Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - XENOPORT INC | Financial_Report.xls |
| EX-12.1 - EX-12.1 - XENOPORT INC | d849886dex121.htm |
| EX-31.1 - EX-31.1 - XENOPORT INC | d849886dex311.htm |
| EX-32.1 - EX-32.1 - XENOPORT INC | d849886dex321.htm |
| EX-31.2 - EX-31.2 - XENOPORT INC | d849886dex312.htm |
| EX-23.1 - EX-23.1 - XENOPORT INC | d849886dex231.htm |
| EX-10.31 - EX-10.31 - XENOPORT INC | d849886dex1031.htm |
| EX-10.3.3 - EX-10.3.3 - XENOPORT INC | d849886dex1033.htm |
Table of Contents
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2014
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number 000-51329
XenoPort, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 94-3330837 | |
| (State or other jurisdiction of incorporation or organization) |
(IRS employer identification no.) | |
| 3410 Central Expressway, Santa Clara, California |
95051 (Zip code) | |
| (Address of principal executive offices) |
(408) 616-7200
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common stock, par value $0.001 per share |
The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes ¨ No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes þ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ¨ | Accelerated filer þ | Non-accelerated filer ¨ | Smaller reporting company ¨ | |||
| (Do not check if a smaller reporting company) | ||||||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No þ
As of June 30, 2014 (the last business day of the registrant’s most recently completed second quarter), the aggregate market value of the registrant’s common stock held by non-affiliates of the registrant was approximately $250.4 million based on the closing sale price as reported on The NASDAQ Global Select Market for such date, which excludes an aggregate of 10,256,800 shares of the registrant’s common stock held by officers, directors and stockholders that the registrant has concluded are affiliates of the registrant. Exclusion of such shares should not be construed to indicate that the holder of any such shares possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the registrant or that such person is controlled by, or under common control with, the registrant.
Indicate the number of shares outstanding of each of the registrant’s classes of common stock, as of the latest practicable date.
| Class |
Outstanding at February 13, 2015 | |
| Common stock, par value $0.001 per share |
62,756,809 shares |
DOCUMENTS INCORPORATED BY REFERENCE
| Document |
Parts Into Which Incorporated | |
| Portions of the Definitive Proxy Statement for the Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission pursuant to Regulation 14A not later than 120 days after the end of the fiscal year covered by this Form 10-K are incorporated by reference. |
Part III, Items 10-14 |
Table of Contents
XENOPORT, INC.
| Page | ||||||
| PART I. | ||||||
| Item 1. | 3 | |||||
| 3 | ||||||
| 4 | ||||||
| 5 | ||||||
| 7 | ||||||
| 15 | ||||||
| 16 | ||||||
| 18 | ||||||
| 20 | ||||||
| 20 | ||||||
| 22 | ||||||
| 26 | ||||||
| 27 | ||||||
| 28 | ||||||
| 28 | ||||||
| 29 | ||||||
| 29 | ||||||
| 30 | ||||||
| 30 | ||||||
| Item 1A. | 30 | |||||
| Item 1B. | 68 | |||||
| Item 2. | 68 | |||||
| Item 3. | 68 | |||||
| Item 4. | 68 | |||||
| PART II. | ||||||
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 69 | ||||
| Item 6. | 71 | |||||
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
73 | ||||
| Item 7A. | 89 | |||||
| Item 8. | 90 | |||||
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
90 | ||||
| Item 9A. | 90 | |||||
| Item 9B. | 92 | |||||
| PART III. | ||||||
| Item 10. | 92 | |||||
| Item 11. | 92 | |||||
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 93 | ||||
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
95 | ||||
| Item 14. | 95 | |||||
| PART IV. | ||||||
| Item 15. | 96 | |||||
| 102 | ||||||
| 104 | ||||||
HORIZANT, REGNITE, Transported Prodrug, XENOPORT and the XenoPort logo are trademarks of XenoPort, Inc.
2
Table of Contents
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, which are subject to the “safe harbor” created by those sections. Forward-looking statements are based on our management’s beliefs and assumptions and on information currently available to our management. All statements other than statements of historical facts are “forward-looking statements” for purposes of these provisions, including any projections or earnings. In some cases, you can identify forward-looking statements by terms such as “anticipate,” “believe,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “will,” “would” and similar expressions intended to identify forward-looking statements. These statements involve known and unknown risks, uncertainties and other factors, which may cause our actual results, performance, time frames or achievements to be materially different from any future results, performance, time frames or achievements expressed or implied by the forward-looking statements. We discuss many of these risks, uncertainties and other factors in this Annual Report on Form 10-K in greater detail under the heading “Risk Factors.” Given these risks, uncertainties and other factors, you should not place undue reliance on these forward-looking statements. Also, these forward-looking statements represent our estimates and assumptions only as of the date of this filing. You should read this Annual Report on Form 10-K completely and with the understanding that our actual future results may be materially different from what we expect. We hereby qualify our forward-looking statements by these cautionary statements. Except as required by law, we assume no obligation to update these forward-looking statements publicly, or to update the reasons actual results could differ materially from those anticipated in these forward-looking statements, even if new information becomes available in the future.
XenoPort, Inc. is a biopharmaceutical company focused on developing and commercializing a portfolio of internally discovered product candidates for the potential treatment of neurological and other disorders. We are currently commercializing HORIZANT® (gabapentin enacarbil) Extended-Release Tablets in the United States and REGNITE® (gabapentin enacarbil) Extended-Release Tablets is being marketed in Japan by Astellas Pharma Inc.
HORIZANT has been approved by the U.S. Food and Drug Administration, or FDA, for the treatment of moderate-to-severe primary restless legs syndrome, or RLS, in adults and for the management of postherpetic neuralgia, or PHN, in adults. RLS, also known as Willis-Ekbom Disease, is a neurological disorder characterized by an urge to move the legs, usually caused or accompanied by uncomfortable and unpleasant sensations in the legs. PHN is a neuropathic (nerve) pain syndrome that can follow the healing of an outbreak of herpes zoster, commonly known as shingles. REGNITE has been approved by the Japanese Ministry of Health, Labor and Welfare, or MHLW, as a treatment for patients with moderate-to-severe primary RLS.
Our development-stage product candidates include: XP23829, a novel fumaric acid ester product candidate that we are developing as a potential treatment for psoriasis and potentially for relapsing forms of multiple sclerosis, or MS; gabapentin enacarbil, which the National Institute on Alcohol Abuse and Alcoholism, or NIAAA, plans to evaluate as a potential treatment for alcohol use disorder, or AUD; and arbaclofin placarbil, or AP, which was licensed to Reckitt Benckiser Pharmaceuticals, Inc. (now doing business as Indivior PLC) for the development and commercialization worldwide in all indications. In addition, XP21279, which is a potential treatment for patients with advanced idiopathic Parkinson’s disease, has completed Phase 2 development and could be developed as resources permit or if we engage a partner to develop and commercialize this product candidate.
The potential for gabapentin enacarbil, XP23829 and/or XP21279 could be enhanced through third-party strategic arrangements that would further development and/or enhance commercialization efforts of these assets. As such, we plan to enter into partnerships, collaborations or other strategic arrangements with pharmaceutical companies: (1) when access to a primary care physician or an expanded sales force is necessary to maximize the commercial potential of gabapentin enacarbil, XP23829 and/or XP21279 in the United States; (2) for the
3
Table of Contents
development and commercialization of gabapentin enacarbil, XP23829 and/or XP21279 outside the United States or that require resources or expertise beyond our current capabilities; or (3) to develop and commercialize product candidates that fall outside our core focus or our core development capabilities.
The conventional approach to designing new oral drugs is to rely on the drug’s ability to passively diffuse through the intestinal wall to enter the bloodstream and reach the targeted tissue. However, this can be a difficult task, because the chemical and physical properties that allow a drug to bind to its cellular target and cause the intended therapeutic effect frequently impair the drug’s ability to passively diffuse through the wall of the intestines. Critical to the success of any drug is its ability to access the targeted tissues, achieve and maintain effective concentrations at the site of therapeutic action for an appropriate period of time and have minimal side effects. In addition, convenient administration is frequently necessary to ensure patient compliance. Many marketed drugs do not possess all of these attributes, leading to limitations in their therapeutic benefit and commercial potential.
If the medical need is high, drugs with poor absorption from the gastrointestinal, or GI, tract are still developed and marketed, but often with suboptimal therapeutic benefit. In some cases, drugs that are poorly absorbed from the GI tract are marketed as injectable medicines, which are inconvenient for patients. Another problem frequently encountered by drug designers occurs when a drug is well-absorbed from the intestines but does not last in the bloodstream for a sufficient period of time to maintain a therapeutic benefit. In this situation, frequent oral dosing is required, which is inconvenient for patients and can lead to poor compliance. In addition, drugs requiring frequent dosing often exhibit unwanted side effects when the drug is present in high concentration and then ineffectiveness when the concentration of the drug is insufficient. Sustained-release formulations that deliver medicine slowly as a pill travels through the entire GI tract can sometimes improve the utility of drugs that exhibit suboptimal therapeutic properties. However, drugs absorbed only in the upper GI tract do not benefit from sustained-release formulations.
The human body contains specific membrane proteins, known as transporters, which are responsible for carrying nutrients into cells and across cell barriers. Active transport refers to cellular transporter mechanisms that interact with substrates such as nutrients and use energy to carry them across membranes. One aspect of our expertise and know-how utilizes the body’s natural mechanisms for actively transporting nutrients through cellular barriers to permit certain parent drugs with suboptimal oral absorption to be effectively and efficiently delivered into the body after the oral administration. We have identified specific, high-capacity nutrient transporter proteins in the intestines and chemically modified the structure of the parent drug to create what we call a Transported Prodrug that utilizes these transporters to achieve absorption across the intestinal cell barrier through active transport. Our Transported Prodrugs are designed to split apart, releasing the parent drug and natural substances that generally have well-studied, known safety characteristics. Gabapentin enacarbil, AP and XP21279 are all prodrugs that target transporter proteins that are present throughout the entire GI tract, including the colon.
Another aspect of our expertise and know-how is our prodrug chemistry knowledge, including control of the kinetics of cleavage of prodrugs in certain tissues. XP23829 is a prodrug that we believe to be passively absorbed from the GI tract, but it has been designed to selectively split apart in a specific way, releasing monomethyl fumarate, or MMF. Our preclinical studies of XP23829 have demonstrated reduced GI irritation and higher achievable MMF levels than dimethyl fumarate, or DMF, which is another prodrug of MMF.
We have designed our prodrugs to be absorbed in the lower GI tract, which enables formulation using sustained-release technology and thereby may be used to maintain appropriate blood concentrations for an extended period after dosing. As a result of the improved oral absorption of our prodrugs, they may potentially have enhanced therapeutic benefits compared to the parent drugs, such as improved clinical efficacy, reduced side effects and less frequent dosing, which may result in increased patient convenience and compliance.
4
Table of Contents
Gabapentin Enacarbil (Known as HORIZANT in the United States and REGNITE in Japan)
Gabapentin enacarbil is our first approved product. It was approved in the United States in April 2011 for the treatment of moderate-to-severe primary RLS in adults and was approved in June 2012 for the management of PHN in adults. Glaxo Group Limited, or GSK, held commercialization rights for HORIZANT in the United States from March 2007 to April 2013. On May 1, 2013, pursuant to a termination and transition agreement with GSK, we completed the reacquisition of the exclusive rights to commercialize, promote, manufacture and distribute HORIZANT in the United States and assumed all responsibilities for any further development, manufacturing and commercialization of HORIZANT in the United States. REGNITE was approved by the Japanese MHLW in January 2012 for the treatment of moderate-to-severe primary RLS, and Astellas holds exclusive rights to commercialize REGNITE in Japan. For more information on our relationships with GSK and Astellas, see “Current and Former Partnerships and Collaborations” below.
Approved Indications
RLS
Background on RLS. RLS is a neurological condition that causes an irresistible urge to move the legs. This urge is usually caused or accompanied by unpleasant sensations of burning, creeping, tugging or tingling inside the patients’ legs, ranging in severity from uncomfortable to painful. These RLS-related symptoms typically begin or worsen during periods of rest or inactivity, particularly when lying down or sitting, and may be temporarily relieved by movement such as walking or massaging the legs. Symptoms often worsen at night, and disturbed sleep is a common result of RLS. Left untreated, RLS may cause exhaustion, daytime fatigue, inability to concentrate and impaired memory.
Potential Market. Although the exact prevalence rate of moderate-to-severe primary RLS is uncertain, a study published in Movement Disorders in 2010 indicated that approximately 2% to 3% of people in the United States are afflicted with moderate-to-severe primary RLS.
Although the exact prevalence is uncertain, Astellas estimates that there are approximately 2.1 million patients in Japan with RLS.
Competing Treatments for RLS. In the United States, the currently approved and most widely prescribed treatments for RLS belong to a class of drugs called dopamine agonists and include: MIRAPEX (pramipexole) from Boehringer Ingelheim GmbH and generic pramipexole; REQUIP (ropinirole) from GSK and generic ropinirole; and NEUPRO (a rotigotine transdermal system), a dopamine agonist patch from UCB S.A., all of which compete with HORIZANT. In Japan, we believe that REGNITE competes with SIFROL (pramipexole) and the UCB rotigotine transdermal system that was launched in February 2013 by Otsuka Pharmaceutical Co., Ltd., which has exclusive rights to market it in Japan.
PHN in the United States
Background on PHN. Neuropathic pain is pain that results from damage to nerves. One form of chronic neuropathic pain is PHN. PHN is a complication of shingles, a painful outbreak of rash or blisters on the skin caused by a reactivation of the same virus that causes chicken pox. PHN is often characterized as constant stabbing, burning or electric shock-like sensations in the area affected by shingles after the rash has cleared. Approximately 10% to 15% of all patients with shingles develop PHN, which can persist for many years.
Potential Market. We estimate that the prevalence of PHN is approximately 150,000 patients in the United States. In May 2006, Merck & Co., Inc. received FDA approval for ZOSTAVAX, a live attenuated vaccine to help prevent shingles. In October 2006, the U.S. Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices voted unanimously to recommend that adults 60 years of age and older be vaccinated with ZOSTAVAX for the prevention of shingles. In 2011, ZOSTAVAX was approved by the FDA for use in adults between the ages of 50 to 59 years. While ZOSTAVAX is not a treatment for shingles or PHN, the availability of this vaccine could impact the market for therapies for PHN.
5
Table of Contents
Competing Treatments for PHN. Products that we believe compete with HORIZANT in the United States for the management of PHN include drugs that act on the same target as HORIZANT, such as LYRICA (pregabalin) and NEURONTIN (gabapentin) from Pfizer Inc., GRALISE (once-daily formulation of gabapentin) from Depomed, Inc. and generic gabapentin. HORIZANT could also experience competition from a capsaicin patch (marketed as QUTENZA by Acorda Therapeutics, Inc.) and transdermal patches containing the anesthetic known as lidocaine (marketed as LIDODERM by Endo Pharmaceuticals, Inc.).
Commercialization
United States
We hold all commercialization rights for HORIZANT in the United States. We acquired the exclusive rights to commercialize, promote, manufacture and distribute HORIZANT in the United States on May 1, 2013, following the expiration of a transition period under a termination and transition agreement we entered into with GSK. We initiated our commercialization efforts in approximately 40 territories and, beginning in the third quarter, expanded our sales force to approximately 70 territories in the United States. We expect to further expand our sales force to up to approximately 120 sales representatives by mid-2015. Our sales representatives primarily focus their promotional efforts on specialty doctors in each geographic territory. Our sales organization consists of both XenoPort-employed sales representatives and contract sales representatives who are solely dedicated to HORIZANT education and promotion. We expect to continue to rely on our contract sales organization to provide back office logistical support for the XenoPort-employed sales representatives. In addition, we rely upon third-party service providers for manufacturing and distribution of HORIZANT. We will continue to monitor our promotion and commercialization of HORIZANT, and we may consider potential partnering opportunities to enable greater access to the primary care physician market.
During the year ended December 31, 2014, our net product sales of HORIZANT were $20.2 million, which increased from $6.4 million during the year ended December 31, 2013. Net product sales of HORIZANT during the year ended December 31, 2013 reflected our sales of HORIZANT after the transition period ended on April 30, 2013.
Japan
Astellas holds exclusive rights to commercialize REGNITE in Japan. We receive high-teen royalties on net sales of REGNITE.
Intellectual Property and Data Exclusivity
In the United States, the patent rights relating to the HORIZANT prodrug and its synthesis, as well as formulations and methods of its use, are owned by us and include a number of issued U.S. patents with expiration dates ranging from 2022 to 2029, as well as a number of pending U.S. patent applications with similar potential expiration dates. A patent term extension request has been filed with the U.S. Patent and Trademark Office, or USPTO, seeking to extend the expiration date of our granted composition-of-matter patent for the HORIZANT prodrug, which could provide product-specific exclusivity to mid-2025. We have six issued U.S. patents listed in the Patent and Exclusivity Information Addendum of the FDA’s publication, Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book, for HORIZANT. Two of the Orange Book-listed patents have expiration dates in 2022, one has an expiration date in 2025, two have expiration dates in 2026 and one has an expiration date in 2029. Additionally, the FDA granted new chemical entity data exclusivity for HORIZANT that extends to April 2016, and orphan drug (data) exclusivity for use of HORIZANT in the management of PHN that extends to June 2019. In Japan, the patent rights relating to the REGNITE prodrug, its synthesis, formulations and methods of use are also owned by us. The Japanese Patent Office granted a term extension on our composition-of-matter patent, which now extends to 2025 for REGNITE. We also hold patents or pending patent applications outside the United States and Japan that are directed to the gabapentin enacarbil composition of matter and its synthesis, as well as formulations and methods of use thereof.
6
Table of Contents
HORIZANT Post-Marketing Commitments and Post-Marketing Requirements
The FDA approval of HORIZANT for the treatment of moderate-to-severe primary RLS was associated with the requirement to conduct a program of post-marketing commitments, or PMCs, and post-marketing requirements, or PMRs, in adults, including: a 12-week, double-blind, placebo-controlled efficacy study evaluating 300 mg, 450 mg and 600 mg tablets of HORIZANT dosed once per day (“adult low-dose efficacy study”); two simulated driving studies; a drug-drug interaction study with morphine; and a cardiovascular safety, or QTc, study. Additionally, the RLS approval included a commitment to conduct a pediatric program for subjects 13 years and older. The pediatric clinical program, which was scheduled to commence after the adult low-dose efficacy data was obtained, includes: a pharmacokinetics, or PK, study; a double-blind, parallel group, efficacy and safety study; an open-label long-term safety extension study; and a simulated driving study. The specific protocol submission and trial completion dates for these PMCs/PMRs range from April 2011 through July 2024. Many of these PMCs/PMRs, including conduct of the adult low-dose efficacy study, have been conducted and fulfilled by GSK; however, we are responsible for fulfilling the remaining, and any additional future, post-marketing commitments and/or requirements, which will be expensive and time-consuming and may divert management time and resources away from our commercialization efforts or the development of our product candidates.
In January 2014, we reported top-line results from the adult low-dose efficacy study. The study assessed the efficacy, safety and tolerability of three doses of gabapentin enacarbil (300 mg, 450 mg or 600 mg) versus placebo dosed once per day in 487 subjects with normal renal function. The comparisons of the 600 mg and 450 mg dose groups to placebo reached statistical significance (p<0.05) on both of the co-primary endpoints of the study (reduction from baseline in the International Restless Legs Syndrome Rating Scale Total Score (IRLS), and the percentage of responders as determined by the Clinical Global Impression of Improvement (CGI-I)). The difference between the 300 mg dose group and placebo was statistically significant for CGI-I but not for the IRLS score. Gabapentin enacarbil was generally well tolerated in the study. The two most common treatment emergent adverse events reported in the study that were greater than or equal to 5% and at least the rate of placebo in any gabapentin enacarbil treatment arm were: somnolence (7%, 10%, 16% and 11% for placebo, 300 mg, 450 mg and 600 mg dose groups, respectively) and dizziness (4%, 4%, 7% and 12% for placebo, 300 mg, 450 mg and 600 mg dose groups, respectively). The study report for this study is in preparation and will be submitted to the FDA upon its completion.
XP23829 — A Prodrug of Monomethyl Fumarate
We are actively developing XP23829 as a potential treatment of patients with psoriasis and we may potentially develop XP23829 for relapsing forms of MS. We hold a composition-of-matter patent and a formulation patent in the United States on XP23829 that expire at the earliest in 2029 and hold patents or pending patent applications directed to the XP23829 methods of synthesis and use in the United States. We have also filed applications directed to the XP23829 composition of matter and methods of synthesis and use in other jurisdictions.
Prodrug Background
XP23829 is a fumaric acid ester compound and a patented prodrug of MMF. We are developing XP23829 as a potential treatment for psoriasis and potentially for relapsing forms of MS. Fumaric acid ester compounds have shown immuno-modulatory and neuroprotective effects in cell-based systems and preclinical models of disease. A product containing a combination of fumaric acid ester compounds, known as FUMADERM, from Biogen Idec Inc. is approved in Germany for the treatment of psoriasis. TECFIDERA (dimethyl fumarate), also from Biogen, is another fumaric acid ester prodrug that converts to MMF in the body. TECFIDERA was approved in March 2013 by the FDA for patients with relapsing forms of MS and by the European Commission in February 2014 for the treatment of relapsing-remitting MS.
7
Table of Contents
Our Prodrug
XP23829 is a novel prodrug of MMF that we believe may provide improved tolerability and efficacy compared to other fumaric acid ester compounds, including DMF. In preclinical studies that compared molar equivalent doses of XP23829 to DMF, XP23829 provided higher blood levels of the biologically active molecule MMF and a similar or greater degree of efficacy in MS and psoriasis animal models. Toxicology studies conducted in three species showed that XP23829 caused less stomach irritation when compared to DMF.
Development Program
We have conducted three Phase 1 clinical trials of XP23829 that included a total of approximately 160 healthy volunteers. The trials evaluated the PK profile of different formulations of XP23829, as well as the safety, tolerability and pharmacodynamics of XP23289 compared with TECFIDERA, which was also evaluated in one of our three Phase 1 clinical trials. XP23289 was associated with expected levels of exposure to MMF in blood with different formulations providing differing levels of exposure. XP23829 was generally well tolerated, with no serious adverse events reported in these trials.
In October 2012, we announced preliminary results from our first Phase 1 clinical trial of XP23829 in healthy adults designed to assess the pharmacokinetics, safety and tolerability of single doses of four different formulations of XP23829. The trial was a randomized, double-blind, two-period crossover, food effect comparison clinical trial of XP23829. Sixty subjects were assigned to five cohorts of 12, with each cohort receiving one of four different formulations of XP23829 or placebo. The trial demonstrated that administration of XP23829 resulted in the expected levels of MMF in the blood. As anticipated, the four formulations produced different pharmacokinetic, or PK, profiles of MMF, including one formulation that could potentially be dosed twice a day and at least one formulation that may be suitable for once-a-day dosing. XP23829 was generally well-tolerated in the trial. All 12 subjects in each cohort completed both dosing periods.
In September 2013, we announced preliminary results from two additional Phase 1 clinical trials of XP23829. One of the Phase 1 trials was a randomized, double-blind, placebo-controlled, multiple ascending dose study of the pharmacokinetics, safety and tolerability of two formulations of XP23829 in healthy adult subjects. Five separate cohorts (12 active and three placebo subjects per cohort) were enrolled sequentially and received one of two formulations of XP23829 dosed with or without food. Following up-titration of up to five days, dosing for each cohort ranged from 400 to 1,000 mg XP23829 per day, administered either once a day, or QD, or twice a day, or BID, with dosing for an additional six days. On the seventh day of target dose administration, PK evaluations were conducted following the morning dose. Similarly, a sixth cohort of healthy subjects received 240 mg BID TECFIDERA, which is also a prodrug of MMF. As a consequence of its higher molecular weight, 400 mg of XP23829 is approximately equimolar to 240 mg of DMF (i.e., these amounts of each compound can potentially be enzymatically converted to approximately the same amount of MMF).
The preliminary results of this trial showed that both formulations of XP23829 produced MMF in the plasma, but with different PK profiles. The 400 mg BID XP23829 Formulation 1 dosed without food provided an MMF PK profile that was nearly identical to that of 240 mg BID TECFIDERA dosed without food. As expected, XP23829 Formulation 2, administered as 800 mg QD and 500 mg BID dosed with food, provided more extended exposure to MMF. These cohorts had a lower mean maximal plasma concentration, or Cmax, of MMF, while providing a mean daily area under the concentration versus time curve, or AUC, of MMF that was approximately 25% less than that seen after dosing 240 mg BID TECFIDERA. In addition, pharmacodynamic data was also accessed in this study. Compared to pre-dosing baseline blood levels, XP23829 dosed once or twice a day selectively reduced blood lymphocytes and increased blood eosinophils when assessed after 12 days of dosing. Most post-treatment lymphocyte counts and all eosinophil cell counts were within normal limits, and no subjects had reduction of lymphocytes below 500 per microliter. Lymphocyte counts returned to baseline levels when assessed eight to ten days after the last dose. These effects were not observed in placebo-dosed subjects, and there was no effect of XP23829 on other monitored blood cell populations. Other fumaric acid ester compounds, including TECFIDERA and FUMADERM, reduce blood lymphocytes with maximal effect occurring after many months of dosing. Both products are also known to cause transient minor increases in blood eosinophils. XP23829 was generally well tolerated in the study. The most common adverse events were flushing and gastrointestinal-related events, which were generally mild to moderate in severity.
8
Table of Contents
The other Phase 1 study was an open-label, PK and mass balance study in healthy male subjects. One group of six subjects received a single dose of XP23829 that incorporated a radiolabel in the fumarate portion of the molecule and a separate group of six subjects received XP23829 with the radiolabel in the promoiety portion of the molecule. As expected, the metabolism and disposition of radioactivity from the fumarate-labeled version of XP23829 was similar to what has been publicly reported for dosing healthy subjects with DMF labeled in the same position. For the promoiety-labeled version of XP23829, 98% of the recovered radioactivity was in urine. The major metabolites of the promoiety found in humans were the same as those observed in animal studies.
In addition to human clinical studies, we have also completed 13-week toxicology studies of XP23829 in three animal species. Each study included a dose of DMF chosen to provide a similar MMF exposure to that of the highest dose of XP23829. The adverse effects of XP23829 were similar to those of DMF, except for fewer and less severe adverse effects in the stomach after dosing XP23829. The target organs for XP23829 were identical to DMF, and there was no increase in severity of any adverse effects for XP23829 compared with DMF. We believe that these preliminary results support a conclusion that all observed adverse effects of XP23829 were attributable to exposure to MMF, the common active metabolite of DMF and XP23829.
In June 2014, we announced the initiation of a Phase 2 multi-center, randomized, double-blind, placebo-controlled study designed to assess the efficacy and safety of XP23829 as a potential treatment of patients with moderate-to-severe chronic plaque-type psoriasis. We expect to enroll approximately 200 subjects in this trial, which is being conducted in the United States. The trial will include a screening and washout phase of up to four weeks, a 12-week treatment phase and a four-week post-treatment phase. Eligible study subjects will be randomized to placebo or one of three treatment arms of XP23829: 400 mg or 800 mg once daily; or 400 mg twice daily. The primary endpoint of the study will examine the percent change in Psoriasis Area and Severity Index (PASI) score from baseline at the end of week 12. Secondary endpoints will include the proportion of subjects who achieve a reduction of 75% or greater from baseline in PASI (PASI-75) score and subjects who achieve a Static Physicians Global Assessment (sPGA) score of “clear” or “almost clear”. We expect to report top-line results of this clinical trial in the third quarter of 2015.
Potential Target Indications
Psoriasis
Background on Psoriasis. Psoriasis is a chronic, systemic, inflammatory disease that manifests in the skin and/or joints. It typically manifests as thick scaling red plaques, with variable morphology and distribution, resulting from an unusually high rate of skin cell growth. There is no cure for psoriasis, and treatment often requires complex medical intervention. The main cause of psoriasis is uncertain, but it is thought to be caused by autoimmunity, genetic predisposition and environmental factors.
Potential Market. Psoriasis is the most prevalent autoimmune disease in the United States with as many as 7.5 million Americans suffering from the condition. It is estimated that approximately 1.5 million adults in the United States are considered to have moderate-to-severe psoriasis and between 150,000 and 260,000 new cases of psoriasis are diagnosed each year.
Current Treatments. In the United States, products that are currently approved for the treatment of psoriasis include topical therapies, oral systemic therapies and biological therapies. Topical therapies include corticosteroids, anthrolin and synthetic vitamin D and vitamin A. Oral systemic therapies include generic agents, such as acitretin, cyclosporine and methotrexate, and a branded agent known as OTEZLA (apremilast), marketed by Celgene Corporation, which was approved in September 2014 for the treatment of patients with moderate-to-severe plaque psoriasis who are candidates for phototherapy or systemic therapy. Biological therapies that are approved to treat psoriasis include: COSENTYX (secukinumab), marketed by Novartis AG; ENBREL (etanercept), marketed by Amgen Inc.; HUMIRA (adalimumab), marketed by AbbVie, Inc.; REMICADE (infliximab), marketed by Johnson & Johnson and Merck & Co., Inc.; and STELARA (ustekinumab), marketed by Johnson & Johnson.
9
Table of Contents
Relapsing Forms of MS
Background on MS and Relapsing Forms of MS. MS is a chronic and progressive neurodegenerative disease in which the body’s immune system attacks the myelin protein that wraps around nerve fibers. The disease typically strikes between the ages of 20 to 40 years, and because it is progressive in nature, disability accumulates over time and can lead to permanent impairment of mobility, cognition and the ability for self-care. After a subsequent attack, followed by a remission of symptoms, the condition is diagnosed as relapsing MS. This classification represents approximately two-thirds of the patients with a diagnosis of MS. A typical course of the disease involves progressively more frequent relapses of symptoms resulting in greater levels of disability after each relapse.
Potential Market. Although the exact prevalence is not known, it is estimated that approximately 250,000 to 350,000 people in the United States have been diagnosed with MS and that approximately one million people worldwide suffer from MS.
Current Treatments. Products that could compete with XP23829 for the treatment of relapsing forms of MS include oral and injectable agents that are approved in the United States for the treatment of relapsing forms of MS. These include oral agents such as AUBAGIO (teriflunomide), marketed by Genzyme Corporation, a Sanofi company; GILENYA (fingolimod), marketed by Novartis; and TECFIDERA (dimethyl fumarate), marketed by Biogen. In addition, several injectable products are approved in the United States and include AVONEX (interferon beta 1a), PLEGRIDY (peginterferon beta 1a) and TYSABRI (natralizumab), marketed by Biogen; BETASERON (interferon beta 1b) and EXTAVIA (interferon beta 1b), marketed by Bayer AG/Novartis; COPAXONE (glatiramer acetate), marketed by Teva Pharmaceutical Industries Ltd.; REBIF (interferon beta 1a), marketed by Merck Serono S.A.; and LEMTRADA (alemtuzumab), marketed by Genzyme/Sanofi-Aventis.
Further Clinical Development of XP23829
Based on the favorable preliminary results of XP23829 Phase 1 and toxicology studies, and pending the Phase 2 clinical trial results in patients with moderate-to-severe chronic plaque-type psoriasis, we intend to continue to advance the development of XP23829. We have received feedback from the Division of Dermatology and Dentistry Products of the FDA for XP23829 as a potential treatment for moderate-to-severe chronic plaque-type psoriasis. We believe that a positive outcome from the Phase 2 clinical trial of XP23829 in patients with moderate-to-severe chronic plaque-type psoriasis could enable us to request and End-of-Phase 2 meeting with the FDA and prepare to enter Phase 3 development of XP23829 as a potential treatment for patients with moderate-to-severe chronic plaque-type psoriasis.
We have also received feedback from the Division of Neurology Products of the FDA regarding proposed development plans for XP23829 as a potential treatment for patients with relapsing forms of MS. Based on the feedback we have received, we believe that the FDA would allow us to initiate potential Phase 3 clinical development for this indication using XP23829 doses that produce MMF exposure similar to that produced by the approved dose of TECFIDERA. The FDA encouraged the exploration of multiple doses of XP23829 to assess the potential advantages of doses that produced either higher or lower MMF exposure.
We are also in discussions with the FDA concerning proposed development strategies for XP23829 that could support a New Drug Application, or NDA, submission under Section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act, as amended, or FDCA. Section 505(b)(2) of the FDCA permits the submission of an NDA where at least some of the information required for approval comes from studies not conducted by, or for, the applicant and for which the applicant has not obtained a right of reference. As a consequence, the preclinical and clinical development programs leading to the submission of an NDA under Section 505(b)(2) may be less expensive to carry out and could potentially be concluded in a shorter period of time than programs required for an NDA submitted under Section 505(b)(1) of the FDCA. However, at this time, we believe that the FDA will require additional information with respect to XP23829 before making any determination on the applicability of the Section 505(b)(2) pathway. In addition, given that our strategy is to develop XP23829 with the objective of differentiating XP23829 from other products for psoriasis and relapsing forms of MS, we believe it is more likely that we will pursue a full development pathway for XP23829 under Section 505(b)(1) of the FDCA.
10
Table of Contents
XP23829 Development, Commercialization and Partnering Strategy
We plan to continue development of XP23829 and are also in discussions with potential partners regarding the development and commercialization of XP23829.
Gabapentin Enacarbil — A Transported Prodrug of Gabapentin
Gabapentin enacarbil is a Transported Prodrug of gabapentin. We signed a clinical trial agreement with the NIAAA under which the NIAAA plans to a conduct a clinical trial evaluating gabapentin enacarbil as a potential treatment of patients with AUD. In the United States, we hold patent rights relating to the gabapentin enacarbil prodrug and its synthesis, as well as formulations and methods of its use, and in Japan we also hold patent rights relating to the prodrug and its synthesis, as well as formulations and methods of its use. We also hold patents or pending patent applications outside of the United States and Japan that are directed to gabapentin enacarbil and its synthesis, as well as formulations and methods of its use.
Parent Drug Background
Gabapentin enacarbil is metabolized by the body to release gabapentin, a drug that has been sold by Pfizer as Neurontin since 1993 and is currently sold as a generic drug by a number of companies. Gabapentin is approved for marketing in the United States as adjunctive therapy in the treatment of partial seizures in patients with epilepsy and for the management of PHN. Data from clinical trials indicate that oral gabapentin may reduce alcohol craving and alcohol withdrawal symptoms. In addition, single-site clinical trials have also indicated that in alcohol dependent patients who are abstinent at treatment initiation, orally administered gabapentin may delay the onset to heavy drinking and may relieve symptoms of insomnia. In these studies, gabapentin was reported to be generally well-tolerated.
Gabapentin absorption is highly variable among patients, and there is a limit on the gabapentin exposure that can be achieved by direct oral administration of the parent drug. Published results from clinical trials of gabapentin in epilepsy patients indicated that, for the same dose level, some patients absorbed as little as 10% of the dose of gabapentin administered while others absorbed more than 70%. In addition, the short duration of gabapentin circulating in blood after oral dosing has required that it be administered up to three times a day, which could lead to poor compliance with dosing regimens and, therefore, may result in reduced efficacy in some patients.
We believe that these suboptimal characteristics of orally administered gabapentin result from the mechanism responsible for the absorption of gabapentin from the GI tract. Gabapentin is actively transported across the GI tract after administration. However, the specific transporter mechanism responsible for gabapentin absorption appears to have limited capacity, which seems to vary among individuals, and which is predominantly expressed in the upper GI tract. Due to gabapentin’s poor absorption in the lower GI tract, the use of traditional sustained-release formulations to correct the frequent dosing requirement has not been possible.
Our Transported Prodrug
Gabapentin enacarbil is designed to address the limitations of gabapentin by targeting high-capacity nutrient transporter mechanisms expressed throughout the length of the GI tract. We believe that this approach can address the variable and suboptimal exposure to gabapentin as experienced by some patients. By targeting transporters expressed throughout the length of the GI tract, we have been able to develop a sustained-release oral formulation of gabapentin enacarbil that we believe provides more consistent absorption of gabapentin and has overcome the need for frequent dosing of gabapentin.
Gabapentin enacarbil is designed to rapidly convert to gabapentin once absorbed from the GI tract, resulting in limited systemic exposure to the intact Transported Prodrug. In addition to producing gabapentin, gabapentin enacarbil is metabolized to release other components with well-studied, favorable safety characteristics. Gabapentin enacarbil has demonstrated a favorable safety profile in clinical trials. The most common side effects of gabapentin enacarbil are somnolence/sedation and dizziness, somnolence and headache.
11
Table of Contents
Target Indication
AUD
Background on AUD. In May 2013, the American Psychiatric Association issued the 5th edition of the Diagnostic and Statistical Manual of Mental Disorders, or DSM–5. The DSM-5 defines a person with AUD as anyone meeting any two of the 11 criteria summarized below during the same 12-month period, with the severity of AUD defined as mild, moderate or severe based on the number of criteria met:
| • | alcohol is often taken in larger amounts or over a longer period than was intended; |
| • | there is a persistent desire or unsuccessful efforts to cut down or control alcohol use; |
| • | a great deal of time is spent in activities necessary to obtain alcohol, use alcohol or recover from its effects; |
| • | craving, or a strong desire or urge to use alcohol; |
| • | recurrent alcohol use resulting in a failure to fulfill major role obligations at work, school or home; |
| • | continued alcohol use despite having persistent or recurrent social or interpersonal problems caused or exacerbated by the effects of alcohol; |
| • | important social, occupational or recreational activities are given up or reduced because of alcohol use; |
| • | recurrent alcohol use in situations in which it is physically hazardous; |
| • | alcohol use is continued despite knowledge of having a persistent or recurrent physical or psychological problem that is likely to have been caused or exacerbated by alcohol.; |
| • | tolerance, as defined by either of the following: |
| • | a need for markedly increased amounts of alcohol to achieve intoxication or desired effect, or |
| • | markedly diminished effect with continued use of the same amount of alcohol; and |
| • | withdrawal, as manifested by either of the following: |
| • | the characteristic withdrawal syndrome for alcohol, or |
| • | alcohol (or a closely related substance, such as a benzodiazepine) is taken to relieve or avoid withdrawal symptoms. |
Potential Market. It is estimated that AUD affects approximately 17 million people in the United States.
Current Treatments. In the United States, there are three FDA-approved drugs for the treatment of AUD: ANTABUSE (disulfiram), marketed by TEVA; REVIA and VIVITROL, which are oral and injectable formulations of naltrexone, marketed by Barr Pharmaceuticals, Inc. and Alkermes, Inc., respectively; and CAMPRAL (acamprosate), marketed by Actavis Inc. Disulfiram works by increasing the body’s concentration of acetaldehyde, which is a toxic byproduct of alcohol breakdown and, in excess quantities, causes symptoms such as nausea and flushing. These unpleasant side effects are a deterrent to drinking for some people. Naltrexone is an opioid antagonist. Its mechanism of action in alcoholism is not understood, but it is thought to work through the blockage of endogenous opioid receptors involved in the pleasant sensations associated with drinking and thus can reduce alcohol cravings. Acamprosate’s mechanism of action in alcoholism is also not completely understood. It is thought to interact with glutamate and GABA neurotransmitter systems, restoring the balance of neuronal excitation and inhibitions that are altered by chronic alcohol exposure.
Planned Clinical Development by the NIAAA of Gabapentin Enacarbil in Patients with AUD. Under our clinical trial agreement with the NIAAA, the NIAAA plans to conduct a clinical trial evaluating HORIZANT in patients with AUD. The clinical trial is planned to be a randomized, double-blind, placebo-controlled clinical trial of the safety and efficacy of HORIZANT in patients with AUD but who are abstinent at treatment initiation. The proposed clinical trial is expected to have a treatment period of six months and to enroll approximately 350 patients. In the fourth quarter of 2014, representatives from XenoPort and the NIAAA conducted a pre-
12
Table of Contents
investigational new drug, or IND, meeting with the FDA. Based on this meeting, we believe that it may be possible for the proposed clinical trial to support a potential supplemental new drug application, or sNDA, for HORIZANT as a potential treatment for AUD if the results from the clinical trial are robust and compelling, and if there is additional confirmatory evidence from the literature or other sources to support the findings. In connection with the clearance by the FDA of the NIAAA’s IND application, the NIAAA has indicated that the proposed clinical trial will begin in the first half of 2015 and that the results may be available by the end of 2016. Under the terms of our agreement with the NIAAA, we will supply clinical trial material and the NIAAA will conduct and pay all other expenses associated with the proposed clinical trial of HORIZANT. We will have access to study results generated under the proposed clinical trial to support any potential regulatory filings, including a potential sNDA, for HORIZANT as a potential treatment for AUD.
XP21279 — A Transported Prodrug of Levodopa
XP21279 is a Transported Prodrug of levodopa that may be a potential treatment of patients with idiopathic Parkinson’s disease. We hold a composition-of-matter patent and a formulation patent in the United States on XP21279 that expire at the earliest in 2025 and hold patents or pending patent applications directed to the XP21279 methods of synthesis and use in the United States. We have also filed applications directed to the XP21279 composition of matter and methods of synthesis and use in other jurisdictions. At this time, we plan to continue development of XP21279 to the extent our resources permit or we enter into a partnership or collaboration with a third party.
Parent Drug Background
Patients with Parkinson’s disease have a deficiency of the neurotransmitter dopamine resulting from neuronal degeneration within certain nerve cells in an area of the brain collectively known as the substantia nigra. Levodopa is an immediate precursor of dopamine that, unlike dopamine, readily crosses the blood-brain barrier. When administered in conjunction with carbidopa (and, in some cases, with benzerazide or carbidopa and entacapone), levodopa is protected from rapid metabolism by enzymes that are found throughout the body outside of the brain. Once levodopa crosses the blood-brain barrier it is able to be converted to dopamine at its desired site of action in the brain. Levodopa is widely viewed as one of the most effective treatments of Parkinson’s disease, and virtually all patients with Parkinson’s disease ultimately require it. However, levodopa has many undesirable pharmacokinetic characteristics, including its rapid breakdown by gastric and other peripheral enzymes, a narrow absorption window within the GI tract and a short duration of exposure in blood after oral dosing that leads to the fluctuation of drug plasma concentrations upon frequent dosing. The poor colonic absorption of levodopa has precluded the development of a satisfactory sustained-release formulation that would prolong absorption beyond the small intestine.
Our Transported Prodrug
We believe that XP21279 has the potential to improve upon the limitations of levodopa. XP21279 is designed to engage natural nutrient transport mechanisms located throughout the length of the GI tract and then be rapidly converted to levodopa by the body’s naturally occurring enzymes. In addition to levodopa, the metabolic breakdown products of XP21279 are substances with favorable safety characteristics. Because XP21279 is designed to be well absorbed from the lower GI tract, we believe that it can be formulated for sustained release, thus reducing fluctuations of levodopa levels in the bloodstream. From December 2002 to December 2004, we were engaged in a collaboration with ALZA, now a subsidiary of Johnson & Johnson, to jointly develop Transported Prodrugs of levodopa. In March 2005, ALZA relinquished all rights to such Transported Prodrugs, subject to a low single-digit royalty upon net sales of certain product candidates if they are ultimately commercialized.
Development Program
We have conducted three Phase 1 clinical trials of XP21279 that included a total of 82 healthy volunteers. The trials evaluated the PK profile of different formulations of XP21279 administered with carbidopa compared
13
Table of Contents
to a combination of levodopa/carbidopa. The results of these Phase 1 clinical trials indicated that XP21279/carbidopa was well absorbed and rapidly converted to levodopa. Exposure to the intact Transported Prodrug was negligible. Data from the trials indicated that compared to the PK data of levodopa/carbidopa, XP21279/carbidopa was associated with a decreased peak-to-trough ratio of levodopa blood levels over 24 hours compared to levodopa/carbidopa. XP21279 was generally well tolerated, with no serious adverse events reported in these trials.
In 2010, we reported preliminary results from an open-label, crossover, Phase 1 clinical trial of XP21279 administered with carbidopa in ten Parkinson’s disease patients who were sequentially administered levodopa/carbidopa three or four times per day for 14 days followed by administration of XP21279/carbidopa three times per day for 14 days. Efficacy assessments at the end of each treatment period showed improvements with XP21279 over levodopa. However, because the trial was not blinded (i.e., subjects knew what treatment was administered), the results of the efficacy analyses must be viewed with caution. XP21279 was well tolerated.
In 2011, we reported preliminary results of a Phase 2, randomized, crossover clinical trial of XP21279 that compared optimized treatment with either SINEMET (immediate-release levodopa/carbidopa) or XP21279 co-formulated with carbidopa (XP21279/CD) in advanced Parkinson’s disease patients with motor fluctuations. The trial enrolled patients with Parkinson’s disease at 12 U.S. sites who were on a stable regimen of SINEMET dosed four or five times per day. Subjects were required to have “off time” in at least half of the inter-dose intervals between the first and last daily doses of SINEMET and an average daily “off time” greater than or equal to two hours during the three-day baseline assessment period. The trial consisted of an open-label, crossover optimization phase followed by a double-blind, crossover treatment phase. Thirty-five subjects entered the open-label phase of the trial, during which doses of SINEMET and XP21279/CD were each optimized for two weeks in a random order using the same protocol-specified guidelines. Results of the PK analysis from the trial showed that subjects had significantly lower variation in levodopa blood levels over a 16-hour time period while taking XP21279/CD as compared to SINEMET. However, in the primary efficacy endpoint of the trial, the improvement with XP21279/CD was not statistically better than the improvement seen with optimized SINEMET dosed four or five times per day during the double-blind phase of the trial. All treatment-emergent adverse events were mild to moderate in severity. There were no serious adverse events.
Target Indication
Parkinson’s Disease
Background on Parkinson’s Disease. Parkinson’s disease is a motor system disorder that results from the loss of dopamine-producing nerve cells in the brain. Dopamine is a chemical that is naturally produced by the body. It is responsible for smooth, coordinated function of the body’s muscles and movement. When approximately 80% of dopamine-producing cells are damaged, the symptoms of Parkinson’s disease appear. The primary symptoms of Parkinson’s disease are tremor or shaking, slowness of movement, rigidity or stiffness and difficulty with balance.
Potential Market. It is estimated at least 500,000 people in North America are living with Parkinson’s disease. According to the National Institute of Neurological Disorders and Stroke, Parkinson’s disease usually affects people over 50 years of age.
Current Treatments. In the United States, products approved for the treatment of advanced idiopathic Parkinson’s Disease include DUOPA (levodopa/carbidopa suspension) from AbbVie and RYTARY, an extended-release oral capsule formulation of levodopa/carbidopa from Impax Laboratories, Inc., which were both recently FDA approved; SINEMET (levodopa/carbidopa) that is distributed in the United States by Bristol Meyers Squibb Company; STALEVO, a combination therapy of levodopa/carbidopa/entacapone that is marketed in the United States by Novartis; and generic levodopa/carbidopa drugs. In addition, there are a number of products that are approved for idiopathic Parkinson’s disease and administered as adjunctive therapy to levodopa/carbidopa in patients with advanced idiopathic Parkinson’s disease. These include AZILECT (rasagiline mesylate), marketed by TEVA, and dopamine agonists such as NEUPRO (a rotigotine transdermal system) marketed by UCB, MIRAPEX (pramipexole) marketed by Boehringer Ingelheim, REQUIP (ropinirole) and REQUIP XL (ropinirole extended-release tablets) marketed by GSK and generic dopamine agonists.
14
Table of Contents
Potential Clinical Development of XP21279 in Parkinson’s Disease. In 2012, we conducted an End-of-Phase 2 meeting with the FDA in which we received feedback that a proposed development program for XP21279 could support a potential NDA submission under Section 505(b)(2) of the FDCA. Based on our discussions with the FDA, we believe that a single, pivotal, Phase 3 clinical trial comparing optimized doses of XP21279 to SINEMET, along with an open-label safety study, could form the basis for an NDA submission as a potential treatment for advanced idiopathic Parkinson’s disease. The FDA provided specific guidance on the proposed design of the pivotal trial and confirmed that efficacy and safety data from this study could be included in the product label.
XP21279 Development, Commercialization and Partnering Strategy
To the extent that our resources permit, we may develop XP21279 further as a potential treatment for patients with idiopathic Parkinson’s disease, or we may enter into an agreement with a third party for such development.
Current and Former Partnerships and Collaborations
Indivior PLC
In May 2014, we entered into an exclusive license agreement with Reckitt, which became effective on June 19, 2014. Subsequent to our agreement, the pharmaceutical division of Reckitt was divested into a new business entity, Indivior PLC, which is now our exclusive licensee. Under the terms of our agreement, we granted exclusive, world-wide rights to develop and commercialize pharmaceutical products containing AP and other prodrugs of baclofen or R-baclofen, or AP Products, for all indications, subject to our right of first negotiation to collaborate to develop and commercialize AP Products for non-addiction indications. In exchange for these rights, we received an upfront, non-refundable cash payment of $20.0 million in June 2014 and also received an additional $5.0 million in July 2014 after delivery of specified materials to our exclusive licensee, both of which were recognized in the third quarter of 2014. We are also eligible to receive aggregate cash payments of up to $120.0 million upon Indivior’s achievement of certain contingent event-based payments, of which $70.0 million are regulatory and development-based and $50.0 million are commercialization-based. In addition, we are entitled to receive tiered double-digit royalty payments of up to the mid-teens on a percentage basis on potential future net sales of AP Products in the United States, and high single-digit royalty payments on potential future net sales of AP Products outside the United States. Indivior has indicated that it intends to conduct a Phase 2 clinical trial of AP as a potential treatment for patients with AUD in the second quarter of 2015.
Astellas Pharma Inc.
In December 2005, we entered into an agreement in which we licensed to Astellas exclusive rights to develop and commercialize gabapentin enacarbil in Japan, Korea, the Philippines, Indonesia, Thailand and Taiwan. Under the terms of this agreement, we received an initial license payment of $25.0 million and have subsequently received $40.0 million in milestone payments as of December 31, 2014. As of February 2015, we remain eligible to receive potential clinical and regulatory contingent payments totaling up to an additional $20.0 million. In May 2013, Astellas informed us that they did not have plans for commercialization of REGNITE in the countries other than Japan in their territory. As a result, all rights to gabapentin enacarbil in Korea, the Philippines, Indonesia, Thailand and Taiwan reverted to us. Gabapentin enacarbil, known as REGNITE in Japan, has been approved by the MHLW and is being commercialized by Astellas in Japan. We are entitled to receive percentage-based high-teen royalties on net sales of REGNITE in Japan, with the royalties recognized when royalty payments are received by us. We have received and recognized $0.6 million in royalties for the year ended December 31, 2014 for net sales of REGNITE in Japan. Astellas is solely responsible for the manufacturing of REGNITE to support its development and commercialization in Japan. Astellas may terminate the agreement at its discretion; in such event, all REGNITE product rights would revert to us and we would be entitled to specified transition assistance from Astellas.
15
Table of Contents
Glaxo Group Limited
In February 2007, we entered into an exclusive collaboration agreement with GSK to develop and commercialize gabapentin enacarbil in all countries of the world excluding the Astellas territory. In November 2010, we amended and restated our collaboration agreement with GSK, pursuant to which we acquired all rights to gabapentin enacarbil outside of the United States previously granted to GSK and obtained the right, but not the obligation, to pursue development of HORIZANT for: (i) the potential treatment of diabetic peripheral neuropathy; (ii) the potential treatment of PHN to the extent that a product label would reflect a superiority claim over a currently approved drug; and (iii) any additional indications in the United States. GSK remained responsible for further development and regulatory matters with respect to HORIZANT for the potential management of PHN and manufacturing and commercialization of HORIZANT in the United States for all indications. In November 2012, we reached an agreement with GSK to terminate our collaboration agreement pursuant to a termination and transition agreement. The termination and transition agreement also provided for a mutual release of claims and resolved all ongoing litigation between the parties.
Under the terms of the termination and transition agreement, during a transition period that ended on April 30, 2013, GSK continued to exclusively commercialize, promote, manufacture and distribute HORIZANT in the United States. We were not responsible for any losses associated with the terminated collaboration agreement, were no longer eligible to receive any further milestone payments from GSK and did not receive any revenue or incur any losses from GSK’s sales of HORIZANT during the transition period. GSK continued to fully fund the costs associated with the management and conduct of clinical studies initiated by GSK prior to the date of the termination and transition agreement. In addition, prior to the end of the transition period, GSK provided to us inventory of gabapentin enacarbil in GSK’s possession that was not required for use by GSK in the manufacture of HORIZANT. In exchange for such inventory, we will make annual payments to GSK of $1.0 million for six years beginning in 2016. Following the transition period, we assumed all responsibilities for further development, manufacturing and commercialization of HORIZANT in the United States. GSK also continued to supply us with HORIZANT through October 2013 on pricing terms established under the termination and transition agreement, and also performed certain validation work with respect to the 300 mg dosage form of HORIZANT and other services for us through December 2013. In connection with the termination and transition agreement, pursuant to a separate stock purchase agreement entered into between us and GSK, GSK purchased an aggregate of $40.0 million of our common stock in November and December of 2012.
Patents and Proprietary Rights
We will be able to protect our technology from unauthorized use by third parties only to the extent that our technology is covered by valid and enforceable patents or effectively maintained as trade secrets and able to be utilized without infringing the proprietary rights of others. Our success in the future will depend in part on obtaining and maintaining patent protection for our technologies, product candidates and marketed products. Accordingly, patents and other proprietary rights are essential elements of our business. Our policy is to actively seek in the United States and selected foreign countries patent protection for novel technologies and compositions of matter that are commercially important to the development of our business.
Issued U.S. and foreign patents generally expire 20 years after filing. We hold a number of issued patents in the United States, including composition-of-matter patents for HORIZANT/gabapentin enacarbil, and for our XP23829, AP and XP21279 product candidates. We also have a number of pending patent applications in the United States relating to HORIZANT/gabapentin enacarbil, XP23829, AP and XP21279. Of the issued U.S. patents that we hold, many are related to compounds, pharmaceutical compositions containing the compounds and therapeutic methods of using the compounds and compositions. We further hold a number of issued U.S. patents related to methods of synthesis, proteomics methodology and screening methodology that could be useful in connection with HORIZANT and our product candidates. In addition, we hold a number of issued foreign national patents and pending patent applications, and we have pending Patent Cooperation Treaty international patent applications that preserve our rights to pursue national patent rights outside of the United States and pending European regional patent applications that permit us to pursue patent rights in a
16
Table of Contents
number of European member countries. The claims in these various patents and patent applications are directed to compositions of matter, including claims covering product candidates, lead compounds and key intermediates, pharmaceutical compositions, methods of use and processes for making our compounds, along with methods of design, synthesis, selection and use of Transported Prodrugs in general and to our research and development programs in particular.
The patent rights relating to the HORIZANT prodrug (gabapentin enacarbil) and its synthesis, as well as formulations and methods of its use are owned by us and include a number of issued U.S. patents with expiration dates ranging from 2022 to 2029, as well as a number of pending U.S. patent applications with similar potential expiration dates. We have petitioned the USPTO to grant a U.S. patent term extension on a composition-of-matter patent for the HORIZANT prodrug, which could extend product-specific exclusivity under that patent from 2022 to mid-2025. In addition, we hold a granted U.S. patent directed to the crystalline form of the HORIZANT prodrug that has an expiration date of 2026. Six of our issued U.S. patents are listed in the Orange Book for HORIZANT. Two of the Orange Book-listed patents have expiration dates in 2022, one has an expiration date in 2025, two have expiration dates in 2026 and one has an expiration date in 2029. Additionally, the FDA granted new chemical entity data exclusivity for HORIZANT that extends to April 2016, and orphan drug (data) exclusivity for use of HORIZANT in the management of PHN that extends to June 2019. In Japan, the patent rights relating to the REGNITE prodrug, its synthesis, formulations and methods of use are also owned by us. The Japanese Patent Office granted a term extension on our composition-of-matter patent, which now extends to 2025 for REGNITE. We believe that in all countries in which we hold or have licensed rights to patents or patent applications related to HORIZANT, REGNITE and gabapentin enacarbil, the composition-of-matter patents on the gabapentin active pharmaceutical ingredient, or API, itself have all expired. For XP23829, we own an issued U.S. composition-of-matter patent that has an expiration date of 2029. For AP, we hold issued U.S. composition-of-matter patents that have expiration dates ranging from 2024 to 2025. For XP21279, we own an issued U.S. composition-of-matter patent that has an expiration date of 2025. Although third parties may challenge our rights to, or the scope or validity of, our patents, to date, other than the European opposition described below, we have not received any communications from third parties challenging our patents or patent applications covering HORIZANT, REGNITE or our product candidates.
In 2008, a law firm, on behalf of an undisclosed client, filed an opposition against the grant of one of our European patent applications covering gabapentin enacarbil. The European Patent Office, or EPO, in opposition proceedings in 2010, undertook a formal review of the issues raised by the opponent and decided to maintain the grant of our European patent covering the composition of matter of gabapentin enacarbil. While the law firm that filed the opposition initially appealed the ruling on behalf of the undisclosed client, that appeal was withdrawn in 2010. The base composition-of-matter patent on gabapentin, the parent drug of HORIZANT/REGNITE/gabapentin enacarbil, expired in 2000; however, through September 2004, Pfizer continued to offer a proprietary gabapentin product (NEURONTIN) under a patent covering a process to make gabapentin with fewer contaminants. This formulation patent, which has an expiration date of 2017 and is listed in the Orange Book, has been the subject of ongoing litigation between Pfizer and several generic manufacturers, including Actavis, Ivax Pharmaceuticals, Inc. and Teva. Pfizer now markets generic gabapentin through its Greenstone Ltd. Subsidiary. Actavis and Teva, along with many other generic companies, also currently market generic versions of gabapentin. In 2011, the litigation was settled, and Pfizer granted a license to Teva, Ivax and Actavis (Purepac unit) to make and sell gabapentin under the formulation patent. We have not been a party to this litigation, and we believe that the manufacturing process for gabapentin enacarbil does not infringe the patent that was the subject of this litigation. Since the Pfizer settlement apparently did not prevent the sale of gabapentin by other generic manufacturers, we and/or Astellas are not limited in our choices of potential suppliers for the gabapentin parent drug.
Certain product candidates that we develop may be submitted to the FDA for approval under Section 505(b)(2) of the Food, Drug and Cosmetic Act, or FDCA, which was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act. Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from studies not conducted by, or for, the applicant and for which the applicant has not obtained a right of reference. In accordance with the Hatch-Waxman Act, such NDAs may be required to include certifications, known as
17
Table of Contents
Paragraph IV certifications, that certify that any patents listed in the Orange Book, with respect to any product referenced in the Section 505(b)(2) application, are invalid, unenforceable or will not be infringed by the manufacture, use or sale of the product that is the subject of the Section 505(b)(2) application. Under the Hatch-Waxman Act, the holder(s) of any Orange Book patent(s) that the Section 505(b)(2) application certifies over may file a patent infringement lawsuit after receiving notice of the Paragraph IV certification. Filing of a patent infringement lawsuit within 45 days of an Orange Book patent owner’s receipt of notice triggers a one-time, automatic, up to 30-month stay of the FDA’s ability to approve the 505(b)(2) application. Accordingly, we may invest a significant amount of time and expense in the development of one or more products only to be subject to significant delay and patent litigation before such products may be commercialized, if at all. We are not aware of any unexpired Orange Book-listed patents covering MMF, R-baclofen or levodopa, which molecules are the pharmaceutically active metabolites of XP23829, AP and XP21279, respectively. However, we are aware that there are six patents listed in the Orange Book for TECFIDERA that have expiration dates ranging from 2018 to 2028. If we file a 505(b)(2) application referencing TECFIDERA data, we will have to provide a Paragraph IV certification against any such listed patents if they have not expired by that time. Although we believe that an approved XP23829 product would not infringe the claims in the listed patents, that the listed patents would expire before any such potential 505(b)(2) submission would otherwise occur based on our current anticipated product development timelines, and/or that the claims in the listed patents are invalid and/or unenforceable, if we provide a Paragraph IV certification, the patent holder may file a patent infringement lawsuit, triggering up to a 30-month stay on the FDA’s ability to approve any 505(b)(2) application submitted by us.
We also rely on trade secret protection and confidentiality agreements to protect our proprietary know-how that is not patentable, processes for which patents are difficult to enforce and any other elements of our drug discovery process that involve proprietary know-how and technology that is not covered by patent applications, especially where patent protection is not believed to be appropriate or obtainable. We require all of our employees, consultants and advisors to enter into confidentiality agreements. Where it is necessary to share our proprietary information or data with outside parties, our policy is to make available only that information and data required to accomplish the desired purpose and only pursuant to a duty of confidentiality on the part of those parties.
We do not own or operate manufacturing facilities for the production of clinical or commercial quantities of HORIZANT, REGNITE or any of our product candidates. We also have limited management expertise in commercial supply operations. For our product candidates, we have relied on, and we expect to continue to rely on, a limited number of third-party drug substance and drug product manufacturers. We do not have commercial supply agreements with any of these third parties for our product candidates, and our agreements with these parties are generally terminable at will by either party at any time. For HORIZANT, GSK supplied us with HORIZANT through October 2013, and also performed certain validation work with respect to the 300 mg dosage form of HORIZANT and other services for us through December 2013. Currently, we rely on Patheon Pharmaceuticals Inc. as our single source supplier of clinical and commercial product pursuant to a commercial supply agreement. Our purchase price for the manufacture and supply of HORIZANT is volume-based. Both parties have early termination rights with notice periods of varying lengths for various causes, including for the counterparty’s uncured breach. Patheon also has the right to terminate the supply agreement upon 18 months prior notice if we sell, assign or otherwise transfer rights to HORIZANT to a competitor of Patheon. If, for any reason, Patheon or these third-party drug substance and drug product manufacturers are unable or unwilling to perform under our agreements or enter into new agreements, we may not be able to locate alternative manufacturers or enter into favorable agreements with them. In addition, qualifying and then successfully registering a new HORIZANT manufacturer with the FDA is time consuming and could require at least about 18 months to carry out. Our inability to acquire sufficient quantities of HORIZANT could result in future stockouts and existing patients not having access to drug product. Any inability to acquire sufficient quantities of our product candidates in a timely manner from these third parties could delay clinical trials and prevent us from developing and commercializing these product candidates in a cost-effective manner or on a timely basis, if at all.
18
Table of Contents
If we are unable to manufacture or contract to manufacture sufficient quantities of HORIZANT, the commercialization of HORIZANT could be impaired or interrupted. For example, manufacturing delays resulted in a stockout of HORIZANT in 2013. As a result of the manufacturing delays, our full commercial promotion of HORIZANT was delayed for approximately one month. If we or Patheon experience other delays or issues, our ability to commercialize HORIZANT could be further impaired. As part of the termination and transition agreement, GSK provided its inventory of gabapentin enacarbil drug substance to us. Although the inventory of drug substance has reached the end of its specified shelf life, we believe that such inventory will remain in specification and will be usable, or in the alternative, we believe the drug substance can be re-crystallized into usable form. GSK relied on a single source supplier of gabapentin enacarbil drug substance, and its agreement with such manufacturer has expired. If we are incorrect about the usability of the gabapentin enacarbil drug substance, are unable to have it meet specifications upon re-crystallization, or are unable to enter into an agreement with the contract manufacturer or qualify an alternative manufacturer, we may be limited in the amount of HORIZANT we could manufacture and the commercialization of HORIZANT could be impaired or interrupted. Under the terms of our license agreement with Astellas, Astellas is solely responsible for the manufacture of REGNITE to support its commercialization in Japan. To our knowledge, Astellas is currently relying on single source suppliers for commercial supplies of REGNITE. As a result, if Astellas fails to manufacture or contract to manufacture sufficient quantities of REGNITE, commercialization of REGNITE could be impaired in Japan.
Under the terms of our license agreement with Indivior, Indivior is solely responsible for the manufacture of AP to support its development and commercialization of AP Products. If Indivior fails to establish AP manufacturing capabilities itself and/or to identify and qualify one or more suitable manufacturers of AP, or if such manufacturers terminate their agreements with Indivior and Indivior is otherwise unable to manufacture or contract to manufacture sufficient quantities of AP, development and commercialization of AP Products could be impaired or delayed.
We rely on a single source supplier of MMF, which is used to make XP23829, under purchase orders issued from time to time. We are aware of several alternative suppliers of MMF, and we believe at least one alternative manufacturer could potentially supply MMF in the event that our supplier determines to not sell MMF to us at a price that is commercially attractive. If we are unable to qualify an alternative supplier of MMF, this could delay the development of, and impair our ability to commercialize, XP23829.
We rely on both domestic and ex-U.S. third-party suppliers of XP23829 active pharmaceutical ingredient and finished drug product through a three-step chain of manufacturing services and supply agreements, and we have purchased XP23829 formulated in different forms from multiple suppliers at specified transfer prices under quotations agreed upon by the parties as part of master services agreements. In the event that such suppliers or manufacturers terminate our respective agreements under specified circumstances, we would not be able to manufacture drug substance or XP23829 until alternative suppliers are identified and qualified, which could also delay the clinical development of, and impair our ability to commercialize, XP23829.
We rely on a single source supplier of levodopa, which is used to make XP21279, under purchase orders issued from time to time. We are aware of several alternative suppliers of levodopa, and we believe at least one alternative manufacturer could potentially supply levodopa in the event that our supplier determines to not sell levodopa to us at a price that is commercially attractive. If we are unable to qualify an alternative supplier of levodopa, this could further delay the development of, and impair our ability to commercialize, XP21279.
We rely on a single source supplier of XP21279 drug substance under a manufacturing services and product supply agreement. In the event that such supplier terminates the agreement under specified circumstances, we would not be able to manufacture drug substance until a qualified alternative supplier is identified and qualified, which could also further delay the development of, and impair our ability to commercialize, this product candidate.
We have purchased XP21279 formulated in sustained-release tablets from a single source supplier at specified transfer prices under quotations agreed upon by the parties as part of a master services agreement. We have also qualified another supplier for the manufacture of XP21279 with carbidopa bi-layer tablets to be supplied under quotations agreed upon by the parties as part of a master services agreement. In the event that
19
Table of Contents
either supplier terminates its agreement under specified circumstances for the manufacture of XP21279 sustained-release tablets or carbidopa bi-layer tablets, we would not be able to manufacture XP21279 until an alternative supplier is qualified. This could further delay the development of, and impair our ability to commercialize, XP21279.
Since inception, we have devoted a significant amount of resources to develop our product candidates. For the years ended December 31, 2014, 2013 and 2012, we recorded $23.7 million, $33.3 million and $42.9 million, respectively, in research and development expenses. As part of a restructuring that we implemented in March 2010, we eliminated our discovery research department, which prevents us from being able to discover additional product candidates at this time. In addition, we have implemented subsequent reductions in force as a result of our suspension of our clinical development activities for XP21279 and our previous decision to terminate further investment in AP, which limits the number of product candidates that we can develop.
The pharmaceutical and biotechnology industries are intensely competitive. HORIZANT/REGNITE compete, and our product candidates that may obtain approval will likely compete, with existing drugs and therapies. There are many pharmaceutical companies, biotechnology companies, public and private universities, government agencies and research organizations actively engaged in research and development of products targeting the same markets as HORIZANT/REGNITE and our product candidates. Many of these organizations have substantially greater financial, technical, manufacturing and marketing resources than we have. Several of them have developed or are developing therapies that could be used for treatment of the same diseases as HORIZANT/REGNITE or diseases that we are targeting in our clinical development programs. In addition, many of these competitors have significantly greater commercial infrastructures than we have.
We also face competition from manufacturers of generic drugs. Generic competition often results in decreases in the prices at which branded products can be sold, particularly when there is more than one generic available in the marketplace as in the case with generics competing with HORIZANT. In addition, legislation enacted in the United States allows for, and in a few instances in the absence of specific instructions from the prescribing physician mandates, the dispensing of generic products rather than branded products where a generic version is available.
HORIZANT/REGNITE and our product candidates may also compete in the future with new products currently under development by others. In this regard, there may be other compounds of which we are not aware or that are at an earlier stage of development and may compete directly with HORIZANT/REGNITE or our product candidates. Any products that we develop are likely to be in a highly competitive market, and many of our competitors may succeed in developing products that may render our products obsolete or noncompetitive. In particular, our marketed products and product candidates face competition as described below:
| • | HORIZANT/REGNITE. Products that we believe compete with HORIZANT in the United States include the following drugs approved for the treatment of moderate-to-severe primary RLS: MIRAPEX (pramipexole) from Boehringer Ingelheim and generic pramipexole; REQUIP (ropinirole) from GSK and generic ropinirole; and NEUPRO (a rotigotine transdermal system), a dopamine agonist patch from UCB. In Japan, we believe that REGNITE competes with SIFROL (pramipexole) and the UCB rotigotine transdermal system, which was launched in February 2013 by Otsuka, which has exclusive rights to market in Japan. Products that we believe compete with HORIZANT in the United States for the management of PHN include drugs that act on the same target as HORIZANT, such as GRALISE (once-daily formulation of gabapentin) from Depomed, LYRICA (pregabalin) and NEURONTIN (gabapentin) from Pfizer and generic gabapentin. HORIZANT could also experience competition from a capsaicin patch (marketed as QUTENZA by Acorda Therapeutics, Inc.) and transdermal patches containing the anesthetic known as lidocaine (marketed as LIDODERM by Endo). In addition, while not a treatment for shingles or PHN, HORIZANT could experience competition from a shingles vaccine, known as |
20
Table of Contents
| ZOSTAVAX by Merck & Co., that is a live attenuated vaccine to help prevent shingles. There is also a shingles vaccine in late-stage development, known as HZ/su, which is being developed by GSK. |
| • | XP23829. Products that could compete with XP23829, our product candidate that is a prodrug of MMF for the treatment of psoriasis include topical therapies, oral systemic therapies and biological therapies. Topical therapies include corticosteroids, anthrolin and synthetic vitamin D and vitamin A. Oral systemic agents include acitretin, cyclosporine, methotrexate and OTEZLA (apremilast) from Celgene Corporation. Biological therapies that are approved to treat psoriasis include COSENTYX (secukinumab), marketed by Novartis; ENBREL (etanercept), marketed by Amgen Inc.; HUMIRA (adalimumab), marketed by AbbVie; REMICADE (infliximab), marketed by Johnson & Johnson and Merck & Co.; and STELARA (ustekinumab), marketed by Johnson & Johnson. There are also a number of possible competitive products that are in late-stage product development in the United States, including brodalumab, CF-101, ixekizumab, tildrakizumab and tofacitinib that are being developed by Amgen, Can-Fite BioPharma Ltd., Eli Lilly and Company, Sun Pharmaceutical Industries Ltd./Merck & Co. and Pfizer, respectively. |
Products that could compete with XP23829 for the treatment of relapsing forms of MS include oral and injectable agents that are approved in the United States for the treatment of relapsing forms of MS. These include oral agents such as AUBAGIO (teriflunomide), marketed by Genzyme Corporation, a Sanofi company; GILENYA (fingolimod), marketed by Novartis; and TECFIDERA (dimethyl fumarate), marketed by Biogen. In addition, several injectable products are approved in the United States and include AVONEX (interferon beta 1a), PLEGRIDY (peginterferon beta-1a) and TYSABRI (natralizumab), marketed by Biogen; BETASERON (interferon beta 1b) and EXTAVIA (interferon beta 1b), marketed by Bayer AG/Novartis; COPAXONE (glatiramer acetate), marketed by Teva; REBIF (interferon beta 1a), marketed by Merck Serono S.A.; and LEMTRADA (alemtuzumab), marketed by Genzyme/Sanofi-Aventis. There are also potentially competitive products in late-stage product development in the United States, including daclizumab high-yield process (DAC HYP) from AbbVie/Biogen, laquinimod from Teva, RPC1063 from Receptos, FP187 from Forward Pharma A/S, and ocrelizumab from Roche Holding AG.
| • | Gabapentin enacarbil. Products that could compete with gabapentin enacarbil for the treatment of AUD include ANTABUSE (disulfiram), marketed by TEVA, REVIA and VIVITROL, oral and injectable formulations of naltrexone, marketed by Barr Pharmaceuticals, Inc. and Alkermes, Inc., respectively, and CAMPRAL (acamprosate), marketed by Actavis Inc. |
| • | XP21279. If resources permit and we choose to develop XP21279 as a potential treatment for advanced idiopathic Parkinson’s Disease, products that could compete with XP21279, our product candidate that is a Transported Prodrug of levodopa, include: DUOPA (levodopa/carbidopa suspension) from AbbVie; RYTARY, an extended-release oral capsule formulation of levodopa/carbidopa from Impax, which were both recently FDA approved; SINEMET (levodopa/carbidopa) that is distributed in the United States by Bristol Meyers Squibb Company; STALEVO, a combination therapy of levodopa/carbidopa/entacapone that is marketed in the United States by Novartis and generic levodopa/carbidopa drugs. In addition, there are a number of products that are approved for idiopathic Parkinson’s disease and administered as adjunctive therapy to levodopa/carbidopa in patients with advanced idiopathic Parkinson’s disease. These include AZILECT (rasagiline mesylate), marketed by TEVA and dopamine agonists such as NEUPRO (a rotigotine transdermal system) marketed by UCB, MIRAPEX (pramipexole) marketed by Boehringer Ingelheim and REQUIP (ropinirole) and REQUIP XL (ropinirole extended-release tablets) marketed by GSK and generic dopamine agonists. Other therapies under development in the United States include extended-release formulations of levodopa/carbidopa such as DM-1992 and OS-320 being developed by Depomed and Osmotica, respectively. |
With respect to HORIZANT/REGNITE and our product candidates, we believe that our ability to successfully compete will depend on, among other things:
| • | the existence of competing or alternative products in the marketplace, including generic competition, and the relative price of those products; |
21
Table of Contents
| • | the efficacy, safety and reliability of HORIZANT/REGNITE and our product candidates compared to competing or alternative products; |
| • | product acceptance by physicians, other health care providers and patients, including, with respect to HORIZANT, our ability to overcome any negative market perception due to GSK’s prior promotional efforts and a lack of physician awareness; |
| • | our ability to obtain patent and/or other proprietary protection for our products and technologies; |
| • | obtaining reimbursement for our products in approved indications; |
| • | our ability to complete clinical development and obtain regulatory approvals for our product candidates, and the timing and scope of regulatory approvals; |
| • | our ability to comply with applicable laws, regulations and regulatory requirements and restrictions with respect to the commercialization of our products, including with respect to any changed or increased regulatory restrictions; |
| • | our ability to supply commercial quantities of a product to the market; |
| • | our ability to attract and retain qualified product development and commercial personnel; and |
| • | our ability to successfully collaborate with pharmaceutical companies and/or third-party vendors in the development and commercialization of new products. |
With respect to our product candidates, we expect to compete on, among other things, product efficacy and safety, time to market, price, extent of adverse side effects experienced and convenience of treatment procedures. Our objective is to develop and commercialize new medicines with superior efficacy, convenience, tolerability and/or safety. To the extent that we are able to commercialize and develop new medicines, they are likely to compete with existing drugs that have long histories of effective and safe use and with new therapeutic agents. We expect that any product candidates that we commercialize with current or potential business partners or on our own will compete with existing, market-leading medicines.
Product Approval Process
The clinical testing, manufacturing, labeling, storage, distribution record keeping, advertising, promotion, import, export and marketing, among other things, of our product and product candidates are subject to extensive regulation by governmental authorities in the United States and other countries. The FDA, under the FDCA, regulates pharmaceutical products in the United States. The steps required before a drug may be approved for marketing in the United States generally include:
| • | preclinical laboratory tests and animal tests; |
| • | the submission to the FDA of an IND application for human clinical testing, which must become effective before human clinical trials commence; |
| • | adequate and well-controlled human clinical trials to establish the safety and efficacy of the product; |
| • | the submission to the FDA of an NDA; |
| • | FDA acceptance, review and approval of the NDA; and |
| • | satisfactory completion of an FDA inspection of the manufacturing facilities at which the product is made to assess compliance with current Good Manufacturing Practices, or cGMPs. |
The testing and approval process requires substantial time, effort and financial resources, and the receipt and timing of any approval is uncertain. The FDA may suspend clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
Preclinical studies include laboratory evaluations of the product candidate, as well as animal studies to assess the potential safety and efficacy of the product candidate. The results of the preclinical studies, together
22
Table of Contents
with manufacturing information and analytical data, are submitted to the FDA as part of the IND, which must become effective before clinical trials may be commenced. The IND will become effective automatically 30 days after receipt by the FDA, unless the FDA raises concerns or questions about the conduct of the trials as outlined in the IND prior to that time. In this case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can proceed.
Clinical trials involve the administration of the product candidates to healthy volunteers or patients with the disease to be treated under the supervision of a qualified principal investigator. Further, each clinical trial must be reviewed and approved by an independent institutional review board, or IRB, either centrally or individually at each institution at which the clinical trial will be conducted. The IRB will consider, among other things, ethical factors, the safety of human subjects and the possible liability of the institution.
Clinical trials typically are typically conducted in three sequential phases prior to approval, but the phases may overlap. These phases generally include the following:
Phase 1. Phase 1 clinical trials represent the initial introduction of a product candidate into human subjects, frequently healthy volunteers. In Phase 1, the product candidate is usually tested for safety, including adverse effects, dosage tolerance, absorption, distribution, metabolism, excretion and pharmacodynamics.
Phase 2. Phase 2 clinical trials usually involve studies in a limited patient population to (1) evaluate the efficacy of the product candidate for specific indications, (2) determine dosage tolerance and optimal dosage and (3) identify possible adverse effects and safety risks.
Phase 3. If a product candidate is found to be potentially effective and to have an acceptable safety profile in Phase 2 studies, the clinical trial program will be expanded to Phase 3 clinical trials to further demonstrate clinical efficacy, optimal dosage and safety within an expanded patient population at geographically dispersed clinical study sites.
Phase 4 clinical trials are conducted after approval to gain additional experience from the treatment of patients in the intended therapeutic indication and to document a clinical benefit in the case of drugs approved under accelerated approval regulations, or when otherwise requested by the FDA in the form of post-market requirements or commitments. Failure to promptly conduct any required Phase 4 clinical trials could result in withdrawal of approval.
The results of preclinical studies and clinical trials, together with detailed information on the manufacture, composition and quality of the product, are submitted to the FDA in the form of an NDA requesting approval to market the product. The NDA must be accompanied by a significant user fee payment. The FDA has substantial discretion in the approval process and may refuse to accept any application or decide that the data is insufficient for approval and require additional preclinical, clinical or other studies.
Generally, regulatory approval of a new drug by the FDA may follow one of three routes. The most traditional of these routes is the submission of a full NDA under Section 505(b)(1) of the FDCA. A second route, which is possible where an applicant chooses to rely in part on data generated or approvals obtained previously by other parties and/or on data described in published literature, is to submit an NDA consisting of both sponsor conducted studies and data from other sources described in Section 505(b)(2) of the FDCA. The final route is the submission of an Abbreviated New Drug Application for products that are shown to be pharmaceutically and therapeutically equivalent to previously approved drug products as permitted under Section 505(j) of the FDCA.
Both Section 505(b)(1) and Section 505(b)(2) applications are required by the FDA to contain full reports of investigations of safety and effectiveness. However, in contrast to a traditional NDA submitted pursuant to Section 505(b)(1) in which the applicant submits all of the data demonstrating safety and effectiveness, an application submitted pursuant to Section 505(b)(2) can rely upon previous findings by the FDA that the parent drug is safe and effective in that indication, and/or upon data described in published literature. As a consequence, the preclinical and clinical development programs leading to the submission of an NDA under Section 505(b)(2) may be less expensive to carry out and could potentially be concluded in a shorter period of time than programs required for a Section 505(b)(1) application. In its review of any NDA submission, however, the FDA has broad
23
Table of Contents
discretion to require an applicant to generate additional data related to safety, efficacy, or quality, and it is impossible to predict the number or nature of the additional data requirements or studies that may be necessary before the FDA will grant approval.
As a result of the termination and transition agreement with GSK, we are now the sponsor of the NDA for HORIZANT for the treatment of moderate-to-severe primary RLS in adults, the sponsor of the sNDA for HORIZANT for the management of PHN in adults and are responsible for leading the registration of HORIZANT for any additional indications in the United States. For our other product candidates that are undergoing clinical trials, we intend to follow the development pathway permitted under the FDCA that will maximize the commercial opportunities for these proprietary prodrugs. We are evaluating both Section 505(b)(1) and Section 505(b)(2) NDA routes for our proprietary prodrugs. In the event that we decide to utilize Section 505(b)(2) of the FDCA to pursue an approval of our proprietary prodrugs in indications for which the relevant parent drug has previously been approved, we will engage in discussions with the FDA to determine which, if any, portions of our development program can be modified.
In addition, for any XenoPort NDAs submitted under Section 505(b)(2) of the FDCA, the patent certification and related provisions of the Hatch-Waxman Act apply. In accordance with the Hatch-Waxman Act, such NDAs may be required to include Paragraph IV certifications that certify that any patents listed in the Orange Book with respect to any product referenced in the Section 505(b)(2) application are invalid, unenforceable or will not be infringed by the manufacture, use or sale of the product that is the subject of the Section 505(b)(2) application. Under the Hatch-Waxman Act, the holder of patents that the Section 505(b)(2) application references may file a patent infringement lawsuit after receiving notice of the Paragraph IV certification. Filing of a patent infringement lawsuit within 45 days of the patent owner’s receipt of notice triggers a one-time, automatic, stay of up to 30-months of the FDA’s ability to approve the 505(b)(2) application. A Section 505(b)(2) application may also not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired. For example, we are aware that there are six patents listed in the Orange Book for TECFIDERA that have expiration dates ranging from 2018 to 2028. If we file a 505(b)(2) application referencing TECFIDERA data, we will have to provide a Paragraph IV certification against any such listed patents if they have not expired by that time. Additionally, the FDA may require us to perform one or more additional clinical studies or measurements to support differences between our development products and the approved products being referenced in the application. The FDA may also reject our future Section 505(b)(2) submissions and require us to file such submissions under Section 505(b)(1) of the FDCA, which could cause delay and be considerably more expensive and time consuming.
Once the NDA submission has been accepted for filing, which occurs, if at all, 60 days after submission of the NDA, the FDA sets a PDUFA date that informs the applicant of the specific date by which the FDA intends to complete its review. This is typically 12 months from the date of submission of the NDA application. The review process is often extended by FDA requests for additional information or clarification. Before approving an NDA, the FDA may inspect the facilities at which the product is manufactured and will not approve the product unless the manufacturing facility complies with cGMPs and will also inspect clinical trial sites for integrity of data supporting safety and efficacy. The FDA may delay approval of an NDA if applicable regulatory criteria are not satisfied and/or the FDA requires additional testing or information. The FDA may require post-marketing testing and surveillance to monitor safety or efficacy of a product. The FDA will issue either an approval of the NDA or a complete response letter, detailing the deficiencies and information required in order for reconsideration of the NDA.
Federal and State Fraud and Abuse and Data Privacy and Security and Transparency Laws and Regulations
In addition to FDA restrictions on marketing of pharmaceutical products, federal and state healthcare laws restrict certain business practices in the biopharmaceutical industry. These laws include, but are not limited to, anti-kickback, false claims, data privacy and security, and transparency statutes and regulations. We are subject to these laws in association with the commercialization our products.
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration, directly or indirectly, to induce, or in return for, purchasing, leasing,
24
Table of Contents
ordering or arranging for the purchase, lease or order of any good, facility, item or service reimbursable under Medicare, Medicaid or other federal healthcare programs. The term “remuneration” has been broadly interpreted to include anything of value, including for example, gifts, discounts, the furnishing of supplies or equipment, credit arrangements, payments of cash, waivers of payment, ownership interests and providing anything at less than its fair market value. The Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers and formulary managers on the other. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly, and our practices may not in all cases meet all of the criteria for a statutory exception or safe harbor protection. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all of its facts and circumstances. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the statute has been violated. The intent standard under the Anti-Kickback Statute was amended by the Patient Protection and Affordable Care Act as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, PPACA, to a stricter intent standard such that a person or entity no longer needs to have actual knowledge of this statute or the specific intent to violate it in order to have committed a violation. In addition, PPACA codified case law that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act (discussed below). Further, the civil monetary penalties statute imposes penalties against any person or entity who, among other things, is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent.
The federal false claims laws prohibit, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment or approval to the federal government or knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. As a result of a modification made by the Fraud Enforcement and Recovery Act of 2009, a claim includes “any request or demand” for money or property presented to the U.S. government. Recently, several pharmaceutical and other healthcare companies have been prosecuted under these laws for, among other things, allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the companies’ marketing of the product for unapproved, and thus non-reimbursable, uses. The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created new federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payers and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of, or payment for, healthcare benefits, items or services.
In addition, we may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and its implementing regulations, imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to business associates — independent contractors or agents of covered entities that receive or obtain protected health information in connection with providing a service on behalf of a covered entity. HITECH also created four new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
25
Table of Contents
Additionally, the federal Physician Payments Sunshine Act within the PPACA, and its implementing regulations, require that certain manufacturers of drugs, devices, biologicals and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report information related to certain payments or other transfers of value made or distributed to physicians and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, the physicians and teaching hospitals and to report annually certain ownership and investment interests held by physicians and their immediate family members.
Also, many states have similar healthcare statutes or regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer. Some states require the posting of information relating to clinical studies. In addition, California requires pharmaceutical companies to implement a comprehensive compliance program that includes a limit on expenditures for, or payments to, individual medical or health professionals. We have adopted a comprehensive compliance program that we believe complies with California law. If our operations are found to be in violation of any of the health regulatory laws described above or any other laws that apply to us, we may be subject to penalties, including potentially significant criminal, civil and/or administrative penalties, damages, fines, disgorgement, individual imprisonment, exclusion of products from reimbursement under government programs, contractual damages, reputational harm, administrative burdens, diminished profits and future earnings and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations. To the extent that any of our products are sold in a foreign country, we may be subject to similar foreign laws and regulations, which may include, for instance, applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws and implementation of corporate compliance programs and reporting of payments or transfers of value to healthcare professionals.
Pharmaceutical Coverage, Pricing and Reimbursement
In both domestic and foreign markets, sales of HORIZANT/REGNITE and any approved products will depend in part on the availability of coverage and adequate reimbursement from third-party payers. Third-party payers include government health administrative authorities, managed care providers, private health insurers and other organizations. Patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third-party payers to reimburse all or part of the associated healthcare costs. Patients are unlikely to use our products unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our products. Sales of our products will therefore depend substantially, both domestically and abroad, on the extent to which the costs of our products will be paid by third-party payers. These third-party payers are increasingly focused on containing healthcare costs by challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the coverage and reimbursement status of newly approved healthcare product candidates. The market for our products and product candidates for which we may receive regulatory approval will depend significantly on access to third-party payers’ drug formularies, or lists of medications for which third-party payers provide coverage and reimbursement. The industry competition to be included in such formularies often leads to downward pricing pressures on pharmaceutical companies. Also, third-party payers may refuse to include a particular branded drug in their formularies or otherwise restrict patient access to a branded drug when a less costly generic equivalent or other alternative is available. Because each third-party payer individually approves coverage and reimbursement levels, obtaining coverage and adequate reimbursement is a time-consuming, costly and sometimes unpredictable process. We may be required to provide scientific and clinical support for the use of any product to each third-party payer separately with no assurance that approval would be obtained, and we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our products. This process could delay the market acceptance of any product and could have a negative effect on our future revenues and operating results. We cannot be certain that our products and our product candidates will be considered cost-effective. Because coverage and reimbursement determinations are made on a payer-by-payer basis, obtaining acceptable coverage and reimbursement from one payer does not guarantee we will obtain similar acceptable coverage or reimbursement from another payer. If we are unable to obtain coverage of, and adequate reimbursement and payment levels for, our product candidates from third-party payers, physicians may limit how much or under what circumstances they will prescribe or administer them and patients may decline to
26
Table of Contents
purchase them. This in turn could affect our ability to successfully commercialize our products and impact our profitability, results of operations, financial condition and future success.
In addition, in many foreign countries, particularly the countries of the European Union, the pricing of prescription drugs is subject to government control. In some non-U.S. jurisdictions, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the EU provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. We may face competition for our product candidates from lower-priced products in foreign countries that have placed price controls on pharmaceutical products. In addition, there may be importation of foreign products that compete with our own products, which could negatively impact our profitability.
In the United States and foreign jurisdictions, there have been, and we expect there will continue to be, a number of legislative and regulatory changes to the healthcare system that could affect our future results of operations as we begin to directly commercialize our products. In particular, there have been and continue to be a number of initiatives at the United States federal and state level that seek to reduce healthcare costs.
The recently enacted federal Drug Supply Chain Security Act, imposes new obligations on manufacturers of pharmaceutical products, among others, related to product tracking and tracing. Among the requirements of this new federal legislation, manufacturers will be required to provide certain information regarding the drug product to individuals and entities to which product ownership is transferred, label drug product with a product identifier, and keep certain records regarding the drug product. Further, under this new legislation, manufacturers will have drug product investigation, quarantine, disposition, and notification responsibilities related to counterfeit, diverted, stolen, and intentionally adulterated products, as well as products that are the subject of fraudulent transactions or which are otherwise unfit for distribution such that they would be reasonably likely to result in serious health consequences or death.
Furthermore, political, economic and regulatory influences are subjecting the healthcare industry in the United States to fundamental change. Initiatives to reduce the federal deficit and to reform healthcare delivery are increasing cost-containment efforts. We anticipate that Congress, state legislatures and the private sector will continue to review and assess alternative benefits, controls on healthcare spending through limitations on the growth of private health insurance premiums and Medicare and Medicaid spending, the creation of large insurance purchasing groups, price controls on pharmaceuticals and other fundamental changes to the healthcare delivery system. Any proposed or actual changes could limit or eliminate our spending on development projects and affect our ultimate profitability. For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care Education Reconciliation Act, collectively PPACA, was signed into law. PPACA has the potential to substantially change the way healthcare is financed by both governmental and private insurers. Among other cost containment measures, PPACA:
| • | established an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents; |
| • | revised the methodology by which rebates owed by manufacturers to the state and federal government for covered outpatient drugs under the Medicaid Drug Rebate Program are calculated; |
| • | increased the statutory minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program to 23.1% and 13% of the average manufacturer price for most branded and generic drugs, respectively; |
| • | extended the Medicaid Drug Rebate program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations; and |
27
Table of Contents
| • | established the Medicare Part D coverage gap discount program by requiring manufacturers to provide a 50% point-of-sale-discount off the negotiated price of applicable brand drugs to eligible beneficiaries during their coverage gap period as a condition for the manufacturers’ outpatient drugs to be covered under Medicare Part D. |
Other legislative changes have been proposed and adopted in the United States since the Affordable Care Act was enacted. In August 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers up to 2% per fiscal year, which went into effect in April 2013 and will remain in effect through 2024 unless additional Congressional action is taken. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, further reduced Medicare payments to several providers.
In addition, applicable reforms also occur on the local level, including, for example, two counties (King County, Washington and Alameda County, California) have enacted pharmaceutical manufacturer take-back ordinances/regulations that place primary responsibility for end-of-life management of drug products on drug producers. These new take-back programs will require the creation, administration, promotion and payment of costs including county administrative costs of administering and enforcing the programs.
In the future, there may continue to be additional proposals relating to the reform of the U.S. healthcare system, some of which could further limit the prices we are able to charge for our products, or the amounts of reimbursement available for our products. If future legislation were to impose direct governmental price controls and access restrictions, it could have a significant adverse impact on our business. Managed care organizations, as well as Medicaid and other government agencies, continue to seek price discounts. Some states have implemented, and other states are considering, price controls or patient access constraints under the Medicaid program, and some states are considering price-control regimes that would apply to broader segments of their populations that are not Medicaid-eligible. Due to the volatility in the current economic and market dynamics, we are unable to predict the impact of any unforeseen or unknown legislative, regulatory, payer or policy actions, which may include cost containment and healthcare reform measures.
Customers and Financial Information About Geographic Areas
In the United States, HORIZANT is sold to distributors who primarily distribute the product to pharmacies. In 2014, the principal distributors for HORIZANT were Cardinal Health, Inc., McKesson Corporation and AmerisourceBergen Corporation. In 2014, 95% of the HORIZANT product sales were derived from these three distributors. We have standard industry agreements made in the ordinary course of business with these distributors, which include prompt payment discounts and various standard fee or rebate arrangements. Purchases are made on a purchase order basis.
Further information on our total revenues other than HORIZANT product sales is included in Note 2 to our financial statements.
We lease approximately 103,000 square feet of office and laboratory space in an office building in Santa Clara, California. In October 2012, we entered into a Second Amendment to Lease with SI 34 LLC, or Sobrato, with respect to our current office space at 3410 Central Expressway, Santa Clara, California, or, as amended, the 3410 Lease. In November 2014, we entered into a Third Amendment to Lease with Sobrato, extending the term of the 3410 Lease for an additional nine months, so that the 3410 Lease will expire on May 31, 2016. We had also leased approximately 59,000 square feet at an adjacent building at 3400 Central Expressway, Santa Clara, California, but we terminated the lease in February 2013, and the rent obligations expired in August 2013.
28
Table of Contents
As of December 31, 2014, we had 152 full-time employees, 43 of whom were engaged in product development activities. Sixty employees hold post-graduate degrees, including three with M.D. degrees, 13 with Ph.D. degrees and four with Pharm.D. degrees. Our employees are not represented by a collective bargaining agreement. We believe our relations with our employees are good.
Executive Officers of the Registrant
The following sets forth certain information regarding our executive officers as of February 13, 2015:
| Name |
Age | Position | ||||
| Ronald W. Barrett, Ph.D. |
59 | Chief Executive Officer and Director | ||||
| Vincent J. Angotti |
47 | Executive Vice President, Chief Operating Officer | ||||
| Gregory T. Bates, D.V.M. |
56 | Senior Vice President of Regulatory Affairs and Quality | ||||
| Gianna M. Bosko. |
45 | Senior Vice President, Chief Legal Officer and Secretary | ||||
| William G. Harris |
56 | Senior Vice President of Finance and Chief Financial Officer | ||||
| Richard K. Kim, M.D. |
44 | Senior Vice President, Clinical Development & Medical Affairs, Chief Medical Officer | ||||
Ronald W. Barrett is one of our founders and has served as our chief executive officer since September 2001. He served as our chief scientific officer from 1999 to 2001. Dr. Barrett has been a director since August 1999. From 1989 to 1999, he held various positions at Affymax Research Institute, a company employing combinatorial chemistry and high-throughput target screening for drug discovery, the most recent of which was senior vice president of research. Glaxo Wellcome plc, a pharmaceutical company, acquired Affymax Research Institute in 1995. Glaxo Wellcome subsequently merged with SmithKline Beecham plc, a pharmaceutical company, in 2000 to form GlaxoSmithKline plc, a pharmaceutical company. Prior to Affymax Research Institute, Dr. Barrett was a molecular pharmacologist in the Neuroscience Group at Abbott Laboratories, a healthcare company, from 1986 to 1989. Dr. Barrett received a B.S. from Bucknell University and a Ph.D. in pharmacology from Rutgers University. Dr. Barrett is a member of the board of directors of Concert Pharmaceuticals, Inc., a publicly-traded clinical-stage biopharmaceutical company.
Vincent J. Angotti has been our executive vice president, chief operating officer since June 2012. He was previously our senior vice president and chief commercialization officer from 2008 to 2012. From 2001 to 2008, he held several positions with Reliant Pharmaceuticals, Inc., a pharmaceutical company, the most recent of which was senior vice president of sales and marketing. GlaxoSmithKline acquired Reliant Pharmaceuticals in 2008. Prior to Reliant Pharmaceuticals, from 1991 to 2001, Mr. Angotti held several positions at Novartis Pharmaceuticals Corporation, a pharmaceutical company, most recently as executive director, field operations. Mr. Angotti received a B.S. from Cornell University and an M.B.A. from Columbia University.
Gregory T. Bates has been our senior vice president of regulatory affairs and quality since June 2012. He was previously our vice president of regulatory affairs from 2006 to June 2012. From 1998 to 2006, Dr. Bates held various positions at Pharmacyclics, Inc, a biopharmaceutical company, the most recent of which was senior director of regulatory affairs. Prior to Pharmacyclics, in 1998, Dr. Bates was director of regulatory affairs and quality at Otsuka America Pharmaceutical, Inc. From 1995 to 1998, he was manager of regulatory affairs at Genentech, Inc., a biotechnology company, and from 1990 to 1995, he was senior manager of agribusiness regulatory affairs at Syntex (USA), Inc., a pharmaceutical company. Dr. Bates received a B.A. from the University of California, Berkeley and a Doctor of Veterinary Medicine from the University of California, Davis.
Gianna M. Bosko has been our senior vice president, chief legal officer and secretary since April 2013. She was previously our senior vice president, chief administrative officer, general counsel and secretary from August 2010 to April 2013. Prior to that, she was vice president, general counsel and secretary from 2007 to 2010, and senior corporate counsel from 2005 to 2007. From 2004 to 2005, Ms. Bosko was a legal consultant, providing general corporate and in-house legal consulting services for private and public companies, including XenoPort. From 1996 to 2004, she was an associate at Cooley LLP, a law firm, practicing general corporate and securities law, with an emphasis on securities transactions and mergers and acquisitions. Ms. Bosko received an A.B. from Stanford University and a J.D. from the University of Chicago Law School.
29
Table of Contents
William G. Harris has been our senior vice president of finance and chief financial officer since November 2001. From 1996 to 2001, he held several positions with Coulter Pharmaceutical, Inc., a biotechnology company engaged in the development of novel therapies for the treatment of cancer and autoimmune diseases, the most recent of which was senior vice president and chief financial officer. Corixa Corp., a developer of immunotherapeutic products, acquired Coulter Pharmaceutical in 2000. Prior to Coulter Pharmaceutical, from 1990 to 1996, Mr. Harris held several positions at Gilead Sciences, Inc., the most recent of which was director of finance. Mr. Harris received a B.A. from the University of California, San Diego and an M.B.A. from Santa Clara University, Leavey School of Business and Administration. Mr. Harris is a member of the board of directors of Capnia, Inc., a publicly-traded commercial stage medical diagnostics and therapeutics company.
Richard K. Kim has been our senior vice president, clinical development and medical affairs, and chief medical officer since January 2014. He was previously our vice president of medical affairs from July 2013 to January 2014. From 2012 to 2013, Dr. Kim was vice president of clinical development and head of the multiple sclerosis therapeutic area, and from 2010 to 2012, he was senior director, clinical development at Elan Pharmaceuticals, Inc. Prior to that, from 2006 to 2010, Dr. Kim was global medical director, medical affairs at Biogen Idec Inc. From 2004 to 2006, he was medical director, clinical development for EMD Serono, Inc. Prior to that, he was associate medical director from 2002 to 2004 at Purdue Pharma L.P. Dr. Kim received a B.S. degree from University of California at Los Angeles and his M.D. from Tulane University. He completed his neurology residency training at Stanford University, and is currently a diplomat of the American Board of Psychiatry and Neurology.
We were incorporated in Delaware in May 1999. Our principal offices are located at 3410 Central Expressway, Santa Clara, California 95051, and our telephone number is (408) 616-7200. Our Web site address is www.XenoPort.com. Information found on, or accessible through, our Web site is not a part of, and is not incorporated into, this Annual Report on Form 10-K. HORIZANT, REGNITE, Transported Prodrug, XENOPORT and the XenoPort logo are our trademarks. Service marks, trademarks and trade names appearing in this Annual Report on Form 10-K are the property of their respective owners. Unless the context requires otherwise, references in this Annual Report on Form 10-K to “the Company,” “we,” “us” and “our” refer to XenoPort, Inc.
We file electronically with the U.S. Securities and Exchange Commission, or SEC, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended. We make available on our Web site at www.XenoPort.com, free of charge, copies of these reports as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. Further, copies of these reports are located at the SEC’s Public Reference Room at 100 F Street, NE, Washington, D.C. 20549. Information on the operation of the Public Reference Room can be obtained by calling the SEC at 1-800-SEC-0330. The SEC maintains a Web site that contains reports, proxy and information statements, and other information regarding our filings, at www.sec.gov.
30
Table of Contents
The following risks and uncertainties may have a material adverse effect on our business, financial condition or results of operations. Investors should carefully consider the risks described below before making an investment decision. The risks described below are not the only ones we face. Additional risks not presently known to us or that we believe are immaterial may also significantly impair our business operations. Our business could be harmed by any of these risks. The trading price of our common stock could decline due to any of these risks, and investors may lose all or part of their investment.
Risks Related to our Business and Industry
We have incurred cumulative operating losses since inception, we expect to continue to incur losses for the foreseeable future and we may never achieve or sustain profitability on an annual basis.
We have incurred cumulative losses of $586.8 million since our inception in May 1999 through December 31, 2014. We have made, and expect to continue to make, substantial expenditures in connection with our commercialization of HORIZANT and to further develop and potentially commercialize our product candidates. We continue to expect substantial costs and expenses associated with maintaining sales, marketing and commercial capabilities, as well as those associated with research, development, clinical trials, manufacturing and potential regulatory approvals and commercialization of XP23829. In addition, we do not believe that collaboration revenues and revenues generated from sales of HORIZANT and REGNITE will be sufficient alone to sustain our operations, at least in the near term, and we therefore expect to continue to experience negative cash flows for the foreseeable future as we fund our operating losses and capital expenditures. Annual losses have had, and will continue to have, an adverse effect on our stockholders’ equity.
Because of the numerous risks and uncertainties associated with drug development and commercialization, we are unable to predict the timing or amount of increased expenses or when, or if, we will be able to achieve or sustain profitability. HORIZANT is approved by the U.S. Food and Drug Administration, or FDA, for two indications: the treatment of moderate-to-severe primary restless legs syndrome, or RLS, in adults; and management of postherpetic neuralgia, or PHN, in adults. With the exception of the proposed clinical trial in patients with alcohol use disorder, or AUD, planned to be conducted by the National Institute on Alcohol Abuse and Alcoholism, or NIAAA, we are solely responsible for all HORIZANT commercialization and development activities, including all post-marketing requirements and commitments. REGNITE has been approved by the Japanese Ministry of Health, Labour and Welfare, or MHLW, as a treatment for patients with moderate-to-severe RLS, and Astellas Pharma Inc. is responsible for promoting REGNITE in Japan.
To date, we have not generated revenue from sales of HORIZANT or REGNITE that is sufficient to fund our operations. We have financed our operations primarily through the sale of equity and debt securities, non-equity payments from collaboration, partnership, licensing and other similar forms of revenue-generating transactions with third-party business partners, and interest earned on investments. Prior to 2013, we devoted substantially all of our efforts to research and development, including clinical trials. Since the beginning of 2013, we have devoted substantial efforts to the initiation, maintenance and growth of our HORIZANT commercial operations, and we expect our selling, general and administrative expenses to continue to increase year-over-year primarily due to the effect of expenses incurred in maintaining and growing marketing, distribution and other commercial capabilities for HORIZANT, largely through third-party service providers, as well as the internal and external expenses of supporting and maintaining our XenoPort-employed sales representatives and those arising from our contract sales representatives. If sales-related revenue from HORIZANT, REGNITE or any product candidate that receives marketing approval is insufficient, if we or our business partners are unable to develop and commercialize our product candidates or if development is delayed, we may never become profitable on an annual basis. Even if we do become profitable on an annual basis, we may not be able to sustain or increase our profitability.
31
Table of Contents
Our success depends substantially on the success of HORIZANT. If we do not successfully market and sell HORIZANT, or if Astellas does not effectively market and sell REGNITE in Japan, we may be unable to generate significant product revenue and may be unable to achieve sustained profitability.
For the year ended December 31, 2014, we recorded net sales in the United States of HORIZANT of only $20.2 million. To achieve sustained profitability, we will need to generate substantially more product revenue from HORIZANT, REGNITE or our product candidates that may receive approval.
Our ability to generate significant revenue from HORIZANT depends on our ability to achieve market acceptance of, and to otherwise effectively market, HORIZANT for the treatment of moderate-to-severe primary RLS and for the management of PHN in adults. We may not be able to devote sufficient resources to the advertising, promotion and sales efforts for HORIZANT. We also need to continue to expend time and resources to train our sales team to be credible, persuasive and compliant in discussing HORIZANT with physicians. We also need to continue to train and monitor our sales team to ensure that a consistent and appropriate message about HORIZANT is being delivered. If we are unable to effectively educate physicians and potential customers about the benefits and risks of HORIZANT, we could face significant pressure from branded and/or unbranded (generic) competition, and negative market perception due to Glaxo Group Limited’s, or GSK’s, prior promotional efforts as well as a lack of physician awareness, third-party reimbursement and differentiation from currently approved treatments. Due to our limited resources, we have made decisions on how best to allocate our resources to efficiently promote HORIZANT. We may not be correct on our decisions, which would limit the sales we could achieve from HORIZANT and could lead to our failure to capitalize on its market opportunity. For example, we have chosen where to locate our XenoPort-employed sales representatives and contract sales representatives based on certain characteristics of the relevant territories where we promote HORIZANT. If other territories where we have chosen not to locate our sales team could have generated greater HORIZANT sales than the territories where we are promoting HORIZANT, we will be unable to capture the full potential value of HORIZANT. Further, new or future territories may not perform as well as the initial territories. In addition, our ability to grow HORIZANT sales in future periods is also dependent on price increases, and we have periodically increased the price of HORIZANT, most recently in January 2015. We cannot assure you that price increases we have taken or may take in the future will not negatively affect HORIZANT sales volumes. We could also fail to comply with applicable regulatory guidelines with respect to the marketing and manufacturing of HORIZANT or with post-marketing commitments or requirements mandated by the FDA, which could result in administrative or judicially imposed sanctions, including warning letters, civil and criminal penalties, injunctions, product seizures or detention, product recalls and total or partial suspension of production. In addition, if we are unable to effectively train sales representatives and equip them with effective materials, including medical and sales literature to help inform and educate potential customers about the benefits and risks of HORIZANT and its proper administration, our efforts to successfully commercialize HORIZANT could be put in jeopardy, which could have a material adverse effect on our financial condition, stock price and operations.
Other factors that may inhibit or delay our efforts to commercialize HORIZANT or any other approved product candidates include:
| • | the inability of our contract sales organization to provide our sales team with appropriate technical expertise or our inability to recruit and retain adequate numbers of effective sales and marketing personnel; |
| • | our inability to successfully retain the XenoPort-employed sales representatives and our contract sales representatives; |
| • | our inability to effectively manage and maintain third-party logistical support for both our XenoPort-employed sales representatives and our contract sales representatives; |
| • | the inability of our sales team personnel to obtain access to adequate numbers of physicians to provide appropriate information on the advantages and risks of prescribing HORIZANT or other products that may result from our product candidates; |
| • | inability to obtain coverage and adequate reimbursement from third-party payers; |
32
Table of Contents
| • | the lack of complementary products offered by our sales team personnel, which may put us at a competitive disadvantage compared to companies with more extensive product lines; and |
| • | unforeseen costs and expenses associated with maintaining the XenoPort-employed sales representatives, our contract sales representatives and/or contracting for additional sales team personnel as part of any potential future sales team expansions or to add or replace current sales representatives. |
The competition for qualified personnel in the pharmaceutical and biotechnology field is intense, and we may experience difficulties in recruiting, hiring and retaining qualified individuals. Prior to our commercial launch of HORIZANT, we had no experience commercializing products on our own, and we have only limited management expertise in developing and maintaining a commercial organization. Due to our limited internal resources, we have contracted, and anticipate that we will continue to contract, with third-party vendors to manage and support much of our growth and sales infrastructure. We will be at risk to the extent we rely on such third parties without effective oversight. In addition, such third-party contractors may not be the most efficient allocation of resources if we could implement such infrastructure internally in a more cost-effective manner. If we are not successful in recruiting and retaining qualified sales and marketing personnel or in otherwise maintaining sales and marketing infrastructure, we will have difficulty commercializing HORIZANT or our product candidates, which would adversely affect our business and financial condition.
Astellas initiated sales of REGNITE in Japan in July 2012. We have limited control over the amount and timing of resources that Astellas dedicates to the marketing of REGNITE, and Astellas could fail to effectively commercialize, market and distribute REGNITE. In May 2013, Astellas informed us that they did not have plans for commercialization of REGNITE in the five other Asian countries in their licensed territory. As a result, in May 2013, all rights to gabapentin enacarbil in Korea, the Philippines, Indonesia, Thailand and Taiwan reverted to us, and we will not receive any revenues from Astellas for the development and commercialization of gabapentin enacarbil in these territories.
HORIZANT or REGNITE may not achieve significant sales, even if we or Astellas devote substantial resources to its commercialization. Even if we achieve significant levels of sales of HORIZANT, the costs of maintaining sales and marketing capabilities and a distribution and supply chain infrastructure will be substantial and may outweigh any sales of HORIZANT, preventing us from achieving or maintaining profitability. The success of HORIZANT and REGNITE is dependent on a number of factors, which include competition from alternative treatments for RLS and, in the case of HORIZANT, for the management of PHN, including branded and unbranded (generic) treatments in the United States, pricing pressures and whether HORIZANT can obtain sufficient third-party coverage or reimbursement, among other factors that are described in this section.
In 2013, due to manufacturing delays, there was a stockout of HORIZANT, meaning that an insufficient amount of HORIZANT was in the supply chain to meet the demand of orders by pharmacies and wholesalers. As a result, starting in March 2013, certain patients who had been prescribed HORIZANT were not able to have such prescriptions filled. Although this stockout was resolved in May 2013, the implementation of our full commercial promotion of HORIZANT was delayed for approximately one month. If we or Patheon Pharmaceuticals Inc., our contract manufacturer of HORIZANT, or our third-party distribution, warehousing and inventory control service providers experience further delays or issues or otherwise fail to maintain adequate supplies of HORIZANT, our ability to fully commercialize HORIZANT could be hindered, the future demand for, and product sales of, HORIZANT could be further reduced and our business could be harmed, all of which could accelerate our need for additional capital.
The historical and pro forma financial statements we have filed with the SEC relating to HORIZANT may not be an indication of our ability to commercialize HORIZANT.
On May 1, 2013, pursuant to our termination and transition agreement with GSK, we completed the reacquisition of the exclusive rights to commercialize, promote, manufacture and distribute HORIZANT in the United States from GSK and assumed all responsibilities for any further development, manufacturing and commercialization of HORIZANT in the United States beginning on that date. In July 2013, we filed historical financial statements and pro forma financial information relating to the HORIZANT business, and the SEC stated that it would not object to our conclusion that the filing of the historical financial statements relating to the
33
Table of Contents
HORIZANT business represents substantial compliance with the requirements of Rule 3-05 of Regulation S-X, or Rule 3-05. However, we were advised by GSK that the HORIZANT product line had not been a separate legal entity of GSK and was never operated as a stand-alone business, division or subsidiary. GSK also advised us that it had never prepared full stand-alone or full carve-out financial statements for the HORIZANT business, and that GSK has never maintained the distinct and separate accounts necessary to prepare financial statements that fully comply with the requirements of Rule 3-05. As a result, these historical statements may not be an indication of the performance of HORIZANT under GSK for the periods indicated. In addition, the assumptions used in preparing the pro forma financial information we filed in July 2013 and subsequently updated through December 31, 2013 may not prove to be accurate or relevant to the HORIZANT business, in particular on a go-forward basis, and therefore should not be relied upon either as a measure of our ability to commercialize HORIZANT or future HORIZANT performance.
We have only limited experience in maintaining a commercial sales force on our own and, to the extent we do not successfully maintain a commercial sales force, sales of HORIZANT and our business will be harmed.
We converted our initial HORIZANT contract sales team to XenoPort-employed sales representatives, we have expanded our promotional efforts into new sales territories and we have added additional contract sales representatives through our contract sales organization. We expect to further expand our sales force to up to approximately 120 sales representatives by mid-2015, comprised of both XenoPort-employed sales representatives and contract sales representatives. We will also continue to rely on our contract sales organization to provide back office logistical support for the XenoPort-employed sales representatives. We have only limited experience in maintaining a commercial sales force on our own, and we may not be able to successfully maintain the XenoPort-employed sales representatives in a cost-effective or timely manner, or otherwise realize a positive return on our investment. We will also need to commit additional resources to maintain the XenoPort-employed sales representatives, and maintaining such sales personnel will impose significant added responsibilities on members of management, including the need to recruit, maintain and integrate additional employees. We will also have to compete with other pharmaceutical and life sciences companies to recruit and retain our dedicated sales team. Our ability to commercialize HORIZANT will depend, in part, on our ability to establish and maintain our dedicated sales team effectively. Because of the numerous risks and uncertainties involved with establishing and maintaining a commercial sales force, we may experience difficulties commercializing HORIZANT, which would adversely affect our business and financial results. In addition, we continue to rely on our current contract sales organization to provide us with our contract sales representatives, and thus we will need to continue to rely on our contract sales organization to provide us with and maintain contract pharmaceutical sales professionals with appropriate technical expertise to effectively commercialize HORIZANT.
Our success also depends substantially on our product candidates that are still under development. If we or our business partners are unable to bring any of these product candidates to market, or experience significant delays in doing so, our ability to generate revenue and our likelihood of success will be reduced.
Our ability to generate revenue in the future will depend heavily on the successful development and commercialization of our product candidates. Any of our product candidates could be unsuccessful if it:
| • | does not demonstrate acceptable safety and efficacy in preclinical studies or clinical trials or otherwise does not meet applicable regulatory standards for approval; |
| • | does not offer therapeutic or other improvements over existing or future drugs used to treat the same conditions; |
| • | is not capable of being produced in commercial quantities at acceptable costs; |
| • | is not accepted in the medical community; or |
| • | is not reimbursed by third-party payers or is reimbursed only at limited levels. |
For example, in 2011, following our preliminary results of a Phase 2 clinical trial of XP21279 for the potential treatment of patients with Parkinson’s disease who were experiencing motor fluctuations, we announced that we were deferring further investment in the program pending the outcome of discussions with
34
Table of Contents
regulatory authorities and availability of resources. Based on our subsequent End-of-Phase 2 meeting with the FDA, we plan to continue development of XP21279 to the extent additional resources permit or we enter into an arrangement with a third-party business partner, which we may be unable to do. If our resources prove insufficient or we are unable to establish a third-party arrangement for the development and commercialization of XP21279, we may be unable to advance its development. Our resources may also prove insufficient to develop XP23829 without a business partner. If we are unable to make additional product candidates commercially available, we may not be able to generate substantial product revenues, which would adversely affect our business and financial condition.
In May 2013, following our preliminary results of a Phase 3 clinical trial to evaluate arbaclofen placarbil, or AP, for the potential treatment of spasticity in multiple sclerosis, or MS, patients, we announced that we were terminating further investment in AP for such treatment. In May 2014, we announced that we had entered into a license agreement with Reckitt Benckiser Pharmaceuticals, Inc. (now Indivior PLC), which became effective in June 2014, pursuant to which we granted to Indivior exclusive world-wide rights to develop and commercialize pharmaceutical products containing AP and other prodrugs of baclofen or R-baclofen, or collectively, AP Products, for all indications, subject to our right of first negotiation to collaborate to develop and commercialize AP Products for non-addiction indications. Accordingly, we are now wholly dependent on Indivior to develop and commercialize AP Products, and if Indivior is unable to bring any AP Products to market, or experiences significant delays in doing so, our ability to generate any additional revenue from AP will be substantially impaired and our business may be harmed.
We currently depend, and may in the future depend, on third-party strategic arrangements to complete the development, regulatory approval and commercialization of some of our product candidates. These arrangements may place the development of our product candidates outside our control, may require us to relinquish important rights, may otherwise be on terms unfavorable to us and may ultimately not be successful.
As a result of our license agreement with Indivior, we are wholly dependent on Indivior to develop and commercialize AP Products. Future financial returns to us, if any, under our license agreement with Indivior depend on the achievement of development, regulatory and commercialization contingent-event based payments, plus royalties on potential future net product sales. Therefore, any associated future financial returns to us and our stockholders under the Indivior license agreement depend entirely on the efforts of, and funding by, Indivior for the development and commercialization of AP Products. If Indivior is not successful in developing and bringing AP Products to market, our ability to generate revenue from AP will be substantially diminished, the development or commercialization of AP could be delayed or terminated, and our business and financial condition could be harmed. Further, if Indivior fails to establish itself and/or identify and qualify one or more suitable manufacturers of AP, or if such manufacturers terminate their agreement with Indivior and Indivior is otherwise unable to manufacture or contract to manufacture sufficient quantities of AP, development and commercialization of AP Products could be impaired or delayed. In addition, if the Indivior license terminates in such a way that AP reverts to us and we seek alternative arrangements with one or more other third parties to develop and commercialize AP, we may not be able to enter into such an agreement with another suitable third-party or third-parties on acceptable terms, or at all. Moreover, although we have a retained a right of first negotiation to collaborate to develop and commercialize AP Products for non-addiction indications, this right is limited and Indivior is under no obligation to us to enter into a collaboration with us for AP Products. Accordingly, we could fail to capitalize on, or share in, any successful development of AP by Indivior apart from the contingent-event based and royalty payment obligations to us under the Indivior license agreement.
In December 2005, we entered into a license agreement with Astellas for the development and commercialization of gabapentin enacarbil, also known as REGNITE, in Japan, Korea, the Philippines, Indonesia, Thailand and Taiwan. In 2007, we entered into an exclusive collaboration arrangement with GSK to develop and commercialize gabapentin enacarbil worldwide, excluding the original Astellas territory. Following a significant dispute in 2012, including the filing of lawsuits in California and Delaware, we terminated our collaboration with GSK pursuant to a termination and transition agreement, under which the product rights to HORIZANT returned to us in May 2013. We cannot control the amount and timing of resources that Astellas
35
Table of Contents
devotes to the development or commercialization of REGNITE or to its marketing and distribution. For example, in May 2013, Astellas informed us that they did not have plans for commercialization of REGNITE in the countries other than Japan in their licensed territory. As a result, all rights to gabapentin enacarbil in Korea, the Philippines, Indonesia, Thailand and Taiwan reverted to us. In addition, Astellas may abandon further commercialization of REGNITE in Japan, or terminate their license agreement with us at any time, which could delay or impair the development and commercialization of REGNITE and harm our business.
In addition to our partnerships with Astellas and Indivior, we may enter into future strategic arrangements with third parties to further develop and commercialize HORIZANT/gabapentin enacarbil and/or to develop and commercialize our product candidates. For example, we have entered into a clinical trial agreement with the NIAAA pursuant to which the NIAAA plans to conduct a clinical trial of HORIZANT in patients with AUD. Our dependence on current and future partners or collaborators for the development, regulatory approval and commercialization of some of our products and product candidates subjects us to a number of risks, including:
| • | we are not able to control the amount and timing of resources that our business partners devote to the development or commercialization of our products and product candidates, or to their marketing and distribution; |
| • | disputes may arise between us and our business partners, such as the litigation proceedings with GSK in 2012, that result in the delay or termination of the research, development or commercialization of our products and product candidates or that result in costly litigation or arbitration that diverts management’s attention and resources; |
| • | our business partners may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing; |
| • | if we do not receive timely and accurate information from any business partner or our third-party vendors regarding sales activities, expenses and resulting operating profits and losses, our estimates at a given point in time could be incorrect and we could be required to record adjustments in future periods or restate our financial results for prior periods; |
| • | our business partners may not be successful in their efforts to obtain regulatory approvals in a timely manner, or at all; |
| • | our business partners may receive regulatory sanctions relating to other aspects of their business that could adversely affect the approval or commercialization of our products or product candidates; |
| • | our business partners may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our proprietary information or expose us to potential litigation; |
| • | business combinations or significant changes in a business strategy may also adversely affect a business partner’s willingness or ability to complete its obligations under any arrangement; |
| • | a business partner could independently move forward with a competing product candidate developed either independently or in collaboration with others, including our competitors; |
| • | our business partners may experience financial difficulties; and |
| • | strategic arrangements may be terminated or allowed to expire, which would delay development or commercialization and may also increase the cost of developing or commercializing our products or product candidates. |
For example, in 2007, we entered into a business relationship with Xanodyne Pharmaceuticals, Inc. for the development and commercialization of XP21510 in the United States. In 2009, Xanodyne terminated the arrangement. Likewise, our collaboration with GSK was not successful and was terminated following a significant dispute and litigation related to GSK’s performance under the collaboration.
36
Table of Contents
We have a significant amount of indebtedness. We may not be able to generate enough cash flow from our operations to service our indebtedness, and we may incur additional indebtedness in the future, which could adversely affect our business, financial condition and results of operations.
In February 2015, we issued $115.0 million aggregate principal amount of our 2.50% convertible senior notes due 2022, or the 2022 Notes. Our ability to make payments on, and to refinance, the 2022 Notes, and to fund planned capital expenditures, sales and marketing efforts, research and development efforts, working capital and other general corporate purposes depends on our ability to generate cash in the future. This, to a certain extent, is subject to general economic, financial, competitive, legislative, regulatory and other factors, some of which are beyond our control. If we do not generate sufficient cash flow from operations or if future borrowings are not available to us in an amount sufficient to repay our indebtedness, including any amounts due under the 2022 Notes at their maturity, or to fund our liquidity needs, we may be forced to refinance all or a portion of the 2022 Notes, on or before the maturity thereof, sell assets, reduce or delay capital expenditures, seek to raise additional capital or take other similar actions. We may not be able to affect any of these actions on commercially reasonable terms or at all. Our ability to refinance our indebtedness will depend on our financial condition at the time, the restrictions in the instruments governing our present and potential future indebtedness and other factors, including market conditions. In addition, in the event of a default with respect to the 2022 Notes, the holders of the 2022 Notes and/or the trustee under the indenture governing the 2022 Notes may accelerate the payment of our obligations under 2022 Notes, which could have a material adverse effect on our business, financial condition and results of operations. A default under the indenture governing the 2022 Notes could also lead to a default under agreements governing future indebtedness, which could also have a material adverse effect on our business, financial condition and results of operations. Our inability to generate sufficient cash flow to satisfy our debt service obligations, or to refinance or restructure our obligations on commercially reasonable terms or at all, would likely have a material adverse effect on our business, financial condition and results of operations.
In addition, our significant indebtedness combined with our other financial obligations and contractual commitments could have other important consequences. For example, it could:
| • | make us more vulnerable to adverse changes in general U.S. and worldwide economic, industry and competitive conditions and adverse changes in government regulation; |
| • | limit our flexibility in planning for, or reacting to, changes in our business and our industry; |
| • | require the dedication of a substantial portion of our cash flow from operations to service our indebtedness, thereby reducing the amount of our cash flow available for other purposes; |
| • | result in dilution to our stockholders in the event of conversions of the 2022 Notes; |
| • | place us at a possible competitive disadvantage with less leveraged competitors and competitors that may have better access to capital resources; and |
| • | limit our ability to obtain additional financing or borrow additional amounts for working capital, capital expenditures, debt service requirements, execution of our business strategy or other purposes. |
Any of these factors could materially and adversely affect our business, financial condition and results of operations. In addition, if we incur additional indebtedness, which we are not prohibited from doing under the terms of the indenture governing the 2022 Notes, the risks related to our business and our ability to service our indebtedness would increase.
We may need additional funding and may be unable to raise capital when needed, which would force us to delay, reduce or eliminate our commercialization efforts or our current product development programs.
We may need to raise additional capital to fund our operations, to maintain a sales infrastructure and marketing and distribution capabilities for HORIZANT and to continue the development of XP23829, as well as to fund any required repurchases of the 2022 Notes and/or our repayment obligations under the 2022 Notes. Our future funding requirements will depend on many factors, including:
| • | the timing, receipt and amount of sales or royalties from HORIZANT, REGNITE and our potential products; |
37
Table of Contents
| • | the costs of our sales and marketing, supply chain, distribution, pharmacovigilance, compliance and safety infrastructure to promote and distribute HORIZANT, including the internal and external expenses of supporting and maintaining our XenoPort-employed sales representatives and expenses associated with our contract sales representatives and our planned and any other potential future expansions to our dedicated sales team; |
| • | the scope, rate of progress, results and costs of our preclinical testing, clinical trials and other research and development activities, including the timing and costs of further development of XP23829; |
| • | the costs of manufacturing clinical and commercial supplies of HORIZANT and our product candidates; |
| • | the timing and costs of complying with the remaining post-marketing commitments and post-marketing requirements established in connection with the approval of HORIZANT, and any future additional commitments or requirements imposed on us by the FDA; |
| • | the number and characteristics of product candidates that we pursue, including any additional potential indications for HORIZANT such as the treatment of patients with AUD, or the extent to which we pursue any further development of XP21279; |
| • | the costs, timing and outcomes of regulatory approvals, if any; |
| • | the timing of any contingent-event based and royalty payments under our licensing arrangement with Indivior; |
| • | the terms and timing of any collaboration or partnership arrangements that we may establish or modify; |
| • | the costs associated with any potential litigation; |
| • | the costs of preparing, filing, prosecuting, maintaining, defending and enforcing any patent claims and other intellectual property rights; and |
| • | the extent to which we acquire or invest in businesses, products or technologies that complement our business, although we have no commitments or agreements relating to any of these types of transactions. |
In addition, we are required to make periodic interest payments to the holders of our 2022 Notes and to make payments of principal upon maturity. In this regard, if holders of the 2022 Notes do not convert their 2022 Notes prior to the maturity date, we will be required to repay the principal amount of all then outstanding 2022 Notes plus accrued and unpaid interest. We may also be required to repurchase the 2022 Notes for cash upon the occurrence of a change of control or certain other fundamental changes involving us. If our capital resources are insufficient to satisfy our debt service obligations, we will be required to seek to sell additional equity or debt securities or to obtain debt financing.
Until we can generate a sufficient amount of product revenues, if ever, we expect to finance future cash needs through public or private equity or equity-linked offerings, debt financings or third-party business transactions. If we raise additional funds by issuing our common stock, or securities convertible into, exchangeable or exercisable for common stock, our stockholders will experience dilution. Any debt financing or additional equity that we raise may contain terms that are not favorable to our stockholders or us. To the extent that we raise additional capital through partnering, collaborative or other strategic arrangements, we may be required to relinquish, on terms that are not favorable to us, rights to some of our technologies or product candidates that we would otherwise seek to develop or commercialize ourselves. Our ability to raise additional funds and the terms upon which we are able to raise such funds may be adversely impacted by the uncertainty regarding our financial condition, the commercial prospects of HORIZANT and REGNITE based on their respective sales to date and our relative lack of experience in commercializing products, and/or current economic conditions, including the effects of disruptions to, and volatility in, the credit and financial markets in the United States, Asia, the European Union and other regions of the world.
We believe that our existing capital resources, including proceeds we received from our convertible debt offering that closed in February 2015, together with interest income from our investments and anticipated product revenue, will be sufficient to meet our projected operating requirements through at least the first quarter of 2018. We have based our cash sufficiency estimate on assumptions that may prove to be incorrect, and we could utilize our available capital resources sooner than we expect. Further, our operating plan may change, and
38
Table of Contents
we may need additional funds to meet operational needs and capital requirements for product development and commercialization sooner than planned. We have no credit facility or committed sources of capital other than potential contingent event-based and royalty payments that we are eligible to receive under our partnerships with Astellas and Indivior. With the exception of the proposed clinical trial in patients with AUD that the NIAAA plans to conduct, we are solely responsible for all HORIZANT commercialization and development activities, including all post-marketing requirements and commitments. Such costs could be greater than we anticipate, or sales of HORIZANT may be less than we anticipate, either or both of which could accelerate our need for additional capital.
Additional funds may not be available to us on terms that are acceptable to us, or at all. If adequate funds are not, or we anticipate that they may not be, available on a timely basis, we may, among other things:
| • | reduce the amount of resources devoted to medical affairs, advertising, promotion or sales of HORIZANT; |
| • | curtail or delay further XP23829 development; or |
| • | conduct additional workforce or other expense reductions. |
For example, in 2012, we suspended clinical development activities for XP21279 to focus our resources on development of our other product candidates. In addition, in May 2013, we terminated further investment in AP as a treatment for spasticity in patients with MS to focus our resources on the commercialization of HORIZANT and continued development of XP23829. If we are required to reduce the amount of resources devoted to medical affairs, advertising, promotion or sales of HORIZANT, curtail or delay further XP23829 development or conduct additional workforce or other expense reductions, our business and prospects could be materially and adversely affected. In addition, we may face substantial financial costs in order to comply with the remaining post-marketing commitments and post-marketing requirements required by the FDA in connection with the approval of HORIZANT, and our inability to carry out and fund such efforts to comply with these requirements could have material adverse consequences to us, including the loss of marketing approval for HORIZANT.
We rely on third parties to perform many essential services for HORIZANT, including services related to warehousing and inventory control, distribution, customer service, government price reporting, recording of sales, accounts receivable management, cash collection and adverse event reporting, and if such third parties fail to provide us with accurate information, perform as expected or comply with legal and regulatory requirements, our efforts to commercialize HORIZANT may be significantly impacted and/or we may be subject to regulatory sanctions.
We intend to continue to rely on third-party service providers to perform a variety of functions related to the sale and distribution of HORIZANT, key aspects of which are out of our direct control. The services provided by these third parties include warehousing and inventory control, distribution, customer service, government price reporting, recording of sales, accounts receivable management and cash collection. If these third-party service providers fail to comply with applicable laws and regulations, fail to meet expected deadlines or otherwise do not carry out their contractual duties to us, or if supplies of HORIZANT encounter physical or natural damage at their facilities, our ability to deliver HORIZANT to meet commercial demand would be significantly impaired. If these third parties do not provide us with timely and accurate information, it could impact our ability to comply with our financial reporting, state and federal healthcare professional aggregate spend reporting, government price reporting and securities laws obligations, which could expose us to the risk of stockholder lawsuits and/or regulatory actions and adversely affect our business. In addition, we have engaged third parties to perform various other services for us relating to adverse event reporting, safety database management, fulfillment of requests for medical information regarding HORIZANT and related services. If the quality or accuracy of the data maintained or services performed by these third parties is insufficient, we could be subject to regulatory sanctions.
39
Table of Contents
The commercial success of HORIZANT, REGNITE or any other products that we or our business partners may develop will depend upon the degree of market acceptance among physicians, patients, healthcare payers and the medical community.
HORIZANT, REGNITE or any other products that may result from our product candidates may not gain market acceptance among physicians, patients, healthcare payers and the medical community. If these products do not achieve an adequate level of acceptance, we may not generate material product or collaboration revenues and we may not become profitable. The degree of market acceptance of HORIZANT, REGNITE or any products resulting from our product candidates will depend on a number of factors, including:
| • | the ability to offer such products for sale at competitive prices; |
| • | sufficient third-party payer coverage or reimbursement for such products; |
| • | the product labeling required by the FDA, the Japanese MHLW or any other regulatory authorities; |
| • | demonstrated efficacy and safety in clinical trials; |
| • | the prevalence and severity of any side effects; |
| • | potential or perceived advantages over alternative treatments; |
| • | perceptions about the relationship or similarity between our products or product candidates and the parent drug upon which each product or product candidate is based; |
| • | the timing of market entry relative to competitive treatments; |
| • | relative convenience and ease of administration; and |
| • | the strength of marketing and distribution support. |
For example, as HORIZANT is a prodrug of an already approved drug, gabapentin, and is indicated for the treatment of conditions that also have been treated by branded and unbranded (generic) competitors, there could be a perception among physicians and payers that HORIZANT may not offer a significant clinical advantage or be sufficiently differentiated from competitive treatments to justify its price, thereby limiting the market acceptance and sales that we may achieve with HORIZANT. In addition, HORIZANT’s limited past sales performance under GSK and a HORIZANT inventory stockout in 2013 may have created a negative market perception that could be difficult to overcome in our marketing efforts.
Our ability to generate revenue from HORIZANT, REGNITE or any other products that we or our business partners may develop will depend on the availability of coverage and adequate reimbursement from third-party payers and drug pricing policies and regulations.
In both U.S. and foreign markets, our ability and that of our business partners to successfully commercialize HORIZANT, REGNITE or any other products that we or our business partners may develop, and our ability to attract strategic partners for these products, depends in significant part on the availability of financial coverage and adequate reimbursement from third-party payers, including, in the United States, governmental payers such as the Medicare and Medicaid programs, managed care organizations and private health insurers. Many patients may be unable to pay for HORIZANT, REGNITE or any other products that we or our business partners may develop. We cannot be sure that coverage and adequate reimbursement in the United States, Japan, Europe or elsewhere will be available for HORIZANT, REGNITE or any other products that may result from our product candidates, and any reimbursement that may become available may be decreased or eliminated in the future. Additionally, coverage and reimbursement determinations are made on a payer-by-payer basis. Therefore, obtaining acceptable coverage and reimbursement from one payer does not guarantee we will obtain similar acceptable coverage or reimbursement from another payer. Third-party payers increasingly are challenging prices charged for medical products and services, and many third-party payers may refuse to provide reimbursement for particular drugs when an equivalent generic drug or suitable alternative treatment is available. Although we believe HORIZANT, REGNITE and any other products that may result from our product candidates represent an improvement over the parent drugs upon which they are based and should be considered unique and not subject to substitution by a generic parent drug, it is possible that a third-party payer may consider HORIZANT,
40
Table of Contents
REGNITE or our product candidates and the respective generic parent drug as equivalents and only offer to reimburse patients for the generic drug. Even if we show improved efficacy or improved convenience of administration with HORIZANT, REGNITE or our product candidates, pricing of the existing parent drug may limit the amount we or our business partners will be able to charge for HORIZANT, REGNITE or our product candidates. If reimbursement is not available or is available only at limited levels, we or our business partners may not be able to successfully commercialize HORIZANT, REGNITE or any of our product candidates that obtain marketing approval, and may not be able to obtain a satisfactory financial return on such products.
Such reimbursement pricing pressures have increased as a result of the Medicare Prescription Drug Improvement and Modernization Act of 2003, or the 2003 MMA, due to the enhanced purchasing power of the private sector plans that negotiate on behalf of Medicare beneficiaries. Furthermore, managed care organizations, as well as Medicaid and other government agencies, continue to seek price discounts. Some states have implemented, and other states are considering, price controls or patient access constraints under the Medicaid program, and some states are considering price-control regimes that would apply to broader segments of their populations that are not Medicaid-eligible. If legislation were enacted to mandate rebates or provide for direct government negotiation in prescription drug benefits, access and reimbursement for HORIZANT or our product candidates upon commercialization could be restricted.
The trend toward managed healthcare in the United States and the changes in health insurance programs, as well as the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively, PPACA, enacted in 2010, may result in lower prices for pharmaceutical products, including HORIZANT or any other products that may result from our product candidates. In addition, if the 2003 MMA or the PPACA were amended to impose direct governmental price controls and access restrictions, these could have a significant adverse impact on our business, including on any product sales revenue from HORIZANT. Any future regulatory changes regarding the healthcare industry or third-party coverage and reimbursement may affect demand for HORIZANT or any other products that may result from our product candidates and could harm our sales and profitability.
If competitors are able to develop and market products that are more effective, safer or less costly than HORIZANT, REGNITE or any other products that we or our business partners may develop, our commercial opportunity will be reduced or eliminated.
We face competition from established pharmaceutical and biotechnology companies, including generic competitors, as well as from academic institutions, government agencies and private and public research institutions. Our commercial opportunities will be reduced or eliminated if competitors develop and commercialize products that are safer, more effective, have fewer side effects or are less expensive than HORIZANT, REGNITE or any other products that may result from our product candidates. In addition, significant delays in the development of our product candidates could allow competitors to bring products to market before us and impair our ability to effectively commercialize our product candidates.
Products that we believe compete with HORIZANT in the United States include the following drugs approved for the treatment of moderate-to-severe primary RLS: MIRAPEX (pramipexole) from Boehringer Ingelheim GmbH, and generic pramipexole; REQUIP (ropinirole) from GSK and generic ropinirole; and NEUPRO (a rotigotine transdermal system), a dopamine agonist patch from UCB S.A. In Japan, we believe that REGNITE competes with SIFROL (pramipexole) and the UCB rotigotine transdermal system, which was launched in February 2013 by Otsuka Pharmaceutical Co., Ltd., which has exclusive rights to market in Japan. Products that we believe compete with HORIZANT in the United States for the management of PHN include drugs that act on the same target as HORIZANT, such as LYRICA (pregabalin) and NEURONTIN (gabapentin) from Pfizer Inc., GRALISE (once-daily formulation of gabapentin) from Depomed, Inc. and generic gabapentin. HORIZANT could also experience competition from a capsaicin patch (marketed as QUTENZA by Acorda Therapeutics, Inc.) and transdermal patches containing the anesthetic known as lidocaine (marketed as LIDODERM by Endo Pharmaceuticals, Inc.). In addition, while not a treatment for shingles or PHN, HORIZANT could experience competition from a shingles vaccine, known as ZOSTAVAX marketed by Merck & Co., Inc., that is a live attenuated vaccine to help prevent shingles. There is also a shingles vaccine in late stage development, known as HZ/su, which is being developed by GSK.
41
Table of Contents
Products that could compete with XP23829, our product candidate that is a prodrug of monomethyl fumarate, or MMF, for the treatment of psoriasis include topical therapies, oral systemic therapies and injectable biological therapies. Topical therapies include corticosteroids, anthrolin and synthetic vitamin D and vitamin A. Oral systemic therapies include acitretin, cyclosporine, methotrexate and OTEZLA (apremilast) from Celgene Corporation. Biological therapies that are approved to treat psoriasis include COSENTYX (secukinumab) marketed by Novartis AG; ENBREL (etanercept), marketed by Amgen Inc.; HUMIRA (adalimumab), marketed by AbbVie, Inc; REMICADE (infliximab), marketed by Johnson & Johnson and Merck & Co.; and STELARA (ustekinumab), marketed by Johnson & Johnson. There are also a number of possible competitive products that are in late-stage product development in the United States, including brodalumab, CF-101, ixekizumab, tildrakizumab and tofacitinib that are being developed by Amgen, Can-Fite BioPharma Ltd., Eli Lilly and Company, Sun Pharmaceutical Industries Ltd./Merck & Co., and Pfizer, respectively.
Products that could compete with XP23829 for the treatment of relapsing forms of MS include oral and injectable agents that are approved in the United States for the treatment of relapsing forms of MS. These include oral agents such as AUBAGIO (teriflunomide), marketed by Genzyme Corporation, a Sanofi company; GILENYA (fingolimod), marketed by Novartis; and TECFIDERA (dimethyl fumarate), marketed by Biogen Idec Inc. In addition, several injectable products are approved in the United States and include AVONEX (interferon beta 1a), PLEGRIDY (peginterferon beta-1a) and TYSABRI (natralizumab), marketed by Biogen; BETASERON (interferon beta 1b) and EXTAVIA (interferon beta 1b), marketed by Bayer AG/Novartis; COPAXONE (glatiramer acetate), marketed by Teva Pharmaceutical Industries Ltd.; REBIF (interferon beta 1a), marketed by Merck Serono S.A.; and LEMTRADA (alemtuzumab) from Genzyme/Sanofi-Aventis. There are also potentially competitive products in late-stage product development in the United States, including daclizumab high-yield process (DAC HYP) from AbbVie/Biogen, laquinimod from Teva, RPC1063 from Receptos, Inc., FP187 from Forward Pharma A/S, and ocrelizumab from Roche Holding AG.
Products that could compete with gabapentin enacarbil for the treatment of AUD include ANTABUSE (disulfiram) marketed by TEVA; REVIA and VIVITROL, oral and injectable formulations of naltrexone, marketed by Barr Pharmaceuticals, Inc. and Alkermes, Inc., respectively; and CAMPRAL (acamprosate), marketed by Actavis Inc.
If resources permit and we choose to develop XP21279 as a potential treatment for advanced idiopathic Parkinson’s Disease, products that could compete with XP21279, our product candidate that is a Transported Prodrug of levodopa, include: DUOPA (levodopa/carbidopa suspension) from AbbVie; RYTARY, an extended-release oral capsule formulation of levodopa/carbidopa from Impax Laboratories, Inc., which were both recently FDA approved; SINEMET (levodopa/carbidopa) that is distributed in the United States by Bristol Meyers Squibb Company; STALEVO, a combination therapy of levodopa/carbidopa/entacapone that is marketed in the United States by Novartis and generic levodopa/carbidopa drugs. In addition, there are a number of products that are approved for idiopathic Parkinson’s disease and administered as adjunctive therapy to levodopa/carbidopa in patients with advanced idiopathic Parkinson’s disease. These include AZILECT (rasagiline mesylate), marketed by TEVA and dopamine agonists such as NEUPRO (a rotigotine transdermal system) marketed by UCB, MIRAPEX (pramipexole) marketed by Boehringer Ingelheim and REQUIP (ropinirole) and REQUIP XL (ropinirole extended-release tablets) marketed by GSK and generic dopamine agonists. Other therapies under development in the United States include extended-release formulations of levodopa/carbidopa such as DM-1992 and OS-320 being developed by Depomed and Osmotica Pharmaceutical Corp., respectively.
There may be other compounds of which we are not aware that are at an earlier stage of development and may compete with our products or product candidates. If any of those compounds are successfully developed and approved, they could compete directly with our products or product candidates.
Many of our and our business partners’ competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing, distributing and selling approved products than we do. Established pharmaceutical companies may invest heavily to quickly discover and develop novel compounds that could make HORIZANT, REGNITE or our product candidates obsolete. Larger pharmaceutical companies also may have significantly greater sales forces, distribution capabilities and marketing expertise, which may result in more effective communication and awareness of their products. Smaller or early-stage companies may also prove to be significant competitors,
42
Table of Contents
particularly through strategic arrangements with large and established companies. In addition, these third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies and technology licenses complementary to our programs or advantageous to our business. Accordingly, competitors may succeed in obtaining patent protection, receiving FDA approval or discovering, developing and commercializing medicines before we or our business partners do. We are also aware of other companies that may currently be engaged in the discovery of medicines that will compete with HORIZANT, REGNITE or products that may result from our product candidates. In addition, in the markets that we are targeting, we expect to compete against current market-leading medicines. If we are not able to compete effectively against our current and future competitors, our business will not grow and our financial condition will suffer.
Off-label sale or use of gabapentin products could lead to pricing pressure or decrease sales of HORIZANT.
U.S. physicians are permitted to prescribe legally available drugs for uses that are not described in the drug’s labeling and that differ from those uses tested and approved by the FDA. The occurrence of such off-label uses in the practice of medicine could significantly reduce our ability to market and sell HORIZANT or any other products that we may develop.
Off-label prescriptions written for gabapentin for indications for which we are marketing or may develop HORIZANT could adversely affect our ability to generate revenue from the sales of HORIZANT. This could result in reduced sales and increased pricing pressure on HORIZANT, which in turn would reduce our ability to generate meaningful revenue and have a negative impact on our results of operations.
If product liability lawsuits are brought against us, we will incur substantial liabilities and may be required to limit commercialization of HORIZANT or any other products that we may develop.
We face an inherent risk of product liability exposure related to the commercial use of HORIZANT and the testing of HORIZANT or our product candidates in human clinical trials. If we cannot successfully defend ourselves against claims that HORIZANT, our product candidates or products that may result from our product candidates caused injuries, we will incur substantial liabilities.
Regardless of merit or eventual outcome, liability claims may result in:
| • | decreased demand for HORIZANT or any product candidates or products that may result from our product candidates; |
| • | injury to our reputation; |
| • | costly recalls of HORIZANT or other products that may result from our product candidates; |
| • | withdrawal of clinical trial participants; |
| • | costs to defend the related litigation; |
| • | substantial monetary awards to clinical trial participants or patients; |
| • | loss of revenue; and |
| • | the inability to commercialize any future products that may result from our product candidates. |
We have product liability insurance that covers our commercial use and clinical trials up to a $15.0 million annual aggregate limit. Insurance coverage is increasingly expensive, and we may not be able to maintain insurance coverage at a reasonable cost and we may not be able to obtain insurance coverage that will be adequate to satisfy any liability that may arise.
If third parties do not manufacture HORIZANT, REGNITE or our product candidates in sufficient quantities or at an acceptable cost, commercialization of HORIZANT and REGNITE and clinical development and commercialization of our product candidates would be harmed or delayed.
We do not own or operate manufacturing facilities for the production of clinical or commercial quantities of HORIZANT, REGNITE or any of our product candidates. We also have limited management expertise in
43
Table of Contents
commercial supply operations. For our product candidates, we have relied on, and we expect to continue to rely on, a limited number of third-party drug substance and drug product manufacturers. We do not have commercial supply agreements with any of these third parties for our product candidates, and our agreements with these parties are generally terminable at will by either party at any time. For HORIZANT, GSK supplied us with HORIZANT through October 2013, and also performed certain validation work with respect to the 300 mg dosage form of HORIZANT and other services for us through December 2013. Currently, we rely on Patheon as our single source supplier of clinical and commercial product pursuant to a commercial supply agreement. Our purchase price for the manufacture and supply of HORIZANT is volume-based. Both parties have early termination rights with notice periods of varying lengths for various causes, including for the counterparty’s uncured breach. Patheon also has the right to terminate the supply agreement upon 18 months prior notice if we sell, assign or otherwise transfer rights to HORIZANT to a competitor of Patheon. If, for any reason, Patheon or these third–party drug substance and drug product manufacturers are unable or unwilling to perform under our agreements or enter into new agreements, we may not be able to locate alternative manufacturers or enter into favorable agreements with them. In addition, qualifying and then successfully registering a new HORIZANT manufacturer with the FDA is time consuming and could require at least about 18 months to carry out. Our inability to acquire sufficient quantities of HORIZANT could result in future stockouts and existing patients not having access to drug product. Any inability to acquire sufficient quantities of our product candidates in a timely manner from these third parties could delay clinical trials and prevent us from developing and commercializing these product candidates in a cost-effective manner or on a timely basis, if at all.
If we are unable to manufacture or contract to manufacture sufficient quantities of HORIZANT, the commercialization of HORIZANT could be impaired or interrupted. For example, manufacturing delays resulted in a stockout of HORIZANT in 2013. As a result of the manufacturing delays, our full commercial promotion of HORIZANT was delayed for approximately one month. If we or Patheon experience other delays or issues, our ability to commercialize HORIZANT could be further impaired. As part of the termination and transition agreement, GSK provided its inventory of gabapentin enacarbil drug substance to us. Although the inventory of drug substance has reached the end of its specified shelf life, we believe that such inventory will remain in specification and will be usable, or in the alternative, we believe the drug substance can be re-crystallized into usable form. GSK relied on a single source supplier of gabapentin enacarbil drug substance, and its agreement with such manufacturer has expired. If we are incorrect about the usability of the gabapentin enacarbil drug substance, are unable to have it meet specifications upon re-crystallization or are unable to enter into an agreement with the contract manufacturer or qualify an alternative manufacturer, we may be limited in the amount of HORIZANT we could have manufactured and the commercialization of HORIZANT could be impaired or interrupted. Under the terms of our license agreement with Astellas, Astellas is solely responsible for the manufacture of REGNITE to support its commercialization in Japan. To our knowledge, Astellas is currently relying on single source suppliers for commercial supplies of REGNITE. As a result, if Astellas fails to manufacture or contract to manufacture sufficient quantities of REGNITE, commercialization of REGNITE could be impaired in Japan.
Under the terms of our license agreement with Indivior, Indivior is solely responsible for the manufacture of AP to support its development and commercialization of AP Products. If Indivior fails to establish AP manufacturing capabilities itself and/or to identify and qualify one or more suitable manufacturers of AP, or if such manufacturers terminate their agreements with Indivior and Indivior is otherwise unable to manufacture or contract to manufacture sufficient quantities of AP, development and commercialization of AP Products could be impaired or delayed.
We rely on a single source supplier of MMF, which is used to make XP23829, under purchase orders issued from time to time. We are aware of several alternative suppliers of MMF, and we believe at least one alternative manufacturer could potentially supply MMF in the event that our supplier determines to not sell MMF to us at a price that is commercially attractive. If we are unable to qualify an alternative supplier of MMF, this could delay the development of, and impair our ability to commercialize, XP23829.
We rely on both domestic and ex-U.S. third-party suppliers of XP23829 active pharmaceutical ingredient and finished drug product through a three-step chain of manufacturing services and supply agreements, and we have purchased XP23829 formulated in different forms from multiple suppliers at specified transfer prices under
44
Table of Contents
quotations agreed upon by the parties as part of master services agreements. In the event that such suppliers or manufacturers terminate our respective agreements under specified circumstances, we would not be able to manufacture drug substance or XP23829 until alternative suppliers are identified and qualified, which could also delay the clinical development of, and impair our ability to commercialize, XP23829.
We rely on a single source supplier of levodopa, which is used to make XP21279, under purchase orders issued from time to time. We are aware of several alternative suppliers of levodopa, and we believe at least one alternative manufacturer could potentially supply levodopa in the event that our supplier determines to not sell levodopa to us at a price that is commercially attractive. If we are unable to qualify an alternative supplier of levodopa, this could further delay the development of, and impair our ability to commercialize, XP21279.
We rely on a single source supplier of XP21279 drug substance under a manufacturing services and product supply agreement. In the event that such supplier terminates the agreement under specified circumstances, we would not be able to manufacture drug substance until a qualified alternative supplier is identified and qualified, which could also further delay the development of, and impair our ability to commercialize, this product candidate.
We have purchased XP21279 formulated in sustained-release tablets from a single source supplier at specified transfer prices under quotations agreed upon by the parties as part of a master services agreement. We have also qualified another supplier for the manufacture of XP21279 with carbidopa bi-layer tablets to be supplied under quotations agreed upon by the parties as part of a master services agreement. In the event that either supplier terminates its agreement under specified circumstances for the manufacture of XP21279 sustained-release tablets or carbidopa bi-layer tablets, we would not be able to manufacture XP21279 until an alternative supplier is qualified. This could further delay the development of, and impair our ability to commercialize, XP21279.
If we or our business partners are required to obtain alternate third-party manufacturers, it could delay or prevent the clinical development and commercialization of HORIZANT or our product candidates.
We or our business partners may not be able to maintain or renew existing, or obtain new, third-party manufacturing arrangements on acceptable terms, if at all. If we or our business partners are unable to continue relationships with suppliers for HORIZANT/gabapentin enacarbil, XP23829, AP and XP21279, or to continue relationships at an acceptable cost or if these suppliers fail to meet our requirements for HORIZANT or these product candidates for any reason, we or our business partners would be required to obtain alternative suppliers. Any inability to obtain qualified alternative suppliers, including an inability to obtain, or a delay in obtaining, approval of an alternative supplier from the FDA, would delay or prevent the clinical development and commercialization of HORIZANT or our product candidates.
Use of third-party manufacturers may increase the risk that we or our business partners will not have adequate supplies of HORIZANT, REGNITE or our product candidates.
Reliance on third-party manufacturers by us or our business partners will create exposure that could result in disruptions to the supply chain, product stockouts, patients not having access to their regular treatment, higher costs or lost product revenues, or it could delay or prevent:
| • | the commercialization of our products or products that may result from our product candidates; |
| • | the initiation or completion of clinical trials; |
| • | the submission of applications for regulatory approvals; and |
| • | the approval of our product candidates by the FDA or foreign regulatory authorities. |
In particular, our or our business partners’ contract manufacturers:
| • | could encounter difficulties in achieving volume production, quality control and quality assurance or suffer shortages of qualified personnel, which could result in their inability to manufacture sufficient |
45
Table of Contents
| quantities of drugs to meet commercial needs or clinical supplies of HORIZANT, REGNITE or our product candidates; |
| • | could terminate or choose not to renew manufacturing agreements, based on their own business priorities, at a time that is costly or inconvenient for us or our business partners; |
| • | could fail to establish and follow FDA-mandated current good manufacturing practices, or cGMPs, which are required for FDA approval of our product candidates, or fail to document their adherence to cGMPs, either of which could require costly recalls of products that have already received approval, lead to significant delays in the availability of material for clinical study or delay or prevent marketing approval for our product candidates; |
| • | could encounter financial difficulties that would interfere with their obligations to supply HORIZANT, REGNITE or our product candidates; and |
| • | could breach, or fail to perform as agreed under, manufacturing agreements. |
For example, in 2013, due to manufacturing delays by GSK and Patheon, there was an insufficient amount of HORIZANT in the supply chain to meet the demand of orders of HORIZANT by pharmacies and wholesalers, leading to approximately a one-month delay in our full commercial promotion of HORIZANT.
If we or our business partners are not able to obtain and maintain adequate supplies of HORIZANT, REGNITE or our product candidates, it will have a significant impact on the commercialization efforts for HORIZANT or REGNITE, and will make it more difficult to develop our product candidates. HORIZANT, REGNITE, our product candidates and any products that we may develop may compete with other products and product candidates for access to manufacturing facilities.
In addition, the manufacturing facilities of certain of our suppliers, as well as a storage facility of drug substance, are located outside of the United States. This may give rise to difficulties in importing our products or product candidates or their components into the United States or other countries as a result of, among other things, shipping losses, regulatory agency import inspections, incomplete or inaccurate import documentation, customs detention or seizures or defective packaging.
If our or our business partners’ preclinical studies do not produce successful results or clinical trials do not demonstrate safety and efficacy in humans, we or our business partners will not be able to commercialize our product candidates.
To obtain the requisite regulatory approvals to market and sell any of our product candidates, we and our business partners must demonstrate, through extensive preclinical studies and clinical trials, that the product candidate is safe and effective in humans. Preclinical and clinical testing is expensive, can take many years and has an uncertain outcome. A failure of one or more of our or our business partners’ clinical trials could occur at any stage of testing. For example, in July 2010, GSK announced top-line results from a 30-week, double-blind, placebo-controlled, Phase 2 clinical trial of HORIZANT as a potential prophylactic treatment for migraine headaches in which HORIZANT did not demonstrate a statistically significant improvement on the primary endpoint when compared to placebo. In addition, long-term safety concerns may prevent the approval of any of our product candidates by a regulatory authority. For example, in February 2010, safety concerns related to a preclinical finding of pancreatic acinar cell tumors in rats delayed FDA approval of the HORIZANT NDA at that time. Furthermore, success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and interim results of a clinical trial do not necessarily predict final results. In addition, the results of clinical trials by third parties evaluating a prodrug sharing the same parent drug as our prodrug candidates, including prodrugs of MMF such as TECFIDERA, developed by Biogen and approved by the FDA in March 2013, may not be indicative of the results in clinical trials that we may conduct with our prodrug candidates, including XP23829. Further, unfamiliarity with novel patient-reported outcome tools, trial assessments or endpoints or with certain patient populations, including related subject drop-out rates, could result in additional cost, delay or failure of our clinical trials. For example, in 2013, we announced that our first Phase 3 clinical trial in MS patients with spasticity was unsuccessful in demonstrating that AP provided statistically significant improvement relative to placebo in the co-primary endpoints of the study.
46
Table of Contents
We or our business partners may experience numerous unforeseen events during, or as a result of, preclinical testing and the clinical trial process, which could delay or prevent our or our business partners’ ability to commercialize our product candidates, including:
| • | regulators or institutional review boards may not authorize us to commence a clinical trial at a prospective trial site; |
| • | preclinical testing or clinical trials may produce negative or inconclusive results, which may require the conduct of additional preclinical or clinical testing or abandonment of projects that we expect to be promising; |
| • | we or our business partners may suspend or terminate our clinical trials if the participating patients are being exposed to unacceptable health risks; |
| • | risks associated with clinical trial design may result in a failure of the clinical trial to show statistically significant results even if the product candidate is effective; |
| • | regulators or institutional review boards may suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements; and |
| • | the effects of our product candidates may not be the desired effects or may include undesirable side effects. |
Any failure or delay in commencing or completing clinical trials for our product candidates could severely harm our business.
The commencement and completion of clinical trials for our product candidates may be delayed or terminated as a result of many factors, including:
| • | a clinical hold on an investigational new drug application; |
| • | delays in enrollment, unanticipated high drop-out rates and variability in the number and types of subjects available for clinical trials, all of which we have experienced in the past; |
| • | our or our business partners’ inability to manufacture or obtain from third parties materials sufficient for use in preclinical studies and clinical trials; |
| • | difficulty in maintaining contact with patients after treatment, resulting in incomplete data; |
| • | poor effectiveness of product candidates during clinical trials; |
| • | unforeseen safety issues or side effects; and |
| • | governmental or regulatory delays and changes in regulatory requirements, policies and guidelines. |
Any delay in commencing or completing clinical trials for our product candidates would delay commercialization of our product candidates and severely harm our business and financial condition. In addition, unforeseen safety issues or side effects could result from our business partners’ current or future clinical trials, which could delay or negatively impact the commercialization of our product candidates. It is also possible that none of our product candidates will complete clinical trials in any of the markets in which we or our business partners intend to sell those product candidates. Accordingly, we or our business partners would not receive the regulatory approvals needed to market our product candidates, which would severely harm our business and financial condition.
We rely on third parties to conduct preclinical and clinical trials on our behalf. If these third parties do not perform as contractually required or expected, we may not be able to obtain regulatory approval for, or commercialize, our product candidates.
We do not have the ability to independently conduct clinical trials, and we must rely on third parties, such as contract research organizations, medical institutions, clinical investigators, collaborators, partners and contract
47
Table of Contents
laboratories, to conduct our clinical trials. We have, in the ordinary course of business, entered into agreements with such third parties, and we will continue to do so. Nonetheless, we, as the sponsor, are responsible for confirming that each of our clinical trials is conducted in accordance with its general investigational plan and protocol. Moreover, the FDA requires us to comply with regulations and standards, commonly referred to as good clinical practices, for conducting and recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the trial participants are adequately protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements. For example, we need to prepare, and ensure our compliance with, various procedures required under good clinical practices, even though third-party contract research organizations have prepared and are complying with their own, comparable procedures. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if the third parties need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our preclinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval for our product candidates, or successfully commercialize HORIZANT.
As an illustrative example, in 2011, the FDA announced that certain bioanalytical studies conducted by a contract research organization may need to be repeated or confirmed by the pharmaceutical company sponsors of the marketing applications that included such studies. The FDA’s decision was the result of two inspections and an internal audit at a facility that identified significant instances of misconduct and violations of federal regulations, including falsification of documents and manipulation of samples. Although we have not contracted with this contract research organization for any studies or clinical trials, if one of the contract research organizations that conducted trials on our behalf were found to have similar or other violations, the FDA may require such trials to be repeated or it may affect the approvability of our product candidates and harm our business.
HORIZANT and REGNITE remain, and future products, if any, will remain, subject to ongoing regulatory review. If we or our business partners fail to comply with continuing regulations, these approvals could be rescinded and the sale of our products could be suspended.
Any regulatory approval to market a product could be conditioned on conducting additional, costly, post-approval studies or implementing a risk evaluation and mitigation strategy or could contain strict limits on the indicated uses included in the labeling. For example, the FDA approval of HORIZANT for the treatment of moderate-to-severe primary RLS included requirements for GSK to conduct a program of post-marketing commitments, or PMCs, and post-marketing requirements, or PMRs, in adults, including: a 12-week, double-blind, placebo-controlled efficacy study evaluating 300 mg, 450 mg and 600 mg tablets of HORIZANT dosed once per day (“low-dose efficacy study”); two simulated driving studies; a drug-drug interaction study with morphine; and a cardiovascular safety, or QTc, study. GSK also agreed to conduct a pediatric program for subjects 13 years and older. The pediatric clinical program, which was not completed by GSK and is scheduled to commence after requested adult data is obtained and reviewed by the FDA, includes: a pharmacokinetics, or PK, study; a parallel, fixed-dose response efficacy study; a long-term safety study; and a simulated driving study. The specific protocol submission and trial completion dates for these PMCs/PMRs range from April 2011 through July 2024. Although GSK completed some of these PMCs/PMRs, including conduct of the low-dose efficacy study, we are responsible for fulfilling the remaining, and any additional future, post-marketing study requirements, which will be expensive and time-consuming and may divert management time and resources away from our commercialization efforts or the development of our product candidates.
HORIZANT has certain warnings and precautions in the label, including information that HORIZANT may cause significant driving impairment. A medication guide, which contains information about the labeling intended for the patient, is also required to be distributed with HORIZANT. Moreover, a product may later cause adverse effects that limit or prevent its widespread use, force us to withdraw it from the market or impede or delay our ability to obtain regulatory approvals in additional countries or indications. In addition, the contract manufacturer of a product and its facilities will continue to be subject to FDA review and periodic inspections to ensure adherence to applicable regulations, and the manufacturing, labeling, packaging, adverse event reporting,
48
Table of Contents
storage, advertising, promotion and record keeping related to HORIZANT, REGNITE and any future products remain subject to extensive regulatory requirements. Furthermore, the NIAAA plans to initiate a clinical trial evaluating HORIZANT in patients with AUD that will be subject to statutory reporting of safety information and subject to FDA review and potential inspections to ensure adherence to applicable regulations. There is always the risk that new data generated from the proposed NIAAA clinical trial, or from investigator-initiated clinical trials of HORIZANT, could be perceived in a negative fashion by either the physician community or the health authorities. Likewise, adverse events or safety concerns involving HORIZANT observed in the proposed NIAAA clinical trial or in investigator-initiated clinical trials could result in regulatory authorities denying or withdrawing approval of HORIZANT for any or all indications, including the use of HORIZANT for the treatment of patients in its currently-approved indications.
We and our business partners are also subject to regulation by regional, national, state and local agencies, including the Department of Justice, the Federal Trade Commission, the Office of Inspector General of the U.S. Department of Health and Human Services and other regulatory bodies, as well as governmental authorities in those foreign countries in which we may commercialize our products. The FDCA, the Public Health Service Act and other federal and state statutes and regulations govern to varying degrees the research, development, manufacturing and commercial activities relating to prescription pharmaceutical products, including preclinical testing, approval, production, labeling, sale, distribution, import, export, post-market surveillance, advertising, dissemination of information and promotion. These statutes and regulations include, but are not limited to, anti-kickback statutes and false claims statutes.
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration, directly or indirectly, to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare good, facility, item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical companies on the one hand and prescribers, purchasers and formulary managers on the other. Although there are a number of statutory exceptions and regulatory safe harbors protecting identified common activities from prosecution, the exceptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exception or safe harbor.
Federal false claims laws prohibit, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment or approval to the federal government, or knowingly making, or causing to be made, a false statement to get a false claim paid.
Recently, several pharmaceutical and other healthcare companies have been prosecuted under these laws for, among other things, allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. Other companies have been prosecuted for causing false claims to be submitted because of the company’s marketing of the product for unapproved, and thus non-reimbursable, uses. Pharmaceutical and other healthcare companies have also been prosecuted on other legal theories of Medicare fraud. Federal law requires applicable manufacturers of covered drugs to report payments and other transfers of value to physicians and teaching hospitals, and several states have similar healthcare professional aggregate spend reporting obligations or prohibitions. Other states require the posting of information relating to clinical studies. In addition, California requires pharmaceutical companies to implement a comprehensive compliance program that includes a limit on expenditures for, or payments to, individual medical or health professionals. We have adopted a comprehensive compliance program that we believe complies with California law. Several additional states are considering similar proposals.
In addition to the federal Anti-Kickback Statute and federal false claims laws, the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and its implementing regulations, also imposes certain requirements relating to the privacy, security and transmission of individually identifiable personal information, including health information. State and foreign laws governing the privacy and security of health information in certain circumstances differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
49
Table of Contents
The majority of states also have statutes or regulations similar to the federal healthcare laws that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer.
It is possible that some of our business activities could be subject to challenge under one or more of such laws. Such a challenge could have a material adverse effect on our business, financial condition, results of operations and growth prospects. If we or our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including potentially significant criminal, civil and/or administrative penalties, damages, fines, individual imprisonment, exclusion of products from reimbursement under U.S. federal or state healthcare programs, contractual damages, reputational harm, administrative burdens, diminished profits and future earnings and the curtailment or restructuring of our operations, any of which could materially adversely affect our ability to operate our business and our financial results.
Although compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, the risks cannot be entirely eliminated. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business. Moreover, achieving and sustaining compliance with these laws may prove costly.
We have contracted with a contract sales organization that currently employs our contract sales representatives who help promote HORIZANT as part of our dedicated sales team. In addition, although we have established the XenoPort-employed sales force by hiring sales representatives from our contract sales organization, we continue to rely on that organization to provide back office support for the XenoPort-employed sales representatives. However, we expect that government and regulatory agencies will hold us responsible for any actions by any member of our sales team regardless of whether they are employed directly by XenoPort or by our contract sales organization. If we or our contract sales organization fails to comply with the regulatory requirements of the FDA and other applicable U.S. and foreign regulatory authorities or if previously unknown problems with our products, manufacturers or manufacturing processes are discovered, we and our business partners could be subject to administrative or judicially imposed sanctions, including:
| • | restrictions on our products, manufacturers or manufacturing processes; |
| • | warning letters; |
| • | civil or criminal penalties or fines; |
| • | injunctions; |
| • | product seizures, detentions or import bans; |
| • | voluntary or mandatory product recalls and publicity requirements; |
| • | suspension or withdrawal of regulatory approvals; |
| • | total or partial suspension of production; and |
| • | refusal to approve pending applications for marketing approval of new drugs or supplements to approved applications. |
If we or our business partners are not able to obtain or maintain required regulatory approvals, we or our business partners will not be able to commercialize HORIZANT, REGNITE or our product candidates, our ability to generate revenue will be materially impaired and our business will not be successful.
Our product candidates and the activities associated with their development and commercialization are subject to comprehensive regulation by the FDA and other agencies in the United States and by comparable authorities in other countries. The inability to obtain or maintain FDA approval or approval from comparable authorities in other countries would prevent us and our business partners from commercializing HORIZANT, REGNITE or our product candidates in the United States or other countries. Although HORIZANT and
50
Table of Contents
REGNITE have been approved for commercial sale in the United States and Japan, respectively, we may never receive regulatory approval for the commercial sale of our product candidates. In addition, even if a product candidate ultimately receives regulatory approval, the regulatory process may include significant delays that could harm our business. For example, in 2010, our former collaborator GSK received a Complete Response letter from the FDA in which a preclinical finding of pancreatic acinar cell tumors in rats precluded approval of the HORIZANT NDA for the treatment of moderate-to-severe primary RLS at that time. GSK responded to questions raised by the FDA in the Complete Response letter with an NDA resubmission and amended the NDA from a Section 505(b)(1) to a 505(b)(2) application in order for the FDA to be able to consider published gabapentin nonclinical data in their assessment of HORIZANT. HORIZANT subsequently received approval from the FDA in 2011. However, our business was harmed due to the delay in obtaining approval for HORIZANT as a treatment for moderate-to-severe primary RLS. Moreover, if the FDA requires that any of our products or product candidates be scheduled by the U.S. Drug Enforcement Agency, or DEA, we or our business partners will be unable to continue or begin commercial sale of that product until the DEA completes scheduling proceedings. If any of our products or product candidates is classified as a controlled substance by the DEA, we or our business partners would have to register annually with the DEA and those products or product candidates would be subject to additional regulation.
We have only limited experience in preparing and filing the applications necessary to gain regulatory approvals. The process of applying for regulatory approval is expensive, often takes many years and can vary substantially based upon the type, complexity and novelty of the product candidates involved. The application process begins with the submission of an NDA that the FDA initially reviews and either accepts or rejects for filing. NDA submissions are complex electronic filings, which include vast compilations of data sets, integrated documents and data calculations. The FDA has substantial discretion in the submission process and may refuse to accept an NDA submission for any reason, including insufficient information or if there are errors or omissions relating to the electronic transmittal process, data entry, data compilation or formatting. For example, in 2008, GSK withdrew a previously submitted NDA for HORIZANT for the treatment of moderate-to-severe primary RLS in connection with the FDA’s request that the data from a single study be reformatted.
Changes in the regulatory approval policy during the development period, changes in, or the enactment of, additional regulations or statutes or changes in regulatory review for each submitted product application may cause delays in the approval or rejection of an NDA. If the FDA were to miss a Prescription Drug User Fee Act, or PDUFA, timing goal for one of our product candidates, the development and commercialization of the product candidate could be delayed or impaired. For example, in 2009, the FDA notified GSK that it was extending the PDUFA timing goal for HORIZANT for the treatment of moderate-to-severe primary RLS for three months. In addition, the FDA may convene an advisory committee at any time during the review process. The advisory committee review process can be a lengthy and uncertain process that could delay the FDA’s NDA approval and delay or impair the development and commercialization of our product candidates.
The FDA has substantial discretion in the approval process and may refuse to approve any application or decide that our data is insufficient for approval and require additional preclinical, clinical or other studies. Varying interpretations of the data obtained from preclinical and clinical testing or other studies could delay, limit or prevent regulatory approval of any of our product candidates. As part of their review process, the FDA could require additional studies or trials to satisfy particular safety concerns. For example, although we had discussions with the FDA in 2012 regarding the studies required by the FDA to support an NDA submission for XP21279 for the potential treatment of advanced idiopathic Parkinson’s disease, when or if we decide to pursue these studies and the approval of XP21279, the FDA could change their guidance or require additional studies, causing delay or the expenditure of additional resources. Even if the FDA or other regulatory agency approves a product candidate, the approval may impose significant restrictions on the indicated uses, conditions for use, labeling, advertising, promotion, marketing and/or production of such product and may impose ongoing commitments or requirements for post-approval studies, including additional research and development and clinical trials, and we or our business partners may be unable to maintain regulatory approvals for our products. In addition, the FDA and other agencies also may impose various civil or criminal sanctions for failure to comply with regulatory requirements, including withdrawal of product approval.
51
Table of Contents
We and our business partners will need to obtain regulatory approval from authorities in foreign countries to market our product candidates in those countries. Approval by one regulatory authority does not ensure approval by regulatory authorities in other jurisdictions. If we or our business partners fail to obtain approvals from foreign jurisdictions, the geographic market for our product candidates would be limited.
An NDA submitted under Section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act subjects us to the risk that we may be subject to a patent infringement lawsuit that would delay or prevent the review and approval of our product candidate.
Certain product candidates that we develop, including XP23829, could potentially be submitted to the FDA for approval under Section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act, as amended, or FDCA, which was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act. Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from studies not conducted by, or for, the applicant and for which the applicant has not obtained a right of reference.
For NDAs submitted under Section 505(b)(2) of the FDCA, the patent certification and related provisions of the Hatch-Waxman Act apply. In accordance with the Hatch-Waxman Act, such NDAs may be required to include certifications, known as Paragraph IV certifications, that certify that any patents listed in the Patent and Exclusivity Information Addendum of the FDA’s publication, Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book, with respect to any product referenced in the Section 505(b)(2) application, are invalid, unenforceable or will not be infringed by the manufacture, use or sale of the product that is the subject of the Section 505(b)(2) application. Under the Hatch-Waxman Act, the holder of patents that the Section 505(b)(2) application references may file a patent infringement lawsuit after receiving notice of the Paragraph IV certification. Filing of a patent infringement lawsuit within 45 days of the patent owner’s receipt of notice triggers a one-time, automatic, up to 30-month stay of the FDA’s ability to approve the 505(b)(2) application. For example, we are aware that there are seven patents listed in the Orange Book for TECFIDERA that have expiration dates between April 2018 and February 2028. If we file a Section 505(b)(2) application referencing TECFIDERA data, we will have to provide a Paragraph IV certification against any such listed patents if they have not expired by that time. Although we believe that an approved XP23829 product would not infringe the claims in the listed patents, that the listed patents would expire before any such potential Section 505(b)(2) submission would otherwise occur based on our current anticipated product development timelines, and/or that the claims in the listed patents are invalid and/or unenforceable, if we provide a Paragraph IV certification, the patent holder may file a patent infringement lawsuit, triggering up to a 30-month stay on the FDA’s ability to approve our Section 505(b)(2) application. Accordingly, we may invest a significant amount of time and expense in the development of one or more products only to be subject to significant delay and patent litigation before such products may be commercialized, if at all. A Section 505(b)(2) application may also not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, or NCE, listed in the Orange Book for the referenced product has expired. The FDA may also require us to perform one or more additional clinical studies or measurements to support the change from the approved product. To avoid such potential delay or for other reasons, we may pursue a full development pathway of such product candidate under Section 505(b)(1) of the FDCA, which could be more expensive and time consuming. In this regard, while we are in discussions with the FDA about potentially pursuing XP23829 development under the Section 505(b)(2) pathway, we believe at this time that it is likely that we will pursue a full development pathway for XP23829 under Section 505(b)(1) of the FDCA. Even if we submit an NDA under Section 505(b)(2), the FDA may reject our future Section 505(b)(2) submissions and require us to file such submissions under Section 505(b)(1). These factors, among others, may limit our ability to successfully commercialize our product candidates.
Current healthcare laws and regulations and future legislative or regulatory reforms to the healthcare system may affect our ability to profitably sell any products that we may develop.
The United States and some foreign jurisdictions are considering, or have enacted, a number of legislative and regulatory proposals designed to change the healthcare system in ways that could affect our ability to sell our
52
Table of Contents
products profitably. Among policy makers and payers in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
In 2010, PPACA became law in the United States. PPACA has the potential to substantially change healthcare financing and delivery by both governmental and private insurers, and significantly impact the pharmaceutical industry. Among the provisions of PPACA of importance to the pharmaceutical industry are the following:
| • | an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs; |
| • | an increase in the rebates a manufacturer must pay under the Medicaid Drug Rebate Program, retroactive to January 1, 2010, to 23.1% and 13% of the average manufacturer price for branded (with certain exceptions) and generic drugs, respectively; |
| • | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to reimburse Medicare Part D sponsors 50% off the wholesale acquisition cost of applicable brand drugs utilized by eligible beneficiaries during their coverage gap period, as a condition for the manufacturers’ outpatient drugs to be covered under Medicare Part D. The coverage discount is subject to increase; |
| • | extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| • | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for certain individuals with income at or below 133% of the federal poverty level that took effect in 2014, thereby potentially increasing manufacturers’ Medicaid rebate liability; |
| • | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; |
| • | new requirements to report annually certain payments or “transfers of value” provided to physicians and teaching hospitals, as each is defined in PPACA and its implementing regulations, and to report any ownership and investment interests held by physicians and their immediate family members during the preceding calendar year, with data collection requirements that began on August 1, 2013 and to report to the Centers for Medicare & Medicaid Services on an annual basis; |
| • | a new requirement to track and annually report certain drug samples that manufacturers and distributors provide to licensed practitioners; |
| • | expansion of healthcare laws, including the False Claims Act and the Anti-Kickback Statute, new government investigative powers and enhanced penalties for noncompliance under certain healthcare laws; |
| • | a licensure framework for follow-on biologic products; and |
| • | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
In addition, other legislative changes have been proposed and adopted since PPACA was enacted. In 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, or the ATRA, which, among other things, reduced
53
Table of Contents
Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on our customers and accordingly, our financial operations.
Additionally, other recent federal legislation imposes new obligations on manufacturers of pharmaceutical products, among others, related to product tracking and tracing. Among the requirements of this new legislation, manufacturers are required to provide certain information regarding the drug product to individuals and entities to which product ownership is transferred, label drug product with a product identifier, and keep certain records regarding the drug product. Further, under this new legislation, manufacturers will have drug product investigation, quarantine, disposition, notification and purchaser license verification responsibilities related to counterfeit, diverted, stolen, and intentionally adulterated products, as well as products that are the subject of fraudulent transactions or which are otherwise unfit for distribution such that they would be reasonably likely to result in serious health consequences or death. In addition, two counties (King County, Washington and Alameda County, California) have enacted pharmaceutical manufacturer take-back ordinances/regulations that place primary responsibility for end-of-life management of drug products on drug producers. These new take-back programs will require the creation, administration, promotion and payment of costs including county administrative costs of administering and enforcing the programs.
We anticipate that PPACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and an additional downward pressure on the price that we receive for HORIZANT and any other approved product, and could seriously harm our business. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payers. Insurers may also refuse to provide any coverage of uses of approved products for medical indications other than those for which the FDA has granted market approvals. As a result, significant uncertainty exists as to whether and how much third-party payers will reimburse for HORIZANT and any newly-approved drugs, which in turn will put pressure on the pricing of drugs.
We also cannot be certain that HORIZANT or any other products that may result from our product candidates will successfully be placed on the list of drugs covered by particular health plan formularies, nor can we predict the negotiated price for such products, which will be determined by market factors. Many states have also created preferred drug lists and include drugs on those lists only when the manufacturers agree to pay a supplemental rebate. If HORIZANT or other products that may result from our product candidates are not included on these preferred drug lists, physicians may not be inclined to prescribe them to their patients, thereby diminishing the potential market for such products. Astellas will face similar pricing and reimbursement restrictions in Japan for REGNITE, and further efforts to reform the Japanese healthcare system may increase such restrictions.
If some or all of our patents expire, are invalidated or are unenforceable, or if some or all of our patent applications do not yield issued patents or yield patents with narrow claims, competitors may develop competing products using our intellectual property and our business will suffer.
Our success will depend in part on our ability to obtain and maintain patent and trade secret protection for our technologies, HORIZANT, REGNITE and our product candidates both in the United States and other countries. We have a number of U.S. and foreign patents, patent applications and rights to patents related to our compounds, product candidates, products and technology, but we cannot guarantee that issued patents will be enforceable or that pending or future patent applications will result in issued patents. Alternatively, a third-party may successfully circumvent our patents. Our rights under any issued patents may not provide us with sufficient protection against competitive products or otherwise cover commercially valuable products or processes.
The degree of future protection for our proprietary technologies, HORIZANT, REGNITE and our product candidates is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
| • | it is possible that we will fail to identify patentable aspects of inventions made in the course of development and commercialization activities before it is too late to obtain patent protection on them; |
54
Table of Contents
| • | we might not have been the first to make the inventions covered by each of our pending patent applications and issued patents; |
| • | we might not have been the first to file patent applications for these inventions; |
| • | others may independently develop similar or alternative technologies or duplicate any of our technologies; |
| • | it is possible that any one or more of our pending patent applications will not result in issued patents; |
| • | it is possible that the claims of our pending patent applications will be narrowed during prosecution, which may limit the scope of patent protection that may be obtained; |
| • | any patents issued to us or our business partners may not provide a basis for commercially viable products or may be challenged by third parties; or |
| • | the patents of others may have an adverse effect on our ability to do business. |
Even if valid and enforceable patents cover HORIZANT, REGNITE or our product candidates and technologies, the patents will provide protection only for a limited amount of time.
Our and our business partners’ ability to obtain patents is highly uncertain because, to date, some legal principles remain unresolved, there has not been a consistent policy regarding the breadth or interpretation of claims allowed in patents in the United States and the specific content of patents and patent applications that are necessary to support and interpret patent claims is highly uncertain due to the complex nature of the relevant legal, scientific and factual issues. Furthermore, the policies governing biotechnology patents outside the United States are even more uncertain. Changes in either patent laws or interpretations of patent laws in the United States and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection. For example, in 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to United States patent law. These changes include provisions that affect the way patent applications are being filed, prosecuted and litigated. The United States Patent Office has developed new and untested regulations and procedures to govern the full implementation of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, only became effective in March 2013. While we cannot predict with certainty the impact the Leahy-Smith Act will have on the operation of our business, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations and growth prospects. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we and our business partners may not be able to prevent third parties from practicing our and our business partners’ inventions in countries outside the United States, or from selling or importing products made using our and our business partners’ inventions in and into the United States or other jurisdictions. Competitors may use our and our business partners’ technologies in jurisdictions where we have not obtained patent protection to develop their own products and may export otherwise infringing products to territories where we and our business partners have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our products and our and our business partners’ patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Our issued patents regarding HORIZANT and REGNITE and any future patents that may issue regarding these products or our product candidates or methods of using them can be challenged by our competitors who can argue such patents are invalid and/or unenforceable. For example, in 2008, a law firm on behalf of an undisclosed client filed an opposition against the patent grant of one of our European patent applications covering gabapentin enacarbil. The European Patent Office in opposition proceedings in 2010, undertook a formal review of the issues raised by the opponent and decided to maintain the grant of our European patent covering the composition of matter of gabapentin enacarbil. While the law firm that filed the opposition initially appealed the ruling on behalf of the undisclosed client, that appeal was withdrawn in 2010. Patents also may not protect HORIZANT,
55
Table of Contents
REGNITE or our product candidates if competitors devise ways of making them or similar products without legally infringing our patents. The FDCA and FDA regulations and policies provide incentives to manufacturers to challenge patent validity and these same types of incentives encourage manufacturers to submit NDAs that rely on literature and clinical data not prepared for or by the drug sponsor.
We may obtain patents for certain product candidates many years before marketing approval is obtained for those products. Because patents have a limited life, which may begin to run prior to the commercial sale of the related product, the commercial value of the patent may be limited. However, we may be able to apply for patent term extensions.
As part of the approval process of our product candidates in the United States, the FDA may determine that the product candidates be granted an exclusivity period during which other manufacturers’ applications for approval of generic versions of our products will not be accepted and/or approved. Generic manufacturers can submit an abbreviated new drug application, or ANDA, referencing an approved product at any time after the acceptance exclusivity period has tolled for the approved product, and thereby initiate a challenge to the approved product’s listed patents under a Paragraph IV certification. In addition, the FDA can approve an ANDA any time after the approval exclusivity period (if different from the acceptance exclusivity) has tolled. For example, the FDA granted HORIZANT five years of data exclusivity based on it being an NCE. Thus, after April 2015, the end of the first four years of the exclusivity period, the FDA could accept an ANDA that contains unauthorized reference to HORIZANT, and in April 2016, after the full five-year exclusivity period has tolled, the FDA could approve such ANDA submissions. It is possible that generic manufacturers are considering attempts to seek FDA approval for a similar or identical drug as HORIZANT through an ANDA, which would require a Paragraph IV certification, thereby triggering challenges to our Orange Book-listed patents. If our patents are subject to such challenges, we may need to spend significant resources to defend such challenges and we may not be able to defend our patents successfully. We expect that the approval of an ANDA that results in the launch of a similar or identical drug as HORIZANT could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
In June 2012, HORIZANT was granted orphan drug designation by the FDA for treatment of PHN. As a result of this designation, we are entitled to seven years of orphan drug exclusive approval of HORIZANT for the management of PHN in adults. As the holder of this orphan drug exclusivity, we are required to assure the availability of sufficient quantities of HORIZANT to meet the needs of patients. Failure to do so could result in withdrawal of our orphan drug exclusivity.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements. Periodic maintenance and annuity fees on any issued patent are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we or our business partners fail to maintain the patents and patent applications covering HORIZANT, REGNITE or our product candidates, our competitors might be able to enter the market, which would have a material adverse effect on our business.
We also rely on trade secrets to protect our technology, especially where we do not believe that patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. Our employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our confidential information to competitors. Enforcing a claim that a third-party illegally obtained and is using our trade secrets is expensive and time-consuming, and the outcome is unpredictable. Failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
56
Table of Contents
Our research and development collaborators may have rights to publish data and other information in which we have rights. In addition, we sometimes engage individuals or entities to conduct research that may be relevant to our business. The ability of these individuals or entities to publish or otherwise publicly disclose data and other information generated during the course of their research is subject to certain contractual limitations. In most cases, these individuals or entities are, at the least, precluded from publicly disclosing our confidential information and are only allowed to disclose other data or information generated during the course of the research after we have been afforded an opportunity to consider whether patent and/or other proprietary protection should be sought. If we do not apply for patent protection prior to such publication or if we cannot otherwise maintain the confidentiality of our technology and other confidential information, then our ability to receive patent protection or protect our proprietary information may be jeopardized.
We may become involved in lawsuits and other proceedings to protect, defend or enforce our patents or other intellectual property, which could be expensive, time consuming and unsuccessful.
Competitors and other third parties may misappropriate, infringe or otherwise violate our intellectual property rights including our patent, trademark, copyright and know-how rights. To counter such infringement or other violations, we may be required to file legal actions such as claims, suits, proceedings and/or litigation, which can be expensive, distracting and time consuming. Any such legal actions could provoke the relevant parties to assert counterclaims against us, including claims alleging that we infringe their patents, have misappropriated their proprietary know-how or otherwise violated their own intellectual property rights. In addition, in a patent infringement proceeding, a court may decide that one or more of the patents we assert against a third-party is invalid or unenforceable, in whole or in part, construe the patent’s claims narrowly or refuse to prevent the other party from using the technology at issue on the grounds that our patents do not cover the activities. Similarly, if we assert trademark infringement claims, a court may determine that the marks we have asserted are invalid or unenforceable or that the party against whom we have asserted trademark infringement has superior rights to the marks in question. In such a case, we could ultimately be forced to cease our use of such marks. We may also become involved in other disputes relating to our intellectual property rights, such as opposition, post-grant review, derivation, interference or re-examination proceedings before the USPTO or its foreign counterparts and, if we are unable to successfully resolve any such disputes, we could lose rights in our valuable intellectual property. In any intellectual property disputes, even if we are successful, any award of monetary damages, injunction or other remedy we receive may not be commercially valuable. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
Third-party claims of intellectual property infringement would require us to spend significant time and money and could prevent us from developing or commercializing our products.
Our commercial success depends in part on not infringing the patents and proprietary rights of other parties and not breaching any licenses that we have entered into with regard to our technologies and products. Because others may have filed, and in the future are likely to file, patent applications covering products or other technologies of interest to us that are similar or identical to ours, patent applications or issued patents of others may have priority over our patent applications or issued patents. For example, we are aware of a family of third-party patent applications relating to prodrugs of gabapentin. We believe the applications have been abandoned in the United States, the European Patent Office, Canada, Australia and the United Kingdom. Additionally, with respect to the development of XP23829, we are aware of third-party patents and patent applications relating to the use of fumarates in the treatment of MS and psoriasis, including several such patents held by Biogen and included in the TECFIDERA Orange Book listing. With respect to the claims contained in these patent applications and patents, we believe that an approved XP23829 product would not infringe the claims of the patents at issue, that the patents would expire before any infringement would otherwise occur based on our current anticipated product development timelines, and/or that the claims in the third-party patents or patent applications are invalid and/or unenforceable. In addition, we believe that in all countries in which we hold or have licensed rights to patents or patent applications related to HORIZANT, REGNITE and gabapentin
57
Table of Contents
enacarbil, the composition-of-matter patents on gabapentin, the metabolite of gabapentin enacarbil, have all expired. However, it is possible that a judge or jury will disagree with our conclusions regarding non-infringement, invalidity and/or expiration of these patents that may relate to XP23829 and HORIZANT, and we could incur substantial costs in litigation if we are required to defend against patent suits brought by third parties or if we initiate these suits. In addition, there could be other third-party patents or patent applications covering certain aspects of our planned development or commercialization activities that we are not yet aware of. Any legal action against our current or potential future business partners or us claiming damages and seeking to enjoin commercial activities relating to the affected products and processes could, in addition to subjecting us to potential injunction that could prevent us from selling the affected products, expose us to potential liability for damages and require our current or potential future business partners or us to obtain a license to continue to manufacture or market the affected products and processes. Licenses required under any of these patents may not be available on commercially acceptable terms, if at all. Failure to obtain such licenses could materially and adversely affect our ability to develop, commercialize and sell HORIZANT, REGNITE or our product candidates. Such legal actions against us could also include the theory of contributory infringement, or claiming that because our prodrugs are broken down in the body into an active metabolite and other substances, that we have infringed on patents that cover the use of the active metabolite. We believe that there may continue to be significant litigation in the biotechnology and pharmaceutical industry regarding patent and other intellectual property rights. If we become involved in litigation, it could consume a substantial portion of our management and financial resources and we may not prevail in any such litigation.
Furthermore, our commercial success will depend, in part, on our and our business partners’ ability to develop additional product candidates in current indications of interest or opportunities in other indications. Some of these activities may involve the use of genes, gene products, screening technologies and other research tools that are covered by third-party patents. Court decisions have indicated that the exemption from patent infringement afforded by the Hatch-Waxman Act does not encompass all research and development activities associated with product development. In some instances, we may be required to obtain licenses to such third-party patents to conduct our development activities, including activities that may have already occurred. It is not known whether any license required under any of these patents would be made available on commercially acceptable terms, if at all. Failure to obtain such licenses could materially and adversely affect our ability to maintain a pipeline of potential product candidates and to bring new products to market. If we are required to defend against patent suits brought by third parties relating to third-party patents that may be relevant to our development activities, or if we initiate such suits, we could incur substantial costs in litigation. Moreover, an adverse result from any legal action in which we are involved could subject us to damages and/or prevent us from conducting some of our development activities.
We may expend our limited resources to market or sell a product or pursue the development of a particular candidate or indication and fail to capitalize on other products or product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we must focus on products and product candidates for the specific indications that we believe are the most promising. As a result, we may forego or delay pursuit of opportunities with other product candidates or other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products, including HORIZANT, or profitable market opportunities. In addition, we may spend valuable time and managerial and financial resources on marketing or promoting a product that is not commercially viable or developing product candidates for specific indications that ultimately do not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product or product candidate, we may relinquish valuable rights to that product or product candidate through strategic arrangements in situations where it would have been more advantageous for us to retain sole rights to development and commercialization.
58
Table of Contents
Safety issues with HORIZANT, REGNITE or our product candidates, or the parent drugs or other components of HORIZANT, REGNITE or our product candidates, or with approved products of third parties that are similar to HORIZANT, REGNITE or our product candidates, could decrease sales of HORIZANT and REGNITE, or give rise to delays in the regulatory approval process, restrictions on labeling or product withdrawal.
Discovery of previously unknown problems, or increased focus on a known problem, with an approved product may result in restrictions on its permissible uses, including withdrawal of the medicine from the market. The label for HORIZANT currently includes warnings and precautions related to driving impairment, somnolence/sedation and dizziness, lack of interchangeability with gabapentin, suicidal behavior or ideation, multiorgan hypersensitivity, discontinuation and tumorigenic potential. Under our clinical trial agreement with the NIAAA, the NIAAA plans to conduct a new clinical trial evaluating HORIZANT in patients with AUD which will be subject to statutory reporting of safety information and subject to FDA review. If we, our collaborator or others later identify undesirable side effects caused by HORIZANT or any of our other product candidates that receive marketing approval:
| • | regulatory authorities may require the addition of labeling statements, specific warnings, contraindications or field alerts to physicians and pharmacies; |
| • | regulatory authorities may withdraw their approval of the product and require us to take our approved drug off the market; |
| • | we may be required to change the way the product is administered, conduct additional clinical trials, change the labeling of the product or conduct a Risk Evaluation and Mitigation Strategies, or REMS, program; |
| • | we may have limitations on how we promote our drugs; |
| • | sales of products may decrease significantly; |
| • | we may be subject to litigation or product liability claims; and |
| • | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product or could substantially increase our commercialization costs and expenses, which in turn could delay or prevent us from generating significant revenues from its sale.
HORIZANT, REGNITE and our product candidates may also be affected by the safety of the parent drugs or drugs related to our products or product candidates. Although gabapentin, baclofen (which includes the R-isomer of baclofen) and levodopa, the parent drugs of HORIZANT/REGNITE/gabapentin enacarbil, AP and XP21279, respectively, have been used successfully in patients for many years, newly observed toxicities or worsening of known toxicities, in preclinical studies of, or in patients receiving, gabapentin, baclofen or levodopa, or reconsideration of known toxicities of gabapentin, baclofen or levodopa in the setting of new indications, could result in increased regulatory scrutiny of HORIZANT/REGNITE/gabapentin enacarbil, AP and XP21279, respectively. For example, the label for baclofen, the R-isomer of which is the parent drug of AP, includes a warning that hallucinations and seizures have occurred on abrupt withdrawal of baclofen dosing without proper tapering in spasticity patients. As another example, although a product called FUMADERM, which contains fumaric acid ester compounds, including dimethyl fumarate, or DMF (another prodrug of MMF), has been approved and used in Germany for the treatment of psoriasis, FUMADERM has not been approved in the United States. In addition, in March 2013, the FDA approved Biogen’s TECFIDERA as a treatment for relapsing forms of MS. Any safety concerns or other problems noted by regulators with respect to DMF, TECFIDERA or MMF could increase the risk of regulatory scrutiny of XP23829, possibly delaying or preventing any regulatory approval of XP23829. For example, it has been reported that some patients taking FUMADERM developed progressive multifocal leukoencephalopathy, or PML, a progressive brain disease. Furthermore, in October 2014, Biogen announced that an MS patient developed PML after having taken TECFIDERA for four and a half years as part of Biogen’s ENDORSE clinical trial, during which time the patient apparently experienced severe lymphopenia for over three and a half years, and the patient died from
59
Table of Contents
complications of pneumonia. In its SEC filings, Biogen also notes the potential risk of PML cases that may develop in patients previously treated with TYSABRI who switched to therapy with TECFIDERA. If a link between PML and TECFIDERA, DMF or MMF is established in the future, it could increase regulatory scrutiny of XP23829 or delay or prevent its approval.
The FDA has substantial discretion in the NDA approval process and may refuse to approve any application if the FDA concludes that the risk/benefit analysis of a potential drug treatment for a specific indication does not warrant approval. For example, in 2010, safety concerns related to a preclinical finding of pancreatic acinar cell tumors in rats precluded FDA approval of the HORIZANT NDA in RLS in its form at that time. Although there were similar findings of rat pancreatic acinar cell tumors following treatment with gabapentin, the parent drug of HORIZANT, the FDA has, to date, not prevented the use of gabapentin. In the 2010 Complete Response letter, the FDA noted that they had concluded that the seriousness and severity of refractory epilepsy and the benefit to patients provided by gabapentin justified the potential risk at that time. Thus, although the parent drug for, or a drug related to, one of our product candidates may be approved by the FDA in a particular indication, the FDA may conclude that our product candidate’s risk/benefit profile does not warrant approval in a different indication, and the FDA may delay or refuse to approve our product candidate. Such conclusion and refusal would prevent us from developing and commercializing our product candidates and severely harm our business and financial condition. For example, even with the approval of TECFIDERA for relapsing forms of MS, the FDA may not agree that the risk/benefit profile of XP23829 for the treatment of psoriasis or other potential indications, if established, would warrant approval in such indications. HORIZANT, REGNITE and our product candidates are engineered to be broken down by the body’s natural metabolic processes and to release the active drug and other substances. While these breakdown products are generally regarded as safe, it is possible that there could be unexpected toxicity associated with these breakdown products that will cause any or all of HORIZANT/REGNITE/gabapentin enacarbil, XP23829, XP21279 and AP to be poorly tolerated by, or toxic to, humans. Any unexpected toxicity of, or suboptimal tolerance to, our product or product candidates could reduce sales of HORIZANT and REGNITE, and delay or prevent commercialization of our product candidates.
Additionally, problems with approved products marketed by third parties that utilize the same therapeutic target or that belong to the same therapeutic class as the parent drug of HORIZANT, REGNITE or our product candidates could adversely affect the commercialization of HORIZANT or REGNITE or the development of our product candidates. For example, if either gabapentin or pregabalin, drugs from Pfizer that are marketed as NEURONTIN and LYRICA, respectively, encounters unexpected toxicity problems in humans, the FDA may restrict the use of HORIZANT since it is believed to share the same therapeutic target as gabapentin and pregabalin. In 2008, the FDA added warnings to 11 antiepileptic drugs, including gabapentin, regarding an increased risk of suicide or suicidal thoughts. In 2009, the FDA approved safety label changes for all approved antiepileptic drugs, except those indicated only for short-term use, to include a warning about an increased risk of suicidal thoughts or actions. In addition, in 2011, the FDA added warnings to the labels of antiepileptic drugs regarding an increased risk of drug reaction with Eosinophilia and Systemic Symptoms, or DRESS, also known as multiorgan hypersensitivity, which has been reported in patients taking antiepileptic drugs. HORIZANT, as a compound that is believed to share the same therapeutic target as antiepileptic drugs such as gabapentin and pregabalin, has similar warnings regarding suicidality and DRESS in its label. Additional scrutiny could be placed on HORIZANT if it is found to have an increased risk of suicides or suicidal behavior. In 2010, the FDA released draft guidance recommending that prospective suicidality assessments be performed in clinical trials of any drug with central nervous system activity. We expect that the FDA will follow this guidance, and we or our business partners will be required to perform suicidality assessments in all clinical trials, including Phase 1 trials, of any of our product candidates with central nervous system activity. Finally, if the FDA determines that a drug may present a risk of substance abuse, it can recommend to the DEA that the drug be scheduled under the Controlled Substances Act. While gabapentin is not a scheduled drug at the present time, pregabalin has been scheduled as a controlled substance. Since pregabalin is a scheduled drug, it is possible that the FDA may require additional testing of HORIZANT in the future, the results of which could lead the FDA to conclude that HORIZANT should be scheduled as well. Scheduled substances are subject to DEA regulations relating to manufacturing, storage, distribution and physician prescription procedures, and the DEA regulates the amount of a scheduled substance that is available for clinical trials and commercial distribution. Accordingly, any scheduling action that the FDA or DEA may take with respect to HORIZANT may limit HORIZANT’s
60
Table of Contents
marketing approval. In addition, any failure or delay in commencing or completing clinical trials or obtaining regulatory approvals for our product candidates would delay commercialization of our product candidates, and severely harm our business and financial condition.
If we do not establish partnerships, collaborations or other strategic arrangements for our product candidates, we may have to alter our development and commercialization plans.
Our strategy includes selectively partnering, collaborating or otherwise entering into strategic arrangements with leading pharmaceutical and biotechnology companies to assist us in furthering development and potential commercialization of some of our product candidates. We intend to do so, especially for indications that involve a large, primary care market that must be served by large sales and marketing organizations or to develop and commercialize product candidates that fall outside our core focus or our core development capabilities. We face significant competition in seeking appropriate business partners, and these arrangements are complex and time consuming to negotiate and document. We may not be able to negotiate additional strategic arrangements on acceptable terms, or at all. We are unable to predict when, if ever, we will enter into any additional third-party strategic arrangements because of the numerous risks and uncertainties associated with establishing these arrangements. If we are unable to negotiate additional partnerships, collaborations or other strategic arrangements, we may not be able to maximize the market opportunity of a product, or we may have to curtail the development of a particular product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization, reduce the scope of our sales or marketing activities or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms, or at all. If we do not have sufficient funds, we will not be able to bring our product candidates to market and generate product revenues. For example, we do not intend to pursue any further development of XP21279 unless we have sufficient funds to do so or we enter into partnerships, collaborations or other strategic arrangements with third parties for such development. Accordingly, if we are unable to establish third-party strategic arrangements for the development and commercialization of these product candidates, we may determine to abandon these product candidates entirely in which case we would not receive any return on our investments in these product candidates.
We may not develop additional prodrug product candidates.
As part of a restructuring in 2010, we eliminated our discovery research department, which prevents our ability to discover additional product candidates at this time. If we are unable to develop suitable product candidates from our internal efforts, we may pursue additional product candidates through in-licensing. Any growth through in-licensing would depend upon the availability of suitable product candidates at favorable prices and upon advantageous terms and conditions. To obtain additional product candidates, we may also reconstitute our discovery research department, which would require the expenditure of significant resources and the identification and hiring of a number of highly-skilled employees. Such efforts could divert the time and resources from the later-stage development or commercialization of HORIZANT or our product candidates.
If we are unable to develop or obtain suitable product candidates, we will not be able to increase our revenues in future periods, which could result in significant harm to our financial position and adversely impact our stock price.
If we fail to attract and keep senior management and key scientific personnel, we may be unable to successfully develop or commercialize HORIZANT or our product candidates.
Our success depends on our continued ability to attract, retain and motivate highly qualified management, commercial, clinical and scientific personnel and on our ability to develop and maintain important relationships with leading clinicians. If we are not able to retain our key personnel, we may not be able to successfully commercialize HORIZANT, or develop or commercialize our product candidates. Competition may limit our
61
Table of Contents
ability to hire and retain highly qualified personnel on acceptable terms. Moreover, none of our employees have employment commitments for any fixed period of time and could leave our employment at will. We do not carry “key person” insurance covering members of senior management or key scientific personnel. If we fail to identify, attract and retain qualified personnel, we may be unable to continue our development and commercialization activities.
If we use biological and hazardous materials in a manner that causes contamination or injury or violates laws, we may be liable for damages.
Our development activities involve the use of potentially harmful biological materials as well as hazardous materials, chemicals and various radioactive compounds. We cannot completely eliminate the risk of accidental contamination or injury from the use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for damages that result, and any liability could exceed our resources. We, our business partners, the third parties that conduct clinical trials on our behalf and the third parties that manufacture HORIZANT or our product candidates are subject to federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and waste products. The cost of compliance with these laws and regulations could be significant. The failure to comply with these laws and regulations could result in significant fines and work stoppages and may harm our business.
Our facility is located in California’s Silicon Valley, in an area with a long history of industrial activity and use of hazardous substances, including chlorinated solvents. Environmental studies conducted prior to our leasing of the site found levels of metals and volatile organic compounds in the soils and groundwater at our site. While these constituents of concern predated our occupancy, certain environmental laws, including the U.S. Comprehensive, Environmental Response, Compensation and Liability Act of 1980, impose strict, joint and several liabilities on current operators of real property for the cost of removal or remediation of hazardous substances. These laws often impose liability even if the owner or operator did not know of, or was not responsible for, the release of such hazardous substances. As a result, while we have not been, we cannot rule out the possibility that we could in the future be held liable for costs to address contamination at the property beneath our facility, which costs could be material.
Our facility is located near known earthquake fault zones, and the occurrence of an earthquake, extremist attack or other catastrophic disaster could cause damage to our facilities and equipment, which could require us to cease or curtail operations.
Our facility is located near known earthquake fault zones and, therefore, is vulnerable to damage from earthquakes. In 1989, a major earthquake struck this area and caused significant property damage and a number of fatalities. We are also vulnerable to damage from other types of disasters, including power loss, attacks from extremist organizations, fire, floods and similar events. If any disaster were to occur, our ability to operate our business could be seriously impaired. We may not have adequate insurance to cover our losses resulting from disasters or other similar significant business interruptions, and we do not plan to purchase additional insurance to cover such losses due to the cost of obtaining such coverage. Any significant losses that are not recoverable under our insurance policies could seriously impair our business and financial condition.
Data breaches and cyber-attacks could compromise our intellectual property or other sensitive information and cause significant damage to our business and reputation.
In the ordinary course of our business, we maintain sensitive data on our networks, including our intellectual property, personally identifiable information and proprietary or confidential business information relating to our business and that of our customers and business partners. The secure maintenance of this information is critical to our business and reputation. We believe that companies have been increasingly subject to a wide variety of security incidents, cyber-attacks and other attempts to gain unauthorized access. These threats can come from a variety of sources, all ranging in sophistication from an individual hacker to a state-sponsored attack. Cyber threats may be generic, or they may be custom-crafted against our information systems. Over the past year,
62
Table of Contents
cyber-attacks have become more prevalent and much harder to detect and defend against. Our network and storage applications may be subject to unauthorized access by hackers or breached due to operator error, malfeasance or other system disruptions. It is often difficult to anticipate or immediately detect such incidents and the damage caused by such incidents. These data breaches and any unauthorized access or disclosure of our information or intellectual property could compromise our intellectual property and expose sensitive business information. In addition, a data breach or privacy violation that leads to disclosure or modification of or prevents access to patient information, including personally identifiable information or protected health information, could harm our reputation, compel us to comply with federal and/or state breach notification laws, subject us to mandatory corrective action, require us to verify the correctness of database contents and otherwise subject us to liability under laws and regulations that protect personal data, resulting in increased costs or loss of product revenue. Cyber-attacks could also cause us to incur significant remediation costs, result in product development or commercialization delays, disrupt key business operations, cause us to incur financial loss and other regulatory penalties because of lost or misappropriated information, including sensitive patient data, and divert attention of management and key information technology resources. These incidents could also subject us to liability, expose us to significant expense and cause significant harm to our reputation and business.
Our ability to use our net operating loss carryforwards and certain other tax attributes is uncertain and may be limited.
Our ability to use our federal and state net operating loss, or NOL, carryforwards to offset potential future taxable income and related income taxes that would otherwise be due is dependent upon our generation of future taxable income before the expiration dates of the NOL carryforwards, and we cannot predict with certainty when, or whether, we will generate sufficient taxable income to use all of our NOL carryforwards. In addition, utilization of NOL carryforwards to offset potential future taxable income and related income taxes that would otherwise be due is subject to annual limitations under the “ownership change” provisions of Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, or the Code, and similar state provisions, which may result in the expiration of NOL carryforwards before future utilization. In general, under the Code, if a corporation undergoes an “ownership change,” generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period, the corporation’s ability to use its pre-change NOL carryforwards and other pre-change tax attributes (such as research and development tax credits) to offset its post-change taxable income or taxes may be limited. Our prior equity offerings, conversions of the 2022 Notes, and other changes in our stock ownership, some of which are outside of our control, may have resulted or could in the future result in an ownership change. If a limitation were to apply, utilization of our domestic NOL and tax credit carryforwards could be limited in future periods and a portion of the carryforwards could expire before being available to reduce future income tax liabilities.
Risks Related to Our Common Stock
Our stock price historically has been volatile and is likely to continue to be volatile in the future, and purchasers of our common stock could incur substantial losses.
The market price of our common stock, and the market prices of securities of biopharmaceutical companies in general, have been highly volatile. In this regard, the market price of our common stock is likely to continue to be subject to wide fluctuations. The market price of our common stock may be influenced by many factors, including:
| • | the commercial sales of HORIZANT, REGNITE or any of our other products that may result from our product candidates that may in the future be approved by the FDA or its foreign counterparts; |
| • | the costs to maintain adequate sales, marketing and commercial capabilities to commercialize HORIZANT; |
| • | adverse results or delays in clinical trials; |
| • | the timing of achievement of our clinical, regulatory, collaborating, partnering and other milestones, such as the commencement of clinical development, the completion of a clinical trial, the filing for regulatory |
63
Table of Contents
| approval or the establishment of strategic arrangements for the development and commercialization of one or more of our product candidates; |
| • | announcement of FDA approvability, approval or non-approval of our product candidates, and the timing of the FDA review process; |
| • | actions taken by regulatory agencies with respect to HORIZANT, REGNITE or our product candidates, our clinical trials, our business partners’ clinical trials or our sales and marketing activities; |
| • | actions taken by regulatory agencies with respect to products or drug classes related to HORIZANT, REGNITE or our product candidates; |
| • | problems in our manufacturing or supply chain that limit or exhaust the quantity of supplies of HORIZANT that are available to patients; |
| • | changes in our business partners’ business strategies; |
| • | developments in our business relationships with Astellas, Indivior and/or the NIAAA, including potential disputes or the termination or further modification of our agreements with Astellas, Indivior and/or the NIAAA; |
| • | regulatory developments in the United States and foreign countries; |
| • | changes in the structure of healthcare payment systems; |
| • | any intellectual property matter involving us, including infringement lawsuits; |
| • | actions taken by regulatory agencies with respect to our or our business partners’ compliance with regulatory requirements; |
| • | announcements or actions by stockholder activists; |
| • | possible sales of our common stock by holders of the 2022 Notes who may view the 2022 Notes as a more attractive means of equity participation in our company; |
| • | the conversion of some or all of the 2022 Notes and any sales in the public market of shares of our common stock issued upon conversion of the 2022 Notes; |
| • | hedging or arbitrage trading activity involving our common stock, including in connection with arbitrage strategies employed or that may be employed by investors in our 2022 Notes; |
| • | announcements of technological innovations or new products by us or our competitors; |
| • | market conditions for equity investments in general, or the biotechnology or pharmaceutical industries in particular; |
| • | changes in financial estimates or recommendations by securities analysts; |
| • | sales of large blocks of our common stock; |
| • | sales of our common stock by our executive officers, directors and significant stockholders; |
| • | restatements of our financial results and/or material weaknesses in our internal controls; and |
| • | the loss of any of our key scientific or management personnel. |
The stock markets in general and the markets for biotechnology stocks in particular, have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. These broad market fluctuations may adversely affect the trading price of our common stock. In the past, purported class action lawsuits have often been instituted against companies, including our company, whose securities have experienced periods of volatility in market price. Any such litigation brought against us could result in substantial costs, which would hurt our financial condition and results of operations, divert management’s attention and resources and possibly delay our clinical trials or commercialization efforts.
64
Table of Contents
Fluctuations in our operating results could cause our stock price to decline.
Our operating results will be difficult to predict and will likely fluctuate from quarter to quarter and year to year. Due to our acquisition of the HORIZANT business in May 2013 and the relative lack of our own historical sales data, HORIZANT sales may be difficult to predict from period to period and as a result, you should not rely on HORIZANT sales results in any period as being indicative of future performance, and sales of HORIZANT may be below the expectation of securities analysts or investors in the future.
The following factors are likely to result in fluctuations of our operating results from quarter to quarter and year to year:
| • | the level and timing of commercial sales of HORIZANT, REGNITE or any of our other products that may result from our product candidates approved by the FDA or its foreign counterparts; |
| • | the costs to maintain adequate sales, marketing and commercial capabilities to commercialize HORIZANT, and the costs associated with fulfilling the remaining, and any additional future, PMCs and PMRs for HORIZANT; |
| • | changes in the amount of deductions from gross sales, including government-mandated rebates, chargebacks and discounts that can vary because of changes to the government discount percentage or due to different levels of utilization by entities entitled to government rebates and discounts and changes in patient demographics; |
| • | adverse results or delays in our or our business partners’ clinical trials; |
| • | the timing and achievement of our clinical, regulatory, strategic and other milestones, such as the commencement of clinical development, the completion of a clinical trial, the filing for regulatory approval or the establishment of strategic arrangements for one or more of our product candidates; |
| • | announcement of FDA approvability, approval or non-approval of our product candidates and the timing of the FDA review process; |
| • | actions taken by regulatory agencies with respect to HORIZANT, REGNITE or our product candidates, our clinical trials, our business partners’ clinical trials or our sales and marketing activities; |
| • | actions taken by regulatory agencies with respect to products or drug classes related to HORIZANT, REGNITE or our product candidates; |
| • | problems in our manufacturing or supply chain that limit or exhaust the quantity of supplies of HORIZANT that are available to patients; |
| • | changes in our business partners’ business strategies; |
| • | developments in our relationship with our business partners, including potential disputes or the termination or modification of our agreements; |
| • | actions taken by regulatory agencies with respect to our or our business partners’ compliance with regulatory requirements; |
| • | regulatory developments in the United States and foreign countries; |
| • | changes in the structure of healthcare payment systems; |
| • | any intellectual property matter involving us, including patent infringement lawsuits; and |
| • | announcements of technological innovations or new products by us or our competitors. |
Due to these fluctuations in our operating results, a period-to-period comparison of our results of operations may not be a good predictor of our future performance. For example, due primarily to the recognition of revenues from upfront, milestone and contingent event-based payments from our agreements with Astellas, GSK and/or Indivior, we were profitable for the year ended December 31, 2007, in the three months ended June 30, 2011 and in the three months ended September 30, 2014. However, while recognition of these revenues resulted in a
65
Table of Contents
profitable year or quarter for those periods, we incurred net losses in each full year since 2007. In any particular financial period, the actual or anticipated fluctuations could be below the expectations of securities analysts or investors and our stock price could decline.
We may become subject to increased stockholder activism efforts that each could cause a material disruption to our business.
Certain influential institutional investors and hedge funds have taken steps to involve themselves in the governance and strategic direction of certain companies due to governance or strategic-related disagreements between such companies and such stockholders. For example, Clinton Relational Opportunity Master Fund, L.P. conducted a proxy contest with respect to matters voted upon at our 2014 annual meeting of stockholders, which contest followed multiple communications to us urging a change in our capital allocation strategy away from our HORIZANT commercialization efforts and a change in our management. Responding to this proxy contest was time-consuming and was a significant distraction for our board of directors, management and employees, and diverted the attention of our board of directors and senior management from the pursuit of our business strategy. The expenses for legal and advisory fees and expenses and associated costs incurred in connection with this proxy contest were substantial and increased our operating expenses for the first half of 2014 by approximately $1.2 million. Increased or future stockholder activism efforts could result in substantial costs and a further diversion of management’s attention and resources, which could harm our business and adversely affect the market price of our common stock.
Failure to maintain effective internal controls in accordance with Section 404 of the Sarbanes-Oxley Act of 2002 could have a material adverse effect on our stock price.
Section 404 of the Sarbanes-Oxley Act of 2002 and the related rules and regulations of the SEC require annual management assessments of the effectiveness of our internal control over financial reporting and a report by our independent registered public accounting firm attesting to, and reporting on, the effectiveness of our internal control over financial reporting. If we fail to maintain the adequacy of our internal control over financial reporting, as such standards are modified, supplemented or amended from time to time, or if we fail to maintain effective internal control over financial reporting around our commercial promotion of HORIZANT, we may not be able to ensure that we can conclude on an ongoing basis that we have effective internal control over financial reporting in accordance with Section 404 of the Sarbanes-Oxley Act of 2002 and the related rules and regulations of the SEC. If we cannot favorably assess, or our independent registered public accounting firm is unable to provide an unqualified attestation report on, the effectiveness of our internal control over financial reporting, investor confidence in the reliability of our financial reports may be adversely affected, which could have a material adverse effect on our stock price.
Because a small number of existing stockholders own a large percentage of our voting stock, they may be able to exercise significant influence over our affairs, acting in their best interests and not necessarily those of other stockholders.
As of February 13, 2015, our executive officers, directors and holders of 5% or more of our outstanding common stock, based upon information known to us and derived from Schedules 13G filed with the SEC, beneficially owned approximately 57% of our common stock. The interests of this group of stockholders may not always coincide with our interests or the interests of other stockholders. This concentration of ownership could also have the effect of delaying or preventing a change in our control or otherwise discouraging a potential acquirer from attempting to obtain control of us, which in turn could reduce the market price of our common stock.
66
Table of Contents
Our stockholder rights plan, anti-takeover provisions in our charter documents and under Delaware law, and provisions in the indenture governing the 2022 Notes could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our amended and restated certificate of incorporation and bylaws may delay or prevent an acquisition of us, a change in our management or other changes that stockholders may consider favorable. These provisions include:
| • | a classified board of directors; |
| • | a prohibition on actions by our stockholders by written consent; |
| • | the ability of our board of directors to issue preferred stock without stockholder approval, which could be used to make it difficult for a third party to acquire us; |
| • | notice requirements for nominations for election to the board of directors; and |
| • | limitations on the removal of directors. |
Moreover, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
We have adopted a rights agreement under which certain stockholders have the right to purchase shares of a new series of preferred stock, at an exercise price of $140.00 per one one-hundredth of a share, if a person acquires more than 15% of our common stock. The rights plan could make it more difficult for a person to acquire a majority of our outstanding voting stock. The rights plan could also reduce the price that investors might be willing to pay for shares of our common stock and result in the market price of our common stock being lower than it would be without the rights plan. In addition, the existence of the rights plan itself may deter a potential acquirer from acquiring us. As a result, either by operation of the rights plan or by its potential deterrent effect, mergers and acquisitions of us that our stockholders may consider in their best interests may not occur.
Furthermore, the indenture governing the 2022 Notes requires us to repurchase the 2022 Notes for cash if we undergo certain fundamental changes and, in certain circumstances, to increase the conversion rate for a holder of 2022 Notes. A takeover of us may trigger the requirement that we repurchase our 2022 Notes and/or increase the conversion rate, which could make it more costly for a potential acquirer to engage in a business combination transaction with us.
These provisions, alone or together, could delay or prevent hostile takeovers and changes in control or changes in our management, and could under certain circumstances, reduce the market price of our common stock.
If there are large sales of our common stock, the market price of our common stock could drop substantially.
If our existing stockholders sell a large number of shares of our common stock or the public market perceives that existing stockholders might sell shares of our common stock, the market price of our common stock could decline significantly. As of February 13, 2015, we had 62,756,809 outstanding shares of common stock, substantially all of which may be sold in the public market without restriction. In addition, future issuances by us of our common stock upon the exercise or settlement of equity-based awards and conversions of the 2022 Notes would dilute existing stockholders’ ownership interest in our company and any sales in the public market of these shares of common stock, or the perception that these sales might occur, could also adversely affect the market price of our common stock.
67
Table of Contents
Item 1B. Unresolved Staff Comments.
Not applicable.
We lease approximately 103,000 square feet of office and laboratory space in an office building in Santa Clara, California. In October 2012, we entered into a Second Amendment to Lease with SI 34 LLC, or Sobrato, with respect to our current office space at 3410 Central Expressway, Santa Clara, California, or, as amended, the 3410 Lease. In November 2014, we entered into a Third Amendment to Lease with Sobrato, extending the term of the 3410 Lease for an additional nine months, so that the 3410 Lease will expire on May 31, 2016. We had also leased approximately 59,000 square feet at an adjacent building at 3400 Central Expressway, Santa Clara, California, but we terminated the lease in February 2013, and the rent obligations expired in August 2013.
We are not a party to any material legal proceedings at this time. From time to time, we may be involved in litigation relating to claims arising out of our ordinary course of business.
Item 4. Mine Safety Disclosures.
Not applicable.
68
Table of Contents
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. |
Market for Registrant’s Common Equity; Dividends
Our common stock is traded on The NASDAQ Global Select Market under the symbol “XNPT.” As of February 13, 2015, there were approximately 73 holders of record of our common stock. No cash dividends have been paid on our common stock to date, and we intend to utilize any earnings for development of our business. The following table sets forth, for the periods indicated, the range of high and low intraday sales prices of our common stock as quoted on The NASDAQ Global Select Market for the two most recent fiscal years.
| High | Low | |||||||
| 2014 |
||||||||
| 4th Quarter |
$ | 9.24 | $ | 5.17 | ||||
| 3rd Quarter |
5.65 | 4.12 | ||||||
| 2nd Quarter |
5.38 | 3.15 | ||||||
| 1st Quarter |
7.20 | 4.94 | ||||||
| 2013 |
||||||||
| 4th Quarter |
$ | 6.50 | $ | 4.72 | ||||
| 3rd Quarter |
6.11 | 4.24 | ||||||
| 2nd Quarter |
7.59 | 4.61 | ||||||
| 1st Quarter |
9.31 | 6.61 | ||||||
The closing price for our common stock as reported by The NASDAQ Global Select Market on February 13, 2015 was $6.69 per share.
Issuer Purchases of Equity Securities
None.
69
Table of Contents
Performance Measurement Comparison(1)
The following graph shows the total stockholder return of an investment of $100 in cash on December 31, 2009 for: (i) our common stock; (ii) the NASDAQ Composite Index; and (iii) the NASDAQ Biotechnology Index for the five-year period ended December 31, 2014. Pursuant to applicable SEC rules, all values assume reinvestment of the full amount of all dividends; however, no dividends have been declared on our common stock to date. The stockholder return shown on the graph below is not necessarily indicative of future performance, and we do not make or endorse any predictions as to future stockholder returns.
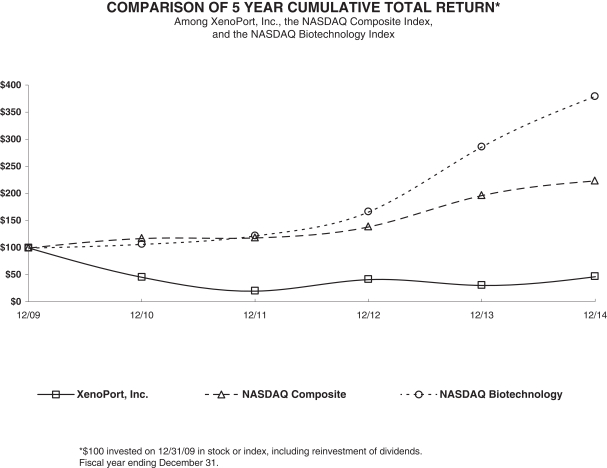
| (1) | This section is not “soliciting material,” is not deemed “filed” with the SEC and is not to be incorporated by reference into any filing of XenoPort under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing. |
70
Table of Contents
Item 6. Selected Financial Data.
You should read the following selected financial data together with our audited financial statements and related notes and the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section and other financial information included in this Annual Report on Form 10-K.
| Year Ended December 31, | ||||||||||||||||||||
| 2014(1) | 2013(2) | 2012 | 2011 | 2010 | ||||||||||||||||
| (In thousands, except per share amounts) | ||||||||||||||||||||
| Statement of Operations Data: |
||||||||||||||||||||
| Revenues: |
||||||||||||||||||||
| Product sales, net |
$ | 20,173 | $ | 6,414 | $ | — | $ | — | $ | — | ||||||||||
| Collaboration revenue |
26,134 | 1,137 | 11,515 | 8,515 | 1,515 | |||||||||||||||
| Royalty revenue |
561 | 400 | 109 | — | — | |||||||||||||||
| Net revenue from unconsolidated joint operating activities |
— | — | 10,000 | 35,000 | 1,364 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total revenues |
46,868 | 7,951 | 21,624 | 43,515 | 2,879 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Operating expenses (gains): |
||||||||||||||||||||
| Cost of product sales |
2,094 | 1,170 | — | — | — | |||||||||||||||
| Research and development |
23,679 | 33,325 | 42,947 | 43,788 | 52,546 | |||||||||||||||
| Selling, general and administrative |
70,194 | 59,084 | 30,244 | 30,427 | 28,323 | |||||||||||||||
| Gain on litigation settlement |
— | — | (20,499 | ) | — | — | ||||||||||||||
| Restructuring charges |
— | — | — | 2,923 | 5,275 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Total operating expenses |
95,967 | 93,579 | 52,692 | 77,138 | 86,144 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss from operations |
(49,099 | ) | (85,628 | ) | (31,068 | ) | (33,623 | ) | (83,265 | ) | ||||||||||
| Interest and other income |
253 | 213 | 254 | 243 | 796 | |||||||||||||||
| Interest and other expense |
(487 | ) | (468 | ) | — | — | — | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss before income taxes |
(49,333 | ) | (85,883 | ) | (30,814 | ) | (33,380 | ) | (82,469 | ) | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss |
$ | (49,333 | ) | $ | (85,883 | ) | $ | (30,814 | ) | $ | (33,380 | ) | $ | (82,469 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Basic and diluted net loss per share |
$ | (0.81 | ) | $ | (1.81 | ) | $ | (0.78 | ) | $ | (0.94 | ) | $ | (2.68 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Shares used to compute basic and diluted net loss per share |
60,856 | 47,545 | 39,434 | 35,400 | 30,813 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance Sheet Data:(4) |
||||||||||||||||||||
| Cash, cash equivalents and short-term investments |
$ | 102,056 | $ | 58,658 | $ | 139,002 | $ | 94,442 | $ | 108,595 | ||||||||||
| Working capital |
91,806 | 53,616 | 141,317 | 83,922 | 99,314 | |||||||||||||||
| Restricted investments |
1,725 | 1,725 | 1,955 | 1,954 | 1,948 | |||||||||||||||
| Total assets |
123,061 | 78,541 | 159,048 | 104,036 | 121,229 | |||||||||||||||
| Other noncurrent liability(3) |
3,269 | 2,782 | 2,314 | — | — | |||||||||||||||
| Accumulated deficit |
586,816 | 537,483 | 451,600 | 420,786 | 387,406 | |||||||||||||||
| Total stockholders’ equity |
91,140 | 53,693 | 130,210 | 75,135 | 93,959 | |||||||||||||||
| (1) | The increase in total revenues in 2014 was primarily from the recognition of $25.0 million in collaboration revenue resulting from our license agreement with Indivior (formerly Reckitt Benckiser Pharmaceuticals, Inc.). |
| (2) | On November 8, 2012, we executed a termination and transition agreement with Glaxo Group Limited, or GSK, that terminated our development and commercialization agreement with respect to HORIZANT, and also provided for a mutual release of claims and resolved all ongoing litigation between the parties. Pursuant to the termination and transition agreement, during a transition period that ended on April 30, 2013, GSK continued to exclusively commercialize, promote, manufacture and distribute HORIZANT in the United States. We did not receive any revenue nor incur any losses from GSK’s sales of HORIZANT during the transition period. On May 1, 2013, we assumed all responsibilities for further development, |
71
Table of Contents
| manufacturing and commercialization of HORIZANT in the United States. The results of operations of the acquired HORIZANT business, along with the fair values of the assets acquired in the transaction, have been included in our financial statements since May 1, 2013. |
| (3) | In connection with our reacquisition of the HORIZANT business on May 1, 2013, GSK provided us with inventory of gabapentin enacarbil in GSK’s possession that was not required for use by GSK in the manufacture of HORIZANT and related manufacturing property and equipment. In exchange for such inventory, we agreed to make annual payments to GSK of $1.0 million for six years beginning in 2016, which was recorded at its present value as “other noncurrent liability” on our balance sheet and is being accreted to its contractual value over the repayment period. |
| (4) | On February 3, 2015, we completed a private placement of $115.0 million aggregate principal amount of 2.50% Convertible Senior Notes due 2022, or the 2022 Notes, to the investment bank acting as the initial purchaser who subsequently resold the 2022 Notes to qualified institutional buyers. The 2022 Notes sold in the offering include $15.0 million aggregate principal amount of 2022 Notes sold to the initial purchaser pursuant to its option to purchase additional 2022 Notes, which was exercised in full on January 29, 2015. The estimated net proceeds from the offering of the 2022 Notes is approximately $111.4 million, after deducting the initial purchaser’s discount and estimated offering expenses payable by us. |
72
Table of Contents
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
Overview
XenoPort, Inc. is a biopharmaceutical company focused on developing and commercializing a portfolio of internally discovered product candidates for the potential treatment of neurological and other disorders. We are currently commercializing HORIZANT® (gabapentin enacarbil) Extended-Release Tablets in the United States and developing our novel fumaric acid ester product candidate, XP23829, as a potential treatment for psoriasis and potentially for relapsing forms of multiple sclerosis, or MS. In addition, REGNITE® (gabapentin enacarbil) Extended-Release Tablets are being marketed in Japan by Astellas Pharma Inc, or Astellas. We currently depend, and may in the future depend, on collaboration, partnership, licensing and other strategic arrangements with third-party business partners.
HORIZANT has been approved by the U.S. Food and Drug Administration, or FDA, for the treatment of moderate-to-severe primary restless legs syndrome, or RLS, in adults and for the management of postherpetic neuralgia, or PHN, in adults. RLS, also known as Willis-Ekbom Disease, is a neurological disorder characterized by an urge to move the legs, usually caused or accompanied by uncomfortable and unpleasant sensations in the legs. PHN is a neuropathic (nerve) pain syndrome that can follow the healing of an outbreak of herpes zoster, commonly known as shingles. In September 2014, we announced an agreement with the National Institute on Alcohol Abuse and Alcoholism, or NIAAA, under which the NIAAA would conduct a clinical trial evaluating the use of HORIZANT as a potential treatment for alcohol use disorder, or AUD. The study is planned to be a randomized, double-blind, placebo-controlled clinical trial of the safety and efficacy of HORIZANT in patients with AUD but are abstinent at treatment initiation. The study is expected to have a treatment period of six months and to enroll approximately 350 patients in the first half of 2015, and the NIAAA anticipates that the results may be available by the end of 2016. Under the terms of the agreement, we will supply clinical trial material and the NIAAA will conduct and pay all other expenses associated with the proposed clinical trial of HORIZANT. We will have access to study results generated under the proposed clinical trial to support any potential regulatory filings for HORIZANT as a potential treatment for AUD.
We reacquired development and commercialization rights to HORIZANT from Glaxo Group Limited, or GSK, our former collaborator for this product, on May 1, 2013. We have focused our promotional and sales efforts on specialty doctors in specific geographic territories. We initiated our commercialization efforts in approximately 40 territories and, beginning in the third quarter, expanded the sales force to approximately 70 territories in the United States. We expect to further expand our sales force to up to approximately 120 sales representatives by mid-2015. Our sales organization consists of both XenoPort-employed sales representatives and contract sales representatives who are solely dedicated to HORIZANT education and promotion. We expect to continue to rely on our contract sales organization to provide back office logistical support for the XenoPort-employed sales representatives.
Our future product sales of HORIZANT will be dependent upon the success of our strategies for commercialization, promotion and distribution, as well as our ability to successfully execute on these activities and to comply with applicable laws, regulations and regulatory requirements. Our commercialization efforts may ultimately not be successful. In this regard, we rely on third parties to perform a variety of functions related to the sale and distribution of HORIZANT, key aspects of which are out of our direct control. If these third-party service providers fail to comply with applicable laws and regulations, fail to meet expected deadlines or otherwise do not carry out their contractual duties to us, or if HORIZANT encounters physical or natural damage at their facilities, our ability to deliver HORIZANT to meet commercial demand would be significantly impaired.
XP23829 is a fumaric acid ester compound and a patented prodrug of monomethyl fumarate, or MMF. Fumaric acid ester compounds have shown immuno-modulatory and neuroprotective effects in cell-based systems and preclinical models of disease. A fumaric acid ester product is approved in Germany for the treatment of psoriasis, and a fumaric acid ester compound, known as TECFIDERA (dimethyl fumarate), was approved by the FDA in March 2013 for the treatment of relapsing forms of MS and by the European Commission in February 2014 for the treatment of relapsing-remitting MS. We have evaluated XP23829 in Phase 1 studies with healthy subjects to determine its safety and pharmacokinetic profile. In June 2014, we initiated a Phase 2 clinical trial of XP23829 in patients with moderate-to-severe chronic plaque-type psoriasis. We are evaluating 400 mg and 800 mg of XP23829 dosed once a day, and 400 mg of XP23829 dosed twice a day in this trial.
73
Table of Contents
We also have two other product candidates: arbaclofen placarbil, or AP, which is licensed to Indivior PLC, a new business entity that was formed by divestment of the pharmaceutical division from Reckitt Benckiser Group plc, and XP21279. With respect to AP, in May 2014, we entered into the exclusive license agreement with Indivior, which became effective on June 19, 2014. Under the terms of this agreement, we granted exclusive, world-wide rights to develop and commercialize pharmaceutical products containing AP and other prodrugs of baclofen or R-baclofen, or AP Products, for all indications, subject to our right of first negotiation to collaborate to develop and commercialize AP Products for non-addiction indications. In exchange for these rights, we received an upfront, non-refundable cash payment of $20.0 million in June 2014 and also received an additional $5.0 million in July 2014 after our delivery of specified materials, both of which were recognized in the third quarter of 2014. We are also eligible to receive aggregate cash payments of up to $120.0 million upon the achievement by Indivior of certain contingent event-based payments, of which $70.0 million are regulatory and development-based and $50.0 million are commercialization-based. In addition, we are entitled to receive tiered double-digit royalty payments of up to the mid-teens on a percentage basis on potential future net sales of AP Products in the United States, and high single-digit royalty payments on potential future net sales of AP Products outside the United States. XP21279 is a potential treatment for patients with advanced idiopathic Parkinson’s disease. We may develop it further, to the extent that our resources permit, or we may enter into third-party business relationships in order to carry out such development activities.
The potential for gabapentin enacarbil, XP23829 and/or XP21279 could be enhanced through third-party strategic arrangements that would further development and/or enhance commercialization efforts of these assets. As such, we plan to enter into partnerships, collaborations or other strategic arrangements with pharmaceutical companies: (1) when access to a primary care physician or an expanded sales force is necessary to maximize the commercial potential of gabapentin enacarbil, XP23829 and/or XP21279 in the United States; (2) for the development and commercialization of gabapentin enacarbil, XP23829 and/or XP21279 outside the United States or that require resources or expertise beyond our current capabilities; or (3) to develop and commercialize product candidates that fall outside our core focus or our core development capabilities.
Historically, revenues recognized through May 1, 2013 were primarily comprised of upfront, milestone and contingent event-based payments from our collaboration and partnership arrangements. While a significant portion of our revenues in 2014 consisted of revenues from the recognition of cash payments we received under our license agreement with Indivior and we may in the future recognize revenues from contingent-event based payments from Indivior, we expect the future composition of our revenues to consist primarily of revenues from HORIZANT product sales. Although we are recognizing revenue from HORIZANT product sales in the United States, our relative lack of commercialization experience, as an organization and with respect to HORIZANT product sales, will make future operating results difficult to predict. In this regard, our product sales revenue may vary significantly from period to period as our commercialization efforts progress, and we may be unable to meaningfully increase HORIZANT product sales or otherwise successfully commercialize HORIZANT in a timely manner or at all. We also expect that the expenses of increasing and maintaining our sales and marketing capabilities, including with respect to the internal and external costs of supporting and maintaining our XenoPort-employed sales representatives and our contract sales representatives, and maintaining a distribution and supply chain infrastructure will continue to be substantial, and these costs may exceed the revenues that we are able to generate from HORIZANT product sales. We recorded $20.2 million in net sales from HORIZANT in 2014.
Gabapentin enacarbil is licensed to Astellas in Japan. We are entitled to receive percentage-based high-teen royalties on net sales of REGNITE in Japan, and the royalties are recognized as revenue when royalty payments are received. In 2014, the royalty revenue from net sales of REGNITE in Japan was $0.6 million. We expect royalty revenues from our partnership with Astellas to fluctuate based on the results of their commercialization, marketing and distribution efforts for REGNITE in Japan and, to a lesser extent, foreign currency fluctuations; however, we do not expect that royalty revenues from our partnership with Astellas will be meaningful. We expect our research and development expenses to increase in 2015 compared to 2014 levels primarily due to increased development expenses for XP23829, including our Phase 2 clinical trial in patients with moderate-to-severe chronic plaque-type psoriasis that was initiated in June 2014, as well as increased costs in anticipation of potential Phase 3 clinical activities. The timing and amount of research and development expenses incurred will primarily depend upon the extent of current or future clinical trials for XP23829 as well as the related expenses
74
Table of Contents
associated with our development organization, regulatory requirements for our product candidates, product candidate manufacturing costs and our ability to raise additional funds. Our future research and development expenses are subject to numerous assumptions that may prove to be wrong and also are subject to risks related to the difficulty and uncertainty of clinical success and regulatory approvals of our product candidates. In 2015, we expect our selling, general and administrative expenses to increase compared to 2014 levels primarily due to expenses incurred in maintaining marketing, distribution and other commercial capabilities for HORIZANT, largely through third-party service providers, as well as internal and external expenses of supporting and maintaining our XenoPort-employed sales representatives and our contract sales representatives, which we expect to further expand in 2015.
On February 3, 2015, we completed a private placement of $115.0 million aggregate principal amount of 2.50% Convertible Senior Notes due 2022, or the 2022 Notes, to the investment bank acting as the initial purchaser who subsequently resold the 2022 Notes to qualified institutional buyers. The 2022 Notes sold in the offering include $15.0 million aggregate principal amount of 2022 Notes sold to the initial purchaser pursuant to its option to purchase additional 2022 Notes, which was exercised in full on January 29, 2015. The estimated net proceeds from the offering of the 2022 Notes is approximately $111.4 million, after deducting the initial purchaser’s discount and estimated offering expenses payable by us. For more information on the 2022 Notes, see “Liquidity and Capital Resources” below.
On January 29, 2014, we completed an underwritten public offering of 12,000,000 shares of our common stock at a price to the public of $6.00 per share. Net cash proceeds from the public offering were $67.3 million, after deducting the underwriting discounts and commissions and offering expenses payable by us. On February 21, 2014, the underwriters exercised in full their option to purchase 1,800,000 additional shares resulting in additional net cash proceeds of $10.1 million, after deducting the underwriting discounts and commissions and offering expenses payable by us.
We believe that our existing capital resources, including proceeds we received from our convertible debt offering that closed in February 2015, together with interest income from our investments and anticipated product revenue, will be sufficient to meet our projected operating requirements through at least the first quarter of 2018. We have based our cash sufficiency estimate on assumptions that may prove to be incorrect, and we could utilize our available capital resources sooner than we expect. Further, our operating plan may change, and we may need additional funds to meet operational needs and capital requirements for product development and commercialization sooner than planned. We have no credit facility or committed sources of capital other than potential contingent event-based and royalty payments that we are eligible to receive under our partnerships with Astellas and Indivior. With the exception of the proposed clinical trial in patients with AUD that the NIAAA plans to conduct, we are solely responsible for all HORIZANT commercialization and development activities, including all post-marketing requirements and commitments. Such costs could be greater than we anticipate, or sales of HORIZANT may be less than we anticipate, either or both of which could accelerate our need for additional capital. In addition, we do not believe that revenues generated from HORIZANT and REGNITE sales and other revenues generated from our partnerships with Astellas and Indivior will be sufficient alone to sustain our operations, at least in the near term, and we therefore expect to continue to experience negative cash flows for the foreseeable future as we fund our operating losses and capital expenditures. Additional funds may not be available to us on terms that are acceptable to us, or at all. If adequate funds are not, or we anticipate that they may not be, available on a timely basis, we may be required to, among other things, reduce the amount of resources devoted to medical affairs, advertising, promotion or sales of HORIZANT, curtail or delay further XP23829 development or conduct additional workforce or other expense reductions, any of which could have a material adverse effect on our business and prospects.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at
75
Table of Contents
the date of the financial statements, as well as the reported revenues and expenses during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments related to each of our critical accounting areas. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Revenue Recognition
Product Sales
We began selling HORIZANT to wholesalers in May 2013. We recognize revenue from HORIZANT product sales when there is persuasive evidence that an arrangement exists, delivery to the customer has occurred, the price is fixed or determinable and collectability is reasonably assured. We record estimated reductions to revenues for customer incentives such as cash discounts for prompt payment, distributor fees, expected returns, government rebates such as Medicaid reimbursements and our patient assistance program. These estimates are deducted from gross product sales at the time such revenues are recognized. If future actual results vary from our estimates, we may need to adjust these estimates, which could have an effect on product sales and earnings in the period of adjustment.
Items Deducted from Gross Product Sales
Prompt Pay Discount
We offer cash discounts to our customers, generally 2% of the sales price, as an incentive for prompt payment. Based on our and GSK’s commercialization experience, we expect our customers to continue to comply with the prompt payment terms to earn the cash discount, as we are selling HORIZANT to the same customers under similar prompt payment terms and conditions. We estimate cash discounts for prompt payment based on contractual terms, GSK’s historical customer utilization rates and our customer utilization rates. We account for cash discounts by reducing accounts receivable by the full amount and recognizing the discount as a reduction of revenue in the same period the related revenue is recognized.
Distributor Fees
Under our inventory management agreements with our significant wholesalers, we pay the wholesalers a fee for distribution services as well as the maintenance of inventory levels. These distributor fees are based on a contractually determined fixed percentage of sales. We accrue the contractual amount and recognize the discount as a reduction of revenue in the same period the related revenue is recognized.
Product Returns
We do not provide our customers with a general right of product return, but permit returns if the product is damaged or defective when received by the customer, or if the product has or is nearly expired. HORIZANT tablets currently have a shelf-life of 36 months from date of manufacture. We will accept returns for products that will expire within six months or that have expired up to one year after their expiration dates. We obtained actual return history by type from GSK since GSK’s product launch in 2011, which provides a basis to reasonably estimate our future product returns. The sales returns accrual is estimated principally based on taking into consideration our specific adjustments to GSK’s returns history, our returns history, the shelf life of product, shipment and prescription trends and estimated distribution channel inventory level. Such estimates require significant judgment by management.
Government Rebates and Chargebacks
We participate in a number of government rebate programs, such as the Medicaid Drug Rebate Program that provides assistance to eligible low-income patients based on each individual state’s guidelines regarding
76
Table of Contents
eligibility and services; Public Health Services or 340b programs, and the Medicare Part D Coverage Gap Discount Program, which provides rebates on prescriptions that fall within the “donut hole” coverage gap; and the Department of Veterans Affairs that offers discounts to authorized users of HORIZANT. HORIZANT is also listed on the Federal Supply Schedule, or FSS, of the General Services Administration, which provides a discount to the Department of Defense, Department of Veterans Affairs and TriCare. We estimate reductions to our revenues for government rebate programs based on product pricing, current rebates, GSK’s historical utilization rates, our utilization rates, new information regarding changes in these programs’ regulations and guidelines that would impact the amount of the actual rebates, our expectations regarding future rebates for these programs and estimated levels of inventory in the distribution channel. Such estimates require significant judgment by management.
Patient Assistance
We offer a co-pay card program to assist commercially insured patients with the cost of their HORIZANT-related co-payments. Participating retail pharmacies get reimbursed by us for the amount of the co-pay assistance provided to eligible patients. We estimate and accrue the cost of our co-pay program based on historical redemption activity for this program. We reimburse the participating pharmacies approximately one month after the prescriptions subject to co-pay assistance are filled. These estimates require significant judgment by management and could vary based on program acceptance.
A rollforward of our product sales allowances for the period from May 1, 2013, the date we assumed all responsibilities for further development, manufacturing and commercialization of HORIZANT in the United States, through December 31, 2014 is as follows (in thousands):
| Prompt Pay Discount |
Distributor Fees |
Product Returns |
Government Rebates and Chargebacks |
Patient Assistance |
Total | |||||||||||||||||||
| Balance at December 31, 2012 |
$ | — | $ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||||
| Revenue Allowances: |
||||||||||||||||||||||||
| Provision |
152 | 554 | 80 | 227 | 181 | 1,194 | ||||||||||||||||||
| Payments and credits related to sales made during the period |
(130 | ) | (378 | ) | (3 | ) | (52 | ) | (108 | ) | (671 | ) | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2013 |
22 | 176 | 77 | 175 | 73 | 523 | ||||||||||||||||||
| Revenue Allowances: |
||||||||||||||||||||||||
| Provision (recovery), net |
509 | 1,873 | (58 | ) | 1,909 | 1,043 | 5,276 | |||||||||||||||||
| Payments and credits related to sales made during the period |
(463 | ) | (1,628 | ) | (2 | ) | (1,270 | ) | (928 | ) | (4,291 | ) | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2014 |
$ | 68 | $ | 421 | $ | 17 | $ | 814 | $ | 188 | $ | 1,508 | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
The increase in the product sales allowances during 2014 compared to 2013 was primarily due to the 215% increase in HORIZANT product sales during 2014.
Multiple-Element Arrangements
Revenue arrangements are accounted for in accordance with the provisions of the Revenue Recognition-Multiple-Element Arrangements topic of the Financial Accounting Standards Board Accounting Standards Codification, or the Codification.
In evaluating arrangements with multiple elements, we considered whether components of the arrangement represent separate units of accounting based upon whether certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered items. This evaluation required subjective determinations and required management to make judgments about the fair value of individual elements and whether such elements are separable from other aspects of the contractual relationship. The consideration received in such arrangements is
77
Table of Contents
allocated among the separate units of accounting based on the relative selling price method under which the selling price for each deliverable is determined using vendor-specific objective evidence of selling price, if it exists; otherwise, third-party evidence of selling price. If vendor-specific objective evidence and third-party evidence of selling price are not available for a deliverable, we used our best estimate of the selling price for that deliverable when applying the relative selling price method. The applicable revenue recognition criteria are applied to each of the separate units.
Revenues from multiple deliverables combined as a single unit of accounting are deferred and recognized over the period during which we remain obligated to perform services. The specific methodology for the recognition of the revenue (e.g., straight-line or according to specific performance criteria) is determined on a case-by-case basis according to the facts and circumstances applicable to a given agreement.
Payments received in excess of revenues recognized are recorded as deferred revenue until such time as the revenue recognition criteria have been met.
Collaboration revenue consisted of the recognition of revenues from upfront and milestone payments from our partnership with Astellas and from upfront and additional payments from our partnership with Indivior. Net revenue from unconsolidated joint operating activities included all revenue that resulted solely from our terminated collaboration agreement with GSK. We account for the revenue-related activities of these collaboration agreements as follows:
| • | Up-front, licensing-type payments. Up-front, licensing-type payments are assessed to determine whether or not the licensee is able to obtain any stand-alone value from the license. Where this is not the case, we do not consider the license deliverable to be a separate unit of accounting, and the revenue is deferred with revenue recognition for the license fee being assessed in conjunction with the other deliverables that constitute the combined unit of accounting. |
| • | Milestones. Revenue recognition for milestone payments will occur only if the consideration earned from the achievement of a milestone meets all the criteria for the milestone to be considered substantive at the inception of the arrangement, such that it: (i) is commensurate with either our performance to achieve the milestone or the enhancement of the value of the item delivered as a result of a specific outcome resulting from our performance to achieve the milestone; (ii) relates solely to past performance; and (iii) is reasonable relative to all deliverables and payment terms in the arrangement. |
We will assess the nature of, and appropriate accounting for, contingent payments, on a case-by-case basis in accordance with the provisions of the Revenue Recognition topic of the Codification.
| • | Profit and loss sharing. This represented our share of the profits and losses from the co-promotion of HORIZANT with GSK until the termination of our collaboration agreement. Amounts were recognized in the period in which the related activities occurred, and their financial statement classification was based on our assessment that these activities constituted part of our ongoing central operations. |
| • | Product royalties. We are entitled to receive royalties on net sales of REGNITE in Japan. Astellas initiated sales of REGNITE in Japan in July 2012, and we recognize the associated product royalties when they can be reliably measured and collectability is reasonably assured (generally upon receipt of the royalty payment). |
Inventories
Inventories are stated at the lower of cost or market. Cost is determined on a first-in, first-out, or FIFO, basis. Inventories that are not expected to be consumed within 12 months of our normal operating cycle following the balance sheet date are classified as long-term inventories. We regularly evaluate our inventories for excess quantities and obsolescence (expiration), taking into account such factors as historical and anticipated future sales compared to quantities on hand and the remaining shelf life of HORIZANT. We evaluate our long-range demand for HORIZANT and expected consumption of the gabapentin enacarbil active pharmaceutical ingredient, or API, based on projected sales of HORIZANT. If future demand for HORIZANT is less than projected sales, we could incur an inventory write-down, which would have an adverse effect on earnings in the
78
Table of Contents
period of the write-down. We are also subject to manufacturing delays or disruptions, which could result in us incurring inventory-related losses, as well as delay the development of and/or impair our ability to commercialize products that we develop.
Accrued Expenses
As part of the process of preparing financial statements, we are required to estimate accrued expenses. This process involves communicating with our applicable personnel and service providers to identify services that have been performed on our behalf and estimating the level of services performed and the associated costs incurred for the services when we have not yet been invoiced or otherwise notified of actual costs. The majority of our service providers invoice us monthly in arrears for services performed. We make estimates of our accrued expenses as of each balance sheet date in our financial statements based on facts and circumstances known to us. We periodically confirm the accuracy of our estimates with selected service providers and make adjustments, if necessary. To date, we have not adjusted our estimates at any particular balance sheet date by any material amount. Examples of estimated accrued expenses include:
| • | fees paid to contract research organizations in connection with preclinical and toxicology studies and clinical trials; |
| • | fees paid to investigative sites in connection with clinical trials; |
| • | fees paid to contract manufacturers in connection with the production of clinical trial materials; and |
| • | professional service fees, such as marketing fees and sales operations fees. |
We base our expenses related to clinical trials on our estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and clinical research organizations that conduct and manage clinical trials on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If we do not identify costs that we have begun to incur or if we underestimate or overestimate the level of services performed or the costs of these services, our actual expenses could differ from our estimates.
Fair Value Measurements
The carrying amounts of certain of our financial instruments, including cash and cash equivalents and short-term investments, restricted investments, accounts receivables and accounts payable, approximate their fair value due to their short-term maturities. We account for the fair value of our financial instruments in accordance with the provisions of the Fair Value Measurement topic of the Codification.
Fair value is the price that would be received to sell an asset or paid to transfer a liability (an exit price) in an orderly transaction between market participants at the measurement date. We apply the market approach valuation technique for fair value measurements on a recurring basis and attempt to maximize the use of observable inputs and minimize the use of unobservable inputs. The fair value hierarchy prioritizes the inputs to valuation techniques used to measure fair value into three broad levels. All of our cash equivalents and short-term investments are measured using inputs classified at Level 1 or Level 2 within the fair value hierarchy. Level 1 inputs are quoted prices in active markets for identical assets. Level 2 inputs are based upon quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active and model-based valuation techniques for which all significant inputs are observable in the market or can be corroborated by observable market data for substantially the full term of the assets. Level 3 inputs are unobservable inputs that are supported by little or no market activity and are significant to the fair value of the assets or liabilities. Where applicable, these models project future cash flows and discount the future amounts to a present value using market-based observable inputs obtained from various third-party data providers, including but not limited to, benchmark yields, interest rate curves, reported trades, broker/dealer quotes and market reference data.
79
Table of Contents
Stock-Based Compensation
The provisions of the Compensation — Stock Compensation topic of the Codification establish accounting for stock-based awards exchanged for employee services. In accordance with the topic, for stock options, awards and stock purchase rights, stock-based compensation cost is measured on the grant date, based on the fair value of the award, and is recognized as expense over the requisite employee service period.
We estimate the fair value of stock options and stock purchase rights using a Black-Scholes valuation model which requires the input of highly subjective assumptions, including the option’s expected life and the price volatility of the underlying stock. We derive the expected life assumption using data obtained from similar entities, taking into consideration factors such as industry, stage of life cycle, size and financial leverage. We use our historical volatility to derive the expected stock price volatility assumption. The fair value of each option grant is estimated on the date of grant using the Black-Scholes option valuation model, and the resulting charge is expensed using the straight-line attribution method over the vesting period. Restricted stock units, or RSUs, are measured at the fair value of our common stock on the date of grant and expensed over the period of vesting using the straight-line attribution approach. The Black-Scholes option-pricing model was developed for use in estimating the fair value of short-lived, exchange-traded options that have no vesting restrictions and are fully transferable.
We account for stock compensation arrangements to non-employees in accordance with the Equity-Based Payments to Non-Employees topic of the Codification, using a fair value approach. The compensation costs of these arrangements are subject to remeasurement over the vesting terms as earned.
Research and Development Expenses
Research and development expenses consisted of costs associated with both partnered and unpartnered research activities, as well as costs associated with our drug discovery efforts, conducting preclinical studies and clinical trials, manufacturing development efforts and activities related to regulatory filings. Research and development expenses specifically consisted of: external research and development expenses incurred under agreements with (i) third-party contract research organizations and investigative sites, where a substantial portion of our preclinical studies and all of our clinical trials are conducted, (ii) third-party manufacturing organizations, where a substantial portion of our preclinical supplies and all of our clinical supplies are produced, and (iii) consultants; employee-related expenses, which include salaries and benefits; and facilities, depreciation and amortization and other allocated expenses, which include direct and allocated expenses for rent and maintenance of facilities, depreciation of leasehold improvements and equipment and laboratory and other supplies. We use our employee and infrastructure resources across multiple research projects, including our drug development programs. We do not allocate our employee and infrastructure costs on a project-by-project basis.
Our current portfolio of proprietary product candidates is summarized in the table below. The table summarizes development initiatives, including the related stages of development and the direct, third-party research and development expenses recognized in connection with each of our product candidates. The information in the column labeled “Estimated Completion of Current Phase” is our current estimate of the timing of completion of the current phase of development. The actual timing of completion could differ materially from the estimates provided in the table. For a discussion of the risks and uncertainties associated with the timing of completing a product development phase, see the “Our success also depends substantially on our product candidates that are still under development. If we or our business partners are unable to bring any of these product candidates to market, or experience significant delays in doing so, our ability to generate product revenue and our likelihood of success will be reduced;” “If third parties do not manufacture HORIZANT, REGNITE or our product candidates in sufficient quantities or at an acceptable cost, commercialization of HORIZANT and REGNITE and clinical development and commercialization of our product candidates would be harmed or delayed;” “If we or our business partners are required to obtain alternate third-party manufacturers, it could delay or prevent the clinical development and commercialization of HORIZANT or our product candidates;” “Use of third-party manufacturers may increase the risk that we or our business partners will not have adequate supplies of HORIZANT, REGNITE or our product candidates;” “If our or our business partners’ preclinical studies do not produce successful results or our clinical trials do not demonstrate safety and efficacy in humans, we or our
80
Table of Contents
business partners’ will not be able to commercialize our product candidates;” “Any failure or delay in commencing or completing clinical trials for our product candidates could severely harm our business;” “We rely on third parties to conduct our preclinical and clinical trials. If these third parties do not perform as contractually required or expected, we may not be able to obtain regulatory approval for, or commercialize, our product candidates;” “If we or our business partners are not able to obtain or maintain required regulatory approvals, we or our business partners will not be able to commercialize HORIZANT, REGNITE or our product candidates, our ability to generate revenue will be materially impaired and our business will not be successful;” and “If we do not establish partnerships, collaborations or other strategic arrangements for our product candidates, we may have to alter our development and commercialization plans” sections of “Risk Factors.”
| Product Candidate |
Description | Phase of Development | Estimated Completion of Current Phase |
Related R&D Expenses Year Ended December 31, |
||||||||||||||
| 2014 | 2013 | 2012 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||
| XP23829 |
Psoriasis/ MS |
Phase 2 Under evaluation |
2015 Under evaluation |
$ | 8,540 | $ | 4,775 | $ | 3,502 | |||||||||
| AP* |
Spasticity | Discontinued in 2013 |
N/A - Partnered | 116 | 9,380 | 13,224 | ||||||||||||
| XP21279 |
Parkinson’s disease |
Phase 2 completed | Pending resources | 32 | 74 | 208 | ||||||||||||
| Other(1) |
14,991 | 19,096 | 26,013 | |||||||||||||||
|
|
|
|
|
|
|
|||||||||||||
| Total research and development |
$ | 23,679 | $ | 33,325 | $ | 42,947 | ||||||||||||
|
|
|
|
|
|
|
|||||||||||||
| * | We discontinued our development of AP in 2013 and in June 2014, we granted exclusive, world-wide rights to Indivior to develop and commercialize AP Products for all indications. |
| (1) | “Other” constituted research and development costs for our marketed product and product candidates that are not directly allocated to XP23829, AP or XP21279. For the year ended December 31, 2014, “other” expenses consisted primarily of personnel costs of $9.9 million and office and facilities overhead costs of $3.8 million. For the year ended December 31, 2013, “other” expenses consisted primarily of personnel costs of $11.6 million and office and facilities overhead costs of $4.2 million. For the year ended December 31, 2012, “other” expenses consisted primarily of personnel costs of $17.0 million and office and facilities overhead costs of $6.0 million. |
A significant component of our total operating expenses is our ongoing investment in research and development activities, including the clinical development of XP23829. The process of conducting the clinical research necessary to obtain FDA approval is costly and time consuming. We consider the active management and development of XP23829 to be critical to our long-term success. The actual probability of success for each product candidate and clinical program may be impacted by a variety of factors, including, among others, the quality of the product candidate, early clinical data, investment in the program, competition, manufacturing capability and commercial viability. Furthermore, our strategy includes entering into additional partnerships, collaborations or other strategic arrangements with third parties to participate in the development and commercialization of at least some of our product candidates. In situations in which third parties have control over the preclinical development or clinical trial process for a product candidate, such as under our license agreement with Indivior, the estimated completion date is largely under the control of that third party and not under our control. We cannot forecast with any degree of certainty which of our other product candidates, if any, will be subject to future partnerships, collaborations or other strategic arrangements, or how such arrangements would affect our development plans or capital requirements.
As a result of the uncertainties discussed above, we are unable to determine the duration and completion costs of our research and development projects or when and to what extent we will generate revenues from the commercialization and sale of any of our product candidates.
81
Table of Contents
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board, or FASB, issued Accounting Standards Update, or ASU, No. 2014-09, Revenue from Contracts with Customers: Topic 606 (ASU 2014-09), which creates a single source of revenue guidance under U.S. generally accepted accounting principles, or GAAP, for all companies in all industries. The core principle of ASU 2014-09 is that revenue should be recognized in a manner that depicts the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. ASU 2014-09 defines a five-step process in order to achieve this core principle, which may require the use of judgment and estimates. ASU 2014-09 also requires expanded qualitative and quantitative disclosures relating to the nature, amount, timing and uncertainty of revenue and cash flows arising from contracts with customers, including significant judgments and estimates used. ASU 2014-09 will be effective for us in the first quarter of fiscal 2017, and early adoption is not permitted. We may adopt ASU 2014-09 either by way of a full retrospective approach for all periods presented in the period of adoption or a modified retrospective approach. We are currently evaluating the impact that ASU 2014-09 will have on our financial statements and have not yet determined which transition method we will apply.
In August 2014, the FASB issued ASU No. 2014-15, Presentation of Financial Statements—Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern (ASU 2014-15). This update is intended to define management’s responsibility to evaluate whether there is substantial doubt about an organization’s ability to continue as a going concern and to provide related footnote disclosures. ASU 2014-15 requires management to assess an entity’s ability to continue as a going concern by incorporating and expanding upon certain principles that are currently in U.S. auditing standards. Specifically, the amendments: (1) provide a definition of the term substantial doubt; (2) require an evaluation every reporting period including interim periods; (3) provide principles for considering the mitigating effect of management’s plans; (4) require certain disclosures when substantial doubt is alleviated as a result of consideration of management’s plans; (5) require an express statement and other disclosures when substantial doubt is not alleviated; and (6) require an assessment for a period of one year after the date that the financial statements are issued (or available to be issued). ASU 2014-15 will be effective for annual periods ending after December 15, 2016, and interim periods within annual periods beginning after December 15, 2016, with early adoption permitted. ASU 2014-15 will be effective for us beginning with our annual report for fiscal 2016 and interim periods thereafter. We are currently evaluating the impact that ASU 2014-15 will have on our financial statements.
82
Table of Contents
Results of Operations
Years Ended December 31, 2014, 2013 and 2012
Revenues
Our net product sales revenue was from the sales of HORIZANT after the transition period ended on April 30, 2013. Our collaboration revenue consisted of the recognition of revenues from upfront and milestone payments from our partnership with Astellas and from upfront and additional payments from our partnership with Indivior. Our royalty revenue was from Astellas and was based on its net sales of REGNITE in Japan. Our net revenue from unconsolidated joint operating activities consisted of the recognition of a contingent event-based payment under our terminated collaboration with GSK.
| Year Ended December 31, | 2013 to 2014 Change |
2012 to 2013 Change |
||||||||||||||||||||||||||
| 2014 | 2013 | 2012 | $ | % | $ | % | ||||||||||||||||||||||
| (In thousands, except percentages) | ||||||||||||||||||||||||||||
| Product sales, net |
$ | 20,173 | $ | 6,414 | $ | — | $ | 13,759 | 215 | % | $ | 6,414 | 100 | % | ||||||||||||||
| Collaboration revenue |
26,134 | 1,137 | 11,515 | 24,997 | 2199 | % | (10,378 | ) | (90 | )% | ||||||||||||||||||
| Royalty revenue |
561 | 400 | 109 | 161 | 40 | % | 291 | 267 | % | |||||||||||||||||||
| Net revenue from unconsolidated joint operating activities |
— | — | 10,000 | — | 0 | % | (10,000 | ) | (100 | )% | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
| Total revenues |
$ | 46,868 | $ | 7,951 | $ | 21,624 | $ | 38,917 | 489 | % | $ | (13,673 | ) | (63 | )% | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
The increase in total revenues for 2014 compared to 2013 was primarily due to the recognition of $25.0 million in collaboration revenue resulting from the Indivior licensing agreement during the third quarter of 2014. The increase in HORIZANT net product sales in 2014 compared to 2013 was primarily due to the full year effect of HORIZANT net product sales in 2014 compared to only eight months in 2013, as well an increase in sales volume due to enhanced promotional efforts and, to a lesser extent, from an increase in average net selling prices due to price increases initiated during the first half of 2014.
The increase in net product sales for 2013 compared to 2012 was due to the acquisition of the HORIZANT business on May 1, 2013 and our first commercial sales in 2013. The decrease in collaboration revenue for 2013 compared to 2012 was due to the recognition of a $10.0 million milestone payment from Astellas in connection with the approval of REGNITE in Japan in 2012. The decrease in net revenue from unconsolidated joint operating activities for 2013 compared to 2012 was due to the recognition of a $10.0 million contingent payment from GSK in connection with the first commercial sale of HORIZANT by GSK for the management of PHN in adults in 2012.
While our revenues for 2014 included revenues from the recognition of cash payments received under our license agreement with Indivior and we may in the future recognize revenues from contingent-event based payments from Indivior, we expect the future composition of our revenues to consist primarily of revenues from HORIZANT net product sales. We expect our HORIZANT net product sales to increase in 2015 compared to 2014 levels and that they will be dependent upon the success of our strategies for commercialization, promotion, manufacturing and distribution, as well as our ability to successfully execute on these activities and to comply with applicable laws, regulations and regulatory requirements. We expect royalty revenue from our partnership with Astellas to fluctuate based on the results of its commercialization, marketing and distribution efforts for REGNITE in Japan; however, we do not expect that royalty revenues from our partnership with Astellas will be meaningful. We expect collaboration revenue to fluctuate based on Indivior’s timing and achievement of AP contingent-event based payments and to the extent we enter into new partnership, collaboration or other strategic arrangements for our marketed product or any of our other product candidates.
Cost of Product Sales
Cost of product sales consisted of the direct and indirect costs to manufacture product sold, including tableting, packaging, storage, and shipping and handling costs related to our product sales of HORIZANT. Cost
83
Table of Contents
of product sales also included raw materials, specifically gabapentin enacarbil, acquired from GSK as part of the acquisition of the HORIZANT business and recorded at fair value. The fair value of the inventory acquired from GSK represents a lower cost than if new raw materials were purchased at current third-party costs.
| Year
Ended December 31, |
2013 to 2014 Change |
2012 to 2013 Change |
||||||||||||||||||||||||||
| 2014 | 2013 | 2012 | $ | % | $ | % | ||||||||||||||||||||||
| (In thousands, except percentages) | ||||||||||||||||||||||||||||
| Cost of Product Sales |
$ | 2,094 | $ | 1,170 | $ | — | $ | 924 | 79 | % | $ | 1,170 | 100 | % | ||||||||||||||
The increase in cost of product sales in 2014 compared to 2013 was primarily from an increase in volume of HORIZANT product sales due to the full period effect of HORIZANT product sales in 2014 compared to 2013 and, to a lesser extent, due to enhanced promotional efforts. The increase in cost of product sales in 2013 compared to 2012 was due to HORIZANT product sales that commenced in May 2013.
We expect cost of product sales of HORIZANT to remain relatively constant as a percentage of product sales in 2015.
Research and Development Expenses
| Year Ended December 31, | 2013 to 2014 Change |
2012 to 2013 Change |
||||||||||||||||||||||||||
| 2014 | 2013 | 2012 | $ | % | $ | % | ||||||||||||||||||||||
| (In thousands, except percentages) | ||||||||||||||||||||||||||||
| Research and development |
$ | 23,679 | $ | 33,325 | $ | 42,947 | $ | (9,646 | ) | (29 | )% | $ | (9,622 | ) | (22 | )% | ||||||||||||
The decrease in research and development expenses for 2014 compared to 2013 was principally due to:
| • | decreased net costs for AP of $9.3 million primarily due to decreased clinical, manufacturing and consulting costs as a result of terminating further development of AP as a potential treatment for spasticity in patients with MS in 2013; and |
| • | decreased personnel costs of $1.8 million primarily due to decreased headcount and corresponding decreased non-cash stock-based compensation; partially offset by |
| • | increased net costs for XP23829 of $3.8 million primarily due to increased toxicology and clinical costs. |
The decrease in research and development expenses for 2013 compared to 2012 was principally due to:
| • | decreased net costs for AP of $3.8 million primarily due to decreased clinical costs as a result of the termination of further development of AP as a potential treatment for spasticity in patients with MS in 2013, partially offset by increased manufacturing costs; and |
| • | decreased personnel costs of $5.4 million primarily due to decreased headcount and decreased non-cash stock-based compensation; partially offset by |
| • | increased net costs for XP23829 of $1.3 million primarily due to increased clinical, manufacturing and consulting costs, partially offset by decreased toxicology costs. |
We expect our research and development expenses to increase in 2015 compared to 2014 levels primarily due to increased development expenses for XP23829, including the cost of our Phase 2 clinical trial in patients with moderate-to-severe chronic plaque-type psoriasis that we initiated in June 2014, as well as increased costs in anticipation of our potential XP23829 Phase 3 clinical activities, which will be partially offset by a lack of development expenses for AP, which was exclusively licensed to Indivior in June 2014. The timing and amount of research and development expenses incurred will primarily depend upon the extent of current or future clinical trials for XP23829 as well as the related expenses associated with our development organization, regulatory requirements for our product candidate and product candidate manufacturing costs.
84
Table of Contents
Selling, General and Administrative Expenses
Selling, general and administrative expenses consisted primarily of compensation for executive, sales, marketing, finance, legal, compliance and administrative personnel. Other sales, general and administrative expenses included facility costs not otherwise included in research and development expenses, the cost of market research activities, legal and accounting services, other professional services and consulting fees.
| Year Ended December 31, | 2013 to 2014 Change |
2012 to 2013 Change |
||||||||||||||||||||||||||
| 2014 | 2013 | 2012 | $ | % | $ | % | ||||||||||||||||||||||
| (In thousands, except percentages) | ||||||||||||||||||||||||||||
| Selling, general and administrative |
$ | 70,194 | $ | 59,084 | $ | 30,244 | $ | 11,110 | 19 | % | $ | 28,840 | 95 | % | ||||||||||||||
The increase in selling, general and administrative expenses in 2014 compared to 2013 was principally due to costs related to the continued commercialization and promotion of HORIZANT; specifically, increased professional fees of $4.6 million (which included contract sales force costs) and personnel costs of $5.9 million.
The increase in selling, general and administrative expenses in 2013 compared to 2012 was principally due to costs related to the commercialization and promotion of HORIZANT; specifically, increased professional fees of $12.5 million (which included contract sales force costs), marketing costs of $12.2 million and personnel costs of $3.1 million.
Since May 1, 2013, we have been responsible for the commercialization and promotion of HORIZANT in the United States. Accordingly, we expect continued increases in selling, general and administrative expenses in 2015 compared to 2014 levels due to expenses to be incurred in maintaining and expanding marketing, distribution and other commercial capabilities largely through third-party service providers, as well as the internal and external expenses of supporting, maintaining and increasing our XenoPort-employed sales representatives and expenses arising from maintaining and increasing our contract sales representatives.
Gain on Litigation Settlement
As a result of the termination and transition agreement that we entered into with GSK that provided for a mutual release of claims and resolved all ongoing litigation between the parties, we recorded a gain of $20.5 million in the fourth quarter of 2012.
Interest Income/Expense
Interest income consisted primarily of interest earned on our cash equivalents and short-term investments, and interest expense consisted primarily of the accretion of our other noncurrent liability.
| Year Ended December 31, | 2013 to 2014 Change |
2012 to 2013 Change |
||||||||||||||||||||||||||
| 2014 | 2013 | 2012 | $ | % | $ | % | ||||||||||||||||||||||
| (In thousands, except percentages) | ||||||||||||||||||||||||||||
| Interest income |
$ | 253 | $ | 213 | $ | 254 | $ | 40 | 19 | % | $ | (41 | ) | (16 | )% | |||||||||||||
| Interest expense |
487 | 468 | — | 19 | 4 | % | 468 | 100 | % | |||||||||||||||||||
Interest income for each of the years ended December 31, 2014, 2013 and 2012 remained relatively constant.
The increase in interest expense for the years ended December 31, 2014 and 2013 related to annual payments of $1.0 million to GSK in connection with the 2012 termination and transition agreement for six years beginning in 2016, recorded at its present value, which is being accreted to its contractual value over the repayment period. We expect interest expense to increase in 2015 compared to 2014 due to interest expense associated with our 2022 Notes that we issued in February 2015.
85
Table of Contents
Liquidity and Capital Resources
| As of December 31, | ||||||||
| 2014 | 2013 | |||||||
| Cash and cash equivalents and short-term investments |
$ | 102,056 | $ | 58,658 | ||||
| Working capital |
91,806 | 53,616 | ||||||
| Restricted investments |
1,725 | 1,725 | ||||||
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| (In thousands) | ||||||||||||
| Cash provided by (used in): |
||||||||||||
| Operating activities |
$ | (32,696 | ) | $ | (77,903 | ) | $ | (27,630 | ) | |||
| Investing activities |
(53,699 | ) | 63,509 | (35,192 | ) | |||||||
| Financing activities |
77,769 | (1,156 | ) | 73,570 | ||||||||
| Capital expenditures (included in investing activities above) |
493 | 256 | 123 | |||||||||
Due to our significant research and development and, more recently, sales and marketing expenditures, we have generated cumulative operating losses since we incorporated in 1999. As such, we have funded our operations primarily through sales of our equity and debt securities, non-equity payments from our business partners and interest earned on investments. At December 31, 2014, we had available cash and cash equivalents and short-term investments of $102.1 million. Our cash and investment balances are held in a variety of interest-bearing instruments, including corporate debt securities and money market accounts. Cash in excess of immediate requirements is invested with regard to liquidity and capital preservation, and we seek to minimize the potential effects of concentration and degrees of risk.
Net cash used in operating activities was $32.7 million, $77.9 million and $27.6 million in the years ended December 31, 2014, 2013 and 2012, respectively. The net cash used in operating activities in 2014 and 2013 primarily reflected our net loss, partially offset by non-cash stock-based compensation. The net cash used in operating activities in 2012 primarily reflected our net loss and, to a lesser extent, a gain on litigation settlement, net of stock premium in connection with the stock purchase agreement that we entered into with GSK in connection with the termination of our development and commercialization agreement, partially offset by non-cash stock-based compensation.
Net cash provided by (used in) investing activities primarily reflected the timing of purchases of investments and proceeds from maturities of investments.
Net cash provided by (used in) financing activities primarily reflected net cash provided by (used in) the issuance of common stock in the years ended December 31, 2014, 2013 and 2012. In 2014, we also received $77.4 million in net proceeds from our public offering of common stock. Additionally, in 2012, we recorded proceeds from the issuance of common stock to GSK in connection with the stock purchase agreement that we entered into with GSK in connection with the termination of our development and commercialization agreement of $30.3 million.
On February 3, 2015, we completed a private placement of $115.0 million aggregate principal amount of 2022 Notes to the investment bank acting as the initial purchaser who subsequently resold the 2022 Notes to qualified institutional buyers. The 2022 Notes sold in the offering include $15.0 million aggregate principal amount of 2022 Notes sold to the initial purchaser pursuant to its option to purchase additional 2022 Notes, which was exercised in full on January 29, 2015. The estimated net proceeds from the offering of the 2022 Notes is approximately $111.4 million, after deducting the initial purchaser’s discount and estimated offering expenses payable by us. For more information on the 2022 Notes, see “2022 Notes” below.
On January 29, 2014, we completed an underwritten public offering of 12,000,000 shares of our common stock at a price to the public of $6.00 per share. Net cash proceeds from the public offering were $67.3 million, after deducting the underwriting discounts and commissions and offering expenses payable by us. On February 21, 2014, the underwriters exercised in full their over-allotment option of 1,800,000 shares resulting
86
Table of Contents
in cash proceeds of $10.1 million, after deducting the underwriting discounts and commissions and offering expenses payable by us.
We believe that our existing capital resources, including proceeds we received from our convertible debt offering that closed in February 2015, together with interest income from our investments and anticipated product revenue, will be sufficient to meet our projected operating requirements through at least the first quarter of 2018. We have based our cash sufficiency estimate on assumptions that may prove to be incorrect, and we could utilize our available capital resources sooner than we expect. Further, our operating plan may change, and we may need additional funds to meet operational needs and capital requirements for product development and commercialization sooner than planned. We have no credit facility or committed sources of capital other than potential contingent event-based and royalty payments that we are eligible to receive under our partnerships with Astellas and Indivior.
With the exception of the proposed clinical trial in patients with AUD that the NIAAA plans to conduct, we are solely responsible for all HORIZANT commercialization and development activities, including all post-marketing requirements and commitments. Such costs could be greater than we anticipate, or sales of HORIZANT may be less than we anticipate, either or both of which could accelerate our need for additional capital. In addition, we do not believe that revenues generated from HORIZANT and REGNITE sales and other revenues generated from our partnerships with Astellas and Indivior will be sufficient alone to sustain our operations, at least in the near term, and we therefore expect to continue to experience negative cash flows for the foreseeable future as we fund our operating losses and capital expenditures.
Our forecast of the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors, including the factors discussed in “Risk Factors.” Because of the numerous risks and uncertainties associated with the development and commercialization of our marketed product and product candidates, and the extent to which we enter into additional partnerships, collaborations or other strategic arrangements with third parties to participate in their development and commercialization, we are unable to estimate the amounts of increased capital outlays and operating expenditures associated with our current and anticipated clinical trials. Our future funding requirements will depend on many factors, including:
| • | the timing, receipt and amount of sales or royalties from HORIZANT, REGNITE and our potential products; |
| • | the costs of our sales and marketing, supply chain, distribution, pharmacovigilance, compliance and safety infrastructure to promote and distribute HORIZANT, including the internal and external expenses of supporting and maintaining our XenoPort-employed sales representatives and expenses associated with our contract sales representatives and our planned and any other potential future expansions to our dedicated sales team; |
| • | the scope, rate of progress, results and costs of our preclinical testing, clinical trials and other research and development activities, including the timing and costs of further development of XP23829; |
| • | the costs of manufacturing clinical and commercial supplies of HORIZANT and our product candidates; |
| • | the timing and costs of complying with the remaining post-marketing commitments and post-marketing requirements established in connection with the approval of HORIZANT, and any future additional commitments or requirements imposed on us by the FDA; |
| • | the number and characteristics of product candidates that we pursue, including any additional potential indications for HORIZANT such as the treatment of patients with AUD, or the extent to which we pursue any further development of XP21279; |
| • | the costs, timing and outcomes of regulatory approvals, if any; |
| • | the timing of any contingent-event based and royalty payments under our licensing arrangement with Indivior; |
| • | the terms and timing of any collaboration or partnership arrangements that we may establish or modify; |
87
Table of Contents
| • | the costs associated with any potential litigation; |
| • | the costs of preparing, filing, prosecuting, maintaining, defending and enforcing any patent claims and other intellectual property rights; and |
| • | the extent to which we acquire or invest in businesses, products or technologies that complement our business, although we have no commitments or agreements relating to any of these types of transactions. |
In addition, we are required to make periodic interest payments to the holders of our 2022 Notes and to make payments of principal upon maturity. In this regard, if holders of the 2022 Notes do not convert their 2022 Notes prior to the maturity date, we will be required to repay the principal amount of all then outstanding 2022 Notes plus accrued and unpaid interest. We may also be required to repurchase the 2022 Notes for cash upon the occurrence of a change of control or certain other fundamental changes involving us. If our capital resources are insufficient to satisfy our debt service obligations, we will be required to seek to sell additional equity or debt securities or to obtain debt financing.
Until we can generate a sufficient amount of product revenues, if ever, we expect to finance future cash needs through public or private equity or equity-linked offerings, debt financings or third-party business transactions. If we raise additional funds by issuing our common stock, or securities convertible into, exchangeable or exercisable for common stock, our stockholders will experience dilution. Any debt financing or additional equity that we raise may contain terms that are not favorable to our stockholders or us. To the extent that we raise additional capital through partnering, collaborative or other strategic arrangements, we may be required to relinquish, on terms that are not favorable to us, rights to some of our technologies or product candidates that we would otherwise seek to develop or commercialize ourselves. Our ability to raise additional funds and the terms upon which we are able to raise such funds may be adversely impacted by the uncertainty regarding our financial condition, the commercial prospects of HORIZANT and REGNITE based on their respective sales to date and our relative lack of experience in commercializing products, and/or current economic conditions, including the effects of disruptions to, and volatility in, the credit and financial markets in the United States, Asia, the European Union and other regions of the world.
Additional funds may not be available to us on terms that are acceptable to us, or at all. If adequate funds are not, or we anticipate that they may not be, available on a timely basis, we may be required to, among other things, reduce the amount of resources devoted to medical affairs, advertising, promotion or sales of HORIZANT, curtail or delay further XP23829 development or conduct additional workforce or other expense reductions, any of which could have a material adverse effect on our business and prospects. In addition, we may face substantial financial costs in order to comply with the remaining post-marketing commitments and post-marketing requirements established in connection with the approval of HORIZANT, and our inability to carry out and fund such efforts to comply with these requirements could have material adverse consequences to us, including the loss of marketing approval for HORIZANT.
2022 Notes
In February 2015, we completed a private placement of $115.0 million principal amount of 2022 Notes. Interest on the 2022 Notes will be payable semi-annually in cash in arrears on February 1 and August 1 of each year, beginning on August 1, 2015, at a rate of 2.50% per year. The 2022 Notes mature on February 1, 2022, unless earlier converted or repurchased. The 2022 Notes are not redeemable prior to the maturity date.
The 2022 Notes are convertible at an initial conversion rate of 93.2945 shares of our common stock per $1,000 principal amount of the 2022 Notes, subject to adjustment, which is equal to an initial conversion price of approximately $10.72 per share of common stock. Upon conversion, the 2022 Notes may be settled in shares of our common stock, together with a cash payment in lieu of delivering any fractional share. The conversion rate will be subject to adjustment in some events but will not be adjusted for any accrued and unpaid interest. In addition, following certain corporate events that occur prior to the maturity date, we will increase the conversion rate for a holder who elects to convert its 2022 Notes in connection with such a corporate event in certain circumstances.
88
Table of Contents
If we undergo a fundamental change, which would include specified change of control transactions, or liquidation or dissolution or our common stock ceasing to be listed or quoted on specified national securities exchanges, and the fundamental change occurs prior to the maturity date of the 2022 Notes, holders of the 2022 Notes may require us to repurchase all or part of their 2022 Notes at a repurchase price equal to 100% of the principal amount of the 2022 Notes to be repurchased, plus accrued and unpaid interest to, but excluding, the fundamental change repurchase date.
Off-Balance Sheet Arrangements
We have no material off-balance sheet arrangements as defined in Regulation S-K 303(a)(4)(ii).
Contractual Obligations
Our future contractual obligations at December 31, 2014 were as follows (in thousands):
| Payments Due by Period | ||||||||||||||||||||
| Contractual Obligations (1) |
Total | Less Than 1 Year |
1-3 Years |
3-5 Years |
Greater Than 5 Years |
|||||||||||||||
| Purchase commitments (2) |
$ | 2,888 | $ | 2,888 | $ | — | $ | — | $ | — | ||||||||||
| Long-term payable (3) |
6,000 | — | 2,000 | 2,000 | 2,000 | |||||||||||||||
| Operating lease obligations (4) |
3,354 | 2,368 | 986 | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| $ | 12,242 | $ | 5,256 | $ | 2,986 | $ | 2,000 | $ | 2,000 | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| (1) | Does not include our obligations under the 2022 Notes since they were issued in February 2015. For more information on the 2022 Notes, please see “2022 Notes” above. |
| (2) | At December 31, 2014, we had non-cancelable purchase orders and minimum purchase obligations of approximately $2.9 million under our commercial supply agreement with Patheon Pharmaceuticals Inc. and GSK. |
| (3) | On November 8, 2012, we executed a termination and transition agreement with GSK that terminated our development and commercialization agreement with respect to HORIZANT. GSK provided us inventory of gabapentin enacarbil in GSK’s possession that was not required for use by GSK in the manufacture of HORIZANT. In exchange for such inventory, we will make annual payments to GSK of $1.0 million for six years beginning in 2016. |
| (4) | In the fourth quarter of 2014, we entered into an amendment to the lease with respect to our current office space at 3410 Central Expressway, Santa Clara, California, which extended the term of the lease for an additional nine months so that the lease will expire on May 31, 2016. |
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
The primary objective of our investment activities is to preserve our capital to fund operations. We also seek to maximize income from our investments without assuming significant risk. To achieve our objectives, we maintain a portfolio of cash equivalents and investments in a variety of securities of high-credit quality. As of December 31, 2014, we had cash and cash equivalents and short-term investments of $102.1 million consisting of cash and highly liquid investments deposited in highly rated financial institutions in the United States. A portion of our investments may be subject to interest rate risk and could fall in value if market interest rates increase. However, because our investments are short-term in duration, we believe that our exposure to interest rate risk is not significant and a 1% movement in market interest rates would not have a significant impact on the total value of our portfolio. We actively monitor changes in interest rates.
In February 2015, we completed a private placement of $115.0 million aggregate principal amount of 2.50% convertible senior notes due 2022, or the 2022 Notes. The 2022 Notes have a fixed annual interest rate of 2.50% and we, therefore, do not have economic interest rate exposure on the 2022 Notes. However, the fair value of the
89
Table of Contents
2022 Notes will be exposed to interest rate risk. Generally, the fair value of the 2022 Notes will increase as interest rates fall and decrease as interest rates rise. The fair value of the 2022 Notes will also be affected by volatility in our stock price.
From time to time, we are subject to exposure to fluctuations in foreign exchange rates in connection with agreements with certain foreign contract manufacturers and royalty payments from our collaboration partner. To date, the effect of the exposure to these fluctuations in foreign exchange rates has not been material, and we do not expect it to be material in the foreseeable future. We do not hedge our foreign currency exposures. We have not used derivative financial instruments for speculation or trading purposes.
Item 8. Financial Statements and Supplementary Data.
The information required by this item is incorporated herein by reference to the financial statements and schedule listed in Item 15 (1) and (2) of Part IV of this Annual Report on Form 10-K.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
Not applicable.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
Based on their evaluation as required by paragraph (b) of Rules 13a-15 or 15d-15 of the Securities Exchange Act of 1934, as amended, or the Exchange Act, as of December 31, 2014, our chief executive officer and chief financial officer concluded that our disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) or 15d-15(e)) were effective.
Limitations on the Effectiveness of Controls. A control system, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Because of inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues, if any, within an organization have been detected. Accordingly, our disclosure controls and procedures are designed to provide reasonable, not absolute, assurance that the objectives of our disclosure control system are met and, as set forth above, our chief executive officer and chief financial officer have concluded, based on their evaluation as of December 31, 2014, that our disclosure controls and procedures were effective to provide reasonable assurance that the objectives of our disclosure control system were met.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Our internal control system is designed to provide reasonable assurance regarding the preparation and fair presentation of financial statements for external purposes in accordance with generally accepted accounting principles. All internal control systems, no matter how well designed, have inherent limitations and can provide only reasonable assurance that the objectives of the internal control system are met.
Under the supervision and with the participation of our management, including our chief executive officer and chief financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control — 2013 Framework. Based on our evaluation, management concluded that our internal control over financial reporting was effective as of December 31, 2014.
Ernst & Young LLP, an independent registered public accounting firm, has audited our financial statements included herein and has issued an audit report on the effectiveness of our internal control over financial reporting, which report is included below.
90
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of XenoPort, Inc.
We have audited XenoPort, Inc.’s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). XenoPort, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, XenoPort, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of XenoPort, Inc. as of December 31, 2014 and 2013, and the related statements of comprehensive loss, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2014 of XenoPort, Inc., and our report dated February 27, 2015 expressed an unqualified opinion thereon.
| /s/ ERNST & YOUNG LLP |
Redwood City, California
February 27, 2015
91
Table of Contents
Changes in Internal Controls Over Financial Reporting
There were no changes in our internal controls over financial reporting during the fourth quarter of 2014 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
On February 24, 2015, Ernest Mario, Ph.D., a current director of XenoPort, informed the nominating and corporate governance committee of the board of directors of XenoPort of his decision to not stand for re-election as a Class 1 director of the company at the 2015 annual meeting of stockholders, due to a desire to reduce his professional responsibilities related to public company boards. Dr. Mario indicated that his decision to resign was not a result of any disagreement with XenoPort on any matter relating to its operations, policies or practices. Dr. Mario will resign as a director of the company effective upon the expiration of the current Class 1 term at the 2015 annual meeting of stockholders.
Certain information required by Part III is omitted from this Annual Report on Form 10-K because we intend to file our definitive proxy statement for our 2015 annual meeting of stockholders, or the Proxy Statement, pursuant to Regulation 14A of the Exchange Act, not later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, and certain information to be included in the Proxy Statement is incorporated herein by reference.
Item 10. Directors, Executive Officers and Corporate Governance.
The information required by this item with respect to our executive officers may be found under the caption, “Executive Officers of the Registrant” in Item 1 of this Annual Report on Form 10-K. The information required by this item relating to our directors and nominees, including information with respect to our audit committee, audit committee financial experts and procedures by which stockholders may recommend nominees to our board of directors, may be found under the section entitled “Proposal 1 — Election of Directors” appearing in the Proxy Statement. Such information is incorporated herein by reference. Information regarding compliance with Section 16(a) of the Exchange Act may be found under the section entitled “Section 16(a) Beneficial Ownership Reporting Compliance” appearing in our Proxy Statement. Such information is incorporated herein by reference.
We have adopted a code of ethics that applies to our employees, officers and directors and incorporates guidelines designed to deter wrongdoing and to promote honest and ethical conduct and compliance with applicable laws and regulations. In addition, the code of ethics incorporates our guidelines pertaining to topics such as conflicts of interest and workplace behavior. We have posted the text of our code of ethics on our Web site at www.XenoPort.com in connection with “Investor Relations/Corporate Governance” materials. In addition, we intend to promptly disclose (1) the nature of any amendment to our code of ethics that applies to our principal executive officer, principal financial officer, principal accounting officer or controller or persons performing similar functions and (2) the nature of any waiver, including an implicit waiver, from a provision of our code of ethics that is granted to one of these specified officers, the name of such person who is granted the waiver and the date of the waiver on our Web site in the future.
Item 11. Executive Compensation.
The information required by this item is included in our Proxy Statement under the sections entitled “Executive Compensation,” “Director Compensation,” “Compensation Committee Interlocks and Insider Participation” and “Compensation Committee Report” and is incorporated herein by reference.
92
Table of Contents
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. |
Equity Compensation Plan Information
The following table provides certain information regarding our equity compensation plans in effect as of December 31, 2014:
| Plan Category |
Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants and Rights (a) |
Weighted-Average Exercise Price of Outstanding Options, Warrants and Rights (b) |
Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans (Excluding Securities Reflected in Column (a)) |
|||||||||
| Equity compensation plans approved by security holders: |
||||||||||||
| 1999 Stock Plan(1) |
107,433 | $ | 6.02 | — | ||||||||
| 2005 Equity Incentive Plan(2) |
5,807,106 | $ | 12.74 | — | ||||||||
| 2005 Non-Employee Directors’ Stock Option Plan(3) |
633,750 | $ | 15.83 | — | ||||||||
| 2005 Employee Stock Purchase Plan(4) |
— | $ | — | 277,882 | ||||||||
| 2014 Equity Incentive Plan(5) |
485,310 | $ | 4.54 | 4,479,789 | ||||||||
| Equity compensation plans not approved by security holders: |
||||||||||||
| New Hire Option Agreement with Vincent J. Angotti(6) |
140,612 | $ | 42.59 | — | ||||||||
| 2010 Inducement Award Plan(7) |
475,745 | $ | 6.64 | — | ||||||||
|
|
|
|
|
|||||||||
| Total |
7,649,956 | $ | 12.55 | 4,757,671 | ||||||||
|
|
|
|
|
|
|
|||||||
| (1) | In December 1999, we adopted the 1999 Stock Plan, or the 1999 Plan, which was terminated in June 2005 in connection with our initial public offering so that no further awards may be granted under the 1999 Plan. Although the 1999 Plan has terminated, all outstanding options under the 1999 Plan will continue to be governed by their existing terms. |
| (2) | In January 2005, we adopted the 2005 Equity Incentive Plan, or the 2005 Plan, which became effective in June 2005 in connection with our initial public offering. A total of 2,000,000 shares of common stock were initially authorized for issuance under the 2005 Plan. Our board of directors could increase the share reserve of the 2005 Plan as of each January 1, from January 1, 2006 through January 1, 2015, by an amount determined by our board; provided, however, that the increase for any year could not exceed the lesser of (1) 2.5% of the total number of shares of our common stock outstanding on the December 31st of the preceding calendar year or (2) 2,000,000 shares. During the year ended December 31, 2014, the annual increase to the 2005 Plan reserve was 1,195,009 shares. Restricted stock unit awards have been granted under the 2005 Plan and are included in column (a). The weighted-average exercise price in column (b) does include the effect of the restricted stock unit awards under the 2005 Plan, which awards do not carry an exercise price. At December 31, 2014, the weighted-average exercise price of outstanding options under the 2005 Plan was $15.04, excluding the restricted stock unit awards. In June 2014, in connection with our adoption of the 2014 Equity Incentive Plan, or the 2014 Plan, described below, no further awards may be granted under the 2005 Plan. Although the 2005 Equity Incentive Plan has terminated, all outstanding options under the 2005 Plan will continue to be governed by their existing terms. |
| (3) | In January 2005, we adopted the 2005 Non-Employee Directors’ Stock Option Plan, or the 2005 Directors’ Plan, which became effective in June 2005 in connection with our initial public offering. The 2005 Directors’ Plan provided for the automatic grant of options to purchase shares of our common stock to non-employee directors. A total of 150,000 shares of our common stock were initially authorized for issuance under the 2005 Directors’ Plan. Our board of directors could increase the share reserve of the 2005 |
93
Table of Contents
| Directors’ Plan as of each January 1, from January 1, 2006 through January 1, 2015, by an amount determined by our board; provided, however, that the increase for any year could not exceed the excess of (1) the number of shares of our common stock subject to options granted under the 2005 Directors’ Plan during the preceding calendar year over (2) the number of shares added back to the share reserve of the 2005 Directors’ Plan during the preceding calendar year from cancellations. During the year ended December 31, 2014, the annual increase to the 2005 Directors’ Plan reserve was 120,000 shares. In June 2014, in connection with our adoption of the 2014 Plan described below, no further awards may be granted under the 2005 Directors’ Plan. Although the 2005 Directors’ Plan has terminated, all outstanding options under the 2005 Directors’ Plan will continue to be governed by their existing terms. |
| (4) | In January 2005, we adopted the 2005 Employee Stock Purchase Plan, or ESPP, which became effective in June 2005 in connection with our initial public offering. The ESPP allows for qualified employees (as defined in the ESPP) to purchase shares of our common stock at a price equal to the lower of 85% of the closing price of our common stock at the beginning of the offering period or 85% of the closing price of our common stock on the date of purchase. A total of 250,000 shares of our common stock were initially authorized for issuance under the ESPP. Our board of directors could increase the share reserve of the ESPP as of each January 1, from January 1, 2006 through January 1, 2015, by an amount determined by our board; provided, however that the increase for any year could not exceed the lesser of (1) 1% of the total number of shares of our common stock outstanding on the December 31st of the preceding calendar year or (2) 250,000 shares. During the year ended December 31, 2014, the annual increase to ESPP reserve was 250,000. |
| (5) | In June 2014, the 2014 Plan was approved by our stockholders. The 2014 Plan is intended to be the successor to the 2005 Plan, 2005 Directors’ Plan and the Company’s 2010 Inducement Award Plan, collectively referred to as the Prior Plans. All outstanding stock awards granted under the Prior Plans will continue to be subject to the terms and conditions as set forth in the agreements evidencing such stock awards. The terms of the 2014 Plan provide for the grant of incentive stock options, nonstatutory stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards, other stock awards and performance awards that may be settled in cash, stock or other property. The total number of shares of our common stock available for issuance under the 2014 Plan is initially 4,471,059 shares plus up to an additional 7,847,852 Returning Shares (as defined below) as such shares become available from time to time. “Returning Shares” means the shares subject to outstanding awards granted under the Prior Plans and the 1999 Plan that, from and after the effective date of the 2014 Plan: (a) expire or terminate for any reason prior to exercise or settlement; (b) are forfeited, cancelled or otherwise returned to us because of the failure to meet a contingency or condition required for the vesting of such shares; or (c) other than with respect to outstanding stock options and stock appreciation rights granted under the Prior Plans or the 1999 Plan with an exercise or strike price of at least 100% of the fair market value of the underlying common stock on the date of grant, are reacquired or withheld (or not issued) by us to satisfy a tax withholding obligation in connection with a stock award. Eligible participants under the 2014 Plan include our employees and directors, including our executive officers and consultants. Restricted stock unit awards have been granted under the 2014 Plan and are included in column (a). The weighted-average exercise price in column (b) does include the effect of the restricted stock unit awards under the 2014 Plan, which awards do not carry an exercise price. At December 31, 2014, the weighted-average exercise price of outstanding options under the 2014 Plan was $5.38, excluding the restricted stock unit awards. |
| (6) | On May 1, 2008, Mr. Angotti was granted a new employee inducement stock award outside of our stockholder approved equity plans consisting of nonqualified stock options to purchase 140,612 shares of our common stock. The stock options have a per share exercise price of $42.59, the closing trading price of our common stock on the NASDAQ Global Market on May 1, 2008. The stock options have a ten-year term and vested over four years, with 25% cliff vesting on the first anniversary of the May 1, 2008 grant date, and 1/48th of the shares subject to the options vesting monthly thereafter. |
| (7) | In May 2010, the 2010 Inducement Award Plan, or the 2010 Inducement Plan, was adopted by our board of directors and became effective. We granted awards under the 2010 Inducement Plan to persons not previously employees or directors of ours (or following bona fide periods of non-employment by us and our affiliates) as inducements material to such individuals entering into employment with us and to provide |
94
Table of Contents
| incentives for such persons to exert maximum efforts for our success. A total of 350,000 shares of common stock were initially authorized for issuance under the 2010 Inducement Plan and an additional 625,000 shares were authorized for issuance in 2011. The 2010 Inducement Plan provided for the grant of stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards and other stock awards. Restricted stock unit awards have been granted under the 2010 Inducement Plan and are included in column (a). The weighted-average exercise price in column (b) includes the effect of the restricted stock unit awards under the 2010 Inducement Plan, which awards do not carry an exercise price. At December 31, 2014, the weighted-average exercise price of outstanding options under the 2010 Inducement Plan was $7.56, excluding the restricted stock unit awards. In June 2014, in connection with our adoption of the 2014 Plan described above, no further awards may be granted under the 2010 Inducement Plan. Although the 2010 Inducement Plan has terminated, all outstanding options under the 2010 Inducement Plan will continue to be governed by their existing terms. |
Security Ownership of Certain Beneficial Owners and Management
The information required by this item relating to security ownership of certain beneficial owners and management is included in our Proxy Statement under the section entitled “Security Ownership of Certain Beneficial Owners and Management” and is incorporated herein by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
The information required by this item is included in our Proxy Statement under the sections entitled “Transactions with Related Persons” and “Proposal 1 — Election of Directors” and is incorporated herein by reference.
Item 14. Principal Accountant Fees and Services.
The information required by this item is incorporated herein by reference to the information included in our Proxy Statement under the section entitled “Proposal 2 — Ratification of Selection of Independent Registered Public Accounting Firm.”
95
Table of Contents
Item 15. Exhibits, Financial Statement Schedules.
1. Index to Financial Statements
The following Financial Statements are included herein:
2. Index to Financial Statement Schedules
None.
All schedules are omitted because they are not applicable, or not required, or because the required information is included in the financial statements or notes thereto.
96
Table of Contents
3. Exhibits — The following exhibits are included herein or incorporated herein by reference:
| Exhibit Number |
Description of Document | |
| 3.1 | Amended and Restated Certificate of Incorporation(1) | |
| 3.2 | Certificate of Amendment of Amended and Restated Certificate of Incorporation(2) | |
| 3.3 | Amended and Restated Bylaws(3) | |
| 3.4 | Certificate of Designation of Series A Junior Participating Preferred Stock(4) | |
| 4.1 | Specimen Common Stock Certificate(5) | |
| 4.2 | Form of Right Certificate(6) | |
| 4.3 | Rights Agreement, dated as of December 15, 2005, by and between the Company and Mellon Investor Services LLC(7) | |
| 4.4 | Amendment No. 1 to Rights Agreement, dated as of January 29, 2015, by and between the Company and Computershare Inc., successor rights agent to Computershare Shareowner Services LLC (f/k/a Mellon Investor Services LLC(8) | |
| 4.5 | Indenture, dated as of February 3, 2015, between XenoPort, Inc. and U.S. Bank National Association, including the form of 2.50% Convertible Senior Note due 2022(9) | |
| 10.1* | Form of Indemnification Agreement between the Company and its officers and directors(10) | |
| 10.2* | Form of Employee Proprietary Information Agreement between the Company and its executive officers(11) | |
| 10.3 | Lease Agreement, dated September 24, 2001, by and between the Company and Sobrato Interests(11) | |
| 10.3.1 | First Amendment to Lease Agreement, dated February 29, 2008, by and between the Company and Sobrato Interests(12) | |
| 10.3.2 | Second Amendment to Lease Agreement, dated October 24, 2012, by and between the Company and SI 34 LLC(13) | |
| 10.3.3 | Third Amendment to Lease Agreement, dated November 24, 2014, by and between the Company and SI 34 LLC | |
| 10.4 | Lease, dated February 29, 2008, by and between the Company and Sobrato Interests(14) | |
| 10.4.1 | Lease Amendment and Termination Agreement, dated February 12, 2013, by and between the Company and SI 34, LLC(15) | |
| 10.5* | 1999 Stock Plan(11) | |
| 10.6* | Form of Stock Option Agreement under the 1999 Stock Plan(11) | |
| 10.7* | 2005 Equity Incentive Plan(10) | |
| 10.8* | Form of Option Agreement under the 2005 Equity Incentive Plan(10) | |
| 10.9* | Form of Stock Unit Award Agreement under the 2005 Equity Incentive Plan(16) | |
| 10.10* | Form of Non-Employee Director Stock Unit Award Agreement under the 2005 Equity Incentive Plan (17) | |
| 10.11* | 2005 Non-Employee Directors’ Stock Option Plan, as amended May 1, 2012(18) | |
| 10.12* | Form of Stock Option Agreement under the 2005 Non-Employee Directors’ Stock Option Plan(19) | |
97
Table of Contents
| Exhibit Number |
Description of Document | |
| 10.13* | 2005 Employee Stock Purchase Plan(20) | |
| 10.14* | Form of 2005 Employee Stock Purchase Plan Offering Document(21) | |
| 10.15* | 2010 Inducement Award Plan, as amended effective August 1, 2011(22) | |
| 10.16* | Form of Option Agreement under the 2010 Inducement Award Plan(23) | |
| 10.17* | Form of Stock Unit Award Agreement under the 2010 Inducement Award Plan(24) | |
| 10.18* | 2014 Equity Incentive Plan(25) | |
| 10.19* | Form of Stock Option Grant Notice and Option Agreement for Employees under the 2014 Equity Incentive Plan(26) | |
| 10.20* | Form of Restricted Stock Unit Grant Notice and Restricted Stock Unit Award Agreement for Employees under the 2014 Equity Incentive Plan(27) | |
| 10.21* | Forms of Stock Option Grant Notice and Option Agreement for Non-Employee Directors under the 2014 Equity Incentive Plan(28) | |
| 10.22* | Form of Restricted Stock Unit Grant Notice and Restricted Stock Unit Award Agreement for Non-Employee Directors under the 2014 Equity Incentive Plan(29) | |
| 10.23* | New Hire Option Agreement between Vincent J. Angotti and the Company(30) | |
| 10.24* | Performance Stock Unit Agreement, dated May 13, 2010, between the Company and Ronald W. Barrett, Ph.D.(31) | |
| 10.25* | Severance Rights Agreement, dated February 9, 2012, between the Company and Ronald W. Barrett, Ph.D.(32) | |
| 10.26* | Form of Severance Rights Agreement, between the Company and each of Vincent J. Angotti, Gregory T. Bates, D.V.M., Gianna M. Bosko, William G. Harris and Richard Kim, M.D.(33) | |
| 10.27* | Form of Change of Control Agreement between the Company and certain of its officers(34) | |
| 10.28* | XenoPort, Inc. Corporate Bonus Plan, amended and restated effective January 1, 2015(35) | |
| 10.29* | Term Sheet for Non-Employee Director Compensation(36) | |
| 10.30* | 2014 Executive Compensation Information(37) | |
| 10.31* | 2015 Executive Compensation Information | |
| 10.32† | Amended and Restated Distribution and License Agreement, dated as of October 31, 2009, between the Company and Astellas Pharma Inc.(38) | |
| 10.33† | Termination and Transition Agreement, dated November 8, 2012, by and between the Company and Glaxo Group Limited (39) | |
| 10.34 | Stock Purchase Agreement, dated November 8, 2012, by and between the Company and Glaxo Group Limited (40) | |
| 10.36† | Master Manufacturing and Supply Agreement, dated as of September 25, 2013, by and between the Company and Patheon Pharmaceuticals Inc. (41) | |
| 10.37† | License Agreement, dated as of May 14, 2014, between XenoPort, Inc. and Reckitt Benckiser Pharmaceuticals, Inc.(42) | |
| 12.1 | Statement of Computation of Ratio of Earnings to Fixed Charges and Ratio of Combined Fixed Charges and Preferred Stock Dividends | |
98
Table of Contents
| Exhibit Number |
Description of Document | |
| 23.1 | Consent of Independent Registered Public Accounting Firm | |
| 24.1 | Power of Attorney (included in the signature page hereto) | |
| 31.1 | Certification of the Chief Executive Officer pursuant to Securities Exchange Act Rules 13a-14(a) and 15d-14(a) as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
| 31.2 | Certification of the Chief Financial Officer pursuant to Securities Exchange Act Rules 13a-14(a) and 15d-14(a) as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
| 32.1 | Certification required by Rule 13a-14(b) or Rule 15d-14(b) and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. §1350)(43) | |
| 101.INS | XBRL Instance Document | |
| 101.SCH | XBRL Taxonomy Extension Schema Document | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | |
| 101.LAB | XBRL Taxonomy Extension Labels Linkbase Document | |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document | |
| * | Represents a management contract or compensation plan or arrangement. |
| † | Confidential treatment has been granted for portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission pursuant to Rule 24b-2 of the Securities Exchange Act of 1934, as amended. |
| (1) | Incorporated herein by reference to the same numbered exhibit of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2005, as filed with the SEC on August 11, 2005. |
| (2) | Incorporated herein by reference to Exhibit 3.4 of our current report on Form 8-K (File No. 000-51329), as filed with the SEC on May 18, 2012. |
| (3) | Incorporated herein by reference to Exhibit 3.2 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2005, as filed with the SEC on August 11, 2005. |
| (4) | Incorporated herein by reference to Exhibit 3.1 of our current report on Form 8-K (File No. 000-51329), as filed with the SEC on December 16, 2005. |
| (5) | Incorporated herein by reference to the same numbered exhibit of our registration statement on Form S-1, as amended (File No. 333-122156), as filed with the SEC on April 13, 2005. |
| (6) | Incorporated herein by reference to Exhibit 4.1 of our current report on Form 8-K (File No. 000-51329), as filed with the SEC on December 16, 2005. |
| (7) | Incorporated herein by reference to Exhibit 4.2 of our current report on Form 8-K (File No. 000-51329), filed with the SEC on December 16, 2015. |
| (8) | Incorporated herein by reference to Exhibit 4.2 of our current report on Form 8-K (File No. 000-51329), filed with the SEC on February 3, 2015. |
| (9) | Incorporated herein by reference to Exhibit 4.1 of our current report on Form 8-K (File No. 000-51329), as filed with the SEC on February 3, 2015. |
| (10) | Incorporated herein by reference to the same numbered exhibit of our registration statement on Form S-1, as amended (File No. 333-122156), as filed with the SEC on March 2, 2005. |
| (11) | Incorporated herein by reference to the same numbered exhibit of our registration statement on Form S-1 (File No. 333-122156), as filed with the SEC on January 19, 2005. |
| (12) | Incorporated herein by reference to the same numbered exhibit of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended March 31, 2008, as filed with the SEC on May 8, 2008. |
99
Table of Contents
| (13) | Incorporated herein by reference to the same numbered exhibit of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended September 30, 2012, as filed with the SEC on October 25, 2012. |
| (14) | Incorporated herein by reference to Exhibit 10.32 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended March 31, 2008, as filed with the SEC on May 8, 2008. |
| (15) | Incorporated herein by reference to the same numbered exhibit of our annual report on Form 10-K (File No. 000-51329) for the year ended December 31, 2012, as filed with the SEC on March 14, 2013. |
| (16) | Incorporated herein by reference to the same numbered exhibit of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2008, as filed with the SEC on August 7, 2008. |
| (17) | Incorporated herein by reference Exhibit 10.35 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended March 31, 2012, as filed with the SEC on May 8, 2012. |
| (18) | Incorporated herein by reference Exhibit 10.33 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended March 31, 2012, as filed with the SEC on May 8, 2012. |
| (19) | Incorporated herein by reference Exhibit 10.34 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended March 31, 2012, as filed with the SEC on May 8, 2012. |
| (20) | Incorporated herein by reference to Exhibit 10.11 of our registration statement on Form S-1, as amended (File No. 333-122156), as filed with the SEC on March 2, 2005. |
| (21) | Incorporated herein by reference to Exhibit 10.12 of our registration statement on Form S-1, as amended (File No. 333-122156), as filed with the SEC on March 2, 2005. |
| (22) | Incorporated herein by reference to Exhibit 10.17 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2011, as filed with the SEC on August 5, 2011. |
| (23) | Incorporated herein by reference to Exhibit 99.3.2 of our registration statement on Form S-8 (File No. 333-166760), as filed with the SEC on May 12, 2010. |
| (24) | Incorporated herein by reference to Exhibit 99.3.3 of our registration statement on Form S-8 (File No. 333-166760), as filed with the SEC on May 12, 2010. |
| (25) | Incorporated herein by reference to Exhibit 10.1 of our current report on Form 8-K (File No. 000-51329), as filed with the SEC on June 16, 2014. |
| (26) | Incorporated herein by reference to Exhibit 10.2 of our current report on Form 8-K (File No. 000-51329), as filed with the SEC on June 16, 2014. |
| (27) | Incorporated herein by reference to Exhibit 10.3 of our current report on Form 8-K (File No. 000-51329), as filed with the SEC on June 16, 2014. |
| (28) | Incorporated herein by reference to Exhibit 10.29 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2014, as filed with the SEC on August 7, 2014. |
| (29) | Incorporated herein by reference to Exhibit 10.30 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2014, as filed with the SEC on August 7, 2014. |
| (30) | Incorporated herein by reference to Exhibit 99.4 of our registration statement on Form S-8 (File No. 333-150730), as filed with the SEC on May 8, 2008. |
| (31) | Incorporated herein by reference to Exhibit 10.41 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2010, as filed with the SEC on August 6, 2010. |
| (32) | Incorporated herein by reference to Exhibit 10.39 of our current report on Form 8-K (File No. 000-51329), filed with the SEC on February 10, 2012. |
| (33) | Incorporated herein by reference to Exhibit 10.40 of our current report on Form 8-K (File No. 000-51329), filed with the SEC on February 10, 2012. |
| (34) | Incorporated herein by reference to Exhibit 10.22 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2014, as filed with the SEC on August 7, 2014. |
| (35) | Incorporated herein by reference to Exhibit 10.28 of our current report on Form 8-K (File No. 000-51329) as filed with the SEC on January 16, 2015. |
| (36) | Incorporated herein by reference to Exhibit 10.32 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2014, as filed with the SEC on August 7, 2014. |
100
Table of Contents
| (37) | Incorporated herein by reference to Exhibit 10.26 of our annual report on Form 10-K (File No. 000-51329) for the year ended December 31, 2013, as filed with the SEC on February 28, 2014. |
| (38) | Incorporated herein by reference to Exhibit 10.35 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended September 30, 2009, as filed with the SEC on November 4, 2009. |
| (39) | Incorporated herein by reference to Exhibit 10.31 of our annual report on Form 10-K (File No. 000-51329) for the year ended December 31, 2012, as filed with the SEC on March 14, 2013. |
| (40) | Incorporated herein by reference to Exhibit 10.32 of our annual report on Form 10-K (File No. 000-51329) for the year ended December 31, 2012, as filed with the SEC on March 14, 2013. |
| (41) | Incorporated herein by reference to Exhibit 10.35 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended September 30, 2013, as filed with the SEC on November 8, 2013 |
| (42) | Incorporated herein by reference to Exhibit 12.1 of our current report on Form 8-K (File No. 000-51329) as filed with the SEC on June 24, 2014. |
| (43) | This certification accompanies the annual report on Form 10-K to which it relates, is not deemed filed with the Securities and Exchange Commission and is not to be incorporated by reference into any filing of the Registrant under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended (whether made before or after the date of the Form 10-K), irrespective of any general incorporation language contained in such filing. |
101
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| XenoPort, Inc. (Registrant) | ||||||
| February 27, 2015 | /s/ Ronald W. Barrett | |||||
| Ronald W. Barrett Chief Executive Officer and Director | ||||||
| February 27, 2015 | /s/ William G. Harris | |||||
| William G. Harris Senior Vice President of Finance and Chief Financial Officer (Principal Financial and Accounting Officer) | ||||||
102
Table of Contents
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Ronald W. Barrett and William G. Harris, and each of them, as his or her true and lawful attorneys-in-fact and agents, each with the full power of substitution for him or her, and in his or her name and in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that each of said attorneys-in-fact and agents, and any of them or his or her substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
| Signature |
Title |
Date | ||
| /s/ Ronald W. Barrett Ronald W. Barrett |
Chief Executive Officer and Director (Principal Executive Officer) |
February 27, 2015 | ||
| /s/ William G. Harris William G. Harris |
Senior Vice President of Finance and Chief Financial Officer (Principal Financial and Accounting Officer) |
February 27, 2015 | ||
| /s/ Paul L. Berns Paul L. Berns |
Director | February 27, 2015 | ||
| /s/ Dennis M. Fenton Dennis M. Fenton |
Director | February 27, 2015 | ||
| /s/ John G. Freund John G. Freund |
Director | February 27, 2015 | ||
| /s/ Catherine J. Friedman Catherine J. Friedman |
Director | February 27, 2015 | ||
| /s/ Jeryl L. Hilleman Jeryl L. Hilleman |
Director | February 27, 2015 | ||
| /s/ Ernest Mario Ernest Mario |
Director | February 27, 2015 | ||
| /s/ William J. Rieflin William J. Rieflin |
Director | February 27, 2015 | ||
| /s/ Wendell Wierenga Wendell Wierenga |
Director | February 27, 2015 | ||
103
Table of Contents
| Exhibit Number |
Description of Document | |
| 3.1 | Amended and Restated Certificate of Incorporation(1) | |
| 3.2 | Certificate of Amendment of Amended and Restated Certificate of Incorporation(2) | |
| 3.3 | Amended and Restated Bylaws(3) | |
| 3.4 | Certificate of Designation of Series A Junior Participating Preferred Stock(4) | |
| 4.1 | Specimen Common Stock Certificate(5) | |
| 4.2 | Form of Right Certificate(6) | |
| 4.3 | Rights Agreement, dated as of December 15, 2005, by and between the Company and Mellon Investor Services LLC(7) | |
| 4.4 | Amendment No. 1 to Rights Agreement, dated as of January 29, 2015, by and between the Company and Computershare Inc., successor rights agent to Computershare Shareowner Services LLC (f/k/a Mellon Investor Services LLC(8) | |
| 4.5 | Indenture, dated as of February 3, 2015, between XenoPort, Inc. and U.S. Bank National Association, including the form of 2.50% Convertible Senior Note due 2022(9) | |
| 10.1* | Form of Indemnification Agreement between the Company and its officers and directors(10) | |
| 10.2* | Form of Employee Proprietary Information Agreement between the Company and its executive officers(11) | |
| 10.3 | Lease Agreement, dated September 24, 2001, by and between the Company and Sobrato Interests(11) | |
| 10.3.1 | First Amendment to Lease Agreement, dated February 29, 2008, by and between the Company and Sobrato Interests(12) | |
| 10.3.2 | Second Amendment to Lease Agreement, dated October 24, 2012, by and between the Company and SI 34 LLC(13) | |
| 10.3.3 | Third Amendment to Lease Agreement, dated November 24, 2014, by and between the Company and SI 34 LLC | |
| 10.4 | Lease, dated February 29, 2008, by and between the Company and Sobrato Interests(14) | |
| 10.4.1 | Lease Amendment and Termination Agreement, dated February 12, 2013, by and between the Company and SI 34, LLC(15) | |
| 10.5* | 1999 Stock Plan(11) | |
| 10.6* | Form of Stock Option Agreement under the 1999 Stock Plan(11) | |
| 10.7* | 2005 Equity Incentive Plan(10) | |
| 10.8* | Form of Option Agreement under the 2005 Equity Incentive Plan(10) | |
| 10.9* | Form of Stock Unit Award Agreement under the 2005 Equity Incentive Plan(16) | |
| 10.10* | Form of Non-Employee Director Stock Unit Award Agreement under the 2005 Equity Incentive Plan (17) | |
| 10.11* | 2005 Non-Employee Directors’ Stock Option Plan, as amended May 1, 2012(18) | |
| 10.12* | Form of Stock Option Agreement under the 2005 Non-Employee Directors’ Stock Option Plan(19) | |
104
Table of Contents
| Exhibit Number |
Description of Document | |
| 10.13* | 2005 Employee Stock Purchase Plan(20) | |
| 10.14* | Form of 2005 Employee Stock Purchase Plan Offering Document(21) | |
| 10.15* | 2010 Inducement Award Plan, as amended effective August 1, 2011(22) | |
| 10.16* | Form of Option Agreement under the 2010 Inducement Award Plan(23) | |
| 10.17* | Form of Stock Unit Award Agreement under the 2010 Inducement Award Plan(24) | |
| 10.18* | 2014 Equity Incentive Plan(25) | |
| 10.19* | Form of Stock Option Grant Notice and Option Agreement for Employees under the 2014 Equity Incentive Plan(26) | |
| 10.20* | Form of Restricted Stock Unit Grant Notice and Restricted Stock Unit Award Agreement for Employees under the 2014 Equity Incentive Plan(27) | |
| 10.21* | Forms of Stock Option Grant Notice and Option Agreement for Non-Employee Directors under the 2014 Equity Incentive Plan(28) | |
| 10.22* | Form of Restricted Stock Unit Grant Notice and Restricted Stock Unit Award Agreement for Non-Employee Directors under the 2014 Equity Incentive Plan(29) | |
| 10.23* | New Hire Option Agreement between Vincent J. Angotti and the Company(30) | |
| 10.24* | Performance Stock Unit Agreement, dated May 13, 2010, between the Company and Ronald W. Barrett, Ph.D.(31) | |
| 10.25* | Severance Rights Agreement, dated February 9, 2012, between the Company and Ronald W. Barrett, Ph.D.(32) | |
| 10.26* | Form of Severance Rights Agreement, between the Company and each of Vincent J. Angotti, Gregory T. Bates, D.V.M., Gianna M. Bosko, William G. Harris and Richard Kim, M.D.(33) | |
| 10.27* | Form of Change of Control Agreement between the Company and certain of its officers(34) | |
| 10.28* | XenoPort, Inc. Corporate Bonus Plan, amended and restated effective January 1, 2015(35) | |
| 10.29* | Term Sheet for Non-Employee Director Compensation(36) | |
| 10.30* | 2014 Executive Compensation Information(37) | |
| 10.31* | 2015 Executive Compensation Information | |
| 10.32† | Amended and Restated Distribution and License Agreement, dated as of October 31, 2009, between the Company and Astellas Pharma Inc.(38) | |
| 10.33† | Termination and Transition Agreement, dated November 8, 2012, by and between the Company and Glaxo Group Limited (39) | |
| 10.34 | Stock Purchase Agreement, dated November 8, 2012, by and between the Company and Glaxo Group Limited (40) | |
| 10.36† | Master Manufacturing and Supply Agreement, dated as of September 25, 2013, by and between the Company and Patheon Pharmaceuticals Inc. (41) | |
| 10.37† | License Agreement, dated as of May 14, 2014, between XenoPort, Inc. and Reckitt Benckiser Pharmaceuticals, Inc.(42) | |
| 12.1 | Statement of Computation of Ratio of Earnings to Fixed Charges and Ratio of Combined Fixed Charges and Preferred Stock Dividends | |
105
Table of Contents
| Exhibit Number |
Description of Document | |
| 23.1 | Consent of Independent Registered Public Accounting Firm | |
| 24.1 | Power of Attorney (included in the signature page hereto) | |
| 31.1 | Certification of the Chief Executive Officer pursuant to Securities Exchange Act Rules 13a-14(a) and 15d-14(a) as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
| 31.2 | Certification of the Chief Financial Officer pursuant to Securities Exchange Act Rules 13a-14(a) and 15d-14(a) as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
| 32.1 | Certification required by Rule 13a-14(b) or Rule 15d-14(b) and Section 1350 of Chapter 63 of Title 18 of the United States Code (18 U.S.C. §1350)(43) | |
| 101.INS | XBRL Instance Document | |
| 101.SCH | XBRL Taxonomy Extension Schema Document | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | |
| 101.LAB | XBRL Taxonomy Extension Labels Linkbase Document | |
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document | |
| * | Represents a management contract or compensation plan or arrangement. |
| † | Confidential treatment has been granted for portions of this exhibit. Omitted portions have been filed separately with the Securities and Exchange Commission pursuant to Rule 24b-2 of the Securities Exchange Act of 1934, as amended. |
| (1) | Incorporated herein by reference to the same numbered exhibit of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2005, as filed with the SEC on August 11, 2005. |
| (2) | Incorporated herein by reference to Exhibit 3.4 of our current report on Form 8-K (File No. 000-51329), as filed with the SEC on May 18, 2012. |
| (3) | Incorporated herein by reference to Exhibit 3.2 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2005, as filed with the SEC on August 11, 2005. |
| (4) | Incorporated herein by reference to Exhibit 3.1 of our current report on Form 8-K (File No. 000-51329), as filed with the SEC on December 16, 2005. |
| (5) | Incorporated herein by reference to the same numbered exhibit of our registration statement on Form S-1, as amended (File No. 333-122156), as filed with the SEC on April 13, 2005. |
| (6) | Incorporated herein by reference to Exhibit 4.1 of our current report on Form 8-K (File No. 000-51329), as filed with the SEC on December 16, 2005. |
| (7) | Incorporated herein by reference to Exhibit 4.2 of our current report on Form 8-K (File No. 000-51329), filed with the SEC on December 16, 2015. |
| (8) | Incorporated herein by reference to Exhibit 4.2 of our current report on Form 8-K (File No. 000-51329), filed with the SEC on February 3, 2015. |
| (9) | Incorporated herein by reference to Exhibit 4.1 of our current report on Form 8-K (File No. 000-51329), as filed with the SEC on February 3, 2015. |
| (10) | Incorporated herein by reference to the same numbered exhibit of our registration statement on Form S-1, as amended (File No. 333-122156), as filed with the SEC on March 2, 2005. |
| (11) | Incorporated herein by reference to the same numbered exhibit of our registration statement on Form S-1 (File No. 333-122156), as filed with the SEC on January 19, 2005. |
| (12) | Incorporated herein by reference to the same numbered exhibit of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended March 31, 2008, as filed with the SEC on May 8, 2008. |
106
Table of Contents
| (13) | Incorporated herein by reference to the same numbered exhibit of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended September 30, 2012, as filed with the SEC on October 25, 2012. |
| (14) | Incorporated herein by reference to Exhibit 10.32 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended March 31, 2008, as filed with the SEC on May 8, 2008. |
| (15) | Incorporated herein by reference to the same numbered exhibit of our annual report on Form 10-K (File No. 000-51329) for the year ended December 31, 2012, as filed with the SEC on March 14, 2013. |
| (16) | Incorporated herein by reference to the same numbered exhibit of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2008, as filed with the SEC on August 7, 2008. |
| (17) | Incorporated herein by reference Exhibit 10.35 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended March 31, 2012, as filed with the SEC on May 8, 2012. |
| (18) | Incorporated herein by reference Exhibit 10.33 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended March 31, 2012, as filed with the SEC on May 8, 2012. |
| (19) | Incorporated herein by reference Exhibit 10.34 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended March 31, 2012, as filed with the SEC on May 8, 2012. |
| (20) | Incorporated herein by reference to Exhibit 10.11 of our registration statement on Form S-1, as amended (File No. 333-122156), as filed with the SEC on March 2, 2005. |
| (21) | Incorporated herein by reference to Exhibit 10.12 of our registration statement on Form S-1, as amended (File No. 333-122156), as filed with the SEC on March 2, 2005. |
| (22) | Incorporated herein by reference to Exhibit 10.17 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2011, as filed with the SEC on August 5, 2011. |
| (23) | Incorporated herein by reference to Exhibit 99.3.2 of our registration statement on Form S-8 (File No. 333-166760), as filed with the SEC on May 12, 2010. |
| (24) | Incorporated herein by reference to Exhibit 99.3.3 of our registration statement on Form S-8 (File No. 333-166760), as filed with the SEC on May 12, 2010. |
| (25) | Incorporated herein by reference to Exhibit 10.1 of our current report on Form 8-K (File No. 000-51329), as filed with the SEC on June 16, 2014. |
| (26) | Incorporated herein by reference to Exhibit 10.2 of our current report on Form 8-K (File No. 000-51329), as filed with the SEC on June 16, 2014. |
| (27) | Incorporated herein by reference to Exhibit 10.3 of our current report on Form 8-K (File No. 000-51329), as filed with the SEC on June 16, 2014. |
| (28) | Incorporated herein by reference to Exhibit 10.29 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2014, as filed with the SEC on August 7, 2014. |
| (29) | Incorporated herein by reference to Exhibit 10.30 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2014, as filed with the SEC on August 7, 2014. |
| (30) | Incorporated herein by reference to Exhibit 99.4 of our registration statement on Form S-8 (File No. 333-150730), as filed with the SEC on May 8, 2008. |
| (31) | Incorporated herein by reference to Exhibit 10.41 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2010, as filed with the SEC on August 6, 2010. |
| (32) | Incorporated herein by reference to Exhibit 10.39 of our current report on Form 8-K (File No. 000-51329), filed with the SEC on February 10, 2012. |
| (33) | Incorporated herein by reference to Exhibit 10.40 of our current report on Form 8-K (File No. 000-51329), filed with the SEC on February 10, 2012. |
| (34) | Incorporated herein by reference to Exhibit 10.22 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2014, as filed with the SEC on August 7, 2014. |
| (35) | Incorporated herein by reference to Exhibit 10.28 of our current report on Form 8-K (File No. 000-51329) as filed with the SEC on January 16, 2015. |
| (36) | Incorporated herein by reference to Exhibit 10.32 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended June 30, 2014, as filed with the SEC on August 7, 2014. |
107
Table of Contents
| (37) | Incorporated herein by reference to Exhibit 10.26 of our annual report on Form 10-K (File No. 000-51329) for the year ended December 31, 2013, as filed with the SEC on February 28, 2014. |
| (38) | Incorporated herein by reference to Exhibit 10.35 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended September 30, 2009, as filed with the SEC on November 4, 2009. |
| (39) | Incorporated herein by reference to Exhibit 10.31 of our annual report on Form 10-K (File No. 000-51329) for the year ended December 31, 2012, as filed with the SEC on March 14, 2013. |
| (40) | Incorporated herein by reference to Exhibit 10.32 of our annual report on Form 10-K (File No. 000-51329) for the year ended December 31, 2012, as filed with the SEC on March 14, 2013. |
| (41) | Incorporated herein by reference to Exhibit 10.35 of our quarterly report on Form 10-Q (File No. 000-51329) for the period ended September 30, 2013, as filed with the SEC on November 8, 2013 |
| (42) | Incorporated herein by reference to Exhibit 12.1 of our current report on Form 8-K (File No. 000-51329) as filed with the SEC on June 24, 2014. |
| (43) | This certification accompanies the annual report on Form 10-K to which it relates, is not deemed filed with the Securities and Exchange Commission and is not to be incorporated by reference into any filing of the Registrant under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended (whether made before or after the date of the Form 10-K), irrespective of any general incorporation language contained in such filing. |
108
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of XenoPort, Inc.
We have audited the accompanying balance sheets of XenoPort, Inc. as of December 31, 2014 and 2013, and the related statements of comprehensive loss, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2014. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of XenoPort, Inc. at December 31, 2014 and 2013, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2014, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), XenoPort, Inc.’s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated February 27, 2015, expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Redwood City, California
February 27, 2015
109
Table of Contents
XENOPORT, INC.
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| (In thousands, except per share amounts) |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 11,958 | $ | 20,584 | ||||
| Short-term investments |
90,098 | 38,074 | ||||||
| Accounts receivable |
2,895 | 939 | ||||||
| Inventories |
1,458 | 1,262 | ||||||
| Prepaids and other current assets |
3,185 | 2,826 | ||||||
|
|
|
|
|
|||||
| Total current assets |
109,594 | 63,685 | ||||||
| Property and equipment, net |
2,422 | 2,552 | ||||||
| Long-term inventories |
9,098 | 10,185 | ||||||
| Restricted investments and other assets |
1,947 | 2,119 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 123,061 | $ | 78,541 | ||||
|
|
|
|
|
|||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 2,835 | $ | 1,432 | ||||
| Accrued compensation |
7,148 | 2,741 | ||||||
| Accrued preclinical and clinical costs |
1,554 | 824 | ||||||
| Other accrued liabilities |
5,117 | 3,938 | ||||||
| Deferred revenue |
1,134 | 1,134 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
17,788 | 10,069 | ||||||
| Deferred revenue |
10,864 | 11,997 | ||||||
| Other noncurrent liability |
3,269 | 2,782 | ||||||
| Commitments and contingencies |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock, $0.001 par value; 5,000 shares authorized; no shares issued or outstanding |
— | — | ||||||
| Common stock, $0.001 par value; 100,000 shares authorized; 62,475 and 47,800 shares issued and outstanding, at December 31, 2014 and 2013, respectively |
62 | 48 | ||||||
| Additional paid-in capital |
677,924 | 591,128 | ||||||
| Accumulated other comprehensive loss |
(30 | ) | — | |||||
| Accumulated deficit |
(586,816 | ) | (537,483 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
91,140 | 53,693 | ||||||
|
|
|
|
|
|||||
| Total liabilities and stockholders’ equity |
$ | 123,061 | $ | 78,541 | ||||
|
|
|
|
|
|||||
The accompanying notes are an integral part of these financial statements.
110
Table of Contents
XENOPORT, INC.
STATEMENTS OF COMPREHENSIVE LOSS
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| (In thousands, except per share amounts) |
||||||||||||
| Revenues: |
||||||||||||
| Product sales, net |
$ | 20,173 | $ | 6,414 | $ | — | ||||||
| Collaboration revenue |
26,134 | 1,137 | 11,515 | |||||||||
| Royalty revenue |
561 | 400 | 109 | |||||||||
| Net revenue from unconsolidated joint operating activities |
— | — | 10,000 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total revenues |
46,868 | 7,951 | 21,624 | |||||||||
|
|
|
|
|
|
|
|||||||
| Operating expenses: |
||||||||||||
| Cost of product sales |
2,094 | 1,170 | — | |||||||||
| Research and development |
23,679 | 33,325 | 42,947 | |||||||||
| Selling, general and administrative |
70,194 | 59,084 | 30,244 | |||||||||
| Gain on litigation settlement |
— | — | (20,499 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
95,967 | 93,579 | 52,692 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(49,099 | ) | (85,628 | ) | (31,068 | ) | ||||||
| Interest income |
253 | 213 | 254 | |||||||||
| Interest expense |
(487 | ) | (468 | ) | — | |||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
(49,333 | ) | (85,883 | ) | (30,814 | ) | ||||||
| Other comprehensive loss: |
||||||||||||
| Unrealized gains (losses) on available-for-sale securities |
(30 | ) | (22 | ) | 38 | |||||||
|
|
|
|
|
|
|
|||||||
| Comprehensive loss |
$ | (49,363 | ) | $ | (85,905 | ) | $ | (30,776 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per share |
$ | (0.81 | ) | $ | (1.81 | ) | $ | (0.78 | ) | |||
|
|
|
|
|
|
|
|||||||
| Shares used to compute basic and diluted net loss per share |
60,856 | 47,545 | 39,434 | |||||||||
|
|
|
|
|
|
|
|||||||
The accompanying notes are an integral part of these financial statements.
111
Table of Contents
XENOPORT, INC.
STATEMENTS OF STOCKHOLDERS’ EQUITY
| Common Stock | Additional Paid in Capital |
Accumulated Other Comprehensive Income (Loss) |
Accumulated Deficit |
Total Stockholders’ Equity |
||||||||||||||||||||
| Shares | Amount | |||||||||||||||||||||||
| (In thousands, except share amounts) | ||||||||||||||||||||||||
| Balance at December 31, 2011 |
35,514,636 | $ | 35 | $ | 495,902 | $ | (16 | ) | $ | (420,786 | ) | $ | 75,135 | |||||||||||
| Issuance of common stock upon exercise of options and vesting of restricted stock units |
259,650 | 1 | (481 | ) | — | — | (480 | ) | ||||||||||||||||
| Issuance of common stock in connection with Employee Stock Purchase Plan |
185,249 | — | 763 | — | — | 763 | ||||||||||||||||||
| Employee stock-based compensation expense |
— | — | 12,281 | — | — | 12,281 | ||||||||||||||||||
| Issuance of common stock upon public offering, net of offering costs |
7,076,922 | 7 | 43,012 | — | — | 43,019 | ||||||||||||||||||
| Issuance of common stock to Glaxo Group Limited, or GSK |
4,031,212 | 4 | 30,264 | — | — | 30,268 | ||||||||||||||||||
| Change in unrealized gains (losses) on investments |
— | — | — | 38 | — | 38 | ||||||||||||||||||
| Net loss |
— | — | — | — | (30,814 | ) | (30,814 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2012 |
47,067,669 | 47 | 581,741 | 22 | (451,600 | ) | 130,210 | |||||||||||||||||
| Issuance of common stock upon exercise of options and vesting of restricted stock units |
590,242 | 1 | (1,876 | ) | — | — | (1,875 | ) | ||||||||||||||||
| Issuance of common stock in connection with Employee Stock Purchase Plan |
142,455 | — | 719 | — | — | 719 | ||||||||||||||||||
| Employee stock-based compensation expense |
— | — | 10,544 | — | — | 10,544 | ||||||||||||||||||
| Change in unrealized gains (losses) on investments |
— | — | — | (22 | ) | — | (22 | ) | ||||||||||||||||
| Net loss |
— | — | — | — | (85,883 | ) | (85,883 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2013 |
47,800,366 | 48 | 591,128 | — | (537,483 | ) | 53,693 | |||||||||||||||||
| Issuance of common stock upon exercise of options and vesting of restricted stock units |
646,806 | — | (395 | ) | — | — | (395 | ) | ||||||||||||||||
| Issuance of common stock in connection with Employee Stock Purchase Plan |
227,720 | — | 714 | — | — | 714 | ||||||||||||||||||
| Employee stock-based compensation expense |
— | — | 9,041 | — | — | 9,041 | ||||||||||||||||||
| Issuance of common stock upon public offering, net of offering costs |
13,800,000 | 14 | 77,436 | — | — | 77,450 | ||||||||||||||||||
| Change in unrealized gains (losses) on investments |
— | — | — | (30 | ) | — | (30 | ) | ||||||||||||||||
| Net loss |
— | — | — | — | (49,333 | ) | (49,333 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Balance at December 31, 2014 |
62,474,892 | $ | 62 | $ | 677,924 | $ | (30 | ) | $ | (586,816 | ) | $ | 91,140 | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
The accompanying notes are an integral part of these financial statements.
112
Table of Contents
XENOPORT, INC.
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| (In thousands) | ||||||||||||
| Operating activities |
||||||||||||
| Net loss |
$ | (49,333 | ) | $ | (85,883 | ) | $ | (30,814 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation and amortization |
623 | 1,199 | 2,516 | |||||||||
| Accretion of investment discounts and amortization of investment premiums, net |
1,151 | 1,007 | 1,295 | |||||||||
| Stock-based compensation expense |
9,041 | 10,544 | 12,281 | |||||||||
| Gain on litigation settlement, net of stock premium in connection with the stock purchase agreement with GSK |
— | — | (11,243 | ) | ||||||||
| Changes in assets and liabilities: |
||||||||||||
| Accounts receivable |
(1,956 | ) | (939 | ) | — | |||||||
| Prepaids and other current and noncurrent assets |
(186 | ) | 16 | 712 | ||||||||
| Inventories |
891 | 286 | — | |||||||||
| Accounts payable |
1,403 | 865 | (465 | ) | ||||||||
| Accrued compensation |
4,407 | (2,134 | ) | 699 | ||||||||
| Accrued restructuring charges |
— | (993 | ) | (1,737 | ) | |||||||
| Accrued preclinical and clinical costs |
730 | (3,573 | ) | (36 | ) | |||||||
| Other accrued liabilities, current and noncurrent |
1,666 | 2,839 | 677 | |||||||||
| Deferred revenue, current and noncurrent |
(1,133 | ) | (1,137 | ) | (1,515 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(32,696 | ) | (77,903 | ) | (27,630 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Investing activities |
||||||||||||
| Purchases of investments |
(155,885 | ) | (91,000 | ) | (147,645 | ) | ||||||
| Proceeds from maturities of investments |
102,679 | 154,765 | 112,576 | |||||||||
| Purchases of property and equipment |
(493 | ) | (256 | ) | (123 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) investing activities |
(53,699 | ) | 63,509 | (35,192 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Financing activities |
||||||||||||
| Net cash proceeds provided by (used in) issuance of common stock and exercise of stock options |
77,769 | (1,156 | ) | 43,302 | ||||||||
| Proceeds from issuance of common stock to GSK in connection with the stock purchase agreement |
— | — | 30,268 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by (used in) financing activities |
77,769 | (1,156 | ) | 73,570 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net increase (decrease) in cash and cash equivalents |
(8,626 | ) | (15,550 | ) | 10,748 | |||||||
| Cash and cash equivalents at beginning of period |
20,584 | 36,134 | 25,386 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents at end of period |
$ | 11,958 | $ | 20,584 | $ | 36,134 | ||||||
|
|
|
|
|
|
|
|||||||
| Non-cash investing activity |
||||||||||||
| Right to the HORIZANT business |
$ | — | $ | — | $ | 13,557 | ||||||
| Inventories |
$ | — | $ | 11,733 | $ | — | ||||||
| Manufacturing property and equipment |
$ | — | $ | 1,967 | $ | — | ||||||
The accompanying notes are an integral part of these financial statements.
113
Table of Contents
XENOPORT, INC.
1. Organization and Summary of Significant Accounting Policies
Nature of Operations
XenoPort, Inc., or the Company, was incorporated in the state of Delaware on May 19, 1999. The Company is a biopharmaceutical company focused on developing and commercializing a portfolio of internally discovered product candidates for the potential treatment of neurological and other disorders. The Company is currently commercializing HORIZANT® (gabapentin enacarbil) Extended-Release Tablets in the United States and developing its novel fumaric acid ester product candidate, XP23829, as a potential treatment for psoriasis and potentially for relapsing forms of multiple sclerosis, or MS. The Company’s other product candidates are: XP21279 and arbaclofen placarbil, or AP, which is licensed to Indivior PLC (formerly Reckitt Benckiser Pharmaceuticals, Inc.) Effective June 2014, the Company granted to Indivior exclusive, world-wide rights to develop and commercialize pharmaceutical products containing AP and other prodrugs of baclofen or R-baclofen, or collectively, the AP Products, for all indications, subject to the Company’s right of first negotiation with Indivior to collaborate to develop and commercialize AP Products for non-addiction indications (See Note 2 for more information). XP21279 is a potential treatment for patients with advanced idiopathic Parkinson’s disease. The Company may develop it further, to the extent that its resources permit, or it may enter into collaboration, partnership, licensing and other similar forms of revenue-generating transactions with third-party business partners for such development. HORIZANT and the Company’s product candidates are prodrugs that are typically created by modifying the chemical structure of currently marketed drugs, referred to as parent drugs, and are designed to correct limitations in the oral absorption, distribution and/or metabolism of the parent drug. HORIZANT and each of the Company’s product candidates are orally-available, patented molecules that address potential markets with clear unmet medical needs. The Company’s facilities are located in Santa Clara, California.
On November 8, 2012, the Company reached an agreement with Glaxo Group Limited, or GSK, to terminate the Amended and Restated Development and Commercialization Agreement dated November 7, 2010, between the Company and GSK regarding HORIZANT and resolved all ongoing litigation between the parties. Under the terms of the November 2012 termination and transition agreement, during a transition period that ended on April 30, 2013, GSK continued to exclusively commercialize, promote, manufacture and distribute HORIZANT in the United States. The Company was not eligible to receive any milestone payments from GSK and did not receive any revenue or incur any losses from GSK’s sales of HORIZANT during the transition period. GSK continued to fully fund the costs associated with the management and conduct of required post-marketing clinical studies initiated by GSK prior to the date of the termination and transition agreement. In addition, GSK provided to the Company inventory of gabapentin enacarbil in GSK’s possession that was not required for use by GSK in the manufacture of HORIZANT and related manufacturing property and equipment. In exchange for such inventory, the Company will make annual payments to GSK of $1,000,000 for six years beginning in 2016, which was recorded at its present value as “Other noncurrent liability” on the Company’s balance sheet for the periods ended December 31, 2014 and 2013 and is being accreted at an effective interest rate of 17.5% to its contractual value over the payment period. On May 1, 2013, the Company completed the acquisition of the HORIZANT business in accordance with the termination and transition agreement and assumed all responsibilities and associated rights for the further development, manufacturing and commercialization of HORIZANT in the United States on that date. GSK was responsible for the commercial manufacture and supply of HORIZANT during the transition period. The Company did not assume any liabilities of GSK’s HORIZANT business as of the acquisition date.
Basis of Preparation
The Company’s financial statements are prepared in accordance with the Financial Accounting Standards Board, or FASB, Accounting Standards Codification, or the Codification, which is the single source of authoritative U.S. generally accepted accounting principles, or GAAP.
114
Table of Contents
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from these estimates.
Fair Value of Financial Instruments
The carrying amounts of certain of the Company’s financial instruments, including cash and cash equivalents and short-term investments, restricted investments, accounts receivables and accounts payables, approximate their fair value due to their short-term maturities. The Company accounts for the fair value of its financial instruments in accordance with the provisions of the Fair Value Measurement topic of the Codification.
Fair value is the price that would be received to sell an asset or paid to transfer a liability (an exit price) in an orderly transaction between market participants at the measurement date. The Company applies the market approach valuation technique for fair value measurements on a recurring basis and attempts to maximize the use of observable inputs and minimize the use of unobservable inputs. The fair value hierarchy prioritizes the inputs to valuation techniques used to measure fair value into three broad levels. All of the Company’s cash equivalents and short-term investments are measured using inputs classified at Level 1 or Level 2 within the fair value hierarchy. Level 1 inputs are quoted prices in active markets for identical assets. Level 2 inputs are based upon quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active and model-based valuation techniques for which all significant inputs are observable in the market or can be corroborated by observable market data for substantially the full term of the assets. Level 3 inputs are unobservable inputs that are supported by little or no market activity and are significant to the fair value of the assets or liabilities. Where applicable, these models project future cash flows and discount the future amounts to a present value using market-based observable inputs obtained from various third-party data providers, including but not limited to, benchmark yields, interest rate curves, reported trades, broker/dealer quotes and market reference data.
Cash Equivalents and Short-Term Investments
The Company considers all highly liquid investments with original maturities of 90 days or less at the time of purchase to be cash equivalents, which primarily consist of money market funds, U.S. government-sponsored agencies and corporate debt securities.
Management determines the appropriate classification of securities at the time of purchase. All investments have been designated as available-for-sale. The Company views its available-for-sale portfolio as available for use in current operations. Accordingly, the Company has classified all investments as short-term, even though the stated maturity may be one year or more beyond the current balance sheet date. Available-for-sale securities are carried at estimated fair value with unrealized gains and losses reported as a component of other comprehensive loss in the statements of comprehensive loss.
The cost of securities is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization and accretion is included in interest income. Realized gains and losses and declines in value judged to be other-than-temporary on available-for-sale securities, if any, are recorded in interest income and expense. The cost of securities sold is based on the specific-identification method. Interest and dividends are included in interest income.
Restricted Investments
Under a facilities operating lease agreement, the Company is required to secure a letter of credit with cash or securities. At December 31, 2014 and 2013, the Company recorded $1,500,000 of restricted investments related to the letter of credit (see Note 7 for more information). The $1,500,000 was classified as restricted investments in the accompanying balance sheets at both December 31, 2014 and 2013.
115
Table of Contents
In connection with the Company’s license to use radioactive materials in its research facilities, it must maintain a $225,000 letter of credit with the Radiological Health Branch of the State of California. This requirement has been fulfilled through certificates of deposit with a financial institution. The fair value of the secured amount of $225,000 was classified as restricted investments in the accompanying balance sheets at both December 31, 2014 and 2013.
Segment Information
The Company operates in one operating segment, which is the development and commercialization of product candidates for the potential treatment of neurological and other disorders, and has operations solely in the United States. To date, all of the Company’s revenues from product sales are related to sales of HORIZANT in the United States. The Company also has recognized upfront and milestone payments and royalty revenue from its partnership with Astellas Pharma Inc. and recognized upfront and additional payments from its partnership with Indivior (see Note 2 for more information on these arrangements).
Concentrations of Risk
The Company invests cash that is not being used for operational purposes. This exposes the Company to credit risk in the event of default by the institutions holding the cash and cash equivalents and available-for-sale securities to the extent of the amounts on its balance sheets. The credit risk is mitigated by the Company’s investment policy, which allows for the purchase of low risk debt securities issued by the U.S. government, U.S. government-sponsored agencies and highly rated banks and corporations, subject to certain concentration limits. The maturities of these securities are maintained at no longer than 16 months. The Company believes its established guidelines for investment of its excess cash enhances safety and liquidity through its policies on diversification and investment maturity.
Financial instruments that potentially subject the Company to concentrations of credit risk consist principally of cash and cash equivalents and available-for-sale investment securities in high-credit quality debt securities issued by the U.S. government, U.S. government-sponsored enterprises and highly rated banks and corporations. The carrying amounts of cash equivalents and available-for-sale investment securities are stated at fair value.
The Company is subject to credit risk from its accounts receivable related to product sales. The Company’s trade accounts receivable arises from product sales in the United States. Three wholesale distributors represented 41%, 28% and 26%, respectively, of product sales for the year ended December 31, 2014. These three customers individually comprised 48%, 26% and 22%, respectively, of accounts receivable as of December 31, 2014. Three wholesale distributors represented 47%, 33% and 16%, respectively, of product sales for the year ended December 31, 2013. These three customers individually comprised 45%, 29% and 22%, respectively, of product sales related to accounts receivable as of December 31, 2013. To date, the Company has not experienced any losses with respect to the collection of its accounts receivable and believes that its accounts receivable are collectible.
The Company relies on a single third-party contract manufacturer organization to manufacture HORIZANT.
Property and Equipment
Property and equipment are stated at cost, less accumulated depreciation and amortization. Depreciation and amortization is computed using the straight-line method over the estimated useful lives of the respective assets, which is generally five to ten years for the Company’s laboratory equipment, furniture and fixtures and manufacturing property and equipment and generally three years for the Company’s computer equipment and software. Leasehold improvements are amortized over their estimated useful lives or the remaining lease term, whichever is shorter.
116
Table of Contents
Revenue Recognition
Product Sales
The Company began selling HORIZANT to wholesalers in May 2013 following the return of the Company’s commercial rights from GSK. The Company recognizes revenue from HORIZANT product sales when there is persuasive evidence that an arrangement exists, delivery to the customer has occurred, the price is fixed or determinable and collectability is reasonably assured. Revenue from product sales is recorded net of estimated allowances for customer incentives such as cash discounts for prompt payment, distributor fees, expected returns, as appropriate (which are based on an analysis of historical return rates and deductions of HORIZANT, since its commercial launch by GSK in the second quarter of 2011), government rebates such as Medicaid reimbursements and patient assistance programs. Calculating certain of these items involves estimates and judgments based on contractual terms, historical utilization rates data for HORIZANT and new information regarding changes in these programs’ regulations and guidelines that would impact the amount of the actual rebates, the Company’s expectations regarding future utilization rates for these programs and channel inventory data. If future actual results vary from the Company’s estimates, the Company may need to adjust these estimates, which could have an effect on product sales and earnings in the period of adjustment.
Items Deducted from Gross Product Sales
Prompt Pay Discount
The Company offers cash discounts to its customers, generally 2% of the sales price, as an incentive for prompt payment. Based on the Company’s and GSK’s commercialization experience, the Company expects customers to continue to comply with the prompt payment terms to earn the cash discount as the Company is selling HORIZANT to the same customers under similar prompt payment terms and conditions. The Company estimates cash discounts for prompt payment based on contractual terms, GSK’s historical customer utilization rates and the Company’s historical customer utilization rates. The Company accounts for cash discounts by reducing accounts receivable by the full amount and recognizing the discount as a reduction of revenue in the same period the related revenue is recognized.
Distributor Fees
Under the Company’s inventory management agreements with significant wholesalers, the Company pays the wholesalers a fee primarily for distribution services as well as the maintenance of inventory levels. These distributor fees are based on a contractually determined fixed percentage of sales. The Company accrues the contractual amount and recognizes the discount as a reduction of revenue in the same period the related revenue is recognized.
Product Returns
The Company does not provide its customers with a general right of product return, but permits returns if the product is damaged or defective when received by the customer, or if the product has or is nearly expired. HORIZANT tablets currently have a shelf-life of 36 months from the date of manufacture. The Company will accept returns for products that will expire within six months or that have expired up to one year after their expiration dates. The Company obtained actual return history classified by the reasons for returns from GSK since GSK’s product launch in 2011, which provides a basis to reasonably estimate the Company’s future product returns. The Company estimates returns taking into consideration Company-specific adjustments to GSK’s returns history, the Company’s returns history, the shelf life of product, shipment and prescription trends and estimated distribution channel inventory levels.
Government Rebates and Chargebacks
The Company participates in a number of government rebate programs, such as the Medicaid Drug Rebate Program that provides assistance to eligible low-income patients based on each individual state’s guidelines
117
Table of Contents
regarding eligibility and services, Public Health Services or 340b programs, and the Medicare Part D Coverage Gap Discount Program, which provides rebates on prescriptions that fall within the “donut hole” coverage gap; and the Department of Veterans Affairs that offers discounts to authorized users of HORIZANT. HORIZANT is also listed on the Federal Supply Schedule, or FSS, of the General Services Administration which provides a discount to the Department of Defense, Department of Veterans Affairs, and TriCare. The Company estimates reductions to the Company’s revenues for government rebate programs based on product pricing, current rebates, GSK’s historical utilization rates, the Company’s actual utilization rates, new information regarding changes in these programs’ regulations and guidelines that would impact the amount of the actual rebates, the Company’s expectations regarding future rebates for these programs and estimated levels of inventory in the distribution channel.
Patient Assistance
The Company offers a co-pay card program to assist commercially insured patients with the cost of their HORIZANT related co-payments. Participating retail pharmacies get reimbursed by the Company for the amount of the co-pay assistance provided to eligible patients. The Company estimates and accrues the cost of the co-pay program based on historical redemption activity for this program. The Company reimburses the participating pharmacies approximately one month after the prescriptions subject to co-pay assistance are filled.
Multiple-Element Arrangements
Revenue arrangements are accounted for in accordance with the provisions of the Revenue Recognition-Multiple-Element Arrangements topic of the Codification.
In evaluating arrangements with multiple elements, the Company considers whether components of the arrangement represent separate units of accounting based upon whether certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered items. This evaluation requires subjective determinations and requires management to make judgments about the fair value of individual elements and whether such elements are separable from other aspects of the contractual relationship. The consideration received in such arrangements is allocated among the separate units of accounting based on the relative selling price method under which the selling price for each deliverable is determined using vendor-specific objective evidence of selling price, if it exists; otherwise, third-party evidence of selling price. If vendor-specific objective evidence and third-party evidence of selling price are not available for a deliverable, the Company will use its best estimate of the selling price for that deliverable when applying the relative selling price method. The applicable revenue recognition criteria are applied to each of the separate units.
Revenues from multiple deliverables combined as a single unit of accounting are deferred and recognized over the period during which the Company remains obligated to perform services. The specific methodology for the recognition of the revenue (e.g., straight-line or according to specific performance criteria) is determined on a case-by-case basis according to the facts and circumstances applicable to a given agreement.
Payments received in excess of revenues recognized are recorded as deferred revenue until such time as the revenue recognition criteria have been met.
Collaboration revenue consisted of the recognition of revenues from upfront and milestone payments from the Company’s partnership with Astellas Pharma Inc. and from upfront and additional payments from its license agreement with Indivior. Net revenue from unconsolidated joint operating activities included all revenue that resulted solely from the Company’s terminated collaboration agreement with GSK. The Company accounts for the revenue-related activities of these collaboration agreements as follows:
| • | Up-front, licensing-type payments. Up-front, licensing-type payments are assessed to determine whether or not the licensee is able to obtain any stand-alone value from the license. Where this is not the case, the Company does not consider the license deliverable to be a separate unit of accounting, and the revenue is deferred with revenue recognition for the license fee being assessed in conjunction with the other deliverables that constitute the combined unit of accounting. |
118
Table of Contents
| • | Milestones. Revenue recognition will occur only if the consideration earned from the achievement of a milestone meets all the criteria for the milestone to be considered substantive at the inception of the arrangement, such that it: (i) is commensurate with either the Company’s performance to achieve the milestone or the enhancement of the value of the item delivered as a result of a specific outcome resulting from the Company’s performance to achieve the milestone; (ii) relates solely to past performance; and (iii) is reasonably relative to all deliverables and payment terms in the arrangement. |
The Company will assess the nature of contingent payments, and appropriate accounting for, these payments on a case-by-case basis in accordance with the provisions of the Revenue Recognition topic of the Codification.
| • | Profit and loss sharing. This represented the Company’s share of the profits and losses from the co-promotion of HORIZANT with GSK under the terminated collaboration with GSK. Amounts were recognized in the period in which the related activities occurred, and their financial statement classification was based on the Company’s assessment that these activities constituted part of the Company’s ongoing central operations. |
| • | Product royalties. The Company is entitled to receive royalties on net sales of gabapentin enacarbil (known as REGNITE) in Japan. Astellas initiated sales of REGNITE in Japan in July 2012, and the Company recognizes the associated product royalties when they can be reliably measured and collectability is reasonably assured (generally upon receipt of the royalty payment). |
Cost of Product Sales
Cost of product sales includes direct and indirect costs to manufacture product sold, including tableting, packaging, storage, shipping and handling costs and inventory write-downs, if any.
Accounts Receivable
Trade accounts receivable are recorded net of allowances for cash discounts for prompt payment, wholesaler discounts and chargebacks. The need for bad debt allowance is evaluated each reporting period based on the Company’s assessment of the credit worthiness of its customers.
Inventories
Inventories are stated at the lower of cost or market. Cost is determined on a first-in, first-out, or FIFO, basis. Inventories include active pharmaceutical ingredient, or API, and contract manufacturing costs. The Company regularly evaluates the Company’s inventories for excess quantities and obsolescence (expiration), taking into account such factors as historical and anticipated future sales compared to quantities on hand and the remaining shelf life of HORIZANT. Write-downs of inventories are considered to be permanent reductions in the cost basis of inventories.
Inventories that are not expected to be consumed within 12 months following the balance sheet date are classified as long-term inventories.
Inventories as of December 31, 2014 and 2013 were summarized as follows (in thousands):
| Year Ended December 31, | ||||||||
| 2014 | 2013 | |||||||
| Raw materials |
$ | 9,273 | $ | 10,258 | ||||
| Work in progress |
517 | 76 | ||||||
| Finished goods |
766 | 1,113 | ||||||
|
|
|
|
|
|||||
| Total inventory |
10,556 | 11,447 | ||||||
| Less: Long-term inventories |
9,098 | 10,185 | ||||||
|
|
|
|
|
|||||
| Total inventory classified as current |
$ | 1,458 | $ | 1,262 | ||||
|
|
|
|
|
|||||
119
Table of Contents
Long-term inventories consisted of gabapentin enacarbil API used for production of HORIZANT. The Company evaluates demand for HORIZANT and expected consumption of the API based on long-term projected sales of HORIZANT.
Research and Development
All research and development costs, including those funded by third parties, are expensed as incurred. Research and development costs consisted of salaries, employee benefits, laboratory supplies, costs associated with clinical trials, including amounts paid to clinical research organizations, other professional services and facility costs.
Clinical Trials
The Company accrues and expenses the costs for clinical trial activities performed by third parties based upon estimates of the percentage of work completed over the life of the individual study in accordance with agreements established with contract research organizations and clinical trial sites. The Company determines the estimates through discussions with internal clinical personnel and external service providers as to progress or stage of completion of trials or services and the agreed upon fee to be paid for such services. Costs of setting up clinical trial sites for participation in the trials are expensed immediately as research and development expenses. Clinical trial site costs related to patient visits are accrued as patients’ progress through the trial and are reduced by any payments made to the clinical trial site. Non-refundable advance payments for research and development goods or services are recognized as expense as the related goods are delivered or the related services are provided in accordance with the provisions of the Research and Development Arrangements topic of the Codification.
Nonretirement Postemployment Benefits
On May 31, 2012, the Company adopted the XenoPort Amended and Restated 2012 Severance Plan, or the 2012 Severance Plan, for the benefit of the Company’s non-executive employees. Under the terms of the 2012 Severance Plan, a non-executive employee terminated by the Company because of elimination of his or her position is eligible to receive continuation of medical insurance under COBRA and specified severance payments based on the employee’s level and years of service with the Company. The Company accounts for employee termination benefits in accordance with the provisions of the Compensation-Nonretirement Postemployment Benefits topic of the Codification and records employee termination liabilities once they are both probable and estimable for severance provided under the Company’s existing severance program.
On June 14, 2013, the Company implemented a reduction in force that included the elimination of certain non-executive positions as the Company completed certain work projects on its AP development program. As a result, the Company recorded severance benefits charges of $703,000 in 2013, which were included in the “Research and development” line of the “Operating expenses” section of the Company’s statements of comprehensive loss. As of December 31, 2014 and 2013, there were no associated liability balances.
Stock-Based Compensation
The Compensation — Stock Compensation topic of the Codification establishes accounting for stock-based awards exchanged for employee services. In accordance with this topic, for stock options, awards and stock purchase rights under the Company’s employee stock purchase plan, or ESPP, stock-based compensation cost is measured at grant date, based on the fair value of the award, and is recognized as expense over the requisite employee service period.
120
Table of Contents
The effect of recording stock-based compensation under the Compensation — Stock Compensation topic was as follows:
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| (In thousands, except | ||||||||||||
| per share amounts) | ||||||||||||
| Stock-based compensation by type of award: |
||||||||||||
| Employee stock options and awards |
$ | 8,531 | $ | 10,083 | $ | 11,956 | ||||||
| ESPP |
510 | 461 | 325 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total stock-based compensation |
$ | 9,041 | $ | 10,544 | $ | 12,281 | ||||||
|
|
|
|
|
|
|
|||||||
| Effect on basic and diluted net loss per share |
$ | (0.15 | ) | $ | (0.22 | ) | $ | (0.31 | ) | |||
|
|
|
|
|
|
|
|||||||
The Company’s employee non-cash stock-based compensation was reported as follows:
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| (In thousands) | ||||||||||||
| Research and development |
$ | 2,062 | $ | 3,059 | $ | 4,364 | ||||||
| Selling, general and administrative |
6,979 | 7,485 | 7,917 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 9,041 | $ | 10,544 | $ | 12,281 | |||||||
|
|
|
|
|
|
|
|||||||
Valuation Assumptions
The Company estimates the fair value of all of its stock options and stock purchase rights on the date of grant using a Black-Scholes valuation model that requires the input of highly subjective assumptions, including the option’s expected life and the price volatility of the underlying stock. The Company derived the expected life assumptions using data obtained from similar entities, taking into consideration factors such as industry, stage of life cycle, size and financial leverage. The Company has determined that its historical volatility can be used to derive the expected stock price volatility assumption. The Company expenses the resulting charge using the straight-line attribution method over the vesting period. Restricted stock units are measured at the fair value of the Company’s common stock on the date of grant and expensed over the period of vesting using the straight-line attribution approach. The calculation of the Black-Scholes valuations used the following weighted-average assumptions:
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Dividend yield |
0 | % | 0 | % | 0 | % | ||||||
| Volatility for options |
0.81 | 0.82 | 0.81 | |||||||||
| Volatility for ESPP |
0.55 | 0.59 | 0.65 | |||||||||
| Weighted-average expected life of options (years) |
5.25 | 5.25 | 5.09 | |||||||||
| Weighted-average expected life of ESPP rights (years) |
0.75 | 0.75 | 0.75 | |||||||||
| Risk-free interest rate for options |
1.52-1.77 | % | 0.70-1.60 | % | 0.62-1.02 | % | ||||||
| Risk-free interest rate for ESPP rights |
0.05-0.17 | % | 0.07-0.17 | % | 0.07-0.27 | % | ||||||
Income Taxes
Income taxes are accounted for in accordance with the Income Taxes topic of the Codification using the liability method. Deferred tax assets and liabilities are determined based on the difference between the financial statement and tax bases of assets and liabilities as measured by the enacted tax rates that will be in effect when these differences reverse. The Company provides a valuation allowance against net deferred tax assets if, based upon the available evidence, it is more-likely-than-not that the deferred tax assets will not be realized.
121
Table of Contents
The recognition, derecognition and measurement of a tax position is based on management’s best judgment given the facts, circumstances and information available at the reporting date.
As of December 31, 2014, the Company had unrecognized tax benefits and expects no significant changes in unrecognized tax benefits in the next 12 months (see Note 10 for more information).
The Company’s policy is to recognize interest and penalties related to the underpayment of income taxes as a component of income tax expense. To date, there have been no interest or penalties charged to the Company in relation to the underpayment of income taxes.
Comprehensive Loss
The Company presents all non-owner changes in stockholders’ equity in a single continuous statement of comprehensive loss and shows: (i) each component of net loss along with total net loss; (ii) each component of other comprehensive loss along with a total for other comprehensive loss; and (iii) a total amount for comprehensive loss. The Company’s other comprehensive loss is comprised of unrealized gains (losses) on available-for-sale securities.
Advertising Costs
Advertising costs, included in selling, general and administrative expenses, are charged to expense as incurred. Advertising expenses for the year ended December 31, 2014 and 2013, was $9,568,000 and $8,244,000, respectively. The Company did not have advertising costs for the year ended December 31, 2012.
Net Loss Per Share
Basic net loss per share is calculated by dividing the net loss by the weighted-average number of common shares outstanding for the period without consideration for potential common shares. Diluted net loss per share is computed by dividing the net loss by the weighted-average number of common shares outstanding for the period plus any dilutive potential common shares for the period determined using the treasury-stock method. For purposes of this calculation, restricted stock units, options to purchase stock and warrants are considered to be potential common shares and are only included in the calculation of diluted net loss per share when their effect is dilutive.
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| (In thousands, except | ||||||||||||
| per share amounts) | ||||||||||||
| Numerator: |
||||||||||||
| Net loss |
$ | (49,333 | ) | $ | (85,883 | ) | $ | (30,814 | ) | |||
|
|
|
|
|
|
|
|||||||
| Denominator: |
||||||||||||
| Weighted-average common shares outstanding |
60,856 | 47,545 | 39,434 | |||||||||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per share |
$ | (0.81 | ) | $ | (1.81 | ) | $ | (0.78 | ) | |||
|
|
|
|
|
|
|
|||||||
| Outstanding securities at period end not included in the computation of diluted net loss per share as they had an anti-dilutive effect: |
||||||||||||
| Restricted stock units and options to purchase common stock |
7,650 | 6,824 | 6,339 | |||||||||
| Warrants outstanding |
— | 283 | 283 | |||||||||
|
|
|
|
|
|
|
|||||||
| 7,650 | 7,107 | 6,622 | ||||||||||
|
|
|
|
|
|
|
|||||||
On January 29, 2014, the Company completed an underwritten public offering of 12,000,000 shares of its common stock at a price to the public of $6.00 per share. On February 21, 2014, the underwriters exercised in full their option to purchase 1,800,000 additional shares.
122
Table of Contents
On February 3, 2015, the Company completed a private placement of $115,000,000 aggregate principal amount of 2.50% Convertible Senior Notes due 2022, or the 2022 Notes, to the investment bank acting as the initial purchaser who subsequently resold the 2022 Notes to qualified institutional buyers. The 2022 Notes are convertible at an initial conversion rate of 93.2945 shares of the Company’s common stock per $1,000 principal amount of the 2022 Notes, which is equal to an initial conversion price of approximately $10.72 per share of common stock (see Note 12 for more information).
Recent Accounting Pronouncements
In May 2014, the FASB issued Accounting Standards Update, or ASU, No. 2014-09, Revenue from Contracts with Customers: Topic 606 (ASU 2014-09), which creates a single source of revenue guidance under GAAP for all companies in all industries. The core principle of ASU 2014-09 is that revenue should be recognized in a manner that depicts the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. ASU 2014-09 defines a five-step process in order to achieve this core principle, which may require the use of judgment and estimates. ASU 2014-09 also requires expanded qualitative and quantitative disclosures relating to the nature, amount, timing and uncertainty of revenue and cash flows arising from contracts with customers, including significant judgments and estimates used. ASU 2014-09 will be effective for the Company in the first quarter of fiscal 2017, and early adoption is not permitted. The Company may adopt ASU 2014-09 either by way of a full retrospective approach for all periods presented in the period of adoption or a modified retrospective approach. The Company is currently evaluating the impact that ASU 2014-09 will have on its financial statements and has not yet determined which transition method it will apply.
In August 2014, the FASB issued ASU No. 2014-15, Presentation of Financial Statements — Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern (ASU 2014-15). This update is intended to define management’s responsibility to evaluate whether there is substantial doubt about an organization’s ability to continue as a going concern and to provide related footnote disclosures. ASU 2014-15 requires management to assess an entity’s ability to continue as a going concern by incorporating and expanding upon certain principles that are currently in U.S. auditing standards. Specifically, the amendments: (1) provide a definition of the term substantial doubt; (2) require an evaluation every reporting period including interim periods; (3) provide principles for considering the mitigating effect of management’s plans; (4) require certain disclosures when substantial doubt is alleviated as a result of consideration of management’s plans; (5) require an express statement and other disclosures when substantial doubt is not alleviated; and (6) require an assessment for a period of one year after the date that the financial statements are issued (or available to be issued). ASU 2014-15 will be effective for annual periods ending after December 15, 2016, and interim periods within annual periods beginning after December 15, 2016, with early adoption permitted. ASU 2014-15 will be effective for the Company beginning with its annual report for fiscal 2016 and interim periods thereafter. The Company is currently evaluating the impact that ASU 2014-15 will have on its financial statements.
2. License and Collaboration Agreements
Indivior PLC
In May 2014, the Company entered into an exclusive license agreement with Indivior, which became effective on June 19, 2014. Under the terms of this agreement, the Company granted to Indivior exclusive, world-wide rights to develop and commercialize AP Products for all indications, subject to its right of first negotiation with Indivior to collaborate to develop and commercialize AP Products for non-addiction indications. In exchange for these rights, the Company received an upfront, non-refundable cash payment of $20,000,000 in June 2014 and also received an additional $5,000,000 in July 2014 after delivery of specified materials to Indivior. The Company is also eligible to receive aggregate cash payments of up to $120,000,000 upon the achievement by Indivior of certain contingent event-based payments, of which $70,000,000 are regulatory and development-based and $50,000,000 are commercialization-based. In addition, the Company is entitled to receive tiered double-digit royalty payments of up to the mid-teens on a percentage basis on potential future net
123
Table of Contents
sales of the products in the United States, and high single-digit royalty payments on potential future net sales of the products outside the United States. The Company also agreed to transfer its existing AP Investigational New Drug applications, or INDs, know-how, and to provide supply transition assistance to facilitate the establishment of Indivior’s AP Product manufacturing capabilities pursuant to a mutually agreed supply transition plan.
The Company assessed its transfer and assistance obligations under the Indivior agreement using the multiple-element arrangements revenue recognition guidance since those obligations involved more than one deliverable to Indivior. This analysis involved identification of the deliverables, determining if they qualify as separate units of accounting, arriving at fair value estimates for each separate unit of accounting, and allocating total consideration to each unit of accounting. The Company identified the following non-contingent deliverables under the license agreement: (1) grant of exclusive rights, or the license; (2) transfer of know-how necessary for the manufacture and development of AP Products; (3) transfer of INDs; (4) transfer of specified materials; and (5) supply of transition assistance to facilitate the establishment of Indivior’s AP Product manufacturing capabilities pursuant to a mutually agreed upon supply transition plan. The license, know-how, INDs and supply of transition assistance deliverables were combined into one unit of accounting since each of these deliverables was dependent on, and not separate from each other, and accordingly did not have stand-alone value. The materials represented the other unit of accounting. The total of the upfront cash payment of $20,000,000 and the additional cash payment of $5,000,000 were allocated into two units of accounting using the relative estimated selling price method. The Company developed its best estimate of selling prices for each deliverable in order to allocate the non-contingent arrangement consideration to the two units of accounting. For the license, know-how, INDs and supply of transition assistance, the Company used the discounted cash flow method to estimate the price at which it could sell them on a stand-alone basis. Embedded in the estimate were significant assumptions including: (1) probabilities of success during the development process; (2) potential customer market for the drug; (3) selling price for the drug; (4) costs to develop, maintain and manufacture the developed drug; and (5) discount rate. For the materials, the Company estimated the selling price based on the cost to purchase such materials from third party suppliers. Both the initial upfront cash payment of $20,000,000 and the additional cash payment of $5,000,000 were recognized as collaboration revenue upon the transfer of the deliverables to Indivior, which occurred in the third quarter of 2014.
Astellas Pharma Inc.
In December 2005, the Company entered into an agreement pursuant to which it licensed to Astellas Pharma Inc., exclusive rights to develop and commercialize gabapentin enacarbil in Japan, Korea, the Philippines, Indonesia, Thailand and Taiwan. Astellas commenced commercialization of gabapentin enacarbil in Japan, and began selling gabapentin enacarbil in July 2012 under the trade name of REGNITE® (gabapentin enacarbil) Extended-Release Tablets. In May 2013, Astellas notified the Company that it did not have plans for the commercialization of gabapentin enacarbil in the five other Asian countries in its territory. As a result, in May 2013, the rights to gabapentin enacarbil in Korea, the Philippines, Indonesia, Thailand and Taiwan reverted to the Company. In the years ended December 31, 2014, 2013 and 2012, the Company recognized collaboration revenue of $1,134,000, $1,137,000 and $1,515,000, respectively, representing amortization of the up-front license payment under this agreement. In the year ended December 31, 2012, the Company also recognized a $10,000,000 milestone payment in connection with the approval of REGNITE in Japan. For the years ended December 31, 2014, 2013 and 2012, the Company recognized $561,000, $400,000 and $109,000, respectively, in royalty revenue. As of December 31, 2014, the Company had recognized an aggregate of $54,073,000 of revenue pursuant to this agreement. At December 31, 2014, $11,998,000 of revenue was deferred under this agreement and was being recognized on a straight-line basis over a period that the Company expects to remain obligated to provide services, of which $1,134,000 was classified within current liabilities and the remaining $10,864,000 was recorded as a noncurrent liability.
Glaxo Group Limited
Prior to the November 2012 termination and transition agreement (see Note 1 for more information), in January 2012, the Company provided notice to GSK of the Company’s belief that, among other matters, GSK had materially breached its contractual obligation. In February 2012, the Company and GSK commenced
124
Table of Contents
litigation. The November 2012 termination and transition agreement provided for a mutual release of claims and resolved all ongoing litigation between the parties (see Note 3 for more information).
In February 2007, the Company entered into an exclusive collaboration agreement with GSK to develop and commercialize gabapentin enacarbil in all countries of the world excluding the Astellas territory. In November 2010, the Company amended and restated its collaboration agreement with GSK, pursuant to which the Company reacquired all rights to gabapentin enacarbil outside of the United States previously granted to GSK (which excludes the Astellas territory) and obtained the right, but not the obligation, to pursue development of HORIZANT for: (i) the potential treatment of diabetic peripheral neuropathy; (ii) the potential treatment of postherpetic neuralgia, or PHN, to the extent that a product label would reflect a superiority claim over a currently approved drug; and (iii) any additional indications in the United States. In April 2011, the FDA approved HORIZANT for the treatment of RLS in adults. Shipments of HORIZANT to wholesalers commenced in June 2011, and HORIZANT was commercially launched in July 2011. In June 2012, the FDA approved HORIZANT for the management of PHN in adults. Under the collaboration agreement, GSK remained responsible for further development and regulatory matters with respect to HORIZANT and manufacturing and commercialization of HORIZANT in the United States for all indications.
In March 2007, GSK made an up-front, non-refundable license payment of $75,000,000. Under the terms of the amended and restated collaboration agreement, the Company received $130,000,000 in aggregate clinical and regulatory event-based payments that have been fully recognized through December 31, 2012, including $10,000,000 received and fully recognized in June 2012 in connection with the first commercial sale of HORIZANT for the management of PHN in adults. The Company concluded that the up-front license payment did not have value to GSK on a stand-alone basis without the benefit of the specified development activities that the Company performed in connection with HORIZANT and that $85,000,000 of milestones paid for clinical trial and pre-clinical activities were either not sufficiently substantive or not sufficiently at risk to be accounted for using the “when-earned” model. Accordingly, these milestones and the up-front payment were combined into one unit of accounting that was recognized over the development period to commercialization of the product, during which time delivery of substantially all of the efforts required for the completion of the Company’s contractual responsibilities under the GSK agreement occurred, and the Company determined that no additional performance obligations resulted from the amended agreement. As of December 31, 2012, the Company had recognized an aggregate of $205,000,000 of up-front license, milestone and contingent event-based payments pursuant to this agreement and no revenue was deferred under this agreement.
The Company’s net revenue from unconsolidated joint operating activities from the GSK collaboration agreement was $0, $0 and $10,000,000 for the years ended December 31, 2014, 2013 and 2012, respectively.
The following table presents the Company’s total revenues that have been recognized during the year indicated pursuant to its current license agreement with Astellas and Indivior and its terminated collaboration agreement with GSK (in thousands):
| Year Ended December 31, | ||||||||||||
| 2014 | 2013 | 2012 | ||||||||||
| Astellas |
$ | 1,695 | $ | 1,537 | $ | 11,624 | ||||||
| GSK |
— | — | 10,000 | |||||||||
| Indivior |
25,000 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | 26,695 | $ | 1,537 | $ | 21,624 | |||||||
|
|
|
|
|
|
|
|||||||
3. Acquisitions and Acquisition-Related Items
On November 8, 2012, the Company reached an agreement with GSK to terminate the collaboration agreement dated November 7, 2010 between the Company and GSK (see Note 1 for more information on the termination and transition agreement and see Note 2 for more information on the terminated collaboration agreement).
Pursuant to a separate stock purchase agreement entered into between the parties on November 8, 2012, GSK purchased $20,000,000 of common stock of the Company, or an aggregate of 1,841,112 shares at $10.863
125
Table of Contents
per share, which per share price represented a 12.5 percent premium to the average of the closing prices of the Company’s common stock for the ten trading days prior to October 31, 2012. In addition, the Company was granted a Put Option that was exercisable for six months from November 9, 2012 to require GSK to purchase up to an additional $20,000,000 of common stock of the Company at a 12.5 percent premium to the average of the closing prices of the Company’s common stock for the ten trading days prior to the day the Company exercises this option. On November 9, 2012, the Company exercised the Put Option in full. The value of the Put Option was the difference between the price paid by GSK for the shares put, $20,000,000, and the value of the shares based on the Company’s closing stock price on the day of exercise. On November 9, 2012, pursuant to the exercise of the Put Option, GSK purchased an additional 2,190,100 shares at $9.132 per share.
The Company accounted for the termination and transition agreement and the stock purchase agreement with GSK in accordance with the provisions of the Business Combinations topic of the Codification. Under the provisions of the Business Combinations topic, the acquisition date for a business is the date on which the Company obtains control of the acquiree. The Company obtained control of the HORIZANT business at the end of the transition period, or May 1, 2013. Accordingly, on November 8, 2012, the transaction did not meet the definition of a business combination, and the Company accounted for the transaction as a multiple-element arrangement consisting of the settlement of a pre-existing relationship in exchange for the right to acquire the HORIZANT business at the end of a defined transition period, the sale of common stock at a premium and the obligation by the Company to make certain cash payments to GSK in the future. The value of the HORIZANT business was based on an estimated discounted cash flow analysis attributable to the HORIZANT business.
The following table summarizes the fair value of consideration exchanged as part of the termination and transition agreement (in thousands):
| Cash payable to GSK (recorded as “Other noncurrent liability” on the Company’s Balance Sheet for the year ended December 31, 2012) |
$ | 2,314 | ||
| Issuance of common shares to GSK |
30,268 | |||
| Cash received for common shares issued to GSK |
(40,000 | ) | ||
| Settlement of litigation with GSK |
20,499 | |||
| Transaction costs |
476 | |||
|
|
|
|||
| $ | 13,557 | |||
|
|
|
The components of the consideration transferred are described below:
| • | The cash payable to GSK represents the net present value of the annual payments to GSK of $1,000,000 for six years beginning in 2016. |
| • | The Company recorded the issuance of common shares to GSK based on the Company’s closing stock prices on the respective stock issuance dates. The Company issued 1,841,112 and 2,190,100 common shares to GSK on November 8, 2012 and November 9, 2012, respectively. |
| • | The termination and transition agreement provided for a mutual release of claims and resolved all ongoing litigation between the Company and GSK and effectively settled a preexisting relationship (see Note 1 for more information). As a result, the Company recorded a gain on the settlement of litigation, which represented foregone potential future monetary damages. This amount was valued based on a probability weighted scenario analysis that took into consideration the probability of each potential future alternative outcomes of the litigation between the parties. |
| • | The transaction costs represent direct external costs incurred by the Company in connection with the November 2012 transaction. |
The consideration exchanged was recorded in the Company’s financial statements for the year ended December 31, 2012.
On May 1, 2013, the Company obtained control of, and thus completed the acquisition of, the HORIZANT business. The acquisition constituted a business acquisition as defined under the provisions of the Business Combinations topic. Accordingly, the acquisition reflected the removal of the right to the HORIZANT business
126
Table of Contents
(which was recorded at fair value, as determined by a discounted cash flow analysis, that remained unchanged as of May 1, 2013), and the recording of the acquisition date fair values of the acquired inventory and manufacturing property and equipment assets. Assets acquired as a result of this transaction were as follows (in thousands):
| Inventories |
$ | 11,733 | ||
| Manufacturing property and equipment |
1,967 | |||
|
|
|
|||
| Total assets acquired |
$ | 13,700 | ||
|
|
|
Inventories were comprised of acquired raw materials, specifically gabapentin enacarbil, and were valued using the cost approach. The cost approach provides a framework for estimating value based on the principle of substitution. The estimated fair value was calculated based on the costs to replace or reproduce the asset, including the cost of recrystallization of the raw material received, while considering any impact on the value of any obsolescence.
Manufacturing property and equipment acquired included certain equipment and a dedicated manufacturing suite for use in the manufacture of HORIZANT and was recorded at fair value using the replacement cost method. The replacement cost method uses an estimate of the cost of replacing or reproducing an asset to measure value. This amount is then reduced to reflect the amount of depreciation that has accrued given the asset’s age. Other factors and adjustments were considered in concluding on the fair value, including obsolescence of the acquired assets. Depreciation and amortization is computed using the straight-line method over the estimated useful lives of the respective assets, which is ten years.
In 2013, the Company incurred $476,000 in transaction costs related to the HORIZANT business acquisition, which primarily consisted of legal, accounting and valuation-related expenses. These expenses were recorded in the “Selling, general and administrative” line of the “Operating expenses” section of the Company’s statement of comprehensive loss.
The following unaudited supplemental pro forma information summarizes the combined results of operations of the Company and the HORIZANT business as though the acquisition had occurred on January 1, 2012 (in thousands):
| Year Ended December 31, |
||||||||
| 2013 | 2012 | |||||||
| Revenues |
$ | 9,128 | $ | 28,119 | ||||
| Net loss |
$ | 92,791 | $ | 84,695 | ||||
These amounts have been calculated after applying the Company’s accounting policies and adjusting the results of the HORIZANT business to reflect the following adjustments:
| • | An amendment to depreciation expense included in costs of goods sold to reflect the depreciation of the fair value of the acquired manufacturing property and equipment over the estimated remaining useful lives of the respective assets. |
| • | The difference between the cost of API that GSK incurred and the Company’s recorded fair value of the API. |
| • | The difference in selling expense between GSK direct selling expenses and the Company’s third party contractual costs. |
The pro forma information presented above is for informational purposes only and is not indicative of the results of operations that would have been achieved if the acquisition had taken place at the beginning of January 2012, nor is it indicative of any future results.
127
Table of Contents
4. Cash and Cash Equivalents, Short-Term Investments and Restricted Investments
The following are summaries of cash and cash equivalents, short-term investments and restricted investments (in thousands):
| Cost | Gross Unrealized Gains |
Gross Unrealized Losses |
Estimated Fair Value |
|||||||||||||
| As of December 31, 2014: |
||||||||||||||||
| Cash |
$ | 3,383 | $ | — | $ | — | $ | 3,383 | ||||||||
| Money market funds |
8,576 | — | — | 8,576 | ||||||||||||
| U.S. government-sponsored agencies |
2,000 | — | (2 | ) | 1,998 | |||||||||||
| Corporate debt securities |
88,127 | 12 | (40 | ) | 88,099 | |||||||||||
| Certificates of deposit |
1,725 | — | — | 1,725 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 103,811 | $ | 12 | $ | (42 | ) | $ | 103,781 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Reported as: |
||||||||||||||||
| Cash and cash equivalents |
$ | 11,958 | ||||||||||||||
| Short-term investments |
90,098 | |||||||||||||||
| Restricted investments |
1,725 | |||||||||||||||
|
|
|
|||||||||||||||
| $ | 103,781 | |||||||||||||||
|
|
|
|||||||||||||||
| Cost | Gross Unrealized Gains |
Gross Unrealized Losses |
Estimated Fair Value |
|||||||||||||
| As of December 31, 2013: |
||||||||||||||||
| Cash |
$ | 2,281 | $ | — | $ | — | $ | 2,281 | ||||||||
| Money market funds |
10,046 | — | — | 10,046 | ||||||||||||
| U.S. government-sponsored agencies |
2,500 | — | — | 2,500 | ||||||||||||
| Corporate debt securities |
43,831 | 6 | (6 | ) | 43,831 | |||||||||||
| Certificates of deposit |
1,725 | — | — | 1,725 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| $ | 60,383 | $ | 6 | $ | (6 | ) | $ | 60,383 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Reported as: |
||||||||||||||||
| Cash and cash equivalents |
$ | 20,584 | ||||||||||||||
| Short-term investments |
38,074 | |||||||||||||||
| Restricted investments |
1,725 | |||||||||||||||
|
|
|
|||||||||||||||
| $ | 60,383 | |||||||||||||||
|
|
|
|||||||||||||||
At December 31, 2014 and 2013, the contractual maturities of all investments held were less than 16 months and one year, respectively. All investments have been designated as available-for-sale, and changes to unrealized gains and losses are reflected as the only component of accumulated other comprehensive loss. The Company views its available-for-sale portfolio as available for use in current operations. Accordingly, the Company has classified all investments as short-term, even though the stated maturity may be one year or more beyond the current balance sheet date.
No gross realized gains or losses were recognized in 2014, 2013 and 2012, and accordingly, there were no amounts reclassified out of accumulated other comprehensive loss to earnings in 2014, 2013 or 2012.
128
Table of Contents
The Company’s available-for-sale investments, which include cash equivalents and short-term investments, are measured at fair value on a recurring basis and are classified at the following fair value hierarchy (see Note 1 for the Company’s accounting policy on measuring fair value of financial instruments) (in thousands):
| Total As of December 31, 2014 |
Fair Value Measurements at Reporting Date Using | |||||||||||||||
| Description |
Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
|||||||||||||
| Money market funds |
$ | 8,576 | $ | 8,576 | $ | — | $ | — | ||||||||
| U.S. government-sponsored agencies |
1,998 | — | 1,998 | — | ||||||||||||
| Corporate debt securities |
88,099 | — | 88,099 | — | ||||||||||||
| Certificates of deposit |
1,725 | — | 1,725 | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
$ | 100,398 | $ | 8,576 | $ | 91,822 | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total As of December 31, 2013 |
Fair Value Measurements at Reporting Date Using | |||||||||||||||
| Description |
Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
|||||||||||||
| Money market funds |
$ | 10,046 | $ | 10,046 | $ | — | $ | — | ||||||||
| U.S. government-sponsored agencies |
2,500 | — | 2,500 | — | ||||||||||||
| Corporate debt securities |
43,831 | — | 43,831 | — | ||||||||||||
| Certificates of deposit |
1,725 | — | 1,725 | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total |
$ | 58,102 | $ | 10,046 | $ | 48,056 | $ | — | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
5. Property and Equipment, net
Property and equipment at December 31, 2014 and 2013 consisted of the following (in thousands):
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Laboratory equipment |
$ | 10,561 | $ | 10,701 | ||||
| Manufacturing property and equipment |
1,975 | 1,967 | ||||||
| Furniture and fixtures |
1,076 | 1,107 | ||||||
| Computer equipment and software |
5,656 | 5,475 | ||||||
| Leasehold improvements |
3,405 | 3,344 | ||||||
|
|
|
|
|
|||||
| 22,673 | 22,594 | |||||||
| Less: Accumulated depreciation and amortization |
(20,251 | ) | (20,042 | ) | ||||
|
|
|
|
|
|||||
| Property and equipment, net |
$ | 2,422 | $ | 2,552 | ||||
|
|
|
|
|
|||||
129
Table of Contents
6. Other Accrued Liabilities
Other accrued liabilities at December 31, 2014 and 2013 were as follows (in thousands):
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Accrued product costs |
$ | 2,009 | $ | 1,903 | ||||
| Accrued selling and marketing expenses |
1,210 | 883 | ||||||
| Accrued general and administrative expenses |
466 | 632 | ||||||
| Accrued rebates, allowances and returns |
986 | 315 | ||||||
| Other liabilities |
446 | 205 | ||||||
|
|
|
|
|
|||||
| Total other accrued liabilities |
$ | 5,117 | $ | 3,938 | ||||
|
|
|
|
|
|||||
7. Commitments and Contingencies
Operating Leases
In February 2008, the Company entered into a lease for approximately 59,000 square feet of office space at 3400 Central Expressway, Santa Clara, California, the 3400 Lease. The term of the 3400 Lease extended to August 2013, which was 60 months from the date the premises were considered ready for occupation by the Company.
Also in February 2008, the Company amended its lease with respect to the Company’s current office space at 3410 Central Expressway, Santa Clara, California, or, as amended, the 3410 Lease, that commenced in December 2001. This amendment extended the term of the 3410 Lease for approximately two years from the original expiration date of December 10, 2011, so that the 3410 Lease would expire in August 2013, on the same date as the 3400 Lease.
In October 2012, the Company further amended the 3410 Lease to extend the term for an additional two years, so that the 3410 Lease would expire on August 27, 2015. In November 2014, the Company further amended the 3410 Lease to extend the term for an additional nine months, so that the 3410 Lease will expire on May 31, 2016.
In connection with the 3410 Lease, the Company entered into a letter of credit agreement of $1,500,000 in December 2006. The fair value of the certificate of deposit is presented as restricted investments on the balance sheet at $1,500,000 as of December 31, 2014 and 2013. This letter of credit is required until the termination of the lease.
The Company recognized rent expense on a straight-line basis over the applicable lease terms. Rent expense was $2,615,000, $2,626,000 and $3,185,000 for the years ended December 31, 2014, 2013 and 2012, respectively. Deferred rent asset of $180,000 and $477,000 at December 31, 2014 and 2013, respectively, represented the difference between rent expense recognized and actual cash payments related to the Company’s operating leases. At December 31, 2014, deferred rent was comprised of a current deferred rent asset of $127,000 and a noncurrent deferred rent asset of $53,000. At December 31, 2013, deferred rent was comprised of a current deferred rent asset of $308,000 and a noncurrent deferred rent asset of $169,000.
At December 31, 2014, future minimum payments under the Company’s non-cancelable operating lease were as follows (in thousands):
| Year ending December 31: |
||||
| 2015 |
$ | 2,368 | ||
| 2016 |
986 | |||
|
|
|
|||
| Total minimum lease payments |
$ | 3,354 | ||
|
|
|
130
Table of Contents
Guarantees and Indemnifications
The Company, as permitted under Delaware law and in accordance with its bylaws, indemnifies its officers and directors for certain events or occurrences, subject to certain limits, while the officer or director is or was serving at the Company’s request in such capacity. The term of the indemnification period is for the officer’s or director’s lifetime. The Company may terminate the indemnification agreements with its officers and directors upon 90 days’ written notice, but termination will not affect claims for indemnification relating to events occurring prior to the effective date of termination. The maximum amount of potential future indemnification is unlimited; however, the Company has a director and officer insurance policy that limits its exposure and may enable it to recover a portion of any future amounts paid. The Company believes the fair value of these indemnification agreements is minimal. Accordingly, the Company had not recorded any liabilities for these agreements as of December 31, 2014.
8. Stockholders’ Equity
Common Stock
At December 31, 2014 and 2013, the Company was authorized to issue 100,000,000 shares of common stock. On January 29, 2014, the Company completed an underwritten public offering of 12,000,000 shares of its common stock at a price to the public of $6.00 per share. Net cash proceeds from the public offering were $67,300,000, after deducting the underwriting discounts and commissions and offering expenses payable by the Company. On February 21, 2014, the underwriters exercised in full their option to purchase 1,800,000 additional shares resulting in additional net cash proceeds of $10,100,000, after deducting the underwriting discounts and commissions and offering expenses payable by the Company.
Stockholders’ Rights Plan
On December 16, 2005, the Company adopted a preferred stock rights plan pursuant to which each share of the Company’s common stock outstanding has attached to it a preferred stock purchase right, or a right, that confers the right to purchase one one-hundredth of a share of Series A junior participating preferred stock at an exercise price of $140.00 per right, subject to adjustment, and will be exercisable only if a person or group acquires beneficial ownership of 15% or more of the Company’s common stock or announces a tender offer for 15% or more of the Company’s common stock. If such a person acquires 15% or more of the Company’s common stock, all rights holders, except such person, will be entitled to acquire the Company’s common stock at a discount through the exercise of the right. The rights plan has been designed to discourage acquisitions of more than 15% of the Company’s common stock without negotiations with the board of directors. The rights expire on January 13, 2016. The rights trade with the Company’s common stock, unless and until they are separated upon the occurrence of certain future events. The board of directors may terminate the rights plan at any time or redeem the rights prior to the time the rights are triggered.
Equity Incentive Plans
On June 11, 2014, the Company’s stockholders approved the 2014 Equity Incentive Plan, or the 2014 Plan. The 2014 Plan is intended to be the successor to the 2005 Equity Incentive Plan, the 2005 Non-Employee Directors’ Stock Option Plan and the 2010 Inducement Award Plan, collectively referred to as the Prior Plans. All outstanding stock awards granted under the Prior Plans will continue to be subject to the terms and conditions as set forth in the agreements evidencing such stock awards.
1999 Stock Plan
Under the terms of the 1999 Stock Plan, or the 1999 Plan, options or stock purchase rights were granted by the board of directors to employees, directors and consultants. Options granted were either incentive stock options or non-statutory stock options. Incentive stock options were granted to employees with exercise prices of no less than the fair value, and non-statutory options were granted to employees, directors or consultants at exercise prices of no less than 85% of the fair value of the common stock on the grant date as determined by the
131
Table of Contents
board of directors. Options vested as determined by the board of directors, generally at the rate of 25% at the end of the first year, with the remaining balance vesting ratably over the next three years for initial employee grants and ratably over four years for subsequent grants. Options granted under the 1999 Plan expire no more than ten years after the date of grant. All options granted under the 1999 Plan have vested.
2005 Equity Incentive Plan
In January 2005, the Company’s board of directors adopted the 2005 Equity Incentive Plan, or the 2005 Plan. Under the terms of the 2005 Plan, options vest as determined by the board of directors, generally at the rate of 25% at the end of the first year, with the remaining balance vesting ratably over the next three years for initial employee grants and ratably over four years for subsequent grants. Options granted under the 2005 Plan expire no more than ten years after the date of grant.
In January 2007, the Company’s board of directors approved the use of grants of restricted stock units to employees, directors or consultants under the 2005 Plan as part of the Company’s long-term incentive compensation program. Restricted stock units have no exercise price, are valued using the closing market price on the date of grant and vest as determined by the board of directors, typically either: (i) in annual tranches over a four-year period at the rate of 25% at the end of each year; (ii) in annual tranches over a three-year period at the rate of 25%, 25% and 50%, respectively, at the end of each year; (iii) in annual tranches over a three-year period at 50% on the second anniversary and 50% on the third year anniversary; or (iv) in one tranche on the one-year anniversary of the grant date. Employees can elect to have the Company withhold a portion of shares to pay for their payroll taxes in connection with the vesting of restricted stock units, where the Company would then make a cash payment for the associated payroll taxes on behalf of the employees, or employees can elect to make the cash payment for the associated payroll taxes.
In May 2010, the Company granted performance stock unit awards to two executive employees under the 2005 Plan. Each performance stock unit award was scheduled to vest three years from the grant date, with the actual number of shares of common stock of the Company subject to issuance to be between 0% and 200% of the target amount, based on the performance of the Company’s total shareholder return as compared to the total shareholder returns of a group of pre-selected pharmaceutical companies over a performance period ending on the third anniversary of the grant date. The target amount of shares of common stock of the Company that were subject to issuance under the performance stock unit awards was 140,000, and the grant date fair value using a lattice valuation model of these performance stock unit awards was $2,675,000. In 2010, a performance stock unit award representing a target amount of 40,000 shares was cancelled due to the departure of one of the two executive employees. In 2013, 97,300 shares were cancelled based on the performance of the Company’s total shareholder return as compared to the total shareholder returns of a group of pre-selected pharmaceutical companies over the three year performance period. At December 31, 2014 and 2013, there were no shares subject to these performance stock unit awards, and the associated expense recognized in the year ended December 31, 2014, 2013 and 2012 was $0, $232,000 and $638,000, respectively.
In February 2014, the Company granted performance stock options to its executive officers under the 2005 Plan. Each performance stock option is subject to two, equally weighted, performance conditions. One performance condition relates to the achievement of a pre-determined development criteria and the other relates to achievement of a pre-determined revenue criteria. Each performance stock option vests, if at all, upon achievement of the respective performance condition during a performance period ending on December 31, 2015. Shares associated with each performance condition vest 50% upon achievement of the performance condition, with the remaining 50% to vest on the one-year anniversary date of the initial vest date. The target amount of shares of common stock that are subject to issuance under these performance stock options was 316,000, and the grant date fair value using a Black-Scholes valuation model of these performance stock options was $1,334,000. During the year ended December 31, 2014, the Company recognized $445,000 in expense associated with these performance stock options.
Under the terms of the 2005 Plan, the maximum number of shares that may be issued shall not exceed the total of 2,000,000, plus any shares issuable from options previously granted from the 1999 Plan at the date of the Company’s initial public offering, plus an annual increase equal to the lesser of (i) 2.5% of the total number of
132
Table of Contents
common shares outstanding at the end of the preceding calendar year and (ii) 2,000,000 common shares. During the year ended December 31, 2014, the annual increase to the 2005 Plan reserve was 1,195,009 shares. In June 2014, in connection with the Company’s adoption of the 2014 Equity Incentive Plan, or the 2014 Plan, described below, no further awards may be granted under the 2005 Plan. Although the 2005 Plan has terminated, all outstanding options under the 2005 Plan will continue to be governed by their existing terms.
2005 Non-Employee Directors’ Stock Option Plan
In January 2005, the Company’s board of directors adopted the 2005 Non-Employee Directors’ Stock Option Plan, or the 2005 Directors’ Plan, under which non-statutory options are automatically granted to non-employee directors.
Before May 1, 2012, any individual who first became a non-employee director automatically received an option to purchase 25,000 shares subject to vesting in four equal successive annual installments. From May 1, 2012 to June 10, 2014, any individual who first became a non-employee director automatically received an option to purchase 30,000 shares subject to vesting in 24 successive equal monthly installments. Prior to May 1, 2012, non-employee directors serving on the date of each annual meeting of stockholders received an option to purchase 10,000 shares subject to vesting in 12 successive equal monthly installments measured from the grant date. From May 1, 2012 to June 10, 2014, non-employee directors serving on the date of each annual meeting of stockholders received an option to purchase 15,000 shares subject to vesting in 12 successive equal monthly installments measured from the grant date. Stock options were granted at exercise prices no less than the fair value on the grant date and expired no more than ten years after the date of grant. In addition, from May 1, 2012 to June 10, 2014, any individual who served as a non-employee director on the date of each annual meeting received a restricted stock unit award of 5,000 shares. These restricted stock unit awards vest in full on the one-year anniversary of the grant date.
Under the terms of the 2005 Directors’ Plan, the maximum number of shares that may be issued shall not exceed the total of 150,000, plus an annual increase equal to the excess of (i) the number of shares subject to options granted in the preceding calendar year, over (ii) the number of shares added back to the share reserve from cancellations, provided that such increase shall not exceed 150,000 shares. During the year ended December 31, 2014, the annual increase to the 2005 Directors’ Plan reserve was 120,000 shares. In June 2014, in connection with the Company’s adoption of the 2014 Plan described below, no further awards may be granted under the 2005 Directors’ Plan. Although the 2005 Directors’ Plan has terminated, all outstanding options under the 2005 Directors’ Plan will continue to be governed by their existing terms.
New Employee Inducement Stock Awards
In May 2008, the Company’s then Senior Vice President and Chief Commercialization Officer was granted a new employee inducement stock award outside of the Company’s stockholder-approved equity plans consisting of nonqualified stock options to purchase 140,612 shares of the Company’s common stock. The stock options have a per share exercise price of $42.59, the closing trading price of the Company’s common stock on the NASDAQ Global Market on the May 1, 2008 grant date. The stock options have a ten-year term and vested over four years, with 25% cliff vesting on the first anniversary of the May 1, 2008 grant date, and 1/48th of the shares subject to the options vesting monthly thereafter. The Company also granted to the Company’s Senior Vice President and Chief Commercialization Officer a new employee inducement stock award outside of the Company’s stockholder-approved equity plans consisting of restricted stock units for 10,000 shares of the Company’s common stock. The restricted stock units vested in four equal annual installments on each anniversary of the May 1, 2008 grant date.
2010 Inducement Award Plan
In May 2010, the Company’s board of directors adopted the 2010 Inducement Award Plan, or the 2010 Inducement Plan. Under the terms of the 2010 Inducement Plan, options vest as determined by the board of directors or the compensation committee of the board of directors, generally at the rate of 25% at the end of the first year, with the remaining balance vesting ratably over the next three years. Options granted under the 2010
133
Table of Contents
Inducement Plan expire no more than ten years after the date of grant. Restricted stock units have no exercise price, are valued using the closing market price on the date of grant and vest as determined by the board of directors or the compensation committee of the board of directors, typically in annual tranches over a four-year period at the rate of 25% at the end of each year.
A total of 350,000 shares of common stock were initially authorized for issuance under the 2010 Inducement Plan, and an additional 625,000 shares were authorized for issuance in 2011. Under the terms of the 2010 Inducement Plan, the maximum number of shares that may be issued shall not exceed the total of 975,000. In June 2014, in connection with the Company’s adoption of the 2014 Plan described below, no further awards may be granted under the 2010 Inducement Plan. Although the 2010 Inducement Plan has terminated, all outstanding options under the 2010 Inducement Plan will continue to be governed by their existing terms.
2014 Equity Incentive Plan
The terms of the 2014 Plan provide for the grant of incentive stock options, nonstatutory stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards, other stock awards and performance awards that may be settled in cash, stock or other property, that may be granted by the board of directors or the independent compensation committee of the board of directors. Options granted may be either incentive stock options or non-statutory stock options. Incentive stock options may be granted to employees with exercise prices of no less than the fair value, and non-statutory options may be granted to employees, directors or consultants at exercise prices of no less than fair value, of the common stock on the grant date. Options vest as determined by the board of directors or the compensation committee of the board of directors, generally at the rate of 25% at the end of the first year, with the remaining balance vesting ratably over the next three years. Options granted under the 2014 Plan expire no more than ten years after the date of grant. The terms of the 2014 Plan also provides for the grant of restricted awards, which have no exercise price, are valued using the closing market price on the date of grant and vest as determined by the board of directors or the compensation committee of the board of directors, typically in annual tranches over a four-year period at the rate of 25% at the end of each year. The 2014 Plan also provides for the grant of performance stock or cash awards that may vest or become payable upon the attainment of pre-determined performance goals during a performance period.
Under the terms of the 2014 Plan, any individual who first becomes a non-employee director automatically receives an option to purchase 30,000 shares subject to vesting in 24 successive equal monthly installments. Non-employee directors serving on the date of each annual meeting of stockholders receives an option to purchase 15,000 shares subject to vesting in 12 successive equal monthly installments measured from the grant date. Stock options are granted at exercise prices no less than the fair value on the grant date and expire no more than ten years after the date of grant. In addition, any individual who serves as a non-employee director on the date of each annual meeting receives a restricted stock unit award of 5,000 shares. These restricted stock unit awards vest in full on the earlier of: (1) the one-year anniversary of the grant date or (2) the date of the next annual meeting following the grant date.
The total number of shares of the Company’s common stock available for issuance under the 2014 Plan is initially 4,471,059 shares plus up to an additional 7,847,852 Returning Shares (as defined below) as such shares become available from time to time. “Returning Shares” means the shares subject to outstanding awards granted under the Prior Plans and the 1999 Stock Plan that, from and after the effective date of the 2014 Plan: (a) expire or terminate for any reason prior to exercise or settlement; (b) are forfeited, cancelled or otherwise returned to the Company because of the failure to meet a contingency or condition required for the vesting of such shares; or (c) other than with respect to outstanding stock options and stock appreciation rights granted under the Prior Plans or the 1999 Stock Plan with an exercise or strike price of at least 100% of the fair market value of the underlying Company common stock on the date of grant, are reacquired or withheld (or not issued) by the Company to satisfy a tax withholding obligation in connection with a stock award. Eligible participants under the 2014 Plan include the Company’s employees and directors, including the Company’s executive officers and consultants. At December 31, 2014, there were 4,479,789 shares remaining and available for future grant under the 2014 Plan.
134
Table of Contents
A summary of option activity as of and for the year ended December 31, 2014 is presented below:
| Shares | Weighted- Average Exercise Price |
Weighted- Average Remaining Contractual Term |
Aggregrate Intrinsic Value |
|||||||||||||
| (In Thousands) | ||||||||||||||||
| Outstanding at January 1, 2014 |
5,368,964 | $ | 16.81 | |||||||||||||
| Options granted |
1,986,620 | 6.12 | ||||||||||||||
| Options cancelled |
(437,453 | ) | 11.60 | |||||||||||||
| Options exercised |
(289,519 | ) | 4.49 | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2014 |
6,628,612 | 14.49 | 5.86 | $ | (8,283 | ) | ||||||||||
|
|
|
|
|
|
|
|||||||||||
| Exercisable at December 31, 2014 |
4,504,585 | $ | 18.26 | 4.45 | $ | (3,427 | ) | |||||||||
|
|
|
|
|
|
|
|
|
|||||||||
A summary of restricted stock unit activity for the year ended December 31, 2014 is presented below:
| Shares | Weighted- Average Grant Date Fair Value |
|||||||
| Outstanding at January 1, 2014 |
1,455,190 | $ | 7.15 | |||||
| Awards granted |
394,790 | 6.07 | ||||||
| Awards cancelled |
(188,500 | ) | 6.87 | |||||
| Awards vested |
(640,136 | ) | 7.27 | |||||
|
|
|
|||||||
| Outstanding at December 31, 2014 |
1,021,344 | $ | 6.70 | |||||
|
|
|
|
|
|||||
The weighted-average grant date fair values of options granted in the years ended December 31, 2014, 2013 and 2012 were $4.05, $5.22 and $3.12 per share, respectively. The weighted-average grant date fair values of restricted stock units granted in the years ended December 31, 2014, 2013 and 2012 were $6.07, $8.13 and $5.22 per share, respectively.
The aggregate intrinsic value of all options outstanding and exercisable at December 31, 2014 was based on a closing stock price of $8.77.
The total intrinsic value of options exercised in the years ended December 31, 2014, 2013 and 2012 was $758,000, $304,000 and $137,000, respectively. The total fair value of options that vested in the years ended December 31, 2014, 2013 and 2012 was $4,572,000, $4,786,000 and $8,107,000, respectively. The total fair value of restricted stock units that vested in the years ended December 31, 2014, 2013 and 2012 was $4,656,700, $6,202,000 and $4,372,000, respectively.
As of December 31, 2014, the total compensation cost related to 2,124,027 unvested options and unvested awards covering 1,021,344 shares not yet recognized was $11,616,000. This amount will be recognized over an estimated weighted-average amortization period of 2.41 years.
Employee Stock Purchase Plan
As of December 31, 2014, the Company had reserved a total of 1,445,555 shares of common stock for issuance under the ESPP. In addition, the board of directors may increase the share reserve as of each January 1 through January 1, 2015, by an amount not to exceed the lesser of (i) 1% of the total number of shares of common stock outstanding on December 31 of the preceding calendar year or (ii) 250,000 shares. The ESPP permits eligible employees to purchase common stock at a discount through payroll deductions during defined offering periods. The price at which the stock is purchased is equal to the lower of 85% of the fair market value of the common stock at the beginning of an offering period or after a purchase period ends. During the years ended December 31, 2014, 2013 and 2012, 227,720, 142,455 shares and 185,249 shares, respectively, were purchased under the ESPP. During the year ended December 31, 2014, the annual increase to the ESPP was 250,000 shares. At December 31, 2014 and 2013, there were 277,882 and 255,602 shares, respectively, remaining and available for future grant under the ESPP.
135
Table of Contents
Warrants
At December 31, 2014 and 2013, 0 and 283,420 warrants were outstanding and exercisable for shares of common stock at $25.40 per share, respectively.
9. Preferred Stock
At December 31, 2014 and 2013, the Company was authorized to issue 5,000,000 shares of preferred stock, of which 1,000,000 shares were authorized for issuance as Series A junior participating preferred stock.
10. Income Taxes
Deferred income taxes reflect the net tax effects of net operating loss, or NOL, and tax credit carryovers and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s net deferred tax assets were as follows (in thousands):
| December 31, | ||||||||
| 2014 | 2013 | |||||||
| Net operating loss carryforwards |
$ | 182,791 | $ | 161,663 | ||||
| Research credit carryforwards |
31,249 | 30,111 | ||||||
| Capitalized research and development |
3,760 | 5,579 | ||||||
| Deferred revenue |
4,824 | 4,700 | ||||||
| Stock awards |
16,901 | 14,672 | ||||||
| Other |
3,043 | 1,605 | ||||||
|
|
|
|
|
|||||
| Total net deferred tax assets |
242,568 | 218,330 | ||||||
| Valuation allowance |
(242,568 | ) | (218,330 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax assets |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
Realization of net deferred tax assets is dependent upon the Company generating future taxable income, the timing and amount of which are uncertain. Accordingly, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance increased by $24,238,000, $27,210,000 and $6,259,000 during 2014, 2013 and 2012, respectively.
As of December 31, 2014, the Company had NOL carryforwards for federal income tax purposes of $463,798,000, which expire in the years 2022 through 2034, and federal research and development tax credits of $24,022,000, which expire in the years 2021 through 2034.
As of December 31, 2014, the Company had NOL carryforwards for state income tax purposes of $367,492,000, which expire in the years 2015 through 2034, and state research and development tax credits of $12,507,000, which do not expire.
Approximately $529,000 of the valuation allowance for net deferred tax assets relates to benefits of stock option deductions that, when recognized, will be allocated directly to additional paid-in capital.
The Company files income tax returns in the U.S. federal jurisdiction and various state jurisdictions. To date, the Company has not been audited by the Internal Revenue Service or any state income tax jurisdiction. Tax years 2000 to 2014 remain subject to examination by the U.S. federal jurisdiction and various state jurisdictions.
Utilization of the Company’s NOL and credit carryforwards may be subject to substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code and similar state provisions. Such annual limitation could result in the expiration of the NOL and credit carryforwards before utilization. As of December 31, 2014, based on the analyses performed on annual limitation as a result of ownership changes that may have occurred from inception through December 2014, the Company expects to be able to use all of its NOL and tax credit carryforwards.
136
Table of Contents
The Company’s unrecognized tax benefits relate to state research and development tax credits claimed on the Company’s state tax returns. The state research and development tax credits have not been utilized, are fully offset by a valuation allowance and currently have no tax impact.
A reconciliation of the Company’s beginning and ending amount of unrecognized tax benefits is as follows (in thousands):
| January 1, 2013 |
$ | — | ||
| Increases related to prior year tax positions |
1,340 | |||
|
|
|
|||
| December 31, 2013 |
1,340 | |||
| Increases related to currrent year tax positions |
50 | |||
|
|
|
|||
| December 31, 2014 |
$ | 1,390 | ||
|
|
|
11. Quarterly Financial Data (Unaudited)
The following table summarizes the unaudited quarterly financial data for the last two fiscal years (in thousands, except per share data):
| Quarter Ended | ||||||||||||||||||||||||||||||||
| Dec. 31, 2014 |
Sept. 30, 2014(1) |
June 30, 2014 |
March 31, 2014 |
Dec. 31, 2013 |
Sept. 30, 2013 |
June 30, 2013 |
March 31, 2013 |
|||||||||||||||||||||||||
| Selected Quarterly Data: |
||||||||||||||||||||||||||||||||
| Total revenues |
$ | 7,091 | $ | 31,068 | $ | 5,334 | $ | 3,375 | $ | 2,879 | $ | 2,527 | $ | 2,086 | $ | 459 | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Gross profit(2) |
$ | 6,143 | $ | 5,088 | $ | 4,321 | $ | 2,527 | $ | 2,121 | $ | 1,732 | $ | 1,391 | $ | — | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Net income (loss) |
$ | (17,657 | ) | $ | 8,259 | $ | (19,387 | ) | $ | (20,548 | ) | $ | (19,138 | ) | $ | (18,816 | ) | $ | (24,381 | ) | $ | (23,548 | ) | |||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| Basic and diluted net income (loss) per share |
$ | (0.28 | ) | $ | 0.13 | $ | (0.31 | ) | $ | (0.36 | ) | $ | (0.40 | ) | $ | (0.39 | ) | $ | (0.51 | ) | $ | (0.50 | ) | |||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
| (1) | Total revenues and net income was primarily from the recognition of $25,000,000 in collaboration revenue resulting from the Indivior licensing agreement (see Note 2 for more information). |
| (2) | Gross profit relates solely to HORIZANT product sales. |
12. Subsequent Events
In February 2015, the Company completed a private placement of $115,000,000 principal amount of 2022 Notes. Interest on the 2022 Notes will be payable semi-annually in cash in arrears on February 1 and August 1 of each year, beginning on August 1, 2015, at a rate of 2.50% per year. The 2022 Notes mature on February 1, 2022, unless earlier converted or repurchased. The 2022 Notes are not redeemable prior to the maturity date. The 2022 Notes are convertible at an initial conversion rate of 93.2945 shares of our common stock per $1,000 principal amount of the 2022 Notes, subject to adjustment, which is equal to an initial conversion price of approximately $10.72 per share of common stock. Upon conversion, the 2022 Notes may be settled in shares of the Company’s common stock, together with a cash payment in lieu of delivering any fractional share. The conversion rate will be subject to adjustment in some events but will not be adjusted for any accrued and unpaid interest. In addition, following certain corporate events that occur prior to the maturity date, the Company will increase the conversion rate for a holder who elects to convert its 2022 Notes in connection with such a corporate event in certain circumstances. If the Company undergoes a fundamental change, which would include specified change of control transactions, or liquidation or dissolution or its common stock ceasing to be listed or quoted on specified national securities exchanges, and the fundamental change occurs prior to the maturity date of the 2022 Notes, holders of the 2022 Notes may require the Company to repurchase all or part of their 2022 Notes at a repurchase price equal to 100% of the principal amount of the 2022 Notes to be repurchased, plus accrued and unpaid interest to, but excluding, the fundamental change repurchase date.
137
Table of Contents
On January 29, 2015, the Company entered into Amendment No. 1 to Rights Agreement, originally entered into on December 15, 2005, with Computershare Inc., successor rights agent to Computershare Shareowner Services LLC (f/k/a Mellon Investor Services LLC), as rights agent. This Amendment is entered in connection with the 2022 Notes offering described above and accommodates the structure of a “blocker” in the 2022 Notes such that no holder of the 2022 Notes will be entitled to receive shares of the Company’s common stock deliverable upon conversion of the 2022 Notes to the extent that such receipt would cause the holder to become, directly or indirectly, the “beneficial owner” (as defined in the 2022 Notes) of more than 14.99% of the shares of the Company’s common stock outstanding at such time.
138