesses whether the fee is fixed or determinable based on the payment terms associated with the transaction and whether the sales price is subject to refund or adjustment. Service revenues are recognized as the services are provided.
The Company assesses cash collectability based on a number of factors, including past collection history with the client and the client's creditworthiness. If the Company determines that collectability is not reasonably assured, it defers revenue recognition until collectability becomes reasonably assured, which is generally upon receipt of the cash. The Company's arrangements are generally non-cancellable, though clients typically have the right to terminate their agreement for cause if the Company materially fails to perform. Cell manufacturing services are generally distinct arrangements whereby the Company is paid for time and materials or for fixed monthly amounts. Revenue is recognized when efforts are expended or contractual terms have been met.
For service agreement contracts where the Company is delivering services by executing more than one act, revenue is recognised based on the proportional performance method. Under this method, the costs are recognised in the income statement as incurred and the revenue recognized will be a proportion of the total contract consistent with the costs proportion of total costs. Any amounts invoiced to clients as a result of contractual terms are recognized as deferred income to the extent it exceeds the performance completed.
The Company also incurs revenue of some consumables which are incidental to the services provided as foreseen in the clinical services contracts. The Company bills customers for reimbursable expenses and immediately recognizes these billings in revenue, as the revenue is deemed earned.
n. Financial Liabilities Measured at Fair Value
F-10
1) Fair Value Option
Topic 815 provides entities with an option to report certain financial assets and liabilities at fair value with subsequent changes in fair value reported in earnings. The election can be applied on an instrument by instrument basis. The Company elected the fair value option to its convertible bonds. The liability is measured both initially and in subsequent periods at fair value, with changes in fair value charged to finance expenses, net (See also Note 14).
2) Warrants and Price Protection Mechanism Derivative Classified as a Liability
Warrants that entitle the holder to down-round protection (through ratchet and anti-dilution provisions) and price protection mechanism derivatives in respect of shares entitled to down-round protection are classified as liabilities on the balance sheet. The liability is measured both initially and in subsequent periods at fair value, with changes in fair value charged to finance expenses, net (See Note 14).
3) Derivatives
Embedded derivatives are separated from the host contract and carried at fair value when (1) the embedded derivative possesses economic characteristics that are not clearly and closely related to the economic characteristics of the host contract and (2) a separate, standalone instrument with the same terms would qualify as a derivative instrument. The derivative is measured both initially and in subsequent periods at fair value, with changes in fair value charged to finance expenses, net. As to embedded derivatives arising from the issuance of convertible debentures, see Note 14.
o. Income Taxes
1) With respect to deferred taxes, income taxes are computed using the asset and liability method. Under the asset and liability method, deferred income tax assets and liabilities are determined based on the differences between the financial reporting and tax bases of assets and liabilities and are measured using the currently enacted tax rates and laws. A valuation allowance is recognized to the extent that it is more likely than not that the deferred taxes will not be realized in the foreseeable future.
2) The Company follows a two-step approach to recognizing and measuring uncertain tax positions. The first step is to evaluate the tax position for recognition by determining if the available evidence indicates that it is more likely than not that the position will be sustained on examination. If this threshold is met, the second step is to measure the tax position as the largest amount that is greater than 50% likely of being realized upon ultimate settlement.
3) Taxes that would apply in the event of disposal of investment in Subsidiaries have not been taken into account in computing the deferred income taxes, as it is the Company’s intention to hold these investments and not realize them.
p. Stock-based Compensation
The Company accounts for employee stock-based compensation in accordance with the guidance of ASC Topic 718, Compensation - Stock Compensation, which requires all share based payments to employees, including grants of employee stock options, to be recognized in the financial statements based on their grant date fair values. The fair value of the equity instrument is charged to compensation expense and credited to additional paid in capital over the period during which services are rendered. The Company recorded stock based compensation expenses using the straight line method.
The Company follows ASC Topic 505-50, Equity-Based Payments to Non-Employees, for stock options issued to consultants and other non-employees. In accordance with ASC Topic 505-50, these stock options issued as compensation for services provided to the Company are accounted for based upon the fair value of the options. The fair value of the options granted is measured on a final basis at the end of the related service period and is recognized over the related service period using the straight line method.
F-11
q. Redeemable Common Stock
Common stock with embedded redemption features, such as an unwind option, whose settlement is not at the Company’s discretion, are considered redeemable common stock. Redeemable common stock is considered to be temporary equity and are therefore presented as a mezzanine section between liabilities and equity on the Company's consolidated balance sheets. Subsequent adjustment of the amount presented in temporary equity is required only if the Company's management estimates that it is probable that the instrument will become redeemable.
r. Loss per Share of Common Stock
Net loss per share, basic and diluted, is computed on the basis of the net loss for the period divided by the weighted average number of common shares outstanding during the period. Diluted net loss per share is based upon the weighted average number of common shares and of common shares equivalents outstanding when dilutive. Common share equivalents include: (i) outstanding stock options under the Company’s Global Share Incentive Plan (2012) and warrants which are included under the treasury share method when dilutive, and (ii) common shares to be issued under the assumed conversion of the Company’s outstanding convertible debentures, which are included under the if-converted method when dilutive. The computation of diluted net loss per share for the year ended November 30, 2015 includes common share equivalents due to warrants (See Note 11).
s Concentration of Credit Risk
Financial instruments that potentially subject the Company to concentration of credit risk consist of principally cash and cash equivalents, restricted cash and certain receivables. The Company held these instruments with highly rated financial institutions and the Company has not experienced any credit losses in these accounts and does not believe the Company is exposed to any significant credit risk on these instruments. There is no bad debt allowance provided to date.
t. Beneficial Conversion Feature (“BCF”)
When the Company issues convertible debt, if the stock price is greater than the effective conversion price (after allocation of the total proceeds) on the measurement date, the conversion feature is considered "beneficial" to the holder. If there is no contingency, this difference is treated as issued equity and reduces the carrying value of the host debt; the discount is accreted as deemed interest on the debt (See Note 7).
u. Other Comprehensive Loss
Other comprehensive loss represents adjustments of foreign currency translation.
v. Newly Issued Accounting Pronouncements
In August 2014, the FASB issued ASU No. 2014-15, “Presentation of Financial Statements— Going Concern (Subtopic 205-40), Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern”. Continuation of a reporting entity as a going concern is presumed as the basis for preparing financial statements unless and until the entity’s liquidation becomes imminent. Preparation of financial statements under this presumption is commonly referred to as the going concern basis of accounting. Prior to this, there was no guidance under U.S. GAAP about management’s responsibility to evaluate whether there is substantial doubt about an entity’s ability to continue as a going concern or to provide related footnote disclosures. The amendments in this update provide that guidance. In doing so, the amendments reduce diversity in the timing and content of footnote disclosures. The amendments require management to assess an entity’s ability to continue as a going concern by incorporating and expanding upon certain principles that are currently in U.S. auditing standards. Specifically, the amendments (1) provide a definition of the term “substantial doubt”, (2) require an evaluation every reporting period including interim periods, (3) provide principles for considering the mitigating effect of management’s plans, (4) require certain disclosures when substantial doubt is alleviated as a result of consideration of management’s plans, (5) require an express statement and other disclosures when substantial doubt is not alleviated, and (6) require an assessment for a period of one year after the date that the financial statements are issued (or available to be issued). For the period ended November 30, 2015, management evaluated the Company’s ability to continue as a going concern and concluded that substantial doubt has not been alleviated about the Company’s ability to continue as a going concern. While the Company continues to explore further significant sources of financing, management’s assessment was based on the uncertainty related to the availability amount and nature of such financing over the next twelve months.
F-12
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update No. 2014-09 (ASU 2014-09) "Revenue from Contracts with Customers." ASU 2014-09 will supersede most current revenue recognition guidance, including industry-specific guidance. The underlying principle is that an entity will recognize revenue upon the transfer of goods or services to customers in an amount that the entity expects to be entitled to in exchange for those goods or services. The guidance provides a five-step analysis of transactions to determine when and how revenue is recognized. Other major provisions include capitalization of certain contract costs, consideration of the time value of money in the transaction price, and allowing estimates of variable consideration to be recognized before contingencies are resolved in certain circumstances. The guidance also requires enhanced disclosures regarding the nature, amount, timing and uncertainty of revenue and cash flows arising from an entity’s contracts with customers. The guidance is effective for the interim and annual periods beginning on or after December 15, 2016 (early adoption is not permitted). The guidance permits the use of either a retrospective or cumulative effect transition method. On July 9, 2015, the FASB decided to delay the effective date of the new revenue standard by one year. The FASB also agreed to allow entities to choose to adopt the standard as of the original effective date. The Company is currently evaluating the impact of this standard.
In July 2015, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update No. 2015-11 (ASU 2015-11), Simplifying the Measurement of Inventory. According to ASU 2015-11, an entity should measure inventory within the scope of this update at the lower of cost and net realizable value. Net realizable value is the estimated selling prices in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. Subsequent measurement is unchanged for inventory measured using LIFO or the retail inventory method. The amendments in ASU 2015-11 more closely align the measurement of inventory in GAAP with the measurement of inventory in International Financial Reporting Standards (IFRS). The Board has amended some of the other guidance in Topic 330 to more clearly articulate the requirements for the measurement and disclosure of inventory. However, the Board does not intend for those clarifications to result in any changes in practice. Other than the change in the subsequent measurement guidance from the lower of cost or market to the lower of cost and net realizable value for inventory within the scope of ASU 2015-11, there are no other substantive changes to the guidance on measurement of inventory. For public business entities, the amendments in ASU 2015-11 are effective for fiscal years beginning after December 15, 2016, including interim periods within those fiscal years. The amendments in ASU 2015-11 should be applied prospectively with earlier application permitted as of the beginning of an interim or annual reporting period. The Company elected to early adopt the above. The adoption doesn’t have a significant impact on the Company’s consolidated financial position or results of operations.
During November 2015, the FASB issued ASU 2015-17, Balance Sheet Classification of Deferred Taxes, which simplifies the presentation of deferred income taxes. ASU 2015-17 provides presentation requirements to classify deferred tax assets and liabilities as noncurrent in a classified statement of financial position. The standard is effective for fiscal years beginning after December 15, 2016, including interim periods within that reporting period. Early adoption is permitted for any interim and annual financial statements that have not yet been issued. We early adopted ASU 2015-17 effective November 30, 2015 on a prospective basis. The adoption did not have a significant impact on the Company’s consolidated financial position or results of operations.
In January 2016, the FASB issued ASU 2016-01, Financial Instruments – Overall: Recognition and Measurement of Financial Assets and Financial Liabilities. The pronouncement requires equity investments (except those accounted for under the equity method of accounting, or those that result in consolidation of the investee) to be measured at fair value with changes in fair value recognized in net income. ASU 2016-01requires public business entities to use the exit price notion when measuring the fair value of financial instruments for disclosure purposes, requires separate presentation of financial assets and financial liabilities by measurement category and form of financial asset, and eliminates the requirement for public business entities to disclose the method(s) and significant assumptions used to estimate the fair value that is required to be disclosed for financial instruments measured at amortized cost. These changes become effective for the Company's fiscal year beginning January 1, 2018. The expected adoption method of ASU 2016-01 is being evaluated by the Company and the adoption is not expected to have a significant impact on the Company’s consolidated financial position or results of operations.
In February 2016, the FASB issued ASU 2016-02, Leases (Topic 842), which supersedes the existing guidance for lease accounting, Leases (Topic 840). ASU 2016-02 requires lessees to recognise leases on their balance sheets, and leaves lessor accounting largely unchanged. The amendments in this ASU are effective for fiscal years beginning after December 15, 2018 and interim periods within those fiscal years. Early application is permitted for all entities. ASU 2016-02 requires a modified retrospective approach for all leases existing at, or entered into after, the date of initial application, with an option to elect to use certain transition relief. The Company is currently evaluating the impact of this new standard on its consolidated financial statements.
F-13
NOTE 3 – ACQUISITION OF MASTHERCELL
Description of the Transaction
The Company entered into a share exchange agreement (the "Share Exchange Agreement") dated November 3, 2014 and addendum dated March 2, 2015 with MaSTherCell SA, Cell Therapy Holding SA (collectively “MaSTherCell”). According to the Share Exchange Agreement, in exchange for all of the issued and outstanding shares of MaSTherCell, the Company issued to the shareholders of MaSTherCell an aggregate of 42,401,724 shares (the “Consideration Shares”) of common stock at a price of $0.58 per share for an aggregate price of $24,593 thousand (the “Consideration Share Pricing”). Out of the Consideration Shares, 8,173,483 shares will be allocated to the bondholders of MaSTherCell in case of conversion (See "MaSTherCell convertible bonds" below).
MaSTherCell SA and Cell Therapy Holding SA are companies incorporated in Belgium. All MaSTherCell shares were purchased either through Cell Therapy Holding SA or directly through MaSTherCell.
MaSTherCell is a technology-driven, customer-oriented CDMO specializing in cellular therapy development for advanced medicinal products. To perform its services, MaSTherCell has a production and laboratory facility (the “GMP Unit”) that received the good manufacturing practice (GMP) authorization for production of advanced therapy medicinal products (ATMP) by the Belgian drug agency (AFMPS).
The MaSTherCell acquisition is accounted for as a business combination. The results of operations of MaSTherCell have been included in the Company’s consolidated statements of operations starting from March 2, 2015, the date on which the Company obtained effective control of MaSTherCell. The revenue and net loss from operations of MaSTherCell for the period from March 2, 2015, the acquisition date, to November 30, 2015 was approximately $3 million and $2.3 million, respectively.
As part of the agreement the parties agreed on certain post closing conditions, mainly related to a post closing financing of $10 million and a valuation which meets a threshold of $45 million. In the event that the Company did not achieve those conditions within eight (8) months of the closing date, MaSTherCell had an option to unwind the transaction (the “Unwind Option”) by delivering to the Company all of the Consideration Shares plus any amount that the Company has advanced or invested in MaSTherCell.
On November 12, 2015, the Company and MaSTherCell and each of the shareholders of MaSTherCell (the “MaSTherCell Shareholders”), entered into an amendment (“Amendment No. 2”) to the Share Exchange Agreement. Under Amendment No. 2, the conditions under which the MaSTherCell Shareholders under the original agreement could unwind the transaction was extended to November 30, 2015. Under Amendment No. 2, the Company agreed to remit to MaSTherCell, by way of an equity investment, the sum of EUR 3.8 million by November 30, 2015 (the “Initial Investment”), to be followed by a subsequent equity investment by December 31, 2015 in MaSTherCell of EUR 1.2 million. These equity investments were to be made out of the proceeds from equity or equity-linked investments and/or credit facilities that the Company was required to have in place on or before November 30, 2015 in the aggregate amount of at least $10 million (the “Post Closing Financing”). The extended right of the MaSTherCell Shareholders to unwind the transaction could have been exercised by them only if the Company had not achieved the Post Closing Financing and/or completed the Initial Investment by November 30, 2015. As of November 30, 2015, the Company did not meet the above mentioned conditions and therefore the shares are presented as redeemable common stock.
On December 10, 2015, the Company entered into definitive agreements with accredited investors relating to a private placement (the “Private Placement”) of (i) 8,227,647 shares (the “Shares”) of the Company’s common stock, par value $0.0001 per share (the “Common Stock”) and (ii) three year warrants (the “Investor Warrants”) to purchase up to an additional 8,227,647 shares of the Company’s Common Stock at a per share exercise price of $0.52. The purchased securities were issued pursuant to subscription agreements (each the “Subscription Agreements”) between the Company and the purchasers for aggregate proceeds to the Company of $4,278 thousand. From the proceeds of the Private Placement, on December 10, 2015 the Company remitted to MaSTherCell the Initial Investment of € 3.8 million or $4,103 thousand (out of the original obligation for investment of €5 million), in compliance with its obligations as required under the Share Exchange Agreement. As a result, the right of the former MaSTherCell shareholders to unwind the merger with the Company was terminated.
F-14
In connection with the Initial Investment, the Company agreed to invest € 2.2 million in MaSTherCell equity in addition to the Initial Investment. The Company’s agreement represents an increase of € 1 million over the amount which the Company was previously obligated to invest in MaSTherCell under Amendment No. 2 as additional equity and replaces any funding obligation that the Company had under the Share Exchange Agreement, as amended.
MaSTherCell Convertible Bonds
On September 18, 2014, MaSTherCell entered into convertible bond agreements with certain of MaSTherCell’s existing and new investors raising €1.6 million (the “Convertible Bonds”). The bonds bear interest at an annual rate of 3% and will mature on September 18, 2016. As part of the original terms, each bond can be converted into 0.36 class A common shares of MaSTherCell (the “Conversion Shares”). As part of the Share Exchange Agreement, the parties agreed that, in case of conversion of the Convertible Bonds upon Uplisting (listing of the Company’s shares on NASDAQ or any other national exchange in the United States of America which provides at least the same level of liquidity) within 14 months of the closing date, the bondholders will be entitled to convert into a total of 8,173,483 out of the Consideration Shares.
In case the bondholders elect not to convert the convertible bonds, or in case they are not allowed to convert in the absence of Uplisting within 14 months from the closing date and the convertible bonds remain a liability of MaSTherCell, then the Consideration Shares will be reduced by the amount remaining outstanding to the bondholders. To that effect, the number of Consideration Shares to be released back to the Company, shall be determined by dividing the subscription amount of the outstanding convertible bonds plus interest owed thereunder (converted into USD according to the currency exchange rate applicable on the day of conversion) by the consideration and by applying the resulting quotient to actual total number of Consideration shares.
The Company records the convertible bonds on its consolidated balance sheet at their fair value (See Note 13).
Fair Value of Consideration Transferred
On the acquisition date, the fair value of the total consideration transferred to acquire MaSTherCell was as follows (in thousands):
|
Total purchase consideration: |
|||
|
Redeemable common stock |
$ | 24,592 | |
|
Less convertible bonds |
3,134 | ||
|
Total fair value of consideration transferred |
$ | 21,458 |
The following table summarizes the allocation of purchase price to the fair values of the assets acquired and liabilities assumed as of the acquisition date (in thousands):
|
Total assets acquired: |
|||
|
Cash and cash equivalents |
$ | 305 | |
|
Property and equipment |
4,236 | ||
|
Inventory |
231 | ||
|
Other current assets |
1,664 | ||
|
Other intangible assets |
18,977 | ||
|
Goodwill |
10,106 | ||
|
Total assets |
35,519 |
Total liabilities assumed:
F-15
|
Deferred income |
947 | ||
|
Deferred taxes |
4,440 | ||
|
Loan payables |
6,998 | ||
|
Other liabilities |
1,676 | ||
|
Total liabilities |
14,061 | ||
|
Total consideration transferred |
$ | 21,458 |
The allocation of the purchase price to the net assets acquired and liabilities assumed resulted in the recognition of other intangible assets which comprised of: Customer Relationships of $349 thousand, Know-How of $17,037 thousand, Backlog of $250 thousand and Brand Name of $1,341 thousand. These other intangible assets have a useful life between 1.75 and 11.75 years. The useful life of the other intangible assets for amortization purposes was determined considering the period of expected cash flows generated by the assets used to measure the fair value of the intangible assets adjusted as appropriate for the entity-specific factors, including legal, regulatory, contractual, competitive, economic or other factors that may limit the useful life of intangible assets.
The fair value of the Know How was estimated using a relief of royalties approach. Under this method, the fair value of the Know How is equal to the royalty fee that the owner of the Know How could profit from if he was to license the Know How out.
The fair value of the Backlog was estimated using the income approach. An income and expense forecast was built based upon Backlog revenue estimates and the cost to perform each contract. On this basis, a free cash flow for the asset was derived, under several assumptions.
Customer Relationships and Brand Name were estimated using a discounted cash flow method with the application of the multi-period excess earnings method. Under this method, an intangible asset’s fair value is equal to the present value of the incremental after-tax cash flows attributable only to the subject intangible asset after deducting contributory asset charges. An income and expenses forecast was built based upon specific intangible asset revenue and expense estimates.
Acquired goodwill is not amortized unless impaired. Goodwill isn't amortized for tax purposes.
Pro forma Impact of Business Combination
The unaudited pro forma financial results have been prepared using the acquisition method of accounting and are based on the historical financial information of the Company and MaSTherCell. The unaudited pro forma condensed financial results have been prepared for illustrative purposes only and do not purport to be indicative of the results of operations that actually would have resulted had the acquisition of MaSTherCell occurred at the beginning of the fiscal year, or of future results of the combined entities. The unaudited pro forma condensed financial information does not reflect any operating efficiencies and expected realization of cost savings or synergies associated with the acquisition.
Unaudited supplemental pro forma combined results of operations:
|
|
Year Ended November 30, | |||||
|
|
2015 | 2014 | ||||
|
|
(in thousands) | |||||
|
Revenue |
$ | 3,886 | $ | 1,606 | ||
|
Net loss |
$ | 5,558 | $ | 9,573 | ||
|
Net loss per common share: |
||||||
|
Basic |
$ | 0.10 | $ | 0.18 | ||
|
Diluted |
$ | 0.13 | $ | 0.21 | ||
Adjustments for the unaudited supplemental pro forma combined results of operations are as follows:
F-16
|
|
Year Ended November 30, | |||||
|
|
2015 | 2014 | ||||
|
|
(in thousands) | |||||
|
Amortization of intangibles |
$ | 471 | $ | 2,124 | ||
|
Deferred income |
88 | 400 | ||||
|
Deferred tax |
(183 | ) | (354 | ) | ||
|
Transaction costs |
(258 | ) | 258 | |||
|
Interest on convertible bonds |
(21 | ) | (1,483 | ) | ||
|
Total |
$ | 97 | $ | 945 | ||
Acquisition-related Costs
Acquisition-related expenses consist of transaction costs which represent external costs directly related to the acquisition of MaSTherCell and primarily include expenditures for professional fees such as legal, accounting and other directly related incremental costs incurred to close the acquisition by both the Company and MaSTherCell.
Acquisition-related expenses for the year ended November 30, 2015 were $258 thousand. These expenses were recorded to selling and general administrative expense in the consolidated statements of comprehensive loss.
NOTE 4 - SEGMENT INFORMATION
The Chief Executive Officer ("CEO") is the Company’s chief operating decision-maker ("CODM"). At November 30, 2015, following the acquisition of MaSTherCell, management has determined that there are two operating segments, based on the Company's organizational structure, its business activities and information reviewed by the CODM for the purposes of allocating resources and assessing performance.
CDMO
The Contract Development and Manufacturing Organization (“CDMO”) activity is operated by MaSTherCell, which specializes in cell therapy development for advanced medicinal products. MaSTherCell is providing two types of services to its customers: (i) process and assay development services and (ii) GMP contract manufacturing services. The CDMO segment includes only the results of MaSTherCell.
CTB
The Cellular Therapy Business (“CTB”) activity is based on our technology that demonstrates the capacity to induce a shift in the developmental fate of cells from the liver and differentiating (converting) them into “pancreatic beta cell-like” insulin producing cells for patients with Type 1 Diabetes. This segment is comprised of all entities aside from MaSTherCell.
The Company assesses the performance based on a measure of "Adjusted EBIT" (earnings before financial expenses and tax, and excluding share-based compensation expenses and non-recurring income or expenses). The measure of assets has not been disclosed for each segment.
Prior to the acquisition of MaSTherCell, the Company operated as one reporting segment. For this reason, the Company does not disclose comparative data for the year ended November 30, 2014.
Segment data for the year ended November 30, 2015 is as follows:
|
|
Corporate | |||||||||||
|
|
and | |||||||||||
|
|
CDMO | CTB | Eliminations | Consolidated | ||||||||
|
|
(in thousands) | |||||||||||
|
Net revenues from external customers |
$ | 3,320 | $ | $ | (346 | ) | $ | 2,974 | ||||
|
Cost of revenues |
(3,099 | ) | (3,099 | ) | ||||||||
|
Research and development expenses, net |
(1,279 | ) | 346 | (933 | ) | |||||||
|
Operating expenses |
(1,304 | ) | (1,799 | ) | (3,103 | ) | ||||||
|
Depreciation and amortization expense |
(1,984 | ) | (5 | ) | (1,989 | ) | ||||||
|
Adjusted EBIT |
$ | (3,067 | ) | $ | (3,083) | (6,150 | ) | |||||
|
|
||||||||||||
|
Share-based compensation |
(803 | ) | (803 | ) | ||||||||
|
Acquistion costs |
(258 | ) | (258 | ) | ||||||||
|
Financial income |
1,850 | 1,850 | ||||||||||
|
Loss before income tax |
(5,361 | ) |
F-17
Geographic, Product and Customer Information
Substantially all of the Company's revenues and long lived assets are located in Belgium.
Net revenues from single customers from the CDMO segment that exceed 10% of total net revenues are:
|
|
Year Ended | ||
|
|
November 30, | ||
|
|
2015 | ||
|
|
(in thousands) | ||
|
Customer A |
$ | 1,921 | |
|
Customer B |
$ | 626 |
NOTE 5 – PROPERTY AND EQUIPMENT
The following table represents the components of property and equipment:
|
|
November 30, | |||||
|
|
2015 | 2014 | ||||
|
|
(in thousands) | |||||
|
Cost: |
||||||
|
Production facility |
$ | 3,638 | $ | - | ||
|
Office furniture and computers |
120 | 16 | ||||
|
Lab equipment |
1,200 | 6 | ||||
|
|
4,958 | 22 | ||||
|
Less – accumulated depreciation |
(662 | ) | (9 | ) | ||
|
Total |
$ | 4,296 | $ | 13 | ||
Depreciation expense for the years ended November 30, 2015 and 2014 was $681 thousand and $4 thousand, respectively.
NOTE 6 – INTANGIBLE ASSETS AND GOODWILL
Changes in the carrying amount of the Company’s goodwill for the year ended November 30, 2015 is as follows:
|
|
November 30, | ||
|
|
2015 | ||
|
|
(in thousands) | ||
|
Goodwill as of December 1, 2014 |
$ | - | |
|
Goodwill as acquired |
10,106 | ||
|
Translation differences |
(571 | ) | |
|
Goodwill as of November 30,2015 |
$ | 9,535 |
F-18
Goodwill Impairment
The Company reviews goodwill for impairment annually and whenever events or changes in circumstances indicate the carrying amount of goodwill may not be recoverable. The Company performed a quantitative two-step assessment for goodwill impairment for the CDMO unit.
As part of the first step of the two-step impairment test, the Company compared the fair value of the reporting units to their carrying values and determined that the carrying amount of the units do not exceed their fair values. The Company estimated the fair value of the unit by using an income approach based on discounted cash flows. The assumptions used to estimate the fair value of the Company’s reporting units were based on expected future cash flows and an estimated terminal value using a terminal year growth rate based on the growth prospects for each reporting unit. The Company used an applicable discount rate which reflected the associated specific risks for the CDMO unit future cash flows.
Key assumptions used to determine the estimated fair value include: (a) expected cash flow for the five-year period following the testing date (including market share, sales volumes and prices, costs to produce and estimated capital needs); (b) an estimated terminal value using a terminal year growth rate of 3% determined based on the growth prospects ; and (c) a discount rate of 17.2%. Based on the Company’s assessment as of November 30, 2015, the carrying amount of its reporting unit does not exceeds its fair value.
A decrease in the terminal year growth rate of 1% or an increase of 1% to the discount rate would reduce the fair value of the reporting unit by approximately $2.9 million and $3.4 million, respectively. These changes would not result in an impairment.
Other Intangible Assets
Other intangible assets consisted of the following:
|
|
November 30, | ||
|
|
2015 | ||
|
|
(In thousands) | ||
|
Gross Carrying Amount: |
|||
|
Know How |
$ | 16,073 | |
|
Backlog |
237 | ||
|
Customer relationships |
330 | ||
|
Brand name |
1,266 | ||
|
|
17,906 | ||
|
Accumulated amortization |
1,253 | ||
|
Net carrying amount of other intangible assets |
$ | 16,653 |
Intangible asset amortization expenses were approximately $1.3 million for the year ended November 30, 2015. Estimated aggregate amortization expenses for each of the five succeeding years ending November 30th are as follows:
|
|
2016 | 2017 to 2020 | ||||
|
|
(in thousands) | |||||
|
Amortization expenses |
$ | 1,744 | $ | 1,608 | ||
NOTE 7 – CONVERTIBLE LOAN AGREEMENTS
a. Nine Investments Limited
F-19
On May 29, 2014, the Company entered into a convertible loan agreement with Nine Investments Limited, a Hong Kong company (“Nine Investments”), pursuant to which Nine Investments loaned the Company $1.5 million. The Company received the funds on June 4, 2014 (the “Closing Date”). Interest is calculated at 8% semiannually and was payable, along with the principal on or before December 31, 2014.
Nine Investments may convert all or part of the loan into shares of the Company's common stock at $0.40 per share. The conversion price and the number of shares of common stock deliverable upon the conversion of the loan shall be subject to adjustment in the event and in the manner following: (i) if and whenever the Company’s common shares at any time outstanding shall be subdivided into a greater or consolidated into a lesser number of common shares, or in case of any capital reorganization or of any reclassification of the capital of the Company or in case of the consolidation, merger or amalgamation of the Company with or into any other company or of the sale of the assets of the Company as or substantially as an entirety or of any other company, the conversion price shall be decreased or increased proportionately; and (ii) in the event the Company issues any shares of common stock or securities convertible into shares at a price less than the conversion price, the conversion price shall be reduced for any unpaid or unconverted loan amount to the new issuance price.
In addition, a consideration for entering into the loan agreement, on June 5, 2014, the Company issued to Nine Investments 500,000 shares of its common stock.
The Company allocated the proceeds from Nine Investments between the shares and the convertible loan based on the relative fair value. In addition, the conversion right is detachable from the loan and classified as a derivative due to down round protection (full ratchet and anti-dilution provisions). Therefore, the Company attributed, to the conversion right derivative, out of the proceeds allocated to the convertible loan based on its fair value. The allocation of conversion right and shares represents a discount to the loan that will be accreted until the maturity date of the loan.
The table below presents the fair value of the instruments issued as of the Closing Date and the allocation of the proceeds:
|
|
Total Fair | Allocation of | ||||
|
|
Value | Proceeds | ||||
|
|
(in thousands) | |||||
|
Loan component |
$ | 1,262 | $ | 746 | ||
|
Shares component |
250 | 180 | ||||
|
Embedded derivative component |
574 | 574 | ||||
|
Total |
$ | 2,086 | $ | 1,500 | ||
The Company estimated the fair value of the embedded derivative by using the Black-Scholes formula for option pricing using the following parameters: Share price $0.50; Exercise price $0.40; Volatility 94%; Dividend yield 0; Risk-free interest 0.05% and 80% likelihood for conversion. The bonus shares component was recorded as additional paid-in-capital and the fair value of the embedded derivative component is classified as a financial liability because the conversion price and the number of shares of common stock deliverable upon the conversion of the loan shall be subject to adjustment and will be measured in subsequent periods at fair value with changes in fair value charged to financial expenses or income, net.
On December 31, 2014, the Company executed an amendment to the convertible loan agreement with Nine Investments Limited to extend the due date of the loan from December 31, 2014 to January 31, 2015. As of the date of the approval of these financial statements, the Company has not finalized the terms and revised maturity date of this loan, although it believes it will be successful in extending the agreement upon mutually agreeable terms as soon as practicable. The Company continues to accrete interest until the terms are agreed upon.
b. Other Non U.S. Investors – Convertible Loans without Anti-Dilution
In September 2014, the Company entered into convertible loans agreements for $150 thousand. The loans bear an annual interest rate of 6% and mature in six months, unless converted earlier. The lenders shall have the right to convert all or any portion of the outstanding principal amount and all accrued but unpaid interest thereon into shares of common stock of the Company at a conversion price of $0.40 per share. Since the stock price is greater than the effective conversion price (after allocation of the total proceeds) on the measurement date, the conversion features is considered "beneficial" to the holders and equal to $135 thousand. The difference is treated as issued equity and reduces the carrying value of the host debt; the discount is accreted as deemed interest on the debt. As of the date of this report, the Company has not finalized the terms and revised maturity date for these loans, although it believes it will be successful in extending the agreement upon mutually agreeable terms as soon as practicable. The Company continues to accrete interest until the terms are agreed upon.
F-20
c. Other U.S. Investors and Non-U.S. Investors – Convertible Loans with Anti-Dilution
During the year ended November 30, 2015, the Company entered into five convertible loan agreements with new investors for a total amount of $950 thousand as follows (the “2015 Convertible Notes”):
| Grant Date | Maturity Date | November 30, | |
| 2015 | |||
| (in thousands) | |||
| Convertible loan a | June 9, 2015 | December 9, 2015 | $ 50 |
| Convertible loan b | June 16, 2015 | December 16, 2015 | 250 |
| Convertible loan c | June 24, 2015 | December 31, 2015 | 350 |
| Convertible loan d | October 15, 2015 | October 15, 2016 | 50 |
| Convertible loan e | October 27, 2015 | April 27, 2016 | 250 |
| $ 950 |
Interest is calculated at 6% annually and is payable, along with the principal on or before the maturity date.
The lenders have the right to convert all or part of the 2015 Convertible Notes (principal and the accrued interest) into shares of the Company’s common stock at a price per share equal to 75% of the Market Price (as defined below) (the "Conversion Price"), provided that the Conversion Price will not be less than $0.40 (the “Floor Price”). Market Price is defined as the average closing trading price for the Company’s common shares on the Over The Counter Market ("OTCQB") for the five trading days prior to the lender’s notice of conversion being delivered to the Company.
The principal under the 2015 Convertible Notes shall automatically convert into units that include one common share and one warrant exercisable into one additional common share, upon the Qualified Offering (as defined below) at the same terms as the Qualified Offering. A Qualified Offering is defined as an offering of securities of the Company with gross proceeds equal to or greater than $5 to 6 million and with a price per share of the common stock underlying such Qualified Offering equal to or greater than the Floor Price.
In the event the Company issues any common shares or securities convertible into common shares at a price less than the Floor Price (the "New Issuance Price"), the Floor Price shall be reduced for any unpaid or unconverted principal amount to the New Issuance Price.
The conversion right is detachable from each of the 2015 Convertible Notes and classified as a derivative due to down-round protection (full ratchet and anti-dilution provisions). Therefore, the Company allocated the conversion derivative from each of the 2015 Convertible Notes based on their respective fair value. The allocation of conversion right represents a discount to the principal amount and will be accreted until the maturity date of each of the 2015 Convertible Notes.
The table below presents the fair value as of the grant date:
| Embedded | ||||||
| Loan | Derivative | |||||
| Component | Component | |||||
| (in thousands) | ||||||
| Convertible loan a | $ | 46 | $ | 4 | ||
| Convertible loan b | 232 | 18 | ||||
| Convertible loan c | 323 | 27 | ||||
| Convertible loan d | 47 | 3 | ||||
| Convertible loan e | 237 | 13 | ||||
| $ | 885 | $ | 65 |
F-21
On December 23, 2015, the holders of all the 2015 Convertible Notes and the Company agreed to convert the 2015 Convertible Notes and accrued interest into units of the Company’s common stock, each unit comprising one share of the Company’s common stock and one three-year warrant to purchase an additional share of the Company’s common stock at an exercise price of $0.52. Each holder of 2015 Convertible Notes will receive a number of units equal to that holder’s investment in the 2015 Convertible Notes plus the accrued interest divided by $0.52. Furthermore, in the event the Company issues any common shares or securities convertible into common shares in a private placement for cash at a price less than $0.52 (the “New Issuance Price”) before December 23, 2016, the Company will issue, for no additional consideration, additional common shares to subscribers, according to the mechanism defined in the agreements. This provision does not apply to issuance of shares under options, issuance of shares under existing rights to acquire shares, nor issuance of shares for non-cash consideration.
NOTE 8 – LOANS
a. Terms of Long-term Loans
|
|
Interest Rate | ||||||||||||||
|
|
Loan Amount | November 30, | Year of | November 30, | |||||||||||
|
|
(in thousands) | Grant Date | 2015 | Maturity | 2015 | ||||||||||
|
|
(in thousands) | ||||||||||||||
|
Long-term loan a (*) |
€1,400 | August 1, 2012 | 4.05% | 2022 | $ | 1,086 | |||||||||
|
Long-term loan b |
€1,000 | August 13, 2012 | 6%-7.5% | 2023 | 1,089 | ||||||||||
|
Long-term loan c |
€250 | August 6, 2012, | 6% | 2022 | 205 | ||||||||||
|
Long-term loan d |
€250 | February 10, 2014 | 5.5% | 2024 | 247 | ||||||||||
|
Long-term loan e |
€290 | April 23, 2015 | 5.5% | 2020 | 350 | ||||||||||
|
Long-term loan f |
€800 | February 21, 2014 | Euribord + 2% | 2016 | 529 | ||||||||||
|
|
$ | 3,506 | |||||||||||||
|
Current portion of loans payable |
966 | ||||||||||||||
|
|
$ | 2,540 |
(*) For the loan from ING Bank in Belgium (“ING”), ING requested a business pledge on the Company assets for a value of € 1.4 million.
b. Terms of Short-term Loans and Current Portion of Long Term Loans
|
|
Amount in the | Interest Rate | |||||||
|
|
Currency of | November 30, | November 30, | ||||||
|
|
Loan | 2015 | 2015 | ||||||
|
|
(in thousands) | ||||||||
|
Current portion of loans payable a |
Euro | 4.05% | $ | 139 | |||||
|
Current portion of loans payable b |
Euro | 6%-7.5% | 166 | ||||||
|
Current portion of loans payable c |
Euro | 6% | 54 | ||||||
|
Current portion of loans payable d |
Euro | 5.5% | 35 | ||||||
|
Current portion of loans payable e |
Euro | 5.5% | 43 | ||||||
|
Current portion of loans payable f |
Euro | Euribord + 2% | 529 | ||||||
|
|
$ | 966 | |||||||
|
Short term-loans* |
Euro | 7% | 1,334 | ||||||
|
Short term-loan** |
Euro | 6.3% | 529 | ||||||
|
|
$ | 2,829 |
F-22
| * |
On various dates from September 14, 2015 through the year 2015, MaSTherCell received short term loans from management and shareholders for a total amount of €1,247 thousand, which bear an annual interest rate of 7%. No maturity dates were defined. |
| ** |
On October 30, 2015, MaSTherCell received from ING bank in Belgium a short term credit facility for a maximum amount of €500 thousand. The credit facility bears an interest rate of libor plus a margin defined by the bank. The maturity date of the loan is December 15, 2015. |
NOTE 9 - COMMITMENTS
a. Tel Hashomer Medical Research, Infrastructure and Services Ltd.
On February 2, 2012, the Company’s Israeli Subsidiary entered into a licensing agreement with THM Research, Infrastructure and Services Ltd (the “Licensor”). According to the agreement, the Israeli Subsidiary was granted a worldwide, royalty bearing, exclusive license to transdifferentiation of cells to insulin producing cells, including the population of insulin producing cells, methods of making this population, and methods of using this population of cells for cell therapy or diabetes treatment developed by Dr. Sarah Ferber of THM..
As consideration for the license, the Israeli Subsidiary will pay the following to the Licensor:
| 1) |
A royalty of 3.5% of net sales; | |
| 2) |
16% of all sublicensing fees received; | |
| 3) |
An annual license fee of $15 thousand, which commenced on January 1, 2012 and shall be paid once every year thereafter (the “Annual Fee”). The Annual Fee is non-refundable, but it shall be credited each year due, against the royalty noted above, to the extent that such are payable, during that year; and | |
| 4) |
Milestone payments as follows: | |
| a) |
$50 thousand on the date of initiation of phase I clinical trials in human subjects; | |
| b) |
$50 thousand on the date of initiation of phase II clinical trials in human subjects; | |
| c) |
$150 thousand on the date of initiation of phase III clinical trials in human subjects; | |
| d) |
$750 thousand on the date of initiation of issuance of an approval for marketing of the first product by the FDA; and | |
| e) |
$2 million when worldwide net sales of Products (as defined in the agreement) have reached the amount of $150 million for the first time, (the “Sales Milestone”). | |
As of November 30, 2015, the Israeli Subsidiary has not reached any of these milestones.
In the event of closing of an acquisition of all of the issued and outstanding share capital of the Israeli Subsidiary and/or consolidation of the Israeli Subsidiary or the Company into or with another corporation (“Exit”), the Licensor shall be entitled to choose whether to receive from the Israeli Subsidiary a one-time payment based, as applicable, on the value of either 5,563,809 shares of common stock of the Company at the time of the Exit or the value of 1,000 shares of common stock of the Israeli Subsidiary at the time of the Exit.
In May, 2014, the Israeli Subsidiary entered into a research service agreement with the Licensor. According to the agreement, the Licensor will perform a study at the facilities and use the equipment and personnel of the Chaim Sheba Medical Center (the “Hospital”), for the consideration of approximately $92 thousand for a year. In May 2015, the Israeli Subsidiary renewed the research agreement for an additional year with annual consideration of approximately $110 thousand.
b. Pall Life Science Belgium BVBA
On May 6, 2013, the Company entered into a Process Development Agreement with Pall Life Science Belgium BVBA (formerly ATMI BVBA), a Belgian Company that is a wholly owned Subsidiary of Pall Corporation (“Pall”), a U.S. publicly-traded company. According to the agreement, Pall will provide services in cell research. The Company will use Pall’s unique technology while the Company will provide to Pall the required materials for purpose of the study. According to the agreement, the Company will pay per achieved phase, as defined in the agreement, with a total consideration of approximately (€607) $642 thousand for all services. As of November 30, 2015, the Company received services in total value of $460 thousand.
F-23
c. Maryland Technology Development Corporation
On June 30, 2014, the Company’s U.S. Subsidiary entered into a grant agreement with Maryland Technology Development Corporation (“TEDCO”). TEDCO was created by the Maryland State Legislature in 1998 to facilitate the transfer and commercialization of technology from Maryland’s research universities and federal labs into the marketplace and to assist in the creation and growth of technology based businesses in all regions of the State. TEDCO is an independent organization that strives to be Maryland’s lead source for entrepreneurial business assistance and seed funding for the development of startup companies in Maryland’s innovation economy. TEDCO administers the Maryland Stem Cell Research Fund to promote State funded stem cell research and cures through financial assistance to public and private entities within the State. Under the agreement, TEDCO has agreed to give the U.S Subsidiary an amount not to exceed approximately $406 thousand (the “Grant”). The Grant will be used solely to finance the costs to conduct the research project entitled “Autologous Insulin Producing (AIP) Cells for Diabetes” during a period of two years.
On July 22, 2014, the U.S Subsidiary received an advance payment of $203 thousand on account of the grant. Through November 30, 2015, the Company spent the full amount of the grant. On September 21, 2015 the U.S Subsidiary received the second advance payment in amount of $203 thousand. Through November 30, 2015, the Company utilized $11 thousand. The amount of grant that was utilized through November 30, 2015, was recorded as a deduction of research and development expenses in the statement of comprehensive loss.
d. Department De La Gestion Financiere Direction De L’analyse Financiere (“DGO6”)
On November 17, 2014, the Company's Belgian Subsidiary, received the formal approval from the Walloon Region, Belgium (Service Public of Wallonia, DGO6) for a €2.015 million ($2.4 million) support program for the research and development of a potential cure for Type 1 Diabetes. The financial support is composed of a €1,085 thousand (70% of budgeted costs) grant for the industrial research part of the research program and a further recoverable advance of €930 thousand (60% of budgeted costs) of the experimental development part of the research program. The grants will be paid to the Belgian Subsidiary over a period of approximately 3 years. The grants are subject to certain conditions with respect to the Belgian Subsidiary’s work in the Walloon Region.
In addition, the DGO6 is also entitled to a royalty upon revenue being generated from any commercial application of the technology. On December 9 and 16, 2014, the Belgian Subsidiary received €651 thousand and €558 thousand under the grant, respectively. Up through November 30, 2015, an amount of $1.4 million (€1.1 million) was recorded as deduction of research and development expenses and an amount of $114 thousand was recorded as advance payments on account of grant.
On March 20, 2012, MaSTherCell had been granted an investment grant from the DGO6 for an amount of €1,421 thousand. This grant is related to the investment in the production facility with a coverage of 32% of the investment planned. A first payment of €568 thousand has been received in August 2013. The remaining part is expected to be paid by the end of fiscal 2016.
e. Israel-U.S Binational Industrial Research and Development Foundation (“BIRD”)
On September 9, 2015, the Israeli Subsidiary entered into a pharma Cooperation and Project Funding Agreement (CPFA) with BIRD and Pall Corporation, a U.S. company. BIRD will give a conditional grant of $400 thousand each (according to terms defined in the agreement), for a joint research and development project for the use Autologous Insulin Producing (AIP) Cells for the Treatment of Diabetes (the “Project”). The Project started on March 1, 2015. Upon the conclusion of product development, the grant shall be repaid at the rate of 5% of gross sales. The grant will be used solely to finance the costs to conduct the research of the project during a period of 18 months starting on March 1, 2015.
Up through November 30, 2015, an amount of $153 thousand was recorded as deduction of research and development expenses and receivable on account of grant. On September 21, 2015, the Israeli Subsidiary received $100 thousand under the grant.
F-24
f. Lease Agreement
MaSTherCell has an operational lease agreement for the rent of offices for a period of 12 years expiring on November 30, 2027. The costs per year are €28 thousand ($30 thousand).
In January 2015, the Israeli subsidiary signed an operational lease agreement for the rent of labs and office for three years, which will be used for the research and development activities in Israel. The costs per year are NIS 120 thousand ($31 thousand).
g. Collaboration agreement
On November 12, 2015, the Company, through its wholly owned Israeli subsidiary, entered into a Collaboration Agreement (the “Collaboration Agreement) with Biosequel LLC, a company incorporated under the laws of Russia (“Biosequel”) to collaborate, on a non-exclusive basis, in carrying out clinical trials and eventually marketing the Company’s products in Russia, Belarus and Kazakhstan. The collaboration is divided into two stages, with the first focused on obtaining the requisite regulatory approvals for conducting clinical trials, as well as performing all clinical and other testing required for market authorization in the defined territory. The second stage will focus on marketing the products and will be subject to successful market acceptance. Biosequel will fund the costs for the first stage, which is expected to last for five or more years, but may terminate earlier if the necessary regulatory approvals are not obtained by the second anniversary of the agreement. The Collaboration Agreement is also terminable under certain limited conditions relating to a party’s insolvency or bankruptcy related event or breach of a material term of the agreement and force majeure events. The Company shall be the sole and exclusive owner of any and all results of the pre-marketing approval R&D and clinical trials. As of November 30, 2015, none of the requisite regulatory approvals for conducting clinical trials had been obtained.
NOTE 10 – CAPITAL DEFICIENCY
a. Share Capital
The Company’s common shares are traded on the OTC Market Group’s OTCQB under the symbol “ORGS”.
b. Financings
1) During the year 2014, the Company issued 1,773,079 units at a purchase price of $0.52 per unit to private investors in a non-brokered private placement for total consideration of $922 thousand. Each unit consisted of one share of the Company’s common stock and one non-transferable common share purchase warrant, with each warrant entitling the holder to acquire one additional share of the Company’s common stock at an exercise price of $0.52 per share for a period of three years. The fair value of these warrants as of the date of issuance was $625 thousand using the Black-Scholes valuation model based on the following assumptions: dividend yield of 0% for all years; expected volatility of 101%-117%; risk free interest of 0.68% -0.95%, and an expected life of three years.
2) On June 30, 2014, the Company entered into a debt settlement agreement with one creditor, whereby it settled a debt in the amount of $24 thousand by the issuance of 46,175 share of its common stock at a price per share of $0.52. On July 14, 2014, the Company entered into a debt settlement agreement with another creditor, whereby it settled a debt in the amount of $13 thousand by the issuance of 25,759 shares of its common stock at a price per share of $0.52.
3) In July 2014, one of the investors exercised warrants to purchase 96,154 shares of the Company’s common stock at an exercise price of $0.52 for a total consideration of $50 thousand. As an inducement to the investor to exercise the warrants, the Company issued the investor twice the number of warrants exercised, which was 192,308 new warrants, with each warrant entitling the holder to acquire one additional share of the Company’s common stock at an exercise price of $0.52 per share for a period of three years. The fair value of these warrants as of the date of issuance was $60 thousand using the Black-Scholes valuation model based on the following assumptions: dividend yield of 0% for all years; expected volatility of 103%; risk free interest of 0.98%, and an expected life of three years.
F-25
4) On various dates from October 27, 2015 up through November 30, 2015, the Company entered into definitive agreements with accredited investors relating to a private placement (the “Private Placement”) of (i) 8,083,416 shares of the Company’s common stock and (ii) three year warrants to purchase up to an additional 8,083,416 shares of the Company’s Common Stock at a per share exercise price of $0.52. The purchased securities were issued pursuant to subscription agreements between the Company and the purchasers for aggregate proceeds to the Company of $4,203 thousand. Furthermore, in the event the Company issues any common shares or securities convertible into common shares in a private placement for cash at a price less than $0.52 (the “New Issuance Price”) before November 30, 2016, the Company will issue, for no additional consideration, additional common shares to subscribers in the $0.52 per share which total each subscriber’s subscription proceeds divided by the New Issuance Price, minus the number of shares already issued to such subscriber. This provision does not apply to issuance of shares under options, issuance of shares under existing rights to acquire shares, nor issuance of shares for non-cash consideration (See also Note 14).
The Company allocated the proceeds from the private placement based on the fair value of the warrants and the price protection derivative components. The residual amount was allocated to the shares.
The table below presents the fair value of the instruments issued as of the Closing Date and the allocation of the proceeds (for the fair value as of November 30, 2015, see Note 14):
|
|
Total Fair | ||
|
|
Value | ||
|
|
(in thousands) | ||
|
Warrants component |
$ | 1,390 | |
|
Price protection derivative component |
1,529 | ||
|
Shares component |
1,284 | ||
|
Total |
$ | 4,203 |
c. Credit Facilities
On October 30, 2015, the Company entered into securities purchase agreements with two accredited investors pursuant to which these lenders (”Lenders”) furnished to the Company access to a $5 million credit line (collectively, the “Credit Facility Agreements”). Pursuant to the Credit Facility Agreements, upon request the Company is entitled to receive $500 thousand or such lesser amount as may then be available under the credit facility (the “Advance Amount”), pro-rata from the credit providers under the Credit Facility Agreements, in consideration of which, it will issue to such persons, promissory notes for the amount advanced (each a “Credit Note”). The Company may draw down on the credit facility as needed until the entire $5.0 million is exhausted. Unless extended by mutual arrangement, the credit facility terminates on the earlier to occur of (i) November 30, 2016 and (ii) such time as the Company shall have raised in excess of $10 million in an equity investment. In consideration of the funding commitment under the Credit Facility Agreements, the Company issued to these Lenders 2,358,000 warrants to purchase up to an aggregate of shares of the Company’s Common Stock at a per share exercise price of $0.53 per share (the “Commitment Warrants”). The Commitment Warrants become first exercisable upon the scheduled expiration or termination of the credit facility through the third anniversary thereof. Additionally, upon the issuance of Credit Notes, the Lender is entitled to three year warrants (“Drawdown Warrants”) to purchase additional shares of the Company’s Common Stock in an amount equal to the quotient of: 0.50 X Advance Amount / $0.53. If the entire $5 million were drawn down by the Company, it would issue to the Lenders a total of 4,716,980 Drawdown Warrants. The fair value of the Commitment Warrants as of the date of issuance was $0.09 using the Black-Scholes valuation model based on the following assumptions: dividend yield of 0% for all years; expected volatility of 80%; risk free interest of 0.34% and an expected life of one year.
All Credit Notes that may be issued under the Credit Facility Agreements mature on November 30, 2016. Interest on the outstanding principal amount of the Credit Notes accrues at a per annum rate of 12%, payable at maturity or upon an event of default. The Credit Notes contain customary events of default for transactions of this nature. Upon an event of default, the Lender has the right to require the Company to prepay the outstanding principal amount of the Credit Notes plus all accrued and unpaid interest. In addition, the Lender may require prepayment of the Credit Notes at par in connection with certain major transactions and the occurrence of certain other triggering events.
F-26
d. Warrants
As part of the Company’s private placements as described in Notes 10b, the Company issued warrants as follows:
(1) Warrants which are subject to exercise price adjustments - presented as a financial liabilty as of November 30, 2015
| Exercise | ||||||||||||
| Price / | ||||||||||||
| Number of | Adjusted | Number of | ||||||||||
| Issuance | Warrants | Exercise | Expiration | Warrants | ||||||||
| Date | Issued | Price | Date | Outstanding | ||||||||
| October 27, 2015 | 192,308 | $0.40 | March 27, 2018 | 192,308 | ||||||||
| November 30, 2015 | 7,891,108 | $0.40 | November 30, 2018 | 7,891,108 | ||||||||
| 8,083,416 | 8,083,416 |
For the fair value calculation of these warrants, see Note 14.
(2) Warrants which are not subject to exercise price adjustments – presented in equity as of November 30, 2015
|
|
Exercise Price / | |||||||||||
|
|
Number of | Adjusted | Number of | |||||||||
|
Grant |
Warrants | Exercise | Expiration | Warrants | ||||||||
|
Date |
Issued | Price | Date | Outstanding | ||||||||
|
March 2014 |
713,023 | $0.52 | March 2017 | 713,023 | ||||||||
|
April 2014 |
384,615 | $0.52 | April 2017 | 384,615 | ||||||||
|
July 2014 |
192,308 | $0.52 | July 2017 | 192,308 | ||||||||
|
July 2014 |
144,230 | $0.52 | July 2017 | 144,230 | ||||||||
|
August 2014 |
115,385 | $0.52 | August 2017 | 115,385 | ||||||||
|
October 2015 |
2,358,490 | $0.53 | October 2018 | 2,358,490 | ||||||||
|
|
3,908,051 | 3,908,051 |
NOTE 11 – LOSS PER SHARE
The following table sets forth the calculation of basic and diluted loss per share for the periods indicated:
|
|
Year Ended | |||||
|
|
November 30, | |||||
|
|
2015 | 2014 | ||||
|
|
(in thousands, | |||||
|
|
except per share data) | |||||
|
Basic: |
||||||
|
Loss for the year |
$ | 4,461 | $ | 5,504 | ||
|
Weighted average number of common shares outstanding |
55,798,416 | 54,162,596 | ||||
|
Loss per common share |
$ | 0.08 | $ | 0.10 | ||
|
Diluted: |
||||||
|
Loss for the year |
$ | 4,461 | 5,504 | |||
|
Changes in fair value of embedded derivative and interest expenses on convertible bonds |
1,272 | |||||
|
Change in fair value of warrants |
559 | 598 | ||||
|
Loss for the year |
$ | 6,292 | 6,102 | |||
|
|
||||||
|
Weighted average number of shares used in the computation of basic loss per share |
55,798,416 | 54,162,596 | ||||
|
Number of dilutive shares related to convertible bonds |
873,380 | |||||
|
Number of dilutive shares related to warrants |
249,116 | 559,373 | ||||
|
Weighted average number of common shares outstanding |
56,920,912 | 54,721,969 | ||||
|
|
||||||
|
Loss per common share |
$ | 0.11 | $ | 0.11 | ||
F-27
Basic loss per share does not include 42,401,724 of redeemable common stock since the contingent criteria regarding the Unwind Option had not been met as of November 30, 2015.
Diluted loss per share does not include 42,401,724 redeemable common stock, 12,899,314 shares underlying outstanding options, 7,546,750 shares issuable upon exercise of warrants and 1,100,000 shares upon conversion of convertible notes for the year ended November 30, 2015, because the effect of their inclusion in the computation would be anti-dilutive.
Diluted loss per share does not include 15,267,559 shares underlying outstanding options, 400,000 shares due to stock-based compensation to service providers, 2,682,256 shares issuable upon exercise of warrants and 701,796 shares upon conversion of loans for the year ended November 30, 2014, because the effect of their inclusion in the computation would be anti-dilutive.
NOTE 12 – STOCK-BASED COMPENSATION
a. Global Share Incentive Plan
On May 23, 2012, the Company's board of directors adopted the global share incentive plan (2012) ("Global Share Incentive Plan (2012)"). Under the Global Share Incentive Plan (2012), 12,000,000 shares of common stock have been reserved for the grant of options, which may be issued at the discretion of the Company's board of directors from time to time. Under this plan, each option is exercisable into one share of common stock of the Company. The options may be exercised after vesting and in accordance with the vesting schedule that will be determined by the Company's board of directors for each grant. The maximum contractual life term of the options is 10 years.
b. Options Granted to Employees and Directors
1) On August 22, 2014, the Company approved an aggregate of 2,762,250 stock options to the Company’s Chief Executive Officer that are exercisable at $0.0001 per share. Out of the total approved, 414,304 options vested immediately with a fair value as of the date of grant of $261 thousand using the Black-Scholes valuation model, 1,242,996 options will vest quarterly over 4 years, with a fair value as of the date of grant of $783 thousand using the Black-Scholes valuation model, and 1,104,950 options will be vested pursuant to performance milestones that will be determined by the Compensation Committee of the Company's Board. Up to the date of this report, no performance milestones have been determined. All the options will expire on August 22, 2024.
2) On June 18, 2015, the Company approved an aggregate of 500,000 stock options to two directors that are exercisable at the market price on date of grant, or $0.53 per share. The options vest immediately and expire on June 18, 2020. The fair value of those options as of the date of grant was $136 thousand using the Black-Scholes valuation model.
F-28
The volatility of stock-based compensation is based on historical volatility of the Company for the last two years. The expected term is the mid-point between the vesting date and the maximum contractual term for each grant equal to the contractual life. The fair value of each option grant is based on the following assumptions:
|
|
Year Ended November 30, | ||
|
|
2015 | 2014 | |
|
Value of one common share |
$0.53 | $0.53 - 0.63 | |
|
Dividend yield |
0% | 0% | |
|
Expected stock price volatility |
85.7% | 100.5-100.6% | |
|
Risk free interest rate |
1.68% | 1.67-2.52% | |
|
Expected term (years) |
2.5 | 5 - 10 | |
A summary of the Company's stock options granted to employees and directors as of November 30, 2015 and 2014 and changes for the years then ended is presented below:
|
|
2015 | 2014 | ||||||||||
|
|
Weighted | Weighted | ||||||||||
|
|
Average | Average | ||||||||||
|
|
Exercise | Exercise | ||||||||||
|
|
Number of | Price | Number of | Price | ||||||||
|
|
Options | $ | Options | $ | ||||||||
|
Options outstanding at the beginning of the year |
12,809,455 | 0.27 | 12,294,765 | 0.265 | ||||||||
|
Changes during the year: |
||||||||||||
|
Granted |
500,000 | 0.53 | 2,707,300 | 0.194 | ||||||||
|
Exercised |
(623,806 | ) | 0.001 | |||||||||
|
Expired |
(2,440,120 | ) | 0.68 | |||||||||
|
Forfeited |
(528,125 | ) | 0.5 | (1,568,804 | ) | 0.205 | ||||||
|
|
||||||||||||
|
Options outstanding at end of the year |
10,341,210 | 0.16 | 12,809,455 | 0.27 | ||||||||
|
Options exercisable at end of the year |
8,696,162 | 0.09 | 9,661,548 | 0.568 | ||||||||
Costs incurred with respect to stock-based compensation for employees and directors for the years ended November 30, 2015 and 2014 were $713 thousand and $1,200 thousand, respectively. As of November 30, 2015, there was $984 thousand of unrecognized compensation costs related to non-vested employees and directors stock options, to be recorded over the next 2.73 years.
The following table presents summary information concerning the options granted to employees and directors outstanding as of November 30, 2015:
| Weighted | Weighted | |||||||||||
| Average | Average | Aggregate | ||||||||||
| Exercise | Number of | Remaining | Exercise | Intrinsic | ||||||||
| Prices | Outstanding | Contractual | Price | Value | ||||||||
| $ | Options | Life | $ | $ | ||||||||
| (in thousands) | ||||||||||||
| 0.0001 | 4,439,205 | 7.1 | 0.0001 | 1,464 | ||||||||
| 0.001 | 3,338,285 | 6.2 | 0.001 | 1,098 | ||||||||
| 0.50 | 400,000 | 8.7 | 0.5 | |||||||||
| 0.53 | 500,000 | 9.5 | 0.53 | |||||||||
| 0.75 | 250,000 | 7.6 | 0.75 | |||||||||
| 0.79 | 942,520 | 6.6 | 0.79 | |||||||||
| 0.85 | 471,200 | 6.5 | 0.85 | |||||||||
| 0.17 | 10,341,210 | 6.9 | 1.0.16 | 2,562 |
F-29
The following table presents summary of information concerning the options exercisable as of November 30, 2015:
| Exercise | Number of | |||||
| Prices | Exercisable | |||||
| $ | Options | Total | ||||
| (in thousands) | ||||||
| 0.0001 | 3,584,645 | 0.3 | ||||
| 0.001 | 3,338,285 | 3 | ||||
| 0.50 | 325,000 | 163 | ||||
| 0.53 | 500,000 | 265 | ||||
| 0.75 | 100,000 | 75 | ||||
| 0.79 | 565,512 | 447 | ||||
| 0.85 | 282,720 | 240 | ||||
| 8,696,162 | 1,193 |
c. Options Granted to Consultants and Service Providers
1) On April 3, 2014, the Company entered into a consulting agreement with Aspen Agency Limited, a Hong Kong corporation (“Aspen”), pursuant to which Aspen has agreed to provide investment banking, investor relations and business development services to the Company. In consideration for Aspen’s services, the Company agreed to issue to Aspen 3,000,000 stock options in two separate tranches of 1,000,000 and 2,000,000, with the second tranche vesting if they exercise the first tranche, to acquire shares of the Company’s common stock at an exercise price of $0.52 per share, for a period of three years. The fair value of the options was $744 thousand and was recorded as additional paid in capital in the balance sheet with a corresponding expense in general and administrative expenses. On October 23, 2014, the Company entered into a termination agreement with Aspen in which both parties agreed to terminate the consulting agreement and to cancel the first tranche of options. By way of cancellation of the first tranche of options, the second tranche was cancelled as well. The fair value of each option grant is estimated on the date of grant using a hybrid model combining a Monte Carlo simulation and Black-Scholes option pricing model with the following assumptions:
|
|
Year Ended |
|
|
November 30, |
|
|
2014 |
|
Value of one common share |
$0.51 |
|
Dividend yield |
0% |
|
Expected stock price volatility |
100% |
|
Risk free interest rate |
0.11-0.95% |
|
Expected term (years) |
1-3 |
2) On August 1, 2014, the Company granted an aggregate of 1,080,000 stock options to a consultant that are exercisable at $0.50, with 216,000 vesting immediately and 216,000 for each of the next four years. The options expire on August 1, 2018. The fair value of these options as of the date of grant was $403 thousand using the Black-Scholes option valuation model.
The fair value of each stock option grant is estimated at the date of grant using the Black-Scholes valuation model. The volatility is based on historical volatilities of companies in comparable stages as well as the historical volatility of companies in the industry and, by statistical analysis of the daily share-pricing model. The volatility of stock-based compensation granted after November 30, 2013 is based on historical volatility of the Company for the last two years. The expected term is equal to the contractual life, based on management estimation for the expected dates of exercising of the options.
The fair value of each option grant is based on the following assumptions:
F-30
|
|
Year Ended November 30, | ||
|
|
2015 | 2014 | |
|
Value of one common share |
$0.65,0.53 | $0.53 | |
|
Dividend yield |
0% | 0% | |
|
Expected stock price volatility |
86%,89% | 101% | |
|
Risk free interest rate |
1.34%,1.42% | 1.31% | |
|
Expected term (years) |
5 | 4 | |
A summary of the status of the stock options granted to consultants and service providers as of November 30, 2015, and 2014 and changes for the years then ended is presented below :
|
|
2015 | 2014 | ||||||||||
|
|
Weighted | Weighted | ||||||||||
|
|
Average | Average | ||||||||||
|
|
Exercise | Exercise | ||||||||||
|
|
Number of | Price | Number of | Price | ||||||||
|
|
Options | $ | Options | $ | ||||||||
|
Options outstanding at the beginning of the year |
2,458,104 | 0.75 | 1,378,104 | 0.95 | ||||||||
|
Changes during the year: |
||||||||||||
|
Granted |
200,000 | 0.51 | 2,080,000 | 0.51 | ||||||||
|
Expired |
(1,000,000 | ) | 0.52 | |||||||||
|
Options outstanding at end of the year |
2,658,104 | 0.75 | 2,458,104 | 0.75 | ||||||||
|
Options exercisable at end of the year |
1,521,624 | 0.65 | 1,171,384 | 0.77 | ||||||||
The following table presents summary information concerning the options granted to consultants and service providers outstanding as of November 30, 2015 (in thousands, except per share data):
| Weighted | Weighted | |||||||||||
| Average | Average | Aggregate | ||||||||||
| Exercise | Number of | Remaining | Exercise | Instrinsic | ||||||||
| Prices | Outstanding | Contractual | Price | Value* | ||||||||
| $ | Options | Life | $ | $ | ||||||||
| 0.50 | 1,080,000 | 2.67 | 0.50 | |||||||||
| 0.52 | 100,000 | 4.55 | 0.52 | |||||||||
| 0.61 | 100,000 | 6.98 | 0.61 | |||||||||
| 0.65 | 100,000 | 4.2 | 0.65 | |||||||||
| 0.69 | 706,904 | 6.17 | 0.69 | |||||||||
| 0.96 | 100,000 | 7.35 | 0.96 | |||||||||
| 1.40 | 471,200 | 6.38 | 1.40 | |||||||||
| 2,658,104 | 4.72 | 0.75 | - |
* There is no instrinsic value, as all options are “out of the money”
The following table presents summary of information concerning the options exercisable as of November 30, 2015 (in thousands, except per share data):
F-31
| Exercise | Number of | Total | ||||
| Prices | Exercisable | Exercise | ||||
| $ | Options | Value $ | ||||
| 0.50 | 432,000 | 216 | ||||
| 0.61 | 60,000 | 37 | ||||
| 0.69 | 706,904 | 488 | ||||
| 0.96 | 40,000 | 38 | ||||
| 1.40 | 282,720 | 396 | ||||
| 1,521,624 | 1,175 |
Costs incurred with respect to stock-based compensation for consultants and service providers for the year ended November 30, 2015 and 2014 was $90 and $923, respectively. As of November 30, 2015, there was $260 thousand of unrecognized compensation costs related to non-vested consultants and service providers, to be recorded over the next 4.55 years.
NOTE 13 – TAXES
a. The Company and the US Subsidiary
The Company and the US Subsidiary are taxed according to tax laws of the United States. The income of the Company is taxed in the United States at a rate of up to 35%.
b. The Israeli Subsidiary
The Israeli Subsidiary is taxed according to Israeli tax laws. The regular corporate tax rate in Israel for 2014 and 2015 is 26.5%. As of 2016, the tax rate has decreased to 25%.
c. The Belgian Subsidiaries
The Belgian Subsidiaries are taxed according to Belgian tax laws. The regular corporate tax rate in Belgium for 2014 and 2015 is 34%.
d. Tax Loss Carryforwards
1) As of November 30, 2015, the Company had net operating loss (NOL) carry forwards equal to $4.3 million that is available to reduce future taxable income. The Company’s NOL carry forward is equal to $138 thousand, and may be restricted under Section 382 of the Internal Revenue Code (“IRC”). IRC Section 382 applies whenever a corporation with an NOL experiences an ownership change. As a result of Section 382, the taxable income for any post change year that may be offset by a pre-change NOL may not exceed the general Section 382 limitation, which is the fair market value of the pre-change entity multiplied by the long term tax exempt rate.
2) U.S. Subsidiary - As of November 30, 2015, the U.S. Subsidiary had approximately $494 thousand of NOL carry forwards that are available to reduce future taxable income with no limited period of use.
3) Israeli Subsidiary - As of November 30, 2015, the Israeli Subsidiary had approximately $3.2 million of NOL carry forwards that are available to reduce future taxable income with no limited period of use.
4) Belgian Subsidiaries - As of November 30, 2015, the Belgian Subsidiaries had approximately $8.3 million (€7.8 million) of NOL carry forwards that are available to reduce future taxable income with no limited period of use.
e. Deferred Taxes
The following table presents summary of information concerning the Company’s deferred taxes as of the periods ending November 30, 2015 and 2014 (in thousands):
F-32
|
|
November 30, | |||||
|
|
2015 | 2014 | ||||
|
|
(U.S dollars in thousands) | |||||
|
Net operating loss carry forwards |
$ | 5,658 | $ | 1,626 | ||
|
Research and development expenses |
(178 | ) | 230 | |||
|
Employee benefits |
31 | 14 | ||||
|
Property and equipment |
268 | |||||
|
Convertible bonds |
45 | |||||
|
Deferred income |
(508 | ) | ||||
|
Intangible assets |
(5,661 | ) | ||||
|
Less: Valuation allowance |
(2,982 | ) | (1,870 | ) | ||
|
Net deferred tax liabilities |
$ | (3,327 | ) | $ | - | |
Realization of deferred tax assets is contingent upon sufficient future taxable income during the period that deductible temporary differences and carry forwards losses are expected to be available to reduce taxable income. As the achievement of required future taxable income is not considered more likely than not achievable, the Company and all of its subsidiaries except MaSTherCell have recorded full valuation allowance.
The changes in valuation allowance are comprised as follows:
|
|
Year Ended November 30, | |||||
|
|
2015 | 2014 | ||||
|
|
(U.S dollars in thousands) | |||||
|
Balance at the beginning of year |
$ | (1,870 | ) | $ | (1,212 | ) |
|
Additions during the year |
(1,112 | ) | (658 | ) | ||
|
Balance at end of year |
$ | (2,982 | ) | $ | (1,870 | ) |
f. Reconciliation of the Theoretical Tax Expense to Actual Tax Expense
The main reconciling item between the statutory tax rate of the Company and the effective rate is the provision for full valuation allowance with respect to tax benefits from carry forward tax losses.
g. Tax Assessments
1) The Company - As of November 30, 2015, the Company has received a final tax assessment up to the year 2009.
2) U.S. Subsidiary and the Israeli Subsidiary - As of November 30, 2015, the U.S. Subsidiary and the Israeli Subsidiary have not received any final tax assessment.
3) Belgian Subsidiary - As of November 30, 2015, the Belgian Subsidiary has received a final tax assessment for the year 2014.
4) MaSTherCell - As of November 30, 2015, MaSTherCell has received a final tax assessment for the years 2012 to 2014.
h. Uncertain Tax Provisions
As of November 30, 2015, the Company has not accrued a provision for uncertain tax positions.
NOTE 14 - FAIR VALUE PRESENTATION
The Company measures fair value and discloses fair value measurements for financial assets and liabilities. Fair value is based on the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. The accounting standard establishes a fair value hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below:
F-33
| • |
Level 1: Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs. |
| • |
Level 2: Observable inputs that are based on inputs not quoted on active markets, but corroborated by market data. |
| • |
Level 3: Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs. |
In determining fair value, the Company utilizes valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs, to the extent possible, and considers credit risk in its assessment of fair value.
As of November 30, 2015 and 2014, the Company’s liabilities that are measured at fair value and classified as level 3 fair value are as follows (in thousands):
|
|
November 30, | November 30, | ||||
|
|
2015 | 2014 | ||||
|
|
Level 3 | Level 3 | ||||
|
Warrants (1) |
$ | 1,382 | $ | 560 | ||
|
Price protection derivative (1) |
1,533 | |||||
|
Embedded derivatives*(1) |
289 | 992 | ||||
|
Convertible bonds (2) |
$ | 1,888 | $ |
| * |
The embedded derivative is presented in the Company's balance sheets on a combined basis with the related host contract (the convertible loans). |
(1) The fair value of the warrants, price protection derivatives and embedded derivatives is determined by using a Monte Carlo Simulation Model. This model, in contrast to the closed form model, such as the Black-Scholes Model, enables the Company to take into consideration the conversion price changes over the conversion period of the loan, and therefore is more appropriate in this case.
(2) The fair value of the convertible bonds described in Note 7 is determined by using a binomial model for the valuation of the embedded derivative and the fair value of the bond was calculated based on the effective rate on the valuation date (6%). The binomial model used the forecast of the Company share price during the convertible bond's contractual term. Since the convertible bond is in Euro and the model is in USD, the Company has used the Euro/USD forward rates for each period. In order to solve for the embedded derivative fair value, the calculation was performed as follows:
| • |
Stage A - The model calculates a number of potential future share prices of the Company based on the volatility and risk-free interest rate assumptions. | |
| • |
Stage B - the embedded derivative value is calculated "backwards" in a way that takes into account the maximum value between holding the bonds until maturity or converting the bonds. |
The following table presents the assumptions that were used for the models as of November 30, 2015:
|
|
Price Protection | ||||||||
|
|
Derivative and | Embedded | Convertible | ||||||
|
|
Warrants | Derivative | Bonds | ||||||
|
Fair value of shares of common stock |
$ | 0.33 | $ | 0.33 | $ | 0.33 | |||
|
Expected volatility |
87%-98% | 87% | 88% | ||||||
|
Discount on lack of marketability |
14% | - | 18% | ||||||
|
Risk free interest rate |
0.44%-1.24% | 0.11%-0.49% | 0.42% | ||||||
|
Expected term (years) |
2.9-3 | 0.08-0.87 | 0.8 | ||||||
|
Expected dividend yield |
0% | 0% | 0% | ||||||
|
Expected capital raise dates |
Q2 2016-Q4 2016, Q4 2017 |
F-34
The following table presents the assumptions that were used for the models as of November 30, 2014:
|
|
Embedded | ||
|
|
Warrants | Derivative | |
|
Value of one common share |
$0.65 | $0.65 | |
|
Dividend yield |
0% | 0% | |
|
Expected stock price volatility |
100% | 100% | |
|
Risk free interest rate |
0.03 – 0.11% | 0.04% | |
|
Expected term (years) |
0.3 – 0.8 | 0.08 | |
|
Expected capital raise dates |
March 2015 | March 2015 |
The table below sets forth a summary of the changes in the fair value of the Company’s financial liabilities classified as Level 3 for the year ended November 30, 2015 :
|
|
Price | |||||||||||
|
|
Embedded | Convertible | Protection | |||||||||
|
|
Warrants | Derivatives | Bonds | Derivative | ||||||||
|
|
(in thousands) | |||||||||||
|
Balance at beginning of the year |
$ | 560 | $ | 992 | $ | - | $ | - | ||||
|
Additions |
1,390 | 112 | 3,234 | 1,526 | ||||||||
|
Changes in fair value related to warrants expired* |
(525 | ) | - | 7 | ||||||||
|
Changes in fair value during the period |
(43 | ) | (815 | ) | (1,221 | ) | ||||||
|
Translation adjustments |
- | - | (125 | ) | ||||||||
|
Balance at end of the year |
$ | 1,382 | $ | 289 | $ | 1,888 | $ | 1,533 |
(*) During the twelve months ended November 30, 2015, 1,826,718 warrants have expired. There were no transfers to Level 3 during the twelve months ended November 30, 2015.
The Company has performed a sensitivity analysis of the results for the warrants fair value to changes in the assumptions for expected volatility with the following parameters:
|
|
Base -10% | Base | Base+10% | |||||||
|
|
(in thousands) | |||||||||
|
As of November 30, 2015 |
$ | 1,263 | $ | 1,382 | $ | 1,486 |
The Company has performed a sensitivity analysis of the results for the price protection derivative fair value to changes in the assumptions expected volatility with the following parameters:
|
|
Base -10% | Base | Base+10% | |||||||
|
|
(in thousands) | |||||||||
|
As of November 30, 2015 |
$ | 1,502 | $ | 1,533 | $ | 1,552 |
The Company has performed a sensitivity analysis of the results for the convertible bonds fair value to changes in the assumptions expected volatility with the following parameters:
|
|
Base -5% | Base | Base+5% | |||||||
|
|
(in thousands) | |||||||||
|
As of November 30, 2015 |
$ | 1,885 | $ | 1,888 | $ | 1,920 |
The table below sets forth a summary of the changes in the fair value of the Company’s financial liabilities classified as Level 3 for the year ended November 30, 2014 :
F-35
|
|
Embedded | |||||
|
|
Warrants | Derivatives | ||||
|
|
(in thousands) | |||||
|
Balance at beginning of the year Additions |
$ | 1,158 | $ | 574 | ||
|
Changes in fair value during the year |
(348 | ) | 418 | |||
|
Changes in fair value related to warrants expired |
(250 | ) | ||||
|
Balance at end of the year |
$ | 560 | $ | 992 |
There were no transfers to Level 3 during the twelve months ended November 30, 2014.
NOTE 15 – RESEARCH AND DEVELOPMENT EXPENSES, NET
|
|
Year Ended November 30, | |||||
|
|
2015 | 2014 | ||||
|
|
(in thousands) | |||||
|
Total expenses |
$ | 1,860 | $ | 2,478 | ||
|
Less grant |
(793 | ) | (929 | ) | ||
|
Total |
$ | 1,067 | $ | 1,549 | ||
NOTE 16 – FINANCIAL EXPENSES (INCOME), NET
|
|
Year Ended November 30, | |||||
|
|
2015 | 2014 | ||||
|
|
(in thousands) | |||||
|
Decrease in fair value of warrants and financial liabilities measured at fair value |
$ | (2,596 | ) | $ | (180 | ) |
|
Interest expense on convertible loans |
726 | 691 | ||||
|
Funding fees |
135 | |||||
|
Foreign exchange loss, net |
50 | 10 | ||||
|
Issuance of warrants as induced conversion |
260 | |||||
|
Other expenses |
(30 | ) | 11 | |||
|
Total |
$ | (1,850 | ) | $ | 927 | |
NOTE 17 - RELATED PARTY TRANSACTIONS
|
|
November 30, | |||||
|
|
2015 | 2014 | ||||
|
|
(in thousands) | |||||
|
Management and consulting fees to the Chairman of the Board |
$ | 57 | $ | 34 | ||
|
Compensation to the nonexecutive directors |
||||||
|
(except the Chairman of the Board) |
$ | 71 | $ | 39 | ||
|
Convertible loan from a related Fund* |
$ | 350 | $ | |||
* The convertible loan was granted with the same terms as the convertibles loans from third parties
NOTE 18 - SUBSEQUENT EVENTS
a. On December 7, 2015 and December 21, 2015, the Company entered into definitive agreements with accredited investors relating to a private placement of (i) 240,385 shares of the Company’s common stock and (ii) three year warrants to purchase up to an additional 240,385 shares of the Company’s common stock at a per share exercise price of $0.52. The purchased securities were issued pursuant to subscription agreements between the Company and the purchasers for aggregate proceeds to the Company of $125 thousand.
F-36
b. On December 10, 2015, the Company remitted to MaSTherCell the Initial Investment of $4,103 thousand (€ 3.8 million, out of original obligation for investment of €5 million), in compliance with its obligations as required under the Share Exchange Agreement. As a result, the right of the former MaSTherCell shareholders to unwind the merger with the Company was terminated (See note 3).
c. On February 18, 2016, the Israeli subsidiary entered into a Collaboration Agreement with Grand China Energy Group Limited with headquarters in Beijing, China (“Grand China”) to collaborate in carrying out clinical trials and marketing the Company’s autologous insulin producing cell therapy product (“API”) in the Peoples Republic of China, Hong Kong and Macau (the “Territory”), based on achieving certain pre-market development milestones. Upon achieving the pre-market development milestones by Grand China, the parties will collaborate on marketing the products in the Territory. Grand China will bear all costs associated with the pre-marketing development efforts in the Territory. Subject to the completion of the pre-marketing development milestones, the Israeli subsidiary has agreed to grant to Grand China, or a fully owned subsidiary thereof, under a separate sub-license agreement to the intellectual property underlying the API solely for commercialization of the Company’s products in the Territory. Grand China has agreed to pay annual license fees, ongoing royalties based on net sales generated by Grand China and its sublicensees, milestone payments and sublicense fees.
F-37