Attached files
| file | filename |
|---|---|
| EX-31.1 - EXHIBIT 31.1 - GenMark Diagnostics, Inc. | gen-20141231ex311.htm |
| EX-32.2 - EXHIBIT 32.2 - GenMark Diagnostics, Inc. | gen-20141231ex322.htm |
| EX-21.1 - EXHIBIT 21.1 - GenMark Diagnostics, Inc. | gen-20141231ex211.htm |
| EX-23.1 - EXHIBIT 23.1 - GenMark Diagnostics, Inc. | gen-20141231ex231.htm |
| EX-32.1 - EXHIBIT 32.1 - GenMark Diagnostics, Inc. | gen-20141231ex321.htm |
| EX-23.2 - EXHIBIT 23.2 - GenMark Diagnostics, Inc. | gen-20141231ex232.htm |
| EX-31.2 - EXHIBIT 31.2 - GenMark Diagnostics, Inc. | gen-20141231ex312.htm |
| EXCEL - IDEA: XBRL DOCUMENT - GenMark Diagnostics, Inc. | Financial_Report.xls |
| XML - IDEA: XBRL DOCUMENT - GenMark Diagnostics, Inc. | R9999.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
____________________________________________
FORM 10-K
____________________________________________
(Mark One) |
ý | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the year ended December 31, 2014
¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number: 001-34753
____________________________________________
GenMark Diagnostics, Inc.
(Exact name of registrant as specified in its charter)
____________________________________________
Delaware | 27-2053069 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
5964 La Place Court, Carlsbad, California | 92008-8829 | |
(Address of principal executive offices) | (Zip code) | |
Registrant’s telephone number, including area code: 760-448-4300
Securities registered pursuant to Section 12(b) of the Act
Title of Each Class: | Name of Each Exchange on which Registered: | |
Common Stock, par value $0.0001 per share | The NASDAQ Stock Market LLC (NASDAQ Global Market) | |
Securities registered pursuant to Section 12(g) of the Act: None
____________________________________________
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act of 1933, as amended. YES ¨ NO x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Securities Exchange Act of 1934, as amended. YES ¨ NO x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ¨ Accelerated filer x Non-accelerated filer ¨ Smaller reporting company ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
As of June 30, 2014, the last business day of the registrant’s most recent completed second quarter, the aggregate market value of the common stock held by non-affiliates of the registrant was approximately $518,324,000 based on the closing sale price for the registrant’s common stock on the NASDAQ Global Market on that date of $13.53 per share. This number is provided only for the purpose of this report on Form 10-K and does not represent an admission by either the registrant or any such person as to the status of such person.
The number of outstanding shares of the registrant’s common stock on February 20, 2015 was 41,881,986. The common stock is listed on the NASDAQ Global Market (trading symbol “GNMK”).
____________________________________________
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive Proxy Statement to be filed with the Securities and Exchange Commission within 120 days after the close of the fiscal year are incorporated by reference into Part III of this report.
TABLE OF CONTENTS
Page | ||
Item 1. | ||
Item 1A. | ||
Item 1B. | ||
Item 2. | ||
Item 3. | ||
Item 4. | ||
Item 5. | ||
Item 6. | ||
Item 7. | ||
Item 7A. | ||
Item 8. | ||
Item 9. | ||
Item 9A. | ||
Item 9B. | ||
Item 10. | ||
Item 11. | ||
Item 12. | ||
Item 13. | ||
Item 14. | ||
Item 15. | ||
1
Forward-Looking Statements
This Annual Report on Form 10-K, or Annual Report, particularly in Item 1. “Business” and Item 7. “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and the documents incorporated herein by reference, include forward-looking statements within the meaning of Section 27A of the Securities Act and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act. All statements, other than statements of historical fact, are statements that could be deemed to be forward-looking statements, including, but not limited to, statements regarding our future financial position, business strategy, research and development efforts, and plans and objectives of management for future operations. When used in this Annual Report, the words “believe,” “may,” “could,” “will,” “estimate,” “continue,” “intend,” “expect,” “target,” “anticipate,” “aim,” “plan” and similar expressions, including their use in the negative, are intended to identify forward-looking statements.
These forward-looking statements are based on current expectations, estimates, forecasts and projections about our business and the industry in which we operate and management’s beliefs and assumptions. They are not guarantees of future performance or development and involve known and unknown risks, uncertainties and other factors that are in some cases beyond our control. As a result, any or all of our forward-looking statements in this Annual Report may turn out to be inaccurate. Risks and other factors that may cause such differences include, but are not limited to, those described under the heading “Risk Factors” in Item 1A of Part I of this Annual Report.
In light of these risks, uncertainties and assumptions, actual results and timing of events could differ materially and adversely from those anticipated or implied in the forward-looking statements. Accordingly, readers are cautioned not to place undue reliance on such forward-looking statements.
Except as required by law, we do not intend to update these forward-looking statements publicly or to update the reasons actual results could differ materially from those anticipated in these forward-looking statements, even if new information becomes available in the future.
Trademarks and Trade Names
GenMark®, eSensor®, XT-8™ and ePlex™ and our other logos and trademarks are the property of GenMark Diagnostics, Inc. or its subsidiaries. All other brand names or trademarks appearing in this Annual Report are the property of their respective holders. Our use or display of other parties’ trademarks, trade dress or products in this Annual Report does not imply that we have a relationship with, or the endorsement or sponsorship of, the trademark or trade dress owners.
Use of External Estimates
This Annual Report includes market share and industry data and forecasts that we obtained from industry publications and surveys. Industry publications, surveys and forecasts generally state that the information contained therein has been obtained from sources believed to be reliable, but there can be no assurance as to the accuracy or completeness of included information. We have not independently verified any of the data from third-party sources nor have we ascertained the underlying economic assumptions relied upon therein. While we are not aware of any misstatements regarding the industry and market data presented herein, the data involve risks and uncertainties and are subject to change based on various factors.
2
PART I.
Item 1. | BUSINESS |
GenMark Diagnostics, Inc., or GenMark, is a molecular diagnostics company focused on developing and commercializing our proprietary eSensor® detection technology. References herein to “we,” “us” or “our” refer to GenMark Diagnostics, Inc. and its wholly owned subsidiaries, unless the context specifically requires otherwise.
Overview
Our proprietary electrochemical technology enables fast, accurate and highly sensitive detection of multiple distinct biomarkers in a single sample. Our XT-8 system received 510(k) clearance from the United States Food and Drug Administration, or FDA, and is designed to support a broad range of molecular research and diagnostic tests with a compact and easy-to-use workstation and disposable test cartridges. Our XT-8 system supports up to 24 independent test cartridges, each of which can be run independently, resulting in a highly convenient and flexible workflow for our target customers, which are primarily hospitals and reference laboratories. As of December 31, 2014, we had an installed base of 540 XT-8 analyzers, or placements, with our customers.
We have a menu of eight tests for use with our XT-8 system. Four of our diagnostic tests have received FDA clearance, our Cystic Fibrosis Genotyping Test, our Warfarin Sensitivity Test, our Thrombophilia Risk Test, and our Respiratory Viral Panel. In addition to these four FDA cleared tests, we have developed two hepatitis C virus, or HCV, genotyping tests, a 3A4/3A5 genotyping test, and a 2C19 genotyping test, versions of which are available for research use only (RUO).
In addition, we have recently completed the development stage of our ePlex system, which integrates automated nucleic acid extraction and amplification with our eSensor® detection technology to enable operators using the ePlex system to place a raw or a minimally prepared patient sample directly into our test cartridge and obtain results without any additional steps. This sample-to-answer capability is enabled by the robust nature of our eSensor® detection technology, which is not impaired by sample impurities that we believe hinder competing technologies. We have designed our ePlex system to further simplify workflow and provide powerful, cost-effective molecular diagnostics solutions to a significantly expanded group of hospitals and reference laboratories. We are currently developing seven assays for our ePlex system: which include gram-positive (GP) and gram-negative (GN) sepsis panels, a respiratory panel (RP), a gastrointestinal (GI) pathogen panel, an HCV genotyping test (HCVg), a central nervous system (CNS) panel and a fungal panel (FP). We intend to continue investing in our ePlex system and its related test menu for the foreseeable future. We expect to initiate the European launch of the system in the middle of 2015, and launch the system in the United States in the first half of 2016.
Since inception, we have incurred net losses from operations each year, and we expect to continue to incur losses for the foreseeable future. Our losses attributable to operations for the fiscal year ended December 31, 2014 and 2013 were approximately $38.3 million and $33.6 million, respectively. As of December 31, 2014, we had an accumulated deficit of $262.5 million. Our operations to date have been funded principally through sales of capital stock, borrowings and cash from operations. We expect to incur increasing expenses over the next several years, principally to develop and commercialize our ePlex system and additional diagnostic tests, as well as to further increase our manufacturing capabilities and domestic and international commercial organization.
3
Our Strategy
Our goal is to become the market leading provider of automated, multiplex molecular diagnostic testing systems. We intend to expand the use of our XT-8 system and diagnostic tests in the United States and expand our business globally with the commercialization of the ePlex system starting with its European launch in the middle of 2015. To achieve these objectives, we intend to:
• | Grow our Installed Base of Customers. We have identified those laboratories and hospitals that we believe will benefit from our eSensor technology. We intend to leverage our commercial organization to drive placements of our XT-8 system in the U.S. and our ePlex system both domestically and internationally. We anticipate that the expansion of our installed base of customers will drive sales of our test cartridges, from which we anticipate generating the majority of our revenues for the foreseeable future. |
• | Increase Utilization of Tests. We intend to increase the use of our diagnostic tests by developing and offering tools and support tailored to our products such as accredited physician education programs and seminars, product training for our customers and reimbursement support. These activities are designed to aid in establishing the clinical utility of multiplex molecular diagnostic tests, which we believe will increase adoption of our products. |
• | Launch our ePlex System. We have recently completed the development stage of our ePlex system. We believe the ePlex system will be attractive to a broader range of hospitals and laboratories that lack the technical or economic resources to perform molecular diagnostic testing with existing products and technology. We believe the ePlex system will expand our current target user base from approximately 1,000 domestic customers to approximately 12,000 potential customers globally. |
• | Expand our Menu of Clinical Diagnostic Products. We intend to develop a broad menu of molecular diagnostic tests for our ePlex system that we believe will satisfy important medical needs and present attractive commercial opportunities. We are currently developing seven assays for our ePlex system and anticipate continuing to invest in expanding our assay test menu for the foreseeable future. |
• | Execute International Expansion and Explore Out-Licensing Opportunities. We plan to offer our molecular diagnostic products in European and other international markets commencing in 2015. We intend to utilize a direct sales and technical support team in certain key European countries, which we expect will be augmented by marketing partners and distributors in other geographies as we expand internationally. We also intend to explore opportunities to leverage our intellectual property position in detection technologies through licensing or the establishment of partnerships. |
Revenues, net loss and total assets for the past three years are contained in our consolidated financial statements in Part II of this Annual Report. Substantially all revenues for the periods reported in our consolidated financial statements in Part II of this Annual Report were derived from customers located within the United States.
Our Technology
Our eSensor® Technology
Our proprietary eSensor technology is based on the principles of DNA hybridization and electrochemical detection. DNA naturally forms a double-stranded structure, with each strand binding with high affinity, or hybridizing, only to a complementary strand. Our technology takes advantage of this highly specific binding by first creating two types of single-stranded DNA, the capture probe and the signal probe. The capture probe and signal probe are each complementary to a different segment of the target DNA that is the focus of the particular diagnostic test. Using our proprietary technology and processes, we attach our capture probes to a proprietary monolayer on the surface of a gold electrode within our proprietary test cartridges. We separately attach ferrocene, a proprietary label, to our signal probes.
Before placing the sample into our XT-8 test cartridge, the technician mixes the amplified DNA sample with our signal probe. If the target biomarker is present in the prepared patient sample, a segment of the biomarker DNA will hybridize with a solution containing our signal probe. This solution is then run past an electrode, against which our capture probes have been immobilized. The as-yet unbound segment of the target biomarker binds to our capture probe, creating a target DNA, signal probe, capture probe complex at the surface of the electrode. This complex produces an electrochemical signal which is analyzed and interpreted by our XT-8 system.
4
With our ePlex sample-to-answer test cartridges, the operator adds a patient sample directly or with minimal preparation into the sample chamber, closes the lid, and inserts the test cartridge into the ePlex instrument. Within the instrument, the same steps performed by a technician with the XT-8 system are performed within the ePlex test cartridge, resulting in delivery of target DNA and signal probes to the eSensor electrodes within the ePlex cartridge. As with XT-8, when a complex forms as a result of a target match, the complex produces an electrochemical signal that is interpreted by the ePlex system.
Our XT-8 and ePlex test cartridges utilize the combination of distinct electrodes and multiple signal probes to detect more than 200 target biomarkers from a single sample, enabling highly multiplex testing. Our eSensor® technology is highly specific for the target biomarker, and is not based on optical or fluorescent detection. As a result, our diagnostic tests are less prone to sample contamination risk and do not require many of the time-consuming washing and preparation steps required by competing technologies. The sample preparation steps required before using our XT-8 test cartridges are DNA/RNA purification and a polymerase chain reaction, or PCR, amplification, which involves amplifying, or generating billions of copies of, the target DNA molecules, followed by transfer of the sample to our test cartridge and insertion of the test cartridge into any open slot in our XT-8 system. In some tests, amplified DNA is subject to an additional enzymatic treatment to produce a single-stranded-DNA. In contrast, the ePlex system generally requires no pre-analytic steps to be performed by the user, except, in limited cases, minimal up-front sample handling is required.
We believe our proprietary electrochemical signal detection technology has several advantages over other signal detection platforms, including:
Key Advantages of eSensor | Description |
Robust Signal | Our capture probes are highly target specific, reducing the binding of non-target DNA which minimizes potential interference from components in a patient’s sample, such as blood, saliva or urine. |
High Sensitivity and Accuracy | Each electrode can routinely detect approximately one nanomolar of target DNA, and a sensitivity of 10 picomolar of target DNA has been achieved. Such concentrations are readily produced from patient samples using several commercially-validated amplification technologies such as PCR. |
Streamlined Sample Preparation | Our technology directly detects the target DNA sequence with highly specific signal probes and electrode-bound capture probes without the need for many of the washing steps typically required to remove unbound target DNA and labels. |
Efficient Multiplexing | Each of the electrodes in our test cartridge acts independently of the others and produces a comprehensive and informative signal eliminating the need for averaging of multiple measurements commonly required by competing technologies. |
Small Footprint with Low Maintenance | Our eSensor® technology enables our systems to be low cost and have relatively few moving parts compared to competitive systems. Our XT-8 system, for example, requires no calibration and minimal maintenance, with a small footprint of approximately 16-by-16 inches in its standard configuration. |
Cost-Effective Development of New Tests | The use of electrochemical technology allows our systems to leverage third-party advances in microelectronics such as miniaturization and manufacturing efficiencies. Many electronic components associated with our core processes are produced in large volumes at low cost and size for use in numerous fields. Our technology is also highly flexible which allows us to add new diagnostic tests to our menu or to add new content to existing diagnostic tests without modifying the system platform. |
Functionality Outside of Molecular Diagnostics | Our eSensor® technology has broad applicability to detect a range of biomolecules including proteins and small molecule drugs. This versatility opens the possibility of developing mixed analyte sensors, such as tests that can detect antibodies to a certain pathogen plus the pathogen itself. |
Digital Microfluidics
Digital microfluidics is another technology included within our ePlex system which we have exclusively licensed within a defined field of use from Advanced Liquid Logic Inc., or ALL, an affiliate of Illumina, Inc. Digital microfluidics is a technique for moving small droplets of liquid using electrowetting, a process for making a surface hydrophobic or hydrophilic based on the application of a voltage to a surface. The ePlex printed circuit board contains eSensor electrodes capable of nucleic acid detection along with electrowetting electrodes capable of digital microfluidics. The ePlex system uses numerous choreographed digital inputs to perform the fluid manipulations associated with sample-to-answer molecular diagnostics. Drops are dispensed, mixed, merged, heated, cooled, split and delivered, all under precise and programmable digital control. In
5
this manner, standard procedures of the molecular diagnostics lab (e.g., DNA purification, polymerase chain reaction, exonuclease digestion etc.) can be performed automatically within our ePlex cartridge.
Our Platforms
Our XT-8 Test Cartridges. Our XT-8 test cartridges are devices specifically configured for a particular diagnostic test to be used within our XT-8 instrument. Each test cartridge includes a sample compartment and a plastic cover that forms a hybridization chamber. The test cartridge is fitted with a diaphragm pump and valves that circulate the hybridization solution, including the signal probe and prepared patient sample, when inserted into the XT-8 system. The test cartridge also includes a printed circuit board consisting of an array of gold-plated electrodes, a silver/silver chloride reference electrode, and two auxiliary electrodes. Each electrode is customized with a proprietary monolayer that immobilizes the DNA capture probes specific for each target of a test panel. The test cartridge also contains an electrically erasable, programmable read-only memory (EEPROM) component that stores information related to the cartridge such as an assay identifier, cartridge lot number and expiration date.
Our XT-8 Instrument. Our XT-8 instrument is a multiplex workstation that has a modular design consisting of an integrated touch screen and up to three analyzers. Each analyzer contains eight modules into which individual test cartridges are placed. The test cartridge slots operate independently of each other allowing up to 24 independent test cartridges to be loaded at one time, with the remaining slots available for use at any future time while the system is running. Each slot contains a test cartridge connector, a precision-controlled heater, an air pump and electronics. The air pumps drive the diaphragm pump and valve system in the test cartridge, eliminating fluid contact between the system and the cartridge. The pneumatic pumping enables recirculation of the hybridization solution allowing the target DNA and the signal probes to efficiently hybridize with the complementary capture probes on the electrodes. The diaphragm pump in the test cartridge is connected to a pneumatic source from the XT-8 system and provides unidirectional pumping of the hybridization mixture through the cartridge during hybridization.
We believe that our XT-8 system and related diagnostic tests offer reference laboratories and hospitals the following benefits:
Key Features & Benefits | Description |
Broad Test Menu | Eight tests available on our XT-8 system including 4 FDA cleared assays across a number of areas in molecular diagnostics. |
Fast Turnaround | Approximately 30 minutes to result from amplified DNA sample with minimal technician time needed. |
Accurate Results | Our Cystic Fibrosis Genotyping Test, our Warfarin Sensitivity Test and our Thrombophilia Risk Test demonstrated 100% accuracy in clinical studies compared to DNA sequencing and other standards. |
Ease of Use | Minimal manual processing steps, intuitive touch-screen interface and clear result reports. |
Random Access | Each of up to 24 test cartridge slots can be accessed independently resulting in a highly flexible workflow. |
Minimal Maintenance | No routine maintenance or calibration required. |
Multiplex Capability | Detects numerous distinct biomarkers in a single sample reducing the need for reflex testing. |
Our ePlex Sample-To-Answer Test Cartridges. Our ePlex test cartridges are single use, fully-integrated consumables containing all necessary reagents for a particular diagnostic test to be used within our ePlex instrument. Each test cartridge includes a sample compartment with a plastic cover into which a raw or minimally prepared sample is placed. The test cartridge contains liquid reagents in proprietary blister packs, optimized for long term reagent storage and delivery into the appropriate reagent reservoirs. The test cartridge also contains biologic reagents such as enzymes and oligonucleotides, stabilized with proprietary additives designed to support long term room temperature stability and dried onto various internal surfaces where they can be rehydrated for use during the sample-to-answer diagnostic test. The ePlex test cartridge also includes a printed circuit board consisting of an array of gold-plated electrodes. Using the same eSensor technology contained in XT-8 cartridges, each electrode is similarly customized with a proprietary monolayer that immobilizes the DNA capture probes specific for each target of a test panel. The ePlex test cartridge also contains an EEPROM that stores information related to the cartridge such as an assay identifier, cartridge lot number and expiration date. The ePlex EEPROM and associated workflow and programming also supports positive patient identification, helping to ensure error-free linking of the ePlex test report to the correct patient.
6
Our ePlex Instrument. Our ePlex instrument is a multiplex platform that has a modular design consisting of an integrated touch screen and up to four analyzers. Each tower contains six bays into which individual ePlex sample-to-answer test cartridges are placed. The test cartridge bays operate independently supporting continuous random access. Each bay contains a test cartridge connector, several precision-controlled heaters, a mix motor, an air pump and mechanisms for dispensing liquid from blister packs and positioning a small magnet. Each bay also contains all of the necessary electronics hardware and firmware for controlling the bay, performing real-time monitoring, calibration testing and generating test results. The air pump, blister/magnet mechanisms and motor drive the nucleic acid extraction and reagent dispensing initial phase of the sample-to-answer diagnostic tests and avoid fluid contact between the ePlex bay and the cartridge. All subsequent steps are driven by a proprietary electrowetting technology.
We believe our ePlex system and related diagnostic tests offer reference laboratories and hospitals the following benefits:
Key Features & Benefits | Description |
Broad Test Menu | Seven tests are in development on our ePlex system. |
Ease of Use | Minimal manual processing steps, intuitive touch-screen interface and clear result reports. |
True Sample-to-Answer | The user simply adds raw or minimally prepared sample to the test cartridge and inserts the cartridge into the instrument. The test cartridge is self-contained, with all required reagents stored in room-temperature stable formats. |
Fast Turnaround | Results are produced in approximately 60-90 minutes from sample input. |
Random Access | Each of up to 24 test cartridge bays can be accessed independently resulting in a highly flexible workflow. |
Minimal Routine Maintenance | Minimal maintenance or calibration required. |
Multiplex Capability | Detects numerous distinct biomarkers in a single sample reducing the need for reflex testing. |
License Agreements
California Institute of Technology. We have a license from the California Institute of Technology to patents and patent applications related to nucleic acid-mediated electron transfer technology. We license certain of these patents on an exclusive basis. The license grant is worldwide, fully paid-up, and extends until the last of the underlying patents expires. The agreement is also conditioned on us paying all associated patent maintenance and prosecution fees. Either party may terminate the license agreement upon a material breach by the other party, subject to a cure period. We may terminate the license agreement for any reason upon 60 days written notice.
Roche Molecular Systems, Inc. We have a non-exclusive license from Roche Molecular Systems, Inc. to utilize a form of chemically modified thermostable DNA polymerase that is a component in some of our commercial products. We paid a one-time upfront fee for this license and are obligated to pay quarterly running royalties on net sales. The agreement remains in effect until the last to expire of the underlying patents. Either party may terminate the license agreement upon a material breach of the license agreement by the other party, subject to a cure period, or upon the filing for bankruptcy of the other party.
Caliper Life Sciences Inc. In March 2012, we entered into a license agreement with Caliper Life Sciences Inc., or Caliper, pursuant to which we obtained a non-exclusive license under Caliper’s microfluidics patent portfolio. In consideration for the license, we agreed to pay Caliper certain up-front and sales-based milestone payments, as well as a royalty on the sale of certain products. In addition, we obtained an unconditional release from any and all claims based upon any alleged infringement of the licensed patents prior to the effective date of the agreement.
Advanced Liquid Logic, Inc. (ALL) In July 2012, we entered into a development collaboration and license agreement with ALL, which was acquired by Illumina. in July 2013. Under the terms of the agreement, we established a collaborative program to develop in-vitro diagnostic products incorporating ALL’s proprietary electro-wetting technology in conjunction with our electrochemical detection. We paid ALL an upfront license payment of $250,000 and agreed to pay up to $1,750,000 in potential additional milestone payments. Pursuant to the agreement, the parties agreed to enter into a supply agreement relating to the manufacture and supply of certain ALL components. The agreement also provides that we would, upon the occurrence of certain events, be obligated to pay to ALL a royalty consisting of a low- to mid-single digit percent of net sales of designated licensed products containing ALL components which we manufacture or are otherwise not manufactured and supplied by ALL.
7
Market Opportunity
We believe the global market for molecular diagnostics is currently approximately $5.0 billion and will experience a growth rate of approximately 15% per year over the course of the next several years based on research published by leading market research firms. Although we believe the global market for molecular diagnostics is approximately $5.0 billion, we believe the aggregate global market for the tests we currently offer, are actively developing on ePlex, or will consider developing in the near term is approximately $2.3 billion and is expected to grow by more than 20% per year for the next several years. Many factors are driving growth of this market, including increased demand for infectious disease diagnostics panels, the expansion of genetic testing for disease predisposition, and advances in personalized medicine, such as the tailoring of therapies to those individuals most likely to respond.
Research and Development
In addition to developing our ePlex system and expanding our diagnostic test menus, our research and development team is focused on the following initiatives:
• | On Market Product Support. A role of our research and development team is to assist our manufacturing and quality assurance teams in ensuring high product quality and thorough complaint handling and investigation. In addition, we work to improve quality control methods and metrics and are active participants in the continuous improvement processes utilized by our product manufacturing teams. |
• | Improving the Clinical and Practical Utility of our Tests. Our research and development organization also helps establish the clinical utility and value of our molecular diagnostic tests. We have previously and intend to continue to partner with academic and reference laboratories to perform validation and clinical studies on our tests. Key aspects of our efforts are aimed at improving workflow in the laboratory setting, positively comparing our tests to historical or “gold standard” tests and demonstrating that our tests can help improve patient care and lower diagnostic and medical treatment costs. We intend to publish the results from these clinical studies in peer-reviewed or trade journals, submit them to regulatory bodies and present them at industry conferences in support of our commercialization strategy. |
• | Developing New Test Capabilities. We may develop capabilities for utilizing our eSensor® technology in protein and small molecule detection, through research collaborations or otherwise. These capabilities may enhance our future menu offerings or provide us with out-licensing opportunities. |
Manufacturing
We manufacture our proprietary test cartridges and ancillary reagents at our headquarters in Carlsbad, California. We perform reagent formulation, test cartridge manufacturing and packaging of final components and test cartridges in accordance with applicable guidelines for medical device manufacturing. We currently lease an approximately 53,000 square foot office and manufacturing facility to support our manufacturing operations.
We outsource manufacturing of our XT-8 instrument to Leica Biosystems Melbourne Pty Ltd., or Leica. We also intend to outsource the manufacture of our ePlex instrument. We rely on third party suppliers, including in certain instances, sole source suppliers, for oligonucleotide and other raw materials used in our products and much of the disposable component molding and sub-component assembly for our test cartridges.
We have implemented a quality management system designed to comply with FDA regulations and ISO standards governing diagnostic medical device products. These regulations control the design, manufacture, testing and release of diagnostics products, as well as raw material receipt and control. In 2012, our Carlsbad, California facility obtained ISO 13485 certification. We also have controlled methods for the consistent manufacturing of our proprietary test cartridges and reagents at our facilities. Our key outsourcing partners are regularly audited to help ensure a continual supply of high quality components.
We plan to continue to manufacture components that we determine are highly proprietary or highly customized, while outsourcing more commodity-like components. We are likely to establish additional outsourcing partnerships as we manufacture additional products.
Sales and Marketing
Our current sales and marketing strategy is to continue to expand the placement and use of our XT-8 system and diagnostic tests in the United States and expand our business globally with the commercialization of the ePlex system starting
8
with its European launch in the middle of 2015. Our products are sold in the United States through a geographically dispersed direct sales and technically specialized service organization, which is supported by a centralized team of product managers and marketing, customer support, and technical support personnel. We intend to utilize a direct sales and technical support team in certain key European countries, which we expect will be augmented by marketing partners and distributors in other geographies as we further expand internationally.
Our sales representatives typically have experience in molecular diagnostics and a network of laboratory contacts within their respective territories. We utilize our representatives’ knowledge along with market research databases to target and qualify our customers. We execute a variety of sales campaigns and strategies to meet the buying criteria of the different customer segments we serve. To support our expanding molecular test menu, growth in our customer base and our launch plans for our ePlex system, we continue to make investments in these customer facing organizations.
Our sales cycle typically includes customer evaluations and validations of our products. Upon successful validation, a customer may generally acquire our XT-8 system and consumables in the following ways:
• | Reagent Rental: A reagent rental agreement requires a customer commitment to purchase a minimum number of test cartridges over the term of the agreement, and a portion of the charge for each cartridge equates to a usage fee for the equipment. Our reagent rental agreements do not typically provide for any cancellation rights by the customer. |
• | Capital Purchase: The XT-8 instrument is paid for upfront and in its entirety by the customer. Customers are also eligible to receive structured pricing incentives if they enter into an optional annual minimum cartridge commitment. |
Customers
In 2014, no single customer represented more than 10% of our total revenue. In 2013, Natural Molecular Testing Corporation, or NMTC, represented approximately 30% of our total revenue. NMTC filed for bankruptcy in October 2013.
Instrument placements are defined in terms of the number of analyzers sold to or placed with a customer, reflecting a direct correlation between the reagent test revenue opportunity and the number of test cartridges that can be analyzed at any one time. As of December 31, 2014, we had placed 540 XT-8 analyzers at 204 unique customer sites, or approximately 2.6 analyzers per customer. This compares with 413 analyzer placements at 173 unique customer sites, or approximately 2.4 analyzers per customer, as of December 31, 2013.
The increase in analyzers placed and related revenue generated in 2014 over the prior year is due to an increase in the number of new customers buying our products and growth in the sale of consumables to existing customers.
Competition
We primarily face competition in the molecular diagnostic testing markets with testing products and systems developed by public and private companies such as Cepheid, Siemens, Hologic, Inc., Luminex Corporation, Nanosphere, Inc., Seegene, Roche Diagnostics, a division of F. Hoffmann-La Roche Ltd., bioMerieux (which recently acquired Biofire Diagnostics, Inc.) and Abbott Molecular Diagnostics, a division of Abbott Laboratories. Our diagnostic tests also face competition with laboratory developed tests, or LDTs, developed by national and regional reference laboratories and hospitals. We believe that our testing systems compete largely on the basis of accuracy and reliability, enhanced laboratory workflow, multiplex capability, ease-of-use and return on investment for customers.
Many of our competitors have substantially greater financial, technical, research and other resources and larger, more established marketing, sales and distribution organizations than we do. Many of our competitors also offer broader product lines and have greater brand recognition than we do. Moreover, our existing and new competitors may make rapid technological developments that may result in our technologies and products becoming obsolete before we recover the expenses incurred to develop them or before they generate significant revenue.
Intellectual Property
To establish and protect our proprietary technologies and products, we rely on a combination of our patents, copyrights, trademarks, and trade secrets, as well as other intellectual property rights in our technology and business information. Our intellectual property portfolio for our core electrochemical technology was initially built through the combination of our
9
acquisition of the Clinical Micro Sensors business from Motorola and licensing patents from the California Institute of Technology.
We believe that our patent portfolio, which includes approximately 130 owned and exclusively licensed U.S. and foreign patents and approximately 25 pending applications, provides us with robust protection of our electrochemical detection techniques, chemical insulators and attachment points on electrode surfaces and other technology that, collectively, form the staple of our eSensor® platform. We continue to pursue the issuance of new patents to protect our ongoing research, development and commercial activities, including with respect to our ePlex system and related consumables. In general, patents have a term of at least 20 years from the application filing date or earlier claimed priority date. A majority of our issued and exclusively licensed patents are scheduled to expire by 2021, with approximately one half of the patents expiring by 2018. Several of our pending applications have the potential to mature into patents that may expire between 2028 and 2034. Our success depends to a significant degree upon our ability to police infringement, derive licensing revenues and continue to develop proprietary products and technologies without infringing the intellectual property rights of others.
We also rely in part on trade-secret protection of our intellectual property. We attempt to protect our trade secrets by entering into confidentiality agreements with third parties, employees and consultants. Our employees and consultants also sign agreements requiring that they assign to us their interests in intellectual property, such as patents and copyrights arising from their work for us. All employees sign an agreement not to compete unfairly with us during their employment and upon termination of their employment through the misuse of confidential information.
We also have filed for registration, or obtained registration, in the U.S. and other countries for marks used with our products and technology. Our issued and pending trademark registrations in the U.S. include eSensor®, GenMark®, GenMark DX®, ePlex™ and GenMark ePlex™, among others.
Trademark protection continues in some countries for as long as the mark is used and, in other countries, for as long as it is registered. Registrations generally are for fixed but renewable terms.
Government Regulation
The design, development, manufacture, testing and sale of our molecular diagnostic products are subject to regulation by numerous governmental authorities, principally the FDA, and corresponding state and foreign regulatory agencies.
Regulation by the FDA
In the United States, the Federal Food, Drug, and Cosmetic Act, or FDCA, FDA regulations and other federal and state statutes and regulations govern, among other things, medical device design and development, preclinical and clinical testing, premarket clearance or approval, registration and listing, manufacturing, labeling, storage, advertising and promotion, sales and distribution, export and import, and post-market surveillance. The FDA regulates the design, manufacturing, servicing, sale and distribution of medical devices, including molecular diagnostic test kits and instrumentation systems. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending applications, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution.Unless an exemption applies, each medical device we wish to distribute commercially in the United States will require marketing authorization from the FDA prior to distribution.
The two primary types of FDA marketing authorization required applicable to a device are premarket notification, also called 510(k) clearance, and premarket approval, also called PMA. We have obtained 510(k) clearance from the FDA for the following molecular diagnostic tests on our XT-8 system: the eSensor ® Warfarin Sensitivity Test, the Cystic Fibrosis Genotyping Test, the Thrombophilia Risk Test, and the Respiratory Viral Panel.
Proposed Regulation of Labortatoy Developed Tests (LDTs). In October 2014, the FDA promulgated draft guidance which describes a new proposed regulatory framework for LDTs. Based on this proposal, clinical laboratories that develop and use LDTs would be required to comply with specific regulatory requirements (e.g., adverse even reporting, quality system regulation, premarket submission, and FDA review) prior to the use of LDTs for clinical diagnostic purposes. The timeline for phasing in the proposed regulatory requirements would begin upon finalization of the FDA guidance document. The ultimate impact of this draft guidance on our customers remains uncertain.
Regulation after FDA Clearance or Approval. Any devices we manufacture or distribute pursuant to clearance or approval by the FDA are subject to pervasive and continuing regulation by the FDA and certain state agencies. We are required to adhere to applicable regulations setting forth detailed GMP requirements, as set forth in the QSR, which includes testing, control and documentation requirements. Non-compliance with these standards can result in fines, injunctions, civil penalties,
10
recalls or seizures of products, total or partial suspension of production, refusal of the government to grant 510(k) clearance or PMA of devices, withdrawal of marketing approvals and criminal prosecutions. We have designed and implemented quality system processes within our manufacturing facilities in order to comply with FDA’s GMP requirements.
Because we are a medical device manufacturer, we must also comply with FDA’s medical device reporting requirements whenever there is evidence that reasonably suggests that one of our products may have caused or contributed to a death or serious injury. We must also report any incident in which our product has malfunctioned if that malfunction would likely cause or contribute to a death or serious injury if it were to recur.
Labeling, advertising, and promotional activities are subject to scrutiny by the FDA and, in certain circumstances, by the Federal Trade Commission. Medical devices approved or cleared by the FDA may not be promoted for unapproved or uncleared uses, otherwise known as “off-label” promotion. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including substantial monetary penalties and criminal prosecution. We have implemented quality system processes and advertising/promotional policies designed to comply with these requirements.
Environmental Regulations. We are also subject to numerous federal, state and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control and disposal of hazardous or potentially hazardous substances. Some of these laws require us to obtain licenses or permits to conduct our operations. We have numerous policies and quality system procedures in place to ensure compliance with these laws and to minimize the risk of occupational exposure to hazardous materials. We do not expect the operations of our products to produce significant quantities of hazardous or toxic waste or radiation that would require the use of extraordinary disposal practices. Although the costs to comply with these applicable laws and regulations have not been material, we cannot predict the impact on our business of new or amended laws or regulations or any changes in the way existing and future laws and regulations are interpreted or enforced, nor can we ensure we will be able to obtain or maintain any required licenses or permits.
Export of Our Products. Medical devices that are legally marketed in the U.S. may be exported anywhere in the world without prior FDA notification or approval. Devices that have not been approved or cleared in the U.S. must follow the export provisions of the FDCA. Depending on which section of the FDCA we may export under, we may need to request an export permit letter or export certificate, or we may need to submit a simple notification. Export certificates may be requested by foreign customers or foreign governments to provide proof of the products’ status as regulated by the FDA. The export certificate is prepared by FDA and contains information about a product’s regulatory or marketing status in the United States.
Clinical Laboratory Improvement Amendments of 1988. The use of our products is also affected by the Clinical Laboratory Improvement Amendments of 1988, or CLIA, and related federal and state regulations, which provide for regulation of laboratory testing. Any customers using our products for clinical use in the United States will be regulated under CLIA, which establishes quality standards for all laboratory testing to ensure the accuracy, reliability and timeliness of patient test results regardless of where the test was performed. In particular, these regulations mandate that clinical laboratories must be certified by the federal government or a federally approved accreditation agency, or must be located in a state that has been deemed exempt from CLIA requirements because the state has in effect laws that provide for requirements equal to or more stringent than CLIA requirements. Moreover, these laboratories must meet quality assurance, quality control and personnel standards, and they must undergo proficiency testing and inspections. The CLIA standards applicable to clinical laboratories are based on the complexity of the method of testing performed by the laboratory, which range from “waived” to “moderate complexity” to “high complexity.” We expect that most of our products will be categorized as “high complexity,” since most molecular diagnostic tests are currently FDA-cleared as CLIA “high complexity” devices.
Foreign Government Regulation. We intend to market our products in European and other select international markets. The regulatory pre-market requirements for IVD devices vary from country to country. Some countries impose product standards, packaging requirements, labeling requirements and import restrictions on devices. Each country has its own tariff regulations, duties and tax requirements. Failure to comply with applicable foreign regulatory requirements may subject us to fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Fraud and Abuse Regulations
We are subject to numerous federal and state health care anti-fraud laws, including the federal anti-kickback statute and False Claims Act that are intended to reduce waste, fraud and abuse in the health care industry. These laws are broad and subject to evolving interpretations. They prohibit many arrangements and practices that are lawful in industries other than health care, including certain payments for consulting and other personal services, some discounting arrangements, the provision of gifts and business courtesies, the furnishing of free supplies and services, and waivers of payments. In addition,
11
many states have enacted or are considering laws that limit arrangements between medical device manufacturers and physicians and other health care providers and require significant public disclosure concerning permitted arrangements. These laws are vigorously enforced against medical device manufacturers and have resulted in manufacturers paying significant fines and penalties and being subject to stringent corrective action plans and reporting obligations. We must operate our business within the requirements of these laws and, if we were accused of violating them, we could be forced to expend significant resources on investigation, remediation and monetary penalties.
Patient Protection and Affordable Care Act
Our operations are affected by the federal Patient Protection and Affordable Care Act of 2010, as modified by the Health Care and Education Reconciliation Act of 2010, which we refer to as the Health Care Act. The Health Care Act imposes a 2.3% excise tax on sales of medical devices by manufacturers. Taxable devices include any medical device defined in section 201(h) of the FDCA and intended for use by humans, with limited exclusions for devices purchased by the general public at retail for individual use. There is no exemption for small companies, and we began paying the tax in January 2013. The Health Care Act also requires manufacturers to report to the Department of Health and Human Services detailed information about financial arrangements with physicians and teaching hospitals. These reporting provisions preempt state laws that require reporting of the same information, but not those that require reports of different or additional information. Failure to comply subjects the manufacturer to significant civil monetary penalties.
Employees
As of December 31, 2014, we had 195 employees. Approximately 105 employees were involved in research and development, 26 in operations, manufacturing and quality assurance, 42 in sales and marketing, and 22 in general and administrative functions. Our success will depend in large part upon our ability to attract and retain employees. We face competition in this regard from other companies, research and academic institutions, government entities and other organizations. None of our employees are covered by a collective bargaining agreement.
Corporate and Available Information
Our principal corporate offices are located at 5964 La Place Court, Carlsbad, California.
We make available, free of charge, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and any amendments to those reports, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities and Exchange Commission, or the SEC. We also make these documents and certain public financial information available on our website, which is www.genmarkdx.com. Our SEC reports and other financial information can be accessed through the investor relations section of our website. Some of the information found on our website is not part of this or any other report we file with or furnish to the SEC.
Item 1A. | RISK FACTORS |
You should consider each of the following factors as well as the other information in this Annual Report in evaluating our business and our prospects. The risks and uncertainties described below are not the only ones we face. If any of the following risks actually occur, our business and financial results could be harmed. In that case, the trading price of our common stock could decline. You should also refer to the other information set forth in this Annual Report, including our financial statements and the related notes.
We may not successfully commercialize our ePlex system and its related test menu.
Our current plan for achieving positive cash flow and our future growth projections relies upon the successful development and commercialization of our ePlex system and its related test menu.We have designed our ePlex system to integrate automated nucleic acid extraction and amplification with our eSensor® technology to allow operators to place raw or minimally prepared patient samples directly into our test cartridges and obtain clinically relevant results. Based on extensive market research and input received from our customers, we believe that our ePlex system will offer advantages over competitive systems, including in respect of multiplexing capability, reduced hands-on processing time, testing capacity and flexibility, and other factors. However, the commercial success of ePlex will depend on a number of factors, including, but not limited to, timely market introduction, overall market acceptance, our ability to offer a broad test menu at a competitive price, our ability to manage the risks associated with scaling up new product production, the effective management of purchase and supply commitments and inventory levels in line with anticipated product demand, the availability of products in appropriate quantities and at expected costs to meet anticipated demand and drive market adoption, and the risk that new products may not have the level of quality in the early stages of introduction that our customers expect. If we are unsuccessful in effectively
12
commercializing our ePlex system within our expected time frame, or at all, our business and future prospects may be adversely affected.
We and our key suppliers may have difficulties scaling manufacturing operations for our anticipated future growth.
To date, we have produced our products in limited quantities relative to the quantities necessary to achieve our desired revenue growth. Developing the necessary manufacturing and quality procedures internally and in conjunction with our key suppliers for a significant number of newly developed, unique products is a complex process. We or our suppliers may not be prepared to produce sufficient quantities of, or may have difficulty maintaining consistency and quality among, our products. If we or our key suppliers encounter difficulties in scaling manufacturing operations as a result of, among other things, process transfer complexities, quality control and quality assurance issues, and availability of components and raw material supplies, our reputation may be harmed and we may not achieve our anticipated financial results within the time frame we expect, or at all.
To manage our anticipated future growth effectively, we must enhance our manufacturing and supply chain capabilities and operations, information technology infrastructure, and financial and accounting systems and controls. Organizational growth and scale-up of operations could strain our existing managerial, operational, financial and other resources. If our management is unable to effectively prepare for our expected future growth, our expenses may increase more than anticipated, our revenue could grow more slowly than expected, and we may not be able to achieve our commercialization goals. Our failure to effectively implement the necessary processes and procedures and otherwise prepare for our anticipated growth could have a material adverse effect on our future financial condition and prospects.
Our financial results will depend on the acceptance and increased demand among reference laboratories, hospitals and the medical community of our molecular diagnostic technology and products.
Our future success depends on the belief by our target customers and the medical community that our molecular diagnostic products are a reliable, medically-relevant, accurate and cost-effective replacement for other diagnostic testing methods. Medical offices and many hospitals outsource their diagnostic testing needs to national or regional reference laboratories. Our business success depends on our ability to convince these target laboratories and hospitals to perform these tests internally with our products if they have historically outsourced their testing needs or have historically used non-molecular methods to perform such testing, or to replace their current molecular testing platforms with our system and its related test offerings.
Many other factors may affect the market acceptance and commercial success of our molecular diagnostic technology and products, including:
• | the relative convenience, ease of use, accuracy, scalability, and time-to-result of our diagnostic products over competing products; |
• | the introduction of new technologies and competing products that may make our technologies and products a less attractive solution for our target customers; |
• | the breadth of our menu of available diagnostic tests relative to our competitors; |
• | our success in training reference and hospital-based laboratories in the proper use of our products; |
• | the acceptance in the medical community and key opinion leaders of our molecular diagnostic technology and products; |
• | the extent and success of our marketing and sales efforts; and |
• | general economic conditions. |
Professional societies, government agencies, practice management groups, private health/science foundations and organizations involved in healthcare issues may publish guidelines, recommendations or studies for the healthcare and patient communities. Recommendations of government agencies or these other organizations may relate to such matters as cost-effectiveness and use of related products. Organizations like these have in the past made recommendations about our competitors’ products, such as the need for less frequent screening tests, which could result in reduced product sales. Moreover, the perception by the investment community or stockholders that recommendations, guidelines or studies will result in decreased use of our products could adversely affect the prevailing market price for our common stock.
13
Disruptions in the supply of raw materials, consumable goods or other key product components, or issues associated with their quality from our single source suppliers, could result in delays or difficulties successfully launching our ePlex system or a significant disruption in sales and profitability.
We must manufacture or engage third parties to manufacture components of our products in sufficient quantities and on a timely basis, while maintaining product quality, acceptable manufacturing costs and complying with regulatory requirements. Our instrument systems and certain critical components are custom-made by only a few outside suppliers. In certain instances, we and our customers have a sole source supply for certain key products, product components and ancillary items used to run our tests. If we are unable to satisfy our forecasted demand from existing suppliers for our products, or we or our customers are unable to find alternative suppliers for key product components or ancillary items at reasonably comparable prices, it could have a material adverse effect on our financial condition and results of operations. Additionally, we have entered into supply agreements with most of our suppliers of strategic reagents and parts to help ensure component availability and flexible purchasing terms with respect to the purchase of such components. If our suppliers discontinue production of a key component for one or more of our products, we may be unable to identify or secure a viable alternative on reasonable terms, or at all, which could limit our ability to manufacture our products.
In determining the required quantities of our products and the manufacturing schedule, we must make significant judgments and estimates, based on seasonality, inventory levels, current market trends and other related factors. Because of the inherent nature of estimates and our limited experience in marketing our products, there could be significant differences between our estimates and the actual amounts of products we require. This can result in shortages if we fail to anticipate demand, or excess inventory and write-offs if we order more than we need.
Reliance on third-party manufacturers entails risk to which we would not be subject if we manufactured these components ourselves, including:
• | reliance on third parties for regulatory compliance and quality assurance; |
• | possible breaches of manufacturing agreements by the third parties because of factors beyond our control; |
• | possible regulatory violations or manufacturing problems experienced by our suppliers; |
• | possible termination or non-renewal of agreements by third parties, based on their own business priorities, at times that are costly or inconvenient for us; |
• | the potential obsolescence and/or inability of our suppliers to obtain required components; |
• | the potential delays and expenses of seeking alternate sources of supply or manufacturing services; |
• | the inability to qualify alternate sources without impacting performance claims of our products; |
• | reduced control over pricing, quality and timely delivery due to the difficulties in switching to alternate suppliers or assemblers; and |
• | increases in prices of raw materials and key components. |
The manufacturing operations for our test cartridges use highly technical processes involving unique, proprietary techniques. In addition, the manufacturing equipment we use would be costly to repair or replace and could require substantial lead time to repair or replace. Any interruption in our operations or decrease in the production capacity of our manufacturing facility or the facilities of any of our suppliers because of equipment failure, natural disasters such as earthquakes, tornadoes and fires, or otherwise, would limit our ability to meet customer demand for our products and would have a material adverse effect on our business, financial condition and results of operations. In the event of a disruption, we may lose customers and we may be unable to regain those customers thereafter. Our insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all.
Our quarterly revenue and operating results may vary significantly and we may experience constraints or inefficiencies caused by unanticipated acceleration and deceleration of customer demand.
Revenue from our infectious disease products fluctuates based upon the occurrence of related outbreaks and changes in testing recommendations. Influenza and other respiratory-related outbreaks are usually more concentrated in the first and fourth quarters of the year. New information or the introduction of advanced treatment options with respect to a particular disease may also affect related testing. Although certain infectious disease outbreaks tend to occur each year, the timing, severity and length of these incidents varies from one year to another and can vary across different patient populations. In addition, we may not
14
accurately predict changes to infectious disease testing recommendations affecting our products. As a result, we may not be able to accurately forecast sales from our infectious disease products.
Also, unanticipated changes in customer demand for our products may result in constraints or inefficiencies related to our manufacturing, sales force and implementation resources and administrative infrastructure. These constraints or inefficiencies may adversely affect us as a result of delays, lost potential product sales or loss of current or potential customers due to their dissatisfaction. Similarly, over-expansion or investments in anticipation of growth that does not materialize, or develops more slowly than we expect, could harm our financial results and result in overcapacity.
We face intense competition from established and new companies in the molecular diagnostics field and expect to face increased competition in the future.
The markets for our technologies and products are highly competitive and we expect the intensity of competition to increase. We compete with many companies in the United States engaged in the development, commercialization and distribution of similar products intended for clinical molecular diagnostic applications. Categories of our competitors include:
• | companies developing and marketing multiplex molecular diagnostics systems, including: Luminex; Nanosphere, Inc.; bioMerieux, which recently acquired BioFire Diagnostics, Inc.; Qiagen NV; Abbott Molecular Diagnostics, a division of Abbott Laboratories; Hologic, Inc. and Cepheid; |
• | large hospital-based laboratories and reference laboratories who provide large-scale testing using their own proprietary testing methods, including Quest Diagnostics Incorporated and Laboratory Corporation of America; and |
• | companies that manufacture laboratory-based tests and analyzers, including: Cepheid; Siemens; Hologic, Inc.; Qiagen NV; bioMérieux; Roche Diagnostics, a division of F. Hoffmann-La Roche Ltd.; and Abbott Molecular Diagnostics. |
Our diagnostic tests also face competition from laboratory developed tests, or LDTs, developed by national and regional reference laboratories and hospitals. Although the FDA has issued draft guidance related to LDTs, LDTs may not currently be subject to the same regulatory requirements, including those requiring clinical trials and FDA review and clearance or approval that may apply to our diagnostic products.
We anticipate that we will face increased competition in the future as new companies enter the market with new technologies, our competitors improve their current products and expand their menu of diagnostic tests, and as we expand our operations internationally. Many of our current competitors, as well as many of our potential competitors, have greater name recognition, more substantial intellectual property portfolios, longer operating histories, significantly greater resources to invest in new technologies, more substantial experience in new product development, greater regulatory expertise, and more extensive manufacturing and distribution capabilities. It is critical to our success that we anticipate changes in technology and customer requirements and successfully introduce enhanced and competitive technology to meet our customers’ and prospective customers’ needs on a timely basis.
We may be unsuccessful in expanding sales of our product offerings outside the United States.
Assuming we receive the applicable regulatory approvals, we plan to offer our molecular diagnostic products in European and other international markets in the near future. We intend to utilize a direct sales and technical support team in certain key European countries, which we expect will be augmented by marketing partners and distributors in other strategic areas as we expand internationally. We have introduced our XT-8 system to key opinion leader sites in certain countries as we establish our technology and certain tests within these markets in preparation for the international launch of our ePlex instrument system. If we are unable to establish the infrastructure or recruit highly qualified personnel to support our direct sales and support organization, or if we are unsuccessful in developing awareness and acceptance of our products and technology internationally, our future financial performance would be adversely affected. Furthermore, any distributors we establish may not commit the necessary resources to market and sell our products to meet our expectations. If distributors do not perform adequately or in compliance with applicable laws and regulations in particular geographic areas, or if we are unable to locate distributors in particular geographic areas, our ability to realize revenue growth based on sales outside the United States would be harmed.
In order to market our products in the European Union and many other foreign jurisdictions, we, or our distributors or partners, may be required to meet and/or comply with numerous and varying regulatory requirements before selling our products in such jurisdiction, including safety and efficacy and governing, among other things, clinical studies and commercial sales and distribution of our products. The approval procedure varies among countries and can involve additional testing. The regulatory approval process outside the United States may include all of the risks associated with obtaining FDA approval, as
15
well as additional risks. We may not obtain approvals from regulatory authorities outside the United States on a timely basis, if at all, which could harm our ability to expand into markets outside the United States.
The regulatory clearance or approval process for certain products is expensive, time consuming and uncertain, and the failure to obtain and maintain required clearances or approvals could prevent us from commercializing our products.
We are investing significantly in the research and development of our ePlex instrument and its related molecular diagnostic tests to expand our future product offerings. Our molecular diagnostic products, including our ePlex system, will require 510(k) clearance or pre-market approval by the FDA prior to their marketing for commercial use in the United States. For international commercialization, the classification of, and the regulatory pre-market requirements for, our molecular diagnostic products vary from country to country. There are a number of potential risks associated with the regulatory review processes for our products in development. For example, regulatory authorities may require that we conduct additional studies that could impact the cost associated with product development and could potentially delay commercial launch of the product. In addition, we may be unsuccessful in obtaining regulatory clearance for all of our desired intended uses for our products or product approval or clearance within certain jurisdictions.
The regulatory environment is constantly evolving. For example, the FDA conducted a review of the pre-market clearance process in response to internal and external concerns regarding the 510(k) program and, in January 2011, announced 25 action items designed to make the process more rigorous and transparent. Some of these proposals, if enacted, could impose additional regulatory requirements for device manufacturers which could delay our ability to obtain new 510(k) clearances, increase the costs of compliance or restrict our ability to maintain our current clearances. Similarly, the European Union, or EU, is proposing to update the European Directive 98/79/EC on in vitro diagnostic medical device, or IVD Directive (IVDD), that could impact the classification of our molecular diagnostic products and result in additional regulatory requirements, which could delay our ability to CE Mark our products. Delays in receipt of, or failure to obtain, clearances or approvals for future products, including our ePlex instrument and products that are currently in design or development, would result in delayed, or no, realization of revenues from such products and in substantial additional costs, which could decrease our profitability.
We must also comply with the applicable FDA and foreign regulatory agency post-market requirements. Any failure to maintain post-market compliance with FDA or foreign regulatory requirements could harm our business, operations, and/or financial condition.
We derive revenues from the sale of research use only, or RUO, tests, which are not intended for diagnostic purposes. Clinical laboratories are regulated under the Clinical Laboratory Improvement Amendments of 1988, or CLIA, and may validate the clinical diagnostic use of a laboratory developed test, or LDT, specifically for use in their laboratory using any labeled products. FDA has traditionally practiced enforcement discretion regarding the use of the LDTs for clinical diagnostic purposes. However, the FDA has recently promulgated draft guidance which outlines stringent regulatory requirements for CLIA labs in order to use LDTs for clinical diagnostic application. These proposed requirements, if implemented, may result in a significant reduction in the sale of our RUO products, which could reduce our revenues and adversely affect our operations and/or financial condition.
Our products could infringe patent rights of others, which may require costly litigation and, if we are not successful, could cause us to pay substantial damages or limit our ability to commercialize our products.
Our commercial success depends on our ability to develop, manufacture and market our systems and tests and use our proprietary technology without infringing the patents and other proprietary rights of third parties. As the molecular diagnostic industry expands and more patents are issued, the risk increases that there may be patents issued to third parties that relate to our products and technology of which we are not aware or that we must challenge to continue our operations as currently contemplated. Our products may infringe or may be alleged to infringe these patents.
The patent positions of medical device companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in patents in these fields has emerged to date in the United States or in many foreign jurisdictions. Both the U.S. Supreme Court and the Court of Appeals for the Federal Circuit have made, and will likely continue to make, changes in how the patent laws of the U.S. are interpreted. For example, three recent Supreme Court cases, Association for Molecular Pathology et al. v. Myriad Genetics, Inc., et al., Mayo Collaborative Services v. Prometheus Laboratories and Alice v. CLS Bank, have introduced additional questions regarding the patentability of isolated naturally occurring genes and gene fragments, proteins, peptides, natural products, and related diagnostic and therapeutic methods which are likely to be resolved only through continued litigation. The overall impact of these decisions and others on the molecular diagnostics industry remains uncertain and our interpretation of the scope of these rulings on existing or future patents may be inaccurate.
16
There is a significant amount of uncertainty regarding the extent of patent protection and infringement. Companies may have filed pending patent applications that cover technologies we incorporate in our products. As a result, we could be subjected to substantial damages for past infringement or be required to modify our products or stop selling them if it is ultimately determined that our products infringe a third party’s proprietary rights. Even if we are successful in defending against potential intellectual property infringement claims, we could incur substantial costs in doing so. Any litigation related to such claims could consume our resources and lead to significant damages, royalty payments or an injunction on the sale of certain products. Any additional licenses to patented technology could obligate us to pay substantial additional royalties, which could adversely impact our product costs and harm our business.
If our customers are not adequately reimbursed or compensated for the use of our products we may have difficulty selling our products.
Our ability to sell our products depends in part on the extent to which reimbursement related to performing tests using our products is available from governmental authorities, such as Medicare and other governmental programs, private insurance plans, managed care organizations and other organizations. There are ongoing efforts by governmental and third-party payers to contain or reduce the costs of healthcare coverage. In addition, efforts to reform the healthcare delivery system in the United States and Europe has increased pressure on healthcare providers to reduce costs, which has, in turn, increased pressure on medical device manufacturers to decrease prices charged for their products. If purchasers or users of our products are not able to obtain adequate reimbursement for the cost of using our products, either directly or indirectly, they may forego or reduce their purchase and use of our products.
Obtaining coverage and reimbursement approval for a product from each government or third-party payor is a time consuming and costly process that could require us to provide supporting scientific, clinical and cost-effectiveness data for the use of our product to each government or third-party payor. We may not be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement. For example, Medicare and Medicaid generally do not reimburse providers who use our Warfarin Sensitivity Test. In addition, eligibility for coverage does not imply that any product will be covered and reimbursed in all cases or reimbursed at a rate that allows our potential customers to make a profit or even cover their costs. Further, third-party payors may choose to reimburse our customers per test based on individual biomarker detection, rather than on the basis of the number of results given by the test. This may result in reference laboratories, public health institutions and hospitals electing to use separate tests to screen for each disease or condition so that they can receive reimbursement for each test they conduct. In that event, these entities may purchase separate tests for each disease, rather than products, such as ours, that can be used to return highly multiplexed test results.
Our credit facility requires that we satisfy certain milestones in order to access funding and contains restrictions that limit our flexibility in operating our business.
In January 2015, we entered into a loan and security agreement with General Electric Capital Corporation and certain other lenders. Pursuant to the agreement, we may borrow up to $10 million by March 31, 2015 and up to an additional $25 million if we timely satisfy certain regulatory requirements related to our ePlex system. In addition, we have access to up to $5 million under a revolving credit facility, subject to certain conditions and a defined borrowing base. If we fail to satisfy the conditions to funding under our credit facility, including, but not limited to, as a result of the failure to timely achieve the identified regulatory milestones, we may not have the ability to borrow certain amounts under our credit facility.
In addition, we must comply with certain affirmative and negative covenants under our credit facility, including covenants that limit or restrict our ability to, among other things:
• | incur additional indebtedness or issue certain preferred shares, |
• | pay dividends on, repurchase or make distributions in respect of, our capital stock or make other restricted payments, |
• | make certain investments or acquisitions, |
• | sell certain assets, |
• | create liens, or |
• | enter into certain transactions with our affiliates. |
If we default under the agreement, because of a covenant breach or otherwise, the outstanding amounts thereunder could become immediately due and payable and the lenders could terminate all commitments to extend further financing.
17
If we are unable to obtain, maintain and enforce intellectual property protection covering our products, others may be able to make, use or sell products substantially the same as ours, which could adversely affect our ability to compete in the market.
Our commercial success is dependent in part on obtaining, maintaining and enforcing intellectual property rights, including our patents and other intellectual property rights. If we are unable to obtain, maintain and enforce intellectual property protection covering our products, others may be able to make, use or sell products that are substantially the same as ours without incurring the sizeable development and licensing costs that we have incurred, which would adversely affect our ability to compete in the market.
We seek to obtain and maintain patents and other intellectual property rights to restrict the ability of others to market products that compete with our products. Currently, our patent portfolio is comprised on a worldwide basis of approximately 130 owned and exclusively licensed patents and approximately 25 additional pending applications. In general, patents have a term of at least 20 years from the application filing date or earlier claimed priority date. A majority of our issued and exclusively licensed patents are scheduled to expire by 2021, with approximately one half of the patents expiring by 2018. Several of our pending applications have the potential to mature into patents that may expire between 2028 and 2034. However, not all of the pending or future patent applications owned by or licensed to us are guaranteed to mature into patents, and, moreover, issued patents owned by or licensed to us now or in the future may be found by a court to be invalid or otherwise unenforceable. Also, even if our patents are determined by a court to be valid and enforceable, they may not be sufficiently broad to prevent others from marketing products similar to ours or designing around our patents, despite our patent rights, nor provide us with freedom to operate unimpeded by the patent rights of others.
We also rely on trade-secret protection to protect our interests in proprietary know-how and for processes for which patents are difficult to obtain or enforce. We may not be able to protect our trade secrets adequately. We have limited control over the protection of trade secrets used by our licensors, collaborators and suppliers. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third party illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets. We rely, in part, on non-disclosure and confidentiality agreements with our employees, consultants and other parties to protect our trade secrets and other proprietary technology. These agreements may be breached and we may not have adequate remedies for any breach. Moreover, others may independently develop equivalent proprietary information, and third parties may otherwise gain access to our trade secrets and proprietary knowledge. Any disclosure of confidential data into the public domain or to third parties could allow our competitors to learn our trade secrets and use the information in competition against us.
We are subject to various federal and state laws pertaining to health care fraud and abuse, including anti-kickback, self-referral, false claims and fraud laws, and any violations by us of such laws could result in fines or other penalties.
Our commercial, research and other financial relationships with healthcare providers and institutions are subject to various federal and state laws intended to prevent health care fraud and abuse. The federal anti-kickback statute prohibits the knowing offer, receipt or payment of remuneration in exchange for or to induce the referral of patients or the use of products or services that would be paid for in whole or part by Medicare, Medicaid or other federal health care programs. Remuneration has been broadly defined to include anything of value, including cash, improper discounts, and free or reduced price items and services. Many states have similar laws that apply to their state health care programs as well as private payors. Violations of the anti-kickback laws can result in exclusion from federal health care programs and substantial civil and criminal penalties.
The federal False Claims Act, or the FCA, imposes liability on persons who, among other things, present or cause to be presented false or fraudulent claims for payment by a federal health care program. The FCA has been used to prosecute persons submitting claims for payment that are inaccurate or fraudulent, that are for services not provided as claimed, or for services that are not medically necessary. We have implemented procedures designed to ensure our compliance with relevant legal requirements. Nevertheless, if our marketing, sales or other arrangements, including our reagent rental arrangements, were determined to violate anti-kickback or related laws, including the FCA, then our revenues could be adversely affected, which would likely harm our business, financial condition and results of operations.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or the PPACA, also imposes new reporting and disclosure requirements on device manufacturers for payments to healthcare providers and ownership of their stock by healthcare providers. Further, the PPACA, among other things, amends the intent requirement of the federal anti-kickback and criminal healthcare fraud statutes. A person or entity can now be found guilty under the PPACA without actual knowledge of the statute or specific intent to violate it. In addition, the PPACA provides that the government
18
may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the false claims statutes. In February 2013, CMS released the final rule implementing the federal Physician Payments Sunshine Act, or the Sunshine Act. The law requires certain pharmaceutical, biologic, and medical device manufacturers to annually report to CMS payments or other transfers of value they furnish to physicians and teaching hospitals. These new reporting requirements took effect on August 1, 2013. Failure to submit required information may result in significant civil monetary penalties. We expect compliance with the PPACA and Sunshine Act to impose significant administrative and financial burdens on us.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians for marketing. Some states, such as California, Massachusetts and Vermont, mandate implementation of corporate compliance programs, along with the tracking and reporting of gifts, compensation and other remuneration to physicians. The shifting commercial compliance environment and the need to build and maintain robust and expandable systems to comply with different compliance and/or reporting requirements in multiple jurisdictions increase the possibility that a healthcare company may run afoul of one or more of the requirements.
State and federal authorities have aggressively targeted medical device companies for alleged violations of these anti-fraud statutes, based on improper research or consulting contracts with doctors, certain marketing arrangements that rely on volume-based pricing, off-label marketing schemes and other improper promotional practices. Companies targeted in such prosecutions have paid substantial fines in the hundreds of millions of dollars or more, have been forced to implement extensive corrective action plans, and have often become subject to consent decrees severely restricting the manner in which they conduct their business. If we become the target of such an investigation or prosecution based on our contractual relationships with providers or institutions, or our marketing and promotional practices, we could face similar sanctions which would materially harm our business.
Once we commence commercial operations outside the United States, we will be subject to the U.S. Foreign Corrupt Practices Act, or the FCPA, and other countries’ anti-corruption/anti-bribery regimes, such as the U.K. Bribery Act. The FCPA prohibits improper payments or offers of payments to foreign governments and their officials for the purpose of obtaining or retaining business. Safeguards we implement to discourage improper payments or offers of payments by our employees, consultants, sales agents or distributors may be ineffective, and violations of the FCPA and similar laws may result in severe criminal or civil sanctions, or other liabilities or proceedings against us, any of which would likely harm our reputation, business, financial condition and results of operations.
We are currently reliant on the commercial success of our XT-8 system and its related test menu to partially fund our current operations and development programs.
We currently market our XT-8 instrument system and four FDA-cleared diagnostic tests. In addition, we have several diagnostic tests in the research, development or design stage. We have primarily placed our XT-8 systems with customers at no initial charge through reagent rental agreements, under which customers generally commit to purchase minimum quantities of test cartridges and reagents (consumables) over a typical period of one to three years, with a component of the cartridge and reagent price allocated to recover the instrument price. We also offer our XT-8 systems for sale. We intend to continue to dedicate a significant portion of our resources to the commercialization of our XT-8 system and its related test menu, while also dedicating significant resources to the commercialization of our ePlex system and the development of its related test menu. As a result, to the extent that our XT-8 system and our existing and future diagnostic and research products are not commercially successful or are withdrawn from the market for any reason, our operating results, financial condition and critical development programs would be harmed and we may be required to seek additional funding to support our ongoing operations.
In addition, we have limited marketing, sales and distribution experience and capabilities. Our ability to achieve profitability depends on attracting customers for our products and building brand loyalty. To successfully perform sales, marketing, distribution and customer support functions ourselves, we face a number of risks, including:
• | our ability to attract and retain the skilled support team, marketing staff and sales force necessary to commercialize and gain market acceptance for our technology and our products; |
• | the ability of our sales and marketing team to identify and penetrate the potential customer base, including hospitals and national and regional reference laboratories; and |
• | the difficulty of establishing brand recognition and loyalty for our products. |
Some hospital-based and reference laboratories may not consider adopting our XT-8 system unless we offer a broader menu of diagnostic tests or may choose not to convert from competitive products unless and until we are able to offer a sample-
19
to-answer instrument solution, such as our ePlex instrument. In addition, in order to commercialize our products, we are required to undertake time consuming and costly development activities, including clinical studies for which the outcome is uncertain. Products that appear promising during early development and preclinical studies may, nonetheless, fail to demonstrate the results needed to support regulatory approval or, if approved, may not generate the demand we expect. If we are unable to effectively compete with our XT-8 system and its related test menu, our revenues and our ability to achieve profitability will be significantly impaired.
Legislative or regulatory healthcare reforms may have a material adverse effect on our business and results of operations.
Federal and state governments in the United States are undertaking efforts to control growing health care costs through legislation, regulation and voluntary agreements with medical care providers and third-party payors. In March 2010, Congress enacted the PPACA. While the PPACA involves expanding coverage to more individuals, it includes new regulatory mandates and other measures designed to constrain medical costs. Among other requirements, the PPACA imposes a 2.3% excise tax on sales of medical devices by manufacturers that is expected to cost the medical device industry up to $20 billion over the decade following its effectiveness. Taxable devices include any medical device defined in Section 201(h) of the FDCA and intended for use by humans, with limited exclusions for devices purchased by the general public at retail for individual use. There is no exemption for small companies, and we began paying the tax in 2013. Complying with PPACA may significantly increase our tax liabilities and costs, which could adversely affect our business and financial condition.
In August 2011, President Obama signed into law the Budget Control Act of 2011, which among other things, created automatic reductions to several government programs, including aggregate reductions to Medicare payments to providers of up to 2% per fiscal year. In January 2013, the American Taxpayer Relief Act of 2012, or the ATRA, delayed for another two months the budget cuts mandated by these sequestration provisions of the Budget Control Act of 2011. In March 2013, President Obama signed an executive order implementing sequestration, and in April 2013, the 2% Medicare payment reductions went into effect. The ATRA also, among other things, reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our products or additional pricing pressure.
We have a history of net losses, and we may never achieve or maintain profitability.
We have a history of significant net losses and a limited history commercializing our molecular diagnostic products. We obtained FDA clearance for our first generation molecular diagnostic system in 2006, and commenced a limited marketing effort for this system. We initially offered our XT-8 system and our Warfarin Sensitivity Test in July 2008, our Cystic Fibrosis Genotyping Test in July 2009, our Thrombophilia Risk Test in April 2010, and our Respiratory Viral Panel in September 2012. Our net losses were approximately $38.3 million and $33.6 million for the years ended December 31, 2014 and 2013, respectively. As of December 31, 2014, we had an accumulated deficit of $262.5 million. We expect to continue to incur significant expenses for the foreseeable future in connection with our ongoing operations, primarily related to our commercial organization (sales and marketing), research and development and regulatory activities, maintaining our existing intellectual property portfolio, obtaining additional intellectual property rights and investing in corporate infrastructure. We cannot provide any assurance that we will achieve profitability and, even if we achieve profitability, that we will be able to sustain or increase profitability on a quarterly or annual basis. Further, because of our limited commercialization history and the rapidly evolving nature of our target market, we have limited insight into the trends that may emerge and affect our business. We may make errors in predicting and reacting to relevant business trends, which could harm our business and financial condition.
We may need to raise additional funds in the future, and such funds may not be available on a timely basis, or at all.
Until such time, if ever, as we can generate positive cash flows from operations, we will be required to finance our operations with our cash resources. We may need to raise additional funds in the future to support our operations. We cannot be certain that additional capital will be available as needed, on acceptable terms, or at all. If we require additional capital at a time when investment in our company, in molecular diagnostics companies, or the marketplace in general is limited, we may not be able to raise such funds at the time that we desire, or at all. If we do raise additional funds through the issuance of equity or convertible securities, the percentage ownership of holders of our common stock could be significantly diluted. In addition, newly issued securities may have rights, preferences or privileges senior to those of holders of our common stock. If we raise additional funds through collaborations and licensing arrangements, we could be required to relinquish significant rights to our technologies and products, or grant licenses on terms that are not favorable to us.
20
If we are unable to retain key employees or hire additional skilled employees, we may be unable to achieve our goals.
Our performance is substantially dependent on the performance of our senior management. Competition for top management personnel is intense and we may not be able to recruit and retain the personnel we need. Our senior managers can terminate their relationship with us at any time. The loss of services of any of these key personnel could significantly reduce our operational effectiveness and investor confidence and our stock price could decline. We do not maintain key-man life insurance on any of our employees.
In addition, our product development and marketing efforts could be delayed or curtailed if we are unable to attract, train and retain highly skilled technical employees and scientific advisors. To expand our research, product development and commercial efforts, we will need to retain additional people skilled in areas such as electrochemical and molecular science, information technology, manufacturing, sales, marketing and technical support. Because of the complex and technical nature of our systems and the dynamic market in which we compete, any failure to attract and retain a sufficient number of qualified employees could materially harm our ability to develop and commercialize our technology. We may not be successful in hiring or retaining qualified personnel, and any failure to do so could have a material adverse effect on our business, financial condition and results of operations.
We and our suppliers, contract manufacturers and customers are subject to various governmental regulations, and we may incur significant expenses to comply with, and experience delays in our product commercialization as a result of, these regulations.
Our manufacturing processes and facilities and those of some of our contract manufacturers must comply with the federal Quality System Regulation, or QSR, which covers the procedures and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of our devices. The FDA enforces the QSR through periodic announced and/or unannounced inspections of manufacturing facilities. We and our contract manufacturers have been, and anticipate in the future being, subject to such inspections, as well as to inspections by other federal and state regulatory agencies.
We must also file reports of device corrections and removals and adhere to the FDA’s rules on labeling and promotion. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including substantial monetary penalties and criminal prosecution.
Failure to comply with applicable FDA requirements, or later discovery of previously unknown problems with our products or manufacturing processes, including our failure or the failure of one of our contract manufacturers to take satisfactory corrective action in response to an adverse QSR inspection, can result in, among other things:
• | administrative or judicially imposed sanctions; |
• | injunctions or the imposition of civil penalties; |
• | recall or seizure of our products; |
• | total or partial suspension of production or distribution; |
• | withdrawal or suspension of marketing clearances or approvals; |
• | clinical holds; |
• | warning letters; |
• | refusal to permit the import or export of our products; and |
• | criminal prosecution. |
Any of these actions, in combination or alone, could prevent us from marketing, distributing or selling our products and would likely harm our business.
In addition, a product defect or regulatory violation could lead to a government-mandated or voluntary recall by us. We believe that the FDA would request that we initiate a voluntary recall if a product was defective or presented a risk of injury or gross deception. Regulatory agencies in other countries have similar authority to recall devices because of material deficiencies or defects in design or manufacture that could endanger health. Any recall would divert management attention and financial resources, could cause the price of our shares of common stock to decline and expose us to product liability or other claims,
21
including contractual claims from parties to whom we sold products, and harm our reputation with customers. A recall involving our XT-8 system or our diagnostic tests would be particularly harmful to our business and financial results.
The use of our diagnostic products by our customers is also affected by CLIA and related federal and state regulations that provide for regulation of laboratory testing. CLIA is intended to ensure the quality and reliability of clinical laboratories in the United States by mandating specific standards in the areas of personnel qualifications, administration, participation in proficiency testing, patient test management, quality assurance, quality control and inspections. Current or future CLIA requirements or the promulgation of additional regulations affecting laboratory testing may prevent some laboratories from using some or all of our diagnostic products.
If our products do not perform as expected or the reliability of the technology on which our products are based is questioned, our operating results and business would suffer.
Our success depends on the market’s confidence that we can provide reliable, high quality, molecular diagnostic products. We believe that customers in our target markets are likely to be particularly sensitive to product defects and errors. As a result, our reputation and the public image of our products and technologies will be significantly impaired if our products fail to perform as expected. Although our diagnostic systems are designed to be user friendly, the functions they perform are complex and our products may develop or contain undetected defects or errors.
We currently manufacture our proprietary test cartridges at our Carlsbad, California manufacturing facility. We outsource manufacturing of our XT-8 system and much of the disposable component molding for our test cartridges. In 2012, we formalized our relationship with Leica Biosystems Melbourne Pty Ltd., or Leica, the contract manufacturer of our XT-8 instrument system. Leica specializes in manufacturing of electronic and electromechanical devices for medical use. While we work closely with Leica to ensure continuity of supply while maintaining high quality and reliability, we cannot guarantee that these efforts will be successful. We currently anticipate manufacturing the proprietary test cartridges for our ePlex system, and outsourcing the manufacture of our ePlex system to a third party manufacturing partner.
If we experience a material defect or error in any of our current or future products, it could result in the loss or delay of revenues, increased costs, delayed or reduced market acceptance, damaged reputation, diversion of development and management resources, legal and/or regulatory claims, recalls, increased insurance costs or increased service and warranty costs, any of which could materially harm our business, financial condition and results of operations.
We also face the risk of product liability exposure related to the sale of our products. We currently carry product liability insurance that covers us against specific product liability claims. We also carry a separate general liability and umbrella policy that covers us against certain claims but excludes coverage for product liability. Any claim in excess of our insurance coverage, or for which we do not have insurance coverage, would need to be paid out of our cash reserves, which would harm our financial condition. We cannot assure you that we have obtained sufficient insurance or broad enough coverage to cover potential claims. Also, we cannot assure you that we can or will maintain our insurance policies on commercially acceptable terms, or at all. A product liability claim could significantly harm our business, financial condition and results of operations.
We may not be able to correctly estimate or control our future operating expenses, which could lead to cash shortfalls.
Our operating expenses may fluctuate significantly in the future as a result of a variety of factors, many of which may be outside of our control. These factors include, but are not limited to:
• | the time and resources required to develop, and conduct clinical studies and obtain regulatory clearances for, additional diagnostic tests; |
• | the expenses we incur for research and development required to maintain and improve our technology, including developing our ePlex system; |
• | the costs of preparing, filing, prosecuting, defending and enforcing patent claims and other patent related costs, including litigation costs and the results of such litigation; |
• | the expenses we incur in connection with commercialization activities, including product marketing, sales and distribution expenses; |
• | the expenses we incur in licensing technologies from third parties to expand the menu of diagnostics tests we plan to offer; |
• | our sales strategy and whether the revenues from sales of our test cartridges or XT-8 system will be sufficient to offset our expenses; |
22
• | the costs to attract and retain personnel with the skills required for effective operations; and |
• | the costs associated with being a public company. |
Our budgeted expense levels are based in part on our expectations concerning future revenues from sales of our XT-8 system and its related test menu, as well our assessment of the future investments needed to expand our commercial organization and support research and development activities in connection with our ePlex system. We may be unable to reduce our expenditures in a timely manner to compensate for any unexpected events or a shortfall in revenue. Accordingly, a shortfall in demand for our products or other unexpected events could have an immediate and material impact on our business and financial condition.
We incur costs and demands upon management as a result of complying with the laws and regulations affecting public companies in the United States, and failure to comply with these laws could harm our business and the price of our common stock.
As a public company listed in the United States, we incur significant legal, accounting and other expenses. In addition, changing laws, regulations and standards relating to corporate governance and public disclosure, including regulations implemented by the SEC, the Public Company Accounting Oversight Board (PCAOB), and The NASDAQ Global Market, may increase our legal and financial compliance costs and make some activities more time consuming. These laws, regulations and standards are subject to varying interpretations and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. We intend to invest resources to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expenses and a diversion of management’s time and attention from revenue-generating activities to compliance activities. If we nevertheless fail to comply with new laws, regulations and standards, regulatory authorities may initiate legal proceedings against us and our business may be harmed.
Economic conditions and an uncertain economic outlook may adversely impact our business, results of operations, financial condition or liquidity.
Global economic conditions may remain challenging and uncertain for the foreseeable future. These conditions not only limit our access to capital but also make it extremely difficult for our customers, our vendors and us to accurately forecast and plan future business activities, and they could cause U.S. and foreign businesses and consumers to slow spending on our products and services, which would delay and lengthen sales cycles. Some of our customers rely on government research grants to fund technology purchases. If negative trends in the economy affect the government’s allocation of funds to research, there may be less grant funding available for certain of our customers to purchase technologies from us. Certain of our customers may face challenges gaining timely access to sufficient credit or may otherwise be faced with budget constraints, which could result in decreased purchases of our products or in an impairment of their ability to make timely payments to us. If our customers do not make timely payments to us, we may be required to assume greater credit risk relating to those customers, increase our allowance for doubtful accounts, and our days sales outstanding would be negatively impacted. Although we maintain allowances for doubtful accounts for estimated losses resulting from the inability of our customers to make required payments, we may not continue to experience the same loss rates that we have in the past. Additionally, these economic conditions and market turbulence may also impact our suppliers, causing them to be unable to supply in a timely manner sufficient quantities of customized components, thereby impairing our ability to manufacture on schedule and at commercially reasonable costs.
We are exposed to risks associated with long-lived and intangible assets that may become impaired and result in an impairment charge.
The carrying amounts of long-lived and intangible assets are affected whenever events or changes in circumstances indicate that the carrying amount of any asset may not be recoverable. These events or changes might include an inability to successfully deliver an instrument to the marketplace and attain customer acceptance, a change in the rights or use of licensed intellectual property, adjustments to our depreciation assumptions, or other matters. Adverse events or changes in circumstances may affect the estimated discounted future cash flows expected to be derived from long-lived and intangible assets. If at any time we determine that an impairment has occurred, we will be required to reflect the impaired value as a charge, resulting in a reduction in earnings in the quarter such impairment is identified and a corresponding reduction in our net asset value. In the past we have incurred, and in the future we may incur, impairment charges. For example, during the year ended December 31, 2013, we recorded an impairment charge of $1.6 million related to previously capitalized payments made under the terms of a license agreement, which we terminated in December 2013. A material reduction in earnings resulting from such a charge could cause us to fail meet the expectations of investors and securities analysts, which could cause the price of our stock to decline.
23
Providing XT-8 systems to our customers through reagent rental agreements may harm our liquidity.
The majority of our XT-8 systems are provided to customers via “reagent rental” agreements, under which customers are afforded the right to use the XT-8 system in return for a commitment to purchase minimum quantities of reagents and test cartridges over a period of time. Accordingly, we must either incur the expense of manufacturing XT-8 systems well in advance of receiving sufficient revenues from test cartridges to recover our expenses or obtain third party financing sources for the purchase of our XT-8 systems. The amount of capital required to provide these systems to customers depends on the number of systems placed. Our ability to generate capital to cover these costs depends on the amount of our revenues from sales of reagents and test cartridges sold through our reagent rental agreements. We do not currently sell enough reagents and test cartridges to recover all of our fixed expenses, and therefore we currently have a net loss. If we cannot sell a sufficient number of reagents and test cartridges to offset our fixed expenses, our liquidity will continue to be adversely affected.
We use hazardous chemicals, biological materials and infectious agents in our business. Any claims relating to improper handling, storage or disposal of these materials could be time consuming and costly.
Our research, product development and manufacturing processes involve the controlled use of hazardous materials, including chemicals, biological materials and infectious disease agents. Our operations produce hazardous waste products. We cannot eliminate the risk of accidental contamination or discharge and any resulting injury from these materials. We may be sued for any injury or contamination that results from our use or the use by third parties of these materials, and our liability may exceed our insurance coverage and our total assets. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of these hazardous materials and specified waste products, as well as the discharge of pollutants into the environment and human health and safety matters. Our operations are regulated and may require that environmental permits and approvals be issued by applicable government agencies. Compliance with environmental laws and regulations may be expensive and may impair our research, development and production efforts. If we fail to comply with these requirements, we could incur substantial costs, including civil or criminal fines and penalties, clean-up costs or capital expenditures for control equipment or operational changes necessary to achieve and maintain compliance. In addition, we cannot predict the impact on our business of new or amended environmental laws or regulations or any changes in the way existing and future laws and regulations are interpreted and enforced.
Our ability to use our net operating loss carryforwards may be limited.
As of December 31, 2014, we had net operating loss, or NOL, carryforwards available of approximately $128.4 million million for U.S. federal income tax purposes. These loss carryforwards will expire in varying amounts through 2034. Section 382 of the U.S. Internal Revenue Code of 1986, as amended, or the Code, generally imposes an annual limitation on the amount of NOL carryforwards that may be used to offset taxable income when a corporation has undergone significant changes in stock ownership. We have determined that we have experienced multiple ownership changes under Section 382 of the Code. Our ability to use the current NOL carryforwards may also be limited by the issuance of common stock in the future. To the extent our use of NOL carryforwards is limited, our income may be subject to corporate income tax earlier than it would if we were able to use NOL carryforwards. We have recorded a full valuation allowance against our net deferred tax assets.
We also had non-U.S. NOL carryforwards as of December 31, 2014. As a result of the liquidation of Osmetech plc in the fourth quarter of 2013, our expectation is that the non-U.S. NOL carryforwards will not be utilized and, therefore, we have not accounted for them as a deferred tax asset.
Information technology systems implementation issues or security threats could disrupt our internal operations and adversely affect our financial results.
Portions of our information technology infrastructure may experience interruptions, delays or cessations of service or produce errors in connection with ongoing systems implementation work. In particular, we have implemented an enterprise resource planning software system. To more fully realize the potential of this system, we are continually reassessing and upgrading processes and this may be more expensive, time consuming and resource intensive than planned. Any disruptions that may occur in the operation of this system or any future systems or any unauthorized access to our information systems could increase our expenses and adversely affect our ability to report in an accurate and timely manner the results of our consolidated operations, our financial position and cash flows and to otherwise operate our business in a secure environment, all of which could adversely affect our financial results, stock price and reputation.
24
Provisions of our certificate of incorporation, our bylaws and Delaware law could make an acquisition of our Company, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove the current members of our board and management.
Certain provisions of our certificate of incorporation and bylaws could discourage, delay or prevent a merger, acquisition or other change of control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. Furthermore, these provisions could prevent or frustrate attempts by our stockholders to replace or remove members of our Board of Directors. These provisions also could limit the price that investors might be willing to pay in the future for our common stock, thereby depressing the market price of our common stock. Stockholders who wish to participate in these transactions may not have the opportunity to do so. These provisions:
• | allow the authorized number of directors to be changed only by resolution of our Board of Directors; |
• | provide that our stockholders may remove our directors only for cause; |
• | establish a classified board of directors, such that not all members of the Board of Directors may be elected at one time; |
• | authorize our Board of Directors to issue without stockholder approval up to 100,000,000 shares of common stock, that, if issued, would dilute our stock ownership and could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that is not approved by our Board of Directors; |
• | authorize our Board of Directors to issue without stockholder approval up to 5,000,000 shares of preferred stock, the rights of which will be determined at the discretion of the Board of Directors that, if issued, could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that is not approved by our Board of Directors; |
• | require that stockholder actions must be effected at a duly called stockholder meeting or by unanimous written consent; |
• | establish advance notice requirements for stockholder nominations to our Board of Directors or for stockholder proposals that can be acted on at stockholder meetings; |
• | limit who may call stockholder meetings; and |
• | require the approval of the holders of 80% of the outstanding shares of our capital stock entitled to vote in order to amend certain provisions of our certificate of incorporation and bylaws. |
In addition, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which may, unless certain criteria are met, prohibit large stockholders, in particular those owning 15% or more of the voting rights on our common stock, from merging or combining with us for a prescribed period of time.
Item 1B. | UNRESOLVED STAFF COMMENTS |
None.
Item 2. | PROPERTIES |
We currently operate from a facility located in Carlsbad, California. We do not own any real property. In February 2010, we entered into a lease for an approximately 31,000 square foot facility in Carlsbad, California, the term of which originally ran through September 2017. The facility is part of a three-building office and research and development project located at 5964 La Place Court, Carlsbad, California. In January 2012, we signed a lease amendment which expanded our executive and administrative office, research and development, and manufacturing space by approximately an additional 22,000 square feet and extended the term of the lease through June 2021. We believe that our current leased facilities are adequate to meet our needs for the foreseeable future.
Item 3. | LEGAL PROCEEDINGS |
We are from time to time subject to various claims and legal actions in the ordinary course of our business. We believe that there are currently no claims or legal actions that would reasonably be expected to have a material adverse effect on our results of operations or financial condition.
25
Item 4. | MINE SAFETY DISCLOSURES |
Not applicable.
PART II.
Item 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our common stock has been quoted on The NASDAQ Global Market under the symbol “GNMK” since May 28, 2010. Prior to that time, our stock traded under the ticker symbol “OMH” on the London Stock Exchange. The following table sets forth for the indicated periods the high and low sales prices per share of our common stock as reported on The NASDAQ Global Market.
High | Low | ||||||
Year Ended Year Ended December 31, 2014 | |||||||
First Quarter | $ | 14.18 | $ | 9.28 | |||
Second Quarter | $ | 13.63 | $ | 8.48 | |||
Third Quarter | $ | 13.87 | $ | 8.64 | |||
Fourth Quarter | $ | 14.18 | $ | 8.62 | |||
Year Ended Year Ended December 31, 2013 | |||||||
First Quarter | $ | 13.03 | $ | 8.86 | |||
Second Quarter | $ | 16.00 | $ | 9.25 | |||
Third Quarter | $ | 12.59 | $ | 8.75 | |||
Fourth Quarter | $ | 13.37 | $ | 10.78 | |||
Stock Performance Graph
The graph below compares the cumulative total stockholder returns on our common stock for the period indicated with the cumulative total stockholder returns on the NASDAQ Composite Index and the NASDAQ Biotechnology Index for the same period. The graph assumes that $100 was invested on May 28, 2010 in our common stock and in each index and that all dividends were reinvested. No cash dividends have been declared on our common stock. Stockholder returns over the indicated period should not be considered indicative of future stockholder returns.
26
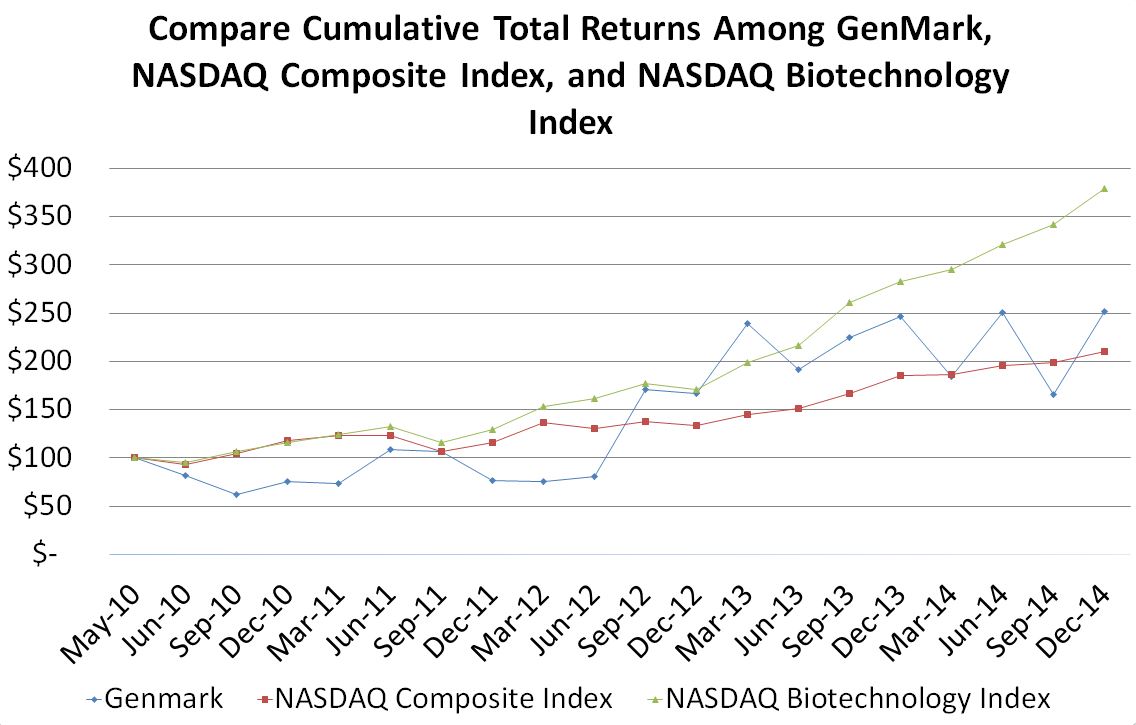
Stockholders
The last reported sale price of common stock on February 20, 2015 as reported on the NASDAQ Global Market was $13.22. As of February 20, 2015, there were 2,770 holders of record of our common stock.
Dividend Policy
We have never declared or paid any cash dividends on our common stock and do not expect to pay any dividends for the foreseeable future. In addition, our loan agreement with General Electric Capital Corporation contains a negative covenant which may limit our ability to pay dividends. We currently intend to retain any future earnings to fund the operation, development and expansion of our business. Any future determination to pay dividends will be at the sole discretion of our Board of Directors and will depend upon a number of factors, including our results of operations, capital requirements, financial condition, future prospects, contractual arrangements, restrictions imposed by applicable law, any limitations on payments of dividends present in our current and future debt arrangements, and other factors our Board of Directors may deem relevant.
Item 6. | SELECTED CONSOLIDATED FINANCIAL DATA |
The following selected consolidated financial data relates to GenMark Diagnostics, Inc. and its consolidated subsidiaries. The selected consolidated statement of comprehensive loss data presented below of GenMark Diagnostics, Inc. for the years ended December 31, 2014, 2013, and 2012 and the selected consolidated balance sheet data of GenMark Diagnostics, Inc. as of December 31, 2014, and 2013 have been derived from the audited consolidated financial statements of GenMark Diagnostics, Inc., which have been prepared in accordance with generally accepted accounting principles in the United States, or U.S. GAAP, included elsewhere in this Annual Report. The selected consolidated statement of comprehensive loss data presented for the years ended December 31, 2011 and 2010 and the selected consolidated balance sheet data as of December 31, 2012, 2011, and 2010 have been derived from the audited financial statements not included in this Annual Report.
The results for the periods shown below are not necessarily indicative of the results to be expected for any future periods. The selected consolidated financial data should be read together with the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section and with the consolidated financial statements and condensed consolidated
27
financial statements of GenMark Diagnostics, Inc. and related notes included elsewhere in this Annual Report.
FIVE YEAR SELECTED FINANCIAL DATA | |||||||||||||||||||
Years ended December 31, | |||||||||||||||||||
2014 | 2013 | 2012 | 2011 | 2010 | |||||||||||||||
Consolidated Statements of Comprehensive Loss Data: | (In thousands, except per share data) | ||||||||||||||||||
Revenue | |||||||||||||||||||
Product revenue | $ | 30,328 | $ | 27,204 | $ | 20,211 | $ | 4,700 | $ | 2,341 | |||||||||
License and other revenue | 266 | 200 | 258 | 309 | 223 | ||||||||||||||
Total revenue | 30,594 | 27,404 | 20,469 | 5,009 | 2,564 | ||||||||||||||
Cost of revenue(1) | 13,127 | 15,570 | 11,640 | 6,206 | 3,979 | ||||||||||||||
Gross profit (loss) | 17,467 | 11,834 | 8,829 | (1,197 | ) | (1,415 | ) | ||||||||||||
Operating expenses | |||||||||||||||||||
Sales and marketing | 12,629 | 12,818 | 6,378 | 4,969 | 4,555 | ||||||||||||||
General and administrative (1) | 12,069 | 11,836 | 10,806 | 8,960 | 7,415 | ||||||||||||||
Research and development | 31,823 | 22,060 | 13,536 | 8,737 | 6,646 | ||||||||||||||
Total operating expenses | 56,521 | 46,714 | 30,720 | 22,666 | 18,616 | ||||||||||||||
Loss from operations | (39,054 | ) | (34,880 | ) | (21,891 | ) | (23,863 | ) | (20,031 | ) | |||||||||
Other income (expense): | |||||||||||||||||||
Interest income (expense), net (2) | 224 | 384 | (48 | ) | (74 | ) | — | ||||||||||||
Therapeutic discovery credit | — | — | — | — | 1,644 | ||||||||||||||
Other income (expenses) (2) | (6 | ) | 897 | (16 | ) | 19 | (1 | ) | |||||||||||
Total other income (expense) | 218 | 1,281 | (64 | ) | (55 | ) | 1,643 | ||||||||||||
Loss before income taxes | (38,836 | ) | (33,599 | ) | (21,955 | ) | (23,918 | ) | (18,388 | ) | |||||||||
Income tax expense (benefit) | (573 | ) | 44 | 148 | 52 | 15 | |||||||||||||
Net loss | $ | (38,263 | ) | $ | (33,643 | ) | $ | (22,103 | ) | $ | (23,970 | ) | $ | (18,403 | ) | ||||
Net loss per share, basic and diluted | $ | (0.93 | ) | $ | (0.95 | ) | $ | (0.84 | ) | $ | (1.45 | ) | $ | (1.88 | ) | ||||
Weighted average number of shares outstanding, basic and diluted | 41,346 | 35,253 | 26,215 | 16,572 | 9,797 | ||||||||||||||
As of December 31, | |||||||||||||||||||
2014 | 2013 | 2012 | 2011 | 2010 | |||||||||||||||
(In thousands) | |||||||||||||||||||
Consolidated Balance Sheet Data: | |||||||||||||||||||
Cash and cash equivalents and marketable securities(3)(4)(5)(6) | $ | 70,506 | $ | 105,589 | $ | 51,250 | $ | 30,320 | $ | 18,329 | |||||||||
Total assets | 91,970 | 121,754 | 68,016 | 38,186 | 26,314 | ||||||||||||||
Long-term liabilities | 1,653 | 2,349 | 2,392 | 1,171 | 1,307 | ||||||||||||||
Total liabilities | 13,946 | 12,586 | 11,566 | 7,552 | 5,247 | ||||||||||||||
Accumulated deficit | (262,472 | ) | (224,209 | ) | (190,566 | ) | (168,463 | ) | (144,493 | ) | |||||||||
Total stockholders’ equity (3)(4)(5)(6) | 78,024 | 109,168 | 56,450 | 30,634 | 21,067 | ||||||||||||||
_____________________
(1) | Certain 2013 costs, totaling $324,000, previously reported as Cost of sales have been reclassified as General and administrative expenses to better align those costs with the functional areas that benefit from those expenditures. |
(2) | Certain amounts included in other income and expense in 2013 of $314,000 were reclassified into interest income (expense) to better align the true nature of the expenses. |
(3) | In August 2013, we issued approximately 8.7 million shares of common stock at a price of $9.84 per share. We raised approximately $81.0 million in net proceeds. |
(4) | In June 2012, we issued approximately 11.5 million shares of common stock at a price of $4.20 per share. We raised approximately $45.1 million in net proceeds. |
28
(5) | In June 2011, we issued approximately 8.1 million shares of common stock at a price of $4.25 per share. We raised approximately $31.7 million in net proceeds. |
(6) | In June 2010, we closed our initial public offering, in which we sold approximately 4.6 million shares of common stock at a price to the public of $6.00 per share. We raised approximately $22.6 million in net proceeds. |
Item 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
You should read the following in conjunction with the “Selected Consolidated Financial Data” and the consolidated financial statements of GenMark and the related notes thereto that appear elsewhere in this Annual Report. In addition to historical information, the following discussion and analysis includes forward looking information that involves risks, uncertainties and assumptions. Actual results and the timing of events could differ materially from those anticipated by these forward looking statements as a result of many factors, including those discussed under the heading “Risk Factors” included elsewhere in this Annual Report. See also “Forward Looking Statements” included elsewhere in this filing.
Overview
GenMark was formed by Osmetech as a Delaware corporation in February 2010. GenMark had no operations prior to its initial public offering, which was completed in June 2010. Immediately prior to the closing of the initial public offering, GenMark acquired all of the outstanding ordinary shares of Osmetech in a reorganization under the applicable laws of the United Kingdom. As a result of the reorganization, all of the issued ordinary shares in Osmetech were cancelled in consideration of: (i) the issuance of common stock of GenMark to the former shareholders of Osmetech; and (ii) the issuance of new shares in Osmetech to GenMark. Following the reorganization, Osmetech became a wholly-owned subsidiary controlled by GenMark, and the former shareholders of Osmetech received shares of GenMark. Once the reorganization became effective, all stock options granted under the Osmetech plc 2003 U.S. Equity Compensation Plan, long term incentive awards and all warrants issued by Osmetech were exchanged for options and warrants exercisable for the common stock of the GenMark. Any historical discussion of GenMark relates to Osmetech and its consolidated subsidiaries prior to the reorganization. In September 2012, GenMark placed Osmetech into liquidation to simplify its corporate structure. The liquidation of Osmetech was competed in the fourth quarter of 2013.
We are a molecular diagnostics company focused on developing and commercializing our proprietary eSensor® detection technology. Our proprietary electrochemical technology enables fast, accurate and highly sensitive detection of multiple distinct biomarkers (e.g. up to 80) in a single sample. Our XT-8 system received 510(k) clearance from the United States Food and Drug Administration, or FDA, and is designed to support a broad range of molecular diagnostic tests with a compact and easy-to-use workstation and disposable test cartridges. Our XT-8 system supports up to 24 independent test cartridges, each of which can be run independently, resulting in a highly convenient and flexible workflow for our target customers, which are primarily hospitals and reference laboratories. As of December 31, 2014, we had an installed base of 540 XT-8 analyzers, or placements, with our customers.
Since inception, we have incurred net losses from operations each year, and we expect to continue to incur losses for the foreseeable future. Our losses attributable to operations for the years ended December 31, 2014, 2013, and 2012 were approximately $38.3 million, $33.6 million, and $22.1 million, respectively. As of December 31, 2014, we had an accumulated deficit of $262.5 million. Our operations to date have been funded principally through sales of capital stock, borrowings and cash from operations. We expect to incur increasing expenses over the next several years, principally to develop and commercialize our ePlex™ system and additional diagnostic tests, as well as to further increase our spending to manufacture, sell and market our products.
Our Products and Technology
We have a menu of eight tests for use with our XT-8 system. Four of our diagnostic tests have received FDA clearance, including our Cystic Fibrosis Genotyping Test, our Warfarin Sensitivity Test, our Thrombophilia Risk Test, and our Respiratory Viral Panel. In addition to these four FDA cleared tests, we have developed two hepatitis C virus, or HCV, genotyping tests, a 3A4/3A5 genotyping test, and a 2C19 genotyping test, versions of which are available for research use only (RUO).
In addition, we have recently completed the development stage of our ePlex system, which integrates automated nucleic acid extraction and amplification with our eSensor® detection technology to enable operators using the ePlex system to place a raw or a minimally prepared patient sample directly into our test cartridge and obtain results without any additional steps. This sample-to-answer capability is enabled by the robust nature of our eSensor® detection technology, which is not impaired by sample impurities that we believe hinder competing technologies. We have designed our ePlex system to further simplify
29
workflow and provide powerful, cost-effective molecular diagnostics solutions to a significantly expanded group of hospitals and reference laboratories. We are currently developing seven assays for our ePlex system, which include gram-positive (GP) and gram-negative (GN) sepsis panels, a respiratory panel (RP), a gastrointestinal (GI) pathogen panel, an HCV genotyping test (HCVg), a central nervous system (CNS) panel and a fungal panel (FP). We intend to continue investing in our ePlex system and its related test menu for the foreseeable future. We expect to initiate the European launch of the system in the middle of 2015, and launch the system in the United States in the first half of 2016.
Revenue
Revenue from operations includes product sales, principally of our diagnostic tests for use with our XT-8 system. We primarily place our XT-8 system with customers through a reagent rental agreement, under which we retain title to the instrument and customers commit to purchasing minimum quantities of reagents and test cartridges over a period of one to three years. We also offer our XT-8 system for sale.
Revenue also includes licensing revenue from the out-licensing of our electrochemical detection technology. We may enter into additional sub-licenses of our technology generating additional revenue, but do not anticipate that this will provide a significant portion of our future revenue.
Cost of Revenues
Cost of revenues includes the cost of materials, direct labor and manufacturing overhead costs used in the manufacture of our consumable test kits for our XT-8 system, including royalties on product sales. Cost of revenues also includes depreciation on revenue generating systems that have been placed with our customers under a reagent rental agreement, cost of systems sold to customers, amortization of licenses related to our products and other costs such as warranty, royalty and customer technical support. We manufacture our test cartridges in our facility and have recently invested in significant capacity for expansion. This potential underutilized capacity may result in a high cost of revenues relative to revenue, if manufacturing volumes are not able to fully absorb operating costs. Our XT-8 systems are procured from a contract manufacturer and are generally capitalized as fixed assets and depreciated on a straight line basis over their useful life as a charge to cost of revenues. We expect our cost of revenues to increase as we place additional XT-8 systems and manufacture and sell an increasing menu of accompanying diagnostic tests; however, we expect our gross margins related to our XT-8 system to increase as manufacturing efficiencies, improved procurement practices, instrument reliability increases and other improvements decrease costs as a percentage of sales.
Sales and Marketing Expenses
Sales and marketing expenses include costs associated with our direct sales force, sales management, marketing, technical support and business development activities. These expenses primarily consist of salaries, commissions, benefits, stock-based compensation, travel, advertising, promotions, samples and trade shows. We expect sales and marketing costs to increase as we scale-up our domestic and international commercial efforts to expand our customer base.
Research and Development Expenses
Research and development expenses primarily include expenses related to the development of our ePlex instrument and related test menu. These expenses also include certain clinical study expenses incurred in preparation for FDA clearance for these products, intellectual property prosecution and maintenance costs, and quality assurance expenses. The expenses primarily consisted of salaries, benefits, stock-based compensation costs, outside design and consulting services, laboratory supplies, contract research organization expenses, clinical study supplies and facility costs. We expense all research and development costs in the periods in which they are incurred.
General and Administrative Expenses
Our general and administrative expenses include expenses related to our executive, accounting and finance, compliance, information technology, legal, facilities, human resource, administrative and investor relations activities. These expenses consist primarily of salaries, benefits, stock-based compensation costs, independent auditor costs, legal fees, consultants, travel, insurance, and public company expenses, such as stock transfer agent fees and listing fees for NASDAQ. For the year ended December 2013, an impairment charge of $1.6 million related to previously capitalized license payments was included in generl and administrative expenses as a result of the termination of a license agreement.
Foreign Exchange Gains and Losses
30
Transactions in currencies other than our functional currency are translated at the prevailing rates on the dates of the applicable transaction. Foreign exchange gains and losses arise from differences in exchange rates during the period between the date a transaction denominated in a foreign currency is consummated and the date on which it is settled or translated. Prior to our initial public offering in 2010, exchange gains and losses included those arising on cash balances held by Osmetech denominated in currencies other than its functional currency, the British Pound. Since the initial public offering, the functional currency of GenMark has been the U.S. dollar. Since the initial public offering, foreign exchange gains and losses have been primarily related to amounts due under a single license agreement, which were denominated in Euros. In connection with the liquidation of Osmetech plc in the fourth quarter of 2013, we realized a translation loss of $450,000 to eliminate accumulated other comprehensive loss.
Interest Income and Interest Expense
Interest income includes interest earned on our cash and cash equivalents and investments. Interest expense represents interest incurred on our loan payable and on other liabilities.
Provision for Income Taxes
We make certain estimates and judgments in determining income tax expense for financial statement purposes. These estimates and judgments occur in the calculation of certain tax assets and liabilities, which arise from differences in the timing of recognition of revenue and expense for tax and financial statement purposes.
We assess the likelihood that we will be able to recover our deferred tax assets. We consider all available evidence, both positive and negative, including historical levels of income, expectations and risks associated with estimates of future taxable income, and ongoing prudent and feasible tax planning strategies in assessing the need for the valuation allowance. If it is more likely than not that we will not recover our deferred tax assets, we will increase our provision for income taxes by recording a valuation allowance against the deferred tax assets that we estimate will not ultimately be recoverable.
Our income tax returns are based on calculations and assumptions that are subject to examination by the Internal Revenue Service and other tax authorities. In addition, the calculation of our tax liabilities involves dealing with uncertainties in the application of complex tax regulations. We recognize liabilities for uncertain tax positions based on a two-step process. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates that it is more likely than not that the position will be sustained on audit, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit as the largest amount that is more than 50% likely of being realized upon settlement. While we believe we have appropriate support for the positions taken on our tax returns, we regularly assess the potential outcomes of examinations by tax authorities in determining the adequacy of our provision for income taxes. We continually assess the likelihood and amount of potential adjustments and adjust the income tax provision, income taxes payable and deferred taxes in the period in which the facts that give rise to a revision become known.
Critical Accounting Policies and Significant Judgments and Estimates
Revenue
We recognize revenue from product sales and contractual arrangements, net of discounts and sales related taxes. We recognize revenue from product sales when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed or determinable and collectability is reasonably assured. Where applicable, all revenue is stated net of sales taxes and trade discounts.
We offer customers the choice to either purchase a system outright or to receive possession of a system free of charge in exchange for a commitment to purchase an annual minimum amount of molecular diagnostic test cartridges.
When a system is sold, revenue is generally recognized upon shipment of the unit consistent with contract terms. When a system is placed free of charge under a "reagent rental" agreement, we retain title to the equipment and it remains capitalized on our balance sheet under property and equipment. Under our reagent rental agreements, our customers pay a system usage fee, which is included in the price of each test cartridge purchased. Our reagents and diagnostic test cartridges (consumables) are priced to include the expense of system usage and maintenance of the system and are included in product revenue in our consolidated financial statements.
We sell our durable systems and disposable test cartridges through a direct sales force in the United States. The system price is not dependent upon the purchase of any amount of disposable test cartridges. Revenue on system and test cartridge sales is generally recognized upon shipment consistent with contract terms, which is when title and the risk of loss and rewards of ownership have been transferred to the customer and there are no other post-shipment obligations.
31
Revenue related to royalties received from licenses is generally recognized evenly over the contractual period to which the license relates.
In those cases where we bill shipping and handling costs to customers, the amounts billed are classified as other revenue.
Allowance for Doubtful Accounts Receivable
We maintain an allowance for doubtful accounts for estimated losses resulting from the inability of our customers to make required payments. Our allowance for doubtful accounts is based on our assessment of the collectability of specific customer accounts, the aging of accounts receivable, and the general condition of the economy. Changes to the allowance for doubtful accounts are charged to sales and marketing expense.
Inventory
We value inventories at the lower of cost or market on a part-by-part basis and provide an inventory reserve for estimated obsolescence and excess inventory based upon historical turnover and assumptions about future demand for our products and market conditions. We determine excess and obsolete inventories based on an estimate of the future demand for our products within a specified time horizon, generally 12 months. The estimates we use for demand are also used for near-term capacity planning and inventory purchasing and are consistent with our revenue forecasts. If our actual demand is less than our forecast demand, we may be required to take additional excess inventory charges, which would decrease gross margin and adversely impact net operating results in the future.
Property and Equipment — net
Property, equipment and leasehold improvements are recorded at cost and depreciated using the straight-line method over the assets’ estimated useful lives, which are noted below. We generally capitalize our XT-8 instruments, and provide these to customers for no charge. Each category of property and equipment is analyzed to determine its useful life. We look at the manufacturers’ estimates of useful life and adjust these for actual experience in our operating environment. Useful lives are reviewed periodically and occasionally changed as circumstances dictate.
Machinery and laboratory equipment | 3 - 5 years |
XT-8 Instruments | 4 years |
Office equipment | 5 years |
Leasehold improvements | over the shorter of the remaining life of the lease or the useful economic life of the asset |
Repair and maintenance costs are expensed as incurred. During 2014, we disposed of certain assets no longer in use with a net book value of $102,000, recorded in operations. During 2013, we recorded a one-time impairment charge of $302,000 related to production equipment which had been built for NMTC.
Impairment of Long-Lived Assets
We assess the recoverability of long-lived assets, including intangible assets and systems at customer locations by periodically evaluating the carrying value of such assets whenever events or changes in circumstances indicate that the carrying amount of these assets may not be recoverable. If impairment is indicated, we write down the carrying value of the asset to the estimated fair value. During the year ended December 31, 2013, we recorded an impairment charge of $1,624,000 related to previously capitalized payments made under a license agreement, which we terminated in December 2013.
Stock-Based Compensation
We grant stock options with an exercise price equal to the closing price of our common stock on the NASDAQ Global Market on each grant date. We use the Black-Scholes option-pricing model as the method for determining the estimated fair value of stock options and we use the grant date fair value of our common stock for valuing restricted stock awards. The estimated fair value of stock-based awards exchanged for employee and non-employee director services are expensed over the requisite service period. The stock-based compensation expense related to shares issued under our 2013 Employee Stock Purchase Plan is also estimated using the Black-Scholes option-pricing model. The Black-Scholes model requires the use of highly subjective and complex assumptions which determine the fair value of stock-based awards, including the stock option’s expected term and the price volatility of the underlying stock. These assumptions include:
32
• | Expected Term. The expected term represents the period that our stock-based awards are expected to be outstanding and is determined by using the simplified method. |
• | Expected Volatility. Expected volatility represents the volatility in our stock price expected over the expected term of the stock option. |
• | Expected Dividend. The Black-Scholes option-pricing model calls for a single expected dividend yield as an input. We assumed no dividends as we have never paid dividends and have no plans to do so. |
• | Risk-Free Interest Rate. The risk-free interest rate used in the Black-Scholes option-pricing model is based on published government rates in effect at the time of grant for periods corresponding with the expected term of the option. |
Income Taxes
Our income tax expense, deferred tax assets and liabilities and reserves for unrecognized tax benefits reflect management’s best assessment of estimated future taxes to be paid. We are currently subject to income taxes only in the United States but have been subject to income taxes in both the United States and the United Kingdom in previous years. Significant judgments and estimates are required in determining our consolidated income tax expense.
We believe that it is more likely than not that the benefit from our deferred tax assets will not be realized. In recognition of this risk, we have provided a full valuation allowance on the net deferred tax assets relating to our net operating loss carryforwards and other deferred tax assets. If our assumptions change and we determine that we will be able to realize our deferred tax assets, the tax benefits relating to any reversal of the valuation allowance on deferred tax assets will be accounted for as a reduction of income tax expense.
Changes in tax laws and rates could also affect recorded deferred tax assets and liabilities in the future. Management is not aware of any such changes that would have a material effect on our results of operations, cash flows or financial position.
We recognize tax liabilities in accordance with ASC Topic 740 and we adjust these liabilities when our judgment changes as a result of the evaluation of new information not previously available. Due to the complexity of some of these uncertainties, the ultimate resolution may result in a payment that is materially different from our current estimate of the tax liabilities. These differences will be reflected as increases or decreases to income tax expense in the period in which they are determined.
Recent Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standard Board, or the FASB, or other standard setting bodies that the Company adopts as of the specified effective date. The Company believes that the impact of recently issued standards that are not yet effective will not have a material impact on its financial position or results of operations upon adoption.
In May 2014, the FASB issued Accounting Standards Update (ASU) 2014-09, Revenue from Contracts with Customers. The new standard is based on the principle that revenue should be recognized to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. ASU 2014-09 will be effective for the Company beginning in the first quarter of fiscal 2017 and allows for a full retrospective or a modified retrospective adoption approach. The Company is currently evaluating the impact of ASU 2014-09 on its consolidated financial statements.
Comparison of Years Ended December 31, 2014, 2013 and 2012 (in thousands):
Years ended December 31, | 2014 vs 2013 | 2013 vs 2012 | |||||||||||||||||||||||
2014 | 2013 | 2012 | $ Change | % Change | $ Change | % Change | |||||||||||||||||||
Revenue | $ | 30,594 | $ | 27,404 | $ | 20,469 | $ | 3,190 | 12 | % | $ | 6,935 | 34 | % | |||||||||||
Our revenue consists primarily of revenue from the sale of test cartridges and reagents (consumables) with a small component from our sale of instruments and other revenue.
A former customer, Natural Molecular Testing Corporation, or NMTC, filed for bankruptcy during 2013. During the year ended December 31, 2013, revenue from this former customer, NMTC, was $8,162,000. Our base business excludes revenues this former customer.
33
For the year ended December 31, 2014, excluding this former customer our base business grew 59%, or $11,353,000, compared to the same period of 2013. Consumables revenue from our base business during the year ended December 31, 2014 increased by 71% to $29,235,000 compared to $17,123,000 in the prior year period. This increase in consumable revenue was primarily driven by increases in purchases of our infectious disease assays and a 31% increase in the number of XT-8 analyzers installed at customer locations to 540 at December 31, 2014 from 413 as of December 31, 2013. Pricing changes did not have an impact on revenue during the current period.
During the year ended December 31, 2013, our base business grew 120% to approximately $19,242,000 compared with $8,756,000 for the same period ended December 31, 2012. Consumables revenue from our base business during the year ended December 31, 2013 increased by 117% to $17,123,000 compared to $7,906,000 in the same period of 2012. This increase in consumable revenue was primarily driven by a 39% increase in the number of XT-8 analyzers installed at customer locations to 413 at December 31, 2013 from 297 as of December 31, 2012. The increase in consumables revenue was not attributable to any one assay. Pricing changes did not have an impact on revenue. In addition to the increase in reagent revenue, higher instrument sales during the twelve months ended December 31, 2013 resulted in an additional $1,124,000 of revenue over the same period in 2012.
Years ended December 31, | 2014 vs 2013 | 2013 vs 2012 | ||||||||||||||||||||||
2014 | 2013 | 2012 | $ Change | % Change | $ Change | % Change | ||||||||||||||||||
Cost of Revenue | $ | 13,127 | $ | 15,570 | 11,640 | $ | (2,443 | ) | (16 | )% | $ | 3,930 | 34 | % | ||||||||||
Gross Profit | $ | 17,467 | $ | 11,834 | 8,829 | $ | 5,633 | 48 | % | $ | 3,005 | 34 | % | |||||||||||
The decrease in cost of revenue for the twelve months ended December 31, 2014 compared to the twelve months ended December 31, 2013 was primarily related to the one-time inventory reserve expense recorded in 2013, of $1,192,000 related to the NMTC bankruptcy, in addition to a $748,000 decrease in personnel related costs and a $404,000 decrease in amortization and depreciation expense. These decreases were partially offset by increased royalties expense of $227,000. The improvement to gross profit during the year ended December 31, 2014, compared to December 31, 2013, was due to reduced excess and obsolete expense related to the NMTC bankruptcy, increased sales of higher margin products and a reduction in manufacturing personnel costs.
The increase in cost of revenue for the year ended December 31, 2013 compared to the year ended December 31, 2012 was primarily the result of the increase in consumable revenues, specifically a $2,097,000 increase in standard product costs and the one-time inventory reserve expense recorded in 2013, of $1,192,000 related to NMTC. The increase was also due to higher facility and depreciation expenses of $832,000 due to our expanded production infrastructure and impairment of production equipment related to NMTC, in addition to increased royalties and license amortization expense of $564,000 and instrument depreciation, warranty and repair of $365,000. We also incurred higher personnel related costs of $361,000 to support our increased manufacturing volumes, all of which were partially offset by higher absorption and manufacturing efficiencies of $187,000. The improvement to gross profit during the year ended December 31, 2013 compared to the year ended December 31, 2012 was primarily due to increased sales volumes and manufacturing and absorption efficiencies.
Years ended December 31, | 2014 vs 2013 | 2013 vs 2012 | ||||||||||||||||||||||
2014 | 2013 | 2012 | $ Change | % Change | $ Change | % Change | ||||||||||||||||||
Sales and Marketing | $ | 12,629 | $ | 12,818 | 6,378 | $ | (189 | ) | (1 | )% | $ | 6,440 | 101 | % | ||||||||||
The decrease in sales and marketing expense for the year ended December 31, 2014, compared to the year ended December 31, 2013, was primarily driven by a one-time charge to bad debt expense of $2,702,000 related to amounts owed by NMTC which was recorded in 2013, which was offset by an increase in salaries and employee related expenses of $2,187,000, including increased stock-based compensation expense of $625,000, an increase in marketing communications and trade show expenses of $323,000 and travel expenses of $207,000, in each case incurred in connection with expanding our domestic and international organization.
The increase in sales and marketing expense for the year ended December 31, 2013, compared to the year ended December 31, 2012, was primarily driven by a one-time charge to bad debt expense of $2,702,000, related to amounts owed by NMTC which was recorded in 2013. A significant portion of the additional increase in expense was related to our commitment to expanding our domestic and international organization, including, an increase in salary expense of $1,326,000, an increase in commissions and bonus expense of $548,000, additional stock-based compensation of $623,000, higher samples expense of $153,000 for new customers, consulting expense of $482,000, and travel expenses of $166,000.
34
Years ended December 31, | 2014 vs 2013 | 2013 vs 2012 | ||||||||||||||||||||||
2014 | 2013 | 2012 | $ Change | % Change | $ Change | % Change | ||||||||||||||||||
General and Administrative | $ | 12,069 | $ | 11,836 | 10,806 | $ | 233 | 2 | % | $ | 1,030 | 10 | % | |||||||||||
The increase in general and administrative expense for the year ended December 31, 2014 compared to the year ended December 31, 2013 was primarily due to employee related expenses of $1,377,000, including increased stock-based compensation expense of $786,000, in addition to increased bank and payroll fees of $221,000 and building lease and utilities expenditures of $208,000, partially offset by a $1,622,000 decrease in license expense resulting from a one-time impairment charge from the termination of a license agreement in 2013.
The increase from December 31, 2013, compared to the year ended December 31, 2012, was primarily due to the one-time impairment charge of $1,624,000 noted above and a medical device tax due to new legislation of $324,000, which was partially offset by lower professional services of $439,000, lower consulting fees of $274,000 and lower legal fees $158,000.
Years ended December 31, | 2014 vs 2013 | 2013 vs 2012 | ||||||||||||||||||||||
2014 | 2013 | 2012 | $ Change | % Change | $ Change | % Change | ||||||||||||||||||
Research and Development | $ | 31,823 | $ | 22,060 | 13,536 | $ | 9,763 | 44 | % | $ | 8,524 | 63 | % | |||||||||||
The increase in research and development expense for the year ended December 31, 2014, compared to the year ended December 31, 2013, was primarily due to an increase in costs associated with the development of our ePlex system of $6,109,000 and an increase in ePlex assay development costs of $3,556,000, comprised largely of employee related expenses, outside services and prototype materials.
The increase in research and development expense for the year ended December 31, 2013, compared to the year ended December 31, 2012, was primarily due to an increase in costs associated with the development of our ePlex system of $5,476,000 and an increase in ePlex assay development costs of $2,894,000, largely as a result of increases in research and development personnel.
Years ended December 31, | 2014 vs 2013 | 2013 vs 2012 | ||||||||||||||||||||||
2014 | 2013 | 2012 | $ Change | % Change | $ Change | % Change | ||||||||||||||||||
Other Income (Expense) | $ | 218 | $ | 1,281 | (64 | ) | $ | (1,063 | ) | (83 | )% | $ | 1,345 | (2,102 | )% | |||||||||
The change in other income (expense) for the year ended December 31, 2014, compared to the same period of the prior year, was due primarily to the sale of our preferred stock investment in Advanced Liquid Logic, Inc., or ALL, in connection with ALL's acquisition by Illumina, Inc. resulting in a $1,392,000 realized gain in 2013 and an increase in amortization of premiums on marketable securities of $388,000, partially offset by the absence of the $450,000 loss realized upon liquidation of our U.K. subsidiary Osmetech in the fourth quarter of 2013.
The change for the year ended December 31, 2013, compared to the year ended December 31, 2012, was primarily due to the sale of our preferred stock investment in ALL resulting in a $1,392,000 realized gain in 2013, and an increase in interest income earned on investments of $722,000. These gains were partially offset by amortization of premiums on marketable securities of $314,000 and $450,000 of accumulated other comprehensive loss, which was realized upon the liquidation of our U.K. subsidiary Osmetech in the fourth quarter of 2013.
Years ended December 31, | 2014 vs 2013 | 2013 vs 2012 | ||||||||||||||||||||||
2014 | 2013 | 2012 | $ Change | % Change | $ Change | % Change | ||||||||||||||||||
Income Tax Expense (Benefit) | $ | (573 | ) | $ | 44 | 148 | $ | (617 | ) | (1,402 | )% | $ | (104 | ) | (70 | )% | ||||||||
Due to net losses incurred, we have only recorded tax provisions associated with uncertain tax positions, interest on uncertain tax positions and minimum tax payments. The decrease in income tax expense for the year ended December 31, 2014 is primarily due to a reduction in the unrecognized tax benefits associated with uncertain tax positions of $610,000, as a result of the expiration of applicable statutes of limitations.
Liquidity and Capital Resources
35
To date we have funded our operations primarily from the sale of our common stock, borrowings and cash from operations. We have incurred net losses from operations each year and have not yet achieved profitability. At December 31, 2014, we had $65,814,000 of working capital, including $70,506,000 in cash, cash equivalents, and marketable securities.
Cash Flows
The following table shows cash flow information for the years ended December 31, 2014, 2013 and 2012:
Years Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Cash used in operating activities | $ | (29,572 | ) | $ | (23,796 | ) | $ | (16,243 | ) | ||
Cash provided by (used in) investing activities | 29,417 | (72,564 | ) | (2,146 | ) | ||||||
Cash provided by financing activities | 1,287 | 80,833 | 44,319 | ||||||||
Net increase (decrease) in cash and cash equivalents | $ | 1,132 | $ | (15,527 | ) | $ | 25,930 | ||||
Cash flows used in operating activities
In 2014, net cash used in operating activities was $29,572,000, primarily driven by a net loss of $38,263,000, adjusted for non-cash charges of $9,789,000 and changes in operating assets and liabilities resulting in a net decrease in cash of $1,098,000 during the period. The non-cash charges in operating activities, the primarily comprised of stock-based compensation of $5,796,000 an increase from prior year due to increases in personnel, depreciation and amortization of $3,358,000 as a result of increases in property and equipment and intangible licenses and accretion of premiums on investments of $702,000. The decrease in cash due to changes in operating assets and liabilities was primarily attributable to a $2,030,000 increase in accounts receivable resulting from stronger fourth quarter revenues in the current year compared to prior year, an increase in other liabilities of $537,000 and an increase in inventories of $229,000 to meet increasing demands, partially offset by a $1,797,000 increase in accrued compensation.
Cash flows used in investing activities
Net cash provided by investing activities of $29,417,000 was primarily due to the maturity of marketable securities of $56,050,000 and proceeds from the sale of marketable securities of $7,497,000. These cash inflows were partially offset by $28,054,000 in purchases of marketable securities and $5,726,000 in property, plant and equipment acquired in the year ended December 31, 2014.
Cash flows provided by financing activities
Net cash provided by financing of $1,287,000 for the twelve months ended December 31, 2014, was primarily due to proceeds from the issuance of stock under our employee stock purchase plan of $812,000, and proceeds from the exercise of employee stock options of $531,000.
We have prepared cash flow forecasts which indicate, based on our current cash resources available, that we will have sufficient resources to fund our business for at least the next 12 months. We expect capital outlays and operating expenditures to increase over the next several years as we grow our customer base and revenues, and expand our research and development, commercialization and manufacturing activities. Factors that could affect our capital requirements, in addition to those previously identified, include, but are not limited to:
• | the level of revenues and the rate of our revenue growth; |
• | change in demand from our customers; |
• | the level of expenses required to expand our U.S. and international commercial (sales and marketing) activities; |
• | the level of research and development investment required to and develop and commercialize our ePlex system and related test menu and maintain our XT-8 system; |
• | our need to acquire or license complementary technologies; |
• | the costs of filing, prosecuting, defending and enforcing patent claims and other intellectual property rights; |
• | competing technological and market developments; and, |
• | changes in regulatory policies or laws that affect our operations. |
36
In September 2012, we entered into a term loan with Banc of California, consisting of the following two loans.
1) | We increased the letter of credit provided to our landlord of our Carlsbad, California facility to $758,000 from the previous letter of credit of $500,000. The increase in the letter of credit was required by our landlord pursuant to our second and third amendments to the lease for our Carlsbad, California location, in connection with our lease of additional space at this facility. This letter of credit was secured with $758,000 of restricted cash at December 31, 2014. |
2) | We obtained a variable rate term loan from Banc of California in the amount of $836,000 with an initial interest rate of 3.75%. This term loan replaced the Square 1 equipment loan of the same amount with an interest rate of 6.75%. We repaid all outstanding amounts under this loan in the third quarter of 2013. |
On August 19, 2013, we completed the public offering of 8,765,000 shares of our common stock at a price of $9.84 per share and raised approximately $80,672,000 in net proceeds.
In January 2015, we entered into a loan agreement with General Electric Capital Corporation and certain financial institutions, as lenders. Pursuant to the agreement, the lenders have agreed to provide us with (a) up to $35,000,000 in a series of term loans and (b) a revolving loan in the maximum amount of $5,000,000, each of which is subject to certain terms and conditions as set forth in the agreement.
Although we do not currently anticipate requiring additional capital, if additional capital is required, we cannot be certain that it will be available when needed or that our actual cash requirements will not be greater than anticipated. If we raise additional funds through the issuance of equity or convertible debt securities, the percentage ownership of our stockholders could be significantly diluted, and these newly issued securities may have rights, preferences or privileges senior to those of existing stockholders. If we raise additional funds through collaborations and licensing arrangements, we might be required to relinquish significant rights to our technologies or products, or grant licenses on terms that are not favorable to us.
Contractual Obligations
As of December 31, 2014, we had the following contractual obligations (in thousands):
Payments due by period | |||||||||||||||||||
Total | Less than 1 Year | 1-3 Years | 4-5 Years | After 5 Years | |||||||||||||||
Operating lease obligations (1) | $ | 7,522 | $ | 1,084 | $ | 2,239 | $ | 2,346 | $ | 1,853 | |||||||||
Licensing payment obligation | 1,446 | 1,200 | 166 | 80 | — | ||||||||||||||
Instrument purchase obligations | 669 | 669 | — | — | — | ||||||||||||||
Total obligations | $ | 9,637 | $ | 2,953 | $ | 2,405 | $ | 2,426 | $ | 1,853 | |||||||||
_____________________
(1) | We enter into operating leases in the ordinary course of business with respect to facilities. Our lease agreements have fixed payment terms based on the passage of time. Certain facility leases require payment of maintenance and real estate taxes. Our future operating lease obligations could change if we terminate certain contracts or if we enter into additional operating leases. |
In February 2011, we entered into a 36-month operating lease for office equipment with total lease payments of $85,000. In conjunction with the lease, the lessor paid us approximately $27,000 to payoff previous contracts for similar equipment leased from a different vendor.
In January 2012, we entered into a lease amendment with the landlord of our Carlsbad, California facility to rent an additional 22,000 square feet. The lease amendment required an additional security deposit of $22,000 and an increase in our standby letter of credit to $758,000. We took possession of the additional space on January 1, 2013, at which time the rent increased by approximately $35,000 per month, subject to annual increases of between 3% and 4%. The term of the lease was also extended to June 30, 2021.
In August 2012, we entered into a three year supply agreement with Leica for the purchase of our XT-8 instrument. Amounts reported in the table above reflect minimum purchase commitments under this supply agreement which we can satisfy through instrument purchases or the payment of a designated fee for each instrument we fail to purchase under the prescribed minimum amounts, subject to certain permitted exclusions.
37
Impact of Inflation
The effect of inflation and changing prices on our operations was not significant during the periods presented.
Off-Balance Sheet Arrangements
We have no off-balance sheet arrangements. We have provided a $758,000 standby letter of credit to our landlord as security for future rent in connection the lease of our Carlsbad, California facility, which is recorded as restricted cash on the balance sheet.
Item 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Quantitative and Qualitative Disclosures about Market Risk
Our exposure to market risk is limited to our cash and cash equivalents, all of which have maturities of less than three months, and marketable securities, which have maturities of less than one year. The goals of our investment policy are preservation of capital, fulfillment of liquidity needs and fiduciary control of cash and investments. We also seek to maximize income from our investments without assuming significant risk. To achieve our goals, we may in the future maintain a portfolio of cash equivalents and investments in a variety of securities that management believes to be of high credit quality. We currently do not hedge interest rate exposure. Because of the short-term maturities of our cash equivalents and short-term investments, we do not believe that an increase in market rates would have a material negative impact on the value of our portfolio.
Interest Rate Risk
As of December 31, 2014, based on current interest rates and total borrowings outstanding, a hypothetical 100 basis point increase or decrease in interest rates would have an insignificant pre-tax impact on our results of operations.
Foreign Currency Exchange Risks
All of our operating facilities are located within the United States. We are a U.S. entity and our functional currency is the U.S. dollar. Virtually all of our revenues are based in the United States. We currently have no material operations outside of the United States, which significantly diminishes the extent of any foreign currency exchange risk we currently face.
38
Item 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
REPORT OF ERNST & YOUNG LLP, INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of GenMark Diagnostics, Inc.
We have audited the accompanying consolidated balance sheet of GenMark Diagnostics, Inc. as of December 31, 2014 and 2013, and the related consolidated statements of comprehensive loss, stockholders' equity and cash flows for the years then ended. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of GenMark Diagnostics, Inc. at December 31, 2014 and 2013, and the consolidated results of its operations and its cash flows for the years then ended, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), GenMark Diagnostics, Inc.'s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated February 24, 2015 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
San Diego, California
February 24, 2015
39
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
GenMark Diagnostics, Inc.
Carlsbad, California
We have audited the accompanying consolidated statements of comprehensive loss, stockholders’ equity, and cash flows for the year ended December 31, 2012 of GenMark Diagnostics, Inc. and subsidiaries (the “Company”) (formerly Osmetech plc and subsidiaries). These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provides a reasonable basis for our opinion.
In our opinion, such consolidated financial statements present fairly, in all material respects, the consolidated results of GenMark Diagnostics, Inc. and subsidiaries’ operations and its cash flows for the year ended December 31, 2012, in conformity with accounting principles generally accepted in the United States of America.
/s/ DELOITTE & TOUCHE LLP
San Diego, California
March 14, 2013
40
GENMARK DIAGNOSTICS, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except par value)
As of December 31, | |||||||
2014 | 2013 | ||||||
Current assets | |||||||
Cash and cash equivalents | $ | 36,855 | $ | 35,723 | |||
Marketable securities | 33,651 | 69,866 | |||||
Accounts receivable, net of allowances of $2,702 and $2,736, respectively | 4,889 | 2,859 | |||||
Inventories | 2,137 | 2,102 | |||||
Prepaid expenses and other current assets | 575 | 552 | |||||
Total current assets | 78,107 | 111,102 | |||||
Property and equipment, net | 11,052 | 8,591 | |||||
Intangible assets, net | 1,870 | 1,197 | |||||
Restricted cash | 758 | 758 | |||||
Other long-term assets | 183 | 106 | |||||
Total assets | $ | 91,970 | $ | 121,754 | |||
Current liabilities | |||||||
Accounts payable | $ | 3,468 | $ | 3,863 | |||
Accrued compensation | 5,172 | 3,375 | |||||
Loan payable | — | 37 | |||||
Other current liabilities | 3,653 | 2,962 | |||||
Total current liabilities | 12,293 | 10,237 | |||||
Long-term liabilities | |||||||
Deferred rent | 1,445 | 1,601 | |||||
Other non-current liabilities | 208 | 748 | |||||
Total liabilities | 13,946 | 12,586 | |||||
Commitments and contingencies—See note 7 | |||||||
Stockholders’ equity | |||||||
Preferred stock, $0.0001 par value; 5,000 authorized, none issued | — | — | |||||
Common stock, $0.0001 par value; 100,000 authorized; 41,859 and 41,520 shares issued and outstanding as of December 31, 2014 and December 31, 2013, respectively | 4 | 4 | |||||
Additional paid-in capital | 340,502 | 333,363 | |||||
Accumulated deficit | (262,472 | ) | (224,209 | ) | |||
Accumulated other comprehensive income (loss) | (10 | ) | 10 | ||||
Total stockholders’ equity | 78,024 | 109,168 | |||||
Total liabilities and stockholders’ equity | $ | 91,970 | $ | 121,754 | |||
See accompanying notes to consolidated financial statements.
41
GENMARK DIAGNOSTICS, INC.
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(In thousands, except per share data)
Years ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Revenue | |||||||||||
Product revenue | $ | 30,328 | $ | 27,204 | $ | 20,211 | |||||
License and other revenue | 266 | 200 | 258 | ||||||||
Total revenue | 30,594 | 27,404 | 20,469 | ||||||||
Cost of revenue | 13,127 | 15,570 | 11,640 | ||||||||
Gross profit | 17,467 | 11,834 | 8,829 | ||||||||
Operating expenses | |||||||||||
Sales and marketing | 12,629 | 12,818 | 6,378 | ||||||||
General and administrative | 12,069 | 11,836 | 10,806 | ||||||||
Research and development | 31,823 | 22,060 | 13,536 | ||||||||
Total operating expenses | 56,521 | 46,714 | 30,720 | ||||||||
Loss from operations | (39,054 | ) | (34,880 | ) | (21,891 | ) | |||||
Other income (expense) | |||||||||||
Interest income | 244 | 403 | 42 | ||||||||
Interest expense | (20 | ) | (19 | ) | (90 | ) | |||||
Other income (expense) | (6 | ) | 897 | (16 | ) | ||||||
Total other income (expense) | 218 | 1,281 | (64 | ) | |||||||
Loss before provision for income taxes | (38,836 | ) | (33,599 | ) | (21,955 | ) | |||||
Income tax expense (benefit) | (573 | ) | 44 | 148 | |||||||
Net loss | $ | (38,263 | ) | $ | (33,643 | ) | $ | (22,103 | ) | ||
Net loss per share, basic and diluted | $ | (0.93 | ) | $ | (0.95 | ) | $ | (0.84 | ) | ||
Weighted average number of shares outstanding basic and diluted | 41,346 | 35,253 | 26,215 | ||||||||
Other comprehensive loss | |||||||||||
Net loss | $ | (38,263 | ) | $ | (33,643 | ) | $ | (22,103 | ) | ||
Net unrealized losses on marketable securities, net of tax | (20 | ) | (4 | ) | — | ||||||
Comprehensive loss | $ | (38,283 | ) | $ | (33,647 | ) | $ | (22,103 | ) | ||
See accompanying notes to consolidated financial statements.
42
GENMARK DIAGNOSTICS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(In thousands)
Common Stock | Additional paid-in capital | Accumulated other comprehensive loss | Accumulated deficit | Total stockholders' equity | ||||||||||||||||||
Shares | Par Value | |||||||||||||||||||||
Balance—December 31, 2011 | 20,478 | $ | 2 | $ | 199,531 | $ | (436 | ) | $ | (168,463 | ) | $ | 30,634 | |||||||||
Issuance of stock in lieu of accrued bonuses | 93 | — | 255 | — | — | 255 | ||||||||||||||||
Stock-based compensation expense | — | — | 2,352 | — | — | 2,352 | ||||||||||||||||
Shares issued under stock-based compensation plans, net of cancellations | 682 | — | 223 | — | — | 223 | ||||||||||||||||
Issuance of common stock, net of offering expenses | 11,500 | 1 | 45,088 | — | — | 45,089 | ||||||||||||||||
Net loss | — | — | — | — | (22,103 | ) | (22,103 | ) | ||||||||||||||
Balance—December 31, 2012 | 32,753 | 3 | 247,449 | (436 | ) | (190,566 | ) | 56,450 | ||||||||||||||
Issuance of stock in lieu of accrued bonuses | — | — | 653 | — | — | 653 | ||||||||||||||||
Stock-based compensation expense | — | — | 3,893 | — | — | 3,893 | ||||||||||||||||
Issuance of employee stock purchase plan shares | 33 | — | 300 | — | — | 300 | ||||||||||||||||
Restricted stock awards issued, net of cancellations | (122 | ) | — | — | — | — | — | |||||||||||||||
Shares issued under stock-based compensation plans | 91 | — | 396 | — | — | 396 | ||||||||||||||||
Issuance of common stock, net of offering expenses | 8,765 | 1 | 80,672 | — | — | 80,673 | ||||||||||||||||
Elimination of cumulative foreign currency translation adjustments upon liquidation of foreign subsidiary | — | — | — | 450 | — | 450 | ||||||||||||||||
Net loss | — | — | — | — | (33,643 | ) | (33,643 | ) | ||||||||||||||
Unrealized loss on marketable securities | — | — | — | (4 | ) | — | (4 | ) | ||||||||||||||
Balance—December 31, 2013 | 41,520 | 4 | 333,363 | 10 | (224,209 | ) | 109,168 | |||||||||||||||
Stock-based compensation expense | — | — | 5,796 | — | — | 5,796 | ||||||||||||||||
Issuance of employee stock purchase plan shares | 89 | — | 812 | — | — | 812 | ||||||||||||||||
Restricted stock awards issued, net of cancellations | 149 | — | — | — | — | — | ||||||||||||||||
Shares issued under stock-based compensation plans | 101 | — | 531 | — | — | 531 | ||||||||||||||||
Net loss | — | — | — | — | (38,263 | ) | (38,263 | ) | ||||||||||||||
Unrealized loss on marketable securities | — | — | — | (20 | ) | — | (20 | ) | ||||||||||||||
Balance—December 31, 2014 | 41,859 | $ | 4 | $ | 340,502 | $ | (10 | ) | $ | (262,472 | ) | $ | 78,024 | |||||||||
See accompanying notes to consolidated financial statements.
43
GENMARK DIAGNOSTICS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
Years ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Operating activities: | |||||||||||
Net loss | $ | (38,263 | ) | $ | (33,643 | ) | $ | (22,103 | ) | ||
Adjustments to reconcile net loss to net cash used in operating activities: | |||||||||||
Depreciation and amortization | 2,656 | 2,530 | 1,198 | ||||||||
Amortization of premiums on marketable securities | 702 | 314 | — | ||||||||
Stock-based compensation | 5,796 | 3,893 | 2,352 | ||||||||
Provision for bad debt | — | 2,721 | (24 | ) | |||||||
Non-cash inventory adjustments | 450 | 1,779 | (482 | ) | |||||||
Gain on sale of investment in preferred stock | — | (1,392 | ) | — | |||||||
Elimination of cumulative foreign currency translation adjustments upon liquidation of foreign subsidiary | — | 450 | — | ||||||||
Impairment of intangible asset | — | 1,624 | — | ||||||||
Other non-cash adjustments | 185 | — | — | ||||||||
Changes in operating assets and liabilities: | |||||||||||
Accounts receivable | (2,030 | ) | (2,390 | ) | (2,068 | ) | |||||
Inventories | (229 | ) | (1,313 | ) | 880 | ||||||
Prepaid expenses and other assets | (184 | ) | (119 | ) | 68 | ||||||
Accounts payable | 85 | 1,343 | 728 | ||||||||
Accrued compensation | 1,797 | 951 | 1,811 | ||||||||
Other liabilities | (537 | ) | (544 | ) | 1,397 | ||||||
Net cash used in operating activities | (29,572 | ) | (23,796 | ) | (16,243 | ) | |||||
Investing activities | |||||||||||
Change in restricted cash | — | 585 | (1,343 | ) | |||||||
Payments for intellectual property licenses | (350 | ) | (882 | ) | (1,327 | ) | |||||
Purchases of property and equipment | (5,726 | ) | (4,270 | ) | (3,476 | ) | |||||
Purchases of marketable securities | (28,054 | ) | (76,190 | ) | (1,000 | ) | |||||
Proceeds from sales of marketable securities | 7,497 | 6,643 | — | ||||||||
Maturities of marketable securities | 56,050 | 1,550 | 5,000 | ||||||||
Net cash provided by (used in) investing activities | 29,417 | (72,564 | ) | (2,146 | ) | ||||||
Financing activities | |||||||||||
Proceeds from issuance of common stock | 812 | 86,547 | 48,300 | ||||||||
Costs incurred in conjunction with public offering | — | (5,510 | ) | (3,211 | ) | ||||||
Principal repayment of borrowings | (56 | ) | (766 | ) | (1,984 | ) | |||||
Proceeds from borrowings | — | 166 | 991 | ||||||||
Proceeds from stock option exercises | 531 | 396 | 223 | ||||||||
Net cash provided by financing activities | 1,287 | 80,833 | 44,319 | ||||||||
Net increase (decrease) in cash and cash equivalents | 1,132 | (15,527 | ) | 25,930 | |||||||
Cash and cash equivalents at beginning of year | 35,723 | 51,250 | 25,320 | ||||||||
Cash and cash equivalents at end of year | $ | 36,855 | $ | 35,723 | $ | 51,250 | |||||
Non-cash investing and financing activities: | |||||||||||
Property and equipment purchased with capital lease | $ | — | $ | — | $ | 109 | |||||
Transfer of systems from property and equipment into inventory | $ | 256 | $ | 575 | $ | 223 | |||||
Property and equipment costs incurred but not paid included in accounts payable | $ | 124 | $ | 603 | $ | 592 | |||||
Leasehold improvements related to lease incentives | $ | — | $ | — | $ | 1,359 | |||||
Intellectual property acquisition included in accrued expenses | $ | 550 | $ | 450 | $ | — | |||||
Offering costs incurred but not paid included in other liabilities | $ | — | $ | 65 | $ | — | |||||
Supplemental cash flow information: | |||||||||||
Cash paid for interest | $ | 20 | $ | 19 | $ | 90 | |||||
Cash received for interest | $ | 244 | $ | 403 | $ | 42 | |||||
Cash paid for income taxes, net | $ | 24 | $ | 21 | $ | 91 | |||||
See accompanying notes to consolidated financial statements.
44
GENMARK DIAGNOSTICS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Organization and basis of presentation
Organization
GenMark Diagnostics, Inc., the Company or GenMark, was formed by Osmetech plc, or Osmetech, as a Delaware corporation in February 2010, and had no operations prior to its initial public offering, or the IPO, which was completed in June 2010. Immediately prior to the closing of the IPO, GenMark acquired all of the outstanding ordinary shares of Osmetech in a reorganization under the applicable laws of the United Kingdom. As a result of the reorganization, all of the issued ordinary shares in Osmetech were cancelled in consideration of (i) the issuance of common stock of GenMark to the former shareholders of Osmetech and (ii) the issuance of new shares in Osmetech to GenMark. Following the reorganization, Osmetech became a subsidiary controlled by GenMark, and the former shareholders of Osmetech received shares of GenMark. Any historical discussion of GenMark relates to Osmetech and its consolidated subsidiaries prior to the reorganization. In September 2012, GenMark placed Osmetech into liquidation to simplify its corporate structure. The liquidation of Osmetech was completed in the fourth quarter of 2013.
As the reorganization was deemed to be a transaction under common control, GenMark accounted for the reorganization in a manner similar to a pooling-of-interests. Once the reorganization became effective, all stock options granted under the Osmetech plc 2003 U.S. Equity Compensation Plan, long term incentive awards and all warrants issued were exchanged for options and warrants exercisable for the common stock of the Company.
Corporate Reorganization
During the quarter ended June 30, 2011, the Company underwent a corporate reorganization intended to simplify its U.S. entity structure. As part of the reorganization, Osmetech Technologies, Inc. merged into Clinical Micro Sensors, Inc., or CMS, with CMS surviving. Additionally, Osmetech plc converted to a U.K. limited company for U.K. legal and tax purposes, and made an entity classification election to be treated as an entity disregarded from GenMark Diagnostics, Inc. for U.S. federal income tax purposes. The reorganization did not trigger any material U.S. federal or U.K. income tax expense. Additionally, the post-reorganization structure allowed GenMark Diagnostics, Inc. to elect to file a consolidated U.S. federal income tax return with its remaining U.S. subsidiaries, CMS and Osmetech, Inc. The liquidation of Osmetech plc was competed in the fourth quarter of 2013.
Segment Reporting
The Company currently operates in one reportable business segment, which encompasses the development, manufacturing, sales and support of instruments and molecular tests based on its proprietary eSensor® detection technology. Substantially all of the Company’s operations and assets are in the United States of America.
Use of Estimates
The preparation of consolidated financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the amounts reported in the financial statements and the notes thereto. The Company’s significant estimates included in the preparation of the financial statements are related to accounts receivable, inventories, property and equipment, intangible assets, employee related compensation accruals, warranty liabilities, tax valuation accounts and share-based compensation. Actual results could differ from those estimates.
Basis of Presentation
The accompanying financial statements have been prepared on a going-concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The Company has incurred net losses from operations since its inception and has an accumulated deficit of $262,472,000 at December 31, 2014. Management expects operating losses to continue through the foreseeable future. The Company's ability to transition to attaining profitable operations is dependent upon achieving a level of revenues adequate to support its cost structure through expanding its product offerings and consequently increasing its product revenues. Cash, cash equivalents, restricted cash, and investments at December 31, 2014 totaled $70,506,000. The Company has prepared cash flow forecasts which indicate, based on the
45
Company’s current cash resources available, that the Company will have sufficient resources to fund its business for at least the next 12 months.
The accompanying consolidated financial statements have been prepared in accordance with United States generally accepted accounting principles, or U.S. GAAP, and applicable regulations of the Securities and Exchange Commission, or the SEC.
Principles of Consolidation
The consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries. All intercompany accounts and transactions have been eliminated in consolidation.
Reclassifications
Certain costs previously reported as cost of revenue, in 2013 totaling $324,000, have been reclassified as general and administrative expenses to better align those costs with the functional areas that benefit from those expenditures. In addition, the Company also reclassified certain amounts included in other income and expense in 2013 of $314,000 into interest income to better align the true nature of the expenses. Reclassifications of prior year financial information have been made to conform to the current year presentation. None of the changes impacts the Company’s previously reported consolidated net revenue, earnings from operations, net earnings or net earnings per share.
2. Summary of Significant Accounting Policies and Significant Accounts
Cash and Cash Equivalents and Marketable Securities
Cash and cash equivalents consist of cash on deposit with banks, money market instruments and certificates of deposit with original maturities of three months or less at the date of purchase. Marketable securities consist of certificates of deposits that mature in greater than three months. Marketable securities are accounted for as "available-for-sale" with the carrying amounts reported in the balance sheets stated at cost, which approximates their fair market value, with unrealized gains and losses, if any, reported as a separate component of stockholders' equity and included in comprehensive loss.
Restricted Cash
Restricted cash represents amounts designated for uses other than current operations and includes $758,000 at December 31, 2014 held as security for the Company’s letter of credit with Banc of California.
Fair Value of Financial Instruments
The Company uses a fair value hierarchy with three levels of inputs, of which the first two are considered observable and the last unobservable, to measure fair value:
• | Level 1 — Quoted prices in active markets for identical assets or liabilities. |
• | Level 2 — Inputs, other than Level 1, that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. |
• | Level 3 — Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. |
The carrying amounts of financial instruments such as accounts receivable, prepaid expenses and other current assets, accounts payable, and accrued liabilities approximate the related fair values due to the short-term maturities of these instruments.
Receivables
Accounts receivable consist of amounts due to the Company for sales to customers and are recorded net of an allowance for doubtful accounts. The allowance for doubtful accounts is determined based on an assessment of the collectability of specific customer accounts, the aging of accounts receivable, and a reserve for unknown items based upon the Company’s historical experience.
The allowance for doubtful accounts as of December 31, 2014, is as follows (in thousands):
46
Allowance for doubtful accounts | |||
Balance December 31, 2012 | $ | 30 | |
Provision for doubtful accounts | 2,721 | ||
Write-off and recoveries, net | (15 | ) | |
Balance December 31, 2013 | $ | 2,736 | |
Provision for doubtful accounts | — | ||
Write-off and recoveries, net | (34 | ) | |
Balance December 31, 2014 | $ | 2,702 | |
The Company has included $2,702,000 in the allowance for doubtful accounts as of December 31, 2014 for past due amounts from former customer, Natural Molecular Testing Corporation, or NMTC.
Inventories
Inventories are stated at the lower of cost (first-in, first-out) or market and include direct labor, materials, and manufacturing overhead. The Company periodically reviews inventory for evidence of slow-moving or obsolete parts, and writes inventory down to market value, as needed. This write down is based on management’s review of inventories on hand, compared to estimated future usage and sales, shelf-life assumptions, and assumptions about the likelihood of obsolescence. If actual market conditions are less favorable than those projected by the Company, additional inventory write-downs may be required. Inventory impairment charges establish a new cost basis for inventory and charges are not reversed subsequently to income, even if circumstances later suggest that increased carrying amounts are recoverable.
Property and Equipment-net
Property, equipment and leasehold improvements are recorded at cost and depreciated using the straight-line method over the estimated useful lives of the assets, which are:
Machinery and laboratory equipment | 3 – 5 years |
XT-8 Instruments | 4 years |
Office equipment | 5 years |
Leasehold improvements | over the shorter of the remaining life of the lease or the useful economic life of the asset |
Property and equipment includes diagnostic instruments used for sales demonstrations or placed with customers under several types of arrangements, including performance evaluation programs, or PEPs, and rentals. PEPs are placed with customers for evaluation periods of up to six months. The customer is generally required to purchase a minimum quantity of reagents and, at the end of the evaluation period, must purchase or return the instrument or sign a reagent rental agreement. Maintenance and repair costs are expensed as incurred. During the year ended December 31, 2013 the Company recorded an impairment charge included in depreciation expense of $302,000 related to production equipment which had been built for NMTC.
Intangible Assets
Intangible assets are comprised of licenses or sublicenses to technology covered by patents owned by third parties, and are amortized on a straight-line basis over the expected useful lives of these assets, which is generally 10 years. Amortization of licenses typically begins upon the Company obtaining access to the licensed technology and is recorded in cost of revenues for licenses supporting commercialized products. The amortization of licenses to technology supporting products in development is recorded in research and development expenses.
Impairment of Long-Lived Assets
The Company assesses the recoverability of long-lived assets, including intangible assets, by periodically evaluating the carrying value whenever events or changes in circumstances indicate that the carrying amount may not be recoverable. If impairment is indicated, the Company writes down the carrying value of the asset to its estimated fair value. This fair value is primarily determined based on estimated discounted cash flows. During 2013, the Company recorded an impairment charge of
47
$1,624,000 to general and administrative expenses related to previously capitalized payments made under a license agreement, which the Company terminated in December 2013. The Company also recorded an impairment charge of $302,000 related to production equipment built for NMTC during 2013. The Company did not recognize any impairment charges during the years ended December 31, 2014 and 2012.
Revenue Recognition
The Company recognizes revenue from product sales and contract arrangements, net of discounts and sales related taxes. The Company recognizes revenue from product sales when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed or determinable and collectability is reasonably assured.
The Company offers customers the choice to either purchase a system outright or to receive a system free of charge in exchange for an annual minimum purchase commitment for diagnostic test cartridges. When a system is sold, the Company generally recognizes revenue upon shipment of the unit, however, if the end user already has the instrument being purchased installed at its location, revenue is recognized when the revenue recognition terms other than delivery have been satisfied. When a system is placed free of charge under a “reagent rental” agreement, the Company retains title to the equipment and it remains capitalized on the balance sheet under property and equipment. Under reagent rental agreements, the Company’s customers pay an additional system rental fee for each test cartridge purchased which varies based on the monthly volume of test cartridges purchased. The system rental fee and diagnostic test cartridges are recognized as contingent rental payments and are included in product revenue in the Company’s consolidated financial statements.
The Company has not had significant product returns and is not contractually obligated to accept returns unless such returns are related to warranty provisions. The Company does not accept reagent product returns, mainly due to FDA regulations, and does not offer volume rebates or provide price protection.
The Company enters into performance evaluation program, or PEP, agreements pursuant to which a system is installed on the premises of a pre-qualified customer for the purpose of allowing the customer to evaluate the system’s functionality over an extended trial period. The customer generally agrees to purchase a starter kit at the time of installation and agrees to purchase a minimum volume of reagents over the life of the trial period.
Revenues related to royalties received from licenses are recognized evenly over the contractual period to which the license relates.
In those cases where the Company bills shipping and handling costs to customers, the amounts billed are included in product revenue.
Product Warranties
The Company generally offers a one-year warranty for its systems sold to customers and up to a sixty day warranty for reagents and provides for the estimated cost of the product warranty at the time the system sale is recognized. Factors that affect the Company’s warranty reserves include the number of units sold, historical and anticipated rates of warranty repairs and the cost per repair. The Company periodically assesses the adequacy of the warranty reserve and adjusts the amount as necessary.
Product warranty reserve activity for the years ended December 31, 2014, 2013 and 2012 is as follows (in thousands):
2014 | 2013 | 2012 | |||||||||
Beginning balance | $ | 226 | $ | 217 | $ | 92 | |||||
Warranty expenses incurred | (608 | ) | (649 | ) | (305 | ) | |||||
Provisions | 577 | 658 | 430 | ||||||||
Ending balance | $ | 195 | $ | 226 | $ | 217 | |||||
Research and Development Costs
The Company expenses all research and development costs in the periods in which they are incurred unless there is alternative future use that supports the capitalization of an asset.
48
Income Taxes
Current income tax expense is the amount of income taxes expected to be payable for the current year. A deferred income tax liability or asset is established for the expected future tax consequences resulting from the differences in financial reporting and tax bases of assets and liabilities. A valuation allowance is provided if it is more likely than not that some or all of the deferred tax assets will not be realized. A full valuation allowance has been recorded against the Company’s net deferred tax assets due to the uncertainty surrounding the Company’s ability to utilize these assets in the future. The Company provides for uncertain tax positions when such tax positions do not meet the recognition thresholds or measurement standards prescribed by the authoritative guidance on income taxes. Amounts for uncertain tax positions are adjusted in periods when new information becomes available or when positions are effectively settled. The Company recognizes accrued interest related to uncertain tax positions as a component of income tax expense.
A tax position that is more likely than not to be realized is measured at the largest amount of tax benefit that is greater than 50% likely of being realized upon settlement with the taxing authority that has full knowledge of all relevant information. Measurement of a tax position that meets the more likely than not threshold considers the amounts and probabilities of the outcomes that could be realized upon settlement using the facts, circumstances and information available at the reporting date.
Stock-Based Compensation
The Company recognizes stock-based compensation expense related to stock options, shares purchased under the Company's 2013 Employee Stock Purchase Plan, or ESPP, restricted stock awards, and restricted stock units granted to employees and directors in exchange for services. The compensation expense is based on the fair value of the applicable award utilizing various assumptions regarding the underlying attributes of the award. The stock-based compensation expense is recorded in cost of revenues, sales and marketing, research and development, and general and administrative expenses based on the employee's respective function.
The estimated fair value of stock options granted, net of forfeitures expected to occur during the vesting period, is amortized as compensation expense on a straight-line basis to reflect vesting as it occurs. The expense is derived from the Black-Scholes Option Pricing Model that uses several judgment-based variables to calculate the expense. The inputs include the expected term of the stock option, the expected volatility and other factors.
• | Expected Term. Expected term represents the period that the stock-based awards are expected to be outstanding and is determined by using the simplified method. |
• | Expected Volatility. Expected volatility represents the volatility in the Company’s stock price expected over the expected term of the option and is determined by review of the Company’s and similar companies’ historical experience. |
• | Expected Dividend. The Black-Scholes Option Pricing Model calls for a single expected dividend yield as an input. The Company assumed no dividends as it has never paid dividends and has no current plans to do so. |
• | Risk-Free Interest Rate. The risk-free interest rate used in the Black-Scholes Option Pricing Model is based on published U.S. Treasury rates in effect at the time of grant for periods corresponding with the expected term of the option. |
The compensation expense related to the grant of restricted stock awards or units is calculated as the fair market value of the stock on the grant date as further adjusted to reflect expected forfeitures.
Foreign Currency Translation
In 2010, the Company changed its functional currency from the British Pound to the U.S. Dollar and assets and liabilities of the Company’s entities outside of the U.S. were translated into U.S. dollars based on foreign currency exchange rates in effect at the end of each period, and revenues and expenses were translated at weighted average exchange rates during the periods. Gains or losses resulting from these foreign currency translations of the Company’s assets and liabilities were recorded in accumulated other comprehensive loss in the consolidated balance sheets. Upon the liquidation of Osmetech in the fourth quarter of 2013, $450,000 of accumulated other comprehensive loss was realized in other expense during 2013.
Transactions in foreign currencies were recognized using the rate of exchange prevailing at the date of the transaction. Foreign exchange gain (loss), which is included in the accompanying consolidated statements of operations, totaled $19,000 $19,000 and $6,000 for the years ended December 31, 2014, 2013 and 2012, respectively, and relate primarily to transactions denominated in U.S. dollars which were paid in Euros.
49
Net Loss per Common Share
Basic net loss per share is calculated by dividing loss available to stockholders of our common stock (the numerator) by the weighted average number of shares of the Company's common stock outstanding during the period (the denominator). Shares issued during the period and shares reacquired during the period are weighted for the portion of the period that they were outstanding. Diluted loss per share is calculated in a similar way to basic loss per share except that the denominator is increased to include the number of additional shares that would have been outstanding if the dilutive potential shares had been issued unless the effect would be anti-dilutive.
The computations of diluted net loss per share for the years ended December 31, 2014, 2013 and 2012 did not include the effects of the following stock options to acquire stock which were outstanding as of the end of each year because the inclusion of these securities would have been anti-dilutive (in thousands).
Year Ended December 31, | ||||||||
2014 | 2013 | 2012 | ||||||
Options outstanding to purchase common stock | 2,479 | 1,821 | 1,539 | |||||
Unvested restricted stock | 916 | 976 | 966 | |||||
Unvested performance stock | 32 | — | — | |||||
Total | 3,427 | 2,797 | 2,505 | |||||
Concentration of Risk
Sales to individual customers representing greater than 10% of the total revenues for the years ended December 31, 2014, 2013 and 2012 were as follows:
Year Ended December 31, | ||||||||
2014 | 2013 | 2012 | ||||||
Natural Molecular Testing Corporation | — | % | 30 | % | 58 | % | ||
Companion Dx Reference Labs, LLC | — | — | % | 10 | % | |||
Comprehensive Loss
The Company has the option to present the total of comprehensive income, the components of net income, and the components of other comprehensive income either in a single continuous statement of comprehensive income or in two separate but consecutive statements. The Company’s comprehensive loss is comprised of net losses, unrealized gains and losses on available for sale securities and foreign currency translation.
Recent Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standard Board, or the FASB, or other standard setting bodies that the Company adopts as of the specified effective date. The Company believes that the impact of recently issued standards that are not yet effective will not have a material impact on its financial position or results of operations upon adoption.
In May 2014, the FASB issued Accounting Standards Update (ASU) 2014-09, Revenue from Contracts with Customers. The new standard is based on the principle that revenue should be recognized to depict the transfer of promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. ASU 2014-09 will be effective for the Company beginning in the first quarter of fiscal 2017 and allows for a full retrospective or a modified retrospective adoption approach. The Company is currently evaluating the impact of ASU 2014-09 on its consolidated financial statements.
3. Intangible Assets, net
Intangible assets as of December 31, 2014 and 2013 consisted of the following (in thousands):
50
December 31, 2014 | December 31, 2013 | ||||||||||||||||||||||
Gross carrying amount | Accumulated amortization | Net carrying amount | Gross carrying amount | Accumulated amortization | Net carrying amount | ||||||||||||||||||
Licensed intellectual property | $ | 2,750 | $ | (880 | ) | $ | 1,870 | $ | 2,409 | $ | (1,212 | ) | $ | 1,197 | |||||||||
In March 2012, the Company entered into a license agreement with Caliper Life Sciences Inc., or Caliper, pursuant to which the Company obtained a non-exclusive license under Caliper’s microfluidics patent portfolio. In consideration for the license, the Company agreed to pay Caliper $400,000 in up-front payments recorded as an intangible asset on the Company’s balance sheet plus certain sales-based milestone payments, as well as a royalty on the sale of certain products. As part of the agreement, the Company obtained an unconditional release from any and all claims based upon any alleged infringement of the licensed patents prior to the effective date of the agreement. The Company met sales-based milestones in March 2013 and March 2014 triggering a payment of $450,000 and $550,000, respectively, which were made after the fiscal year during which the respective milestone was achieved.
In July 2012, the Company entered into a development collaboration and license agreement with Advanced Liquid Logic, Inc., or ALL, which was acquired by Illumina, Inc. in July 2013. Under the terms of the agreement, the Company established a collaborative program to develop in-vitro diagnostic products incorporating ALL’s proprietary electro-wetting technology in conjunction with the Company’s electrochemical detection. The Company paid ALL an upfront license payment of $250,000 and agreed to pay up to $1,750,000 in potential additional milestone payments. Pursuant to the agreement, the parties agreed to enter into a supply agreement relating to the manufacture and supply of certain ALL components. The agreement also provides that the Company would, upon the occurrence of certain events, be obligated to pay to ALL a royalty consisting of a low- to mid-single digit percent of net sales of designated licensed products containing ALL components which the Company manufactures or are otherwise not manufactured and supplied by ALL. The Company met certain milestones in August 2013 and June 2014 resulting in payment of $200,000 and $350,000, respectively.
In October 2010, the Company entered into an intellectual property license agreement under which the Company previously made license fee payments totaling $2.1 million to the licensor. The Company terminated this license agreement in December 2013 and recorded an impairment charge to general and administrative expenses for the remaining net book value of $1.6 million at that time.
Intellectual property licenses had a weighted average remaining amortization period of 7.36 years as of December 31, 2014. Amortization expense for intangible assets amounted to $227,000, $342,000 and $200,000 for the years ended December 31, 2014, 2013 and 2012, respectively. Estimated future amortization expense for these licenses is as follows (in thousands):
Years Ending December 31, | Future Amortization Expense | ||
2015 | $ | 257 | |
2016 | 254 | ||
2017 | 254 | ||
2018 | 254 | ||
2019 | 254 | ||
Thereafter | 597 | ||
Total | $ | 1,870 | |
4. Stockholders’ Equity
Follow-on Stock Offering
In August 2013, the Company completed a public follow-on stock offering of 8,765,000 shares of its common stock which generated net cash proceeds of approximately $80,672,000.
5. Stock-Based Compensation
51
In 2010, the Company adopted the 2010 Equity Incentive Plan, or the 2010 Plan, which provides for the grant of incentive and nonstatutory stock options, restricted stock, stock appreciation rights, restricted stock units, restricted stock bonuses and other stock-based awards. Employee participation in the 2010 Plan is at the discretion of the compensation committee of the board of directors of the Company. All options granted under the 2010 Plan are exercisable at a price equal to the closing quoted market price of the Company’s shares on the NASDAQ Global Market on the date of grant and generally vest over a period of between one and four years.
Options are generally exercisable for a period up to 10 years after grant and are forfeited if employment is terminated before the options vest. As of December 31, 2014, there were 437,127 shares available for future grant of awards under the 2010 Plan.
The following table summarizes stock option activity during the year ended December 31, 2014:
Number of shares | Weighted average exercise price | |||||
Outstanding December 31, 2013 | 1,821,216 | $ | 6.89 | |||
Granted | 946,916 | $ | 11.89 | |||
Exercised | (101,403 | ) | $ | 5.30 | ||
Forfeitures | (187,294 | ) | $ | 9.57 | ||
Outstanding December 31, 2014 | 2,479,435 | $ | 8.66 | |||
Vested and expected to vest at December 31, 2013 | 2,269,316 | $ | 6.25 | |||
Exercisable at December 31, 2014 | 1,282,634 | $ | 6.25 | |||
The weighted average fair value of options granted during the years ended December 31, 2014, 2013 and 2012 was $7.45, $7.30 and $3.80 per share, respectively. Options that were exercisable as of December 31, 2014 had a remaining weighted average contractual term of 5.96 years and an aggregate intrinsic value of $9,466,000. As of December 31, 2014, there was $7,417,000 of unrecognized compensation cost related to stock options. That cost is expected to be recognized over a weighted average-period of 2.9 years. The intrinsic value of options exercised during the years ended December 31, 2014, 2013 and 2012 was $584,000, $730,000 and $277,000, respectively. As of December 31, 2014, there were 2,479,435 stock options outstanding, which had a remaining weighted average contractual term of 7.37 years and an aggregate intrinsic value of $12,320,000.
Valuation of Stock-Based Awards
The assumptions used in the valuation of stock-based awards for the years ended December 31, 2014, 2013 and 2012, are summarized in the following table:
Years Ended December 31, | ||||||||
2014 | 2013 | 2012 | ||||||
Expected volatility (%) | 69 | % | 74 | % | 75 | % | ||
Expected life (years) | 6.08 | 6.08 | 5.92 | |||||
Risk free rate (%) | 1.82 | % | 1.17 | % | 0.97 | % | ||
Expected dividend yield (%) | — | % | — | % | — | % | ||
The Company is required to estimate potential forfeitures of restricted stock grants and adjust compensation cost recorded accordingly. The estimate of forfeitures is based on historical forfeiture experience and will be adjusted over the requisite service period to the extent that actual forfeitures differ, or are expected to differ, from such estimates. Changes in estimated forfeitures will be recognized through a cumulative catch-up adjustment in the period of evaluation and will also impact the amount of stock compensation expense to be recognized in future periods.
Share Warrants
During 2009, the Company issued warrants to purchase 88,317 of Osmetech’s ordinary shares with an exercise price of £6.90 per share to a director for services to the Company in connection with a stock offering completed in 2009. Pursuant to the terms of the warrant, this warrant was converted in connection with the Company’s reorganization into a warrant to purchase
52
88,317 shares of the Company’s common stock at an exercise price of $9.98. These warrants were fully vested and exercisable upon issuance, and expired unexercised on June 30, 2012.
Restricted Stock Awards and Units
In March 2013, the Company transitioned to granting restricted stock units under the 2010 Plan in lieu of granting restricted stock awards. The Company’s restricted stock activity for the year ended December 31, 2014 was as follows:
Restricted Stock Awards | Restricted Stock Units | ||||||||||||
Number of shares | Weighted Average Grant Date Fair Value | Number of shares | Weighted Average Grant Date Fair Value | ||||||||||
Unvested at December 31, 2013 | 508,606 | $ | 4.54 | 474,847 | $ | 11.51 | |||||||
Granted | — | $ | — | 548,569 | $ | 11.62 | |||||||
Vested | (295,231 | ) | $ | 4.36 | (189,193 | ) | $ | 11.94 | |||||
Forfeitures | (39,765 | ) | $ | 5.77 | (92,231 | ) | $ | 11.70 | |||||
Unvested as December 31, 2014 | 173,610 | $ | 4.56 | 741,992 | $ | 11.46 | |||||||
Restricted stock awards or units may be granted at the discretion of the compensation committee of the board of directors under the 2010 Plan in connection with the hiring or retention of personnel and are subject to certain conditions. Restrictions expire at certain dates after the grant date in accordance with specific provisions in the applicable award agreement.
As of December 31, 2014, there was $695,000 of unrecognized compensation cost related to restricted stock awards. That cost is expected to be recognized over a weighted average-period of 1.22 years. The total fair value of restricted stock awards that vested during the years ended December 31, 2014, 2013 and 2012 was $3,466,000, $4,201,000 and $724,000, respectively.
As of December 31, 2014, there was $6,404,000 of unrecognized compensation cost related to restricted stock units. That cost is expected to be recognized over a weighted average period of 2.79 years. The total fair value of restricted stock units that vested during the years ended December 31, 2014, 2013 and 2012 was $2,121,000, $72,000 and zero, respectively.
The Company granted 43,200 performance-based restricted stock units in March 2014 with a grant date fair value of $12.30. The vesting and issuance of Company stock pursuant to these awards depends on obtaining regulatory clearance of various products within a defined time. Stock-based compensation expense for performance-based awards is recognized when it is probable that the applicable performance criteria will be satisfied. The probability of achieving the relevant performance criteria is evaluated on a quarterly basis. On December 31, 2014, 10,800 units were earned and vested with a total fair value of $147,000. As of December 31, 2014, there was $399,000 in unrecognized stock-based compensation expense related to the remaining unvested awards.
Stock-Based Compensation Expense Recognition
Stock-based compensation was recognized in the consolidated statements of comprehensive loss as follows (in thousands):
Years Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Cost of revenue | $ | 73 | $ | 162 | $ | 125 | |||||
Sales and marketing | 1,848 | 1,223 | 558 | ||||||||
Research and development | 1,194 | 613 | 509 | ||||||||
General and administrative | 2,681 | 1,895 | 1,160 | ||||||||
Stock-based compensation expense | $ | 5,796 | $ | 3,893 | $ | 2,352 | |||||
No stock-based compensation was capitalized during the periods presented, and there was no unrecognized tax benefit related to stock-based compensation for the years ended December 31, 2014, 2013 and 2012, respectively.
Employee Stock Purchase Plan
53
Following the adoption of the ESPP by the Company’s board of directors in March 2013, the Company's stockholders approved the ESPP in May 2013 at the Company's Annual Meeting of Stockholders. A total of 650,000 shares of the Company’s common stock are reserved for issuance under the ESPP, which permits eligible employees to purchase common stock at a discount through payroll deductions.
The price at which stock is purchased under the ESPP is equal to 85% of the fair market value of the common stock on the first or the last day of the offering period, whichever is lower. Generally, each offering under the ESPP will be for a period of six months as determined by the Company's board of directors; provided that no offering period may exceed 27 months. Employees may invest up to 10% of their gross compensation through payroll deductions. In no event may an employee purchase more than 1,500 shares of common stock during any six-month offering period. As of December 31, 2014, there were 528,142 shares of common stock available for issuance under the ESPP. As the ESPP is a compensatory plan as defined by the authoritative guidance for stock compensation, stock-based compensation expense, calculated using the Black-Scholes model at the beginning of each six-month offering period, related to the ESPP has been recorded during the year ended December 31, 2014.
A summary of ESPP activity for the year ended December 31, 2014 and 2013 is as follows (in thousands, except share, and per share data):
Year Ended December 31, | ||||||
2014 | 2013 | |||||
Shares issued | 88,419 | 33,439 | ||||
Weighted average fair value of shares issued | $ | 11.20 | $ | 11.90 | ||
Employee purchases | $ | 812 | $ | 299 | ||
6. Income Taxes
For the years ended December 31, 2014, 2013, and 2012, all pretax earnings and losses were generated in the United States.
The components of income tax expense (benefit) were as follows for the years ended December 31, 2014, 2013, and 2012, respectively (in thousands):
Years Ended December 31, | |||||||||||
2014 | 2013 | 2012 | |||||||||
Current expense: | |||||||||||
U.S. federal | $ | — | $ | — | $ | — | |||||
State | (573 | ) | 44 | 103 | |||||||
Foreign (non-U.S. entities) | — | — | 45 | ||||||||
Total current expense (benefit) | $ | (573 | ) | $ | 44 | $ | 148 | ||||
The components of net deferred income taxes consisted of the following at December 31, 2014 and 2013, respectively (in thousands):
54
As of December 31, | |||||||
2014 | 2013 | ||||||
Deferred income tax assets (liabilities): | |||||||
NOL and credit carryforwards | $ | 44,768 | $ | 31,893 | |||
Compensation accruals | 3,078 | 1,912 | |||||
Accruals and reserves | 2,551 | 2,394 | |||||
State tax provision | 8 | 5 | |||||
Federal benefit of state UTP | — | 172 | |||||
Inventory adjustments | 513 | 538 | |||||
Intangible assets | 355 | 1,316 | |||||
Mark to market of marketable securities | — | 111 | |||||
Other | 10 | — | |||||
Subtotal: Deferred tax assets | 51,283 | 38,341 | |||||
Depreciation | (635 | ) | (494 | ) | |||
Valuation allowance | (50,648 | ) | (37,847 | ) | |||
Net deferred income taxes | $ | — | $ | — | |||
A reconciliation of income tax expense to the amount computed by applying the statutory federal income tax rate to the loss from operations is summarized for the years ended December 31, 2014, 2013, and 2012, respectively, as follows:
Years Ended December 31, | ||||||||
2014 | 2013 | 2012 | ||||||
U.S. Federal statutory income tax rate | 34.0 | % | 34.0 | % | 34.0 | % | ||
Permanent differences | (0.3 | )% | (0.6 | )% | (0.9 | )% | ||
State taxes | 2.6 | % | (1.0 | )% | 5.3 | % | ||
Executive compensation limitation | (0.7 | )% | (0.4 | )% | — | % | ||
Stock-based compensation | (1.5 | )% | (0.9 | )% | — | % | ||
Effect of non-U.S. operations | — | % | — | % | (0.2 | )% | ||
Other | 0.3 | % | (0.9 | )% | (0.2 | )% | ||
Valuation allowance | (32.9 | )% | (30.3 | )% | (38.9 | )% | ||
Total tax provision | 1.5 | % | (0.1 | )% | (0.9 | )% | ||
The Company had federal net operating loss (NOL) carryforwards available of approximately $128,400,000 as of December 31, 2014 after consideration of limitations under Section 382 of the Internal Revenue Code, or Section 382, as further described below. Additionally, the Company had state NOL carryforwards available of $122,400,000 as of December 31, 2014. These federal and state NOLs may be used to offset future taxable income and will begin to expire in 2025 and 2016, respectively.
Of the $128,400,000 and $122,400,000 of federal and state NOL carryforwards at December 31, 2014, $4,710,000 represents excess tax benefits related to equity compensation which will result in an increase in equity if and when such excess benefits are ultimately realized.
The future utilization of the Company’s NOL carryforwards to offset future taxable income may be subject to a substantial annual limitation as a result of changes in ownership by stockholders that hold 5% or more of the Company’s common stock. An assessment of such ownership changes under Section 382 was completed through December 31, 2014. As a result of this assessment, the Company determined that it experienced multiple ownership changes through 2014 which will limit the future utilization of NOL carryforwards. The Company has reduced its deferred tax assets related to NOL carryovers that are anticipated to expire unused as a result of ownership changes. These tax attributes have been excluded from deferred tax assets with a corresponding reduction of the valuation allowance with no net effect on income tax expense or the effective tax rate. Additionally, future ownership changes may further impact the utilization of existing NOLs.
The Company has established a full valuation allowance for its deferred tax assets due to uncertainties that preclude it from determining that it is more likely than not that the Company will be able to generate sufficient taxable income to realize
55
such assets. Management assesses the available positive and negative evidence to estimate if sufficient future taxable income will be generated to utilize the existing deferred tax assets. A significant piece of objective negative evidence evaluated was the cumulative loss incurred over the three year period ended December 31, 2014. Such objective evidence limits the ability to consider other subjective evidence such as the Company's projections for future growth. Based on this evaluation, as of December 31, 2014, a valuation allowance of $50,648,000 has been recorded in order to measure only the portion of the deferred tax asset that more likely than not will be realized. The amount of the deferred tax asset considered realizable, however, could be adjusted if objective negative evidence in the form of cumulative losses is no longer present and additional weight may be given to subjective evidence, such as estimates of future taxable income during carryforward periods and the Company's projections for growth.
The Company applies the two-step approach to recognizing and measuring uncertain tax positions. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates it is more likely than not that the position will be sustained on audit, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit as the largest amount, which is more than 50% likely of being realized upon ultimate settlement. Income tax positions must meet a more likely than not recognition threshold at the effective date to be recognized upon the adoption of ASC 740 and in subsequent periods. This interpretation also provides guidance on measurement, derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition.
The following table summarizes the changes to unrecognized tax benefits for the years ended December 31, 2014, 2013 and 2012, respectively (in thousands):
Years Ended December 31, | ||||||
2014 | 2013 | 2012 | ||||
Beginning balance of unrecognized tax benefits | 382 | 382 | 382 | |||
Lapses in the statute of limitations | (382 | ) | ||||
Ending balance of unrecognized tax benefits | — | 382 | 382 | |||
At December 31, 2014 and December 31, 2013, the Company has accrued interest and penalties of zero and $228,000, respectively. The Company does not anticipate that there will be a significant change in the amount of unrecognized tax benefits over the next twelve months. The Company recognizes interest and penalties related to uncertain tax positions in income tax expense.
Prior to 2013, the Company was subject to taxation in the United Kingdom, the United States and various states jurisdictions. After 2013, the Company is subject to taxation in the United States and various state jurisdictions. As of December 31, 2014, the Company’s tax years from 2010 through 2012 are subject to examination by the United Kingdom tax authorities. Except for net operating losses generated in prior years carrying forward to the current year, as of December 31, 2014, the Company is no longer subject to U.S. federal, state, local or foreign examinations by tax authorities for years before 2010.
7. Commitments and Contingencies
Leases
The Company has lease agreements for its office, manufacturing, warehousing and laboratory space and for office equipment. Rent and operating expenses charged were $1,147,000, $1,049,000 and $1,323,000 for the years ended December 31, 2014, 2013, and 2012, respectively. Pursuant to the Company’s lease agreements, a portion of the monthly rental has been deferred. The balance deferred at December 31, 2014 and 2013 was $1,601,000 and $1,725,000, respectively.
Annual future minimum obligations for leases as of December 31, 2014 are as follows (in thousands):
56
Years Ending December 31, | Amount | ||
2015 | $ | 1,084 | |
2016 | 1,116 | ||
2017 | 1,123 | ||
2018 | 1,155 | ||
2019 | 1,191 | ||
Thereafter | 1,853 | ||
Total minimum lease payments | $ | 7,522 | |
Legal Proceedings
From time to time, the Company is party to litigation and other legal proceedings in the ordinary course, and incidental to the conduct of its business. While the results of any litigation or other legal proceedings are uncertain, the Company does not believe the ultimate resolution of any pending legal matters is likely to have a material effect on its financial position or results of operations.
8. Inventories
Inventory on hand as of December 31, 2014 and 2013 was comprised of the following (in thousands):
December 31, | |||||||
2014 | 2013 | ||||||
Raw materials | $ | 593 | $ | 713 | |||
Work-in-process | 660 | 437 | |||||
Finished goods | 884 | 952 | |||||
$ | 2,137 | $ | 2,102 | ||||
9. Property and Equipment, net
Property and equipment comprised of the following as of December 31, 2014 and 2013 (in thousands):
December 31, | |||||||
2014 | 2013 | ||||||
Property and equipment—at cost: | |||||||
Plant and machinery | $ | 6,737 | $ | 3,260 | |||
XT-8 Instruments | 6,611 | 7,013 | |||||
Office equipment | 1,441 | 1,325 | |||||
Leasehold improvements | 4,050 | 3,755 | |||||
Total property and equipment—at cost | 18,839 | 15,353 | |||||
Less accumulated depreciation | (7,787 | ) | (6,762 | ) | |||
Property and equipment, net | $ | 11,052 | $ | 8,591 | |||
Depreciation expense was $2,429,000, $2,187,000 and $998,000 for the years ended December 31, 2014, 2013 and 2012, respectively. During the year ended December 31, 2014, the Company disposed of certain assets no longer in use with a net book value of $102,000, recorded in operations. During the year ended December 31, 2013 the Company recorded an impairment charge included in depreciation expense of $302,000 related to production equipment which had been built for NMTC.
10. Loan payable
In September 2012, the Company entered into a new term loan with Banc of California, consisting of the following two loans.
57
1) The Company increased the letter of credit provided to its landlord of its Carlsbad, California facility to $758,000 from the previous letter of credit of $500,000. The increase in the letter of credit was required by the Company’s landlord in connection with the Company’s lease of additional space at this facility. The letter credit was secured with $758,000 of restricted cash at December 31, 2014.
2) The Company obtained a variable rate term loan from Banc of California in the amount of $836,000 with an initial interest rate of 3.75% that expired in July 2013. This term loan replaced the Square 1 Bank equipment loan of the same amount with an interest rate of 6.75%. As of December 31, 2014, the Company had repaid all outstanding amounts under this loan.
Pursuant to the terms of the Banc of California business loan agreement, the Company was required to maintain restricted cash, honor certain representations and warranties (including, but not limited to, organization, financial information and taxes), affirmative covenants (including, but not limited to, financial records, insurance and environmental compliance and reports), negative covenants (including, but not limited to, indebtedness and liens, continuity of operations and loans, acquisitions and guaranties) and other provisions; however, the Company was not required to maintain liquidity ratios, restrictive covenants or other limitations, to which it was subject under the Square 1 Bank loan and security agreement.
Principal repayment obligations under the Loan Agreement as of December 31, 2014 was $0.
In January 2015, the Company entered into a loan and security agreement with General Electric Capital Corporation. See Footnote 16, Subsequent events, for more information.
11. Employee benefit plan
The Company has a 401(k) tax-deferred savings plan, whereby eligible employees may contribute a percentage of their eligible compensation. The Company may make matching contributions under the 401(k) plan; however, the Company has not made any such contributions to date.
12. Other current liabilities
Other current liabilities as of December 31, 2014 and 2013 consisted of the following (in thousands):
December 31, | |||||||
2014 | 2013 | ||||||
Accrued royalties | $ | 1,210 | $ | 1,020 | |||
Outside services and consulting | 1,029 | 907 | |||||
Accrued warranties | 195 | 226 | |||||
Other accrued liabilities | 1,219 | 809 | |||||
Total | $ | 3,653 | $ | 2,962 | |||
13. Fair value of financial instruments
The following table presents the financial instruments measured at fair value on a recurring basis on the financial statements of the Company and the valuation approach applied to each class of financial instruments at December 31, 2014 and 2013, respectively, (in thousands):
58
December 31, 2014 | |||||||||||||||
Quotes Prices in Active Markets for Identical Assets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | Total | ||||||||||||
Money market funds (cash equivalents) | $ | 31,412 | $ | — | $ | — | $ | 31,412 | |||||||
Corporate notes and bonds | — | 18,387 | — | 18,387 | |||||||||||
U.S. government and agency securities | — | 15,264 | — | 15,264 | |||||||||||
$ | 31,412 | $ | 33,651 | $ | — | $ | 65,063 | ||||||||
December 31, 2013 | |||||||||||||||
Quotes Prices in Active Markets for Identical Assets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | Total | ||||||||||||
Money market funds (cash equivalents) | $ | 19,910 | $ | — | $ | — | $ | 19,910 | |||||||
Corporate notes and bonds | 22,954 | 22,954 | |||||||||||||
U.S. government and agency securities | 43,115 | 43,115 | |||||||||||||
Commercial paper | — | 3,797 | — | 3,797 | |||||||||||
$ | 19,910 | $ | 69,866 | $ | — | $ | 89,776 | ||||||||
At December 31, 2014, the carrying value of the financial instruments measured and classified within Level 1 was based on quoted prices and marked to market. Level 2 inputs for the valuations are limited to quoted prices for similar assets or liabilities in active markets and inputs other than quoted prices that are observable for the asset or liability.
14. Investments
The following table summarizes the Company’s available-for-sale investments at December 31, 2014 (in thousands) :
Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated Fair Value | ||||||||||||
Corporate notes and bonds | $ | 18,408 | $ | 1 | $ | (22 | ) | $ | 18,387 | ||||||
U.S. government and agency securities | 15,267 | 2 | (5 | ) | 15,264 | ||||||||||
Total | $ | 33,675 | $ | 3 | $ | (27 | ) | $ | 33,651 | ||||||
During 2013, the Company sold its preferred stock investment in Advanced Liquid Logic, Inc., or ALL, in connection with ALL's acquisition by Illumina, Inc., resulting in a $1,392,000 realized gain.
The following table summarizes the maturities of the Company’s available-for-sale securities at December 31, 2014 (in thousands):
Amortized Cost | Estimated Fair Value | ||||||
Due in one year or less | $ | 31,425 | $ | 31,403 | |||
Due after one year through two years | 2,250 | 2,248 | |||||
Total | $ | 33,675 | $ | 33,651 | |||
59
15. Quarterly financial data (unaudited)
Year Ended December 31, 2014 | |||||||||||||||
(In thousands, except per share data) | |||||||||||||||
First Quarter | Second Quarter | Third Quarter | Fourth Quarter | ||||||||||||
Total revenue | $ | 7,913 | $ | 6,555 | $ | 6,300 | $ | 9,826 | |||||||
Gross profit | $ | 4,322 | $ | 3,455 | $ | 3,691 | $ | 5,999 | |||||||
Loss from operations | $ | (8,860 | ) | $ | (11,265 | ) | $ | (10,272 | ) | $ | (8,657 | ) | |||
Net loss | $ | (8,783 | ) | $ | (11,210 | ) | $ | (9,658 | ) | $ | (8,612 | ) | |||
Per share data: | |||||||||||||||
Net loss per common share—basic and diluted | $ | (0.22 | ) | $ | (0.27 | ) | $ | (0.23 | ) | $ | (0.21 | ) | |||
Year Ended December 31, 2013 | |||||||||||||||
(In thousands, except per share data) | |||||||||||||||
First Quarter | Second Quarter | Third Quarter | Fourth Quarter | ||||||||||||
Total revenue | $ | 11,101 | $ | 5,215 | $ | 4,637 | $ | 6,451 | |||||||
Gross profit | $ | 6,186 | $ | 2,062 | $ | 555 | $ | 3,032 | |||||||
Loss from operations | $ | (4,231 | ) | $ | (8,087 | ) | $ | (12,291 | ) | $ | (10,271 | ) | |||
Net loss | $ | (4,175 | ) | $ | (8,019 | ) | $ | (10,817 | ) | $ | (10,632 | ) | |||
Per share data: | |||||||||||||||
Net loss per common share—basic and diluted | $ | (0.13 | ) | $ | (0.25 | ) | $ | (0.30 | ) | $ | (0.26 | ) | |||
16. Subsequent events
The Company has completed an evaluation of all subsequent events through the issuance date of these Consolidated Financial Statements and the following represents subsequent events for disclosure.
On January 12, 2015, the Company entered into a Loan and Security Agreement with General Electric Capital Corporation, or GECC, pursuant to which the Company obtained (a) up to $35,000,000 in a series of term loans and (b) a revolving loan in the maximum amount of $5,000,000.
The term loans will accrue interest at a rate equal to a) the greater of 1.00% or the 3-year treasury rate in effect at the time of funding, plus (b) an applicable margin between 4.95% and 5.90% per annum. The Company is only required to make interest payments on amounts borrowed pursuant to the term loans from the applicable funding date until March 1, 2017 (the “Interest Only Period”). Following the Interest Only Period, monthly installments of principal and interest under the term loans will be due until the original principal amount and applicable interest is fully repaid by January 12, 2019 (the “Maturity Date”).
Borrowings under the agreement may be used to satisfy the Company’s future working capital needs, including funding the commercial launch of the Company’s ePlex system internationally and domestically. The Company has not yet borrowed any amounts under the agreement.
Borrowings under the revolving loan will accrue interest at a rate equal to (a) the greater of 1.25% per annum or GECC’s base rate as determined by a three-month LIBOR-based formula, plus (b) an applicable margin between 2.95% and 3.95% based on certain criteria as set forth in the agreement. All principal and interest outstanding under the Revolving Loan is due and payable on the Maturity Date. Following the revolving credit facility activation date, the Company would be required to pay GECC a commitment fee equal to 0.75% per annum of the amounts made available but unborrowed under the Revolving Loan.
As of December 31, 2014, the Company capitalized costs related to securing the debt of $90,000 which were capitalized and will be amortized over the life of the loans. The Company is required to pay GECC an annual management fee which will be expensed.
Item 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
60
Item 9A. | CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures designed to provide reasonable assurance that information required to be disclosed in reports we file under the Exchange Act is recorded, processed, summarized and reported within the specified time periods and accumulated and communicated to management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. The design of any system of controls is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions; over time, controls may become inadequate because of changes in conditions, or the degree of compliance with policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
As of the end of the period covered by this Annual Report on Form 10-K, we carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act. Based on this evaluation, our management, with the participation of our Chief Executive Officer and Chief Financial Officer, concluded that, as of December 31, 2014, our disclosure controls and procedures were effective.
Changes in Internal Control Over Financial Reporting
There has been no change in our internal control over financial reporting that occurred in the quarter ended December 31, 2014 that materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) of the Exchange Act). Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States and includes those policies and procedures that:
• | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; |
• | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and |
• | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements. |
Because of inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risks that controls may become inadequate because of changes in conditions or that the degree of compliance with policies or procedures may deteriorate.
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2014 based on the framework in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (COSO). Based on our evaluation under this framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2014.
Management’s assessment of the effectiveness of our internal control over financial reporting as of December 31, 2014 has been audited by Ernst & Young LLP, an independent registered public accounting firm, as stated in their report which is included herein.
61
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of GenMark Diagnostics, Inc.
We have audited GenMark Diagnostics, Inc.’s internal control over financial reporting as of December 31, 2014, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). GenMark Diagnostics, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, GenMark Diagnostics, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2014, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the 2014 consolidated financial statements of GenMark Diagnostics, Inc. and our report dated February 24, 2015 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
San Diego, California
February 24, 2015
62
Item 9B. | OTHER INFORMATION |
None.
PART III.
Item 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
The information required by this Item is incorporated in this Annual Report by reference from the information under the captions “Board of Directors Information,” “Executive Officers” and “Section 16(a) Beneficial Ownership Reporting Compliance” contained in the Proxy Statement to be filed in connection with our 2015 Annual Meeting of Stockholders, or the Proxy Statement.
Code of Business Conduct and Ethics
We have adopted a code of business conduct and ethics for our directors, officers and employees, which is available on our website at www.genmarkdx.com in the Investor Relations section under “Corporate Governance.” If we make any substantive amendments to the code of business conduct and ethics or grant any waiver from a provision of the code of business conduct and ethics to any executive officer or director, we will promptly disclose the nature of the amendment or waiver on our website. The information on, or that can be accessed from, our website is not incorporated by reference into this Annual Report.
Item 11. | EXECUTIVE COMPENSATION |
The information required by this Item is incorporated in this Annual Report by reference from the information under the captions “Executive Compensation,” “Compensation Committee Interlocks and Insider Participation” and “Report of the Compensation Committee” contained in the Proxy Statement.
Item 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this Item is incorporated in this Annual Report by reference from the information under the captions “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information” contained in the Proxy Statement.
Item 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required by this Item is incorporated in this Annual Report by reference from the information under the captions “Certain Relationships and Related Transactions,” and “Board of Directors Information” contained in the Proxy Statement.
Item 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information required by this Item is incorporated in this Annual Report by reference from the information under the captions “Principal Accountant Fees and Services” and “Report of the Audit Committee” contained in the Proxy Statement.
Item 15. | EXHIBITS, FINANCIAL STATEMENTS AND SCHEDULES |
(a) | Documents filed as part of this Annual Report. |
1. | The following financial statements of GenMark Diagnostics, Inc. and Report of Independent Registered Public Accounting Firm, are included in this report: |
Report of Ernst & Young LLP, Independent Registered Public Accounting Firm
63
Report of Deloitte & Touche LLP, Independent Registered Public Accounting Firm
Consolidated Balance Sheets at December 31, 2014 and 2013
Consolidated Statements of Comprehensive Loss for the years ended December 31, 2014, 2013 and 2012
Consolidated Statements of Stockholders’ Equity for the years ended December 31, 2014, 2013 and 2012
Consolidated Statements of Cash Flows for the years ended December 31, 2014, 2013 and 2012
Notes to Consolidated Financial Statements
2. | List of financial statement schedules. All schedules are omitted because they are not applicable or the required information is shown in the financial statements or notes thereto. |
3.
4. | List of Exhibits required by Item 601 of Regulation S-K. See Item 15(b) below. |
(b) | Exhibits. |
The exhibits listed in the accompanying “Exhibit Index” are filed, furnished or incorporated by reference as part of this Annual Report, as indicated.
64
SIGNATURES
Pursuant to the requirements of the Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized, on February 24, 2015.
GENMARK DIAGNOSTICS, INC. | |||
By: | /s/ HANY MASSARANY | ||
Name: | Hany Massarany | ||
Title: | Chief Executive Officer, President and Director (principal executive officer) | ||
February 24, 2015
By: | /s/ SCOTT MENDEL | ||
Name: | Scott Mendel | ||
Title: | Chief Financial Officer (principal financial and accounting officer) | ||
February 24, 2015
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Hany Massarany and Scott Mendel, jointly and severally, his attorneys-in-fact, each with the power of substitution, for him or her in any and all capacities, to sign any amendments to this Report on Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact, or his substitute or substitutes may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report on Form 10-K has been signed below by the following persons on behalf of the registrant in the capacities and on the dates indicated.
Signature | Title | Date |
/S/ HANY MASSARANY | President, Chief Executive Officer and Director (principal executive officer) | 2/24/2015 |
Hany Massarany | ||
/S/ SCOTT MENDEL | Chief Financial Officer (principal financial and accounting officer) | 2/24/2015 |
Scott Mendel | ||
/S/ JAMES FOX | Chairman of the Board | 2/24/2015 |
James Fox | ||
/S/ DARYL J. FAULKNER | Director | 2/24/2015 |
Daryl J. Faulkner | ||
/S/ KEVIN C. O’BOYLE | Director | 2/24/2015 |
Kevin C. O’Boyle | ||
/S/ MICHAEL S. KAGNOFF | Director | 2/24/2015 |
Michael S. Kagnoff | ||
Lisa M. Giles | Director | |
INDEX TO EXHIBITS
65
Exhibit | Description |
3.1 | Certificate of Incorporation (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010). |
3.2 | Amended and Restated By-Laws (incorporated by reference to our Current Report on 8-K filed on October 31, 2014). |
10.1 | Lease between The Campus Carlsbad, LLC and Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, dated February 8, 2010 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010). |
10.2 | Settlement and Release Agreement and First Amendment to Lease between The Campus Carlsbad, LLC and Clinical Micro Sensors, Inc., dated July 1, 2010 (incorporated by reference herein form our Form 10-K as filed with the SEC on March 14, 2013). |
10.3 | Settlement and Release Agreement and Second Amendment to Lease, dated January 19, 2012, by and between the Campus Carlsbad, LLC and Clinical Micro Sensors, Inc. d.b.a. GenMark Diagnostics, Inc. (incorporated by reference to our Annual Report on Form 10-K filed with the Commission on March 21, 2012). |
10.4 | Third Amendment to Lease agreement dated August 28, 2012, by and between The Campus Carlsbad, LLC and Clinical Micro Sensors, Inc. dba GenMark Diagnostics, Inc. (incorporated by reference herein from our Form 10-Q as filed with the SEC on November 8, 2012). |
10.5 | Second Amendment to License Agreement dated June 20, 2000 by and between California Institute of Technology and Clinical Micro Sensors, Inc. (incorporated by reference herein form our Form 10-K/A as filed with the SEC on April 18, 2013). † |
10.6 | Amended and Restated Chemically Modified Enzymes Kit Patent License Agreement by and between Roche Molecular Systems, Inc., F. Hoffman-La Roche Ltd., and Clinical Micro Sensors, Inc. dba Osmetech Molecular Diagnostics, dated February 27, 2008 (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on May 21, 2010). |
10.7 | Non-Exclusive License Agreement by and between Clinical Micro Sensors, Inc. d.b.a. GenMark Diagnostics, Inc. and Caliper Life Sciences Inc. dated effective as of March 27, 2012 (incorporated by reference herein from our Form 10-Q as filed with the SEC on May 10, 2012). |
10.8 | Development Collaboration and License Agreement, dated July 26, 2012, by and between Advanced Liquid Logic, Inc. and Clinical Micro Sensors, Inc. dba GenMark Diagnostics, Inc. (incorporated by reference herein from our Form 10-Q/A as filed with the SEC on March 22, 2013). † |
10.9 | GenMark Diagnostics, Inc. 2010 Equity Incentive Plan, as amended (incorporated by reference to our Definitive Proxy Statement on Schedule 14A filed with the SEC on April 17, 2014).* |
66
Exhibit | Description |
10.10 | Form of Stock Option Agreement (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on April 20, 2010).* |
10.11 | Form of Restricted Stock Agreement (incorporated by reference herein to our Form 10-Q as filed with the SEC on November 9, 2010).* |
10.12 | Form of Restricted Stock Units Grant Notice and Agreement (incorporated by reference herein to our Form 8-K as filed with the SEC on March 12, 2013).* |
10.13 | Form of Amendment of Restricted Stock, Restricted Stock Unit and/or Stock Option Agreement(s)(incorporated by reference herein to our Form 10-Q as filed with the SEC on May 6, 2014).* |
10.14 | The GenMark Diagnostics, Inc. 2014 Bonus Plan (incorporated by reference herein to our Form 8-K as filed with the SEC on March 11, 2014).* |
10.15 | GenMark Diagnostics, Inc. 2013 Employee Stock Purchase Plan (incorporated by reference to our Definitive Proxy Statement on Schedule 14A filed with the Commission on April 5, 2013).* |
10.16 | Form of Director and Officer Indemnification Agreement (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010).* |
10.17 | Executive Employment Agreement, dated January 1, 2010, by and between Osmetech Technology Inc. and Jon Faiz Kayyem, Ph.D. (incorporated by reference to our Registration Statement on Form S-1 (File No. 333-165562) filed with the Commission on March 19, 2010).* |
10.18 | Executive Employment Agreement, dated as of April 5, 2011, by and between GenMark Diagnostics, Inc. and Hany Massarany (incorporated by reference herein from our Form 10-Q as filed with the SEC on May 13, 2011).* |
10.19 | Employment Offer Letter effective May 7, 2014 by and between GenMark Diagnostics, Inc. and Scott Mendel (incorporated by reference to our Current Report on Form 8-K filed with the SEC on May 12, 2014).* |
10.20 | GenMark Diagnostics, Inc. Non-Plan Stock Option Agreement with Scott Mendel (incorporated by reference to our Registration Statement on Form S-8 (File No. 333-195924) filed with the SEC on May 13, 2014).* |
10.21 | GenMark Diagnostics, Inc. Non-Plan Restricted Stock Units Agreement with Scott Mendel (incorporated by reference to our Registration Statement on Form S-8 (File No. 333-195924) filed with the SEC on May 13, 2014).* |
10.22 | Separation Agreement and General Release dated May 9, 2014 by and between GenMark Diagnostics, Inc. and Richard Slansky (incorporated by reference to our Form 10-Q filed with the SEC on August 11, 2014).* |
10.23 | Executive Employment Agreement dated March 1, 2010, by and between Clinical Micro Sensors, Inc. d.b.a. GenMark Diagnostics, Inc. and Jeffrey Hawkins (incorporated by reference herein from our Form 10-Q as filed with the SEC on May 13, 2011).* |
10.24 | Executive Employment Agreement dated April 13, 2010 by and between Osmetech Molecular Diagnostics and Jennifer Williams (incorporated by reference herein from our Form 10-K as filed with the SEC on March 14, 2013).* |
10.25 | Employment Offer Letter dated April 1, 2013 by and between Clinical Micro Sensors, Inc., d.b.a. GenMark Diagnostics and Ingo Chakravarty (incorporated by reference from our Form 10-Q filed on May 6, 2014).* |
10.26 | XT-8 Instrument Supply Agreement, dated August 3, 2012, by and between Leica Biosystems Melbourne Pty Ltd and Clinical Micro Sensors, Inc. dba GenMark Diagnostics, Inc. (incorporated by reference herein from our Form 10-Q/A as filed with the SEC on March 22, 2013).† |
21.1 | List of Subsidiaries ü |
23.1 | Consent of Deloitte & Touche LLP, Independent Registered Public Accounting Firm ü |
23.2 | Consent of Ernst & Young LLP, Independent Registered Public Accounting Firm ü |
67
Exhibit | Description |
24.1 | Power of Attorney (included on the signature page hereto). ü |
31.1 | Certification of principal executive officer pursuant to Rule 13a-14(a) and 15d-14(a) of the Securities Exchange Act of 1934, as amended. ü |
31.2 | Certification of principal financial officer pursuant to Rule 13a-14(a) and 15d-14(a) of the Securities Exchange Act of 1934, as amended. ü |
32.1 | Certification of the principal executive officer pursuant to Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended, and 18 U.S.C. section 1350. ü |
32.2 | Certification of the principal financial officer pursuant to Rule 13a-14(b) of the Securities Exchange Act of 1934, as amended, and 18 U.S.C. section 1350. ü |
101 | XBRL Instance Document |
101 | XBRL Taxonomy Extension Schema Document |
101 | XBRL Taxonomy Calculation Document |
101 | XBRL Taxonomy Definition Linkbase Document |
101 | XBRL Taxonomy Label Linkbase Document |
101 | XBRL Taxonomy Presentation Linkbase Document |
_____________________
* | Indicates a management contract or compensatory plan or arrangement in which any director or named executive officer participates. |
ü | Included in this filing. |
† | Confidential treatment has been granted with respect to certain portions of this exhibit. |
68