Attached files
| file | filename |
|---|---|
| EXCEL - IDEA: XBRL DOCUMENT - Islet Sciences, Inc. | Financial_Report.xls |
| EX-14.1 - CODE OF ETHICS - Islet Sciences, Inc. | once_ex141.htm |
| EX-31.2 - CERTIFICATION - Islet Sciences, Inc. | once_ex312.htm |
| EX-31.1 - CERTIFICATION - Islet Sciences, Inc. | once_ex311.htm |
| EX-32.1 - CERTIFICATION - Islet Sciences, Inc. | once_ex321.htm |
| EX-21.1 - SUBSIDIARIES OF THE REGISTRANT - Islet Sciences, Inc. | once_ex211.htm |
U.S. SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
|
þ
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the Fiscal Year Ended April 30, 2014
|
o
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the transition period from ________________ to ________________
Commission File Number 001-34048
Islet Sciences, Inc.
(Exact name of registrant as specified in its charter)
|
Nevada
|
87-0531751
|
|
|
(State or other jurisdiction of
|
(IRS Employer
|
|
|
incorporation or organization)
|
Identification No.)
|
8601 Six Forks Rd, Suite 400
Raleigh, NC 27615
(Address of principal executive offices)
Issuer’s telephone number, including area code: 919.480.1518
Securities registered pursuant to Section 12(b) of the Act: None.
Securities registered pursuant to Section 12(g) of the Act: Common Stock, $0.001 Par Value.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was Required to submit and post such files). þ Yes ¨ No
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. (as defined in Rule 12b-2 of the Exchange Act). Check one:
|
Large accelerated filer
|
¨
|
Non-accelerated filer
|
¨
|
|
Accelerated Filer
|
¨
|
Smaller reporting company
|
þ
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes¨ No þ
As of October 31, 2013, the last day of the Registrant’s most recently completed second fiscal quarter; the aggregate market value of the shares of the Registrant’s common stock held by non-affiliates (based upon the closing stock price of $0.25 as reported on the Over-the-Counter Bulletin Board) was approximately $9,107,409. Shares of the Registrant’s common stock held by each executive officer and director and by each person who owns 10 percent or more of the outstanding common stock have been excluded in that such persons may be deemed to be affiliates of the Registrant. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
As of July 22, 2014, there were 68,273,253 outstanding shares of the registrant’s common stock, $0.001 par value.
Documents incorporated by reference: None.
Islet Sciences, Inc.
Form 10-K
Table of Contents
|
Page
|
||||
|
PART I
|
||||
|
Item 1.
|
Business
|
3
|
||
|
Item 1A.
|
Risk Factors
|
15
|
||
|
Item 2.
|
Properties
|
22
|
||
|
Item 3.
|
Legal Proceedings
|
22
|
||
|
PART II
|
||||
|
Item 5.
|
Market for Registrant’s Common Equity and Related Stockholder Matters and Issuer Purchases of Equity Securities
|
23
|
||
|
Item 7.
|
Management's Discussion and Analysis of Financial Condition and Results of Operations
|
23
|
||
|
Item 8.
|
Financial Statements and Supplementary Data
|
27
|
||
|
Item 9.
|
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
|
27
|
||
|
Item 9A
|
Controls and Procedures
|
27
|
||
|
Item 9B.
|
Other Information
|
29
|
||
|
PART III
|
||||
|
Item 10.
|
Directors, Executive Officers and Corporate Governance
|
29
|
||
|
Item 11.
|
Executive Compensation
|
34
|
||
|
Item 12.
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
|
36
|
||
|
Item 13.
|
Certain Relationships and Related Transactions, and Director Independence
|
38
|
||
|
Item 14.
|
Principal Accountant Fees and Services
|
39
|
||
|
PART IV
|
||||
|
Item 15.
|
Exhibits
|
40
|
||
|
Signatures
|
41
|
|||
|
Financial Statements
|
F-1
|
|||
2
PART I
Corporate History
Islet Sciences, Inc. (the “Company”, “we”, or “our”) was incorporated under the name One E-Commerce Corporation on September 14, 1994 in the State of Nevada. Effective February 23, 2012, the Company changed its name to Islet Sciences, Inc.
On December 30, 2011, the Company entered into an Agreement and Plan of Merger (the “Merger Agreement”) with ONCE, Inc., a Delaware corporation which was wholly-owned by the Company (“Merger Sub”), and Islet Sciences, Inc., a Delaware corporation (“ISI”). Pursuant to the Merger Agreement, on December 30, 2011, Merger Sub was merged with and into ISI, the holders of common stock of ISI received an aggregate of 38,005.87 shares of the Company’s Series B preferred stock, $.001 par value per share (“Series B Preferred”) in exchange for the cancellation of all of the shares of common stock of ISI formerly owned by them, and the holders of Series A preferred stock of ISI received an aggregate of 1,173 shares of the Company’s Series A preferred stock, $.001 par value per share (“Series A Preferred”) in exchange for the cancellation of all of the shares of Series A preferred stock of ISI formerly owned by them (the “Merger”).
Effective February 23, 2012, the Company completed a 1-for-45 reverse stock split (the “Reverse Split”) of shares of its common stock. Upon effectiveness of the Reverse Split, shares of Series A Preferred were automatically converted into an aggregate of 1,173,000 shares of common stock at a conversion ratio of one share of Series A Preferred for one thousand shares of common stock, and shares of Series B Preferred were automatically converted into an aggregate of 38,005,870 shares of common stock, at a conversion ratio of one share of Series B Preferred for one thousand shares of common stock. Upon conversion of Series A Preferred, the holders received the Company’s warrants to purchase an aggregate of 586,500 shares of Common Stock at an exercise price of $1.00 per share.
Merger with BHV Pharma
In March 2014 the Company announced it had signed a binding letter of intent to enter into a merger agreement with and acquire Brighthaven Ventures, LLC, a North Carolina limited liability company, d/b/a BHV Pharma (“BHV”) a privately held pharmaceutical company developing the SGLT2 inhibitor remogliflozin etabonate (“remogliflozin”) for type 2 diabetes and non-alcoholic steatohepatitis (“NASH”). Remogliflozin is currently in phase II clinical development. In exchange for 100% ownership of BHV, the Company will issue 30 million shares of its common stock to the holders of BHV units. Additional shares of common stock will be issued upon successful completion of development, regulatory and commercial milestones associated with the remogliflozin program. James Green and William Wilkison, the current members of BHV, are the Chief Executive Officer and the Chief Operating Officer of the Company, respectively, and are the sole members of BHV. The Company anticipates it will sign the merger agreement within the next 30 days and complete the merger upon successful completion of regulatory review.
Acquisition of DiaKine Therapeutics, Inc.
On March 14, 2012, the Company completed its acquisition of DiaKine Therapeutics, Inc., a Delaware corporation (“DTI”), and shareholders of DTI, whereby the Company issued to the DTI shareholders an aggregate of 200,000 shares of its newly designated shares of Series C convertible preferred stock, par value $0.001 per share (“Series C Preferred”) in exchange for all issued and outstanding shares of DTI. The Company also issued to DTI 100,000 shares of its common stock for no additional consideration in satisfaction of DTI’s liabilities outstanding at the closing under the agreement. As a result, DTI became a wholly-owned subsidiary of the Company. Subsequent to the closing of the DTI acquisition, the preferred stock was converted into 2,000,000 shares of common stock.
DTI’s compounds are small-molecule drugs that block the destructive, inflammatory actions of immune agents called cytokines. Cytokines have been shown in numerous studies to be components in the inflammation pathway that destroy insulin producing beta cells found in the pancreatic islets – a hallmark of type 1 diabetes and Latent Autoimmune Diabetes of Adults (LADA). Additionally, there is evidence that lipotoxicity, glucotoxicity, and other inflammatory factors induces progressive beta cell drop out in type 2 diabetes.
3
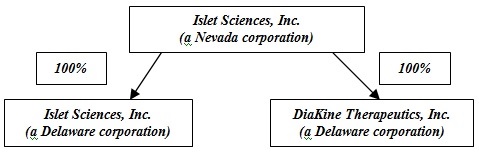
Company’s Corporate Structure
Below is the Company’s current corporate structure:
Our Business
Overview
We are a biotechnology company engaged in the research, development, and commercialization of new medicines and technologies for the treatment of metabolic diseases and related indications where there is a significant measurable unmet medical need. The rising incidence of obesity is associated with many obesity-related health complications, including cardiovascular disease, diabetes, hyperlipidemia, hypertension, nonalcoholic fatty liver disease/steatohepatitis (NAFLD/NASH). This constellation is also recognized as the metabolic syndrome and is characterized by underlying insulin resistance. These various diseases have interrelated risk factors and markers, such that often treatment of one disease may allow new therapies and opportunities for treatment in one of these related indications. Our focused effort to develop new therapies and related diagnostics for metabolic related diseases establishes us as a recognized leader in a large and growing market.
Metabolism is the ability of your body and its organs to process energy derived from the intake of nutrition. Metabolic diseases typically involve a derangement or malfunction of the normal processing of such metabolism. Some of the more prevalent metabolic diseases include obesity (excessive storage of nutrients); diabetes (loss of storage capability); NAFLD/NASH (excessive storage of lipids and fibrotic accumulation in the liver); dyslipidemia (inability to process fat from the blood); hypertension (excessive elevation of blood pressure).
We are developing the following therapeutic product candidates, which will include BHV’s SGLT2 inhibitor remogliflozin etabonate once the BHV merger is completed, to address the needs of patients suffering from metabolic disease:
|
●
|
Remogliflozin, a selective SGLT2 inhibitor in phase II clinical development for type 2 diabetes and NASH (BHV Pharma);
|
|
●
|
ISLT-P, an implantable suspension of encapsulated insulin-producing porcine islet cells, for the treatment of insulin-dependent diabetes;
|
|
●
|
ISLT-2669, a novel lead IL-12 small molecule inhibitor selected for preclinical development for treatment of type 2 diabetes. This IL12 inhibitor blocks the auto-immune and inflammatory cascade initiated by IL-12 receptor activation; and
|
|
●
|
ISLT-LSF Analogs, a library of small molecule lisofylline analogs that block inflammatory actions of cytokines that destroy insulin-producing beta cells, for diabetes and diabetes-related complications.
|
We are also developing the following diagnostic product candidate to better enable patients and their physicians to understand disease diagnosis and progression.
|
●
|
ISLT-Bdx, a PCR based molecular diagnostic measuring hypomethylated beta cell-derived DNA as a biomarker of beta cell loss for the early detection of type 1 diabetes or onset of insulin dependent type 2 diabetes;
|
4
Remogliflozin etabonate (“Remo”) is a selective O-linked glycoside SGLT2 inhibitor. In the kidney, glucose is filtered in the glomerulus and is reabsorbed through active transport mechanisms in the proximal tubule. Both sodium-glucose co-transporters, SGLT1 and SGLT2, are responsible for glucose reabsorption. SGLT2 is expressed exclusively in the proximal tubule and accounts for approximately 90% of glucose reabsorption. SGLT1, which is also expressed in GI and cardiac tissues, among others, accounts for the remaining approximate 10% of reabsorption. SGLT1 inhibition is known to cause significant GI distress; its effect on cardiac tissue is not completely known but it has been demonstrated that SGLT1 plays an active metabolic role in cardiac tissue.
Treating hyperglycemia with drugs that block renal glucose reabsorption through the SGLT2 transporter represents a validated approach to the treatment of type 2 diabetes. In non-diabetics, plasma glucose is filtered by the kidneys, but virtually all of it is reabsorbed. Inhibiting functional SGLT2 in the kidney reduces glucose reabsorption and therefore, more excess blood glucose is excreted into the urine which results in reduced circulating glucose, weight loss and increased insulin sensitivity via a beta-cell sparing insulin independent mechanism of action.
Pre-Clinical Summary: The Drug Metabolism and Pharmacokinetics (“DPMK”) package, as well as long term safety studies in rodents and dog, including two-year oncology studies, are complete. Toxicology studies in dog (52 week), rat (26 week) and mouse (13 week) demonstrate a very wide therapeutic window. Multiple diabetic rodent models also demonstrated significant plasma glucose, as well as reduction of fatty liver disease and weight loss.
Clinical Summary: Remo has been administered in sixteen phase I, three phase IIa, and two phase IIb clinical studies. Approximately 850 subjects, have been dosed with Remo. Remo has demonstrated dose-related glycosuria consistent with SGLT2 inhibition with best in class lowering of HbA1c, plasma glucose, and body weight observed in subjects with T2DM over 12 weeks of treatment. Remo has also proven to be well tolerated with low incidences of urogenital infections over 12 weeks of treatment in subjects with T2DM.
Phase I Clinical Studies: Remo has proven to be safe and well tolerated. Sixteen phase I studies, with doses ranging up to 4000 mg/day, have been performed in order to measure the PK, safety and drug-drug interaction of Remo. Remogliflozin etabonate (prodrug) was rapidly absorbed and extensively converted to its active entity, remogliflozin. Drug exposures were proportional to the dose administered and there were no apparent age or sex effect on the PK. There were no clinically significant changes in vital signs or ECGs and a thorough QtC study showed no effects of the drug on cardiac function. Subjects with mild and moderate renal impairment did not demonstrate any decrease in pharmacodynamic efficacy (as measured by UGE) or alteration in plasma PK. Drug-drug interaction studies with ketaconazole, bupropion, and metformin demonstrated no adverse effects of Remo on these compounds or vice versa. Formulation studies evaluated a number of formulations, with the biphasic formulation demonstrating the ideal characteristics for delivering remogliflozin primarily during the waking hours and minimizing remogliflozin exposure during the sleep period. The biphasic formulation was validated in healthy volunteers and demonstrated significant inhibition of SGLT2 during the waking hours while minimizing remogliflozin exposure during the sleep period.
Phase IIa Clinical Studies: Three phase IIa studies were conducted in subjects with T2DM. One study (KG219017) demonstrated that Remo caused FPG lowering in type II diabetics. All patients taking Remo also had clinically significant increases in urinary glucose excretion. Another study (KGW108201) utilizing echo MRI demonstrated that the weight loss observed with patients taking Remo was due to adipose (fat) loss and not fluid or lean mass loss. A third study (KG2110243) demonstrated that Remo was safe and effective when co-dosed with metformin. Fifty poorly controlled T2DM subjects taking metformin were treated with Remo for 13 days. The weighted mean glucose and insulin values following an oral glucose tolerance test (75 g glucose) were decreased in all treatment groups.
Phase IIb Clinical Studies: Two 12-week phase IIb studies of Remo were completed. The studies were multi-center, randomized, double blind, placebo-controlled, parallel-group, and dose-ranging with pioglitazone as an active comparator. The primary objective of both studies was to determine the dose response and efficacy of a range of doses of Remo versus placebo on the change from baseline in HbA1c over 12 weeks. Secondary objectives included additional markers of glycemic control, body weight and waist circumference. In addition, the safety and tolerability of Remo were characterized. Efficacy measurements included HbA1c, fasting plasma glucose, weight change and blood pressure. Safety measurements included clinical chemistry, UTI, GFI, and incidence of hypoglycemia. HbA1c entry criterion for both studies was ≥7.0% to ≤9.5%. In all doses, there were clinically significant reductions in HbA1c compared to placebo. The decreases in HbA1c were rapid, and were seen as early as Week 4 and continuing through to Week 12.
5
Study KG2110375 (“Remo QD”) was conducted to determine the dose response and efficacy of a range of once-daily doses with 252 subjects randomized across 14 countries. Clinically significant decreases from baseline in FPG were observed at Week 12 compared with placebo in all Remo once daily doses but were not as robust as the twice daily doses. Changes in FPG were seen as early as Week 2 and were maintained throughout to Week 12. In addition, the 24-hour UGE level increased from baseline in all remogliflozin etabonate treatment groups. Statistically significant decreases in body weight from baseline compared with placebo were observed at Week 12.
Study KG2105255 (“Remo BID”) was conducted to determine the dose response and efficacy of a range of twice-daily doses with 336 subjects randomized across 18 countries. A clinically significant dose response for the change from baseline in HbA1c at week 12 and significant decreases in HbA1c versus placebo were seen for all Remo doses. The decrease in HbA1c was generally rapid, beginning as early as Week 4. Significant dose-related decreases from baseline in FPG were observed at week 12 in all Remo doses compared with placebo, with changes seen as early as week 2. Similarly, 24-hour urinary glucose excretion (“UGE”) increased from baseline in all Remo treatment groups at week 12. Statistically significant decreases in body weight from baseline compared with placebo were observed at week 12.
Remogliflozin etabonate was efficacious for Hba1c lowering in both twice daily and once daily dosing, although the magnitude of decrease in Hba1c was greater with twice daily dosing. The compound also caused weight loss in both studies. Remogliflozin was well tolerated in both studies, with low incidences of hypoglycemia, urinary tract infection and genital infection.
Phase IIb Safety Profile: In both the Remo QD and Remo BID phase IIb studies, Remo was safe and well tolerated with no drug related SAEs. The overall rate of AEs across the treatment groups was low and the AE’s were all considered mild. The incidence of reported hypoglycemic events was low and were mild to moderate in intensity. There was also a low incidence of renal/urinary AEs. There were no significant differences from baseline in ECG. There were no significant changes in clinical chemistry parameters related to bone metabolism.
ISLT-P is an implantable suspension of encapsulated insulin-producing porcine islet cells, for the treatment of insulin-dependent diabetes. We believe that ISLT-P has significantly more commercial potential than human-to-human (allotransplantation) islet replacement approaches, due to the high cost and inherently limited supply of human islets. These implanted porcine islets are expected to produce insulin in response to increases in blood glucose, replacing the function of the patient’s destroyed pancreatic islets. Our near term development plans include initiation of an IND-enabling second species preclinical safety and efficacy study. After successful filing of an investigational new drug (“IND”) application, we anticipate initiating an open label phase I / II clinical study in patients with type 1 diabetes.
We expect ISLT-Bdx, our PCR based molecular diagnostic measuring beta-cell loss, to be the first commercially available diagnostic capable of recognizing active beta cell loss through measurement of hypomethalated beta-cell DNA circulating in bodily fluids. The diagnostic has been shown to predict the onset of type 1 diabetes as early as 625 days in advance of definitive diagnosis in populations with familial predisposition to type 1 diabetes. Early detection may provide for more effective disease management. We believe the diagnostic may also prove useful in measuring the progression of type 2 diabetics into insulin dependent type 2 diabetics. Our near term development strategy for ISLT-Bdx is the analysis of clinical serum samples from prospective type I diabetics and type II diabetics in clinical trials to confirm the ability of the assay to predict the onset of diabetes. In addition, we are establishing a state-of-the-art laboratory that will be certified under the Clinical Laboratory Improvement Amendments of 1988, or CLIA, to perform diagnostic testing of patient serum samples. We are dedicated to further validating and commercializing our diagnostic capabilities and making our diagnostic services available to all patients who need them through sales and distribution partnerships.
ISLT-2669 is a first-in-class immune-modulating IL-12 inhibitor shown to protect insulin-producing beta-cells from cytokines responsible for cell destruction. IL-12 is a pro-inflammatory cytokine important for immune responses leading to type 1 diabetes and atherosclerosis and involves the JAK/STAT4 signaling system to induce genes linked to chronic inflammatory disorders. It has been demonstrated that hyperglycemia, obesity and diabetes markedly increase IL-12/STAT4 expression in key tissues. We identified this compound from our proprietary library of orally bioavailable immune modulators. These compounds were screened in vitro for their ability to provide beta cell protection against cytokines and preservation of insulin secretion. From this initial screening effort, we identified ISLT-2669 as a potent inhibitor of STAT4 activation. ISLT-2669 had an EC50~10nM for inhibition of cytokine-induced apoptosis of a pancreatic beta cell line and produced insulin secretion responses very similar to LSF in vitro. Importantly, basal insulin secretion was not stimulated by increasing concentrations of ISLT-2669. This compound was also very stable in rat liver microsomes and showed little inhibitory effects on various CYP enzymes. The compound appeared to have good oral bioavailability (F=77%) and a rapid tmax (~30 min). An additional study in a rat model of NASH showed that ISLT-2669 may directly reduce liver fibrosis.
6
We continue to create and screen another series of small molecule analogs of lisofylline (“LSF”). LSF is a novel compound shown to have anti-inflammatory properties and has been the subject of three clinical studies. In these trials, LSF was well tolerated with no subject experiencing a serious adverse event. IL-12 and STAT 4 activation are important pathways linked to inflammatory damage to insulin producing cells. It has been shown that LSF and LSF analogs reduce activation of IL-12/STAT4, leading to preservation of islet function and viability and reduction of insulin resistance. IL-12 is made directly in insulin producing beta cells of the pancreas and can directly lead to reduced insulin secretion and cell death while Stat4 deletion reduces insulin resistance. In the Phase I/II trial, LSF significantly reduced the plasma levels of several major inflammatory cytokines linked to autoimmunity. LSF however has poor pharmaceutical characteristics. Therefore, these next generation compounds have better oral bioavailability and efficacy than LSF, offering the promise of providing a new therapeutic approach to treat type 1 or type 2 diabetes. We are developing a structure/activity relationship assay as well as creating new LSF analogs based on our previously generated data.
We intend to develop and commercialize these product candidates ourselves or in conjunction with appropriate business partners.
Market Opportunity
Diabetes is a chronic disease that occurs when the pancreas does not produce enough insulin or when the body cannot effectively use the insulin it produces. Insulin is a hormone secreted by beta-cells in the pancreas to regulate blood sugar. Hyperglycemia (high blood sugar) is a common effect of uncontrolled diabetes and over time leads to serious damage to many of the body's systems, especially the nerves and blood vessels. According to the World Health Organization, 347 million people worldwide have diabetes with an estimated 3.4 million deaths attributable to the disease in 2010. The American Diabetes Association (“ADA”) estimates that 29 million Americans have diabetes, 9.3% of the population, with another 86 million prediabetics that account for a $176 billion direct medical expenditure. Also according to the ADA, diabetes represents the 7th leading cause of death in the United States. Standard & Poor's has estimated the annual market for diabetes medications will hit $58 billion by 2018, from about $35 billion today (http://www.fiercepharma.com/special-reports/10-top-selling-diabetes-drugs-2012).
Type 1 diabetes (previously known as insulin-dependent or juvenile diabetes) is characterized by an autoimmune attack leading to destruction of insulin producing pancreatic beta-cells. The islets of Langerhans are invaded by mononuclear cells in an inflammatory reaction termed ‘insulitis’, leading to loss of up to 70% -80% of β cells by the time of onset of clinical symptoms (9). The destruction of β cells commences years before diagnosis. The result is an inability by the pancreas to produce insulin thereby requiring daily administration of exogenous insulin. The cause of type 1 diabetes is unknown, it is not preventable and there is currently no cure for the disease. Type 2 diabetes (formerly known as adult-onset diabetes) results from the body’s ineffective use of insulin. Type 2 diabetes comprises 90% of the global diabetic population and is largely the result of excess body weight and reduced physical activity.
Over time, diabetes can lead to complications in heart, blood vessels, eyes, kidneys, and nerves. Diabetes is associated with increased risk of heart disease and stroke. 50% of people with diabetes die of cardiovascular disease (primarily heart disease and stroke) ( Morrish NJ, Wang SL, Stevens LK, Fuller JH, Keen H. Mortality and causes of death in the WHO Multinational Study of Vascular Disease in Diabetes. Diabetologia 2001, 44 Suppl 2:S14–S21.). Combined with diabetic induced reductions in blood flow, neuropathy (nerve damage) in the feet increases incidence rates of foot ulcers, infections and limb amputation. Diabetic retinopathy is a common cause of blindness and is the result of long-term accumulated damage to blood vessels in the retina. Diabetes is among the leading causes of kidney failure (Global status report on noncommunicable diseases 2010. Geneva, World Health Organization, 2011.).
7
The overall risk of dying among people with diabetes is at least double the risk of their peers without diabetes (Roglic G, Unwin N, Bennett PH, Mathers C, Tuomilehto J, Nag S et al. The burden of mortality attributable to diabetes: realistic estimates for the year 2000. Diabetes Care, 2005, 28(9):2130–2135). The overall risk of dying among people with diabetes is at least double the risk of their peers without diabetes (Roglic G, Unwin N, Bennett PH, Mathers C, Tuomilehto J, Nag S et al. The burden of mortality attributable to diabetes: realistic estimates for the year 2000. Diabetes Care, 2005, 28(9):2130–2135).
In parallel with the increasing incidence of obesity and insulin resistance in the general population is the occurrence of fatty liver. Non-Alcoholic Fatty Liver Disease (“NAFLD”) is a chronic hepatic disorder that may affect up to 25% of the general population; including a significant portion of pediatric cases. NAFLD can progress to Nonalcoholic steatohepatitis (“NASH”) which is thought to affect 2%-5% of the U.S. population. Non-alcoholic fatty liver disease (NAFLD) is a condition defined by excessive fat accumulation in the form of triglycerides (steatosis) in the liver. A subgroup of NAFLD patients have liver cell injury and inflammation in addition to excessive fat called non-alcoholic steatohepatitis (NASH). NASH dramatically increases the risks of cirrhosis, liver failure, and hepatocellular carcinoma (HCC). The prevalence of NAFLD (40-90%) and NASH (10-20%) is increasing globally (Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis, World Gastroenterology Organization Global Guidelines, June 2012).
NASH is characterized by increased fibrosis, cirrhosis and chronic inflammation which can lead to end-stage liver disease. Current predictions estimate 40% of the population will be obese by 2025, and that NAFLD/NASH will become the leading cause of liver disease and liver transplantation. Even now the demand for livers suitable for transplant and non-transplantable livers for research far exceeds availability, leaving an urgent need for alternative human cell-based models.
NAFLD/NASH is a common, often “silent” liver disease. It resembles alcoholic liver disease, but occurs even in people who drink little or no alcohol. The major feature of NAFLD/NASH is fat in the liver, along with inflammation and damage. Most people with NAFLD/NASH generally feel well and are not aware of their liver problem. Left untreated NAFLD/NASH leads to liver fibrosis and ultimately cirrhosis of the liver, in which the liver is permanently damaged and no longer able to function properly. While the underlying reason for NAFLD/NASH is not known, two key factors thought to be responsible include insulin resistance and oxidative stress inside liver cells.
Although NAFLD/NASH has become more common, its underlying cause is still not clear. It most often occurs in persons who are middle-aged and overweight or obese. Many patients with NAFLD/NASH have elevated blood lipids, such as cholesterol and triglycerides, and many have diabetes or pre-diabetes, but not every obese person or every patient with diabetes has NAFLD/NASH. Furthermore, some patients with NAFLD/NASH are not obese, do not have diabetes, and have normal blood cholesterol and lipids. NAFLD/NASH can occur without any apparent risk factor and can even occur in children. Thus, NAFLD/NASH is not simply obesity that affects the liver. While the underlying reason for the liver injury that causes NASH is not known, several factors are possible candidates:
● insulin resistance
● release of toxic inflammatory proteins by fat cells (cytokines)
● oxidative stress (deterioration of cells) inside liver cells
Currently, no specific therapies for NAFLD/NASH exist. The most important recommendations given to persons with this disease are to reduce their weight (if obese or overweight), follow a balanced and healthy diet, increase physical activity and avoid alcohol. Experimental approaches under evaluation in patients with NAFLD/NASH include antioxidants, such as vitamin E, selenium, and betaine. These medications act by reducing the oxidative stress that appears to increase inside the liver in patients with NAFLD/NASH. Another experimental approach to treating NAFLD/NASH is the use of newer antidiabetic medications—even in persons without diabetes. Most patients with NAFLD/NASH have insulin resistance, meaning that the insulin normally present in the bloodstream is less effective for them in controlling blood glucose and fatty acids in the blood than it is for people who do not have NAFLD/NASH. The newer antidiabetic medications make the body more sensitive to insulin and may help reduce liver injury in patients with NAFLD/NASH.NASH (Non-alcoholic steatohepatitis) is characterized by inflammation and concurrent fat accumulation in the liver when not associated with excessive alcohol intake. Steatohepatitis is the progressive form of non-alcoholic fatty liver disease and can progress to cirrhosis. NASH is a frequent cause of unexplained cirrhosis and is associated with lysosomal acid lipase deficiency. While the underlying reason for the liver injury that causes NASH is not known, several factors are possible candidates including insulin resistance, release of toxic inflammatory proteins by fat cells (cytokines), or oxidative stress (deterioration of cells) inside liver cells.
8
Our Strategy
Our strategy is to transition into a clinical-stage growth company and become a leading innovator, developer and provider of therapeutics and diagnostics for patients suffering from metabolic related disease. As part of that transition we will continue to rebrand the Company to better reflect our broader focus on metabolic related disease. We believe that completion of our announced merger with BHV Pharma will successfully complete our transition into a clinical stage growth company. After the completion of our merger with BHV Pharma, we intend to change the Company’s name. We also intend to seek additional investment in the Company to support the development of our pipeline of therapeutic and diagnostic product candidates. Additionally, the Company intends to develop strategic relationships with other companies in order to accelerate, where possible, product development and to develop distribution channels for our product candidates. The key elements of our strategy are as follows:
|
●
|
Initiate two phase IIb clinical studies for remogliflozin etabonate, one for the type 2 diabetes indication and the other for the NASH indication, to measure safety and efficacy as well as to select doses for the respective phase III development programs.
|
|
●
|
Validate and commercialize ISLT-Bdx for type 1 diabetes using a CLIA certified diagnostic laboratory and a sales and distribution partner. Once the commercial market is established and validation for type 2 diabetes complete, we expect to bring forward a diagnostic product for type 2 diabetes;
|
|
●
|
Continue preclinical development of ISLT-2669 and LSF analogs for diabetes and other autoimmune and/or inflammatory related diseases
|
|
●
|
Advance ISLT-P into a second species IND enabling study targeting T1DM
|
|
●
|
Form development and commercialization partnerships where appropriate; and
|
|
●
|
Selectively acquire rights to and develop complementary product candidates.
|
Intellectual Property
The Company seeks patents and other intellectual property rights to protect and preserve our proprietary technology and our right to capitalize on the results of its research and development activities. The Company is pursuing patents in the US and in foreign countries. The Company may also rely on trade secrets, know-how, continuing technological innovations and licensing opportunities to provide competitive advantages for its eventual products in various markets and to accelerate new product introductions.
Below is a list of the US patent applications held by the Company.
Islet Sciences, Inc.
|
Title
|
Patent Application Number
|
Status
|
||
|
Ex Vivo Maturation of Islet Cells
|
W02103/049693
|
Pending
|
||
| US/14,346,454 | ||||
| PCT/US2012/058087 | ||||
|
Compositions And Methods For Detecting Hypo-Methylated DNA In Body Fluids
|
PCT/US2014/020431
|
Pending
National Phase begins September 2015
|
9
The maturation patent comprises a novel method for isolating islets cells from a porcine animal model in which the pancreas is excised, intact, from the porcine animal model which is then severed into a series of appropriate sizes and volume. The isolated and severed exocrine tissue and islet cells are then treated in-vitro with a non-specific collagenase enzyme exposure for a specific period that results in a partial digestion of the exocrine tissue comprising islets cells that are embedded within and protected by the partial digested exocrine tissue. It is the partial digestion followed by in vitro maturation that allows islets to function and be able to survive long term. In addition, the present invention further comprises a novel method for maturing a plurality of isolated porcine islet cells that are functional and can respond to glucose stimulation early in the maturation process.
The international patent application PCT/US2014/010431 provides for methods of detecting hypomethylated DNA in a process similar to that described for the beta cell loss diagnostic but in bodily fluids such as saliva and urine as opposed to blood/serum. The obvious advantage is the non-invasive nature of this testing.
Patent applications licensed by the Company from Winthrop University Hospital
|
Title
|
Patent Application Number
|
Status
|
||
|
Method For Using Probe Based PCR Detection To Measure The Levels Of Circulating Demethylated Beta Cell-Derived DNA as a Measure Of Beta Cell Loss In Diabetes
|
PCT/US2013/028862
|
Pending
Enters National
Phase September 2014
|
||
|
Method For Using Probe Based PCR Detection To Measure The Levels Of Circulating Demethylated Beta Cell-Derived DNA as a Measure Of Beta Cell Loss In Diabetes
|
US Patent Application 13/279,261
|
Pending
|
Patent Applications that are licensed by Islet Sciences, Inc. from Yale University
|
Title
|
Patent Application Number
|
Status
|
||
|
Compositions and Methods of Diagnosing Diseases and Disorders associated with b cell death
|
EP 2723901
|
Pending
|
||
| US 14/127,906 | ||||
| These applications correspond to WO2012/178007 |
The family of patent applications corresponding to WO2012/178007 and PCT/US2013/028862 relate to compositions and methods for detecting cell death by detecting β-cell insulin gene DNA in a biological sample based on the discovery that the presence of extrapancreatic hypomethylated β-cell DNA is indicative of β-cell death. Thus, in one embodiment, the invention is a method of detecting hypomethylated β-cell insulin DNA in a biological sample of a subject including the steps of: obtaining a biological sample from the subject, where the biological sample is obtained from outside of the subject's pancreas, and where the biological sample contains β-cell insulin DNA; determining the methylation status of at least one of the CpG dinucleotides in the β-cell insulin DNA, where when at least one of the CpG dinucleotides in the β-cell insulin DNA is determined to be unmethylated, the hypomethylated β-cell insulin DNA is detected.
BHV Pharma
|
Title
|
Patent (Application) Number
|
Status
|
||
|
Glucopyranosyloxypyrazole derivatives, medicinal compositions containing the same and intermediates in the production thereof
|
US 6,972,283; 7,056,892; 7,115,575;
|
Patent
|
||
|
Glucopyranosyloxypyrazole Derivates and Use Thereof in Medicines
|
US 7,084,123; 7,393,838; 7,465,713
|
Patent
|
||
|
Progression Inhibitor For Disease Attributed To Abnormal Accumulation Of Liver Fat
|
US 12/511,654
|
Pending
|
||
| WO2006/0091149 | ||||
|
Process For Production Of Glucopyranosyloxypyrazole Derivative
|
US 8,022,192
|
Patent
|
||
| WO2006/098413 | ||||
|
Combination immediate/delayed release delivery system for short half-life pharmaceuticals including remogliflozin
|
PCT/US2011/043143
|
Pending
|
||
| WO2012/006398 |
10
The first two sets of issued patents relate to composition of matter patents for glucopyranosyloxypyrazole derivatives representing remogliflozin (the active circulating species) and remogliflozin etabonate (the prodrug) which have inhibitory activity in human SGLT2 and are useful as agents for the prevention or treatment of diabetes, diabetic complications or obesity, and to pharmaceutical compositions comprising the same and intermediates thereof. The patent application US 12/511,654 and WO2006/0091149 provides a pharmaceutical composition characterized by containing a sodium/glucose cotransporter 2 inhibitor as an active ingredient. This pharmaceutical composition is capable of inhibiting any abnormal accumulation of fat in the liver and is highly suitable for use as a progression inhibitor for not only general fatty liver, but also non-alcoholic fatty liver (NAFL), non-alcoholic steatohepatitis (NASH), hyperalimentation-induced fatty liver, diabetic fatty liver, alcohol-induced fatty liver or toxic fatty liver. US 8,022,192 and WO2006/098413 discloses a process for producing a glucopyranosyloxypyrazole derivative, such as remogliflozin etabonate, which is useful as a prophylactic or therapeutic agent for a disease induced by hyperglycemia, such as diabetes, a diabetes complication and obesity. This process is quite useful as the process for producing a pharmaceutical. The family of patent applications corresponding to WO2012/006398 (as well as South African patent ZA 2013/00877) relate to a combination immediate/delayed release delivery system for compounds which have short half-lives, such as the antidiabetic remogliflozin etabonate. This delivery system provide a dosage form that has two distinct phases of release, a formulation that promotes immediate release of the compound upon ingestion and another formulation which delays the release of the compound so that a once-daily dosing regimen of remogliflozin etabonate may be achieved while providing effective control of plasma glucose and minimizing the nighttime exposure of this compound. Methods for forming the so-described immediate/delayed release delivery system and using such delivery system for treating diabetes are also provided.
DiaKine Therapeutics, Inc.
DiaKine has licensed the rights for next generation small molecule immune modulator-anti-inflammatory drugs from the University of Virginia Patent Foundation.
US Patents licensed by DTI from the University of Virginia Patent Foundation
|
Title
|
Patent Application Number
|
Status
|
||
|
Lisofylline analogs and methods for use
|
US 8,481,580
US 13/928691
|
Patent
Pending
|
||
| Lisofylline analogs and methods for use | US 13/477,613 | Pending |
Foreign Patents and Patent Applications that are licensed by DTI from the University of Virginia Patent Foundation
|
Title
|
Patent Application Number
|
Status
|
||
|
Lysofylline Analogs and Methods for Use
|
WO2007027719A2
|
Pending
|
11
There can be no assurance that the Company will succeed in obtaining any patent protection from its pending patent applications. No assurance can be given that any patent will be issued or that the scope of any patent protection will exclude competitors or that any patent, if issued, will be held valid if subsequently challenged. There can be no assurance that any steps the Company takes in this regard will be adequate to deter misappropriation of its proprietary rights or independent third parties developing functionally equivalent products. Despite precautions, unauthorized parties may attempt to engineer, reverse engineer, copy, or obtain and use the Company’s products or other information. Although management believes that the Company’s products do not infringe on the intellectual property rights of others, there can be no assurance that an infringement claim will not be asserted in the future. The prosecution or defense of any intellectual property litigation can be extremely expensive and would place a material burden upon the Company’s working capital.
License and Supply Agreements
On May 2, 2012, the Company, entered into a license agreement with the Yale University (“Yale”). Under the agreement, the Company received exclusive license to technology directed to compositions and methods for detecting cell death by detecting β-cell insulin gene DNA in a biological sample based on the discovery that the presence of extrapancreatic hypomethylated β-cell DNA is indicative of β-cell death and supports our efforts with the ISLT-BDx which is patented by Yale. In consideration of the license granted under the agreement, the Company paid Yale a license issue royalty of $10,000 (plus a $10,000 annual renewal fee) and issued 20,000 shares of its common stock, and agreed to pay certain milestones royalties by issuing an aggregate of 160,000 shares of common stock. The Company also agreed to pay to Yale a royalty on net sales. The agreement will expire automatically, on a country-by-country basis, on the date on which the last of the claims of the subject patents expires. The agreement can be terminated by Yale if the Company defaults on its obligations under the agreement and fails to cure such default within 60 days of a written notice by the university. The Company can terminate the agreement upon a six month notice subject to payment of all amounts due Yale under the agreement.
On July 23, 2012, the Company entered into a licensing agreement with the Winthrop University Hospital (“Winthrop”) to license certain patents and technology directed to compositions and methods for detecting cell death by detecting β-cell insulin gene DNA in a biological sample based on the discovery that the presence of extrapancreatic hypomethylated β-cell DNA is indicative of β-cell death and supports our efforts with the ISLT-BDx. In consideration of the license granted under the agreement, the Company agreed to pay to Winthrop a license issue royalty of $10,000 (plus a $10,000 annual renewal fee) and issue 20,000 shares of its common stock, and to pay certain milestones royalties by issuing an aggregate of 160,000 shares of common stock. The Company also agreed to pay to Winthrop a royalty on net sales. The agreement will expire automatically, on a country-by-country basis, on the date on which the last of the claims of the subject patents expires. It can be terminated by Winthrop if the Company defaults on its obligations under the agreement and fails to cure such default within 60 days of a written notice by the university. The Company can terminate the agreement upon a six month notice subject to payment of all amounts due Winthrop under the agreement. The Company is currently in default regarding its payment obligations under the foregoing agreements and is in active discussions with Winthrop University Hospital.
On January 10, 2012, the Company entered into a letter agreement with Progenitor Cell Therapy, LLC (“PCT”), a subsidiary of NeoStem, Inc. (“NeoStem”), which was amended by an agreement dated May 15, 2012 by and between the Company and NeoStem, PCT’s parent company. Under the agreement, PCT was to, among other things, generate protocols, develop procedures for testing and quality control and manufacturing to support the development and submission of an investigational new drug application to the FDA for the Company’s encapsulated porcine islet cells for the treatment of diabetes. The Letter Agreement estimated the duration of services to span approximately six months beginning on January 17, 2012. The Letter Agreement required the parties to enter into a mutually agreeable Services Agreement that would more particularly set forth the services to be provided by PCT and include customary representations, warranties, covenants, indemnities and agreements between the parties. The Services Agreement was never entered into by the parties. As compensation for the services of PCT, the Company agreed to pay to PCT a non-refundable monthly fee of $63,000 and a non-refundable monthly charge of between $33,000 and $54,000. NeoStem was entitled to receive shares and warrants of the Company’s common stock, as well as additional shares for no consideration so that NeoStem’s ownership was not less than 1% of outstanding shares on a fully diluted basis. PCT was also given the exclusive right for a period of ten years to manufacturer any product involved in the services to be provided under the agreement. With respect to commercial production of such products, PCT will be entitled to a royalty on gross sales and a percentage of sublicensing fees, royalties, milestone fees or profit sharing payments.
12
On February 7, 2013, the services had not yet been completed and the Company provided to NeoStem Inc. notice of termination of the agreement with PCT dated January 10, 2012, as amended by the agreement dated May 15, 2012 by and between the Company and NeoStem.
On April 25, 2014, LLC, PCT filed a lawsuit against the Company in the United States District Court for the District of New Jersey (Case No. 2:14-cv-02658-SDW-MCA). PCT’s complaint asserts various claims, including breach of contract and unjust enrichment, based on the alleged failure of the Company to pay for services and goods provided by PCT under a January 10, 2012 letter agreement. See Item 3 for legal proceedings.
On July 23, 2012, the Company entered into a long-term supply agreement with Spring Point Project, a source animal facility to purchase pigs for use in the Company’s xenotransplantation research. On August 12, 2013, the Company received from Spring Point Project a notice of termination of the supply agreement, effective November 10, 2013, unless the Company cures the default before that date. The default was not cured and the agreement with Spring Point Project has been terminated.
In August 2012, the Company entered into an agreement with the Regents of the University of California, Los Angles, (“UCLA’), whereby UCLA was provided work dealing with small molecule mediated porcine islet proliferation. Work under this agreement will be performed for a period of six months with an estimated cost of $23,100. The Company had continued to work with UCLA on a month to month basis following the six month period. On February 25, 2014, the Company terminated its licensing agreement with UCLA.
In May 2013, the Company entered into a sales and services agreement with the Regents of the University of California, Irvine (“UCI”) to provide materials consisting of isolated islets to be supplied to the Spring Point Project. The Company terminated this agreement on March 24, 2014 for settlement payments totaling $150,000 on total liabilities of approximately $305,000 for expenses incurred in this and last fiscal year.
In September 2013, the Company entered into a consulting agreement with American Capital Ventures, Inc. to provide consulting services related to investor relations and corporate communications. The Company terminated this agreement on January 23, 2014.
Competition
The biotechnology and pharmaceutical industries are highly competitive and characterized by rapid technological change. We face significant competition in each of the aspects of our business from other pharmaceutical and biotechnology companies. Many of our competitors have substantially greater research and development capabilities and financial, scientific, marketing and human resources than we do. As a result, our competitors may succeed in developing products earlier than we do, obtaining approvals from the Food and Drug Administration or FDA or other regulatory agencies for those products more rapidly than we do, or developing products that are more effective than those we propose to develop. Similarly, our collaborators face similar competition from other competitors who may succeed in developing products more quickly, or developing products that are more effective, than those developed by our collaborators. Any products that we may develop or discover are likely to be in highly competitive markets.
Government Regulation
Overview
The development and commercialization of our products will be subject to extensive regulation in the U.S. by a number of regulatory authorities, including the FDA, and by comparable regulatory authorities in foreign countries. These regulatory authorities and other federal, state and local entities will regulate, among other things, the preclinical and clinical testing, safety, effectiveness, approval, manufacturing, labeling, packaging, export, storage, recordkeeping, adverse event reporting, and promotion and advertising of our products. We will require FDA approval of our products in the United States, including a review of the manufacturing processes and facilities used to produce our products, before we may market the products in the U.S.
13
Remogliflozin etabonate and ISLT-2669 will be subject to approval by CDER (Center for Drug Evaluation and Research), a division in the FDA while ISLT maintains an active IND for remogliflozin etabonate and will plan to file an IND for 2669 after completion of IND-enabling studies. ISLT-P will be classified as combination biological and device products by the CBER (Center for Biologicals Evaluation and Research), another division within the FDA. The islets comprise the biological component and the microencapsulation material comprises the device component. The FDA Center for Biologics (“CBER”) has jurisdiction, meaning the ISLT-P product will be reviewed primarily as a biological product. Finally, ISLTDX will be subject to approval by the Medical Device division of the FDA.
Clinical Trial Process
Remogliflozin etabonate is being developed with a new formulation. The next step in the development process will be to examine the safety and efficacy of multiple doses of remogliflozin etabonate in our biphasic formulation in two phase IIb clinical studies; one for T2DM and the other for NASH. This study will be very similar in design to the studies previously run with remogliflozin etabonate. Pending completion of this study, ISLT will have a formal end of phase II meeting with the FDA to obtain guidance and permission for the phase III studies.
For the ISLT-P and ISLT-2669 and LSF inhibitory analogs, the development of a therapeutic product for human use under applicable laws and regulations is a multi-step process. First, in vitro and/or animal testing must be conducted in a manner consistent with good laboratory practices to establish the potential safety and effectiveness of the experimental product with regard to a given disease. Before human clinical trials may begin for new drugs and biologics, an IND application containing, among other things, preclinical data, chemistry, manufacturing and control information, an investigative plan, must be submitted to the FDA. The Clinical trials following approval of an IND will also require the approval and oversight by an Institutional Review Board (“IRB”) to assure proper patient protection. Clinical trials of certain medical devices generally require the same sort of submission in the form of an application for an investigational device exemption (“IDE”). We believe that the submission of an IND will be adequate for ISLT-P, and that a separate IDE submission will not be required for either product. Once a trial begins, changes to the investigational product or study protocol may require prior approval before implementation. There can be no assurance that submission of an IND application or an IDE will result in the ability to commence clinical trials. In addition, the FDA may place a clinical trial on hold or terminate it at any phase if, among other reasons, it concludes that clinical subjects are being exposed to an unacceptable health risk. Finally, pursuant to the FDA's Bioresearch Monitoring (“BIMO”) program, the FDA conducts on-site inspections and data audits of the conduct and reporting of all FDA-regulated research. The FDA's BIMO compliance programs address inspections of non-clinical testing labs, clinical investigators, clinical trial sponsors/monitors and IRBs.
Clinical trials of pharmaceuticals or biologics typically involve three phases, although those phases can overlap.
● Phase I is conducted to evaluate the basic safety, metabolism, and pharmacology, and pharmacokinetics, and to identify potential side effects with escalating doses, the maximum tolerated dose of the experimental product in humans, and if possible, to begin to evaluate various routes, dosages, and schedules of product administration. These studies are often conducted in healthy subjects. Phase I trials are not intended to find early indications of effectiveness; however, it is not uncommon to evaluate these endpoints.
● Phase I/II clinical trials are conducted to evaluate safety and initial efficacy indications in the patient population afflicted with a specific disease or condition for which the product is intended for use.
● Phase II clinical trials are conducted in groups of patients afflicted with a specific disease or condition for which the product is intended for use in order to further test safety, begin evaluating effectiveness, optimize dosage amounts and determine dose schedules and routes of administration.
● Phase III studies are usually randomized, double blind studies testing for product safety and effectiveness in an expanded patient population in order to evaluate the overall risk/benefit relationship of the product and to provide an adequate basis for product labeling. These studies also may compare the safety and effectiveness of the product with currently available products.
Employees
As of July 22, 2014, the Company had 3 fulltime and 2 part time employees. The Company also utilizes the services of consultants and advisors on an appropriate as needed basis.
14
Emerging Growth Company
The Company is an “emerging growth company”, as defined in the Jumpstart Our Business Startups Act of 2012 (“JOBS Act”), and may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of section 404(b) of the Sarbanes-Oxley Act, and exemptions from the requirements of Sections 14A(a) and (b) of the Securities Exchange Act of 1934 to hold a nonbinding advisory vote of shareholders on executive compensation and any golden parachute payments not previously approved.
The Company has elected to use the extended transition period for complying with new or revised accounting standards under Section 102(b)(1) of the JOBS Act. This election allows us to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies. As a result of this election, our financial statements may not be comparable to companies that comply with public company effective dates.
We will remain an “emerging growth company” until the earliest of (1) the last day of the fiscal year during which our revenues exceed $1 billion, (2) the date on which we issue more than $1 billion in non-convertible debt in a three year period, (3) the last day of the fiscal year following the fifth anniversary of the date of the first sale of our common equity securities pursuant to an effective registration statement filed pursuant to the Securities Act of 1933, as amended, or (4) when the market value of our common stock that is held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter.
To the extent that we continue to qualify as a “smaller reporting company”, as such term is defined in Rule 12b-2 under the Securities Exchange Act of 1934, after we cease to qualify as an emerging growth company, certain of the exemptions available to us as an emerging growth company may continue to be available to us as a smaller reporting company, including: (1) not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes Oxley Act; (2) scaled executive compensation disclosures; and (3) the requirement to provide only two years of audited financial statements, instead of three years.
ITEM 1A. RISK FACTORS
An investment in our common stock involves a high degree of risk. You should carefully consider the risks described below, together with all of the other information included in this report, before making an investment decision. If any of the following risks actually occurs, our business, financial condition or results of operations could suffer. In that case, the trading price of our common stock could decline, and you may lose all or part of your investment.
We have a limited operating history and are a development stage company.
We started our operations in 2010 with the formation of ISI. To date, we have had no sales and profits. We cannot assure investors that we will ever become or remain profitable. An investment in our securities is subject to all of the risks involved in a newly established business venture. Potential investors should be aware of the problems, delays, expenses, and difficulties experienced by companies in the early developmental stage, which generally include unanticipated problems and additional costs relating to the commencement of operation and implementation of a business plan.
Future funding requirement
To date, we have not generated any revenue from product sales. We do not know when, or if, we will generate any revenue from product sales. We do not expect to generate any revenue from product sales unless and until we obtain regulatory approval of our product candidates and our companion diagnostic products or any of our other product candidates. At the same time, we expect our expenses to increase in connection with our ongoing development activities, particularly as we continue the research, development and clinical trials of, and seek regulatory approval for, our product candidates and companion diagnostic. In addition, subject to obtaining regulatory approval of any of our product candidates and companion diagnostic, we expect to incur significant commercialization expenses for product sales, marketing, manufacturing and distribution. We anticipate that we will need significant additional funding, which may not be available to us on acceptable terms, or at all, and if not so available, may require us to delay, limit, reduce, or cease our operations.
15
Until such time that we can generate meaningful revenue from product sales, if ever, we expect to finance our operating activities through public or private equity or debt financings, government or other third-party funding, marketing and distribution arrangements, and other collaborations, strategic alliances and licensing arrangements or a combination of these approaches. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of our common stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of our common stockholders. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through government or other third-party funding, marketing and distribution arrangements or other collaborations, or strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us.
There are risks associated with the Company’s pending merger with BHV.
In March 2014, the Company announced it had signed a binding letter of intent to enter into a merger agreement with and acquire BHV. The Company anticipates it will sign the merger agreement within the next thirty days and complete the merger upon successful regulatory review. The addition of BHV's remogliflozin to our pipeline is a key element of our strategy to transition into a clinical stage company and attract additional funding. If this transaction is not completed or its completion is delayed the Company’s strategy, results of operations and consolidated financial condition may be adversely impacted.
If we cease to continue as an operational entity, due to lack of funding or otherwise, you may lose your entire investment in the Company.
Our current plans indicate that we will need substantial additional capital for research and development, including costs associated with developing our technology and conducting clinical trials of our product candidates, before we have any anticipated revenue generating products.
When we require additional funds, general market conditions or the then-current market price of our common stock may not support capital raising transactions such as additional public or private offerings of our common stock. If we require additional funds and we are unable to obtain them on a timely basis or on terms favorable to us, we may be required to scale back our development of new products, sell or license some or all of our technology or assets, or curtail or cease operations.
You may experience dilution of your ownership interests due to the future issuance of additional shares of our common stock.
We may in the future issue our previously authorized and unissued securities, resulting in the dilution of the ownership interests of our common stockholders. We are currently authorized to issue one hundred million shares of common stock and ten million shares of preferred stock with such designations, preferences and rights as determined by our board of directors. Issuance of additional shares of common stock may substantially dilute the ownership interests of our existing stockholders. We may also issue additional shares of our common stock or other securities that are convertible into or exercisable for common stock in connection with the hiring of personnel, future acquisitions, future public or private placements of our securities for capital raising purposes, or for other business purposes. Any such issuance would further dilute the interests of our existing stockholders.
There has been no active public market for the Company's securities.
There has been no active public market for the Company common stock. An active public market for the Company's common stock may not develop or be sustained. The market price of the common stock may fluctuate significantly in response to factors, some of which are beyond the Company's control, such as product liability claims or other litigation, the announcement of new pharmaceuticals or pharmaceutical enhancements by the Company’s competitors, developments concerning intellectual property rights and regulatory approvals, quarterly variations in competitors' results of operations, changes in earnings estimates or recommendations by securities analysts, developments in our industry, and general market conditions and other factors, including factors unrelated to our operating performance.
16
The Company’s common stock is considered a "penny stock" and may be difficult to sell.
The SEC has adopted regulations which generally define "penny stock" to be an equity security that has a market or exercise price of less than $5.00 per share, subject to specific exemptions. The market price of the Company’s common stock is currently below $5.00 per share and therefore is designated as a "penny stock" according to SEC rules. This designation requires any broker or dealer selling these securities to disclose certain information concerning the transaction, obtain a written agreement from the purchaser and determine that the purchaser is reasonably suitable to purchase the securities. These rules may restrict the ability of brokers or dealers to sell such shares and may affect the ability of investors to sell their shares. In addition, since the Company’s common stock is currently quoted on the OTC Bulletin Board, investors may find it difficult to obtain accurate quotations of the stock and may find few buyers to purchase the stock or a lack of market makers to support the stock price.
No assurance of future successful development.
Prospects for companies in the medical industry generally are uncertain given the nature of the industry and, accordingly, investments in medical companies should be regarded as highly speculative.
Our products may not be successfully developed or commercialized, which would harm us and force us to curtail our operations.
We may not be able to obtain regulatory approvals for product candidates we develop, to enter clinical trials for any of our product candidates, or to commercialize any products, on a timely basis or at all. If we are unable, for technological or other reasons, to complete the development, introduction or scale-up of manufacturing of potential products, or if our products do not achieve a significant level of market acceptance, we would be forced to curtail or cease operations.
Any product candidate we advance into clinical trials may cause undesirable side effects that could delay or prevent its regulatory approval or commercialization.
Undesirable side effects caused by any product candidate we advance into clinical trials could interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by the FDA or other regulatory authorities for any or all targeted indications. This, in turn, could prevent us from commercializing these or any other product candidate we advance into clinical trials.
Any one or a combination of these events could prevent us from achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing the product, which in turn could delay or prevent us from generating significant revenues from the sale of the product.
If we receive regulatory approval we will also be subject to ongoing FDA obligations and continued regulatory review such as continued safety reporting requirements, and we may also be subject to additional FDA post-marketing obligations. FDA and corresponding foreign regulatory requirements could adversely affect our ability to generate revenue and require additional expenditures to bring our products to market.
Any regulatory approvals that we receive for our products may also be subject to limitations on the indicated uses for which the product may be marketed or contain requirements for potentially costly post-marketing follow-up studies. In addition we or our third party manufacturers may be required to undergo a pre-approval inspection of manufacturing facilities by the FDA and foreign authorities before obtaining marketing approval and will be subject to periodic inspection by the FDA and corresponding foreign regulatory authorities under reciprocal agreements with the FDA. Such inspections may result in compliance issues that could prevent or delay marketing approval or require the expenditure of money or other resources to correct noncompliance.
17
If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product, our collaborators, or us, including requiring withdrawal of the product from the market. Our product candidates will also be subject to ongoing FDA requirements for the labeling, packaging, storage, advertising, promotion, record-keeping, and submission of safety and other post-market information on the drug. If our product candidates fail to comply with applicable regulatory requirements, a regulatory agency may:
- issue warning letters;
- impose civil or criminal penalties;
- withdraw regulatory approval;
- suspend any ongoing clinical trials;
- refuse to approve pending applications or supplements to approved applications filed by us or our collaborators;
- impose restrictions on operations, including costly new manufacturing requirements; or
- seize or detain products or require a product recall.
Moreover, in order to market any products outside of the United States, we and our collaborators must establish and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from that required to obtain FDA approval. The regulatory approval process in other countries may include all of the risks described above regarding FDA approval in the United States. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others. Failure to obtain regulatory approval in other countries or any delay or setback in obtaining such approval could have the same adverse effects detailed above regarding FDA approval in the United States. As described above, such effects include the risk that our product candidates may not be approved for all indications requested, which could limit the uses of our product candidates and adversely impact potential royalties and product sales, and that such approval may be subject to limitations on the indicated uses for which the product may be marketed or require costly, post-marketing follow-up studies.
If we or our collaborators fail to comply with applicable domestic or foreign regulatory requirements, we and our collaborators may be subject to fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions, and criminal prosecution.
The transplantation of animal cells into humans involves risks which have resulted in additional FDA oversight and which in the future may result in additional regulation that may prevent or delay approval of our potential products and require additional expenditures to bring these products to market.
Our business involves the transplantation of animal cells into humans, a process known as xenotransplantation. Xenotransplantation poses a risk that viruses or other animal pathogens may be unintentionally transmitted to a human patient. The FDA will require testing to determine whether infectious agents, including porcine endogenous retroviruses, also known as PERV, are present in patients who have received cells, tissues, or organs from porcine sources. While PERV has not been shown to cause any disease in pigs, it is not known what effect, if any, PERV may have on humans.
Other companies are currently conducting clinical trials involving the transplantation of porcine cells into humans. The FDA requires lifelong monitoring of all recipients of xenotransplantation products. If PERV or any other virus or infectious agent is detected in tests or samples from these transplant recipients, the FDA may require that we not initiate or halt our clinical trials and perform additional tests to assess the risk of infection to potential patients. This could result in additional costs to us and delay in the trials of our products under development.
The FDA has published guidelines for development of xenotransplantation products and is continuing to monitor closely the development of such products to determine if additional guidelines are required as more data is obtained. Failure to comply with FDA guidelines may result in fines and civil penalties, suspension of production, suspension or delay in product approval, product seizure or recall, or withdrawal of product approval.
18
If we are unable to protect effectively our intellectual property, third parties may use our technology, which could impair our ability to compete in our markets.
Our success will depend on our ability to obtain and protect patents on our technology and to protect our trade secrets. The patents we currently license and any future patents we may obtain or license, may not afford meaningful protection for our technology and products. Others may challenge our patents and, as a result, our patents could be narrowed, invalidated, or unenforceable. In addition our current and future patent applications may not result in the issuance of patents in the United States or foreign countries. Competitors might develop products similar to ours that do not infringe our patents. In order to protect or enforce our patent rights, we may initiate interference proceedings, oppositions, or patent litigation against third parties, such as infringement suits. These lawsuits could be expensive, take significant time, and divert management's attention from other business concerns. The patent position of biotechnology firms generally is highly uncertain, involves complex legal and factual questions, and has recently been the subject of much litigation. No consistent policy has emerged from the U.S. Patent and Trademark Office or the courts regarding the breadth of claims allowed or the degree of protection afforded under biotechnology patents. In addition there is a substantial backlog of biotechnology patent applications at the U.S. Patent and Trademark Office, and the approval or rejection of patent applications may take several years.
In addition to patent protection we require our employees, consultants, advisors, and collaborators to execute confidentiality agreements. However, these agreements may not provide us with adequate protection against improper use or disclosure of confidential information. In addition, in some situations, these agreements may conflict with, or be subject to, the rights of third parties with whom our employees, consultants or advisors have prior employment or consulting relationships. Further, others may gain access to our trade secrets or independently develop substantially equivalent proprietary information and techniques.
Our success will depend partly on our ability to operate without infringing on or misappropriating the proprietary rights of others. If we are sued successfully for infringement or misappropriation of another’s proprietary rights, our ability to generate revenue could be substantially reduced or eliminated.
Any of our anticipated products may infringe patent and other proprietary rights of third parties. In addition, our competitors, many of which have substantially greater resources than us and have made significant investments in competing technologies or products, may seek to apply for and obtain patents that will prevent, limit, or interfere with our ability to make, use, and sell products either in the U.S. or international markets. Intellectual property litigation is costly and even if we prevail, the cost of such litigation could adversely affect our business, financial condition, and results of operations. In addition, litigation is time consuming and could divert management attention and resources away from our business. If we do not prevail in any litigation, we could be required to stop the infringing activity and/or pay substantial damages. Under some circumstances in the United States, these damages could be triple the actual damages the patent holder incurs. If we have supplied infringing products to third parties for marketing or licensed third parties to manufacture, use, or market infringing products, we may be obligated to indemnify these third parties for any damages they may be required to pay to the patent holder and for any losses the third parties may sustain themselves as the result of lost sales or damages paid to the patent holder.
If a third party holding rights under a patent successfully asserts an infringement claim with respect to any of our products, we may be prevented from manufacturing or marketing our infringing product in the country or countries covered by the patent we infringe, unless we can obtain a license from the patent holder. Any required license may not be available to us on acceptable terms, or at all. Some licenses may be non-exclusive, and therefore, our competitors may have access to the same technology licensed to us. If we fail to obtain a required license or are unable to design around a patent, we may be unable to market some of our anticipated products, which would adversely affect our ability to generate and grow revenues.
Some jurisdictions may require us to grant licenses to third parties. Such compulsory licenses could be extended to include some of our product candidates, which may limit our potential revenue opportunities.
Many countries, including certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition most countries limit the enforceability of patents against government agencies or government contractors. In these countries the patent owner may be limited to monetary relief and may be unable to enjoin infringement, which could materially diminish the value of the patent. Compulsory licensing of life-saving products is also becoming increasingly popular in developing countries, either through direct legislation or international initiatives. Such compulsory licenses could be extended to include some of our product candidates, which may limit our potential revenue opportunities.
19
We have no commercial production capability and we may encounter production problems or delays, which could result in lower revenues.
To date, we have not produced any commercially available products. To produce product to anticipated customer demand levels we will need to develop our commercial production capability and maintain adequate levels of inventory. We may not be able to produce sufficient quantities to meet market demand. We may not be able to maintain acceptable quality standards if we ramp up production. If we cannot achieve the required level and quality of production, we may need to outsource production or rely on licensing and other arrangements with third parties. We may not be able to successfully outsource our production or enter into licensing or other arrangements under acceptable terms with these third parties, which could adversely affect our business. Our inability to identify potential manufacturers, or to enter into or maintain agreements with them on acceptable terms, could delay or prevent the commercialization of our products, which would adversely affect our ability to generate revenues and could prevent us from achieving or maintaining profitability. In addition reliance on third-party manufacturers could reduce our gross margins and expose us to the risks inherent in relying on others. We may also encounter problems with production yields, shortages of qualified personnel, production costs, and the development of advanced manufacturing techniques and process controls.
We will be required to comply with good manufacturing practice requirements, and our failure to do so may subject us to fines and other penalties that will delay or prevent us from marketing and selling our products.
We, our collaborators, or other third party manufacturers of our products must comply with current good manufacturing practice, or cGMP, requirements demanded by customers and enforced by the FDA through its facilities inspection program. These requirements include quality control, quality assurance, and the maintenance of records and documentation. We, our collaborators, or other third party manufacturers of our products may be unable to comply with these cGMP requirements and with other FDA, state, and foreign regulatory requirements. These requirements may change over time and we, or third party manufacturers, may be unable to comply with the revised requirements. A failure to comply with these requirements may result in criminal and civil penalties, including fines, suspension of production, suspension or delay in product approval, product seizure or recall, or withdrawal of product approval. If the safety of any quantities supplied by third-parties is compromised due to their failure to adhere to applicable laws or for other reasons, we may not be able to obtain regulatory approval for, or successfully commercialize, product candidates that we may develop.
We may incur substantial liabilities from any product liability claims, including claims made against third parties that we have agreed to indemnify. Our insurance coverage for those claims may be unavailable or inadequate.
We face an inherent risk of product liability exposure related to the testing of our product candidates in human clinical trials, and will face an even greater risk if we sell our product candidates commercially. An individual may bring a liability claim against us if one of our product candidates causes, or merely appears to have caused, an adverse effect or injury. These risks will exist even for products developed that may be cleared for commercial sale. If we cannot successfully defend ourselves against any product liability claims, we may incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in any one or a combination of the following:
- injury to our reputation;
- withdrawal of clinical trial participants;
- costs of related litigation;
- substantial monetary awards to patients or other claimants;
- decreased demand for our product candidates;
- loss of revenues; and
- the inability to commercialize our product candidates.
20
We intend to secure limited product liability insurance coverage, but we may not be able to obtain such insurance on acceptable terms with adequate coverage, or at reasonable or affordable costs. The amount of insurance coverage we obtain may not be adequate to protect us from all liabilities. We may not have sufficient resources to pay for any liabilities resulting from a claim beyond the limit of, or excluded from, our insurance coverage.
We use biological and hazardous materials in our business. If we are subject to claims relating to improper handling, storage, or disposal of these materials, our financial condition would suffer.
Our research and development processes involve the storage, use, and disposal of hazardous materials, including biological hazardous materials that could be dangerous to human health and safety or the environment. We are subject to federal, state, and local regulations governing the use, manufacture, storage, handling, and disposal of materials and waste products. Compliance with applicable environmental laws and regulations may be expensive, and current or future environmental laws and regulations may impair our product development efforts.
In the event of an accident we could be held liable for any damages that result and any liability could exceed the limits or fall outside the coverage of our insurance. We may not be able to maintain insurance on acceptable terms or at all. We could be required to incur significant costs to comply with current or future environmental laws and regulations.
If we experience significant fluctuations in our rate of anticipated growth and fail to balance our expenses with our revenue forecasts, our results could be harmed and the Company’s value may decline without advance notice.
Due to the unpredictability of new markets that we enter and deteriorating general economic and financial market conditions, we may not be able to accurately forecast our rate of growth. We plan our expense levels and investment on estimates of future revenue and future anticipated rate of growth. We may not be able to adjust our spending quickly enough if the rate of new or renewed orders falls short of our expectations. As a result, we expect that our revenues, operating results and cash flows may fluctuate significantly on a quarterly basis. We believe that period-to-period comparisons of our revenues, operating results and cash flows may not be meaningful and should not be relied upon as an indication of future performance.
Inability to manage growth
Although no assurance can be given, the Company contemplates that growth will occur as the Company implements its business strategies. The Company expects the expansion of its business to place a significant strain on its limited managerial, operational, and financial resources. The Company will be required to expand its operational and financial systems significantly and to expand, train, and manage its work force in order to manage the expansion of its operations. The Company’s failure to fully integrate new employees into its operations could have a material adverse effect on its business, prospects, financial condition, and results of operations. The Company’s ability to attract and retain highly skilled personnel in connection with its growth is critical to its operations and expansion. The Company faces competition for these types of personnel from other biotechnology companies and more established organizations, many of which have significantly larger operations and greater financial, marketing, human, and other resources than does the Company. The Company may not be successful in attracting and retaining qualified personnel on a timely basis, on competitive terms, or at all. If the Company is not successful in attracting and retaining these personnel, its business, prospects, financial condition, and results of operations will be materially adversely affected.
Failure to achieve and maintain effective internal controls in accordance with Section 404 of the Sarbanes-Oxley Act of 2002 could prevent the Company from producing reliable financial reports or identifying fraud. In addition, current and potential stockholders could lose confidence in the Company's financial reporting, which could have an adverse effect on the Company's stock price.
Effective internal controls are necessary for the Company to provide reliable financial reports and effectively prevent fraud, and a lack of effective controls could preclude the Company from accomplishing these critical functions. We are required to document and test our internal control procedures in order to satisfy the requirements of Section 404 of the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”), which requires annual management assessments of the effectiveness of the Company's internal controls over financial reporting.
21
If we fail to maintain the adequacy of our internal accounting controls, as such standards are modified, supplemented or amended from time to time, we may not be able to ensure that we can conclude on an ongoing basis that we have effective internal controls over financial reporting in accordance with Section 404. Failure to achieve and maintain an effective internal control environment could cause investors to lose confidence in our reported financial information, which could have an adverse effect on our stock price.
Our principal place of business is a shared executive suite at 8601 Six Forks Rd, Suite 400 Raleigh, NC 27615.
From time to time, we may become involved in various lawsuits and legal proceedings which arise in the ordinary course of business. However, litigation is subject to inherent uncertainties, and an adverse result in these or other matters may arise from time to time that may harm our business. We are currently not aware of any such legal proceedings or claims that we believe will have a material adverse effect on our business, financial condition or operating results.
In April 2012, Sand Dollar Partners, LLC, (“Sand Dollar”), a shareholder of the Company filed a complaint in the Superior Court of Arizona, Pima County against, among other parties, ISI, our wholly-owned subsidiary, John Steel, our former CEO and director, and Jonathan Lakey. In 2010, Sand Dollar invested $357,000 in ISI through the purchase of a convertible promissory note which was converted into 3,591,729 shares of the Company’s common stock. The plaintiff contends that it was entitled to issuance of additional shares and nomination of one board member.
In July 2012, a complaint was filed against the Company, and John Steel in the United States District Court for the District of Utah, Central Division for infringement and misappropriation of a patent. The plaintiffs contend that they were the actual purchasers of the MicroIslet patent out of MicroIslet’s bankruptcy proceedings in 2009 and that the respective intellectual property rights have been never assigned to either ISI or the Company. As a result, they allege that the Company’s claim to the ownership of the MicroIslet patent based on the assignment of the patent by its founders is baseless. The complaint sought monetary damages including punitive damages of at least $12 million, costs, attorneys' fees, and declaratory judgment. On January 8, 2013 the Court dismissed the plaintiff’s action for lack of recoverable damages. The plaintiffs refiled their claim and the Company has filed a motion with the Court for dismissal. We believe the plaintiffs’ claims to be without merit and will continue to vigorously defend against this action.
On October 25, 2013, the Company entered into a settlement agreement with Sand Dollar. At a hearing on February 21, 2014, the Company and Sand Dollar agreed to amend the settlement agreement whereby, Sand Dollar placed in escrow all of the Company’s common stock it held and retained a broker dealer to sell sufficient shares to receive $500,000 in cash and to pay fees to the broker dealer. Additionally, the Company agreed to issue 130,000 warrants that will vest over the next three years and make a $30,000 payment on May 16, 2014. The Company issued these warrants and made the $30,000 payment on May 16, 2014 fully completing the settlement and will receive back the remaining 1,344,529 shares of common stock. The broker dealer sold 2,247,200 shares of the Company’s escrowed stock to settle the obligation and the Company will receive back the remaining 1,344,529 shares of its common stock.
On April 25, 2014, Progenitor Cell Therapy, LLC (“PCT”), a subsidiary of NeoStem, Inc. (“NeoStem”), filed a lawsuit against the Company in the United States District Court for the District of New Jersey (Case No. 2:14-cv-02658-SDW-MCA). PCT’s complaint asserts various claims, including breach of contract and unjust enrichment, based on the alleged failure of the Company to pay for services and goods provided by PCT under a January 10, 2012 letter agreement. PCT seeks an unspecified amount of compensatory and other damages, plus interest and costs. The Company filed an answer to the complaint on June 24, 2014 denying all liability, including on the grounds that the January 10, 2012 letter agreement is unenforceable and PCT failed to provide the goods and services it stated it would provide. In connection with its answer to PCT’s complaint, the Company filed a counterclaim against PCT and a third-party complaint against NeoStem to seek, among other things, (i) a declaration that the January 10, 2012 letter agreement is unenforceable, (ii) monetary damages and (iii) rescission of equity securities the Company previously issued to NeoStem. The Company intends to vigorously defend the claims by PCT and prosecute its claims against PCT and NeoStem. As the lawsuit is at an early stage, the Company cannot at this time estimate the possible loss or range of loss, if any, that may result from this lawsuit.
22
Market for Our Common Stock
Our common stock, $0.001 par value, is quoted on the OTC Bulletin Board under the symbol “ISLT.” The following table shows the high and low closing prices for the periods indicated. The quotations provided below reflect inter-dealer prices without retail mark-up, markdown, or commissions, and may not represent actual transactions.
|
Year
|
High
|
Low
|
||||||
|
Fiscal 2014
|
||||||||
|
Quarter Ended April 30, 2014
|
$
|
0.50
|
$
|
0.29
|
||||
|
Quarter Ended January 31, 2014
|
0.98
|
0.18
|
||||||
|
Quarter Ended October 31, 2013
|
0.51
|
0.20
|
||||||
|
Quarter Ended July 31, 2013
|
0.51
|
0.18
|
||||||
|
Fiscal 2013
|
||||||||
|
Quarter Ended April 30, 2013
|
$
|
4.75
|
$
|
0.15
|
||||
|
Quarter Ended January 31, 2013
|
5.00
|
1.99
|
||||||
|
Quarter Ended October 31, 2012
|
3.10
|
3.00
|
||||||
|
Quarter Ended July 31, 2012
|
6.00
|
3.00
|
||||||
As of July 22, 2014, we had 323 shareholders of record. The holders of common stock are entitled to one vote for each share held of record on all matters submitted to a vote of stockholders. Holders of the common stock have no preemptive rights and no right to convert their common stock into any other securities. There are no redemption or sinking fund provisions applicable to the common stock.
Dividend
We have not paid dividends on our common stock and do not anticipate paying such dividends in the foreseeable future.
Securities authorized for issuance under equity compensation plans
As of the date of this Annual Report, we do not have any securities authorized for issuance under any equity compensation plans and we do not have any equity compensation plans.
SPECIAL NOTE OF CAUTION REGARDING FORWARD-LOOKING STATEMENTS
CERTAIN STATEMENTS IN THIS REPORT, INCLUDING STATEMENTS IN THE FOLLOWING DISCUSSION, ARE WHAT ARE KNOWN AS "FORWARD-LOOKING STATEMENTS", WHICH ARE BASICALLY STATEMENTS ABOUT THE FUTURE. FOR THAT REASON, THESE STATEMENTS INVOLVE RISK AND UNCERTAINTY SINCE NO ONE CAN ACCURATELY PREDICT THE FUTURE. WORDS SUCH AS "PLANS", "INTENDS", "WILL", "HOPES", "SEEKS", "ANTICIPATES", "EXPECTS "AND THE LIKE OFTEN IDENTIFY SUCH FORWARD-LOOKING STATEMENTS, BUT ARE NOT THE ONLY INDICATION THAT A STATEMENT IS A FORWARD-LOOKING STATEMENT. SUCH FORWARD-LOOKING STATEMENTS INCLUDE STATEMENTS CONCERNING OUR PLANS AND OBJECTIVES WITH RESPECT TO THE PRESENT AND FUTURE OPERATIONS OF THE COMPANY, AND STATEMENTS WHICH EXPRESS OR IMPLY THAT SUCH PRESENT AND FUTURE OPERATIONS WILL OR MAY PRODUCE REVENUES, INCOME OR PROFITS. NUMEROUS FACTORS AND FUTURE EVENTS COULD CAUSE THE COMPANY TO CHANGE SUCH PLANS AND OBJECTIVES OR FAIL TO SUCCESSFULLY IMPLEMENT SUCH PLANS OR ACHIEVE SUCH OBJECTIVES, OR CAUSE SUCH PRESENT AND FUTURE OPERATIONS TO FAIL TO PRODUCE REVENUES, INCOME OR PROFITS. THEREFORE, THE READER IS ADVISED THAT THE FOLLOWING DISCUSSION SHOULD BE CONSIDERED IN LIGHT OF THE DISCUSSION OF RISKS AND OTHER FACTORS CONTAINED IN THIS REPORT ON FORM 10-K AND IN THE COMPANY'S OTHER FILINGS WITH THE SECURITIES AND EXCHANGE COMMISSION. NO STATEMENTS CONTAINED IN THE FOLLOWING DISCUSSION SHOULD BE CONSTRUED AS A GUARANTEE OR ASSURANCE OF FUTURE PERFORMANCE OR FUTURE RESULTS.
23
Unless the context otherwise requires, the "Company", "we," "us," and "our," refer to (i) Islet Sciences, Inc., a Nevada corporation; (ii) Islet Sciences, Inc., a Delaware corporation (“ISI”), and (iii) DiaKine Therapeutics, Inc. (“DTI”).
Overview
We are a biotechnology company engaged in the research, development, and commercialization of new medicines and technologies for the treatment of metabolic diseases and related indications where there is a significant measurable unmet medical need. The rising incidence of obesity is associated with many obesity-related health complications, including cardiovascular disease, diabetes, hyperlipidemia, hypertension, nonalcoholic fatty liver disease/steatohepatitis (NAFLD/NASH). This constellation is also recognized as the metabolic syndrome and is characterized by underlying insulin resistance.
Metabolism is the ability of your body and its organs to process energy derived from the intake of nutrition. Metabolic diseases typically involve a derangement or malfunction of the normal processing of such metabolism. Some of the more prevalent metabolic diseases include obesity (excessive storage of nutrients); diabetes (loss of storage capability); NAFLD/NASH (excessive storage of lipids and fibrotic accumulation in the liver); dyslipidemia (inability to process fat from the blood); hypertension (excessive elevation of blood pressure). Since the primary risk factor for metabolic disease is often tied to obesity, the primary patient populations tend be concentrated in first-world nations where there is broad access to healthcare. However, due to the influx of “Western” diets and more sedentary lifestyles, more and more developing nations, such as China, India, and Brazil are seeing significant increases in the number of patients being diagnosed with metabolic disease.
We are developing the following therapeutic product candidates, which will include BHV’s SGLT2 inhibitor remogliflozin etabonate once the BHV merger is completed, to address the needs of patients suffering from metabolic disease:
|
●
|
Remogliflozin, a highly selective SGLT2 inhibitor in phase II clinical development for type 2 diabetes and NASH (BHV Pharma);
|
|
●
|
ISLT-P, an implantable suspension of encapsulated insulin-producing porcine islet cells, for the treatment of insulin-dependent diabetes;
|
|
●
|
ISLT-2669, a novel lead IL-12 small molecule inhibitor selected for preclinical development for treatment of type 2 diabetes. This IL12 inhibitor blocks the auto-immune and inflammatory cascade initiated by IL-12 receptor activation; and
|
|
●
|
ISLT-LSF Analogs, a library of lisofylline small molecule analogs that block inflammatory actions of cytokines that destroy insulin-producing beta cells, for diabetes and diabetes-related complications.
|
We are also developing the following diagnostic product candidate to better enable patients and their physicians to understand disease diagnosis and progression.
|
●
|
ISLT-Bdx, a PCR based molecular diagnostic measuring hypomethylated beta cell-derived DNA as a biomarker of beta cell loss for the early detection of type 1 diabetes or onset of insulin dependent type 2 diabetes;
|
We intend to develop these technologies in partnership with large pharmaceutical or biotechnology companies.
24
Going Concern
The financial statements included elsewhere in this current report on Form 10-K have been prepared assuming we will continue as a going concern. We incurred operating losses and negative operating cash flows through April 30, 2014, and as of that date our cash position was approximately $1.1 million and our current liabilities were approximately $3.8 million. We have incurred net losses of approximately $3.0 million and negative operating cash flows of approximately $1.0 million for the fiscal year ended April 30, 2014. Currently, management has projected that additional cash will be needed to allow us to continue our operations through April 30, 2015. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Our future cash requirements will depend on many factors, including continued scientific progress in our research and development programs, the scope and results of pre-clinical and clinical trials, the time and costs involved in obtaining regulatory approvals, the costs involved in filing, prosecuting and enforcing patents, competing technological and market developments, and the cost of product commercialization. We do not expect to generate a positive cash flow from operations at least until the commercial launch of our first product and possibly later given the expected spending for research and development programs and the cost of commercializing product candidates. During fiscal year ended April 30, 2014, we completed private placements of equity securities for aggregate gross proceeds of approximately $2.2 million. Although we have had success in raising this capital, there cannot be any assurance that this success will continue in future periods which is necessary to fund our future capital requirements, research and operations.
Results of Operations
Fiscal Years Ended April 30, 2014 and 2013
There were no revenues for the fiscal years ended April 30, 2014 and 2013.
During the fiscal year ended April 30, 2014, general and administrative expenses totaled approximately $2.2 million, compared to approximately $8.1 million for the fiscal year ended April 30, 2013. Beginning in October 2013, management implemented a strategy to significantly reduce expenses, raise additional interim capital and to add additional product candidates by signing a binding letter of intent to acquire BHV Pharma. As a result of this plan and other actions, the primary reason for the $5.9 million decrease in general and administrative expenses was due to a reduction of expense of $2.4 million associated with stock issuance in the settlement with a placement agent, a reduction of stock compensation of $2.2 million and a reduction in travel related expenses of $0.5 million.
During the fiscal year ended April 30, 2014, research and development expenses totaled approximately $0.6 million, compared to approximately $4.1 million for the fiscal year ended April 30, 2013. The primary reason for the $3.5 decrease in research and development expenses was the substantial completion or curtailment of various supply agreements, studies and subcontractors such as the $1.4 million decrease in the Progenitor Cell Therapy agreement to provide the protocols, procedures, systems, equipment, testing, quality controls, and manufacturing and distribution services to support the development and commercialization of the Company’s encapsulated porcine islet cells for the treatment of diabetes. There was also a $0.7 million decrease in the Spring Point Project, a source animal facility to purchase pigs for use in the Company’s xenotransplantation research. Additionally, the Company completed or cancelled various agreements with universities and subcontractors decreasing research and development expenses by an additional $0.9 million.
Liquidity and Capital Resources
We have historically financed our operations primarily through the issuance of common stock and debt and by relying on other financing. We have not generated revenues from sales of products and have had losses since inception. We anticipate that we will incur substantial additional operating losses in future years as we progress in our research and development programs. We do not expect to produce revenues from product sales for the foreseeable future so our revenues will be limited to research grants or licensing agreements we are able to obtain.
We will need significant additional funding to fully implement our strategy, either through equity or debt financings or partnering arrangements, or we will be forced to curtail or cease some or all of our operations. As of April 30, 2014, we had $1,141,380 cash on hand. The Company has no available or negotiated credit facilities with third party lenders.
25
Operating Activities
During the fiscal years ended April 30, 2014 and 2013, cash used in operating activities was approximately $1.0 million and approximately $3.9 million, respectively. The decrease in use of cash is primarily attributable to a decrease in our research and development efforts and overall general operating expenses which resulted in net losses of approximately $3.0 million offset by the accumulation of approximately $1.0 million of payables for delayed payments to our vendors due to our cash management, non-cash expenses from the issuance of common stock issued as compensation and for services of approximately $0.9 million.
Financing Activities
During the fiscal year ended April 30, 2014, cash provided by financing activities was approximately $2.1 million compared to approximately $2.0 million during the fiscal year ended April 30, 2013. Of the total net cash provided by financing activities, approximately $2.0 million was from the net proceeds from the private placement of common stock that has been issued at year end.
Critical Accounting Policies
Our significant accounting policies are disclosed in Note 2 to our consolidated financial statements. Certain of our policies require the application of management judgment in making estimates and assumptions which affect the amounts reported in the consolidated financial statements and disclosures made in the accompanying notes. Those estimates and assumptions are based on historical experience and various other factors deemed to be applicable and reasonable under the circumstances. The use of judgment in determining such estimates and assumptions is by nature, subject to a degree of uncertainty. Accordingly, actual results could differ from the estimates made.
Intangible Assets
Intangible assets represents a patent acquired from a third party, which is recorded at cost and amortized over the remaining life of the patent. This patent was fully impaired and written off to expense during the year ended April 30, 2014. Intangible assets also include the purchase of DiaKine Therapeutics, Inc. patent portfolio and know-how as in-process research and development. The intangible assets with estimable useful lives are amortized on a straight line basis over their respective estimated useful lives to their estimated residual values. This method of amortization approximates the expected future cash flow generated from their use. Definite lived intangibles are reviewed for impairment in accordance with FASB ASC 360, Property, Plant and Equipment.
Goodwill
Goodwill represents the excess of the purchase price over the estimated fair value of the identifiable assets acquired and liabilities assumed in business acquisitions. Goodwill is reviewed at least annually for impairment in the fourth quarter of the fiscal year, at the Company level, which is the sole reporting unit, and at any other time at which events occur or circumstances indicate that the carrying amount of goodwill may exceed its fair value. Such indicators would include a significant reduction in the Company’s market capitalization, a decrease in operating results or a deterioration in the Company’s financial position.
Research and Development
Research and development expenses consist of expenses incurred in performing research and development activities including salaries and benefits, facilities and other overhead expenses, contract services and other outside expenses. Research and development costs are charged to operations when incurred.
26
Stock Based Compensation
Stock awards
FASB ASC 718, Compensation-Stock Compensation (FASB ASC 718), requires the fair value of all share-based payments to employees, including grants of stock options, to be recognized in the statement of operations over the requisite service period. FASB ASC-718 requires all share based payments to employees, including grants of employee stock option, to be recognized in operating results as compensation expense based on fair value over the requisite service period of the awards. The Company determines the fair value of share-based awards using the Black-Scholes option-pricing model which uses both historical and current market data to estimate fair value. The method incorporates various assumptions such as the risk-free interest rate, expected volatility, expected dividend yield, expected forfeiture rate and expected life of the options. As allowed by FASB ASC 718, for companies with a short period of publicly traded stock history, the Company’s estimate of expected volatility is based on the average volatilities of a sampling of three companies with similar attributes to the Company, including industry, stage of life cycle, size and financial leverage. The risk-free rate for periods within the contractual life of the stock options is based on the U.S. Treasury yield curve in effect at the time of grant valuation. The expected term of options granted is derived using the “simplified method” which computes expected term as the average of the sum of the vesting term plus the contract term as historically the Company had limited activity surrounding its options. The Company has not issued stock options for nonemployees.
Warrants
Warrants granted to service providers are normally valued at the fair value of the instrument on the date of the grant (grant date) and are recognized in the statement of operations over the requisite service period or when they vest. When the requisite service period precedes the grant date and a market condition exists in the warrant, the Company values the warrant using the Black-Scholes option-pricing model. Warrants issued in connection with capital raises are normally valued at the fair value of the instrument on the date of the grant (grant date) and valued for disclosure purposes if they meet all the criteria under FASB ASC 718. The Company values these warrant using the Black-Scholes Method as well. As allowed by FASB ASC 718, for companies with a short period of publicly traded stock history, the Company’s estimate of expected volatility is based on the average volatilities of a sampling of companies with similar attributes to the Company, including industry, stage of life cycle, size and financial leverage. The risk-free rate for periods within the contractual life of the warrant is based on the U.S. Treasury yield curve in effect at the time of grant valuation.
Off-Balance Sheet Arrangements
We do not have off-balance sheet arrangements, financings, or other relationships with unconsolidated entities or other persons, also known as "special purpose entities" (SPEs).
The Company's consolidated audited financial statements for the fiscal years ended April 30, 2014 and 2013, together with the report of the independent certified public accounting firm thereon and the notes thereto, are presented beginning at page F-1.
None.
Disclosure Controls and Procedures
The Securities and Exchange Commission defines the term “disclosure controls and procedures” to mean controls and other procedures of an issuer that are designed to ensure that information required to be disclosed in the reports that it files or submits under the Securities Exchange Act of 1934 is recorded, processed, summarized and reported, within the time periods specified in the Securities and Exchange Commission’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by an issuer in the reports that it files or submits under the Securities Exchange Act of 1934 is accumulated and communicated to the issuer’s management, including its principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure. The Company maintains such a system of controls and procedures in an effort to ensure that all information which it is required to disclose in the reports it files under the Securities Exchange Act of 1934 is recorded, processed, summarized and reported within the time periods specified under the SEC's rules and forms and that information required to be disclosed is accumulated and communicated to principal executive and principal financial officers to allow timely decisions regarding disclosure.
27
As of the end of the period covered by this report, we carried out an evaluation, under the supervision and with the participation of our Chief Executive Officer (“CEO”) and Chief Financial Officer (“CFO”), of the effectiveness of the design and operation of our disclosure controls and procedures. Based on this evaluation, our CEO and CFO concluded that our disclosure controls and procedures were not effective as of the end of the period covered by this report.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act. Our internal control over financial reporting is designed to provide reasonable assurance regarding the (i) effectiveness and efficiency of operations, (ii) reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles, and (iii) compliance with applicable laws and regulations.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies and procedures may deteriorate.
Management assessed the effectiveness of our internal control over financial reporting as of the end of the period covered by this report. In making this assessment, we used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control - Integrated Framework - 1992. Based on our assessment, we determined that, as of the end of the period covered by this report, our internal control over financial reporting was not effective based on those criteria.
During our assessment of the effectiveness of internal control over financial reporting as of the end of the period covered by this report, management identified the following material weaknesses:
|
1.
|
Lack of Internal Audit Function – We lack qualified resources to perform the internal audit functions properly as well as oversight of recording and reporting of information. In addition, the scope and effectiveness of the internal audit function are yet to be developed.
|
|
2.
|
Review of Financial Information and Financial Reporting – We do not have adequate levels of review of financial information necessary to ascertain the accounting for complex transactions as well as review of financial information presented.
|
|
3.
|
Lack of Segregation of Duties – We do not have segregation of duties between recording, authorizing and testing.
|
Remediation Initiative
We are developing a plan to ensure that all information will be recorded, processed, summarized and reported accurately, and as of the date of this report, we have taken the following steps to address the above-referenced material weakness in our internal control over financial reporting:
|
1.
|
We will continue to educate our management personnel to increase its ability to comply with the disclosure requirements and financial reporting controls; and
|
|
2.
|
We will increase management oversight of accounting and reporting functions in the future; and
|
|
3.
|
As soon as we can raise sufficient capital or our operations generate sufficient cash flow, we will hire additional personnel to handle our accounting and reporting functions.
|
While the first two steps of our remediation process are ongoing, we do not expect to remediate the weaknesses in our internal controls over financial reporting until the time when we start to commercialize our products (and, therefore, may have sufficient cash flow for hiring sufficient personnel to handle our accounting and reporting functions).
28
A material weakness (within the meaning of PCAOB Auditing Standard No. 5) is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis. A significant deficiency is a deficiency, or a combination of deficiencies, in internal control over financial reporting that is less severe than a material weakness, yet important enough to merit attention by those responsible for oversight of the company's financial reporting.
This annual report does not include an attestation report of our registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by our registered public accounting firm because as a smaller reporting company and an emerging growth company we are not subject to Section 404(b) of the Sarbanes-Oxley Act of 2002.
Changes in Internal Controls over Financial Reporting
No change in our system of internal control over financial reporting occurred during the fourth quarter of the fiscal year ended April 30, 2014 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Mr. Joel Perlin, VP and Director for the Company loaned the Company $75,000 earning an interest rate of 6.5% during the fiscal year ending April 30, 2014. The Company has issued promissory notes to Mr. Perlin.
Mr. John Steel, Director and former CEO had agreed to advance to the Company up to $160,000 during any fiscal year. In May 2013, Mr. Steel increased his loan to the Company by $17,500 for a total of $25,880. The Company has issued promissory notes to the CEO in the aggregate principal amount of $25,880 of which $12,405 has been paid.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE.
The following table sets forth certain information as of July 28, 2014 concerning our directors and executive officers:
|
Name
|
Age
|
Position
|
||
|
James Green
|
45
|
Chief Executive Officer and Director
|
||
|
William Wilkison, Ph.D.
|
53
|
Chief Operating Officer and Director
|
||
|
Joel D. Perlin
|
68
|
Vice-President and Director
|
||
|
Steve Delmar
|
58
|
Chief Financial Officer
|
||
|
Dr. Eric Barnett
|
51
|
Director
|
||
|
Dr. Michael Luther, Ph.D.
|
58
|
Director
|
||
|
John Steel
|
55
|
Director
|
29
John Steel was the Chairman and Chief Executive Officer of the Company at April 30, 2013. Effective July 9, 2013, Mr. Steel resigned as an officer and Chairman of the Company. On July 15, 2013, Mr. Michael Earley was appointed as the Chairman and Chief Executive Officer of the Company. On July 30, 2013, Mr. Earley resigned from his positions at the Company. Dr. Jerry Nadler resigned from his position as Director of the Company on August 4, 2013. Dr. Jonathan Lakey resigned from his position as Director at the Company on April 12, 2014.
James Green, age 45, joined Islet Sciences as CEO in October 2013 and in January 2014 was appointed to the position of Chairman. Mr. Green joined Islet from BHV Pharma where he served as Chairman and CEO since 2009. BHV Pharma is a clinical stage pharmaceutical company focused on developing novel therapeutics for metabolic diseases. Prior to BHV, James spent several years at GlaxoSmithKline (“GSK”) in business development and finance roles. During that time he was responsible for license, acquisition, divestiture, and collaboration agreements related to preclinical and clinical development programs as well as equity investments in portfolio companies and special projects related to corporate growth. Prior to GSK, Mr. Green advised Boards and management teams on M&A, financings, and other strategic alternatives while at Citigroup, Bank of America, and Ernst & Young. Mr. Green was also a trustee for corporate, municipal, and securitized bond issues while at BNY Mellon. James earned an undergraduate business degree in business administration and marketing from University of North Florida and an MBA with a concentration in corporate finance from the Katz Graduate School of Business at The University of Pittsburgh.
William Wilkison, Ph.D., age 53, joined Islet Sciences as Chief Operating Officer in October 2013 and was appointed as the Corporate Secretary in January 2014 and was appointed as a Director of the Company in April 2014. Dr. Wilkison was previously at BHV Pharma where he was a founder, Director, and Chief Scientific Officer since its inception. BHV Pharma is a clinical stage pharmaceutical company focused on developing novel therapeutics for metabolic diseases. Prior to founding BHV in 2009, Dr. Wilkison spent several years at GSK in business development roles where he was responsible for managing key relationships between GSK and partner companies and participating on therapeutic project teams. Prior to GSK, Dr. Wilkison was a founder and Chief Operating Officer at Artecel Sciences, Inc. an adipose-derived stem cell discovery company. Artecel Sciences, Inc. was spun off from Zen-Bio, Inc. a biotech company founded by Dr. Wilkison, focused on the development and commercialization of human adipose cells. Prior to Zen-Bio, Dr. Wilkison spent six years at Glaxo/GlaxoWellcome studying metabolic disease and was responsible for target/lead identification and development. Dr. Wilkison has published numerous peer-reviewed manuscripts in the metabolic disease area and is an inventor on 17 approved patents. Dr. Wilkison holds a Ph.D. from Duke University Medical Center and did his postdoctoral work at Harvard Medical School.
Steve Delmar, age 58, joined Islet Sciences as Chief Financial Officer in 2014 and brings more than 35 years of financial and operational executive leadership experience to the company, serving most recently as Director of Business Operations at Computer Sciences Corporation (CSC). Prior to his role at CSC, Mr. Delmar served as Chief Financial Officer of Integrity Management Consulting, a provider of major systems acquisition and program management support services to government agencies. He also served as Senior Vice President and Chief Financial Officer of ACE*COMM Corporation, a publicly held telecommunications company where he successfully completed multiple acquisitions and facilitated the sale of the company to a private equity investor. Mr. Delmar spent 22 years at Microlog Corporation, a publicly held internet software and services telecommunications company, including 15 years as an executive officer, serving most recently as Chief Financial Officer. While at Microlog, Steve successfully led the initial public offering and secondary offerings as well as multiple acquisitions. Steve earned a Bachelor of Science in Accounting from Clemson University and is a member of the AICPA.
Joel D. Perlin, age 68, has been the President of H.S. Perlin Co., Inc. since 2002. After his graduation from San Diego State University in 1969, he began to pursue a career in the gold and rare coin industry. He has since become a renowned professional numismatist and a recognized expert in international gold trade and advisory. Mr. Perlin combines his coin expertise with a keen insight into factors affecting the precious metals trade, resulting in highly successful and lucrative gold investment market trading for his clients. For more than 40 years, he has been instrumental in developing personalized investment portfolios using both rare coins and U.S. gold coins for high net worth investors, corporate pension plans and financially private investors. Mr. Perlin receive his B.S. degree in marketing from San Diego State University in 1969. We believe that Mr. Perlin's expertise in financial matters based on his extensive experience in international gold trade and advisory positions him well as our director.
30
Dr. Eric Barnett, age 51, Executive Vice President at Piedmont Pharmaceuticals, is a doctor of medicine, and chartered accountant. At Piedmont Pharmaceuticals, Eric is responsible for Business Development and Marketing, to develop and execute the company’s long-term growth strategies including launching new brand initiatives. Dr. Barnett was previously Vice President at GSK and member of the clinical executive committee where he was responsible for financial capital management of the global research and development clinical portfolio. After an early career in medicine, Dr. Barnett joined a predecessor of PricewaterhouseCoopers (“PWC”) in London where he was a member of an advisory team instrumental in streamlining the United Kingdom’s National Health Service. After his work with PWC and prior to joining GSK, Eric was the CFO at one of London’s National Health Service hospital groups. Dr. Barnett earned his medical degree at the University of Bristol in Bristol, England. He also is a qualified chartered accountant (PwC alumnus) and a member of the Institute of Chartered Accountants of England and Wales.
Dr. Michael Luther, age 58, is Senior Vice President of Discovery and Development at Albany Molecular Research, Inc. (“AMRI”). At AMRI, Dr. Luther is responsible for the strategic, operational, and business development activities for AMRI’s global discovery and development divisions including scientific oversight as well as P/L responsibility. Prior to his current position at AMRI, Dr. Luther was Corporate Vice President of Global Discovery Research Services at Charles River Labs. Dr. Luther has also previously served as the founding President of the David H. Murdock Research Institute, Vice President and Site Head at Merck-Frosst, and Vice President of High Throughput Biology at GSK. Dr. Luther earned a Ph.D. in biophysical chemistry from Washington & St. Louis University School of Medicine and was a MDA Post-Doctoral Fellow at the Salk Institute. Dr. Luther also holds a Master of Business Administration from the Duke University Fuqua School of Business.
John Steel, age 55, was the President and CEO of Islet Sciences, Inc. from June 2010 until July 9, 2013. Mr. Steel brings over twenty years of senior management and investment experience in the healthcare services and biotechnology sector. In 1998, Mr. Steel founded MicroIslet, Inc., the firm that pioneered and developed the technology that comprises Islet Sciences, Inc., and served as its Chairman and Chief Executive Office from September 1998 to 2002. In 2002 MicroIslet, Inc. became public trough a merger with ALD Services, Inc. and Mr. Steel became Chairman and CEO of the public company, in which capacity he served until 2007. From January 1996 to December 1997, Mr. Steel was Chief Executive Officer of AKESIS Pharmaceuticals, Inc., a company that developed a patented treatment for insulin resistance for Type II diabetes. From 1996 to 2007 Mr. Steel served as a director of AKESIS Pharmaceuticals, Inc., which became a public company in December 2004 through a merger with Liberty Mint, Ltd. From January 1987 to June 1990, Mr. Steel served as the Vice President of Defined Benefit Inc., a company he founded in 1986 that provided financial services to health care professionals. After Defined Benefit Inc., Mr. Steel was an active investor and consultant within numerous areas including early-stage biotechnology and device companies through Steel Management. Mr. Steel himself is diagnosed with type 1 diabetes and is actively involved in fund raising for various diabetes research-related charities. Mr. Steel has recently chaired panels regarding the future of diabetes for the California Insurance Commissioner. Mr. Steel is also a noted speaker on the topic of diabetes - including its management, economics, and future opportunities for improvement in therapeutic modalities. Mr. Steel received his M.B.A. degree with an emphasis in finance from the University of Southern California and a Bachelor of Arts degree from Dartmouth College. We believe that Mr. Steel's qualifications and his extensive experience with biotechnology companies developing treatments for diabetes provide a unique perspective for our board.
Our directors hold their positions on the board until our next annual meeting of the shareholders, and until their successors have been qualified after being elected or appointed. Officers serve at the discretion of the board of directors.
There are no family relationships among our directors and executive officers. There is no arrangement or understanding between or among our executive officers and directors pursuant to which any director or officer was or is to be selected as a director or officer, and there is no arrangement, plan or understanding as to whether non-management shareholders will exercise their voting rights to continue to elect the current board of directors.
Our directors and executive officers have not, during the past ten years:
|
●
|
had any bankruptcy petition filed by or against any business of which was a general partner or executive officer, either at the time of the bankruptcy or within two years prior to that time,
|
|
●
|
been convicted in a criminal proceeding and is not subject to a pending criminal proceeding,
|
|
●
|
been subject to any order, judgment or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining, barring, suspending or otherwise limiting his involvement in any type of business, securities, futures, commodities or banking activities; or
|
|
●
|
been found by a court of competent jurisdiction (in a civil action), the Securities Exchange Commission or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended or vacate
|
31
Board Committees
Our board of directors has established an audit committee, a compensation committee and a nominating and corporate governance committee. Each committee will operate under a charter approved by our board of directors. The composition and function of each of these committees are described below.
Audit Committee
Our audit committee is comprised of Dr. Barnett and Mr. Perlin. Dr. Barnett is the chairperson of the committee. Our board of directors has determined that Dr. Barnett is an audit committee financial expert, as defined by the rules of the Securities and Exchange Commission, and satisfies the financial sophistication requirements of applicable NASDAQ rules.
Under the applicable NASDAQ rules, we are permitted to phase in our compliance with the independent audit committee requirements set forth in NASDAQ Marketplace Rule 5605(c)(2)(A)(ii) on the same schedule as we are permitted to phase in our compliance with the independent audit committee requirement pursuant to Rule 10A-3(b)(1)(iv)(A) under the Exchange Act, which require (1) one independent member at the time of listing; (2) a majority of independent members within 90 days of listing; and (3) all independent members within one year of listing.
Our audit committee is authorized to:
- approve and retain the independent auditors to conduct the annual audit of our financial statements;
- review the proposed scope and results of the audit;
- review and pre-approve audit and non-audit fees and services;
- review accounting and financial controls with the independent auditors and our financial and accounting staff;
- review and approve transactions between us and our directors, officers and affiliates;
- recognize and prevent prohibited non-audit services;
- establish procedures for complaints received by us regarding accounting matters;
- oversee internal audit functions, if any; and
- prepare the report of the audit committee that the rules of the Securities and Exchange Commission require to be included in our annual meeting proxy statement.
Compensation Committee
Our compensation committee is comprised of Dr. Luther, Dr. Barnett and Mr. Perlin. Dr. Luther is the Chair of the Compensation Committee. Our compensation committee is authorized to:
32
- review and recommend the compensation arrangements for management, including the compensation for our president and chief executive officer;
- establish and review general compensation policies with the objective to attract and retain superior talent, to reward individual performance and to achieve our financial goals;
- administer our stock incentive plans; and
- prepare the report of the compensation committee that the rules of the Securities and Exchange Commission require to be included in our annual meeting proxy statement.
Nominating and Governance Committee
Our nominating and governance committee is comprised of Dr. Wilkison, Dr. Barnett, and Mr. Perlin. Dr. Wilkison is the chairman of the committee. Our nominating and governance committee is authorized to:
- identify and nominate members of the board of directors;
- develop and recommend to the board of directors a set of corporate governance principles applicable to our company; and
- oversee the evaluation of our board of directors.
Compensation Committee Interlocks and Insider Participation
No member of our compensation committee has at any time been an employee of ours. None of our executive officers serves as a member of another entity’s board of directors or compensation committee that has one or more executive officers serving as a member of our board of directors or compensation committee.
Code of Business Conduct and Ethics
We have adopted a code of business conduct and ethics that will apply to all of our employees, officers and directors, including those officers responsible for financial reporting. The code of business conduct and ethics is attached as Exhibit 14.1 to this report and available on our website at www.isletsciences.com. Any amendments to the code, or any waivers of its requirements, will be disclosed on our website.
Section 16(a) Beneficial Reporting Compliance
Directors, executive officers and holders of more than 10% of our outstanding common stock are required to comply with Section 16(a) of the Securities Exchange Act of 1934, which requires generally that such persons file reports regarding ownership of transactions in securities of the Company on Forms 3, 4, and 5. Below is the information with respect to failures of directors, officers and/or beneficial owners of more than ten percent of any class of equity securities of the Company to timely file reports under Section 16(a):
|
Name
|
Form
|
Date of
Reporting Event
|
Required
Filing Date
|
Date of Filing
|
||||
|
James Green
|
3
|
10/30/2013(1)
|
11/9/2013
|
6/11/2014
|
||||
|
William Wilkison
|
3
|
10/30/2013(2)
|
11/9/2013
|
6/11/2014
|
||||
|
Eric Barnett
|
3
|
1/3/2014(3)
|
1/13/2014
|
3/25/2014
|
||||
|
Michael Luther
|
3
|
3/7/2014(3)
|
3/16/2014
|
Not filed
|
||||
|
Richard Schoninger
|
3
|
3/14/2014(4)
|
3/23/2014
|
3/27/2014
|
(1) Date of appointment as the CEO and a director of the Company.
(2) Date of appointment as the COO of the Company.
(3) Date of appointment as a director of the Company.
(4) Date of acquisition of 10% of common stock.
33
The following is a summary of the compensation we paid to our executive officers, for the two fiscal years ended April 30, 2014 and 2013.
Summary Compensation Table
|
Name and Position
|
Year
|
Salary ($) | Bonus ($) |
Stock Awards ($)
|
Option Awards ($)
|
All Other Compensation ($)
|
Total ($)
|
|||||||||||||||||||
|
James Green, CEO1
|
2014
|
12,500 | 62,500 | - | 205,815 | - | 280,815 | |||||||||||||||||||
|
2013
|
- | - | - | - | - | |||||||||||||||||||||
|
William Wilkison, COO1
|
2014
|
12,500 | 62,500 | - | 205,815 | - | 280,815 | |||||||||||||||||||
|
2013
|
- | - | - | - | - | |||||||||||||||||||||
|
Richard Egan, CFO2
|
2014
|
88,944 | - | - | - | - | 88,944 | |||||||||||||||||||
|
2013
|
122,990 | - | 280,000 | - | - | 402,990 | ||||||||||||||||||||
|
Dr. Jonathon Lakey, CSO3
|
2014
|
152,579 | - | 137,344 | - | - | 289,923 | |||||||||||||||||||
|
2013
|
238,605 | - | 350,000 | - | 187,481 | 776,086 | ||||||||||||||||||||
|
John Steel4
|
2014
|
180,000 | - | 92,813 | - | 10,000 | 282,813 | |||||||||||||||||||
|
2013
|
212,500 | - | 1,074,149 | - | 15,274 | 1,301,923 | ||||||||||||||||||||
|
(1)
|
James Green was appointed the CEO of the Company on October 30, 2013. Dr. William Wilkinson was appointed the COO of the Company on October 30, 2013.
|
|
(2)
|
Richard Egan resigned as CFO on May 22, 2014.
|
|
(3)
|
On March 24, 2014, the Company notified Dr. Jonathan Lakey that it was terminating the agreement between the parties dated June 26, 2012 related to Dr. Lakey’s position as Chief Scientific Officer and Chair of the Company’s Scientific Advisory Board. Dr. Lakey resigned from his position as a director on April 14, 2014
|
|
(4)
|
John Steel resigned his position as the Chairman and the CEO of the Company on July 9, 2013.
|
Compensation Discussion and Analysis
Overview
We intend to provide our named executive officers (as defined in Item 402 of Regulation S-K) with a competitive base salary that is in line with their roles and responsibilities when compared to peer companies of comparable size in similar locations.
Employment Agreements
James Green
On October 30, 2013, Mr. James Green accepted the Company’s appointment as the Chief Executive Officer and Director of the Company, and the Company entered into an employment agreement with Mr. Green, dated October 30, 2013. Mr. Green is compensated as follows:
|
1.
|
Initially, no annual salary until the Company’s financial condition improves to allow sufficient cash flow;
|
|
2.
|
Bonus of $12,500 per each month of service payable within 10 days following the completion by the Company of a financing with proceeds of at least $1,500,000;
|
|
3.
|
Options to purchase 1,500,000 shares of the Common Stock of the Company, exercisable at $0.265 per share, as follows: (i) 500,000 shares vesting on the 91st day from the option grant date, (ii) 1,000,000 shares vesting in equal installments of 200,000 on the last day of each 90 day period starting from the 91st day after the option grant date. If Mr. Green’s employment terminates on account of a termination by the company without cause, or a termination by executive for good reason or in conjunction with a change of control, then the Company agrees that all outstanding unvested stock options shall immediately be 100% vested upon the date that his employment terminates.
|
34
Dr. William Wilkison
On October 30, 2013, Dr. William Wilkison accepted the Company’s appointment as the Chief Operating Officer and as the Company’s board of directors Observer, and the Company entered into an employment agreement with Dr. Wilkison dated October 30, 2013. Dr. Wilkison is compensated as follows:
|
1.
|
Initially, no annual salary until the Company’s financial condition improves to allow sufficient cash flow;
|
|
2.
|
Bonus of $12,500 per each month of service payable within 10 days following the completion by the Company of a financing with proceeds of at least $1,500,000;
|
|
3.
|
Options to purchase 1,500,000 shares of the Common Stock of the Company, exercisable at $0.265 per share, as follows: (i) 500,000 shares vesting on the 91st day from the option grant date, (ii) 1,000,000 shares vesting in equal installments of 200,000 on the last day of each 90 day period starting from the 91st day after the option grant date. If Dr. Wilkison’s employment terminates on account of a termination by the company without cause, or a termination by executive for good reason, or in the event of a change of control, then the Company agrees that all outstanding unvested stock options shall immediately be 100% vested upon the date that his employment terminates.
|
On May 22, 2014, the Board of Directors approved the following changes to the compensation for Mr. Green and Mr. Wilkison: (i) combined annual salary budget of $300,000 payable monthly pro rata (up to $25,000 per month) to be divided between Messrs. Green and Wilkison at the discretion of the CEO; the salary budget will be increased to $500,000 upon raising of additional capital by the Company and approval by the Board of a respective corporate budget, and (ii) full health insurance benefits.
Richard Egan
On April 1, 2012, the Company and Richard Egan entered into a consulting agreement (the “CFO Agreement”) for his service as the Company’s Chief Financial Officer for a term of two years. Under the CFO Agreement, Mr. Egan was compensated on an hourly basis at the rate of $150 per hour, payable monthly in equal installments in arrears. He also received a stock award of 200,000 shares of common stock of which 150,000 shares of common stock were subject to forfeiture if Mr. Egan’s employment was terminated before January 31, 2013. Mr. Egan resigned as the Chief Financial Officer as of May 22, 2014.
Steven Delmar
On May 22, 2014, Mr. Delmar was appointed as the Company’s Treasurer and Chief Financial Officer. Mr. Delmar will receive an initial annual salary of $100,000, a grant of 300,000 stock options to purchase common shares of the Company, a conditional grant of 100,000 stock options upon the effective date of an S-4 filing and full health insurance benefits.
Outstanding Equity Awards at Fiscal Year End
The following table reflects the unexercised options, stock that has not vested and equity incentive plan awards for each named executive officer outstanding as of the end of the fiscal year ended April 30, 2014:
|
Option Awards
|
|||||||||||||||||
|
Name
|
Number of Securities Underlying Unexercised Options
(#)
Exerciseable
|
Number of Securities Underlying Unexercised Options
(#)
Unexerciseable
|
Equity Incentive Plan Awards: Number of Securities Underlying Unexercised Unearned Options
(#)
|
Option Exercise Price
($)
|
Option Expiration Date
|
||||||||||||
|
James Green
|
700,000 | (1) | 800,000 | — | $ | 0.265 |
3/31/2020
|
||||||||||
|
William Wilkison
|
700,000 | (1) | 800,000 | — | $ | 0.265 |
3/31/2020
|
||||||||||
|
(1)
|
James Green and Dr. William Wilkison were issued options to purchase 1,500,000 shares of the Company’s common stock at $0.265 per share as part of their employment agreement with the Company. As of April 30, 2014, both Mr. Green and Dr. Wilkison have vested 700,000 options each.
|
35
Additional Narrative Disclosure
We currently have no plans that provide for the payment of retirement benefits, or benefits that will be paid primarily following retirement, including, but not limited to, tax qualified defined benefit plans, supplemental executive retirement plans, tax qualified defined contribution plans and non-qualified defined contribution plans.
Director Compensation
The following table reflects the compensation of the directors (other than the named executive officers) including director fees, consulting fees and scientific advisory board fees for the Company’s fiscal year ended April 30, 2014:
|
Fees Earned or Paid in Cash
|
Stock Awards
|
Total
|
||||||||||
| Name of Director |
($)
|
($)(1)
|
($)
|
|||||||||
|
Joel D. Perlin(2)
|
- | 92,813 | 92,813 | |||||||||
|
John Steel(2)
|
190,000 | 92,813 | 285,906 | |||||||||
|
Jerry Nadler(2)
|
- | 24,750 | 24,750 | |||||||||
|
Jonathan Lakey(2)
|
152,579 | 137,344 | 289,923 | |||||||||
|
Dr. Eric Barnett
|
- | 22,400 | 22,400 | |||||||||
|
Dr. Michael Luther, Ph.D.
|
- | 20,000 | 20,000 | |||||||||
|
James Green
|
- | - | - | |||||||||
|
Dr. William Wilkison, Ph.D.
|
- | - | - | |||||||||
|
(1)
|
The amounts in these columns represent the compensation cost of stock awards granted during the fiscal year ended April 30, 2014, except that these amounts do not include any estimate of forfeitures. The amount recognized for these awards was calculated based on the value of the stock awards at the time of vesting.
|
|
(2)
|
As of April 30, 2014, Messrs. Perlin, Steel, Nadler, and Lakey each earned stock awards of 247,500, 247,500, 61,875 and 335,625 shares of common stock respectfully.
|
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth information regarding beneficial ownership of our common stock as of the date of this report by (i) any person or group with more than 5% of any class of voting securities, (ii) each director, (iii) our chief executive officer and each other executive officer whose cash compensation for the most recent fiscal year exceeded $100,000, and (iv) all such executive officers and directors as a group. Unless otherwise specified, the address of each of the officers and directors set forth below is in care of the Company, 8601 Six Forks Rd., Suite 400, Raleigh, North Carolina 27615. Except as indicated in the footnotes to this table and subject to applicable community property laws, the persons named in the table to our knowledge have sole voting and investment power with respect to all shares of securities shown as beneficially owned by them.
36
|
Name
|
Office
|
Shares Beneficially Owned (1)
|
Percent of
Class (2) |
|||||||
|
Officers and Directors
|
||||||||||
|
John Steel (7)
|
Director
|
7,233,127 | 10.7 | % | ||||||
|
Richard Egan(5)
|
CFO
|
250,000 | 0.4 | % | ||||||
|
James Green(6)
|
CEO and Director
|
900,000 | 1.3 | % | ||||||
|
William Wilkison, Ph.D.(6)
|
COO and Director
|
900,000 | 1.3 | % | ||||||
|
Steve Delmar(8)
|
CFO
|
- | - | |||||||
|
Joel D. Perlin(3)(4)
|
Vice-President and Director
|
5,515,000 | 8.1 | % | ||||||
|
Jonathan Lakey(4)(7)
|
Director
|
2,821,250 | 4.2 | % | ||||||
|
Eric Barnett
|
Director
|
70,000 | 0.1 | % | ||||||
|
Dr. Michael Luther
|
Director
|
50,000 | 0.1 | % | ||||||
|
All officers and directors as a
|
17,739,377 | 25.0 | % | |||||||
|
group (9 persons named above)
|
||||||||||
|
5% Securities Holders
|
||||||||||
|
Charles Dupont
|
5,980,685 | 8.9 | % | |||||||
|
13740 Nob Avenue
|
||||||||||
|
Belmar, California 92014
|
||||||||||
|
Andrew K. Boszhardt
|
5,066,666 | 7.4 | % | |||||||
|
C/O Great Oaks Capital - 660 Madison Ave.
|
||||||||||
|
New York, NY 10065
|
||||||||||
|
Richard Schoninger
|
7,200,000 | 10.4 | % | |||||||
|
31 West 11th St., Apt 4A
|
||||||||||
|
New York, NY 10011
|
||||||||||
| (1) | Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to securities. | |
| (2) | Based on 67,516,253 shares of the Company’s common stock outstanding as of April 30, 2014. | |
| (3) | Includes shares and warrants held by entities affiliated with or controlled by Mr. Perlin as follows: | |
| 2,640,000 shares held by H.S. Perlin Co., Inc., Defined Benefit Pension Plan | ||
| 2,001,875 shares of common stock and 120,000 warrants held by The Perlin Family Trust (DTD 12/27/95) | ||
| 61,875 shares held by Joel Perlin. | ||
| (4) | Includes a stock grant of 495,000 shares of common stock vesting over two years in equal quarterly installments of 61,875 shares at the end of each fiscal quarter starting with the first full fiscal quarter after June 26, 2012 of which a stock grant of 433,125 shares has vested as of the date of this report. | |
| (5) | Richard Egan resigned as Treasurer and Chief Financial Officer of the Company as of May 22, 2014. | |
| (6) | James Green and Dr. William Wilkison were issued options to purchase 1,500,000 shares of the Company’s common stock at $0.265 per share as part of their employment agreement with the Company. As of April 30, 2014, both Mr. Green and Dr. Wilkison have vested 700,000 options each. | |
| (7) | John Steel resigned as CEO of the Company on July 1, 2013. | |
| (8) | On March 24, 2014, the Company notified Dr. Jonathan Lakey that it was terminating the agreement between the parties dated June 26, 2012 related to Dr. Lakey’s position as Chief Scientific Officer and Chair of the Company’s Scientific Advisory Board. Dr. Lakey resigned from his position as a director on April 14, 2014 |
37
Change in Control
As of the date of this report, there were no arrangements which may result in a change in control of the Company.
Securities Authorized for Issuance under Equity Compensation Plan
None.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Transactions with related persons
Mr. John Steel, our Director at April 30, 2014, has a loan to ISI in an aggregate amount of $13,475 bearing no interest and payable on demand.
Mr. Joel Perlin, our Director at April 30, 2014, has a loan to ISI an aggregate amount of $62,543 at an interest rate of 6.5% and payable on demand.
James Green and William Wilkison, our current Chief Executive Officer and Chief Operating Officer, respectively, are the sole members of BHV.
Other than the above transactions or as otherwise set forth in this report or in any reports filed by the Company with the SEC, there have been no related party transactions, or any other transactions or relationships required to be disclosed pursuant to Item 404 of Regulation S-K. The Company is currently not a subsidiary of any company.
The Company’s Board conducts an appropriate review of and oversees all related party transactions on a continuing basis and reviews potential conflict of interest situations where appropriate. The Board has not adopted formal standards to apply when it reviews, approves or ratifies any related party transaction. However, the Board believes that the related party transactions are fair and reasonable to the Company and on terms comparable to those reasonably expected to be agreed to with independent third parties for the same goods and/or services at the time they are authorized by the Board.
Director Independence
Each of Eric Barnett and Michael Luther qualifies as an independent director pursuant to the definition of “independent director” under the Rules of NASDAQ, Marketplace Rule 5605(a)(2).
38
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The following lists fees billed by the auditors for the Company, for the years ended April 30, 2014 and 2013:
|
|
2014
|
2013
|
||||||
|
Audit Fees
|
$
|
71,600
|
$
|
45,750
|
||||
|
Audit Related Fees
|
-
|
-
|
||||||
|
Tax Fees
|
9,300
|
14,061
|
||||||
|
All Other Fees
|
-
|
8,859
|
||||||
|
●
|
Audit Fees. Represents fees for professional services provided for the audit of the Company’s annual financial statements and review of its quarterly financial statements, and for audit services provided in connection with other statutory or regulatory filings.
|
|
●
|
Audit-Related Fees. Represents fees for assurance and other services related to the audit of Company’s financial statements.
|
|
●
|
Tax Fees. Represents fees for professional services provided primarily for tax compliance and advice. These fees include the tax returns for Islet Sciences, Inc., One E-Commerce Corporation and DiaKine Therapeutics, Inc. for multiple periods.
|
|
●
|
All Other Fees. Represents fees for products and services not otherwise included in the categories above.
|
In the event that we should require substantial non-audit services, the audit committee would pre-approve such services and fees.
39
PART IV
|
Number
|
|
Description
|
|
2.1
|
Certificate of Merger (1)
|
|
|
2.2
|
Agreement and Plan of Merger (1)
|
|
|
3.1
|
Articles of Incorporation of the Company
|
|
|
3.2
|
Certificate of Amendment to Articles of Incorporation of the Company dated March 30, 1999
|
|
|
3.3
|
Certificate of Amendment to Articles of Incorporation of the Company dated February 16, 2012
|
|
|
3.4
|
Amended and Restated By-laws of the Company
|
|
|
4.1
|
Certificate of Designations of Preferences, Rights and Limitations of Series A Convertible Preferred Stock (1)
|
|
|
4.2
|
Certificate of Designations of Preferences, Rights and Limitations of Series B Convertible Preferred Stock (1)
|
|
|
4.3
|
Certificate of Designations of Preferences, Rights and Limitations of Series C Convertible Preferred Stock (2)
|
|
|
4.4
|
Form of Warrant (3)
|
|
|
4.5
|
Form of Warrant issued to NeoStem Inc.
|
|
|
4.6
|
Specimen of Common Stock Certificate
|
|
|
10.1
|
Stock Purchase Agreement by and between Islet Sciences, Inc. and John Welch (1)
|
|
|
10.2
|
Agreement dated January 10, 2012 by and between ISI and Progenitor Cell Therapy (4)
|
|
|
10.3
|
Share Exchange Agreement dated February 23, 2012 by and among the Company, ISI and DiaKine Therapeutics, Inc. (5)
|
|
|
10.4
|
Form of Lock-Up Agreement (5)
|
|
|
10.5
|
Employment Agreement dated March 1, 2012 by and between the Company and John Steel
|
|
|
10.6
|
Consulting Agreement dated April 1, 2012 by and between the Company and Richard Egan
|
|
|
10.7
|
Form of Subscription Agreement (3)
|
|
|
10.8
|
Long-Term Supply Agreement with Spring Point Project (6)
|
|
|
10.9
|
Separation Agreement and Release dated July 1, 2013 by and between the Company and John Steel (7)
|
|
|
10.10
|
Consulting Agreement dated July 1, 2013 by and between the Company and John Steel (7)
|
|
| 14.1 | Code of Ethics | |
| 21.1 | List of Subsidiaries | |
| 31.1 | Certifications of Joel Perlin pursuant to Exchange Act Rules 13a-14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
| 31.2 | Certifications of Richard Egan pursuant to Exchange Act Rules 13a-14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
| 32.1 | Certification pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |
| 101 | Interactive data files pursuant to Rule 405 of Regulation S-T |
Footnotes: - Need to be updated
|
(1)
|
Incorporated by reference to our Current Report on Form 8-K filed with the SEC on January 6, 2012.
|
|
(2)
|
Incorporated by reference to our Current Report on Form 8-K filed with the SEC on March 16, 2012.
|
|
(3)
|
Incorporated by reference to our Current Report on Form 8-K filed with the SEC on March 21, 2012.
|
|
(4)
|
Incorporated by reference to our Current Report on Form 8-K filed with the SEC on January 13, 2012.
|
|
(5)
|
Incorporated by reference to our Current Report on Form 8-K filed with the SEC on February 29, 2012.
|
|
(6)
|
Incorporated by reference to our Current Report on Form 8-K filed with the SEC on July 27, 2012.
|
|
(7)
|
Incorporated by reference to our Current Report on Form 8-K filed with the SEC on July 16, 2013.
|
40
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
Islet Sciences, Inc.
|
|||
|
Date: July 28, 2014
|
By:
|
/s/ James Green
|
|
|
James Green
|
|||
|
Chief Executive Officer
|
|||
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant in the capacities and on the dates indicated.
|
Name and Title
|
Date
|
||
|
/s/ James Green
|
July 28, 2014
|
||
|
James Green, CEO
|
|||
|
(Principal Executive Officer)
|
|||
|
/s/ Joel Perlin
|
July 28, 2014
|
||
|
Joel Perlin
|
|||
|
Vice-President and Director
|
|||
|
/s/ Steve Delmar
|
July 28, 2014
|
||
|
Steve Delmar
|
|||
|
Chief Financial Officer
|
|||
|
(Principal Financial Officer and Principal Accounting Officer)
|
|||
|
/s/ John Steel
|
July 28, 2014
|
||
|
John Steel, Director
|
|||
|
/s/ Dr. Eric Barnett
|
July 28, 2014
|
||
|
Dr. Eric Barnett, Director
|
|||
| /s/ Michael Luther, Ph.D. | July 28, 2014 | ||
| Michael Luther, Ph.D., Director | |||
| /s/ William Wilkison, Ph.D. | July 28, 2014 | ||
| William Wilkison Ph.D., Director |
41
Report of Independent Registered Public Accounting Firm
Board of Directors and Stockholders of
Islet Sciences, Inc. and Subsidiaries
We have audited the accompanying consolidated balance sheets of Islet Sciences, Inc. and Subsidiaries (A Development Stage Company) (the “Company”) as of April 30, 2014 and 2013 and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the two years then ended, and for the period from May 4, 2010 (Inception) through April 30, 2014. These consolidated financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We have conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal controls over financial reporting. Our audit includes consideration of internal controls over financial reporting as a basis for designing audit procedures that are appropriate in the circumstance, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Islet Sciences, Inc. and Subsidiaries (A Development Stage Company) as of April 30, 2014 and 2013, and the results of its operations and cash flows for each of the two years then ended, and for the period from May 4, 2010 (Inception) through April 30, 2014, in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has incurred losses since inception resulting in an accumulated deficit of approximately $20.4 million as of April 30, 2014 and further losses are anticipated in the development of its business that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
|
July 28, 2014
|
/s/ PKF
|
||
|
San Diego, California
|
PKF
|
||
|
Certified Public Accountants
|
|||
|
A Professional Corporation
|
F-1
Islet Sciences, Inc. and Subsidiaries
(A Development Stage Company)
Consolidated Balance Sheets
|
April 30,
|
April 30,
|
|||||||
|
2014
|
2013
|
|||||||
|
ASSETS
|
||||||||
|
CURRENT ASSETS
|
||||||||
|
Cash
|
$ | 1,141,380 | $ | 3,589 | ||||
|
Prepaid expenses
|
700 | 50,000 | ||||||
|
Advance to related party
|
- | 2,405 | ||||||
|
Total current assets
|
1,142,080 | 55,994 | ||||||
|
OTHER ASSETS
|
||||||||
|
Intangible assets, net (Note 3)
|
1,367,000 | 1,475,788 | ||||||
|
Goodwill (Note 3)
|
2,111,107 | 2,111,107 | ||||||
|
Total other assets
|
3,478,107 | 3,586,895 | ||||||
|
TOTAL ASSETS
|
$ | 4,620,187 | $ | 3,642,889 | ||||
|
LIABILITIES & STOCKHOLDERS' EQUITY
|
||||||||
|
CURRENT LIABILITIES
|
||||||||
|
Accounts payable
|
$ | 3,271,174 | $ | 2,233,314 | ||||
|
Subscribed shares - not issued
|
- | 96,600 | ||||||
|
Accrued stock compensation expenses (Note 4)
|
433,346 | 63,702 | ||||||
|
Notes payable - related parties
|
91,641 | 11,880 | ||||||
|
Derivative liability
|
- | 58,588 | ||||||
|
Total current liabilities
|
3,796,161 | 2,464,084 | ||||||
|
Deferred income taxes
|
547,000 | 547,000 | ||||||
|
Total liabilities
|
4,343,161 | 3,011,084 | ||||||
|
Commitments and Contingencies (Note 5)
|
||||||||
|
STOCKHOLDERS' EQUITY
|
||||||||
|
Preferred stock, $0.001 par value, 10,000,000 shares authorized; no shares
|
||||||||
|
issued and outstanding at April 30, 2014 and April 30, 2013
|
- | - | ||||||
|
Common stock, $0.001 par value, 100,000,000 shares authorized;
|
||||||||
|
67,516,253 and 56,715,117 shares issued and outstanding at April 30, 2014 and April 30, 2013, respectively
|
67,517 | 56,716 | ||||||
|
Additional paid-in capital
|
20,598,242 | 18,017,392 | ||||||
|
Deficit accumulated during the developments stage
|
(20,388,733 | ) | (17,442,303 | ) | ||||
|
Total stockholders' equity
|
277,026 | 631,805 | ||||||
|
TOTAL LIABILITIES & STOCKHOLDERS' EQUITY
|
$ | 4,620,187 | $ | 3,642,889 | ||||
See accompanying notes to the consolidated financial statements
F-2
Islet Sciences, Inc. and Subsidiaries
(A Development Stage Company)
Consolidated Statements of Operations
|
For the period from May 4, 2010 (Inception) through
|
||||||||||||
|
Year ended April 30,
|
April 30, | |||||||||||
|
2014
|
2013
|
2014 | ||||||||||
|
REVENUE
|
$ | - | $ | - | $ | - | ||||||
|
OPERATING EXPENSES
|
||||||||||||
|
General and administrative
|
2,195,198 | 8,058,684 | 12,562,136 | |||||||||
|
Research and development
|
646,250 | 4,063,790 | 6,795,160 | |||||||||
|
Impairment loss
|
93,586 | - | 93,586 | |||||||||
|
Total operating expenses
|
2,935,034 | 12,122,474 | 19,450,882 | |||||||||
|
LOSS FROM OPERATIONS
|
(2,935,034 | ) | (12,122,474 | ) | (19,450,882 | ) | ||||||
|
OTHER INCOME (EXPENSE)
|
||||||||||||
|
Other income
|
- | 430,000 | 430,000 | |||||||||
|
Other expenses
|
- | - | (1,345,710 | ) | ||||||||
|
Interest expense
|
(11,397 | ) | (10,125 | ) | (22,143 | ) | ||||||
|
Total other expense
|
(11,397 | ) | 419,875 | (937,853 | ) | |||||||
|
LOSS BEFORE INCOME TAXES
|
(2,946,430 | ) | (11,702,599 | ) | (20,388,734 | ) | ||||||
|
INCOME TAX EXPENSE
|
- | - | - | |||||||||
|
NET LOSS
|
$ | (2,946,430 | ) | $ | (11,702,599 | ) | $ | (20,388,734 | ) | |||
|
NET LOSS PER COMMON SHARE,
|
||||||||||||
|
BASIC AND DILUTED
|
$ | (0.05 | ) | $ | (0.21 | ) | ||||||
|
WEIGHTED AVERAGE NUMBER OF
|
||||||||||||
|
COMMON SHARES OUTSTANDING BASIC AND DILUTED
|
58,214,535 | 54,511,585 | ||||||||||
See accompanying notes to the consolidated financial statements
F-3
Islet Sciences, Inc. and Subsidiaries
(A Development Stage Company)
Consolidated Statements of Stockholders' Equity
|
Deficit
|
||||||||||||||||||||||||||||||||||||||||||||
|
Accumulated
|
||||||||||||||||||||||||||||||||||||||||||||
|
Series A Preferred
|
Series B Preferred
|
Series C Preferred
|
Common Stock
|
Additional
|
During the
|
Total
|
||||||||||||||||||||||||||||||||||||||
|
Number of
|
Number of
|
Number of
|
Number of
|
Paid-In
|
Development
|
Shareholders’
|
||||||||||||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Shares
|
Amount
|
Shares
|
Amount
|
Shares
|
Amount
|
Capital
|
Stage
|
Equity/(Deficit)
|
||||||||||||||||||||||||||||||||||
|
Balance, May 4, 2010
|
- | $ | - | - | $ | - | - | $ | - | - | $ | - | $ | - | $ | - | $ | - | ||||||||||||||||||||||||||
|
Issuance of founders' common stock
|
- | - | - | - | - | - | 13,430,000 | 13,430 | (13,430 | ) | - | - | ||||||||||||||||||||||||||||||||
|
Issuance of common stock in exchange for intangible asset
|
- | - | - | - | - | - | 3,000,000 | 3,000 | 197,000 | - | 200,000 | |||||||||||||||||||||||||||||||||
|
Issuance of common stock to reduce accounts payable – related party
|
- | - | - | - | - | - | 100,000 | 100 | 9,900 | - | 10,000 | |||||||||||||||||||||||||||||||||
|
Conversion of debt and accrued interest into common stock
|
- | - | - | - | - | - | 3,591,729 | 3,592 | 353,408 | - | 357,000 | |||||||||||||||||||||||||||||||||
|
Stock-based compensation
|
- | - | - | - | - | - | 7,031 | - | 7,031 | |||||||||||||||||||||||||||||||||||
|
Issuance of common stock for services
|
- | - | - | - | - | - | 1,200,000 | 1,200 | 118,800 | - | 120,000 | |||||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | - | (701,566 | ) | (701,566 | ) | |||||||||||||||||||||||||||||||
|
Balance, April 30, 2011
|
- | - | - | - | - | - | 21,321,729 | 21,322 | 672,709 | (701,566 | ) | (7,535 | ) | |||||||||||||||||||||||||||||||
|
Sale of common stock at $0.10/share
|
- | - | - | - | - | - | 8,090,000 | 8,090 | 780,910 | - | 789,000 | |||||||||||||||||||||||||||||||||
|
Issuance of common stock for services
|
- | - | - | - | - | - | 4,931,668 | 4,932 | 488,236 | - | 493,168 | |||||||||||||||||||||||||||||||||
|
Sale of common stock at $0.12/share
|
- | - | - | - | - | - | 2,474,997 | 2,475 | 287,525 | - | 290,000 | |||||||||||||||||||||||||||||||||
|
Issuance of common stock for purchase consideration in reverse merger
|
- | - | - | - | - | - | 1,187,476 | 1,187 | 533,178 | - | 534,365 | |||||||||||||||||||||||||||||||||
|
Conversion of shares during merger
|
- | - | 38,006 | 38 | - | - | (38,005,870 | ) | (38,006 | ) | 37,968 | - | - | |||||||||||||||||||||||||||||||
|
Assumption of One E Commerce Shares upon reverse merger
|
- | - | - | - | - | - | 187,063 | 187 | (187 | ) | - | - | ||||||||||||||||||||||||||||||||
|
Sale of preferred A shares at $450/share
|
1,173 | 1 | - | - | - | - | 464,638 | - | 464,639 | |||||||||||||||||||||||||||||||||||
|
Purchase of Diakine with shares
|
- | - | - | - | 200,000 | 200 | 100,000 | 100 | 2,829,523 | - | 2,829,823 | |||||||||||||||||||||||||||||||||
|
Conversion of shares upon completion of merger with One E Commerce
|
(1,173 | ) | (1 | ) | (38,006 | ) | (38 | ) | - | - | 39,178,870 | 39,179 | (39,140 | ) | - | - | ||||||||||||||||||||||||||||
|
Automatic conversion of shares
|
- | - | - | - | (200,000 | ) | (200 | ) | 2,000,000 | 2,000 | (1,800 | ) | - | - | ||||||||||||||||||||||||||||||
|
Sale of common stock at $0.45/share
|
- | - | - | - | - | - | 3,118,922 | 3,119 | 1,346,876 | - | 1,349,995 | |||||||||||||||||||||||||||||||||
|
Stock-based compensation
|
- | - | - | - | - | - | - | - | 4,761 | - | 4,761 | |||||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | (5,038,138 | ) | (5,038,138 | ) | ||||||||||||||||||||||||||||||||
|
Balance, April 30, 2012
|
- | - | - | - | - | - | 44,584,855 | 44,585 | 7,405,197 | (5,739,704 | ) | 1,710,078 | ||||||||||||||||||||||||||||||||
|
Sale of common stock at $0.45 a share
|
- | - | - | - | - | - | 7,041,667 | 7,042 | 3,008,405 | - | 3,015,447 | |||||||||||||||||||||||||||||||||
|
Issuance of common shares for services
|
- | - | - | - | - | - | 4,563,855 | 4,564 | 6,685,689 | - | 6,690,253 | |||||||||||||||||||||||||||||||||
|
Issuance of common stock for purchase consideration in reverse merger and settlement of litigation
|
- | - | - | - | - | - | 524,740 | 525 | 734,112 | - | 734,637 | |||||||||||||||||||||||||||||||||
|
Issuance of warrants for services
|
- | - | - | - | - | - | - | - | 183,989 | - | 183,989 | |||||||||||||||||||||||||||||||||
|
Net loss
|
- | - | - | - | - | - | - | - | - | (11,702,599 | ) | (11,702,599 | ) | |||||||||||||||||||||||||||||||
|
Balance, April 30, 2013
|
- | - | - | - | - | - | 56,715,117 | 56,716 | 18,017,392 | (17,442,303 | ) | 631,805 | ||||||||||||||||||||||||||||||||
|
Sale of common stock at $0.41 a share
|
- | - | - | - | - | - | 233,333 | 233 | 96,367 | - | 96,600 | |||||||||||||||||||||||||||||||||
|
Stock-based compensation
|
- | - | - | - | - | - | - | - | 192,100 | - | 192,100 | |||||||||||||||||||||||||||||||||
|
Sale of common stock at $0.25 a share
|
- | - | - | - | - | - | 9,210,000 | 9,210 | 2,021,090 | - | 2,030,300 | |||||||||||||||||||||||||||||||||
|
Issuance of common shares for services
|
- | - | - | - | - | - | 1,357,803 | 1,358 | 251,345 | - | 252,703 | |||||||||||||||||||||||||||||||||
|
Issuance of warrants for settlement of litigation
|
- | - | - | - | - | - | - | - | 19,948 | - | 19,948 | |||||||||||||||||||||||||||||||||
|
Net Loss
|
- | - | - | - | - | - | - | - | - | (2,946,430 | ) | (2,946,430 | ) | |||||||||||||||||||||||||||||||
|
Total Balance, April 30, 2014
|
- | $ | - | - | $ | - | - | $ | - | 67,516,253 | $ | 67,517 | $ | 20,598,242 | $ | (20,388,733 | ) | $ | 277,026 | |||||||||||||||||||||||||
See accompanying notes to the consolidated financial statements
F-4
Islet Sciences, Inc. and Subsidiaries
(A Development Stage Company)
Consolidated Statements of Cash Flows
|
For the period
|
||||||||||||
|
from May 4, 2010
|
||||||||||||
|
(Inception)
|
||||||||||||
|
Year ended April 30,
|
through April 30,
|
|||||||||||
|
2014
|
2013
|
2014
|
||||||||||
|
Cash flows from operating activities:
|
||||||||||||
|
Net loss
|
$ | (2,946,430 | ) | $ | (11,702,599 | ) | $ | (20,388,734 | ) | |||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
||||||||||||
|
Equity issued for acquisition of One E-Commerce Corporation
|
- | - | 534,365 | |||||||||
|
Equity issued for payment of accounts payable - related party
|
- | - | 10,000 | |||||||||
|
Stock based compensation for services and other
|
459,393 | 5,635,053 | 6,622,805 | |||||||||
|
Derivative liabilities
|
(58,588 | ) | 72,174 | 838,461 | ||||||||
|
Accrued stock compensation expenses
|
373,644 | 63,702 | 1,572,711 | |||||||||
|
Amortization of intangible asset
|
15,202 | 30,404 | 106,414 | |||||||||
|
Impairment loss
|
93,586 | - | 93,586 | |||||||||
|
Change in operating assets and liabilities:
|
||||||||||||
|
Advances to related party
|
2,405 | (2,405 | ) | - | ||||||||
|
Prepaid expense
|
50,000 | (50,000 | ) | - | ||||||||
|
Accounts payable
|
1,037,860 | 2,064,736 | 3,191,622 | |||||||||
| Accounts payable - related party | - | (14,228 | ) | (20,000 | ) | |||||||
|
Net cash used in operating activities
|
(972,928 | ) | (3,903,163 | ) | (7,438,770 | ) | ||||||
|
Cash flows from investing activities:
|
||||||||||||
|
Net cash provided by investing activities
|
- | - | - | |||||||||
|
Cash flows from financing activities:
|
||||||||||||
|
Proceeds from issuance of stock
|
1,950,958 | 1,891,182 | 6,548,375 | |||||||||
|
Subscribed shares - not issued
|
80,000 | 96,600 | 1,584,865 | |||||||||
|
Proceeds from notes payable - related parties
|
107,165 | 13,500 | 507,666 | |||||||||
|
Payments on notes payable - related parties
|
(27,404 | ) | (3,062 | ) | (60,756 | ) | ||||||
|
Net cash provided by financing activities
|
2,110,719 | 1,998,220 | 8,580,150 | |||||||||
|
Net increase (decrease) in cash
|
1,137,791 | (1,904,943 | ) | 1,141,380 | ||||||||
|
Cash at beginning period
|
3,589 | 1,908,532 | - | |||||||||
|
Cash at end period
|
$ | 1,141,380 | $ | 3,589 | $ | 1,141,380 | ||||||
|
SUPPLEMENTAL DISCLOSURES OF CASH FLOW INFORMATION:
|
||||||||||||
|
Cash paid during the period for:
|
||||||||||||
|
Interest
|
$ | - | $ | - | $ | - | ||||||
|
Income taxes
|
$ | - | $ | - | $ | - | ||||||
|
SUPPLEMENTAL DISCLOSURE OF NON-CASH INVESTING AND FINANCING INFORMATION:
|
||||||||||||
|
Shares issued for settlement of accrued expenses
|
$ | 4,000 | $ | 1,135,365 | $ | 1,139,365 | ||||||
|
Shares issued for settlement of derivative liabilities
|
$ | - | $ | 838,461 | $ | 838,461 | ||||||
|
Common stock issued for subscribed shares liability
|
$ | 177,300 | $ | 1,124,265 | $ | 1,504,865 | ||||||
|
Shares isssued to escrow as security for legal expenses
|
$ | 700 | $ | - | $ | 700 | ||||||
|
Shares issued for acquisition of Diakine Therapeutics, Inc.
|
$ | - | $ | - | $ | 2,829,823 | ||||||
|
Net liabilities assumed in acquisition of Diakine Theapeutics, Inc.
|
$ | - | $ | - | $ | 101,284 | ||||||
|
Deferred income tax liability and goodwill associated with the acquisition of Diakine Therapeutics, Inc.
|
$ | - | $ | - | $ | 547,000 | ||||||
|
Common stock issued in exchange for convertible notes
|
$ | - | $ | - | $ | 357,000 | ||||||
|
Common stock issued in exchange for intangible asset
|
$ | - | $ | - | $ | 200,000 | ||||||
|
Common stock issued in exchange for accounts payable - related party
|
$ | - | $ | - | $ | 10,000 | ||||||
See accompanying notes to the consolidated financial statements
F-5
ISLET SCIENCES, INC. AND SUBSIDIARIES
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
NOTE 1. DESCRIPTION OF BUSINESS AND GOING CONCERN
Description of Business
We are a biotechnology company engaged in the research, development, and commercialization of new medicines and technologies for the treatment of metabolic disease and related indications where there is significant measurable unmet medical need. The rising incidence of obesity is associated with many obesity-related health complications, including cardiovascular disease, diabetes, hyperlipidemia, hypertension, nonalcoholic fatty liver disease/steatohepatitis (NAFLD/NASH). This constellation is also recognized as the metabolic syndrome and is characterized by underlying insulin resistance. These various diseases have interrelated risk factors and markers, such that often treatment of one disease may allow new therapies and opportunities for treatment in one of these related indications. Our focused effort to develop new therapies and related diagnostics for metabolic related diseases establishes us as a recognized leader in a large and growing market.
In March 2014, the Company announced it had signed a binding letter of intent to enter into a merger agreement with and acquire Brighthaven Ventures, LLC d/b/a BHV Pharma (“BHV”) a privately held pharmaceutical company developing the SGLT2 inhibitor remogliflozin etabonate (“remogliflozin”) for type 2 diabetes and non-alcoholic steatohepatitis (“NASH”). Remogliflozin is currently in phase II clinical development. The Company anticipates it will sign the merger agreement within the next thirty days.
Islet Sciences was incorporated under the name One E-Commerce Corporation on September 14, 1994 in the State of Nevada. Effective February 23, 2012, the Company changed its name to Islet Sciences, Inc. On March 14, 2012, Islet Sciences acquired DiaKine Therapeutics, Inc., a Delaware corporation (“DTI”). Islet Sciences together with its subsidiaries, Islet Sciences Inc., a Delaware corporation (“ISI”), and DTI are referred to as the Company.
Going Concern
The consolidated financial statements have been prepared assuming the Company will continue as a going concern. This contemplates the continuity of operations, realization of assets, and liquidation of liabilities in the normal course of business. Since inception, the Company has incurred operating losses of $20,388,734 and has had negative operating cash flows of $7,438,771. As of April 30, 2014, the Company had cash of $1,141,380. Further, the Company has incurred net losses of $2,946,430 and negative operating cash flows of $972,928 for the fiscal year ended April 30, 2014. The consolidated financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might be necessary should the Company be unable to continue as a going concern.
The Company’s future cash requirements will depend on many factors, including continued scientific progress in its research and development programs, the scope and results of pre-clinical and clinical trials, the time and costs involved in obtaining regulatory approvals, the costs involved in filing, prosecuting and enforcing patents, competing technological and market developments, and the cost of product commercialization. The Company does not expect to generate a positive cash flow from operations at least until the commercial launch of its first product and possibly later given the expected spending for research and development programs and the cost of commercializing product candidates. The Company’s continued operations will depend on its ability to raise funds through various potential sources such as debt and equity financing. There can be no assurance that such capital will be available on favorable terms or at all. If the Company is unable to raise additional capital, the Company will likely be forced to curtail its desired development activities, which would delay the development of its product candidates.
F-6
Reverse Merger Agreement
On September 15, 2011, Mr. John Welch, shareholder, director and Chief Executive Officer of One-E Commerce Corporation, entered into a stock purchase agreement, pursuant to which Mr. Welch sold to ISI, an aggregate of 9,902,180 shares of One E-Commerce Corporation common stock, which shares then represented approximately 54.06% of the issued and outstanding shares of common stock, and certain convertible promissory notes in the aggregate principal amount of $514,458 and accrued interest, previously issued by One E-Commerce Corporation, for an aggregate purchase price of $250,000. Additionally, under the stock purchase agreement, ISI agreed to cause One E-Commerce Corporation to enter into a reverse merger transaction at a future date whereby the Company was to acquire all of the outstanding equity interests of ISI in consideration for the issuance of its shares to the shareholders of ISI (“Reverse Merger Transaction”). The closing that took place on September 22, 2011, resulted in the change of control of One E-Commerce Corporation. Immediately after the closing, the shares together with the notes, acquired by ISI comprised 54.06% of the then issued and outstanding common stock of the One-E Commence Corporation on a non-diluted basis and 82.8% on a fully-diluted basis.
On December 30, 2011, ONCE, Inc., a Delaware corporation wholly-owned by the Company (the “Merger Sub”), ISI and Islet Sciences consummated the Reverse Merger Transaction, whereby the Merger Sub was merged with and into ISI, and the holders of common stock of ISI received an aggregate of 38,005.87 shares of Islet Sciences’ Series B preferred stock, $0.001 par value per share (“Series B Preferred”) in exchange for the cancellation of all of the shares of common stock of ISI formerly owned by them, and the holders of Series A preferred stock of ISI received an aggregate of 1,173 shares of the Series A preferred stock, $0.001 par value per share (“Series A Preferred”) in exchange for the cancellation of all of the shares of Series A preferred stock of ISI formerly owned by them. The issuance of Series A and B Preferred, with each share of preferred stock having voting rights equal to 1,000 shares of the Company’s common stock, resulted in ISI’s shareholders having obtained control of the combined Company. ISI is deemed to be the accounting acquirer (legal acquiree) and One E-Commerce Corporation to be the accounting acquiree (legal acquirer). The financial statements before the date of the Reverse Merger Transaction are those of ISI with the results of the Company being consolidated from the date of the Reverse Merger Transaction. The equity section and earnings per share have been retroactively restated to reflect the reverse acquisition. The merger of a private operating company into a non-operating public shell corporation with nominal net assets is considered to be a capital transaction, in substance, rather than a business combination, for accounting purposes. Accordingly, ISI treated this transaction as a capital transaction without recording goodwill or adjusting any of its other assets or liabilities. The consideration in the amount of $1,345,710 for the Company, consisting of $250,000 paid in cash and $1,095,710 paid in the form of common stock, was recorded as another expense item in the accompanying consolidated statements of operations during the year ended April 30, 2012. Effective February 23, 2012, Islet Sciences completed a 1-for-45 reverse stock split of its issued and outstanding common stock. Upon effectiveness of the reverse stock split, all outstanding shares of Series A Preferred and Series B Preferred were converted into common shares based on their respective conversion ratios. In connection with the closing of the Reverse Merger Transaction, ISI agreed to cancel the shares and the outstanding notes of One E-Commerce Corporation purchased from Mr. Welch, and the interest accrued thereon effective upon the effectiveness of the reverse stock split.
NOTE 2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
The accompanying consolidated financial statements are prepared in accordance with accounting principles generally accepted in the United States of America (“GAAP”). All shares and per share amounts in these consolidated financial statements and notes thereto have been retroactively adjusted to give effect to the reverse stock split.
The accompanying consolidated financial statements include the accounts of Islet Sciences and its wholly-owned subsidiaries, ISI and DTI. All significant intercompany balances have been eliminated.
The Company's planned principal operations have not yet commenced. Accordingly, the Company's activities have been accounted for as those of a development stage enterprise in accordance with Financial Accounting Standards Board ("FASB") Accounting Standards Codification (“ASC”) 915-10, Accounting and Reporting by Development Stage Enterprises (FASB ASC 915-10). All losses since inception have been considered as part of the Company's development stage activities.
F-7
Emerging Growth Company
The Company is an “emerging growth company” and has elected to use the extended transition period for complying with new or revised accounting standards under Section 102(b)(1) of the JOBS Act. This election allows us to delay the adoption of new or revised accounting standards that have different effective dates for public and private companies until those standards apply to private companies.
Use of Estimates
The preparation of consolidated financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Significant estimates include the recoverability of long-lived assets, the valuation of intangible assets and goodwill, the valuation of common stock, warrants and stock options and the valuation of deferred tax assets. Actual results could differ from those estimates.
Concentration of Credit Risk
The Company maintains its cash balances at a credit-worthy financial institution and management believes the risk of loss of cash balances to be low. The Company’s cash balances were not fully insured at April 30, 2014.
Intangible Assets
Intangible assets represent a patent acquired from a third party, which is recorded at cost and amortized over the remaining life of the patent. This patent was fully impaired and written off during the year ended April 30, 2014. Intangible assets also include the purchase of DiaKine Therapeutics, Inc. patent portfolio and know-how as in-process research and development (“IPR&D”). IPR&D has an indefinite life and is not amortized until completion and development of the project, at which time the IPR&D becomes an amortizable asset. If the related project is not completed in a timely manner or the project is terminated or abandoned, the Company may have an impairment related to the IPR&D, calculated as the excess of the asset’s carrying value over its fair value. The intangible assets with estimable useful lives are amortized on a straight line basis over their respective estimated useful lives to their estimated residual values. This method of amortization approximates the expected future cash flow generated from their use. Definite lived intangibles are reviewed for impairment in accordance with FASB ASC 360, Property, Plant and Equipment (FASB ASC 360).
Goodwill
Goodwill represents the excess of the purchase price over the estimated fair value of the identifiable assets acquired and liabilities assumed in business acquisitions. Goodwill is reviewed at least annually for impairment in the fourth quarter of the fiscal year, at the Company level, which is the sole reporting unit, and at any other time at which events occur or circumstances indicate that the carrying amount of goodwill may exceed its fair value. Such indicators would include a significant reduction in the Company’s market capitalization, a decrease in operating results or a deterioration in the Company’s consolidated financial position.
Impairment of Long-Lived Assets
The Company applies the provisions of FASB ASC 360-10, Property, Plant and Equipment (FASB ASC 360-10), where applicable to all long lived assets. FASB ASC 360-10 addresses accounting and reporting for impairment and disposal of long-lived assets. The Company periodically evaluates the carrying value of long-lived assets to be held and used in accordance with FASB ASC 360-10. FASB ASC 360-10 requires impairment losses to be recorded on long-lived assets used in operations when indicators of impairment are present and the undiscounted cash flows estimated to be generated by those assets are less than the assets’ carrying amounts. In that event, a loss is recognized based on the amount by which the carrying amount exceeds the fair market value of the long-lived assets. Loss on long-lived assets to be disposed of is determined in a similar manner, except that fair market values are reduced for the cost of disposal.
F-8
Loss Per Share Data
Basic loss per share is calculated based on the weighted average common shares outstanding during the period. Diluted earnings per share also give effect to the dilutive effect of restricted stock. The Company does not present diluted earnings per share for years in which it incurred net losses as the effect is anti-dilutive.
At April 30, 2014, 123,750 unvested shares of restricted common stock, warrants to exercise 12,430,798 shares of common stock and 3,000,000 shares of stock options were outstanding, but were not included in the computation of diluted earnings per share as their effect would be anti-dilutive. At April 30, 2013, 2,275,625 unvested shares of restricted common stock and warrants to exercise 6,366,798 shares of common stock were outstanding, but were not included in the computation of diluted earnings per share as their effect would be anti-dilutive
Fair Value of Financial Instruments
The Company adopted FASB ASC 820, Fair Value Measurements and Disclosures (FASB ASC 820), which provides a framework for measuring fair value under GAAP. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. The standard also expands disclosures about instruments measured at fair value and establishes a fair value hierarchy which requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. The standard describes three levels of inputs that may be used to measure fair value:
Level 1 — Quoted prices for identical assets and liabilities in active markets;
Level 2 — Quoted prices for similar assets and liabilities in active markets; quoted prices for identical or similar assets and liabilities in markets that are not active; and model-derived valuations in which all significant inputs and significant value drivers are observable in active markets.
Level 3 — Valuations derived from valuation techniques in which one or more significant inputs or significant value drivers are unobservable.
Research and Development
Research and development expenses consist of expenses incurred in performing research and development activities including salaries and benefits, facilities and other overhead expenses, contract services and other outside expenses. Research and development costs are charged to operations when incurred.
Stock Based Compensation
Stock awards
FASB ASC 718, Compensation-Stock Compensation (FASB ASC 718), requires the fair value of all share-based payments to employees, including grants of stock options, to be recognized in the statement of operations over the requisite service period. FASB ASC 718 requires all share based payments to employees, including grants of employee stock option, to be recognized in operating results as compensation expense based on fair value over the requisite service period of the awards. The Company determines the fair value of share-based awards using the Black-Scholes option-pricing model which uses both historical and current market data to estimate fair value. The method incorporates various assumptions such as the risk-free interest rate, expected volatility, expected dividend yield, expected forfeiture rate and expected life of the options. As allowed by FASB ASC 718, for companies with a short period of publicly traded stock history, the Company’s estimate of expected volatility is based on the average volatilities of a sampling of three companies with similar attributes to the Company, including industry, stage of life cycle, size and financial leverage. The risk-free rate for periods within the contractual life of the stock options is based on the U.S. Treasury yield curve in effect at the time of grant valuation. The expected term of options granted is derived using the “simplified method” which computes expected term as the average of the sum of the vesting term plus the contract term as historically the Company had limited activity surrounding its options. The Company has not issued stock options for nonemployees.
F-9
The Company used the following assumptions in valuing the stock options issued:
|
April 30,
|
April 30,
|
|||||||
|
2014
|
2013
|
|||||||
|
Price per share of common stock
|
$ | 0.25 | $ | - | ||||
|
Exercise price per share
|
$ | 0.265 | $ | - | ||||
|
Expected volatility
|
75.5 | % | - | % | ||||
|
Risk-free interest rate
|
1.30 | % | - | % | ||||
|
Dividend yield
|
- | - | ||||||
Warrants
Warrants granted to service providers are normally valued at the fair value of the instrument on the date of the grant (grant date) and are recognized in the consolidated statement of operations over the requisite service period or when they vest. When the requisite service period precedes the grant date and a market condition exists in the warrant, the Company values the warrant using the Black-Scholes option pricing model. Warrants issued in connection with capital raises are normally valued at the fair value of the instrument on the date of the grant (grant date) and valued for disclosure purposes if they meet all the criteria under FASB ASC 718. The Company values these warrant using the Black-Scholes option pricing model as well. As allowed by FASB ASC 718, for companies with a short period of publicly traded stock history, the Company’s estimate of expected volatility is based on the average volatilities of a sampling of three companies with similar attributes to the Company, including industry, stage of life cycle, size and financial leverage. The risk-free rate for periods within the contractual life of the warrant is based on the U.S. Treasury yield curve in effect at the time of grant valuation.
The Company used the following assumptions in valuing the warrants issued:
|
April 30,
|
April 30,
|
|||||||
|
2014
|
2013
|
|||||||
|
Price per share of common stock
|
$ | 0.18 - $0.98 | $ | 0.45 - $1.40 | ||||
|
Exercise price per share
|
$ | 0.25 - $0.50 | $ | 0.45 - $2.00 | ||||
|
Expected volatility
|
79.1 - 97.1 | % | 80.0 - 102.7 | % | ||||
|
Risk-free interest rate
|
0.65 - 1.67 | % | 0.65 - 1.30 | % | ||||
|
Dividend yield
|
- | - | ||||||
Income Taxes
The Company accounts for income taxes under an asset and liability approach. This process involves calculating the temporary and permanent differences between the carrying amounts of the assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The temporary differences result in deferred tax assets and liabilities, which would be recorded on the Company’s consolidated balance sheets in accordance with FASB ASC 740, Income Taxes, which established financial accounting and reporting standards for the effect of income taxes. The Company must assess the likelihood that its deferred tax assets will be recovered from future taxable income and, to the extent the Company believes that recovery is not likely, the Company must establish a valuation allowance. Changes in the Company’s valuation allowance in a period are recorded through the income tax provision on the consolidated statements of operations.
FASB ASC 740 clarifies the accounting for uncertainty in income taxes recognized in an entity’s consolidated financial statements and prescribes a recognition threshold and measurement attributes for financial statement disclosure of tax positions taken or expected to be taken on a tax return. Under FASB ASC 740-10, the impact of an uncertain income tax position on the income tax return must be recognized at the largest amount that is more-likely-than-not to be sustained upon audit by the relevant taxing authority. An uncertain income tax position will not be recognized if it has less than a 50% likelihood of being sustained. Additionally, FASB ASC 740 provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition. As a result of the implementation of FASB ASC 740, the Company recognized no material adjustment in the liability for unrecognized income tax benefits.
F-10
The Company’s policy is to record interest and penalties on uncertain tax positions as income tax expense. As of April 30, 2014 and 2013, the Company has no accrued interest or penalties related to uncertain tax positions.
Segment Reporting
The Company currently operates in a single operating segment. In addition, financial results are prepared and reviewed by management as a single operating segment. The Company continually evaluates its operating activities and the method utilized by management to evaluate such activities and will report on a segment basis if and when appropriate to do so.
Recent Pronouncements
In May 2014, the FASB issued ASU No. 2014-09 “Revenue from Contracts with Customers,” which will supersede nearly all existing revenue recognition guidance under GAAP. ASU No. 2014-09 provides that an entity recognize revenue when it transfers promised goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. This update also requires additional disclosure about the nature, amount, timing and uncertainty of revenue and cash flows arising from customer contracts, including significant judgments and changes in judgments, and assets recognized from costs incurred to obtain or fulfill a contract. ASU No. 2014-09 allows for either full retrospective or modified retrospective adoption and will become effective for the Company in the first quarter of 2018. The Company is a development stage entity and will evaluate the effects of this update on its consolidated financial statements when it generates revenues.
In June 2014, the FASB issued ASU No. 2014-10, which eliminated certain financial reporting requirements of companies previously identified as “Development Stage Entities” (Topic 915). The amendments in this ASU simplify accounting guidance by removing all incremental financial reporting requirements for development stage entities. The amendments also reduce data maintenance and, for those entities subject to audit, audit costs by eliminating the requirement for development stage entities to present inception-to-date information in the statements of income, cash flows, and shareholder equity. Early application of each of the amendments is permitted for any annual reporting period or interim period for which the entity’s financial statements have not yet been issued (public business entities) or made available for issuance (other entities). Upon adoption, entities will no longer present or disclose any information required by Topic 915. The Company has adopted this standard and will commence in future presentations.
Other Income
In the year ended April 30, 2013, the Company received an indemnification payment of $430,000 from a third party in connection with an agreement to indemnify the Company against litigation and other expenses incurred in connection with certain litigation.
NOTE 3. INTANGIBLE ASSETS AND GOODWILL
On May 4, 2010, the Company was assigned the intellectual property rights for a patent that was issued on December 1, 1999. The rights to this patent were purchased out of the bankruptcy proceedings of MicroIslet, Inc. for $200,000 and then assigned to ISI in exchange for the issuance of 3,000,000 shares of common stock.
On March 14, 2012, the Company acquired the IPR&D from DiaKine Therapeutics, Inc. As of April 30, 2014, the $1.3 million of acquired IPR&D is classified as indefinite life asset and is not being amortized. In conjunction with this acquisition, the Company recognized $2.1 million of goodwill. Initially, the residual purchase price of $1,564,107 was recorded as goodwill related to the acquisition of DTI. However, none of the goodwill recognized or amortization of the IPR&D acquired is expected to be deductible for income tax purposes. Accordingly, in conjunction with the valuation of intangible assets acquired, it was determined that a deferred income tax liability of $547,000 was required to reflect the book to tax differences of the acquisition. This same amount was added to the goodwill balance. The Company performed its annual impairment test of goodwill during the fiscal fourth quarter using an analysis of the present value of future discounted cash flows for its valuation. The Company evaluated its goodwill for impairment during the year ended April 30, 2014 and determined that the fair value of the reporting unit is in excess of its carrying value including goodwill.
F-11
The following is a summary of goodwill and intangible assets for the years ended April 30, 2014 and 2013:
|
April 30, 2014
|
April 30, 2013
|
|||||||||||||||||||||||||||
|
Gross
|
Net
|
Gross
|
Net
|
|||||||||||||||||||||||||
|
Carrying
|
Accumulated
|
Carrying
|
Carrying
|
Accumulated
|
Carrying
|
|||||||||||||||||||||||
|
Amount
|
Amortization
|
Impairment
|
Amount
|
Amount
|
Amortization
|
Amount
|
||||||||||||||||||||||
|
Identifiable intangibles:
|
||||||||||||||||||||||||||||
|
Patents
|
$ | 200,000 | $ | (106,414 | ) | $ | (93,586 | ) | $ | - | $ | 200,000 | $ | (91,212 | ) | $ | 108,788 | |||||||||||
|
In-process technology
|
1,367,000 | - | - | 1,367,000 | 1,367,000 | - | 1,367,000 | |||||||||||||||||||||
|
Subtotal of identifiable intangibles
|
1,567,000 | (106,414 | ) | (93,586 | ) | 1,367,000 | 1,567,000 | (91,212 | ) | 1,475,788 | ||||||||||||||||||
|
Goodwill
|
2,111,107 | - | - | 2,111,107 | 2,111,107 | - | 2,111,107 | |||||||||||||||||||||
|
Total goodwill and intangibles
|
$ | 3,678,107 | $ | (106,414 | ) | $ | (93,586 | ) | $ | 3,478,107 | $ | 3,678,107 | $ | (91,212 | ) | $ | 3,586,895 | |||||||||||
The patent is being amortized based on the remaining life of the patent, which was 6.5 years at May 4, 2010, the date of assignment. For the year ended April 30, 2014, the amount amortized to expense was $15,202 and the Company did not expect to realize economic benefit from the claims associated with this intellectual property, therefore, the Company incurred an impairment of this asset of $93,586 during the year ended April 30, 2014. For the year ended April 30, 2013, the amount amortized to expense was $30,404.
NOTE 4. STOCKHOLDERS’ EQUITY
As of April 30, 2014, the Company had 10,000,000 authorized shares of preferred stock, par value $0.001 per share; no shares were issued and outstanding at April 30, 2014 and 2013. Shares of Series A and Series B preferred stock issued at the closing of the Reverse Merger Transaction were automatically converted, upon the effectiveness of the reverse split on February 23, 2012, into common stock at a conversion rate of one thousands shares of common stock for one share of preferred stock. On March 14, 2012, the Company issued shares of Series C Preferred stock at $0.001 par value, in connection with the DTI acquisition. These shares were converted on April 26, 2012 into common stock at a conversion rate of ten shares of common stock for one share of preferred stock. At the conversion of all issued and outstanding shares of Series A, Series B and Series C preferred stock, designations of all three series of preferred stock were cancelled.
As of April 30, 2014, the Company had 100,000,000 authorized shares of common stock, par value $0.001 per share. Holders of common stock are entitled to one vote for each share held on all matters submitted to a vote of stockholders. Holders of common stock are entitled to receive dividends ratably, if any, as may be declared by the Board of Directors out of legally available funds, subject to any preferential dividend rights of any outstanding preferred stock. Upon liquidation, dissolution or winding up, the holders of common stock are entitled to receive ratably net assets available after the payment of all debts and other liabilities and subject to the prior rights of any outstanding preferred stock.
Holders of common stock have no preemptive, subscription, redemption or conversion rights. The outstanding shares of common stock are fully paid and non-assessable. The rights, preferences and privileges of holders of the common stock are subject to, and may be adversely affected by, the rights of holders of shares of any series of preferred stock.
F-12
Grants
At the inception, the Company issued 13,430,000 founder shares of common stock in consideration for intellectual property assignments related to the Company’s purpose, mission and contributions to the business plan and strategy.
On May 4, 2010, the Company issued 3,000,000 shares of common stock in consideration for the assignment of the intellectual property rights purchased from a company in bankruptcy proceedings for $200,000.
In May 2010, the Company issued a convertible note payable to an investor, in the amount of $357,000. In September 2010, the note payable was converted to 3,591,729 shares of common stock.
In July 2010, a Scientific Advisor Board member, and in October 2010, the CFO, received a restricted stock grant of 100,000 shares of common stock for services provided to the Company. The shares vest 50% on the first anniversary of the date of the agreement and the remaining shares vest on the second anniversary of the date of the agreement. For the years ended April 30, 2012 and 2011, the Company recognized stock-based compensation expense of $7,031, and $4,761, respectively. In June 2012, the grant was replaced with a grant of 300,000 shares of common stock with 50% vesting at the first anniversary date and 50% vesting at the second anniversary date. The shares were not issued as of April 30, 2014. The share price of the shares issued was valued at $0.10 per share, which equated to the then share price of common stock being sold to other investors.
In March 2011, as part of a consultant agreement for consulting services to be provided for a one year period, the Company issued 1,200,000 shares of common stock. The common shares were valued at $120,000 at the grant date, and were fully expensed as stock-based compensation upon issuance as the shares were not subject to forfeiture. The share price of the shares issued was valued at $0.10 per share, which equated to the then share price of common stock being sold to other investors.
In April 2011, the Company converted an accounts payable balance with a related party in the amount of $10,000 into 100,000 shares of common stock.
From June to September of 2011, the Company consummated a private placement of its common stock to certain accredited investors whereby the Company issued 8,090,000 shares of common stock for cash at a purchase price of $0.10 per share and 2,474,997 shares of common stock for a cash purchase price of $0.12 per share for total gross proceeds of $1,106,000. Originally, the shares sold were those of ISI’s common stock, which were ultimately converted into common stock of Islet Sciences as a result of the Reverse Merger Transaction and following the reverse stock split of Islet Sciences’ common stock.
In October 2011, the Company granted Mr. John Steel, the Company’s CEO, 1,190,000 shares of common stock, for the services that he has provided in previous periods as part of his employment agreement; 866,668 shares of common stock, as a signing bonus as part of his employment agreement; and 866,668 shares of common stock, as a deferred stock compensation with a vesting schedule of 50% on the first anniversary date and 50% on the second anniversary date. The total number of shares issued was 2,056,668 and were valued at $205,667 at the grant date, and was expensed ratably over the corresponding service period. The share price of the shares issued was valued at $0.10 per share, which equated to the then share price of shares being sold to other investors. The Company has agreed to issue Mr. Steel and additional 200,000 of common shares upon the completion of the contemplated DiaKine Therapeutics, Inc. acquisition. For the year ending April 30, 2012, the Company accrued $280,000 as a stock-based compensation expense related to the 200,000 shares which were to be granted as the result of the DiaKine Therapeutics, Inc. purchase. The shares were valued at $1.40 per share based on the valuation performed at the acquisition date and were issued during the year ended April 30, 2013, except for 433,334 shares which were forfeited because Mr. Steel resigned as the CEO of the Company on July 1, 2013 before these shares vested.
In October 2011, the Board of Directors granted certain consultants to the Company 2,775,000 shares of common stock as part of consultant agreements for services to be provided for multiple year periods. For the year ended April 30, 2012, the Company recognized $277,542 of stock-based compensation expense related to these grants. The share price of the shares issued was valued at $0.10 per share, which equated to the then share price of common stock being sold to other investors.
F-13
In October 2011, the Board of Directors allocated 400,000 shares of common stock for future issuance to members of the Company’s Scientific Advisory Board once Scientific Advisory Board members have been selected, other than Dr. Lakey. The members of the Scientific Advisory Board will have a term of two years.
In December 2011, the Company consummated a private placement of its preferred stock to certain accredited investors whereby the Company issued 1,173 shares of Series A preferred stock for cash at a purchase price of $450 per share for total net proceeds of $464,640. Originally, the shares sold were those of ISI’s Series A preferred stock, which were ultimately converted into common stock of Islet Sciences as a result of the Reverse Merger Transaction and following the reverse stock split of Islet Sciences’ common stock. In conjunction with the issuance of preferred stock, each share of issued preferred stock included the right to receive a warrant to purchase 500 shares of common stock of Islet Sciences at a price of $1.00 per share upon conversion of the preferred stock. The warrants were valued at approximately $147,000.
As per the DTI acquisition agreement, the Company was required to issue to Mr. John Welch shares of common stock representing 3% of the outstanding shares, after giving effect to the issuance of shares of common stock in the financing. On December 30, 2011, the Company issued 1,187,476 shares of common stock valued at $534,365 and was included in other expenses. During the year ended April 30, 2012, the Company computed the remaining number of shares issuable under the anti-dilutive provision of the contract in the amount of 375,398 shares, valued at $525,557 and are included as derivative liabilities in the consolidated balance sheets. The shares were valued at $1.40 per common share based on the stock valuation performed at the acquisition date. On June 6, 2012, the Company issued 375,398 shares of common stock to John Welch pursuant to the DTI acquisition.
On March 14, 2012, in exchange for all issued and outstanding stock of DTI, the Company issued to DTI shareholders 200,000 shares of Series C Preferred Stock with each share of preferred stock convertible into ten shares of the Company’s common stock. The Company also issued 100,000 shares of its common stock as part of the acquisition. On April 24, 2012, the Company issued a total of 2,000,000 shares of common stock to holders of 200,000 shares Series C Preferred Stock upon conversion of Series C Preferred Stock.
In March and April of 2012, the Company consummated a private placement of its common stock at a per share price of $0.45 pursuant to a series of subscription agreements with a number of accredited investors including one of the Company’s directors. The investors in the private placement were also issued, for no additional consideration, warrants to purchase shares of the Company’s common stock. The warrants granted the subscribers the right to purchase a number of shares of common stock, par value $.001 per share, of the Company’s common stock equal to fifty percent (50%) of the number of shares of common stock subscribed for. The warrants have an initial exercise price equal to $1.00 per share and shall be exercisable for a five year period. The warrants were valued at approximately $392,000 based on the assumptions used in Note 2. In April 2012, 3,118,922 shares of common stock and 1,559,461 warrants were issued for total net proceeds of $1,349,995. The remaining shares of common stock and warrants were subsequently issued in May 2012 and were part of additional capital raises on May 2, 2012 and May 9, 2012 described below.
On April 1, 2012, as compensation for future services, the CFO was awarded a grant of 150,000 shares of common stock which shall be subject to forfeiture if his consulting engagement is terminated before January 31, 2013. These shares vested during the year ended April 30, 2013 and were valued at $210,000, which was based on a previous appraisal at a share price of $1.40 and were included as general and administrative expenses in the consolidated statements of operations for the year ended April 30, 2013.
In May 2, 2012, the Company issued an aggregate of 4,552,222 shares of common stock at $0.45 per share and warrants to purchase 2,276,111 shares of common stock at an exercise price of $1.00 per share in connection with two closings of the private placement of its common stock of $2,048,500 that occurred in April 2012. The warrants were valued at approximately $492,000 based on the assumptions used in Note 2.
On May 9, 2012, the Company consummated a private placement of an aggregate of 1,711,667 shares its common stock at a per share price of $0.45, for gross proceeds of $770,250 pursuant to a series of subscription agreements with a number of accredited investors. The investors in the private placement were also issued for no additional consideration, warrants to purchase 855,833 shares of common stock at an exercise price of $1.00 per share. The warrants were valued at approximately $185,000 based on the assumptions used in Note 2.
F-14
In May 2012, the Company, entered into a license agreement with the Yale University (“Yale”). As part of this agreement, the Company issued 20,000 shares of its common stock valued at $28,000 which is included in the consolidated statement of operations for the year ended April 30, 2013.
On May 23, 2012, the Company issued a total of 270,000 shares of common stock as compensation to its employee and consultants valued at $378,000 of which 200,000 shares (or $280,000). The shares issued were valued using a previous appraisal of $1.40 per share.
On June 21, 2012, the Company consummated a private placement of an aggregate of 717,778 shares of common stock at a per share price of $0.45, for gross proceeds of $323,000 pursuant to a series of subscription agreements with a number of accredited investors. The investors in the private placement were also issued for no additional consideration warrants to purchase 358,889 shares of common stock at an exercise price of $1.00 per share. The warrants were valued at approximately $78,000 based on the assumptions used in Note 2.
In August 2012, the Company agreed to assume certain liabilities in connection with claims and liens on certain patents acquired for approximately $17,000 in cash and issued 144,342 shares of its common stock. The shares issued were valued using a previous appraisal of $1.40 per share or approximately $202,000. These costs have been expensed to general and administrative expenses during the year ended April 30, 2013.
In August 2012, the Company issued 1,750,000 shares of its common stock to one of its placement agents pursuant to an agreement dated as of April 21, 2011, as amended on June 29, 2012. The initial performance conditions in the agreement were not met by the placement agent, however, both parties agreed to settle and amend the agreement by issuing a total of 1,750,000 shares of the Company’s common stock to the placement agent. These shares were issued and the expense in the amount of $2,450,000 during the year ended April 30, 2013, which was based on a share price of $1.40 based on a stock valuation performed, is included as general and administrative expense in the consolidated statements of operations for the year ended April 30, 2013.
In August of 2012, the Company issued 3,000 shares of common stock to The Regents of the University of California as part of an exclusive licensing agreement. The Company valued these shares at approximately $4,200 and included the cost as research and development expense within the consolidated statements of operations for the year ended April 30, 2013.
In August 2012, the Company issued 60,000 shares of common stock that were previously designated as subscribed shares – not issued. The Company initially received proceeds of approximately $27,000 in December 2011. The investor was also issued for no additional consideration, a warrant to purchase 30,000 shares of common stock at an exercise price of $1.00 per share. The warrant was valued at approximately $6,000 based on the assumptions used in Note 2.
In September 2012, the Company issued an aggregate of 309,375 shares of its common stock to members of the Board of Directors which were considered to be vested. The Company valued these shares based on an earlier appraisal of $1.40 per share or approximately $433,000, and included the costs as general and administrative expenses within the consolidated statements of operations for the year ended April 30, 2013.
In October 15, 2012, the Board of Directors approved a grant of 250,000 shares of common stock to Dr. Jonathan Lakey for his role in the ongoing process with Yale University, PCT, and Spring Point Project which vested on January 31, 2013. These shares were issued and the expense in the amount of $350,000, which was based on a previous appraisal at a share price of $1.40, is included as research and development expenses in the consolidated statements of operations for the year ended April 30, 2013.
On October 19, 2012, the Company issued 375,000 shares of its common stock to a consultant to provide financial services. The shares are not subject to any vesting conditions or forfeiture. These shares were issued in the amount of $525,000, which was based on a previous appraisal at a share price of $1.40 and is included as general and administrative expenses in the consolidated statements of operations for the year ended April 30, 2013.
F-15
On November 21, 2012, the Company issued 20,000 shares of common stock to Winthrop University Hospital as part of the agreement to license certain patents and technology. These shares were valued at an earlier appraisal of $1.40 per share or $28,000. The Company accounted for these costs as research and development expenses within the consolidated statements of operations for the year ended April 30, 2013.
On December 19, 2012, the Company issued a total of 433,334 shares of common stock as compensation to its CEO, John Steel, valued at $606,667 based on a previous appraisal at a share price of $1.40 and included in general and administrative expenses within the consolidated statements of operations for the year ended April 30, 2013.
On December 26, 2012, an aggregate of 309,375 shares of its common stock issuable to members of the Board of Directors fully vested. These shares were issued during the year ended April 30, 2013 in the amount of approximately $433,000, which was based on a previous appraisal at a share price of $1.40 and is included as general and administrative expenses in the consolidated statements of operations for the year ended April 30, 2013.
On January 25, 2013, the Company issued 30,000 shares to a consultant to provide investor relations services. The shares are not subject to any vesting conditions or forfeiture. These shares were expensed at a value of $44,000. An additional $4,000 was accrued for the balance of the amount due under this agreement. A total of $48,000 is included in general and administrative expenses within the consolidated statements of operations for these services for the year ended April 30, 2013.
On January 28, 2013, the Company received net proceeds of $96,600 for the sale of 70,000 shares of common stock at $1.40 per share as part of a private offering. The Company had not issued these shares as of April 30, 2013, and has recorded these proceeds as subscribed shares not issued at April 30, 2013. On November 11, 2013, the Company issued these common shares.
On February 5, 2013, the Company issued 20,000 shares of common stock to certain related parties (Jerry L. Nadler, David Taylor-Fishwick, Anca Dobrian and Swarup Chalrabarti - “12LO Licensors”) as part of an exclusive licensing agreement. The Company valued these shares using a previous appraisal of $1.40 per share or $28,000 and included the cost as research and development expense within the consolidated statements of operations for the year ended April 30, 2013.
On March 19, 2013, an aggregate of 247,500 shares of its common stock issuable to members of the Board of Directors fully vested. The Company valued these shares based on the April 30, 2013 share price of $0.23 per share, or $56,925, and recorded it to accrued expense until the common stock is issued. The Company has included the costs as general and administrative expenses within the consolidated statements of operations for the year ended April 30, 2013. On April 10, 2014, the Company issued 61,875 common shares to one of its Board Members.
On April 1, 2013, the Company issued 5,000 shares of common stock to Southridge Partners II. LLP as part of a settlement agreement dated April 1, 2013 regarding an investment made by Southridge Partners II, LLP in the Company. The Company valued these shares at $7,000 which was based on a previous appraisal at a share price of $1.40 and is included as general and administrative expenses in the consolidated statements of operations for the year ended April 30, 2013.
At April 30, 2013, the Company had an indemnification agreement with a third-party, whereby, the Company received $80,000 in cash for shares of common stock of the Company that were to be provided by the same third party. On October 31, 2013, the Company was notified that the third-party did not transfer shares of Company common stock in accordance with the terms of the agreement. The Board of Directors has approved the issuance of Company common stock to fulfill the requirements of the indemnification agreement. On November 19, 2013, the Company issued 320,000 shares of common stock to satisfy its subscribed shares valued at $80,000. The Company has recorded $80,000 in general and administrative expense within the consolidated statements of operations for the year ended April 30, 2014.
During the year ended April 30, 2013, the Company initially issued 400,000 shares as part of its agreement with PCT along with a warrant to purchase 350,000 shares at an exercise price of $1.00 per share. This initial issuance was valued at approximately $855,000 and accrued at April 30, 2012. In addition and as part of the anti-dilution provision, the Company issued an additional 223,771 shares of common stock to maintain PCT’s 1% ownership of the Company. These additional issuances were valued at an earlier appraisal of $1.40 per share or approximately $314,000.
F-16
On July 31, 2013, an aggregate of 247,500 shares of its common stock issuable to members of the Board of Directors fully vested. The Company valued these shares based on the July 31, 2013 share price of $0.40 per share, or $99,000, and recorded it to accrued stock compensation expense until the common stock is issued. The Company has included these costs as general and administrative expenses within the consolidated statements of operations for the year ended April 30, 2014. On April 10, 2014, the Company issued 61,875 common shares to one of its Board Members.
On July 31, 2013, an aggregate of 350,000 shares of its common stock issuable to members of the Scientific Advisory Board fully vested. The Company valued these shares based on the July 31, 2013 share price of $0.40 per share, or $140,000, and recorded it to accrued stock compensation expense until the common stock is issued. The Company has included these costs as research and development expenses within the consolidated statements of operations for the year ended April 30, 2014. On April 1, 2014, the Company issued 200,000 of common shares to the Scientific Advisory Board Members.
On August 20, 2013, an aggregate of 200,000 shares of its common stock issuable to members of the Scientific Advisory Board fully vested. The Company valued these shares based on the October 31, 2013 share price of $0.40 per share, or $80,000. The Company has included these costs as research and development expenses within the consolidated statements of operations for the year ended April 30, 2014. On April 1, 2014, the Company issued 200,000 of common shares to the Scientific Advisory Board Members.
On October 30, 2013, as part of the employee agreements with the new CEO and COO, the Board of Directors granted each of them stock options to purchase 1.5 million shares of Company common stock at an exercise price of $0.265. The stock options will vest as follows: (i) 500,000 shares vesting on the 91st day from the option grant date, (ii) 1,000,000 shares vesting in equal installments of 200,000 on the last day of each 90 day period starting from the 91st day after the option grant date. At April 30, 2014, 700,000 stock options vested for each the CEO and COO. The Company recorded stock-based compensation of $192,100 for the vested stock options as general and administrative expenses within the consolidated statements of operations for the year ended April 30, 2014.
On October 31, 2013, an aggregate of 185,625 shares of its common stock issuable to members of the Board of Directors fully vested. The Company valued these shares based on the October 31, 2013 share price of $0.25 per share, or $46,406, and recorded it to accrued stock compensation expense until the common stock is issued. The Company has included these costs as general and administrative expenses within the consolidated statements of operations for the year ended April 30, 2014.
Beginning in December 2013 and concluding in March 2014, the Company received net proceeds of $1,950,300 for the sale of 8,890,000 shares of Company common stock as part of a private placement to multiple investors. The investors in the private placement were also issued for no additional consideration warrants to purchase 4,445,000 shares of Company common stock at an exercise price of $0.35 per share. The warrants were valued at approximately $1,302,000 based on the assumptions used in Note 2. The Company issued these common stock shares during the fourth quarter ending April 30, 2014. Additionally, the Company is obligated to issue 1,489,000 warrants at an exercise price of $0.25 to the investment banker for the private placement. These warrants were valued at approximately $266,000, however, as the cost of these warrants would have been a capital raise cost, there was no financial impact to the Company for the issuance of these warrants.
On January 8, 2014, the Company issued 700,000 shares of Company common stock as part of a collateral agreement for the benefit of the legal firms that are representing the Company in the Sand Dollar Partners, LLC complaint in the Superior Court of Arizona and the complaint that was filed against the Company and John Steel for infringement and misappropriation of MicroIslet patent in the United States District Court for the District of Utah, Central Division (see Note 6). These shares are to be held in escrow to secure payment of the legal fees incurred in the defense of the complaint. Since these shares are being held in escrow until a future date to potentially satisfy the Company’s obligations, the Company has recorded these shares at par value or $700 to prepaid expenses. If common stock shares are issued from escrow to pay the legal fees, the common stock will be valued on the date of settlement. Legal fees incurred relating to this matter are included in accounts payable on the accompanying consolidated balance sheet. On May 16, 2014, the Company completed the settlement with Sand Dollar Partners, LLC. (see Note 6).
F-17
On January 24, 2014, the Board of Directors approved the termination of the existing Scientific Advisory Board (“SAB”). The SAB members will not be entitled to the remaining 550,000 unvested shares.
On January 31, 2014, an aggregate of 185,625 shares of its common stock issuable to members of the Board of Directors fully vested. The Company valued these shares based on the January 31, 2014 share price of $0.60 per share, or $111,375, and recorded it to accrued stock compensation expense until the common stock is issued. The Company has included these costs as general and administrative expenses within the consolidated statements of operations for the year ended April 30, 2014.
On February 10, 2014, as part of a settlement agreement the Company issued 25,000 shares of its common stock to American Capital Ventures. The Company valued these shares based on the February 10, 2014 share price of $0.60 per share, or $15,000, The Company has included these costs as general and administrative expenses within the consolidated statements of operations for the year ended April 30, 2014.
On February 13, 2014, as part of an agreement for Dr. Eric Barnett to join the Board of Directors the Company granted 70,000 shares of its common stock issuable and fully vested. The Company valued these shares based on the February 13, 2014 share price of $0.32 per share, or $22,400. The Company has included these costs as general and administrative expenses within the consolidated statements of operations for the year ended April 30, 2014. On March 5, 2014 the Company issued these common shares.
On March 12, 2014, as part of compensation to join the Company’s Board of Directors the Company granted 50,000 shares of its common stock issuable and fully vested to Dr. Michael Luther. These shares based on the February 13, 2014 share price of $0.40 per share, or $20,000, and recorded it to accrued stock compensation expense until the common stock is issued. The Company has included these costs as general and administrative expenses within the consolidated statements of operations for the year ended April 30, 2014.
On April 30, 2014, as part of settling a consulting agreement, the Company granted 14,500 shares of its common stock issuable and fully vested to a consultant. The Company valued these shares at $48,000, and recorded it to accrued stock compensation expense until the common stock is issued. The Company has included these costs as general and administrative expenses within the consolidated statements of operations for the year ended April 30, 2014.
On April 30, 2014, an aggregate of 123,750 shares of its common stock issuable to members of the Board of Directors fully vested. The Company valued these shares based on the April 30, 2014 share price of $0.30 per share, or $37,125, and recorded it to accrued stock compensation expense until the common stock is issued. The Company has included these costs as general and administrative expenses within the consolidated statements of operations for the year ended April 30, 2014.
F-18
The following table summarizes information regarding the warrants outstanding as of April 30, 2014 and 2013:
|
Number of Warrants
|
Weighted Average Exercise Price
|
Expiry Dates
|
|||||||||
|
Warrants at April 30, 2011
|
- | $ | - | n/a | |||||||
|
Granted
|
2,145,961 | $ | 1.00 |
February 2017 to March 2017
|
|||||||
|
Expired
|
- | $ | - | n/a | |||||||
|
Warrants at April 30, 2012
|
2,145,961 | $ | 1.00 |
February 2017 to March 2017
|
|||||||
|
Granted
|
4,420,837 | $ | 1.08 |
April 2017 to January 2018
|
|||||||
|
Expired
|
(200,000 | ) | $ | 2.00 |
August 20, 2017
|
||||||
|
Warrants at April 30, 2013
|
6,366,798 | $ | 1.02 |
February 2017 to January 2018
|
|||||||
|
Granted
|
6,064,000 | $ | 0.33 |
September 2015 to June 2019
|
|||||||
|
Expired
|
- | $ | - | n/a | |||||||
|
Warrants at April 30, 2014
|
12,430,798 | $ | 0.68 |
September 2015 to June 2019
|
|||||||
On October 30, 2013, the Company issued 3,000,000 Nonqualified Stock Options as part of the employment agreements for James Green, CEO and William Wilkison Ph.D, COO. These options were issued with a strike price of $0.265. Options vest over eighteen months from the grant date and expire six and one half years from grant date.
The following table summarizes information regarding the stock options outstanding as of April 30, 2014 and 2013:
|
Number of Stock Options
|
Weighted Average Exercise Price
|
Weighted Average Contractual Life in Years
|
||||||||||
|
Stock Options at April 30, 2013
|
- | $ | - | - | ||||||||
|
Granted
|
3,000,000 | $ | 0.265 | 6.5 | ||||||||
|
Exercised
|
- | - | - | |||||||||
|
Expired or canceled
|
- | $ | - | - | ||||||||
|
Stock Options at April 30, 2014
|
3,000,000 | $ | 0.265 | 6.0 | ||||||||
|
Options Exercisable
|
1,400,000 | $ | 0.265 | 6.0 | ||||||||
The weighted average fair value of options granted during fiscal 2014 was $0.14. The intrinsic value of options outstanding, vested and exercisable, and exercised is $0. Stock-based compensation expense is $192,100 for the year ended April 30, 2014.
The following table summarizes information regarding non-vested stock options as of April 30, 2014 and 2013.
F-19
|
Number of Stock Options
|
Stock Options Weighted Average Grant Date Fair Value
|
|||||||
|
Nonvested options at April 30, 2013
|
- | $ | - | |||||
|
Granted
|
3,000,000 | $ | 0.14 | |||||
|
Vested/Issued
|
(1,400,000 | ) | $ | 0.14 | ||||
|
Forfeited
|
- | - | ||||||
|
Nonvested stock options at April 30, 2014
|
1,600,000 | $ | 0.14 | |||||
At April 30, 2014, total compensation costs related to non-vested stock option awards not yet recognized totaled $219,430. The weighted average period over which this amount is expected to be recognized is one year.
NOTE 5. COMMITMENTS AND CONTINGENCIES
Contracts
On January 10, 2012, the Company entered into an agreement with Progenitor Cell Therapy, LLC (PCT), a subsidiary of NeoStem, Inc. (NeoStem), which was amended by an agreement dated May 15, 2012 by and between the Company and NeoStem, PCTs parent company. Under the agreements, PCT will be providing the protocols, procedures, systems, equipment, testing, quality controls, and manufacturing and distribution services to support the development and commercialization of the Companys encapsulated porcine islet cells for the treatment of diabetes. As compensation for the services of PCT, the Company agreed to pay to PCT a non-refundable monthly fee of $63,000 and a non-refundable monthly charge of between $33,000 and $54,000. NeoStem was entitled to receive shares and warrants of the Companys common stock, as well as additional shares for no consideration so that NeoStems ownership is not less than 1% of outstanding shares on a fully diluted basis. PCT has the right for a period of ten years to be the exclusive manufacturer of any product involved in the services to be provided under the agreement. With respect to commercial production of such products, PCT will be entitled to a royalty of 2.85% of gross sales and 5% of any sublicensing fees, royalties, milestone fees or profit sharing payments. On February 7, 2013, the Company provided to NeoStem Inc. notice of termination of the agreement with Progenitor Cell Therapy, Inc. dated January 10, 2012, as amended by the agreement dated May 15, 2012 by and between the Company and NeoStem. During the years ended April 30, 2014, and 2013, the Company expensed as research and development expense within the consolidated statements of operations of approximately $0 and $936,000, respectively, related to this contract.
On July 23, 2012, the Company entered into a long-term supply agreement with Spring Point Project, a source animal facility to purchase pigs for use in the Company’s xenotransplantation research. During the years ended April 30, 2014 and 2013, the Company expensed for the year as research and development expense within the consolidated statements of operations of approximately $350,000 and $971,000, respectively, related to this contract. On August 12, 2013, the Company received from Spring Point Project a notice of termination of the supply agreement, effective November 10, 2013, unless the Company cured the default before that date. The default was not cured and the agreement with Spring Point Project has been terminated.
In August 2012, the Company entered into an agreement with the Regents of the University of California, Los Angles, (“UCLA’), whereby UCLA will provide work dealing with small molecule mediated porcine islet proliferation. The Company had a net credit to research and development expense of approximately $43,000 for the year ended April 30, 2014 within the consolidated statements of operations related to this contract. On February 25, 2014, the Company terminated its agreement with UCLA. The Company incurred $71,000 of expense during the fiscal year ending April 30, 2013.
F-20
In May 2013, the Company entered into a sales and services agreement with the Regents of the University of California, Irvine (“UCI”) to provide materials consisting of isolated islets to be supplied to the Spring Point Project. For the year ended April 30, 2014, the Company expensed as research and development expense within the consolidated statements of operations of approximately $110,000, related to this contract. The Company terminated this agreement on March 24, 2014 for settlement payments totaling $150,000 on total liabilities of approximately $305,000 for expenses incurred in this and last fiscal year. The Company incurred $276,000 of expense during the fiscal year ending April 30, 2013.
In September 2013, the Company entered into a consulting agreement with American Capital Ventures, Inc. to provide consulting services for implementation of the Company’s corporate and business development plan and to plan review and create corporate communications. On September 12, 2013, 200,000 shares vested and were valued at $0.29 per share, or $58,000. The Company terminated this agreement on January 23, 2014 and agreed to issue 25,000 shares valued at $0.60 per share of common stock as part of the termination agreement. The Company issued the shares on February 11, 2014 and expensed $15,000 related to this consulting agreement within the consolidated statements of operations for the year ended April 30, 2014.
Licenses
On May 2, 2012, the Company, entered into a license agreement with the Yale University (“Yale”). Under the agreement, the Company received exclusive license to the technology patented by Yale. In consideration of the license granted under the agreement, the Company paid Yale a license issue royalty of $10,000 (plus a $10,000 annual renewal fee) and issued 20,000 shares of its common stock, and agreed to pay certain milestones royalties by issuing an aggregate of 160,000 shares of common stock. The Company also agreed to pay to Yale a royalty on net sales. The agreement will expire automatically, on a country-by-country basis, on the date on which the last of the claims of the subject patents expires. The agreement can be terminated by Yale if the Company defaults on its obligations under the agreement and fails to cure such default within 60 days of a written notice by Yale. The Company can terminate the agreement upon a six month notice subject to payment of all amounts due to Yale under the agreement. During years ended April 30, 2014 and 2013, the Company expensed approximately $100,000 net a credit of approximately $156,510 for current and past year services and $144,000, respectively, within the consolidated statements of operations.
On July 23, 2012, the Company entered into a licensing agreement with the Winthrop University Hospital (“Winthrop”) to license certain patents and technology. In consideration of the license granted under the agreement, the Company agreed to pay to Winthrop a license issue royalty of $10,000 (plus a $10,000 annual renewal fee) and issue 20,000 shares of its common stock, and to pay certain milestones royalties by issuing an aggregate of 160,000 shares of common stock. The Company also agreed to pay to Winthrop a royalty on net sales. The agreement will expire automatically, on a country-by-country basis, on the date on which the last of the claims of the subject patents expires. It can be terminated by Winthrop if the Company defaults on its obligations under the agreement and fails to cure such default within 60 days of a written notice by the university. The Company can terminate the agreement upon a six month notice subject to payment of all amounts due Winthrop under the agreement. During the year ended April 30, 2014 and 2013, the Company expensed as research and development expense within the consolidated statements of operations of approximately $62,000 and $200,000 respectively, related to this contract. The Company is currently in default regarding its payment obligations under the foregoing license and research agreements.
On January 22, 2014, the Company terminated its licensing agreement dated, July 25, 2012, with The Regents of The University of California, Los Angeles.
Legal Proceedings
In April 2012, Sand Dollar Partners, LLC, a shareholder of the Company filed a complaint in the Superior Court of Arizona, Pima County against, among other parties, ISI, our wholly-owned subsidiary, John Steel, our former CEO and director, and Jonathan Lakey, our former director. In 2010, Sand Dollar invested $357,000 in ISI through the purchase of a convertible promissory note which was converted into 3,591,729 shares of the Company’s common stock. The plaintiff contends that it was entitled to issuance of additional shares and nomination of one board member.
F-21
On October 25, 2013, the Company entered into a settlement agreement with Sand Dollar Partners, LLC. At a hearing on February 21, 2014, the Company and Sand Dollar agreed to amend the settlement agreement whereby, Sand Dollar placed in escrow all of the Company’s common stock it held and retained a broker dealer to sell sufficient shares to receive $500,000 in cash and to pay fees to the broker dealer. Additionally, the Company agreed to issue 130,000 warrants that will vest over the next three years and make a $30,000 payment on May 15, 2014. The Company issued these warrants and made the $30,000 payment on May 15, 2014 fully completing the settlement. The broker dealer sold 2,247,200 shares of the Company’s escrowed stock to settle the obligation and the Company will receive back the remaining 1,344,529 shares of its common stock. The sale of these shares and the return of the remaining shares had no impact on the consolidated financial statements.
On April 25, 2014, PCT, filed a lawsuit against the Company in the United States District Court for the District of New Jersey (Case No. 2:14-cv-02658-SDW-MCA). PCT’s complaint asserts various claims, including breach of contract and unjust enrichment, based on the alleged failure of the Company to pay for services and goods provided by PCT under a January 10, 2012 letter agreement. PCT seeks an unspecified amount of compensatory and other damages, plus interest and costs. The Company filed an answer to the complaint on June 24, 2014 denying all liability, including on the grounds that the January 10, 2012 letter agreement is unenforceable and PCT failed to provide the goods and services it stated it would provide. In connection with its answer to PCT’s complaint, the Company filed a counterclaim against PCT and a third-party complaint against NeoStem to seek, among other things, (i) a declaration that the January 10, 2012 letter agreement is unenforceable, (ii) monetary damages and (iii) rescission of equity securities the Company previously issued to NeoStem. The Company intends to vigorously defend the claims by PCT and prosecute its claims against PCT and NeoStem. As the lawsuit is at an early stage, the Company cannot at this time estimate the possible loss or range of loss, if any, that may result from this lawsuit.
In July 2012, a complaint was filed against the Company and John Steel in the United States District Court for the District of Utah, Central Division for infringement and misappropriation of a patent. The plaintiffs contend that they were the actual purchasers of the MicroIslet patent out of MicroIslet’s bankruptcy proceedings in 2009 and that the respective intellectual property rights have been never assigned to either ISI or the Company. As a result, they allege that the Company’s claim to the ownership of the MicroIslet patent based on the assignment of the patent by its founders is baseless. The complaint sought monetary damages including punitive damages of at least $12 million, costs, attorneys' fees, and declaratory judgment. On January 8, 2013, the Court dismissed the plaintiff’s action for lack of recoverable damages. The plaintiffs refiled their claim and the Company has filed a motion with the Court for dismissal. The Company believes the plaintiffs’ claims to be without merit and will continue to vigorously defend against this action and has determined that it is unlikely any damages will be paid.
NOTE 6. RELATED PARTY TRANSACTIONS NOT INCLUDED ELSEWHERE
The Company borrowed $25,880 from its former CEO. Promissory notes were issued for these amounts. Repayments of $12,405 were made during the year. The remaining balance at April 30, 2014 is $13,475, repayments of $5,000 were made subsequent to year end. The balance at April 30, 2013 was $11,880.
During the year ended April 30, 2014, one of the Company’s Board Members loaned the Company a total of $74,531 at a 6.5% interest rate. A promissory note was issued for this amount. Repayments of $15,000 were made during the year. The remaining balance at April 30, 2014 was $62,542, repayments of $20,000 were made subsequent to year end.
A contractor of the Company loaned the Company $15,624. No repayments have been made as of April 30, 2014.
On May 10, 2010, the Company entered into a consulting agreement with John Steel to provide executive management services to the Company for a monthly fee of $15,000. On July 1, 2013, as part of Mr. Steel’s separation agreement, Mr. Steel and the Company entered into a new consulting agreement, whereby, Mr. Steel will provide business development services to the Company as may be directed by the CEO. Under the consulting agreement, Mr. Steel will receive a monthly consulting fee of $15,000. The consulting agreement expired on June 30, 2014. The outstanding amounts due to John Steel for his consulting services is approximately $208,000 and $18,000 at April 30, 2014 and 2013, respectively, which is included in accounts payable on the consolidated balance sheets.
F-22
NOTE 7. INCOME TAXES
Temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes give rise to the Company's deferred income taxes. The components of the Company's deferred tax assets as of April 30 are as follows:
|
2014
|
2013
|
|||||||
|
Deferred Tax Assets:
|
||||||||
|
Net operating loss carryforwards
|
$ | 7,288,600 | $ | 6,133,500 | ||||
|
Tax credits
|
355,400 | 428,200 | ||||||
|
Accrued expenses
|
- | 42,500 | ||||||
|
Derivatives
|
- | 23,300 | ||||||
|
Share-based compensation
|
178,500 | 2,000 | ||||||
|
Depreciation and amortization
|
62,900 | 19,600 | ||||||
|
Total deferred tax asset
|
7,885,400 | 6,649,100 | ||||||
|
Valuation Allowance
|
(7,885,400 | ) | (6,649,100 | ) | ||||
|
Deferred Tax Liabilities - Intangibles
|
(547,000 | ) | (547,000 | ) | ||||
| (547,000 | ) | (547,000 | ) | |||||
|
Total deferred tax liability
|
$ | (547,000 | ) | $ | (547,000 | ) | ||
FASB ASC 740, Income Taxes, requires that a valuation allowance be established when it is more likely than not that its net deferred tax asset will not be realized. In determining whether a valuation allowance is required, a company must take into account all positive and negative evidence with regard to the utilization of a deferred tax asset. FASB ASC 740 further states that it is difficult to conclude that a valuation allowance is not needed when there is negative evidence such as cumulative losses in recent years.
The Company plans to continue to provide a full valuation allowance on future tax benefits until it can sustain an appropriate level of profitability and until such time, the Company would not expect to recognize any significant tax benefits in its future results of operations. The valuation allowance increased by $1,236,300 and $4,304,000 for the years ended April 30, 2014 and 2013, respectively.
As of April 30, 2014, the Company had net operating loss carry forwards for Federal and state income tax purposes of approximately $18,270,000 and $18,457,000, respectively. These Federal and state net operating loss carry forwards will begin to expire in 2031. The Company had research and development credits carry forwards for Federal and state income tax purposes of approximately $284,000 and $72,000 respectively. Pursuant to the Internal Revenue Code, Sections 382 and 383, use of the Company’s net operating loss and credit carry forward could be limited if a cumulative change in ownership of more than 50% occurs within a three-year period. This analysis has not been performed as of April 30, 2014. Currently, the 2010 through 2013 tax years are open and subject to examination.
The provision for income taxes differs from the amount computed by applying the US statutory Federal income tax rate of 34% to income before income taxes. The reconciliation of statutory and effective taxes for the years ended April 30, 2014 and 2013 are presented below:
F-23
|
2014
|
2013
|
|||||||
|
Provision computed at statutory rate
|
$ | (1,005,000 | ) | $ | (3,929,000 | ) | ||
|
State income tax, net of Federal tax benefit
|
(172,000 | ) | (674,000 | ) | ||||
|
Research and development credit
|
(18,000 | ) | (325,000 | ) | ||||
|
Change in valuation allowance
|
1,236,300 | 4,304,000 | ||||||
|
Permanent differences
|
13,000 | 625,000 | ||||||
|
Other
|
(54,300 | ) | (1,000 | ) | ||||
|
Income tax expense
|
$ | - | $ | - | ||||
NOTE 8. SUBSEQUENT EVENTS
On May 22, 2014, Mr. Delmar was appointed as the Company’s Chief Financial Officer. Mr. Delmar will receive, a grant of 300,000 stock options to purchase common shares of the Company and a conditional grant of 100,000 stock options upon the effective date of an S-4 filing.
On May 9, 2014, the Company issued 742,500 common shares to the members of the Board of Directors as part of the approved compensation plan.
On June 4, 2014, the Company issued 14,500 common shares to a consultant part of a settlement agreement.
F-24